
WIRKSTOFFKOMBINATIONEN - mm.wiwi.uni-due.de · Primäres Ziel des AMNOG ist es, dem Prinzip...
37
1 WIRKSTOFFKOMBINATIONEN qualitative und monetäre Herausforderungen Ein aktueller Diskussionsbeitrag mit konkreten Lösungsansätzen 14. Juli 2015 Autoren (alphabetisch) Dr. Jürgen Bausch Dr. Johannes Bruns Wolfgang Kaesbach Dr. Ulf Maywald Peter Schmidt Prof. Volker Ulrich Prof. Dr. Jürgen Wasem Einzelaspekte wurden mit Sachverständigen aus dem BMG, der Politik, dem GBA, den Schiedsinstanzen und der pharmazeutischen Industrie diskutiert
Transcript of WIRKSTOFFKOMBINATIONEN - mm.wiwi.uni-due.de · Primäres Ziel des AMNOG ist es, dem Prinzip...

1
WIRKSTOFFKOMBINATIONEN
qualitative und monetäre Herausforderungen
Ein aktueller Diskussionsbeitrag mit konkreten Lösungsansätzen
14. Juli 2015
Autoren (alphabetisch) Dr. Jürgen Bausch Dr. Johannes Bruns Wolfgang Kaesbach Dr. Ulf Maywald Peter Schmidt Prof. Volker Ulrich Prof. Dr. Jürgen Wasem
Einzelaspekte wurden mit Sachverständigen aus dem BMG, der Politik, dem
GBA, den Schiedsinstanzen und der pharmazeutischen Industrie diskutiert

2
Vorbemerkung:
Die freie Kombination von zugelassenen Wirkstoffen zur Lösung von
Patienten-Problemen ist selbstverständlicher Bestandteil ärztlichen
Handelns.
Zugelassene Fixkombinationen sind nicht selten
Versorgungsstandard.
In der Onkologie jedoch eröffnen freie Wirkstoffkombinationen
mituner bessere Heilungschancen. Sie werden häufig verordnet,
verfügen nicht über eine arzneimittelrechtliche Zulassung und sind
verantwortlich für relevante Ausgabensteigerungen der GKV und der
privaten Krankenkassen.
Der vorliegende Diskussionsbeitrag beleuchtet im vorderen,
allgemeinen Teil die Problematik und bringt im speziellen
Nachfolgeteil Lösungsansätze.
Ziel ist:
Die Versorgungsqualität zu verbessern und Kosten zu reduzieren.

3
- Allgemeiner Teil -
„Kombitherapien in der Onkologie und Virologie
Denkbare Alternativen zur Flexibilisierung der Erstattungsmodelle,
insbesondere von Kombinationsbehandlungen“
Einleitung in die Problematik
Ein konkretes Beispiel aus der Onkologie erleichtert den Zugang zu
diesem sperrigen Thema:
„Hochmaligne Non Hodgkin-Lymphome (NHL) lassen sich gut
chemotherapeutisch behandeln und können sogar komplett geheilt
werden.“ So kann es der Laie bei Wikipedia nachlesen, wenn man „NHL“
in das System eingibt. Es ist keine zwei Jahrzehnte her, da war diese
schwerwiegende Diagnose ein Todesurteil. Die moderne Behandlung
erfolgt in der Regel durch eine Kombinationstherapie nach dem CHOP-
Schema. Ein Cocktail aus Cyclophosphamid (C), Doxorubizin (D),
Vincristin (Onkovin® (O)) und Prednisolon (P). Diese Chemotherapie wird
seit einiger Zeit mit dem monoklonalen CD-20-Antikörper Rituximab
kombiniert. Eine Fünferkombination mit dem Achronym „R-CHOP“.
Niemand kommt auf die Idee, diesen internationalen Therapie-Standard in
die Nähe des „off-label-use“ zu lokalisieren, obwohl es eigentlich keine
arzneimittelrechtliche Zulassung für dieses bewährte therapeutische
Vorgehen gibt.

4
Kombitherapien entwickeln sich zur Regelversorgung
Dieses sehr spezielle, aber typische Beispiel aus der Haemato-Onkologie
würde unter Kostenträgern, Arzneimittel-Experten, Ökonomen und
Gesundheitspolitikern niemanden interessieren, wenn nicht nach Aussage
vieler Onkologen aus der Versorgungsebene übereinstimmend berichtet
wird, dass eine freie Wirkstoffkombination in ihrem Fachgebiet in mehr als
der Hälfte aller Fälle die Regel geworden ist. Zwar nicht regelhaft mit so
vielen Wirkstoffkomponenten wie beim NHL, aber fast immer ohne eine
arzneimittelrechtliche Zulassung und ohne das Vorliegen von
prospektiven belastbaren Daten zu Nutzen und Schaden in der
Kombinationsbehandlung. Vorherrschend ist anekdotische Evidenz.
Allerdings gestützt auf Zulassung und Nutzenbewertung der
Komponenten und die gewachsenen molekularbiologischen Erkenntnisse
der Wissenschaft.
Die Einzel-Wirkstoffe verfügen seit 2011 in Deutschland nach der
europäischen Zulassung auf der Basis einer frühen Nutzenbewertung
über einen ausgehandelten Erstattungspreis für die zugelassene
Monotherapie. Bei den älteren zugelassenen Bestandsarzneimitteln fehlt
die Nutzenbewertung und die Preissetzung ist in der Verantwortung des
Unternehmens. Generischer Wettbewerb hat jedoch Preissenkungen
bewirkt.
In der Kombination addieren sich die Preise der
Wirkstoffkomponenten unbehindert.
Fest steht allerdings: Kombinationsbehandlungen führen mehrheitlich zu
besseren Therapie-ergebnissen. Und sie bewirken bemerkenswerte
Ausgabensteigerungen, was erklärt, warum z.B. im
Arzneimittelausgabensegment der Onkologica Jahr für Jahr zweistellige
Steigerungsraten die Regel sind.
Wenn man in dieses Problemfeld der Kombitherapien mehr Transparenz
und neue Lösungsansätze bringen möchte, macht es Sinn, sich zu
vergegenwärtigen:
Seit es bewusstes medizinisches Handeln gibt, verfügt der Arzt
grundsätzlich über die Freiheit in der Wahl seiner Therapie.

5
Daran haben auch einengende Vorschriften des Sozialgesetzbuches V
nichts entscheidend ändern können. Patient und Arzt sind letztendlich
starke Verbündete in dem Ziel: ein bestmögliches Therapie-Ergebnis
erzielen zu wollen. Voraussetzung ist allerdings der indikationsgerechte
Einsatz im Zulassungsgebiet des Arzneimittels. Eine formale Zulassung
einer vom Arzt frei gewählten Wirkstoffkombination ist nicht zwingend.
Es liegt allerdings in der Natur der Dinge, dass nicht alles, was Ärzte
dabei tun und Patienten haben wollen, zielführend ist und bezahlbar
bleibt. Aber auch die Kostenträger müssen ein Interesse daran haben,
dass ihre Versicherten schnellen Zugang zum medizinischen Fortschritt
und neuen Therapie-Optionen haben, bei denen eine „therapierelevante
Verbesserung der Behandlung erwartbar ist“ (§35 c, Abs. 2 SGB V).
Auf eine knappe Formel gebracht:
Kombinationsbehandlungen, insbesondere in der Krebstherapie, aber
auch bei Hepatitis C und HIV sind wichtiger Therapie-Bestandteil. In der
Regel - bezogen auf den erlaubten zeitgleichen Einsatz - außerhalb einer
arzneimittelrechtlichen Zulassung. Sie haben in vielen Fällen die Potenz,
das Therapie-Ergebnis zu optimieren. Eine Trennung von Spreu und
Weizen ist jedoch nicht der Regelfall.
Und jede Kombitherapie verteuert add on – unabhängig vom klinischen
Nutzen - den Behandlungs-Aufwand ungeschmälert im Vergleich zur
Mono-Therapie.
Vorausschauende Arzneimittelpolitik bedeutet, diese qualitativ ungefilterte
Versorgungssituation mit einem Treibsatz auf der Ausgabenseite
einzufangen.
„Mehr Qualität zu überschaubaren Kosten“ könnte die Devise sein.
Systemeingriffe zeichnen sich ab
Das Ziel wird sein: Sinnvolle Kombinationsbehandlungen allen Patienten
zugänglich zu machen, wenn sie zu einer erwartbaren therapierelevanten
Verbesserung führen können. Der davon ausgehende Ausgabenanstieg
ist durch geeignete Eingriffe abzubremsen. Das Industrie-Interesse an
einem unverändert bleibenden ausgehandelten Erstattungspreis auf der
Ebene der Monotherapie ist zu berücksichtigen.

6
Der einfachste Weg für die Lösung dieses Problems wäre, wenn die
pharmazeutischen Hersteller freiwillig bei allen Wirkstoffen im
Kombinationsfall auf den vollen ausgehandelten Monotherapiepreis
verzichten, indem sie den Krankenkassen für diese Umsatzsteigerung
einen General-Rabatt einräumen, der nicht öffentlich gemacht wird.
Es bedarf keiner Vertiefung der Diskussion, dass ein freiwilliger Verzicht
kein zukunftsfähiges Modell darstellt.
Eckpunkte für einen konstruktiven Lösungsansatz
Trennung der Spreu vom Weizen
Der GBA erhält eine Schlüsselrolle.
Kombitherapien, die üblich sind, obwohl keine arzneimittelrechtliche
Zulassung vorliegt, werden auf Antrag vom GBA geprüft.
Liegt aufgrund der aktuellen Erkenntnislage ein Anhaltspunkt, ein
Hinweis oder ein Beleg für einen mehr als geringfügigen klinischen
Nutzen vor, oder ist eine therapierelevante Verbesserung der
Behandlung erwartbar, dann listet der GBA diese Kombination
positiv in seiner Arzneimittelrichtlinie.
Gewogen und zu leicht befunden
Kombitherapien, die diese Hürde nicht überspringen, sind obsolet.
Sie verweisen Ärzte im Falle von Verordnungen in eine
Begründungspflicht, sofern eine Wirtschaftlichkeitsprüfung erfolgt.
Freie Kombinationen von zugelassenen Wirkstoffen sind
grundsätzlich zulässig, wenn sie indikationsgerecht eingesetzt
werden. Ohne positive Listung durch den GBA trägt der Arzt voll
das Wirtschaftlichkeitsrisiko.
Die wichtigsten Antragsteller
Kassen, KVen, onkologische Kompetenzzentren und Hersteller sind beim GBA antragsberechtigt.

7
Normsetzung
Die Entscheidung des GBA hat im positiven Fall normsetzenden
Charakter. Denn eine Kombinationsbehandlung, die nach Prüfung
durch den GBA bezüglich ihrer Nutzenpotenz für den Patienten
positiv gelistet wurde, wird in gewisser Weise „amtlich zertifiziert“,
ohne arzneimittelrechtlich zugelassen zu sein.
Herstellerzustimmung
Hersteller von Komponenten einer Kombinationstherapie müssen
analog den Regelungen für den „off label use“ ihre Zustimmung
inklusive der Unternehmerhaftung bekennen.
Kombipreisverhandlungen
Wer positiv gelistet wurde, geht verpflichtend in erneute
Preisverhandlungen mit dem Spitzenverband. Die ausgehandelten
Abschläge (Rabatte) bleiben verdeckt. Eine Schiedslösung ist
vorzunehmen, sofern die Verhandlungen erfolglos geblieben sind.
Schlussbemerkung
Das vorliegende Konzept zielt darauf ab, die Versorgungsqualität mit
freien Kombinationsbehandlungen zu verbessern und den Preisauftrieb,
der von dem zunehmenden Trend zur kombinierten Behandlung ausgeht,
durch zwingende Rabattverhandlungen abzubremsen.
Dadurch wird das Verhandlungsergebnis des Erstattungspreises nach
AMNOG im monotherapeutischen Einsatz nicht tangiert.
Einzelheiten des Konzepts und Detailregelungen zur Umsetzung finden
sich im anschließenden „speziellen Teil“ des Positionspapiers.

8
- Spezieller Teil -
WIRKSTOFFKOMBINATIONEN
qualitative und monetäre Herausforderungen
Ein aktueller Diskussionsbeitrag mit konkreten Lösungsansätzen
20. Juli 2015
Autoren (alphabetisch) Dr. Jürgen Bausch Dr. Johannes Bruns Wolfgang Kaesbach Dr. Ulf Maywald Peter Schmidt Prof. Volker Ulrich Prof. Dr. Jürgen Wasem

9
Inhalt
Glossar
I. Die Ausgangslage
II. Vorausbemerkungen zu Lösungsansätzen
III. Lösungsansätze
„Einfache Lösungen“
IV. Die optimale Lösung
Anforderungen
V. Vorschlag zur Umsetzung „Kombinationstherapie“
1. Aufruf zur Verhandlung
2. Verhandlung
3. Änderungsbedarf im §130b Verfahren
VI. Konfliktlösungsmechanismus
VII. „Sequentielle Kombinationen“
VIII. Datengrundlagen als Voraussetzung für Flexibilisierung
IX. Datenverfügbarkeit und Datenschutz
X. Handlungsempfehlung für die Politik

10
Glossar Fixe Kombination Im Kontext dieses Papiers bedeutet „fixe
Kombination“ nicht die Kombination zweier Wirkstoffe in einer Tablette, sondern die per Zulassung erforderliche Kombination zweier Wirkstoffe (um die Wirkung zu erreichen),
Freie Kombination
Im Kontext dieses Papiers bedeutet „freie Kombination“ die Kombination zweier Arzneimittel, die in keiner der beiden Zulassungen so vorgesehen ist, sondern vom Arzt aus individuellen Erwägungen heraus eingesetzt wird (z.B. synergistischer Wirkmechanismus)
Sequentielle (Tumor)Therapie
Sequentielle Therapie bedeutet, dass ein Tumor in verschiedenen Stadien der Erkrankung mit verschiedenen Arzneimitteln (auch in jeweils fixen und freien Kombinationen) behandelt wird. Im Kontext dieses Papieres wird, wenn von Kombination die Rede ist, immer eine fixe oder freie Kombination im gleichen Krankheitsstadium verstanden, auch wenn die Verordnung der Kombinationspartner zeitlich, je nach Therapieschema, nicht am selben Tag erfolgen muss.
Off-label-use
Off-label-use ist die Anwendung von Arzneimitteln außerhalb Ihrer zugelassenen Anwendungsgebiete („echter off-label-use). Der Sonderfall bei der o.g. „freien“ Kombination besteht jedoch darin, dass jeder einzelne Partner der Kombination für das Anwendungsgebiet zugelassen ist, aber die Kombination weder systematisch geprüft noch zugelassen wurde, sondern sich „in der Realität ergibt“ („unechter off-label-use“). Daher sind die bisherigen Regularien des Systems zum off-label-use, auf die in diesem Papier Bezug genommen wird, nur Analogien, eine 1:1-Übertragung ist nicht möglich. Fehlt einem Kombinationspartner die Zulassung für die Erkrankung, die mit der Kombinationstherapie behandelt werden soll, gelten die Regelungen des „echten“ off-label-use bzw. §35c SGB V uneingeschränkt.

11
Die Ausgangslage:
1. Die Versicherten sollen auch in Zukunft nach ihrem objektiven
Bedarf und nicht nach ihrer Zahlungsfähigkeit Zugang zu den
Versorgungsleistungen der GKV haben, die für die Behandlung
ihrer jeweiligen Erkrankung medizinisch notwendig und
zweckmäßig sind. Das schließt auch sehr teure Leistungen ein.
2. Das bestehende Solidarsystem, das den Zugang zu allen im
Einzelfall medizinisch notwendigen Leistungen ermöglicht,
muss allerdings bezahlbar bleiben, es darf nicht überfordert
werden. Kosten und Nutzen müssen in einem angemessenen
Verhältnis stehen.
Die Versorgung der Patienten mit teuren Spezialprodukten, zu denen
auch die Biopharmazeutika gehören, erfordert einen stetig wachsenden
Anteil der GKV-Arzneimittelausgaben (ein Drittel der Ausgaben für nur 5%
der Verordnungen). Nach derzeitigem Stand wird dieser Kostendruck in
absehbarer Zeit weiter deutlich zunehmen. Denn die Entwicklungspipeline
der forschenden Industrie ist mit fast 600 Biopharmazeutika prall gefüllt.
Allein die darin steckende Ausgabendynamik wird die GKV trotz
verhandelten Erstattungsbeträgen mit großen Herausforderungen
konfrontieren. Mit dem Arzneimittelmarktneuordnungsgesetz (AMNOG)
wurde ein bemerkenswerter und folgenreicher Paradigmenwechsel in der
Versorgung Deutschlands mit patentgeschützten Arzneimitteln eingeleitet.
Primäres Ziel des AMNOG ist es, dem Prinzip „Preis folgt dem Nutzen“
bei neuen Arzneimitteln im Rahmen der GKV stärker Geltung zu
verschaffen. Das AMNOG ist aber kein reines „Kostendämpfungsgesetz“,
es verfolgt ebenso industrie- und sozialpolitische Belange. Dem
Gesetzentwurf zufolge sollen den Patienten im Krankheitsfall die „besten
und wirksamsten Arzneimittel zur Verfügung stehen“, müssen die
Arzneimittelpreise und -verordnungen „wirtschaftlich und kosteneffizient
sein“ und werden „verlässliche Rahmenbedingungen für Innovationen, die
Versorgung der Versicherten und die Sicherung von Arbeitsplätzen“
geschaffen (Deutscher Bundestag 2010).

12
3. Vorhersehbare Finanzierungsprobleme u.a. bei onkologischen
Therapien, bei denen Wirkstoffkombinationen zu besseren
therapeutischen Ergebnissen führen als die Monotherapie mit
nur einem Wirkstoff.
Zunehmend sind in der Onkologie neue (und teure) Wirkstoffe mit
unterschiedlichen Angriffspunkten besser wirksam als etablierte
Standardtherapien. Gestiegene Überlebenszeiten zu vergleichsweise
vertretbarer Lebensqualität sind beispielhaft ein Merkmal dieser
Entwicklung. Die „Onkologie wird zunehmend chronisch“, neue Wirkstoffe
werden regelhaft ergänzend und selten ersetzend eingesetzt. Noch
deutlicher und aktueller zeigt sich der Trend zu Kombinationsschemata in
der Hepatitis C-Behandlung.
Im Falle einer Kombination solcher Einzelwirkstoffe addieren sich die
Behandlungskosten je Patient jedoch in relevante sechsstellige
Größenordnungen. Die Ausgaben für Krebstherapeutika steigen regelhaft
mit deutlich zweistelligen Wachstumsraten. Bei der Preisbildung solcher
Wirkstoffe vor AMNOG und bei der Preisverhandlung nach AMNOG sind
– trotz Horizon Scanning - die Einsatzmöglichkeiten der Einzelsubstanzen
in der Kombitherapie in der Regel nicht bekannt. Insbesondere, wenn
Arzneistoffe sowohl in der Monotherapie zugelassen als auch in
Kombination nicht explizit kontraindiziert sind, versucht jeder Hersteller,
den maximalen Preis in beiden Anwendungen zu erreichen. Die freie
Kombination von Einzelwirkstoffen ist dann quasi eine Indikations- und
Markterweiterung, ohne dass es dafür einer Neuzulassung bedarf.
Beispiele dazu sind weiter unten angeführt.
Gesucht wird also –vereinfacht ausgedrückt - ein Verfahren,
welches bei Monotherapie den Preis 1,0 garantiert, bei Kombination
aber anstelle 2,0 z.B. nur Faktor 1,5.
4. Mangelnde Flexibilität der Rahmenbedingungen zur Findung
eines adäquaten §130b-Erstattungsbetrags.
Neben dem „Kombinationstherapieproblem“ tritt ein weiteres Problem auf,
wenn z.B. Zulassungserweiterungen mit neuen Dosierungen, aber
unveränderten Warenzeichen und Packungsgrößen einhergehen. So wird
Ipilimumab, derzeit mit 3mg/kg Körpergewicht beim Melanom für ca. 80T€
Jahrestherapiekosten erhältlich, auch in der Indikation NSCLC geprüft.
Allerdings mit 10mg/kg Körpergewicht, was zu 270T€
Jahrestherapiekosten oder – zumindest mittelbar - einem für den
Hersteller ggf. nicht annehmbar niedrigen Preis beim Melanom führen

13
würde. In Fällen, in denen der Hersteller eine neue Indikation nicht als
gesondertes Warenzeichen (was durch EU- und/oder EMA-Vorgaben
limitiert sein kann), sondern als Erweiterung der Fachinformation des
bestehenden Präparates in den Markt bringt, gerät die „Mischpreislogik“
des AMNOG, die in den allermeisten Fällen richtig ist, an ihre Grenzen,
weil die eine Indikation zu teuer und die andere „zu preiswert“ wäre.
Ebenso problematisch ist, wenn die EMA so genannte
„Evidenztransferentscheidungen“ trifft, also ein Arzneimittel mit mehr
Indikationen bzw. breiter zulässt, als klinische Daten vorliegen, weil eine
Wirkung auch in Patientengruppen, die nicht geprüft wurden, plausibel ist
und erwartet werden kann, z.B.. bei Daltegravir (HIV) und Secukinumab
(Psoriasis). Dies führt unter den aktuellen AMNOG-Regelungen
zwangsläufig zu Verwerfungen im AMNOG-Prozess. Da der Hersteller für
die „unverhofft“ zugelassene Population keine Daten vorlegen kann, gilt
nicht etwa aus dem Grund, dass Wirkungs-/Nutzenverhältnis nicht
plausibel wäre, sondern allein wegen fehlender Belege ein Zusatznutzen
als nicht belegt. Betrifft dies ggf. eine relevant große Subpopulation, sind
Verhandlungen über einen Mischpreis „belastet“ mit der Folge einer
möglichen Marktrücknahme, die auch die wichtigen Subpopulationen trifft.
Z.B. hat Secukinumab in einer direkt vergleichenden Studie Überlegenheit
gegen den TNF-alpha-Hemmer Etanercept, den Biological-
Therapiestandard bei schwerer Psoriasis gezeigt. Die EMA hat jedoch
nicht nach Versagen von DMARDs zugelassen, sondern nach Versagen
topischer Therapie. Leitliniengerecht wird Etanecerpt erst nach Versagen
von MTX und Ciclosporin eingesetzt. Für diesen Vergleich gibt es aber
keine Daten. Weder Ärzte noch Hersteller kämen jedoch (derzeit) auf die
Idee, mit einem neuen Biologikum die DMARDs abzulösen, würden sich
aber genau diesem Szenario in Nutzenbewertung und
Erstattungsbetragsverhandlung ausgesetzt sehen.
Auch hier wird ein Verfahren gesucht, einzelne Subgruppen nicht zu
verhandeln bzw. ggf. nicht zu erstatten (Teilerstattungsausschluss),
auch wenn sie formal verordnungsfähig bleiben könnten).
.

14
II. Vorausbemerkungen zu Lösungsansätzen
1. Auch (freie) Kombinationsbehandlungen müssen
verbesserten patientenrelevanten Nutzen belegen.
Onkologen berichten, dass bereits mehr als die Hälfte ihrer
Chemotherapiepatienten zwei- und dreifachkombiniert behandelt werden.
Gründe sind nationale und internationale Erkenntnisse durch erste
Studien, Fallberichte, Kongressberichte, Zentrenentscheidungen,
Vermeidung von Resistenzbildungen oder unterschiedlicher Wirkansatz
der Wirkstoffe an der Krebszelle. Belastbare Evidenz ist hier jedoch häufig
nicht vorhanden.
Die Indikationsstellung für eine „freie“ (also per Zulassung nicht
ausgeschlossene, aber auch nicht in Studien geprüfte)
Kombinationstherapie erfordert daher eine strenge Auswahl, vornehmlich
durch ein Tumorboard oder Zweitmeinungsverfahren. Die Durchführung
ist nur unter kontrollierten Bedingungen wirtschaftlich und sollte eine
engmaschige Evaluation der Ergebnisse durch lückenlose
Verlaufsmeldung an klinische Krebsregister voraussetzen, um den
dringend notwendigen Erkenntnisgewinn zu generieren.
2. Neue Ansätze sind zum Umgang mit dem Problem
erforderlich
Da alle Einzelkomponenten einer Kombinationstherapie entweder nach
AMNOG verhandelte Erstattungsbeträge haben oder bei einer Zulassung
vor 2011 frei bepreist sind, addieren sich die Kosten im Falle der
kombinierten Anwendung mit dem Ziel eines besseren
Therapieergebnisses formal korrekt zu einer hohen Ausgabenbelastung
pro Patient und auch zu einem hohen Budget-Impact für das System.
Alle bisherigen Instrumente der Begrenzung des Ausgabenanstieges
nach SGB V sind nicht geeignet, die Belastung des GKV-Systems durch
Kombinationstherapien zu bremsen. Die derzeit im Rahmen des §130b
möglichen Modelle sind für einen sachgerechten Umgang mit den vielen
Fallgestaltungen (fixe und freie Kombinationen, EMA-Zulassungen weiter
oder enger als die untersuchte Population, …) zu wenig flexibel. Es bedarf
ergänzender Instrumente, die bei wenig neuem Aufwand sachgerechtere
Lösungen ermöglichen.

15
3. Das AMNOG soll mit jeder der denkbaren und hier
skizzierten Lösungen lediglich sachgerecht ergänzt, aber
nicht in Grundsätzen verändert oder gar ersetzt werden.
III. Lösungsansätze
„Einfache“ Lösungen
Sucht man nach Lösungsansätzen, ergibt sich keine einfache Lösung.
Ein Lösungsansatz in Analogie des früheren RSA-Risikopools, der vor
Einführung des Morbi-RSA hochpreisige Fälle ab einem Schwellenwert
ausgeglichen hat, schein politisch nicht durchsetzbar und würde nur die
Einzelkasse von Risiken entlasten, jedoch nicht die GKV insgesamt. Er
löst weder das oben skizzierte „Ipilimumab-Problem“ noch die Probleme
des Mischpreisansatzes.
§130c-Verträge sind zur Lösung ebenfalls ungeeignet, denn diese
würden erstens vom guten Willen des Herstellers abhängen (den nicht
jeder Hersteller mitbringt) und zweitens würde es lange dauern, bis alle
Kassen unter Vertrag sind. Vom Verwaltungsaufwand bei >100 Kassen
ganz zu schweigen. Auch ist die Onkologie ein denkbar ungeeignetes
Feld für den Krankenkassenwettbewerb.
Gesetzliche Abschläge (d.h. Abschlag nur bei tatsächlichem Einsatz in
Kombination) über die Apothekenrechenzentren - in etwa analog des
Generikaabschlags-Rückabwicklungsverfahrens bis 2011 - welche die
Vorleistung der Einzelkasse bis zur „Kombinations-
ausgleichsrabattzahlung“ auf 8-12 Wochen limitieren würde, scheiden
auch aus. Denn erstens wären dann bislang unbeteiligte Dritte (die
Apothekenrechenzentren) zu beteiligen, die heterogen aufgestellt sind
und eigene wirtschaftliche Interessen anmelden würden. Und schließlich
würde ein solches Modell z.B. keine §130b-Budget-Impact-
Überschreitung auf Ebene Hersteller-GKV-Spitzenverband abwickeln
können und so würde die Chance vergeben, bestehende andere
AMNOG- Reibungspunkte gleich mit einer sachgerechte(re)n Lösung
zuzuführen.

16
Überhaupt ist das Problem „Budget-Impact-Überschreitung“ derzeit nicht
zufriedenstellend lösbar, denn bei Überschreitung von vorab festgelegten
Mengen folgt derzeit maximal eine Kündigung der
Erstattungsbetragsvereinbarung. Bei erweiterter Datenverfügbarkeit (vgl.
später) ließen sich auch Staffeln anhand von abgesetzten Packungen,
therapierten Patienten o.ä. verhandeln und der Erstattungsbetrag könnte
ohne Neuverhandlung zur Laufzeit der Vereinbarung in Abhängigkeit des
Erreichungsgrades „floaten“, egal ob als Listenpreisänderung oder per
nachträglichem Rückerstattungsverfahren. Auch Kapitationen wären
abrechenbar, egal ob auf Packungs-, Umsatz- oder gar Patientenebene.
Letzteres zum Beispiel, um das Risiko unterschiedlicher Therapiedauern
auf den Hersteller verlagern zu können, denn Sovaldi® kann z.B. - je
nach Kombination und Genotyp - 8, 12 oder 24 Wochen gegeben werden,
was die Kosten extrem schwanken lässt.

17
IV. Die optimale Lösung
Anforderungen
Die optimale Lösung muss mehrere Bedingungen erfüllen, sie soll:
Den Budget-Impact von Kombinationstherapien (v.a. in der
Onkologie) wirksam begrenzen, ohne „unfair“ gegenüber
einzelnen Kombinationspartnern zu sein
(zumindest teilweise) evidenzbasiert (und nicht rein finanzbasiert)
sein
den Grundmechanismus des AMNOG – den transparenten §130b-
Preis als Bezugspreis für weitere Steuerungsmechanismen und
Preisreferenzierung - nicht „konterkarieren“, aber sachgerecht
weiterentwickeln
den Partnern der §130b-Vereinbarungen dennoch mehr Flexibilität
verschaffen, wie z.B. Kapitation, „nachgelagerte“ Preis-Volumen-
Vereinbarungen mit „verdecktem Rabatt bei
Volumenüberschreitung“, „Kombinationstherapierabatte“,
nachgelagerte Rabatte für Subgruppen ohne Zusatznutzen etc.
mit wenig Aufwand auf Routinedaten umsetzbar sein (kein neuer
Aufwand beim Verordner, wenig neuer Aufwand bei den Kassen)
mit einem Eskalationsmechanismus (Schiedsamt) versehen sein
auch funktionieren, wenn die Kombinationspartner von
verschiedenen Herstellern stammen
auch funktionieren, wenn ein Bestandsmarktarzneimittel mit einem
neuen (AMNOG-) Produkt kombiniert wird (z.B. Lenalidomid +anti-
PD1 oder Pertuzumab + Trastuzumab)
auch funktionieren, wenn es sich um freie Kombinationen handelt,
die vom Arzt (weil pharmakologisch naheliegend und per
Fachinformation zumindest nicht ausgeschlossen) eingesetzt
werden und nicht durch den/die Hersteller explizit so zur
Zulassung gebracht worden sind (z.B. Crizotinib + antiVEGF,
Nivolumab+BRAF600-Inhibitor)
idealerweise auch mit den NUBs im stationären Sektor
synchronisiert sein

18
Hier wird schon ersichtlich, dass es kaum möglich ist, eine Lösung zu
erschaffen, die allen Anforderungen gleichermaßen gerecht wird. Gewisse
(politische) Kompromisse werden (zu Lasten der einen oder anderen
Anforderung) nötig sein um eine Lösung zu schaffen, die alle
Anforderungen summarisch bestmöglich erfüllt.

19
V. Vorschlag zur Umsetzung „Kombinationstherapie“
Es wird weiterhin „Produkt für Produkt“ ein transparenter §130b-
(Monotherapie-)Erstattungsbetrag verhandelt. Die Idee des AMNOG bleibt
im ersten Schritt unangetastet. Die oben geschilderten Grenzen bei fixer
oder freier Kombinationstherapie werden folgendermaßen
überwunden:
Erste Stufe: Aufruf zur Verhandlung
Für neu zugelassene fixe Kombinationen wird der AMNOG-Prozess
nicht verändert, lediglich die Verhandlungen werden mittels des
neuen Modells zur Datenverfügbarkeit und Verrechnung (siehe unten)
flexibilisiert.
Freie Kombinationen, die nicht explizit zugelassen, aber auch nicht
explizit ausgeschlossen sind, werden ohne hinreichende Evidenz
angewendet, erzeugen hohe Kosten und Behandlungsergebnisse
bleiben regelhaft unbekannt. Krankenkassen könnten zukünftig zur
Budgetsteuerung einen solchen Einsatz über Off-label-use-Prüfanträge
sanktionieren, was sich insbesondere in der Onkologie nicht anbietet und
verhindert werden werden soll.
Andererseits könnte als Voraussetzung für die Erstattungsfähigkeit freier
Kombinationen eine ordnungsgemäße Zulassung verlangt werden, der
regelhaft eine „normale“ frühe Nutzenbewertung folgt. Diese Hürde
erscheint aber zu hoch, da Therapien sich auch aus klinischer Evidenz
oder Investigator initiated Trials entwickeln können. Außerdem haben
pharmazeutische Unternehmer (z.B. wegen zu kurzer Restlaufzeit des
Patentes) u.U. kein Interesse, notwendige Studien zu finanzieren und
entsprechende Zulassungserweiterungen zu betreiben. Weiterhin ist die
dafür notwendige Zeitschiene in der Regel so erheblich, dass das System
dabei regelhaft „von der (Verordnungs-)Realität überholt“ wird und die
normative Kraft des Faktischen zur Erstattung der Kombinationen führt.

20
Daher bietet sich folgende Lösung an:
1. Alles nachfolgend Gesagte gilt nur, wenn mindestens ein
Arzneimittel, für das eine frühe Nutzenbewertung vorliegt, Partner in
der freien Kombination und das in der freien Kombination
verwendete Bestandsmarktprodukt noch patentgeschützt ist. Reine
Bestandsmarktkombinationen (die in der Regel generisch sind), sind
nicht vom Regelungskontext dieses Papiers umfasst.
„Kombinationsaufruf“ durch den GBA und nachfolgende Listung
in der AM-RL
Bei freie Kombinationen erscheint der Ressourceneinsatz mangels
Evidenz nicht gerechtfertigt. In welchen Therapiegebieten „freie
Kombinationen“ in der Regel unwirtschaftlich sind, wird in der
Arzneimittelrichtlinie nach §92 Absatz 1 Satz 2 Nr. 6 klargestellt.
Dem G-BA wird die Aufgabe zugewiesen, freie Kombinationen, die
für die Versorgung von Bedeutung sind (Aufgreifkriterium nach §35
Abs. 6 Satz 1 alt), - „nach Aufrufen“ und einer den praktischen
Gegebenheiten angepassten Bewertung der bestverfügbaren
Evidenz als in dieser Kombination verordnungsfähig in einer neuen
Anlage der AM-RL zu listen. Zum Aufruf antragsbedingt sind
Trägerorganisationen des GBA, Hersteller und zertifizierte
Tumorzentren (ggf. einfach „Dritte“). Das Nähere zu den Kriterien
der Anträge und der Bewertung regelt der G-BA in seiner
Verfahrensordnung.
Die Aussicht auf ein Potential für einen mehr als nur
geringfügigen therapeutischen Nutzen, gemessen am Ausmaß
des erzielbaren therapeutischen Effekts (z.B. in Analogie zu §33a
Abs. 7 SGB V alt), sollte das wesentliche Kriterium sein. Es muss
zumindest Anhaltspunkte geben, dass klinisch relevante Endpunkte
besser beeinflusst werden als bei einer Monotherapie. Rein
pharmakologische Erwägungen reichen nicht. Eine durch
bestverfügbare Evidenz belegte positive Nutzen-Schadens-Bilanz
ist Voraussetzung für die Listung. Auch §35c Abs. 2 SGB V
(Anspruch auf Versorgung mit zugelassenen Arzneimitteln in
klinischen Studien), stellt darauf ab, dass „eine therapierelevante
Verbesserung der Behandlung einer schwerwiegenden Erkrankung
im Vergleich zu bestehenden Behandlungsmöglichkeiten zu
erwarten ist“, was ebenfalls ein Weniger an Evidenz als
normalerweise erforderlich bedeutet. In der Regel sollte die Hürde
für sinnvolle Kombinationen nicht zu hoch sein, denn die

21
Produkte sind ja alle in der Monotherapie für die in Rede stehende
Erkrankung zugelassen.
Die bei der zulassungsüberschreitenden Anwendung von
Arzneimitteln nach §35c Absatz 1 und 2 bewährten Regelungen
werden hier analog angewendet. Bei Bedarf kann der G-BA eine
Expertengruppe „freie Kombination“ vor einer Listung in der AM-RL
hinzuziehen. Voraussetzung für eine Listung ist die Erklärung jedes
pharmazeutischen Unternehmers, (pU) auf Anfrage des G-BA die
Haftung nach §84 AMG für die Anwendung seines Produktes in
freier Kombination zu übernehmen. Der G-BA verzichtet auf den
Aufruf und veröffentlicht das Ergebnis der Anfrage, wenn -
mindestens ein beteiligter - pU die Haftung nach §84 AMG ablehnt,
damit die Ärzte über das haftungsrechtliche Risiko Ihrer
Therapieentscheidungen informiert sind.
Die Listung der freien Kombination in der Anlage der AM-RL
erlischt, sobald diese Kombination eine arzneimittelgesetzliche
Zulassung erhält.
Bei der Kostenbelastung der Solidargemeinschaft auf Basis
geringer Evidenz sind mit der Listung verbundene Auflagen zur
Qualitätssicherung gerechtfertigt, um regelhaft zusätzliche
Evidenz zu generieren. Dazu zählen verpflichtend die lückenlose
elektronische (Verlaufs-)Meldung der Ärzte an die klinischen
Krebsregister, aber auch weitere Auflagen zur Sicherung von
Prozess-, Struktur- und Ergebnisqualität, wie z.B. die Beschränkung
des Einsatzes auf Zentren mit begleitenden Investigator Initiated
Trials (in Analogie zu §92 Abs. 2a). Dadurch würde der
Leistungsanspruch des Versicherten nicht verkürzt, denn die
Verordnung der gelisteten Kombination bleibt ja formal für jeden
Arzt weiter möglich.
Allerdings setzen sich Ärzte, die die Qualitätskriterien nicht erfüllen
oder nicht beurteilte freie Kombinationen verordnen, einem höheren
Risiko der Wirtschaftlichkeitsprüfung aus. Dass GBA-
„Empfehlungen“ zum Setting von Behandlungen wirken, zeigt auch
die Erfolgsgeschichte der – zertifizierten – Brustzentren, in denen
inzwischen über 90% aller Mammakarzinome behandelt werden.

22
Die nach Haftungsübernahmeerklärung, Aufruf und positiver
Evidenzbewertung erfolgte Listung der Kombination in der
Arzneimittelrichtlinie mündet in einer neuen, (ggf. trilateralen)
Erstattungsbetragsverhandlung für die Fälle des Einsatzes der
Produkte in Kombination, vgl. unten.
Hier wäre in §92 Abs. 2b (neu) vorzusehen, dass verordnungsfähige
freie Kombinationen mit einem mehr als nur geringfügigen
therapeutischen Nutzen in der AM-Richtlinie gelistet werden. In
§130B wäre zu regeln, dass nach erfolgter Listung neue
Rabattverhandlungen für die Fälle von Kombinationstherapien
eröffnet sind.
Vorschlag: § 92 wird um Abs. 2b (neu) ergänzt:
(1)Die Richtlinien nach Abs. 2 Satz 1 Nr. 6 enthalten Arzneimittel mit
patentgeschützten Wirkstoffen und einem mehr als nur
geringfügigen therapeutischen Nutzen gemessen am Ausmaß des
erzielbaren therapeutischen Effekts, die als Partner von nicht
explizit zugelassenen Kombinationstherapien verordnungsfähig
sind. (2)Das Nähere zur Benennung der Arzneimittel und zu den
Voraussetzungen der Verordnung regelt der Gemeinsame
Bundesausschuss in seiner Verfahrensordnung. §35c Abs. 1 und 2
sowie §92 (2a) gelten entsprechend.
Noch zu prüfen ist, ob die Ermächtigungsnorm in §92 (2) SGB V
ausreicht, dass der GBA die angesprochenen Auflagen zur
Qualitätssicherung in der entsprechenden Anlage
„verordnungsfähige Kombinationen“ der Arzneimittelrichtlinie
rechtssicher beschließen kann.
Exkurs: In diesem Zusammenhang wird nachdrücklich eine
Konkretisierung des §65c SGB V angeregt, da das
Krebsfrüherkennungs- und Registergesetz noch nicht zu für die
klinische Krebsregistrierung in Deutschland ausreichend
einheitlichen und zukunftsfähigen Strukturen geführt hat. So
scheitert ebenso die (Verlaufs)Meldung der verordnenden Ärzte an
der fehlenden Standardisierung der Umsetzung des
Meldeprozesses in die Praxisverwaltungssysteme wie auch an den
notwendigen Standards des Datenaustausches zwischen den
Registern.

23
Wenn der G-BA zum Beispiel aus den Daten der klinischen
Krebsregistrierung ableiten soll, ob eine Therapie in freier
Kombination sinnvoll ist oder nicht, müssen diese lückenlos und
national einheitlich verfügbar sein.
Eine nationale Zusammenführung der Krebsregisterdaten zum
Zwecke des Erkenntnisgewinns für G-BA, IQWIG und IQTIG ist
derzeit nicht vorgesehen. Die Zusammenarbeit des G-BA nach §65c
Abs. 7 mit einer Vielzahl von mit unterschiedlichen
Softwaresystemen arbeitenden klinischen Krebsregistern ist nicht
umsetzbar. Erforderlich sind eine Standardisierung der Technik (von
Arztsoftware zum Register z.B. durch die Vorgabe einer
Spezifikation durch die GKV, von Register zu Register z.B. durch
die Vorgabe einer Spezifikation durch die Krebsgesellschaft), die
Errichtung einer bundesweiten Register(daten)-Kopfstelle und die
„Beendigung des Föderalismus“ hinsichtlich länderspezifischer
Meldegesetze und Datenschutzregelungen.
An dieser Stelle sei angemerkt, dass andere europäische Länder
erweiterte Qualitätsanforderungen an Verordnungen bereits
eingeführt haben. So sind z.B. in Italien begleitende Register
ebenfalls Voraussetzung für die Verordnung und in UK entscheidet
nicht der Arzt über die Verordnung einer freien Kombination,
sondern die Clinical Commissioning Group.
2. Zweite Stufe: „Rabattvereinbarung: Erstattungsbetrag,
Kombinationen“
Es wird unterstellt, dass auch die pharmazeutischen Unternehmer
Kombinationsrabattverhandlungen“ als Chance begreifen, eine
gesetzliche „Rasenmähermethode“ zur Kostensenkung zu
vermeiden.
Hersteller entweder explizit als in fixer Kombination zugelassener
oder vom G-BA zur Verwendung in freier Kombination gelisteter
Arzneimittel verhandeln mit dem GKV-Spitzenverband einen Rabatt
auf den Erstattungsbetrag, sofern das Arzneimittel im Rahmen einer
Kombinationstherapie verordnet ist.

24
3. Änderungsbedarf im §130bVerfahren
Die bei Kombinationstherapien auftretenden unterschiedlichen
Fallkonstellationen können aber auf demselben
gesetzestechnischen Weg geregelt werden:
Fixe explizit zugelassene Kombinationen können von dem selben
Hersteller (z.B: Dabrafenib + Trametinib) oder von verschiedenen
Herstellern (z.B. demnächst Crizotinib + Ceritinib) ausgeboten
werden und auch patentgeschützte Kombinationspartner aus dem
Bestandsmarkt enthalten (derselbe Hersteller: z.B. Trastuzumab +
Pertuzumab, verschiedene Hersteller: z.B. Lenalidomid +
Pembrolizumab).
Freie Kombinationen können ebenso von demselben Hersteller
(Pertuzumab + Trastuzumab-Emtansin) oder von verschiedenen
Herstellern (Anti-PD1 + BRAF600-Inhinbitor) stammen und
ebenfalls auch patentgeschützte Partner aus dem Bestandsmarkt
beinhalten (Trastuzumab + Anti-PD1).
Im SGB V müsste klargestellt werden, dass für diese
Kombinationsanwendungen explizit kein neuer Erstattungsbetrag,
sondern ein Rabatt auf den §130b-(Monotherapie)
Erstattungsbetrag zu verhandeln ist, solange die explizite Zulassung
der Kombination aussteht. Die Listung der Kombination durch den
G-BA mündet in ein neues Verhandlungsverfahren zum
Kombinationsrabatt, auch wenn die vereinbarte Geltungsdauer des
initialen Erstattungsbetrages für die Monotherapie noch nicht
ausgelaufen ist. Der Kombinationsrabatt ist – anders als der initiale
§130b-(Monotherapie)Erstattungsbetrag immer nachgelagert und
verdeckt und wird mit den u.g. Daten/Technik abgewickelt.
Ebenso müsste klar gestellt werden, dass betroffene
Bestandsmarktarzneimittel für die Fälle, in denen diese in
Kombination eingesetzt werden, ebenso in eine Rabattverhandlung
nach §130b einzubeziehen sind, ohne dass zuvor eine Bewertung
nach §35a durchgeführt wurde.

25
Dies erscheint gerechtfertigt, da das neue Arzneimittel in seiner
Zulassung die Kombination mit dem Bestandsmarktarzneimittel
quasi „mitbringt“, ohne dass sich dessen Zulassung oder
Fachinformation ändert und die Verordnungen „ohne eigenes Zutun“
des Herstellers deutlich zunehmen.
Vorschlag:
In 130b Abs. 1 werden die Sätze 8 und 9 (neu) angehängt:
(8) Der Spitzenverband Bund der Krankenkassen vereinbart mit
den pharmazeutischen Unternehmern der als Kombinationspartner
zugelassenen Arzneimittel einen Rabatt auf den
Erstattungsbetrag sowie für Arzneimittel nach § 92 Abs. 2b einen
Rabatt auf den Abgabepreis des pharmazeutischen Unternehmers
ohne Mehrwertsteuer für vor dem 31.12.2010 in den Markt
eingeführte Arzneimittel für die Fälle der Verordnung im Rahmen
einer Kombinationstherapie. (9) Abs. 1 Sätze 5 und 7, Abs. 4 unter
Bezugnahme auf die Veröffentlichung des Beschlusses nach § 92
Abs. 2b , Abs. 7, Abs. 9 Sätze 1 und 4 ff sowie § 217f Abs. 7
gelten entsprechend.
In §130b Abs. 5 wird folgender neuer Satz 3 eingefügt:
Satz 2 gilt in Fällen des Abs. 1 Satz 8 entsprechend
§130b Abs. 9 ist entsprechend zu ergänzen
Noch zu prüfen ist, ob weitergehende Verweise und / oder Präzisierungen
erforderlichsind.

26
VI. Konfliktlösungsmechanismus für die
„Kombinationsrabattverhandlungen“ mittels
Schiedsstellenverfahren
Die Regelungen zur Schiedsstelle (§130b Abs. 5 bis 8) finden
entsprechende Anwendung.
a) Schiedsverfahren mit nur einem Hersteller, der alle
Kombinationspartner anbietet
Für fixe explizit zugelassene Kombinationen bestehend aus „AMNOG-
und Bestandsmarktprodukt“ gilt der status quo unverändert, wie z.B.
bereits bei der Kombination Pertuzumab + Trastuzumab) implizit
geschehen Der Kombinationsrabatt sowie die Zuweisung des Rabattes
oder von Teilen davon auf die jeweiligen Kombinationspartner wird im
Rahmen des normalen §130b-Schiedsverfahrens des AMNOG-Produktes
festgesetzt.
Bei freier Kombination entscheidet die Schiedsstelle im Rahmen Ihrer
(dann neuen) Geschäftsordnung.
a) Schiedsverfahren mit mehr als einem Hersteller
Es gelten dieselben vorstehend dargestellten Regelungen. Allerdings ist
die Besetzung der Schiedsstelle neu zu fassen. Bei zwei betroffenen
Herstellern gäbe es weiterhin drei Unparteiische (allerdings mit jeweils
zwei Stimmen), weiterhin zwei Mitglieder des GKV-Spitzenverbandes
(dann auch mit jeweils zwei Stimmen) und je zwei Mitglieder eines
Herstellers (4 Vertreter der Herstellerseite insgesamt). Bei drei oder mehr
betroffenen Herstellern müssten die Sitze proportional erweitert werden.
Die Entscheidungsgrundlagen der Schiedsstelle ändern sich insoweit, als
ein Nutzenbeschluss des G-BA nach §35a für die freie Kombination nicht
vorliegt. An dessen Stelle sind die zum Beschluss des G-BA zur Listung
der freien Kombination veröffentlichten „Tragenden Gründe“
heranzuziehen. Hält die Schiedsstelle weitere Gutachten für erforderlich,
wird das Verfahren für die Dauer des / der Gutachten unterbrochen
(clockstop).

27
Die Schiedsstelle hat weiterhin zu prüfen, ob auch für die
verfahrensgegenständliche freie Kombination die Preise vergleichbarer
Monotherapeutika Berücksichtigung finden. Größere Bedeutung erhalten
die tatsächlichen europäischen Preise. Da die Listung durch den G-BA
erst nach Detektion der freien Kombination in den
Verordnungsdatenerfolgen kann, liegen bedingt durch den zeitlichen
Versatz auch regelhaft Vergleichspreise aus den Ländern nach Anlage 2
der Rahmenvereinbarung nach §130b Abs. 9 vor. Trotz anderslautender
Behauptungen werden neue Arzneimittel nach erfolgter europäischer
Zulassung regelhaft in Deutschland zuerst eingeführt. Insbesondere für
Bestandsmarktarzneimittel sind die tatsächlichen europäischen Preise ein
geeigneter Vergleichsmaßstab.

28
VII. „Sequentielle Kombinationen“
Ein weiteres, bisher nicht adressiertes Problem, sind sequentielle
Therapien, d.h. zeitlich aufeinanderfolgende (und ggf. jeweils kombinierte)
medikamentöse Therapien. In der Onkologie müssen wegen der
zunehmenden Anzahl therapeutischer Optionen und Therapielinien
zukünftig im Grunde Therapieschemata und –sequenzen anstelle
einzelner Arzneimittel in einzelnen Therapielinien verglichen werden. Das
ist in randomisierten Studien extrem schwer, auch hier ruhen die
Hoffnungen auf einer lückenlosen Dokumentation durch die klinischen
Krebsregister, die gesetzgeberisch nochmals unterstützt werden
sollte, da der Föderalismus hier eine schnelle Entwicklung konvergenter
Strukturen eher behindert. . Auch hier addiert sich der Budgetimpact
durch in der jeweiligen Therapielinie - und bei Wechsel der Therapielinie
aufeinanderfolgend angewendete (neue) Kombinationen - zu erheblichen
Größenordnungen. In der Indikation NSCLC ist das bereits zu sehen.
Dieses Problem ist finanziell problematischer zu fassen, weshalb es im
Rahmen dieses Papieres nicht thematisiert werden soll. Allerdings ist die
Grundlage jedweder zukünftigen Lösung für dieses Problem die
lückenlose Krebsregistrierung und ebenfalls die Nutzbarkeit der Morbi-
RSA-Daten, die ja – patientenpseudonymbezogen – auch die zeitliche
Komponente enthalten.

29
VIII. Datengrundlagen als Voraussetzung für
Flexibilisierung
Derzeit verfügt der GKV-Spitzenverband nur über die
Arzneimittelmarktdaten nach §84 Abs. 5 SGB V. Diese enthalten im
Prinzip nur die Anzahl Packungen eines Arzneimittels pro Zeiteinheit und
sind für flexible Abrechnungsmodelle daher zu hoch aggregiert.
Um die o.g. Ziele zu erreichen, werden daher die im Rahmen der
Durchführung des Risikostrukturausgleiches ohnehin anfallenden Daten
nach §267 Abs. 7 Nr. 1 und 2 SGB V bzw. nach §30 Risikostruktur-
Ausgleichsverordnung zur nachgelagerten, aber zeitnahen und vor allem
aufwandsarm zentralen Abrechnung von „Patient Access Schemes“
genutzt. Durch deren Granularität eignen sie sich sehr gut zum Ausgleich
von Mehrkosten durch z.B. Kombinationstherapien, Dosiserhöhungen
durch Zulassungserweiterung während der Laufzeit einer §130b-
Vereinbarung (Bsp. Ipilimumab), überschrittener Preis-Volumen-Klauseln
aus den §130b-Verträgen.
Auch zukünftige Gentherapien könnten so „performance based“ oder in
Annuitätenmodellen erstattet werden und würden keine potentiell
siebenstelligen Einmalzahlungen (z.B. Glybera®) pro Patient erfordern.
Es müsste ein neuer zulässiger Verwendungszweck für die Morbi-RSA-
Daten ins SGB V eingefügt werden, bzw. müsste in §217f (7) die
zwingende Anonymisierung und das Verbot des Kassenbezugs
gestrichen werden. Die RSA-Daten sollen laut AMNOG ohnehin schon für
die Zwecke des §130b nutzbar sein (geregelt in §217f (7) SGB V, der mit
dem AMNOG neu kam), allerdings ist die damals gewählte
Aggregationsebene „Anonymisierung und ohne Kassenbezug“ hier zu
hoch, um noch Nutzen stiften zu können. Damals wurde auf Intervention
einzelner Kassen die Anonymisierung im §217f (7) verpflichtend, da die
Sorge bestand, das Arzneimittelmanagement der Kassen würde
transparent. Diese Sorge sollte heute aus mehreren Gründen heraus
unberechtigt sein: die GKV will gemeinsam - auch zum Zwecke
sachgerechterer §130b-Verhandlungen – das GAmSi-Verfahren nach §84
Absatz 5 neu gestalten, incl. der Nutzung von Patientenpseudonymen
und Kassenzugehörigkeit (pseudonymisiert). Außerdem kaufen alle
großen Kassen regelhafte die Verordnungsdaten der eigenen GKV-

30
Konkurrenz bei Dienstleistern wie IMS und Insight Health und haben so
mittlerweile ohnehin Transparenz zum Geschehen bei der Konkurrenz.
Vorschlag: § 217f Abs. 7 wird neu gefasst:
Der Spitzenverband Bund der Krankenkassen kann zur
Durchführung seiner gesetzlichen Aufgaben nach § 130b Absatz
1 die Daten nach § 268 Absatz 3 Satz 14 in Verbindung mit Satz 1
Nr. 1 bis 7 nutzen und verarbeiten.
Alle Strukturen zur Nutzung der Daten für diesen Zweck jedenfalls
existieren bereits. Sie liegen auch bereits im GKV-Spitzenverband vor, er
darf sie aber nicht benutzen.
Auf Basis dieser Daten könnten Rechnungen entweder zentral durch
den GKV-Spitzenverband selbst an die Hersteller gestellt werden,
genauso könnte der GKV-SV auch seine Mitgliedskassen über die zu
fordernden Beträge informieren. Einzelkasse bzw. deren
Dienstleister fordern selbst. Die Kassen würden also (am Tag der
Abgabe in der Apotheke zum vollen Monotherapiepreis) weiter in
Vorleistung gehen, jedoch später aufwandsarm Erstattungen erhalten.
Damit bleiben die Monotherapiepreise und der AMNOG-Prozess formal
intakt, nur „Sonderkonstellationen“ werden extra abgewickelt.
Denkbar wäre eine solche Lösung auch über das derzeitige,
kassenindividuelle Nacherstattungsverfahren nach §130b (wenn Tag 366
nicht mit dem verhandelten Preis erreicht werden kann), aber erstens
wäre der Verwaltungsaufwand dafür extrem und zweitens würde der
GKV-Spitzenverband weiterhin nicht die (für lösungsorientierte
Verhandlungen ohne Vorurteile) nötige Datentransparenz erhalten. Dass
der Bedarf bereits heute existiert und der Weg mangels besserer
Alternativen genutzt wird, belegt das Beispiel „Erstattungsbetrag
Xigduo®“.
Man kann im Rahmen der §130b-Verhandlungen verschätzte
Patientenanteile oder Mengen „preisunschädlich und zur Laufzeit
glattziehen“. Das Modell eignet sich für einen themenbezogenen,
„listenpreisunschädlichen“ und damit „leisen“ Interessenausgleich, ohne
dafür unnötigerweise zur völligen Vertraulichkeit des Erstattungsbetrages
zurückzukehren.
Mit einer solchen Prinziplösung hätte z.B. auch der aus Versorgungssicht
zumindest zu diskutierende opt-out der Antiepileptika Trobalt® und
Fycompa® verhindert werden können. Bei den Antiepileptika hätte man
etwa festlegen können, dass für (lt. RSA-Daten) jede Erstlinienverordnung
100% Abschlag fällig werden, bei einem Einsatz nach Versagen von

31
mindestens x vorhergehenden Antiepileptika aber 0% Abschlag. Auch
„Fehlverordnungen“, also die Verordnung neuer Arzneimittel an andere
als die im Rahmen von §130b verhandelten Patientenpopulationen,
könnten finanziell ausgeglichen werden.
Ein weiteres Beispiel ist Sovaldi®. Poltisch wurde kontrovers diskutiert,
dass die GKV bereits Rabattverträge abschließt, bevor der §130b-Preis
verhandelt war. Triebfeder dafür war nicht etwa die Rabatthöhe, sondern
die Kapitation auf 12 Wochen Therapiedauer (und damit eine gewisse
Planungssicherheit für die vertragsschließenden Krankenkassen), die
ohne Nutzung der RSA-Daten derzeit im Rahmen des §130b nicht
möglich ist, mit der beschriebenen Änderung aber bereits auf zentraler
Ebene als Komponente des §130b-Erstattungspreises möglich wäre.
Auch die „Rückwirkung des Erstattungsbetrages“ bis zum Zeitpunkt x
nach der Markteinführung könnte so abgebildet werden. Ein
(gesetzlicher?) Budget-Cap für das Jahr eins am Markt (d.h. vor
Preisverhandlung), als Absicherung der Solidargemeinschaft bei Erhalt
der freien Preisbildung und des sofortigen Marktzuganges wäre möglich.
Ordnungspolitisch und AMNOG-denklogisch korrekt – auch wenn das hier
nur angesprochen und nicht gefordert werden soll - könnte man ein
solches Verfahren/eine solche Möglichkeit „flexiblerer“ §130b-
Vereinbarungen ausschließlich auf Produkte mit Zusatznutzen in
mindestens einer Subgruppe beschränken, denn völlig zusatznutzenfreie
Produkte werden AMNOG-denklogisch auch nicht gebraucht bzw. sollten
nicht den Vorteil eines weiterhin hohen Listenpreises (mit nachgelagerten
Rabatten) gewährt bekommen.

32
Datenverfügbarkeit und Datenschutz
Alle oben genannten Möglichkeiten und darüber hinaus die weiteren
„Financial-Based“-Vertragsmodelle“ (aus Anhang 1) wären unter Nutzung
der pseudonymisierten Morbi-RSA-Satzart 400 (und ggf. 500 und 600),
und zwar ohne jegliche erneute Datenlieferungen bzw. diesbezügliche
Prozesse zwischen Einzelkassen, GKV-Spitzenverband und BVA
abrechenbar.
Alle Daten zur Erkennung von Kombinationstherapien sind in den
Satzarten des Morbi-RSA vorhanden, die entsprechende IT steht beim
GKV-Spitzenverband und dem BVA (mit Einschränkungen seit §303 [neu]
auch beim DIMDI) zur Verfügung.
In der Rahmenvereinbarung nach §130b Abs. 9 zwischen GKV und
Herstellerverbänden müsste auch festgelegt werden, wie eine
Kombinationstherapie „datentechnisch“ detektiert wird. Eine Möglichkeit
wäre hier z.B. eine Überlappung von „definierten-Tagesdosen-basierten
Packungsreichdauern“ der Kombinationspartner zu detektieren, da das
Verordnungsdatum je Patientenpseudonym bekannt ist. Die Regelung der
Details zur Datennutzung ist schon heute in der §130b-
Rahmenvereinbarung vorgesehen, da lt. §130b Abs. 9 dort auch das
Nähere zu Inhalt, Form und Verfahren der jeweils erforderlichen
Auswertung der Daten nach § 217f Absatz 7 und der Übermittlung der
Auswertungsergebnisse an den pharmazeutischen Unternehmer sowie
zur Aufteilung der entstehenden Kosten zu vereinbaren ist.
Der Datenschutz bliebe vollständig gewahrt. Eine Depseudonymisierung
wäre an keiner Stelle erforderlich, die Verordnungen eines einzelnen
Arztes wären auch nicht zentral prüfbar, da sie Satzart 400 keinen
Arztbezug enthält:
33
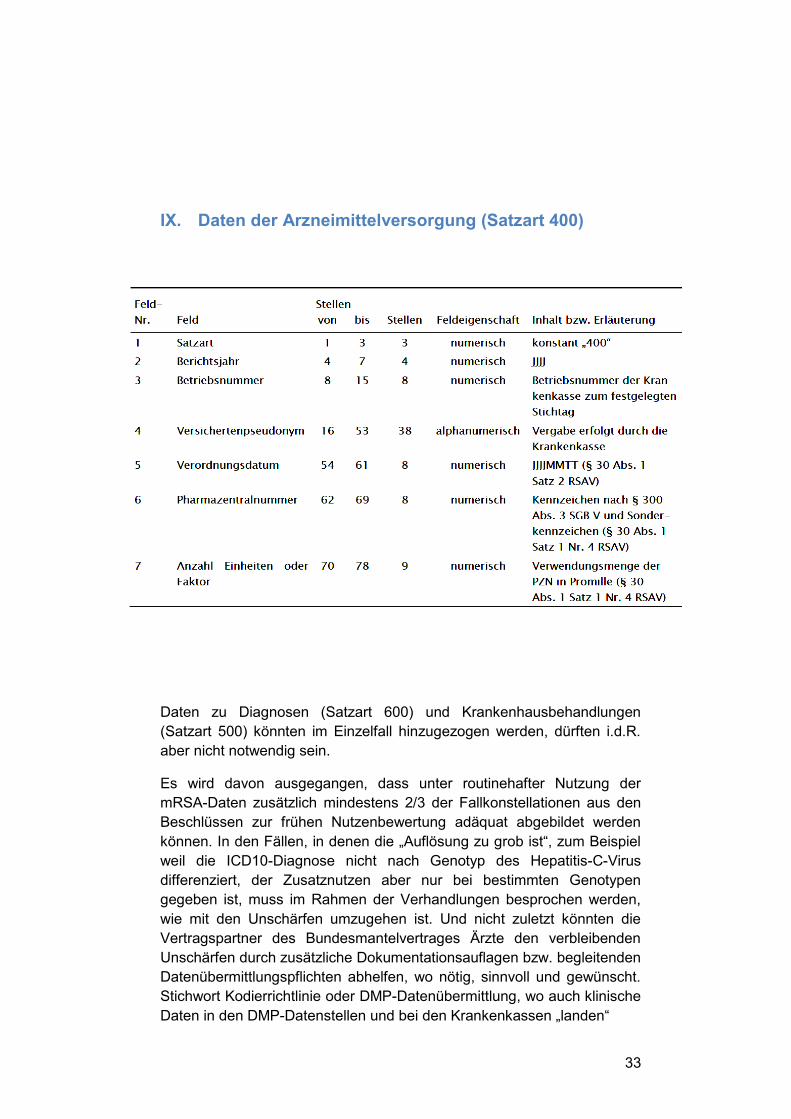
IX. Daten der Arzneimittelversorgung (Satzart 400)
Daten zu Diagnosen (Satzart 600) und Krankenhausbehandlungen
(Satzart 500) könnten im Einzelfall hinzugezogen werden, dürften i.d.R.
aber nicht notwendig sein.
Es wird davon ausgegangen, dass unter routinehafter Nutzung der
mRSA-Daten zusätzlich mindestens 2/3 der Fallkonstellationen aus den
Beschlüssen zur frühen Nutzenbewertung adäquat abgebildet werden
können. In den Fällen, in denen die „Auflösung zu grob ist“, zum Beispiel
weil die ICD10-Diagnose nicht nach Genotyp des Hepatitis-C-Virus
differenziert, der Zusatznutzen aber nur bei bestimmten Genotypen
gegeben ist, muss im Rahmen der Verhandlungen besprochen werden,
wie mit den Unschärfen umzugehen ist. Und nicht zuletzt könnten die
Vertragspartner des Bundesmantelvertrages Ärzte den verbleibenden
Unschärfen durch zusätzliche Dokumentationsauflagen bzw. begleitenden
Datenübermittlungspflichten abhelfen, wo nötig, sinnvoll und gewünscht.
Stichwort Kodierrichtlinie oder DMP-Datenübermittlung, wo auch klinische
Daten in den DMP-Datenstellen und bei den Krankenkassen „landen“

34
Es wird hiermit klar, dass eine Nutzung der mRSA-Daten für Zwecke der
Rückabwicklung von Preisabschlägen - ergänzend zum §130b-
Erstattungsbetrag - kein Datenschutzproblem darstellen würde. Es geht
hier auch um keinerlei Änderung an den Mechanismen des Morbi-
RSA, lediglich Teile der Datengrundlage des RSA würden einer
zusätzlichen Nutzung verfügbar gemacht.

35
X. Handlungsempfehlungen für die Politik
Anspruch dieses Lösungsvorschlages ist, den Budget-Impact derzeitiger
und zukünftiger Kombinationstherapien zu begrenzen und dabei den
Interessenausgleich zwischen Solidargemeinschaft und Herstellern zu
sichern, , vorhandene Datenstrukturen und –techniken nutzbar zu machen
und allenfalls wenige neue Prozesse zwischen den Beteiligten
einzuführen und insoweit das AMNOG problemorientiert
weiterzuentwickeln .
Wenn den obigen Vorschlägen gefolgt wird, sind wenige Ergänzungen im
SGB V erfoderlich:
1. Freie nicht explizit zugelasse Kombinationen, müssen vom GBA
dahingehend bewertet werden, ob sie grundsätzlich
verordnungsfähig sind oder ob an verordnende Ärzte weitergehende
Qualitätsanforderungen (Prozess-, Struktur-, Dokumentations-
qualität) zu stellen sind. Dazu bedarf es im Rahmen der
Arzneimittelrichtlinie nach §92 Abs. 1 Satz 6 einer
Ermächtigungsnorm in §92 Abs. 2b (neu).Zur Unterstützung des
Schaffens von Transparenz zur Ergebnisqualität in der Onkologie
sollte über eine Konkretisierung des §65c SGB V nachgedacht
werden, sodass die Professionalisierung und Standardisierung der
klinischen Krebsregistrierung unterstützt wird.
2. Wenn – wie hier als als Kern der Überlegungen vorgeschlagen - die
Solidargemeinschaft mit geringeren Kosten als der Summe der
Preise der Monotherapien für den Fall des nicht explizit
zugelassenen zeitgleichen Einsatzes in Kombination ) belastet
werden soll (nachgelagerter Rabatt auf den §130b-
Erstattungsbetrag bzw. den Preis des Bestandsmarktarzneimittels)
sind noch folgende Änderungen erforderlich:
a. Der Verwendungszweck der Daten nach §267 SGB V muss
durch eine Streichung in §217f (7) SGB V maßvoll erweitert
werden, sodass die Daten pseudonymisiert genutzt werden
können, um Verläufe zu detektieren.

36
b. In §130b müssen „Kombinationsrabattverhandlungen“
ermöglicht und mit einem Konfliktlösungsmechanismus
versehen werden-.
3. Sollte die (gegenüber der Monotherapie) veränderte Preisbildung
von Kombinationen , also die Änderungen in §92b, politisch nicht
gewünscht oder nicht durchsetzbar sein, ist auch die
Verfügbarmachung der Daten nach § 268 Absatz 3 Satz 14 in
Verbindung mit Satz 1 Nr. 1 bis 7 für die Zwecke des §130b (und
damit implizit die Ermöglichung zentraler, aber nachgelagerter
Rabatte, die den initialen Erstattungsbetrag ergänzen) immer noch
ein Wert an sich. Minimallösung wäre demnach die alleinige
Anpassung des §217f Absatz 7.

37
Zusammenfassung
Zur Lösung der meisten Patientenprobleme bedienen sich die Ärzte seit
Hippokrates der Möglichkeit, unterschiedliche Wirkprinzipien miteinander
zu kombinieren.
In den zurückliegenden Jahren hat sowohl der Trend, Fixkombinationen
bis zur Zulassungsreife zu entwickelnm als auch freie Kombinationen
unterschiedlich wirkender Arzneimittel in breitem Umfang
einzusetzen,ganz erheblich zugenommen.
Vornehmlich in der Behandlung viraler Erkrankungen (HIV, Hepatitis C)
und vor allem in der Onkologie.
Dieser unumkehrbare Trend in der Medizin führt nicht zwangsläufig und in
jedem Fall zu einem verbesserten Therapieergebnis. Und hat immer zur
Folge, dass sich die Behandlungen erheblich verteuern (add on!).
Im vorliegenden Diskussionspapier wird eine aktuelle Situationsanalyse
vorgenommen und es werden konkrete Vorschläge präsentiert, wie man
unter Fortentwicklung der gesetzlichen Vorgaben durch geringe
Änderungen im SGB V die Problematik einer Lösung näherbringen kann.
Ganz verkürzt dargestellt geht es darum, die Versorgungsqualität mit
freien, nicht zugelassenen Kombinationsbehandlungen zu verbessern und
den Kostenauftrieb im Kombinationsfall durch nachgelagerte Rabatte der
Hersteller an die Krankenkassen einzubremsen, ohne das Ergebnis der
Erstattungspreisverhandlung für die Monotherapie gemäß AMNOG zu
tangieren.
Der ohnehin bereits jetzt bestehende große politische Handlungsbedarf
insbesondere in der Onkologie dürfte sich in den nächsten Jahren noch
deutlich verschärfen, wenn die Krebsimmuntherapeutika weitere
Fortschritte präsentieren werden. Da dieses Wirkprinzip in der Regel
zusätzlich zu laufenden Behandlungen kombiniert werden wird.
Die Autoren dieses Diskussionspapiers erwarten in Fachkreisen einen
lebhaften Diskurs. Konstruktive Vorschläge zur Weiterentwicklung sind
ausdrücklich erwünscht.
(Postadresse: Dr. Jürgen Bausch, Eichgrabenstraße 17, 63628 Bad
Soden-Salmünster)

















