
Wirkprofil und zytotoxisches Potential von Trofosfamid im ... · Aus der Medizinischen Klinik I der...
82
Aus der Medizinischen Klinik I der Universität zu Lübeck Direktor: Prof. Dr. med. H.L. Fehm Wirkprofil und zytotoxisches Potential von Trofosfamid im Vergleich zu den Oxazaphosphorinanaloga Cyclophosphamid und Ifosfamid Inauguraldissertation zur Erlangung der Doktorwürde der Universität zu Lübeck - aus der Medizinischen Fakultät - vorgelegt von Carola Letsch aus Bad Segeberg Lübeck 2004
Transcript of Wirkprofil und zytotoxisches Potential von Trofosfamid im ... · Aus der Medizinischen Klinik I der...

Aus der Medizinischen Klinik I der Universität zu Lübeck
Direktor: Prof. Dr. med. H.L. Fehm
Wirkprofil und zytotoxisches Potential von Trofosfamid im Vergleich zu den
Oxazaphosphorinanaloga Cyclophosphamid und Ifosfamid
Inauguraldissertation zur
Erlangung der Doktorwürde der Universität zu Lübeck
- aus der Medizinischen Fakultät -
vorgelegt von Carola Letsch
aus Bad Segeberg
Lübeck 2004

1. Berichterstatter: Prof. Dr. med. Thomas Wagner
2. Berichterstatter: Prof. Dr. med. Heiko Iven
Tag der mündlichen Prüfung: 29.04.05
Zum Druck genehmigt: Lübeck, den 29.04.05
Gez. Prof. Dr. med. Peter Dominiak
- Dekan der Medizinischen Fakultät -

Inhaltsverzeichnis
Inhaltsverzeichnis Seite
I. Einleitung 5
II. Material und Methoden
1. Material und Geräte 13
2. Methoden
2.1 Zellkultur 18
2.2 Zytostatika-Aufbereitung 19
2.3 MTT-Assay 20
2.4.1 Trypanblau-Färbung 22
2.4.2 Comet-Assay 23
2.5 DNA-Isolation 27
2.6 Fluorometrischer Cross-link-Assay 30
2.7 Biometrie und Statistik 33
III. Ergebnisse 1. MTT-Assay 34
2.1 Trypanblau-Färbung 38
2.2 Comet-Assay (Einzelzellgelelektrophorese) 39
3.1 Validierung der Messung am Spectralfluorophotometer 44
3.2 Fluorometrischer Cross-link-Assay 47
IV. Diskussion 49
1. Zytotoxisches Potential von 4-OH-Trofosfamid 51
2. Detektion von DNA-Schäden 54
3. klinische Relevanz der Arbeit 59
V. Zusammenfassung 60
VI. Literaturverzeichnis 62

Inhaltsverzeichnis
VII. Anhang
1. Abbildungsverzeichnis 74
2. Tabellenverzeichnis 75
3. Tabellen 76
4. Abkürzungsverzeichnis 78
VIII. Danksagung 80
IX. Lebenslauf 81

Einleitung
5
I. Einleitung
Trotz des großen Fortschrittes in der Therapie der Tumorerkrankungen sind die
Krebserkrankungen neben den Herz-Kreislauferkrankungen zu den häufigsten
Todesursachen geworden (Becker, 1998). In der Behandlung von Krebs nimmt die
Chemotherapie neben der Operation, der Strahlentherapie und der Immuntherapie
eine wichtige Rolle ein. Die derzeit gebräuchlichen Chemotherapeutika, die
Zytostatika, führen zur Apoptose oder zur Schädigung von Tumorzellen. Je nach
Art und Stadium der Krebserkrankung erfolgt der Einsatz der Chemotherapie mit
verschiedenen Zielsetzungen. Bei der Behandlung von malignen Lymphomen, des
Morbus Hodgkin, der akuten Leukämien im Kindesalter oder der Karzinome des
Hodens wird eine kurative Therapie angestrebt. Zudem kann die Chemotherapie
auch eingesetzt werden, um die Lebensqualität betroffener Patienten zu
verbessern oder ihr Leiden zu mindern, also unter palliativen Gesichtspunkten.
Um sekundäre Resistenzen zu verhindern und die Wirksamkeit zu erhöhen,
werden häufig Polychemotherapien angewendet (Teicher, 1997).
Von den unterschiedlichen Gruppen der Chemotherapeutika, die zur Anwendung
kommen, sind die Alkylantien die ältesten. Sie gehören zusammen mit den
Antimetaboliten, wie zum Beispiel dem Methotrexat, zu denen, die nach wie vor
am häufigsten angewendet werden (Teicher, 1997). Alkylantien werden sowohl in
Kombinationschemotherapien, als auch als Hochdosistherapeutika bei
Knochenmark- oder Stammzelltransplantationen eingesetzt (Colvin, 1999; Teicher,
1997).
Die Entdeckung der Chemotherapie kann zurückverfolgt werden bis in Zeiten des
ersten Weltkrieges, als nach dem Einsatz von Gelbkreuzkampfstoff (z.B.
Dichlordiethylsulfid = Lost = Senfgas) bei der Autopsie der Gefallenen neben
schweren Reizungen der Haut und des Respirationstraktes eine Hypoplasie des
Knochenmarks und des lymphatischen Gewebes feststellt wurde (Kohn und
Bruce, 1996; DeVita 1997; Teicher, 1997). Diese Erkenntnisse wurden erst nach
dem zweiten Weltkrieg veröffentlicht, weil es sich bei dem Senfgas um ein
Kampfgas handelte (Goodman et al., 1946). Da sich Senfgas als zu toxisch erwies

Einleitung
6
und man eine stabilere Verbindung verwenden wollte, wurde das Stickstofflost (s.
Abb.1) entwickelt.
Abb.1: Diese beiden Verbindungen bildeten die Grundlage bei der Entwicklung der Oxazaphosphorine.
Die heute verwendeten Stickstofflost-Derivate zeichnen sich dadurch aus, dass sie
wesentlich weniger toxisch sind, kaum noch zu lokalen Reizwirkungen führen und
somit auch oral gegeben werden können ( Blomqvist et al., 1995; Gunsilius et al.,
2001). Die Oxazaphosphorine stellen heute die bedeutendsten Wirkstoffe aus der
Gruppe der alkylierenden Zytostatika dar. Die bekannteste und zugleich am
meisten verwendete Substanz dieser Reihe ist das Cyclophosphamid (Brock,
1989).
Cyclophosphamid wurde 1958 von Arnold, Bourseaux und Brock synthetisiert und
ist eines der am gründlichsten untersuchten Alkylantien (Arnold et. al; 1958;
Sladeck, 1988). Neben seinen tumorreduzierenden Eigenschaften besitzt es auch
eine hämatopoetische Toxizität, die eine Leukopenie und unter Hochdosistherapie
eine Knochenmarksaplasie nach sich ziehen kann. Auf der Suche nach einem
nebenwirkungsärmeren Zytostatikum entdeckten Arnold und Bourseaux zwei
weitere interessante Oxazaphosphorine (Arnold und Bourseaux, 1958). Zunächst
das Trofosfamid, das eng mit Cyclophosphamid verwandt ist, sich aber durch die
dritte Chlorethylgruppe, die am Stickstoffatom des Sechseringes liegt,
unterscheidet (siehe Abb.2). Als weitere Verbindung entdeckten sie das Ifosfamid,
welches nicht mehr die typische N-Mustard-Struktur aufweist. Seine beiden
Chlorethylgruppen liegen nicht am selben Stickstoffatom, sondern befinden sich
die eine Gruppe intra- und die andere extrazyklisch (Semont et al., 1981).
Cl CH2 CH2
S
Cl CH2 CH2
a) Lost
Cl CH2 CH2 N - CH3
Cl CH2 CH2
b) Stickstofflost

Einleitung
7
Alle drei Oxazaphosphorine sind so genannte Prodrugs und benötigen eine
Biotransformation, um zytotoxisch zu wirken. In vivo geschieht dieses mit Hilfe des
multifunktionellen Oxidase-Systems der Leber (Cytochrom P450 Isoenzyme),
welches den Ring an Position 4 hydroxyliert. Für die Hydroxylierung von
Trofosfamid wurde das Cytochrom P450 3A4 Isoenzym identifiziert (May-Manke et
al., 1999). Die 4-OH-Verbindungen stehen im Gleichgewicht mit ihrer Aldoform,
die spontan zu Acrolein und einer alkylierenden Mustardverbindung zerfällt.
Weiterhin können Ifosfamid und Cyclophosphamid auch an den
Chlorethylseitenketten oxidiert werden. Hierbei entstehen Chloracetaldehyd (CAA)
und die pharmakologisch inaktiven Verbindungen 2- /3- Dechloroethyl-Ifosfamid
bzw. 3-Dechloroethyl-Cyclophosphamid (siehe Abb. 2).
Die eigentlich alkylierenden Substanzen, die Mustardverbindungen, entstehen aus
den 4-OH-Metaboliten unter Abspaltung von Acrolein, das für die Nebenwirkungen
an der Blase (hämorrhagische Zystitis) verantwortlich gemacht wird (Brock et al.,
1979; Cox und Abel, 1979; Wagner, 1994). Um Folgen wie zum Beispiel
Blutungen der Blasenwand zu verhindern, wird bei dem Einsatz der
Oxazaphosphorine den Patienten Mesna zugeführt. Mesna ist ein
Sulfhydrylgruppendonator, der 1981 von Brock et al. als ein klinisch gut
einsetzbarer Uroprotektor entdeckt wurde (Brock et al. 1981; Brock und Pohl
1983). Als anionische Verbindung wird Mesna einerseits von den meisten Zellen
nicht aufgenommen und andererseits im Blut schnell zum inaktiven Dimesna
oxidiert. Dieses wird in der Niere glomerulär filtriert, tubulär reabsorbiert, in den
Nierenzellen erneut zu Mesna reduziert und schließlich über den Urin
ausgeschieden. Es gelangt so in die Blase, wo seine freie Sulfhydrylgruppe mit
Acrolein reagiert und dieses somit inaktiviert. Darüber hinaus wird aber auch die
Umwandlung der 4-Hydroxymetabolite zu Acrolein blockiert (Brock et al 1981;
Dorr, 1991).
Die drei verschiedenen Mustardverbindungen mit ihren chemischen Strukturen
haben je nach Substrat unterschiedliche alkylierende Aktivitäten. Trotz dieser
Unterschiede in der chemischen Struktur wird die unterschiedliche Aktivität dem
spezifischen metabolischen Abbau zugeschrieben (Zalupski und Baker, 1988;
Wagner, 1997). Alkylierende Substanzen sind hoch reaktive Verbindungen, die in

Einleitung
8
der Lage sind, Hydrogengruppen von organischen Substanzen durch
Alkylgruppen zu ersetzen (Wagner, 1997). Die zytotoxische Wirkung beruht
vornehmlich auf der Alkylierung der Nucleinsäuren, insbesondere der DNA. In vitro
Ergebnisse haben gezeigt, dass die primären Angriffspunkte von Trofosfamid die
Phosphodiester-Bindungen in der DNA und die Orthophosphatgruppen der freien
Nukleotide sind (Lindemann und Harbers, 1980). Aus der Lokalisation des
alkylierenden Angriffspunktes an der DNA wurde der Schluss gezogen, dass der
Hauptmechanismus die intermolekulare Bildung von so genannten „Cross-links“
sei (Wagner, 1997). Hierbei handelt es sich um kovalente Bindungen zwischen
einem komplementären Basenpaar der DNA, die zum Zelltod führen können, da
die DNA an diesen Stellen nicht mehr abgelesen werden kann.
R.F. Struck hat die Bildung von Cross-links an Oligonucleotiden für
Isophosphoramid Mustard, Nitrogen Mustard und Phosphoramid Mustard
nachgewiesen (Struck et al., 2000). Darüber hinaus deckte Hartley an
Patientenleukozyten, die mit Ifosfamid behandelt worden waren, mit Hilfe des
Comet-Assays Cross-links auf (Hartley et al., 1999). Mit Hilfe des „alkalischen
Elution-Assays“ wurden neben der Fähigkeit von Cyclophosphamid zur Bildung
von Cross-links auch DNA-Strangbrüche an Lungenfibroblasten nachgewiesen
(Hengstler et al., 1997). Bisher konnte noch niemand nachweisen, dass auch
Trofosfamid in der Lage ist, DNA-Schäden hervorzurufen.
Obwohl Trofosfamid 1973 zugelassen wurde, ist nur wenig über dieses
Oxazaphosphorin bekannt. Im Vergleich mit seinen Verwandten Cyclophosphamid
und Ifosfamid konnte Trofosfamid bisher in der klinischen Praxis kaum Akzeptanz
gewinnen. Die Therapie mit Trofosfamid ist ausschließlich oral möglich. Dies
wurde von den behandelten Patienten als weniger belastend empfunden im
Vergleich zur intravenösen Chemotherapie (Mross et al., 1998; Hartmann, 2003).
Insbesondere wird Trofosfamid z. Zt. bei palliativen Behandlungsansätzen oder in
Langzeittherapien eingesetzt (Blomqvist et al., 1995; Salminen et al., 1995;
Salminen et al., 1997; Helsing, 1996; Enk und Knop, 2000; Reichardt et al., 2002;
Andersson et al., 2002; Hartmann et al., 2003;). Anwendungsgebiete für
Trofosfamid sind unter anderem die Erhaltungstherapie bei lymphoretikulären
Tumoren, Hämoblastosen und malignen soliden Tumoren mit disseminiertem

Einleitung
9
Wachstum sowie bei Karzinomen der Ovarien, der Mammae, beim kleinzelligen
Bronchialkarzinom und beim Seminom. Die antineoplastische Wirkung wurde
zuerst am Yoshida ascites sarcoma bei der Ratte nachgewiesen (Brock und
Hohorst, 1967). Bei der Festlegung der kurativen Dosis und der LD50 (letale Dosis,
bei der 50% der Tiere sterben) zeigte sich, dass Trofosfamid toxischer ist als
Cyclophosphamid.
Lange wurde angenommen, dass der Hauptmetabolit von Trofosfamid das
Ifosfamid sei, weshalb Trofosfamid oft als orales Ifosfamid eingesetzt wurde.
Tatsächlich konnte unsere Arbeitsgruppe beweisen, dass sich das Trofosfamid
strukturell (s.Abb2), pharmakokinetisch und in seinen zytotoxischen Eigenschaften
von Ifosfamid unterscheidet (Brinker et al., 2002). Klinische Studien über die
Therapie mit Ifosfamid zeigten die erheblichen Nebenwirkungen, wie zum Beispiel
die Neuro- und Nephrotoxizität auf (van Dyk et al., 1972; Goren et al., 1987; Bern
et al., 1995; Goren et al., 1996). In der Studie von Brinker konnte festgestellt
werden, dass sich dagegen unter der Therapie mit Trofosfamid (160mg/m²) nur
geringe Nebenwirkungen einstellten. In einer früheren Arbeit wurde eine Dosis von
50mg/kg (1700mg/m²) appliziert, aber es konnte auch hier keine Neurotoxizität
festgestellt werden. (Flakson und Falkson, 1978).
Da es keine komplette Kreuzresistenz zwischen Ifosfamid und Cyclophosphamid
gibt, erschien es K. Mross möglich, Patienten mit Trofosfamid zu therapieren,
auch wenn sie bereits mit Cyclophosphamid vorbehandelt wurden (Becher et al.,
1996; Mross et al., 1998). Er konnte belegen, dass Trofosfamid auch bei mehrfach
vorbehandelten Patientinnen mit metastasiertem Mammakarzinom eine sinnvolle
Therapieoption darstellt. Wirkungen am Tumor und unerwünschte
Nebenwirkungen stehen insgesamt in einem guten Verhältnis zueinander (Mross
et al., 1998). Die Tatsache, dass gegen Cyclophosphamid resistente Tumoren
noch sensibel auf Trofosfamid reagierten, könnte darin begründet sein, dass die
aktiven Metabolite des Trofosfamids wegen ihrer höheren Lipophilie besser durch
die Zellmembranen permeieren (Wagner, 1997). Selbst bei Patientinnen, die mit
drei und mehr Chemotherapien vorbehandelt worden waren, erwies sich
Trofosfamid noch als wirksam (Mross et al., 1998). Des Weiteren beschrieben die
Patientinnen die orale Darreichungsform als weniger belastend als eine

Einleitung
10
intravenöse Therapie. Schon andere Arbeitsgruppen beschrieben den palliativen
Einsatz der oralen Trofosfamidtherapie als eine interessante Therapiemöglichkeit
mit milden Nebenwirkungen, zum Beispiel bei Knochen- oder Weichteilsarkomen,
bei Lymphomen und Ovarialkarzinomen (Salminen et al., 1997; Gunsilius et al.,
2001; Hartmann et al., 2003). Um die Nebenwirkungen des CMF-Schemas
(Cyclophosphamid, Methotrexat und 5-Fluorouracil) zu mindern, tauschten
Albrecht und Mitarbeiter Cyclophosphamid gegen Trofosfamid aus (Albrecht et al.,
1984). Der zytotoxische Effekt auf Stammzellen wurde bereits untersucht. Es hat
sich gezeigt, dass Trofosfamid im Gegensatz zu Ifosfamid und Cyclophosphamid,
die lediglich toxisch auf proliferierende Stammzellen wirken, auch auf ruhende
Stammzellen Einfluss nimmt (Wagner, 1997).
Da in der Studie von Brinker nachgewiesen wurde, dass nach oraler Gabe von
Trofosfamid nicht nur ein Abbau zu Ifosfamid und Cyclophosphamid (Boos et al.,
1993), sondern auch eine direkte Hydroxylierung zu 4-OH-Trofosfamid erfolgt
(Brinker et al., 2002), erscheint die genauere Untersuchung der Wirkung von 4-
OH-Trofosfamid auf die Tumorzelle noch bedeutsamer.
Pharmakokinetische Untersuchungen ergaben, dass bei der Metabolisierung von
Trofosfamid durch den ersten Dechlorethylierungsschritt hauptsächlich Ifosfamid
und zu einem geringeren Teil Cyclophosphamid entsteht (Boos et al., 1993). In
beiden Fällen wird Chloracetaldehyd (CAA) freigesetzt (s. Abb.2), und bei dem
weiteren Abbau von Ifosfamid entsteht nochmals CAA. Jüngere Ergebnisse
belegen eine eigene zytotoxische Aktivität bzw. Antitumorwirkung von CAA
(Brüggemann et al, 1997; Börner et al., 2000). Dadurch bekommt diese quantitativ
erhebliche CAA-Freisetzung bei der Metabolisierung von Trofosfamid eine
besondere Bedeutung.
Bislang wurden in verschiedenen Studien die zytotoxischen und klinischen
Wirkungen von Cyclophosphamid und Ifosfamid untersucht (Kurowski et al., 1991;
Hengstler et al., 1997; Schlenke et al., 1999; Johnstone et al., 2000). Obwohl das
Trofosfamid immer größere klinische Akzeptanz gewinnt, gibt es bisher keine
vergleichenden Studien zur Zytotoxizität zwischen Trofosfamid, Ifosfamid und
Cyclophosphamid. Vor diesem Hintergrund wird in der vorliegenden Arbeit die

Einleitung
11
halbmaximale letale Dosis (IC50) von 4-OH-Trofosfamid im direkten Vergleich zu 4-
OH-Cyclophosphamid und 4-OH-Ifosfamid in vitro an Tumorzelllinien ermittelt. Da
die Oxazaphosphorine im klinischen Alltag stets in Kombination mit Mesna
eingesetzt werden und da in Studien ermittelt wurde, dass Mesna selbst einen
tumorprotektiven Effekt besitzen kann, wurden die Versuchsreihen jeweils
zusätzlich in Gegenwart von Mesna durchgeführt (Dissertation Brüggemann,
1999; Kisro et al.,2000; Kisro et al., 2001).
Ein weiteres Ziel der vorliegenden Arbeit ist es, die Hypothese zu überprüfen,
dass Trofosfamid die DNA alkyliert, die in Folge dessen durch DNA-Strangbrüche
und Cross-links geschädigt wird. Zum Nachweis von DNA-Strangbrüchen und
DNA-DNA-Cross-links wurde eine Einzelzellgelelektrophorese bzw. ein
fluorometrischer Assay an zwei etablierten humanen Tumorzelllinien angewendet.
Um einen Bezug zur Klinik zu erhalten, wurden die Untersuchungen mit
Dosierungen im Bereich der von Brinker ermittelten Plasmaspitzenspiegel des 4-
Hydroxymetaboliten durchgeführt.
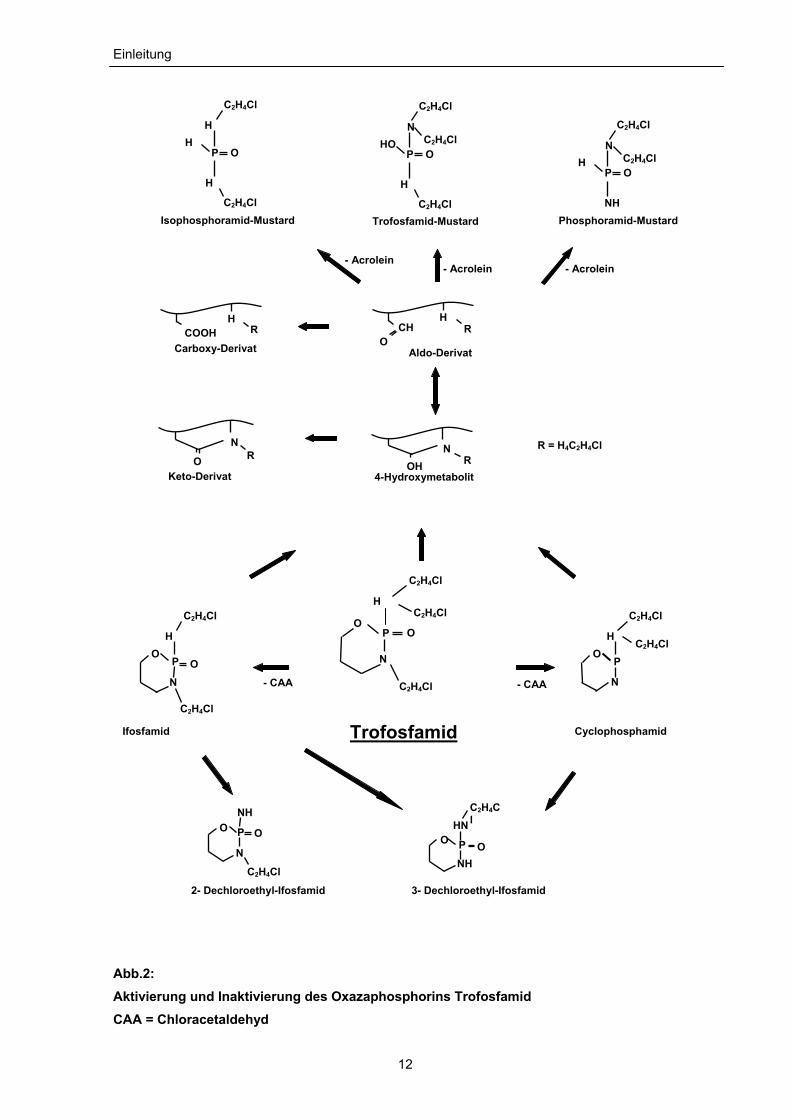
Einleitung
12
Abb.2: Aktivierung und Inaktivierung des Oxazaphosphorins Trofosfamid CAA = Chloracetaldehyd
C2H4Cl
H
P O
H
C2H4Cl
H
Isophosphoramid-Mustard
C2H4Cl
N
P O
H
C2H4Cl
HO C2H4Cl
Trofosfamid-Mustard
C2H4Cl
N
P O
NH
H C2H4Cl
Phosphoramid-Mustard
COOH H
R
Carboxy-Derivat Aldo-Derivat
CH H
RO
4-Hydroxymetabolit
NOH R
Keto-Derivat
N
O R R = H4C2H4Cl
C2H4Cl
H
P O N
C2H4Cl
O
Ifosfamid Trofosfamid
C2H4Cl
C2H4Cl
H
P O
N
OC2H4Cl C2H4Cl
H
P
N
O
Cyclophosphamid
C2H4Cl
- Acrolein - Acrolein - Acrolein
- CAA - CAA
3- Dechloroethyl-Ifosfamid
C2H4Cl HN
P ONH
O
2- Dechloroethyl-Ifosfamid
NH
P O
N
C2H4Cl
O

Material und Methoden
13
II.1 Material und Geräte II.1.1 Zytostatika und Reagenzien II.1.1.1 Mafosfamid: cis-2s-(bis (-2chlorethyl) amino) tetrahydro-2-oxid-2H-1,3,2-
oxazaphosphorin-4yl-thio-ethansulfonsäure. Mafosfamid zerfällt im Medium
spontan zu 4-OH-Cyclophosphamid und Mesna (Mercaptoethansulfonsäure). Es
wurde als Mafosfamid L-Lysine (50 mg) mit einem Molekulargewicht von 547,46
freundlicherweise von Asta Medica AG (Frankfurt) zur Verfügung gestellt und bei
4°C trocken gelagert.
II.1.1.2. 4-Hydroxyperoxy-Ifosfamid (4-OOH-Ifosfamid): 3-/2-chlorethyl)-2-( (2-chlorethyl)
amino) tetrahydro-2-oxid-2H-1,3,2-oxazaphosphorin-4-yl-hydroperoxid. Das 4-
OOH-Ifosfamid mit einem Molekulargewicht von 293,1 wurde freundlicherweise
von Asta Medica AG (Frankfurt) zur Verfügung gestellt und bei –70°C trocken
gelagert. Bei der Lösung im Medium kommt es zu einem spontanen Zerfall zu 4-
OH-Ifosfamid, dem eigentlich in vivo vorkommenden Metaboliten.
II.1.1.3. 4-Hydroxyperoxy-Trofosfamid (4-OOH-Trofosfamid): cis-(+-)-3-(2-chloroethyl)-2-
(bis(2-chloroethyl) amino) tetrahydro-2-oxide-2H-1,3,2-oxazaphosphorine-4-yl-
hydroperoxide. Das 4-OOH-Trofosfamid mit einem Molekulargewicht von 355,6
wurde freundlicherweise von Asta Medica zur Verfügung gestellt und bei –70°C
trocken gelagert. Bei der Lösung im Medium kommt es zu einem spontanen Zerfall
zu 4-OH-Trofosfamid, dem eigentlich in vivo vorkommenden Metaboliten.
II.1.1.4 MMS: Methylmetansulfonat (Merck)

Material und Methoden
14
II.1.1.5 Cisplatin: Platinex-Lösung (10mg / 20ml) Bristol-Meyers Squibb GmbH
II.1.1.6 Mesna: Mercaptoethansulfonsäure-Natrium. Es wurde von Asta Medica in Form
von „Uromitexan 400mg“ freundlicherweise zur Verfügung gestellt. Gelagert wurde
es bei Raumtemperatur. Das Molekulargewicht beträgt 164,18 g/mol.
II.1.2 Material der Zellkultur 1. Zelllinien: Weichteilsarkom der Schilddrüse S117
Mammakarzinom MX1
Beide Tumorzelllinien wurden vom Deutschen Krebsforschungszentrum in
Heidelberg (Deutschland) bezogen.
2. Tumorzellmedium: RMPI (Roswell Memorial Park Institute) 1640 (25mM
Hepes, L Glutamine) (Bio Whittaker Europe)
3. fetales Kälberserum (Biochrom KG Berlin)
4. Penicillin-Streptomycin (Roche Diagnostiks GmbH)
5. Trypsin – EDTA (GIBCO)
6. Zellkulturflaschen: (Nunclon™)
7. Einfriermedium: cryo-safe 1 (c-c-pro GmbH)
II.1.2.1Geräte der Zellkultur: 1. Hämocytometer: (Sysmex KX21)
2. Neubauer Zählkammer
3. Zentrifuge: (Heraeus Sepatech, Megafuge 1,0)
4. Brutschrank (Heraeus) (37°C, pH2O 47mmHg, 95%O2, 5% CO2)

Material und Methoden
15
II.1.3 Material des MTT-Assays
1. MTT: (Sigma) 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl Tetrazolium Bromid. 2. Mikrotiterplatten: NUNC Brand Products, NUNCLEON TM Surface
3. DMSO (Dimethylsulfoxid) (Sigma)
II.1.3.1 Geräte des MTT-Assays: 1. Plattenreader Spectralphotometer (Titertek Multiscan Plus, Flow, Irvine, UK)
II.1.4 Material des Comet-Assays 1. Kulturflaschen, 25 cm², Nunclon
2. Falkon 50ml
3. Objektträger, aufgeraut: Labkraft, Dakin Fully Frosted (Curtin Metheson
Scientific Inc., Houston, Texas)
4. Deckgläser: 24 x 40mm IDL (Marienfeld)
5. Phosphat-Natriumchlorid (PBS)-Puffer: Dulbecco Biochrom KG w/o Ca2+, Mg2+
6. Lysepuffer: pH11,5-12 400ml PBS-Puffer
73g NaCl (Pulver) (Sigma)
18,6g EDTA (Ethylenediaminetertaacetic acid) (Pulver)
(Sigma)
0,6g Trizma Base (tris(Hydroymethyl)aminomethan)
(Sigma)
5g N-Lauroyl-Sarcosinat (Sigma)
Vor Gebrauch Zugabe von 1% TritonX (Sigma) und
10% DMSO (Sigma)
Einstellung des pH-Wertes mit NaOH bzw. HCl
7. Elektrophoresepuffer: pH10 2l aqua dest (Apotheke UKL)
24g NaOH (Merck) (Natriumhydroxidplätzchen)
Vor Gebrauch Zugabe von 10ml EDTA (1mM) (Sigma)
8. Ethidiumbromid (Sigma)
9. NMA: Normal Meltingpoint Agarose (Sigma)
10. LMA: Low Meltingpoint Agarose (Sigma)
11. Trypanblau (Sigma) 0,4%ig

Material und Methoden
16
II.1.4.1 Geräte 1. Wärmeblock: Eppendorf Thermomixer 5436
2. Elektrophoresekammer: Werner Hassa GmbH Maxicell Primo EC340
3. Zentrifuge: Heraeus Sepatech, Megafuge 1,0
4. Biofuge A (Heraeus Sepatech)
5. Fluoreszenzmikroskop Axioskop (Zeiss)
6. Software: isis 3 (in situ imaging system) (Meta systems)
II.1.5 DNA-Isolation 1. Kulturflaschen, 25 cm², Nunclon
2. Reaktionsgefäß 50ml (Falkon)
3. Reaktionsgefäß 1,5ml (Eppendorf) Safe-Lock Tubes 1,5ml
4. Reagenz B: pH8 500ml aqua dest.
24,23g Tris (Trishydroxymethylaminomethan) (Sigma)
11,17g EDTA (Sigma)
4,38g NaCl (Sigma)
5g SDS (Biomol) Dodecylsulfate sodium salt
HCl
NaOH
5. Polypropylenröhrchen 5ml (Becton Dickinson) Polypropylene round bottom
tube 12x75mm
6. Filterpapier (Schleicher & Schüll)
7. Ethanol 96% (Apotheke UKL)
8. Ethanol 70% hergestellt aus Ethanol 96%
9. NaClO4 (Merck) Natriumperchlorat-Monohydrat
10. Chloroform (Merck)
11. TE-Puffer: Tris/EDTA-Puffer pH8:
10mM Tris-HCl
1mM EDTA

Material und Methoden
17
II.1.5.1 Geräte 1. DNA-RNA-Calculator: Spektralfluorophotometer zur Quantifizierung der DNA
(Gene Quant II - Photometer)
2. Wasserbad
II.1.6 Fluorometrischer Assay zur Aufdeckung von DNA-Interstrang-Cross-links 1. Höchstfarbstoff 33258 (Bisbenzimide H) (Sigma)
2. Reaktionsgefäße 1,5ml (Lichtgeschützt, ambra) (Eppendorf) Safe-lock Tubes
3. TE-Puffer: Tris/EDTA-Puffer pH8:
5mM Tris (Trishydroxymethylaminomethan)
0,5mM EDTA
4. Tris (Trishydroxymethylaminomethan) (Sigma)
5. EDTA (Sigma)
6. Analysepuffer: TE-Hoechst-Puffer:
TE-Puffer 5mM
Hoechstfarbstoff 33258 0,1µg/ml
II.1.6.1 Geräte 1. Wasserbad
2. Spektralfluorophotometer (Shimadzu RF1501)
3. Software: Hyper RF 1,57

Material und Methoden
18
II. 2. Zellkultur Prinzip:
Es wurden die humanen Zelllinien MX1, eine Mammakarzinomzelllinie, und S117,
ein polymorphkerniges Weichteilsarkom aus der Schilddrüse, verwendet. Beide
Zelllinien wurden aus dem Deutschen Krebsforschungszentrum in Heidelberg
bezogen. Die Tumorzellen wurden als Aliquots von je ca. 2 Mio. Zellen in
flüssigem Stickstoff gelagert. Um ausreichend Zellen für die Versuchsansätze zu
erhalten, mußten sie zunächst aufgetaut und in Kulturflaschen subkultiviert
werden. Zur Vermehrung wurden die Zellen ab einer Konfluenz von 95 bis 100%
von einer Kulturflasche auf mehrere aufgeteilt.
Durchführung:
Die in flüssigem Stickstoff kryokonservierten Tumorzellen wurden in einem 37°C
warmen Wasserbad aufgetaut und in einem speziellen Medium (RPMI 1640
Kulturmedium (25 mM Hepes, L-Glutamin) (Bio Whittaker Europe) mit 10%igem
Zusatz von fetalem Kälberserum (fKS) (Biochrom KG Berlin) und 0,2%igem
Zusatz von Penicillin - Streptomycin (Roche)) aufgenommen. Um die Tumorzellen
von dem Gefriermedium (cryo-safe, c-c-pro) zu befreien, wurden sie nach ihrer
Suspension zentrifugiert und das Pellet im oben genannten Medium in 80 cm²
Zellkulturflaschen (Nunclon) im Brutschrank (Heraeus) bei 37°C in
wasserdampfgesättigter Atmosphäre mit 5% CO2 kultiviert. Während die MX1-
Zellen alle vier Tage subkultiviert wurden, reichten für die Vermehrung der S117 –
Zellen drei Tage aus. Für die Umsetzung der Zellen wurde zunächst das
Nährmedium abgesaugt, der Boden der Zellkulturflasche einmalig mit 10 ml NaCl
– Lösung gewaschen und nach Zugabe einer 10%igen Trypsin-Lösung für 5
Minuten bei 37°C inkubiert. Durch Beklopfen der Kulturflasche lösten sich die
Zellen vom Boden. Nach Zugabe von 5 ml Antitrypsinmedium (RPMI 1640
Kulturmedium (25 mM Hepes, L-Glutamin) (Bio Whittaker Europe)) mit 10%igem
Zusatz von fetalem Kälberserum (fKS) (Biochrom KG Berlin) und 0,2%igem
Zusatz von Penicillin - Streptomycin (Roche) wurden die Zellen in einem 50 ml
Falkonröhrchen mit 1000 U/min (S117) bzw. mit 1500 U/min (MX1) für 3 Minuten
zentrifugiert. Das Pellet wurde in Tumorzellmedium resuspendiert, anschließend
die Zellzahl bestimmt und pro Kulturflasche 4–6x106 Tumorzellen eingesät.

Material und Methoden
19
II.2.2 Zytostatika-Aufbereitung:
Die pulverförmigen Zytostatika wurden im Medium (RMPI 1640, 10% fetales
Kälberserum, 0,2% Penicillin - Streptomycin) suspendiert und für 2 Minuten im
verschlossenen Reagenzglas in ein Ultraschallbad gestellt. 4-Hydroxyperoxy-
Ifosfamid und 4-Hydroxyperoxy-Trofosfamid zerfällt im Medium spontan zu 4-OH-
Ifosfamid bzw. 4-OH-Trofosfamid. Im Medium spaltet sich Mafosfamid spontan zu
4-OH-Cyclophosfamid und Mesna im äquimolaren Verhältnis (Pohl, 1983). Es
wurden verschiedene Zytostatikakonzentrationen durch Verdünnung mit Medium
hergestellt.

Material und Methoden
20
II. 2.3 MTT-Assay Prinzip:
Der MTT-Assay wurde erstmals von Mosman beschrieben und in einer von
unserer Arbeitsgruppe erarbeiteten Modifikation durchgeführt (Wiedemann et al.,
1993; Mosman, 1983). Mit dem MTT-Assay ist eine kolorimetrische Messung
lebender Zellen aufgrund ihrer Stoffwechselaktivität in Korrelation zur Zellzahl
möglich. Stoffwechselaktive Zellen reduzieren mit ihrer NADH-abhängigen
Succinatdehydrogenase (SDH) der Mitochondrien das gelbe Tetrazoliumsalz MTT
(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl Tetrazolium Bromid) zu einem
blauvioletten Formazanprodukt. Die Menge des gebildeten Formazans steht in
direkter Beziehung mit der Anzahl an lebenden Zellen. Durch Zugabe von DMSO
werden die Zellen lysiert, das gebildete Formazan wird dabei gelöst und kann
spektralphotometrisch bei 540nm gemessen werden. Hierbei ist die Absorption
direkt proportional zur Zahl der lebenden Zellen. Die Zellüberlebensrate wurde
nach folgender Formel berechnet:
100 * OD Probe = % Zellüberleben OD Kontrolle OD = optische Dichte
Es handelt sich bei dem MTT-Assay um eine bereits für verschiedene Zelllinien
etablierte Methode. Es wurden von unserer Arbeitsgruppe Wachstumsstudien
durchgeführt, um die optimale Zellzahl und Dauer der Inkubation zu ermitteln und
so eine ausreichende Extinktion zu erhalten. So erzielte man eine Optimierung der
Meßergebnisse und stellte Standardbedingungen für die Versuche mit den
verschiedenen Zelllinien auf (Wössmann et al. 1996). Konzentrationen von 5*104
Zellen / ml hatten sich für beide Zelllinien als optimal erwiesen. Die ermittelte
Inkubationszeit für das Weichteilsarkom betrug drei Tage, die des
Mammakarzinoms vier Tage.

Material und Methoden
21
Abb.3: Das gelbe Tetratrazoliumsalz MTT wird in vitalen Zellen im Mitochondrium durch die Succinatdehydrogenase (SDH) zu einem blauvioletten Formazankristall umgewandelt.
Durchführung:
Wie unter (II.2.1) beschrieben, wurden die Zellen aus den Kulturflaschen
gewonnen, die Zellzahl bestimmt und auf 5*104 Zellen / ml eingestellt. Pro
Versuchsansatz wurden sechs Vertiefungen einer 96-Loch-Mikrotiterplatte
(Nuncleon) befüllt. Um eine Verdunstung aus den mit Testansätzen befüllten
Vertiefungen zu verhindern, wurden zunächst in die randständigen Vertiefungen
100µl Medium gegeben. Anschließend wurden 100µl Zellsuspension in die
Vertiefungen pipettiert und je 50µl Zytostatika - Lösung und 50µl Mesna – Lösung
in äquimolarer Konzentration nach Belegplan befüllt. Die Mikrotiterplatten wurden
im Brutschrank (37°C, pH2O 47mmHg: 95% O2, 5% CO2) inkubiert. Während die
MX1-Zelllinie vier Tage inkubiert wurde, reichten für das Weichteilsarkom S117
entsprechend dem Vermehrungsverhalten drei Tage Inkubationzeit aus. Als
Kontrollansatz wurden bei einer weiteren Mikrotiterplatte fünf Reihen à sechs
Vertiefungen mit 100µl Zellsuspension und 100µl Medium bestückt. Nach der
Inkubation wurde zu jeder Vertiefung 25µl MTT (2mg / ml) pipettiert. Nach weiterer
vierstündiger Inkubation im Brutschrank wurde die flüssige Phase aus den
Vertiefungen vorsichtig bis auf 10µl aspiriert, ohne die Formazankristalle vom
Boden abzulösen. Unter Zugabe von 100µl DMSO und nach fünfminütigem
seitlichen Beklopfen der Platten wurde die Lösung der Kristalle erreicht. Die
Absorption wurde direkt im Anschluß mittels des
Mikrotiterplattenspektralphotometers (Titertek Multiscan Plus, Flow, Irvine, UK) bei
einer Wellenlänge von 540nm gemessen.
MTT Formazankristall
SDH

Material und Methoden
22
II.2.4.1 Trypanblau - Färbung Prinzip:
Für Routineuntersuchungen auf die Vitalität von Zellen haben sich Tests bewährt, die
darauf basieren, dass bei lebenden Zellen bestimmte Farbstoffe nicht in das
Zellinnere gelangen können, während tote Zellen sich mit dem entsprechenden
Farbstoff anfärben. Der am weitesten verbreitete Test auf Lebensfähigkeit von Zellen
ist der sogenannte „Trypanblaufärbetest“, der als Routinetest einfach und schnell
anzuwenden ist. Trypanblau ist ein saurer Farbstoff, der als Anion sehr leicht
Proteine binden kann. Die Farbstoffaufnahme ist stark abhängig von der Färbedauer
sowie temperatur- und pH-abhängig. Es ist weiterhin zu berücksichtigen, dass mit
möglichst wenig Serum im Medium gearbeitet werden sollte, da sich die Anzahl der
gefärbten Zellen bei hoher Serumkonzentration drastisch vermindert und so eine
vorhandene Lebensfähigkeit vorgetäuscht wird.
Durchführung:
Es wurden 2,5x105 Zellen der jeweiligen Testansätze in Eppendorfreaktionsgefäße
(1,5ml) pipettiert und in der Biofuge A (Heraeus Sepatech) zenrifugiert (S117: 3 min
1000U (174xg), MX1 4 min 1500U (390xg)). Anschließend wurde der Überstand
dekantiert und die Zellen in 50µl PBS resuspendiert. Die vorgewärmte 0,4%ige
Trypanblaulösung wurde nun in einem Verhältnis von 1:1 mit der Zellsuspension
vermengt und für 3 Minuten bei 37°C inkubiert, um danach mit der Auszählung in der
Neubauer-Zählkammer zu beginnen. Hierbei wurden die nicht gefärbten Zellen als
lebendig und durchgängig blau gefärbte Zellen als tot betrachtet. Der Prozentsatz an
toten Zellen wurde nach folgender Formel ermittelt:
tote Zellen in % = Anzahl blau gefärbter Zellen x 100
Gesamtzellzahl

Material und Methoden
23
II.2.4.2 Comet – Assay Prinzip:
Der Comet-Assay, auch Einzelzellgelelektrophorese genannt, hat sich als eine
sensitive und zuverlässige Methode bewährt, um DNA-Schäden wie DNA-
Strangbrüche oder DNA-Cross-links an einzelnen Zellen nachzuweisen. Erstmals
wurde er von Östling und Johannson beschrieben (Östling und Johannson, 1984).
In einer modifizierten Version nach Singh wurde der Assay in einem alkalischen
Milieu durchgeführt (Singh et al., 1988). Dieser erleichtert die Denaturierung und
Entspiralisierung der DNA. Die Zellen werden in einer Agaroseschicht auf einen
Objektträger gegeben und lysiert. In der Elektrophorese wandert die negativ
geladene DNA zur Anode. Je kürzer die DNA-Einzelstränge sind, um so weiter
und schneller wandern sie, d.h. je kleiner die Bruchstücke sind, um so länger ist
die zurückgelegte Strecke und damit der Cometschweif. Nach Schweiflänge
wurden die Cometen in verschiedene Klassen eingeteilt. In der „Klasse 1“ stellt
sich nur der Kern der Tumorzelle dar, welcher als „Kopf“ bezeichnet wird. Bei der
„Klasse 2“ sind wenige DNA-Bruchstücke gewandert, der Schweif ist kürzer als
der Kopf. In der „Klasse 3“ sind die Cometschweife länger als der Kopf, und bei
der „Klasse 4“ liegt eine maximale Schweifbildung vor (siehe Abb. 4-7).
Neben den Versuchsansätzen mit verschiedenen Konzentrationen von 4-OH-
Trofosfamid wurde stets eine Probe ohne jegliche Zugabevon Zytostatika - Lösung
und eine weitere mit 200µM Methylmethansulfonat (MMS) angesetzt. MMS ist ein
bekannter direkter DNA-Alkylierer, der als Standardreagenz zur
Strangbruchbildung eingesetzt wird (Pfuhler, 1996; Speit, 1999).
Bei jeder Probe wurde die Lebensfähigkeit der Zellen mit dem
„Trypanblaufärbetest“ ermittelt. Der Anteil der toten Zellen sollte 15% (bezogen auf
die unbehandelte Kontrollgruppe) nicht überschreiten, da ansonsten nicht zu
differenzieren wäre, ob die Cometschweifbildung auf eine DNA-Fragmentation im
Verlauf der Zellapoptose zurückzuführen ist oder ob in der lebenden Tumorzelle
DNA-Strangbrüche durch das Zytostatikum entstanden sind.

Material und Methoden
24
Abb.4: Klasse 1 – Comet ohne erkenn-bare Schweifbildung (400fache Vergrößerung)
Abb.5: Klasse 2 – Comet mit beginnender Schweifbildung (400fache Vergrößerung)
Abb.6: Klasse 3 – Comet mit fort-geschrittener Schweifbildung(400fache Vergrößerung)
Abb.7: Klasse 4 – Comet mit maximal ausgebildetem Schweif (400fache Vergrößerung)

Material und Methoden
25
Durchführung:
Es wurden 2x106 Tumorzellen pro Kulturflasche (25cm2 Nunclon) eingesät. Nach
24 Stunden erfolgte ein Mediumwechsel und die Zugabe der Zytostatika. Hierzu
wurde das Medium und die nicht adhärenten Zellen abgesaugt, die Kulturflaschen
mit 5ml NaCl – Lösung gespült und 9ml frisches Medium auf die Zellen gegeben.
Im Anschluss wurde jeweils 1ml Zytostatika - Lösung hinzupipettiert. Die Kontrolle
erhielt entsprechend 1ml Medium. Die Zellkulturansätze wurden vier Stunden im
Zellkulturschrank (Hera Cell, Heraeus) inkubiert (37°C, pH2O 47mmHg, 95%O2,
5%CO2). Anschließend wurden die Zellen (wie unter II.2.1 beschrieben) trypsiniert
und in Falkonröhrchen (50ml, Sarstedt) bei 1000 bzw. 1500 U/min (174xg bzw.
390xg) für 3 Minuten zentrifugiert (Megafuge 1,0 Heraeus). Nach der
Resuspension der Zellen im Medium wurde die Zellzahl, wie unter (II.2.1)
beschrieben, bestimmt. Es wurden jeweils 175 000 Zellen in ein Reaktionsgefäß
(Eppendorf 1,5ml) pipettiert, nochmals zenrifugiert, der Überstand dekantiert und
die Zellen in 50µl PBS resuspendiert.
Abb.8: Objektträgerbeschichtung: schematische Darstellung des so genannten Sandwichgels LMP = low melting point = niedriger Schmelzpunkt NMP = normal melting point = normaler Schmelzpunkt
Objektträgerbeschichtung:
NMP-Agarose (Sigma) wurde in PBS 0,75%ig aufgelöst und bei 60°C im
Wärmeblock in 1,5ml-Reaktionsgefäßen warm gehalten, um das Aushärten zu
verhindern. Aufgeraute Objektträger (Dakin Fully Frosted) wurden mit 250µl der
flüssigen Agarose beschichtet, mit einem Deckglas (24x60mm) luftblasenfrei
eingedeckelt und 15 Minuten im Kühlschrank (4°C) ausgehärtet. Anschließend
wurde das Deckglas vorsichtig entfernt. 333µl 0,5%iger LMP-Agarose (40°C, in
PBS) wurden mit 50µl Zellsuspension (s.o.) vermischt, hiervon wurden 75µl als 2.
aufgerauter Objektträger
Grundschicht (250µl NMP-Agarose)
Zellschicht (75µl LMP-Agarose)
Deckschicht (100µl LMP-Agarose)

Material und Methoden
26
Schicht auf die Objektträger pipettiert und mit einem Deckglas bedeckt, damit sich
die Zellen gleichmäßig auf der ersten Schicht verteilten. Wieder erhärtete das Gel
innerhalb von 15 Minuten im Kühlschrank. Die letzte Schicht, 100µl LMP-Agarose,
wurde nach vorsichtiger Entfernung des Deckglases aufgetragen, eingedeckt und
weitere 15 Minuten im Kühlschrank gefestigt. Nach erneuter Entfernung des
Deckglases wurden die Objektträger in eine Küvette gestellt und mit frisch fertig
gestelltem Lysepuffer (4°C) (2,5M NaCl, 100mM EDTA, 10mM Tris, 1% Na-
Sarcosinat, in PBS gelöst, pH 11, mit Zugabe von 1%Triton x 100, 10% DMSO
direkt vor Gebrauch) im Kühlschrank bei 4°C für eine Stunde inkubiert. Im
Anschluss folgte eine 20 minütige Inkubation mit Elektrophoresepuffer (4°C)
(300mM NaOH, 1mM EDTA in H2O, pH10). Danach wurde die
Elektrophoresekammer (Werner Hassa GmbH Maxicel Primo EC 340) mit den
Objektträgern bestückt und anschließend mit dem Elektrophoresepuffer aufgefüllt,
bis die Objektträger ca. 2mm hoch bedeckt waren. Die elektrische Spannung (U)
wurde auf 25 Volt und die Stromstärke auf 300 mA eingestellt. Schwankungen in
der Stromstärke wurden durch Zugabe oder Abnahme von Elektrophoresepuffer
ausgeglichen. Hierbei wurde Widerstand durch Zugabe von Elektrophoresepuffer
vermindert, d.h. Stromstärke bei konstanter Spannung entsprechend vergrößert.
Nach 20 minütiger Elektrophorese wurden die Objektträger wieder in die Küvette
gestellt und dreimal gewaschen. Dazu wurde die Küvette mit 4°C - kaltem PBS
gefüllt und fünf Minuten lang im Kühlschrank inkubiert. Die DNA wurde mit
Ethidiumbromid (EB) in einem abgedunkelten Raum gefärbt, da EB UV-instabil ist.
40µl EB-Lösung (Sigma, 4%ig in PBS gelöst) wurden auf die 3. Agaroseschicht
pipettiert und mit einem Deckglas abgedeckt und am Fluoreszensmikroskop
(Zeiss) mit einem CY-3 Filter ausgewertet. Pro Versuchsansatz wurden zwei
Objektträger manuell durch mäanderförmiges Absuchen unter dem Mikroskop
ausgewertet. Hierbei wurden ca. 100 Cometen ausgezählt und klassifiziert. Dieses
wurde durch beispielhafte Fotos dokumentiert (Software: „isis3“ Meta systems).

Material und Methoden
27
II.2.5 DNA-Isolation Prinzip:
Aus den beiden Tumorzelllinien S117 und MX1 wurde die DNA durch eine
modifizierte Version des Nuncleon Protokolls (Scotlab GmbH) isoliert, um sie auf
DNA-DNA-Cross-links zu untersuchen. Mit dieser Methode wird sowohl eine hohe
Quantität als auch ein hoher Reinheitsgrad der DNA erreicht. Dafür wurden die
Zellen in einem Puffer lysiert und anschließend in 3 Phasen aufgetrennt. Die DNA
wurde mittels Ethanol ausgefällt und in einem Puffer gelöst. Die DNA-Menge
wurde photometrisch ermittelt.
Abb.9: Die DNA-haltige Phase wird in ein Falkonröhrchen pipettiert, die DNA durch Ethanol ausgefällt und so makroskopisch sichtbar gemacht, um sie für die nachfolgende Analyse in ein weiteres Reaktionsgefäß überführen zu können.
DNA
Ethanol 96%ig
DNA
Ethanol 96%ig
Chl
orof
orm P
rote
in
DNA

Material und Methoden
28
Durchführung:
24 Stunden nach der Einsaat von mindestens 2x106 Tumorzellen pro Kulturflasche
(Nunclon 25 cm2) folgte nach Entfernung des Mediums die 4 stündige Inkubation
mit dem Zytostatikum im Brutschrank (Heraeus) (37°C, pH2O 47mmHg; 95% O2,
5% CO2). Anschließend wurden die Zellen (wie unter II.2.1 beschrieben) aus den
Kulturflaschen entfernt, jede Probe in ein Falkonröhrchen (50ml) überführt und bei
1000 bzw. 1500 U/min (174xg bzw. 390xg) drei Minuten zentrifugiert. Das so
entstandene Pellet wurde durch Resuspension in NaCl-Lösung (0,9%) und
Zentrifugation gewaschen. Die Tumorzellen wurden in 2ml Reagenz B (400mM
Tris-HCl, pH 8, 60mM EDTA, 150mM NaCl, 1% SDS in H2O) lysiert und in ein
Polypropylen-Röhrchen (Becton-Dickinson) (5ml Falkon) überführt.
Nach Zugabe von 500µl NaClO4 und 15 minütigem Invertieren erfolgte eine 25
minütige Inkubation im Wasserbad bei 65°C, währenddessen die Reagenzgefäße
alle 5 Minuten geschwenkt wurden. Nach Zugabe von 2ml Chloroform (-20°C)
folgte ein Inversionsvorgang von 10 Minuten. Durch 4 minütige Zentrifugation bei
1400xg entstanden 3 Phasen. Die obere DNA–haltige Phase wurde in ein
Falkonröhrchen (50ml) pipettiert. Nach Zugabe von 5ml eiskaltem 96%igen
Ethanol fiel die DNA aus, so dass sie makroskopisch sichtbar wurde. Durch
Schütteln des Röhrchens bildete sich aus dem DNA-Faden ein Knäuel, welches
aus dem Alkohol entfernt und in ein Reaktionsgefäß (1,5 ml Eppendorf) überführt
wurde. Nach dreimaligem Waschen der DNA mit eiskaltem 70%igen Ethanol
wurde der Alkohol dekantiert. Nach ca. 15 minütiger Verdunstung letzter
Ethanoltropfen wurde die DNA in 150µl TE-Puffer (10mM Tris-HCl, 1mM EDTA,
pH 8,0) gelöst und über Nacht im Kühlschrank bei 4°C inkubiert. Am nächsten Tag
wurden die Proben je nach Viskosität bis zur Pipettierfähigkeit durch Zugabe von
TE-Puffer verdünnt.
Der DNA-Gehalt wurde mit einem DNA-RNA-Kalkulator (Gene Quant II-
Photometer) spektralphotometrisch bestimmt, um sicherzustellen, dass eine
möglichst geringe Verunreinigung vorliegt. Dazu wurde die DNA-Probe in einem
Verhältnis 50:1 mit 10 molarem TE-Puffer verdünnt, gut durchmischt und in die
Küvette des Kalkulators gegeben. Bei einer Wellenlänge von 260nm wurde der
Gehalt an doppelsträngiger DNA bestimmt, dabei wurde ein Abgleich von 260nm

Material und Methoden
29
gegen 280nm durchgeführt. Alle Proben wurden doppelt gemessen, und anhand
der Ergebnisse wurde die DNA-Konzentration in 1,5 ml-Reaktionsgefäßen auf
75µg pro ml eingestellt.

Material und Methoden
30
II.2. 6. Fluorometrischer Cross-link-Assay Prinzip:
Der Nachweis von Interstrang-Cross-links erfolgte nach dem Prinzip von P.G.
Penketh, der 1997 einen Assay beschrieb, der bei einem physiologischen pH-Wert
durchführbar ist (Penketh et al., 1997). Interstrang-Cross-links sind kovalente
Bindungen zwischen einem komplementären Basenpaar der DNA. Um die aus
den Tumorzellen gewonnene DNA auf Cross-links zu untersuchen, wird sie mit
einem blau fluoreszierenden Bisbenzimidfarbstoff Hoechst 33258 gefärbt, der
vornehmlich an doppelsträngiger DNA (ds DNA) bindet. Der Nachweis von DNA-
DNA-Interstrang-Cross-links basiert auf den unterschiedlichen
Reaktionsmechanismen von DNA, die durch Cross-links verbunden ist, im
Vergleich zu Cross-link-freier DNA. Bei einem Erhitzungsvorgang wird die
doppelsträngige DNA in Einzelstränge aufgetrennt. Bei der anschließenden
Abkühlung der DNA unterliegt die Cross-link-freie DNA einer Fehlpaarung der
Basen, so dass die komplementären DNA-Stränge nicht mehr zusammenfinden
können. Im Gegensatz dazu finden die DNA-Stränge der durch Cross-links
teilweise verbundenen DNA sehr schnell wieder zusammen. An diesen Stellen
kann nun der Farbstoff wieder binden. Die so angefärbte DNA wird von Licht der
Wellenlänge 365nm angeregt. Die Emission wird mit einem
Spektralfluorophotometer (Shimadzu RF-1501) vor und nach dem Erhitzen bei
460nm gemessen. Bei Negativkontrolle werden die Tumorzellen ohne Zytostatika
inkubiert und bei der Positivkontrolle mit Cisplatin vorbehandelt, das nachweislich
Cross-links hervorruft (Roberts und Pascoe, 1972; Sullivan et al., 2002). Bei der
Negativprobe sinkt die Fluoreszenzintensität nach dem Erhitzen auf ein Minimum.
Bei den behandelten Proben, die Cross-links aufweisen, fällt die
Fluoreszenzintensität weniger stark ab. Um die erhobenen Daten zu normalisieren
und das Ausmaß der Crosslinkbildung zu verdeutlichen, wurde die folgende
Formel angewendet:

Material und Methoden
31
(E N / E V) – (K N / K V) = %-Anteil Cross-links
( 1- (K N / K V))
E N = Fluoreszenzintensität der Probe nach dem Hitze-Kälte-Zyklus
E V = Fluoreszenzintensität der Probe vor dem Hitze-Kälte-Zyklus
K N = Fluoreszenzintensität der Kontrollprobe nach dem Hitze-Kälte-Zyklus
K V = Fluoreszenzintensität der Kontrollprobe vor dem Hitze-Kälte-Zyklus
DNA ohne Cross-links: DNA mit Cross-links:
Abb.10: Auftrennung der DNA in Einzelstränge durch den Erhitzungs- und Abkühlungsvorgang aufgezeigt an DNA mit und ohne DNA-Interstrang-Cross-links
dsDNA vor dem Erhitzen
ssDNA nach dem Erhitzen
ssDNA nach dem Abkühlen
dsDNA nach dem Erhitzen
dsDNA vor dem Erhitzen
dsDNA nach dem Abkühlen

Material und Methoden
32
Durchführung:
Die DNA aus den Tumorzellen wurde in TE-Puffer (10mM Tris-HCl, 1mM EDTA,
pH 8,0 in H2O) gelöst und auf eine Konzentration von 75 µg/ml eingestellt. 15µl
aus jedem Ansatz wurden nun in 1,5 ml der Farbstofflösung (0,1µg/ml Höchst
33258 in 1,5 ml 5 mM Tris-HCL, 0,5mM EDTA) transferiert, um eine DNA-
Konzentration von 0,75 µg/ml zu erhalten. Da der verwendete Fluoreszenzfarbstoff
(2´-(4-hydroxyphenyl)-5-(4-methyl-1piperazinyl)-2,5´bi-1H-benzimidazol, Sigma),
bei UV-Licht instabil wird, mussten die Gefäße mit Aluminiumfolie umwickelt
werden. Zunächst wurde die Fluoreszenz mit dem Spektralfluorophotometer
gemessen, dann wurden die Proben für jeweils fünf Minuten im Wasserbad bei
96°C erhitzt, in ein Eiswasserbad gestellt und anschließend in einem Wasserbad
bei 21°C inkubiert. Danach folgte die zweite Fluoreszenzmessung aller Proben am
Spektralfluorophotometer, und der prozentuale Crosslinkanteil wurde durch die
oben genannte Formel ermittelt.

Material und Methoden
33
II.7 Biometrie und Statistik
Die Erstellung der Graphiken und die statistische Analyse folgender Parameter
erfolgte mit dem Computerprogramm ExcelTM: Mittelwert, Median,
Standardabweichung, Standardfehler. Zum Vergleich der Mittelwerte der zu
untersuchenden Größen zweier unabhängiger Gruppen wurde der Mann-Whitney
U-Test verwendet. Ein Signifikanzniveau (p) von <0,05 wurde als statistisch
signifikant gewertet. Sämtliche Daten wurden zuvor per Hand in den Computer
eingegeben.

Ergebnisse
34
III. Ergebnisse
III.1. MTT-Assay Beide Tumorzelllinien, das Weichteilsarkom aus der Schilddrüse S117 und das
Mammakarzinom MX1, wurden mit den drei verschiedenen aktiven
Oxazaphosphorinmetaboliten Mafosfamid, 4-OH-Ifosfamid und 4-OH-Trofosfamid
drei bzw. vier Tage im Brutschrank inkubiert. Da Mafosfamid im Tumorzellmedium
im äquimolaren Verhältnis zu 4-OH-Cyclofosfamid und Mesna zerfällt, wurde auch
zu 4-OH-Trofosfamid und 4-OH-Ifosfamid Mesna im äquimolaren Verhältnis
zugefügt.
Im Vergleich der drei aktiven 4-OH-Metabolite der Oxazaphosphorine läßt sich bei
beiden Zelllinien beobachten, dass 4-OH-Trofosfamid (+Mesna) ein signifikant
höheres zytotoxisches Potential aufweist als 4-OH-Ifosfamid (+Mesna). Besonders
ausgeprägt ist dieser Effekt auf die Zelllinie MX1. Hier liegt die IC50 (inhibierende
Konzentration, bei der 50% der Zellen sterben) von 4-OH-Ifosfamid (+Mesna) fast
doppelt so hoch wie die von 4-OH-Trofosfamid (+Mesna) (IC50: 4-OH-
Ifosfamid+Mesna = 3,2µM, 4-OH-Trofosfamid + Mesna = 1,7µM). Zudem wird
deutlich, dass - verglichen mit Mafosfamid - sogar noch geringere Konzentrationen
von 4-OH-Trofosfamid (+Mesna) genügen, um die Zellzahl des Mammakarzinoms
auf die Hälfte zu reduzieren (IC50 Mafosfamid = 2,8µM).
Bei allen Oxazaphosphorinmetaboliten ist zu beobachten, dass die IC50–Werte
des Mammakarzinoms wesentlich niedriger sind als für das Weichteilsarkom der
Schilddrüse. Hier liegt die IC50 für 4-OH-Trofosfamid (+Mesna) um das Achtfache
höher als beim Mammakarzinom (IC50 = 13,99µM) und damit über dem Wert von
Mafosfamid (IC50 = 11,05), das an der Zelllinie S117 am stärksten zytotoxisch
wirkt. Auch hier ist Ifosfamid (+Mesna) mit einer IC50 von 19,95µM am wenigsten
zytotoxisch wirksam (siehe Abb.11 und 12).
Die gleichzeitige Gabe von Mesna in äquimolaren Konzentrationen ergab bei der
Gabe von 4-OH-Trofosfamid in den Konzentrationen von 5 bis 50µM (S117) bzw.
0,5 bis 30µM (MX1) und der IC50 keinen signifikanten Unterschied bezüglich der
Zytotoxizität auf beide Zelllinien (P>0,5). Ebenfalls ergibt sich keine Signifikanz

Ergebnisse
35
(P>0,05) beim Vergleich der Zytotoxizität von 4-OH-Ifosfamid als Einzelsubstanz
bzw. in Kombination mit Mesna. Im Verlauf des Zellüberlebens der S117
Tumorzellen wurde bei einer einzigen Konzentration (20µM 4-OH-Ifosfamid) ein
signifikanter Unterschied gemessen (P = 0,034). Ansonsten konnte durch Zugabe
von Mesna bei beiden Zelllinien kein signifikanter Unterschied in der Zytotoxizität
bestimmt werden (siehe Abb.13 und 14).
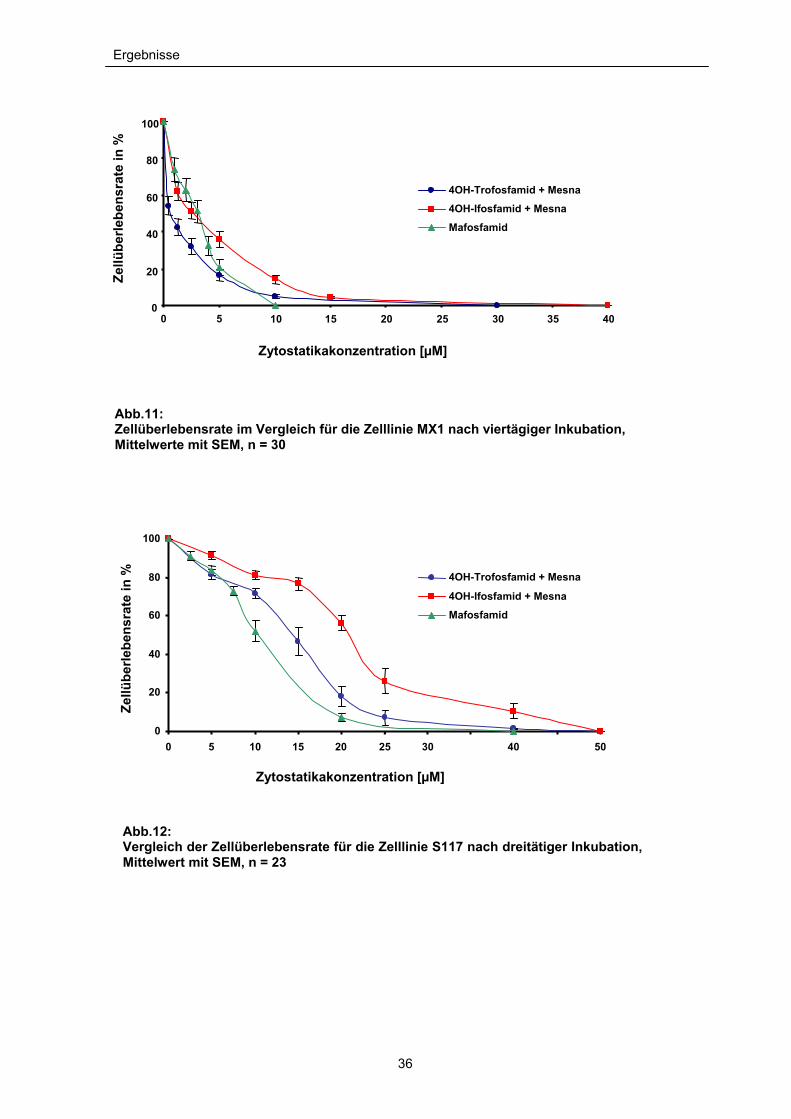
Ergebnisse
36
0 20 40 60 80
100
0 5 10 15 20 25 30 40 50
4OH-Trofosfamid + Mesna
4OH-Ifosfamid + Mesna
Mafosfamid
Zellü
berle
bens
rate
in %
Zytostatikakonzentration [µM]
0
20
40
60
80
100
0 5 10 15 20 25 30 35 40
4OH-Trofosfamid + Mesna
4OH-Ifosfamid + Mesna
Mafosfamid
Zellü
berle
bens
rate
in %
Zytostatikakonzentration [µM]
Abb.11: Zellüberlebensrate im Vergleich für die Zelllinie MX1 nach viertägiger Inkubation, Mittelwerte mit SEM, n = 30
Abb.12: Vergleich der Zellüberlebensrate für die Zelllinie S117 nach dreitätiger Inkubation, Mittelwert mit SEM, n = 23
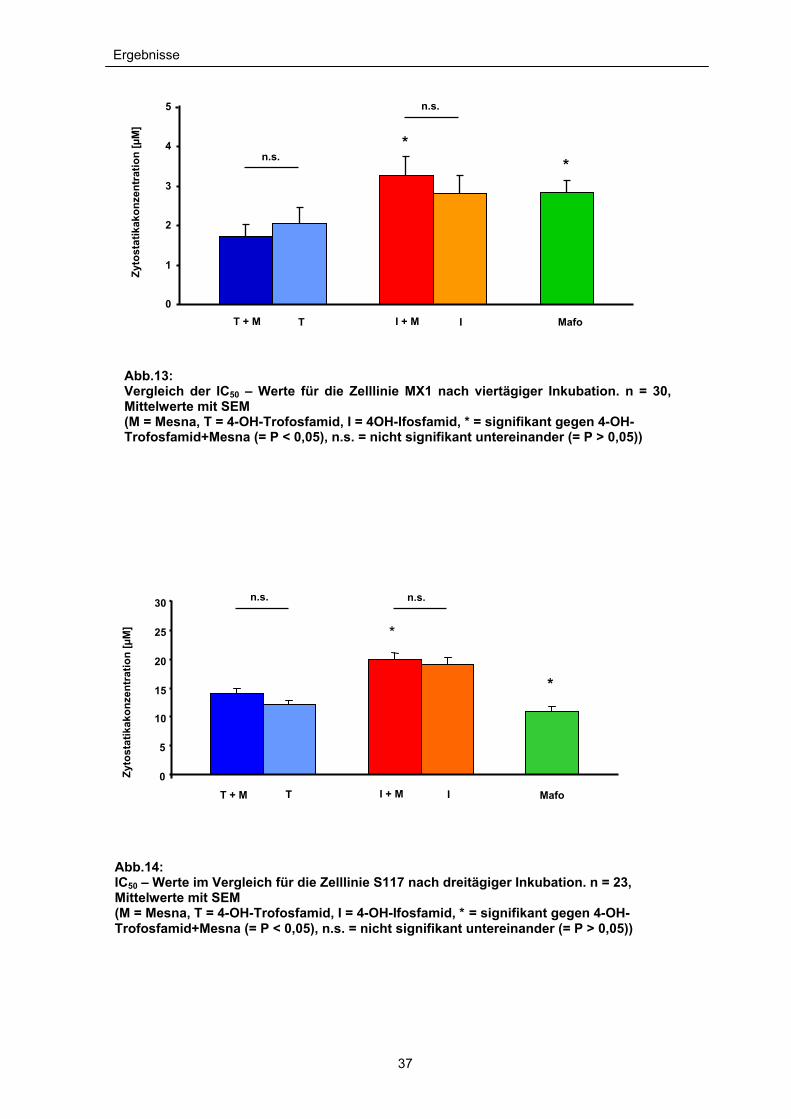
Ergebnisse
37
Abb.13: Vergleich der IC50 – Werte für die Zelllinie MX1 nach viertägiger Inkubation. n = 30, Mittelwerte mit SEM (M = Mesna, T = 4-OH-Trofosfamid, I = 4OH-Ifosfamid, * = signifikant gegen 4-OH-Trofosfamid+Mesna (= P < 0,05), n.s. = nicht signifikant untereinander (= P > 0,05))
0 1 2 3 4
T + M T I + M I Mafo
Zyto
stat
ikak
onze
ntra
tion
[µM
] *
*
5 n.s.
n.s.
Abb.14: IC50 – Werte im Vergleich für die Zelllinie S117 nach dreitägiger Inkubation. n = 23, Mittelwerte mit SEM (M = Mesna, T = 4-OH-Trofosfamid, I = 4-OH-Ifosfamid, * = signifikant gegen 4-OH-Trofosfamid+Mesna (= P < 0,05), n.s. = nicht signifikant untereinander (= P > 0,05))
0
5
10
15
20
25
30
I + M T I Mafo T + M
*
*
n.s. n.s.
Zyto
stat
ikak
onze
ntra
tion
[µM
]

Ergebnisse
38
III.2.1 Trypanblau-Färbung
Diese Färbung erfolgte zur Überprüfung der Überlebensrate der verwendeten
Zellen des Comet-Assays. Vollständig blau gefärbte Zellen wurden als tot
betrachtet. In den Versuchsansätzen ergaben sich die in den Tabellen 3 und 4 (s.
VII.3 im Anhang) aufgeführten Überlebensraten. Es wurden niemals Werte über
15% erreicht, so dass davon ausgegangen werden kann, dass die Bildung der
Cometen auf die Wirkung der Zytostatika zurückzuführen ist. So konnte vermieden
werden, dass die beim Zelltod auftretende DNA-Fragmentation die
Beobachtungen verfälscht.

Ergebnisse
39
III.2.2 Comet – Assay - Einzelzellgelelektrophorese
Jeder Objektträger wurde manuell mäanderförmig unter dem
Fluoreszensmikroskop betrachtet und die Anzahl der verschiedenen Arten der
Cometen - Klasse eins bis vier bestimmt (Klassifizierung siehe unter II.2.4).
Als Kontrollgruppen wurden unbehandelte Zellen der jeweiligen Tumorzelllinie
sowie mit 200µM Methylmethansulfonat (MMS) behandelte Zellen untersucht. Wie
bereits mehrfach nachgewiesen (Miyamae et al., 1998; Pfuhler und Wolf, 1996,
Speit et al. 1999), zeigte sich durch MMS auch in diesen Versuchsreihen eine
maximale Strangbruchbildung. Die mit MMS inkubierten Proben wiesen zu 100%
Klasse 4–Cometen auf. Bei beiden Tumoren stellte sich heraus, dass fast 90% der
Zellen in der unbehandelten Gruppe keinen DNA-Schaden aufwiesen, d.h.
hauptsächlich Cometen der Klasse 1 zu sehen waren. Im Mittel zeigten sich im
Kontrollansatz ohne Zugabe eines Zytostatikums zu etwa 7% Klasse 4–Cometen
(siehe Abb.15 und 16).
Die verwendeten Konzentrationen an 4-OH-Trofosfamid lagen bei dem MX1-
Tumor zwischen dem 0,4 und dem 94 fachen IC50 (inhibiting concentration, durch
den MTT-Assay ermittelt, siehe III.2). Bei der S117 Zelllinie ergab sich ein Bereich
zwischen der 0,04 und 5,7 fachen Konzentration der IC50. Es zeigte sich, dass 4-
OH-Trofosfamid mit steigender Konzentration mehr Strangbrüche verursachte. Die
Anzahl der Zellen, bei denen Schweife sichtbar wurden, stieg an und es
vergrößerte sich zudem die Schweiflänge. Das heißt, der Anteil an Klasse 1-
Cometen nahm zugunsten der Klasse 2- und Klasse 3-Cometen stetig ab, wobei
die Klasse 4–Cometen meist unter 20% blieben.
Bei dem Mammakarzinom MX1 waren die ersten Cometschweife ab einer
Konzentration von 2,5µM zu beobachten. Dies entspricht in etwa dem
anderthalbfachen der IC50. Bei der höchsten Konzentration von 160µM hatten sich
aus der DNA von über 85% der Zellen Cometschweife der Klasse 2 und 3 gebildet
(siehe Abb. 17 und 19).
Das Sarkom S117 zeigte schon bei einer Konzentration von 80µM einen
maximalen Anteil der Cometschweif-Bildung in der Klasse 2 und 3 von über 95%.

Ergebnisse
40
Ein erster deutlicher Anstieg der Cometen der Klasse 2 und 3 ergab sich schon ab
dem 0,4 fachen der im MTT-Assay ermittelten IC50 bei 5µM (siehe Abb.18 und 20).
Abb.15 zeigt eine repräsentative Aufnahme der Cometen am Beispiel des Weichteilsarkoms S117, wie sie in der unbehandelten Kontrollgruppe hauptsächlich zu beobachten waren (400fache Vergrößerung).
Abb.16 zeigt ein beispielhaftes Bild der Cometen nach Inkubation der S117 Zellen mit MMS (200µM), das hauptsächlich Klasse 4 – Cometen aufweist (200fache Vergrößerung).
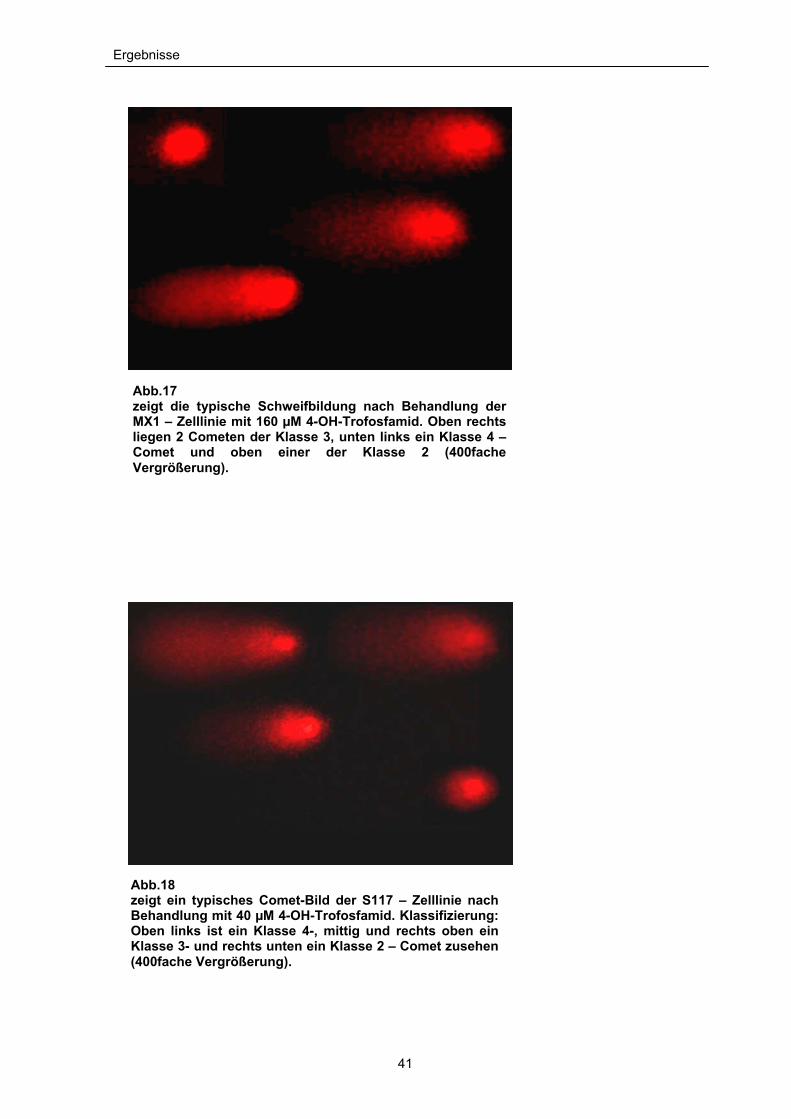
Ergebnisse
41
Abb.17 zeigt die typische Schweifbildung nach Behandlung der MX1 – Zelllinie mit 160 µM 4-OH-Trofosfamid. Oben rechtsliegen 2 Cometen der Klasse 3, unten links ein Klasse 4 –Comet und oben einer der Klasse 2 (400fache Vergrößerung).
Abb.18 zeigt ein typisches Comet-Bild der S117 – Zelllinie nach Behandlung mit 40 µM 4-OH-Trofosfamid. Klassifizierung: Oben links ist ein Klasse 4-, mittig und rechts oben ein Klasse 3- und rechts unten ein Klasse 2 – Comet zusehen (400fache Vergrößerung).

Ergebnisse
42
Darüber hinaus konnte bei dem Vergleich von 4-OH-Trofosfamid mit der
Kombination aus 4-OH-Trofosfamid und Mesna (im äquimolaren Verhältnis) bei
beiden Tumoren kaum ein Unterschied in der Schweifbildung erkannt werden.

Ergebnisse
43
C
omet
- K
lass
en -
Ant
eil i
n %
Klasse 2 Klasse 1
Klasse 3 Klasse 4
4-OH-Trofosfamid + Mesna (äquimolar) [µM]
0
20
40
60
80
100
Kontrolle 0,6 1,25 2,5 5 10 20 40 80 160
Abb.19: Mammakarzinom MX1 Prozentualer Anstieg der Cometenbildung unter ansteigender Dosis von 4-OH-Trofosfamid + Mesna (n = 3) im Vergleich zur unbehandelten Kontrollgruppe (n = 12), Mittelwerte
Klasse 2 Klasse 1
Klasse 3 Klasse 4
Com
et -
Kla
ssen
- A
ntei
l in
%
4-OH-Trofosfamid + Mesna (äquimolar) [µM]
0
20
40
60
80
100
Kontrolle 0,6 1,25 2,5 5 10 20 40 80
Abb.20: Weichteilsarkom der Schilddrüse S117 Prozentualer Anstieg der Cometenbildung unter ansteigender Dosis von 4-OH-Trofosfamid + Mesna (n = 3) im Vergleich zur unbehandelten Kontrollgruppe (n = 8), Mittelwerte

Ergebnisse
44
III.3.1 Validierung des Cross-link-Assays am Spectralfluorophotometer
Zunächst wurde ermittelt, wie stark die Schwankungen innerhalb eines
Versuchsansatzes, d.h. innerhalb einer Zellpassage sind (Tab.1). Dazu wurde die
DNA aus den Tumorzellen (MX1) nach dem Protokoll von II.2.5 isoliert und
hieraus sechs verschiedene Proben angesetzt. Die Extinktionen wurden, wie unter
II.2.6 beschrieben, vor und nach dem Hitze-Kälte-Zyklus ermittelt. Anschließend
wurden mit Hilfe der Computersoftware ExcelTM die Mittelwerte, die
Standardabweichungen, der Standardfehler und die Variationskoeffizienten
berechnet.
Tabelle 1: ermittelter Extinktionsabfall der unbehandelten Kontrolle
Extinktion vor dem
Erhitzungszyklus
Extinktion nach dem
Erhitzungszyklus
Extinktion nach dem
Erhitzungszyklus in %
803,55 82,55 10,27
781,4 63,95 8,18
814,8 68,1 8,36
830,35 70,7 8,51
773,2 92,15 11,91
758,15 74,9 9,88
Mittelwert 793,58 75,39 9,52
Standardabweichung 24,9 9,95 1,328
Standardfehler 9,96 3,79 0,53
Variationskoeffizient 3,1 % 12,6 % 13,96 %
Die Fluoreszenz nicht behandelter DNA fällt nach dem Erhitzungsvorgang stark ab
(auf 9,52% des Ausgangswertes), da sich ohne Zugabe von Zytostatika keine
Cross-links gebildet haben. Die Standardabweichung, der Standardfehler und der
Variationskoeffizient (Präzision) sind sehr gering und entsprechen somit unseren
Akzeptanzkriterien.
Tabelle 1 zeigt den Extinktionsabfall und dessen Streuungen innerhalb eines Versuchansatzes anhand unbehandelter DNA der Zelllinie MX 1. (n = 6)
Ergebnisse
45
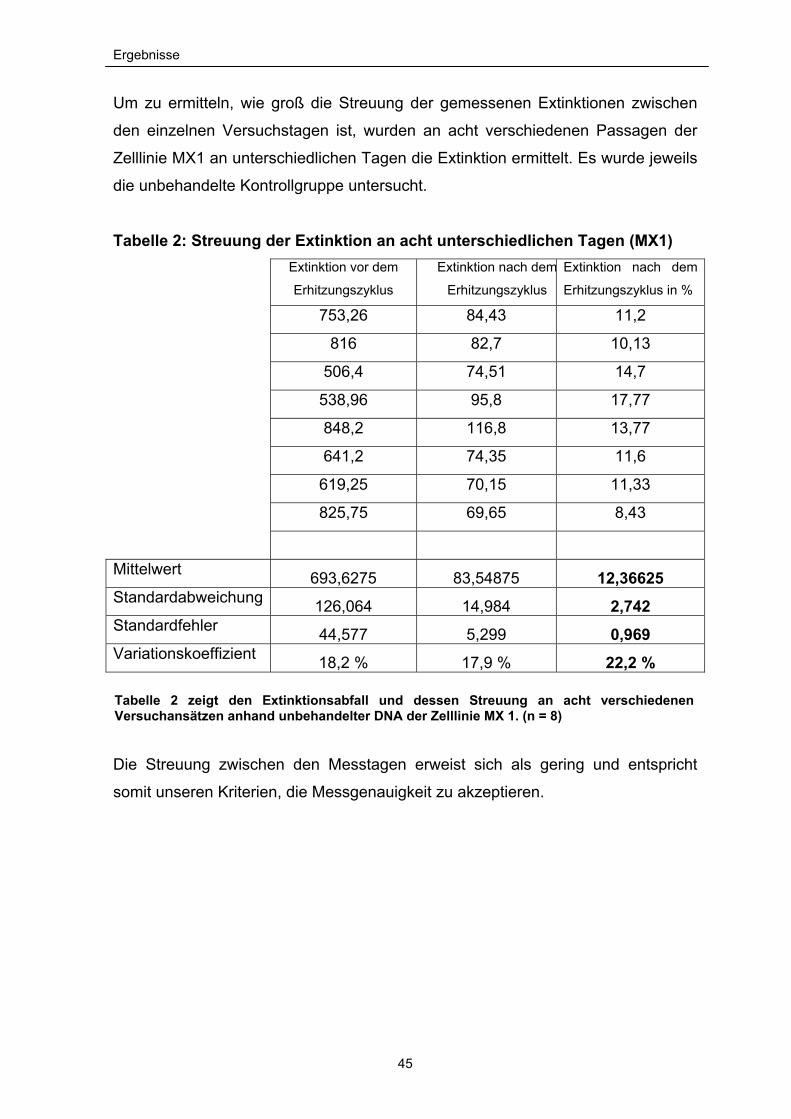
Um zu ermitteln, wie groß die Streuung der gemessenen Extinktionen zwischen
den einzelnen Versuchstagen ist, wurden an acht verschiedenen Passagen der
Zelllinie MX1 an unterschiedlichen Tagen die Extinktion ermittelt. Es wurde jeweils
die unbehandelte Kontrollgruppe untersucht.
Tabelle 2: Streuung der Extinktion an acht unterschiedlichen Tagen (MX1)
Extinktion vor dem
Erhitzungszyklus
Extinktion nach dem
Erhitzungszyklus
Extinktion nach dem
Erhitzungszyklus in %
753,26 84,43 11,2
816 82,7 10,13
506,4 74,51 14,7
538,96 95,8 17,77
848,2 116,8 13,77
641,2 74,35 11,6
619,25 70,15 11,33
825,75 69,65 8,43
Mittelwert 693,6275 83,54875 12,36625 Standardabweichung 126,064 14,984 2,742 Standardfehler 44,577 5,299 0,969 Variationskoeffizient 18,2 % 17,9 % 22,2 %
Die Streuung zwischen den Messtagen erweist sich als gering und entspricht
somit unseren Kriterien, die Messgenauigkeit zu akzeptieren.
Tabelle 2 zeigt den Extinktionsabfall und dessen Streuung an acht verschiedenen Versuchansätzen anhand unbehandelter DNA der Zelllinie MX 1. (n = 8)
Ergebnisse
46
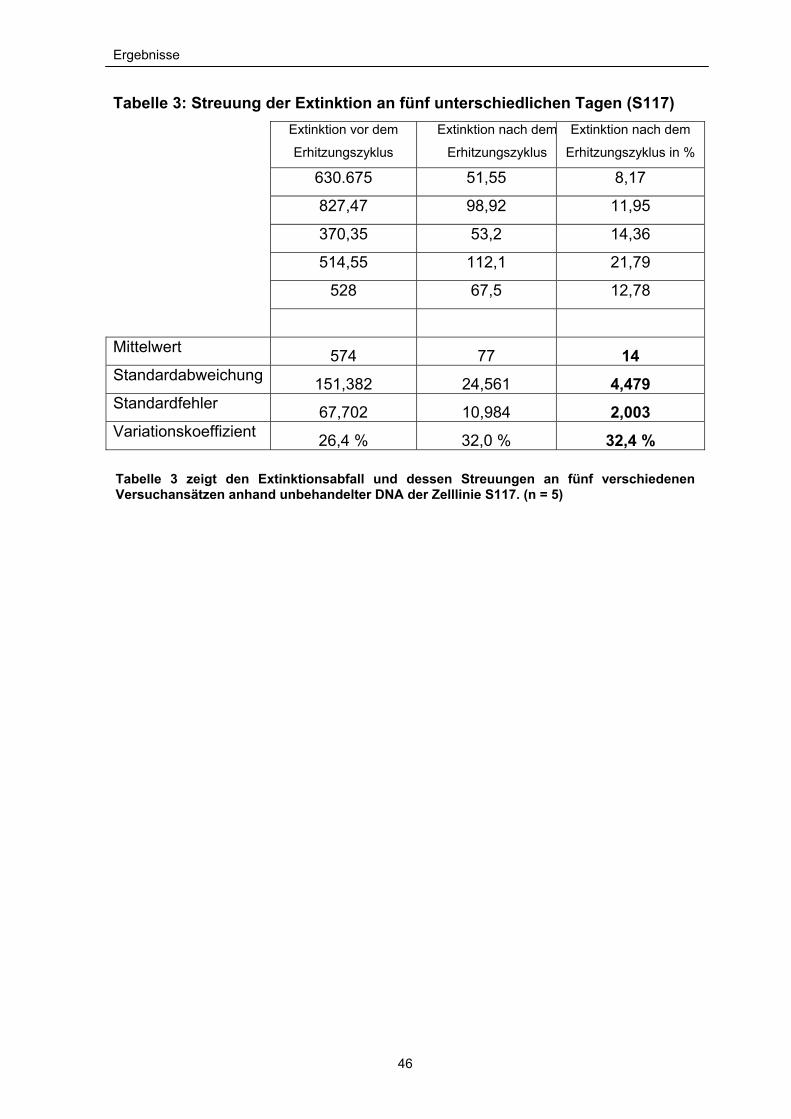
Tabelle 3: Streuung der Extinktion an fünf unterschiedlichen Tagen (S117)
Extinktion vor dem
Erhitzungszyklus
Extinktion nach dem
Erhitzungszyklus
Extinktion nach dem
Erhitzungszyklus in % 630.675 51,55 8,17
827,47 98,92 11,95
370,35 53,2 14,36
514,55 112,1 21,79
528 67,5 12,78
Mittelwert 574 77 14 Standardabweichung 151,382 24,561 4,479 Standardfehler 67,702 10,984 2,003 Variationskoeffizient 26,4 % 32,0 % 32,4 %
Tabelle 3 zeigt den Extinktionsabfall und dessen Streuungen an fünf verschiedenen Versuchansätzen anhand unbehandelter DNA der Zelllinie S117. (n = 5)

Ergebnisse
47
III.3.2 Fluorometrischer Cross-link-Assay
Als Kontrollgruppen wurden unbehandelte Zellen der jeweiligen Tumorzelllinie
sowie mit 100µM Cisplatin behandelte Zellen untersucht. Bei der unbehandelten
Kontrollgruppe fiel die Extinktion nach dem Erhitzen bei MX1 im Mittel auf 12,36 %
und bei S117 auf 14 % ab. (siehe Validierung unter III.3.1)
(EN / EV) – (KN / KV)
= %-Anteil Cross-links
(1- KN / KV)
EN = Fluoreszenzintensität der Probe nach dem Hitze-Kälte-Zyklus
EV = Fluoreszenzintensität der Probe vor dem Hitze-Kälte-Zyklus
KN = Fluoreszenzintensität der Kontrolle nach dem Hitze-Kälte-Zyklus
KV = Fluoreszenzintensität der Kontrolle vor dem Hitze-Kälte-Zyklus
Bei beiden Tumoren kam es nach Gabe von Cisplatin zu einer deutlichen Bildung
von Cross-links, da die Fluoreszenzintensität nur auf ca. 40% der
Ausgangsfluoreszenz abfiel. Dies ergibt unter Einbezug der unbehandelten
Kontrollgruppe durch die oben genannte Formel einen prozentualen Cross–link–
Anteil von über 30% (siehe Abb.21 und 22).
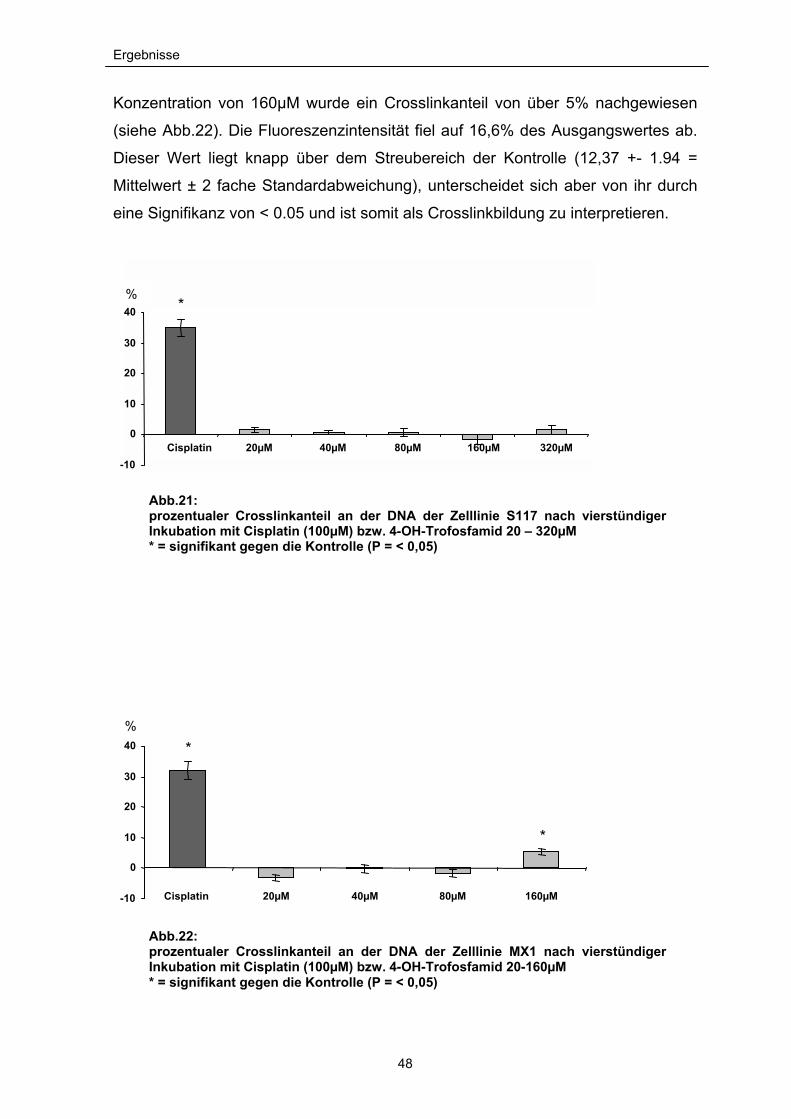
4-OH-Trofosfamid konnte nach der vierstündigen Inkubationszeit mit dem
Weichteilsarkom S117 in den Konzentrationen von 20 bis 320µM keine
nachweisbaren Cross-links erzeugen (siehe Abb.21). Die gemessenen Werte
überschreiten nicht den Bereich der zu erwartenden Streuungen eines
Kontrollansatzes. Da in der Literatur bereits die Bildung von Cross-links durch
Metabolite der Oxazaphosphorine beschrieben wurde, sollten sehr hohe
Konzentrationen verwendet und die Induktion von Cross-links nochmals überprüft
werden. Selbst bei diesen Einzelversuchen mit extrem hohen zytotoxischen
Konzentrationen von 3,2mM und 6,5mM 4-OH-Trofosfamid wurde nur ein geringer
prozentualer Cross-link-Anteil gemessen (6,8% und 5,9%).
Auch bei dem Mammakarzinom konnte in den Konzentrationen von 20 bis 80 µM
keine Bildung von Cross-links gemessen werden. Lediglich bei einer
Ergebnisse
48
Konzentration von 160µM wurde ein Crosslinkanteil von über 5% nachgewiesen
(siehe Abb.22). Die Fluoreszenzintensität fiel auf 16,6% des Ausgangswertes ab.
Dieser Wert liegt knapp über dem Streubereich der Kontrolle (12,37 +- 1.94 =
Mittelwert ± 2 fache Standardabweichung), unterscheidet sich aber von ihr durch
eine Signifikanz von < 0.05 und ist somit als Crosslinkbildung zu interpretieren.
-10
0
10
20
30
40
Cisplatin 20µM 40µM 80µM 160µM 320µM
% *
Abb.21: prozentualer Crosslinkanteil an der DNA der Zelllinie S117 nach vierstündiger Inkubation mit Cisplatin (100µM) bzw. 4-OH-Trofosfamid 20 – 320µM * = signifikant gegen die Kontrolle (P = < 0,05)
-10 0
10 20 30 40
Cisplatin 20µM 40µM 80µM 160µM
*
*
Abb.22: prozentualer Crosslinkanteil an der DNA der Zelllinie MX1 nach vierstündiger Inkubation mit Cisplatin (100µM) bzw. 4-OH-Trofosfamid 20-160µM * = signifikant gegen die Kontrolle (P = < 0,05)
%

Diskussion
49
IV. Diskussion
Die Oxazaphosphorine verfügen über ein breites Indikationsspektrum und gehören
zu den am häufigsten verwendeten und wirkungsvollsten Zytostatika. Trotz
jahrzehntelanger effektiver Anwendung dieser Substanzen sind ihr molekularer
Wirkmechanismus sowie die Antitumorwirkung noch immer nicht genau bekannt.
Trofosfamid ist seit über 30 Jahren als Arzneimittel zugelassen. Zunächst wurde
davon ausgegangen, dass Trofosfamid zu seinen beiden Schwestersubstanzen
abgebaut wird, hauptsächlich zu Ifosfamid und in einem geringeren Teil zu
Cyclophosphamid (Hempel et al., 1997; Kollmannsberger et al., 1999). Da
Trofosfamid nur als orale Formulierung in Tablettenform verfügbar ist, wurde es
lediglich als eine Art orales Ifosfamid betrachtet. Somit schien es nicht notwendig
zu sein, Trofosfamid weiter und genauer zu untersuchen. Jüngste Ergebnisse
unserer Arbeitsgruppe identifizierten allerdings 4-OH-Trofosfamid als
eigenständigen Metaboliten im Abbau von Trofosfamid (Brinker et al., 2002).
Trofosfamid hat bis jetzt im klinischen Einsatz und in experimentellen Arbeiten
wenig Beachtung gefunden, obwohl es erhebliche Vorteile gegenüber anderen
Chemotherapien besitzt. Zum einen ist es für die ambulante Behandlung
besonders gut geeignet, da es zu den wenigen Zytostatika gehört, die oral
appliziert werden. Bei Versuchen, Trofosfamid intravenös zu applizieren, wurden
neben Phlebitis auch andere Nebenwirkungen beobachtet (Drings et al., 1970).
Zum anderen zeigt sich unter der oralen Therapie eine gute Verträglichkeit mit
relativ wenigen Nebenwirkungen (Wist und Risberg, 1991; Wolff et al., 1991;
Mross et al., 1998; Marschner et al., 1999; Brinker et al., 2002). Erst jüngste
Untersuchungen unserer Arbeitsgruppe an mit Trofosfamid behandelten Patienten
beschrieben bei dem weitaus überwiegenden Teil der Patienten ein Fehlen von
Nebenwirkungen (Dissertation Brinker, 2004; Preiss et al., 2004). Darüber hinaus
konnten Tumorpatienten, die bereits auf die üblicherweise verwendeten
Chemotherapeutika resistente Tumoren aufwiesen, mit Trofosfamid erfolgreich
behandelt werden (Blomquist et al., 1995; Reichardt et al., 2002; Hartmann et al.,
2003).

Diskussion
50
Vor dem Hintergrund dieser außergewöhnlichen Eigenschaften von Trofosfamid
und der Tatsache, dass es noch keine direkt vergleichenden Studien der drei
Oxazaphosphorine gab, war es Ziel dieser Arbeit, die Toxizität von 4-OH-
Trofosfamid, 4-OH-Ifosfamid und 4-OH-Cyclophosphamid zu vergleichen.
Weiterhin sollte das Wirkprofil von 4-OH-Trofosfamid in Bezug auf DNA-Schäden
untersucht werden. Es galt festzustellen, inwiefern DNA-Strangbrüche bzw.
Interstrang-Cross-links für die Toxizität verantwortlich sind.
Da die Oxazaphosphorine so genannte Prodrugs sind, die in vivo erst durch die
mischfunktionellen Oxidasen der Leber zu den 4-OH-Verbindungen hydroxyliert
werden, wurden in den Versuchen die 4-OH-Metabolite eingesetzt. 4-OH-
Cyclophosphamid ist aufgrund seiner Instabilität nur in Form von Mafosfamid
erhältlich. Dies zerfällt nach Lösung im Medium in äquimolarem Verhältnis zu 4-
OH-Cyclophosphamid und Mesna (Mercaptoethansulfonsäure-Natrium) (Pohl,
1983). Um die drei Oxazaphosphorine miteinander vergleichen zu können, wurde
zu den 4-OH-Metaboliten von Trofosfamid und Ifosfamid Mesna in einem
äquimolaren Verhältnis zugefügt.

Diskussion
51
IV.1 Zytotoxisches Potential von 4-OH-Trofosfamid
1995 forderte Barton A. Kamen, einen unmittelbaren Vergleich der verschiedenen
Oxazaphosphorine durchzuführen. Das Ziel sollte es sein, festzustellen, inwiefern
sich die Oxazaphosphosphorine Ifosfamid und Cyclophosphamid in der Stärke
ihrer Zytotoxizität und im Wirkprofil voneinander unterscheiden (Kamen et al.,
1995). Da für Trofosfamid ein eigenständiger Metabolit - das 4-OH-Trofosfamid -
ermittelt wurde, galt es das Wirkspektrum und die Wirkstärke von diesem zu
ermitteln und mit den aktiven Metaboliten von Ifosfamid und Cyclophosphamid zu
vergleichen (Brinker et al., 2002; Preiss et al., 2004).
Die im MTT-Assay ermittelten Daten zeigten deutlich, dass 4-OH-Trofosfamid
wesentlich toxischer auf beide untersuchten Tumorzelllinien wirkt als der 4-OH-
Metabolit von Ifosfamid. Beim Mammakarzinom zeigte sich, dass die Toxizität von
4-OH-Trofosfamid sogar doppelt so hoch ist wie die von 4-OH-Ifosfamid und
ebenfalls stärker ist als die von Mafosfamid. Da die ermittelte benötigte
Konzentration im Bereich der Plasmaspitzenspiegel liegt, ist davon auszugehen,
dass 4-OH-Trofosfamid auch in vivo einen zytotoxischen Effekt gegenüber
Karzinomzellen ausübt. Klinische Studien, die den Einsatz von Trofosfamid an
Patientinnen mit Mammakarzinom untersuchten, bestätigen die von mir in vitro
erhobene Empfindlichkeit dieses Tumors für Trofosfamid (Mross, 1998).
Klinische Studien belegen, dass Weichteilsarkome im Allgemeinen zu den weniger
sensiblen Tumoren gehören. Bei den angewendeten Chemotherapien hat sich
bisher gezeigt, dass Ifosfamid neben Doxorubicin zu den wirksamsten Substanzen
zählt. Diese Arbeit bestätigte die geringere Empfindlichkeit des Weichteilsarkoms
im Vergleich zu dem Mammakarzinom. Die benötigte zytotoxisch wirksame
Konzentration von 4-OH-Trofosfamid liegt deutlich höher als bei dem
Mammakarzinom. In vitro wird etwa die achtfache Menge der gemessenen
Spitzenspiegel im Blut benötigt, um die Sarkomzellen auf die Hälfte zu reduzieren.
Es stellt sich die Frage, ob 4-OH-Trofosfamid auch bei Sarkomen in vivo einen
effektiven zytotoxischen Effekt ausüben kann. Die bisher veröffentlichten Studien
über die Therapie bei Weichteilsarkomen belegen durchaus eine Empfindlichkeit
dieser Tumoren für Trofosfamid (Kollmannberger et al., 1999; Hartmann et al.,

Diskussion
52
2003). Im Vergleich der Zytotoxizität erweist sich Cyclophosphamid am
Weichteilsarkom am wirkungsvollsten, wobei Trofosfamid seiner Wirkung sehr
nahe kommt. Ifosfamid wirkt hingegen am wenigsten zytotoxisch. Es ergibt sich
nach diesen in vitro Ergebnissen ein großer Bedarf, in Studien in vivo zu
untersuchen, ob Trofosfamid nicht sogar dem Ifosfamid vorzuziehen wäre. Es hat
sich in klinischen Studien mit palliativen Therapiezielen bereits gezeigt, dass
Trofosfamid das Überleben der Patienten signifikant verlängern kann, wenn sie
bereits chemotherapeutisch vorbehandelt sind bzw. ein Sarkom schon Metastasen
ausgebildet hat (Blomquist et al., 1995; Reichardt et al., 2002; Hartmann et al.,
2003; Laws et al., 2003).
Die stärkere Wirksamkeit von Trofosfamid auf die Tumorzelle lässt sich vermutlich
zum einen dadurch erklären, dass es lipophiler ist als Cyclophosphamid. Zum
anderen entstehen bei der Metabolisierung drei verschiedene 4-OH-Metabolite,
die alle zytotoxisch wirksam werden, wobei die Menge an 4-OH-Trofosfamid am
größten ist (Brinker et al., 2002; Preiss et al., 2004). Dieser Metabolit hat im
Gegensatz zu den 4-OH-Metaboliten von Ifosfamid und Cyclophosphamid eine
dritte Chlorethylgruppe. Studien konnten belegen, dass es keine komplette
Kreuzresistenz zwischen den Oxazaphosphorinen Ifosfamid und
Cyclophosphamid gibt. Es wurde gezeigt, dass sie mit unterschiedlichen
Abschnitten der DNA reagieren, was vermutlich an der unterschiedlichen
Anordnung der Chlorethylgruppen innerhalb der Moleküle liegt (Burchenal und
Riley, 1949; Dong et al., 1995; Struck et al., 2000). Die dritte Chlorethylgruppe des
Trofosfamids könnte erklären, warum bereits vorbehandelte Tumoren noch
sensibel auf Trofosfamid reagieren. Darüber hinaus könnte dies auch der Grund
für das unterschiedliche Wirkprofil der verschiedenen Oxazaphosphorine sein. 4-
OH-Trofosfamid erwies sich zwar an beiden Tumorzelllinien als zytotoxischer als
Ifosfamid, bei dem Weichteilsarkom war Mafosfamid allerdings etwas wirksamer.
Ein weiterer Grund für den Unterschied hinsichtlich Wirkstärke und Wirkprofil der
4-OH-Metabolite liegt in der anfallenden Menge an Chloracetaldehyd während der
Metabolisierung. Beim Abbau von Trofosfamid entstehen große Mengen an
Chloracetaldehyd, dem eine ganz eigene Zytotoxizität nachgewiesen wurde
(Dissertation Brüggemann 1999; Börner et al., 2000). Im Gegensatz dazu kommen

Diskussion
53
beim Cyclophosphamid nur quantitativ zu vernachlässigende Mengen an
Chloracetaldehyd vor.
Es zeigte sich, dass sich die im MTT-Assay ermittelte halbmaximale inhibierende
Konzentration (IC50) von 4-OH-Trofosfamid durch die Kombination mit Mesna in
äquimolaren Konzentrationen nicht signifikant verändert - weder am
Weichteilsarkom noch am Mammakarzinom. Der von einigen Autoren
angenommene zytoprotektive Effekt von Mesna auf die Tumorzelle scheint erst
bei mehrfach molarem Überschuss aufzutreten (Dissertation Brüggemann, 1999;
Kisro et al., 2000; Kisro et al., 2001). Bei den Untersuchungen von S. Brüggemann
ist außerdem zu bedenken, dass die Tumorzellen dem Mesna längere Zeit
ausgesetzt waren, als dies in vivo zuträfe, da es relativ schnell renal eliminiert
wird. Eine mögliche Erklärung, weshalb Mesna die zytotoxische Wirkung der
Oxazaphosphorinen nicht beeinflusst, ist, dass es aufgrund seines anionischen
Charakters die meisten Zellmembranen nicht durchdringen kann, wobei die
eigentlich wirksamen Metabolite erst intrazellulär entstehen. Allerdings bleibt zu
bedenken, dass Mesna den endogenen Thiolmetabolismus indirekt beeinflusst,
indem es die Freisetzung von Cystein aus Cystin steigert. Dieses kann die
Zellmembran durchdringen und damit intrazelluläre Reaktionen wie zum Beispiel
die Wirkung der Oxazaphosphorine beeinflussen (Kempgens et al., 2003).
Bei der höheren toxischen Wirkung von Trofosfamid im Gegensatz zu Ifosfamid
und Cyclophosphamid besteht zwangsläufig die Gefahr, dass es bei einer in vivo-
Therapie auch auf den ganzen Organismus toxischer wirkt, das heißt mehr oder
schwerwiegendere Nebenwirkungen erzeugen könnte. Als Brock die
halbmaximale letale Dosis (LD50) für Ratten mit einem Yoshida Aszites Sarkom
ermittelte, erwies sich Trofosfamid im Vergleich zu Cyclophosphamid als toxischer
(Brock, 1967). Im Gegensatz dazu wurde in den bisher veröffentlichten Studien
eine gute Verträglichkeit von Trofosfamid mit wenigen unerwünschten Wirkungen
beschrieben (Strumberg et al., 1997; Mross, 1998; Schmidt-Sandte et al., 1999;
Brinker et al., 2002). Da Trofosfamid ein stärkeres tumortoxisches Potential
aufweist, können geringere Dosierungen eingesetzt werden als bei seinen
Schwestersubstanzen, was wiederum weniger Nebenwirkungen nach sich zieht.

Diskussion
54
IV.2 Detektion von DNA-Schäden
Bisher wurde angenommen, dass Ifosfamid der Hauptmetabolit im
Trofosfamidabbau sei. Da erst seit kurzem bekannt ist, dass Trofosfamid einen
eigenen wirksamen 4-OH-Metaboliten besitzt, gab es noch keine Untersuchungen
über seine Wirkung auf die DNA. So ist es zur Lehrmeinung geworden, dass
Trofosfamid genauso wie Ifosfamid als alkylierendes Zytostatikum DNA-
Strangbrüche und Cross-links verursacht, obwohl dies nie untersucht wurde (Boos
et al., 1993; Hartley et al., 1999; Struck et al., 2000).
4-OH-Trofosfamid erwies sich im MTT-Assay als wesentlich zytotoxischer als
seine beiden Schwestersubstanzen. Aus diesem Grunde wurden weitere
Untersuchungen durchgeführt, um zu ermitteln, auf welchem Mechanismus diese
Zytotoxizität beruht. Dazu dienten als Methode der Comet-Assay
(Einzelzellgelelektrophorese) und ein fluorometrischer Assay zur Detektion von
Interstrang-Cross-links. Dabei beschränkte sich die Arbeit auf die Untersuchung
von 4-OH-Trofosfamid, da die Wirkung von 4-OH-Ifosfamid und 4-OH-
Cyclophosphamid auf die DNA bereits in unserer Arbeitsgruppe untersucht wurde
(Dissertation Kistner, 2003). Es konnte hierbei eine konzentrationsabhängige
Strangbruchbildung, jedoch keine Entstehung von Cross-links nachgewiesen
werden.
Es stellte sich die Aufgabe zu untersuchen, ob 4-OH-Trofosfamid in den klinisch
relevanten Konzentrationen bzw. im Bereich der halbmaximal inhibierenden
Konzentration (IC50) an der DNA der Tumorzellen Cross-links bzw. Strangbrüche
verursacht. Die verwendeten Konzentrationen und Inkubationszeiten orientierten
sich an den in unserer Arbeitsgruppe ermittelten Plasmaspitzenspiegeln und der
Halbwertzeit von 4-OH-Trofosfamid in vivo nach der Gabe von täglich 450mg
Trofosfamid über 7 Tage (Brinker et al., 2002). Zum Nachweis von DNA-
Strangbrüchen hat sich der Comet-Assay (II.2.4.2) als eine sensitive und
zuverlässige Methode bewährt. Es ließ sich eine konzentrationsabhängige
Fragmentierung der DNA in Form von Strangbrüchen durch 4-OH-Trofosfamid
nachweisen. Die DNA-Strangbrüche werden über die Ausbildung von
Cometschweifen detektiert. Im Comet-Assay zeigten sich beim Mammakarzinom

Diskussion
55
die ersten Strangbrüche etwa ab dem anderthalbfachen der gemessenen
Plasmaspitzenspiegel bei 2,5µM. Zur maximalen Cometausbildung kam es bei
160µM. Im Vergleich mit den Ergebnissen unserer Arbeitsgruppe zeigt sich auch
hier eine stärkere Wirksamkeit von Trofosfamid, da bei der Behandlung mit 4-OH-
Ifosfamid bzw. 4-OH-Cyclophosphamid sich die ersten Cometschweife erst ab
einer Konzentration von 20µM bildeten (Dissertation Kistner, 2003).
Beim Weichteilsarkom der Schilddrüse traten im Bereich der dreifachen Höhe der
gemessenen Plasmakonzentrationen von 4-OH-Trofosfamid die ersten DNA-
Strangbrüche auf (5µM). Bei einer Konzentration von 80µM (ca. 47-fache der
Plasmawerte) waren über 95% der Zellkerne zerstört. Wiederum zeigt sich 4-OH-
Trofosfamid im Vergleich mit den 4-OH-Metaboliten seiner beiden
Schwestersubstanzen als wirksamer, da diese erst ab einer Konzentration von
20µM eine geringe Zunahme der Schweiflänge bewirkten (Dissertation Kistner
2003).
Um die Vermutung von Hartley zu untersuchen (Hartley et al., 1999), dass Mesna
die von Acrolein induzierte Strangbruchbildung verhindern könnte, wurden stets
zusätzlich Proben mit Mesna im äquimolaren Verhältnis zu 4-OH-Trofosfamid
angesetzt. Es ist zwar bekannt, dass Mesna das Acrolein inaktiviert (Brock et al.,
1982), jedoch konnten keine Unterschiede in den verschiedenen Ansätzen
ermittelt werden. Dies begründet sich vermutlich darin, dass Mesna aufgrund
seines anionischen Charakters nicht in die Zelle aufgenommen wird. Acrolein
entsteht jedoch erst innerhalb der Zelle beim Zerfall von 4-OH-Trofosfamid über
seine Aldo-Form zu Trofosfamid-Mustard und Acrolein (siehe Abb.2). In den
verwendeten äquimolaren Konzentrationen kann Mensa die Strangbruchbildung
auch nicht durch die intrazelluläre Steigerung der Cysteinkonzentration
beeinflussen.
In dieser Arbeit wurde zur Detektion von Interstrang-Cross-links der unter II.2.6
beschriebene fluorometrische Assay nach P.G. Penketh angewendet (Penketh et
al., 1997). Er ist unter physiologischen pH-Bedingungen durchführbar und kommt
so den Verhältnissen in vivo am nächsten. Viele Assays zur Aufdeckung von
Cross-links finden unter alkalischen Bedingungen statt. In diesem Milieu alkylieren

Diskussion
56
z.B. Nitrogenmustard-Verbindungen wesentlich leichter, weil das Nitrogenatom
unprotoniert ist. Die so gewonnenen Ergebnisse sind jedoch wegen dieses
unphysiologischen pH-Wertes nur bedingt auf die Verhältnisse in vivo übertragbar.
Die Versuchsbedingungen wurden so gewählt, dass ein Bezug zur Klinik möglichst
ersichtlich bleibt.
Es konnte keine konzentrationsabhängige Crosslinkbildung gemessen werden.
Lediglich bei einer Konzentration von 160µM 4-OH-Trofosfamid (+Mesna) konnten
an der DNA des Mammakarzinoms 16,6% Cross-links festgestellt werden. Beim
Weichteilsarkom wurden bis 320µM keine Cross-links entdeckt. Selbst in den
durchgeführten Einzelmessungen mit extrem hohen zytotoxischen Dosierungen in
Bereichen mehrerer Millimolar konnte keine signifikante Crosslinksbildung
beobachtet werden. Dies bedeutet, dass die Entstehung von Cross-links nicht
entscheidend zur Antitumorwirkung von Trofosfamid im Rahmen einer Therapie
beiträgt, da hierzu Konzentrationen nötig wären, welche die gemessenen
Plasmaspitzenspiegel um ein Vielfaches übersteigen würden.
In verschiedenen Studien wurde bewiesen, dass Cisplatin Cross-links verursacht
(Costa et al., 1997; Samuel et al., 1998). Roberts und Pascoe wiesen nach, dass
Cisplatin kovalente Brücken zwischen zwei komplementären DNA-Strängen
hervorruft, die als Interstrang-Crosslinks bezeichnet werden (Roberts und Pascoe,
1972; Marzilli et al., 2001; Sullivan et al., 2002). Auch in meinen Versuchsreihen
konnte nach der Inkubation mit Cisplatin (100µM) ein deutlicher Cross-link-Anteil
(ca.30%) an beiden Zelllinien detektiert werden. Dieser Versuchsteil galt für diesen
Assay als interne Kontrolle und beweist, dass gebildete Interstrang-Cross-links
prinzipiell erfasst werden.
Es stellt sich die Frage, ob durch 4-OH-Trofosfamid tatsächlich keine Cross-links
hervorgerufen wurden, ob sie eventuell labil waren oder von der Zelle innerhalb
der Inkubationszeit von vier Stunden repariert werden konnten. Dies müsste
allerdings den Schluss nach sich ziehen, dass die von 4-OH-Trofosfamid
verursachten Cross-links sich von denen, die Cisplatin hervorruft, unterscheiden.
Es ist zu bedenken, dass in der von mir verwendeten Methode nur DNA-
Interstrang-Cross-links detektiert werden. Zwischen Protein und DNA gebildete

Diskussion
57
Cross-links werden in diesem Test nicht ermittelt. Dies liegt unter anderem daran,
dass bei der DNA-Gewinnung (siehe II.2.5) die Proteine von der DNA getrennt
werden. Dem steht jedoch entgegen, dass sich im Comet-Assay im Rahmen von
Einzelversuchen auch keine Cross-links zeigten. Die Zellen wurden mit MMS
vorbehandelt, um Strangbrüche zu erzeugen. Substanzen, die Cross-links
ausbilden, führen zu einer Verkürzung des Cometschweifes im Vergleich zu der
Kontrollgruppe, die nur mit MMS behandelt wurde. Der Cometschweif verlängerte
sich trotz Zugabe von 4-OH-Trofosfamid konzentrationsabhängig, was Cross-links
verhindert hätten. Es zeigte sich sogar ein additiver Effekt bei der
Strangbruchbildung, da der Cometschweif noch länger wurde als bei der mit MMS
behandelten Kontrollgruppe. Angenommen, die Anzahl der Strangbrüche
überschreitet die der Interstrang-Cross-links, so könnten diese sehr geringen
Mengen an Cross-links laut Penketh nicht mehr detektiert werden, da mehr als ein
Cross-link benötigt wird, um die beiden DNA-Stränge wieder zusammen zu fügen.
Hartley et al. untersuchten mittels Comet-Assay und dem alkalischen
Elutionsassay die Fähigkeit von Ifosfamid, Cross-links bzw. DNA-Strangbrüche zu
bilden (Hartley et al., 1999). Sie entdeckten in den Lymphozyten der mit Ifosfamid
therapierten Patienten Cross-links, jedoch keine Strangbrüche. Darüber hinaus
behandelten sie Patientenlymphozyten ex vivo mit 4-OH-Ifosfamid bzw. mit der
eigentlich alkylierenden Verbindung aus dem Ifosfamidstoffwechsel, dem
Isophosphoramidmustard. Nach Behandlung mit 4-OH-Ifosfamid konnten keine
Cross-links, jedoch ein hoher Anteil an Strangbrüchen gemessen werden. Im
gegensatz dazu wurden nach Isophosphoramidmustard-Gabe Cross-links
detektiert. So machten sie das Acrolein, das beim Zerfall von 4-OH-Ifosfamid
entsteht, für die Strangbrüche verantwortlich. Die Fähigkeit von Acrolein,
Strangbrüche zu verursachen, wurde schon 1986 von Crook et al. entdeckt
(Crook, 1986). Das Hartley et al. keine Strangbrüche in den Patientenlymphozyten
fanden, begründeten sie mit dem gleichzeitigen Einsatz des Uroprotektors Mesna,
der Acrolein inaktiviert (Brock et al., 1982). Leider sind dem Artikel von Hartley et
al. keine Konzentrationsangaben von 4-OH-Ifosfamid bzw.
Isophosphoramidmustard zu entnehmen (Hartley et al., 1999). Zudem ist es
fraglich, ob Mesna tatsächlich die Strangbruchbildung verhindern kann, da es als
anionische Verbindung in die meisten Zellen nicht aufgenommen wird, Acrolein

Diskussion
58
aber erst innerhalb der Zelle entsteht. Meine Ergebnisse zeigten dagegen, dass
die Menge an Strangbrüchen durch Mesna nicht beeinträchtigt wird. Vielmehr
kommt die Möglichkeit in Betracht, dass die von Acrolein induzierten Strangbrüche
den Cross-links mengenmäßig so sehr überlegen sind, dass die Sensitivität der
Messmethoden zu gering ist, um die Cross-links zu erfassen.

Diskussion
59
IV. 3 Klinische Relevanz der Arbeit In der Therapie der malignen Tumorerkrankungen ist man auf der Suche nach
weiteren Behandlungsmöglichkeiten. Insbesondere gilt dies für Patientengruppen
mit vorbehandelten Tumorrezidiven, mit Metastasen oder für Krebserkrankungen
im hohen Alter mit Sekundärkomplikationen, wie zum Beispiel Nieren- oder
Herzinsuffizienz, welche eine herkömmliche Chemotherapie nicht mehr zulassen.
Eine gute palliative Behandlungsmöglichkeit bietet die Therapie mit Trofosfamid,
da es in Form von Tabletten verabreicht wird und nicht - wie zum Beispiel
Ifosfamid - intravenös injiziert werden muss. Auch Cyclophosphamid ist in Form
von Dragees verfügbar, es wird jedoch im klinischen Alltag in der Tumortherapie
insbesondere bei der Kombination mit anderen Zytostatika hauptsächlich
intravenös appliziert. Ein weiterer großer Vorteil liegt darin, dass Trofosfamid bei
einer hohen Zytotoxizität gegenüber den Tumorzellen nur relativ wenige
Nebenwirkungen aufzeigt (Strumberg et al., 1997; Schmidt-Sandte et al., 1999;
Wiedemann et al., 1999; Brinker et al., 2002).
Die vorliegende Arbeit konnte zum einen zeigen, welche Arten von DNA-Schäden
durch Trofosfamid hervorgerufen werden. Zum anderen wurde belegt, dass der 4-
OH-Metabolit von Trofosfamid insbesondere im Vergleich mit den eng verwandten
Verbindungen 4-OH-Ifosfamid und 4-OH-Cyclophosphamid sowohl ein anderes
Wirkprofil als auch ein hohes zytotoxisches Potential aufweist. Vor dem
Hintergrund dieser Ergebnisse ist das Interesse an Trofosfamid für die palliative
Krebstherapie gestiegen, so dass noch weitere Untersuchungen zur
Aufschlüsselung des Wirkprofils unternommen worden sind. Wie neuere
Ergebnisse zeigen, lässt sich für Trofosfamid bei oraler Dauergabe, der so
genannten metronomischen Therapie, auch ein antiangiogentisches Potential
nachweisen (Vogt et al., 2003; Klink et al., 2004). Sollte sich ein weiterer
Angriffspunkt an Tumoren mit maligner Vaskularisierung herauskristallisieren,
würde sich Trofosfamid wegen seiner guten Verträglichkeit als ausgezeichnet
anwendbare Substanz anbieten.
Es erscheint daher sehr wahrscheinlich, dass Trofosfamid in Zukunft eine höhere
klinische Akzeptanz erlangen und einer wachsenden Patientenzahl zur Verfügung
stehen wird.

Zusammenfassung
60
V. Zusammenfassung
Trofosfamid gehört - wie seine beiden Schwestersubstanzen Ifosfamid und
Cyclophosphamid - zu den Oxazaphosphorinen, die in vivo durch
mischfunktionelle Oxidasen der Leber zu ihren 4-OH-Metaboliten aktiviert werden.
Diese Arbeit beschäftigt sich mit der Zytotoxizität und dem Wirkprofil von 4-OH-
Trofosfamid auf die DNA von Tumorzellen im Vergleich mit den 4-OH-Metaboliten
von Ifosfamid und Cyclophosphamid. Dies wurde an zwei humanen
Tumorzelllinien untersucht, zum einen am Weichteilsarkom der Schilddrüse
(S117) und zum anderen an einem Mammakarzinom (MX1).
Die Zytotoxizität wurde mittels MTT-Assay ermittelt, bei dem die Aktivität der
mitochondrialen Succinatdehydrogenase gemessen wird und auf das
Zellüberleben geschlossen werden kann. Die Toxizität von 4-OH-Trofosfamid war
deutlich höher als die von 4-OH-Ifosfamid, kam der von 4-OH-Cyclophosphamid
nahe und übertraf diese sogar an der MX1 Zelllinie (Mammakarzinom). Es konnte
im Rahmen der in vivo zu erwartenden Plasmaspiegel ein
konzentrationsabhängiger Zelltod gemessen werden. Es zeigten sich Differenzen
im Wirkprofil und der Wirkstärke von 4-OH-Trofosfamid auf der einen und 4-OH-
Ifosfamid bzw. 4-OH-Cyclophosphamid auf der anderen Seite. Durch äquimolare
Konzentrationen von Mesna, einem Uroprotektor, der bei der in vivo Therapie
zusammen mit Trofosfamid eingesetzt wird, wurde die Zytotoxizität nicht
beeinträchtigt.
Mit Hilfe des Comet-Assays, auch Einzelzellgelelektrophorese genannt, konnte
zum ersten Mal gezeigt werden, dass 4-OH-Trofosfamid konzentrationsabhängig
DNA-Strangbrüche verursacht. Mesna konnte auch hier die Wirkung nicht
beeinflussen. Im Vergleich mit den Ergebnissen unserer Arbeitsgruppe zeigt sich,
dass 4-OH-Trofosfamid schon in wesentlich geringeren Konzentrationen DNA-
Strangbrüche induziert als die 4-OH-Metabolite von Ifosfamid und
Cyclophosphamid.
Mittels fluorometrischem Assay wurde die Fähigkeit von 4-OH-Trofosfamid
untersucht, kovalente Brücken zwischen zwei komplementären DNA-Strängen zu
bilden. Diese so genannten Interstrang-Cross-links konnten nicht nachgewiesen
werden, so dass davon auszugehen ist, dass der Untergang der Tumorzellen am
ehesten durch Brüche in der DNA und nicht durch irreversible Verschmelzung

Zusammenfassung
61
(Cross-links) zustande kommt. Zusammenfassend kann festgestellt werden, dass
die vorliegenden Daten dazu beitragen, Trofosfamid hinsichtlich der Wirkstärke
und dem Wirkprofil von seinen beiden Schwestersubstanzen zu unterscheiden.
Die Ergebnisse beweisen, dass Trofosfamid nicht nur die orale Form des
Ifosfamids darstellt. Gerade vor dem Hintergrund der guten Verträglichkeit im
klinischen Einsatz, insbesondere bei resistenten Tumoren, bei palliativen
Therapieansätzen oder bei Langzeittherapien, scheint Trofosfamid nicht mehr nur
eine Alternative zu sein.

Literaturverzeichnis
62
VI. Literaturverzeichnis Albrecht M, Kleinhauf-Houken A, Trams G, Thomsen K: 5 years experiences with
adjuvant chemotherapy in primary breast cancer.
Geburtsh. u. Frauenheilk. 44: 550-556 (1984)
Andersson PO, Braide I, Nilsson-Ehle H: Trofosfamide as a salvage therapy for
anaplastic large cell lymphoma relapsing after high-dose chemotherapy.
Leuk Lymphoma 43: 2351-2353 (2002)
Arnold H, Bourseaux F: Synthese und Abbau zytostatisch wirksamer zyklischer N-
Phosphoramidester des Bis-(β-chloräthyl)-amins
Angew. Chem. 70: 539-544 (1958)
Arnold H, Bourseaux F, Brock N: Neuartige Krebs-Chemotherapeutika aus der
Gruppe der zyklischen N-Lostphosphoramidester.
Naturwiss.45: 64-66 (1958)
Becher R, Kloke O, Hyungs J, Hartwich G, Bartels H, Szanto J, Wolf E, Illinger HJ,
Halabi S, Rieche K, Hering KG, Ohl S, DeDycker R, Huhn R, Hofeler H, Pielken
HJ, Hawig I, Hirche H, Seeber S: Epirubicin and ifosfamide in metastatic breast
cancer.
Semin Oncol 23: 28-33 (1996)
Becker N: Cancer mortality and prevention in the European Union.
Eur. J. Surg. Oncol. 24: 370-374 (1998)
Berns JS, Haghighat A, Staddon A, Cohen RM, Schmidt R, Fisher S, Rudnick MR,
Tomaszewski JE: Severe, irreversible renal failure after ifosfamide treatment. A
clinicopathologic report of two patients.
Cancer Res. 76: 497-500 (1995)

Literaturverzeichnis
63
Blomqvist C, Wiklund T, Pajunen M, Virolainen M, Elomaa I: Oral trofosfamide: an
active drug in the treatment of soft-tissue sarcoma.
Cancer Chemother. Pharmacol. 36: 263-265 (1995)
Boos J, Küpker F, Blaschke G, Jürgens H: Trofosfamide metabolism in different
species-ifosfamide is the predominant metabolite.
Cancer Chemother. Pharmacol. 33: 71-76 (1993)
Börner K, Kisro J, Brüggemann SK, Hagenah W, Peters SO, Wagner T:
Metabolism of ifosfamide to chloroacetaldehyde contributes to antitumor activity in
vivo.
Drug Metab. Dispos. 28: 573-576 (2000)
Brinker A, Kisro J, Letsch C, Brüggemann SK und Wagner T: New insights into the
clinical pharmacokinetics of trofosfamide.
Int. J. Clin. Pharm. and Ther. 40: 376-381 (2002)
Brinker A. Pharmakokinetik und Toxizität von oralem Trofosfamid bei Patienten mit
soliden Tumoren und Non Hodgkin Lymphom
Med. Diss. Lübeck (2004)
Brock N: Oxazaphosphorine cytostatics: past-present-future. seventh cain
memorial award lecture.
Cancer Res. 49: 1-7 (1989)
Brock N, Stekar J, Pohl J; Antidote against the urotoxic effects of the
oxazaphosphorine derivates cyclophosphamide, ifosfamide and trofosfamide.
Naturwissenschaften: 66 (1): 60-61 (1979)
Brock N, Hohorst HJ: Metabolism of cyclophosphamide.
Cancer 20: 900-904 (1967)

Literaturverzeichnis
64
Brock N, Pohl J, Steckar J, Scheef W: Studies on the urotoxizity of
oxazaphosphorine cytostatics and its prevention III. Profile of action of sodium 2-
mercaptoethane sulfonate (Mesna).
Eur. J. Cancer Clin. Oncol. 18: 1377-1387 (1982)
Brock N, Pohl J, Stekar J: Detoxification of urotoxic oxazaphosphorines by
sulfhydryl compounds.
J. Cancer Res. and Clin. Oncol. 100: 311-320 (1981)
Brock N, Pohl J: The development of mesna for regional detoxification.
Cancer Treatment Reviews 10: 33-43 (1983)
Brock N, Steckar J, Pohl J, Niemeyer U, Scheffler G: Acrolein, the causative factor
of urotoxic side-effects of cyclophosphamide, ifosfamide, trofosfamide and
sufosfamide.
Arzneimittelforschung 29: 659-661 (1979)
Brüggeman SK: Chloracetaldehyd – Entdeckung einer eigenständigen
Tumorzelltoxizität des Ifosfamidmetaboliten.
Med. Diss. Lübeck (1999)
Brüggemann SK, Kisro J, Wagner T: Ifosfamide cytotoxicity on human tumor and
renal cells: role of chloroacetaldehyde in comparison to 4-hydroxyifosfamide.
Cancer Res. 57: 2676-2680 (1997)
Burchenal JH and Riley JB: Nitrogen mustards: the relationship between chemical
structure and chemotherapeutic activity.
Cancer Res. 9: 553-554 (1949)
Colvin OM: An overview of Cyclophosphamid development and clinical
applications.
Curr. Pharm. Des.5: 555-560 (1999)

Literaturverzeichnis
65
Costa M, Zhitkovich A, Harris M, Paustenbach D, Gargas M: DNA-Protein Cross-
links produced by various chemicals in cultured human lymphoma cells.
J. Toxicol. Envir. Health 50: 433-449 (1997)
Cox PJ, Abel G: Cyclophosphamide cystitis studies aimed at its minimisation.
Biochem. Pharmacol. 28: 3499-3502 (1979)
Cox PJ; Cyclophosphamide cystitis and bladder cancer. A hypothesis.
Eur. J. Cancer 15: 1071-1072 (1979)
Cox PJ; Cyclophosphamide cystitis—identification of acrolein as the causative
agent
Biochem. Pharmacol. 28: 2045-2049 (1979)
Crook TR, Souhami RL, Mc Lean AEM: Cytotoxicity, DNA crosslinking and single
strand brake induced by activated cyclophosphamide and acrolein in human
leukemia cells.
Cancer Res. 46: 5029-5034 (1986)
DeNeve W, Valeriote F, Edlestein M, Everett C, Bischoff M: In vivo DNA cross-
linking by cyclophosphamide: comparison of human chronic lymphatic leukemia
cells with mouse L1210 leukemia and normal bone marrow cells.
Cancer Res. 49: 3452-3456 (1989)
DeVita V Jr: Principles of Cancer management: chemotherapy. In: Vincent T.
DeVita Jr, Samuel Hellman, Steven a. Rosenberg.
Cancer. Principles & Practice of Oncology. 5th Edition (17): 333-347, Lippincott-
Raven, Philadelphia, New York (1997)
Dong Q, Barsky D, Colvin ME, Melius CF, Ludeman SM, Moravek JF, Colvin OM,
Bigner DD, Modrich P, Friedman HS: A structural basis for a phosphoramide
mustard-induces DNA interstrand cross-link at 5´-d(GAC).
Poc. Natl. Acad. Sci. USA 92: 12170-12174 (1995)

Literaturverzeichnis
66
Dorr RT: Chemoprotectants for cancer chemotherapy.
Semin.Oncol. 18: 48-58 (1991)
Drings P, Allner R, Brock N, Burkert H, Fischer M, Folsch E, Gerhartz H, Gotzky P,
Hoppe I, Kanzler G, Klein HO, Mainzer K, Martin H, Obrecht P, Palme G, Paulisch
H, Riegg H, Schubert JC, Treske U, Weise W, Willems D, Wilmanns H, Witte S,
Wohlenberg H: Experiences with N-Lost phosphoramide esters.
Dtsch. Med. Wochenschr. 95: 491-497 (1970)
Enk AH, Knop J: Stabilizing the course of patients with stage IV advanced
malignant melanoma by trofosfamide treatment.
Der Hautarzt 51: 486-489 (2000)
Falkson G, Falkson HC: Trofosfamide in the treatment of patients with cancer. A
pilot trial.
S. Afr. Med. J. 53: 886-888 (1978)
Goodman LS, Wintrobe MM, Damashek W, Goodman MJ, Gilman MA, McLennan
MT: Nitrogen mustard therapy. Use of methyl-bis(beta-chloroethyl)amine
Hydrochloride and Tris(Beta-Chlorethyl)amine Hydrochloride for Hodgkin´s
Disease, Lymphosarcoma, Leukemia and certain allied and miscellaneous
disorders.
J.A.M.A. 132: 126-132 (1946)
Goren MP, Wright RK, Prat CB, Horowitz ME, Doge RK, Viar MJ, Kovnar EH:
Potentiation of ifosfamide neurotoxicity, hematotoxicity and tubular nephrotoxicity
by prior cis-diamminedichloroplatinum(II)therapy.
Cancer Res. 47: 1457-1460 (1987)
Goren MP, Wright RK, Prat CB, Pell FE: Dechloroethylation of ifosfamide and
neurotoxicity (letter).
Lancet 2: 1219-1220 (1986)

Literaturverzeichnis
67
Gunsilius E, Gierlich T, Mross K, Gastl G, Unger C: Palliative chemotherapy in
pretreated patients with advanced cancer: oral trofosfamide is effective in ovarian
carcinoma.
Cancer Invest. 19: 808-811 (2001)
Hartley JM, Spanswick VJ, Gander M, Giacomini G,Whelan J, Souhami RL,
Hartley AJ: Measurement of DNA cross-linking in patients on ifosfamide therapy
using the single cell electrophoresis (comet) assay.
Clin. Cancer Res. 5: 507-512, (1999)
Hartmann JT, Oechsle K, Mayer F, Kanz L, Bokemeyer C: Phase II trial of
trofosfamide in patients with advanced pretreated soft tissue sarcomas.
Anticancer Res. 23: 1899-1901 (2003)
Helsing MD: Trofosfamide as a salvage treatment with low toxicity in malignant
lymphoma. A phase II study.
Eur. J. Cancer 33: 500-502 (1997)
Hempel G, Krumpelmann S, May-Manke A, Hohenlochter B, Blaschke G,
Juergens H und Boos J: Pharmacokinetics of trofosfamide and its dechlorethylated
metabolites.
Cancer Chemother. Pharmacol. 40: 45-50 (1997)
Hengstler JG, Hengst A, Fuchs J, Tanner B, Pohl J, Oesch F: Induction of DNA
crosslinks and DNA strand lesions by cyclophosphamide after activation by
cytochrome P450 2B1.
Mutation Res. 373: 215-223 (1997)
Johnstone CJ, Lind MJ, Griffin MJ, Boddy AV: Ifosfamide metabolism and DNA
damage in tumor and peripheral blood lymphocytes of breast cancer patients.
Cancer Chemother. Pharmacol. 46: 433-441 (2000)

Literaturverzeichnis
68
Kamen BA, Frenke lE, Colvin OM: Ifosfamide: Should the honeymoon be over? J.
Clin. Oncol. 13: 307-309 (1995)
Kempgens B, Kisro J, GruberY, Bahrs H, Wagner Th: Two schedules of
application for mesna and their influence on the thiol-metabolism.
Int. J. Clin. Pharm. and Ther. 41: 608-609 (2003)
Kisro J, Brueggemann SK, Thiede S, Kim S, Wiedemann GJ, Wagner Th:
Reduction of ifosfamide cytotoxicity by Mesna depense on different cysteine
uptake capacities of human tumor cells.
Proceedings of the American Assiciation for Cancer Research 41 (2000)
Kisro J, Brueggemann SK, Thiede S, Kuepper S, Wagner Th: Mechanism of
Mesna interference with oxazaphosphorines in vitro. Correlation to
pharmacokinetics of mesna and other thiols in patients with ifosfamide or
cyclophosphamide chemotherapy.
Proceedings of the American Assiciation for Cancer Research 42 (2001)
Kistner K: Wirkung der Oxazaphosphorinmetaboliten Chloracetaldehyd, 4-OH-
Ifosfamid und 4-OH-Cyclohosphamid auf die DNA humaner Tumorzellen in vitro.
Med. Diss. Lübeck (2003)
Klink T,Bela C, Engels C, Brüggemann S, Marxsen J, Stölting S, Kisro J, Vollmert
M, Wagner T: Metronomic scheduling of trofosfamide chemotherapy in human
NSCL xenografts highly increases therapeutic efficacy compared to conventinal
scheduling by inhibition of angionesis.
Cancer Res. 45 Abstract No. 62 (2004)
Kohn KW, Bruce F: Beyond DNA Cross-linking: history and prospects of DNA
targeted cancer treatment – fifteenth cain memorial award lecture.
Cancer Res. 56: 5533-5546 (1996)

Literaturverzeichnis
69
Kollmannberger C, Brugger W, HartmannJT, Maurer F, Bohm P, Kanz L,
Bokemeyer C: Phase II study of oral trofosfamide as palliative therapy in
pretreated patients with metastatic soft-tissue sarcoma.
Anticancer Drugs. 10: 453-456 (1999)
Kurowski V, Cerny T, Küpfer A, Wagner T: Metabolism and pharmacokinetics of
oral and intravenous ifosfamide.
J. Cancer Res. Clin. Oncol. 117: 148-153 (1991)
Laws HJ, van Kaick B, Pape H, Paulussen M, Gobel U: Relapse after high-dose
therapy in relapsed Ewing´s tumor patients: effects of maintenance chemotherapy
in two selected patient?
Onkologie 26: 573-577 (2003)
Lindemann H, Harbers E: Interaction of the three alkylating drugs,
cyclophosphamide, ifosfamide and trofosfamide, with DNA and DNA-constituents
in vitro.
Arzneim.-Forsch./Drug. Res. 30: 2075-2080 (1980)
Marzilli LG, Saad JS, Kuklenyik Z, Keating KA, Xu Y: Relationship of solution and
protein-bound structures of DNA duplexes with the major intrastrand cross-link
lesion formed on cisplatin binding to DNA.
J. A. Chem. Soc. 123: 2764-2770 (2001)
May-Manke A, Kroemer H, Hempel G, Bohnenstenge lF, Hohenlöchter B,
Blaschke G, Boos J: Investigation of the major human hepatic cytochrome P450
involved in 4-hydroxylation and N-dechloroethylation of trofosfamide.
Cancer Chemother. Pharmacol. 44: 327- 334 (1999)
Miyamae Y, Iwasaki K, Kinae N, Tsuda S, Murakami M, Tanaka M, Sasaki YF:
Detection of DNA lesions induced by chemical mutagens using the single cell gel
electrophoresis (comet) assay, 2. Relationship between DNA migration and
alkaline condition.

Literaturverzeichnis
70
Mutat. Res. 393: 109-113 (1997)
Miyamae Y, Zaizen K, Ohara K, Mine Y, Saski YF: Detection of DNA lesions
induced by chemical mutagens using the single cell gel electrophoresis (comet)
assay, 2.Relationship between the onset of DNA damage and characteristics of
mutagens.
Mutat. Res. 393: 999-1006 (1997)
Mross K, Rüter A, Gierlich T und Unger C: Tumor growth control by oral
trofosfamide in patients with metastatic breast cancer.
Onkologie 21: 52-56 (1998)
Östling O, Johannson KJ: Microelectrophoretic study of radiatio induced DNA
damage in individual mammalian cells.
Biochem. Biophys. Res. Comun. 123: 291-298 (1984)
Pfuhler S, Wolf HU: Detection of DNA-crosslinking agents with the alkaline comet
assay.
Environ. Mol. Mutagen. 27: 196-201 (1996)
Pohl J: ASTA Z7557, Summary of Investigators.
Asta-Werke, DeGussa Pharma Gruppe, Bielefeld (1983)
Preiss R, Baumann F, Stefanovic D, Niemeyer U, Ponisch W, Niederwieser D:
Investigations on the pharmacokinetics of trofosfamide and its metabolites-first
report of 4-hydroxy-trofosfamide kinetics in humans.
Cancer Chemother. Pharmacol. 29: (2004)
Reichardt P, Pink D, Tilgner J, Kretschmar A, Thuss-Patience PC, Dorken B: Oral
trofosfamide: an active and well tolerated maintenance therapy for adult patients
with advanced bone and soft tissue sarcomas. Results of a retrospective analysis.
Onkologie 25: 541-546 (2002)

Literaturverzeichnis
71
Roberts JJ, Pascoe JM: Crosslinking of complementary strand of DNA in
mammalian cells by antitumor platinum compounds.
Nature: 235 (5336): 282-284 (1972)
Samuel SK, Spencer VA, Bajno L, Sun JM, Holth LT, Oesterreich S, Davie JR: In
situ cross-linking by cisplatin of nuclear matrix-bound transcription factors to
nuclear DNA of human breast cancer cells.
Cancer Res. 58: 3004-3008 (1998)
Salminen E, Nikkanen V, Lindholm L: Trofosfamide is effective in refractory non-
Hodgkin´s lymphoma.
Eur. J. Cancer 31A : 2419-2420 (1995)
Salminen E, Nikkanen V, Lindholm L: Palliative chemotherapy in non-Hodgkin´s
lymphoma.
Oncology 54: 108-111 (1997)
Schlenke P, Kisro J, Deeken M, Zajac S, Klich S, Wagner T: The cytotoxicity of
mafosfamide on G-CSF mobilized hematopoietic prognitors is reduced by SH
groups of albumin- implications for further purging strategies
Bone Marrow Transplantation 23: 157-161 (1999)
Schmidt-Sandte W, Dageforde J, Klapdor R, Wagner T, Wiedemann GJ:
Trofosfamide in patients with pancreatic cancer.
Anticancer Res. 19: 2485-2487 (1999)
Semont H, Hecquet C, Adolphe M, Deysson G: Effect of three alkylating antimitotic
agents and their metabolites on in vitro monogranulocytic colony forming cells
from mouse bone marrow.
Drug Res.31(I): (1981)

Literaturverzeichnis
72
Singh NP, Mc Coy MT, Tice RR, Schneider EL: A simple technique of
quantification of low levels of DNA damage in individual cells.
Exp. Cell. Res. 175: 184-191 (1988)
Sladeck NE. Metabolism of oxazaphosphorines.
Pharmacol. Ther. 37: 301-355 (1988)
Speit G, Trenz K, Schutz P, Rothfuss A, Merk O: The influence of temperature
during alkaline treatment and electrophoresis and results obtained with the comet
assay.
Toxicol. Lett. 110: 73-78 (1999)
Struck RF, Davis RL Jr, Berardini MD, Loechler EL: DNA guanine-guanine
crosslinking sequence specificity of isophosphoramide mustard, the alkylating
metabolite of the clinical antitumor agent ifosfamide.
Cancer Chemother. Pharmacol. 45: 59-62 (2000)
Strumberg D, Harstrick A, Klaasen U, Muller C, Eberhardt W, Korn MW, Wilke H,
Seeber S: Phase II trial of continuous oral trofosfamide in patients with advanced
colorectal cancer refractory to 5-fluorouracil.
Anticancer Drugs. 8: 293-295 (1997)
Sullivan ST, Saad JS, Fanizzi FP, Marzilli LG: Dramatic 5´-residue effect on
conformer distribution of short olinucleotide retro models of the cisplatin-DNA
cross-link: Implications for the lippard and cross-link distorted base pair steps
present in cisplatin-DNA duplex adducts.
J. Am. Chem. Soc. 124: 1558-1559 (2002)
Teicher BA: Pharmacology of cancer chemotherapy. Antitumor alkylating agents.
In: Vincent T. DeVitaJr, Samuel Hellman, Steven A. Rosenberg.
Cancer Principles & Practice of oncology. 5th Edition (19.4): 405-418, Lippincott-
Raven, Philadelphia, New York. (1997).

Literaturverzeichnis
73
van Dyk JJ, Falkson HC, Van-der MA, Falkson G: Unexpected toxicity in patients
treated with ifosfamide.
Cancer Res. 32: 921-924 (1972)
Vogt T, Hafner C, Bross K, Bataille F, Jauch KW, Berand A, Landthaler M,
Andreesen R, reichle A: Antiangiogenetic therapy with pioglitazone, rofecoxib, and
metronomic trofosfamide in patients with advanced malignant vascular tumors.
Cancer 98: 2251-2256 (2003)
Vrzoc M, Petras M; Comparison of DNA damage in peripheral blood and spleen
lymphocytes using the single-cell gel assay.
Mutation Res., 379: 263-269 (1997)
Wagner A, Hempel G, Boos J: Trofosfamide: a review of its pharmacodynamic and
pharmacocinetic properties and therapeutic potential in the oral treatment of
cancer.
Anticancer Drugs 8: 419-431 (1997)
Wagner Th: Ifosfamide clincal pharmacokinetics.
Clin. Pharmacokinet. 26: 439-456 (1994)
Wist E, Riberg T: Trofosfamide in Non-Hodgkin´s lymphoma.
Acta Oncologica 30: 819-821 (1991)
Wolff JEA, Mölenkamp G, Westphal S, Pietsch T, Gnekow A, Kortmann RD, Kuehl
J: Oral trofosfamide and etoposide in pediatric patients with glioblastoma
multiforme.
Cancer 89: 2131-2137 (2000)
Zalupsi M, Baker LH: Ifosfamide.
J. Natl. Cancer Inst. 80: 556-566 (1988)

Anhang
74
VII. Anhang
VII.1 Abbildungsverzeichnis Seite
Abb.1 Strukturformeln Lost und Stickstofflost 6
Abb.2 Metabolismus der Oxazaphosphorine 12
Abb.3 MTT-Metabolismus im Mitochondrium 21
Abb.4 Comet Klasse 1 24
Abb.5 Comet Klasse 2 24
Abb.6 Comet Klasse 3 24
Abb.7 Comet Klasse 4 24
Abb. 8 Objektträger mit Agarose-Sandwichgel 25
Abb.9 DNA-Isolation 27
Abb.10 DNA im Hitze-Kälte-Zyklus 31
Abb.11 Zellüberlebensrate MX1 36
Abb.12 Zellüberlebensrate S177 36
Abb.13 IC50 MX1 37
Abb.14 IC50 S117 37
Abb.15 Typische Cometenkonstellation unbehandelter S117 Zellen 40
Abb.16 Typische Cometenkonstellation nach Inkubation mit MMS 40
Abb.17 Typische Cometenkonstellation nach Inkubation mit 160µM
4-OH-Trofosfamid (MX1) 41
Abb.18 Typische Cometenkonstellation nach Inkubation mit 40µM
4-OH-Trofosfamid (S117) 41
Abb.19 Verteilung der Cometklassen MX1 43
Abb.20 Verteilung der Cometklassen S117 43
Abb.21 Prozentualer Crosslinkanteil S117 48
Abb.22 Prozentualer Crosslinkanteil MX1 48

Anhang
75
VII.2 Tabellenverzeichnis Seite
Tab.1 Extinktionsabfall und dessen Schwankungen innerhalb eines
Versuchansatzes anhand unbehandelter DNA der Zelllinie MX 1 44
Tab.2 Extinktionsschwankungen an acht unterschiedlichen Tagen (MX1) 45
Tab.3 Extinktionsschwankungen an fünf unterschiedlichen Tagen (S117) 46
Tab.4 Zellüberleben S117 Trypanblau-Färbung 76
Tab.5 Zellüberleben MX1 Trypanblau-Färbung 77
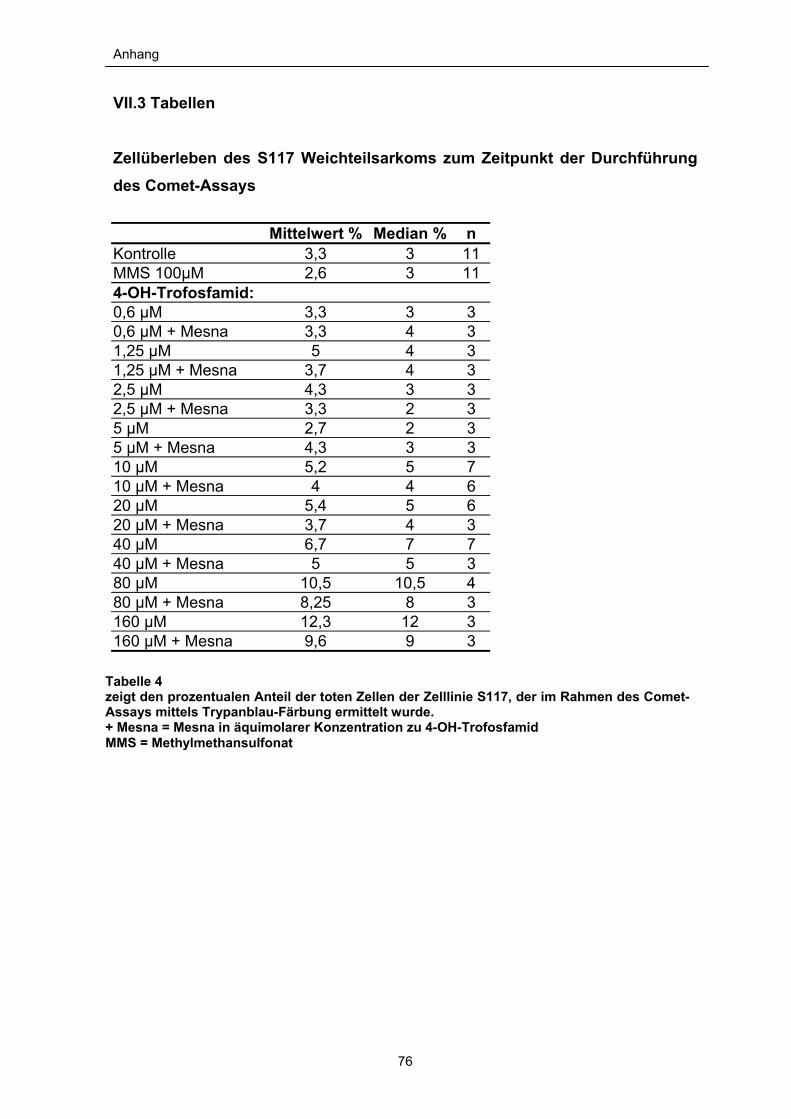
Anhang
76
VII.3 Tabellen
Zellüberleben des S117 Weichteilsarkoms zum Zeitpunkt der Durchführung des Comet-Assays
Mittelwert % Median % n Kontrolle 3,3 3 11 MMS 100µM 2,6 3 11 4-OH-Trofosfamid: 0,6 µM 3,3 3 3 0,6 µM + Mesna 3,3 4 3 1,25 µM 5 4 3 1,25 µM + Mesna 3,7 4 3 2,5 µM 4,3 3 3 2,5 µM + Mesna 3,3 2 3 5 µM 2,7 2 3 5 µM + Mesna 4,3 3 3 10 µM 5,2 5 7 10 µM + Mesna 4 4 6 20 µM 5,4 5 6 20 µM + Mesna 3,7 4 3 40 µM 6,7 7 7 40 µM + Mesna 5 5 3 80 µM 10,5 10,5 4 80 µM + Mesna 8,25 8 3 160 µM 12,3 12 3 160 µM + Mesna 9,6 9 3
Tabelle 4 zeigt den prozentualen Anteil der toten Zellen der Zelllinie S117, der im Rahmen des Comet-Assays mittels Trypanblau-Färbung ermittelt wurde. + Mesna = Mesna in äquimolarer Konzentration zu 4-OH-Trofosfamid MMS = Methylmethansulfonat

Anhang
77
Zellüberleben des MX1 Mammakarzinoms zum Zeitpunkt der Durchführung des Comet-Assays
Mittelwert % Median % n Kontrolle 3,6 4,0 8 MMS 100µM 4,4 5,0 8 4-OH-Trofosfamid: 0,6 µM 1,7 0,0 3 0,6 µM + Mesna 3,7 3,0 3 1,25 µM 3,3 3,0 3 1,25 µM + Mesna 3,7 4,0 3 2,5 µM 2,3 3,0 3 2,5 µM + Mesna 6 6 3 5 µM 2,7 3,0 3 5 µM + Mesna 4,3 3,0 3 10 µM 5,3 5,0 3 10 µM + Mesna 6,3 7,0 3 20 µM 8,8 8,5 4 20 µM + Mesna 8 8 2 40 µM 9,7 10,0 3 40 µM + Mesna 9,3 10,0 2 80 µM 6,7 8,0 2 80 µM + Mesna 7,0 6,0 3
Tabelle 5 zeigt den prozentualen Anteil der toten Zellen der Zelllinie MX1, der im Rahmen des Comet-Assays mittels Trypanblau-Färbung ermittelt wurde. + Mesna = Mesna in äquimolarer Konzentration zu 4-OH-Trofosfamid MMS = Methylmethansulfonat

Abkürzungsverzeichnis
78
VII.4 Abkürzungsverzeichnis
CAA Chloracetaldehyd
C max Maximal - Konzentration
CMF Cyclophosphamid + Methotrexat + 5- Fluorouracil
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure
EB Ethidiumbromid
EDTA Ethylene Diamine Tetra-Acetat =
Äthylendiamintetraessigsäure
fKS fetales Kälberserum
IC 50 inhibiting concentration = inhibierende Konzentration
auf 50%
LD 50 letale Dosis mit 50% Zellüberleben
LMP low melting point = niedriger Schmelzpunkt
Mesna Mercaptoethansulfonsäure
MMS Methylmethansulfonat
MTT 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl Tetrazolium
Bromid
NaCl Natriumchlorid
NaOH Natriumhydroxid
NMP normal melting point = normaler Schmelzpunkt
n.s. nicht signifikant OD optische Dichte PBS Phosphat-Natriumchlorid Puffer
RMPI Roswell Memorial Park Institute
RNA Ribonukleinsäure
SDH Succinatdehydrogenase
SDS Dodecylsulfate sodium salt
SEM standard error of mean = Standardabweichung
TE Tris-HCl / EDTA
Tris Trishydroxymethylaminomethan

Abkürzungsverzeichnis
79
U/min Umdrehungen pro Minute
UV ultraviolett
WHO World health organization
(Weltgesundheitsorganisation)

Danksagung
80
VIII. Danksagung
Mein besonderer Dank gilt zunächst Herrn Prof. Dr. med. Thomas Wagner für
die Überlassung des Themas zur Promotion sowie für die hervorragende
Unterstützung und Betreuung während der gesamten Arbeit.
Für die besonders freundliche Einarbeitung in die verschiedenen Tätigkeiten im
Labor möchte ich Frau Kerstin Kistner sowie den medizinisch technischen
Assistentinnen der Arbeitsgruppe danken.
Herrn Sven Süffke danke ich, für seine Beratung und Unterstützung in Software –
Angelegenheiten.
Insbesondere möchte ich mich bei meinen Eltern für anhaltende herzliche
Unterstützung und Förderung bedanken, ohne die mir die Durchführung der Arbeit
sicher nicht gelungen wäre.
Dank gilt auch Frau Andrea Brinker für die freundschaftliche Zusammenarbeit
insbesondere bei der Durchsicht des Manuskriptes.
Für die liebevolle Geduld und für die lebhaften Diskussionen von methodischen
und inhaltlichen Fragen möchte ich mich von ganzem Herzen bei Jan Lindenkamp bedanken.

Lebenslauf
81
IX. Lebenslauf
Persönliche Daten: Name: Carola Letsch
Geburtstag: 10.12.1974
Geburtsort: Bad Segeberg
Familienstand: ledig
Staatsangehörigkeit: deutsch
Mutter: Helma Letsch, geb. Pein
Geschäftsführerin
Geb. am 27.07.1949
Vater: Rudolf Letsch
Kfz-Elektriker-Meister
Geb. am 12.08.1945
Schulbildung: 1981 - 1985 Theodor Storm-Schule in Bad Segeberg
1985 - 1991 Realschule am Seminarweg in Bad Segeberg
1991 - 1994 Kreisberufsschule Bad Oldesloe
05 / 1994 Abitur
Ausbildung: 1994 - 1997 Ausbildung zur Diätassistentin am Universitäts-
krankenhaus Eppendorf in Hamburg
Studium: 10 / 1997 Studium der Humanmedizin in Lübeck
08 / 1999 Ärztliche Vorprüfung
08 / 2000 erster Abschnitt der ärztlichen Prüfung
03 / 2003 zweiter Abschnitt der ärztlichen Prüfung
05 / 2004 dritter Abschnitt der ärztlichen Prüfung

Lebenslauf
82
Dissertation: 11/ 2000 – 05 / 2002 praktische Untersuchungen zur Dissertation
Publikation: Brinker A., Kisro J., Letsch C., Brüggemann S.K. und
Wagner T.: New insights into the clinical
pharmacokinetics of Trofosfamide, Int. J. clin. pharm.
and ther. 40/8 376 – 381 (2002)
Beruflicher Werdegang: 07/ 2004 – 10/ 2004 Gynäkologie im Marien-Krankenhaus Lübeck
11/2004 Gynäkologie AK Segeberger Kliniken












