
WS 2010/11 Prof. Lindhorst Organische Chemie für...
47
WS 2010/11 Prof. Lindhorst Organische Chemie für Nebenfachstudierende Audimax, Hörsaal G Einzeltermin am 3.2.2011, 9:15 - 10:00, OHP5 vom 14.12.2010 bis zum 3.2.2011 MNF-chem0001 Allgemeine Chemie II (Agarwiss. u. Ökotroph.) und Biol-prop300 Allgemeine Chemie II Di, 8:15 - 10:00, Do, 9:15 - 10:00 Allgemeine Chemie II (Humanmedizin, Zahnmedizin) vom 14.12.2010 bis zum 3.2.2011; Di, 8:15 - 9:00 Do, 9:15 - 10:00
-
Upload
nguyenthuan -
Category
Documents
-
view
214 -
download
0
Transcript of WS 2010/11 Prof. Lindhorst Organische Chemie für...

WS 2010/11
Prof. Lindhorst
Organische Chemie für Nebenfachstudierende
Audimax, Hörsaal G Einzeltermin am 3.2.2011, 9:15 - 10:00, OHP5 vom 14.12.2010 bis zum 3.2.2011
MNF-chem0001 Allgemeine Chemie II (Agarwiss. u. Ökotroph.) und Biol-prop300 Allgemeine Chemie II Di, 8:15 - 10:00,
Do, 9:15 - 10:00 Allgemeine Chemie II (Humanmedizin, Zahnmedizin) vom 14.12.2010 bis zum 3.2.2011; Di, 8:15 - 9:00
Do, 9:15 - 10:00

Verlaufsplan
1 14.12.10
Beginn
di 1. Teil • Kohlenwasserstoffe
• Homologe Reihen und Nomenklatur: Alkane, Alkene, Alkine
• Bindung und Molekülgeometrie
2. Teil Vertiefung: 3D-Zeichenweisen, Tetraeder-struktur, cis-trans-Isomerie, π- und σ-Bindungen, Elektronendelokalisation, Aromatizität
2 16.12.10
do • Funktionelle Gruppen
• Stoffklassen
• Isomerie an Beispielen
3 21.12.10
di 1. Teil • Physikalische Eigenschaften org. Moleküle und intermolekulare WW
• Polarisierung von Bindungen (Elektronegativität)
• Substituenteneffekte und Reaktivität (+ und –I; + und –M-Effekte)
• Einfluss funktioneller Gruppen auf Stoffeigenschaften: Elektrophilie und Nukleophilie
2. Teil Vertiefung: Prinzipielle Reaktionsmechanismen: Substitution, Addition, Eliminierung
WEIHNACHTSPAUSE 4 11.01.11
di 1. Teil • Organische Säuren
• Organische Basen
• Aminosäuren, Peptidbindung
2. Teil Vertiefung: Mono-, Oligo-, Polymere; Naturstoffe
5 13.01.11
do • Chiralität; Enantiomere, Diastereomere
• Optische Aktivität

6 18.01.11
di 1. Teil • Carbonsäurederivate, Acylierungsvermögen
• Säurekatalysierte Veresterung; Verseifung
• Natürliche Ester: Aromen, Lipide, Amphiphilie, Detergentien, Micellen
2. Teil Vertiefung: Substanzbeispiele, Zellmembran, Überstrukturen
7 20.01.11
do • Carbonylverbindungen: Aldehyde und Ketone
• grundlegende Reaktivität, CH-Acidität
• Nukleophile Addition an C=O-Doppelbindung
8 25.01.11
di 1. Teil • Hydroxycarbonylverbindungen; intramolekulare Halbacetalbildung
• Kohlenhydrate
• Glycosidische Bindung
2. Teil Vertiefung: Saccharide
9 27.01.11
do • Peptide, Proteine, Enzyme, Katalyse
• Biosynthese und molekulare Diversität
10 01.02.11
di 1. Teil • Oligonukleotide, DNA, RNA
• Translation
2. Teil Vertiefung: Chemische Synthese von Polymeren und Biopolymeren
11 03.02.11
do • Molekulare Wechselwirkungen
• Wirkstoffe, Wirkstoffsuche, kombinatorische Chemie
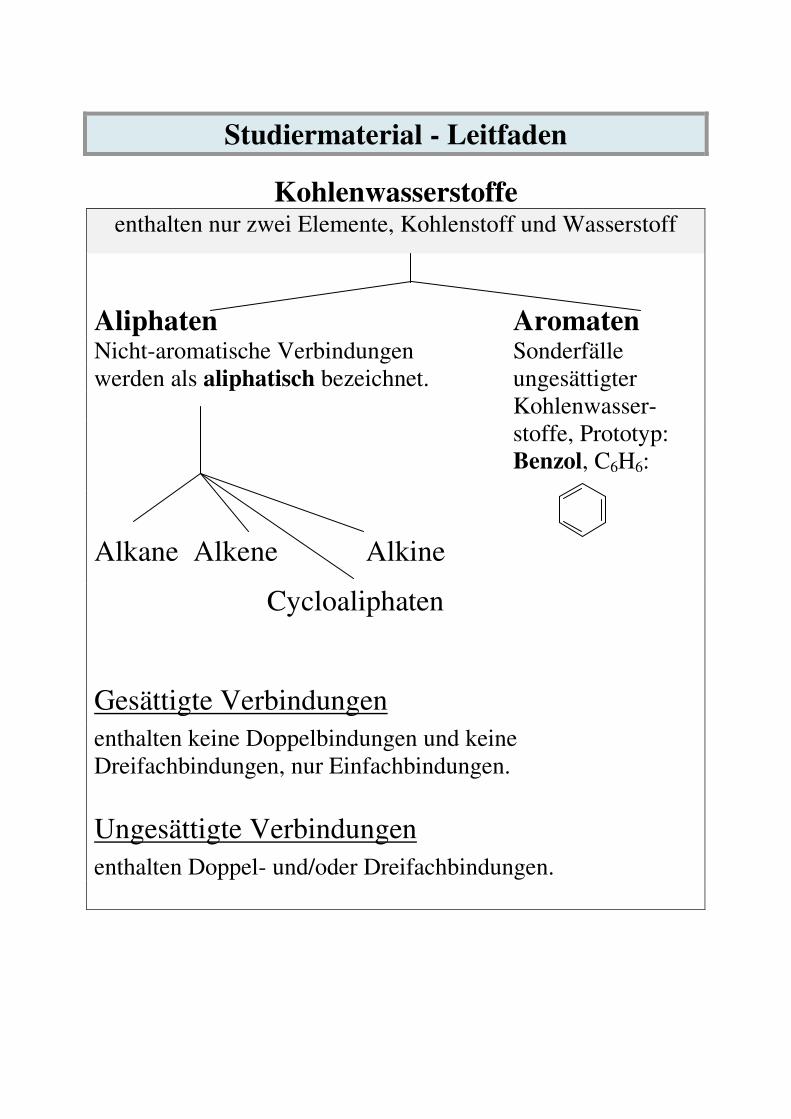
Studiermaterial - Leitfaden
Kohlenwasserstoffe enthalten nur zwei Elemente, Kohlenstoff und Wasserstoff
Aliphaten Aromaten Nicht-aromatische Verbindungen Sonderfälle werden als aliphatisch bezeichnet. ungesättigter Kohlenwasser- stoffe, Prototyp: Benzol, C6H6:
Alkane Alkene Alkine
Cycloaliphaten
Gesättigte Verbindungen enthalten keine Doppelbindungen und keine Dreifachbindungen, nur Einfachbindungen.
Ungesättigte Verbindungen enthalten Doppel- und/oder Dreifachbindungen.
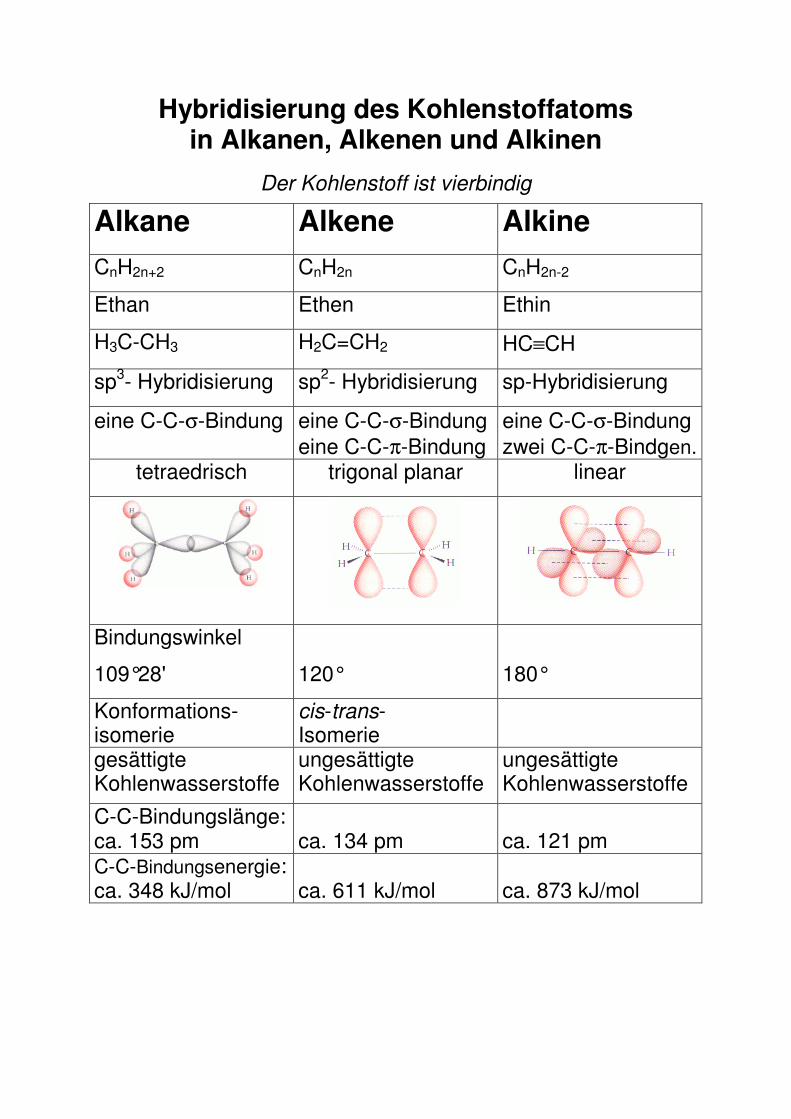
Hybridisierung des Kohlenstoffatoms in Alkanen, Alkenen und Alkinen
Der Kohlenstoff ist vierbindig
Alkane Alkene Alkine
CnH2n+2 CnH2n CnH2n-2
Ethan Ethen Ethin
H3C-CH3 H2C=CH2 HC≡CH
sp3- Hybridisierung sp2- Hybridisierung sp-Hybridisierung
eine C-C-σ-Bindung eine C-C-σ-Bindung eine C-C-π-Bindung
eine C-C-σ-Bindung zwei C-C-π-Bindgen.
tetraedrisch trigonal planar linear
Bindungswinkel
109°28'
120°
180°
Konformations-isomerie
cis-trans- Isomerie
gesättigte Kohlenwasserstoffe
ungesättigte Kohlenwasserstoffe
ungesättigte Kohlenwasserstoffe
C-C-Bindungslänge: ca. 153 pm
ca. 134 pm
ca. 121 pm
C-C-Bindungsenergie: ca. 348 kJ/mol
ca. 611 kJ/mol
ca. 873 kJ/mol

Molekül- bzw. Formeldarstellungen
am Beispiel des Ethans
Summenformel: C2H6
ausführliche Formeldarstellungen: H3C-CH3
Linienformel: Strichenden (bzw. Ecken) bedeuten C-Atome, die Wasserstoffatome, die für die Absättigung der Kohlenstoffvalenzen erforderlich sind, werden nicht gezeichnet (Heteroatome werden eingetragen).
Dreidimensionale Formeldarstellungen: ♦ Keilstrichformel: fette Bindungsstriche weisen aus der Papierebene heraus, gestrichelte weisen hinter die Papierbene
♦ Sägebockformel:
Newman-Projektion Eine C-C-Bindung wird senkrecht zur Papierbene gelegt, das vordere C-Atom wird als Punkt, das hintere als Kreis dargestellt (bzw. das hintere ist verdeckt und der Kreis symbolisiert die Elektronendichte der σ-Bindung).
→→→→ Konformationsanalyse
C C
H
HH
H
HH
C C
H
H
H
H
H
H
H
H
H H
H H
H
H H
H
HH

Die homologe Reihe der Alkane
Homologe sind strukturell sehr eng verwandte Substanzen. Die Verbindungen der homologen Reihe der Alkane besitzen die allgemeine Summenformel CnH
2n+2 und unterscheiden sich jeweils durch eine Methylengruppe (eine CH2-Einheit).
Methan CH4
Ethan H3C-CH3
Propan H3C-CH2-CH3
Butan H3C-(CH2)2-CH3
Pentan H3C-(CH2)3-CH3
Hexan H3C-(CH2)4-CH3
Heptan H3C-(CH2)5-CH3
Octan H3C-(CH2)6-CH3
Nonan H3C-(CH2)7-CH3
Decan H3C-(CH2)8-CH3
Undecan H3C-(CH2)9-CH3
Dodecan H3C-(CH2)10-CH3
Tridecan H3C-(CH2)11 -CH3
Tetradecan H3C-(CH2)12-CH3
Pentadecan H3C-(CH2)13-CH3
Hexadecan H3C-(CH2)14-CH3
Heptadecan H3C-(CH2)15-CH3
Octadecan H3C-(CH2)16-CH3
Nonadecan H3C-(CH2)17-CH3
Eicosan H3C-(CH2)18-CH3
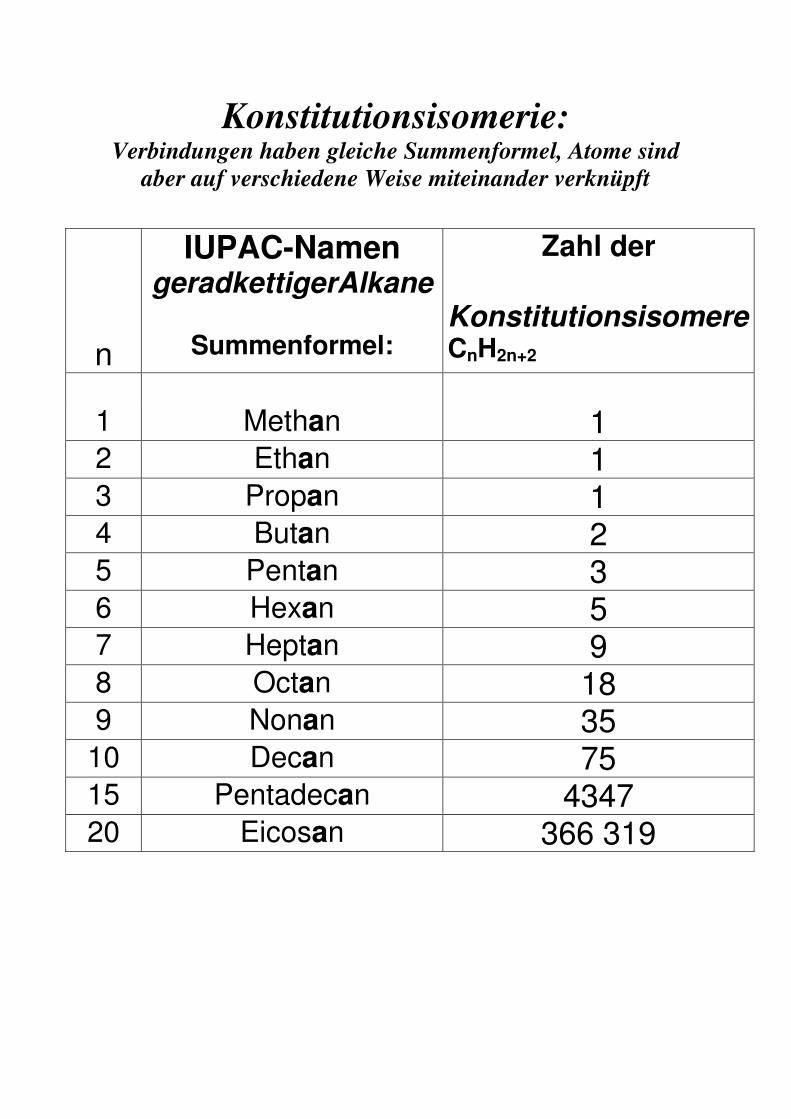
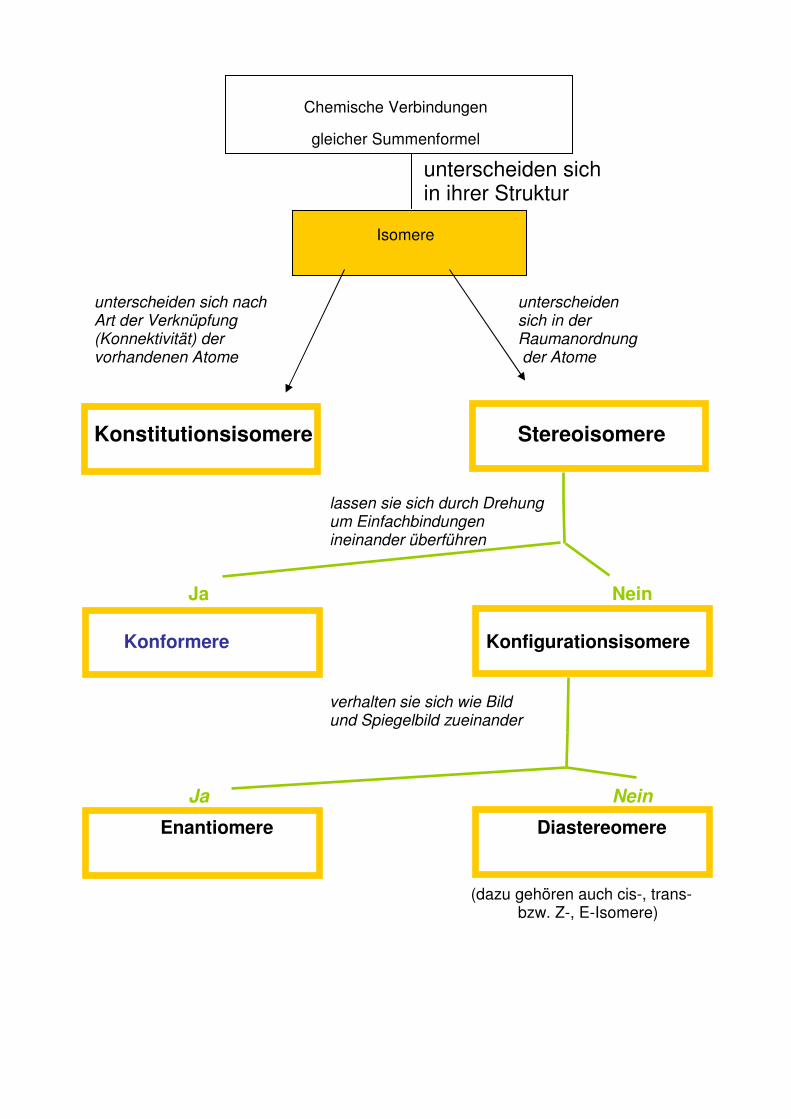
Konstitutionsisomerie: Verbindungen haben gleiche Summenformel, Atome sind
aber auf verschiedene Weise miteinander verknüpft
IUPAC-Namen geradkettigerAlkane
Zahl der
n
Summenformel:
Konstitutionsisomere CnH2n+2
1 Methan 1 2 Ethan 1 3 Propan 1 4 Butan 2 5 Pentan 3 6 Hexan 5 7 Heptan 9 8 Octan 18 9 Nonan 35
10 Decan 75 15 Pentadecan 4347 20 Eicosan 366 319
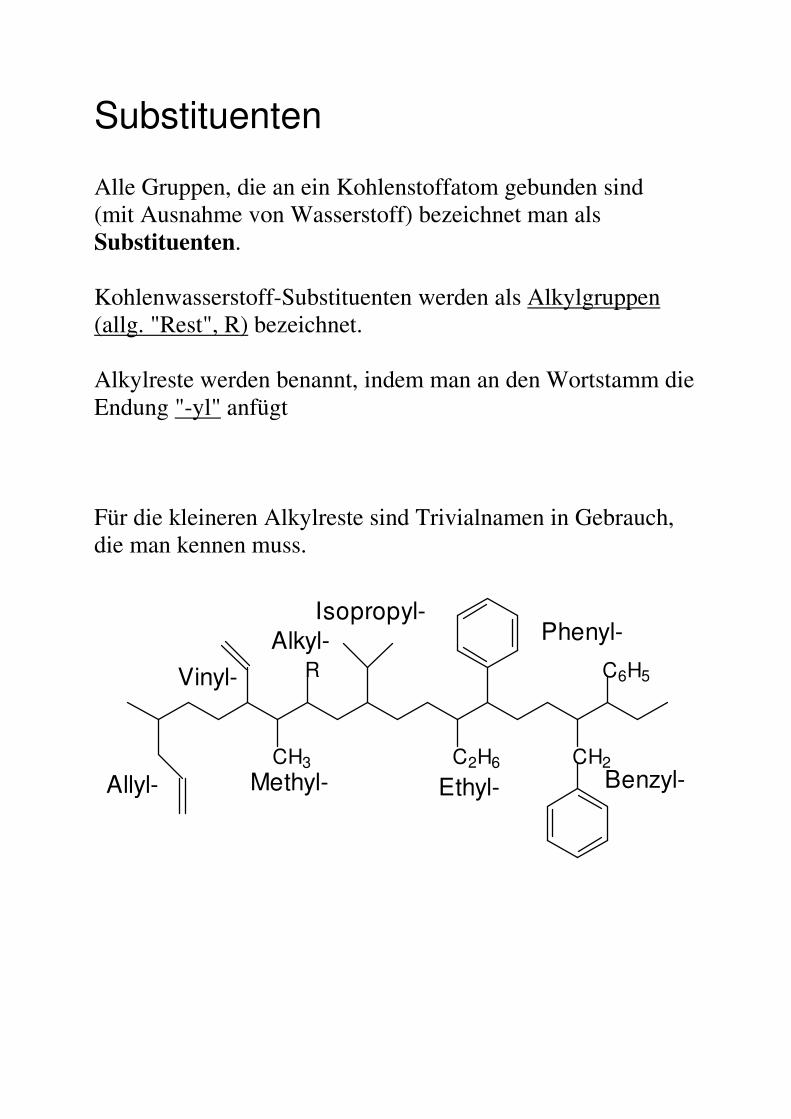
Substituenten Alle Gruppen, die an ein Kohlenstoffatom gebunden sind (mit Ausnahme von Wasserstoff) bezeichnet man als Substituenten. Kohlenwasserstoff-Substituenten werden als Alkylgruppen (allg. "Rest", R) bezeichnet. Alkylreste werden benannt, indem man an den Wortstamm die Endung "-yl" anfügt Für die kleineren Alkylreste sind Trivialnamen in Gebrauch, die man kennen muss.
CH3
R
C2H6
C6H5
CH2
Methyl-
Alkyl-Isopropyl-
Phenyl-
Benzyl-
Vinyl-
Allyl- Ethyl-

Nomenklatur: Benennung von Molekülen nach IUPAC (=International Union of Pure and Applied Chemistry)
Einige Trivialnamen bleiben offiziell in Gebrauch
PRÄFIX – STAMMNAME – SUFFIX
Substituenten (und deren entsprechend bezeichnet Anzahl und Position) in Stammalkan Hauptfunktion alphabetischer Reihenfolge
� Der Stammname ist meist lateinischen oder griechischen Ursprungs und gibt die Anzahl der Kohlenstoffatome der Kette an. Zur Bennennung einer Verbindung sucht man die längste lineare Kette im Molekül, die Bezeichnung für das entsprechende Stammalkan liegt dann dem Namen der Verbindung zu Grunde. (siehe homologe Reihe der Alkane).
� Besitzt eine Verbindung mehrere Ketten gleicher Länge, wird diejenige zu Grunde gelegt, welche die meisten Substituenten enthält.
� Die Namen der einzelnen Substituenten werden bestimmt, mit dem Suffix –yl versehen und in alphabetischer Reihenfolge als Präfixe vor den Namen geordnet [praefigere (lat.) = vorne anheften]
� Die längste Kette wird von dem Ende her nummeriert, das einem der Substituenten am nächsten ist. Wenn zwei Substituenten vom jeweiligen Kettenende gleich weit entfernt sind, dann ist derjenige für die Nummerierung relevant, dessen Anfangsbuch-stabe im Alphabet vorne steht.
� Der IUPAC-Name der Verbindung ergibt sich nun, indem man zunächst die Namen der Substituenten in alphabetischer Reihenfolge mit der Nummer des C-Atoms, an das er gebunden ist, auflistet und dann den Stammnamen zufügt.
� Treten Substituenten mehrfach auf, werden die Präfixe Di-, Tri-, Tetra- usw. verwendet. Bei cyclischen Verbindungen wird das Präfix Cyclo- verwendet. Diese Präfixe werden bei der alphabetischen Reihung nicht berücksichtigt. Verzweigte Seitenketten: dort trägt dasjenige C-Atom, die Nummer 1, das mit der Hauptkette verbunden ist.
Der Stammname enthält eine Endung [Suffix von suffigere (lat.) = anhängen], die auf die Stoffklasse hinweist um die es sich handelt: -an für Alkane -en für Alkene -in für Alkine -ol für Alkohole usw.
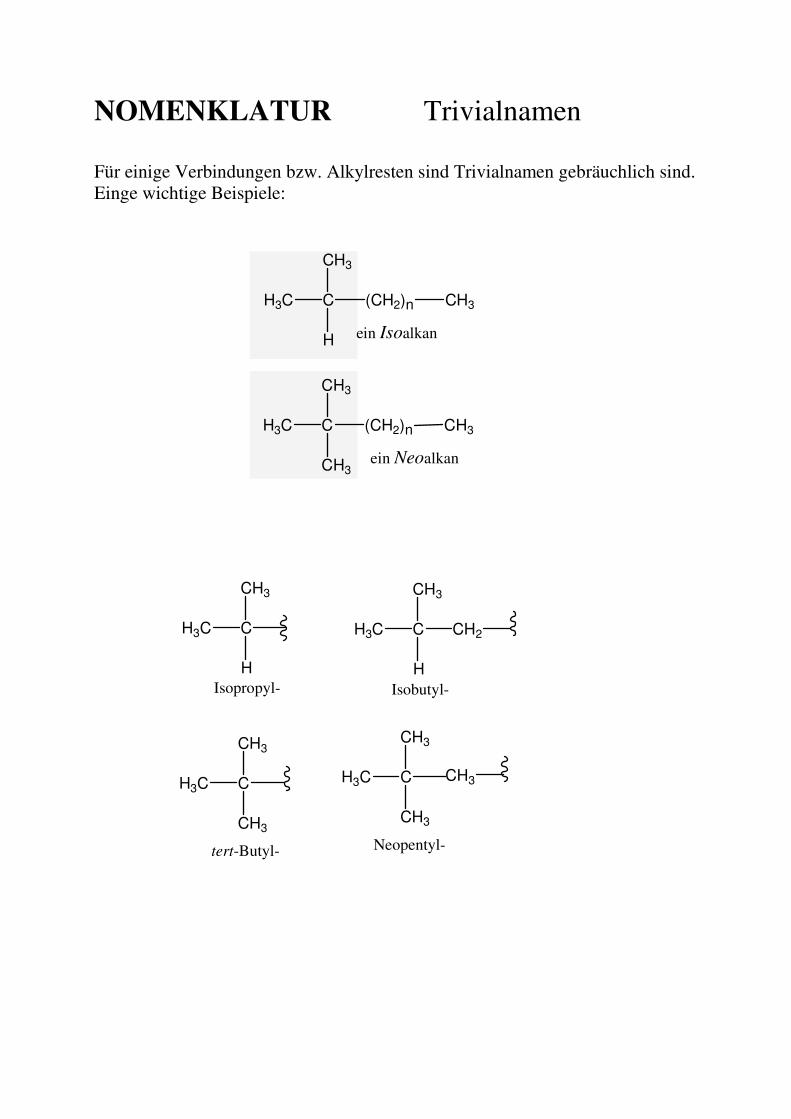
NOMENKLATUR Trivialnamen
Für einige Verbindungen bzw. Alkylresten sind Trivialnamen gebräuchlich sind. Einge wichtige Beispiele:
H3C C
CH3
H
(CH2)n CH3
H3C C
CH3
CH3
(CH2)n CH3
ein Isoalkan
ein Neoalkan
H3C C
CH3
H
H3C C
CH3
CH3
Isopropyl-
H3C C
CH3
H
CH2
Isobutyl-
tert-Butyl-
H3C C
CH3
CH3
Neopentyl-
CH3
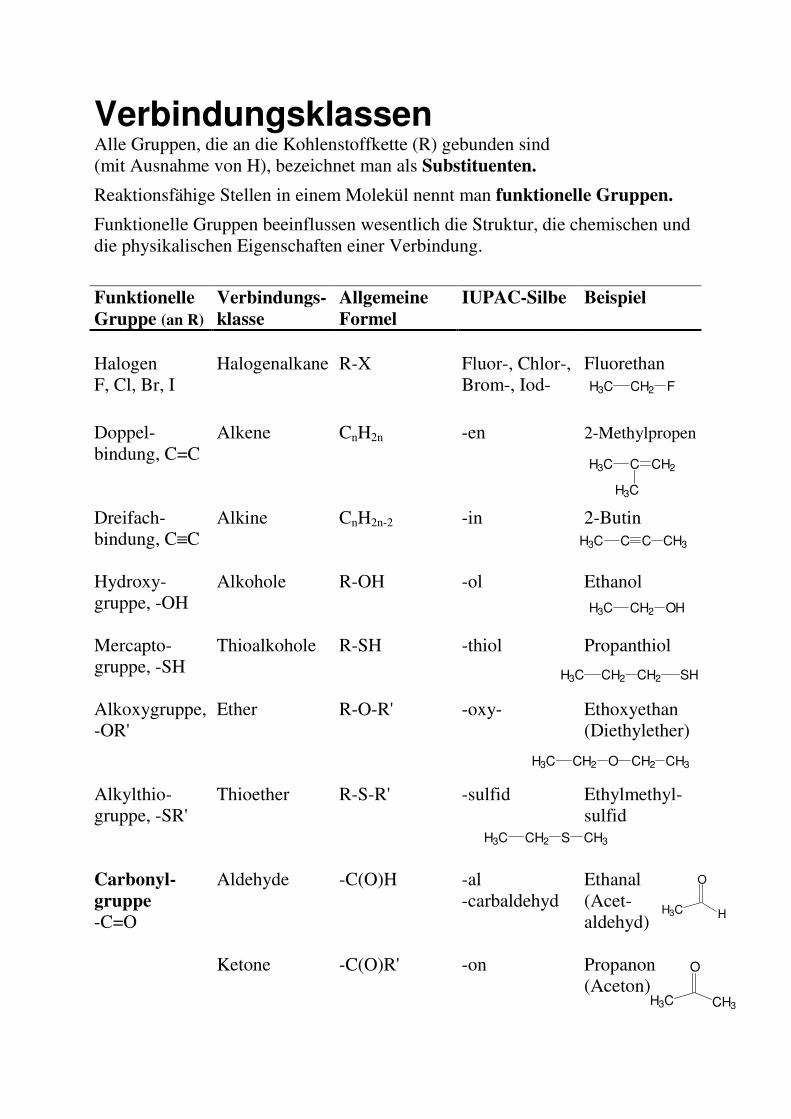
Verbindungsklassen Alle Gruppen, die an die Kohlenstoffkette (R) gebunden sind (mit Ausnahme von H), bezeichnet man als Substituenten.
Reaktionsfähige Stellen in einem Molekül nennt man funktionelle Gruppen.
Funktionelle Gruppen beeinflussen wesentlich die Struktur, die chemischen und die physikalischen Eigenschaften einer Verbindung.
Funktionelle
Gruppe (an R)
Verbindungs-
klasse
Allgemeine
Formel
IUPAC-Silbe Beispiel
Halogen F, Cl, Br, I
Halogenalkane R-X Fluor-, Chlor-, Brom-, Iod-
Fluorethan H3C CH2 F
Doppel-bindung, C=C
Alkene CnH2n -en 2-Methylpropen
Dreifach-bindung, C≡C
Alkine CnH2n-2 -in 2-Butin
Hydroxy-gruppe, -OH
Alkohole R-OH -ol Ethanol
Mercapto-gruppe, -SH
Thioalkohole R-SH -thiol Propanthiol
Alkoxygruppe, -OR'
Ether R-O-R' -oxy- Ethoxyethan (Diethylether)
Alkylthio-gruppe, -SR'
Thioether R-S-R' -sulfid Ethylmethyl-sulfid
Carbonyl-
gruppe
-C=O
Aldehyde -C(O)H -al -carbaldehyd
Ethanal (Acet- aldehyd)
Ketone -C(O)R' -on Propanon (Aceton)
H3C C CH2
H3C
H3C C C CH3
H3C CH2 OH
H3C CH2 CH2 SH
H3C CH2 O CH2 CH3
H3C CH2 S CH3
H3C
O
H
H3C
O
CH3
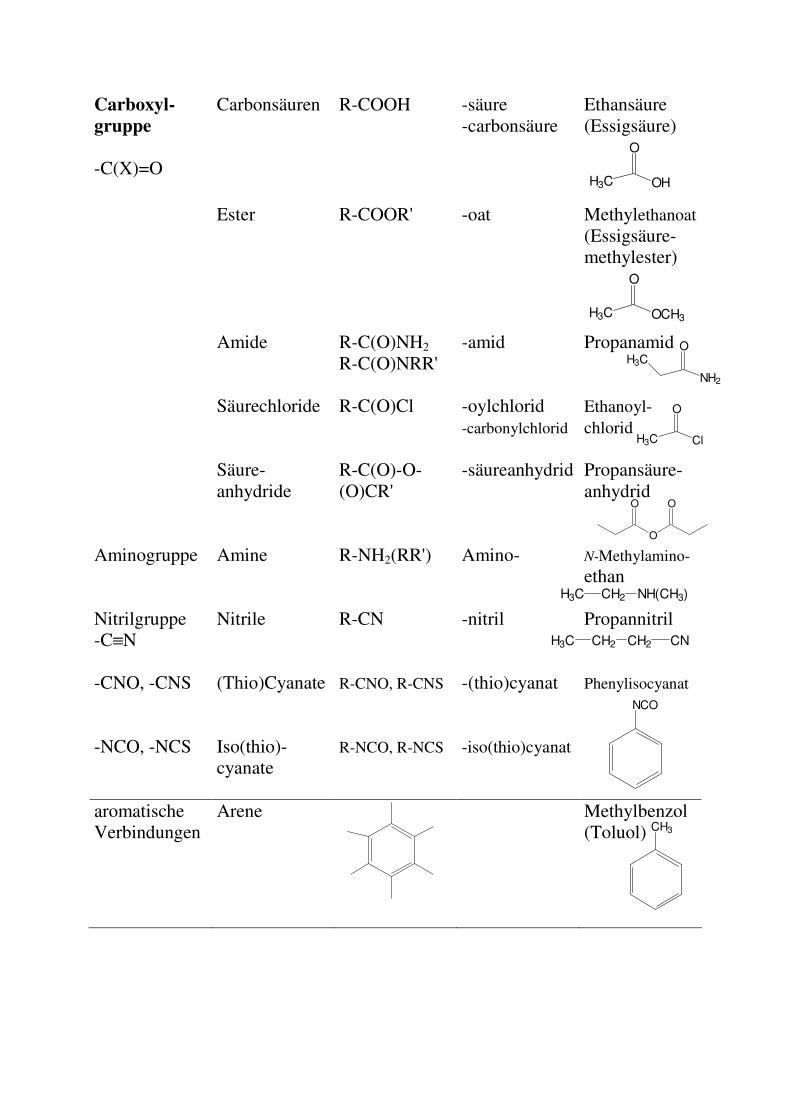
Carboxyl-
gruppe
-C(X)=O
Carbonsäuren
R-COOH -säure -carbonsäure
Ethansäure (Essigsäure)
H3C
O
OH
Ester
R-COOR' -oat Methylethanoat (Essigsäure-methylester)
Amide
R-C(O)NH2
R-C(O)NRR' -amid Propanamid
Säurechloride R-C(O)Cl -oylchlorid -carbonylchlorid
Ethanoyl-chlorid
Säure-anhydride
R-C(O)-O-(O)CR'
-säureanhydrid Propansäure-anhydrid
Aminogruppe Amine R-NH2(RR') Amino- N-Methylamino-
ethan
Nitrilgruppe -C≡N
Nitrile R-CN -nitril Propannitril
-CNO, -CNS (Thio)Cyanate R-CNO, R-CNS -(thio)cyanat Phenylisocyanat
-NCO, -NCS Iso(thio)-cyanate
R-NCO, R-NCS -iso(thio)cyanat
aromatische Verbindungen
Arene Methylbenzol (Toluol)
O
NH2
H3C
O
O O
H3C CH2 CH2 CN
NCO
H3C CH2 NH(CH3)
CH3
H3C
O
OCH3
H3C
O
Cl
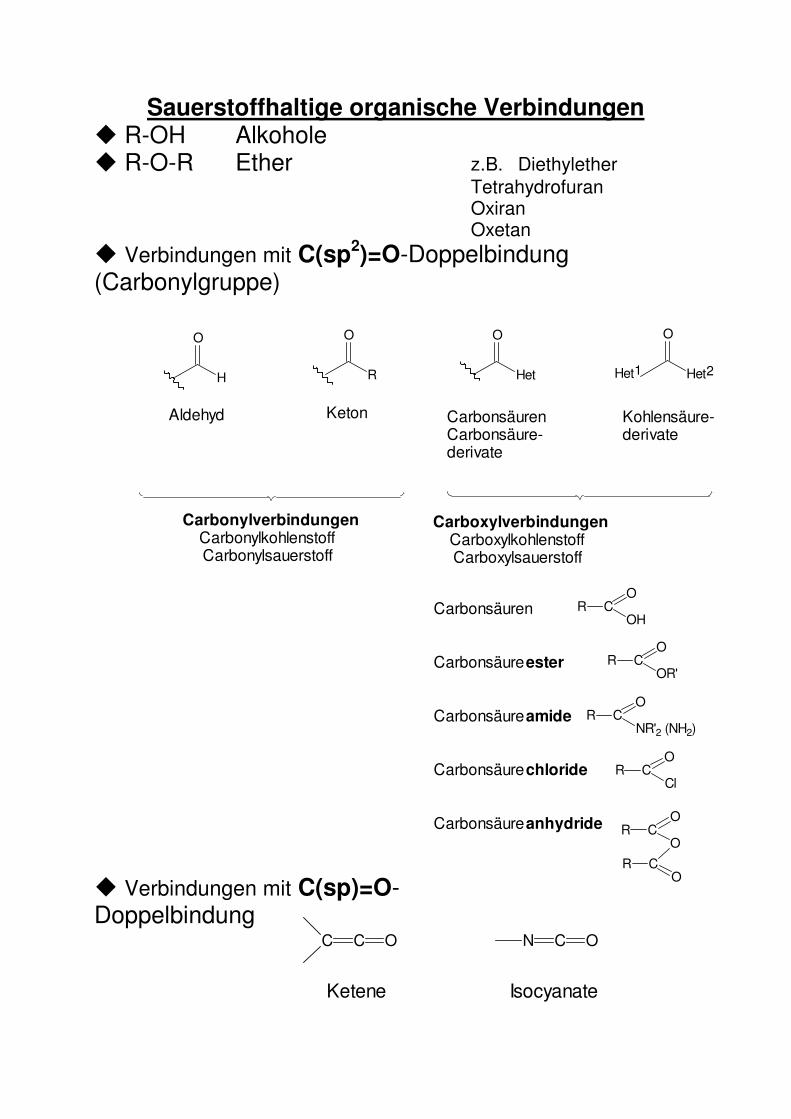
Sauerstoffhaltige organische Verbindungen � R-OH Alkohole � R-O-R Ether z.B. Diethylether Tetrahydrofuran Oxiran Oxetan � Verbindungen mit C(sp2)=O-Doppelbindung (Carbonylgruppe) � Verbindungen mit C(sp)=O-Doppelbindung
C C O
Ketene Isocyanate
N C O
O
H
O
R Het1
O
Het2
O
Het
Aldehyd Keton CarbonsäurenCarbonsäure-derivate
Kohlensäure-derivate
CarbonylverbindungenCarbonylkohlenstoffCarbonylsauerstoff
CarboxylverbindungenCarboxylkohlenstoffCarboxylsauerstoff
Carbonsäuren
Carbonsäureester
Carbonsäureamide
Carbonsäurechloride
Carbonsäureanhydride
R CO
OH
R CO
Cl
R CO
NR'2 (NH2)
R CO
OR'
R CO
O
OCR

Physikalische Eigenschaften von Alkanen
Alkanstrukturen sind regelmäßig gebaut und nehmen unter anderem eine Zickzackanordnung ein.
Alkanmoleküle sind unpolar (die Elektronen sind gleichmäßig im Molekül verteilt) und werden nur von den schwachen van der Waals-
Kräften zusammengehalten, deren Energie mit der 6. Potenz des Molekülabstandes abnimmt. Van der Waals-Kräfte wirken zwischen den Moleküloberflächen und sind daher um so stärker, je größer das Molekül ist. Also nehmen die Schmelz- und Siedepunkte und die Dichte der Alkane mit steigender Molmasse aufgrund der zunehmenden Anziehungskräfte zwischen den Molekülen zu. Bei verzweigten Alkanen sind wegen der kleineren Oberfläche die van der Waals-Kräfte geringer als bei geradkettigen Isomeren. Die Höhe der Schmelzpunkte wird auch von der Packungsdichte im kristallinen Zustand beeinflusst. Alkane mit geradzahliger Kohlenstoffzahl sind besser gepackt und schmelzen daher relativ etwas höher als Alkane mit ungerader C-Zahl.
H
C
C
C
C
C
C
H
H
H H HH H H
H HH HH

Elektronegativität ist eine Bezeichnung für die Fähigkeit der an chemischen Bindungen beteiligten Atome, gemeinsame Elektronen von benachbarten Atomen innerhalb des Moleküls unterschiedlich stark anzuziehen.
Die Elektronegativität bestimmt wesentlich den Charakter der Bindung!
Der Begriff Elektronegativität geht auf Pauling zurück, der 1932 die erste empirische Elektronegativitäts-Skala aufstellte und später etwas modifizierte. An den verschiedenartigen Definitionen und der willkürlichen Wahl der Skala erkennt man, dass die Elektronegativität keine wohl definierte physikalische Größe ist.
Die Elektronegativität eines Atoms ist um so größer, je höher die Kernladung ist und je stärker sie über die Elektronenhülle hinaus wirken kann. Die Elektronegativität nimmt im Periodensystem von links nach rechts innerhalb der Periode und normalerweise von unten nach oben innerhalb einer Gruppe zu. Also:
Im Periodensystem stehen die elektronegativsten Elemente oben und rechts.
Von der Elektronegativität ist die Elektronenaffinität zu unterscheiden, die sich auf die Aufnahme eines Elektrons durch ein freies, ungebundenes Atom oder ein Molekül bezieht.

Induktive und mesomere Effekte
Durch die unterschiedliche Elektronegativität der Elemente sind viele Bindungen polarisiert.
Die Bindungspartner tragen dadurch Partialladungen: δδδδ+und δδδδ-
♦ Diese wirken sich auch auf weitere Bindungen polarisierend
aus, mit zunehmendem Abstand der betrachteten Bindung von dem/r polarisierenden Atom/Gruppe in immer geringerem Maße:
=> Induktiver (I-) Effekt Man unterscheidet Substituenten mit +I-Effekt: erhöhen die Elektronendichte am substituierten C-Atom; (gebundenes Atom hat geringere Elektronegativität als C-Atom) -I-Effekt: erniedrigen die Elektronendichte am substituierten C-Atom; (gebundenes Atom hat höhere Elektronegativität als C-Atom) Kohlenstoffreste, die stark elektronegative Elemente tragen, wie eine CCl3- oder eine CF3-Gruppe haben -I-Effekt! Alkylsubstituenten wirken einen +-I-Effekt aus! => Hyperkonjugation

♦ Ein zweiter Effekt kommt bei sp2-hybridisierten C-Atomen zum
Tragen. Er kommt durch Konjugation zwischen π-Systemen oder π-Systemen mit freien Elektronenpaaren zustande und kann durch mesomere Grenzstrukturen (Resonanzstrukturen) beschrieben werden: => Mesomerer (M-) Effekt
Je nachdem ob ein Substituent durch Mesomerie
Elektronen aufnehmen oder abgeben kann, spricht man von -M- Effekt oder +M-Effekt Auch hier beschreibt das Vorzeichen (+) die Erhöhung, (-) die Erniedrigung der Elektronendichte des Zentrums an dem ein M-Substituent gebunden ist. Substituenten mit freien Elektronenpaaren sind +M-Substituenten, Substituenten mit π-Systemen, vor allem, wenn elektronegative Elemente beteiligt sind, besitzen meist -M-Charakter.

Dissoziationsenergien Wenn sich Atome zu Molekülen vereinigen, wird Energie frei. Zur Spaltung eines Moleküls bzw. einer Bindung muss eine äquivalente Menge Energie aufgebracht werden. Die Energie, die verbraucht oder freigesetzt wird, wenn eine Bindung gespalten oder gebildet wird, bezeichnet man als Dissoziationsenergie DH0
Eine Bindung kann homolytisch oder heterolytisch gespalten werden. Dissoziationsenergien beziehen sich
auf homolytische Spaltungen!
homolytische Spaltung: A-B � A. + B. heterolytische Spaltung: A-B � A- + :B+
Teilchen mit ungepaarten Elektronen am C:
kohlenstoffzentrierte Radikale R.
sp2-hybridisiert Teilchen mit positiver Ladung am Kohlenstoff:
Carbeniumionen RC+
sp2-hybridisiert Teilchen mit negativer Ladung am Kohlenstoff:
Carbanionen RC-
(sp2) sp3-hybridisiert Je stabiler ein Radikal, umso kleiner ist seine Dissoziationsenergie.

Die Stärke von C-H- und C-C-Bindungen ist von der Molekülstruktur abhängig.
Dissoziationsenergien einiger Alkane
Verbindung DH0 [kJ/mol] Verbindung DH0 [kJ/mol]
H3C-H 440 H3C-CH3 377 H5C2-H 410 H5C2-CH3 360
(H3C)2HC-H 396 H5C2-C2H5 343 (H3C)3C-H 389 (CH3)C-CH3 352
(CH3)C-C(CH3)3 301
Stabilität von Radikalen und Carbeniumionen
CH3-Radikal/Kation < primäres < sekundäres < tertiäres
STABILITÄT ⇒
⇐ENERGIEGEHALT ⇐DISSOZIATIONSENERGIE
Begründung der Stabilitätsreihung durch HYPERKONJUGATION Hierunter versteht man die Wechselwirkung zwischen einer α-C-H-Bindung und dem p-Orbital des sp2-C-Atoms (des Radikals oder Carbeniumions), die in bestimmten Konformationen möglich ist. Dadurch wird der Elektronenbedarf des elektronenärmeren sp2-C-Atoms ausgeglichen. (⇒ +I -Effekt von Alkylgruppen)

Begriffe zur Reaktivität
Polarität: ungleiche Verteilung von Elektronendichte Polarisierbarkeit: ein Maß für die Fähigkeit der Elektronenhülle eines Atoms auf die Änderung des elektrischen Feldes zu reagieren. Nucleophile: Elektronendonoren; haben eine negative Ladung oder ein freies Elektronenpaar; elektronenreiche Verbindungen oder Teilchen, die bevorzugt mit elektrophilen Zentren reagieren. Elektrophile: Elektronenpaarakzeptoren; haben Elektronenmangel; elektronenarme Verbindungen oder Teilchen, die bevorzugt mit nucleophilen Zentren reagieren.
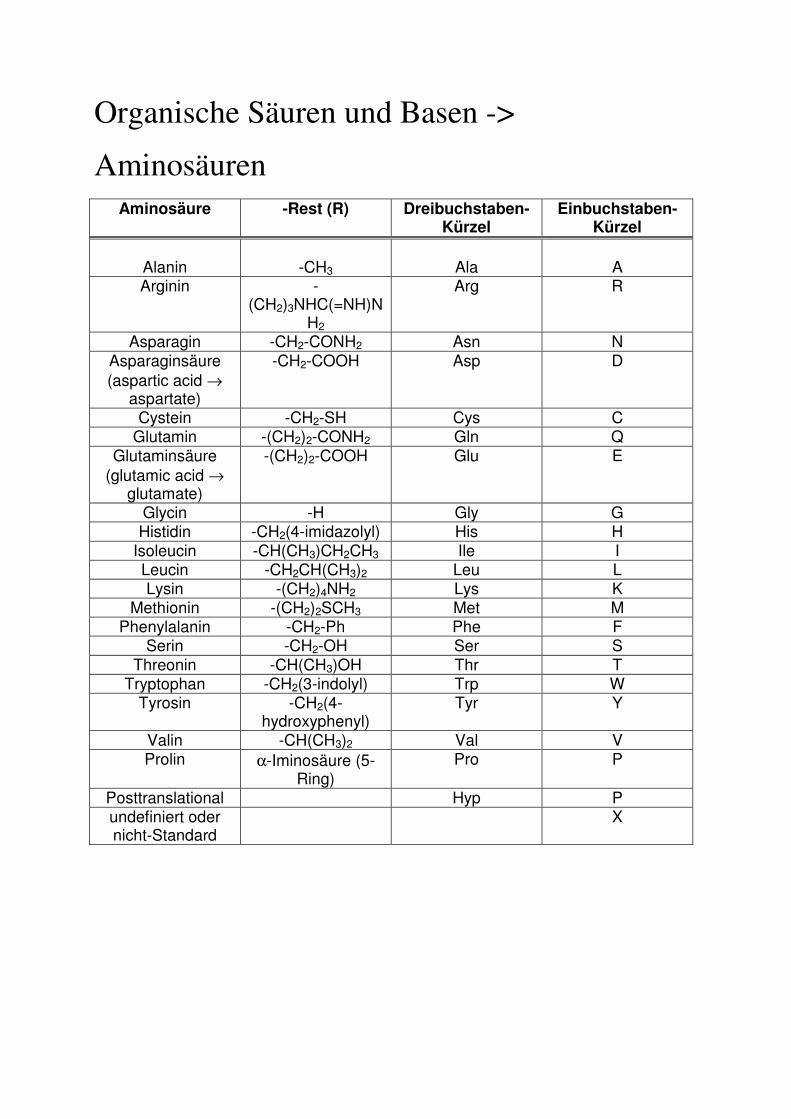
Organische Säuren und Basen ->
Aminosäuren
Aminosäure
-Rest (R) Dreibuchstaben- Kürzel
Einbuchstaben-Kürzel
Alanin
-CH3
Ala
A
Arginin -(CH2)3NHC(=NH)N
H2
Arg R
Asparagin -CH2-CONH2 Asn N Asparaginsäure (aspartic acid →
aspartate)
-CH2-COOH Asp D
Cystein -CH2-SH Cys C Glutamin -(CH2)2-CONH2 Gln Q
Glutaminsäure (glutamic acid →
glutamate)
-(CH2)2-COOH Glu E
Glycin -H Gly G Histidin -CH2(4-imidazolyl) His H
Isoleucin -CH(CH3)CH2CH3 Ile I Leucin -CH2CH(CH3)2 Leu L Lysin -(CH2)4NH2 Lys K
Methionin -(CH2)2SCH3 Met M Phenylalanin -CH2-Ph Phe F
Serin -CH2-OH Ser S Threonin -CH(CH3)OH Thr T
Tryptophan -CH2(3-indolyl) Trp W Tyrosin -CH2(4-
hydroxyphenyl) Tyr Y
Valin -CH(CH3)2 Val V Prolin α-Iminosäure (5-
Ring) Pro P
Posttranslational Hyp P undefiniert oder nicht-Standard
X
L-Aminosäure
CO2H
H
R
CO2H
H
R
D-Aminosäureα-C-Atom:
stereogenes Zentrumwenn R H
H2N CO2H
R
α α
=/
Darstellung in derFischer-Projektion
H2N NH2
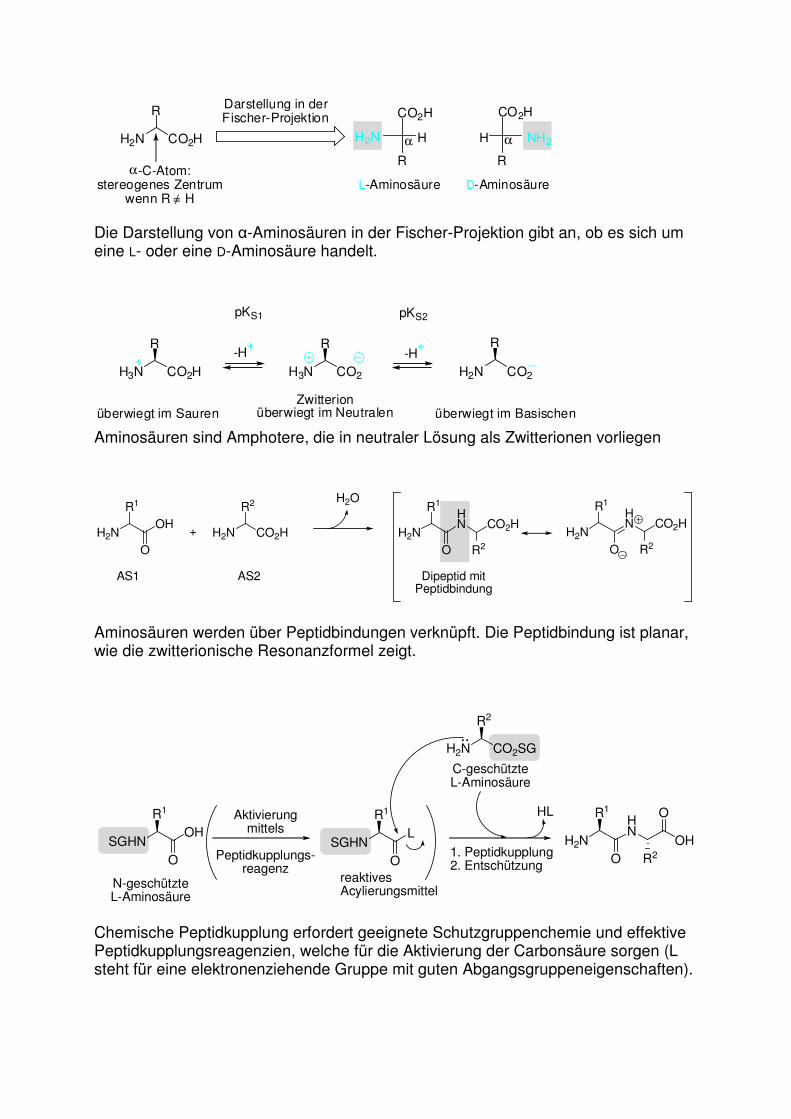
Die Darstellung von α-Aminosäuren in der Fischer-Projektion gibt an, ob es sich um eine L- oder eine D-Aminosäure handelt.
H3N CO2
R
H2N CO2
R
H3N CO2H
R-H
überwiegt im BasischenZwitterion
überwiegt im Neutralenüberwiegt im Sauren
+-H
+
pKS1 pKS2
+ _
Aminosäuren sind Amphotere, die in neutraler Lösung als Zwitterionen vorliegen
H2N
R1
H2N CO2H
R2
O
OH
AS1 AS2
+ H2N
HN CO2H
R1
O R2
Dipeptid mitPeptidbindung
H2O
H2N
HN CO2H
R1
O R2
Aminosäuren werden über Peptidbindungen verknüpft. Die Peptidbindung ist planar, wie die zwitterionische Resonanzformel zeigt.
SGHN
R1
H2N CO2SG
R2
O
OHSGHN
R1
O
L
N-geschützteL-Aminosäure
1. Peptidkupplung2. Entschützung
C-geschützteL-Aminosäure
H2N
HN
OH
R1
reaktivesAcylierungsmittel
HL
O R2
OAktivierungmittels
Peptidkupplungs-reagenz
Chemische Peptidkupplung erfordert geeignete Schutzgruppenchemie und effektive Peptidkupplungsreagenzien, welche für die Aktivierung der Carbonsäure sorgen (L steht für eine elektronenziehende Gruppe mit guten Abgangsgruppeneigenschaften).
Chemische Verbindungen
gleicher Summenformel unterscheiden sich in ihrer Struktur
Isomere
unterscheiden sich nach unterscheiden Art der Verknüpfung sich in der (Konnektivität) der Raumanordnung vorhandenen Atome der Atome
Konstitutionsisomere Stereoisomere
lassen sie sich durch Drehung um Einfachbindungen ineinander überführen
Ja Nein
Konformere Konfigurationsisomere
verhalten sie sich wie Bild und Spiegelbild zueinander
Ja Nein
Enantiomere Diastereomere
(dazu gehören auch cis-, trans- bzw. Z-, E-Isomere)
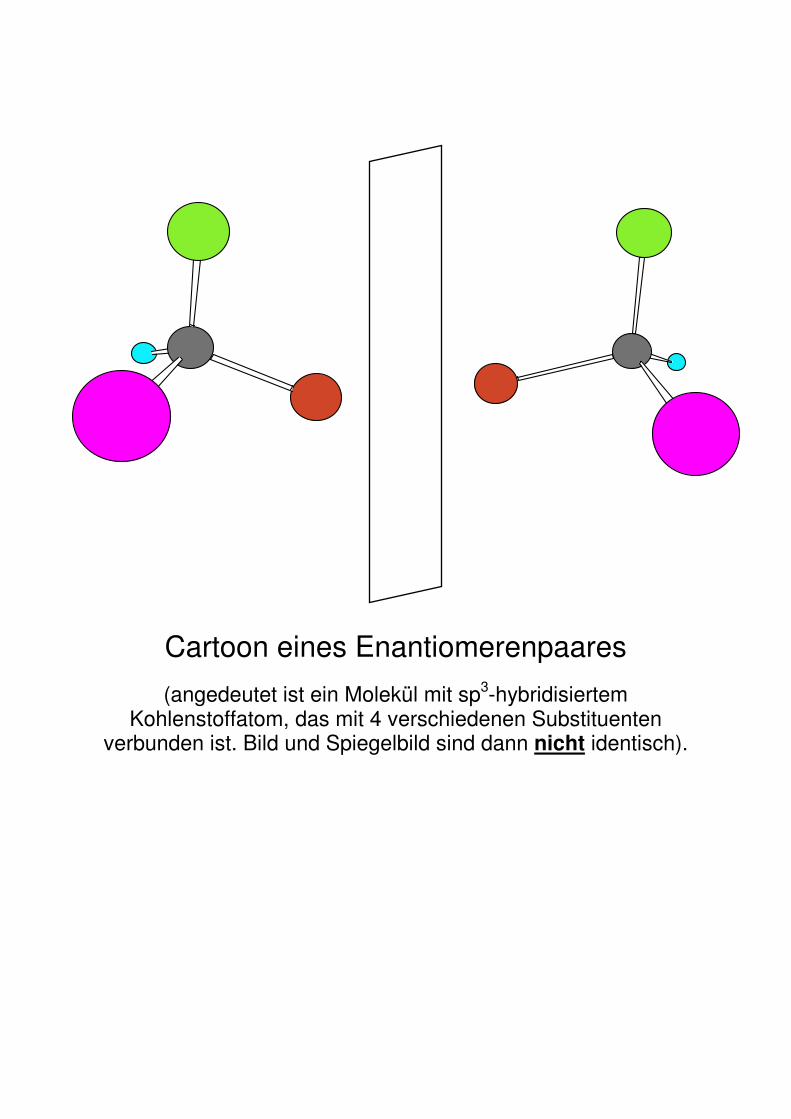
Cartoon eines Enantiomerenpaares
(angedeutet ist ein Molekül mit sp3-hybridisiertem Kohlenstoffatom, das mit 4 verschiedenen Substituenten
verbunden ist. Bild und Spiegelbild sind dann nicht identisch).

cis-trans-Isomerie (geometrische Isomerie)
Diese Form der Isomerie tritt im Zusammenhang mit Doppelbindungen auf,
wo es keine freie Drehbarkeit um die C-C-Bindung gibt.
Dadurch kann es bei der Substitution von H-Atomen in Alkenen räumlich unterschiedliche Formen geben,
die cis- ([lat.], „diesseits") und die trans-([lat.], „jenseits") Form.
cis-Konfiguration trans-
Konfiguration
Benennung cis-2,3-Dichlor-
buten trans-2,3-Dichlor-
buten
Strukturformel
Schmelztemperatur [°C]
-80.5 -49.8
Siedetemperatur [°C]
+60.3 +47.7
Dipolmoment [Debey]
1.89*1018 0.0
Abstand zwischen den C-Atomen [nm]
0.34 0.47

Begriffe zur Stereochemie
Chiralität: z.B. Zentrochiralität: ein C-Atom ist ein stereogenes Zentrum, wenn es sp3-hybridisiert und an vier verschiedene Substituenten gebunden ist. Damit ein Molekül chiral ist, darf es keine Drehspiegelachsen als Symmetrieelement besitzen.
Konfiguration: verschiedene räumliche Anordnung der Gruppen in einem chiralen Molekül
Optische Aktivtität: die Fähigkeit chiraler Moleküle, die Schwingungsebene linear polarisierten Lichtes zu drehen.
Drehwert: der Winkel, um den eine optisch aktive Substanz die Ebene linear polarisierten Lichtes dreht; nach rechts => (+), nach links => (-).
Inversion der Konfigration: Umkehr der Konfiguration
Retention der Konfiguration: deren Erhalt
Racemisierung: aus einem enantiomerenreinen Edukt entstehen beide Isomere
Racemat: ein 1:1-Gemisch von zwei Enantiomeren
Epimerisierung: nur eine einziges von mehreren stereogenen Zentren wird in seiner Konfiguration invertiert.

Optische Aktivität
Elektromagnetische Wellen, zu denen auch das sichtbare Licht gehört, sind
Transversalwellen, d.h. sie schwingen senkrecht zur Ausbreitungsrichtung (vergleichbar mit Wasserwellen). In einem "normalen" Lichtstrahl kommen alle
Schwingungsrichtungen vor. Durch einen Polarisationsfilter können bis auf eine alle Schwingungsrichtungen absorbiert werden, man erhält linear polarisiertes Licht.
Leitet man linear polarisiertes Licht durch eine Lösung einer chiralen Substanz, wird die Polarisationsebene gedreht. Diese Eigenschaft nennt man optische Aktivität. Ein Enantiomer dreht die Ebene nach rechts - dies wird mit (+) bezeichnet - das andere Enantiomer um den gleichen Wert nach links: (-).
Beispielsweise ist die D-Glucose rechtsdrehend (+), die L-Glucose entsprechend linksdrehend (-). Bei der Fructose ist die D-Form dagegen linksdrehend!
Der Drehwinkel (α) hängt dabei von der eingesetzten Verbindung, der
Konzentration (c), der Schichtdicke (d) und dem Vorhandensein weiterer optisch aktiver Substanzen ab. Als substanzspezifischer Wert wird die "Spezifische
Drehung" definiert, bei c = 1 g/ml und d = 10 cm.
Bei einem 1:1-Gemisch beider Enantiomere heben sich die Drehungen auf, ein
solches Gemisch nennt man racemisches Gemisch oder Racemat.
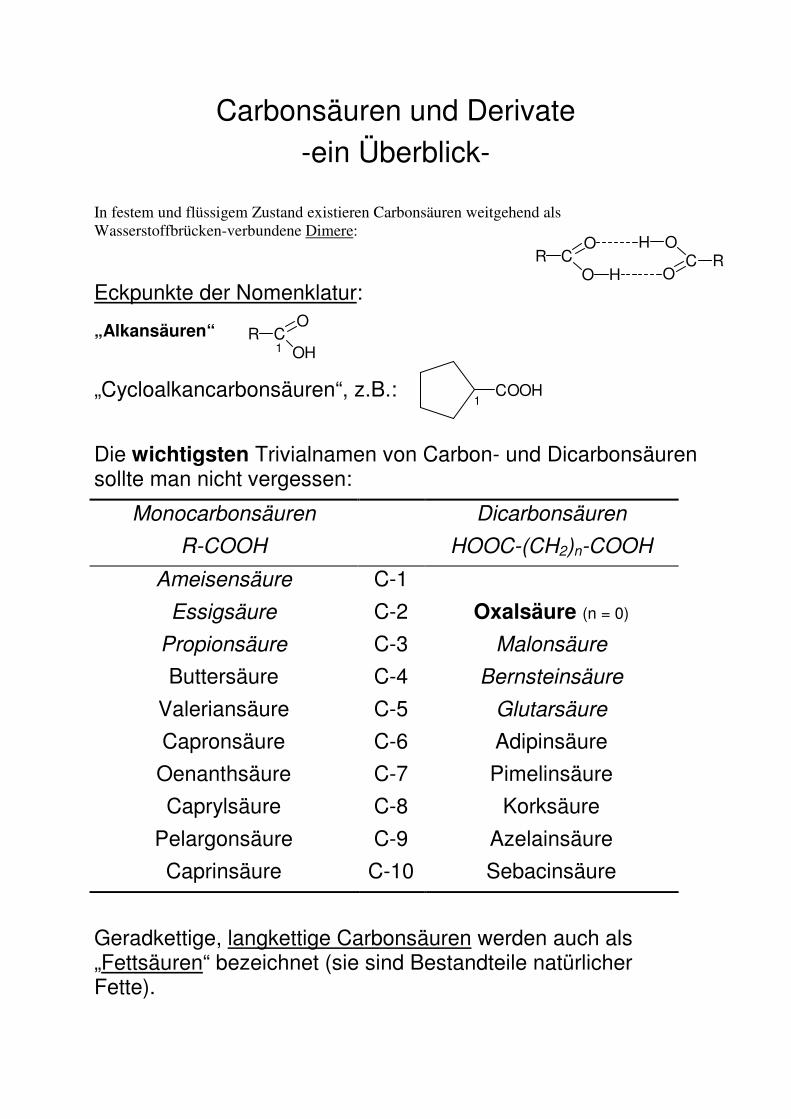
Carbonsäuren und Derivate
-ein Überblick-
In festem und flüssigem Zustand existieren Carbonsäuren weitgehend als Wasserstoffbrücken-verbundene Dimere:
Eckpunkte der Nomenklatur:
„Alkansäuren“
„Cycloalkancarbonsäuren“, z.B.:
Die wichtigsten Trivialnamen von Carbon- und Dicarbonsäuren sollte man nicht vergessen:
Monocarbonsäuren
R-COOH
Dicarbonsäuren
HOOC-(CH2)n-COOH
Ameisensäure C-1
Essigsäure C-2 Oxalsäure (n = 0)
Propionsäure C-3 Malonsäure
Buttersäure C-4 Bernsteinsäure
Valeriansäure C-5 Glutarsäure
Capronsäure C-6 Adipinsäure
Oenanthsäure C-7 Pimelinsäure
Caprylsäure C-8 Korksäure
Pelargonsäure C-9 Azelainsäure
Caprinsäure C-10 Sebacinsäure
Geradkettige, langkettige Carbonsäuren werden auch als „Fettsäuren“ bezeichnet (sie sind Bestandteile natürlicher Fette).
R CO
O H
H O
OC R
R CO
OH1
COOH1
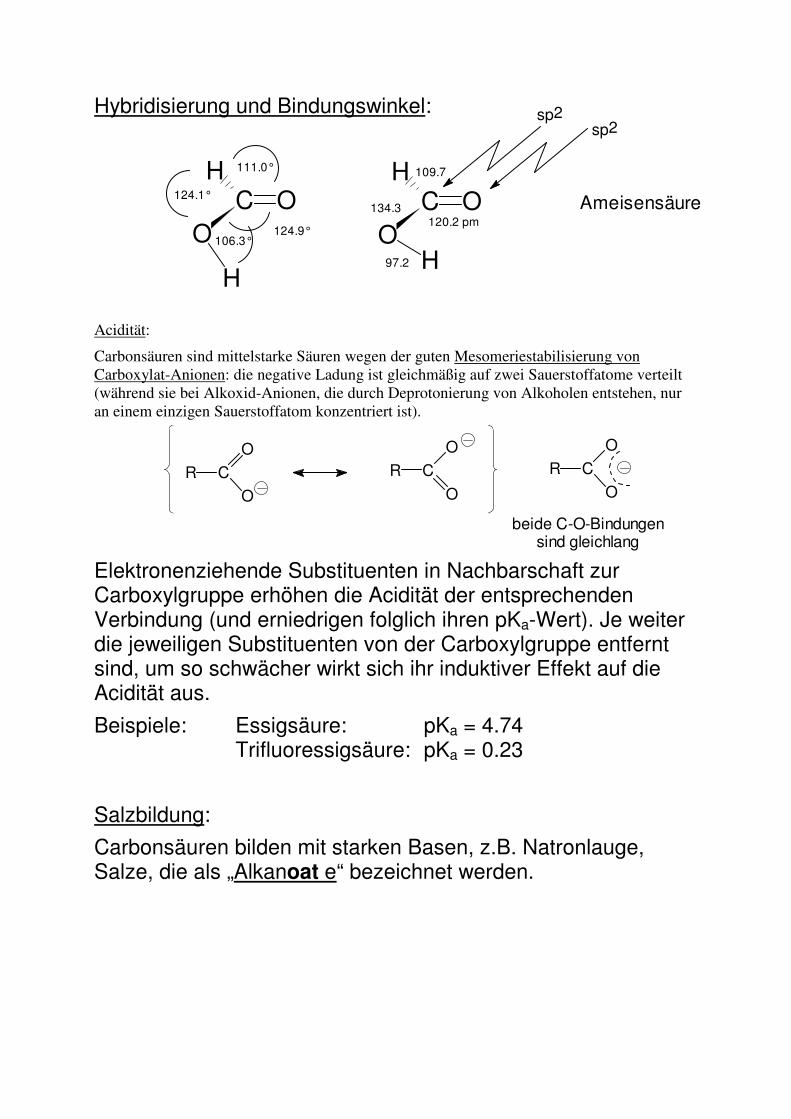
Hybridisierung und Bindungswinkel:
Acidität:
Carbonsäuren sind mittelstarke Säuren wegen der guten Mesomeriestabilisierung von Carboxylat-Anionen: die negative Ladung ist gleichmäßig auf zwei Sauerstoffatome verteilt (während sie bei Alkoxid-Anionen, die durch Deprotonierung von Alkoholen entstehen, nur an einem einzigen Sauerstoffatom konzentriert ist).
Elektronenziehende Substituenten in Nachbarschaft zur Carboxylgruppe erhöhen die Acidität der entsprechenden Verbindung (und erniedrigen folglich ihren pKa-Wert). Je weiter die jeweiligen Substituenten von der Carboxylgruppe entfernt sind, um so schwächer wirkt sich ihr induktiver Effekt auf die Acidität aus.
Beispiele: Essigsäure: pKa = 4.74 Trifluoressigsäure: pKa = 0.23
Salzbildung:
Carbonsäuren bilden mit starken Basen, z.B. Natronlauge, Salze, die als „Alkanoat e“ bezeichnet werden.
124.1°
124.9°
111.0°
106.3°
C OH
O
H
C OH
OH
Ameisensäure
sp2sp2
120.2 pm134.3
109.7
97.2
R C
O
O
R C
O
O
beide C-O-Bindungensind gleichlang
R C
O
O
COOH
COO- Na+
Natriumdodecanoat
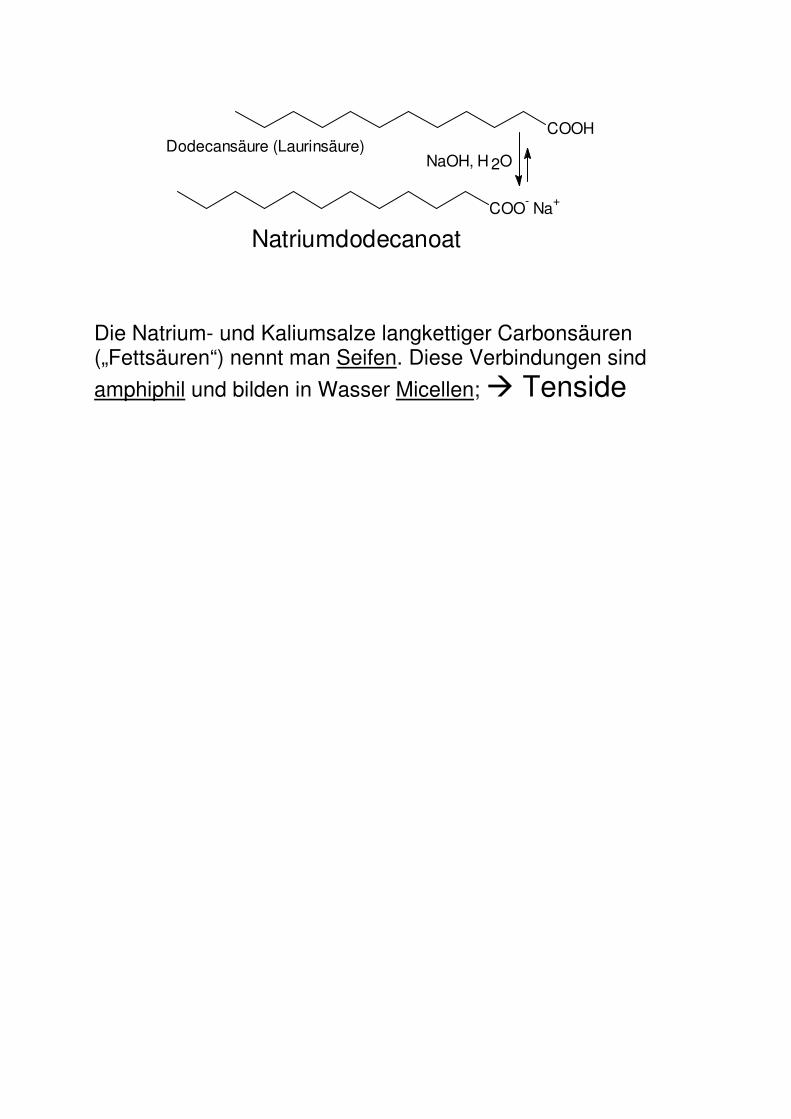
NaOH, H 2ODodecansäure (Laurinsäure)
Die Natrium- und Kaliumsalze langkettiger Carbonsäuren („Fettsäuren“) nennt man Seifen. Diese Verbindungen sind
amphiphil und bilden in Wasser Micellen; � Tenside
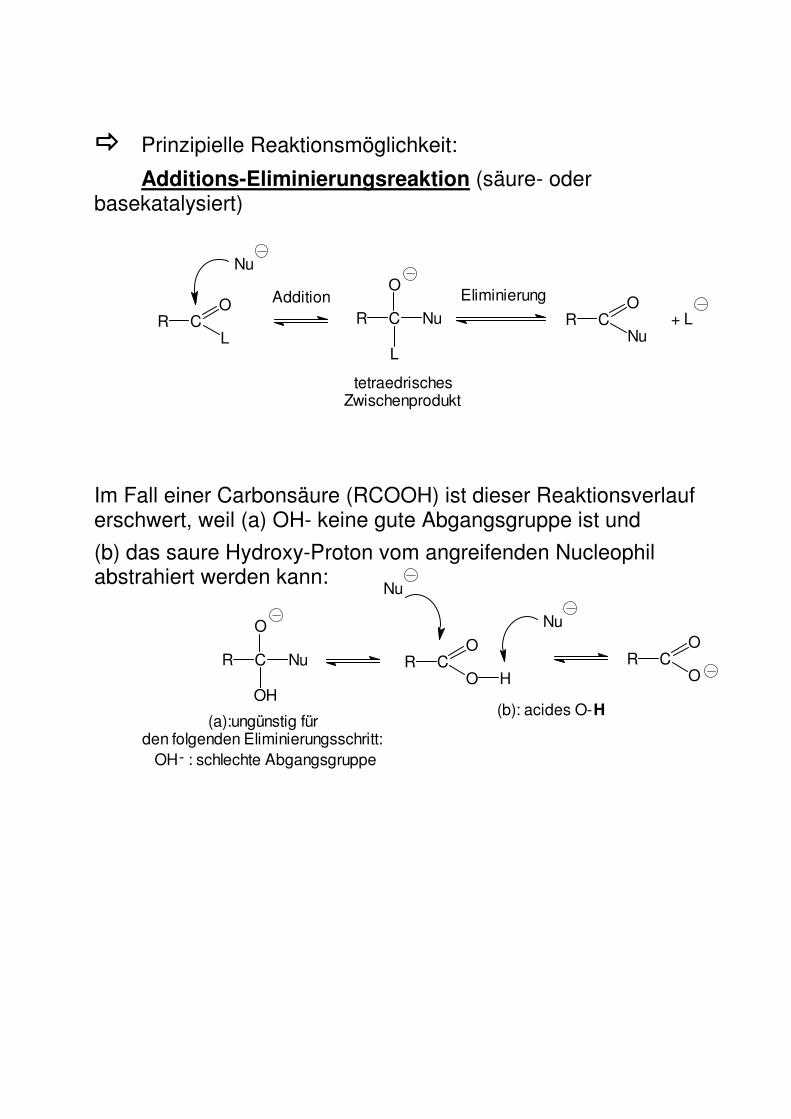
���� Prinzipielle Reaktionsmöglichkeit:
Additions-Eliminierungsreaktion (säure- oder basekatalysiert)
Im Fall einer Carbonsäure (RCOOH) ist dieser Reaktionsverlauf erschwert, weil (a) OH- keine gute Abgangsgruppe ist und
(b) das saure Hydroxy-Proton vom angreifenden Nucleophil abstrahiert werden kann:
R CO
NuR C
O
L
Nu
tetraedrischesZwischenprodukt
R CO
L
Nu
+ LAddition Eliminierung
R CO
O H
Nu
Nu
R C
O
OH
Nu R CO
O
(a):ungünstig fürden folgenden Eliminierungsschritt: OH- : schlechte Abgangsgruppe
(b): acides O-H
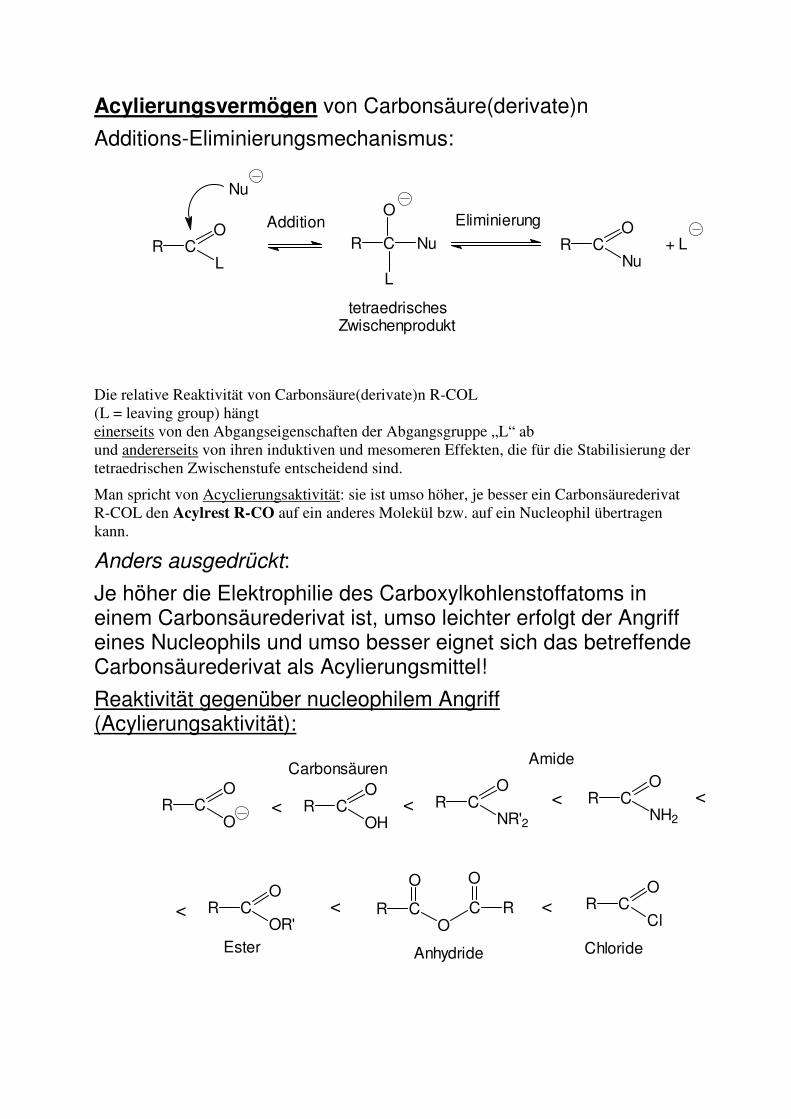
Acylierungsvermögen von Carbonsäure(derivate)n
Additions-Eliminierungsmechanismus:
Die relative Reaktivität von Carbonsäure(derivate)n R-COL (L = leaving group) hängt einerseits von den Abgangseigenschaften der Abgangsgruppe „L“ ab und andererseits von ihren induktiven und mesomeren Effekten, die für die Stabilisierung der tetraedrischen Zwischenstufe entscheidend sind.
Man spricht von Acyclierungsaktivität: sie ist umso höher, je besser ein Carbonsäurederivat R-COL den Acylrest R-CO auf ein anderes Molekül bzw. auf ein Nucleophil übertragen kann.
Anders ausgedrückt:
Je höher die Elektrophilie des Carboxylkohlenstoffatoms in einem Carbonsäurederivat ist, umso leichter erfolgt der Angriff eines Nucleophils und umso besser eignet sich das betreffende Carbonsäurederivat als Acylierungsmittel!
Reaktivität gegenüber nucleophilem Angriff (Acylierungsaktivität):
R CO
OR'R C
O
Cl
R CO
NR'2R C
O
OH
RC
O
O
O
CR
< < < <
< <
R CO
O
R CO
NH2
<
Ester
Amide
ChlorideAnhydride
Carbonsäuren
R CO
NuR C
O
L
Nu
tetraedrischesZwischenprodukt
R CO
L
Nu
+ LAddition Eliminierung
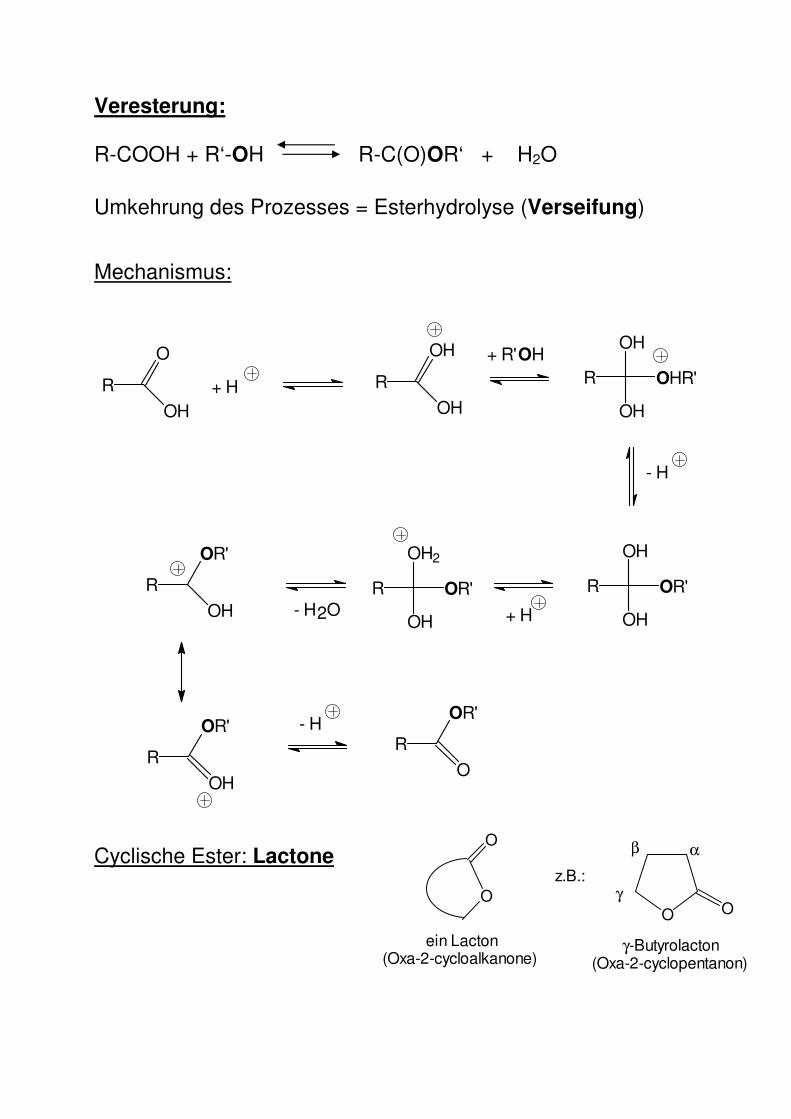
Veresterung:
R-COOH + R‘-OH R-C(O)OR‘ + H2O
Umkehrung des Prozesses = Esterhydrolyse (Verseifung)
Mechanismus:
Cyclische Ester: Lactone
R
O
OH
R
OR'
OH
+ H R
OH
OH
+ R'OHR
OH
OH
OHR'
- H
R
OH
OH
OR'
+ H
R
OH2
OH
OR'- H2O
R
OR'
OH
- HR
OR'
O
O
O
O O
αβ
γz.B.:
γ-Butyrolacton(Oxa-2-cyclopentanon)
ein Lacton(Oxa-2-cycloalkanone)

Carbonylverbindungen
Aldehyde werden als Alkanale, Ketone als Alkanone benannt.
Aldehyde haben die allgemeine Strukturformel R-C(O)H, Ketone folgen
der Formel R-C(O)R’. Aldehyde werden als Alkanale, Ketone als
Alkanone bezeichnet. Die kurzkettigen Vertreter der homologen Reihen
der Carbonylverbindungen besitzen Trivialnamen, die viel gängiger als
die IUPAC-Bezeichnungen sind. Der kleinste Vertreter der Aldehyde ist
Formaldehyd mit der Formel H-C(O)H und wird nach IUPAC-Regeln als
Methanal bezeichnet. Ethanal, der nächst größere Vertreter der
Aldehyde, ist unter dem Namen Acetaldehyd gut bekannt. Der kleinste
Vertreter der Ketone ist ein C3-Körper und heißt nach systematischer
Nomenklatur Propanon, ist aber besser unter der Bezeichnung Aceton
bekannt. Auch viele andere wichtige und gut bekannte
Carbonylverbindungen werden mit ihren Trivialnamen benannt,
darunter Acetophenon und Benzophenon. Der Geruch von Benzaldehyd
aus Bittermandelöl ist vielen vertraut, ähnlich verhält es sich mit Muscon
aus Moschus und mit Anisol, das in Anis und Fenchel vorkommt.
Bekannte Duftstoffe sind außerdem Campher und die α,β-ungesättigte
Verbindung Zimtaldehyd. Crotonaldehyd (But-2-en-al) ist ein anderer
wichtiger α,β-ungesättigter Aldehyd. Die kleinste
Dicarbonylverbindungen heißt Glyoxal (Ethandial), der Dialdehyd
Glutaraldehyd (1,5-Pentandial) ist als Desinfektionsmittel und als
molekularer Quervernetzer gut bekannt.
In den Aldehyden nimmt das Carbonyl-C-Atom naturgemäß immer die
1-Position der Kohlenstoffkette ein, im Fall der Benennung eines Ketons
muss jedoch angegeben werden, an welcher Position in der Kette sich die
Carbonylgruppe befindet. So kann man beispielsweise Pentan-2-on von
Pentan-3-on unterscheiden. Die Position direkt neben der
Carbonylgruppe wird als α-Position gekennzeichnet, eine Nomenklatur,
die auch für andere Verbindungen mit (C=O)-Doppelbindungen, z. B.
den Carbonsäuren, verwendet wird. Die nachfolgenden Positionen im
Kohlenstoffgerüst einer Carbonylverbindung werden mit β, γ und δ
bezeichnet.

Die Natur der Carbonylgruppe bedingt die typische elektrophile Reaktivität am Carbonyl-C-
Atom und die α-CH-Acidität.
Die Carbonylgruppe in Aldehyden und Ketonen ist trigonal planar von ihren Substituenten umgeben. Sowohl das Carbonyl-C-Atom als auch das Carbonyl-Sauerstoffatom sind sp2-hydridisiert. Die (C=O)-Doppelbindung ist stark polarisiert, so dass sich am Carbonyl-C-Atom weniger Elektronendichte, am elektronegativeren O-Atom mehr Elektronendichte befindet. Man sagt, das C-Atom trägt eine positive Partialladung δ+, das O-Atom eine negative, δ-. Zwei Effekte führen zu dieser sehr typischen Polarisierung der (C=O)-Doppelbindung: der –I-Effekt, den das elektronegative Carbonyl-Sauerstoffatom ausübt, sowie der –M-Effekt, der durch die Resonanz der π-Elektronen der (C=O)-Doppelbindung mit den freien Elektronenpaaren des Carbonyl-O-Atoms zustande kommt. Es ist wichtig, diese elektronischen Verhältnisse zu kennen, sie gelten mehr oder weniger immer gleich, wo immer sich eine (C=O)-Doppelbindung in einem organischen Molekül befindet. Das Carbonyl-C-Atom ist demnach elektrophil, das Carbonyl-O-Atom nucleophil und basisch.
Die Resonanzverhältnisse, die in einer Carbonylgruppe herrschen, wirken sich auch auf ihre Nachbarschaft aus. Ganz wesentlich führen sie dazu, dass die H-Atome in α-Stellung eine deutliche Acidität zeigen. Für die Deprotonierung von Acetaldehyd in α-Stellung beträgt der pKS-Wert 17, während der eines normalen Alkans im Bereich von 50 liegt. Sehr gebräuchliche Basen wie Natronlauge, Natriumethanolat oder Triethylamin reichen aus, um eine Carbonylverbindung in α-Stellung zu deprotonieren. Dabei entsteht ein resonanzstabilisiertes Anion, in dem die negative Ladung zwischen dem α-C-Atom und dem Carbonyl-O-Atom verteilt ist. Dieses Anion heißt Enolat. Achtung: Das Wasserstoff-Atom, das direkt an das Carbonyl-C-Atom gebunden ist, wird unter diesen Bedingungen nicht deprotoniert!
HC
CH3
O
150 pm112 pm
120 pm
HC
CH3
O
elektrophil
nucleophilund basisch
Base B_
BH
HC
CH2
O
_H
CCH2
O_
α
CH-Acidität inα-Position
Enolat-Ion
δ-
δ-
δ+δ-
δ-δ-
Die drei prominenten reaktiven Stellen in Carbonylverbindungen sind das elekrophile Carbonyl-C-Atom, das elektronenreiche Carbonyl-O-Atom und die aciden H-Atome in α-Position.

RC
R
O
Nu-H(H)+OH
R Nu(H)
R
AdditionEliminierungvon Wasser
RC
R
Nu
+ H2O
H2O
OH
R OH
RHydrate
meistens nichtisolierbar
R'OH
OH
R OR'R
Halbacetale
acyclische instabil,cyclische stabil
R'OH, H+
OR'
R OR'R
Acetale
isolierbar,säurelabil
CN
OH
R CN
RCyanhydrine
Edukte fürα-Hydroxycarbonsäurenund β-Hydroxyamine
_
Wasser
Alkohol
Alkohol,Säurekatalyse
labil gegenHgCl2
BlausäureCyanide
R'SH, H+
SR'
R SR'
RThiolLewis-Säure-Katalyse
OH
R NR'(H)
RHalbaminal
H2N-NH2
R'NH2 , NH3 instabil,eliminiert Wasser R
CR
NR'(H)
Imin(Schiff'sche Base)
primäre Amineoder Ammoniak
Hydroxylamin
RC
R
N
Hydrazon
NH2
H2N-OH
RC
R
N
Oxim
OH
RC
R
N
Semicarbazon
NH
Hydrazin
Semicarbazid
H2N-NHO
NH2
O
NH2
Thioacetal
Bei der Addition von Nucleophilen an die Carbonylgruppe entstehen je nach der Natur des angreifenden Nucleophils unterschiedliche Stoffklassen, wobei typischerweise zwei Heteroatome an das ehemalige Carbonyl-C-Atom gebunden sind. Die Addition von Stickstoffnucleophilen ist von Wassereliminierung begleitet, es entstehen Produkte mit (C=N)-Doppelbindung.

RC
H
O
+ R'OH
OH
R OR'
H
OH2
R OR'
H
+
R OR'
H
+R OR'
H
+ OR'
R OR'
H
CH
O
OH
C H
OH
O
acyclisches Halbacetalinstabil
acyclisches Acetalstabil
cyclisches Halbacetalstabil
Hydroxycarbonyl-verbindung
intermolekulareReaktion
intramolekulareReaktion
-H2O
R'OH, -H+
H+
Acyclische Halbacetale sind instabil, das Gleichgewicht liegt links, cyclische Halbacetale sind stabil, das Gleichgewicht liegt rechts; Säurekatalyse führt zu Acetalbildung.
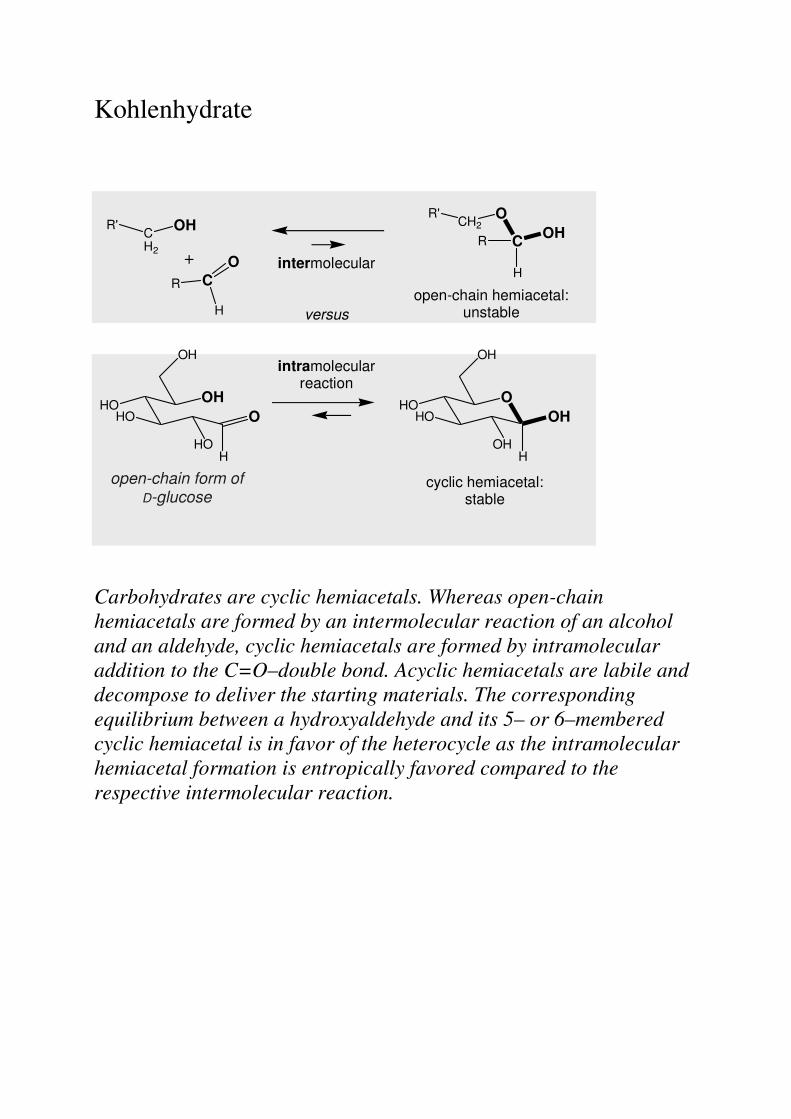
Kohlenhydrate
Carbohydrates are cyclic hemiacetals. Whereas open-chain hemiacetals are formed by an intermolecular reaction of an alcohol and an aldehyde, cyclic hemiacetals are formed by intramolecular addition to the C=O–double bond. Acyclic hemiacetals are labile and decompose to deliver the starting materials. The corresponding equilibrium between a hydroxyaldehyde and its 5– or 6–membered cyclic hemiacetal is in favor of the heterocycle as the intramolecular hemiacetal formation is entropically favored compared to the respective intermolecular reaction.
O
OH
HOHO
OH
OH
OH
HO
HOHO
OH
O
R C
OCH2
OH
R C
OHCH2
O
H
R'R'
H
H H
+
open-chain hemiacetal:unstable
cyclic hemiacetal:stable
intermolecular
versus
intramolecularreaction
open-chain form of D-glucose

OH
HO
HOHO
OH
O
H
open-chain formof D-glucose
as zig-zagrepresentation
as Fischerprojection
HO
O
OH
HO
OH
HO
OH
HO
OH
OH
OH
O
1
2
4
5
6
1
2
3
4
5
6
1
36
The open–chain form of D–glucose as zig–zag and as Fischer projection. Numbering of the carbon chain is indicated. To compare glucose with its stereoisomers it is most suitably represented as a Fischer projection.
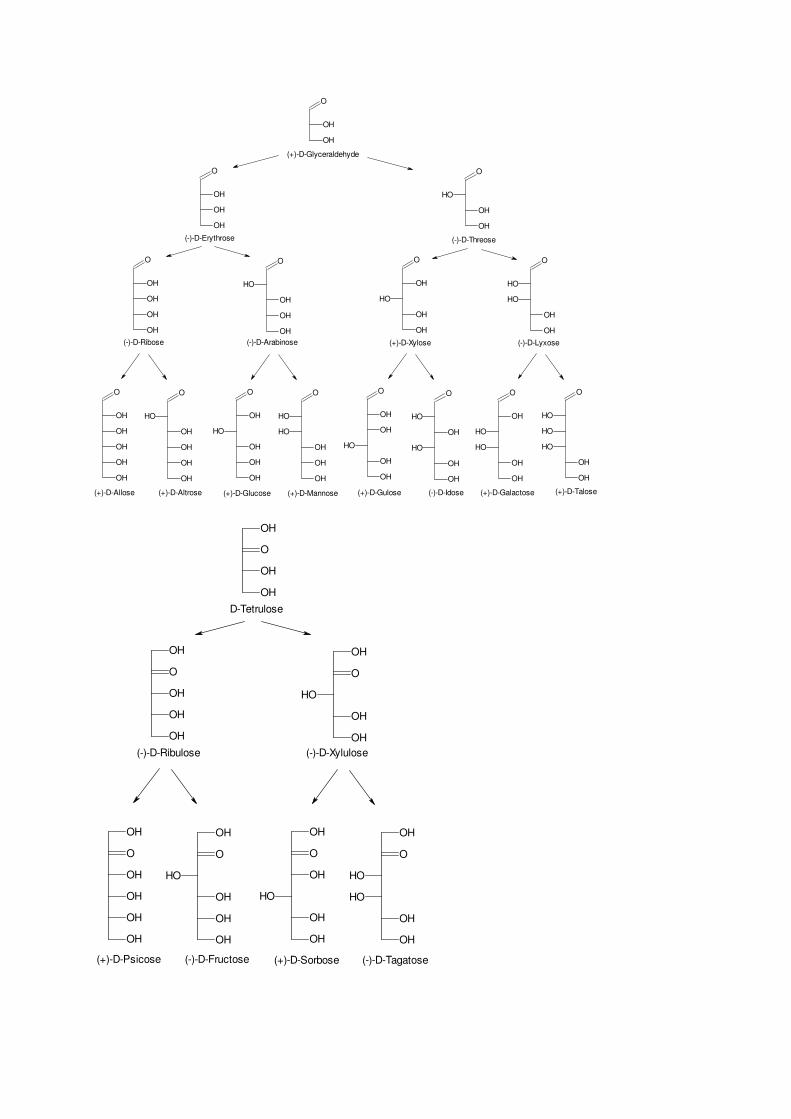
The group of aldoses is derived from glyceraldehyde by the formal insertion of stereogenic (HCOH)–groups, ketoses can be built up from 1,3–dihydroxyacetone.
O
OHOH
O
OH
HO
OH
OH
O
OH
O
OH
OH
OH
O
HO
OH
O
(HCOH) n
OHOH
OH
O
(HOCH) n
OH
D-Glyceraldehyde L-Glyceraldehyde 1,3-Dihydroxyacetone
D-TetruloseL-Tetrulose
D-Aldoses
1
2
3+n
1
2+n
3+n
1
2
3
1
2
3
O
(HCOH) n
OH
HO
L-Aldoses
1
2+n
3+n
1
2
34
HO
4+n
D-Ketoses
OH
O
(HOCH) n
OH
1
2
3+n OH
4+n
L-Ketoses
OH
O
OH
OH
O
OH
OH
OH
O
OH
OH
OH
O
OH
OH
HO
O
OH
OH
OH
OH
HOOH
O
OH
OH
OH
OH
O
OH
OH
OH
HO
O
OH
OH
HO
HO
OH
O
OH
OH
HO
OH
O
OH
OH
OH
HO
O
OH
OH
OH
HO
HO
O
HO
OH
OH
HO
HO
OH
O
HO
OH
OH
HO
OH
O
OH
HO
OH
OH
O
OH
HO
OH
OH
HO
(+)-D-Glyceraldehyde
(-)-D-Erythrose (-)-D-Threose
(-)-D-Ribose (-)-D-Arabinose (+)-D-Xylose (-)-D-Lyxose
(+)-D-Allose (+)-D-Altrose (+)-D-Glucose (+)-D-Mannose (+)-D-Gulose (-)-D-Idose (+)-D-Galactose (+)-D-Talose
O
OH
OH
OH
O
OH
OH
OH
OH
OH
OH
OH
OH
O
HO
O
OH
OH
OH
OH
OH
OH
OH
OH
O
HO
O
OH
OH
OH
OH
HO
OH
OH
HO
OH
O
HO
D-Tetrulose
(-)-D-Ribulose (-)-D-Xylulose
(+)-D-Psicose (-)-D-Fructose (+)-D-Sorbose (-)-D-Tagatose
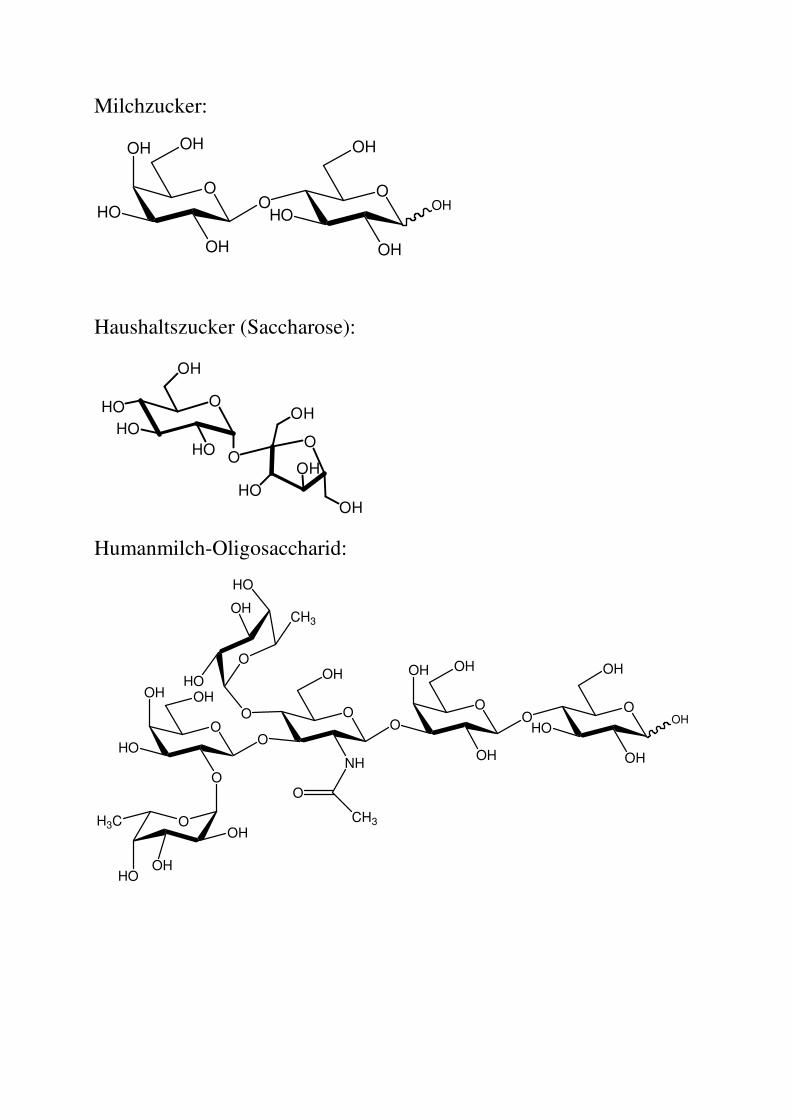
Milchzucker:
O
OH
HO
OH OH
OO
OH
HO
OH
OH
Haushaltszucker (Saccharose):
Humanmilch-Oligosaccharid:
O
OH
OH OH
OO
OH
HO
OH
OH
O
CH3
HO
OH
HO
O
OH3C
HOOH
OH
O
O
NH
OH
O
CH3
O
HO
OH OH
OO
O
HOHO
OH
HO
O
OH
OH
OH
O
HO
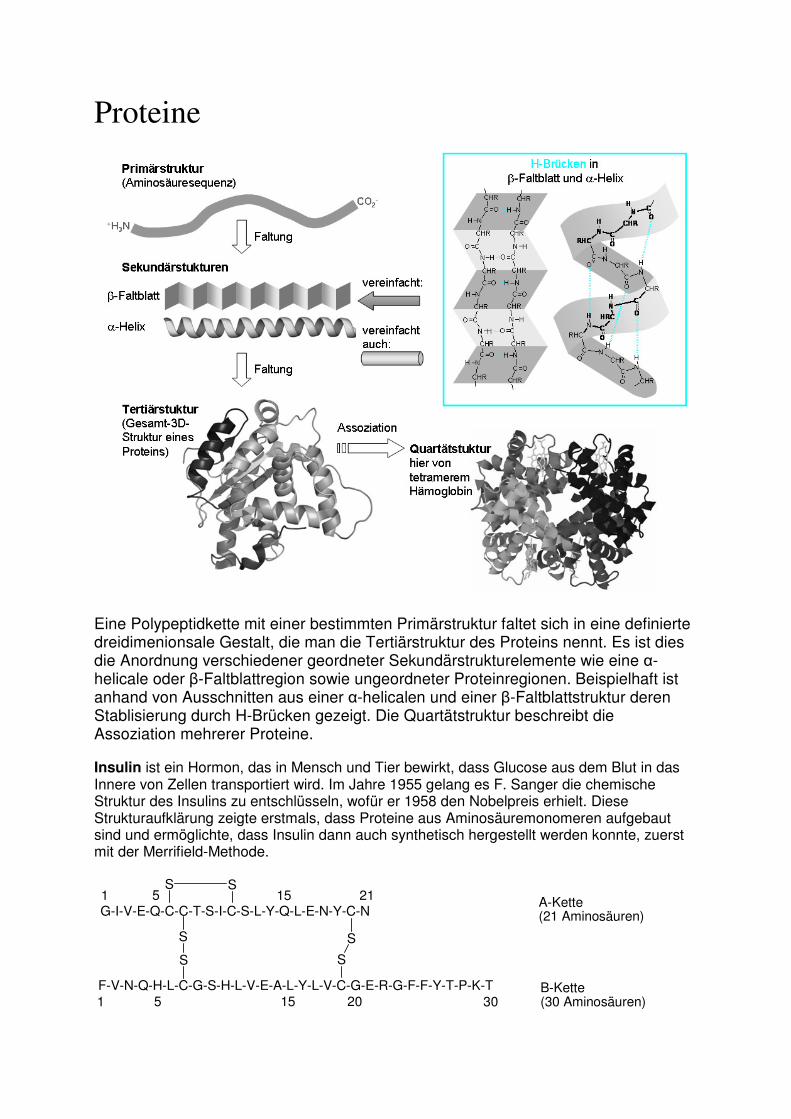
Proteine
Eine Polypeptidkette mit einer bestimmten Primärstruktur faltet sich in eine definierte dreidimenionsale Gestalt, die man die Tertiärstruktur des Proteins nennt. Es ist dies die Anordnung verschiedener geordneter Sekundärstrukturelemente wie eine α-helicale oder β-Faltblattregion sowie ungeordneter Proteinregionen. Beispielhaft ist anhand von Ausschnitten aus einer α-helicalen und einer β-Faltblattstruktur deren Stablisierung durch H-Brücken gezeigt. Die Quartätstruktur beschreibt die Assoziation mehrerer Proteine.
Insulin ist ein Hormon, das in Mensch und Tier bewirkt, dass Glucose aus dem Blut in das Innere von Zellen transportiert wird. Im Jahre 1955 gelang es F. Sanger die chemische Struktur des Insulins zu entschlüsseln, wofür er 1958 den Nobelpreis erhielt. Diese Strukturaufklärung zeigte erstmals, dass Proteine aus Aminosäuremonomeren aufgebaut sind und ermöglichte, dass Insulin dann auch synthetisch hergestellt werden konnte, zuerst mit der Merrifield-Methode.
G-I-V-E-Q-C-C-T-S-I-C-S-L-Y-Q-L-E-N-Y-C-NA-Kette (21 Aminosäuren)
1 21
F-V-N-Q-H-L-C-G-S-H-L-V-E-A-L-Y-L-V-C-G-E-R-G-F-F-Y-T-P-K-T1 5 15 20 30
B-Kette (30 Aminosäuren)
5 15S S
S S
S S

Naturstoffe, z. B.: Alkaloide ursprünglich: Substanzen mit alkali-ähnlichem Charakter heute: eine Gruppe verschiedenster Verbindungen pflanzlichen Ursprungs, die Stickstoff-Heterocyclen enthalten
z.B. Morphin Der Chemiker Ludwig Knorr (1859-1921, Jena) spekulierte 1889 über die chemische Struktur des Alkaloids Morphin: „Meine Studien über das Morphin haben es wahr-scheinlich gemacht, dass diese wichtige Base und wohl auch andere dem Morphin nahe verwandte Alkaloide als Oxazine aufgefasst werden müssen. Die mir bis jetzt bekannten Thatsachen scheint die in der folgenden Formel ausgedrückte Auffassung des Morphins am besten zu erklären“:
N
H
HO
O
HO
CH3
H
CH
CH
O
N
CH2
CH2
CH3
CH
(C10H5OH)
CH2
OH
Die Wurzel von Tollkirsche (A. belladonna) enthält als Hauptalkaloid Atropin. Je nach der Menge eingenommener Tollkirsche, die wohlschmeckend ist und leicht mit der Kirsche verwechselt werden kann, werden beim Menschen Trockenheit des Mundes, weite Pupillen (pupillenerweiternde Wirkung!), Gesichtsrötung, Erregung, Verwirrung, Halluzinationen, Bewusstlosigkeit und Krämpfe beobachtet. Die Vergiftung kann auch tödlich verlaufen.
KOKASTRAUCH: Das Alkaloid Cocain blockiert die periphere Schmerzempfindung. Die dauernde Einnahme von Cocain macht einen gesunden Menschen nach einem kurzen euphorischen Einstieg im Laufe weniger Monate zu einem geistigen wie körperlichen Wrack. Das Hauptalkaloid des Tabaks ist das Nikotin. Alle Tabakarten gehören zur Gattung “Nicotiana”. Diesen Namen trägt die Pflanze seit 1565 zu Ehren von Jean NICOT, der die Tabakpflanze in Frankreich populär machte. Nicotin ist ein starkes Gift. Bei Dosen zwischen 50 und 100 mg erfolgt der Tod rasch durch Lähmung des Atemzentrums im Gehirn. Struktur?
N
O
O
HOHH3C
NH3CO
O
COOH

RNA, DNA
O
O
O
P
OO
HO
Nukleobase
O
O
O
P
O
O
Nukleobase
O
O
O
P
O
O
Nukleobase
N
NO
NH2
HN
N N
N
O
NH2
HN
NO
O
CH3
N
N N
N
NH2
Genetischer Code:
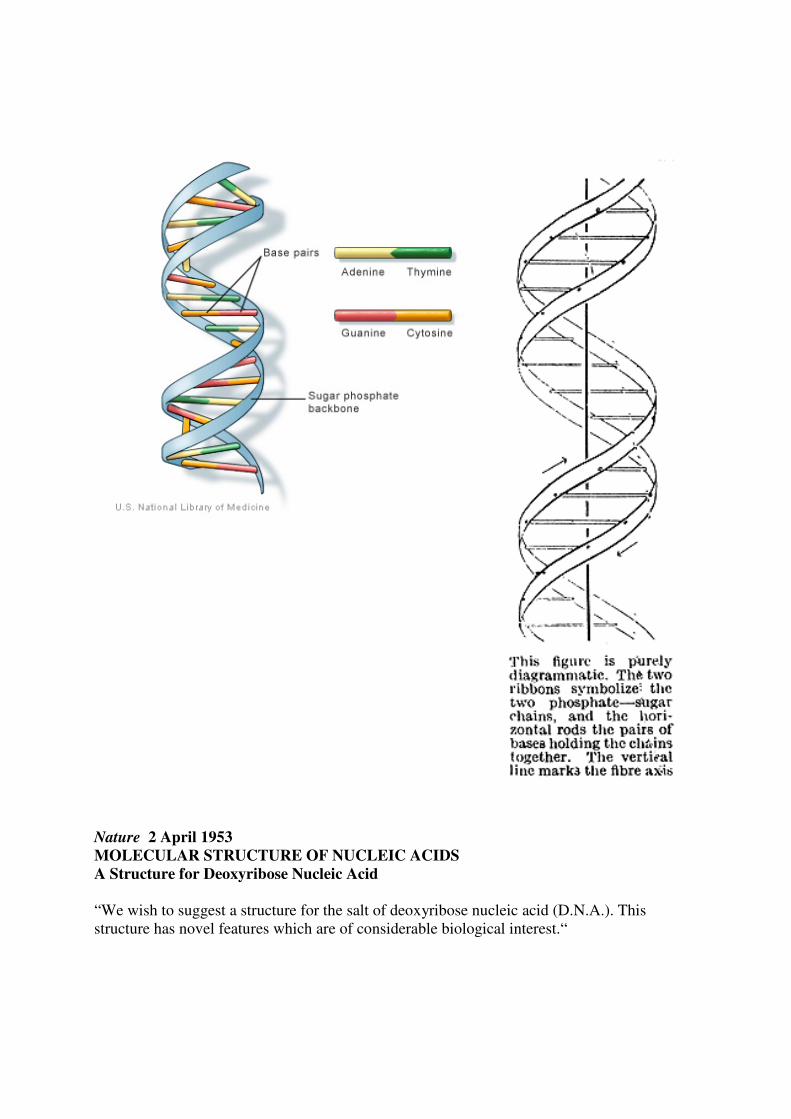
Nature 2 April 1953
MOLECULAR STRUCTURE OF NUCLEIC ACIDS
A Structure for Deoxyribose Nucleic Acid “We wish to suggest a structure for the salt of deoxyribose nucleic acid (D.N.A.). This structure has novel features which are of considerable biological interest.“
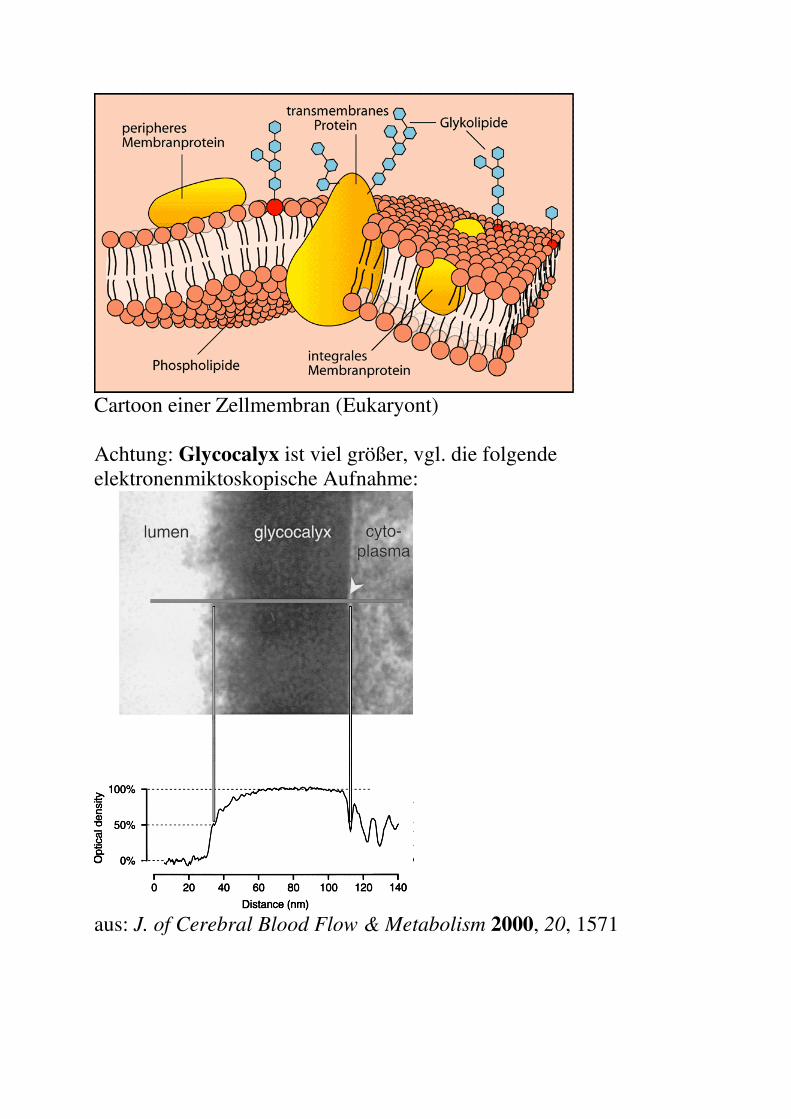
Cartoon einer Zellmembran (Eukaryont) Achtung: Glycocalyx ist viel größer, vgl. die folgende elektronenmiktoskopische Aufnahme:
aus: J. of Cerebral Blood Flow & Metabolism 2000, 20, 1571


















