![Synthese, Charakterisierung und Reaktivität von ... · x oder 2 K Cp3 Cp4 (D) (E) [{Cp4(OC) (C) 2 Fe} 2] I. Einleitung 2 2. Substituentenfreie P 4-Komplexe des Eisens Ausgehend vom](https://static.fdokument.com/doc/165x107/5e0e2b48ca79a779e3491070/synthese-charakterisierung-und-reaktivitt-von-x-oder-2-k-cp3-cp4-d-e.jpg)
Synthese, Struktur, Reaktivität und Bindungsverhältnisse
171
DISSERTATION zur Erlangung der Doktorwürde der Fakultät für Chemie der Ruhr-Universität Bochum vorgelegt von Diplom-Chemiker und Diplom-Arbeitswissenschaftler Holger Fölsing aus Ettingshausen Tag der mündlichen Prüfung: Juni 2000
Transcript of Synthese, Struktur, Reaktivität und Bindungsverhältnisse

DISSERTATION
zur
Erlangung der Doktorwürde
der
Fakultät für Chemie
der
Ruhr-Universität Bochum
vorgelegt von
Diplom-Chemiker und Diplom-Arbeitswissenschaftler
Holger Fölsing
aus Ettingshausen
Tag der mündlichen Prüfung:
Juni 2000

Neue Carbonylmetall-Komplexe von Aluminium, Gallium und
Indium:
Synthese, Struktur, Reaktivität und Bindungsverhältnisse.
Gutachter: Prof. Dr. R. A. Fischer

Prof. Dr. W. S. Sheldrick

Die vorliegende Arbeit entstand in der Zeit von November 1997 bis Mai 2000 am Institut für
Anorganische Chemie II der Ruhr-Universität in Bochum.
Meinem hochverehrten Lehrer
Herrn Professor Dr. Roland Augustinus Fischer

danke ich für die herausfordernde Aufgabenstellung und sein aktives Interesse an dieser Arbeit zu
besonderem Dank verpflichtet. Die mir gewährten großzügigen Freiheiten und das in mich gesetzte
Vertrauen waren für mich von unschätzbarem Wert.

Mein Dank gilt ferner:
Den Kolleginnen Frau Sabine Bendix, Frau Dipl.-Chem. Julia Hambrock, Frau Heike Kampschulte,
Frau Dr. Anjana Devi, Frau Dipl.-Chem. Maria Diaz, Frau Dr. Alissa Frank, Frau Sylvia Grum,
Frau Dipl.-Chem. Nicola Oberbeckmann, Frau Dr. Pia Wenneck, Frau Dipl.-Chem. Ulrike
Weckenmann, Frau Dipl.-Bio. Dana Weiß und den Kollegen Herrn Dipl.-Chem. Ralf Becker, Herrn
Dr. Qingmin Cheng, Herrn Rolf Deibert, Herrn Dipl.-Chem. Frank Hipler, Herrn Dr. Holger
Hoffmann, Herrn Dipl.-Chem. Thorsten Johann, Herrn Jürgen Klußmann, Herrn Dr. André Manz,
Herrn Dr. Jens Müller, Herrn Dipl.-Phys. Harish Parala, Herrn Dr. Wolfram Rogge, Herrn Matthias
Ruttert, Herrn Dipl.-Chem. Oliver Segnitz, Herrn Dipl.-Chem. Oliver Stark, Herrn Dipl.-Min. Frank
Stowasser, Herrn Tobias Steinke, Herrn Dr. Harald Sussek, Herrn Dr. Jurij Weiß, Herrn Dipl.-Ing.
Holger Winkler, Herrn Dipl.-Chem. Carl Winter und Herrn Dipl.-Min. Andreas Wohlfahrt für ihre
Beiträge, die zum Gelingen dieser Arbeit beigetragen haben, für ihre Geduld und für die angenehme
Zeit im Labor.
Herrn Dr. Klaus Merz und Frau Manuela Winter für die Durchführung der Kristallstrukturanalysen.
Ganz besonders bedanke ich mich bei Frau Sabine Masukowitz und bei Frau Ursula Bossek die
einen wesentlichen Anteil zum Gelingen dieser Arbeit beigetragen haben.
Prof. Dr. Matthias Drieß und allen Mitgliedern seines Arbeitskreises für die freundliche Aufnahme
und die Unterstützung beim Neuanfang in Bochum.
Prof. Dr. mult. Dr. h. c. Alois Haas und allen Mitgliedern seines Arbeitskreises für die Unterstützung
beim Aufbau in Bochum.
Meinem Studienkollegen in Heidelberg Meik Ranft für die gemeinsame Zeit im Studium.
Meiner Frau Kathrin und meinen Eltern für moralische und finanzielle Unterstützung.
Sowie allen ungezählten Mitarbeitern der Institute und Werkstätten der Ruhr-Universität Bochum,
die mich bei dieser Arbeit unterstützt haben.

Meinen Eltern Ingrid und HorstFölsingin Liebe und Dankbarkeit

„Ich kann wirklich nicht behaupten, daß ich gerne zur Schule ging,aber der Unterricht von Professor Nachtigaller besaß eineeinzigartige Qualität: Hatte er einmal begonnen, vergaß man allesrund um die Nachtschule. War Nachtigaller endlich in denKlassenraum gewackelt (er verspätete sich regelmäßig) und hatteseine fünf Doktorhüte (die er einerseits aus Eitelkeit, andererseitszur Wärmung seiner Außengehirne trug) abgelegt, begann erumgehend den Unterricht.In diesem Augenblick fiel auch seine Zittrigkeit und seineHinfälligkeit von ihm ab, leichtfüßig wie eine Ballerina tänzelte ervor der Klasse auf und ab und präsentierte seinen Lehrstoff mitGebärden, Mimik und einer Stimmakrobatik, die jedem Schauspieler,Tänzer oder Sänger zur Weltkarriere verholfen hätten. Er hatte dieBegabung, jeden Lehrstoff, den er gerade präsentierte, körperlichdarstellen zu können: Vor unseren staunenden Augen verwandelteer sich nur durch ein paar Grimassen und Verrenkungen in ein Zebraoder in eine Glockenblume, in einen Bergkristall oder eine Mikrobe,in ein Atom oder in den Satz des Pythagoras. Professor Nachtigallerhatte den Beruf der Lehrkraft in den Bereich der Kunst überführt.Und auch als Künstler war er – wie in allen anderen Disziplinen, dieer beherrschte – ein Genie.

aus: „Die 13 1/2 Leben des Käpt´n Blaubär“


Inhaltsverzeichnis I
Inhaltsverzeichnis
1 EINLEITUNG UND PROBLEMSTELLUNG 2
1.1 ÜBERGANGSMETALL-SUBSTITUIERTE ALANE, GALLANE UND INDANE, STAND DER
FORSCHUNG 2
1.2 BINDUNGSVERHÄLTNISSE DER GRUPPE-13 METALL-ÜBERGANGSMETALL-KOMPLEXE 9
1.3 PROBLEMSTELLUNG DIESER ARBEIT 14
2 SYNTHESE UND STRUKTUR CARBONYLHALTIGER ÜBERGANGSMETALL-
SUBSTITUIERTER ALANE 18
2.1 SYNTHESE DONORSTABILISIERTER ÜBERGANGSMETALL-ALUMINIUM-SYSTEME 19
2.2 STRUKTURCHEMIE DONORSTABILISIERTER ÜBERGANGSMETALL-ALUMINIUM-SYSTEME21
2.3 SPEKTROSKOPISCHE EIGENSCHAFTEN DER ALUMINIUM-KOMPLEXE 25
2.3.1 NMR-SPEKTROSKOPIE 25
2.3.2 INFRAROT-SPEKTROSKOPIE 26
3 SYNTHESE UND STRUKTUR CARBONYLHALTIGER ÜBERGANGSMETALL-
SUBSTITUIERTER GALLANE 28
3.1 SYNTHESE DONORSTABILISIERTER ÜBERGANGSMETALL-GALLIUM-SYSTEME 29
3.2 STRUKTURCHEMIE DONORSTABILISIERTER ÜBERGANGSMETALL-GALLIUM-SYSTEME 31
3.3 SPEKTROSKOPISCHE EIGENSCHAFTEN DER GALLIUM-KOMPLEXE 45
3.3.1 NMR-SPEKTROSKOPIE 45
3.3.2 INFRAROT-SPEKTROSKOPIE 47
4 SYNTHESE UND STRUKTUR CARBONYLHALTIGER ÜBERGANGSMETALL-
SUBSTITUIERTER INDANE 50

Inhaltsverzeichnis II
4.1 SYNTHESE DONORSTABILISIERTER ÜBERGANGSMETALL-INDIUM-SYSTEME 50
4.2 STRUKTURCHEMIE DONORSTABILISIERTER ÜBERGANGSMETALL-INDIUM-SYSTEME 51
4.3 SPEKTROSKOPISCHE EIGENSCHAFTEN DER INDIUM-KOMPLEXE 55
4.3.1 NMR-SPEKTROSKOPIE 55
4.3.2 INFRAROT-SPEKTROSKOPIE 56
4.4 EXKURS: REAKTIVITÄT UND FUNKTIONALISIERUNG 58
5 INTERMETALLISCHE SCHICHTEN 63
5.1 MOCVD INTERMETALLISCHER SCHICHTEN 63
5.2 ÜBERGANGSMETALL-ERDMETALL-KOMPLEXE ALS EINKOMPONENTEN-PRECURSOREN 65
5.3 [(CP*AL)CR(CO)5] ALS EINKOMPONENTEN-PRECURSOR FÜR MOCVD 67
5.3.1 SYNTHESE VON [(CP*AL)CR(CO)5] 67
5.3.2 EXPERIMENTALANORDNUNG ZUR VAKUUM-MOCVD 70
5.3.3 ABSCHEIDUNG EINES AL-CR-FILMS 71
5.3.4 SYNTHESE UND STRUKTUR VON [(CO)5M(η2-C8H14)] (M = MO, W) 73
6 EXPERIMENTELLER TEIL 79
6.1 ALLGEMEINE ARBEITSTECHNIKEN 79
6.1.1 SCHENKELFRITTENTECHNIK 79
6.1.2 LÖSUNGSMITTEL 80
6.2 ROUTINEANALYSENMETHODEN 81
6.2.1 ELEMENTARANALYTIK 81
6.2.2 KERNRESONANZSPEKTROSKOPIE 81
6.2.3 INFRAROTSPEKTROSKOPIE 82
6.2.4 SIEDEPUNKTE UND SUBLIMATIONSPUNKTE 84
6.2.5 EINKRISTALL-RÖNTGENSTRUKTURANALYSEN 84
6.3 ARBEITSVORSCHRIFTEN UND ANALYTISCHE DATEN 84
6.3.1 AUSGANGSVERBINDUNGEN UND REAGENZIEN 84
6.3.2 NEUE VERBINDUNGEN 86
6.4 DURCHFÜHRUNG UND ANALYTIK DES MOCVD-EXPERIMENTS 98

Inhaltsverzeichnis III
6.4.1 ANALYTISCHE CHARAKTERISIERUNG DÜNNER INTERMETALLISCHER FILME 98
6.5 RÖNTGENDIFFRAKTOMETRIE 98
6.5.1 RUTHERFORD-RÜCKSTREU-SPEKTROMETRIE (RBS) 99
6.5.2 ATOMEMISSIONSPEKTROMETRIE MIT INDUKTIV GEKOPPELTEN PLASMA (ICP) 99
7 TABELLEN 102
8 ZUSAMMENFASSUNG DER ERGEBNISSE 131
8.1 CARBONYLHALTIGE ÜBERGANGSMETALL-SUBSTITUIERTE ALANE, GALLANE UND INDANE
131
8.2 INTERMETALLISCHE SCHICHTEN 135
9 LITERATURVERZEICHNIS 137

Abbildungsverzeichnis IV
Abbildungsverzeichnis
ABBILDUNG 1: MOLEKÜLSTRUKTUR VON CP*AL-FE(CO)4 IM KRISTALL (ORTEP-
DARSTELLUNG).......................................................................................................4
ABBILDUNG 2: MOLEKÜLSTRUKTUR VON FE2(CO)6[µ-INC(SIME3)3]3 IM KRISTALL
(ORTEP-DARSTELLUNG)........................................................................................5
ABBILDUNG 3: MOLEKÜLSTRUKTUR VON (CO)3RU[GACL(THF)2][GACL2(THF)]2 IM KRISTALL
...............................................................................................................................6
ABBILDUNG 4: SCHEMATISCHE DARSTELLUNG DER M←E-σ-BINDUNG UND M→E-π-
RÜCKBINDUNG IN M-EIR-BINDUNGEN....................................................................9
ABBILDUNG 5: SCHEMATISCHE DARSTELLUNG DER MOLEKÜLORBITALE AM ZENTRUM E VON
EIR-FRAGMENTEN (E = AL, GA, IN; R= CP*, SI(SIME3)3, C(SIME3)3). .............11
ABBILDUNG 6: MOLEKÜLSTRUKTUR VON (CO)5CR-AL[(CL)(TMPDA)] (1) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................21
ABBILDUNG 7: MOLEKÜLSTRUKTUR VON (CO)4FE-AL[(CL)(TMPDA)] (2) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................23
ABBILDUNG 8: IR-SPEKTREN VON (2) UND (4) (ν(CO)-BEREICH)................................................27
ABBILDUNG 9: MOLEKÜLSTRUKTUR VON (CO)5CR-GA[(CL)(TMPDA] (5) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................32
ABBILDUNG 10: MOLEKÜLSTRUKTUR VON (CO)4FE-GA[(CL)(TMPDA)] (11) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................36
ABBILDUNG 11: MOLEKÜLSTRUKTUR VON (CO)4FE-GA[(BR)(TMPDA)] (12) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................37
ABBILDUNG 12: MOLEKÜLSTRUKTUR VON (CO)4FE-GA[(I)(TMPDA)] (13) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................39
ABBILDUNG 13: MOLEKÜLSTRUKTUR VON (CO)4FE-GA[(ME)(TMPDA)] (14) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................40
ABBILDUNG 14: MOLEKÜLSTRUKTUR VON (CO)4FE-GA[(PMDETA)]+ (15) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................42

Abbildungsverzeichnis V
ABBILDUNG 15: MOLEKÜLSTRUKTUR VON {(CO)4FE-GA[(ET)2NCH2CH2N(ET)]}2 (16) IM
KRISTALL (ORTEP-DARSTELLUNG). ....................................................................45
ABBILDUNG 16: IR-SPEKTREN VON (12) UND (14) (ν(CO)-BEREICH)..........................................48
ABBILDUNG 17: IR-SPEKTREN VON (13) UND (15) (ν(CO)-BEREICH)..........................................48
ABBILDUNG 18: MOLEKÜLSTRUKTUR VON (CO)4FE-IN[(BR)(TMPDA)] (17) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................52
ABBILDUNG 19: MOLEKÜLSTRUKTUR VON (CO)4FE-IN[(BR)(PMDETA)] (18) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................54
ABBILDUNG 20: IR-SPEKTREN VON (17) UND (18) (ν(CO)-BEREICH)..........................................57
ABBILDUNG 21: SCHEMA EINER POTENTIELLEN STABILISIERUNG VON (CO)NM-E[(LN)]+ DURCH
KRONENETHER-LIGANDEN UND ÜBERTRAGUNGSREAKTIONEN. .............................59
ABBILDUNG 22: MOLEKÜLSTRUKTUR VON [(CO)4FE-(INBR3)2][K(12-KRONE-4)2]2 (19) IM
KRISTALL (ORTEP-DARSTELLUNG) – (ZUR VEREINFACHUNG NUR DAS ANION
[(CO)4FE-(INBR3)2]2-). ......................................................................................60
ABBILDUNG 23: MOLEKÜLSTRUKTUR VON [(CO)4FE-(INBR3)2][K(12-KRONE-4)2]2 (19) IM
KRISTALL (ORTEP-DARSTELLUNG) – (ZUR VEREINFACHUNG NUR DAS KATION
[K(12-KRONE-4)2]2)+.........................................................................................61
ABBILDUNG 24: VAKUUM-MOCVD-APPARATUR ZUR ABSCHEIDUNG EINES AL-CR-FILMS......70
ABBILDUNG 25: RUTHERFORD-RÜCKSTREU-SPEKTRUM DER ALCR-PROBE................................72
ABBILDUNG 26: MOLEKÜLSTRUKTUR VON [(CO)5MO(η2-C8H14] (20) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................75
ABBILDUNG 27: MOLEKÜLSTRUKTUR VON [(CO)5W(η2-C8H14] (21) IM KRISTALL
(ORTEP-DARSTELLUNG)......................................................................................76
ABBILDUNG 28: SCHEMATISCHE DARSTELLUNG EINER SCHENKELFRITTENAPPARATUR..............80
ABBILDUNG 29: VERWENDETE TROCKNUNGS- UND LÖSUNGSMITTEL.........................................81
ABBILDUNG 30: PROTONENRESTSIGNALE BZW. LÖSUNGSMITTELSIGNALE DER JEWEILIGEN
SOLVENTIEN. ........................................................................................................82
ABBILDUNG 31: SCHEMA DER ICP-APPARATUR ......................................................................100

Abbildungsverzeichnis VI
ABBILDUNG 32: MOLEKÜLSTRUKTUREN EINIGER NEUER VERBINDUNGEN IM KRISTALL. LINKS
OBEN: (CO)4FE-IN[(BR)(TMPDA)]; RECHTS OBEN: (CO)4FE-GA[(CL)(TMPDA)];
MITTE LINKS: (CO)4FE-IN[(BR)(PMDETA)]; MITTE RECHTS: (CO)4FE-
GA[(BR)(TMPDA)]; LINKS UNTEN: (CO)5CR-AL[(CL)(TMPDA)]; RECHTS UNTEN:
(CO)4FE-GA[(I)(TMPDA)] ...............................................................................132
ABBILDUNG 33: VERDRÄNGUNG EINES HALOGENID-LIGANDEN DURCH MEHRZÄHNIGE
CHELATLIGANDEN AM BEISPIEL DES KOMPLEXES (CO)4FE-GA[(PMDETA)]+I- IM
KRISTALL...........................................................................................................133
ABBILDUNG 34: MOLEKÜLSTRUKTUR DER NEUEN VERBINDUNG
[(CO)4FE-(INBR3)2][K(12-KRONE-4)2]2..........................................................134

Tabellenverzeichnis VII
Tabellenverzeichnis
TABELLE 1: ZUSAMMENSTELLUNG UND NUMERIERUNGSSCHEMA DER NEUEN
ÜBERGANGSMETALL-SUBSTITUIERTEN ALANE (1-4). ............................................20
TABELLE 2: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (1)..........................22
TABELLE 3: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (2)..........................23
TABELLE 4: 1H-CHEMISCHE VERSCHIEBUNGEN DER KOMPLEXE (1-4). ........................................25
TABELLE 5: 13C-CHEMISCHE VERSCHIEBUNGEN DER KOMPLEXE (1-4). ......................................26
TABELLE 6: ZUSAMMENSTELLUNG DER ν(CO)-ABSORPTIONEN [CM-1] DER KOMPLEXE (1-4)....27
TABELLE 7: ZUSAMMENSTELLUNG UND NUMERIERUNGSSCHEMA DER NEUEN
ÜBERGANGSMETALL-SUBSTITUIERTEN GALLANE (5-16). ......................................31
TABELLE 8: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (5)..........................32
TABELLE 9: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (11)........................36
TABELLE 10: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (12)......................37
TABELLE 11: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (13)......................39
TABELLE 12: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (14)......................40
TABELLE 13: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (15)......................43
TABELLE 14: STRUKTURELL CHARAKTERISIERTE EISEN-GALLIUM-KOMPLEXE: σ(FE-GA)-
BINDUNGSLÄNGEN UND IR-DATEN. [A] NUJOL. [B] N-HEXAN. [C]
METHYLENCHLORID..............................................................................................44
TABELLE 15: 1H-CHEMISCHE VERSCHIEBUNGEN DER KOMPLEXE (5-15). ....................................46
TABELLE 16: 13C-CHEMISCHE VERSCHIEBUNGEN DER KOMPLEXE (5-15). ..................................47
TABELLE 17: ZUSAMMENSTELLUNG DER ν(CO)-ABSORPTIONEN [CM-1] DER KOMPLEXE (5-16).
.............................................................................................................................49
TABELLE 18: ZUSAMMENSTELLUNG UND NUMERIERUNGSSCHEMA DER NEUEN
ÜBERGANGSMETALL-SUBSTITUIERTEN INDANE (17) UND (18)...............................51
TABELLE 19: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (17)......................53
TABELLE 20: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (18)......................54

Tabellenverzeichnis VIII
TABELLE 21: STRUKTURELL CHARAKTERISIERTE EISEN-INDIUM-KOMPLEXE: σ(FE-IN)-
BINDUNGSLÄNGEN UND IR-DATEN. [A] NUJOL. [B] METHYLENCHLORID. [C]
METHYLENCHLORID. [D] PARAFFIN. [E] TOLUOL. [F] N-PENTAN. .........................55
TABELLE 22: 1H-CHEMISCHE VERSCHIEBUNGEN DER KOMPLEXE (17) UND (18). [124] ...............56
TABELLE 23: 13C-CHEMISCHE VERSCHIEBUNGEN DER KOMPLEXE (17) UND (18). [125] .............56
TABELLE 24: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (19)......................61
TABELLE 25: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (20)......................75
TABELLE 26: AUSGEWÄHLTE BINDUNGSLÄNGEN [PM] UND -WINKEL [°] VON (21)......................77
TABELLE 27: ALLE IN DIESER DISSERTATION RÖNTGENOGRAPHISCH CHARAKTERISIERTEN
VERBINDUNGEN ..................................................................................................102
TABELLE 28: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)5CR-AL[(CL)(TMPDA)] (1).......................................................................103
TABELLE 29: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)5CR-AL[(CL)(TMPDA)] (1).......................................................................104
TABELLE 30: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-AL[(CL)(TMPDA)] (2). ......................................................................105
TABELLE 31: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-AL[(CL)(TMPDA)] (2). ......................................................................106
TABELLE 32: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)5CR-GA[(CL)(TMPDA)] (5)......................................................................107
TABELLE 33: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)5CR-GA[(CL)(TMPDA)] (5)......................................................................108
TABELLE 34: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-GA[(CL)(TMPDA)] (11).....................................................................109
TABELLE 35: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-GA[(CL)(TMPDA)] (11).....................................................................110

Tabellenverzeichnis IX
TABELLE 36: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-GA[(BR)(TMPDA)] (12).....................................................................111
TABELLE 37: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-GA[(BR)(TMPDA)] (12).....................................................................112
TABELLE 38: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-GA[(I)(TMPDA)] (13)........................................................................113
TABELLE 39: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-GA[(I)(TMPDA)] (13)........................................................................114
TABELLE 40: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-GA[(ME)(TMPDA)] (14)....................................................................115
TABELLE 41: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-GA[(ME)(TMPDA)] (14)....................................................................116
TABELLE 42: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-GA[(PMDETA)]+(I-) (15)..................................................................117
TABELLE 43: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-GA[(PMDETA)]+(I-) (15)..................................................................119
TABELLE 44: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-IN[(BR)(TMPDA)] (17). .....................................................................120
TABELLE 45: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-IN[(BR)(TMPDA)] (17). .....................................................................121
TABELLE 46: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
(CO)4FE-IN[(BR)(PMDETA)] (18). ...................................................................122
TABELLE 47: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
(CO)4FE-IN[(BR)(PMDETA)] (18). ...................................................................123

Tabellenverzeichnis X
TABELLE 48: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
[(CO)4FE-(INBR3)2][K(12-KRONE-4)2]2 (19). .................................................124
TABELLE 49: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
[(CO)4FE-(INBR3)2][K(12-KRONE-4)2]2 (19). .................................................125
TABELLE 50: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
[(CO)5MO(η2-C8H14] (20)..............................................................................126
TABELLE 51: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
[(CO)5MO(η2-C8H14] (20)..............................................................................127
TABELLE 52: KRISTALLOGRAPHISCHE DATEN UND MEßPARAMETER FÜR
[(CO)5W(η2-C8H14] (21)................................................................................128
TABELLE 53: ATOMKOORDINATEN UND ÄQUIVALENTE ISOTROPE ATOMARE
AUSLENKUNGSMUSTER FÜR NICHT-WASSERSTOFFATOME VON
[(CO)5W(η2-C8H14] (21)................................................................................129

Verwendete Abkürzungen XI
Verwendete Abkürzungen
tBu tert.-Buthyl-Rest (-C(CH3)3)
Cp Cyclopentadienyl-Rest (η5-C5H5)
Cp* Pentamethylcyclopentadienyl-Rest (η5-C5(CH3)5)
CVD Chemical Vapour Deposition (Chemische Dampfabscheidung)
coe cis-Cycloocten
δ Chemische Verschiebung in ppm
Do Donor
E Element der dritten Hauptgruppe (Al, Ga, In)
EA Elementaranalyse (klassische Verbrennungsanalyse)
EI Elektronenstoßionisation
Et Ethyl-Rest (-CH2CH3)
ICP Atomemissionspektrometrie mit induktiv gekoppelten Plasma
IR Spektroskopie mit Licht im infraroten Bereich (4000-500 cm-1)
L Ligand
Me Methyl-Rest (-CH3)
MS Massenspektrometrie
MOCVD Metal Organic Chemical Vapour Deposition
NMR Nuclear Magnetic Resonance (Kernspinresonanz)
TMPDA Tetramethylpropylendiamin
PMDETA Pentamethyldiethylentriamin
R Alkyl- oder Arylgruppen
RBS Rutherford-Rückstreu-Spektrometrie
RT Raumtemperatur
THF Tetrahydrofuran (C4H8O)

Intensitäts- und Multiplizitätsangaben XII
Intensitäts- und Multiplizitätsangaben
Infrarotspektroskopie:
vs sehr stark
s stark
m mittel
w schwach
vw sehr schwach
sh Schulter
NMR:
s Singulett
d Dublett
t Triplett
q Quartett
quin Quintett

A
EINLEITUNG UND PROBLEMSTELLUNG

Einleitung und Problemstellung 2
1 Einleitung und Problemstellung
1.1 Übergangsmetall-substituierte Alane, Gallane und Indane, Stand der
Forschung
Komplexverbindungen mit Metall-Metall-Bindungen M-E zwischen d-Elementen (M) und den
Gruppe-13 Elementen Aluminium, Gallium und Indium (E) haben eine lange Tradition. Schon W.
Hieber, Wegbereiter der systematischen Metallcarbonyl-Chemie, berichtete im Jahre 1942 über die
Hochdruckcarbonylierung einer Mischung von feinverteiltem Indium- und Kobalt-Metall zu
In[Co(CO)4]3. [1] Das Vorliegen direkter, unverbrückter Zweizentren-Zweielektronenbindungen
(In-Co) wurde später von Robinson und Schussler kristallographisch bestätigt. [2] Im Verlauf der
zurückliegenden Jahrzehnte erfüllten unterschiedlichste Ziele diese Chemie immer wieder mit neuem
Leben. In direkter Beziehung zur Olefinpolymerisation mit Ziegler-Natta-Katalysatoren gewannen
zuerst Metall/Aluminium-Komplexe an Bedeutung. Schon im Jahre 1960 berichteten Natta et al.
über ein rotes, diamagnetisches Produkt, das sie bei der Umsetzung von AlEt3 mit Cp2TiCl2
erhielten [3] und ursprünglich als Komplex mit einer direkten Ti-Al Bindung aufgefaßt wurde. [4] Die
charakteristischen Strukturelemente für Metall/Aluminium-Komplexe sind jedoch Brückenstrukturen
M-X-Al (X = Cl, H, CO, CRn). [5-7] Aus dem Umfeld der Metallbasen-Chemie, in der Gruppe-
13 Organyle als Sonden für Lewis-basische Zentren dienten, [8-10] stammt tatsächlich der erste
Komplex, der eine M-Al Bindung aufwies, das Lewis-Säure-Base Addukt {Cp(CO)2Fe-
Al[(C6H5)3]-}. [11] Erheblichen Einfluß auf die allgemeine Entwicklung der Übergangsmetall-
Hauptgruppenelement Chemie hatte die Entdeckung der Fischer-Carben- und Carbin-
Komplexe. [12] Seitdem gilt der Komplexchemie und den Bindungsverhältnissen carbenanaloger
bzw. „isovalenzelektronischer“ Fragmente der höheren Homologen des Kohlenstoffs sowie anderer
Hauptgruppenelemente im Periodensystem ungebrochene Beachtung.

Einleitung und Problemstellung 3
Zwei illustre, jüngste Beispiele dafür sind die FeGe- und FeGa-Komplexe {[Cp*(CO)2Fe-
GeMe2(DMAP)]+} (DMAP = Dimethylaminopyridin) [13] und (CO)4Fe-GaAr* (Ar* = 2,6-
(2,4,6- Triisopropylphenyl)phenyl). [70] Ogino et al. beschreiben ihr System als „Germylen“-
Komplex mit einer Fe-Ge Dreifachbindung, Robinson et al. diskutierten ein „Ferrogallyne“ mit einer
Fe-Ga Dreifachbindung. Das kationische Fe-Ge-System ist analog zum neutralen Gallium-Komplex
Cp(CO)2Fe-GaR2(py) (R = η1-C5H4Me), [14] das neutrale Ferrogallin entspricht formal einem
kationischen Fischer-Carbinkomplex. [15-20]
Ungewöhnliche Mehrfachbindungsverhältnisse prägen seit Mitte der 80er Jahre die Chemie von
Hauptgruppenelement-Übergangsmetall Komplexen, die u. a. etabliert wurde durch die Arbeiten von
G. Huttner und W. A. Herrmann über „Iniden“-Komplexe, [21] Komplexe bzw. Metallkumulene mit
substituentenfreien Hauptgruppenelementen [22] etabliert wurde und in einer Reihe von
Übersichtsartikeln und Monographien behandelt ist. [23-28] Im Vergleich zu den Elementen der
Gruppen 14, 15 und 16 fand die Chemie der Gruppe-13 Metalle dabei aber kaum Beachtung, [29]
wichtige allgemeine Zusammenfassungen der synthetischen und strukturchemischen Ergebnisse der
Gruppe-13 Metall-Übergangsmetallchemie bis etwa zum Jahre 1989/90 stammen von M. J. Taylor,
A. T. T. Hsieh und N. C. Norman et al.. [30-32] Erst vor drei Jahren erschien ein wichtiger Beitrag
von P. P. Power et al. dazu. [33]
Der gegenwärtige Aufschwung der Koordinationschemie von Aluminium, Gallium und Indium an
Übergangsmetall-Fragmenten geht auf Impulse zurück, die erst seit Beginn der 90er Jahre allmählich
wirksam wurden und zwei sehr verschiedenen Motivationen entstammen. Die eine ist, daß
metallorganischen Verbindungen ein interessantes Potential als Vorstufen für eine Fülle von
Anwendungen in der Materialwissenschaft zukommen könnte. Prototypisch dafür ist die Suche nach
alternativen Vorstufen für die Erzeugung von III/V-Halbleiterschichten nach den Verfahren der
Chemischen Dampfabscheidung (CVD). [153,154,159]
Ein wichtiger Leitgedanke ist dabei das Konzept der sogenannten Einkomponentenvorstufe (single
source precursor). [160,34,35] Dies führten u. a. auch Kaesz et al. durch, die eine Abscheidung der
β-CoGa-Phase aus dem Komplex (CO)4Co-GaCl2(THF) nach einem MOCVD-Verfahren
beschrieben haben. [36,37] Eine Serie von Arbeiten in unserer Arbeitsgruppe schloß sich dieser
Konzeption an. [38-42]

Einleitung und Problemstellung 4
Unabhängig davon erlebte die Chemie niedervalenter bzw. niedrigkoordinierter Gruppe-13
Metallverbindungen einen starken Aufschwung. Prominente Beispiele hierfür sind der Cluster
[Cp*Al]4, [43,91] seine Alkylanaloga [ER´]4 und R´´2E-ER´´2 (R´ = TMS3C; R´´ = TMS2CH; E =
Al, Ga, In) [44-49,95] und die anionischen Systeme [R´´2Al-AlR´´2]- [50,51] oder [Ar*Ga-
GaAr*]2-. [52] So geriet auch hier mehr und mehr die Koordinationschemie der
Organometallspezies [RE] an Übergangsmetallen in den Vordergrund, wobei E in der niedrigen
Oxidationsstufe +I vorliegt und diese Verbindungen als Ausgangsverbindungen für
Komplexsynthesen von Gruppe-13 Übergangsmetall-Verbindungen herangezogen wurden.
Ein besonderes Highlight ist sicher die von Uhl et al. stammende Verbindung Ni(InR´)4, [53] ein
erster Beleg für die Existenz homoleptischer, Gruppe-13 metallreicher Komplexe, die gemeinsam mit
den Komplexen [CpNi-AlCp*2] [92] und Cp*Al-Fe(CO)4 [54] (Abbildung 1) repräsentativ sind
für die Weiterentwicklung und aktuellen Trends des Gebiets der Gruppe-13 Metall-Übergangsmetall
Chemie. Übergangsmetall-substituierte Alane, Gallane und Indane wurden mittlerweile in einer
großen Vielzahl synthetisiert und auch strukturell charakterisiert.
FeAl
Abbildung 1: Molekülstruktur von Cp*Al-Fe(CO)4 im Kristall (ORTEP-Darstellung)

Einleitung und Problemstellung 5
Uhl et al. stellten 1997 den trigonal-bipyramidalen Fe2In3-Cluster Fe2(CO)6[µ-InC(SiMe3)3]3
durch Substitutionsreaktion von COT im Komplex Fe(CO)3(COT) durch „In[C(SiMe3)3]“, das im
Festkörper wie auch in Lösung als Tetramer In4[C(SiMe3)3]4 vorliegt, dar (Abbildung 2). [55,56]
Auch ein entsprechendes Galliumderivat ist bekannt, es liegt als Dimer (Me3Si)3SiGa-GaSi(SiMe3)3
vor und entsteht bei der Reaktion von mindestens 2 Äquivalenten Li(THF)3Si(SiMe3)3 mit
Ga[GaCl4]. [57] Auf ähnliche Weise konnten durch CO-Substitution an Übergangsmetallcarbonyl-
Dimeren die verwandten Systeme M2(CO)n[µ-InC(SiMe3)3]2 (M = Co, Mn; n = 6, 8) erhalten
werden. [58,59]
Abbildung 2: Molekülstruktur von Fe2(CO)6[µ-InC(SiMe3)3]3 im Kristall (ORTEP-Darstellung).
In der gleichen Arbeitsgruppe gelang auch die Darstellung eines Co4Ga4-Clusters aus einer Ga(I)-
Spezies (RGa)4 und Jonas´Reagens CpCo(C2H4)2 [60], indem die beiden Ethylenmoleküle als labile
Liganden fungieren.

Einleitung und Problemstellung 6
Insgesamt erscheint diese Chemie zu viele Reaktionskanäle zu erlauben, so daß eine gezielte und
ausbeutestarke Synthese einzelner Produkte nicht möglich ist. [61,62] In einer ähnlich
unübersichtlichen Reaktion von Ru3(CO)12 mit Ga2Cl4 und in Anwesenheit von metallischem Gallium
konnten Whittlesey et al. 1997 die Komplexe (CO)3Ru[GaCl(THF)2][GaCl2(THF)]2·0.5 THF und
(CO)8Ru2[GaCl2(THF)]2 isolieren und röntgenographisch charakterisieren. [63]
Abbildung 3: Molekülstruktur von (CO)3Ru[GaCl(THF)2][GaCl2(THF)]2 im Kristall
Nur wenige Arbeiten beschäftigen sich bisher mit der Reaktivität höhergeladener Metallcarbonylate
gegenüber dreiwertigen Aluminium-, Gallium- und Indium-Verbindungen. Komplexe der Art
[(CO)5M-Al(R)(Do2)] (M = Cr, Mo, W; R = Cl, Me, Et, iBu; Do2 = TMEDA, TMPDA) wurden
bereits in unserer Arbeitsgruppe synthetisiert [64,65] und waren Gegenstand theoretischer
Betrachtungen. [64,189,190]

Einleitung und Problemstellung 7
R. D. Ernst et al. beschrieben 1980, gestützt durch IR- und NMR-Daten verschiedene Lewis-Base-
Addukte des Typs (CO)4Fe-Ga[(C2H5)(Do)n] (Do = Lewis-Base; n = 1, 2) und publizierten zwei
Jahre später die Röntgenstrukturanalyse von (CO)4Fe-Ga[(C2H5)(THF)]2. [66,67] Beide
Komplexe wurden aus der Umsetzung von EtGaCl2 mit Na2[Fe(CO)4] erhalten. Ungeklärt blieb die
Herkunft der Vinyl-Gruppe (C2H3) am Gallium im Produktkomplex. 1994 konnte in unserer
Arbeitsgruppe gezeigt werden, daß die Umsetzung von 2 Mol K2[Fe(CO)4] mit Cl2GaMe und
Abfangen der dianionischen Zwischenstufe mit [PPN]Cl das zweifach übergangsmetallsubstituierte
Gallan {[(CO)4Fe]2(µ-GaMe)}[PPN]2 ergibt. [65,68] In der gleichen Arbeit wurde auch über
Verbindungen des Typs {(CO)4Fe-Ga[(CH2)3NMe2](R)}[PPN] (R = Cl, tBu) und {(CO)5Cr-
Ga[(CH2)3NMe2](R)}[PPN] (R = Cl, Me, tBu) berichtet. Zwei Jahre später konnte mit der
Röntgenstrukturanalyse von (CO)5Cr-Ga[(Me)(TMEDA)] ein weiterer Nachweis für eine direkte
Cr-Ga-Bindung erbracht werden. [69] Robinson et al. konnten 1997 durch Reaktion von
Na2[Fe(CO)4] mit Cl2Ga(C6H3Trip2) (Trip = 2,4,6-Triisopropylphenyl), die donorfreie Verbindung
(CO)4Fe-Ga(C6H3Trip2) darstellen, der seitens der Autoren eine Fe-Ga-Dreifachbindung
zugeschrieben wird. [70] Bereits kurze Zeit später wurde dieses Postulat von Cotton et al. mit Hilfe
von Dichtefunktionalitätsrechnungen widerlegt. [71] Eaborn et al. gelang es durch Umsetzung von
Na2[Fe(CO)4] mit zwei Äquivalenten Cl2InC(SiMe3)3 die erste Bis-Indium-substituierte
Übergangsmetallverbindung {[(CO)4Fe][(µ-Cl)InC(SiMe3)3]2} darzustellen. [72] 1993 konnten
Huttner et al. ausgehend von Indiumtribromid und zwei Äquivalenten K2[Cr(CO)5] den
dianionischen „Iniden“-Komplex {[(CO)5Cr]2(µ-InBr)}2- synthetisieren und durch eine
Röntgenstrukturanalyse zu verifizieren. [73] Die gleiche Arbeitsgruppe erhielt durch Reaktion von
drei Äquivalenten K2[Cr(CO)5] mit TlCl3 die Verbindung
[(CO)5Cr=Tl=Cr(CO)5][K+-(2.2.2)Kryptand] mit linear zweifach koordiniertem Thallium. [74]
Behrens et al. konnten durch Umsetzung von InX3 (X = Cl, Br, I) mit Decacarbonyldimetallaten
[M2(CO)10]2- (M = Cr, Mo, W) die Komplexe (CO)5M-In[(X)(THF)] isolieren. [75]

Einleitung und Problemstellung 8
Für die Darstellung von Übergangsmetall-Erdmetall-Komplexen gibt es eine Reihe unterschiedlicher
Synthesestrategien, die in nachfolgender Auflistung kurz dargestellt sind: [76-97]
(1) Protolyse von Erdmetall-Kohlenstoff-Bindungen mittels Übergangsmetall-Hydriden
(2) Oxidative Addition von Erdmetall-Kohlenstoff-Bindungen an ungesättigte
Übergangsmetallfragmente
(3) Salzeliminierung zwischen Carbonylmetallaten und Erdmetallhalogeniden
(4) Insertion niedervalenter Erdmetallhalogenide in Metall-Metall- oder Metall-Halogen-Bindungen
(5) Insertion von elementarem Gallium und Indium
(6) Substitution labiler Liganden an Übergangsmetall-Komplexen durch CO-analoge Indium(I)-,
Gallium(I)- und Aluminium(I)organyle

Einleitung und Problemstellung 9
1.2 Bindungsverhältnisse der Gruppe-13 Metall-Übergangsmetall-Komplexe
Durch die Synthese neuer Erdmetall-Übergangsmetall-Komplexe ist die Frage nach den
Bindungsverhältnissen innerhalb dieser Cluster aufgeworfen worden, die zum Teil recht umstritten ist.
Dieses gilt insbesondere für den verstärkt diskutierten Aspekt des Ausmaßes von Rückbindungen
des Typs M(dπ)-E(pπ). [170] Um dieser Fragestellung nachzugehen, wurden Gruppe-13 Metall-
Übergangsmetall-Modellkomplexe herangezogen, die eine Vereinfachung zu den experimentell
synthetisierten Verbindungen darstellen und somit leichter quantentheoretischen Untersuchungen, wie
DFT (density functional theory), CDA (charge decomposition analysis) und NBO (natural bond
orbital), zugänglich sind. Mit Hilfe dieser Theorien ist es möglich, mehr über die Eigenschaften von
niedervalenten Erdmetallverbindungen als Liganden zu erfahren und die M-E-Bindung genauer zu
charakterisieren.
Eine einheitliche Behandlung, die alle Typen bislang dargestellter M-E-Bindungssysteme umfaßt, ist
aufgrund der Komplexität derzeit noch nicht möglich. Allerdings kann zumindest ein allgemein
anerkanntes Bindungsmodell für Übergangsmetall-Erdmetall-Komplexe mit einwertigen
metallorganischen Verbindungen der Gruppe-13 vom Typ EIR angeführt werden (Abbildung 4).
M E Rσ
π
E RM
Abbildung 4: Schematische Darstellung der M←E-σ-Bindung und M→E-π-Rückbindung in M-EIR-Bindungen.

Einleitung und Problemstellung 10
Es existiert eine starke σ-Donor-Bindung von dem Elektronenpaar des elektropositiven
Erdmetallatoms E in die leeren σ-Atomorbitale des Übergangsmetallatoms, während der M→E-π-
Rückbindung aufgrund der geringen Elektronegativität am Erdmetallzentrum E eine geringere
Bedeutung zukommt. Die M→E-π-Rückbindung konkurriert mit der E←R-π-Donorbindung, wenn
der Substituent R ein oder zwei besetzte p(π)-Orbitale aufweist. [98] Aufgrund des großen
Repertoires an möglichen Substituenten an beiden Metallzentren sowie in Abhängigkeit von der
Koordinationszahl am Zentrum E können die M-E-Bindungen über einen sehr weiten Bereich
systematisch variieren, wobei die sehr unterschiedlichen Elektronegativitäten der Metallatome M und
E eine polare M-E-Bindung (Eδ+- Mδ-) vermuten lassen. Das hat zur Folge, daß die Beschreibungen
für die Metall-Metall-Bindung in derartigen Komplexen deutlich voneinander abweichen.
Schnöckel et al. haben für den Komplex [(CO)4FeAlCp*] eine Beschreibung als Kontaktionenpaar
[(CO)4Fe]2- [AlCp*]2+ den Vorzug vor einer kovalenten Auffassung im Sinne einer
Komplexstabilisierung gegeben, da Modellrechnungen gezeigt haben, daß in dem Komplex eine
höhere Ladungspolarisierung herrscht, als es für die hypothetische Vergleichsverbindung [Cp*Al=O]
gefunden wurde. Die Übergangsmetall-Erdmetall-Bindung setzt sich demnach aus einem negativ
geladenen Übergangsmetall- und einem positiv geladenen Gruppe-13 Metallatom zusammen. Die
Autoren argumentieren, daß der carbenoide Charakter des [Cp*Al]-Teilchens im Komplex nicht
mehr vorhanden ist, weil die Elektronendichte auf das Übergangsmetall-Fragment übertragen wurde.
Daher wird die Beschreibung der Fragmente Cp*E in den Komplexen im Sinne einer formal
niedrigen Oxidationstufe +1 kritisiert. Es läge vielmehr ein AlIII-Zentrum vor. [99]
Demgegenüber favorisieren Fischer et al. die Beschreibung der M-E-Bindung als zwar stark polare,
aber durchaus kovalente Donor-Akzeptor-Bindung gegenüber der extremen Formulierung als
Kontaktionenpaar ([Cp*Al→Fe(CO)4]). Die monomeren Erdmetallfragmente EIR besitzen formal
ein freies Elektronenpaar (Lewis-Base) am Metallzentrum und zwei unbesetzte p-Orbitale senkrecht
zur E-C-Bindung (oder im Fall von Cp-Liganden, zum Schwerpunkt des C5-Rings), wodurch sie in
der Metallorganik als nützliche Zweielektronen-Donor-Liganden fungieren können. [100] Daneben
weisen die EIR-Fragmente π-Akzeptor-Eigenschaften auf und ähneln den Liganden des Amin- und
Phosphan-Typs. Eine Analogie der EIR-Liganden zum CO-Ligand besteht in strukturchemischer
bzw. koordinationschemischer Sicht. [189,170]

Einleitung und Problemstellung 11
R E
Abbildung 5: Schematische Darstellung der Molekülorbitale am Zentrum E von EIR-Fragmenten (E= Al, Ga, In; R= Cp*, Si(SiMe3)3, C(SiMe3)3).
Seit der Entdeckung der Fischer-Carben- und Carbinkomplexe [101] ist die Erforschung
carbenanaloger oder isovalenzelektronischer Fragmente der höheren Homologen des Kohlenstoffs
und benachbarter Hauptgruppenelemente forciert worden, wobei das Studium ungewöhnlicher
Mehrfachbindungen seit Mitte der 80er Jahre die Chemie von Komplexen aus
Hauptgruppenelementen und d-Metallen prägt. [170] Hierbei sind zwei von Robinson et al.
synthetisierte Beispiele hervorzuheben, in denen eine Metall-Metall-Dreifachbindung postuliert
wurde. Dies ist der Komplex Na2[Mes*2C6H3Ga-GaC6H3Mes*2] (Mes* = 2,4,6-iPr3C6H2), [52]
der durch die Reduktion von [(Mes*2C6H3)GaCl2] mit Natrium gebildet wird, sowie die sehr
kontrovers diskutierte Verbindung [(CO)4Fe-GaC6H3Mes*2], [70] in der nach Aussage der
Autoren eine Fe-Ga-Dreifachbindung vorliegen soll (Schema 1).
Schema 1: Synthese von [(CO)4Fe-GaC6H3Mes*2] (Mes* = 2,4,6- iPr3C6H2).
GaCl
ClGa
CO
Fe
OC CO
CONa2[Fe(CO) 4]
- 2 NaCl

Einleitung und Problemstellung 12
Die Auffassung einer Dreifachbindung begründet sich zum einen darin, daß der neutrale Fe-Ga-
Komplex formal einem kationischen Fischer-Carbin entspricht. [15-17]

Einleitung und Problemstellung 13
Zum anderen hat die Charakterisierung der Molekülstruktur ergeben, daß eine sehr kurze Fe-Ga-
Bindung verbunden mit einer zweifach-koordinierten linearen Anordnung um das Ga-Atom
vorliegt. [70] Neben diesen zuletzt angeführten klassischen Kriterien gelten hohe Bindungsenergien
und eine große Kraftkonstante als Beleg für eine Mehrfachbindung. Allerdings treffen die genannten
Kriterien auf die Elemente der ersten Achterperiode zu. Die Übertragbarkeit auf Metall-Metall-
Bindungen sowie auf die schweren Hauptgruppenelemente ist fragwürdig. [33, 102, 103]
Die Annahme eines „Ferrogallins“ wurde von Cotton angefochten, der zuerst eine Einfachbindung,
später anhand von DFT-Berechnungen höchstens eine Zweifachbindung postulierte. [71, 104]
Power et al. behandeln dieses Problem für Fe-Ga-Komplexe mit dreifach koordinierten Zentrum E
und kommen zu dem Schluß, daß π-Wechselwirkungen zwischen den Eisen- und Gallium-Zentren
kaum Bedeutung haben. [33, 137] Zu einem ähnlichen Ergebnis kommen auch Linti und Mitarbeiter,
die die Ga-Fe-Bindung in Derivaten von Fe2(CO)9-Clustern mit [GaSi(SiMe3)3] in verbrückender
Position mittels DFT-Analysen untersucht haben. Die Ga-Fe-Bindung läßt sich als eine
Donorbindung vom Gallium- zum Eisenfragment charakterisieren und die kurze Bindungslänge
resultiert vorwiegend aus der geringen Koordinationszahl am Gallium-Zentrum. [105]
Frenking et al. haben durch quantentheoretische Untersuchungen an den Modellkomplexen
[(CO)4Fe-Ga(η5-Cp)] und [(CO)4FeGa(η1-Ph)] für die Robinson-Verbindung allgemeine
Aussagen über die Bindungsverhältnisse in M-E-Bindungen abgeleitet: [98, 106]
1. Die σ-Donor und π-Akzeptor-Eigenschaften von EIR-Fragmenten hängen von dem
Substituenten R ab. Demnach ist bei starken π-Donoren die M←E-σ-Bindung deutlich größer,
als die M→E-π-Rückbindung, während schwache π-Donoren zu einer größeren M→E-π-
Rückbindung führen. Die M→E-π-Rückbindung folgt dem Trend B > Al > Ga > In > Tl.
2. Ähnliche Verhältnisse lassen sich im Hinblick auf die Bindungsenergien zwischen
Übergangsmetallen und EIR-Fragmenten verfolgen. Die Bindungsenergien in M-E-Bindungen sind
groß und folgen dem Trend B > Al > Ga, In > Tl. Wenn der Substituent R ein schwacher π-
Donor ist (R = Ph, CH3), liegen kürzere und stärkere M-E-Bindungen vor. Bei stärkeren π-
Donor-Gruppen (R = Cp, N(SiH3)2) liegt der umgekehrte Fall vor.
3. Die Übergangsmetall-Erdmetall-Bindung läßt sich vorwiegend als ionisch charakterisieren. Der
kovalente Beitrag ist als gering einzustufen.

Einleitung und Problemstellung 14
Bezogen auf [(CO)4Fe-GaC6H3Mes*2] bedeutet dieses, daß die M→E-π-Rückbindung zwar
ebenso wichtig wie die M←E-σ-Bindung ist, jedoch ist die Bindungsordnung im Gegensatz zu
Fischer-Carbin-Komplexen kleiner als 1, d.h. es liegt weder eine Einfach- noch eine
Dreifachbindung vor. Das Problem liegt in dem unangemessenen Bindungsmodell. Die Beschreibung
der Bindung anhand einfacher VB-Modelle ist nicht auf die Bindungssituation in schwereren Atomen
übertragbar. Dieses gilt vor allem für die Bindungsverhältnisse in Donor-Akzeptor-Komplexe der
Übergangsmetalle. [106]
Zusammenfassend läßt sich feststellen, daß keine experimentellen Anhaltspunkte für die Auffassung
einer Fe-Ga-Mehrfachbindung in [(CO)4Fe-GaC6H3Mes*2] existieren. Es wurde gezeigt, daß der
dominierende Faktor bei der Beschreibung der Bindungsverhältnisse in M-E-Komplexen die
Bindungspolarität ist. Diese läßt sich gemäß der Reihe E(R)L2 > E(X)L2 > ER und Al > Ga, In in
Abhängigkeit von der Koordinationszahl von E und den daran gebundenen Substituenten R, X und L
bestimmen. [170] Die M-E-Bindungen sind am besten als stark polare Lewis-Säure-Base-Addukte
aufzufassen, wobei in einigen M-E-Komplexen ein gewisser M→E-π-Rückbindungsanteil zu
verzeichnen ist, der aus den Donor- bzw. Akzeptoreigenschaften der Substituenten R am
Erdmetallzentrum E resultiert.
1.3 Problemstellung dieser Arbeit
Die wesentliche Neuentwicklung der letzten Jahre betrifft die Etablierung der Chemie von RaE-
Fragmenten (E = Al, Ga und In; a = 1 und 2) in der Koordinationsspähre von d-Metallen, wobei
sich die sterische Abschirmung niedervalenter Zentren E und ihre intramolekulare
Adduktstabilisierung als erfolgreiche Leitlinien erwiesen haben. Beide Konzepte sind in anderen
Zusammenhängen lange erprobt und Stand des Lehrbuchwissens, wurden aber erst in der letzten Zeit
verstärkt für die Koordinationschemie der Erdmetalle umgesetzt.
Offensichtlich gibt es zwei verschiedene Typen von Übergangsmetall-Erdmetall-Komplexen. Zum
einen die niedrigkoordinierten LnM-ER (R = Ar*, KZ = 2; R = Cp*, C(SiMe3)3, Si(SiMe3)3, KZ =
4) zum anderen die höherkoordinierten Komplexe wie L´nM-E(X)aLb.

Einleitung und Problemstellung 15
Bei den höher koordinierten Komplexen unterscheidet man zwischen den Verbindungen mit a) einem
größeren Anteil an Übergangsmetall-Fragmenten am Erdmetall und b) verschiedenen Donoren und
mehreren Resten R (EL3, E(X)L2, E(X)2L, EX3, X = Cl, Br, I, Alkyl, Ph). In der Literatur wird aber
immer noch differenziert zwischen den niedrigkoordinierten Komplexen, in denen formal dem
Erdmetallzentrum E die Oxidationszahl +I zugewiesen wird (E(I)) und den höherkoordinierten
Komplexen wobei hier das Erdmetallzentrum mit E(III) klassifiziert wird. Außerdem gelten in diesen
Komplexen die ER-Fragmente als σ-Donoren wogegen die Fragmente E(X)aLb eher als
Akzeptoren (σ) für die negative Ladung verstanden werden. Es zeigt sich, daß sich die M-E-
Bindungen in den verschiedenen Komplextypen gut in das Lewis-Donor-Akzeptor-Konzept für
Metall-Liganden-Bindungen einfügen. Während in M/E-Komplexen inzwischen als recht gut
charakterisiert und verstanden gelten dürfen- wenn auch laufend neue quantentheoretische Arbeiten
dazu erscheinen- leiten sich aus den obengenannten Betrachtungen die folgenden Fragen ab:
(1) Wie steht es mit der Reaktivität der M-E-Bindungen: z. B. in welchem Umfang lassen sich
metallkoordinierte Zentren E chemisch derivatisieren; oder lassen sich Liganden mit sehr stark
elektronendonierenden Ligatoren E für die Chemie an elektronenreichen d-Metallzentren nutzen
?
(2) Wie wirkt sich die Ligandenvariation am Zentrum E für Komplexe des Typs ER3 (L3, XL2, X2L,
X3) bzw. ER (Cp*, Ar*, C(SiMe3)3, Si(SiMe3)3) aus, in Bezug auf die Bindungslänge der M-E-
Bindung ?
(3) Was bedeutet dies für die Diskussion E(I) versus E(III) in diesen Komplexen bzw. für das σ-
Donor-π-Akzeptor-Verhältnis des Komplexfragments E, auch in Hinblick auf die, in den Reger-
Komplexen postulierten Ga(I)- und In(I)-Verbindungen ?
Ausgehend von diesen Fragestellungen sollte im Rahmen dieser Arbeit, aufbauend auf den
grundlegenden Arbeiten zur Übergangsmetall-Erdmetallchemie von M. M. Schulte [65,69], J.
Weiß [64,107,189] und O. Segnitz [190] aus unserer Arbeitsgruppe, die Reaktivität höhergeladener
Metallcarbonyle des Eisens, Chroms, Molydäns uns Wolframs gegenüber dreiwertigen Aluminium-,
Gallium- und Indiumverbindungen untersucht und die erhaltenen Verbindungen eingehend
charakterisiert werden.

Einleitung und Problemstellung 16
Ausgehend von Untersuchungen zur Reaktivität der M-E-Bindung (M = Fe, Cr, Mo, W; E = Al,
Ga, In) sollte eine mögliche Verwendbarkeit der Komplexe als „E(I)-Syntheseäquivalente“
experimentell ausgelotet und systematisch erweitert werden.
Weitere Bemühungen galten der Synthese von Verbindungen der Art (CO)nM-E(Cp*) (M = Fe,
Cr, Mo, W; E = Al, Ga, In; n = 4, 5) und deren potentielle Nutzung in MOCVD-Prozessen.
B
SYNTHESE UND STRUKTUR

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 18
2 Synthese und Struktur carbonylhaltiger Übergangsmetall-
substituierter Alane
Übergangsmetall-Aluminium-Komplexe sind nicht selten, jedoch sind in fast allen Fällen die Metalle
über Brückenliganden (Hydrid, Alkyl) verbunden, z. B. in {(µ-H)3[(MePh2P)3(H)-
Re(AlMe2)]}. [108] Dies trifft vor allem dann zu, wenn das Übergangsmetall keine harten Lewis-
basischen Liganden, insbesondere keine Carbonylliganden trägt. Sind Carbonylliganden anwesend,
bevorzugt das harte Lewis-saure und oxophile Aluminium die Ausbildung von Isocarbonyl-
Strukturen M-CO-Al, gegenüber direkten M-Al-Bindungen. [109-116] Die Tendenz zur
Isocarbonylbildung ist dabei so groß, daß Aluminiumamide mit Übergangsmetallcarbonylen unter
Carbonylinsertion in die Al-N-Bindung reagieren und die entsprechenden Übergangsmetall-
Aluminium-Carbamoylkomplexe bilden. Ein Beispiel ist der röntgenographisch charakterisierte
Komplex {(CO)4Fe(µ-Me2NCO)Al[(NMe2)(µ-NMe2)]2}. [117]
Die Liste von strukturell charakterisierten Komplexen mit direkten Übergangsmetall-Aluminium-
Bindungen ohne jegliche weitere Verbrückung ist dagegen sehr kurz. Um solche Bindungen zu
realisieren, bieten sich unterschiedliche Strategien an. Einerseits sollte das Übergangsmetall-Fragment
keine Carbonylliganden tragen, um die Isocarbonylstrukturen zu vermeiden. Dieser Strategie folgend
konnten überwiegend Cyclopentadienylübergangsmetall-Aluminium-Komplexe der frühen und späten
Übergangsmetalle, wie die dimeren [Cp2Ti-AlEt2]2, [4] {[(Cp(η2-C2H4)]Co-AlEt2}2 [118] oder
[(CpNi)2(Cp*Al)2] [92] erhalten werden. Die alternative Strategie ist der Einsatz von starken
Übergangsmetallcarbonylnukleophilen wie z. B. [CpFe(CO)2]- oder [(Me3P)Co(CO)3]- anstatt
[Co(CO)4]- und gleichzeitiger geeigneter Lewis-Basen-Donorstabilisierung des Aluminiumzentrums.
Diesem Konzept folgend konnten eine ganze Reihe von Übergangsmetall-Aluminium-Komplexen
synthetisiert werden. Strukturell charakterisiert sind die Komplexe Cp(CO)2Fe-
Al(iBu)[(CH2)3NMe2] [183], [CpFe(CO)2]2AlAr (Ar = 2-[(Dimethylamino)methyl]phenyl), [119]
und Cp(CO)2Fe-Al(tmp)2 (tmp = 2,2,6,6-Tetramethylpiperidin). [120]

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 19
Dieses Konzept war auch für andere harte, oxophile Metalle erfolgreich und so konnte der erste
Übergangsmetall-Lanthanid-Komplex mit direkter M-Ln-Bindung im Komplex Cp(CO)2Ru-
Lu[(Cp)2(THF)] erhalten werden. [121]
Anionische Lewis-Säure-Base-Addukte wurden durch Umsetzung von Carbonylmetallat-anionen mit
Triphenylaluminium erhalten. Mit dem starken Nukleophil [CpFe(CO)2]- konnte so der anionische
Komplex [Cp(CO)2Fe-AlPh3][Et4N] gewonnen werden. Schwächere Nukleophile wie [Co(CO)4]-,
[Mn(CO)5]- oder [CpW(CO)3]- lieferten dagegen wieder Isocarbonylkomplexe [L(CO)nM-CO-
AlPh3]- oder Mischungen aus beiden, wie durch IR-spektroskopische Untersuchungen belegt
wurde. [11]
Die Pionierarbeiten auf dem Gebiet der donorstabilisierten Übergangsmetall-Aluminium und -Gallium
Komplexe leistete jedoch M. M. Schulte et al.. Ihnen gelang 1996 durch Umsetzung von
K2[Cr(CO)5] mit Cl2ER (E = Al, Ga; R = Cl, Alkyl) die Synthese der Systeme (CO)5Cr-
E[(R)(TMEDA)]. [65,69] Aufbauend auf diesen grundlegenden Arbeiten sollte diese
Verbindungsklasse (Al, Ga und In) erweitert und die Reaktivität, sowie die Bindungsverhältnisse
dieser Systeme genauer untersucht werden.
2.1 Synthese donorstabilisierter Übergangsmetall-Aluminium-Systeme
Es werden zu Suspensionen der Carbonylmetallate K2[Fe(CO)4] und K2[M(CO)5] (M = Cr, Mo,
W; in situ aus den Hexacarbonylen und KC8 hergestellt) in THF äquimolare Mengen von AlX3 (X
= Cl, I) oder Cl2AlMe als Lösung in THF zugegeben. Die primären Reaktionsprodukte sind
monoanionische Übergangsmetall-substituierte Alane, die in guten Ausbeuten isoliert werden
können. [64,65] Nach Zusatz der chelatisierenden Base TMPDA liegen in der THF-
Reaktionslösung schon nach kurzer Reaktionszeit die neutralen Produkte (1-4) einer doppelten
Salzeliminierung vor. Offensichtlich bevorzugt das Aluminium die Koordination von neutralen harten
Donoren, im Gegensatz zum weicheren Chlorid- oder Iodidion.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 20
N
Al N
X
N
Al N
Me
(CO)nM
(CO)nM
K2[M(CO)n]
1. THF + AlX3
2. CH2Cl2 + TMPDA
- 2 KX
1. THF + Cl2AlMe
2. CH2Cl2 + TMPDA
- 2 KCl
(1-3)
(4)
Schema 2: Synthese der Verbindungen (1-4) (M = Fe, Cr; n = 4, 5; X = Cl, I)
Eine Zusammenstellung und das Numerierungsschema der synthetisierten Übergangsmetall-
substituierten Alane (1-4) ist in Tabelle 1 gegeben. Werden die Verbindungen in einkristalliner Form
erhalten, sind sie gegenüber der Laboratmosphäre und Feuchtigkeit über einige Tage stabil. Die
mikrokristallinen Pulver dagegen zeigen innerhalb von wenigen Stunden an der Oberfläche
Zersetzung.
Nr. Verbindung M n R oder X Donor
1 (CO)5Cr-Al[(Cl)(TMPDA)] Cr 5 Cl TMPDA
2 (CO)4Fe-Al[(Cl)(TMPDA)] Fe 4 Cl TMPDA
3 (CO)4Fe-Al[(I)(TMPDA)] Fe 4 I TMPDA
4 (CO)4Fe-Al[(Me)(TMPDA)] Fe 4 Me TMPDA
Tabelle 1: Zusammenstellung und Numerierungsschema der neuen Übergangsmetall-substituiertenAlane (1-4).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 21
2.2 Strukturchemie donorstabilisierter Übergangsmetall-Aluminium-Systeme
Geeignete Kristalle von (CO)5Cr-Al[(Cl)(TMPDA)] (1) und (CO)4Fe-Al[(Cl)(TMPDA)] (2)
wurden Einkristall-Röntgenstrukturanalysen unterzogen. Die Ergebnisse sind in Abbildung 6 und
Abbildung 7 dargestellt. Die dazugehörigen ausgewählten Bindungslängen [pm] und Bindungswinkel
[°] sind in Tabelle 2 und Tabelle 3 zusammengestellt.
Abbildung 6: Molekülstruktur von (CO)5Cr-Al[(Cl)(TMPDA)] (1) im Kristall(ORTEP-Darstellung).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 22
Tabelle 2: Ausgewählte Bindungslängen [pm] und -winkel [°] von (1).
Bindungslängen
Cr(1)-Al(1)1 248.2(1) Cr(1)-C(9) 188.2(4)
Al(1)-N(1) 207.0(3) Cr(1)-C(10) 184.5(3)
Al(1)-N(2) 204.1(3) Cr(1)-C(11) 187.1(3)
Al(1)-Cl(1) 219.8(1) Cr(1)-C(12) 186.2(3)
Cr(1)-C(8) 189.1(4) C(8)-O(8) 115.1(4)
Bindungswinkel
C(8)-Cr(1)-Al(1) 91.64(10) Cl(1)-Al(1)-Cr(1) 123.63(5)
C(9)-Cr(1)-Al(1) 89.52(11) N(1)-Al(1)-Cr(1) 119.16(9)
C(10)-Cr(1)-Al(1) 174.62(12) N(1)-Al(1)-N(2) 118.57(9)
C(11)-Cr(1)-Al(1) 81.93(10) N(2)-Al(1)-Cl(1) 96.08(8)
C(12)-Cr(1)-Al(1) 84.23(10) N(2)-Al(1)-Cr(1) 118.57(9)
Die röntgenstrukturanalytische Untersuchung eines gelblichen Kristalls von (1) zeigte das Vorliegen
einer direkten und unverbrückten Cr-Al-Bindung. Nur wenige weitere Beispiel mit einer derartigen
Struktureinheit sind bekannt, nämlich die von M. M. Schulte et al. synthetisierte Komplexe wie
(CO)5Cr-Al[(Cl)(TMEDA)]. [65] Der Einsatz von TMPDA gegenüber dem von M. Schulte
verwendeten Liganden TMEDA bietet hier den Vorteil, daß die erhaltenen einkristallinen Chargen
von (1) kein Methylenchlorid mehr im Kristall enthalten, was bei der Präparation für die
Einkristallröntgenstrukturanalyse erhebliche Vorteile bietet. Sind nämlich Lösungsmittelmoleküle in
den Kristall eingebaut (wie Beispielsweise (CO)4Fe-Al[(Cl)(TMPDA)] (2)), werden diese aufgrund
des hohen Dampfdrucks von CH2Cl2 sehr schnell trübe und brüchig, da das Solvens aus dem
Kristall hinausdiffundiert, und sind somit ungeeignet für eine röntgenographische Charakterisierung.
Ein weiterer Vorteil liegt in der relativ starren Konformation des Al(TMPDA)-Sechsrings im
Vergleich zum flexibleren „envelope“-Al(TMEDA)-Fünfring. Flexiblere Systeme erhöhen in der
Regel die Gefahr von Fehlordnungen der Moleküle im Kristallverband und erniedrigen somit die
Güte der Strukturbestimmung.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 23
Das Aluminium-Zentrum in (1) ist tetraedrisch von zwei Stickstoffliganden, dem Chlor- und dem
Chrom-Zentrum koordiniert. Die Al-N(1)/N(2)-Abstände sind nahezu gleichlang (207.0(3) und
204.1(3) pm). Der Al-Cl-Abstand von 219.8(1) pm ist bedingt durch die Vierfachkoordination des
Aluminiums geringfügig länger als der durch Hochtemperatur-Mikrowellen-Spektroskopie bestimmte
Abstand in gasförmigen AlCl von 212.983 pm. [122] Das Chrom-Zentrum ist oktaedrisch von fünf
Carbonylliganden und dem Aluminium-Zentrum koordiniert. Der Winkel Al(1)-Cr(1)-C(10) liegt mit
174.6(1)° nahe am Wert von 180° für einen idealen Oktaeder. Die äquatorialen Carbonylliganden
sind nur leicht zum Aluminium-Zentrum hingeneigt (Al-Cr-Ceq: 81.9(1) - 91.6(1)°). Der Cr-Al-
Bindungsabstand von 248.2(1) pm liegt im Bereich der Summe der Kovalenzradien von Aluminium
(125 pm) und Chrom (125 pm), [151] ist aber um 10 pm länger als im Komplex (CO)5Cr-AlCp*
von Schnöckel et al. (237.6 pm). Der Komplex 1 ist die vierfach adduktstabilisierte und (CO)5Cr-
AlCp* die lineare Variante für terminal koordinierte Erdmetallfragmente. Dieser Sachverhalt und die
Beobachtung, daß M-E-Bindungen in Abhängigkeit der Substituenten an beiden Metallzentren und in
Abhängigkeit von der Koordinationszahl am Zentrum E über einen sehr weiten Bereich systematisch
variieren, werden im „Gallium-Kapitel“ ausführlicher behandelt.
Die Verbindung (2) kristallisiert in Form farbloser Nadeln aus einer Methylenchlorid-Lösung aus.
Abbildung 7: Molekülstruktur von (CO)4Fe-Al[(Cl)(TMPDA)] (2) im Kristall(ORTEP-Darstellung).
Tabelle 3: Ausgewählte Bindungslängen [pm] und -winkel [°] von (2).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 24
Bindungslängen
Fe(1)-Al(1)1 236.0(5) Fe(1)-C(4) 174.2(13)
Al(1)-N(1) 201.4(12) Fe(1)-C(6) 175.7(16)
Al(1)-N(2) 200.3(11) Fe(1)-C(14) 176.3(16)
Al(1)-Cl(1) 216.2(6) Fe(1)-C(17) 180.0(18)
C(8)-C(9) 152.6(3) C(6)-O(2) 116.0(18)
Bindungswinkel
C(4)-Fe(1)-Al(1) 81.95(5) Cl(1)-Al(1)-Fe(1) 117.66(2)
C(6)-Fe(1)-Al(1) 175.89(5) N(1)-Al(1)-Fe(1) 118.65(4)
C(14)-Fe(1)-Al(1) 82.82(5) N(1)-Al(1)-N(2) 97.63(5)
C(17)-Fe(1)-Al(1) 87.12(5) N(2)-Al(1)-Cl(1) 98.92(4)
C(8)-C(9)-C(7) 115.21(13) N(2)-Al(1)-Fe(1) 119.91(4)
Die röntgenstrukturanalytische Untersuchung dieser farblosen Nadeln von (2) zeigt das Vorliegen
einer direkten und unverbrückten Fe-Al-Bindung. Der Einsatz von TMPDA gegenüber TMEDA
bietet auch hier den Vorteil, daß die erhaltenen einkristallinen Chargen von (2) kein Methylenchlorid
mehr im Kristall enthalten. Ein weiterer Vorteil liegt wie auch bei Verbindung (1) in der relativ starren
Konformation des Al(TMPDA)-Sechsrings im Vergleich zum flexibleren „envelope“-Al(TMEDA)-
Fünfring.
Das Aluminium-Zentrum in (2) ist tetraedrisch von zwei Stickstoffliganden, dem Chlor- und dem
Eisen-Zentrum koordiniert. Die Al-N(1)/N(2)-Abstände sind nahezu gleichlang (200.3(11) und
201.4(12) pm) und vergleichbar mit den in Verbindung {[(η5-C5H5)(CO)2]Fe-
Al[(CH2)3NMe2](iBu)} gemessenen Abständen. [183] Der Al-Cl-Abstand beträgt 216.2(6) pm.
Das Eisen-Zentrum ist trigonal-bipyramidal von vier Carbonylliganden und dem Aluminium-Zentrum
koordiniert. Der Winkel Al(1)-Fe(1)-C(6) liegt mit 175.89(5)° nahe am Wert von 180° für eine
ideale lineare Anordnung. Die äquatorialen Carbonylliganden sind nur leicht zum Aluminium-Zentrum
hingeneigt (Al-Fe-Ceq: 81.95(5)° - 87.12(5)°) (Regenschirmeffekt).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 25
Der Fe-Al-Bindungsabstand von 236.0(5) pm ist kleiner als in den „typischen“ Al(III)-Komplexen,
z. B. 245.6(1) pm für [(iBu){Me2N-(CH2)3}Al-FeCp(CO)2] [183] und 251.0(2) pm für das
Lewis-Base-/Lewis-Säure-Addukt [(Ph)3Al-FeCp(CO)2]- [11] In Legierungen (z. B.) Al3Fe [123])
ist der mittlere Fe-Al-Abstand (255 pm) um ca. 20 pm größer. Der Bindungsabstand in Verbindung
(CO)4Fe-AlCp* [54] ist um rund 13 pm kürzer. Ähnliche Al-M-Bindungslängen wie für (CO)4Fe-
AlCp* liegen in mehrkernigen Komplexen mit donorfreien Al-Zentren wie [(CpNi)2(Cp*Al)2] [11]
(228 pm) und [{EtAl-CoCp*(η2-C2H4)}2] [123] (233 pm) vor.
2.3 Spektroskopische Eigenschaften der Aluminium-Komplexe
2.3.1 NMR-Spektroskopie
In den 1H-NMR-Spektren [124] der Verbindungen (1-3) können jeweils zwei Signale für die
Methylprotonen und ein bei Raumtemperatur schlecht aufgelöstes, komplexes Kopplungsmuster für
die Methylprotonen des TMPDA-Liganden beobachtet werden. Die Koordination des Liganden
scheint also recht starr zu sein, offenbar liegt der AlN2C3-Sechsring in der Sesselkonformation vor
und somit können axiale und äquatoriale Positionen unterschieden werden. Für Verbindung (4) findet
man noch ein zusätzliches Singulett für die Methylgruppe. Die 13C-NMR-Spektren zeigen die
erwarteten zwei Signale für die axialen, bzw. äquatorialen Methylgruppen und zwei Resonanzen für
die jeweiligen Methylengruppen. Die chemischen Verschiebungen der Protonenresonanzen für die
TMPDA-Komplexe (1-4) sind in Tabelle 4 aufgelistet.
Nr. Verbindung N-CH3 N-CH2 CH2CH2CH2 Al-CH3
1 (CO)5Cr-Al[(Cl)(TMPDA)] 2.72; 2.69 3.52 2.19 -
2 (CO)4Fe-Al[(Cl)(TMPDA)] 2.88; 2.78 3.24 2.44 -
3 (CO)4Fe-Al[(I)(TMPDA)] 2.71; 2.68 3.30 2.17 -
4 (CO)4Fe-Al[(Me)(TMPDA)] 2.79; 2.58 3.18 2.22 0.17
Tabelle 4: 1H-chemische Verschiebungen der Komplexe (1-4).
Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 26
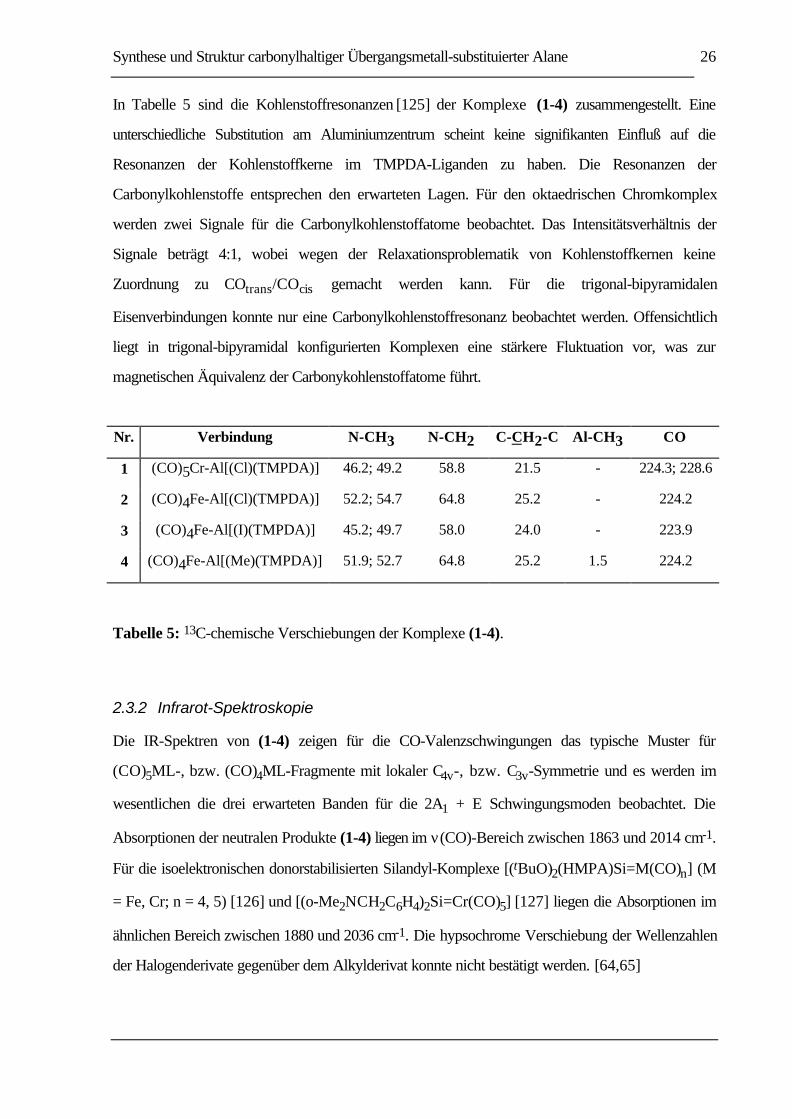
In Tabelle 5 sind die Kohlenstoffresonanzen [125] der Komplexe (1-4) zusammengestellt. Eine
unterschiedliche Substitution am Aluminiumzentrum scheint keine signifikanten Einfluß auf die
Resonanzen der Kohlenstoffkerne im TMPDA-Liganden zu haben. Die Resonanzen der
Carbonylkohlenstoffe entsprechen den erwarteten Lagen. Für den oktaedrischen Chromkomplex
werden zwei Signale für die Carbonylkohlenstoffatome beobachtet. Das Intensitätsverhältnis der
Signale beträgt 4:1, wobei wegen der Relaxationsproblematik von Kohlenstoffkernen keine
Zuordnung zu COtrans/COcis gemacht werden kann. Für die trigonal-bipyramidalen
Eisenverbindungen konnte nur eine Carbonylkohlenstoffresonanz beobachtet werden. Offensichtlich
liegt in trigonal-bipyramidal konfigurierten Komplexen eine stärkere Fluktuation vor, was zur
magnetischen Äquivalenz der Carbonykohlenstoffatome führt.
Nr. Verbindung N-CH3 N-CH2 C-CH2-C Al-CH3 CO
1 (CO)5Cr-Al[(Cl)(TMPDA)] 46.2; 49.2 58.8 21.5 - 224.3; 228.6
2 (CO)4Fe-Al[(Cl)(TMPDA)] 52.2; 54.7 64.8 25.2 - 224.2
3 (CO)4Fe-Al[(I)(TMPDA)] 45.2; 49.7 58.0 24.0 - 223.9
4 (CO)4Fe-Al[(Me)(TMPDA)] 51.9; 52.7 64.8 25.2 1.5 224.2
Tabelle 5: 13C-chemische Verschiebungen der Komplexe (1-4).
2.3.2 Infrarot-Spektroskopie
Die IR-Spektren von (1-4) zeigen für die CO-Valenzschwingungen das typische Muster für
(CO)5ML-, bzw. (CO)4ML-Fragmente mit lokaler C4v-, bzw. C3v-Symmetrie und es werden im
wesentlichen die drei erwarteten Banden für die 2A1 + E Schwingungsmoden beobachtet. Die
Absorptionen der neutralen Produkte (1-4) liegen im ν(CO)-Bereich zwischen 1863 und 2014 cm-1.
Für die isoelektronischen donorstabilisierten Silandyl-Komplexe [(tBuO)2(HMPA)Si=M(CO)n] (M
= Fe, Cr; n = 4, 5) [126] und [(o-Me2NCH2C6H4)2Si=Cr(CO)5] [127] liegen die Absorptionen im
ähnlichen Bereich zwischen 1880 und 2036 cm-1. Die hypsochrome Verschiebung der Wellenzahlen
der Halogenderivate gegenüber dem Alkylderivat konnte nicht bestätigt werden. [64,65]
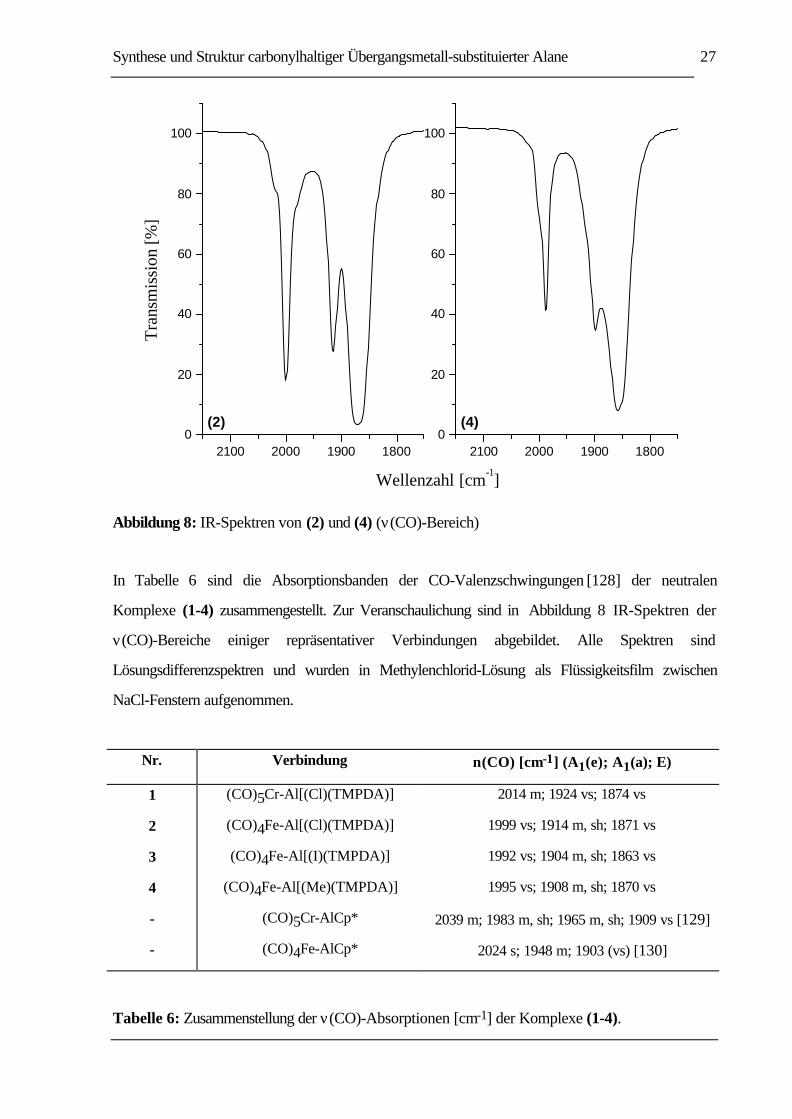
Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Alane 27
2100 2000 1900 18000
20
40
60
80
100
(2)
Tran
smis
sion
[%]
Wellenzahl [cm-1]
2100 2000 1900 18000
20
40
60
80
100
(4)
Abbildung 8: IR-Spektren von (2) und (4) (ν(CO)-Bereich)
In Tabelle 6 sind die Absorptionsbanden der CO-Valenzschwingungen [128] der neutralen
Komplexe (1-4) zusammengestellt. Zur Veranschaulichung sind in Abbildung 8 IR-Spektren der
ν(CO)-Bereiche einiger repräsentativer Verbindungen abgebildet. Alle Spektren sind
Lösungsdifferenzspektren und wurden in Methylenchlorid-Lösung als Flüssigkeitsfilm zwischen
NaCl-Fenstern aufgenommen.
Nr. Verbindung ν(CO) [cm-1] (A1(e); A1(a); E)
1 (CO)5Cr-Al[(Cl)(TMPDA)] 2014 m; 1924 vs; 1874 vs
2 (CO)4Fe-Al[(Cl)(TMPDA)] 1999 vs; 1914 m, sh; 1871 vs
3 (CO)4Fe-Al[(I)(TMPDA)] 1992 vs; 1904 m, sh; 1863 vs
4 (CO)4Fe-Al[(Me)(TMPDA)] 1995 vs; 1908 m, sh; 1870 vs
- (CO)5Cr-AlCp* 2039 m; 1983 m, sh; 1965 m, sh; 1909 vs [129]
- (CO)4Fe-AlCp* 2024 s; 1948 m; 1903 (vs) [130]
Tabelle 6: Zusammenstellung der ν(CO)-Absorptionen [cm-1] der Komplexe (1-4).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 28
3 Synthese und Struktur carbonylhaltiger Übergangsmetall-
substituierter Gallane
Im Jahre 1980 beschrieben R. D. Ernst et al., gestützt durch IR- und NMR Daten verschiedene
Lewis-Base-Addukte des Typs (CO)4Fe-Ga[(C2H5)(Do)n] (Do = Lewis-Base, n = 1, 2) und zwei
Jahre später die Röntgenstrukturanalyse von (CO)4Fe-Ga[(C2H3)(THF)]2. [66,67] Beide
Komplexe wurden aus der Umsetzung von EtGaCl2 mit Na2[Fe(CO)4] erhalten. Durch Reaktion
von Ru3(CO)12 mit Ga2Cl4 in Anwesenheit von metallischem Gallium konnten Whittlesey et al. 1997
die Komplexe (CO)3Ru[GaCl(THF)2][GaCl2(THF)]2·0.5 THF und (CO)8Ru2[GaCl2(THF)]2
isolieren und röntgenographisch charakterisieren.
1994 konnte in unserer Arbeitsgruppe gezeigt werden, daß die Umsetzung von 2 Mol K2[Fe(CO)4]
mit Cl2GaMe und Abfangen der dianionischen Zwischenstufe mit [PPN]Cl das zweifach
übergangsmetallsubstituierte Gallan {[(CO)4Fe]2(µ-GaMe)}[PPN]2 ergibt. [65,68] In der gleichen
Arbeit wurde auch über Verbindungen des Typs {(CO)4Fe-Ga[(CH2)3NMe2](R)}[PPN] (R = Cl,
tBu) und {(CO)5Cr-Ga[(CH2)3NMe2](R)}[PPN] (R = Cl, Me, tBu) berichtet. Zwei Jahre später
konnte mit der Röntgenstrukturanalyse von (CO)5Cr-Ga[(Me)(TMEDA)] ein weiterer Nachweis für
eine direkte Cr-Ga-Bindung erbracht werden. [69] Robinson et al. gelang es 1997 durch Reaktion
von Na2[Fe(CO)4] mit Cl2Ga(C6H3Trip2) (Trip = 2,4,6-Triisopropylphenyl), die donorfreie
Verbindung (CO)4Fe-Ga(C6H3Trip2) darzustellen, der seitens der Autoren eine Fe-Ga-
Dreifachbindung zugeschrieben wird. [70] Bereits kurze Zeit später wurde dieses Postulat von
Cotton et al. mit Hilfe von Dichtefunktionalitätsrechnungen widerlegt. [71]
Die Pionierarbeiten auf dem Gebiet der donorstabilisierten Aluminium- und Gallium-Komplexe
leisteten jedoch M. M. Schulte et al.. Ihnen gelang 1996 durch Umsetzung von K2[Cr(CO)5] mit
Cl2EMe (E = Al, Ga; R = Cl, Alkyl) die Synthese der Systeme (CO)5Cr-E[(R)(TMEDA)]. [65]
Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 29
3.1 Synthese donorstabilisierter Übergangsmetall-Gallium-Systeme
Analog zur Darstellung der Aluminium-Komplexe (1-4) werden zu Suspensionen der
Carbonylmetallate K2[Fe(CO)4] und K2[M(CO)5] (M = Cr, Mo, W; in situ aus den
Hexacarbonylen und KC8 hergestellt) in THF äquimolare Mengen von EX3 (E = Al, Ga, In; X = Cl,
Br, I) oder Cl2GaMe (alternativ 2 Äquivalente von ClGaMe2) als Lösung in THF zugegeben.
N
Ga N
X
N
Ga N(CO)4Fe
(CO)nM
K2[M(CO)n]
1. THF + GaX3
2. CH2Cl2 + TMPDA
- 2 KX
1. THF + Cl2GaMe
2. CH2Cl2 + TMPDA
- 2 KCl
(5, 6, 8, 9, 11-13)
(7, 10, 14)
N
Ga N
Me
(CO)nM
1. THF + GaI3
2. CH2Cl2 + PMDETA
- 2 KI
1. Toluol + Cl2Ga[(Et 2)NCH2CH2N(Et)]
- 2 KCl
(16)
(15)
N
(CO)4Fe
Fe(CO)4
Ga
N
Ga
NN
N
I
Et
Et
Et
Et Et
Et
Schema 3: Syntheseweg der Verbindungen (5-16) (M = Fe, Cr, Mo, W; n = 4, 5; X = Cl, Br, I).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 30
Nachfolgend wird direkt die chelatisierende Base (TMPDA, PMDETA) zugegeben und nach einer
Stunde Reaktionszeit das Lösungsmittel gegen Methylenchlorid ausgetauscht. Die THF-Lösungen
hellen im allgemeinen nach Zugabe der Base leicht auf. Im Falle von Eisen zeigen die vorher rosa-
roten bis roten Lösungen eine orange bis schwache Rosafärbung. Die Lösungen der Chromspezies
wechseln von grün nach gelb, die seiner höheren Homologen Molybdän und Wolfram von rot nach
orange-gelb. Nach dem Austausch des Lösungsmittels verändern sich die Farben nur unwesentlich.
Die Reaktionsverläufe wurden mittels IR-Spektroskopie verfolgt. Schema 3 zeigt den allgemeinen
Syntheseweg.
Die Isolierung der Übergangsmetall-substituierten Gallane (5-16) gelingt problemlos in Form
mikrokristalliner Pulver. Nach Abkondensieren des Methylenchlorids werden die Rückstände mit
kaltem Pentan gewaschen und am Hochvakuum getrocknet. Im Fall der Chloride kann auch mit
Diethylether gewaschen werden. Die Eisenverbindungen fallen als weiße Pulver, die
Chromverbindungen als blaßgelbe und die Molybdän- und Wolframverbindungen als orange Pulver
an.
Vorteilhafter hat sich die Isolierung der Verbindungen in einkristalliner Form gezeigt, wodurch fast
perfekte Elementaranalysen erhalten werden konnten. Filtrierte Methylenchlorid-Lösungen werden
auf etwa 15 ml eingeengt (Ansatzgröße 2 mmol) und im Schlenkrohr in waagrechter Position auf –
30°C gekühlt. Die Kristallisation setzt über Nacht ein und wohlgeformte Nadeln. Quader oder
Blättchen der Verbindungen (5, 11-16) werden erhalten. Je nach Kristallisationsdauer konnten
Kristalle mit bis zu 10 mm Kantenlänge gezogen werden. Trennt man die Mutterlauge von den
Kristallen ab und engt weiter ein kann durch Nachkristallisation die Ausbeute um ca. 20% gesteigert
werden. Die Ausbeuten an makrokristallinem Material liegen dann durchweg über 90%.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 31
Eine Zusammenstellung und das Numerierungsschema der synthetisierten Übergangsmetall-
substituierten Gallane (5-16) ist in Tabelle 7 gegeben. Werden die Verbindungen in einkristalliner
Form erhalten, sind sie gegenüber der Laboratmosphäre und Feuchtigkeit über Tage stabil. Die
mikrokristallinen Pulver dagegen zeigen innerhalb von ein bis zwei Stunden an der Oberfläche
Zersetzung.
Nr. Verbindung M n R oder X Donor
5 (CO)5Cr-Ga[(Cl)(TMPDA)] Cr 5 Cl TMPDA
6 (CO)5Cr-Ga[(I)(TMPDA)] Cr 5 I TMPDA
7 (CO)5Cr-Ga[(Me)(TMPDA)] Cr 5 Me TMPDA
8 (CO)5Mo-Ga[(I)(TMPDA)] Mo 5 I TMPDA
9 (CO)5W-Ga[(I)(TMPDA)] W 5 I TMPDA
10 (CO)5W-Ga[(Me)(TMPDA)] W 5 Me TMPDA
11 (CO)4Fe-Ga[(Cl)(TMPDA)] Fe 4 Cl TMPDA
12 (CO)4Fe-Ga[(Br)(TMPDA)] Fe 4 Br TMPDA
13 (CO)4Fe-Ga[(I)(TMPDA)] Fe 4 I TMPDA
14 (CO)4Fe-Ga[(Me)(TMPDA)] Fe 4 Me TMPDA
15 (CO)4Fe-Ga[(PMDETA)]+I- Fe 4 - PMDETA
16 {(CO)4Fe-Ga[(Et)2NCH2CH2N(Et)]} Fe 4 - -
Tabelle 7: Zusammenstellung und Numerierungsschema der neuen Übergangsmetall-substituiertenGallane (5-16).
3.2 Strukturchemie donorstabilisierter Übergangsmetall-Gallium-Systeme
Von den Verbindungen (CO)5Cr-Ga[(Cl)(TMPDA] (5), (CO)4Fe-Ga[(Cl)(TMPDA)] (11),
(CO)4Fe-Ga[(Br)(TMPDA)] (12), (CO)4Fe-Ga[(I)(TMPDA)] (13), (CO)4Fe-
Ga[(Me)(TMPDA)] (14), (CO)4Fe-Ga[(PMDETA)]+I- (15) und {(CO)4Fe-
Ga[(Et)2NCH2CH2N(Et)]} (16) konnten geeignete und wohlgeformte Kristalle für die
Einkristallröntgenstrukturanalyse durch Kristallisation aus einer Methylenchlorid- oder Toluol-Lösung
gewonnen werden.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 32
Die Verbindung (CO)5Cr-Ga[(Cl)(TMPDA] (5) kristallisiert in Form gelblicher Quader.
Abbildung 9 zeigt eine ORTEP-Darstellung von (5). In Tabelle 8 sind relevante Bindungslängen [pm]
und Bindungswinkel [°] aufgelistet.
Abbildung 9: Molekülstruktur von (CO)5Cr-Ga[(Cl)(TMPDA] (5) im Kristall(ORTEP-Darstellung).
Tabelle 8: Ausgewählte Bindungslängen [pm] und -winkel [°] von (5).
Bindungslängen
Ga(1)-Cr(2) 248.6(2) N(1)-C(21) 147.6(14)
Ga(1)-N(1) 217.2(8) N(2)-C(8) 146.6(15)
Ga(1)-N(2) 217.6(9) N(2)-C(20) 148.6(14)
Ga(1)-Cl(99) 224.0(30) N(2)-C(22) 149.5(13)
Ga(1)-C(99) 206.0(40) O(2)-C(17) 116.5(16)
Bindungswinkel
N(1)-Ga(1)-Cr(2) 118.3(2) C(17)-Cr(2)-Ga(1) 177.2(4)
N(2)-Ga(1)-Cr(2) 118.0(3) C(14)-Cr(2)-Ga(1) 88.8(4)
Cl(99)-Ga(1)-Cr(2) 130.5(6) C(15)-Cr(2)-Ga(1) 82.9(4)

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 33
C(99)-Ga(1)-Cr(2) 121.6(9) O(2)-C(17)-Cr(2) 176.3(11)
N(1)-Ga(1)-N(2) 94.2(3) C(8)-C(4)-C(18) 115.6(10)
Die röntgenstrukturanalytische Untersuchung eines gelblichen Kristalls von (5) zeigte das Vorliegen
einer direkten und unverbrückten Cr-Ga-Bindung. Nur wenige weitere Beispiel mit einer derartigen
Struktureinheit sind bekannt, nämlich die von M. M. Schulte et al. synthetisierte Komplexe wie
(CO)5Cr-Ga[(Cl)(TMEDA)]. [65]
Das Gallium-Zentrum in (5) ist tetraedrisch von zwei Stickstoffliganden, dem Chlor- und dem
Chrom-Zentrum koordiniert. Die Ga-N(1)/N(2)-Abstände sind nahezu gleichlang (217.2(8) und
217.6(9) pm). Auffällig lang ist der Bindungsabstand Ga-Cl mit 224.0(30) pm. Für vierfach
koordinierte Galliumverbindungen liegen die Bindungslängen für terminale Ga-Cl-Bindungen bei etwa
216(±)5 pm. Das Chrom-Zentrum ist oktaedrisch von fünf Carbonylliganden und dem Gallium-
Zentrum koordiniert. Der Winkel Ga(1)-Cr(2)-C(17) liegt mit 177.2(4)° nahe am Wert von 180° für
einen idealen Oktaeder. Die äquatorialen Carbonylliganden sind nur leicht zum Gallium-Zentrum
hingeneigt (Ga-Cr-Ceq: 82.9(4) – 88.8(4)°). Der Cr-Ga-Bindungsabstand von 248.6(2) pm liegt im
Bereich der Summe der Kovalenzradien von Gallium (120 pm) und Chrom (125 pm). [151] Der
Vergleich mit den Literaturdaten zeigt den Trend einer gewissen Bindungsverkürzung in der Reihe
der Liganden X = Alkyl, H, Halogenid: (CO)5Cr-Ga[(Et)(TMEDA)] 251.7(4) pm und (CO)5Cr-
Ga[(Cl)(TMEDA)] 245.6 (1) pm. Diese Bindungslängenvariationen werden noch signifikanter, wenn
man auch die Systeme wie (CO)5Cr-GaCp* [133] (240.5(7)) mit einbezieht. Die Bindungslänge der
Cr-Ga-Bindung wird bestimmt durch a) die Koordinationszahl am Ga (KZ = 2 oder 4) und b) die
σ-Donor-Stärke der Liganden L (TMPDA oder PMDETA) am Gallium-Zentrum. Dabei
stabilisieren starke und harte Donoren das Gallium-Zentrum (Gaδ+). Dies führt zu einer „ionischeren“
Bindung und zu einer Kontraktion der Bindungsorbitale und damit zu einem kürzeren Cr-Ga-
Abstand. Ebenso wirkt sich der Einfluß der Liganden Cl, Br, I und CH3 auf den Abstand der Cr-
Ga-Bindung aus. Durch den leicht elektronenziehenden Effekt der Halogenide im Vergleich zu
Alkylsubstituenten kommt es, wie auch oben schon beschrieben, zu einer Kontraktion der
Bindungsorbitale.
Die röntgenstrukturanalytischen Untersuchungen der Kristalle der Verbindungen (11-14) werden in
der folgenden Diskussion wegen ihrer Ähnlichkeit zusammengefaßt. Alle vier Verbindungen zeigen
das Vorliegen einer direkten und unverbrückten Fe-Ga-Bindung. Die Gallium-Zentren der

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 34
Verbindungen (11-14) sind tetraedrisch je von zwei Stickstoffliganden, dem jeweiligen Liganden
Chlor, Brom, Iod oder Methyl und dem Eisen-Zentrum koordiniert.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 35
Die Ga-N(1)/N(2)-Abstände sind jeweils nahezu gleichlang. Das Eisen-Zentrum ist trigonal-
bipyramidal koordiniert, wobei der Gallium-Ligand eine axiale Position besetzt. Die
Koordinationsgeometrie der äquatorialen Carbonylliganden erfolgt in einer Weise, daß die Winkel
CO-Ga-Fe deutlich kleiner als 90° sind, welches auch als sogenannter Regenschirmeffekt
bezeichnet wird. Die jeweiligen Winkel (Ga-Fe-C)eq liegen mit 176.5(7)° nahe am Wert von 180°.
Die Ga-Cl-Bindungslänge von 224.9(1) pm in (11) entspricht fast denen für [(η5-C5H5)(CO)2Fe-
GaCl2(NMe3)] gefundenen Werten (225.1 und 224.0 pm). [188] Die Fe-Ga-Bindungsabstände
liegen alle im Bereich der Summe der Kovalenzradien von Gallium (120 pm) und Eisen (124
pm). [151] Die Fe-Ga-Bindungsabstände sind typisch für diese Art von Molekülverbindungen sehr
kurz. Auch hier kann wieder die Beobachtung gemacht werden, daß die Länge der Fe-Ga-
Bindungen in Abhängigkeit der Substituenten an beiden Metallzentren und in Abhängigkeit von der
Koordinationszahl am Zentrum E über einen sehr weiten Bereich systematisch variieren (Tabelle 14).
Die Bindungsverkürzung in der Serie (CO)4Fe-Ga[(X)(TMPDA)] (X = Cl, Br, I; alle um 233 pm)
und (CO)4Fe-Ga[(CH3)(TMPDA)] (241.6(3) pm) belegen dies eindeutig. Auch in der Literatur
findet man entsprechende Serien, wie z. B. [Cp(CO)2Fe-GaX2(NR3)], deren Fe-Ga-Abstände
zwischen 236.2(3) pm für X = Cl und 245.7(4) pm für X = Alkyl variieren. Für (CO)4Fe-GaAr* mit
linear zweifach koordiniertem Gallium-Atom ermittelt man eine ungewöhnlich kurze Fe-Ga-
Bindungslänge von 222.5(7) pm. Dies findet seine Parallele für die vorher beschriebene (CO)5M-
Serie im Vergleich von (CO)5Cr-GaCp* (240.5(7) pm) mit (CO)5Cr-Ga[(Et)(TMEDA)]. Um
weitere Aussagen über M-E-Bindungslängen zu treffen wurden in diversen Publikationen
Bindungsdissoziationsenergien auf MP2/II-Niveau an Modellkomplexen ((CO)5W(L) L =
Erdmetallfragment) berechnet. [170] Auffällig an den quantentheoretischen Ergebnissen ist, daß die
Fragmente EXL2 (z. B. Ga(Cl)(TMPDA)) gegenüber den Varianten EX (z. B. GaCp*) signifikant
höhere Dissoziationsenergien aufweisen. Dies steht der oben diskutierten Bindungslängenverkürzung
für die Komplexe des Typs MEX und MER gegenüber, wenn man kürzere Bindungen als Indiz für
stärkere Bindungen ansieht und auf entsprechende π-Wechselwirkungen schließt, die für MEX
möglich, aber für MEXL2 unmöglich sein sollten. [52, 105]

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 36
Der Begriff Mehrfachbindung impliziert oft eine höhere Bindungsstärke und kürzere Bindungen, diese
Sicht ist jedoch problematisch. [33, 102] Die auch der Interpretation der Fe-Ga-Bindung in
(CO)4Fe-GaAr* zugrundeliegende Auffassung, daß auffällig kurze Bindungen für erhöhte
Bindungsordnung steht, wurde in der Literatur wiederholt kritisch diskutiert. [102]
Abbildung 10: Molekülstruktur von (CO)4Fe-Ga[(Cl)(TMPDA)] (11) im Kristall(ORTEP-Darstellung).
Tabelle 9: Ausgewählte Bindungslängen [pm] und -winkel [°] von (11).
Bindungslängen
Ga(1)-Fe(1) 234.03(9) N(1)-C(12) 149.5(7)
Ga(1)-N(1) 208.2(4) N(1)-C(2) 149.6(7)
Ga(1)-N(5) 209.8(4) N(1)-C(11) 150.7(6)
Ga(1)-Cl(1) 224.95(13) C(2)-C(3) 151.8(8)
Fe(1)-C(61) 176.4(6) O(61)-C(61) 115.7(6)
Bindungswinkel
N(1)-Ga(1)-Fe(1) 120.86(14) C(61)-Fe(1)-Ga(1) 83.03(16)
N(5)-Ga(1)-Fe(1) 118.83(13) C(62)-Fe(1)-Ga(1) 85.59(18)
Cl(1)-Ga(1)-Fe(1) 121.53(4) C(63)-Fe(1)-Ga(1) 175.6(2)

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 37
N(5)-Ga(1)-Cl(1) 96.17(12) O(61)-C(61)-Fe(1) 176.2(4)
N(1)-Ga(1)-N(5) 97.21(17) C(4)-C(3)-C(2) 115.0(5)
Abbildung 11: Molekülstruktur von (CO)4Fe-Ga[(Br)(TMPDA)] (12) im Kristall(ORTEP-Darstellung).
Tabelle 10: Ausgewählte Bindungslängen [pm] und -winkel [°] von (12).
Bindungslängen
Ga(1)-Fe(1) 233.3(5) N(1)-C(112) 149.9(14)
Ga(1)-N(1) 212.4(10) N(1)-C(2) 148.4(13)
Ga(1)-N(5) 207.3(9) N(1)-C(111) 148.1(13)
Ga(1)-Br(1) 241.3(5) C(2)-C(3) 146.4(16)
Fe(1)-C(13) 176.1(13) O(13)-C(13) 115.2(16)
Bindungswinkel
N(1)-Ga(1)-Fe(1) 120.0(2) C(13)-Fe(1)-Ga(1) 87.5(4)
N(5)-Ga(1)-Fe(1) 120.8(2) C(14)-Fe(1)-Ga(1) 87.9(4)
Br(1)-Ga(1)-Fe(1) 120.27(17) C(11)-Fe(1)-Ga(1) 175.5(3)
N(5)-Ga(1)-Br(1) 97.0(2) O(13)-C(13)-Fe(1) 178.4(10)
N(1)-Ga(1)-N(5) 95.9(3) C(4)-C(3)-C(2) 112.7(10)

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 38

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 39
Abbildung 12: Molekülstruktur von (CO)4Fe-Ga[(I)(TMPDA)] (13) im Kristall(ORTEP-Darstellung).
Tabelle 11: Ausgewählte Bindungslängen [pm] und -winkel [°] von (13).
Bindungslängen
Ga(1)-Fe(1) 233.71(8) Fe(1)-C(2) 177.1(5)
Ga(1)-N(1) 209.4(4) C(2)-O(2) 115.0(6)
Ga(1)-N(2) 210.9(4) N(1)-C(8) 148.9(6)
Ga(1)-I(1) 264.21(6) N(1)-C(5) 149.3(6)
Fe(1)-C(1) 176.8(5) C(5)-C(6) 151.3(7)
Bindungswinkel
N(1)-Ga(1)-Fe(1) 120.50(11) C(2)-Fe(1)-Ga(1) 177.17(18)
I(1)-Ga(1)-Fe(1) 117.18(3) C(3)-Fe(1)-Ga(1) 84.06(16)
I(1)-Ga(1)-N(1) 99.18(10) C(4)-Fe(1)-Ga(1) 88.30(16)
C(11)-N(2)-C(10) 106.6(4) O(2)-C(2)-Fe(1) 178.0(5)
N(1)-Ga(1)-N(2) 95.59(15) C(5)-C(6)-C(7) 115.9(4)
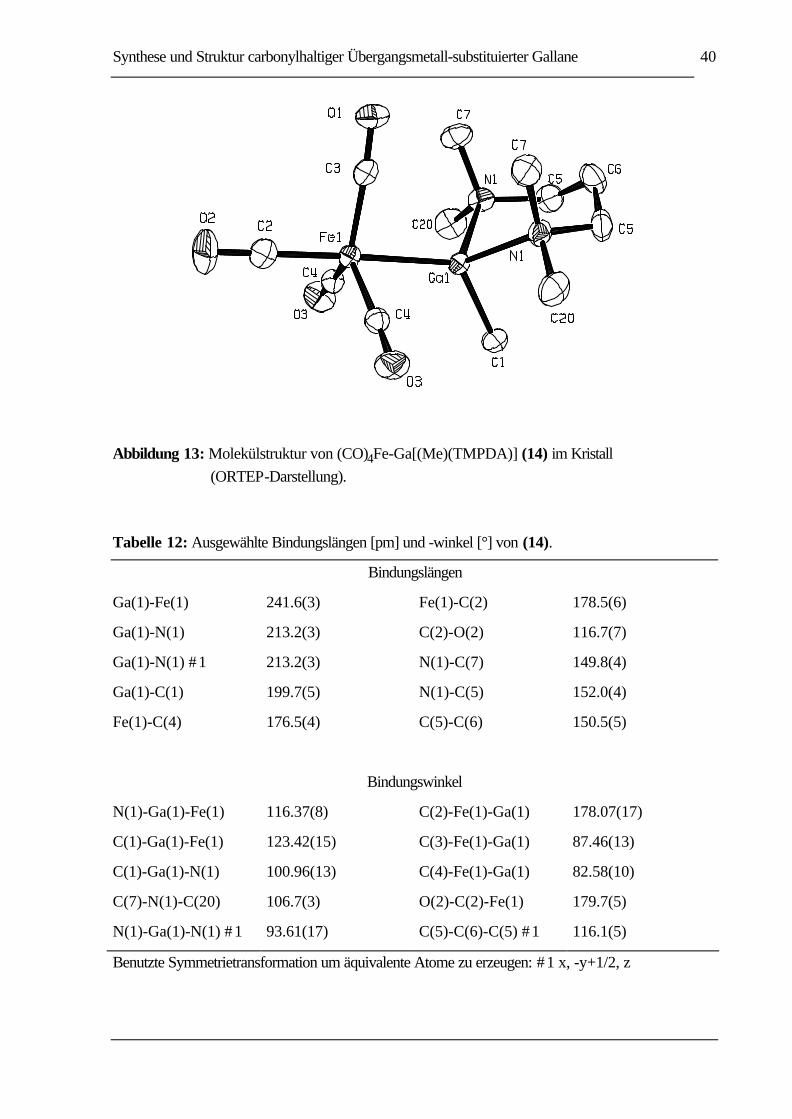
Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 40
Abbildung 13: Molekülstruktur von (CO)4Fe-Ga[(Me)(TMPDA)] (14) im Kristall(ORTEP-Darstellung).
Tabelle 12: Ausgewählte Bindungslängen [pm] und -winkel [°] von (14).
Bindungslängen
Ga(1)-Fe(1) 241.6(3) Fe(1)-C(2) 178.5(6)
Ga(1)-N(1) 213.2(3) C(2)-O(2) 116.7(7)
Ga(1)-N(1) #1 213.2(3) N(1)-C(7) 149.8(4)
Ga(1)-C(1) 199.7(5) N(1)-C(5) 152.0(4)
Fe(1)-C(4) 176.5(4) C(5)-C(6) 150.5(5)
Bindungswinkel
N(1)-Ga(1)-Fe(1) 116.37(8) C(2)-Fe(1)-Ga(1) 178.07(17)
C(1)-Ga(1)-Fe(1) 123.42(15) C(3)-Fe(1)-Ga(1) 87.46(13)
C(1)-Ga(1)-N(1) 100.96(13) C(4)-Fe(1)-Ga(1) 82.58(10)
C(7)-N(1)-C(20) 106.7(3) O(2)-C(2)-Fe(1) 179.7(5)
N(1)-Ga(1)-N(1) #1 93.61(17) C(5)-C(6)-C(5) #1 116.1(5)
Benutzte Symmetrietransformation um äquivalente Atome zu erzeugen: #1 x, -y+1/2, z

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 41
Die Verbindung (CO)4Fe-Ga[(PMDETA)]+ (15) kristallisiert aus einer Methylenchlorid-Lösung in
wohlgeformten orangen Quadern in der monoklinen Raumgruppe P21/c aus. Die
Röntgeneinkristallstrukturuntersuchung an dem ausgewählten orangefarbenen Kristallen von (15)
zeigte das Vorliegen einer direkten und unverbrückten Fe-Ga-Bindung. Das Eisen-Zentrum ist
trigonal-bipyramidal koordiniert, wobei der Gallium-Ligand eine axiale Position besetzt. Der Winkel
Ga(1)-Fe(2)-C(40) liegt mit 174.9(2)° nahe am Wert von 180° für eine ideale lineare Anordnung in
einer trigonalen Bipyramide. Die Bindungswinkel der axialen Carbonylgruppen (C-Fe-C) liegen im
Bereich von 94.1(4) bis 95.0(3)°, die der äquatorialen Carbonylgruppen im Bereich 114.0(4) bis
122.5(4)°. Die Koordinationsgeometrie der äquatorialen Carbonylliganden erfolgt in einer Weise,
daß die Ga-Fe-C-Winkel (80.5(3) to 89.1(3)°) deutlich kleiner als 90°C sind, welches auch als
„Regenschirmeffekt“ bezeichnet wird. Hierbei handelt es sich um eine stärkere Neigung der
äquatorialen Carbonylgruppen hin zum großen „[(PMDETA)Ga]-Ligand“ als es für andere
Fe(CO)4L-Komplexe (L = PPh3, PPh2H, C5H5N, C3Ph2S) bekannt ist. [131] Diese ähnliche
Anordnung wurde auch schon in den Verbindungen [HB(3,5-Me2pz)3Ga-Fe(CO)4, [132]
[HB(3,5-Me2pz)3In-Fe(CO)4, [152k] und (η5C5Me5)GaFe(CO)4 beschrieben. [133] Diese
Beobachtung kann als struktureller Hinweis auf eine polare M-E-Bindung gedeutet
werden. [134,135]
Das Gallium-Zentrum in (15) ist verzerrt-tetraedrisch von drei Stickstoffliganden und dem Eisen-
Zentrum koordiniert. Die Ga-N(4)/N(5)/N(6)-Abstände sind nahezu gleichlang (209.5(6), 206.4(6)
und 207.9(6) pm) und stimmen mit den durchschnittlichen Bindungslängen von 209.0 pm in der
Literatur überein. [136,189] Die N-Ga-N-Winkel im Chelatring befinden sich zwischen 85.8(2) und
107.5(3)°. Die interessanteste Tatsache der Verbindung (15) ist die kurze Ga-Fe-Bindungslänge von
231.0(1) pm (die durchschnittliche Bindungslänge in der Literatur ist 243.0 pm). [137,188]
Vergleicht man diese Einfachbindung mit vier Komplexen die bereits publiziert wurden, läßt sich
folgendes feststellen: Es existieren zwei kürzere Fe-Ga-Bindungen mit 222.5(7) pm und 227.3(4) pm
in den Verbindungen (η5C5Me5)GaFe(CO)4, [133] und Ar*Ga-Fe(CO)4 und zwei unwesentlich
längere Bindungen mit 231.5(3) pm ([HB(3,5-Me2pz)3Ga-Fe(CO)4) und 233.8(2) pm ([(CO)4Fe-
Ga(Cl)(TMPDA)]).
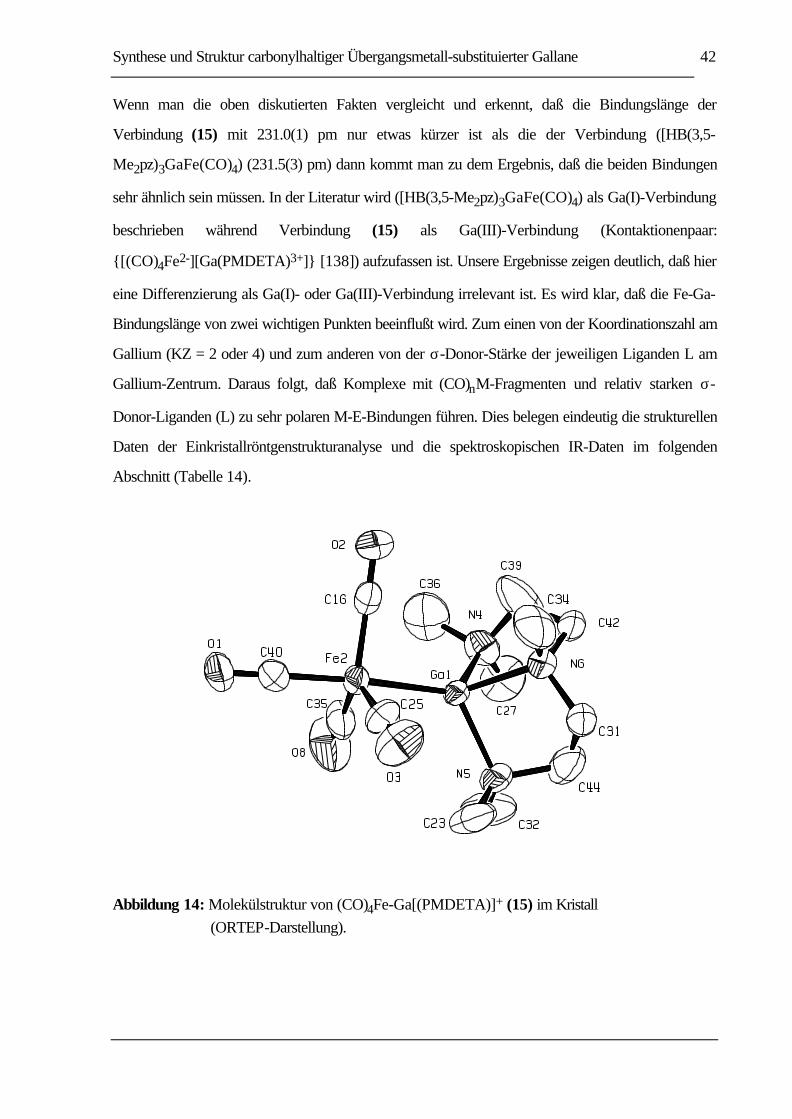
Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 42
Wenn man die oben diskutierten Fakten vergleicht und erkennt, daß die Bindungslänge der
Verbindung (15) mit 231.0(1) pm nur etwas kürzer ist als die der Verbindung ([HB(3,5-
Me2pz)3GaFe(CO)4) (231.5(3) pm) dann kommt man zu dem Ergebnis, daß die beiden Bindungen
sehr ähnlich sein müssen. In der Literatur wird ([HB(3,5-Me2pz)3GaFe(CO)4) als Ga(I)-Verbindung
beschrieben während Verbindung (15) als Ga(III)-Verbindung (Kontaktionenpaar:
{[(CO)4Fe2-][Ga(PMDETA)3+]} [138]) aufzufassen ist. Unsere Ergebnisse zeigen deutlich, daß hier
eine Differenzierung als Ga(I)- oder Ga(III)-Verbindung irrelevant ist. Es wird klar, daß die Fe-Ga-
Bindungslänge von zwei wichtigen Punkten beeinflußt wird. Zum einen von der Koordinationszahl am
Gallium (KZ = 2 oder 4) und zum anderen von der σ-Donor-Stärke der jeweiligen Liganden L am
Gallium-Zentrum. Daraus folgt, daß Komplexe mit (CO)nM-Fragmenten und relativ starken σ-
Donor-Liganden (L) zu sehr polaren M-E-Bindungen führen. Dies belegen eindeutig die strukturellen
Daten der Einkristallröntgenstrukturanalyse und die spektroskopischen IR-Daten im folgenden
Abschnitt (Tabelle 14).
Abbildung 14: Molekülstruktur von (CO)4Fe-Ga[(PMDETA)]+ (15) im Kristall(ORTEP-Darstellung).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 43
Tabelle 13: Ausgewählte Bindungslängen [pm] und -winkel [°] von (15).
Bindungslängen
Ga(2)-Fe(2) 231.0(1) N(6)-C(34) 151.5(11)
Ga(2)-N(4) 209.5(6) C(16)-O(2) 115.2(10)
Ga(1)-N(5) 206.4(6) N(4)-C(36) 143.9(13)
Ga(1)-N(6) 207.9(6) N(4)-C(39) 154.8(13)
Fe(2)-C(16) 175.9(9) C(31)-C(44) 154.8(14)
Bindungswinkel
C(25)-Fe(2)-Ga(2) 80.5(3) N(5)-Ga(2)-Fe(2) 127.49(18)
C(16)-Fe(2)-Ga(2) 89.1(2) N(6)-Ga(2)-Fe(2) 120.41(17)
C(40)-Fe(2)-Ga(2) 174.9(2) N(5)-Ga(2)-N(6) 85.8(2)
C(35)-Fe(2)-Ga(2) 87.0(2) N(5)-Ga(2)-N(4) 107.5(3)
N(4)-Ga(2)-Fe(2) 120.7(2) N(6)-Ga(2)-N(4) 86.2(3)
Als Fazit ist festzuhalten, daß in Abhängigkeit vom Niveau der quantenchemischen Berechnungen
und dem gewählten Beschreibungsmodell sich mehr oder weniger detaillierte Bilder der
Bindungsverhältnisse ergeben, deren gemeinsamer und die Verhältnisse dominierender Faktor die
Bindungspolarität ist. Diese läßt sich gemäß der Reihe E(R)L2 > E(X)L2 > ER und Al > Ga, In in
Abhängigkeit von der Koordinationszahl von E und den daran gebundenen Substituenten R, X und L
bestimmen. Insbesondere die koordinativ gesättigten Fragmente E(X)L2 entsprechen weitgehend den
Amin- oder Phosphor-Liganden (py, NR3, PR3). Sie erlauben aber aufgrund des elektopositiveren
Charakters des Zentrums E deutlich erhöhte negative Ladungsdichten am d-Metall. Die M-E-
Bindungen in den diskutierten Komplexen sind daher am besten als stark polare Lewis Säure/Base
Addukte aufzufassen. Eine Analogie der RE-Liganden zum CO-Ligand besteht in strukturchemischer
bzw. koordinationschemischer Sicht, insbesondere, was die Bindungsmodi terminal und µ2- bzw.
µ3-verbrückend angeht. Die Antwort auf die Frage, ob es eine ausgeprägte π-Akzeptorkapazität
der Fragmente RE, Ar*Ga, (Me3Si)3SiGa oder (Me3Si)3CIn gibt oder nicht, ist sehr stark vom
Niveau der theoretischen Betrachtungsweise und den zugrundeliegenden Modellen abhängig.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 44
Experimentelle Evidenzen dafür fehlen jedenfalls noch und es ist fraglich, ob sie gefunden werden
können, da das klassische Bild und Konzept „Mehrfachbindung“ hier versagt. Auch die Serie der
zum Typ [(CO)nM-EX(L2)] isovalenzelektronischen, koordinativ gesättigten Verwandten [(CO)nM-
EX2(L)] (X = H, Alkyl, Halogenid; L = neutraler 2e--Lewis-Donor Ligand) wurde
bindungstheoretisch am Beispiel einer homologen Reihe von Co-Ga Komplexen untersucht. [139]
Verbindung Abstand Fe-Ga
[pm]
ν (CO) [cm-1]
(CO)4Fe-GaAr* 222.5(7) 2032 s; 1959 s; 1941 vs; 1929 vs [a]
(CO)4Fe-GaCp* 227.3(4) 2037 s; 1966 s; 1942 vs [b]
(CO)4Fe-GaCp [140] 232.7 2040, 1983, 1959
(CO)4Fe-Ga[(PMDETA)]+I- 231.0(1) 1998 vs; 1920 vs, sh; 1887 vs [c]
(CO)4Fe-Ga[(HB(3,5-Me2pz)3)] 231.5(3) 2008, 1926, 1878 [a]
(CO)4Fe-Ga[(Br)(TMPDA)] 233.3(5) 2013 vs; 1931 m, sh; 1893 vs [c]
(CO)4Fe-Ga[(I)(TMPDA)] 233.7(8) 2012 vs; 1932 m, sh; 1893 vs [c]
(CO)4Fe-Ga[(Cl)(TMEDA)] 233.8(2) 2011 vs; 1928 vs; 1881 vs [c]
(CO)4Fe-Ga[(Cl)(TMPDA)] 234.0(9) 2013 vs; 1930 m, sh; 1893 vs [c]
(CO)4Fe-Ga[(Me)(TMPDA)] 241.6(3) 1994 vs; 1907 m, sh; 1870 vs [c]
Tabelle 14: Strukturell charakterisierte Eisen-Gallium-Komplexe: σ(Fe-Ga)-Bindungslängen undIR-Daten. [a] Nujol. [b] n-Hexan. [c] Methylenchlorid.
Die Verbindung (16) kristallisiert in Form gelblicher dünner Blättchen aus einer Toluol-Lösung aus.
Da die Blättchen der Verbindung (16) zu dünn waren können an dieser Stelle keine Bindungswinkel
und –abstände diskutiert werden, da die Werte mit einem zu großen Fehler behaftet sind (kein
ausreichender Datensatz).
{(CO)4Fe-Ga[(Et)2NCH2CH2N(Et)]}2 ist ein enger Verwandter der auch
einkristallröntgenstrukturanalytisch gesicherten Verbindung {(CO)4Fe-Ga[(C2H3)(THF)]}2 [67,
186, 188] und kann als das Dimerisierungsprodukt angesehen werden.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 45
Abbildung 15 zeigt die ORTEP-Darstellung der Verbindung (16). Man erkennt, daß das
Eisenzentrum trigonal-bipyramidal koordiniert ist, wobei auch hier der Gallium-Ligand eine axiale
Position besetzt. Die beiden Gallium-Zentren sind verzerrt tetraedrisch koordiniert. Die zwei
Galliumatome und zwei Stickstoffatome bilden einen planaren Vierring.
Abbildung 15: Molekülstruktur von {(CO)4Fe-Ga[(Et)2NCH2CH2N(Et)]}2 (16) im Kristall(ORTEP-Darstellung).
3.3 Spektroskopische Eigenschaften der Gallium-Komplexe
3.3.1 NMR-Spektroskopie
In den 1H-NMR-Spektren [124] der Verbindungen (5-14) können jeweils zwei Signale für die
Methylprotonen und ein bei Raumtemperatur schlecht aufgelöstes, komplexes Kopplungsmuster für
die Methylprotonen des TMPDA-Liganden beobachtet werden. Die Koordination des Liganden
scheint also recht starr zu sein, offenbar liegt der GaN2C3-Sechsring in der Sesselkonformation vor
und somit können axiale und äquatoriale Positionen unterschieden werden. Für Verbindung (15)
findet man ein ebenso komplexes Spektrum für den PMDETA-Liganden wie auch in (16) für den
dimeren Komplex.
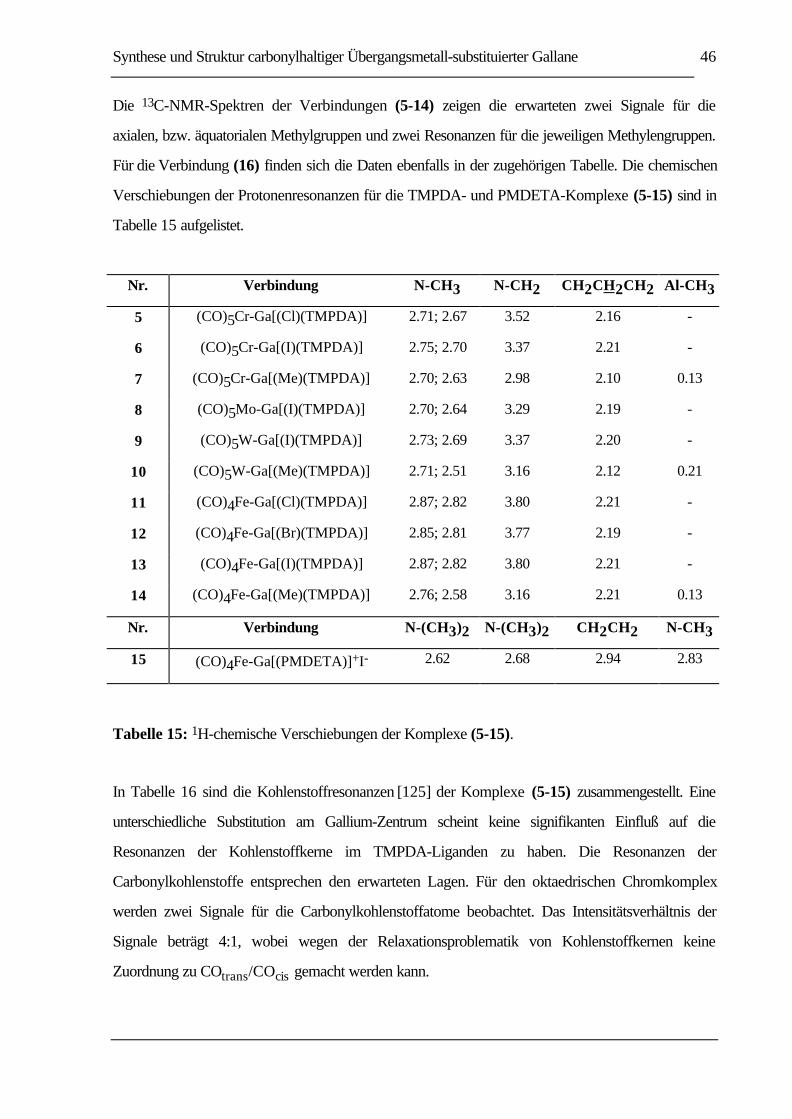
Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 46
Die 13C-NMR-Spektren der Verbindungen (5-14) zeigen die erwarteten zwei Signale für die
axialen, bzw. äquatorialen Methylgruppen und zwei Resonanzen für die jeweiligen Methylengruppen.
Für die Verbindung (16) finden sich die Daten ebenfalls in der zugehörigen Tabelle. Die chemischen
Verschiebungen der Protonenresonanzen für die TMPDA- und PMDETA-Komplexe (5-15) sind in
Tabelle 15 aufgelistet.
Nr. Verbindung N-CH3 N-CH2 CH2CH2CH2 Al-CH3
5 (CO)5Cr-Ga[(Cl)(TMPDA)] 2.71; 2.67 3.52 2.16 -
6 (CO)5Cr-Ga[(I)(TMPDA)] 2.75; 2.70 3.37 2.21 -
7 (CO)5Cr-Ga[(Me)(TMPDA)] 2.70; 2.63 2.98 2.10 0.13
8 (CO)5Mo-Ga[(I)(TMPDA)] 2.70; 2.64 3.29 2.19 -
9 (CO)5W-Ga[(I)(TMPDA)] 2.73; 2.69 3.37 2.20 -
10 (CO)5W-Ga[(Me)(TMPDA)] 2.71; 2.51 3.16 2.12 0.21
11 (CO)4Fe-Ga[(Cl)(TMPDA)] 2.87; 2.82 3.80 2.21 -
12 (CO)4Fe-Ga[(Br)(TMPDA)] 2.85; 2.81 3.77 2.19 -
13 (CO)4Fe-Ga[(I)(TMPDA)] 2.87; 2.82 3.80 2.21 -
14 (CO)4Fe-Ga[(Me)(TMPDA)] 2.76; 2.58 3.16 2.21 0.13
Nr. Verbindung N-(CH3)2 N-(CH3)2 CH2CH2 N-CH3
15 (CO)4Fe-Ga[(PMDETA)]+I- 2.62 2.68 2.94 2.83
Tabelle 15: 1H-chemische Verschiebungen der Komplexe (5-15).
In Tabelle 16 sind die Kohlenstoffresonanzen [125] der Komplexe (5-15) zusammengestellt. Eine
unterschiedliche Substitution am Gallium-Zentrum scheint keine signifikanten Einfluß auf die
Resonanzen der Kohlenstoffkerne im TMPDA-Liganden zu haben. Die Resonanzen der
Carbonylkohlenstoffe entsprechen den erwarteten Lagen. Für den oktaedrischen Chromkomplex
werden zwei Signale für die Carbonylkohlenstoffatome beobachtet. Das Intensitätsverhältnis der
Signale beträgt 4:1, wobei wegen der Relaxationsproblematik von Kohlenstoffkernen keine
Zuordnung zu COtrans/COcis gemacht werden kann.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 47
Für die trigonal-bipyramidalen Eisenverbindungen konnte nur eine Carbonylkohlenstoffresonanz
beobachtet werden. Offensichtlich liegt in trigonal-bipyramidal konfigurierten Komplexen eine
stärkere Fluktuation vor, was zur magnetischen Äquivalenz der Carbonykohlenstoffatome führt.
Nr. Verbindung N-CH3 N-CH2 C-CH2-C Ga-CH3 CO
5 (CO)5Cr-Ga[(Cl)(TMPDA)] 46.7; 49.2 58.3 22.1 - 223.4; 226.1
6 (CO)5Cr-Ga[(I)(TMPDA)] 46.7; 49.7 59.4 22.7 - 225.3; 228.6
7 (CO)5Cr-Ga[(Me)(TMPDA)] 46.7; 48.6 57.4 23.6 6.5 227.9; 229.3
8 (CO)5Mo-Ga[(I)(TMPDA)] 46.2; 49.1 58.3 23.1 - 211.8; 215.0
9 (CO)5W-Ga[(I)(TMPDA)] 46.7; 49.6 59.4 22.9 - 201.7; 204.7
10 (CO)5W-Ga[(Me)(TMPDA)] 47.7; 50.6 58.8 20.9 8.8 206.4; 208.2
11 (CO)4Fe-Ga[(Cl)(TMPDA)] 49.9; 54.2 63.7 25.4 - 217.7
12 (CO)4Fe-Ga[(Br)(TMPDA)] 49.2; 53.9 62.5 24.9 - 219.8
13 (CO)4Fe-Ga[(I)(TMPDA)] 49.9; 54.2 63.7 25.4 - 221.7
14 (CO)4Fe-Ga[(Me)(TMPDA)] 51.9; 52.9 64.4 25.5 2.4 224.8
Nr. Verbindung N-CH3 N-(CH3)2 N-(CH3)2 N-CH2 CO
15 (CO)4Fe-Ga[(PMDETA)]+I- 48.6 53.9 54.1 55.8 217.9
Tabelle 16: 13C-chemische Verschiebungen der Komplexe (5-15).
3.3.2 Infrarot-Spektroskopie
Die IR-Spektren von (5-14) zeigen für die CO-Valenzschwingungen das typische Muster für
(CO)5ML-, bzw. (CO)4ML-Fragmente mit lokaler C4v-, bzw. C3v-Symmetrie und es werden im
wesentlichen die drei erwarteten Banden für die 2A1 + E Schwingungsmoden beobachtet. Die
Absorptionen der neutralen und ionischen Produkte (5-16) liegen im ν(CO)-Bereich zwischen 1877
und 2048 cm-1.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 48
2100 2000 1900 18000
20
40
60
80
100
120
(12)
Tran
smis
sion
[%
]
Wellenzahl [cm-1]
2100 2000 1900 18000
20
40
60
80
100
120
(14)
Abbildung 16: IR-Spektren von (12) und (14) (ν(CO)-Bereich)
2100 2000 1900 18000
20
40
60
80
100
(13)
Tra
nsm
issi
on [%
]
Wellenzahl [cm-1]
2100 2000 1900 18000
20
40
60
80
100
(15)
Abbildung 17: IR-Spektren von (13) und (15) (ν(CO)-Bereich)

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Gallane 49
In Tabelle 17 sind die Absorptionsbanden der CO-Valenzschwingungen [128] aller neuen neutralen
Komplexe (5-14), (16) und dem ionischen Komplex (15) zusammengestellt. Zur Veranschaulichung
sind in Abbildung 16 und Abbildung 17 IR-Spektren der ν(CO)-Bereiche einiger repräsentativer
Verbindungen abgebildet. Alle Spektren sind Lösungsdifferenzspektren und wurden in
Methylenchlorid-Lösung als Flüssigkeitsfilm zwischen NaCl-Fenstern aufgenommen.
Nr. Verbindung ν(CO) [cm-1] (A1(e); A1(a); E)
5 (CO)5Cr-Ga[(Cl)(TMPDA)] 2048 m; 1961 vs; 1913 vs
6 (CO)5Cr-Ga[(I)(TMPDA)] 2033 m; 1943 vs; 1917 vs
7 (CO)5Cr-Ga[(Me)(TMPDA)] 2013 m, 1935 vs; 1887 vs
8 (CO)5Mo-Ga[(I)(TMPDA)] 2030 m; 1942 vs; 1914 vs
9 (CO)5W-Ga[(I)(TMPDA)] 2034 m; 1949 vs; 1919 vs
10 (CO)5W-Ga[(Me)(TMPDA)] 2030 m; 1933 vs; 1894 vs
11 (CO)4Fe-Ga[(Cl)(TMPDA)] 2013 vs; 1930 m, sh; 1893 vs
12 (CO)4Fe-Ga[(Br)(TMPDA)] 2013 vs; 1931 m, sh; 1893 vs
13 (CO)4Fe-Ga[(I)(TMPDA)] 2012 vs; 1932 m, sh; 1893 vs
14 (CO)4Fe-Ga[(Me)(TMPDA)] 1994 vs; 1907 m, sh; 1870 vs
15 (CO)4Fe-Ga[(PMDETA)]+I- 1998 vs; 1920 vs, sh; 1887 vs
16 [(CO)4Fe-Ga(Et2NCH2CH2Et)]2 1998 vs; 1941 s, sh; 1916 vs; 1877 m, sh
Tabelle 17: Zusammenstellung der ν(CO)-Absorptionen [cm-1] der Komplexe (5-16).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 50
4 Synthese und Struktur carbonylhaltiger Übergangsmetall-
substituierter Indane
Literaturbekannte Beispiele für niedervalente donorstabilisierte E(I)X-Verbindungen (E = Al, Ga, In,
Tl; X = Halogen, Alkyl) in der Koordinationssphäre von Übergangsmetallcarbonyl-Fragmenten sind
sehr selten. Im Falle des Indiums konnten Behrens et al. 1977 durch Reaktion von InX3 mit
Na2M2(CO)10 (X = Cl, Br, I; M = Cr, Mo, W) die Komplexe (CO)5M-In[(X)(THF)] isolieren und
über die Kristallstruktur von (CO)5Cr-In[(Br)(THF)] berichten. [75] Verwandte
Übergangsmetallcarbonyl-Komplexe von Cr, Mo, W und Fe, die koordinierte In(I)-Spezies
enthalten sind ebenso bekannt. [141] Reger et al. veröffentlichten 1994 die Synthesen und
Strukturen von (CO)nM-In[HB(3,5-Me2pz)3] (M = Fe, W; n = 4, 5), die durch doppelte
Salzeliminierung zwischen den entsprechenden Dianionen [M(CO)n]2- und der
Pyrazolylboratoverbindung Cl2In[HB(3,5-Me2pz)3] erhalten wurden. [142] Über eine Reihe von
verwandten Verbindungen der allgemeinen Formel L(CO)nM-InX2(Do) wurde kürzlich berichtet.
[31,32,85,143-149]
4.1 Synthese donorstabilisierter Übergangsmetall-Indium-Systeme
Die Darstellung der Komplexe (CO)4Fe-In[(Br)(TMPDA)] (17) und (CO)4Fe-In[(Br)(PMDETA)]
(18) erfolgt prinzipiell nach der einheitlichen Vorgehensweise wie bei den analogen Aluminium- und
Galliumverbindungen: Aus der Carbonylverbindung Fe(CO)5 wird mittels der Reagenz K-
Selectrid [150] in THF das Eisenmetallatdianion K2[Fe(CO)4] erzeugt und für weitere Reaktionen
isoliert. Nach Zugabe von InBr3 und der Lewis-Base TMPDA (N,N,N´,N´-
Tetramethylpropylendiamin) oder PMDETA (N,N,N´,N´,N´´-Pentamethyldiethylentriamin) bilden
sich nach zweifacher Salzeliminierung die Neutralkomplexe (17) und (18).
Die Darstellung der Komplexe (17) und (18) verliefen durchweg glatt. Es wurden stets gelbe
Methylenchlorid-Lösungen erhalten, aus denen die gewünschten Produkte in sehr guten Ausbeuten in
Form gelber Quader auskristallisierten. War für weitere Umsetzungen eine hohe Reinheit nicht
vonnöten, dann konnte das nach Abdestillieren des Lösungsmittels verbleibende hellgelbe Pulver
abgetrennt werden.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 51
Die Produkte sind sehr gut in Methylenchlorid löslich, mäßig in Toluol und THF, unlöslich in Pentan
und in trockener Form unter Schutzgas unbegrenzt lagerfähig.
N
In N
Br
N
In N
Br
(CO) 4Fe
(CO) 4Fe
K2[Fe(CO)4]
1. THF + InBr 3
2. CH2Cl2 + TMPDA
- 2 KBr
1. THF + InBr 3
2. CH2Cl2 + PMDETA
- 2 KBr
(17)
(18)
N
Schema 4: Synthese der Verbindungen (17) und (18)
Nr. Verbindung M n X Donor
17 (CO)4Fe-In[(Br)(TMPDA)] In 4 Br TMPDA
18 (CO)4Fe-In[(Br)(PMDETA)] In 4 Br PMDETA
Tabelle 18: Zusammenstellung und Numerierungsschema der neuen Übergangsmetall-substituiertenIndane (17) und (18).
4.2 Strukturchemie donorstabilisierter Übergangsmetall-Indium-Systeme
Die röntgenstrukturanalytische Untersuchung eines gelben quaderförmigen Kristalls von (CO)4Fe-
In[(Br)(TMPDA)] (17) zeigt das Vorliegen einer direkten und unverbrückten Fe-In-Bindung. Das
Indium-Zentrum in (17) ist tetraedrisch von zwei Stickstoffliganden, dem Brom- und dem Eisen-
Zentrum koordiniert.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 52
Die In-N(1)/N(2)-Abstände sind nahezu gleichlang (230.3(1) und 230.7(8) pm) und erheblich länger
als die In-N-Abstände in der Verbindung (CO)4Fe-In[HB(3,5-Me2pz)3] gemessenen ((219.2(9),
219.5(10) und 221.1(10) pm). Der In-Br-Abstand beträgt 258.4(2) pm. Das Eisen-Zentrum ist
trigonal-bipyramidal von vier Carbonylliganden und dem Indium-Zentrum koordiniert. Der Winkel
In(1)-Fe(1)-C(2) liegt mit 176.6(4)° nahe am Wert von 180° für eine ideale lineare Anordnung. Die
äquatorialen Carbonylliganden sind nur leicht zum Indium-Zentrum hingeneigt (In-Fe-Ceq: 83.6(3)° -
86.9(2)°), was auch im verwandten Komplex (CO)4Fe-In[(Br)(PMDETA)] (In-Fe-Ceq: 82.5(2)° -
90.8(1)°) beobachtet wird (Regenschirmeffekt). Der Fe-In-Bindungsabstand von 249.7(2) pm ist
mit einer der kürzesten in der Literatur bekannten Abstände. Ein Vergleich mit den Literaturdaten
liefert folgende Daten: {InCl[Fe(CO)2Cp]2}2 255.3(1) und 255.8(1) pm,
{InCl(PMe2Ph)[Fe(CO)2Cp]2}2 258.2(1) und 256.9(2) pm,
[CpFe(CO)2][In(CH2CH2CH2NMe2)2] 263.9(8) pm, [In{Fe(CO)4}3]3- 263.3 pm, und
[Et4N][(2,2´-Bipyridin)InFe2(CO)8 254.5(3) und 255.2(3) pm. Da die Summe der Kovalenzradien
von Eisen und Indium 271 pm beträgt kann man hier von einer sehr kurzen Fe-In-Bindung
sprechen. [151] Die Bindung in den Komplexen (17) und (18) kann als Lewis-Säure/Base Addukt
verstanden werden. Das In(Br)TMPDA- oder In(Br)PMDETA-Fragment fungiert hier als Lewis-
Base in Gegensatz zum Lewis-Säure-Fragment (CO)4Fe (In→Fe).
Abbildung 18: Molekülstruktur von (CO)4Fe-In[(Br)(TMPDA)] (17) im Kristall(ORTEP-Darstellung).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 53
Tabelle 19: Ausgewählte Bindungslängen [pm] und -winkel [°] von (17).
Bindungslängen
In(1)-Fe(1) 249.7(2) Fe(1)-C(2) 175.9(12)
In(1)-N(1) 230.3(1) C(2)-O(2) 117.9(16)
In(1)-N(2) 230.7(8) N(1)-C(6) 149.1(14)
In(1)-Br(1) 258.4(2) N(1)-C(7) 150.2(14)
Fe(1)-C(1) 179.1(2) C(7)-C(8) 154.2(17)
Bindungswinkel
C(2)-Fe(1)-In(1) 176.6(4) N(1)-In(1)-Fe(1) 119.7(2)
C(3)-Fe(1)-In(1) 83.6(3) Br(1)-Inl(1)-N(1) 94.5(2)
C(4)-Fe(1)-In(1) 86.0(4) N(1)-In(1)-N(2) 91.4(3)
O(2)-C(2)-Fe(1) 176.10(15) C(11)-N(2)-C(10) 108.1(4)
Br(1)-In(1)-Fel(1) 126.50(7) C(7)-C(8)-C(9) 115.2(1)
Kristalle von geeigneter Qualität für eine röntgenstrukturanalytische Untersuchung von (CO)4Fe-
In[(Br)(PMDETA)] (18) konnten durch Kristallisation einer kleinen Substanzmenge aus einer
gesättigten Methylenchlorid-Lösung bei -30°C innerhalb weniger Tage erhalten werden. Die
Diffraktion eines hellgelben Kristalls von (18) zeigte auch hier das Vorliegen einer direkten und
unverbrückten Fe-In-Bindung. Die Länge der Fe-In-Bindung von 254.3(1) pm liegt im Bereich der
in der Literatur bekannten Bindungslängen. [152] Sie entspricht dabei gut der Summe der
Kovalenzradien der beiden Metalle (271.0 pm). Das Eisen-Zentrum ist trigonal-bipyramidal
koordiniert, wobei der Indium-Ligand eine axiale Position besetzt. Die Bindungswinkel der axialen
Carbonylgruppen (C-Fe-C) liegen im Bereich von 89.4(5) bis 98.3(4)°, die der äquatorialen
Carbonylgruppen im Bereich 112.8(4) bis 126.9(4)°. Die Koordinationsgeometrie der äquatorialen
Carbonylliganden erfolgt in einer Weise, daß die In-Fe-C-Winkel (85.7(3) to 88.2(3)°) deutlich
kleiner als 90°C sind, welches auch als „Regenschirmeffekt“ bezeichnet wird. Die In-Br-Bindung ist
mit 267.2(1) pm um 9 pm länger als in Verbindung (17). Diese Tatsache kann durch die Erhöhung
der Koordination am Indium-Zentrum erklärt werden. Die eigentliche Absicht, war die Darstellung
einer Indium analogen Verbindung von (15). Auch hier sollte das Halogenid über den Chelateffekt
der Base PMDETA aus der Verbindung verdrängt werden.
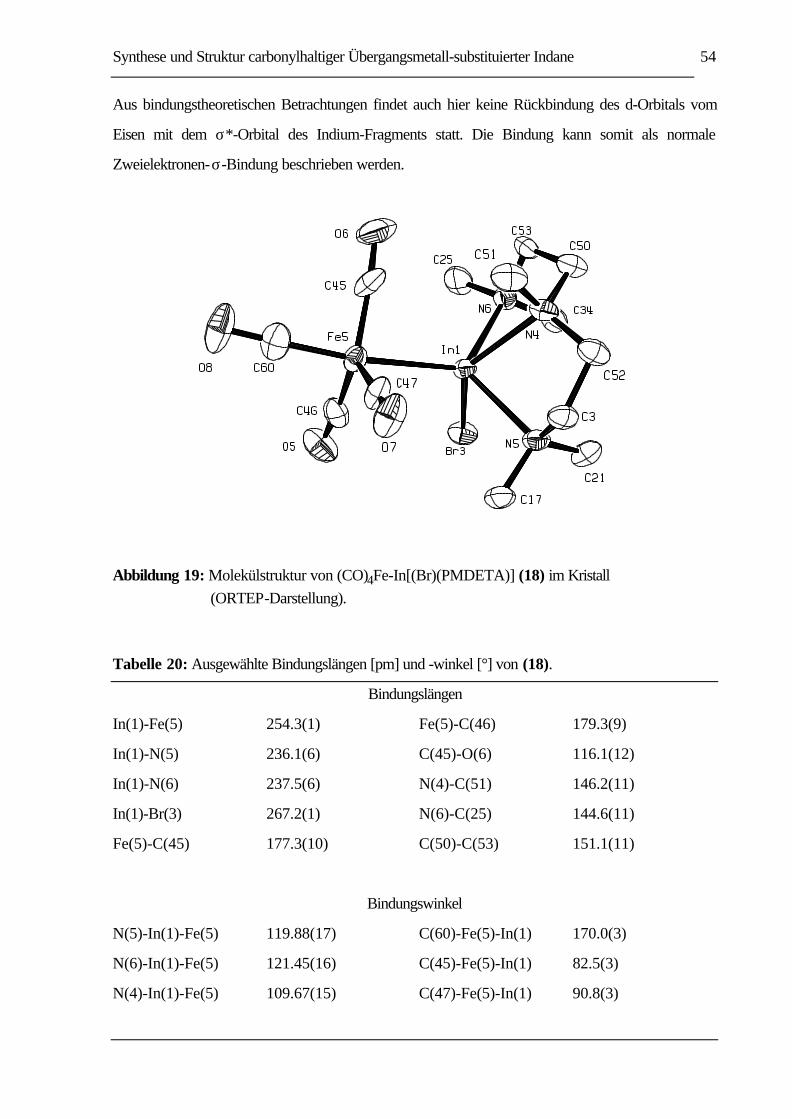
Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 54
Aus bindungstheoretischen Betrachtungen findet auch hier keine Rückbindung des d-Orbitals vom
Eisen mit dem σ*-Orbital des Indium-Fragments statt. Die Bindung kann somit als normale
Zweielektronen-σ-Bindung beschrieben werden.
Abbildung 19: Molekülstruktur von (CO)4Fe-In[(Br)(PMDETA)] (18) im Kristall(ORTEP-Darstellung).
Tabelle 20: Ausgewählte Bindungslängen [pm] und -winkel [°] von (18).
Bindungslängen
In(1)-Fe(5) 254.3(1) Fe(5)-C(46) 179.3(9)
In(1)-N(5) 236.1(6) C(45)-O(6) 116.1(12)
In(1)-N(6) 237.5(6) N(4)-C(51) 146.2(11)
In(1)-Br(3) 267.2(1) N(6)-C(25) 144.6(11)
Fe(5)-C(45) 177.3(10) C(50)-C(53) 151.1(11)
Bindungswinkel
N(5)-In(1)-Fe(5) 119.88(17) C(60)-Fe(5)-In(1) 170.0(3)
N(6)-In(1)-Fe(5) 121.45(16) C(45)-Fe(5)-In(1) 82.5(3)
N(4)-In(1)-Fe(5) 109.67(15) C(47)-Fe(5)-In(1) 90.8(3)

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 55
Fe(5)-In(1)-Br(3) 110.54(4) C(46)-Fe(5)-In(1) 85.9(3)
O(7)-C(47)-Fe(5) 177.0(7) N(4)-C(52)-C(3) 109.6(7)
Verbindung Abstand Fe-In
[pm]
ν(CO) [cm-1]
(CO)4Fe-In[(HB(3,5-Me2pz)3)] 246.3(2) 2011 s; 1919 vs; 1890 s; 1864 sh [a]
(CO)4Fe-In[(Br)(TMPDA)] 249.7(2) 2013 vs; 1959 w, sh; 1932 m, sh; 1895 vs [b]
µ-(CO){µ-
In[(CH2)3N(CH3)2]}[(Cp)(CO)Fe]2
253.6(1) 1920 vs; 1883 m; 1735 s [c]
(CO)4Fe-In[(Br)(PMDETA)] 254.3(1) 2004 vs; 1925 m; 1890 vs; 1866 vs [b]
(CO)6Fe2-[In(C(SiMe3)3)]3 258.2(1) 1987 vw; 1960 m; 1921 s; 1910 m [d]
[{(Me3Si)3C}In(µ-Cl)2{µ-
Fe(CO)4}In{C(SiMe3)3}]
262.4(2)
263.1(1)
2030; 2000; 1990; 1980 [e]
Cp(CO)2Fe-In[(CH2)3N(CH3)2]2 263.9(1) 1958 vs; 1904 vs [f]
Tabelle 21: Strukturell charakterisierte Eisen-Indium-Komplexe: σ(Fe-In)-Bindungslängen und IR-Daten. [a] Nujol. [b] Methylenchlorid. [c] Methylenchlorid. [d] Paraffin. [e] Toluol. [f]n-Pentan.
4.3 Spektroskopische Eigenschaften der Indium-Komplexe
4.3.1 NMR-Spektroskopie
In den 1H-NMR-Spektrum von (17) kann man zwei Signale für die Methylprotonen und ein bei
Raumtemperatur schlecht aufgelöstes, komplexes Kopplungsmuster für die Methylenprotonen des
TMPDA-Liganden beobachtet werden. Die Koordination des Liganden scheint also recht starr zu
sein, offenbar liegt der InN2C3-Sechsring in der Sesselkonformation vor (wie auch bei den
Aluminium- und Galliumkomplexen) und somit können axiale und äquatoriale Positionen
unterschieden werden. Das 13C-NMR-Spektrum von (17) zeigt die erwarteten zwei Signale für die
axialen, bzw. äquatorialen Methylgruppen und zwei Resonanzen für die jeweiligen Methylengruppen.
Die chemischen Verschiebungen der Protonenresonanzen für die TMPDA- und PMDETA-
Komplexe (17) und (18) sind in Tabelle 22 aufgelistet.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 56
Nr. Verbindung N-CH3 N-CH2 CH2-CH2-CH2
17 (CO)4Fe-In[(Br)(TMPDA)] 2.69; 2.62 3.58 2.20
18 (CO)4Fe-In[(Br)(PMDETA)] 2.71; 2.63 3.18 -
Tabelle 22: 1H-chemische Verschiebungen der Komplexe (17) und (18). [124]
In Tabelle 23 sind die Kohlenstoffresonanzen der Komplexe (17) und (18) zusammengestellt. Eine
unterschiedliche Substitution oder Veränderung der chelatisierenden Base scheint keinen signifikanten
Einfluß auf die Resonanzen der Kohlenstoffkerne zu haben. Die Resonanzen der
Carbonylkohlenstoffatome entsprechen den erwarteten Lagen. Für die trigonal-bipyramidalen
Eisenverbindungen konnte eine Carbonylkohlenstoffresonanz beobachtet werden. Im Gegensatz zu
den oktaedrischen Chrom-, Molybdän- und Wolfram-Komplexen (siehe Aluminium und Gallium),
wo zwei Signale für die Carbonylkohlenstoffatome beobachtet werden, liegt offensichtlich in trigonal-
bipyramidal konfigurierten Komplexen eine stärkere Fluktuation vor, welche zur magnetischen
Äquivalenz der Carbonylkohlenstoffatome führt.
Nr. Verbindung N-CH3 N-CH2 C-CH2-C CO
17 (CO)4Fe-In[(Br)(TMPDA)] 45.9; 49.2 60.1 22.4 217.1
18 (CO)4Fe-In[(Br)(PMDETA)] 49.2; 53.6; 54.7 60.3 - 217.1
Tabelle 23: 13C-chemische Verschiebungen der Komplexe (17) und (18). [125]
4.3.2 Infrarot-Spektroskopie
Die IR-Spektren von (17) und (18) zeigen für die CO-Valenzschwingungen das typische Muster für
(CO)4ML-Fragmente mit lokaler C3v-Symmetrie und es werden im wesentlichen die drei erwarteten
Banden für die 2A1 + E Schwingungsmoden beobachtet. Die Absorptionen der neutralen Produkte
(17) und (18) liegen im ν(CO)-Bereich zwischen 1866 und 2013 cm-1. Auch die isoelektronischen
donorstabilisierten Reger-Komplexe liegen exakt in diesem Wellenzahlenbereich.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 57
2100 2000 1900 180020
40
60
80
100
(17)
Tra
nsm
issi
on [%
]
Wellenzahl [cm-1]
2100 2000 1900 180020
40
60
80
100
(18)
Abbildung 20: IR-Spektren von (17) und (18) (ν(CO)-Bereich)

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 58
4.4 Exkurs: Reaktivität und Funktionalisierung
Die Salzeliminierung ist eine geeignete Methode zur Darstellung von Übergangsmetallsubstituierten
Alanen, Gallanen und Indanen. Die Anzahl der dargestellten Verbindungen in dieser Arbeit belegen
dies. Die thermodynamische Triebkraft dieser Reaktion ist die Alkalihalogenidabspaltung. Wie durch
Arbeiten in unserer Arbeitsgruppe gezeigt wurde, ist die Salzeliminierung eine sehr flexible, da
allgemein anwendbare Darstellungsmethode für übergangsmetallsubstituierte Organo-
Erdmetallkomplexe. Bei meist recht hohen Ausbeuten finden i. a. in der Gallium-Chemie keine
Mehrfachsubstitutionen oder die Bildung stabiler ionischer „At-Komplexe“ statt. Die Bildung von
Isocarbonyl-Strukturen wird für die übergangsmetallsubstituierten Gallane ebenfalls nicht beobachtet.
Ähnliches gilt für die Indium-Chemie, jedoch können sich dort durchaus Nachteile durch
Mehrfachsubstitutionen am Indiumtrihalogenid oder durch „At-Komplex-Bildung“ ergeben.
Grundsätzlich ist die Salzeliminierung jedoch auf Carbonylmetallate beschränkt und war auch im
Rahmen der vorliegenden Arbeit ausschließlich angewandte Methode zur Darstellung der
gewünschten Komplexe.
Ein Teilziel dieser Arbeit war es, eine mögliche Syntheseroute zu finden, die es erlaubt durch den
leicht zugänglichen Verbindungstypus (CO)nM-E[(X)(Ln)] (M = Fe, Cr, Mo, W; n = 4, 5; X = Cl,
Br, I, CH3; L = TMPDA, PMDETA; n = 2, 3) einen alternativen Zugang zu den Verbindungen der
Klasse (CO)nM-ER (R = Cp*, N(SiMe3)3, C(SiMe3)3) zu erhalten.
Im Einklang mit den theoretischen Analysen zu den Bindungsverhältnissen der M-E-Bindung lassen
sich die komplexgebundenen Fragmente E(X)L2 thermisch oder photochemisch nicht so glatt und
selektiv freisetzen wie erhofft. Die sehr polaren Bindungen M-E sind dafür zu stark. Wichtig für die
angestrebte Chemie am komplexgebundenen Zentrum E ist die leichte Abspaltung der
stabilisierenden Lewis-Base-Liganden und die gleichzeitige Einführung eines Restes R durch ein
geeignetes nucleophiles Übertragungsreagenz um zu niedrig koordinierten Zentren zu gelangen.
Anhand dieser Überlegungen erschien es ideal als Liganden Ln einen geeigneten Kronenether
einzusetzen. Eine mögliche Verbindung mit der allgemeinen Formel (CO)nM-E[(I)(Kronenether)]
oder (CO)nM-E[(Kronenether)]+I-, die in ähnlicher Form erstmals in Verbindung (15) vorlag, sollte
der 1. Schritt einer neuen Syntheseroute darstellen. Das Resultat dieser Bemühungen zeigt die
folgende Kristallstruktur der Verbindung (19).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 59
Die Idee war der Einsatz eines spezifischen Kronenethers (z. B. 12-Krone-4 für Li+) um bei
geeigneter Kombination im 2. Schritt der Synthese den Kronenether gegen einen anderen Rest (R =
Cp*, N(SiMe3)3, C(SiMe3)3) austauschen zu können (Abbildung 21). Dies kann nur im Sinne einer
thermodynamisch bevorzugten Lewis-Säure/Base-Reaktion erfolgen. Vielversprechende Hinweise
dafür liegen im Falle der Kronenether-Derivate vor (19). Für nachfolgende Arbeiten muß geprüft
werden, ob der nucleophile Angriff von sterisch anspruchsvollen Amid-Liganden oder auch von Cp*
mit den entsprechenden Kalium- oder Lithium-Reagenzien unter Übertragung des K+- oder Li+-
spezifischen Kronenethers in unpolaren Medien gelingt. Das Hauptaugenmerk sollte dabei auf der
Vermeidung von freien K+- oder Li+-Ionen liegen. Zwei sehr vielversprechende Ansätze liefern die
Schulte-Komplexe (CO)4Fe-Ga[(Cl)(THF)2] (mögliche Verdrängung des THF durch Kronenether)
und [(CO)5Cr-In[(Br)(THF)]∞ (in Substanz isoliert).
Die Möglichkeit der Verdrängung von Halogenid-Liganden an E(X)Ln-Fragmenten durch
mehrzähnige Chelatliganden wurde für eine Reihe von Übergangsmetall-Erdmetall-Verbindungen
untersucht, gelingt jedoch nicht in allen untersuchten Fällen (siehe (18)). Jedoch zeigt das Beispiel des
darstellten Komplexes (19) die prinzipielle Zugänglichkeit der kationischen Intermediate (CO)nM-
E[(Ln)]+. Entscheidenden Einfluß hat dabei die Wahl der mehrzähnigen Lewis-Base-Liganden und
des Solvens sowie die Größe des Halogenid- sowie des Erdmetallatoms.
(OC)4Fe GaO O
OO(OC)4Fe GaR
OO
O
OOO
+
(OC)4Fe
I + KR
[K(12-Krone-4)]+I-
+
I- (OC)4Fe GaR+ KR, Toluol
- [K(18-Krone-6)]+I-Ga
-
Abbildung 21: Schema einer potentiellen Stabilisierung von (CO)nM-E[(Ln)]+ durch Kronenether-Liganden und Übertragungsreaktionen.

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 60
Die röntgenstrukturanalytische Untersuchung eines gelblichen Kristalls von (19) zeigte das Vorliegen
einer ionischen Struktur. Wegen der größeren Übersichtlichkeit wurden Kation und Anion separat
als ORTEP-Bilder auf den nachfolgenden Seiten abgebildet.
Das Indium-Zentrum in (19) ist tetraedrisch von drei Bromliganden und den (CO)4Fe-InBr3-
Fragment umgeben. Die In-Br-Bindungslänge liegt mit den Werten 251.3(2) pm, 255.2(2) pm und
255.4(2) pm exakt in Bereich vergleichbarer Komplexe wie z. B. Cp(PPh3)Ni-InBr2(OPPh3)
(255.4(1) pm, 255.7(1) pm) oder Cp(CO)Ni-InBr2(Quinuclidin) (255.0(1) pm, 255.8(1) pm). Das
Eisen-Zentrum stark verzerrt-oktaedrisch von vier Carbonylliganden und den beiden Indium-Zentren
koordiniert. Der Fe-In-Bindungsabstand von 262.4(2) pm entspricht denjenigen Abständen die in
Tabelle 21 diskutiert wurden. Der Abstand von K+ im Kronenether entspricht mit durchschnittlich
274 pm den jeweiligen Literaturdaten.
Das Kation in dieser Struktur ist ungewöhnlich, da meist das Erdmetall von (CO)nM-Fragmenten
koordiniert wird wie in den Arbeiten von Huttner et al. gezeigt wurde ({[(CO)5Cr]2(µ-InBr)}2- [73]
und [(CO)5Cr=Tl=Cr(CO)5][K+-(2.2.2)Kryptand] [74]).
Abbildung 22: Molekülstruktur von [(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2 (19) im Kristall
(ORTEP-Darstellung) – (zur Vereinfachung nur das Anion [(CO)4Fe-(InBr3)2]2-).

Synthese und Struktur carbonylhaltiger Übergangsmetall-substituierter Indane 61
Abbildung 23: Molekülstruktur von [(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2 (19) im Kristall
(ORTEP-Darstellung) – (zur Vereinfachung nur das Kation [K(12-Krone-4)2]2)+.
Tabelle 24: Ausgewählte Bindungslängen [pm] und -winkel [°] von (19).
Bindungslängen
In(1)-Fe(1) 262.4(2) Fe(1)-C(1) 183.6(23)
In(1)-Br(1) 251.3(2) C(1)-O(1) 116.4(22)
In(1)-Br(2) 255.2(2) C(2)-O(2) 108.3(22)
In(1)-Br(3) 255.4(2) K(1)-O(29) 273.9(17)
Fe(1)-C(2) 182.6(21) K(1)-O(11) 274.3(19)
Bindungswinkel
Br(1)-In(1)-Fe(1) 111.57(7) In(1)-Fe(1)-In(1) 106.58(11)
Br(2)-In(1)-Fe(1) 111.13(6) In(1)-Fe(1)-C(1) 74.73(6)
Br(3)-In(1)-Fe(1) 120.81(7) In(1)-Fe(1)-C(2) 80.07(6)

C
INTERMETALLISCHE SCHICHTEN

Intermetallische Schichten 63
5 Intermetallische Schichten
5.1 MOCVD intermetallischer Schichten
Die Entwicklung neuer Materialien für die Mikroelektronik, bzw. der Halbleiterindustrie erfordert
zunehmend die interdisziplinäre Zusammenarbeit von Molekülchemikern, Festkörperchemikern und
Materialwissenschaftlern. Ein Anwachsen der Zahl von Publikationen zu diesem Thema in
einschlägigen materialwissenschaftlich orientierten Journalen und fachübergreifenden Tagungen
belegen diese Entwicklung. Gerade im Bereich der Dünnschicht-Technologie profitiert die
Mikroelektronik durch neue schonende Beschichtungsmethoden wie MOCVD (Metal Organic
Chemical Vapour Deposition) oder MOVPE (Metal Organic Vapour Phase Epitaxy) zum Beispiel
zur Darstellung von III/V-Halbleitermaterialien wie InP, InAs, GaAs, GaP, InGaAs und
InGaAsP. [153-155] Das grundlegende Kennzeichen der MOCVD- und MOVPE-
Beschichtungsmethoden ist das Konzept der molekularen Vorstufe (Precursor). Insbesondere
metallorganische Quellenmoleküle bergen hier ein großes Variationspotential. Dieses auf
Pionierarbeiten von Manasevit [156-158] basierende, erst während des zurückliegenden Jahrzehnts
erkannte technologische Potential metallorganischer Verbindungen stimulierte auch bestimmte
Bereiche der zugehörigen Grundlagenchemie beträchtlich. [159-170]Dem Molekülchemiker stehen
prinzipiell zwei Wege offen, um die Abscheidung binärer, ternärer und multinärer dünner Schichten
für Halbleiter, Metalle, Legierungen, Keramiken und Hartstoffen zu bewerkstelligen. Der eine Weg,
er auch bisher in der Technik ausschließlich gegangen wurde, umfaßt die Nutzung zweier oder
mehrerer unabhängiger Quellen zur Abscheidung anorganischer Schichten [171], wobei die
Kontrolle der Filmstöchiometrie durch Einstellung eines für die jeweilige Anwendung spezifischen
Molenbruchverhältnisses der Komponenten in der Gasphase erfolgt. Die Optimierung des
Abscheidungsprozesses hinsichtlich der Stöchiometrie und Reinheit der Schichten ist hierbei vor
allem ein meß- und regeltechnisches Problem. Die präzise Kenntnis und gezielte Variation von
Systemparametern wie Stoffströme, Prozeßdruck und Substrattemperatur sind dabei für den Erhalt
reproduzierbarer Wachstumsraten unerläßlich.

Intermetallische Schichten 64
Das auf Manasevits Arbeiten zurückgehende Lehrbuchbeispiel für diese Mehrkomponentenstrategie
kann die Erzeugung des III/V-Halbleiters Galliumarsenid durch MOVPE oder auch OMMBE
(Organo Metallic Molecular Beam Epitaxy) angeführt werden. [159,172]
Ga(CH3)3 + AsH3 GaAs + 3 CH4
550 - 650ºC
H2
Schema 5: Zielkomponentenstrategie zur Homo- bzw. Heteroepitaxie von GaAs
Der zweite molekülchemische Weg zu anorganischen Schichten verfolgt die Strategie des „single-
source-precursors“. Die zugrundeliegende Idee ist, zwei oder mehrere schichtkonstituierende Atome
in einem Quellenmolekül zu vereinigen, um somit Kontrolle über die Schichtstöchiometrie durch
Anpassung der Molekülstruktur des Precursors zu erhalten. Ein Vorteil dieses Konzeptes wäre, daß
der Systemparameter Stoffstromführung mehrerer Komponenten hier wegfällt und lediglich Druck-
und Temperaturführung von Bedeutung sind. Allerdings verhindern einige Nachteile die technische
Anwendung der Einkomponenten-Strategie. Diese sind vor allem der oft schwierige Zugang zu
geeigneten, flüchtigen Precursoren und anderseits der mit anwachsender Strukturkomplexität des
Precursors auch zunehmende Kontaminationsgrad an Fremdatomen in den Schichten. Eine Reihe
von Metallphasen, insbesondere Elementkombinationen aus Übergangsmetallen und schweren
Elementen der 3. Hauptgruppe sind gegenüber III/V-Halbleitern wie GaP, GaAs und GaSb
thermodynamisch stabil. [173]

Intermetallische Schichten 65
5.2 Übergangsmetall-Erdmetall-Komplexe als Einkomponenten-Precursoren
Um als potentielle Einkomponenten-Precursoren zur MOCVD intermetallischer Phasen in Betracht
zu kommen, muß eine metallorganische Komplexverbindung eine Reihe von Kriterien erfüllen. [64,
170, 183, 187] Die Erzeugung reiner intermetallischer Phasen aus Einkomponenten-Quellen verlangt
daher folgende Vorgaben:
a) heteroatomare Moleküle
b) passende Metallkombinationen
c) in der Ligandsphäre präformierte, chemisch inerte, leicht flüchtige Abgangsgruppen
d) niedrige Molekülmasse (akzeptabler Dampfdruck)
e) hoher Massenteil der Metalle in der Verbindung
f) gute Löslichkeit
g) einfacher synthetischer Zugang
h) langfristig unzersetzt lagerfähig
i) effiziente Reinigung des Precursors
Daß Precursoren der allgemeinen Formel L(CO)nM-E[(R)2(Do)] (L = Cp, CO, PMe3; n = 1-3; M
= Co, Fe, Cr, Mo, W, Ni; E = Al, Ga, In; R = Alkyl; Do (Donor) = N, O, P) [174] grundsätzlich
als Einkomponenten-Vorstufen für Kontaktmetallisierungen von III/V-Halbleitern geeignet sind,
wurde unter anderem durch Arbeiten von R. A. Fischer, M. Schulte, A. Miehr, J. Weiß und O.
Segnitz gezeigt. [174-190]
Dieses umfassende Anforderungsprofil schränkt die Zahl geeigneter Gruppen und Reste zum Aufbau
der Ligandensphäre um die jeweiligen Metallatome im erheblichen Umfang ein. Auf der d-Metall-
Seite sind dies vornehmlich π-Liganden wie CO, η2-C2H
4, η3-C
3H
5, η5-C
5H
5, und Lewis-Basen wie
PR3. Für die Metalle der 13. Gruppe (Al, Ga und In) kommen neben Zweielektronen-Lewis-
Donoren (OR2, NR
3, etc.) nur Alkyl- oder H-Substituenten in Betracht. Alle anderen M-X-
Bindungen, wie z.B. in Alkoxiden, β-Diketonaten, Amiden oder Halogeniden sind für eine
niederenergetische Pyrolyse auszuschließen. [166]

Intermetallische Schichten 66
Als ein weiteres Kriterium für einen erfolgreichen Einsatz in MOCVD-Anwendungen zeigt sich, daß
der β-H-Eliminierungsmechanismus bei der Molekülfragmentierung ein kinetisch bevorzugter
Reaktionsweg ist. So sind Alkylreste mit β-H-Atomen gegenüber allen anderen Alkylresten und
damit auch Aryl-Resten als Substituenten vorzuziehen. Darüber hinaus sollten die beiden
Metallzentren direkte M-E-Bindungen aufweisen und nicht über Brückenliganden miteinander
verknüpft sein, denn der Brückenligand wird mit großer Wahrscheinlichkeit in die wachsende Schicht
inkorporiert. [164, 166].
Die besten Aussichten auf Einsetzbarkeit in MOCVD-Prozessen zur Generierung von
intermetallischen Übergangsmetall-Erdmetall-Schichten hätten demnach Verbindungen, in denen
Organoerdmetallreste RmE (m = 1, 2) an Übergangsmetalle bzw. Übergangsmetall-Carbonyl-
Fragmente über direkte M-E-Bindungen fixiert sind. Demnach können bestimmte intermetallische
Phasen der Gruppe 13 Metalle mit Übergangsmetallen als epitaktische und gegen unerwünschte
Grenzflächenreaktionen inerte Metallisierungen für Schottky-Barieren und Ohmsche Kontakte in der
III/V-Halbleitertechnik eingesetzt werden. [173, 191, 192]
Die Abscheidung eines stabilen Metallfilms auf einem III/V-Material ist von Interesse für die
Entwicklung einer Vielzahl neuartiger elektronischer und photonischer Bauelemente, wie zum Beispiel
Heterobipolartransistoren (HEMT), resonanten Tunneldioden bzw. Transistoren (DBRT, RTBTs)
und Quantenkopf-Heterostrukturen. [166] Dünne Schichten aus den Aluminiden NiAl(GaAs) [193-
195] und MnAl(AlAs) [196] sowie CoGa(GaAs) [197] und PtGa(GaAs) [198] stellen hier ein
Anwendungspotential dar. Die meisten elementaren Metalle erweisen sich, im Gegensatz zu
Erdmetall-Übergangsmetall-Verbindungen als chemisch reaktiv gegenüber III/V-Halbleiter-
Oberflächen. Dieses führt zu Veränderungen an den Grenzschichten und somit zum Verlust der
Funktionsfähigkeit des elektronischen Bauteils. [199]
Eine große Problematik innerhalb der Precursorchemie resultiert aus den gegenläufigen Effekten der
Stabilität des Precursors und den kinetischen Verhältnissen der Zerfallschemie unter MOCVD-
Bedingungen. Übergangsmetall-Erdmetall-Komplexe (vor allem EIR-Fragmente) weisen häufig
sterisch anspruchsvolle organische Reste R (u.a. Cp*, C(SiMe3)3) auf, die der Stabilisierung des
Precursors dienen.

Intermetallische Schichten 67
Diese Liganden erschweren die Einsatzmöglichkeiten für MOCVD-Anwendungen, denn durch die
hohen Molekülmassen wird nur ein inakzeptabler Dampfdruck erreicht und/oder es kommt zur
Ausbildung von flüchtigen und thermisch stabilen Teilchen (wie z.B. Cp*Ga), die den Reaktor
unfragmentiert verlassen. Das Fragment [GaCp*] weist eine sehr hohe Stabilität auf, so daß ein
Erdmetallverlust bei der Abscheidung der galliumhaltigen intermetallischen Phase die Folge wäre. Es
können auch Umlagerungen oder Umverteilungsvorgänge bei den Liganden während der Pyrolyse
auftreten, die einen ähnlichen Effekt, nämlich den Verlust an Erdmetall, bewirken. [187]
Während [(CO)4CoIn(RN)2] (RN = CH2CH2CH2NMe2) schon bei niedrigen Temperaturen um
200°C Co1In1-Filme liefert, bildet sich im Fall der eng verwandten NiIn-Quelle [Cp(CO)NiIn(RN)2]
unter denselben Bedingungen flüchtiges und pyrolysestabiles [CpIn], das abtransportiert wird. Die
Cp-Übertragung auf das In-Atom hat die Folge, daß nickelreiche Ni1In1-x-Filme gebildet werden. Es
wird deutlich, daß die Metallzusammensetzung der Vorstufen in den abgeschiedenen Filmen nicht
unbedingt reproduziert wird, wobei aber aus Vorstufen mit einem Verhältnis E/M > 1 auch
erdmetallreiche Schichten erhalten werden. [170] Das Verhältnis von Ligandenabspaltung oder –
reorganisation, während des Molekülzerfalls an der heißen Oberfläche ist bestimmend für die
Schichtzusammensetzung, d.h. durch die Vorgabe der Liganden im Precursormolekül können
kinetisch bevorzugte Abbauwege vorgegeben und die gewünschten intermetallischen Phasen erzielt
werden. Diese Konstitutions- und Strukturvielfalt öffnet auch für die Zukunft eine große Palette von
Variationsmöglichkeiten zur Erzeugung von neuen Materialien mit Hilfe der MOCVD-Technik.
5.3 [(Cp*Al)Cr(CO)5] als Einkomponenten-Precursor für MOCVD
5.3.1 Synthese von [(Cp*Al)Cr(CO)5]
Die folgenden Arbeiten (Synthese und Auswertung) wurden in Kooperation mit Tobias Steinke
durchgeführt, [200] da die Herstellung der Edukte sehr zeitaufwendig waren. Durch diese
Zusammenarbeit war es möglich schnell aussagefähige Resultate zu erzielen.
Die Synthese von [(Cp*Al)Cr(CO)5] nach Schnöckel et al. 1999 [201] erfolgt in zwei
Reaktionsschritten. Im ersten Teil wird eine geeignete, leicht substituierbare Abgangsgruppe, in
diesem Fall Cycloocten, unter CO-Abspaltung an Chrom gebunden.

Intermetallische Schichten 68
Die Photolyse von Cr(CO)6 in Gegenwart des Olefins cis-Cycloocten (coe) liefert den Olefin-
Komplex [(η2-C8H14)Cr(CO)5] gemäß Schema 6 in einer Ausbeute von 24.1 %. [227]
Schema 6: Synthese von [(η2-C8H14)Cr(CO)5].
Die Ursache für die relativ geringe Produktmenge beruht auf die Einstellung eines Gleichgewichtes,
welches auf die Seite der Edukte verlagert ist. So tritt bei Behandlung von [(η2-C8H14)Cr(CO)5] mit
Kohlenmonoxid in einer Alkanlösung die schnelle und vollständige Rückreaktion zu C8H14 und
Cr(CO)6 ein. Deshalb ist es während der Durchführung notwendig, das gebildete Kohlenmonoxid
durch zeitweiliges Einleiten von Argon in die Reaktionslösung aus dem Gleichgewicht zu entfernen.
Dennoch wird bei der Kristallisation aus Pentan bei –78°C ein Gemisch des Produkts
[(coe)Cr(CO)5] und des Edukts Chromhexacarbonyl erhalten. Das gelblich kristalline Produkt kann
durch anschließende Sublimation bei 0°C und 10-2 Torr in reiner Form isoliert werden. Die Reinheit
von Pentacarbonyl(η2-cis-Cycloocten)chrom wurde mittels IR-Spektroskopie untersucht. Es zeigen
sich im wesentlichen drei Absorptionen im CO-Valenzschwingungsbereich (2070.8 (m), 1953.8 (s)
und 1948.8 cm-1 (sh)), die mit den angegebenen Literaturwerten gut überein stimmen und für
terminale Carbonylgruppen und Moleküle mit C4v-Symmetrie charakteristisch sind. [227]
Wie bereits eingangs erwähnt ist [(η2-C8H14)Cr(CO)5] geeignet Cr(CO)5 zu übertragen und somit
unterschiedliche LCr(CO)5-Derivate (L = THF, P(OMe)3, Piperidin, etc.) zugänglich zu
machen. [227] Im Hinblick auf Übergangsmetall-Erdmetall-Komplexe ist es von Interesse, ob auch
andere Elektronendonorliganden, wie zum Beispiel elektronenreiche EIR-Fragmente an die Chrom-
Einheit gebunden werden können.
+ Cr(CO)6hν 25ºCn-Pentan
Cr(CO)5 + CO

Intermetallische Schichten 69
Nachdem Fischer et al. 1997 den ersten Komplex mit einer terminalen Cp*Al-Einheit [(CO)4Fe-
AlCp*] synthetisiert hatten, [54] ist nun ein weiteres Beispiel für diese Verbindungsklasse publiziert
worden. Nach der Syntheseroute von Schnöckel et al. wird hierfür [(AlCp*)4] mit
[Cr(CO)5(C8H14)] bei 65°C in Toluol umgesetzt. Durch nachfolgendes langsames Abkühlen der
Reaktionslösung auf -30°C werden gelblich-grüne Kristalle von [Cp*AlCr(CO)5] erhalten. [201]
Schema 7: Synthese von [Cp*AlCr(CO)5].
Das Infrarot-Spektrum der Reaktionslösung zeigt die Abnahme der charakteristischen CO-
Valenzschwingungen für [(η2-C8H14)Cr(CO)5] und die Existenz neuer Carbonyl-Banden, die sich
dem Cr-Al-Komplex zuschreiben lassen (2038.7 (m), 1981.4 (m), 1913.8 cm-1 (vs)). Daneben
wurden kernresonanzspektroskopische Untersuchungen durchgeführt, anhand derer sich die
Substitutionsreaktion, durch Vergleich mit den in der Literatur genannten Daten, belegen läßt. [201]
Das 1H-Spektrum weist ein prägnantes Singulett bei 1.73 ppm auf, das im Vergleich zu [(Cp*Al)4]
zum höheren Feld hin verschoben ist. Die Resonanzen im 13C-Spektrum (8.9 und 116.0 ppm) sind
für die Methyl-C- und Ring-C-Atome charakteristisch und ebenfalls hochfeldverschoben. Das den
CO-Gruppen zugeordnete Signal erscheint bei 221.3 ppm. Besonders aussagekräftig und
charakteristisch für die Bildung des Cr-Al-Komplexes ist das 27Al-NMR-Spektrum. Hier zeigt sich
ein verbreitertes Signal, dessen chemische Verschiebung mit –25.2 ppm, im Vergleich zu [(Cp*Al)4]
(δ = -80.8 ppm) einen deutlichen Tieffeldshift erfährt.
Schnöckel et al. postulieren aufgrund röntgenstrukturanalytischer Ergebnisse für diesen Komplex eine
stark polare Bindung vom Typ Cp*Al2+⋅⋅⋅Cr2-(CO)5, die es ermöglichen soll, das Molekül unzersetzt
in die Gasphase zu überführen.
65ºC, Toluol
- C8H14Cr(CO)5 + 1/4 (Cp*Al)4 Cr
OC CO
COOC
COAl

Intermetallische Schichten 70
Massenspektroskopische Untersuchungen bestätigen dieses und lassen auf eine potentielle Eignung
des Komplexes [Cp*Al-Cr(CO)5] zur Generierung von Al/Cr-Filmen durch chemische
Dampfabscheidung schließen. Basierend auf den Ergebnissen von Schnöckel und Mitarbeitern sollte
dieser Aspekt in der vorliegenden Arbeit geprüft werden. Dazu wurden 140 mg des Precursors
hergestellt und wie nachfolgend beschrieben in einem MOCVD-Prozeß zur Abscheidung eines
intermetallischen Films eingesetzt.
5.3.2 Experimentalanordnung zur Vakuum-MOCVD
Zur Routineuntersuchung der Precursoren unter Vakuum-MOCVD Bedingungen wurde ein mit
Si/SiO2-Substrat (oberflächlich oxidierte Si-Wafer mit einer Fläche von ca. 100 mm2) beschickter,
horizontaler Heißwandrohrreaktor verwendet. Der Reaktorausgang war über eine Kühlfalle über
eine Öldrehschieberpumpe mit einer Hochleistungsvakuumpumpe verbunden. Auf inerte oder
reaktive Trägergase wurde verzichtet. Nach Stabilisierung des Basisvakuums (PBasis = 10-3 Torr)
und der Reaktortemperatur auf 450°C wurde die Temperatur des Precursorreservoirs langsam zum
Erreichen der gewünschten Sublimation bei 70°C hochgeregelt.
Abbildung 24: Vakuum-MOCVD-Apparatur zur Abscheidung eines Al-Cr-Films

Intermetallische Schichten 71
5.3.3 Abscheidung eines Al-Cr-Films
Der Precursor [(Cp*Al)Cr(CO)5] sublimierte im Vakuum durch das geheizte Rohr des verwendeten
horizontalen Heißwand-Rohrreaktors und ergab eine metallisch glänzende Abscheidung auf den
eingesetzten Si/SiO2-Substraten, die mittels Röntgendiffraktometrie und Rutherford-Rückstreu-
Spektrometrie (RBS) analysiert wurde. Das Metallverhältnis n(Cr)/n(Al), des im gesamten
Rohrinnenwand abgeschiedenen Films (ohne Substrate) wurde nach quantitativem Lösen in
verdünnter Salpetersäure mittels Atomemissionspektrometrie mit induktiv gekoppelten Plasma (ICP)
bestimmt. Zusätzlich wurde ein 1H-NMR-Spektrum des Precursorrückstands aufgenommen. Die
Zusammensetzung der kondensierten Abgase konnte aufgrund unzureichender Abgasmenge nicht
kernresonanzspektroskopisch analysiert werden (-196°C).
Das Röntgendiffraktogramm der Probe kann nur als erster Hinweis auf die Kristallinität und
Phasenzusammensetzung des Materials gewertet werden. Das Signal/Rauschverhältnis war schlecht
und die Reflexe sind breit, so daß der röntgenamorphe Anteil des erhaltenen Films wohl noch sehr
hoch ist bzw. die geringe Schichtdicke des Films als Ursache gelten dürfte.
Die Probe zeigt nur den Reflex 111 bei 2Θ = 38.194°, der im Bereich der Reflexlage für
elementares kubisches Aluminium liegt (2Θ = 38.472°). Aus der Reflexlage wurde der
Netzebenenabstand d = 235.4 pm und die Gitterkonstante a = 407.7 pm bestimmt, die mit den
Referenzdaten für Cr-Al-Phasen (u.a. Al9Cr4, Al17Cr9, Al13Cr2, Al8Cr5 etc.) aus der Datenbank
PDF des International Centers for Diffraction Data (ICDD) verglichen wurden. [202] Hierbei konnte
keine Übereinstimmung der erhobenen Werte mit den Vergleichsdaten festgestellt werden, so daß
die entsprechenden Angaben für Al herangezogen wurden. Die synthetisierte Phase weist eine im
Vergleich zu elementaren Aluminium vergleichbare, geringfügig größere Gitterkonstante auf
(Literaturwerte für Al: d = 233.8 pm; a = 404.9 pm), was durch geringe Anteile an Chrom in der
kubischen Aluminiumphase oder auch durch Defekte im Film zu erklären wäre.
Die Schichtzusammensetzung wurde auch mittels Rutherford-Rückstreu-Spektroskopie (RBS)
bestimmt. RBS ist eine kernphysikalische Standardmethode zur Materialcharakterisierung. Mit ihrer
Hilfe können Schichtdicken und Oberflächenbelegungungen quantitativ und standardfrei untersucht
werden.
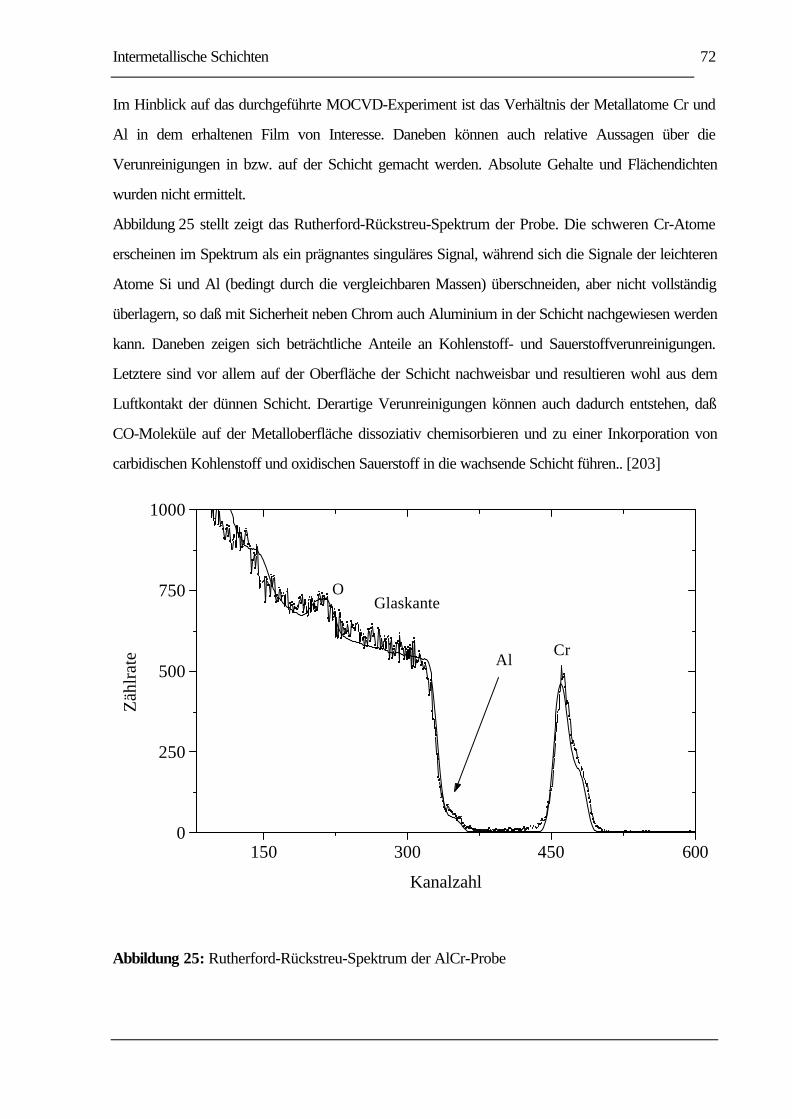
Intermetallische Schichten 72
Im Hinblick auf das durchgeführte MOCVD-Experiment ist das Verhältnis der Metallatome Cr und
Al in dem erhaltenen Film von Interesse. Daneben können auch relative Aussagen über die
Verunreinigungen in bzw. auf der Schicht gemacht werden. Absolute Gehalte und Flächendichten
wurden nicht ermittelt.
Abbildung 25 stellt zeigt das Rutherford-Rückstreu-Spektrum der Probe. Die schweren Cr-Atome
erscheinen im Spektrum als ein prägnantes singuläres Signal, während sich die Signale der leichteren
Atome Si und Al (bedingt durch die vergleichbaren Massen) überschneiden, aber nicht vollständig
überlagern, so daß mit Sicherheit neben Chrom auch Aluminium in der Schicht nachgewiesen werden
kann. Daneben zeigen sich beträchtliche Anteile an Kohlenstoff- und Sauerstoffverunreinigungen.
Letztere sind vor allem auf der Oberfläche der Schicht nachweisbar und resultieren wohl aus dem
Luftkontakt der dünnen Schicht. Derartige Verunreinigungen können auch dadurch entstehen, daß
CO-Moleküle auf der Metalloberfläche dissoziativ chemisorbieren und zu einer Inkorporation von
carbidischen Kohlenstoff und oxidischen Sauerstoff in die wachsende Schicht führen.. [203]
150 300 450 6000
250
500
750
1000
AlCr
OGlaskante
Zäh
lrat
e
Kanalzahl
Abbildung 25: Rutherford-Rückstreu-Spektrum der AlCr-Probe

Intermetallische Schichten 73
Das Ergebnis der Röntgendiffraktometrie legte nahe, daß sich bei der Abscheidung des Precursors
selektiv ein aluminiumreicher Film mit der Zusammensetzung Al1Cr1-x gebildet hat. Die Analyse der
Rutherford-Rückstreu-Spektrometrie (RBS) ist dazu gegensätzlich, wobei hier jedoch eine höhere
Meßgenauigkeit erreicht wird und vergleichende Aussagen über die relativen Anteile der Metall-
Atome in dem Film erlaubt. Diese Ergebnisse können für die Interpretation der
Schichtzusammensetzung herangezogen werden. Demnach haben sich auf den Si-Substraten
chromreiche Phasen Cr2-xAl1-x (-0.2 < x > 0.2) abgeschieden, die im hohen Maße durch C- und O-
Anteile verunreinigt sind.
Zusammenfassend läßt sich feststellen, daß ein intermetallischer Film während des MOCVD-
Experiments auf den Substraten abgeschieden wurde, der nicht die im Precursor vorgegebene Al:Cr
Stöchiometrie von 1:1 aufweist. Der offenbare Verlust an Aluminium ergibt sich aus der thermischen
Stabilität des Cp*-Al-Fragments, das die heiße Reaktionszone unfragmentiert verläßt und schließlich
abtransportiert wird, aber nicht nachgewiesen werden konnte. Dieses Ergebnis stimmt qualitativ mit
der ICP-Analyse überein, wo ein Metallverhältnis n(Cr) : n(Al) von 4.5 : 1 ermittelt wurde.
Eine eingehendere Betrachtung der Schichtzusammensetzung, des Precursorrückstands und der
damit verbundenen Reaktionsabläufe ist im Rahmen dieser Arbeit nicht möglich gewesen. Diese
notwendigen Analysen und die Generierung neuer Phasen sind für zukünftige Arbeiten vorgesehen.
5.3.4 Synthese und Struktur von [(CO)5M(η2-C8H14)] (M = Mo, W)
Um weitere MOCVD-Experimente durchführen zu können, sollten weitere Einkomponenten-
Precursoren hergestellt werden. Das Hauptinteresse lag dabei in der Darstellung der Zielmoleküle
[(Cp*Al)Mo(CO)5] und [(Cp*Al)W(CO)5]. Die Ausgangsverbindungen [(CO)5Mo(η2-C8H14]
und [(CO)5W(η2-C8H14] wurden analog der bekannten Chromverbindung hergestellt und
charakterisiert.
Die seit langem bekannte und mit hoher Quantenausbeute ablaufende Photosubstitution der
Hexacarbonyle von Metallen der 6. Nebengruppe ist mit einer Vielzahl von n-Donoren-Liganden
durchgeführt worden. [204-210]

Intermetallische Schichten 74
Nur vereinzelt wurde über die Einführung olefinischer Liganden berichtet; einige (η2-
Olefin)nM(CO)6-n-Komplexe (n = 1, 2) sind infrarotspektroskopisch in Reaktionslösungen
nachgewiesen worden, [211-213] doch nur wenige wurden in Substanz isoliert oder
röntgenstrukturanalytisch bestimmt. [214-216] Die geringe Stabilität der Komplexe (20) und (21)
äußert sich unter anderem darin, daß der olefinische Ligand bereits bei Raumtemperatur leicht durch
n-Donor-Liganden verdrängt wird. So reagieren verwandte olefinische Verbindungen z. B. mit
Triphenylphosphin oder Piperidin zu den entsprechenden LM(CO)5-Komplexen, die anhand ihrer
IR-Spektren [217] identifiziert wurden.
Durch Photolyse von Mo(CO)6 mit einem Überschuß an cis-Cycloocten in Pentan erhält man
Verbindung (20). Das Produkt kristallisiert, zusammen mit dem Edukt Mo(CO)6, nach etwa 20
Stunden bei –78°C aus. Das Hexacarbonyl wird mittels Sublimation von dem Produkt getrennt,
wobei ein nachfolgendes umkristallisieren nicht nötig war. Verbindung 20 kristallisiert in Form großer
farbloser Quader aus und ist als Feststoff uneingeschränkt unter einer Argonatmosphäre haltbar. In
Lösung dagegen zerfällt (20) ohne einen entsprechenden Reaktionspartner bei Raumtemperatur in
das Hexacarbonyl.
Mo(CO)6 + Pentan + hυ
W(CO)6 + Pentan + hυ
Mo(CO)6
W(CO)6
(20)
(21)
- CO
- CO
Schema 8: Synthese der Verbindungen (20) und (21).

Intermetallische Schichten 75
Geeignete Kristalle von [(CO)5Mo(η2-C8H14] (20) und [(CO)5W(η2-C8H14] (21) wurden
Einkristallröntgenstrukturen unterzogen. Die Ergebnisse der Analysen sind in den folgenden
Abbildungen und Tabellen zusammengestellt.
Abbildung 26: Molekülstruktur von [(CO)5Mo(η2-C8H14] (20) im Kristall (ORTEP-Darstellung).
Tabelle 25: Ausgewählte Bindungslängen [pm] und -winkel [°] von (20).
Bindungslängen
Mo(1)-C(1) 254.1(3) C(1)-C(2) 137.2(5)
Mo(1)-C(2) 252.7(3) C(12)-O(12) 115.1(4)
Mo(1)-C(11) 205.5(4) C(3)-C(4) 152.2(5)
Mo(1)-C(12) 197.6(4) C(4)-C(5) 154.0(6)
Mo(1)-C(14) 205.2(5) C(5)-C(6) 152.4(7)
Bindungswinkel
C(14)-Mo(1)-C(2) 88.91(14) C(2)-C(1)-Mo(1) 73.7(2)
C(11)-Mo(1)-C(2) 76.65(14) C(8)-C(1)-Mo(1) 114.5(2)
C(14)-Mo(1)-C(1) 94.85(15) C(3)-C(2)-Mo(1) 115.2(2)
C(11)-Mo(1)-C(1) 107.62(13) C(1)-C(8)-C(7) 113.0(3)

Intermetallische Schichten 76
C(2)-C(1)-C(8) 125.0(4) O(12)-C(12)-Mo(1) 179.0(4)
Das Molybdän-Zentrum in 20 ist oktaedrisch von vier Carbonyl- und zwei Kohlenstoffliganden
(Cycloolefin) koordiniert. Der Bindungsabstand Mo-C(12) des axialen Carbonylliganden liegt bei
197.6(4) pm. Die äquatorialen Carbonylliganden liegen im Bereich von 205.2(5) bis 205.5(4) pm.
Der Winkel O(12)-C(12)-Mo ist mit 179.0(4) pm als linear zu bezeichnen.
Abbildung 27: Molekülstruktur von [(CO)5W(η2-C8H14] (21) im Kristall (ORTEP-Darstellung).
Der olefinische Ligand in Verbindung (21) besetzt eine axiale Position am oktaedrisch koordinierten
Wolframatom. Dabei steht die Kohlenstoff-Kohlenstoff-Doppelbindung parallel zu den
Carbonylliganden C(5)-O(5) und C(1)-O(1) und senkrecht zu den Carbonylliganden C(3)-O(3) und
C(2)-O(2). Die Bindungsabstände (W-C(1), W-C(2), W-C(3), W-C(5)) der äquatorialen
Carbonylliganden sind mit durchschnittlich 202.3(7) pm nahezu gleich mit den entsprechenden
Abständen in (η2-Acrylsäuremethylester)2W(CO)4 (204.5(8) pm). Die Bindungsabstände W-C(11)
und W-C(88) liegen bei 249(2) pm und 251(2) pm. Diese Abstände sind etwa 20 pm länger als die
von vergleichbaren Bis(η2-olefin)tetracarbonylmolybdän- und -wolfram-Komplexe (230 pm). [213]

Intermetallische Schichten 77
Tabelle 26: Ausgewählte Bindungslängen [pm] und -winkel [°] von (21).
Bindungslängen
W(1)-C(11) 249(2) C(11)-C(88) 144(4)
W(1)-C(88) 251(2) C(4)-O(4) 124(3)
W(1)-C(1) 198(4) C(22)-C(33) 151(4)
W(1)-C(3) 202.3(7) C(33)-C(44) 153(5)
W(1)-C(4) 190(3) C(44)-C(55) 150(4)
Bindungswinkel
C(3)-W(1)-C(11) 97.8(8) C(88)-C(11)-W(1) 73.9(15)
C(3)-W(1)-C(88) 90.3(8) C(77)-C(88)-W(1) 113.6(16)
C(1)-W(1)-C(11) 76.7(10) C(22)-C(11)-W(1) 117.7(15)
C(1)-W(1)-C(88) 109.7(13) O(4)-C(4)-W(1) 178.0(19)
C(88)-C(11)-C(22) 124.6(19) C(88)-C(77)-C(66) 109.0(20)
Die Syntheseroute zur Darstellung der neuen Precursormoleküle wurde exakt zu der der
Chromverbindung eingehalten. Es gelang jedoch nicht die Zielmoleküle [(Cp*Al)Mo(CO)5] und
[(Cp*Al)W(CO)5] in dieser Arbeit herzustellen. Diese Moleküle aber in Zukunft ein Thema für
nachfolgende Arbeiten sein.

D
EXPERIMENTELLER TEIL

Experimenteller Teil 79
6 Experimenteller Teil
6.1 Allgemeine Arbeitstechniken
Alle nachfolgend beschriebenen Umsetzungen mit metallorganischen Verbindungen wurden unter
sorgfältigem Ausschluß von Luft und Feuchtigkeit in ausgeheizten Glasapparaturen (Schlenkrohr-,
Vakuum-Linie- und Glove-Box-Techniken) durchgeführt. Alle Apparaturen wurden vor Benutzung
im Hochvakuum ausgeheizt und mit Inertgas gespült. Als Inertgase wurden nachgereinigter Stickstoff
und Argon verwendet (Reinigung und Trocknung über BASF-Kupferkatalysator und Molekularsieb
4Å getrocknet. Geschlossene Apparaturen wurde über Quecksilberrückschlagventile an die
Abgasleitung angeschlossen. Tieftemperaturreaktionen wurden im Aceton/Trockeneis-, bzw. Im
Isopropanol/N2-Bad durchgeführt.
Die Trocknung, Sublimation und Destillation aller Verbindungen erfolgte im Hochvakuum
(Öldrehschieberpumpe der Fa. Edwards, p < 1.5·10-2 mbar bzw. Turbomolekularpumpe der Fa.
Edwards, p < 10-5 mbar). Weitere Details zur Arbeitstechnik finden sich in der Literatur. [218]
Die Kristallisation der Organometallverbindungen erfolgte, sofern nicht anders erwähnt, aus möglichst
konzentrierten Lösungen bei -30°C aus den jeweils angegebenen Lösungsmitteln. Mutterlaugen
wurden ein- bis zweimal nachkristallisiert, die Ausbeuten beziehen sich auf die vereinigten
Kristallfraktionen.
6.1.1 Schenkelfrittentechnik
Zur Synthese einiger hochempfindlicher Verbindungen wurde die Schenkelfrittentechnik eingesetzt.
Ein schematischer Aufbau einer solchen Apparatur ist in Abbildung 28 dargestellt. Der
Reaktionsschenkel A sowie die seitlich angesetzten Rundkolben mit Schnabelansatz B werden in der
Glovebox befüllt. Die Apparatur wird dann über das Anschlußstück C an die Hochvakuumanlage
angesetzt und sofort evakuiert. Werden leicht flüchtige Feststoffe oder Flüssigkeiten eingesetzt,
müssen diese vorher in der unter Argon-Überdruck stehenden Apparatur eingefroren werden. In die
evakuierte Anlage wird dann in den Reaktionsschenkel A oder auch in einen der Rundkolben B das
gewünschte Lösungsmittel direkt vom frischen Trocknungsmittel einkondensiert.

Experimenteller Teil 80
Durch Drehen der Apparatur um das Anschlußstück C bzw. der Schnabelkolben werden die
Reagentien der Vorratskolben B in den Reaktionsschenkel A gegeben. Ein eventuell erforderlicher
Lösungsmittelaustausch wird durch Ab- und Einkondensieren bewerkstelligt. Nach vollendeter
Reaktion kann die Reaktionslösung durch Drehen der Apparatur über die Fritte D in den Schenkel E
filtriert werden. Ein Rückstand kann durch Umkondensieren des Lösungsmittels gewaschen werden.
Die filtrierte Reaktionslösung wird in den seitlich angesetzten Kolben F überführt. Dieser wird unter
Argon-Schutzatmosphäre von der Apparatur abgehängt. Durch diese Arbeitstechnik kann das
Einschleppen von Luft oder Feuchtigkeit wesentlich minimiert werden.
E
FD
A
B
C
Abbildung 28: Schematische Darstellung einer Schenkelfrittenapparatur
6.1.2 Lösungsmittel
Die für präparative Arbeiten verwendeten Lösungsmittel wurden durch mehrtägiges Kochen unter
Rückfluß in Umlaufapparaturen unter Verwendung der üblichen Trocknungsmittel Abbildung 29
absolutiert, mit Stickstoff oder Argon gesättigt und über Molekularsieb 4Å aufbewahrt. Den
Lösungsmitteln Toluol, Tetrahydrofuran und Diethylether wurde Benzophenon als H2O/O2-Indikator
zugesetzt. Der Trocknungsgrad aller Lösungsmittel wurde durch Titration nach Karl Fischer
überprüft. Nur Lösungsmittel mit einem Wassergehalt von < 3 ppm kamen zum Einsatz.

Experimenteller Teil 81
Lösungsmittel Reinheitsgrad Trocknungsmittel
Diethylether chem. rein 1. KOH
2. Na/K-Legierung oder Na-Staub in Paraffin
Methylenchlorid chem. rein 1. Phosphorpentaoxid
2. Na/Pb-Legierung
n-Pentan chem. rein Na/K-Legierung oder Na-Staub in Paraffin
Tetrahydrofuran z. Synthese 1. KOH
2. Na/K-Legierung oder Na-Staub in Paraffin
Toluol z. Synthese Na/K-Legierung
Abbildung 29: Verwendete Trocknungs- und Lösungsmittel
6.2 Routineanalysenmethoden
6.2.1 Elementaranalytik
Die Elementzusammensetzung der Verbindungen wurden im mikroanalytischen Labor der Fakultät
für Chemie der Ruhr-Universität Bochum (Leitung: Frau Bartholomäus) durchgeführt, wobei ein C-,
H-, N-Analysenautomat vom Typ CHN-O-Rapid der Firma Heraeus mit
Wärmeleitfähigkeitsdetektion eingesetzt wurde.
6.2.2 Kernresonanzspektroskopie
Die Proben zur Aufnahme von Kernresonanzspektren wurden in hochreinen, deuterierten
Lösungsmitteln der Firma Merck (Uvasol®) und der Firma Deutero gelöst, die zuvor auf die oben
erwähnte Art und Weise getrocknet und mit Stickstoff bzw. Argon gesättigt wurden. Zur
Probenpräparation luft- oder feuchtigkeitsempfindlicher Substanzen wurden die Lösungsmittel direkt
in die NMR-Rohre einkondensiert und diese am Hochvakuum abgeschmolzen.

Experimenteller Teil 82
Zur Aufnahme der Spektren wurden folgende Geräte benutzt:
• Bruker NMR-Spektrometer DPX-400
• Bruker NMR-Spektrometer AC-200
• Bruker NMR-Spektrometer DPX-200
• Bruker NMR-Spektrometer DRX-400
Die Angabe der chemischen Verschiebung δ erfolgt in ppm relativ zur eingestrahlten Frequenz,
wobei bei 1H-NMR-Spektren das 1H-Restsignal des deuterierten Lösungsmittels als interner
Standard verwendet wird (
Abbildung 30). Alle 13C-NMR-Spektren sind, sofern nicht anders angegeben,
protonenbreitbandentkoppelt.
1H 13C
CDCl3 7.24 77.0
CD2Cl2 5.32 53.5
C6D6 7.16 128.0
d6-Aceton 2.04 29.8, 206.3
d8-THF 1.73/3.58 25.5, 67.7
Abbildung 30: Protonenrestsignale bzw. Lösungsmittelsignale der jeweiligen Solventien.
Für die Angabe der Signalmultiplizitäten werden folgende Abkürzungen verwendet: s = Singulett, d =
Dublett, t = Triplett, q = Quartett, m = Multiplett, br = breit. Bei den 1H-NMR-Spektren ist
außerdem die Anzahl der Protonen pro Signal angegeben. Die Werte der Kopplungskonstanten
werden in Hz angegeben und sind als Beträge zu verstehen. Die Vorzeichen der
Kopplungskonstanten wurden nicht bestimmt.
6.2.3 Infrarotspektroskopie
Die IR-Spektren wurden als Lösung der Verbindung zwischen CaF2- oder NaCl-Fenstern an einem
Perkin-Elmer 1720X FT-IR-Spektrometer aufgenommen. Die abgebildeten Spektren sind

Experimenteller Teil 83
Lösungsdifferenzspektren. Die Küvette wurde im Trockenschrank bei 120°C ausgeheizt und vor der
Befüllung intensiv mit Inertgas gespült.

Experimenteller Teil 84
Für die Angabe der Signalintensitäten werden folgende Abkürzungen verwendet: vs = sehr stark, s =
stark, m = mittel, w = schwach, vw = sehr schwach, sh = Schulter.
6.2.4 Siedepunkte und Sublimationspunkte
Angegebene Siede- und Sublimationspunkte sind entsprechend der Ungenauigkeit der
Vakuummessung an einer präparativen Anlage mit einem relativ großen Fehler behaftet
(Fehlerschätzung: 5°C, ± 100 m Torr).
6.2.5 Einkristall-Röntgenstrukturanalysen
Die Einkristall-Strukturanalysen der vorliegenden Verbindungen wurden von Herrn Dr. Klaus Merz,
Lehrstuhl für Anorganische Chemie I der Ruhr-Universität Bochum und von Frau Manuela Winter,
Lehrstuhl für Analytische Chemie der Ruhr-Universität Bochum durchgeführt.
Die jeweiligen Meßgeräte, Meßparameter, die kristallographischen Daten und Einzelheiten zur
Lösung und Verfeinerung des Datensatzes sind in Abschnitt E (Tabellen) aufgeführt.
6.3 Arbeitsvorschriften und analytische Daten
6.3.1 Ausgangsverbindungen und Reagenzien
Zur Darstellung der metallorganischen Ausgangsprodukte wurden handelsübliche Basischemikalien
ohne weitere Reinigung eingesetzt. Kommerziell nicht erhältliche Laborpräparate sind, versehen mit
den Literaturzitaten bzw. den angewandten Arbeitsvorschriften, nachfolgend aufgelistet und wurden
diesen Vorschriften entsprechend synthetisiert. Anschließend wird die präparative Vorgehensweise
zur Synthese der neuen Verbindungen ausführlich mit den analytischen Daten beschrieben.
Dikalium-tetracarbonylferrat(2-), K2[Fe(CO)4] [219]
Dikalium-pentacarbonylchromat(2-), K2[Cr(CO)5] [65,220]
Dikalium-pentacarbonylmolybdat(2-), K2[Mo(CO)5] [65,220]
Dikalium-pentacarbonylwolframat(2-), K2[W(CO)5] [65,220]
Pentamethylcyclopentadienylaluminiumchlorid, [Cp*AlCl2]2 [221]
Pentamethylcyclopentadienylgalliumiodid, [Cp*GaI2]2
Methylgalliumdichlorid, Cl2GaMe [222]

Experimenteller Teil 85
Dimethylgalliumchlorid, ClGaMe2 [222,223]
Dimethylaluminiumchlorid, ClAlMe2 [222,223]
GaBr3 [224-226]
GaI3 [224-226]
[(CO)5Cr(η2-C8H14] [227]
Cp*K [228,229]
Cp*SiMe3 [230]
Cp*Al [231,232]
Cp*Ga [233]
KC8 [234]
VORSICHT: Galliumtrichlorid (GaCl3) ist stark Lewis-azide und reagiert heftig mit jedem Donor
unter beträchtlicher Wärmeentwicklung, so auch beim Lösen in Donorsolventien. Die Lösung von
Galliumtrichlorid sollte daher stets unter Kühlung (-78°C) erfolgen. Beim Arbeiten mit
Galliumtrichlorid sind mehrere zusätzliche Punkte zu beachten: 1. Galliumtrichlorid greift NEOPREN-
Handschuhe der Glove-Boxen an. 2. Das Abwiegen von Galliumtrichlorid kann nicht auf Alufolie
erfolgen, da auch diese sofort zersetzt wird. 3. Für die Lagerung von Galliumtrichlorid haben sich
Teflongefäße mit Schraubverschluß bewährt. Andere Kunststoffe werden ebenso angegriffen, wie
alle gebräuchlichen Arten von Schliff-Fett, welches im allgemeinen schon nach kurzer Dauer zum
„Fressen“ der Glasschliffe führt.

Experimenteller Teil 86
6.3.2 Neue Verbindungen
Übergangsmetall-Aluminium-Komplexe
Allgemeine Arbeitsvorschrift:
Zu einer auf -78°C gekühlten Suspension von 2 mmol des Übergangscarbonylmetallats K2[M(CO)5]
(M = Cr, Mo) oder K2[Fe(CO)4] in 40 ml THF wird eine auf -78°C vorgekühlte THF-Lösung von
2 mmol eines Methylaluminiumdichlorids oder Aluminiumtrihalogenids (AlCl3, AlI3) gegeben.
Alternativ können auch 4 mmol eines Dimethlyaluminiumchlorids zugegeben werden. Nach der
Zugabe wird das Kühlbad entfernt, die Reaktionslösung auf Raumtemperatur erwärmt und 1 h
gerührt bevor die chelatisierende Base (TMPDA) im leichten Überschuß (2.1 mmol) zugegeben
wird. Nach einer halben Stunde Reaktionszeit wird das Lösungsmittel im Hochvakuum eingeengt,
der Rückstand mit 40 ml Methylenchlorid aufgenommen und intensiv gerührt. Die Methylenchlorid-
Lösung wird filtriert (meist 2 mal) und das Lösungsmittel wiederum im Hochvakuum bis zur Trockne
eingeengt. Es werden weiße, gelbliche, grünliche oder auch rötliche mikrokristalline Pulver erhalten.
Die Ausbeuten liegen durchweg bei 92-95% analysenreiner Substanz.
Kristallisation:
Bei den Tetracarbonyleisen-Verbindungen hat sich die Diffusionskristallisation bewährt. Eine
konzentrierte Methylenchlorid-Lösung (2 mmol/10 ml) wird mit 10 ml Toluol vorsichtig überschichtet
und bei Kühlschranktemperatur erschütterungsfrei gelagert. Bei den Pentacarbonylmetall-
Verbindungen war die Tieftemperaturkristallisation bei -30°C aus konzentrierten Methylenchlorid-
Lösungen (2 mmol/10 ml) äußerst erfolgreich. Das Schlenkrohr sollte dabei möglichst waagrecht
liegen. Innerhalb eines Tages bilden sich meist nadel- oder quaderförmige Kristalle mit Kantenlängen
bis zu 8 mm. Das Aufbewahren der Kristalle in der Mutterlauge für kristallographische Zwecke ist
sinnvoll. Bei vollständigem Entfernen des Lösungsmittels werden die Kristalle trüb und verlieren das
koordinierende Lösungsmittelmolekül (THF, CH2Cl2). Hier sollte eine Kristallpräparation bei tiefen
Temperaturen und unter einer Methylenchlorid-Atmosphäre oder THF-Atmosphäre vorgenommen
werden. Ansonsten wird die Mutterlauge abkanüliert, weiter eingeengt und zur Nachkristallisation
erneut gekühlt. Insgesamt liegen die Ausbeuten an einkristallinem Material durchweg über 88%.

Experimenteller Teil 87
Chloro(pentacarbonylchrom)(tetramethylpropylendiamin)aluminum (1)
[(CO)5Cr-Al(Cl)(TMPDA)]: Aus 2 mmol K2[Cr(CO)5] (540 mg), 2 mmol AlCl3 (267 mg) und
2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 692 mg (90%) gelbliche
nadelförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C12H18AlClCrN2O5: M = 384.42 (berechnet) C 37.45, H 4.68, N 7.28; (gefunden) C
37.12, H 4.49, N 7.18. IR (CH2Cl2, cm-1): ν(CO) = 2014 (m), 1924 (vs), 1874 (vs). 1H-NMR
(250 MHz, CD2Cl2, 298 K, δ ppm): 3.52 (m, 4H, NCH2), 2.72 (s, 6H, N(CH3)2), 2.69 (s, 6H,
N(CH3)2), 2.19 (m, 2H, CH2CH2CH2). 13C-NMR (63 MHz, CD2Cl2, 298 K, δ ppm): 21.5
(CH2CH2CH2), 46.2 (NMe2), 49.2 (NMe2), 58.8 (NCH2), 224.3, 228.6 (Cr-CO).
Chloro(tetracarbonyleisen)(tetramethylpropylendiamin)aluminum (2)
[(CO)4Fe-Al(Cl)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol AlCl3 (267 mg) und
2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 634 mg (88%) farblose
nadelförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C11H18ClFeAlN2O4: M = 360.27 (berechnet) C 36.64, H 4.99, N 7.77; (gefunden) C
36.44, H 4.77, N 7.54. IR (CH2Cl2, cm-1): ν(CO) = 1999 (vs), 1914 (m, sh), 1871 (vs). 1H-
NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.24 (t, 3J = 23.7 Hz, 4H, NCH2), 2.88 (s, 6H,
N(CH3)2), 2.78 (s, 6H, N(CH3)2), 2.44 (m, 2H, CH2CH2CH2). 13C-NMR (100 MHz,
CD2Cl2, 298 K, δ ppm): 25.2 (CH2CH2CH2), 52.2 (NMe2), 54.7 (NMe2), 64.8 (NCH2),
224.2 (Fe-CO).
Iodo(tetracarbonyleisen)(tetramethylpropylendiamin)aluminum (3)
[(CO)4Fe-Al(I)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol AlI3 (900 mg) und 2.1
mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 804 mg (89%) farblose nadelförmige
Kristalle aus Methylenchlorid bei -30°C.
EA für C11H18FeAlIN2O4: M = 451.72 (berechnet) C 29.22, H 3.98, N 6.20; (gefunden) C
29.14, H 3.77, N 6.04. IR (CH2Cl2, cm-1): ν(CO) = 1992 (vs), 1904 (m, sh), 1863 (vs). 1H-
NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.30 (t, 3J = 23.7 Hz, 4H, NCH2), 2.71 (s, 6H,

Experimenteller Teil 88
N(CH3)2), 2.68 (s, 6H, N(CH3)2), 2.17 (m, 2H, CH2CH2CH2). 13C-NMR (100 MHz,
CD2Cl2, 298 K, δ ppm): 24.0 (CH2CH2CH2), 45.2 (NMe2), 49.7 (NMe2), 58.0 (NCH2),
223.9 (Fe-CO).
Methyl(tetracarbonyleisen)(tetramethylpropylendiamin)aluminum (4)
[(CO)4Fe-Al(Me)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol MeAlCl2 (226 mg)
und 2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 611 mg (90%) farblose
nadelförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C12H21FeAlN2O4: M = 339.82 (berechnet) C 42.36, H 6.18, N 8.24; (gefunden) C
42.04, H 6.01, N 7.94. IR (CH2Cl2, cm-1): ν(CO) = 1995 (vs), 1908 (m, sh), 1870 (vs). 1H-
NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.18 (t, 3J = 22.7 Hz, 4H, NCH2), 2.79 (s, 6H,
N(CH3)2), 2.58 (s, 6H, N(CH3)2), 2.22 (m, 2H, CH2CH2CH2), 0.17 (s, 3H, AlCH3). 13C-
NMR (100 MHz, CD2Cl2, 298 K, δ ppm): 1.5 (AlCH3), 25.2 (CH2CH2CH2), 51.9 (NMe2),
52.7 (NMe2), 64.8 (NCH2), 224.2 (Fe-CO).

Experimenteller Teil 89
Übergangsmetall-Gallium-Komplexe
Allgemeine Arbeitsvorschrift:
Zu einer auf -78°C gekühlten Suspension von 2 mmol des Übergangscarbonylmetallats K2[M(CO)5]
(M = Cr, Mo, W) oder K2[Fe(CO)4] in 40 ml THF wird eine auf -78°C vorgekühlte THF-Lösung
von 2 mmol eines Methylgalliumdichlorid oder Galliumtrihalogenid (GaCl3, GaBr3, GaI3) gegeben.
Alternativ können auch 4 mmol eines Dimethlygalliumchlorids zugegeben werden. Nach der Zugabe
wird das Kühlbad entfernt, die Reaktionslösung auf Raumtemperatur erwärmt und 1 h gerührt bevor
die chelatisierende Base (TMPDA, PMDETA) im leichten Überschuß (2.1 mmol) zugegeben wird.
Nach einer halben Stunde Reaktionszeit wird das Lösungsmittel im Hochvakuum eingeengt, der
Rückstand mit 40 ml Methylenchlorid aufgenommen und intensiv gerührt. Die Methylenchlorid-
Lösung wird filtriert (meist 2 mal) und das Lösungsmittel wiederum im Hochvakuum bis zur Trockne
eingeengt. Es werden weiße, gelbliche oder auch grünliche mikrokristalline Pulver erhalten. Die
Ausbeuten liegen durchweg bei 90-95% analysenreiner Substanz.
Kristallisation:
Bei einigen der Tetracarbonyleisen-Verbindungen hat sich die Diffusionskristallisation bewährt. Eine
konzentrierte Methylenchlorid-Lösung (2 mmol/10 ml) wird mit 10 ml Toluol vorsichtig überschichtet
und bei Kühlschranktemperatur erschütterungsfrei gelagert. Bei den Pentacarbonylmetall-
Verbindungen war die Tieftemperaturkristallisation bei -30°C aus konzentrierten Methylenchlorid-
Lösungen (2 mmol/10 ml) äußerst erfolgreich. Das Schlenkrohr sollte dabei möglichst waagrecht
liegen. Innerhalb eines Tages bilden sich meist nadel- oder quaderförmige Kristalle mit Kantenlängen
bis zu 10 mm. Das Aufbewahren der Kristalle in der Mutterlauge für kristallographische Zwecke ist
sinnvoll. Ansonsten wird die Mutterlauge abkanüliert, weiter eingeengt und zur Nachkristallisation
erneut gekühlt. Insgesamt liegen die Ausbeuten an einkristallinem Material durchweg über 85%.
Chloro(pentacarbonylchrom)(tetramethylpropylendiamin)gallium (5)
[(CO)5Cr-Ga(Cl)(TMPDA)]: Aus 2 mmol K2[Cr(CO)5] (540 mg), 2 mmol GaCl3 (352 mg) und
2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute 777 mg (91%) farblose
quaderförmige Kristalle aus Methylenchlorid bei -30°C.

Experimenteller Teil 90
EA für C12H18ClCrGaN2O5: M = 427,16 (berechnet) C 33.71, H 4.21, N 6.55; (gefunden) C
33.58, H 4.09, N 6.54. IR (CH2Cl2, cm-1): ν(CO) = 2048 (m), 1961 (vs), 1913 (vs). 1H-NMR
(300 MHz, CD2Cl2, 298 K, δ ppm): 3.52 (t, 3J = 7.1 Hz, 4H, NCH2), 2.71 (s, 6H, N(CH3)2),
2.67 (s, 6H, N(CH3)2), 2.16 (m, 2H, CH2CH2CH2). 13C-NMR (75 MHz, CD2Cl2, 298 K, δ
ppm): 22.1 (CH2CH2CH2), 46.7 (NMe2), 49.2 (NMe2), 58.3 (NCH2), 223.4, 226.1 (Cr-CO).
Iodo(pentacarbonylchrom)(tetramethylpropylendiamin)gallium (6)
[(CO)5Cr-Ga(I)(TMPDA)]: Aus 2 mmol K2[Cr(CO)5] (540 mg), 2 mmol GaI3 (900 mg) und 2.1
mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 933 mg (90%) farblose quaderförmige
Kristalle aus Methylenchlorid bei -30°C.
EA für C12H18CrGaIN2O5: M = 518,61 (berechnet) C 27.76, H 3.47, N 5.40; (gefunden) C
27.68, H 3.29, N 5.24. IR (CH2Cl2, cm-1): ν(CO) = 2033 (m), 1943 (vs), 1917 (vs). 1H-NMR
(300 MHz, CD2Cl2, 298 K, δ ppm): 3.37 (t, 3J = 6.2 Hz, 4H, NCH2), 2.75 (s, 6H, N(CH3)2),
2.70 (s, 6H, N(CH3)2), 2.21 (m, 2H, CH2CH2CH2). 13C-NMR (75 MHz, CD2Cl2, 298 K, δ
ppm): 22.7 (CH2CH2CH2), 46.7 (NMe2), 49.7 (NMe2), 59.4 (NCH2), 225.3, 228.6 (Cr-CO).
Methyl(pentacarbonylchrom)(tetramethylpropylendiamin)gallium (7)
[(CO)5Cr-Ga(Me)(TMPDA)]: Aus 2 mmol K2[Cr(CO)5] (540 mg), 4 mmol ClGaMe2 (540 mg)
und 2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 748 mg (92%) farblose
quaderförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C13H21CrGaN2O5: M = 406,71 (berechnet) C 38.36, H 5.16, N 6.88; (gefunden) C
38.18, H 5.09, N 6.54. IR (CH2Cl2, cm-1): ν(CO) = 2013 (m), 1935 (vs), 1887 (vs). 1H-NMR
(300 MHz, CD2Cl2, 298 K, δ ppm): 2.98 (t, 3J = 7.1 Hz, 4H, NCH2), 2.70 (s, 6H, N(CH3)2),
2.63 (s, 6H, N(CH3)2), 2.10 (m, 2H, CH2CH2CH2), 0.13 (s, 3H, GaCH3). 13C-NMR (75
MHz, CD2Cl2, 298 K, δ ppm): 6.5 (GaCH3), 23.6 (CH2CH2CH2), 46.7 (NMe2), 48.6
(NMe2), 57.4 (NCH2), 227.9, 229.3 (Cr-CO).

Experimenteller Teil 91
Iodo(pentacarbonylmolybdän)(tetramethylpropylendiamin)gallium (8)
[(CO)5Mo-Ga(I)(TMPDA)]: Aus 2 mmol K2[Mo(CO)5] (628 mg), 2 mmol GaI3 (900 mg) und
2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: mg (%) farblose quaderförmige
Kristalle aus Methylenchlorid bei -30°C.
EA für C12H18MoGaIN2O5: M = 544.83 (berechnet) C 26.43, H 3.30, N 5.14; (gefunden) C
26.21, H 3.19, N 5.02. IR (CH2Cl2, cm-1): ν(CO) = 2030 (m), 1942 (vs), 1914 (vs). 1H-NMR
(300 MHz, CD2Cl2, 298 K, δ ppm): 3.29 (t, 3J = 7.1 Hz, 4H, NCH2), 2.70 (s, 6H, N(CH3)2), ),
2.64 (s, 6H, N(CH3)2), 2.19 (m, 2H, CH2CH2CH2). 13C-NMR (75 MHz, CD2Cl2, 298 K, δ
ppm): 23.1 (CH2CH2CH2), 46.2 (NMe2), 49.1 (NMe2), 58.3 (NCH2), 211.8, 215.0 (Mo-CO).
Iodo(pentacarbonylwolfram)(tetramethylpropylendiamin)gallium (9)
[(CO)5W-Ga(I)(TMPDA)]: Aus 2 mmol K2[W(CO)5] (804 mg), 2 mmol GaI3 (900 mg) und 2.1
mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 1126 mg (89%) farblose quaderförmige
Kristalle aus Methylenchlorid bei -30°C.
EA für C12H18GaIWN2O5: M = 632.74 (berechnet) C 22.76, H 2.84, N 4.43; (gefunden) C
22.66, H 2.69, N 4.24. IR (CH2Cl2, cm-1): ν(CO) = 2034 (m), 1949 (vs), 1919 (vs). 1H-NMR
(300 MHz, CD2Cl2, 298 K, δ ppm): 3.37 (t, 3J = 7.3 Hz, 4H, NCH2), 2.73 (s, 6H, N(CH3)2),
2.69 (s, 6H, N(CH3)2), 2.20 (m, 2H, CH2CH2CH2). 13C-NMR (75 MHz, CD2Cl2, 298 K, δ
ppm): 22.9 (CH2CH2CH2), 46.7 (NMe2), 49.6 (NMe2), 59.4 (NCH2), 201.7, 204.7 (W-CO).
Methyl(pentacarbonylwolfram)(tetramethylpropylendiamin)gallium (10)
[(CO)5W-Ga(Me)(TMPDA)]: Aus 2 mmol K2[W(CO)5] (804 mg), 2 mmol MeGaCl2 (311 mg)
und 2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 948 mg (88%) farblose
quaderförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C13H21GaWN2O5: M = 538.57 (berechnet) C 28.96, H 3.89, N 5.20; (gefunden) C
28.78, H 3.73, N 5.14. IR (CH2Cl2, cm-1): ν(CO) = 2030 (m), 1933 (vs), 1894 (vs). 1H-NMR
(300 MHz, CD2Cl2, 298 K, δ ppm): 3.16 (t, 3J = 6.1 Hz, 4H, NCH2), 2.71 (s, 6H, N(CH3)2),
2.51 (s, 6H, N(CH3)2), 2.12 (m, 2H, CH2CH2CH2), 0.21 (s, 3H, GaCH3). 13C-NMR (75

Experimenteller Teil 92
MHz, CD2Cl2, 298 K, δ ppm): 8.8 (GaCH3), 20.9 (CH2CH2CH2), 47.7 (NMe2), 50.6
(NMe2), 58.8 (NCH2), 206.4, 208.2 (W-CO).
Chloro(tetracarbonyleisen)(tetramethylpropylendiamin)gallium (11)
[(CO)4Fe-Ga(Cl)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol GaCl3 (352 mg) und
2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute 725 mg (90%) farblose
nadelförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C11H18ClFeGaN2O4: M = 403.01 (berechnet) C 26.70, H 3.90, N 5.66; (gefunden) C
26.41, H 3.87, N 5.64. IR (CH2Cl2, cm-1): ν(CO) = 2013 (vs), 1930 (m, sh), 1893 (vs). 1H-
NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.80 (t, 3J = 26.1 Hz, 4H, NCH2), 2.87 (s, 6H,
N(CH3)2), 2.82 (s, 6H, N(CH3)2), 2.21 (m, 2H, CH2CH2CH2). 13C-NMR (100 MHz,
CD2Cl2, 298 K, δ ppm): 25.4 (CH2CH2CH2), 49.9 (NMe2), 54.2 (NMe2), 63.7 (NCH2),
217.7 (Fe-CO).
Bromo(tetracarbonyleisen)(tetramethylpropylendiamin)gallium (12)
[(CO)4Fe-Ga(Br)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol GaBr3 (710 mg) und
2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 814 mg (91%) farblose
nadelförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C11H18BrFeGaN2O4: M = 447.46 (berechnet) C 29.50, H 4.02, N 6.26; (gefunden) C
29.41, H 3.97, N 6.08. IR (CH2Cl2, cm-1): ν(CO) = 2013 (vs), 1931 (m, sh), 1893 (vs). 1H-
NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.77 (t, 3J = 24.1 Hz, 4H, NCH2), 2.85 (s, 6H,
N(CH3)2), 2.81 (s, 6H, N(CH3)2), 2.19 (m, 2H, CH2CH2CH2). 13C-NMR (100 MHz,
CD2Cl2, 298 K, δ ppm): 24.9 (CH2CH2CH2), 49.2 (NMe2), 53.9 (NMe2), 62.5 (NCH2),
219.8 (Fe-CO).
Iodo(tetracarbonyleisen)(tetramethylpropylendiamin)gallium (13)
[(CO)4Fe-Ga(I)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol GaI3 (900 mg) und 2.1
mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 910 mg (92%) farblose nadelförmige
Kristalle aus Methylenchlorid bei -30°C.

Experimenteller Teil 93
EA für C11H18FeGaIN2O4: M = 494.46 (berechnet) C 26.70, H 3.90, N 5.66; (gefunden) C
26.41, H 3.87, N 5.64. IR (CH2Cl2, cm-1): ν(CO) = 2012 (vs), 1932 (m, sh), 1893 (vs). 1H-
NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.80 (t, 3J = 26.1 Hz, 4H, NCH2), 2.87 (s, 6H,
N(CH3)2), 2.82 (s, 6H, N(CH3)2), 2.21 (m, 2H, CH2CH2CH2). 13C-NMR (100 MHz,
CD2Cl2, 298 K, δ ppm): 25.4 (CH2CH2CH2), 49.9 (NMe2), 54.2 (NMe2), 63.7 (NCH2),
221.7 (Fe-CO).
Methyl(tetracarbonyleisen)(tetramethylpropylendiamin)gallium (14)
[(CO)4Fe-Ga(Me)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol Cl2GaMe (311 mg)
und 2.1 mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute: 696 mg (91%) farblose
nadelförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C12H21FeGaN2O4: (berechnet) C 37.64, H 5.90, N 7.32; (gefunden) C 37.44, H 5.73, N
7.12. IR (CH2Cl2, cm-1) ν(CO) = 1994 (vs), 1907 (vs, sh), 1870 (vs). 1H-NMR (250 MHz,
CD2Cl2, 298 K, δ ppm): 3.16 (t, 3J = 13.6 Hz, 4H, NCH2), 2.76 (s, 6H, N(CH3)2), 2.58 (s, 6H,
N(CH3)2), 2.21 (m, 2H, CH2CH2CH2), 0.13 (s, 3H, GaCH3). 13C-NMR (100 MHz, CD2Cl2,
298 K, δ ppm): 2.4 (GaCH3), 25.5 (CH2CH2CH2), 51.9 (NMe2), 52.9 (NMe2), 64.4 (NCH2),
224.8 (Fe-CO).
(tetracarbonyleisen)(pentamethyldiethylentriamin)galliumiodid (15)
[(CO)4Fe-Ga(PMDETA)]+I-: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol GaI3 (900 mg) und
2.1 mmol PMDETA (0.35 ml) (270 mg, d = 0.77 g/ml). Ausbeute: 487 mg (90%) orange
quaderförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C14H25Cl2FeGaN3O4: M = 540.56 (berechnet) C 31.08, H 4.62, N 7.77; (gefunden) C
31.01, H 4.55, N 7.68. IR (CH2Cl2, cm-1): ν(CO) = 1998 (vs), 1920 (vs, sh), 1887 (vs). 1H-
NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 2.62 (s, 6H, N(CH3)2), 2.68 (s, 6H, N(CH3)2),
2.83 (m, 3H, NCH3), 2.94 (m, 8H, CH2CH2). 13C-NMR (63 MHz, CD2Cl2, 298 K, δ ppm):
48.6 (NMe), 53.9 (NMe2), 54.1 (NMe2), 55.8 (NCH2), 217.9 (Fe-CO).

Experimenteller Teil 94
Bis-Tetracarbonyleisen-bis-[N, N , N´-Triethylethylendiamin]gallium (16)
Verwendung von Schenkelfrittentechnik. Zu einer auf –78°C gekühlten Suspension von 492 mg (2
mmol) K2Fe(CO)4 in 40 ml THF wird eine Lösung von mg (2 mmol) Cl2Ga[(Et)2NCH2CH2N(Et)]
in 20 ml THF gegeben. Nach 1 h wird das Lösungsmittel im Vakuum abkondensiert und der
Rückstand in 30 ml Toluol aufgenommen. Die klare Lösung wird vom weißen Niederschlag
abfiltriert, der Niederschlag noch einmal mit 10 ml Toluol gewaschen und die vereinigten
Toluolextrakte auf etwa 15 ml eingeengt. Nach Kühlung auf -30°C kristallisiert (16) in Form
gelblicher dünner Rauten aus. Die Verbindung ist extrem empfindlich gegen Luft und Feuchtigkeit.
Ausbeute: einige Kristalle
EA für C24H38Fe2Ga2N4O8: M = 760.68 (berechnet) C 37.86, H 4.99, N 7.36; (gefunden) C
37.66, H 4.65, N 7.08. IR (CH2Cl2, cm-1): ν(CO) = 1998 (vs), 1941 (s, sh), 1916 (vs), 1877
(m, sh). 1H-NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 0.39 (t, 3H, NCH2CH3), 2.09 (m, 2H,
NCH2CH3), 2.26 (m, 4H, NCH2CH2N), 2.59 (m, 10H, N(CH2CH3)2). 13C-NMR (63 MHz,
CD2Cl2, 298 K, δ ppm): 11.5 (NCH2CH3), 41.8 (NCH2CH3), 44.1 (NCH2CH2N), 55.3
(N(CH2CH3)2), 216.9 (Fe-CO).

Experimenteller Teil 95
Übergangsmetall-Indium-Komplexe
Allgemeine Arbeitsvorschrift:
Zu einer auf -78°C gekühlten Suspension von 2 mmol des Übergangscarbonylmetallats
K2[Fe(CO)4] in 40 ml THF wird eine auf -78°C vorgekühlte THF-Lösung von 2 mmol
Indiumtribromid (InBr3) gegeben. Nach der Zugabe wird das Kühlbad entfernt, die Reaktionslösung
auf Raumtemperatur erwärmt und 1 h gerührt bevor die chelatisierende Base (TMPDA, PMDETA)
im leichten Überschuß (2.1 mmol) zugegeben wird. Nach einer halben Stunde Reaktionszeit wird das
Lösungsmittel im Hochvakuum eingeengt, der Rückstand mit 40 ml Methylenchlorid aufgenommen
und intensiv gerührt. Die Methylenchlorid-Lösung wird filtriert (meist 2 mal) und das Lösungsmittel
wiederum im Hochvakuum bis zur Trockne eingeengt. Es werden gelbliche mikrokristalline Pulver
erhalten. Die Ausbeuten liegen durchweg bei 94-97% analysenreiner Substanz.
Kristallisation:
Bei den Tetracarbonyleisen-Verbindungen war die Tieftemperaturkristallisation bei -30°C aus
konzentrierten Methylenchlorid-Lösungen (2 mmol/10 ml) äußerst erfolgreich. Das Schlenkrohr
sollte dabei möglichst waagrecht liegen. Innerhalb eines Tages bilden sich quaderförmige Kristalle mit
Kantenlängen bis zu 15 mm. Das Aufbewahren der Kristalle in der Mutterlauge für
kristallographische Zwecke ist sinnvoll. Ansonsten wird die Mutterlauge abkanüliert, weiter eingeengt
und zur Nachkristallisation erneut gekühlt. Insgesamt liegen die Ausbeuten an einkristallinem Material
durchweg über 91%.
Bromo(tetracarbonyleisen)(tetramethylpropylendiamin)indium (17)
[(CO)4Fe-In(Br)(TMPDA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol InBr3 (710 mg) und 2.1
mmol (0.3 ml) TMPDA (230 mg, d = 0.77 g/ml). Ausbeute 877 mg (89%) gelbe quaderförmige
Kristalle aus Methylenchlorid bei -30°C.
EA für C11H18BrFeInN2O4: M = 492.56 (berechnet) C 26.80, H 3.72, N 5.75; (gefunden) C
27.02, H 3.85, N 5.83. IR (CH2Cl2, cm-1): ν(CO) = 2013 (vs), 1959 (w, sh), 1932 (m, sh),
1895 (vs). 1H-NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.58 (t, 3J = 24.9 Hz, 4H, NCH2),
2.69 (s, 6H, N(CH3)2), 2.62 (s, 6H, N(CH3)2), 2.20 (m, 2H, CH2CH2CH2). 13C-NMR (63

Experimenteller Teil 96
MHz, CD2Cl2, 298 K, δ ppm): 22.4 (CH2CH2CH2), 45.9 (NMe2), 49.2 (NMe2), 60.1
(NCH2), 217.1 (Fe-CO).
Bromo(tetracarbonyleisen)(pentamethyldiethylentriamin)indium (18)
[(CO)4Fe-In(Br)(PMDETA)]: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol InBr3 (710 mg) und
2.1 mmol (0.35 ml) PMDETA (270 mg, d = 0.77 g/ml). Ausbeute 996 mg (93%) gelbe
quaderförmige Kristalle aus Methylenchlorid bei -30°C.
EA für C13H23BrFeInN3O4: M = 535.56 (berechnet) C 29.10, H 4.29, N 7.83; (gefunden) C
29.28, H 4.15, N 7.93. IR (CH2Cl2, cm-1): ν(CO) = 2004 (vs), 1925 (m), 1890 (vs), 1866 (vs).
1H-NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.18 (m, 8H, CH2CH2), 2.89 (m, 3H, NCH3),
2.71 (s, 6H, N(CH3)2), 2.63 (s, 6H, N(CH3)2). 13C-NMR (63 MHz, CD2Cl2, 298 K, δ ppm):
49.2 (NMe), 53.6 (NMe2), 54.7 (NMe2), 60.3 (NCH2), 217.1 (Fe-CO).
[(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2 (19)
[(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2: Aus 2 mmol K2[Fe(CO)4] (492 mg), 2 mmol InBr3 (710
mg) und 2.1 mmol (0.40 ml) 12-Krone-4-Ether (270 mg, d = 0.77 g/ml). Ausbeute 745 mg (80%)
gelbliche quaderförmige Kristalle aus einer Mischung Toluol/Methylenchlorid bei -30°C.
EA für C36H64Br6FeIn2K2O20: M = 1660.02 (berechnet) C 26.02, H 3.86, O 19.28; (gefunden)
C 26.38, H 4.11, O 19.53. IR (CH2Cl2, cm-1): ν(CO) = 2062 (m), 2013 (vs), 1977 (s, sh), 1957
(vs). 1H-NMR (250 MHz, CD2Cl2, 298 K, δ ppm): 3.48 (s, 16H, CH2CH2).

Experimenteller Teil 97
M(CO)5-Transferreagenzien [235236,237]
Pentacarbonyl(η2-cis-Cycloocten)molybdän (20)
5.28 g Mo(CO)6 und 20 ml (20 mmol) cis-Cycloocten (16.6 g, d = 0.83 g/ml) werden in 400 ml
Pentan suspendiert. Die Reaktionslösung wird in einen UV-Reaktorsystem (Heraeus UV TQ 150
W) ca. 20 Stunden bei Raumtemperatur bestrahlt. Aus der anfangs farblosen Suspension wird eine
hellgraue Lösung. Die Lösung wird aus dem UV-Reaktorsystem in ein ausgeheiztes und mit Argon
beschicktes Schlenkrohr umkanüliert. Das Rohprodukt wird über Nacht oder mindestens 5 Stunden
auf Trockeneis (-78°C) gestellt. Das Produkt kristallisiert in Form farbloser Nadeln aus die mittels
Sublimation noch vom restlichen Mo(CO)6 gereinigt werden muß. Ausbeute: 2.42 g, 35% bezogen
auf Mo.
EA für C13H14MoO5: M = 345.94 (berechnet) C 45.10, H 4.05; (gefunden) C 45.08, H 4.01.
IR (CH2Cl2, cm-1): ν(CO) = 2074 (m), 1957 (vs), 1945 (vs). 1H-NMR (200 MHz, CD2Cl2,
298 K, δ ppm): 3.95 (m, 2H, CH=CH), 2.33 (m, 4H, CHCH2CH2), 1.66 (m, 4H,
CHCH2CH2CH2), 1.35 (m, 4H, CHCH2CH2CH2CH2).
Pentacarbonyl(η2-cis-Cycloocten)wolfram (21)
7.04 g W(CO)6 und 20 ml (20 mmol) cis-Cycloocten (16.6 g, d = 0.83 g/ml) werden in 400 ml
Pentan suspendiert. Die Reaktionslösung wird in einen UV-Reaktorsystem (Heraeus UV TQ 150
W) ca. 16 Stunden bei Raumtemperatur bestrahlt. Aus der anfangs farblosen Suspension wird eine
hellgelbe Lösung. Die Lösung wird aus dem UV-Reaktorsystem in ein ausgeheiztes und mit Argon
beschicktes Schlenkrohr umkanüliert. Das Rohprodukt wird über Nacht auf Trockeneis (-78°C)
gestellt. Das Produkt kristallisiert in Form gelber Quader aus die mittels Sublimation noch vom
restlichen W(CO)6 gereinigt werden muß. Ausbeute: 3.21 g, 37% bezogen auf W.
EA für C13H14WO5: M = 433.85 (berechnet) C 35.96, H 3.23; (gefunden) C 35.87, H 3.19. IR
(CH2Cl2, cm-1): ν(CO) = 2079 (m), 1960 (vs), 1946 (vs). 1H-NMR (200 MHz, CD2Cl2, 298
K, δ ppm): 3.99 (m, 2H, CH=CH), 2.38 (m, 4H, CHCH2CH2), 1.70 (m, 4H, CHCH2CH2CH2),
1.41 (m, 4H, CHCH2CH2CH2CH2).

Experimenteller Teil 98
6.4 Durchführung und Analytik des MOCVD-Experiments
Das Vakuum-MOCVD-Experiment wurde in einem Heißwandrohrreaktor durchgeführt. Nachdem
der Reaktor mit den gereinigten Si-Substraten bestückt war, wurde das gesamte System für 5 h auf
einen Basisdruck von 10-3 Torr evakuiert. Der Reaktor wurde während dieser Zeit auf 400°C
geheizt, um Restfeuchtigkeit zu desorbieren. Die Precursorquelle wurde in einer Glove-Box mit 140
mg der Vorstufe [(Cp*Al)Cr(CO)5] befüllt und unter Schutzgas an den Reaktor angesetzt. Das
gesamte System wurde nochmals für einige Stunden auf 10-3 Torr evakuiert. Danach wurde der
Ofen auf die vorgesehene Abscheidetemperatur (400°C) eingestellt. Nachdem sich die
Ofentemperatur stabilisiert hatte, wurde das Precursor-Reservoir auf 70°C geheizt. Der Precursor
sublimierte in der heißen Zone des Reaktors und ergab eine metallisch glänzende Beschichtung auf
den Si-Substraten und den Wänden des Reaktors. Es wurden keine Trägergase eingesetzt.
6.4.1 Analytische Charakterisierung dünner intermetallischer Filme
Im Rahmen dieser Arbeit standen für die zerstörungsfreie instrumentelle Analyse der erzeugten
dünnen Schicht drei Verfahren zur Verfügung: Röntgendiffraktometrie, Rutherford-Rückstreu-
Spektrometrie (RBS) und Atomemissionspektrometrie mit induktiv gekoppelten Plasma (ICP).
Routineanalysemethoden, wie Rasterelektronenmikroskopie mit energiedispersiven
Röntgenanalysator (REM-EDX) und AUGER-Elektronenspektroskopie (AES) konnten nicht
durchgeführt werden. [238-240]
6.5 Röntgendiffraktometrie
Die Aufnahme des Röntgendiffraktogramms der Schicht erfolgte mit einem hochauflösenden
HRXRD Bruker axs D8 Discover mit primärseitigen Göbel-Spiegel (Cu-Kα-Strahlung, λ = 154.056
pm und Schrittweite 0.005°). Genaue Angaben über den Versuchsaufbau und die Auswertung der
Ergebnisse sind in der Literatur beschrieben. [241, 242]

Experimenteller Teil 99
6.5.1 Rutherford-Rückstreu-Spektrometrie (RBS)
Die Rutherford-Rückstreu-Spektrometrie (RBS) beruht auf der Coulombstreuung des positiv
geladenen Kerns eines schnellen Ions (z.B. He+) und des positiven Kerns eines ruhenden Atoms. Die
Coulombabstoßung zwischen den positiven Kernen von Projektil und Targetatom kann klassisch als
Streuung in einem Zentralkräftefeld beschrieben werden.
Die RBS-Analyse besitzt gegenüber anderen Oberflächenanalyse-Techniken (u.a.
Rasterelektronenspektroskopie (REM), AUGER-Elektronenspektroskopie) den Vorteil, daß
quantitative Aussagen über die Zusammensetzung des Films sowie über das Tiefenprofil gemacht
werden können, ohne dabei Standards zu gebrauchen. Jedoch können durch diese Methode keine
Informationen über die chemische Bindung innerhalb der Schicht erhalten werden. Daneben ist es
schwierig zwischen Targetatomen mit vergleichbaren Massen zu unterscheiden (dieses gilt besonders
für schwere Atome), da die Rückstreuenergie im erheblichen Maße von der Masse der betreffenden
Atomsorte abhängig ist.
Die RBS-Spektren wurden am 4MV Dynamitron-Tandem mit einem He+-Strahl der Energie 2.0
MeV aufgenommen. Der Detektor stand unter einem Winkel von 170° zum Strahl. Die Probe war
zur Vermeidung von Channeling Effekten um einen Winkel von 7° gekippt. Die Spektren wurden mit
dem Programm RBX analysiert. Details zum Versuchsaufbau und Auswertung der Ergebnisse sind in
der Literatur beschrieben. [243-245]
6.5.2 Atomemissionspektrometrie mit induktiv gekoppelten Plasma (ICP)
Zusätzlich zu den genannten Bestimmungen wurde das Metallverhältnis in dem erzeugten dünnen Film
mittels Atomemissionspektrometrie mit induktiv gekoppelten Plasma (ICP) der Firma Philipps
Unicam PU 7000 durch das Chemielaboratorium der Petrologie der Ruhr-Universität Bochum
bestimmt (Abbildung 31). Dazu wurde von der Reaktorwand der Metallfilm quantitativ mit
verdünnter Salpetersäure gelöst.
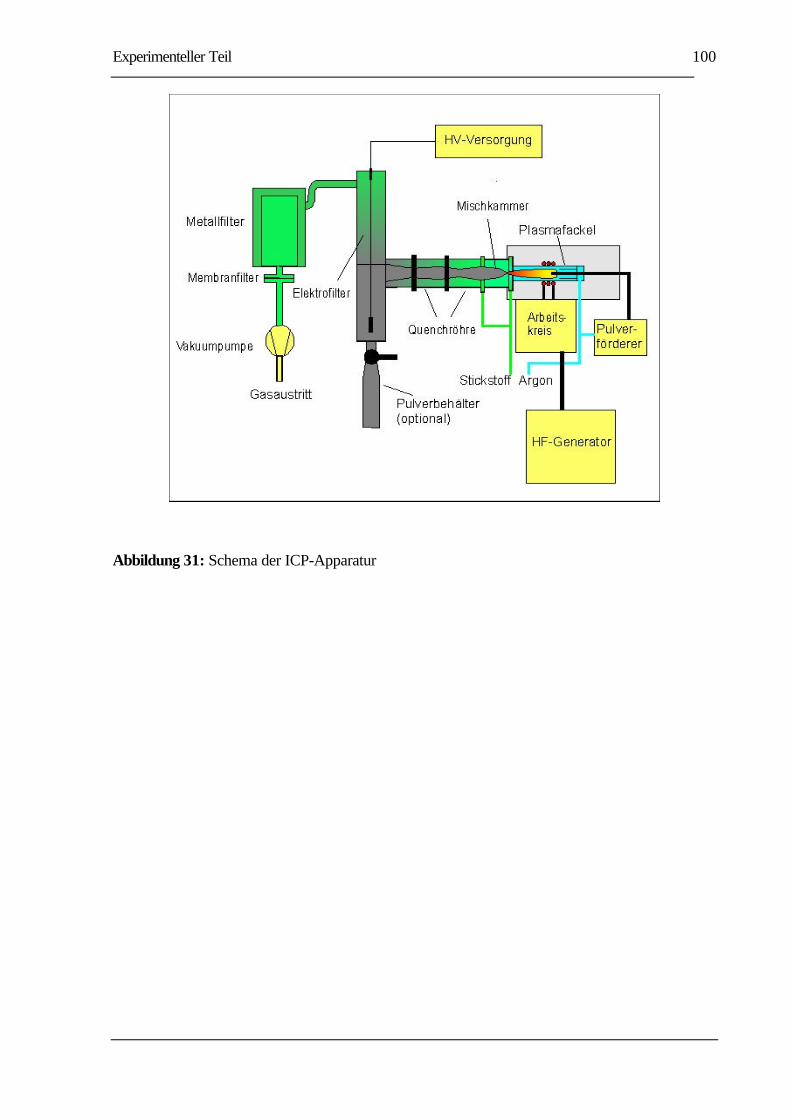
Experimenteller Teil 100
Abbildung 31: Schema der ICP-Apparatur

E
TABELLEN

Tabellen 102
7 Tabellen
Die folgenden Tabellen liefern ergänzende kristallographische Daten zu den Verbindungen (1), (2),
(5), (11-15) und (17-21) (Tabelle 27). Für jede einzelne Verbindung wird hier jedoch nur die
entsprechenden Meßparameter und die Atomkoordinaten und äquivalente isotrope atomare
Auslenkungsmuster für Nicht-Wasserstoffatome dargestellt. Alle weiteren Daten wie z. Bsp.
Bindungslängen, Bindungswinkel, Torsionswinkel, anisotrope Temperaturfaktoren und
Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Wasserstoffatome sind
wegen ihres Umfangs der elektronischen Version dieser Dissertation als txt- oder lst-files angefügt.
Nr. Verbindung
1 (CO)5Cr-Al[(Cl)(TMPDA)]
2 (CO)4Fe-Al[(Cl)(TMPDA)]
5 (CO)5Cr-Ga[(Cl)(TMPDA)]
11 (CO)4Fe-Ga[(Cl)(TMPDA)]
12 (CO)4Fe-Ga[(Br)(TMPDA)]
13 (CO)4Fe-Ga[(I)(TMPDA)]
14 (CO)4Fe-Ga[(Me)(TMPDA)]
15 (CO)4Fe-Ga[(PMDETA)]+I-
17 (CO)4Fe-In[(Br)(TMPDA)]
18 (CO)4Fe-In[(Br)(PMDETA)]
19 [(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2
20 [(CO)5Mo(η2-C8H14]
21 [(CO)5W(η2-C8H14]
Tabelle 27: Alle in dieser Dissertation röntgenographisch charakterisierten Verbindungen

Tabellen 103
Ergänzende kristallographische Daten
Tabelle 28: Kristallographische Daten und Meßparameter für (CO)5Cr-Al[(Cl)(TMPDA)] (1).
Formel C12H18AlClCrN2O5
Formelmasse 384.42 a. m. u.
Kristallfarbe und -form gelbe Nadeln; 0.30×0.28×0.14 mm
Meßtemperatur [K] 193(2)
Raumgruppe Monoklin, C2/c
a [pm] 2645.8(5)
b [pm] 899.8(5)
c [pm] 1556.7(3)
α [°] 90
β [°] 115.83(5)
γ [°] 90
V [106×pm3] 3336(2)
Z 10
ρber. [g×cm-3] 1.546
F(000) 1540
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 1.131
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 3016
Anzahl der unabhängigen Reflexe 2948 [R(int) = 0.0586]
Meßbereich 2.42° < Θ < 25.00°; h(-8/31), k(-10/10), l(-18/16)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0343
wR2 0.0650
Goodness-of-fit auf F2 0.738
Restelektronendichte [eÅ-3] max. +0.267, min. -0.285

Tabellen 104
Tabelle 29: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)5Cr-Al[(Cl)(TMPDA)] (1).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
Cr(1) -869(1) 2127(1) -611(1) 22(1)
Al(1) -1477(1) 2635(1) -2322(1) 20(1)
Cl(1) -2383(1) 3084(1) -2899(1) 36(1)
N(1) -1300(1) 4443(3) -2967(2) 26(1)
N(2) -1538(1) 1058(3) -3304(2) 21(1)
C(1) -1476(2) 5837(4) -2652(3) 42(1)
C(2) -686(1) 4605(4) -2683(2) 34(1)
C(3) -1606(2) 4419(4) -4036(2) 32(1)
C(4) -1521(1) 2979(4) -4492(2) 30(1)
C(5) -1802(1) 1628(4) -4318(2) 28(1)
C(6) -984(1) 358(4) -3103(2) 25(1)
C(7) -1913(1) -167(4) -3271(2) 30(1)
C(8) -987(1) 57(5) -800(2) 28(1)
O(8) -1055(1) -1208(3) -897(2) 43(1)
C(9) -751(1) 4182(5) -393(2) 30(1)
O(9) -654(1) 5420(3) -217(2) 51(1)
C(10) -380(1) 1910(4) 662(2) 29(1)
O(10) -60(1) 1831(3) 1457(2) 49(1)
C(11) -1557(1) 2234(4) -543(2) 30(1)
O(11) -1989(1) 2255(3) -514(2) 47(1)
C(12) -297(1) 2014(4) -1001(2) 24(1)
O(12) 61(1) 1932(3) -1232(2) 34(1)

Tabellen 105
Ergänzende kristallographische Daten
Tabelle 30: Kristallographische Daten und Meßparameter für (CO)4Fe-Al[(Cl)(TMPDA)] (2).
Formel C11H18AlClFeN2O4
Formelmasse 360.28 a. m. u.
Kristallfarbe und -form farblose Nadeln; 0.20×0.20×0.30 mm
Meßtemperatur [K] 203(2)
Raumgruppe Monoklin, P2(1)/c
a [pm] 781.8(1)
b [pm] 1200.0(1)
c [pm] 2061.7(3)
α [°] 90
β [°] 108.35(3)
γ [°] 90
V [106×pm3] 1934(6)
Z 4
ρber. [g×cm-3] 1.530
F(000) 912
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 1.260
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 2495
Anzahl der unabhängigen Reflexe 2423 [R(int) = 0.0832]
Meßbereich 2.72° < Θ < 22.50°; h(-8/8), k(-12/12), l(-19/22)
Verfeinerung Full-matrix least-squares auf F2
R1 0.1185
wR2 0.2717
Goodness-of-fit auf F2 1.172
Restelektronendichte [eÅ-3] max. +1.36, min. -0.85

Tabellen 106
Tabelle 31: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-Al[(Cl)(TMPDA)] (2).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
Fe(1) 8842(11) 117(13) 6364(11) 24(2)
O(1) 11348(11) -1395(12) 6187(12) 42(3)
Al(1) 10411(13) 1572(15) 6554(16) 25(2)
O(3) 7202(5) 1637(16) 5381(15) 48(3)
N(1) 12113(3) 1952(18) 5855(15) 28(3)
C(1) 10262(12) -780(11) 6247(14) 32(6)
N(2) 12064(3) 1462(12) 7293(17) 29(7)
C(2) 6873(17) -886(12) 6230(18) 29(6)
C(7) 13395(15) 2804(15) 6086(13) 33(3)
C(9) 13368(15) 2031(11) 7298(12) 35(3)
C(8) 14403(8) 2441(17) 6674(17) 37(1)
O(2) 5790(9) -153(15) 6119(18) 57(2)
O(4) 7182(12) 637(13) 7659(12) 61(1)
C(6) 11195(4) 2493(15) 5304(14) 43(5)
C(3) 7746(12) 1032(12) 5749(13) 34(1)
C(5) 12974(14) 975(17) 5571(16) 45(8)
C(11) 11096(14) 1531(18) 7929(11) 39(1)
C(4) 7757(12) 454(11) 7151(11) 35(2)
C(10) 12947(19) 352(13) 7323(17) 34(7)
C(12) 3433(12) 6433(15) 5827(15) 46(2)
Cl(1) 9391(17) 3212(13) 6750(13) 43.47

Tabellen 107
Ergänzende kristallographische Daten
Tabelle 32: Kristallographische Daten und Meßparameter für (CO)5Cr-Ga[(Cl)(TMPDA)] (5).
Formel C12.70H20.10Cl0.30CrGaN2O5
Formelmasse 413.16 a. m. u.
Kristallfarbe und -form gelbliche Quader; 0.20×0.20×0.30 mm
Meßtemperatur [K] 293(2)
Raumgruppe Triklin, P-1
a [pm] 889.00(4)
b [pm] 909.63(5)
c [pm] 1187.64(6)
α [°] 92.15(4)
β [°] 96.55(4)
γ [°] 114.82(3)
V [106×pm3] 862.0(8)
Z 2
ρber. [g×cm-3] 1.592
F(000) 421
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 2.261
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 4349
Anzahl der unabhängigen Reflexe 2983 [R(int) = 0.0275]
Meßbereich 2.55° < Θ < 25.18°; h(-10/10), k(-10/10), l(-10/9)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0783
wR2 0.2194
Goodness-of-fit auf F2 1.083
Restelektronendichte [eÅ-3] max. +0.646, min. -0.777

Tabellen 108
Tabelle 33: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)5Cr-Ga[(Cl)(TMPDA)] (5).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
Ga(1) 1604(1) 3346(1) 7031(1) 24(1)
Cr(2) -563(2) 1830(2) 8230(2) 28(1)
O(3) 2254(11) 2753(11) 10188(7) 41(2)
N(1) 3671(11) 2683(12) 6950(7) 29(2)
N(2) 3219(11) 5898(11) 7565(8) 32(2)
O(4) -1095(12) 4843(11) 8739(8) 49(2)
C(9) 4752(13) 6524(13) 6992(9) 28(2)
O(1) -169(12) -1272(11) 7690(10) 60(3)
C(12) 4237(15) 2241(16) 8049(10) 40(3)
O(2) -3210(12) 219(12) 9726(10) 64(3)
O(5) -2888(13) 1121(15) 6017(9) 70(3)
C(5) -2002(16) 1386(17) 6860(12) 48(3)
C(4) -830(14) 3738(14) 8540(10) 34(3)
C(3) 1192(14) 2432(14) 9451(10) 32(2)
C(1) -306(14) -98(15) 7891(11) 40(3)
C(2) -2196(16) 790(15) 9133(13) 46(3)
C(11) 5086(14) 3929(14) 6491(10) 35(3)
C(7) 3649(15) 6171(14) 8807(11) 42(3)
C(13) 2988(15) 1193(14) 6127(10) 38(3)
C(8) 2227(17) 6812(15) 7208(13) 50(3)
C(10) 5812(14) 5601(15) 7142(11) 40(3)
Cl(1) 1360(3) 3750(3) 5190(2) 36(5)
C(6) 1000(4) 3390(4) 5310(3) 21(7)

Tabellen 109
Ergänzende kristallographische Daten
Tabelle 34: Kristallographische Daten und Meßparameter für (CO)4Fe-Ga[(Cl)(TMPDA)] (11).
Formel C11H18ClFeGaN2O4
Formelmasse 403.29 a. m. u.
Kristallfarbe und -form farblose Nadeln; 0.20×0.20×0.30 mm
Meßtemperatur [K] 203(2)
Raumgruppe Orthorhombisch, P212121
a [pm] 732.0(5)
b [pm] 1463.8(3)
c [pm] 1491.4(3)
α [°] 90
β [°] 90
γ [°] 90
V [106×pm3] 1598.0(6)
Z 4
ρber. [g×cm-3] 1.676
F(000) 816
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 2.772
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 5044
Anzahl der unabhängigen Reflexe 3472 [R(int) = 0.0476]
Meßbereich 2.78° < Θ < 29.43°; h(-9/9), k(-20/7), l(-16/8)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0421
wR2 0.0884
Goodness-of-fit auf F2 0.997
Restelektronendichte [eÅ-3] max. +0.749, min. -0.806
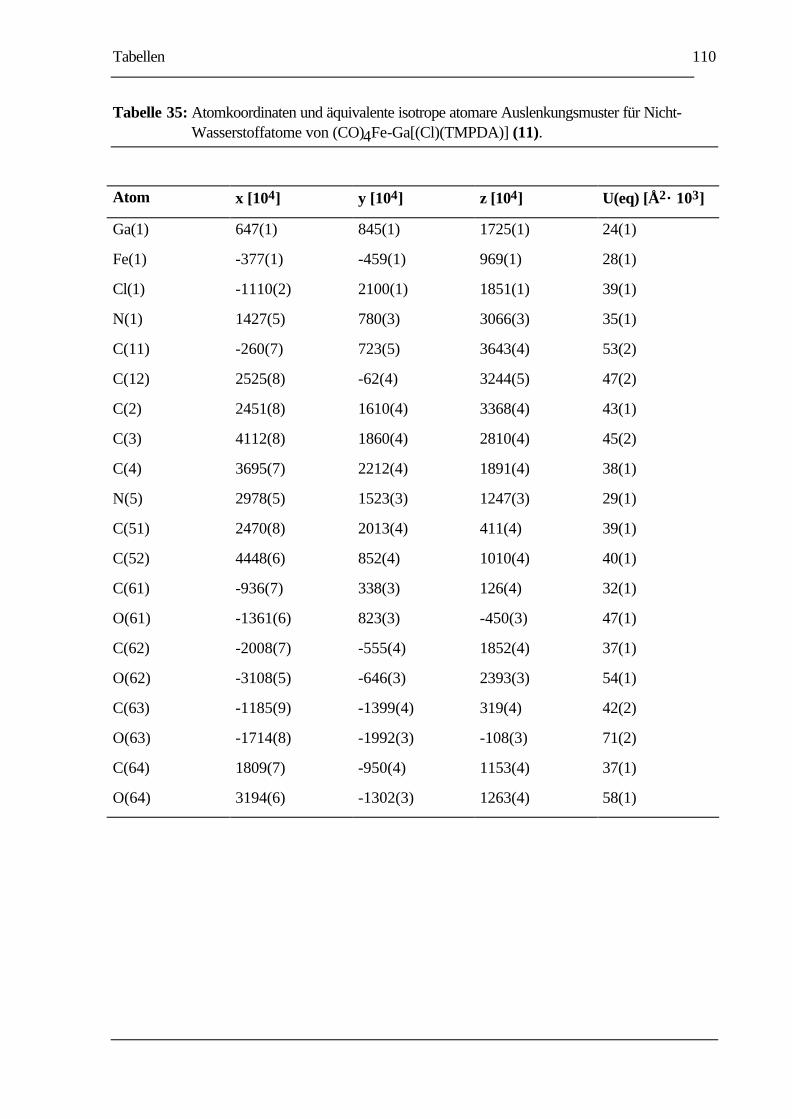
Tabellen 110
Tabelle 35: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-Ga[(Cl)(TMPDA)] (11).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
Ga(1) 647(1) 845(1) 1725(1) 24(1)
Fe(1) -377(1) -459(1) 969(1) 28(1)
Cl(1) -1110(2) 2100(1) 1851(1) 39(1)
N(1) 1427(5) 780(3) 3066(3) 35(1)
C(11) -260(7) 723(5) 3643(4) 53(2)
C(12) 2525(8) -62(4) 3244(5) 47(2)
C(2) 2451(8) 1610(4) 3368(4) 43(1)
C(3) 4112(8) 1860(4) 2810(4) 45(2)
C(4) 3695(7) 2212(4) 1891(4) 38(1)
N(5) 2978(5) 1523(3) 1247(3) 29(1)
C(51) 2470(8) 2013(4) 411(4) 39(1)
C(52) 4448(6) 852(4) 1010(4) 40(1)
C(61) -936(7) 338(3) 126(4) 32(1)
O(61) -1361(6) 823(3) -450(3) 47(1)
C(62) -2008(7) -555(4) 1852(4) 37(1)
O(62) -3108(5) -646(3) 2393(3) 54(1)
C(63) -1185(9) -1399(4) 319(4) 42(2)
O(63) -1714(8) -1992(3) -108(3) 71(2)
C(64) 1809(7) -950(4) 1153(4) 37(1)
O(64) 3194(6) -1302(3) 1263(4) 58(1)

Tabellen 111
Ergänzende kristallographische Daten
Tabelle 36: Kristallographische Daten und Meßparameter für (CO)4Fe-Ga[(Br)(TMPDA)] (12).
Formel C11H18BrFeGaN2O4
Formelmasse 447.75 a. m. u.
Kristallfarbe und -form farblose Nadeln; 0.20×0.20×0.30 mm
Meßtemperatur [K] 203(2)
Raumgruppe Orthorhombisch, P212121
a [pm] 754(2)
b [pm] 1466(4)
c [pm] 1461(4)
α [°] 90
β [°] 90
γ [°] 90
V [106×pm3] 1616(7)
Z 4
ρber. [g×cm-3] 1.841
F(000) 888
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 5.049
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 4166
Anzahl der unabhängigen Reflexe 2831 [R(int) = 0.1590]
Meßbereich 2.78° < Θ < 26.17°; h(-8/8), k(-14/17), l(-9/18)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0727
wR2 0.1896
Goodness-of-fit auf F2 1.052
Restelektronendichte [eÅ-3] max. +1.630, min. -1.963

Tabellen 112
Tabelle 37: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-Ga[(Br)(TMPDA)] (12).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
Ga(1) 664(1) 9157(1) 4249(1) 20(1)
Br(1) -1195(1) 7825(1) 4403(1) 35(1)
N(1) 2972(12) 8472(5) 3777(5) 27(2)
C(111) 2478(17) 7989(7) 2925(7) 38(2)
C(112) 4398(16) 9133(7) 3511(8) 37(2)
C(2) 3658(16) 7783(7) 4433(7) 36(2)
C(3) 4103(17) 8148(8) 5336(8) 41(3)
C(4) 2430(17) 8410(8) 5905(8) 39(3)
N(5) 1448(12) 9237(5) 5606(5) 29(2)
C(511) -206(19) 9316(10) 6177(8) 49(3)
C(512) 2544(18) 10101(8) 5737(8) 45(3)
Fe(1) -374(2) 10443(1) 3478(1) 25(1)
C(11) -1163(19) 11356(7) 2823(7) 39(3)
O(11) -1730(19) 11968(6) 2377(7) 70(3)
C(12) -929(14) 9639(6) 2634(6) 28(2)
O(12) -1367(14) 9152(5) 2068(5) 47(2)
C(13) -1983(16) 10551(7) 4344(8) 38(2)
O(13) -3054(13) 10637(6) 4900(6) 47(2)
C(14) 1797(17) 10946(7) 3639(8) 39(3)
O(14) 3186(13) 11287(6) 3735(7) 56(2)

Tabellen 113
Ergänzende kristallographische Daten
Tabelle 38: Kristallographische Daten und Meßparameter für (CO)4Fe-Ga[(I)(TMPDA)] (13).
Formel C11H18FeGaIN2O4
Formelmasse 494.74 a. m. u.
Kristallfarbe und -form farblose Quader; 0.30×0.20×0.30 mm
Meßtemperatur [K] 293(2)
Raumgruppe Triklin, P-1
a [pm] 820.59(13)
b [pm] 827.83(13)
c [pm] 1434.8(2)
α [°] 75.15(3)
β [°] 83.95(3)
γ [°] 62.11(3)
V [106×pm3] 832.7(2)
Z 2
ρber. [g×cm-3] 1.973
F(000) 480
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 4.354
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 4369
Anzahl der unabhängigen Reflexe 2904 [R(int) = 0.0494]
Meßbereich 2.81° < Θ < 25.07°; h(-8/9), k(-9/9), l(-16/17)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0346
wR2 0.0828
Goodness-of-fit auf F2 0.998
Restelektronendichte [eÅ-3] max. +0.758, min. -0.761

Tabellen 114
Tabelle 39: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-Ga[(I)(TMPDA)] (13).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
I(1) 2896(1) 4638(1) 4222(1) 33(1)
O(1) 7426(5) 119(5) 3430(3) 40(1)
Ga(1) 2746(1) 3430(1) 2713(1) 21(1)
O(3) 4610(6) 6575(6) 1526(3) 54(1)
Fe(1) 5279(1) 2708(1) 1694(1) 25(1)
O(4) 3416(6) 1634(6) 501(3) 53(1)
O(2) 8493(6) 1971(6) 459(3) 57(1)
N(2) 1921(5) 1363(5) 3407(3) 27(1)
C(3) 4796(7) 5083(7) 1583(4) 33(1)
N(1) 116(5) 5488(5) 2180(3) 26(1)
C(5) -1369(6) 5562(7) 2890(4) 33(1)
C(1) 6523(6) 1125(7) 2753(3) 29(1)
C(8) -295(7) 5216(7) 1261(3) 34(1)
C(9) 94(7) 7363(6) 1935(4) 35(1)
C(4) 4117(7) 2081(7) 972(4) 34(1)
C(10) 3334(7) -48(7) 4167(4) 35(1)
C(2) 7216(7) 2249(7) 935(4) 36(1)
C(6) -1482(7) 3733(7) 3258(4) 35(1)
C(11) 1868(8) 317(7) 2716(4) 38(1)
C(7) 102(7) 2166(7) 3900(4) 33(1)

Tabellen 115
Ergänzende kristallographische Daten
Tabelle 40: Kristallographische Daten und Meßparameter für (CO)4Fe-Ga[(Me)(TMPDA)] (14).
Formel C12H21FeGaN2O4
Formelmasse 382.88 a. m. u.
Kristallfarbe und -form farblose Quader; 0.20×0.20×0.30 mm
Meßtemperatur [K] 203(2)
Raumgruppe Orthorhombisch, Pnma
a [pm] 1324.0(2)
b [pm] 1162.3(1)
c [pm] 1031.6(1)
α [°] 90
β [°] 90
γ [°] 90
V [106×pm3] 1588(3)
Z 4
ρber. [g×cm-3] 1.602
F(000) 784
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 2.623
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 3512
Anzahl der unabhängigen Reflexe 1417 [R(int) = 0.0520]
Meßbereich 2.50° < Θ < 24.97°; h(-11/15), k(-13/13), l(-12/4)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0364
wR2 0.1037
Goodness-of-fit auf F2 0.874
Restelektronendichte [eÅ-3] max. +0.646, min. -0.777

Tabellen 116
Tabelle 41: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-Ga[(Me)(TMPDA)] (14).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
Ga(1) 3553(1) 2500 5642(1) 20(1)
Fe(1) 3711(1) 2500 7975(1) 21(1)
C(2) 3874(4) 2500 9693(5) 35(1)
O(1) 1512(3) 2500 8180(3) 36(1)
O(2) 3975(4) 2500 10817(4) 64(1)
N(1) 2656(2) 1163(2) 4821(2) 28(1)
O(3) 4848(2) 387(2) 7540(2) 41(1)
C(1) 4735(4) 2500 4440(4) 31(1)
C(3) 2379(4) 2500 8048(4) 25(1)
C(7) 1721(3) 890(3) 5578(3) 38(1)
C(4) 4393(2) 1221(3) 7677(3) 27(1)
C(5) 2404(3) 1401(3) 3408(3) 39(1)
C(6) 1828(5) 2500 3185(5) 47(1)
C(20) 3296(3) 109(3) 4846(4) 42(1)

Tabellen 117
Ergänzende kristallographische Daten
Tabelle 42: Kristallographische Daten und Meßparameter für(CO)4Fe-Ga[(PMDETA)]+(I-) (15).
Formel C14H25Cl2Fe2Ga2IN3O4
Formelmasse 622.74 a. m. u.
Kristallfarbe und -form orange Quader; 0.20×0.20×0.30 mm
Meßtemperatur [K] 293(2)
Raumgruppe Monoklin, P21/c
a [pm] 1415.5(1)
b [pm] 2439.5(2)
c [pm] 1354.7(1)
α [°] 90
β [°] 97.86(2)
γ [°] 90
V [106×pm3] 4633.9(8)
Z 4
ρber. [g×cm-3] 1.785
F(000) 2448
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 3.374
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 23953
Anzahl der unabhängigen Reflexe 8199 [R(int) = 0.0421]
Meßbereich 1.67° < Θ < 25.10°; h(-14/16), k(-29/29), l(-16/9)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0522
wR2 0.1334
Goodness-of-fit auf F2 1.032
Restelektronendichte [eÅ-3] max. +2.404, min. -1.160

Tabellen 118
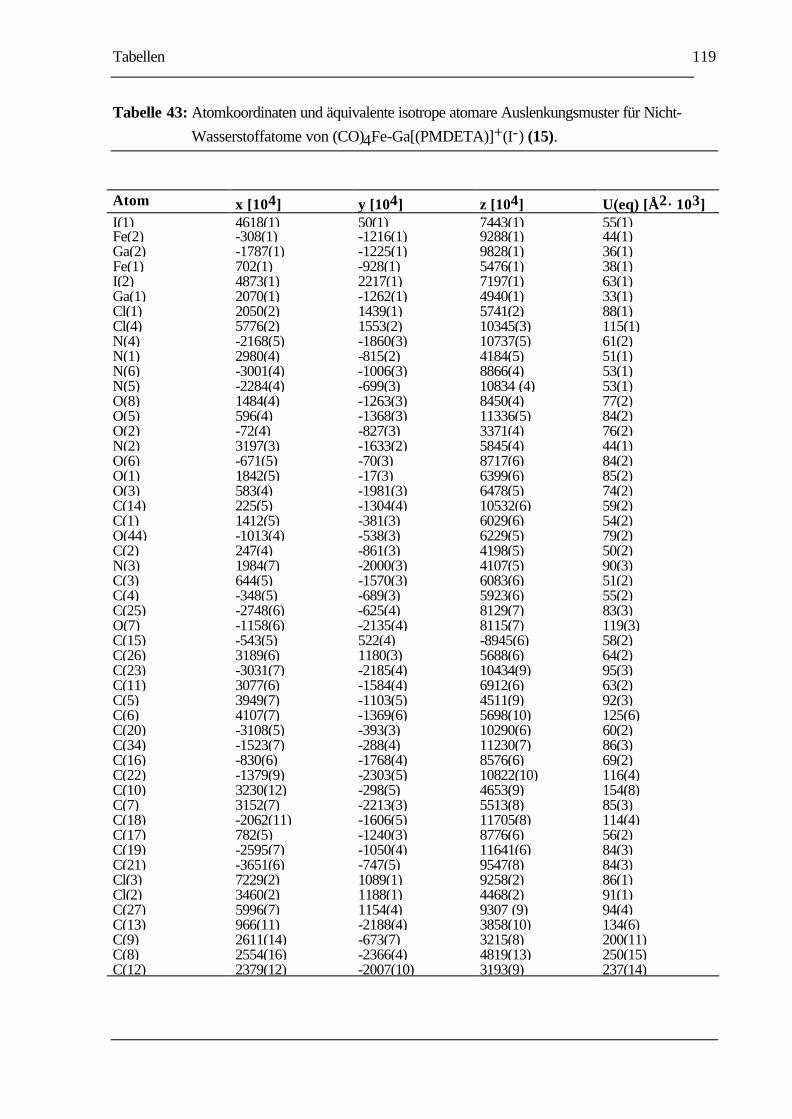
Tabellen 119
Tabelle 43: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-Ga[(PMDETA)]+(I-) (15).
Atom x [104] y [104] z [104] U(eq) [Å2×103]I(1) 4618(1) 50(1) 7443(1) 55(1)Fe(2) -308(1) -1216(1) 9288(1) 44(1)Ga(2) -1787(1) -1225(1) 9828(1) 36(1)Fe(1) 702(1) -928(1) 5476(1) 38(1)I(2) 4873(1) 2217(1) 7197(1) 63(1)Ga(1) 2070(1) -1262(1) 4940(1) 33(1)Cl(1) 2050(2) 1439(1) 5741(2) 88(1)Cl(4) 5776(2) 1553(2) 10345(3) 115(1)N(4) -2168(5) -1860(3) 10737(5) 61(2)N(1) 2980(4) -815(2) 4184(5) 51(1)N(6) -3001(4) -1006(3) 8866(4) 53(1)N(5) -2284(4) -699(3) 10834 (4) 53(1)O(8) 1484(4) -1263(3) 8450(4) 77(2)O(5) 596(4) -1368(3) 11336(5) 84(2)O(2) -72(4) -827(3) 3371(4) 76(2)N(2) 3197(3) -1633(2) 5845(4) 44(1)O(6) -671(5) -70(3) 8717(6) 84(2)O(1) 1842(5) -17(3) 6399(6) 85(2)O(3) 583(4) -1981(3) 6478(5) 74(2)C(14) 225(5) -1304(4) 10532(6) 59(2)C(1) 1412(5) -381(3) 6029(6) 54(2)O(44) -1013(4) -538(3) 6229(5) 79(2)C(2) 247(4) -861(3) 4198(5) 50(2)N(3) 1984(7) -2000(3) 4107(5) 90(3)C(3) 644(5) -1570(3) 6083(6) 51(2)C(4) -348(5) -689(3) 5923(6) 55(2)C(25) -2748(6) -625(4) 8129(7) 83(3)O(7) -1158(6) -2135(4) 8115(7) 119(3)C(15) -543(5) 522(4) -8945(6) 58(2)C(26) 3189(6) 1180(3) 5688(6) 64(2)C(23) -3031(7) -2185(4) 10434(9) 95(3)C(11) 3077(6) -1584(4) 6912(6) 63(2)C(5) 3949(7) -1103(5) 4511(9) 92(3)C(6) 4107(7) -1369(6) 5698(10) 125(6)C(20) -3108(5) -393(3) 10290(6) 60(2)C(34) -1523(7) -288(4) 11230(7) 86(3)C(16) -830(6) -1768(4) 8576(6) 69(2)C(22) -1379(9) -2303(5) 10822(10) 116(4)C(10) 3230(12) -298(5) 4653(9) 154(8)C(7) 3152(7) -2213(3) 5513(8) 85(3)C(18) -2062(11) -1606(5) 11705(8) 114(4)C(17) 782(5) -1240(3) 8776(6) 56(2)C(19) -2595(7) -1050(4) 11641(6) 84(3)C(21) -3651(6) -747(5) 9547(8) 84(3)Cl(3) 7229(2) 1089(1) 9258(2) 86(1)Cl(2) 3460(2) 1188(1) 4468(2) 91(1)C(27) 5996(7) 1154(4) 9307 (9) 94(4)C(13) 966(11) -2188(4) 3858(10) 134(6)C(9) 2611(14) -673(7) 3215(8) 200(11)C(8) 2554(16) -2366(4) 4819(13) 250(15)C(12) 2379(12) -2007(10) 3193(9) 237(14)

Tabellen 120
Ergänzende kristallographische Daten
Tabelle 44: Kristallographische Daten und Meßparameter für (CO)4Fe-In[(Br)(TMPDA)] (17).
Formel C11H18BrFeInN2O4
Formelmasse 492.85 a. m. u.
Kristallfarbe und -form gelbliche Quader; 0.20×0.20×0.20 mm
Meßtemperatur [K] 293(2)
Raumgruppe Orthorhombisch, P212121
a [pm] 753.4(3)
b [pm] 1497.8(9)
c [pm] 1519.2(9)
α [°] 90
β [°] 90
γ [°] 90
V [106×pm3] 1714(2)
Z 4
ρber. [g×cm-3] 1.909
F(000) 960
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 4.533
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 2720
Anzahl der unabhängigen Reflexe 2199 [R(int) = 0.0379]
Meßbereich 1.91° < Θ < 25.28°; h(-6/5), k(-5/17), l(-18/2)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0409
wR2 0.1363
Goodness-of-fit auf F2 1.076
Restelektronendichte [eÅ-3] max. +0.731, min. -0.980
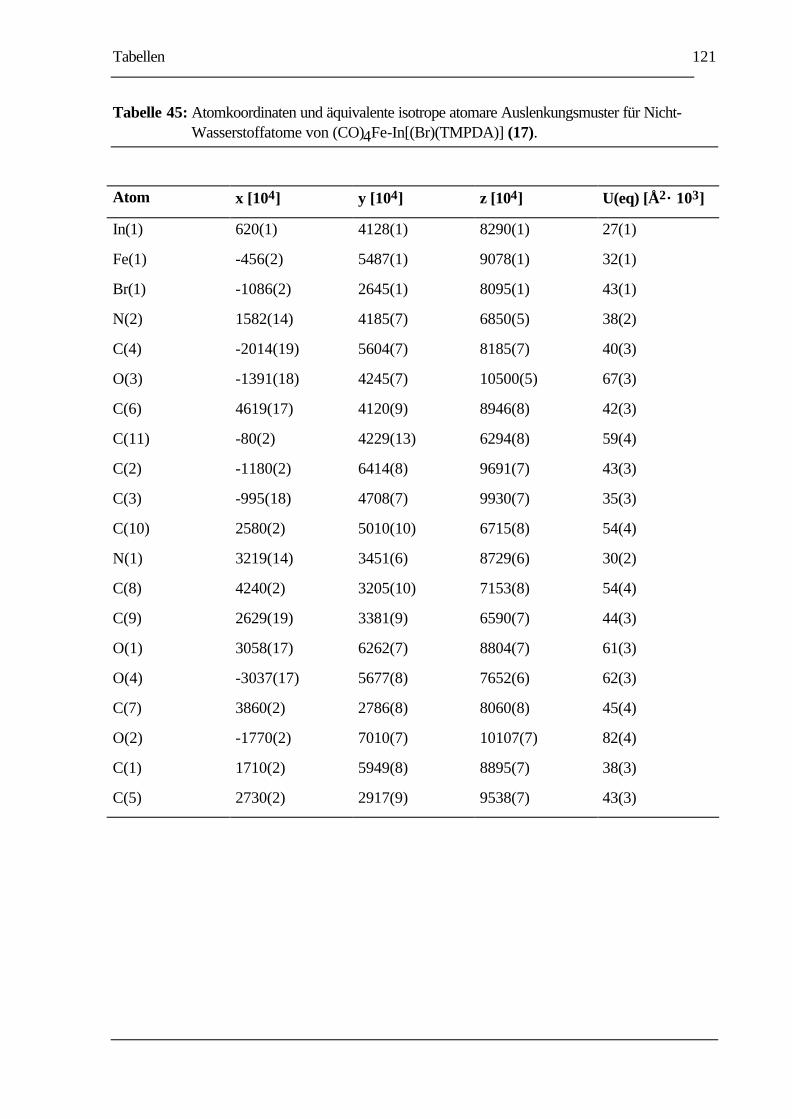
Tabellen 121
Tabelle 45: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-In[(Br)(TMPDA)] (17).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
In(1) 620(1) 4128(1) 8290(1) 27(1)
Fe(1) -456(2) 5487(1) 9078(1) 32(1)
Br(1) -1086(2) 2645(1) 8095(1) 43(1)
N(2) 1582(14) 4185(7) 6850(5) 38(2)
C(4) -2014(19) 5604(7) 8185(7) 40(3)
O(3) -1391(18) 4245(7) 10500(5) 67(3)
C(6) 4619(17) 4120(9) 8946(8) 42(3)
C(11) -80(2) 4229(13) 6294(8) 59(4)
C(2) -1180(2) 6414(8) 9691(7) 43(3)
C(3) -995(18) 4708(7) 9930(7) 35(3)
C(10) 2580(2) 5010(10) 6715(8) 54(4)
N(1) 3219(14) 3451(6) 8729(6) 30(2)
C(8) 4240(2) 3205(10) 7153(8) 54(4)
C(9) 2629(19) 3381(9) 6590(7) 44(3)
O(1) 3058(17) 6262(7) 8804(7) 61(3)
O(4) -3037(17) 5677(8) 7652(6) 62(3)
C(7) 3860(2) 2786(8) 8060(8) 45(4)
O(2) -1770(2) 7010(7) 10107(7) 82(4)
C(1) 1710(2) 5949(8) 8895(7) 38(3)
C(5) 2730(2) 2917(9) 9538(7) 43(3)

Tabellen 122
Ergänzende kristallographische Daten
Tabelle 46: Kristallographische Daten und Meßparameter für (CO)4Fe-In[(Br)(PMDETA)] (18).
Formel C13H23BrFeInN3O4
Formelmasse 535.92 a. m. u.
Kristallfarbe und -form gelbliche Quader; 0.30×0.30×0.30 mm
Meßtemperatur [K] 293(2)
Raumgruppe Monoklin, C2/c
a [pm] 3071.7(7)
b [pm] 874.6(2)
c [pm] 2867.7(6)
α [°] 90
β [°] 94.99(5)
γ [°] 90
V [106×pm3] 7675(3)
Z 16
ρber. [g×cm-3] 1.855
F(000) 4224
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 4.059
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 8778
Anzahl der unabhängigen Reflexe 6215 [R(int) = 0.0419]
Meßbereich 2.03° < Θ < 24.98°; h(-23/35), k(-10/9), l(-24/33)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0526
wR2 0.1591
Goodness-of-fit auf F2 1.102
Restelektronendichte [eÅ-3] max. +1.653, min. -1.620

Tabellen 123
Tabelle 47: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von (CO)4Fe-In[(Br)(PMDETA)] (18).
Atom x [104] y [104] z [104] U(eq) [Å2×103]In(1) 1506(1) 9778(1) 1316(1) 23(1)In(2) 938(1) 4958(1) 3935(1) 25(1)N(1) 498(2) 3483(7) 4389(2) 27(1)Br(3) 1636(1) 7615(1) 685(1) 36(1)C(3) 517(3) 10298(10) 1448(3) 34(2)Br(4) 644(1) 7406(1) 4399(1) 41(1)Fe(5) 1860(1) 12278(1) 1098(1) 30(1)N(6) 1860(2) 8087(8) 1875(2) 31(1)Fe(6) 753(1) 5181(1) 3053(1) 31(1)O(2) 1690(2) 4879(9) 2960(3) 59(2)C(8) 1660(3) 5743(10) 4802(3) 38(2)O(1) 552(3) 5262(9) 2039(2) 64(2)C(11) 583(2) 1840(9) 4303(3) 30(2)N(3) 1631(2) 5527(7) 4286(2) 31(1)O(7) 1016(2) 13886(7) 1039(3) 54(2)N(5) 752(2) 9384(8) 1120(2) 32(1)O(5) 2049(3) 10849(8) 214(2) 58(2)O(6) 2346(3) 11983(10) 2020(3) 78(3)C(17) 651(3) 9976(9) 637(3) 36(2)C(18) 1073(2) 1515(8) 4404(3) 30(2)C(21) 614(3) 7768(10) 1123(3) 40(2)C(22) 625(3) 5243(10) 2440(3) 36(2)N(4) 1133(2) 10132(7) 2060(2) 33(2)C(25) 2320(3) 8049(12) 1802(3) 48(2)C(26) 526(3) 6988(11) 3150(3) 50(2)C(27) 1330(3) 5003(9) 3010(3) 37(2)N(2) 1334(2) 2410(7) 4092(2) 28(1)O(3) 370(3) 8195(10) 3179(3) 82(3)C(34) 1686(3) 6503(9) 1847(3) 39(2)C(37) 1932(2) 4293(10) 4159(3) 36(2)C(39) 1779(2) 2732(9) 4310(3) 28(2)O(4) 221(2) 2431(8) 3097(2) 55(2)C(44) 33(2) 3834(10) 4226(3) 32(2)C(50) 1331(3) 9055(11) 2417(3) 37(2)C(40) 427(3) 3514(11) 3100(3) 37(2)C(41) 546(3) 3830(10) 4904(3) 36(2)C(52) 655(3) 9884(10) 1960(3) 39(2)C(47) 1343(3) 13216(9) 1061(3) 38(2)C(42) 1770(3) 6987(11) 4087(4) 50(2)C(51) 1212(3) 11702(10) 2221(3) 41(2)C(45) 2150(3) 12041(11) 1656(4) 49(2)C(43) 1363(3) 1624(10) 3640(3) 38(2)C(46) 1966(3) 11337(10) 565(3) 40(2)C(53) 1805(3) 8730(10) 2351(3) 37(2)O(8) 2436(2) 14846(8) 934(4) 73(3)C(60) 2183(3) 13854(11) 1000(4) 49(2)

Tabellen 124
Ergänzende kristallographische Daten
Tabelle 48: Kristallographische Daten und Meßparameter für[(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2 (19).
Formel C36H64Br6FeIn2K2O20
Formelmasse 1660.02 a. m. u.
Kristallfarbe und -form gelbliche Quader; 0.25×0.20×0.15 mm
Meßtemperatur [K] 203(2)
Raumgruppe Orthorhombisch, Pba2
a [pm] 1176.3(1)
b [pm] 2285.2(3)
c [pm] 1085.1(1)
α [°] 90
β [°] 90
γ [°] 90
V [106×pm3] 2916.8(6)
Z 4
ρber. [g×cm-3] 1.890
F(000) 1624
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 5.345
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 6856
Anzahl der unabhängigen Reflexe 4483 [R(int) = 0.0421]
Meßbereich 1.78° < Θ < 25.08°; h(-11/13), k(-27/9), l(-12/12)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0533
wR2 0.1289
Goodness-of-fit auf F2 0.977
Restelektronendichte [eÅ-3] max. +1.919, min. -1.148

Tabellen 125
Tabelle 49: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von [(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2 (19).
Atom x [104] y [104] z [104] U(eq) [Å2×103]In(1) 10195(1) -915(1) 5519(1) 35(1)Br(1) 8472(1) -1586(1) 5328(1) 47(1)Fe(1) 10000 0 4083(2) 33(1)Br(2) 11771(1) -1601(1) 4796(1) 49(1)Br(3) 10727(1) -797(1) 7753(1) 51(1)O(1) 12407(8) 49(4) 4851(10) 58(3)O(2) 9713(10) -879(4) 2214(9) 65(3)C(1) 11444(14) 34(5) 4576(12) 44(3)C(2) 9909(11) -576(6) 2901(15) 49(3)K(1) 5184(2) -1402(1) 109(3) 47(1)O(11) 3442(12) -2070(6) 996(15) 101(5)C(12) 2351(15) -1690(9) 730(30) 139(13)C(13) 2420(20) -1127(12) 850(30) 133(11)O(14) 3257(13) -840(7) 791(11) 91(4)C(15) 3390(20) -433(10) 1813(16) 127(11)C(16) 4150(20) -726(14) 2860(30) 169(15)O(17) 5104(11) -836(6) 2377(12) 87(4)C(18) 5839(14) -1201(7) 3282(13) 63(4)C(19) 5417(19) -1794(9) 3370(20) 95(6)O(110) 5393(11) -2055(6) 2320(11) 87(4)C(111) 4538(16) -2553(6) 2220(16) 71(5)C(112) 3300(30) -2306(13) 1970(30) 182(19)C(21) 7530(30) -673(12) -1480(20) 137(10)C(22) 6579(17) -302(8) -1653(19) 82(6)O(23) 5362(14) -699(7) -1969(14) 115(6)C(24) 5550(30) -801(10) -2950(20) 181(18)C(25) 4409(17) -1264(9) -3015(15) 83(6)O(26) 4814(10) -1889(7) -2216(12) 99(5)C(27) 5370(40) -2067(19) -2980(20) 290(30)C(28) 6000(30) -2506(7) -1914(15) 118(10)O(29) 6741(11) -2125(6) -921(12) 91(4)C(210) 7730(30) -1988(17) -1330(30) 230(30)C(211) 8069(16) -1525(10) -390(20) 92(6)O(212) 7286(14) -948(6) -449(12) 113(6)

Tabellen 126
Ergänzende kristallographische Daten
Tabelle 50: Kristallographische Daten und Meßparameter für [(CO)5Mo(η2-C8H14] (20).
Formel C13H14MoO5
Formelmasse 346.18 a. m. u.
Kristallfarbe und -form farblose Quader; 0.45×0.40×0.30 mm
Meßtemperatur [K] 203(2)
Raumgruppe Monoklin, P2(1)/c
a [pm] 1236.8(6)
b [pm] 994.2(4)
c [pm] 1225.0(5)
α [°] 90
β [°] 108.35(3)
γ [°] 90
V [106×pm3] 1429.7(11)
Z 4
ρber. [g×cm-3] 1.608
F(000) 696
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 0.929
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 3441
Anzahl der unabhängigen Reflexe 2078 [R(int) = 0.0776]
Meßbereich 2.68° < Θ < 25.58°; h(-11/8), k(-5/12), l(-13/14)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0357
wR2 0.0902
Goodness-of-fit auf F2 1.067
Restelektronendichte [eÅ-3] max. +0.518, min. -0.853

Tabellen 127
Tabelle 51: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von [(CO)5Mo(η2-C8H14] (20).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
Mo(1) 3504(1) 1141(1) 1819(1) 26(1)
C(1) 2773(4) -52(3) 3282(3) 30(1)
C(2) 2856(4) -1016(3) 2511(3) 29(1)
C(3) 1827(4) -1670(4) 1672(3) 40(1)
C(5) 1375(4) -2800(5) 3382(4) 48(1)
C(4) 1494(5) -2946(4) 2173(4) 52(1)
C(6) 583(5) -1692(5) 3535(4) 55(1)
C(7) 1148(5) -398(5) 4098(4) 53(1)
C(8) 1678(4) 499(4) 3378(4) 43(1)
C(11) 3913(4) -308(4) 818(3) 35(1)
O(11) 4145(4) -1053(3) 228(3) 56(1)
C(12) 4100(3) 2472(3) 955(3) 29(1)
O(12) 4463(3) 3245(3) 464(2) 41(1)
C(13) 3122(4) 2743(4) 2695(3) 37(1)
O(13) 2942(4) 3667(3) 3146(3) 63(1)
C(14) 1932(5) 1295(4) 595(4) 35(1)
O(14) 1084(3) 1398(3) -112(3) 55(1)
C(15) 5119(5) 1035(3) 3002(4) 35(1)
O(15) 6013(4) 1015(3) 3603(3) 56(1)

Tabellen 128
Ergänzende kristallographische Daten
Tabelle 52: Kristallographische Daten und Meßparameter für [(CO)5W(η2-C8H14] (21).
Formel C13H14WO5
Formelmasse 434.09 a. m. u.
Kristallfarbe und -form gelbe Quader; 0.48×0.35×0.25 mm
Meßtemperatur [K] 203(2)
Raumgruppe Monoklin, P21/(c)
a [pm] 1221.5(2)
b [pm] 989.1(1)
c [pm] 1234.3(2)
α [°] 90
β [°] 108.5(4)
γ [°] 90
V [106×pm3] 1414.2(3)
Z 4
ρber. [g×cm-3] 2.039
F(000) 824
λ [Mo(Kα), pm] 71.073
Absorptionskoeffizient [mm-1] 8.180
Gerät Bruker AXS Flächenzähler
Meßmodus ω-scan
Anzahl der Reflexe 3331
Anzahl der unabhängigen Reflexe 2310 [R(int) = 0.1029]
Meßbereich 1.76° < Θ < 25.27°; h(-14/8), k(-11/11), l(-14/12)
Verfeinerung Full-matrix least-squares auf F2
R1 0.0871
wR2 0.2363
Goodness-of-fit auf F2 1.246
Restelektronendichte [eÅ-3] max. +2.863, min. -5087
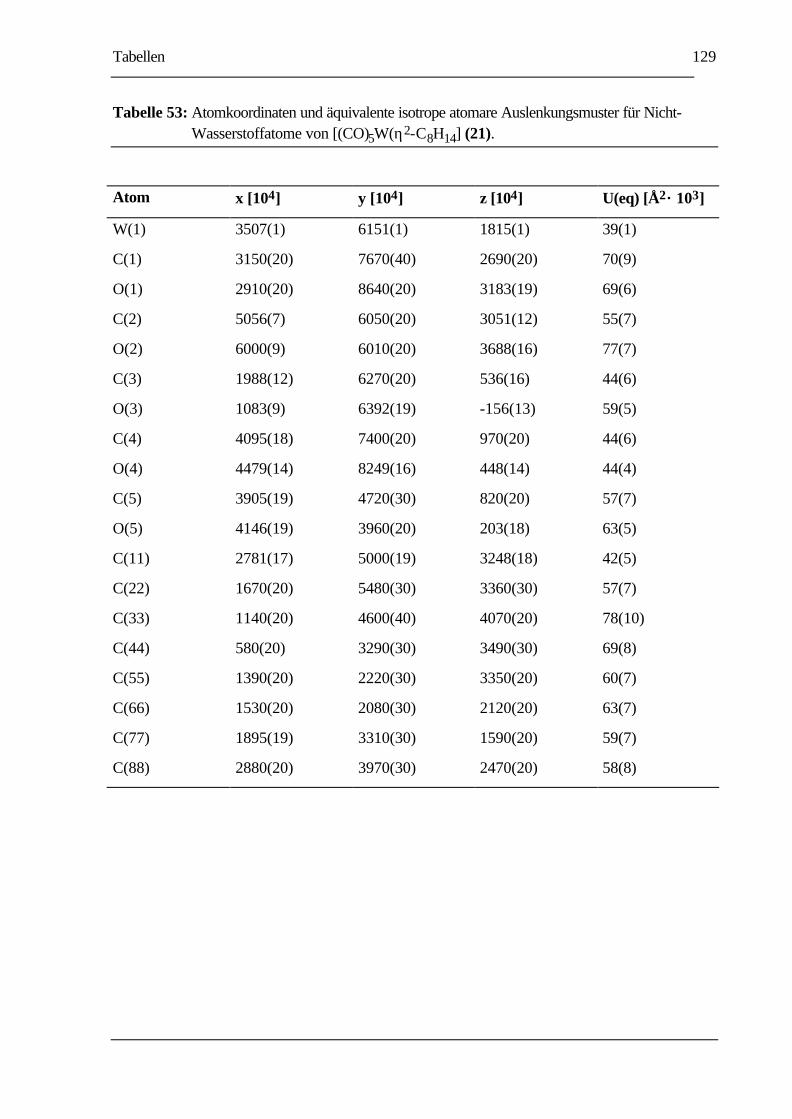
Tabellen 129
Tabelle 53: Atomkoordinaten und äquivalente isotrope atomare Auslenkungsmuster für Nicht-Wasserstoffatome von [(CO)5W(η2-C8H14] (21).
Atom x [104] y [104] z [104] U(eq) [Å2×103]
W(1) 3507(1) 6151(1) 1815(1) 39(1)
C(1) 3150(20) 7670(40) 2690(20) 70(9)
O(1) 2910(20) 8640(20) 3183(19) 69(6)
C(2) 5056(7) 6050(20) 3051(12) 55(7)
O(2) 6000(9) 6010(20) 3688(16) 77(7)
C(3) 1988(12) 6270(20) 536(16) 44(6)
O(3) 1083(9) 6392(19) -156(13) 59(5)
C(4) 4095(18) 7400(20) 970(20) 44(6)
O(4) 4479(14) 8249(16) 448(14) 44(4)
C(5) 3905(19) 4720(30) 820(20) 57(7)
O(5) 4146(19) 3960(20) 203(18) 63(5)
C(11) 2781(17) 5000(19) 3248(18) 42(5)
C(22) 1670(20) 5480(30) 3360(30) 57(7)
C(33) 1140(20) 4600(40) 4070(20) 78(10)
C(44) 580(20) 3290(30) 3490(30) 69(8)
C(55) 1390(20) 2220(30) 3350(20) 60(7)
C(66) 1530(20) 2080(30) 2120(20) 63(7)
C(77) 1895(19) 3310(30) 1590(20) 59(7)
C(88) 2880(20) 3970(30) 2470(20) 58(8)

F
ZUSAMMENFASSUNG

Zusammenfassung der Ergebnisse und Ausblick 131
8 Zusammenfassung der Ergebnisse
Die vorliegende Arbeit beschreibt die Synthese, Charakterisierung und beschichtungstechnische
Anwendung von Übergangsmetall-substituierten und –stabilisierten Aluminium-, Gallium- und
Indiumverbindungen. Die Reaktivität höhervalenter Carbonylmetallate gegenüber dreiwertigen
(Alkyl-) Erdmetallhalogeniden führt zu der Stoffklasse der Übergangsmetall-substituierten Alanen,
Gallanen und Indanen. In dieser Arbeit wurde anhand der Metallkomplexe (CO)nM-E[(X)(Lm)] die
Reihe für E = Al, Ga, In, für X = Cl, Br, I, CH3, für L = N-Donoren und für Cr, Mo ,W sowie Fe
systematisch vervollständigt.
8.1 Carbonylhaltige Übergangsmetall-substituierte Alane, Gallane und Indane
Durch Umsetzungen der Carbonylmetallat-Dianionen [M(CO)n]2- (M = Cr, Mo, W, n = 5; M = Fe,
n = 4) mit (Alkyl-)Erdmetallhalogeniden (Al, Ga, In) konnten nach doppelter Salzeliminierung die
neutralen und ionischen donorstabilisierten Systeme (CO)nM-E[(X)(TMPDA)], (CO)4Fe-
In[(Br)(PMDETA) und (CO)4Fe-Ga[(PMDETA)]+I- (M = Cr, Mo, W, n = 5; M = Fe, n = 4; X =
Cl, Br, I, CH3; TMPDA = Tetramethylpropylendiamin, PMDETA = Pentamethyldiethylentriamin)
isoliert und röntgenstrukturanalytisch charakterisiert werden. Alle oben beschriebenen neuen
Verbindungen sind überraschend stabil und ohne Schutzgasatmosphäre längere Zeit haltbar. Auf der
Basis von strukturellen und spektroskopischen Daten lassen sich die neuen Komplexe in die schon
bekannte Basis unseres Arbeitskreises einordnen und klassifizieren. Zusammenfassend läßt sich
sagen, daß die Erdmetall-Systeme (Al, Ga, In) am besten als donorstabilisierende Spezies
aufzufassen sind. Eine Unterteilung in E(I)- und E(III)-Komplexe ist irrelevant und entspricht nicht
mehr dem Stand der Technik. Es handelt sich bei allen Verbindungen um Donor-Akzeptor-Systeme,
in denen ein E(X)L2-, E(X)L3- oder EL3-Fragment mit seinem freien Elektronenpaar an ein
16e-[M(CO)n]-Fragment koordiniert. Die E(X)L2-, E(X)L3- oder EL3-Einheit verhält sich dabei
wie ein starker σ-Donor mit sehr schwachen Akzeptoreigenschaften. Die M-E-Bindung ist somit
polar-kovalent, mit hohen ionischen Anteilen. In Abbildung 32 sind einige der neuen Komplexe in
zusammengefaßter Form dargestellt.

Zusammenfassung der Ergebnisse und Ausblick 132
Abbildung 32: Molekülstrukturen einiger neuer Verbindungen im Kristall. Links oben: (CO)4Fe-In[(Br)(TMPDA)]; Rechts oben: (CO)4Fe-Ga[(Cl)(TMPDA)]; Mitte links:(CO)4Fe-In[(Br)(PMDETA)]; Mitte rechts: (CO)4Fe-Ga[(Br)(TMPDA)]; Linksunten: (CO)5Cr-Al[(Cl)(TMPDA)]; Rechts unten: (CO)4Fe-Ga[(I)(TMPDA)]
In Abhängigkeit vom Niveau der quantenmechanischen Rechnungen, in unserer Arbeitsgruppe und
der Literatur, und dem gewählten Beschreibungsmodell (insbesondere der Methodik und den
Konzepten, nach denen Ladungsverteilungen berechnet werden) ergeben sich mehr oder weniger
detaillierte Bilder der Bindungsverhältnisse, deren gemeinsamer und dominierender Faktor die
Bindungspolarität ist.

Zusammenfassung der Ergebnisse und Ausblick 133
Diese läßt sich gemäß der Reihe E(R)L2 > E(X)L2 > ER und Al > Ga, In in Abhängigkeit von der
Koordinationszahl von E (KZ = 2, 4) und den daran gebundenen Substituenten R (CH3, Cp*), X
(Cl, Br, I) und L (TMPDA, PMDETA) bestimmen. Insbesondere die koordinativ gesättigten
Fragmente E(X)L2 entsprechen hier weitgehend den Amin- oder Phosphanliganden (py, NR3, PR3),
erlauben aufgrund des elektropositiven Charakters des Zentrums E deutlich erhöhte negative
Ladungsdichten am d-Metall. Die M-E-Bindungen in den diskutierten Komplexen ((1), (2), (5), (11-
15), (17) und (18)) sind daher am besten als stark polare Lewis-Säure-Base-Addukte aufzufassen.
Eine Analogie der ER-Liganden (R = Cp*, Ar*, C(SiMe3)3, Si(SiMe3)3) zum CO-Ligand besteht
gewiß, in strukturchemischer bzw. koordinationschemischer Sicht.
Ein weiteres Ergebnis dieser Arbeit war die Möglichkeit der Verdrängung von Iodid als schwach
gebundenes Anion als Abgangsgruppe durch dreizähnige, tripodale Chelatliganden (Abbildung 33).
Abbildung 33: Verdrängung eines Halogenid-Liganden durch mehrzähnige Chelatliganden amBeispiel des Komplexes (CO)4Fe-Ga[(PMDETA)]+I- im Kristall.
Ein Teilziel dieser Arbeit war es am komplexgebundenen Zentrum E eine leichte Abspaltung der
stabilisierenden Lewis-Base-Liganden und gleichzeitige Einführung eines Restes R zu erreichen.

Zusammenfassung der Ergebnisse und Ausblick 134
Wichtig für die angestrebte Chemie war ein geeignetes nucleophiles Übertragungsreagenz zu finden,
um zu niedrig koordinierten Zentren E zu gelangen. Dabei wurden Kronenether hinsichtlich ihrer
Eignung für einen diesbezüglichen Einsatz untersucht. Dies kann nur im Sinne einer thermodynamisch
bevorzugten Lewis-Säure/Base-Reaktion erfolgen. Auf dem Weg zu einer erfolgreichen
Syntheseroute konnte ein Kronenetherderivat gemäß Abbildung 34 erhalten werden. Die
Möglichkeit der Verdrängung von Halogenid-Liganden an E(X)L2-Fragmenten durch mehrzähnige
Chelatliganden wurde für eine Reihe von Übergangsmetall-Erdmetall-Verbindungen untersucht,
gelingt jedoch nicht in allen untersuchten Fällen. Jedoch zeigt das Beispiel in Abbildung 33 die
prinzipielle Zugänglichkeit der kationischen Intermediate [(CO)nM-ELm]+. Entscheidenden Einfluß
wird dabei die Wahl der mehrzähnigen Lewis-Base-Liganden und des Solvens sowie die Größe des
Halogenid- sowie des Erdmetallatoms haben.
Abbildung 34: Molekülstruktur der neuen Verbindung [(CO)4Fe-(InBr3)2][K(12-Krone-4)2]2

Zusammenfassung der Ergebnisse und Ausblick 135
8.2 Intermetallische Schichten
Die Synthese des Carbonylmetall-Komplexes [(Cp*Al)Cr(CO)5] nach Schnöckel et al. 1999
konnte innerhalb dieser Arbeit nachvollzogen werden. Die Annahme, daß dieser Komplex unzersetzt
in die Gasphase überführbar ist, wurde bestätigt. Demzufolge ist [(Cp*Al)Cr(CO)5] zur Generierung
von Al/Cr-Filmen mittels MOCVD-Technik geeignet, wobei sich bei der Abscheidung des
Precursors auf den Si-Substraten chromreiche Filme der Zusammensetzung Cr2-xAl1-x (-0.2 < x >
0.2) gebildet haben.
Darüber hinaus sind weitere Synthesen von M-E-Komplexen mit einer terminalen Cp*Al-Einheit
vom Typ Cp*Al-M(CO)5 (M = Mo, W) geplant, die ebenfalls als Precursoren für MOCVD-
Anwendungen dienen sollen.
Zusammenfassend läßt sich feststellen, daß Cp*Al ein geeigneter Komplexligand zur Synthese von
Erdmetall-Übergangsmetall-Komplexen darstellt. Die Koordinationschemie von niederkoordinierten
Organoaluminium-Fragmenten an elektronenreichen Übergangsmetallen mit π-aciden CO-Gruppen
stellt aus strukturchemischer und bindungstheoretischer Sicht ein vielversprechender, ausbaufähiger
Sektor der Chemie dar. Bemerkenswert wenig ist noch immer über die Reaktivität der M-E-
Komplexe bekannt. Es besteht nach wie vor die Frage, inwieweit sich die vielen neuen Verbindungen
als Bausteine für weitere Synthesen nutzen lassen.

G
LITERATURVERZEICHNIS

Literaturverzeichnis 137
9 Literaturverzeichnis
[1] W. Hieber, U. Teller, Z. Anorg. Allg. Chem. 1942, 249, 43.
[2] W. R. Robinson, D. P. Schussler, Inorg. Chem. 1973, 12, 848.
[3] G. Natta, G. Mazzanti, Tetrahedron 1960, 8, 86.
[4] P. Corradini, A. Sirigu, Inorg. Chem. 1967, 6, 601.
[5] F. N. Tebbe, L. J. Guggenberger, J. Chem. Soc., Chem. Commun. 1973, 227.
[6] G. Erker, R. Zwettler, C. Krüger, R. Noe, S. Werner, J. Am. Chem. Soc. 1990, 112,
9620.
[7] G. Erker, M. Albrecht, C. Krüger, S. Werner, P. Binger, F. Langhauser,
Organometallics 1992, 11, 3517.
[8] H. Brunner, P. C. Wailes, H. D. Kaesz, Inorg. Nucl. Chem. Lett. 1965, 1, 125.
[9] R. Kumar, H. Rahbarnoohi, M. J. Heeg, D. G. Dick, J. P. Oliver, Inorg. Chem. 1994,
33, 1103.
[10] A. Alich, N. J. Nelson, D. Strope, D. F. Shriver, Inorg. Chem. 1972, 11, 2976.
[11] J. M .Burlitch, M. E. Leonowicz, R. B. Petersen, R. E. Hughes, Inorg. Chem. 1979,
18, 1097.
[12] E. O. Fischer, Angew. Chem. 1974, 86, 651.
[13] H. Tobita, K. Ishiyama, Y. Kawano, S. Inomata, H. Ogino, Organometallics 1998,
17, 789.
[14] M. L. H. Green, P. Mountford, G. J. Smount, S. R. Speel, Polyhedron 1990, 9, 2763.
[15] E. O. Fischer, D. Wittmann, D. Himmelreich, U. Schubert, K. Ackermann, Chem. Ber.
1982, 115, 3141.
[16] H. Fischer, A. Montsch, U. Schubert, D. Neugebauer, Angew. Chem. 1981, 93, 483.
[17] U. Schubert, E. O. Fischer, D. Wittmann, Angew. Chem. 1980, 92, 662.
[18] M. R. Terry, C. Kelley, N. Lugan, G. L. Geoffreoy, B. S. Haggerty, A. L. Rheingold,
Organometallics 1993, 12, 3607.
[19] M. Bottrill, M. Green, A. G. Orpen, D. R. Saunders, I. D. Williams, J. Chem. Soc.,
Dalton Trans. 1989, 511.
[20] M. Green, A. G. Orpen, I. D. Williams, J. Chem. Soc., Chem. Commun. 1982, 493.
[21] G. Huttner, Acc. Chem. Res. 1986, 19, 406.

Literaturverzeichnis 138
[22] W. A. Herrmann, Angew. Chem. 1986, 98, 57.
[23] T. P. Fehlner, Comments Inorg. Chem. 1988, 7, 307.
[24] O. J. Scherer, Angew. Chem. 1985, 97, 905.
[25] O. J. Scherer, Comments Inorg. Chem. 1987, 6, 1.
[26] M. D. Vaira, P. Stoppioni, M. Perzzini, Polyhedron 1987, 6, 351.
[27] K. H. Whitmire, J. Coord. Chem. B 1988, 17, 95.
[28] W. A. Nugent, Metal-Ligand Multiple Bonds, Wiley, New York 1988.
[29] S. K. Boocock, S. G. Shore, Comprehensive Organometallic Chemistry, Pergamon,
Oxford 1982.
[30] M. J. Taylor, Metal to Metal Bonding, Academic Press, London 1975.
[31] A. T. T: Hsieh, Inorg. Chim. Acta 1975, 14, 87.
[32] N. A. Compton, R. J. Errington, N. C. Norman, Adv. Organomet. Chem. 1990, 31, 91.
[33] P. J. Brothers, P. P. Power, Adv. Organomet. Chem. 1996, 39, 1.
[34] A. H. Cowley, B. L. Benac, J. G. Ekerdt, R. A. Jones, K. B. Kidd, J. Y. Lee, J. E.
Miller, J. Am. Chem. Soc. 1988, 110, 6248.
[35] V. Lakhotia, D. A: Neumayer, A. H. Cowley, R. A. Jones, J. G. Ekerdt, Chem. Mater.
1995, 7, 546.
[36] F. Maury, L. Brandt, H. D. Kaesz, J. Organomet. Chem. 1993, 449, 159.
[37] Y.-J. Chen, H. D. Kaesz, Y. K. Kim, H.-J. Müller, R. S. Williams, Z. Xue, Appl. Phys.
Lett. 1989, 55, 2760.
[38] R. A. Fischer, W. Rogge, Electrochemical Society Proceedings 1997, 25, 1034.
[39] R. A. Fischer, A. Miehr, T. Metzger, Thin Solid Films 1996, 289, 147.
[40] R. A. Fischer, A. Miehr, Chem. Mater. 1996, 8, 497.
[41] R. A. Fischer, A. Miehr, M. M. Schulte, E. Herdtweck, J. Chem. Soc., Chem.
Commun. 1995, 337.
[42] R. A. Fischer, E. Herdtweck, T. Priermeier, Inorg. Chem. 1994, 33, 934.
[43] S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke, A. Kuhn, Angew.
Chem. 1993, 105, 1828.
[44] C. Schnitter, H. W. Roesky, C. Röpken, R. Herbst-Irmer, H.-G. Schmidt, M.
Noltemeyer, Angew. Chem. 1998, 110, 2059.

Literaturverzeichnis 139
[45] W. Uhl, A. Janzschak, J. Organomet. Chem. 1998, 555, 263.
[46] W. Uhl, W. Hiller, M. Layh, W. Schwarz, Angew. Chem. 1992, 104, 1378.
[47] W. Uhl, S. U. Keimling, K. W. Klinkhammer, W. Schwarz, Angew. Chem. 1997, 109,
64.
[48] C. Dohmeier, D. Loos, H. Schnöckel, Angew. Chem. 1996, 108, 141.
[49] W. Uhl, Angew. Chem. 1993, 105, 1449.
[50] R. J. Wehmschulte, K. Ruhlandt-Senge, M. M. Olmstead, H. Hope, B. E. Sturgeon, P.
P. Power, Inorg. Chem. 1993, 32, 2983.
[51] W. Uhl, A. Vester, W. Kaim, J. Poppe, J. Organomet. Chem. 1993, 454, 9.
[52] J. Su, X.-W. Li, R. C. Crittendon, G. H. Robinson, J. Am. Chem. Soc. 1997, 119,
5471.
[53] W. Uhl, M. Pohlmann, R. Wartchow, Angew. Chem. 1998, 110, 1007.
[54] J. Weiß, D. Stetzkamp, B. Nuber, R. A. Fischer, C. Boehme, G. Frenking, Angew.
Chem. 1997, 109, 95.
[55] W. Uhl, R. Graupner, M. Layh, U. Schütz, J. Organomet. Chem. 1995, 493, C1-C5.
[56] W. Uhl, M. Pohlmann, Organometallics 1997, 16, 2478.
[57] G. Linti, W. Koestler, Angew. Chem., Int. Ed. Engl. 1996, 35, 550.
[58] W. Uhl, S. U. Keimling, W. Hiller, M. Neumayer, Chem. Ber. 1995, 128, 1137.
[59] W. Uhl, S. U. Keimling, W. Hiller, M. Neumayer, Chem. Ber. 1996, 129, 400.
[60] W. Uhl, persönliche Mitteilung.
[61] U. Flörke, P. Balsaa, H.-J. Haupt, Acta Crystallogr., Sect. C 1986, 42, 275.
[62] H.-J. Haupt, P. Balsaa, U. Flörke, Z. Anorg. Allg. Chem. 1988, 557, 81.
[63] G. N. Harakas, B. R. Whittlesey, Inorg. Chem. 1997, 36, 2707.
[64] J. Weiß, Promotionsarbeit, Ruprecht-Karls-Universität Heidelberg, Heidelberg, 1997.
[65] M. M. Schulte, Promotionsarbeit, TU München, München, 1996.
[66] T. H. Cymbaluk, R. D. Ernst, Inorg. Chem. 1980, 19, 2381.
[67] J. C. Vanderhooft, R. D. Ernst, R. J. Neustadt, T. H. Cymbaluk, Inorg. Chem. 1982,
21, 1876.
[68] R. A. Fischer, M. M. Schulte, E. Herdtweck, M. R. Mattner, Inorg. Chem. 1997, 36,
2010.

Literaturverzeichnis 140
[69] M. M. Schulte, E. Herdtweck, G. Raudaschl-Sieber, R. A. Fischer, Angew. Chem.
1996, 108, 489.
[70] J. Su, X.-W. Li, R. C. Crittendon, C. F. Campana, G. H. Robinson, Organometallics
1997, 16, 4511.
[71] F. A. Cotton, X. Feng, Organometallics 1998, 17, 128.
[72] J. L. Atwood, S. G. Bott, P. B. Hitchcock, C. Eaborn, R. S. Shariffudin, J. D. Smith,
A. C. Sullivan, J. Chem. Soc. Dalton Trans. 1987, 747.
[73] O. J. Curnow, B. Schiemenz, G. Huttner, L. Zsolnai, J. Organomet. Chem. 1993, 459,
17.
[74] B. Schiemenz, G. Huttner, Angew. Chem. 1993, 105, 1840.
[75] H. Behrens, M. Moll, E. Sixtus, G. Thiele, Z. Naturforsch., Teil B 1977, 32, 1109.
[76] S. K. Boocock, S. G. Shore, Comprehensive Organometallic Chemistry Vol. 6,
Pergamon, Oxford, 1982.
[77] N. A. Compton, R. J. Errington, N. C. Norman, Adv. Organomet. Chem. 1990, 31, 91.
[78] K. Jonas, G. Wilke, Angew. Chem. 1969, 81, 534.
[79] A. T. T. Hsieh, M. J. Mays, J. Organomet. Chem. 1972, 37, 9.
[80] D. L. Thorn, R. L. Harlow, J. Am. Chem. Soc. 1989, 111, 2575.
[81] D. J. Patmore, W. A. G. Graham, Chem. Comm. 1965, 1.
[82] D. J. Patmore, W. A. G. Graham, Inorg. Chem. 1966, 5, 1586.
[83] J. Chatt, C. Eaborn, P. N. Kapoor, J. Organomet. Chem. 1970, 23, 109.
[84] J. Hoyano, D. J. Patmore, W. A. G. Graham, Inorg. Nucl. Chem. Letters 1968, 4, 201.
[85] A. T. T. Hsieh, M. J. Mays, Inorg. Nucl. Chem. Letters 1971, 7, 223.
[86] A. T. T. Hsieh, Ph. D. Thesis, University of Cambridge, Cambridge, 1971.
[87] A. T. T. Hsieh, M. J. Mays, J. Chem. Soc., Chem. Commun. 1971, 1234.
[88] A. T. T. Hsieh, Inorg. Chim. Acta 1975, 14, 87.
[89] M. L. H. Green, P. Mountford, G. J. Smout, S. R. Speel, Polyhedron 1990, 9, 2763.
[90] M. Tacke, H. Schnöckel, Inorg. Chem. 1989, 28, 2895.
[91] C. Dohmeier, C. Robl, M. Tacke, H. Schnöckel, Angew. Chem. 1991, 103, 594.
[92] C. Dohmeier, H. Krautscheid, H. Schnöckel, Angew. Chem. 1994, 106, 2570.
[93] A. T. T. Hsieh, M. J. Mays, J. Organomet. Chem. 1972, 37, 9.

Literaturverzeichnis 141
[94] H.-J. Haupt, F. Neumann, J. Organomet. Chem. 1974, 74, 185.
[95] W. Uhl, R. Graupner, M. Layh, U. Schütz, J. Organomet. Chem. 1995, 493, C1-C5.
[96] W. Uhl, M. Pohlmann, Organometallics 1997, 16, 2478.
[97] G. Linti, W. Koestler, Angew. Chem., Int. Ed. Engl. 1996, 35, 550.
[98] J. Uddin, C. Boehme, G. Frenking. submitted to Chem. Eur. J.
[99] C. Üffing, A. Ecker, R. Köppe, H. Schnöckel, Organometallics 1998, 17, 2373.
[100] R. Murugavel, V. Chandrasekhar, Angew. Chem. 1999, 111, 1289.
[101] E. O. Fischer, Angew. Chem. 1974, 86, 651.
[102] K. H. Klinkhammer, Angew. Chem. 1997, 109, 2414.
[103] W. Kutzelnigg, Angew. Chem. 1984, 96, 262.
[104] F. A. Cotton, A. H. Cowley, X. Feng, J. Am. Chem. Soc. 1998, 120, 1795.
[105] G. Linti, W. Köstler, Chem. Eur. J. 1998, 4, 942.
[106] C. Boehme, G. Frenking, Chem. Eur. J. 1999, 5, 2184.
[107] J. Weiß, R. A. Fischer, Y Pelletier, C. Reber, Inorg. Chem. 1997,
[108] W. A. Skupinski, J. C. Huffmann, J. W. Bruno, K. G. Caulton, J. Am. Chem. Soc.
1984, 106, 8128.
[109] R. B. Peterson, J. J. Stezowski, C. Wan, J. M. Burlith, R. E. Hughes, J. Am. Chem.
Soc. 1971, 93, 3532.
[110] S. B. Butts, E. M. Holt, S. H. Strauss, N. W. Alcock, R. E. Stimson, D. F. Shriver,
J. Am. Chem. Soc. 1979, 101, 5864.
[111] D. L. Grimmett, J. A. Labinger, J. N. Bonfiglio, S. T. Masuo, E. Shearin, J. S. Miller,
Organometallics 1983, 2, 1325.
[112] C. M. Lukehart, G. P. Torrence, J. V. Zeile, J. Am. Chem. Soc. 1975, 97, 6903.
[113] N. E. Kim, N. J. Nelson, D. F. Shriver, Inorg. Chim. Acta 1973, 7, 393.
[114] A. J. Conway, G. J. Gainsford, R. R. Schrieke, J. D. Smith, J. Chem. Soc., Dalton
Trans. 1975, 2499.
[115] J. D. Protasiewicz, A. Masschelein, S. J. Lippard, J. Am. Chem. Soc. 1993, 115, 808.
[116] J. D. Protasiewicz, B. S. Bronk, A. Masschelein, S. J. Lippard, Organometallics 1994,
13, 1300.
[117] J. F. Janik, E. N. Duesler, R. T. Paine, J. Organomet. Chem. 1987, 323, 149.

Literaturverzeichnis 142
[118] J. J. Schneider, C. Krüger, M. Nolte, I. Abraham, T. S. Ertel, H. Bertagnolli, Angew.
Chem. 1994, 106, 2537.
[119] H. Braunschweig, J. Müller, B. Ganter, Inorg. Chem. 1996, 35, 7443.
[120] B. N. Anand, I. Krossing, H. Nöth, Inorg. Chem. 1997, 36, 1979.
[121] I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Chuklanova, N. I. Kirillova, K.
Shestakova, I. N. Parshina, A. I. Gusev, G. K.-I. Magomedov, J. Am. Chem. Soc.
1993, 115, 3156.
[122] D. R. Lide, J. Chem. Phys. 1965, 42, 1013.
[123] P. J. Black, Acta Crystallogr. 1955, 8, 175.
[124] Alle Spektren wurden in CD2Cl2 bei 250 MHz und 298K aufgenommen. Die
chemischen Verschiebungen sind δ-Werte bezogen auf das Protonenrestsignal des
Lösungsmittels und gegen TMS (δ = 0) korrigiert.
[125] Alle Spektren wurden in CD2Cl2 bei 100 MHz und 298K aufgenommen. Die
chemischen Verschiebungen sind δ-Werte bezogen auf das Lösungsmittelsignal und
gegen TMS (δ = 0) korrigiert.
[126] C. Leis, D. L. Wilkinson, H. Handwerker, C. Zybill, G. Müller, Organometallics 1992
11, 514.
[127] R. Probst, C. Leis, S. Gamper, E. Herdtweck, C. Zybill, N. Auner, Angew. Chem.
1991, 103, 1155.
[128] Aufgenommen in CH2Cl2-Lösung als Film zwischen NaCl-Fenstern. Die Zuordnung
erfolgt in der Reihenfolge A1(e), A1(a) und E beginnend bei der höchsten Wellenzahl.
[129] IR: ν(CO)-Absorptionen [cm-1] KBr-Preßling
[130] IR: ν(CO)-Absorptionen [cm-1] Toluol
[131] (a) P. E. Riley, R. E. Davis, Inorg. Chem. 1980, 19, 159. (b) B. T. Kilbourn, U.
A. Reaburn, D. T. Thompson, J. Chem. Soc. A 1969, 1906. (c) F. A. Cotton
B. M. Troup, J. Am. Chem. Soc. 1974, 96, 3438. (d) G. Dettlaf, U. Behrens, E.
Weiss, J. Organomet. Chem. 1978, 152, 95.
[132] D. L. Reger, D. G. Garza, Organometallics 1998, 17, 3624.
[133] P. Jutzi, B. Neumann, G. Reumann, H.-G. Stammler, Organometallics 1998
17, 1305.

Literaturverzeichnis 143
[134] M. Stürmann, W. Saak, H. Marsmann, M. Weidenbruch, Angew. Chem. Int. Ed.
1999, 115, 187.
[135] (a) B.E. Bursten, K.J. Novo-Gradac, J. Am. Chem. Soc. 1987, 109, 904.
(b) B.E. Bursten, R.J. Strittmatter, Angew. Chem. Int. Ed. Engl. 1991, 30, 1069.
(c) G.S. Ferguson, P.T. Wolczanski, L. Parkanyi, M. Zonnevyelle,
Organometallics 1988, 7, 1967. (d) D. Selent, M. Ramm, C. Janiak, J.
Organomet. Chem. 1995, 501, 235. (e) G. Jansen, M. Schubart, B. Findeis,
L. H. Gade, I. J. Scowen, M. Mc. Partlin, J. Am. Chem. Soc. 1998, 120, 7239.
(f) J.E. Bender IV, A.J. Shusterman, M.M. Banaszak Holl, J.W. Kempf,
Organometallics 1999, 18, 1547.
[136] D. L. Reger, Y. Ding, Organometallics 1993, 12, 4485.
[137] (a) X. He, R. A. Bartlett, P. P. Power, Organometallics 1994, 13, 548. (b) R. M.
Campell, L. M. Clarkson, W. Clegg, D. C. R. Hockless, N. L. Pickett, N. C.
Norman, Chem. Ber. 1992, 124, 55.
[138] (a) F. A. Cotton, R. V. Parish, J. Chem. Soc. 1960, 1440. (b) P. M. Treichel, W. K.
Dean, W. M. Douglas, Inorg. Chem. 1972, 11, 1609. (c) D. J. Darensbourg, H. H.
Nelson, C. L. Hyde, Inorg. Chem. 1974, 13, 2135.
[139] R. A. Fischer, A. Miehr, H. Hoffmann, W. Rogge, C. Boehme, G. Frenking, E.
Herdtweck, Z. Anorg. Allg. Chem. 1999, im Druck, angenommen 24.04.1999.
[140] Auf Basis von DFT-Rechnungen ermittelte Daten für Bindungslängen und
IR-Frequenzen.
[141] M. Veith, K. Kunze, Angew. Chem. 1991, 103, 92.
[142] D. L. Reger, S. S. Mason, A. L. Rheingold, B. S. Haggerty, F. P. Arnold,
Organometallics 1994, 13, 5049.
[143] L. M. Clarkson, W. Clegg, N. C. Norman, P. W. Tucker, Inorg. Chem. 1988, 27, 2653.
[144] L. M. Clarkson, K. McCrudden, N. C. Norman, Polyhedron 1990, 9, 2533.
[145] L. M. Clarkson, W. Clegg, D. C. R. Hockless, N. C. Norman, L. J. Farrugia, S. G.
Bott, J. L. Atwood, J. Chem. Soc. Dalton Trans. 1991, 2241.
[146] L. M. Clarkson, N. C. Norman, L. J. Farrugia, Organometallics 1991, 10, 1286.
[147] F. P. Gabbai, A. Schier, J. Riede, H. Schmidbaur, Inorg. Chem. 1995, 34, 3855.

Literaturverzeichnis 144
[148] P. Lange, A. Schier, H. Schmidbaur, Inorg. Chem. 1996, 35, 637.
[149] K. Bader, Doktorarbeit; Humboldt-Universität Berlin, Berlin, 1997.
[150] K(s-C4H9)3BH wird von der Firma Aldrich Chemical Co. als 0.5 M THF-Lösung
unter dem Markennamen K-Selectride verkauft.
[151] J. E. Huheey, E. A. Kreiter, R. L. Kreiter (Hrsg.), Anorganische Chemie: Prinzipien
von Struktur und Reaktivität; de Gruyter-Verlag, Berlin, New York, 1995.
[152] (a) H. Preut, H.-J. Haupt, Acta Crystallogr., Sect. B 35 1979, 35, 2191. (b) J. L.
Atwood, S. G. Bott, P. B. Hitchcock, C. Eaborn, R. S. Shariffundin, J. D. Smith,
A. C. Sullivan, J. Chem. Soc., Dalton Trans. 1987, 747. (c) J. M. Cassidy, K.
H. Whitmire, Acta Crystallogr., Sect. C 46 1990, 1781. (d) V. G. Albano, M.
Cane, M. C. Iapalucci, G. Longoni, M. Monari, J. Organomet. Chem. 1991
407, C9. (e) L. M. Clarkson, N. C. Norman, L. J. Farrugia, Organometallics
1991, 10, 1286. (f) K. Merzweiler, F. Rudolph, L. Brands, Z. Naturforsch. B
1992, 47, 470. (g) R. A. Fischer, J. Behm, T. Priermeier, W. Scherer, Angew.
Chem., Int. Ed. Engl. 1993, 32, 746. (h) C.-C. Lin, G. Kong, H. Cho, B. R.
Whittlesey, Inorg. Chem. 1993, 32, 2705. (i) R. A. Fischer, E. Herdtweck, T.
Priermeier, Inorg. Chem. 1994, 33, 934. (j) P. Braunstein, M. Knorr, M.
Strampfer, A. DeCian, J. Fischer, J. Chem. Soc., Dalton Trans. 1994, 117.
(k) D. L. Reger, S. S. Mason, A. L. Rheingold, B. S. Haggerty, F. P. Arnold,
Organometallics 1994, 13, 5049. (l) C. J. Carmalt, N. C. Norman, R. F.
Pember, L. J. Farrugia, Polyhedron 1995, 14, 417.
[153] G. B. Stringfellow, Organometallic Vapor-Phase Epitaxy, Theory and Practice;
Academic Press; San Diego, 1989.
[154] M. L. Hitchman, K. F. Jensen, Chemical Vapor Deposition; Academic Press,
San Diego, 1993.
[155] D. Widmann, H. Mader, H. Friedrich, Technologie hochintegrierter Schaltungen
Band 19; Springer Verlag, Berlin, Heidelberg, 1988.
[156] H. M. Manasevit, W. I. Simpson, J. Electrochem. Soc. 1968, 12, 156.
[157] H. M. Manasevit, Appl. Phys. Lett. 1969, 116, 1725.
[158] H. M. Manasevit, J. Cryst. Growth 1972, 13/14, 306.

Literaturverzeichnis 145
[159] A. H. Cowley, R. A. Jones, Angew. Chem. 1989, 101, 1235.
[160] J. E. Miller, J. G. Ekerdt, Chem. Mater. 1992, 4, 7.
[161] E. Yablonovith, Science 1989, 246, 347.
[162] D. Bradley, New Scientist 1988, 14, 38.
[163] J. Hampden-Smith, T. T. Kodas, Chemical Vapour Deposition 1995, 1, 8.
[164] T. T. Kodas, J. Hampden-Smith, The Chemistry of Metal CVD, VCH, Weinheim,
1994.
[165] R. M. Fix, G. Gordon, D. M. Huffmann, J. Am. Chem. Soc. 1990, 112, 7833.
[166] R. A. Fischer, Habilitationsschrift, Technische Universität München, 1994.
[167] J. W. Orton, Chemtronics 1988, 3, 130.
[168] M. L. Hitchman, K. F. Jensen, Chemical Vapour Deposition, Academic Press, London,
1993.
[169] P. Zanella, G. Rossetto, J. O. Williams, Chem. Mater. 1991, 3, 225.
[170] R. A. Fischer, J. Weiß, Angew. Chem. 1999, 111, 6000.
[171] P. Zanella, G. Rossetto, J. O. Williams, Chem. Mater. 1991, 3, 225.
[172] G. B. Springfeld, J. Cryst. Growth 1968, 77, 1.
[173] H. D. Kaesz, R. S. Williams, R. F. Hicks, J. I. Zink, Y.-J. Chen, H.-J. Müller, Z. Xue,
D. Xu, D. K. Shuh, Y. K. Kim, New J. Chem. 1990, 14, 527.
[174] R. A. Fischer, J. Behm, T. Priermeier, W. Scherer, Angew. Chem. 1993, 105, 776.
[175] R. A. Fischer, H. D. Kaesz, S. I. Khan, H.-J. Müller, Inorg. Chem. 1990, 29, 1601.
[176] R. A. Fischer, W. Scherer, M. Kleine, Angew. Chem. 1993, 105, 778.
[177] R. A. Fischer, J. Behm, J. Organomet. Chem. 1991, 413, C10-C14.
[178] R. A. Fischer, J. Behm, E. Herdtweck, C. Kronseder, J. Organomet. Chem. 1992, 437,
C29-C34.
[179] R. A. Fischer, J. Behm, Chem. Ber. 1992, 125, 37.
[180] R. A. Fischer, J. Behm, T. Priermeier, J. Organomet. Chem. 1992, 429, 275.
[181] R. A. Fischer, T. Priermeier, W. Scherer, J. Organomet. Chem. 1993, 459, 65.
[182] R. A. Fischer, E. Herdtweck, T. Priermeier, Inorg. Chem. 1994, 33, 934.
[183] R. A. Fischer, T. Priermeier, Organometallics 1994, 13, 4306.
[184] R. A. Fischer, M. Kleine, O. Lehmann, M. Stuke, Chem. Mater. 1995, 7, 1863.

Literaturverzeichnis 146
[185] R. A. Fischer, A. Miehr, M. M. Schulte, T. Priermeier, Adv. Mater. 1995, 7, 58.
[186] R. A. Fischer, M. M. Schulte, T. Priermeier, J. Organomet. Chem. 1995, 493, 139.
[187] R. A. Fischer, Chem. unserer Zeit 1995, 29, 141.
[188] R. A. Fischer, A. Miehr, T. Priermeier, Chem. Ber. 1995, 128, 831.
[189] R. A. Fischer, M. M. Schulte, J. Weiß, L. Zsolnai, A. Jacobi, G. Huttner, G. Frenking,
C. Boehme, S. F. Vyboishchikov, J. Am. Chem. Soc.
[190] O. Segnitz, Diplomarbeit, Ruprecht-Karls-Universität Heidelberg, Heidelberg, 1998.
[191] K. M. Yu, W. Walukiewicz, J. M. Jaklevic, E. E. Haller, Appl. Phys. Lett. 1987, 51,
189.
[192] J. S. Solomon, S. R. Smith, J. Vac. Sci. Technol. 1987, A5, 1809.
[193] J. P. Harbison, T. Sands, N. Tabatabaie, W. K. Chan, L. T. Florez, V. G. Keramidas,
Appl. Phys. Lett. 1988, 53, 1717.
[194] N. Tabatabaie, T. Sands, J. P. Harbison, H. L. Gilchrist, V. G. Keramidas, Appl. Phys.
Lett. 1988, 53, 2528.
[195] T. Sands, J. P. Harbison, W. K. Chan, S. A. Schwarz, C. C. Chang, C. J. Palmstrom,
V. G. Keramidas, Appl. Phys. Lett. 1988, 52, 1216.
[196] J. P. Harbison, T. Sands, R. Ramesh, L. T. Florez, B. J. Wilkens, V. G. Keramidas, J.
Chryst. Growth 1991, 111, 978.
[197] D. A. Baugh, A. A. Talin, R. S. Williams, T.-C. Kuo, K. L. Wang, J. Vac. Sci.
Technol. 1991, B9, 2154.
[198] Y. K. Kim, D. A. Baugh, D. K. Shuh, R. S. Williams, L. P. Sadwick, K. L. Wang, J.
Mater. Res. 1990, 5, 2139.
[199] J. R. Arthur, Surf. Sci. 1974, 43, 449.
[200] T. Steinke, Hausarbeit im Rahmen der Ersten Staatsprüfung für das Lehramt für die
Sekundarstufe I und für die Sekundarstufe II, Ruhr-Universität Bochum, Bochum,
2000.
[201] Q. Yu, A. Purath, A. Donchev, H. Schnöckel, Organometallics 1999, 584, 94.
[202] Datenbank PDF, Set Nr. 48, International Center for Diffraction Data (ICDD).
[203] K. A. Singmaster, F. A. Houle, R. J. Wilson, J. Phys. Chem. 1990, 94, 6864.
[204] W. Strohmeier, Angew. Chem. 1964, 76, 873.

Literaturverzeichnis 147
[205] J. Nasielski, A. Colas, Inorg. Chem. 1978, 17, 237.
[206] W. Strohmeier, D. von Hobe, Chem. Ber. 1961, 94, 761.
[207] E. Koerner von Gustorf, F.-W. Grevels, Top. Curr. Chem. 1969, 13, 366.
[208] G. L. Geoffroy, M. S. Wrighton, Organometallic Photochemistry, Academic Press,
New York, 1979.
[209] L. Moggi, A. Juris, D. Sandrini, M. F. Manfrin, Rev. Chem. Intermed. 1981, 4, 171.
[210] M. S. Wrighton, J. L. Graff, C. L. Reichel, R. D. Sauner, Ann. N. Y. Acad. Sci. 1980,
333, 188.
[211] I. W. Stolz, G. R. Dobson, R. K. Sheline, Inorg. Chem. 1963, 2, 1264.
[212] M. Wrighton, G. S. Hammond, H. B. Gray, J. Organomet. Chem. 1974, 70, 283.
[213] F.-W. Grevels, M. Lindemann, R. Benn, R. Goddard, C. Krüger, Z. Naturforsch.,
Teil B 1980, 35, 1298.
[214] M. Herberhold, Angew. Chem. 1968, 80, 314.
[215] F.-W. Grevels, D. Schulz, E. Koerner von Gustorf, Angew. Chem. 1974, 86, 558.
[216] B. E. Foulger, F.-W. Grevels, D. Hess, E. Koerner von Gustorf, J. Leitich, J. Chem.
Soc. Dalton Trans. 1979, 1451.
[217] F.-J. Müller, Dissertationsarbeit, Universität Würzburg, Würzburg, 1968.
[218] D. F. Shriver, M. A. Dredzon, The Manipulation of Air-Sensitive Compounds;
Wiley-Interscience, New York, 1986.
[219] J. A. Gladysz, W. Tam, J. Org. Chem. 1978, 43, 2279.
[220] M. A. Schwindt, T. Lejon, L. S. Hegedus, Organometallics 1990, 9, 2814.
[221] H.-J. Koch, S. Schulz, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt, A. Heine, R.
Herbst-Irmer, D. Stalke, G. M. Sheldrick, Chem. Ber. 1992, 125, 1107.
[222] B. Armer, H. Schmidbaur, Chem. Ber. 1967, 100, 1521.
[223] H. Schmidbaur in Handbuch der Präparativen Anorganischen Chemie (Hrsg.:
G. Bauer), Enke, Stuttgart, 1978, 848.
[224] W. Klemm, W. Tilk, Z. Anorg. Allgem. Chem 1932, 207, 161.
[225] W. Fischer, O. Jübermann, Z. Anorg. Allgem. Chem. 1936, 227, 227.
[226] I. D. Corbett, R. K. McMullan, J. Amer. Chem. Soc. 1955, 77, 4217.
[227] F.-W. Grevels, V. Skibbe, J. Chem. Soc., Chem. Commun. 1984, 681.

Literaturverzeichnis 148
[228] G. Rabe, H. W. Roesky, D. Stalke, F. Pauer, G. M. Sheldrick, J. Organomet. Chem.
1991, 403, 11.
[229] G. H. Llinás, M. Mena, F. Palacios, P. Royo, R. Serrano, J. Organomet. Chem. 1988,
340, 37.
[230] P. Jutzi, H. Saleske, D. Bühl, H. Grohe, J. Organomet. Chem. 1983, 252, 29.
[231] C. Dohmeier, C. Robl, M. Tacke, H. Schnöckel, Angew. Chem. 1991, 103, 594.
[232] S. Schulz, H. W. Roesky, H. J. Koch, G. M. Sheldrick, D. Stalke, A. Kuhn, Angew.
Chem. 1993, 105, 1828.
[233] D. Loos, H. Schnöckel, J. Organomet. Chem. 1993, 463, 37.
[234] M. A. Schmidt, T. Lejon, L. S. Hegedus, Organometallics 1990, 9, 2814.
[235] F.-W. Grevels, V. Skibbe, J. Chem. Soc., Chem. Commun. 1984, 681.
[236] M. Wrighton, G. S. Hammond, H. B. Gray, J. Organomet. Chem. 1974, 70, 283.
[237] (a) I. W. Stolz, G. R. Dobson, R. K. Sheline, Inorg. Chem. 1963, 2, 1264. (b) F.-W.
Grevels, M. Lindemann, R. Benn, R. Goddard, C. Krüger, Z. Naturforsch., Teil B
1980, 35, 1298.
[238] L. E. Davis, N. C. McDonald, P. M. Palmberg, G. E. Riach, R. E. Weber, Handbook of
AUGER Electron Spectroscopy, Physical Electronics Industries Inc., Eden Prairie,
Minnesota, 1972.
[239] T. Sekine, Y. Nagasawa, M. K. Sakai, A. S. Parkes, J. D. Geller, A. Mogami, K.
Hirata, Handbook of AUGER Electron Spectroscopy, JEOL, Tokyo, 1982.
[240] G. E. McGuire, AUGER Electron Spectroscopy Reference Manual, Plenum Press,
New York, 1979.
[241] W. Massa, Kristallstrukturbestimmung, Teubner Studienbücher Chemie, Stuttgart,
1996.
[242] H. Krischner, B. Koppelhuber-Bitschnau, Röntgenstrukturanalyse und
Rietveldmethode: Eine Einführung, Braunschweig, 1994.
[243] L. C. Feldman, J. W. Mayer, Fundamentals of survace and thin film analysis, Elsevier,
New York, 1986.
[244] T. G. Finstadt, W.-K. Chu, Rutherford backscattering spectrometry on thin solid films,
Academic Press, San Diego, 1988.

Literaturverzeichnis 149
[245] A. Sherman, Chemical vapour deposition for microelectronics, New Jersey, 1987.