
Si CP MAS NMR
152
Untersuchung der Kristallinität oberflächennaher Bereiche mikroporöser Materialien mittels NMR-Spektroskopie Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften der Fakultt für Geowissenschaften der Ruhr-Universitt Bochum vorgelegt von Cristina Osterhoff Betreuer: Prof. Dr. Hermann Gies Bochum, im November 2000
Transcript of Si CP MAS NMR

Untersuchung der Kristallinität
oberflächennaher Bereiche mikroporöser Materialien
mittels NMR-Spektroskopie
Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften
der Fakultät für Geowissenschaften der Ruhr-Universität Bochum
vorgelegt von
Cristina Osterhoff
Betreuer: Prof. Dr. Hermann Gies
Bochum, im November 2000

Inhaltsverzeichnis i
Inhalt 1. Theoretische Grundlagen 1
1.1 Zeolithe und zeolithartige Materialien 1 1.1.1 Einführung 1 1.1.2 Nomenklatur und Zeolith-Klassifikation 2
1.1.2.1 Clathrasile 7 1.1.2.1.1 Dodecasil 3C (MTN) 9
1.1.3 Zeolitheigenschaften und Anwendungen 12 1.1.4 Entstehung von natürlichen und synthetischen Zeolithen und zeolithartigen Materialien 15 1.1.4.1 Natürliche Zeolithe 15 1.1.4.2 Synthetische Zeolithe 16
1.2 Zeolithoberfläche und Reaktivität 21
1.2.1 Definition und Charakterisierung 21 1.2.2 Der Zusammenhang zwischen Oberfläche und katalytischer Aktivität 24
2. Ziel der Arbeit 27 3. Synthese und angewandte Methoden 29
3.1 Allgemeines über die Synthesedurchführung 29
3.2 Versuchsreihe I [SiO2:M(C5H11N):H2O = 1:1,5:55,5] 31 3.2.1 Syntheseplanung 31 3.2.2 Syntheseergebnisse der Versuchsreihe I 33
3.3 Versuchsreihe II [SiO2:M(C5H11N):H2O = 1:4:55,5] 35
3.3.1 Syntheseplanung und Synthesedurchführung 35 3.3.1.1 Variation der Syntheseparameter: Synthesetemperatur, chemische Zusammensetzung, Arbeitsweise, Synthesedauer, Füllhöhe 35
3.3.2 Syntheseergebnisse der Versuchsreihe II 37

Inhaltsverzeichnis ii
3.4 Zusätzliche Behandlungen, die an der ZSM-39
mikrokristallinen Probe durchgeführt worden sind 41 3.4.1 Calcinierung 41 3.4.2 Behandlung mit einer starken Base (NaOH) 42 3.4.3 Deuterierung 43 3.4.4 Fluorierung 43 3.5 Apparatives 45 3.5.1 Mikroskopische Methoden 45 3.5.1.1 Lichtmikroskopie 45 3.5.1.2 Rasterelektronenmikroskopie 45 3.5.2 Röntgenographische Methoden 46 3.5.2.1 Pulverdiffraktometrie 46 3.5.3 Spektroskopische Metoden 47 3.6 Zusammenfassung der Synthesedurchführung 48 4. Festkörper NMR-Spektroskopische Untersuchungen
an externen Clathrasil-Oberflächen 49 4.1 Einführung in die Theorie 49 4.1.1 Das grundlegende Prinzip der NMR 50 4.1.2 Das Kreuzpolarisationsexperiment (CP MAS) 55 4.1.2.1 Der Einfluss der Probenrotation unter
MAS auf die Hartmann-Hahn-Bedingung 59
4.2 Experimentelle Ergebnisse 61 4.2.1 1H/29Si CP MAS-Experimente, durchgeführt an �grobkörnigen� Dodecasil 3C-Substanzen mit einer mittleren Korngrösse zwischen 1 und 300 µm 61 4.2.2 1H/29Si CP MAS-Experimente, durchgeführt an
�feinkörnigen� Dodecasil 3C-Substanzen mit einer mittleren Korngrösse zwischen 0,01 µm und 0,1 µm 65 4.2.2.1 Die as-synthesized Form 65 4.2.2.2 Die mit NaOH behandelte Form 69 4.2.2.3 Die calcinierte Form 70 4.2.2.4 Die deuterierte Form 72
4.2.3 NMR-Untersuchungen an fluorierten �fein-körnigen� Dodecasil 3C-Materialien 74

Inhaltsverzeichnis iii
4.2.3.1 19F MAS NMR-Experimente, durchgeführt an einer mit Flusssäuere HF behandelte Dodecasil 3C-Probe (mittlere Korngrösse: 0,1 µm) 74 4.2.3.2 19F/29Si CP MAS NMR-Experimente, durchgeführt an einer mit HF beladene Dodecasil 3C-Probe (mittlere Korngrösse: 0,1 µm) 77
4.3 Zusammenfassung der Synthese-Experimente und der Kreuzpolarisations-Untersuchungen 1H/29Si CP MAS NMR und 19F/29Si CP MAS NMR 80
5. Zusätzliche NMR-Untersuchungen an externen
Clathrasil-Oberflächen 82
5.1 Theoretische Betrachtung der Relaxationszeiten 82 5.1.1 Der Zusammenhang zwischen Blochschen Gleichungen und Relaxationszeiten 82
5.1.1.2 Der Zusammenhang zwischen der Kreuzpolarisationszeit und den heteronuklearen Abständen 86
5.2 Experimentelle Daten 88
5.2.1 Berechnung der Kreuzpolarisationszeiten 88 5.2.1.1 Kontaktzeitvariationsmessungen, durchgeführt
an der as-syntheiszed Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm 88
5.2.1.2 Kontaktzeitvariationsmessungen, durchgeführt an der deuterierten Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm 92
5.2.1.3 Vergleich zwischen den Kontaktzeitvariationsdaten, erhalten aus der as-synthesized, der deuterierten, der calcinierten und der fluorierten Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm 97
5.2.2 Berechnung der Spin-Gitter T1ρ - Relaxationszeiten und der Kreuzrelaxationszeiten Tcp für die as-synthesized-, deuterierte-, calcinierte- und fluorierte-Form der feinkörnigen Dodecasil 3C-Proben 99
5.2.3 Relaxationszeitmessungen 101

Inhaltsverzeichnis iv
5.2.3.1 Berechnung der Spin-Gitter T1 � Relaxationszeiten und der Spin-Spin T2 � Relaxationszeiten für die as-synthesized Form des Dodecasil 3C (mittlere Korngrösse 0,1 µm) 101
5.3 Zusammenfassung der Relaxations- und Kontaktzeitvariationsmessungen 106
6. Untersuchungen an Quarzmehl 108
6.1 Klassierung durch die Fallgeschwindigkeit (Schlämmung) 108 6.1.1 Grundlagen und allgemeine Vorraussetzungen
der Sedimentationsanalyse 108 6.1.2 Probenvorbereitung 110
6.1.3 Sedimentationsmethoden 110 6.1.3.1 Die Sedimentation im Atterberg-Zylinder 110 6.1.3.2 Die Zentrifugen-Methode 112
6.2 Experimentelle Untersuchungen an Quarzmehl 113
6.2.1 Zielsetzung 113 6.2.2 Probenvorbereitung und Durchführung des
Trennverfahrens 114 6.2.3 Korngrössebestimmung durch
Rasterelektronenmikroskopie 116 6.2.4 NMR-Untersuchungen an Quarzmehl 118
6.2.4.1 29Si MAS NMR-Experimente 118 6.2.4.2 1H/29Si MAS NMR-Experimente 121 6.3 Zusammenfassung der Quarzmehl-Untersuchungen 123 7. Zusammenfassung 124 8. Literatur 129 9. Abkürzungen und Symbole 133

Inhaltsverzeichnis v
Anhang Danksagung Erklärung Lebenslauf

1. Zeolithoberfläche und Reaktivität 1
1. Theoretische Grundlagen
1.1 Zeolithe und zeolithartige Materialien
1.1.1 Einführung Der Name Zeolith hat als Ursprung zwei griechische Wörter mit der Bedeutung siedende (�zeo“) Steine (�lithos“). Die Bezeichnung wurde 1756 von einem schwedischen Mineralogen namens Cronstedt einer neuen Klasse von silikatischen Mineralien zugewiesen. Cronstedts Entdeckung beruhte auf einem ungewöhnlichen physikalisch-chemischen Verhalten von verschiedenen Mineralien, die durch Erhitzen in der Lage waren, sehr leicht Wasser freizusetzen. Im klassischen Sinne gelten die Zeolithe als �Einzigartig in ihrem strukturellen Aufbau mit [SiO4]4--und/oder [AlO4]5-Tetraedern, die über alle vier Ecken miteinander zu einer drei dimensionalen porösen Gerüststruktur verknüpft sind� (Dyer, 1988). In Zeolithen sind diese Gerüststrukturen offen, so dass Kationen und Wassermoleküle in den Kavitäten und Kanälen angelagert werden und sich dabei noch frei bewegen können. Gegenwärtig gibt es 46 Strukturtypen, die als natürliche Minerale bekannt sind. Die Regelmässigkeit im strukturellen Aufbau und die genau definierten Porenöffnungen von Zeolithstrukturen verleiht ihnen besondere Eigenschaften und ermöglicht es, dadurch die industriellen und kommerziellen Anwendungen zu erweitern. Das ist der Grund, warum in den letzten Jahren eine Vielzahl von verwandten mikroporösen Materialien im Labor synthetisch hergestellt und strukturell charakterisiert worden sind. Die Entdeckung von zeolithartigen Materialien erforderte eine Erweiterung der klassischen Zeolith-Definition zu: �Jede dreidimensionale Gerüststruktur aus tetraedrisch koordinierten T-Atomen (T = Si, Al, Fe, Co, P, Ga, B, Be, Zn u. a.), die über gemeinsame Sauerstoffatome miteinander verknüpft sind� (Meier und Ohlson, 1992). Zu den zeolithartigen Materialien gehören sowohl reine SiO2-Modifikationen, wie z. B. Porosile, als auch Gallophosphate, Alumosilikate, Alumophosphate und Beryllophosphate.

1. Zeolithoberfläche und Reaktivität 2
Die Structure Comission of the International Zeolite Association beschloss, jede neue Gerüststruktur, wie z. B. natürliche und synthetische zeolithartige Materialien, zu denen:
- Silikatphasen mit einer Gerüststruktur ähnlich denen des natürlichen Minerals Melanophlogit und des synthetischen Dodecasils,
- Materialien mit Gerüststrukturen, die OH-Spezies enthalten, z. B. Partheit Ca8[Al16Si16O60(OH)8].16H2O,
- strukturelle Varianten des Sodaliths und Cancriniths, wie z. B. Feldspatoide, gehören, in den �Atlas of Zeolite Structure Types� aufzunehmen.
1.1.2 Nomenklatur und Zeolith-Klassifikation Für die natürlichen Zeolithe und zeolithartigen Substanzen wurde eine einfache Nomenklatur, basierend auf der Topologie des Wirtsgerüstes, vorgeschlagen und von der IUPAC genehmigt. So werden die meisten synthetischen Zeolithe durch eine Kombination von drei Grossbuchstaben und Zahlen, dem s.g. �Strukturtyp-Code� bezeichnet. Die Buchstabenfestlegung für den Strukturtyp-Code einer neuen Gerüststrukturtopologie basiert im normalen Fall auf dem Namen, der für die erstmalige Beschreibung des Materials benutzt wurde. Während sich z. B. der Strukturtyp-Code VFI vom zeolithartigen hydratisierten Allumophosphat VPI-5 (Davies et al., 1988) ableitet, das am Virginia Polytechnic Institute als Nummer 5 synthetisiert worden ist, entspricht der Strukturtyp-Code RSN dem kationenausgetauschten Zinkosilikat RUB-17 (Röhrig und Gies, 1995a, b), das an der Ruhr-Universität Bochum als Nummer 17 hergestellt worden ist. Alle diese Strukturtypen mit ihrem Codenamen, ihrer Bezeichnung, der chemischen Zusammensetzung und der Raumgruppe, den Gitterparametern, Atompositionen und Referenzen wurden im �Atlas of Zeolite Structure Types� von Meier und Olson 1992 (Meier und Ohlson, 1992) zusammengefasst, und ausführlich charakterisiert. Zu den aufgeführten Strukturtypen zählen auch die unterbrochenen Gerüststrukturen („interrupted frameworks“), wenn die Gerüststrukturatome (ausser Sauerstoff) zumindest in der calcinierten Form tetraedrisch koordiniert sind. Da so viele Zeolithe und zeolithartige Materialien existieren, hat sich die Klassifizierung nach dem Strukturtyp-Code als sehr nützlich erwiesen.

1. Zeolithoberfläche und Reaktivität 3 In Abb. 1.1 wird gezeigt, wie die unterschiedlichen Verknüpfungen der T-Atome zur Ausbildung von kanalartigen-, käfigartigen-, bzw. einer Kombination von beiden Hohlräumen führen können.
AEI AFI EMT
DOH MFI LTA Abb. 1.1 Ausgewählte Beispiele zeolithischer Gerüststrukturtopologien mit den dazugehörigen Strukturtyp-Codes, wobei jeder Knotenpunkt ein T-Atom repräsentiert, während die O-Atome aus Übersichtsgründen nicht dargestellt sind.
Es wurden mehrere Kriterien für die Klassifikation der Silikate im allgemeinen und der Zeolithe im besonderen benutzt. Eines davon basiert auf der Einteilung der Silikate nach der Zahl der Tetraeder pro 1000Å, der s.g. FD, �framework density� (Brunner und Meier, 1989). Die FD wurde als Äquivalent zu der Dichte eingeführt und beschreibt die Porosität der Materialien. Für die vorliegende Arbeit sind Substanzen mit einer FD niedriger als 21 von Bedeutung, die als poröse Materialien bezeichnet werden. Zu dieser Kategorie zählen die Porosile. Ein zweites Kriterium hat als Basis die Klassifizierung der Silikate nach der Kanal/Käfiggeometrie der Poren. So werden die Porosile nach Liebau et al. (Liebau et al., 1986) in zwei Kategorien eingeteilt: in Clathra-Verbindungen mit käfigartigen Hohlräumen und Zeo-Verbindungen mit kanalartigen Hohlräumen.
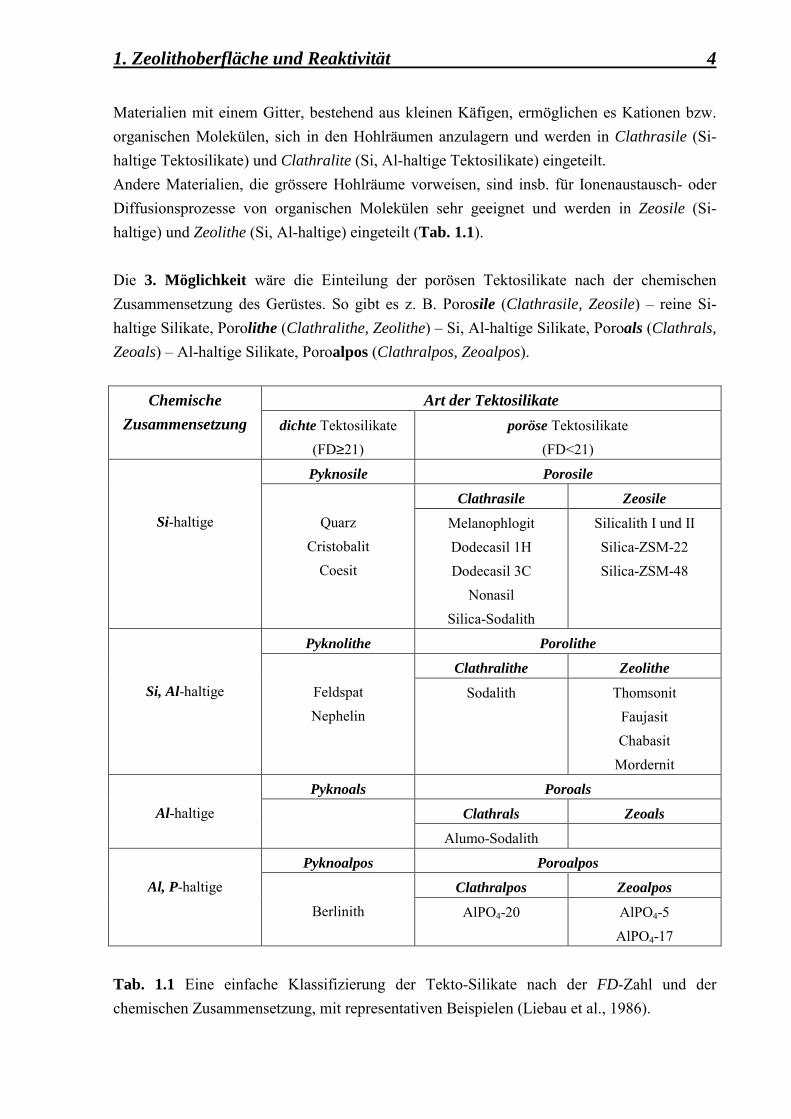
1. Zeolithoberfläche und Reaktivität 4 Materialien mit einem Gitter, bestehend aus kleinen Käfigen, ermöglichen es Kationen bzw. organischen Molekülen, sich in den Hohlräumen anzulagern und werden in Clathrasile (Si-haltige Tektosilikate) und Clathralite (Si, Al-haltige Tektosilikate) eingeteilt. Andere Materialien, die grössere Hohlräume vorweisen, sind insb. für Ionenaustausch- oder Diffusionsprozesse von organischen Molekülen sehr geeignet und werden in Zeosile (Si-haltige) und Zeolithe (Si, Al-haltige) eingeteilt (Tab. 1.1). Die 3. Möglichkeit wäre die Einteilung der porösen Tektosilikate nach der chemischen Zusammensetzung des Gerüstes. So gibt es z. B. Porosile (Clathrasile, Zeosile) � reine Si-haltige Silikate, Porolithe (Clathralithe, Zeolithe) � Si, Al-haltige Silikate, Poroals (Clathrals, Zeoals) � Al-haltige Silikate, Poroalpos (Clathralpos, Zeoalpos).
Art der Tektosilikate Chemische Zusammensetzung dichte Tektosilikate
(FD≥21) poröse Tektosilikate
(FD<21)
Pyknosile Porosile
Clathrasile Zeosile
Si-haltige
Quarz Cristobalit
Coesit
Melanophlogit Dodecasil 1H Dodecasil 3C
Nonasil Silica-Sodalith
Silicalith I und II Silica-ZSM-22 Silica-ZSM-48
Pyknolithe Porolithe
Clathralithe Zeolithe
Si, Al-haltige
Feldspat Nephelin
Sodalith Thomsonit Faujasit Chabasit
Mordernit
Pyknoals Poroals
Clathrals Zeoals
Al-haltige
Alumo-Sodalith
Pyknoalpos Poroalpos
Clathralpos Zeoalpos
Al, P-haltige
Berlinith AlPO4-20 AlPO4-5 AlPO4-17
Tab. 1.1 Eine einfache Klassifizierung der Tekto-Silikate nach der FD-Zahl und der chemischen Zusammensetzung, mit representativen Beispielen (Liebau et al., 1986).
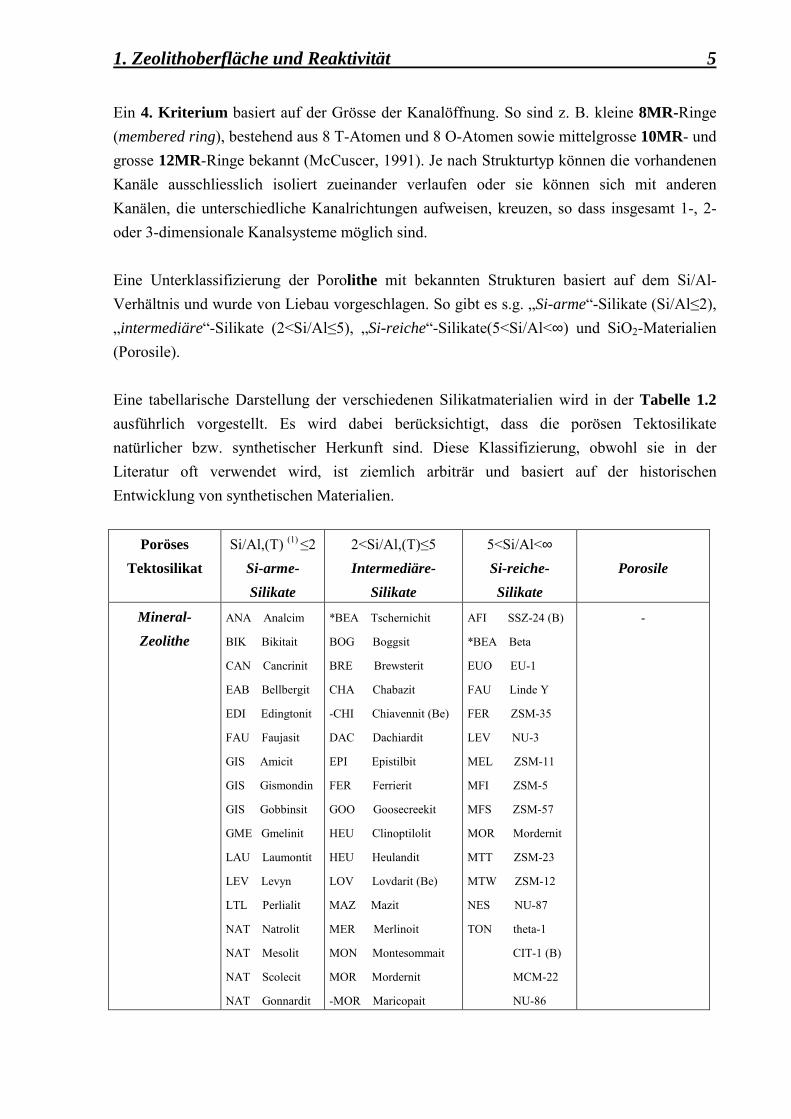
1. Zeolithoberfläche und Reaktivität 5 Ein 4. Kriterium basiert auf der Grösse der Kanalöffnung. So sind z. B. kleine 8MR-Ringe (membered ring), bestehend aus 8 T-Atomen und 8 O-Atomen sowie mittelgrosse 10MR- und grosse 12MR-Ringe bekannt (McCuscer, 1991). Je nach Strukturtyp können die vorhandenen Kanäle ausschliesslich isoliert zueinander verlaufen oder sie können sich mit anderen Kanälen, die unterschiedliche Kanalrichtungen aufweisen, kreuzen, so dass insgesamt 1-, 2- oder 3-dimensionale Kanalsysteme möglich sind. Eine Unterklassifizierung der Porolithe mit bekannten Strukturen basiert auf dem Si/Al-Verhältnis und wurde von Liebau vorgeschlagen. So gibt es s.g. �Si-arme�-Silikate (Si/Al≤2), �intermediäre�-Silikate (2<Si/Al≤5), �Si-reiche�-Silikate(5<Si/Al<∞) und SiO2-Materialien (Porosile). Eine tabellarische Darstellung der verschiedenen Silikatmaterialien wird in der Tabelle 1.2 ausführlich vorgestellt. Es wird dabei berücksichtigt, dass die porösen Tektosilikate natürlicher bzw. synthetischer Herkunft sind. Diese Klassifizierung, obwohl sie in der Literatur oft verwendet wird, ist ziemlich arbiträr und basiert auf der historischen Entwicklung von synthetischen Materialien.
Poröses Tektosilikat
Si/Al,(T) (1) ≤2 Si-arme-Silikate
2<Si/Al,(T)≤5 Intermediäre-
Silikate
5<Si/Al<∞ Si-reiche- Silikate
Porosile
Mineral-Zeolithe
ANA Analcim
BIK Bikitait
CAN Cancrinit
EAB Bellbergit
EDI Edingtonit
FAU Faujasit
GIS Amicit
GIS Gismondin
GIS Gobbinsit
GME Gmelinit
LAU Laumontit
LEV Levyn
LTL Perlialit
NAT Natrolit
NAT Mesolit
NAT Scolecit
NAT Gonnardit
*BEA Tschernichit
BOG Boggsit
BRE Brewsterit
CHA Chabazit
-CHI Chiavennit (Be)
DAC Dachiardit
EPI Epistilbit
FER Ferrierit
GOO Goosecreekit
HEU Clinoptilolit
HEU Heulandit
LOV Lovdarit (Be)
MAZ Mazit
MER Merlinoit
MON Montesommait
MOR Mordernit
-MOR Maricopait
AFI SSZ-24 (B)
*BEA Beta
EUO EU-1
FAU Linde Y
FER ZSM-35
LEV NU-3
MEL ZSM-11
MFI ZSM-5
MFS ZSM-57
MOR Mordernit
MTT ZSM-23
MTW ZSM-12
NES NU-87
TON theta-1
CIT-1 (B)
MCM-22
NU-86
-
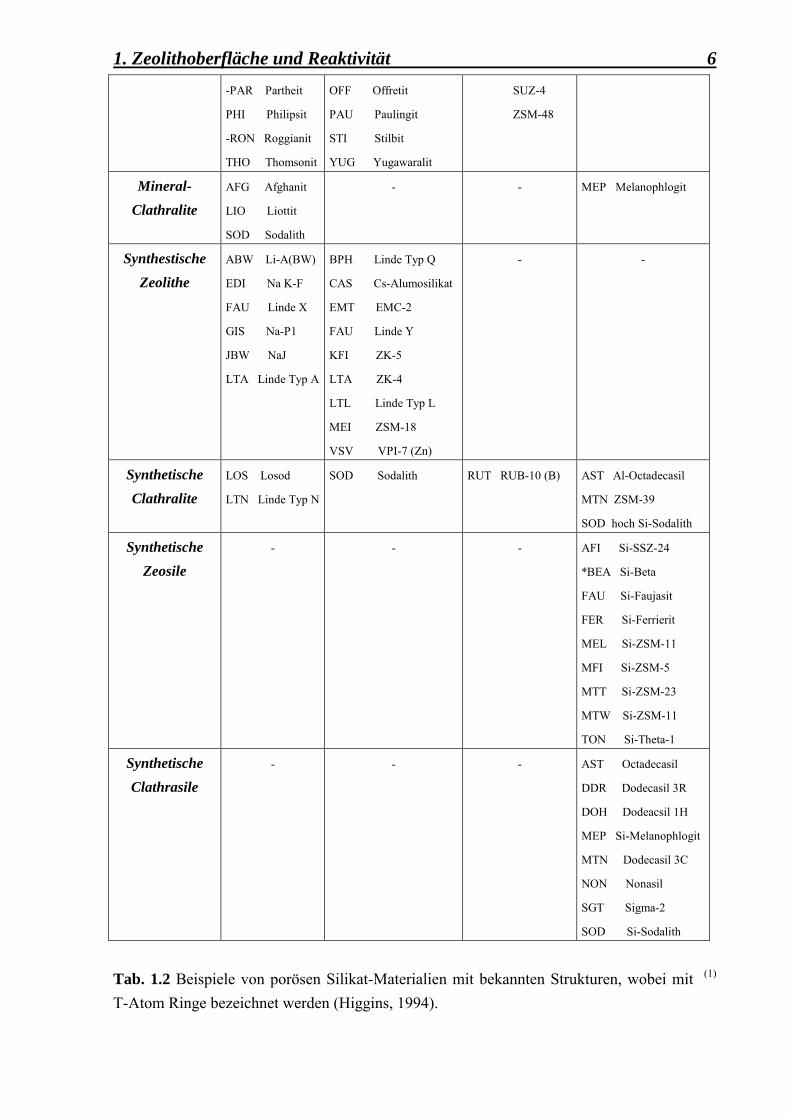
1. Zeolithoberfläche und Reaktivität 6
-PAR PartheitPHI Philipsit
-RON Roggianit
THO Thomsonit
OFF Offretit
PAU Paulingit
STI Stilbit
YUG Yugawaralit
SUZ-4
ZSM-48
Mineral-Clathralite
AFG Afghanit
LIO Liottit
SOD Sodalith
- - MEP Melanophlogit
Synthestische Zeolithe
ABW Li-A(BW)
EDI Na K-F
FAU Linde X
GIS Na-P1
JBW NaJ
LTA Linde Typ A
BPH Linde Typ Q
CAS Cs-Alumosilikat
EMT EMC-2
FAU Linde Y
KFI ZK-5
LTA ZK-4
LTL Linde Typ L
MEI ZSM-18
VSV VPI-7 (Zn)
- -
Synthetische Clathralite
LOS Losod
LTN Linde Typ N
SOD Sodalith RUT RUB-10 (B) AST Al-Octadecasil
MTN ZSM-39
SOD hoch Si-Sodalith
Synthetische Zeosile
- - - AFI Si-SSZ-24
*BEA Si-Beta
FAU Si-Faujasit
FER Si-Ferrierit
MEL Si-ZSM-11
MFI Si-ZSM-5
MTT Si-ZSM-23
MTW Si-ZSM-11
TON Si-Theta-1
Synthetische Clathrasile
- - - AST Octadecasil
DDR Dodecasil 3R
DOH Dodeacsil 1H
MEP Si-Melanophlogit
MTN Dodecasil 3C
NON Nonasil
SGT Sigma-2
SOD Si-Sodalith
Tab. 1.2 Beispiele von porösen Silikat-Materialien mit bekannten Strukturen, wobei mit (1) T-Atom Ringe bezeichnet werden (Higgins, 1994).

1. Zeolithoberfläche und Reaktivität 7
1.1.2.1 Clathrasile Die Clathrasile gehören zu der Klasse der porösen Tektosilikate. In ihrer as-synthesized Form bestehen die Clathrasilgitter aus Käfigen, die durch sogenannte Gastmoleküle besetzt sind (Gies und Marler, 1992). Die grossen Käfige enthalten meist organische Moleküle, während in den kleineren Käfigen Gase (z. B. F, N2, CO2, Ar, CH4) angelagert sind. Die kleinsten Käfige sind in der Regel unbesetzt. Die Wirtsgerüste der Clathrasile sind elektrisch neutral und benötigen für ihre Synthese als Template neutrale Gastmoleküle. Zwischen den Gast-Molekülen und dem Wirt-System existieren schwache Van der Waals-Bindungen, die dazu führen, dass das Kristallgitter erhalten bleibt. In der Tabelle 1.3 werden die Clathrasile anhand von Nomenklatur, Strukturtyp-Code, kristallographischen Informationen und Käfigtyparten zusammengestellt. Die Basiskäfige werden benutzt, um die Struktur der Clathrasile zu beschreiben. Durch die Verknüpfung über Ecken, Kanten und/oder Flächen, via Sauerstoffatome, werden vollständige Gerüststrukturen gebildet (Gies, 1991). Die Clathrasile DDR, DOH, MEP, MTN haben z. B. als Basiskäfig ein Pentagondodecaeder, gekennzeichnet durch den polyhedralen Käfig [512], wie es aus der Tabelle 1.3 zu entnehmen ist. Um aus der as-synthesized Form der Materialien gastmolekülfreie Porosile zu erhalten, müssen die in den Käfigen angelagerten Templat-Moleküle durch die Calcinierung bei hohen Temperaturen in einer oxidativen Atmosphäre eliminiert werden. Die meisten polyhedralen Clathrasil-Käfige bestehen aus relativ engen Ringen mit 4 bis 8 T-Atomen, so dass für eine komplette Eliminierung der Templat-Moleküle eine lange Calcinierungszeit benötigt wird. In den calcinierten Clathrasilen ist die Zersetzung des Templates erforderlich, da nur die Templatreste durch die Porenöffnung nach aussen dringen können, um den Käfig bzw. Kanal für die Adsorption von anderen Molekülen freizugeben. Bei einer �kurzen� und partiellen Calcinierung von ein paar Stunden, kommt es zu einer partiellen Oxidation, so dass noch Reste von nichtoxidierten karbonhaltigen Materialien in den Käfigen übrig bleiben. Ein Teil der Bindungen in den organischen Molekülen werden gebrochen, aber die Kohlenwasserstoffe werden nicht vollständig zu CO2 oxidiert. Nach einer �langen� und kompletten Calcinierung wird das Silikat-Gitter frei von Gastmolekülen sein, wobei die Kristallinität der Probe noch erhalten bleibt.
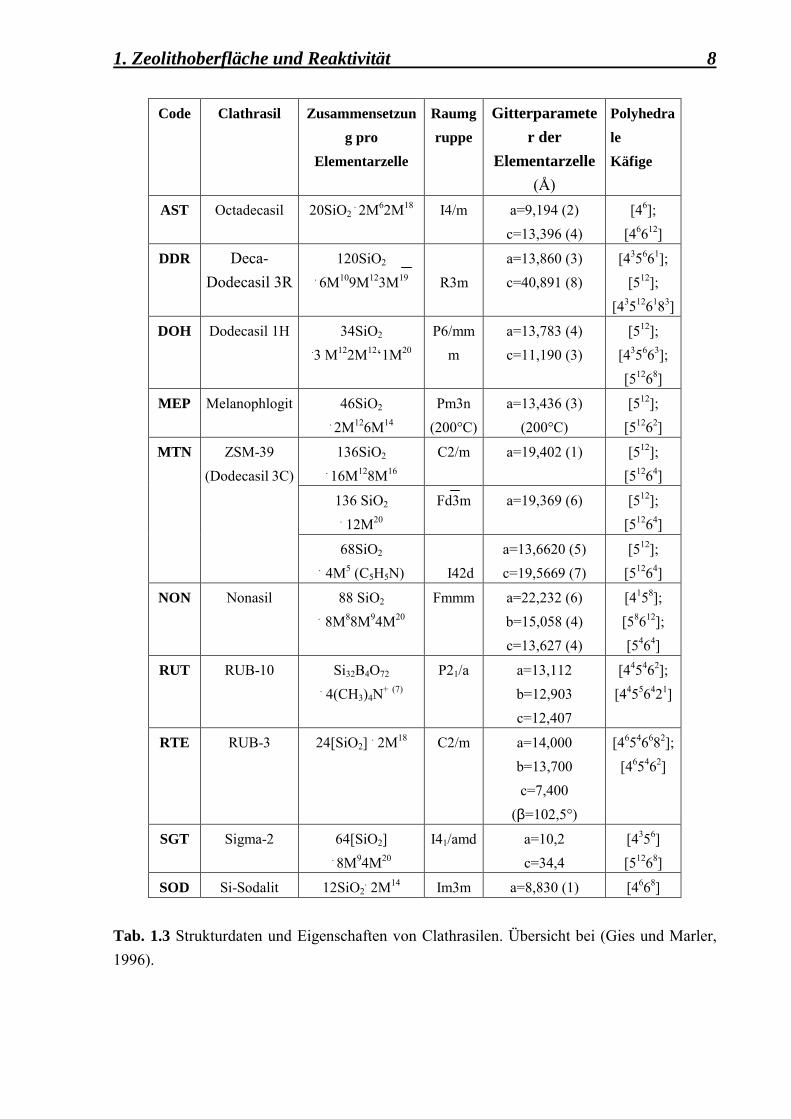
1. Zeolithoberfläche und Reaktivität 8
Code Clathrasil Zusammensetzung pro
Elementarzelle
Raumgruppe
Gitterparameter der
Elementarzelle (Å)
Polyhedrale Käfige
AST Octadecasil 20SiO2 . 2M62M18 I4/m a=9,194 (2)
c=13,396 (4) [46];
[46612]
DDR Deca-Dodecasil 3R
120SiO2 . 6M109M123M19
R3m
a=13,860 (3) c=40,891 (8)
[435661]; [512];
[435126183]
DOH Dodecasil 1H 34SiO2 .3 M122M12�1M20
P6/mmm
a=13,783 (4) c=11,190 (3)
[512]; [435663]; [51268]
MEP Melanophlogit 46SiO2 . 2M126M14
Pm3n (200°C)
a=13,436 (3) (200°C)
[512]; [51262]
136SiO2 . 16M128M16
C2/m a=19,402 (1) [512]; [51264]
136 SiO2 . 12M20
Fd3m a=19,369 (6) [512]; [51264]
MTN ZSM-39 (Dodecasil 3C)
68SiO2 . 4M5 (C5H5N)
I42d
a=13,6620 (5) c=19,5669 (7)
[512]; [51264]
NON Nonasil 88 SiO2 . 8M88M94M20
Fmmm a=22,232 (6) b=15,058 (4) c=13,627 (4)
[4158]; [58612]; [5464]
RUT RUB-10 Si32B4O72 . 4(CH3)4N+ (7)
P21/a a=13,112 b=12,903 c=12,407
[445462]; [44556421]
RTE RUB-3 24[SiO2] . 2M18 C2/m a=14,000 b=13,700 c=7,400
(β=102,5°)
[46546682]; [465462]
SGT Sigma-2 64[SiO2]
. 8M94M20 I41/amd a=10,2
c=34,4 [4356] [51268]
SOD Si-Sodalit 12SiO2. 2M14 Im3m a=8,830 (1) [4668]
Tab. 1.3 Strukturdaten und Eigenschaften von Clathrasilen. Übersicht bei (Gies und Marler, 1996).

1. Zeolithoberfläche und Reaktivität 9 Die Clathrasile sind, wie die ganze Familie der Porosile, bekannt für ihren hydrophoben Charakter. Im Gegensatz zu Quarz und anderen natürlichen Silikaten, zeigen Zeosile lipophile Eigenschaften und adsorbieren organische Moleküle sogar in einem wässrigen Medium. Im Gegensatz zu den Zeolithen, die als starke Säuren bekannt sind, zeigen die Clathrasile einen sehr schwach aziden Charakter, vergleichbar mit einer Brönstedt-Säure.
1.1.2.1.1 Dodecasil 3C (MTN) Die Vorsilbe Dodeca aus dem Namen Dodecasil 3C verweist erstens auf den Polyeder des Wirtsgitters, ein Dodecahedron, der als Basiskäfig ([512]-Käfig) des betrachteten Porosils dient. Die Endung sil stammt von Clathrasil, einem porösen Tektosilikat mit einer framework density FD unter 21. Die 3 gibt die Zahl der Schichten in der Elementarzelle an, während das c für die ideale Symetrie des Gerüstes (kubisch) steht. Dodecasil 3C (MTN) (hergestellt unabhängig voneinander von Gies, 1982 (Gies et al., 1982) und Schlenker, 1981 (Schlenker et al., 1981)) ist das bis jetzt am meisten untersuchte Clathrasil. Ein Modell der MTN-Struktur wurde erstmals von Schlenker et al. (Schlenker et al., 1981) vorgeschlagen. Dabei diente die bei 800°C eine halbe Stunde lang calcinierte MTN-Probe als Modell.
Abb. 1.2 Schematische Darstellung der zwei verschiedenen Käfigtyparten: kleine [512]- und grosse [51264]-Käfige, die in Dodecasil 3C vorkommen.[51264]-Käfig [512]-Käfig

1. Zeolithoberfläche und Reaktivität 10 Die MTN-Struktur besteht aus eckenverknüpften [SiO4]-Tetraedern. Diese bilden eine Wirtstruktur, die aus flächenverknüpften Pentagondodecahedra (Schlenker et al., 1981), aufgebaut wird. Bei Verknüpfung solcher Schichten in der Reihenfolge ABC ergeben sich zwei Arten von käfigförmigen Hohlräumen, wiederum kleinere [512]- und grössere [51264]-Käfige, in denen Gastmoleküle eingelagert werden können (Abb. 1.2). Der kleine [512]- Käfig hat ein Volumen von 0,080 nm3, während der grosse [51264]- Käfig 0,250 nm3 gross ist. Aus der Tabelle 1.4 kann entnommen werden, welche Gase und organische Verbindungen in den unterschiedlich grossen Käfigen aufgenommen werden können.
[51264]-Käfig [512]-Käfig Symmetrie/Eigenschaften
Kr Kr kubisch
Xe Xe kubisch
N(CH3)3 Trimethylamin, CO2 N2, CH4, Ar kubisch
C4H8O Tetrahydrofuran ? optisch anisotrop
C4H8S Tetrahydrothiophan ? optisch anisotrop
C5H10NH Piperidin optisch anisotrop
Tab. 1.4 Käfiginhalt und Symmetrie der Dodecasil 3C-Materialien (Gies, 1987).
Die Elementarzelle enthält 136 SiO2.nM-Moleküle, wobei mit M das Templat-Molekül bezeichnet ist. MTN kristallisiert bei Raumtemperatur kubisch und besitzt die Symmetrie Fd3m (Gies et al., 1982). Durch Abkühlen ist eine Reihe von Phasenumwandlung zu beobachten (Knorr und Depmeier, 1998, Könnecke et al., 1992). So sind z. B. bei Dodecasil 3C tetragonale, orthorombische und monokline Symetrien bekannt, was gut unter dem Polarisationsmikroskop zu erkennen ist, anhand der Isotropie bzw. der Anisotropie der Kristalle.
Abb. 1.3 Die Pentagondodecaeder-Grundschicht nach (111)-Richtung.

1. Zeolithoberfläche und Reaktivität 11
200 µm grosse aluminiumfreie Einkristalle wurden von (Gies, 1987) gezüchtet. Das Silika-Endglied wird als Dodecasil 3C bezeichnet, um dadurch die Verwandtschaft zu den anderen Mitgliedern einer polytypen Reihe, die aus Dodecaeder-Schichten aufgebaut ist, zu zeigen. Gleichzeitig wurde von Gies et al. beobachtet, dass es durch Erhitzen der Probe bei 100-120°C zu einer displaziven Umwandlung der Probe in eine kubische Phase kommt. Durch Abkühlen findet bei ~ 95-105°C ein Phasenübergang statt Die Strukturverfeinerung des Trimethylamin-haltigen Materials lieferte Daten über die kurzen Si-O Abstände (1,566 Å), die grossen Si-O-Si Winkel (174,5°) und die hohen Temperaturfaktoren für Sauerstoff. Daraus ergeben sich Informationen über die statische bzw. dynamische Unordnung des Silikatgitters. Es wurden auch 3-mm grosse Piperidin-haltige Dodecasil 3C-Einkristalle gezüchtet. Durch differential scanning calorimetry-Messungen (DSC) wurden Phasenübergänge zwischen 46°C und 161°C beobachtet. Die hoch-Temperaturform (161°C) weist eine kubische Symmetrie (I-42d) auf, während bei der tief-Temperaturform die Symmetrie noch nicht bestimmt worden ist. Es wurden auch andere Besonderheiten der MTN-Struktur beobachtet, wie z. B. die Strukturänderung, die durch die schnelle Aufheizung auf 700°C, gefolgt von einer Abkühlung auf ca. 159°C, zustande kommt. Der s.g �memory effect� wird dadurch neutralisiert, bleibt aber bei der tief-Temperaturform erhalten. Könnecke et al. (Könnecke et al., 1992) calcinierte 150 µm grosse D3C Kristalle 48 Stunden lang bei 900 °C, um aus den Käfigen das Pyrrolidin und die anderen noch existierenden Hilfsgase zu eliminieren. Durch diese partielle Calcinierung gelang es, eine schwarze Probe zu bekommen, in der noch Reste von kohlenstoffhaltigen Materialien vorhanden waren. Die DSC-Messungen zeigten einen Phasenübergang bei ca. 451 K. Durch die Strukturverfeinerung für den bei 523±15 K calcinierten Kristall wurde eine Gitterkonstante von a = 19,369(6) Å ermittelt. Die erhaltene Symmetrie war Fd3m. Die Verfeinerung des anisotropen Temperaturfaktors und die Fourier-Analyse zeigten aufgespaltene Positionen von drei von vier Sauerstoffatomen. Die kurzen Si-O-Abstände (1,581 Å) und die grossen Si-O-Si-Winkel (163,5 °) deuten aber auf eine ungeordnete Struktur mit einer mittleren Fd3m-Symmetrie des Käfigs hin. Die geringen Unterschiede der thermischen Parameter dieser Verfeinerung und einer anderen, die bei 623 K durchgeführt wurde, brachten die Autoren auf die Idee, dass die Unordnung in Dodecasil 3C statischer Natur sei.

1. Zeolithoberfläche und Reaktivität 12
1.1.3 Zeolitheigenschaften und Anwendungen Zeolithe werden sowohl in der Industrie, als auch für den privaten Nutzen eingesetzt. Sie finden überwiegend Anwendungen als Ionenaustauscher (als Enthärtungsmittel oder für die Dekontaminierung flüssiger nuklearer Abwässer), in der Landwirtschaft und dem Gartenbau, als Trocknungsmittel, als Molekularsiebe und nicht zuletzt als formselektive Katalysatoren. Die Ionenaustauscheigenschaft von Zeolithen, wie z. B. Enthärtung von Waschwasser, basiert auf einem kontrollierten Kationen-Diffusionsprozess. Dabei werden Ca2+- und Mg2+-Kationen, durch Na+-Kationen der zugefügten Zeolithe ersetzt, so dass die Härte des Wasser erniedrigt wird. Daher werden umweltschädigende Phosphate, wie z.B. Na-Tripolyphosphate, die früher im Waschmittel zu finden waren, durch umweltverträgliche Zeolithe ersetzt. Zeolithe werden auch bei der Dekontaminierung flüssiger nuklearer Abwässer erfolgreich eingesetzt. Radioaktive Isotope, wie z. B. 137Cs und 90Sr, die in geringen Konzentrationen in nuklearen Abfällen zu finden sind, werden in den Hohlräumen von Zeolith-Gerüststrukturen aufgenommen. Die am meisten für die Radioisotopenkontrolle eingesetzte Zeolithe gehören zu der Gruppe der Clinoptilolithe und Mordernite, die nach der Tschernobyl-Katastrophe erfolgreich eingesetzt worden sind, um die Radioaktivität des Trink- und Waschwassers messen zu können (Dyer, 1995). Zur Tiernahrung von verschiedenen Tierarten (Schafe, Schweine, Rinder, Pferde) werden Zeolithe zugesetzt um den pH-Wert kontrollieren zu können und dadurch eine Gewichtszunahme der Tiere und gleichzeitig eine Minderung der Krankheiten zu bewirken (Dyer, 1995). Im Gartenbau wird durch die Zufuhr von Zeolithen die Bodenqualität um 5-10% erhöht, so dass die Ernten verbessert werden. Der Prozess beruht auf der Dissoziation von H2O-Molekülen, die an der Zeolithoberfläche angelagert sind, in Protonen H+ und Hydroxylionen OH-. Dabei werden H3O+-Ionen erzeugt, die danach selektiv von den Zeolithen aufgenommen werden. Das ermöglicht anderen Kationen wie z. B. NH4
+ die frei gewordenen Plätze zu besetzen. Der Zeolith übernimmt in diesem Fall die Rolle eines s. g. �controlled-released fertilizer“, kontrolliert-freigesetzter Dünger. Der am besten dafür geeignete natürliche Zeolith ist der Clinoptilolith (Dyer, 1995).

1. Zeolithoberfläche und Reaktivität 13
Der Erfolg, solche Materialien als molecular sieves (Molekularsiebe) einzusetzen, basiert auf der Fähigkeit eines Zeolithes andere flüssige oder gasförmige Moleküle aufgrund ihrer effektiven Molekülgrössen auszuschliessen, der starken Affinität des Wassers zur inneren Zeolithoberfläche, der leichten Regenerierung und dem langen Nutzungsleben. Alle diese Eigenschaften ermöglichen es, die Palette der Zeolith-Anwendungen zu erweitern. So werden sowohl synthetische, als auch natürliche Zeolithe, wie z. B. Chabasite, Mordernite, Clinoptilolite, für die: • Reinigung von verschiedenen Gasströmen (CO2, H2S und Stickoxide), • Trennung der n-Paraffine von iso-Paraffinen, • Gewinnung von Sauerstoff, durch die PSAs-Methode, aus dem englischen �pressure Swing applications�, wobei eine Luftsäuberung von toxischen Gasen stattfindet, • Behandlung umweltschädlicher Chemikalien, wie z. B. Vulkanisierungsagentien, • Einführung von CO2 in kohlensäurehaltige Getränke, benutzt. Vor ca. 30 Jahren wurde entdeckt, dass synthetische Faujasite als formselektive Katalysatoren erfolgreich eingesetzt werden können. Seitdem gab es eine regelrechte Explosion von Patenten, Vorträgen und Publikationen über die Vielfalt von Reaktionen, bei denen die Zeolithe als Katalysatoren eingesetzt werden können. Das Cracken (�Knacken�) von Rohöl bei der Benzinherstellung, bei der langkettige Kohlenwasserstoffe in C1-C6-Fraktionen umgewandelt werden, ist eine der wichtigsten industriellen katalytischen Nutzungen der Zeolithe (Higgins, 1994). Die Zeolithe besitzen eine hohe katalytische Aktivität, da die Zeolith-Struktur einen grossen Anteil an s.g. aktiven Zentren (�active sites�) pro interner Oberflächeneinheit besitzt. Diese Zentren sind bekannt unter dem Namen Brönsted- bzw. Lewis-Zentren. Die Brönsted-Zentren entstehen durch die Bildung von Hydroxyl-Gruppen, was durch einen der drei folgenden Wege erfolgen kann: 1. Ammonium-Ionenaustausch (wie z. B. bei der Herstellung von USY):
Na-Zeolith(s) + NH4+
(aq) → NH4-Zeolith(s) + Na+(aq)
NH4-Zeolith(s) → NH3(g) ↑ + H-Zeolith(s) (calcinieren) 2. Polyvalenter-Ionenaustausch (wie z. B. in den ausgetauschten seltenen Erden Y-ReY):

1. Zeolithoberfläche und Reaktivität 14
nNa-Zeolith (s) + M(H2O)n+
(aq) M(H2O)-(Zeolith)n (s) + nNa+(aq)
M(H2O)-(Zeolith)n (s) → Mn+(OH)-1n (s) + (H-Zeolith)+1 (s) (calcinieren)
3. Direkter Ionenaustausch mit Mineralsäure (wie z. B. mit ZSM-5). 4. In allen diesen Fällen wird die H-Form des Zeolithes hergestellt. Brönsted-Zentren existieren auch an der Alumosilikatoberfläche und verlieren durch Erhitzen bei 550°C das Wasser, um Lewis-Zentren zu bilden. Aluminium (Al*) ist in diesem Fall dreifach koordiniert:
Die Lewis-Zentren sind insbesondere in Anwesenheit von H2O instabil. Ein sog. Stearning-Prozess führt zu der Stabilisierung der Zeolith-Struktur, so dass durch die Eliminierung der Aluminium-Spezies aus der Gerüststruktur �reelle� Lewis-Zentren entstehen:
O O O O O O O O O
Si Al Si Al Si Si Al Si Al * - -+
+
+H O2
(AlO)
Sintern

1. Zeolithoberfläche und Reaktivität 15
1.1.4 Entstehung von natürlichen und
synthetischen Zeolithen und zeolithartigen
Materialien
1.1.4.1 Natürliche Zeolithe
Lange Zeit wurde vermutet, dass Zeolithe vulkanischer Herkunft seien. Vor 30 Jahren wurden in Japan, Korea, Indonesien, Südafrika, Russland, Bulgarien, Ungarn, Slovakien, Italien, Kuba, USA Ablagerungen von reinen Zeolithen (>90%) in den Sediment- und den niedriggradigen metamorphen Gesteinen gefunden. Die Untersuchungen, die danach durchgeführt werden konnten, ergaben, dass Zeolithe durch unterschiedliche Bedingungen und über verschiedene Wege �geformt� werden können. Der erste Weg ist der sog. diagenetische Weg. Durch einen Verdampfungsprozess, der in relativ trockenen, basischen Erden stattfindet, wird Na2CO3 produziert. Dieses reagiert mit den im Boden existierenden Tonmineralien und führt zu Bildung von Zeolithen. Durch diesen Weg sind z. B. die Analcim-Lager im San Joaquin Valley, Kalifornien (USA) entstanden (Garces, 1999). Zeolithe sind auch in hydrologisch offenen Systemen durch das Eindringen des Wassers in feste Bodenschichten zu bekommen, so dass Vertikalzonen entstehen. Die oberen Schichten enthalten Quarz und Tonmineralien, während in den unteren Schichten Clinoptilolithe, Philipsite, Chabasite, und Analcime zu finden sind. Dieses Phänomen wurde sowohl in den John Day Formationen in Oregon (USA), als auch in Oahü (Hawaii) beobachtet (Garces, 1999). In hydrologisch geschlossenen Systemen, wie z. B. in wasserarmen, geschlossenen Becken, kann es durch die Interaktion von basischem, salzhaltigem Wasser mit Silikaquellen zur Entstehung von Zeolithen kommen. Als Beispiele dafür können der Tecosa-See in Kalifornien (USA) und der Magadi-See (Kenia) erwähnt werden (Garces, 1999). Durch die Versenkungsdiagenese entstehen Zonen mit einem unterschiedlichen Gehalt an Zeolithen. Diese haben eine Struktur und eine Zusammensetzung ähnlich den Zeolithen, die

1. Zeolithoberfläche und Reaktivität 16
in den hydrologisch offenen Systemen entstanden sind. Der Hauptanteil der Prozesse, die dabei stattfinden sind niedriggradig metamorpher Natur. Die bekanntesten durch Diagenese entstandenen Zeolith-Lager sind diejenigen in Japan (Garces, 1999). Der zweite Weg ist der s.g. hydrothermale Weg. Es wurde beobachtet, dass durch die hydrothermale Zirkulation in den vulkanischen Aschen Zeolithe entstehen können. In den sog. �kalten� Aschen sind die Clinoptilolite und die Mordernite zu finden, während die �wärmeren� Aschen Analcime, Heulandite, Laumonite und Wairakite enthalten. Zu den frühhydrothermalen Pegmatitlagerungen zählen diejenigen aus Andreasburg (Deutschland) und Elba (Italien) (Dyer, 1988). Ein anderer Weg ist die sog. �Genese in Geoautoklaven�. Diese findet bei hoher Temperatur und hohem Dampfdruck statt. Sie kommt in porösen Gussgesteinen, wie z. B. Basalten, vor und führt zu einer �Zeolisation�, wie es in Italien, Bulgarien und Japan beobachtet worden ist (Garces, 1999).
1.1.4.2 Synthetische Zeolithe Die am meisten benutze Methode zur Herstellung synthetischer Zeolithe und zeolithartiger Materialien ist die s.g. hydrothermale Synthese, die in der Abbildung 1.5 schematisch dargestellt wird. In der hydrothermalen Synthese kommt es zu gegenseitigen Wechselwirkungen zwischen den Molekülen der Kationen-Quelle (Si, Al) und den Templat-Molekülen. Als Si-Quelle werden Substanzen wie z. B. Aerosil, Silikagel, Kieselsäure, Tetramethoxysilan (TMOS) etc. verwendet. Als Al-Quelle werden Substanzen wie z. B. Aluminiumhydroxid, Aluminiumoxid, Aluminate und Alkoxide eingesetzt. Diese Reagenzien sind für den Aufbau des Wirt-Systems verantwortlich. Als Template werden oft Moleküle eingesetzt, die einen amphiphilen Charakter besitzen. Diese Templat-Moleküle werden in den Wirt-Käfigen angedockt und wirken dadurch als Gast-Systeme. Zwischen Wirt- und Gast-System existieren sowohl ionische Bindungen als auch schwache van der Waals-Wechselwirkungen, Kräfte die dafür sorgen, dass das neu entstandene System elektrisch stabil ist. Zu bemerken ist, dass für eine gezielte Synthese die Bildung der Käfige von der Art, der Form und der Grösse der Templat-Moleküle abhängt. Die Template sind überwiegend organischer
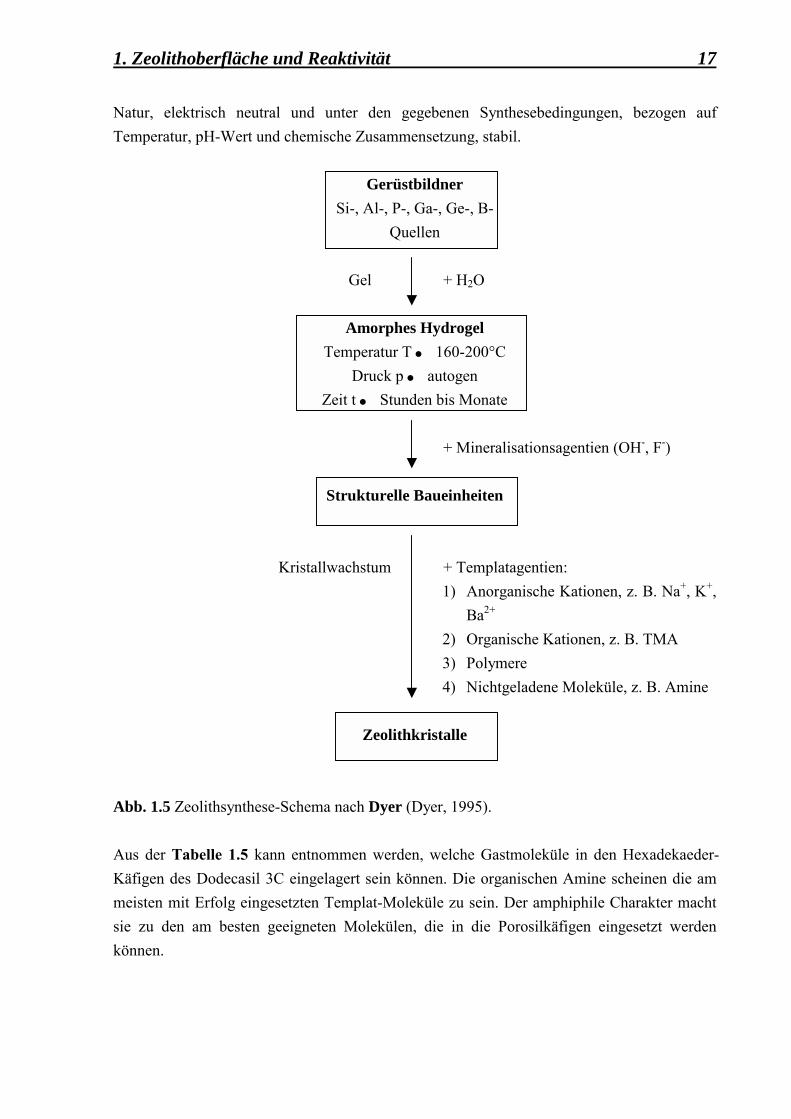
1. Zeolithoberfläche und Reaktivität 17 Natur, elektrisch neutral und unter den gegebenen Synthesebedingungen, bezogen auf Temperatur, pH-Wert und chemische Zusammensetzung, stabil.
Gerüstbildner Si-, Al-, P-, Ga-, Ge-, B-
Quellen
Gel + H2O
Amorphes Hydrogel Temperatur T ! 160-200°C
Druck p ! autogen Zeit t ! Stunden bis Monate
+ Mineralisationsagentien (OH-, F-)
Strukturelle Baueinheiten
Kristallwachstum + Templatagentien: 1) Anorganische Kationen, z. B. Na+, K+,
Ba2+ 2) Organische Kationen, z. B. TMA 3) Polymere 4) Nichtgeladene Moleküle, z. B. Amine
Zeolithkristalle
Abb. 1.5 Zeolithsynthese-Schema nach Dyer (Dyer, 1995).
Aus der Tabelle 1.5 kann entnommen werden, welche Gastmoleküle in den Hexadekaeder-Käfigen des Dodecasil 3C eingelagert sein können. Die organischen Amine scheinen die am meisten mit Erfolg eingesetzten Templat-Moleküle zu sein. Der amphiphile Charakter macht sie zu den am besten geeigneten Molekülen, die in die Porosilkäfigen eingesetzt werden können.

1. Zeolithoberfläche und Reaktivität 18
Anzahl der Strukturatom
e der Gastmoleküle
Azyklische Moleküle
Zyklische Moleküle
1 Kr, Xe -
2 MeNH2 -
3 EtNH2 -
4 Me2CHNH2, Me3N, PriSH -
5 ButNH2, Et2NH, MeCH(NH2)CH2NH2, Me2Net, MeCH(NH2)Et
monozyklisch
6 ButCH2NH2
monozyklisch
7 SF6 -
8 -
bizyklisch
Tabelle 1.5 Beispiele von Templat-Molekülen, die in die Hexadekaeder-Käfigen des Dodecasil 3C eingebaut werden können.
O S N
S
NH2
O NH
N
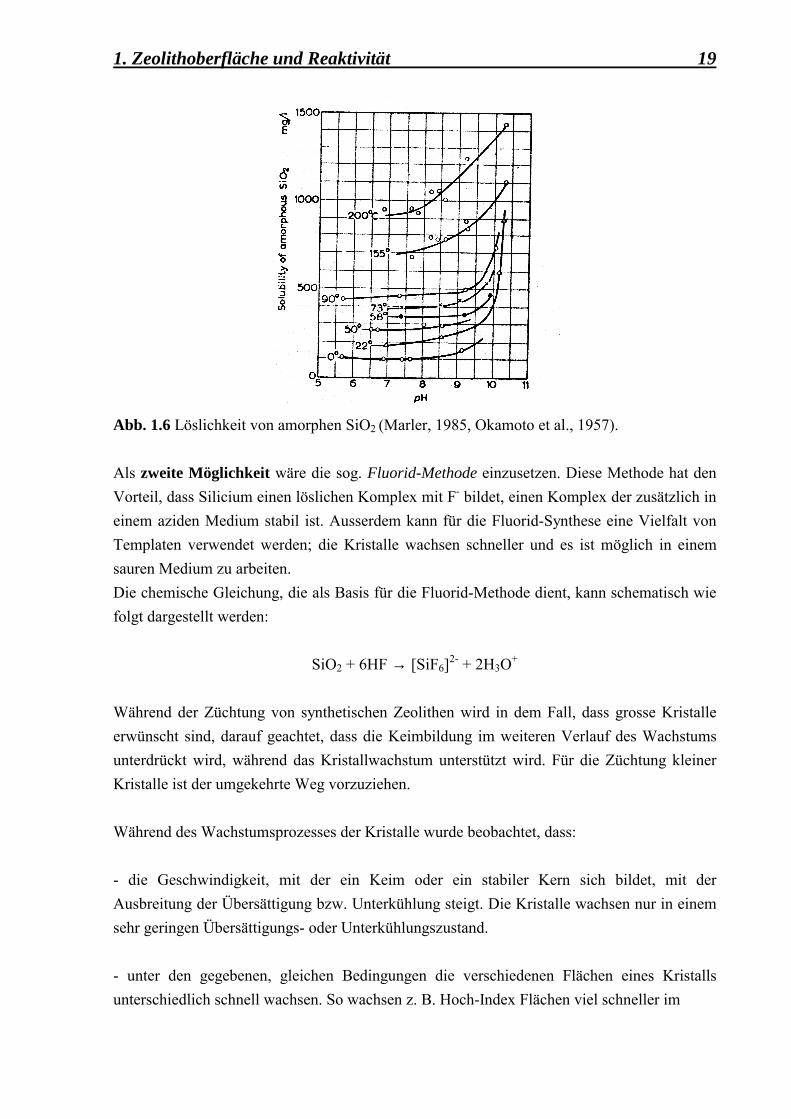
Zwei Wege werden benutzt, um das Kristallwachstum von mikroporösen Zeolithen zu unterstützen. Der erste Weg führt über die Löslichkeit der (Si, Al)-Quelle, die so hoch wie möglich gehalten wird, indem der pH-Wert der Syntheselösung in einen basischen Bereich gebracht wird (Abb. 1.6). So werden für die Synthese von Zeolithen überwiegend zyklische und azyklische Amine, wie z. B. Tetraalkylammoniumhydroxid, als OH--Quelle benutzt und gegenüber den Alkali- oder Erdalkalihydroxiden bevorzugt.
1. Zeolithoberfläche und Reaktivität 19
Abb. 1.6 Löslichkeit von amorphen SiO2 (Marler, 1985, Okamoto et al., 1957).
Als zweite Möglichkeit wäre die sog. Fluorid-Methode einzusetzen. Diese Methode hat den Vorteil, dass Silicium einen löslichen Komplex mit F- bildet, einen Komplex der zusätzlich in einem aziden Medium stabil ist. Ausserdem kann für die Fluorid-Synthese eine Vielfalt von Templaten verwendet werden; die Kristalle wachsen schneller und es ist möglich in einem sauren Medium zu arbeiten. Die chemische Gleichung, die als Basis für die Fluorid-Methode dient, kann schematisch wie folgt dargestellt werden:
SiO2 + 6HF → [SiF6]2- + 2H3O+ Während der Züchtung von synthetischen Zeolithen wird in dem Fall, dass grosse Kristalle erwünscht sind, darauf geachtet, dass die Keimbildung im weiteren Verlauf des Wachstums unterdrückt wird, während das Kristallwachstum unterstützt wird. Für die Züchtung kleiner Kristalle ist der umgekehrte Weg vorzuziehen. Während des Wachstumsprozesses der Kristalle wurde beobachtet, dass: - die Geschwindigkeit, mit der ein Keim oder ein stabiler Kern sich bildet, mit der Ausbreitung der Übersättigung bzw. Unterkühlung steigt. Die Kristalle wachsen nur in einem sehr geringen Übersättigungs- oder Unterkühlungszustand. - unter den gegebenen, gleichen Bedingungen die verschiedenen Flächen eines Kristalls unterschiedlich schnell wachsen. So wachsen z. B. Hoch-Index Flächen viel schneller im

1. Zeolithoberfläche und Reaktivität 20
Vergleich zu den Niedrig-Index Flächen und streben nach einer Weile sogar zu verschwinden, wie es aus der Abb. 1.7 zu entnehmen ist. - wenn an der Oberfläche Defekte vorhanden sind, dann streben diese Oberflächen dazu, viel schneller zu wachsen, als die entsprechenden defektfreien Oberflächen. - die Zeit, die ein Keim braucht um zu kristallisieren, umgekehrt proportional zu der Wachstumsgeschwindgkeit der verschiedenen Flächen ist.

1. Zeolithoberfläche und Reaktivität 21
1.2 Zeolithoberfläche und Reaktivität
1.2.1 Definition und Charakterisierung Die Grenzfläche wird nach Van Hove als �eine Region im Raum, begrenzt durch eine geringe Zahl von Atomen, die sich auf jeder Seite der planaren Fläche befinden�, definiert (Van Hove und Tong, 1979). Atkins versteht unter Grenzfläche �eine Region, in der eine Phase endet und eine andere beginnt� (Atkins, 1990). Nach dem Roempp Chemie Lexikon wird �die Grenzfläche im allgemeinen Sinne als die Fläche bezeichnet, die zwei nichtmischbare Phasen voneinander trennt� (Roempp, 1995). Im engeren Sinne ist unter Grenzfläche �die trennende Fläche zwischen kondensierten Phasen (flüssig-fest, flüssig-flüssig, fest-fest)� zu verstehen. Ist dagegen die eine Phase ein Gas und die angrenzenden Phasen sind entweder Flüssigkeiten oder Feste Stoffe, so werden üblicherweise die Grenzflächen Oberflächen genannt. Die beobachteten Gesetzmässigkeiten gelten prinzipiell nicht nur für kristalline, sondern auch für amorphe Festkörper, da im Nahbereich fast alle amorphen Stoffe eine ähnliche Ordnung wie ihre kristalline Modifikation zeigen. Nach dem aktuellen Wissen sind die in der Oberfläche kristalliner Stoffe befindlichen Atome und Ionen valenzmässig nicht abgesättigt. Die Existenz der Restvalenzen bewirkt, dass sich weitere Atome oder Ionen von der gleichen Art oder auch fremde Teilchen anlagern. Es werden Oberflächen von unterschiedlichen Dicken betrachtet, von weniger als einer Monolage bis zu ungefähr 8 Monolagen (~1015 Atome/1 cm2). In Bezug auf zeolithartige Materialien wird die äussere Oberfläche als die Phasengrenzfläche zwischen der einen Phase, Luft oder Flüssigkeit, und der anderen Phase, in dem gegebenen Fall Festkörper, definiert. Das deutet auf eine Diskontinuität der Struktur hin. Für die innerhalb eines geometrisch begrenzten Teilchens durch Hohlräume, Kapillaren, Käfige gebildeten Oberflächen, wird der Ausdruck innere Oberfläche verwendet. Die Atome sind in diesem Fall ladungsabgesättigt und mit der kontinuerlichen Struktur verankert. Im Falle feindisperser Silikate, die durch den Zerteilungsgrad eine bedingte grosse Oberfläche aufweisen, spielt die äussere Oberfläche eine bedeutende Rolle. Je mehr sich das Verhältnis
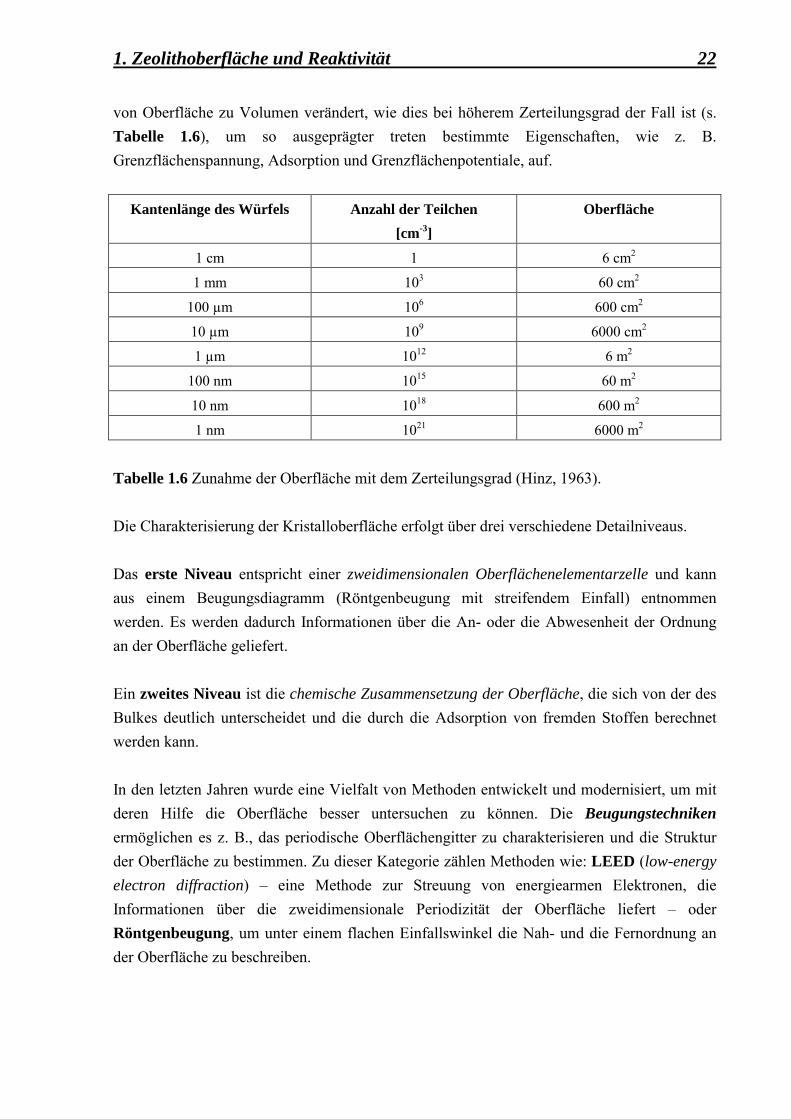
1. Zeolithoberfläche und Reaktivität 22 von Oberfläche zu Volumen verändert, wie dies bei höherem Zerteilungsgrad der Fall ist (s. Tabelle 1.6), um so ausgeprägter treten bestimmte Eigenschaften, wie z. B. Grenzflächenspannung, Adsorption und Grenzflächenpotentiale, auf.
Kantenlänge des Würfels Anzahl der Teilchen [cm-3]
Oberfläche
1 cm 1 6 cm2
1 mm 103 60 cm2
100 µm 106 600 cm2
10 µm 109 6000 cm2
1 µm 1012 6 m2
100 nm 1015 60 m2
10 nm 1018 600 m2
1 nm 1021 6000 m2
Tabelle 1.6 Zunahme der Oberfläche mit dem Zerteilungsgrad (Hinz, 1963).
Die Charakterisierung der Kristalloberfläche erfolgt über drei verschiedene Detailniveaus. Das erste Niveau entspricht einer zweidimensionalen Oberflächenelementarzelle und kann aus einem Beugungsdiagramm (Röntgenbeugung mit streifendem Einfall) entnommen werden. Es werden dadurch Informationen über die An- oder die Abwesenheit der Ordnung an der Oberfläche geliefert. Ein zweites Niveau ist die chemische Zusammensetzung der Oberfläche, die sich von der des Bulkes deutlich unterscheidet und die durch die Adsorption von fremden Stoffen berechnet werden kann. In den letzten Jahren wurde eine Vielfalt von Methoden entwickelt und modernisiert, um mit deren Hilfe die Oberfläche besser untersuchen zu können. Die Beugungstechniken ermöglichen es z. B., das periodische Oberflächengitter zu charakterisieren und die Struktur der Oberfläche zu bestimmen. Zu dieser Kategorie zählen Methoden wie: LEED (low-energy electron diffraction) � eine Methode zur Streuung von energiearmen Elektronen, die Informationen über die zweidimensionale Periodizität der Oberfläche liefert � oder Röntgenbeugung, um unter einem flachen Einfallswinkel die Nah- und die Fernordnung an der Oberfläche zu beschreiben.

1. Zeolithoberfläche und Reaktivität 23
Für die Bestimmung der chemischen Zusammensetzung der Oberfläche werden Ionisations-methoden eingesetzt. Ziel dabei ist es, die eventuellen Verunreinigungen, die an der Oberfläche angelagert werden können, nachzuweisen oder dünne Schichten, die im Verlaufe eines Experimentes adsorbiert wurden, zu analysieren (Atkins, 1990). Zu diesen Methoden zählen die Photoelektronen-Spektroskopie (XPS), Photoelektronen-Spektromikroskopie (PESM) und Auger-Spektroskopie (AES). Die Interferenztechniken (ARUPS � angle-resolved ultraviolet photoelectron spectroscopy, ARXPD � angle-resolved X-ray photoelectron difraction, NPD � normal photoelectron difraction, NEXAFS � near-edge X-ray absorption fine structure) werden eingesetzt, um strukturelle Informationen, wie z. B. molekulare Orientierung und Bindungslängen zu bekommen. Ausserdem werden Streuungstechniken, wie LEIS � low energy ion-scattering verwendet, um Daten über die Geometrie der Oberfläche zu erhalten. Die mikroskopischen und topographischen Techniken (AFM � atomic force microscopy, FIM � field-ion microscopy, FEM � field-emission microscopy, STM-scanning tunneling microscopy, SAM-scanning Auger microscopy, PEM-photoelectron microscopy, LEEM-low-energy electron microscopy) ermöglichen es, eine topographische Karte der Oberfläche zu erstellen und dadurch auch Informationen über die Zusammensetzung und Struktur der Oberfläche zu bekommen. Die am meisten benutzten Techniken sind die Vakuum- bzw. Ultrahochvakuum-Techniken und die mikroskopischen Techniken. Der Nachteil dabei ist, dass keine realistischen katalytischen Bedingungen erzeugt werden können. Alle diese Methoden ergeben ein mehr oder weniger vollständiges Bild der Oberfläche, deren Struktur, Geometrie und chemischer Zusammensetzung.
1. Zeolithoberfläche und Reaktivität 24
1.2.2 Der Zusammenhang zwischen Oberfläche
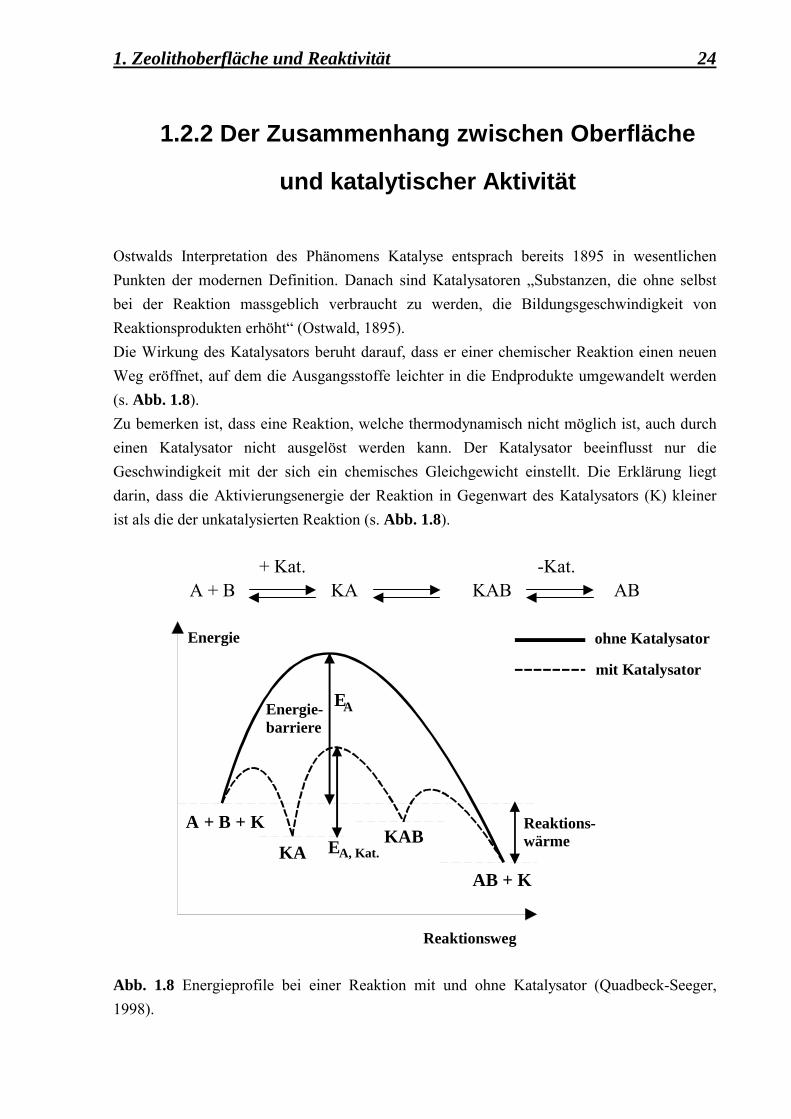
und katalytischer Aktivität Ostwalds Interpretation des Phänomens Katalyse entsprach bereits 1895 in wesentlichen Punkten der modernen Definition. Danach sind Katalysatoren �Substanzen, die ohne selbst bei der Reaktion massgeblich verbraucht zu werden, die Bildungsgeschwindigkeit von Reaktionsprodukten erhöht� (Ostwald, 1895). Die Wirkung des Katalysators beruht darauf, dass er einer chemischer Reaktion einen neuen Weg eröffnet, auf dem die Ausgangsstoffe leichter in die Endprodukte umgewandelt werden (s. Abb. 1.8). Zu bemerken ist, dass eine Reaktion, welche thermodynamisch nicht möglich ist, auch durch einen Katalysator nicht ausgelöst werden kann. Der Katalysator beeinflusst nur die Geschwindigkeit mit der sich ein chemisches Gleichgewicht einstellt. Die Erklärung liegt darin, dass die Aktivierungsenergie der Reaktion in Gegenwart des Katalysators (K) kleiner ist als die der unkatalysierten Reaktion (s. Abb. 1.8).
+ Kat. -Kat. A + B KA KAB AB
Abb. 1.8 Energieprofile bei einer Reaktion mit und ohne Katalysator (Quadbeck-Seeger, 1998).
Reaktionsweg
Energie
Energie-barriere
A + B + K
AB + KKA
E
E
A
A, Kat.
Reaktions-wärme
ohne Katalysator
mit Katalysator
KAB

1. Zeolithoberfläche und Reaktivität 25 Der Unterschied zwischen einem homogenen und einem heterogenen Katalysator besteht darin, dass der homogene Katalysator der gleichen Phase angehört wie das Reaktionssystem, während der heterogene Katalysator und die Reaktanden (Flüssigkeiten oder Gase) sich einander berühren, jedoch verschiedene Phasen sind. Von grosser Wichtigkeit ist die heterogene Katalyse. Diese zeichnet sich durch die Reaktionen, die an der Oberfläche des Katalysators stattfinden, aus. Dabei werden Edukte bei der Adsorption an der Katalysatoroberfläche in einen aktiven Zustand versetzt. Die meisten technischen Katalysatoren sind Misch- oder Mehrstoffkatalysatoren, d. h. neben der kataytisch wirksamen Substanz enthalten sie noch weitere Zusätze (Promotoren). Diese Zusätze können auf einer katalytisch wirksame Schicht aufgebracht werden und dadurch als Trägerkatalysatoren dienen.
Abb. 1.9 Ortep-Darstellung der externen Dodecasil 3C-Oberfläche mit kleineren [512]- und grösseren [51264]- halbkäfigförmigen Hohlräumen.

1. Zeolithoberfläche und Reaktivität 26
Zu den wichtigsten Eigenschaften eines technischen Katalysators zählen, im zeitlichen Verlauf, neben der Aktivität und der Selektivität, die Form, die chemische und mechanische Stabilität. Dafür gibt es eine Vielzahl von wichtigen Faktoren, über die sich die Eigenschaften eines heterogenen Katalysators beeinflussen lassen. Als wichtige Einflussgrössen haben sich erwiesen: • die Stabilität der aktiven Phase; • die chemische Zusammensetzung des Festkörpers mit den katalytsich aktiven • die Oberflächenzusammensetzung; • die Porenstruktur des Katalysators; • der Stoff- und Wärmetransport an einem Katalysator. Zu bemerken ist, dass bei Gas/Festkörper Reaktionen besonders die Porosität und damit auch die Oberflächen eine entscheidende Rolle spielen. Auch ein geometrischer Faktor ist von Bedeutung da ein Molekül, um leicht aktiviert zu werden, sich in geeigneter Weise an die Katalysatoroberfläche �anpassen� muss. Das Ergebnis ist eine erhöhte Aktivität. Von den vielen Einflussgrössen gewann die Oberflächenzusammensetzung in den letzten Jahren immer mehr an Bedeutung. Eine definierte chemische Zusammensetzung der aktiven Oberfläche, und zwar nicht nur der äusseren, sondern auch der inneren, an den tief in den Festkörper hineinreichenden Poren, ist für die Funktionsfähigkeit des aktiven Zentrums sehr wichtig.

2. Ziel der Arbeit 27
2. Ziel der Arbeit Es ist schon längst bekannt, dass mikroporöse Tektosilikate, zu der Kategorie auch die Clathrasile gehören, in der heterogenen Katalyse als formselektive Katalysatoren eingesetzt werden können. Diese Eigenschaft ist auf die selektive Zugänglichkeit des Porensystems und die Einschränkungen im Reaktionsvolumen des Zeolithen zurückzuführen. Die Analyse der Wachstumsflächen von Zeolithkristallmodellen lässt erkennen, dass die äussere Oberfläche Kavitäten aufweist, die durchaus einen Beitrag zu der Formselektivität liefern könnten. Das erste Ziel der vorliegenden Arbeit war es, eine perfekt kristalline D3C-Probe herzustellen, die eine Kristallitgrösse von 10-100 nm aufweisen sollte. Es ist bekannt, dass die mittlere Korngrösse des Clathrasils D3C (Schlenker et al., 1981) normalerweise 100-800 µm beträgt. Durch die Züchtung von D3C-Kristallen im nm-Bereich würde das Verhältnis Oberfläche/Volumen 100 bis 1000 fach vergrössert, um mit Hilfe der Kernresonanzspektroskopie die äussere Oberfläche �sichtbar� zu machen. Die Popularität der Festkörper NMR-Spektroskopie und insbesondere der Kreuzpolarisationstechnik CP MAS ist in den letzten Jahren wegen der Informationen, die über die lokale Struktur, die Ordnung und die Dipol-Dipol Wechselwirkungen zu bekommen sind, erheblich gestiegen. Der Vorteil des Kreuzpolarisationsexperimentes, im englischen auch cross-polarisation genannt, liegt darin, dass die Probleme, die durch die niedrige Sensitivität der so genannten �seltenen Kerne� entstehen können, durch die Verstärkung der Empfindlichkeit beseitigt werden. Gleichzeitig findet ein Magnetisierungstransfer von einem Kern mit einer grossen natürlichen Häufigkeit, wie z. B. 1H, 19F, auf einen Kern mit einer geringeren, wie z. B. 13C, 15N, 29Si, statt. In Anbetracht dessen, dass die Strukturuntersuchungen mit Beugungsmethoden keinen Aufschluss über die Beschaffenheit der Oberfläche liefern können und die Kraftfeldmikroskopie keine überzeugenden experimentellen Ergebnisse in der Oberflächenabbildung erbringen konnte, wurde überlegt, die NMR-Spektroskopie dafür einzusetzen. Man sollte jedoch nicht vergessen, dass die Kernresonanzspektroskopie als eine nicht-oberflächensensitive Untersuchungsmethode bekannt ist.
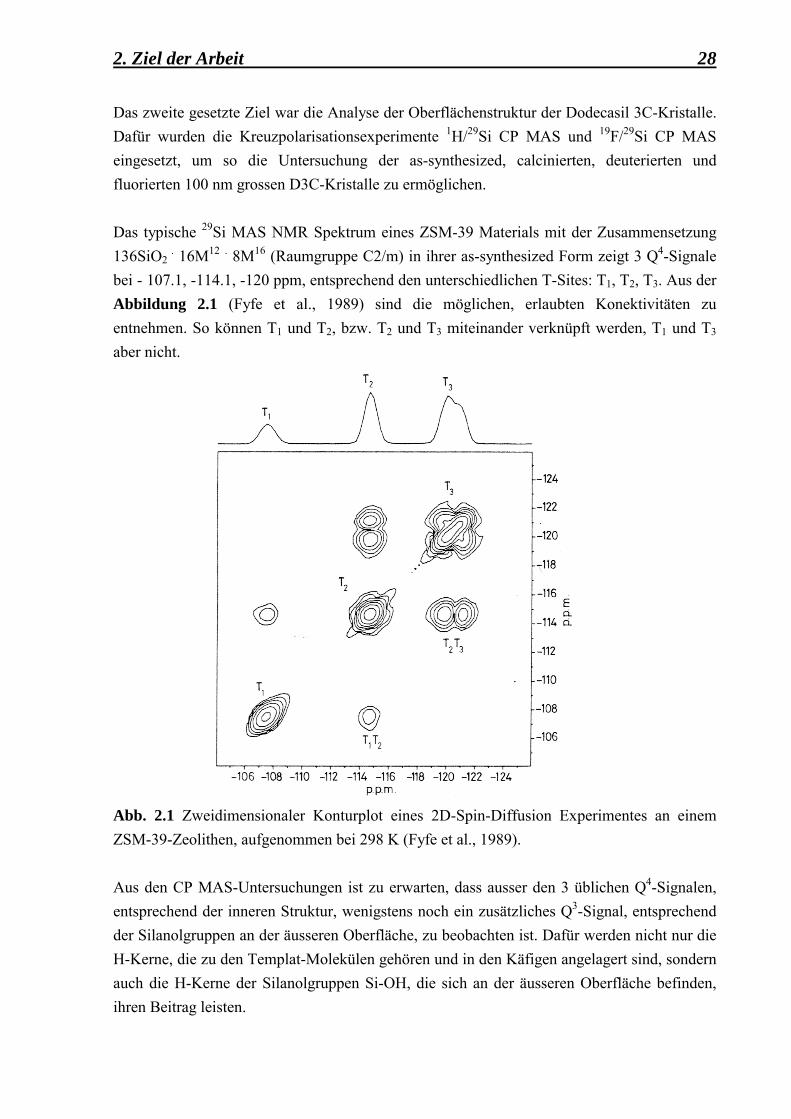
2. Ziel der Arbeit 28 Das zweite gesetzte Ziel war die Analyse der Oberflächenstruktur der Dodecasil 3C-Kristalle. Dafür wurden die Kreuzpolarisationsexperimente 1H/29Si CP MAS und 19F/29Si CP MAS eingesetzt, um so die Untersuchung der as-synthesized, calcinierten, deuterierten und fluorierten 100 nm grossen D3C-Kristalle zu ermöglichen. Das typische 29Si MAS NMR Spektrum eines ZSM-39 Materials mit der Zusammensetzung 136SiO2
. 16M12 . 8M16 (Raumgruppe C2/m) in ihrer as-synthesized Form zeigt 3 Q4-Signale bei - 107.1, -114.1, -120 ppm, entsprechend den unterschiedlichen T-Sites: T1, T2, T3. Aus der Abbildung 2.1 (Fyfe et al., 1989) sind die möglichen, erlaubten Konektivitäten zu entnehmen. So können T1 und T2, bzw. T2 und T3 miteinander verknüpft werden, T1 und T3 aber nicht.
Abb. 2.1 Zweidimensionaler Konturplot eines 2D-Spin-Diffusion Experimentes an einem ZSM-39-Zeolithen, aufgenommen bei 298 K (Fyfe et al., 1989).
Aus den CP MAS-Untersuchungen ist zu erwarten, dass ausser den 3 üblichen Q4-Signalen, entsprechend der inneren Struktur, wenigstens noch ein zusätzliches Q3-Signal, entsprechend der Silanolgruppen an der äusseren Oberfläche, zu beobachten ist. Dafür werden nicht nur die H-Kerne, die zu den Templat-Molekülen gehören und in den Käfigen angelagert sind, sondern auch die H-Kerne der Silanolgruppen Si-OH, die sich an der äusseren Oberfläche befinden, ihren Beitrag leisten.

3. Synthese und angewandte Methoden 29
3. Synthese und angewandte Methoden
3.1 Allgemeines über die Synthesedurchführung
Es ist seit langem bekannt, dass Kristallwachstum und Keimbildung zwei wichtige Prozesse sind, die die Kristallzüchtung positiv bzw. negativ beeinflussen können. Der Wachstumsprozess basiert im Wesentlichen auf der Anlagerung von atomaren bzw. molekularen Wachstumseinheiten (Bausteinen) an der Oberfläche eines Kristalls (Wilke und Bohm, 1988). Der Keimbildungsprozess beruht in erster Linie, nach der Überschreitung des Gleichgewichts, auf der Entstehung einer Ausgangsphase, die sich in einem metastabilen Zustand der Unterkühlung bzw. Übersättigung befindet. Dem folgt eine spontane Bildung von submikroskopischen Kristallpartikeln, die als Keime der neuen Phase betrachtet werden können. In manchen Fällen erfolgt die Kristallisation sogar durch Fremdpartikel, Staub, Erschütterungen oder Ladungen. Sie kann gleichzeitig an der Gefässwand oder an der Oberfläche ausgelöst werden, so dass nie die Gewissheit besteht, ob in den einzelnen Fällen eine wirklich spontane, homogene Keimbildung beobachtet werden kann. Bei der Untersuchung der Kinetik der Keimbildung ist zu beobachten, dass sich aus einer übersättigten Phase nicht unbedingt die stabilste Phase mit der geringsten freien Enthalpie ∆GK
* abscheidet, sondern zuerst die Phase mit der schnellsten Bildungskinetik auftritt. Diese wird meistens durch die Keimbildungsgeschwindigkeit bestimmt, die ihrerseits eine negative Änderung der freien Enthalpie der Keimbildung ∆GK
* < 0 vorweist. Für die Züchtung kleiner Zeolithkristalle ist es erforderlich, die Keimbildung zu stimulieren und das Wachstum der Kristalle zu unterdrücken. Dies wird durch die Optimierung der Syntheseparameter: ► Temperatur, ► chemische Zusammensetzung,

3. Synthese und angewandte Methoden 30
► Konzentration der Si-Quelle, ► Druck, ► pH-Wert, ► Synthesedauer, ► Arbeitsweise (statisch, rotiert, gerührt), erreicht. Verknüpft mit der geringen Grösse der Partikel, die unter bestimmten Bedingungen gezüchtet werden, ist eine sehr grosse spezifische Oberfläche. So werden z. B. kugelförmige Kristalle, die eine Korngrösse von 100 nm vorweisen, eine äussere spezifische Oberfläche von ungefähr 50 m2/cm3 besitzen. Je kleiner die Kristalle, desto grösser wird die äussere Oberfläche sein. Der Anteil der äusseren Oberfläche an der Gesamtoberfläche, wozu die innere Oberfläche zählt, wird dadurch vergrössert. Die für die Zeolithsynthese am meisten benutzte Kristallzuchtmethode ist die s.g. Hydrothermalsynthese. Die Kristallisation erfolgt in diesem Fall aus einer wässrigen Lösung bei relativ hohen Temperaturen (130-250 °C) und unter autogenen Drücken (~25 bar). Als Apparatur zur hydrothermalen Kristallzüchtung wird ein Stahlautoklav, der nach dem Laudise- und Nielsen-Modell gebaut worden ist, benutzt (Wilke und Bohm, 1988). Dieser ist mit einem Teflonbehälter versehen, der ein Volumen von 30 ml fasst und in den die Syntheselösung eingebracht wird. Viele der Zeolithe werden aus Kieselgellösungen in Anwesenheit von Gastmolekülen, die als Template dienen, synthetisiert. Die in dieser Arbeit verwendete Methode für die Polykondensation ist die der alkalinen Zeolithsynthese OH- - Ionen, wobei mono- bzw. polyzyklische Amine, wie z. B. Piperidin, als OH- - Quelle dienen. Bei der Zeolithsynthese spielen die organischen Gastmoleküle drei wichtige Rollen: 1. sie füllen die Kavitäten der porösen Struktur, was zu einer Erhöhung der Stabilität des
Materials führt. 2. sie liefern ein Bild der Grösse und der Form des Porensystems der gebildeten
Gerüststrukturen. Kugelförmige Template bilden bevorzugt käfigartige Kavitäten, während kettenförmige Template überwiegend zum Aufbau von Kanalsysteme führen.
3. sie beeinflussen die Endstruktur des Zeolithes.

3. Synthese und angewandte Methoden 31
3.2 Versuchsreihe I [SiO2:M(C5H11N):H2O = 1:1,5:55,5]
3.2.1 Syntheseplanung
Der erste Schritt der Arbeit bestand darin, die Synthese der rein Si-haltigen ZSM-39 Kristalle (Dodecasil 3C) mit einer mittleren Korngrösse zwischen 50 und 800 µm zu reproduzieren. Als Grundlage für die Synthesevorschriften, die zu der Herstellung der gewünschten Clathrasile führten, dienten die früheren Arbeiten von Gies (Gies, 1987). Eine ausführliche Angabe über die Syntheseparameter, Synthesespezies und Synthesebedingungen kann aus der Tabelle 3.1 entnommen werden. Der Synthesevorgang wurde wie folgt durchgeführt: Das Ausgangsgel bestand aus einer Si-Quelle, in diesem Fall Aerosil, einem Templat (Piperidin) und destilliertem Wasser in einem Molverhältnis von 1:1,5:55,5. Piperidin, eine farblose, Fisch- bis Ammoniak-artig riechende Flüssigkeit ist mit Wasser mischbar und kann in Alkohol, Ether, Benzol oder Chloroform gelöst werden (Roempp, 1995). Aerosil, eine hochdisperse �pyrogene� Kieselsäure von über 99,8% SiO2-Gehalt, wird durch Hydrolyse von Siliciumtetrachlorid in einer Knallgasflamme (2H2 + O2 + SiCl4 → SiO2 + 4HCl, �Flammenhydrolyse“) hergestellt. Aerosil ist aus amorphen, kugelförmigen Teilchen aufgebaut, die einen Durchmesser von 10-20 nm vorweisen. Die Oberfläche von 1 g Aerosil, mit einem Volumen von ca. 15 ml, beträgt zwischen 100-400 m2. Die Aerosil-Teilchen besitzen auf ihrer Oberfläche Silanol (Si-OH) - Gruppen, die über relativ schwache Wasserstoff-Brückenbindungen miteinander verknüpft sind, wodurch es zur Gerüstbildung kommt (Roempp, 1995).
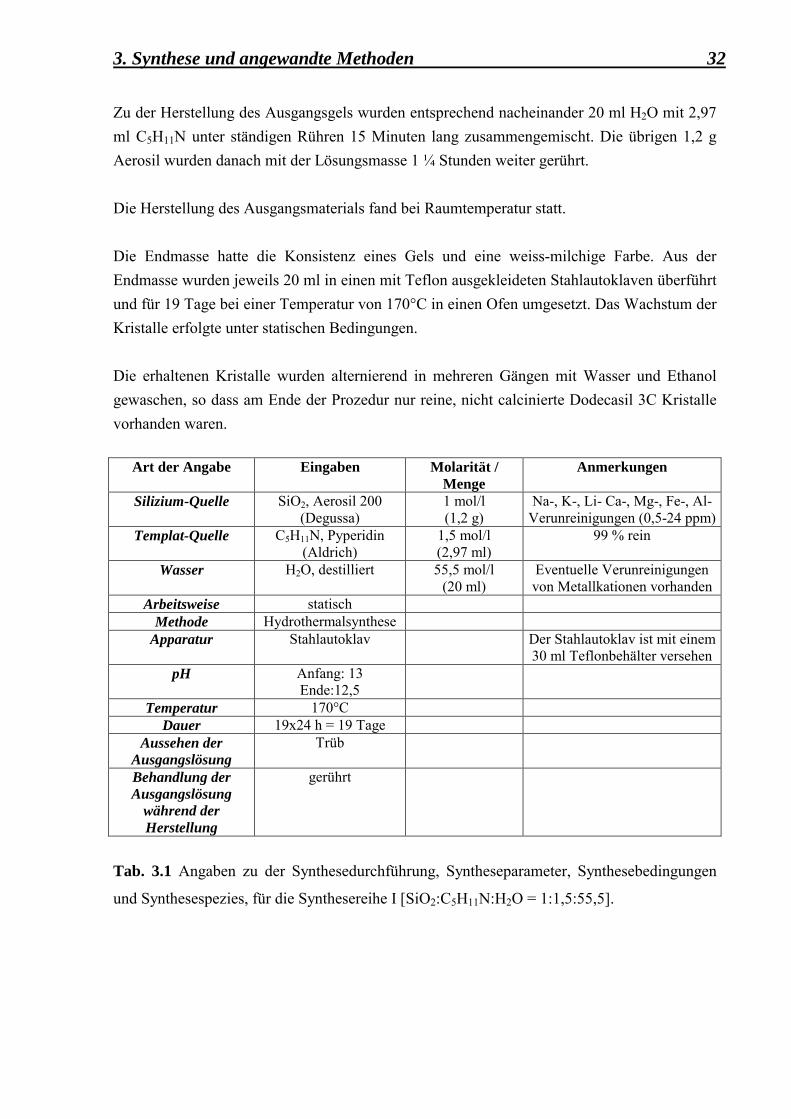
3. Synthese und angewandte Methoden 32 Zu der Herstellung des Ausgangsgels wurden entsprechend nacheinander 20 ml H2O mit 2,97 ml C5H11N unter ständigen Rühren 15 Minuten lang zusammengemischt. Die übrigen 1,2 g Aerosil wurden danach mit der Lösungsmasse 1 ¼ Stunden weiter gerührt. Die Herstellung des Ausgangsmaterials fand bei Raumtemperatur statt. Die Endmasse hatte die Konsistenz eines Gels und eine weiss-milchige Farbe. Aus der Endmasse wurden jeweils 20 ml in einen mit Teflon ausgekleideten Stahlautoklaven überführt und für 19 Tage bei einer Temperatur von 170°C in einen Ofen umgesetzt. Das Wachstum der Kristalle erfolgte unter statischen Bedingungen. Die erhaltenen Kristalle wurden alternierend in mehreren Gängen mit Wasser und Ethanol gewaschen, so dass am Ende der Prozedur nur reine, nicht calcinierte Dodecasil 3C Kristalle vorhanden waren.
Art der Angabe Eingaben Molarität / Menge
Anmerkungen
Silizium-Quelle SiO2, Aerosil 200 (Degussa)
1 mol/l (1,2 g)
Na-, K-, Li- Ca-, Mg-, Fe-, Al-Verunreinigungen (0,5-24 ppm)
Templat-Quelle C5H11N, Pyperidin (Aldrich)
1,5 mol/l (2,97 ml)
99 % rein
Wasser H2O, destilliert 55,5 mol/l (20 ml)
Eventuelle Verunreinigungen von Metallkationen vorhanden
Arbeitsweise statisch Methode Hydrothermalsynthese
Apparatur Stahlautoklav
Der Stahlautoklav ist mit einem 30 ml Teflonbehälter versehen
pH Anfang: 13 Ende:12,5
Temperatur 170°C Dauer 19x24 h = 19 Tage
Aussehen der Ausgangslösung
Trüb
Behandlung der Ausgangslösung
während der Herstellung
gerührt
Tab. 3.1 Angaben zu der Synthesedurchführung, Syntheseparameter, Synthesebedingungen
und Synthesespezies, für die Synthesereihe I [SiO2:C5H11N:H2O = 1:1,5:55,5].
3. Synthese und angewandte Methoden 33
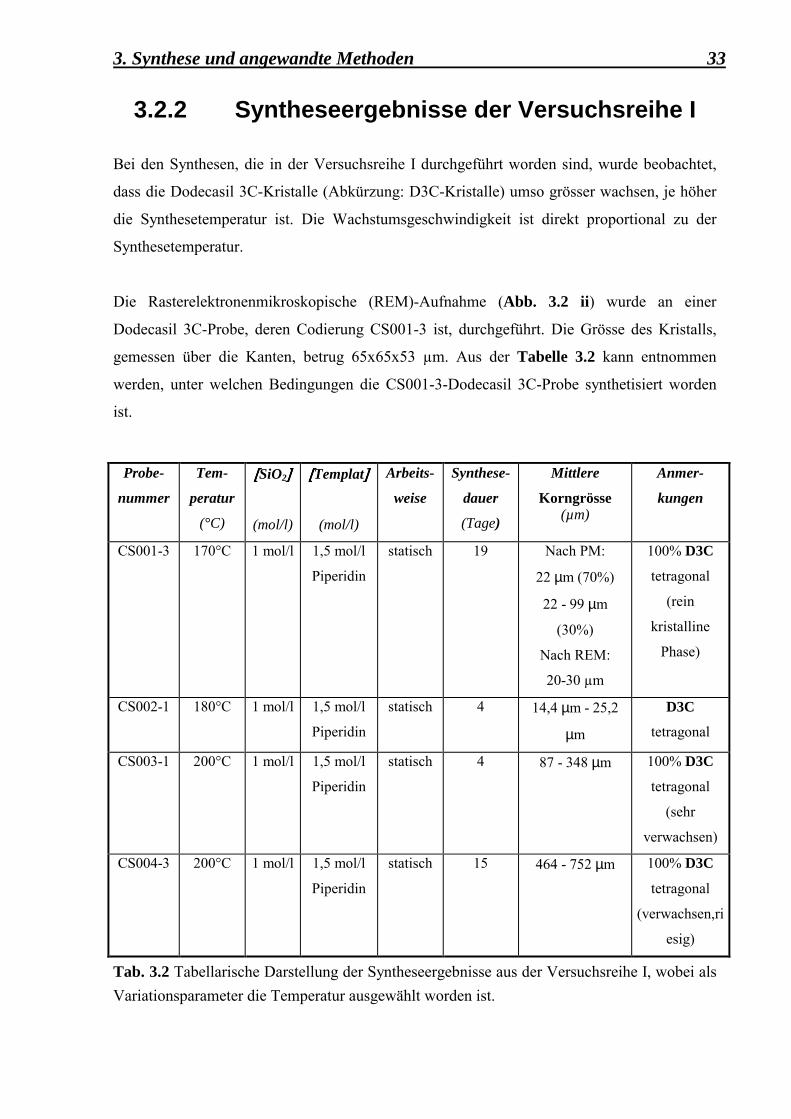
3.2.2 Syntheseergebnisse der Versuchsreihe I Bei den Synthesen, die in der Versuchsreihe I durchgeführt worden sind, wurde beobachtet,
dass die Dodecasil 3C-Kristalle (Abkürzung: D3C-Kristalle) umso grösser wachsen, je höher
die Synthesetemperatur ist. Die Wachstumsgeschwindigkeit ist direkt proportional zu der
Synthesetemperatur.
Die Rasterelektronenmikroskopische (REM)-Aufnahme (Abb. 3.2 ii) wurde an einer
Dodecasil 3C-Probe, deren Codierung CS001-3 ist, durchgeführt. Die Grösse des Kristalls,
gemessen über die Kanten, betrug 65x65x53 µm. Aus der Tabelle 3.2 kann entnommen
werden, unter welchen Bedingungen die CS001-3-Dodecasil 3C-Probe synthetisiert worden
ist.
Probe-
nummer
Tem-
peratur
(°C)
[[[[SiO2]]]]
(mol/l)
[[[[Templat]]]]
(mol/l)
Arbeits-
weise
Synthese-
dauer
(Tage)
Mittlere
Korngrösse (µm)
Anmer-
kungen
CS001-3 170°C 1 mol/l 1,5 mol/l
Piperidin
statisch 19 Nach PM:
22 µm (70%)
22 - 99 µm
(30%)
Nach REM:
20-30 µm
100% D3C
tetragonal
(rein
kristalline
Phase)
CS002-1 180°C 1 mol/l 1,5 mol/l
Piperidin
statisch 4 14,4 µm - 25,2
µm
D3C
tetragonal
CS003-1 200°C 1 mol/l 1,5 mol/l
Piperidin
statisch 4 87 - 348 µm
100% D3C
tetragonal
(sehr
verwachsen)
CS004-3 200°C 1 mol/l 1,5 mol/l
Piperidin
statisch 15 464 - 752 µm 100% D3C
tetragonal
(verwachsen,ri
esig)
Tab. 3.2 Tabellarische Darstellung der Syntheseergebnisse aus der Versuchsreihe I, wobei als Variationsparameter die Temperatur ausgewählt worden ist.
3. Synthese und angewandte Methoden 34
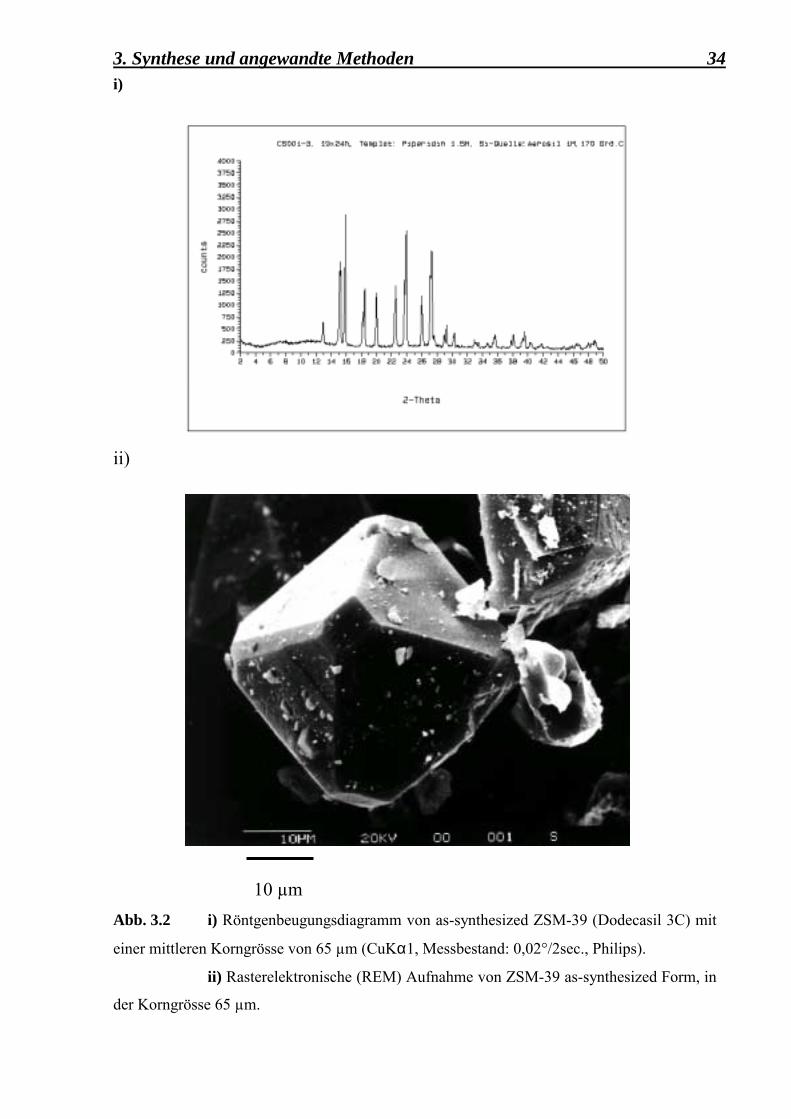
i)ii)
10 µm Abb. 3.2 i) Röntgenbeugungsdiagramm von as-synthesized ZSM-39 (Dodecasil 3C) mit
einer mittleren Korngrösse von 65 µm (CuKα1, Messbestand: 0,02°/2sec., Philips).
ii) Rasterelektronische (REM) Aufnahme von ZSM-39 as-synthesized Form, in
der Korngrösse 65 µm.

3. Synthese und angewandte Methoden 35
Das Röntgenbeugungsdiagramm (Abb. 3.2 i) zeigt, dass keine andere Phase ausser Dodecasil 3C vorhanden ist. Ausserdem wurde durch die Indizierung der Reflexe bemerkt, dass die Kristalle eine tetragonale Symmetrie aufweisen und nicht in der üblichen, für das System charakteristischen kubischen Symmetrie auftreten. In dem 2θ-Bereich zwischen 5° und 12° sind zwei breite Reflexe zu beobachten, Reflexe die auf das Röntgendiffraktometer zurückzuführen sind und die bei jeder Messung in dem gegebenen Bereich auftreten, unabhängig davon, welche Substanz für die Untersuchung bereit gestellt wurde. Die CS001-3-Dodecasil 3C-Probe wurde weiterhin für die NMR-Untersuchungen, die ausführlicher im Kapitel 4 beschrieben werden, verwendet. Sie wurde unterschiedlichen Experimenten unterzogen und diente als Vergleich zu der feinkristallinen, mikroporösen, 0,1 µm kleinen Dodecasil 3C-Substanz.
3.3 Versuchsreihe II
[SiO2:M(C5H11N):H2O = 1:4:55,5]
3.3.1 Syntheseplanung und Synthesedurchführung
3.3.1.1 Variation der Syntheseparameter: Synthesetemperatur, chemische Zusammensetzung,
Arbeitsweise, Synthesedauer, Füllhöhe
Von grosser Bedeutung für die vorliegende Arbeit war die Züchtung von Dodecasil 3C-Kristallen, die eine mittlere Korngrösse zwischen 10 und 200 nm vorweisen. Die Ausgangsbedingungen waren die der Versuchsreihe I.

3. Synthese und angewandte Methoden 36
Der erste Parameter, der optimiert werden musste, war die Synthesetemperatur. Nach
mehreren Versuchen wurde beobachtet, dass eine Synthesetemperatur von 160°C im
gegebenen Fall die optimale Temperatur ist, um die Dodecasil 3C-Kristalle in der
gewünschten Kristallitgrösse herzustellen (Tab. 3.3-Anhang). Gleichermassen wurde es
vermieden, die Synthesetemperatur unter 160°C einzustellen, da in dem Bereich zwischen
110°C und 160°C auch andere, fremde Phasen, wie z. B. Schichtsilikate, auftreten.
Die Konzentration der Silicium-Quelle wurde so hoch wie möglich gewählt, um dadurch das
Wachstum der Dodecasil 3C-Kristalle zu unterdrücken. Eine Aerosil-Konzentration von
4 mol/l erwies sich als ideal. Über die Grenze von 4 mol/l hinaus wurde beobachtet, dass
keine Dodecasil 3C-Kristalle mehr wachsen (Tab. 3.3-Anhang).
Durch die Auswahl anderer Silicium-Quellen bzw. Template wurden unbekannte Phasen
hergestellt, wie aus der Tabelle 3.5-Anhang zu entnehmen ist.
Die Arbeitsweise war gleichermassen ein wichtiger Faktor, der bei der Synthese von kleinen
Dodecasil-3C-Kristallen eine entscheidende Rolle gespielt hat. Gegen alle Erwartungen
wurde festgestellt, dass unter ständigem Rühren (während der Synthese) die Kristalle viel
grösser wachsen als unter statischen Bedingungen (Vergleich Tab. 3.3-Anhang und Tab.
3.5-Anhang). Ein ähnliches Verhalten des Kristallwachstums wurde auch im Falle einer
Rotation des Autoklaven, der die Syntheselösung enthielt, beobachtet.
Unter statischen Bedingungen wurden Kristalle mit der kleinsten Korngrösse (φ~0,1 µm)
synthetisiert (Probe CS024).
Die Synthesedauer wurde auf drei Tage reduziert. Zwei Tage Synthesezeit erwiesen sich als
unzureichend, um eine perfekt kristalline Probe zu erhalten. Unter drei Tagen Synthesezeit
war der Hauptteil der erhaltenen Proben amorph (s. Tab. 3.6-Anhang).
Die Füllhöhe war, neben den o. g. vier Parametern (Synthesetemperatur, chemische
Zusammensetzung, Arbeitsweise, Synthesedauer), der entscheidende Faktor, der dazu geführt
hat, dass am Ende der Synthese die hergestellten Kristalle eine mittlere Korngrösse von ca.
0,1 µm betrug. So wurde beobachtet, dass durch die partielle Füllung des Teflonbehälters (1/3

3. Synthese und angewandte Methoden 37
Menge des Ausgangsgels ~ 8 ml), die erhaltenen Kristalle die gewünschte Korngrösse
zeigten (Probe CS024as).
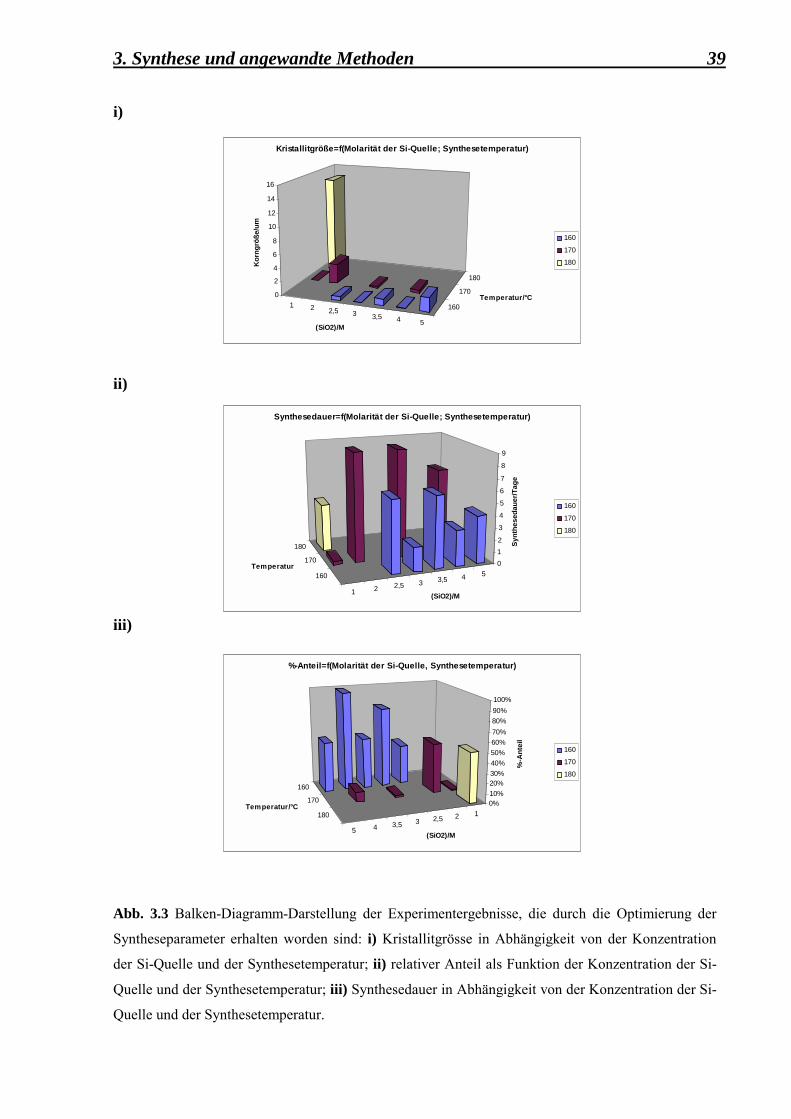
3.3.2 Syntheseergebnisse der Versuchsreihe II
Die Probe mit dem Codenamen CS024as wurde für die weiteren Untersuchungen ausgewählt,
da sie in der Korngrösse und der Kristallinität den gewünschten Bedingungen entsprach. Dies
ist sehr deutlich in der Abbildung 3.3 i), ii), iii) zu beobachten.
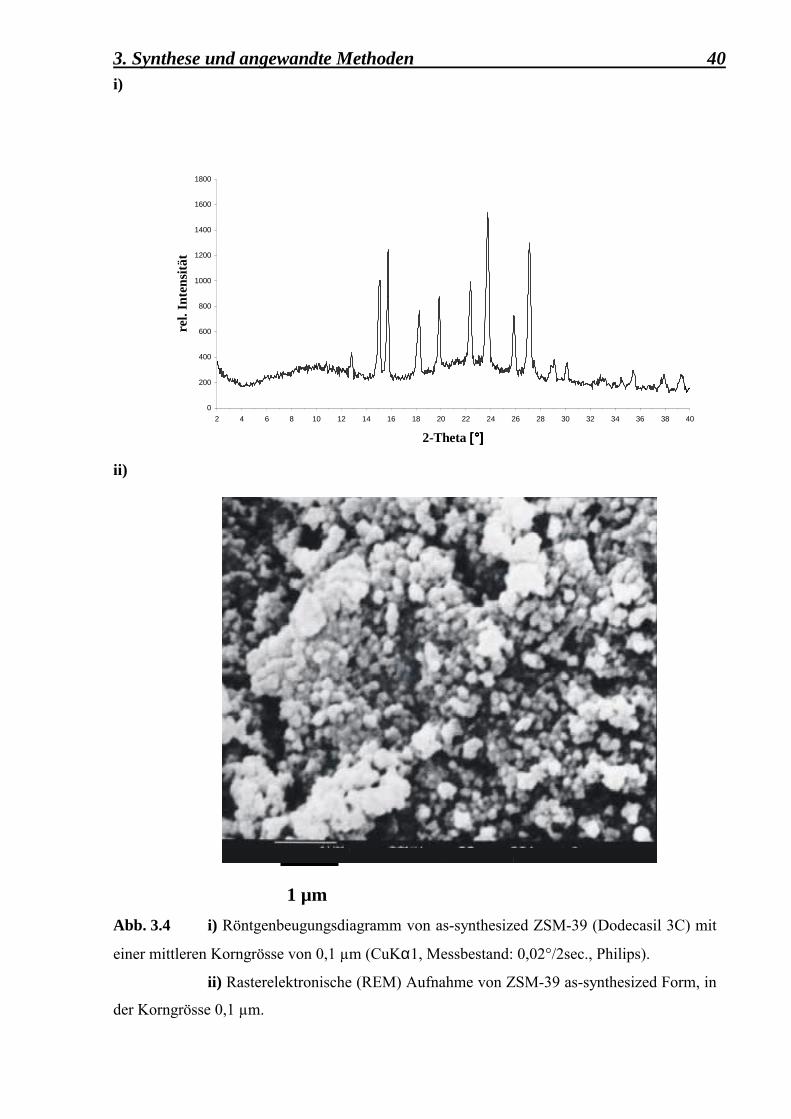
Aus der Abb. 3.4 ii) kann die mittlere Korngrösse des Dodecasil 3C-Kristalls auf 0,1 µm
geschätzt werden. Ausserdem zeigt das Pulverdiagramm (Abb. 3.4 i)) alle typischen Reflexe
des zu untersuchenden Clathrasils.
Die Synthese unter den gegebenen Bedingungen: 160°C-Synthestemperatur, 4 mol/l-Aerosil
und 1,5 mol/l Piperidin-Konzentration, 3 Tage-Synthesedauer, statische Arbeitsbedingungen
und eine Füllhöhe von 1/3 des Teflonbehälters, liess sich mehrmals reproduzieren. Die
erhaltenen Substanzen in ihrer as-synthesized Form zeigten identische Eigenschaften wie die
Dodecasil 3C- Probe CS024as.
Zusätzlich ist zu bemerken, dass das Vorheizen der Stahlautoklaven zu keinen befriedigenden
Ergebnissen geführt hat. Der Hauptteil der Ausbeute ist in den meisten Fällen amorph
gewesen, wie es aus den gemessenen, den Proben entsprechenden Pulverdiagrammen zu
entnehmen war.
Durch die Zugabe einer starken Base, wie z. B. NaOH (0,1 mol/l), wachsen die Kristalle sehr
gross (s. Tab. 3.6 Anhang, Probe: CS065). Ein ähnliches Verhalten ist sowohl im Falle eines
HF-Zuschusses (s. Tab. 3.5-Anhang, Probe: CS048), als auch durch Zugeben von
Tetramethylammoniumhydroxid TMAOH (s. Tab. 3.5-Anhang, Probe: CS049) zu
beobachten.

3. Synthese und angewandte Methoden 38
Eine Vorbehandlung der Syntheselösung, während des Rührens des Ausgangsmaterials,
scheint das Wachstum der Kristalle zu fördern (s. Tab. 3.6-Anhang, Proben: CS061 und
CS063).
Die hohen Autoklaven sind mit Teflonbehältern, deren Füllmenge 100 ml beträgt und 3 mal
soviel an Ausgangsgel aufnehmen können, versehen. Es zeigte sich, wie es aus der Tabelle
3.3-Anhang zu entnehmen ist, dass die Kristalle in den hohen Autoklaven viel schneller
wachsen als in solchen mit 30 ml Volumen.
Eine geringere Füllhöhe als 1/3 des Teflonbehälters scheint zu sehr kleinen Kristallen zu
führen; der Hauptteil der Synthesemasse ist aber amorph und so von geringerer Bedeutung (s.
Tab. 3.6-Anhang, Probe: CS051).
An Proben, die eine Korngrösse über 1 µm vorweisen, wurden keine Röntgenaufnahmen
durchgeführt.
3. Synthese und angewandte Methoden 39
i)
1 2 2,5 3 3,5 4 5
160
170
180
0
2
4
6
8
10
12
14
16
Kor
ngrö
ße/u
m
(SiO2)/M
Temperatur/°C
Kristallitgröße=f(Molarität der Si-Quelle; Synthesetemperatur)
160
170
180
ii)
1 2 2,5 3 3,5 4 5160
170
180
012
3
4
5
6
7
8
9
Synt
hese
daue
r/Tag
e
(SiO2)/M
Temperatur
Synthesedauer=f(Molarität der Si-Quelle; Synthesetemperatur)
160
170
180
iii)
Abb. 3.3 Balken-Diagramm-Darstellung der Experimentergebnisse, die durch die Optimierung der
Syntheseparameter erhalten worden sind: i) Kristallitgrösse in Abhängigkeit von der Konzentration
der Si-Quelle und der Synthesetemperatur; ii) relativer Anteil als Funktion der Konzentration der Si-
Quelle und der Synthesetemperatur; iii) Synthesedauer in Abhängigkeit von der Konzentration der Si-
Quelle und der Synthesetemperatur.
122,533,545
160
170
1800%10%20%30%40%50%60%70%80%90%100%
%-A
ntei
l
(SiO2)/M
Temperatur/°C
%-Anteil=f(Molarität der Si-Quelle, Synthesetemperatur)
160
170
180
3. Synthese und angewandte Methoden 40
i)0
200
400
600
800
1000
1200
1400
1600
1800
2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 34 36 38 40
2-Theta [°][°][°][°]
rel.
Inte
nsitä
t
ii)
1 µm
Abb. 3.4 i) Röntgenbeugungsdiagramm von as-synthesized ZSM-39 (Dodecasil 3C) mit
einer mittleren Korngrösse von 0,1 µm (CuKα1, Messbestand: 0,02°/2sec., Philips).
ii) Rasterelektronische (REM) Aufnahme von ZSM-39 as-synthesized Form, in
der Korngrösse 0,1 µm.
3. Synthese und angewandte Methoden 41
3.4 Zusätzliche Behandlungen, die an der ZSM-39 mikrokristallinen Probe durchgeführt
worden sind
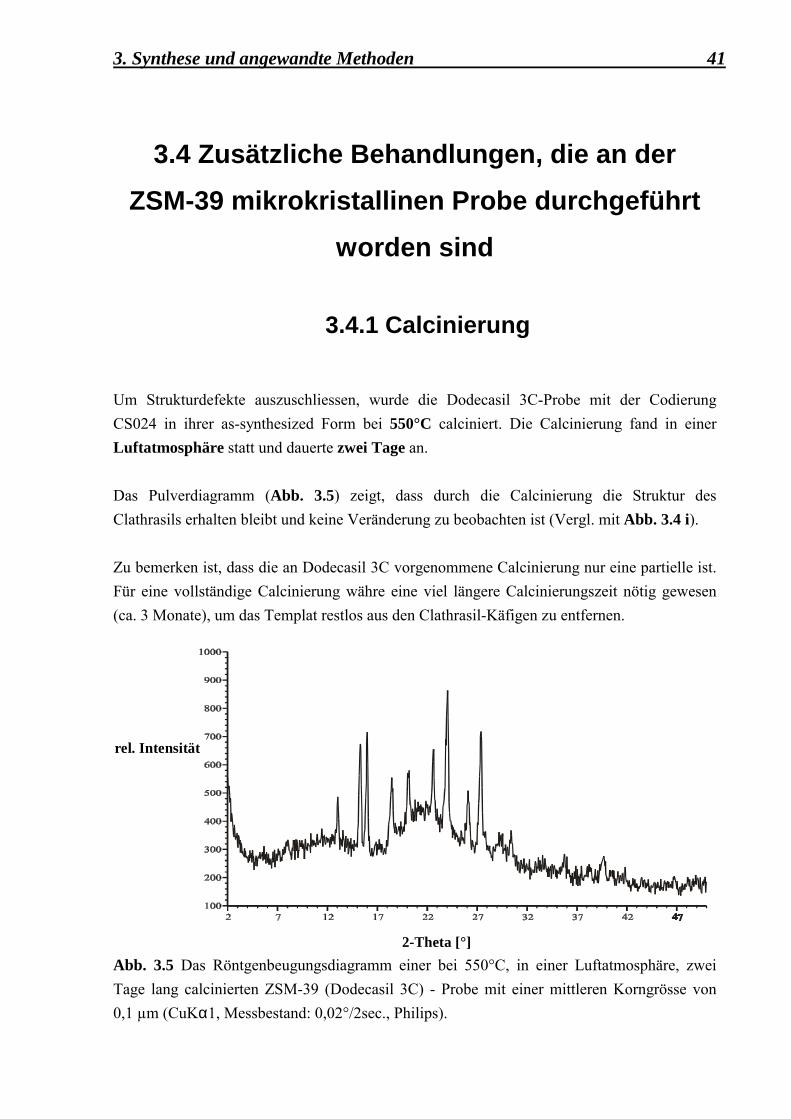
3.4.1 Calcinierung Um Strukturdefekte auszuschliessen, wurde die Dodecasil 3C-Probe mit der Codierung CS024 in ihrer as-synthesized Form bei 550°C calciniert. Die Calcinierung fand in einer Luftatmosphäre statt und dauerte zwei Tage an. Das Pulverdiagramm (Abb. 3.5) zeigt, dass durch die Calcinierung die Struktur des Clathrasils erhalten bleibt und keine Veränderung zu beobachten ist (Vergl. mit Abb. 3.4 i). Zu bemerken ist, dass die an Dodecasil 3C vorgenommene Calcinierung nur eine partielle ist. Für eine vollständige Calcinierung währe eine viel längere Calcinierungszeit nötig gewesen (ca. 3 Monate), um das Templat restlos aus den Clathrasil-Käfigen zu entfernen.
Abb. 3.5 Das Röntgenbeugungsdiagramm einer bei 550°C, in einer Luftatmosphäre, zwei Tage lang calcinierten ZSM-39 (Dodecasil 3C) - Probe mit einer mittleren Korngrösse von 0,1 µm (CuKα1, Messbestand: 0,02°/2sec., Philips).
rel. Intensität
2-Theta [°]
3. Synthese und angewandte Methoden 42
3.4.2 Behandlung mit einer starken Base (NaOH)
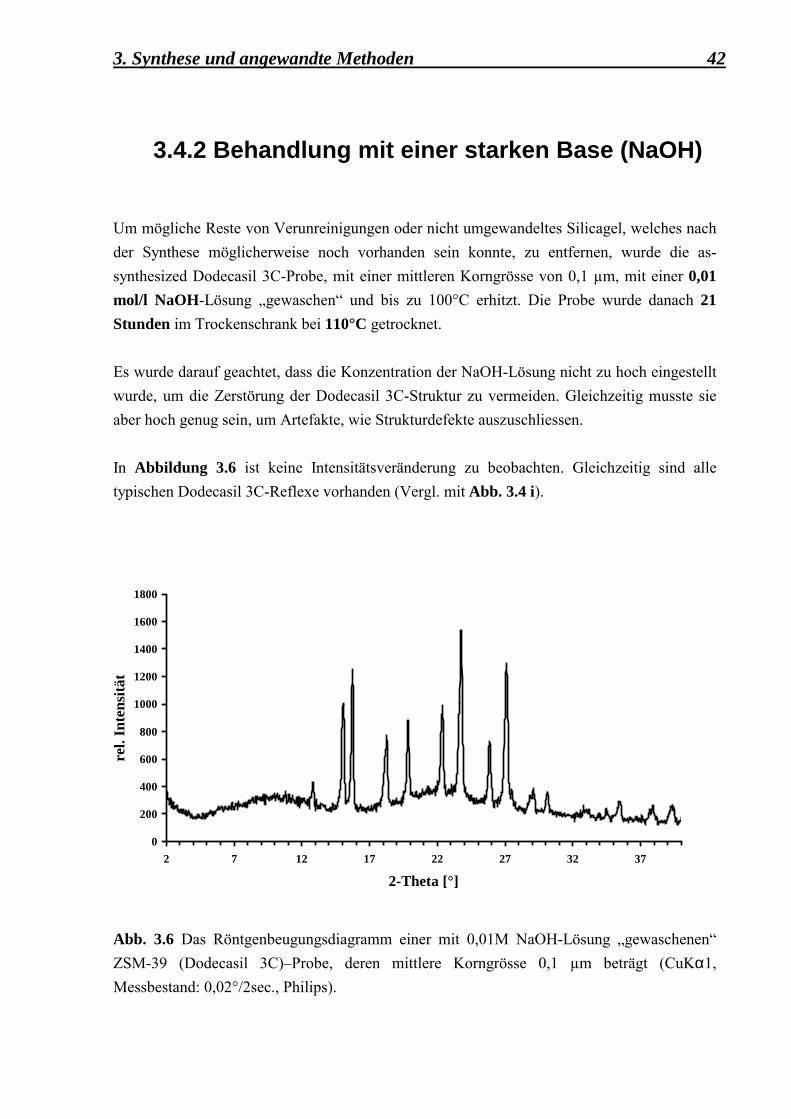
Um mögliche Reste von Verunreinigungen oder nicht umgewandeltes Silicagel, welches nach der Synthese möglicherweise noch vorhanden sein konnte, zu entfernen, wurde die as-synthesized Dodecasil 3C-Probe, mit einer mittleren Korngrösse von 0,1 µm, mit einer 0,01 mol/l NaOH-Lösung �gewaschen� und bis zu 100°C erhitzt. Die Probe wurde danach 21 Stunden im Trockenschrank bei 110°C getrocknet. Es wurde darauf geachtet, dass die Konzentration der NaOH-Lösung nicht zu hoch eingestellt wurde, um die Zerstörung der Dodecasil 3C-Struktur zu vermeiden. Gleichzeitig musste sie aber hoch genug sein, um Artefakte, wie Strukturdefekte auszuschliessen. In Abbildung 3.6 ist keine Intensitätsveränderung zu beobachten. Gleichzeitig sind alle typischen Dodecasil 3C-Reflexe vorhanden (Vergl. mit Abb. 3.4 i).
Abb. 3.6 Das Röntgenbeugungsdiagramm einer mit 0,01M NaOH-Lösung �gewaschenen� ZSM-39 (Dodecasil 3C)�Probe, deren mittlere Korngrösse 0,1 µm beträgt (CuKα1, Messbestand: 0,02°/2sec., Philips).
0
200
400
600
800
1000
1200
1400
1600
1800
2 7 12 17 22 27 32 37
2-Theta [°]
rel.
Inte
nsitä
t

3. Synthese und angewandte Methoden 43
3.4.3 Deuterierung Für die weiteren NMR-Untersuchungen wurde eine Dodecasil 3C-Probe mit einer mittleren Korngrösse von 0,1 µm benötigt, die einer Deuterierung unterzogen werden konnte. Als Ziel wurde gesetzt, die Protonen H+, gehörend zu den Oberflächensilanolgruppen Si-OH, durch Deuterium zu ersetzen. Die as-synthesized CS024-Probe wurde drei mal mit deuteriertem Wasser D2O imprägniert. Während der NMR-Messung wurde das deuterierte Dodecasil 3C die ganze Zeit feucht gehalten und sogar im Rotor mit D2O durchtränkt. Zwischen dem Röntgenbeugungsdiagramm der deuterierten ZSM-39 (Dodecasil 3C) - Probe mit einer mittleren Korngrösse von 0,1 µm (CuKα1, Messbestand: 0,02°/2sec., Philips PW1050/25) und dem Röntgenbeugungsdiagramm der as-synthesized-Form des Dodecasil 3C bestand kein Unterschied.
3.4.4 Fluorierung
Um eine Bestätigung der Richtigkeit der durchgeführten NMR-Untersuchungen zu bekommen, wurde die 0,1 µm grosse Dodecasil-Probe fluoriert. Durch die Fluorierung erhoffte man sich, die Hydroxyl-Gruppen, gehörend zu den externen Silanolgruppen Si-OH, und automatisch die dazu gehörenden H+ durch Fluor F- zu ersetzen, um SiF-Verbindungen zu erzeugen. Dadurch wurde ein Magnetisierungstransfer von einem Kern mit einer hohen natürlichen Häufigkeit F- auf einen benachbarten Si-Kern, der eine niedrige natürliche Häufigkeit aufweist, ermöglicht. So wurde bewiesen, dass die erhaltenen Signale nur von der Oberfläche stammen. Die Dodecasil 3C-Probe mit der Codierung CS024 as-synthesized, wurde bei 60°C mit einer 2mM Kaliumfluorid KF- und 0,1M Schwefelsäure H2SO4-Lösung zusammengemischt. Neben dem gewünschten Endprodukt HF, das als Fluorierungsagenz benutzt worden ist, entstand noch zusätzlich Kaliumsulfat K2SO4. Dieses Nebenprodukt führte aber zu keiner Veränderung der Struktur des Dodecasil 3C und beeinflusste durch nichts den Fluorierungsprozess.
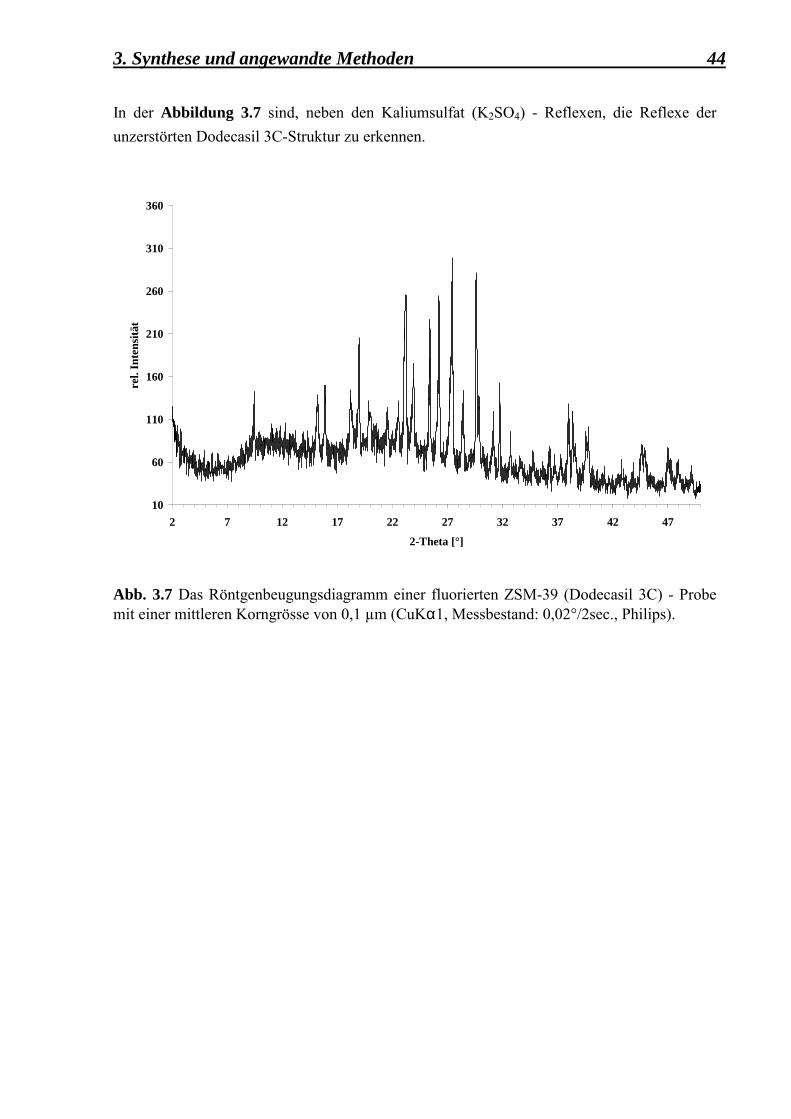
3. Synthese und angewandte Methoden 44 In der Abbildung 3.7 sind, neben den Kaliumsulfat (K2SO4) - Reflexen, die Reflexe der unzerstörten Dodecasil 3C-Struktur zu erkennen.
Abb. 3.7 Das Röntgenbeugungsdiagramm einer fluorierten ZSM-39 (Dodecasil 3C) - Probemit einer mittleren Korngrösse von 0,1 µm (CuKα1, Messbestand: 0,02°/2sec., Philips).10
60
110
160
210
260
310
360
2 7 12 17 22 27 32 37 42 47
2-Theta [°]
rel.
Inte
nsitä
t

3. Synthese und angewandte Methoden 45
3.5 Apparatives
3.5.1 Mikroskopische Methoden
3.5.1.1 Lichtmikroskopie Die synthetisierten kristallinen Phasen wurden mit einem Lichtmikroskop des Typs Stereomikroskop WILD M8 auf ihrer Reinheit, Kristallinität und ihrer Qualität untersucht. Gleichzeitig erlaubte das Gerät die Bestimmung der Kristallitgrösse bis zu 1 µm mit einer grosser Genauigkeit. Das dafür verwendete Objektiv Pol Planeofluar 63/0,90 zeigte unter 1 µm einen zu grossen Messfehler, so dass die Kristallitgrösse nur geschätzt werden konnte. Für eine genauere Bestimmung der Korngrösse wurden andere mikroskopische Methoden eingesetzt, wie z. B. die Rasterelektronenmikroskopie.
3.5.1.2 Rasterelelktronenmikroskopie Da für die weiteren Kernresonanzspektroskopischen Untersuchungen nur Kristallite mit einer mittleren Korngrösse unter 1 µm von Bedeutung waren, liessen sich diese sogenannten �feinkörnigen� Materialien mittels eines Rasterelektronenmikroskop (REM) auf ihre Grössen hin untersuchen. Ausserdem war die Kristallform für Kristalle grösser als 1 µm sehr gut zu erkennen. Die REM-Untersuchungen an den in dieser Arbeit synthetisierten kristallinen Phasen wurden an einem Rasterelektronenmikroskop des Typs Cambridge 250, MK 3D, am Institut für Geologie durchgeführt.

3. Synthese und angewandte Methoden 46
3.5.2 Röntgenographische Methoden
3.5.2.1 Pullverdiffraktometrie Für die Überprüfung der Qualität der synthetisierten Materialien und zugleich für die Identifizierung der Materialien wurden Röntgenaufnahmen mit Hilfe eines Pulverdiffraktometers PW 1050 der Firma Philips registriert. Das Pulverdiffraktometer wurde mittels eines Si-Standard-Präparates geeicht. Die Geräteparameter des Pulverdiffraktometers können aus der Tabelle 3.3 entnommen werden.
Nutzstrahlung Cu Kα mit der Wellenlänge λ = 1,5418 Å Heizstrohm 20 mA
Beschleunigungsspannung 40 kV Beugungsgeometrie Bragg-Brentano
Detektor Szintillationszähler Monochromatisierung Ni-Filter
Probenträger Kunstoffträger mit Quarzplättchen Messmodus Schrittweise Schrittweite 0,02
Zähler pro Schritt 2 Tab. 3.3 Tabellarische Auflistung der Geräteparameter eines Pulverdiffraktometers Philips des Typs PW 1050.

3. Synthese und angewandte Methoden 47
3.5.3 Spektroskopische Methoden Die NMR-Untersuchungen wurden mit Hilfe eines 400 MHz ASX Spektrometers der Firma Bruker durchgeführt. Für das Intervall zwischen 91 und 165 MHz wurde ein 4 mm Bruker (HP WB 73 A MAS 4 BL CP BB VTN) � Probenkopf benötigt, bei dem die Rotationsgeschwindigkeit, die erreicht werden konnte, 15 kHz betrug. Mit Hilfe dieser Art von Probenkopf wurden unter MAS-Bedingungen Kerne wie 1H gemessen. Die verwendeten Rotoren haben einen Durchmesser von 4 mm gehabt. Für das Intervall zwischen 40 und 120 MHz wurde ein 7 mm Bruker (HPWB 73 A MAS 7 BL CP BB VTN) � Probenkopf eingesetzt, bei dem die maximale Rotationsgeschwindigkeit 7 kHz betrug. Für die 29Si MAS-, 1H/29Si CP MAS- und 1H/29Si HPDEC NMR Messungen wurden 7 mm Rotoren verwendet. Die komplementären 19F MAS- und 19F/29Si CP MAS NMR Messungen wurden mit Hilfe eines 4 mm Bruker (HP WB 73 B MAS 4BL CP BB WVT) � Probenkopf durchgeführt. Für die Kontakzeitvariationsmessungen diente als Untersuchungsmethode die Kreuzpolarisationstechnik 1H/29Si CP MAS, wobei die Kontaktzeit zwischen 1 und 100 msec variiert worden ist.

3. Synthese und angewandte Methoden 48
3.6 Zusammenfassung der Synthesedurchführung
Die optimalen Synthesebedingungen für die Herstellung eines mikrokristallinen Clathrasils (Dodecasil 3C) mit einer mittleren Korngrösse von 0,1 µm sind: 4M Aerosil (als Silicium-Quelle), 1,5M Pyperidin (als Templat-Quelle), relativer Anteil : 0,00666%, Synthesedauer: 3 Tage, Synthesetemperatur: 160°C, Füllhöhe: 1/3 des Teflonbehälters. Durch die Veränderung der o. g. Syntheseparameter wurde die Oberfläche des Dodecasil 3C auf 50-100 m2/g erhöht, so dass dadurch die Bedingungen der Oberflächenuntersuchungen mit Hilfe der Kernresonanzspektroskopie erfüllt worden sind. Der Synthesedurchgang der Dodecasil 3C-Probe mit der Codierung CS024 wurde mehrmals wiederholt, und die Synthese lies sich reproduzieren (Probe CS134). Die zusätzlichen Behandlungen, die an der as-synthesized Form des Dodecasil 3C unternommen worden sind, wurden als Hilfe für den Ausschluss eventueller Artefakte eingesetzt. Durch das Waschen mit einer schwach konzentrierten NaOH-Lösung (0,01M) wurde ausgeschlossen, dass noch Reste von Silicagel oder nach der Synthese übrig gebliebenen nicht umgewandelten Templates, vorhanden sind. Die partielle Calcinierung ( 2 Tage bei 550°C, in einer oxidativen Atmosphäre) erlaubte es, Strukturdeffekte zu eliminieren und führte zugleich zu der Gewissheit, dass durch die NMR-Untersuchungen, die an der mikrokristallinen CS024 - Dodecasil 3C-Probe unternommen wurden, die externe Oberfläche und nicht die Strukturdefekte charakterisiert werden. Die Deuterierung war ein zusätzlicher Schritt in Richtung der Beschreibung der externen Dodecasil 3C-Oberfläche. Dadurch wurden Protonen an der externen Oberfläche, gehörend zu den Silanolgruppen Si-OH, durch Deuterium ersetzt, um nur ein Magnetisierungstransfer von den Templat (Pyperidin) � Protonen aus dem Inneren des Käfigs zu ermöglichen. Durch die Fluorierung wurde die externe Oberfläche des Clathrasils Dodecasil 3C mit Fluor bedeckt. Die Hydroxyl-Gruppen OH- wurden durch Fluorid-Gruppen F- ersetzt.

4. Festkörper NMR-Spektroskopie 49
4. Festkörper NMR-Spektroskopische Untersuchungen an externen Clathrasil-
Oberflächen
4.1 Einführung in die Theorie Die Kernresonanzspektroskopie, auch NMR genannt (aus dem englischen Nuclear Magnetic Resonanz), zählt zu den wichtigsten und aussagekräftigsten analytischen Methoden der modernen Chemie. Sie ist in einem weiten Temperaturbereich (zwischen �190°C und +300°C) anwendbar. Ausserdem wird das zu untersuchende Material durch die Messung nicht zerstört. Die Kernresonanzspektroskopie ist eine Untersuchungsmethode, die in einer Vielzahl von Bereichen angewendet wird, um Informationen über Details der Molekularstruktur und über Wechselwirkungen zwischen benachbarten Kernen zu bekommen, um Informationen über die Gast- bzw. Wirt-Moleküle zu erhalten sowie um den Verlauf einer Reaktion zu bestimmen und dadurch mehr über die Kinetik eines Prozesses zu erfahren. Die Kernresonanzspektroskopie hat als physikalische Grundlage den Kernmagnetismus. Das Prinzip basiert auf Wechselwirkungen zwischen magnetischen Kernmomenten einer Substanz und einem äusseren Magnetfeld B0. Dies führt zu Veränderung der Populationen der diskreten Energiezustände � den sog. Eigenzuständen -, die gemessen werden können. Jeder einzelne Kern, der ein magnetisches Moment besitzt, kann mit einer charakteristischen Radiofrequenz angeregt werden, so dass anschliessend die Relaxation von Energie als Spektrallinie - Resonanzlinie - registriert wird. Das Resultat ist ein NMR-Spektrum, bei dem die individuelle chemische Umgebung der Kerne eine charakteristische Verschiebung der Resonanzposition verursacht. Diese sogenannte chemische Verschiebung (δδδδ) wird immer in Bezug auf die Resonanzposition einer Standardsubstanz angegeben und lässt sich somit bequem in chemische Informationen umsetzen.

4. Festkörper NMR-Spektroskopie 50
In Festkörpern treten eine Reihe von lokalen Wechselwirkungen auf, wie zum Beispiel Dipol-Dipol-Wechselwirkungen, die Anisotropie der chemischen Verschiebung und Quadrupol-Wechselwirkungen, die von der Orientierung der Kristalle im äusseren Magnetfeld abhängen. Daraus resultieren für die in der Festkörper-NMR übliche Messung an Pulverproben starke Verbreiterungen der Resonanzsignale. Um diese Auswirkung zu eliminieren, wird die sogenannte MAS-Technik verwendet (MAS = magic angle spinning), bei der die Probe um den magischen Winkel von 54,74° in Bezug auf B0 schnell rotiert. Für die Untersuchung der Kristallstruktur von Festkörpern ist die am meisten verwendete Technik die Röntgenbeugung. Sie wird eingesetzt, um die periodische Ordnung, d.h. die Fernordnung, in Festkörpern zu untersuchen. Im Gegensatz dazu wird die Festkörper NMR-Spektroskopie angewendet, um Informationen über die unmittelbare Kern-Umgebung, d.h. die Nahordnung zu bekommen. Die Kernresonanzspektroskopie an Festkörpern stellt somit eine komplementäre Methode zu Röntgenanalytik dar und ermöglicht es, wichtige strukturelle Informationen über die untersuchten Substanzen zu gewinnen.
4.1.1 Das grundlegende Prinzip der NMR Jeder Kern mit einem von Null verschiedenen Eigendrehimpuls oder auch Spin besitzt ein magnetisches Moment µ, das durch einen konstanten Faktor γγγγ - das sog. gyromagnetische Verhältnis - proportional zu dem Kernspin I ist:
µ = γhI Ein typisches NMR-Experiment kann in 3 Schritten durchgeführt werden: i) erstens ist die Vorbereitung des Systems erforderlich. Unter dem Einfluss eines externen, konstanten magnetischen Feldes:
B = (0, 0, B0) werden die Zeeman-Energieniveaus entartet, so dass sich eine energetische Aufspaltung ergibt, wie es in der Abbildung 4.1 zu erkennen ist:
(4.1)
(4.2)

4. Festkörper NMR-Spektroskopie 51
Abb. 4.1 Schematische Darstellung der Aufspaltung des Zeeman-Energieniveaus eines Kerns mit halbzahligem Spin.
∆ ∆ ∆ ∆ E = h v L
m = - 1 / 2I
m = + 1 / 2I
E
B 00
Em = - γhmB0 mit m = -I, ..., +I.
Im thermischen Gleichgewicht stellt sich eine Gleichgewichtsmagnetisierung M0 ein:
wobei: N = Zahl der Spins / m3; k = Boltzmann-Konstante; T = Temperatur; µ0 = magnetische Konstante des Vakuums (4.10-7 VsA-1m-1). Zunächst ist die Bewegung eines Spins I, der sich in einem Magnetfeld B0 parallel zur z-Richtung eines Koordinatensystems befindet, zu beobachten (Abb. 4.2). Das magnetische Moment präzediert um das B0-Feld mit einer konstanten Kreisfrequenz, der sog. Larmorfrequenz,
ωL = -γB0 oder: ωL = 2πνL
,3 0
02
0 BkT
NM µµ ><=
(4.3)
(4.4)
(4.5)
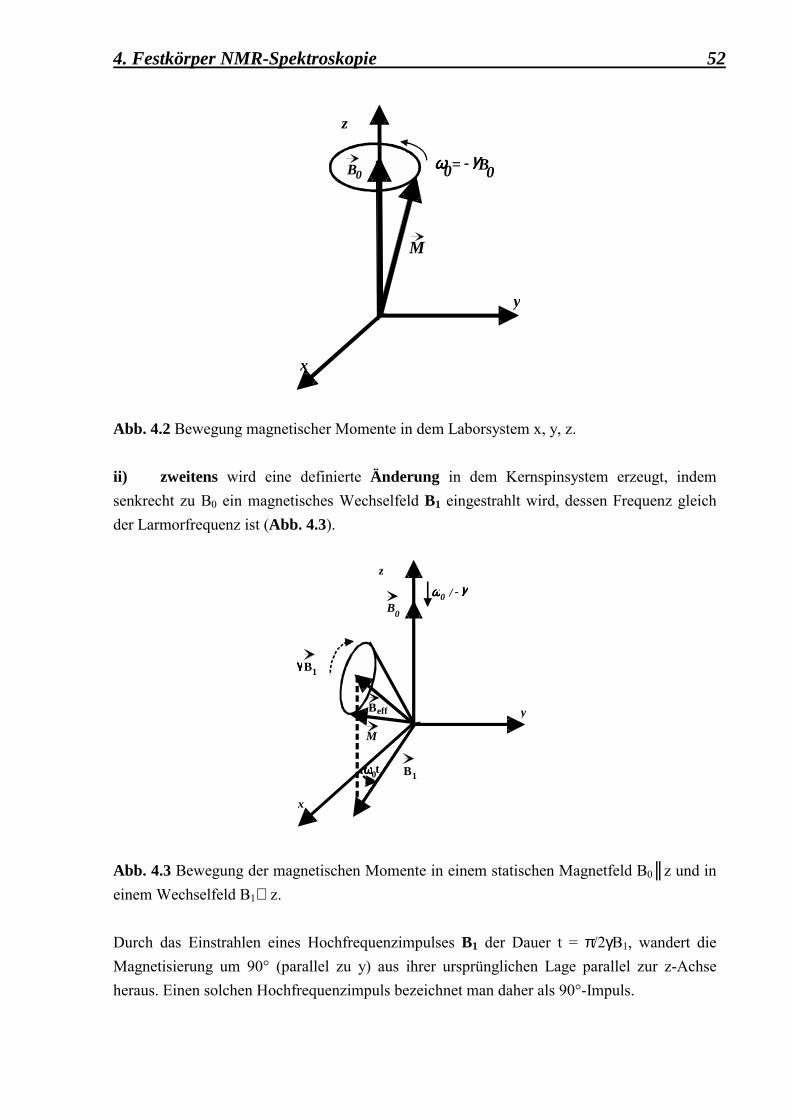
4. Festkörper NMR-Spektroskopie 52
Abb. 4.2 Bewegung magnetischer Momente in dem Laborsystem x, y, z.
z
B0
M
y
x
ωωωω0= γγγγB0-
ii) zweitens wird eine definierte Änderung in dem Kernspinsystem erzeugt, indem senkrecht zu B0 ein magnetisches Wechselfeld B1 eingestrahlt wird, dessen Frequenz gleich der Larmorfrequenz ist (Abb. 4.3).
Abb. 4.3 Bewegung der magnetischen Momente in einem statischen Magnetfeld B0║z und in einem Wechselfeld B1⊥ z.
z
B0
M
y
x
ωωωω0 / γγγγ-
Beff
γγγγB1
ωωωω0t B1
Durch das Einstrahlen eines Hochfrequenzimpulses B1 der Dauer t = π/2γB1, wandert die Magnetisierung um 90° (parallel zu y) aus ihrer ursprünglichen Lage parallel zur z-Achse heraus. Einen solchen Hochfrequenzimpuls bezeichnet man daher als 90°-Impuls.
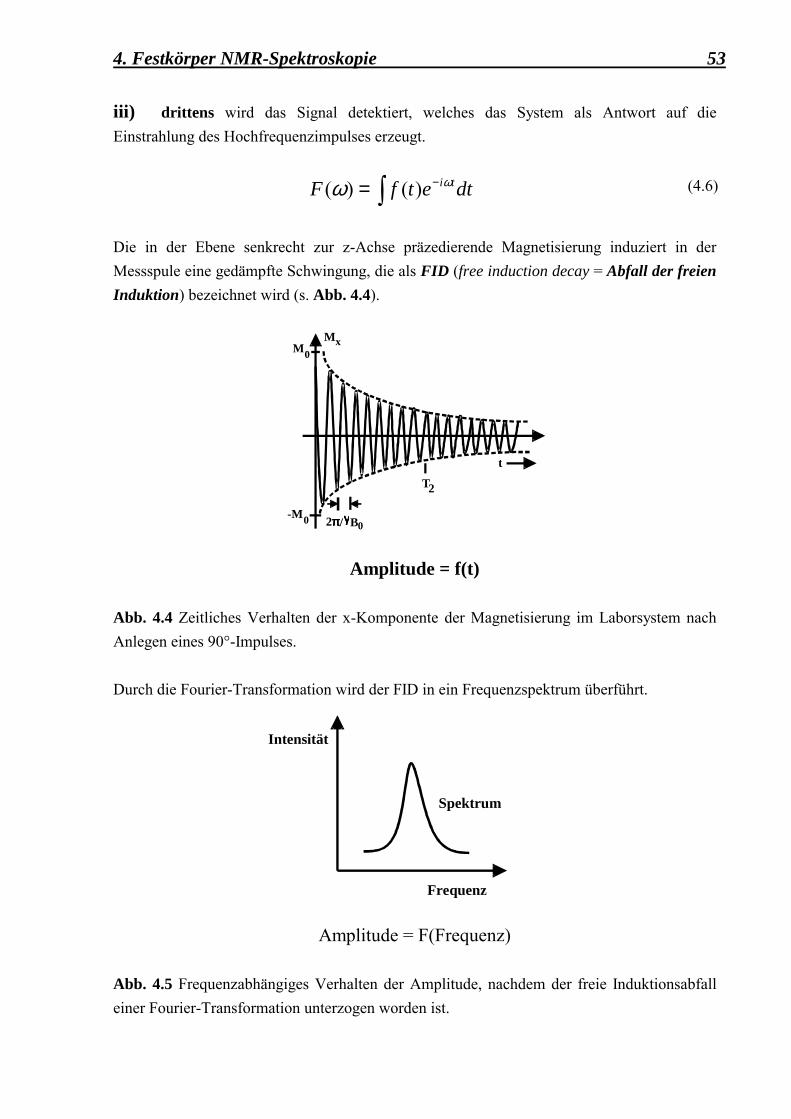
4. Festkörper NMR-Spektroskopie 53 iii) drittens wird das Signal detektiert, welches das System als Antwort auf die Einstrahlung des Hochfrequenzimpulses erzeugt.
Die in der Ebene senkrecht zur z-Achse präzedierende Magnetisierung induziert in der Messspule eine gedämpfte Schwingung, die als FID (free induction decay = Abfall der freien Induktion) bezeichnet wird (s. Abb. 4.4).
dtetfF ti∫ −= ωω )()( (4.6)
Amplitude = f(t)
Abb. 4.4 Zeitliches Verhalten der x-Komponente der Magnetisierung im Laborsystem nach Anlegen eines 90°-Impulses.
-M0
M0
t
T2
Mx
2ππππ/γγγγB0
Durch die Fourier-Transformation wird der FID in ein Frequenzspektrum überführt.
Amplitude = F(Frequenz)
Abb. 4.5 Frequenzabhängiges Verhalten der Amplitude, nachdem der freie Induktionsabfall einer Fourier-Transformation unterzogen worden ist.
Intensität
Frequenz
Spektrum

4. Festkörper NMR-Spektroskopie 54
Durch die Fourier-Transformation wird das akkumulierte Zeit-Domäne-Signal in ein resultierendes Frequenz-Domäne-NMR-Spektrum umgewandelt, mit einem Signal zu Rausch-Verhältnis, welches deutlich reduziert ist im Vergleich zu einem Spektrum, das durch konventionelle Techniken der Continous Waves (CW) aufgenommen wurde. Das Signal zu Rausch-Verhältnis wird signifikant durch eine längere Messung verbessert. Im Anschluss an den 90°-Impuls relaxieren die z-Komponenten des Systems mit der Spin-Gitter Relaxationszeit T1 und die x-,y-Komponenten durch die Spin-Spin Wirkung und aufgrund der Probeninhomogenität mit der Spin-Spin Relaxationszeit T2 wieder in ihren Ausgangszustand. Die Rückkehr des Spinsystems zur Gleichgewichtsmagnetisierung wird durch die Blochschen Gleichungen beschrieben:
wobei mit T1 = Spin-Gitter-Relaxationszeit und T2 = Spin-Spin-Relaxationszeit bezeichnet werden. Zusammengefasst basiert die Beobachtung eines magnetischen Kernresonanzsignals auf drei grundlegenden Erfordernissen: einem polarisierenden magnetischen Feld B0, um die Zeemanenergieniveaus aufzuspalten, einem resonanten Radio-Frequenz Feld B1, um die Übergangsprozesse zu bewirken, und der Detektion eines Induktionssignal.
( )1/0 1 Tt
z eMM −−=
2/0, Tt
yx eMMM −=
(4.7)
(4.8)

4. Festkörper NMR-Spektroskopie 55
4.1.2 Das Kreuzpolarisationsexperiment (CP MAS) Das Akronym CP MAS steht für Cross Polarisation, im deutschen Kreuzpolarisation, und Magic Angle Spinning. MAS wurde schon E. R. Andrew vorgeschlagen (Andrew, 1971). Das CP-Experiment gewann viel später an Bedeutung und zwar 1972. Die kombinierte Anwendung von CP-MAS kam gleich danach (1975/1976) durch die Wissenschaftler J. Schaefer und E. O. Stejskal zu Sprache (Schaefer J. und O., 1976). Das CP-Experiment basiert auf einem Magnetisierungstransfer von einem Kernspinsystem I (mit einer hohen natürlichen Häufigkeit), wie z. B. 1H, 19F, zu einem Kernspinsystem S (mit einer niedrigen natürlichen Häufigkeit), wie z. B. 13C, 15N, 29Si etc. Dabei ist die Hartmann-Hahn Bedingung für die Amplituden der gleichzeitig eingestrahlten Hochfrequenzimpulse für die beiden beteiligten Kerne einzustellen: γγγγIBI = γγγγSBS Dadurch kommt es zu einer Steigerung der relativen Empfindlichkeit für S-Kerne mit geringer natürlichen Häufigkeit, wie es für 29Si-Kerne zutrifft, um den Faktor γI/γS. Von grosser Wichtigkeit bei einem CP-Experiment ist die Wiederholungsrate für die Spektrenakkumulation. Diese ist nicht mehr von der Relaxationszeit T1(nS) des �schwachen� Kerns abhängig, sondern von der des stärkeren Kerns, wie z. B. T1(1H). Die Werte für T1(1H) sind oft kürzer als T1(S). So besteht die Möglichkeit, ein störendes Hindernis für die Festkörper-NMR-Messungen zu überwinden, zumindest für die Substanzen, bei denen S-Kerne sich in räumlicher Nähe von Protonen H+ oder F--Ionen befinden. Für ein besseres Verständnis wird der Verlauf des Kreuzpolarisationsexperimentes in der Abbildung 4.6 i) und ii) schematisch dargestellt.
(4.8)
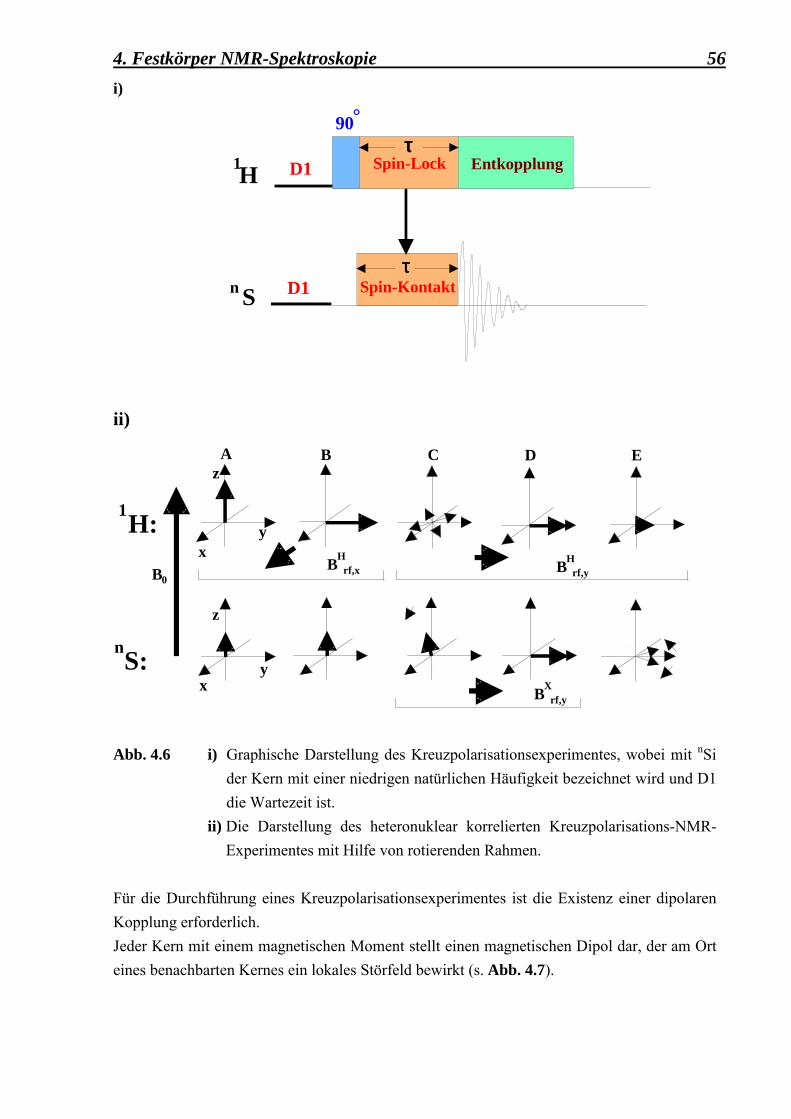
4. Festkörper NMR-Spektroskopie 56
i)1H
Sn
D1
D1
90°
Spin-Lock
Spin-Kontaktττττ
Entkopplungττττ
ii) Abb. 4.6 i) Graphische Darstellung des Kreuzpolarisationsexperimentes, wobei mit nSi
der Kern mit einer niedrigen natürlichen Häufigkeit bezeichnet wird und D1 die Wartezeit ist.
ii) Die Darstellung des heteronuklear korrelierten Kreuzpolarisations-NMR-Experimentes mit Hilfe von rotierenden Rahmen.
A B C D E
xy
z
z
yx
B0B rf,x
HB rf,y
H
B rf,yX
H:1
S:n
Für die Durchführung eines Kreuzpolarisationsexperimentes ist die Existenz einer dipolaren Kopplung erforderlich. Jeder Kern mit einem magnetischen Moment stellt einen magnetischen Dipol dar, der am Ort eines benachbarten Kernes ein lokales Störfeld bewirkt (s. Abb. 4.7).

4. Festkörper NMR-Spektroskopie 57
Abb. 4.7 Schematische Darstellung der dipolaren Kopplung, die als Wechselwirkung, die zwischen zwei lokalen Feldern der Kerne I und S auftritt, dargestellt wird.
B
z
I
Sββββr
0000
IS
IS
Für zwei Spins, die durch die Operatoren der Drehimpulse I und S und den internuklearen Vektor rIS charakterisiert sind, kann die dipolare Kopplung durch die folgende Formel beschrieben werden:
Aus der Formel ist zu entnehmen, dass die dipolare Kopplung mit dem internuklearen Abstand in dritter Potenz abnimmt:
HD ∝∝∝∝ rIS -3
Da anisotrope Dipol-Dipol Wechselwirkungen mit Protonen insbesondere zu der Verbreiterung der S-Resonanzsignale in Festkörper-NMR-Spektren führen, wird deren Beseitigung angestrebt. Dies ist durch die sog. �Entkopplung� der beiden Kerne 1H und nS zu erreichen. Andere Kerne als 1H oder 19F beeinflussen die Linienbreite meist wenig. Ein wichtiger Aspekt der CP-Technik sind die Abstandsinformationen, die durch ein Kreuzpolarisationsexperiment zu bekommen sind. Aus theoretischer Sicht wird die Signalintensität des S-Spins IS(t) als eine Funktion der Kontaktzeit (ττττ) beschrieben (s. Abb. 4.6 i)).
ZZIS
ISSID SI
rH
−
−= 3
220
21cos3
2βγγ
πµ
h (4.9)
(4.10)

4. Festkörper NMR-Spektroskopie 58
wobei: TIS = TCP = die Kreuzrelaxationszeit zwischen den I- und den S-Spins;
T1ρρρρ = die Spin-Gitter Relaxationszeit im eingestrahlten hochfrequenten Magnetfeld für die I-Spins
Zwischen der Kreuzrelaxationszeit und dem 2. Moment besteht eine umgekehrt proportionale Abhängigkeit, beschrieben durch die Formel:
Bekannt ist, dass das 2. Moment ein Mass für die quadratische Linienbreite ist:
Nach Slichter (Slichter, 1990) wird das 2. Moment durch die folgende Formel beschrieben:
wobei: rIS = der Abstand zwischen den I- und den S-Spins θIS = der Winkel zwischen dem internuklearen Vektor und der externen Feldrichtung und h = 6,6262 x 10-34 J.s µ0 = 1,2566 x 10-6 V.s.A-1.m-1 ist. Daraus folgt, dass die r-6 Abhängigkeit des 2. Momentes auf die nahe gelegenen Nachbarn limitiert werden kann.
( )
( )
,
1
expexp
)(
1
1
−
−−
−
=
STT
TSTC
IIS
IS
ρ
ρ
ττ
τ
>∆∝< 21IS
ISTω
62 −>∝∆< ISIS rω
2MFWHM ∝
( ) ,1415
4,
62
02222 ∑ −+
>=∆<
SIISSI rIIh
πµγγω
(4.11)
(4.12)
(4.13)
(4.14)
(4.15)
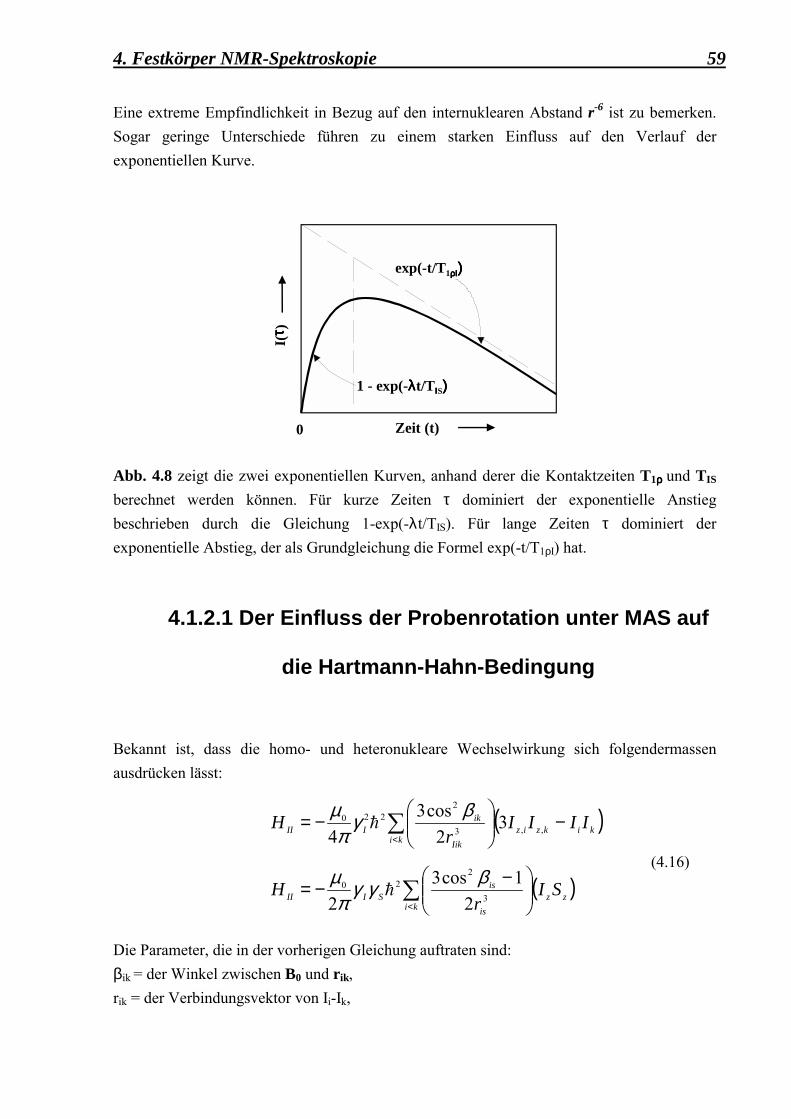
4. Festkörper NMR-Spektroskopie 59 Eine extreme Empfindlichkeit in Bezug auf den internuklearen Abstand r-6 ist zu bemerken. Sogar geringe Unterschiede führen zu einem starken Einfluss auf den Verlauf der exponentiellen Kurve.
Abb. 4.8 zeigt die zwei exponentiellen Kurven, anhand derer die Kontaktzeiten T1ρρρρ und TIS berechnet werden können. Für kurze Zeiten τ dominiert der exponentielle Anstieg beschrieben durch die Gleichung 1-exp(-λt/TIS). Für lange Zeiten τ dominiert der exponentielle Abstieg, der als Grundgleichung die Formel exp(-t/T1ρI) hat.
exp(-t/T1ρΙρΙρΙρΙ))))
1 - exp(-λλλλt/TΙΙΙΙS))))
Zeit (t)
I(ττ ττ )
0
4.1.2.1 Der Einfluss der Probenrotation unter MAS auf
die Hartmann-Hahn-Bedingung
Bekannt ist, dass die homo- und heteronukleare Wechselwirkung sich folgendermassen ausdrücken lässt:
Die Parameter, die in der vorherigen Gleichung auftraten sind: βik = der Winkel zwischen B0 und rik, rik = der Verbindungsvektor von Ii-Ik,
( )kikzizki Iik
ikIII IIII
rH −
−= ∑
<,,3
2220 3
2cos3
4βγ
πµ
h
( )zzki is
isSIII SI
rH ∑
<
−−= 3
220
21cos3
2βγγ
πµ
h
(4.16)

4. Festkörper NMR-Spektroskopie 60
γI = das gyromagnetische Verhältnis des Spins I, γS = das gyromagnetische Verhältnis des Spins S, Iz = der I-Spin, entsprechend der z-Komponente, Sz = der S-Spin, entsprechend der z-Komponente. Unter MAS-Bedingungen werden die b-Terme zeitabhängig. Es gilt:
Für die Beobachtung zahlreicher Kerne mit einen Spin I=1/2 bleibt die CP-Technik eine der bevorzugten Methoden. Die Überlegung, die NMR-Methode für die Untersuchung der externen Oberfläche einzusetzen rührte daher, dass die Kernresonanzspektroskopie keine typisch oberflächensensitive Methode ist. Die einzigen Experimente, die in dieser Richtung durchgeführt worden sind, waren die 1H MAS NMR-Messungen, bei denen aber zu viele andere Signale, die schwer zugeordnet werden können, auftraten. In den folgenden Kapitelabschnitten werden die Ergebnisse von verschiedenen NMR-Messungen dargestellt, wobei die am meisten benutzte Methode die der Kreuzpolarisations-Technik sein wird. Es werden 1H/29Si CP MAS-, 19F/29Si CP MAS-Untersuchungen, ergänzt durch Kontaktzeitvariationsmessungen und Relaxationszeitberechnungen, durchgeführt, um dadurch ein vollständiges Bild von der externen Oberfläche zu erhalten.
ijij
JIij
ijijij
JIij
ijSijijSijij
rb
rb
tbtbtb
γγγ
γγγ
δωδω
23
)2(
3)1(
)2()1(
sin2
cossin2
)2sin()cos()(
h
h
=
=
−+−=
(4.17)

4. Festkörper NMR-Spektroskopie 61
4.2 Experimentelle Ergebnisse
4.2.1 1H/29Si CP MAS-Experimente, durchgeführt
an „grobkörnigen“ Dodecasil 3C-Substanzen mit
einer mittleren Korngrösse zwischen 1 und 300 µm Da die dipolaren Wechselwirkungen abstandsabhängig sind, lassen sich Aussagen über die lokale Struktur und Ordnung anhand der Kreuzpolarisationsexperimente machen. Dodecasil 3C enthält drei symmetrisch inäquivalente Lagen, sog. sites T1, T2, T3 in einem relativen Verhältnis zueinander von 8:32:96 und wird schematisch in der Abbildung 4.9 dargestellt. Für die unterschiedlichen Positionen wurden anhand der NMR-Spektroskopie mit Hilfe von 29Si MAS- und 29Si 2D NMR Spin-Diffusions-Experimenten erstens die Lage der Signale und zweitens die Konnektivitäten zwischen den Sites T1 und T2, bzw. T2 und T3, bestimmt (Fyfe et al., 1989).
Abb. 4.9 Schematische Darstellung des Gittergerüstes eines ZSM-39-Zeolithen (Fyfe et al., 1989). Die drei unterschiedlichen Gitterpositionen sind durch T1, T2, T3 (eingekreist) gekennzeichnet. In der Abbildung treten auch die möglichen Konnektivitäten in Erscheinung.

4. Festkörper NMR-Spektroskopie 62
Beim 1H/29Si CP MAS NMR-Experiment an Dodecasil 3C kann ein Magnetisierungstransfer sowohl von Protonen, die zu den Silanolgruppen Si-OH gehören, die sich an der externen Oberfläche des Clathrasils befinden, als auch von Protonen, die zu den organischen Templatmolekülen gehören und die in den Pentagondodecaeder-Käfigen angelagert sind, stattfinden (Abb. 4.10). Die Protonen, die von Si-O-H-Gruppen stammen, zeigen eine feste Orientierung. Das Q3-Si wird in diesem Fall bevorzugt polarisiert. Die Protonen, die zu den Templat-Molekülen gehören, zeigen durch die Templat-Molekül-Dynamik keine festen Wechselwirkungen zu dem Käfig-Kanal. Das Spektrum kann einer quantitativen Auswertung unterzogen werden, da alle Si-Kerne an dem Transfer-Prozess beteiligt sind. In der Abbildung 4.10 werden graphisch die Möglichkeiten, die im Falle des Clathrasils Dodecasil 3C bei dem Magnetisierungstransfer von 1H-Kernen auf 29Si-Kerne auftreten können, dargestellt. Ausser den zwei schon vorher erwähnten Möglichkeiten: Magnetisierungstransfer von �Templat-Protonen� und Magnetisierungstransfer von �Oberflächen-Protonen�, können auch Artefakte, wie z.B. nicht umgewandeltes Silicagel oder Strukturdefekte das Spektrum beeinflussen. Diese Artefakte können gleichzeitig ihren Beitrag zu der Veränderung des 1H/29Si CP MAS NMR-Spektrum leisten und Signale in dem entsprechenden Bereich von -90 bis -102 ppm liefern, so dass die Trennung zwischen einem Q3-Oberflächensignal und Q3-Artefaktsignal erschwert wird. Um die Artefakte auszuschliessen, wurden an der as-synthesized Form des Dodecasil 3C verschiedene Behandlungen durchgeführt, wie z.B. das Calcinieren in einer oxidativen Atmosphäre oder das Waschen mit einer NaOH-Lösung. Durch das Calcinieren wurden die eventuell vorhandenen Strukturdefekte eliminiert, während die NaOH-Behandlung dazu führte, dass mögliche Reste an nicht umgewandeltem Templat ausgewaschen worden sind. Die Möglichkeit, eine amorphe Clathrasil-Masse zu untersuchen, wurde durch diese zusätzlichen Behandlungen ausgeschlossen. Diesen Behandlungen folgten 1H/29Si Kreuzpolarisationsmessungen, deren Ergebnisse in den Abschnitten 4.2.2.2 bzw. 4.2.2.3 dargestellt werden. Das von der Dodecasil 3C-Substanz aufgenommene 1H/29Si CP MAS NMR-Spektrum zeigt, wie es auch aus der Arbeit von C. Fyfe, H. Gies und Y. Feng (Fyfe et al., 1989) zu entnehmen ist, drei verschiedene Q4-Signale, entsprechend der drei unterschiedlichen T�Sites-Positionen: T1(Si3): -107,3 ppm, T2(Si2): -115,1 ppm, T3(Si1): -120,9 ppm, mit einem relativen Verhältnis zueinander von 1:4:12. Durch die Phasenumwandlung, von einem
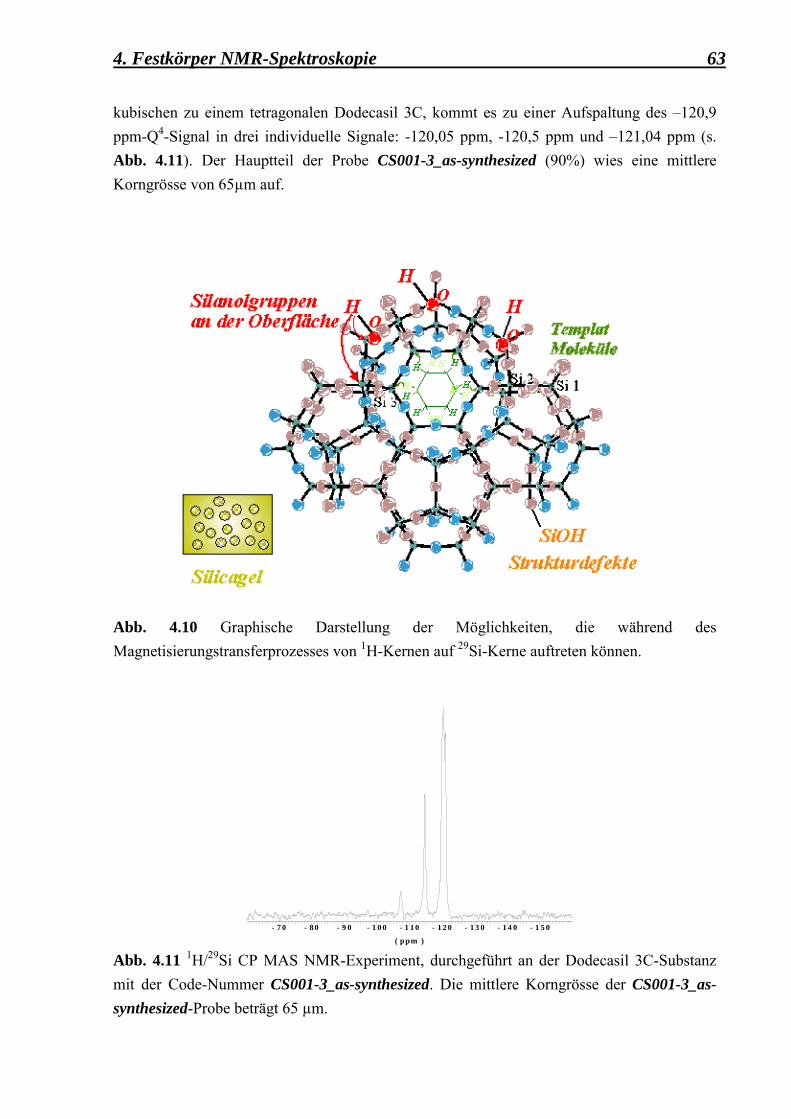
4. Festkörper NMR-Spektroskopie 63 kubischen zu einem tetragonalen Dodecasil 3C, kommt es zu einer Aufspaltung des �120,9 ppm-Q4-Signal in drei individuelle Signale: -120,05 ppm, -120,5 ppm und �121,04 ppm (s. Abb. 4.11). Der Hauptteil der Probe CS001-3_as-synthesized (90%) wies eine mittlere Korngrösse von 65µm auf.
Abb. 4.10 Graphische Darstellung der Möglichkeiten, die während des Magnetisierungstransferprozesses von 1H-Kernen auf 29Si-Kerne auftreten können.
Abb. 4.11 1H/29Si CP MAS NMR-Experiment, durchgeführt an der Dodecasil 3C-Substanz mit der Code-Nummer CS001-3_as-synthesized. Die mittlere Korngrösse der CS001-3_as-synthesized-Probe beträgt 65 µm.
( ppm )
- 1 5 0- 1 4 0- 1 3 0- 1 2 0- 1 1 0- 1 0 0- 9 0- 8 0- 7 0
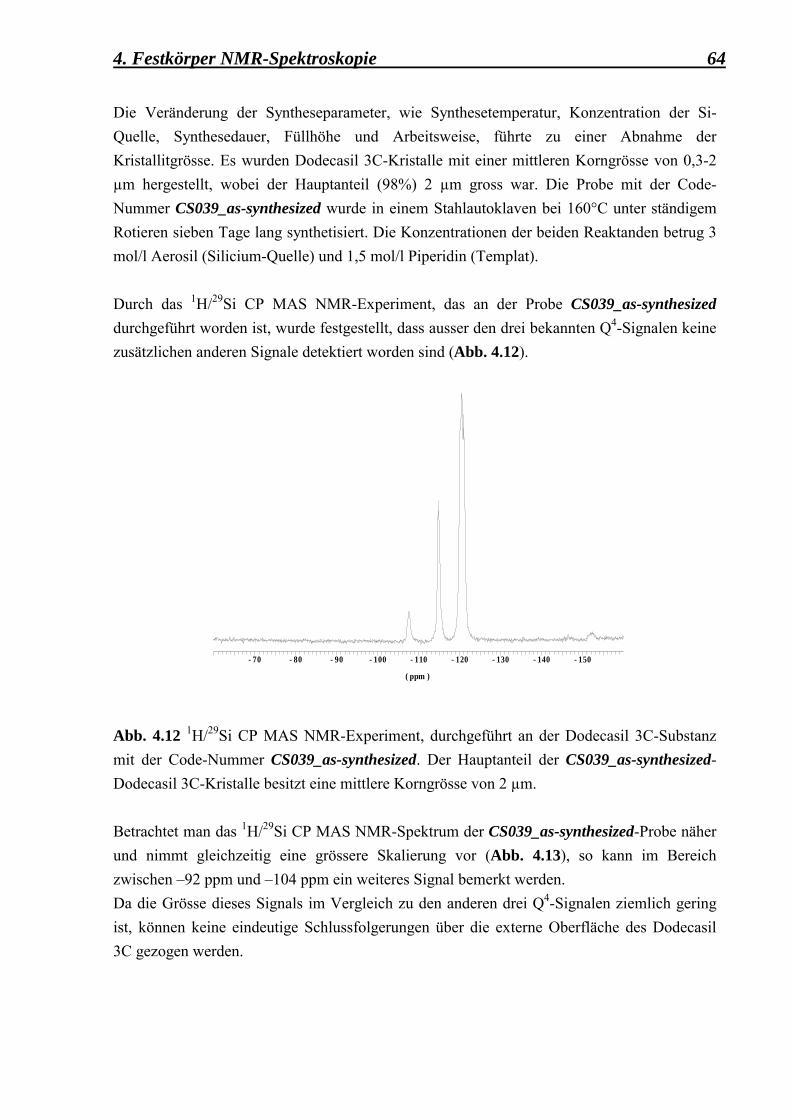
4. Festkörper NMR-Spektroskopie 64 Die Veränderung der Syntheseparameter, wie Synthesetemperatur, Konzentration der Si-Quelle, Synthesedauer, Füllhöhe und Arbeitsweise, führte zu einer Abnahme der Kristallitgrösse. Es wurden Dodecasil 3C-Kristalle mit einer mittleren Korngrösse von 0,3-2 µm hergestellt, wobei der Hauptanteil (98%) 2 µm gross war. Die Probe mit der Code-Nummer CS039_as-synthesized wurde in einem Stahlautoklaven bei 160°C unter ständigem Rotieren sieben Tage lang synthetisiert. Die Konzentrationen der beiden Reaktanden betrug 3 mol/l Aerosil (Silicium-Quelle) und 1,5 mol/l Piperidin (Templat). Durch das 1H/29Si CP MAS NMR-Experiment, das an der Probe CS039_as-synthesized durchgeführt worden ist, wurde festgestellt, dass ausser den drei bekannten Q4-Signalen keine zusätzlichen anderen Signale detektiert worden sind (Abb. 4.12).
Abb. 4.12 1H/29Si CP MAS NMR-Experiment, durchgeführt an der Dodecasil 3C-Substanz mit der Code-Nummer CS039_as-synthesized. Der Hauptanteil der CS039_as-synthesized-Dodecasil 3C-Kristalle besitzt eine mittlere Korngrösse von 2 µm.( ppm )
- 150- 140- 130- 120- 110- 100- 90- 80- 70
Betrachtet man das 1H/29Si CP MAS NMR-Spektrum der CS039_as-synthesized-Probe näher und nimmt gleichzeitig eine grössere Skalierung vor (Abb. 4.13), so kann im Bereich zwischen �92 ppm und �104 ppm ein weiteres Signal bemerkt werden. Da die Grösse dieses Signals im Vergleich zu den anderen drei Q4-Signalen ziemlich gering ist, können keine eindeutige Schlussfolgerungen über die externe Oberfläche des Dodecasil 3C gezogen werden.
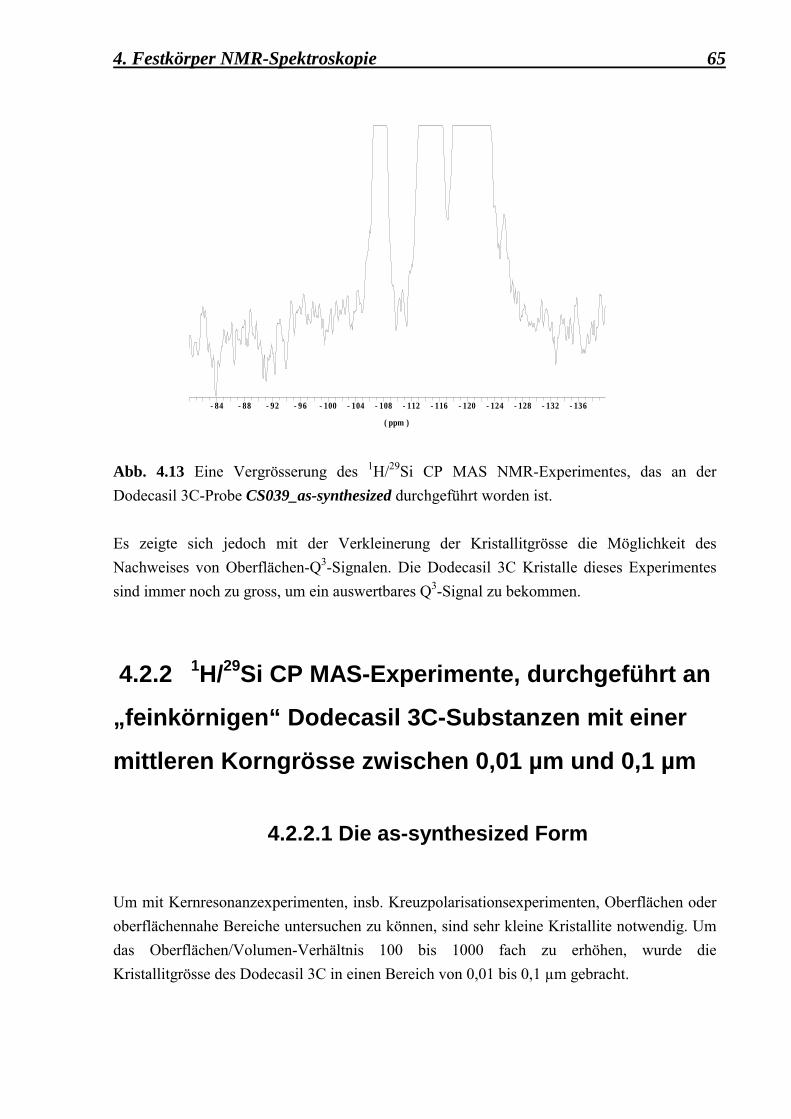
4. Festkörper NMR-Spektroskopie 65
Abb. 4.13 Eine Vergrösserung des 1H/29Si CP MAS NMR-Experimentes, das an der Dodecasil 3C-Probe CS039_as-synthesized durchgeführt worden ist.( ppm )
- 136- 132- 128- 124- 120- 116- 112- 108- 104- 100- 96- 92- 88- 84
Es zeigte sich jedoch mit der Verkleinerung der Kristallitgrösse die Möglichkeit des Nachweises von Oberflächen-Q3-Signalen. Die Dodecasil 3C Kristalle dieses Experimentes sind immer noch zu gross, um ein auswertbares Q3-Signal zu bekommen.
4.2.2 1H/29Si CP MAS-Experimente, durchgeführt an
„feinkörnigen“ Dodecasil 3C-Substanzen mit einer
mittleren Korngrösse zwischen 0,01 µm und 0,1 µm
4.2.2.1 Die as-synthesized Form Um mit Kernresonanzexperimenten, insb. Kreuzpolarisationsexperimenten, Oberflächen oder oberflächennahe Bereiche untersuchen zu können, sind sehr kleine Kristallite notwendig. Um das Oberflächen/Volumen-Verhältnis 100 bis 1000 fach zu erhöhen, wurde die Kristallitgrösse des Dodecasil 3C in einen Bereich von 0,01 bis 0,1 µm gebracht.

4. Festkörper NMR-Spektroskopie 66 Das gemessene 1H/29Si CP MAS NMR-Spektrum (Abb. 4.14) zeigte bei der Probe CS024_as-synthesized, ausser den drei üblichen Q4-Signalen, zusätzlich noch zwei breite Q3-Signale, entsprechend der externen Oberfläche: Q3(Si2-OH): -99,1 ppm und Q3(Si3-OH): -111,2 ppm. Zur Vereinfachung wurden die Bezeichnungen: Q3(Si3-OH) und Q3(Si1-OH) durch S2(Si2) und S3(Si1) ersetzt. Sie repräsentieren die unterschiedlichen �S-Sites-Positionen� der Si-Atome an der externen Oberfläche oder anders gesagt �die an der Oberfläche kristallographisch inäquivalente Lagen�. Für ein besseres Verständnis wurde in der Tabelle 4.1 ein Vergleich zwischen den chemischen Verschiebungen, die für die Dodecasil 3C-Substanz aus den Literaturangaben (Abkürzung L. A.) zu entnehmen sind, und den chemischen Verschiebungen der Dodecasil 3C-Proben CS001-3_as-synthesized und CS024_as-synthesized gezogen. Die Dodecasil 3C-Probe mit der Code-Nummer CS001-3_as-synthesized und der mittleren Korngrösse φ ≅ 2 µm zeigt, im Vergleich zu den Literaturwerten aus C. Fyfe, H. Gies und Y. Feng (Fyfe et al., 1989), eine Aufspaltung des dritten Signals, das üblicherweise bei �120,9 ppm auftritt, in drei individuelle Signale: �120,05 ppm, -120,5 ppm, -121,04 ppm . Diese Aufspaltung kommt dadurch zustande, dass während der Synthese von CS001-3_as-synthesized eine Phasenumwandlung bei ca. 80°C vom kubischen zum tetragonalen Kristallsystem stattgefunden hat.
Abb. 4.14 1H/29Si CP MAS NMR-Experiment, durchgeführt an der Dodecasil 3C-Substanz mit der Code-Nummer CS024_as-synthesized. Die Kristallitgrösse der CS024_as-synthesized-Dodecasil 3C-Probe beträgt 0,01 µm bis 0,1 µm.
( ppm )
- 1 3 5- 1 3 0- 1 25- 1 2 0- 1 15- 1 1 0- 1 0 5- 1 0 0- 9 5- 9 0- 8 5- 80- 7 5
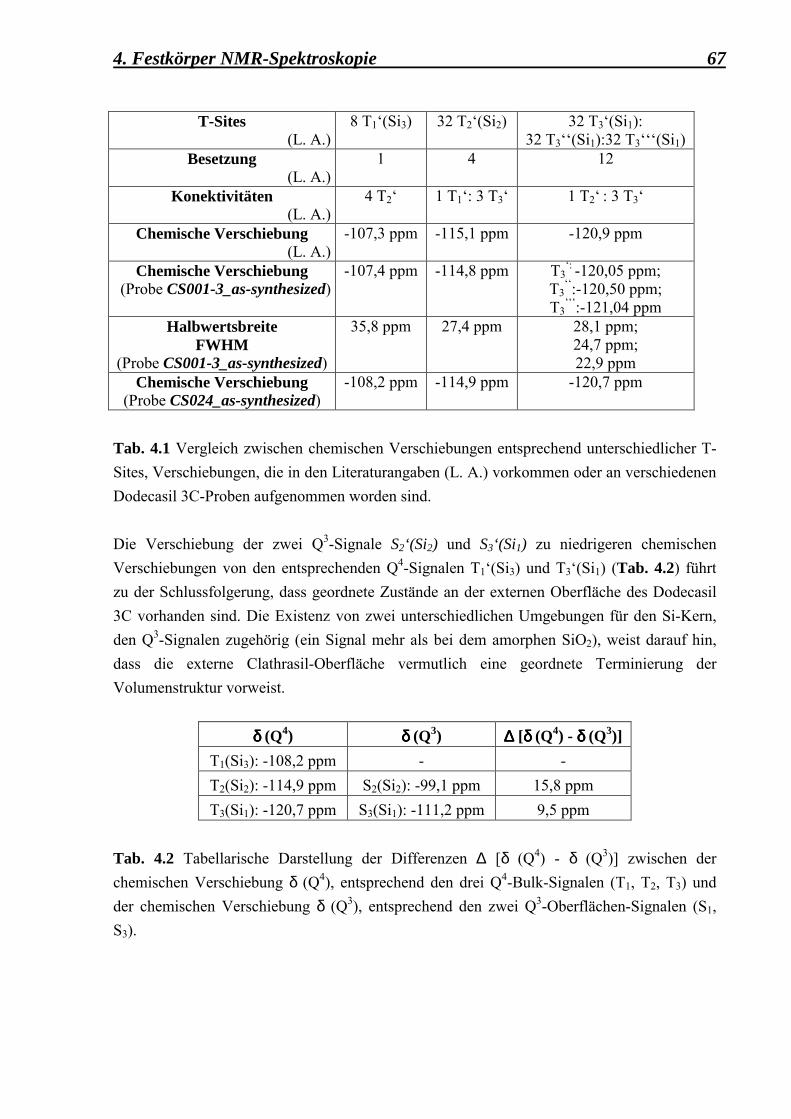
4. Festkörper NMR-Spektroskopie 67
T-Sites (L. A.)
8 T1�(Si3) 32 T2�(Si2) 32 T3�(Si1): 32 T3��(Si1):32 T3���(Si1)
Besetzung (L. A.)
1 4 12
Konektivitäten (L. A.)
4 T2� 1 T1�: 3 T3� 1 T2� : 3 T3�
Chemische Verschiebung (L. A.)
-107,3 ppm -115,1 ppm -120,9 ppm
Chemische Verschiebung (Probe CS001-3_as-synthesized)
-107,4 ppm -114,8 ppm T3�: -120,05 ppm;
T3��:-120,50 ppm;
T3���:-121,04 ppm
Halbwertsbreite FWHM
(Probe CS001-3_as-synthesized)
35,8 ppm 27,4 ppm 28,1 ppm; 24,7 ppm; 22,9 ppm
Chemische Verschiebung (Probe CS024_as-synthesized)
-108,2 ppm -114,9 ppm -120,7 ppm
Tab. 4.1 Vergleich zwischen chemischen Verschiebungen entsprechend unterschiedlicher T-Sites, Verschiebungen, die in den Literaturangaben (L. A.) vorkommen oder an verschiedenen Dodecasil 3C-Proben aufgenommen worden sind.
Die Verschiebung der zwei Q3-Signale S2‘(Si2) und S3‘(Si1) zu niedrigeren chemischen Verschiebungen von den entsprechenden Q4-Signalen T1�(Si3) und T3�(Si1) (Tab. 4.2) führt zu der Schlussfolgerung, dass geordnete Zustände an der externen Oberfläche des Dodecasil 3C vorhanden sind. Die Existenz von zwei unterschiedlichen Umgebungen für den Si-Kern, den Q3-Signalen zugehörig (ein Signal mehr als bei dem amorphen SiO2), weist darauf hin, dass die externe Clathrasil-Oberfläche vermutlich eine geordnete Terminierung der Volumenstruktur vorweist.δδδδ (Q4) δδδδ (Q3) ∆∆∆∆ [δδδδ (Q4) - δδδδ (Q3)] T1(Si3): -108,2 ppm - - T2(Si2): -114,9 ppm S2(Si2): -99,1 ppm 15,8 ppm T3(Si1): -120,7 ppm S3(Si1): -111,2 ppm 9,5 ppm
Tab. 4.2 Tabellarische Darstellung der Differenzen ∆ [δ (Q4) - δ (Q3)] zwischen der chemischen Verschiebung δ (Q4), entsprechend den drei Q4-Bulk-Signalen (T1, T2, T3) und der chemischen Verschiebung δ (Q3), entsprechend den zwei Q3-Oberflächen-Signalen (S1, S3).
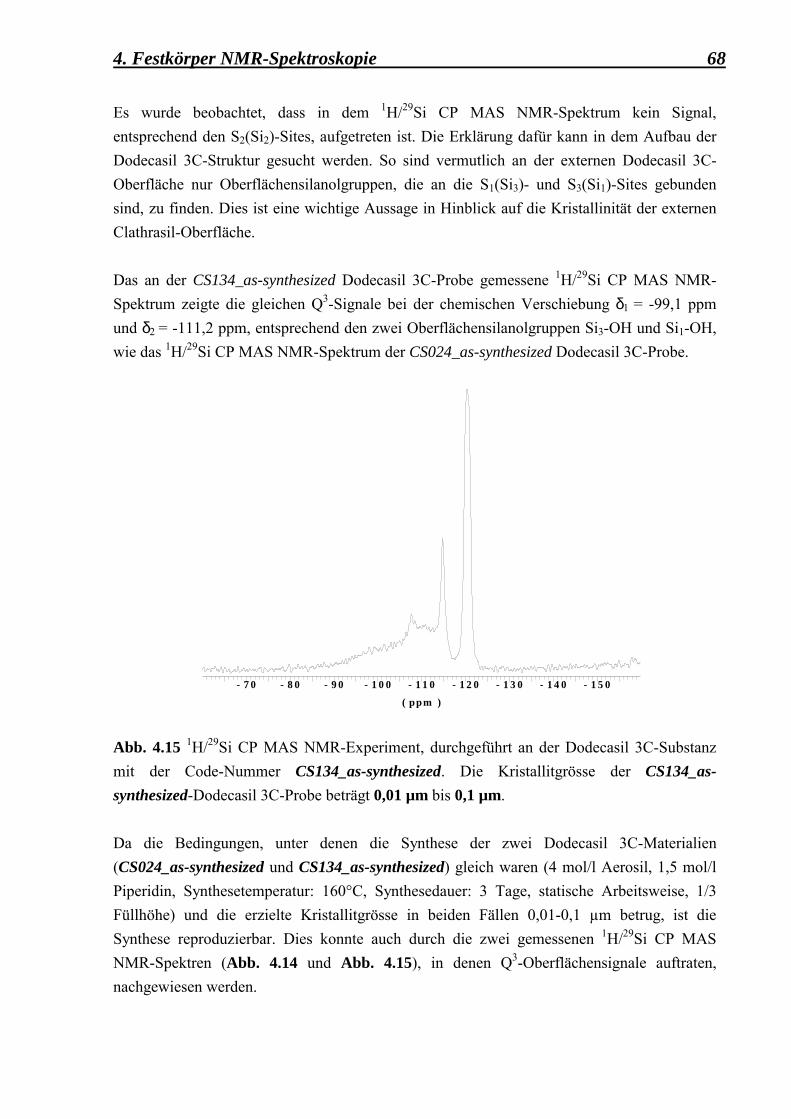
4. Festkörper NMR-Spektroskopie 68 Es wurde beobachtet, dass in dem 1H/29Si CP MAS NMR-Spektrum kein Signal, entsprechend den S2(Si2)-Sites, aufgetreten ist. Die Erklärung dafür kann in dem Aufbau der Dodecasil 3C-Struktur gesucht werden. So sind vermutlich an der externen Dodecasil 3C-Oberfläche nur Oberflächensilanolgruppen, die an die S1(Si3)- und S3(Si1)-Sites gebunden sind, zu finden. Dies ist eine wichtige Aussage in Hinblick auf die Kristallinität der externen Clathrasil-Oberfläche. Das an der CS134_as-synthesized Dodecasil 3C-Probe gemessene 1H/29Si CP MAS NMR-Spektrum zeigte die gleichen Q3-Signale bei der chemischen Verschiebung δ1 = -99,1 ppm und δ2 = -111,2 ppm, entsprechend den zwei Oberflächensilanolgruppen Si3-OH und Si1-OH, wie das 1H/29Si CP MAS NMR-Spektrum der CS024_as-synthesized Dodecasil 3C-Probe.
Abb. 4.15 1H/29Si CP MAS NMR-Experiment, durchgeführt an der Dodecasil 3C-Substanz mit der Code-Nummer CS134_as-synthesized. Die Kristallitgrösse der CS134_as-synthesized-Dodecasil 3C-Probe beträgt 0,01 µm bis 0,1 µm.
( ppm )- 1 5 0- 1 4 0- 1 3 0- 1 2 0- 1 1 0- 1 0 0- 9 0- 8 0- 7 0
Da die Bedingungen, unter denen die Synthese der zwei Dodecasil 3C-Materialien (CS024_as-synthesized und CS134_as-synthesized) gleich waren (4 mol/l Aerosil, 1,5 mol/l Piperidin, Synthesetemperatur: 160°C, Synthesedauer: 3 Tage, statische Arbeitsweise, 1/3 Füllhöhe) und die erzielte Kristallitgrösse in beiden Fällen 0,01-0,1 µm betrug, ist die Synthese reproduzierbar. Dies konnte auch durch die zwei gemessenen 1H/29Si CP MAS NMR-Spektren (Abb. 4.14 und Abb. 4.15), in denen Q3-Oberflächensignale auftraten, nachgewiesen werden.
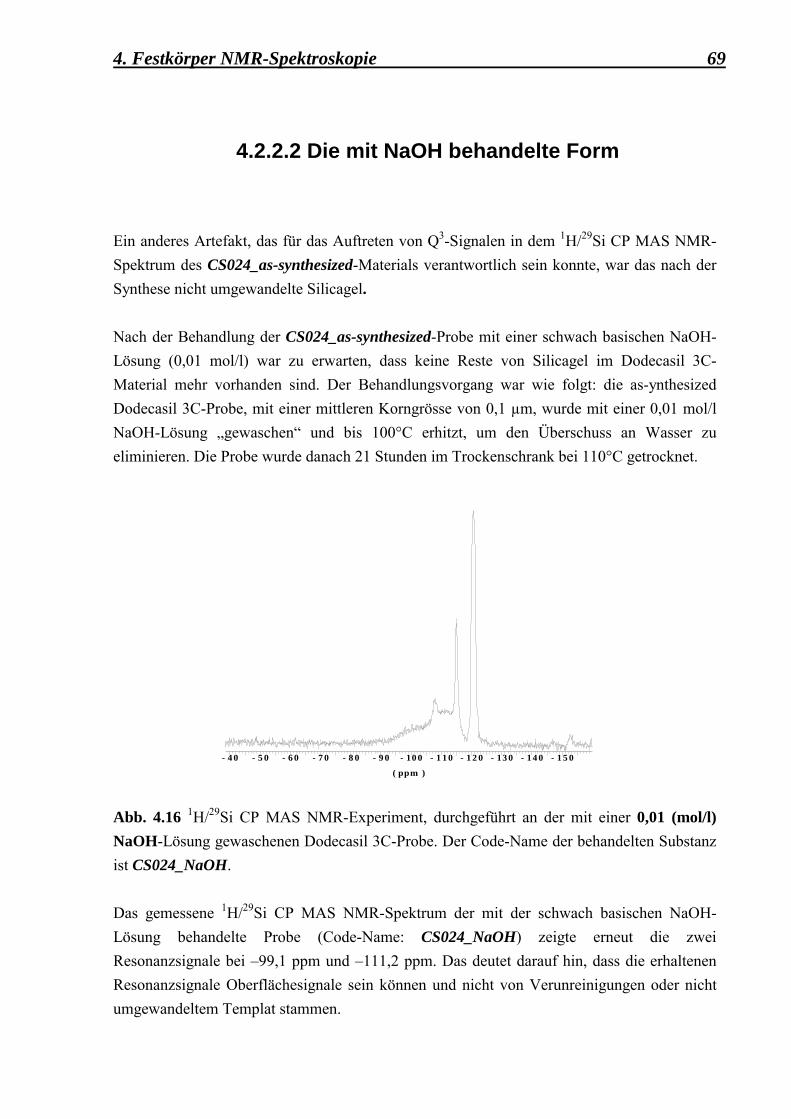
4. Festkörper NMR-Spektroskopie 69
4.2.2.2 Die mit NaOH behandelte Form
Ein anderes Artefakt, das für das Auftreten von Q3-Signalen in dem 1H/29Si CP MAS NMR-Spektrum des CS024_as-synthesized-Materials verantwortlich sein konnte, war das nach der Synthese nicht umgewandelte Silicagel. Nach der Behandlung der CS024_as-synthesized-Probe mit einer schwach basischen NaOH-Lösung (0,01 mol/l) war zu erwarten, dass keine Reste von Silicagel im Dodecasil 3C-Material mehr vorhanden sind. Der Behandlungsvorgang war wie folgt: die as-ynthesized Dodecasil 3C-Probe, mit einer mittleren Korngrösse von 0,1 µm, wurde mit einer 0,01 mol/l NaOH-Lösung �gewaschen� und bis 100°C erhitzt, um den Überschuss an Wasser zu eliminieren. Die Probe wurde danach 21 Stunden im Trockenschrank bei 110°C getrocknet.
Abb. 4.16 1H/29Si CP MAS NMR-Experiment, durchgeführt an der mit einer 0,01 (mol/l) NaOH-Lösung gewaschenen Dodecasil 3C-Probe. Der Code-Name der behandelten Substanz ist CS024_NaOH.
( ppm )
- 1 5 0- 14 0- 13 0- 1 2 0- 1 1 0- 10 0- 9 0- 8 0- 7 0- 6 0- 5 0- 4 0
Das gemessene 1H/29Si CP MAS NMR-Spektrum der mit der schwach basischen NaOH-Lösung behandelte Probe (Code-Name: CS024_NaOH) zeigte erneut die zwei Resonanzsignale bei �99,1 ppm und �111,2 ppm. Das deutet darauf hin, dass die erhaltenen Resonanzsignale Oberflächesignale sein können und nicht von Verunreinigungen oder nicht umgewandeltem Templat stammen.

4. Festkörper NMR-Spektroskopie 70
4.2.2.3 Die calcinierte Form Zu den Artefakten, die zu der Entstehung (Bildung) von Q3-Signalen führen können, zählen unter anderem Strukturdefekte (Abb. 4.10). Während der vorherigen Experimente ging man davon aus, dass die untersuchten Kristalle perfekt waren. Die ganze Zeit bestand aber die Möglichkeit, dass in der Struktur Defekte vorhanden waren. Die einfachste Art von Strukturdefekten sind die sogenannten „Schottky“- oder „Schottky-Wagner-Defekte“. Wenn in einem neutralen Gitter eines der Atome (z. B. ein Kation) fehlt - d.h. es wurde aus dem Gitter eliminiert -, dann entsteht ein Loch, das als Schottky-Defekt bezeichnet wird. In einem Kristall der elektrisch neutral ist, muss für einen Ausgleich gesorgt werden, so dass die fehlende Ladung kompensiert wird. Das kann erreicht werden, indem ein anderer Schottky-Defekt erzeugt wird. So kann an einer anderen Stelle in der Struktur ein Anion fehlen. Ein anderer Weg die Ladung auszugleichen wäre das Einführen von fremden Ionen mit einer höheren elektrischen Ladung. Es besteht noch eine zweite Möglichkeit, Strukturdefekte zu erzeugen und zwar durch eine einfache Verschiebung von Ionen in eine benachbarte interstitialen Lage (Huheey, 1983). Solche Defekte sind unter dem Namen „Frenkel-Defekte“ bekannt. In solchen Fällen werden Ionen, die klein genug sind, eine interstitiale Lage, wie z. B. eine tetraedrische Lücke in einem oktaedrischen Gitter, besetzen. Die dritte Möglichkeit, die besteht, ist eine Kombination der beiden Defekte. So kann ein Ion in eine interstitialen Lage wandern, während ein anderes den frei gewordene Platz ausfüllt. Um diesen unerwünschten Effekt auszuschliessen, wurde die as-synthesized Form des Dodecasil 3C-Materials (Probe CS024_as-synthesized) 2 Tage lang bei 550°C in einer oxidativen Atmosphäre einer partiellen Calcinierung unterzogen. Durch die Calcinierung und die damit verbundene Erhöhung der Temperatur kommt es zu einer Unordnung im Gitter wodurch die Atome eine hohe Mobilität zeigen. Durch das Kühlen besetzen die Atome die eventuellen Lücken, die vorhanden waren, und werden rigid. Die Atome haben wieder feste Plätze eingenommen. Man bekommt eine geordnete Struktur, der Kristall ist perfekt und keine Defekte sind mehr vorhanden. Mit dem erhaltenen Material (Probe CS024_calciniert) wurde ein 1H/29Si CP MAS NMR-Experiment durchgeführt. Zu beobachten war, dass die vorher in dem 1H/29Si CP MAS NMR-Spektrum der CS024_as-synthesized-Probe existierenden Q3-Oberflächensignale immer noch vorhanden waren. Gleichzeitig kam es zu keiner Intensitätsveränderung der Q3-Signale der calcinierten im Vergleich zu der as-synthesized Substanz. So war es durch die NMR-
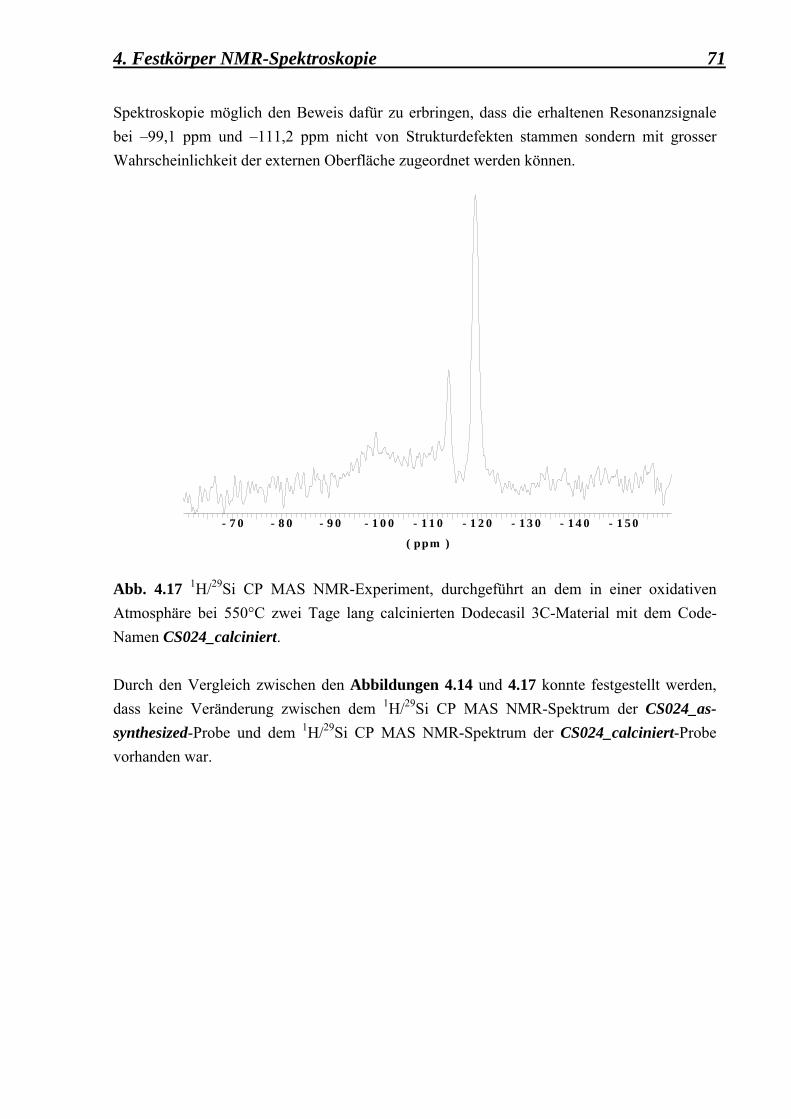
4. Festkörper NMR-Spektroskopie 71 Spektroskopie möglich den Beweis dafür zu erbringen, dass die erhaltenen Resonanzsignale bei �99,1 ppm und �111,2 ppm nicht von Strukturdefekten stammen sondern mit grosser Wahrscheinlichkeit der externen Oberfläche zugeordnet werden können.
Abb. 4.17 1H/29Si CP MAS NMR-Experiment, durchgeführt an dem in einer oxidativen Atmosphäre bei 550°C zwei Tage lang calcinierten Dodecasil 3C-Material mit dem Code-Namen CS024_calciniert.
( ppm )
- 1 5 0- 1 4 0- 1 3 0- 1 2 0- 1 1 0- 1 0 0- 9 0- 8 0- 7 0
Durch den Vergleich zwischen den Abbildungen 4.14 und 4.17 konnte festgestellt werden, dass keine Veränderung zwischen dem 1H/29Si CP MAS NMR-Spektrum der CS024_as-synthesized-Probe und dem 1H/29Si CP MAS NMR-Spektrum der CS024_calciniert-Probe vorhanden war.
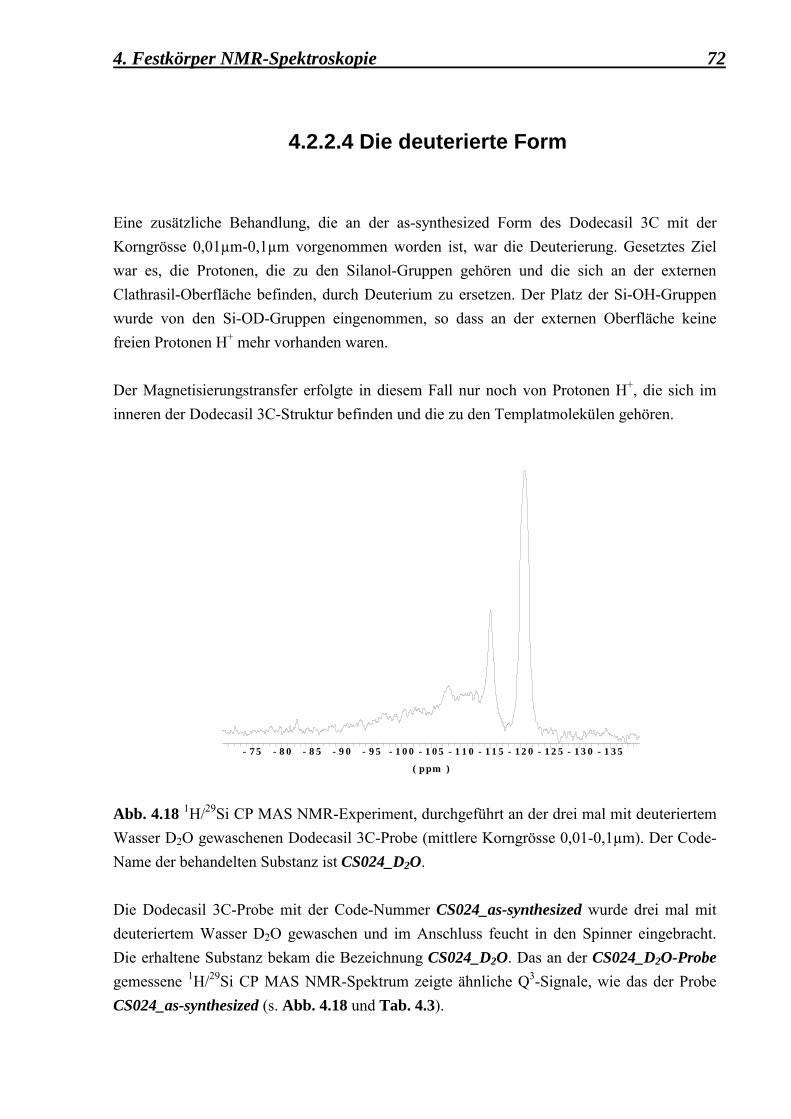
4. Festkörper NMR-Spektroskopie 72
4.2.2.4 Die deuterierte Form
Eine zusätzliche Behandlung, die an der as-synthesized Form des Dodecasil 3C mit der Korngrösse 0,01µm-0,1µm vorgenommen worden ist, war die Deuterierung. Gesetztes Ziel war es, die Protonen, die zu den Silanol-Gruppen gehören und die sich an der externen Clathrasil-Oberfläche befinden, durch Deuterium zu ersetzen. Der Platz der Si-OH-Gruppen wurde von den Si-OD-Gruppen eingenommen, so dass an der externen Oberfläche keine freien Protonen H+ mehr vorhanden waren. Der Magnetisierungstransfer erfolgte in diesem Fall nur noch von Protonen H+, die sich im inneren der Dodecasil 3C-Struktur befinden und die zu den Templatmolekülen gehören.
Abb. 4.18 1H/29Si CP MAS NMR-Experiment, durchgeführt an der drei mal mit deuteriertem Wasser D2O gewaschenen Dodecasil 3C-Probe (mittlere Korngrösse 0,01-0,1µm). Der Code-Name der behandelten Substanz ist CS024_D2O.
( ppm )
- 1 3 5- 1 3 0- 1 2 5- 1 2 0- 1 1 5- 1 1 0- 1 0 5- 1 0 0- 9 5- 9 0- 8 5- 8 0- 7 5
Die Dodecasil 3C-Probe mit der Code-Nummer CS024_as-synthesized wurde drei mal mit deuteriertem Wasser D2O gewaschen und im Anschluss feucht in den Spinner eingebracht. Die erhaltene Substanz bekam die Bezeichnung CS024_D2O. Das an der CS024_D2O-Probe gemessene 1H/29Si CP MAS NMR-Spektrum zeigte ähnliche Q3-Signale, wie das der Probe CS024_as-synthesized (s. Abb. 4.18 und Tab. 4.3).
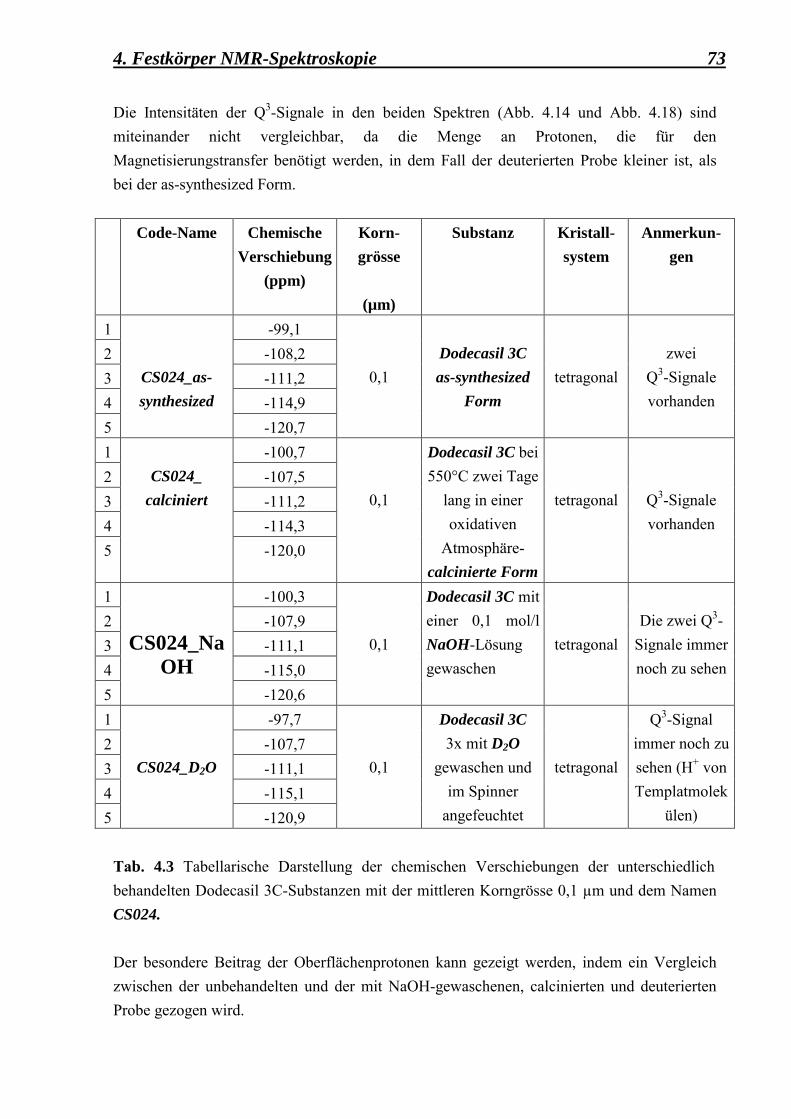
4. Festkörper NMR-Spektroskopie 73 Die Intensitäten der Q3-Signale in den beiden Spektren (Abb. 4.14 und Abb. 4.18) sind miteinander nicht vergleichbar, da die Menge an Protonen, die für den Magnetisierungstransfer benötigt werden, in dem Fall der deuterierten Probe kleiner ist, als bei der as-synthesized Form.
Code-Name Chemische Verschiebung
(ppm)
Korn-grösse
(µm)
Substanz Kristall- system
Anmerkun- gen
1 -99,1 2 -108,2 3 -111,2 4 -114,9 5
CS024_as-synthesized
-120,7
0,1
Dodecasil 3C as-synthesized
Form
tetragonal
zwei
Q3-Signale vorhanden
1 -100,7 2 -107,5 3 -111,2 4 -114,3 5
CS024_
calciniert
-120,0
0,1
Dodecasil 3C bei 550°C zwei Tage
lang in einer oxidativen
Atmosphäre-calcinierte Form
tetragonal
Q3-Signale vorhanden
1 -100,3 2 -107,9 3 -111,1 4 -115,0 5
CS024_NaOH
-120,6
0,1
Dodecasil 3C mit einer 0,1 mol/l NaOH-Lösung gewaschen
tetragonal
Die zwei Q3-
Signale immer noch zu sehen
1 -97,7 2 -107,7 3 -111,1 4 -115,1 5
CS024_D2O
-120,9
0,1
Dodecasil 3C 3x mit D2O
gewaschen und im Spinner
angefeuchtet
tetragonal
Q3-Signal immer noch zu sehen (H+ von Templatmolek
ülen)
Tab. 4.3 Tabellarische Darstellung der chemischen Verschiebungen der unterschiedlich behandelten Dodecasil 3C-Substanzen mit der mittleren Korngrösse 0,1 µm und dem Namen CS024.
Der besondere Beitrag der Oberflächenprotonen kann gezeigt werden, indem ein Vergleich zwischen der unbehandelten und der mit NaOH-gewaschenen, calcinierten und deuterierten Probe gezogen wird.

4. Festkörper NMR-Spektroskopie 74
Jedes durchgeführte Experiment hat etwas typisches und gibt eine für das Oberflächen-Problem charakteristische Antwort. So verfügt die as-synthesized Probe sowohl über Oberflächen-Protonen, als auch über Templat-Protonen. Das NMR-Spektrum zeigt 29Si-Signale von der Oberfläche und 29Si-Signale von dem Bulk. Die mit einer schwach basischen Lösung behandelte Probe verfügt, genauso wie die as-synthesized Probe, über Oberflächen-Protonen und Templat-Protonen. Das NMR-Spektrum zeigt ähnliche Signale wie vorher. Die Bestätigung der Oberflächendetektion mit Hilfe der Kernresonanzspektroskopie wurde dadurch erbracht. Die calcinierte Probe besitzt überwiegend Oberflächenprotonen, da die SDA-Moleküle (Templat-Moleküle) im Inneren der Käfige zerstört wurden. Die erhaltenen Q3-Oberflächensignale zeigen eine erhöhte Intensität im Vergleich zu den Bulk-Signalen. Durch die Deuterierung der Probe existieren nur noch SDA-Protonen, die für den Magnetisierungstransfer sorgen können. Die Oberflächen-Protonen wurden durch Deuterium ersetzt und die Q4-Signale sind im Vergleich zu den Q3-Signale ausgeprägter.
4.2.3 NMR-Untersuchungen an fluorierten „fein-
körnigen“ Dodecasil 3C-Materialien
4.2.3.1 19F MAS NMR-Experimente, durchgeführt an einer
mit Flusssäure HF behandelte Dodecasil 3C-Probe
(mittlere Korngrösse: 0,1 µm)
Die Lösung ergab sich durch das Mischen von 0,06734 g Dodecasil 3C-Kristallen (mittlere Korngrösse 0,1 µm) mit 0,03845 g (2 mmol) Kaliumfluorid KF, 0,111 ml H2SO4 (98%) und 19,889 ml H2O. Die erhaltene Masse wurde bei 100°C eine Stunde lang gerührt und anschliessend im Trockenschrank bei 110°C getrocknet. Die Fluorierungsreaktionen sind im folgenden Reaktionsschema dargestellt:
2KF + H2SO4 → K2SO4 + 2HF
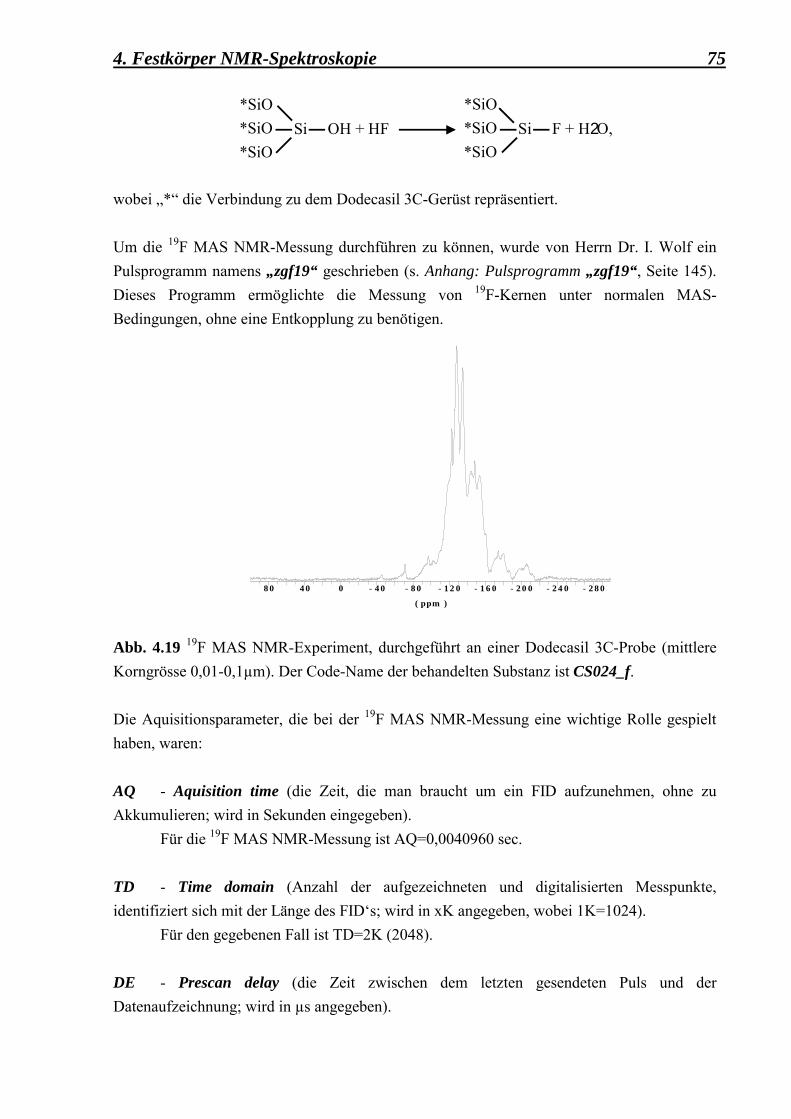
4. Festkörper NMR-Spektroskopie 75
wobei �*� die Verbindung zu dem Dodecasil 3C-Gerüst repräsentiert. Um die 19F MAS NMR-Messung durchführen zu können, wurde von Herrn Dr. I. Wolf ein Pulsprogramm namens „zgf19“ geschrieben (s. Anhang: Pulsprogramm „zgf19“, Seite 145). Dieses Programm ermöglichte die Messung von 19F-Kernen unter normalen MAS-Bedingungen, ohne eine Entkopplung zu benötigen.
*SiO*SiO*SiO
Si OH + HF *SiO*SiO*SiO
Si F + H2O,
Abb. 4.19 19F MAS NMR-Experiment, durchgeführt an einer Dodecasil 3C-Probe (mittlere Korngrösse 0,01-0,1µm). Der Code-Name der behandelten Substanz ist CS024_f.
( ppm )
- 2 8 0- 2 4 0- 2 0 0- 1 6 0- 1 2 0- 8 0- 4 004 08 0
Die Aquisitionsparameter, die bei der 19F MAS NMR-Messung eine wichtige Rolle gespielt haben, waren: AQ - Aquisition time (die Zeit, die man braucht um ein FID aufzunehmen, ohne zu Akkumulieren; wird in Sekunden eingegeben). Für die 19F MAS NMR-Messung ist AQ=0,0040960 sec. TD - Time domain (Anzahl der aufgezeichneten und digitalisierten Messpunkte, identifiziert sich mit der Länge des FID�s; wird in xK angegeben, wobei 1K=1024). Für den gegebenen Fall ist TD=2K (2048). DE - Prescan delay (die Zeit zwischen dem letzten gesendeten Puls und der Datenaufzeichnung; wird in µs angegeben).

4. Festkörper NMR-Spektroskopie 76
DE betrug 4,5 µs. D1 - Relaxationszeit (die Zeit, die ein Spin braucht, um in den ursprünglichen Gleichgewichtszustand zurückzukehren; wird in Sekunden eingegeben). In diesem Fall war die D1=5 s. P1 - Pulslänge (entspricht unter normalen Bedingungen dem 90°-Puls; wird in µs gemessen). Für die 19F MAS NMR-Messung war der 90°-Puls P1=P90°=10µs lang. NS - Number of scans (die Zahl der aufgenommenen Scans). NS=1000 Zu den Aquisitionsparametern kamen noch Prozessparameter hinzu, wie z. B.: LB - Line-Broadning (die resultierende Linienverbreiterung ist normalerweise grösser als Null und ersetzt die Sensitivität; wird in Hertz eingegeben). Für den gegebenen Fall wurde keine Linienverbreiterung vorgenommen LB=0 Hz. SI - Size (die Zahl der Datenpunkte, mit deren Hilfe die Fourier-Transformation berechnet werden konnte; muss immer eine Potenz von 2 sein). SI=2K (2048). Das 19F MAS NMR-Spektrum zeigte scharfe, deutlich voneinander getrennte Resonanzlinien bei den chemischen Verschiebungen: -123 ppm, -128,5 ppm, -135,1 ppm, -144,3 ppm, -149,8 ppm, �154,1 ppm und den dazu gehörenden Seitenbanden. Da es in der Literatur keine Referenzdaten über die chemischen Verschiebungen verschieden fluorierter Si-O-Fx-Verbindungen gibt, konnte man die erhaltenen Resonanzsignale nicht entsprechend zuordnen. Es besteht die Möglichkeit, dass nur ein Teil der externen Oberfläche fluoriert wurde, was durch die Existenz der beidseitig paarweise auftretenden Seitenbanden angedeutet wird. Des weiteren können auch Nebenprodukte, wie z. B. Natriumtetrafluorosiloxan NaSiF2-O-SiF2Na (Hollemann und Wiberg, 1985) oder Natriumhexafluorosilikat Na2SiF6 (Wilmes, 1993) zu dem Auftreten der entsprechenden Signale führen. Schlussfolgerung ist, dass die Beladung erfolgreich verlaufen ist, da durch die selektive Zugänglichkeit des Porensystems und die Einschränkungen im Reaktionsvolumen die HF-Moleküle nicht in den Pentagondodecaeder-Käfige eingedrungen sind, sondern an der externen Clathrasil-Oberfläche haften geblieben sind.

4. Festkörper NMR-Spektroskopie 77
4.2.3.2 19F/29Si CP MAS NMR-Experimente, durchgeführt
an einer mit HF beladene Dodecasil 3C-Probe
(mittlere Korngrösse: 0,1 µm)
Da die innere Oberfläche des Dodecasil 3C nur über 6er-Ringe verfügt, die einen freien Durchmesser von ca. 2Å aufweisen und für Flusssäure-Moleküle bei der Fluorierung nicht zugänglich ist, können 19F/29Si Cross-Polarisationsexperimente durchgeführt werden. So besteht die Möglichkeit, einen Magnetisierungstransfer von 19F-Kernen der fluorierten D3C-Oberfläche auf die benachbarten 29Si-Kerne durchzuführen. Dadurch wird die Nahordnung der Oberflächensiliziumatome eindeutig identifiziert und analysiert. Basierend auf den Erfahrungen, die die Arbeitsgruppen aus Mulhouse (Hoffner, F. M., Delmotte, L. und Kessler, H.) und Münster über das 19F/29Si CP MAS NMR-Experimentgesammelt haben, wurde die relativ neue Cross-Polarisations-Technik eingesetzt, um Infor-mationen über den Aufbau der externen Oberfläche zu bekommen. Für die Einstellung der Hartmann-Hahn-Bedingungen wurde als Standard Natriumhexafluorosilikat benutzt. Die NMR-Spektren wurden mit Hilfe eines Bruker ASX 400-Spektrometers aufgenommen. Die chemische Verschiebung der 19F und 29Si-Kerne wurde in Bezug auf CFCl3 bzw. TMS festgelegt. Für die CP-Sequenz wurde von Dr. I. Wolf ein Programm namens „cp19f.3“ erstellt (s. Anhang: Pulsprogramm „cp19f.3“, Seite 146). Die wichtigsten Aquisitionsparameter, die bei der 19F/29Si CP MAS NMR-Messung eine Rolle gespielt haben, waren: Aquisition time AQ: 0,0512 s Time domain TD: 1024 (1K) Prescan delay DE: 71,4 µs Relaxationszeit D1=V9: 5 s Number of scans NS: 30000 Die für die Messung angewandten Prozessparameter waren: Line-Broadning LB: 160 Hz Size SI: 1024 (1K)

4. Festkörper NMR-Spektroskopie 78 Das an der Dodecasil 3C-Substanz mit dem Code-Namen CS024_f gemessene 19F/29Si CP MAS NMR-Spektrum zeigte bei den chemischen Verschiebungen �97,6 ppm und �115,7 ppm zwei gut voneinander getrennte Resonanzsignale (Abb. 4.20).
Abb. 4.20 19F/29Si CP MAS NMR-Experiment, durchgeführt an einer Dodecasil 3C-Probe mit dem Code-Namen CS024_f (mittlere Korngrösse 0,01-0,1µm).
( ppm )
- 1 5 0- 1 4 0- 1 3 0- 1 2 0- 1 1 0- 1 0 0- 9 0- 8 0- 7 0- 6 0- 5 0
Durch den an dem 19F/29Si CP MAS NMR-Spektrum vorgenommenen Plot war es möglich, ein zusätzliches Resonanzsignal bei der chemischen Verschiebung von �107,5 ppm zu beobachten. Die Schwierigkeit bestand darin, die drei erhaltenen Resonanzsignale den entsprechenden T- bzw. S-Sites Positionen zuzuordnen. Das es keine Literaturangaben zu ähnlichen Verbindungen gab, besteht die Möglichkeit, dass nicht nur eine der OH-Gruppen an der externen Oberfläche durch Fluor ersetzt wurde und eine Struktur der Art:
ergab, sondern mehrere (zwei oder drei) F-Atome sich an die externe Oberfläche angelagert haben und an die Oberflächensiliziumatome banden:
*SiO*SiO*SiO
Si F

4. Festkörper NMR-Spektroskopie 79
Fazit ist, dass die an einem nahezu perfekt kristallinen Clathrasil-Material durchgeführte 19F/29Si CP MAS NMR-Messung es ermöglicht hat, die externe Oberfläche �sichtbar� zu machen und gleichzeitig die Theorie bekräftigt hat, dass die in dem 1H/29Si CP MAS NMR-Experiment erhaltenen Q3-Signale der Oberfläche zugeordnet werden können.
F*SiO
*SiOSi
F
F*SiO Si
FFund

4. Festkörper NMR-Spektroskopie 80
4.3 Zusammenfassung der Synthese-
Experimente und der Kreuzpolarisations-
Untersuchungen 1H/29Si CP MAS NMR und 19F/29Si CP MAS NMR
1. Die Synthese-Experimente der vorliegenden Arbeit lieferten Dodecasil 3C-
Kristalle mit einer mittlern Korngrösse von ca. 0,1 µm, wie die Rasterelektronenmikroskopischen Aufnahmen zeigen (Abb. 3.4 ii).
2. Die Synthesebedingungen wurden in solcher Weise optimiert, dass das Verhältnis
Oberfläche zu Volumen (Bulk) 100 bis 1000 fach vergrössert worden ist, um so die externe Clathrasiloberfläche für das NMR-Experiment �sichtbar� zu machen. Die Konzentration der Si-Quelle, im gegebenen Fall Aerosil, wurde so hoch wie möglich gebracht (4 mol/l), um möglichst kleine Dodecasil 3C-Kristalle zu synthetisieren. Oberhalb von 4 mol/l-Aerosil entstanden keine Dodecasil 3C-Kristalle. Die Synthesetemperatur wurde auf 160°C eingestellt, so niedrig wie möglich, um die Keimbildung zu unterstützen und den Einfluss des Kristallwachstums zu reduzieren. Bei Temperaturen über 160°C wachsen die Kristalle zu gross. Bei Temperaturen unter 160°C entstehen fremde Phasen aus der Reihe der Schichtsilikate wie z. B. RUB-15. Die Arbeitsweise zeigte sich gegen alle Erwartungen als ein Faktor, der wenig Einfluss auf die Kristallitgrösse ausgeübt hat. Unter ständigem Rühren oder Rotieren während der Synthese ist die Keimbildung unterdrückt, und die Kristalle wachsen viel schneller und viel grösser als erwartet. Unter statischen Synthesebedingungen waren die hergestellten Dodecasil 3C-Kristalle die kleinsten. Die Synthesedauer reduzierte sich auf 3 Tage. Über diese Synthesezeit hinaus wuchsen die Dodecasil 3C-Kristalle zu gross. Die Füllhöhe war der entscheidende Faktor, der die Züchtung von Kristallen mit Korngrössen von 0,01µm-0,1µm ermöglicht hat. Durch das Füllen des Teflon-Behälters auf 1/3 zeigten die Kristalle am Ende der Synthese die bis dahin kleinste Korngrösse.

4. Festkörper NMR-Spektroskopie 81
3. Das Röntgenbeugungsdiagramm der CS024_as-synthesized-Verbindung mit der
Korngrösse 0,01µm-0,1µm zeigte starke, scharfe Reflexe, die tetragonal indiziert wurden.
4. Die an der CS024_as-synthesized-Probe durchgeführten 1H/29Si CP MAS NMR-
Untersuchungen zeigten, ausser den drei gewöhnlichen Bulk-Q4-Signalen, entsprechend den verschiedenen T-Sites-Positionen (T1(Si3):-108,2 ppm, T2(Si2): -114,9 ppm, T3(Si1): -120,7 ppm) noch zwei zusätzliche Oberflächen-Q3-Signale, entsprechend den an der externen Oberfläche symmetrisch inäquivalenten Lagen (S2(Si2): -99,1 ppm und S3(Si1): -111,2 ppm).
5. Die zwei in dem 1H/29Si CP MAS NMR-Spektrum erhaltenen Q3-Signale wurden
in dem reproduzierten Dodecasil 3C-Material wiedergefunden. 6. Die Existenz von zwei verschiedenen Q3-Signalen, die bei niedrigeren
chemischen Verschiebungen im Vergleich zu den entsprechenden Q4-Signalen auftreten, erlaubte es, die Hypothese aufzustellen, dass die externe Oberfläche des Clathrasils Dodecasil 3C eine geordnete Terminierung der Volumenstruktur vorweist und somit kristallin sein kann.
7. Die zusätzlichen Behandlungen, die an der CS024_as-synthesized-Verbindung
vorgenommen worden sind, wie z.B. Calcinieren oder das Waschen mit einer NaOH-Lösung, haben es ermöglicht, Artefakte wie Strukturdefekte oder nicht umgewandeltes Silicagel auszuschliessen. Fazit ist, dass die Q3-Signale, die in dem 1H/29Si CP MAS NMR-Spektrum der as-synthesized-Form der Dodecasil 3C-Substanz zu beobachten sind, der externen Clathrasil-Oberfläche zugeordnet werden konnten. Die Auswertung der Spektren ergab, dass die Oberfläche ein H-terminierter Ausschnitt der Volumenstruktur ist, die mit grosser Wahrscheinlichkeit von der Pentagondodecaeder-Schicht gebildet wird.
8. Die Präsenz der drei Resonanzsignale, die in dem 19F/29Si CP MAS NMR-
Spektrum bei �97,6 ppm, -107,5 ppm und �115,7 ppm zu beobachten waren, bekräftigt die Theorie, dass die externe Dodecasil 3C-Oberfläche mit Hilfe der NMR-Spektroskopie detektierbar ist, obwohl die Kernresonanzspektroskopie keine typisch oberflächensensitive Methode ist.

5. Relaxations- und Kontaktzeitvariationsmessungen 82
5. Zusätzliche NMR-Untersuchungen an externen Clathrasil-Oberflächen
5.1 Theoretische Betrachtung der Relaxationszeiten
5.1.1 Der Zusammenhang zwischen Blochschen
Gleichungen und Relaxationszeiten Unter Relaxationszeit ist die Zeit zu verstehen, die ein Kernspinsystem braucht, um in den Gleichgewichtszustand zurückzukehren. So vergrössert sich die mit der Zeit t variierende Komponente Mz wieder auf M0, während die beiden anderen Komponenten Mx und My gegen null gehen.
Abb. 5.1 �Der makroskopische Magnetisierungsvektor M0 wurde unter der Einwirkung eines Impulses um den Winkel θ aus seiner Gleichgewichtslage herausgedreht und präzediert anschliessend mit der Larmor-Frequenz νL. Im ortsfesten Koordinatensystem hat M0 zur Zeit t die Koordinaten Mx, My und Mz� (Friebolin, 1992).
x
y
z
My
Mx
Mz
M0
ϕϕϕϕ
θθθθ

5. Relaxations- und Kontaktzeitvariationsmessungen 83
Die quantitative Beschreibung des Relaxationsphänomens erfolgt durch die Blochschen Gleichungen. Nach der klassischen Theorie gilt für die Bewegung eines magnetischen Momentes M, das sich in dem Magnetfeld B befindet, die Gleichung (5.1):
Mit γ wird das gyromagnetische Verhältnis bezeichnet. Das Feld B besteht aus zwei Magnetfeldern: einem statischen, bezeichnet durch B0, und einem rotierenden, bezeichnet durch B1. Im kartesischen Koordinatensystem nimmt B0 die z-Richtung an, während B1 in der x,y-Ebene im Uhrzeigersinn mit der Frequenz ω rotiert :
Bx = B1cos(ωt) By = -B1sin(ωt) (5.2) Bz = B0
Durch das Einsetzen der oberen Gleichungen (5.5) in die Gleichung (5.4), erhält man:
Für die zeitliche Änderung der Magnetisierung muss zusätzlich die Relaxation mit den damit verbundenen Relaxationszeiten berücksichtigt werden. Hierfür wurden von Felix Bloch empirisch die beiden Zeitkonstanten T1 und T2 eingeführt, die die Relaxation der Magnetisierung parallel zum äusseren Magnetfeld, dass heisst entlang der z-Richtung, bzw. senkrecht dazu, als voneinander unabhängige Grössen beschreiben.
[ ]BMdt
dM ×= γ
( )[ ]tBMBMdt
dMzy
x ωγ sin10 +=
( )[ ]tBMBMdt
dMzx
y ωγ cos10 −−=
( ) ( )[ ]tBMtBMdt
dMyx
z ωωγ cossin 11 −−=
(5.1)
(5.3)

5. Relaxations- und Kontaktzeitvariationsmessungen 84
Der vollständige Satz der Blochschen Gleichungen wird dann durch folgende Formeln beschrieben:
In einem simplifizierten Fall, für
M(0) = (0, M0, 0), (5.5) bei der Zeit t = 0 wird die Änderung der Magnetisierung M(0) nur durch die y-Koordinate beeinflusst. So ergibt sich als Lösung:
Für den Abbau der transversalen Magnetisierung nach einem eingestrahlten HF-Impuls in der (x,y)-Ebene ist die Relaxationszeit T2 verantwortlich. Die für diesen Prozess charakteristische Zeitkonstante ist unter dem Namen Spin-Spin-Relaxation, auch transversale Relaxationszeit genannt, bekannt. Der Aufbau der Gleichgewichtsmagnetisierung parallel zur z-Achse wird durch die T1-Zeit, die sogenannte Spin-Gitter-Relaxation, beschrieben. Deshalb wird die Spin-Gitter-Relaxationszeit auch longitudinale Relaxationszeit genannt. Zu bemerken ist, dass der Resonanzeffekt nicht bei einer einzigen scharfen Frequenz ω = ω0 auftritt, sondern in einem Bereich ∆ω0 um die Larmor-Frequenz ω0. Zwischen Linienbreite ∆ω0 und Spin-Spin-Relaxationszeit T2 besteht der Zusammenhang:
( )[ ]
( )[ ]
( ) ( )[ ]1
011
201
210
cossin
cos
sin
TMMtBMtBM
dtdM
TM
BMtBMdt
dMTMtBMBM
dtdM
zyx
z
yxz
y
xzy
x
−−−−=
−−=
−+=
ωωγ
ωγ
ωγ
( )
( )
−−=
−=
−=
10
20
20
exp1)(
cosexp)(
expsin)(
TtMtM
tTtMtM
TttMtM
z
y
x
ω
ω
(5.4)
(5.6)

5. Relaxations- und Kontaktzeitvariationsmessungen 85
vorausgesetzt, dass keine zusätzlichen Einflüsse zu einer vergrösserten Linienbreite führen. In Festkörpern ist die Spin-Spin-Relaxation T2 im allgemeinen um mehrere Grössenordnungen kürzer als die Spin-Gitter-Relaxation T1. Deshalb wird die Linienbreite allein durch die T2-Relaxationszeit bestimmt, solange keine zusätzlichen Effekte, wie z. B. Dipol- oder elektrische Quadrupolwechselwirkungen für Energieübergänge sorgen und somit die T1-Relaxationszeit stark beeinflussen (verkürzen). Bekannt ist, dass anstelle des äusseren Magnetfeldes im rotierenden Koordinatensystem nur noch die magnetische Induktion der Resonanzabweichung
wirkt. Dieser auch unter dem Namen �Resonanzoffset� bekannte Effekt verschwindet, wenn die für die untersuchten Spins eingestrahlte Hochfrequenz mit der Larmor-Frequenz
ωL=γB0 (5.9) identisch ist. Das im rotierenden Koordinatensystem effektiv wirkende Feld ist eine vektorielle Addition von Hochfrequenzfeld und Offset:
B=(BHF, 0, B0-ω/γ) Die Untersuchung der kernmagnetischen Relaxation hat besonders beim Studium dynamischer Prozesse eine gewisse Eigenständigkeit erlangt, so dass teilweise zwischen Untersuchungen der kernmagnetischen Resonanz und Untersuchungen der kernmagnetischen Relaxation unterschieden werden kann. Ausserdem können die Relaxationszeiten im Prinzip bestimmt werden, indem man die Breite einzelner Spektrallinien misst. Eine andere Möglichkeit wäre, die Spin-Echo-Techniken einzusetzen, um wertvolle Informationen über die Bewegung der Moleküle zu erhalten.
2
2T
=∆ω
( )γ
ωω −L
(5.7)
(5.8)

5. Relaxations- und Kontaktzeitvariationsmessungen 86
5.1.1.2 Der Zusammenhang zwischen der
Kreuzpolarisationszeit und den heteronuklearen
Abständen Die reziproken Relaxationszeiten T1
-1 und T2-1 entsprechen den Geschwindigkeitskonstanten
für die beiden Relaxationsprozesse Spin-Gitter- und Spin-Spin-Relaxation. Analog dazu wird im Falle eines Kreuzpolarisationsexperimentes eine sogenannte Kreuzpolarisationszeit TIS als Parameter eingeführt, um den zeitlichen Aufbau von Magnetisierung durch Polarisationstransfer vom Spinsystem I zum Spinsystem S zu beschreiben. Die Kreuzpolarisationsrate TIS
-1 hängt folgendermassen mit den zweiten Momenten der heteronuklearen und homonuklearen Dipol-Dipol Wechselwirkungen zusammen:
Die Konstante CIS, die dabei auftritt, wird folgendermassen definiert:
wobei Ni der Anzahl der vorhandenen I-Spins entspricht und die Terme bi, bim und aij durch die folgenden Formeln:
beschrieben werden, wobei P2(cosθ) der Formel entspricht aus der der magische Winkel berechnet werden kann.
III
ISSIS
ISIS C
TT
⟩∆⟨
⟩∆⟨==−
2
21 1
ωω
( ) ( )[ ] 21
122222 223
−
≠
−
≠
+++×= ∑ ∑ ∑
ji i jijijiijiiijIS bbbbaNbaC π
( )imimSIimi Prbb θγγ cos2 232 −== h
( )ijijIij Pra θγ cos2322 −−= h
( ) ( )1cos321cos 2
2 −= θθP
(5.10)
(5.11)
(5.12)

5. Relaxations- und Kontaktzeitvariationsmessungen 87
So werden im Falle einer homonuklearen bzw. heteronuklearen Kopplung, die zweiten Momente durch die folgenden Formeln: homonuklear:
heteronuklear:
definiert. Für ein 1H/29Si CP MAS NMR-Experiment wird das heteronukleare 2. Moment durch die Formel:
beschrieben, wobei: I (der Kernspin I), γ(29Si) = -5,38188x107 s-1T-1 (das gyromagnetische Verhältnis entsprechend des 29Si-Kerns), γ(1H) = 2,67520x108 s-1T-1 (das gyromagnetische Verhältnis entsprechend des 1H-Kerns), h = 6,6262x10-34 Js (Boltzmann Konstante), µ0 = 1,2566x10-6 VsA-1m-1 (die magnetische Konstante des Vakuums). Aus den vorherigen Gleichungen folgt, dass zwischen der Kreuzpolarisationszeit TIS, dem heteronuklearen zweiten Moment <∆ω2
IS> und dem internuklearen Abstand rIS folgende Relation existiert:
aus der entnommen werden kann, dass die Kreuzpolarisationsrate umgekehrt proportional zu dem internuklearen Abstand in der 6-ten Potenz ist.
∑=∆j ij
IIII r 6242 1
209
hγω
∑=∆i im
SIISS r 62222 1
51
hγγω
( ) ∑+=∆ 6222 11
154
rNhII HSiSiH γγω
62 11
rT ISIS
∝∆∝ ω
(5.13)
(5.14)
(5.15)
, (5.16)

5. Relaxations- und Kontaktzeitvariationsmessungen 88
5.2 Experimentelle Daten
5.2.1 Berechnung der Kreuzpolarisationszeiten
5.2.1.1 Kontaktzeitvariationsmessungen, durchgeführt an der
as-syntheiszed Form des Dodecasil 3C, mit einer
mittleren Korngrösse von 0,1 µm Kontaktzeitvariationsexperimente ermöglichen es, Informationen über das Verhalten der Oberfläche, bzw. des Bulkes zu gewinnen. Bekannt ist, dass die as-synthesized Probe über chemisch an Si-OH gebundene Protonen verfügt. Diese sind starr und befinden sich nahe der Oberfläche. Zusätzlich enthält die as-synthesized-Probe auch Templat-Protonen. Diese sind mobil und befinden sich weiter von der Oberfläche entfernt. Die kurzen Abstände zwischen H+, die als Magnetisierungsquelle dienen, und Si-Kernen deuten auf eine schnelle Relaxation hin. Zu erwarten wäre, dass die aus den Kontaktzeitvariationsmessungen erhaltenen Kreuzpolarisationszeiten in diesem Fall kurz sein sollten. Im Vergleich dazu verfügt die deuterierte Probe ausschliesslich über Templat-Protonen, da die Silanol-Protonen durch Deuterium-Atome ersetzt worden sind. Die Abstände zwischen Templat-Protonen und Si-Atomen sind länger, als im Falle der as-synthesized-Probe. Zu erwarten wäre, dass die Relaxation entsprechend länger ist, während die aus den Kontaktzeitvariationsexperimenten erhaltenen Kreuzpolarisationszeiten gross sind. Eine Reihe von 1H/29Si CP MAS NMR-Experimenten mit einer variablen Kontaktzeit zwischen 1 und 100 ms wurde an der as-synthesized Form des Dodecasil 3C mit einer mittleren Korngrösse von 0,1 µm durchgeführt (Probe CS024_as-synthesized). Für die as-synthesized Form des Dodecasil 3C erfolgte der Magnetisierungstransfer sowohl von Protonen H+, die zu den Silanol-Gruppen Si-OH gehören und an der externen Oberfläche angelagert sind, als auch von H+, die sich im Inneren der Pentagondodecaeder�Käfige befinden und die zu den Templatmolekülen gehören.
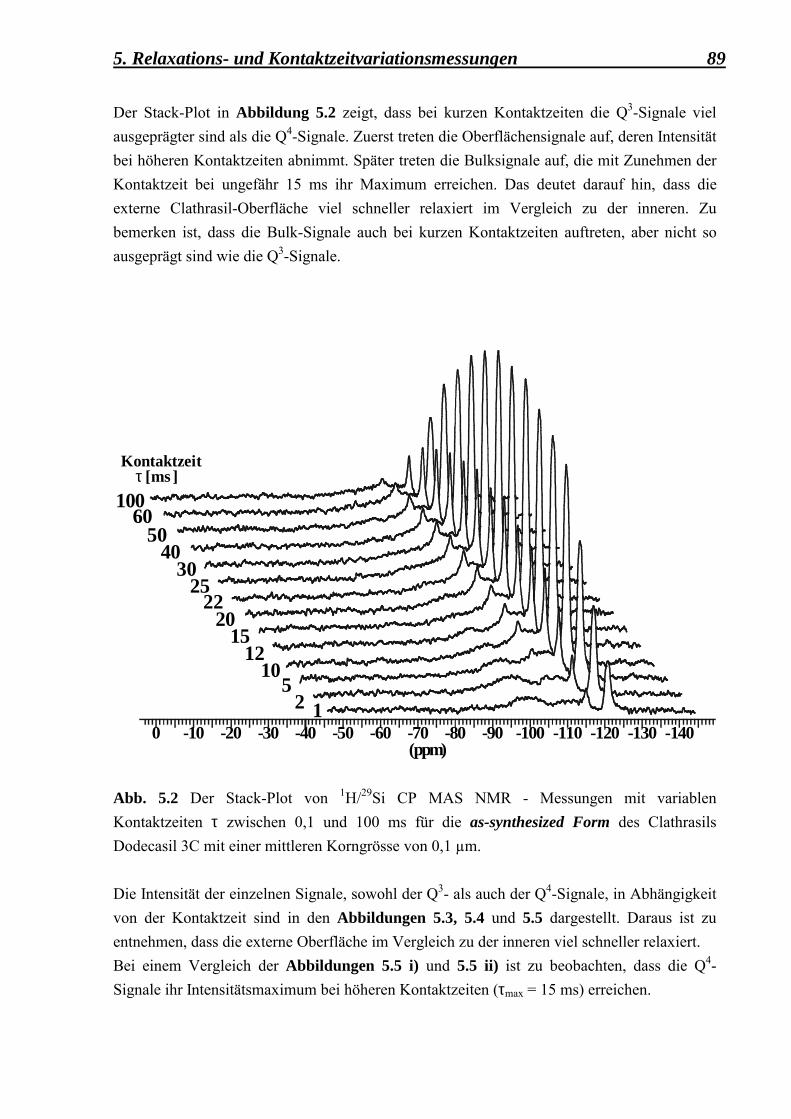
5. Relaxations- und Kontaktzeitvariationsmessungen 89 Der Stack-Plot in Abbildung 5.2 zeigt, dass bei kurzen Kontaktzeiten die Q3-Signale viel ausgeprägter sind als die Q4-Signale. Zuerst treten die Oberflächensignale auf, deren Intensität bei höheren Kontaktzeiten abnimmt. Später treten die Bulksignale auf, die mit Zunehmen der Kontaktzeit bei ungefähr 15 ms ihr Maximum erreichen. Das deutet darauf hin, dass die externe Clathrasil-Oberfläche viel schneller relaxiert im Vergleich zu der inneren. Zu bemerken ist, dass die Bulk-Signale auch bei kurzen Kontaktzeiten auftreten, aber nicht so ausgeprägt sind wie die Q3-Signale.
Abb. 5.2 Der Stack-Plot von 1H/29Si CP MAS NMR - Messungen mit variablen Kontaktzeiten τ zwischen 0,1 und 100 ms für die as-synthesized Form des Clathrasils Dodecasil 3C mit einer mittleren Korngrösse von 0,1 µm.
1
(ppm)-140-130-120-110-100-90-80-70-60-50-40-30-20-100
125
1012
1520
2225
3040
5060
00
Kontaktzeit τ [ms ]
Die Intensität der einzelnen Signale, sowohl der Q3- als auch der Q4-Signale, in Abhängigkeit von der Kontaktzeit sind in den Abbildungen 5.3, 5.4 und 5.5 dargestellt. Daraus ist zu entnehmen, dass die externe Oberfläche im Vergleich zu der inneren viel schneller relaxiert. Bei einem Vergleich der Abbildungen 5.5 i) und 5.5 ii) ist zu beobachten, dass die Q4-Signale ihr Intensitätsmaximum bei höheren Kontaktzeiten (τmax = 15 ms) erreichen.
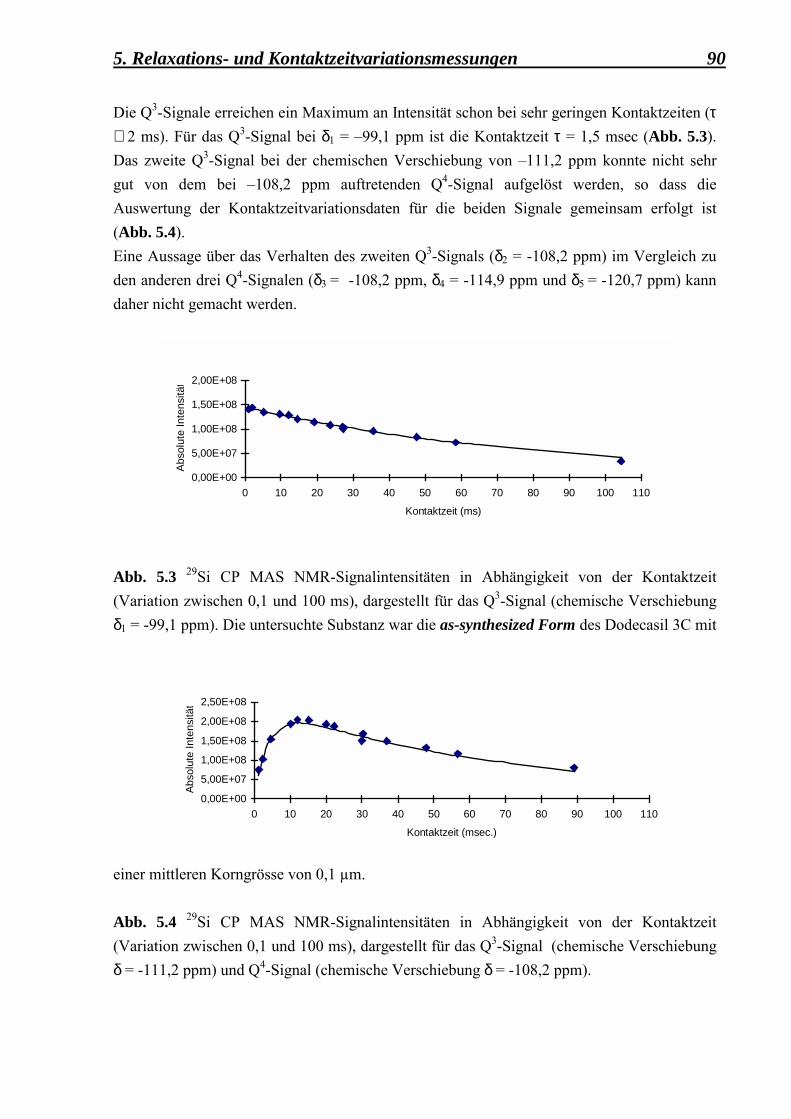
5. Relaxations- und Kontaktzeitvariationsmessungen 90 Die Q3-Signale erreichen ein Maximum an Intensität schon bei sehr geringen Kontaktzeiten (τ ≅ 2 ms). Für das Q3-Signal bei δ1 = �99,1 ppm ist die Kontaktzeit τ = 1,5 msec (Abb. 5.3). Das zweite Q3-Signal bei der chemischen Verschiebung von �111,2 ppm konnte nicht sehr gut von dem bei �108,2 ppm auftretenden Q4-Signal aufgelöst werden, so dass die Auswertung der Kontaktzeitvariationsdaten für die beiden Signale gemeinsam erfolgt ist (Abb. 5.4). Eine Aussage über das Verhalten des zweiten Q3-Signals (δ2 = -108,2 ppm) im Vergleich zu den anderen drei Q4-Signalen (δ3 = -108,2 ppm, δ4 = -114,9 ppm und δ5 = -120,7 ppm) kann daher nicht gemacht werden.
Abb. 5.3 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das Q3-Signal (chemische Verschiebung δ1 = -99,1 ppm). Die untersuchte Substanz war die as-synthesized Form des Dodecasil 3C mit
0,00E+00
5,00E+07
1,00E+08
1,50E+08
2,00E+08
0 10 20 30 40 50 60 70 80 90 100 110
Kontaktzeit (ms)
Abso
lute
Inte
nsitä
t
einer mittleren Korngrösse von 0,1 µm. Abb. 5.4 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das Q3-Signal (chemische Verschiebung δ = -111,2 ppm) und Q4-Signal (chemische Verschiebung δ = -108,2 ppm).
0,00E+00
5,00E+07
1,00E+08
1,50E+08
2,00E+08
2,50E+08
0 10 20 30 40 50 60 70 80 90 100 110
Kontaktzeit (msec.)
Abso
lute
Inte
nsitä
t
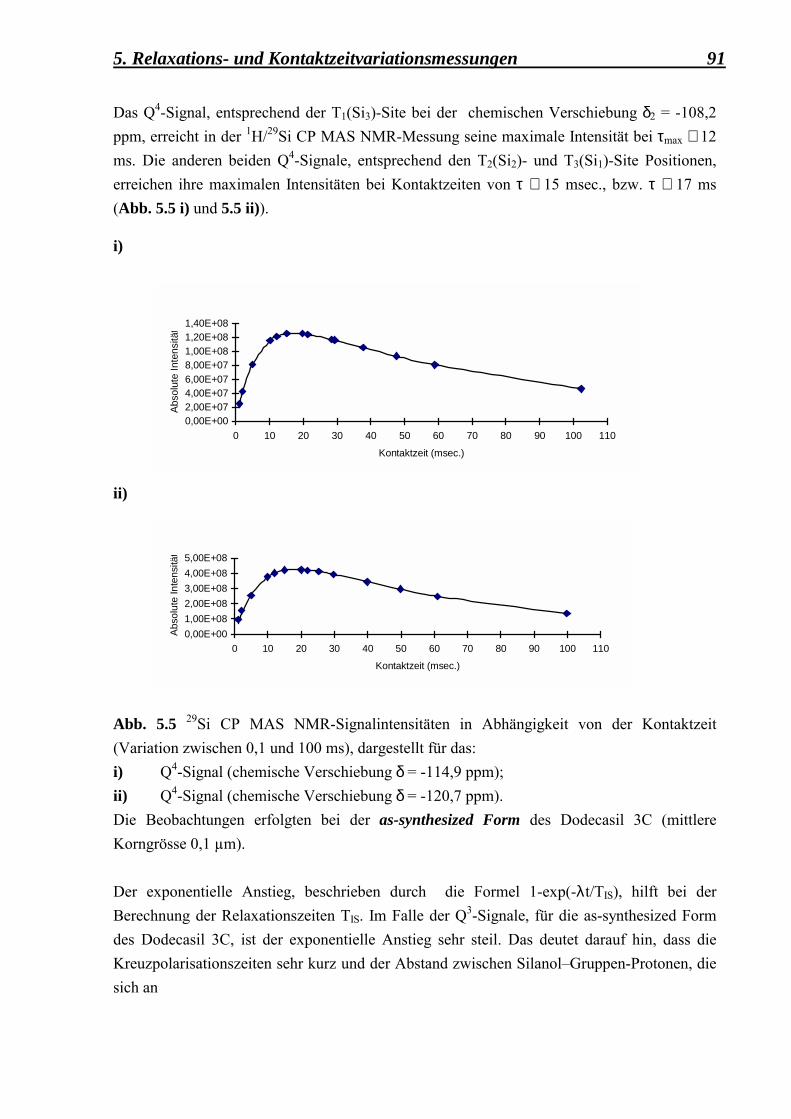
5. Relaxations- und Kontaktzeitvariationsmessungen 91 Das Q4-Signal, entsprechend der T1(Si3)-Site bei der chemischen Verschiebung δ2 = -108,2 ppm, erreicht in der 1H/29Si CP MAS NMR-Messung seine maximale Intensität bei τmax ≅ 12 ms. Die anderen beiden Q4-Signale, entsprechend den T2(Si2)- und T3(Si1)-Site Positionen, erreichen ihre maximalen Intensitäten bei Kontaktzeiten von τ ≅ 15 msec., bzw. τ ≅ 17 ms (Abb. 5.5 i) und 5.5 ii)).
i)
0,00E+002,00E+074,00E+076,00E+078,00E+071,00E+081,20E+081,40E+08
0 10 20 30 40 50 60 70 80 90 100 110
Kontaktzeit (msec.)
Abso
lute
Inte
nsitä
t
ii)
Abb. 5.5 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das: i) Q4-Signal (chemische Verschiebung δ = -114,9 ppm); ii) Q4-Signal (chemische Verschiebung δ = -120,7 ppm). Die Beobachtungen erfolgten bei der as-synthesized Form des Dodecasil 3C (mittlere Korngrösse 0,1 µm).
0,00E+001,00E+082,00E+083,00E+084,00E+085,00E+08
0 10 20 30 40 50 60 70 80 90 100 110
Kontaktzeit (msec.)
Abso
lute
Inte
nsitä
t
Der exponentielle Anstieg, beschrieben durch die Formel 1-exp(-λt/TIS), hilft bei der Berechnung der Relaxationszeiten TIS. Im Falle der Q3-Signale, für die as-synthesized Form des Dodecasil 3C, ist der exponentielle Anstieg sehr steil. Das deutet darauf hin, dass die Kreuzpolarisationszeiten sehr kurz und der Abstand zwischen Silanol�Gruppen-Protonen, die sich an

5. Relaxations- und Kontaktzeitvariationsmessungen 92
der äusseren Clathrasil-Oberfläche befinden und die zusammen mit den Bulk-Protonen für den Magnetisierungstransfer sorgen, und den �externen� Si-Atome klein ist. Im Falle der Q4-Signale ist der exponentielle Anstieg flach, was sich auch in den längeren Kreuzpolarisationszeiten widerspiegelt. Der exponentielle Abfall, beschrieben durch die Formel exp(-t/T1ρI), zeigt keinen deutlichen Unterschied zwischen dem Verlauf der exponentiellen Kurven für die Q3-Signale und den exponentiellen Kurven für die Q4-Signale. Aus diesem Grund wird der exponentielle Abstieg nur für die weitere Berechnung der Relaxationszeiten T1ρI wichtig sein. Fazit ist, dass die Kontaktzeitvariationsmessungen, die an der as-synthesized Form des Dodecasil 3C durchgeführt worden sind, es ermöglicht haben, Schlussfolgerungen über das Verhalten der externen Clathrasil-Oberfläche zu ziehen, so dass behauptet werden kann, dass die äussere Oberfläche viel schneller im Vergleich zu der inneren relaxiert.
5.2.1.2 Kontaktzeitvariationsmessungen, durchgeführt an der
deuterierten Form des Dodecasil 3C, mit einer
mittleren Korngrösse von 0,1 µm Eine andere Reihe von 1H/29Si CP MAS NMR-Experimenten mit einer variablen Kontaktzeit zwischen 1 und 100 ms wurde an der deuterierten Form des Dodecasil 3C mit einer mittleren Korngrösse von 0,1 µm durchgeführt (Probe CS-024_deuteriert). In diesem Fall erfolgte der Magnetisierungstransfer für die deuterierte Form des Dodecasil 3C nur von H+, die sich im Inneren der Pentagondodecaeder�Käfige befinden und zu den Templatmolekülen gehören. Durch die Deuterierung wurde erreicht, dass die Protonen H+, gehörend zu den Silanol-Gruppen Si-OH an der externen Oberfläche, durch Deuterium ersetzt worden sind. Der Stack-Plot in der Abbildung 5.6 zeigt, dass bei kurzen Kontaktzeiten zuerst die Q4-Signale auftreten, denen viel später die Q3-Signale folgen. Bei sehr kurzen Kontaktzeiten sind überhaupt keine Q3-Signale zu beobachten. Je grösser die Kontaktzeit wird, desto deutlicher treten die Q3-Signale auf, so dass das Intensitätsmaximum nur sehr spät, bei der Kontaktzeit τ = 12 ms, erreicht wird.
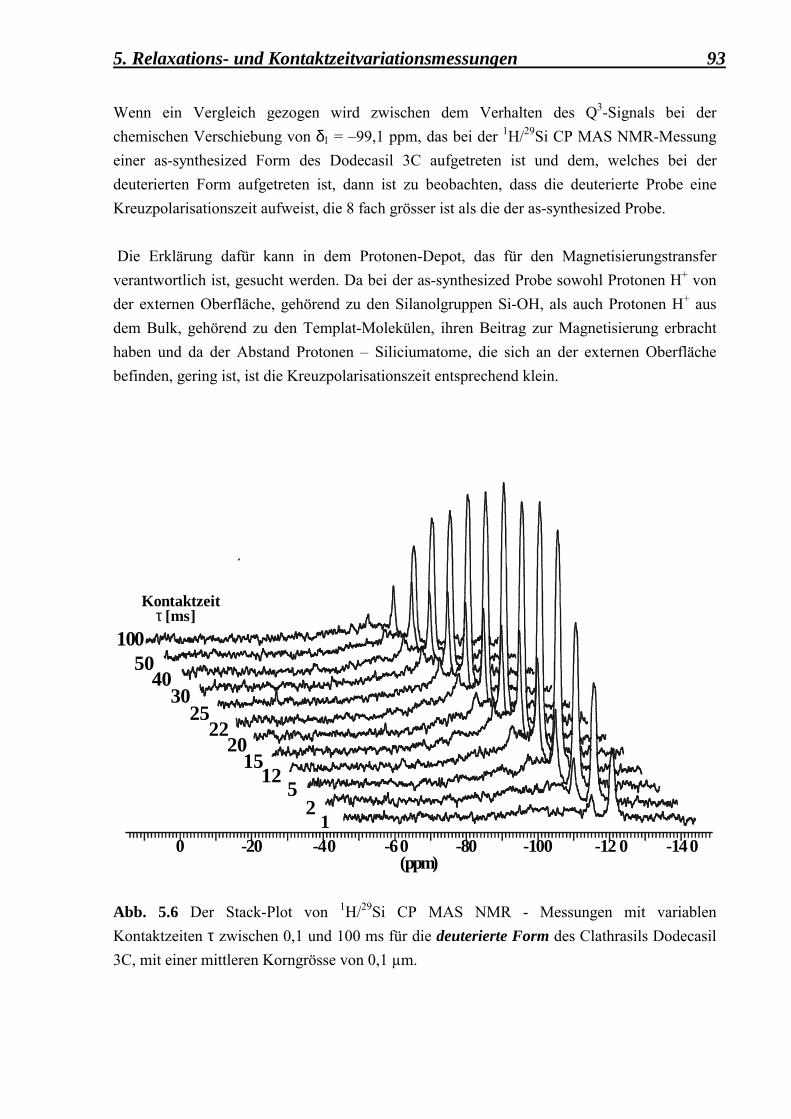
5. Relaxations- und Kontaktzeitvariationsmessungen 93 Wenn ein Vergleich gezogen wird zwischen dem Verhalten des Q3-Signals bei der chemischen Verschiebung von δ1 = �99,1 ppm, das bei der 1H/29Si CP MAS NMR-Messung einer as-synthesized Form des Dodecasil 3C aufgetreten ist und dem, welches bei der deuterierten Form aufgetreten ist, dann ist zu beobachten, dass die deuterierte Probe eine Kreuzpolarisationszeit aufweist, die 8 fach grösser ist als die der as-synthesized Probe. Die Erklärung dafür kann in dem Protonen-Depot, das für den Magnetisierungstransfer verantwortlich ist, gesucht werden. Da bei der as-synthesized Probe sowohl Protonen H+ von der externen Oberfläche, gehörend zu den Silanolgruppen Si-OH, als auch Protonen H+ aus dem Bulk, gehörend zu den Templat-Molekülen, ihren Beitrag zur Magnetisierung erbracht haben und da der Abstand Protonen � Siliciumatome, die sich an der externen Oberfläche befinden, gering ist, ist die Kreuzpolarisationszeit entsprechend klein.
Abb. 5.6 Der Stack-Plot von 1H/29Si CP MAS NMR - Messungen mit variablen Kontaktzeiten τ zwischen 0,1 und 100 ms für die deuterierte Form des Clathrasils Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm.
(pp )
1
-140-12 0-100-80-60-40-200m
12
51215
2022
2530
4050
00
Kontaktzeit τ [ms]
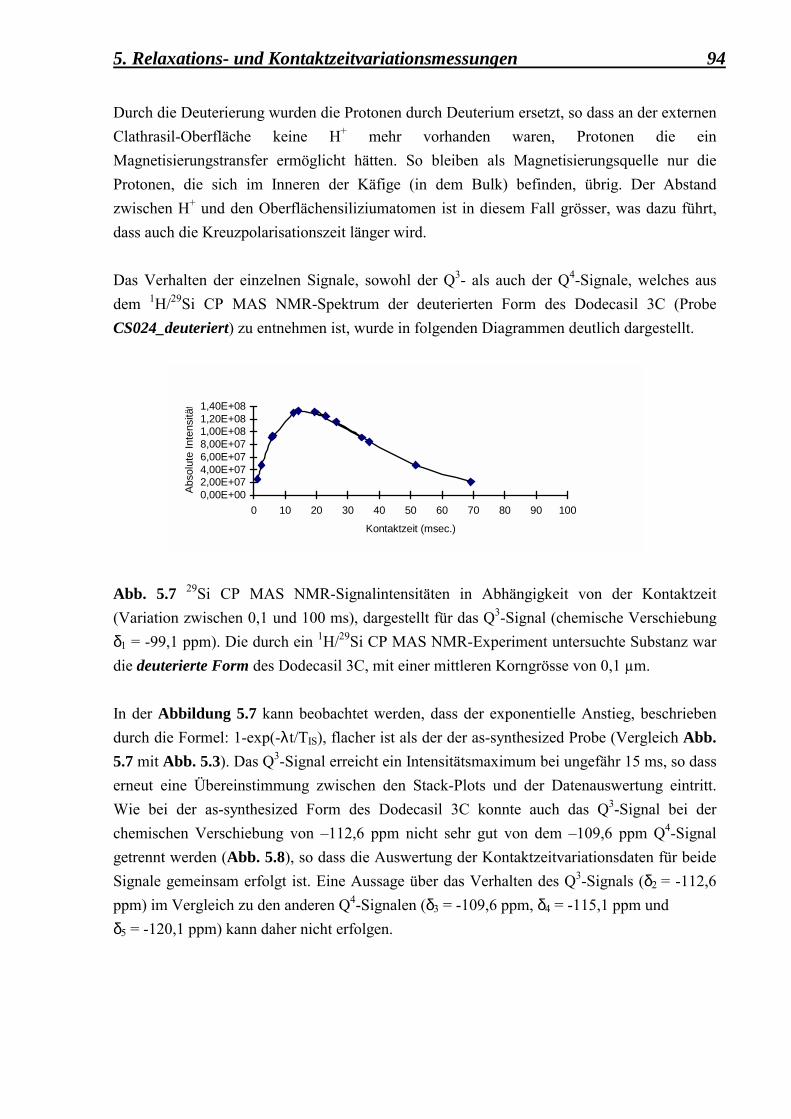
5. Relaxations- und Kontaktzeitvariationsmessungen 94 Durch die Deuterierung wurden die Protonen durch Deuterium ersetzt, so dass an der externen Clathrasil-Oberfläche keine H+ mehr vorhanden waren, Protonen die ein Magnetisierungstransfer ermöglicht hätten. So bleiben als Magnetisierungsquelle nur die Protonen, die sich im Inneren der Käfige (in dem Bulk) befinden, übrig. Der Abstand zwischen H+ und den Oberflächensiliziumatomen ist in diesem Fall grösser, was dazu führt, dass auch die Kreuzpolarisationszeit länger wird. Das Verhalten der einzelnen Signale, sowohl der Q3- als auch der Q4-Signale, welches aus dem 1H/29Si CP MAS NMR-Spektrum der deuterierten Form des Dodecasil 3C (Probe CS024_deuteriert) zu entnehmen ist, wurde in folgenden Diagrammen deutlich dargestellt.
Abb. 5.7 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das Q3-Signal (chemische Verschiebung δ1 = -99,1 ppm). Die durch ein 1H/29Si CP MAS NMR-Experiment untersuchte Substanz war die deuterierte Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm.
0,00E+002,00E+074,00E+076,00E+078,00E+071,00E+081,20E+081,40E+08
0 10 20 30 40 50 60 70 80 90 100
Kontaktzeit (msec.)
Abso
lute
Inte
nsitä
t
In der Abbildung 5.7 kann beobachtet werden, dass der exponentielle Anstieg, beschrieben durch die Formel: 1-exp(-λt/TIS), flacher ist als der der as-synthesized Probe (Vergleich Abb. 5.7 mit Abb. 5.3). Das Q3-Signal erreicht ein Intensitätsmaximum bei ungefähr 15 ms, so dass erneut eine Übereinstimmung zwischen den Stack-Plots und der Datenauswertung eintritt. Wie bei der as-synthesized Form des Dodecasil 3C konnte auch das Q3-Signal bei der chemischen Verschiebung von �112,6 ppm nicht sehr gut von dem �109,6 ppm Q4-Signal getrennt werden (Abb. 5.8), so dass die Auswertung der Kontaktzeitvariationsdaten für beide Signale gemeinsam erfolgt ist. Eine Aussage über das Verhalten des Q3-Signals (δ2 = -112,6 ppm) im Vergleich zu den anderen Q4-Signalen (δ3 = -109,6 ppm, δ4 = -115,1 ppm und δ5 = -120,1 ppm) kann daher nicht erfolgen.
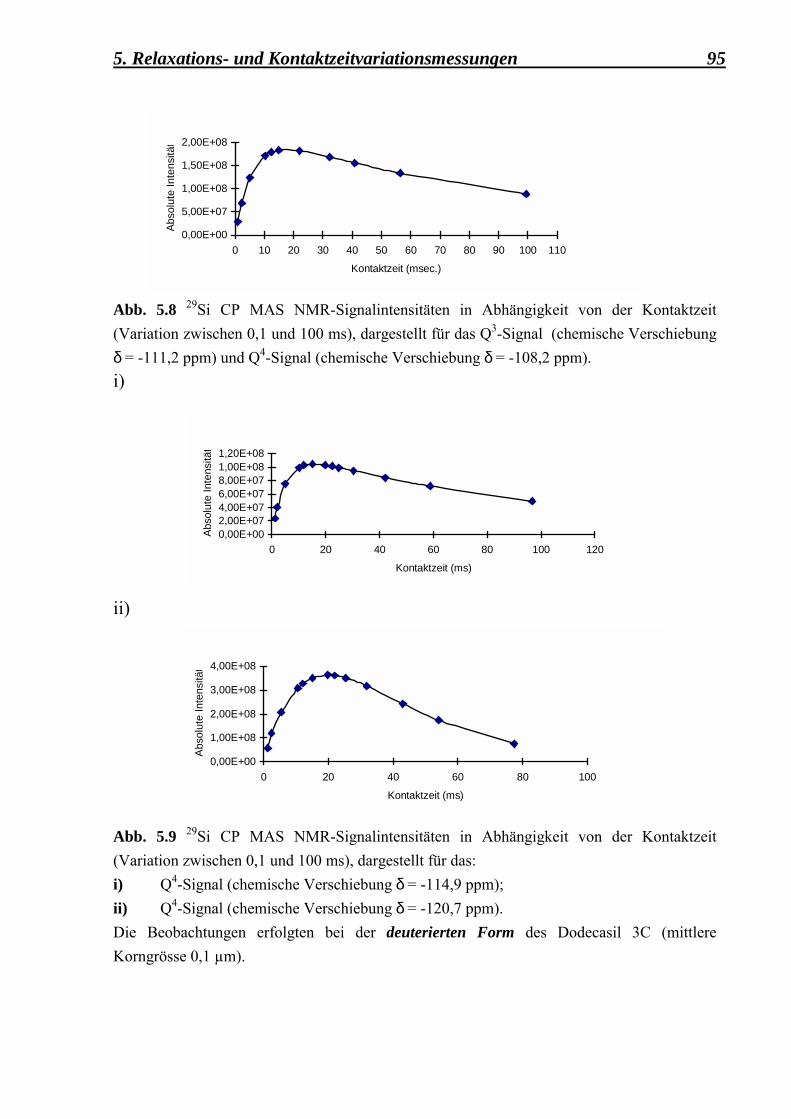
5. Relaxations- und Kontaktzeitvariationsmessungen 95
Abb. 5.8 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das Q3-Signal (chemische Verschiebung δ = -111,2 ppm) und Q4-Signal (chemische Verschiebung δ = -108,2 ppm).
0,00E+00
5,00E+07
1,00E+08
1,50E+08
2,00E+08
0 10 20 30 40 50 60 70 80 90 100 110
Kontaktzeit (msec.)
Abso
lute
Inte
nsitä
t
i)
0,00E+002,00E+074,00E+076,00E+078,00E+071,00E+081,20E+08
0 20 40 60 80 100 120
Kontaktzeit (ms)
Abso
lute
Inte
nsitä
t
ii)
Abb. 5.9 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das: i) Q4-Signal (chemische Verschiebung δ = -114,9 ppm); ii) Q4-Signal (chemische Verschiebung δ = -120,7 ppm). Die Beobachtungen erfolgten bei der deuterierten Form des Dodecasil 3C (mittlere Korngrösse 0,1 µm).
0,00E+00
1,00E+08
2,00E+08
3,00E+08
4,00E+08
0 20 40 60 80 100
Kontaktzeit (ms)
Abso
lute
Inte
nsitä
t

5. Relaxations- und Kontaktzeitvariationsmessungen 96
Das Q4-Signal, entsprechend der T2(Si2)-Site bei der chemischen Verschiebung δ4 = -115,1 ppm, erreicht in der 1H/29Si CP MAS NMR-Messung die maximale Kreuzpolarisation τmax = 17 ms (Abb. 5.9 i)). Das Q4-Signal, entsprechend der T3(Si1)-Site bei der chemischen Verschiebung δ5 = -120,7 ppm, weist eine maximale Kontaktzeit von τmax = 20 ms auf (Abb. 5.9 ii)). Wenn man eine Parallele zwischen den Kreuzpolarisationszeiten, entsprechend den Q4-Signalen für die as-synthesized Form, und den Kreuzpolarisationszeiten, entsprechend den Q4-Signalen für die deuterierte Form, zieht, beobachtet man, dass die deuterierte Form eine grössere Kontaktzeit vorweist. Der exponentielle Abfall, beschrieben durch die Formel exp(-t/T1ρI), zeigt keinen deutlichen Unterschied zwischen dem Verlauf der exponentiellen Kurven für die Q3-Signale und dem der exponentiellen Kurven für die Q4-Signale. Aus diesem Grund wird der exponentielle Abfall nur für die weitere Berechnung der Relaxationszeiten T1ρI wichtig sein. Die Templatmoleküle im Inneren der Käfige verfügen über eine grosse Mobilität im Vergleich zu den Silanolgruppen, die an der externen Oberfläche ziemlich rigid verteilt sind. Diese Beweglichkeit der Template spiegelt sich auch in dem Kreuzpolarisationsexperiment wider. So braucht eine deuterierte Substanz eine längere Zeit um zu relaxieren, als eine Substanz in ihrer as-synthesized Form. Diese Mobilität der Moleküle verleiht gleichzeitig einem CP-Experiment einen näherungsweise quantitativen Charakter, was unter normalen Bedingungen nicht möglich wäre.
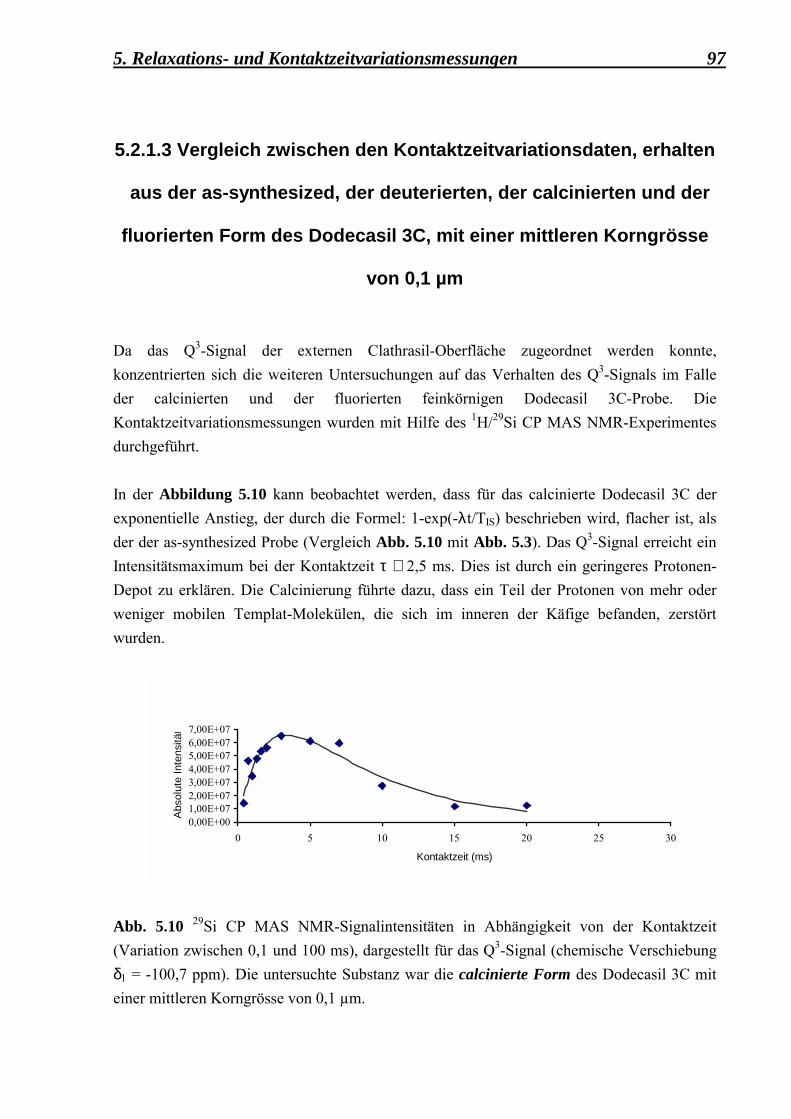
5. Relaxations- und Kontaktzeitvariationsmessungen 97
5.2.1.3 Vergleich zwischen den Kontaktzeitvariationsdaten, erhalten
aus der as-synthesized, der deuterierten, der calcinierten und der
fluorierten Form des Dodecasil 3C, mit einer mittleren Korngrösse
von 0,1 µm Da das Q3-Signal der externen Clathrasil-Oberfläche zugeordnet werden konnte, konzentrierten sich die weiteren Untersuchungen auf das Verhalten des Q3-Signals im Falle der calcinierten und der fluorierten feinkörnigen Dodecasil 3C-Probe. Die Kontaktzeitvariationsmessungen wurden mit Hilfe des 1H/29Si CP MAS NMR-Experimentes durchgeführt. In der Abbildung 5.10 kann beobachtet werden, dass für das calcinierte Dodecasil 3C der exponentielle Anstieg, der durch die Formel: 1-exp(-λt/TIS) beschrieben wird, flacher ist, als der der as-synthesized Probe (Vergleich Abb. 5.10 mit Abb. 5.3). Das Q3-Signal erreicht ein Intensitätsmaximum bei der Kontaktzeit τ ≅ 2,5 ms. Dies ist durch ein geringeres Protonen-Depot zu erklären. Die Calcinierung führte dazu, dass ein Teil der Protonen von mehr oder weniger mobilen Templat-Molekülen, die sich im inneren der Käfige befanden, zerstört wurden.
Abb. 5.10 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das Q3-Signal (chemische Verschiebung δ1 = -100,7 ppm). Die untersuchte Substanz war die calcinierte Form des Dodecasil 3C mit einer mittleren Korngrösse von 0,1 µm.
0,00E+001,00E+072,00E+073,00E+074,00E+075,00E+076,00E+077,00E+07
0 5 10 15 20 25 30
Kontaktzeit (ms)
Abso
lute
Inte
nsitä
t
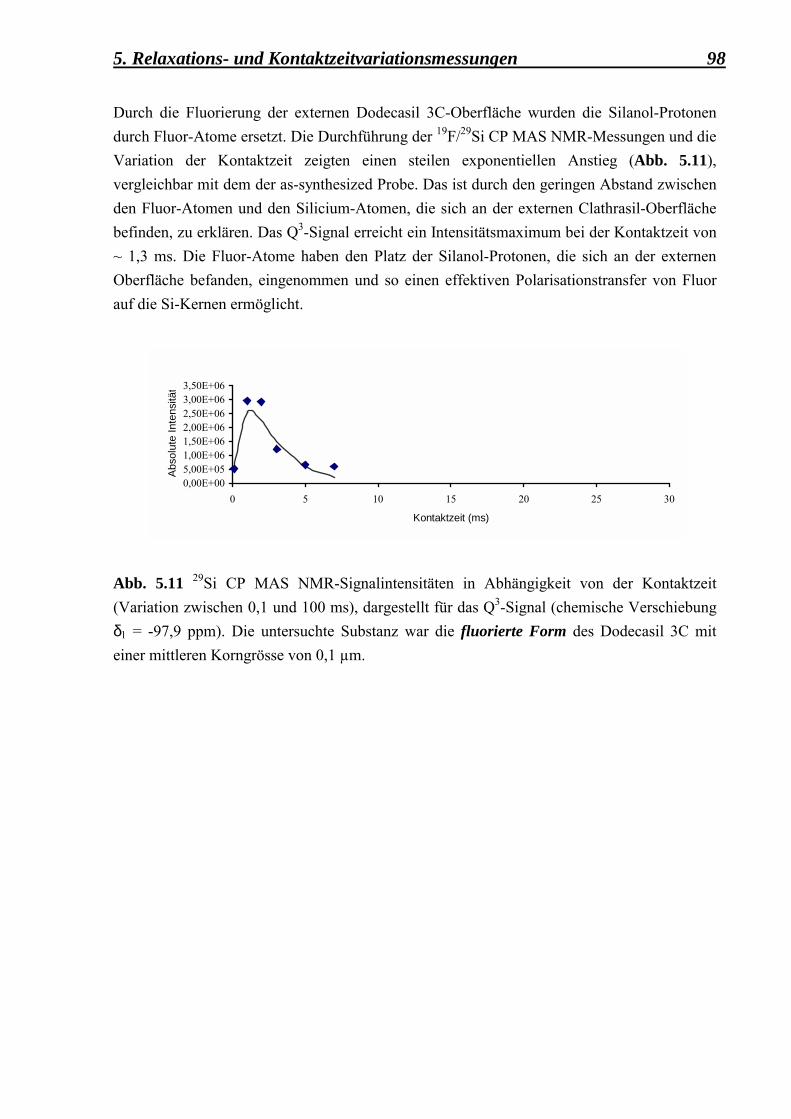
5. Relaxations- und Kontaktzeitvariationsmessungen 98 Durch die Fluorierung der externen Dodecasil 3C-Oberfläche wurden die Silanol-Protonen durch Fluor-Atome ersetzt. Die Durchführung der 19F/29Si CP MAS NMR-Messungen und die Variation der Kontaktzeit zeigten einen steilen exponentiellen Anstieg (Abb. 5.11), vergleichbar mit dem der as-synthesized Probe. Das ist durch den geringen Abstand zwischen den Fluor-Atomen und den Silicium-Atomen, die sich an der externen Clathrasil-Oberfläche befinden, zu erklären. Das Q3-Signal erreicht ein Intensitätsmaximum bei der Kontaktzeit von ~ 1,3 ms. Die Fluor-Atome haben den Platz der Silanol-Protonen, die sich an der externen Oberfläche befanden, eingenommen und so einen effektiven Polarisationstransfer von Fluor auf die Si-Kernen ermöglicht.
Abb. 5.11 29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit (Variation zwischen 0,1 und 100 ms), dargestellt für das Q3-Signal (chemische Verschiebung δ1 = -97,9 ppm). Die untersuchte Substanz war die fluorierte Form des Dodecasil 3C mit einer mittleren Korngrösse von 0,1 µm.
0,00E+005,00E+051,00E+061,50E+062,00E+062,50E+063,00E+063,50E+06
0 5 10 15 20 25 30
Kontaktzeit (ms)
Abso
lute
Inte
nsitä
t

5. Relaxations- und Kontaktzeitvariationsmessungen 99
5.2.2 Berechnung der Spin-Gitter T1ρρρρ - Relaxationszeiten und der
Kreuzrelaxationszeiten Tcp für die as-synthesized-, deuterierte-,
calcinierte- und fluorierte-Form der feinkörnigen Dodecasil 3C-
Proben Die Berechnung der Relaxationszeiten Tcp und T1ρ erfolgte für die as-synthesized, calcinierte, deuterierte und fluorierte Form des Dodecasil 3C aus den Kontaktzeitvariationsmessungen über die Kreuzpolarisationsexperimente 1H/29Si CP MAS NMR, bzw. 19F/29Si CP MAS NMR. Aus Tabelle 5.2 kann entnommen werden, dass die Kreuzrelaxationszeit Tcp zwischen den I- und den S-Spins für das Q3-Signal der as-synthesized Dodecasil 3C-Probe (mittlere Korngrösse 0,1µm) sehr kurz (Tcp = 1,5 ms) und gleichzeitig vergleichbar mit der der fluorierten Form (Tcp = 1,3 ms) ist. In beiden Fällen sind die Abstände zwischen den Oberflächenprotonen und den Oberflächen-Siliciumatomen sehr kurz, was sich auch in den kurzen Relaxationszeiten widerspiegelt. Die calcinierte Dodecasil 3C-Probe weist eine längere Tcp-Relaxationszeit auf (Tcp = 2,6 msec.), die vergleichbar mit den Ergebnissen aus der τmax-Berechnung ist. Die deuterierte Form der feinkörnigen Probe zeigte eine viel längere Kreuzrelaxationszeit (Tcp = 7,3 msec.) als die as-synthesized Form.
Tcp T1ρρρρ ττττmax Chemische Verschiebung
Probe
[ms] [ms]
I0 Standard Abweichung
[ms] [ppm] CS024_as-synthesized
1,5 1,47 2,10E+08 3,10E+06 1,5 -99,1
CS024_calciniert 2,6 6,8 1,50E+06 1,82E+06 2,5 -100,7 CS024_deuteriert 7,3 39,2 6,06E+09 3,06E+06 15 -97,7 CS024_fluoriert 1,3 2 7,70E+06 1,57E+05 1,25 -97,6 Tab. 5.2 Tabellarische Zusammenfassung der Relaxationszeiten Tcp und T1ρ, berechnet für das Oberflächen Q3-Signal der as-synthesized, calcinierten, deuterierten und fluorierten Form des Dodecasil 3C (mittlere Korngrösse 0,1 µm).
Die Spin-Gitter-Relaxationszeiten T1ρ zeigen ein ähnliches Verhalten wie die Kreuzrelaxationszeiten. Die einzige Ausnahme konnte an dem Wert der Spin-Gitter-Relaxationszeit für die as-synthesized Form beobachtet werden, der über dem Wert der
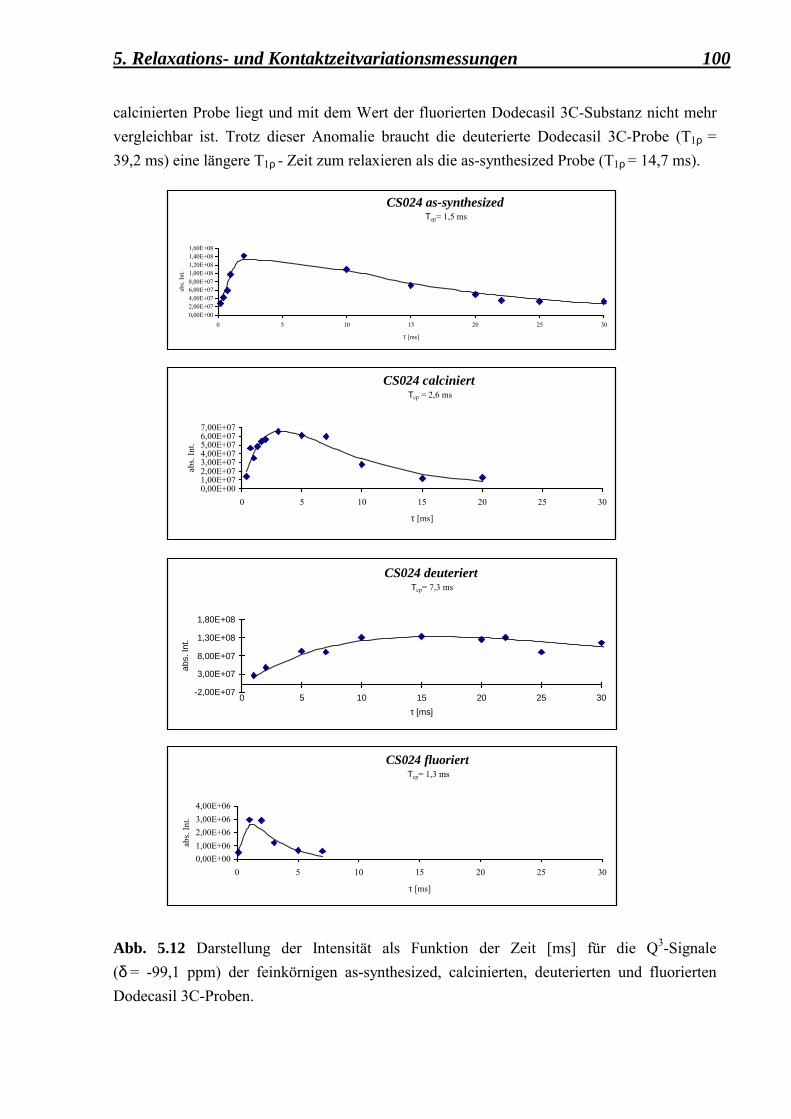
5. Relaxations- und Kontaktzeitvariationsmessungen 100 calcinierten Probe liegt und mit dem Wert der fluorierten Dodecasil 3C-Substanz nicht mehr vergleichbar ist. Trotz dieser Anomalie braucht die deuterierte Dodecasil 3C-Probe (T1ρ = 39,2 ms) eine längere T1ρ - Zeit zum relaxieren als die as-synthesized Probe (T1ρ = 14,7 ms).
Abb. 5.12 Darstellung der Intensität als Funktion der Zeit [ms] für die Q3-Signale (δ = -99,1 ppm) der feinkörnigen as-synthesized, calcinierten, deuterierten und fluorierten Dodecasil 3C-Proben.
CS024 as-synthesizedΤcp= 1,5 ms
0,00E+002,00E+074,00E+076,00E+078,00E+071,00E+081,20E+081,40E+081,60E+08
0 5 10 15 20 25 30
τ [ms]
abs.
Int.
CS024 calciniertΤcp = 2,6 ms
0,00E+001,00E+072,00E+073,00E+074,00E+075,00E+076,00E+077,00E+07
0 5 10 15 20 25 30
τ [ms]
abs.
Int.
CS024 deuteriertΤcp= 7,3 ms
-2,00E+07
3,00E+07
8,00E+07
1,30E+08
1,80E+08
0 5 10 15 20 25 30τ [ms]
abs.
Int.
CS024 fluoriert Τcp= 1,3 ms
0,00E+001,00E+062,00E+063,00E+064,00E+06
0 5 10 15 20 25 30
τ [ms]
abs.
Int.

5. Relaxations- und Kontaktzeitvariationsmessungen 101
5.2.3 Relaxationszeitmessungen
5.2.3.1 Berechnung der Spin-Gitter T1 - Relaxationszeiten und der
Spin-Spin T2 - Relaxationszeiten für die as-synthesized Form des
Dodecasil 3C (mittlere Korngrösse 0,1 µm) Die Berechnung sowohl der Spin-Gitter- als auch der Spin-Spin-Relaxationszeiten erfolgte für die as-synthesized Form des feinkörnigen Dodecasil 3C (Probe CS024_as-synthesized). Die T1-Relaxationszeit, bekannt auch unter dem Namen longitudinale Relaxation, ist eine charakteristische Zeitkonstante, die für den Temperaturausgleich zwischen Spin-System und Gitter sorgt. Die Rückkehr in den Gleichgewichtszustand erfolgt durch die Relaxation entlang der z-Achse (Abb. 5.1) und wird durch die 1. Blochsche Gleichung beschrieben:
Mz = M0[1-exp(-t/T1)] (5.17) Da Dipol-Dipol Wechselwirkungen zwischen dem Bulk und der Oberfläche auftreten, können Aussagen gemacht werden, wie schnell die Oberfläche im Vergleich zu dem Volumen relaxiert. Für den Fall, dass die Oberfläche gross ist, sind kurze T1-Zeiten zu erwarten. Experimentell wurde anhand eines 1H/29Si HPDEC-Experimentes ein sogenanntes multi-zg - Programm gestartet, wobei die D1-Zeit, eine Programmgrösse, die der Relaxationszeit entspricht, zwischen 100 und 1600 s variiert wurde. Für jedes einzelne Signal wurde bei den entsprechenden D1-Zeiten die absolute Intensität gemessen. Die simulierte Intensität ergab sich aus der Formel:
Isim. = I0[1-exp(-D1/T1x)] + C (Konstante) (5.18)
Die graphischen Darstellungen (Abb. 5.13) zeigen die Genauigkeit der Anpassungen der theoretischen Kurve an die experimentellen Daten (Punkte).
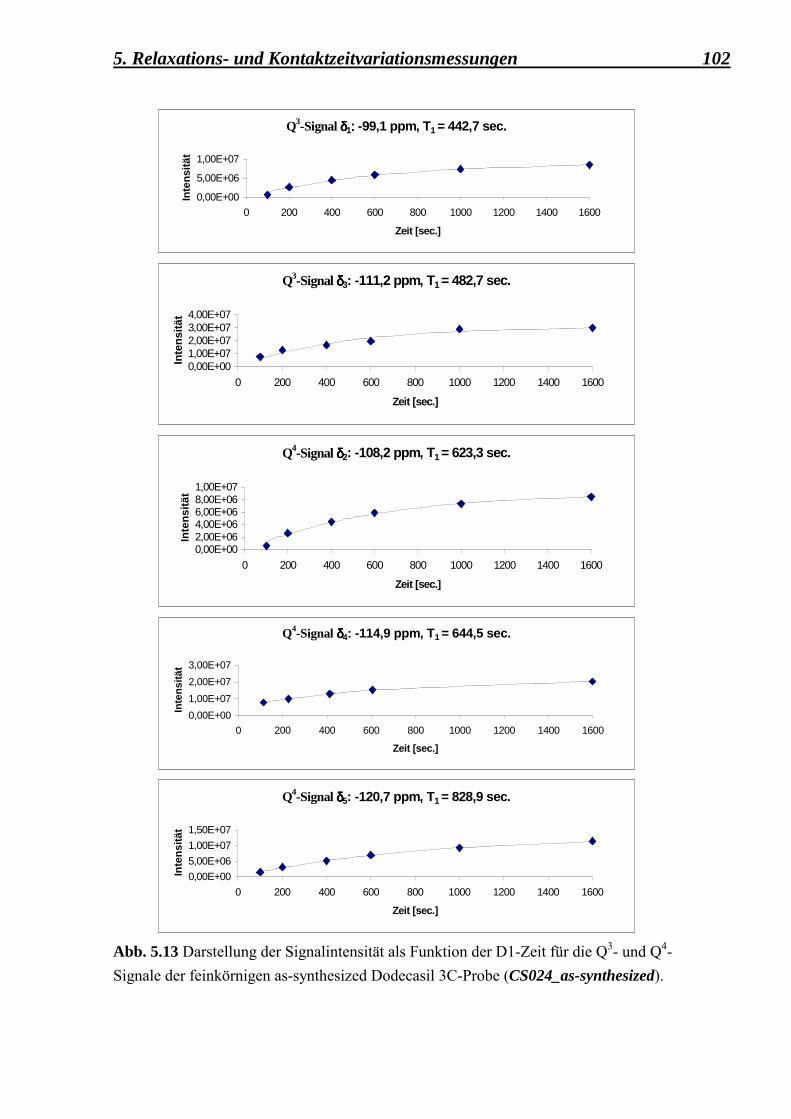
5. Relaxations- und Kontaktzeitvariationsmessungen 102
Abb. 5.13 Darstellung der Signalintensität als Funktion der D1-Zeit für die Q3- und Q4- Signale der feinkörnigen as-synthesized Dodecasil 3C-Probe (CS024_as-synthesized).
Q3-Signal δδδδ1:::: -99,1 ppm, T1 = 442,7 sec.
0,00E+00
5,00E+06
1,00E+07
0 200 400 600 800 1000 1200 1400 1600
Zeit [sec.]
Inte
nsitä
t
Q4-Signal δδδδ2: -108,2 ppm, T1 = 623,3 sec.
0,00E+002,00E+064,00E+066,00E+068,00E+061,00E+07
0 200 400 600 800 1000 1200 1400 1600
Zeit [sec.]
Inte
nsitä
t
Q3-Signal δδδδ3: -111,2 ppm, T1 = 482,7 sec.
0,00E+001,00E+072,00E+073,00E+074,00E+07
0 200 400 600 800 1000 1200 1400 1600
Zeit [sec.]
Inte
nsitä
t
Q4-Signal δ δ δ δ4: -114,9 ppm, T1 = 644,5 sec.
0,00E+001,00E+072,00E+073,00E+07
0 200 400 600 800 1000 1200 1400 1600
Zeit [sec.]
Inte
nsitä
t
Q4-Signal δδδδ5: -120,7 ppm, T1 = 828,9 sec.
0,00E+005,00E+061,00E+071,50E+07
0 200 400 600 800 1000 1200 1400 1600
Zeit [sec.]
Inte
nsitä
t

5. Relaxations- und Kontaktzeitvariationsmessungen 103
Die experimentellen Daten zeigen eine kurze T1-Relaxationszeit (442,7 s) für das Q3-Signal bei der chemischen Verschiebung von -99,1 ppm, im Vergleich zu den anderen Q4-Signalen (623, 3 s, 644,5 s, 828,9 s). Die longitudinale Relaxation T1 des zweiten Q3-Signals (δ3 = -111,2 ppm) weist gleichermassen eine kürzere Relaxationszeit (482,7 s) relativ zu den Q4-Signalen auf. Das deutet darauf hin, dass die äussere Oberfläche viel schneller im Vergleich zu dem Bulk relaxiert. Die T2-Relaxationszeit ist eine charakteristische Zeitkonstante, die für den �Temperaturausgleich� zwischen zwei Spin-Systemen sorgt. Die Rückkehr in den Gleichgewichtszustand erfolgt durch die Relaxation in der (x, y)-Fläche und wird durch die 2. und 3. Blochschen Gleichungen beschrieben:
Mx� = M0exp(-t/T2) und My� = M0exp(-t/T2) (5.19)
Über die T2-Relaxationszeit kann die natürliche Linienbreite bestimmt werden:
T2 = 1/π∆ν1/2 (5.20)
Für eine breite Resonanzlinie sind zusätzlich auch noch andere Einflüsse, die zu einer chemischen Verschiebung führen, verantwortlich. Das deutet darauf hin, dass sich die Homogenität der chemischen Umgebung verändert. Daraus resultiert, dass die Oberfläche amorph sein muss, wenn die T2-Zeit, die mit Hilfe eines Echo-Experimentes bestimmt worden ist, kleiner ist, als die direkte T2-Zeit, die aus der Halbwertsbreite FWHM des 29Si MAS-Signales berechnet wurde. Das erhaltene Signal ist in diesem Fall breit. Wenn die Oberfläche perfekt geordnet ist, dann verfügt jedes Si-Atom über eine gleiche Umgebung. In diesem Fall wäre zu erwarten, dass es zu einer langsamen Relaxation kommt und die direkte T2-Relaxation gross ist. Das erhaltenen Signal ist in diesem Fall schmal, was auf eine homogene, gut kristalline Probe hindeutet. Experimentell wurde anhand eines zweidimensionalen Experimentes ein cpt2 - Pulsprogramm (s. Anhang) gestartet, wobei die T2-Zeiten in eine sogenannten vdlist - Liste eingetragen worden sind (2 ms, 4 ms, 8 ms, 16 ms, 32 ms, 64 ms, 128 ms, 256 ms, 512 ms). Für jedes einzelne Signal wurde bei den entsprechenden T2-Zeiten die absolute Intensität gemessen. Die simulierte Intensität ergab sich aus der Formel:
Isim. = I0exp(-D2/T2x)] + C (Konstante) (5.21)
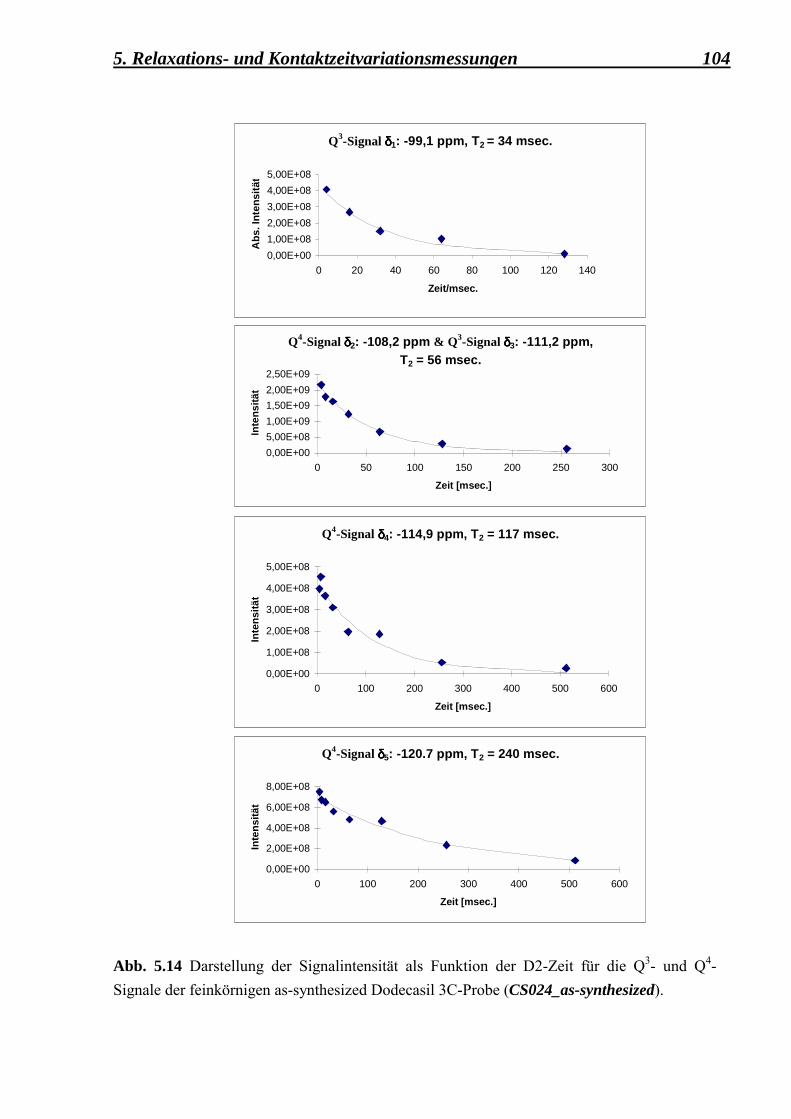
5. Relaxations- und Kontaktzeitvariationsmessungen 104
Abb. 5.14 Darstellung der Signalintensität als Funktion der D2-Zeit für die Q3- und Q4-Signale der feinkörnigen as-synthesized Dodecasil 3C-Probe (CS024_as-synthesized).
Q3-Signal δδδδ1: -99,1 ppm, T2 = 34 msec.
0,00E+001,00E+082,00E+083,00E+084,00E+085,00E+08
0 20 40 60 80 100 120 140
Zeit/msec.
Abs
. Int
ensi
tät
Q4-Signal δδδδ2: -108,2 ppm & Q3-Signal δδδδ3: -111,2 ppm,T2 = 56 msec.
0,00E+005,00E+081,00E+091,50E+092,00E+092,50E+09
0 50 100 150 200 250 300
Zeit [msec.]
Inte
nsitä
t
Q4-Signal δ δ δ δ4: -114,9 ppm, T2 = 117 msec.
0,00E+00
1,00E+08
2,00E+08
3,00E+08
4,00E+08
5,00E+08
0 100 200 300 400 500 600
Zeit [msec.]
Inte
nsitä
t
Q4-Signal δδδδ5: -120.7 ppm, T2 = 240 msec.
0,00E+00
2,00E+08
4,00E+08
6,00E+08
8,00E+08
0 100 200 300 400 500 600
Zeit [msec.]
Inte
nsitä
t
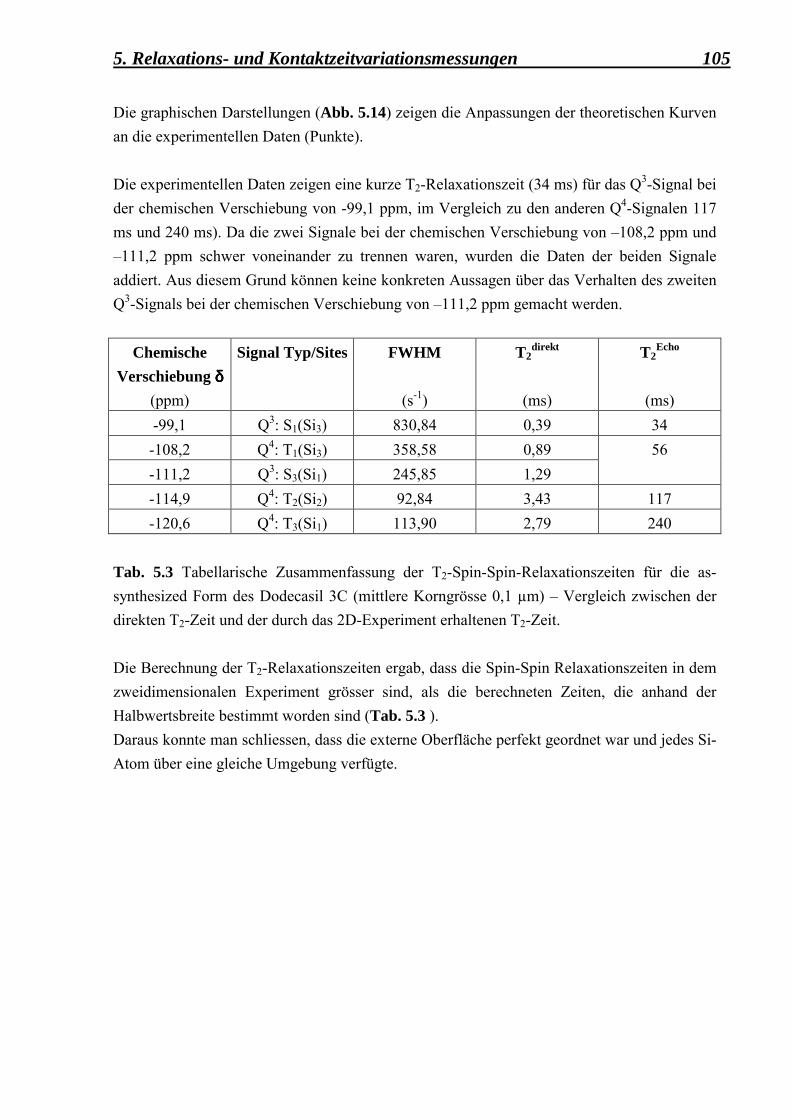
5. Relaxations- und Kontaktzeitvariationsmessungen 105 Die graphischen Darstellungen (Abb. 5.14) zeigen die Anpassungen der theoretischen Kurven an die experimentellen Daten (Punkte). Die experimentellen Daten zeigen eine kurze T2-Relaxationszeit (34 ms) für das Q3-Signal bei der chemischen Verschiebung von -99,1 ppm, im Vergleich zu den anderen Q4-Signalen 117 ms und 240 ms). Da die zwei Signale bei der chemischen Verschiebung von �108,2 ppm und �111,2 ppm schwer voneinander zu trennen waren, wurden die Daten der beiden Signale addiert. Aus diesem Grund können keine konkreten Aussagen über das Verhalten des zweiten Q3-Signals bei der chemischen Verschiebung von �111,2 ppm gemacht werden.
Chemische Verschiebung δδδδ
(ppm)
Signal Typ/Sites FWHM
(s-1)
T2direkt
(ms)
T2Echo
(ms) -99,1 Q3: S1(Si3) 830,84 0,39 34 -108,2 Q4: T1(Si3) 358,58 0,89 -111,2 Q3: S3(Si1) 245,85 1,29
56
-114,9 Q4: T2(Si2) 92,84 3,43 117 -120,6 Q4: T3(Si1) 113,90 2,79 240
Tab. 5.3 Tabellarische Zusammenfassung der T2-Spin-Spin-Relaxationszeiten für die as-synthesized Form des Dodecasil 3C (mittlere Korngrösse 0,1 µm) � Vergleich zwischen der direkten T2-Zeit und der durch das 2D-Experiment erhaltenen T2-Zeit.
Die Berechnung der T2-Relaxationszeiten ergab, dass die Spin-Spin Relaxationszeiten in dem zweidimensionalen Experiment grösser sind, als die berechneten Zeiten, die anhand der Halbwertsbreite bestimmt worden sind (Tab. 5.3 ). Daraus konnte man schliessen, dass die externe Oberfläche perfekt geordnet war und jedes Si-Atom über eine gleiche Umgebung verfügte.

5. Relaxations- und Kontaktzeitvariationsmessungen 106
5.3 Zusammenfassung der Relaxations- und Kontaktzeitvariationsmessungen
Durch die Variation der Kontaktzeiten zwischen 0,1 und 100 ms zeigte sich ein deutlicher Unterschied in der Relaxation des Q3-Signals, entsprechend der as-synthesized- bzw. der deuterierten Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm (Vergleich Abb. 5.2 Und Abb. 5.6). Zu beobachten war, dass das Q3-Signal, das bei der chemischen Verschiebung von �99,1 ppm auftritt, im Falle der as-synthesized-Probe, ein Maximum an Intensität schon bei sehr geringen Kontaktzeiten (1,5 ms) erreicht. Das gleiche Q3-Signal (-99,1 ppm) wird in dem 1H/29Si CP MAS NMR-Stack-Plot-Spektrum bei sehr kurzen Kontaktzeiten nicht beobachtet. Mit zunehmender Kontaktzeit werden die Q3-Signale sichtbar und erreichen ein Intensitätsmaximum sehr spät, bei einer Kontaktzeit von 12 ms. Das zweite Q3-Signal, bei der chemischen Verschiebung von �111,2 ppm, konnte nicht sehr gut von dem bei �108,2 ppm auftretenden Q4-Signal aufgelöst werden, so dass die Auswertung der Kontaktzeitvariationsdaten für die beiden Signale gemeinsam erfolgt ist und so für ein Vergleich nicht von Bedeutung ist. Die Kontaktzeitvariationsmessungen haben es erlaubt, die Berechnung der Kreuzrelaxationszeiten TIS für die as-synthesized, die deuterierte, die calcinierte und die fluorierte Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm, durchzuführen. Bekannt ist, dass die as-synthesized Probe über chemisch an Si-OH gebundene Protonen verfügt. Diese sind starr und befinden sich nahe der Oberfläche. Zusätzlich enthält die as-synthesized-Probe auch Templat-Protonen, die mobil sind und sich weiter von der Oberfläche entfernt befinden. Durch die graphische Darstellung der 1H/29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit wurde ein steiler exponentieller Anstieg sowohl im Falle der as-synthesized als auch der fluorierten Probe ermittelt. Die Erklärung dafür liegt in dem geringen Abstand zwischen den Protonen, bzw. den Fluor-Atomen und den Silizium Atomen, die sich an der externen Oberfläche befinden.

5. Relaxations- und Kontaktzeitvariationsmessungen 107
Die kurzen Abstände zwischen H+, bzw. F-, die als Magnetisierungsquelle dienen, und Si-Kernen deuten auf eine schnelle Relaxation hin. Die aus den Kontaktzeitvariationsmessungen erhaltenen Kreuzpolarisationszeiten sind in diesem Fall sehr kurz. Die Deuterierung führt dazu, dass die Oberflächen-Protonen durch Deuterium ersetzt werden und so als Magnetisierungsquelle nur Protonen, die sich im Inneren der Käfigen befinden, übrig bleiben. Die Abstände zwischen Templat-Protonen und Si-Atomen sind länger, als im Falle der as-synthesized-Probe. Die Relaxation ist auch entsprechend länger, während die aus den Kontaktzeitvariationsexperimenten erhaltenen Kreuzpolarisationszeiten gross sind. Die Berechnung der Spin-Gitter- T1 und Spin-Spin T2-Relaxationszeiten ergab, dass die äussere Oberfläche viel schneller relaxiert im Vergleich zu der inneren.

6. Untersuchungen an Quarzmehl 108
6. Untersuchungen an Quarzmehl
6.1 Klassierung durch die Fallgeschwindigkeit (Schlämmung)
Bei den meisten sedimentpetrographischen Untersuchungen, die vorgenommen werden, wird eine Klassierung durch die Fallgeschwindigkeit fast ausnahmslos in Flüssigkeiten durchgeführt. So können zur Vereinfachung Sedimentier- und Spülverfahren auch unter dem Begriff „Schlämmung“ zusammengefasst werden.
6.1.1 Grundlagen und allgemeine Voraussetzungen
der Sedimentationsanalyse Nach W. V. Engelhardt, H. Füchtbauer und G. Müller (Engelhardt et al., 1964), die sich mit Methoden der Sediment-Untersuchung beschäftigt haben, �beruhen die verschiedenen Methoden der Sedimentationsanalyse auf der Formel von Stokes�. Mit Hilfe dieser Formel ist es möglich, den �Reibungswiederstand einer ruhenden Sedimentationsflüssigkeit (oder eines Gases) gegenüber absinkenden kugelförmigen Teilchen zu berechnen.� Nach Stokes ist die Teilchengeschwindigkeit V, gemessen in cm/s, direkt proportional zum Quadrat des Kugelradius r (cm), zu der Gravitationskonstante g (cm/s2), zu der Dichtedifferenz von fallenden Kugeln und Sedimentationsflüssigkeit (g/cm3) und umgekehrt proportional zu der Viskosität der Flüssigkeit (g/cms).
Wird dieser Ausdruck nacher aufgelöst, so erhält man:
221
92 rDDgV
η−=
( ) VDDg
r ⋅−
=212
9η
(6.1)
(6.2)

6. Untersuchungen an Quarzmehl 109
Da die Teilchengeschwindigkeit V einem Verhältnis zwischen der Fallhöhe h (cm) und Fallzeit (s) entspricht, kann der Kugelradius r (cm) durch die folgende Formel ermittelt werden:
Bei der Sedimentationsanalyse wird die Korngrösse überwiegend in µm und die Fallzeit in Minuten ausgedrückt. Durch das Einsetzen des Zahlenwertes für g (=981 cm/s2) und mit dem Durchmesser (d = 2r) als Grösse für die Partikel, ergibt sich für d:
wobei d in µm und t in Minuten angegeben sind. Aus der letzten Gleichung kann die Zeit berechnet werden, �die eine Kugel bestimmter Grösse und Dichte in einem bestimmten Medium zum Durchfallen einer bestimmten Höhe benötigt� (Engelhardt et al., 1964). Für die Gültigkeit der Stokes-Gleichung bei der Sedimentation in Flüssigkeiten sind eine Reihe von Voraussetzungen nötig: 1. Die Teilchen der Flüssigkeit müssen im Vergleich zu den fallenden Teilchen sehr
klein sein (104 bis 107 mal kleiner); 2. Die Flüssigkeit muss im Verhältnis zu den fallenden Teilchen von unendlicher
Ausdehnung sein, was erreicht werden kann, wenn der Zylinderdurchmesser wenigstens 5 cm beträgt;
3. Die fallenden Teilchen müssen unbedingt starr aber nicht immer glatt sein; 4. Zwischen Flüssigkeit und Teilchen darf kein Gleiten erfolgen; 5. Die Fallgeschwindigkeit muss gering sein, und die Teilchen dürfen nicht die 50 µm-
Korndurchmessergrenze (Oseen, 1929) überschreiten; 6. Die Teilchen müssen kugelförmig sein.
( ) th
DDgr ⋅
−=
2129η
( ) tDDhd
⋅−⋅⋅=
21
5,174 η
(6.3)
(6.4)

6. Untersuchungen an Quarzmehl 110
Eine der wichtigsten Voraussetzungen für die Durchführung einer Schlämmanalyse ist somit das Arbeiten unter thermokonstanten Bedingungen. Die wichtigsten und am meisten benutzten Schlämmflüssigkeiten sind Wasser, Benzol und Methanol. Als Schlämmflüssigkeit wird im Normalfall destilliertes oder entmineralisiertes Wasser mit einem Zusatz von Natriumpyrophosphat (Na4P2O7
. 10H2O; 4,5 g/l, was eine 0,01 mol/l Lösung ergibt) oder Ammoniak (ebenfalls 0,01 mol/l) verwendet. Diese Zusätze sind von grosser Wichtigkeit, da sie als Stabilisatoren wirken und weitgehend die Koagulation verhindern.
6.1.2 Probenvorbereitung Bei der Probenvorbereitung muss beachtet werden, dass die Substanz aus einer reinen Phase (s. Röntgenpulverdiagramm von Quarzmehl, Abb. 6.1) besteht, die gleichzeitig feinkörnig ist (<63 µm). Diese Bedingung wird erreicht, indem die zu untersuchende Substanz in einer Achat-Kugelmühle zerkleinert wird. Die Substanz wird erstens gewogen und in einem Rundkolben in der Sedimentierflüssigkeit mindestens 12 Stunden lang in einer Schüttelapparatur, die in einem Raum mit konstanter Temperatur aufgestellt ist, geschüttelt. Unmittelbar danach wird die Aufschlämmung in Sedimentationsgefässe gefüllt, so dass die optimale Konzentration der Suspension bei ca. 5-20 g Festsubstanz pro Liter liegt.
6.1.3 Sedimentationsmethoden
6.1.3.1 Die Sedimentation im Atterberg-Zylinder Eine Vielfalt von Methoden, wie z. B. die Sedimentation im Atterberg-Zylinder, die Pipetten-Methode, die Sedimentationswaagen, die Aräometermethode, die Zentrifugen-Methode, die photometrische Methode u.s.w., werden im Falle einer Schlämmung von feinkörnigen Sedimenten eingesetzt. Für die vorliegende Arbeit wurden davon zwei verwendet und zwar die Sedimentation im Atterberg-Zylinder und die Zentrifugen-Methode.

6. Untersuchungen an Quarzmehl 111
Die Sedimentation im Atterberg-Zylinder ist bei den sedimentologischen Untersuchungen die am häufigsten angewandte Methode. Sie erlaubt im Gegensatz zu den meisten anderen Sedimentationsmethoden die vollständige Gewinnung der einzelnen Kornfraktionen. Der Atterberg-Zylinder, der bei der Schlämmung benötigt wird, weist einen inneren Durchmesser von mindestens 5 cm und eine Höhe von 25 bis 35 cm auf. Er besitzt ca. 3 bis 4 cm über dem Boden einen Siphon, an den ein Stück Schlauch mit Quetschhahn angeschlossen werden kann. Der Zylinder wird durch einen eingeschliffenen Glasstöpsel verschlossen. Es werden normalerweise für die Sedimentationsanalyse ungraduierte Zylinder eingesetzt, da eine Nacheichung und eine genaue Bestimmung der Null-Linie ohnehin häufig erforderlich ist. Für das weitere Trennen der Partikel werden Zylinder eingesetzt, bei denen die Null-Linie mindestens 3 cm über den Boden liegt. Nur so kann vermieden werden, dass bereits sedimentierte Teilchen durch die beim Ablassen der Flüssigkeit entstehende Saugwirkung mitgerissen werden. Nach dem Einfüllen der Aufschlämmung bei geschlossenem Siphon wird aus einem Vorratsbehälter Schlämmflüssigkeit, im gegebenen Fall Wasser, gemischt mit einem Stabilisator (Antikoagulant), bis zu der gewünschten Fallhöhe eingefüllt, der Zylinder verschlossen und mehrmals in horizontaler Lage mit dem Siphon nach oben kräftig geschüttelt, bis eine gute Durchmischung stattgefunden hat. Nach Ablauf der für die Sedimentation von Partikeln eines bestimmter Durchmessers benötigten Fallzeit wird der Glasstöpsel abgenommen, der Quetschhahn geöffnet, die abfliessende Trübe aufgefangen und in ein Filtriergerät geschüttet. Wenn z. B. eine Fallzeit für einen Durchmesser von 1 µm gewählt wurde, dann haben alle in der abgelassenen Trübe erhaltenen Körner einen Durchmesser der kleiner als 1 µm ist. Die Anzahl von Schlämmungen, die bis zur vollständigen Abtrennung eines bestimmten Korngrössenbereiches erforderlich sind, richtet sich nach der Teilchengrösse, der Art des zu schlämmenden Materials und dem Dispergierungsgrad. Sie ist bei den feinkörnigen Proben grösser als bei den grobkörnigen. Nach Engelhardt, W. V., Füchtbauer, H. und Müller, G. (Engelhardt et al., 1964) sind in der Regel für die Abtrennung einer Fraktion unter 2 µm 10 bis 25 Schlämmungen erforderlich. Das bedeutet, dass die Abtrennung dieser Kornklassen 10 bis 15 Tage dauern kann, da durch die lange Fallzeit pro Tag oft nur eine einzige Schlämmung durchgeführt werden kann.

6. Untersuchungen an Quarzmehl 112
Ein abgekürztes Verfahren, bei dem nur ein Teil der abgeschlämmtem Fraktionen gewonnen wird, besteht darin, dass jeweils nur die ersten zwei oder drei Abschlämmungen, die normalerweise mehr als 50% des Materials einer Fraktion enthalten, filtriert werden. Trotz des langen Arbeitsprozesses ist der Substanzverlust bei sauberem Arbeiten sehr gering und übersteigt selten 4%. Das Filtrieren von Fraktionen mit Teilchengrössen < 2 µm bringt den Verlust der Hauptmenge des Untersuchungsgutes mit sich. Deswegen ist es von Vorteil, für feinkörnige Partikel die Sedimentation im Atterberg-Zylinder mit der Zentrifugen-Methode zu koppeln.
6.1.3.2 Die Zentrifugen-Methode Die Abtrennung von Korngrössen kleiner als 2 µm durch Schlämmanalyse führt oft zu Ungenauigkeiten, die durch die lange Schlämmdauer, durch Erschütterungen oder ähnliches hervorgerufen werden. So können im Atterberg-Zylinder bei längerem Stehen trotz Konstanthaltung der Temperatur oft Schichtungsphänomene innerhalb der Suspension beobachtet werden, die derartige Störungen im Ablauf der Sedimentation anzeigen. Im Falle einer Sedimentation in einer Zentrifuge kann bei Verwendung einer Ultrazentrifuge die durch die Zentrifugalbeschleunigung auf die Teilchen einwirkende Kraft im Vergleich zur Schwerkraft bis auf das 750000-fache gesteigert werden (Svedberg, 1988). Durch den Einfluss der Zentrifugation auf die Sedimentation, wird das Stokes�sche Gesetz entsprechend geändert:
wobei ω der Winkelgeschwindigkeit ( = Umdrehungen/s), s1 der Entfernung der Rotationsachse zum Meniskus der Suspension, s2 der Entfernung der Rotationsachse bis zu dem Boden des Zentrifugengefässes entspricht. Die Methode schliesst damit lückenlos an das Atterberg-Verfahren an.
( ) tDDss
d⋅−
⋅=21
1
2
2
ln6
η
ω(6.5)

6. Untersuchungen an Quarzmehl 113
6.2 Experimentelle Untersuchungen an Quarzmehl
6.2.1 Zielsetzung Ziel der an Quarzmehl durchgeführten Untersuchungen war es, eine Parallele zwischen dem Verhalten des Quarzmehls, bei dem durch konventionellen Methoden (Mahlen, Fraktionieren, Sedimentieren, Zentrifugieren, Filtrieren) die Partikelgrösse verkleinert wurde, und dem Clathrasil Dodecasil 3C, einem Silikat, das in vergleichbarer Korngrösse hergestellt wurde (φ~0,1 µm), zu ziehen. Bekannt ist, dass Quarz ein perfekt geordnetes Siliziumoxid ist. Von grossem Interesse war es festzustellen, ob durch Kernresonanzspektroskopische Methoden und insbesondere durch die Kreuzpolarisation die externe Quarz-Oberfläche auch sichtbar wird. Wichtig dabei war, die Quarz-Partikel zu verkleinern, um die Silanol-Gruppen an der äusseren Oberfläche durch NMR beobachten zu können. Das Pulverdiagramm von Quarzmehl Dörentrup Nummer 10 wurde mit dem des α-Quarzes Si3O6 verglichen.
Abb. 6.1 Röntgenpulverdiagramm von Quarzmehl Dörentrup mit einer mittleren Korngrösse von unter 40 µm.

6. Untersuchungen an Quarzmehl 114 Es zeigten sich in beiden Fällen identische Reflexe, wobei die drei intensivsten bei 2θ-Werten von 26,65; 20,86 und 39,49 auftraten. Anhand des Fallzeitdiagramms nach Riedel wurde mit grosser Genauigkeit die Berechnung der Fallzeit von Quarzkugeln in einer Mischung von Wasser und Antikoagulant (in diesem Fall eine Natriumpyrophosphatlösung Na4P2O7
. 10H2O, 0,01 mol/l) bei 17°C durchgeführt (Abb. 6.2).
Abb. 6.2 Fallzeitdiagramm von Quarzkugeln in einer Mischung von Wasser und Stabilisator, wobei als Antikoagulant eine Natriumpyrophosphatlösung Na4P2O7
. 10H2O, 0,01 mol/l verwendet wurde (Engelhardt et al., 1964).
6.2.2 Probenvorbereitung und Durchführung des
Trennverfahrens Die Sedimentation erfolgte nach der Atterberg-Methode. Die Quarzmehl-Probe wurde erstens in einer Kugelmühle 65 Minuten lang gemahlen, wobei ein Achat-Behälter und dreizehn unterschiedlich grosse Achatkugeln (mit einem Durchmesser zwischen 1 und 5 cm) benutzt wurden. Die nach dem Mahlen erhaltene Masse wurde mit Hilfe mehrerer Siebe mit Maschenweiten von 400 µm, 300 µm, 200 µm, 100 µm und 40 µm fraktioniert. Nach der

6. Untersuchungen an Quarzmehl 115
Schlämmung (Fallzeit je nach gewünschter Korngrösse) wurde die Mischung aus Proben und Schlämmflüssigkeit durch Zentrifugieren (15 Minuten je Durchgang) voneinander getrennt. Nach den ersten 24 Stunden wurden die groben Teilchen aus der Sedimentationskolonne entfernt. Es wurde drauf geachtet, dass der Atterberg-Zylinder, der bei der Schlämmung benötigt wurde, über einen inneren Durchmesser von mindestens 5 cm und eine Höhe von 25 bis 35 cm verfügte und dass der Siphon sich ca. 3 bis 4 cm über dem Boden befand. Der ungraduierte Zylinder, der bei der Sedimentationsanalyse eingesetzt wurde, verfügte über einen eingeschliffenen Glasstöpsel, mit dem er verschlossen werden konnte. Die genaue Bestimmung der Null-Linie wurde vor jedem Experiment vorgenommen. Nach dem Einfüllen der Aufschlämmung bei geschlossenem Siphon wurde aus einem Vorratsbehälter die Schlämmflüssigkeit (Wasser und Natriumpyrophosphat als Stabilisator), bis zu der gewünschten Fallhöhe eingefüllt, der Zylinder verschlossen und mehrmals in horizontaler Lage mit dem Siphon nach oben kräftig geschüttelt, bis eine gute Durchmischung stattgefunden hatte. Nach Ablauf der für die Sedimentation eines bestimmten Durchmessers benötigten Fallzeit wurde der Glasstöpsel abgenommen, der Quetschhahn geöffnet, die abfliessende Trübe aufgefangen und in mehrere Zentrifugenröhrchen gefüllt. Für Partikel mit einem Durchmesser unter 2 µm wurde beispielsweise 69 Stunden Fallzeit gewählt. Die genauen Fallzeiten, die für die unterschiedlichen Quarzmehl-Korngrössen benötigt wurden sind in der Tabelle 6.1 zusammengefasst. Alle Voraussetzungen, die bei der Sedimentation in Flüssigkeiten nötig sind, wie z. B. sehr kleine fallende Teilchen im Vergleich zu den Flüssigkeitsteilchen, ein Zylinderdurchmesser von wenigstens 5 cm, um eine Flüssigkeit von unendlicher Ausdehnung im Verhältnis zu den fallenden Teilchen zu erzielen, starre Teilchen, kein Gleiten zwischen Flüssigkeit und Teilchen, eine geringe Fallgeschwindigkeit und kugelförmige Partikel, wurden erfüllt. Die Durchführung der Schlämmanalyse wurde unter thermokonstanten Bedingungen durchgeführt. Da die erzielte Korngrösse sehr gering war (~0,1 µm), wurden mehrere Sedimentationskollonen verwendet, um so viel wie möglich an Quarzmehl-Fraktion zu gewinnen.

6. Untersuchungen an Quarzmehl 116
Proben-Code Teilchengrösse nach dem Falldiagramm
Vorgehensweise Fallzeit
QMD3 < 40 µm 65 Minuten gemahlen, fraktioniert (40 µm-Sieb)
keine
QMD5 < 2 µm 65 Minuten gemahlen, fraktioniert (40 µm-Sieb), 69 Stunden sedimentiert, 10 Minuten zentrifugiert, 2 Tage im
Trockenschrank bei 110°C gelagert.
69 Stunden
QMD6 < 1 µm 65 Minuten gemahlen, fraktioniert (40 µm-Sieb), 10 Tage sedimentiert, 10 Minuten zentrifugiert, 2 Tage im Trockenschrank bei 110°C gelagert.
10 Tage
QMD10 < 0,1 µm 65 Minuten gemahlen, fraktioniert (40 µm-Sieb), 19 Tage sedimentiert, 10 Minuten zentrifugiert, 2 Tage im Trockenschrank bei 110°C gelagert.
19 Tage
Tab. 6.1 Zusammenhang zwischen den Teilchengrössen, die anhand des Falldiagramms bestimmt worden sind, und Fallzeit.
6.2.3 Korngrössebestimmung durch
Rasterelektronenmikroskopie Für die weiteren Untersuchungen wurden drei Quarzmehl-Proben ausgewählt, QMD3 (grobkörnig), QMD5 (kleinkörnig) und QMD10 (feinkörnig), die unterschiedliche Korngrössen aufweisen (s. Tabelle 6.1). Die REM-Untersuchungen wurden an diesen drei Materialien vorgenommen, um die Bestätigung zu bekommen, dass die nach dem Falldiagramm berechneten Korngrössen richtig sind. Die REM-Untersuchungen an den in dieser Arbeit verwendeten Quarzmehlproben unterschiedlicher Korngrösse wurden an einem Rasterelektronenmikroskop des Typs Cambridge 250, MK3D am Institut für Geologie durchgeführt.

6. Untersuchungen an Quarzmehl 117 Es zeigte sich, dass die Korngrössen, die anhand des Falldiagramms berechnet wurden und die Werte, die durch die REM-Aufnahme bestimmt wurden (s. Tabelle 6.2), vergleichbar sind.
i)ii)
iii)
Abb. 6.3 Rasterelektronenmikroskopische Aufnahmen von Quarzmehl unterschiedlicher Korngrösse: i) Probe QMD3, < 40 µm; ii) Probe QMD5, < 2 µm; iii) Probe QMD10, < 0,1 µm.

6. Untersuchungen an Quarzmehl 118
6.2.4 NMR-Untersuchungen an Quarzmehl
6.2.4.1 29Si MAS NMR-Experimente
Die drei Quarzmehl-Proben, die vorher einer REM-Untersuchung unterzogen worden waren, wurden auch für die 29Si MAS NMR-Experimente ausgewählt, QMD3 (grobkörnig), QMD5 (kleinkörnig) und QMD10 (feinkörnig), um Aufschluss über den Anteil der Oberflächensilanolgruppen an dem Gesamtsignal zu bekommen. Die 29Si MAS NMR-Experimente wurden unter identischen Bedingungen durchgeführt: Raumtemperatur, Rotationsgeschwindigkeit: 2,5 kHz, Aquisitionszeit: 102,5 ms, Pulsprogramm: zg, Pulslänge P1: 5 µs. Nur die Zahl der aufgenommenen Scans war von Experiment zu Experiment unterschiedlich. In allen drei 29Si MAS NMR-Experimenten war je ein Q4-Signal zu beobachten. Bei der QMD3-Probe (< 40 µm), die 65 Minuten gemahlen und durch eine Siebkolonne fraktioniert wurde, trat das Q4-Signal bei �107,2 ppm auf. Die QMD5-Quarzmehl-Probe (< 2 µm), die zusätzlich noch 69 Stunden sedimentiert wurde, zeigte ein vergleichbares Q4-Signal bei der gleichen chemischen Verschiebung von �107,2 ppm. Die QMD10-Quarzmehl-Probe (<0,1 µm), die noch länger sedimentiert wurde, und zwar 19 Tage, wies auch ein Resonanzsignal bei �107,2 ppm auf. Aus der Tabelle 6.2 können die Halbwertsbreiten FWHM des �107,2 ppm-Signals, entsprechend der jeweiligen Proben QMD3 (<40 µm), QMD5 (< 2 µm) und QMD10 (<0,1 µm), entnommen werden. Es war zu beobachten, dass je kleiner die Korngrösse der Quarz-Partikel, desto grösser die Halbwertsbreite des gemessenen Q4-Resonanzsignals wurde. So weist die QMD10-Fraktion (< 0,1 µm), die am längsten sedimentiert wurde (19 Tage), eine Halbwertsbreite von 30,51 Hz auf im Vergleich zu der QMD3-Fraktion, die keiner Sedimentierung unterzogen worden ist und eine FWHM von 22,88 Hz zeigt.

6. Untersuchungen an Quarzmehl 119
Proben-CodeTeilchengrö-sse nach dem Falldiagramm
Teilchengrösse nach REM
29Si MAS-Experiment
1H/29Si CP MAS-Experiment
FWHM nach NMR
Fallzeit
QMD3 < 40 µm 1-20 µm -107,21 ppm Kein Signal 22,88 Hz keine QMD5 < 2 µm 0,1-2 µm -107,28 ppm Q2: -90,6 ppm
Q3: -97,7 ppm Q4: -107,1 ppm
25,63 Hz 69 Stunden
QMD6 < 1 µm 0,01-1 µm
-107,26 ppm Q2: -90,9 ppm Q3: -97,7 ppm Q4: -107,2 ppm
27,76 Hz 10 Tage
QMD10 < 0,1 µm ~ 0,1 µm -107,30 ppm Q2: -91,2 ppm Q3: -98,3 ppm Q4: -107,7 ppm
30,51 Hz 19 Tage
Tab. 6.2 Tabellarischer Vergleich zwischen den Teilchengrössen unterschiedlicher Quarzmehlfraktionen, die aus dem Falldiagramm berechnet bzw. mit Hilfe des Rasterelektronenmikroskopes bestimmt wurden. Die Tabelle enthält gleichzeitig die Halbwertsbreiten-Werte und die chemischen Verschiebungen, die aus den 29Si MAS- bzw. 1H/29Si CP MAS NMR-Experiment resultierten.
Abb. 6.4 29Si MAS NMR-Experiment, durchgeführt an der QMD3-Quarzmehl-Fraktion (< 40 µm), die keiner Sedimentation unterzogen worden ist.
( pp m )
- 1 1 8- 1 1 6- 1 1 4- 1 1 2- 1 1 0- 1 0 8- 1 0 6- 1 0 4- 1 0 2- 1 0 0- 9 8- 9 6- 9 4- 9 2

6. Untersuchungen an Quarzmehl 120
Abb. 6.5 29Si MAS NMR-Experiment, durchgeführt an der QMD5-Quarzmehl-Fraktion (< 2 µm), die 69 Stunden lang sedimentiert wurde.
( ppm )
- 11 8- 1 16- 1 1 4- 1 1 2- 1 1 0- 10 8- 1 06- 1 0 4- 1 0 2- 1 0 0- 9 8- 96- 9 4- 92
Abb. 6.6 29Si MAS NMR-Experiment, durchgeführt an der QMD10-Quarzmehl-Fraktion (< 0,1 µm), die 19 Tage lang sedimentiert wurde.
( ppm )
- 1 1 8- 1 1 6- 1 1 4- 1 1 2- 1 1 0- 1 0 8- 1 0 6- 1 0 4- 1 0 2- 1 0 0- 9 8- 9 6- 9 4- 9 2
Die drei 29Si MAS NMR-Spektren (Abb. 6.4, Abb. 6.5, Abb. 6.6), die von unterschiedlichen Quarzmehl-Fraktionen aufgenommen worden sind, zeigten ein ähnliches Q4-Signal entsprechend des Bulkes. Der Unterschied bestand nur darin, dass die Halbwertsbreiten (FWHM) verschieden waren, wie man aus der Tabelle 6.2 entnehmen kann. Je kleiner die Korngrössen der Quarzmehlpartikel sind, desto grösser ist die Halbwertsbreite des Volumen-Signals.
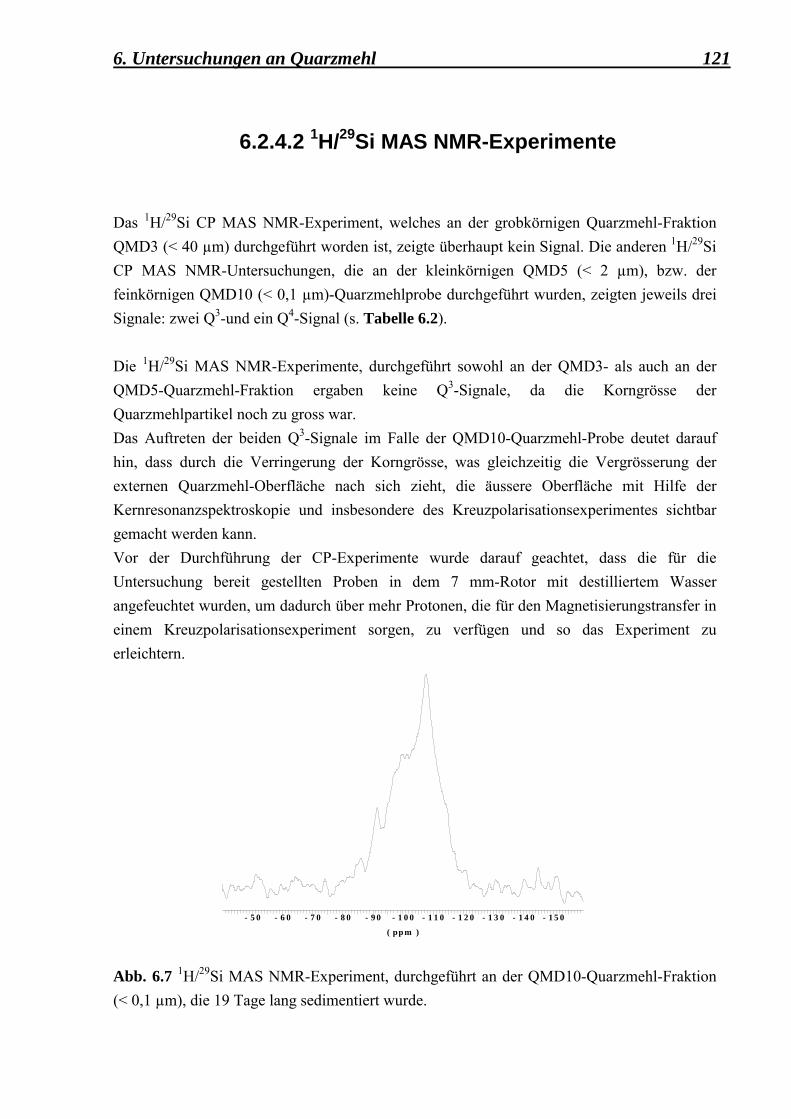
6. Untersuchungen an Quarzmehl 121
6.2.4.2 1H/29Si MAS NMR-Experimente
Das 1H/29Si CP MAS NMR-Experiment, welches an der grobkörnigen Quarzmehl-Fraktion QMD3 (< 40 µm) durchgeführt worden ist, zeigte überhaupt kein Signal. Die anderen 1H/29Si CP MAS NMR-Untersuchungen, die an der kleinkörnigen QMD5 (< 2 µm), bzw. der feinkörnigen QMD10 (< 0,1 µm)-Quarzmehlprobe durchgeführt wurden, zeigten jeweils drei Signale: zwei Q3-und ein Q4-Signal (s. Tabelle 6.2). Die 1H/29Si MAS NMR-Experimente, durchgeführt sowohl an der QMD3- als auch an der QMD5-Quarzmehl-Fraktion ergaben keine Q3-Signale, da die Korngrösse der Quarzmehlpartikel noch zu gross war. Das Auftreten der beiden Q3-Signale im Falle der QMD10-Quarzmehl-Probe deutet darauf hin, dass durch die Verringerung der Korngrösse, was gleichzeitig die Vergrösserung der externen Quarzmehl-Oberfläche nach sich zieht, die äussere Oberfläche mit Hilfe der Kernresonanzspektroskopie und insbesondere des Kreuzpolarisationsexperimentes sichtbar gemacht werden kann. Vor der Durchführung der CP-Experimente wurde darauf geachtet, dass die für die Untersuchung bereit gestellten Proben in dem 7 mm-Rotor mit destilliertem Wasser angefeuchtet wurden, um dadurch über mehr Protonen, die für den Magnetisierungstransfer in einem Kreuzpolarisationsexperiment sorgen, zu verfügen und so das Experiment zu erleichtern.
Abb. 6.7 1H/29Si MAS NMR-Experiment, durchgeführt an der QMD10-Quarzmehl-Fraktion (< 0,1 µm), die 19 Tage lang sedimentiert wurde.
( pp m )
- 1 5 0- 1 4 0- 1 3 0- 1 2 0- 1 1 0- 1 0 0- 9 0- 8 0- 7 0- 6 0- 5 0

6. Untersuchungen an Quarzmehl 122
Die zwei Resonanzsignale, die im Falle der QMD10-Quarzmehl-Fraktion (< 0,1 µm) zu beobachten sind und die bei der chemischen Verschiebung von �91,17 ppm (Q2-Signal) bzw. -98,32 ppm (Q3-Signal) auftraten, können als Oberflächen-Signale betrachtet werden. Schon bei Quarzmehl-Partikeln, die Korngrössen zwischen 0,1 und 2 µm aufwiesen (QMD5), sind diese zwei Q3-Signale in dem 1H/29Si CP MAS NMR-Spektrum sichtbar. Die zwei verschiedenen Q3-Signale zeigen, dass an der externen Quarzmehl-Oberfläche zwei verschiedene Positionen vorhanden sind. Die 1H/29Si CP MAS NMR-Experimente, die an Quarzmehl-Fraktionen unterschiedlicher Korngrösse durchgeführt worden sind, haben bestätigt, dass durch die Verkleinerung der Partikelgrösse die äussere Oberfläche einer Substanz sichtbar gemacht werden kann. Es treten in beiden Fällen, sowohl bei Dodecasil 3C als auch bei Quarzmehl, zwei Q3-Signale auf, die den Silanolgruppen, die an der externen Oberfläche angelagert sind, zugeordnet werden können. Die 1H/29Si CP MAS NMR-Untersuchungen, die an Quarzmehl durchgeführt wurden, sind der Beweis dafür, dass die Theorie der Anwendbarkeit der Kernresonanzspektroskopie in der Oberflächencharakterisierung über eine reale Basis verfügt.

6. Untersuchungen an Quarzmehl 123
6.3 Zusammenfassung der Quarzmehl-Untersuchungen
Die strukturellen Eigenschaften der Quarzmehl-Oberfläche zeigten sich ähnlich, wie die der Clathrasil-Oberfläche. Durch die Verkleinerung der Korngrösse ergab sich bei den Quarzmehl-Proben genau wie bei den Clathrasil-Substanzen die Möglichkeit, spektroskopisch die externe Oberfläche nachzuweisen. Mit Hilfe der Kernresonanzspektroskopie und insbesondere der Kreuzpolarisationstechnik war es möglich, zu dem für Quarzmehl charakteristischen Q4-Volumen-Signal, das bei der chemischen Verschiebung von �107,7 ppm auftritt, zwei zusätzliche Resonanzsignale zu erhalten. Diese waren bei den chemischen Verschiebungen von �91,17 ppm, bzw. bei �98, 32 ppm zu beobachten (s. Abb. 6.7). Das erste Resonanzsignal entspricht einem Q2-Signal und befindet sich ungefähr 16,5 ppm von dem Q4-Volumen-Signal zu niedrigeren chemischen Verschiebungen entfernt. Das zweite Resonanzsignal ist um 9,3 ppm von dem Q4-Volumen-Signal verschoben und entspricht einem Q3-Signal. Anhand der Ergebnisse der Kreuzpolarisations-Experimente ist zu vermuten, dass die externe Quarzmehloberfläche morphologisch mit Strukturelemente der Art:
bedeckt ist. In den NMR-Experimenten war zu beobachten, dass es durch die mechanische Zerkleinerung des Quarzmehls und der damit verbundenen Oberflächenvergrösserung zu einem ähnlichen Verhalten wie im Falle von synthetisch hergestellten kleinen Dodecasil-Kristallen kam. Das Erzeugen einer grossen Oberfläche ist die entscheidende Bedingung, um die externe Oberfläche von verschiedenen Substanzen kernresonanzspektroskopisch nachzuweisen.
*SiO*SiO*SiO
Si OH
Q3-Signal
OH*SiO
*SiOSi
OH
Q2-Signal

7. Zusammenfassung 124
7. Zusammenfassung Im ersten Teil der vorliegenden Arbeit wurde der Einfluss von Syntheseparametern wie Silicium-Quelle, Art des Templates, Synthesedauer, Synthesetemperatur und Füllhöhe auf die Kristallitgrösse vorgestellt. Die Synthesebedingungen wurden in solcher Weise optimiert, dass das Verhältnis der Atome im Kristall Oberfläche zu Volumen (Bulk) 100 bis 1000 fach vergrössert worden ist. Als optimale Synthesebedingungen für die Herstellung eines mikrokristallinen Clathrasils Dodecasil 3C (Code-Name CS024_as-sythesized) mit einer mittleren Korngrösse zwischen 0,01 und 0,1 µm wurden folgende Bedingungen ermittelt: 4 mol/l Aerosil (als Silicium-Quelle), 1,5 mol/l Piperidin (als Templat-Quelle), relativer Anteil : 0,00666%, Synthesedauer: 3 Tage, Synthesetemperatur: 160°C, Füllhöhe: 1/3 des Teflonbehälters. Der grundlegende Ansatz dieser Synthese-Strategie bestand darin, die Syntheseparameter so zu verändern, dass die externe Oberfläche des Clathrasils Dodecasil 3C auf 50-100 m2 erhöht und so für das Kernresonanzexperiment sichtbar gemacht wurde. So wurde die Konzentration der Silicium-Quelle bis zu der Grenze der Synthese von noch-kristallinen Phasen erhöht, die Synthesetemperatur so niedrig wie möglich gehalten, um das Kristallwachstum zu unterdrücken und die Keimbildung zu stimulieren, die Synthesedauer kurz eingestellt, um kleinstmögliche Kristallite zu erzeugen. Es stellte sich heraus, dass einer der entscheidenden Faktoren, der für eine erfolgreiche Synthese gesorgt hat, die Füllhöhe war. Entgegen aller Erwartungen zeigte sich die Arbeitsweise als ein Faktor, der einen geringen Einfluss auf die Kristallitgrösse ausgeübt hat. Unter ständigem Rühren oder Rotieren während der Synthese wuchsen die Kristalle viel schneller und viel grösser als erwartet. Durch die Synthese unter statischen Bedingungen hatten die hergestellten Dodecasil 3C-Kristalle die kleinste Kristallitgrösse. Die Analyse der Syntheseprodukte erfolgte in erster Linie durch die XRD- und Rasterelektronenmikroskopischen Aufnahmen (Abbildungen 3.3ii und 3.4 ii). Das Röntgenbeugungsdiagramm der CS024_as-synthesized-Verbindung mit der Korngrösse 0,01µm-0,1µm zeigte starke, scharfe Reflexe, die tetragonal indiziert werden konnten. Der Synthesedurchgang der Dodecasil 3C-Probe mit der Kodierung CS024 wurde mehrmals wiederholt um die Reproduzierbarkeit des Ergebnisses abzusichern (Probe CS134).

7. Zusammenfassung 125
Zusätzlich wurden unterschiedliche Behandlungen an der as-synthesized Form des Dodecasil 3C vorgenommen. Durch das Waschen mit einer schwach konzentrierten NaOH-Lösung (0,01mol/l) wurde ausgeschlossen, dass noch Reste von Silicagel oder nach der Synthese übrig gebliebenem nicht umgewandeltem Templat, vorhanden sind. Die partielle Calcinierung (2 Tage bei 550°C, in einer oxidativen Atmosphäre) erlaubte es, Strukturdeffekte zu eliminieren, und führte zugleich zu der Gewissheit, dass durch die NMR-Untersuchungen, die an der mikrokristallinen CS024 - Dodecasil 3C-Probe unternommen wurden, die externe Oberfläche und nicht die Strukturdeffekte charakterisiert werden. Die Deuterierung wurde durchgeführt, um die Protonen, die sich an der externen Oberfläche befinden (Silanolgruppen Si-OH), durch Deuterium zu ersetzten. Dadurch bestand die Möglichkeit, einen Magnetisierungstransfer nur von den Templat (Pyperidin) � Protonen aus dem inneren des Käfigs- zu ermöglichen und die Relaxation der Oberfläche zu beobachten. Die Fluorierung diente dazu, die externe Oberfläche des Clathrasils Dodecasil 3C mit Fluor zu bedecken und so die Hydroxyl-Gruppen OH- durch Fluorid-Gruppen F- zu ersetzten. Der zweite Teil der Arbeit stellt die Ergebnisse der Kernresonanzspektroskopischen Untersuchungen der im ersten Teil erhaltenen feinkörnigen Dodecasil 3C-Kristalle zusammen. Die an der CS024_as-synthesized-Probe durchgeführten 1H/29Si CP MAS NMR-Untersuchungen zeigten ausser den drei gewöhnlichen Bulk-Q4-Signalen, die den verschiedenen T-Sites-Positionen (T1(Si3):-108,2 ppm, T2(Si2): -114,9 ppm, T3(Si1): -120,7 ppm) entsprechen, noch zwei zusätzliche Oberflächen-Q3-Signale, die den an der externen Oberfläche kristallographisch inäquivalenten Lagen (S2(Si2): -99,1 ppm und S3(Si1): -111,2 ppm) zugeordnet wurden. Die zwei in dem 1H/29Si CP MAS NMR-Spektrum erhaltenen Q3-Signale konnten in dem reproduzierten Dodecasil 3C-Material wieder beobachtet werden. Da die zwei zu niedrigeren chemischen Verschiebungen von den entsprechenden Q4-Signalen verschoben sind, konnte die Hypothese aufgestellt werden, dass die externe Oberfläche des Clathrasils Dodecasil 3C einen regelmässigen Aufbau vorweist und somit kristallin sein kann. Die zusätzlichen Behandlungen, die an der CS024_as-synthesized-Verbindung vorgenommen worden sind, wie z.B. Calcinieren oder die Probe mit einer NaOH-Lösung waschen, haben es ermöglicht, Artefakte wie Strukturdefekte oder nicht umgewandeltes Silicagel

7. Zusammenfassung 126
auszuschliessen. In jedem der aufgenommenen 1H/29Si CP MAS NMR-Spektren waren die Q3-Signale vorhanden, so dass ein Einfluss durch die oben erwähnten Möglichkeiten, die ebenfalls zum Auftreten solcher Signale hätten führen können, ausgeschlossen werden konnte. Das 19F/29Si CP MAS NMR-Spektrum zeigte bei den chemischen Verschiebung �97,6 ppm, -107,5 ppm und �115,7 ppm drei Signale, die die Theorie bekräftigten, dass bei entsprechender Probevorbereitung die externe Dodecasil 3C-Oberfläche mit Hilfe der NMR-Spektroskopie detektierbar ist, obwohl die Kernresonanzspektroskopie keine typisch oberflächensensitive Methode ist. Im dritten Teil der Arbeit wurden zusätzliche NMR-Untersuchungen durchgeführt, um Informationen über die Relaxation an der externen Clathrasil-Oberfläche zu bekommen. Durch die Variation der Kontaktzeiten zwischen 0,1 und 100 ms zeigte sich ein deutlicher Unterschied in der Relaxation des Q3-Signals, entsprechend der as-synthesized- bzw. der deuterierten Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm. Zu beobachten war, dass das Q3-Signal, das bei der chemischen Verschiebung von �99,1 ppm auftritt, im Falle der as-synthesized-Probe, ein Maximum an Intensität schon bei sehr geringen Kontaktzeiten (1,5 ms) erreicht. Das gleiche Q3-Signal (-99,1 ppm) wird in dem 1H/29Si CP MAS NMR-Stack-Plot-Spektrum bei sehr kurzen Kontaktzeiten nicht beobachtet. Mit zunehmender Kontaktzeit werden die Q3-Signale sichtbar und erreichen ein Intensitätsmaximum sehr spät, bei einer Kontaktzeit von 12 ms. Das zweite Q3-Signal bei der chemischen Verschiebung von �111,2 ppm konnte nicht sehr gut von dem bei �108,2 ppm auftretenden Q4-Signal aufgelöst werden, so dass die Auswertung der Kontaktzeitvariationsdaten für die beiden Signale erfolgt ist und so für ein Vergleich nicht von Bedeutung ist. Die Kontaktzeitvariationsmessungen haben es erlaubt, die Berechnung der Kreuzrelaxationszeiten TIS für die as-synthesized, die deuterierte, die calcinierte und die fluorierte Form des Dodecasil 3C, mit einer mittleren Korngrösse von 0,1 µm, durchzuführen. Bekannt ist, dass die as-synthesized Probe über chemisch an Si-OH gebundene Protonen verfügt. Diese sind starr und befinden sich nahe an der Oberfläche. Zusätzlich enthält die as-synthesized-Probe auch Templat-Protonen, die mobil sind und sich weiter von der Oberfläche entfernt befinden. Durch die graphische Darstellung der 1H/29Si CP MAS NMR-Signalintensitäten in Abhängigkeit von der Kontaktzeit wurde ein steiler exponentieller Anstieg sowohl im Falle der as-synthesized als auch der fluorierten Probe ermittelt. Die Erklärung dafür liegt in dem geringen Abstand zwischen den Protonen, bzw. den Fluor-Atomen und den Silicium Atomen, die sich an der externen Oberfläche befinden.

7. Zusammenfassung 127
Die kurzen Abstände zwischen H+, bzw. F-, die als Magnetisierungsquelle dienen, und Si-Kernen deuten auf eine schnelle Relaxation hin. Die aus den Kontaktzeitvariationsmessungen erhaltenen Kreuzpolarisationszeiten sind in diesem Fall sehr kurz. Die Deuterierung führt dazu, dass die Oberflächen-Protonen durch Deuterium ersetzt werden und so als Magnetisierungsquelle nur Protonen, die sich im Inneren der Käfigen befinden, übrig bleiben. Die Abstände zwischen Templat-Protonen und Si-Atomen sind länger, als im Falle der as-synthesized-Probe. Die Relaxation ist auch entsprechend länger, während die aus den Kontaktzeitvariationsexperimenten erhaltenen Kreuzpolarisationszeiten gross sind. Die Berechnung der Spin-Gitter- T1 und Spin-Spin T2-Relaxationszeiten ergab, dass die äussere Oberfläche viel schneller relaxiert im Vergleich zu der inneren. Der letzte Teil der Arbeit zeigte das NMR-Verhalten anderen Substanzen, wie z. B. Quarzmehl, die durch konventionelle Methoden (Mahlen, Fraktionieren, Sedimentieren, Zentrifugieren) zerkleinert worden sind. Es traten in dem 1H/29Si CP MAS NMR-Spektrum einer 19 Tage sedimentierten Quarzmehl-Fraktion (< 0,1 µm), neben dem üblichen Q4-Bulk-Signal bei der chemischen Verschiebung von �107,2 ppm, noch zwei zusätzliche Q3-Signale (-91,2 ppm und �98,3 ppm) auf. Das erste Resonanzsignal entspricht einem Q2-Signal und befindet sich ungefähr 16,5 ppm von dem Q4-Volumen-Signal entfernt, bei niedrigeren chemischen Verschiebungen. Das zweite Resonanzsignal ist um 9,3 ppm von dem Q4-Volumen-Signal verschoben und entspricht einem Q3-Signal. Daraus ist zu entnehmen, dass die externe Quarzmehloberfläche morphologisch mit Strukturelemente der Art: Si-(OH)2 und Si-(OH) bedeckt ist. Durch die mechanische Zerkleinerung der Quarzmehl-Partikel wurde eine grosse externe Oberfläche erzeugt, so dass es zu einem ähnlichen Verhalten wie im Falle der synthetisch hergestellten kleinen Dodecasil-Kristalle kam. Das Erzeugen einer grossen Oberfläche ist die entscheidende Bedingung, um die externe Oberfläche von verschiedenen Substanzen kernresonanzspektroskopisch nachzuweisen. Diese Kreuzpolarisationsmessungen, die an unterschiedlich grossen Quarzmehl-Fraktionen durchgeführt worden sind, beweisen, dass die externe Oberfläche verschiedener Substanzen mit Hilfe einer nicht oberflächensensitive Methode, wie es die NMR-Spektroskopie ist, detektierbar ist. Die Bedingung, die dabei erfüllt werden muss, ist, die Korngrösse, entweder durch die Synthese von kleinen Kristallen oder durch nachträgliche Behandlungen, in den µm-Bereich zu bringen und dadurch die äussere Oberfläche zu vergrössern.

7. Zusammenfassung 128
Fazit ist, dass die Q3-Signale, die in dem 1H/29Si CP MAS NMR-Spektrum der as-synthesized-Form der Dodecasil 3C-Substanz zu beobachten sind, der externen Clathrasil-Oberfläche zugeordnet werden konnten. Die Auswertung der Spektren ergab, dass im Falle des Dodecasil 3C die Oberfläche kristallin ist und mit grosser Wahrscheinlichkeit von der Pentagondodecaeder Schicht terminiert wird. Durch den Vergleich zwischen typischen Oberflächenuntersuchungsmethoden wie z. B. AFM, XPS, LEED etc. und der NMR-Spektroskopie, wurde in dieser Arbeit gezeigt, dass die Kernresonanzspektroskopie eine gute oberflächensensitive Methode ist. Während die erste Kategorie der Oberflächenuntersuchungsmethoden grosse, glatte Oberflächen, die im Vakuum untersucht werden müssen, benötigen, gelingt es mit Hilfe der NMR-Spektroskopie, die Materialien so zu untersuchen, wie sie in der Katalyse auch eingesetzt werden. Dadurch bekommt man Informationen über die reelle Struktur, so wie sie auch im Wirklichkeit ist.

8. Literatur 129
8. Literatur Andrew, E.R. (1971): Prog. Nucl. Magn. Reson. Spectrosc. 8: S. 1. Atkins, P.W. (1990): Physikalische Chemie, Weinheim, VCH-Verlag. S. 782. Benn, R., Günther, H. (1983): Moderne Pulsfolgen in der hochauflösenden NMR-
Spektroskopie, Angew. Chem. 95: S. 381. Brunner, E. (1995): The use of small and weakly basic probe molecules for the investigation
of Brönstd acid sites in zeolites by NMR spctroscopy, Zeolithes: A Refined Tool for Designing Catalytic Sites.
Brunner, G.O. und Meier, W.M. (1989): Framework density distribution of zeolite-type
tetrahedral nets. Nature. 337: S. 146. Communications (1978): The Measuremnent of Proton-Enhanced Carbon-13 T1 Values by a
Method Wich Suppresses Artifacts, Journal of Magnetic Resonance. 30: S. 613. Davies, M.E., Saldarriaga, C., Montes, C., Garces, J. und Crowder, C. (1988): A
molecular sieve with eighteen-membered rings. Nature. 331: S. 698. Derome, A. E. (1993): Modern NMR Techniques for Chemistry Research, Pergamon Press
Oxford, New York, Seoul, Tokyo. 4.4: S. 85. Dyer, A. (1988): An introduction to zeolite molecular sieves. John Wiley & Sons. Dyer, A. (1995): Zeolite surface and reactivity,, Vaughan, D.J. und Pattrick, R.A.D., Editors.
Chapmann & Hall: London. S. 333. Engelhardt, W.F., Füchtbauer, H. und Müller, G. (1964): Sediment-Petrologie. Stuttgart,
Schweizerbart. Eric, J. D., MacNicol, D. D., Vögtle, F., Lehn, J.-M. (1995): Solid-State Supramolecular Chemistry: Crystal Engineering, Volum-Editoren: MacNicol, David D., Toda, Fumio, Bishop, Roger, Elsevier Science Ltd., Oxford. 6: S. 851.

8. Literatur 130
Feng, Y. (1991): One and Two-Dimensional High-Resolution Solid-State NMR Investigationof zeolite Structures, A thesis submitted in partial fulfillment of requirements for the degree of doctor of philosophy, The faculty of graduate studies (Department of Chemistry), The University of British Columbia.
Friebolin, H. (1992): Ein und zweidimensionale NMR-Spektroskopie. zweite Auflage. Eine
Einführung. Weinheim, VCH-Verlag. Fyfe, C. A., Gies, H. und Feng, Y. (1989): Demonstration of Three-dimensional Lattice
Connectivities in Zeolites by Two-dimensional High Resolution Solid State N.M.R. Spectroscopy. J. Chem Soc. 17: S. 1240.
Fyfe, C. A., Diaz, A., Lewis, A., Grondey, H., Feng, Y., Gies, H., Kokotailo, G. T.,
Chezeau, J. M. (1997): Investigation of the Three Dimensional Struktures of Zeolite Molecular Sieve-Sorbate Complexes by One- and Two-Dimensional Solid State NMR Techniques, Vortrag, AK-Treffen �NMR-Spektroskopie�, Institut für Mineralogie der Ruhr-Universität Bochum.
Fyfe, C. A., Grondey, H., Diaz, A. C., Kokotailo, G. T., Feng, Y., Huang, Y., Wong-
Moon, K. C., Mueller, K. T., Strobl, H., Lewis, A. R. (1995): One and two-dimensional solid-state NMR investigations of the three-dimensional structures of zeolite-organic sorbate complexes, Zeolithes: A Refined Tool for Designing Catalytic Sites.
Garces, J. M. (1999): Observations on Zeolite Applications, Ed. in Proceedings of the 12th
International Zeolite Conference von Treacy, M. M. J., Marcus, B. K., Bisher, M. E., Higgins, J. B., Conference held July 5-10, 1998, Baltimore, Maryland, USA, Materials Research Socety, Warrendale, Pennsylvania. I: S. 551.
Gies, H., Liebau, F. und Gerke, H. (1982): Dodecasile - eine neue Reihe polytyper
Einschlussverbindungen von SiO2. Angew. Chem. 94: S. 214. Gies, H. (1987): Einfluss der Gastmoleküle auf die Struktur und Stabilität von Porosilen, in
Mathematisch-Naturwwissenschaftliche Fakultät. Christian-Albrechts-Universität Kiel.

8. Literatur 131
Gies, H. (1991): Clathrasils zeosils: inclusion compounds with silica host frameworks.Inclusion Compounds, in Inorganic and Physical Aspects of Inclusion, Atwood, J.L.e.a., Editor. Oxford Univ. Press: Oxford. S. 1.
Gies, H. und Marler, B. (1992): The structure-controlling role of organic templates for the
synthesis of porosils in the system SiO2/template/H2O. Zeolites. 12(655): S. 42. Gies, H. und Marler, B. (1996): Crystalline Microporous Silicas as Host-Guest Systems, in
Comprehensive Supramolecular Chemistry, Atwood, J.L., Editor. Elsevier/Pergamon: Oxford. S. 851.
Groennen, E. J. J., Alma, N. C. M., Dorrepaal, A. J., Hays, G. R., Kortbeek, A. G. T. G.
(1985): Solid-state 29Si n.m.r. spectroscopy of clathrasil; the relationship betweeen the chemical shift and the structure of silicates, Zeolites. 5.
Günther, Harald (1992): NMR-Spektroskopie. Grundlagen, Konzepte und Anwendungen der
Protonen und Kohlenstoff-13-Kernresonanz-Spektroskopie in der Chemie, 3., neubearbeitete und erweiterte Auflage, Georg Thieme Verlag Stuttgart New York. 11.8: S. 469.
Higgins, J.B. (1994): Silica Zeolites and Clathrasils, in SILICA Physical Behavior,
Geochemistry and Materials Applications, Heaney, P.J., Prewitt, C.T. und Gibbs, G.V., Editors. Mineralogical Society of America: Washington D. C. S. 507.
Hinz, W. (1963): SILIKATE. Einführung in Theorie und Praxis. Berlin, VEB Verlag für
Bauwesen. S. 188. Hoffner, F. Marcucilli, Delmotte, L., Kessler, H., (1993): A 19F/29Si CP MAS n.m.r. study of
microporous solids synthesized in fluoride medium, Zeolites. 13: S. 60. Hollemann, A.F. und Wiberg, E. (1985): Lehrbuch der anorganischen Chemie. 91. - 100.
Auflage, ed. Wiberg, N., Walter de Gruyter-Verlag. S. 74. Huheey, J.E. (1983): Inorganic Chemistry. Principles of structure and reactivity. Third
Edition ed., Harper Collins. Kalinowski, Hans-Otto; Berger, Stefan; Braun, Sigmar (1984): 13C-NMR-Spektroskopie,
Georg Thieme Verlag Stuttgart.

8. Literatur 132
Kessler, H., Gehrke, Matthias, Griesinger, C. (1988): Zweidimensionale NMR-Spektroskopie, Grundlagen und Übersicht über die Experimente, Angew. Chem.. 100: S. 507.
Knorr, K. und Depmeier, W. (1988): The Cubic/Tetragonal Phase Transition in D3C-THF:
an Optical and X-ray Powder Diffraction Study. Journal of Solid State Chemistry. 137: S. 87.
Knorr, K. (1994): Untersuchungen zur Struktur und zu strukturellen Phasenumwandlumgen
am Clathrasil Dodecasil-3C, Dissertation. Kolodziejski, W., Klinowski, J. (1994): New NMR Techniques for the Study of Catalysis,
University of Cambridge, Cambridge, England. Könnecke, M., G., M. und Fuess, H. (1992): Static disorder of dodecasil 3C. A single-
crystal study with synchrotron radiation. Zeitschrift für Kristallographie. 201: S. 147. Liebau, F., Gies, H., Gunawardane, R.P. und Marler, B. (1986): Classification of
tectosilicates and systematic nomenclature of clathrate type tectosilicates: a proposal. Zeolites. 6: S. 373.
Marler, B. (1985): Synthesen von Clathrasilen mit optisch aktiven Gastmolekülen. McCuscer, L.B. (1991): Zeolite crystallography. Structure determination in the absence of
conventional single crystal data, Acta Cryst. A 47: S. 297. Meier, W.M. und Ohlson, D.H. (1992): Atlas of zeolite structure types. Butterworth-
Heinemann. Okamoto, G., Okura, T. und Goto, K. (1957): Properties of silica in water. Geochim. et
Cosmochim. Acta. 12: S. 123. Oseen, K.W. (1929): Die anisotropen Flüssigkeiten; Tatsachen u. Theorien. Fortschritte d.
Chemie, Physik u. physikal. Chemie ; 20, 2 ed., Berlin. Borntraeger. S. 87. Ostwald, A. (1895): I. Ueber Trimethylbernsteinsäuren verschiedener Herkunft ; II. Ueber
Anilsäuren di- und trisubstituierter Bernsteinsäuren .... Heidelberg, Naturw.-math. Fak.: Heidelberg. S. 63.

8. Literatur 133
Pines, A., Gibby, M. G., Waugh, I. S., (1973): J. Chem. Phys.. 59: S. 569. Quadbeck-Seeger, H.-J. (1998): Katalyse. Ruhr-Universität Bochum, Fakultät für Chemie:Bochum. Ripmeester, John A., Ratcliffe, Christopher I. (1996): Solid-state NMR Spectroscopy, Ed.
in Comprehensive Supramolecular Chemistry von Atwood, J. L., Davies, J., Eric, D., MacNicol, D. D., Vögtle, F., Lehn, J.-M., Physical Methods in Supramolecular Chemistry, Volum-Editoren: Davies, J., Eric, D., Ripmeester, J. A. 8: S. 323.
Roempp (1995): Chemielexikon. CD-ROM, 9. ed. Georg Thieme Verlag, Stuttgart, New
York. Röhrig, C. und Gies, H. (1995a): Ein neuer Zincosilicat-Zeolith mit Neuner-Ring Knälen.
Angew. Chem. 107 (1)(Germ. Ed.): S. 125. Röhrig, C. und Gies, H. (1995b): A new Zincosilcate Zeolite with nine-ring Channels.
Angew. Chem Int. 34 (1)(Ed. Engl.,): S. 63. Röhrig, Carsten (1995): Synthese und Charakterisierung zeolithartiger Zinkosilikate,
Dissertation. S. 8. Schaefer J. und O., S.E. (1976): J. Am. Chem. Soc. 98: S. 1031. Schlenker, J.L., Dweyer, F.G., Jenkins, E.E., Rohrbaugh, W.J. und Kokotailo, G.T.
(1981): Crystal structure of a synthetic high silica zeolite-ZSM-39. Nature. 294: S. 340.
Slichter, C.P. (1990): Principles of magnetic resonance. 3. enl. and updated ed., Berlin,
Springer. S. 655. Svedberg (1988): Uppsala accelerator news, in Department of Radiation Sciences. Uppsala
University, Uppsala. Van Hove, M.A. und Tong, S.Y. (1979): Surface crystallography by LEED : theory,
computation and structural results. Berlin, Springer. S. 286.

8. Literatur 134
West, Anthony R. (1992): Grundlagen der Festkörperchemie, VCH-Verlag, Weinheim. 4: S.
175. Wilke, K.-T. und Bohm, J. (1988): Kristallzüchtung. Vol. 1., Frankfurt/Main, Verlag Harri
Deutsch. S. 153. Wilmes, A. (1993): Taschenbuch Chemische Substanzen. Frankfurt/Main, Verlag Harri
Deutsch. S. 651. Wolf, I. (1997): Über die Magnetische Kernresonanz, Seminarvortrag, Ruhr-Universität
Bochum, Institut für Mineralogie, insb. Kristallographie. Wrackmeyer, B. (1988): Chemie in unserer Zeit, VCH-Verlag, 22. Jahrgang, Nr. 3: S. 100. Yannoni, C. S. (1982): Acc. Chem. Res.. 15: S. 201.

9. Abkürzungen und Symbole 135
9. Abkürzungen und Symbole ADC analog to digital converter AEI Strukturtyp-Code von AlPO4-18 AES Auger-Spektroskopie AFI Strukturtyp-Code von AlPO4-5 ARUPS angle-resolved ultraviolet photoelectron spectroscopy ARXPD angle-resolved X-ray photoelectron diffraction B0 äusseres polarisierendes Magnetfeld B1 Hochfrequenzimpuls CP cross polarization, Kreuzpolarisation δiso isotrope chemische Verschiebung DDR Strukturtyp-Code von Deca-Dodecasil 3R DOH Strukturtyp-Code von Dodecasil 1H EMT Strukturtyp-Code von EMC-2 FEM photoelectron microscopy FID free induction decay FIM field-ion microscopy FWHM full width at half maximum, Halbwertsbreite γ gyromagnetisches Verhältnis χ0 statische Suszeptibilität I Kernspin k Boltzmann-Konstante LEED low-energy electron diffraction, Niedrig Energie Elektronenstreuung LEEM low-energy electron microscopy, Niedrig Energie Mikroskopie LEIS low energy ion-scattering LTA Strukturtyp-Code von Linde A MAS magic angle spinning MEP Strukturtyp-Code von Melanophlogite MFI Strukturtyp-Code von ZSM-5 MTN Strukturtyp-Code von ZSM-39 (D3C) NEXAFS near-edge X-ray absorption fine structure NPD normal photoelectron difraction µ magnetisches Moment µ0 magnetische Konstante des Vakuums

9. Abkürzungen und Symbole 136
ν Rotationsfrequenz NMR nuclear magnetic resonance, Kernresonanzspektroskopie PEM photoelectron microscopy PM Polarisationsmikroskopie PESM Photoelektronen-Spektromikroskopie PSAs pressure Swing applications r12 internuklearer Kernabstand RSN Strukturtyp-Code von RUB-17 REM Raster-Elektronenmikroskopie SAM scanning Auger microscopy STM scanning tunneling microscopy T Temperatur VFI Strukturtyp-Code von VPI-5 ω Angularfrequenz XPS X-ray Photoelektronen-Spektroskopie XRD X-ray diffraction, Pulverdiffraktometrie

Anhang Pulsprogramm: cp19f.3
Pulsprogramm: „cp19f.3“ ;cp19f.3 ;cross-polarization sequence ;for use with high power X-amplifier (standard: AMT) ;set: ;p3 = 90 degree 19F pulse ;p15 = contact pulse (typ. 1ms) define pulse p90=p3+p17 ;p17 correction pulse 1 ze 2 2u setf2^11 d1 dblo tlo 10u:c4 hl1 3 (p90 ph1):f2:f3:c4 ;90 degree 1H pulse (p15 ph3):f2:f3 (p15 ph2):t:c4 ;cross polarization p4:f2:f3:c4 hl2 2u:e ph0:r 1u:e adc p7:f2:f3 aq rcyc=2 ph31 wr #0 exit ph1=1 3 ph2=0 0 1 1 2 2 3 3 ph3=0 ph0=0 ph31=0 2 1 3 2 0 3 1 ;TL(0) set to drive HP X amplifier ;DBL(0) set to drive HP 19F amplifier ;rd=pw=0 ;ns=8*n

Anhang Pulsprogramm: cp19f.3
Pulsprogramm: „zgf19“ ;zgf19 ;1D sequence 1 ze 2 d1 hi1 p1 ph1 go=2 ph31 wr #0 exit ph1=0 2 1 3 ph31=0 2 1 3 ;tl0: ecoupler high power level ;p1 : 90 degree transmitter high power pulse ;d1 : relaxation delay; 1-5 * T1
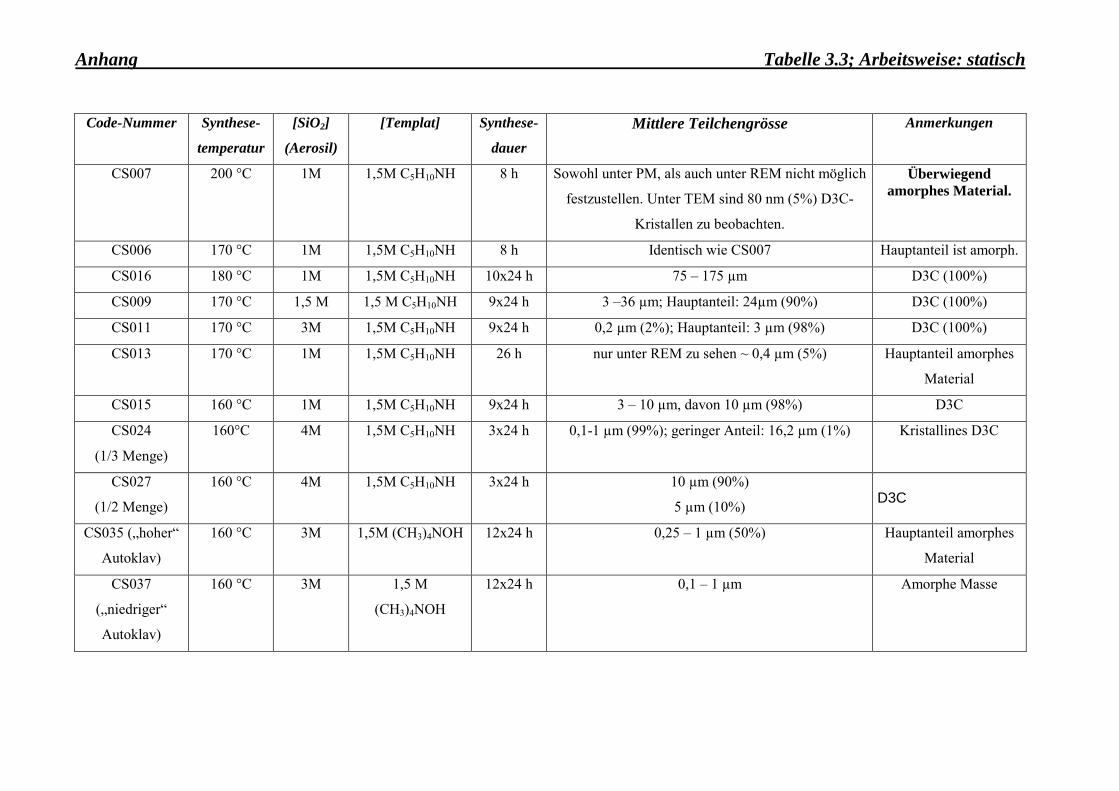
Anhang Tabelle 3.3; Arbeitsweise: statisch
Code-Nummer Synthese-
temperatur
[SiO2]
(Aerosil)
[Templat] Synthese-
dauer Mittlere Teilchengrösse Anmerkungen
CS007 200 °C 1M 1,5M C5H10NH 8 h Sowohl unter PM, als auch unter REM nicht möglich
festzustellen. Unter TEM sind 80 nm (5%) D3C-
Kristallen zu beobachten.
Überwiegend amorphes Material.
CS006 170 °C 1M 1,5M C5H10NH 8 h Identisch wie CS007 Hauptanteil ist amorph.
CS016 180 °C 1M 1,5M C5H10NH 10x24 h 75 � 175 µm D3C (100%)
CS009 170 °C 1,5 M 1,5 M C5H10NH 9x24 h 3 �36 µm; Hauptanteil: 24µm (90%) D3C (100%)
CS011 170 °C 3M 1,5M C5H10NH 9x24 h 0,2 µm (2%); Hauptanteil: 3 µm (98%) D3C (100%)
CS013 170 °C 1M 1,5M C5H10NH 26 h nur unter REM zu sehen ~ 0,4 µm (5%) Hauptanteil amorphes
Material
CS015 160 °C 1M 1,5M C5H10NH 9x24 h 3 � 10 µm, davon 10 µm (98%) D3C
CS024
(1/3 Menge)
160°C 4M 1,5M C5H10NH 3x24 h 0,1-1 µm (99%); geringer Anteil: 16,2 µm (1%) Kristallines D3C
CS027
(1/2 Menge)
160 °C 4M 1,5M C5H10NH 3x24 h 10 µm (90%)
5 µm (10%) D3C
CS035 (�hoher�
Autoklav)
160 °C 3M 1,5M (CH3)4NOH 12x24 h 0,25 � 1 µm (50%) Hauptanteil amorphes
Material
CS037
(�niedriger�
Autoklav)
160 °C 3M 1,5 M
(CH3)4NOH
12x24 h 0,1 � 1 µm Amorphe Masse
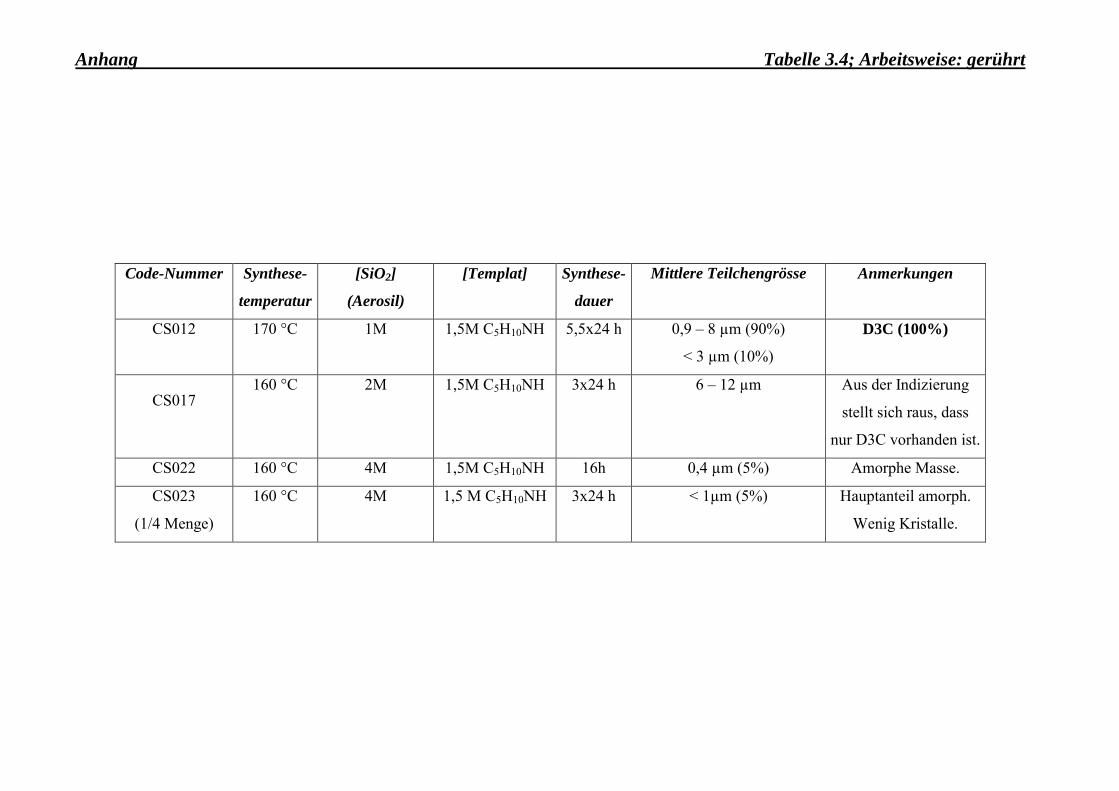
Anhang Tabelle 3.4; Arbeitsweise: gerührt
Code-Nummer Synthese-
temperatur
[SiO2]
(Aerosil)
[Templat] Synthese-
dauer
Mittlere Teilchengrösse Anmerkungen
CS012 170 °C 1M 1,5M C5H10NH 5,5x24 h 0,9 � 8 µm (90%)
< 3 µm (10%)
D3C (100%)
CS017 160 °C 2M 1,5M C5H10NH 3x24 h 6 � 12 µm Aus der Indizierung
stellt sich raus, dass
nur D3C vorhanden ist.
CS022 160 °C 4M 1,5M C5H10NH 16h 0,4 µm (5%) Amorphe Masse.
CS023
(1/4 Menge)
160 °C 4M 1,5 M C5H10NH 3x24 h < 1µm (5%) Hauptanteil amorph.
Wenig Kristalle.
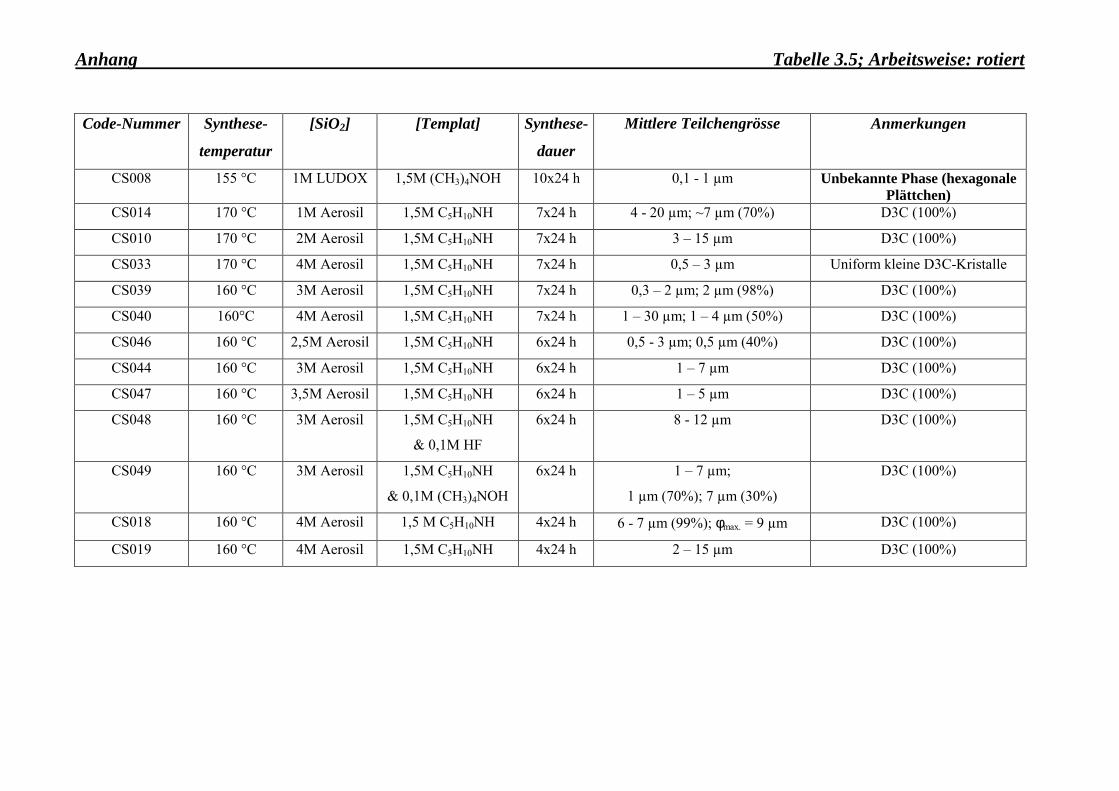
Anhang Tabelle 3.5; Arbeitsweise: rotiert
Code-Nummer Synthese-
temperatur
[SiO2]
[Templat] Synthese-
dauer
Mittlere Teilchengrösse Anmerkungen
CS008 155 °C 1M LUDOX 1,5M (CH3)4NOH 10x24 h 0,1 - 1 µm Unbekannte Phase (hexagonale Plättchen)
CS014 170 °C 1M Aerosil 1,5M C5H10NH 7x24 h 4 - 20 µm; ~7 µm (70%) D3C (100%)
CS010 170 °C 2M Aerosil 1,5M C5H10NH 7x24 h 3 � 15 µm D3C (100%)
CS033 170 °C 4M Aerosil 1,5M C5H10NH 7x24 h 0,5 � 3 µm Uniform kleine D3C-Kristalle
CS039 160 °C 3M Aerosil 1,5M C5H10NH 7x24 h 0,3 � 2 µm; 2 µm (98%) D3C (100%)
CS040 160°C 4M Aerosil 1,5M C5H10NH 7x24 h 1 � 30 µm; 1 � 4 µm (50%) D3C (100%)
CS046 160 °C 2,5M Aerosil 1,5M C5H10NH 6x24 h 0,5 - 3 µm; 0,5 µm (40%) D3C (100%)
CS044 160 °C 3M Aerosil 1,5M C5H10NH 6x24 h 1 � 7 µm D3C (100%)
CS047 160 °C 3,5M Aerosil 1,5M C5H10NH 6x24 h 1 � 5 µm D3C (100%)
CS048 160 °C 3M Aerosil 1,5M C5H10NH
& 0,1M HF
6x24 h 8 - 12 µm D3C (100%)
CS049 160 °C 3M Aerosil 1,5M C5H10NH
& 0,1M (CH3)4NOH
6x24 h 1 � 7 µm;
1 µm (70%); 7 µm (30%)
D3C (100%)
CS018 160 °C 4M Aerosil 1,5 M C5H10NH 4x24 h 6 - 7 µm (99%); φmax. = 9 µm D3C (100%)
CS019 160 °C 4M Aerosil 1,5M C5H10NH 4x24 h 2 � 15 µm D3C (100%)
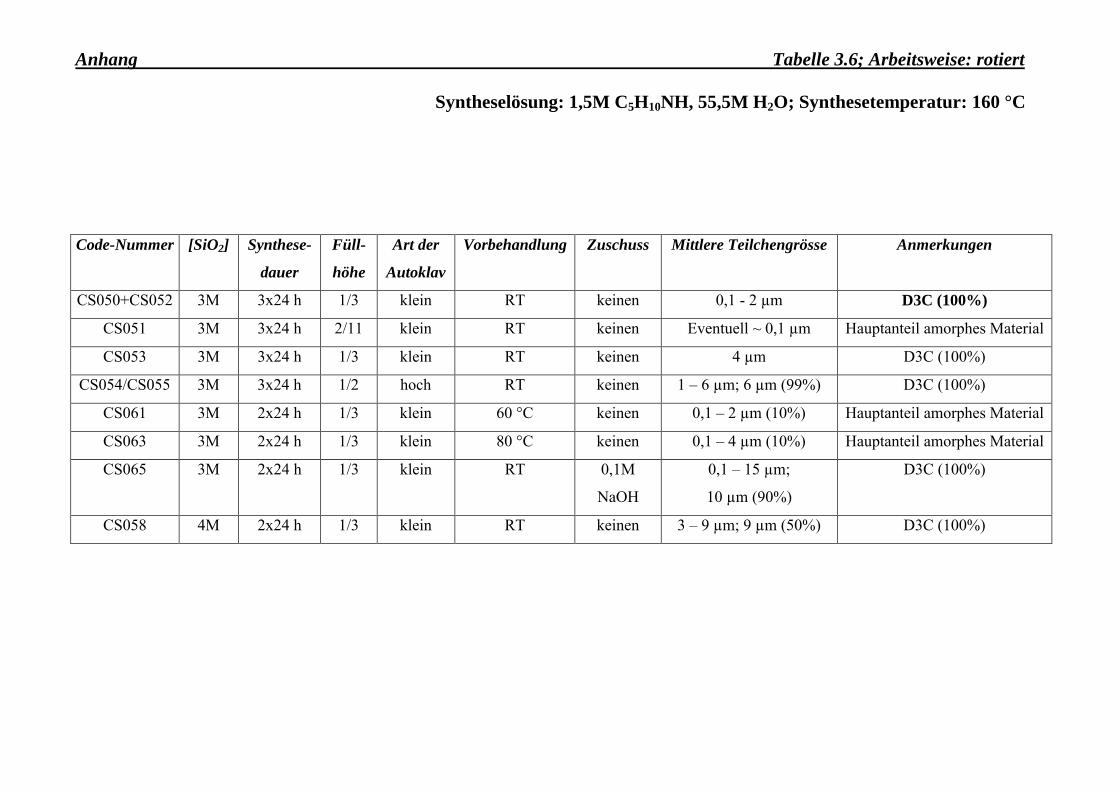
Anhang Tabelle 3.6; Arbeitsweise: rotiert
Syntheselösung: 1,5M C5H10NH, 55,5M H2O; Synthesetemperatur: 160 °C
Code-Nummer [SiO2]
Synthese-
dauer
Füll-
höhe
Art der
Autoklav
Vorbehandlung Zuschuss Mittlere Teilchengrösse Anmerkungen
CS050+CS052 3M 3x24 h 1/3 klein RT keinen 0,1 - 2 µm D3C (100%)
CS051 3M 3x24 h 2/11 klein RT keinen Eventuell ~ 0,1 µm Hauptanteil amorphes Material
CS053 3M 3x24 h 1/3 klein RT keinen 4 µm D3C (100%)
CS054/CS055 3M 3x24 h 1/2 hoch RT keinen 1 � 6 µm; 6 µm (99%) D3C (100%)
CS061 3M 2x24 h 1/3 klein 60 °C keinen 0,1 � 2 µm (10%) Hauptanteil amorphes Material
CS063 3M 2x24 h 1/3 klein 80 °C keinen 0,1 � 4 µm (10%) Hauptanteil amorphes Material
CS065 3M 2x24 h 1/3 klein RT 0,1M
NaOH
0,1 � 15 µm;
10 µm (90%)
D3C (100%)
CS058 4M 2x24 h 1/3 klein RT keinen 3 � 9 µm; 9 µm (50%) D3C (100%)

Danksagung
Danksagung Mein Dank gilt folgenden Personen: Herrn Prof. Dr. Hermann Gies für die interessante und sehr aktuelle Themenstellung, die Überlassung des Arbeitsplatzes und für die Bereitschaft meine Arbeit zu betreuen, Herrn Dr. Ingo Wolf für die freundliche Hilfsbereitschaft, die Begleitung auf dem interessanten wissenschaftlichen Terrain sowie die zahlreichen wichtigen Tips und Hilfestellungen, Herrn Dr. Bernd Marler für das Interesse an dieser Arbeit und seine unermüdliche Korrektur- und Diskussionsbereitschaft, Herrn Dr. Peter Daniels für die Hilfe bei der Korrektur der Arbeit und das Beibringen des logischen Denkens, Frau Renate Sczesny für ihre freundliche und freundschaftliche Art und die Versorgung mit den aktuellsten wissenschaftlichen Publikationen,
Frau Sandra Grabowski für ihre ständige Hilfsbereitschaft und die vielen guten Ratschläge, was das Leben im Chemielabor betrifft, Herr Dr. Heribert Graetsch für die engagierte Hilfe bei den Quarzmehluntersuchungen und die Bereitschaft, sein Wissen weiterzugeben, Herr Dr. Walter Gebert für die hochkompetente Art sein Wissen über die Röntgenpulverdiffraktometrie zu vermitteln, meinem Ehemann, Dipl. Chem. Martin Osterhoff, der in jedem schweren Moment an meiner Seite war und zu jeder Zeit bereit war die Korrekturen an dieser Arbeit durchzuführen, allen Kolleginnen und Kollegen für die freundliche Aufnahme in der Arbeitsgruppe, die Hilfestellung, die sie mir jedes Mal gegeben haben, wenn ein wissenschaftliches Problem aufgetreten ist,

Danksagung
allen meinen Siedler-Spielkameraden, die vermutlich anonym bleiben wollen und die in der Mittagspause für eine angenehme Entspannung gesorgt haben, meinen Eltern die viel Geld und Hoffnung in mein Studium investiert haben, der Deutschen Forschungsgemeinschaft (DFG) für die finanzielle Unterstützung, ohne die die vorliegende Arbeit nicht durchgeführt werden konnte.

Erklärung
Erklärung Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig und ohne unerlaubte Hilfe ausgeführt und verfasst habe, und dass die Arbeit in dieser oder ähnlicher Form bei keiner Fakultät oder anderen Hochschule eingereicht wurde.
Cristina Osterhoff

Lebenslauf
Lebenslauf Persönliche Angaben Geburtsdatum 30.04.1970 Geburtsort Mühlbach/Rumänien Staatsangehörigkeit deutsch Familienstand verheiratet Eltern Mathias und Rafila, geb. Morariu Bildungsgang Schulausbildung 1976 - 1984 Allgemeinschule Nr. 2, Mühlbach/Rumänien 1984 - 1986 Erste Lyzealstufe: Industriallyzeum Mühlbach, Fach:
Industrielle Chemie 1986 - 1988 Zweite Lyzealstufe - Chemielyzeum "Terapia",
Klausenburg, Fach: Organische Chemie Juni 1988 Erlangung der allgemeinen Hochschulreife am
Chemielyzeum "Terapia", Klausenburg Berufsausbildung 1989-1994 Studium an der Chemie-Hochschule der Universität "Babeş-
Bolyai", Klausenburg; Fachrichtung: Chemie-Physik Juli 1994 Abschluss: Diplom, Lyzenzprüfung-Gesamtnote: 9,36 (1,3) November1994 Anerkennung als Dipolm-Chemikerin vom Ministerium für
Wissenschaft und Forschung des Landes Nordrhein-Westfalen
März-August1995 Zusammenarbeit mit Prof. Dr. Baerns und Herrn Dr. rer.
nat. Grünert, Lehrstuhl für technische Chemie an der Fakultät für Chemie, Ruhr-Universität Bochum
Seit August1996 Promotionsstudium am Institut für Mineralogie, insb.
Kristallographie der Ruhr-Universität Bochum, Betreuer Prof. Dr. Hermann Gies
Hobbys Malen, Fotografieren, Lesen, ,Gesellschaftsspiele.













![13C CP/MAS NMR Study of the Layered Compounds [C ...zfn.mpdl.mpg.de/data/Reihe_A/51/ZNA-1996-51a-0910.pdf912 T. Ueda et al. 13C CP/MAS NMR Study of [C 6H5CH2CH2NH3]2[CH3NH3]n_ ,Pb](https://static.fdokument.com/doc/165x107/6068be4a5994db3c4a31fd51/13c-cpmas-nmr-study-of-the-layered-compounds-c-zfnmpdlmpgdedatareihea51zna-1996-51a-0910pdf.jpg)




