Synthese der Oligonukleotide - Carell group...Die stereoselektive Synthese von -anomeren Zuckern...
19
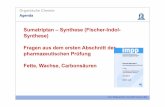
39 2. Synthese der Oligonukleotide 2.1 Synthese der Nukleoside 2.1.1 Allgemeines Effiziente Syntheserouten zu Nukleosiden werden zur Synthese verschiedenster Analoga benötigt. Diese Nukleosid Analoga konkurrieren im zellulären Geschehen mit den natürlichen Nukleosiden T, U, C, G und A. Sie stören daher die zellulären Prozesse. Viele derartiger Verbindungen haben anti-virale oder anti-proliferative Eigenschaften. Die t-RNA enthält eine ganze Reihe modifizierter Nukleoside. Das Studium wieso die Natur für die Übersetzung des genetischen Codes auf diese Substanzen zurückgreift bedingt die Synthese dieser modifizierten Nukleoside und deren Einbau in DNA und RNA. Prinzipiell werden heute drei Wege (A, B und C) zur Synthese von Nukleosiden beschritten. Auf dem Weg A, findet eine nukleophile Substitution statt. Die Abgangsgruppe befindet sich am anomeren Zentrum des Zuckers. Weg B liegt ebenfalls eine nukleophile Substitution zugrunde. Hier ist das Nukleophil allerdings der Zucker. Weg C bedeutet Aufbau des Heterocyclus am Zuckergerüst. HO NH N N O NH 2 N O OH HO O OH X NH N N O NH 2 N H HO O OH NH 2 NH N YC N O NH 2 Z HO NH 2 CY N N O OH B C A C A B
Transcript of Synthese der Oligonukleotide - Carell group...Die stereoselektive Synthese von -anomeren Zuckern...
39
2. Synthese der Oligonukleotide
2.1 Synthese der Nukleoside
2.1.1 Allgemeines
Effiziente Syntheserouten zu Nukleosiden werden zur Synthese verschiedenster
Analoga benötigt. Diese Nukleosid Analoga konkurrieren im zellulären Geschehen
mit den natürlichen Nukleosiden T, U, C, G und A. Sie stören daher die zellulären
Prozesse. Viele derartiger Verbindungen haben anti-virale oder anti-proliferative
Eigenschaften. Die t-RNA enthält eine ganze Reihe modifizierter Nukleoside. Das
Studium wieso die Natur für die Übersetzung des genetischen Codes auf diese
Substanzen zurückgreift bedingt die Synthese dieser modifizierten Nukleoside und
deren Einbau in DNA und RNA.
Prinzipiell werden heute drei Wege (A, B und C) zur Synthese von Nukleosiden
beschritten. Auf dem Weg A, findet eine nukleophile Substitution statt. Die
Abgangsgruppe befindet sich am anomeren Zentrum des Zuckers. Weg B liegt
ebenfalls eine nukleophile Substitution zugrunde. Hier ist das Nukleophil allerdings
der Zucker. Weg C bedeutet Aufbau des Heterocyclus am Zuckergerüst.
HO
NH
N
N
O
NH2N
O
OH
HO
O
OH
X
NH
N
N
O
NH2N
H
HO
O
OH
NH2 NH
N
YCN
O
NH2Z
HO NH2
CYN
NO
OH
B
C
A
C
A
B

40
2.1.2 Synthesen basierend auf Weg A
Ältere Methoden der Nukleosidsynthese beruhen auf der Umsetzung der
Schwermetallsalze der Heterocyclen mit einem Chlor- oder Bromzucker. Diese als
Fischer-Helferich oder Koenigs-Knorr Synthesen bekannte Methoden werden heute
nur noch vereinzelt angewendet. Als Schwermetallsalze fungieren die weichen HgII+
oder AgI+ Salze. Für die Synthese müssen alle anderen potentiell nukleophilen
Stellen geschützt werden. Nachteilig ist auch die oft schlechte Löslichkeit der
Schwermetallsalze. Die Halogenzucker müssen unter wasserfreien Bedingungen
gehandhabt werden, da sie sehr hydrolyseanfällig sind.
Die Methode liefert in der Regel die richtige regiochemische Verknüpfung d.h. N1 für
die Pyrimidine und N9 für die Purine. Die Reaktion verläuft nach einem SN2-
Mechanismus.
Statt der Schwermetallsalze lassen sich auch die alkylierten Basen verwenden.
Deren N-Atome im Heterocyclus sind in der Regel bereits nukleophil genug. Die
Methode heißt Hilbert-Johnson Nukleosidierung.
HO
O
HO OH
N
N
N
NH2
N
AcO
O
AcO
Br
OAc
N
N
N
HN
N
ClHg
O
O
AcO
O
AcO OAc
N
N
N
HN
N
O
O
AcO
O
AcO
Br
OAc
N
N
N
Cl
N
ClHgNH
Ac
HO
O
HO OH
NH
N
N
N
O
NH2
Xylen, 120oC
NH3/MeOH
Alternativ
1. Xylen, 120oC
2. NaOH, H2O

41
Eine moderne Variante der Hilbert-Johnson Methode ist die Silyl-Hilbert-Johnson
Methode, die auch Nukleosidierung nach Vorbrüggen genannt wird. Hierbei wird ein
stabilerer Zuckervorläufer, z.B. mit einer schlechteren Abgangsgruppe wie dem
Acetat am anomeren Zentrum eingesetzt. Vor der Kupplung wird der Zucker mit einer
Lewissäure zur „Aktivierung“ umgesetzt. In situ werden die nukleophilen Zentren
geschaffen.

42
Es entsteht eine Oxycarbenium-Ion Zwischenstufe. Die Nukleosidierung erfolgt daher
mehr nach einem SN1-Mechanismus und liefert daher meist Gemische des und
des Produktes. Befindet sich am C2‘ eine weitere Acetat oder Benzoat-Gruppe so
kann das Oxycarbeniumion von dieser Gruppe unter Nachgruppenbeteiligung
stabilisiert werden. Die Base kann entweder direkt nach Deprotonierung mit Base
(regiochemisch oft uneinheitliche Produkte) oder nach Silylierung (Vorbrüggen)
eingesetzt werden. Durch die Silylierung (mit HMDS) nimmt die Nukleophilie der N-
Atome im Heterocyclus zu. Das freie Elektronenpaar ist nicht mehr an der
aromatischen Stabilisierung beteiligt.
Es ist allerdings schwer abzuschätzen wo die Reaktion am Heterocyclus erfolgt und
oft werden keine einheitlichen Produkte erhalten. Der Angriff erfolgt immer vom
elektronenreichsten d.h. basischsten N im Heterocyclus. Durch den SN1-Charakter
bedingt ist auch die Bildung eines -/-Anomerengemisches. Zwei weitere Beispiele:
AcO
O
AcO
OAc
O O
AcO
O
AcO
OAc
OAc
AcO
O
AcO OO
AcO
O
AcO OO
AcO
O
AcO OAc
N
N
O-Si
OAcO
O
AcO OAc
N
NH
O
O
N
N
O
O
Si
Si
HN
NH
O
O
Me3SiCl
H2O
TfO-
Si OTf
N
N
O
O
Si
Si
TMS-Tf oder
SnCl4, Hg(OAc)2
Acetonitril oder1,2-Dichlormethan
-20oC - 50
oC

43
Mechanistisch erfolgt also zunächst die Bildung eines elektrophilen
Oxycarbeniumions. Dann bildet sich ein -Komplex aus der silylierten Base und der
Lewis-Säure. Dann erfolgt die Kupplungsreaktion.
Kritisch ist die -Komplexbildung. Bei schwachen Lewissäuren liegt nur wenig
Komplex vor. Es reagiert dann das basischste N im Heterocyclus also z.B. das N1
bei Pyrimidinen. Starke Lewissäuren (SnCl4) führen zur totalen Komplexierung dann
reagiert unter Umständen das weniger elektronenreiche Zentrum. Hier hilft dann das
Arbeiten in nukleophilen Lösungsmitteln. Das Nukleosidierungen oft Gleichgewichts-
reaktionen sind, kann durch Rühren bei leicht erhöhter Temperatur noch ein
Umlagern des primär gebildeten kinetischen Produktes zum thermodynamisch
günstigeren Produkt erfolgen.
AcO
O
AcO OAc
N
N
S
NHTMS
O
AcO
O
AcO
OAc
OAc
AcO
O
AcO OAc
N
N
S
NH2
O
BzO
O
AcO
Cl
N3
N
NN
HN
Cl
BzO
O
AcO
N3
N
NN
N
Cl
BzO
O
AcO
N3 N
NN
N
Cl
NH
N
S
NH2
O
(Me3Si)2NH
N
N
S
NHTMS
OTMS
SnCl4
NH3, MeOH
+
+

44
2.1.3 Kontrolle der Stereochemie am anomeren Zentrum
Wie oben bereits erwähnt, führen nukleophile Gruppen am C2‘ zu einer
Koordinierung des Oxycarbeniumions. Das ist besonders bei 2‘-Acyloxy-oder 2‘-
benzoylocygruppen der Fall. Das Oxycarbeniumion wird durch Verbrückung, unter
Nachbargruppenbeteiligung stabilisiert. Die Reaktion mit dem Nukleophil erfolgt dann
trans zur Nachbargruppe. Diese Gesetzmäßigkeit trägt den Namen Bakers-1,2-trans
Regel.
Aus der Regel folgt, dass die Reaktion der 1‘-Chlor-D-Arabinose und 1‘-Chlor-D-
Lyxose mit einer Base hauptsächlich die -Anomeren ergibt. Aus der 1‘-Chlor-D-
Ribose und der 1‘-Chlor-D-Xylose entstehen als Hauptprodukte die -Anomeren.
Der Nachbargruppeneffekt ist augenfällig wenn die -Verteilung bestimmt wir mit
einer p-Nitrobenzoylschutzgruppe an der C2‘-OH Gruppe. Der elektronenziehende
Effekt der p-Nitrogruppe reduziert die Basizität der einsamen Elektronenpaare am
Carboxyl-O-Atom, so dass die Stabilisierung des Oxycarbeniumions schlechter sein
N
NN
N
Si
NBz Si
N
N
O
O
Si
Si
Me3SiOX
N
NN
N
Si
NBz Si
Si
N
N
O
O
Si
SiSi
N
NN
N
NBz Si
Si
Reaktion an verschiedenenStellen
N
N
NH
HN O
O
Alloxazin
N
N
N
N O
O
Si
Si
1
2
3
3

45
sollt. Genau das wird beobachtet. Es entsteht mehr vom nicht Baker-Regel Produkt
als vorausgesagt wird. Die Reaktion wird deutlich weiter ins SN1-Regime verlagert.
2.1.3 Synthesen basierend auf Strategie B
Prinzipiell sind die Basen elektronenarme Heterocylen, in denen durch Alkylierung
oder Silylierung nukleophile Zentren geschaffen werden müssen. Den
elektronenarmen Charakter der Basen kann man ausnutzen, wenn ein
Zuckernukleophil eingesetzt wird. Die Base enthält als Substrukturelement quasi ein
„vinyloges Säurechlorid“. Dieses wird mit dem Aminozucker umgesetzt. Diese Art der
BzO
O
BzO
Cl
O O
BzO
O
BzO OBz
BzO
O
BzO O O
H
BzO
O
BzO O O
Base
BzO
O
BzO
Cl
OAc BzO
OCl
OAcOBzO
Base
BzO
O
BzO
Cl
OAcBzO
O
BzO
Cl
OAc
OBase
HgCl+
O O
NO2
Base-HgCl
D-Arabinose D-Lyxose Anomere
AnomereD-Ribose D-Xylose
p-Nitrobenzoat
46
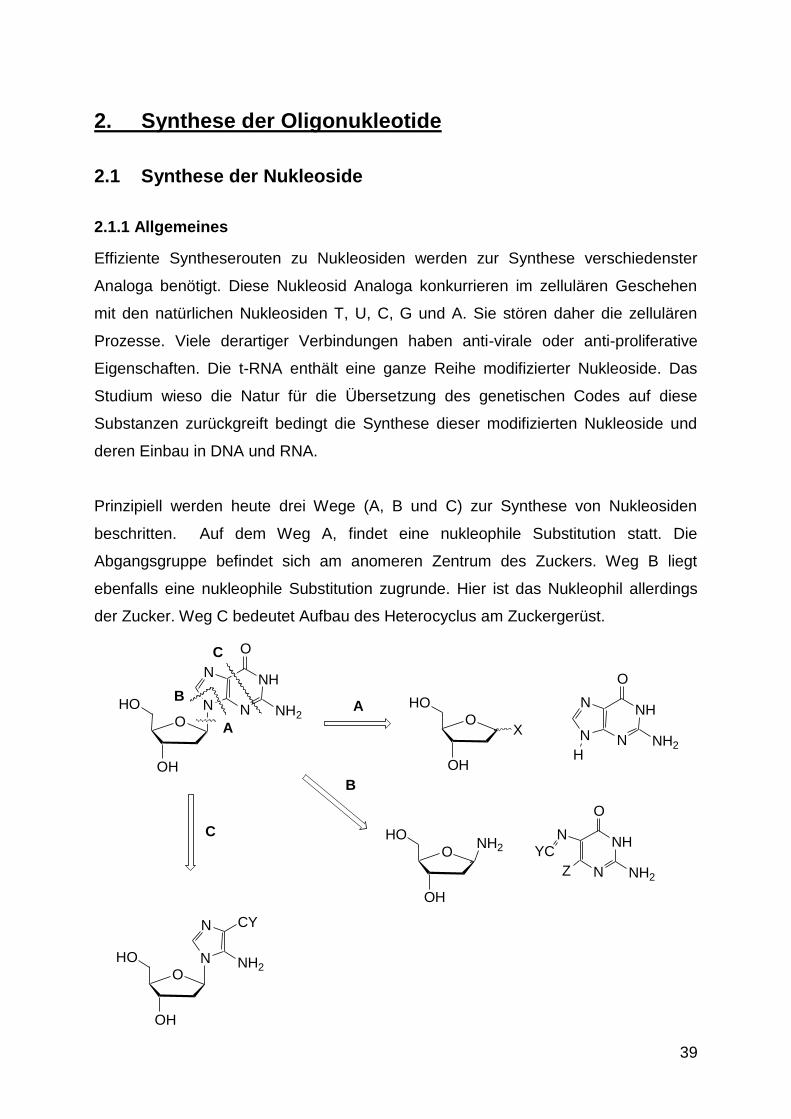
Nukleosidierung wird relativ selten angewendet. Vom Reaktionstyp her handelt es
sich um eine nukleophile Substitution an einem Heteroaromaten, ähnlich wie bei der
Umsetzung von Peptiden mit dem Saenger‘s-Reagenz zur Endgruppenbestimmung.
2.1.4 Aufbau des Heterocyclus am Zucker
Führen alle Nukleosidierungen nur zur Bildung des falschen Regioisomeren oder
reagiert die Base gar nicht, so muss der Heterocyclus am Zuckergerüst aufgebaut
werden. Dieser Weg führte z.B. zur erfolgreichen Synthese von Flavin-Basen, die
aus den Alloxazinen nicht hergestellt werden können, da andere, im Alloxazin
basischere Zentren, bevorzugt reagieren.
AcO
O
AcO OAc
NH2AcO
O
AcO OAc
HN NH
NHO2N
O
NHAc
NH
NHO2N
O
NHAcCl
N
N
NH
HN O
O
Alloxazin
N
N
N
N O
O
Si
Si
1
2
3
3
AcO
O
AcO
NH2
OAc
NO2
NO2
NH4Cl
AcO
O
AcO OAc
HN
NO2
1. H2, Pd/C
NH
NH
O
O
O O
AcO
O
AcO OAc
N
N
N
NH
O
O
2.

47
Ganz ähnlich gelangen auch die Totalsynthesen, der in t-RNA vorkommenden Basen
Pseudouridin und Wyosin. Bei Pseudouridin handelt es sich um ein C-Nukleosid.
Diese Verbindungen sind aufgrund des Fehlens einer Acetal-Substruktur wesentlich
resistenter gegenüber Hydrolysen. C-Nukleoside werden daher als Glycosylase-
Inhibitoren eingesetzt. Glycosylasen sind Enzyme die glycosidische Bindungen
spalten.
2.1.5 Stereoselektiver Zugang zu - und -Nukleosiden
Für die stereoselektive Synthese wurden eigene Synthesen entwickelt. Die selektive
Darstellung von -Anomeren gelingt mit Hilfe der Oxazilidin-Methode. Gezeigt ist die
Synthese von -Arabinofuranosyl-Adenin. Zur Reduktion von OH-Gruppen, wie hier
zur Darstellung der Desoxyribose aus dem Ribosevorläufer wird häufig die Barton-
McCombi Reaktion angewendet.
Die stereoselektive Synthese von -anomeren Zuckern gelingt mit Hilfe des 2-Fluor-
1-methylpyridiniumtosylats. Reaktion mit dem Zucker unter thermodynamischer
Kontrolle fixiert das anomere Zentrum, so dass der große Pyridiniumrest die sterisch
HO
O
HO OH
NHHN
O
O
AcO
O
AcO OAc
N
C
O
AcO
O
AcO OAc
N
NNH2
O
NHMeHO
O
HO OH
N
N
N
N
N
O
O O
O O
O
Ph
Ph
N
NO
O
t-Bu
t-Bu
Li O O
O O
OH
Ph
Ph N N
Ot-Bu
O t-Bu
THF, -78oC
Swern Oxidation
H+ Mild

48
günstigere -Position einnimmt. SN2-Substitution durch die silylierte Base erfolgt
hauptsächlich unter inversion der Konfiguration zum -Anomeren.
BzO
O
O
OH
O
BzO
O
O O
O N
BzO
O
O OBase
O
RO
ROO
NH
S
ORO
ROO
N
S
N
N
H2N
O2N
O
RO
RO OH
N
HN
N
NS
NH2
ORO
RO O
N
N
N
N
NH2
S
SMe
NaH
DMF
HgBr2
Ra-Ni
H2O, NH4OH
Bu3SnH, AIBN
O
RO
ROO
N
S
HgBr
NF
ORO
ROO
N
S
N
N
H2N
H2N
ORO
RO OH
N
N
N
N
NH2
N
N
Cl
O2N
NH2
ORO
RO
N
N
N
N
NH2
, H2O
N
N
OSi(Me3)
(Me3)SiO
NH
O
Al/Hg
NaH,
Cs2
MeJ
Ts-

49
2.2 Synthese der Nukleotide
2.2.1 Die Chemie der Phosphor- und Phosphorigsäureester
Der in der Biologie relevante Phosphor (P) hat die Oxidationsstufe P(V) und ist
vierfach koordiniert. Zur Synthese biologisch relevanter Strukturen ist deshalb die
Kenntnis der Chemie dieser Phosphorverbindungen notwendig.
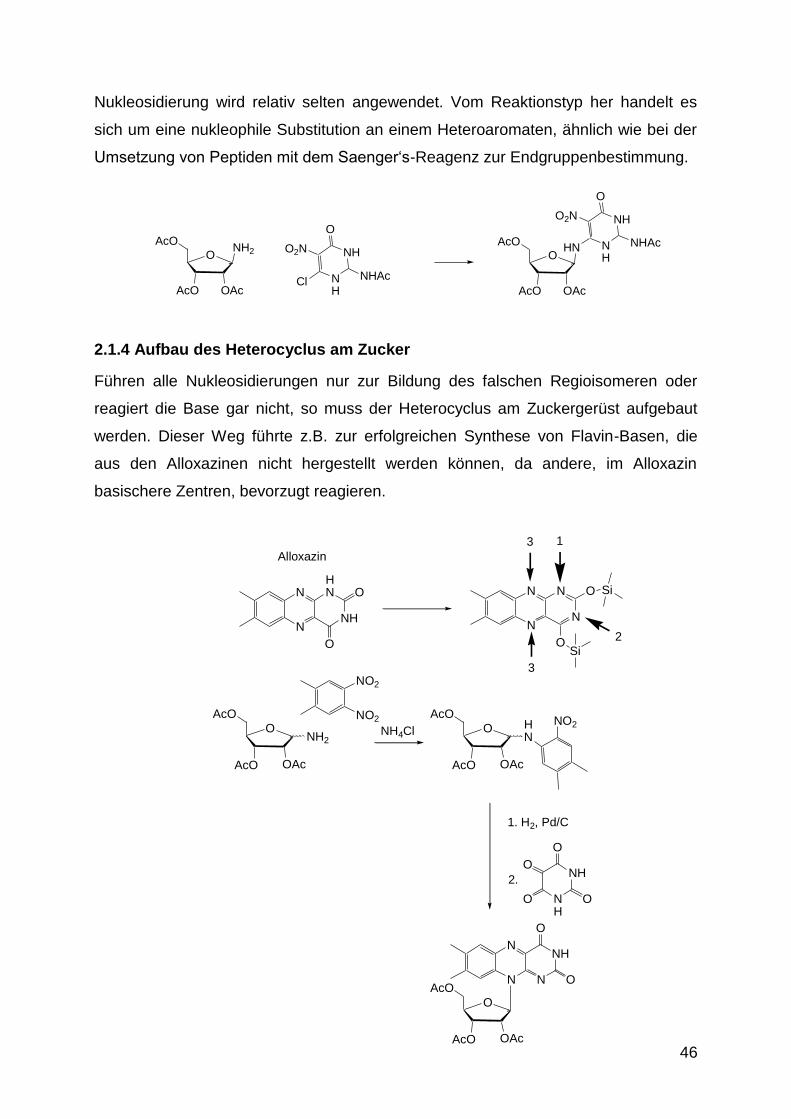
Die P-O Einfachbindungen sind sp3 Hybridorbitale und ca. 1.6 Å lang.
In den Triestern und in anderen Verbindungen sind die P=O Bindungen 1.46 Å lang.
Es handelt sich um pd Hybridorbitale. Der Phosphor weitet also seine Schale unter
Beteiligung von d-Orbitalen auf.
Die Triester wie PO(OEt)(OBu)(OPh) sind löslich in vielen organischen
Lösungsmitteln und leicht an Kieselgel chromatographierbar. Der P bildet ein stabiles
chirales Zentrum.
Die Diester wie PO(OEt)(OBu)(OH) sind wasserlösliche Verbindungen und mit einem
pKa-Wert von 1.5 wesentlich stärker sauer als Essigsäure (pKa = 4.75). Sie liegen in
Wasser deprotoniert vor. Die Ladung ist zwischen den zwei O-Atomen am P
delokalisiert (deshalb der niedrige pKa-Wert).
PO
OH
HO
HOP O
PO
O
O
O PhBu
EtP
O
OH
O
OBu
EtP
O
OH
O
HO
Et
PO
O
O
OBu
Et
50
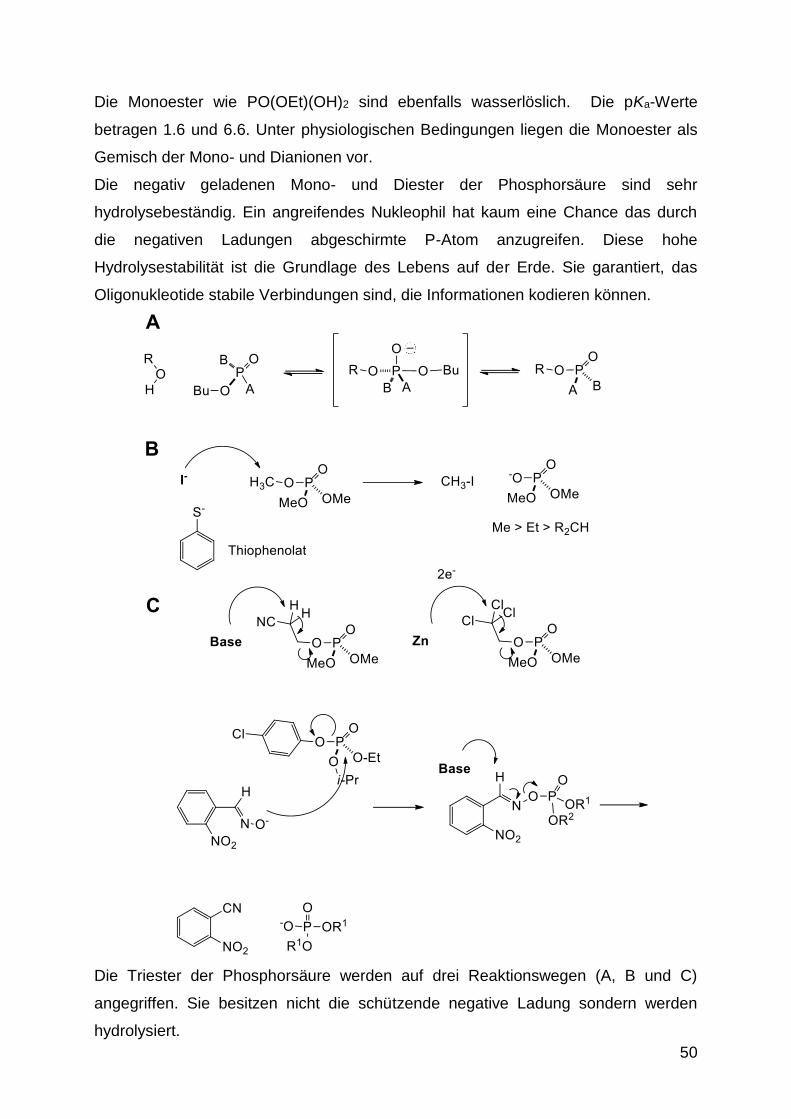
Die Monoester wie PO(OEt)(OH)2 sind ebenfalls wasserlöslich. Die pKa-Werte
betragen 1.6 und 6.6. Unter physiologischen Bedingungen liegen die Monoester als
Gemisch der Mono- und Dianionen vor.
Die negativ geladenen Mono- und Diester der Phosphorsäure sind sehr
hydrolysebeständig. Ein angreifendes Nukleophil hat kaum eine Chance das durch
die negativen Ladungen abgeschirmte P-Atom anzugreifen. Diese hohe
Hydrolysestabilität ist die Grundlage des Lebens auf der Erde. Sie garantiert, das
Oligonukleotide stabile Verbindungen sind, die Informationen kodieren können.
Die Triester der Phosphorsäure werden auf drei Reaktionswegen (A, B und C)
angegriffen. Sie besitzen nicht die schützende negative Ladung sondern werden
hydrolysiert.

51
Reaktionsweg A beschreibt eine Substitution nach dem SN2P-Mechanismus. Es ist
ein assoziativer Prozess bei dem das Nukleophil zuerst an das P-Atom addiert. Erst
anschließend zerfällt das Zwischenprodukt. In diesem Prozess ist die Pseudorotation
oft langsamer als der Zerfall des Zwischenproduktes, so dass chirale Information
teilweise erhalten bleibt. Dieser Hydrolysemechanismus ist besonders schnell, wenn
es sich um Arylester handelt (s.o.) Der niedrigere pKa-Wert der Phenolate (pKa-Wert
ca. 10) im Vergleich zu nichtaromatischen Alkoholen (pKa-Wert ca. 15) zeigt bereits,
dass Phenolate wesentlich bessere Abgangsgruppen sind, die die negative Ladung
besser, durch Delokalisation in den Ring, stabilisieren können. Zur Abspaltung von
Arylestern als Schutzgruppen verwendet man meist Oxinat-Anionen als Nukleophile,
die dann in einer -Eliminierung (s.u.) in einem zweiten Schritt abgespalten werden.
Als Arylester-Schutzgruppe werden meist p-Chlorphenolester verwendet (warum?).
Reaktionsweg B beschreibt den Angriff sehr weicher Nukleophile. Sie greifen nicht
am P-Atom sondern an den Alkylgruppen eines Esterrestes an. Das sind typische
SN2-Reaktionen in denen der Phosphorsäuremonoester als gute Abgangsgruppe
fungiert. Diese Reaktion wird zum entschützen von Phosphorsäuretriestern
verwendet. Sie funktioniert phantastisch bei Methylestern, ist bei Ethyl- oder
sekundären Zentren allerdings schon relativ schlecht. Me > Et > R2CH.
Ausgenutzt wird diese Reaktivität bei der Entschützung von Phosphorsäure-
methylestern mit Thiophenolat.
Reaktionsweg C beschreibt eine -Eliminierung von Alkylphosphat-Triestern. Diese
Reaktion ist dann besonders gut wenn am -C-Atom noch eine elektronenziehende
Gruppe vorhanden ist, die die H-Atome acidifiziert. Hier hat sich die Cyanoethyl-
Gruppe sehr bewährt. Sie wir heute als Schutzgruppe in der Oligonukleotidchemie
angewendet. Ein weiteres Beispiel ist die Trichlorethylester-Schutzgruppe, die
Reduktiv mit Zn entfernt werden kann. Durch -Eliminierung werden auch die
Oximat-Ester abgespalten.
Die Phosphorsäuredi- und -monoester sind wie schon gesagt sehr
hydrolysebeständig. Das st im Fall der DNA sehr wichtig. RNA muss jedoch,
nachdem die Information in ein Protein translatiert wurde abgebaut werden, sonst
würde die Zelle nach der Produktion eines m-RNA Stranges ewig das kodierte

52
Protein erzeugen. Zum Abbau der DNA durch Enzyme (Ribonukleasen) nutzt man
aus, dass Aktivierungsenergien durch Proximitätseffekte und Ringspannung
(Aktivierungsentropie) stark reduziert werden können. Fünfring enthaltende, cyclische
Phosphorsäurediester werden aufgrund der Spannung im 5-Ring 107 mal schneller
hydrolysiert als die nicht zyklischen Diester. Das entspricht einer Verringerung der
freien Aktivierungsenthalpie G# um 36 kJ/mol. Genau dieses Prinzip nutzen die
Ribonukleasen, die RNA nach dem unten gezeigten Mechanismus hydrolysieren.
Die intrinsische Stabilität der Mono- und Diester wird durch die 5-Ring
Zwischenstruktur überwunden. Hydrolyse kann so erfolgen.
2.2.2 Kondensierte Phosphate und Synthese von Phosphorsäureesrtern
Die Synthese von Phosphorsäureester kann mit Hilfe von Dicyclohexylcarbodiimid
(DCC) analog einer Carbonsäureester Synthese erfolgen. Hierzu wird die
Phosphorsäure durch Umsetzung mit DCC aktiviert. Dann erfolgt der Angriff des
Alkohols. DCC erlaubt hauptsächlich Synthese von Diestern aus Monoestern.
Triester werden nicht gebildet. Hierzu reicht die Aktivität des DCC nicht aus. Zur
Triestersynthese verwendet man meistens Mesitylensulfonylchlorid oder die
entsprechenden Tetrazolide und Nitrotriazolide. Die Azolid-Anionen haben den
Vorteil, dass sie sehr wenig nukleophil sind. Schon das Cl- bereitet manchmal
Probleme und führt zur Phosphorsäureester Spaltung. Das Mesitylen wird auf Grund
O
O
O OH
Py
PO
O
O
O
O
O O
Py
PHO O
P
RO
OR
O O-
H2O
OP
O OH
O
B
BH
OP
O OH
OH
OH OP
O
OH
OH
HO
Nu

53
der erzeugten sterischen Abschirmung des S-Zentrum verwendet. So wird der
nukleophile Angriff des Alkoholats auf das P-Atom gefördert.
Viele heute verwendete Synthesen laufen über den P in der Oxidationsstufe P(III),
also über die Phosphit-Trister. Hier nutzt man die größere Reaktivität der P(III)-
Spezies aus (Vgl. PCl3 versus POCl3). Bahnbrechend war die Entwicklung der
Phosphoramidit-Chemie durch Caruthers und Matteucci (Abbildung A unten).
Hier werden die Diamide der Phosphorigsäure mit Tetrazol umgesetzt. Dabei reicht
die Acidität des Tetrazol aus um das N-Atom im Phosphorigsäureamid zu
protonieren. Erst nach der Protonierung werden die N-Substituenten zu sehr guten
Abgangsgruppen. Man kann also die stabilen Amide einsetzen und so das Arbeiten
mit den Hydrolyseanfälligen Halogenverbindungen vermeiden. Intermediär entstehen
die Tetrazolide der P(III)-Spezies, als Reaktivester. Diese reagieren mit Nukleophilen
wie z.B. Alkoholen z. B. zu den Triestern. Unter bestimmten Umständen
(Stöchiometrie) kann auf der Stufe der Diester die Reaktion gestoppt werden. Die
Phosphit-triester sind in der Regel auch instabil und werden nach erfolgter Bildung
mit Iod oder tert-Butylhydroperoxid zu den Phosphat-triestern aufoxidiert.
Basierend auf alten Arbeiten der Todd-Gruppe hat sich in den letzten Jahren auch
die H-Phosphonat Chemie (Abbildung B oben) stark entwickelt. Durch Umsetzung
von PCl3 mit Imidazol und nachfolgender milder Hydrolyse entstehen H-Phosphonate
P
OR
OH
O OR
P
HO
HO
O ORN
C
N
S
O
O
NN
NN
N
C
NH
P
O
HO
ROO
P
OR
OR
O NN
N
NP
OR
OR
O OR'
NH
C
NH
O
P
OR
OR
O O S
O
O
P
O
OH
RO OR'
R'-OH
HOR'

54
als Tautomere der Phosphit-Monoester. Diese H-Phosphonate sind im Gegensatz zu
den Phosphit-Monoester relativ stabil. Sie lassen sich durch Umsetzung mit einem
sperrigen Säurechlorid wie Pivaloylsäurechlorid oder Adamantylsäurechlorid in
gemischte Anhydride als aktivierte H-Phosphonate überführen. Diese reagieren dann
mit Alkoholen zu den H-Phosphonat Diestern. Erst ganz am Ende werden auch die
H-Phosphonate durch Umsetzung mit Iod oder tert-Butylhydroperoxid zu den
Phosphorsäurediestern aufoxidiert.
2.2.3 Synthese biologisch wichtiger Phosphat-Monoester
Zur Synthese der Phosphat-Monoester bedient man sich zweier Methoden. Sie sind
aus den Triestern durch Hydrolyse erhältlich. Zur Synthese der Monoester wurde das
Catechol-Phosphorsäurechlorid entwickelt. Es reagiert mit Alkoholen zum Triester,
der dann selektiv zum Monoester aufgrund der hohen Reaktivität des Catechol-
System (phenolisch und cyclisch) hydrolysiert werden kann. Darüber hinaus wurden

55
Reagenzien entwickelt, die Auf Grund ihres großen Raumbedarfs nur mit primären
Alkoholen zu den Phosphorsäuremonoestern reagieren können. Das ist wichtig, da in
der Natur zumeist die primäre 5‘-Position der Ribose phosphoryliert vorliegt. Die
gebräuchlichsten Reagienzen sind das Bis(2-tert-butylphenyl)phosphorchloridat
(Hydrolyse mit H2O) oder das Bis(2,2,2-trichlor-1,1-dimethylethyl)phosphorchloridat
(Hydrolyse mit Zn-Salzen).
Besonders gut funktionieren auch Methoden, die von Yoshikawa und Sowa-Ouchi
entwickelt wurden. Hierbei wird Phosphorylchlorid in einem Phosphorsäuretriester
Lösungsmittel mit dem Alkohol zur Reaktion gebracht (Yoshikawa). Nachfolgende
Hydrolyse gibt die Monoester. Wichtig hier sind die Reaktionsbedingungen.
Umsetzung von Phosphorylchlord mit dem Alkohol in Acetonitril/Pyridin/H2O (Sowa-
Ouchi) führt ebenfalls in 80%-iger Ausbeute und >90%-iger Selektivität zur Synthese
der Monoester.

56
2.2.4 Synthese kondensierter Phosphate, Pyrophosphate
Einige Beispiele zeigen wie verbreitet und in der Natur wichtig kondensierte
Phosphate sind. Das ATP ist der wichtigste Energiespeicher in unsere Zellen.
Verbindungen wie das NAD+ oder das FAD sind wichtige Coenzyme, die
entscheidend an der Zellatmung teilnehmen. Die Biosynthese der komplexen Zucker
auf den Oberflächen unserer Zellen geschieht im Wesentlichen mit Hilfe der UDP-
Zucker Vorläufer z.B. UDP-Glucose oder UDP-Galaktose. cADP-Ribose ist an der
Regulierung des Calciumhaushaltes wesentlich als second messenger beteiligt.
Die Synthese derartiger Verbindungen im Laboratorium erfolgt über die Aktivierung
der Monoester. So können Monoester mit DCC aktiviert werden. Umsetzung mit
Morpholin oder Diisopropylamin führt zur Bildung der Amidate, die mit Diphosphaten
zu den Triphosphaten umgesetzt werden können. Direkte Reaktion der
Monophosphate mit dem Diphenylphosphorchloridat führt nach Hydrolyse zur
Bildung der Diphosphate. Das intermediär entstehende Diphenyloxy-geschützte
Diphosphat kann mit Phosphat in einer SN2-Reaktion zum Triphosphat umgesetzt
werden.
OO
HO OH
N
N
N
N
NH2
PO
PO
PO
O O O O O O
OO
HO OH
N
N
N
N
NH2
PO
PO
O O O O
R
ROO
HO OH
N
NH2
O
OO
HO OH
NPO
PO
O O O O
NH
O
O
O
OHHO
HO
OHH
O
O
HO OH
N
N
N
N
NH2
P
OO
O
O
OHHO
P
O
O
ON
N
NH
N O
O
OH
HO OH
ORATP
NAD+ FAD
(OH)
(H)
UDP-Glucose
(UDP-Galactose)
cADP-Ribose

57
Wichtig bei der Synthese ist, dass sichergestellt wird, das jedes Phosphatzentrum
am Besten schon in der Synthese eine negative Ladung trägt, weil sonst rasche
Hydrolyse erfolgt. Alle Synthese von kondensierten Phosphaten vermeiden die
Bildung von Triphosphat Zwischenstufen.
Poulter hat z.B. auch die 5‘-OH Tosylate direkt mit Diphosphat oder sogar
Triphosphat umgesetzt, was eine direkte Synthese ermöglichte.
OHP
OP
HO
O O O O
TsO
O
AcO OAc
Ad
O
AcO OAc
ThyOP
O
O O
O
AcO OAc
AdOP
OP
HO
O O O O
O
AcO OAc
ThyOP
OP
PhO
PhO O O O
O
AcO OAc
ThyOP
O
N O
O
OHP
OP
HO
O O O O O
AcO OAc
ThyOP
OP
O
O O O OP
O
O O
DCC,NH
O
PO
Cl
PhO
PhO
dTTP