
18 - Stoffwechsel Von Phosphoglyceriden Sphingolipiden Und Cholesterin

Block 13, WS 08/09J.Graf
Pathophysiologie der Leber / Ausgewählte Kapitel.
Inhalte: A: Leber und Fettstoffwechsel
einschl. Cholesterin- und Gallensäurestoffwechsel / CholestaseB: Alkoholische Fettleber, NASH, C: Häm-Synthese – Porphyrien
angeboren / erworben D: Bilirubinstoffwechsel – Hyperbilirubinämien:
konjugiert / unkonjugiert, angeboren / erworbenE: Genetische Defekte und Lebererkrankungen:
Überblick; ausgewählte Krankheitsbilder:M. Wilson, Hämochromatose, α1-Antitrypsin-Mangel, Störungen des Membrantransports
Teilweise korrespondierende Kapitel in Böcker-Denk-Heitz: Pathologie: Kap. 32.3, 32.5.3, 32.6

xx
Leber und Fettstoffwechsel
LipoproteineCholesterin
Gallensäuren
(Patho-) Biochemiez.T. Wiederholung

Zählung der C-Atome im Steroidring: CHOLESTERIN

Aufbau der Lipoproteine
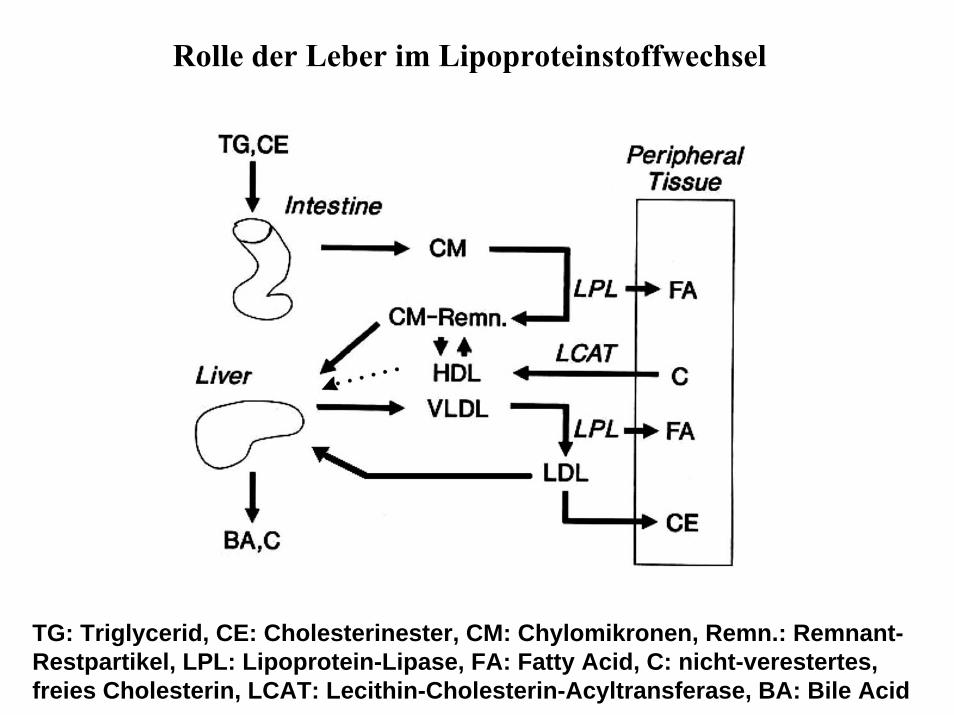
Rolle der Leber im Lipoproteinstoffwechsel
TG: Triglycerid, CE: Cholesterinester, CM: Chylomikronen, Remn.: Remnant-Restpartikel, LPL: Lipoprotein-Lipase, FA: Fatty Acid, C: nicht-verestertes, freies Cholesterin, LCAT: Lecithin-Cholesterin-Acyltransferase, BA: Bile Acid

Lipoproteine
Dichte:
CM: 0.95VLDL: <1.006 - IDL – LDL: 1.006-1.063HDL: 1.063-1.2( HDL3: >1.12)
Elektrophoretische Wanderung:
α1 (HDL) - Prae-ß (VLDL) - ß (LDL)

Wichtige Funktionen einiger Apoproteine:
A1 - Bindung von Cholesterin in der Peripherie- Aktivierung der LCAT( C + PL → CE + Lyso-Lecithin)
B100 - Bindung an LDL-Rezeptor ( CE-Aufnahme in Leber und Peripherie)
C2 - Aktivierung der LPLase(Abgabe von Fettsäuren aus Chylomikronenund VLDL)
E - Bindung an hepatischen Rezeptor
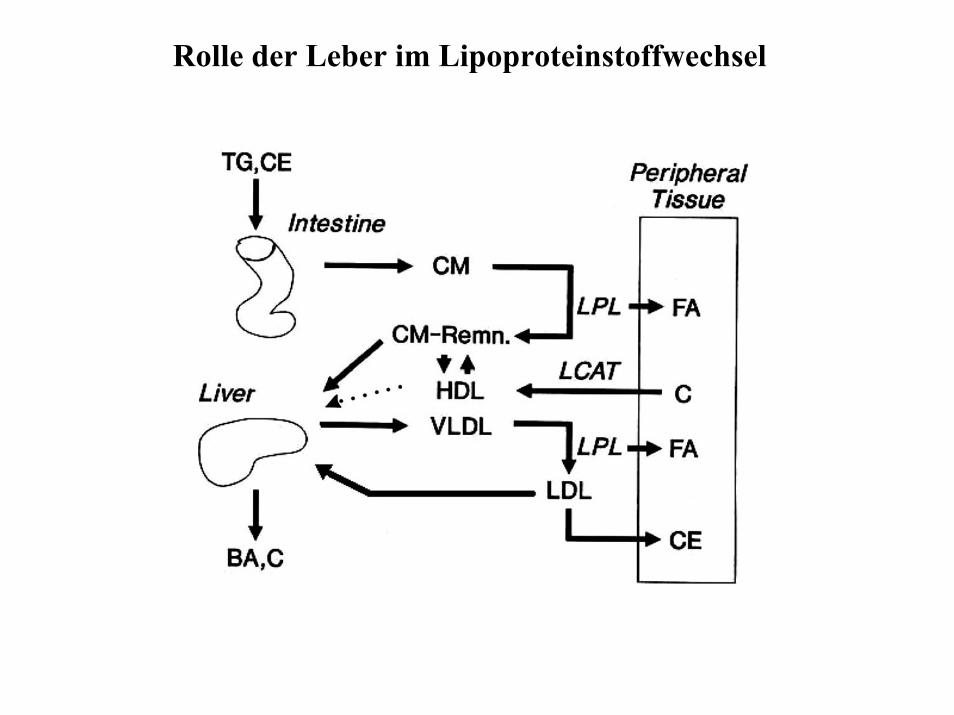
Rolle der Leber im Lipoproteinstoffwechsel

Cholesterin-Umsatz: pro Tag
Zufuhr:Nahrung: 150-400 mg/TagNeusynthese: 600-800 mg/Tag
Ges.: ca. 1g / Tag
Abgabe:Haut 80 mg/TagStoffwechselprodukte der Steroidhormone 50 mg/TagGallensäuren im Stuhl 400 mg/TagMit der Galle ausgeschiedenes Cholesterin 600 mg/Tag
Ges.: ca. 1g / Tag

Umsatz von Cholesterin in der Leber:
Aufnahme und Bildung:
Aufnahme von CE, weniger C:CM-RemnantsLDLVLDL-Remnants (Ratte) bzw. IDL
Bildung von C aus CE über lysosomale saure CE-Hydrolase (Esterase)
Neubildung von C aus AcetylCoA über HMGCoA-ReductaseRückkoppelungshemmung durch freies C und CDCA
Abgabe:CE in VLDL (synthetisiert über ACAT)C in nHDLCholesterin in der GalleGallensäuren in der Galle
Rückkoppelung an geschwindigkeitsbestimmenden Schritten - Beziehung zur Gallesekretion:
AcCoAHMGCoA-Reductase >< freies C (CDCA)
Cholesterin7α-Hydroxylierung >< CDCA und auch andere Gallensäuren
Gallensäuren

Stoffwechsel von Fettsäuren undCholesterinim Bezug zum Glukosestoffwechsel der Leber

Stoffwechsel von Fettsäuren:
Malonyl-CoA hat eine zentrale Bedeutung:
1. seine Synthese wird durch Glukagon gehemmt (z.B. im Hunger), 2. Malonyl-CoA hemmt die Aufnahme von Fettsäuren in die Mitochondrien (Carnitin-Acyl-Transferase) und damit deren ß-Oxydation →
Fettsäuresynthese hemmt die Fettsäureoxydation:
Ac-CoA → Malonyl-CoA → FS-Synthese┴ (Hemmung)
Mitochondrien: Ac-CoA ← ß-Oxydation ← FS-Aufnahme in Mitochondrien

Synthese von Cholesterin:
AcAc-CoA →Hydroxymethylglutaryl-CoA →Mevalonsäure →Isoprenoide →schrittweise Verknüpfung zum Cholesterin
HMG-CoA-Reductaseist dabei das geschwindigkeitsbestimmende Enzym:
COOH-CH2-(CH3)C(OH)-CH2-COOH →COOH-CH2-(CH3)C(OH)-CH2-CH2OH

Cholsäure
Amphiphile „Napfstruktur“

Synthese von Gallensäuren:
Verkürzung und Oxydation an der Seitenkette von Cholesterol (C24) und Hydrierung der Doppelbindung (C5)
2. 7α-Hydroxylierung als gesteuerter (Produkthemmung) und geschwindigkeitsbestimmender Schritt
3. weitere Ringhydroxylierung→ Chenodesoxycholsäure und Cholsäure
(3α,7α-Dihydroxycholansäure) (3α,7α,12α-Trihydroxycholansäure)
4. Konjugation der Seitenkette mit Glycin (NH2-CH2-COOH) und Taurin (NH2-(CH2)2-SO3H) Bildung von z.B. TCA (taurocholic acid), GCA, GCDCA, ......Sekretion von Gallensäuren in die Gallekanalikuli
5. Bildung sekundärer Gallensäuren: 7α-Dehydroxylierung im Darm bzw. 7-Epimerisierung↓ ↓Lithocholsäure (LCA) Desoxycholsäure (DCA)(3α-Hydroxycholansäure) (3α,12α-Di...)Ursodesoxycholsäure (UDCA)(3α,7ß-Dihydroxycholansäure)

Rolle der Leber im Cholesterinstoffwechsel

Gallen-Mizellen und -Vesikel:
Gallensäuremoleküle sind amphiphil und lagern sich mit Cholesterin (wie in der Zellmembran) zu Vesikeln und Stapeln von Doppelschichten (Micellen) aneinander. In dieser Form wird Cholesterin in Lösung gehalten, wozu ein Mischungsverhältnis von ca.
70 % Gallensäure20 % Lecithin10 % Cholesterin
günstig ist. Insbes. bei Mangel an Gallensäuren kann Cholesterin ausfallen → Bildung von speckig glänzenden
Cholesterinsteinen.
Rolle vonCDCAUDCA
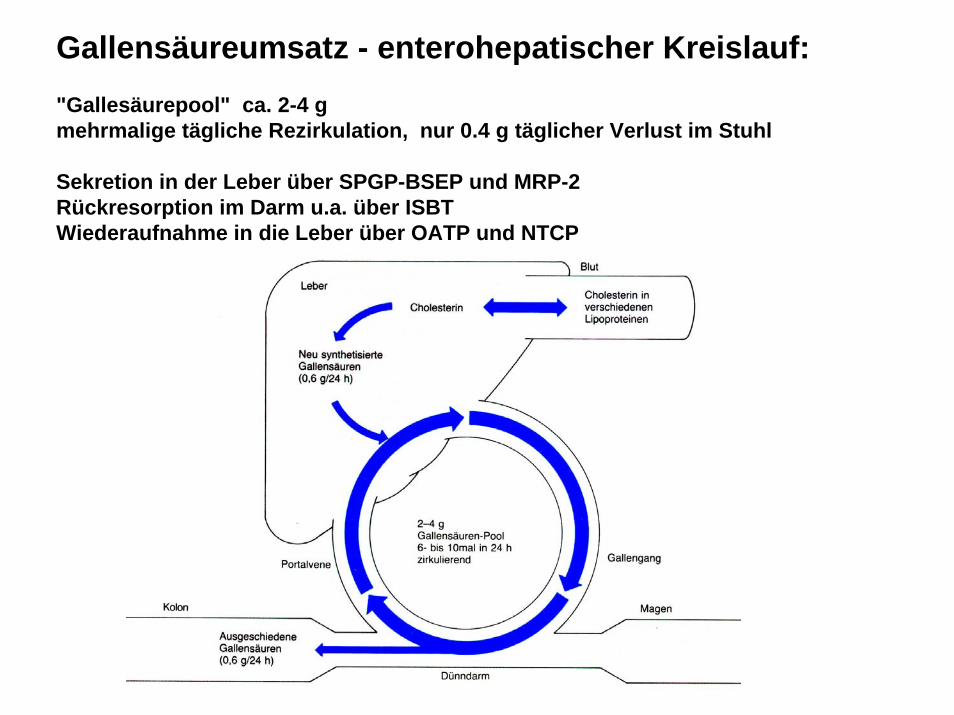
Gallensäureumsatz - enterohepatischer Kreislauf:"Gallesäurepool" ca. 2-4 g mehrmalige tägliche Rezirkulation, nur 0.4 g täglicher Verlust im Stuhl
Sekretion in der Leber über SPGP-BSEP und MRP-2Rückresorption im Darm u.a. über ISBTWiederaufnahme in die Leber über OATP und NTCP

Cholestase verschiedener Ursachen erhöhte Plasma-Gallensäuren und Plasma-Cholesterin (Juckreiz, Xanthelasmen...)
Steatorrhoe mit Vitaminmangel (A,D,E,K)
Gallensteine
genetische Erkrankungen (z.B. PFIC 2, s.u.)Gallensäure-Malabsorptionchologene Diarrhoeu.a.m.
Gallensäureumsatz - enterohepatischer Kreislauf:
Störungen des Gallensäurestoffwechsels sind an der Entstehung vieler Symptome beteiligt:

xx
Alkoholische Fettleber,Fettleberhepatitis
undNASH
Non-alcoholic Steatosis Hepatis

Die durchschnittliche Ernährung in der "Wohlstandsgesellschaft" ist unausgewogen:
3000 kcal
Protein 13-15% (ca. 100g)Kohlehydrate 40% (ca. 300g)Fett 40% (ca. 130g)Äthanol 7% (ca. 30g)
Akuter und chronische Alkoholkonsum kann durch Laborbefunde nachgewiesen werden: γ-GT CDT (carbohydrate deficient transferrin)
Chronischer Alkoholkonsum erhöht das Risiko an Lebercirrhose zu erkranken:
Frauen: < 30 g / Tag durch 5 Jahre Männer: < 60 g / Tag

Alkohol verursacht auch extrahepatische Schäden z.B.:
Hirnatrophie Periphere Neuropathie Hypertrophe Gastritis HodenatrophiePankeatitis:
akute Pankreasnekrosechronische Pankreatitis

Alkoholstoffwechsel in der Leber
(Entstehung evtl. toxischer Produkte)
1. Alkoholdehydrogenase (ADH) Bildung von Acetaldehyd, Acetat, NADH↑Lactat-Produktion evtl. gesteigert
2. PeroxysomenHOOH
3. MEOS (microsomal ethanol oxidizing system) Cytochrome (CYP2E1), .O2
--Bildung
2 und 3 sind induzierbar !

Bezug des Alkoholstoffwechsels zum Fettstoffwechsel:
Fettsäuresynthese ↑ : AcCoA↑ → MalonyCoA → Elongation
Ketogenese ↑
ß-Oxydation ↓, Carnitin-Acyl-Transferase ↓, NADH ↑
Glycerin-3-Phosphat ↑, Triglyceridsynthese ↑

Leber-schädigende Komponenten bei Alkoholkonsum (z.T. fraglich)
Acetaldehyd: bindet an SH- und NH2-Gruppen → Proteine verändert
Glutathion vermindert Mikrotubuli gestörtMitochondrien morphologisch verändert und Zitratzyklus ↓Zellvergrößerung ("Ballonierung") mit SinusoidkompressionO2-Metabolite (.O2
-, H2O2)→ Lipidoxydation
Änderung der Membranfluiditätu.a.

Entstehung der Fettleber und Störung des Lipoproteinstoffwechsels
1. Lipolyse im Fettgewebegefördert durch Catecholamine und andere Stress-Hormone, Leber nimmt freie Fettsäuren über eigenen Membran-Transporter auf
2. Erhöhte Zufuhr von Nahrungsfetten3. Gesteigerte Fettsäuresynthese in der Leber und verminderter Abbau
→ gesteigerte Triglyceridsynthese, aber 4. Abgabe von Triglyceriden, Phopholipiden und Cholesterinestern als VLDL
eingeschränkt, daa. die Apoproteinsynthese gestört ist (Apo B und C)b. Mikrotubuli und damit Sekretion von VLDL gestört sind - VLDL bleiben im Post-Golgi "stecken"
5. Periphere Lipoproteinverwertung gestörtLPLase ↓ (ApoC-Mangel, Insulinresistenz?)→ Hyperlipoproteinämie Typ V = VLDL und CM ↑
6. kleine HDL3 erhöht: HDL nehmen bei mangelnder LPLase-Aktivität (= verzögerte postprandiale "Klärung" des Plasmas) TG aus CM auf. TG werden dann durch hepatische Lipase extrahiert und es entstehen kleine HDL3)

Weitere durch Alkohol bedingte Stoffwechselstörungen:
metabolische Acidose (Lactat-, Keto-)Hyperuricämie
Purinabbau bei Alkoholexzeß erhöht; renale Harnsäure-Ausscheidung durchAcidose vermindert. - säure-hemmbare tubuläre Sekretion
evtl. Hypoglykämiegesteigerte hepatische Glykolyse bei Alkoholoxydation bei gleichzeitig glykogen-verarmter Leber (bei Hunger)
evtl. Hyperglykämiebei "Satten"; sympathische Aktivierung der Glykogenolyse, Insulinresistenz
Induktion von MEOS bzw. CYP2E1Oxydation via MEOS verbraucht NADPH ohne ATP zu produzieren(C2H5OH + NADPH + H+ + O2 → Acetaldehyd + NADP+ + H2O); NADPH-oxidase bildet ohne anderem Substrat reaktive O-Metabolite (ROS) Akute Alkoholgabe hemmt Barbiturat- und Phenothiazin-AbbauNach Induktion aber beschleunigter Medikamentenabbau
Lipidperoxydation

Folgen der Fettleber:
Fettleberhepatitis die langsamer oder rascher in Cirrhose übergeht*)
Nekrose und Entzündungszeichen, Leukozyteninfiltration (Granulo, Lympho)Mallory-Bodies (Zytokeratin) oder alk. Hyalin in Zellen Fibrose beginnt um Venolen und perisinusoidalBildung von Myofibroblasten (aus "stellate cells")
aus Ito-Zellen durch IL-1 und TGFßCollagensynthese Typ I u. III Stimulation über PDGF-Rezeptoren
klinisch evtl. Fieber(cholestatischer) Ikterus, Splenomegaliearterielle Spinnen-Angiome, Ascites, Oedeme, Blutungen, Encephalopathie
*) Fettleber kann nach 1 Monat Alkoholkarenz reversibel seinAlkoholhepatitis ist prognostisch ungünstiger

Cirrhotischer Umbau
Ausbildung von PseudolobuliBindegewebssepten von Periportalfeld zu Zentralvene dazwischen Ausbildung von Pseudolobuli
kleinknotige Laennec'sche Cirrhose
Die klinischen Symptome können dabei immer noch mild sein oder fehlen: Müdigkeit, Schwäche, Verletzlichkeit der Haut, Appetitlosigkeit, Gewichtsverlust, Muskelschwund, Palmarerythem, Vergrößerung der Parotis und Tränendrüsen, Männer: verminderte Rumpfbehaarung, Gynäkomastie, Hodenatrophie
Progredienter Verlauf mit diversen Komplikationsmöglichkeiten, die schließlich zum Tod führen können:
postsinusoidale portale HypertensionAscites mit allen seinen Ursachen portocavale Anastomosen, Blutungen Kreislauf- und Nierenbelastung hepatorenales Syndrom (HRS) hepatische Encephalopathie, Leber-coma

Laborbefunde :
Fettleber: oft leichte AST(SGOT)↑ ALT(SGPT)↑ (Bei alk. Zirrhose AST/ALT >2) evtlaP↑ BR↑
Bei fortgeschrittenem Umbau:Anämie (GI-Blutungen, Folsäure- und B12 Mangel) Splenomegalie, Acanthozytose durch Hypercholesterinämie, Thrombopenie (Hypersplenismus, Thrombopoetinmangel) evtl. Leukozytose (Hepatitis) oder Leukopenie (Hypersplenismus),PTZ verlängert, Albumin ↓, γ-Globulin ↑, Hyponatriämie, Hypokaliämie (Aldo↑)NH3↑, evtl. prärenale AzotämieMg und Pi evtl ↓


Fettleber
CYPs - ROS
ZytokineApoptose/NekroseEntzündungFibrose

Alcoholic liver damage and fibrogenesis
MPT: mitochondrial permeability transition

Alkoholische Fettleber

Alkoholische Fettleber

Mallory-Körper

Alkoholische Zirrhose

Atrophie des Cortex

Atrophie des Kleinhirns

xx
Porphyrien
Störungen der Häm-Syntheseund Bildung
toxischer Zwischenprodukte

HÄM-Synthese:
Bildung des Porphyrinringes aus 4 Pyrrolringen
Lichtabsorption und Fluoreszenz durch konjugierte Doppelbindungen
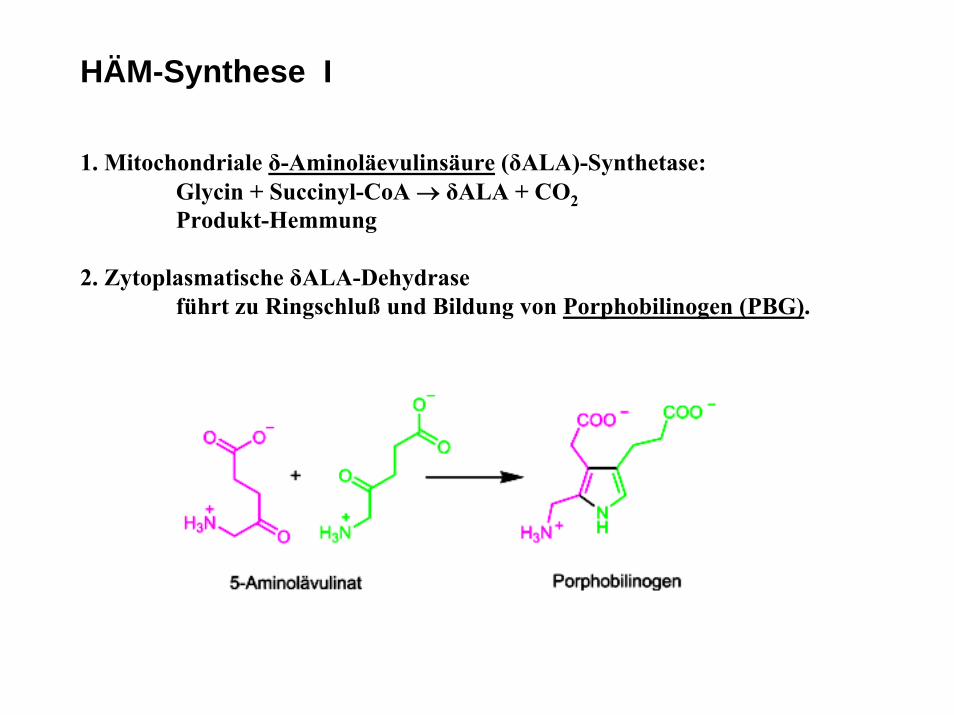
HÄM-Synthese I
1. Mitochondriale δ-Aminoläevulinsäure (δALA)-Synthetase:Glycin + Succinyl-CoA → δALA + CO2Produkt-Hemmung
2. Zytoplasmatische δALA-Dehydraseführt zu Ringschluß und Bildung von Porphobilinogen (PBG).

HÄM-Synthese II
Synthese vonUroporphyrinogen III
und über Zwischenstufen
schließlich vonProtoporphyrin IX,in das Fe eingebaut wird

HÄM-Synthese
1. Mitochondriale δ-Aminoläevulinsäure (δALA)-Synthetase:Glycin + Succinyl-CoA → δALA + CO2Produkt-Hemmung (COOH-CH2-CH2-CO-CH2-NH2)
2. Zytoplasmatische δALA-Dehydraseführt zu Ringschluß und Bildung von Porphobilinogen (PBG).
3. PBG-Desaminase und Uroporphyrinogen (III) - SynthaseAc P Ac P P Ac Ac P 4 mögliche Isomere , praktisch nur Nr. III" " " " " " " "
4. Koproporphyrinogen III via UG-DecarboxylaseM P M P P M M P
5. Protoporphyrinogen IX via KG-OxydaseM V M P P M M V 15 mögliche Isomere
6. Protoporphyrin IX via PG-Oxydasegeeignetes Arrangement der Doppelbindungen
7. Häm via Fe-Chelatase

Formen der Porphyrien und zugeordnete Enzymstörungen
Alternativprodukte der Hämsynthese entstehen bei Störung des Folgeschrittes:
δALA Porhyrien:4a ↓ 1. erythropoetische P
PBG →UGI, UPI 2. erythrohepatische ProtoP1,3a ↓ 3. hepatische
UGIII → UPIII, UGI, UPI 3a. akut intermittierende P3b ↓ 3b. P. cutanea tarda
KGIII → KPIII 3c. hered. Kopro-Porphyrie3c ↓ 3d. P. variegata
PGIX 4. erworbene P3d ↓ 4a. Pb
PPIX 4b. chlor. Phenole2,4a ↓ 4c. symptomatische P
Häm
Definition der Porphyrien:Krankheiten mit vermehrter Ausscheidung von Porphyrinen in Harn und Stuhl

Hereditäre erythropoetische Porphyrie:
Erythrodontie

Porphyria cutanea tarda

Symptome der Porphyrien (allgemein):
1. Photosensibilität(fehlt, wenn nur δALA und PBG erhöht sind)Excitation der Porphyrine bei ca 400 nm, Emission ca 600 nm→ Urticaria, Oedeme, Ulcera, Hämolyse
2. neurologische Störungen(insbes. neurotoxisch durch δALA und PBG, auch durch Häm-Mangel)zentral - peripher - vegetativ
psych. Veränderungen, LähmungenKoliken oder ObstipationTachycardie und HypertoniePolyurie (ADH-Störung)

Formen der Porphyrien:
1. Erythropoetische P (sehr selten) ARStörung der Erythroblasten → Anämie, Blasen an Haut, VerstümmelungenFluoreszierende Erythrodontie
2. Erythrohepatische ProtoporphyrieGestörte Ferrochelatase, PPIX ↑ in Haut und Leber →Leberinsuffizienz und Ikterus, Cholestyramin hilft
3. Hepatische Porphyrien..-gene wasserlöslich, vor allem im Harn..-ine schlecht wasserlöslich, vor allem im Stuhl
3 a, c, d sind klinisch ähnlich:: Anfälle von kolikartigen Schmerzen, Erbrechen, Obstipation, Parästhesien, Polyurie, Hypertonie ausgelöst durch Medikamente (und Alkohol) die P450 induzieren (Häm-Verbrauch/Mangel)Barbiturate, Meprobamat, Hydantoine, Halothan, Chloramphenicol, Sulfonamidekeine Leberstörungen: Therapie: Hämatin, Noxe vermeiden
3 b: P. cutanea tardavor allem Haut-Symptome, kaum neurologisch, da kein Häm-Defizitα) genetisch bedingter UGIII-Decarboxylase-Mangelß) Enzymstörung bei alkoholischer Leberschädigung und bei erhöhtem Serum- und Leber-Eisen(gefördert durch Blei und Benzol)Therapie: Aderlässe um Eisen zu reduzieren, Chloroquin kann UIII komplexieren → Harn
4. Erworbene4 a: Blei hemmt die δALA-Dehydrase (→ δALA↑ ; neurolog. Schäden) und die Ferrochelatase (alle P's ↑)
Symptome aber besonders abdominell und neurologisch4 b: chlorierte Phenole (z.B. bei Insektizidvergiftungen), Smptome ähnlich der P. cutanea tarda4 c: symptomatisch: Uro- und Koproporphyrin bei vielen Krankheiten erhöht:
Hepatitis, Cirrhose, Fettleber, Cholestase, Anämien, Infektionskrankheiten, Diabetes, Hämochromatose, hereditäre Hyperbilirubinämien (Dubin Johnson, Rotor), Tumore

Ikterus Skleren
Ikterus / Hyperbilirubinämie

Abbau von Häm / Bilirubinstoffechsel
Häm↓ 1. Hämoxygenase (verbraucht O2 und NADPH, Bildung von Fe3+ und CO)Biliverdin↓ 2. Biliverdinreduktase (NADPH→NADP)Bilirubin IXα (BR)
Albumin-Bindung Aufnahme in Leber (OATP)Ligandin-Bindung
↓ 3. UDP-glucuronyltransferase / KonjugationBilirubin-glucuronid (BR-G)
Ausscheidung in die Galle (MRP2)↓ 4. teilweise bakterielle Dekonjugation und ReduktionBilirubin → Urobilinogen → Stercobilinogen
↓ ↓ 5. OxydationUrobilin Stercobilin
Entsprechende Einteilung der Hyperbilirubinämien bzw. des Ikterusvorwiegend konjugiert / vorwiegend nicht konjugiertprä-, intra- und posthepatisch

Unterschiede zwischen nicht-konjugiertem und konjugiertem Bilirubin
Nicht konjugiert KonjugiertBR BR-G
Diazoreaktion indirekt direktLöslichkeit fettlöslich wasserlöslichBindung an Albumin +++ +Renale Ausscheidung 0 +ZNS-Toxizität (Neugeb.) ++
Urobilinogen und Stercobilinogensind farblos, erst Urobilin, Stercobilin und weitere Abbauprodukte sind gefärbt(Dipyrrole, z.B. Mesobilifuscin) und geben dem Stuhl die dunkle Farbe.
Urobilinogen macht den enterohepatischen Kreislauf durch →.fehlt bei Gallegangsverschluß = wird nicht gebildet.passiert die Leber bei Leber-Schädigung und tritt in den Harn über.vermehrt im Stuhl bei Hämolyse

Systematik der Hyperbilirubinämien
I. Vorwiegend unkonjugierte Hyperbilirubinämien
bei vermehrtem Erythroytenabbaubei Störung der Bilirubinaufnahme in die Leberbei Störung der Konjugation in der Leber
II. Vorwiegend konjugierte Hyperbilirubinämien
bei gestörter Sekretion von konjugiertem Bilirubin bei intrahepatischer Cholestasebei Behinderung des Galleflusses („chirurgische Cholestase“)

Systematik der Hyperbilirubinämien (Teil I)
I. Vorwiegend unkonjugierte Hyperbilirubinämienvermehrte Produktion und/oder verminderte Konjugation
1. intravaskuläre und extravaskuläre Hämolyse(vestärkt bei gleichzeitig beeinträchtigter Leberfunktion: Fieber, Sepsis,Schock, Hypoxie, Cirrhose, Lebertumoren....)
a. erworben:Splenomegalie, Immunhämolyse
Wärme-AK (IgG; dir.Coombstest); z.T.Sphärozytenbildung : (idiopathisch, Lymphome, SLE, Tumore, α-Methydopa, Penicillin, Chinidine)Kälteagglutinine (IgM><I-Ag der Ery): idiopathisch, Lymphom, Mycoplasmenparoxysmale Kältehämoglobinurie (IgG-Donath-Landsteiner, früher bei Lues)
mechanisch (Mikroangiopathie), barfüssiges Joggen, Karate, künstl. (Aorten)klappen, maligne Hypertonie,Eklampsie, DIC, thrombotisch-thrombozytopenische Purpura, u.a.
Infektion (Malaria, Clostridien) Membranschäden
Stachelzellen bei Cirrhose: spur: C↑; target: C+PL↑paroxysmale nächtliche Hämoglobinurie(fehlender Decay Accelerating Factor: fehlende C3b Inaktivierung zu C3d)

Systematik der Hyperbilirubinämien (Teil II)
I. Vorwiegend unkonjugierte Hyperbilirubinämien
1. intravaskuläre und extravaskuläre Hämolyse
a. erworben (s.o.):
b. hereditäre Erythrozytendefekte:
MembranschädenSphärozytose (Spectrin-Mangel); Elliptozytose (band 4.1: bindet Actin an Glycophorin)Pyropoikilozytose (Spektrin-Störung, Verformung und Lyse >44oC)Stomatocytose ("ausgetrocknet" oder über-hydriert - gestörter Na-Transport)
EnzymdefekteGlykolyse (Hexose6P-Isomerase, Pyr-Kinase u.a.)Pentose-Shunt (G6P-Dehydrogenase, Phosphogluconat-Reductase → GSH-Reductase) (empfindlich gegenüber Medikamenten: Antimalaria (Chloroquin, Primaquin, Sulfonamide, etc)
HämoglobinopathienChromosom 16: zeta, alpha; Chromosom 11: epsilon, gamma, delta, betaAlpha- und Beta-Thalassämien, SichelzellanämieMethämoglobinämie (stabiles M-Hb oder Cytb5reductase-Mangel)

Systematik der Hyperbilirubinämien (Teil III)I. Vorwiegend unkonjugierte Hyperbilirubinämien1. intravaskuläre und extravaskuläre Hämolyse
a. erworben (s.o.):b. hereditäre Erythrozytendefekte (s.o.):
2. "Ineffektive Erythropoiese"Thalassämieen, B12-Mangel, erythropoietische Porphyrie
3. Infarzierungen und massive BlutungenLungenembolie, Ruptur von Aneurysmen, postoperativer Ikterus
4. Gestörte Aufnahme von Bilirubin in die LeberArzneimittel (Flavaspidic acid - Kompetion mit Ligandin / Kontrastmittel, Novobiocin)Unterform von Gilbert-Syndrom ? (s.u.)
5. Gestörte Konjugation in der LeberNeugeborene, insbes. unreife (in utero : Plazenta als Ausscheidungsorgan,
Ikterus, < 5mg/dl, entwickelt sich nach der Geburt)Fetale Erythroblastose (Kombination mit Nr.1)
> 20 mg/dl Gefahr des KernikterusErbliche nicht-konjugierte Hyperbilirubinämien
Gilbert (TATA-Störung der UDP-Glukuronyltransferase, BR < 6, BR-MG in Galle, Phenobarbital wirksam, kein Kernikterus)(z.T. mit geringer Hämolyse oder BR-Anstieg bei Fasten)
Crigler-Najjar II (dominant, BR < 20, BR-MG in Galle, Phenobarbital zurInduktion der UDP-Glukuronyltransferase wirksam, ± Kernikterus)
Crigler-Najjar I (rezessiv, BR 20-45, Phenobarbital unwirksam, Kernikterus)Erworbene Konjugationsstörungen
Arzneimittel (insbes. bei Neugeborenen: Chloramphenicol, Vit. K)Brustmilchikterus (Pregnan-3ß,20α-diol)Hypothyreose (Kretinismus)z.T. bei parenchymatösen Lebererkrankungen (Hepatitis, Cirrhose...)

Systematik der Hyperbilirubinämien (Teil IV)
I. Vorwiegend unkonjugierte HyperbilirubinämienNr 1-5 s.o.
II. Vorwiegend konjugierte HyperbilirubinämieGestörte Ausscheidung von BR-DG (Harn BR+)
a. Familiäre Störungen:Erblicher Dubin-Johnson-Ikterus
(BR-DG↑, BR↑, MRP-2-Defekt, Melanin-artiges Pigment in Leberzellen, BSP-Rezirkulation, Koproporphyrin -I (statt III) im Urin)
Rotor-Syndrom (wie DJ, aber kein Pigmentbildung und keine BSP-Rezirkulation)BRIC: Benigne familiäre recurrente intrahepatische Cholestase (Episoden mit Pruritus und Ikterus)PFIC: Progressive familiäre intrahepatische recurrente Cholestase
(BRIC und PFIC: Störungen von Transportern der Zellmembran. s.u.)Recurrenter Schwangerschaftsikterus (-Cholestase) (Östrogen-Empfindlichkeit)
b. Erworbene Störungen:Orale Kontrazeptiva17αOH-Androgene (Methyltestosteron)Arzneimittel-Cholestase (Erythromycin, Chloramphenicol + Entzündung /tox-allerg.)Hepatitis (virale u.a. Infektionen, toxisch - arzneimittelinduziert, AIH)intrahepatisches Abflußhindernis (z.B. granulomatöse Erkrankungen)Primär biliäre CirrhoseAutoimmuncholangitis / TransplantatPrimär sklerosierende Cholangitismechanische Abflußbehinderung in den extrahepatischen Gallenwegen
("chirurgische Cholestasen")Steine, Kompression durch Tumore, Narbenstrikturen ......

Genetische Defekte und Lebererkrankungen:
Auswahl:M. Wilson, Hämochromatose, α1-Antitrypsin-Mangel, Störungen des Membrantransports
Viele andere genetische Störungen betreffen die LeberBeispiele sind:
Galaktosämie, Fruktoseintoleranz, Glykogenosenlysosomale Speichererkrankumgen, peroxysomale Störungen (Zellweger´s cerebrohepatorenales Syndrom), Störungen des LipoproteinstoffwechselsStörungen der Proteinsekretion (z.B. Afibrinogenämie) Störungen der Harnstoffsynthese

xx
MorbusWilson
Kupferspeicherung

M. Wilson – Kupferspeichererkrankung - hepatolentikuläre Degeneration
Die Kupferausscheidung erfolgt durch eine Transport-ATPase in die Gallenkanalikuli. ATP7B:im endoplasmatischen Retikulum synthetisiert, im Golgi modifiziertin Membranen des Transgolgi-Netzwerks und in perikanalikulären Vesikeln, Exozytose in das Lumen der Kanalikuli
Pathologie und KlinikAR, am Chromosom 13 kodiertVerschiedene Mutationen von ATP7B führen zu veränderter Expression / Lokalisatioon
z.B. H1069Q-ATP7B im endoplasmatischen Retikulumunvollstädiges (verkürztes) Protein diffus granulär im Zytoplasma
Die Kupferausscheidung über die Galle ist damit herabgesetzt und nur unvollständig durch renale Ausscheidung kompensiert, sodass Kupfer in verschiedenen Geweben abgelagert wird und zu Störungen führt. In ca. 95% der Fälle ist der Coeruloplasminspiegel des Plasmas vermindert.
Leberzellschädigung mit Nekrosen und Übergang in Zirrhose, Zellen sind balloniert, Lipofuscin, Lochkerne (intranukleäres Glykogen), Mallory Körper. Periportalfelder mit lymphozytäre Infiltration. Die Leberschädigung steht vor allem bei jugendlichen Patienten im Vordergrund.Charakteristisch: Kayser-Fleischer´scher Kornealring. Evtl. Katarakt.Degeneration der Stammganglien (Linsenkern) führt zu extrapyramidal-motorischen Störungen ( Rigor, Bewegungsarmut, Intentionstremor)Schädigung des proximalen Nierentubulus führen zu vielfachen Transportstörungen

M. Wilson: Verfärbungen an der Leber, endoskopisch
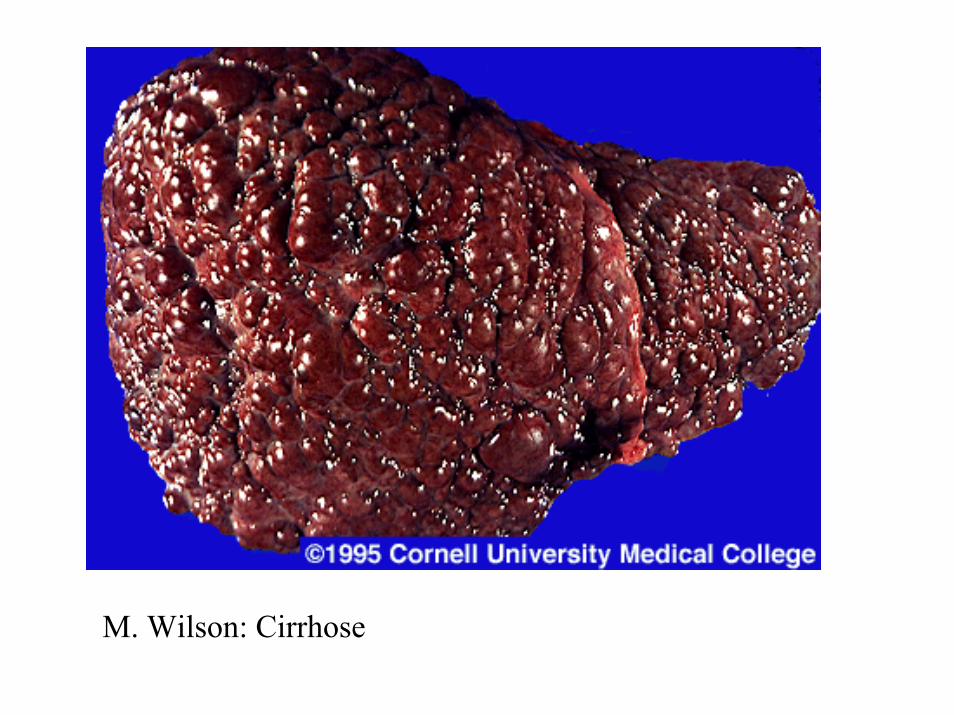
M. Wilson: Cirrhose

M. Wilson
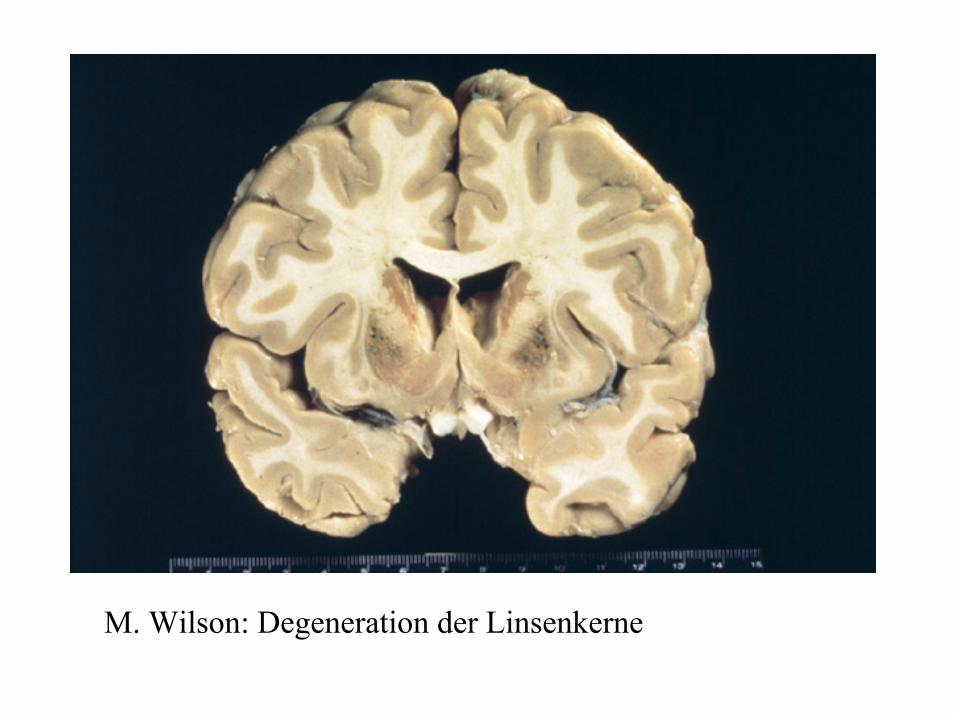
M. Wilson: Degeneration der Linsenkerne

xx
HereditäreHämochromatose
EisenspeicherungBronzediabetes

Hereditäre (idiopathische) Hämochromatose (HHC) – Bronzediabetes (I)
EisenresorptionFe2+ an der luminalen Membran der Enterozyten über drei Mechanismen aufgenommen:
Endozytose von luminalem Transferrin (Tf)-Eisennach Bindung an den Transferrin-Rezeptor (TfR),
Cotransport von Fe2+ mit H+ mittels DCT1 (=DMT1; divalent cation (metal) transporter)
Transport vom Häm-Eisen (Mechanismus ?). Intrazellulär bindet Fe2+ an Mobilferrin und wird durch die basolaterale Membran durch Ferroportin (= IREG1, iron regulated gene 1) an das Blut abgegeben.
Bei Eisenmangel ist die Expression von DCT1 und IREG1 erhöht. Dabei beträgt das Serum-Eisen <0.4 mg/l, das ungesättigtes Serum-Transferrin (= ungesättigte Eisenbindungskapazität, UEBK) ist erhöht, und Serum-Ferritin erniedrigt. Es besteht also eine inverse Korrelation zwischen der Expression der Transportproteine und dem Serum-Eisen bzw. Serum-Ferritin.
Bei hereditärer Hämochromatose verhalten sich die Enterozyten des Duodenums so, als ob Eisenmangel bestünde, d.h.: trotz Eisenüberladung des Organismus ist die Eisenresorption gesteigert.

„MUCOSA-BLOCK“ der Eisenresorption

Hereditäre (idiopathische) Hämochromatose (HHC) – Bronzediabetes (II)
HFE:
HFE, früher HLA-H, ist verwandt mit MHC Klasse I Molekülen.85% der Patienten homozygot für die Cys282Tyr Mutation (Cys durch Tyr ersetzt) an HFE. Das Gen ist auch an Chromosom 6 lokalisiert; die Mutation liegt in der ß2-Mikroglobulin-Bindungsdomäne.Manche Patienten sind heterozygot mit C282Y und H63D Mutationen.
Die Beziehung der seit 1975 bekannten Mutation von HFE zur gesteigerten Eisenresorption ist noch nicht vollständig geklärt:
Hypothesen der Pathogenese:
1. nicht mutiertes HFE an der apikalen Zellmembran ist assoziiert mit dem Transferrin-Rezeptor (TfR).HFE hemmt die Bindung von Fe-Tf Komplexen.C292Y-HFE wird vemindert exprimiert, die Blockade fällt weg.
2. In den Krypten vermindert das mutierte C292Y-HFE die Aufnahme von Fe aus dem, Blut.Bei HHC ist das intrazelluläre Eisen vermindert →Enterozyten an der Zottenspitze exprimieren an der apikalen Membran vermehrt DCT1 und IREG1
seltenere genetischer Defekte, die zu Eisenüberladung führen:(b) Juvenile Hämochromatose – HFE2-Hämochromatose, Gen an Chromosom 1, Funktion unbekannt.(c) HFE3-Hämochromatose mit Mutation an Transferrinrezeptor 2(d) HFE4-Hämochromatose: Mutation an Ferroportin, das auch an Makrophagen exprimiert wird.
An überladenen Zellen wird Eisen als Ferritin und Hämosiderin gespeichert. Die schädigende Wirkung von Fe beruht vor allem auf gesteigerter Lipidperoxydation.

Hereditäre (idiopathische) Hämochromatose (HHC) – Bronzediabetes (III)
Symptomatik
HHC äußert sich vor allem bei Männern (♂ : ♀ = 10 : 1) meist erst nach dem 40 Lj.Lebervergrößerung mit Übergang zu Zirrhose erhöhtes Risiko für Leberkarzinome braun-graue Hautpigmentierung ("Bronzefärbung") endokrine Störungen (Diabetes mellitus, Hypopituitarismus mit HodenatrophieSchädigung quergestreifter Muskulatur (Herzinsuffizienz) SplenomegalieLeukopenie, ThrombopenieKnochenmarksveränderungen (Sideroblasten) Arthritis.
Für die Therapie durch regelmäßige Aderlässe ist eine frühzeitige Diagnose von Bedeutung. Da die Penetranz der Erkrankung gering ist (> 1/10 der Bevölkerung ist heterozygot für die C282Y Mutation !) ist der Wert genetischer Analysen fraglich. Es sollte bei Verdacht bzw. familiärer Häufung daher auch auf unspezifische Frühsymptome geachtet werden wie Müdigkiet ohne erklärbare Ursache, Gelenksschmerzen, Herklopfen, Bauchschmerzen, erhöhte Leberwerte, Lebervergrößerung und erhöhtes Serum-Ferritin, um eine rechtzeitige Therapie einzuleiten.

Zelluläre Eisenablagerungen bei Hämochromatose

Hämochromatose

xx
α1-Antitrypsin-Mangel
Beteiligung von Leber und Lunge

Entstehung eines Lungenemphysems und einer chronisch obstruktiven Lungenfunktionsstörung (COPD, chronic obstructive pulmonarydisease), insbesondere bei Rauchern, bei denen Neutrophile und Makrophagen aktiviert sind: Vermehrte Bildung von Proteasen und Inaktivierung der Inhibitoren

α1-Antitrypsin-Mangelα1-Antitrypsin (AAT)
52-kDa Glykoprotein, zur Familie der Serpine (Serinprotease-Inhibitoren). Von der Leber sezerniert (Akute-Phase-Protein). Steuert Gleichgewicht zwischen Aufbau und Abbau von Matrixproteinen
In der Lunge wirkt AAT als Inhibitor der Neutrophilen-Elastase und von Matrix-abbauenden Proteasen der Makrophagen. Es schützt vor dem Abbau von Elastin und verhindert die Entstehung eines Lungenemphysems und auch einer chronisch obstruktiven Lungenfunktionsstörung (COPD, chronic obstructive pulmonary disease), insbesondere bei Rauchern, bei denen Neutrophile und Makrophagen aktiviert sind.
AATD (AAT-Deficiency)autosomal rezessiv Mehr als 75 verschiedene Mutationen der normalen AAT-Allele (MM) bekannt homozygote ZZ Mutation (342 Glu → Lys) weitaus am häufigsten (1/2500-1/6000)
Folgen:Hemmung der Sekretion und folgende Leberschäden
(intrazelluläre Polymerisation der ZZ-Form)panlobulären Emphysems (early onset emphysema).
Sowohl der Zeitpunkt und Grad der Leberschädigung als auch die Ausbildung eines Lungenemphysems sind sehr variabel – nur ca. 10-15% der Homozygoten (ZZ) zeigen klinische Symptome einer Leberinsuffizienz (evtl. schon als Neugeborene), Lungenemphysem im Erwachsenenalter ist häufiger. Vorhersagen über den Krankheitsverlauf sind kaum möglich. Besondere Gefahr des Rauchens !

Einschlußkörper in Leberzellen bei AATD

xx
Selteneangeborene Störungen
des Membrantransportes
mit Cholestaseund / oder Ikterus

Basolaterale Transporter

Canaliculäre Transporter

Duktuläre Transporter

Transport-Proteine an Membranen der Leberzellen und ihre genetischen Störungen
Abkürzung Funktion ErkrankungCanalikuläre Membran
MDR1 amphiphile, organische ?(PGP) CationenMDR3 Phospholipid Transport PFIC-3 (gGT )
"Flippase"MRP2 multispezifischer Dubin-Johnson Syndrom
org. Anionen-TransportSPGP Gallensäuren PFIC-2
(Byler-Subtyp)FIC1 (Lokalisation ?) ? PFIC-1, BRIC
(Byler-Subtyp)ATP7B (vesiculär) Cu-Transport M Wilson
Basolaterale (sinusoidale) Membran
NTCP Na-abhängiger ?Taurocholat Transport
OATP org. Anionen ?OCT1 org. Cationen ?
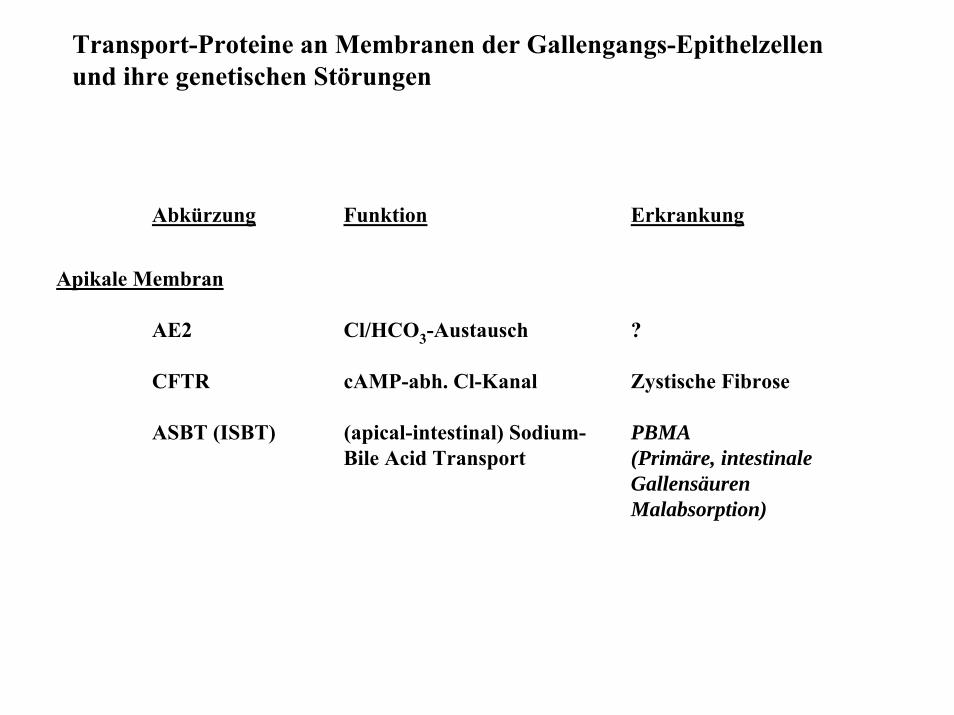
Transport-Proteine an Membranen der Gallengangs-Epithelzellen und ihre genetischen Störungen
Abkürzung Funktion Erkrankung
Apikale Membran
AE2 Cl/HCO3-Austausch ?
CFTR cAMP-abh. Cl-Kanal Zystische Fibrose
ASBT (ISBT) (apical-intestinal) Sodium- PBMABile Acid Transport (Primäre, intestinale
GallensäurenMalabsorption)

Lernunterlagen zur Vorlesung und Prüfungsstoff:
www/meduniwien.ac.at/graf/B13
(2 MB)
Ausführliche Lernunterlagen in Englisch z.B.:Univ. Washington: Integrierter Kurs zum Gastrointestinaltrakthttp://www.uwgi.org/gut/liver_01.asp


















