Aus der Medizinischen Klinik I · Aldosteron wird in der Leber überwiegend in...
49
1 Aus der Medizinischen Klinik I des Marienhospitals Herne - Universitätsklinik - der Ruhr-Universität Bochum Direktor: Prof. Dr. med. L. C. Rump Glukokortikoidsupprimierbarer Hyperaldosteronismus in einer deutschen Familie Inaugural-Dissertation zur Erlangung des Doktorgrades der Medizin einer Hohen Medizinischen Fakultät der Ruhr-Universität Bochum vorgelegt von Christian Altenhenne aus Bochum 2007
Transcript of Aus der Medizinischen Klinik I · Aldosteron wird in der Leber überwiegend in...

1
Aus der Medizinischen Klinik I
des Marienhospitals Herne - Universitätsklinik -
der Ruhr-Universität Bochum Direktor: Prof. Dr. med. L. C. Rump
Glukokortikoidsupprimierbarer Hyperaldosteronismus in einer deutschen Familie
Inaugural-Dissertation zur
Erlangung des Doktorgrades der Medizin einer
Hohen Medizinischen Fakultät der Ruhr-Universität Bochum
vorgelegt von Christian Altenhenne
aus Bochum 2007

2
Dekan: Prof. Dr. med. G. Muhr Referent: Prof. Dr. med. L. C. Rump Koreferent: Prof. Dr. med. J. T. Epplen Tag der Mündlichen Prüfung: 15.04.2008

3
In Dankbarkeit meinen Eltern und meinem Zwillingsbruder gewidmet!

1
Seite Inhaltsverzeichnis 1 Verzeichnis der Abkürzungen 3 1. Einleitung 4 2. Methodik 6 2.1. Allgemeines 6 2.2. Testerklärung und –interpretation 6 2.2.1. Screeningtest 6 2.2.2. Bestätigungstest 6 2.2.3. Bildgebende Verfahren 8 2.2.4. Seitengetrennte Nebennierenvenenblutentnahme 8 2.2.5. Orthostasetest 9 2.3. Spezielle Untersuchungen bei Verdacht auf GSH 10 2.3.1. Langzeit-Dexamethasontest 10 2.3.2. Molekularbiologische Untersuchungen 11 3. Ergebnisse 12 3.1. Allgemeine Daten und Befunde der Patienten 12 3.2. Ergebnisse der Testverfahren 15 3.2.1. Screeningtest 15 3.2.2. Bestätigungstest 16 3.2.3. Bildgebende Verfahren 16

2
3.2.4. Seitengetrennte Nebennierenvenenblutentnahme 17 3.2.5. Orthostasetest 18 3.3. Spezielle Ergebnisse bei GSH 18 3.3.1. Langzeit-Dexamethasontest 18 3.3.2. Molekularbiologische Untersuchungen 20 3.3.3. Blutdruck nach einwöchiger Dexamethasongabe 23 3.3.4. Blutdruckverlauf unter medikamentöser Therapie 23 3.3.5. ARQ und Mikroalbumin unter verschiedenen Medikamenten 26 4. Diskussion 29 4.1. Klinik 29 4.2. Diagnostik 30 4.3. Genetik 31 4.4. Therapie – Beeinflussung der Aldosteroneffekte 33 4.5. Ausblick 35 5. Zusammenfassung 36 6. Literaturverzeichnis 38 7. Danksagung 44 8. Lebenslauf 45

3
Verzeichnis der Abkürzungen A Aldosteron ACE angiotensin converting enzyme ACTH adrenocorticotropes Hormon APA Aldosteron-produzierendes Adenom ARQ Aldosteron/Renin-Quotient bp Basenpaare C Cortisol CT Computertomographie DNA Desoxyribonukleinsäure EDTA Ethylendiamintetraessigsäure GSH Glukokortikoidsupprimierbarer Hyperaldosteronismus IBNH idiopathische bilaterale Nebennierenrindenhyperplasie kb Kilobasen MRT Magnetresonanztomographie mmHg Millimeter Quecksilber NaCl Natriumchlorid NNR Nebennierenrinde NNV Nebennierenvene PCR Polymerase Chain Reaction PHA Primärer Hyperaldosteronismus V. Vena

4
1. Einleitung
In dieser Arbeit werden Familienmitglieder einer deutschen Familie
charakterisiert, die an einem glukokortikoidsupprimierbaren
Hyperaldosteronismus (GSH) leiden.
Bisher wurde der autosomal-dominant vererbte GSH in Deutschland noch in
keiner Familie beschrieben [42]; lediglich in den USA, China, Japan und
Italien [8, 28, 32, 45]. Der Gendefekt liegt auf Chromosom 8 [20].
Der GSH weist unterschiedliche Phänotypen [12] auf, obwohl er eine
monogenetische Form des primären Hyperaldosteronismus (PHA) ist [23].
Für die Diagnose eines GSH ist daher das Vorhandensein eines PHA
erforderlich. Das Krankheitsbild des PHA wurde erstmalig 1955 von Jerome
Conn beschrieben und seitdem in der Literatur als Conn-Syndrom
bezeichnet. Bei dem PHA kommt es zu einer autonomen
Aldosteronproduktion und -sekretion in der Zona glomerulosa der
Nebennierenrinde. Das klassische von Conn beschriebene Symptomtrias
bestehend aus Hypertonie, metabolischer Alkalose und Hypokaliämie [5]
wurde bei den ersten Patientenuntersuchungen zu 100% nachgewiesen [6].
Die Prävalenz des PHA hat in den letzten Jahren in der hypertensiven
Bevölkerung deutlich zugenommen und ist inzwischen die häufigste
Ursache der sekundären arteriellen Hypertonie. Eine kürzlich durchgeführte
Literaturrecherche (12 Studien mit 6649 Patienten aus 5 Kontinenten) zeigte
eine Prävalenz des PHA von 6.5% in der hypertensiven Bevölkerung [4].
Dieses Ergebnis lässt sich auf gezielte Untersuchungen [15, 36] und die
Anwendung des Aldosteron/Renin-Quotienten (ARQ) zurückführen.
Inzwischen ist erwiesen, dass viele Patienten mit PHA einen normalen
Kaliumspiegel haben [35]. Der Aldosteron/Renin-Quotient ist dabei der
effektivste Screeningtest.
Die Kombination aus supprimierter Plasma-Renin-Konzentration und
erhöhtem Serum-Aldosteron ist typisch für den PHA [31].
Bei einem positiven Screeningtest ist ein weiterer Test erforderlich, der die
Diagnose des PHA bestätigt. Als Ursache des PHA können die
idiopathische bilaterale Nebennierenrindenhyperplasie (> 70%), das
Aldosteron-produzierende Adenom (< 30%) und der GSH (~ 1%) bei

5
chimärem 11ß-Hydroxylase/Aldosteronsynthase-Gen in Frage kommen. Zur
genaueren Differenzierung sind bildgebende Verfahren, Orthostasetest und
selektive Nebennierenvenenblutentnahme erforderlich. Dennoch kann die
Differentialdiagnose in einigen Fällen schwierig sein [30].
Das klassische Symptomtrias von Conn wird nur noch selten beobachtet
und ist somit keine Voraussetzung für die Diagnose PHA. Da bis zu 90% der
Patienten normokaliämisch sind, schließt das Fehlen einer Hypokaliämie
einen PHA nicht aus [31].
Unter physiologischen Bedingungen wird die Aldosteronproduktion
hauptsächlich durch ACTH, Angiotensin II und dem Kalium-Spiegel reguliert.
Die Synthese des Steroidhormons Aldosteron erfolgt ausgehend von
Cholesterin unter Mithilfe von verschiedenen Enzymen über die
Zwischenstufen Pregnenolon, Progesteron, 11-Desoxycorticosteron und
Corticosteron. Die 11ß-Hydroxylase (CYP11B1) sorgt für die Umwandlung
von 11-Desoxycorticosteron in Corticosteron, und die Aldosteronsynthase
(CYP11B2) katalysiert die Umwandlung von Corticosteron in Aldosteron. Die
11ß-Hydroxylase wird durch ACTH aktiviert, während die
Aldosteronsynthase durch Angiotensin II und den Kaliumspiegel beeinflusst
wird. In der Niere ist Aldosteron verantwortlich für die Regulation des
Elektrolyt- und Wasserhaushalts, wobei Natrium und Wasser rückresorbiert
sowie Kalium und Protonen exkretiert werden.
Die Nebennieren eines gesunden Erwachsenen produzieren etwa 100-175
µg/Tag (277 – 485 nmol/Tag) Aldosteron. Während einer einzigen
Leberpassage werden ca. 75% des zirkulierenden Hormons inaktiviert.
Aldosteron wird in der Leber überwiegend in Tetrahydro-Glucuronid-Derivate
und zu einem geringen Teil – in Leber und Niere – in ein 18-Glucuronid
umgewandelt. Die Ausscheidung erfolgt über den Harn [44]. Unverändert
werden über den Urin ca. 5% des Aldosterons ausgeschieden [31].

6
2. Methodik
2.1. Allgemeines
Um das Vorliegen eines PHA und seine Unterformen festzustellen, sind die
im folgenden beschriebenen Testverfahren erforderlich.
Des Weiteren dürfen Aldosteron-Rezeptor-Antagonisten (Spironolakton,
Eplerenon) und Kortison während der Durchführung der Testverfahren nicht
gegeben werden. Andere blutdrucksenkende Medikamente wie ACE-
Hemmer oder Betablocker beeinflussen den ARQ nur wenig [16, 21, 43].
2.2. Testerklärung und -interpretation
2.2.1. Screeningtest
Als Screeningtest für den PHA wird der Aldosteron-Renin-Quotient
verwendet, der schon 1981 beschrieben wurde [18].
Hierzu erfolgt eine venöse Blutentnahme von EDTA-Blut und Serum-Blut bei
dem Patienten um 8.00 Uhr im Liegen. Der Patient muss hierfür vorher
mindestens eine Stunde liegen. Das EDTA-Blut wird zur späteren
Bestimmung der Plasma-Renin-Konzentration (ng/l) eingefroren. Im Serum
wird Aldosteron bestimmt (pg/ml).
ARQ-Werte > 50 ergeben einen Verdacht auf einen PHA [38].
Die Blutbestimmungen wurden im Labor Dr. Eberhard u. Partner in
Dortmund durchgeführt.
2.2.2. Bestätigungstest
Für die Diagnose des PHA ist ein Bestätigungstest notwendig. Bei den
möglichen Testverfahren wird der NaCl-Belastungstest hier zugrunde gelegt,
da er einfach durchzuführen ist.

7
Dem Patienten wird im Liegen um 8.00 Uhr venöses Blut zur
Aldosteronbestimmung entnommen. Anschließend erhält der Patient 2 l
NaCl (0.9%) intravenös über 4 Stunden im Liegen. Während dieser Zeit
müssen regelmäßige Blutdruckkontrollen erfolgen. Nach der Gabe erfolgt
eine erneute Blutabnahme im Liegen. Hierbei führt das NaCl zu einer
Zunahme des intravasalen Volumens und physiologischerweise zu einer
Verminderung von Renin, Angiotensin II und letztendlich Aldosteron.
Ein PHA liegt vor, wenn sich das Aldosteron nicht unter 70 pg/ml
supprimieren lässt [41].
Zur differentialdiagnostischen Abklärung ist zunächst eine radiologische
Bildgebung der Nebennieren erforderlich (s. Abbildung 1).
Abbildung 1: Modifizierte Darstellung zur Differentialdiagnose bei
Vorliegen eines PHA (A=Aldosteron, C=Cortisol, MRT=Magnetresonanz-
tomographie, CT=Computertomographie) nach [40]
MRT / CT
Lateralisierung?
Idiopathischebilaterale NNR-
Hyperplasie
Therapie mitMineralokortikoid-
Rezeptor-Antagonisten
Aldosteron-produzierendes
Adenomwahrscheinlich
EinseitigelaparoskopischeAdrenalektomie
SeitengetrennteNebennierenvenen-
Blutentnahme
DifferentialdiagnoseAdenom/bilaterale
Hyperplasie(Vergleich A/C-QuotientVena cava inferior und
Orthostasetest)
Einseitiger Knoten>1cm Radiolog. kein Hinw eis auf Knoten
nein
ja beidseitig selektiveKatheterisierung
Nur einseitig selektiveKatheterisierung

8
2.2.3 Bildgebende Verfahren
Dünnschicht-Computertomographie oder Kernspintomographie der
Nebennieren werden zur Identifizierung eines Nebennierenrindentumors
eingesetzt. Bei diesen bildgebenden Verfahren liegt die Sensitivität und
Spezifität allerdings nur bei ca. 60% [17]. Bei einem
Nebennierenrindentumor > 1 cm liegt sehr wahrscheinlich ein Aldosteron-
produzierendes Adenom (APA) vor.
2.2.4 Seitengetrennte Nebennierenvenenblutentnahme
Bei einem nicht eindeutigen Befund der bildgebenden Verfahren wird zur
Differenzierung zwischen Aldosteron-produzierendem Adenom und
idiopathischer bilateraler Nebennierenrindenhyperplasie eine seiten-
getrennte Nebennierenvenenblutentnahme durchgeführt (s. Abbildung 1).
Hierbei wird in beiden Nebennierenvenen sowie in der Vena cava inferior
Blut selektiv abgenommen zur individuellen Bestimmung von Aldosteron und
Cortisol. Das Ergebnis der seitengetrennten Nebennierenvenen-
blutentnahme ist besonders aussagefähig, wenn beide Quotienten aus den
Cortisolwerten der betreffenden Nebennierenvene und der Vena cava
inferior > 1.1 betragen [33]. Charakteristisch für Patienten mit Aldosteron-
produzierendem Adenom ist ein Gradient des Aldosteron/Cortisolquotienten
zur tumortragenden Seite von mehr als 3:1 [41].
Die selektive Katheterisierung der rechten Nebennierenvene gelingt aber
nur in 74% der Untersuchungen [46]. Ursächlich ist hierfür der direkte
Abgang der rechten Nebennierenvene aus der Vena cava inferior.
Bei nur selektiver einseitiger Nebennierenvenenblutentnahme kann das
Ergebnis dennoch zur weiteren Abklärung herangezogen werden (s.
Abbildung 2).

9
Abbildung 2: Differentialdiagnose bei selektiver einseitiger Nebennieren-
venenblutentnahme (A=Aldosteron, C=Cortisol, IBNH=idiopathische
bilaterale Nebennierenrindenhyperplasie) nach [11]
Falls sich aus den bildgebenden Verfahren und der seitengetrennten
Nebennierenvenenblutentnahme eine Differenzierung zwischen einem
Aldosteron-produzierendem Adenom und einer idiopathischen bilateralen
Nebennierenrindenhyperplasie nicht eindeutig ergibt, ist der Orthostasetest
erforderlich.
2.2.5 Orthostasetest
Beim Orthostasetest wird Aldosteron im Serum um 8.00 Uhr im Liegen
bestimmt. Danach soll der Patient umhergehen; um 12.00 Uhr erfolgt dann
eine erneute Aldosteronbestimmung im Stehen. Physiologisch wird bei
diesem Test ein Anstieg des Aldosteronwertes erwartet. Ein Abfall des
Aldosteronwertes – im Tagesverlauf von 8.00 Uhr (liegend) und 12.00 Uhr
(stehend) – ist jedoch charakteristisch für ein Aldosteron-produzierendes
Adenom. Erklärt wird dies durch eine ACTH-Abhängigkeit der
Aldosteronsekretion.
Verhältnis zwischenA/C-Quotient auf der selektiv entnommenen Seite
und A/C-Quotient der Vena cava inferior
keine Unterscheidung zwischenAldosteron-produzierendem Adenom
und IBNH
Aldosteron-produzierendes Adenomder Gegenseite liegt vor
< 1 ≥ 55

10
Bei der idiopathischen bilateralen Nebennierenrindenhyperplasie ist der
noch vorhandene Einfluss von Angiotensin II auf die Aldosteronsekretion mit
Anstieg des Aldosterons unter Orthostase typisch.
Es ist allerdings zu beachten, dass auch 30% der Aldosteron-
produzierenden Adenome durch Angiotensin II beeinflusst werden und einen
Aldosteronanstieg bei aufrechter Körperhaltung zeigen [31].
Bei nicht übereinstimmenden Befunden oder bei häufigem Auftreten einer
arteriellen Hypertonie in der Familienanamnese sind weitere
Untersuchungen auf einen GSH notwendig.
2.3. Spezielle Untersuchungen bei Verdacht auf GSH
2.3.1. Langzeit-Dexamethasontest
Hierbei werden Aldosteron und Renin im Blut und im 24 Stunden-
Sammelurin Aldosteron und Cortisol sowie deren Metabolite vor und nach
einwöchiger Gabe von Dexamethason (2 mg/d) gemessen. Dexamethason
hat dabei keine mineralokortikoide Wirkung.
Beim GSH soll die Dexamethasongabe zu einer verminderten Expression
des ACTH-abhängigen chimären Gens führen und damit zu einer
verminderten Produktion von Aldosteron.
Erhöhte Werte von 18-Hydroxycortisol und 18-Hydroxycorticosteron im
Sammelurin, die sich nach einwöchiger Gabe von Dexamethason
normalisieren, sprechen für einen hochgradigen Verdacht auf einen GSH.
Des Weiteren soll sich unter Dexamethasongabe eine Normalisierung der
Aldosteron- und Reninwerte zeigen.
Die Urinuntersuchungen wurden im Steroidlabor am Pharmakologischen
Institut der Universität Heidelberg durchgeführt [39].

11
2.3.2. Molekularbiologische Untersuchungen
Der Gendefekt (chimäres Gen) wird mit Southern-Blot Hybridisierung (Exon
1, 3-4 der CYP11B1/2-Gene) nach BamHI (Restriktionsenzym) Spaltung der
genomischen DNA, Agarosegelelektrophorese und Transfer auf eine
Nylonmembran identifiziert. Für die Bestimmung des Bruchpunkts wird mit
long range-PCR ein 3256 bp langes Fragment spezifisch für das CYP11B1
Gen sowie das chimäre Gen amplifiziert und sequenziert.
Die Untersuchungen wurden im Institut für Humangenetik an der Universität
Bochum durchgeführt [19].

12
3. Ergebnisse
3.1. Allgemeine Daten und Befunde der Patienten
Patient 1 ist 18 Jahre alt, 182 cm groß mit einem Gewicht von 68 kg. In der
24-Stunden-Blutdruckmessung wurde ein Blutdruck von 156/98 mmHg und
eine Herzfrequenz von 65/min gemessen. Es zeigte sich keine
Nachtabsenkung (s. Tabelle 1). Ein erhöhter Blutdruck war bereits seit 4
Jahren bekannt.
Patient 2, der 23-jährige Bruder von Patient 1, ist 177 cm groß und wiegt
85 kg. Trotz blutdrucksenkender Medikamente zeigte er in der 24-Stunden-
Blutdruckmessung einen Durchschnittswert von 201/116 mmHg und eine
Herzfrequenz von 59/min (s. Tabelle 1). Ein erhöhter Blutdruck ist auch seit
4 Jahren bekannt. Aufgrund eines exzessiven Blutdruckanstiegs mit der
Folge einer idiopathischen Faszialisparese wurde der Patient bereits in
einem anderen Krankenhaus behandelt und eine antihypertensive Therapie
eingeleitet. Ein Schlaganfall konnte damals im MRT ausgeschlossen
werden. Eine vermutete Linksherzhypertrophie im Elektrokardiogramm
(Sokolow I-Index von 3.6 mV; Norm <3.5) bestätigte sich in der
echokardiographischen Untersuchung nicht.
Patient 3 ist der 51–jährige Vater von Patient 1 und 2 (Körpergröße: 181
cm, Gewicht: 73 kg). Bei ihm ist seit ca. 10 Jahren ein Bluthochdruck mit
Werten > 200/100 mmHg bekannt. In der Anamnese findet sich lediglich
eine rechtsseitige Nierenkolik vor ca. 20 Jahren. Ohne Medikamente zeigte
sich ein durchschnittlicher Blutdruck von 183/113 mmHg in der 24-Stunden-
Blutdruckmessung und eine Herzfrequenz von 71/min (s. Tabelle 1).
Aufgrund des lange bestehenden hohen Blutdrucks wies das
Elektrokardiogramm auf eine Linksherzhypertrophie (Sokolow I-Index von
5.0 mV; Norm <3.5) hin. Im Rahmen einer echokardiographischen
Abklärung hatte sich diese Linksherzhypertrophie mit einer intraventrikulären
Septumdicke von 1.41 cm bestätigt.
13
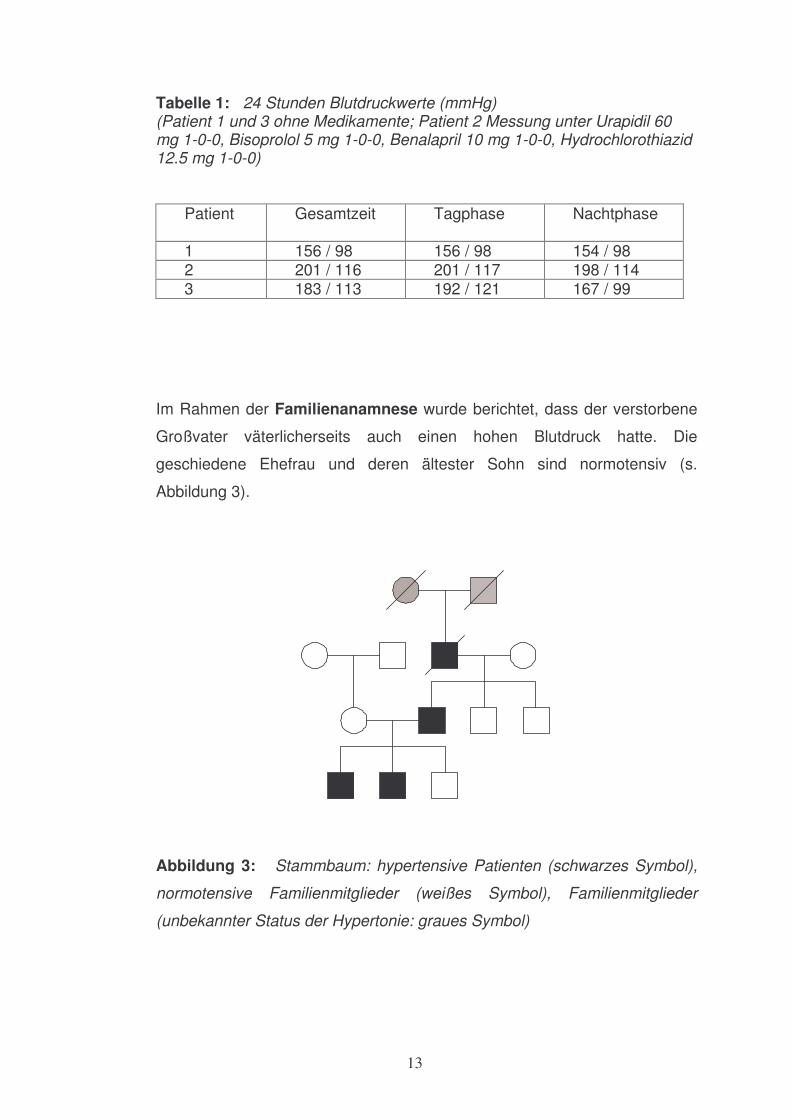
Tabelle 1: 24 Stunden Blutdruckwerte (mmHg) (Patient 1 und 3 ohne Medikamente; Patient 2 Messung unter Urapidil 60 mg 1-0-0, Bisoprolol 5 mg 1-0-0, Benalapril 10 mg 1-0-0, Hydrochlorothiazid 12.5 mg 1-0-0)
Patient
Gesamtzeit Tagphase Nachtphase
1 156 / 98 156 / 98 154 / 98 2 201 / 116 201 / 117 198 / 114 3 183 / 113 192 / 121 167 / 99
Im Rahmen der Familienanamnese wurde berichtet, dass der verstorbene
Großvater väterlicherseits auch einen hohen Blutdruck hatte. Die
geschiedene Ehefrau und deren ältester Sohn sind normotensiv (s.
Abbildung 3).
Abbildung 3: Stammbaum: hypertensive Patienten (schwarzes Symbol),
normotensive Familienmitglieder (weißes Symbol), Familienmitglieder
(unbekannter Status der Hypertonie: graues Symbol)
14
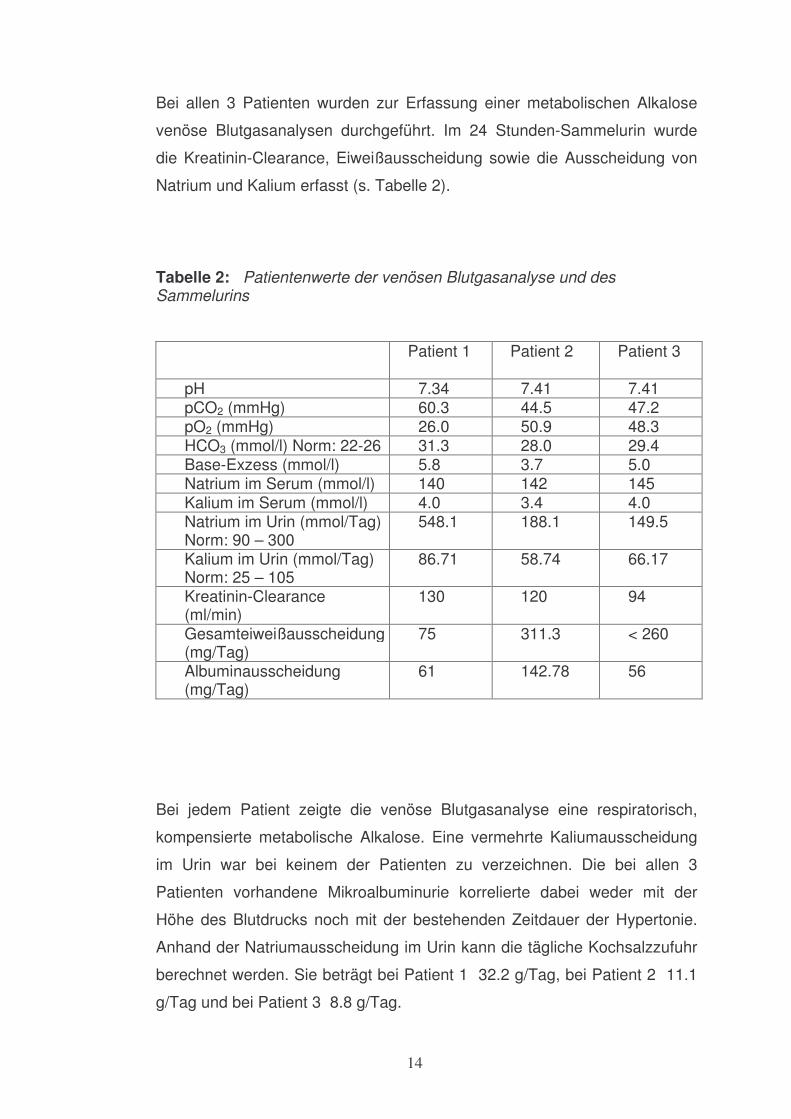
Bei allen 3 Patienten wurden zur Erfassung einer metabolischen Alkalose
venöse Blutgasanalysen durchgeführt. Im 24 Stunden-Sammelurin wurde
die Kreatinin-Clearance, Eiweißausscheidung sowie die Ausscheidung von
Natrium und Kalium erfasst (s. Tabelle 2).
Tabelle 2: Patientenwerte der venösen Blutgasanalyse und des Sammelurins
Patient 1
Patient 2 Patient 3
pH 7.34 7.41 7.41 pCO2 (mmHg) 60.3 44.5 47.2 pO2 (mmHg) 26.0 50.9 48.3 HCO3 (mmol/l) Norm: 22-26 31.3 28.0 29.4 Base-Exzess (mmol/l) 5.8 3.7 5.0 Natrium im Serum (mmol/l) 140 142 145 Kalium im Serum (mmol/l) 4.0 3.4 4.0 Natrium im Urin (mmol/Tag) Norm: 90 – 300
548.1 188.1 149.5
Kalium im Urin (mmol/Tag) Norm: 25 – 105
86.71 58.74 66.17
Kreatinin-Clearance (ml/min)
130 120 94
Gesamteiweißausscheidung (mg/Tag)
75 311.3 < 260
Albuminausscheidung (mg/Tag)
61 142.78 56
Bei jedem Patient zeigte die venöse Blutgasanalyse eine respiratorisch,
kompensierte metabolische Alkalose. Eine vermehrte Kaliumausscheidung
im Urin war bei keinem der Patienten zu verzeichnen. Die bei allen 3
Patienten vorhandene Mikroalbuminurie korrelierte dabei weder mit der
Höhe des Blutdrucks noch mit der bestehenden Zeitdauer der Hypertonie.
Anhand der Natriumausscheidung im Urin kann die tägliche Kochsalzzufuhr
berechnet werden. Sie beträgt bei Patient 1 32.2 g/Tag, bei Patient 2 11.1
g/Tag und bei Patient 3 8.8 g/Tag.
15
3.2. Ergebnisse der Testverfahren
3.2.1. Screeningtest
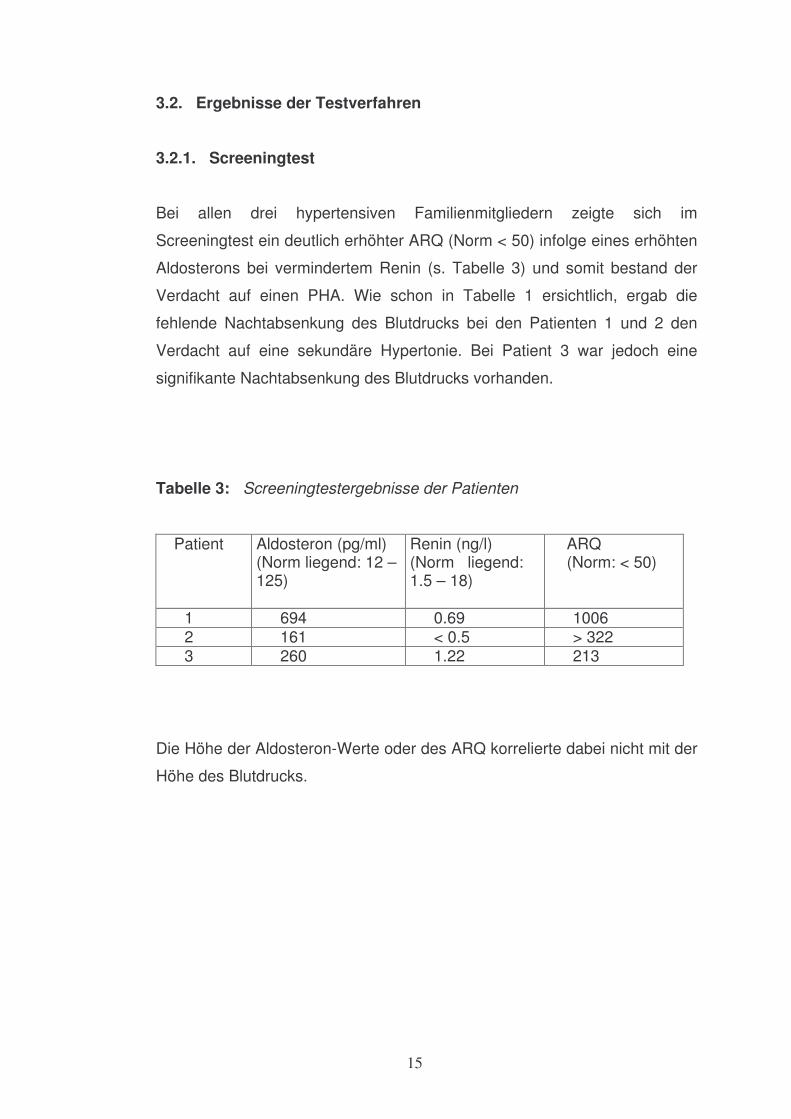
Bei allen drei hypertensiven Familienmitgliedern zeigte sich im
Screeningtest ein deutlich erhöhter ARQ (Norm < 50) infolge eines erhöhten
Aldosterons bei vermindertem Renin (s. Tabelle 3) und somit bestand der
Verdacht auf einen PHA. Wie schon in Tabelle 1 ersichtlich, ergab die
fehlende Nachtabsenkung des Blutdrucks bei den Patienten 1 und 2 den
Verdacht auf eine sekundäre Hypertonie. Bei Patient 3 war jedoch eine
signifikante Nachtabsenkung des Blutdrucks vorhanden.
Tabelle 3: Screeningtestergebnisse der Patienten Patient
Aldosteron (pg/ml) (Norm liegend: 12 – 125)
Renin (ng/l) (Norm liegend: 1.5 – 18)
ARQ (Norm: < 50)
1 694 0.69 1006 2 161 < 0.5 > 322 3 260 1.22 213
Die Höhe der Aldosteron-Werte oder des ARQ korrelierte dabei nicht mit der
Höhe des Blutdrucks.
16
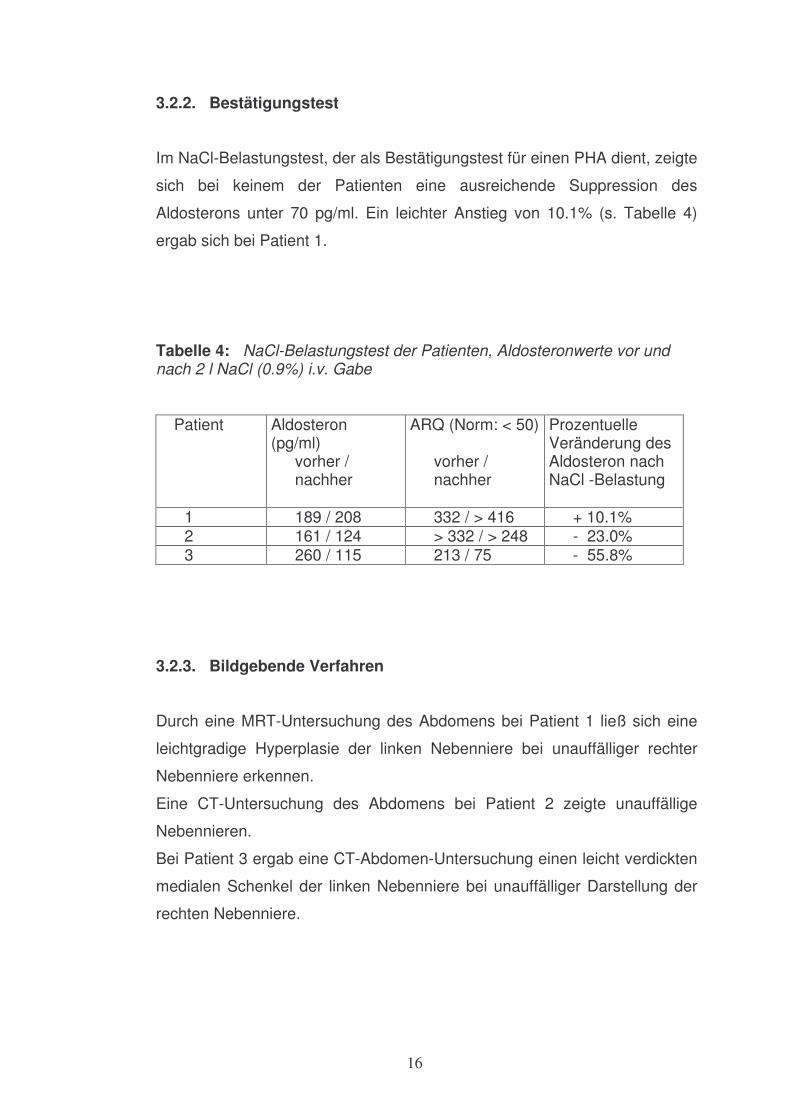
3.2.2. Bestätigungstest
Im NaCl-Belastungstest, der als Bestätigungstest für einen PHA dient, zeigte
sich bei keinem der Patienten eine ausreichende Suppression des
Aldosterons unter 70 pg/ml. Ein leichter Anstieg von 10.1% (s. Tabelle 4)
ergab sich bei Patient 1.
Tabelle 4: NaCl-Belastungstest der Patienten, Aldosteronwerte vor und nach 2 l NaCl (0.9%) i.v. Gabe Patient
Aldosteron (pg/ml)
vorher / nachher
ARQ (Norm: < 50) vorher / nachher
Prozentuelle Veränderung des Aldosteron nach NaCl -Belastung
1 189 / 208 332 / > 416 + 10.1% 2 161 / 124 > 332 / > 248 - 23.0% 3 260 / 115 213 / 75 - 55.8%
3.2.3. Bildgebende Verfahren
Durch eine MRT-Untersuchung des Abdomens bei Patient 1 ließ sich eine
leichtgradige Hyperplasie der linken Nebenniere bei unauffälliger rechter
Nebenniere erkennen.
Eine CT-Untersuchung des Abdomens bei Patient 2 zeigte unauffällige
Nebennieren.
Bei Patient 3 ergab eine CT-Abdomen-Untersuchung einen leicht verdickten
medialen Schenkel der linken Nebenniere bei unauffälliger Darstellung der
rechten Nebenniere.

17
3.2.4. Seitengetrennte Nebennierenvenenblutentnahme
Nur bei Patient 1 wurde eine seitengetrennte Nebennierenvenen-
blutentnahme durchgeführt (s. Tabelle 5).
Tabelle 5: Seitengetrennte Nebennierenvenenblutentnahme von Patient 1
(Eine selektive Abnahme liegt vor, wenn der Quotient aus dem Cortisolwert
der NNV und der V. cava inferior > 1.1 beträgt.)
Die Werte zeigen, dass nur eine selektive Abnahme aus der linken
Nebennierenvene durchgeführt wurde und dass eine Unterscheidung
zwischen APA und IBNH nicht möglich war (s. Abbildung 2). Die
seitengetrennte Nebennierenvenenblutentnahme war somit nicht verwertbar.
Vena cava inferior Aldosteron absolut [pg/ml] 572 links 52300 rechts 588
Cortisol absolut [µg/dl] 29 links 582 rechts 25 Aldosteron / Cortisol 19.7 links 89.9 rechts 23.5
Verhältnis Aldosteron / Cortisol
der Venae suprarenales -
Cortisol absolut Vena suprarenalis / Vena cava inferior
3.83
links 20.07 rechts 0.86
Vena suprarenalis

18
3.2.5. Orthostasetest
Beim Orthostasetest zeigte Patient 1 einen Aldosteron-Anstieg von
34.2%. Hingegen fiel der Aldosteronwert bei den Patienten 2 und 3 ab
(s. Tabelle 6).
Tabelle 6: Orthostasetest, Aldosteronwerte (pg/ml) vorher und nachher
Patient Aldosteron vorher
Aldosteron nachher
Prozentuelle Veränderung des Aldosterons
1 199 267 + 34.2% 2 155 89 - 42.6% 3 157 102 - 35.0%
Die Ergebnisse bei Patient 2 und 3 deuteten auf ein Aldosteron-
produzierendes Adenom hin. Bei Patient 1 konnte der Anstieg des
Aldosterons als eine idiopathische bilaterale Nebennierenrindenhyperplasie
interpretiert werden.
3.3. Spezielle Ergebnisse bei GSH
3.3.1. Langzeit-Dexamethasontest
Beim 24 Stunden-Sammelurin zur Bestimmung von Aldosteron und Cortisol,
sowie deren Vorläuferprodukte und Metabolite zeigte sich bei allen
Patienten ein deutlich erhöhtes freies 18-Hydroxycortisol, was auf einen
GSH schließen ließ. Nach einwöchiger Therapie mit täglicher Gabe von
2 mg Dexamethason verringerten sich freies 18-Hydroxycortisol, freies
Aldosteron sowie seine Hauptmetabolite Aldosteron-18-Glucuronid und

19
Tetrahydroaldosteron bis zum Normbereich und teilweise sogar unterhalb
des Normbereichs. Die Werte sind getrennt für Mineralokortikoide und
Glukokortikoide aufgeführt (s. Tabellen 7a und 7b).
Tabelle 7a: Urinausscheidungen der Mineralokortikoide (SU=Sammelurin; 18-OHF=18-Hydroxycortisol, frei; 18-OHB=18-Hydroxycorticosteron, frei; Aldo=Aldosteron, frei; Aldo-ER=Aldosteron-18-Glucuronid; TH-Aldo=Tetrahydroaldosteron; 18-OHDOC=18-Hydroxy-Desoxycorticosteron, frei; DOC=11-Desoxycorticosteron, frei). 24 h SU
Patient 1 vor / nach
Patient 2 vor / nach
Patient 3 vor / nach
Normwerte in µg / 24 h
18-OHF 1830 / 70 785 / 50 541 / 38 40 – 145 18-OHB 28.0 / 2.0 8.8 / 1.2 13.2 / 0.9 1.5 – 6.5 Aldo 1.69 / < 0.1 1.0 / 0.1 0.66 / 0.12 0.1 – 0.4 Aldo-ER 64.0 / 2.0 28.7 / 3.0 28.0 / 4.0 3.5 – 17.5 TH-Aldo 184 / < 10 113 / 10 126 / 12 10 – 70 18-OHDOC
3.3 / < 0.1 2.6 / < 0.1 2.2 / 0.1 0.2 – 1.8
DOC 0.34 / < 0.1 0.33 / < 0.1 0.55 / 0.12 0.1 – 0.4
Tabelle 7b: Urinausscheidungen der Glukokortikoide (SU=Sammelurin; Freies B=Corticosteron, frei; THE=Tetrahydrocortison; Freies F=Cortisol, frei; Freies E=Cortison, frei; THF=Tetrahydrocortisol). 24 h SU
Patient 1 vor / nach
Patient 2 vor / nach
Patient 3 vor / nach
Normwerte pro 24 h
Freies B 6.1 / 0.95 1.14 / 0.2 1.45 / 0.2 0.1 – 2.5 µg THE 6.7 / 0.1 4.2 / 0.1 2.2 / 0.2 0.5 – 5.5 mg Freies F 69 / < 10 29 / < 10 27 / < 10 10 – 60 µg Freies E 146 / < 10 101 / < 10 122 / < 10 20 – 140 µg THF 3.6 / 0.08 3.5 / 0.1 1.9 / 0.1 0.5 – 3.5 mg

20
Als weiteren Hinweis auf einen GSH zeigte sich nach Dexamethasongabe
bei allen Patienten ein normaler Aldosteronwert und eine Steigerung des
Reninwertes. Bei Patient 3 ergab sich sogar ein normaler Reninwert. Durch
die Veränderung der Aldosteron- und Reninwerte normalisierte sich bei allen
Patienten der ARQ (s. Tabelle 8).
Tabelle 8: Aldosteron, Renin, ARQ nach Dexamethasongabe Patient
Aldosteron (pg/ml)
Renin (ng/l) ARQ
1 12 0.97 12.4 2 12 0.60 20 3 39 5.26 7.4
3.3.2. Molekularbiologische Untersuchungen
Zur Diagnosesicherung des GSH wurden molekularbiologische
Untersuchungen durchgeführt.
Nach Spaltung der genomischen DNA mit Hilfe des Restriktionsenzyms
BamHI und Southern-Blot Hybridisierung des Exon 1, 3-4 der CYP11B1/2-
Gene zeigte sich bei den drei Patienten ein 6.5 kb großes Fragment,
welches auf das chimäre Gen hindeutete. Bei der normotensiven Mutter und
deren ältester Sohn ließ sich dieses nicht nachweisen (s. Abbildung 4).
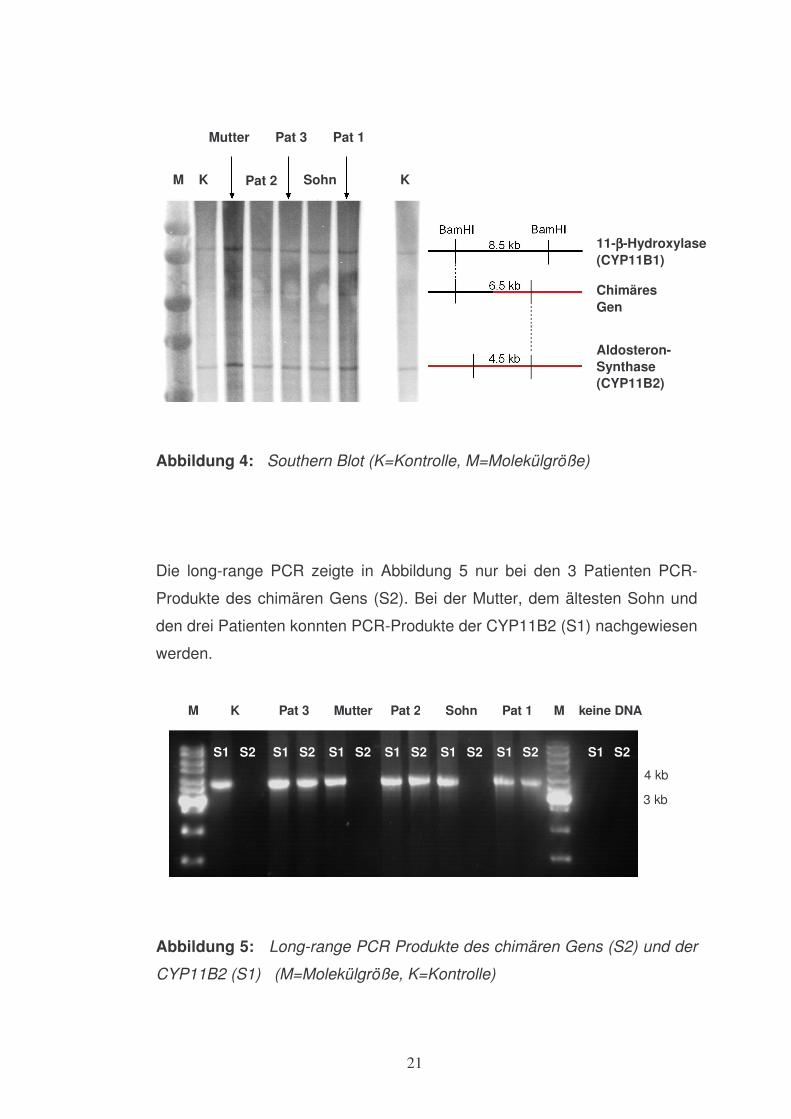
21
Abbildung 4: Southern Blot (K=Kontrolle, M=Molekülgröße)
Die long-range PCR zeigte in Abbildung 5 nur bei den 3 Patienten PCR-
Produkte des chimären Gens (S2). Bei der Mutter, dem ältesten Sohn und
den drei Patienten konnten PCR-Produkte der CYP11B2 (S1) nachgewiesen
werden.
Abbildung 5: Long-range PCR Produkte des chimären Gens (S2) und der
CYP11B2 (S1) (M=Molekülgröße, K=Kontrolle)
M K Pat 3 Mutter Pat 2 Sohn Pat 1 M keine DNA
S1 S2 S1 S2 S1 S2 S1 S2 S1 S2 S1 S2 S1 S2
3 kb
4 kb
M K
Pat 2
Sohn
K
Mutter Pat 3 Pat 1
11-ββββ-Hydroxylase (CYP11B1)
Chimäres Gen
Aldosteron- Synthase (CYP11B2)

22
Um den Bruchpunkt des chimären Gens zu identifizieren, wurden die PCR-
Produkte sequenziert (Ausschnitte hierzu zeigt Abbildung 6). Die Nukleotid-
Nummern 108 bis 162 entsprechen dabei der CYP11B1; die
Bruchpunktregion spiegelt sich in den Nummern 162 bis 247 und das
chimäre Gen (CYP11B1/2) in den Nummern 248 bis 272 wider.
Abbildung 6: Sequenzierung der PCR-Produkte (K=Kontrolle)
Durch die molekularbiologischen Verfahren konnte bei den drei Patienten
das chimäre Gen in der heterozygoten Form nachgewiesen werden.
K
Sohn
Mutter
Pat 3
Pat 1
Pat 2
CYP11B1CYP11B2

23
3.3.3. Blutdruck nach einwöchiger Dexamethasongabe
Trotz Senkung des Aldosteronwertes (s. Tabelle 8) nach einwöchiger
Dexamethasongabe bei allen drei Patienten blieb die erwartete Reduktion
des Blutdrucks bei den Patienten 1 und 3 aus. Bei Patient 2 zeigte sich eine
deutliche Absenkung des Blutdrucks auf 157/95 mmHg (s. Tabelle 9).
Tabelle 9: Blutdruck nach einer Woche Dexamethason (2 mg/Tag)
Patient
Blutdruck (mmHg) Herzfrequenz (pro min)
1 163 / 90 67 2 157 / 95 86 3 190 / 116 80
3.3.4. Blutdruckverlauf unter medikamentöser Therapie
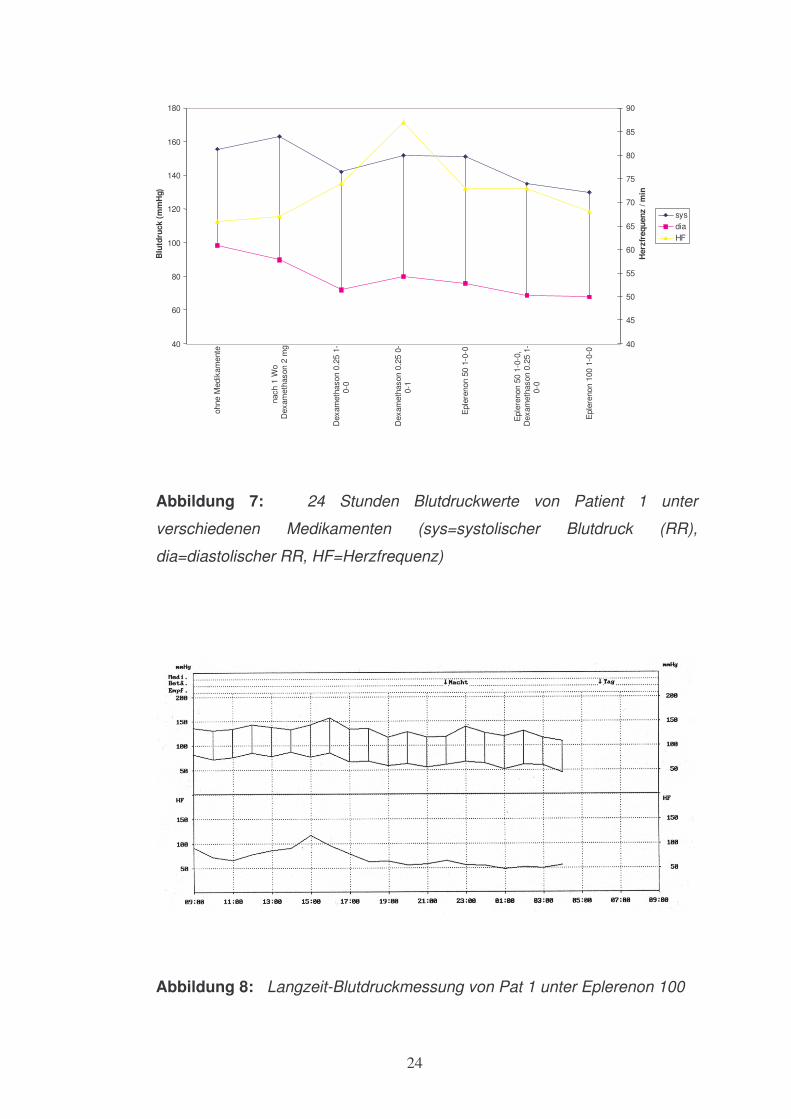
Nach 4-wöchiger Therapie mit Dexamethason 0.25 mg verbesserte sich der
Blutdruckwert bei Patient 1 (s. Abbildung 7). Nach Umstellung der 0.25 mg
Dexamethasontherapie auf eine abendliche Gabe zeigte der systolische
Blutdruck wieder eine Erhöhung bis fast auf den Ausgangswert. Unter
Therapie mit 50 mg Eplerenon stellte sich keine relevante
Blutdruckbesserung im Vergleich zum Ausgangswert ein. Erst die
Kombination aus 50 mg Eplerenon und 0.25 mg Dexamethason führte zu
einer deutlichen Reduktion des Blutdrucks.
Ein normaler Blutdruck wurde erst unter 100 mg Eplerenon erreicht (s.
Abbildung 8).
24
Abbildung 7: 24 Stunden Blutdruckwerte von Patient 1 unter
verschiedenen Medikamenten (sys=systolischer Blutdruck (RR),
dia=diastolischer RR, HF=Herzfrequenz)
Abbildung 8: Langzeit-Blutdruckmessung von Pat 1 unter Eplerenon 100
40
60
80
100
120
140
160
180
ohne
Med
ikam
ente
nach
1 W
o D
exam
etha
son
2 m
g
Dex
amet
haso
n 0.
25 1
- 0-
0
Dex
amet
haso
n 0.
25 0
- 0-
1
Epl
eren
on 5
0 1-
0-0
Epl
eren
on 5
0 1-
0-0,
Dex
amet
haso
n 0.
25 1
- 0-
0
Epl
eren
on 1
00 1
-0-0
Blu
tdru
ck (
mm
Hg)
40
45 50 55 60 65
70 75 80 85
90
Her
zfre
quen
z / m
in
sys dia HF
25
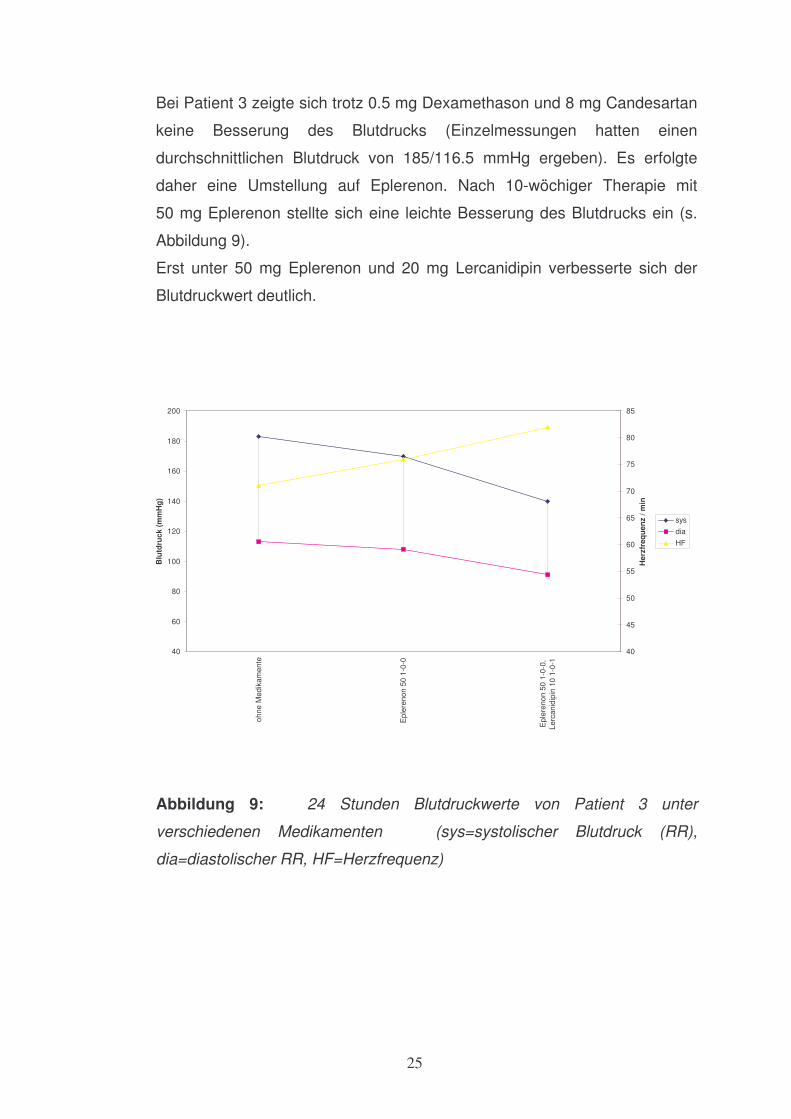
Bei Patient 3 zeigte sich trotz 0.5 mg Dexamethason und 8 mg Candesartan
keine Besserung des Blutdrucks (Einzelmessungen hatten einen
durchschnittlichen Blutdruck von 185/116.5 mmHg ergeben). Es erfolgte
daher eine Umstellung auf Eplerenon. Nach 10-wöchiger Therapie mit
50 mg Eplerenon stellte sich eine leichte Besserung des Blutdrucks ein (s.
Abbildung 9).
Erst unter 50 mg Eplerenon und 20 mg Lercanidipin verbesserte sich der
Blutdruckwert deutlich.
Abbildung 9: 24 Stunden Blutdruckwerte von Patient 3 unter
verschiedenen Medikamenten (sys=systolischer Blutdruck (RR),
dia=diastolischer RR, HF=Herzfrequenz)
40
60
80
100
120
140
160
180
200
ohne
Med
ikam
ente
Epl
eren
on 5
0 1-
0-0
Epl
eren
on 5
0 1-
0-0,
Lerc
anid
ipin
10
1-0-
1
Blu
tdru
ck (m
mH
g)
40
45
50
55
60
65
70
75
80
85
Her
zfre
quen
z / m
in
sysdiaHF

26
Bei Patient 2 zeigte sich im Verlauf unter 0.5 mg Dexamethason, 10 mg
Benazepril und 12.5 mg Hydrochlorothiazid in der 24-Stunden-
Blutdruckmessung ein Durchschnittswert von 145/76 mmHg.
Zusammenfassend lässt sich feststellen, dass nicht alle Patienten unter
Dexamethasontherapie eine Blutdrucksenkung aufwiesen. Des Weiteren
zeigte sich auch eine unterschiedliche Latenzzeit, mit der sich unter
Dexamethasontherapie eine Blutdruckreduktion einstellte. Eine
morgendliche Gabe von Dexamethason senkte den Blutdruck, eine
abendliche Gabe jedoch nicht.
3.3.5. ARQ und Mikroalbumin unter verschiedenen Medikamenten
Unter verschiedenen Medikamenten wurden Aldosteron- und Reninwerte
und der ARQ bestimmt (s. Tabelle 10 und 11).
Tabelle 10: Werte von Aldosteron, Renin und ARQ unter verschiedenen Medikamenten und deren Einfluss auf den Blutdruck bei Patient 1
Medikament
Aldosteron (Norm: 12 - 125 pg/ml)
Renin (Norm: 1.5 – 18 ng/l)
ARQ (Norm: < 50)
Blutdruck (mmHg)
Ohne Medikation 694 0.69 1006 156 / 99 Dexamethason 0.25 1-0-0
107 10.0 11 142 / 72
Eplerenon 50 1-0-0 107 1.25 86 151 / 76 Eplerenon 50 1-0-0, Dexamethason 0.25 1-0-0
296 30.8 9.6 135 / 69
Eplerenon 100 1-0-0
137 12.0 11 130 / 68

27
Die oben genannten Medikamente wurden für mindestens 4 Wochen oder
länger eingenommen. Weder geringe Dosierungen von Dexamethason bzw.
Eplerenon oder die Kombinationstherapie führten zu einer adäquaten
Blutdruckreduktion. Erst durch die Therapie mit 100 mg Eplerenon konnte
ein normaler Blutdruck erreicht werden.
Tabelle 11: Werte von Aldosteron, Renin und ARQ unter verschiedenen Medikamenten und deren Einfluss auf den Blutdruck bei Patient 3
Medikament
Aldosteron (Norm: 12 - 125 pg/ml)
Renin (Norm: 1.5 – 18 ng/l)
ARQ (Norm: < 50)
Blutdruck (mmHg)
Ohne Medikation 260 1.22 213 183 / 113 Eplerenon 50 1-0-0, Lercanidipin 10 1-0-1
65 4.81 14 140 / 91
Unter Therapie mit Eplerenon 50 mg und Lercanidipin 20 mg zeigte sich
nach einem Zeitraum von 4 Monaten ein normaler Aldosteron- und
Reninwert und ferner eine deutliche Besserung des Blutdrucks.
Zusätzlich wurde unter verschiedenen Medikamenten die
Albuminausscheidung gemessen. Eine Reduktion der Albuminausscheidung
hat dabei einen protektiven Einfluss auf die Nierenfunktion (s. Tabelle 12
und 13).

28
Tabelle 12: Werte von Mikroalbumin im Spontanurin unter verschiedenen Medikamenten bei Patient 1
Medikament Mikroalbumin / Kreatinin im
Spontanurin (Norm: < 24 mg / g)
Blutdruck (mmHg)
Eplerenon 50 1-0-0 10.7 151 / 76 Eplerenon 50 1-0-0, Dexamethason 0.25 mg 1-0-0
7.6 135 / 69
Tabelle 13: Werte von Mikroalbumin im Spontanurin unter verschiedenen Medikamenten bei Patient 3
Medikament Mikroalbumin / Kreatinin im
Spontanurin (Norm: < 24 mg / g)
Blutdruck (mmHg)
Eplerenon 50 1-0-0, Lercanidipin 10 1-0-1
9.3 140 / 91
Unter Eplerenon zeigte sich bei Patient 1 und 3 keine Mikroalbuminurie
mehr.
Da der Patient 2 aus persönlichen Gründen nicht zu den Kontrollen
erschien, können hierzu keine Werte angegeben werden.

29
4. Diskussion
4.1. Klinik
Bei den hier vorgestellten Patienten einer deutschen Familie konnte mit dem
NaCl-Belastungstest ein PHA nachgewiesen werden. Dieser PHA war im
Rahmen der Hypertonieabklärung aufgefallen. Neben einer unterschiedlich
ausgeprägten Hypertonie, respiratorisch kompensierten metabolischen
Alkalose wies nur Patient 2 eine sehr leichte Hypokaliämie auf. Wie auch
schon in der Literatur beschrieben, schließt das Fehlen einer Hypokaliämie
einen PHA nicht mehr aus. Denn zehn Jahre nach seiner Erstbeschreibung
zeigte Conn, dass die arterielle Hypertonie beim PHA viele Jahre bestehen
kann, bevor eine Hypokaliämie auftritt. Die Existenz des normokaliämischen
Conn-Syndroms bewies er 1968 [7]. Wie bei Patient 3 in der 24-Stunden-
Blutdruckmessung sichtbar, kann auch eine signifikante Nachtabsenkung
vorliegen. Aufgrund der Pathophysiologie wird eine vermehrte
Kaliumausscheidung erwartet, die sich aber bei keinem Patienten zeigte.
Wenn Patienten mit klassischem PHA phasenweise normokaliämisch sind,
kann dies ein Hinweis auf eine kochsalzarme Diät sein. Das reduzierte
Natriumangebot am Nierentubulussystem kann nämlich zu einer
erniedrigten Kaliumclearance führen. Durch erhöhte Kochsalzzufuhr kann
die renale Kaliumclearance gesteigert werden [3]. Trotz hoher errechneter
Kochsalzzufuhr bei Patient 1 zeigte sich sowohl im Serum als auch im Urin
ein normaler Kaliumwert.
Die Höhe des Blutdrucks war nicht abhängig von der Höhe des
Aldosteronwertes, des ARQ oder des Alters.
Obwohl Patient 2 seit einer idiopathischen Faszialisparese bereits
zahlreiche blutdrucksenkende Medikamente einnahm, zeigten sich weiterhin
deutlich erhöhte Blutdruckwerte.
Kardiologischerseits wies Patient 3 eine Linksherzhypertrophie auf, die sich
infolge der langjährigen Hypertonie entwickelt hatte.

30
4.2. Diagnostik
Um die Ursache des PHA bei diesen Patienten abzuklären, wurden weitere
Untersuchungen durchgeführt.
Die Bildgebung bei Patient 1 erbrachte keinen eindeutigen Hinweis auf ein
APA. Außerdem war die seitengetrennte Nebennierenvenenblutentnahme
nur auf der linken Seite selektiv. Eine weitere Klärung bezüglich Adenom
oder Hyperplasie war auch nicht unter Hinzunahme des Quotienten aus dem
A/C-Wert der linken Nebennierenvene und dem A/C-Wert der Vena cava
inferior möglich. Des Weiteren konnte auch durch den Orthostasetest nicht
sicher zwischen einem APA und einer IBNH unterschieden werden.
Aufgrund der Familienanamnese und der nicht eindeutigen Befunde wurde
eine weitere Abklärung bezüglich eines GSH durchgeführt. Als
Screeningtest für den GSH kam der Langzeit-Dexamethasontest zur
Anwendung [22], wo sich eine Normalisierung der Aldosteronwerte und des
ARQ einstellte.
Dass ein Aldosteronwert kleiner 40 pg/ml nach Langzeit-Dexamethasontest
die Diagnose GSH mit Sicherheit stellen würde [22], bestätigte sich in
nachfolgenden Untersuchungen nicht. Auch Patienten mit PHA ohne
Nachweis des chimären Gens wiesen nämlich Aldosteronwerte von kleiner
20 pg/ml nach Langzeit-Dexamethasontest [13, 27] auf.
Pathophysiologisch wird bei Patienten mit GSH durch die Kortisontherapie
eine Verminderung des ACTH und letztendlich eine Verminderung des
Blutdrucks erwartet.
Bei diesem Test kann jedoch auch die Blutdruckreduktion ausbleiben, was
sich auch in dieser Familie herausstellte. Lediglich Patient 2 zeigte eine
signifikante Blutdruckreduktion nach einwöchiger Dexamethasongabe (2 mg
täglich).
Ferner kann bei Patienten mit IBNH und APA eine Senkung des
Aldosteronwertes auftreten [26]. Ein aussagekräftiges Ergebnis bezüglich
des GSH ist durch den Langzeit-Dexamethasontest nicht zu erzielen.
Die Bewertung der Urinausscheidung und genetische Testverfahren müssen
somit für die Diagnose GSH herangezogen werden.

31
Bei der Untersuchung des Sammelurins auf Ausscheidungen von
Mineralokortikoiden und Glukokortikoiden jeweils vor und nach Langzeit-
Dexamethasontest waren u.a. deutlich erhöhte Werte von 18-
Hydroxycortisol und 18-Hydroxycorticosteron zu verzeichnen, die sich nach
Dexamethasongabe normalisierten. Dabei gilt 18-Hydroxycortisol als
deutlicher Hinweis für den GSH. Leicht erhöhte Werte sind jedoch auch für
ein APA festgestellt worden.
Als weiterer Hinweis auf einen GSH ist alternativ eine Bestimmung von 18-
Hydroxycortisol im Blut möglich [26].
Um letztendlich den Beweis eines GSH zu erbringen, sind
molekularbiologische Untersuchungen erforderlich. Hierbei konnte der GSH
eindeutig bei den hier vorgestellten Patienten nachgewiesen werden.
4.3. Genetik
Der GSH wurde erstmalig 1966 beschrieben [37], wo es unter
Dexamethasontherapie zu einer Verminderung des Blutdrucks, des
Aldosteronwertes und zur Normalisierung des Kaliumwertes kam. Aber erst
1992 wurde die genetische Ursache aufgedeckt.
Der GSH ist eine autosomal dominant vererbte Erkrankung, wobei der
Defekt auf Chromosom 8 liegt. Durch ungleiches crossing-over entsteht ein
chimäres Gen, welches aus Teilen des Gens für die Aldosteronsynthase
(CYP11B2) und aus Teilen des Gens für die 11ß-Hydroxylase (CYP11B1)
besteht [20]. Die Aldosteronsynthase ist das limitierende Enzym der
Aldosteronsynthese, wobei Aldosteron in der gesunden Nebenniere unter
dem Einfluss von Angiotensin II vermehrt gebildet wird. Die in der Zona
fasciculata lokalisierte 11ß-Hydroxylase ist in der Cortisol-Biosynthese
involviert und wird durch ACTH reguliert. Beim GSH hat das aus dem
chimären Gen resultierende Enzym einerseits Aldosteronsynthase-Aktivität,
wird jedoch andererseits in seiner Expression durch ACTH reguliert. Dabei
enthält das chimäre Gen die ACTH-abhängige Promoterregion der 11ß-
Hydroxylase. Dadurch kommt es zu einer ektopen Aldosteronsynthase-
Aktivität in der Zona fasciculata, die nicht mehr durch den physiologischen

32
Regulator Angiotensin II, sondern durch ACTH gesteuert wird. Hierbei wird
die Aldosteronsynthese eng an die Cortisolsynthese gekoppelt. Der erhöhte
Aldosteronwert verursacht durch das vermehrte Plasmavolumen letztendlich
den hohen Blutdruck.
Der GSH wird mit einer Häufigkeit von 0.5 – 1% des PHA vermutet [29].
Eine genaue Angabe wird erst möglich sein, wenn gezielte Untersuchungen
durchgeführt werden.
Der GSH wurde bisher in mehreren Familien in Nordamerika, Asien und
Italien beschrieben [8, 28, 32, 45]. Eine genetische Untersuchung in der
polnischen Bevölkerung bei 129 Patienten mit PHA und 132 Patienten mit
essentieller Hypertonie konnte den GSH nicht nachweisen [1]. Das deutet
darauf hin, dass wahllose Untersuchungen nicht ratsam sind [14, 29]. Es
müssen also die Familienanamnese, das frühe Auftreten der Hypertonie,
fehlende Risikofaktoren und natürlich der Nachweis eines PHA
berücksichtigt werden. Da der PHA inzwischen die häufigste, monokausale
Ursache der arteriellen Hypertonie ist [31] und zunehmend weltweit
diagnostiziert wird [4], wird die Prävalenz des GSH bei entsprechenden
Untersuchungen vermutlich höher sein.
Der GSH tritt bereits in der Jugend durch Hypertonie in Erscheinung. Im 13.
Lebensjahr haben viele betroffene Patienten bereits eine arterielle
Hypertonie entwickelt [9, 10]. Dies deckt sich mit dem jugendlichen Auftreten
bei Patient 1 und 2.
Wie auch in der Literatur beschrieben, zeigen die GSH-Patienten ein
unterschiedliches Erscheinungsbild. Die Ursache hierfür ist jedoch nicht
geklärt. Denn obwohl Patient 1 den höchsten Aldosteronwert aufwies, hatte
er den niedrigsten Blutdruck im Gegensatz zu den anderen hier
vorgestellten Patienten.
Patienten mit GSH haben zwei normale Kopien der Gene, die für die
Aldosteronsynthase und die 11ß-Hydroxylase kodieren. Aber durch
ungleiches crossing-over wird die Aldosteronsynthese jetzt unter dem
Einfluss von ACTH (Promoterregion der 11ß-Hydroxylase) beeinflusst [25].
Gründe der unterschiedlichen Ausprägung der Hypertonie können
möglicherweise im Kochsalzverbrauch, in der Vererbung von

33
blutdrucksenkenden Genen oder in der verminderten Ausprägung des
chimären Gens liegen [10].
In Deutschland wurde bisher kein GSH beschrieben. Bei den Patienten der
hier vorgestellten Familie zeigte sich die unterschiedliche Ausprägung des
GSH, was sich auch mit den Aussagen in der Literatur deckt, dass der
Phänotyp des GSH sehr weit variiert [12]. Die Patienten wiesen verschieden
hohe Blutdruckwerte, Aldosteronwerte und ARQ-Werte auf. Im
Orthostasetest zeigte sich zusätzlich eine unterschiedliche Reaktion. Bei
diesem Test wird beim GSH, wie auch beim APA, durch die ACTH-
Abhängigkeit eine Abnahme des Aldosteronwertes erwartet. Dies trat aber
nur bei Patient 2 und 3 ein, wohingegen bei Patient 1 ein Anstieg des
Aldosteronwertes zu verzeichnen war.
Im NaCl-Belastungstest kam es bei Patient 1 ferner zu einem Anstieg des
Aldosteronwertes. Vermutlich zeigen Patienten mit einem GSH in diesem
Fall auch einen Anstieg des Aldosteronwertes im Orthostasetest. Dies sollte
bei weiteren GSH-Fällen Beachtung finden. Vielleicht hätte hier auch eine
Bestimmung der ACTH-Werte zu einer Klärung beitragen können.
4.4. Therapie – Beeinflussung der Aldosteroneffekte
Unter einwöchiger Dexamethasongabe (2mg/Tag) reduzierte sich der
Blutdruck jedoch nur bei Patient 2, obwohl alle Patienten nun einen
normalen Aldosteronwert aufwiesen. Während Patient 1 unter längerer
Therapie mit 0.25 mg Dexamethason eine Besserung des Blutdrucks zeigte,
stellte sich bei Patient 3, vermutlich aufgrund des schon lange bestehenden
Bluthochdrucks, keine Reduktion des Blutdrucks ein.
Auch nach Entfernung eines APA kommt es meist nicht zur unmittelbaren
Senkung des Blutdrucks [15].
Unter Therapie mit dem selektiven Aldosteron-Rezeptor-Antagonisten
Eplerenon konnte bei Patient 1 erst unter 100 mg ein normaler Blutdruck
erreicht werden. Hierbei wurde Eplerenon wegen der geringeren
Nebenwirkungen dem Spironolakton vorgezogen [24].

34
Bei Patient 3 zeigte sich unter Eplerenon 50 mg und zusätzlicher Gabe von
Lercanidipin 20 mg eine deutliche Besserung des Blutdrucks. Bei der
medikamentösen Therapie sollten bevorzugt Calcium-Antagonisten zum
Einsatz kommen, da sie zur Verminderung der Aldosteronsynthese führen
können. In vitro Untersuchungen deuten darauf hin [9].
Unter oben genannter Medikation normalisierte sich bei Patient 3 der
Aldosteronwert und nahezu der auch bei Patient 1.
In der Literatur sind zahlreiche genomische und nicht-genomische
Aldosteroneffekte, wie u.a. myokardiale Fibrose, Beeinflussung der
Albuminausscheidung, Vasokonstriktion in der Niere, usw. beschrieben
worden. Durch die Aldosteron-Rezeptor-Antagonisten sind aber nur die
genomischen Aldosteroneffekte, die über die Blockade des klassischen
zytoplasmatischen Aldosteron-Rezeptors vermittelt sind, beeinflussbar [44].
Es sollten daher mehrere Therapieziele verfolgt werden:
Zu allererst muss der Blutdruck normalisiert werden, ferner sollten der
Aldosteronwert reduziert bzw. die negativen Effekte des Aldosterons
antagonisiert werden.
Hinsichtlich der Albuminausscheidung wiesen die Patienten nahezu gleiche
Werte auf, obwohl sie verschieden hohe Blutdruckwerte hatten, und die
Hypertonie unterschiedlich lange bestand. Unter Therapie mit Eplerenon
zeigte sich bei Patient 1 und 3 ein Verschwinden der Mikroalbuminurie. Dies
lässt sich nicht allein auf eine Blutdrucksenkung zurückführen. Bei Patient 1
kam es unter Eplerenon 50 mg zwar zu einer Normalisierung der
Mikroalbuminurie, aber zu keiner relevanten Blutdrucksenkung. Dies deutet
somit auf einen direkten genomischen Effekt von Aldosteron, nämlich auf die
Beeinflussung der Albuminausscheidung in der Niere, hin [34].
Bei experimentellen Untersuchungen bei Kaninchen führt Aldosteron zur
Konstriktion der afferenten und efferenten Arteriole des Glomerulums. Diese
Konstriktion ist auf nicht-genomische Aldosteroneffekte zurückzuführen, da
sie schnell eintrat. Die Konstriktion konnte durch Neomycin (spezifischer
Inhibitor der Phospholipase C) aufgehoben werden, sodass diese
Aldosteronwirkung auf die Beteiligung der Phospholipase C hinweist. Durch
Anwendung von L-Typ-Calciumantagonisten konnte die Konstriktion der

35
afferenten Arteriole verhindert werden. Eine kurzfristige Einwirkzeit von
Spironolakton hatte keinen Einfluss auf den Gefäßtonus [2].
4.5. Ausblick
Die medikamentöse Therapie bei Patienten, die an GSH leiden, sollte
vorzugsweise kausal durch Dexamethason – unter Beachtung der
Nebenwirkungen – erfolgen. Falls sich Nebenwirkungen durch Kortison
ergeben, oder eine ausreichende Blutdruckeinstellung durch alleinige
Kortisontherapie nicht möglich ist, sollte zusätzlich mit einem Aldosteron-
Rezeptor-Antagonist therapiert werden. Hierbei ist Eplerenon wegen der
geringeren Nebenwirkungen dem Spironolakton der Vorzug zu geben.
Da nicht alle negativen Aldosteroneffekte durch Aldosteron-Rezeptor-
Antagonisten verhindert werden können, ist eine Monotherapie mit
Spironolakton oder Eplerenon vermutlich nicht ausreichend.
Von entscheidender Bedeutung ist aber die Normalisierung des Blutdrucks,
wobei dann auch Calcium-Antagonisten zum Einsatz kommen sollten.

36
5. Zusammenfassung
In dieser Arbeit wurden erstmalig bei einer deutschen Familie
Untersuchungen und Ergebnisse dargestellt, die zur Diagnose eines GSH
führten. Bisher wurde der GSH nur in Nordamerika, Asien und Italien
beschrieben.
Obwohl alle drei betroffenen Patienten dieser Familie den gleichen
Gendefekt haben, wiesen sie deutlich unterschiedlich erhöhte Werte
hinsichtlich des Blutdrucks, des Aldosterons und des ARQ – bei nahezu
gleichen Mikroalbuminuriewerten – auf, so dass eine entsprechende
spezifische medikamentöse Therapie notwendig war. Die Höhe des
Blutdrucks war dabei nicht von der Höhe des Aldosteronwertes abhängig.
Der GSH ist eine seltene monogenetische Form (vermutete Prävalenz 0.5 –
1%) des PHA. Zunehmend bei normokaliämischen Patienten wird der PHA
nachgewiesen, und er ist die häufigste Ursache der sekundären arteriellen
Hypertonie. Der ARQ ist der effektivste Screeningtest für den PHA.
Klinisch wurde der GSH erstmalig 1966 beschrieben, aber erst 1992 die
genetische Ursache aufgedeckt. Der GSH ist eine autosomal dominant
vererbte Erkrankung, wobei der Defekt auf Chromosom 8 liegt. Durch
ungleiches crossing-over entsteht ein chimäres Gen, welches aus Teilen
des Gens für die Aldosteronsynthase und aus Teilen des Gens für die 11ß-
Hydroxylase besteht. Dabei enthält das chimäre Gen die ACTH-abhängige
Promoterregion der 11ß-Hydroxylase. Unter dem Einfluss von ACTH kommt
es daher zu einer ektopen Aldosteronsynthese in der Zona fasciculata.
Im Rahmen der Ursachenabklärung des PHA bei den Patienten hatten sich
– als Hinweis auf einen GSH – im Sammelurin deutlich erhöhte Werte für
18-Hydroxycortisol und 18-Hydroxycorticosteron gezeigt, die sich nach
einwöchiger Gabe von 2 mg/d Dexamethason (Suppression von ACTH)
normalisierten. Des Weiteren konnten normale Aldosteronwerte erreicht
werden.
Molekularbiologisch konnte der GSH bei den Patienten eindeutig
nachgewiesen werden.
Obwohl sich die Aldosteronwerte nach einwöchiger Dexamethasongabe
normalisiert hatten, zeigte zunächst nur ein Patient eine Blutdruckreduktion.

37
Erst nach längerer Dexamethasontherapie reagierte ein weiterer Patient mit
einer Besserung des Blutdrucks, während der dritte Patient überhaupt keine
Blutdruckreduktion zeigte.
Im Verlauf der weiteren Therapie konnte in der 24-Stunden-
Blutdruckmessung – unter verschiedenen Dosierungen und Medikamenten
(Eplerenon; Eplerenon und Dexamethason; Dexamethason, Benazepril und
Hydrochlorothiazid; Eplerenon und Lercanidipin) – bei allen Patienten eine
deutliche Besserung, ja sogar bei einem Patienten eine Normalisierung des
Blutdrucks erreicht werden.
Unter medikamentöser Therapie mit Eplerenon zeigte sich keine
Mikroalbuminurie mehr, was auf einen direkten genomischen Effekt des
Aldosterons hinweist. Da die nicht-genomischen Aldosteroneffekte nicht
durch Aldosteron-Rezeptor-Antagonisten beeinflusst werden können, sollte
vorzugsweise mit Dexamethason unter Beachtung der Nebenwirkungen
therapiert werden. Eine ausreichende Blutdrucksenkung war bei den
Patienten unter alleiniger Dexamethasontherapie nicht zu erzielen, jedoch
sowohl unter Eplerenon als auch unter Lercanidipin.

38
6. Literaturverzeichnis
[1] Adler G, Widecka K, P�czkowska M, Dobrucki T, Placha G, Drozd R,
Parczewski M, Januszewicz A, Gaciong Z, Ciechanowicz A (2005).
Genetic screening for glucocorticoid-remediable aldosteronism (GRA):
experience of three clinical centres in Poland. J Appl Genet 46, 329-
332
[2] Arima S, Kohagura K, Xu HL, Sugawara A, Abe T, Satoh F, Takeuchi
K, Ito S (2003). Nongenomic vascular action of aldosterone in the
glomerular microcirculation. J Am Soc Nephrol 14, 2255-2263
[3] Bravo EL, Tarazi RC, Dustan HP, Fouad FM, Textor SC, Gifford RW,
Vidt DG (1983). The changing clinical spectrum of primary
aldosteronism. Am J Med 74, 641-651
[4] Büchner N, Vonend O, Rump LC (2006). Pathophysiologie der
Hypertonie: Was gibt es Neues? Herz 31, 294-302
[5] Conn JW (1955). Primary aldosteronism, a new clinical syndrome. J
Lab Clin Med 45, 6-17
[6] Conn JW, Cohen EL, Rovner DR (1964). Suppression of plasma renin
activity in primary aldosteronism. J Am Med Assoc 190, 213-221
[7] Conn JW, Cohen EL, Rovner DR, Nesbit RM (1965). Normokalemic
primary aldosteronism. A detectable cause of curable “essential”
hypertension. JAMA 193, 200-206
[8] Ding W, Liu L, Hu R, Xu M, Chen J (2002). Clinical and gene mutation
studies on a Chinese pedigree with glucocorticoid-remediable
aldosteronism. Chin Med J (Engl) 115, 979-982

39
[9] Dluhy RG, Lifton RP (1999). Glucocorticoid-remediable aldosteronism.
J Clin Endocrinol Metab 84, 4341-4344
[10] Dluhy RG, Anderson B, Harlin B, Ingelfinger J, Lifton R (2001).
Glucocorticoid-remediable aldosteronism is associated with severe
hypertension in early childhood. J Pediatr 138, 715-720
[11] Espiner EA, Ross DG, Yandle TG, Richards AM, Hunt PJ (2003).
Predicting surgically remedial primary aldosteronism: role of adrenal
scanning, posture testing, and adrenal vein sampling. J Clin Endocrinol
Metab 88, 3637-3644
[12] Fallo F, Pilon C, Williams TA, Sonino N, Morra di Cella S, Veglio F, De
Iasio R, Montanari P, Mulatero P (2004). Coexistence of different
phenotypes in a family with glucocorticoid-remediable aldosteronism. J
Hum Hypertens 18, 47-51
[13] Fardella CE, Mosso L, Gómez-Sánchez C, Cortés P, Soto J, Gómez L,
Pinto M, Huete A, Oestreicher E, Foradori A, Montero J (2000). Primary
hyperaldosteronism in essential hypertensives: prevalence,
biochemical profile, and molecular biology. J Clin Endocrinol Metab 85,
1863-1867
[14] Gates LJ, Benjamin N, Haites NE, MacConnachie AA, McLay JS
(2001). Is random screening of value in detecting glucocorticoid-
remediable aldosteronism within a hypertensive population? J Hum
Hypertens 15, 173-176
[15] Gordon RD (2004). Primary aldosteronism-actual epidemics or false
alarm? Arq Bras Endocrinol Metabol 48, 666-673
[16] Hamlet SM, Tunny TJ, Woodland E, Gordon RD (1985). Is
aldosterone/renin ratio useful to screen a hypertensive population for
primary aldosteronism? Clin Exp Pharmacol Physiol 12, 249-252

40
[17] Harper R, Ferrett CG, McKnight JA, McIlrath EM, Russell CF, Sheridan
B, Atkinson AB (1999). Accuracy of CT scanning and adrenal vein
sampling in the pre-operative localization of aldosterone-secreting
adrenal adenomas. QJM 92, 643-650
[18] Hiramatsu K, Yamada T, Yukimura Y, Komiya I, Ichikawa K, Ishihara
M, Nagata H, Izumiyama T (1981). A screening test to identify
aldosterone-producing adenoma by measuring plasma renin activity.
Results in hypertensive patients. Arch Intern Med 141, 1589-1593
[19] Jonsson JR, Klemm SA, Tunny TJ, Stowasser M, Gordon RD (1995). A
new genetic test for familial hyperaldosteronism type I aids in the
detection of curable hypertension. Biochem Biophys Res Commun 207,
565-571
[20] Lifton RP, Dluhy RG, Powers M, Rich GM, Cook S, Ulick S, Lalouel JM
(1992). A chimaeric 11 beta-hydroxylase/aldosterone synthase gene
causes glucocorticoid-remediable aldosteronism and human
hypertension. Nature 355, 262-265
[21] Lins PE, Adamson U (1986). Plasma aldosterone-plasma renin activity
ratio. A simple test to identify patients with primary aldosteronism. Acta
Endocrinol (Copenh) 113, 564-569
[22] Litchfield WR, New MI, Coolidge C, Lifton RP, Dluhy RG (1997).
Evaluation of the dexamethasone suppression test for the diagnosis of
glucocorticoid-remediable aldosteronism. J Clin Endocrinol Metab 82,
3570-3573
[23] Luft FC (2003). Mendelian forms of human hypertension and
mechanisms of disease. Clin Med Res 1, 291-300
[24] Magni P, Motta M (2005). Aldosterone receptor antagonists: biology
and novel therapeutic applications. Curr Hypertens Rep 7, 206-211

41
[25] McMahon GT, Dluhy RG (2004). Glucocorticoid-remediable
aldosteronism. Arq Bras Endocrinol Metabol 48, 682-686
[26] Mosso L, Gómez-Sánchez CE, Foecking MF, Fardella C (2001). Serum
18-hydroxycortisol in primary aldosteronism, hypertension, and
normotensives. Hypertension 38, 688-691
[27] Mulatero P, Veglio F, Pilon C, Rabbia F, Zocchi C, Limone P, Boscaro
M, Sonino N, Fallo F (1998). Diagnosis of glucocorticoid-remediable
aldosteronism in primary aldosteronism: aldosterone response to
dexamethasone and long polymerase chain reaction for chimeric gene.
83, 2573-2575
[28] Mulatero P, di Cella SM, Williams TA, Milan A, Mengozzi G, Chiandussi
L, Gomez-Sanchez CE, Veglio F (2002). Glucocorticoid remediable
aldosteronism: low morbidity and mortality in a four-generation italian
pedigree. J Clin Endocrinol Metab 87, 3187-3191
[29] Pizolla F, Trabetti E, Guarini P, Mulatero P, Ciacciarelli A, Blengio GS,
Corrocher R, Olivieri O (2005). Glucocorticoid remediable
aldosteronism (GRA) screening in hypertensive patients from a primary
care setting. J Hum Hypertens 19, 325-327
[30] Plouin PF, Amar L, Chatellier G (2004). Trends in the prevalence of
primary aldosteronism, aldosterone-producing adenomas, and
surgically correctable aldosterone-dependent hypertension. Nephrol
Dial Transplant 19, 774-777
[31] Reincke M, Seiler L, Rump LC (2003). Normokaliämischer primärer
Hyperaldosteronismus. Deutsches Ärzteblatt 100, A 184-190
[32] Rich GM, Ulick S, Cook S, Wang JZ, Lifton RP, Dluhy RG (1992).
Glucocorticoid-remediable aldosteronism in a large kindred: clinical

42
spectrum and diagnosis using a characteristic biochemical phenotype.
Ann Intern Med 116, 813-820.
[33] Rossi GP, Sacchetto A, Chiesura-Corona M, De Toni R, Gallina M,
Feltrin GP, Pessina AC (2001). Identification of the etiology of primary
aldosteronism with adrenal vein sampling in patients with equivocal
computed tomography and magnetic resonance findings: results in 104
consecutive cases. J Clin Endocrinol Metab 86, 1083-1090
[34] Rump LC (2007). Secondary rise of albuminuria under AT1-receptor
blockade – what is the potential role of aldosterone escape? Nephrol
Dial Transplant 22, 5-8
[35] Seiler L, Rump LC, Schulte-Mönting J, Slawik M, Borm K, Pavenstädt
H, Beuschlein F, Reincke M (2004). Diagnosis of primary
aldosteronism: value of different screening parameters and influence of
antihypertensive medication. Eur J Endocrinol 150, 329-337
[36] Stowasser M, Gordon RD, Gunasekera TG, Cowley DC, Ward G,
Archibald C, Smithers BM (2003). High rate of detection of primary
aldosteronism, including surgically treatable forms, after ´non-selective`
screening of hypertensive patients. J Hypertens 21, 2149-2157
[37] Sutherland DJ, Ruse JL, Laidlaw JC (1966). Hypertension, increased
aldosterone secretion and low plasma renin activity relieved by
dexamethasone. Can Med Assoc J 95, 1109-1119
[38] Trenkel S, Seifarth C, Schobel H, Hahn EG, Hensen J (2002). Ratio of
serum aldosterone to plasma renin concentration in essential
hypertension and primary aldosteronism. Exp Clin Endocrinol Diabetes
110, 80-85

43
[39] Vecsei P, Abdelhamid S, Mittelstadt GV, Lichtwald K, Haack D,
Lewicka S (1983). Aldosterone metabolites and possible aldosterone
precursors in hypertension. J Steroid Biochem 19, 345-351
[40] Vonend O, Rump LC (2006). Normokaliämischer primärer
Hyperaldosteronismus. Dtsch Med Wochenschr 131, H24-27
[41] Vonend O, Kokulinsky P, Laufer U, Adams S, Liermann D, Hahn KU,
Rump LC (2006). Wertigkeit von Funktionstests und seitengetrennter
Nebennierenvenenblutentnahme für die Diagnose des primären
Hyperaldosteronismus.
http://www.egms.de/en/meetings/hoch2005/05hoch165.shtml (Zugriff
vom 10.9.2006)
[42] Vonend O, Altenhenne C, Büchner NJ, Dekomien G, Maser-Gluth C,
Weiner SM, Sellin L, Hofebauer S, Epplen JT, Rump LC (2007). A
German family with glucocorticoid remediable aldosteronism. Nephrol
Dial Transplant 22, 1123-1130
[43] Weinberger MH, Fineberg NS (1993). The diagnosis of primary
aldosteronism and separation of two major subtypes. Arch Intern Med
153, 2125-2129
[44] Willenbrock R, Philipp S, Dietz R (Hrsg.) (2005). Inhibition des
Aldosterons: Neue Aspekte. UNI-MED Verlag Bremen
[45] Yokota K, Ogura T, Kishida M, Suzuki J, Otsuka F, Mimura Y, Oishi T,
Hirata M, Tobe K, Makino H (2001). Japanese family with
glucocorticoid-remediable aldosteronism diagnosed by long-
polymerase chain reaction. Hypertens Res 24, 589-594
[46] Young WF, Klee GG (1988). Primary aldosteronism. Diagnostic
evaluation. Endocrinol Metab Clin North Am 17, 367-395

44
7. Danksagung
Herrn Professor Dr. L. C. Rump danke ich herzlich für die Überlassung des
Themas dieser Dissertation, seine hilfreiche Kritik sowie für die gute und
freundliche Betreuung.
Zu Dank bin ich auch den Mitarbeitern der Medizinischen Klinik I des
Marienhospitals Herne (Jun.-Prof. Dr. O. Vonend, Dr. N. J. Büchner, PD Dr.
S. M. Weiner und Dr. L. Sellin) und Dr. S. Hofebauer verpflichtet, die mir bei
Fragen zur Seite standen.
Ein besonderer Dank gilt dem Institut für Humangenetik an der Universität
Bochum (Prof. Dr. J. T. Epplen und Frau Dr. G. Dekomien), das die
molekularbiologischen Untersuchungen aufgebaut und durchgeführt hat.
Dem Steroidlabor am Pharmakologischen Institut der Universität Heidelberg
– insbesondere Frau Dr. C. Maser-Gluth – bin ich dankbar für die
Bestimmung der Mineralokortikoide und Glukokortikoide im Urin.

45
8. Lebenslauf
Am 08.06.1976 wurde ich, Christian Altenhenne, in Bochum geboren. Meine
Eltern sind Karl Altenhenne, Gymnasiallehrer und Heidrun Altenhenne,
Realschullehrerin i.K..
1982 – 86 Besuch der Heckerschule, Grundschule in Essen
1986 – 95 Besuch des Gymnasiums Essen-Werden
1995 Abitur, Leistungskurse: Chemie und Erdkunde
1995 – 96 Grundwehrdienst in Budel / Niederlande und Rheine
ab WS 96/97 Studium der Medizin an der Georg-August-Universität
Göttingen
September 98 Ärztliche Vorprüfung
August 1999 Erster Abschnitt der Ärztlichen Prüfung
1999 – 2001 Famulaturen in den Fächern: Innere Medizin, Dermatologie,
Nephrologie / Rheumatologie und Kardiologie in Essen und
Göttingen
März 2002 Zweiter Abschnitt der Ärztlichen Prüfung
2002 – 03 Praktisches Jahr:
1. Tertial - Universitätsklinikum Göttingen (Dermatologie)
2. Tertial - Kantonales Spital Altstätten / Schweiz (Chirurgie)
3. Tertial - Universitätsklinikum Göttingen (Innere Medizin)
28. Mai 2003 Dritter Abschnitt der Ärztlichen Prüfung
1. Juli 2003 Arzt im Praktikum in der Medizinischen Klinik I des
Marienhospitals Herne, Klinikum der Ruhr-Universität
Bochum
seit 1.10.04 Assistenzarzt am Marienhospital Herne (s.o.)
10/04 – 3/06 Untersuchungen der Familienmitglieder mit der Diagnose
GSH

46