
Aus der Medizinischen Universitätsklinik und Poliklinik
153
I Aus der Medizinischen Universitätsklinik und Poliklinik der Albert-Ludwigs-Universität Freiburg im Breisgau &KDUDNWHULVLHUXQJKXPRUDOHUXQG]HOOXOlUHU,PPXQDQWZRUWHQ JHJHQGDV+HSDWLWLV%9LUXV;$QWLJHQ PLWWHOV'1$,PPXQLVLHUXQJLP0DXVPRGHOO INAUGURAL-DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg im Breisgau Vorgelegt 2002 von Herrn Stephan Meckel geboren in Karlsruhe
Transcript of Aus der Medizinischen Universitätsklinik und Poliklinik

I
Aus der Medizinischen Universitätsklinik und Poliklinik
der Albert-Ludwigs-Universität Freiburg im Breisgau
��������������� �������� �� ��������� �������������
����� �� �������������� ���������
������ �������������� �� ��������
INAUGURAL-DISSERTATION
zur Erlangung des Medizinischen Doktorgrades
der Medizinischen Fakultät
der Albert-Ludwigs-Universität
Freiburg im Breisgau
Vorgelegt 2002 von Herrn Stephan Meckel
geboren in Karlsruhe

II
Dekan: Prof. Dr. rer. nat. M. Schumacher
1. Gutachter: Prof. Dr. med. Dr. h.c. H.E. Blum
2. Gutachter: Prof. Dr. sc. nat. H. Pircher
Jahr der Promotion: 2002

III
Meinen Eltern

IV
���������������
���������� ���������������������������������������������������������
! �������� �� �������������� """"""""""""""""""""""""""""""""""""""
!# $������������% &��������� �� '����� ��� ������(������ """"""""""""" )
!) �������������� ��� �������� � """"""""""""""""""""""""""""""""" *
!* �� �������������� ��&������ """""""""""""""""""""""""""""""""""" +
1.4.1 Funktionen des X-Proteins ___________________________________________ 9
1.4.2 Immunologische Charakterisierung des X-Proteins _______________________ 12
!, ��������������""""""""""""""""""""""""""""""""""""""""""""" )
1.5.1 Allgemeines Prinzip _______________________________________________ 13
1.5.2 Modulation und Verstärkung der Effizienz ______________________________ 15
!- .����/������ �������������� """""""""""""""""""""""""""""""""""" 0
1.6.1 Das Vakzinia-Expressionssystem _____________________________________ 19
1.6.2 Immunologischer Einsatz rekombinanter Vakzinia-Viren __________________ 22
!1 �(��/��������"""""""""""""""""""""""""""""""""""""""""""""""" #)
������� ��� ������� ����������������������������������������� ��
#! ��������� ��� &�������������� """"""""""""""""""""""""""""""""" #*
2.1.1 Das adw-Expressionsplasmid pRc/CMV-X _____________________________ 24
2.1.2 Klonierung des Vakzinia Shuttle–Plasmids pSc11-X ______________________ 24
2.1.3 Klonierung des Expressionsplasmids pcDNA3-X_________________________ 27
2.1.4 Transformation kompetenter E.coli-Bakterien ___________________________ 27
2.1.5 Mini- und Maxi-Präparation der Plasmid DNA __________________________ 29
#!# $2����������� �� 3���� """"""""""""""""""""""""""""""""""""""" )4
2.2.1 Zellen___________________________________________________________ 30
2.2.2 Liposomaler Gen-Transfer___________________________________________ 31
2.2.3 Calcium-Phosphat Transfektion ______________________________________ 32
2.2.4 Bestimmung der Transfektionseffizienz mit dem Luziferase-Assay___________ 32

V
#!) 5����� �������� �� 6�������3�� �������� ��� ���������2������ "" ))
2.3.1 Lyse der transfizierten Zellen ________________________________________ 33
2.3.2 SDS-PAGE ______________________________________________________ 34
2.3.3 Immunoblot ______________________________________________________ 34
#!* ��������� �����/������� ��������������"""""""""""""""""""""""""" ),
2.4.1 Allgemeines zum Vakzinia-Virus _____________________________________ 35
2.4.2 Sicherheitsmaßnahmen im Umgang mit Vakzinia-Viren ___________________ 37
2.4.3 Herstellung von Vakzinia-Virussuspensionen im kleinen und großen Maßstab __ 38
2.4.4 Titrierung von Vakzinia-Virussuspensionen im Plaquetest__________________ 40
2.4.5 Homologe Rekombination und Selektion rekombinanter Vakzinia-Viren ______ 44
2.4.6 Charakterisierung rekombinanter Vakzinia-Viren und ihrer Genprodukte ______ 49
2.4.7 UV-Inaktivierung der Replikation rekombinanter Vakzinia-Viren____________ 51
#!, �� 3�3� $2��������� """"""""""""""""""""""""""""""""""""""""""""" ,#
2.5.1 DNA-Immunisierung_______________________________________________ 52
2.5.2 Gewinnung des Serums, Präparation der Milz und Erstellung einer Einzel-
Zellsuspension __________________________________________________________ 53
2.5.3 ELISA zum Nachweis von HBx-spezifischen Antikörpern _________________ 54
2.5.4 In vitro Proliferationsanalyse von T-Zellen______________________________ 55
2.5.5 In vitro Stimulation und Nachweis von zytotoxischen T-Zellen (CTLs) _______ 56
���������� �������������������������������������������������������� ��
)! ��������� �� ��������������� ��� &�������������� """"""""""""" ,+
3.1.1 Konstruktion des Expressionsplasmids pcD/3-X-ayw______________________ 59
3.1.2 Klonierung des Vakzinia Transferplasmids______________________________ 61
3.1.3 Präparation der Plasmid-DNA________________________________________ 63
3.1.4 Optimierung der Transfektionseffizienz mit dem Reportergen Luziferase ______ 64
3.1.5 Expression von HBx-adw und HBx-ayw________________________________ 66
)!# ��������� �����/������� ��������������"""""""""""""""""""""""""" -+
3.2.1 Rekombination und Selektion der Virusklone____________________________ 70
3.2.2 Präparationen rekombinanter Virusklone _______________________________ 70
3.2.3 Charakterisierung rekombinanter Vakzinia-Viren und ihrer Genprodukte ______ 71
3.2.4 UV-Inaktivierung rekombinanter Vakzinia-Viren_________________________ 76
)!) �� 3�3� $���/��� """"""""""""""""""""""""""""""""""""""""""""""" 1+
3.3.1 Anti-HBx Antikörper nach DNA-Immunisierung_________________________ 79
3.3.2 Die T-Zellproliferationsantwort nach DNA-Immunisierung_________________ 82

VI
3.3.3 Zytotoxische T-Zellaktivität _________________________________________ 86
������������������������������������������������������������������� ��
*! ������������ ��� 7�����7��� �������������� ��(������ """"""""""" 0+
4.1.1 HBx als Zielantigen der spezifischen Immuntherapie______________________ 93
*!# $2������������� ����� """""""""""""""""""""""""""""""""""""""""" +1
*!) �� 3���� $���/��� """""""""""""""""""""""""""""""""""""""""""""" +0
*!* �� 3�3� $���/��� """""""""""""""""""""""""""""""""""""""""""""" 44
4.4.1 HBx-spezifische Antikörperproduktion________________________________ 100
4.4.2 T-Zellproliferationsantworten _______________________________________ 102
4.4.3 CTL-Antworten __________________________________________________ 105
*!, 8��(���������3�� """"""""""""""""""""""""""""""""""""""""""" 41
������������� ����������������������������������������������� ��
�������������������� ������������������������������������������� �
���������� ������������������������������������������������������ ��

1
����� ��
� � !"!# $ %$& �$'() ) &*�*� +,&
Das Hepatitis-B-Virus (HBV) ist der Prototyp der Familie der Hepadnaviren. Es handelt
sich um kleine umhüllte DNA-Viren, die primär die Leber infizieren. Hierzu werden
auch das Woodchuck-Hepatitis-Virus (WHV), das Ground-Squirrel-Hepatitis-Virus
(GSHV), das Enten-Hepatitis-B-Virus (DHBV) und das Reiher-Hepatitis-B-Virus
(HHBV) gezählt.
Die infektiösen Viruspartikel haben elektronenmikrospisch eine sphärische Gestalt mit
einem Durchmesser von 42 nm und werden auch nach ihrem Entdecker als Dane-Partikel
benannt. Die Konzentration zirkulierender Dane-Partikel im Serum HBV-infizierter
Patienten variiert zwischen 103 und 109 Viruspartikel/ml. Die Virushülle besteht aus
verschiedenen HBV-Hüllproteinen, welche ein ikosaedrisches Kapsid mit einem
Durchmesser von 22 bis 25 nm umgeben. Dieses besteht aus 240 Einheiten des HBcAg
(Hepatitis-B-Virus Core-Antigen) und enthält das Virus-Genom. Im Serum infizierter
Patienten werden außerdem noch 22 nm große, nicht infektiöse sphärische und
fadenförmige Partikel gefunden, welche überwiegend aus dem kleinen HBV-Hüllprotein
(HBsAg) bestehen, jedoch keine virale DNA enthalten. Diese Partikel kommen während
einer Infektion in Konzentrationen von 106 bis 1014/ml Serum vor und sind sehr
immunogen, weshalb sie zur Entwicklung einer HBV-Vakzine genutzt werden konnten
(261).
Das je nach Subtyp 3100 bis 3300 Basenpaare lange Genom besteht aus nur partiell
doppelsträngiger DNA. Der vollständige Minusstrang wird im Replikationsverlauf in
mRNA transkribiert und enthält 4 offene Leserahmen, welche in verschiedenen
Leserastern liegen und sich teilweise überlappen. Von diesen kodiert jeweils einer für die
verschiedenen Formen des HBsAg (kleines, mittleres und großes Hüllprotein), für die
Nukleokapsidantigene (HBcAg und sezerniertes HBeAg), für die DNA-Polymerase (P-

2
Protein) sowie für das X-Protein (siehe Abb. 1). In vivo hat man bisher 4 verschiedene
mRNAs gefunden: die 3.3 kb große prägenomische RNA dient zur Translation der
Polymerase, des Core-Antigens sowie des HBeAg. Das große Hüllprotein (präS1-
HBsAg) wird von der 2.4-kb-RNA synthetisiert, das mittlere (präS2) und das kleine
Hüllprotein von der heterogenen 2.1-kb-RNA. Das X-Antigen wird durch eine 0,8 kb
große mRNA kodiert. Bei der viralen Replikation dient die 3.3-kb mRNA gleichzeitig als
prägenomisches RNA-Intermediat, welches mit dem Polymerase-Protein über das ε-
Signal interagieren kann. Die Polymerase besitzt eine Reverse-Transkriptase-Aktivität,
die ein kurzes Stück genomischer DNA synthetisiert, um dann mit dem RNA-Transkript
zu hybridisieren. Dieses fungiert dann als Primer zur Synthese des gesamten
genomischen DNA-Stranges (296). Mit Hilfe der RNase-H-Aktivität der Polymerase
wird der Abbau der RNA am RNA/DNA-Hybrid vollzogen und schließlich zuletzt der
komplementäre DNA-Strang synthetisiert. Das X-Protein scheint für die Infektiosität von
HBV in vivo, nicht jedoch für den Replikationszyklus essentiell zu sein (19, 32, 186,
198).
�//! ! 9���������� �� ����:����!

3
�� �' %$- !"!# $. /()0!#$1$&$ ,1% �" 1 2 %$+ ���*�13$2) !1
Das HBV wird parenteral übertragen. Dabei ist der Mensch das einzige Reservoir für die
Erreger, wobei eine experimentelle Übertragung auf den Schimpansen möglich ist. Eine
Infektion mit dem Virus führt zu einer akuten Hepatitis, welche in 5-10% chronisch
verläuft. Die Infektion des Neugeborenen durch eine chronisch infizierte Mutter kann
pränatal, aber vor allem perinatal, selten auch über die Muttermilch erfolgen und führt in
90% zu einer chronischen Verlaufsform. Dieser Infektionsmodus spielt eine wesentliche
Rolle in Hochendemiegebieten wie Ostasien, Zentral- und Westafrika mit einer
Durchseuchung der Bevölkerung von 20-80%. Die akute Verlaufsform hat eine
Inkubationszeit von 2-6 Monaten und verläuft in bis zu 65% asymptomatisch. Die
symptomatische Form geht mit Ikterus, Hepatomegalie evtl. auch Fieber und
Krankheitsgefühl einher und dauert in der Regel 2-3 Wochen. Die chronische HBV-
Infektion wird histopathologisch abhängig vom Entzündungsgrad und Ausprägung einer
Leberfibrose/-zirrhose in unterschiedliche Schweregrade unterteilt. Mit der Dauer einer
chronischen Infektion und Ausbildung einer Leberzirrhose steigt das Risiko, ein
hepatozelluläres Karzinom (HCC) zu entwickeln. Weltweit rechnet man insgesamt mit
ca. 350 Millionen chronisch infizierten Personen und etwa 250 000 sterben pro Jahr am
HCC. Neben der kontinuierlichen Hepatozytenregeneration und der genetischen
Instabilität scheint auch die Integration des HBV-Genoms an zufälliger Stelle in die
chromosomale DNA des Hepatozyten eine Rolle bei der Entstehung des HCC zu spielen
(202).
Zur serologischen Diagnose einer akuten HBV-Infektion bestimmt man das HBsAg. Eine
Ausheilung wird durch durch den Verlust von HBsAg und das Auftreten von anti-HBs
und anti-HBe Antikörpern angezeigt. Eine chronische Verlaufsform zeigt sich durch die
Persistenz von HBsAg über einen Zeitraum von mehr als 6 Monaten. Zur Bestimmung
der Höhe der viralen Replikation wird die virale DNA-Menge im Serum mittels
quantitativer Polymerasekettenreaktion gemessen. Der Durchseuchungsgrad einer
Bevölkerung lässt sich am besten mittels anti-HBc Antikörper ermitteln, da diese sowohl
nach einer akuten als auch bei einer chronischen Infektion lebenslang persistieren (siehe
Abb. 2).

4
�//! #! ;�������7�� &�������� �� �����( ����� �����% ��/�������������� ���������������
��(������ (entnommen aus Modrow S, Falke D, Molekulare Virologie (186)).
�4 �--,1'()0!#$1$&$ %$+ �$'() ) & �
In verschiedenen Studien konnte gezeigt werden, daß das HBV nicht direkt zytopathisch
für die infizierten Hepatozyten ist (46). Untersuchungen zur Pathogenese der Hepatitis B
im humanen System und im Tiermodell erbrachten eindeutige Hinweise, daß die
Hepatitis durch zelluläre HBV-Antigen-spezifische und Antigen-unspezifische
Immunreaktionen initiiert wird (29,107). Bei der Viruselimination sind sowohl zelluläre
als auch humorale Immunmechanismen beteiligt. Es konnte gezeigt werden, daß neben
der Virus-spezifischen, T-Zell vermittelten Lyse infizierter Zellen auch inflammatorische
Zytokine, welche von aktivierten Antigen-spezifischen und -unspezifischen
lymphomononukleären Zellen sezerniert werden, eine wichtige Rolle spielen. Letztere
können direkt und auf nicht-zytopathischem Wege eine Inhibition der Virusreplikation
und der viralen Genexpression induzieren (5, 47).

5
Bei einem selbstlimitierenden Verlauf einer akuten Hepatitis B beobachtet man eine
besonders starke, multispezifische und polyklonale T-Zellantwort gegen verschiedene
HBV-Antigene. Im Falle einer chronischen HBV-Infektion gelingt die vollständige
Viruselimination nicht und es findet sich eine bedeutend schwächere und antigenetisch
stärker restringierte Immunantwort im Blut der Patienten. Es konnten zahlreiche MHC-
Klasse-I und -Klasse-II restringierte T-Zell-Epitope im Bereich der HBV-
Strukturproteine identifiziert werden, wobei die CD4+ T-Helferzellantworten vor allem
gegen das Core-Antigen gerichtet sind, während die HBV-Hüllproteine, das Core-
Antigen und das Polymerase-Antigen häufig Ziel einer CD8+ zytotoxischen T-
Zellantwort (CTL) sind (47). Auch nach Ablauf einer selbstlimitierenden akuten
Hepatitis können Spuren der HBV-DNA im Serum in sehr niedrigen Konzentrationen
mittels PCR trotz kompletter klinischer und serologischer Ausheilung über Jahrzehnte
nachweisbar bleiben. Diese „low-level“-Persistenz der viralen DNA tritt in Kombination
mit nachweisbaren HBV-spezifischen CTL-Antworten auf. Dies könnte darauf
hinweisen, daß die HBV-DNA transkriptionell aktiv ist und somit wenige virale Partikel
bzw. Antigene lebenslang produziert werden, welche eine protektive CTL-Antwort
ständig unterhalten (175, 221).
Im transgenen Mausmodell wurden weitere Untersuchungen zu HBV-spezifischen
zytotoxischen T-Zellen und den Mechanismen der nicht-zytopathischen Immunantwort
durchgeführt. Transgene Mäuse, welche das HBV-Hüllprotein in ihren Hepatozyten
exprimieren, entwickelten nach dem intravenösen, adoptiven Transfer von CD8+ MHC-
Klasse-I restringierten, HBsAg-spezifischen CTL-Klonen eine akute Hepatitis (5, 106,
190, 301). Es wurden typische Zeichen der Apoptose mit der Bildung von azidophilen
Councilman Bodies, welche charakterischerweise auch bei einer humanen, akuten viralen
Hepatitis auftreten, beobachtet. Bei diesem Prozess des CTL-induzierten Killings
infizierter Hepatozyten werden simultan Fas-vermittelte und Perforin abhängige
Apoptose-Wege aktiviert (196).
Die CTL-induzierte Apoptose betrifft jedoch insgesamt nur 5% aller Hepatozyten und es
kommt konsekutiv durch das Einwandern weiterer Entzündungszellen zu einer
nekroinflammatorischen Reaktion mit dem klinischen Bild des Ikterus. Hierbei spielt
auch die Antigen-unspezifische Zerstörung von Hepatozyten mittels Interferon-γ (IFN-γ)
und Tumor-Nekrose-Faktor-α (TNF-α) Sekretion durch aktivierte Leber-Makrophagen

6
und T-Zellen eine wichtige Rolle (4). Neben dem Killing von Hepatozyten kommt es
vermittelt durch die CTLs zu einer nicht-letalen Suppression der viralen Replikation und
Expression in allen Hepatozyten (104, 106). Dieser nicht-zytopathische Prozess ist
Zytokin vermittelt und kann durch spezifische anti-IFN-γ und anti-TNF-α Antikörper
blockiert werden.
Außerdem wurde untersucht, daß ein solcher Prozess auch durch den Transfer von
HBsAg-spezifischen MHC-Klasse-II restringierten T-Zellklonen in transgene Mäuse
initiiert werden kann. Ein weiterer starker Inhibitor der HBV-Replikation in der Leber ist
Interleukin-12 (IL-12), welches über die Induktion von IFN-γ wirksam wird (40). Die
beiden Zytokine IFN-γ und IL-2 wirken über die Aktivierung von Lebermakrophagen,
welche das antiviral wirksame TNF-α sezernieren (99, 105). Dieses hemmt konsekutiv
die Expression von HBV-Proteinen durch eine posttranskriptionelle Degradation der
HBV-RNA in der infizierten Zelle.
Eine ungeklärte Frage bleibt jedoch weiterhin der genaue Mechanismus der HBV-
Persistenz bei einer chronischen HBV-Infektion. Der immunologische Toleranz-Effekt
ist wahrscheinlich entscheidend für die fehlende antivirale Immunantwort und Persistenz
der neonatalen HBV-Infektion nach einer Mutter-zu-Kind-Transmission des Virus. Es
wurde wiederholt gezeigt, daß die CTL-Antwort chronisch infizierter Patienten sehr
schwach im Vergleich zu einer akuten, selbstlimitierenden HBV-Infektion ist (185, 207,
220).
Es gibt mehrere Hypothese, die eine virale Persistenz und Suppression der zellulären
Immunität erklären können:
♦ Induktion einer peripheren immunologischen Toleranz.
♦ Erschöpfung der T-Zellantwort durch eine sehr hohe Viruslast und Antigenämie.
♦ CTL-Inaktivierung durch Antigenpräsentation ohne den Kontext kostimulatorischer
Signale in der Leber.
♦ Inhibition der Antigenpräsentation durch virale Mechanismen.
♦ „Down“- Regulation der viralen Gen-Expression.
♦ HBV-Infektion extrahepatischer, peripherer immunologischer Organe (253).

7
♦ Umgekehrte Expression des Fas-Liganden in der infizierten Leberzelle und
Destruktion der CTLs über den Fas Ligand–Fas Rezeptor-Mechanismus (93, 102).
♦ Eine verminderte Zytokinproduktion vom TH1-Subtyp oder das Vorliegen einer
TH0/TH2 Polarisierung könnte die komplette Virus-Elimination verhindern.
Um diese Hypothesen belegen zu können, bedarf es weiterer, intensiver
immunologischer Untersuchungen in Hepadnavirus Tiermodellen und im humanen
System.
Eine Selektion von CTL-Escape-Mutanten kann im Rahmen einer chronischen HBV-
Infektion auftreten. Sie spielt jedoch wahrscheinlich keine so bedeutende Rolle für die
Viruspersistenz wie z.B. bei der HIV-Infektion (7, 27, 28, 130). Die Mutanten werden
charakteristischerweise durch eine starke und besonders restringierte Immunantwort
selektioniert – bei der chronischen HBV-Infektion kein häufig anzutreffender Zustand
(siehe oben). Außerdem entstehen sie meist sekundär auf dem Boden einer bereits
persistierenden HBV-Infektion. Inwieweit solche Mutanten als primäre Ursache für die
HBV-Persistenz in Frage kommen, ist bisher ungeklärt.
Die CD4+ MHC-Klasse-II restringierte T-Zellhilfe ist ein wichtiger Faktor für die Stärke,
Spezifität und Polarisierung der CTL-Antwort während einer akuten und auch
chronischen HBV-Infektion. Eine Assoziation bestimmter MHC-Klasse-II Allelen
(DRB1*1302) mit einem protektiven Effekt gegen die chronische HBV-Infektion konnte
in Fall-Kontroll-Studien nachgewiesen werden (268).
Die Differenzierung von CD4+ T-Zellen in TH1- (inflammatorische) und TH2-
Effektorzellen entscheidet, ob eine humorale oder zelluläre Immunantwort vorherrschend
sein wird (231). TH1-Zellen zeichnen sich durch Sekretion großer Mengen der Zytokine
IL-2, IFN-γ, TNF-α, TNF-β, Granulozyten-Makrophagen-Colony Stimulating Factor
(GM-CSF) und IL-3 aus und sind wichtig für die Initiation einer Antigen-spezifischen,
zellulären Immunantwort. Weiterhin erkennen TH1-Zellen Peptide im Kontext mit
MHC-Klasse-II Molekülen auf der Oberfläche von professionellen Antigen-
präsentierenden Zellen (APCs), wie dendritischen Zellen, Makrophagen und aktivierten
B-Zellen (184, 187). Weitere wichtige Funktionen der TH1-Zellen sind die Rekrutierung
von Makrophagen an den Infektionsort durch eine IL-3- und GM-CSF-vermittelte

8
Stimulation der Produktion im Knochenmark sowie die Aktivierung der Makrophagen
über die Zytokine IFN-γ und TNF-α (254).
Die klassischen T-Helferzellen vom TH2-Typ werden zumeist durch extrazelluläre
Erreger, wie die meisten Bakterien, aktiviert (206). Mit einem Zytokinprofil zur B-Zell
Stimulation sezernieren sie insbesondere IL-4, IL-5, IL-6 und IL-10. Durch die
Produktion von Antikörpern gegen virale Oberflächenantigene durch aktivierte B-Zellen
können auch die Viruspartikel in der Blutbahn gebunden werden. Somit kann initial
Schutz gegen eine Infektion erzeugt und ein sekundäres Fortschreiten der Viren von
Zelle zu Zelle verhindert werden.
Patienten mit einer akuten, selbstlimitierenden HBV-Infektion zeigen vorwiegend ein
TH1-spezifisches Zytokinmuster. Dagegen produzieren die intrahepatischen T-
Helferzellen von chronisch infizierten Patienten ein variables Muster mit einzelnen TH1-
und TH2-Klonen und Kombinationen von Zytokinen (TH0) (15, 123, 128, 129). Dieses
Phänomen könnte eine Erklärung für die Insuffizienz der Immunantwort bei chronisch
infizierten Patienten darstellen.
In ersten experimentellen Ansätzen einer HBV-Immuntherapie konnte gezeigt werden,
daß sowohl rekombinantes HBsAg nach i.m. Administration als auch DNA-
Immunisierung mit verschiedenen Formen der HBV-Hüllantigene in Mäusen und
Primaten eine potente T-Zellantwort erzeugen können (41, 64, 94, 126, 145, 170, 238).
Insbesondere mit der Methode der DNA-Immunisierung ließen sich im murinen System
besonders starke humorale und zelluläre Immunantworten gegen Epitope der Hüll- und
HBV-Core-Antigene induzieren. Mit der adjuvanten Gabe von Zytokin kodierenden
Plasmiden wie IL-2, GM-CSF, IL-12 und IFN-γ konnten stärkere CTL-Antworten
induziert und eine Differenzierung in TH1- und TH2-Subtypen durch Koimmunisierung
der entsprechenden Zytokin-Plasmide gezeigt werden (49, 96, 300). Durch die
Konstruktion von Polytop-Minigen-Plasmiden, welche mehrere humane HLA
restringierte Epitope der HBV-Polymerase-, Hüll- und Core-Antigene kodieren, konnte
die Entwicklung und Optimierung von Epitop-spezifischen DNA-Vakzinen in HLA-
transgenen Mäusen getestet werden (124, 158). Zum jetzigen Zeitpunkt werden bereits
die ersten klinischen Studien der Phase I zur therapeutischen DNA-Immunisierung
chronischer HBV-infizierter Patienten durchgeführt (262).

9
��� �(& �$'() ) &*�*� +,& 5*/+!)$ 1
Der kleinste ORF des HBV-Genoms wurde erstmalig von Galibert et al (92) beschrieben
und kodiert für das lösliche, intrazelluläre X-Protein mit einer Länge von 145 bis 154
Aminosäuren (17 kD). Seine Rolle bei der HBV-Replikation und Pathogenese der
Hepatitis-B-Infektion ist bis dato unklar, zumal bisher kein Protein der vorhergesagten
Größe in infizierten Zellen oder Viruspartikeln sicher in vivo nachweisbar war. Ursache
dafür ist wahrscheinlich die sehr schnelle Degradation des Proteins bzw. eine sehr
schwache intrazelluläre Expression unterhalb der experimentellen Nachweisgrenze. In
vitro Transfektionsstudien mit heterologen Promotoren konnten ein 17 kD großes Protein
nachweisen, welches sich immunhistochemisch in humanen Lebergewebe primär im
Zytoplasma, in einigen Zellen jedoch ausschließlich im Nukleus lokalisieren läßt (61, 92,
247, 318). Es besteht eine Homologie von 80% zwischen den 4 unterschiedlichen HBV-
Subtypen, darüber hinaus eine 71 bzw. 31%ige Homologie zu WHV und GSHV.
Hochkonservierte Sequenzen befinden sich im Bereich des Amino- und Karboxy-
Terminus des ORF. Im Gegensatz dazu enthält das DHBV und das HHBV keinen X-
ORF. Weitere Untersuchungen konnten zeigen, daß das X-Protein nicht essentiell für die
Replikation von HBV in vitro zu sein scheint (32, 310). Allerdings ist es im Woodchuck-
Modell notwendig für eine erfolgreiche Infektion und Etablierung einer chronischen
Hepatitis (43, 321).
!*! <�������� �� ��&������
Das X-Protein in seiner Rolle als Transaktivator zur Transkription zahlreicher viraler und
zellulärer Gene ist schon seit längerem bekannt. Es aktiviert in vitro und in vivo u.a. die
Promotor/Enhancer Elemente der HBV-Core und –Surface Gene (55, 223, 229) sowie
heterologe Promotoren, wie den SV-40 early-Promotor, den Rous-Sarcoma-Virus (RSV)
Promotor (251), den Herpes-Simplex-Virus Thymidine-Kinase Promotor (316), das
Long-Terminal-Repeat (LTR) des humanen T-lymphotrophen Virus (HTLV-1), HIV-
Promotoren (278) und den β-Interferon Promotor (277). Ausserdem konnte gezeigt
werden, daß das X-Protein die Aktivität der RNA-Polymerasen II und III über die
Transkriptionsfaktoren NF-κB und AP-1 und AP-2 steigern kann und als Ko-Aktivator

10
über Protein-Protein Interaktionen u.a. mit den Aktivator-Molekülen CREB (cAMP
responsive element binding protein), ATF-2 (activating transcription factor-2) und TBP
(TATA-binding protein) zu wirken scheint (8, 165, 214, 245, 278). Ob alle diese in vitro
Interaktionen auch unter in vivo Bedingungen, bei sehr geringer Expression des X-
Antigens, relevant sind, ist allerdings eher unwahrscheinlich.
Unklarheit besteht weiterhin über die Rolle des X-Proteins bei der Karzinogenese des
HCC trotz zahlreicher Erkenntnisse über seine pleiotropen Transaktivator-Funktionen
und zellulären Ziel-Moleküle (78). Sequenzen des X-ORF werden häufig an zufälliger
Stelle chromosomal integriert in HCC-Zellen gefunden und oft kann eine gesteigerte
Expression von HBx im Vergleich zu den übrigen HBV-Antigenen beobachtet werden
(69, 169, 203, 205, 256, 291). Häufig kommt es dabei zur Fusion von Teilen des X-ORF
mit zellulären, kodierenden Sequenzen bei intakter Transaktivator-Funktion des X-
Proteins (305). Die Selektion HBxAg-positiver Hepatozyten ist möglicherweise ein erster
Schritt bei der Multi-Step Karzinogenese des HCC. In Transfektionsstudien und in HBx-
transgenen Mäusen konnte bereits eine transformierende Aktivität des X-Proteins
nachgewiesen werden (119, 135, 141). Andere Arbeitsgruppen konnten diese Ergebnisse
aber nicht bestätigen (151, 222). Weitere Schritte sind die Alteration von Signal-
Transduktionsmolekülen durch HBx wie NF-κB, den ras- und raf-abhängigen
Proteinkinasen (MAP) und der Proteinkinase C (PKC), mit nachfolgender Störung der
Wachstumsregulation und Resistenz der Zellen gegen Apoptose-Stimuli durch Zytokine
bzw. einem immunologischen „Escape“ der Zellen (21, 22, 133, 161, 199, 245, 294).
Insbesondere aktiviertes NF-kB ist ein sehr potenter Inhibitor der TNF-α induzierten
Apoptose und könnte den infizierten und mutierten Leberzellen während der chronischen
Infektion einen Selektionsvorteil verschaffen (20).
Eine wichtige Rolle könnte auch die Inaktivierung des Tumor-Suppressor-Gens p53
durch eine Komplex-Bildung mit HBx spielen (77, 273, 279, 293). p53 besitzt eine
zentrale Bedeutung bei der Reparatur von DNA-Schäden und Verhinderung
unkontrollierter Zellproliferation und Tumortransformation durch Induktion der
Apoptose in der geschädigten Zelle; daher sind Alterationen der Aktivität des Proteins
oder Mutationen dieses Gens assoziiert mit zahlreichen Tumorformen (70, 120, 280, 287,
317). In neueren Studien konnte aber auch gezeigt werden, daß eine HBx-Expression mit
der Induktion von Apoptose assoziiert sein kann (45, 255).

11
�//! )! &���������� ������������� �� ��&������ ��� �������������� ;������=����������������!
Die Veränderungen der Gen-Expressionsmuster können zur Resistenz gegen Apoptose-Stimuli z.B. durch
Zytokine führen. Der Selektionsvorteil HBxAg-positiver Zellen führt über weitere Schritte im Sinne einer
Multi-Step Karzinogenese durch die Akkumulation von Mutationen zur HCC-Transformation der Zelle.
Modifiziert nach MA Feitelson und LX Duan (78).
Es ist allerdings klar, daß das X-Protein kein akut transformierendes Onkogen sein kann,
da durchschnittlich 30-60 Jahre bei einer chronischen HBV-Infektion bis zum Auftreten
eines HCC vergehen. Vielmehr scheint es unterschiedliche Schritte im Rahmen einer
Multi-Step Karzinogenese zu modulieren, deren Bedeutung in vivo Gegenstand der
weiteren Forschung ist (78).

12
Neuere Untersuchungen zeigen eine Assoziation von HBx mit der 19S- und auch mit der
20S- Untereinheit des Proteasom-Komplexes der Zelle (121, 122, 250, 319). Das X-
Protein scheint sowohl als Substrat als auch als Inhibitor des 26S-Proteasom-Komplexes
zu wirken, ausserdem ist diese Interaktion möglicherweise essentiell für dessen
Transaktivator-Funktionen. Der Proteasom-Komplex greift als zentrale Schaltstelle der
Degradation Ubiquitin gekoppelter Proteine in zahlreiche zelluläre Prozesse wie u.a. die
Signal-Transduktion, die Kontrolle des Zellzyklus, Stress-Antworten der Zelle,
Aktivierung der Transkription, DNA-Reparatur, Apoptose und Antigenpräsentation ein
(60, 118). Eine Hemmung der Degradation viraler Proteine im Proteasom könnte u.a. zur
Suppression der viralen Antigenpräsentation in Assoziation mit MHC-Klasse-I
Molekülen führen und so den immunologischen „Escape“ der Virus-infizierten Zelle
fördern (226). Für diese Hypothese fehlen aber noch eindeutige experimentelle in vivo
Daten.
!*!# ���������7�� ��������������� �� ��&������
Nachweisbare Antikörper gegen das X-Protein finden sich bei HBV-infizierten Personen
in 7-54% (115, 132, 174, 189). Es besteht keine Korrelation zwischen anti-HBx Titern
und einer chronischen HBV-Infektion bzw. dem Vorliegen eines HCC. Ein bestimmter
Prozentsatz HBV-infizierter Patienten produziert überhaupt kein anti-HBx, ein Indiz für
eine sehr schwache Immunogenität des Proteins und /oder eine hohe quantitative und
qualitative Heterogenität der humoralen Immunantworten. Peptid-spezifische ELISA-
Untersuchungen zur Bestimmung der Epitop-Feinspezifität chronisch infizierter
Patienten zeigten eine kurze immundominante Region im Bereich des Karboxy-Terminus
des X-Proteins (252). Weiterhin konnten bisher HLA-Klasse-II restringierte T-
Zellantworten gegen das X-Protein bei akut und teilweise auch bei chronisch infizierten
Patienten nachgewiesen werden, wobei mit Peptiden mehrere T-Zell-Epitope, ebenfalls
hauptsächlich im Bereich des Karboxy-Terminus liegend, charakterisiert werden konnten
(127). Im Vergleich mit den immunogenen Core- und Hüll-Antigenen fallen die T-
Zellantworten aber weitaus schwächer aus.
In einer neueren Studie konnten HLA-A2.1 restringierte CD8+ T-Zell-Epitope des X-
Proteins mit synthetischen Peptiden bei chronisch HBV-Infizierten nachgewiesen

13
werden. Die Ergebnisse dieser Studie sollten aber noch durch weitere Experimente,
welche insbesondere eine endogene Prozessierung des HBxAg durch die Targetzelle
nachweisen können, bestätigt werden (51).
��� ���*�--,1 & $+,1#
!,! ���������� &������
Die DNA-Immunisierung, auch bekannt als genetische oder Polynukleotid-
Immunisierung, ist eine effiziente Methode zur spezifischen Aktivierung des
Immunsystems. Obwohl schon zu Beginn der 90er Jahre bekannt war, daß die Injektion
von nackter Plasmid-DNA in eine Maus zur Expression des auf dem Plasmid kodierten
Genes in vivo führt (303), konnte erst 1992/1993 in ersten Studien die Bedeutung dieses
Ansatzes für die Entwicklung von Vakzinen und Immuntherapeutika gezeigt werden (89,
264, 281, 289). Das Prinzip dieser Methode beruht auf der Applikation von gereinigter,
nackter Plasmid-DNA, welche aus einem nicht-replikationsfähigen Expressions-Vektor
mit den Antigen-kodierenden Sequenzen und entsprechenden regulatorischen Elementen
besteht, in den Empfänger-Organismus. Dort wird die DNA von den Wirtszellen
aufgenommen und das Antigen exprimiert. Das endogen produzierte Protein kann so
zytosolisch prozessiert und adäquat auf der Zelloberfläche präsentiert werden, um eine
spezifische Immunantwort des Empfängers zu induzieren. Auf diesem Wege der
endogenen Synthese von Antigenen durch die Wirtszelle wird eine Infektion durch ein
virales Pathogen imitiert. Durch die Präsentation von Peptidfragmenten des Antigens im
Kontext von MHC-Klasse-I Molekülen kommt es zur Aktivierung von spezifischen
CD8+ zytotoxischen T-Zellen (CTLs) über den T-Zellrezeptor. Je nach Proteintyp
werden die produzierten Antigene auch von der transfizierten Zelle sezerniert und
können extrazellulär das Priming einer humoralen und CD4+ T-Zell Immunantwort
induzieren. Die freigelassenen Proteine können aber auch von Antigen-präsentierenden
Zellen (APC) aufgenommen und über MHC-Klasse-II und –Klasse-I restringierte
Antigenpräsentation eine T-Helferzell- und CTL-Antwort (Cross-Priming) generieren.
Die DNA-Immunisierung bietet die gleichen immunologischen Vorteile wie die
Immunisierung mit lebenden, attenuierten Vektoren, jedoch ist die applizierte Plasmid-

14
DNA nicht replikationsfähig und das Risiko einer unkontrollierten Ausbreitung des
Vektors in immundefizienten Wirtsorganismen oder einer Übertragung auf weitere
Personen vergleichsweise minimal.
Tang et al. (264) konnten zum ersten Mal zeigen, daß die Mäuse, denen mit einer Gene-
Gun Plasmid-DNA injiziert worden war, eine Antikörperantwort gegen das kodierte
Protein entwickelten. Bald entdeckte man auch, daß durch diese Technik der
Immunisierung breite zelluläre und humorale Immunantworten gegen verschiedene virale
Antigene, wie z.B. Proteine des Influenza-Virus (89, 281), des HIV-1 (289) und des
HBV (64), induziert werden konnten.
�//! *! ;7������7�� ��������� �� &������ ��� ������������ �������������� ��
�������� 3�� �������� �� ��������� �������������! Modifiziert nach M. Geißler.
Seitdem wurden in zahlreichen Infektions- und Tiermodellen DNA-Vakzine gegen
weitere virale, bakterielle und auch Protozoon-Erreger, wie HIV-2 (312), das Herpes-
Simplex (34, 167), Rabies (307), HCV (74, 95), Tuberkulose (159, 265, 266), Malaria-

15
Sporozoiten (188), Mykoplasmen (18), Leishmanien (308), Zytomegalieviren (100),
Toxoplasmen (6), Rotaviren (114) und zuletzt auch Ebola (309) und Listeriose (81)
untersucht. Der Vorteil dieser Immunisierungstechnik gegenüber der herkömmlichen
Immunisierung mit rekombinanten Antigenen ist die Induktion einer zytotoxischen,
CD8+ T-Zell- und inflammatorischen T-Helferzellantwort, welche für die Eradizierung
verschiedenster viraler Erreger unerlässlich ist; teilweise ist es sogar möglich, eine
partielle oder komplette Protektion gegen die Pathogene im Tiermodell zu
demonstrieren.
!,!# ��������� �� ��������� ��� $((������
Es gibt zahlreiche Möglichkeiten, die durch eine DNA-Vakzine induzierten
Immunantworten quantitativ und qualitativ in ihrer Effizienz zu modulieren. In der Regel
sind multiple Applikationen notwendig, um maximale Immunantworten zu induzieren.
Es gibt jedoch noch keine verläßlichen Daten, wie ein optimales Immunisierungs-
Protokoll (DNA-Menge, Zahl und Frequenz der Einzeldosen) aussehen soll. Eine
mögliche Optimierungsstrategie ist die Wahl größerer zeitlicher Abstände zwischen den
DNA-Einzeldosen (88, 152).
Antigen-spezifische Immunantworten konnten nach intravenöser, intramuskulärer,
intranasaler, intraepidermaler, intradermaler, intravaginaler und neuerdings auch
intrasplenischer und intrahepatischer Applikation von DNA gezeigt werden (11, 98, 290,
304). In den meisten Studien wurden die Plasmide allerdings intradermal oder
intramuskulär injiziert (56, 65, 285). Zur epidermalen Applikation der DNA wird häufig
die Genkanone (Gene Gun) eingesetzt. Bei dieser Methode werden Gold-Mikropartikel
mit Plasmiden beladen und durch Beschleunigung in die Haut injiziert. Auf diese Weise
werden die Zellen der Epidermis (u.a. Langerhans-Zellen) direkt durch die Mikropartikel
transfiziert. Im Vergleich zur Nadelinjektion ist die Genkanone deutlich effizienter, da
man gleiche Antikörper- und CTL-Antworten mit 100- bis 5000-fach geringerer DNA-
Menge erzielen kann (80, 209). Trotz dieser Unterschiede in der Effizienz sind die
induzierten Immunantworten weder von längerer Dauer noch konnte man eine bessere
Protektion gegen Pathogene in vivo erreichen. Im Gegenteil, während nach
intramuskulärer DNA-Injektion überwiegend TH1-Antworten mit einem hohen IgG2a :

16
IgG1 Verhältnis, IFN-γ Produktion und geringer IL-4 Produktion induziert werden,
überwiegen nach Immunisierung mit der Genkanone TH2-Antworten mit vorherrschend
IgG1 Antikörpern, weniger IFN-γ und mehr IL-4 Produktion (80, 87, 210). Wird die
DNA intradermal mittels Nadel injiziert, so können beide TH1- und TH2-Profile
generiert werden (210, 219).
Neuere Ansätze der Applikation wie die Beladung von synthetischen Mikrosphären mit
Plasmid-DNA (14, 36, 113), der Einsatz selbst-replizierender RNA-Vakzine oder
artifizieller, bakterieller Chromosomen (BAC-VAC) als replizierende Vakzine eröffnen
erfolgsversprechende Perspektiven zur Entwicklung hochimmunogener DNA-und RNA-
Impfstoffe (72, 153, 257, 314). Die Stärke und Qualität der induzierten Immunantworten
muß noch in weiteren Studien mit diesen neuen Methoden genauer charakterisiert
werden.
Zytokine sind essentielle Moleküle für die Regulation und Koordination der zellulären
und humoralen Immunantwort. Bereits 1993 konnte gezeigt werden, daß durch die
Injektion von Zytokin-Plasmiden in den Muskel die Immunantwort auf ein
rekombinantes Protein durch die charakteristische Aktivität des Zytokins in vivo
verstärkt wurde (218). Durch eine Ko-Immunisierung mit Zytokin kodierenden
Plasmiden können die durch ein Antigenplasmid induzierten Immunantworten nicht nur
verstärkt, sondern auch in Richtung einer TH1- oder TH2-Antwort gelenkt werden. So
stimuliert beispielsweise die adjuvante Injektion eines IL-2 Plasmids eine TH1-
polarisierte humorale und zelluläre Immunität gegen ein Antigen (48, 95).
Das Zytokin GM-CSF fördert die Produktion von Granulozyten und Makrophagen,
sowie die Reifung und Aktivierung von APCs und dendritischen Zellen (116).
Intramuskulär verabreicht kann die Koexpression von GM-CSF sowohl die humorale wie
die zelluläre Immunantwort gegen Plasmid kodierte Antigene verstärken (95, 125, 248,
306).
IL-12 ist der Prototyp eines TH1-Zytokins mit einer sehr potenten Induktion der CTL-
und NK-Aktivität sowie einer erhöhten IFN-γ Produktion (272). Die Ko-Immunisierung
mit einem IL-12 Plasmid auf intranasalem oder intramuskulärem Wege kann eine

17
Induktion der zellulären Immunität vom TH1-Typ gegen verschiedene Antigene deutlich
steigern, vor allem mit einer verstärkten CTL-Antwort und T-Zellproliferation bei
gleichzeitig verminderter Antikörperproduktion (49, 125, 136, 249, 275, 276). Ein
weiteres TH1-Zytokin, das IL-18, scheint dagegen nur zu einer moderaten Steigerung der
CTL-Aktivität, aber deutlicher Erhöhung der Antikörpertiter und T-Helferzellantwort zu
führen (137).
Auch der Zeitpunkt der Ko-Administration von Zytokin-Plasmiden scheint sich auf die
Effizienz der Immunantworten auszuwirken. Sudien mit GM-CSF und einem
Fusionsprotein aus IL-2 und dem Fc-Teil eines murinen Antikörpers zeigten bei Injektion
vor oder mit der Gabe des Antigenplasmids eine schwächere bzw. eher TH2-dominante
Immunantwort, während die Gabe 3 bis 5 Tage nach DNA-Immunisierung zu einer
potenten CTL-Antwort bzw. TH1-dominanten Zytokinproduktion führte (17, 147).
Das Prinzip der Zytokin-Koimmunisierung wurde in dieser Arbeit angewandt, um
Immunantworten gegen das HBxAg zu verstärken.
Durch die Verwendung nicht-methylierter, palindromischer DNA-Sequenzen, welche
CpG-Oligodinukleotide (CpG-ODN) enthalten, können unabhängig von einem Antigen
Monozyten, NK-Zellen und B-Zellen aktiviert werden und als immunstimulatorisches
Adjuvans zur Induktion einer Immunreaktion vom TH1-Subtyp bei der DNA-
Immunisierung eingesetzt werden (143, 144, 297).
Die Wahl unterschiedlicher Carrier bei der initialen Immunisierung und den
nachfolgenden Booster-Immunisierungen bietet sich als weitere Methode zur
Optimierung und effektiven Steigerung von Antigen-spezifischen Immunantworten an.
Benutzt man zwei Carrier-Systeme, welche keine immunologische Kreuzreaktivität
besitzen, wie beispielsweise Plasmid-DNA und rekombinantes Protein oder Plasmid-
DNA und rekombinante Vakzinia-Viren, so kann man in vielen Fällen sehr effiziente
Immunantworten induzieren (225). In einem Malaria-Modell konnte durch ein
heterologes Protokoll mit Plasmid-Priming und rekombinanten Vakzinia-Viren als
Boost-Impfstoff eine komplette Protektion gegen Plasmodium berghei erreicht werden
(242, 244).

18
�6 �$2!-7 1(1)$ �(28 1 (*� +$1
Dem englischen Landarzt Edward Jenner gelang im Jahre 1776 zum ersten Mal die
erfolgreiche Vakzinierung eines 8jährigen Jungen gegen die Pocken (Variola) mit dem
Kuhpockenvirus. Dieses Impfverfahren wurde später durch die Verwendung des
engverwandten Vakzinia-Virus, welches aus dem Pferd isoliert worden war und eine
mildere Impfreaktion zeigte, abgelöst. Wie schon damals von Jenner prophezeit, wurde
dank dieser Vakzine der letzte Fall einer Pocken-Infektion im Jahre 1977 beschrieben
und 1980 erklärte die WHO offiziell die Eradizierung dieser Infektionskrankheit.
Das Vakzinia-Virus ist ein 300-400 nm großes Viruspartikel aus der Familie der
Poxviren, dessen Hülle aus Lipoprotein-haltigen Membranen die komplexe
Nukleokapsidstruktur umschließt. Dieses beherbergt wiederum das ca. 200.000
Basenpaare große, virale Genom aus linearer, doppelsträngiger DNA. Zahlreiche Virus
kodierte Enzyme, u.a. eine DNA-abhängige RNA-Polymerase, ein Transkriptionsfaktor
und Capping-Enzyme, liegen verpackt innerhalb des Kapsides vor und können nach dem
Eintritt in die Wirtszelle mRNA-Transkripte mit typischen eukaryontischen
Eigenschaften synthetisieren. Initial werden nur die sogenannten „early“ Gene
transkribiert, welche das Wachstum der benachbarten Zellen stimulieren, die Replikation
des viralen Genoms und die Transkription der „intermediate“ Gene und „late“ Gene
steuern. Nach der Synthese der „late“ Struktur-Gene kommt es zum Assembly der
viralen Partikel, welche dann durch den Golgi-Apparat transportiert und über die
Plasmamembran aus der Zelle geschleust werden. Jede der drei Gen-Klassen besitzt
spezifsche Promotorsequenzen, die von viralen Proteinen erkannt werden und bilden die
Basis für den programmierten, Kaskaden-artigen Ablauf der viralen Genregulation (193).
In den frühen 80er Jahren wurden die Methoden zur Herstellung rekombinanter
Vakzinia-Viren maßgeblich in der Arbeitsgruppe von Bernard Moss entwickelt (302).
Die meisten Strategien zur Fremdgen-Insertion funktionieren nach dem Prinzip der
homologen Rekombination (siehe unten).

19
�//! ,! ��� .�����������>�� �� ������������ (modifiziert nach Current Protocols in Molecular
Biology (9)).
!-! �� ���������$2������>���
Die gentechnische Rekombination von Vakzinia-Viren bietet eine hervorragende
Möglichkeit zur Expression von Fremdgenen in eukaryontischen Zellen. Wegen des sehr
vielfältigen Wirt-Spektrums im Bereich eukaryontischer Zellen und permissiver
Versuchstiere und einer durchschnittlich sehr starken Expression durch die viralen
Promotoren ist das System für zahlreiche Anwendungen geeignet: Analyse von Protein-
Protein Interaktionen, Epitop-Charakterisierung monoklonaler Antikörper, Zellfusion,
Expression von Ionenkanälen sowie funktionelles Screening von cDNA-Bibliotheken
(193).
Bei der Rekombination werden zumeist nicht-essentielle Gene des viralen Genoms wie
z.B. die Thymidinkinase (TK) durch die Insertion des Fremdgens deletiert, wobei Gene
bis zu einer Größe von 25.000 Basenpaaren im viralen Genom toleriert werden (173). Da

20
das Vakzinia-Virus im Zytoplasma der Wirtszelle repliziert und einen eigenen
Transkriptions-Apparat verwendet, müssen kontinuierliche ORFs und virale Promotoren
zur Fremdgenexpression eingesetzt werden. Der Expressionszeitpunkt im viralen
Replikationszyklus und die Stärke der Expression können durch die Wahl eines „early“,
„intermediate“ oder „late“ Promotors bestimmt werden. Ein häufig verwendeter
Promotor ist der kombinierte „early/ late“ Promotor, mit dem sich eine sehr starke
Expression erreichen lässt (52, 204).
Zur Herstellung eines rekombinanten Vakzinia-Virus verwendet man ein Plasmid als
Transfervektor (shuttle vector) mit einer Expressionskassette, bestehend aus dem
entsprechenden Vakzinia Promotor und einer Klonierungsstelle (multiple cloning site)
zur Insertion des Fremdgens. Diese wird von homologen Sequenzen genomischer
Vakzinia-DNA am 5´-und am 3´-Ende flankiert, welche den Ort der Integration im
viralen Genom determinieren (42, 75, 163). Die Rekombination findet mit einer
Wahrscheinlichkeit von 0.1% als doppeltes Cross-Over zwischen den homologen
Sequenzen der infizierten viralen und der transfizierten Plasmid-DNA in der Wirtszelle
statt (Abb. 6, Plasmid-Karte siehe Kap. 2.4.5).
Bei der Selektion rekombinanter Klone können unterschiedliche Methoden angewendet
werden. Wurde die Fremdgen-Sequenz in den viralen TK-Lokus inseriert, so können die
Rekombinanten über ihren TK-negativen Phänotyp in TK-defizienten Zellen selektioniert
werden (163). Alternativ können über den Transfervektor Antibiotikumresistenz-Gene
oder Reporter-Gene ko-inseriert werden, die ein Farb-Screening wie z.B. durch ß-
Galaktosidase (lacZ Gen) oder β-Glukuronidase Synthese ermöglichen (39, 42, 75). Eine
neuere Methode funktioniert über die Komplementierung eines Defektes in der
extrazellulären Virusproduktion, so daß alle Plaques rekombinante Viren enthalten (31).
Auch eine direkte in vitro Ligation des Fremdgenes in das Vakzinia-Genom konnte
erfolgreich durchgeführt werden (235).

21
�//! -! �������� .����/������� ���7��� ����(�������� &����� �� ������������ :����! Das
Fremdgen wird von den Vakzinia-DNA-Sequenzen TKL und TKR flankiert. Das Fremdgen steht unter der
Kontrolle des Vakzinia „early/late“-Promotors p7.5. Das ko-inserierte Selektionsgen lazZ wird durch den
Vakzinia-„late“-Promotor p11 kontrolliert. Modifiziert nach (9).
Eine weitere Modifikation ist der Einsatz der hocheffizienten RNA-Polymerase des
Bakteriophagen T7 zur Expression von Fremdgenen in Vakzinia-Virus Vektoren.
Entweder wird die T7-RNA-Polymerase und das Fremdgen unter der Kontrolle eines T7-
Promotors über getrennte Vektoren ko-infiziert und exprimiert oder beide Proteine
werden über ein induzierbares System im gleichen Vakzinia-Virus synthetisiert (86,
295).
Ein erhöhtes Maß an Sicherheit im Umgang mit den Vakzinia-Viren und der Verzicht auf
potenzielle Impfrisiken (siehe Kap. 2.4.2) konnte durch die Entwicklung neuer

22
attenuierter, replikations-inkompetenter Vakzinia-Stämme erreicht werden; so ist unter
anderem das modifizierte Vakzinia-Virus Ankara (MVA) durch eine mehr als 500-fache
Passage in Hühnerembryonen-Zellen entstanden. MVA ist ein hocheffizienter und
immunogener Expressionsvektor, mit dem unter S1-Sicherheitsstandard gearbeitet
werden darf (117, 242).
!-!# ���������7��� $����� �����/������� ��������������
Zur Produktion monoklonaler Antikörper oder zur Immunisierung von Versuchstieren
können rekombinante Vakzinia-Viren äquivalent zu gereinigtem, rekombinanten Protein
mit sehr hoher Effizienz eingesetzt werden (193). Daneben werden Vakziniavektoren
aber vor allem zur Induktion von CD8+ zytotoxischen T-Lymphozyten in vivo, sowie zur
Generierung von Antigen-präsentierenden Targetzellen in vitro genutzt. Die
intrazelluläre Expression eines Antigens durch das Virus erlaubt eine „natürliche“
Prozessierung und Präsentation von Peptidfragmenten desselben im Kontext von MHC-
Klasse-I Molekülen auf der Oberfläche der infizierten Zelle. Zur Determinierung der
Targetzellen werden autologe oder MHC-syngene Zellen mit den rekombinanten
Vakzinia-Viren infiziert, welche das gewünschte Antigen unter der Kontrolle eines
„early“ oder „early/late“ Promotors exprimieren (271). Die Targetzellen können mit51Chromium markiert werden und in einem Chrom-Release-Assay mit
Effektorlymphozyten von immunisierten Versuchstieren oder humanen T-Lymphozyten
verwendet werden (24, 288). Mit dem Epstein-Barr-Virus immortalisierte B-Zellen
dienen u.a. als autologe Targetzellen zur Untersuchung humaner CTLs.
Mit der Immunisierung von Versuchstieren, von der Maus bis zum Schimpansen, können
rekombinante Vakzinia-Viren, welche eines oder mehrere Gene von RNA- oder DNA-
Viren exprimieren, eine partielle oder sogar komplette Protektion gegen die
entsprechende Infektion erzeugen (192). Der protektive Effekt stützt sich in den meisten
Fällen auf neutralisierende Antikörper gegen Hüllproteine der Erreger sowie auf die
Induktion einer potenten CTL-Antwort gegen die infizierten Zellen (146, 191). Auch bei
der Herstellung von prophylaktischen und therapeutischen Vakzinen gegen
experimentelle Tumoren im Tiermodell konnten rekombinanten Vakzinia-Viren

23
zusammen mit Zytokinen (IL-2 und IL-12) und kostimulatorischen Molekülen (B7-1 und
B7-2) erfolgreich eingesetzt werden (217, 230).
In der Veterinärmedizin zeigten erste erfolgversprechende Freiland-Immunisierungen mit
Fallen, welche mit einem rekombinaten Vakzinia-Virus zur Expression eines Rabies-
Virus Glykoproteins ausgestattet waren, eine protektive Immunität und verminderte
Inzidenz der Tollwut bei Wildtieren (37). In humanen, klinischen Studien der Phase I mit
HIV-1 Hüllproteinen und Glykoproteinen konnten allerdings bisher nur niedrig-potente
humorale und zelluläre Immunantworten mit dem Vakziniavektor-System induziert
werden (57, 58). Eine Optimierung könnte eventuell durch die adjuvante Administration
von proinflammatorischen Zytokinen wie IL-2, IL-12, IFN-γ oder eine Booster-Strategie
in Kombination mit der DNA-Immunisierung (s.o.) erreicht werden (193, 242).
�� �,3#(7$1&)$"",1#
1. Es soll ein rekombinantes Vakzinia-Virus zur Expression des X-Proteins vom
adw-Subtyp hergestellt werden.
2. Zur Analyse von CTL-Antworten gegen das HBxAg soll mit dem Vakzinia-
Virus Expressionssystem die Präsentation des HBxAgs in syngenen Stimulator-
und Zielzellen etabliert werden, da eine HBx stabil-exprimierende Zellinie nicht
zur Verfügung stand bzw. nicht konstruiert werden konnte
3. Die Immunantworten gegen das X-Protein sollen im murinen System mittels
DNA-Immunisierung auf humoraler und zellulärer Ebene charakterisiert
werden. Das Ziel ist die Testung einer potenziellen DNA-Vakzine gegen das
HBxAg zur Immuntherapie der chronischen HBV-Infektion im Tiermodell.

24
������� ���������
�� �$+&)$"",1# %$+ /"(&- %*�$2)!+$1
#! ! �� ����$2������������ �.7?�����
Dieser Vektor wurde uns freundlicherweise von Dr. M. Melegari, Molecular Hepatology
Laboratory, MGH, Boston überlassen (172). Hierbei wurde die kodierende Sequenz des
X-ORF aus dem HBV-Überlängenkonstrukt adwR9 (32) in das Plasmid pGEM-7
subkloniert. Anschließend wurde es über die Restriktionsstellen NcoI, welche sich direkt
vor dem Startcodon des X-ORF befindet, sowie NsiI (schneidet ca +200 Basenpaare
unterhalb des X-ORF-Stopcodons) in die HindIII-Klonierungsstelle des
Expressionsvektors pRcCMV-2 (#V750-20, Invitrogen, Carlsbad CA) ligiert. Der offene
Leserahmen des HBV X-Proteins vom Subtyp adw2 wird durch einen CMV-Promotor
kontrolliert. Der Vektor enthält ein Ampicillinresistenz-Gen zur Vermehrung in E.coli-
Bakterien.
#! !# '�������� �� �������� ;�����@&����� �;7 ��
Die für das X-Protein kodierende Sequenz vom adw-2 Subtyp wurde aus dem Plasmid
pRc/CMV-X durch gezielte Mutagenese mittels PCR zur Subklonierung in den Vakzinia-
Virus Shuttle-Vektor pSc11 vorbereitet. Über den sense-Primer AGG-27 wurde vor dem
Startcodon ATG eine EcoRV-Restriktionsstelle eingeführt. Ebenso wurde eine EcoRV-
Schnittstelle hinter dem Stopcodon des X-ORF durch den antisense-Primer GTC-27
eingefügt. Die PCR des 555-Basenpaare-langen X-ORF-Fragments wurde mit dem
Expand High Fidelity PCR-System (Boehringer, Mannheim, #1732641) durchgeführt.
In diesem System wird ein Enzym-Mix, bestehend aus der Taq-Polymerase und der Pwo

25
DNA-Polymerase (16), verwendet. Letztere enthält eine 3´-5´-Exonukleaseaktivität,
durch die eine dreimal niedrigere Mutationsrate der PCR erzielt wird.
�//! 1! ;�������� ��� '�������� �� ����$2������������ �.7?����� (nach M. Melegari).
Die Primer wurden von der Firma Birsner + Grob (BIG Biotech GmbH, Freiburg) nach
Vorgaben synthetisch hergestellt und als gefriergetrocknetes Pellet angeliefert. Nach
Resuspension in dH2O wurden jeweils 100 pmol pro Primer eingesetzt.
Sequenz der Primer:
EcoRV X-ORF Startcodon
AGG-27 X-sense: AGG GAT ATC CAA GCT CAT GGC TGC TAG
GTC-27 X-antisense: GTC GAT ATC CAT GCC CCA AAG CCA CCC
EcoRV
26
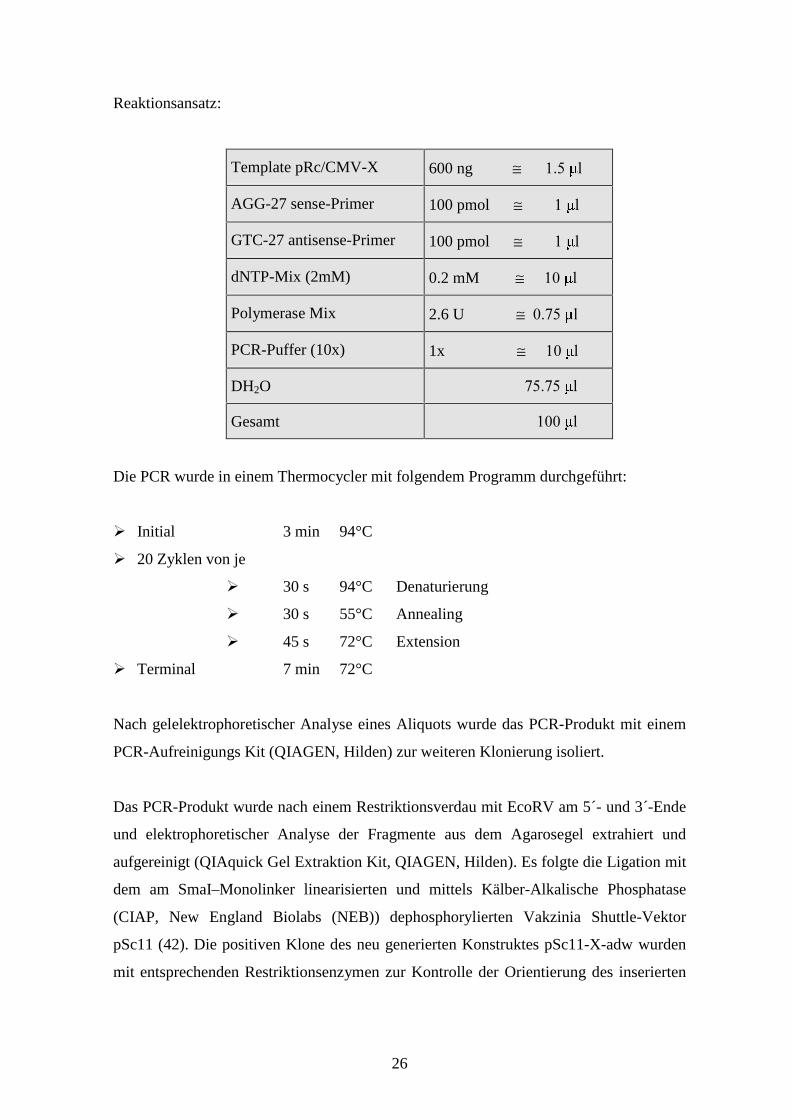
Reaktionsansatz:
Template pRc/CMV-X 600 ng ≅���������� �
AGG-27 sense-Primer 100 pmol ≅���������� �
GTC-27 antisense-Primer 100 pmol ≅���������� �
dNTP-Mix (2mM) 0.2 mM ≅�������� �
Polymerase Mix 2.6 U ≅������� �
PCR-Puffer (10x) 1x ≅�������� �
DH2O ������������������������������ �
Gesamt ������������������������������� �
Die PCR wurde in einem Thermocycler mit folgendem Programm durchgeführt:
� Initial 3 min 94°C
� 20 Zyklen von je
� 30 s 94°C Denaturierung
� 30 s 55°C Annealing
� 45 s 72°C Extension
� Terminal 7 min 72°C
Nach gelelektrophoretischer Analyse eines Aliquots wurde das PCR-Produkt mit einem
PCR-Aufreinigungs Kit (QIAGEN, Hilden) zur weiteren Klonierung isoliert.
Das PCR-Produkt wurde nach einem Restriktionsverdau mit EcoRV am 5´- und 3´-Ende
und elektrophoretischer Analyse der Fragmente aus dem Agarosegel extrahiert und
aufgereinigt (QIAquick Gel Extraktion Kit, QIAGEN, Hilden). Es folgte die Ligation mit
dem am SmaI–Monolinker linearisierten und mittels Kälber-Alkalische Phosphatase
(CIAP, New England Biolabs (NEB)) dephosphorylierten Vakzinia Shuttle-Vektor
pSc11 (42). Die positiven Klone des neu generierten Konstruktes pSc11-X-adw wurden
mit entsprechenden Restriktionsenzymen zur Kontrolle der Orientierung des inserierten

27
X-ORF in Bezug auf den Vakzinia „early-late“ Promotor p7.5 untersucht. Das Plasmid
enthält eine Ampicillinresistenz zur Vermehrung in E.coli-Bakterien.
#! !) '�������� �� $2������������ �7���)��
Um den Einfluß des HBV-Subtyps auf die Immunogenität des X-Antigens zu
untersuchen, wurde ein weiterer DNA-Expressionsvektor kloniert. Der HBx-ORF vom
Subtyp ayw wurde aus dem HBV-Überlängenkonstrukt pCH-9/3091 (von Prof. M.
Nassal, Abteilung Medizin II, Uniklinik Freiburg freundlicherweise zur Verfügung
gestellt) mit den Enzymen NcoI (Schnittstelle liegt direkt innerhalb des Startcodons ATG
des X-ORF) und SmaI (Schnittstelle liegt +153 bp hinter dem X-ORF Stopcodon)
ausgeschnitten. Das DNA-Fragment wurde mit dem Klenow-Fragment (Gibco BRL,
Eggenstein) an der NcoI-Schnittstelle in ein „blunt-end“ umgewandelt und aufgereinigt.
Danach wurde es in das mit EcoRV linearisierte und mit CIAP vorbehandelte
Expressionsplasmid pcDNA3.1(+) (#V790-20, Invitrogen, Carlsbad CA) ligiert. Der
Vektor pcDNA3.1(+) ist bis auf die MCS identisch mit pRc/CMV-2, was wichtig für
vergleichende Studien mit pRc/CMV-X-adw und pcDNA3-X-ayw war. Die gewonnenen
und aufgereinigten Klone der transformierten E.coli wurden mittels geeignetem
Restriktionsverdau auf eine korrekte Orientierung der inserierten Sequenz überprüft. Das
so entstandene Expressionskonstrukt wurde pcD/3-X genannt.
#! !* =���(�������� ����������� $!7�������������
Die Transformation von kompetenten Bakterien dient der effektiven Vermehrung von
Plasmid DNA in E.coli-Bakterien. In dieser Arbeit wurden ausschließlich INVαF´One
Shot Zellen (#C2020-03, Invitrogen, Carlsbad CA) zur Plasmid-Vermehrung
eingesetzt. Diese Zellen besitzen eine besonders hohe Transformationseffizienz. Die
Transformation von 1-10 µl Ligationsprodukt (5-50 ng DNA) erfolgte nach dem
mitgelieferten Protokoll. Anschließend wurden 50-100 µl der transformierten
Bakteriensuspension auf Ampicillin-haltige Agarplatten (15 g / l Bactoagar in LB-
Medium, 50 µg / ml Ampicillin) ausgestrichen und bei 37°C im Brutschrank über Nacht
28
inkubiert. Die Selektion der positiven Klone im Ampicillin-haltigem Agar erfolgte über
das Resistenzgen gegen das Antibiotikum Ampicillin, welches von den transformierten
Vektoren koexprimiert wird. Die ausgebildeten Kolonien wurden unter sterilen
Bedingungen zur weiteren Vermehrung in Mini-Präp-Kulturen und Analyse in
Polystyrolröhrchen mit LB-Medium überführt.
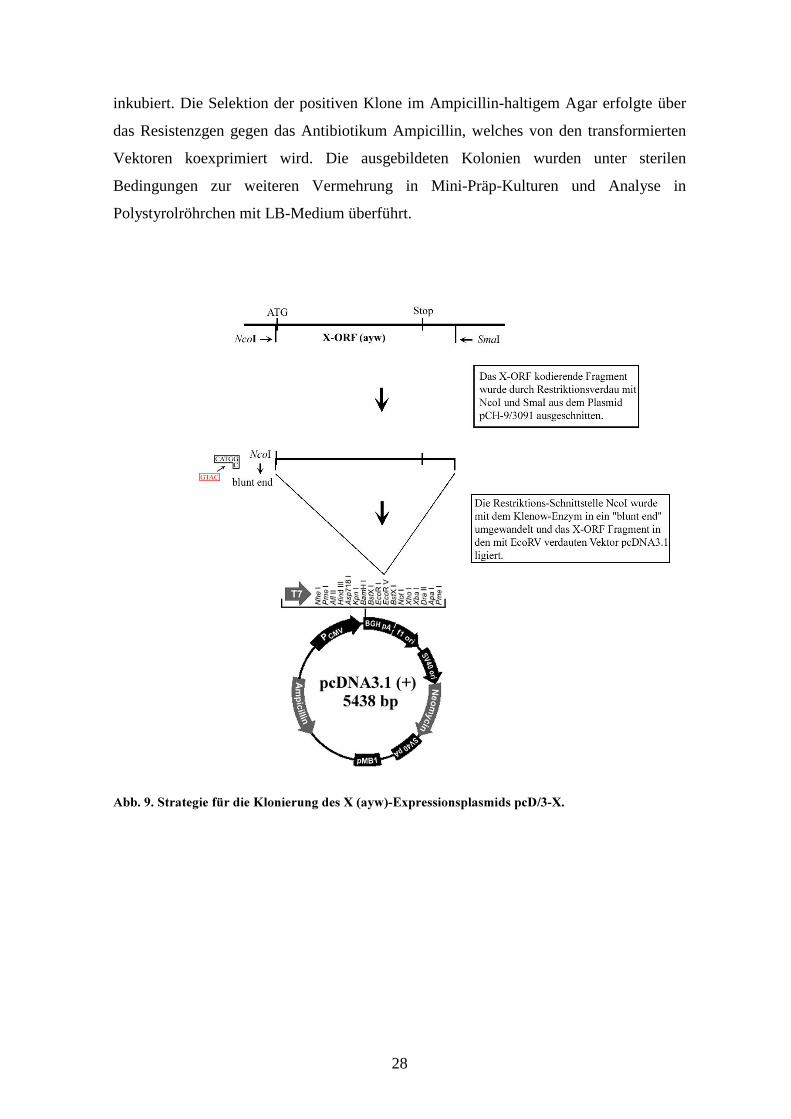
�//! +! ;�������� (A� ��� '�������� �� � B�>�C�$2������������ �7�?)��!

29
#! !, ����� �� ��2��&���������� ��� &����� ���
Zur Erzeugung größerer Mengen an Plasmid DNA müssen E.coli-Flüssigkulturen
angelegt werden. Die Bakterien werden nach Transformation oder aus bereits angelegten
Glycerol-Stocks in LB-Medium (10 g bacto-tryptone, 10 g NaCl, 5 g Yeastextrakt pro
Liter dH2O) unter Zugabe des Selektionsantibiotikums Ampicillin (100 µg / ml) bei 37°C
in einem Rotationsinkubator (250 U / min) über Nacht kultiviert. Zur Analyse wurden 5
ml LB-Kulturen (Mini-Präp) in Polystyrolröhrchen angelegt und die vermehrte Plasmid-
DNA nach der Methode der alkalischen Lyse isoliert (30). Die bei 4°C abzentrifugierten
Bakterienpellets wurden mit dem Spin Miniprep Kit (#27104, QIAGEN, Hilden)
alkalisch lysiert und die Plasmid-DNA nach Adsorption an eine mit Silika-Matrix
beschichtete Säule mit dH2O oder TE-Puffer (pH 8.5) eluiert.
Zur anschließenden DNA-Immunisierung wurde die DNA der Expressionsplasmide in
großen Mengen aus mehreren 500 ml LB-Übernachtkulturen extrahiert. Die DNA wurde
mit dem QIAGEN Plasmid Mega bzw. Giga Kit (#12181 und #12191, QIAGEN, Hilden)
aus den Bakterien isoliert und nach der Isopropanol-/Ethanolpräzipitation in einem
Volumen von 400-600 µl 0.15 M Tris-Cl (pH 8.5) gelöst. Nach gelelektrophoretischer
Analyse und spektrophotometrischer Konzentrations- und Reinheitsbestimmung wurde
die Plasmid-DNA bei –20°C bis zu ihrer weiteren Verwendung tiefgefroren. Es wurden
Konzentrationen von 4-8 µg / µl und Plasmid-Mengen von 2-4 mg / 500ml
Bakterienkultur erzielt. Bei einem Überwiegen der „supercoiled“ Form (>90 %) in der
gelelektrophoretischen Analyse wurde auf den folgenden Schritt der CsCl-Gradienten
Reinigung der Plasmid-DNA verzichtet. Aus den bei -80°C angelegten Glycerol-Stocks
(Bakteriensuspension in 20 % Glycerol) der positiven Klone konnten die Plasmide später
jederzeit wieder amplifiziert werden.

30
&���������������7�� ��������>�D
� Konzentrationsbestimmung
� C = 50 µg / ml x VF x OD260 (doppelsträngige DNA)
� C = 20 µg / ml x VF x OD260 (einzelsträngige DNA)
� Reinheitsanalyse
� Ratio OD280 : OD260 > 1.8
��� �9'+$&& !1&&),% $1 1 : )+!
#!#! 8�����
Bei den Transfektions- und Expressionsstudien in dieser Arbeit wurde mit folgenden
Zellinien gearbeitet:
:�0 8�����% �=�� ��! �.E� *,-
Murine fetale Myoblasten, welche aus der Muskulatur der hinteren Maus-Extremität
gewonnen wurden. Die Zellen lassen sich mit sehr niedriger Effizienz (ca. 1-5%)
transfizieren. Im Rahmen der HBx-Transfektionsstudien dienen sie als in vitro Modell
für die intramuskuläre DNA-Applikation bei der genetischen Immunisierung von
Mäusen. Medium: DMEM-10 (Gibco BRL, Eggenstein) mit 10%igem fetalem
Kälberserum (FCS), Zusatz von Penicillin / Streptomycin (Gibco BRL, Eggenstein) und
nicht-essentiellen Aminosäuren (NEAA, GIBCO BRL, Eggenstein) (50).
�F��1 8�����
Es handelt sich hierbei um eine humane Hepatomzellinie (freundlicherweise zur
Verfügung gestellt von Dr. M. Geißler, Universitätsklinikum Freiburg, (195)) welche für
die HBx-Transfektionsstudien zum Nachweis einer Expression des X-Proteins in
Lebergewebe verwendet wurde. Kultiviert wurden diese Zellen in DMEM-10 +
Pen/Strep und NEAA.

31
&0 , 8�����% �=�� ��! =���-*
Murine Mastozytomzellen vom Haplotyp H-2d, d. h. syngen zur Balb/c Maus (216). Die
Transfektion dieser Zellinie wurde zur Expression des X-Proteins in potenziellen
Targetzellen für die Zytotoxizitätsassays durchgeführt. Versuch der Etablierung einer
stabilen Zellinie, welches das X-Protein exprimiert. Medium : DMEM-10 + Pen
/Strep/NEAA.
$E* 8�����% �=�� ��! =���)+
Murine T-Zell Lymphomlinie, welche mittels 9,10-dimethyl-1,2-Benzanthracene in einer
C57BL/6 Maus induziert wurde (Haplotyp H-2b). Auch mit dieser Zellinie wurde
versucht, eine syngene Targetzellinie, welche das X-Protein stabil exprimiert, für die
Charakterisierung der CTL-Aktivität in C57BL/6 Mäusen zu erzeugen. Medium DMEM-
10 + Pen./Step./NEAA (215).
#!#!# E��������� :���=���(��
Zur Charakterisierung der Expression des X-Proteins wurden G-8, P815 und EL4 Zellen
mit den Plasmiden pRc/CMV-X (adw2) und pcDNA3-X (ayw) nach der Methode des
Liposomen vermittelten Gentransfers transfiziert (79). Die kritischen Parameter dieses
hocheffizienten Transfektionssytems sind das Liposom–DNA Verhältnis, die absolute
DNA-Menge und die Inkubationszeit, welche in dieser Arbeit für die G-8 Zellen, die nur
schlecht transfizierbar sind, mittels des Enzyms Luziferase als Reportergen-Assay
optimiert wurden (s.u.). Die Transfektion erfolgte mit dem LipofectAMINE Reagenz
(Gibco BRL, Eggenstein, #18324-012) in Serum-freien Medium Opti-MEM (Gibco
BRL, Eggenstein, #31985). Die Zellen wurden am Vortag auf eine 6-Well-Platte
umgesetzt, so daß sie zum Zeitpunkt der Transfektion gerade die optimale Konfluenz von
ca. 70% erreicht hatten. Pro Transfektionsansatz wurden 4 µg Plasmid-DNA mit dem
Liposom-Komplex in dem optimierten Liposom-DNA Verhältnis von 5:1 sorgfältig
durchmischt und ca. 35 min. bei Raumtemperatur in Opti-MEM Medium inkubiert.
Anschließend wurden die Zellen in jeweils 2 ml Opti-MEM Medium überführt und
vorsichtig mit dem Liposomen-DNA Komplexen beträufelt, um den gesamten
Monolayer damit abzudecken. Nach 2 h Inkubation bei 37°C wurden die Zellen mit

32
MEM (GIBCO BRL, Eggenstein) gewaschen und mit frischen, Serum-haltigem Medium
versorgt. Die Lyse der Zellen zur Proteinextraktion erfolgte nach 12- bis 72-stündiger
Inkubation im 37°C CO2-Begasungsbrutschrank.
#!#!) ���7���&������ =���(������
Die transiente Transfektion von HUH-7 Zellen wurde nach der Methode der
Calciumphosphatpräzipitation durchgeführt (101). Es wurden 4 µg der gereinigten
Plasmid DNA mit einer 1 M CaCl2-Lösung (Endkonzentration 0.125 M) und sterilem
dH2O in einem Endvolumen von 75 µl vorgegeben. Diese Lösung wurde mit 75 µl
2xBBS-Puffer sorgfältig durchmischt und für 15 Minuten bei Raumtemperatur inkubiert.
Die Zellen waren am Vortag auf eine Six-Well-Platte umgesetzt worden. Die
Transfektionslösung wurde nun vorsichtig tropfenweise auf das Medium der Zellen
gegeben und für ca. 15 h bis 24 h im Brutschrank bei 35°C und 2% CO2 darauf belassen.
Anschließend wurden die Zellen zweimal mit MEM gewaschen und mit frischem
Medium bei 37°C und 5% CO2 für weitere 24h bis 48h inkubiert. Zur Beurteilung der
Transfektionseffizienz wurden die Zellen mit dem Plasmid pEGFP-N1 (CLONTECH
Labs Inc., Palo Alto, CA, #6085-1) kotransfiziert, so daß nach 24-48h die Stärke des
grün fluoreszierenden EGFP-Signals mit dem Fluoreszenz-Mikroskop beurteilt werden
konnte.
#!#!* �������� ��� =���(�������((������ ��� ��� E��(�������>
Zur Optimierung der Transfektionsbedingungen von G-8 Zellen und CV-1 Zellen mit
dem liposomalen System (siehe Kap. 2.2.2) wurde die Luziferaseaktivität (68) als Marker
für die Transfektionseffizienz in den lysierten Zellen bestimmt. Bei der Transfektion des
Reportergen-Plasmids pCMV-Luc+ wurden das Liposom-DNA-Verhältnis (1:5 bis 1:20)
und die Inkubationszeit der Zellen mit dem DNA/Liposomen-Komplexen (2 h, 4 h und 6
h) variiert. Die Zellen wurden nach 24 h mit jeweils 500 µl Lysepuffer (25 mM
Glyzylglyzin, pH 7.8, 4 mM EGTA, 15 mM MgSO4, 15 mM KH2PO4, 1 mM DTT und
1% Triton X-100) pro well für 20 min. auf Eis lysiert. Direkt vor Beginn der Messung

33
wurden jeweils 20 µl des Zellysats in Duplikate mit 300 µl des Analyse-Puffers (25 mM
Glyzylglyzin, pH 7.8, 4 mM EGTA, 15 mM MgSO4, 15 mM KH2PO4, 1 mM DTT und 1
mM ATP) in Luminometerröhrchen vermischt. Die Luziferaseaktivität wurde für 10 s in
einem Luminometer (Lumat LB 9705, Fa EG&G Berthold) nach automatischer Injektion
von jeweils 100 µl Luziferinlösung (250 mM D-Luciferin (Sigma, Taufkirchen, #L-
9504), 25 mM Glyzylglyzin, pH 7.8, 10 mM DTT) in relativen Light-Units (RLA)
gemessen. Hierbei kommt es zu einer schnellen, ATP-abhängigen Oxidation des
Substrates Luziferin, begleitet von einer Lichtemission, welche proportional zur Menge
des Enzyms Luziferase ausfällt. Als Positivkontrolle diente ein Lysat mit sehr hohen
Light-Units aus einer früheren Messung, die Negativkontrolle wurde mit Mock-DNA
transfizierten Zellen ermittelt.
��4 ;$&)$+1 �"!)) 1# 8,+ <,(" )() :$1 �$&) --,1# %$+ �1) #$1*
$9'+$&& !1
#!)! E>� ��� ����(�������� 8�����
Da die potenziellen intrazellulären Zielstrukturen, mit denen das X-Protein interagiert,
noch nicht genau bekannt sind, wurden verschiedene Lysepuffer mit unterschiedlichem
Salz- und Detergentiengehalt zur optimalen Extraktion des Proteins getestet. Die
zentrifugierten Zellpellets wurden mit RIPA-Puffer (0.15 M NaCl, 1% NP-40, 50 mM
Tris, 0.5% DOC und 1% SDS), mit Core-Lysepuffer (0.15 M NaCl, 1% NP-40) sowie
mit dem 1x SDS-Gelladepuffer [50 mM Tris -Cl pH 6.8, 10% Glycerol, 2% SDS, 100
mM DTT, 5% 2-ME (2-Merkaptoethanol), 0.1% Bromphenol-Blau] für jeweils 8-10 min.
bei 4°C lysiert und anschließend bei –80°C sofort tiefgefroren.

34
#!)!# ;�;�&�:$
Bei der diskontinuierlichen SDS-PAGE (149) werden die aufzutrennenden Proteine in
Anwesenheit von SDS (Natriumdodecylsulfat) von einer einheitlichen negativen Ladung
umgeben. Die Auftrennung erfolgt bei der Polyacrylamid Gelelektrophorese mit
konstanter Porengröße auschließlich entsprechend der Molmasse der Proteine. Zunächst
wurden die Lysate in 1x SDS-Gelladepuffer (siehe Kap. 2.3.1) 10 Minuten bei 100°C
denaturiert und anschließend in einem 5%igen Sammelgel fokussiert und dann im
12%igem Trenngel (Running-Gel) aufgetrennt. Die Lösung der Gele wurde nach
Maniatis „Molecular Cloning“ (233) angerichtet, die Polymerisation des Acrylamids
(#A-3574, Sigma, Taufkirchen) wurde durch Zugabe von APS und TEMED (#T-7024,
Sigma, Taufkirchen) induziert, dann wurde die Lösung schnell zwischen zwei
Glasplatten gegossen und mit dH2O überschichtet. Das Sammelgel wurde auf das
Trenngel gegossen und ein Kamm eingesetzt. Nach vollständiger Polymerisation wurden
die denaturierten Proteinproben auf das Sammelgel geladen und in einer mit Laufpuffer
(250 mM Glycin, pH 8.3, 25 mM Tris-Base, 0.1% SDS) gefüllten Elektrophoresekammer
(BIORAD, München) für 90 min. mit einer Spannung von 140 mV aufgetrennt. Als
Größenstandard wurde der Protein-Molekulargewichtsmarker Low Standard (2.85 –43
kD, GIBCO BRL, Eggenstein, #16040-016) verwendet.
#!)!) �����/���
Beim Western-Blotting werden die elektrophoretisch separierten Proteine auf einer
Membran mit spezifischen Antikörpern nachgewiesen (270). Zunächst wurden die mit
der SDS-PAGE aufgetrennten Proteine durch einen Transfer vom Gel auf eine
Nitrozellulose-Membran (Immobilon-P Transfer Membrane, Millipore, Bedford MA,
#IPVH 09120) übertragen. Das Gel wurde hierzu luftblasenfrei auf die Membran plaziert
und zwischen zwei Whatman 3MM-Papieren in einer Transferapparatur (BIORAD,
München) eingespannt. Bei einer Spannung von 100 mV, mit der Membran der Anode
zugewandt, wurden die Proteine im Transferpuffer (48 mM Tris-Base, 39 mM Glycin pH
8.3, 0.037% SDS, 20% Methanol) für 1 h auf der Membran immobilisiert. Um die freien
Proteinbindungsstellen der Membran abzusättigen, wurde die Membran anschließend

35
über Nacht bei 4°C in einem Blockierungspuffer [3% fettfreies Milchpulver, 1% bovines
Serumalbumin (BSA) in PBS] auf einer Schwenkvorrichtung inkubiert. Die Inkubation
mit dem monoklonalen Maus-Antikörper gegen das HBV-X-Protein α-X36 (dieser
Antikörper wurde uns freundlicherweise von Prof. J. Wands, Brown University,
Providence, RI, USA überlassen) erfolgte in einer Verdünnung von 1:500 in
Blockierungspuffer für eine Stunde bei Raumtemperatur unter kontinuierlichem
Schwenken. Nach dreimaligem Waschen mit PBS / 0,02% Tween (BIORAD,
München) für jeweils 10 Minuten wurde die Membran mit dem Meerrettich-Peroxidase
konjugierten 2. Antikörper anti-Mouse Ig (#NXA931, Amersham, Freiburg) in einer
Verdünnung von 1:3000 wiederum für 1 Stunde bei Raumtemperatur inkubiert. Der
Bindungsnachweis der Antikörper erfolgte nach erneutem dreimaligem Waschen mit
PBS / 0,02% Tween mit dem ECL-Chemolumineszenzsystem (Amersham, Freiburg,
#RPN-2106). Hierzu wurde die Membran für eine Minute in der Substratlösung
geschwenkt und zur Autoradiographie in einer Dunkelkammer auf einen Kodak-BMR-
Röntgenfilm mit verschiedenen Belichtungszeiten (10 s bis 1 h) plaziert.
��� �$+&)$"",1# +$2!-7 1(1)$+ �(28 1 (*� +$1
#!*! � ���������� �� ������������
In der vorliegenden Arbeit wurden Vakzinia-Viren vom WR-(Western Reserve) Stamm
verwendet. Diese wurden uns freundlicherweise von Dr. J. Hausmann (Abteilung
Virologie am Institut für Mikrobiologie und Hygiene, Universität Freiburg) überlassen.
Die Versuchsprotokolle wurden in Anlehnung an Standardmethoden modifiziert (9, 163).

36
#!*! ! � �������� 8�������� �� ��������(�������% .����/�������% ;�������� ��
��������2������
��� % �(��7�� ����� �����> �����> 7���% �=�� ��! ��E�14
Diese Zellinie wurde 1964 zur Untersuchung der malignen in vitro Transformation mit
dem Rous Sarkom-Virus etabliert (139). Die Zellen wachsen adhärent mit typischer
Fibroblasten-artiger Morphologie im Monolayer und eigenen sich in besonderem Masse
zum Studium des zytopathischen Effektes (CPE), welcher durch die Infektion mit dem
Vakzinia-Virus hervorgerufen wird. Sie wurden zur Stock-Amplifikation der Viren, für
die homologe Rekombination, für den Protein-Expressionsnachweis und zur Plaquetest-
Titerbestimmung eingesetzt. Medium: DMEM-5 (Gibco BRL, Eggenstein) mit 5%igem
fetalem Kälberserum (FCS), Zusatz von Penicillin/Streptomycin (Gibco BRL,
Eggenstein) und nicht-essentiellen Aminosäuren (NEAA, GIBCO BRL, Eggenstein).
�='� *) �% =�>������ ����� ��� B='�C ���� ������7���% �=�� ��!
�.E�0)4)!
Adhärent im Monolayer, Fibroblasten-artig wachsende Zellen, bei denen eine Deletion
des Thymidinkinase (TK) Gens vorliegt. Sie werden in einem Selektionsmedium unter
Zusatz von 5´-Bromodesoxyuridin (BrdU) kultiviert. Dieses Zytostatikum wird durch das
intakte Enzym TK phosphoryliert und in die DNA eingebaut, was zum Abbruch der
Replikation und schließlich zum Zelltod führt. Daher eignet sich diese Zellinie besonders
zur Selektion rekombinanter Vakzinia-Viren, welche keinen intakten TK-Lokus mehr
besitzen und so ungehindert in diesen Zellen in Anwesenheit von BrdU replizieren und
Plaques ausbilden können. Vakzinia-Viren vom Wildtyp mit intakter TK werden jedoch
bei der Replikation gehemmt. Medium: DMEM-10 (Gibco BRL, Eggenstein) mit
10%igem fetalem Kälberserum (FCS), Zusatz von Penicillin / Streptomycin (Gibco BRL,
Eggenstein), nicht-essentiellen Aminosäuren (NEAA, GIBCO BRL, Eggenstein) und 50
µg / ml 5´-BrdU.

37
#!*!# ;�7���������G������ �� F����� ��� ��������������
Alle Arbeiten mit Vakzinia-Viren unterliegen nach dem Anhang III der Gentechnik-
Sicherheits- Verordnung (GenTSV) Sicherheitsmaßnahmen der Stufe 2 (S2). Daher muß
offenes Arbeiten mit den Vakzinia-Viren ausschließlich unter einer Laminar-Flow Bank
(Hood) durchgeführt werden. Dabei kommen nur sterile Wegwerf-Plastik-Pipetten und –
gefäße sowie kein gläsernes Material/Gerät, insbesondere keine Pasteurpipetten zur
Anwendung. Eine Aerosolbildung muß bei Pipettierarbeiten unter allen Umständen
vermieden werden. Alle anfallenden flüssigen Abfälle werden unter Zusatz von 3%igem
Buraton inaktiviert und anschließend sofort autoklaviert. Feststoffabfälle müssen
innerhalb der Sicherheitswerkbank in Polyamid-Beuteln doppelt verschlossen, in speziell
dafür gekennzeichneten Behältern gesammelt und ebenfalls sofort nach Abschluß der
Arbeiten autoklaviert werden. Als besondere Maßnahme gegen eine akzidentielle
Exposition werden die Hände und Unterarme des Experimentators durch den Gebrauch
von zwei Lagen Handschuhen und Schutzkitteln mit verlängerten Armbündchen
geschützt. Eine Exposition der Augen soll durch das Tragen einer Schutzbrille vermieden
werden, da eine Infektion des Auges (Keratitis) eine sehr ernsthafte Komplikation des
Vakzinia-Virus darstellt. Eine Impfung zur Prophylaxe bei ungeimpften
Wissenschaftlern wird in einer Stellungnahme des ZKBS (19.6790-10-14, Mai 1997,
Seiten 80-82) sehr kontrovers diskutiert. Bei einer Abwägung von Risiko und Nutzen
einer Impfung wird hier auf die seltenen, aber gelegentlich sehr gravierenden
Impfkomplikationen hingewiesen. Im Gegensatz dazu ist die Wahrscheinlichkeit einer
akzidentiellen Infektion unter S2-Schutzmaßnahmen als sehr gering anzusehen. Ein
Impfstoff kann derzeit nur über die „Centers of Disease Control“ (CDC, Atlanta, USA)
bezogen werden. Personen mit schweren dermatologischen Erkrankungen (Ekzemen),
sowie Schwangere und Immunsupprimierte dürfen nicht mit Vakzinia-Viren arbeiten.
Ungeimpfte Personen dürfen nicht mit Vakzinia-Virus infizierten Versuchstieren arbeiten
sowie nicht an „Large-Scale“ Virusproduktionen (Impfstoffproduktion) beteiligt sein.
Bei Kontaminationen der Arbeitsfläche bzw. Geräten/ Materialien muß eine
Inaktivierung mit 3%igem Buraton durchgeführt werden, und die Hood bzw. das Gerät
nachfolgend mit UV-Licht bestraht werden.

38
#!*!) ��������� 3�� �������������������� �� ������� �� ���G��
��G��/
#!*!)! ��(������ �� ��������(�������
Bei allen Infektionsexperimenten muß auf eine exakte Konzentration der
Virussuspension, sowie auf eine angemessene Dosierung des Inokulums in bezug auf die
Zahl der infizierten Zellen geachtet werden. Die Konzentration einer Virussuspension
wird in pfu/ml angegeben, wobei pfu für „plaque forming unit“ steht. Diese Einheit ist
ein Maß für die absolute Menge an infektiösen Viruspartikeln in einer titrierten
Virussuspension. Dagegen bezeichnet die „Multiplicity of infection“ (MOI) das
Verhältnis zwischen der Anzahl infektiöser Viruspartikel und der Zahl der infizierten
Zellen. Die Zellzahlen werden mit Hilfe einer Neubauer-Zählkammer bestimmt und man
rechnet bei einer Infektion eines konfluenten Monolayers unter Standardbedingungen mit
Durchschnittswerten je nach Größe der entsprechenden Kulturschalen. Nach mehreren
Auszählungen wurde bei einer konfluenten 5cm∅-Petrischale mit CV-1 Zellen von einer
Zellzahl von 1 x 106 ausgegangen. Die Fläche einer 5cm∅-Schale beträgt ca. 20 cm2
(genau 19,63 cm2), so daß zur Umrechnung auf die Fläche einer 75cm2- Flasche der
Faktor 3,75, einer 6-Well-Platte der Faktor 0.5 bzw. einer 150 cm2-Flasche der Faktor 7.5
eingesetzt werden kann.
Zur Virusvermehrung wurden 80-90% konfluente CV-1 Zellen im Monolayer infiziert.
Die Infektion erfolgte mit einer MOI von 0.2 (0.05-0.5 Variationsbreite), d.h. bei einem
Titer des Wildtyp-Virus WR-Stammes von 107 pfu / ml wurden 20 µl der
Virussuspension pro 5cm∅-Schale (106 Zellen), bzw. 75 µl pro 75cm2-Flasche
eingesetzt. Das Inokulum sollte ein Volumen von ca 0,04 ml / cm2 haben, also 800 µl auf
eine 5cm∅-Schale bzw. 3 ml auf eine 75cm2-Flasche. Es wurde durch Verdünnung des
entsprechenden Volumens der Virussuspension in DMEM-0 hergestellt. Die Infektion
wurde in Serum-freiem Medium (DMEM-0, Gibco BRL, Eggenstein) durchgeführt, da
Serumproteine Viruspartikel binden und so inaktivieren können. Nach guter
Durchmischung des Inokulums wurde das Kulturmedium von den Zellen abgenommen,
die Zellen einmal mit MEM gewaschen und schließlich mit dem Inokulum für 1 h bei
37°C im Begasungsbrutschrank (5% CO2) inkubiert. Während der Inkubation wurden die

39
Zellen alle 10-15 Minuten kurzzeitig geschwenkt, um eine optimale Verteilung des
Inokulums auf dem Zellrasen zu gewährleisten. Nach der Abnahme des Inokulums
wurden die Zellen mit dem Erhaltungsmedium DMEM-5 bei 37°C im CO2-Inkubator
inkubiert. Eine optimale Virusausbeute wurde je nach MOI nach ca. 56 h (24-72 h)
erreicht. Zur Kontrolle wurden die Zellen täglich auf sichtbare Zeichen des
zytopathischen Effektes (CPE) untersucht und entsprechend die Inkubationszeit
individuell angepaßt (siehe Abb. 11-14).
#!*!)!# �������� 3�� ����� � ��(�������� ��� 8�����
Nach Ablauf der Inkubation wurden die infizierten Zellen mit einem sterilen Zellschaber
abgelöst und im Kulturmedium in sterile 15 ml oder 50 ml Polypropylen-Röhrchen
überführt. Nach 5-minütiger Abzentrifugation bei 1200 rpm und 4°C wurde der
Überstand verworfen und die Zellen in 0.25-0.5 ml DMEM-0 pro 5cm∅-Schale bzw. 1-
1.5 ml pro 75cm2-Flasche resuspendiert. Da 80-90% der Viren Zell-assoziert sind,
wurden sie durch 3 Zyklen Frieren und Tauen aus den Zellen herausgelöst. Dies geschah
durch abwechselndes Überführen der Röhrchen mit der Virus-/Zellsuspension von einem
Ethanol-Trockeneisbad in ein 37°C -Wasserbad. Anschließend wurden sie noch zur
weiteren Ablösung der Viren vom Zelldebris für 3-5 Minuten in einem Ultraschall-
Wasserbad behandelt. Da sich die Viruspartikel nun in Suspension befanden, wurde der
zelluläre Debris bei 1000 rpm für 2 Minuten abzentrifugiert und der Viren-haltige
Überstand bei -20°C bzw. –80°C tiefgefroren.
#!*!)!) ��������� 3�� ����� �� ��2��&������������ �� �������� ���
Die Herstellung von Mini-Stocks erfolgte durch die Infektion einer 5cm∅-Schale 80%
konfluenter CV-1 Zellen (siehe Abb. 10) mit einem Inokulum, welches aus einem
aufgearbeiteten Plaque oder Virussuspensionen mit sehr niedrigem Titer hergestellt
wurde. Nach 48-96 h Inkubationsdauer (maximaler CPE) bei 37°C im CO2 Brutschrank
wurden die Viruspartikel isoliert (siehe 2.4.1.3). Da der Titer der gewonnenen
Virussuspension unter Umständen noch sehr niedrig war, wurde als Zwischenschritt mit
der Hälfte der aufgearbeiteten Virussuspension eine 75cm2-Flasche infiziert, und die

40
Viren wurden nach 48–96 h in 1-1.5 ml Suspension aufgearbeitet. Hierbei ließen sich
Titer zwischen 1 x 105 und 5 x 106 pfu/ml erreichen. Mit 50-200 µl dieser
Virussuspension wurden 10-15 75cm2-Flaschen CV-1 Zellen zur Herstellung eines Maxi-
Stocks infiziert. Die Zellen wurden bei maximalem CPE (48-72h) aufgearbeitet und die
Viruspartikel in 1-1.2 ml pro 75cm2-Flasche aufgenommen. Die Titer einer solchen
Präparation lagen zwischen 1 x 107 und 5 x 108 pfu / ml (siehe Abb. 13, 14).
#!*!* =�������� 3�� �������������������� �� &��6����
Eine Bestimmung des Titers einer Virus-haltigen Suspension mit dem Plaquetest sollte
immer mit dem selben Zelltyp durchgeführt, der auch anschließend für weitere
Infektionen zur Anwendung kommt, da die Effizienz der Infektion und auch die
Eigenschaften der Zytopathie je nach Zelltyp unterschiedlich ausfallen können. In der
vorliegenden Arbeit wurden die Viren in CV-1 Zellen bzw. teilweise auch in MC57G
Zellen (Fibroblasten) titriert.
#!*!*! ��������� ��� &��6� ��7� '������3���������(��/��
Mit dieser relativ einfach durchführbaren Methode wurden alle Wildtyp WR Vakzinia-
Virus Stocks, bzw. Stocks von rekombinanten Vakzinia-Viren, deren Reinheit zuvor im
doppelten Agarose-Overlay (s.u.) sicher dargestellt wurde, titriert. Dies ist eine wichtige
Voraussetzung, da mit der Kristallviolett-Anfärbung nicht zwischen Plaques
rekombinanter Klone bzw. nicht-rekombinanter, d.h. Wildtyp Klone unterschieden
werden kann.
Zunächst wurde eine Verdünnungsreihe in 10er Potenzschritten der Virussuspension
angerichtet. Pro 15 ml Polystyrol-Röhrchen wurden 1 ml DMEM-0 vorgegeben und in
das erste Röhrchen 110 µl der Virussuspension eingefüllt. Nach sorgfältigem
Durchmischen (Vortexen) wurden 110 µl davon in das nächste Röhrchen übertragen.
Dieser Schritt wurde solange wiederholt, bis eine Verdünnungsreihe von 10-1 bis 10-9
erstellt war.
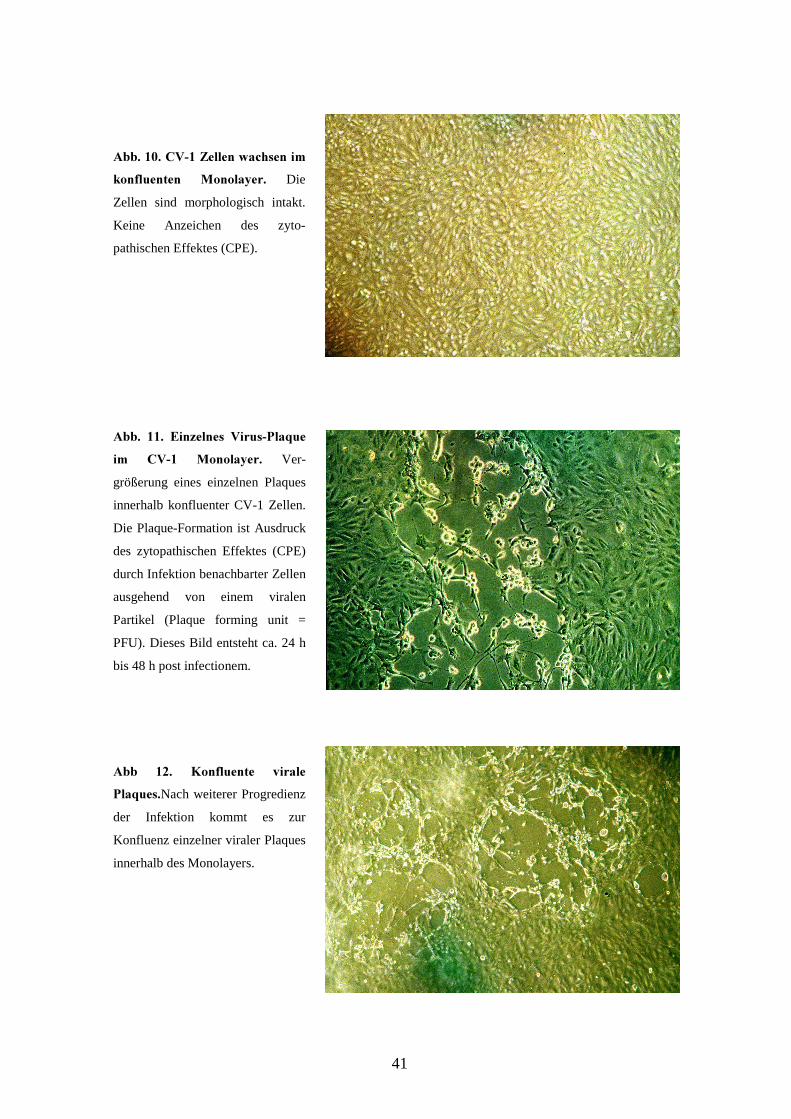
41
�//! 4! ��� 8����� ��7��� ��
���(������ ������>��! Die
Zellen sind morphologisch intakt.
Keine Anzeichen des zyto-
pathischen Effektes (CPE).
�//! ! $������� ����&��6�
�� ��� ������>��! Ver-
größerung eines einzelnen Plaques
innerhalb konfluenter CV-1 Zellen.
Die Plaque-Formation ist Ausdruck
des zytopathischen Effektes (CPE)
durch Infektion benachbarter Zellen
ausgehend von einem viralen
Partikel (Plaque forming unit =
PFU). Dieses Bild entsteht ca. 24 h
bis 48 h post infectionem.
�// #! '��(����� 3�����
&��6�!Nach weiterer Progredienz
der Infektion kommt es zur
Konfluenz einzelner viraler Plaques
innerhalb des Monolayers.

42
�//! )! ��� 8����� ��� /�
������� �>�������7��� $((���!
Vergrößertes Bild konfluierender
Plaques und zahlreicher „blasiger“
Zellen als morphologisches
Korrelat für den massiven CPE.
Dieses Bild entsteht ca. 72 h bis 96
h nach Infektion mit sehr hoher
MOI. Optimaler Zeitpunkt zur
Viruspräparation.
�// *! =������ �&$! Das
Resultat der kompletten Infektion
eines CV-1 Monolayers mit sehr
hoher MOI besteht aus ballonierten,
zytopathischen Zellen und
verklumptem Zell-Debris ca. 96 h
post infectionem.

43
Mit jeweils 1 ml der letzten sechs Verdünnungsschritte (10-3 bis 10-9) wurde je eine
konfluente 5cm∅-Schale der entsprechenden Zellinie, wie in Kap. 2.4.3.1 beschrieben,
infiziert und nach 48h Inkubationsdauer im 37��2-Inkubator erfolgte die Auszählung
der Plaques. Hierzu wurden die Zellen zweimalig mit PBS gewaschen und anschließend
mit jeweils 1 ml einer Kristallviolett-Lösung (0.2 % Kristallviolett (Sigma, Taufkirchen,
#C-3886) in 20 % Ethanol oder in 4 % Paraformaldehyd) überschichtet. Nach 5 bis 10
minütiger Inkubation bei Raumtemperatur wurde die Farblösung abgenommen und der
dünne Farbfilm auf dem Zellrasen an der Raumluft getrocknet. Anschließend zählte man
die im violetten Zellrasen farblos erscheinenden Plaques für jede Verdünnungsstufe aus.
Der Farbstoff wird nur von vitalen Zellen aufgenommen; die Plaques erscheinen farblos,
da es durch den CPE der Viren zur Lyse der infizierten Zellen gekommen ist. Dabei gilt
es zu beachten, daß nur Primärplaques gezählt werden, da es bei einer zu langen
Inkubationsdauer zur Formation von kleineren Sekundärplaques kommen kann. Das
Verhältnis der gezählten Plaques sollte zwischen jedem Verdünnungsschritt ungefähr
dem Faktor 10 entsprechen, damit von einer korrekten Titrierung ausgegangen werden
kann.
Der Wert des Titers T = Anzahl der ausgezählten Plaques
Verdünnungsfaktor der entsprechenden Verdünnungsstufe pfuml
Die Titrierung ist besonders exakt, wenn die vorletzte Verdünnungstufe, auf der Plaques
erkennbar sind, ausgewertet wird.
#!*!*!# ��������� ��� �����/������� &��6� �� ��������� �������93����>
Konfluente 5cm∅-Schalen der entsprechenden Zellinie wurden mit einer
Verdünnungsreihe der Virussuspension, wie in Kap. 2.4.4.1 beschrieben, infiziert. Nach
der Abnahme des Inokulums wurden die Zellen mit jeweils 5 ml des Agarose-
Plaquemediums (Anfertigung, siehe Tabelle unten) überschichtet und für eine Dauer von
48 h im 37��2 -Brutschrank inkubiert. Dann wurde auf die untere eine zweite Schicht
aus je 3 ml Agarose-Plaquemedium, welche das Chromogen X-Gal (400 µl/ml) enthält,
gegeben. Durch die Aktivität des lacZ-Gens, welches für das Enzym Beta-Galaktosidase

44
kodiert, wurde das Substrat X-Gal in den infizierten Zellen in einen blauen Farbstoff
umgewandelt. So kommt es zur Blau-Anfärbung der rekombinanten Plaques innerhalb
der erstarrten Agarose nach weiterer Inkubationzeit von 24 h und die Plaques konnten
nun, wie in Kap. 2.4.4.1 beschrieben, ausgezählt werden. Liegt eine Verunreinigung der
Virussuspension mit nicht-rekombinanten Viren vor, finden sich lichtmikroskopisch
nicht angefärbte Plaques innerhalb des Zellrasens.
#!*!, �������� .����/������� �� ;�������� �����/������� ��������������
Das in der vorliegenden Arbeit verwendete Shuttle-Plasmid pSc11 besitzt zwei
Möglichkeiten zur Selektionierung der rekombinanten Klone (42). Die BrdU-Selektion
über das durch Insertion zerstörte TK-Gen (flankierende TKR -und TKL -Sequenzen) wird
durch das ko-inserierte lacZ-Gen unter der Kontrolle des starken Vakzinia „late“
Promotors p11 (25) in gegenläufiger Orientierung zur Fremdgenkassette ergänzt. Das
Zytostatikum BrdU kann seine letale Wirkung auf die TK-defizienten HuTK-143B Zellen
durch Phosphorylierung mit dem Enzym Thymidinkinase nur dann entfalten, wenn diese
mit nicht-rekombinanten Virusklonen infiziert wurden, welche eine intakte TK besitzen.
Das inserierte Fremdgen wird unter die Kontrolle des compound „early/late“ Promotors
p7.5 (52) gestellt. Da allerdings auch Spontanmutationen im TK-Locus des Wildtyp
Vakzinia-Virus mit einer Häufigkeit von 1:10000 auftreten, können die resultierenden
Plaques von den blau-gefärbten, rekombinanten Plaques nach Zugabe von X-Gal (5-
Bromo-4-Chloro-3-Indolyl-beta-D-Galactopyranosid) zum Selektions-Agarose-Overlay
unterschieden werden. Die Spontanmutanten vermögen zwar unter dem Selektionsdruck
des applizierten BrdU zu replizieren, bilden aber farblose Plaques aus, da sie keine Beta-
Galaktosidase exprimieren können. Die Selektion der rekombinanten Klone erfolgte in
mehreren Runden der sequentiellen Plaque-Aufreinigung im doppelten Agarose-Overlay
(9, 164).

45
��������� 3�� �������93����> �� =�������� ? ;�������� 3�� ���! ��������������
2.5 % LMP-Agarose in dH2O
(Seaplaque Agarose FMC Bioproducts,Rockland ME, #50112)
2x MEM high Glucose
(GIBCO BRL, Eggenstein, #21935) Medium
10 % FCS
2x Penicillin/Streptomycin
2.5 % LMP-Agarose durch Aufkochen (TS = 65°C) verflüssigen
2x MEM und Agarose bei 42°C in Wasserbad halten
Zu gleichen Teilen Agarose und 2x MEM mischen
Endkonzentration: 1x MEM–5, 1.25 % LMP Agarose
H� ��7� �������� 8��� ��� ������7������ ;��((�
! 93����> �� ���F�;�������� �� ='� *)� 8�����
! 93����> ��;��������?=��������
#! 93����> �� ��78 @:��;�������� ������ ��:��
Zusatz von Bromdesoxyuridin(BrdU, Sigma, Taufkirchen,
#B-5002)
1/200 des Endvolumens von200x Stock in dH2O (10
mg/ml) zugeben
Endkonzentration 50 µg/ml
Kein Zusatz
Zusatz von X-Gal (5-Bromo-4-Chloro-3-Indolyl-beta-D-
Galactopyranosid)
(PEQLAB #37-2610)
100x Stock in DMSO (40 mg/ml) 1/100 des Endvolumens
zugeben
Endkonzentration 400 µg/ml
Zellen werden mit jeweils 5ml pro 5cm∅-Schale
überschichtet
Zellen werden mit jeweils 5ml pro 5cm∅-Schale
überschichtet
1. Overlay wird mit jeweils 3ml pro 5cm∅-Schale
überschichtet
Agarose-Overlay für ca. 10-15 Minuten beiRaumtemperatur erstarren lassen
Inkubation der Zellen in 37��2 Brutschrank
�//! ,! ��������� 3�� E�&���������������� ������ �� &��6��;�������� �� =��������
�����/������� ��������������!

46
#!*!,! ��(������ �� =���(������ �� ��������� .����/�������
Die Infektion wurde in zu ca. 85% konfluenten 5cmØ-Schalen mit CV-1 Zellen
durchgeführt. Die Zellen wurden nach einmaligem Waschen mit MEM mit dem Wildtyp
WR Vakzinia-Virus mit einer MOI von 0,02 für 2h unter Schwenken im 37ºC CO2-
Inkubator infiziert (siehe Kap. 2.4.3.1). Nach Abnahme des Inokulums erfolgte die
liposomale Transfektion der Zellen mit dem Vakzinia Shuttle-Vektor pSc11-X bzw. dem
pSc11 Leervektor in einem DNA:Liposom Verhältnis von 1:5. Dieses Verhältnis war in
einer Effizienzanalyse der Transfektion von CV-1 Zellen mit dem Luziferase–Assay
bestimmt worden (siehe Kap. 2.2.4). Jeweils 8 µg der Plasmide pSc11-X und pSc11
wurden mit 40 µl LipofectAMINETM-Reagenz (Gibco BRL, Eggenstein) in insgesamt
600 µl des Serum-freien OptiMEM Medium (Gibco BRL, Eggenstein) gut durchmischt
und 35 Minuten bei Raumtemperatur inkubiert (siehe Kap 2.2.2). Die Infektion und die
Transfektion wurden zeitlich so abgestimmt, daß nach dem Ablauf beider Inkubationen
direkt mit der Transfektion der zuvor mit dem Virus inokulierten Zellen begonnen
werden konnte. Das Inokulum wurde abgenommen und 2.4 ml OptiMEM Medium pro
5cmØ-Schale auf die Zellen gegeben. Anschließend wurden diese mit dem DNA-
Liposom Komplex vorsichtig überträufelt. Nach sorgfältigem Schwenken erfolgte eine
Inkubation von 2 h im 37ºC CO2-Brutschrank. Danach wurde das Medium gewechselt
und die Zellen in DMEM-5 für 48–64 h bis zum maximalen, mikroskopisch sichtbaren
CPE weiter inkubiert. Die Aufarbeitung der Zellen geschah, wie in Kapitel 2.4.3.2
beschrieben, unter Aufnahme der Viren in jeweils 0.5 ml DMEM-0 pro 5cmØ-Schale.
Die gewonnene Virussuspension enthält zu diesem Zeitpunkt ein Gemisch aus
rekombinanten und Wildtyp Vakzinia-Viren in einem sehr hohen Verhältnis zugunsten
der Wildtyp Viren. Diese kann entweder bei –80°C tiefgefroren werden oder sofort zur
weiteren Plaque-Aufreinigung eingesetzt werden.

47
�//! -! &������'���� �� ���������=���(��3����� �;7 !
#!*!,!# ;�������� ��� ���F �� ��������� ������ 93����>����(�����
Die erste Runde der Selektion wurde mit BrdU in HuTK- 143B Zellen durchgeführt, um
die noch in großer Anzahl vorhandenen TK+ Wildtyp Viren zu eliminieren. Die konfluent
gewachsenen Zellen wurden in 5cmØ-Schalen mit einer Verdünnungseihe von 100 bis
105 (siehe Kap. 2.4.4.1) der gewonnen Virussuspension für 1 h infiziert. Nach Abnahme
des Inokulums wurden die Zellen mit jeweils 5 ml des ersten Agarose-Overlays, welcher
den Selektionsmarker BrdU (50 µg / ml) enthält, überschichtet. Die Anfertigung der
Agarose-Overlays erfolgte wie in Abb. 15 beschrieben. Nach Ablauf der Inkubationszeit
von 48 h im 37ºC CO2-Brutschrank wurde über den ersten Agarose-Overlay eine zweite
Lage Agarose/Medium geschichtet, welche das chromogene Substrat der Beta-
Galaktosidase X-Gal (400 µg / ml) enthielt. Nach weiteren 24 Stunden Inkubation zeigte
sich makroskopisch eine Blaufärbung der entstandenen Plaques rekombinanter Viren
innerhalb der Agarose. Die blauen, rekombinanten Plaques wurden mit Hilfe einer 500 µl
Filterpipettenspitze aus der Agarose durch langsames Aspirieren der Zellen abgesaugt

48
und in jeweils 0.5 ml DMEM-0 aufgenommen. Ein blaugefärbtes Plaque besteht aus
Zellen, welche ausgehend von einem einzelnen viralen Klon eine Population
rekombinanter Viruspartikel enthält. Die Suspension sollte kleine Klümpchen der
blaugefärbten Agarose enthalten. Es sollten ca. 5-10 Plaques aus möglichst niedrigen
Verdünnungsstufen mit der Methode des dreimaligen Tauens / Frierens (siehe Kap.
2.4.3.2) aufgearbeitet werden. Dabei gilt es zu beachten, daß sich die Plaques
lichtmikroskopisch gut von eventuell in nächster Nähe vorhandenen farblosen Plaques
abgrenzen lassen, um eine Verunreinigung der gewonnenen Suspension mit Wildtyp
Viren möglichst gering zu halten.
#!*!,!) ;�������� ��7� �6�������� &��6��������� ��� ��� ��������� ��:��
Die aus den infizierten HuTK- 143B Zellen mit der initialen BrdU-Selektion
vorgereinigten rekombinanten Viren mußten in weiteren 3-4 Selektionsschritten von
Kontaminationen mit dem Wildtyp Virus aufgereinigt werden. Erst dann konnte eine
weitere Vermehrung der Klone in Mini- und Maxi-Stocks und eine Analyse des
inserierten Gens erfolgen. In den nachfolgenden Selektionsrunden wurden konfluente
5cmØ-Schalen CV-1 Zellen mit einer Verdünnungsreihe (100 bis 10-2) der
aufgeschlossenen Viren aus Kap. 2.4.5.2. infiziert und mit jeweils 5 ml Agarose /
Medium ohne Zusatz (siehe Abb. 15) für 48 Stunden inkubiert. Der zweite
Agarose/Medium-Overlay enthielt wiederum X-Gal und die nach weiteren 24 Stunden
Inkubation im 37ºC CO2-Brutschrank entstandenen blauen Plaques wurden erneut
abgesaugt und aufgearbeitet (siehe Kap. 2.4.5.2). Pro Runde wurden 2-3 Plaqueklone aus
der vorherigen eingesetzt, die übrigen Klone können bei –80°C tiefgefroren gelagert
werden. Zeigten sich in einer der weiteren Selektionsrunden keine farblosen Plaques
mehr im CV-1-Monolayer, sondern eine reine Population rekombinanter, blaugefärbter
Plaques, so wurde die Selektion an diesem Punkt beendet. Anschließend wurden Mini-
sowie später auch Maxi-Stocks aus den rekombinanten Klonen der aufgearbeiteten
Plaques hergestellt (siehe Kap. 2.4.3.3). Die rekombinanten Viren wurden rVV-X-adw
und rVV-pSc11 genannt.

49
#!*!-� ��������������� �����/������� �������������� �� ����� :���������
#!*!-! &�.�����>� ��� <���������������� �� .������� ��� ���������;��7�
Mit den hergestellten Stocks der rekombinanten Vakzinia Klone rVV-X-adw, rVV-
pSc11 sowie mit Wildtyp Vakzinia-Virus als Negativkontrolle wurden konfluente 5cmØ-
Schalen CV-1 Zellen unter Standard-Bedingungen (siehe Kap. 2.4.3.1) infiziert. Nach
einer Inkubation von 72 h im 37ºC CO2-Brutschrank wurden die Zellen bei maximalem
CPE von den Petrischalen abgekratzt und die DNA mittles Proteinase-K Verdauung mit
dem QIAamp Tissue Kit (QIAGEN, Hilden, #39304) aus den Zellen extrahiert. Nach
Elution wurden die gewonnenen DNA-Proben gelelektrophoretisch analysiert und ihre
Konzentration bestimmt. Jeweils 400 ng der DNA-Proben wurden zur PCR-Analyse
eingesetzt (Protokoll siehe Kap. 2.1.2). Die PCR wurde mit folgendem Programm
durchgeführt: 25 Zyklen à 30 s Denaturieren bei 94°C, 30 s Annealing bei 57°C sowie 5
min. (wegen Fragmentgrößen > 34 kb) Elongation bei 68°C. Die PCR-Produkte wurden
anschließend in einem 0.8% Agarosegel analysiert.
Die beiden Primer wurden innerhalb der flankierenden Regionen des Thymidinkinase-
Lokus (TKR und TKL) des Vakzinia Genoms so gewählt, daß durch Insertion des
Fremdgenes in den TK-Lokus PCR-Produkte der rekombinanten Klone mit
unterschiedlicher Länge entstehen können.
&����� ��� �������� =�>����������D
Sense TK83 – Primer TGG-24 : =:: �=� :=� ��: �=: �=: ��� =�=
Antisense TK1100 – Primer CGG-24 : �:: === �=� =�� �:� ��� ��� �=�
����7���� ��� :�IG�� ��� &�.�&������D
Negativkontrolle Wildtyp Vakzinia-Virus: !4 1 �/ (Position 83–Position 1100)
(mit intaktem TK-Lokus)
Rekombinantes rVV-pSc11: *!,)# �/ (Position 83–Position 4615)
Rekombinantes rVV-X-adw: ,!41+ �/ (Position 83–Position 5159)

50
Gleichzeitig konnte mittels der Agarosegel-Analyse der PCR-Produkte die Reinheit der
Virus-Stocks überprüft werden. Sollten die Stocks der rekombinanten Klone nach der
Selektion mit Spuren des Wildtyp Vakzinia-Virus kontaminiert sein, so würde dies in
zwei unterschiedlichen PCR-Produkten entsprechender Größe resultieren. Diese können
dann anhand ihrer Länge als Banden des rekombinanten Klones und des Wildtyp Virus
in der gelelektrophoretischen Auftrennung dargestellt werden.
'������������7���D
Da schon geringste DNA-Mengen zur Kontamination dieser Nachweis-PCR ausreichen,
mußten besondere Schutzmaßnahmen eingehalten werden:
♦ Die Aufarbeitung der DNA und Durchführung der PCR erfolgte räumlich getrennt
von der Klonierung der Plasmide bzw. Herstellung der rekombinanten Viren.
♦ Es werden neue Aliquots von Puffern, Primern, dNTPs und H2O eingesetzt.
♦ Die Template-DNA wird dem PCR-Mastermix im allerletzten Schritt zugefügt.
♦ Aerosolbildung beim Pipettieren wird strengstens vermieden und die verwendeten
Pipetten werden zuvor mit UV-Licht behandelt, um etwaige DNA-Spuren an der
Pipette zu inaktivieren.
♦ Es werden nur Pipettenspitzen mit Filtereinsatz verwendet.
♦ Um eine mögliche Kontamination der eingesetzten Materialien ausschließen zu
können, wird eine spezielle Negativkontrolle mit H2O an Stelle der Template-DNA
eingesetzt.
#!*!-!# ��7���� ��� :���2������ �����/������� �������������� �� 5������
����
Die HBxAg-Expression durch die Vakzinia-Klone rVV-X-adw und rVV-X-ayw
(freundlicherweise durch Prof. F.V. Chisari, La Jolla, CA, USA überlassen) wurde
zunächst in CV-1 Zellen überprüft. Unter Standardbedingungen wird jeweils eine
konfluente 75cm2 Flasche mit einem Virusklon infiziert (siehe Kap. 2.4.3.1) und die
Zellen bei maximalem CPE nach 72-96 h vom Boden der Flasche abgekratzt und mit 500
µl RIPA-Puffer (s.o.) für 20 Minuten bei 4°C lysiert. Die Zellysate werden unter Zugabe

51
des 6x β-Mercaptoethanol-haltigen Probenpuffers für 10 Minuten bei 100°C im
Wasserbad denaturiert und anschließend auf ein SDS-Polyacrylamidgel geladen. Das
exprimierte X-Protein wurde im Western-Blot (siehe Kap. 2.3) mit dem monoklonalen
Antikörper α-X36 in einer Verdünnung von 1:500 nachgewiesen. Als Negativkontrollen
wurden Zellen mit dem rVV-pSc11 oder Wildtyp Virus infiziert.
Um eine Expression des X-Proteins auch in den Targetzellen für die späteren
Zytotoxizitätsassays nachzuweisen, wurden die adhärent wachsenden Mäuse-
Fibroblasten MC57G (H-2b syngen) und Balb/c-3T3 sowie D2 (beide H-2d syngen) und
die in Suspension kultivierten lymphoiden Zellinien P815 (H-2d syngen ) sowie EL4 (H-
2b syngen ) mit einer MOI von 5-20 pfu / Zelle infiziert. Nach Inkubation wurden die
Zellen bei maximalem CPE mit RIPA-Puffer lysiert und die Lysate im Western-Blot wie
oben beschrieben analysiert.
#!*!1 F��������3����� ��� .���������� �����/������� ��������������
Das Ziel der UV-Inaktivierung rekombinanter Vakzinia-Viren ist die Suppression der
viralen Replikation bei intakter Expression des inserierten Fremdgens in der infizierten
Zelle. Dies ist eine notwendige Voraussetzung zum Einsatz der Viren bei der in vitro
Stimulation muriner Lymphozytenkulturen. Hierbei muß gewährleistet sein, daß keine
viralen Partikel aus lysierten Stimulatorzellen freigesetzt werden und anschließend die
empfindlichen CD8+ T-Lymphozyten durch eine zytopathische Infektion zerstören (10,
76, 134, 313). Um die optimale Bestrahlungsenergie zu ermitteln, wurde eine Titration
der UV-Bestrahlung mit dem UV- Crosslinker-Gerät (BIORAD, München)
durchgeführt.
#!*!1! =�������� ��� ��������� F����� �� ������3�����
Zur UV-Bestrahlung wurde das entsprechende Volumen der Virus-Stammlösung in ca.
800–1000 µl DMEM-0 aufgenommen und so auf dem Boden einer 5cm∅-Schale verteilt,
daß die Virussuspension als dünner Film gerade die gesamte Fläche der Schale bedeckte.
Anschließend wurden in einer Bestrahlungsserie mit Energiestufen von 0 mJ bis 1000 mJ

52
in Schritten zu 100 mJ jeweils gleiche Mengen der Virussuspension im UV-Crosslinker
inaktiviert. Parallel wurden nun CV-1 Zellen in konfluenten 5cm∅-Schalen mit dem UV-
inaktivierten Virus mit einer MOI von 4 zur Kontrolle der Proteinexpression sowie mit
einer niedrigen MOI (0.25) zur Kontrolle der viralen Replikation unter
Standardbedingungen (siehe Kap. 2.4.3) infiziert. Zum Nachweis einer intakten
Proteinexpression nach UV-Bestrahlung wurden die CV-1 Zellen nach 24 h mit RIPA-
Puffer lysiert und im Western-Blot analysiert (siehe Kapitel 2.3). Die virale Replikation
in Form einer sichtbaren Plaque-Bildung wurde durch tägliche mikroskopische
Beobachtung der infizierten CV-1 Zellen bis zum Tag 10 nach der Infektion überprüft.
���� �1 : :! �9'$+ -$1)$
#!,! � ��������������
#!,! ! ���
Es wurden Balb/c- und C57BL/6- Mäuse im Alter von 6-12 Wochen eingesetzt, welche
im Tierstall des Neurozentrums der Universitätsklinik Freiburg gehalten wurden.
#!,! !# =�7���� ��� ����������
Die intramuskuläre Injektion der Plasmid-DNA erfolgte in den rechten oder linken M.
tibialis anterior der Mäuse. Fünf Tage vor Applikation der Expressionsplasmide wurden
je 100 µl einer 0,25%igen Lösung des Lokalanästhetikums Bupivacain (Carbostesin,
Astra GmbH, Wedel) in verschiedene Abschnitte des Muskels injiziert. Diese
Vorinjektion erzeugt eine Nekrose, Inflammation und sukzessive Regeneration des
Muskels, wodurch immunkompetente Zellen wie z.B. Antigen-präsentierende Zellen
(APCs) samt ihres immunmodulatorischen Zytokinrepertoirs in den Muskel einwandern.
In diesem immunologisch hochaktiven Milieu sind dann bei der eigentlichen DNA-
Injektion besonders zahlreiche APCs und aktivierte Effektorzellen erreichbar (63). Fünf
Tage nach der Bupivacain-Gabe wurde die Plasmid-DNA, gelöst in 100 µl einer 0.9%

53
NaCl Lösung, in 4-5 Bereiche desselben Muskels mit einer 27G Nadel injiziert. Je nach
Studiengruppe wurden dabei pro Maus 100-150 µg Plasmid-DNA, bei Zytokin-
Koimmunisierung zusätzlich 25-50 µg des entsprechenden Interleukin-
Expressionsplasmids, eingesetzt. Bei einigen Gruppen wurden ein- oder zweimalige
Booster-Immunisierungen in 2-wöchentlichen Abständen durchgeführt (nach M.
Geißler).
#!,!# :������� �� ;���% &���������� ��� ���� �� $������� ����� $������
8���������
Zehn Tage nach der letzten Immunisierung wurden die Mäuse in einer abgeschlossenen
Kammer mit Isofluran (Forene, Abbott, Wiesbaden, #B506) anästhesiert und
anschließend ca 0,8 ml bis 1 ml Blut durch retrobulbäre Punktion gewonnen. Nach
erfolgter Blutentnahme wurden sie mit Isofluran getötet und sofort in ein 70% Ethanol-
Bad überführt.
Nach 4-6 stündiger Inkubation bei Raumtemperatur wurden die Blutproben zur
Abtrennung der Seren von den korpuskulären Blutbestandteilen zweimal in einer 4°C-
Tischzentrifuge bei 10 000 U / min. für jeweils 10 Minuten zentrifugiert und die
Überstände in sterilen Eppendorf-Cups bei –20°C bis zur weiteren Verwendung gelagert.
Die Mäuse wurden über einen Haut-Längsschnitt und einen Peritonealschnitt längs zur
Milzachse unter sterilen Bedingungen splenektomiert. Mit einem Glasmörser wurden
unter Zugabe von jeweils 10 ml IMDM-10 aus den Milzen Einzel-Zellsuspensionen
erstellt und diese zur Separation von Hilus-Fett- und Bindegewebsbestandteilen über ein
steriles Nylon-Siebgewebe mit einer Maschenweite von 200 µm (Polyamid-Siebgewebe,
Fa Reichelt Chemietechnik, Heidelberg) filtriert. Nach 5-minütiger Abzentrifugation bei
1200 rpm wurden die Zellen mit jeweils 2,5 ml des kurz vor Gebrauch hergestellten
Erythrozyten-Lysepuffers (0.83% NH4Cl / 0,17 M Tris pH 7,5 im Verhältnis 10:1) für 10
min. im 37°C CO2-Begasungsbrutschrank inkubiert. Nach zwei Waschschritten mit
MEM wurden die separierten Leukozyten bei 1200 rpm abzentrifugiert, in jeweils 6 ml
IMDM-10 / 5 x 10-5 M 2-ME (2-Merkaptoethanol) aufgenommen und die

54
Konzentrationen der Lymphozyten in der Neubauer-Zählkammer mit Trypanblaufärbung
(0.02 %) zur Vitalitätsanalyse bestimmt. Pro Maus kann man mit einer Ausbeute von ca
1 x 108 Splenozyten rechnen. Zur weiteren Verwendung in den unten beschriebenen
immunologischen Assays wurde die Konzentration der Einzel-Zellsuspensionen auf 4 x
106 Lymphozyten / ml eingestellt.
#!,!) $E�;� �� ��7���� 3�� ��2�����(�7��� �����I�����
Die Quantifizierung von spezifischen Antikörpern gegen das X-Protein in den Seren
genetisch immunisierter Balb/c und C57BL/6 Mäuse wurde nach dem Prinzip des ELISA
(Enzymimmunoassay) durchgeführt. Das rekombinante HBx zur Bindung der Antikörper
wurde uns freundlicherweise von Dr. S. Urban, Zentrum für Molekulare Biologie,
Universität Heidelberg überlassen (283). Dieses Protein wurde im Baculovirus-System
als 6-His-Tag-Fusionsprotein synthetisiert und in einem Na-Phosphat-Puffer bei pH 5.5
gelöst. Zunächst wurden in einer 96-well „Pro Bind“ Mikrotiterplatte jeweils 1,1 µg
rekombinantes Protein in 50 µl Puffer pro well über Nacht bei 4°C immobilisiert. Diese
Proteinkonzentration hatte sich nach Pilotexperimenten als optimal erwiesen. Nach
zweimaligem Waschen mit PBS 0.02 % Tween wurden die freien Bindungsstellen der
Platten mit 3 % BSA (#A-9647, Sigma, Taufkirchen) in PBS für 2 Stunden abgesättigt.
Anschließend wurden die Seren in verschiedenen Verdünnungsstufen (1:20 bis 1:500 in
PBS/3 % BSA) als Duplikate einpipettiert und für 2 Stunden bei Raumtemperatur
inkubiert. Als Positivkontrolle wurde der monoklonale anti-HBx-Antikörper α-X36
(siehe Kap. 2.3) in einer Verdünnung von 1:2000 eingesetzt. Nach drei weiteren
Waschschritten wurden die Platten mit dem Meerretich-Peroxidase konjugierten anti-
Maus IgG Antikörper (Amersham, 1:1000 in PBS verdünnt) in jeweils 50 µl für eine
Stunde bei Raumtemperatur inkubiert. Die Substratreaktion erfolgte nach 5maligem
Waschen durch Zugabe von jeweils 50 µl der o-Phenyldiamin • 2 HCl (OPD) –Lösung
(#7181E, Abbott, Wiesbaden). Nach Eintritt der Farbentwicklung wurde die Reaktion
mit 1 N Schwefelsäure (100 µl pro well) gestoppt und die Absorptionswerte bei 492 nm
in einem ELISA-Reader bestimmt. Von den errechneten Mittelwerten der Duplikate
wurde die Hintergrundaktivität (Substratreaktion ohne Serum und Antikörper)
subtrahiert.

55
#!,!* �� 3���� &����(�����������>� 3�� =�8�����
Das Prinzip des T-Zellproliferationsassays basiert auf dem Einbau eines leicht messbaren
Markers der DNA –Syntheseleistung der Lymphozyten nach Zugabe eines
unspezifischen (u.a. PMA, PHA, Con A, anti-CD3) oder spezifischen (rekombinantes
Protein oder Peptid) T-Zellstimulus (54, 108, 110). Nach dem in vivo Priming mit
Plasmid-DNA wurden die murinen T-Zellen in der vorliegenden Arbeit mit
rekombinantem X-Protein vom ayw-Subtyp (siehe Kap. 2.5.2) in unterschiedlichen
Konzentrationen in vitro stimuliert. Dabei fungieren MHC-Klasse-II tragende Zellen, u.a.
dendritische Zellen, welche einen Teil der Milz Einzel-Zellsuspension (siehe Kap. 2.5.1)
ausmachen, als Antigen-präsentierende Zellen (APCs) zur Induktion der T-
Zellproliferation. Zur Markierung werden Thymidin-Analoga wie das radioaktive 3H-
Thymidin oder das Bromodesoxyuridin (BrdU) verwendet (212). In dieser Arbeit wurde
mit dem Cell Proliferation ELISA (Boehringer, Mannheim, #1647229) ein BrdU-Assay
zur Proliferationsanalyse eingesetzt.
Zur Stimulation der T-Zellen wurden Triplikate von jeweils 200 µl der Milz Einzel-
Zellsuspension in IMDM-10 in einer Endkonzentration von 5x105 Effektorzellen pro
well auf 96-well Mikrotiterplatten (Round bottom) mit dem HBx-Antigen in
Konzentrationen von 0.1 µg / ml, 1 µg / ml und 10 µg / ml für 48 h inkubiert. Das
Medium enthielt zusätzlich 2-Merkaptoethanol in einer Konzentration von 5 x 10-5 M.
Nach dem Ablauf von 48 h wurden die Zellen mit BrdU in einer Endkonzentration von
100 µM für weitere 12 h markiert. Anschließend wurden die Zellen 10 min. bei 1000
U/min abzentrifugiert, der Überstand verworfen und die Platten abgetrocknet. Nach
einem Fixations/Denaturierungsschritt von 30 min. erfolgte eine 90-minütige Inkubation
mit dem anti-BrdU-POD Fab-Fragment, welches hierbei an das in die zelluläre DNA
inkorporierte BrdU gebunden wurde. Nach mehreren Waschgängen wurde die
eingeleitete Substratreaktion (Tetramethylbenzidine) mit 1 M H2SO4 abgestoppt und die
Extinktionen bei 450 nm quantitativ in einem ELISA-Reader ausgewertet. Es wurden
Mittelwerte der Triplikate berechnet und die Hintergrund-Aktivität (gemessene BrdU-
Bindung an Medium ohne Zellen) von den Extinktionswerten subtrahiert. Um die
spezifische Proliferation zu erfassen, wurden die Differenz-Werte durch Substraktion der
gemessenen Aktivität in Abwesenheit des Antigens gebildet (∆OD).

56
#!,!, �� 3���� ;��������� �� ��7���� 3�� �>����2�7��� =�8����� B�=EC
Mit der DNA-Immunisierung lassen sich Antigen-spezifische CTL-Precursorzellen in
vivo induzieren. Zur Messung einer spezifischen CD8+ zytotoxischen T-Zellantwort
(CTL) der in vivo sensibilisierten murinen Effektorzellen müssen die Precursorzellklone
in vitro expandiert werden. Dies gelingt durch eine Präsentation der prozessierten
Fremdantigene im Kontext von syngenen MHC-Klasse-I Komplexen auf den
Stimulatorzellen. Zu diesem Zweck wurden in Anlehnung an Lefkovits (Hrsg.)
„Immunology Methods Manual“ (10) Milzzellen einer nicht-immunisierten Balb/c- bzw.
C57BL/6- Maus vom identischen Haplotyp (H-2d bzw. H-2b) mit HBx-rekombinanten
Vakzinia-Viren infiziert. Um eine mögliche Proliferation der infizierten Stimulatorzellen
zu supprimieren, werden diese anschließend bestrahlt und schließlich mit den
Effektorzellen in einem fixen Effektor : Stimulator Verhältnis (E:S Ratio) für 5-6 Tage
inkubiert. Zur Quantifizierung der CTL-Aktivität nach erfolgter in vitro Stimulation wird
ein 51Chrom-Release-Assay durchgeführt (38). Zunächst werden syngene Targetzellinien
mit rekombinanten Vakzinia-Viren infiziert, um eine Expression und nachfolgende
Präsentation des HBx-Antigens zu erreichen, und anschließend mit 51Cr markiert.
Während der Inkubation mit den spezifischen zytotoxischen T-Lymphozyten werden
bestimmte Peptidstrukturen der Antigene über die Interaktion der T-Zellrezeptoren mit
MHC-Klasse-I Molekülen erkannt und durch die Lyse der Targetzellen wird 51Cr in den
zellfreien Überstand freigesetzt. Durch eine Analyse der Überstände im Gammacounter
kann die korrigierte prozentuale Lyse berechnet werden.
#!,!,! �� 3���� ;��������� ��� �=E ��� ������������ ��(�������� ;������>���
Zur Stimulation wurden 4 x 107 Effektorzellen einer Maus in einem E:S Verhältnis von 4
: 1 mit den Stimulatorzellen 6 Tage lang inkubiert. Hierzu wurden 1 x 107 syngene
Splenozyten pro Ansatz gepoolt, um dann in einem 15 ml Falcon-Röhrchen zur Infektion
in einem möglichst geringen Volumen von 1 - 2 ml DMEM-0 resuspendiert zu werden.
Die Infektion erfolgte mit einer MOI von 5 für 120 min. auf einer automatischen Roll-
Vorrichtung im 37°C CO2-Begasungsbrutschrank. Die rekombinanten Vakziniaviren
waren zuvor mit 300 mJ UV-Licht inaktiviert worden (siehe Kap. 2.4.7). Nach der
Infektion wurden die Splenozyten zweimal mit jeweils 5 ml MEM gewaschen und

57
anschließend mit 20 Gy (2000 rad) bestrahlt. Die Konzentration der Zellen wurde durch
entsprechende Zugabe von IMDM-10 + 5 x 10-5 M 2-ME (2-Merkaptoethanol) auf 1 x
106 / ml eingestellt und zur Stimulation auf 24-Well-Platten übertragen. Es wurden
jeweils 1 ml (1 x 106 Zellen) der infizierten Stimulatorzellen und 1 ml der Effektoren (4 x
106 Zellen) pro well gemischt und insgesamt 10 wells pro Effektor-Maus angesetzt. Nach
24 Stunden wurden den Ansätzen 10 Units/ml rekombinantes, murines Interleukin-2
(mIL-2 #1271164, Boehringer, Mannheim) zugesetzt.
#!,!,!# ���� ��� �=E�����3���� �� �����.��������>
Als Targetzellinien wurden die syngenen Fibroblasten MC57G für den Haplotyp H-2b
und Balb/3T3 für den Haplotyp H-2d mit rekombinanten Vakzinia-Viren infiziert. Die
Infektion der Targetzellen mit den Vakzinia-Viren rVV-X-ayw bzw. rVV-X-adw sowie
als Negativkontrolle rVV-X-pSc11 erfolgte mit einer MOI von 8 für 150 min. unter
ständigem Schwenken im 37°C-CO2-Inkubator. Anschließend wurden die Zellen
gewaschen, trypsiniert und in jeweils 150 µl IMDM-10 in einem 15 ml Falcon-Tube
aufgenommen. Diese Zellsuspension wurde unter Zugabe von 51Cr mit einer Aktivität
von 5 mCi / ml (Amersham, Freiburg, #CJS1-5mCi) bei einer Endaktivität von 0,4 mCi
für 90 min. markiert. Dabei wurde die Labeling-Suspension auf einer automatischen
Rollvorrichtung im CO2-Begasungsbrutschrank bei 37°C ständig durchmischt. Nach drei
Waschschritten mit MEM wurden die Zellen gezählt und auf eine Konzentration von 1 x
105 / ml eingestellt. Zur Inkubation mit den Effektorzellen wurden diese aus den
Stimulationsansätzen zusammengeführt, gezählt und auf eine Konzentration von 1 x 107 /
ml eingestellt. Die Effektoren wurden nun in Triplikaten in verschiedenen
Effektor:Target (E:T Ratio) Verhältnissen (100:1, 20:1, 5:1) sowohl mit den HBx-
spezifischen Targetzellen als auch mit den unspezifischen rVV-pSc11 infizierten
Targetzellen in einer 96-Well „round-bottom“ Platte inkubiert. Wenn die Ausbeute der
stimulierten Lymphozyten zu gering ausfallen war, wurden niedrigere E:T Verhältnisse
gewählt. Zur Bestimmung der spontanen 51Cr-Freisetzung wurden nur Targetzellen mit
IMDM-10 Medium eingesetzt, für die Maximalfreisetzung wurden die Targetzellen mit 5
%igem Triton X-100 (#T8787, Sigma, Taufkirchen) lysiert.

58
&��������7����D
100 : 1 20 : 1 Spontan Maximal
Effektorzellen (1x107 /ml) 100 µl 20µl - -
Targetzellen (1x 105 / ml) 100 µl 100µl 100µl 100µl
IMDM-10 - 80µl 100µl -
5% Triton X-100 - - - 100µl
Die pipettierten Ansätze wurden bei 1200 rpm kurz „anzentrifugiert“ und anschließend
für eine Dauer von 6 Stunden im 37°C CO2-Begasungsbrutschrank inkubiert. Nach
erneuter Zentrifugation für 4 min. wurden jeweils 100 µl des Überstandes im Gamma-
Counter analysiert. Es wurden Mittelwerte der Triplikate bestimmt und die spezifische
Lyse der zytotoxischen T-Zellen folgendermaßen berechnet:
Spezifische 51Cr-Freisetzung = Effektor-Freisetzung - spontane Freisetzung
Maximale Freisetzung - spontane Freisetzung

59
������
4�� �$+&)$"",1# ,1% �0(+(2)$+ & $+,1# %$+ /"(&- %*�$2)!+$1
)! ! '��������� �� $2������������ �7�?)����>�
Das X-Protein der beiden HBV Subtypen ayw und adw unterscheidet sich innerhalb
seiner Antigenstruktur in 9 von 154 Aminosäuren, dies entspricht einer 82,9 %igen
Homologie der Primärstruktur des Proteins. Zur immunologischen Charakterisierung des
X-Proteins in vivo werden daher Konstrukte beider Virus-Subtypen untersucht. Da das
Plasmid pRc/CMV-X (adw) schon vorlag, sollte das Expressionsplasmid pcD/3-X (ayw)
kloniert werden. Der X-ORF wurde aus dem HBV-ayw Überlängenkonstrukt pCH-
9/3091 (freundlicherweise zur Verfügung gestellt von Prof. Dr. M. Nassal,
Universitätsklinik Freiburg) gewonnen. Ein 617 bp großes Fragment mit dem X-ORF-
Insert wurde durch Restriktionsverdau mit den Enzymen NcoI und SmaI ausgeschnitten.
Der 5´-Basenüberhang an der NcoI-Schnittstelle des Fragmentes wurde mit dem Klenow-
Enzym durch Einbau zusätzlicher Nukleotide in ein „blunt-end“ umgewandelt.
Anschließend wurde der Zielvektor pcDNA/3 mit EcoRV linearisiert, mit dem Enzym
Alkalische Phosphatase (CIAP) dephosphorylisiert und das Insert durch eine „blunt-end“
Ligation eingeführt.
�//! 1! ����>��7�� :�� ��� '��������!
1% Agarosegel. Spur 1: 617 bp X-ORF Insert
mit NcoI und SmaI verdaut, nach Klenow
Reaktion. Spur 2: Zielvektor pcDNA3.1 mit
EcoRV verdaut (5446 bp).

60
Der Nachweis des X-ORF Inserts im Zielvektor pcD/3 erfolgte durch eine DNA-Analyse
der E.coli-Klone, welche mit dem Ligationsprodukt transformiert worden waren, mit der
Mini-Prep Methode. Da das Insert über „blunt“ Schnittstellen in den Vektor ligiert
worden war, mußte auch die korrekte Orientierung des X-ORF in dem positiven pcD/3-X
Klon 1 durch einen geeigneten Restriktionsverdau überprüft werden (siehe Abb. 19). Das
Enzym AvaI schneidet einmal innerhalb des Vektor-Backbones und einmal in der Nähe
�//! 0! ����>� ��� �7�?)�
'����! 1% Agarosegel mit
aufgereinigten Plasmidklonen
aus E.coli. Spur 1: Maxi-Prep-
DNA des Vektors pcD/3 (5446
bp) als Negativ-kontrolle, Spur
2: religierter Vektor pcD/3 als
Negativ-kontrolle, Spur 3:
positiver Klon 1 pc/D3-X (6065
bp), Spuren 4-7: negative
Klone.
�//! +! .���������3���� �� �����3��
&���������! 1% Agarosegel. Spur 1: pcD/3-X
Klon 1 verdaut mit AvaI. Fragmente von 4397 bp,
1119 bp und 549 bp beweisen die korrekte
Insertion des X-ORF. Spur 2: pc/D3-X Klon 1
(6065 bp) linearisiert mit pvuI. Spur 3: pcD/3-X
Klon 1 zirkulär ungeschnitten.

61
des 5´-Endes innerhalb des Inserts. Je nach Orientierung des Inserts resultieren ein
Fragment fixer Größe von 1119 bp und zwei Fragmente unterschiedlicher Größe von
4397 bp und 549 bp bei richtiger Insertion bzw. 4830 bp und 116 bp bei falscher
Insertion.
)! !# '�������� �� �������� =���(��������
Die Herstellung eines HBx rekombinanten Vakzinia-Virus vom adw-Subtyp erforderte
zunächst die Insertion des X-ORF in das Vakzinia Shuttleplasmid pSc11. In einer PCR
wurde der X-ORF aus dem Plasmid pRc/CMV-X amplifiziert. Da der Shuttle-Vektor nur
einen SmaI-Monolinker als Klonierungsstelle besitzt, mußten mittels PCR-Mutagenese
die „blunt“ Restriktionsstellen EcoRV vor dem Start- und hinter dem Stop-Kodon in die
Sequenz eingeführt werden.
Das 555 bp große PCR-Produkt (siehe Abb. 20) wurde aufgereinigt und mit EcoRV
verdaut. Der Zielvektor pSc11 wurde mit SmaI linearisiert und vor der „blunt-end“
Ligation mit CIAP dephosphorylisiert, um die spontane Religation des Vektors zu
begrenzen (siehe Abb. 21).
�//! #4! &�. �� ��9.< B���C!
1% Agarosegel. Spur 1: PCR-
Negativkontrolle. Spur 2: PCR-
Produkt von 555 bp enthält X-ORF
und PCR-Template pRc/CMV-X.

62
Da es bei der „blunt-end“ Ligation zwei Möglichkeiten der Orientierung des Inserts gibt,
wurden die positiven Klone des Plasmids pSc11-X (siehe Abb. 22) mit dem Restriktions-
enzym AatII verdaut. Nur die korrekte Orientierung des X-ORF liefert 3 spezifische
Fragmente mit einer Länge von 5424 bp, 1947 bp und 1055 bp (siehe Abb. 23).
�//! ##! �����&����������
��� &��������� '����. 1%
Agarosegel. Spur 1: Positiv-
kontrolle Vektor pSc11, Spuren
2, 3, 5-8: positive Klone pSc11-
X (8426 bp), Spur 4: doppelte
Insertion (8969 bp), Spur 9:
negativer Klon.
�//! # ! ����>� �� �(�����������
������ �� �����! 1% Agarosegel.
Spur 1: PCR-Fragment EcoRV verdaut
(544 bp). Spur 2: pSc11 Transfervektor
mit SmaI linearisiert (7883 bp).

63
Es wurden schließlich 3 positive Plasmid-DNA Klone mit korrekter Orientierung des X-
ORF weitergeführt und zur Herstellung von rekombinanten Vakzinia-Viren amplifiziert.
)! !) &���������� ��� &���������
Es wurden insgesamt jeweils ca. 15 mg der Plasmide pcD/3-X (ayw) und pRc/CMV-X
(adw) und 2 mg des Plasmids pSc11-X (adw) präpariert. Als Maß für die Reinheit der
Präparation wurde der Quotient aus der spektrophotometrischen Absorption bei 260 nm
und bei 280 nm gebildet. Werte > 1,8 werden als kontaminationsfrei angesehen. Die
hergestellten Präparationen wiesen einen Quotienten zwischen 1.870 und 1.917 auf, was
auf die Gegenwart reiner Plasmid-DNA hinwies. Weiterhin sollte der Anteil
geschlossener zirkulärer DNA in den Präparationen sehr hoch sein, da nur diese für
effiziente Transfektionen in vitro und in vivo geeignet ist. Bakterielle genomische DNA
darf überhaupt nicht nachzuweisen sein, der Anteil relaxierter zirkulärer DNA sollte
nicht größer als 10% sein und der Anteil der geschlossen zirkulären DNA („supercoiled“)
mindestens 90% sein. Bei einzelnen Präparationen wurde der Reinheitsgrad bzw. der
Anteil geschlossen zirkulärer DNA durch anschließende CsCl-Gradienten-Aufreinigung
zusätzlich erhöht.
�//! #)! .�������������>� ���
9����������! 1% Agarosegel.
Plasmid-DNA wurde mit AatII
geschnitten. Spuren 1, 4, 6: negative
Klone von pSc11-X mit einem 5424 bp
Fragment und einem Doppel-Fragment
bei 1508 bp und 1494 bp. Spuren 2, 3,
5: positive Klone pSc11-X mit den
Fragmentlängen 5424 bp, 1947 bp und
1055 bp.

64
)! !* 9��������� ��� =���(�������((������ ��� ��� .���������� E��(����
Die murine Myoblasten-Zellinie G-8 zeigt eine sehr niedrige Transfektionseffizienz mit
der CaPO4-Methode. Mit der Methode des liposomalen Gentransfers lassen sich
hocheffiziente Transfektionen mit zahlreichen Zellinien durchführen. Es gibt jedoch
einige Parameter bei dieser Methode, welche individuell an die jeweiligen Bedingungen
der einzelnen Zellinien angepasst werden müssen. Im Falle der G-8 Zellen wurden die
Parameter DNA:Liposom Verhältnis im Transfektionsansatz und Inkubationszeit der
DNA-Liposom Komplexe während der Transfektion untersucht. Auch die liposomale
Transfektion von CV-1 Zellen mit dem Transferplasmid sollte für die Vakzinia-Virus
Rekombination optimiert werden.
G-8 Zellen wurden in 6-Well-Schalen mit jeweils 1 µg des Plasmids pCMV-Luc+
transfiziert. Dabei wurde in verschiedenen Ansätzen das DNA:Liposom Verhältnis
zwischen 1:5 und 1:12,5 und die Inkubationszeit mit den DNA-Liposom-Komplexen von
2 h, 4 h oder 6 h variiert. Die Luziferaseaktivität als Maß für die Transfektionseffizienz
wurde in den einzelnen Zellysaten 24 h später mit dem Luminometer wie in Kap. 2.2.4
beschrieben, bestimmt und ist in Abb. 25 dargestellt. Die höchste Transfektionseffizienz
wurde mit einem DNA:Liposom Verhältnis von 1:5 und einer kurzen Inkubationszeit von
2 h beobachtet. Gleichwohl zeigt dasselbe DNA:Lipsom Verhältnis auch bei längerer
Inkubation eine effiziente Transfektion, welche jedoch mit zunehmender Dauer
�//! #*! ��2��&������������
����������� &���������! 1%
Agarosegel. Der Anteil an
„supercoiled“ DNA beträgt mehr
als 95%. Spur 1, 2: pcD/3-X (ayw)
Maxi-Präparationen. Spur 3, 4:
pRc/CMV-X (adw) Maxi-
Präparationen.

65
wahrscheinlich als Folge der Zell-Toxizität der Liposomen abnimmt. Daher wurde bei
den weiteren HBx-Expressionsstudien in G-8 Zellen jeweils ein Verhältnis von 1:5 mit
einer Inkubationszeit von 2 h gewählt.
Analog wurden CV-1 Zellen mit DNA:Liposom Verhältnissen von 1:5, 1:10 und 1:15
transfiziert und analysiert. Auch hier zeigte sich noch deutlicher, daß die höchste
Effizienz mit einem Verhältnis von 1:5 erzielt werden kann.
0
2
4
6
8
��������������� ���� ����
2 h 4 h 6 h
���������������
����� �������� ��������!"���##��
1 : 5
1 : 7,5
1 : 10
1 : 12,5
DNA : LiposomVerhältnis
�//! #,! �������� ��� E��(��������3���� �� ��G (A� ��� =���(�������((������
�� ���������� :������(�� �� :�0 8�����! Die Abhängigkeit der Transfektions-
effizienz vom DNA:Liposom Verhältnis und der Inkubationszeit wurde durch Transfektion
mit dem Reportergen pCMV-Luc+ (1 µg) und anschließende Messung der
Luziferaseaktivität in relativen Lichteinheiten (RLU) im Zellysat der einzelnen
Transfektionsansätze gezeigt.

66
0
2
4
6
8
10
12
14
16
18
����
���
����
��$�
�%��&'
�
��
1 : 5 1 : 10 1 : 15
����(���)���*����+%#����
����� �������� ���������!&���##��
)! !, $2������ 3�� ��2���� �� ��2��>�
Die Expression des X-Proteins wurde in der humanen Hepatomzellinie HUH-7 sowie in
der murinen Muskelzellinie G-8 als in vitro Modell für die i.m. Plasmid-DNA
Applikation bei der DNA-Immunsierung überprüft. Bei allen Immunoblotanalysen wurde
der monoklonale Antikörper α-X36 (freundlicherweise von Prof. J. Wands, Brown
University, Providence, RI, USA überlassen) eingesetzt. Das Plasmid pRc/CMV-X
(freundlicherweise von Dr. M. Melegari, Molecular Hepatology Unit, MGH, Boston
überlassen), welches das X-Protein vom adw-Subtyp exprimiert, wurde unter
Standardbedingungen nach der Ca-Phosphat Methode in HUH-7 Zellen transfiziert. Die
G-8 Zellinie wurde liposomal transfiziert. Die Zellen wurden nach 24 Stunden mit
unterschiedlichen Lysepuffern lysiert.
Das X-Protein läßt sich aus beiden Zellinien und mit unterschiedlichen
Transfektionsbedingungen nur mit den stark Detergenzien-haltigen Lysepuffern RIPA
und 1x konzentriertem Western-Blot Ladepuffer oberhalb der Nachweisgrenze des
Immunoblots extrahieren. Dagegen konnte es in den Lysaten des schwachen Core-
Lysepuffers nicht nachgewiesen werden (siehe Abb. 27).
�//! #-! $��(�G �� ����
E����� ��������� �( ���
=���(�������((������ �� ���
8�����! Transfektionsansätze mit
unterschiedlichen DNA:Liposom
Verhältnissen. Messung der
Luziferaseaktivität in relativen
Lichteinheiten (RLU).

67
Die Charakterisierung der zeitabhängigen Expression des X-Proteins wurde in beiden
Zellinien nach transienter Transfektion mit dem Plasmid pRc/CMV-X durchgeführt
(siehe Abb. 28). Der Beginn der Expression in HUH-7 Zellen, welche mit der CaPO4-
Methode transfiziert worden waren, konnte nach 36 h nachgewiesen werden. Ein
Maximum zeigte sich nach 60 h mit durchgängig starker Expression bis 72 h nach der
Transfektion. In G-8 Zellen war die Expression vom ersten Zeitpunkt bei 20 h nach
Transfektion bis 60 h nach Transfektion kontinuierlich stark.
�//! #1! ��7���� ��� ��2 B���C $2������ �� �F��1 �� :�0 8����� ���
����7������7��� E>��((���. Transiente Transfektion (CaPO4-Methode) von HUH-7
Zellen mit pRc/CMV-X (Spur 3-5). Negativkontrolle: untransfizierte HUH-7 Zellen (Spur
1+2). G-8 Zellen liposomal unter optimierten Bedingungen transfiziert (Spur 6-9). Die
Proteinextraktion erfolgte 24h nach Transfektion mit Lysepuffern RIPA, Core-Lysepuffer
(Core) und einem 1x konz. Westernblot-Ladepuffer (Loading). Immunoblot-Analyse der
Proteinextrakte mit einem monoklonalen anti-X Antikörper.

68
Die Expression des X-Proteins vom ayw-Subtyp wurde mit dem subklonierten
Expressionsplasmid pcD/3-X getestet. Hierzu wurden zwei positive Klone des klonierten
Vektors in G-8 Zellen mit dem lipsomalen System unter optimierten Bedingungen
transfiziert. Beide Plasmidklone zeigen funktionierende Synthese des X-Proteins nach 24
h (siehe Abb. 29).
Ebenso wurde die Expression von pcD/3-X in HUH-7 Zellen sowie die zeitabhängige
Expression in beiden Zellinien mit dem Immunoblot-Verfahren getestet (Bilder nicht
gezeigt). Dabei konnten vergleichbare Resultate zu den Expressionsstudien mit dem
Plasmid pRc/CMV-X erzielt werden.
�//! #0! 8����/���������� ���
$2������ 3�� ��2 B���C �� �F��
1 �� :�0 8�����!
�C HUH-7 Zellen mit pRc/CMV-X
nach der Ca-PO4-Methode transient
transfiziert. Nach 20 h bis 72 h wurden
Proteinextrakte gewonnen und im
Immunoblot mit einem monoklonalen,
HBx-spezifischen Antikörper getestet.
�C G-8 Zellen wurden liposomal mit
pRc/CMV-X transfiziert und nach 20 h
bis 60 h geerntet. Die Proteinextrakte
wurden im Immunoblot mit einem
monoklonalen, HBx-spezifischen Anti-
körper getestet.

69
4��� �$+&)$"",1# +$2!-7 1(1)$+ �(28 1 (*� +$1
Als immunologisches Werkzeug zur Charakterisierung von CTL-Antworten gegen HBx
vom ayw-Subtyp sollte ein Vakzinia-Virus, welches das X-Antigen exprimiert,
rekombiniert werden. Die Spezifität einer gegen Epitope des X-Antigens gerichteten
CTL-Antwort kann nur nachgewiesen werden, wenn alle unspezifischen, gegen
Vakzinia-Epitope gerichteten CTLs in einer geeigneten Kontrolle gemessen werden
können. Zu diesem Zweck wurde ein zweites rekombinantes Vakzinia-Virus mit dem
Transfervektor pSc11 ohne Insertion eines Fremdgens hergestellt. Dieses rekombinante
Virus produziert bei ähnlicher Virulenz sämtliche Vakzinia-Epitope und die infizierte
Wirtszelle kann diese der MHC-Klasse-I Präsententation zuführen. So lassen sich bei
Messungen von CTL-Antworten im Chrom-Release-Assay unspezifische CTL-
Aktivitäten quantifizieren.
�//! #+! $2������ �� ��&������ 3�� �>��
;/�>� �� :�0 8�����! Die G-8 Zellen wurden
liposomal unter optimierten Bedingungen mit zwei
Klonen des Plasmids pcD/3-X transfiziert und nach
24 h geerntet. Die extrahierten Proteine wurden im
Immunoblot mit einem monoklonalen, HBx-
spezifischen Antikörper getestet. Spur 1 und 2 zeigen
die Expression von HBx durch jeden der beiden
Plasmidklone. Spur 3 zeigt die Negativkontrolle mit
pcD/3 transfizierten Zellen.

70
)!#! � .����/������� �� ;�������� ��� ��������
Die beiden rekombinanten Vakzinia-Viren rVV-X-ayw und rVV-pSc11 wurden nach der
Methode der homologen Rekombinantion und anschließenden Plaque-Selektion im
doppelten Agarose-Overlay hergestellt. Zunächst wurden CV-1 Zellen mit dem Wildtyp
Vakzinia-Virus bei einer MOI von 0,02 infiziert und direkt im Anschluß mit den
Plasmiden pSc11-X sowie pSc11 mit dem liposomalen Gentransfer unter optimierten
Bedingungen (siehe Kap 2.2.4) transfiziert. Die Zellen wurden nach 2 und 3 Tagen
geerntet und in eine Virussuspension aufgearbeitet. Die rekombinanten Virusklone
wurden in 3 Runden Plaque-Selektion auf TK- 143B Zellen im doppelten Agarose-
Overlay Verfahren aufgereinigt. Die Selektion TK-negativer Klone erfolgte durch das
Zytostatikum Bromo-Desoxyuridine, zusätzlich konnten die positiven Klone durch blaue
Anfärbung der Plaques mit dem Enzym Beta-Galaktosidase optisch sichtbar gemacht
werden. Pro Runde wurden 10 rekombinante Plaques aufgearbeitet. Die folgenden
Infektionen wurden mit Virussuspensionen aus 3 bis 8 dieser Plaques unverdünnt und in
einer 1:10 Verdünnung durchgeführt (siehe Abb. 30). Den Abschluß der Plaque-
Reinigung bildeten 2 Runden einfacher Beta-Galaktosidase Selektion der Klone in CV-1
Zellen. Aus der letzten Runde wurden jeweils 2 Klone von rVV-X-ayw und rVV-pSc-11
amplifiziert und anschließend charakterisiert.
)!#!# &������������ �����/������� ��������
Es wurden Präparationen positiver Klone der rekombinanten Vakzinia-Viren rVV-X-
ayw, rVV-X-adw (Dieses Virus wurde uns freundlicherweise zur Verfügung gestellt von
Prof. F.V. Chisari, Scripps Institute, La Jolla, CA, USA), rVV-pSc11 und des Wildtyp
Vakzinia-Virus WR (Western Reserve Stamm) hergestellt. Solche Präparationen müssen
besonders hohe Titer aufweisen, damit sie zur Antigenexpression in eukaryontischen
Zellen sowie zur Generierung von immunologischen Zielzellen eingesetzt werden
können. Diese Zellen werden mit Multiplizitäten im Bereich von MOI 5 bis MOI 20 bei
einem sehr kleinen Inokulum infiziert, so daß Titer zwischen 107 pfu/ml und 109 pfu/ml
erforderlich sind. Die Titrierung der Präparationen rekombinanter Viren nach
erfolgreicher Selektion wurde zunächst im doppelten Agarose-Overlay Verfahren mit
dem Chromogen X-Gal durchgeführt (siehe Abb. 31). Somit konnte durch

71
lichtmikroskopische Bestimmung des Verhältnisses blauer zu farbloser Plaques der
Reinheitsgrad der Präparationen ermittelt werden. Dieser lag bei den selektionierten
Klonen von rVV-pSc-11 und rVV-X-ayw >99 %. Im nachfolgenden wurden die Titer
reiner Präparationen der rekombinanten Viren und des Wildtyp Vakzinia-Virus mit dem
Kristallviolett-Plaquetest ermittelt (siehe Abb. 32).
)!#!)� ��������������� �����/������� �������������� �� ����� :���������
Die Insertion des Fremdgens rekombinanter Vakzinia-Viren und die Reinheit der
Präparationen konnte mit Hilfe einer analytischen PCR der präparierten Vakzinia-DNA
untersucht werden. Hierzu wurden CV-1 Zellen mit Präparationen der rekombinanten
Vakzinia-Klone und des Wildtyp Virus infiziert und eine DNA-Extraktion der Virus
infizierten Zellen durchgeführt. Diese DNA-Proben wurden in einer PCR mit Primern,
welche im viralen Genom in der Region des Thymidinkinase-Gens binden, analysiert.
Das jeweilige PCR-Produkt konnte durch die Berechnung der Länge der
Insertionssequenz bzw. der Wildtyp Sequenz vorhergesagt werden (siehe Abb. 33).
Die untersuchten Virusklone der rekombinanten Viren zeigten das jeweilige Insert mit
der entsprechenden Länge. Außerdem wiesen die Präparationen einen sehr hohen
Reinheitsgrad auf. Verunreinigungen der Präparationen rekombinanter Viren mit
Wildtyp Viruspartikeln können mit dieser PCR sehr sensitiv aufgespürt werden. In keiner
der DNA-Präparationen ließ sich jedoch eine zusätzliche Bande des Wildtyp Genoms mit
einer Länge von 1,017 kb nachweisen (siehe Abb. 34)

72
��7������ 2.5 x108 pfu / ml
��7����>� 5 x 107 pfu / ml
��7��;7� 4 x 107 pfu /ml
��7�5= 5 x 107 pfu / ml
�//! )4! ;�������� �����/��
������ ��������! Doppelter
Agararose-Overlay mit dem
Substrat X-Gal führt 72 h nach
Infektion zur selektiven Blau-
Färbung der rekombinanten
Virusklone. Die Plaques
werden in der 1:10 Ver-
dünnung (rechts) einzeln abge-
saugt und aufgearbeitet.
�//! ) ! =�������� ����� �����/��
������ ��������������! Die
Original-Suspension wird in 1:10
Verdünnungs-stufen zur Infektion
eingesetzt und im Agarose-Overlay
Verfahren mit X-Gal angefärbt. Die
Zahl der rekombinanten Plaques wird
in der vorletzten Verdünngsstufe
(rechts oben) ausgezählt und daraus
der Virustiter ermittelt.
�//! )#! =�����.������ ���
��2��&������������! Die Höhe
der Titer wurde mit dem
Kristallviolett-Plaquetest bestimmt.

73
Die Expression des X-Antigens durch die rekombinanten Vakzinia-Viren rVV-X-adw
und rVV-X-ayw wurde in Lysaten infizierter CV-1 Zellen nachgewiesen. Die Zellen
wurden mit unterschiedlichen Multiplizitäten zwischen MOI 5 und MOI 20 infiziert. Die
Zellysate wurden im Immunblot mit einem monoklonalen Antikörper gegen das X-
Protein getestet.
�//! ))! :�IG� ���
&�.�&������ /�� ���
����>� �����/�������
��������������!
������������ &�.�&�����
WT Vakzinia-Virus 1.017 kb
rVV-pSc11 4.532 kb
rVV-X-adw 5.079 kb
�//! )*! &�.�&������ 3��
������������ � 3��7��������
����������������! 1% Agarose-
gel. Spur 1: DNA nicht-infizierter
Zellen, Spur 2: PCR-Produkt mit
WT-VV infizierter Zellen, Spur 3:
PCR-Produkt des rekombinanten
VV-pSc11, Spur 4 und Spur 5:
irrelevante PCR-Produkte, Spur 6:
PCR-Produkt des rekombinanten
VV-X-adw. Die Längen
entsprechen dem jeweiligen Insert.

74
Der Western-Blot in Abb. 35 zeigt, daß beide Klone des neu-synthetisierten Vakzinia-
Virus rVV-X-adw ein intaktes X-Protein in der infizierten Zelle synthetisieren können.
Ebenso ist die Expression von HBx Subtyp ayw mit dem rekombinanten Vakzinia-Virus
rVV X-ayw in CV-1 Zellen möglich (Abb. 36). Die Expressionskinetik zeigt, daß eine
Antigenexpression auch schon zu einem sehr frühen Zeitpunkt nach der Infektion (8 h)
schwach nachgewiesen werden kann, was eine wichtige Voraussetzung für den Einsatz
der Vakzinia-Viren zur Herstellung von Zielzellen im Zytotoxizitätsassay war. Eine
erhöhte MOI bei der Infektion scheint keine verstärkte Expression zu bewirken (siehe
Abb. 36). Als Positivkontrolle wurden G-8 Zellen mit dem getesteten Plasmid pcD/3-X
transfiziert, in der Negativkontrolle wurden CV-1 Zellen mit dem Wildtyp Vakzinia-
Virus infiziert.
�//! ),! $2������ ��
�����/������� ������������
���������! Proteine aus CV-1
Zellen im Immunoblot mit HBx-
spezifischem Antikörper. Analyse
90h nach Infektion mit Klonen
rekombinanter Vakzinia-Viren.
Spur 1 und 2: Beide Klone von
rVV-pSc-11 exprimieren kein X-
Protein; Spur 3 und 4: Klon 1 und 2
des rekombinanten Virus rVV-X-
adw sind positiv; Spur 5:
Positivkontrolle HUH-7 Zellen mit
Plasmid pRc/CMV-X transfiziert.

75
Die Antigenexpression wurde weiterhin in verschiedenen syngenen Targetzellinien,
welche in CTL-Assays verwendet werden sollten, untersucht. Die beiden
Fibroblastenlinien MC57G (H-2b) und D2 (H-2d) zeigen eine starke Expression des X-
Proteins nach Infektion mit dem rekombinanten Vakzinia-Virus vom ayw-Subtyp bei
einer MOI von 10. Die Untersuchung der zeitabhängigen Expression konnte schon 9 h
nach Infektion eine deutliche Expression im Immunoblot nachgewiesen werden (siehe
Abb. 37). Diese Resultate sprechen für den Einsatz dieser Zellinien zusammen mit dem
Vakzinia-System zur CTL-Analyse muriner Lymphozyten.
Dagegen ließ sich in den für den CTL-Assay bewährten lymphoiden Zellinien P815 (H-
2d) und EL4 (H-2b) in wiederholten Experimenten mit MOIs zwischen 1 und 20 keine
funktionstüchtige Expression des X-Proteins durch rekombinante Vakzinia-Viren
nachweisen (Resultate nicht gezeigt).
�//! )-! 8����/������� $2������ �� �����/������� ������������ �������>�!
Immunoblot-Analyse von CV-1 Zellen mit HBx-spezifischem Antikörper. Spur 1:
Positivkontrolle G-8 Zellen transfiziert mit Plasmid pcD/3-X. Spur 2: Negativkontrolle CV-1
Lysat infiziert mit WT Vakzinia-Virus. Spur 3 und 4: CV-1 Lysate 8 h nach Infektion mit
rVV X-ayw mit MOI 10 (3) und MOI 20(4). Spur 5 und 6: CV-1 Lysate 22 h nach Infektion
mit rVV X-ayw mit MOI 10 (5) und MOI 20 (6).

76
)!#!*� F��������3����� �����/������� ��������������
Die rekombinanten Vakzinia-Viren sollten u.a. bei der in vitro Stimulation muriner T-
Lymphozytenkulturen eingesetzt werden. Dabei wurden infizierte syngene Splenozyten
als Stimulatorzellen verwendet. Innerhalb der 5- bis 6-tägigen Ko-Kultur-Phase besteht
jedoch das Risiko, daß replikationsfähige virale Partikel aus den Stimulatorzellen
freigesetzt werden und in der Folge zytopathisch auf die empfindlichen Lymphozyten
einwirken.
�//! )1! 8����/������� $2������ �� ��&������ �� =�������������� ��,1: �� �#!
Immunoblot aus Zellysaten von MC57G und D2 Zellen mit HBx-spezifischem Antikörper. Spur 1
und 2: HBx-Expression in D2 9 h bzw. 22 h nach Infektion mit rVV-X-ayw. Spur 3:
Negativkontrolle D2-Zellen mit Wildtyp VV infiziert. Spur 4-6: HBx-Expression in MC57G Zellen
6 h, 9 h und 22 h nach Infektion mit rVV-X-ayw. Spur 7: Negativkontrolle MC57G mit Wildtyp VV
infiziert. Spur 8: Positivkontrolle G-8 Zellen mit Plasmid pcD/3-X (ayw) transfiziert.

77
Aus diesem Grunde sollte die selektive Inaktivierung der viralen Replikation durch eine
minimale UV-Dosis getestet werden. Die optimale UV-Dosis wurde wie oben erwähnt
(siehe Kap. 2.4.7) durch Bestrahlung der Virussuspension von rVV-X-ayw mit
ansteigenden Energiedosen in einem UV-Cross-Linker (BIORAD) ermittelt. Parallel
wurde anschließend die Expression des X-Antigens in CV-1 Zellen und Replikation der
inaktivierten Viren durch lichtmikroskopische Untersuchung der Morphologie des CV-1
Monolayers untersucht. Die CV-1 Zellen wurden 24 h nach der Infektion mit RIPA-
Puffer lysiert und im Immunoblot mit dem monoklonalen, X-spezifischen Antikörper α-
X36 analysiert. Als Positivkontrolle wurde ein Lysat von CV-1 Zellen, welche mit dem
unbestrahlten rVV-X-ayw infiziert worden waren, eingesetzt. Mit Wildtyp Vakzinia-
Virus infizierte CV-1 Zellen wurden als Negativkontrolle getestet.
Wie in Abb. 38 zu sehen ist, fand schon bei der minimalen Inaktivierungs-Dosis von 300
mJ UV-Licht keine intakte virale Replikation der Viren mehr statt. Wurde die Dosis noch
weiter erhöht, liessen sich entsprechend keine Zeichen der Replikation bis zum Tag 10
nach Infektion nachweisen. In der Immunoblot-Analyse ist bei der Minimal-Dosis von
300 mJ eine im Vergleich zur Positivkontrolle schwächere, aber nachweisbare
Expression des X-Antigens vorhanden. Bei höheren Dosen konnte schließlich auch keine
Expression mehr gezeigt werden. In der Folge sollten die rekombinanten Vakzinia-Viren
daher mit einer Dosis von 300 mJ zum Einsatz bei der Lymphozytenstimulation bestrahlt
werden. Damit sollte die Expression des X-Antigens zur optimalen Präsentation auf der
Stimulatorzelle gewährleistet bleiben. Die virale Replikation wird bei dieser Dosis
jedoch wie beabsichtigt vollständig verhindert.

78
�//! )0! F��������3����� ��
�����/������� ������������
�������>�!
�C Immunoblot mit dem HBx-
spezifischen Antikörper α-X36.
Spuren 1-6: Lysate aus mit rVV-
X-ayw infizierten CV-1 Zellen.
Die Viren waren zuvor mit
Intensitäten zwischen 300 mJ und
1000 mJ im UV-Cross-Linker
bestrahlt worden. Spur 7: Positiv-
kontrolle mit unbestrahlten Viren.
Spur 8: Negativkontrolle mit
Wildtyp Vakzinia-Viren.
�C Analyse der Replikation UV-bestrahlter Vakzinia-Viren in CV-1 Zellen. Die im
konfluenten Monolayer infizierten Zellen wurden einmal pro Tag bis Tag 10 nach Infektion
lichtmikroskopisch auf Veränderungen der Morphologie und Plaque-Formation als Folgen
des zytopathischen Effektes replizierender Vakzinia-Viren untersucht.

79
4�4 �1 : :! �+#$71 &&$
)!)! �������2 �����I���� ��7� ��������������
Balb/c (H-2d) und C57BL/6 (H-2b) Mäuse wurden in unterschiedlichen Gruppen mit den
HBx-Expressionsplasmiden pcD/3-X vom ayw-Subtyp und pRc/CMV-X vom adw-
Subtyp mit bzw. ohne adjuvante Applikation der Zytokine IL-12 und IL-18 durch die
Zytokin-Expressionsvektoren pApIL-12p70 (freundlicherweise von Dr. V Zurawski,
Apollon, Malvern, PA, USA zur Verfügung gestellt) und pCI-sIL-18 (freundlicherweise
von Prof. Dr. J Reimann, Ulm zur Verfügung gestellt) immunisiert (siehe Kap 2.5.1).
Dabei wurden jeweils 100 µg DNA des X-Antigen-exprimierenden Plasmids und 50 µg
der Zytokin-Plasmide bzw. des „Leervektors“ pAp021 (Apollon, Malvern PA) pro Maus
intramuskulär injiziert. In der Kontrollgruppe wurden die Mäuse mit dem Plasmid
pcDNA3 immunisiert. Jede Gruppe erhielt 4 Wochen nach der ersten Immunisierung
eine Booster-Immunisierung nach dem gleichen Schema. Die Seren der Mäuse wurden
14 – 21 Tage nach der Booster-Gabe entnommen. In einer Verdünnung von 1 : 20
wurden sie dann auf HBx-spezifische Antikörper mit der ELISA-Methode getestet (siehe
Kap 2.5.3).
Die Balb/c Mäuse, welche mit beiden Subtypen des X-Antigens und adjuvanten
Zytokinen immunisiert wurden, zeigten eine deutliche Antikörperproduktion mit sehr
hohen Serokonversionsraten. Die Gruppen, die mit dem ayw-Plasmid pcD/3-X + pApIL-
12p70 sowie pcD/3-X + pCI-sIL18 und mit dem adw-Plasmid pRc/CMV-X + pCI-sIL-18
immunisiert wurden, zeigten eine Serokonversionsrate von 100 %. Dagegen konnte man
bei den mit X-Antigenplasmid alleine immunisierten Gruppen nur sehr schwache
Antikörperbildung und keine einheitliche Serokonversion beobachten. Das ayw-Plasmid
pcD/3-X induzierte überhaupt keine signifikanten Antikörper, und das adw-Plasmid
Gruppe pRc/CMV-X erzeugte nur teilweise schwache Antikörperantworten (60%
Serokonversionsrate).

80
Bei C57BL/6 Mäusen konnte allgemein nur eine sehr schwache Antikörperproduktion
beobachtet werden. In der Gruppe, welche mit dem ayw-Antigenplasmid pcD/3-X
immunisiert wurde, lag die Rate der Serokonversion bei 60%. Ebenso zeigte die Gruppe
mit dem adw-Plasmid pRc/CMV-X und dem Zytokin IL-18 eine 60%ige Serokonversion
bei überwiegend schwacher Antikörperproduktion. Ansonsten zeigten alle anderen
Gruppen keine signifikante Antikörperbildung, mit der Ausnahme einer einzelnen sehr
hohen anti-X Antwort in der Gruppe, welche mit dem adw-Plasmid pRc/CMV-X +
pApIL-12p70 immunisiert wurden. Insgesamt zeigt sich in diesen Gruppen ein sehr
uneinheitliches Bild, ohne eine eindeutige Aussage über eine tendenzielle Wirkung der
adjuvanten Zytokine. In den Gruppen der Kontrollmäuse wurden keine signifikanten
anti-X Antikörper nachgewiesen.
���������!��,���#�-.��%���
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
pcD/3-X pcD/3-X +IL-12
pcD/3-X +IL-18
pRc/CMV-X
pRc/CMV-X + IL-12
pcD/3 AK X36
���������������� �����
���������
Cut-off 0,95
�//! )+! �������2 �����I���� �� ;���� ��������������� ���/?7 ���! Mäuse-Seren in 5
Gruppen mit spezifischer Immunisierung. 1: 5 Mäuse mit pcD/3-X (ayw). 2: 3 Mäuse mit pcD/3-X
(ayw) + pApIL-12p70. 3: 5 Mäuse mit pcD/3-X (ayw) + pCI-sIL-18. 4: 5 Mäuse mit pRc/CMV-X
(adw). 5: 2 Mäuse mit pRc/CMV-X (adw) + pApIL-12p70. 6: Kontrollgruppe (2 Mäuse) mit
„Leervektor“ pcDNA3 immunisiert. 7: monoklonales Antiserum (α-X36) als Positivkontrolle.

81
In einem anderen Versuch wurden die anti-HBx Antikörpertiter bei verschiedenen
Verdünnungsstufen der Mäuse-Seren im Bereich von 1:20 bis 1:500 untersucht. Balb/c
und C57BL/6 Mäuse wurden zweimal mit jeweils 100 µg des Plasmids pRc/CMV-X
bzw. pcD/3-X i.m. immunisiert. Die Seren wurden 3 Wochen nach der letzten
Immunisierung entnommen. Jeweils eine Balb/c und eine C57BL/6 Maus wurde
einmalig mit rekombinantem X-Protein vom ayw-Subtyp ohne Freund-Adjuvans
subkutan immunsiert. Diese Mäuse zeigen im Vergleich mit den Plasmid-immunisierten
Mäusen keine signifikanten Antikörpertiter und können als Non-Responder unter diesen
Bedingungen angesehen werden. Bei den Plasmid-immunisierten Mäusen sind mit
beiden Subtypvektoren HBx-spezifische Antikörper bis zu einer Serumverdünnung von
1:100 signifikant nachweisbar, was auf niedrige Antikörpertiter hinweist (siehe Abb. 41).
���������!��,��/0��-1��%���
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
1,8
2
2,2
2,4
pcD/3-X pcD/3-X +IL-12
pcD/3-X +IL-18
pRc/CMV-X
pRc/CMV-X + IL-12
pRcCMV-X + IL-18
pcD/3 AK X36
���������������� �����
��
�23'
��*
Cut-off 0,95
�//! *4! �������2 �����I���� �� ;���� ��������������� �,1�E?- ���! Mäuse-Seren
in 6 Gruppen mit spezifischer Immunisierung. 1: 5 Mäuse mit pcD/3-X (ayw). 2: 3 Mäuse mit
pcD/3-X (ayw) + pApIL-12p70. 3: 5 Mäuse mit pcD/3-X (ayw) + pCI-sIL-18. 4: 5 Mäuse mit
pRc/CMV-X (adw). 5: 3 Mäuse mit pRc/CMV-X (adw) + pApIL-12p70. 6: 5 Mäuse mit
pRc/CMV-X (adw) + pCI-sIL-18. 7: Kontrollgruppe (4 Mäuse) mit „Leervektor“ pcDNA3. 8:
monoklonales Antiserum (α-X36) als Positivkontrolle.

82
)!)!#� ��� =�8��������(�������������� ��7� ��������������
Die in vitro T-Zellproliferation der in vivo mit den HBx vom adw -und ayw-Subtyp
sensibilisierten Effektorzellen wurde durch Stimulation mit rekombinantem X-Protein
vom ayw-Subtyp erreicht. Als Maß für die spezifische T-Zellproliferationsantwort wurde
die BrdU-Inkorporation in einem quantitativem ELISA (∆OD bei 450 nm) nach 48 h
Inkubation mit dem X-Antigen ausgewertet.
Zunächst wurde die Proliferationsaktivität nach DNA-Immunisierung mit HBx-
Plasmiden und einmaliger Immunisierung mit rekombinantem X-Protein vom ayw-
Subtyp ohne Freund-Adjuvans verglichen. Sowohl bei der Balb/c wie auch bei der
C57BL/6 Maus zeigte sich kein signifikanter Anstieg der Proliferationsantworten. In
���������!4�����%���!�����
0
0,5
1
1,5
2
2,5
3
3,5
4
1 :20 1 : 40 1 : 100 1 : 500
�������������������
���������
pcD/3-X C57BL/6 Maus 1
pcD3/-X Balb/c Maus 2
pRc/CMV-X C57BL/6 Maus 3
pRc/CMV-X Balb/c Maus 4
rHBx C57BL/6 Maus 5
rHBx Balb/c Maus 6
pc anti-X Serum 1:100
Cut-Off
Cut-Off
�//! * ! ���� ��2�����(�7�� �����I���� �� ����7������7��� ����A�����(��!
Insgesamt 4 C57BL/6 und Balb/c Mäuse wurden mit den Plasmiden pRc/CMV-X (adw) und
pcD/3-X(ayw) zweimal im Abstand von 14 Tagen immunisiert. Jeweils eine Balb/c und eine
C57BL/6 Maus wurde mit 5 µg rHBx(ayw) subkutan ohne Freund-Adjuvans immunisiert. Der
anti-HBxAg-spezifische ELISA wurde 3 Wochen nach der letzten Immunisierung durchgeführt.

83
dieser kleinen Gruppe wurden jeweils nur schwache Proliferationsantworten bei den mit
dem adw-Expressionsvektor pRc/CMV-X immunisierten Mäusen nachgewiesen, wobei
die C57BL/6 Maus einen deutlichen Anstieg bei erhöhter Konzentration des X-Antigens
zeigt (siehe Abb. 42).
In einer weiteren Gruppe wurden die T-Zellproliferationsantworten in C57BL/6 Mäusen
nach dreimaliger i.m. Immunisierung mit den HBx-Expressionsplasmiden beider
Subtypen im Abstand von jeweils 5 Wochen untersucht (siehe Abb. 43). Auch hier
zeigen sich überwiegend sehr schwache T-Zellantworten, wobei bei einer Maus, welche
mit dem ayw-Subtyp immunisiert wurde, ein starker Anstieg der Proliferationsaktivität
gemessen wurde.
0
0,05
0,1
0,15
0,2
0,25
0,3
0,35
0,4
0,45
0,5
���������������� ∆
�� ��� ��
pcD/3-XBalb/c
pRcCMV-XBalb/c
rHBx Balb/c pcD/3-XC57BL/6
pRc/CMV-XC57BL/6
rHBxC57BL/6
5#��*�6����6��%���
�!��##)��#� ��������
1 10
rHBx (µg / ml)
�//! *#! =�8��������(������� ��7� *0 � ;��������� ��� ���2 (c = 1 µg/ml bzw. 10 µg/ml).
2 Balb/c und 2 C57BL/6 Mäuse wurden mit jeweils 100 µg der Plasmide pRc/CMV-X (adw)
und pcD/3-X (ayw) zweimal i.m. immunisiert (6 d nach Bupivacain-Injektion). Zur Kontrolle
wurde jeweils eine Balb/c und C57BL/6 Maus mit 5 µg rekombinantem X-Protein (ayw-
Subtyp) ohne Freund-Adjuvans immunisiert. Die Stimulation der Lymphozyten wurde 3
Wochen nach der Booster-Immunisierung mit 5x105 Zellen pro Effektor angesetzt.

84
In einem weiteren Versuch wurden zwei größere Kohorten bestehend aus Balb/c bzw.
C57BL/6 Mäusen mit HBx-Expressionsplasmiden beider Subtypen und teilweise auch
adjuvant mit Expressionsvektoren der Zytokine IL-12 und IL-18 zweimal im Abstand
von 14 Tagen intramuskulär immunisiert (Schema siehe Kap. 3.3.1). 3 Wochen nach der
Booster-Applikation wurden die Proliferationsaktivitäten nach in vitro Stimulation mit
rekombinantem X-Antigen untersucht. In der Gruppe der Balb/c Mäuse, welche mit dem
ayw-Plasmid pcD/3-X immunisiert worden waren, zeigten sich bei der Mehrzahl der
Mäuse deutliche Proliferationsantworten (siehe Abb. 44). Die adjuvant-applizierten
Zytokin-Plasmide von IL-12 und IL-18 scheinen allerdings keinen signifikant positiven
Effekt auf die T-Zellproliferationsaktivität auszuüben. Weiterhin zeigten mit
„Leervektor“ vakzinierte Mäuse keine spezifische Proliferationsaktivität.
0
0,2
0,4
0,6
0,8
1
1,2
1,4
1,6
1,8
���������������� ∆
�� ��� ��
pcD/3-X pcD/3-X pRc/CMV-X pRc/CMV-X mock
��������
�!��##)��#� ���������/0��-1��%���
1 10
rHBx (µg / ml)
�//! *)! =�8��������(������� �,1�E?- ���! Es wurden jeweils 2 Mäuse mit 100 µg der
Plasmide pRc/CMV-X bzw. pcD/3-X 5 bis 6 d nach Bupivacain-Gabe (0,25%) immunisert.
Die Mäuse erhielten insgesamt 3 DNA-Immunisierungen im Abstand von je 5 Wochen. 14
Tage nach der letzten Immunisierung wurden die Lymphozyten mit rHBx (ayw) für 48 h mit 5
x 105 Zellen pro Ansatz stimuliert.

85
Diese Ergebnisse liessen sich mit dem adw-Plasmid pRc/CMV-X nicht reproduzieren.
Auch in der Gruppe der C57BL/6 Mäuse kam es zu keiner signifikanten HBx-
spezifischen T-Zellproliferationsantwort (Ergebnisse sind nicht abgebildet).
Insgesamt induziert das X-Antigen überwiegend schwache T-Zellproliferations-
antworten. Dabei scheint die Induktion von T-Zellproliferationsantworten gegen das X-
Antigen erfolgreicher in Balb/c Mäusen zu sein, was gut mit den anti-HBx
Antikörpertitern korreliert. Außerdem scheint das ayw-Protein beim Vergleich der
Subtypen auf der Ebene der T-Zellantworten Epitope mit stärkerer Immunogenität zu
besitzen.
0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
��6
!����
�)���
����� ∆
��
�2/'
��*
pcD/3-X pcD/3-X + IL-12 pcD/3-X + IL-18 mock
5#��*�6�
�!��##)��#� ����������#�-.��%���
1 10
rHBx (µg / ml)
3/5 = Anzahl der Responder-Mäuse
2/35/5
2/2
�//! **! =�8��������(������� ���/?7 ���. 3 Gruppen mit spezifischer Immunisierung: Gruppe
1 mit pcD/3-X (ayw); Gruppe 2 mit pcD/3-X (ayw) + pApIL-12p70; Gruppe 3 mit pcD/3-X (ayw)
+ pCI-sIL-18 sowie 1 Kontrollgruppe (2 Mäuse) mit „Leervektor“ pcDNA/3. Die
Proliferationsaktivität von 5 x 105 Zellen pro Effektor wurde nach 48 h Stimulation mit rHBx (c =
1 µg/ml bzw. 10 µg/ml) gemessen; dargestellt sind die Mittelwerte von Respondertieren.

86
)!)!)� 8>����2�7�� =�8�������3����
Nach der DNA-Immunisierung wurden die in vivo aktivierten CTLs durch eine 5- bis 6-
Tage dauernde in vitro Stimulation mit syngenen Splenozyten als Stimulatorzellen
expandiert. Da es bisher nicht gelungen war, eine syngene Targetzellinie mit stabiler
Expression des X-Proteins herzustellen, wurde dieses transient mit rekombinanten
Vakzinia-Viren in den permissiven Targetzellinien exprimiert. Hierzu wurden diese mit
den UV-inaktivierten Vakzinia-Viren rVV-X-adw und rVV-X-ayw infiziert und in einem
Effektor:Stimulator Verhältnis von 4:1 mit den Effektorlymphozyten eingesetzt.
Anschließend wurden die Effektorzellen in einem 6 Stunden 51Cr-Release-Assay auf
zytotoxische Aktivität hin untersucht. Die Lyse der syngenen Targetzellen MC57G (H-
2b) und Balb/3T3 (H-2d), welche mit rekombinanten Vakzinia-Viren infiziert wurden,
wurde mittels 51Cr-Freisetzung im Gamma-Counter gemessen. Es wurden
Effektor:Target (E:T) Verhältnisse von 100:1, 50:1 und 20:1 verwendet. Als Kontrolle
für die unspezifische Lyse wurden Targetzellen mit dem Vakzinia-Virus rVV-pSc11
infiziert.
In einem großen Versuch wurden zwei Kohorten mit unterschiedlichen Gruppen aus
C57BL/6 und Balb/c Mäusen mit den Vektoren pRc/CMV-X und pcD/3-X sowie
adjuvanten Zytokin-Expressionsvektoren für IL-12 und IL-18 vakziniert (Schema der
Immunisierung siehe Kap. 3.3.1). Die Mäuse erhielten insgesamt zwei i.m.
Immunsierungen nach einer Bupivacain-Vorabinjektion (0,25%). Im Chrom-Release-
Assay ließ sich weder bei den Balb/c- noch bei C57BL/6 Mäusen eine spezifische CTL-
Aktivität gegen die Targetzellen nachweisen, welche mit den HBx-rekombinanten
Vakzinia-Viren rVV-X-adw bzw. rVV-X-ayw infiziert worden waren (Daten sind nicht
gezeigt).
Eine weitere Gruppe C57BL/6 Mäuse wurde mit dem adw-Plasmid pRc/CMV-X und den
Expressionsplasmiden der Zytokine IL-12 und GM-CSF (siehe Kap. 3.3.1) intramuskulär
mit Bupivacain (0,25%) ko-immunisiert. 10 Tage nach der Vakzinierung wurde die
Stimulation der Milz-Effektorzellen angesetzt. Im Chrom-Release gegen Vakzinia-Virus
infizierte MC57G Targetzellen zeigte sich wieder keine spezifische CTL-Aktivität (siehe
Abb. 45).

87
Schließlich wurde auch der Versuch einer multiplen Booster-Immunisierung zur
Induktion von HBx-spezifischen CTLs unternommen. Eine Gruppe aus C57BL/6
Mäusen wurde mit den Plasmiden pRc/CMV-X (adw) und pcD/3-X (ayw) dreimal im 5-
wöchigen Abstand nach Bupivacain-Gabe intramuskulär vakziniert. 14 Tage nach der
letzten Boosterung wurden die Splenozyten zur Stimulation entnommen. Die CTL-
Aktivität wurde mit E:T Verhältnissen von 50:1 und 20:1 im Chrom-Release bestimmt
(siehe Abb. 46). Auch in diesem Versuch wurden keine spezifischen CTL-Antworten
gemessen.
0%
2%
4%
6%
8%
10%
12%
14%
16%
18%
������
Maus 1 Maus 2 Maus 3 Maus 4 Maus 5 mock
��������������� �������������������
���!����7��/0��-1��%���
100 : 1 - rVV-X-adw
20 : 1 - rVV-X-adw
100: 1- rVV-pSc11
20 : 1 - rVV-pSc11
E:T Ratio + Targetzellen
�//! *,! �=E�����3���� �� �,1�E?- ���� ��� $D= ����������� 3�� 44D �� #4D !
5 Mäuse wurden mit dem Plasmid pRc/CMV-X und den Zytokinen IL-12 und GM-CSF
einmal i.m. immunisiert. Eine Kontrollmaus wurde mit dem „Leervektor“ pRc/CMV2
immunisiert. Zur Messung der spezifischen CTL-Antwort wurden MC57G Targetzellen mit
dem Vakzinia-Virus rVV-X-adw infiziert. Die unspezifische Aktivität wurde durch die Lyse
rVV-pSc11-infizierter Targetzellen erfasst.

88
0%
2%
4%
6%
8%
10%
12%
14%
16%
������
pcD/3-X Maus 1 pcD/3-X Maus 2 pRc/CMV-X Maus 3 pRc/CMV-X Maus 4
���������������
���!����7��/0��-1��%���
50 : 1 rVV-X (ayw /adw )
20 : 1 rVV-X (ayw /adw )
50 : 1 rVV-pSc11
20: 1 rVV-pSc11
E:T Ratio + Targetzellen
�//! *-! �=E���������� ����� ������������ ��(������� ��2�����(�7�� B������
�>�?���C �� �����(�7�� B�����;7 C ��,1: =�����������! Effektorzellen aus
C57BL/6 Mäusen wurden nach dreimaliger DNA-Immunisierung mit den HBx-
Expressionsplasmiden pcD/3-X und pRc/CMV-X im Chrom-Release-Assay analysiert.

89
���8 �����
�� �--,1)0$+(' $ %$+ =0+!1 &=0$1 �$'() ) &*�*� +,& �13$2) !1
Diese Studie wurde vor dem Hintergrund der Entwicklung einer effizienten
Immuntherapie der chronischen HBV-Infektion (CHB) durchgeführt. Mit einem
Schwerpunkt im asiatischen Subkontinent sind weltweit mehr als 350 Millionen
Menschen chronisch mit HBV infiziert und durch schwerwiegende Komplikationen wie
akute Leber-Insuffizienz, Leberzirrhose und HCC bedroht. Leider hat sich der Pool von
HBV-Trägern, global gesehen, auch durch die Einführung prophylaktischer
Vakzinierungs-Programme in einigen Ländern mit hohem medizinischen Standard nicht
entscheidend verändert (53). Auch die augenblicklich verfügbaren antiviralen Therapien
mit Interferon und Lamivudine sind in ihrer Effizienz nicht zufriedenstellend und nur bei
einem Teil der chronisch infizierten Patienten mit Erfolg einsetzbar. Lamivudine kann
außerdem bei längerem therapeutischem Einsatz die Entstehung viraler Mutanten mit
geringerer Sensitivität für den Wirkstoff fördern (1, 201). Auf der Suche nach neuen,
effizienteren Therapien stehen insbesondere immuntherapeutische Ansätze im
Mittelpunkt, da die Chronizität durch eine insuffiziente Immunantwort gegen die
infizierten Hepatozyten hervorgerrufen wird. Die Entwicklung einer therapeutischen
Vakzine ist daher eine vielversprechende Strategie, um spezifische Immunantworten
gegen virale Partikel und Virus infizierte Zellen zu induzieren. Erste Resultate klinischer
Studien mit einer therapeutische Vakzinierung gegen HPV-Infektionen sind sehr
erfolgsversprechend (148).
Die Aktivität und die antigene Restriktion von CD4+ T-Helferzellen und CD8+
zytotoxischen T-Zellen sind wichtige Faktoren bei der Pathogenese der CHB. Es konnten
zahlreiche CTL-Epitope vor allem in den Hüllantigenen, dem Core-Antigen und der
viralen Polymerase charakterisiert werden (26, 185, 200, 208, 220). Im Verlaufe der
CHB sind die CTL-Antworten nach Isolation der CTLs aus peripherem Blut gegen alle

90
HBV-Proteine sehr schwach bzw. unterhalb der Nachweisgrenze (82), so daß man von
einer partiellen oder totalen Immuntoleranz gegen diese Antigene spricht. Diese „low-
level“ CTL-Aktivität ist nicht ausreichend, um das Virus vollständig zu eliminieren, ist
aber andererseits in der Lage, die Hepatitis zu unterhalten, da eine intrahepatische
Kompartimentalisierung der Effektorzellen besteht. Weiterhin scheint eine starke,
polyklonale, vorrangig gegen das Core- und HBe-Antigen gerichtete CD4+ T-
Helferzellantwort vom TH1-Subtyp zur Virus-Elimination erforderlich zu sein (128, 168,
274). Die komplette Kontrolle der Infektion gelingt sehr wahrscheinlich nur durch die
Induktion einer multispezifischen, polyklonalen humoralen und zellulären Immunantwort
gegen verschiedene HBV-Antigene. So ist das Risiko, eine CHB zu entwickeln, bei
immun-kompromittierten HBV-infizierten Patienten deutlich erhöht (176). Hinzu kommt
auch, daß HBV-Proteine selbst, hauptsächlich das HBeAg, die Polymerase,
wahrscheinlich jedoch auch das X-Antigen immunmodulatorische Funktionen ausüben
können und auch in extrahepatischen, lymphatischen Geweben wie u.a. in Mesenterial-
Lymphknoten, Milz, Niere und Testes nachweisbar sind (84, 299, 315). Andererseits
kann es im Verlaufe der CHB zur akuten Exazerbation der Erkrankung mit Aktivierung
der antiviralen, immunologischen Effektoren kommen. Dies kann dann zur
Viruseradikation mit Serokonversion zu anti-HBe und anti-HBs führen. Weiterhin ist
eine HBs-Serokonversion beim CHB-Patienten auch durch eine
Knochenmarkstransplantation von einem HBV-immunen Spender möglich (150). Diese
Beobachtungen lassen eine aktive Immuntherapie der CHB als einen vielversprechenden
Therapieansatz erscheinen. Theoretisch sollte eine therapeutische HBV-Vakzine eine
starke CD4+ zelluläre T-Helferzellantwort vom TH1-Subtyp und eine Stimulation der
CD8+ CTL-Aktivität induzieren, wobei die Immunantwort gegen Epitope eines oder
mehrerer HBV-Antigene gerichtet sein sollte.
Aufgrund der oben erwähnten Tatsachen und der Erkenntnis, daß die HBe- und HBs-
Serokonversion beide mit einer verbesserten Leber-Histologie, einer höheren
Überlebenswahrscheinlichkeit und einem erniedrigtem Risiko für die Entstehung eines
HCC assoziiert sind, wurden bisher vor allem therapeutische Vakzine, welche auf den
HBV-Hüllproteinen basieren sowie eine Core-CTL-Peptid Vakzine in klinischen Studien
evaluiert. Impfstoffe mit bereits nachgewiesener prophylaktischer Effizienz wie die
Protein-Vakzine Hepagene (MEDEVA Ltd., UK), welche aus den HBV-Hüllantigenen
HBsAg, Teilen des präS1-Antigens, dem präS2-Antigen und dem Adjuvans

91
Aluminiumhydroxid besteht, wurden ebenfalls als therapeutische Vakzine getestet. Es
konnten zwar starke TH1-gewichtete T-Zellantworten in Mäusen und Schimpansen
induziert werden, jedoch ließ sich in einer größeren humanen Phase-II-Studie in
Indonesien keine signifikante Effizienz dieser Vakzine nachweisen (213, 311). Zur Zeit
werden weitere HBV-Hüllprotein-Vakzine mit verschiedenen Adjuvanzien wie z.B. 3D-
MPL (Monophosphoryl Lipid A) oder QS21 (Quillaria Saponaria) in Phase-II-Studien
untersucht (154, 197, 284). Ein weiterer Ansatz ist die kombinierte Gabe von HBsAg und
anti-HBs Antikörpern, welche Komplexe bilden und dadurch humorale und zytotoxische
T-Zellantworten induzieren sollen (298). Darüber hinaus wurde eine Lipopeptid-Vakzine
bestehend aus einem HLA-A2.1 restringierten HBV-Core Peptid als CTL-Epitop und
einem adjuvanten Tetanus-Toxoid T-Helferzellepitop-Peptid untersucht, welches zwar
bei gesunden Individuen sehr starke CTL-Antworten induzieren konnte, jedoch bei CHB-
Patienten keinen therapeutischen Effekt zeigte (112, 286).
Eine neue, sehr effiziente Methode zur Induktion humoraler und zellulärer
Immunantworten stellt die auch in dieser Arbeit verwendete DNA-Immunisierung dar
(siehe Kap. 1.5). Vorteile dieser Methode sind die in vivo Synthese des kodierten
Antigens mit einem potenten Auto-Adjuvans durch CpG-Motive und ein
immunologisches Profil, welches in seiner Kinetik, Spezifität und den Isotypen einer
natürlichen Infektion sehr nahe kommt. Besonders hervorzuheben ist die starke T-
Zellaktivierung mit CTL- und TH1-gewichteten T-Helferzellantworten nach
intramuskulärer Applikation. Nachteilig sieht man das potenzielle Risiko der Plasmid-
Integration und Mutagenese und wahrscheinlich eine geringere Immunogenität der
Impfstoffe bei Primaten und Menschen im Vergleich zu Mäusen an.
In zahlreichen Studien wurde der Einsatz von Plasmiden, welche verschiedene HBV-
Hüllproteine kodieren, in verschiedenen Tiermodellen vor allem in Mäusen und auch in
Schimpansen untersucht (67, 94, 96, 97, 166). Hierbei ließen sich hohe anti-HBs
Antikörpertiter und starke zelluläre Immunantworten induzieren. In der ersten humanen
Studie der Phase I wurde ein HBsAg-kodierendes Plasmid mit der Gene-Gun-Methode
an gesunden Individuen untersucht. Mit einer relativ niedrigen Dosis von 0,25 µg
Plasmid-DNA ließen sich keine effizienten Antikörpertiter induzieren (262). Im
Mittelpunkt anderer präklinischer Studien stand das Core-Protein oder eine Kombination
von HBcAg und Hüllproteinen (94, 96, 145). Auch hier ließen sich im Mausmodell

92
starke Antikörperantworten und potente CTLs sowie eine TH1-Zytokinsekretion
induzieren. Außerdem besteht auch die Möglichkeit, durch Kombiantion mit Zytokin
kodierenden Plasmiden die T-Helferzellprofile selektiv in Richtung TH1 oder TH2 zu
modulieren (49, 241).
Darüber hinaus wurden auch alternative Delivery-Strategien wie der Einsatz retroviraler
oder adenoviraler Vektoren als immuntherapeutische Vakzine in Tiermodellen untersucht
(111, 228, 232). Vielversprechende Ergebnisse lieferte eine Studie, bei der HBV-
transgene Mäuse mit dendritischen Zellen vakziniert wurden. Es gelang hier eine
partielle Toleranzdurchbrechung mit schwacher Induktion von CTL-Antworten und
spezifischen Antikörpern (246). Ein attraktiver Ansatz wäre die Kombination einer
medikamentösen, antiviralen Therapie mit einer Immuntherapie, da sich eine Reduktion
der Viruslast vorteilhaft auf die Durchbrechung der Virus induzierten Immuntoleranz
auswirken kann (33). In einem Tierexperiment konnte bei WHV-infizierten Woodchucks
durch Vorbehandlung mit dem neu-entwickelten antiviralen Wirkstoff L-FMAU (1-(2-
fluoro-5-methyl-b-L-arabinofuranosyl) uracil) und nachfolgender Therapie mit einer
WHs-Antigen-Vakzine ein sehr breites Spektrum von T-Zellantworten gegen sämtliche
WHV-Antigene im Vergleich zur singulären Therapie mit der Antigen-Vakzine
nachgewiesen werden (59). Sollte sich ein ähnlicher Effekt auch bei einer Immuntherapie
von CHB-Patienten nachweisen lassen, wäre dies ein großer Schritt in Richtung der
Entwicklung einer therapeutischen HBV-Vakzine.
Allerdings scheint die Entwicklung einer beim Menschen therapeutisch effizienten
Vakzine sehr viel schwieriger als erwartet zu sein. Bisher konnte mit keiner der klinisch
getesteten Vakzine ein eindeutig positiver Therapie-Effekt bei der CHB nachgewiesen
werden. Gründe dafür sind wahrscheinlich weiterhin Unklarheiten über die ideale
Zusammensetzung einer solchen Vakzine und die große Variabilität virologischer,
klinischer und immunologischer Pathomechanismen der CHB. Auch ergeben sich
Schwierigkeiten, ein optimales Schema zur Administration sowie Therapie-Endpunkte
festzulegen, welche in der längerfristigen Perspektive klinische Relevanz besitzen. Über
die Art der Immunstimulation und die Antigenzusammensetzung einer solchen
therapeutischen Vakzine gibt noch keine allgemeine Übereinstimmung. Daß jedoch die
meisten der bisher getesteten Vakzine auf den Hüll-Antigenen basieren, ist auf die große
Erfahrung mit diesen Antigenen im Bereich prophylaktischer Vakzine und gute

93
Charakterisierung von Antigen-Epitopen auf B- und T-Zellebene (siehe oben)
zurückzuführen. Analog zur Serokonversion bei der natürlichen HBV-Infektion ist
wahrscheinlich auch eine starke CD4+ TH1- und CTL-Antwort für die Virus-Elimination
und Durchbrechung der immunologischen Toleranz bei der CHB erforderlich.
*! ! ��2 �� 8���������� ��� ����(�7��� ������������
Die Immunantworten gegen die beiden Nicht-Strukturantigene HBV-Polymerase und
HBxAg sind bisher noch nicht ausreichend charakterisiert und in ihrer Rolle als
Zielantigene für eine Immunstimulation untersucht worden. Es gibt jedoch einige
Hinweise, daß eine multispezifische, polyklonale Immunantwort mit einem sehr breiten
Antigen-Spektrum zur Toleranzdurchbrechung und Virus-Elimination bei der CHB
notwendig ist (siehe oben). Auch existieren Anhaltspunkte, daß eine
Kombinationstherapie aus antiviraler Therapie und Immuntherapie mit unterschiedlichen
Antigen-Zielstrukturen einer singulären Therapie überlegen sein könnte.
Aus diesen Gründen sollte in dieser Arbeit die Immunogenität des HBx-Antigens auf B-
Zell- und T-Zellebene in einem Tiermodell charakterisiert werden und der Einsatz dieses
Antigens als Zielstruktur einer therapeutischen Vakzine mit der Methode der DNA-
Immunisierung evaluiert werden. Die nachfolgenden Erkenntnisse stellen eine Basis für
die Untersuchung der Immunantworten gegen das X-Antigen im Hinblick auf eine
immuntherapeutische Strategie dar:
♦ In zahlreichen Arbeiten konnte die Expression von HBx im Lebergewebe bei
Patienten mit CHB, zirrhotischen Veränderungen und einem HCC nachgewiesen
werden. Dabei schwanken die quantitativen Angaben der Autoren sehr deutlich
zwischen einer 0 %igen bis über 80 %igen Expression von HBx in HBV-assoziierten
HCCs (109, 131, 291, 292, 318, 320). Um eventuelle Abweichungen aufgrund
unterschiedlicher Spezifitäten der zum Nachweis des X-Proteins in
immunhistologischen Untersuchungen eingesetzten Antikörper auszuschalten, wurde
die Expression in einer neueren Studie mit 11 verschiedenen monoklonalen und
polyklonalen Antiseren im Vergleich analysiert (256). Das Antigen konnte hier in
59% aller HCCs und 53% der zirrhotischen Veränderungen bei CHB mit sämtlichen

94
spezifischen Antikörpern nachgewiesen werden. Darüber hinaus findet man DNA-
Sequenzen des X-ORF innerhalb integrierter HBV-Sequenzen im Genom von HCC-
Zellen (169, 282) sowie intakte RNA-Transkripte des X-ORF in HCCs (69, 205). Die
HBx-Expression bleibt während des Prozesses der Tumortransformation, vom Fokus
präneoplastisch veränderter Hepatozyten über den Nodulus bis zum manifesten HCC
erhalten, während die HBsAg-Expression deutlich herabreguliert wird (13, 256, 269).
Diese Ergebnisse deuten auf eine selektive Expression von HBx im CHB-Patienten
mit einem HCC hin. Sollte es gelingen, spezifische, humorale und zelluläre
Immunantworten gegen Epitope des X-Proteins zu induzieren bzw. zu verstärken, so
könnte auf diesem Wege die Elimination präneoplastischer oder bereits
transformierter Hepatozyten sehr effektiv erfolgen.
♦ Das onkogene Potential von HBx konnte in zahlreichen in vitro Studien mit
verschiedenen Zellinien und auch in HBx-transgenen Mäusen demonstriert werden,
wobei letzteres nicht reproduziert werden konnte (135, 142, 160, 234, 267).
Allerdings ist der genaue Mechanismus bzw. die Rolle von HBx bei der HCC-Genese
bisher unbekannt und es gibt unterschiedliche Hypothesen darüber, wie HBx durch
Interaktionen mit anderen Proteinen und seine pleiotrope transaktivatorische Aktivität
auf zahlreiche Gene entscheidenden Einfluß auf die Karzinogenese ausübt (siehe
Kap. 1.4, 78, 165). Außerdem scheinen die transaktivatorischen Funktionen des
Proteins auch für die HBV-Replikation und Expression und damit auch für die
Aufrechterhaltung einer chronischen HBV-Infektion wichtig zu sein; im Woodchuck-
Modell gelingt die chronische Infektion nur bei intaktem X-ORF (43, 55, 321).
Interessant erscheint weiterhin die Tatsache, daß die transaktivatorische Potenz des
X-Antigens in der genomisch integrierten Form beim HCC vollständig erhalten bleibt
(12, 305). Insbesondere durch Alteration von intrazellulären Signal-
Transduktionskaskaden und Interaktion mit Tumor-Suppressorproteinen wie p53
scheint HBx die unkontrollierte Proliferation HBxAg-positiver Zellen zu stimulieren
und die Resistenz der Zellen gegen Apoptosestimuli zytotoxischer Zytokine zu
fördern (siehe auch Kap 1.2). Ein besonders effektiver Schritt bei der Bekämpfung
der chronischen HBV-Infektion und Durchbrechung der Multi-Step Karzinogenese
des HCCs wäre daher die Elimination HBxAg-exprimierender Hepatozyten durch
gezielte, spezifische Immunstimulation.

95
♦ Über die immunologische Charakterisierung des X-Antigens gibt es bereits einige
aufschlussreiche Studien. Der erste Anhaltspunkt für eine in vivo Expression des X-
Proteins gelang durch den Nachweis von Antikörpern gegen HBx in den Seren HBV-
infizierter Patienten (132). Einige Autoren kommen zu dem Ergebnis, daß die
Induktion einer HBx-Antikörperantwort mit dem HCC assoziiert ist (189, 211).
Andere können nur eine niedrige Inzidenz der anti-X Antikörper beim HCC
nachweisen, dafür aber häufig sehr hohe Titer bei CHB-Patienten ohne HCC (73,
115, 155, 156, 174). In einer Untersuchung zur Epitop-Feinspezifität der B-
Zellantworten gegen HBx konnte eine immundominante Region in der Hälfte des
Karboxy-Terminus des Proteins lokalisiert werden, welche mindestens zwei stärkere
Epitope enthält. Auch hier wurden die höchsten Titer bei Patienten mit CHB
beobachtet, wobei alle Antikörperantworten gegen mehr als ein Epitop gerichtet
waren und insgesamt polyspezifische Antikörperantworten gegen zahlreiche Epitope
gemessen wurden (252) In den Seren akut infizierter Patienten und asymptomatischer
Träger fanden sich auch hier nur Antikörperantworten mit niedrigeren Titern und
weniger zahlreichen Epitopen. Auf welche Weise das hauptsächlich zytoplasmatisch
lokalisierte X-Protein mit seiner sehr kurzen Halbwertszeit von 20-30 Minuten dem
Immunsystem präsentiert wird, bleibt jedoch weiterhin unklar (61, 237). Da es bisher
keine Anhaltspunkte für eine Sekretion des Nicht-Strukturproteins gibt, bleibt noch
die Möglichkeit der Freisetzung aus nekrotischen Hepatozyten. Wegen der
multispezifischen Antikörperantworten mit Epitopen, welche nahezu die gesamte
Peptid-Sequenz abdecken, ist auch eine Prozessierung und anschliessenden Peptid-
Präsentation sehr wahrscheinlich. In einer einzigen Human-Studie mit akut und
chronisch HBV-Infizierten konnten bisher zelluläre Immunantworten vom CD4+ T-
Zelltyp gegen Epitope des X-Proteins nachgewiesen werden (127). Bei 100% der
akut infizierten sowie 35% der chronisch infizierten Patienten wurde eine spezifische
T-Zellproliferation beobachtet, die jedoch im Vergleich mit dem HBcAg deutlich
schwächer ausfiel. Auch konnten verschiedene Epitope, welche wiederum
hauptsächlich in der Hälfte des Karboxy-Terminus der X-Sequenz lokalisiert sind,
durch Stimulation der T-Zellen mit synthetischen Peptiden identifiziert werden. Eine
bedeutende Rolle bei der Elimination Virus infizierter Zellen wird im allgemeinen
den CD8+ zytotoxischen T-Zellen zugeschrieben. Eine einzelne Studie zeigt bisher
das Vorhandensein von 5 HLA-A2.1 restringierten CTL-Epitopen bei chronischen
HBV-Trägern. Es wurde allerdings nicht demonstriert, daß es sich bei diesen

96
Epitopen um intrazellulär prozessierte Peptide handelt (51). Zusammenfassend läßt
sich sagen, daß es sowohl humorale Antikörper- als auch T-Zellantworten gegen
verschiedene Epitope des X-Proteins im Verlaufe einer HBV-Infektion gibt. Es
konnte keine eindeutige Assoziation der Stärke und Spezifität der Immunantwort mit
akuten, chronischen Verlaufsformen bzw. dem HCC nachgewiesen werden, doch gibt
es Hinweise, daß sich häufiger höhere Antikörpertiter bei chronisch Infizierten
beobachten lassen, während die CD4+ T-Zellantworten prozentual häufiger bei einer
akuten HBV-Infektion gemessen werden können. Die Gründe dafür sind unklar, auch
ist bisher nicht eindeutig geklärt, warum die humoralen wie auch die zellulären
Immunantworten im Vergleich mit anderen Strukturantigenen des HBV deutlich
schwächer ausfallen. Die bisher gewonnenen Daten geben aber trotzdem Anlaß zur
Hoffnung, durch eine gezielte, effektive Stimulation spezifische Immunantworten
gegen HBx zu induzieren bzw. bei chronisch infizierten Patienten zu verstärken, um
eine Toleranz des Immunsystems gegenüber diesem Antigen bei der CHB
therapeutisch zu durchbrechen. Dazu bedarf es allerdings weiterhin einer
eingehenden Charakterisierung der HBx-spezifischen Immunantworten, insbesondere
der CTL-Antworten.
♦ In verschiedenen Untersuchungen wurde der 26S Proteasom-Komplex als wichtige
funktionelle Zielstruktur des X-Antigens innerhalb der Wirtszelle identifiziert (83,
121, 122, 250, 319). Interaktionen des X-Proteins mit den Molekülen PSMA7, einer
α-Untereinheit der 20S Proteasomkomponente, und PSMC1, einer ATPase-
Untereinheit der 19S regulatorischen Proteasomkomponente, konnten dabei in vitro
und in vivo demonstriert werden. HBx selbst ist in Ubiquitin-gebundener Form ein
Substrat des Proteasomkomplexes und wird mit einer sehr kurzen Halbwertszeit
hydrolysiert. Weiterhin scheint die intakte Proteasom-Funktion notwendig für die
transaktivatorische Potenz des X-Antigens auf zahlreiche Promotoren zu sein.
Besonders hervorzuheben ist die Erkenntnis, daß HBx alle 3 wichtigen Peptidase-
Aktivitäten des Proteasom-Komplexes zu hemmen vermag und so die
Degradationskapazität Ubiquitin-gekoppelter Proteine als natürliche Substrate des
26S Proteasomes entscheidend reduziert wird; im besonderen ist die chymotrypsin-
artige Aktivität betroffen, welche die wichtigste enzymatische Kapazität der
Proteindegradation innerhalb der Zelle darstellt. Der Proteasom-Komplex ist als
zentrale intrazelluläre Stelle verantwortlich für die nicht-lysosomale

97
Proteindegradation in eukaryontischen Zellen (60). Folglich wird so eine bedeutende
Funktion des Proteasoms bei der Bekämpfung viraler Infektionen in der Zelle negativ
beeinflußt, nämlich die aktive Degradation viraler Proteine sowie die Bereitstellung
von antigenen Peptiden zur Präsentation im Kontext von MHC-Komplexen auf der
Zelloberfläche (226, 243). Insbesondere durch die Induktion von IFN-γ im
Frühstadium einer viralen Infektion wird die zelluläre Synthese von
Proteasomuntereinheiten stimuliert, um schließlich die Peptid-Hydrolysefunktion des
Proteasom-Komplexes zu steigern (85, 90, 162). HBx könnte also auf diesem Wege
die Proteolyse aller HBV-Antigene reduzieren. In der Folge könnte es seine eigene
sowie die Antigenpräsentation aller HBV-Antigene hemmen. Diese Folgerungen
stellen eine mögliche Erklärung für die Beinträchtigung der immunologische
Erkennung HBV-infizierter Zellen dar. In der Konsequenz unterliegen Zellen mit
genomischer Integration des X-ORF und permanenter Expression des X-Proteins
wahrscheinlich einem immunologischen Escaping. Um hier einen Angriffspunkt zu
bieten, könnten diese Hepatozyten durch gezielte Immunstimulation gegen Epitope
des X-Antigens eliminiert werden und so ein Escape Virus-infizierter Zellen bzw.
Zellen mit integriertem HBV-Genom im Stadium der CHB wirkungsvoll bekämpft
werden.
��� �9'$+ -$1)$""$+ �1&()8
Tiermodelle zum experimentellen Studium immunologischer Mechanismen einer HBV-
Infektion sind nicht verfügbar, da HBV nur für Menschen und Schimpansen in vivo
infektiös ist, aber nicht unter in vitro Bedingungen. Artverwandte Hepadnaviren wie
DHBV und WHV lassen nur Untersuchungen an Outbred-Spezies wie der Ente und dem
Woodchuck zu, deren Immunsystem nicht ausreichend charakterisiert ist, und so keine
eindeutige Analyse der zellulären Immunantworten gegen HBV-Antigene erlauben.
Um die Induktion humoraler und zellulärer Immunantworten gegen HBx zu untersuchen
und zu testen, inwieweit das X-Proteins als Zielantigen einer therapeutischen Vakzine
zum Einsatz kommen kann, wurde das Prinzip der DNA-Immunisierung mit
Expressionsvektoren des X-Antigens in einem murinen Inbred-System gewählt. Mehrere

98
Studien zeigten, daß diese Methode sehr effektiv ist, um eine humorale und zelluläre
Immunantwort gegen Antigene verschiedener viraler Pathogene, einschließlich HBV, zu
induzieren (siehe Kap 1.3 und Kap. 4.1). Offenbar scheint die DNA-Immunisierung im
Vergleich mit alternativen Vakzine-Strategien wie exogenem rekombinantem Protein
oder rekombinanten Vakzinia-Viren nicht nur effizienter bei der Stimulation von T-
Zellantworten gegen Antigene mit schwacher Immunogenität zu sein, sondern auch ein
deutlich breiteres Spektrum potenziell immunogener Epitope bei Protein-Antigenen zu
präsentieren (238, 239).
Da es uns bisher nicht gelungen war, stabile Transfektanten mit dem X-Protein Antigen-
präsentierender Zellinien in Haplotyp-syngenen murinen Zellen zu erzeugen, wurden
rekombinante Vakzinia-Viren als Target-System für die immunologische
Charakterisierung der CTL-Antworten gewählt. Dieses System bietet eine sehr hohe
Transfektionseffizienz in eukaryontischen Zellinien und ist wegen der endogenen
Prozessierung der exprimierten Antigene hervorragend zur Antigenpräsentation im
Kontext von MHC-Molekülen geeignet (227, 193).
��4 �1 : )+! �+#$71 &&$
Die erfolgreiche Klonierung des Expressionsplasmids pcD/3-X vom ayw-Subtyp und des
Vakzinia-Transfervektors für den X-ORF vom adw-Subtyp pSc11-X wurde durch
kommerzielle Sequenzierung beider Vektoren bestätigt. Dabei konnte für beide Vektoren
ein intakter ORF ohne Mutationen demonstriert werden. Ein weiterer, entscheidender
Faktor für die Effizienz von Zelltransfektionen in vitro und in vivo ist der Grad der
Plasmid-DNA Präparationen mit einer hohen Reinheit und einem hohen Anteil
geschlossen zirkulärer DNA (98 %) (66). Aus diesem Grunde wurden die Maxi-
Präparationen der Expressionsvektoren pcDNA/3-X und pRc/CMV-X nach erfolgter
Ethanol-/Isopropanolpräzipitation zum Teil einer weiteren Aufreinigung nach der
Methode der zweifachen Caesiumchlorid-Ultrazentrifugation unterzogen.
In mit den Expressionsplasmiden pRc/CMV-X und pcDNA/3-X transfizierten HUH-7
und G-8 Zellen konnte eine starke Expression des X-Proteins vom adw- und ayw-Subtyp

99
bis über 60 h nach Transfektion nachgewiesen werden. Die Selektion stabiler
Transfektanten der murinen, lymphoiden Zellinien P815 und EL4 mit den X-
Expressionsvektoren mittels Gentamycin-Selektion wurde dennoch trotz zahlreicher
Versuche nicht erreicht. Die Ursache ist sehr wahrscheinlich eine toxische Wirkung der
dauerhaft hohen Expression des Transaktivatorproteins HBx mit seinen pleiotropen
Wirkungen auf intrazelluläre Signal-Transduktionsmechanismen und nukleäre
Promotoraktivitäten der Zellen.
Die Herstellung der rekombinanten Vakzinia-Viren rVV-X-adw und rVV-pSc11 erfolgte
durch mehrfache sequentielle Plaque-Reinigung und kombinierte Selektion der Plaques
über das deletierte Thymidinkinase-Gen sowie das Chromogen X-Gal. Die hohe Reinheit
konzentrierter Maxi-Präparationen der rekombinanten Virusklone ist eine wichtige
Voraussetzung für die Infektion von eukaryontischen Zellinien zur Expression des
inserierten Gens. Es können nämlich nicht nur Verunreinigungen durch das
Selektionsverfahren per se, sondern auch Spontanmutanten im viralen Thymidinkinase-
Lokus mit einer veränderten Wachstumskinetik während der Selektion auftreten. Daher
wurden die rekombinanten Virusklone mit dem Plaque-Assay im CV-1-Monolayer und
ergänzend extrahierte virale DNA mit der PCR-Methode analysiert, welche mit hoher
Genauigkeit (99%) die Reinheit der Präparationen demonstrieren konnte (9, 163). Die
Synthese des X-Proteins durch die rekombinanten Vakzinia-Viren beider Subtypen
konnte mittels Infektionsstudien demonstriert werden. Zur Erzeugung von Targetzellen
muß das Antigen in MCH-Klasse-I exprimierenden Zelllinien endogen synthetisiert
werden. In den H-2d und H-2b restringierten, lymphoiden P815 bzw. EL4 Zellen konnte
nach Infektion mit den rekombinanten Vakzinia-Viren keine Expression des X-Proteins
nachgewiesen werden. Eine sehr hohe Synthese des X-Proteins wurde in den murinen
Fibroblasten MC57G (Haplotyp H-2b) und D2 (Haplotyp H-2d) mit den Vakzinia-Viren
schon zu einem sehr frühen Zeitpunkt nach Infektion demonstriert. Dies ist
wahrscheinlich Ausdruck der Aktivität des „compound early/late“ Promotors, unter
dessen Kontrolle das X-Protein in den infizierten Zellen exprimiert wird. Die beiden
Fibroblastenlinien eignen sich sehr gut zur MHC-Klasse-I restringierten
Antigenpräsentation bei der Stimulation muriner T-Lymphozyten mit rekombinanten
Vakzinia-Viren (10).

100
Die Suppression der viralen Replikation rekombinanter Vakzinia-Viren mittels UV-
Bestrahlung konnte entsprechend titriert werden, so daß die Expression des X-Antigens
in ausreichender Menge für die MHC restringierte Präsentation in den Targetzellen
erhalten bleibt. Dies ist eine wichtige Voraussetzung für den Einsatz der Vakzinia-Viren
bei der in vitro Stimulation muriner Lymphozytenkulturen, um eine Kontamination der
empfindlichen T-Zellen zu vermeiden (10, 134, 313). Diese Ergebnisse belegen die
Herstellung und erfolgreiche Testung von zwei rekombinanten Vakzinia-Viren als
effektive Werkzeuge zur immunologischen Untersuchung und Charakterisierung der
beiden häufigsten Subtypen des Hepatitis-B X-Proteins.
���� �1 : :! �+#$71 &&$
Die in vivo Ergebnisse dieser Arbeit unterstützen die von akut und chronisch mit HBV
infizierten Patienten gewonnenen Daten, daß sich das X-Protein auf B- und T-Zellebene
durch eine schwache Immunogenität auszeichnet. Mit der Methode der intramuskulären
DNA-Immunisierung lassen sich im murinen System abhängig vom genetischen
Haplotyp Antikörperantworten und zum Teil auch schwache T-Zellproliferationen
induzieren. Die Stimulation signifikanter CTL-Antworten gegen X-Epitope ist bei
Mäusen des Haplotyps H-2d sowie H-2b mit dieser Methode nicht gelungen.
*!*! ��2�����(�7�� �����I�������������
Die Antikörperproduktion wurde nach zweifacher intramuskulärer Plasmid-
Immunisierung untersucht. Die Daten zeigen signifikante Serokonversionsraten bei
zahlreichen Gruppen im Vergleich zu den Kontrollmäusen. Der Haplotyp der Mäuse
scheint Einfluß auf die Antikörperbildung auszuüben, da es bei den Gruppen mit Balb/c
Mäusen insgesamt zu höheren Serokonversionsraten im Vergleich mit den C57BL/6
Mäusen kommt; ähnliches wurde auch schon nach Vakzinierung von Mäusen mit HBV-
Hüllprotein exprimierenden Plasmiden beobachtet (62, 64). Dieser Effekt wird besonders
deutlich bei den Gruppen, welche adjuvant mit den Zytokinvektoren IL-12 und IL-18
immunisiert wurden. Es wurde kein signifikanter Unterschied bezüglich der beiden

101
Antigensubtypen ayw und adw beobachtet. Jedoch scheinen die beiden adjuvant
applizierten Zytokine IL-12 und IL-18 zu einem positiven Effekt auf die Serokonversion
und Stärke der Antikörperproduktion bei den Balb/c Mäusen zu führen. Die Höhe der
Antikörpertiter nach intramuskulärer Immunisierung mit beiden Subtyp-Plasmiden ohne
adjuvante Zytokinplasmide wurde in einer sehr kleinen Gruppe von Mäusen untersucht.
Hier zeigen sich insgesamt niedrige Antikörpertiter mit signifikanter
Antikörperproduktion bis zu einer Verdünnung der Seren von 1:100. Eine
Vergleichsgruppe wurde einmalig mit rekombinantem X-Antigen ohne Freund´sches
Adjuvans subkutan immunisiert und zeigte darauf keine Antikörperantworten.
Niedrige Antikörpertiter und partielle Serokonversion sind Effekte, welche auch in
verschiedenen Humanstudien beobachtet wurden. Schek et al. berichten, daß ein
bestimmter Prozentsatz HBV-Infizierter keine Antikörperreaktion gegen HBx im
Verlaufe seiner Infektion entwickelt (236). In einer Untersuchung zur Epitop-
Feinspezifität der humoralen Immunantworten konnten zahlreiche Epitope und auch eine
immundominante Region innerhalb der Protein-Sequenz identifziert werden, jedoch
zeigten sich auch sehr heterogene Spezifitäten der Antikörper bei HBV-Patienten. Bei
asymptomatischen HBV-Trägern und akut infizierten Patienten wurden nur sehr niedrige
Titer und wenige Epitope charakterisiert (252). Ursachen für die qualitative und
quantitative Heterogenität der Antikörperantworten könnten die schwache in vivo
Expression von HBx mit seiner kurzen Halbwertszeit, sowie eine schwache
Immunogenität des Proteins aufgrund seiner geringen Größe von nur ca. 17 kD mit
relativ wenigen immunogenen Epitopen sein. HBx wird wahrscheinlich nicht von der
infizierten Zelle sezerniert, sondern ist vielmehr hauptsächlich im Zytosol der Zelle als
Transaktivatorprotein lokalisiert, ein kleiner Anteil scheint auch mit nukleären
Membranen des Zellkerns assoziert zu sein (23, 61, 71, 133, 140, 256).
Über die Mechanismen der Präsentation von HBx bei der Induktion einer humoralen
Immunantwort gibt es keine definitiven Erkenntnisse. Die in dieser Arbeit erzielten
Ergebnisse unterstreichen eine T-Helferzell-abhängige Induktion der humoralen
Immunantwort. Es konnte gezeigt werden, daß bei den Responder-Mäusen vom H-2d-
Haplotyp mit signifikanter Antikörperproduktion auch eine stärkere T-
Zellproliferationsantwort gegen das X-Antigen vom ayw-Subtyp beobachtet werden.
Eine Abhängigkeit der humoralen Immunantworten vom genetischen Haplotyp wurde

102
schon in Studien über humorale Antworten gegen HBV-Hüllproteine und das Core-
Antigen bei verschiedenen murinen „inbred“ Stämmen demonstriert (177, 179). Ebenso
scheint auch beim Menschen eine genetische Prädisposition im Bereich der HLA-Allele
zu existieren, welche bestimmend für die Ausprägung der Antikörperantworten nach
Gabe einer rekombinanten HBV-Vakzine sind (2). Offensichtlich scheint die spezifische
Erkennung eines einzelnen T-Zell-Epitops in Abhängigkeit vom genetischen Haplotyp
zur Aktivierung einer starken T-Helferzellfunktion ausreichend zu sein, welche zu einer
Antikörperproduktion gegen mehrere unterschiedliche B-Zell-Epitope desselben oder
eines anderen HBV-Antigens führen kann (180, 181).
In dieser Arbeit wurde der Effekt einer erhöhten Serokonversionsrate und moderaten
Steigerung der Antikörperproduktion bei den Balb/c Mäusen durch die adjuvante Gabe
von Expressionsplasmiden der TH1-Zytokine IL-12 und IL-18 beobachtet. Kim JJ et al.
fanden eine signifikant erhöhte Antikörperantwort im Zusammenhang mit einer Ko-
Administration des IL-18 Gens, welches sich ansonsten eher durch eine potente
Induktion von IFN-γ sowie der NK-Zellaktivität auszeichnet (138). Hingegen gibt es über
den Einsatz von IL-12-Plasmiden, dem Prototyp der TH1-Zytokine, unterschiedliche
Aussagen über adjuvante Effekte auf die Antikörperproduktion. Chow et al. konnte in
einer Untersuchung mit dem HBsAg eine erhöhte IgM und IgG Produktion mit einer
verstärkten Induktion der IgG2-Subklasse beobachten, was auf eine Aktivierung von
TH1-Zellen hinweist (49). Andere Studien mit viralen Antigenen zeigen demgegenüber
einen negativen Effekt der Ko-Immunisierung mit IL-12 auf spezifische
Antikörperantworten bei simultaner Aktivierung von CD4+ T-Zellantworten vom TH1-
Subtyp und Verstärkung der CTL-Antworten (136, 249).
*!*!# =�8��������(����������������
Ein wichtiges Ergebnis dieser Studie ist die erfolgreiche Stimulation der T-
Zellproliferationsaktivität gegen HBx mittels DNA-Immunisierung im murinen System.
Nach zweimaliger intramuskulärer Immunisierung wurde in der Gruppe der Balb/c
Mäuse ein signifikanter Proliferationsanstieg beobachtet, wenn diese mit dem
rekombinanten X-Antigen vom ayw-Subtyp stimuliert wurden. Dies gelang jedoch nur
bei Mäusen, welche auch mit einem Plasmid vom ayw-Subtyp immunisiert worden

103
waren. In den adw-Gruppen zeigte sich keine signifikante Proliferationsaktivität nach
Stimulation mit dem ayw-Protein, so daß wahrscheinlich keine Kreuz-Reaktivität der
beiden Subtyp-Antigene auf T-Zellebene besteht. Allerdings reagierten in dieser Gruppe
zwischen 60 und 100 % der Balb/c Mäuse mit einer signifikanten Proliferationsantwort,
so daß auch hier Non-Responder vorlagen. Durch die adjuvante Applikation von IL-12-
und IL-18-Plasmiden scheint sich die Responder-Rate leicht zu erhöhen, auf die Stärke
der Proliferationsantworten wurde jedoch kein positiver Einfluß ermittelt.
Möglicherweise liegt diesen Beobachtungen der Effekt der Haplotyp-assozierten
Antigenrestriktion bei der Induktion von CD4+ T-Zellantworten über die MHC-Klasse-II
restringierte Präsentation des X-Antigens zu Grunde. Die Differenzierung der T-
Zellaktivität wurde nicht analysiert, jedoch könnte eine T-Helferzell vermittelte
Aktivierung HBx-spezifischer B-Zellen wahrscheinlich sein, da parallel zur
Proliferationsaktivität in der Gruppe der Balb/c Mäuse auch eine Induktion von
Antikörperantworten gemessen wurde.
Jung et al. berichten in der bis dato einzigen Studie über spezifische T-Zellantworten
vom CD4+ T-Zelltyp gegen HBx-Epitope bei akut und chronisch HBV-infizierten
Patienten. Ihnen gelang es, die T-Zellspezifität mit synthetischen Peptiden und
rekombinantem Antigen sowie durch die Etablierung von T-Zellklonen zu
charakterisieren. Die Stärke der Proliferationsaktivität fiel in dieser Untersuchung im
Vergleich mit dem hochimmunogenen Core-Antigen deutlich schwächer aus. Da keine
Differenzierung der CD4+ T-Zellantworten vorgenommen wurde, läßt sich auch in dieser
Arbeit nur über die Funktion der HBx-spezifischen T-Zellen in vivo spekulieren.
Allerdings scheinen sich einige der T-Zell Epitope mit bereits charakterisierten B-Zell
Epitopen zu überlappen, welche in einer immundominanten Region lokalisiert sind (127,
252). Untersuchungen zur B- und T-Zellfeinspezifität bei den HBV-Hüllproteinen mit
rekombinanten Vakzinen erbrachten aufschlußreiche Erkenntnisse über die Interaktion
von T-Helferzell- und B-Zellepitopen und die Abhängigkeit der T-Zellantworten vom
genetischen Haplotyp sowie vom Subtyp der viralen Antigene (178, 182, 183). So konnte
im murinen System u.a. gezeigt werden, daß die T-Zellerkennung von Epitopen des pre-
S2-Antigens je nach H-2 Haplotyp und adw bzw. ayw-Subtyp der Antigene variiert und
der Responder-Status der T-Zellen davon abhängig ist.

104
Bei der Untersuchung der Proliferationsaktivität weiterer Mäuse-Gruppen mit
modifizierten Immunisierungs-Schemata zeigten sich in der vorliegenden Arbeit nur bei
einzelnen Balb/c und C57BL/6 Mäusen überwiegend schwache Proliferationsantworten.
Zum Vergleich wurde eine kleine Gruppe (n=2) einmalig mit rekombinantem X-Protein
subkutan immunisiert. Es konnte keine signifikante Proliferationsaktivität bei diesen
Mäusen gemessen werden - ein weiterer Hinweis auf die schwache Immunogenität des
X-Proteins bzw. ein schlechtes Ansprechen auf eine singuläre Applikation.
Insgesamt gesehen ist dies die erste Untersuchung einer Induktion von zellulären
Immunantworten gegen HBx in einem murinen inbred-System. Die gemessenen
Resultate zeigen sehr schwache Antigen-spezifische T-Zellantworten nach mehrfacher
DNA-Vakzinierung im Vergleich mit den anderen HBV-Strukturproteinen. Eine
Abhängigkeit der T-Zellantworten vom genetischen Haplotyp und vom Virus-Subtyp
scheint wahrscheinlich zu sein. Darüber hinaus gibt es auch Hinweise auf die Interaktion
von T-Helferzellantworten auf die HBx-spezifische B-Zellaktivität. Um diese
Hypothesen zu bekräftigen, sind weitere Untersuchungen zur Epitop-Feinspezifität und
zum Zytokinexpressionsmuster der spezifischen T-Zelleffektoren erforderlich. Auch
bleibt noch die Klärung der schwachen Intensität der Immunantworten. Zum einen
könnte die in vivo Expression des X-Antigens nicht ausreichend für ein effektives
Priming sein, wobei in dieser Studie alle Expressionsvektoren bei der DNA-
Immunisierung mit starken CMV-Promotoren ausgestattet waren. Vorstellbar ist auch
eine eingeschränkte Immunogenität aufgrund der geringen Größe des X-Proteins mit
potenziell weniger Epitopen als z.B. beim Core-Antigen. Über die Zell-assoziierte
Antigenpräsentation von Epitopen des X-Proteins in HBV-infizierten Zellen gibt es
bisher auch keine Klarheit, so daß durch die zahlreichen intrazellulären Interaktionen und
Transaktivatorfunktionen des X-Proteins auch eine Inhibition von Funktionen der
Antigenpräsentation denkbar wären, wie z.B. der Prozessierung von Peptiden im
Proteasom-Komplex (s.o.).

105
*!*!) �=E����������
Der Nachweis muriner, HBx-spezifischer zytotoxischer T-Zellantworten nach in vitro
Stimulation der Lymphozyten mit Vakzinia infizierten syngenen Splenozyten konnte im
Chrom-Release-Assay nicht gezeigt werden. Es wurden mehrere Versuche mit
unterschiedlichen Prime/Boost Strategien durchgeführt, zunächst erhielten die Mäuse
zweimal i.m. DNA-Immunisierungen nach Vorab-Injektion von 0,25 %igem Bupivacain.
Die Mäuse wurden je nach Kohorte auch mit IL-12 und IL-18 Expressionsplasmiden ko-
immunisiert. Eine weitere Gruppe wurde mit IL-12 und GM-CSF ko-immunisiert.
Schließlich wurde auch eine mehrfache Immunisierung mit 2-maliger Boosterung
getestet. Es wurden jeweils Mäuse beider Haplotypen H-2b und H-2d mit beiden
Antigensubtypen HBx-adw und HBx-ayw untersucht.
In keiner der untersuchten Kohorten zeigte sich eine HBx-spezifische CTL-Aktivität
gegen eines der Antigensubtypen. Allerdings konnten bei einzelnen Individuen
unspezifische Lyse-Aktivitäten mit dem Chrom-Release-Assay dargestellt werden,
welche wahrscheinlich auf eine NK-Zellaktivität (Natürliche Killerzellen) in der
untersuchten Lymphozyten-Population zurückzuführen sind. Dies ist ein deutlicher
Hinweis, daß der Cr-Release-Assay selbst fehlerfrei durchgeführt wurde. Auch wurden
entsprechend hohe Effektor:Target (E:T) Verhältnisse von bis zu 100:1 gewählt, welche
zum Nachweis schwacher spezifischer Aktivitäten notwendig sind.
Eine mögliche Erklärung für den fehlenden Nachweis spezifischer CTL-Aktivitäten in
dieser Arbeit ist der Einsatz des Vakzinia-Virussystems bei der Erzeugung von
spezifischen Stimulator- und Targetzellen zur in vitro Stimulation und Messung der
spezifischen Lyse durch die Effektorzellen. Auch unter Verwendung einer entsprechend
hohen Multiplizität zur Infektion der Stimulator- und Targetzellen kommt es
wahrscheinlich nicht zur Expression der Antigene in der Gesamtzahl der eingesetzten
Zellen, wie es bei einem stabilen Transfektanten der Fall ist. Trotz der sorgfältigen
Titration der UV-Inaktivierung zur Unterdrückung der Virusreplikation kann zudem
nicht 100%ig ausgeschlossen werden, daß während der 5-tägigen Stimulationsphase eine
Virus induzierte Zytotoxizität in der Zellkultur auftritt. Nimmt man diese Faktoren
zusammen mit der bereits oben diskutierten, schwachen Immunogenität des X-Antigens
(siehe Kap. 1.4., Kap. 4.1.1), so läßt sich daraus folgern, daß eventuell schwache CTL-

106
Antworten durch ein nicht optimal sensitives Detektionssystem nicht nachgewiesen
werden konnten. Insbesondere der Nachweis von humoralen und T-Helferzellantworten
gegen das HBx-Antigen in dieser Arbeit konnte demonstrieren, daß ein effizientes
Priming mit der Methode der DNA-Immunisierung zumindest auf der Ebene der B-
Zellen und CD4+ T-Zellen gegen HBx im murinen System möglich ist. Unterstützt wird
diese Hypothese durch den Nachweis von durchgängig sehr schwachen CTL-Antworten
gegen das murine AFP-Antigen in der Maus mit dem Vakzinia-Virussystem bei der
Stimulation der Effektorzellen und beim Einsatz als Targetzellen im 51Cr-Release-Assay
(103). Auch konnte bereits bei anderen HBV-Antigenen gezeigt werden, daß die
Methode der DNA-Immunisierung beim Priming von CTL-Antworten gegen schwach-
immunogene Anitgene im murinen System anderen Vakzine-Strategien durchaus
überlegen sein kann (238, 239). Die Haplotypen-spezifische MHC-Restriktion der HBx-
Epitope könnte ebenfalls ein Problem bei der Antigenerkennung bzw. dem Priming von
CTLs sein. Weitere Studien im Labor von Dr. Geißler mit spezifischen H-2 Konsensus-
Peptiden zur in vitro Stimulation der murinen Lymphozyten konnten aber ebenfalls keine
signifikanten H-2d oder H-2b restringierten CTL-Antworten nachweisen.
Eine weitere mögliche Erklärung für die schlechte Immunogenität von HBx auf der CTL-
Ebene liefert die jüngst bekannt gewordene Interaktion des X-Proteins mit dem
Proteasom-Komplex (Kap. 1.4.1, Kap. 4.1.1, 121, 122, 319). Durch eine Hemmung der
Peptidase-Funktionen des Komplexes könnte das X-Protein die Bereitstellung von
Antigenpeptiden zur Präsentation im Kontext von MHC-Komplexen auf der
Zelloberfläche verhindern. Für diese Hypothese spricht, daß es bisher nicht gelungen ist,
CTL-Antworten im humanen System auf dem Wege der endogenen Prozessierung des X-
Proteins nachzuweisen (51). Die in der vorliegenden Arbeit durchgeführte in vitro
Stimulation und die CTL-Assays funktionieren mit dem Vakzinia-Vektorsystem
ebenfalls auf dem Weg der endogenen Antigenprozessierung. Die postulierte Inhibition
der Peptidase-Aktivitäten des Proteasom-Komplexes könnte so als zentrale Schaltstelle
der Antigenprozessierung die MHC-Klasse-I restringierte Präsentation von
immunogenen Peptiden des X-Proteins ausschalten und damit die spezifische Erkennung
und konsekutive Lyse durch aktivierte zytotoxische T-Zellen verhindern. Zur Erhärtung
dieser Hypothese sind aber weitere Studien in in vivo Modellen notwendig.

107
Daher wäre es ein vielversprechender Ansatz, den kürzlich entdeckten, sogenannten
alternativen Weg der Antigenprozessierung und Peptidbeladung der MHC-Klasse-I
Moleküle („alternate pathway“) zur Präsentation des X-Antigens auszunutzen. Dabei
können Antigene gebunden an das zytoslische 70-kDa schwere Hitzeschockprotein
hsp73 direkt über eine Prozessierung im Post-Golgi-Kompartement auf MHC-Klasse-I
Moleküle geladen werden (224, 240). Auch exogen zugeführtes Antigen bzw.
Peptidfragmente eines Antigens können so der endogenen Prozessierung und MHC-
Klasse-I Präsentation zugeführt werden (258). Mit Hilfe von Fusionsproteinen, bestehend
aus Antigen bzw. immunogenen CTL-Epitopen eines Antigens und dem hsp70-
Hitzeschockprotein aus dem Mycobacterium tuberculosis, entweder in als Protein- oder
DNA-Vakzine bzw. mit anderen Delivery-Systemen appliziert kann die zelluläre, CD8+
Immunität deutlich gesteigert und eine bis zu 30-fache Potenzierung der MHC-Klasse-I
restringierten Präsentation erreicht werden (44, 157, 259, 260). Diese Strategie wird
augenblicklich im Labor von Dr. Geißler in Zusammenarbeit mit Prof. Reimann,
Universität Ulm verfolgt.
��� �,2,13)&'$+&'$2) :$1
Wie bereits oben dargelegt, ist die Induktion einer starken, CD8+ zytotoxischen T-
Zellantwort besonders wichtig für die Entwicklung einer erfolgversprechenden
Immuntherapie der chronischen Hepatitis-B-Virus Infektion. Die in dieser Arbeit
durchgeführten Untersuchungen konnten mit der Methode der DNA-Immunisierung im
Mausmodell keine signifikanten, spezifischen CTL-Antworten gegen das X-Antigen im51Chrom-Release-Assay nach in vitro Restimulation der murinen Lymphozyten
nachweisen. Da dies möglicherweise ein Problem der Sensitivität des Assays per se bzw.
des Vakzinia-Vektorsystems zur Infektion der Stimulator-/Targetzellen sein kann,
könnten sich hier neue, hoch-effiziente Methoden für zukünftige Versuche zur Messung
MHC-Klasse-I restringierter CD8+ T-Zellen anbieten. Solche Systeme können deutlich
sensitiver als der 51Cr-Release-Assay direkt ex vivo Antigen-spezifische CTLs bei sehr
niedrigen Prekursorzell-Zahlen ohne zusätzliche in vitro Restimulation bereits nach
einmaliger i.m. Applikation einer DNA-Vakzine nachweisen. Dazu gehört u.a. der
ELISPOT-Assay, welcher Epitop-spezifische, zytotoxische T-Zellen über die lokale

108
Produktion bestimmter Zytokine, u.a. IFN-γ, die durch einen Antikörper-gekoppelten
Substrat-Chromogen Komplex visualisiert werden, quantifizieren kann (171, 263).
Vergleichende Arbeiten zeigen, daß diese Assays etwa 10- bis 100-fach sensitiver im
Nachweis von CTLs als bisher eingesetzte Methoden sind, da solche nur die Fraktion der
CTLs messen, welche in vitro proliferieren können und wahrscheinlich nur den T-
Gedächtniszellen entsprechen (194).
Ein weiteres, erst kürzlich entwickeltes Verfahren zur quantitativen und funktionellen
Analyse MHC-Klasse-I restringierter T-Zellen ist das System der MHC-Peptid
Tetramere (3, 35, 91). Diese Technik basiert auf der Multimerisierung von MHC-Peptid
Komplexen, welche so eine erhöhte Gesamtavidität zu den T-Zellrezeptoren besitzen.
Diese werden zur Antigen-spezifischen Markierung von zytotoxischen T-Zellen über die
Interaktion MHC-Klasse-I Molekül und T-Zellrezeptor benutzt. Die markierten T-Zell-
Tetramere können schließlich in der Durchflußzytometrie identifiziert und quantifziert
werden und über zusätzliche Antikörpermarkierungen wahlweise phänotypisch und
funktionell (Oberflächenmarker, Zytokinrezeptoren etc) sowie in ihrer klonalen Spezifität
analysiert werden. Mit dieser sensitiven Methode liess sich u.a. zeigen, daß die Zahl
Antigen-spezifischer T-Zellen während einer akuten Infektion viel größer ist als bisher
angenommen wurde; sie umfaßt beispielsweise bei der LCMV-Infektion von Mäusen bis
zu 50% des gesamten Repertoires an CD8+ T-Zellen. Mit dieser Technik können so
elegant und sehr schnell Studien zur T-Zellimmunität an direkt ex vivo entnommenen
Lymphozyten durchgeführt werden. Voraussetzung ist allerdings die Kenntnis
immundominanter Epitope eines Antigens, da die Tetramere Epitop-spezifisch hergestellt
werden müssen. Daher wäre es zunächst sinnvoll, über die Konsensusanalyse bestimmter
Haplotypen solche immundominanten Regionen innerhalb der Peptidsequenz des X-
Proteins zu identifizieren. So könnten dann die T-Zellpopulationen nach Priming mit
einer HBx-Vakzine auf eine Peptid-spezifische T-Zellaktivität untersucht werden. Dies
liesse sich mit einem herkömmlichen 51Cr-Release-Assay nach Peptid-spezifischer in
vitro Stimulation bzw. mit dem ELISPOT-Assay durchführen. Zur Optimierung des T-
Zell Primings könnte man schließlich auch die Vakzine selbst modifizieren, u.a. mit den
oben beschriebenen hsp70-Fusionsvektoren oder mit einer starken, TH1-gewichteten
Induktion durch entsprechende Ko-Immunisierung (siehe Kap. 1.5.2).

109
� �����9��� ��
In der vorliegenden Arbeit wurde die Immunogenität des Hepatitis-B X-Antigens im
murinen System mit der Methode der intramuskulären DNA-Vakzinierung im Hinblick
auf die Entwicklung einer effizienten Immuntherapie der chronischen HBV-Infektion
sowie des HBV-induzierten HCCs untersucht. Das X-Antigen spielt wahrscheinlich über
zahlreiche Interaktionen eine wichtige Rolle bei der Etablierung der chronischen HBV-
Infektion und der HCC-Genese. Im Serum HBV-infizierter Patienten wurden bereits
einzelne Epitope des X-Antigens auf humoraler und T-Helferzell-Ebene in verschiedenen
Studien charakterisiert. Neuerdings gibt es auch Hinweise für HBx-spezifische CTL-
Antworten im humanen System. Zunächst wurden HBx-Expressionsplasmide der beiden
häufigsten HBV-Subtypen ayw und adw kloniert und die entsprechenden Antigene in
eukaryontischen Zellen exprimiert. Um eine effiziente MHC-Klasse-I Präsentation des
X-Antigens zu erreichen, wurden HBx-spezifische Vakzinia-Viren mittels homologer
Rekombination hergestellt und selektioniert. Die Expression der rekombinanten
Vektoren wurde in murinen Haplotyp-syngenen Zellinien getestet. Weiterhin wurde die
optimale UV-Dosis zur Inaktivierung der Replikation rekombinanter Vakziniaviren
titriert. C57BL/6 und Balb/c Mäuse wurden mit den HBx-Expressionsvektoren sowie
adjuvant mit den Zytokin-Vektoren IL-12 und IL-18 immunisiert. Die Antikörper-
Analyse immunisierter Mäuse mit dem ELISA-Verfahren zeigte eine präferentielle
Induktion von HBx-spezifischen Antikörpern in Balb/c Mäusen mit leicht erhöhter
Serokonversionsrate bei Ko-Administration von IL-12- und IL-18. Bei Balb/c Mäusen
wurden überwiegend schwache T-Helferzellantworten nach Immunisierung mit dem
ayw-Subtyp beobachtet. Insgesamt konnte die gleichzeitige Induktion einer signifikanten
Antikörperantwort sowie einer T-Helferzellantwort bei Balb/c Mäusen mittels DNA-
Immunisierung demonstriert werden. Der Nachweis einer signifikanten CD8+
zytotoxischen T-Zellantwort gegen HBx-spezifische Epitope beider Subtypen ist weder
in Balb/c noch in C57BL/6 Mäusen mit der Methode der intramuskulären DNA-
Immunisierung gelungen. Mit der Herstellung rekombinanter Vakzinia-Viren und der
Klonierung von Expressionsvektoren der beiden X-Antigene liefert diese Arbeit wichtige
Grundlagen für zukünftige immunologische Studien mit dem X-Antigen.

110
����� ��������
1. ����� ��% �������� �% ������ �5% �� ��! (1998) Identification and
characterization of mutations in Hepatitis B virus resistant to Lamivudine.
Hepatology #1: 1670-1677.
2. ����� ��% '����� �;% ���7������> �% ���3�� �$% '��� �H% ����� ;H%
������� HE% ����� 8% ��� J�� $H (1989) Genetic prediction of nonresponse
to hepatitis B vaccine. N Engl J Med )# : 708-712.
3. ������ H% �� &% :����� &% ����7� �% �7��>����5������ �% ���� H%
�7��7���� � ��� ��3� � (1996) Phenotypic analysis of antigen-specific T
lymphocytes. Science #1,: 94-96.
4. ���� '% ����>��� =% :������ E:% 5���� ;% ;7����/�� .�% ;7���7�� �H%
���� ;�% ��� ������ <� (1993) Mechanisms of class I restricted
immunopathology. A transgenic mouse model of fulminant hepatitis. J Exp Med
10: 1541-1554.
5. ���� '% :������ E:% 5���� ;% ������� =% ����� :% ����>��� =%
;7����/�� .�% ;7���7�� �H% ���� ;�% ��� ������ <� (1994) Class I-restricted
cytotoxic T lymphocytes are directly cytopathic for their target cells in vivo. J
Immunol ,#: 3245-3253.
6. ��� �5% '��3������ �% 5>��� H% ��� '�3�7 H� (1996) Nucleic acid
vaccination against Toxoplasma gondii in mice. J Eukaryot Microbiol *): 117S.
7. ������ �&% ;7����� �5% ����� ;.% ;���� ;E% '�����% �:% ������ �E%
;��/��� �% �� �H% '��(( $% ��� ;7���> =� (1994) Dominant selection of an
invariant T cell antigen receptor in response to persistent infection by Epstein-Barr
virus. J Exp Med 04: 2335-2340.
8. �(���� �% ��� ;7������� .H (1990) The hepatitis B virus X-gene product trans-
activates both RNA polymerase II and III promoters. EMBO J +: 497-504.

111
9. �/�� <�% ����� .% '������ .$% ����� ��% ;������ H:% ;���� H�% ���
;���� ' (1995) Current Protocols in Molecular Biology. John Wiley & Sons, Inc.,
New York.
10. ��7����� �< (1997) Evaluation of vaccinia virus-specific cytotoxic T cell
responses. In: Immunology Methods Manual (I. Lefkovits, Hrsg.), Academic Press
Ltd, London, Chap 28.3 pp. 1909-1917.
11. ��������� �E% ��>�� H�% H�3����� ��% ����������� �% ����% '% '�� :%
;��� H% 5��� �% ��� 5����� �� (1997) Safety and immunogenicity of
intramuscular and intravaginal delivery of HIV-1 DNA constructs to infant
chimpanzees. J Med Primatol #-: 27-33.
12. ������ �% ������ 9% ������ �% ��3��� �% 8���� �% :���/�� :% ������ :%
������ &% ��� E�3���� � (1994) Hepatitis B virus X gene product acts as a
transactivator in vivo. J Hepatol # : 103-109.
13. �����7� & (1996) Pathogenesis of hepatocellular carcinoma: sequential cellular,
molecular, and metabolic changes. In: Boyer JL, Ockner RK (eds.) Progress in
Liver Diseases. Vol. 14. Philadelphia: Saunders, pp. 161-197.
14. ������3 �% :�����3 &% '����3 �% 9������� 9% ��������3% �% �3�7����� =%
;�/���> �% ��� ������3 � (1999) Local and distant transfection of mdx muscle
fibers with dystrophin and LacZ genes delivered in vivo by synthetic microspheres.
Gene Ther -: 1406-1414.
15. �����/� �% <���7� �% &����� �% ���3���� .% �� &% :% ����� �E% ;������� �%
������ �% ����3��� �;% ��� �������� : (1994) Selective expansion of
cytotoxic T lymphocytes with a CD4+CD56+ surface phenotype and a T helper
type 1 profile of cytokine secretion in the liver of patients chronically infected with
Hepatitis B virus. J Immunol ,#: 3074-3087.
16. ����� 5� (1994) PCR amplification of up to 35-kb DNA with high fidelity and
high yield from lambda bacteriophage templates. Proc Natl Acad Sci USA + :
2216-2220.

112
17. ����7� ��% ;����� ;% ;����/��� =�% 8���� ��% &���> ��% ��3�� �$%
<���� ��% ���� �% ;���� =�% ;��3�� H5% ��� E��3�� �E (1998)
Augmentation and suppression of immune responses to an HIV-1 DNA vaccine by
plasmid cytokine/Ig administration. J Immunol - : 1875-1882.
18. ����> ��% E�� 5�% ��� H������ ;� (1995) Protection against mycoplasma
infection using expression-library immunization. Nature )11: 632-635.
19. ������7������ .% ��� ;7������ � (1992) Hepadnaviral assembly is initiated by
polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J
: 3413-3420.
20. ��� ��% ��� ��������� � (1996) An essential role for NF-kappaB in preventing
TNF-alpha-induced cell death. Science #1*: 782-784.
21. ���� H% ��� ;7������� .H (1994) Hepatitis B virus HBx protein activates Ras-
GTP complex formation and establishes a Ras, Raf, MAP kinase signaling cascade.
Proc Natl Acad Sci USA + : 10350-10354.
22. ���� H% ��� ;7������� .H (1995) Hepatitis B virus HBx protein deregulates cell
cycle checkpoint controls. Proc Natl Acad Sci USA +#: 11215-11219.
23. ���� H% ; <% ����� �% ��� ;7������� .H (1996) Hepatitis B virus HBx protein
induces transcription factor AP-1 by activation of extracellular signal-regulated and
c-Jun N-terminal mitogen-activated protein kinases. J Virol 14: 4978-4985.
24. ������� H.% J������ H5% ;���� :E% ������ �% ��� �� � (1984)
Recombinant vaccinia virus primes and stimulates influenza haemagglutinin-
specific cytotoxic T cells. Nature ) : 578-579.
25. ��������� �% �������� .% ��� 5����� . (1985) One hundred base pairs of 5’
flanking sequence of a vaccinia virus late gene are sufficient to temporally regulate
late transcription. Proc Natl Acad Sci USA 0#: 2096-2100.
26. ���������� �% <������ �% <��77����� <% &���� �% ��������� .% ;7���7�� �H%
<����� &% :����� ;% ��� ������ <� (1991) HLA class I-restricted human

113
cytotoxic T cells recognize endogenously synthesized hepatitis B virus
nucleocapsid antigen. Proc Natl Acad Sci USA 00: 10445-10449.
27. ���������� �% ������� �% ������ <�% E�3���� �% ������% �% ;���� �% &���� �%
:�/���� =% <��77����� <% ��� <������ � (1994) Cytotoxic T lymphocyte
response to a wild type hepatitis B virus epitope in patients chronically infected by
variant viruses carrying substitutions within the epitope. J Exp Med 04: 933-943.
28. ���������� �% ;���� �% ������ <�% &���� �% E�3���� �% �� ����� �% <��77�����
<% ��� <������ � (1994) Natural variants of cytotoxic epitopes are T-cell receptor
antagonists for antiviral cytotoxic T cells. Nature )-+: 407-410.
29. ���������� �% ��� ����� �' (2000) Protection or damage: a dual role for the
virus-specific cytotoxic T lymphocyte response in hepatitis B and C infection. Curr
Opin Microbiol ): 387-392.
30. ����/��� ��% ��� ���> H (1979) A rapid alkaline extraction procedure for
screening recombinant plasmid DNA. Nucleic Acids Res 1: 1513-1517.
31. ���7� .% ��� �� � (1995) Selection of recombinant vaccinia viruses on the
basis of plaque formation. Gene ,0: 157-162.
32. ��� �$% 8���� 8;% :��� $% 3�� 5����7��� <% :����� �% E���� =H% ���
5��� H. (1992) Hepatitis B virus X protein is not central to the viral life cycle in
vitro. J Virol --: 1223-1227.
33. ���� �% ���������� �% &���� �% ��3���� �% &���� �% F�/��� ;% ;7���������� &%
������ .% &���/���7� .% <��77����� <% ��� <������ � (1998) Lamivudine
treatment can restore T cell responsiveness in chronic hepatitis B. J Clin Invest
4#: 968-975.
34. ����� �% ;���/���> E.% �������� ��% ��� E�� � (1996) DNA immunization
against experimental genital herpes simplex virus infection. J Infect Dis 1): 800-
807.
35. ��� & (2000) Generation of MHC-peptide tetramers: a new opportunity for
dissecting T-cell immune responses. Microbes and Infection #: 425-429.

114
36. ������ H�% =����> E% .�7����� H% E��� <J% ��� ���2����� H (1998) Lipid
vesicle size determines the Th1 or Th2 response to entrapped antigen. J Immunol
- : 4000-4007.
37. ���7���� �% ���> <% ��� &������ && (1995) Elimination of fox rabies from
Belgium using a recombinant vaccinia-rabies vaccine: an update. Vet Microbiol *-:
269-279.
38. ������ '=% ���� H% ��������� H�% ��� ����� � (1968) Quantitative assay
of the lytic action of immune lymphoid cells on 51-Cr-labelled allogeneic target
cells in vitro; inhibition by isoantibody and by drugs. Immunology *: 181-196.
39. ������� �5% ��� �� � (356) E. coli beta-glucuronidase (GUS) as a marker for
recombinant vaccinia viruses. Biotechniques +: 352-354.
40. ��3����� �H% :������ E:% ��� ������ <� (1997) Interleukin-12 inhibits
hepatitis B virus replication in transgenic mice. J Virol 1 : 3236-3243.
41. ���� $% 9 �% ��� 9�3� E% H�! (1988) Recognition of hepatitis B surface antigen
by human T lymphocytes. Proliferative and cytotoxic responses to a major
antigenic determinant defined by synthetic peptides. J Immunol *4: 1808-1815.
42. ������/���� ;% ���7����� '% ��� �� � (1985) Vaccinia virus expression
vector: coexpression of beta-galactosidase provides visual screening of
recombinant virus plaques. Mol Cell Biol ,: 3403-3409.
43. ���� �;% '����� ;% :����� .% ������� .5% ����/7���% 5$% =������
��% ���� &H% :���� HE% &�7��� .�% ��� ������ .� (1993) The woodchuck
hepatitis virus X gene is important for establishment of virus infection in
woodchucks. J Virol -1: 1218-1226.
44. ���� ��% 5��� =E% ��� �<% J��� J% J��� .�% &������ ��% ��� 5 =�
(2000) Enhancement of DNA vaccine potency by linkage of antigen gene to an
HSP70 gene. Cancer Res -4: 1035-1042.

115
45. �������� &% &����� ;% ������ :% &�� &E% ����� �E% ������ �% ��� E�3����
� (1997) The hepatitis B virus X gene induces p53-mediated programmed cell
death. Proc Natl Acad Sci USA +*: 8162-8167.
46. ������ <� (1992) Hepatitis B virus biology and pathogenesis. In: Friedmann T
(Hrsg.) Molecular Genetic Medicine, Academic Press, San Diego, pp. 67-104.
47. ������ <� (1999) The immunobiology of viral hepatitis. In: Crispe IN (Hrsg.) T
Lymphocytes in the Liver: Immunbiology, Pathology, and Host Defence, Wiley-
Liss, Inc., pp. 117-138.
48. ���� J�% ���� 5E% ��� 5'% �� J�% ��� =�� �� (1997) Improvement of
hepatitis B virus DNA vaccines by plasmids coexpressing hepatitis B surface
antigen and interleukin-2. J Virol 1 : 169-178.
49. ���� J�% ������ �E% E�� JE% ��� 5'% E�� 5�% ���� J=% ��� =�� ��
(1998) Development of Th1 and Th2 populations and the nature of immune
responses to hepatitis B virus DNA vaccines can be modulated by codelivery of
various cytokine genes. J Immunol -4: 1320-1329.
50. �������� ��% ����� &:% &��7�7� H% ��� �����/��� � (1977) Synapse
formation between two clonal cell lines. Science +-: 995-998.
51. ���� �'% J��� �% ��� ;;% E�� �:% '�� JH% E�� =:% E��% H;% '�� ��%
��� &��� ;� (1999) Induction of cytotoxic T lymphocytes with peptides in vitro:
identification of candidate T-cell epitopes in hepatitis B virus X antigen. J
Immunother ##: 279-287.
52. ��7���� ��% &7���� �% ��� �� � (1985) In vitro mutagenesis of the
promoter region for a vaccinia virus gene: evidence for tandem early and late
regulatory signals. J Virol ,*: 30-37.
53. ������� &H% �7K����� :�% ��>�� E�% E��/��� ;�% ��� ������� �;
(1998) Incidence of hepatitis B virus infection in the United States, 1976-1994:
estimates from the National Health and Nutrition Examination Surveys. J Infect
Dis 10: 954-959.

116
54. ������� H$% '��/��� ��% ������� ��% ;��3�7� $�% ��� ;���/�� 5
(1993) Current Protocols in Immunology, Chap. 3, John Wiley & Sons, Inc., New
York.
55. ������3� .% ;���� :% ��� :���� � (1989) Transcriptional activation of
homologous and heterologous genes by the hepatitis B virus X gene product in
cells permissive for viral replication. J Virol -): 4019-4026.
56. ����> E% 5��� �% F��� '$% ��>�� H% �7���� �% ;�������� �% ����L��>��
�% &�7�� �H% ������ '% ��� ���3� � (1994) Facilitated DNA inoculation
induces anti-HIV-1 immunity in vivo. Vaccine #: 1545-1550.
57. �����> $E% ������� ��% :����/��� &�% ����/ .5% 8������% H% ������� �$%
��((��� ��% � ;E% ��� ����> E (1991) Safety of and immunological response
to a recombinant vaccinia virus vaccine expressing HIV envelope glycoprotein.
Lancet ))1: 567-572.
58. �����> $E% �7$����� �H% ����> E% � ;E% ������� ��% ������� �% ��((���
�% ����/ .5% ;���� :$% ��� :����/��� &� (1993) Enhanced immunity to
human immunodeficiency virus (HIV) envelope elicited by a combined vaccine
regimen consisting of priming with a vaccinia recombinant expressing HIV
envelope and boosting with gp160 protein. Proc Natl Acad Sci USA +4: 1882-
1886.
59. ���� &% '��/� �% ������� �% �� ��! (2000) Induction of antibody to the MHV
surface antigen in chronic WHV carriers by vaccination with alum-adjuvanted
WHsAg alone and in combination with the antiviral drug, 1-(2-fluoro-5-methyl-b-
L-arabinofuranosyl) uracil (L-FMAU). In: Proceedings of the 10th International
Symposium on Viral Hepatitis and Liver Disease, Atlanta, Abstract B113.
60. ��2 9% =����� '% ��� :���/��� �E (1996) Structure and functions of the 20S
and 26S proteasomes. Ann Rev Biochem -,: 801-847.
61. ������ �% &������ H% ;��7���� .H% ����� =�% ��� .����� �$ (1998)
Metabolic labeling of woodchuck hepatitis B virus X protein in naturally infected
hepatocytes reveals a bimodal half-life and association with the nuclear framework.
J Virol 1#: 9359-9364.

117
62. ��3� �E% ��7��� �E% ��� 5����� .: (1993) DNA-based immunization
induces continuous secretion of hepatitis B surface antigen and high levels of
circulating antibody. Hum Mol Genet #: 1847-1851.
63. ��3� �E% 5����� .:% ��� �������2 �� (1993) Direct gene transfer into
skeletal muscle in vivo: factors affecting efficiency to transfer and stability of
expression. Hum Gene Ther *: 151-159.
64. ��3� �E% ��7��� �E% ���7��� �% ;7����( �% ��� 5����� .: (1994) Direct
gene transfer in skeletal muscle: plasmid DNA-based immunization against the
hepatitis B virus surface antigen. Vaccine #: 1503-1509.
65. ��3� �E% ��7��� �E% ��� 5����� .: (1995) Use of plasmid DNA for direct
gene transfer and immunization. Annals of the New York Academy of Sciences
11#: 21-29.
66. ��3� �E% ;7����( �% ������ &% ���7��� �% ;7���� H% ��� 5����� .: (1996)
Comparison of plasmid DNA preparation methods for direct gene transfer and
genetic immunization. Biotechniques # : 92-99.
67. ��3� �E (1999) DNA vaccines for prophylactic or therapeutic immunization
against hepatitis B virus. Mt Sinai J Med --: 84-90.
68. �� 5�� H.% 5��� '�% ��E7� �% ������� �.% ��� ;/������ ; (1987)
Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol
1: 725-737.
69. �������� ��% �7:���> �$% ���� =H% E��� J<% :��� <% ��� ����7�� E
(1992) Hepatitis B X-gene expression in hepatocellular carcinoma. J Hepatol ,:
400-403.
70. ��������� E�% ���3�> �% ;����� �E% �7����� �H% ���������> ��% H�!%
���� H;% ��� ������> � (1992) Mice deficient for p53 are developmentally
normal but susceptible to spontaneous tumours. Nature ),-: 215-221.

118
71. ����� �% '���� �% E7��� .% ��� ;7������� .H (1995) The hepatitis B virus HBx
protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of
transcription factors. EMBO J *: 4747-4757.
72. �/���> =5% H�!% &��� H�% ��� E� �� (1998) Live virus vaccines: something
old, something new, something borrowed. Nat Med *: 1357-1358.
73. $�(�� $% �������� 5�% ��� ������� HE (1986) Detection of hepatitis B virus
X product using an open reading frame Escherichia coli expression vector. Proc
Natl Acad Sci USA 0): 2219-2222.
74. $�7�� H% � &H% :����� �% ��� 5��� H. (1998) Genetic immunization
generates cellular and humoral immune responses against the nonstructural proteins
of the hepatitis C virus in a murine model. J Immunol - : 4917-4923.
75. <������ <:% ��� �� � (1988) Escherichia coli gpt gene provides dominant
selection for vaccinia virus open reading frame expression vectors. J Virol -#:
1849-1854.
76. <�� 8% ���� �E% 8���� E% 5���� �% ������� E% E��/���� H% :��� &%
�������7� H% ��� .������ � (1997) Cultured blood dendritic cells retain HIV-1
antigen-presenting capacity for memory CTL during progressive HIV-1 infection. J
Immunol ,+: 4973-4982.
77. <������� ��% 8� �% ��� E�% ��� E����� 5= (1993) Hepatitis B x antigen
and p53 are associated in vitro and in liver tissues from patients with primary
hepatocellular carcinoma. Oncogene 0: 1109-1117.
78. <������� ��% ��� ��� E� (1997) Hepatitis B virus X antigen in the
pathogenesis of chronic infections and the development of hepatocellular
carcinoma. Am J Pathol ,4: 1141-1157.
79. <������ &E% :���� =.% ���� �% .���� .% ���� �5% 5��� �% ��������
H&% .������:�% ��� �������� � (1987) Lipofection: a highly efficient, lipid-
mediated DNA-transfection procedure. Proc Natl Acad Sci USA 0*: 7413-7417.

119
80. <���6��� ��% �����> ;% 5�/��� .:% ��� .�/���� �E (1997) Different T
helper cell types and antibody isotypes generated by saline and gene gun DNA
immunization. J Immunol ,0: 2278-2284.
81. <������� H% :���� E% �� H% ��� '�(���� ;� (1999) Effective DNA
vaccination against listeriosis by prime/boost inoculation with the gene gun. J
Immunol -): 4510-4518.
82. <������ �% &���� �% ���������� �% ����� �% ������ ��% :�/���� =% ��3���� �%
&���� ��% ��� <��77����� < (1990) Cellular immune response to hepatitis B virus-
encoded antigens in acute and chronic hepatitis B virus infection. J Immunol *,:
3442-3449.
83. <�7��� �% .���� E% ��� ;7������ � (1995) HBx protein of hepatitis B virus
interacts with the C-terminal portion of a novel human proteasome alpha-subunit.
Virus Genes 4: 99-102.
84. <���� :.% �7����� ��% :����� .�% '��� ��% =���� ��% ��� ;���� :.
(1991) Expression of the terminal protein region of hepatitis B virus inhibits
cellular responses to interferons alpha and gamma and double-stranded RNA. Proc
Natl Acad Sci USA 00: 2888-2892.
85. <�� '% :��� �% 5��� '% �L��� �% &������ &�% ��� J��� J (1994)
Displacement of housekeeping proteasome subunits by MHC-encoded LMPs: a
newly discovered mechanism for modulating the multicatalytic proteinase
complex. EMBO J ): 3236-3244.
86. <��� =.% $��� &E% ��� �� � (1987) Use of a hybrid vaccinia virus-T7 RNA
polymerase system for expression of target genes. Mol Cell Biol 1: 2538-2544.
87. <���� ��% ��� ��>�� H. (1994) A qualitative progression in HIV type 1
glycoprotein 120-specific cytotoxic cellular and humoral immune responses in
mice receiving a DNA-based glycoprotein 120 vaccine. AIDS Res Hum Retrovir
4: 1433-1441.

120
88. <���� ��% ���/ ��% ������� ;% ;������ '% ��� ��>�� H. (1997)
Enhancement of immunodeficiency virus-specific immune responses in DNA-
immunized rhesus macaques. Vaccine ,: 924-926.
89. <>��� $<% 5�/��� .:% <���� ��% ��>�� H.% ;������ H�% ��� .�/����
�E (1993) DNA vaccines: protective immunizations by parenteral, mucosal, and
gene-gun inoculations. Proc Natl Acad Sci USA +4: 11478-11482.
90. :�7�>��� �% .�7� 'E% ��� :���/��� �E (1993) Gamma-interferon and
expression of MHC genes regulate peptide hydrolysis by proteasomes. Nature )-,:
264-267.
91. :�L���� =< (2000) Monitoring specific T-cell responses to melanoma vaccines:
ELISPOT, tetramers, and beyond. Guest Commentary. Clin Diagn Laboratory
Immunol 1: 141-144.
92. :���/��� <% ������� $% <���� <% =������ &% ��� ������> & (1979) Nucleotide
sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature
#0 : 646-650.
93. :���� &.% ��(���� 5H% 5��7��� �% ;7������ �% 9��� :% ;������� 5%
'������ &�% ��� .���� E (1995) Involvement of the CD95 (APO-1/Fas)
receptor and ligand in liver damage. J Exp Med 0#: 1223-1230.
94. :����� �% =������ '% ������ ��% 8����� �.% H�!% ��� 5��� H.
(1997) Cellular and humoral immune response to hepatitis B virus structural
proteins in mice after DNA-based immunization. Gastroenterology #: 1307-
1320.
95. :����� �% :���� �% =������ '% ��� 5��� H. (1997) Enhancement of
cellular and humoral immune responses to hepatitis C virus core protein using
DNA-based vaccines augmented with cytokine-expressing plasmids. J Immunol
,0: 1231-1237.
96. :����� �% ;7����/�7� .% .������ H% ��� �$% ��� 5��� H. (1998)
Cytokine and hepatitis B virus DNA co-immunizations enhance cellular and

121
humoral immune responses to the middle but not to the large hepatitis B virus
surface antigen in mice. Hepatology #0 : 202-210.
97. :����� �% �� �% ��7����� ;% ��7���L� �% 9������ �% 9((��������%
5�% 5��� H.% ��� ��� �$ (1999) Intracellular retention of hepatitis B virus
surface proteins reduces interleukin-2 augmentation after genetic immunizations. J
Virol 1): 4284-4292.
98. :������ �% ����� 5.% �������� .% ��� 8������ � (1997) Immunity to
Plasmodium falciparum malaria sporozoites by somatic transgene immunization.
Nature Biotechnol ,: 876-881.
99. :���� &�% <�> :% ��� ������ <� (1992) Tumor necrosis factor alpha negatively
regulates hepatitis B virus gene expression in transgenic mice. J Virol --: 3955-
3960.
100. :������� ���� H�% ������� �;% �������% E�% ��� ;��7��� �� (1996) DNA
immunization confers protection against murine cytomegalovirus infection. J Virol
14: 7921-7928.
101. :����� <E% ��� $/ �H 3� (1973) Transformation of rat cells by DNA of human
adenovirus 5. Virology ,*: 536-539.
102. :��((��� =;% ������ =% <���7��� ;�% :���� �.% ��� <����� =� (1995) Fas
ligand-induced apoptosis as a mechanism of immune privilege. Science #14: 1189-
1192.
103. :���� �<% 9������ �% ���� E% ��7����� ;% '����� =F% ��7��� ;% $���� ;%
$�7�� H% ��� �$% ��� :����� � (2000). Mouse alpha-fetoprotein-specific
DNA-based immunotherapy of hepatocellular carcinoma leads to tumor regression
in mice. Gastroenterology +:1104-12.
104. :������ E:% ���� '% ��// ��% ������� =% .���� E% ;7����/�� .�% ���
������ <� (1994) Cytotoxic T lymphocytes inhibit hepatitis B virus gene
expression by a noncytolytic mechanism in transgenic mice. Proc Natl Acad Sci
USA + : 3764-3768.

122
105. :������ E:% :����� ;% ��� ������ <� (1994) Interleukin-2 and alpha/beta
interferon down-regulate hepatitis B virus gene expression in vivo by tumor
necrosis factor-dependent and -independent pathways. J Virol -0: 1265-1270.
106. :������ E:% ������� =% ��// ��% ������ �% ;7����/��% .% ��� ������ <�
(1996) Intracellular inactivation of the hepatitis B virus by cytotoxic T
lymphocytes. Immunity *: 25-36.
107. :������ E:% .�7�(��� .% ���� H% ;������ �% &�7��� .% ��� ������ <�
(1999) Viral clearance without destruction of infected cells during acute HBV
infection. Science #0*: 825-829.
108. :���� '�% ����� =.% ��� ;��3�7� $� (1984) T cell-activating properties of
an anti-Thy-1 monoclonal antibody. Possible analogy to OKT3/Leu-4. J Exp Med
,+: 716-730.
109. ����� J% ��>��� �% '���>��� '% J�� �% '������ �% ;���� J% <�����
�% ��� '����� = (1991) Expression of X protein and hepatitis B virus replication
in chronic hepatitis. Hepatology ): 417-421.
110. ����7�7� ';% ;���� ��% ��� ���� .H (1989) Activation of Lyt-2+ (CD8+) and
L3T4+ (CD4+) T cell subsets by anti-receptor antibody. J Immunol *#: 2181-
2186.
111. �� �;% ���� �;% �� '% .�3���� �% ��� .�/���� 5; (1996) Costimulatory
protein B7-1 enhances the cytotoxic T cell response and antibody response to
hepatitis B surface antigen. Proc Natl Acad Sci USA +): 7274-7278.
112. �����7��� H% �7��7���� H% E�� ;% =��� �% ������ '% ���� :% 5����� =%
<���H% E�3������ �% ;���� �% ��� ������ . (1999) A pilot study of the CY-
1899 T-cell vaccine in subjects chronically infected with hepatitis B virus. The
CY1899 T Cell Vaccine Study Group. Hepatology )4: 531-536.
113. �����> �E% ����> H% ��� F�/�� . (1998) Microspheres containing plasmid-
encoded antigens elicit cytotoxic T-cell responses. Nat Med *: 365-368.

123
114. �������� H$% ���� ;�% <>��� $<% ;������ H�% :����/���% ��% 5��� ;% ���
.�/���� �E (1996) Protection against rotavirus infections by DNA vaccination. J
Infect Dis 1* ;��� : 93-97.
115. �� H% ;������ �% 5��� �% ;7������ ��% '�� H% ��� ���� . (1988)
Frequent detection of antibodies to hepatitis B virus x-protein in acute, chronic and
resolved infections. Med Microbiol Immunol 11: 195-205.
116. ��(��� �% '�7� <% ��� ;7���� : (1988) Granulocyte/macrophage colony-
stimulating factor and interleukin 1 mediate the maturation of murine epidermal
Langerhans cells into potent immunostimulatory dendritic cells. J Exp Med -1:
700-705.
117. ���7� ��% <��� =.% ;���� :% ������� �5% J��� E�% :������� ;% &�����
�% H�!% $���� 5.% ��3��� 5:% �����(���� ��% ��% �% ��� E�(�� H� (1996)
Patterns of viral replication correlate with outcome in simian immunodeficiency
virus (SIV)-infected macaques: effect of prior immunization with a trivalent SIV
vaccine in modified vaccinia virus Ankara. J Virol 14: 3741-3752.
118. ��7������ � (1996) Ubiquitin-dependent protein degradation. Ann Rev Gen
)4: 405-439.
119. ����� �% ;7���(�� ;% ;��(�� �% <������� ��% &�� �% ��� :����7� 5� (1990)
Malignant transformation of immortalized transgenic hepatocytes after transfection
with hepatitis B virus DNA. EMBO J +: 1137-1145.
120. �������� �% ;������> �% ��������� �% ��� ����� �� (1991) p53 mutations in
human cancers. Science #,): 49-53.
121. � 8% 8���� 8% ��� $% ��2 9% :���/��� �E% ��� E���� =H (1999) Hepatitis
B virus X protein is both a substrate and a potential inhibitor of the proteasome
complex. J Virol 1): 7231-7240.
122. ���� H% '���� H% ;� $�% ��� E���� =H (1996) Proteasome complex as a
potential cellular target of hepatitis B virus X protein. J Virol 14: 5582-5591.

124
123. ���� �% '��� ;% J������ '% =��� J% 5����� =% ��� ���� � (1989)
Hepatitis B core antigen-specific IFN-gamma production of peripheral blood
mononuclear cells in patients with chronic hepatitis B virus infection. J Immunol
*#: 4006-4011.
124. ������ :J% <��� H% �������� :% E�3������ �% ����� �% K�� �% ���
:��7�� �<% 9���(( �% ����/��� �% ���2����� H% ����� .5% ��� ;���� �
(1999) Utilization of MHC class I transgenic mice for development of minigene
DNA vaccines encoding multiple HLA-restricted CTL epitopes. J Immunol -#:
3915-3925.
125. ������ �% ;��������� �H% ���� �'% ��������� �E% ��� ���/�� �� (1997)
Enhanced CTL responses mediated by plasmid DNA immunogens encoding
costimulatory molecules and cytokines. J Immunol ,0: 4591-4601.
126. H�� J% ;��� 5'% ��� �������� � (1988) Human T cell response to the surface
antigen of hepatitis B virus (HBsAg). Endosomal and nonendosomal processing
pathways are accessible to both endogenous and exogenous antigen. J Exp Med
-0: 293-306.
127. H�� ��% ;������ �% 5����� =% ;������� F% �������� H% ��((���� .%
$�7�����/ �% $���/�� H% &��������� :% .��������� :% �� ��! (1991)
Immune response of peripheral blood mononuclear cells to HBx-antigen of
hepatitis B virus. Hepatology ): 637-643.
128. H�� ��% ��������� ��% ;������� F% 5������� $�% 8�7��3��% .% ��((����
.�% $�7�����/ �% <����� :% 5��� �% ��� &��� :. (1995) Activation of a
heterogeneous hepatitis B (HB) core and e antigen-specific CD4+ T-cell population
during seroconversion to anti-HBe and anti-HBs in hepatitis B virus infection. J
Virol -+: 3358-3368.
129. '��� ;% ������� =% 5����� =% J������ '% =���>�����% �% =����� �%
��� '���/� � (1994) Interferon-gamma production specific for hepatitis B
virus antigen by intrahepatic T lymphocytes in patients with acute and chronic
hepatitis B. Am J Gastroenterol 0+: 92-96.

125
130. '���� ;�% H����� .&% =��7�� �'% �>��� �H% ��� �;% �M�6��� .=%
'���7� H=% ��� 5����� �� (1994) Longitudinal analysis of T cell receptor
(TCR) gene usage by human immunodeficiency virus 1 envelope-specific cytotoxic
T lymphocyte clones reveals a limited TCR repertoire. J Exp Med 1+: 1261-1271.
131. '���>��� '% ��>��� �% ;���� J% '������ �% F��� '% <����� �% ;��� �%
������ 9% ���/��� '% ��� '����� = (1989) Detection of hepatitis B virus
X gene protein and antibody in type B chronic liver disease. Gastroenterology +1:
990-998.
132. '�> �% ������� $% =���� �% ��� :���/��� < (1985) The HBV HBx gene
expressed in E. coli is recognised by sera from hepatitis patients. EMBO J *: 1287-
1292.
133. '���� �;% E��� F% 5�� E% E/�� �% ��� ��(7������� &� (1993) Hepatitis
B virus transactivator HBx uses a tumour promoter signalling pathways. Nature
)- : 742-745.
134. '����� .% ����� ;.% �� �H% ��� ;���� ;E (1996) Peptide transporter
(TAP-1 and TAP-2)-independent endogenous processing of Epstein-Barr virus
(EBV) latent membrane protein 2A: implications for cytotoxic T-lymphocyte
control of EBV-associated malignancies. J Virol 14: 5357-5362.
135. '�� ��% '���� '% ;���� �% ��>���� =% ��� H�> : (1991) HBx gene of
hepatitis B virus induces liver cancer in transgenic mice. Nature ), : 317-320.
136. '�� HH% �>>�3�� �% ��������� �E% ����������� ��% ���� '% 5��� �%
��>�� H�% ��� 5����� �� (1997) In vivo engineering of a cellular immune
response by coadministration of IL-12 expression vector with a DNA immunogen.
J Immunol ,0: 816-826.
137. '�� HH% ���������� E'% ;�� H�% =�� �% ������� E% 9�% H% ���� '% � J%
'�����>� '% ������� �% ����7��3 =% 5���� ��% �������% ��% ��>�� H�%
����L��>�� �:% ��� 5����� �� (1998) CD8 positive T cells influence antigen-
specific immune responses through the expression of chemokines. J Clin Invest
4#: 1112-1124.

126
138. '�� HH% =��3��� ��% ���������� E'% ������� E% =�� �% � J% ����������
;% ���� '% ��� E% ��>�� �'% 5���� ��% ����������� ��% ������� ��%
��>�� H�% ����L��>�� �:% ��� 5����� �� (1998) Modulation of amplitude
and direction of in vivo immune responses by co-administration of cytokine gene
expression cassettes with DNA immunogens. Eur J Immunol #0: 1089-1103.
139. '�� ;% �// �.% ��=���� .�% ��� �����7� HE (1965) Enhanced thymidine
kinase activity following infection of green monkey kidney cells by simian
adenoviruses, simian papovavirus SV40, and an adenovirus-SV40 "hybrid".
Virology #1:453-457.
140. '���� �&% ��� ;7������� .H (1997) Activation of Src family kinases by hepatitis
B virus HBx protein and coupled signaling to Ras. Mol Cell Biol 1: 6427-6436.
141. '���� '% ����>� '% ���� ;% J��>����� �% $��� J% ��>���� =% ���
'������ ' (1994) High-level expression of hepatitis B virus HBx gene and
hepatocarcinogenesis in transgenic mice. Hepatology +: 810-819.
142. '���� ' (1995) Hepatitis B virus HBx gene and hepatocarcinogenesis.
Intervirology )0: 134-142.
143. '���� ��% J� �'% ����� ;% 5���7����� =H% ����� :�% =������ .%
'������> :�% ��� '������ �� (1995) CpG motifs in bacterial DNA trigger
direct B-cell activation. Nature )1*: 546-549.
144. '���� �� (1996) Lymphocyte activation by CpG dinucleotide motifs in
prokaryotic DNA. Trends Microbiol *: 73-76.
145. '��/�� �% &������ �&% .��(��/��� '% ������ <�% ;7���7�� �H% .������ H%
��� ;7����/�7� . (1996) DNA immunization induces antibody and cytotoxic T
cell responses to hepatitis B core antigen in H-2b mice. J Immunol ,-: 3687-3695.
146. '������ ��% ������ �% <������� �J% ���� ��% ���% ��� ����> �.
(1993) The cytolytic activity of pulmonary CD8+ lymphocytes, induced by
infection with a vaccinia virus recombinant expressing the M2 protein of
respiratory syncytial virus (RSV), correlates with resistance to RSV infection in
mice. J Virol -1: 1044-1049.

127
147. '���/� '% ��� 'K% '���� �% ;���� '% �������� $% '������� ;% 9���
'% ��>��� J% ���� �% ������� '% '������ �% ��� 9��� ' (2000) The timing
of GM-CSF expression plasmid administration influences the Th1/Th2 response
induced by an HIV-1-specific DNA vaccine. J Immunol -*: 3102-3111.
148. E�7�> �H% =������ �;% �������� $<% 9M����� =% ��3�� �E% ������� <&%
<�����.$% ��� .�/��� H; (1999) Phase IIa safety and immunogenicity of a
therapeutic vaccine, TA-GW, in persons with genital warts. J Infect Dis 1+: 612-
618.
149. E������ F' (1970) Cleavage of structural proteins during the assembly of the
head of bacteriophage T4. Nature ##1: 680-685.
150. E� :'% E���� .% E�� �'% J�� ;=% �� H% E�� 5E% ��� 5������ . (1998)
Clearance of persistent hepatitis B virus infection in Chinese bone marrow
transplant recipients whose donors were anti-hepatitis B core- and anti-hepatitis B
surface antibody-positive. J Infect Dis 10: 1585-1591.
151. E�� =�% <������� �H% ;��� .<% ����>� HE% 5�� ;E% ��� ���� H; (1990)
Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J
Virol -*: 5939-5947.
152. E������ 55% ;���� ��% ����� 5.% ;���� H&% ;7���� ��% ;����> �H% ���
E>�� H� (1997) Immune responses induced by intramuscular or gene gun injection
of protective deoxyribonucleic acid vaccines that express the circumsporozoite
protein from Plasmodium berghei malaria parasites. J Immunol ,+: 6112-6119.
153. E������ 55% J��� �% ��� .���(� �& (1999) DNA and RNA-based vaccines:
principles, progress and prospects. Vaccine 0: 765-777.
154. E���2�.��� :% ����/��� �% '����� �% �� ��! (2000) Characterization of
HBV envelope specific humoral and cellular immune responses induced by
vaccination of healthy adult volunteers. In: Proceedings 10th International
Symposium on Viral Hepatitis And Liver Disease, Atlanta.

128
155. E�3���� �% H����H��� 9% ������ �% 5��� �% ��� &����7���� � (1990)
Hepatitis B virus (HBV) X gene expression in human cells and anti-HBx
antibodies detection in chronic HBV infection. Virology 1*: 299-304.
156. E���� ��% ;������ �% 5��� �% ���� .% =��� 8J% ��� ;7������ �� (1988)
Low incidence and high titers of antibodies to hepatitis B virus X-protein in sera of
Chinese patients with hepatocellular carcinoma. J Med Virol #,: 329-337.
157. E� �5% =�� J&% '�� H=% ���� J�% ;>�� �'% ���� � ��� ���� ;E (2000)
Recombinant adeno-associated virus expressing human papillomavirus type 16 E7
peptide DNA fused with heat shock protein DNA as a potential vaccine for cervical
cancer. J Virol 1*: 2888-2894.
158. E�3������ ��% ���2����� H% ����� �% 9���(( �% ���� $% ���> '% :������
E:% ������ <�% <��� H% ����� .5% ��� ;���� � (1999) Altered helper T
lymphocyte function associated with chronic hepatitis B virus infection and its role
in response to therapeutic vaccination in humans. J Immunol -#: 3088-3095.
159. E����� ��% =�7�� .$% ������ �E% E��� ��% <�77���� E�% ;��3������ $%
������ �H% ������� .:% �������� '% ��� ;��3� �E (1999) Therapy of
tuberculosis in mice by DNA vaccination. Nature *44: 269-271.
160. E/�� �% ������ �% ;���� �% ����� �% ;7�����7��� &% E��� F% 5���/���
H% ��(7������� &�% ��� '���� �; (1996) Hepatoma-derived integrated HBV
DNA causes multi-stage transformation in vitro. Oncogene #: 1597-1608.
161. E7��� .% ��� ;7������� .H (1992) Hepatitis B virus X protein activates
transcription factor NF-kappa B without a requirement for protein kinase C. J Virol
--: 983-991.
162. �� �&% 5���> &H% ;������� ��% ��� ��������� :� (1993) PA28, an activator
of the 20 S proteasome, is inactivated by proteolytic modification at its carboxyl
terminus. J Biol Chem #-0: 22514-22519.
163. ��7���� �% ;���� :E% ��� �� � (1984) General method for production and
selection of infectious vaccinia virus recombinants expressing foreign genes. J
Virol *+: 857-864.

129
164. ��7��� � (1991) Manipulation of vaccinia virus vectors. In: Murray EJ (Hrsg.)
Methods in Molecular Biology, Vol. 7: Gene Transfer and Expression Protocols,
Humana Press Inc., Clifton, NJ, pp. 129-146.
165. ������ �<% ���((��� H&% ��� ;����6� � (1991) HBV X protein alters the DNA
binding specificity of CREB and ATF-2 by protein-protein interactions. Science
#,#: 842-844.
166. ���7��� �% ���7���� �% ��3� �E% 5����� .:% =������ &% ��� ��7��� �E
(1996) DNA-mediated immunization in a transgenic mouse model of the hepatitis
B surface antigen chronic carrier state. Proc Natl Acad Sci USA +): 12496-12501.
167. ����7��� $% .�� .H% J 8% 5��� 5;% ��� .�� �= (1995) Genetic
immunization against herpes simplex virus. Protection is mediated by CD4+ T
lymphocytes. J Immunol ,,: 259-265.
168. ���>��� =% ���� ;% '���� '% J��� '% ��� ����7� �. (1993) Serology of
acute exacerbation in chronic hepatitis B virus infection. Gastroenterology 4,:
1141-1151.
169. ���/��� '% ��� =����� = (1990) Integration of hepatitis B virus DNA and its
implications for hepatocarcinogenesis. Mol Biol Med 1: 243-260.
170. �7����� �H% �������� ������ �E% :�������� .�% .�/���� �E% ;������
H�% <���� H=% 5����� :% ��>�� H.% &�7��� .�% ��� ��3� �E (1999) Route
and method of delivery of DNA vaccine influence immune responses in mice and
non-human primates. Mol Med ,: 287-300.
171. �7'����> ��% ;�3����� .% K�� �% ������ : ��� ;���� � (2000)
Characterization of an in situ IFN-γ ELISA assay which is able to detect specific
peptide responses from freshly isolated splenocytes induced by DNA minigene
immunization. J Immunol Methods #)1: 105-117.
172. �������� �% ;7������� &&% ��� 5��� H. (1998) Cloning and characterization of
a novel hepatitis B virus x binding protein that inhibits viral replication. J Virol 1#:
1737-1743.

130
173. ���7�����> �% ��� �� � (1992) Introduction of foreign DNA into the
vaccinia virus genome by in vitro ligation: recombination-independent selectable
cloning vectors. Virology +4: 522-526.
174. ��>�� �E% =���� E�% ���� �% ��� ;����> HH (1986) Hepatitis B virus
polypeptide X: expression in Escherichia coli and identification of specific
antibodies in sera from hepatitis B virus-infected humans. J Virol ,1: 101-109.
175. ��7����� =�% &�6������ �% :����� ;% ��� ������ <� (1994) Hepatitis B virus
persistence after recovery from acute viral hepatitis. J Clin Invest +): 230-239.
176. ��7��� �E (1997) Prospects for active immunotherapies for hepatitis B virus
chronic carriers. Res Virol *0: 95-99.
177. ����7� �.% E���2�.��� ::% E��� .$% ��� ������ <� (1984) Genetic
regulation of the immune response to hepatitis B surface antigen (HBsAg). IV.
Distinct H-2-linked Ir genes control antibody responses to different HBsAg
determinants on the same molecule and map to the I-A and I-C subregions. J Exp
Med ,+: 41-56.
178. ����7� �.% �7������ �'% �7E�7���� �% =������� :�% ��� ������ <�
(1985) Distinct H-2-linked regulation of T-cell responses to the pre-S and S regions
of the same hepatitis B surface antigen polypeptide allows circumvention of
nonresponsiveness to the S region. Proc Natl Acad Sci USA 0#: 8168-8172.
179. ����7� �.% ��� �7E�7���� � (1986) The nucleocapsid of hepatitis B virus is
both a T-cell-independent and a T-cell-dependent antigen. Science #)*: 1398-1401.
180. ����7� �.% �7E�7���� �% =������� :�% ��� ���� HE (1987) Antibody
production to the nucleocapsid and envelope of the hepatitis B virus primed by a
single synthetic T cell site. Nature )#+: 547-549.
181. ����7� �.% �7E�7���� �% �������> �% ��� =������� :� (1987) A single 10-
residue pre-S(1) peptide can prime T cell help for antibody production to multiple
epitopes within the pre-S(1), pre-S(2), and S regions of HBsAg. J Immunol )0:
4457-4465.

131
182. ����7� �.% H��� H$% �7E�7���� �% ������ :% �������> �% ��� ���� HE
(1990) Importance of subtype in the immune response to the pre-S(2) region of the
hepatitis B surface antigen. II. Synthetic Pre-S(2) immunogen. J Immunol **:
3544-3551.
183. ����7� �.% ���� HE% �7E�7���� �% E�����> '$% =�������% :�% ��� H���
H$ (1990) Importance of subtype in the immune response to the pre-S(2) region of
the hepatitis B surface antigen. I. T cell fine specificity. J Immunol **: 3535-
3543.
184. ����7� �.% ���� �% ;7����� <% &������ �E% H��� H$% ��� ���� HE (1997)
Role of B cells in antigen presentation of the hepatitis B core. Proc Natl Acad Sci
USA +*: 14648-14653.
185. ����� :% .������ �% &���� H% <����� &% :����� ;% ;7���7�� �H% <������ �%
��� ������ <� (1993) HLA-A31- and HLA-Aw68-restricted cytotoxic T cell
responses to a single hepatitis B virus nucleocapsid epitope during acute viral
hepatitis. J Exp Med 11: 751-762.
186. ������ ;% ��� <���� � (1997) Molekulare Virologie, 1. Aufl., Spektrum
Akademischer Verlag, Heidelberg, Berlin, Oxford, pp. 335-357.
187. ������� �% '���� �% ��� H����� �' (1996) The anatomy of T-cell
activation and tolerance. Proc Natl Acad Sci USA +): 2245-2252.
188. ��� :% '������ ��% ;������ ;% �������� $% ;������ �% ������ H�%
��((��� ;E% ��� ;����/��� �� (1995) Complexity of the cytokine and antibody
response elicited by immunizing mice with Plasmodium yoelii circumsporozoite
protein plasmid DNA. J Immunol ,,: 2039-2046.
189. �������> ��% ���2����� �% E����� .�% ��� =������� :� (1985) Antibodies
to peptides detect new hepatitis B antigen: serological correlation with
hepatocellular carcinoma. Science ##1: 429-433.
190. ����>��� =% :����� ;% '���7��� '% �� �% &������ ��% &������� .�%
������� .E% '������� 9% ��� ������ <� (1990) Immunobiology and

132
pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science
#*0: 361-364.
191. �� �% ;���� :E% :���� HE% ��� &�7��� .� (1984) Live recombinant vaccinia
virus protects chimpanzees against hepatitis B. Nature ) : 67-69.
192. �� � (1991) Vaccinia virus: a tool for research and vaccine development.
Science #,#: 1662-1667.
193. �� � (1996) Genetically engineered poxviruses for recombinant gene
expression, vaccination, and safety. Proc Natl Acad Sci USA +): 11341-11348.
194. ������'����� '% ������ H% ;��� �% ;����3� �% 8�L�7 �% ������ H%
;����> H ��� ����� . (1998) Counting antigen-specific CD8 T-cells: a re-
evaluation of bystander activation during viral infection. Immunity 0: 177-187.
195. ����/�>��� �% =����� '% J����� =% ��>����� �% ��>��� '% ��� ;��� H
(1984) Phenotypical stability of a human hepatoma cell line, HuH-7, in long-term
culture with chemically defined medium. Gann 1,: 151-158.
196. �������� J% :������ E:% &�6���� �% ;7����/�� .�% ��� ������ <� (1997)
Differential target cell sensitivity to CTL-activated death pathways in hepatitis B
virus transgenic mice. J Immunol ,0: 5692-5697.
197. ��7��/��� 5% 5��������(( �% ������������N�� &% �� ��! (2000) Effector
function, kinetics and epitope hierarchy of HBV envelope specific, CD8+ T cells
induced by vaccination of healthy, adult volunteers. In: Proceedings 10th
International Symposium on Viral Hepatitis and Liver Disease, Atlanta.
198. ���� �% ��� ;7������ � (1993) Hepatitis B virus replication. Trends Microbiol
: 221-228.
199. ������ :% �3���������� �E% �������� &% &�� &E% ����� �% ������ �% ���
E�3���� � (1994) Ras- and Raf-dependent activation of c-jun transcriptional
activity by the hepatitis B virus transactivator pX. Oncogene +: 2837-2843.
200. ��>����� .% <����� &% :����� ;% ����� :% ����> �% ;7���7�� �H% �������� �%
����� .% &���� HE% ��� .������ �: (1993) HLA A2 restricted cytotoxic T

133
lymphocyte responses to multiple hepatitis B surface antigen epitopes during
hepatitis B virus infection. J Immunol ,4: 4659-4671.
201. ������ �:�% ������� &% ������ $�% �� ��! (1998) Identification of more
than one mutation in the hepatitis B virus polymerase gene arising during
prolonged Lamivudine treatment. J Infect Dis 11: 1382-1385.
202. 9���� �5% H���� :H% .����� �$% ����� ;�% ��� ;���� H (1982) Cloning
and structural analysis of integrated woodchuck hepatitis virus sequences from
hepatocellular carcinomas of woodchucks. Cell #+: 385-394.
203. 9��� ' (1992) Hepatocellular carcinoma: recent progress. Hepatology ,: 948-
963.
204. &���� ��% .�> ��% ��7��� .&% ��� &�7�� �H (1988) A poxvirus-derived
vector that directs high levels of expression of cloned genes in mammalian cells.
Proc Natl Acad Sci USA 0,: 9431-9435.
205. &�������� &% &��� '% '�� �% <���7� �% ��� ���7��� � (1995) Selective
accumulation of the X transcript of hepatitis B virus in patients negative for
hepatitis B surface antigen with hepatocellular carcinoma. Hepatology # : 313-321.
206. &�� 5$% ��� ;���� .� (1994) Lymphocyte responses and cytokines. Cell 1-:
241-251.
207. &���� �% ������ <�% ���������� �% ����� :% <����� &% :�/���� =%
<��77����� <% ��� <������ � (1991) Cytotoxic T lymphocytes recognize an HLA-
A2-restricted epitope within the hepatitis B virus nucleocapsid antigen. J Exp Med
1*: 1565-1570.
208. &���� �% <����� &% ���������� �% :����� ;% �� �% ��������� .<% ��3����
�% ����� �% <��77����� <% ��� ������ <� (1992) Hepatitis B virus (HBV)-
specific cytotoxic T-cell (CTL) response in humans: characterization of HLA class
II-restricted CTLs that recognize endogenously synthesized HBV envelope
antigens. J Virol --: 1193-1198.

134
209. &������ =�% $���/��� ��% �7��/� �% &��>��� ;'% <����% ��% ���
��>�� H. (1995) Gene gun-based nucleic acid immunization: elicitation of
humoral and cytotoxic T lymphocyte responses following epidermal delivery of
nanogram quantities of DNA. Vaccine ): 1427-1430.
210. &������ =�% .�/��� =.% ��� ��>�� H. (1996) Influenza virus nucleoprotein-
specific immunoglobulin G subclass and cytokine responses elicited by DNA
vaccination are dependent on the route of vector DNA delivery. J Virol 14: 6119-
6125.
211. &(�(( $% ;��(��� H% :����� '% ;7������ �% ��� =�������� E (1987) Synthesis of
the X-protein of hepatitis B virus in vitro and detection of anti-X antibodies in
human sera. Virology ,0: 456-460.
212. &������� =% =���>�7� =% ��� �3����� ; (1985) Quantitation of 5-bromo-2-
deoxyuridine incorporation into DNA: an enzyme immunoassay for the assessment
of the lymphoid cell proliferative response. J Immunol Methods 0#. 169-79.
213. &���� �5% �����> �.% �7����� $% ��� =����3��� J (1998) Evaluation of B
and T-cell responses in chimpanzees immunized with Hepagene, a hepatitis B
vaccine containing pre-S1, pre-S2 gene products. Vaccine -: 543-550.
214. K���� �% ������ �<% ��� ;����6� � (1995) Hepatitis B virus transactivator
protein X interacts with the TATA-binding protein. Proc Natl Acad Sci USA +#:
1003-1007.
215. .���� & (1973) Retention of lymphocyte characteristics by myelomas and theta + -
lymphomas: sensitivity to cortisol and phytohemagglutinin. J Immunol 4: 1470-
1475.
216. .���� &% ����� ��% ��� ����� ' (1976) Lysozyme synthesis by established
human and murine histiocytic lymphoma cell lines. J Exp Med *): 1528-1533.
217. .�� H�% ����/������ .;% ������ �% ������� �5% ��3��� '.% �� �%
.���/��� ;�% ��� .���(� �& (1996) IL-12 is an effective adjuvant to
recombinant vaccinia virus-based tumor vaccines: enhancement by simultaneous
B7-1 expression. J Immunol ,-: 3357-3365.

135
218. .�� $% 5�����/� �% ����� ;�% $���/��� .�% &��� =�% E��� �% '��� =H%
��� ����� �� (1993) Systemic immunological effects of cytokine genes injected
into skeletal muscle. Proc Natl Acad Sci USA +4: 4523-4527.
219. .�� $% =���� �% ;��� J% ���� �% ����� H�% .���� �% ;���� ;E%
;������/��� �E% ��� ����� �� (1996) Preferential induction of a Th1 immune
response and inhibition of specific IgE antibody formation by plasmid DNA
immunization. Proc Natl Acad Sci USA +): 5141-5145.
220. .�������� �% <����� &% ;����> H% &���� H% .������ �% ����� �% �� �%
;���� �% ��� ������ <� (1995) The cytotoxic T lymphocyte response to multiple
hepatitis B virus polymerase epitopes during and after acute viral hepatitis. J Exp
Med 0 : 1047-1058.
221. .�������� �% <������ �% &�6������ �% ��� ������ <� (1996) The hepatitis B
virus persists for decades after patients’ recovery from acute viral hepatitis despite
active maintenance of a cytotoxic T-lymphocyte response. Nat Med #: 1104-1108.
222. .��(��/��� '% E����� H% &������ �&% ;7������7���� $% ;������� :% '�7� H%
��� ;7���7�� �H (1997) Long-term expression of the hepatitis B virus core-e- and
X-proteins does not cause pathologic changes in transgenic mice. J Hepatol #-:
119-130.
223. .��(��/��� '% 5��� �% E����� H% ��� &% ������ .% :������ E:% ������
<�% ��� ;7���7�� �H (1999) The hepatitis B virus X protein transactivates viral
core gene expression in vivo. J Virol 1): 10399-10405.
224. .������ H% ��� ;7����/�7� . (1999) Alternative pathways for processing
exogenous and endogenous antigens that can generate peptides for MHC class I-
restricted presentation. Immunol Rev 1#: 131-152.
225. .�/���� �E% �����(���� ��% H����� .&% ����� '�% '���� �E% E�(��
H�% .��3� =�% E ;% � ;E% ������� :&% &���7��� �E% ������� H:% :��7����
.% ������� ��% E>�> ;E% 5>��� �;% ��� �7���� �� (1999) Neutralizing
antibody-independent containment of immunodeficiency virus challenges by DNA
priming and recombinant pox virus booster immunizations. Nat Med ,: 526-534.

136
226. .�7� 'E% :���� �% .������� E% ����� '% ;���� .% ��7�% E% ����� �% ���
:���/��� �E (1994) Inhibitors of the proteasome block the degradation of most
cell proteins and the generation of peptides presented on MHC class I molecules.
Cell 10: 761-771.
227. .���� �;% ��� .����� �� (1997) Recombinant viruses as vaccines and
immunological tools. Curr Opin Immunol +: 517-524.
228. .����3 :% ������� �:% =����� :% �� ��! (1996) Direct injection of a
recombinant retroviral vector expressing HBV core in chronic carrier chimpanzees.
In: Proceedings of the 1996 Meeting on Molecular Biology of Hepatitis B viruses,
Cold Spring Harbour, 164.
229. .���� �= (1992) Review: hepatitis B virus X-gene product: a promiscuous
transcriptional activator. J Med Virol )-: 101-117.
230. .��� H% ������� �% .�� �% =�������� H% &������� $% ��� E�3��� �H (1996) p53 as
a target for cancer vaccines: recombinant canarypox virus vectors expressing p53
protect mice against lethal tumor cell challenge. Proc Natl Acad Sci USA +): 4781-
4786.
231. ;������ &% �/��� H;% ���>/����� �% :������� �% ���3��% H% ������ .E% ���
����� �. (1991) Differing lymphokine profiles of functional subsets of human
CD4 and CD8 T cell clones. Science #,*: 279-282.
232. ;���/��� �% ���� H% H�3����� �% .����3 :% ������� �% =������ '%
������� �:% 9M��� H% ��(��� H% $��� .% ����> ''% H���> �H% ����� ;�%
����� ;H% ����7� �% ��� E�� 5= (1998) Genetic immunization of chimpanzees
chronically infected with the hepatitis B virus, using a recombinant retroviral
vector encoding the hepatitis B virus core antigen. Hum Gene Ther +: 1719-1729.
233. ;��/���� H% <���7� $<% ��� ������� = (1989) Molecular Cloning: A
Laboratory Manual. 2nd ed., Cold Spring Harbor Press.
234. ;7���(�� ;% ��� :����7� 5� (1995) In vitro transformation by hepatitis B virus
DNA. Intervirology )0: 143-154.

137
235. ;7���(������ <% ������ <% ��� <������ <: (1992) Construction of chimeric
vaccinia viruses by molecular cloning and packaging. Proc Natl Acad Sci USA 0+:
9977-9981.
236. ;7��� �% <�7��� �% ��� ;7������ � (1990) The hepadnaviral X protein. In:
Molecular Biology of Hepatitis B Virus, pp. 181-192.
237. ;7��� �% ������7������ .% '�� �% ��� ;7������ � (1991) Phosphorylation
and rapid turnover of hepatitis B virus X-protein expressed in HepG2 cells from a
recombinant vaccinia virus. Oncogene -: 1735-1744.
238. ;7����/�7� .% ���� 5% ���� '% ������ <�% ��� .������ H (1995) Nucleic
acid vaccination primes hepatitis B virus surface antigen-specific cytotoxic T
lymphocytes in nonresponder mice. J Virol -+: 5929-5934.
239. ;7����/�7� .% ���� 5% ��� .������ H (1996) DNA vaccination primes MHC
class I-restricted, simian virus 40 large tumor antigen-specific CTL in H-2d mice
that reject syngeneic tumors. J Immunol ,1: 3550-3558.
240. ;7����/�7� .% ���� 5% ��� .������ H (1997) Stress protein (hsp73)-
mediated, TAP-independent processing of endogenous, truncated SV40 large T
antigen for Db-restricted peptide presentation. Eur J Immunol #1: 2016-2023.
241. ;7����/�7� .% ��� .������ H (1999) Enhancing the immunogenicity of
exogenous hepatitis B surface antigen-based vaccines for MHC-I-restricted T cells.
Biol Chem )04: 285-291.
242. ;7������� H% :��/��� ;�% ����7���� =H% ����� =% .�/��% 'H% ������ ��%
��7��� �% ;����� .% ;���� :E% ��� ���� �� (1998) Enhanced immunogenicity
for CD8+ T cell induction and complete protective efficacy of malaria DNA
vaccination by boosting with modified vaccinia virus Ankara. Nat Med *: 397-402.
243. ;7������ 9% ����7��� �% <����� �% ���������;������ <% ��� ����� H�
(1998) Antiviral activity of the proteasome on incoming human immunodeficiency
virus type 1. J Virol 1#: 3845-3850.

138
244. ;������ �% H��� =.% '�� �% ������� .% ��/��� &% =���% H�% ���
��((��� ;E (1998) Boosting with recombinant vaccinia increases
immunogenicity and protective efficacy of malaria DNA vaccine. Proc Natl Acad
Sci USA +,: 7648-7653.
245. ;��� $% ���7���� &H% ��� J�� =; (1990) Transactivation by the hepatitis B virus
X protein depends on AP-2 and other transcription factors. Nature )**: 72-74.
246. ;����� J% :������ E:% <����� &% ��� ������ <� (1998) Dendritic cell
immunization breaks cytotoxic T lymphocyte tolerance in hepatitis B virus
transgenic mice. J Immunol - : 4520-4529.
247. ;����6� �% H����� ;% ��� ������ H (1987) Expression of the hepatitis B virus
X gene in mammalian cells. Proc Natl Acad Sci USA 0*: 2513-2517.
248. ;�� H�% ;�� H�% ;� J;% E�� ��% ���� H�% ��� ;�� J� (1997) Protective
immunity against heterologous challenge with encephalomyocarditis virus by VP1
DNA vaccination: effect of coinjection with a granulocyte-macrophage colony
stimulating factor gene. Vaccine ,: 1827-1833.
249. ;�� H�% '�� HH% ������ .E% ;���(( '$% �7���� �% &�7�� �% �7$�����> ;&%
5��( �5% &������� ���� ;H% ������ =H% ��77������ .�% ��� 5����� ��
(1999) IL-12 gene as a DNA vaccine adjuvant in a herpes mouse model: IL-12
enhances Th1-type CD4+ T cell-mediated protective immunity against herpes
simplex virus-2 challenge. J Immunol -#: 2912-2921.
250. ;�������� �% E�� =�% &������ ;% =������ &% ���� H;% ��� =���> � (1997)
Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X
protein is conserved among mammalian hepadnaviruses and restricted to
transactivation-proficient X-insertion mutants. J Virol 1 : 6194-6199.
251. ;����� �<% ��� E�� �� (1988) trans-activation of viral enhancers by the
hepatitis B virus X protein. J Virol -#: 427-434.
252. ;������ �% 5����� =% = 8�% 5�� �<% E�3���� �% H�� �% &��� :.% ���
5��� � (1990) Mapping of B-cell epitopes of the human hepatitis B virus X
protein. J Virol -*: 2802-2809.

139
253. ;�������7��� ;% .��� .% :��/� �% ;7���(�� ;% '����� H% '��� �% E������ <%
��� :����7� 5� (1997) Transcription of hepatitis B virus in peripheral blood
mononuclear cells from persistently infected patients. J Virol 1 : 5399-5407.
254. ;��� .�% ��� �������> ' (1989) Antigen-specific activation of effector
macrophages by IFN-gamma producing (TH1) T cell clones. Failure of IL-4-
producing (TH2) T cell clones to activate effector function in macrophages. J
Immunol *#: 760-765.
255. ; <% ��� ;7������� .H (1997) Hepatitis B virus HBx protein sensitizes cells to
apoptotic killing by tumor necrosis factor alpha. Proc Natl Acad Sci USA +*:
8744-8749.
256. ; K% ;7������ ��% ��(���� 5H% 9��� :% &�7����>� .% ��� �����7� &
(1998) Expression of hepatitis B virus X protein in HBV-infected human livers and
hepatocellular carcinomas. Hepatology #1: 1109-1120.
257. ;��� �% E�� ��% :��/ &% ����� :H% �7������� �% ;������� '% ���
<���(�� � (1999) BAC-VAC, a novel generation of (DNA) vaccines: A bacterial
artificial chromosome (BAC) containing a replication-competent, packaging-
defective virus genome induces protective immunity against herpes simplex virus
1. Proc Natl Acad Sci USA +-: 12697-12702.
258. ;�� .% ��� ;��3���3� &' (1995) A mechanism for the specific immunogenicity
of heat shock protein-chaperoned peptides. Science #-+: 1585-1588.
259. ;�� '% ��� J��� .� (1996) Adjuvant-free hsp70 fusion protein system elicits
humoral and cellular immune responses to HIV-1 p24. J Immunol ,-: 873-879.
260. ;�� '% 8�� �% $��� �� ��� J��� .� (1997) Heat shock fusion proteins as
vehicles for antigen delivery into major histocompatibility complex class I
presentation pathway. Proc Natl Acad Sci USA +*: 13146-13151.
261. ;���� 5% ;��3�� �$% $H �����>% 8��� $�% 9����� 5.% 5������ ��%
;���3�> .% ������� H�% ��� '������ � (1980). Hepatitis B vaccine:
demonstration of efficacy in a controlled clinical trial in a high-risk population in
the United States. N Engl J Med. )4): 833-841

140
262. =�7��� �9% .�> �H% 5����� :% ;���� 5<% ������ ;% ��� $������ . (1999)
Phase 1 safety and immune response studies of a DNA vaccine encoding hepatitis
B surface antigen delivered by a gene delivery device. Vaccine 1: 2826-2829.
263. =��7�� =% �7:��� H% ��((��� .% ������> '% $������� H% =���� '% ���
'�>��� � (1990). Detection of individual mouse splenic T-cells producing IFN-γ
an IL-5 using enzyme-linked immunospot (ELISPOT) assay. J Immunol Methods
#0: 65.
264. =��� ��% ����� �% ��� H������ ;� (1992) Genetic immunization is a simple
method for eliciting an immune response. Nature ),-: 152-154.
265. =����� �% E�(�3�� &% ���� 9% �M;��� ;% ����/��� �% E��� $% ;���� �%
���������> �% ������� H% ��� �>��� ' (1999) Immunogenicity and
protective efficacy of tuberculosis DNA vaccines encoding putative phosphate
transport receptors. J Immunol -#: 1113-1119.
266. =�7�� .$% ������ �H% .���� ;% ;��3������ $% :�����>% �% ��� E�����
�� (1996) Vaccination against tuberculosis by DNA injection. Nat Med #: 888-
892.
267. =��������� 9% ������ 9% .����� ��% E�3> .% ������ =% ������ &% ��� ������
�� (1997) The hepatitis B virus X gene potentiates c-myc-induced liver
oncogenesis in transgenic mice. Oncogene *: 395-404.
268. =��� �.% '��������� �% ������ �$% :�������� ��% =���� ��% ���
���� �� (1995) Association between an MHC class II allele and clearance of
hepatitis B virus in the Gambia. N Engl J Med ))#: 1065-1069.
269. =����3 �% ������ <�% ��� �����7� & (1994) Hepatic preneoplasia in hepatitis
B virus transgenic mice. Hepatology #4: 1162-1172.
270. =��/�� �% ;�������� =% ��� :����� H (1979) Electrophoretic transfer of proteins
from polyacrylamide gels to nitrocellulose sheets: procedure and some
applications. Proc Natl Acad Sci USA 1-: 4350-4354.

141
271. =������ �% ����� H% :��� '% �������� :% ������ �% ����� �% ��>�� �%
���� ;% ��� ;���� : (1988) Defective presentation to class I-restricted cytotoxic
T lymphocytes in vaccinia-infected cells is overcome by enhanced degradation of
antigen. J Exp Med -0: 1211-1224.
272. =���7����� : (1995) Interleukin-12: a proinflammatory cytokine with
immunoregulatory functions that bridge innate resistance and antigen-specific
adaptive immunity. Ann Rev Immunol ): 251-276.
273. =���� .% �����3�7 H% :����/���� H% &��3� �% ��� ������� H�! (1995) Direct
interaction of the hepatitis B virus HBx protein with p53 leads to inhibition by HBx
of p53 response element-directed transactivation. J Virol -+: 1851-1859.
274. =�� ;E% ���� &H% E�� �J% J��� &�% ;�� HE% ���� H�% ����� E�% �����
=�% ��� ���� �; (1992) Acute exacerbations of chronic type B hepatitis are
accompanied by increased T cell responses to hepatitis B core and e antigens.
Implications for hepatitis B e antigen seroconversion. J Clin Invest 0+: 87-96.
275. =L� =% ����L��� '% <����� H% ��� 'K% ���� �% ����% �% ������/� J%
=��� '% '������� ;% ����� J% ��>����� H% '�(( 5�% 9�/� =% ��� 9��� '
(1997) Enhancement of cell-mediated immunity against HIV-1 induced by
coinnoculation of plasmid-encoded HIV-1 antigen with plasmid expressing IL-12.
J Immunol ,0: 4008-4013.
276. =���� =% 5���� ��% ������ ��% '����� JE% .���� H% �� ��% ;�����((
�E% H�!% 5����� ;�% 3�� ��� �&% ���� H% E���� �=% ��� ;���� 5H (1998)
Autologous human monocyte-derived dendritic cells genetically modified to
express melanoma antigens elicit primary cytotoxic T cell responses in vitro:
enhancement by cotransfection of genes encoding the Th1-biasing cytokines IL-12
and IFN-alpha. J Immunol -4: 1139-1147.
277. =� H;% ��� ;7������� .� (1987) Transcriptional trans-activating function of
hepatitis B virus. J Virol - : 3448-3453.
278. =� H;% ��� .�/���� 5; (1989) Hepatitis B virus X gene can transactivate
heterologous viral sequences. Proc Natl Acad Sci USA 0-: 2046-2050.

142
279. F��� �% F����7� ;H% :������ H�% '����� ��% ��� E% <������� ��% ��� H�> :
(1995) Functional inactivation but not structural mutation of p53 causes liver
cancer. Nature Genetics +: 41-47.
280. F����7� ;H% ������� �5% ���7�� 5$% ��� ������� $ (1992) The p53 tumor
suppressor protein, a modulator of cell proliferation. J Biol Chem #-1: 15259-
15262.
281. F���� H�% �������> HH% &����� ;$% .���� :�% <������ &E% ������ �H%
:�������� ;�% ��7� ..% ��5��� ��% ��� <������� � (1993) Heterologous
protection against influenza by injection of DNA encoding a viral protein. Science
#,+: 1745-1749.
282. F��� �% 8���7��� �% ���7�� �% '�� ��% ������� �% 8������( �%
���/�7��� '% �� ��! (1994) Genetic heterogeneity of hepatocelullar carcinoma.
Proc Natl Acad Sci USA + : 822-826.
283. F�/�� ;% ����� $% $7������� �% ;���� �% '���� �% ��� ��(7������� &�
(1997) Isolation and molecular characterization of hepatitis B virus X-protein from
a baculovirus expression system. Hepatology #-: 1045-1053.
284. ������������N�� &% ;���� �% E�/�76 $% �� ��! (1996) Vaccine therapy for
chronic hepatitis B: Immunogenicity and safety of two candidate vaccines in
human healthy volunteers. In: Proceedings of the 36th Interscience Conference on
Antimicrobial Agents and Chemotherapy, New Orleans, LO. Abstract #H 20.
285. ��������� �% ;7���((��� ��% &�7��� �% ;7���� �% ��� ;7���((��� ; (1994)
Gene transfer in regenerating muscle. Hum Gene Ther ,: 11-18.
286. �������� �% ������ :% :��> ��% .�� .% <���� &% E�<��� .% J�� E%
������ <�% <��� H% ��� ����������� . (1995) Development of a lipopeptide-
based therapeutic vaccine to treat chronic HBV infection. I. Induction of a primary
cytotoxic T lymphocyte response in humans. J Clin Invest +,: 341-349.
287. ��������� �% ��� '������ '5 (1992) p53 function and dysfunction. Cell 14:
523-526.

143
288. 5����� ��% <��2��� �% &����� =H% <���� =�% ���7� �;% ;7�����> .=% ���
�� � (1988) HIV-1 reverse transcriptase is a target for cytotoxic T lymphocytes
in infected individuals. Science #*4: 64-66.
289. 5��� �% F��� '$% ;�������� �% ����L��>�� �:% ���� '% .�(���� J% ;���
��% ��>�� H% 5������ 5�% ��� 5����� �� (1993) Gene inoculation generates
immune responses against human immunodeficiency virus type 1. Proc Natl Acad
Sci USA +4: 4156-4160.
290. 5��� �% ���� '% ����L��>�� �:% ;�������� �% E� <% F���% '$% ��>�� H%
���3� �% 5������ 5�% ��� 5����� �� (1997) Mucosal immunization with a
DNA vaccine induces immune responses against HIV-1 at a mucosal site. Vaccine
,: 821-825.
291. 5��� 5E% E����� 5=% ��� <������� �� (1991) Hepatitis B x antigen in
hepatitis B virus carrier patients with liver cancer. Cancer Res , : 4971-4977.
292. 5��� 5E% E����� 5=% E��� E% ��� <������� �� (1991) HBxAg in the liver
from carrier patients with chronic hepatitis and cirrhosis. Hepatology *: 29-37.
293. 5��� �5% <������� '% J�� �% <������� ��% : H.% ��� ����� �� (1994)
Hepatitis B virus X protein inhibits p53 sequence-specific DNA binding,
transcriptional activity, and association with transcription factor ERCC3. Proc Natl
Acad Sci USA + : 2230-2234.
294. 5��� �5% :�/�� �'% �������� 5% J�� �% <������� '% ;���/�7��� �5%
����L����� H�% ��� ����� �� (1995) Abrogation of p53-induced apoptosis by
the hepatitis B virus X gene. Cancer Res ,,: 6012-6016.
295. 5��� :�% ;��3�� �'% �� �% ��� <��� =. (1995) Stringent chemical and
thermal regulation of recombinant gene expression by vaccinia virus vectors in
mammalian cells. Proc Natl Acad Sci USA +#: 6773-6777.
296. 5�/�� �% ������� �% ����� �% ������(( �% ������7������ .% ���
;7������ � (1994) Hepadnavirus P protein utilizes a tyrosine residue in the TP
domain to prime reverse transcription. J Virol -0: 2994-2999.

144
297. 5����� :H% E� ��% 5��������� H$% ����� �$% ��� '���� �� (1997)
Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective
as immune adjuvants in tumor antigen immunization. Proc Natl Acad Sci USA +*:
10833-10837.
298. 5�� J�% 5 ��% � ��% 8���� K&% ��� :� ;K (1995) Hepatitis B vaccine
and anti-HBs complex as approach for vaccine therapy. Lancet )*,: 1575-1576.
299. 5������ =�% K�� �=% ��� ;7������� .� (1991) Identification of the hepatitis
B virus factor that inhibits expression of the beta interferon gene. J Virol -,: 4699-
4704.
300. 5��� H% :�/> �H% ;7����/�7� .% ��� .������ H (1999) Priming MHC-I-
restricted cytotoxic T lymphocyte responses to exogenous hepatitis B surface
antigen is CD4+ T cell dependent. J Immunol -): 1880-1887.
301. 5���� ;% :������ E:% ���� '% ;7���7�� �H% ��� ������ <� (1995) Breaking
tolerance leads to autoantibody production but not autoimmune liver disease in
hepatitis B virus envelope transgenic mice. J Immunol ,*: 2504-2515.
302. 5����� .% ������ H�% ���/�� $% ��� �� � (1980) Expression of the vaccinia
virus genome: analysis and mapping of mRNAs encoded within the inverted
terminal repetition. Cell # : 487-493.
303. 5��(( H�% ������ .5% 5������ &% ����� 5% �7��� :% H��� �% ��� <������
&E (1990) Direct gene transfer into mouse muscle in vivo. Science #*1: 1465-
1468.
304. 5��(( H� (1997) Naked DNA transport and expression in mammalian cells.
Neuromuscular Disorders 1: 314-318.
305. 5��������� �% ��/���� F% ��� ��(7������� &� (1988) A transactivating
function encoded in the hepatitis B virus X gene is conserved in the integrated
state. Oncogene ): 545-552.

145
306. ����� 8% ��� $��� �� (1995) Manipulation of the immune response to a plasmid-
encoded viral antigen by coinoculation with plasmids expressing cytokines.
Immunity #: 129-135.
307. ����� 8K% ;�������� ;% =��� �% 5���� 5�% ����� H% ��� $��� �� (1994)
Vaccination with a plasmid vector carrying the rabies virus glycoprotein gene
induces protective immunity against rabies virus. Virology ++: 132-140.
308. � �% ��� E��� <J (1995) Protection against leishmaniasis by injection of DNA
encoding a major surface glycoprotein, gp63, of L. major. Immunology 0*: 173-
176.
309. � E% ;��7��� �% J��� 8% 8��� ;.% ��/�� $:% ��7��� ;=% ��� ��/�� :H
(1998) Immunization for Ebola virus infection. Nat Med *: 37-42.
310. J������ '% ;�������� J% '�/�>��� �% ��� '���� ' (1987) Hepatitis B virus
(HBV) particles are produced in a cell culture system by transient expression of
transfected HBV DNA. Proc Natl Acad Sci USA 0*: 2678-2682.
311. J�� ;&% ;������ �% E����� E% �� ��! (2000) Immunotherapy of chronic
hepatitis B in Asian patients using a novel triple antigen hepatitis B vaccine
(Hepacare). In: Proceedings of the 10th International Symposium on Viral Hepatitis
and Liver Disease, Atlanta. Abstract B124.
312. J����� J% .�/���� �E% E ;% ���(� <% E���� �% ����� H% ����� H�%
�� :% ����� '% 5>��� �% ��� E��3�� �E (1996) Simian immunodeficiency
virus-specific cytotoxic T-lymphocyte induction through DNA vaccination of
rhesus monkeys. J Virol 14: 678-681.
313. J�� �% :��/��� �H% .������ ;.% ���7���� �:% <�(�� �% =������ H�% ���� =%
��� :����/��� &� (1996) Isolation of tyrosinase-specific CD8+ and CD4+ T cell
clones from the peripheral blood of melanoma patients following in vitro
stimulation with recombinant vaccinia virus. J Immunol ,1: 4079-4086.
314. J��� �% 8�� =8% 5��� .<% ��3��� '.% '����� F;% �����7��� <�% E������
55% ��� .���(� �& (1999) Cancer therapy using a self-replicating RNA vaccine.
Nat Med ,: 823-827.

146
315. J�((� �% ��� �'% ����� �;% ��� ���/� � (1990) Extrahepatic hepatitis B
virus DNA sequences in patients with acute hepatitis B infection. Hepatology #:
187-192.
316. 8��� &% ��(7������� &�% ��� '��> . (1988) The HBV X-ORF encodes a
transactivator: a potential factor in viral hepatocarcinogenesis. Oncogene ): 169-
177.
317. 8��/���� :&% ��� E�3��� �H (1993) A comparison of the biological activities of
wild-type and mutant p53. FASEB Journal 1: 855-865.
318. 8������( �% �������� :% '���� .% ;7����� &% E��7���3�7% �% �������� �%
�/��� '% ��� ;7������ �� (1990) Mouse monoclonal antibody directed
against hepatitis B virus X protein synthesized in Escherichia coli: detection of
reactive antigen in liver cell carcinoma and chronic hepatitis. Oncology *1: 143-
148.
319. 8���� 8% =���� �% <���� �% ����>���� �% � 8% ��� E���� =H! (2000)
Structural and functional characterization of interaction between hepatitis B virus
X protein and the proteasome complex. J Biol Chem #1,: 15157-15165.
320. 8� �% E����� 5=% ��� E�% ��� <������� �� (1993) The value of hepatitis
B x antigen as a prognostic marker in the development of hepatocellular carcinoma.
Int J Cancer ,,: 571-576.
321. 8���� <% ;������� H% ��� ;����� � (1994) Woodchuck hepatitis virus X protein
is required for viral infection in vivo. J Virol -0: 2026-2030.

147
���8��� ��
In erster Linie möchte ich mich bei Herrn PD Dr. Geißler für die Überlassung des
Themas, seine intensive praktische und theoretische Betreuung meiner Arbeit und
kritische Korrektur bei der Abfassung der Dissertation bedanken.
Meinem Doktorvater Herrn Prof. Dr. Dr. h.c. Blum danke ich für die herausragenden
Arbeitsbedingungen in seiner Abteilung und seine Begutachtung der Dissertation.
Ebenfalls möchte ich Herrn Prof. Dr. Pircher für die Begutachtung meiner Arbeit danken.
Mein besonderer Dank gilt meinem Freund und Mitstreiter Christian Mauch sowie allen
Mitarbeitern aus dem Labor B3.
Allen übrigen Mitarbeitern der B-Labors in der Medizinischen Universitätsklinik,
Freiburg, namentlich Sabine Bleul, Frau Dr. Dörte Ortmann, Sabine McNally, Dr. zu
Putlitz, PD Dr. von Weizsäcker, Dr. Köck, PD Dr. Moradpour, Prof. Dr. Nassal, Dr.
Baumert danke ich herzlich für die Unterstützung bei meiner Arbeit.
Für die gute Kooperation und methodische Unterstützung bei der Arbeit mit
rekombinanten Vakzinia-Viren möchte ich mich bei Herrn Dr. Jürgen Haussmann,
Institut für Medizinische Mikrobiologie und Hygiene der Universität Freiburg, bedanken.


















