







Sprachen
Seiten
Rechtliche


Abstracts
Chronische Entzündungen des Gastrointestinaltrakts – evidenzbasierte und individualisierte Entscheidungen
Dresden
Samstag, 25. Juni 20118.50 – 16.00 Uhr
Veranstaltungsort:Sächsische Landesbibliothek Staats- und Universitätsbibliothek Zellescher Weg 18 01069 Dresden
Wissenschaftliche Leitung:Prof. Dr. S. Miehlke, HamburgPD Dr. A. Madisch, Hannover
Greifswald18. Juni 2011
Hildesheim 9. April 2011
DresdenDresden25.25. Juni 2011
Stuttgart 16. April 2011
Erlangen 8. Oktober 2011
Trier 5. November 2011
Bonn 26. November 2011
Bielefeld19. März 2011

1
Programm 8.50 Uhr Begrüßung und Einführung
Prof. Dr. S. Miehlke, Hamburg
Session I: Oberer Gastrointestinaltrakt Vorsitz: Prof. Dr. P. Layer, Hamburg PD Dr. A. Madisch, Hannover
9.00 Uhr Gastroösophageale Refluxkrankheit: Manifestationen und ungelöste Probleme Prof. Dr. J. Labenz, Siegen
9.30 Uhr Eosinophile Ösophagitis Dr. P. Hruz, Basel
10.00 Uhr H. pylori Update: Magenlymphom, Magenkarzinom, NSAR/ASS (ohne Abstract) Prof. Dr. W. Fischbach, Aschaffenburg
Session II: Leber, Pankreas Vorsitz: Prof. Dr. J. Labenz, Siegen Prof. Dr. S. Miehlke, Hamburg
10.30 Uhr Therapie der chronischen Hepatitis C – Standards und Perspektiven Prof. Dr. C. Sarrazin, Frankfurt
11.00 Uhr Chronische Hepatitis B: wann und wie behandeln? Prof. Dr. T. Berg, Leipzig
11.30 Uhr Diagnostik und Therapie autoimmuner Leber- und Gallenwegserkrankungen Prof. Dr. C.P. Strassburg, Hannover
12.00 Uhr Akute und chronische Pankreatitis: neue Entwicklungen Prof. Dr. P. Layer, Hamburg
12.30–13.30 Uhr
Mittagspause mit Imbiss

2
Special Lecture
Vorsitz: Prof. Dr. A. Morgner-Miehlke, Dresden
13.30 Uhr Gender-Aspekte in der Viszeralmedizin Prof. Dr. G. Möslein, Bochum
Session III: Dünndarm und Dickdarm Vorsitz: PD Dr. B. Siegmund, Berlin Prof. Dr. T. Berg, Leipzig
14.00 Uhr Morbus Crohn – aktuelle Kontroversen PD Dr. B. Siegmund, Berlin
14.30 Uhr
Colitis ulcerosa – leitliniengerechte Therapiestrategien (ohne Abstract) Prof. Dr. Dr. G. Rogler, Zürich
15.00 Uhr Kollagene und lymphozytäre Kolitis – ein Update Prof. Dr. S. Miehlke, Hamburg
15.30 Uhr Divertikulitis und Clostridium-difficile-Colitis Prof. Dr. V. Groß, Amberg
16.00 Uhr Schlusswort und Verabschiedung PD Dr. A. Madisch, Hannover
Anschriften der Referenten und Vorsitzenden siehe Seite 43

3
Gastroösophageale Refluxkrankheit: Manifestationen und ungelöste Probleme
J. Labenz
Abteilung für Innere Medizin, Ev. Jung-Stilling-Krankenhaus, Siegen
Manifestationen
Die gastroösophageale Refluxkrankheit (GERD) kann sich durch ösophageale und
extraösophageale Syndrome manifestieren (Abb. 1), die allein oder in Kombination
vorkommen können. Als mögliche weitere Manifestation kristallisierte sich der sog.
postnasale Drip heraus, wie durch eine kontrollierte Interventionsstudie gezeigt
werden konnte. Schlafstörungen durch nächtliche Refluxsymptome reihen sich in die
Liste der typischen ösophagealen Syndrome ein. Jüngst wurde aber auch berichtet,
dass Patienten mit Schlafstörungen überzufällig häufig einen asymptomatischen
gastroösophagealen Reflux haben. Auch bei den Symptomen Dysphagie und
epigastrischer Schmerz muss an eine GERD gedacht werden. Ebenso ist die Reflux-
ösophagitis eine mögliche Ursache für gastrointestinale Blutungen, insbesondere
beim älteren Menschen, und eine zur GERD prädisponierende große axiale
Hiatushernie kann über chronische Blutverluste zur Eisenmangelanämie führen.
ÖsophagealeSyndrome
ExtraösophagealeSyndrome
GERD ist eine Erkrankung, bei der Reflux GERD ist eine Erkrankung, bei der Reflux von Mageninhalt belvon Mageninhalt beläästigende Symptome stigende Symptome
und/oder Lund/oder Lääsionen verursacht.sionen verursacht.
Sinusitis
Pharyngitis
Pulmonale Fibrose
Rez. Otitis media
MöglicheAssoziationen
Refluxhusten
Refluxlaryngitis
Refluxasthma
Dentale Erosionen
EtablierteAssoziationen
TypischesRefluxsyndrom
Reflux-Thorax-Schmerzsyndrom
SymptomatischeSyndrome
Refluxösophagitis
Refluxstriktur
Barrett-Ösophagus
Refluxkarzinom
Syndrome mitÖsophagusläsionen
Abb. 1: Montreal-Definition und -Klassifikation der GERD (nach Vakil et al., Am J Gastroenterol. 2006)

4
Ungelöste Probleme
Therapierefraktäre Refluxsymptome: Ca. 70% aller GERD-Patienten leiden an einer
NERD und ca. 30–40% dieser Patienten erfahren durch Protonenpumpeninhibitoren
(PPI) eine unzureichende Symptomkontrolle (fehlende oder partielle Symptom-
response). Das Ursachenspektrum hierfür ist vielfältig (Abb. 2). Kürzlich wurde
berichtet, dass auch die Sprue in die Liste möglicher Differenzialdiagnosen aufge-
nommen werden muss. Allgemeinmaßnahmen, optimierte PPI-Therapie, Antireflux-
operationen bei sorgfältig ausgewählten Patienten und trizyklische Antidepressiva
bzw. Serotonin-Wiederaufnahmehemmer sind Optionen, die einzelnen, sicher aber
nicht allen Patienten mit PPI-refraktärer NERD helfen. Neue Einblicke in die Patho-
genese und Pathophysiologie müssen und werden zu neuen Therapieprinzipien
führen.
PsychischeKomorbidität
MangelndeCompliance
FalscheDiagnose
UnzureichenderPPI-Effekt
FunktionellesSodbrennen
Nicht-saurerReflux
GestörteMagenentleerung
BegleitendesReizdarmsyndrom
Abb. 2: Ursachenspektrum bei therapieresistentem Sodbrennen (nach Labenz & Feußner, Gastroenterologe. 2009)
Nicht-heilende Refluxösophagitis: Ca. 20–30% aller schweren Refluxösophagitiden
(Los-Angeles-Grad C und D) heilen nicht innerhalb von 8 Wochen ab. Allgemein-
maßnahmen (Gewichtsreduktion!), Optimierung der PPI-Therapie und Antireflux-
chirurgie sind die aktuell verfügbaren Optionen.

5
Extraösophageale Syndrome: Extraösophageale Manifestationen der GERD sind
vermutlich seltener als bisher angenommen. Es gibt bisher keine Möglichkeit im
Vorfeld einer Therapie festzustellen, ob eine vermeintliche extraösophageale
Manifestation (z. B. Husten) rein zufällig oder kausal mit einer GERD assoziiert ist.
Ein ursächlicher Zusammenhang lässt sich nur durch eine längerfristige (3 Monate),
hoch dosierte PPI-Therapie nachweisen bzw. ausschließen. Es ist von großer
praktischer Relevanz, dass eine symptomatische GERD (Sodbrennen!) eine conditio
sine qua non für das therapeutische Ansprechen einer extraösophagealen Manifes-
tation ist, d. h. die Suche nach einem asymptomatischen Säurereflux bei Patienten
mit z. B. Laryngitis, Husten oder Asthma ist überflüssig und die HNO-ärztliche
Diagnose einer „Refluxlaryngitis“ nicht akzeptabel.
Barrett-Ösophagus: Die Prognose eines Barrett-Karzinoms ist besonders dann
günstig, wenn der Barrett-Ösophagus zuvor bekannt und der Tumor im Rahmen
eines Überwachungsprogramms detektiert wurde. Allerdings wird weiterhin die große
Mehrzahl aller Barrett-Karzinome im Rahmen der Erstendoskopie entdeckt. Dies
führt zu dem berechtigten Wunsch, dass die Indikationsschwelle für die Erstendo-
skopie besonders niedrig sein sollte. Problematisch erscheinen aus Kosten-Nutzen-
Überlegungen heraus aber die regelmäßigen Überwachungsendoskopien aller
Patienten mit Barrett-Ösophagus. Eine zuverlässige Prädiktion einer Karzinoment-
wicklung ist heute ebenso wenig möglich wie die medikamentöse oder operative
Verhinderung der Progression. Dementsprechend wird man über einen Paradigmen-
wechsel im Management des nicht-dysplastischen Barrett-Ösophagus nachdenken
müssen: Ablation statt Überwachung. Voraussetzungen hierfür wären ein zuverlässig
wirksames, sicheres, breit verfügbares und seitens der Kosten akzeptables Therapie-
verfahren.

6
Eosinophile Ösophagitis
P. Hruz
Abteilung für Gastroenterologie und Hepatologie, Universitätsspital Basel
Die eosinophile Ösophagitis (EoE) ist eine chronisch verlaufende, entzündliche
Erkrankung der Speiseröhre mit zunehmender Inzidenz und Prävalenz. Sie zeigt mit
einem Verhältnis von 3:1 eine deutliche Prädilektion für das männliche Geschlecht.
Etwa 70% der meist jüngeren Patienten sind mit einer atopischen Diathese belastet.
Bei derEoE findet man zelluläre Elemente und Mediatoren einer allergieartigen
Entzündung. Zum gegenwärtigen Zeitpunkt werden 2 pathophysiologische
Mechanismen diskutiert. Neben einer sogenannten TH2-Typ-Immunreaktion spielen
Mastzellen – aktiviert via Immunglobulin E – eine wichtige Rolle bei der initialen
Entzündung wie auch bei der gefürchteten Spätfolge der chronischen Entzündung,
die mit einem Fibrosierungsprozess, dem sogenannten Remodeling, der
ösophagealen Schleimhaut einhergehen kann. Ob es sich beim initialen Trigger um
eine Reaktion auf ein inhalativ oder peroral aufgenommenes Allergen oder um eine
Kombination von unterschiedlichen Allergenen handelt, bleibt allerdings gegenwärtig
unklar.
Eine Anamnese mit den Leitsymptomen „Dysphagie für geformte Speisen“ und
„Bolusimpaktierungen“ ist vor allem bei jüngeren Patienten hoch verdächtig auf das
Vorliegen einer EoE. In der endoskopischen Untersuchung finden sich fakultativ
unspezifische Veränderungen wie rötliche Längsfurchen, weißliche Auflagerungen,
Ringe und Stenosen. Die Diagnose der EoE jedoch beruht – ob mit oder ohne
endoskopische Zeichen – auf dem charakteristischen Nachweis einer dichten
eosinophilen Infiltration der Ösophagusschleimhaut in der Histologie. In den aktuellen
diagnostischen Richtlinien ist der histologische Nachweis von ≥ 15 eosinophilen
Granulozyten pro HPF (= 400-fache Vergrößerung im Mikroskop) für die Diagnose
wegweisend. Da das eosinophile Infiltrat oftmals inhomogen, fleckförmig und
segmentär verteilt ist, sollen multiple Biopsien an verschiedenen Lokalisationen des
Ösophagus entnommen werden. Weiße Auflagerungen sollten noch zusätzlich
biopsiert werden, da diese eosinophilen Mikroabszessen entsprechen können und
die Diagnosestellung erleichtern. Differenzialdiagnostisch muss eine
gastroösophageale Refluxkrankheit in Betracht gezogen werden. Die endoskopische

7
Untersuchung unter ausreichender – mindestens 2 Wochen bestehender –
säuresupprimierender Therapie und segmentale Biopsie-Entnahmen aus dem
proximalen und distalen Ösophagus ermöglichen es aber meist, diese beiden
Krankheiten auseinanderzuhalten. Eine Alternative bietet die Durchführung einer
24h-pH-Metrie. Zurzeit gibt es keine spezifischen biochemischen Marker, die in der
Primärdiagnostik der EoE angewendet werden können.
In der Behandlung werden vorwiegend topische und bei Bedarf systemische
Kortikosteroide eingesetzt, auf welche die Mehrheit der Patienten gut anspricht. Bei
den topischen Kortikosteroiden ist gegenwärtig die optimale ösophageale
Applikationsform in klinischer Erprobung. Bei steroidrefraktärem oder
steroidabhängigem Verlauf ist primär immer die Compliance der Patienten zu
hinterfragen. Immunsuppressiva und Biologika sind mögliche, jedoch für diese
Indikation noch wenig erforschte Therapiealternativen und bevorzugt in
spezialisierten Zentren einzusetzen. Leukotrienantagonisten haben eine Wirkung auf
die Schluckbeschwerden gezeigt, jedoch persistierte die eosinophile Entzündung und
zudem wurden in den erforderlichen hohen Dosierungen erhebliche Nebenwirkungen
registriert. CRTH2-Blocker, eine neue Generation von Antiallergika, welche einen
gemeinsamen Oberflächenrezeptor bei Th2-immunvermittelten Entzündungen
involvierten Zellen blockieren, werden momentan in klinischen Studien erprobt. Bei
Kindern können sowohl die Elementardiät, individuelle Eliminationsdiäten als auch
die rigide 6-Food-Eliminationsdiät zur Behandlung der EoE eingesetzt werden.
Nachteile all dieser Diätformen sind ihr großer Eingriff in den persönlichen Alltag
sowie die Tatsache, dass bei Wiedereinführung der gewohnten Speisen die EoE
meist rezidiviert. Bei adoleszenten und adulten EoE-Patienten hingegen sind die
Verhältnisse komplexer, da hauptsächlich Sensibilisierungen auf Aerogene-Allergene
vorliegen. Diätetische Behandlungen haben bisher keine überzeugenden Resultate
gezeigt.
Eine gefürchtete Spätfolge der unbehandelten EoE ist das sogenannte Remodeling
des Ösophagus, welches mit Fibrosierung und Ausbildung von schweren Strikturen
einhergehen kann. Diese werden mit Ballondilatation behandelt. Neuere Analysen
zeigen, dass diese Behandlung mit einem minimalen und vertretbaren Risiko zu
länger anhaltender Kontrolle der Beschwerden führt. Durch eine frühzeitige und

8
konsequente anti-inflammatorische Therapie kann diese Spätkomplikation
wahrscheinlich vermieden werden.
Um die Betreuung der EoE-Patienten jedoch adäquat zu gewährleisten, müssen
noch viele offene Fragen zu Epidemiologie, Krankheitsverlauf und Therapie
möglichst mittels multizentrisch durchgeführter Studien beantwortet werden.

9
Therapie der chronischen Hepatitis C – Standards und Perspektiven
C. Sarrazin
Medizinische Klinik I, Klinikum der Johann Wolfgang Goethe-Universität, Frankfurt
am Main
Die aktuelle Standardtherapie der chronischen Hepatitis-C-Virus (HCV)-Infektion mit
Peg-Interferon und Ribavirin wird im Laufe des Jahres durch eine Triple-Therapie mit
Proteaseinhibitoren ganz wesentlich erweitert werden.
Im Herbst dieses Jahres wird mit der Zulassung der beiden HCV-Proteaseinhibitoren
Telaprevir und Boceprevir gerechnet.
Beide Substanzen werden als Tabletten dreimal täglich in Kombination mit Peg-
Interferon und Ribavirin als Triple-Therapie verabreicht und beide Substanzen sind
lediglich für Patienten mit einer HCV-Genotyp-1-Infektion zugelassen.
Die abgeschlossenen Phase-III-Studien konnten ebenfalls für beide Substanzen eine
wesentliche Steigerung der dauerhaften virologischen Ansprechraten (SVR)
nachweisen. Bei der Ersttherapie kann unter der Triple-Therapie mit SVR-Raten von
63–75% gerechnet werden, im Vergleich zu 38–44% im Kontrollarm mit der
herkömmlichen Kombinationstherapie. Bei Patienten mit Rückfall (Relaps), partiellem
(partial non-response) oder komplett fehlendem Ansprechen (null-response) in einer
Vortherapie wurden SVR-Raten von 75–86%, 52–57% und 31% erreicht, im
Vergleich zu 24–29%, 7–15% und 5% mit der aktuellen Standardtherapie.
In der praktischen Anwendung der beiden Substanzen gibt es allerdings erhebliche
Unterschiede: Boceprevir wird nach einer sogenannten „lead-in"-Therapie mit Peg-
Interferon und Ribavirin erst nach 4 Wochen zusätzlich und dann für die restliche
Therapiedauer verabreicht, während Telaprevir direkt zum Therapiestart zu Peg-
Interferon und Ribavirin dazugegeben, aber nach 12 Wochen wieder beendet wird
und die weitere Behandlung dann mit Peg-Interferon und Ribavirin allein erfolgt.
Auch die Therapiedauern und Regeln für die Bestimmung der optimalen
Therapiedauer sind unterschiedlich. Wichtig ist dabei der Begriff des anhaltenden
raschen virologischen Ansprechens (extended rapid virologic response, eRVR), der

10
als Abfall der HCV-RNA unter die Nachweisgrenze eines sensitiven Assays nach 4
Wochen Triple-Therapie mit Anhalten zu Woche 12 (Telaprevir) bzw. Woche 24
(Boceprevir) definiert ist. Ist dies der Fall, kann die Behandlung nach 24 (Telaprevir)
bzw. 28 Wochen (Boceprevir) beendet werden. Für Boceprevir ist zusätzlich auch
eine individualisierte Therapie bei vortherapierten Patienten ebenfalls bei Erreichen
eines eRVR über 36 Wochen möglich. Patienten, die kein eRVR erreichen, werden
für 48 Wochen behandelt.
Bei fehlendem Ansprechen zu Woche 4 (> 1000 IU/ml HCV-RNA-Konzentration) wird
die Behandlung mit Telaprevir vorzeitig beendet. Die gesamte Triple-Therapie sollte
bei Patienten mit einem fehlenden 2 log-Abfall zu Woche 12 bzw. bei noch
nachweisbarer HCV-RNA zu Woche 24 vorzeitig beendet werden.
Zusätzliche Nebenwirkungen traten unter beiden Substanzen auf, erfordern ein
spezifisches Management, ggf. die zusätzliche Gabe von Erythropoietin (Boceprevir),
und führten zu etwas höheren Abbruchraten und Dosisreduktionen.
Virusdurchbrüche mit der Selektion von resistenten Virusisolaten wurden bei ca. 5%
der Patienten unter einer Ersttherapie beschrieben und stiegen bei Null-Respondern
auf > 50% der Patienten an. Im Verlauf nach Therapieende zeigen erste Daten, dass
es zu einem langsamen Abfall der Häufigkeit resistenter Varianten kommt. Studien
zur Retherapie von Patienten mit Resistenzen aus der Ersttherapie liegen bisher
nicht vor.
Für Patienten mit einer chronischen Hepatitis C mit anderen HCV-Genotypen gibt es
zunächst keine Änderung und die Kombinationstherapie aus Peg-Interferon und
Ribavirin bleibt der Standard. Mit der Zulassung weiterer Kombinationstherapien mit
und ohne die Gabe von Peg-Interferon und höherer Effektivität beim HCV-Genotyp-1,
insbesondere bei Non-Respondern, aber auch bei Patienten mit HCV-Genotyp-2–6-
Infektion, wird nicht vor 2015 gerechnet.

11
Chronische Hepatitis B: wann und wie behandeln?
T. Berg
Sektion Hepatologie, Klinik und Poliklinik für Gastroenterologie und Rheumatologie,
Universitätsklinikum Leipzig,
Einleitung
Die therapeutischen Möglichkeiten bei chronischer Hepatitis B haben sich in den
letzten Jahren erheblich verbessert. Inzwischen sind in Europa 7 Medikamente zur
Behandlung der chronischen Hepatitis B zugelassen (Standard-Interferon-alfa und
Peg-Interferon-alfa-2a sowie die Nukleos(t)idanaloga Lamivudin, Adefovir,
Telbivudin, Entecavir und Tenofovir) (s. Tab. 1). Die Substanzen unterscheiden sich
in ihrer antiviralen Aktivität, ihrem Nebenwirkungsprofil und im mit ihrem Einsatz
verbundenen Risiko der Resistenzentwicklung. Für die langfristige Kontrolle der
Hepatitis-B-Virus (HBV)-Infektion benötigt die Mehrzahl der HBV-infizierten Patienten
eine antivirale Langzeittherapie über mehrere Jahre. Die Aufrechterhaltung der
Therapieadhärenz und Strategien zur Resistenzvermeidung gehören zu den
besonderen Herausforderungen der Langzeittherapie mit Nukleos(t)idanaloga.
Kriterien für die Indikation zur antiviralen Therapie
Grundsätzlich sollte die Behandlung der HBV-Infektion gemäß den Leitlinien der
Fachgesellschaften erfolgen. Hiernach kommen Patienten mit chronischer Hepatitis B
und quantitativ nachweisbarer Viruslast von > 2000 IU/ml prinzipiell für eine antivirale
Therapie in Betracht. Hintergrund für diese Empfehlung sind Ergebnisse aus
asiatischen Langzeitstudien, in denen ein deutlich erhöhtes Risiko für die
Entwicklung von Leberzirrhose und hepatozellulärem Karzinom (HCC) bereits beim
Vorliegen einer Höhe der HBV-DNA von ≥ 2000 IU/ml gezeigt werden konnte, und
zwar unabhängig von der Höhe der Transaminasen. Die Unterscheidung zwischen
Wildtyp- (HBeAg-positiv, anti-HBe-negativ) und Präcore-Mutanten-HBV-Infektion
(anti-HBe-positiv, HBeAg-negativ) spielt für die Entscheidung über die Durchführung
einer antiviralen Therapie keine Rolle, kann aber hinsichtlich der Auswahl der
Therapieform bzw. der Therapiestrategie von Bedeutung sein.

12
Therapieziele bei chronischer Hepatitis B
Die komplette und anhaltende Suppression der HBV-Replikation wird aufgrund der
bestehenden eindeutigen Korrelation zwischen Höhe der Hepatitis-B-Virämie und
Progression der Erkrankung als wichtigster Therapieendpunkt angesehen.
Nukleos(t)idanaloga-Langzeitstudien konnten eindeutig belegen, dass durch die
langfristige Suppression der Hepatitis-B-Virämie ein signifikanter Rückgang der
histologischen entzündlichen Aktivität und der Fibrose einschließlich Reversion
früher Zirrhosestadien induziert werden kann, verbunden mit einer Reduktion bzw.
Verhinderung der Langzeitkomplikationen (Zirrhose- und HCC-Entwicklung). Eine
Ausheilung der HBV-Infektion mit Verlust des HBsAg und Bildung von anti-HBs-
Antikörpern wird mit den heutzutage zur Verfügung stehenden Medikamenten nur
selten erreicht und stellt daher zwar ein optimales, jedoch bisher wenig realistisches
Therapieziel dar.
Die Monotherapie mit entweder Peg-IFNa oder potenten Nukleos(t)idanaloga bleibt
weiterhin Standard in der Therapie der chronischen Hepatitis B.
Diese beiden Therapieoptionen der chronischen Hepatitis B basieren auf
unterschiedlichen Prinzipien und lassen sich nicht direkt miteinander vergleichen:
Während die (Peg)-IFNa-Therapie aufgrund des Nebenwirkungsprofils nur über
einen begrenzten Zeitraum erfolgen kann, werden die Nukleos(t)idanaloga in der
Regel zur Langzeittherapie eingesetzt, da es bei Kurzzeitanwendung nach Absetzen
der Therapie meist zu einem virologischen Relaps kommt. Das Prinzip der (Peg)-
IFNa-Therapie basiert auf der immunologischen Induktion einer anhaltenden
Remission bei begrenzter Therapiedauer (z. B. über 6–12 Monate). Im Gegensatz
dazu ist das Ziel der Langzeittherapie mit Nukleos(t)idanaloga die anhaltende
Inhibierung der Virusreplikation. Eine primäre Kombinationstherapie von
Nukleos(t)idanaloga oder von (Peg)-IFNa plus Nukleos(t)idanalogon ist bisher nicht
etabliert.
Durch die Einführung der neueren und im Vergleich zu Adefovir und Lamivudin
stärker antiviral wirksamen Inhibitoren der HBV-Polymerase (Entecavir, Tenofovir
und Telbivudin) konnte das Problem der Resistenzentwicklung bei unvorbehandelten
Patienten deutlich reduziert werden. Resistenzentwicklungen gegenüber Entecavir
wurden im Langzeitverlauf über 6 Jahre bei nur 1,2% gefunden. Für Tenofovir sind
bisher keine Resistenzentwicklungen beschrieben worden. Unter einer
Telbivudintherapie liegen die Resistenzraten bei < 5% im Langzeitverlauf, wenn ein

13
komplettes virologisches Ansprechen zur Therapiewoche 24 erreicht worden ist. Im
Gegensatz dazu liegt die Entecavirresistenzrate bei Lamivudin-vorbehandelten
Patienten bei ca. 57% nach 6 Jahren.
Um eine Resistenzbildung gegenüber Nukleos(t)idanaloga zu vermeiden, ist eine
Suppression der HBV-DNA unter die Nachweisgrenze innerhalb von 6–18 Monaten
(in Abhängigkeit von der verwendeten Substanz) zu erreichen, andernfalls sollte die
Therapie umgestellt werden. Kommt es unter der Behandlung zu einem bestätigten
Anstieg der HBV-DNA, der höher als eine logarithmische Stufe ist, liegt mit hoher
Wahrscheinlichkeit eine Resistenz vor und die Therapieform muss angepasst
werden, gegebenenfalls muss eine Kombinationstherapie mit einem Nukleosid- und
einem Nukleotidanalogon begonnen werden. Die Kombination von nicht
kreuzresistenten Nukleos(t)idanaloga führt jedoch nicht zu einer Steigerung der
antiviralen Effektivität. Neuere Studien belegen, dass auch in der Resistenzsituation
eine Monotherapie mit Nukleos(t)idanaloga ebenso effektiv durchgeführt werden
kann, wie bei bisher unvorbehandelten Patienten, wenn die verwendete Substanz
eine volle Suszeptibilität gegenüber der resistenten HBV-Variante besitzt (Beispiel:
Tenofovir bei Lamivudinresistenz).
Die Notwendigkeit einer Kombinationstherapie mit Nukleos(t)idanaloga bei Patienten
mit Resistenzentwicklung oder inkomplettem Ansprechen wird daher zunehmend
kritisch gesehen. Auch die Sicherheit der langfristigen Kombinationstherapie ist
wenig untersucht.
Die Kombination aus Peg-IFNa plus Nukleos(t)idanalogon stellt einen interessanten
Ansatz dar, der in zukünftigen Studien weiter evaluiert werden sollte, da diese
Kombination gegenüber der jeweiligen Monotherapie eine erhöhte antivirale Effizienz
besitzt. Es wird zu prüfen sein, ob auch andere Endpunkte wie der HBsAg-Verlust
langfristig dadurch günstig beeinflusst werden können.
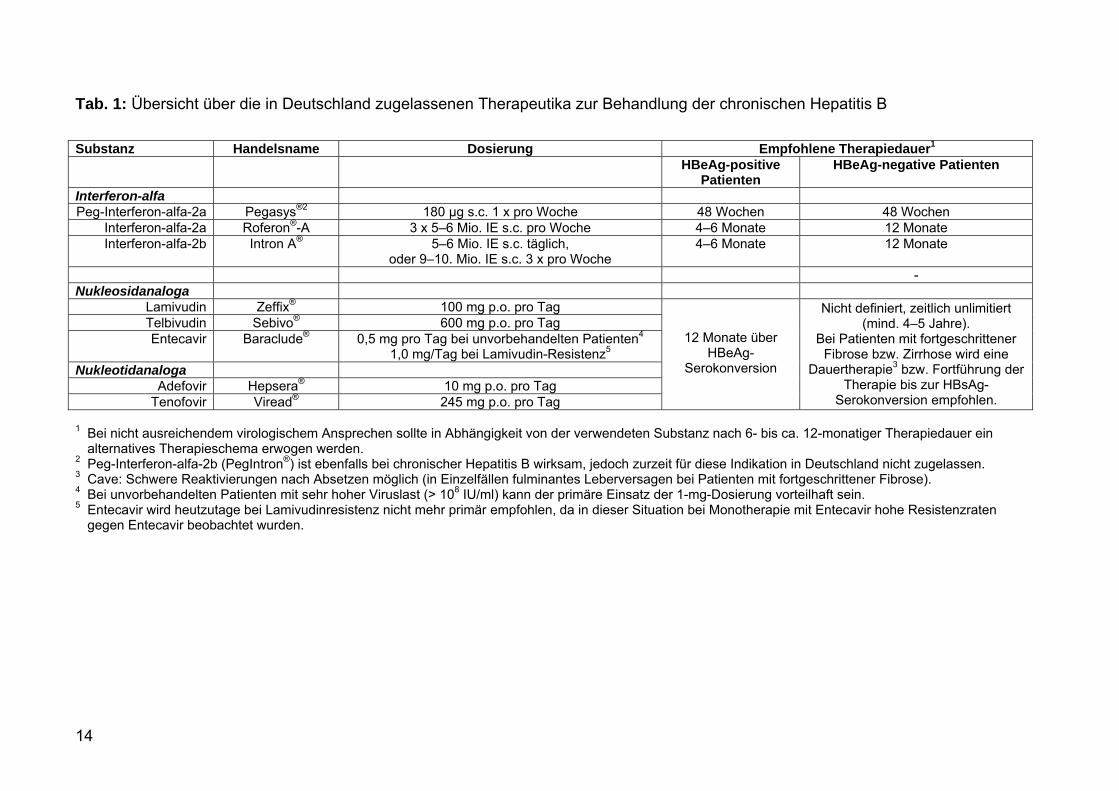
14
Tab. 1: Übersicht über die in Deutschland zugelassenen Therapeutika zur Behandlung der chronischen Hepatitis B Substanz Handelsname Dosierung Empfohlene Therapiedauer1 HBeAg-positive
Patienten HBeAg-negative Patienten
Interferon-alfa Peg-Interferon-alfa-2a Pegasys®2 180 µg s.c. 1 x pro Woche 48 Wochen 48 Wochen
Interferon-alfa-2a Roferon®-A 3 x 5–6 Mio. IE s.c. pro Woche 4–6 Monate 12 Monate Interferon-alfa-2b Intron A® 5–6 Mio. IE s.c. täglich,
oder 9–10. Mio. IE s.c. 3 x pro Woche 4–6 Monate 12 Monate
- Nukleosidanaloga
Lamivudin Zeffix® 100 mg p.o. pro Tag Telbivudin Sebivo® 600 mg p.o. pro Tag Entecavir Baraclude® 0,5 mg pro Tag bei unvorbehandelten Patienten4
1,0 mg/Tag bei Lamivudin-Resistenz5 Nukleotidanaloga
Adefovir Hepsera® 10 mg p.o. pro Tag Tenofovir Viread® 245 mg p.o. pro Tag
12 Monate über HBeAg-
Serokonversion
Nicht definiert, zeitlich unlimitiert (mind. 4–5 Jahre).
Bei Patienten mit fortgeschrittener Fibrose bzw. Zirrhose wird eine
Dauertherapie3 bzw. Fortführung der Therapie bis zur HBsAg-
Serokonversion empfohlen.
1 Bei nicht ausreichendem virologischem Ansprechen sollte in Abhängigkeit von der verwendeten Substanz nach 6- bis ca. 12-monatiger Therapiedauer ein alternatives Therapieschema erwogen werden.
2 Peg-Interferon-alfa-2b (PegIntron®) ist ebenfalls bei chronischer Hepatitis B wirksam, jedoch zurzeit für diese Indikation in Deutschland nicht zugelassen. 3 Cave: Schwere Reaktivierungen nach Absetzen möglich (in Einzelfällen fulminantes Leberversagen bei Patienten mit fortgeschrittener Fibrose). 4 Bei unvorbehandelten Patienten mit sehr hoher Viruslast (> 108 IU/ml) kann der primäre Einsatz der 1-mg-Dosierung vorteilhaft sein. 5 Entecavir wird heutzutage bei Lamivudinresistenz nicht mehr primär empfohlen, da in dieser Situation bei Monotherapie mit Entecavir hohe Resistenzraten
gegen Entecavir beobachtet wurden.

15
Diagnostik und Therapie autoimmuner Leber- und Gallenwegserkrankungen
C.P. Strassburg
Klinik für Gastroenterologie, Hepatologie und Endokrinologie, Medizinische
Hochschule Hannover
Die autoimmunen Lebererkrankungen werden in 3 Krankheitsbilder unterteilt: die
Autoimmunhepatitis (AIH), die primär biliäre Zirrhose (PBC) und die primär
sklerosierende Cholangitis (PSC) (11). Sie zeigen unbehandelt eine Progression zur
Leberzirrhose. Die AIH war die erste chronische Lebererkrankung, bei der eine
konservative Therapie eine dauerhafte Remission erreichen konnte.
Definition und Diagnose der Autoimmunhepatitis (AIH) Die Diagnose der AIH ist gekennzeichnet durch eine Ausschlussdiagnostik anderer
Lebererkrankungen (1). Die serologische Subklassifikation der AIH hat keinen
Einfluss auf die Therapiestrategie. Es sind zu 70–80% Frauen betroffen. Die
Immunglobuline im Serum sind erhöht (7, 12). Hinweisend sind Autoantikörper, die
überlappend auftreten können und auch bei viralen Hepatitiden vorkommen. Die AIH
ist serologisch heterogen und kann prinzipiell in 3 Untergruppen unterteilt werden:
die, die antinukleäre Autoantikörper und Antikörper gegen glatte Muskelzellen (ANA,
SMA) aufweisen (AIH Typ 1), die, die durch Leber-Niere-mikrosomale Autoantikörper
(LKM-1) gekennzeichnet sind (AIH Typ 2), oder die, bei der Antikörper gegen
lösliches Leberantigen/Leber-Pankreas-Antigen (SLA/LP) nachweisbar sind (AIH Typ
3). Am häufigsten ist die ANA-positive AIH (12).
Standardtherapie der AIH Ziel der Therapie ist die Induktion und Erhaltung einer Remission. Sie ist indiziert,
wenn die Aminotransferasen erhöht sind, histologisch multilobuläre oder
Brückennekrosen auftreten oder auch wenn erhebliche hepatische oder
extrahepatische Symptome vorliegen. Gleich effektiv ist die Monotherapie aus
Prednisolon oder die Kombinationstherapie mit Azathioprin. Die Entscheidung zur
Kombinationstherapie orientiert sich am Risikoprofil des Patienten (Schwangerschaft,
metabolisches Syndrom, Diabetes mellitus, Osteoporose). Eine Remissionsinduktion
gelingt in 87% der Fälle innerhalb von 3 Jahren. Allerdings ist die Rückfallrate mit

16
70% innerhalb von 3 Jahren nach Absetzen der Immunsuppression erheblich. Das
10-Jahres-Überleben in Remission beträgt 90%. Ist innerhalb von 4 Jahren keine
Remission erreichbar, bleibt die Lebertransplantation.
Das Problem der Remissionsinduktion
In etwa 10% der Fälle gelingt eine Remissionsinduktion nicht. Hier werden alternative
immunsuppressive Therapeutika eingesetzt: Ciclosporin A, Cyclophosphamid,
Mycophenolsäure, Tacrolimus (FK506). Diese Immunsuppressiva sind wegen ihres
ungünstigen Nebenwirkungsprofils jedoch Studien an hepatologischen Zentren
vorbehalten.
Das Problem der Remissionserhaltung
Mit dem Ziel des Steroidsparens konnte gezeigt werden, dass eine
Remissionserhaltung (aber nicht eine Remissionsinduktion) mit Azathioprin-
Monotherapie (2 mg/kg/KG/Tag p. o.) erreicht werden kann. Eine weitere Möglichkeit
zur potenziellen Verminderung von Steroidnebenwirkungen ist die Anwendung des
topischen Steroids Budesonid. Dessen Vorteile werden in einem über 90%igen
hepatischen First-pass-Metabolismus gesehen, was bei portosystemischen Shunts
und fortgeschrittener Zirrhose eingeschränkt ist. Bei gering ausgeprägter
Leberfibrose und damit eher geringerem Risiko von portosystemischen
Shuntverbindungen kann zur Remissionserhaltung Budesonid eingesetzt werden,
das einen hohen hepatischen First-pass-Metabolismus aufweist und typische
Steroidnebenwirkungen reduzieren oder verhindern helfen kann. Ein erster Bericht
von 10 Patienten, die mit konventioneller Therapie keine Remission erreichten, führt
zu einer zurückhaltenden Beurteilung der Budesonidtherapie. In der Folge wurde in
einer Studie dokumentiert, dass Budesonid zur Remission bei nicht vorbehandelten
Patienten führen kann. In der bislang größten prospektiven randomisierten
Therapiestudie der AIH, in die 207 Patienten aus 30 Zentren eingeschlossen wurden,
zeigte sich, dass Budesonid in der Kombination mit Azathioprin eine komplette
Remissionen erreichte. Die Budesonid-Kombinationsbehandlung wurde gegen die
Kombination aus Prednison und Azathioprin randomisiert, wobei nach 6 Monaten
Behandlungsdauer eine open-label-Weiterbehandlung des Prednisonarms erfolgte.
Eine komplette Remission war als Normalisierung der Aminotransferasen und das
Fehlen von Steroidnebenwirkungen definiert. Ein komplettes Ansprechen wurde im
Budesonidarm nach 12 Monaten in 60,2% beobachtet (Prednison: 49,4%), eine

17
biochemische Remission in 68% (Prednison: 50,6%). Unter den Patienten, die nach
6 Monaten auf Budesonid gewechselt wurden, reduzierten sich die
Steroidnebenwirkungen von 40,2% auf 18,4%. Diese Daten belegen, dass
Budesonid in der Therapie der AIH effektiv ist und künftig eine zunehmend wichtigere
Rolle spielen wird.
Diagnose der primär biliären Zirrhose (PBC) Die Diagnose der PBC erfolgt durch den serologischen Nachweis antimitochondrialer
Antikörper (AMA mit PDH-E2- oder BCKD-E2-Spezifität), das cholestatische
Leberenzymprofil, eine Immunglobulin-M-Erhöhung im Serum sowie durch den
histologischen Nachweis einer entzündlichen Gallenwegsbeteiligung (10).
Sonografisch und in der endoskopisch retrograden Cholangiografie (ERC) sind die
Gallenwege nicht erweitert. Auffällig ist die hohe Anzahl von extrahepatischen
Erkrankungen (Tab. 1).

18
Serologische Befunde Autoantigene
Antimitochondriale Autoantikörper
Pyruvatdehydrogenase (PDH-E2)
Verzweigtkettenketosäuredehydrogenase
(BCKD-E2)
Antinukleäre Autoantikörper
anti-SP100
anti-gp210
anti-Laminin-B-Rezeptor
anti-nucleoporin p62
Extrahepatische Manifestationen
Sicca-Syndrom/Sjögren-Syndrom
rheumatoide Arthritis
Autoimmunthyreoiditis
mixed connective tissue disease (MCTD)
CREST-Syndrom
Polymyalgie
chronisch entzündliche Darmerkrankungen (CED)
systemischer Lupus erythematodes (SLE)
Leberhistologie bei AMA-Negativität: zur Unterstützung der Diagnostik
(Gallenwegsbeteiligung? Granulome?)
bei AMA-Positivität: fakultativ, nur für Fibrosegrad („Staging“) und
entzündliche Aktivität („Grading“)
Die Reihenuntersuchung (Screening) der AMA erfolgt durch Immunfluoreszenz, die durch Western
blot und enzyme-linked immunosorbent assay (ELISA) spezifiziert werden. In 5% treten AMA-nega-
tive Fälle auf, in denen ANA zur weiteren Diagnosefindung beitragen können. Die extrahepatischen
Manifestationen können den Symptomen der PBC zeitlich deutlich vorausgehen.
Tab. 1: Diagnostik der primär biliären Zirrhose
Standardtherapie der PBC
Eine immunsuppressive Behandlung der PBC ist in der überwiegenden Mehrzahl der
Patienten nicht erfolgreich. Die Standardbehandlung besteht aus der oralen Gabe
von 13–15 mg/kg KG/Tag Ursodeoxycholsäure (UDCA). Sie führt zur Besserung der
biochemischen Serumparameter einschließlich des Bilirubins (Mayo-Prognose

19
modell), des Überlebens, aber nicht der portalen Hypertension. Die Datenlage zum
Einfluss von UDCA ist kontrovers, allerdings ist besonders bei früher Behandlung ein
prognostisch günstiger Effekt zu erwarten. Durch UDCA wenig beeinflusst werden
Müdigkeit und Osteoporose (9).
Diagnose der primär sklerosierende Cholangitis (PSC) Die PSC zeichnet sich durch eine progressive Destruktion großer intra- und
extrahepatischer Gallenwege aus und betrifft in 64% der Fälle Männer mit einem
Altersmaximum zwischen 25 und 45 Jahren. Auffallend häufig ist die Colitis ulcerosa
(CU) (England 71%, Schweden 72%, eigene Patienten Hannover 52%) mit der PSC
assoziiert, seltener der Morbus Crohn (eigene Patienten Hannover 11%) (2, 16).
CED-Patienten mit auffälligen Leberwerten (ca. 5%) sollten auf eine PSC untersucht
werden. Die CU bei PSC-Patienten ist häufiger durch eine klinisch inapparente
Pankolitis, eine „backwash ileitis“ und durch rektale Aussparung gekennzeichnet.
Das Dysplasierisiko ist deutlich erhöht. Das Krankheitsbild der PSC ist durch
Oberbauchbeschwerden, Pruritus, Anorexie und Fieber gekennzeichnet, wobei bis
zu 50% der Patienten asymptomatisch sein können. Die Diagnose gründet sich
neben der Cholestase auf die charakteristischen Befunde der ERC sowie der
Leberbiopsie (Ringfibrose der Gallenwege). Serologisch können atypische
antineutrophile zytoplasmatische Autoantikörper (xANCA) bei bis zu 80% der
Patienten nachgewiesen werden, die allerdings zur Diagnosesicherung wenig
beitragen. In einer Untergruppe liegt eine sogenannte „small bile duct PSC“ vor (3),
bei der in der ERC keine Gallenwegsauffälligkeiten nachweisbar sind und die eine
bioptische Sicherung erfordert.
Beurteilung der PSC als Präkanzerose
Anders als bei der AIH ist bei der PSC das Karzinomrisiko erhöht (2). Die Diagnose
des Cholangiokarzinoms (CCC) des PSC-Patienten ist ein unbefriedigendes
klinisches Problem (16), da Stenosen entzündlich bedingt sein können,
biochemische Tests und bioptische Verfahren wenig Sensitivität und Spezifität
aufweisen und bildgebende Verfahren bei intramural wachsenden Tumoren insensitiv
sind. Das CCC-Risiko des PSC-Patienten beträgt 1,5% pro Jahr und ist damit 161-
fach höher als bei Gesunden. Darüber hinaus besteht eine 10-fache Erhöhung des
Kolorektalkarzinomrisikos und eine 14-fache Erhöhung des Pankreaskarzinomrisikos
(2). Die Diagnostik der PSC sollte jährliche koloskopische Untersuchungen und
Ultraschalluntersuchungen des Abdomens einschließen.

20
Standardtherapie der PSC
Die PSC ist durch medikamentöse Maßnahmen nicht heilbar. Die Therapie der Wahl
in Früh- und Spätstadien ist die UDCA in höheren Dosierungen als bei der PBC (15–
30 mg/kg/KG/Tag p. o.) (5). Niedrigere Dosierungen (unter 10 mg/kg/KG) scheinen
weniger wirkungsvoll zu sein. Insgesamt wird die UDCA-Therapie der PSC nach
einer neueren Studie, in der allerdings die Patientenzahl nicht ausreichend war,
kontrovers beurteilt. Überdies erscheint unter UDCA das Risiko einer Kolondysplasie
vermindert (14). Bei rezidivierenden Cholangitisschüben, steigendem Bilirubin und
fortschreitender portaler Hypertension bleibt die Lebertransplantation, wobei die PSC
im seit 2006 eingeführten „model of end-stage liver disease“ (MELD)-Score nur
unzureichend abgebildet wird.
Endoskopische Therapie
Die endoskopische Dilatation könnte die Cholestase verbessern. Die Kombination
mit UDCA-Therapie führt zu einer signifikanten Verlängerung des
transplantationsfreien Zeitraums und des Überlebens (9). UDCA alleine erreicht
diesen Effekt nicht.
Overlap-Syndrome Überlappende Syndrome zwischen PSC und AIH sowie PBC und AIH kommen in ca.
10% der Fälle vor. Verbindliche Diagnostik- oder Therapierichtlinien gibt es nicht. Es
empfiehlt sich eine histologische Evaluation, eine Bestimmung des
Autoantikörperprofils (13). Klinisch wird zunächst die führende Komponente
behandelt: Steroide bei ausgeprägter Hepatitis, UDCA bei Cholestase. Beide
Behandlungen können kombiniert werden.
Lebertransplantation AIH, PBC und PSC sind anerkannte Indikationen für eine Lebertransplantation. Bei
der PSC liegt das 10-Jahres-Überleben bei 70% (4), bei der PBC zwischen 67% und
88% (6) und bei der AIH bei 80–90% (16). Die Rezidivrate aller 3 Krankheitsbilder
nach Lebertransplantation beträgt rund 25%.
Literatur: 1. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL,
Chapman RW, Cooksley WG, Czaja AJ, Desmet VJ, Donaldson PT, Eddleston

21
AL, Fainboim L, Heathcote J, Homberg JC, Hoofnagle JH, Kakumu S, Krawitt EL, Mackay IR, MacSween RN, Maddrey WC, Manns MP, McFarlane IG, Meyer zum Büschenfelde KH, Zeniya M, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999; 31: 929–938.
2. Bergquist A, Ekbom A, Olsson R, Kornfeldt D, Lööf L, Danielsson A, Hultcrantz
R, Lindgren S, Prytz H, Sandberg-Gertzén H, Almer S, Granath F, Broomé U. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol. 2002; 36: 321–327.
3. Broomé U, Glaumann H, Lindstöm E, Lööf L, Almer S, Prytz H, Sandberg-
Gertzén H, Lindgren S, Fork FT, Järnerot G, Olsson R. Natural history and outcome in 32 Swedish patients with small duct primary sclerosing cholangitis (PSC). J Hepatol. 2002; 36: 586–589.
4. Graziadei IW. Recurrence of primary sclerosing cholangitis after liver
transplantation. Liver Transpl. 2002; 8: 575–581. 5. Harnois DM, Angulo P, Jorgensen RA, Larusso NF, Lindor KD. High-dose
ursodeoxycholic acid as a therapy for patients with primary sclerosing cholangitis. Am J Gastroenterol. 2001; 96: 1558–1562.
6. Liermann Garcia RF, Evangelista Garcia C, McMaster P, Neuberger J.
Transplantation for primary biliary cirrhosis: retrospective analysis of 400 patients in a single center. Hepatology. 2001; 33: 22–27.
7. Manns MP, Strassburg CP. Autoimmune hepatitis: clinical challenges.
Gastroenterology. 2001; 120: 1502–1517. 8. Poupon R, Poupon RE. Treatment of primary biliary cirrhosis. Baillieres Best
Pract Res Clin Gastroenterol. 2000; 14: 615–628. 9. Stiehl A, Rudolph G, Klöters-Plachky P, Sauer P, Walker S. Development of
dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome after endoscopic treatment. J Hepatol. 2002; 36: 151–156.
10. Strassburg CP, Manns MP. Autoimmune tests in primary biliary cirrhosis.
Baillieres Best Pract Res Clin Gastroenterol. 2000; 14: 585–599. 11. Strassburg CP, Obermayer-Straub P, Manns MP. Autoimmunity in liver
diseases. Clin Rev Allergy Immunol. 2000; 18: 127–139. 12. Strassburg CP, Manns MP. Autoantibodies and autoantigens in autoimmune
hepatitis. Semin Liver Dis. 2002; 22: 339–352. 13. Strassburg CP, Manns MP. Primär biliäre Leberzirrhose und Overlap-
Syndrome. Diagnostik und Therapie. Internist (Berl). 2004; 45: 16–26. 14. Tung BY, Emond MJ, Haggitt RC, Bronner MP, Kimmey MB, Kowdley KV,
Brentnall TA. Ursodiol use is associated with lower prevalence of colonic

22
neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Ann Intern Med. 2001; 134: 89–95.
15. Tischendorf JJ, Meier PN, Strassburg CP, Klempnauer J, Hecker H, Manns MP,
Krüger M. Characterization and clinical course of hepatobiliary carcinoma in patients with primary sclerosing cholangitis. Scand J Gastroenterol. 2006; 41: 1227–1234.
16. Vogel A, Heinrich E, Bahr MJ, Rifai K, Flemming P, Melter M, Klempnauer J,
Nashan B, Manns MP, Strassburg CP. Long-term outcome of liver transplantation for autoimmune hepatitis. Clin Transplant. 2004; 18: 62–69.

23
Akute und chronische Pankreatitis: neue Entwicklungen
P. Layer
Medizinische Klinik, Israelitisches Krankenhaus in Hamburg
1. Akute Pankreatitis
Ätiopathogenese: Die häufigsten Ursachen der akuten Pankreatitis in der
westlichen Welt sind Gallensteine und Alkoholabusus. Die Mechanismen der
Gallenstein-induzierten Form wurden in den letzten Jahren durch mehrere
experimentelle und klinische Studien weiter aufgehellt, sind aber in den Einzelheiten
weiterhin unklar. Ein wichtiger aktueller Befund ist der Nachweis einer biliären
Genese bei einem großen Teil der akut rezidivierenden „idiopathischen"
Pankreatitiden. Auf zellulärer Ebene mehren sich die Hinweise auf eine wichtige
pathogenetische Rolle freier Radikale bei der Induktion und Progression der
Gewebsschädigung.
Klinisches Bild: Die akute Pankreatitis zeigt typischerweise ein schweres
Krankheitsbild und gehört zu den klassischen Differenzialdiagnosen des akuten
Abdomens. Leitsymptom ist der starke, protrahierte Oberbauchschmerz, in vielen
Fällen mit links-betonter gürtelförmiger Ausstrahlung in Rücken oder Schulter. Fast
regelmäßig bestehen Übelkeit und Erbrechen. Das Abdomen ist prall-elastisch
palpierbar („Gummibauch"). Meist bestehen auch Temperaturen bis 38,5° C; dem-
gegenüber ist hohes Fieber untypisch und muss den Verdacht auf eine andere bzw.
zusätzliche Erkrankung richten. Regelmäßig bestehen Hypotonie und Tachykardie, in
schweren Fällen mit dem Vollbild des Volumenmangelschocks. Einblutungen in die
Bauchwand sind stets Ausdruck einer hämorrhagischen Form.
Die Verdachtsdiagnose kann in vielen Fällen allein aufgrund des klinischen Bildes
gestellt werden. Die wichtigsten Ziele der Frühdiagnostik beinhalten: 1. Diagnose-
sicherung und Ausschluss von Differenzialdiagnosen, 2. Bestimmung des Schwere-
grads (zur Prognoseabschätzung) und 3. Differenzierung der Ätiologie (biliär versus
alkoholisch).
Die Diagnosesicherung erfolgt bei typischer Klinik in der Regel durch den Nachweis
von deutlich erhöhten Pankreasenzymaktivitäten im Serum (Amylase, Lipase). Die
bildgebenden Verfahren, insbesondere die Sonografie, zeigen häufig, aber nicht

24
immer, charakteristische Veränderungen; ihr besonderer Wert liegt im Ausschluss
von wichtigen Differenzialdiagnosen. Diese umfassen unter anderem: akute
Cholezystitis, peptisches Ulkus, Mesenterialinfarkt, Strangulationsileus, Aortenaneu-
rysma, aber auch extraintestinale Ursachen wie basale Pleuritis, Herzhinterwand-
infarkt, Nierenkolik.
Zur Abschätzung des Schweregrads haben sich aufwendige Verfahren wie der
Einsatz von Multiscore-Systemen (Ranson, Glasgow, APACHE II etc.) sowie auch
das initiale Kontrastmittel-CT nicht allgemein durchgesetzt. In den meisten Zentren
wurden sie durch einfachere Serummarker wie CRP oder PMN-Elastase abgelöst,
die mit dem Ausmaß der Nekrosen und somit mit klinischem Schweregrad, Verlauf
und Prognose gut korrelieren. Die bildgebenden Verfahren gewinnen dann in der
weiteren Verlaufsdiagnostik große Bedeutung, insbesondere auch um ggf. infizierte
Nekroseareale, Pseudozysten und Abszesse nachzuweisen, z. B. mittels
sonografisch- oder CT-geführter Feinnadelpunktion. Hinweise auf eine biliäre Genese erzwingen bei schwerer Verlaufsform die unverzügliche ERC, ggf. mit der
Konsequenz der Papillotomie und Steinextraktion.
Therapie: Eine spezifische medikamentöse Behandlung existiert gegenwärtig nicht.
Die Verbesserung der Prognose ist auf die Standardisierung einer intensivmedi-
zinischen Basistherapie zurückzuführen, die gezielt durch problemorientierte
Zusatzmaßnahmen zur Prophylaxe und Therapie von Komplikationen ergänzt wird.
Zum Basisprogramm gehören nach der obligaten stationären Einweisung die orale
Nahrungskarenz, die ausreichende parenterale Flüssigkeits- und Elektrolytsub-
stitution und die Schmerzbekämpfung. Die häufigere leichte Verlaufsform ist hiermit
in wenigen Tagen zu stabilisieren.
Problematischer ist hingegen die Behandlung der schweren Form, bei der sich eine
konsequente interdisziplinäre internistisch-chirurgische Betreuung bewährt. Bei der
schweren biliären Pankreatitis ist früh-elektiv (ggf. nach Endosonografie) eine ERCP
mit Papillotomie und Steinextraktion durchzuführen; hierdurch wird der Verlauf
verbessert.
Bei diesen Patienten sollte – trotz uneinheitlicher Studienlage – eine Antibiotikapro-
phylaxe erwogen werden, da hier ab der 2. Woche mit einer zunehmenden
Infektionsrate der Nekrosen zu rechnen ist. Manifeste Infektionen zählen zu den
wichtigsten Prognose-bestimmenden Faktoren, da sie die typischen schweren
Komplikationen der Pankreatitis begünstigen. Auch in diesen Fällen ist die Therapie

25
initial konservativ. Von der noch oft empfohlenen sofortigen chirurgischen
Nekrosektomie raten wir ab.
Die konsequente Prophylaxe bzw. Frühtherapie dieser systemischen und lokalen
Komplikationen ist für die Prognose von entscheidender Bedeutung. Hierzu zählen
zum einen Schock, Niereninsuffizienz, respiratorische Insuffizienz, metabolische
Entgleisungen und Sepsis, zum anderen Pseudozysten, Blutungen, Abszedierung
bzw. Nekroseninfektion.
Die Kreislaufinsuffizienz entsteht aus dem enormen Volumenverlust in dritte
Räume sowie der massiven systemischen Freisetzung vasoaktiver Mediatoren. Die
Behandlung besteht in hoch dosierter intravenöser Volumensubstitution; mitunter
sind passager Katecholamine notwendig. Ein Nierenversagen kann sowohl bereits
früh als auch erst im weiteren Verlauf auftreten. Therapie der Wahl ist die
Hämodialyse, ggf. als CVVHD.
Eine häufige Komplikation ist die respiratorische Insuffizienz, die meist aus einer
kombinierten Störung der Atemmechanik (reflektorischer Zwerchfellhochstand,
Pleuraerguss) und des Gasaustauschs (Atelektasen, Schocklunge) resultiert. Beim
Auftreten einer arteriellen Hypoxie muss die Indikation zur maschinellen Beatmung
bereits frühzeitig gestellt werden. Mit hoher Letalität behaftet ist die Sepsis, die ihren
Herd meist in Abszessen oder infizierten Nekrosen bzw. Pseudozysten hat und meist
ein chirurgisches Eingreifen erzwingt. Weitere Komplikationen sind Entgleisungen
des Glukose-, Kalium-, Säure-Base- und Kalziumstoffwechsels sowie
Gerinnungsstörungen. Die Problematik der Nekroseninfektion wurde bereits
erwähnt.
Zu den häufigsten lokalisierten Komplikationen zählen Pseudozysten, die eine
große spontane Rückbildungstendenz aufweisen. Sie können aber auch zu
erheblichen Folgekomplikationen führen, z. B. Blutung, Infektion, Thrombose, Ruptur,
Aszites- oder Fistelbildung und mechanischen Problemen wie biliärer oder
duodenaler Obstruktion. Für die Primär- und Sekundärkomplikationen stehen heute
unterschiedliche konservative, interventionelle und chirurgische Therapieverfahren
zur Verfügung, die sich in einem interdisziplinären Vorgehen mit einem
gemeinsamen Therapiekonzept von Gastroenterologen, Chirurgen und Radiologen
optimieren lassen.

26
2. Chronische Pankreatitis
Die Hauptursache der chronischen Pankreatitis ist chronischer Alkoholkonsum, der
sich bei 70–80% der Patienten eruieren lässt. Circa 20% der Patienten haben eine
idiopathische Pankreatitis, entweder eine juvenile Form oder eine Altersform. Andere
definierte Ursachen sind sehr selten.
Das klinische Bild der chronischen Pankreatitis ist in vielen Fällen geprägt durch
einen stadienhaften Verlauf mit Schmerzsyndrom oder Pankreatitisschüben in den
ersten Jahren sowie der Entwicklung einer exokrinen und endokrinen Insuffizienz im
Spätstadium. Entsprechend der variablen klinischen Manifestation sind auch die
Differenzialdiagnosen und das diagnostische Vorgehen abhängig vom
Erscheinungsbild und vom Stadium der Erkrankung. Hierbei ist zu berücksichtigen,
dass das Pankreas eine enorme Reservekapazität aufweist, sodass Funktions-
störungen erst bei > 90%igem Ausfall des Parenchyms fassbar werden.
Entsprechend orientiert sich die internistische konservative Therapie am klinischen
Leitsymptom und am Stadium der Erkrankung: Entscheidend ist die konsequente
Alkoholkarenz, da durch Ausschaltung der ursächlichen Noxe Frequenz und
Heftigkeit der Schmerzschübe reduziert und der Progress der Erkankung verlang-
samt werden können. Auch Nikotinkarenz wird wegen der inzwischen belegten
pathogenetischen Rolle des Rauchens dringend empfohlen.
Ein wesentliches therapeutisches Ziel ist die adäquate medikamentöse
Schmerzbekämpfung. Diese orientiert sich im Wesentlichen an den Empfehlungen
der WHO. Zu beachten ist allerdings, dass bei schweren bzw. refraktären Schmerzen
einerseits die Compliance (Alkohol-/Nikotinkarenz!) hinterfragt werden sollte,
andererseits der chronische Einsatz von Opiaten bei den oft suchtgefährdeten
Patienten immer gegen mögliche endoskopisch-interventionelle oder chirurgische
Ansätze abzuwägen ist.
Ein weiteres Kardinalziel ist die Normalisierung des Ernährungszustands
(Normalisierung der intraluminalen Verdauung, ggf. Behandlung des Diabetes mellitus).
Das Prinzip der Therapie der Maldigestion, die sich meist in der 2. Krankheits-
dekade entwickelt, besteht im Ersatz der Verdauungsenzyme. Hierbei ist die
optimale Substitution der Lipaseaktivität in einer adäquaten galenischen Zubereitung

27
(pH-sensitive Mikropräparationen) von zentraler Bedeutung. Ihre Rolle ergibt sich
aus: 1. dem Fehlen von ausreichenden extrapankreatischen enzymatischen Ersatz-
systemen, 2. dem rascheren Syntheseverlust im Verlauf der Krankheit und 3. aus der
hohen Fragilität gegenüber Pankreasproteasen und Säure. Wichtig sind hierbei
neben einer ausreichenden Dosierung (25–40 kU/Mahlzeit) auch die zeitgerechte,
koordinierte Magenentleerung und die rasche duodenale Freisetzung ins Lumen. Die
Erfolgskontrolle erfolgt primär klinisch (Gewichtsverlauf, Stuhlanamnese). Ein
Therapieversagen erfordert weitergehende differenzialdiagnostische Überlegungen
(unzureichende Dosis, postzibale Asynchronie, Compliance-Probleme, duodenale
Hyperazidität, bakterielle Fehlbesiedelung u. a.).
Der Diabetes mellitus tritt in ca. 30–40% der Fälle im Spätstadium auf und erfordert
meist eine sorgfältige Überwachung infolge der Neigung zu Hypoglykämien; die
Blutzuckerprofile sollten hierbei nicht zu straff eingestellt werden.
3. Autoimmunpankreatitis
In den letzten 10 Jahren hat sich die Autoimmunpankreatitis von einer nur wenig
wahrgenommenen und kaum akzeptierten Entität zu einer wichtigen Differenzial-
diagnose pankreatischer Erkrankungen gewandelt. Das Krankheitsbild ist vor allem
auch wegen seiner enormen differenzialtherapeutischen Implikationen (z. B. konser-
vative Therapie versus Whipple-Operation) von großer klinischer und praktischer
Bedeutung.
Klinik: Die klinische Manifestation lässt oft differenzialdiagnostisch an ein
Pankreaskarzinom denken und ist durch Oberbauchschmerzen, Gewichtsverlust und
häufig durch einen Ikterus charakterisiert. Bei anderen Patienten finden sich eher
Symptome wie bei chronischen, mitunter auch rezidivierenden akuten
(„idiopathischen") Pankreatitiden. In vielen Fällen sind Autoimmunphänomene oder
assoziierte Autoimmunerkrankungen nachweisbar. Es wird vermutet, dass das
Krankheitsbild mehr als ein Drittel der Gruppe der idiopathischen chronischen
Pankreatitiden ausmacht.
Diagnosestellung: Die zur Diagnosesicherung geforderten Kriterien werden in
verschiedenen Ländern unterschiedlich propagiert. Generell gilt aber, dass die
Kombination von mindestens 2 (besser 3!) der folgenden Befunde gefordert wird:

28
– typische bildgebende Befunde (Ultraschall, CT, MR, ERCP);
– Zeichen der Autoimmunität in der Serologie (insbesondere Erhöhung von IgG4);
– suspekte Befunde im zytologischen Aspirat bzw. in der Histologie;
– überzeugendes therapeutisches Ansprechen auf systemische Steroide.
Darüber hinaus gibt es suggestive Begleitbefunde, welche die Diagnose nahelegen
können, so z. B. die cholangitische Beteiligung der extra- und/oder hepatischen
Gallenwege sowie die Assoziation mit Autoimmunerkrankungen wie oben erwähnt.
Dennoch erweist sich die Diagnosesicherung oft als schwieriges Problem; in
unklaren Konstellationen empfiehlt sich dringend die Vorstellung in einem Zentrum,
um dem Patienten einerseits ggf. eine unnötige Operation zu ersparen, aber ihm
andererseits diese nicht fälschlich vorzuenthalten.
Therapie: Obwohl Spontanremissionen auftreten können, sind Kortikosteroide heute
die Therapie der Wahl. Diese sind innerhalb der ersten 2–4 Wochen praktisch immer
wirksam. Zu Dosierungen und Therapiedauer gibt es allerdings keine gesicherten
Empfehlungen. Es gibt Hinweise, dass die Erkrankung nach Absetzen der Steroid-
behandlung in mehr als 50% innerhalb eines Jahres rezidiviert, wobei ein erneuter
Steroidstoß auch zwei Drittel der Rezidive erfolgreich behandeln kann. Umgekehrt
benötigt etwa ein Drittel der Patienten eine Immunsuppression (generell mit
Azathioprin; zu anderen Substanzen liegen nur wenige Berichte vor).
Manifestationen mit primärer Beteiligung der Gallenwege und Cholestase sind mit
einem schweren Verlauf korreliert und erzwingen initial häufig eine intensivere
medikamentöse Therapie sowie nicht selten ein passageres Stenting der großen
Gallengänge.

29
Gender-Aspekte in der Viszeralmedizin
G. Möslein
Allgemein- und Viszeralchirurgie, HELIOS St. Josefs-Hospital Bochum-Linden,
Bochum
Überall wird sie propagiert – die „individualisierte Therapie“. Es gibt genomweite
Assoziationsstudien, die die statistische Signifikanz genetischer Alterationen auf eine
phänotypische Auswirkung untersuchen. Trotz der schier unermesslichen Daten aus
molekulargenetischem Profiling wird oft die wichtigste Genotyp-Phänotyp-Beziehung
unter medizinischen Gesichtspunkten im klinischen Alltag fast regelhaft übersehen.
Männer und Frauen sind verschieden und dies betrifft auch Gesundheit und
Krankheit.
In der jüngsten Vergangenheit mehrten sich die Erkenntnisse über diese
Unterschiede aus dem Bereich der Kardiologie. Von verschiedenen Symptomen,
beispielsweise bei Auftreten eines Herzinfarkts, bis hin zu kompletten – gefährlichen
– Unterschieden im Ansprechen auf Digitalis-Präparate. Man stellte fest, dass sich
diese Arzneimittel in Körpern von Frauen anders verteilen und an Männern getestete
Arzneimittelgaben zu einer Überdosierung mit den entsprechenden Nebenwirkungen
führen können. Gender-Medizin versucht systematisch, die geschlechtsspezifischen
Einflussfaktoren auf die Entstehung und den Verlauf einer Erkrankung auf
Risikofaktoren, Diagnostik und Therapie zu erforschen. Die geschlechterspezifischen
Unterschiede bei Gesundheit und Krankheit werden umso sichtbarer, je älter die
Menschen werden – ein Phänomen unserer Gesellschaft. Höchste Zeit also (wie
geschehen) eine umfassende Datenbank, die an der Charité der Universitätsklinik in
Berlin angesiedelt ist, zu etablieren, um systematisch nach Unterschieden zu
fahnden. Nur so kann eine systematische Analyse, die verschiedene Organsysteme
analysiert auch Frauen eine evidenzbasierte Medizin zuteil werden lassen.
Frauen werden beispielsweise älter als Männer. Zum einen ist die Ursache hierfür
zum Teil verhaltensbedingt, z. B. durch die Bereitschaft, Präventionsmaßnahmen in
Anspruch zu nehmen, oder durch berufliche Risiken der Männer. Zum anderen, so
der Kenntnisstand heute, sind es aber auch reale Unterschiede. Der englische

30
Begriff „gender“ umfasst sowohl die biologischen als auch die psychosozialen
Aspekte der Geschlechtszugehörigkeit.
Frauen werden älter als Männer – lt. statistischem Bundesamt waren 2009 in
Deutschland mehr als 56% der > 60-jährigen Menschen Frauen. Hier spielt neben
der Geschlechterrolle auch eine soziale Rolle eine Bedeutung. Für fast alle
Todesursachen und alle Altersgruppen gilt, dass Männer früher sterben als Frauen.
Ein wesentlicher Grund für das vorzeitige Versterben ist die männliche
Risikobereitschaft, die zu einer wesentlich höheren Sterblichkeit bei jungen Männern
führt. Bei den älter werdenden Männern nehmen Herz-Kreislauf-Erkrankungen einen
wichtigen Platz ein. Weiblichen Geschlechtshormonen kommt offensichtlich eine
schützende Wirkung für ganz verschiedene Erkrankungen zu. Männer neigen bei
Infektionen beispielsweise mehr zu Komplikationen wie Sepsis und
Multiorganversagen. Frauen haben zwar seltener Herzinfarkte, sterben aber häufiger
daran. Autoimmunerkrankungen wie Multiple Sklerose oder rheumatoide Arthritis
betreffen etwa 8% der Bevölkerung: 78% der Betroffenen sind Frauen. Diese hohe
Prävalenz der Autoimmunerkrankungen bei Frauen kann Ergebnis der Interaktion
zwischen Hormonen und dem Immunsystem sein.
Da der Aspekt der biologischen Unterschiede zwischen Männern und Frauen bisher
unbeachtet blieb, fehlen auch gute Zahlen für einen evidenzbasierten
Erkenntnisgewinn. In vielen Studien wird gar nicht aufgeführt, um welches
Geschlecht es sich bei den Probanden handelte. Vor allem früher wurden die
allermeisten Studien mit männlichen Probanden durchgeführt. Um Unterschiede zu
erfassen, sind demnach primär große Datenbanken mit klinischen Informationen
erforderlich, um statistisch relevante Ergebnisse zu erforschen. Ein Beispiel für einen
Erkenntnisgewinn aus solchen Datenbanken liefert die Erfassung der Ergebnisse der
Vorsorgekoloskopie, die in Deutschland auf vorbildliche Weise von den
Krankenkassen empfohlen und finanziert wird. Im Zuge einer Qualitätssicherung
konnten valide Ergebnisse generiert werden.
Kolligs et al. aus München analysierten die Ergebnisse bei 625.918 Patienten, die
sich zwischen 2006 und 2008 einer Dickdarmspiegelung unterzogen hatten.
Fortgeschrittene neoplastische Veränderungen wurden bei 4,6% festgestellt. Die
Daten stammen einerseits aus den Ergebnissen der Vorsorgekoloskopien,

31
andererseits aus Darmspiegelungen, die in allen Altersgruppen wegen Beschwerden
veranlasst wurden und auch aus den Darmspiegelungen, die nach einem positiven
Haemoccult-Test veranlasst wurden. Wenn man nach diesen Unterschieden fahndet
und die Literatur durchforstet, so werden immer wieder Signifikanzen festgestellt, die
einen Einfluss auf die Entstehung, aber auch die Behandlung des Kolonkarzinoms
haben. Bei der familiären Variante des Kolonkarzinoms, dem sogenannte Lynch-
Syndrom, weisen Frauen einen protektiven Effekt auf und erkranken seltener und
später an Kolonkarzinomen als Männer. Bei der Therapie eines fortgeschrittenen
Kolonkarzinoms mit EGFR (epidermal growth factor receptor) führen funktionelle
Polymorphismen zu gegensätzlichen prognostischen Implikationen für männliche und
weibliche Patienten.
Zum Teil fehlen noch die harten evidenzbasierten Daten, um unterschiedliche
Vorsorge- oder Behandlungskonzepte für Patienten mit Erkrankungen in der
Viszeralmedizin festzulegen. Im Sinne einer Prognoseverbesserung beider
Geschlechter, aber auch einer Effizienzsteigerung des kostengeplagten
Gesundheitssystems, muss die Gender-Forschung Eingang in den klinischen Alltag
finden. Konkrete Beispiele für bereits heute einzufordernde unterschiedliche
Behandlungskonzepte werden an konkreten Beispielen vorgestellt.

32
Morbus Crohn – aktuelle Kontroversen
B. Siegmund
Medizinische Klinik I, Charité – Universitätsmedizin, Campus Benjamin Franklin
(CBF), Berlin
Die Erstdiagnose eines Morbus Crohn wird in der Mehrheit bei jüngeren Patienten
gestellt, begleitet sie also für den Rest ihres Lebens und erfordert daher langfristig
ausgerichtete Therapiekonzepte, deren primäre Ziele eine hohe Lebensqualität bei
möglichst geringer Krankheitsaktivität und Komplikationen beinhaltet. Dass wir diese
Ziele mit medikamentöser Therapie erreichen können, verdeutlicht eine kürzlich
publizierte populationsbasierte Kohorte von Patienten aus Cardiff, bei denen der
Verlauf des Morbus Crohn in 3 Zeiträumen untersucht wurde: 1986–1991, 1992–
1997 und 1998–2003. Vergleicht man diese 3 Zeiträume, so nahm der Einsatz
immunsuppressiver Medikamente im Verlauf signifikant zu, wobei die mediane Dauer
bis zur Erstgabe von Thioguaninen abnahm. Gleichzeitig nahm der Anteil der
Patienten mit langfristiger Steroidtherapie ab (1). Die Einflüsse auf die kumulative
Operationswahrscheinlichkeit werden in dieser und einer älteren Arbeit kontrovers
diskutiert (1, 2). Implizieren diese Daten, dass wir alle Patienten möglichst früh
aggressiv immunsuppressiv behandeln sollen, oder welche anderen Faktoren sollten
mit in Betracht gezogen werden, wenn wir bei unseren Patienten differenzierte
Therapieentscheidungen treffen?
Einfache Überlegungen, die auf einer älteren Arbeit basieren, zeigen, dass zu Beginn
der Erkrankung die Entzündung im Vordergrund steht und erst im Verlauf
mutmaßlich als Resultat der Entzündung die Komplikationen wie Stenosen und
Fisteln entstehen (3). Einzelne Risikofaktoren konnten identifiziert werden, die mit
einem hohen Risiko für einen komplizierten Verlauf innerhalb von 5 Jahren assoziiert
sind. Hierzu gehören das junge Alter bei Erstdiagnose, der ileokolische Befall,
perianale Läsionen sowie der Steroidbedarf beim ersten Schub (4).
Wie sollen wir nun behandeln? Die Daten der SONIC-Studie sowie eine retrospektive
französische Beobachtungsstudie zeigen, dass bei Immunsuppressiva-naiven
Patienten auch nach 1 Jahr die Kombinationstherapie Thioguanin plus Infliximab der
jeweiligen Monotherapie für die Remissionsinduktion und die Remissionserhaltung

33
signifikant überlegen ist (5, 6). Aus diesen Daten jedoch die praktische Konsequenz
zu ziehen, alle Patienten, die Immunsuppressiva-naiv sind, kombiniert zu behandeln,
ist nicht sinnvoll, da mit der Kombinationstherapie auch eine Zunahme der
Komplikationen auf lange Sicht zu erwarten ist. Eine frühe Eskalation bei
Therapieversagen der Monotherapie sollte jedoch basierend auf diesen Daten
erfolgen. Grundsätzlich deuten die verfügbaren Studien darauf hin, dass eine
Abheilung der Mukosa einen prognostisch günstigen Faktor darstellt und dies eher
bei Patienten erreicht wird, die eine kürzere Krankheitsdauer haben (7, 8). Kritisch
anzumerken bleibt an dieser Stelle, dass die Mukosaheilung nie der primäre
Endpunkt dieser Studien war und damit diese Schlussfolgerungen durch aktuell
laufende Studien bestätigt werden müssen. Ebenfalls entscheidend wird sein, bei
vorliegender Remission klare Exitstrategien für eine Therapiedeeskalation oder sogar
Therapiepause zu entwickeln.
Eine weitere Kontroverse stellt die postoperative Rezidivprophylaxe vor, die im
Idealfall innerhalb von 2 Wochen nach der Operation eingeleitet werden sollte. Dass
wir diese Therapie bei Patienten mit einem vorherigen komplizierten Verlauf und
entsprechenden Risikofaktoren einleiten, steht nicht zur Diskussion. Soll aber nach
jeder Ileozökalresektion eine medikamentöse Therapie eingeleitet werden? Eine
Metaanalyse zu Azathioprin zeigt, dass dies mit Azathioprin möglich ist (9, 10).
Pilotstudien zeigen, dass auch anti-TNF-Strategien hier effektiv sind (11). Eine
weitere Pilotstudie zeigt aber gleichzeitig, dass wenn ein endoskopisches Rezidiv
postoperativ erkannt wird, der Einsatz von anti-TNF-Strategien ebenfalls wirksam ist
(12). Kontrollierte Studien wären für diese Fragestellung wünschenswert. Bis dahin
könnte man sich für den bislang unkomplizierteren Verlauf auch auf eine
endoskopische Überwachung im Verlauf einigen und davon die weitere
medikamentöse Therapie abhängig machen.
Die Therapie des Morbus Crohn bietet zunehmend mehr Möglichkeiten, erfordert
aber auch das kritische Abwägen von Risikofaktoren, dem zu erwartenden
Krankheitsverlauf und möglichen Nebenwirkungen bei den Therapieentscheidungen.
Wie an den oben diskutierten Punkten zu erkennen, fehlen aktuell für einen Teil der
Kontroversen klare Aussagen. Die Vielzahl der Studien der letzten Jahre zeigt aber
gleichzeitig auf, dass hier viel in Bewegung ist und mit einigen Antworten in naher
Zukunft gerechnet werden darf.

34
Literatur: 1. Ramadas AV, Gunesh S, Thomas GA, Williams GT, Hawthorne AB. Natural
history of Crohn's disease in a population-based cohort from Cardiff (1986–2003): a study of changes in medical treatment and surgical resection rates. Gut. 2010; 59: 1200–1206.
2. Cosnes J, Nion-Larmurier I, Beaugerie L, Afchain P, Tiret E, Gendre JP. Impact
of the increasing use of immunosuppressants in Crohn's disease on the need for intestinal surgery. Gut. 2005; 54: 237–241.
3. Cosnes J, Cattan S, Blain A, Beaugerie L, Carbonnel F, Parc R, Gendre JP.
Long-term evolution of disease behavior of Crohn's disease. Inflamm Bowel Dis. 2002; 8: 244–250.
4. Van Assche G, Dignass A, Panes J, Beaugerie L, Karagiannis J, Allez M,
Ochsenkühn T, Orchard T, Rogler G, Louis E, Kupcinskas L, Mantzaris G, Travis S, Stange E; European Crohn's and Colitis Organisation (ECCO). The second European evidence-based Consensus on the diagnosis and management of Crohn's disease: Definitions and diagnosis. J Crohns Colitis. 2010; 4: 7–27.
5. Colombel JF, Sandborn WJ, Reinisch W, Mantzaris GJ, Kornbluth A,
Rachmilewitz D, Lichtiger S, D'Haens G, Diamond RH, Broussard DL, Tang KL, van der Woude CJ, Rutgeerts P; SONIC Study Group. Infliximab, azathioprine, or combination therapy for Crohn's disease. N Engl J Med. 2010; 362: 1383–1395.
6. Sokol H, Seksik P, Carrat F, Nion-Larmurier I, Vienne A, Beaugerie L, Cosnes J.
Usefulness of co-treatment with immunomodulators in patients with inflammatory bowel disease treated with scheduled infliximab maintenance therapy. Gut. 2010; 59: 1363–1368.
7. Baert F, Moortgat L, Van Assche G, Caenepeel P, Vergauwe P, De Vos M,
Stokkers P, Hommes D, Rutgeerts P, Vermeire S, D'Haens G; Belgian Inflammatory Bowel Disease Research Group; North-Holland Gut Club. Mucosal healing predicts sustained clinical remission in patients with early-stage Crohn's disease. Gastroenterology. 2010; 138: 463–468.
8. Colombel JF, Schreiber S, Rutgeerts P, Sandborn WJ, Yang HJ, Lomax, KG,
Pollak JF, Thakkar RB, Camez A, Huang B, Zhou Q, Mulani P, Chao J. Duration of Crohn's disease affects mucosal healing in adalimumab-treated patients: results from EXTEND. J Crohns Colitis. 2010; 4: S36.
9. Peyrin-Biroulet L, Deltenre P, Ardizzone S, D'Haens G, Hanauer SB, Herfarth H,
Lémann M, Colombel JF. Azathioprine and 6-mercaptopurine for the prevention of postoperative recurrence in Crohn's disease: a meta-analysis. Am J Gastroenterol. 2009; 104: 2089–2096.

35
10. Reinisch W, Angelberger S, Petritsch W, Shonova O, Lukas M, Bar-Meir S, Teml A, Schaeffeler E, Schwab M, Dilger K, Greinwald R, Mueller R, Stange EF, Herrlinger KR; International AZT-2 Study Group. Azathioprine versus mesalazine for prevention of postoperative clinical recurrence in patients with Crohn's disease with endoscopic recurrence: efficacy and safety results of a randomised, double-blind, double-dummy, multicentre trial. Gut. 2010; 59: 752–759.
11. Regueiro M, Schraut W, Baidoo L, Kip KE, Sepulveda AR, Pesci M, Harrison J,
Plevy SE. Infliximab prevents Crohn's disease recurrence after ileal resection. Gastroenterology. 2009; 136: 441–450.
12. Yamamoto T, Umegae S, Matsumoto K. Impact of infliximab therapy after early
endoscopic recurrence following ileocolonic resection of Crohn's disease: a prospective pilot study. Inflamm Bowel Dis. 2009; 15: 1460–1466.

36
Kollagene und lymphozytäre Kolitis – ein Update
S. Miehlke
Magen-Darm-Zentrum, Internistische Kooperation Eppendorf, Hamburg
Die kollagene und die lymphozytäre Kolitis sind Formen der mikroskopischen Kolitis,
die in ihrer klinischen Bedeutung derzeit noch deutlich unterschätzt werden.
Leitsymptom ist die nicht-blutige, wässrige Diarrhö, häufig treten auch nächtliche
Durchfälle, Abdominalschmerzen, Gewichtsverlust und imperativer Stuhldrang auf.
Es besteht eine weibliche Prädominanz, das Hauptmanifestationsalter liegt zwischen
60 und 65 Jahren. Die mikroskopische Kolitis führt bei den betroffenen Patienten zu
einem erheblichen Leidensdruck und zu einer signifikanten Reduktion der
Lebensqualität, vergleichbar mit der Colitis ulcerosa. Der endoskopische Befund ist
üblicherweise unauffällig, die Diagnose kann nur histologisch gesichert werden.
Charakteristisch sind eine lymphoplasmazelluläre Inflammation der Lamina propria,
ein degeneriertes Oberflächenepithel sowie ein verdicktes subepitheliales
Kollagenband (kollagene Kolitis) oder eine Vermehrung intraepithelialer
Lymphozyten (lymphozytäre Kolitis).
Neuere epidemiologische Untersuchungen aus Nordeuropa und Nordamerika
bestätigen, dass die Inzidenz und Prävalenz der mikroskopischen Kolitis zunimmt
und inzwischen ein mit dem Morbus Crohn und der Colitis ulcerosa vergleichbares
Niveau erreicht hat. Durch die klinische Überlappung mit dem Reizdarmsyndrom ist
darüber hinaus von einer relevanten Dunkelziffer auszugehen. Die Ätiologie und
Pathogenese der mikroskopischen Kolitis ist nicht geklärt. Diskutiert wird eine
abnorme immunologische Reaktion auf exogene/luminale Faktoren (Infektionen,
Gallensäuren, Medikamente etc.) bei prädisponierten Personen.
Die Therapie der mikroskopischen Kolitis zielt vor allem auf die rasche
Symptombefreiung, die Normalisierung der Lebensqualität und die Verhütung des
klinischen Rezidivs. Die beste Evidenz für die Therapie der kollagenen und der
lymphozytären Kolitis existiert bisher für Budesonid, das in mehreren randomisierten,
plazebokontrollierten Studien eine hohe Effektivität in der Remissionsinduktion und in
der Remissionserhaltung gezeigt hat. Nach Auffassung der im September 2010
gegründeten Europäischen Expertengruppe (EMCC) ist Budesonid das Mittel der

37
Wahl für alle Patienten mit moderater bis schwerer Symptomatik. Initial sollte mit
9 mg täglich für 6–8 Wochen behandelt werden. Nach einem Auslassversuch sollte
im Fall eines Rezidivs erneut mit Budesonid behandelt und eine dauerhafte
Remissionserhaltung in möglichst niedriger Dosis (3–6 mg täglich) angestrebt
werden. Antidiarrhoika können bei leichter Symptomatik oder additiv bei Bedarf
eingesetzt werden. Der Stellenwert anderer therapeutischer Ansätze
(Aminosalizylate, Bismut, Cholestyramin, Immunsuppressiva, anti-TNFα-Antikörper)
ist bisher nicht gesichert. Das weitere therapeutische Vorgehen bei
Budesonidversagern oder bei Budesonidabhängigkeit ist nicht evidenzbasiert und
muss in Abhängigkeit von der Symptomatik individuell entschieden werden.
In der Zukunft sollten Anstrengungen unternommen werden, das Bewusstsein für die
mikroskopische Kolitis weiter zu schärfen, die Ätiologie und Pathogenese dieser
chronisch entzündlichen Darmerkrankung besser zu verstehen und therapeutische
Alternativen für Budesonidversager und für die Langzeittherapie zu etablieren.

38
Divertikulitis und Clostridium-difficile-Colitis
V. Groß
Medizinische Klinik II, Klinikum St. Marien, Amberg
Die Häufigkeit der Kolondivertikulose steigt mit zunehmendem Alter an. Weitere
Risikofaktoren sind Ballaststoffmangel, hoher Fleischverzehr, Übergewicht sowie
Bewegungsmangel. Circa 75% der Divertikelträger bleiben asymptomatisch, ca. 25%
entwickeln Symptome bzw. Komplikationen. Dazu zählen die Divertikulitis, die
Divertikelblutung, die Divertikel-assoziierte Colitis, sowie die sogenannte
schmerzhafte Divertikelkrankheit. Letztere ist schwierig zu definieren und von einem
Reizdarmsyndrom abzugrenzen.
Die Divertikulitis als wesentliche Komplikation der Divertikulose wird auf der Basis
klinischer Befunde und bildgebender Verfahren (Ultraschall, CT mit rektaler
Kontrastierung) in verschiedene Stadien eingeteilt. Am gebräuchlichsten ist in
Deutschland die Einteilung nach Hansen und Stock.
Divertikulitis: Stadien nach Hansen & Stock*Divertikulitis: Stadien nach Hansen & Stock*
Unkomplizierte leichte Divertikulitis (I)
Akute komplizierte Divertikulitis (II)Peridivertikulitis (Phlegmone) (IIa)Gedeckte Perforation (Abszess) (IIb)Freie Perforation (Peritonitis) (IIc)
Chronisch rezidivierende Divertikulitis (III)
*Nach Hansen & Stock, Langenbecks Arch Chir. 1999; (Suppl. II) 1257
Abb. 1
Die Therapie der akuten Divertikulitis erfolgt stadienadaptiert. Im Stadium I reichen
eine kurzfristige flüssige Ernährung und Kostaufbau nach wenigen Tagen plus eine
orale Antibiotikatherapie, z. B. mit Ciprofloxacin oder Amoxicillin/β-Lactamase-

39
inhibitor. Im Stadium II ist in der Regel eine längerfristige orale Nahrungskarenz und
somit parenterale Ernährung indiziert plus eine i.v.-antibiotische Therapie, z. B. mit
(Acyl)aminopenicillin/β-Lactamaseinhibitor oder Cephalosporin Gruppe II/III plus
Metronidazol oder Carbapenem. Patienten im Stadium IIa können meist konservativ
behandelt werden. Im Stadium IIb erfolgt in der Regel eine konservative
Anbehandlung, in einem Teil der Fälle (ca. 10%) ist eine interventionelle perkutane
Abszessdrainage möglich. Im weiteren Verlauf werden diese Patienten in der Regel
bald operiert. Im Stadium IIc ist eine Notfalloperation erforderlich. Das Stadium III
kann nur operativ kuriert werden. Konservativ anbehandelte Patienten sollten nach
ca. 48 Stunden eine substanzielle Besserung des Lokalbefunds zeigen und nach ca.
einer Woche weitgehend beschwerdefrei sein, ansonsten ist eine frühzeitige OP
indiziert.
Akute Divertikulitis
Stadium IIB Elektiv-OP
Stadium Iunkompliziert
Stadium IIAphlegmonös
Stadium IIBAbszess
Stadium IICPeritonitis
konservativ konservativ konservativ,ggf. Drainage Notfall-OP
substantielle Besserung nach ca. 48 h
ja
weiter konservativ
nein Baldige OP
beschwerdefrei nach ca. 1 Woche weiter konservativ
Therapiealgorithmus akute Divertikulitis
Abb. 2
Die Rezidivrate beträgt nach konservativ behandelter Divertikulitis nach neueren
Studien ca. 13–34%. Die Indikation zur elektiven Operation bei Divertikulitisrezidiven
sollte individuell zusammen mit dem Patienten unter Berücksichtigung der Häufigkeit
und Schwere der Divertikulitisrezidive sowie des Alters und der Begleiterkrankungen
erfolgen. Eine Ausnahme stellen immunsupprimierte Patienten dar, die bei akuter
Divertikulitis ein deutlich erhöhtes Mortalitätsrisiko besitzen und daher in der Regel
bereits nach dem ersten Schub operiert werden sollten.

40
Wodurch können Komplikationen der Divertikulose und Divertikulitisrezidive
verhindert werden? Die Untersuchungen konzentrieren sich auf Ballaststoffe,
Probiotika, das nicht resorbierbare Antibiotikum Rifaximin sowie Mesalazin.
Epidemiologische Untersuchungen zeigten, dass eine ballaststoffreiche Kost mit
einer geringeren Inzidenz einer symptomatischen Divertikelkrankheit assoziiert ist.
Kleinere Fallserien zeigten günstige Effekte einer probiotischen Therapie. Das nicht
resorbierbare Antibiotikum Rifaximin wurde in mehreren offenen Untersuchungen,
zum Teil in Kombination mit Mesalazin eingesetzt. Dabei zeigte sich ein leichter
Vorteil gegenüber den Kontrollen. Mesalazin wird als weiterer Kandidat zur
Verhinderung von Komplikationen der Divertikulose angesehen, da man annimmt,
dass durch eine Stärkung der Mukosabarriere die Entstehung des
Entzündungsprozesses gehemmt wird. Mehrere unkontrollierte Studien weisen auf
eine Wirksamkeit von Mesalazin hin. Eine plazebokontrollierte randomisierte Studie
zur Verhinderung von Divertikulitisrezidiven mit Mesalazin wird aktuell durchgeführt.
Die Häufigkeit der Clostridium-difficile-Colitis hat in den vergangenen 10 Jahren
stark zugenommen. Die klinische Problematik wurde durch die Entstehung eines
Stamms mit erhöhter Virulenz verschärft. C. difficile ist für ca. 15–20% der
Antibiotika-assoziierten Diarrhöen und für ca. 95% der pseudomembranösen
Colitiden verantwortlich. Risikofaktoren für eine C. difficile-Colitis sind eine
vorausgegangene Antibiotikatherapie, höheres Alter (ca. 80% der Fälle bei > 65-
Jährigen), ein längerer Krankenhausaufenthalt, Sondenernährung, eine
gastrointestinale Grunderkrankung, wie z. B. eine chronisch entzündliche
Darmerkrankung, Immunsuppression, eine Protonenpumpenhemmertherapie, sowie
die Unterbringung zusammen mit einem Patienten mit manifester C. difficile-Colitis.

41
Clostridium difficile ColitisClostridium difficile Colitis
C. difficile: obligat anaerob wachsendes grampositives Stäbchenbakterium
Enterotoxin A, Cytotoxin B, binäres Toxin CDTBildung aerotoleranter Sporen
Toleranz gegen Wärme, Austrockunung, verschiedene Chemikalien (einschließlich vieler Desinfektionsmittel)
Vorkommen: ubiquitär (Boden, Wasser, Darmtrakt von Tieren und Menschen)
Häufigkeiten:Im Darm von Erwachsenen: ca. 5%Im Darm von Krankenhauspatienten: ca. 20(-40)%Ursache Antibiotika-assoziierter Diarrhoe: ca. 15-20% Ursache pseudomembranöser Colitis: ca.95%
Epidem. Bulletin 24/2009: 233-239
Abb. 3
Bei Patienten mit über 3 Tage andauernder Diarrhö ohne andere/bekannte Ursache,
insbesondere bei Risikopatienten, wird eine C. difficile-Diagnostik empfohlen. Als
Suchtest eignet sich der Nachweis des „Common Antigens“
(Glutamatdehydrogenase) im EIA (Enzym-gekoppelter Immunadsorptionstest). Der
Test hat eine hohe Sensitivität, jedoch eine niedrige Spezifität. Bei positivem
Testergebnis sollte daher die Diagnose durch den Toxinnachweis bestätigt werden
(Nachweis von Toxin A und B im Stuhl mittels EIA oder Erregeranzüchtung und
Toxinnachweis im Zytotoxizitätstest oder in der PCR). Klinisch kann die
pseudomembranöse Colitis in der Sigmoidoskopie nachgewiesen werden.
Zur Therapie der C. difficile-Colitis stehen als Standardmedikamente Metronidazol
und Vancomycin zur Verfügung. Problematisch ist die Rezidivhäufigkeit der
Erkrankung, die insgesamt bei 10–20% liegt, bei Patienten mit Risikofaktoren (Alter
> 65 Jahre, Notwendigkeit einer weiteren Antibiotikatherapie, schwere Colitis) jedoch
höher ist. Es wird empfohlen, das erste Rezidiv wie die Primärinfektion zu behandeln.
Bei weiteren Rezidiven wird eine ausschleichende Behandlung mit Vancomycin plus
Saccharomyces boulardii empfohlen.

42
Praktische Therapie der C. difficile Colitis Praktische Therapie der C. difficile Colitis
Leichter Verlauf: Metronidazol p.o. 3x500 mg oder 4x250 mg (10 Tage)
Schwerer Verlauf (oder Schwangerschaft oder Alter < 10 Jahre):Vancomycin p.o. 4x125 – 4x500 mg
Sehr schwerer Verlauf:Vancomycon p.o. + Metronidazol i.v.
1. Rezidiv: wie PrimärtherapieWeitere Rezidive: Intervalltherapie mit Vancomycin
1. Woche 4x125 mg2. Woche 3x125 mg3. Woche 2x125 mg (1x125 mg) 4.(-5.) Woche 1x125 mg (125 mg jeden 2. Tag)(6.-7. Woche 125 mg jeden 3. Tag)
plus S. boulardiiNoch Probleme: Rifaximin
Abb. 4
Vor Kurzem wurde von der amerikanischen FDA das makrozyklische Antibiotikum
Fidaxomicin zur Behandlung der C. difficile-Colitis zugelassen. Unmittelbar nach
Ende der Behandlung war Fidaxomicin so wirksam wie Vancomycin, die Rezidivrate
war mit Fidaxomicin jedoch niedriger. Das nicht resorbierbare Antibiotikum Rifaximin
besitzt ebenfalls eine hohe in-vitro-Wirksamkeit gegenüber C. difficile und zahlreiche
Fallberichte sprechen für die klinische Wirksamkeit, auch bei Problempatienten.
Rifaximin ist in verschiedenen Ländern, bisher jedoch nicht in Deutschland zur
Behandlung der C. difficile-Colitis zugelassen. Experimentelle Therapieformen zur
Reduktion der C. difficile-Colitis-Rezidivrate sind Nitazoxanid (z. B. in den USA zur
Behandlung von Protozoen zugelassen), monoklonale Antikörper gegen C. difficile-
Toxine (experimentelle Daten) sowie die „Stuhltransplantation“ (kleine Fallserien).

43
Anschriften der Referenten und Vorsitzenden Prof. Dr. T. Berg Sektion Hepatologie Klinik und Poliklinik für Gastroenterologie und Rheumatologie Universitätsklinikum Leipzig AöR Liebigstr. 20 04103 Leipzig Prof. Dr. W. Fischbach Innere Medizin II Klinikum Aschaffenburg Am Hasenkopf 1 63739 Aschaffenburg Prof. Dr. V. Groß Innere Medizin II Klinikum St. Marien Amberg Mariahilfbergweg 7 92224 Amberg Dr. P. Hruz Kantonsspital Gastroenterologie & Hepatologie Petersgraben 4 4031 Basel Schweiz Prof. Dr. J. Labenz Innere Medizin Ev. Jung-Stilling-Krankenhaus Wichernstr. 40 57074 Siegen Prof. Dr. P. Layer Innere Medizin Israelitisches Krankenhaus in Hamburg Orchideenstieg 14 22297 Hamburg PD Dr. A. Madisch Innere Medizin I Krankenhaus Siloah Klinikum Region Hannover Roesebeckstr. 15 30449 Hannover
Prof. Dr. S. Miehlke Magen-Darm-Zentrum IKE – Internistische Kooperation Eppendorf Eppendorfer Landstr. 42 20249 Hamburg Prof. Dr. A. Morgner-Miehlke Univ.-Klinikum C. Gustav Carus der Technischen Univ. Dresden GB VOU Fetscherstr. 74 01307 Dresden Prof. Dr. G. Möslein Allgemein- und Viszeralchirurgie HELIOS St. Josefs-Hospital Linden Axstr. 35 44879 Bochum Prof. Dr. Dr. G. Rogler Universitätsspital Zürich Klinik für Gastroenterologie & Hepatologie Rämistr. 100 8091 Zürich Schweiz Prof. Dr. C. Sarrazin Innere Medizin I Klinikum der Johann Wolfgang Goethe-Universität Frankfurt Theodor-Stern-Kai 7 60596 Frankfurt PD Dr. B. Siegmund Gastroenterologie Charité Universitätsmedizin Campus Benjamin Franklin (CBF) Hindenburgdamm 30 12203 Berlin Prof. Dr. C.P. Strassburg Klinik für Gastroenterologie, Hepatologie und Endokrinologie Med. Hochschule Hannover Carl-Neuberg-Str. 1 30625 Hannover

Top Related