
Vorlesungsskripte “Physikalisch e Chemie für … · Festkörper, heterogene Stoffgemische)...
29
1 Vorlesungsskripte “Physikalische Chemie für Pharmazeuten” 0. Einführung Wozu brauchen Studierende der Pharmazie Physikalische Chemie ? Die Physikalische Chemie als Bindeglied zwischen Physik und Chemie ist u.a. das Fundament für die Instrumentelle Analytik. 0.1 Gliederung: I. Thermodynamik II. Elektrolyte/Elektrochemie III. Kinetik IV. Elementare Einführung in die “Computational Chemistry” 0.2 Literatur: E. Ehlers, Chemie I, Jungjohann Verlagsgesellschaft, Neckarsulm (6. Aufl. 1994) E. Ehlers, Analytik II, Jungjohann Verlagsgesellschaft, Neckarsulm (7. Aufl. 1993) G. Rücker, M. Neugebauer, G.G. Willems, Instrumentelle Pharmazeutische Analytik, Wiss. Verlagsges. Stuttgart 1992 I. Thermodynamik (Gleichgewichtsthermodynamik) 1. Einführung 1.1 Gegenstand -Quantitative Beschreibung der Zustandsformen der Materie (Gase, Flüssigkeiten, Lösungen, Festkörper, heterogene Stoffgemische) hinsichtlich ihrer Stabilität und Umwandlungsmöglichkeiten. Behandelt Vielteilchensysteme ohne nach der mikroskopischen Natur der Teilchen zu fragen. -Speziell die Chemische Gleichgewichtsthermodynamik beschäftigt sich mit Phasengleichgewichten und Reaktionsgleichgewichten (z.B. Massenwirkungsgesetz), ihrer Wärmetönung, der Möglichkeit der Gewinnung von Arbeit, der Ausbeute von chemischen Reaktionen in Abhängigkeit von Druck, Temperatur, Zusammensetzung,... Untersucht die Triebkräfte chemischer Reaktionen. Macht keine Aussagen zur Zeitabhängigkeit (Kinetik)! Bedeutung für Reaktionsführung in der chemischen Synthese (industriell und biochemisch), Stofftrennung und -reinigung; Umwandlung von Energie verschiedener Formen. 1.2 Grundbegriffe Wieviel Teilchen enthält ein Vielteilchensystem ? Keine exakte Zahl angebbar; Angabe der Stoffmenge (Formelzeichen n) in Mol. Definition : 1 mol = 6.023×10 Teilchen ; N = 6.023×10 mol 23 23 -1 A Phänomenologische Theorie, kommt mit wenigen grundlegenden Postulaten (nicht ableitbaren Axiomen) aus: 0., 1., 2. und 3. Hauptsatz der Thermodynamik. Sie beschreibt makroskopische Eigenschaften von stofflichen Systemen mit Hilfe von Zustandsgrößen: T - Temperatur, U - Innere Energie (Gesamter Energieinhalt von Stoffen) H - Enthalpie (Wärmeenergieinhalt von Stoffen bei konstantem Druck)
Transcript of Vorlesungsskripte “Physikalisch e Chemie für … · Festkörper, heterogene Stoffgemische)...

1
Vorlesungsskripte “Physikalische Chemie für Pharmazeuten”
0. EinführungWozu brauchen Studierende der Pharmazie Physikalische Chemie ?Die Physikalische Chemie als Bindeglied zwischen Physik und Chemie ist u.a. das Fundamentfür die Instrumentelle Analytik.
0.1 Gliederung:I. ThermodynamikII. Elektrolyte/ElektrochemieIII. KinetikIV. Elementare Einführung in die “Computational Chemistry”
0.2 Literatur: E. Ehlers, Chemie I, Jungjohann Verlagsgesellschaft, Neckarsulm (6. Aufl. 1994)E. Ehlers, Analytik II, Jungjohann Verlagsgesellschaft, Neckarsulm (7. Aufl. 1993)G. Rücker, M. Neugebauer, G.G. Willems, Instrumentelle Pharmazeutische Analytik, Wiss.Verlagsges. Stuttgart 1992
I. Thermodynamik (Gleichgewichtsthermodynamik)
1. Einführung1.1 Gegenstand-Quantitative Beschreibung der Zustandsformen der Materie (Gase, Flüssigkeiten, Lösungen,Festkörper, heterogene Stoffgemische) hinsichtlich ihrer Stabilität undUmwandlungsmöglichkeiten. Behandelt Vielteilchensysteme ohne nach der mikroskopischenNatur der Teilchen zu fragen. -Speziell die Chemische Gleichgewichtsthermodynamik beschäftigt sich mitPhasengleichgewichten und Reaktionsgleichgewichten (z.B. Massenwirkungsgesetz), ihrerWärmetönung, der Möglichkeit der Gewinnung von Arbeit, der Ausbeute von chemischenReaktionen in Abhängigkeit von Druck, Temperatur, Zusammensetzung,...Untersucht die Triebkräfte chemischer Reaktionen.Macht keine Aussagen zur Zeitabhängigkeit (�Kinetik)!Bedeutung für Reaktionsführung in der chemischen Synthese (industriell und biochemisch),
Stofftrennung und -reinigung; Umwandlung von Energie verschiedener Formen.
1.2 GrundbegriffeWieviel Teilchen enthält ein Vielteilchensystem ? Keine exakte Zahl angebbar; Angabe derStoffmenge (Formelzeichen n) in Mol.Definition : 1 mol = 6.023×10 Teilchen ; N = 6.023×10 mol23 23 -1
A
Phänomenologische Theorie, kommt mit wenigen grundlegenden Postulaten (nicht ableitbarenAxiomen) aus: 0., 1., 2. und 3. Hauptsatz der Thermodynamik.Sie beschreibt makroskopische Eigenschaften von stofflichen Systemen mit Hilfe vonZustandsgrößen:T - Temperatur,U - Innere Energie (Gesamter Energieinhalt von Stoffen)H - Enthalpie (Wärmeenergieinhalt von Stoffen bei konstantem Druck)

U � U(v,T,n) ; v � v(p,T,n) ; H � H(p,T,n) ;S � S(p,T,n) oder S � S(v,T,n) ; F � F(v,T,n) ; G � G(p,T,n)
�SchmelzH � Hfl. � HEis Schmelzenthalpie�Krist.H � HEis � Hfl. Kristallisationsenthalpie
�RH � Reaktionsenthalpie�RU � Reaktionsenergie
Schmelzen
KristallisierenWasser (flüssig)Eis
�(Z.G.) � Z.G.(Ende) � Z.G.(Anfang)
mv
� � Dichte
vm
� Vspez. Spezifisches Volumen
vn
� V (Molvolumen) ; mn
� M Molmasse ; un
� U molare innere Energie
xi �ni
�i
ni
�
ni
ngesamt
�i �vi
�i
vi
�
vi
vgesamt
2
S - Entropie (Quotient aus reversibel ausgetauschter Wärme und Temperatur, Maß für denUnordnungszustand eine Systems)
F - Freie Energie (maximale Nutzarbeit bei konstantem Volumen)G - Freie Enthalpie (maximale Nutzarbeit bei konstantem Druck)
Diese hängen von Zustandsvariablen (Druck, p, Temperatur T, Stoffmenge, n, ...) ab.Funktionale Zusammenhänge: Zustandsfunktionen, Zustandsgleichungen.
Ziel: Vorausberechnung unbekannter Eigenschaften aus gegebenen Eigenschaften.Zustandsgrößen sind wegunabhängig! Reihenfolge der Änderung von Zustandsvariablen hatkeinen Einfluss. Kreisprozesse verändern Zustandsgrössen nicht.Untersucht werden Zustandsänderungen, hervorgerufen durch Änderung von Zustandsvariablenz.B. Phasenumwandlungen (= “physikalischer Reaktionen” ):
oder chemische Reaktionen: Ausgangsstoffe � Endstoffe
Änderung einer Zustandsgröße (Z.G.) - Definition,Vorzeichengebung:
Extensive und Intensive Größen (Eigenschaften)Intensive Größen (mit Großbuchstaben bezeichnet) hängen im Gegensatz zu den extensiven (mitKleinbuchstaben bezeichnet) nicht von der Stoffmenge (ggf. der Masse, dem Volumen ) ab.Intensive Größen werden werden aus extensiven erhalten durch Division durch- die Stoffmenge: molare Größen, z.B.:
- das Volumen: Dichtegrößen, z.B.:
- die Masse : spezifische Größen
KonzentrationsmaßeMolenbruch
Volumenbruch

�i �mi
�i
mi
�
mi
mgesamt
ci �ni
vmol
l
m�i�
ni
mLM
molkg
0 K � �273.15°C ; 0 °C � 273.15 KT[K] � �[°C] � 273.15
3
Massenbruch
Lösungen:Molarität (molare Konzentration)
Molalität
1.3 System und Umgebunga) Offenes System : Stoff- und Energieaustausch
(Arbeit und/oder Wärme)b) Geschlossenes Systen: nur Energieaustauschc) Adiabatisches System: kein Stoff-, kein
Wärmeaustausch, nur Arbeitsaustauschd) Abgeschlossenes System: weder Energie- noch
Stoffaustausch mit der Umgebung
1.4 Reversible und irreversible ProzesseAlle natürlichen, spontan ablaufenden Prozesse sindirreversibel, sie hinterlassen Spuren. Die Zeit ist nichtumkehrbar! Reversible Prozesse hinterlassen keineSpuren in der Umgebung! Sie sind eine idealisierendeAbstraktion.Die Behandlung an sich irreversibler Prozesse alsGleichgewichtsprozesse mit den Methoden derGleichgewichtsthermodynamik ist eine idealisierendeNäherung. Sie ist einfach und sehr nützlich!
Unterteilung :isotherme Zustandsänderung T = const. ; �T = 0isobare Zustandsänderung p = const. ; �p = 0isochore Zustandsänderung v = const. ; �v = 0adiabatische Z. änderung q = const. ; �q = 0
1.5 Die thermodynamische Temperaturskala
Die bekannte Temperaturskala nach Celsius wurde aus den Fixpunkten Tripelpunkt des Wassersund Siedepunkt abgeleitet. Die allgemeinere thermodynamische Skala verwendet dasselbeIntervall, definiert aber einen anderen, nämlich den absoluten Nullpunkt, bei dem jeglicheWärmebewegung aufhört.
Änderung der Temperatur ist mit Zufuhr oder Abfuhr von Wärmeenergie verbunden.Temperaturmessung nutzt (möglichst) lineare Abhängigkeit einer Meßgröße von T, z.B:Ausdehnung einer Flüssigkeitssäule,....)

Ekin �12
mv 2
E � h�
4
2. Der O. Hauptsatz der ThermodynamikBefinden sich jeweils die Systeme A und B und die Systeme B und C im thermischenGleichgewicht, dann sind auch die Systeme A und C im thermischen Gleichgewicht.Die allen Systemen gemeinsame Eigenschaft ist die Temperatur.
3. Verschiedene Arten der Energie: Arbeit, WärmeEnergie = Vermögen Arbeit zu leisten. Identische Masseinheiten:
1 J = 1 V A s = 1 Nm = 1 kg m s 2 -2
Verschiedene Erscheinungsformen (grundsätzlich ineinander umwandelbar):- Mechanische Arbeit Arbeit = Kraft × Weg Volumenarbeit Arbeit = Druck × Volumen (Druck = Kraft / Fläche)- Elektrische Arbeit Arbeit = Spannung × Ladung
- Wärmeenergie, Wärmemenge: Flussgrösse, Fluss setzt Temperaturdifferenz voraus!Wärmeenergie wird frei gesetzt (gespeichert) bei physikalischen (z.B.Aggregatzustandsänderungen, Modifikationswechsel) und chemischen Veränderungen(chemischen Reaktionen).
- Chemische Energie: Gespeichert in chemischen Bindungen, Oxidationszustand u.a.- Mechanische Energie Kinetische Energie:
(m Masse, v Geschwindigkeit)Potentielle Energie = gespeicherte Energie; Gesamtenergie =kinetische + potentielle Energie
- Energie elektromagnetischer Strahlung: (h=6.626×10 Js : Plancksches Wirkungsquantum , � : Frequenz der Strahlung)-34
4. Aggregatzustände der Materie
Gasförmiger Zustand: ungeordnete, chaotische Translationsbewegung. (Bei Molekülen gibtes zusätzlich Schwingungsbewegungen und Rotationen.) Großer mittler Abstand zwischen denTeilchen, geringe Wechselwirkung (WW) der Teilchen untereinander (Van der Waals-WW.,Dispersions-WW). Keine Nahordnung keine Fernordnung. Alle Gase sind lückenlosmiteinander mischbar. Gar keine WW : Ideales Gas
Flüssiger Zustand: Nahordnung existiert, aber keine Fernordnung. IntermolekulareAnziehungskräfte (Kohäsionskräfte, stärker als in Gasen) schränken die relative Beweglichkeitder Teilchen zueinander ein. Translation und Rotation sind behindert. Nicht alle Flüssigkeitensind lückenlos miteinander mischbar: Phasentrennung.
Fester Zustand: Kristalline Festkörper: Nah- und Fernordnung, i.A. deutlich größereKohäsionsenergie als in Flüssigkeit, abhängig von Bindungsverhältnissen (Ionenkristalle,Molekülkristalle, Atombindungen). Keine Rotation, keine Translation; Schwingungen umRuhelage möglich.Amorphe Festkörper: Nahordnung, keine Fernordnung; hohe Kohäsionsenerghien; eingefroreneFlüssigkeit; GläserNoch weiter eingeschränkte Mischbarkeit, viele feste Phasen.

KristallSchmelzeFlüssigkeit Gas
� T�T
fest
fest/flüssig
flüssig
flüssid/gasförmig
gasförmig
Q
TTF TV
� H
V
F
� H
Verdampfungsenthalpie
Schmelzenthalpie
Verdampfen � Kondensieren: �VH � ��KondHSchmelzen � Kristallisieren: �FH � ��KristH
Cp ��H�T
�
�Qp
�T�
dHdT p
Cv ��U�T
�
�Qv
�T�
dUdT v
5
4.1 Der Phasenbegriff, Phasenumwandlungen: Phase: Vielteilchensysteme mit gleichen (sich lokal nicht sprunghaft ändernden) Eigenschaften;von anderer Phase durch Phasengrenze getrennt. An Phasengrenze sprunghafteEigenschaftsänderung über atomare Distanz.Einphasige Systeme: homogen, Gase immer einphasig, mehrere Komponenten möglich;Mischphase (z.B. Luft)Mehrphasige Systeme: Flüssigkeiten und Feststoffe können ein- (homogene, reine oderMischphase) oder mehrphasig (heterogen) sein.Existenz mehrerer Phasen bei kristallinen Elementen: Allotropie (z.B. bei S, C, P, As, Sb, Sn)Existenz mehrerer Phasen bei kristallinen Verbindungen: Polymorphie (z.B. SiC, SiO , CaCO )2 3
Phasenumwandlungen bei Erhöhung der Temperatur führen von Zustand höherer Ordnung zueinem Zustand niedrigerer Ordnung (undumgekehrt):
Wärmezufuhr bewirkt Überwindung der Kohäsionskräfte.Bei der Schmelztemperatur (T ) bzw. am Siedepunkt (T ) liegen zwei Phasen im GleichgewichtF V
miteinander vor, die Temperatur bleibt so lange konstant bis eine Phase verschwunden ist.Diese Phasenumwandlungen sind reversibel durchführbar:
Energieerhaltung!Enantiotrope Umwandlung: umkehrbare Umwandlung von Modifikationen ineinanderEs existiert eine definierte Umwandlungstemperatur, Monotrope Umwandlung: Umwandlung nur in einer Richtung durchführbarDie “Umwandlungstemperatur” liegt oberhalb des Schmelzpunktes, z.B. Violetter Phosphor
Die Wärmekapazität a) molare ~ bei p= const.
bei v = const.

�q � c�T ; cspez �cm
; cmol �cn
dNv
N� f(v) � Av 24e
�
mv 2
2kT A �m
2�kT
324�
v �8kT�m
ekin �m2
v 2 v 2�
3kTm
� ekin �32
kT k �RNA
Ekin �32
NAkT �32
RT
p� �
NSmv
Ap �
23
Nv
ekin �13�v 2
p �nRT
v; pV � RT
6
Die molare Wärmekapazität ist die Wärmemenge, die ein Mol eines Stoffes um 1 Kerwärmt. C bzw. C sind Stoffkonstanten:p v
b) spezifische Wärmekapazität c :spez
Die spezifische Wärmekapazität ist die Wärmemenge, die ein Gramm eines Stoffes um 1K erwärmt.
Beispiel Wasser: c = 4.184 J K g (14.5 °C � 15.5 °C)spez.-1 -1
Die zugeführte Wärmemenge �q bzw. �Q wird in Form von Translationsenergie,Rotationsenergie, Schwingungsenergie gespeichert.
4.2 Der GaszustandRegellose Translationsbewegung der Gasteilchen (Atome oder Moleküle)Modell: Brownsche Molekularbewegung ( An Pollen unter dem Mikroskop beobachtbar)Maxwell-Boltzmann-Geschwindigkeitsverteilung in Gasen
Häufigkeit, Bruchteil der Teilchen mit derGeschwindigkeit v :
Mittlere Geschwindigkeit:(k : Boltzmannkonstante, m : Teilchenmasse)
mittlere kinetische Energie eines Teilchens:
Für 1 Mol eines idealen Gases (s.u.):
Der Gasdruck p :Summe der auf die Wand übetragenen Impulse pro Flächeneinheit
(Kinetische Gasgleichung, � : Gasdichte)
Aus kinetischer Gasgleichung folgt die thermischeZustandsgleichung für ideale Gase (R= 8.314 JK mol , Gaskonstante):-1 -1
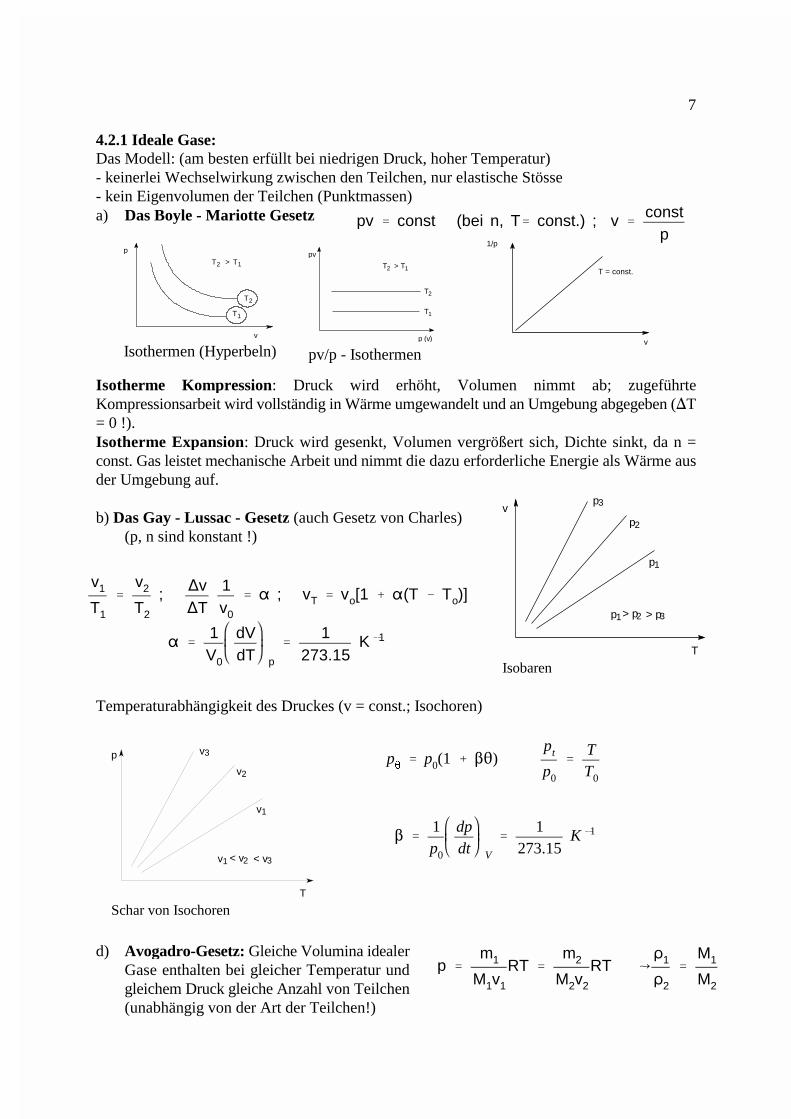
pv � const (bei n, T� const.) ; v �const
p
T1
T2
T2
T1
>
p
v
T1T2 >
pv
p (v)
T1
T2
1/p
v
T = const.
v
T
p1
p2
p3
p1> p2 > p3
v1
T1
�
v2
T2
; �v�T
1v0
� � ; vT � vo[1 � �(T � To)]
� �1V0
dVdT p
�1
273.15K �1
T
v1
v2
v3
v1 < v2 < v3
p
� �1p0
dpdt V
�1
273.15K �1
p�� p0(1 � ��)
pt
p0
�TT0
p �
m1
M1v1
RT �
m2
M2v2
RT �
�1
�2
�
M1
M2
7
Isothermen (Hyperbeln) pv/p - Isothermen
Isobaren
Schar von Isochoren
4.2.1 Ideale Gase:Das Modell: (am besten erfüllt bei niedrigen Druck, hoher Temperatur)- keinerlei Wechselwirkung zwischen den Teilchen, nur elastische Stösse - kein Eigenvolumen der Teilchen (Punktmassen)a) Das Boyle - Mariotte Gesetz
Isotherme Kompression: Druck wird erhöht, Volumen nimmt ab; zugeführteKompressionsarbeit wird vollständig in Wärme umgewandelt und an Umgebung abgegeben (�T= 0 !).Isotherme Expansion: Druck wird gesenkt, Volumen vergrößert sich, Dichte sinkt, da n =const. Gas leistet mechanische Arbeit und nimmt die dazu erforderliche Energie als Wärme ausder Umgebung auf.
b) Das Gay - Lussac - Gesetz (auch Gesetz von Charles)(p, n sind konstant !)
Temperaturabhängigkeit des Druckes (v = const.; Isochoren)
d) Avogadro-Gesetz: Gleiche Volumina idealerGase enthalten bei gleicher Temperatur undgleichem Druck gleiche Anzahl von Teilchen(unabhängig von der Art der Teilchen!)

NA � 6.023×1023
Vmol � 22.414 lmol �1
p ��
MRT � CRT
pgesamt � �i
pi ; xi �pi
pgesamt
�
vi
vgesamt
�
ni
ngesamt
; �i �ni Mi
�i
Ni Mi
�
xi Mi
�i
xi Mi
pN2: pO2
� 4 : 1 � bei pges � 1020 bar: pN2� 816 mbar ; pO2
� 204 mbar
aus pV � RT wird mit p :� p � pbinnen
und V :� V � Veigen
(p � pbinnen)(V � Veigen) � RT
(p �a
V 2)(V � b) � RT
8
Einheiten des Druckes:Pascal: 1Pa = 1 Nm-2
Bar: 1 bar = 10 Pa5
1 bar = 0.1 MPa1 mbar = 1 hPa
Torr: 1 bar = 750.062 torrAtmosphäre: 1 bar = 0.986923 atm
Van der Waals - Isotherme im Vergleich zu Experiment undidealem Gas
bei 273 K (0° C) und 1.013 bar
Alle unter a) bis d) genannte Gesetze sindin der allgemeinen Gasgleichung (s.o.)enthalten; andere Form:
( � : Dichte; C Molarität)Möglichkeit der Molmassebestimmung!
Das Dalton-Partialdruckgesetz für ideale Gase:Allgemeine Gasgleichung gilt für jedes Gas im Gemisch als wäre es alleine vorhanden:Luft:
4.2.2 Reale Gase - thermische Zustandsgleichung
Experimentell bekannt: Alle Gase lassen sich verflüssigen. Ideale Gase sind nicht verflüssigbar!Energetische Wechselwirkung (anziehend, wenn auch schwach!): Ursache für die Möglichkeitder Verflüssigung. Korrektur an Zustandsgleichung für ideale Gase, Berücksichtigung derAnziehung (durch Binnendruck) und des Eigenvolumens (Covolumen):
Van der Waals-Gleichung - thermischeZustandsgleichung für reale Gase, einevon vielen Näherungen:
Die gestrichelten Kurvenstücke um dieschraffierten Flächen A und B werdendurch eine waagerechte Gerade ersetzt.

�dTdp H
�
V � T�V�T p
Cp
; Bei Ti : V � Ti�V�T p
� 0 � Ti �V
�V�T p
VT
��V�T
9
pv/p-Isotherme des Kohlendioxids
Diese Gerade beschreibt dasZweiphasengebiet. Die Flächen Aund B sind gleich groß.
Es existiert eine Temperatur, diekritische Temperatur T , oberhalbk
der keine Verflüssigungdes Gases mehr möglich ist. Vgl.Punkt K, rechte Abbildung! Bei Tk
steht das Gas unter dem kritischenDruck p , und es nimmt das kritischek
(Mol)volumen ein.Charakteristische Grössen für jedesGas/Flüssigkeit!Das nicht ideale Verhalten ist Ursachefür die Abkühlung eines Gases bei(adiabatischer) Expansion. Nurunterhalb einer für jedes Gas charakteristischen Temperatur, der Inversionstemperatur T ,i
möglich! (Technische Gasverflüssigung, Linde-Verfahren)Für T > T : Erwärmung des Gases bei Expansion , keine Verflüssigungi
Für T < T : Abkühlung des Gases bei Expansion , Verflüssigung möglichi
Ideales Gas:
� keine Verflüssigung! (Vgl.: Gay-Lussac!)
Gas T / °C p / barkrit krit
Cl 144 76.92
He -268 2.28
CO 31.1 73.72
Luft -141 37.6
O -118 50.22
H O 174 220.02
Tab.: Kritische Temperaturen undDrücke einiger Gase

u � u(v,T,ni)
�U � 0 ; dU � 0
�u � �w � �q
EKern � 1012 Jmol �1 ; EElektr.�Kern � 106 Jmol �1 ; EVibr. � 103 Jmol �1
T � const. , �T � 0 � q � �w q � const. , �q � 0 � �u � aadiab.
�H � ��U � �(pV) dH � dU � d(pV)
�H � �U��pV � p�V dH � dU � Vdp � pdV
�H � �U � p�V dH � dU � pdVH � H(p,T) H � U �pV
10
3. Der 1. Hauptsatz der ThermodynamikVerschiedene Formulierungen:a) Die innere Energie (0 Summe aller Energien der Teilchen) ist
eine Zustandsfunktion: b) In abgeschlossenem System ist die innere Energie konstant.
(Energieerhaltungssatz)c) In einem geschlossenen System mit seiner Umgebung
ausgetauschte Summe von Arbeit und Wärme ist gleich derÄnderung der inneren Energie des Systems.
d) Unmöglichkeit eines Perpetuum mobile erster Art.
(Äußere Energie = potentielle Energie der Lage im Raum oder kinetische Energie derBewegung durch den Raum des Systems als ganze sind hier uninteressant!)Der Absolutbetrag der inneren Energie des Systems ist nicht bekannt, bzw. abhängig von derDefinition des Nullpunktes. (Bei 0 K bleibt die Kernenergie, Bindungsenergie der Elektronen anKerne; chemische Bindungsenergien; Translations-, Vibrations- und Rotationsenergienverschwinden.)
Achtung: Im Gegensatz zu u sind w und q keine Zustandsgrössen, sondern prozessabhängig!
Vergleich: Isotherme Expansion (Kompression) und Adiabatische Expansion (Kompression)
Expansion: Aus Umgebung entnommene Kein Wärmeaustausch mit UmgebungWärme wird in Expansionsarbeit Expansion: Geleistete Arbeit wird derumgewandelt Inneren Energie entnommen, Abkühlung;Kompression: Zugeführte Arbeit wird als Kompression: Geleistete Arbeit wird derWärme an Umgebung abgegeben Inneren Energie hinzugefügt; Erwärmung.
Adiabatische Expansion Adiabatische KompressionInnere Energie: nimmt ab nimmt zuVolumen: nimmt zu nimmt abTemperatur: sinkt steigtDruck: nimmt ab nimmt zuDichte: sinkt steigt
Die Zustandsfunktion Enthalpie HEingeführt, weil sehr viele Prozesse beikonstantem Druck (isobar) ablaufen(Atmosphärendruck); innere Energie +Volumenarbeit

U � U(V,T) und H � H(p,T)
�AA � �BB � ... � �CC � �DD � ...Ausgangsstoffe � Endstoffe
Edukte � Produkte
�RU � � UProdukte � � UEdukte
�RU � �q � Ckal(TE � TA)
CkalTA
�RH � � HProdukte � � HEdukte�RH � �RU � p�Vr
�RH > 0�RH < 0
�RV �VEnd � VAusg � 0 �RH � �RU
11
Prinzipskizze eines Bombenkalorimeters
Reaktionsenergien, ReaktionsenthalpienDer Zustand (eines Stoffes) ist eindeutig durch die Zustandsgrössen (T, v, p, u, n) bestimmt.Bei konstanter Stoffmenge (hier 1 mol) gilt
Die Angabe von Absolutwerten für U und H (und weitere thermodynamische Zustandsgrößen)erfordert die Definition eines Standardzustandes als Nullpunkt:Standardzustand ist Zustand bei 298 K (25 °C) und p = 101.3 kPa; Kennzeichnung imFormelzeichen durch hochgestelltes Symbol � .
Allgemeine chemischeReaktion:
Reaktionsenergie:(= Reaktionswärme bei konstantem Volumen)Sinnvoll bei isochorer Reaktionsführung, keine Volumenarbeit, da keine Volumenänderung(geschlossenes Reaktionsgefäss)! Messung inBombenkalorimeter (meist Verbrennungsrektionen).
T Endtemperatur; Anfangstemperatur; E
Kalorimeterkonstante, wird durch Kalibrierreaktionmit bekannter Reaktionsenergie, oder elektrischerHeizung bestimmt.
Häufiger werden chemische Reaktionen beikonstantem Druck durchgeführt (isobar, dp=0). Wennsich das Volumen bei der Reaktion ändert (Änderungder Teilchenzahl!), leistet das System Arbeit gegenden Außendruck.
Die Reaktionsenthalpie(= Reaktionswärme bei konstantem Druck)
Endotherme Reaktionen (Abkühlung bei adiabatischer Reaktionsführung )
Exotherme Reaktionen (Erwärmung bei adiabatischer Reaktionsführung)
Beispiel 1 : C(Graphit, fest) + O (gasförmig) � CO (gasförmig) 2 2

C6H12O6(fest) � 6 O2 (gasf.)� 6 CO2 (gasf.) � 6 H2O (gas) ; �RV � 6RT
p�Rv � �RnRT � �End
nEnd � �Ausg
nAusg RT
�AA � �BB � ... � �CC � �DD � ...
�RH � �End
�End�fHEnd � �Ausg
�Ausg�fHAusg
�RH � �C�fHC � �D�fHD � (�A�fHA � �B�fHB)
�H1 � �H2
Ausgangs- zustand
A End-zustand E
Reaktionsweg 1
Reaktionsweg 2
� H
� H 1
2
�VerbrH�
Ausg � H3 � H1
�VerbrH�
Verb � H3 � H2
H2 � H1 � �fH�
H Elemente
Verbindungen
Verbrennungsprodukte
1
2
3�fH�� �
End�End�VerbrH
�� �
Ausg�Ausg�VerbrH
�
12
Beispiel 2 :
Volumenänderung der Feststoffe kann gegenüber der Volumenänderung gasförmiger Stoffevernachlässigt werden:
5.2 Anwendungen des Ersten HauptsatzesBildungsenthalpien und Reaktionsenthalpien - Der Satz von HessBildungsenthalpien: � H (Index f: formation) sind Reaktionsenthalpien einer (gedachten undf
meist nicht durchführbaren) Bildungsreaktion: Reine Elemente � Verbindung
Molare Standardbildungsenthalpien, � H : Reaktionsenthalpie für die Bildungsreaktion vonf �
einem Mol der Verbindung aus den stabilsten Modifikationen der Element beiStandardbedingungen (s.o.). Stabilste Modifikationen unter Standardbedingungen sind z.B.Graphit (nicht Diamant) für Kohlenstoff, H , O , N , F , Cl , im gasförmigen Zustand und Br ,2 2 2 2 2 2
Hg als Flüssigkeiten.Die Standard- Enthalpien der Elemente unter genannten Bedingungen werden willkürlichNull gesetzt (da nicht bekannt).Standard Reaktionsenthalpien beziehen sich auf durch die Reaktionsformel gegebenenStoffumsatz.
5.2.1 Der Satz von HessDie Reaktionsenthalpie (� H, � h) einer beliebigen chemischen Reaktion kann aus denR R
Bildungsenthalpien (� H) der beteiligten Verbindungen berechnet werden, sie ist unabhängigf
davon, ob die Reaktion in einem oder mehreren Schritten abläuft.
5.2.2 Bestimmung derBildungsenthalpien indirekte Bestimmung überKreisprozeß mit Hilfe vonVerbrennungsenthalpien
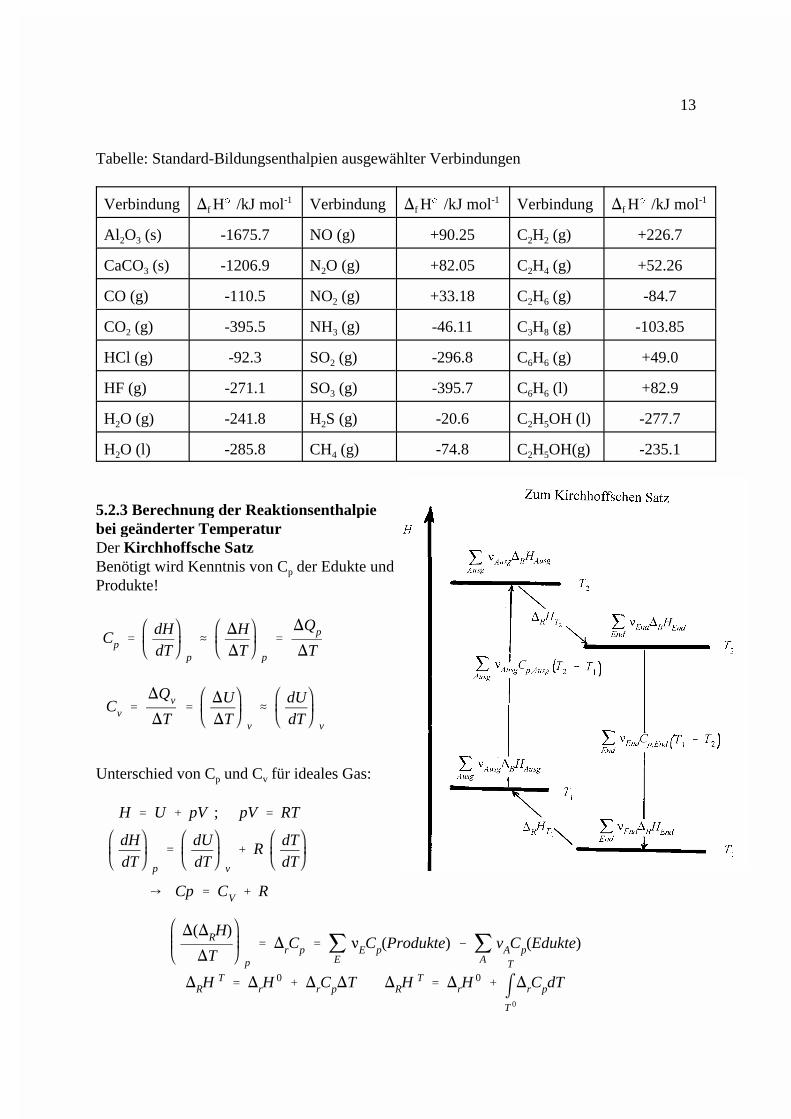
Cp �dHdT p
��H�T p
�
�Qp
�T
Cv �
�Qv
�T�
�U�T v
�dUdT v
�(�RH)
�T p
� �rCp � �E
�ECp(Produkte) � �A
vACp(Edukte)
H � U � pV ; pV � RT
dHdT p
�dUdT v
� RdTdT
� Cp � CV � R
�RH T� �rH
0� �rCp�T �RH T
� �rH0� �
T
T 0
�rCpdT
13
Tabelle: Standard-Bildungsenthalpien ausgewählter Verbindungen
Verbindung � H /kJ mol Verbindung � H /kJ mol Verbindung � H /kJ molf � -1
f � -1
f � -1
Al O (s) -1675.7 NO (g) +90.25 C H (g) +226.72 3 2 2
CaCO (s) -1206.9 N O (g) +82.05 C H (g) +52.263 2 2 4
CO (g) -110.5 NO (g) +33.18 C H (g) -84.72 2 6
CO (g) -395.5 NH (g) -46.11 C H (g) -103.852 3 3 8
HCl (g) -92.3 SO (g) -296.8 C H (g) +49.02 6 6
HF (g) -271.1 SO (g) -395.7 C H (l) +82.93 6 6
H O (g) -241.8 H S (g) -20.6 C H OH (l) -277.72 2 2 5
H O (l) -285.8 CH (g) -74.8 C H OH(g) -235.12 4 2 5
5.2.3 Berechnung der Reaktionsenthalpiebei geänderter TemperaturDer Kirchhoffsche SatzBenötigt wird Kenntnis von C der Edukte undp
Produkte!
Unterschied von C und C für ideales Gas:p v

s � s(p,T,n) ; S � S(p,T) ; S � S(V,T)
Definition: �S �
�Qrev
T
�S > 0dS > 0
�S � 0dS � 0
�Sgesamt � �SSystem � �SUmgebung
�Sgesamt > 0 irreversibler Prozess, spontan oder erzwungen�Sgesamt � 0 reversibler Prozess, System und Umgebung im Gleichgewicht
�Sgesamt < 0 : unmöglich!
14
6. Der Zweite Hauptsatz der ThermodynamikGestattet Aussagen zu:- Richtung chemischer Vorgänge einschließlich Phasenumwandlungen- Freiwilligkeit (Unfreiwilligkeit) von Prozessen- Gleichwichtslage von chemischen Prozessen, mithin zur Ausbeute- Umwandelbarkeit der Wärme in andere Energieformen
Alle natürlichen Vorgänge laufen spontan, d.h. freiwillig in einer Richtung ab. Sie sindirreversibel. Irreversibel bedeutet hier: die erzwungener Umkehrung des Prozesses ist imSystem nur möglich, wenn in der Umgebung bleibende Veränderungen entstehen.Irreversibilität - Zeitachse: Vergangenheit � Zukunft.Reversible Prozesse sind solche, in denen Systen und Umgebung stets im Gleichgewichtverbleiben (Zeitachse ist umkehrbar).
Formulierungen des 2. Hauptsatz der Thermodynamika)Es gibt keine periodisch arbeitende Maschine, die ausschließlich Wärme in eine andereEnergieform (Arbeit) unwandelt, ohne daß gleichzeitig Wärme von einem höherenTemperaturniveau auf ein niedrigeres irreversibel übergeht.
b)In einem abgeschlossenem System gilt:dS = 0 bei reversiblen Prozessen,dS > 0 bei irreversiblen Prozessen.
c)Es existiert kein perpetuum mobile zweiter Art.
Die Entropie ist eine Zustandsgrösse (und eine Zustandsfunktion)
Einheit der Entropie : J mol K auch-1 -1
e.u. (entropy unit)�Q : reversibel zwischen System undrev
Umgebung ausgetauschet Wärme.
Die Entropie ist ein Maß für die thermodynamische Wahrscheinlichkeit (� für die Stabilität )des Systems, weil alle freiwillig ablaufenden Prozesse einem stabilen Gleichgewichtszustandzustreben, der durch ein Maximum der Entropie ausgezeichnet ist. Für abgeschlossene System gilt:
Reversible Prozesse: Irreversible Prozesse:
Für die Gesamtheit System + Umgebung gilt nach Clausius:

�Wrev � �F ; F � F(v,T)
�U � �Qrev � �Wrev � T�S � �Wrev
F � U � TS ; �F � �U � T�S ; dF � dU � TdS
G � F � pV � H � TS ; G � G(p,T)
G � H � TS ; �G � �H � T�S ; dG � dH � TdS
�G < 0
�G > 0
�G � 0
�FS �
Qrev,F
TF
�
�FH
TF
> 0
�VS �
Qrev,V
TV
�
�VH
TV
� 88 JK mol
15
6.1 Triebkraft einer Zustandsänderung (einer chemischen Reaktion)
Maximale Nutzarbeit bei reversibler Prozessführung: Freie Energie F
Maximale Nutzarbeit bei reversibler Prozessführung und konstantem Druck: Freie Enthalpie G
(Gibbs-Helmholtz-Gleichungen)
Bei irreversible Prozessführung wird stets weniger Arbeit gewonnen als maximal möglich!Prozess kann nur unter Arbeitsaufwand rückgängig gemacht werden!
�G legt die Richtung eines isobaren Prozesse fest:
Exergonischer Prozess, kann spontan ablaufen
Endergonischer Prozess, läuft nicht spontan ab
Gleichgewicht
6.2 Die Entropie als Zustandsfunktion
Phasenübergänge: Änderung des Ordnungszustandes:
Schmelzentropie
Verdampfungsentropie
(Allg.: Umwandlungsentropien)
(Kristall: Nahordnung und Fernordnung; Flüssigkeit (Schmelze): Nahordnung, keine
Fernordnung; Gas: Weder Nah- noch Fernordnung, vollständige Unordnung).

dS �
dQrev
T�
Cp
TdT �
�S�T p
�
Cp
T
limT�0
S � 0
limT�0
�S � 0
S �
Elem �
�
T �
0
cpdlnT � �i
�uH
Ti
; S �
Elem � 0, S �
Verbind � 0 !!!
�RS� � �End
�EndS�end � �
AusgvAusgS
�AusgS�Verb � �
ElementeS� � �RS�
S � klnW , k �RNA
16
Entropie des Stickstoffs als Funktion der Temperatur
6.3 Temperaturabhängigkeit der Entropie(hier nur für isobare Prozesse formuliert)
Kenntnis von C als Funktion der Temperatur undp
Kenntnis der Umwandlungsenthalpien gestattet dieBerechnung absoluter Entropien bei beliebigerTemperaur, wenn ein Bezugspunkt definiert ist.Bezugspunkt ist T = 0 K, S = 0 .
7. Der Dritte Hauptsatz der Thermodynamik
-Die Entropie von Idealkristallen wird bei 0 K Null (PlanckscheHypothese). -Die Entropieänderungen von Systemen streben im Gleichgewichtszustandbei Annäherung an den absoluten Nullpunkt gegen Null (NernstschesWärmetheorem)-Der absolute Nullpunkt ist unerreichbar!
7.1 Standard (Normal-)entropien, Reaktionsentropien
Es existieren für alle Elemente und Verbindungen unter Standardbedingungen Normal-(Standard-) entropien � 0. Normalentropien der Elemente sind tabelliert, folglich lassen sichNormalentropien von Verbindungen erhalten nach:
oder allgemein:
7.2 Entropie als UnordnungsmassDie thermodynamische Wahrscheinlichkeit eines Systems gibt an, wievieleRealisierungsmöglichkeiten W (Mikrozustände, Verteilung der Gesamtenergie auf die einzelnenTeilchen, Verteilung der Teilchen im Volumen) dieses System hat.
Statistische Deutung der Entropie nach Boltzmann:
Je geordneter ein System ist, desto weniger Mikrozustände besitzt es und um so niedriger istseine Entropie! Irreversible Vorgänge streben nach maximaler Unordnung.

�G � �H � T�S
1. �H < 0 ; �S > 0 � �G < 0 � exergon2. �H < 0 ; �S � 0 � �G < 0 � exergon3. �H > 0 ; �S � 0 � �G > 0 � endergon4. �H > 0 ; �S > 0 � �G <> 0 � endergon oder exergon
4a) |T�S| < |�H| � endergon4b) |T�S| > |�H| � exergon
�AA � �BB � ...� �CC � �DD � ...
�RG � �RG�� RT ln
a�C
C a�D
D
a�A
A a�B
B
; �RG�� � RT ln
a�C
C a�D
D
a�A
A a�B
B eq
� �RTlnK
ai � fix xi ai � fiC
Ci
C �
� �RG�� �RTlnK ; K � e
�
�RG�
RT�RG � 0
�RG� < 0 ; K > 1
�RG� > 0 ; K < 1
1. �RG�� �
Produkte
�fG�(Produkte) � �
Edukte
�fG�(Edukte)
2. �RG�� �RH�
� T�RS�
17
8. Anwendungen des Zweiten Hauptsatzes8.1 Chemische Reaktionen in geschlossenen Systemen
8.2 Die Freie molare Reaktionsenthalpie und die Gleichgewichtskonstante einer chemischen Reaktion
� G : Molare Freie Standardreaktionsenthalpie; a : Aktivitäten; K : thermodynamischeR i�
Gleichgewichtskonstante; Index eq : equilibriumx : Molenbruchi
C : Molare Konzentrationi
C : Standardkonzentration 1 mol l* -1
f : AktivitätskoeffizienteniC
K und a sind nur von T und bei Gasen zusätzlich von p abhängig.i, eq
8.3 Das Chemische Gleichgewicht
Lage des Gleichgewichtes:
8.4 Ermittlung der freien Standardreaktionsenthalpie:
(Tabellierte � G )f�

CaCO3 � CaO � CO2
C � CO2 � 2 CO
Ag �
gelöst � Cl �gelöst � AgCl�(fest)
�AA � �BB �K1 [�CC � �DD] �
K2 [�EE � �FF] �K3
�GG � �FF
K1 �
a�C
C a�D
D
a�A
A a�B
B
; K2 �
a�E
E a�F
F
a�C
C a�D
D
; K3 �
a�G
G a�H
H
a�E
E a�F
F
Kgesamt � K1 K2 K3
KC �
C�C
C C�D
D
C�A
A C�B
B
�[C]
�C[D]�D
[A]�A[B]
�B
�AA � �BB � �CC � �DD
18
8.5 Das Chemische Gleichgewicht und das MassenwirkungsgesetzIn geschlossenem System läuft eine freiwillige Reaktion vom Anfangszustand nur bis in denGleichgewichtszustand (stabiler Endzustand), d.h. der Prozess verläuft nicht vollständig nacheiner Seite.Gleichgewichte können nur durch äusseren Zwang verschoben werden. (Prinzip vonLeChatelier und Brown: Gleichgewicht weicht dem äusseren Zwang aus, verschiebt sich in dieRichtung die den äußeren Zwang verringert.)Voraussetzung für Einstellung des Gleichgewichtes ist Reversibilität.Chemisches Gleichgewicht ist zugleich ein dynamisches Gleichgewicht, d.h. im Gleichgewichtsind die Geschwindigkeiten der Hin- und der Rückreaktion gleich. Obgleich mikroskopischHin- und Rückreaktion ablaufen, bleiben alle Konzentrationen im Gleichgewicht konstant(Gleichgewichtskonzentrationen).
8.5.1 Einteilung chemischer Gleichgewichtehomogene Gleichgewichte: Nur eine (homogene) Phase, z.B. Gas, Lösungheterogene Gleichgewichte: Zwei oder mehrere Phasen beteiligt, z.B.
Gas- und Festphase:
Flüssig- und Festphase:
LöslichkeitsgleichgewichteVerteilungsgleichgewichtePhasengleichgewichte sind nichtreaktive Gleichgewichte.
8.5.2 Gekoppelte Gleichgewichte
Ausgangsstoffe � Zwischenprodukte � Endprodukte
8.5.3 Das Massenwirkungsgesetz von Guldberg und Waage(Stöchiometrische Gleichgewichtskonstante)
Gilt streng nur in (stark) verdünnten (idealen!) Lösungen!

K � KC Qf ; Qf �f�C
C f�D
D
f�A
A f�B
B
; ai � fi,C
Ci
C �
; limC�0
fi,C � 1
�AA � �BB � �CC � �DD
Kp �
p�C
C p�D
D
p�A
A p�B
B
; pi � CiRT ; Kp � KC (RT)�E A
�i
2H2(g) � O2(g) � 2H2O(g)
Kp �
p 2H2O
p 2H2
pO2
� KC (RT)�1
ai � Ci � 1
CaCO3(fest) � CaO(fest) � CO2(gas)
K � aCO2�
Kp
p �
; p �
� 1 bar
CLM � const. z.B. CH2O � 55.4mol
l
HCl � H2O � H3O�
� Cl �
KC �
[H3O�][Cl �]
[HCl]
�RG�� �RTlnK ; lnK � ln10 logK ; ln10 � 2.303..
�RG�� �2.303RT logK
�logK � pK � K � 10�pK
19
Nur die thermodynamische Gleichgewichtskonstante ist eine echte Konstante:
Besonders starke Abweichungen vom idealen Verhalten (d.h. f =1) bei Elektrolytlösungen!i
8.5.4 Gleichgewichte in der GasphaseAnstelle von Konzentrationen werden meist die Gleichgewichtspartialdrücke verwendet:
Beispiel:
8.5.5 Gleichgewichte unter Beteiligung kondensierter Phasen
a) Reiner Feststoffe Wichtige Konvention:
Beispiel:
Die Lage des Gleichgewichtes wird nicht von der Menge der Feststoffe beeinflusst!
b) Des Lösungsmittels
Beispiel:
8.5.6 Der Gleichgewichtsexponent pK

K > 1 � pK < 0 ; K < 1 � pK > 0
dlnKdT p�const
�
�RH�
RT 2
dlnKdp T�const
� �
�RV
RT; �RV � �Rn RT
p
N2 � 3 H2 � 2 NH3 N2O4 � 2 NO2
�Rn � �1 �Rn � �1
(�G)p,T � 0 ; G(Phase 1)p.T � G(Phase 2)p.T
20
exergonische Reaktion endergonische Reaktion
8.5.7 Temperaturabhängigkeit von Gleichgewichtskonstanten
Van’t Hoffsche Reaktionsisobare
Endotherme Reaktion: � H > 0, lnK (und K) nimmt zu mit steigender TemperaturR�
Exotherme Reaktion: � H < 0, lnK (und K) nimmt ab mit steigender TemperaturR�
Bei Temperaturerhöhung verschiebt sich Gleichgewicht in Richtung des Wärmeverbrauches(Vgl. Prinzip von LeChatelier und Brown !)
8.5.8 Druckabhängigkeit von Gleichgewichtskonstanten
Beispiele:
K steigt mit steigendem Druck K fällt mit steigendem Druck(Erneute Demonstration des Prinzips von LeChatelier und Brown!)
Auswirkungen von Konzentrationsänderungen auf die Lage des Gleichgewichtes:Zufügen von Ausgangsprodukt (Endprodukt) verschiebt Gleichgewicht zugunsten derEndprodukte (Ausgangsprodukte)
8.6 Phasengleichgewichtenichtreaktive Gleichgewichte, keine chemische ReaktionUnverändert gilt imGleichgewicht:
Untergliederung in - Phasengleichgewichte einkomponentiger Systeme Flüssig � Dampf (Verdampfen, Kondensieren)
Fest � Dampf (Sublimieren, Kondensieren)Fest � Flüssig (Schmelzen, Kristallisieren) Zustandsdiagramme reiner Stoffe
- Phasengleichgewichte mehrkomponentiger SystemeLösungsgleichgewichte: Feststoff in Flüssigkeit oder Gas in FlüssigkeitDampfdruck von Lösungen, Vergleich mit reinem LösungsmittelVerteilungsgleichgewichte zwischen zwei flüssigen MischphasenPhasengrenzflächengleichgewichte (Adsorption/Desorption)

dGdT p
� �S� ; dGdp T
� V�
�S�fl dT � V�
fl dp � �S�gasdT � V�
gasdp
dpdT koex
�
S�gas�S�fl
V�
gas�V�
fl
�
�S�U
�V�; �S�U �
�Q�
U
T�
�UH�
T
dpdT koex
�
�UH�
RT 2p ; dp
p� dlnp ; V�
gas �RTp
21
F = K + 2 - P
Einteilung und Beispiele für Zweiphasensysteme
Phase 1 Phase 2 Beispiel
Aerosol gasförmig fest Rauch
gasförmig flüssig Nebel, Spray
Suspension flüssig fest Schlamm, Kosmetika
Emulsion flüssig flüssig Milch, Cremes
Schaum fest, flüssig gasförmig Seifenschaum,Schaumglas
8.6.1 Die Gibbs’sche Phasenregelbestimmt, wieviele Phasen miteinander im Gleichgewicht existieren können, und wievieleFreiheitgrade (=Wahlfreiheiten von Zustandsvariablen p,T,n )idas System bei gegebener Anzahl von Phasen hat.K : Zahl der Komponenten (Chemische Species)F : Anzahl der Freiheitsgrade (T, p, x )i
P : Anzahl der Phasen
Verifizierung für einen reinen Stoff (K=1): F = 3 - P , d.h. wenn drei Phasen (fest, flüssig,gasförmig koexistieren, Tripelpunkt!), dann besteht kein Freiheitsgrad! Druck und Temperatursind festgelegt, vgl. z.B. Phasendiagramm des Wassers!Algemein gilt für einkomponentige Systeme (K = 1):F = 0 � nonvariant (invariant); 3 Phasen im GleichgewichtF = 1 � univariant, entweder p oder T wählbar, 2 Phasen im GleichgewichtF = 2 � divariant, p und T frei wählbar , nur eine Phase existiert.Für zweikomponentige Systeme existiert ein Quadrupelpunkt, d.h. 4 Phasen können imGleichgewicht existieren (2 feste, je eine flüssige und gasförmige)
8.6.2 Phasengleichgewichte einkomponentiger Systeme
Für jede Phase (z.B. fl und gas)gilt:
Verallgemeinerung: U = Umwandlung

dlnpdT koex
� �UH�
RT 2; ln p
p �
�
�UH�
RT� const. ; ln
p1
p2
� �
�UH�
R1T1
�1T2
�SU �
�VH
TdpdT koex
��S�V
Vgas » Vfl � �V �RTp
dpdT koex
�
�UH�
RT 2p ; p � p0e
�
�UH�
RT
SDK
DDK
SubDK
22
Phasendiagramm des Kohlendioxids
Phasendiagramm des WassersTripelpunkt 0.0098 °C, p=6.11mbarSDK Schmelzdruckkurve; DDK:D a m p f d r u c k k u r v e ; S u b D k :Sublimationsdruckkurve;Besonderheit: Schmelzdruckkurve fallend;Druck auf Eis � Schmelzen; � > � !Eis Wasser
Phasendiagramm des Schwefels
gilt allgemein fürP h a s e n g l e i c h g e w i c h t eeinkomponentiger Systeme
U: Verdampfung und Sublimation:
Clausius-Clapeyron-Gleichung:

Aus lnp1
p2
� �
�VerdH�
R1T1
�1T2
� lnp
p �
� const �
�VerdH�
R1T
ln p/p*
1/T
1
2
3
1,2,3: unterschiedliche Flüssigkeiten
�VerdS��
�VerdH�
TVerd
� 88J
K mol
pLM � x flLM p �
LM (pLM � a flLM p �
LM) ; pLösung � p � �i
xip�
i
p �
LM � pLM,L
p �
LM
� 1 � x flLM � xgel
xgel �ngel
ngel � nLM
; ngel « nLM
� xgel �ngel
nLM
; ngel �mgel
Mgel
�p
p �
LM
mLM
MLM
�
mgel
Mgel
Mgel � mgel
p �
LM
�p
MLM
mLM
23
Zusammenhang zwischen Dampfdruckerniedrigung undSiedepunktserhöhung sowie Gefrierpunktserniedrigung
Für nicht assoziierende Flüssigkeiten gilt in guterNäherung diePictet-Troutonsche Regel :
8.6.3 Phasengleichgewichte zweikomponentiger LösungenDampfdruck von Lösungen (flüssige Mischphasen): Vergleich mit reinem LösungsmittelDas Raoultsche Gesetz
(LM : Lösungsmittel, * markiert reines Lösungsmittel)
Nichtflüchtiger gelöster Stoff:
Relative Dampfdruckerniedrigung gleich dem Molenbruch des gelöstem Stoff.
Anwendung: Molmassebestimmung, auch Reinheitsprüfung von Lösungsmittelna) Molmasse aus relativer Dampfdruckerniedrigung

TV � T �
V � �TV � xgel ; Mgel �1000EV
�TV
mgel
mLM
; EV �
RT �2V
�VH�MLM (Ev in K mol �1 kg) ;
TF � T �
F � �TF � EF xgel
EF �
RT �2F
�FH�MLM
Mgel �1000EF
�TF
mgel
mLM
(EF in K mol �1 kg)
�C�x
1A
dndt
� �D�C�x
24
Pfeffersche Zelle
b) aus Siedepunktserhöhung (Ebullioskopie):
c) aus Gefrierpunktserniedrigung (Kryoskopie) :
Beispiele ebullioskopischer und kryoskopischerKonstanten
Lösungsmittel E [K mol kg] E [K mol kg]V-1
F-1
H O 0.52 1.862
CCl 5.07 4.904
Benzen 2.54 5.07
Campher 6.09 40
8.7 Osmose, Dialyse, DiffusionGelöste Teilchen bewegen sich ungeordnet durch das Lösungsmittel - Analogie zum Gas. Beihöherer Temperatur schnellere Bewegung, geringere Behinderung durchLösungsmittelmoleküle.
Räumlicher Konzentrationsunterschied führt zu gerichtetem Stofftransport, der
Konzentrationsunterschiede ausgleicht : DiffusionTriebkraft: Entropiezunahme. Vorgang kommt zum Ende, wenn Konzentrationsgradientenverschwunden sind.Beschreibung des Stofftransportes durch Diffusion bei konstantem Konzentrationsgradienten:Erstes Ficksches Gesetz:
A : Fläche, durch welche Stofftransport erfolgtD : Diffusionskoeffizient
D hängt von Teilchengrösse ab, nimmt mit T zu undsinkt mit der Zähigkeit des Lösungsmittels.
8.7.1 Osmose
Nur Lösungsmittel kann durch Membrandiffundieren, d.h. osmotisches Gleichgewicht isterreicht, wenn die Flüssigkeitssäule der Lösung sohoch gestiegen ist, dass der hydrostatische Druck

�id.v � ngelöstRT �id. � CgelöstRT
� � phydrostat � �hg
Cgelöst �nv
�m
Mgelöstv
H2O
O
O
OO
O OO
O
O
OO
O
O
-
--
---
------
------
--- --- ---
---------
-
--
p � (x1)l p �
1 � (x2)l p �
2
da x1 � x2 � 1 � p � (x1)l (p �
1 � p �
2 ) � p �
2
mit (x1)g �
p1
p� p �
p �
2
1 � 1 �
p �
2
p �
1
(x1)g
25
Dampfdruckdiagramm eines idealenZweistoff-Gemisches
der Säule den osmotischen Druck kompensiert.Für sehr verdünnte Lösungen gilt Van’t Hoffsches Gesetz:� : osmotischer Druck, hängt nicht von Art des Stoffes, sondern nur von Teilchenzahl ab(kolligative Eigenschaft).Im Gleichgewicht wird in der Pfefferschen Zelle der hydrostatische Druck den osmotischenDruck kompensieren:
� : Dichte der Lösungg : Erdbeschleunigungh . Steighöhe
Für reale Lösungen Aktivitäten anstelle Konzentrationen. Möglichkeit der Molmassebestimmung, da (M : Molmasse des Gelösten)gelöst
LM-Moleküle wandern von der verdünnteren zur konzentrierteren Lösung. Wichtig fürLebensvorgänge, Effekt auf biologische Zellen! Physiologische Kochsalzlösung isotonisch zuCytoplasma; hypotonische und hypertonische Lösungen!Wichtige Anwendungen: Molmassebestimmungen, besonders für Makromoleküle (�Dampfdruckosmometrie); Bestimmung von Aktivitätskoeffizienten.Achtung: Kolligative Eigenschaft, Erhöhung der Teilchenzahl bei dissoziierendenVerbindungen! Gesamtkonzentration= Konz. der Anionen + Konz. der Kationen.
8.7.2 DialysePhysikalische Trennung gelöster niedermolekularervon hochmolekularen Verbindungen (Kolloiden)durch semipermeable Membranen.Niedermolekulare Substanzen (kleinere Teilchen)diffundieren durch Membran und werden vomströmenden Lösungsmittel weggeführt, z.B.Hämodialyse, “Blutreinigung”
8.8 FlüssigkeitsgemischeAnwendung des Raoultschen Gesetzes:
Siedekurve: Gerade zwischen Dampfdrücken der reinenKomponentenKondensationskurve: hyperbolischer Verlauf!
pi � fi x fli p �
i
26
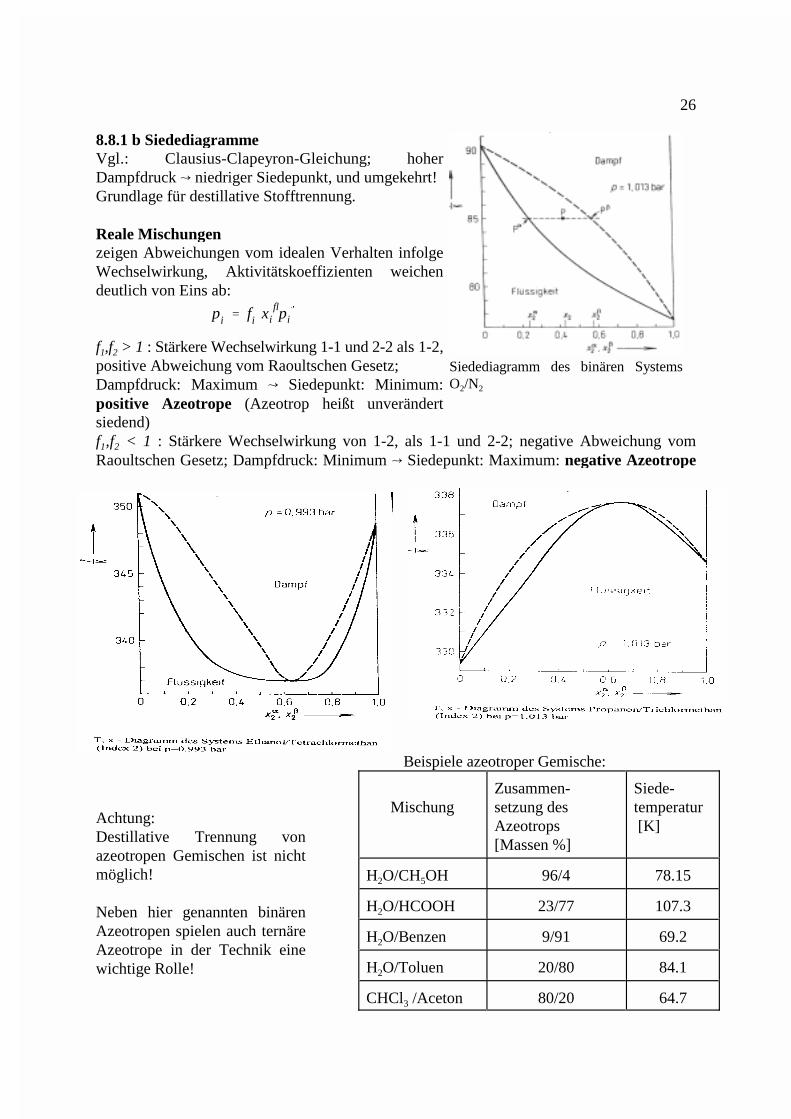
Siedediagramm des binären SystemsO /N2 2
8.8.1 b SiedediagrammeVgl.: Clausius-Clapeyron-Gleichung; hoherDampfdruck � niedriger Siedepunkt, und umgekehrt!Grundlage für destillative Stofftrennung.
Reale Mischungenzeigen Abweichungen vom idealen Verhalten infolgeWechselwirkung, Aktivitätskoeffizienten weichendeutlich von Eins ab:
f ,f > 1 : Stärkere Wechselwirkung 1-1 und 2-2 als 1-2,1 2
positive Abweichung vom Raoultschen Gesetz; Dampfdruck: Maximum � Siedepunkt: Minimum:positive Azeotrope (Azeotrop heißt unverändertsiedend)f ,f < 1 : Stärkere Wechselwirkung von 1-2, als 1-1 und 2-2; negative Abweichung vom1 2
Raoultschen Gesetz; Dampfdruck: Minimum � Siedepunkt: Maximum: negative Azeotrope
Achtung:Destillative Trennung vonazeotropen Gemischen ist nichtmöglich!
Neben hier genannten binärenAzeotropen spielen auch ternäreAzeotrope in der Technik einewichtige Rolle!
Beispiele azeotroper Gemische:
Mischung setzung des temperaturZusammen- Siede-
Azeotrops [K][Massen %]
H O/CH OH 96/4 78.152 5
H O/HCOOH 23/77 107.32
H O/Benzen 9/91 69.22
H O/Toluen 20/80 84.12
CHCl /Aceton 80/20 64.73

ABfest � A �
solv � B �
solv ; K �
aA,eqaB,eq
aAB,fest
� aAaB � KL
27
8.9 Lösungen, Lösevorgänge
Gase, Flüssigkeiten, Festkörper können sich in chemisch von ihnen verschiedenen Flüssigkeiten(Lösungsmitteln, LM) unter Bildung einer homogenen flüssigen Mischphase lösen. LM = Solvens, Gelöster Stoff = SolvatLöslichkeit einer Substanz = maximale Stoffmenge, die sich bei gegebener Temperatur in einerbestimmten LM-Menge löst.Gesättigte Lösung: Bodenkörper, bzw. Gasphase im Gleichgewicht. Übersättigte Lösungen :C > C ; untersättigte Lösungen: C < C .sat sat
Echte Lösungen Kolloidale LösungenSolvat liegt molekulardispers vor Agglomerate von Einzelmolekülen(einzelne Moleküle von LM-Hülle= (Auch Riesenmoleküle: Proteine)Solvathülle umgeben) 1 < Partikelgrösse < 100 nmPartikelgrösse < 1nm (Vgl. Suspensionen Partikelgrösse > 100 nm)
Lösungsvorgang: energetische Wechsewirkung zwischen Solvat und Solvens (Solvatation,speziell für Wasser Hydratation), besonders stark bei geladenen Teilchen (Ionen,elektrolytische Dissoziation) in polaren Lösungsmitteln, führt zu Wärmeeffekt:Lösungsenthalpie (auch Lösungswärme).Zur Lösungsenthalpie tragen bei (Festkörper): - Gitterenergie des Festkörpers muss aufgebracht werden- Solvatationsenergie wird frei - Dissoziationsenthalpie (bei Elektrolyten)- Verdünnungsenthalpie: Dispersion der TeilchenLösungsenthalpie kann positiv oder negativ sein !Entropieeffekt �S > 0; Unordnung wächst.T �S kann positive Lösungsenthalpie überkompensieren ! Entscheidend �G = �H - T �S ! Lösungsprozess findet statt solange �G < 0 !
Löslichkeit hängt ab von Temperatur und der Art des LM und des zu lösenden Stoffes. (“Similissimilibus solvuntur”). Polare Lösungsmittel (hohe Dielektrizitätskonstante, großesDipolmoment) lösen besonders gut polare Substanzen, Elektrolyten; unpolare LM lösenbevorzugt unpolare Substanzen.Löslichkeit von Salzen : Hohe Ladung der Ionen und kleiner Radius führt zu grosserGitterenergie; auch Solvatationsenergie steigt mit Ladungszahl und kleiner werdendem Radius!Effekt der Gitterenergie überwiegt:Beispiele für Löslichkeitsabstufung: LiF < NaF < KF < RbF < CsF
LiF < LiCl < LiBr < LiI
8.9.1 Fest-flüssige SystemeDas Löslichkeitsprodukt KL

Für Elektrolyt AmBn : KL � a m
A ��a n
B ��; KL � KL,C f m
A f nB � [A ��]m[B ��]n f m
A f nB
dlnKL
dT�
�LH 0
RT 2
pKL � �logKL
Cm[mol l �1] �
m�n KL
m mn n
KL � KL,C�fi
Cgas,gelöst
p
ideal
real
p(bar)
O2
N2
x2
0 5 100
10-4Gas � LM � gelöstes Gas
[Gasgelöst]eq � KH p
KH � KH(T)
28
Löslichkeitsexponent: Temperaturabhängigkeit:
Die Löslichkeit hängt auch von der Teilchengröße ab! Je kleiner die Teilchengrösse desto höherdie Löslichkeit! (Übersättigte Lösungen, Keimbildung, Animpfen beim Auskristallisieren!Wichtig für Kinetik, kinetische Hemmungen!)
Tabelle: Löslichkeitsprodukte bei 20° C in Wasser
Verbindung K [mol/l] Verbindung K [mol/l]L,Cn
L,Cn
HgS 2×10 Ca(OH) 8×10-522
-6
Ag S 1.6×10 Mg(OH) 1.2×102-49
2-11
PbS 1×10 CaCO 4.8×10-283
-9
BaSO 1×10 BaCO 5×104-10
3-9
BaCrO 2.4×10 AgCl 1×104-10 -10
Fe(OH) 1.1×10 AgBr 5×103-36 -13
Cr(OH) 6.7×10 AgI 1.5×103-31 -16
Sättigungskonzentration (= molare Löslichkeit) eine Salzes Kat Anm n
Beeinflussbarkeit der Sättigungskonzentration:
- Nur das thermodynamische Löslichkeitprodukt ist echte Konstante:Aktivitätskoeffizienten hängen von Fremdionen ab, sie sinken mit steigenderIonenkonzentration, d.h. K muss steigen : EinsalzeffektL,C
- Gleichionige Zusätze senken die Gleichgewichtskonzentration des Gegenions, Verschiebungder Gleichgewichtslage! Wichtig für Fällungsreaktionen!
8.9.2 Löslichkeit von Gasen in FlüssigkeitenDas Henrysche Absorptionsgesetz:

Ether
Wasser
Wasser
Chloroform
Phase 1
Phase 2
Phase 1
Phase 2oder
XPhase1 � XPhase2
[X]Phase1
[X]Phase2 T
� KN
� �nA
� ��G�A p,T
�
�0
C
grenzflächenaktiv
nicht grenzflächenaktiv
� � �CRT
��
�C p,T
T1 T2
T1 T2<
C
�
�oo
�eq �
�max p
b � p
dlnKads
dT�
�
�adsH0
RT 2
29
8.9.3 Verteilungsgleichgewicht eines Stoffes zwischen zwei flüssigen PhasenDas Nernstsche Verteilungsgesetz:Flüssige Phasen sind (weitestgehend) nicht mischbar.
K : Nernstsche Verteilungskonstante, wichtig für Lösungsmittelextraktion (Ausschütteln) undN
für Verteilungschromatographie!
8.10 Adsorptions-DesorptionsgleichgewichteAnlagerung von Stoffen aus der Gasphase oder Lösung an der Phasengrenze zu einemFestkörper (oder zu einer Flüssigkeit).Wichtig für heterogene Katalyse ( erster Reaktionsschritt), Waschprozesse -Benetzungsphänomene, Löten, Flotation, Stofftrennung (z.B. Gaschromatographie)
8.10.1 Adsorption an GrenzflächenDefinition der Grenzflächenkonzentration(n: Stoffmenge, A : Grenzfläche)
Definion der Grenzflächenspannung, auch (Freie!) OberflächenenergieArbeit, die zu leisten ist, um die Oberfläche zu vergrössern, z.B. für H O: � = 73 mJ m .2
-2
Minimierung von G (hier:�) ist Ursache für die Kugelgestalt von Flüssigkeitstropfen.Je größer die Oberfläche einer gegebenen Stoffmenge, um so energiereicher wird der Stoff,Ursache für höheren Dampfdruck kleinerer Tröpfchen undgrößere Löslichkeit kleiner Festkörperpartikel.
Minimierung von G (hier:�) auch durch Adsorption:
Tenside (grenzflächenaktive Stoffe) wichtig für vielepharmazeutische, besonders kosmetische Bereitungen,Liposomen.
Langmuir-Adsorptionsisotherme:
Temperatur-abhängigkeit:
Adsorption ist stets exotherm, Adsorptionsentropie ist kleiner als Null!



![© SNC Social Networks Consulting GmbH [zurück] [weiter]zurückweiter Die Online-Informationssoftware für heterogene Kooperationsverbände.](https://static.fdokument.com/doc/165x107/55204d7049795902118c17e1/-snc-social-networks-consulting-gmbh-zurueck-weiterzurueckweiter-die-online-informationssoftware-fuer-heterogene-kooperationsverbaende.jpg)














