
Diploma rb eit - mpip-mainz.mpg.de · Dadurc h kann eine V ersorgung der Zelle, ab er auc h die...
180
Orientierungsspezifische Studie der Cytochrom-c-Oxidase in einer biomimetischen Architektur auf Metalloberflächen - Eine kombinierte Untersuchung mit oberflächenverstärkter resonanter Ramanspektroskopie und Elektrochemie Diplomarbeit Markus Alexander Plum Fachbereich Physik Max-Planck-Institut für Polymerforschung Mainz, Mai 2007
Transcript of Diploma rb eit - mpip-mainz.mpg.de · Dadurc h kann eine V ersorgung der Zelle, ab er auc h die...

Orientierungsspezifische Studie derCytochrom-c-Oxidase in einer biomimetischen
Architektur auf Metalloberflächen-
Eine kombinierte Untersuchung mitoberflächenverstärkter resonanter Ramanspektroskopie
und Elektrochemie
Diplomarbeit
Markus Alexander PlumFachbereich Physik
Max-Planck-Institutfür Polymerforschung
Mainz, Mai 2007

Die vorliegende Arbeit wurde amMax-Planck-Institut für Polymerforschung in Mainz
unter Anleitung von
Prof. Dr. K. Kremer, Prof. Dr. W. Knoll und Dr. R. Naumann
in der Zeit von April 2006 bis Mai 2007 angefertigt.
Die Arbeit wurde von Prof. Dr. L. Köpkevon der Universität Mainz mitbetreut.

INHALTSVERZEICHNIS I
Inhaltsverzeichnis1 Einleitung und Motivation 1
1.1 Atmungskette . . . . . . . . . . . . . . . . . . . . . . . . . . . 11.1.1 Mitochondrale Atmungskette . . . . . . . . . . . . . . 31.1.2 Prokaryotische Atmungskette . . . . . . . . . . . . . . 4
1.2 Cytochrom c . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.3 Cytochrom-c-Oxidase . . . . . . . . . . . . . . . . . . . . . . . 7
1.3.1 Struktur der Cytochrom-c-Oxidase . . . . . . . . . . . 71.3.2 Katalytischer Zyklus der Cytochrom-c-Oxidase . . . . . 11
1.4 His-Tag-Technologie . . . . . . . . . . . . . . . . . . . . . . . 121.5 Motivation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2 Theoretische Grundlagen und Messmethoden 172.1 Oberflächenplasmonenresonanz . . . . . . . . . . . . . . . . . 17
2.1.1 Elektromagnetische Beschreibung von Oberflächenplas-monen . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.1.2 Anregung von Oberflächenplasmonen . . . . . . . . . . 222.1.3 Oberflächenplasmonenresonanz-Spektroskopie . . . . . 24
2.2 EIS - Elektrochemische Impedanzspektroskopie . . . . . . . . 272.2.1 Messprinzip der Elektrochemischen Impedanzspektro-
skopie . . . . . . . . . . . . . . . . . . . . . . . . . . . 272.2.2 Graphische Darstellungen der Impedanzspektren . . . . 302.2.3 Schaltkreiselemente . . . . . . . . . . . . . . . . . . . . 312.2.4 Einfache Ersatzschaltkreise . . . . . . . . . . . . . . . . 332.2.5 Spezielle Ersatzschaltkreise . . . . . . . . . . . . . . . . 40
2.3 CV - Cyclovoltammetrie . . . . . . . . . . . . . . . . . . . . . 442.3.1 Cyclovoltammetrie von gelösten Redoxspezies . . . . . 472.3.2 Cyclovoltammetrie von oberflächengebundenen Redox-
spezies . . . . . . . . . . . . . . . . . . . . . . . . . . . 512.3.3 IR-Drop Kompensation . . . . . . . . . . . . . . . . . . 52
2.4 Spektroelektrochemie . . . . . . . . . . . . . . . . . . . . . . . 542.4.1 Potentiostatische Messungen . . . . . . . . . . . . . . . 542.4.2 Zeitaufgelöste Messungen . . . . . . . . . . . . . . . . . 55
2.5 Ramanspektroskopie . . . . . . . . . . . . . . . . . . . . . . . 562.5.1 Darstellung des Raman-Effekts über das Energieterm-
schema . . . . . . . . . . . . . . . . . . . . . . . . . . . 562.5.2 Klassische Deutung des Raman-Effekts . . . . . . . . . 572.5.3 Resonanter Raman-Effekt . . . . . . . . . . . . . . . . 602.5.4 Oberflächenverstärkungseffekt . . . . . . . . . . . . . . 612.5.5 Resonanz Ramanspektroskopie an Metalloporphyrinen 62

INHALTSVERZEICHNIS II
3 Probenherstellung für SPR, EIS und CV 663.1 Herstellung von TSG-Elektroden . . . . . . . . . . . . . . . . 663.2 Funktionalisierung der TSG-Elektrode . . . . . . . . . . . . . 683.3 Anbindung der Cytochrom-c-Oxidase an die Elektrodenober-
fläche . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 693.4 Cytochrom-c-Oxidase in der up- oder down-Konfiguration . . . 733.5 Cytochrom-c-Oxidase N139C . . . . . . . . . . . . . . . . . . . 75
4 Messaufbau für SPR, EIS und CV 76
5 Messergebnisse und Diskussion (Teil 1) 805.1 Überprüfung des ptBLM-Aufbaus . . . . . . . . . . . . . . . . 80
5.1.1 Ergebnisse und Diskussion der Oberflächenplasmonen-resonanz - Spektroskopie . . . . . . . . . . . . . . . . . 80
5.1.2 Ergebnisse und Diskussion der elektrochemischen Impe-danz-Spektroskopie . . . . . . . . . . . . . . . . . . . . 82
5.2 Aktivierung der Cytochrom-c-Oxidase in der up-Konfiguration 845.2.1 Ergebnisse und Diskussion der elektrochemischen Impe-
danz-Spektroskopie . . . . . . . . . . . . . . . . . . . . 855.2.2 Ergebnisse der CV-Messungen . . . . . . . . . . . . . . 875.2.3 Diskussion der CV-Messungen . . . . . . . . . . . . . . 92
5.3 Potentialtitration der Cytochrom-c-Oxidase in der up-Kon-figuration mit und ohne Cytochrom c . . . . . . . . . . . . . . 955.3.1 Ergebnisse und Diskussion der EIS-Potentialtitration . 95
5.4 Aktivierung der Cytochrom-c-Oxidase in der down-Konfigura-tion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 995.4.1 Ergebnisse und Diskussion der CV-Messungen mit der
Mutante N139C . . . . . . . . . . . . . . . . . . . . . . 100
6 Probenherstellung für SERRS 1026.1 Herstellung der Silberelektroden . . . . . . . . . . . . . . . . . 1026.2 Polieren der Silber-Elektroden . . . . . . . . . . . . . . . . . . 1036.3 Elektrochemisches Aufrauhen der Silber-Elektroden . . . . . . 1036.4 Funktionalisierung und Proteinanbindung . . . . . . . . . . . . 104
6.4.1 Untersuchungen mit Cytochrom c . . . . . . . . . . . . 1046.4.2 Untersuchungen mit der Cytochrom-c-Oxidase . . . . . 104
7 Messaufbau für SERRS 1077.1 Messaufbau für die potentiostatischen Messungen . . . . . . . 1077.2 Messaufbau für die zeitaufgelösten Messungen . . . . . . . . . 1087.3 Zelldesign der SERRS-Zelle . . . . . . . . . . . . . . . . . . . 109

INHALTSVERZEICHNIS III
7.3.1 Zelldesign der alten Ramanzelle . . . . . . . . . . . . . 1107.3.2 Zelldesign der neuen Ramanzelle . . . . . . . . . . . . 112
8 Messergebnisse und Diskussion (Teil 2) 1148.1 Oberflächenaufrauhung der Silberelektroden . . . . . . . . . . 114
8.1.1 Durchführung der elektrochemischen Aufrauhung . . . 1148.1.2 Optimierung der verwendeten Aufrauhungszeiten . . . 115
8.2 SERRS-Messungen . . . . . . . . . . . . . . . . . . . . . . . . 1188.2.1 Ergebnisse und Diskussion der SERRS-Messungen mit
Cytochrom c . . . . . . . . . . . . . . . . . . . . . . . 1188.2.2 Ergebnisse und Diskussion der SERRS-Messungen mit
der Cytochrom-c-Oxidase . . . . . . . . . . . . . . . . 123
9 Zusammenfassung und Ausblick 1309.1 Zusammenfassung . . . . . . . . . . . . . . . . . . . . . . . . . 1309.2 Ausblick . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 132
A Anhang 134A.1 Datenmessung und Datenauswertung . . . . . . . . . . . . . . 134
A.1.1 Messung und Auswertung der SPR-Daten . . . . . . . 134A.1.2 Messung und Auswertung der EIS-Daten . . . . . . . . 135A.1.3 Messung und Auswertung der CV-Daten . . . . . . . . 137A.1.4 Auswertung der SERRS-Daten . . . . . . . . . . . . . 139
A.2 Material . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 142A.2.1 Lösungen . . . . . . . . . . . . . . . . . . . . . . . . . 142A.2.2 Chemikalien und Materialien . . . . . . . . . . . . . . 143A.2.3 Geräte . . . . . . . . . . . . . . . . . . . . . . . . . . . 146
A.3 Rotations-Raman-Effekt . . . . . . . . . . . . . . . . . . . . . 148A.4 Konstruktionszeichnung der neuen SERRS-Zelle . . . . . . . . 149A.5 Versuchsaufbau für eine RDE-Messung . . . . . . . . . . . . . 149A.6 Polieren und Aufrauhen der Silberelektroden . . . . . . . . . . 151
A.6.1 Polieren der Silberelektroden . . . . . . . . . . . . . . . 151A.6.2 Neu entwickelte Poliervorschrift . . . . . . . . . . . . . 151A.6.3 AFM-Bilder der polierten und chemisch aufgerauten
Silberelektroden . . . . . . . . . . . . . . . . . . . . . . 152
B Glossar 157
Abbildungsverzeichnis 158
Tabellenverzeichnis 163

INHALTSVERZEICHNIS IV
Literatur 164
Danksagungen 174

1 EINLEITUNG UND MOTIVATION 1
1 Einleitung und MotivationEin Membranprotein ist ein in die Lipidschicht einer Biomembran eingela-gertes Protein. Diese Proteine können als Transmembranproteine die Lipid-doppelschicht der Zellmembran vollständig durchziehen und eine Art Tunnelbilden, um wichtige Transportfunktionen zu übernehmen. Sie bedingen eineselektive Permeabilität der Zelle und sorgen so für einen Stoffaustausch zwi-schen Organellen und der Zellaußenwelt. Der Transport kann dabei aktiv, alsounter Verbrauch von Energie, oder passiv, ohne Energieverbrauch, erfolgen.Dadurch kann eine Versorgung der Zelle, aber auch die Aufrechterhaltungeines bestimmten Zellmilieus oder eines bestimmten Membranpotentials ge-währleistet werden [1].Eine weitere Funktion von Membranproteinen ist die Katalyse wichtiger bio-chemischer Reaktionen, zum Beispiel zur Energiegewinnung. Dies ist vongroßer Bedeutung für die Versorgung und Aufrechterhaltung der Lebens-funktionen eines Organismus [1].Detaillierte Untersuchungen zur Funktion und Kinetik dieser wichtigen Pro-teinklasse sind zur Zeit aufgrund der mangelnden Stabilität der Proteine undder benötigten Membranumgebung nur schwer durchführbar.Die Herstellung von Protein verankerten Lipiddoppelschichtmembranen1 er-öffnet eine Vielzahl von Anwendungsmöglichkeiten zur Beobachtung von Mem-branproteinen. Die ptBLM-Technik ermöglicht ”in-situ” Untersuchungen, diedie Verwendung als Modellmembransysteme zur Erforschung grundlegen-der biophysikalischer Prozesse einschließt. Durch die direkte Verbindung desMembranproteins mit einer Elektrode mit Hilfe der His-Tag-Technologie istes möglich, potentiostatische Messungen vorzunehmen [2].Im Folgenden werden das Transmembranprotein Cytochrom-c-Oxidase undsein Substrat Cytochrom c, die in dieser Arbeit untersucht werden, eingeführtund der derzeitige Forschungsstand erläutert.
1.1 AtmungsketteDie Hydrolyse der Phosphorsäureanhydrid - Bindung des Adenosintriphos-phats (ATP) stellt die wichtigste Energiequelle der endergonischen Abläufein Zellen aller Organismen dar. In aeroben Organismen, von Bakterien bishin zu den Säugetieren, werden 90 % des Adenosintriphosphats durch dieAtmungskette erzeugt [1].
1Eine Protein verankerte Lipiddoppelschichtmembrane wird auch als ptBLM-System(protein tethered bilayer lipid membrane system) bezeichnet.

1 EINLEITUNG UND MOTIVATION 2
Abbildung 1.1: Die Atmungskette.
Die Atmungskette besteht - wie in Abbildung 1.1 gezeigt2 - aus einer Elek-tronentransportkette (Komplex I-IV) und einer ATP-Synthase (Komplex V).Die im Zuge des Abbaus von Nährstoffen gebildeten reduzierten Reduktions-äquivalente FADH2 und NADH werden über die Atmungskette reoxidiert.Die Elektronen werden hierbei unter der Bildung von Wasser auf Sauerstoffals terminalen Elektronenakzeptor übertragen. Die große Differenz der Nor-malpotentiale von NADH/NAD+ (! = ! 0.32 V) zu H2O/O2 (! = + 0.81 V)wird genutzt, um die durch die Enzyme der Atmungskette katalysierte Kaska-de gekoppelter Redoxreaktionen anzutreiben. Zusammenfassend läuft dabeidie exergonische Knallgasreaktion - die Oxidation von Wasserstoff zu Was-ser - ab. Nach der chemiosmotischen Theorie dient die bei diesem Prozessfreiwerdende Energie primär zum Aufbau eines elektrochemischen Protonen-gradienten. Dieses elektrochemische Potential wird zur Phosphorylierung vonADP durch die ATP-Synthase genutzt. Das so erhaltene ATP ist die univer-selle Energiewährung der Zelle [1].Die Atmungskette läuft bei Eukaryoten in den Mitochondrien und bei Pro-karyoten3 im Cytoplasma ab [1].
2In dieser Arbeit wurden alle Röntgenstrukturbilder mit dem frei zugänglichen Pro-gramm ”Chimera” erstellt.
3Als Eukaryoten werden alle Lebewesen mit Zellkern zusammengefasst. Prokaryotendagegen haben keinen Zellkern

1 EINLEITUNG UND MOTIVATION 3
1.1.1 Mitochondrale Atmungskette
Ein Mitochondrium enthält neben seiner äußeren Membran, die an den Inter-membranraum grenzt, noch eine innere Membran, welche direkten Kontaktzur Matrix hat. Den Raum zwischen diesen beiden Membranen nennt manIntermembranraum [1].Die Komplexe der eukaryotischen Atmungskette sind, wie in Abbildung 1.2 zusehen, in der Mitochondrienmembran lokalisiert. Die Atmungskette bestehtaus den Enzym-Komplexen I bis V und den Elektronenüberträgern Cyto-chrom c und Ubichinon, auch Coenzym Q genannt. Die Enzym-KomplexeI bis V sind in der inneren Mitochondrienmembran eingelagert. Das in ver-schiedenen Stoffwechselwegen gewonnene Reduktionsäquivalent NADH wirdan Komplex I (NADH - Ubichinon - Oxidoreduktase) zu NAD+ reoxidiert.Die Elektronen werden daraufhin auf ein in der Membran lösliches Ubichinon(CoQ) übertragen. Ubichinon kann zusätzlich von Komplex II (Succinat-Ubichinon - Oxidoreduktase)4 reduziert werden. Ubichinon überträgt dieElektronen wiederum auf Komplex III (Ubichinon - Cytochrom - c - Oxi-doreduktase, auch bc1-Komplex genannt). Dieser Komplex hat die Aufga-be, das wasserlösliche Cytochrom c zu reduzieren, welches als Elektronen-mediator zwischen Cytochrom bc1 und Komplex IV (Cytochrom-c-Oxidase)fungiert. Die Cytochrom-c-Oxidase katalysiert die Übertragung der Elektro-nen auf molekularen Sauerstoff. Der Elektronentransport über die KomplexeI, III und IV ist an eine Translokation von Protonen durch die Membrangekoppelt und trägt somit zum Aufbau eines Protonengradienten und desMembranpotentials bei. Diese bilden gemeinsam den elektrochemischen Po-tentialgradienten, welcher wiederum gemäß der chemiosmotischen Theoriezur ATP-Synthese aus ADP und Pi (anorganisches Phosphat) genutzt wer-den kann [1].
4Der Komplex II oxidiert FADH2, welches ebenfalls ein in verschiedenen Sto!wech-selwegen gewonnenes Reduktionsäquivalent ist. FADH2 kommt nicht bei allen Lebewesenvor. Dementsprechend fehlt dann dieser Komplex, wie in Abbildung 1.2 zu sehen.
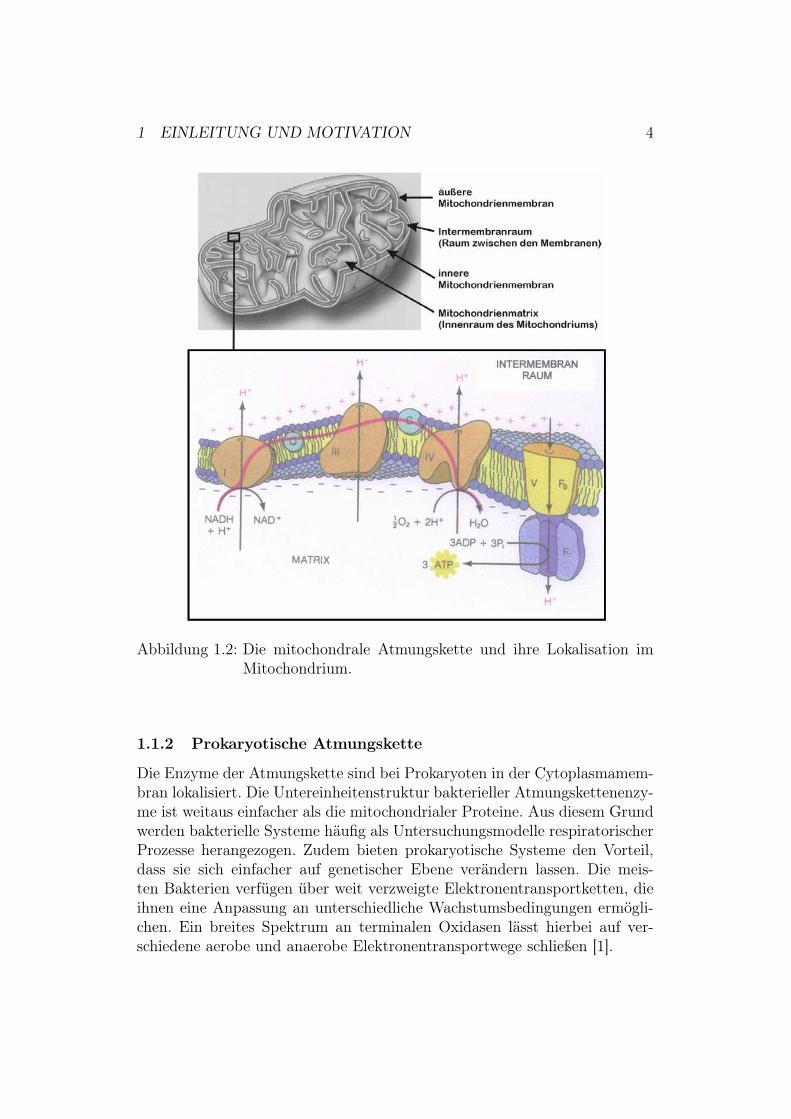
1 EINLEITUNG UND MOTIVATION 4
Abbildung 1.2: Die mitochondrale Atmungskette und ihre Lokalisation imMitochondrium.
1.1.2 Prokaryotische Atmungskette
Die Enzyme der Atmungskette sind bei Prokaryoten in der Cytoplasmamem-bran lokalisiert. Die Untereinheitenstruktur bakterieller Atmungskettenenzy-me ist weitaus einfacher als die mitochondrialer Proteine. Aus diesem Grundwerden bakterielle Systeme häufig als Untersuchungsmodelle respiratorischerProzesse herangezogen. Zudem bieten prokaryotische Systeme den Vorteil,dass sie sich einfacher auf genetischer Ebene verändern lassen. Die meis-ten Bakterien verfügen über weit verzweigte Elektronentransportketten, dieihnen eine Anpassung an unterschiedliche Wachstumsbedingungen ermögli-chen. Ein breites Spektrum an terminalen Oxidasen lässt hierbei auf ver-schiedene aerobe und anaerobe Elektronentransportwege schließen [1].

1 EINLEITUNG UND MOTIVATION 5
1.2 Cytochrom cCytochrom c gehört zu den bestuntersuchten Cytochromen5 überhaupt. De-taillierte Informationen findet man in [3].Die Cytochrom c-Strukturen von Rinderherz und Thunfisch wurden bereitsin den frühen 70er Jahren mittels Röntgenstrukturanalyse erkundet [4][5]. ImJahre 1990 gelang Bushnell die Aufklärung der 3D-Struktur von Rinderherz-Cytochrom c mit einer sehr hohen Auflösung [6].Da der Schwerpunkt dieser Arbeit auf der Cytochrom-c-Oxidase liegt, wirdim Folgenden nur auf die Struktur des Cytochrom c eingegangen. Cytochromc aus Rinderherz (Abbildung 1.3) enthält eine kovalent gebundene Hämgrup-pe, die über zwei Thioetherbrücken zwischen den Thiolgruppen der Cysteine14 und 17 und den Vinylgruppen des Porphyrinrings an das Proteinrückgratgebunden ist (Abbildung 1.3 und 1.4). Wie in Abbildung 1.4 zu sehen, sinddie Imidazolgruppe des Histidins 18 und die Aminosäure Methionin 80 dieLiganden des Häm-Eisens.
Abbildung 1.3: Die Röntgenstruktur des Cytochrom c (Rinderherz) mit Häm- Gruppe.
5Cytochrome sind Proteine, die eine oder mehrere prosthetische Hämgruppen aufwei-sen.
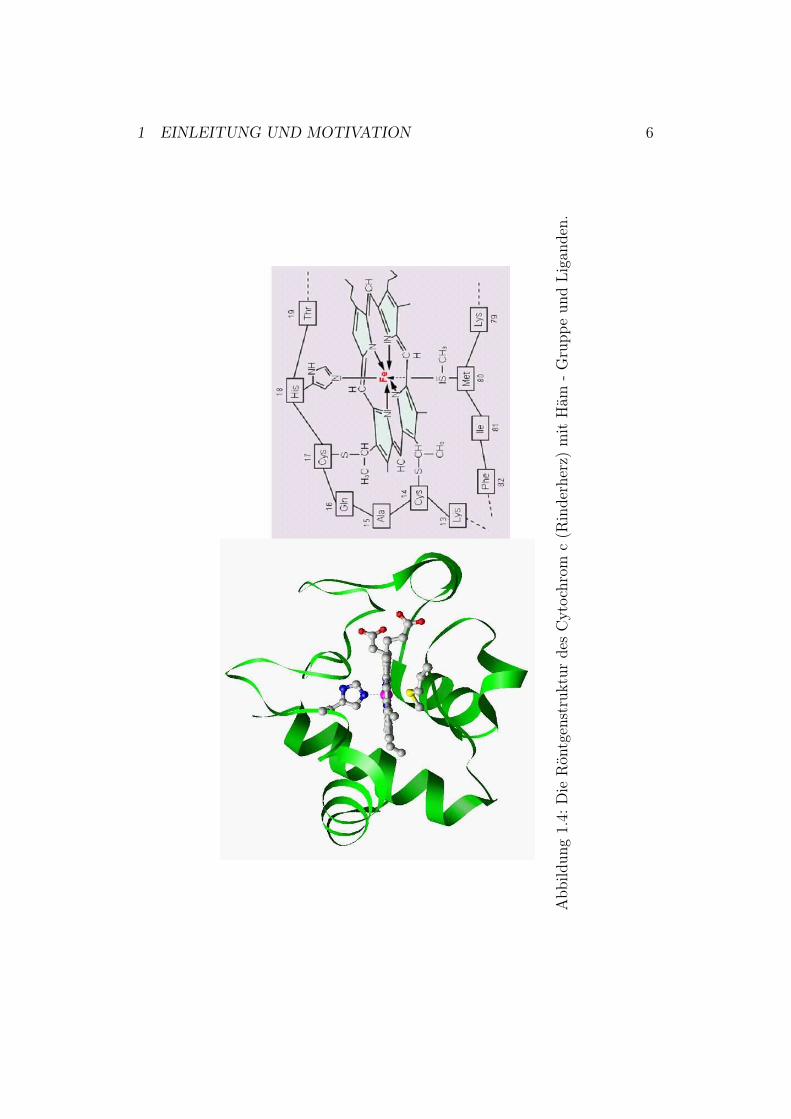
1 EINLEITUNG UND MOTIVATION 6
Abb
ildun
g1.
4:D
ieR
öntg
enst
rukt
urde
sC
ytoc
hrom
c(R
inde
rher
z)m
itH
äm-G
rupp
eun
dLi
gand
en.

1 EINLEITUNG UND MOTIVATION 7
1.3 Cytochrom-c-OxidaseDie Cytochrom-c-Oxidase ist, wie oben beschrieben, das terminale Enzym derAtmungskette. Dieses Enzym katalysiert den Transfer von vier Elektronen,die nacheinander von Cytochrom c geliefert werden, zum Sauerstoffmolekül.Die Reduktion des Sauerstoffmoleküls zu Wasser ist mit einer Verschiebung("Pumpen") von vier Protonen über die innere Mitochondrienmembran ver-bunden [7].
1.3.1 Struktur der Cytochrom-c-Oxidase
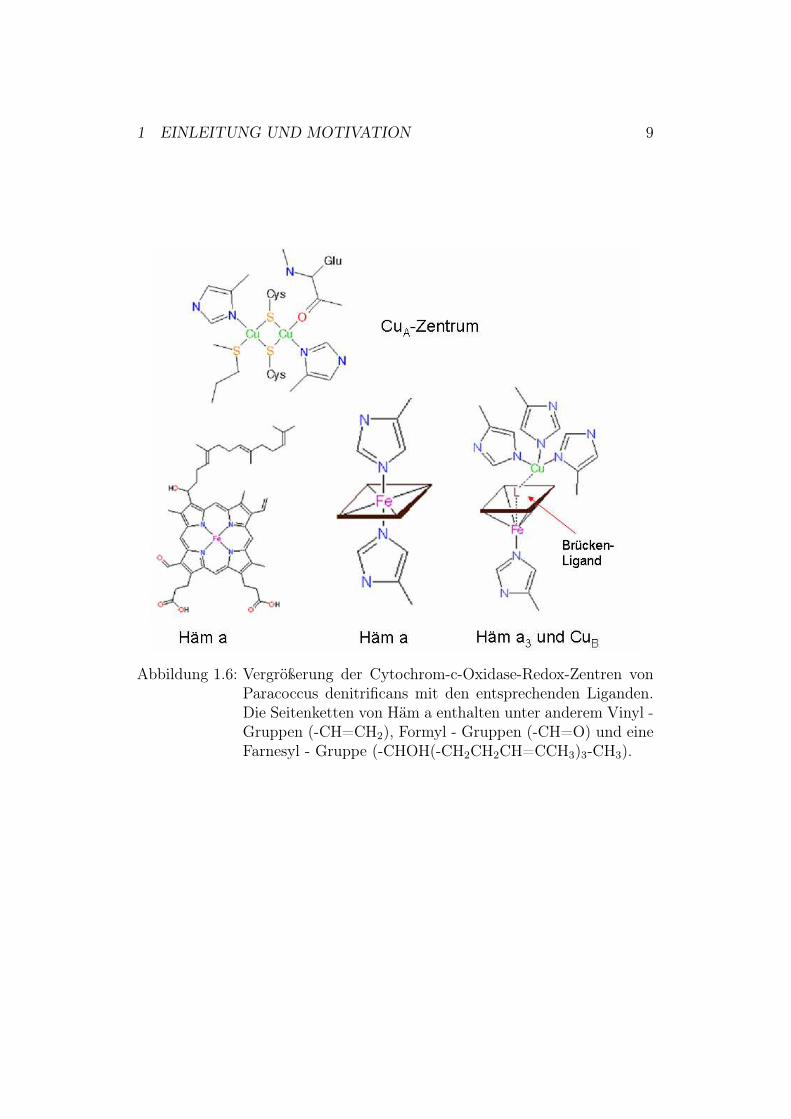
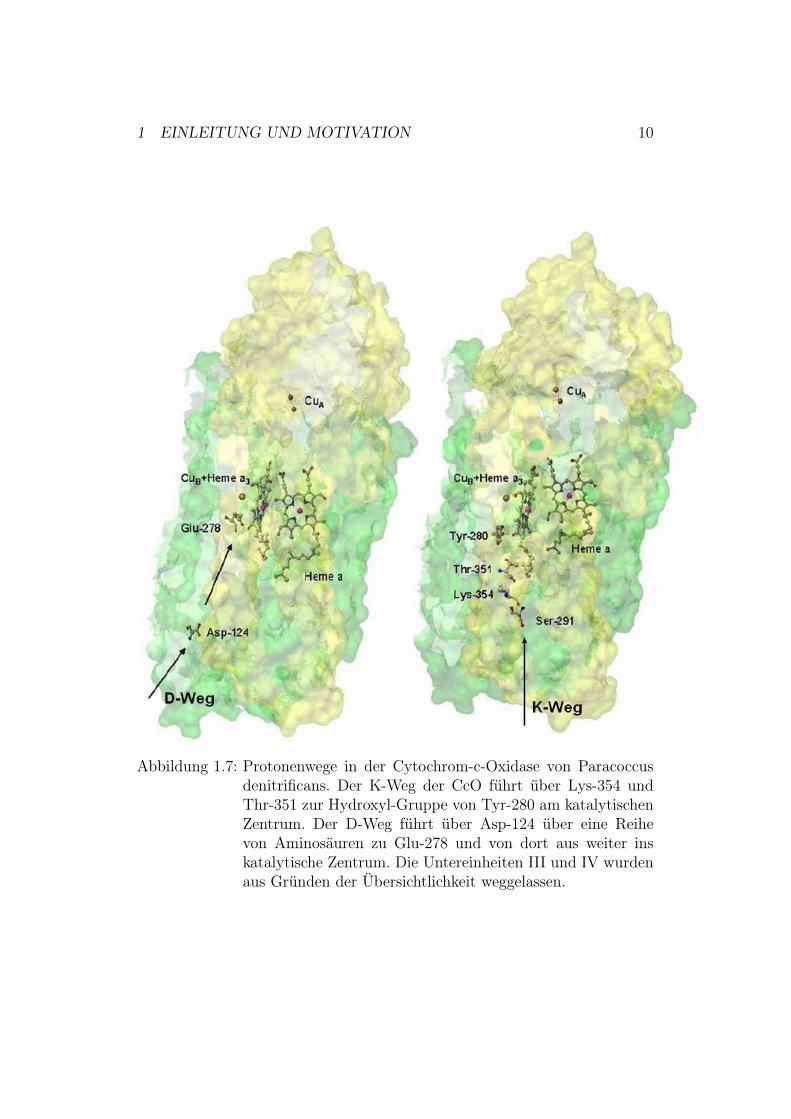
Die atomaren Strukturen der mitochondrialen Cytochrom-c-Oxidase aus Rin-derherz [8] und der bakteriellen Cytochrom-c-Oxidase aus Paracoccus deni-trificans [9] wurden mittels Röntgenkristallographie bestimmt.Cytochrom-c-Oxidase aus Paracoccus denitrificans besteht - wie in Abbil-dung 1.5 zu sehen - aus 3 Kernuntereinheiten (I: grün, II: gelb und III: blau)und einer kleinen, nicht konservierten Untereinheit IV unbekannter Funktion(schwarz) [10].Das Enzym besitzt 4 redox-aktive Metallzentren. Elektronen gelangen vonCytochrom c über das binukleare CuA-Zentrum zum low-spin Häm a undwerden von dort zum aktiven Zentrum, bestehend aus high-spin Häm a3 undCuB, transportiert (Abbildung 1.5 und 1.6)6. High-spin bedeutet hierbei, dassdas Zentralion Eisen aufgrund der Hundschen Regel die größtmögliche Zahlungepaarter d- Elektronen besitzt und mit einer Reihe verschiedener Ligan-den reagiert. Beim low-spin-Zustand liegt dagegen die geringstmögliche Zahlungepaarter d-Elektronen vor, so dass eine verminderte Reaktion mit Ligan-den stattfindet.Basierend auf der Kristallstruktur werden 3 mögliche Protonentransferwege(K-, D- und H-Weg) vorgeschlagen, wobei letzterer zumindest in Prokaryo-ten funktionell keine Rolle zu spielen scheint. Die Bedeutung der anderenbeiden Wege ist durch intensive funktionelle Untersuchungen an Mutanteneindeutig nachgewiesen. Der K-Weg führt über Lys-354 und Thr-351 zurHydroxyl-Gruppe von Tyr-280 am katalytischen Zentrum7. Der D-Weg führtüber Asp-124 über eine Reihe von Aminosäuren zu Glu-278 (siehe Abbildung1.7). Von dort ist der weitere Weg der Protonen nicht genau bekannt [11].
6Ein Brückenligand dient im Allgemeinen als Verbindungsbrücke zweier Metallzentrenin einem Komplex.
7Im Folgenden wird immer Paracoccus denitrificans Nummerierung verwendet.
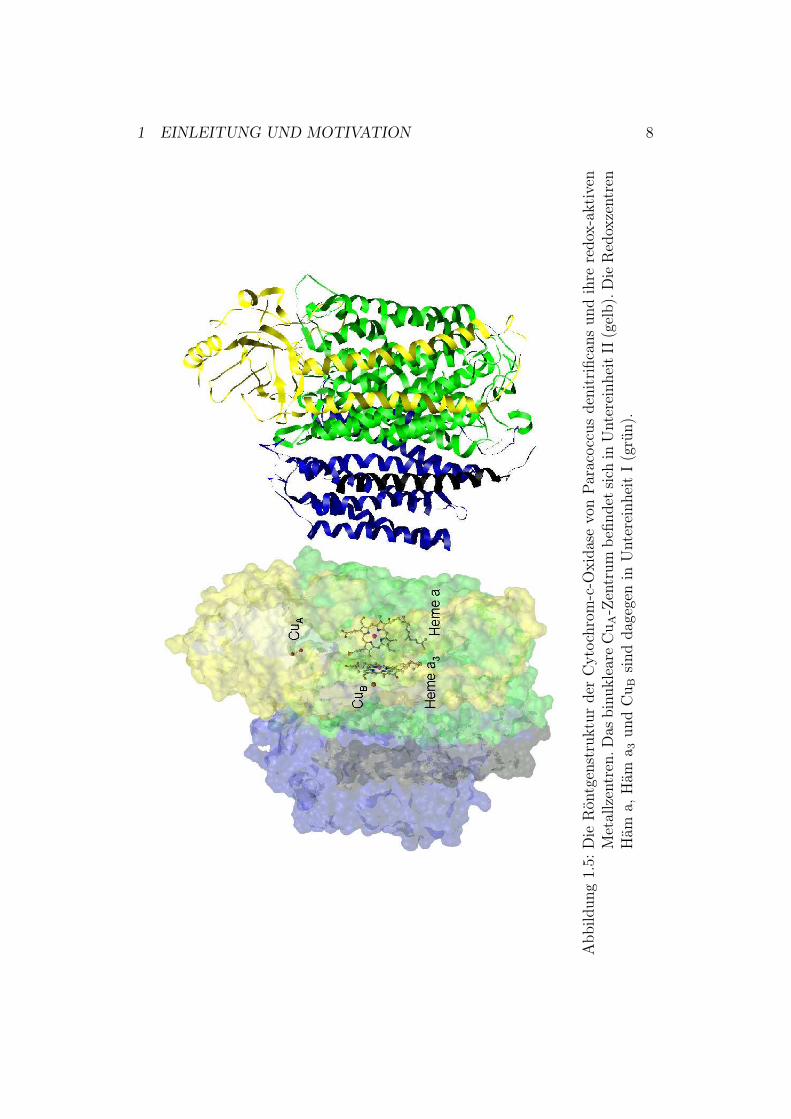
1 EINLEITUNG UND MOTIVATION 8
Abb
ildun
g1.
5:D
ieR
öntg
enst
rukt
urde
rC
ytoc
hrom
-c-O
xida
sevo
nPa
raco
ccus
deni
trifi
cans
und
ihre
redo
x-ak
tiven
Met
allz
entr
en.D
asbi
nukl
eare
Cu A
-Zen
trum
befin
dets
ich
inU
nter
einh
eitI
I(ge
lb).
Die
Red
oxze
ntre
nH
äma,
Häm
a 3un
dC
u Bsin
dda
gege
nin
Unt
erei
nhei
tI
(grü
n).
1 EINLEITUNG UND MOTIVATION 9
Abbildung 1.6: Vergrößerung der Cytochrom-c-Oxidase-Redox-Zentren vonParacoccus denitrificans mit den entsprechenden Liganden.Die Seitenketten von Häm a enthalten unter anderem Vinyl -Gruppen (-CH=CH2), Formyl - Gruppen (-CH=O) und eineFarnesyl - Gruppe (-CHOH(-CH2CH2CH=CCH3)3-CH3).
1 EINLEITUNG UND MOTIVATION 10
Abbildung 1.7: Protonenwege in der Cytochrom-c-Oxidase von Paracoccusdenitrificans. Der K-Weg der CcO führt über Lys-354 undThr-351 zur Hydroxyl-Gruppe von Tyr-280 am katalytischenZentrum. Der D-Weg führt über Asp-124 über eine Reihevon Aminosäuren zu Glu-278 und von dort aus weiter inskatalytische Zentrum. Die Untereinheiten III und IV wurdenaus Gründen der Übersichtlichkeit weggelassen.

1 EINLEITUNG UND MOTIVATION 11
1.3.2 Katalytischer Zyklus der Cytochrom-c-Oxidase
Die Cytochrom-c-Oxidase ist eines der am intensivsten untersuchten Mem-branproteine. Trotzdem wird nach wie vor kontrovers diskutiert, wie vieleProtonen an welcher Stelle im Reaktionszyklus gepumpt werden. Die Pro-tonentransportwege sind bis heute noch ungeklärt. Viele verschiedene Mo-delle wurden hierzu aufgestellt. Abbildung 1.8 zeigt schematisch die Haupt-zwischenprodukte bei der katalytischen Sauerstoffreduktion durch die Cyto-chrom-c-Oxidase nach Michel [12].Ausgehend vom vollständig oxidierten Zustand O (=oxidized) wird nach Auf-nahme des ersten Elektrons der Ein-Elektronen-reduzierte E-Zustand (=elec-tronated) erreicht.Die Zufuhr des zweiten Elektrons führt zur zwei-Elektronen-reduzierten Spe-zies R (=reduced). Sauerstoff kann nun an das reduzierte Häm a3 binden unddas Zwischenprodukt A bilden.
Abbildung 1.8: Das Modell des konventionellen katalytischen Cytochrom-c-Oxidase-Zyklus.

1 EINLEITUNG UND MOTIVATION 12
Eine elektronische Umordnung innerhalb des binuklearen Zentrums führt zurBildung des PM-Zustandes8.Die Zufuhr des dritten Elektrons liefert über den Pr-Zustand den F-Zustand(=oxoferryl). Dabei wird die O-O-Bindung aufgebrochen und Wasser gebil-det. Das führt zu der Translokation von zwei Protonen.Die Aufnahme des vierten Elektrons führt schließlich über den H-Zustandzum vollständig oxidierten Zustand O. Auch hier wird wieder Wasser gebil-det. Es gibt eine erneute Translokation von zwei Protonen.Die von der Cytochrom-c-Oxidase katalysierte Reaktion lässt sich dadurchmit Hilfe der folgenden Nettogleichung beschreiben:
4 ·Fe2+cytc + 8 ·H+
matrix + O2 " 4 ·Fe3+cytc + 2 ·H2O + 4 ·H+
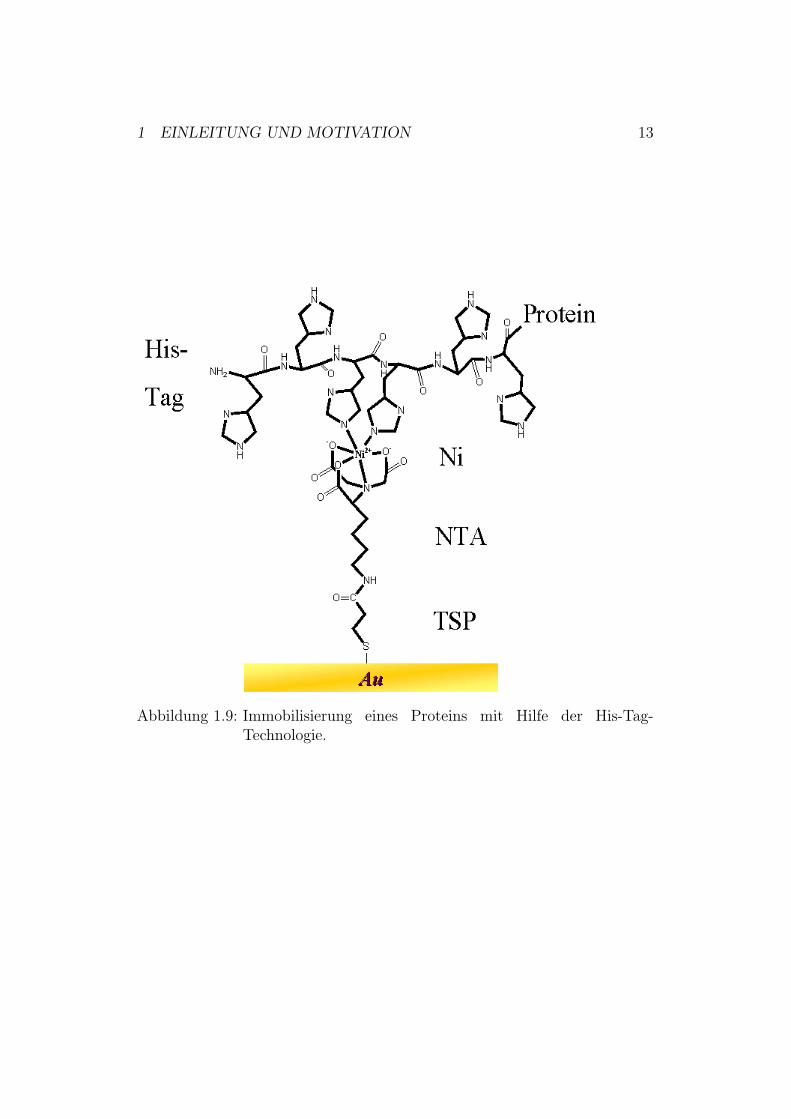
1.4 His-Tag-Technologie1988 berichtete Hochuli erstmals von einem kurzkettigen Affinitätspeptid,bestehend aus wahlweise fünf oder sechs Histidinen, dem so genannten ”His-Tag” [15]. Mit diesem His-Tag fusionierte Proteine zeigten eine bemerkens-werte Affinität zu Ni2+-Metall-Chelat- Komplexen, die ebenfalls von Hochuliein Jahr zuvor entscheidend weiter entwickelt wurden [16].Der His-Tag kann nach Zugabe einer Nickel-Ionen-Lösung spezifisch an zwei-wertige Nickel-Ionen binden. Das Ni2+-Ion ist in einem oktaedrischen Kom-plex mit Wassermolekülen gebunden. Diese können durch Histidin verdrängtwerden, wodurch ein sehr stabiler Chelat-Komplex entsteht. Die Ni2+-Ionenkönnen durch Nitrilo-tri-essigsäure-Reste gebunden werden und mit zweiHistidin-Resten des Proteins im Austausch gegen Wasser interagieren. DieseSpezifität gewährleistet, dass das Protein bindet.Dieser His-Tag kann nicht nur für die Aufreinigung, sondern auch für dieImmobilisierung von Proteinen auf einer TSP-NTA-codierten Oberfläche ge-nutzt werden (Abbildung 1.9)9.
8Dieser Name stammt von der ursprünglichen Annahme, dass das Enzym in dieserForm ein Peroxid (Fe(III)-O-O-Cu2+) oder Hydroperoxid (Fe(III)-OOH Cu2+) am aktivenZentrum gebunden hat. In letzter Zeit verdichten sich aber die Hinweise, dass die O-O-Bindung in dieser Spezies bereits gespalten ist und eine Oxoferryl-Struktur (Fe(IV)=O!
2
HO-Cu2+) vorliegt [13][14].9TSP=thiobis propionic acid (Thiobis-Propionsäure) und NTA=nitrilotriacetic acid
(Nitrilo-tri-essigsäure)
1 EINLEITUNG UND MOTIVATION 13
Abbildung 1.9: Immobilisierung eines Proteins mit Hilfe der His-Tag-Technologie.

1 EINLEITUNG UND MOTIVATION 14
1.5 MotivationMembranproteine konnten bisher aufgrund ihrer Komplexität nur in Lösun-gen oder in vesikulären Systemen studiert werden. Die Untersuchung vonRedoxprozessen gelang häufig nur mit Hilfe von Mediatoren.Das Ziel dieser Arbeit ist die nähere Untersuchung der Redoxkaskade eineskomplexen Membranproteins - der Cytochrom-c-Oxidase - in einem biomime-tischen Membransystem. Hierfür wird das ptBLM-System10 verwendet, mitdessen Hilfe eine Kontrolle über die Orientierung des auf einer Metallober-fläche angebundenen Proteins möglich ist (Abbildung 1.10). Unserer Gruppestehen zwei Orientierungen zur Verfügung. Dabei ist das Enzym entweder ander Untereinheit I oder an der Untereinheit II mit einem His-Tag manipuliert(Abbildung 1.10).
Abbildung 1.10: Die zwei unterschiedlichen Konfigurationsmöglichkeiten deruns zur Verfügung stehenden Cytochrom-c-Oxidase. DerHis-Tag und die Verankerung an die Metalloberfläche sindvergrößert dargestellt. Links im Bild: Das Enzym mit His-Tag an Untereinheit II (orange). Rechts im Bild: Das Enzymmit His-Tag an Untereinheit I (rot). Die Untereinheiten IIIund IV sind gelb bzw. schwarz dargestellt.
10ptBLM: protein tethered bilayer lipid membrane system

1 EINLEITUNG UND MOTIVATION 15
Ein großer Vorteil des ptBLM-Systems besteht darin, definierte elektrischePotentiale an die Oberfläche anzulegen und damit das Protein in einen re-duzierten, oxidierten oder einen beliebigen Zwischenzustand zu bringen. Dashierfür mit einem His-Tag manipulierte Enzym verändert dabei weder seineenzymatischen Eigenschaften noch seine Struktur. Das ermöglicht eine ”in-situ” Analyse, mit deren Hilfe man quantitative Informationen über Konfor-mationsänderungen und den Ladungstransfer dieses schwer zu studierendenProteins erhält.Im Rahmen dieser Diplomarbeit sollen drei Membranproteine unter Ausnut-zung der ptBLM-Technik durch eine Kombination von spektroskopischen undelektrochemischen Messmethoden erforscht werden:
• Erst vor kurzem hat unsere Gruppe die Cytochrom-c-Oxidase mit His-Tag an Untereinheit II erfolgreich durch den direkten Elektronentrans-fer aktiviert. Der Nachweis erfolgte durch Cyclovoltammetrie (CV) undoberflächenverstärkte resonante Ramanspektroskopie (SERRS: Surface-enhanced-resonance Raman Spectroscopy) [17]. Ein Teil dieser Arbeitbesteht in der Fortführung der Untersuchung der Cytochrom-c-Oxidasemit His-Tag an Untereinheit II durch SERRS und durch die zeitaufge-löste oberflächenverstärkte resonante Ramanspektroskopie (TR-SERRS:time-resolved Surface-enhanced-resonance Raman Spectroscopy). Die-ses sehr komplizierte Messverfahren erlaubt detaillierte Einblicke in dieKinetik des Enzym-Mechanismus, der bis heute kontrovers diskutiertwird.
• Studien an durch Mutation veränderten Enzymen fördern das Ver-ständnis über die verwendeten Ladungstransportwege. Deshalb beschäf-tigt sich diese Arbeit auch mit einer Mutation der Cytochrom-c-Oxidasemit His-Tag an Untereinheit II (Mutante N139C).
• Der Schwerpunkt dieser Arbeit besteht im Studium der Cytochrom-c-Oxidase mit His-Tag an Untereinheit I. Dieses Enzym hat genau dieentgegengesetzte Orientierung auf der Oberfläche wie die Cytochrom-c-Oxidase mit His-Tag an Untereinheit II (siehe Abbildung 1.10). Es sollüberprüft werden, ob sich diese Orientierung auch durch den direktenElektronentransfer oder/und mit Hilfe des Elektronendonators Cyto-chrom c aktivieren lässt. Im Falle der erfolgreichen Aktivierung sollenStudien über Elektronentransferprozesse und Protonentranslokationenvorgenommen werden.

1 EINLEITUNG UND MOTIVATION 16
Der ptBLM-Aufbau wird in allen drei Fällen simultan mit Hilfe von Oberflä-chenplasmonenresonanz-Spektroskopie (SPR) und elektrochemischer Impe-danzspektroskopie (EIS) überwacht. Die Funktionsfähigkeit der Proteine wirdmit der elektrochemischen Impedanzspektroskopie, der Cyclovoltammetrieund der oberflächenverstärkten resonanten Ramanspektroskopie beobachtet.Zusätzlich werden in dieser Arbeit die Eigenschaften des natürlichen Sub-strats (Cytochrom c) sowie dessen Wechselwirkung mit dem Enzym geprüft.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 17
2 Theoretische Grundlagen und MessmethodenIn diesem Kapitel werden die Methoden der Oberflächenplasmonenresonanz-Spektroskopie, der elektrochemischen Impedanzspektroskopie, der Cyclovol-tammetrie und der Ramanspektroskopie vorgestellt und ihre theoretischenGrundlagen behandelt. Mit diesen Methoden werden die wichtigsten Eigen-schaften der Proben charakterisiert.
2.1 OberflächenplasmonenresonanzObwohl sich Oberflächenplasmonen bereits durch die Theorie von Maxwellaus dem Jahr 1873 erklären ließen, wurden sie erst 1957 durch Ritchie theo-retisch beschrieben [18]. Die ersten Messungen der Oberflächenplasmonenre-sonanz (SPR, surface plasmon resonance) wurden 1959 von Turbadar veröf-fentlicht [19]. 1968 wurde erstmals von Otto und in einer anderen Messan-ordnung von Kretschmann und Raether die Oberflächenplasmonenresonanzzur Bestimmung des Brechungsindexes verwendet [20][21].
2.1.1 Elektromagnetische Beschreibung von Oberflächenplasmo-nen
An der Grenzfläche zwischen Metall und Dielektrikum kann es zu kollektivenOszillationen des quasi-freien Elektronengases im Metall kommen, welchean elektromagnetische Wellen gekoppelt sind. Diese so genannten Oberflä-chenplasmonen oder auch plasmonischen Oberflächenpolaritoren propagierenentlang der Grenzfläche. Sie werden durch Dissipation ihrer Energie in dasMetall stark gedämpft. Diese Dämpfung geschieht in Ausbreitungsrichtungder Oberflächenplasmonen, wobei fast die gesamte Energie in Wärme umge-wandelt wird [22][23].Zur Herleitung der orts- und zeitabhängigen Feldverteilung der Oberflächen-plasmonen benötigt man die makroskopischen Maxwellgleichungen in Mate-rie [24]:
divD = 4" #ext (2.1)
rotE +1
c
$B$t
= 0 (2.2)
divB = 0 (2.3)
rotH! 1
c
$D$t
=4"
cj ext (2.4)

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 18
Betrachtet man nun ein homogenes isotropes Medium, bei dem sowohl dieLadungs- als auch die Stromdichte Null sind:
D = %E H =Bµ
j ext = 0 #ext = 0 ,
so vereinfachen sich die Maxwell-Gleichungen zu
divE = 0 (2.5)
rotE + µ1
c
$H$t
= 0 (2.6)
divH = 0 (2.7)
rotH! %1
c
$E$t
= 0 (2.8)
Hierbei sind % = %(&) und µ = µ(&) die makroskopischen Responsefunktio-nen. Die meisten Materialien haben eine Permeabilität von µ # 1 [24]. UnterAnnahme von Laserlicht, welches in guter Näherung monochromatisch ist,wird hier dispersionsfrei gerechnet. Im Folgenden wird das magnetische Feldbetrachtet. Berechnungen für das elektrische Feld ergeben sich äquivalent.Mit Hilfe von
rotrotH = !!H µ(&) # 1
und Gleichung (2.6) lässt sich Gleichung (2.8) umformen zu:
!H = %1
c2
$2H$t2
(2.9)
Gesucht wird eine Lösung der Wellengleichung für den monochromatischenFall. Für die Lösung dieses Problems wählt man den folgenden Ansatz [24]:
H(r, t) = H(r) · e(!iωt) (2.10)
Setzt man Gleichung (2.10) in (2.9) ein, so erhält man die zeitunabhängigeHelmholzgleichung:
(! +&2
c2%) ·H(r) = 0 (2.11)
Diese Gleichungen werden durch den Ansatz
H(r) = H0 · eikr (2.12)
mit beliebiger Amplitude H0 gelöst [24].

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 19
Durch Einsetzen und Ausführen des Laplace-Operators erhält man die fol-gende Bedingung:
!k2 +&2%
c2= 0
$ &2 =c2k2
%=
c2k2
n2
Der hier eingeführte Brechungsindex n ist im Allgemeinen komplex:
n =%
% = nr + i'
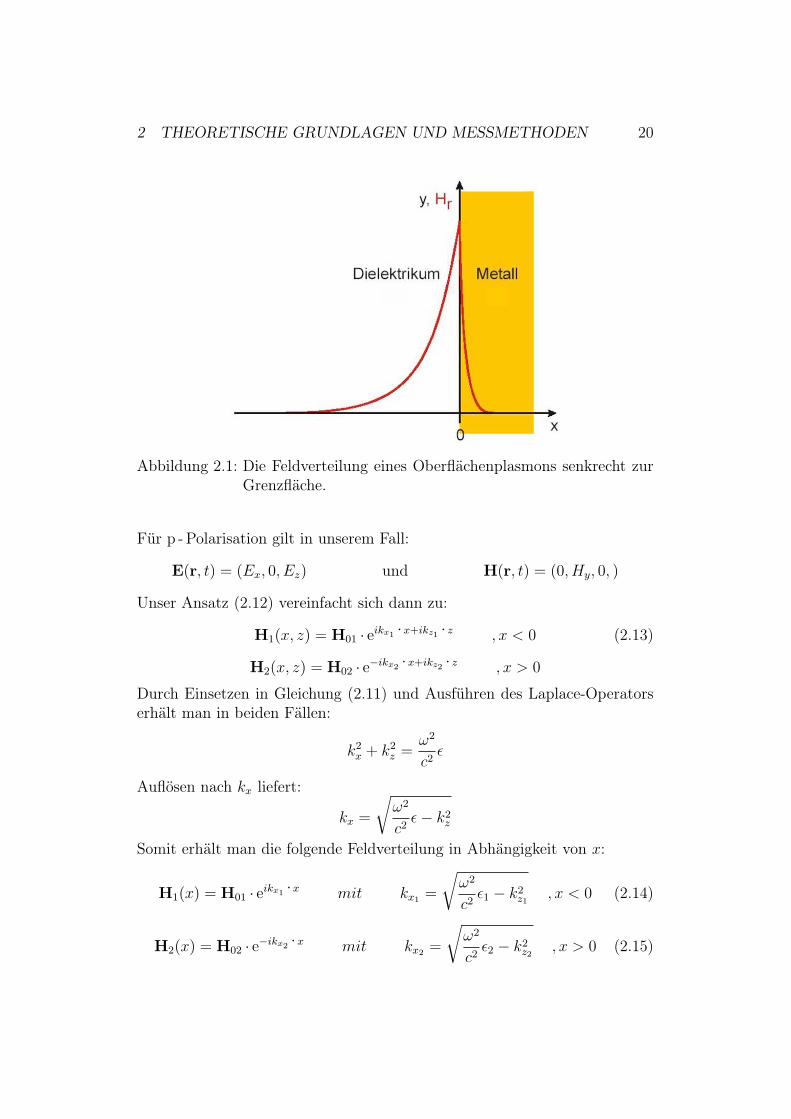
Wie in [24] beschrieben stellt die Lösung von Gleichung (2.11) eine elliptischpolarisierte Welle dar. Wählt man einen festen Wellenvektor k0 (reell, belie-bige Richtung) und eine reelle Amplitude H0, so erhält man den Spezialfalleiner linear polarisierten Welle.Oberflächenplasmonen sind Lösungen an einer Grenzfläche, bei denen daselektromagnetische Feld an der Grenzfläche gebunden ist und an ihr entlang-propagiert.Aus den makroskopischen Maxwellgleichungen kann man die Stetigkeitsbe-dingungen für die Felder an der Grenzfläche unseres Problems bestimmen underhält die Bedingung, dass die Vektorkomponenten von E, H und k parallelzur Grenzfläche kontinuierlich von einem ins andere Medium gehen müssen[24].Trifft nun eine linear polarisierte Welle auf eine Grenzfläche, so müssen dieKontinuitätsbedingungen erfüllt werden. Diese linear polarisierte Welle kannin eine parallel polarisierte (p-polarisierte) und in eine senkrecht polarisierte(s-polarisierte) Welle aufgeteilt werden11 [25]. Die Kontinuitätsbedingungenkönnen für s - und p - Polarisation getrennt betrachtet werden. Jede anderePolarisation ergibt sich als lineare Superposition.Im Folgenden betrachtet man eine Grenzfläche zwischen einem Dielektrikum(1) und einem Metall (2) mit den dazugehörigen Dielektrizitätskonstanten %1
und %2. Die Grenzfläche soll - wie in Abbildung (2.1) zu sehen ist - in dery-z-Ebene lokalisiert sein und durch die Stelle x = 0 gehen.Man kann zeigen, dass man mit s-polarisiertem Licht12 kein Oberflächenplas-mon anregen kann [22].Aus diesem Grund kann man o.B.d.A. die folgende Annahme für die eintref-fende Welle machen:
ky1 = ky2 = ky = 0
11Der Polarisationszustand bezieht sich jeweils auf die Einfallsebene des eingestrahltenLichts. Bei s-polarisiertem Licht liegt der Vektor E in der Ebene der Grenzfläche.
12In diesem Fall hat man lediglich eine elektrische Feldkomponente in y-Richtung, alsoparallel zur Grenzfläche.
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 20
Abbildung 2.1: Die Feldverteilung eines Oberflächenplasmons senkrecht zurGrenzfläche.
Für p - Polarisation gilt in unserem Fall:
E(r, t) = (Ex, 0, Ez) und H(r, t) = (0, Hy, 0, )
Unser Ansatz (2.12) vereinfacht sich dann zu:
H1(x, z) = H01 · eikx1 ·x+ikz1 · z , x < 0 (2.13)
H2(x, z) = H02 · e!ikx2 ·x+ikz2 · z , x > 0
Durch Einsetzen in Gleichung (2.11) und Ausführen des Laplace-Operatorserhält man in beiden Fällen:
k2x + k2
z =&2
c2%
Auflösen nach kx liefert:
kx =
√&2
c2%! k2
z
Somit erhält man die folgende Feldverteilung in Abhängigkeit von x:
H1(x) = H01 · eikx1 ·x mit kx1 =
√&2
c2%1 ! k2
z1, x < 0 (2.14)
H2(x) = H02 · e!ikx2 ·x mit kx2 =
√&2
c2%2 ! k2
z2, x > 0 (2.15)

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 21
Als nächstes soll die Komponente Ez für beide Medien berechnet werden.Dazu kann man Gleichung (2.8) verwenden:
rotH(r, t)! 1
c%$E(r, t)
$t= 0 $ $
$xHy =
1
c%
$
$tEz
Mit Hilfe von Gleichung (2.12) kann man diese Gleichung weiter umformen:
$
$xHy = i&
1
c%Ez $ Ez =
!i c
% &
$
$xHy
Nähert man sich der Stelle x=0 von der Seite des Dielektrikums, so erhältman mit Gleichung (2.14):
Ez1 =!i c
%1 &ikx1Hy1
Ganz analog erhält man:
Ez2 =!i c
%1 &(!i)kx2Hy2
Durch die Kontinuitätsbedingung an der Stelle x = 0
Ez1 = Ez2 Hy1 = Hy2
erhält man die Existenzbedingung für Oberflächenplasmonen:
kx1
kx2
= !%1
%2(2.16)
Diese Gleichung kann nur erfüllt werden, wenn die dielektrischen Konstan-ten %1 und %2 unterschiedliche Vorzeichen haben. Diese Bedingung ist anGrenzflächen zwischen einem Dielektrikum und einem Metall im optischenFrequenzbereich erfüllt [22].Setzt man nun kx1 und kx2 aus den Gleichungen (2.14) und (2.15) ein undverwendet die Kontinuitätsbedingung kz1 = kz2 = kz, so erhält man:
kz =&
c·√
%1 · %2
%1 + %2(2.17)
An dieser Stelle sei nochmals darauf hingewiesen, dass es sich bei %1 und %2
um frequenzabhängige Funktionen handelt. Aus Gleichung (2.10) folgt unter

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 22
Verwendung von (2.18) die gesamte orts- und zeitabhängige Feldverteilungdes magnetischen Feldes in den beiden Medien:
H1(x, z, t) = H01 · ei(kx1 ·x+kz · z!ωt) (2.18)
H2(x, z, t) = H02 · ei(!kx2 ·x+kz · z!ωt)
mit:
kx1 =&
c
√%21
%1 + %2kx2 =
&
c
√%22
%1 + %2kz =
&
c·√
%1 · %2
%1 + %2
Die Eindringtiefen senkrecht zur Grenzfläche, nach denen das Feld in denbeiden Medien auf e!1 abgefallen ist, berechnen sich als Kehrwert des je-weiligen kx. Betrachtet man zum Beispiel die magnetische Feldverteilung aneiner Luft-Gold-Grenzfläche (%Au = !12,1 + i · 1,3), an der ein Oberflächen-plasmon angeregt wird, so erhält man abhängig von der LaserwellenlängeEindringtiefen von ca. 100 nm bis 500 nm [22] [26].
2.1.2 Anregung von Oberflächenplasmonen
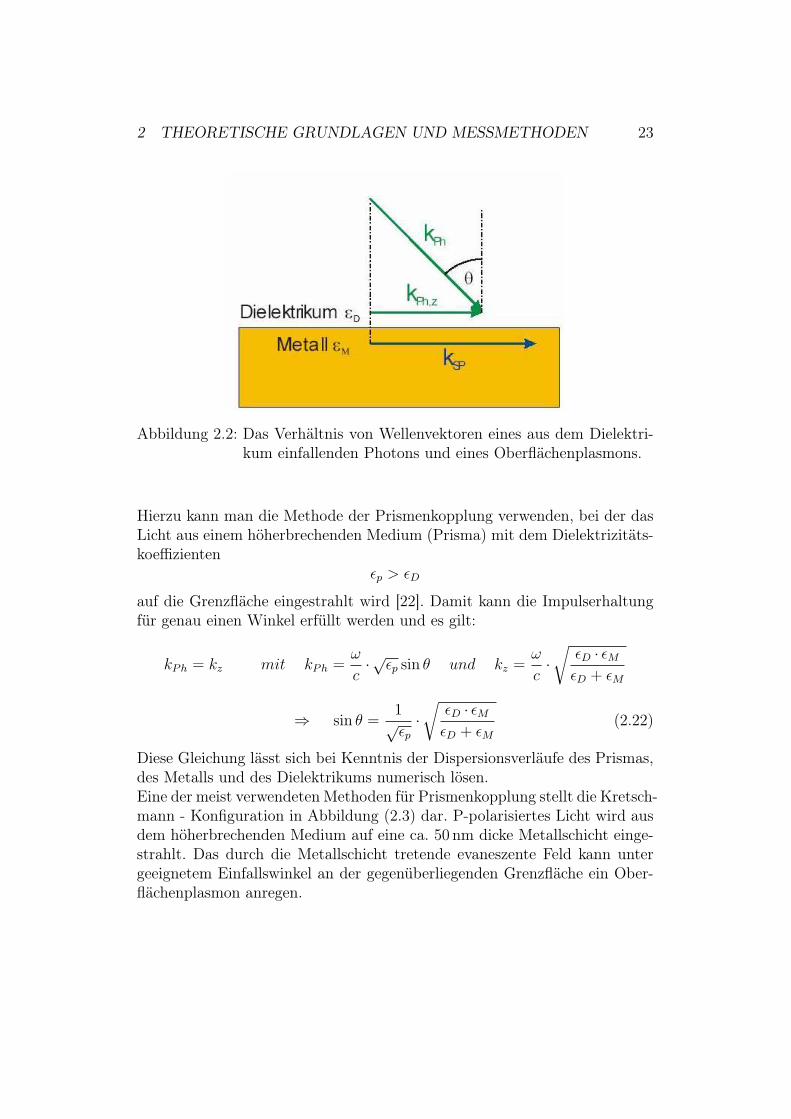
Eine Anregung von Oberflächenplasmonen durch ein einfallendes Photon istnur möglich, wenn die Impulserhaltung erfüllt ist. Im vorliegenden Fall mussdie Wellenvektorkomponente des Photons in Ausbreitungsrichtung kPh,z iden-tisch mit der Wellenzahl des Plasmons kz sein (siehe Abbildung 2.2). Durchden Einfallswinkel ( kann kPh,z verändert werden:
kPh,z =| kPh | · sin ( , (2.19)
wobei der Betrag des Photon-Wellenvektors mit Hilfe der Maxwellgleichun-gen gegeben ist durch [24]:
| kPh |= &
c·%
%D (2.20)
Vergleicht man die Dispersionsrelation des Oberflächenplasmons und dieDispersionsrelation des einfallenden Photons aus den Gleichungen 2.17 und2.20, wird sofort klar, dass es zu keiner Impulsgleichung kommen kann, dafür alle Einfallswinkel gilt:
kPh,z < kz (2.21)
Um eine Kopplung des einfallenden Photons an das Oberflächenplasmon zuerreichen, muss die Impulskomponente des einfallenden Photons in Ausbrei-tungsrichtung kPh,z erhöht werden.
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 23
Abbildung 2.2: Das Verhältnis von Wellenvektoren eines aus dem Dielektri-kum einfallenden Photons und eines Oberflächenplasmons.
Hierzu kann man die Methode der Prismenkopplung verwenden, bei der dasLicht aus einem höherbrechenden Medium (Prisma) mit dem Dielektrizitäts-koeffizienten
%p > %D
auf die Grenzfläche eingestrahlt wird [22]. Damit kann die Impulserhaltungfür genau einen Winkel erfüllt werden und es gilt:
kPh = kz mit kPh =&
c·%%p sin ( und kz =
&
c·√
%D · %M
%D + %M
$ sin ( =1%
%p·√
%D · %M
%D + %M(2.22)
Diese Gleichung lässt sich bei Kenntnis der Dispersionsverläufe des Prismas,des Metalls und des Dielektrikums numerisch lösen.Eine der meist verwendeten Methoden für Prismenkopplung stellt die Kretsch-mann - Konfiguration in Abbildung (2.3) dar. P-polarisiertes Licht wird ausdem höherbrechenden Medium auf eine ca. 50 nm dicke Metallschicht einge-strahlt. Das durch die Metallschicht tretende evaneszente Feld kann untergeeignetem Einfallswinkel an der gegenüberliegenden Grenzfläche ein Ober-flächenplasmon anregen.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 24
Abbildung 2.3: Die Prismenkopplung nach der Kretschmann-Konfiguration.
2.1.3 Oberflächenplasmonenresonanz-Spektroskopie
Die Oberflächenplasmonenresonanz-Spektroskopie ist ein optisches Messver-fahren und erlaubt unter anderem die Detektion ultradünner dielektrischerSchichten von wenigen Nanometern Dicke [22]. Sie beruht auf dem Prinzip,dass Oberflächenplasmonen für eine gegebene Frequenz nur unter bestimmtenEinfallswinkeln angeregt werden können. In der Regel wird die winkelabhän-gige Intensität eines von der Metall-Dielektrikum-Grenzfläche reflektierten,p-polarisierten Laserstrahls verwendet. Der Anregungswinkel des Oberflä-chenplasmons wird durch die Dissipation13 der Energie in die Metallschichtals Intensitätsminimum erkennbar (siehe Abbildung 2.4).Brechungsindexänderungen im Bereich des evaneszenten Feldes eines Ober-flächenplasmons lassen sich als Verschiebung des Anregungswinkels detektie-ren [21].Wird eine dünne dielektrische Schicht auf das Metall aufgebracht, so ändertsich - wie in Abbildung 2.4 gezeigt - die Dispersionsrelation. Deshalb kanndie Adsorption von dielektrischen, dünnen Schichten an der Metalloberflächebeobachtet werden.
13Dissipation bezeichnet in der Physik die kontinuierliche Energieumwandlung eineso!enen Systems in thermische Energie.
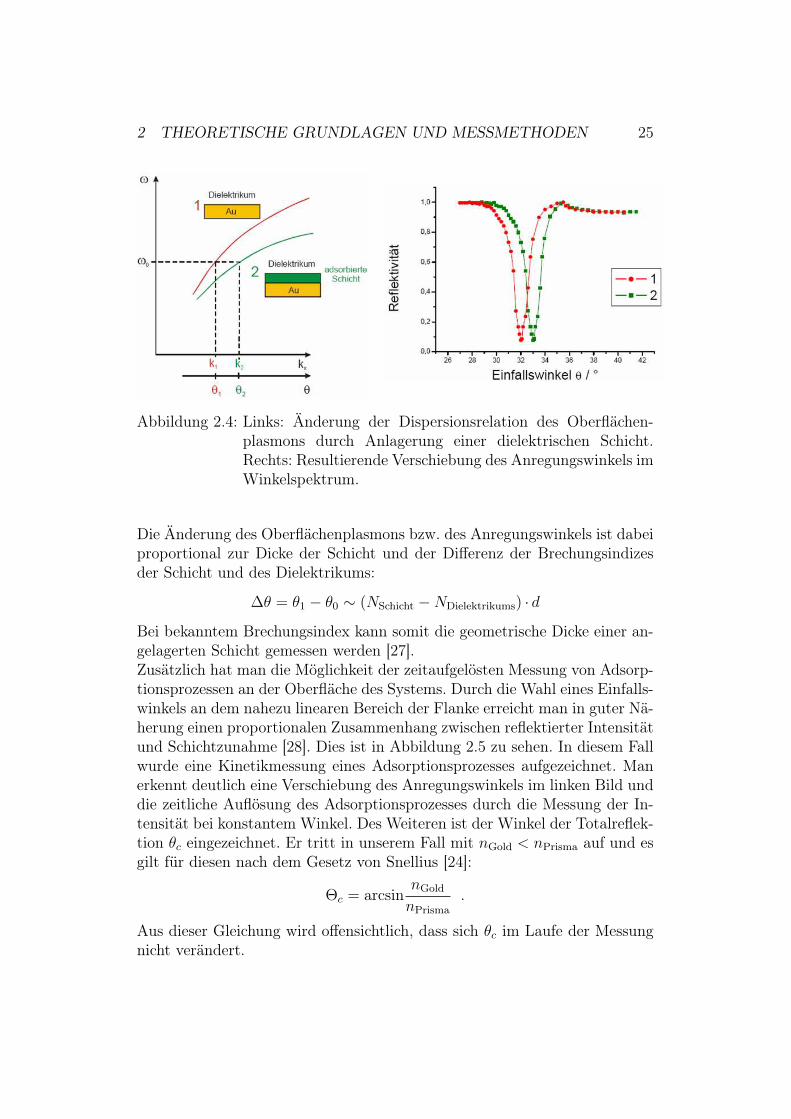
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 25
Abbildung 2.4: Links: Änderung der Dispersionsrelation des Oberflächen-plasmons durch Anlagerung einer dielektrischen Schicht.Rechts: Resultierende Verschiebung des Anregungswinkels imWinkelspektrum.
Die Änderung des Oberflächenplasmons bzw. des Anregungswinkels ist dabeiproportional zur Dicke der Schicht und der Differenz der Brechungsindizesder Schicht und des Dielektrikums:
!( = (1 ! (0 & (NSchicht !NDielektrikums) · d
Bei bekanntem Brechungsindex kann somit die geometrische Dicke einer an-gelagerten Schicht gemessen werden [27].Zusätzlich hat man die Möglichkeit der zeitaufgelösten Messung von Adsorp-tionsprozessen an der Oberfläche des Systems. Durch die Wahl eines Einfalls-winkels an dem nahezu linearen Bereich der Flanke erreicht man in guter Nä-herung einen proportionalen Zusammenhang zwischen reflektierter Intensitätund Schichtzunahme [28]. Dies ist in Abbildung 2.5 zu sehen. In diesem Fallwurde eine Kinetikmessung eines Adsorptionsprozesses aufgezeichnet. Manerkennt deutlich eine Verschiebung des Anregungswinkels im linken Bild unddie zeitliche Auflösung des Adsorptionsprozesses durch die Messung der In-tensität bei konstantem Winkel. Des Weiteren ist der Winkel der Totalreflek-tion (c eingezeichnet. Er tritt in unserem Fall mit nGold < nPrisma auf und esgilt für diesen nach dem Gesetz von Snellius [24]:
"c = arcsinnGold
nPrisma.
Aus dieser Gleichung wird offensichtlich, dass sich (c im Laufe der Messungnicht verändert.
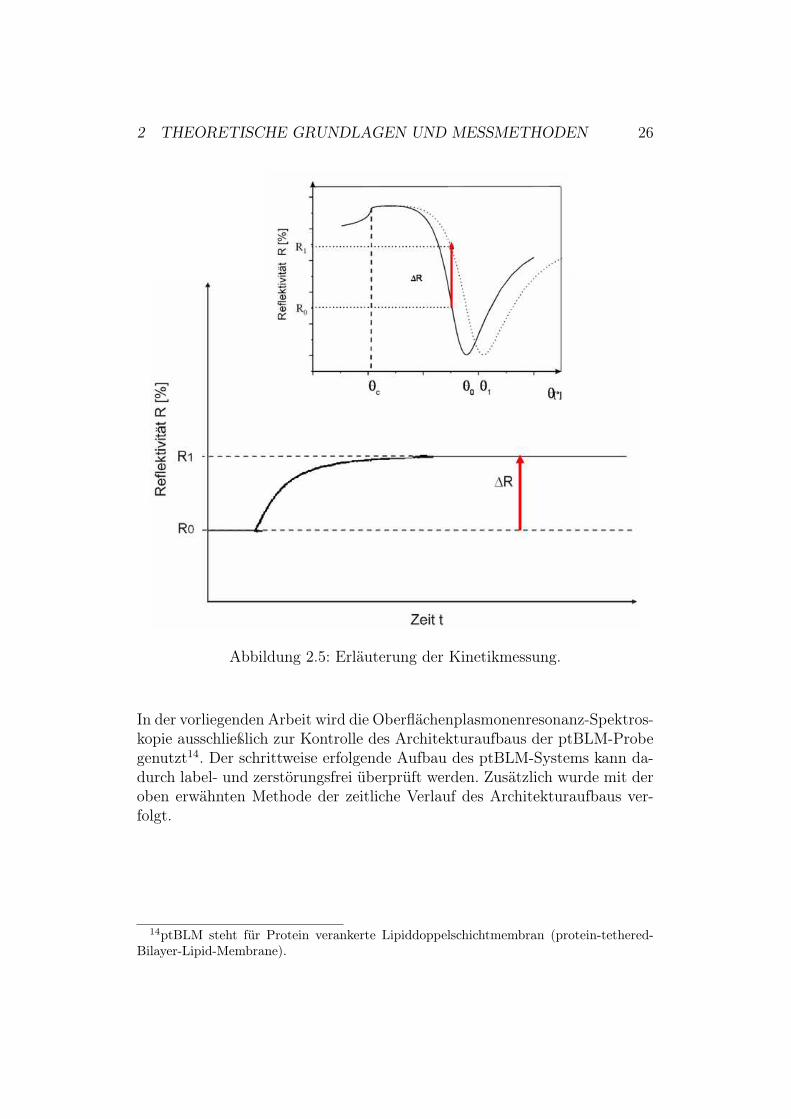
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 26
Abbildung 2.5: Erläuterung der Kinetikmessung.
In der vorliegenden Arbeit wird die Oberflächenplasmonenresonanz-Spektros-kopie ausschließlich zur Kontrolle des Architekturaufbaus der ptBLM-Probegenutzt14. Der schrittweise erfolgende Aufbau des ptBLM-Systems kann da-durch label- und zerstörungsfrei überprüft werden. Zusätzlich wurde mit deroben erwähnten Methode der zeitliche Verlauf des Architekturaufbaus ver-folgt.
14ptBLM steht für Protein verankerte Lipiddoppelschichtmembran (protein-tethered-Bilayer-Lipid-Membrane).

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 27
2.2 EIS - Elektrochemische ImpedanzspektroskopieDa man mit der SPR-Spektroskopie lediglich adsorbierte Schichtdicken nach-weisen kann, wird zur Überprüfung des ptBLM-Aufbaus noch zusätzlich dieImpedanzspektroskopie verwendet. Die Impedanzspektroskopie beruht aufder gleichzeitigen Messung der Beeinflussung von Phase und Amplitude ei-ner Wechselspannung durch die Probe. Die resultierende Antwort beinhaltetInformationen über die elektrischen Eigenschaften des Systems, insbesonde-re an ihren inneren Grenzflächen. Mit diesen Informationen ist es nicht nurmöglich den Systemaufbau zu überprüfen, sondern man kann auch die Dy-namik bewegter Ladungen berechnen [29].Obwohl die ersten elektrochemischen Untersuchungen mit Wechselstrom schonEnde des 19. Jahrhunderts z.B. durch Kohlrausch [30] veröffentlicht wurdenund das Konzept der elektrischen Impedanz 1884 von Heaviside [31] einge-führt wurde, erlebte die Methode erst mit der rasanten Entwicklung elektro-nischer Bauteile, insbesondere der Operationsverstärker, den entscheidendenDurchbruch. Moderne Impedanzanalysatoren bieten heutzutage die Möglich-keit Impedanzen im Frequenzbereich von einigen Millihertz bis zu einigenMegahertz zu bestimmen. Traditionell wird die EIS für Korrosionsbeobach-tungen, Batterien und elektrolytische Abscheidungen sowie die Charakteri-sierung von Halbleitern verwendet. In den letzten Jahren fand sie zusätz-lich einen weit verbreiteten Einsatz in der Biotechnologie. Ein wesentlicherVorteil der Methode ist die zerstörungs- und labelfreie Messung mit rechtgeringem instrumentellen Aufwand. Biologische Systeme können somit in ih-rer natürlichen Umgebung untersucht werden. Auf Grund der hohen Emp-findlichkeit für Membrandefekte bzw. Veränderungen in der Durchlässigkeitder Membran durch funktionelle Membranproteine ist EIS eine besondersgut geeignete Messmethode, um Aussagen über die Membranqualität unddie Funktion von Membranproteinen zu machen. Diese Informationen würdeman auf Grund der Auflösungslimitierung nur sehr kompliziert mit optischenMethoden erhalten [32].
2.2.1 Messprinzip der Elektrochemischen Impedanzspektroskopie
Die Impedanzspektroskopie ist eine Erweiterung der Impedanzanalyse. Hier-bei wird das zu untersuchende System mit einem elektrischen Signal an-geregt15. Das System wird aus seiner Gleichgewichtslage gebracht und dieresultierende Antwort des Systems auf diese Störungen aufgezeichnet.
15Dafür wird meistens eine sinusförmige Wechselspannung in einem Frequenzbereich von10!3 bis 106 Hertz verwendet.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 28
Hierzu benötigt man drei Elektroden:
• An die Arbeitselektrode wird das Störsignal angelegt. Die Proteine sinddirekt mit ihr verbunden.
• Die Gegenelektrode wird verwendet um das Antwortsignal aufzuzeich-nen.
• Die Referenzelektrode wird als Bezugspunkt benötigt, da es nur mög-lich ist Potentialdifferenzen zu messen. Das absolute Potential einereinzelnen Elektrode ist grundsätzlich nicht experimentell messbar [25].
Es gibt verschiedene Möglichkeiten der elektrischen Anregung, die man beider EIS verwenden kann. Die am häufigsten angewendete Störungsart ist die,bei der die Impedanz direkt im Frequenzbereich gemessen wird [29]. Dazuwird dem an der Zelle angelegten Potential eine sinusförmige Wechselspan-nung E(t) überlagert:
E(t) = E0 · sin(& · t)E0 ist die Amplitude der Wechselspannung und ) ihre Frequenz. Der Zu-sammenhang zwischen Winkelgeschwindigkeit & und Frequenz ) ist gegebendurch & = 2").Der resultierende Antwortstrom I(t) ist mit der Phase # verschoben und hateine Amplitude I0:
I(t) = I0 · sin(& · t + #)
Mit der letzten Gleichung wurde ein lineares Antwortverhalten des Systemsvorausgesetzt16. Die Amplitude der angelegten Wechselspannung sollte des-halb möglichst klein gewählt werden, da dann die starke Nichtlinearität ver-nachlässigbar ist. Eine gute Näherung für ein lineares Antwortverhalten er-hält man für E0 < 25 mV bei 25"C [29].Der folgende Ausdruck, analog zum Ohmschen Gesetz, erlaubt uns die Im-pedanz des Systems zu berechnen:
Z(t) =E(t)
I(t)=
E0 · sin(& · t)I0 · sin(& · t + #)
= Z0sin(& · t)
sin(& · t + #)
Mit Hilfe der Relationen [33]:
ei! = cos # + i · sin # sin2 # + cos2 # = 1 tan # =sin #
cos #(2.23)
kann man die Impedanz als komplexe Funktion darstellen.16Bei einem nicht-linearen System würde der Antwortstrom Oberschwingungen der An-
regungsfrequenz enthalten und die Auswertung der Informationen deutlich kompliziertermachen.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 29
Abbildung 2.6: Die Impedanz in der komplexen Gaußschen Zahlenebene.
Die angelegte Spannung und die Stromantwort werden nun komplex beschrie-ben17 (Abbildung 2.6):
E(t) = E0 · eiωt I(t) = I0 · ei(ωt!!)
Hieraus lässt sich der komplexe Widerstand des betrachteten Systems be-rechnen:
Z =E(t)
I(t)=
E0 · eiωt
I0 · ei(ωt!!)=
E0
I0· ei! =| Z | · ei! = Z # + iZ ## (2.24)
Hierbei sind auf Grund der Gleichungen (2.23) der Realteil von Z (Resistanz)und der Imaginärteil von Z (Reaktanz) gegeben durch:
Z # ' Re(Z) =| Z | · cos(#) Z ## ' Im(Z) =| Z | · sin(#)
In Gleichung (2.24) stehen die beiden signifikanten Messgrößen | Z | und #.Der Modul | Z | (Betrag der Impedanz) und das Argument ! (Phasenver-schiebung) sind mit Hilfe der Gleichungen (2.23) gegeben durch:
| Z |=√
(Z #)2 + (Z ##)2 # = arctan
(Z ##
Z #
)(2.25)
Man erkennt in Gleichung (2.24), dass die Zeitabhängigkeit der angelegtenSpannung bzw. des resultierenden Stroms verschwunden ist. Die Impedanz
17Die physikalisch sinnvolle Lösung muss natürlich reell sein. Hier wird lediglich diekomplexe Darstellung aufgrund der einfachen und eleganten Schreibweise verwendet.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 30
ist somit - vorausgesetzt, das System selbst ist zeitunabhängig - zeitinvariantund (normalerweise) frequenzabhängig.Manchmal ist es auch vorteilhaft Messergebnisse mit Hilfe der Admittanz zuanalysieren. Der Zusammenhang zwischen Admittanz Y und Impedanz Z istgegeben durch:
1
Z= Y = Y # + iY ##
Die Admittanz stellt also eine komplexe Leitfähigkeit dar. Den Realteil vonY (Konduktanz) und den Imaginärteil von Y (Suszeptanz) erhält man auffolgende Art:
Y = Y # + iY ## =1
Z=
1
Z # + iZ ## =Z #
(Z #)2 + (Z ##)2! iZ ##
(Z #)2 + (Z ##)2
$ Y # =Z #
(Z #)2 + (Z ##)2und Y ## =
!Z ##
(Z #)2 + (Z ##)2
Im Allgemeinen wird die Impedanz über einen bestimmten Frequenzbereichgemessen, d.h. die Anregungsamplitude E0 bleibt gleich, aber die Frequenzändert sich. Werden nun bei verschiedenen Frequenzen der resultierendeStrom I0 und der dazugehörige Phasenwinkel # gemessen, so ergibt sich zujeder Frequenz ein Wertepaar von Z # und Z ##.Die so erhaltenen Messergebnisse werden mit Hilfe von Ersatzschaltkreisensimuliert, in denen die Schaltkreiselemente die mögliche physikalische Situa-tion im System widerspiegeln sollen. Dadurch erhält man die gewünschtenInformationen über Widerstände und Kapazitäten des Systems [29].Im Folgenden werden die wichtigsten Elemente der Ersatzschaltkreise sowiedie unterschiedlichen Auftragungsarten der Ergebnisse dargestellt.
2.2.2 Graphische Darstellungen der Impedanzspektren
Die Darstellung der Impedanzspektren kann im Bode-Plot, im Nyquist-Plotoder in der frequenzreduzierten Admittanz erfolgen. Welche Informationenman aus den unterschiedlichen Auftragungsarten herauslesen kann, wird inKapitel 2.2.4 erläutert.Der Bode-Plot ist der wichtigste Plot zur Analyse von Impedanzmessungenund wurde zuerst 1938 von Bode verwendet [34]. Der Vorteil dieser Darstel-lung ist die Anschaulichkeit des Frequenzverhaltens im untersuchten System.Im Bode-Plot sind der Betrag der Impedanz und die Phasenverschiebung ge-gen die Frequenz aufgetragen. Die Frequenz wird dabei logarithmisch aufge-tragen. Dadurch wird das Verhalten über den großen verwendeten Frequenz-bereich anschaulich dargestellt.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 31
Der Impedanz-Plot wurde erstmals 1960 von Sluyther theoretisch und expe-rimentell verwirklicht [35][36]. Hierbei wird die einfachste Auftragungsmög-lichkeit gewählt. Der negative Imaginärteil der Impedanz (!Z ##) wird gegenden Realteil (Z #) aufgetragen. Diese so genannte Ortskurve wird auch häufigals Nyquist-Plot bezeichnet.Der Gebrauch der Admittanzebene wurde 1969 von Bauerle eingeführt [37].In der vorliegenden Arbeit wird der frequenzreduzierte Admittanz-Plotverwendet. Die Division der Admittanz durch die Winkelgeschwindigkeit &liefert die frequenzreduzierte Admittanz Y/&. Den Graphen erhält man, in-dem der negative Imaginärteil der frequenzreduzierten Admittanz !Y ##/&gegen den Realteil Y #/& aufgetragen wird.
2.2.3 Schaltkreiselemente
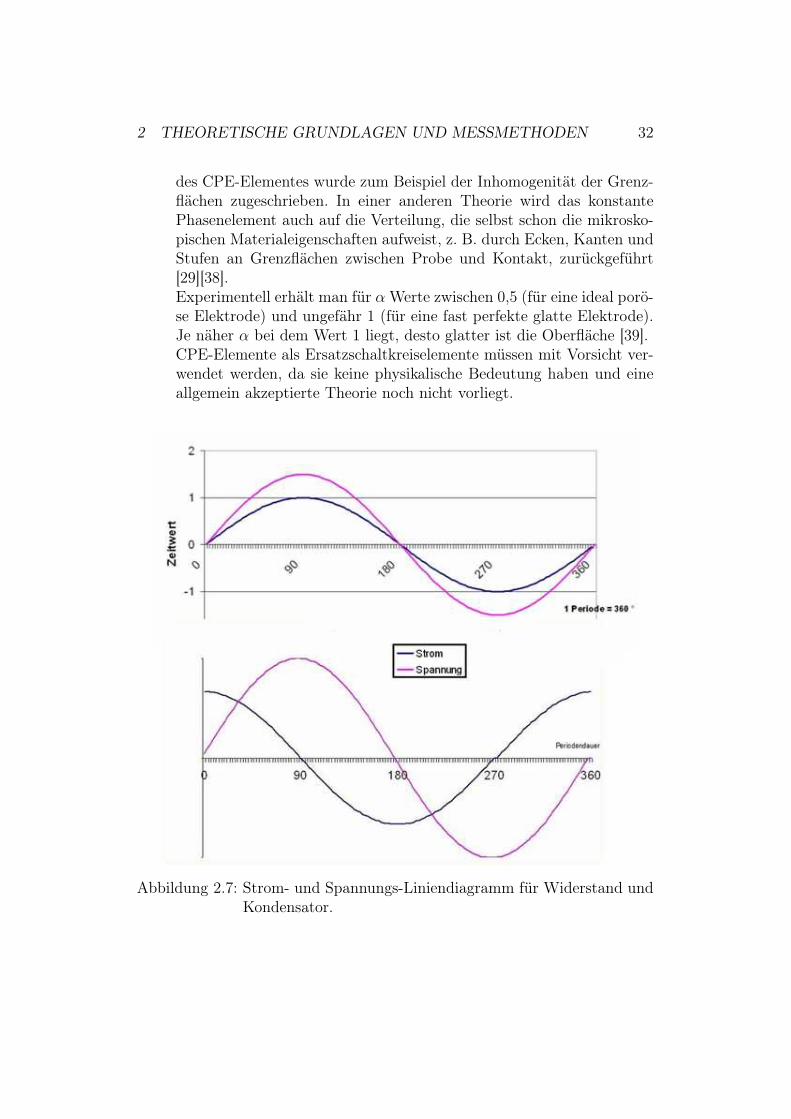
Im Folgenden werden die drei in der vorliegenden Diplomarbeit verwende-ten Schaltkreiselemente Widerstand, Kondensator und konstantes Phasen-element eingeführt. In Abschnitt 2.2.4 wird das Verhalten von Widerstandund Kondensator dann ausführlicher behandelt.Abbildung (2.7) zeigt die Liniendiagramme für Widerstand und Kondensa-tor18.
• Für einen rein ohmschen Widerstand (R) gibt es zwischen Stromund Spannung keine Phasenverschiebung.
• Misst man dagegen ein rein kapazitives Element (C) eilt der Stromder Spannung um "/2 voraus.
• Die untersuchten Systeme verhalten sich nicht immer ideal. Stattdessenverhalten sie sich wie ein konstantes Phasenelement (CPE). DieImpedanz des CPE- Elements ist wie folgt definiert:
ZCPE =A
(i&)α
Diese Gleichung beschreibt ein rein kapazitives Element für den Fall,dass A = 1/C und * = 1 gilt. Ein rein ohmscher Widerstand wirdbeschrieben, falls A = R und * = 0 gilt. Für ein CPE-Element gilt0 <* < 1 und A ist eine frequenz-unabhängige reelle Konstante. Es gibtmehrere Theorien über das nicht-ideale Verhalten der untersuchten Sys-teme, jedoch wurde bisher keine allgemein akzeptiert. Das Verhalten
18Induktivitäten spielen in den zu untersuchenden Systemen dieser Diplomarbeit keinemaßgebliche Rolle und werden hier nicht behandelt.
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 32
des CPE-Elementes wurde zum Beispiel der Inhomogenität der Grenz-flächen zugeschrieben. In einer anderen Theorie wird das konstantePhasenelement auch auf die Verteilung, die selbst schon die mikrosko-pischen Materialeigenschaften aufweist, z. B. durch Ecken, Kanten undStufen an Grenzflächen zwischen Probe und Kontakt, zurückgeführt[29][38].Experimentell erhält man für * Werte zwischen 0,5 (für eine ideal porö-se Elektrode) und ungefähr 1 (für eine fast perfekte glatte Elektrode).Je näher * bei dem Wert 1 liegt, desto glatter ist die Oberfläche [39].CPE-Elemente als Ersatzschaltkreiselemente müssen mit Vorsicht ver-wendet werden, da sie keine physikalische Bedeutung haben und eineallgemein akzeptierte Theorie noch nicht vorliegt.
Abbildung 2.7: Strom- und Spannungs-Liniendiagramm für Widerstand undKondensator.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 33
2.2.4 Einfache Ersatzschaltkreise
Ohmscher Widerstand: Abbildung 2.8 zeigt den Nyquist,den Bode- undden frequenreduzierten Admittanz-Plot19. In diesem Fall handelt es sich umeine Simulationsmessung eines ohmschen Widerstandes (rot: 50 k$ und blau:100 k$).Mit Hilfe des ohmschen Gesetzes erhält man die Impedanz eines rein ohm-schen Widerstandes. Sie besitzt keinen Imaginärteil und ist frequenzunab-hängig:
ZR =E
I=
I ·RI
= R $ Z #R = R und Z ##
R = 0 (2.26)
Der Nyquist–Plot zeigt also nur einen Wert, der dem des Widerstandes ent-spricht.Die frequenzreduzierte Admittanz eines reinen Widerstandes kann man mitHilfe von Gleichung (2.26) berechnen:
YR =1
ZR=
1
R$ Y #
R
&=
1
& ·R undY ##
R
&= 0
Der frequenzreduzierte Admittanz-Plot zeigt demnach eine Gerade parallelzur x-Achse.Über den Bode-Plot erkennt man, dass es sich um ein rein ohmsches Systemhandeln muss. Über den kompletten Frequenzbereich ist die Phasenverschie-bung bei beiden Widerständen 0" und man erhält für den Betrag der Im-pedanz den Widerstand R. Mit Hilfe von Gleichung (2.25) lässt sich diesesVerhalten berechnen:
| Z |=√
(Z #)2 + (Z ##)2 = R # = arctan
(Z ##
Z #
)= 0
19Die Daten für diese und auch die folgenden Abbildungen wurden mit der SoftwareZView simuliert. Die Simulation wurde in dem für uns messbaren Frequenzbereich von10!3 bis 106 Hertz durchgeführt.
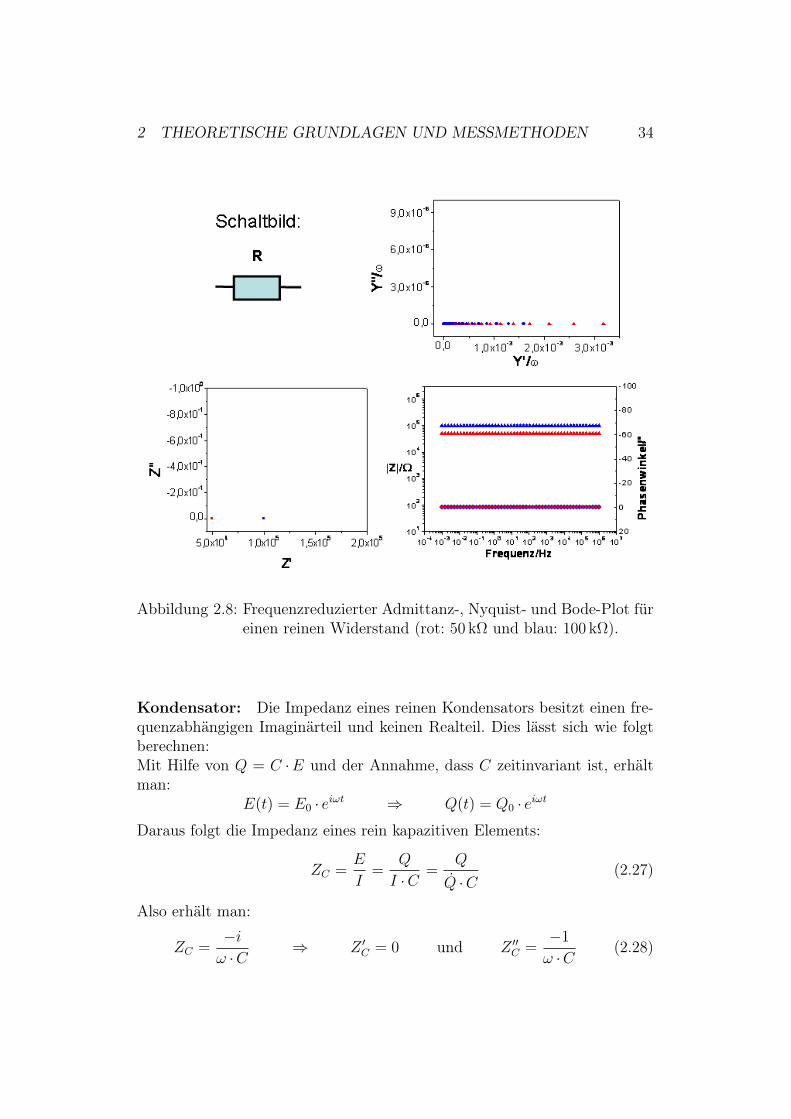
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 34
Abbildung 2.8: Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plot füreinen reinen Widerstand (rot: 50 k$ und blau: 100 k$).
Kondensator: Die Impedanz eines reinen Kondensators besitzt einen fre-quenzabhängigen Imaginärteil und keinen Realteil. Dies lässt sich wie folgtberechnen:Mit Hilfe von Q = C ·E und der Annahme, dass C zeitinvariant ist, erhältman:
E(t) = E0 · eiωt $ Q(t) = Q0 · eiωt
Daraus folgt die Impedanz eines rein kapazitiven Elements:
ZC =E
I=
Q
I ·C =Q
Q ·C(2.27)
Also erhält man:
ZC =!i
& ·C $ Z #C = 0 und Z ##
C =!1
& ·C (2.28)

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 35
Der Nyquist-Plot (Abbildung 2.9) zeigt demnach eine Gerade entlang derY-Achse, welche durch die Stelle Z # = 0 geht.Die frequenzreduzierte Admittanz eines Kondensators kann man mit Hilfevon Gleichung (2.28) berechnen:
YC =1
ZC= i& ·C $ Y #
C
&= 0 und
Y ##R
&= C
Der frequenzreduzierte Admittanz-Plot kann demnach nur einen Wert - dendes untersuchten Kondensators - enthalten.Über den Bode-Plot erkennt man, dass es sich um einen reinen Kondensa-tor handeln muss. Über den kompletten Frequenzbereich ist die Phasenver-schiebung bei beiden Messungen 90" (siehe Abschnitt 2.2.3). Den Betrag derImpedanz und den Phasenwinkel berechnet man mit (2.25):
| Z |=√
(Z #)2 + (Z ##)2 =1
&C# = arctan
(Z ##
Z #
)= !"
2
Aus diesem Grund erhält man für die Impedanz in Abhängigkeit von derFrequenz eine Gerade, aus deren Lage auf die Kapazität zurückgeschlossenwerden kann. Über den kompletten Frequenzbereich ist die Phasenverschie-bung bei beiden Kondensatoren 90". Somit eilt der Strom der Spannung um"/2 voraus (siehe Abbildung 2.7).
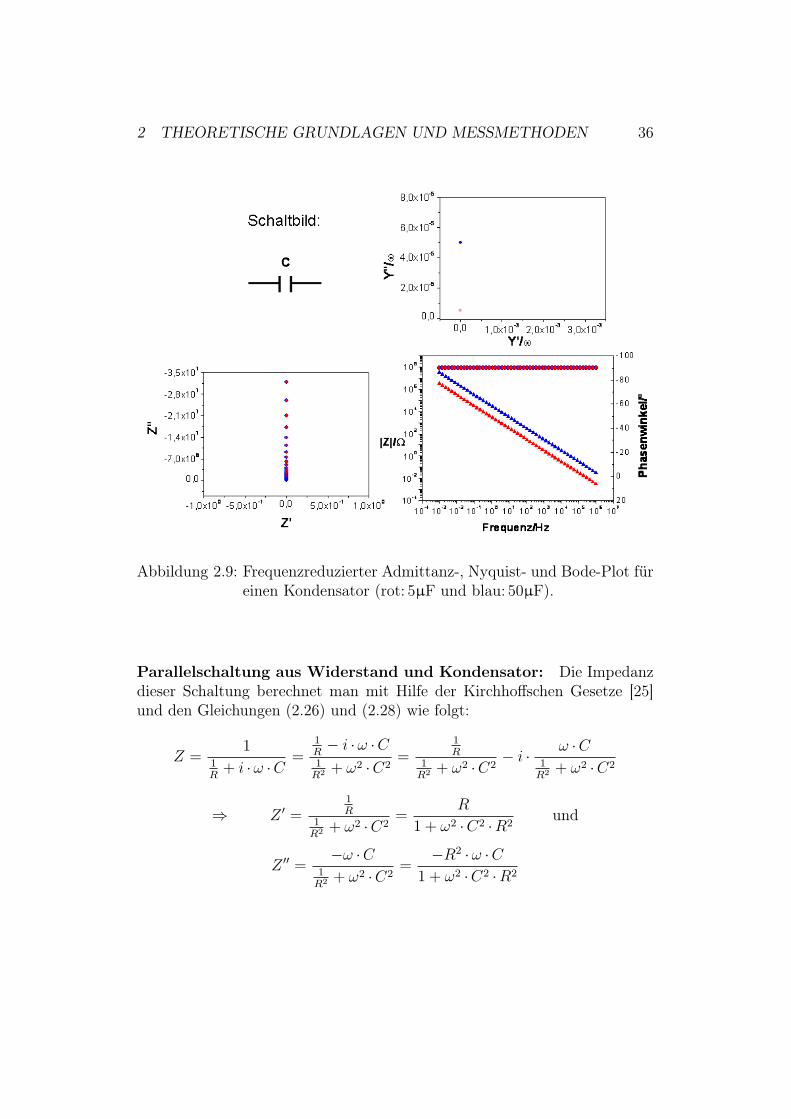
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 36
Abbildung 2.9: Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plot füreinen Kondensator (rot: 5µF und blau: 50µF).
Parallelschaltung aus Widerstand und Kondensator: Die Impedanzdieser Schaltung berechnet man mit Hilfe der Kirchhoffschen Gesetze [25]und den Gleichungen (2.26) und (2.28) wie folgt:
Z =1
1R + i ·& ·C
=1R ! i ·& ·C1
R2 + &2 ·C2=
1R
1R2 + &2 ·C2
! i · & ·C1
R2 + &2 ·C2
$ Z # =1R
1R2 + &2 ·C2
=R
1 + &2 ·C2 ·R2und
Z ## =!& ·C
1R2 + &2 ·C2
=!R2 ·& ·C
1 + &2 ·C2 ·R2

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 37
Man kann zeigen, dass Z # und Z ## die Parameterdarstellung eines Kreises mitRadius R/2 und Mittelpunkt (R/2; 0) erfüllen:
(Z # ! R
2
)2
+ (Z ##)2 =R2
4(Kreisgleichung aus [33])
Da Z ## ( 0, erhält man im Nyquist-Plot in Abbildung 2.10 nur einen Halb-kreis. Der Kreis schneidet - für den Fall, dass man über alle Frequenzen misst- die x-Achse an den Stellen Z # = 0 und Z # = R und eignet sich deshalb sehrgut dazu direkt den Widerstand abzulesen. Die frequenzreduzierte Admittanzdieses Schaltkreises kann man wie folgt berechnen:
Y =1
Z=
1
R+ i ·& ·C $ Y #
&=
1
& ·R undY ##
&= C
Demnach erhält man im frequenzreduzierten Admittanz-Plot eine Geradeparallel zur x-Achse, die die y-Achse im Punkt P(0; C) schneidet.Anhand des Bode-Plots erkennt man, dass sich das System bei hohen Fre-quenzen wie ein Kondensator, bei niedrigen dagegen wie ein Widerstand ver-hält. Dies lässt sich durch Berechnung des Betrages der Impedanz und desPhasenwinkels mit Hilfe von Gleichung (2.25) bestätigen:
| Z |=
√(R
1 + &2 ·C2 ·R2
)2
+
(!R2 ·& ·C
1 + &2 ·C2 ·R2
)2
=
%R2 + R4 ·&2 ·C2
1 + &2 ·C2 ·R2
# = arctan
(! R2 ·ω ·C
1+ω2 ·C2 ·R2
R1+ω2 ·C2 ·R2
)= arctan(!R ·& ·C)
Man erhält also folgende Abhängigkeiten:
& " 0 $ | Z |" R und #" 0
& ") $ | Z |" 1
& ·C " 0 und #" !"
2
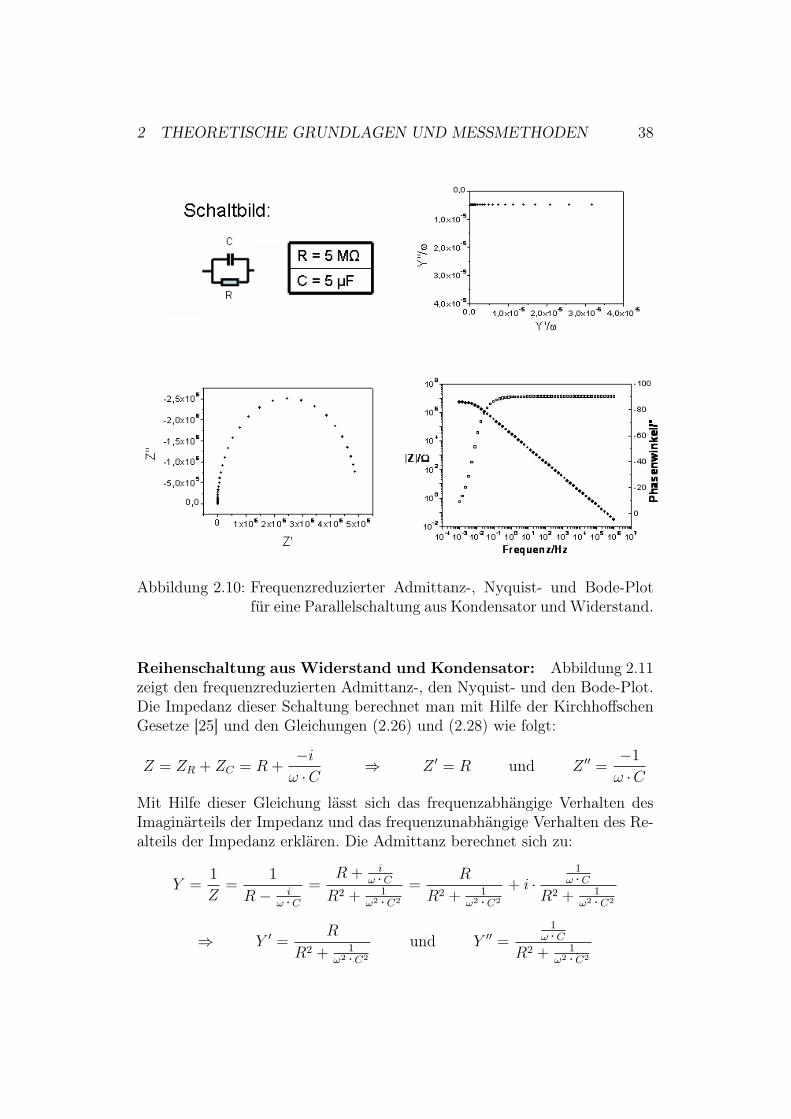
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 38
Abbildung 2.10: Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plotfür eine Parallelschaltung aus Kondensator und Widerstand.
Reihenschaltung aus Widerstand und Kondensator: Abbildung 2.11zeigt den frequenzreduzierten Admittanz-, den Nyquist- und den Bode-Plot.Die Impedanz dieser Schaltung berechnet man mit Hilfe der KirchhoffschenGesetze [25] und den Gleichungen (2.26) und (2.28) wie folgt:
Z = ZR + ZC = R +!i
& ·C $ Z # = R und Z ## =!1
& ·CMit Hilfe dieser Gleichung lässt sich das frequenzabhängige Verhalten desImaginärteils der Impedanz und das frequenzunabhängige Verhalten des Re-alteils der Impedanz erklären. Die Admittanz berechnet sich zu:
Y =1
Z=
1
R! iω ·C
=R + i
ω ·C
R2 + 1ω2 ·C2
=R
R2 + 1ω2 ·C2
+ i ·1
ω ·C
R2 + 1ω2 ·C2
$ Y # =R
R2 + 1ω2 ·C2
und Y ## =1
ω ·C
R2 + 1ω2 ·C2

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 39
Man kann zeigen, dass Y # und Y ## die Parameterdarstellung eines Kreisesmit Radius C/2 und Mittelpunkt (0; C/2) in der frequenzreduzierten Ad-mittanzebene erfüllen20. Da Y #/& ( 0, erhält man im frequenzreduziertenAdmittanz-Plot einen Halbkreis. Der Kreis schneidet - für den Fall, dassman über alle Frequenzen misst - die y-Achse an den Punkten Y ##/& = 0und Y ##/& = C und eignet sich deshalb sehr gut um direkt die Kapazitätabzulesen.Anhand des Bode-Plots erkennt man, dass sich das System bei hohen Fre-quenzen wie ein Widerstand, bei niedrigen dagegen wie ein Kondensator ver-hält. Dies lässt sich durch Berechnung des Betrages der Impedanz und desPhasenwinkels mit Hilfe von Gleichung (2.25) bestätigen:
| Z |=
√
R2 +
(!1
& ·C
)2
=
√R2 +
1
&2 ·C2
# = arctan
( !1ω ·C
R
)= arctan
(!1
& ·C ·R
)
Man erhält also die folgenden Abhängigkeiten:
& " 0 $ | Z |" 1
& ·C und #" !"
2
& ") $ | Z |" R und #" 0
20Dies wurde bereits im vorherigen Abschnitt in ganz ähnlicher Form für eine Parallel-schaltung aus Widerstand und Kondensator in der Impedanz-Ebene dargestellt.
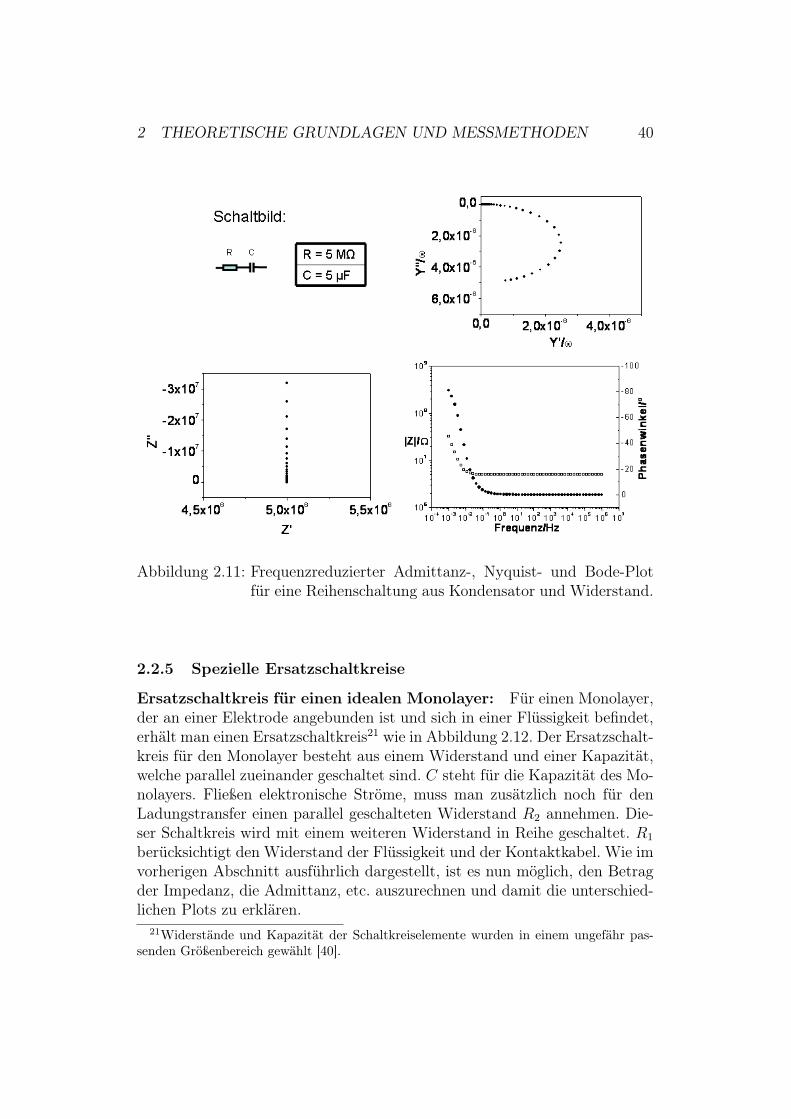
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 40
Abbildung 2.11: Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plotfür eine Reihenschaltung aus Kondensator und Widerstand.
2.2.5 Spezielle Ersatzschaltkreise
Ersatzschaltkreis für einen idealen Monolayer: Für einen Monolayer,der an einer Elektrode angebunden ist und sich in einer Flüssigkeit befindet,erhält man einen Ersatzschaltkreis21 wie in Abbildung 2.12. Der Ersatzschalt-kreis für den Monolayer besteht aus einem Widerstand und einer Kapazität,welche parallel zueinander geschaltet sind. C steht für die Kapazität des Mo-nolayers. Fließen elektronische Ströme, muss man zusätzlich noch für denLadungstransfer einen parallel geschalteten Widerstand R2 annehmen. Die-ser Schaltkreis wird mit einem weiteren Widerstand in Reihe geschaltet. R1
berücksichtigt den Widerstand der Flüssigkeit und der Kontaktkabel. Wie imvorherigen Abschnitt ausführlich dargestellt, ist es nun möglich, den Betragder Impedanz, die Admittanz, etc. auszurechnen und damit die unterschied-lichen Plots zu erklären.
21Widerstände und Kapazität der Schaltkreiselemente wurden in einem ungefähr pas-senden Größenbereich gewählt [40].
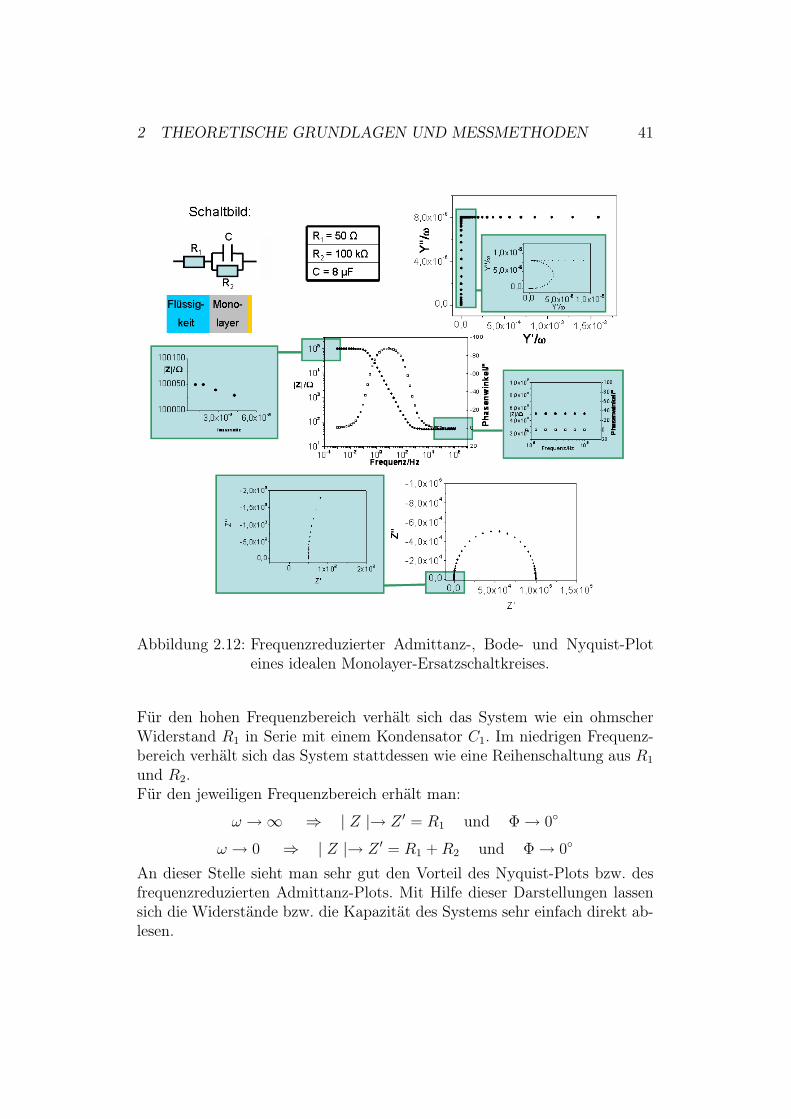
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 41
Abbildung 2.12: Frequenzreduzierter Admittanz-, Bode- und Nyquist-Ploteines idealen Monolayer-Ersatzschaltkreises.
Für den hohen Frequenzbereich verhält sich das System wie ein ohmscherWiderstand R1 in Serie mit einem Kondensator C1. Im niedrigen Frequenz-bereich verhält sich das System stattdessen wie eine Reihenschaltung aus R1
und R2.Für den jeweiligen Frequenzbereich erhält man:
& ") $ | Z |" Z # = R1 und #" 0"
& " 0 $ | Z |" Z # = R1 + R2 und #" 0"
An dieser Stelle sieht man sehr gut den Vorteil des Nyquist-Plots bzw. desfrequenzreduzierten Admittanz-Plots. Mit Hilfe dieser Darstellungen lassensich die Widerstände bzw. die Kapazität des Systems sehr einfach direkt ab-lesen.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 42
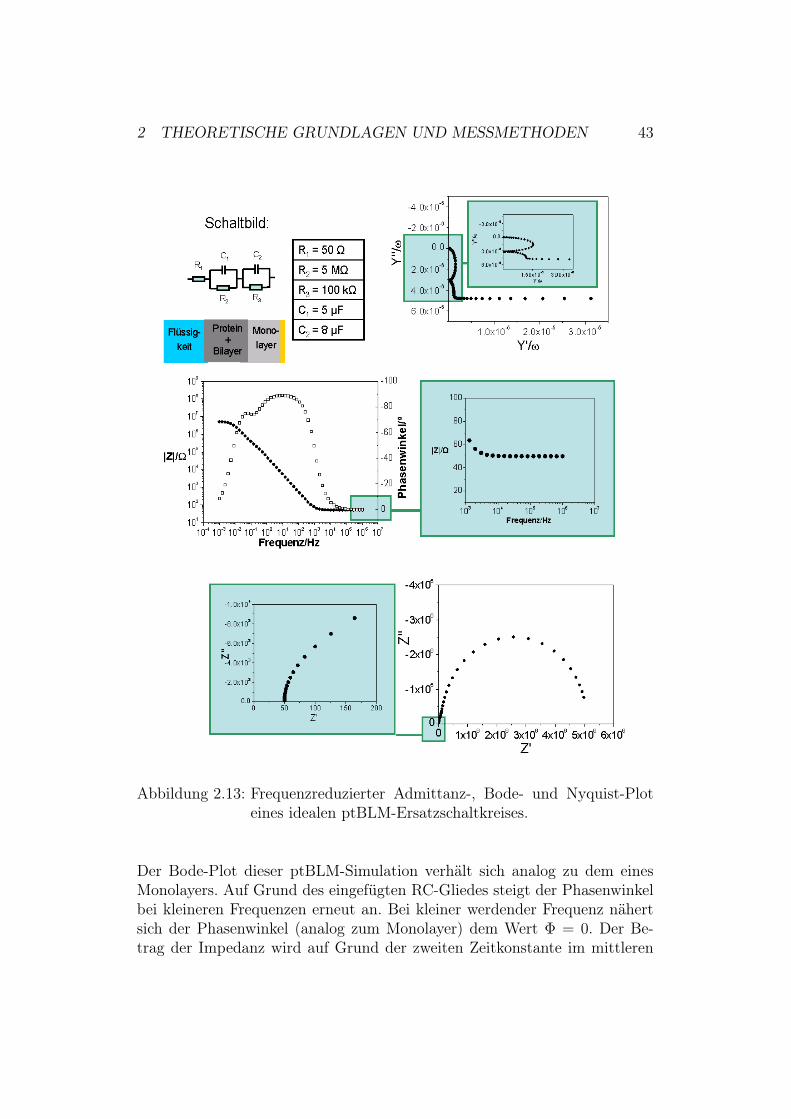
Ersatzschaltkreis für ein ideales ptBLM-System: Ein ptBLM-Systembesteht aus einem Monolayer auf einer Elektrode. An diesem Monolayer wer-den über einen ”His-Tag-Anker” Proteine gebunden. In dieser Arbeit han-delt es sich bei den angebundenen Proteinen um Membranproteine. Mansimuliert mit Hilfe einer Lipiddoppelschicht eine Proteinmembran, um ei-ne proteingerechte Umgebung herzustellen. Lipiddoppelmembranen besitzenauf Grund ihrer Impermeabilität für Ionen einen hohen Widerstand im M$-Bereich. Diese Impermeabilität ist jedoch stark potentialabhängig. Mit Hilfeder Membran ist es somit möglich, Konzentrationsgradienten und damit Po-tentialdifferenzen aufrecht zu erhalten. Die Membran ist also in der Lage,Ladungen zu separieren, weswegen sie auch kapazitive Eigenschaften besitzt.Aus diesem Grund wird unser Monolayerschaltkreis mit einem parallelen RC-Glied erweitert, welches in Reihe geschaltet wird (siehe Abbildung 2.13) [41].Widerstände und Kapazitäten werden in dieser Simulation in dem jeweils pas-senden Größenbereich gewählt [42] [40]. Auch hier ist es wiederum möglich,den Kurvenverlauf der einzelnen Auftragungsarten anhand der im vorheri-gen Abschnitt durchgeführten Prozedur zu erklären. Darauf wird hier jedochverzichtet und lediglich eine qualitative Beschreibung durchgeführt:Der Nyquist-Plot zeigt, sofern die Zeitkonstanten22 der beiden RC-Gliederweit genug auseinander liegen, zwei Halbkreise. In unserem Fall ist dies nichtso. Die Zeitkonstanten liegen relativ dicht beieinander, weshalb sich die Halb-kreise überschneiden. Die Schnittpunkte der Kurve mit der x-Achse ergebensich additiv aus den Einzelwiderständen des Systems.Der Graph des frequenzreduzierten Admittanz-Plots schneidet die y-Achsean dem Punkt, der für den Wert des kleineren Kondensators C1 steht. Mitabnehmender Frequenz bildet sich dann ein Halbkreis. Dieser steht für denKondensator mit höherer Kapazität. Im niedrigen Frequenzbereich verhältsich das System stattdessen wie eine Reihenschaltung aus den Widerstän-den, weshalb der Graph dann in eine Gerade übergeht.
22Die Zeit, die ein System nach einer Anregung benötigt um wieder in seine Gleichge-wichtslage zurückzukehren, bezeichnet man als Relaxationszeit ! = R ·C
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 43
Abbildung 2.13: Frequenzreduzierter Admittanz-, Bode- und Nyquist-Ploteines idealen ptBLM-Ersatzschaltkreises.
Der Bode-Plot dieser ptBLM-Simulation verhält sich analog zu dem einesMonolayers. Auf Grund des eingefügten RC-Gliedes steigt der Phasenwinkelbei kleineren Frequenzen erneut an. Bei kleiner werdender Frequenz nähertsich der Phasenwinkel (analog zum Monolayer) dem Wert # = 0. Der Be-trag der Impedanz wird auf Grund der zweiten Zeitkonstante im mittleren

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 44
Frequenzbereich ebenfalls verändert. Man erhält die folgende Abhängigkeit:
& ") $ | Z |" Z # = R1 und #" 0"
& " 0 $ | Z |" Z # = R1 + R2 + R3 und #" 0"
Die Systeme, die in dieser Diplomarbeit untersucht werden, sind jedoch keineidealen Systeme. Aus diesem Grund müssen die Ersatzschaltkreise den jewei-ligen Bedingungen angepasst werden. Dies geschieht zum Beispiel durch denErsatz eines Schaltkreisgliedes durch ein CPE-Element (Abschnitt 2.2.3).
2.3 CV - CyclovoltammetrieMit den bisher vorgestellten Methoden war es möglich, den Aufbau desptBLM-Systems zu überprüfen. Jedoch sind die Informationen (zum Bei-spiel über kinetische Parameter unseres Systems), die man mit Hilfe von EISund SPR erhalten kann, für unserer Zwecke nicht ausreichend. Da es sichbei den in dieser Diplomarbeit untersuchten Proteinen um Redox-Proteinehandelt, wird deshalb zur näheren Untersuchung der Proben die Cyclovol-tammetrie verwendet. Diese Methode hat sich zur Charakterisierung redox-aktiver Verbindungen etabliert und lässt Rückschlüsse auf den Mechanismuseiner elektrochemischen Reaktion zu.Die Cyclovoltammetrie gehört wie die Impedanzspektroskopie zu den elektro-chemischen Messmethoden. Bei der Impedanzspektroskopie wurde ein kleinesalternierendes Störsignal angelegt und der resultierende Strom gemessen. Beider Cyclovoltammetrie dagegen werden durch eine Dreiecksspannung Poten-tiale jenseits des elektrochemischen Gleichgewichts angelegt. Der Arbeitselek-trode (siehe Abschnitt 2.2.1) und damit der zu untersuchenden Probe wirdausgehend von einem Anfangspotential ein sich zeitlich linear änderndes Po-tential aufgeprägt (siehe Abbildung 2.14). Die dabei in der Messzelle vorsich gehenden Prozesse führen zu einem charakteristischen Stromsignal. DerStromfluss wird, wie in Abbildung 2.14 zu sehen, als Funktion der zwischenArbeitselektrode und Referenzelektrode angelegten Spannung aufgezeichnet.
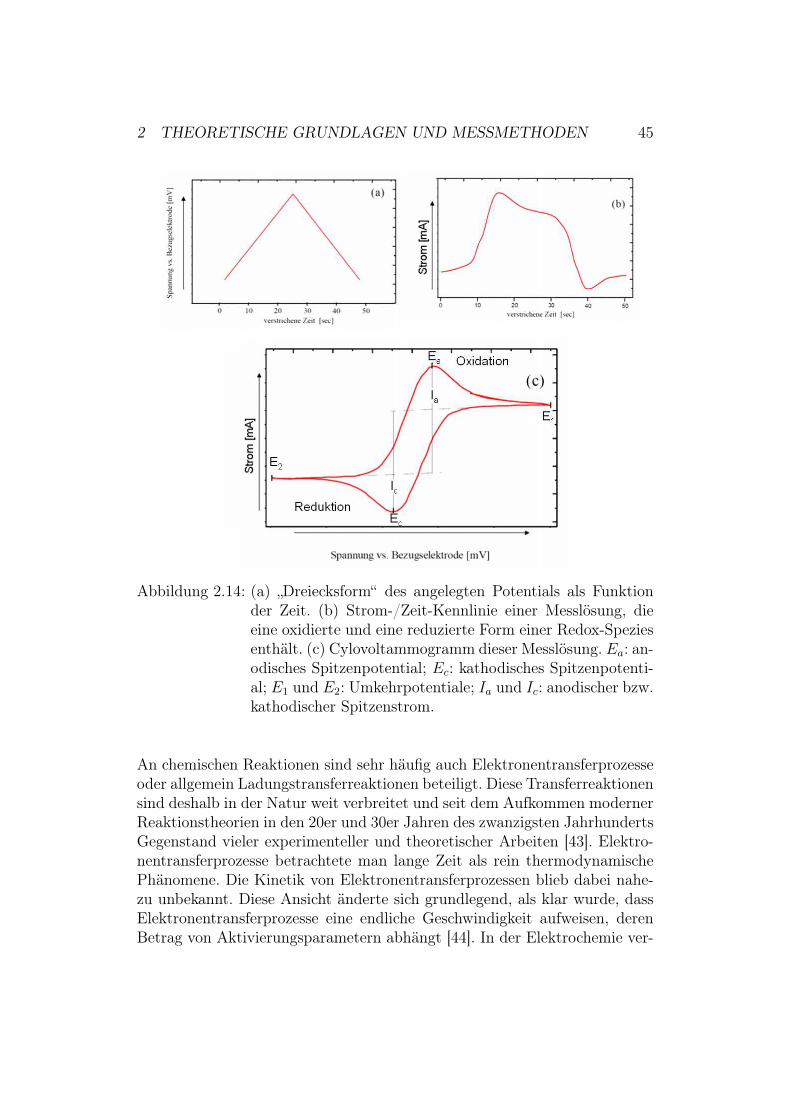
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 45
Abbildung 2.14: (a) „Dreiecksform“ des angelegten Potentials als Funktionder Zeit. (b) Strom-/Zeit-Kennlinie einer Messlösung, dieeine oxidierte und eine reduzierte Form einer Redox-Speziesenthält. (c) Cylovoltammogramm dieser Messlösung. Ea: an-odisches Spitzenpotential; Ec: kathodisches Spitzenpotenti-al; E1 und E2: Umkehrpotentiale; Ia und Ic: anodischer bzw.kathodischer Spitzenstrom.
An chemischen Reaktionen sind sehr häufig auch Elektronentransferprozesseoder allgemein Ladungstransferreaktionen beteiligt. Diese Transferreaktionensind deshalb in der Natur weit verbreitet und seit dem Aufkommen modernerReaktionstheorien in den 20er und 30er Jahren des zwanzigsten JahrhundertsGegenstand vieler experimenteller und theoretischer Arbeiten [43]. Elektro-nentransferprozesse betrachtete man lange Zeit als rein thermodynamischePhänomene. Die Kinetik von Elektronentransferprozessen blieb dabei nahe-zu unbekannt. Diese Ansicht änderte sich grundlegend, als klar wurde, dassElektronentransferprozesse eine endliche Geschwindigkeit aufweisen, derenBetrag von Aktivierungsparametern abhängt [44]. In der Elektrochemie ver-

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 46
schob sich damit allmählich der methodische Schwerpunkt von der statischenzur dynamischen Strommessung.Unter den zahlreichen dynamischen Messmethoden der elektroanalytischenChemie23, die in den letzten Jahrzehnten entwickelt wurden, hat die Cyclo-voltammetrie (CV) eine besonders große Popularität erlangt. Zwei Aspek-te haben zu dieser Entwicklung wesentlich beigetragen: Die theoretischenGrundlagen der Methode wurden unter anderem von Randles und Sevcik be-reits 1948 mathematisch exakt analysiert, so dass alle wichtigen Variantenelektrochemischer Prozesse in ihrer Beziehung zum voltammetrischen Signalquantitativ interpretiert werden können [45] [46]. Moderne numerische Me-thoden ermöglichen außerdem die Berechnung von Cyclovoltammogrammenfür komplexe Elektrodenprozesse [47]. Zusätzlich dient die Cyclovoltamme-trie zur Untersuchung potentialabhängiger Phasengrenzflächenreaktionen wieAdsorptionsprozessen [48]. Entscheidend für den Durchbruch der Methodewar jedoch die Tatsache, dass man in sehr kurzer Zeit Informationen überdie Thermodynamik von Redoxprozessen und die Kinetik von Elektronen-transferreaktionen erhalten kann.Zum Beispiel ist es möglich, anhand der Peakpotentiale das so genannte Mit-telwertpotential auszurechnen:
E1/2 =Epc + Epa
2
Das Mittelwertpotential liegt sehr nahe am thermodynamischen Redox-Poten-tial E0, welches dadurch schnell geschätzt werden kann.In der vorliegenden Arbeit werden die benötigten kinetischen Daten durchVariation der Spannungsvorschubgeschwindigkeit (Scan-Rate) ) gewonnen:
) = dE/dt (2.29)
Man unterscheidet zwei verschiedene Arten von Strömen, faradayische undnicht-faradayische Ströme.Nicht-faradayische Ströme sind von elektrostatischer Natur und in keinsterWeise mit einer elektrochemischen Reaktion verbunden. Sie resultieren ausder Reorganisation der Ionen im Elektrolyten nahe der Elektrodengrenzflächeund werden auch kapazitive Ströme Ic genannt. Für einen SpannungsverlaufE = Einit+) · t, bei konstanter Grenzschichtkapazität Cdl und der Annahme,dass man nur einen Lösungsmittelwiderstand Rs hat, erhält man:
IC =Einit
Rs· e!t/Rs ·Cdl + ) ·Cdl
(1! e!t/Rs ·Cdl
)(2.30)
23Hierzu gehört auch die in Abschnitt 2.2 bereits diskutierte elektrochemische Impe-danzspektroskopie.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 47
Das bedeutet, der kapazitive Strom nimmt mit zunehmender Spannungs-vorschubgeschwindigkeit und zunehmender Grenzschichtkapazität zu. BeimRücklauf des CV bekommt der zweite Summand ein negatives Vorzeichen,so dass sich Hin- und Rückrichtung des CV nach Abklingen des transientenAnteils um !I = 2)Cdl unterscheiden [48].Faradayische Ströme haben ihre Herkunft im heterogenen Ladungstransferzwischen Elektrode und einer Redoxspezies im Elektrolyt, der immer miteinem chemischen Stoffumsatz verbunden ist. Den quantitativen Zusammen-hang liefern die Faradayschen Gesetze [48].
2.3.1 Cyclovoltammetrie von gelösten Redoxspezies
Die einfachste Faradaysche Elektrodenreaktion besteht im heterogenen La-dungstransfer von einer elektroaktiven Spezies (zum Beispiel: Fe) zur Elektro-de (zum Beispiel: Ag) oder umgekehrt, bekannt als Ein-Elektronen-Transfer24.Je nach der Richtung des Ladungstransports unterscheidet man anodischeund kathodische Ströme:Ein anodischer Strom fließt, wenn
• positive Ladungsträger von der (Arbeits-) Elektrode in eine Redoxspe-zies im Elektrolyten übergehen.
• negative Ladungsträger von der Redoxspezies im Elektrolyten in dieElektrode übergehen.
Dieser Vorgang stellt eine Oxidation dar, und die Elektrode wird als Anodebezeichnet.Ein kathodischer Strom fließt, wenn
• positive Ladungsträger von der Redoxspezies im Elektrolyten in dieElektrode übertreten.
• negative Ladungsträger von der Elektrode in eine Redoxspezies imElektrolyten übergehen.
Jetzt stellt der Vorgang eine Reduktion dar, und die Elektrode wird als Ka-thode bezeichnet. Die messbare Gesamtstromdichte j setzt sich additiv ausder anodischen und der kathodischen Teilstromdichte zusammen. Im elektro-chemischen Gleichgewicht E = E0 laufen beide Teilreaktionen gleich schnell
24Kompliziertere Mechanismen werden in dem Übersichtsartikel ”Cyclovoltammetrie -die Spektroskopie des Elektrochemikers” von Jürgen Heinze oder ausführlicher in denÜbersichtswerken von Balzani behandelt [49][50].

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 48
ab und es findet makroskopisch kein Stoffumsatz statt [51]. Nur wenn dasPotential an der Elektrode von E0 verschieden ist, kann auch ein Strom flie-ßen. Die Abweichung des Elektrodenpotentials unter Stromfluss von seinemGleichgewichtspotential bezeichnet man als Überspannung :
+ = E ! E0 (2.31)
Zwei Prozesse prägen die Faradaysche Elektrodenreaktion:
• der heterogene Ladungstransfer zwischen Elektrode und Lösung
• der diffusionsbedingte Massentransport
Der heterogene Ladungstransfer an der Phasengrenzfläche Elektrode/Elektro-lyt wird durch die Butler-Volmer-Gleichung, die Grundgleichung der elektro-chemischen Kinetik, beschrieben [49]:
I = n ·F ·A · k0
(Cox · e!α ·n ·F · ν/R ·T ! Cred · e(1!α) ·n ·F · ν/R ·T
)(2.32)
Hierbei ist * der Durchtritts- oder Symmetriefaktor, ) die Überspannungaus Gleichung (2.31), T die Temperatur, R die molare Gaskonstante und Fdie Faradaykonstante. Des Weiteren steht n für die Anzahl der übertrage-nen Elektronen und A für die aktive Elektrodenoberfläche. Nach der Butler-Volmer-Gleichung hängt der Strom von den Oberflächenkonzentrationen Cox
und Cred der beteiligten Redoxpartner, dem aktuellen ElektrodenpotentialE und der Standardgeschwindigskeitkonstanten des heterogenen Durchtrittsk0 ab. Aufgrund ihres Konzentrationsunterschieds zur übrigen Lösung führendie potentialabhängigen Oberflächenkonzentrationen zu einem diffusionskon-trollierten Massentransport. Die Konzentrationsverteilung in der Diffusions-schicht lässt sich aus dem zweiten Fickschen Gesetz berechnen [49]:
$Cox
$t= Dox ·
$2Cox
$x2und
$Cred
$t= Dred ·
$2Cred
$x2(2.33)
In dieser Gleichung stehen Dox und Dred für den Diffusionskoeffizienten deroxidierten bzw. reduzierten Redox-Spezies. Allerdings gilt das Ficksche Ge-setz nur für eine lineare Diffusion, die jedoch meistens annähernd vorherrscht[49].Der Konzentrationsgradient an der Elektrodenoberfläche ist dem Ladungs-fluss direkt proportional:
j =I
n ·F ·A & !Dox
($Cox
$x
)
x=0
= Dred
($Cred
$x
)
x=0
(2.34)

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 49
Der an der Arbeitselektrode messbare Strom setzt sich aus einem Anteil fürden heterogenen Ladungstransfer und einem Anteil für den Massentransportzusammen. Die jeweiligen Anteile hängen mit den Gleichungen (2.32) und(2.34) zusammen [49].
Man unterscheidet nun drei Fälle voneinander:
Quasireversibler Fall: In diesem allgemeinen Fall bestimmen sowohl La-dungstransfer als auch Massentransport den messbaren Strom
Reversibler Fall: Bei einem reversiblen Prozess ist die Geschwindigkeitdes heterogenen Ladungstransfers so groß, dass sich an der Phasengrenzflächeein thermodynamisches Gleichgewicht einstellt. Die Butler-Volmer-Gleichungvereinfacht sich dadurch zur Nernst-Gleichung.Abbildung 2.14 zeigt ein Cyclovoltammogramm eines reversiblen Redoxsys-tems, das sowohl die oxidierte Form als auch die reduzierte Form einesRedoxpaares enthält. Das Experiment wird bei einem Potential Einit mitEup < Einit < Edown gestartet. Läuft die Spannung zuerst in Richtung posi-tiver Potentiale, so wird entsprechend der Nernst-Gleichung :
E = E0 +R ·Tn ·F · ln
([Cox]
[Cred]
)(2.35)
die reduzierte Form in die oxidierte Form umgesetzt (R: Gaskonstante, T :Temperatur, F : Faradaykonstante, C: Konzentration, n: Anzahl der Elektro-nen, E0: Standardpotential). Das bedeutet, die Oberflächenkonzentrationenhängen nur noch vom Elektrodenpotential ab und werden nicht mehr durchheterogene kinetische Effekte beeinflusst. Der Strom an der Elektrodenober-fläche wird ausschließlich durch den Massentransport als langsamsten Schrittkontrolliert [49].Der Verbrauch der Reaktanden an der Elektrodenoberfläche führt zu einemausgeprägten Konzentrationsgradienten. Dieser Gradient fördert eine schnel-lere Diffusion der Reaktanden und somit den Strom. Der Potentialscan läuftweiter und der Strom wird zunehmend von der endlichen Diffusionsgeschwin-digkeit der Moleküle gehemmt, bis er schließlich ein Maximum erreicht. BeimMaximum am Potential Epc ist die effektive Konzentration der herandiffun-dierenden Moleküle an der Elektrodenoberfläche identisch mit Null. Jedes he-randiffundierte Moleküle wird sofort umgesetzt. Da der Konzentrationsgradi-ent weiter zunimmt, nimmt der Strom I entsprechend der Cotrell-Gleichung
I =n ·F ·A ·
%D ·C
%" ·%
t(2.36)

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 50
mit t!1/2 ab, bis der Umkehrpunkt Edown erreicht ist. In dieser Gleichungsteht A für die elektrochemisch aktive Oberfläche der Elektrode, D und C fürden Diffusionskoeffizienten bzw. die Ausgangskonzentration des Substrates[52][53].Für den Rückweg des Potentialscans gelten obige Ausführungen analog fürden Reduktionsvorgang.Ein ideal reversibler Vorgang hat folgende Charakteristika:
• k0 > 10!1cm/s
• Der Potentialabstand zwischen den Spitzenwerten ist bei 25"C gegebendurch
!Ep = |Epc ! Epa| =59
nmV ,
wobei n für die Anzahl der Elektronen steht, die während eines Redox-prozesses umgesetzt werden.
• Das Peakpotential ist unabhängig von der Spannungsvorschubgeschwin-digkeit.
• Die Peakbreite bei halber Peakstromhöhe ist für alle Spannungsvor-schubgeschwindigkeiten gleich. Sie entspricht 28 mV pro umgesetztemElektron. Je höher die Anzahl der Elektronen, die während eines Re-doxprozesses umgesetzt werden, desto schmaler wird der Peak. Damitist es möglich die Anzahl der Elektronen zu bestimmen.
• Die Beträge der Peakströme nehmen linear mit der Quadratwurzelder Spannungsvorschubgeschwindigkeit zu. Sie sind durch die Randles-Sevcik-Gleichung gegeben:
I # 2,69 · 105 ·n3/2 ·A ·C ·%
D · ) (2.37)
In dieser Gleichung steht I für Ipa bzw. Ipc und n für die Anzahl der Elektro-nen. C steht für die Konzentration der umgesetzten Redox-Spezies Cox bzw.Cred und D für den jeweiligen Diffusionskoeffizienten [48][49].
Irreversibler Fall: Bei diesem Prozess ist der Ladungstransfer an der Elek-trode sehr langsam (k0 < 10!5cm/s). Abhängig vom Potential hat nur dieanodische oder kathodische Durchtrittsreaktion eine messbare Geschwindig-keit [49]. Hierbei wird der Strom also nicht, wie im reversiblen Fall, durch dieDiffusion kontrolliert, sondern durch den Ladungstransfer. Beim irreversiblen

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 51
Ladungsübergang wird der Abstand zwischen dem anodischen und dem ka-thodischen Peakpotential größer. Man benötigt also ein größeres Überpotenti-al um die Redox-Spezies zu reduzieren oder zu oxidieren. Außerdem reduziertsich die Peakhöhe. Total irreversible Systeme zeichnen sich bei Veränderungder Spannungsvorschubgeschwindigkeit durch eine Verschiebung des Peakpo-tentials aus.Ein weiteres mögliches Kriterium für eine irreversible Reaktion ist das Feh-len des Peaks für die Rückreaktion des Produkts. Das ist die Folge des imVergleich zum reversiblen System langsameren Elektronentransfers, der be-wirkt, dass die umgesetzte Redox-Spezies aus dem Elektrodenbereich heraus-diffundiert oder durch Weiterreaktion nicht mehr für die Rückreaktion zurVerfügung steht [48].
2.3.2 Cyclovoltammetrie von oberflächengebundenen Redoxspe-zies
Bis jetzt wurde in der Diskussion der Cyclovoltammetrie immer davon aus-gegangen, dass sich Reaktanden und Produkte frei in Lösung befinden. Indiesem Abschnitt soll nun der Fall von oberflächengebundenen Redoxmole-külen dargestellt werden. Hier spielen Diffusionsphänomene keine Rolle mehr.Im idealen Fall ist die Peakseparation identisch mit 0 mV. Die Peakbreite beihalber Peakstromhöhe beträgt bei 25"C [48]
!E = 3,53R ·Tn ·F = 90,6 mV pro Elektron. (2.38)
In Abbildung 2.15 ist ein Cyclovoltammogramm einer ideal reversiblen ober-flächengebundenen Redoxspezies schematisch dargestellt.Der Peakstrom hängt in diesem Fall ab von der konstanten Oberflächenbele-gung der Redox-Moleküle %tot = %ox +%red, der aktiven ElektrodenoberflächeA, der Spannungsvorschubgeschwindigkeit ), der Faradaykonstanten F , derGaskonstanten R und der Temperatur T . Wieder spielt die Anzahl der über-tragenen Elektronen eine Rolle:
I =n2 ·F 2 · ) ·A ·%tot
4 ·R ·T (2.39)
Im Gegensatz zur diffusionskontrollierten reversiblen Umsetzung von gelös-ten Redoxmolekülen ist der Peakstrom nicht direkt proportional zur Qua-dratwurzel der Spannungsvorschubgeschwindigkeit (Gleichung 2.37), sondernlaut Gleichung 2.39 direkt proportional zu ) [48].

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 52
Abbildung 2.15: Schematisches Cyclovoltammogramm einer ideal oberflä-chengebundenen Redoxspezies.
2.3.3 IR-Drop Kompensation
Wegen der endlichen Leitfähigkeit des Elektrolyten kommt es zu einem Span-nungsabfall, den man als IR-Drop bezeichnet. In einem Cyclovoltammo-gramm macht sich der IR-drop negativ bemerkbar durch eine Vergröße-rung der Peakpotentialdifferenz, durch eine Verbreiterung der Peaks und eineVerringerung des Peakstroms. Diese Verzerrung der experimentellen Kurvenkann, wie in Abbildung 2.16 zu sehen, drastisch ausfallen. Der Effekt ist umsogrößer, je größer der Widerstand der Lösung und je größer der FaradayscheStrom ist. Der Faradaysche Strom wiederum steigt mit zunehmender Konzen-tration des Substrats und zunehmender Spannungsvorschubgeschwindigkeitan.Die wichtigsten Methoden für eine Verkleinerung des IR-Drops sind:
• Verringerung des Lösungsmittelwiderstands durch Erhöhung der Leit-salzkonzentration
• elektronische Kompensation
Auf Grund der Empfindlichkeit unseres ptBLM-Systems25 wurde die elektro-nische Kompensation zur IR-Drop-Kompensation verwendet.
25Membranproteine benötigen eine spezielle Umgebung. Sie würden bei hohen Leitsalz-konzentrationen denaturieren.
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 53
Bei der elektronischen Kompensation - auch ”positive feedback methode” ge-nannt - wird mit einer Korrekturspannung gearbeitet [54]. Die optimale Höhedieser Spannung kann durch eine Impedanzmessung (Messung des Lösungs-mittelwiderstandes) ermittelt werden.
Abbildung 2.16: Cyclovoltammogramm (a) ohne und (b) mit IR-Drop.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 54
2.4 SpektroelektrochemieMit Hilfe von EIS und CV kann man zwar die Aktivität der oberflächenge-bundenen Proteine nachweisen und kinetische Parameter ausrechnen, jedochist keine detaillierte Spezies-Analyse möglich. Wenn ein Protein zum Beispielaus mehreren Redoxzentren besteht26, liefert die Cyclovoltammetrie bzw. dieelektrochemische Impedanzspektroskopie einen Stromwert, welcher dann alleanwesenden Prozesse repräsentiert. So erhält man keine direkte Informationüber die Redoxspezies und deren Redoxzustände. Zudem ist keine Aussageüber die strukturellen Veränderungen des Systems möglich.Die Schwingungs-Spektroskopie ist eine wichtige Methode, um Informationenüber Schwingungs- und Rotationszustände von Molekülen zu erhalten. Mitdiesen Informationen lassen sich Aussagen über den Redox-, Koordinations-und Ligandenzustand von Metallkomplexen machen. Diese Metallkomplexeliegen in allen Hämproteinen vor, wie zum Beispiel beim Cytochrom c oderbei der Cytochrom-c-Oxidase.Durch die Kombination dieser elektrochemischen und spektroskopischen Me-thoden, kurz durch die ”Spektroelektrochemie”, ist es möglich, redoxbedingteund strukturelle Veränderungen von Proteinen nachzuweisen und zuzuord-nen. Die Kombination von elektrochemischen Experimenten mit ”in situ”Spektroskopie wurde in den letzten 25 Jahren mit erhöhter Aufmerksamkeitstudiert und die meisten Spektroskopiearten, wie zum Beispiel UV-visible-,Infrarot- und Ramanspektroskopie miteinbezogen. Aufgrund der Kombina-tion zweier völlig verschiedener Messmethoden sind die Experimente meistsehr komplex aufgebaut und man benötigt spezielle Apparaturen. Die größteSchwierigkeit besteht darin, einen experimentellen Aufbau zu konstruieren,der eine hohe Sensitivität für die spektroskopische und elektrochemische Un-tersuchung aufweist [51].Man kann zwei Messmethoden, die in dieser Diplomarbeit Verwendung fin-den, voneinander unterscheiden:
2.4.1 Potentiostatische Messungen
Das an der Oberfläche gebundene Protein wird durch eine angelegte kon-stante Spannung in einen definierten Zustand (reduziert, oxidiert oder einZwischenzustand) gezwungen. Daraufhin liefert die Schwingungsspektrosko-pie die oben erwähnten Informationen.
26in unserem Fall die vier Redoxzentren der Cytochrom-c-Oxidase

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 55
2.4.2 Zeitaufgelöste Messungen
Ein großer Nachteil der schwingungsspektroskopischen Methoden ist der klei-ne Wirkungsquerschnitt und der damit verbundene Zeitaufwand der Mes-sung. Aus diesem Grund musste ein spezielles Messverfahren für die Zeitauf-lösung entwickelt werden [55]:Eine Rechteckspannung wird an die Elektrode, an die das Protein gebun-den ist, angelegt. Start- und Endpotential (Ei und Ef) werden so gewählt,dass sich das Protein bei den jeweiligen Potentialen in einem vollkommenoxidierten und vollkommen reduzierten Zustand befindet. Nach einer Verzö-gerungszeit , findet eine Schwingungsspektroskopie-Messung im Intervall !tstatt (Abbildung 2.17). Die Zeitauflösung ist damit durch die Wahl von , und!t begrenzt. Nach dem Messvorgang muss das System wieder in den Aus-gangszustand gebracht werden, so dass beim nächsten Messvorgang genaudieselben Voraussetzungen gegeben sind. Man misst also die durchschnittli-che Änderung des Systems zwischen t=, und t=,+!t. Die Potentialsprüngewerden z mal wiederholt, so dass die tatsächliche Messzeit z ·!t beträgt. DieAnzahl der Wiederholungen z muss so gewählt werden, dass man ein vernünf-tiges Signal-Rausch-Verhältnis erhält. Da es in unserem Fall nicht möglichist den Detektor zu triggern, ist er während der kompletten Messzeit z ·Tcaktiv, was leider zu einem verstärkten Hintergundrauschen führt.
Abbildung 2.17: Das Prinzip der zeitaufgelösten Messmethode.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 56
2.5 RamanspektroskopieIm Rahmen dieser Diplomarbeit wurde als Schwingungsspektroskopieart dieRamanspektroskopie gewählt, deren theoretische Grundlagen in diesem Ka-pitel dargelegt werden sollen.Die Ramanspektroskopie beruht auf der als Raman-Effekt bezeichneten in-elastischen Streuung von Licht an Molekülen. Theoretische Grundlagen, un-ter anderem von Smekal [56], führten 1928 zur Entdeckung des Effektes durchRaman und Krishnan [57]. Nur zwei Jahre später erhielt Raman für seineEntdeckung den Nobelpreis für Physik.
2.5.1 Darstellung des Raman-Effekts über das Energietermsche-ma
Eine einfache qualitative Darstellung des Raman-Effekts27, welche schon we-sentliche Züge darlegt, kann mit einem Energietermschema erfolgen.Bestrahlt man ein Molekül mit einem Photon der Energie (E = h · f), dieklein genug ist, um das erste Anregungsniveau nicht zu erreichen28, wird esvom Grundniveau in ein so genanntes virtuelles Niveau angehoben. Virtuellheißt hier, dass es sich nicht um einen stationären Zustand im quantenmecha-nischen Sinne handelt. Es wird weder der nächste elektronische Anregungs-zustand noch ein Vibrationszustand erreicht. Nach der Anregung relaxiertdas Molekül unter Emission eines Photons. Hierbei kann man drei Fälle un-terscheiden (siehe Abbildung 2.18):Bei der Rayleigh-Streuung (a) wechselwirkt das Molekül mit dem einfallen-den Photon und befindet sich anschließend wieder im nicht schwingungsange-regten elektronischen Grundzustand. Die Wellenlängen von einfallender undgestreuter Strahlung sind identisch.Bei der Stokes-Raman-Streuung (b) werden beim Streuprozess Schwingun-gen im Molekül angeregt. Die gestreute Strahlung ist um den Betrag der zurSchwingungsanregung benötigten Energie ärmer und weist somit eine größereWellenlänge als die einfallende Strahlung auf.Bei der Anti-Stokes-Raman-Streuung (c) befindet sich das Molekül vor demStreuprozess in einem schwingungsangeregten Zustand (z.B. v = 1) und rela-xiert nach der Wechselwirkung mit dem Photon in einen schwächer angereg-ten Zustand (z.B. v = 0). Die Energie der Schwingung wird auf das gestreu-te Photon übertragen, welches dadurch eine höhere Energie und damit eine
27In dieser Arbeit wird unter Raman-E!ekt im Allgemeinen immer der Vibrations-Raman-E!ekt bezeichnet. Der Rotations-Raman-E!ekt wird im Abschnitt A.3 kurz er-läutert.
28Wird das erste Anregungsniveau erreicht, kann das Photon absorbiert werden. Danntreten zusätzliche E!ekte auf, wie zum Beispiel Fluoreszenzstrahlung.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 57
kürzere Wellenlänge als das einfallende Photon aufweist. Dieser Streuvorgangtritt üblicherweise mit geringerer Wahrscheinlichkeit als die Stokes-Streuungauf, da er von der Besetzungsdichte des schwingungsangeregten Zustandesabhängt. Die Besetzung folgt der Boltzmann-Verteilung und ist stark tempe-raturabhängig. Der Quotient aus der Anzahl der Moleküle im Zustand ) = 1und der Anzahl der Moleküle im Zustand ) = 2 ist gegeben durch:
n1
n2= e!(E1!E2)/kB ·T
Bei Raumtemperatur befinden sich jedoch die meisten Moleküle im Grund-zustand. Folglich haben die Stokes-Linien eine höhere Intensität als die Anti-Stokes-Linien [58].
Abbildung 2.18: Die Energieniveaus bei der elastischen und inelastischenStreuung.
2.5.2 Klassische Deutung des Raman-Effekts
Die klassische Deutung des Raman-Effekts basiert auf einem Wellenmodell.Im Feld einer elektromagnetischen Welle oszillieren geladene Teilchen. Bei derWechselwirkung zwischen dem Molekül und der elektromagnetischen Wellekommt es zu einer periodischen Verschiebung der Elektronen und Protonenaus den Gleichgewichtslagen. Durch das äußere Feld wird daher ein Dipolmo-ment im Molekül induziert. Dieses Dipolmoment ist proportional zum elektri-schen Feld, das das Dipolmoment induziert hat. Der Proportionalitätsfaktorheißt Polarisierbarkeit [24]:
µ = * ·E

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 58
Das elektrische Feld der elektromagnetischen Welle am Ort des Molekülsverändert sich mit der Zeit:
E = E0 cos(2"f0t) (2.40)
Hierbei ist E0 der maximale Wert des elektrischen Feldes, f die Frequenz derStrahlung und t die Zeit.Die klassische Theorie fordert nun eine Abstrahlung mit der Frequenz f .Man nimmt an, dass die Polarisierbarkeit für eine feste Frequenz konstant ist[24]. In Molekülen können jedoch gewisse Rotationen und Vibrationen dafürsorgen, dass sich die Polarisierbarkeit ändert. Zum Beispiel verändert sich dieForm eines zweiatomigen Moleküls periodisch durch eine Vibration. Wenn dieElektronenwolke nicht mit diesen Vibrationen konstruktiv interferiert, erhältman eine Veränderung der Polarisierbarkeit. Für sehr kleine Abweichungenkann man die Polarisierbarkeit in einer Taylorreihe entwickeln [58]:
* = *0 +$*
$Q·Q + ... (2.41)
*0 ist die Polarisierbarkeit im Gleichgewichtszustand. Q steht für die Nor-malkoordinaten der Kerne29 und ∂α
∂Q ist die Rate, in der die Polarisierbarkeitrelativ zu Q im Gleichgewichtszustand verändert wird. * hat nun die Bedeu-tung eines molekularspezifischen dreidimensionalen Parameters und wird alsPolarisationstensor bezeichnet.Bei kleinen Veränderungen kann die Variation der Kernkoordinaten durcheine harmonische Schwingung dargestellt werden:
Q = Q0 cos(2"ft) (2.42)
Dadurch kann man die Terme höherer Ordnung vernachlässigen. Einsetzenvon Gleichung (2.42) in (2.41) liefert:
* # *0 +$*
$Q·Q0 cos(2"ft) (2.43)
Mit Hilfe von Gleichung (2.40) erhält man damit für das Dipolmoment [58]:
µ = *E = *0E0 cos(2"f0t) +$*
$Q·Q0E0 cos(2"f0t) cos(2"ft) (2.44)
Dies lässt sich unter Verwendung von [33]:
cos - cos . =1
2[cos(- ! .) + cos(- + .)]
29In dem obigen zweiatomigen Spezialfall wäre Q = r ! re.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 59
umschreiben zu:
µ = *0E0 cos(2"f0t)+$*
$Q·Q0E0[cos(2"(f0!f)t)+cos(2"(f0 +f)t)] (2.45)
Die letzte Gleichung zeigt, dass die Streustrahlung drei Anteile mit unter-schiedlichen Frequenzen aufweist. Der erste Term beschreibt die Rayleigh-Streuung, der zweite die Stokes-, der dritte die Anti-Stokes-Signale. Der Po-larisationstensor ändert sich nicht im Term für die Rayleigh-Streuung, dafürjedoch in den Termen für Stokes- und Anti-Stokes-Linien. Hierdurch zeigtsich die fundamentale Bedingung für das Auftreten der Raman-Streuung.Eine Schwingung ist nur dann raman-aktiv, wenn sich in ihrem Verlauf diePolarisierbarkeit ändert [58]:
$*
$Q*= 0 (2.46)
Die Intensität eines Raman-Streuvorgangs ist proportional zum Quadrat dermittleren Polarisierbarkeit |*|2 und als Dipolabstrahlung zur vierten Potenzder reziproken Streuwellenlänge [24]:
IR &|*|2 ·E2
/4
Die vorgestellte klassische Theorie der Raman-Streuung ist zwar sehr an-schaulich, doch kann sie einige der beobachteten Phänomene bei den Raman-Experimenten nicht erklären. Die Theorie wurde im Laufe der Zeit mit denMethoden der Quantenmechanik behandelt. Eine ausführliche Diskussionder quantenmechanischen Beschreibung des Raman-Effekts findet man unter[59]. Diese Theorie erlaubt es bei Molekülen, die sich im Rahmen der Theo-rie genügend genau beschreiben lassen, die Form der Raman-Spektren rechtpräzise zu berechnen.Ein großer Nachteil der Ramanspektroskopie ist der extrem kleine Wirkungs-querschnitt des Streuvorgangs, der 12-14 Größenordnungen unter dem Wir-kungsquerschnitt der Fluoreszenz liegt. Man erhält Wirkungsquerschnittevon 10!31 bis 10!29 cm2/Molekül [60].Durch den kleinen Wirkungsquerschnitt hat man eine extrem kleine Sensiti-vität. Normalerweise kann man dieses Problem mit hohen Konzentrationender zu analysierenden Redox-Spezies oder durch die Erhöhung der Laserleis-tung lösen.Das im Rahmen dieser Diplomarbeit untersuchte System beinhaltet jedocheinen Monolayer von Proteinen, welche auf einer Oberfläche gebunden sind.Dadurch sind sowohl der Konzentration als auch der Laserleistung Grenzengesetzt.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 60
Aber es tritt noch ein weiteres Problem auf. Die Ramanspektroskopie kannverwendet werden um die verschiedenen chemischen Komponenten einer Sub-stanz zu analysieren, da man sie mit den theoretischen Einzelspektren ver-gleichen kann. Allerdings handelt es sich bei den hier untersuchten Proteinenum größere komplizierte Moleküle. Die Raman-Spektren wären also extremkompliziert, da die Proteine aus sehr vielen Moden bestehen, die sich zudemnoch überlagern können. Auch wenn nicht alle Übergänge aufgrund der Be-dingung (2.46) ”raman-aktiv” sind, wäre eine Auswertung der Spektren fastunmöglich.Um dieses System zu untersuchen benötigt man spezielle Verstärkungsfakto-ren, die in den folgenden Kapiteln behandelt werden.
2.5.3 Resonanter Raman-Effekt
Der resonante Raman-Effekt wurde erstmalig von Shorygin erwähnt [61],1961 von Albrecht theoretisch vorhergesagt [62] und konnte 1972 von Spiroan Cytochrom c nachgewiesen werden [63].Die Intensität des Raman-Effekts ist von der Frequenz des Primärlichtesweitgehend unabhängig, sofern sich seine Quantenenergie von der Anregungs-energie eines elektronischen Übergangs hinreichend stark unterscheidet. Wieim vorherigen Abschnitt erwähnt, endet das Lichtquant der Anregung desRaman-Effekts in einem virtuellen Niveau. Nähert sich nun - wie in Abbil-dung 2.19 zu sehen - die Energie des Primärlichtes der Anregungsenergieeines ”reellen” elektronischen Übergangs, wird die Raman - Streuwahrschein-lichkeit größer. Eine solche Verstärkung des Raman-Effekts bezeichnet manals resonanten Raman-Effekt. Die Intensität des Raman-Effekts wird durchdiesen Effekt um mehrere Größenordnungen verstärkt [64]. Man erreicht Ver-stärkungen um einen Faktor von 103 bis 106.Der resonante Raman-Effekt hat eine enorme Bedeutung für die Untersu-chung von großen Molekülen. Mit ihm ist es möglich bestimmte schwingungs-fähige Teile des Moleküls selektiv voneinander zu untersuchen. Dazu wird dieProbe mit Primärlicht angeregt, dessen Frequenz in der Nähe eines reellenAnregungsniveaus der zu untersuchenden Molekülteile liegt. So erhält manein ”Teil-Raman-Spektrum”, das zu diesen Gruppen gehört, das wesentlichintensiver ist als das Raman-Spektrum der restlichen Molekülteile [65].Der große Nachteil der resonanten Raman-Spektroskopie ist das erhöhte Auf-treten von Fluoreszenz und Photodegradation30.
30Bei der direkten Photodegration wird das Licht von der Probe absorbiert, und der soangeregte Molekülteil reagiert unter chemischer Veränderung.
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 61
Abbildung 2.19: Erläuterung des resonanten Ramaneffekts.
2.5.4 Oberflächenverstärkungseffekt
1974 wurde von Fleischmann eine außerordentliche Verstärkung von Raman-Banden des Pyridins in Gegenwart von Chlorid an einer elektrochemischaufgerauhten Silberelektrode beschrieben [66]. Bei diesem Oberflächenver-stärkungseffekt (SERS: Surface Enhanced Raman-Scattering) kann eine Ver-stärkung des Streuquerschnitts gegenüber den freien Molekülen um bis zusechs Größenordnungen auftreten, sofern das Molekül hierbei an der SERS-aktiven Metalloberfläche adsorbiert ist. Als Oberflächenmaterial kommen ei-nige Übergangsmetalle in Betracht. Am gebräuchlichsten sind Silber, Goldund Kupfer [67].Da die Intensität der Raman-Streuung abhängig ist von der Stärke des in-duzierten Dipols, der seinerseits von der Molekülpolarisierbarkeit und demlokalen elektrischen Feld bestimmt wird, werden zwei Teileffekte für die enor-men Verstärkungen verantwortlich gemacht :
• die Verstärkung des lokalen elektrischen Feldes, welches durch den elek-trischen Feldvektor des einfallenden und des abgestrahlten Lichtes ander Oberfläche erzeugt wird. Dieser wird CEME (classical electroma-gnetic enhancement) genannt.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 62
• die Erhöhung der Molekülpolarisierbarkeit a, der sog. chemische Anteil.
Die Meinungen über die Wichtigkeit des letzt genannten Effekts sind un-terschiedlich. Die Abhängigkeit des CEME-Effekts von der Entfernung d ei-nes Streuzentrums von der Oberfläche konnte ohne Näherungen berechnetwerden. Allerdings wird meistens das Ergebnis der dipolaren Plasmonen-Näherung für Silberkugeln verwendet [68]. Dabei erhält man eine Abhängig-keit von
CEME & [r/(r + d)]12
für Moleküle. Für einen Monolayer, der sich in einem Abstand d von derSilberelektrode befindet, erhält man stattdessen:
CEME & [r/(r + d)]10
Demnach nimmt der Effekt sehr schnell ab, je weiter man sich von der Ober-fläche entfernt. Des Weiteren tritt SERS-Aktivität dann am stärksten auf,wenn die Metalloberflächen Nanostrukturen im Bereich von 1-100 nm aufwei-sen [69].
2.5.5 Resonanz Ramanspektroskopie an Metalloporphyrinen
Porphyrine sind unter anderem in häm-basierten Proteinen enthalten, wie imCytochrom c und in der Cytochrom-c-Oxidase. Sie sind organisch-chemischeFarbstoffe und bestehen aus vier Pyrrol-Ringen, die durch vier Methin -Gruppen zyklisch miteinander verbunden sind (Abbildung 2.20).Für die Interpretation spektroskopischer und vieler chemischer Phänomenean Porphyrinen ist es notwendig, den Orbitalaufbau und das resultieren-de elektronische Absorptionsspektrum von Metalloporphyrinen zu verstehen.Die Absorptionseigenschaften von Porphyrinen (z.B.: Cytochrom c in Abbil-dung 2.21) können mit dem so genannten 4-Orbitalmodell von Goutermanbeschrieben werden [70][71]. Das LUMO31 besteht aus einem degenerierten"$-Paar mit einer eg-Symmetrie. Die zwei HOMOs32, a1u und a2u, habendagegen fast dasselbe Energieniveau. Das "-Elektronensystem der Metallo-porphyrine hat einen sehr geringen HOMO-LUMO-Energieabstand von ca.2 eV und absorbiert deshalb sehr stark im sichtbaren Bereich [72].
31LUMO (Lowest Unoccupied Molecular Orbital) ist das niedrigst besetzte Orbital einesMoleküls.
32HOMO (Highest Occupied Molecular Orbital) ist das höchste besetzte Orbital einesMoleküls.

2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 63
Abbildung 2.20: Porphyringerüst eines D4h - Metalloporphyrins mit den Sub-stituenten X und Y. Die zu unterscheidenden Kohlenstoffa-tome sind mit *, - und m gekennzeichnet. M steht für daszentrale Metallatom und N für ein Stickstoffatom.
Auf Grund der ähnlichen Energiedifferenzen von a1u " eg und a2u " eg
können beide elektronischen Energieübergänge nicht voneinander unterschie-den werden. So addieren sich die Übergangsdipole zu der so genannten B-Bande, auch Soret-Bande genannt, während sie sich bei der Q0-Bande gegen-seitig auslöschen. Die Qν-Bande hat ihren Ursprung in Vibrationsübergängen(0" 1), welche durch das Mischen der elektronischen Energieniveaus von Qund B hervorgerufen werden [72].Es gibt also drei mögliche resonante Anregungswellenlängen. Zum einen kannman eine Resonanz mit der Soret-Bande erzeugen:
/ex = 413,1 nm % # (1! 6) · 105M!1cm!1 ,
zum anderen die Q-Bande anregen:
/ex = 520,8 nm bzw. 530,9 nm % # (1! 4) · 104M!1cm!1
So ist es möglich Vibrationsmoden von unterschiedlicher Symmetrie selektivzu untersuchen [73][74].
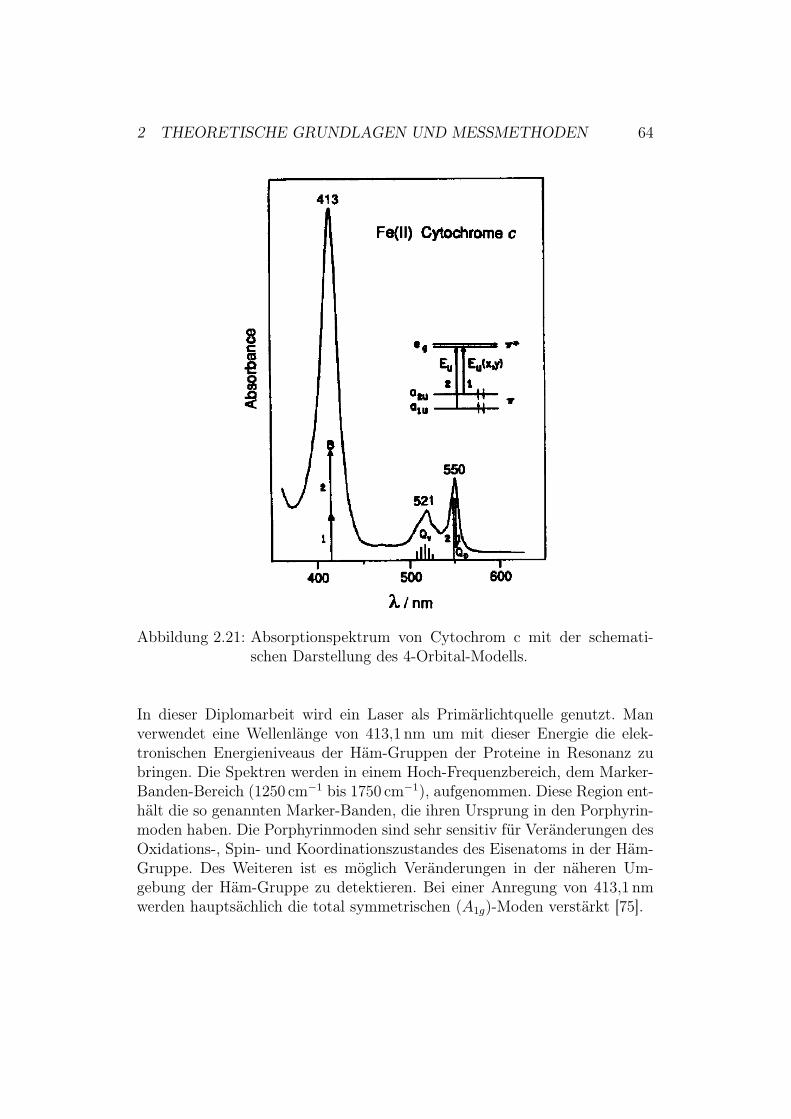
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 64
Abbildung 2.21: Absorptionspektrum von Cytochrom c mit der schemati-schen Darstellung des 4-Orbital-Modells.
In dieser Diplomarbeit wird ein Laser als Primärlichtquelle genutzt. Manverwendet eine Wellenlänge von 413,1 nm um mit dieser Energie die elek-tronischen Energieniveaus der Häm-Gruppen der Proteine in Resonanz zubringen. Die Spektren werden in einem Hoch-Frequenzbereich, dem Marker-Banden-Bereich (1250 cm!1 bis 1750 cm!1), aufgenommen. Diese Region ent-hält die so genannten Marker-Banden, die ihren Ursprung in den Porphyrin-moden haben. Die Porphyrinmoden sind sehr sensitiv für Veränderungen desOxidations-, Spin- und Koordinationszustandes des Eisenatoms in der Häm-Gruppe. Des Weiteren ist es möglich Veränderungen in der näheren Um-gebung der Häm-Gruppe zu detektieren. Bei einer Anregung von 413,1 nmwerden hauptsächlich die total symmetrischen (A1g)-Moden verstärkt [75].
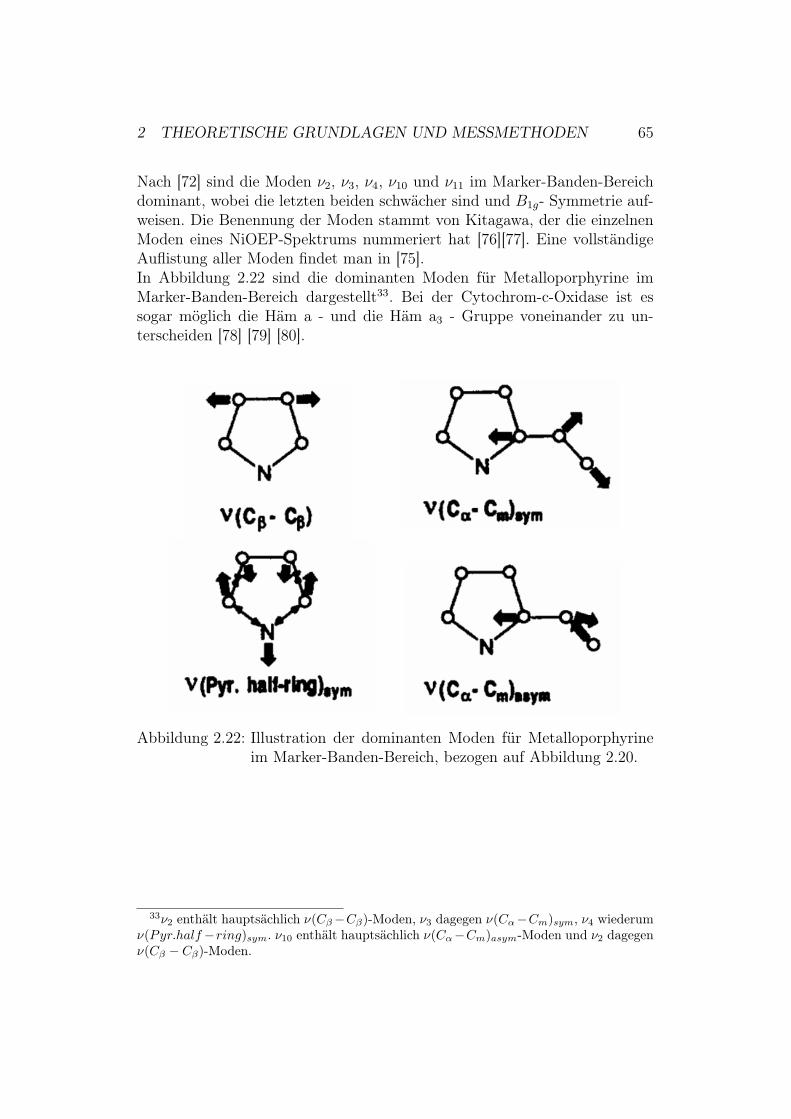
2 THEORETISCHE GRUNDLAGEN UND MESSMETHODEN 65
Nach [72] sind die Moden )2, )3, )4, )10 und )11 im Marker-Banden-Bereichdominant, wobei die letzten beiden schwächer sind und B1g- Symmetrie auf-weisen. Die Benennung der Moden stammt von Kitagawa, der die einzelnenModen eines NiOEP-Spektrums nummeriert hat [76][77]. Eine vollständigeAuflistung aller Moden findet man in [75].In Abbildung 2.22 sind die dominanten Moden für Metalloporphyrine imMarker-Banden-Bereich dargestellt33. Bei der Cytochrom-c-Oxidase ist essogar möglich die Häm a - und die Häm a3 - Gruppe voneinander zu un-terscheiden [78] [79] [80].
Abbildung 2.22: Illustration der dominanten Moden für Metalloporphyrineim Marker-Banden-Bereich, bezogen auf Abbildung 2.20.
33"2 enthält hauptsächlich "(C!!C!)-Moden, "3 dagegen "(C"!Cm)sym, "4 wiederum"(Pyr.half!ring)sym. "10 enthält hauptsächlich "(C"!Cm)asym-Moden und "2 dagegen"(C! ! C!)-Moden.

3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 66
3 Probenherstellung für SPR, EIS und CVDieses Kapitel beschreibt die Konstruktion eines ptBLM-Systems (proteintethered bilayer membrane system) auf einer ultraflachen dünnen Elektro-de34. Als Elektrodenmaterial wird eine TSG-Elektrode (template strippedgold electrode) verwendet. Dies hat mehrere Gründe:Silber hat als Elektrodenmaterial zwar einen höheren Verstärkungsfaktor fürOberflächenplasmonen, jedoch erweist sich die ultraflache Goldschicht als we-sentlich stabiler und langlebiger als die von Silber. Zusätzlich hat man beiGold ein wesentlich größeres Potentialfenster für cyclovoltammetrische Mes-sungen zur Verfügung.Analog zu einer vorausgegangenen Arbeit [42] werden die Proteine auf einerultraflachen funktionalisierten Goldschicht angebunden. Die Herstellung unddie Funktionalisierung einer ultraflachen Goldschicht wird in den folgendenAbschnitten erläutert. Des Weiteren werden die zwei verwendeten His-Tagmanipulierten Cytochrom-c-Oxidasen mit ihrer unterschiedlichen Orientie-rung und die Mutante N139C vorgestellt.
3.1 Herstellung von TSG-ElektrodenDie TSG-Elektroden werden wie in [40] beschrieben hergestellt. Hierbei ver-wendet man Siliziumwafer (26 mm x 45 mm x 0,625 mm), die in einer 78"Cwarmen Lösung (Wasser35, Wasserstoffperoxid und Ammoniak im Verhältnis5:1:1) gereinigt werden. Dazu wird ein Magnetrührer verwendet. Nach einerStunde werden die Siliziumwafer gründlich zuerst mit Wasser und dann mitEthanol gespült. Nach dem Trocknen der Wafer unter einem Argonstromwerden sie mit einer 50 nm dicken Goldschicht bedampft (Abbildung 3.1).Um ein gleichmäßiges Aufdampfen zu gewährleisten, wird der Probenhalterder Aufdampfanlage, in den die Wafer eingebaut werden, rotiert. Die Auf-dampfrate wird kleiner als 0,1 nm/s gehalten. Zusätzlich wird ein Vakuumvon p < 5 · 10!6 mbar verwendet. Nach dem Aufdampfen werden die Probenaus dem Probenhalter der Aufdampfmaschine entfernt.Als nächstes werden LaSFN9-Gläser36 mit Hilfe von EPO-TEK 353ND-4 auf
34Für eine genaue Beschreibung der verwendeten Materialien, Lösungen und Gerätewird auf den Anhang (refmaterial) verwiesen.
35An dieser und an allen anderen Probenvorbereitungspunkten wurde als Wasserquelledeionisiertes Wasser von einer MilliQ - Reinigungsanlage verwendet.
36Die LaSFN9-Gläser (26 mm x 76 mm x 1,5mm) werden zuvor halbiert und unter flie-ßendem Wasser von anhaftenden Glaspartikeln befreit. Gereinigt werden die Objektivträ-ger in einer 2% igen Hellmanex-Lösung, die für 15 Minuten ins Ultraschallbad gestellt wird.Zuletzt werden die Träger gründlich mit Wasser gespült und mit Sticksto!gas getrocknet.
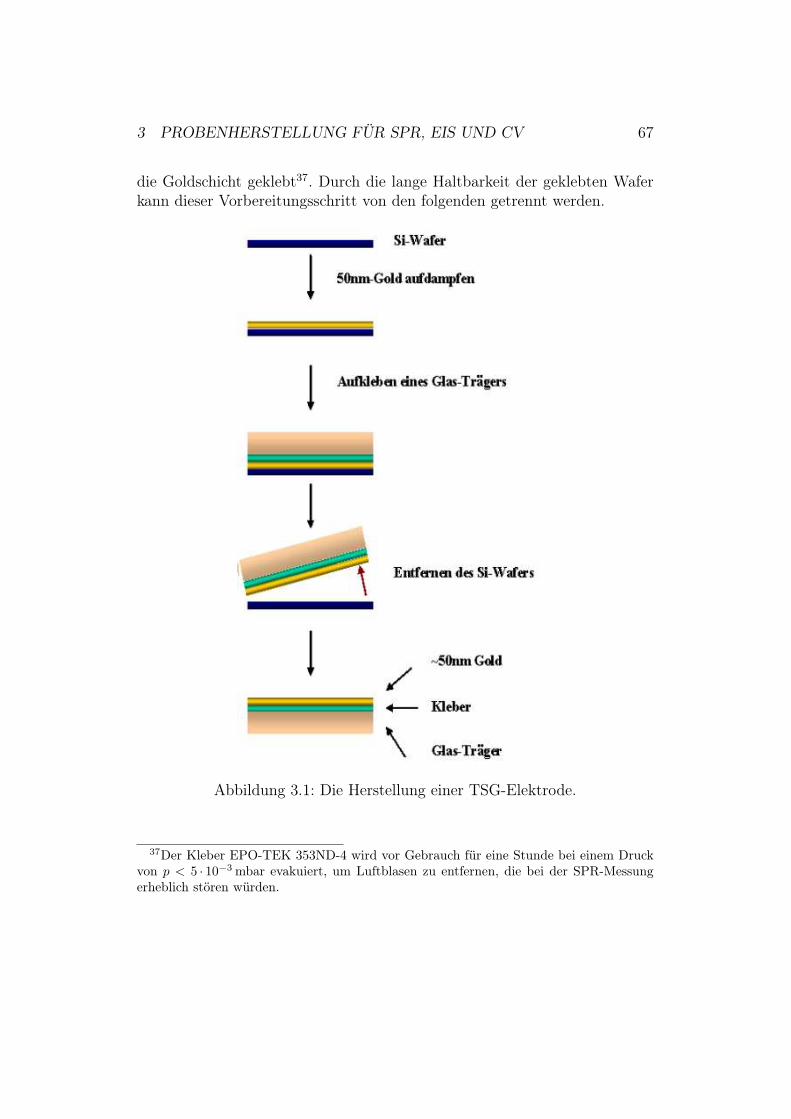
3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 67
die Goldschicht geklebt37. Durch die lange Haltbarkeit der geklebten Waferkann dieser Vorbereitungsschritt von den folgenden getrennt werden.
Abbildung 3.1: Die Herstellung einer TSG-Elektrode.
37Der Kleber EPO-TEK 353ND-4 wird vor Gebrauch für eine Stunde bei einem Druckvon p < 5 · 10!3 mbar evakuiert, um Luftblasen zu entfernen, die bei der SPR-Messungerheblich stören würden.
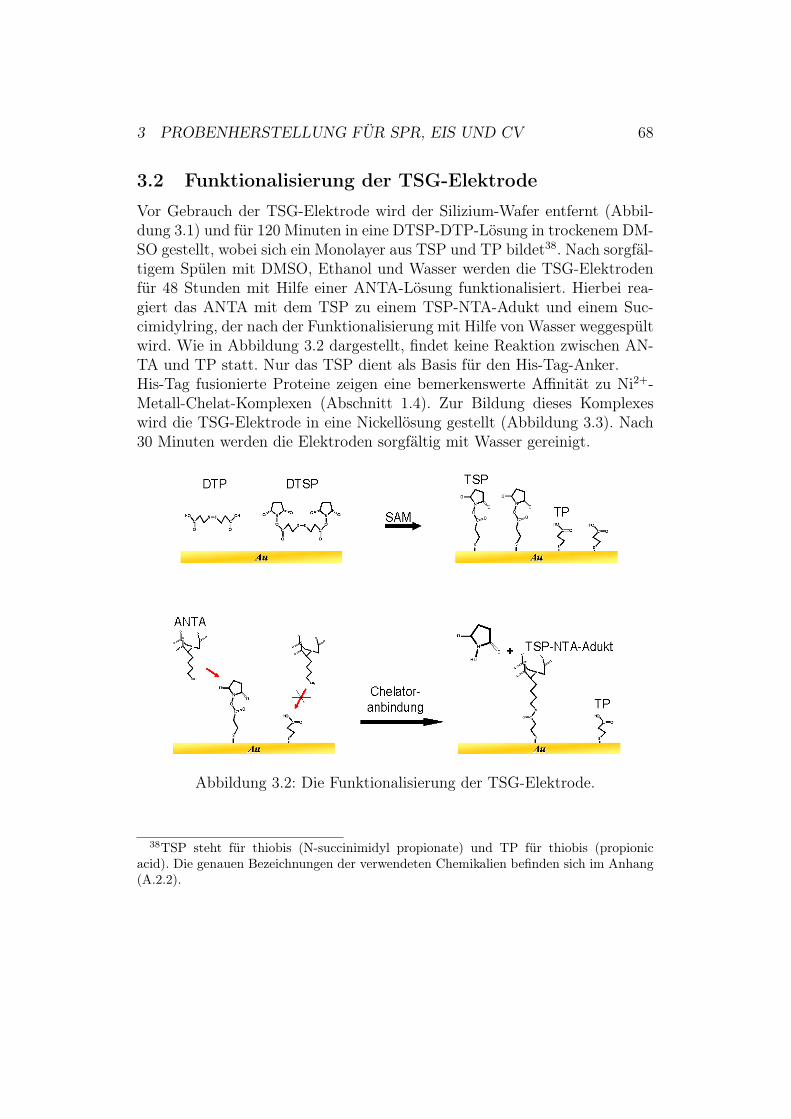
3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 68
3.2 Funktionalisierung der TSG-ElektrodeVor Gebrauch der TSG-Elektrode wird der Silizium-Wafer entfernt (Abbil-dung 3.1) und für 120 Minuten in eine DTSP-DTP-Lösung in trockenem DM-SO gestellt, wobei sich ein Monolayer aus TSP und TP bildet38. Nach sorgfäl-tigem Spülen mit DMSO, Ethanol und Wasser werden die TSG-Elektrodenfür 48 Stunden mit Hilfe einer ANTA-Lösung funktionalisiert. Hierbei rea-giert das ANTA mit dem TSP zu einem TSP-NTA-Adukt und einem Suc-cimidylring, der nach der Funktionalisierung mit Hilfe von Wasser weggespültwird. Wie in Abbildung 3.2 dargestellt, findet keine Reaktion zwischen AN-TA und TP statt. Nur das TSP dient als Basis für den His-Tag-Anker.His-Tag fusionierte Proteine zeigen eine bemerkenswerte Affinität zu Ni2+-Metall-Chelat-Komplexen (Abschnitt 1.4). Zur Bildung dieses Komplexeswird die TSG-Elektrode in eine Nickellösung gestellt (Abbildung 3.3). Nach30 Minuten werden die Elektroden sorgfältig mit Wasser gereinigt.
Abbildung 3.2: Die Funktionalisierung der TSG-Elektrode.
38TSP steht für thiobis (N-succinimidyl propionate) und TP für thiobis (propionicacid). Die genauen Bezeichnungen der verwendeten Chemikalien befinden sich im Anhang(A.2.2).

3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 69
3.3 Anbindung der Cytochrom-c-Oxidase an die Elek-trodenoberfläche
Die Cytochrom-c-Oxidase besitzt als Membranprotein eine Oberfläche mithydrophoben und hydrophilen Teilen. Aus diesem Grund wird als Proteinlö-sungmittel eine DDM-Puffer-Lösung benutzt. Das DDM bildet Mizellen oderbindet direkt an das Protein. So befinden sich die verschiedenen Bereiche desProteins in einer für sie geeigneten Umgebung.Für die Immobilisierung wird die TSG-Elektrode in eine DDM-Lösung ge-bracht, in der sich das Protein mit einer Konzentration von 100 nM/L befin-det. Dadurch binden die His-Tag fusionierten Proteine an die Ni2+-Metall-Chelat-Komplexe. Nach 30 Minuten werden die in dieser Zeit nicht angebun-denen Proteine durch den Austausch der vorhanden Lösung mit einer reinenDDM-Lösung entfernt.
Abbildung 3.3: Die Herstellung einer ptBLM-Probe. In diesem Fall ist dieCytochrom-c-Oxidase mit His-Tag an Untereinheit I (rot)verwendet worden. Die Untereinheiten II, III und IV sindin orange, gelb und schwarz dargestellt.
Als nächstes wird durch eine Dialyse ein Bilayer gebildet, der als künstli-che Proteinmembran fungieren soll. Zuerst wird die DDM-Lösung durch ei-

3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 70
ne DDM-Lipidlösung ersetzt und Biobeads hinzugegeben. Diese nehmen dasDDM in sich auf und das DDM wird durch Lipid ersetzt. Es bildet sich, wiein Abbildung 3.4 zu sehen, die für das Protein benötigte Membranumgebung.Nach ca. 15 Stunden ist die Dialyse abgeschlossen. Die Zelle wird mit einemPBS-Puffer gespült um die Biobeads mit dem aufgesaugten DDM und demüberschüssigen Lipid zu entfernen.Die Architektur eines ptBLM-Systems, bei dem als Protein die Cytochrom-c-Oxidase mit His-Tag an Untereinheit I verwendet wurde, ist in Abbildung3.5 dargestellt [81] [79].
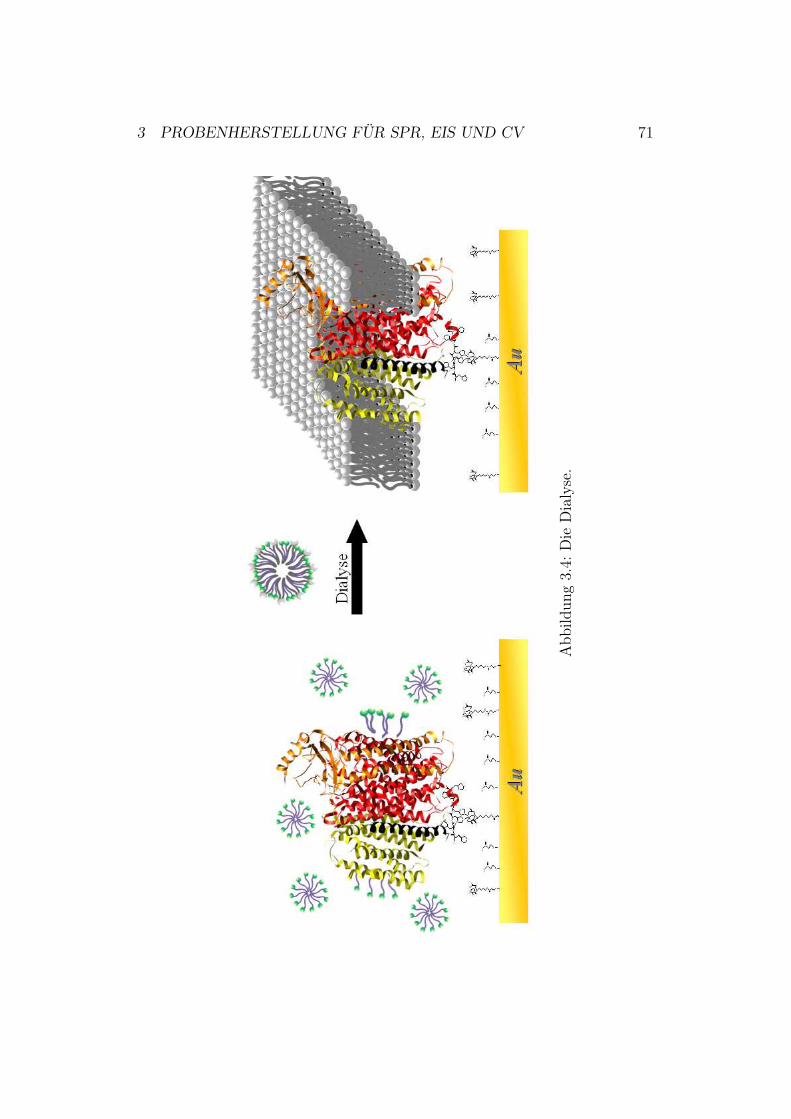
3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 71
Abb
ildun
g3.
4:D
ieD
ialy
se.

3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 72
Abbildung 3.5: Die Architektur einer ptBLM-Probe, bei der als Protein dieCytochrom-c-Oxidase mit His-Tag an Untereinheit I verwen-det worden ist. Die horizontalen Längenangaben beziehensich auf den Teil des Enzyms, der aus dem Bilayer in dieLösung weist. Die funktionalisierte Elektrode ist vergrößertabgebildet.

3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 73
3.4 Cytochrom-c-Oxidase in der up- oder down-Konfi-guration
In dieser Diplomarbeit spielen zwei unterschiedlich His-Tag-manipulierte Pro-teine eine entscheidende Rolle (siehe Abschnitt 1.5). Zur Verfügung stehendie Cytochrom-c-Oxidase von Rhodobacter sphaeroides und die Cytochrom-c-Oxidase von Paracoccus denitrificans. Röntgenkristallstrukturanalysen zei-gen, dass man bei diesen beiden Varianten der Cytochrom-c-Oxidase von ei-nem identischen Elektronen- und Protonenweg ausgehen kann [82] [83] [84].Ein Vorteil des ptBLM-Systems ist der Submembranraum, der als Reservoirfür Ionen dient. Ein weiterer großer Vorteil des ptBLM-Systems ist die strikteKontrolle über die Orientierung des Proteins, je nachdem, an welcher Stellesich der His-Tag am Protein befindet [2]:Bei Rhodobacter sphaeroides ist der His-Tag an den C-Terminus von Unter-einheit II (orange) angebracht. Dagegen ist bei Paracoccus denitrificans derHis-Tag an den C-Terminus von Untereinheit I (rot) befestigt (Abbildung3.6). Auf diese Art und Weise kann das Enzym in zwei verschiedenen Orien-tierungen immobilisiert werden.Bei der down-Konfiguration wird die Cytochrom c-Bindungsseite direkt mitder Elektrode verbunden. Die Elektrode ersetzt dabei das in der Natur fürdie Aktivierung notwendige Cytochrom c. Mit Hilfe von Sauerstoff und Pro-tonen aus der Pufferlösung und den von der Elektrode gelieferten Elektronenwird Wasser gebildet. Die durch diesen Vorgang gewonnene Energie wird fürdas Pumpen von Protonen in den Raum zwischen Membran und Elektrodeverwendet.Bei der up-Konfiguration liegt die Cytochrom c-Bindungsseite der Elektrodegegenüber und zeigt zur Außenseite des ptBLM-Systems. In dieser Konfigu-ration kann das Enzym durch die Zugabe von Cytochrom c aktiviert werden.In Anwesenheit von Sauerstoff sollte die in der Einleitung beschriebene ka-talysierte Reaktion stattfinden:
4 ·Fe2+cytc + 8 ·H+
innen + O2 " 4 ·Fe3+cytc + 2H2O + 4 ·H+
außen
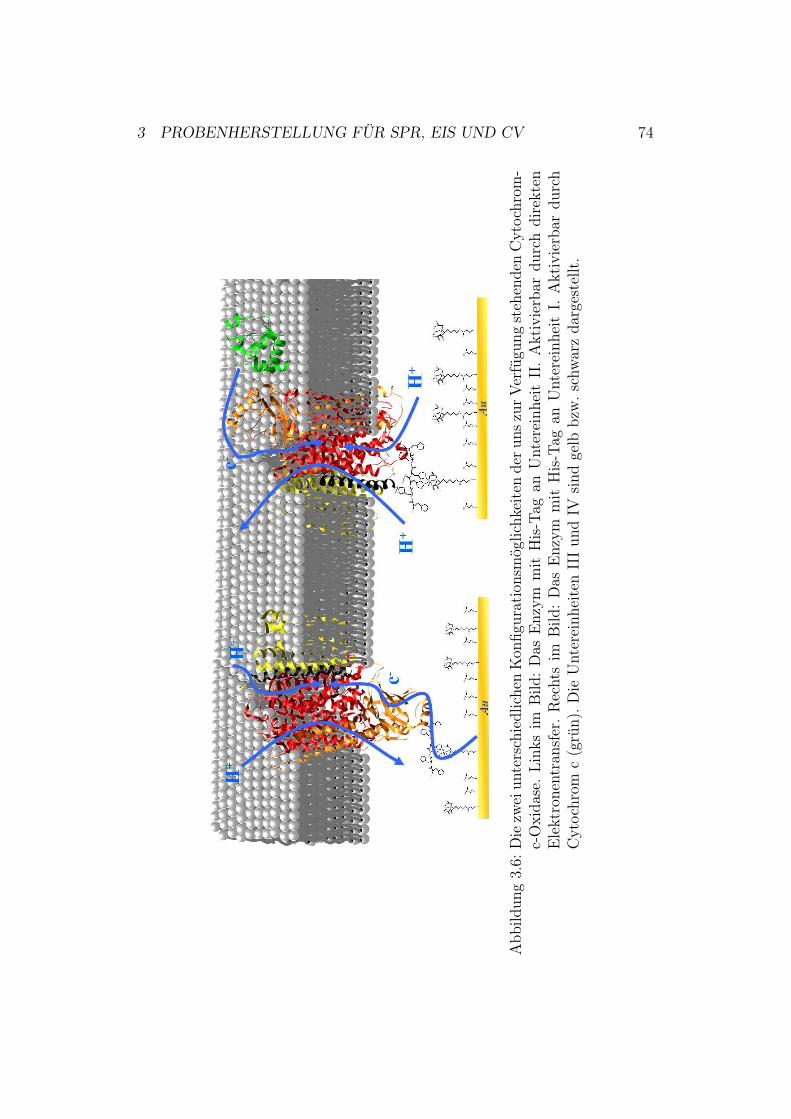
3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 74
Abb
ildun
g3.
6:D
iezw
eiun
ters
chie
dlic
hen
Kon
figur
atio
nsm
öglic
hkei
ten
deru
nszu
rVer
fügu
ngst
ehen
den
Cyt
ochr
om-
c-O
xida
se.
Link
sim
Bild
:D
asE
nzym
mit
His-
Tag
anU
nter
einh
eit
II.
Akt
ivie
rbar
durc
hdi
rekt
enE
lekt
rone
ntra
nsfe
r.R
echt
sim
Bild
:D
asE
nzym
mit
His-
Tag
anU
nter
einh
eit
I.A
ktiv
ierb
ardu
rch
Cyt
ochr
omc
(grü
n).D
ieU
nter
einh
eite
nII
Iun
dIV
sind
gelb
bzw
.sch
war
zda
rges
tellt
.

3 PROBENHERSTELLUNG FÜR SPR, EIS UND CV 75
3.5 Cytochrom-c-Oxidase N139CUm die Protonenpfade zu ermitteln werden verschiedene Mutanten verwen-det (siehe Abschnitt 1.3.1). Die Cytochrom-c-Oxidase N139C ist eine Mutan-te mit His-Tag an Untereinheit II. Demnach handelt es sich um eine Muta-tion der down-Konfiguration. Bei dieser Mutante wird, wie in Abbildung 3.7zu sehen, die Aminosäure Asp-139 (Asparaginsäure) durch die AminosäureCys-139 (Cystein) ersetzt. Durch diese Mutation wird der D-Kanal verän-dert. Damit soll das Pumpen der Protonen verhindert werden. Das Enzymbesitzt jedoch weiterhin eine hohe katalytische Aktivität [85].
Abbildung 3.7: Die Cytochrom-c-Oxidase N139C. Dargestellt sind nur dieUntereinheiten I (rot) und II (orange).

4 MESSAUFBAU FÜR SPR, EIS UND CV 76
4 Messaufbau für SPR, EIS und CVWie bereits in Abschnitt 1.5 erwähnt, wird der Aufbau des ptBLM-Systemssimultan mit Oberflächenplasmonenresonanz-Spektroskopie (SPR-Spektro-skopie) und elektrochemischer Impedanzspektroskopie (EIS) untersucht. Da-für benötigt man eine kombinierte Messapparatur (Abbildungen 4.1 und 4.2)[40][86].Monochromatisches Licht eines He-Ne-Lasers (/ = 632,8 nm) wird mit einemmechanischen Chopper (2) rechteckmoduliert. Die Justage des Laserstrahlserfolgt wie üblich über Irisblenden (1). Mit einem Polarisator (3) wird dasLicht p-polarisiert. Die Laserintensität kann über einen zweiten Polarisator(3) variiert werden. Der Laserstrahl fällt auf ein Prisma an dessen Basismittels Immersionsöl der LaSFN-9-Objektträger der TSG-Elektrode optischgekoppelt ist39. Das Prisma, das Immersionsöl und der Objektträger besitzenden gleichen Brechungsindex. Das von der Probe reflektierte Licht wird durcheine Linse mit Brennweite f = 50 mm auf eine Photodiode fokussiert. DasSignal wird mit Hilfe eines Lock-In Verstärkers gemessen und an den PC(PC1) weitergeleitet. Probe und Detektor sind auf einem Zweikreisgonio-meter montiert und werden mit 5-Phasen-Schrittmotoren, deren Steuerungebenfalls über diesen PC erfolgt, bewegt.
Abbildung 4.1: Der Messaufbau für SPR, EIS und CV.
39Diese Konstruktion wird über einen Reiter zusammengehalten.

4 MESSAUFBAU FÜR SPR, EIS UND CV 77
In Abbildung 4.2 ist das verwendete Zelldesign40 und der Einbau der Zel-le in den Reiter zu sehen, welcher wiederum in die Messapparatur einge-baut wird. Die Abdichtung zwischen Glas und Zelle erfolgt über Dichtringe(nicht eingezeichnet), die als Elektrodenbegrenzung dienen. Die Dichtungs-ringe haben einen Innendurchmesser von 1 cm, so dass man eine Oberflächevon A = " · r2 = 0,785 cm2 zur Verfügung hat.Für die elektrochemischen Messungen (EIS und CV) wird eine Drei - Elek-troden - Anordnung verwendet41. Die aufgedampfte Goldschicht dient alsArbeitselektrode (6). Ein Platindraht (ø = 1 mm, Länge = 5 cm) wird als Ge-genelektrode (5) verwendet. Dieser wird mit Hilfe einer Teflonummantelungabgedichtet. Die Referenzelektrode (4) besteht aus einem chlorierten Silber-draht42, der bis zur Hälfte in ein Glasrohr (Länge = 5 cm) geführt wird43.Das Glasrohr wird mit 3 molarer Kaliumchlorid-Lösung gefüllt. Es handeltsich also um eine Ag/AgCl,KClkonz.-Referenzelektrode. Diese wird ebenfallsdurch eine Teflonummantelung abgedichtet. Mit Hilfe von Ein- (7) und Aus-laufkanälen (8) kann die Flüssigkeit in der Zelle auch während einer Messungausgetauscht oder von Sauerstoff befreit werden. Zum Entfernen des Sauer-stoffs wird die Flüssigkeit in ein anderes, mit Sauerstoffzehrsystem44 gefülltesGefäß geleitet. Zusätzlich wird Argon eingeführt, damit kein Sauerstoff in dieLösung diffundiert.Eine weitere etwas größere Öffnung in der Zelle braucht man, um bei BedarfBiobeads hinzuzugeben. Diese Öffnung wird - außer für den Fall der Zugabevon Biobeads - mit Hilfe eines Teflon-Stopfens (9) abgedichtet.In Abbildung 4.3 ist die komplette Messzelle mit Halterung (Reiter) sowiemit den Anschlüssen für die Elektroden bzw. mit dem Ein- und Auslass fürden Lösungsaustausch zu sehen.Mit einem Autolab-Potentiostaten, den man über einen PC (PC 2) steuert,werden die elektrochemischen Messungen durchgeführt. Die Impedanzspek-tren nimmt man mit einer Anregungsamplitude von 10mV auf. Mit der Wahldieser Amplitude wird eine Störung des Systems weitgehend ausgeschlossen(siehe Abschnitt 2.2.1).
40Die Zelle besteht aus Teflon. Dieses säurebeständige Material lässt sich nach Gebrauchmit Hilfe eines Schwefelsäurebads sehr gut reinigen und wiederverwenden.
41Da sich der Messaufbau von EIS und der Cyclovoltammetrie (CV) nicht unterscheidet,konnten mit derselben Apparatur auch die CV-Experimente gemacht werden.
42Der chlorierte Silberdraht wird mit Hilfe einer Galvanisierungseinheit in Kombinationmit Silberdraht (ø = 1 mm, Länge = 5 cm) und 0,1 M Kaliumchlorid-Lösung hergestellt.
43Ein an der Spitze des Glasröhrchens angebrachter kleiner dünner Platindraht(ø= 0,1mm, Länge = 5mm) sorgt dafür, dass zwischen dem Elektrodeninneren und derPu!erlösung Ladungsträger di!undieren können. Diese Spezialanfertigung wurde vomhauseigenen Glasbläser hergestellt.
44Hierbei handelt es sich um eine spezielle Lösung (siehe Anhang A.2.1).
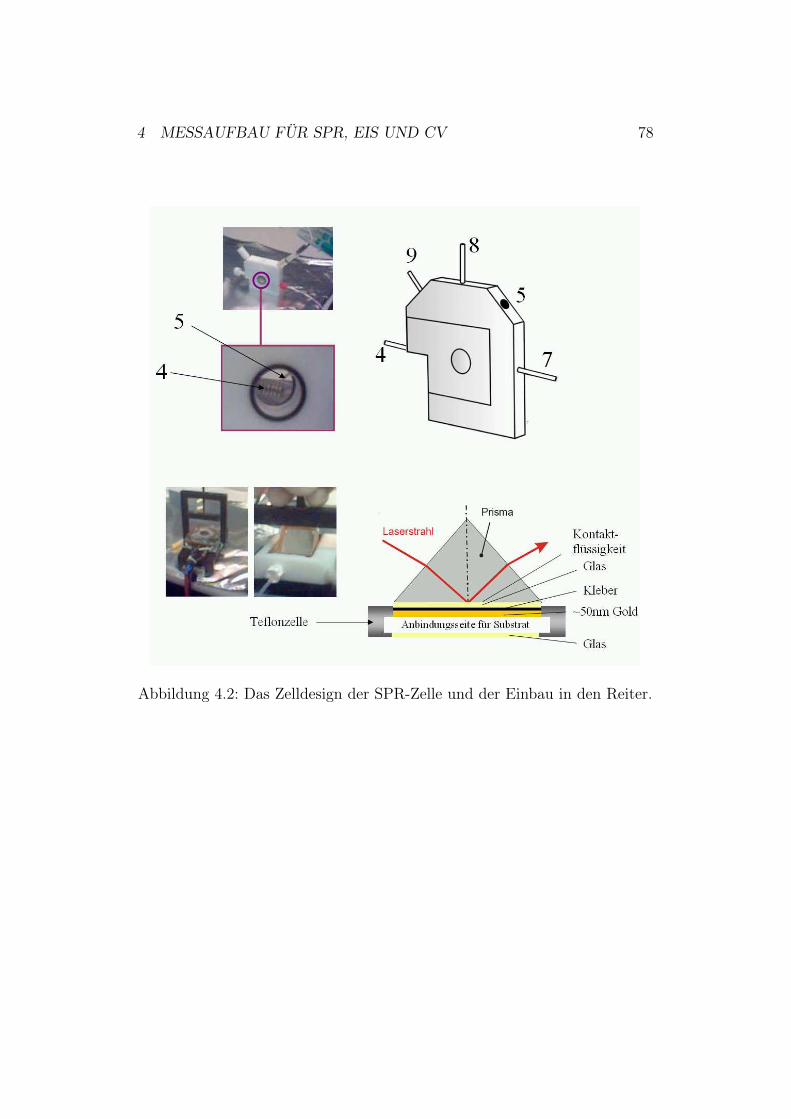
4 MESSAUFBAU FÜR SPR, EIS UND CV 78
Abbildung 4.2: Das Zelldesign der SPR-Zelle und der Einbau in den Reiter.

4 MESSAUFBAU FÜR SPR, EIS UND CV 79
Abb
ildun
g4.
3:K
ompl
ette
Mes
szel
lem
itH
alte
rung
(Rei
ter)
und
den
Ans
chlü
ssen
für
die
Ele
ktro
den
bzw
.dem
Ein
-un
dA
usla
ssfü
rde
nLö
sung
saus
taus
ch.

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 80
5 Messergebnisse und Diskussion (Teil 1)
5.1 Überprüfung des ptBLM-AufbausIn dieser Arbeit wird die Oberflächenplasmonenresonanz - Spektroskopie inKombination mit der Impedanz - Spektroskopie verwendet. Um die Schicht-dickenänderung und die Bestimmung der elektrischen Eigenschaften währenddes ptBLM-Aufbaus an ein und derselben Probe zu verfolgen, wird im Fol-genden die Überprüfung eines ptBLM-Aufbaus gezeigt. Als Beispiel wird dieCytochrom-c-Oxidase mit His-Tag an Untereinheit I (up-Konfiguration) ver-wendet.Da die Änderungen bei der Funktionalisierung der Elektrode (siehe Abschnitt3.2) zu klein sind, wird lediglich die Proteinanbindung und die Dialyse be-obachtet.
5.1.1 Ergebnisse und Diskussion der Oberflächenplasmonenreso-nanz - Spektroskopie
Die in Abschnitt 2.1.3 beschriebene Oberflächenplasmonenresonanz-Spek-troskopie kann zur Detektion von Brechungsindexveränderungen verwendetwerden.Die aufgenommenen SPR-Spektren sind in Abbildung 5.1 dargestellt. Die Re-flektivität wird dabei gegen den Winkel des vom Prisma reflektierten Lichtsaufgetragen.Um sicherzustellen, dass die Probe überhaupt ein Plasmon liefert, wird alsTest ein Spektrum an Luft (schwarz) aufgenommen. Um einen geeignetenWinkel für die Kinetikmessung zu finden, wird die Zelle mit DDM-Lösunggefüllt und ein weiteres Spektrum (rot) aufgenommen45. Man erkennt einepositive Verschiebung des Plasmon-Resonanzwinkels aufgrund des erhöhtenBrechungsindexes. Für die Kinetikmessung ist ein Winkel von 54,5" geeignet.An diesem nahezu linearen Bereich der Flanke erreicht man in guter Nähe-rung einen proportionalen Zusammenhang zwischen reflektierter Intensitätund Schichtzunahme (siehe Abschnitt 2.1.3).Vor (Punkt A) und nach (Punkt B) der Proteinanbindung wird die Kine-
tikmessung unterbrochen und SPR-Spektren aufgenommen (rot bzw. blau).Wenn sich der Proteinlayer anlagert, verschiebt sich der Resonanzwinkel desangeregten Oberflächenplasmons ins Positive. Der Grund hierfür ist der hö-here Brechungsindex, den die angelagerte Schicht, im Vergleich zur Lösung,hat.
45Winkel von mehr als 60" können mit der verwendeten Apparatur nicht gemessen wer-den.
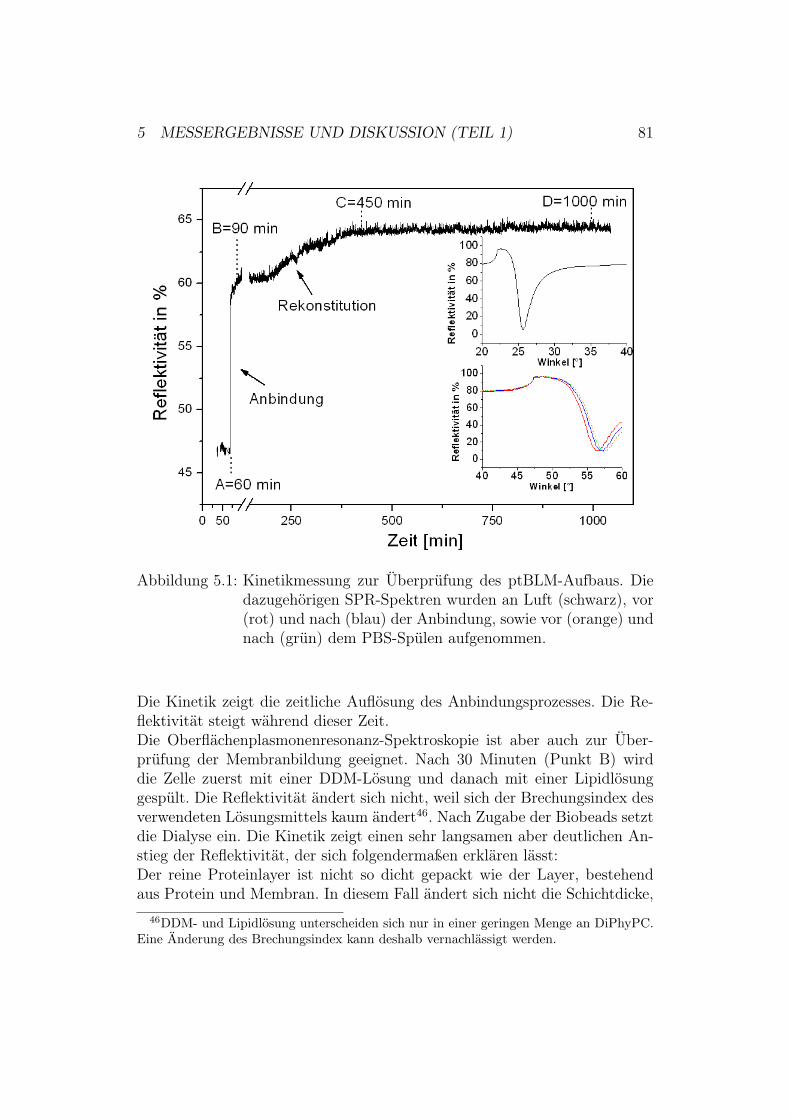
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 81
Abbildung 5.1: Kinetikmessung zur Überprüfung des ptBLM-Aufbaus. Diedazugehörigen SPR-Spektren wurden an Luft (schwarz), vor(rot) und nach (blau) der Anbindung, sowie vor (orange) undnach (grün) dem PBS-Spülen aufgenommen.
Die Kinetik zeigt die zeitliche Auflösung des Anbindungsprozesses. Die Re-flektivität steigt während dieser Zeit.Die Oberflächenplasmonenresonanz-Spektroskopie ist aber auch zur Über-prüfung der Membranbildung geeignet. Nach 30 Minuten (Punkt B) wirddie Zelle zuerst mit einer DDM-Lösung und danach mit einer Lipidlösunggespült. Die Reflektivität ändert sich nicht, weil sich der Brechungsindex desverwendeten Lösungsmittels kaum ändert46. Nach Zugabe der Biobeads setztdie Dialyse ein. Die Kinetik zeigt einen sehr langsamen aber deutlichen An-stieg der Reflektivität, der sich folgendermaßen erklären lässt:Der reine Proteinlayer ist nicht so dicht gepackt wie der Layer, bestehendaus Protein und Membran. In diesem Fall ändert sich nicht die Schichtdicke,
46DDM- und Lipidlösung unterscheiden sich nur in einer geringen Menge an DiPhyPC.Eine Änderung des Brechungsindex kann deshalb vernachlässigt werden.

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 82
sondern der Brechungsindex der bereits angelagerten Schicht. Der Resonanz-winkel des angeregten Oberflächenplasmons verschiebt sich ins Positive. Des-halb steigt die Reflektivität in der Kinetikmessung. Abbildung 5.1 zeigt kei-nen weiteren Anstieg der Reflektivität am Punkt C. Die Dialyse ist jedochan diesem Punkt noch nicht abgeschlossen, da ein vorzeitiges Spülen mitPBS in anderen Versuchen eine Denaturierung der Proteine zur Folge hatte.Wahrscheinlich benötigt die Membran zur Stabilisierung und Reorganisationnoch zusätzlich Zeit. Aus diesem Grund wird die Zelle - wie in Abschnitt3.3 beschrieben - erst nach ungefähr 15 Stunden am Punkt D mit PBS ge-spült. Direkt vor (orange) und nach (grün) der PBS-Spülung wird jeweilsein Spektrum aufgezeichnet. Zwischen den beiden SPR-Spektren stellt mankeinen Unterschied fest. Folglich ändert sich auch nicht die Reflektivität inder Kinetikmessung. Allein der Membranaufbau ist für die zweite Reflektivi-tätsänderung verantwortlich.
5.1.2 Ergebnisse und Diskussion der elektrochemischen Impedanz-Spektroskopie
Auch durch die elektrochemische Impedanz-Spektroskopie soll der Aufbaudes ptBLM-Systems überprüft werden. Die Messungen erfolgen mit den glei-chen Proben, an denen auch mittels SPR der ptBLM-Aufbau beobachtetwird. Dazu wird die Kinetikmessung nicht unterbrochen. Während der EIS-Messung ist keine Veränderung der Reflektivität in der Kinetikmessung zuerkennen. Eine Störung des Systems durch die gewählte Anregungsamplitudevon 10 mV kann somit ausgeschlossen werden (Abschnitt 4).Zur Überprüfung des ptBLM-Aufbaus wird ein Impedanz-Spektrum nach derProteinanbindung aufgenommen. Außerdem wird die Rekonstitution vor undnach dem PBS-Spülen (Abbildung 5.1, Punkt D) überprüft47.Der Vergleich der Fitergebnisse für den Widerstand und die Kapazität vorund nach der Dialyse dient zur Kontrolle des Membranaufbaus. Tabelle 5.1zeigt die Ergebnisse der einzelnen Fits48:
47Alle Messungen werden bei einem Potential von 0 mV vs. Ag/AgCl,KClkonz. aufge-nommen. Dies entspricht einem Potential von 200mV vs. NHE (siehe Abschnitt A.1) [87].Für eine genaue Beschreibung der Datensatzauswertung wird auf den Anhang verwiesen(Abschnitt A.1.2).
48Aufgrund von Kontaktproblemen und eines Gerätedefekts konnte nicht jede Messungin dem gewünschten Intervall von 50 kHz bis 3 mHz durchgeführt werden. Dieser Umstanderhöht den Fehler bei den entsprechenden Fits.
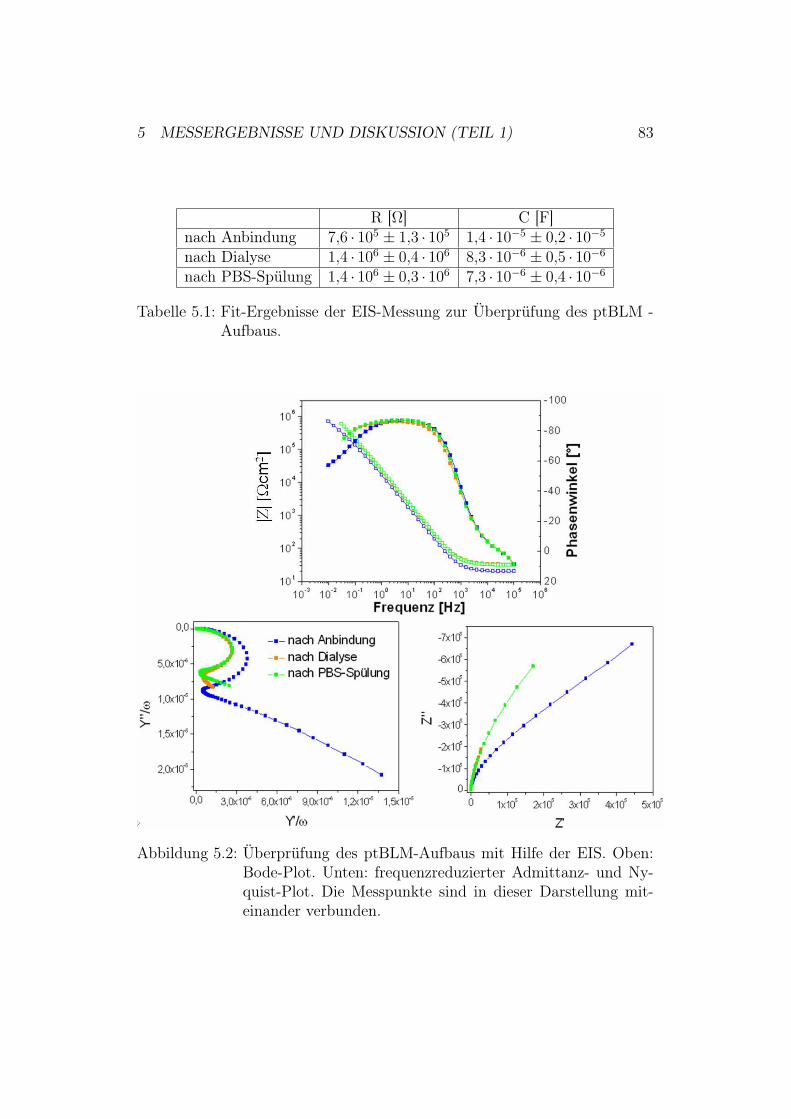
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 83
R [$] C [F]nach Anbindung 7,6 · 105 ± 1,3 · 105 1,4 · 10!5 ± 0,2 · 10!5
nach Dialyse 1,4 · 106 ± 0,4 · 106 8,3 · 10!6 ± 0,5 · 10!6
nach PBS-Spülung 1,4 · 106 ± 0,3 · 106 7,3 · 10!6 ± 0,4 · 10!6
Tabelle 5.1: Fit-Ergebnisse der EIS-Messung zur Überprüfung des ptBLM -Aufbaus.
Abbildung 5.2: Überprüfung des ptBLM-Aufbaus mit Hilfe der EIS. Oben:Bode-Plot. Unten: frequenzreduzierter Admittanz- und Ny-quist-Plot. Die Messpunkte sind in dieser Darstellung mit-einander verbunden.

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 84
Analog zu [42], erhält man nach der Anbindung der Cytochrom-c-Oxidaseeinen Widerstand von mehreren hundert k$ und eine Kapazität von einigenµF. Nach der Bildung der Membran steigt der Membranwiderstand und dieKapazität der Membran fällt drastisch ab. Dies stimmt ebenfalls mit denErgebnissen aus [42] überein. Der Anstieg des Widerstandes lässt sich durchdie Bildung der Membran erklären, die eine abdichtende Wirkung hat.Die Kapazität einer dielektrischen Schicht wird durch ihre Dicke und ih-re dielektrische Konstante bestimmt. Für reine tBLMs (tethered bilayer li-pid membranes), bei denen kein Protein vorhanden ist, erhält man experi-mentell Widerstandswerte von einigen M$ · cm2 und Kapazitäten kleiner als0,5µF/cm2 [88].Bei der Bildung des Bilayers wird das Wasser (zusammen mit dem Detergenz)in der Proteinumgebung durch das Lipid verdrängt. Die dielektrische Kon-stante des verwendeten Lipids ist mit % # 2,2 [42] sehr viel kleiner, als die vonWasser (% # 80, [1]) oder die von Proteinmolekülen (% # 20! 30, [89][90])49.Deshalb erwartet man einen Abfall der Kapazität. Dies wird durch die Mes-sung bestätigt: Die Kapazität geht nach der Dialyse von C = 14µF/cm2 aufC = 8,3µF/cm2 zurück.Nach der Spülung mit PBS verändert sich der Widerstand der Membrannicht. Lediglich die Kapazität verringert sich um einen geringen Faktor. Dieshängt wahrscheinlich mit einer Reorganisation des Bilayers zusammen. Nachder Spülung mit PBS-Puffer befinden sich keine Biobeads mehr in der Mess-zelle. Dadurch kann vielleicht ebenfalls eine Veränderung der Kapazität her-vorgerufen werden.Die gemessenen Werte stimmen mit [17] überein50. Deshalb kann man auchim vorliegenden Fall von einer hohen Oberflächenbedeckung der Proteine vonüber 90 % ausgehen.
5.2 Aktivierung der Cytochrom-c-Oxidase in der up-Konfiguration
Die Cytochrom-c-Oxidase mit His-Tag an Untereinheit I (Cytochrom c-Bin-dungsstelle befindet sich gegenüber der Arbeitselektrode) wird nach der Über-prüfung des ptBLM-Aufbaus mit einer Cytochrom c - Lösung aktiviert.
49Der Wert aus [90] stammt vom Lysozym (ebenfalls ein Enzym) in Lösung und ent-spricht wahrscheinlich am ehesten dem Dielektrizitätswert der Cytochrom-c-Oxidase.
50In [17] wurde die Cytochrom-c-Oxidase in der down-Konfiguration (His-Tag an Unter-einheit II) für die Konstruktion eines ptBLM-Systems verwendet. Bei dieser Konfigurationist es möglich mit Hilfe der Cyclovoltammetrie auf eine Protein-Oberflächenbedeckung vonüber 90 % zurückzuschließen.

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 85
Dazu werden bei verschiedenen Cytochrom c-Konzentrationen sowohl Impe-danz - Spektren aufgenommen, als auch Untersuchungen mit Hilfe der Cyclo-voltammetrie durchgeführt51. Für die Messungen wird reduziertes Cytochromc verwendet, um den in der Natur auftretenden Vorgang zu imitieren. Zusätz-lich werden cyclovoltammetrische Messungen für den physiologisch wenigerinteressanten Fall des oxidierten Cytochrom c durchgeführt.
5.2.1 Ergebnisse und Diskussion der elektrochemischen Impedanz-Spektroskopie
Zur Aktivierung der Cytochrom-c-Oxidase in der up-Konfiguration werdender Lösung verschiedene Cytochrom c Konzentrationen zugesetzt.Die EIS-Spektren werden bei einem Potential von 200 mV vs. NHE als Funk-tion der Cytochrom c-Konzentration aufgezeichnet. In Abbildung 5.3 sinddie Messergebnisse dargestellt. Man erkennt im Nyqist-Plot einen deutlichenRückgang des Widerstandes bei zunehmender Cytochrom c-Konzentration.Die Kapazität dagegen bleibt unverändert, wie der frequenzreduzierte Admit-tanz - Plot zeigt. Die EIS-Daten werden, wie in Abschnitt A.1.2 beschrieben,ausgewertet und der Widerstand gegen die Cytochrom c-Konzentration auf-getragen (Abbildung 5.4). Man kann den Abfall des Membranwiderstandesmit einer Exponentialfunktion darstellen. Eine Sättigung wird nicht erreicht,da man bei weiterer Erhöhung der Cytochrom c-Konzentration das ptBLM-System zerstört52.Durch die Aktivierung der Cytochrom-c-Oxidase werden Protonen aktiv vomSubmembranraum in die Lösung gepumpt. Bedingt durch diesen Protonen-strom wird der Membranwiderstand effektiv verringert53. Diese Annahmewird bestätigt durch die Tatsache, dass der Effekt durch Zusatz einer 0,6 mMKaliumcyanid-Lösung54 ausgeschaltet werden kann [42].
51Auf eine Fehlerrechnung für die jeweils verwendete Cytochrom c-Konzentration, sowiefür die Spannungsvorschubgeschwindigkeit wird im Folgenden verzichtet. Hierfür könnenkeine aussagekräftigen Angaben gemacht werden.
52Dies liegt vermutlich an der Veränderung des pH-Wertes. Das Cytochrom c aktiviertdas Enzym und die Cytochrom-c-Oxidase pumpt Protonen. Dadurch verringert sich derMembranwiderstand. Der verwendete Pu!er sorgt für einen annähernd konstanten pH-Wert während der kompletten Messreihe. Werden jedoch zu viele Protonen gepumpt, soverändert sich der pH-Wert drastisch. Dies verursacht eine Denaturierung der Proteine.
53Der Rückgang des Membranwiderstandes hängt jedoch noch von anderen Faktoren ab(siehe Abschnitt 5.3.1).
54Cyanid-Ionen (CN!) sind Inhibitoren der Cytochrom-c-Oxidase [1].
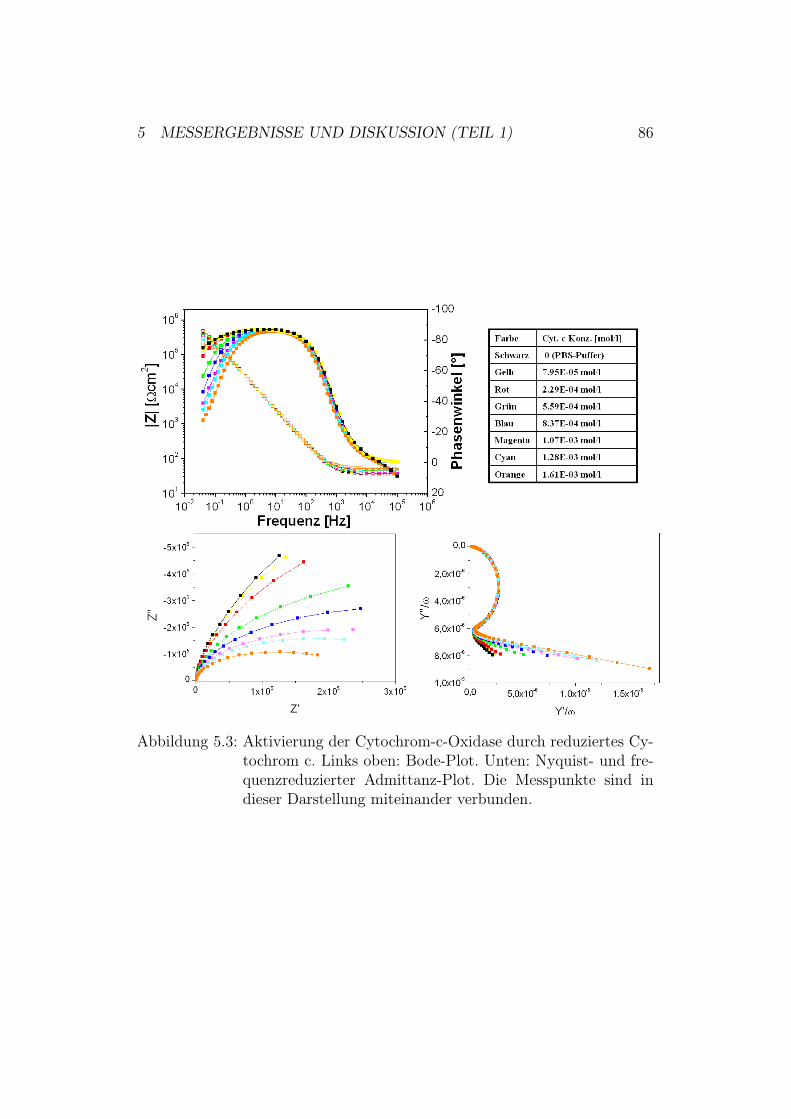
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 86
Abbildung 5.3: Aktivierung der Cytochrom-c-Oxidase durch reduziertes Cy-tochrom c. Links oben: Bode-Plot. Unten: Nyquist- und fre-quenzreduzierter Admittanz-Plot. Die Messpunkte sind indieser Darstellung miteinander verbunden.
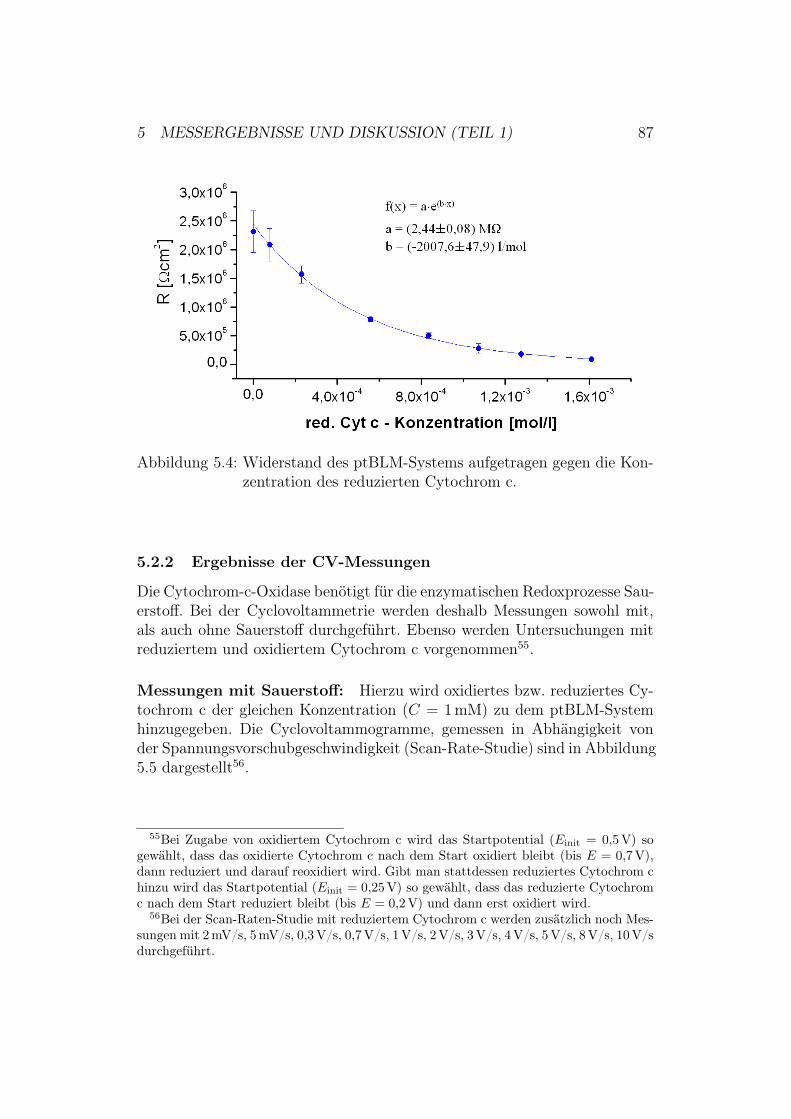
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 87
Abbildung 5.4: Widerstand des ptBLM-Systems aufgetragen gegen die Kon-zentration des reduzierten Cytochrom c.
5.2.2 Ergebnisse der CV-Messungen
Die Cytochrom-c-Oxidase benötigt für die enzymatischen Redoxprozesse Sau-erstoff. Bei der Cyclovoltammetrie werden deshalb Messungen sowohl mit,als auch ohne Sauerstoff durchgeführt. Ebenso werden Untersuchungen mitreduziertem und oxidiertem Cytochrom c vorgenommen55.
Messungen mit Sauerstoff: Hierzu wird oxidiertes bzw. reduziertes Cy-tochrom c der gleichen Konzentration (C = 1 mM) zu dem ptBLM-Systemhinzugegeben. Die Cyclovoltammogramme, gemessen in Abhängigkeit vonder Spannungsvorschubgeschwindigkeit (Scan-Rate-Studie) sind in Abbildung5.5 dargestellt56.
55Bei Zugabe von oxidiertem Cytochrom c wird das Startpotential (Einit = 0,5 V) sogewählt, dass das oxidierte Cytochrom c nach dem Start oxidiert bleibt (bis E = 0,7 V),dann reduziert und darauf reoxidiert wird. Gibt man stattdessen reduziertes Cytochrom chinzu wird das Startpotential (Einit = 0,25 V) so gewählt, dass das reduzierte Cytochromc nach dem Start reduziert bleibt (bis E = 0,2 V) und dann erst oxidiert wird.
56Bei der Scan-Raten-Studie mit reduziertem Cytochrom c werden zusätzlich noch Mes-sungen mit 2mV/s, 5 mV/s, 0,3 V/s, 0,7V/s, 1V/s, 2 V/s, 3 V/s, 4V/s, 5 V/s, 8V/s, 10 V/sdurchgeführt.
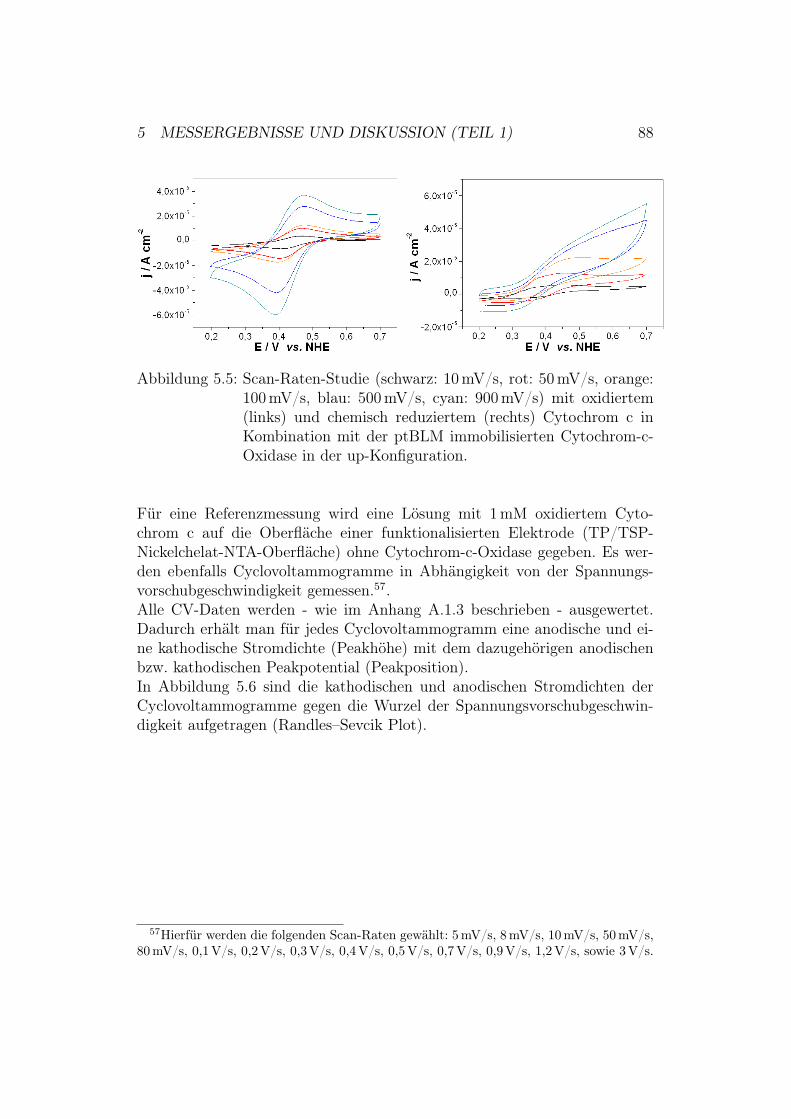
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 88
Abbildung 5.5: Scan-Raten-Studie (schwarz: 10 mV/s, rot: 50 mV/s, orange:100 mV/s, blau: 500 mV/s, cyan: 900 mV/s) mit oxidiertem(links) und chemisch reduziertem (rechts) Cytochrom c inKombination mit der ptBLM immobilisierten Cytochrom-c-Oxidase in der up-Konfiguration.
Für eine Referenzmessung wird eine Lösung mit 1 mM oxidiertem Cyto-chrom c auf die Oberfläche einer funktionalisierten Elektrode (TP/TSP-Nickelchelat-NTA-Oberfläche) ohne Cytochrom-c-Oxidase gegeben. Es wer-den ebenfalls Cyclovoltammogramme in Abhängigkeit von der Spannungs-vorschubgeschwindigkeit gemessen.57.Alle CV-Daten werden - wie im Anhang A.1.3 beschrieben - ausgewertet.Dadurch erhält man für jedes Cyclovoltammogramm eine anodische und ei-ne kathodische Stromdichte (Peakhöhe) mit dem dazugehörigen anodischenbzw. kathodischen Peakpotential (Peakposition).In Abbildung 5.6 sind die kathodischen und anodischen Stromdichten derCyclovoltammogramme gegen die Wurzel der Spannungsvorschubgeschwin-digkeit aufgetragen (Randles–Sevcik Plot).
57Hierfür werden die folgenden Scan-Raten gewählt: 5 mV/s, 8mV/s, 10mV/s, 50 mV/s,80mV/s, 0,1V/s, 0,2V/s, 0,3 V/s, 0,4V/s, 0,5V/s, 0,7V/s, 0,9V/s, 1,2V/s, sowie 3 V/s.
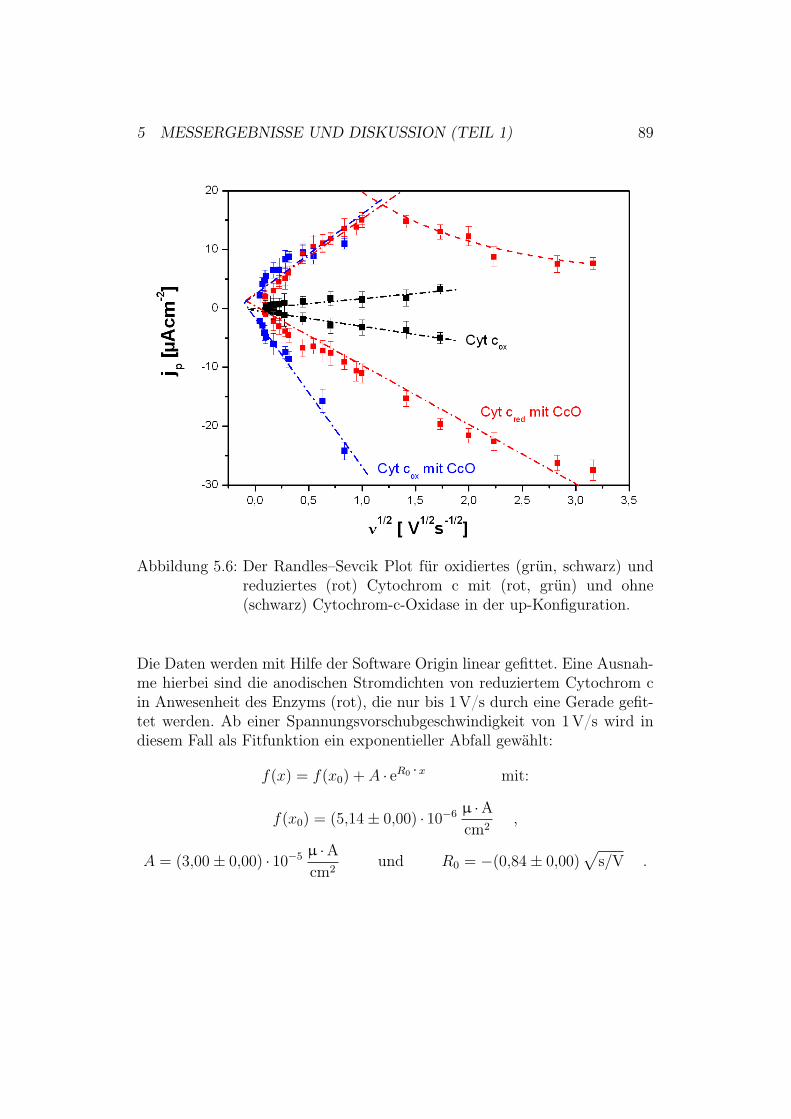
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 89
Abbildung 5.6: Der Randles–Sevcik Plot für oxidiertes (grün, schwarz) undreduziertes (rot) Cytochrom c mit (rot, grün) und ohne(schwarz) Cytochrom-c-Oxidase in der up-Konfiguration.
Die Daten werden mit Hilfe der Software Origin linear gefittet. Eine Ausnah-me hierbei sind die anodischen Stromdichten von reduziertem Cytochrom cin Anwesenheit des Enzyms (rot), die nur bis 1 V/s durch eine Gerade gefit-tet werden. Ab einer Spannungsvorschubgeschwindigkeit von 1 V/s wird indiesem Fall als Fitfunktion ein exponentieller Abfall gewählt:
f(x) = f(x0) + A · eR0 ·x mit:
f(x0) = (5,14 ± 0,00) · 10!6 µ ·Acm2
,
A = (3,00 ± 0,00) · 10!5 µ ·Acm2
und R0 = !(0,84 ± 0,00)√
s/V .

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 90
Die Fitergebnisse für die linearen Fits (f(x) = bx) befinden sich in der fol-genden Tabelle:
banodisch[A s1/2 V!1/2 cm!2] bkathodisch[A s1/2 V!1/2 cm!2]Cyt cox (1,64 ± 0,21) · 10!6 !(3,00 ± 0,15) · 10!6
Cyt cox+CcO (2,00 ± 0,25) · 10!5 !(3,00 ± 0,12) · 10!5
Cyt cred+CcO (1,42 ± 0,10) · 10!5 !(9,90 ± 0,26) · 10!6
Tabelle 5.2: Fit-Ergebnisse des linearen Fits für den Randles–Sevcik Plot.
Um die Standard-Geschwindigkeitskonstante k0 des heterogenen Elektronen-Transfer-Schrittes zum Cytochrom c zu berechnen, werden die Peakpotentialeals Funktion der Scan-Rate aufgetragen (siehe Abbildung 5.7). Die Span-nungsvorschubgeschwindigkeit wird dabei logarithmisch aufgetragen. Gehtman zu immer höheren Spannungsvorschubgeschwindigkeiten über, so steigtdie Abweichung des reduzierten und oxidierten Peakpotentials vom Standard-potential. Dadurch sind quantitative Aussagen über die Kinetik des Elektro-nentransfers möglich [91].Mit Hilfe des speziell für diese Anwendung geschriebenen Programms Jellyfitwerden die Daten gefittet. Der Fit dieses so genannten Trumpet-Plots er-mittelt nicht nur die Standard-Geschwindigkeitskonstante, sondern auch denWert des vorherrschenden Standardpotentials E0 [92] [93]. Tabelle 5.3 zeigtdie von dem Programm gelieferten Fitergebnisse für eine Raumtemperaturvon 20"C (T= 293K).
E0 [V] k0 [1/s]0.409 73.13
Tabelle 5.3: Fit-Ergebniss des Trumpet-Plots.
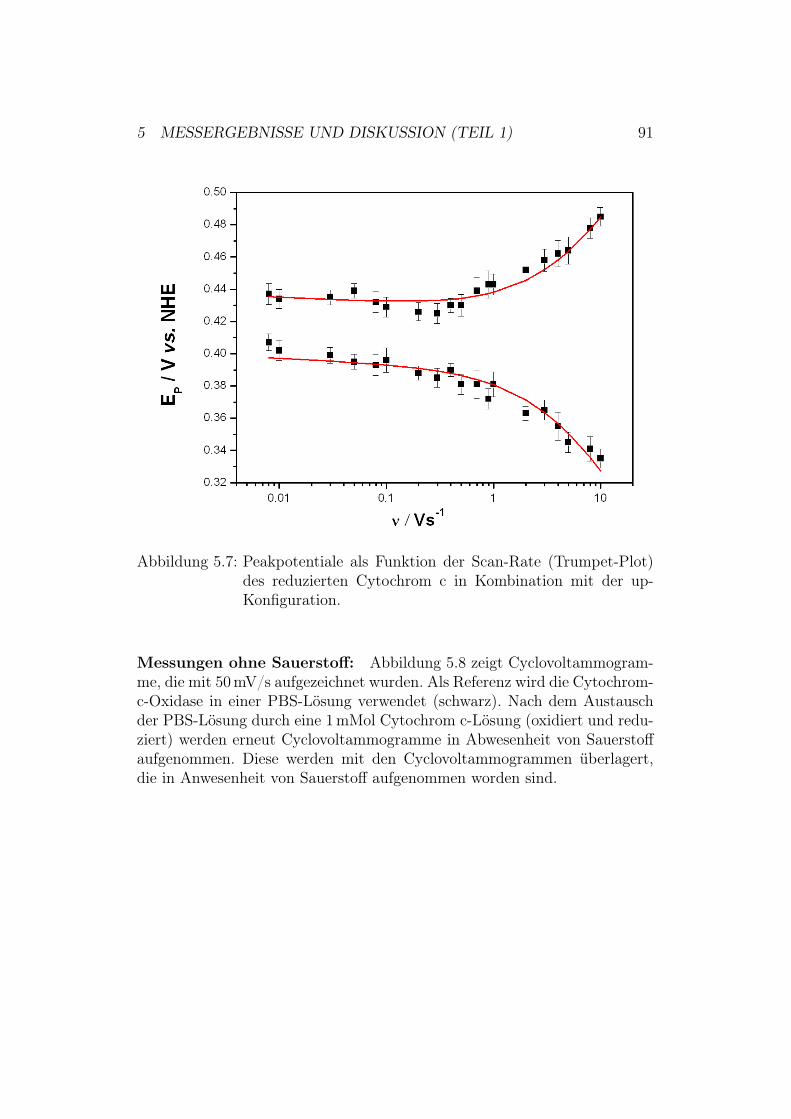
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 91
Abbildung 5.7: Peakpotentiale als Funktion der Scan-Rate (Trumpet-Plot)des reduzierten Cytochrom c in Kombination mit der up-Konfiguration.
Messungen ohne Sauerstoff: Abbildung 5.8 zeigt Cyclovoltammogram-me, die mit 50 mV/s aufgezeichnet wurden. Als Referenz wird die Cytochrom-c-Oxidase in einer PBS-Lösung verwendet (schwarz). Nach dem Austauschder PBS-Lösung durch eine 1 mMol Cytochrom c-Lösung (oxidiert und redu-ziert) werden erneut Cyclovoltammogramme in Abwesenheit von Sauerstoffaufgenommen. Diese werden mit den Cyclovoltammogrammen überlagert,die in Anwesenheit von Sauerstoff aufgenommen worden sind.
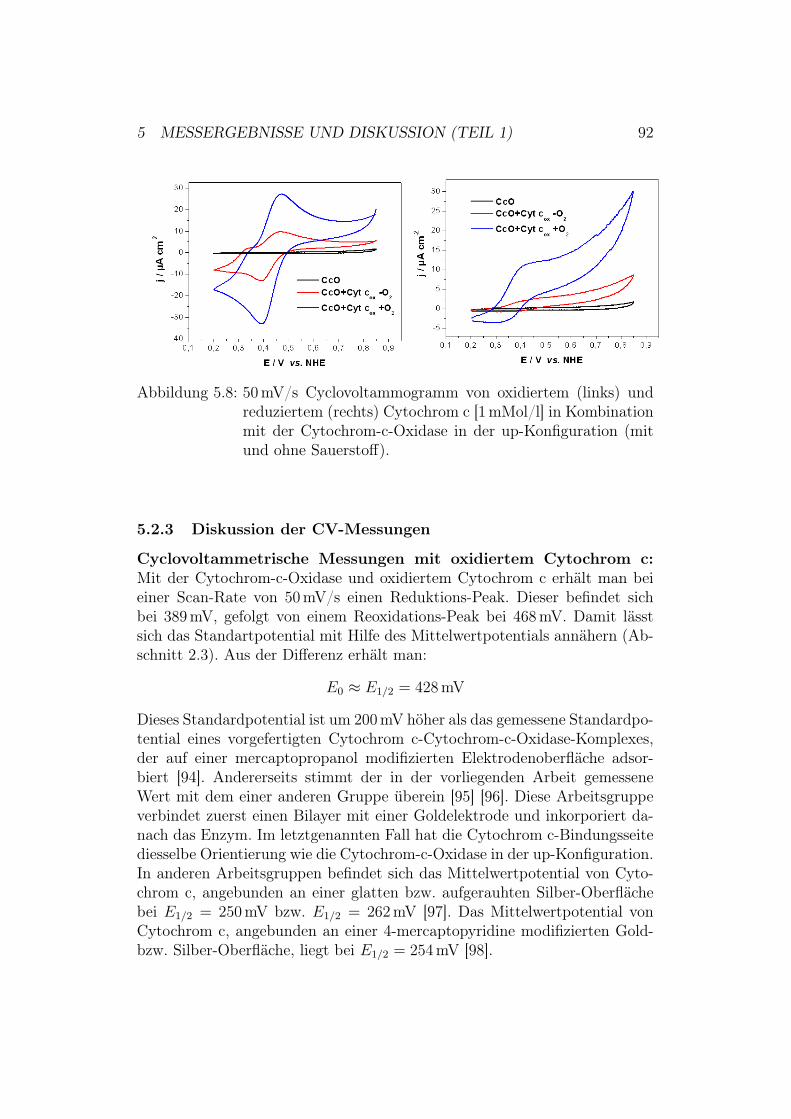
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 92
Abbildung 5.8: 50 mV/s Cyclovoltammogramm von oxidiertem (links) undreduziertem (rechts) Cytochrom c [1 mMol/l] in Kombinationmit der Cytochrom-c-Oxidase in der up-Konfiguration (mitund ohne Sauerstoff).
5.2.3 Diskussion der CV-Messungen
Cyclovoltammetrische Messungen mit oxidiertem Cytochrom c:Mit der Cytochrom-c-Oxidase und oxidiertem Cytochrom c erhält man beieiner Scan-Rate von 50 mV/s einen Reduktions-Peak. Dieser befindet sichbei 389 mV, gefolgt von einem Reoxidations-Peak bei 468 mV. Damit lässtsich das Standartpotential mit Hilfe des Mittelwertpotentials annähern (Ab-schnitt 2.3). Aus der Differenz erhält man:
E0 # E1/2 = 428 mV
Dieses Standardpotential ist um 200 mV höher als das gemessene Standardpo-tential eines vorgefertigten Cytochrom c-Cytochrom-c-Oxidase-Komplexes,der auf einer mercaptopropanol modifizierten Elektrodenoberfläche adsor-biert [94]. Andererseits stimmt der in der vorliegenden Arbeit gemesseneWert mit dem einer anderen Gruppe überein [95] [96]. Diese Arbeitsgruppeverbindet zuerst einen Bilayer mit einer Goldelektrode und inkorporiert da-nach das Enzym. Im letztgenannten Fall hat die Cytochrom c-Bindungsseitediesselbe Orientierung wie die Cytochrom-c-Oxidase in der up-Konfiguration.In anderen Arbeitsgruppen befindet sich das Mittelwertpotential von Cyto-chrom c, angebunden an einer glatten bzw. aufgerauhten Silber-Oberflächebei E1/2 = 250mV bzw. E1/2 = 262mV [97]. Das Mittelwertpotential vonCytochrom c, angebunden an einer 4-mercaptopyridine modifizierten Gold-bzw. Silber-Oberfläche, liegt bei E1/2 = 254mV [98].

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 93
Der Elektronentransfer durch das Enzym könnte eine mögliche Erklärung fürdiese Verschiebung des Cytochrom c- Redoxpotentials um fast 200 mV sein.Deshalb kann man zu dem Schluss kommen, dass das Cytochrom c an derCytochrom c-Bindungsseite der Cytochrom-c-Oxidase reduziert und oxidiertwird. Der Elektronen-Transfer geht also durch das Enzym hindurch. Dadurchist die Tunnel-Barriere mit ungefähr 12 nm (siehe Abbildung 3.5) extremgroß.Der direkte Elektronen-Transfer zwischen der Cytochrom-c-Oxidase und derElektrode ohne Cytochrom c konnte nicht nachgewiesen werden (Abbildung5.8), obwohl er auf Grund der kürzeren Tunnel-Barriere wesentlich verständ-licher wäre.Cyclovoltamogramme von oxidiertem Cytochrom c mit der Cytochrom-c-Oxidase in der up-Konfiguration (mit und ohne Sauerstoff gemessen) sind inAbbildung 5.8 zu sehen. In Anwesenheit von Sauerstoff ist ein deutlicher An-stieg der Peakstromdichte (jp,kathodisch = !4, 9 µAcm!2) im Vergleich zu dersauerstofffreien Messung (jp,kathodisch = !0, 8 µAcm!2) zu sehen. Diesen Ef-fekt kann man als katalytischen Strom interpretieren. Er hat seinen Ursprungin der oft wiederholten Reduktion des Cytochrom c, das mit Sauerstoff immerwieder durch das Enzym katalytisch reoxidiert wird. Diese katalytische Reoxi-dation kann nur mit Sauerstoff und mit der damit verbundenen Aktivität desEnzyms stattfinden. Der Vor-Peak in Abbildung 5.8 resultiert nach [48] ausder Absorption der reduzierten Spezies des Cytochrom c-Redox-Paars.Abbildung 5.6 zeigt die mit Sauerstoff gemessenen anodischen und kathodi-schen Stromdichten von oxidiertem Cytochrom c mit und ohne Cytochrom-c-Oxidase in der up-Konfiguration. Die Stromdichten sind in beiden Fällenlinear von der Wurzel der Spannungsvorschubgeschwindigkeit abhängig. Eshandelt sich also hierbei um einen diffusionskontrollierten Prozess [48]. InAnwesenheit des Enzyms erkennt man für die kathodischen Ströme eine deut-liche Veränderung der Steigung um eine Größenordnung58. Auch hier ist derkatalytische Effekt dafür verantwortlich:Das oxidierte Cytochrom c kann bei entsprechenden Potentialen reduziertwerden. Jedoch erhält man in Anwesenheit des Enzyms einen zusätzlichenStrom. Das Cytochrom c bindet an das Enzym an, wird reduziert und wirddann über das Enzym katalytisch reoxidiert. Das oxidierte Cytochrom c wirddaraufhin auf Grund des angelegten Potentials wieder reduziert und der Vor-gang wiederholt sich.Diese Interpretation ermöglicht es, nicht nur den Anstieg der Stromdichtein Anwesenheit des Enzyms zu erklären, sondern auch den Rückgang derStromdichte bei den sauerstofffreien Messungen.
58Das Verhalten der anodischen Ströme wird im nächsten Abschnitt besprochen.

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 94
Cyclovoltammetrie mit reduziertem Cytochrom c: Auch hier er-kennt man bei der Verwendung von reduziertem Cytochrom c in Kombi-nation mit dem ptBLM-System einen Anstieg der Stromdichte in Gegenwartvon Sauerstoff (siehe Abbildung 5.8). Dieser Effekt konnte bei exakt dem-selben Standartpotential von 430 mV bereits in [99] festgestellt werden. Indieser Referenz beobachtete man jedoch viel geringere Stromdichten. DerAnstieg der anodischen Ströme in Anwesenheit von Sauerstoff wird in [99]der elektrochemischen Oxidation der oxoferryl oder ferredoxyl-Spezies zuge-schrieben. Diese Spezies sind Zwischenprodukte des katalytischen Zyklus undbefinden sich direkt am katalytischen Zentrum (Häm a3/CuB). Dies führt zudem Schluss, dass es sich auch in diesem Fall um einen katalytischen Stromhandelt. Durch das reduzierte Cytochrom c wird die Cytochrom-c-Oxidaseaktiviert. Dabei wird Sauerstoff verbraucht und es bilden sich oxyferryl oderferredoxyl-Spezies, die über die Elektrode zu Sauerstoff regeneriert werden.Durch diesen katalytischen Effekts ist es auch möglich die höheren anodi-schen Stromdichten in Gegenwart des Enzyms im Randles-Plot (Abbildung5.6) zu erklären. In diesem Fall zeigt sich kaum ein Unterschied bei Verwen-dung von reduziertem und oxidiertem Cytochrom. Die Steigung der Geradenfür die anodischen Ströme des oxidierten Cytochrom c ohne Enzym, ist dabeium eine Größenordnung kleiner.In allen drei Fällen erhält man eine lineare Abhängigkeit der anodischen undkathodischen Stromdichten von der Wurzel der Spannungsvorschubgeschwin-digkeiten. Es handelt sich also um diffusionskontrollierte Prozesse, bei denenjeweils ein reversibler Elektronenaustausch mit Hilfe des Cytochrom c statt-findet.Eine nähere Betrachtung der kathodischen Ströme zeigt Folgendes59:In Gegenwart des Enzyms erkennt man deutlich kleinere Stromdichten beiVerwendung von reduziertem statt oxidiertem Cytochrom c (Abbildung 5.6).Das reduzierte Cytochrom c versorgt das Enzym mit einem Elektron undwird oxidiert. Durch das negative Potential der Elektrode wird das Cyto-chrom über das Enzym wieder reduziert und gibt sein Elektron an das Enzymwieder ab. Für den Fall des oxidierten Cytochrom c muss dieses jedoch zuerstreduziert werden, um das Enzym zu aktivieren. Dieser zusätzlich auftretende(faradaischen) Prozess bewirkt eine höhere Stromdichte.Ein weiteres wichtiges Phänomen ist in Abbildung 5.6 zu erkennen. Die an-odische Stromdichte ist für den Fall des reduzierten Cytochrom c nur bisungefähr 1 V/s linear von der Wurzel der Spannungsvorschubgeschwindig-keit abhängig. Danach fällt die Stromdichte exponentiell ab. Dieser Effekt
59Im vorherigen Abschnitt wurde bereits die unterschiedliche Steigung der Geraden vonoxidiertem Cytochrom c mit und ohne Enzym durch einen katalytischen E!ekt erklärt.

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 95
hängt direkt mit dem Umsatz des Enzyms zusammen. Bei höheren Span-nungsvorschubgeschwindigkeiten ist das Enzym nicht mehr in der Lage demsich schnell ändernden Potential zu folgen. Dabei werden immer weniger re-duzierte Cytochrom c-Moleküle regeneriert, was letztendlich zu einem Abfallder Stromdichte führt.Diese Vermutung wird durch den Trumpet-Plot bestätigt, bei dem eine stei-gende Abweichung des reduzierten und oxidierten Peakpotentials vom Stan-dardpotential bei einer Scan-Rate von 1 V/s zu beobachten ist. Die Aus-wertung mit dem Programm Jellyfit ergibt für das verwendete System ei-ne Geschwindigkeitskonstante von k0 = 73 s!1 und ein Standardpotenti-al von E0 = 409 mV. Die berechnete Geschwindigkeitskonstante befindetsich in der selben Größenordnung wie die eines Cytochrom c-Cyctochrom-c-Oxidase-Komplexes (k0 = 20 s!1) [94] und das berechnete Standardpotential(E0 = 409 mV) liegt in demselben Bereich wie in [99] (E0 = 430 mV).
5.3 Potentialtitration der Cytochrom-c-Oxidase in derup-Konfiguration mit und ohne Cytochrom c
Wie in den vorherigen Abschnitten gezeigt, pumpt die Cytochrom-c-Oxidasenach ihrer Aktivierung durch Cytochrom c Protonen. Als Konsequenz desTurnovers60, sollte sich bei der Cytochrom-c-Oxidase in der up-Konfigurationeine positive Potentialdifferenz bilden (bezogen auf die Außenseite gegenüberdem Submembranraum). Messungen des Membranpotentials an Liposomenmit inkorporierter Cytochrom-c-Oxidase (aktiviert durch Cytochrom c) er-geben Werte um die 50 mV [100].
5.3.1 Ergebnisse und Diskussion der EIS-Potentialtitration
Um die Abhängigkeit des ptBLM-Systems vom Potential zu überprüfen wer-den Impedanz-Spektren bei unterschiedlichen Potentialen aufgenommen. Da-zu werden mehrere Messreihen mit unterschiedlichen Cytochrom c - Konzen-trationen aufgestellt61. Der gewählte Potentialbereich liegt dabei zwischen50mV und 400 mV. Die Messergebnisse einer dieser Messreihen sind in Ab-bildung 5.9 dargestellt. Die Auswertung erfolgt analog zu Abschnitt 5.2.1.
60Als Turnover bezeichnet man den Umsatz eines Produktes oder einer Substanz proZeiteinheit.
61Bei diesem Versuch wurde ausschließlich mit reduziertem Cytochrom c gearbeitet.
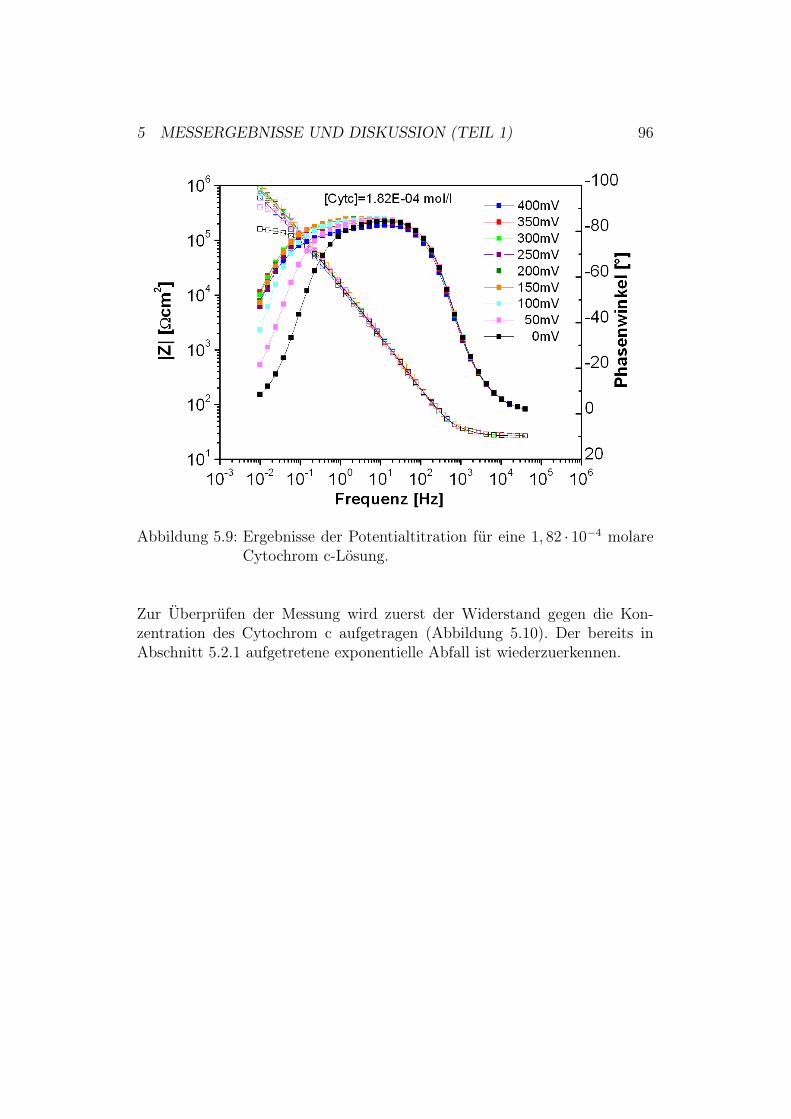
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 96
Abbildung 5.9: Ergebnisse der Potentialtitration für eine 1, 82 · 10!4 molareCytochrom c-Lösung.
Zur Überprüfen der Messung wird zuerst der Widerstand gegen die Kon-zentration des Cytochrom c aufgetragen (Abbildung 5.10). Der bereits inAbschnitt 5.2.1 aufgetretene exponentielle Abfall ist wiederzuerkennen.
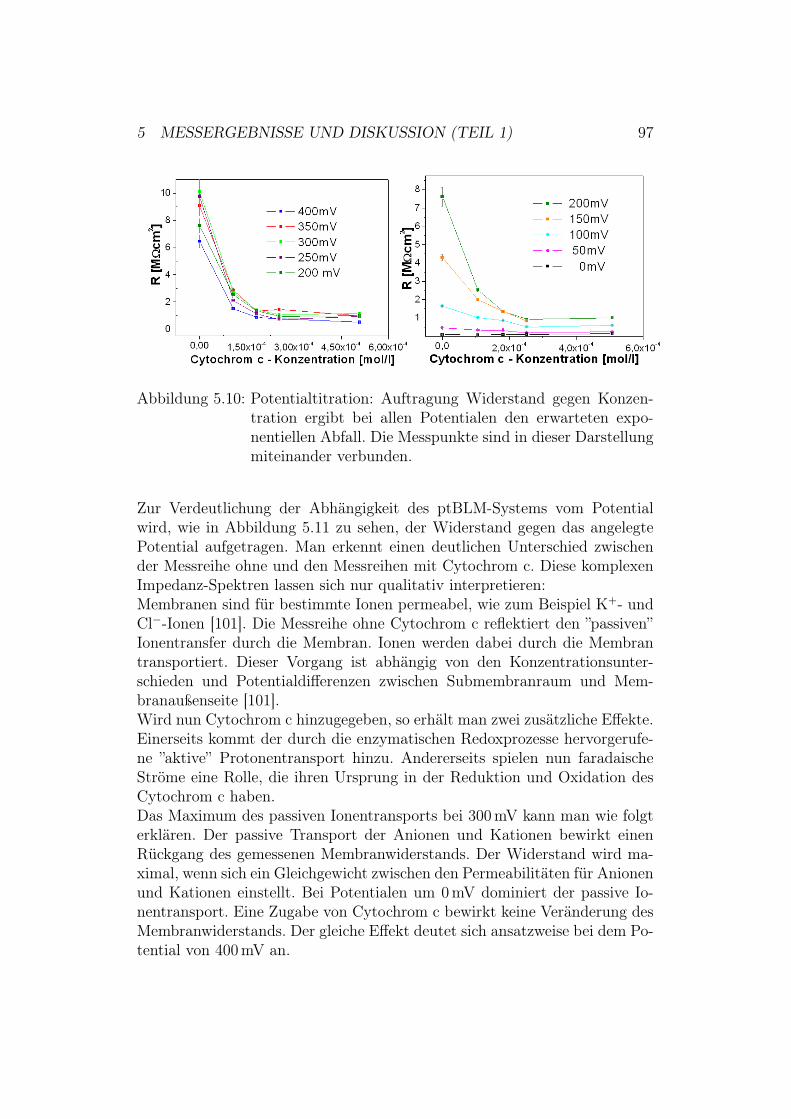
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 97
Abbildung 5.10: Potentialtitration: Auftragung Widerstand gegen Konzen-tration ergibt bei allen Potentialen den erwarteten expo-nentiellen Abfall. Die Messpunkte sind in dieser Darstellungmiteinander verbunden.
Zur Verdeutlichung der Abhängigkeit des ptBLM-Systems vom Potentialwird, wie in Abbildung 5.11 zu sehen, der Widerstand gegen das angelegtePotential aufgetragen. Man erkennt einen deutlichen Unterschied zwischender Messreihe ohne und den Messreihen mit Cytochrom c. Diese komplexenImpedanz-Spektren lassen sich nur qualitativ interpretieren:Membranen sind für bestimmte Ionen permeabel, wie zum Beispiel K+- undCl!-Ionen [101]. Die Messreihe ohne Cytochrom c reflektiert den ”passiven”Ionentransfer durch die Membran. Ionen werden dabei durch die Membrantransportiert. Dieser Vorgang ist abhängig von den Konzentrationsunter-schieden und Potentialdifferenzen zwischen Submembranraum und Mem-branaußenseite [101].Wird nun Cytochrom c hinzugegeben, so erhält man zwei zusätzliche Effekte.Einerseits kommt der durch die enzymatischen Redoxprozesse hervorgerufe-ne ”aktive” Protonentransport hinzu. Andererseits spielen nun faradaischeStröme eine Rolle, die ihren Ursprung in der Reduktion und Oxidation desCytochrom c haben.Das Maximum des passiven Ionentransports bei 300 mV kann man wie folgterklären. Der passive Transport der Anionen und Kationen bewirkt einenRückgang des gemessenen Membranwiderstands. Der Widerstand wird ma-ximal, wenn sich ein Gleichgewicht zwischen den Permeabilitäten für Anionenund Kationen einstellt. Bei Potentialen um 0 mV dominiert der passive Io-nentransport. Eine Zugabe von Cytochrom c bewirkt keine Veränderung desMembranwiderstands. Der gleiche Effekt deutet sich ansatzweise bei dem Po-tential von 400 mV an.
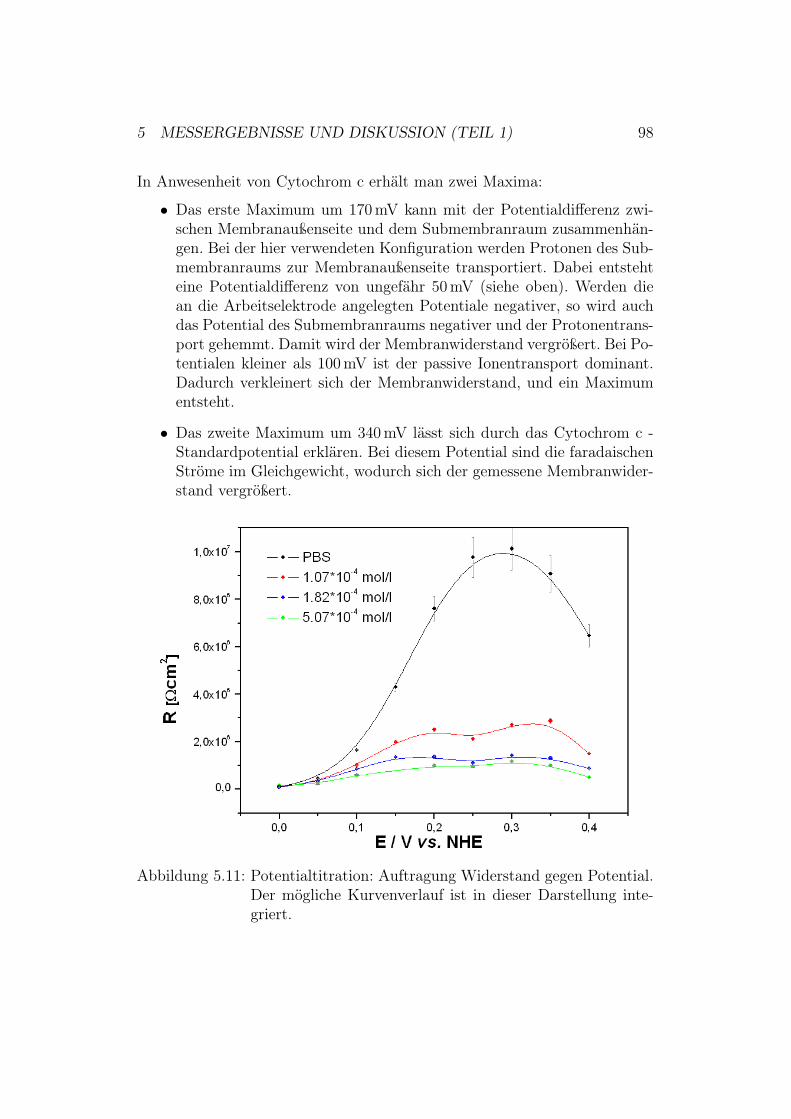
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 98
In Anwesenheit von Cytochrom c erhält man zwei Maxima:
• Das erste Maximum um 170 mV kann mit der Potentialdifferenz zwi-schen Membranaußenseite und dem Submembranraum zusammenhän-gen. Bei der hier verwendeten Konfiguration werden Protonen des Sub-membranraums zur Membranaußenseite transportiert. Dabei entstehteine Potentialdifferenz von ungefähr 50 mV (siehe oben). Werden diean die Arbeitselektrode angelegten Potentiale negativer, so wird auchdas Potential des Submembranraums negativer und der Protonentrans-port gehemmt. Damit wird der Membranwiderstand vergrößert. Bei Po-tentialen kleiner als 100 mV ist der passive Ionentransport dominant.Dadurch verkleinert sich der Membranwiderstand, und ein Maximumentsteht.
• Das zweite Maximum um 340 mV lässt sich durch das Cytochrom c -Standardpotential erklären. Bei diesem Potential sind die faradaischenStröme im Gleichgewicht, wodurch sich der gemessene Membranwider-stand vergrößert.
Abbildung 5.11: Potentialtitration: Auftragung Widerstand gegen Potential.Der mögliche Kurvenverlauf ist in dieser Darstellung inte-griert.

5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 99
5.4 Aktivierung der Cytochrom-c-Oxidase in der down-Konfiguration
In einer vorausgegangen Arbeit konnte bereits der direkte Elektronentransferder Cytochrom-c-Oxidase mit His-Tag an Untereinheit II (down-Konfigurati-on) nachgewiesen werden [17]. In dieser Konfiguration ist das Enzym über dieArbeitselektrode aktivierbar. Werden nun Messungen ohne Sauerstoff vorge-nommen, verliert die Cytochrom-c-Oxidase ihre katalytische Wirkung. Ander Elektrode unterschiedlich angelegten Spannungen zwingen das Enzym inden reduzierten, oxidierten oder einen Zwischenzustand. Mit der Cyclovol-tammetrie konnten zwei Peaks ermittelt werden:
• Der erste Peak bei E # 422 mV konnte man den Protonen zuordnen, diezuerst in den Submembranraum gepumpt und danach an der Elektrodezu Wasserstoff werden (2H+ " H2).
• Der zweite Peak bei E # 202 mV hat seinen Ursprung im direktenElektronentransfer zwischen Enzym und Elektrode (Reduktion der vierRedox-Zentren der Cytochrom-c-Oxidase) [17].
Messungen ohne Sauerstoff zeigten eine starke Verringerung der Stromdichtefür den letztgenannten Bereich. Unter anaeroben Bedingungen entfällt dasProtonenpumpen.Außerdem konnte man eine direkte Aktivierung der Cytochrom-c-Oxidasein der up-Konfiguration in einem Potentialbereich von -600 mV bis +600 mVausschließen (siehe Abbildung 5.12) [17].Im Folgenden wird die Mutanten N139C zur Konstruktion eines ptBLM-System verwendet und mit cyclovoltammetrischen Messungen untersucht.
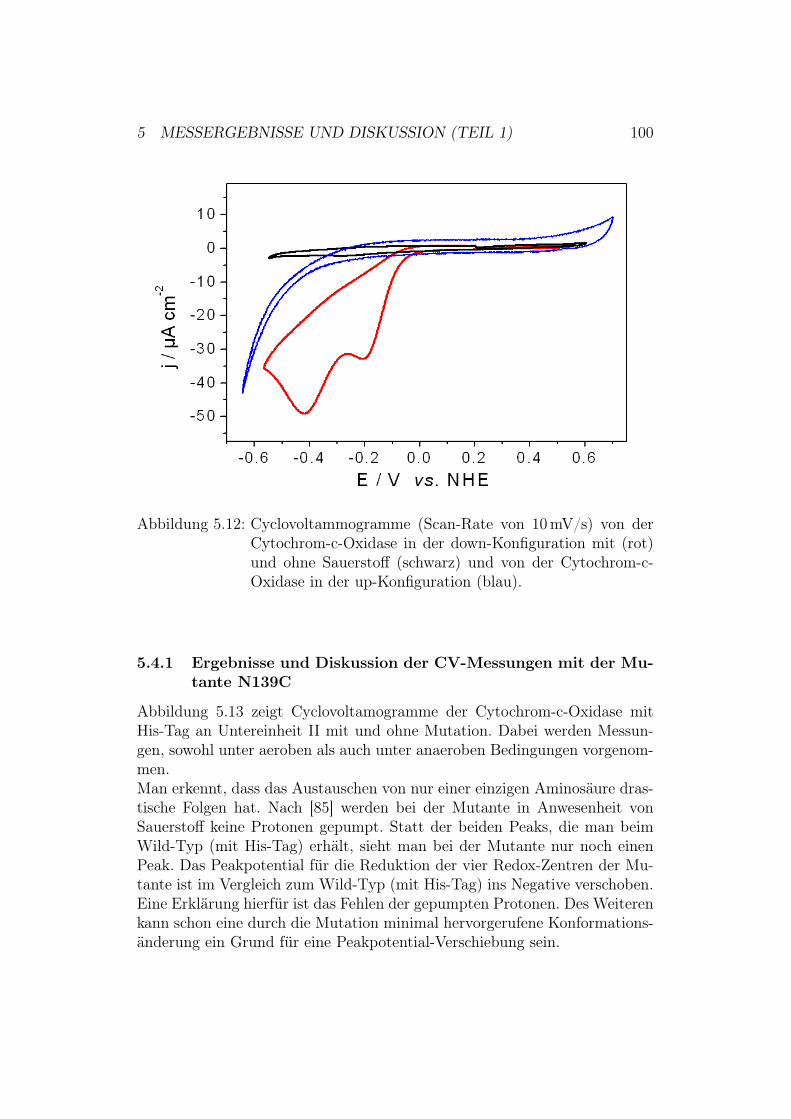
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 100
Abbildung 5.12: Cyclovoltammogramme (Scan-Rate von 10 mV/s) von derCytochrom-c-Oxidase in der down-Konfiguration mit (rot)und ohne Sauerstoff (schwarz) und von der Cytochrom-c-Oxidase in der up-Konfiguration (blau).
5.4.1 Ergebnisse und Diskussion der CV-Messungen mit der Mu-tante N139C
Abbildung 5.13 zeigt Cyclovoltamogramme der Cytochrom-c-Oxidase mitHis-Tag an Untereinheit II mit und ohne Mutation. Dabei werden Messun-gen, sowohl unter aeroben als auch unter anaeroben Bedingungen vorgenom-men.Man erkennt, dass das Austauschen von nur einer einzigen Aminosäure dras-tische Folgen hat. Nach [85] werden bei der Mutante in Anwesenheit vonSauerstoff keine Protonen gepumpt. Statt der beiden Peaks, die man beimWild-Typ (mit His-Tag) erhält, sieht man bei der Mutante nur noch einenPeak. Das Peakpotential für die Reduktion der vier Redox-Zentren der Mu-tante ist im Vergleich zum Wild-Typ (mit His-Tag) ins Negative verschoben.Eine Erklärung hierfür ist das Fehlen der gepumpten Protonen. Des Weiterenkann schon eine durch die Mutation minimal hervorgerufene Konformations-änderung ein Grund für eine Peakpotential-Verschiebung sein.
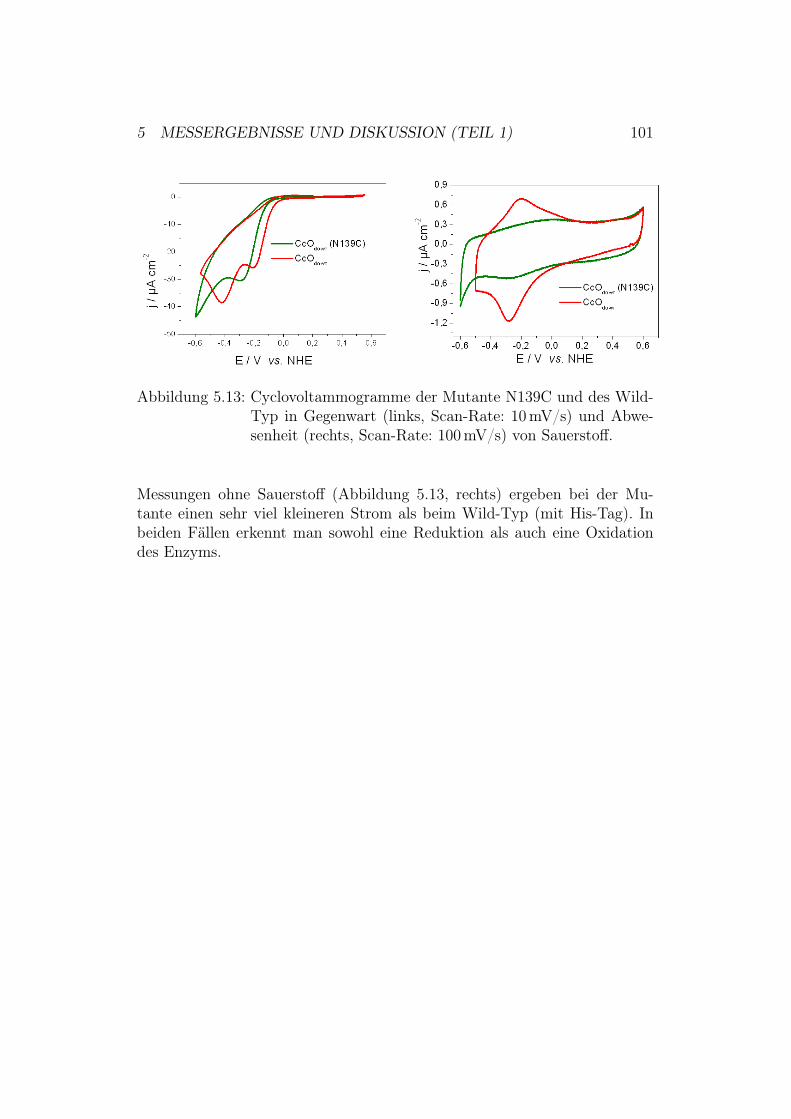
5 MESSERGEBNISSE UND DISKUSSION (TEIL 1) 101
Abbildung 5.13: Cyclovoltammogramme der Mutante N139C und des Wild-Typ in Gegenwart (links, Scan-Rate: 10 mV/s) und Abwe-senheit (rechts, Scan-Rate: 100 mV/s) von Sauerstoff.
Messungen ohne Sauerstoff (Abbildung 5.13, rechts) ergeben bei der Mu-tante einen sehr viel kleineren Strom als beim Wild-Typ (mit His-Tag). Inbeiden Fällen erkennt man sowohl eine Reduktion als auch eine Oxidationdes Enzyms.

6 PROBENHERSTELLUNG FÜR SERRS 102
6 Probenherstellung für SERRSIm Gegensatz zu den in der Elektrochemie verwendeten Goldelektroden müs-sen die verwendeten Silberelektroden für die SERR-Spektroskopie eine be-stimmte Rauhigkeit als Voraussetzung für die Oberflächenverstärkung derRaman-Streuung aufweisen (siehe Abschnitt 2.5.4). Der Oberflächenverstär-kungseffekt hängt stark von dem verwendeten Material ab [60]. Deshalb wirdin diesem Experiment als Elektrodenmaterial Silber verwendet62.Dieses Kapitel beschreibt die Herstellung und Präparation einer chemischaufgerauhten Silberoberfläche63.Analog zu vorausgegangen Arbeiten werden die Proteine auf einer chemischaufgerauhten Silberelektrode angebunden [55] [78]. Die Silberelektroden wer-den vor der Aufrauhung mit einer Poliermaschine poliert. Die aufgerauhteSilberelektrode wird danach für das jeweilige Experiment entweder mit einemreinen TP-Monolayer oder einem DTSP-DTP-Monolayer funktionalisiert.
6.1 Herstellung der SilberelektrodenWeil mit den präparierten Proben keine SPR-Spektroskopie gemacht wird,benötigt man keine ultradünnen Schichten, so dass man auf eine Herstellungvon TSG-Elektroden verzichtet kann. Für die Herstellung der Silberelektro-den wird stattdessen ein Silberstab zersägt. Diese (ø = 10mm, Höhe # 7 mm)haben auf einer Seite ein Gewinde (ø = 4 mm), damit sie in die entsprechendeApparatur eingebaut werden können64. Die Präparation ist für neue und ge-brauchte Elektroden identisch. Die Elektroden sind zwar wieder verwendbar,verlieren jedoch durch die erneute Polierung an Höhe.
62Mehrere Verö!entlichungen zeigen bei Verwendung von Silber-, Gold- oder aufgerauh-ten Silberelektroden in Kombination mit Cytochrom c kaum eine Veränderung im gemes-senen Cyclovoltammogramm [98] [97]. Aus diesem Grund sind die Ergebnisse, die man mitden unterschiedlichen Elektroden erhält, vergleichbar.
63Eine genaue Beschreibung der verwendeten Materialien und Geräte befindet sich imAnhang.
64Da die Elektrode einen Zylinder darstellt, man jedoch lediglich eine Oberfläche vonA = 0,785 cm2 benötigt (analog zur Goldelektrode aus Abschnitt 4), wird ein Teflonmantel(øinnen =10mm, øaußen =11mm, Höhe=7mm) als Begrenzung verwendet. Die Silberelek-troden und die Teflonmäntel wurden von der Feinmechanik-Werkstatt des MPIP Mainzhergestellt.

6 PROBENHERSTELLUNG FÜR SERRS 103
6.2 Polieren der Silber-ElektrodenDie Silberelektroden werden mit einer abgewandelten Form der ”Stewart’sMethode für Edelmetalle” poliert65. Bei allen Polierschritten wird mit einemmöglichst geringen Druck gearbeitet. Dadurch vermeidet man einen allzugroßen Abrieb und das Eindringen von Schleifpartikeln in die Silberlektrode.In der folgenden Tabelle ist die Vorgehensweise dargestellt:
Oberfläche Schmiermittel U/min Zeit [s]CarbiMet P600 SiC wassergekühlt 150 30CarbiMet P1200 SiC wassergekühlt 150 30ChemoMet-Tuch 1µm Al2O3 Suspension 300 30ChemoMet-Tuch 0,3µm Al2O3 Suspension 300 30ChemoMet-Tuch 0,05 µm Al2O3 Suspension 300 30
Tabelle 6.1: Poliervorschrift für Ag-Elektroden
Die Silberelektroden werden zwischen den einzelnen Polierschritten sorgfäl-tig mit Wasser gewaschen oder alternativ für 10 Minuten ins Ultraschall-bad gestellt. Dadurch wird verhindert, dass das verwendete Poliermittel dennächsten Polierschritt kontaminiert. Zum Schluss werden die Silberelektro-den zuerst mit Wasser und dann mit Ethanol für jeweils 30 Minuten insUltraschallbad gestellt.
6.3 Elektrochemisches Aufrauhen der Silber-ElektrodenDas elektrochemische Aufrauhen der Silberelektroden erfolgt in einer im Rah-men der Diplomarbeit speziell angefertigten Zelle. Die Silberelektrode wirdauf einen Stahlkern, der sich in der Mitte eines Teflonzylinders befindet, hin-eingeschraubt und auf diese Weise kontaktiert. Der Teflonzylinder wird mit0,1M KCL-Lösung gefüllt. Referenz- und Gegenelektrode werden von obenin die Lösung gehalten66. Die Vorgehensweise der elektrochemischen Aufrau-hung wird wie in [55] durchgeführt. Nach der Reinigung durch naszierendenWasserstoff entfernt man die entstehenden Luftblasen. Dadurch erhält man
65Dies geschah in Zusammenarbeit mit der Firma Buehler, bei der diese Poliervorschrif-ten erhältlich sind. Der Grund der individualisierten Poliervorschrift ist in Abschnitt A.6zu finden.
66Auch hier wird eine 3 Elektrodenanordnung verwendet (siehe Abschnitt 4).

6 PROBENHERSTELLUNG FÜR SERRS 104
im nächsten Schritt eine gleichmäßige Aufrauhung mit (70/20/20)67. Danachwird die Oberflächenaufrauhung in drei Schritten durchgeführt:
Schritt angelegte Spannung vs. Ag/AgCl,KClkonz. Wdh.Reinigung -2.0 V für & 10 s 1Aufrauhung +0.3 V und -0.3 V für jeweils 70 s 1Aufrauhung +0.3 V und -0.3 V für jeweils 20 s 2
Tabelle 6.2: Protokoll zum Aufrauhen der Ag-Elektroden mit (70/20/20).
6.4 Funktionalisierung und Proteinanbindung6.4.1 Untersuchungen mit Cytochrom c
Für die Untersuchungen mit Cytochrom c wird die aufgerauhte Silberelek-trode mit einer DTP-Lösung für 2 Stunden funktionalisiert (Abbildung 6.1).Danach wird entweder eine oxiderte oder eine chemisch reduzierte Cytochromc-Lösung hinzugegeben. Nach 30 Minuten Anbindungszeit beginnt man mitden Messungen.
6.4.2 Untersuchungen mit der Cytochrom-c-Oxidase
Die Cytochrom-c-Oxidase von Rhodobacter sphaeroides mit His-Tag an Un-tereinheit II (down-Konfiguration) wird mit SERRS untersucht. Die Silber-elektrode wird - wie in Abschnitt 3.2 und 3.3 beschrieben - funktionalisiert.Nach der Proteinanbindung bildet man die Membran durch eine Dialyse. Dasfertige ptBLM-System ist in Abbildung 6.2 dargestellt.
67(70/20/20) steht für die unterschiedlichen Aufrauhungszeiten in den jeweiligen Schrit-ten: +0,3V für 70 s, -0,3 V für 70 s, +0,3V für 20 s, -0,3V für 20 s, +0,3V für 20 s, -0,3 V für20 s. Die angelegte Spannung bezieht sich auf eine Ag/AgCl,KClkonz.-Referenzelektrode.
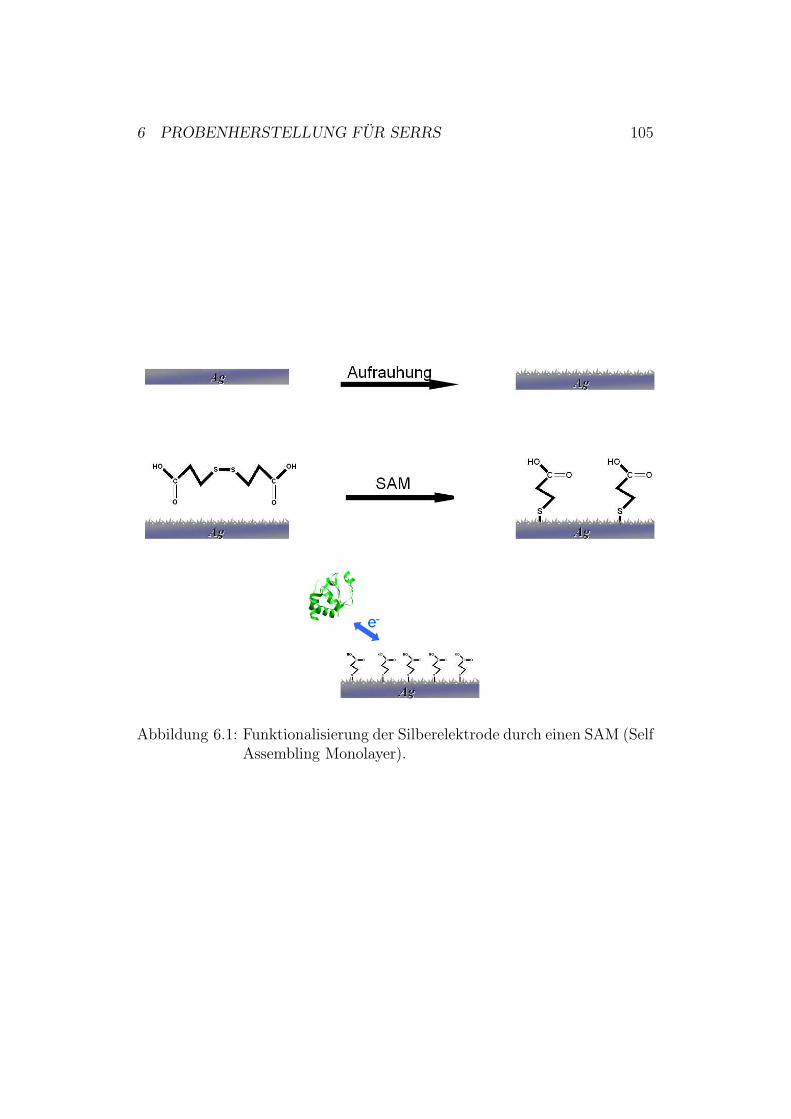
6 PROBENHERSTELLUNG FÜR SERRS 105
Abbildung 6.1: Funktionalisierung der Silberelektrode durch einen SAM (SelfAssembling Monolayer).
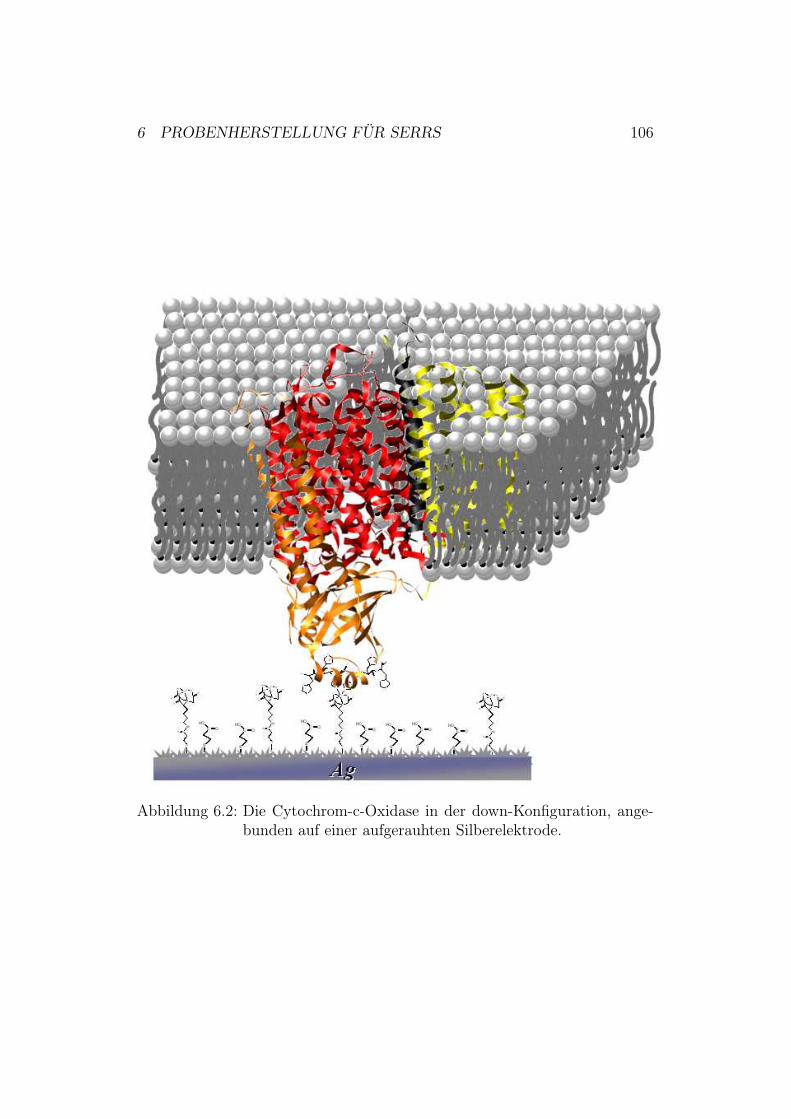
6 PROBENHERSTELLUNG FÜR SERRS 106
Abbildung 6.2: Die Cytochrom-c-Oxidase in der down-Konfiguration, ange-bunden auf einer aufgerauhten Silberelektrode.

7 MESSAUFBAU FÜR SERRS 107
7 Messaufbau für SERRSDurch die Kombination von elektrochemischen Experimenten mit ”in-situ”Raman-Spektroskopie benötigt man einen speziellen experimentellen Aufbau,der zugleich eine hoher Sensitivität für die spektroskopische und elektroche-mische Untersuchung aufweist. Aus diesem Grund wird eine verbesserte Va-riante des Aufbaus von [55] verwendet. Wie bereits in Abschnitt 2.4 erwähnt,unterscheidet man potentiostatische von zeitaufgelösten Messungen. Der ex-perimentelle Aufbau ist dabei gleich und muss lediglich für die zeitaufgelöstenMessungen erweitert werden. In dieser Diplomarbeit finden beide MethodenVerwendung. Daher werden beide Aufbauten in den folgenden Abschnittenerläutert. Die für die Messungen verwendete alte Ramanzelle wird beschrie-ben und die neu konstruierte Ramanzelle vorgestellt.
7.1 Messaufbau für die potentiostatischen MessungenDer Messaufbau für die potentiostatischen Messungen ist schematisch in Ab-bildung 7.1 dargestellt. Die Aufnahme der SERRS-Spektren von der Silbero-berfläche erfolgt durch Anregung bei 413 nm mit einem Kryptonlaser. DerStrahlengang wird mit Hilfe eines Systems aus Irisblenden und Spiegeln jus-tiert. Bevor der Laserstrahl in das Spektrometer geführt wird, wird er durcheinen Monochromator geleitet. Dadurch werden störende Plasmalinien desLasers entfernt. Das gestreute Licht wird im 90"-Winkel zur Strahlrichtungauf den Spektrometerspalt (d = 11µm) mit einem Wasser-Immersionsobjektivfokussiert. Durch den Strahlteiler (ST) kann das gestreute Licht entweder aufeine CCD-Kamera geleitet werden, die man zum Fokussieren verwendet, oderauf das mit flüssigem Stickstoff gekühlte Symphony CCD-Detektor Systemgeführt werden.Bevor das gestreute Licht auf den gekühlten CCD-Detektor trifft, kommtes durch zwei Notchfilter (NF) und einen Spektrographen. Jeder Notchfiltersorgt dafür, dass das gestreute elastische Licht (Raleigh-Signal) unterdrücktwird. Der Spektrograph enthält ein optisches Gitter (1800 Linien/mm) mitdessen Hilfe das Licht dispergiert wird. Bei der verwendeten Apparatur be-trägt die spektrale Breite 3,13 cm!1. Die Laserintensität auf der Probenober-fläche stellt man kleiner als 0,2 mW ein. Dies verhindert die Denaturierungder Proteine und vermindert die Photoreduktion. Der so genannte ”Marker-Banden-Bereich” (siehe Abschnitt 2.5.5) ist in dem typischen Messbereich(1100 cm!1 bis 1900 cm!1) integriert.Die Kalibrierung des Spektrometers erfolgt durch die Silizium - Linie bei520,07 cm!1 und durch die mit Weißlicht hervorgerufene Nulllinie.Die SERRS-Zelle ist mit einem Autolab-Potentiostaten verbunden. Dieser

7 MESSAUFBAU FÜR SERRS 108
legt die gewünschte Spannung an die Probe an und wird über einen PC(PC 1) gesteuert. Die Daten, die man mit Hilfe der stickstoffgekühlten CCD-Kamera erhält, werden an einen PC (PC 2) weitergegeben.
Abbildung 7.1: Der Messaufbau für potentiostatische SERRS-Messungen.
7.2 Messaufbau für die zeitaufgelösten MessungenDas Prinzip der zeitaufgelösten Messung ist bereits in Abschnitt 2.4.2 be-schrieben worden. Der erweiterte Messaufbau für dieses Prinzip ist in Abbil-dung 7.2 dargestellt.Das benötigte Rechteckspannungssignal, dass an die Elektrode gelegt werdensoll, wird von einem Funktionsgenerator (Funkt. 1) erzeugt und an den mitder SERRS-Zelle verbundenen Autolab-Potentiostaten weitergegeben. Einzweiter Funktionsgenerator (Funkt. 2) erzeugt ebenfalls ein Rechteckspan-nungssignal.

7 MESSAUFBAU FÜR SERRS 109
Dieses Signal wird zum Aktivieren und Deaktivieren des Laserstrahls an einen”Akustooptischen Modulator” (AOM) gelegt68. Die Signale werden über einOszilloskop (nicht eingezeichnet) getriggert.
Abbildung 7.2: Der erweiterte Messaufbau für zeitaufgelöste SERRS-Messungen.
7.3 Zelldesign der SERRS-ZelleDie Durchführung von zeitaufgelösten spektroelektrochemischen Messungenverlangt ein spezielles Zelldesign. Die Zelle muss dabei mehrere Eigenschaf-ten besitzen:Die Silberelektrode muss sehr schnell gedreht werden können, damit die durchden Laser hervorgerufene Photoreduktion begrenzt werden kann [55]. Fürsauerstofffreie Messungen muss der Sauerstoff aus der Zelle dauerhaft entferntwerden können. Für zeitaufgelöste Messungen ist es nötig, an der Arbeitselek-trode ein hochfrequentes Rechteckspannungssignal anzulegen. Deshalb muss
68Der AOM ist ein optisches Bauelement, mit dem Laserstrahlen manipuliert werdenkönnen. In einfachster Anwendung kann ein AOM in Kombination mit einer Irisblende(IB) als Schalter eingesetzt werden, der sehr schnell zwischen Blockieren und Durchlassenschalten kann [102]. Der AOM ist dabei um mehrere Größenordnungen schneller, als derin [55] verwendete mechanische Schalter.

7 MESSAUFBAU FÜR SERRS 110
ein möglichst rauschfreier Kontakt zum Autolab-Potentiostaten hergestelltwerden.In den folgenden zwei Abschnitten wird der Aufbau der alten Ramanzellebeschrieben und die Verbesserungen der neu konstruierten Ramanzelle vor-gestellt.
7.3.1 Zelldesign der alten Ramanzelle
Als Arbeitselektrode verwendet man eine mit der Oberfläche nach oben ge-richtete, rotierende Scheiben-Elektrode (RDE) [17]. Die Silberelektrode mitTeflonmantel (siehe Abschnitt 6.1) wird - wie in Abbildung 7.3 gezeigt -auf einen Stahlstift geschraubt. Der Stahlstift dreht sich als Achse in Ku-gellagern und wird erschütterungsarm über einen Zahnriemen durch einenMini-Elektromotor angetrieben. Mit diesem Motor kann man den Stahlstiftund damit auch die Silberelektrode mit bis zu 900 RPM rotieren lassen.Ein mit Puffer-Lösung gefüllter Teflon-Zylinder befindet sich zwischen Elek-trode und Stahlstift. Der Zylinder wird durch die aufgeschraubte Elektrodemit dem Stahlstift fixiert.Die Silberelektrode (Arbeitselektrode) wird über einen Schleifkontakt mitdem Autolab-Potentiostaten verbunden (WEout)69. Gegen- und Referenzelek-trode werden zusammen mit dem Immersionsobjektiv und einem Schlauchzum Entfernen des Sauerstoffs in einem Teflondeckel befestigt und mit Gum-miringen (nicht abgebildet) abgedichtet. Der Teflondeckel wird auf die Mess-zelle aufgeschraubt. Das Objektiv wird mit dem Spektrometer verbundenund kann zum Fokussieren in der Höhe verstellt werden.
69Auch in diesem Fall verwendet man - wie in Abschnitt 4 beschrieben - eine 3-Elektrodenanordnung. Hier jedoch mit dem Unterschied, dass die Arbeitselektrode ausSilber und nicht aus Gold besteht.
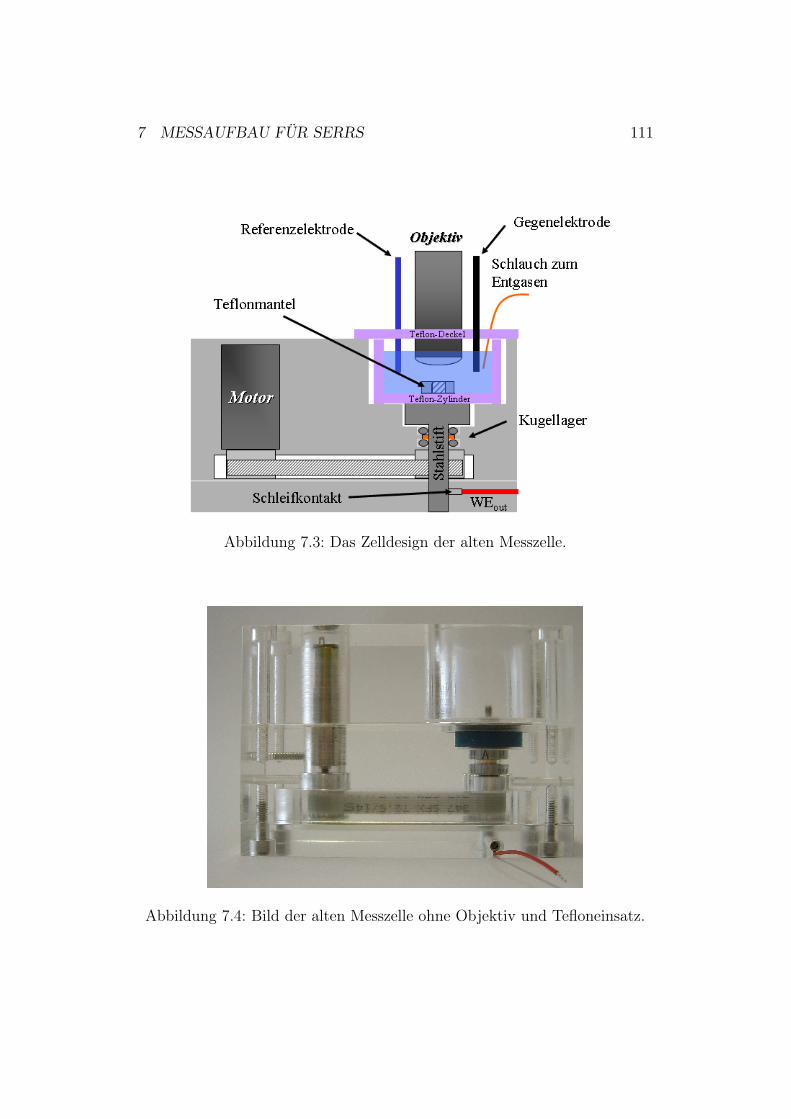
7 MESSAUFBAU FÜR SERRS 111
Abbildung 7.3: Das Zelldesign der alten Messzelle.
Abbildung 7.4: Bild der alten Messzelle ohne Objektiv und Tefloneinsatz.

7 MESSAUFBAU FÜR SERRS 112
7.3.2 Zelldesign der neuen Ramanzelle
Die Konstruktion der alten Messzelle hatte erhebliche Nachteile:
Der Schleifkontakt zeigte ein hohes Untergrundrauschen. Um es zu minimie-ren werden jetzt die rotierende Achse und der Autolab-Potentiostaten übereinen speziell entwickelten Quecksilberkontakt verbunden70.
Es war schwierig die alte Zelle abzudichten. Durch das Rotieren des gesam-ten Teflonzylinders lief die Zelle häufig aus. Dadurch befanden sich Referenz-und Gegenelektrode nicht mehr in der Lösung. Zusätzlich zerstörte der PBS-Puffer die Kugellager. Mit einem nachträglich eingebauten Wellenbrecherkonnte das Auslaufen der Zelle, zwar vermindert, aber nicht völlig verhin-dert werden. Die enormen Strömungen, hervorgerufen durch die Rotationdes kompletten Zylinders, führten dazu, dass sich Gegen- und Referenzelek-trode nicht immer in der Pufferlösung befanden.Aus diesen Gründen rotiert bei dem neuen Zelldesign (Abbildungen 7.5 und7.6) lediglich die Achse mit der aufgeschraubten Silberelektrode. Der Teflon-zylinder (cyan) wird in der Zelle fixiert und dreht sich nicht.71. Das Auslaufender Zelle verhindert eine Gleitringdichtung. Zusätzlich ist ein stärkerer Motorin die Zelle eingebaut72
70Dies geschah in Zusammenarbeit mit Dr. Maarten Van Brussel (Autolab Eco ChemieB.V.).
71Der Teflonzylinder kann jedoch herausgenommen und durch ein Schwefelsäurebad ge-reinigt werden.
72Das Design wurde in Zusammenarbeit mit der Feinmechanik-Werkstatt und der Elek-tronikabteilung des MPIP Mainz erstellt. Eine detaillierte Konstruktionszeichnung ist imAnhang (Abschnitt A.4) zu finden. Die Position des Quecksilberkontaktes, der noch ein-gebaut werden muss, ist angegeben.
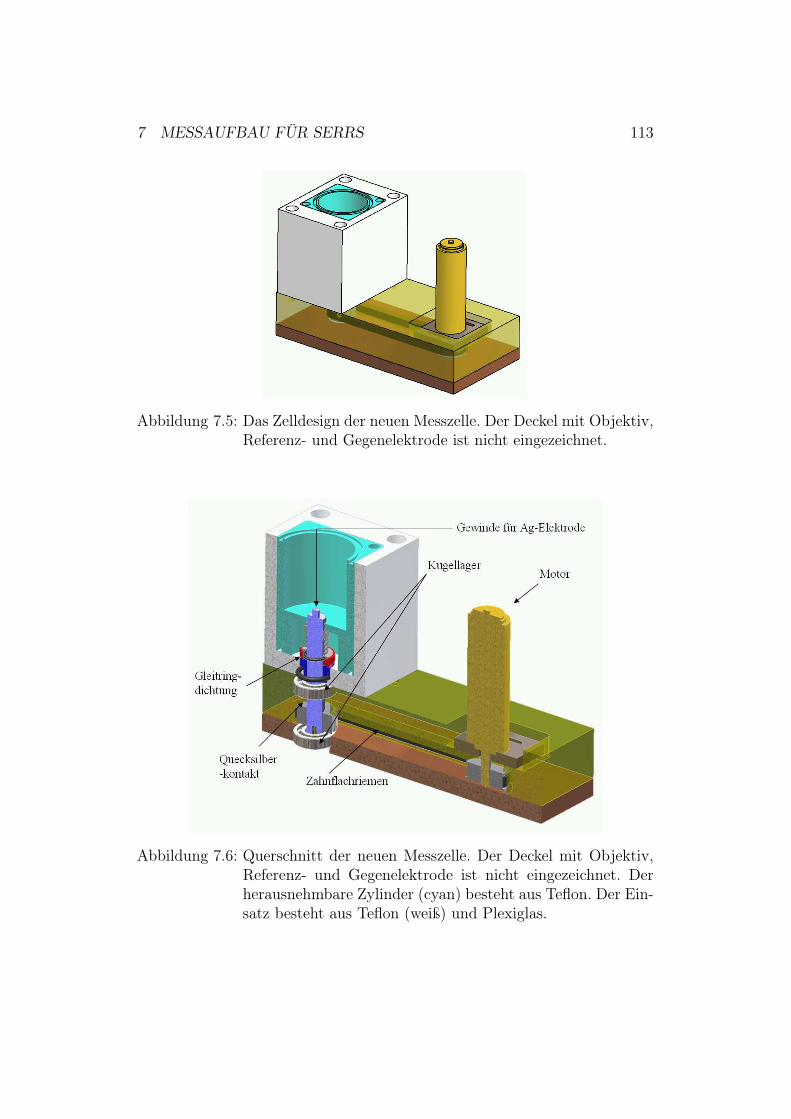
7 MESSAUFBAU FÜR SERRS 113
Abbildung 7.5: Das Zelldesign der neuen Messzelle. Der Deckel mit Objektiv,Referenz- und Gegenelektrode ist nicht eingezeichnet.
Abbildung 7.6: Querschnitt der neuen Messzelle. Der Deckel mit Objektiv,Referenz- und Gegenelektrode ist nicht eingezeichnet. Derherausnehmbare Zylinder (cyan) besteht aus Teflon. Der Ein-satz besteht aus Teflon (weiß) und Plexiglas.
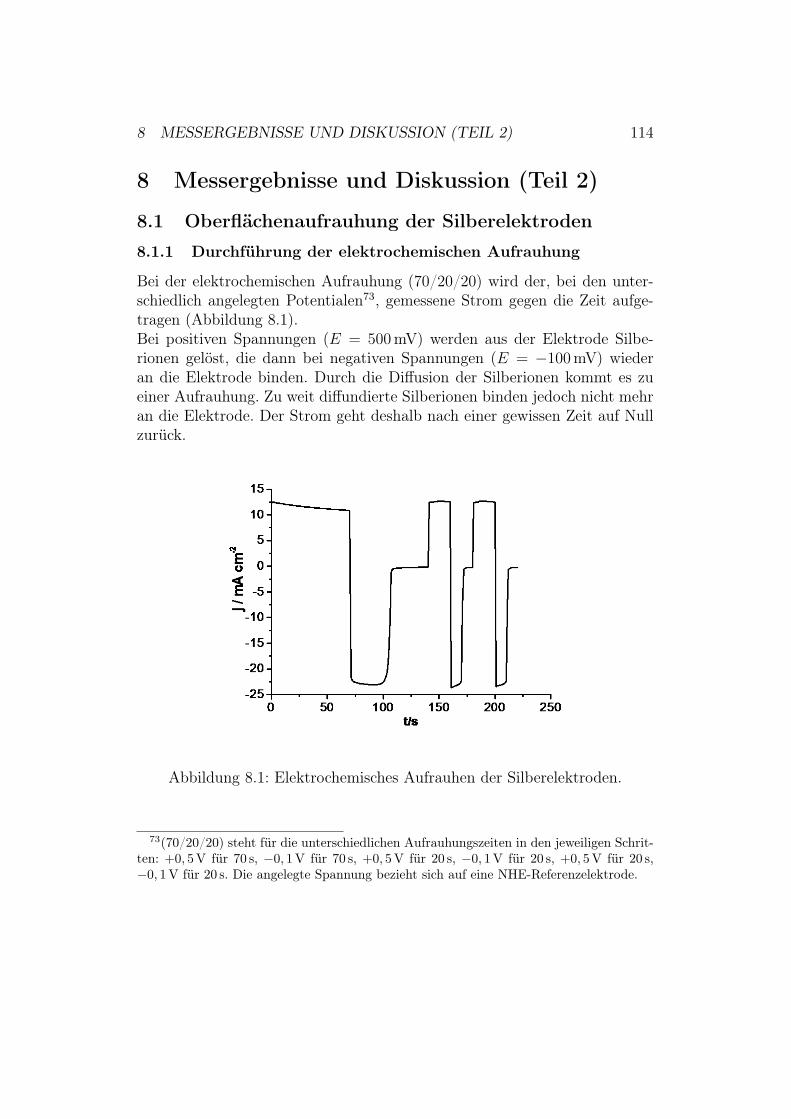
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 114
8 Messergebnisse und Diskussion (Teil 2)
8.1 Oberflächenaufrauhung der Silberelektroden8.1.1 Durchführung der elektrochemischen Aufrauhung
Bei der elektrochemischen Aufrauhung (70/20/20) wird der, bei den unter-schiedlich angelegten Potentialen73, gemessene Strom gegen die Zeit aufge-tragen (Abbildung 8.1).Bei positiven Spannungen (E = 500 mV) werden aus der Elektrode Silbe-rionen gelöst, die dann bei negativen Spannungen (E = !100 mV) wiederan die Elektrode binden. Durch die Diffusion der Silberionen kommt es zueiner Aufrauhung. Zu weit diffundierte Silberionen binden jedoch nicht mehran die Elektrode. Der Strom geht deshalb nach einer gewissen Zeit auf Nullzurück.
Abbildung 8.1: Elektrochemisches Aufrauhen der Silberelektroden.
73(70/20/20) steht für die unterschiedlichen Aufrauhungszeiten in den jeweiligen Schrit-ten: +0, 5 V für 70 s, !0, 1 V für 70 s, +0, 5 V für 20 s, !0, 1 V für 20 s, +0, 5 V für 20 s,!0, 1 V für 20 s. Die angelegte Spannung bezieht sich auf eine NHE-Referenzelektrode.

8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 115
8.1.2 Optimierung der verwendeten Aufrauhungszeiten
Durch den Vergleich der Aufnahmen von verschieden aufgerauhten Silber-elektroden durch das Rasterkraftmikroskop (AFM) soll die Aufrauhungszeitoptimiert werden (Abbildung 8.2). Um weitere Substrukturen auf der aufge-rauhten Oberfläche auszuschließen, werden zudem Bilder mit dem Nanoscope(Abbildung 8.3) aufgenommen74.In Tabelle 8.1 ist die Standardabweichung 0 der jeweiligen Probe für ei-ne 16x16 µm2 -Fläche (Nanoscope-Messung) und für eine 5x5 µm2- Fläche(AFM-Messung) dargestellt. Erwartungsgemäß, nimmt die Oberflächenrau-higkeit mit längeren Aufrauhzeiten zu und führt zu einer Aufrauhung indem gewünschten Größenbereich von mehreren Nanometern. Die AFM- undNanofocus-Bilder lassen erkennen, dass Probe (b) sehr ungleichmäßig aufge-rauht ist. Sind die gebildeten Spitzen - wie bei Probe (d) - zu stark, kannes zu Problemen bei der Anbindung der Proteine und der darauf folgendenMembranbildung kommen. Probe (c) ist am homogensten aufgerauht unddamit am besten geeignet.
Probe 0AFM 0Nanoscope(a) poliert 4,4 nm 11 nm(b) 20/5/5 70 nm 76 nm(c) 70/20/20 80 nm 79 nm(d) 210/60/60 120 nm 117 nm
Tabelle 8.1: Ergebnisse der AFM und Nanoscope-Daten.
74Die AFM und Nanoscope Bilder wurden in Zusammenarbeit mit Helma Burg und An-dreas Best (Arbeitskreis Butt, MPIP-Mainz) angefertigt und ausgewertet. Weitere Bilderbefinden sich im Anhang (A.6).
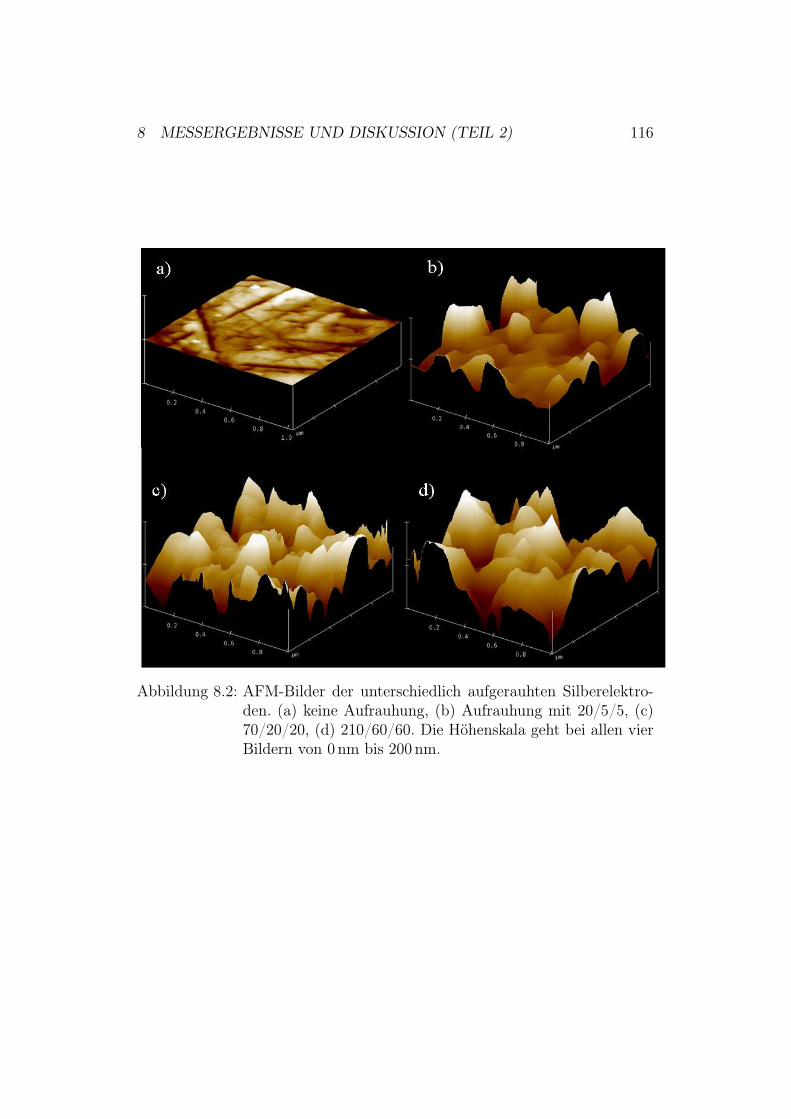
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 116
Abbildung 8.2: AFM-Bilder der unterschiedlich aufgerauhten Silberelektro-den. (a) keine Aufrauhung, (b) Aufrauhung mit 20/5/5, (c)70/20/20, (d) 210/60/60. Die Höhenskala geht bei allen vierBildern von 0 nm bis 200 nm.

8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 117
Abbildung 8.3: Nanoscope-Bilder der unterschiedlich aufgerauhten Sil-berelektroden. (a) keine Aufrauhung, (b) Aufrauhung mit20/5/5, (c) 70/20/20, (d) 210/60/60. Die unterschiedlichenHöhenskalen sind in dem jeweiligen Bild eingezeichnet.

8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 118
8.2 SERRS-MessungenMit cyclovoltammetrischen Messungen lässt sich das Standardpotential vonCytochrom c ermitteln. Das Cytochrom c wird an einer Oberfläche ange-bunden und elektrisch reduziert, bzw. oxidiert. Diese Messungen beinhaltenjedoch keine Angaben über Konformationsänderungen. Durch die oberflä-chenverstärkte resonante Ramanspektroskopie (SERRS) gewinnt man Infor-mationen über die Struktur der Häm-Gruppe und ihrer Umgebung. Dadurchlassen sich Aussagen über den Spin, den Oxidations- und den Konforma-tionszustand des Eisens treffen. Bei der Cytochrom-c-Oxidase ist es sogarmöglich, die beiden vorhandenen Häm-Gruppen voneinander zu unterschei-den (Abschnitt 2.5.5).Alle Spektren werden, wie in den Abschnitten 7.1 und A.1.4 beschrieben,gemessen und ausgewertet. Bei den SERRS-Messungen wird eine Laserinten-sität von circa 0,1 mW auf der Probenoberfläche verwendet. Die Messdauerbeträgt 30 Sekunden.
8.2.1 Ergebnisse und Diskussion der SERRS-Messungen mit Cy-tochrom c
Ergebnisse: Um die elektrochemisch reduzierten und oxidierten Spektrenzu überprüfen, wird jeweils eine Messung mit oxidierter und chemisch redu-zierter Cytochrom c-Lösung durchgeführt. Während der Messungen befindetsich die Cytochrom c-Lösung in der Messzelle. Die Spektren sind in Abbil-dung 8.4 gezeigt.Für die potentiostatischen Messungen wird das an die Silberoberfläche (DTP-modifiziert) angebundene Cytochrom c durch das an die Elektrode angelegtePotential in einen reduzierten, einen oxidierten, oder einen Zwischenzustandgezwungen. Die Spektren werden in einem Potentialbereich von 40 mV bis350 mV aufgenommen (Abbildung 8.5). Zeitaufgelöste Messungen verlangeneine Rückkehr des Systems in den Ausgangszustand (siehe Abschnitt 2.4.2).Deshalb wird eine potentiostatische Rücktitration in einem Potentialbereichvon 350 mV bis -510 mV durchgeführt. Die Ergebnisse sind in Abbildung 8.6dargestellt.
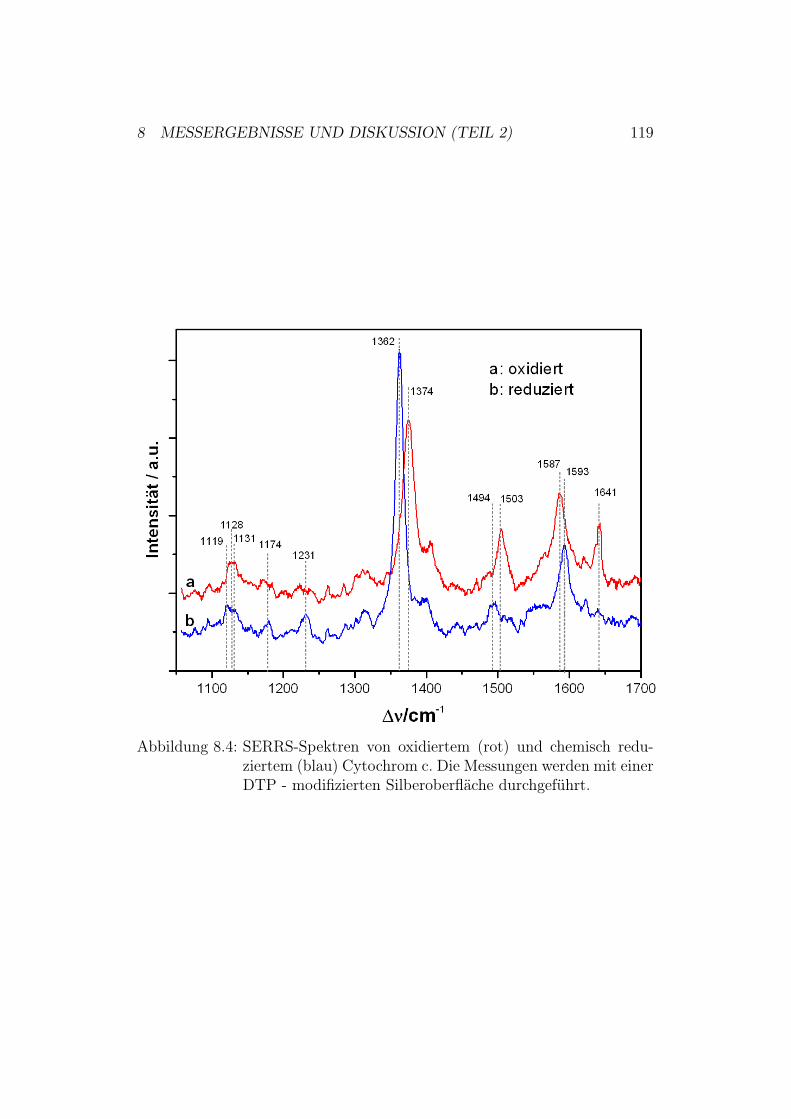
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 119
Abbildung 8.4: SERRS-Spektren von oxidiertem (rot) und chemisch redu-ziertem (blau) Cytochrom c. Die Messungen werden mit einerDTP - modifizierten Silberoberfläche durchgeführt.
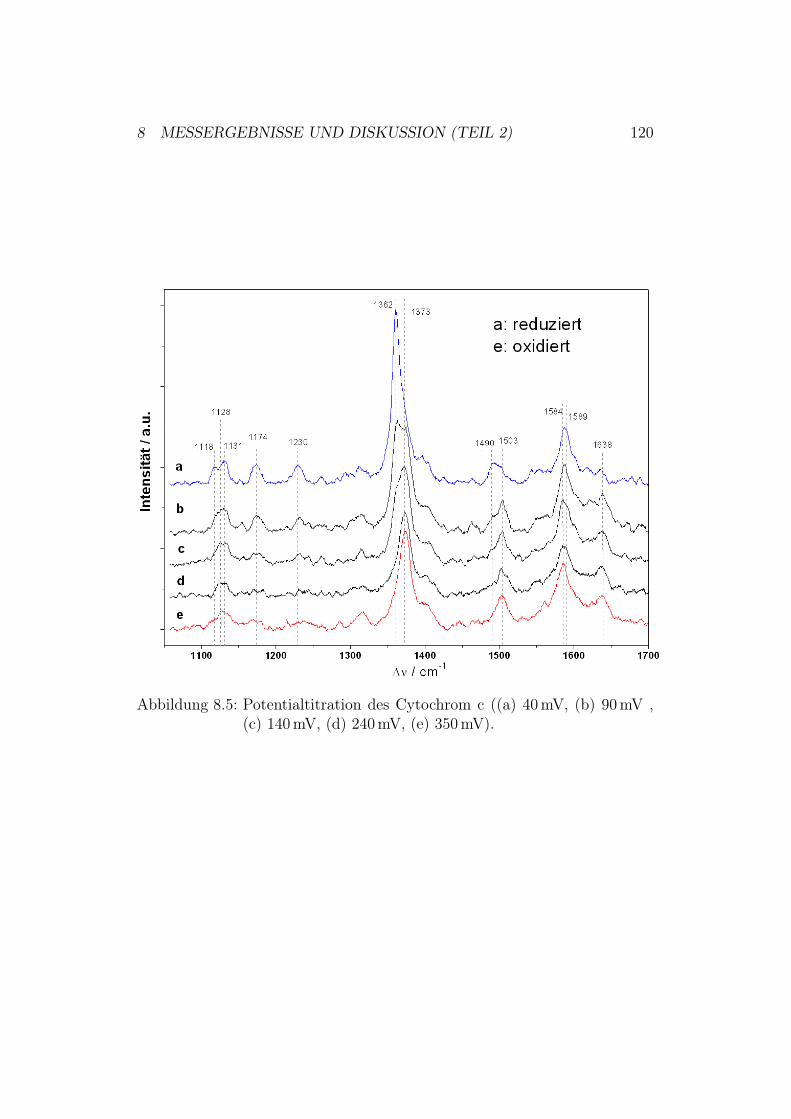
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 120
Abbildung 8.5: Potentialtitration des Cytochrom c ((a) 40 mV, (b) 90 mV ,(c) 140 mV, (d) 240 mV, (e) 350 mV).
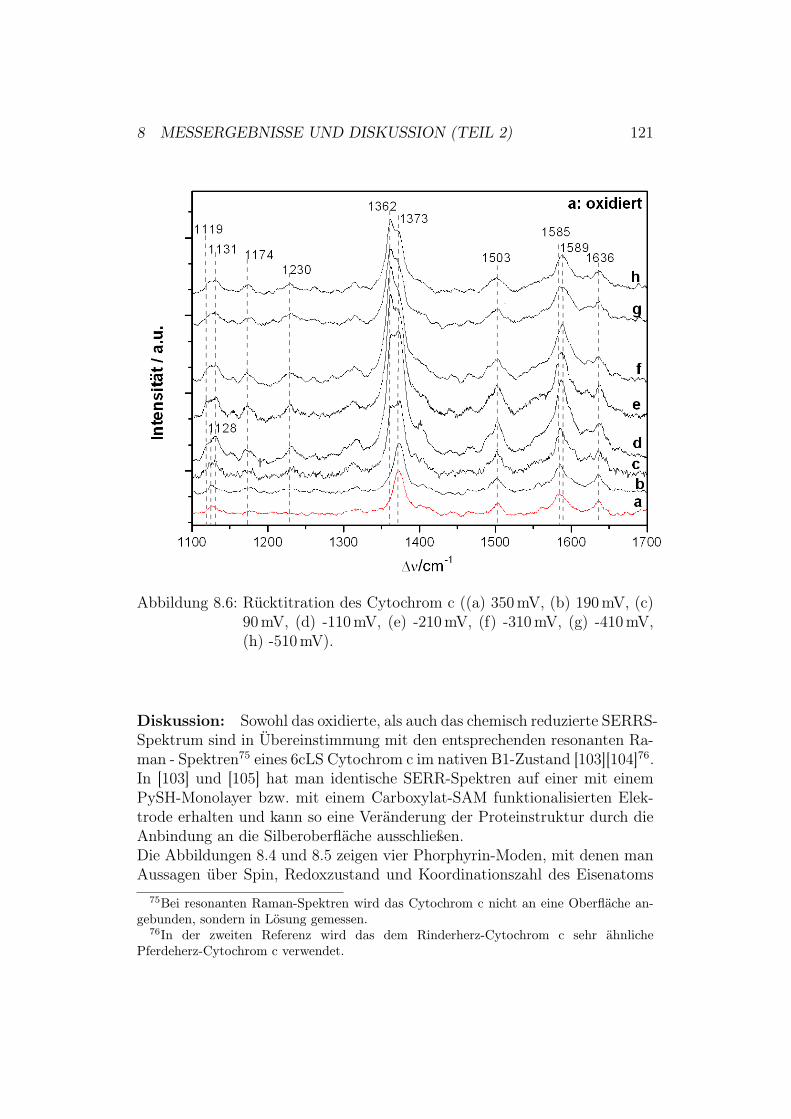
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 121
Abbildung 8.6: Rücktitration des Cytochrom c ((a) 350 mV, (b) 190 mV, (c)90mV, (d) -110 mV, (e) -210 mV, (f) -310 mV, (g) -410 mV,(h) -510 mV).
Diskussion: Sowohl das oxidierte, als auch das chemisch reduzierte SERRS-Spektrum sind in Übereinstimmung mit den entsprechenden resonanten Ra-man - Spektren75 eines 6cLS Cytochrom c im nativen B1-Zustand [103][104]76.In [103] und [105] hat man identische SERR-Spektren auf einer mit einemPySH-Monolayer bzw. mit einem Carboxylat-SAM funktionalisierten Elek-trode erhalten und kann so eine Veränderung der Proteinstruktur durch dieAnbindung an die Silberoberfläche ausschließen.Die Abbildungen 8.4 und 8.5 zeigen vier Phorphyrin-Moden, mit denen manAussagen über Spin, Redoxzustand und Koordinationszahl des Eisenatoms
75Bei resonanten Raman-Spektren wird das Cytochrom c nicht an eine Oberfläche an-gebunden, sondern in Lösung gemessen.
76In der zweiten Referenz wird das dem Rinderherz-Cytochrom c sehr ähnlichePferdeherz-Cytochrom c verwendet.
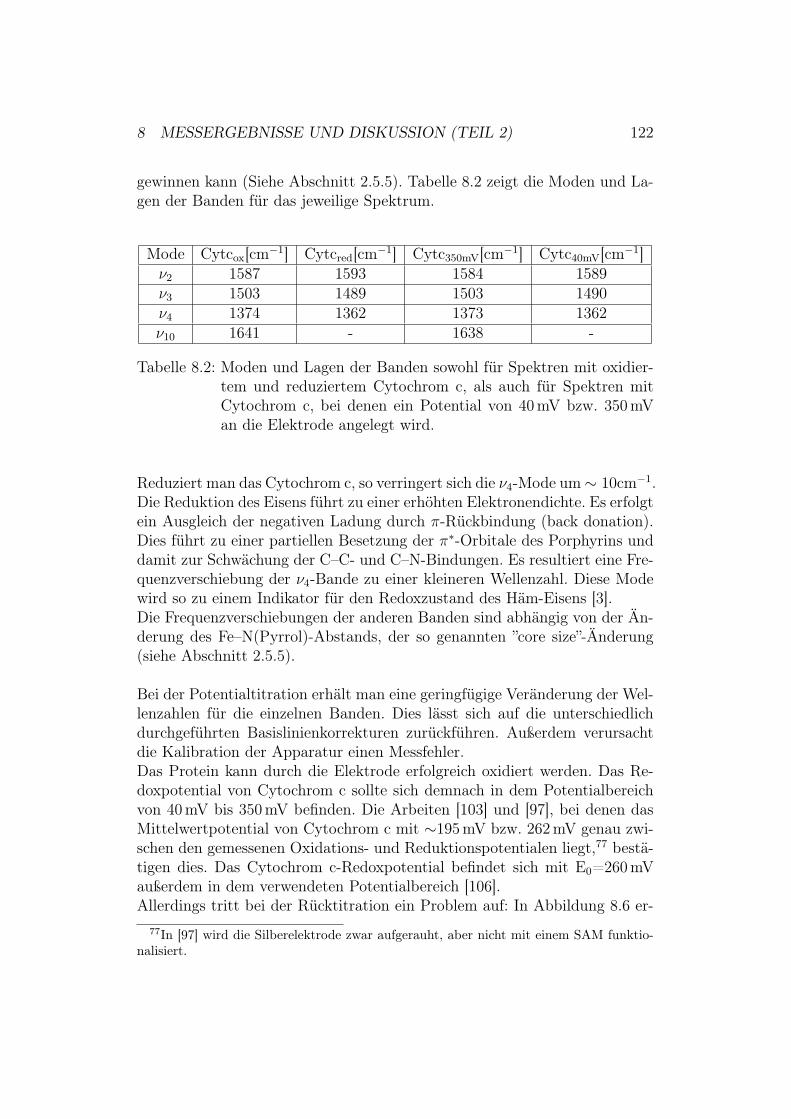
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 122
gewinnen kann (Siehe Abschnitt 2.5.5). Tabelle 8.2 zeigt die Moden und La-gen der Banden für das jeweilige Spektrum.
Mode Cytcox[cm!1] Cytcred[cm!1] Cytc350mV[cm!1] Cytc40mV[cm!1])2 1587 1593 1584 1589)3 1503 1489 1503 1490)4 1374 1362 1373 1362)10 1641 - 1638 -
Tabelle 8.2: Moden und Lagen der Banden sowohl für Spektren mit oxidier-tem und reduziertem Cytochrom c, als auch für Spektren mitCytochrom c, bei denen ein Potential von 40 mV bzw. 350 mVan die Elektrode angelegt wird.
Reduziert man das Cytochrom c, so verringert sich die )4-Mode um& 10cm!1.Die Reduktion des Eisens führt zu einer erhöhten Elektronendichte. Es erfolgtein Ausgleich der negativen Ladung durch "-Rückbindung (back donation).Dies führt zu einer partiellen Besetzung der "$-Orbitale des Porphyrins unddamit zur Schwächung der C–C- und C–N-Bindungen. Es resultiert eine Fre-quenzverschiebung der )4-Bande zu einer kleineren Wellenzahl. Diese Modewird so zu einem Indikator für den Redoxzustand des Häm-Eisens [3].Die Frequenzverschiebungen der anderen Banden sind abhängig von der Än-derung des Fe–N(Pyrrol)-Abstands, der so genannten ”core size”-Änderung(siehe Abschnitt 2.5.5).
Bei der Potentialtitration erhält man eine geringfügige Veränderung der Wel-lenzahlen für die einzelnen Banden. Dies lässt sich auf die unterschiedlichdurchgeführten Basislinienkorrekturen zurückführen. Außerdem verursachtdie Kalibration der Apparatur einen Messfehler.Das Protein kann durch die Elektrode erfolgreich oxidiert werden. Das Re-doxpotential von Cytochrom c sollte sich demnach in dem Potentialbereichvon 40 mV bis 350 mV befinden. Die Arbeiten [103] und [97], bei denen dasMittelwertpotential von Cytochrom c mit &195 mV bzw. 262 mV genau zwi-schen den gemessenen Oxidations- und Reduktionspotentialen liegt,77 bestä-tigen dies. Das Cytochrom c-Redoxpotential befindet sich mit E0=260 mVaußerdem in dem verwendeten Potentialbereich [106].Allerdings tritt bei der Rücktitration ein Problem auf: In Abbildung 8.6 er-
77In [97] wird die Silberelektrode zwar aufgerauht, aber nicht mit einem SAM funktio-nalisiert.

8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 123
kennt man, dass eine vollständige Reduktion durch die Elektrode selbst beiPotentialen von -510 mV nicht möglich ist. Eine denkbare Erklärung ist dieDesorption des Proteinlayers und die damit verbundene Denaturierung desCytochrom c. Das Ablösen des Proteinlayers wurde bereits bei Elektrolytlö-sungen mit hoher Salzkonzentration beobachtet [103]. Nach [94] zeigt Cyto-chrom c nach der Denaturierung einen dramatischen Rückgang des Redox-potentials. Demnach könnten die Proteine während der Messung ihre nativeForm verlieren. Allerdings sollte man für diesen Fall Konformationsänderun-gen im SERRS-Spektrum erkennen. Das Protein befindet sich jedoch immernoch im 6cLS-Zustand.
8.2.2 Ergebnisse und Diskussion der SERRS-Messungen mit derCytochrom-c-Oxidase
Analog zu einer vorausgegangenen Arbeit wird die Cytochrom-c-Oxidase inder down-Konfiguration ohne Sauerstoff untersucht [17]. Dazu wird die Pro-be in die Zelle eingebaut. Das Einleiten von Argongas in die Zelle für (45Minuten) entfernt die Hauptmenge des Sauerstoffs. Als Puffer wird das Sau-erstoffzehrsystem verwendet, das den Restsauerstoff entfernt.
Ergebnisse der potentiostatischen Messung: Für die Potentialtitra-tion wird ein Potentialbereich von -710 mV bis -10 mV gewählt (Abbildung8.7)78. In diesem wird das Enzym oxidiert und reduziert. Dies zeigt ein Ver-gleich mit den cyklovoltammetrischen Messergebnissen aus Abbildung 5.13.Um zu testen, ob sich das System wieder in den Ausgangszustand zurück-bringen lässt, wird - wie in Abbildung 8.9 zu sehen - das Enzym erst oxidiert(rot), dann reduziert (blau) und dann wieder oxidiert (schwarz).
78Die Spektren, die bei -10 und-110mV aufgenommen werden, unterscheiden sich nicht.
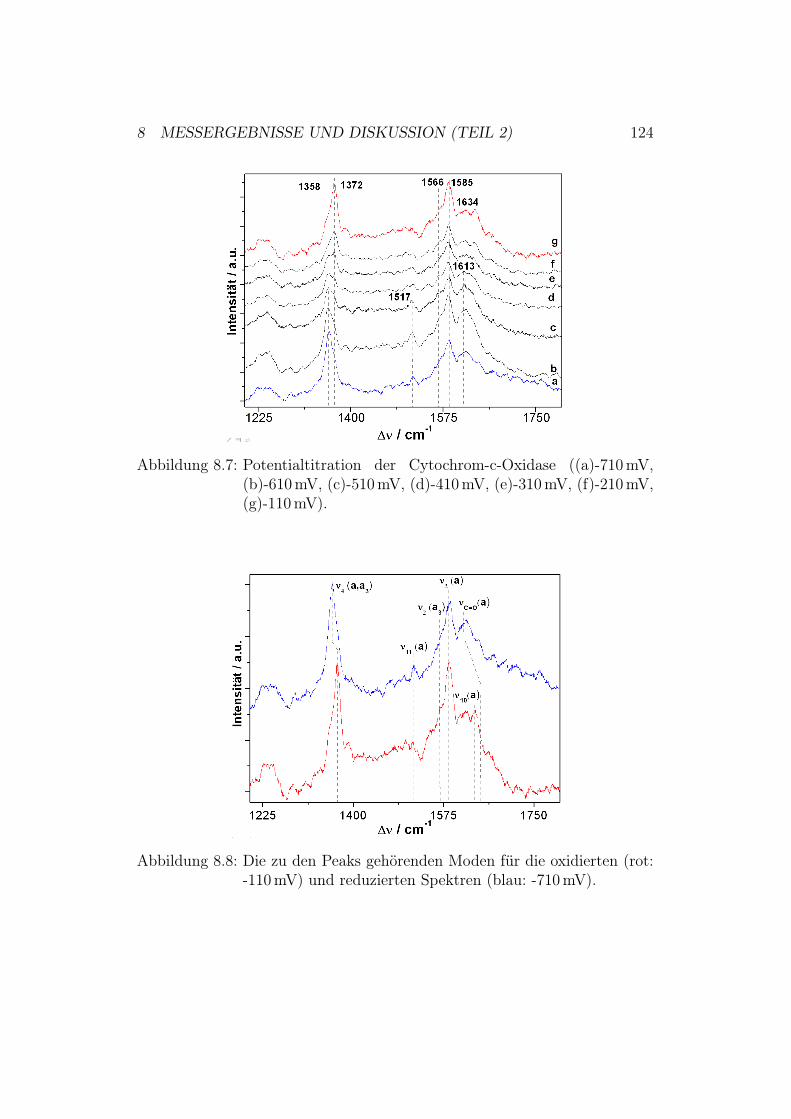
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 124
Abbildung 8.7: Potentialtitration der Cytochrom-c-Oxidase ((a)-710 mV,(b)-610mV, (c)-510 mV, (d)-410 mV, (e)-310 mV, (f)-210 mV,(g)-110 mV).
Abbildung 8.8: Die zu den Peaks gehörenden Moden für die oxidierten (rot:-110 mV) und reduzierten Spektren (blau: -710 mV).
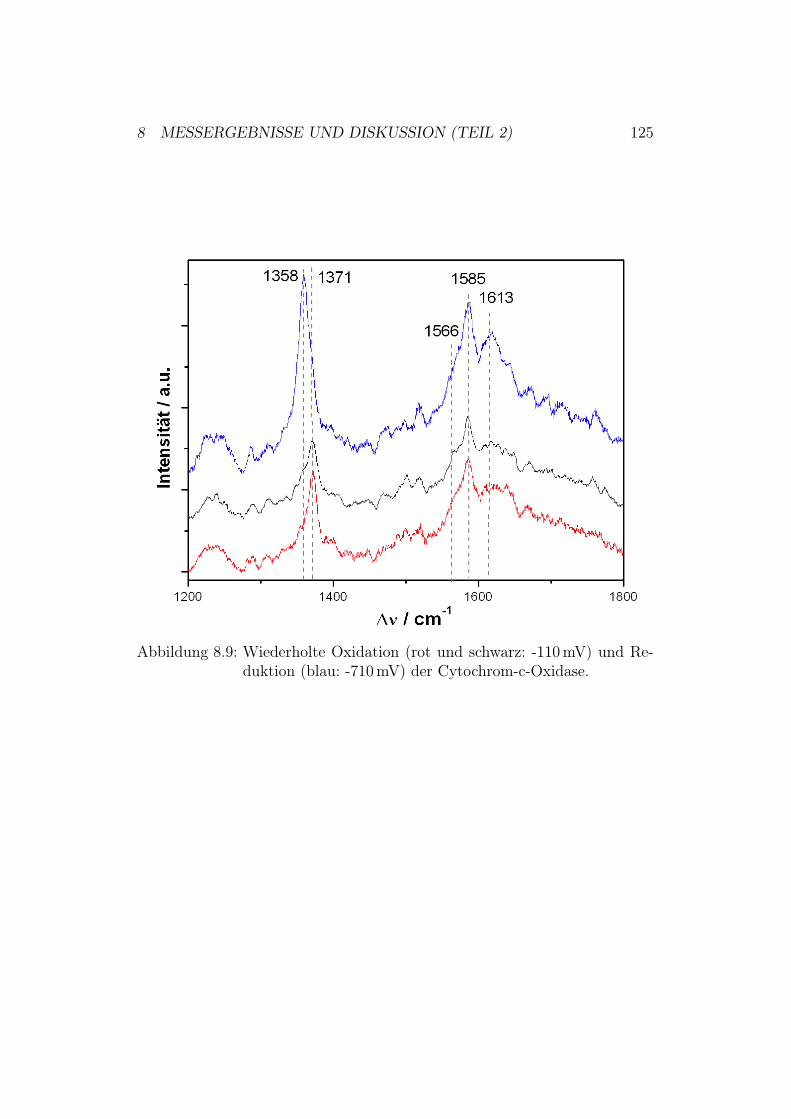
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 125
Abbildung 8.9: Wiederholte Oxidation (rot und schwarz: -110 mV) und Re-duktion (blau: -710 mV) der Cytochrom-c-Oxidase.

8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 126
Ergebnisse der zeitaufgelösten Messung: Für die zeitaufgelöste Mes-sung wird eine Rechteckspannung mit einer Frequenz von 2,5 Hz (= 400 ms)gewählt (Abbildung 8.10). Diese oxidiert (E = !10mV) und reduziert (E =!710mV) das Enzym periodisch. Nach einer Verzögerungszeit , findet ei-ne SERRS-Messung in einem Intervall !t = 20 s statt. Nach dem Mess-vorgang muss das System wieder in den Ausgangszustand gebracht werden,so dass beim nächsten Messvorgang genau dieselben Voraussetzungen vor-handen sind. Man misst also die durchschnittliche Änderung des Systemszwischen t = , und t = , + !t. Die Potentialsprünge werden wiederholt.Aufgrund der durch das Zelldesign bedingten geringen Haltbarkeit der Pro-be ist nur eine Messzeit von 200 s möglich. Die tatsächliche Messzeit beträgtjedoch nur 10 s.Die Wahl dieser Parameter erfordert 20 Messschritte für eine kompletteZeitauflösung.
Um zu testen, ob das gewählte Zeitfenster von 2,5 Hz groß genug ist, werdenzwei Messungen durchgeführt:Bei der ersten Messung wird die Verzögerungszeit , so gewählt, dass sich dasEnzym im oxidierten Zustand befindet (Einstellung a). In diesem Fall wirdvor jeder SERRS-Messung bereits für 180 ms eine Spannung von -10 mV andie Elektrode gelegt.Bei der zweiten Messung wird die Verzögerungszeit , so gewählt, dass sich dasEnzym im reduzierten Zustand befindet (Einstellung b). In diesem Fall wirdvor jeder SERRS-Messung bereits für 180 ms eine Spannung von -710 mV andie Elektrode gelegt.Abbildung 8.11 zeigt die erhaltenen Spektren für die beiden Parameterein-stellungen a (Oxidation) und b (Reduktion).
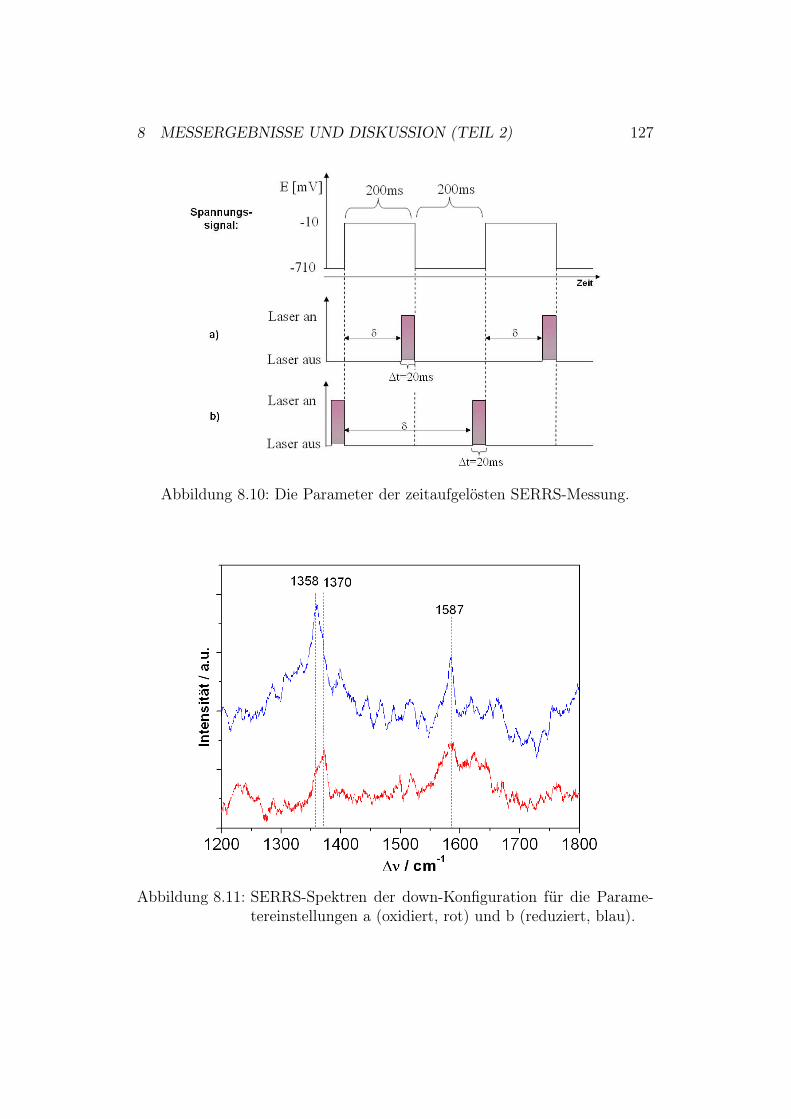
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 127
Abbildung 8.10: Die Parameter der zeitaufgelösten SERRS-Messung.
Abbildung 8.11: SERRS-Spektren der down-Konfiguration für die Parame-tereinstellungen a (oxidiert, rot) und b (reduziert, blau).

8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 128
Diskussion: Die beobachteten Raman-Moden von Häm a und Häm a3 er-lauben die Identifikation von Spin, Oxidations- und Koordinationszustand[107]. Zusätzlich enthalten die Cytochrom-c-Oxidase-Häm-Gruppen vom Typa konjugierte Vinyl- und Formyl-Substituenten. Diese erzeugen resonanz-raman-aktive Streckschwingungen zwischen 1610 cm!1 und 1680 cm!1 [79].In Abschnitt 8.2.1 wird die )4-Bande als ein Indikator für den Redoxzustanddes Häm c-Eisens bezeichnet. Dies trifft jedoch auch auf die Häm-Gruppender Cytochrom-c-Oxidase zu. Wird das Enzym reduziert, resultiert eine Ver-schiebung der )4-Bande zu kleineren Wellenzahlen [107]. Abbildung 8.7 zeigteine solche Verschiebung der )4-Bande von 1372 cm!1 nach 1358 cm!1. Folg-lich lässt sich das Enzym (beide Häm-Gruppen) über die Elektrode reduzieren(blau) und oxidieren (rot).Die SERRS-Spektren stimmen ziemlich gut überein mit den entsprechen-den resonanten Raman-Spektren [107]. Kleine Abweichungen vom resonantenRaman-Spektrum79 sind entweder im Bereich der Messfehler (& 2 cm!1) oderhaben ihren Ursprung im ”elektrochemischen Stark-Effekt” [104]. Der Ver-gleich mit dem resonanten Raman-Spektrum zeigt, dass nicht alle Strukturenaufgelöst werden können. Dazu sind längere Messzeiten, oder das Aufaddie-ren mehrerer Spektren wie in [17] nötig. Das Resonanz-Raman-Spektrumenthält zwei Häm-Gruppen in unterschiedlichen Spin- und Ligandenzustän-den: Einerseits das Häm a (6cLS) und andererseits das Häm a3 (6cHS) [79].
Abbildung 8.7 zeigt ein komplexes Vibrationsspektrum. Aufgrund der un-zureichenden Auflösung lassen sich nur bedingt Aussagen über den Koordi-nationszustand und den Spin machen. In Tabelle 8.3 sind die Moden undLagen der Banden für das oxidierte und reduzierte Spektrum dargestellt.Die hier vorliegenden Spektren zeigen alle eine )2(a)-Mode bei &1585 cm!1.Hierbei handelt es sich um die Low-Spin-Marker Linie des Häm a. Nach [107]befindet sich diese Bande zwischen 1584 cm!1 und 1586 cm!1. Die )2 (a3)-Mode bei &1566 cm!1 ist in den vorliegenden Spektren schwach ausgeprägt.Dies ist die Highspin-Linie des Häm a3, die je nach Redoxzustand zwischen1562 cm!1 und 1569 cm!1 liegt [107].Im oxidierten Zustand erscheint bei 1634 cm!1 die Porphyrins-Mode )10 desHäm a [107]. Dagegen ist die Bande bei )11 = 1515 cm!1 ein eindeutigesMerkmal für eine reduzierte Häm a-Gruppe [79].
79Bei resonanten Raman-Spektren wird das Enzym nicht an eine Oberfläche angebun-den, sondern in Lösung gemessen.
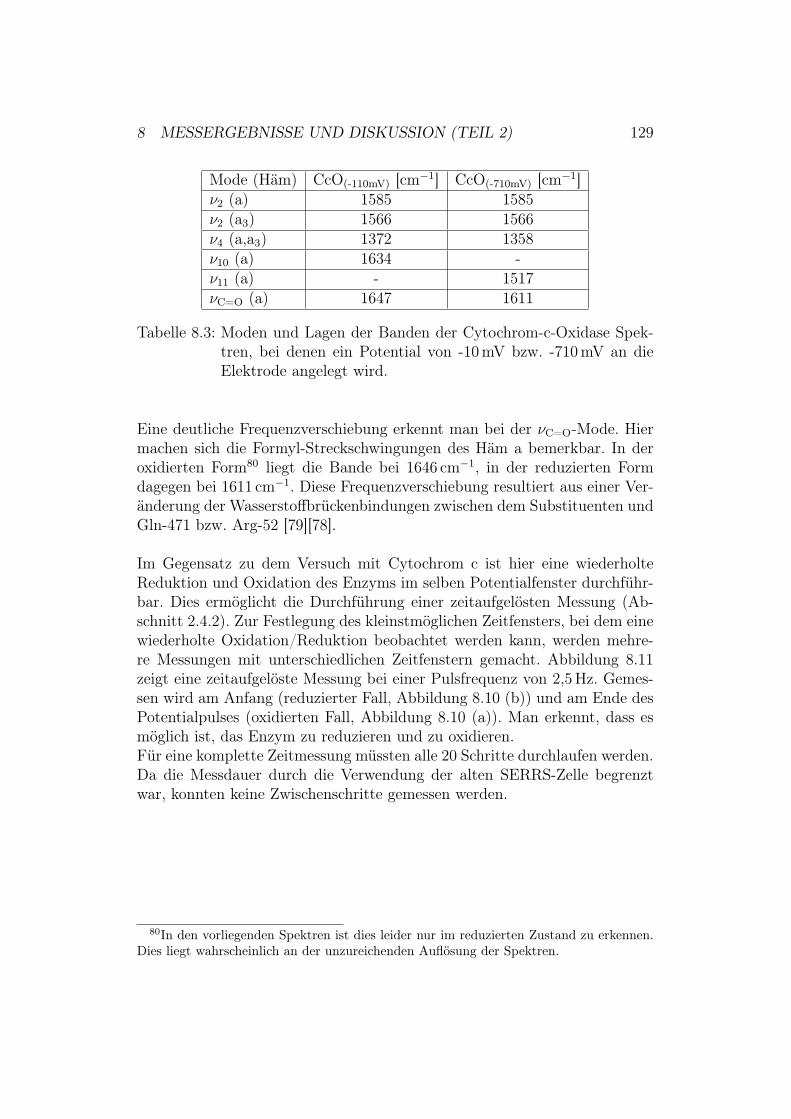
8 MESSERGEBNISSE UND DISKUSSION (TEIL 2) 129
Mode (Häm) CcO(-110mV) [cm!1] CcO(-710mV) [cm!1])2 (a) 1585 1585)2 (a3) 1566 1566)4 (a,a3) 1372 1358)10 (a) 1634 -)11 (a) - 1517)C=O (a) 1647 1611
Tabelle 8.3: Moden und Lagen der Banden der Cytochrom-c-Oxidase Spek-tren, bei denen ein Potential von -10 mV bzw. -710 mV an dieElektrode angelegt wird.
Eine deutliche Frequenzverschiebung erkennt man bei der )C=O-Mode. Hiermachen sich die Formyl-Streckschwingungen des Häm a bemerkbar. In deroxidierten Form80 liegt die Bande bei 1646 cm!1, in der reduzierten Formdagegen bei 1611 cm!1. Diese Frequenzverschiebung resultiert aus einer Ver-änderung der Wasserstoffbrückenbindungen zwischen dem Substituenten undGln-471 bzw. Arg-52 [79][78].
Im Gegensatz zu dem Versuch mit Cytochrom c ist hier eine wiederholteReduktion und Oxidation des Enzyms im selben Potentialfenster durchführ-bar. Dies ermöglicht die Durchführung einer zeitaufgelösten Messung (Ab-schnitt 2.4.2). Zur Festlegung des kleinstmöglichen Zeitfensters, bei dem einewiederholte Oxidation/Reduktion beobachtet werden kann, werden mehre-re Messungen mit unterschiedlichen Zeitfenstern gemacht. Abbildung 8.11zeigt eine zeitaufgelöste Messung bei einer Pulsfrequenz von 2,5 Hz. Gemes-sen wird am Anfang (reduzierter Fall, Abbildung 8.10 (b)) und am Ende desPotentialpulses (oxidierten Fall, Abbildung 8.10 (a)). Man erkennt, dass esmöglich ist, das Enzym zu reduzieren und zu oxidieren.Für eine komplette Zeitmessung müssten alle 20 Schritte durchlaufen werden.Da die Messdauer durch die Verwendung der alten SERRS-Zelle begrenztwar, konnten keine Zwischenschritte gemessen werden.
80In den vorliegenden Spektren ist dies leider nur im reduzierten Zustand zu erkennen.Dies liegt wahrscheinlich an der unzureichenden Auflösung der Spektren.

9 ZUSAMMENFASSUNG UND AUSBLICK 130
9 Zusammenfassung und Ausblick
9.1 ZusammenfassungÜberprüfung des ptBLM-Aufbaus: Bei der Konstruktion eines ptBLM-Systems handelt es sich um eine besonders schwierige und vor allem zeitauf-wendige Probenvorbereitung. Der kleinste Fehler im Aufbau verursacht eineDenaturierung der Proteine und damit den sofortigen Abbruch des Versuchs.Die Aktivität der Enzyme hängt entscheidend von der Qualität der Membranab. Um die Zuverlässigkeit der Messergebnisse zu kontrollieren, ist mit jedemVersuch eine Überprüfung des ptBLM-Aufbaus verknüpft.Die Anbindung der Cytochrom-c-Oxidase (His-Tag an Untereinheit I) aneine DTSP/DTP-Nickelchelat-NTA-modifizierte Goldelektrode ließ sich mitder Oberflächenplasmonenresonanz-Spektroskopie zeitlich auflösen. Mit Hil-fe der Oberflächenplasmonenresonanz-Spektroskopie und der elektrochemi-schen Impedanzspektroskopie gelang der Nachweis einer ptBLM-System -Bildung. Bei der Verwendung der Cytochrom-c-Oxidase mit His-Tag an Un-tereinheit II (Wild-Type und Mutante N139C) erhielt man äquivalente Er-gebnisse, so dass der korrekte ptBLM-Aufbau für alle drei Protein-Typenerfolgreich überprüft werden konnte. Außerdem konnte man in allen Fällenvon einer sehr hohen Protein-Oberflächenbedeckung ausgehen.
Aktivierung der Cytochrom-c-Oxidase mit His-Tag an Unterein-heit I: Die katalytische Aktivität der Cytochrom-c-Oxidase mit His-Tagan Untereinheit I wurde sowohl mit reduziertem, als auch mit oxidiertemCytochrom c effektiv ”in-situ” beobachtet. Dabei handelte es sich jeweilsum einen diffusionskontrollierten Prozess. Es konnte gezeigt werden, dasses bei dieser Konfiguration zu einem Elektronenaustausch zwischen Enzymund Elektrode kommt. Dieser Elektronentransfer trat jedoch nur in Anwe-senheit von Cytochrom c auf. Die Anbindung von Cytochrom c führt vermut-lich zu einer Konformationsänderung. Dadurch kann womöglich ein Öffnendes Elektronendurchgangsweges initialisiert werden. Der Elektronentransferscheint dabei über das Häm a3 stattzufinden [96]. Diese Vermutung bestätigtsich durch resonante Raman-Untersuchungen. Hierbei zeigt sich, nach demAnbinden von Cytochrom c an die Cytochrom-c-Oxidase, eine Strukturver-änderung in der Umgebung von Häm a und Häm a3 [108]. Außerdem ist daskatalytische Zentrum (Häm a3/CuB) das Redox-Centrum, dass sich im Fallder up-Konfiguration der Elektrode am nächsten befindet. Der Mechanismusdieses Prozesses ist jedoch bisher noch nicht verstanden.Die Umsatzrate und das Standardpotential des ptBLM-Systems konnten inKombination mit der up-Konfiguration in Anwesenheit von reduziertem Cy-

9 ZUSAMMENFASSUNG UND AUSBLICK 131
tochrom c berechnet werden. Die Ergebnisse führen zu dem Schluss, dass sichdas Enzym ohne Funktionsverlust in das ptBLM-System einfügen lässt.Die EIS-Potentialtitration zeigt, dass man bei der Interpretation der Ergeb-nisse folgende Effekte berücksichtigen muss:
• Der von den Konzentrationsunterschieden und Potentialdifferenzen zwi-schen Submembranraum und Membranaußenseite abhängige passiveIonentransport.
• Der durch die enzymatischen Redoxprozesse hervorgerufene aktive Pro-tonentransport.
• Die faradaischen Ströme, die ihren Ursprung in der Oxidation und Re-duktion des Cytochrom c haben.
Dadurch erkennt man die Komplexität des verwendeten Systems. Den passi-ven Ionentransport kann man durch Messungen ohne Cytochrom c von denanderen Effekten trennen. Bei der Aktivierung mit Cytochrom c treten diesedrei Effekte gleichzeitig auf.
Mutante N139C: Die cyclovoltammetrischen-Messungen zeigen, dass dieMutante N139C bei einer Spannungsvorschubgeschwindigkeit von 100 mV/skeine Protonen pumpt. Es besteht ein deutlicher Unterschied zwischen denCyclovoltammogrammen von Mutante und Wild-Type. Es ist erstaunlich,dass der Austausch von nur einer einzigen Aminosäure derart drastische Fol-gen für die Funktion eines Enzyms hat.
SERRS- und TR-SERRS Messungen mit Cytochrom c und derCytochrom-c-Oxidase mit His-Tag an Untereinheit II: SERRS -Messungen zeigten eine Absorption des Cytochrom c auf einer aufgerauh-ten funktionalisierten Silberoberfläche. Durch den Vergleich mit ResonanzRaman-Spektren und Cyklovoltammogrammen wurde festgestellt, dass dasProtein bei der Adsorption sowohl seine Form als auch seine Funktion beibe-hält. Das Protein ließ sich durch eine Veränderung des an der Elektrode an-gelegten Potentials oxidieren. Eine Rückreduktion des Proteins gelang selbstbei stark negativen Potentialen nicht.Zeitaufgelöste Messungen waren aufgrund dieser Irreversibilität nicht durch-führbar. Die Cytochrom-c-Oxidase mit His-Tag an Untereinheit II (down -Konfiguration) ließ sich unter Ausschluss von Sauerstoff über die Elektrodereduzieren und oxidieren, wie die Cyclovoltammetrie und die oberflächenver-stärkte resonante Ramanspektroskopie bestätigte. Dieser Vorgang ließ sichzudem wiederholt durchführen.

9 ZUSAMMENFASSUNG UND AUSBLICK 132
In dieser Arbeit wurde gezeigt, dass für eine zeitaufgelöste SERRS-Messungdas Zeitfenster kleiner als 400ms gewählt werden kann. Dazu waren sehrviele Messungen nötig, da die Lebensdauer des Enzyms aufgrund der altenSERRS-Zelle sehr begrenzt war. Dennoch konnte der Ansatz einer Zeitauf-lösung nachgewiesen werden. Die Messdauer betrug 200 Sekunden. In dieserZeit wurde das Enzym 500 mal hintereinander reduziert und wieder reoxi-diert. Die effektive Messzeit betrug dabei jedoch nur 10 Sekunden.Die Auflösung war deshalb sehr viel schlechter als bei der Potentialtitrati-on. Längere Messzeiten oder eine Erhöhung der Laserleistung waren jedochaufgrund der zur Verfügung stehenden Zelle und der auftretenden Photore-duktion nicht möglich. Ein Defekt des Ramanspektrometers machte in derzweiten Hälfte der Diplomarbeit SERRS-Messungen unmöglich. Aus diesemGrund wurde besonderer Wert auf eine Optimierung des Raman-Messsystemsgelegt:Die Ausnutzung des oberflächenverstärkten Ramaneffekts erforderte eine gu-te Qualität der aufgerauhten Silberoberfläche. Im Verlauf der Arbeit wurdedie Technik zum Polieren der Proben geändert. Um das Signal - Rausch-verhältnis der Messungen zu optimieren wurden die Proben nicht mehr vonHand, sondern mit Hilfe einer Maschine und einer im Rahmen der Diplomar-beit speziell entwickelten Vorschrift poliert. Eine erhöhte Stabilität der Mem-bran und damit eine längere Haltbarkeit der Proteine war das Ziel81. Die fürdie Probenpräparation verwendete Poliermethode und die Aufrauhzeit wur-den durch eine Analyse von unterschiedlich aufgerauhten Oberflächen mittelsAFM und Nanoscope optimiert.Mit der im Rahmen der Diplomarbeit neukonstruierten Messzelle erwartetman eine wesentliche Verlängerung der Messzeit und ein deutlich verbesser-tes elektrisches Signal-Rausch-Verhältnis. Gerade der letztgenannte Punktist hierbei von besonderer Bedeutung: Bei einer effektiven Messzeit von 10Sekunden wird das Enzym mit hohen Geschwindigkeiten rotiert und 500 malhintereinander reduziert und wieder reoxidiert. Der Quecksilberkontakt in derneu konstruierten Zelle eignet sich ideal für solche extremen Bedingungen.
9.2 AusblickZeitaufgelöste SERRS- Messungen mit Cytochrom c sind aufgrund der Irre-versibilität in den gemessenen Spektren nicht durchführbar. Nach [103] istes jedoch möglich bei geringen Salzkonzentrationen im verwendeten Pufferein reversibles Cytochrom c-SERRS-Spektrum zu erhalten. Damit würde der
81Die handpolierte Probe enthielt Kratzer im µm-Bereich, die eine Ausbildung der Mem-bran erschwerten (siehe A.6.1).

9 ZUSAMMENFASSUNG UND AUSBLICK 133
zeitaufgelösten Messung nichts mehr im Wege stehen.Im Fall der up-Konfiguration konnte ein exponentieller Abfall bei hohen Scan-rates mit reduziertem Cytochrom c festgestellt werden. Man ist gespannt, obsich mit oxidiertem Cytochrom c ein ähnliches Verhalten bei höheren Scan-rates erkennen lässt. Die Ergebnisse der EIS-Potentialtitration konnten nurqualitativ diskutiert werden. Zudem ist der Mechanismus des katalytischenProzesses bisher noch nicht verstanden. Messungen mit Hilfe der Raman-spektroskopie, die sehr sensitiv auf Strukturveränderungen reagiert, könntenin Kombination mit elektrochemischen Messungen zu einer besseren Einsichtin diese komplexen Prozesse führen.Die Kinetik des Elektronentransfers lässt sich durch TR-SERRS Messun-gen mit der Cytochrom-c-Oxidase mit His-Tag an Untereinheit II (down-Konfiguration) besser verstehen. Nach zeitaufgelösten Messungen ohne Sau-erstoff interessieren auch zeitaufgelöste Messungen mit Spuren von Sauer-stoff. Das Enzym ist dann katalytisch aktiv. Um die Auflösung der TR-SERRS-Messung zu erhöhen wird man versuchen, das Zeitfenster von 400 mszu minimieren, was aber wiederum eine Signaloptimierung erfordert.Die Erforschung des Protonendurchgangsweg erfordert intensive Scanratestu-dien der mutierten Cytochrom-c-Oxidase N139C sowohl mit, als auch ohneSauerstoff. Erkenntnisse über den Protonenpump-Mechanismus gewinnt manauch durch Untersuchungen anderer Mutanten[85].Die RDE-Voltammetrie liefert weitere Informationen über die Kinetik desKatalyseprozesses [109]. Geplant sind Messungen mit einer rotierenden Schei-ben-Elektrode (RDE: rotating disc electode). Die Arbeitselektrode rotiertmitsamt dem ptBLM-System bei genau definierten Umdrehungszeiten. Indieser Arbeit wurde ein neuer Messaufbau für eine geeignete Autolab-RDEerstellt (siehe Anhang A.5).Die Funktionsweisen anderer Membranproteine - wie zum Beispiel des Re-aktionszentrums oder des bc1-Komplexes-, lassen sich mit Hilfe des ptBLM-Systems genauer studieren. Das Reaktionszentrum und der bc1-Komplex sol-len in ein ptBLM-System eingefügt und mit elektrochemischen Methodenuntersucht werden. Dadurch wird die Neugierde geweckt, in einem nächs-ten Schritt zu erkunden, ob sich die SERRS-Spektren von den RR-Spektren[110][111][112][113] nicht unterscheiden.

A ANHANG 134
A Anhang
A.1 Datenmessung und DatenauswertungAlle Datensätze werden nach der jeweiligen Bearbeitung/Auswertung insProgramm Origin importiert und dargestellt.Bei allen Versuchen wird für die Potentialmessung eine Ag/AgCl,KClkonz.-Referenzelektrode verwendet. Um die Spektren mit der Literatur verglei-chen zu können wird das Standardpotential der Normal-Wasserstoffelektrode(NHE) verwendet. Aus diesem Grund werden die Spektren um 200 mV insPositive verschoben [87].
A.1.1 Messung und Auswertung der SPR-Daten
Die Daten werden mit Hilfe eines speziell für diesen Aufbau entwickelten Pro-gramms (Wasplas) aufgenommen. Die Winkelbereiche für Spektren-Messungan Luft und in Flüssigkeit sind in Abbildung A.1 wiedergegeben. Hierbei soll-ten in den beiden wichtigen Bereichen (Totalreflektion und Plasmon) mög-lichst viele Messpunkte aufgenommen werden. Für die Kinetiken wurde einWinkel von 54,5" (siehe Abschnitt 2.1.3) gewählt. Für Spektrenmessungenwährend einer laufenden Kinetik wurde die Kinetikmessung kurz unterbro-chen.Das Programms Waspview ermöglicht eine gute Darstellung der Daten.

A ANHANG 135
Abbildung A.1: Gemessene Winkelbereiche der SPR-Spektren. Links: anLuft. Rechts: in Lösung.
A.1.2 Messung und Auswertung der EIS-Daten
Die Daten werden mit der Software FRA (Eco Chemie, B.V.) aufgenom-men. Das Programm normiert dabei die Daten automatisch auf eine Flächevon 1 cm2. Für alle Impedanzspektren wird ein Messintervall von 50 kHz bis3mHz gewählt. Die Daten werden danach mit Hilfe der Software ZView (Ver-sion 2.6) ausgewertet. Dafür wird ein komplexer nichtlinearer Fitalgorithmusfür die jeweiligen Schaltkreismodelle verwendet. Abbildung A.2 zeigt einenmit diesem Algorithmus gefitteten Datensatz. Hierbei wird mit dem in derAbbildung dargestellten Schaltkreis gefittet (Fitergebnis:rot). Der zum Fit-ten der Daten verwendete Schaltkreis und die Fitergebnisse sind in Abbil-dung A.2 dargestellt. Die Ergebnisse weichen kaum von den in Abschnitt2.2.5 angegebenen Werten für einen idealen ptBLM ab. Der Grund für dieVerwendung eines CPE-Elements sind Inhomogenitäten. Bei dem in der Di-
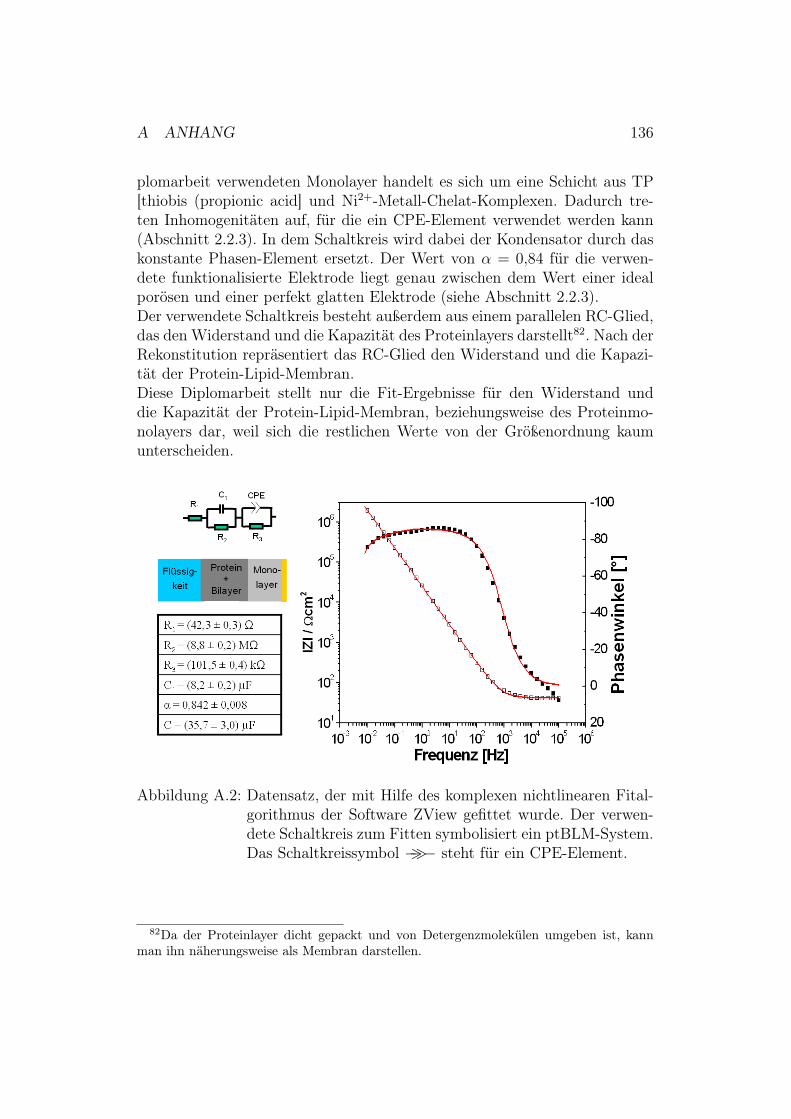
A ANHANG 136
plomarbeit verwendeten Monolayer handelt es sich um eine Schicht aus TP[thiobis (propionic acid] und Ni2+-Metall-Chelat-Komplexen. Dadurch tre-ten Inhomogenitäten auf, für die ein CPE-Element verwendet werden kann(Abschnitt 2.2.3). In dem Schaltkreis wird dabei der Kondensator durch daskonstante Phasen-Element ersetzt. Der Wert von * = 0,84 für die verwen-dete funktionalisierte Elektrode liegt genau zwischen dem Wert einer idealporösen und einer perfekt glatten Elektrode (siehe Abschnitt 2.2.3).Der verwendete Schaltkreis besteht außerdem aus einem parallelen RC-Glied,das den Widerstand und die Kapazität des Proteinlayers darstellt82. Nach derRekonstitution repräsentiert das RC-Glied den Widerstand und die Kapazi-tät der Protein-Lipid-Membran.Diese Diplomarbeit stellt nur die Fit-Ergebnisse für den Widerstand unddie Kapazität der Protein-Lipid-Membran, beziehungsweise des Proteinmo-nolayers dar, weil sich die restlichen Werte von der Größenordnung kaumunterscheiden.
Abbildung A.2: Datensatz, der mit Hilfe des komplexen nichtlinearen Fital-gorithmus der Software ZView gefittet wurde. Der verwen-dete Schaltkreis zum Fitten symbolisiert ein ptBLM-System.Das Schaltkreissymbol !!+! steht für ein CPE-Element.
82Da der Proteinlayer dicht gepackt und von Detergenzmolekülen umgeben ist, kannman ihn näherungsweise als Membran darstellen.

A ANHANG 137
A.1.3 Messung und Auswertung der CV-Daten
In diesem Fall werden die Daten mit Hilfe der beim Autolab-Potentiostatenmitgelieferten Software (Gpes von Eco Chemie, B.V.) ermittelt. Das Pro-gramm normiert dabei die Daten automatisch auf eine Fläche von 1 cm2.Falls möglich wird die in Abschnitt 2.3.3 erwähnte elektronische IR-Drop-Kompensation durchgeführt83.Die Daten werden mit Hilfe der Gpes-Software ausgewertet. Für die Basis-linien-Korrektur des oberen und unteren Kurvenverlaufs verwendet das Pro-gramm eine polynome Funktion (Abbildung A.3). Dadurch ist es möglich,die kapazitive Komponente des Stroms zu subtrahieren (Abbildung A.4).Auf Grund der Potentialauftragung gegen die Normal-Wasserstoffelektrodewerden die Spektren um 200 mV ins Positive verschoben.Durch einen Export des Datensatzes in das Programm Origin ist es möglichmit Hilfe eines Gauss-Fits die Höhe und die Lage der Peaks zu bestimmen.Die Fitergebnisse sind in Abbildung A.4 integriert:
Peak Höhe [µA] Lage [mV]1 26,62 ± 0,07 462,28 ± 0,342 !41,74 ± 0,17 387,94 ± 0,39
Tabelle A.1: Lagen und Höhen der Peaks für das gewählte Beispiel
83Bei der elektronischen Kompensation kann es bei kleinen Scanrates zu zusätzlichenOszillationen kommen. Da der IR-Drop-E!ekt erst bei hohen Scanrates eine wichtige Rollespielt, wurde die Kompensation bei Scanrates ab 1-5 V/s eingeschaltet.
A ANHANG 138
Abbildung A.3: Beispiel einer Basislinien-Korrektur mit Hilfe der Gpes -Software.
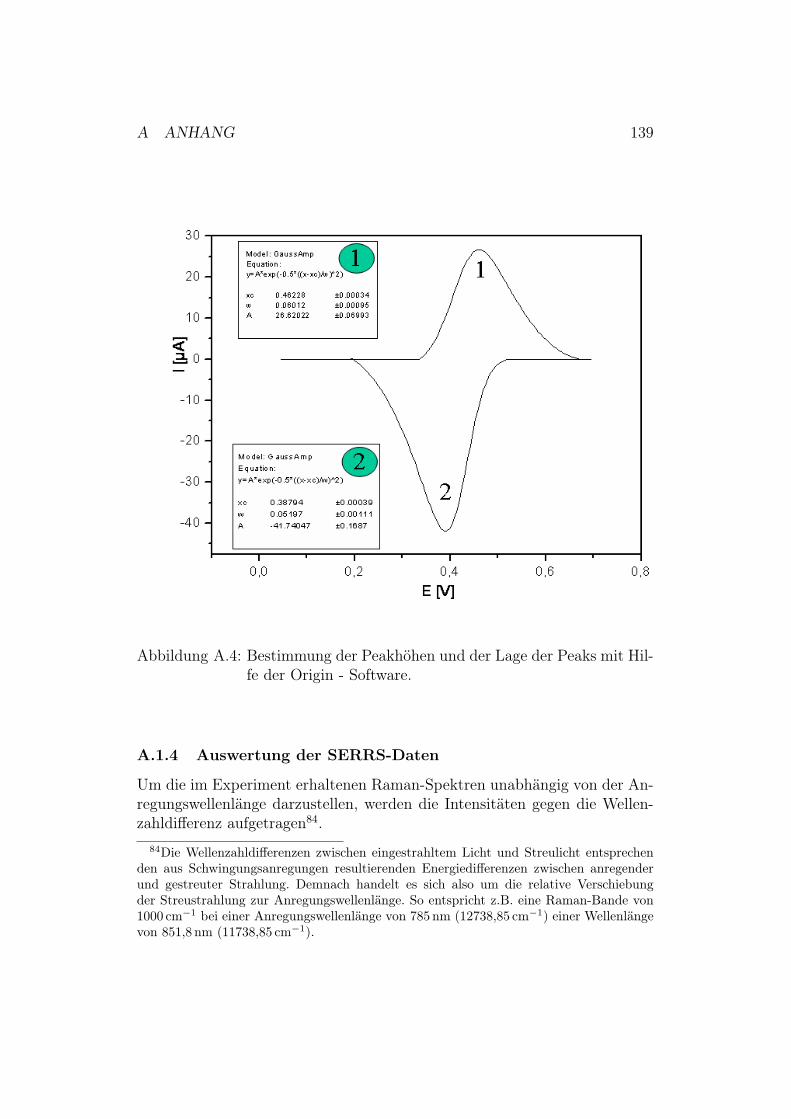
A ANHANG 139
Abbildung A.4: Bestimmung der Peakhöhen und der Lage der Peaks mit Hil-fe der Origin - Software.
A.1.4 Auswertung der SERRS-Daten
Um die im Experiment erhaltenen Raman-Spektren unabhängig von der An-regungswellenlänge darzustellen, werden die Intensitäten gegen die Wellen-zahldifferenz aufgetragen84.
84Die Wellenzahldi!erenzen zwischen eingestrahltem Licht und Streulicht entsprechenden aus Schwingungsanregungen resultierenden Energiedi!erenzen zwischen anregenderund gestreuter Strahlung. Demnach handelt es sich also um die relative Verschiebungder Streustrahlung zur Anregungswellenlänge. So entspricht z.B. eine Raman-Bande von1000 cm!1 bei einer Anregungswellenlänge von 785 nm (12738,85 cm!1) einer Wellenlängevon 851,8 nm (11738,85 cm!1).

A ANHANG 140
Die Daten werden mit Hilfe der bei dem Spektrometer mitgelieferten Labspec-Software erst vorbehandelt und dann ausgewertet:Die Vorbehandlung von Spektren wird definiert als jedwede mathematischeManipulation von Daten vor der eigentlichen Analyse. Sie wird dazu verwen-det, irrelevante, zufällige oder systematische Variationsquellen zu reduzierenoder komplett zu entfernen [114]. Allerdings werden bei der Vorbehandlungdie spektralen Informationen verändert, was entweder positive oder negati-ve Einflüsse auf das Ergebnis haben kann. Deshalb muss immer abgewogenwerden, ob die positiven oder negativen Einflüsse bei der Vorbehandlung vonSpektren überwiegen [114].Die in dieser Diplomarbeit verwendeten Vorbehandlungen sind:
• Basislinienkorrektur: Für die Basislinienkorrektur gibt es mehrereMethoden. Die einfachste Möglichkeit ist, einen konstanten Offset vomSpektrum abzuziehen. Eine weitere Möglichkeit besteht in der Subtrak-tion einer Geraden. Diese Methode wird in dieser Arbeit verwendet undist in Abbildung A.5 (b) dargestellt.
• Entfernen von Spikes: Da geladene Teilchen (z.B.: Myonen) ein Si-gnal in der CCD auslösen können, werden die Spektren zusätzlich von”Spikes” (Spitzen) befreit (Abbildung A.5 (c)).
• Glätten der Spektren: Glättungsmethoden werden zur Reduktiondes willkürliche auftretenden Rauschens verwendet, mit dem Ziel dasSignal/Rausch-Verhältnis zu verbessern. Die grundlegende Annahmedabei ist, dass das Rauschen eine höhere Frequenz besitzt als das be-obachtete Signal. Es gibt mehrere Verfahren zum Glätten von Spektren.Ein falsch gewähltes Verfahren, bzw. ein zu hoher Glättungsfaktor kanndie Situation im Spektrum verfälschen. In dieser Dimplomarbeit wirddie Methode des gleitenden Durchschnitts verwendet. Dabei wird einFenster von n Punkten benutzt, um aus ihnen den Mittelwert zu be-rechnen (Abbildung A.5 (d)).
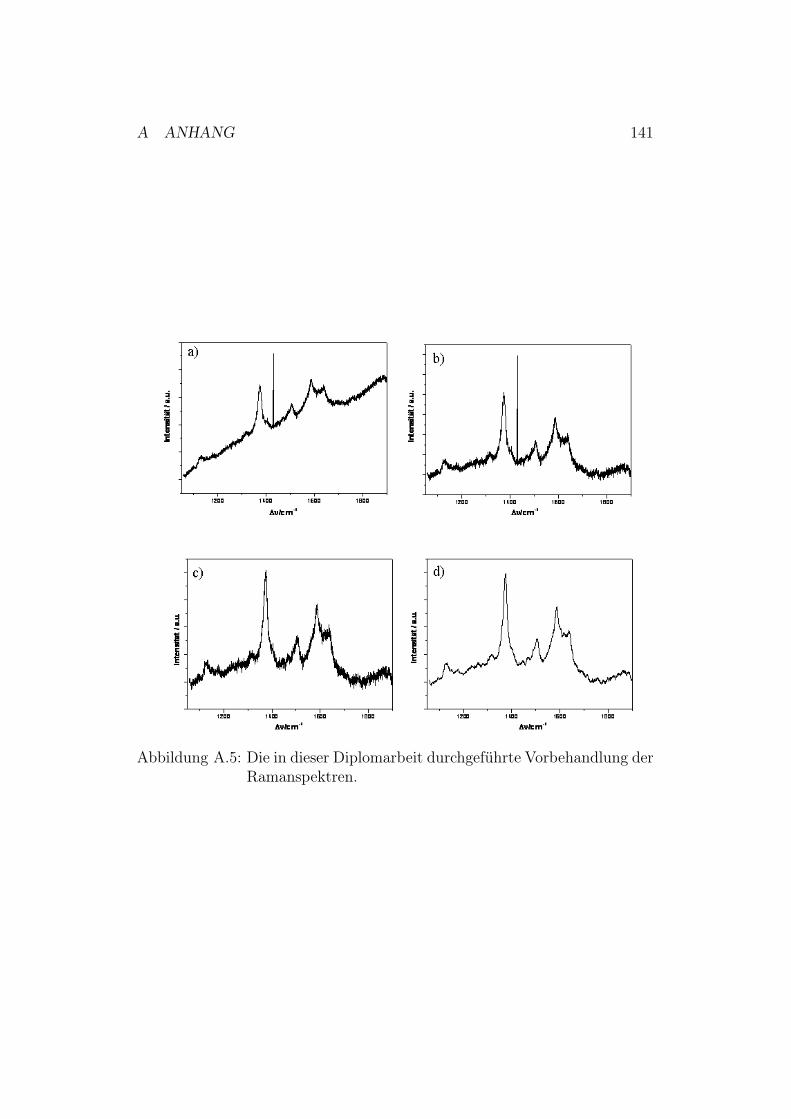
A ANHANG 141
Abbildung A.5: Die in dieser Diplomarbeit durchgeführte Vorbehandlung derRamanspektren.

A ANHANG 142
A.2 MaterialA.2.1 Lösungen
Folgende Lösungen wurden für diese Diplomarbeit selbst hergestellt:
• ANTA-Lösung: Die ANTA-Lösung besteht aus MilliQ-Wasser, in dem0,15 M ANTA und 0,5 M Kaliumcarbonat gelöst sind. Die Lösung wirdmit Hilfe von Kaliumhydroxid auf pH 9.8 titriert.
• Cytochrom c-Lösung (oxidiert): Hierbei werden 50 mg Cytochromc in 1ml PBS-Puffer gelöst. Das verwendete Cytochrom c enthält we-niger als 5% reduziertes Cytochrom c. Ein chemisches Oxidieren wirddaher nicht benötigt.
• Cytochrom c-Lösung (reduziert): Hierbei werden 50 mg Cytochromc in 1 ml PBS-Puffer gelöst und danach mit Natriumdithionit redu-ziert. Nach der Verwendung von Sephadex-Entsalzungssäulen erhältman dann eine salzfreie Lösung mit reduziertem Cytochrom c.
• DDM-Lösung: Die DDM-Lösung besteht aus PBS-Puffer und DDM(1 mg/ml)
• DTSP-DTP-Lösung: DTSP und DTP werden im Verhältnis 60:40 introckenem DMSO (2 mg DTSP/ml) gelöst85.
• DTP-Lösung: Hierfür wird DTP in DMSO (1,33 mg DTP/ml) gelöst.
• Hellmanex-Lösung: Diese Lösung wird mit Hilfe von MilliQ-Wasserund Hellmanex II (20 µl/ml) hergestellt.
• Lipid-Lösung: Die Lipid-Lösung (eigentlich eine Suspension) bestehtaus DDM-Lösung in der DiPhyPC (0,01 mg/ml) gelöst ist. Da das Lipideinige Zeit benötigt, um sich gut zu verteilen, wird diese Suspension 24Stunden vor Gebrauch hergestellt, mit Hilfe eines Reagenzglasschüttlersgemischt (10 Minuten) und dann ins Ultraschallbad (1 Stunde) gestellt.
• Nickellösung: Die Nickellösung besteht aus Nickel(II)-chlorid (40 mM),Essigsäure (50 mM) und MilliQ-Wasser. Sie wird mit Kaliumhydroxidauf pH 5.5 titriert.
85Unsere Gruppe hat herausgefunden, dass sich diese DTSP-DTP-Konzentration alsideal erweist [115].

A ANHANG 143
• PBS-Puffer: Der in der Diplomarbeit verwendete PBS-Puffer86 ent-hält Kaliumphosphat (0.1 M), Kaliumchlorid (0.05 M) und MilliQ-Was-ser. Er wird mit Hilfe von Salzsäure auf pH 8 titriert.
• Sauerstoffzehrsystem: Das Sauerstoffzehrsystem besteht aus PBS-Puffer in dem die drei Komponenten Glukose (3 mg/ml), Glukose-Oxida-se (75 µg/ml) und Katalase (12,5 µ/ml) gelöst sind [116]. Das Prinzipdieses Systems wird im folgenden dargestellt:
Glukose + O2 + H2OGlukose-Oxidase!!!!!!!!!" Glukonsäure + H2O2
2H2O2Katalase!!!!!" 2H2O + O2
• Schwefelsäurebad: Hierfür wird konzentrierte Schwefelsäure verwen-det. Zur Aktivierung wird 0,1 mg/ml Ammoniumperoxodisulfat ver-wendet.
A.2.2 Chemikalien und Materialien
Die verwendeten Chemikalien stammen hauptsächlich von den Firmen Sigma-Aldrich und Fluka Chemie GmbH. Der Hersteller steht in der folgenden Auf-listung in eckigen Klammern. An erster Stelle steht die an manchen Stellender Diplomarbeit verwendete Abkürzung, gefolgt von dem Namen der Sub-stanz:
• Ammoniak (NH3): 32% ig [WTL Laborbedarf GmbH]
• Ammoniumperoxodisulfat (H8N2O8S2) [Fluka]
• ANTA: Nα′ ,Nα′′-bis(carboxymethyl)-L-lysine (C10H18N2O6) [Fluka]
• Argon mit Reinheitsgrad 6.0 [Westfalen AG in Münster]
• Biobeads SM-2 adsorbent 20-50 mesh [Bio-Rad Laboratories, Inc.]
• Cytochrom c: Rinderherz [Sigma]
• Die Cytochrom-c-Oxidase (down-konfiguration87 von Rhodobacter sph-aeroides) und die Cytochrom-c-Oxidase (Mutante N139C, down-Kon-figuration von Rhodobacter sphaeroides) wurden geliefert von RobertB. Gennis, Department of Biochemistry, University of Illinois
86PBS (phosphate bu!ered saline) ist eine Abkürzung für salzartigen Phosphatpu!erund damit ein wenig informative Bezeichnung.
87Der His-Tag befindet sich an der Untereinheit 2

A ANHANG 144
• Die Cytochrom-c-Oxidase (up-Konfiguration88 from Paracoccus deni-trificans) wurde geliefert von Bernd Ludwig University of Frankfurt
• DDM: n-Dodecyl---D-maltoside [Sigma]
• DiphyPC: 1,2 Diphytanoyl-sn-Glycero-3-Phosphocholine [Avanti PolarLipids, Inc]
• DMSO: Dimethylsulfoxid [Acros Organics]
• DTP: dithiobis (propionic acid) [Sigma]
• DTSP: dithiobis (N-succinimidyl propionate) [Sigma]
• Essigsäure (C2H4O2) [Fluka]
• Ethanol (C2H6O): absolut Ethanol [Sigma]
• Gleitringdichtung, bestehend aus zwei Teilen: 921-0100-R-SSE-G4 undREP-T-999 [technico vertriebsges. mbH + Co. KG]
• Glucose Oxidase: Type VII von Asperigillus niger (Schwarzschimmel)[Sigma]
• Glukose: D-(+)-Glucose [Sigma]
• Gold : Feingoldgranulat 99,99% [ESG Edelmetall Service GmbH&Co.K]
• Hellmanex II [Hellma]
• Immersionsöl [Cargille Laboratories, Inc.]
• Kaliumchlorid (KCl) [Sigma]
• Kaliumcarbonat (K2CO3) [Sigma]
• Kaliumhydroxid (KOH) 85%ig [WTL Laborbedarf]
• Kaliumphosphat (K2HPO4:) [Fluka]
• Katalase von der Rinderleber [Sigma]
• Kleber: TSG EPO-TEK, 2 Komponentenkleber (Verhältnis 1:10) [Po-lytec]
88Der His-Tag befindet sich an der Untereinheit 1

A ANHANG 145
• Kleber: Leitender Kleber, nach Erhitzung von 180" für eine Minute er-hält man einen Durchgangswiderstand von 0.00005 $ cm/cm3 und eineScherfestigkeit von 0.091 N/cm2 [Electrade GmbH]
• Kugellager [GRW Miniaturkugellager Germany]
• Natriumdithionit (Na2S2O4) [Fluka]
• Nickel(II)-chlorid Hexahydrat (NiCl2*6H2O) [Fluka]
• Objektträger: BK7 mit Brechungsindex n=1,5 [Menzel-Gläser]
• Objektträger: LaSFN-9 mit Brechungsindex n=1,845 [Hellma]
• O-Ringe: Chemraz-O-Ringe (Dicke: 1,5 mm, innen = 10mm) aus Per-fluorelastomeren mit hoher chemischer Beständigkeit [Green Tweed]
• Platindraht: ø= 1,0 mm und Reinheitsgrad: 99,99% [Chempur]
• Platindraht: ø= 0,1 mm und Reinheitsgrad: 99,99% [Chempur]
• Plexiglas [Cadillac Plastic’s]
• Poliertuch: Chemomet I [Buehler]
• Poliertuch: SiC Grinding paper for metallography wet or dry Grit P600 [Buehler]
• Poliertuch: SiC Grinding paper for metallography wet or dry Grit P1200 [Buehler]
• Prisma LaSFN-9 mit Brechungsindex n=1,845 [Spindler&Hoyer]
• Salzsäure (HCl) 37% ig [WTL Laborbedarf]
• Schwefelsäure (H2SO4) [Acros Organics]
• Sephadex G-25 Medium PD-10 Entsalzungssäule [GE Healthcare]
• Silberdraht:ø= 1,0 mm und Reinheitsgrad 99,99% [Chempur]
• Silberstab: ø= 10 mm und Reinheitsgrad 99,99%
• Siliziumwafer-Spezialanfertigung [CrysTec GmbH]
• Stickstoff (flüssig) und Dewargefäß für Transport [Air Liquide]
• Stickstoff mit Reinheitsgrad 4 [Westfalen AG in Münster]

A ANHANG 146
• Teflon [Cadillac Plastic’s]
• Wasserstoffperoxid (H2O2): 35% ig [Sigma]
• Zahnflachriemen mit Zahnriemenscheiben [Rheinwerkzeug KG]
A.2.3 Geräte
In der folgenden Auflistung befinden sich die verwendeten Geräte. An ersterStelle steht der in der Diplomarbeit verwendete Begriff, gefolgt von demverwendeten Modell. In eckigen Klammern steht der Hersteller:
• AOM (Akustooptischer Modulator): MT200/A0.5, 400 nm [Opto - Elek-tronik]
• Aufdampfanlage: fl 400 mit auto 306 [Edwards]
• Autolab: Autolab Instrument (PGSTAT302) ausgestattet mit einemFRA2-Modul für Impedanzmessungen, einem ECD-Modul-Verstärkerfür niedrige Ströme, einem ADC750 Modul für schnelle Skanmessungenund einem SCAN-GEN Modul [Eco Chemie, B.V.]
• Blaspistole: Elektra Beckum BP 200 [Metabo]
• Chopper: 197 [EG&G]
• Fünf-Phasen-Schrittmotor [Huber]
• Funktionsgenerator: 33250A 80 MHz [Agilent Technologies]
• Galvanisierungseinheit [Labor Elektronik]
• Immersionsobjektiv: Lumplfl100xw/1,00 [Olympus]
• Laser (/=413,1 nm): Innova 90 K [Coherent]
• Laser (/=632,8 nm): 1105P [JDS Uniphase]
• Lock-In Verstärker: 5210 [EG&G]
• Magnetrührer: MR 3001 K [Heidolph]
• Monocromator: LaserspecIII [Spectrolab]
• Motor (1,2 mNm Dauerdrehmoment): 1319T012S in Kombination mitGetriebe: 141 (Übersetzung 14:1) [Faulhaber]

A ANHANG 147
• Motor (16 mNm Dauerdrehmoment): 2342S01C R in Kombination mitGetriebe: 231 (Übersetzung 3.71:1) [Faulhaber]
• Nanofocus: NanoFocus µsurf confocal microscope kombiniert mit 100xObjektiv [Nanoscope]
• Oszilloskop: 9354AM [Le croy]
• pH-Meter: pH-Meter 766 Calimatic [Knick]
• Polarisator [Fa. Halle]
• Poliermaschine: Phoenix 4000 in Kombination mit Probenhalter undNivellierscheibe: 160 MM DMR [Buehler]
• Raman Spektrometer: LabRam, HR800 [HORIBA Jobin Yvon] in Kom-bination mit einem Symphonie CCD Detektor [HORIBA Jobin Yvon],einem Mikroskop: BX41 dimensions [Olympus] und einem Notchfil-ter: Holographic Notch-PlusTM Filter-413,0-1,0 [Kaiser optical systems,Inc.]
• RDE (rotating disc electrode): Autolab, type RDE [Eco Chemie, B.V.]
• Rasterkraftmikroskop: AFM (Atomic Force Microscope) Dimension 3100CL Olympus89 [Veeco]
• Reagenzglasschüttler: Reax 2000 [Heidolph Instruments GmbH]
• Ultraschallbad: Super RK510 H [Sonorex]
• Zweikreisgoniometer [Huber]
89Bei den Messungen wurden folgenden Parameter verwendet: non-contact modeOMCL-AC160TS-W2, K = 42 N/m, Range: 33.5 - 94.1N/m, F0 = 300 kHz, Range: 278 -389 kHz.
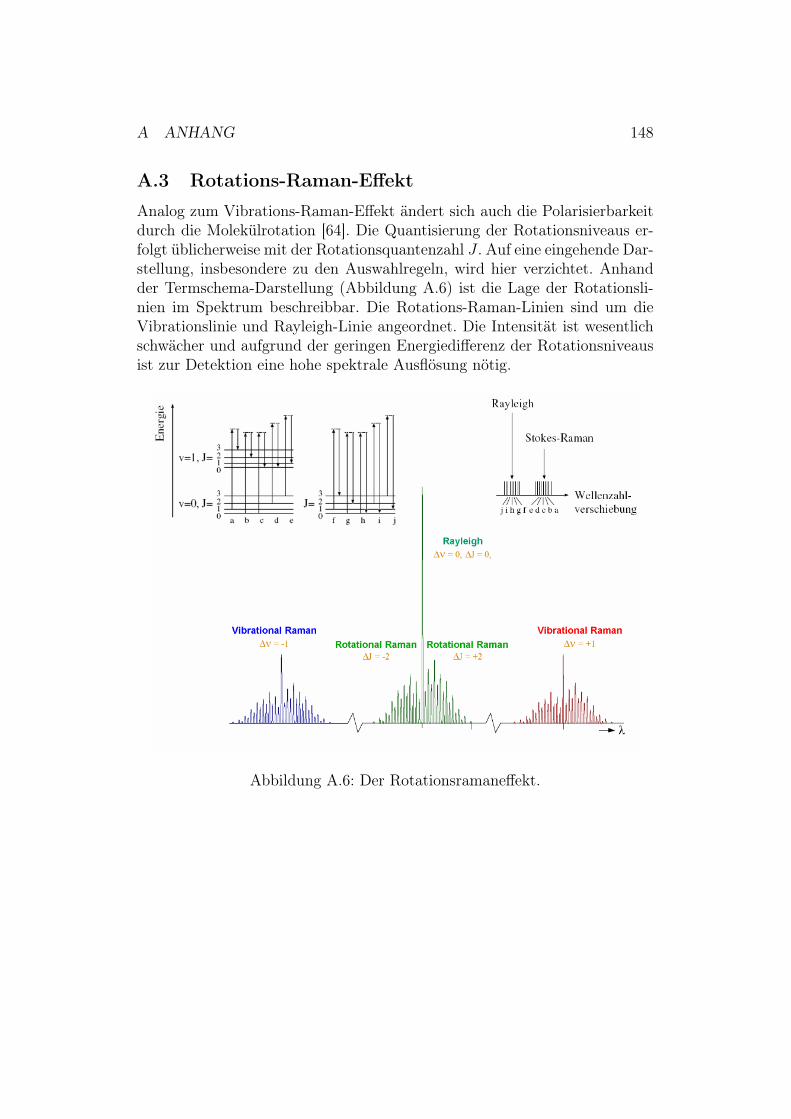
A ANHANG 148
A.3 Rotations-Raman-EffektAnalog zum Vibrations-Raman-Effekt ändert sich auch die Polarisierbarkeitdurch die Molekülrotation [64]. Die Quantisierung der Rotationsniveaus er-folgt üblicherweise mit der Rotationsquantenzahl J . Auf eine eingehende Dar-stellung, insbesondere zu den Auswahlregeln, wird hier verzichtet. Anhandder Termschema-Darstellung (Abbildung A.6) ist die Lage der Rotationsli-nien im Spektrum beschreibbar. Die Rotations-Raman-Linien sind um dieVibrationslinie und Rayleigh-Linie angeordnet. Die Intensität ist wesentlichschwächer und aufgrund der geringen Energiedifferenz der Rotationsniveausist zur Detektion eine hohe spektrale Ausflösung nötig.
Abbildung A.6: Der Rotationsramaneffekt.

A ANHANG 149
A.4 Konstruktionszeichnung der neuen SERRS-Zelle
Abbildung A.7: Konstruktionszeichnung der neuen SERRS-Zelle. Die Län-genangaben sind in Millimeter.
A.5 Versuchsaufbau für eine RDE-MessungHierfür werden die mit Gold bedampften Si-Wafer nicht auf eine Glasplatte,sondern auf einen Bleizylinder (ø= 10mm) mit eingebautem Gewinde ge-klebt. Nach dem Entfernen des Si-Wafers wird der Zylinder auf einen Stahl-stab des gleichen Durchmessers geschraubt. Der Stab besitzt ein weiteresGewinde, mit dem er sich an eine Autolab-RDE befestigen lässt. Damit sichausschließlich die Goldschicht als leitendes Material in der Lösung befindet,wird ein Teflonzylinder über die Probe gestülpt (siehe Abbildung A.8). Umdie Arbeitselektrode (Autolab RDE) mit der Goldschicht zu kontaktieren
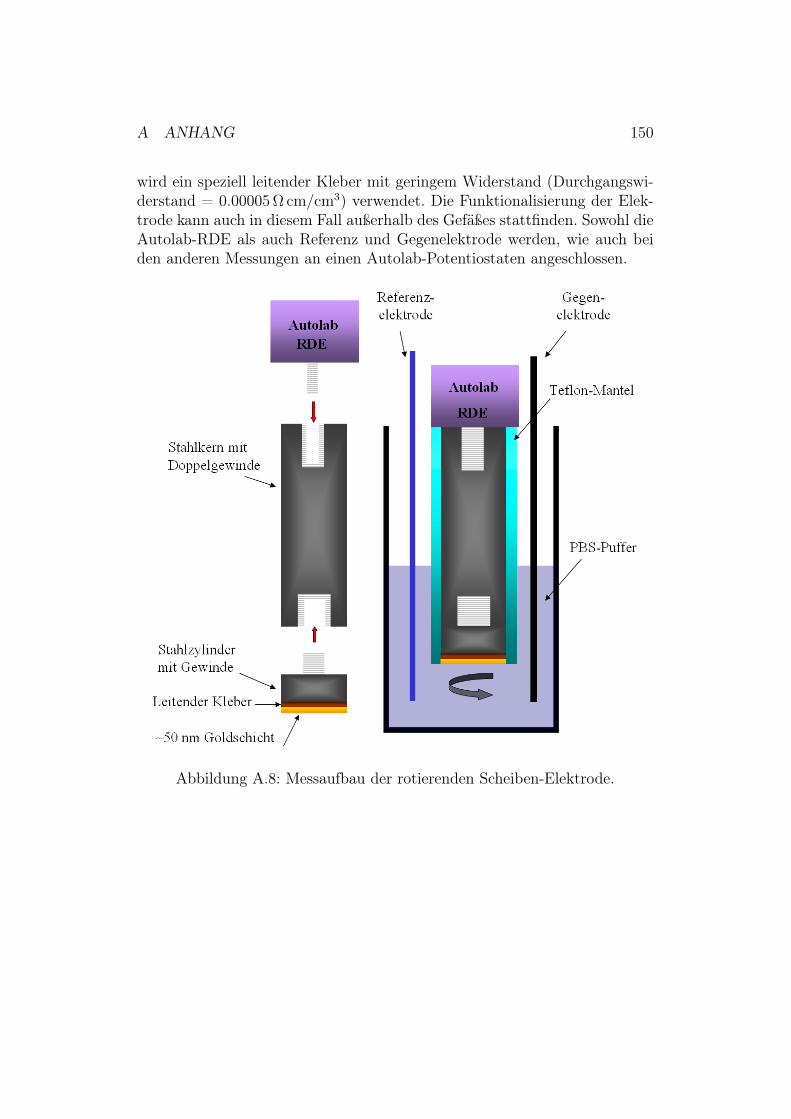
A ANHANG 150
wird ein speziell leitender Kleber mit geringem Widerstand (Durchgangswi-derstand = 0.00005 $ cm/cm3) verwendet. Die Funktionalisierung der Elek-trode kann auch in diesem Fall außerhalb des Gefäßes stattfinden. Sowohl dieAutolab-RDE als auch Referenz und Gegenelektrode werden, wie auch beiden anderen Messungen an einen Autolab-Potentiostaten angeschlossen.
Abbildung A.8: Messaufbau der rotierenden Scheiben-Elektrode.

A ANHANG 151
A.6 Polieren und Aufrauhen der SilberelektrodenA.6.1 Polieren der Silberelektroden
Das Polieren der Silberelektrode wurde am Anfang der Diplomarbeit nochvon Hand gemacht. Hierzu kam ein Poliertuch (SiC Grinding paper for me-tallography wet or dry Grit P 600) in Kombination mit Ethanol zum Einsatz.Der Unterschied zwischen einer handpolierten und einer maschinenpolierten(Poliervorschrift siehe Abschnitt 6.2) Probe ist in Abbildung A.9 zu sehen.Die tiefen Krater der handpolierten Probe können die Bildung einer stabilenMembran verhindern.
Abbildung A.9: Nanofokusbild: Unterschied zwischen einer handpolierten(links) und einer maschinenpolierten Probe (rechts).
A.6.2 Neu entwickelte Poliervorschrift
Bei Stewart’s Methode für Edelmetalle wird nach der Politur mit Silizium-carbit (SiC) Diamantpaste als Poliermittel verwendet. Bei der Präparationmit Diamantpasten können sich jedoch Diamantkörner in die Silberelektro-den eindrücken. Deshalb poliert man stattdessen mit unterschiedlich grobenAluminiumoxidsuspensionen (Tonerde). Die Tonerde ist weniger aggressivund kann durch den hohen Anteil der Oxidpartikel die meisten Kratzer be-seitigen. Nach dem Polieren werden die Silberelektroden zum Entfernen derSchleifpartikel in ein Ultraschallbad gestellt. Abbildung A.10 zeigt ein Bild(Nanofokus), bei dem man eine starke Erhöhung erkennen kann. Vermutlich
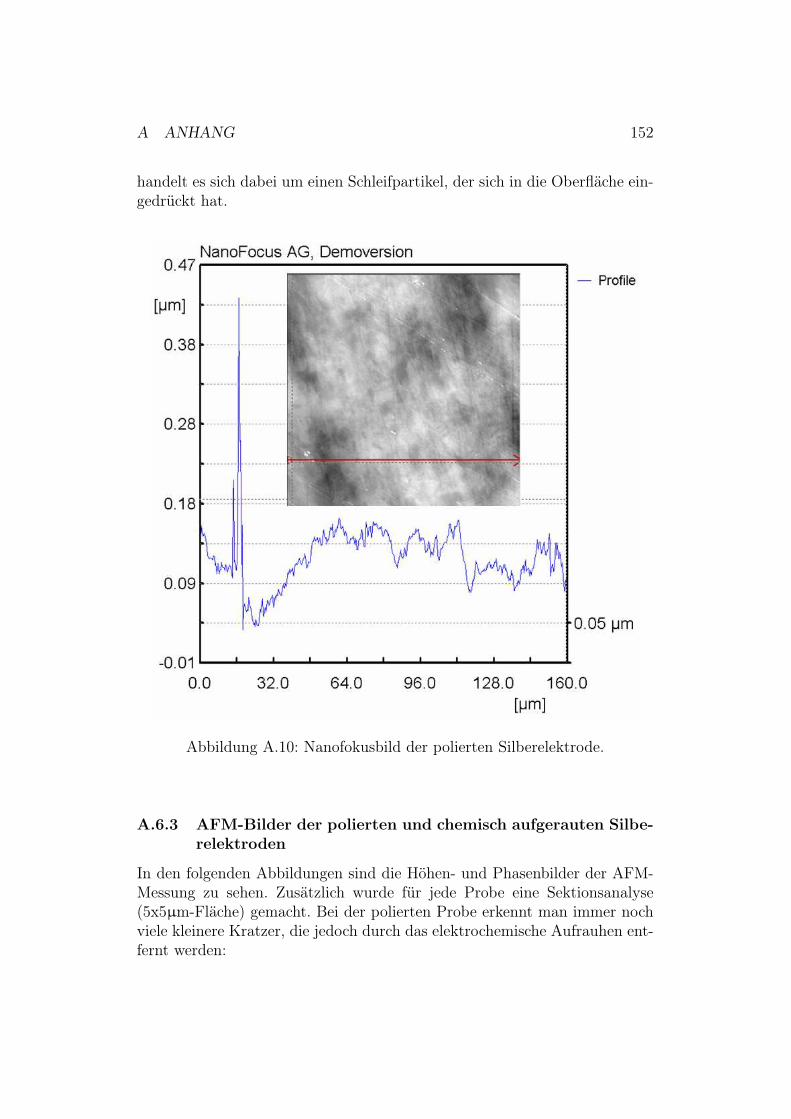
A ANHANG 152
handelt es sich dabei um einen Schleifpartikel, der sich in die Oberfläche ein-gedrückt hat.
Abbildung A.10: Nanofokusbild der polierten Silberelektrode.
A.6.3 AFM-Bilder der polierten und chemisch aufgerauten Silbe-relektroden
In den folgenden Abbildungen sind die Höhen- und Phasenbilder der AFM-Messung zu sehen. Zusätzlich wurde für jede Probe eine Sektionsanalyse(5x5µm-Fläche) gemacht. Bei der polierten Probe erkennt man immer nochviele kleinere Kratzer, die jedoch durch das elektrochemische Aufrauhen ent-fernt werden:
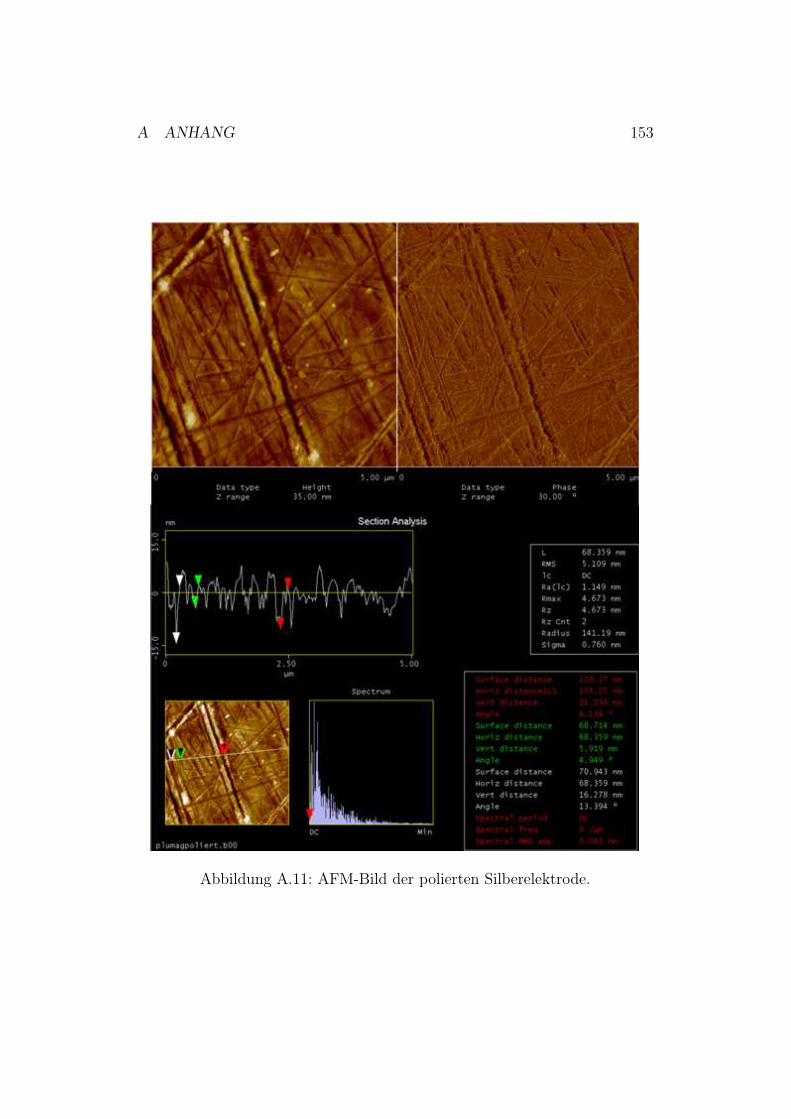
A ANHANG 153
Abbildung A.11: AFM-Bild der polierten Silberelektrode.
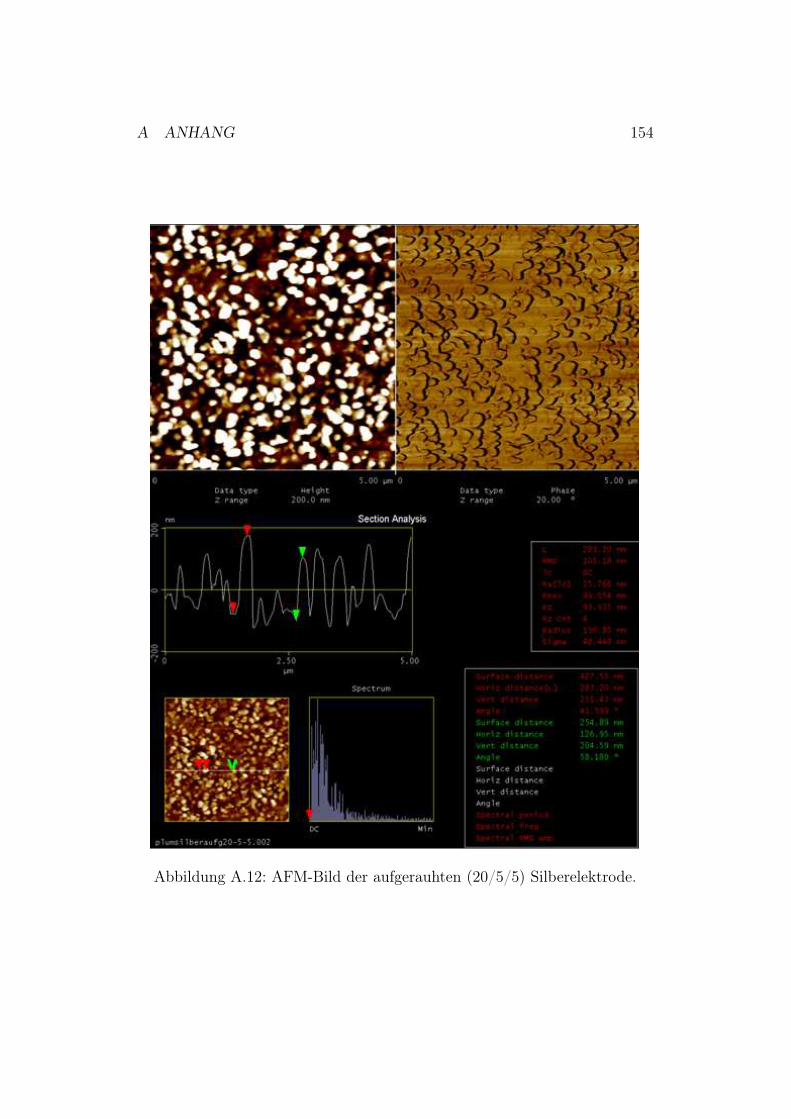
A ANHANG 154
Abbildung A.12: AFM-Bild der aufgerauhten (20/5/5) Silberelektrode.
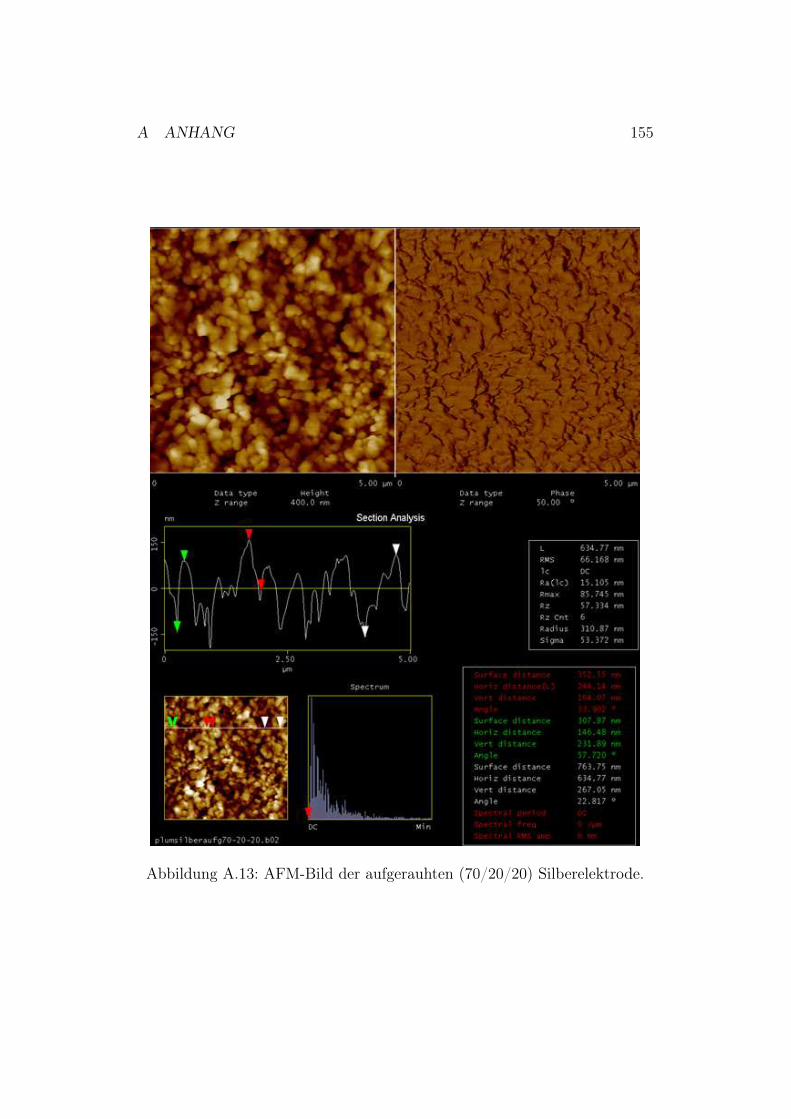
A ANHANG 155
Abbildung A.13: AFM-Bild der aufgerauhten (70/20/20) Silberelektrode.
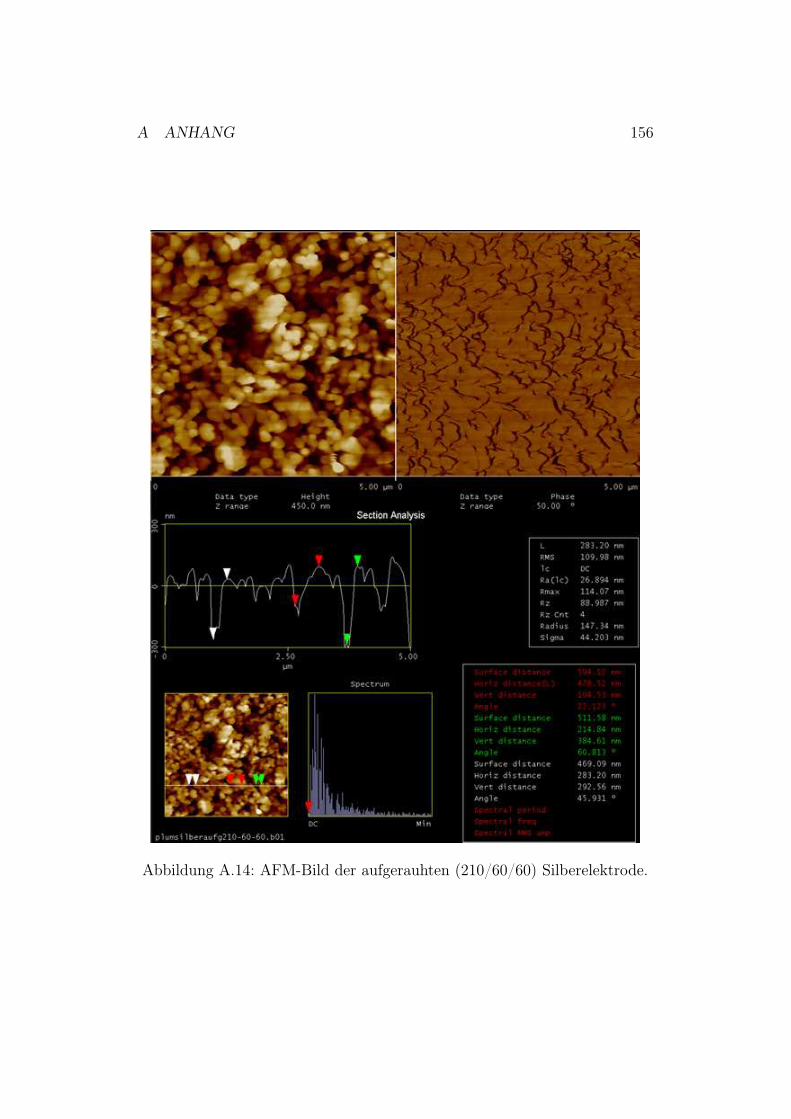
A ANHANG 156
Abbildung A.14: AFM-Bild der aufgerauhten (210/60/60) Silberelektrode.

B GLOSSAR 157
B Glossar• ADP: Adenosine diphosphate
• ATP: Adenosine triphosphate
• AgCl: Silberchlorid
• AFM: Atomic force microscopy (Rasterkraftmikroskop)
• AOM: Akusto-optischer Modulator
• CCD: Charge-coupled device
• CcO: Cytochrom-c-Oxidase
• CV: Cyclovoltammetrie/Cyclovoltammogramm
• Cyt c: Cytochrom c
• EIS: elektrochemische Impedanzspektroskopie
• NHE: Normal hydrogen electrode (Normal-Wasserstoff-Elektrode)
• ptBLM: Protein-tethered bilayer lipid membrane (Protein verankerte Lipid-doppelschichtmembranen)
• pH: pondus Hydrogenii/ -lg[H3O+]
• pH-Wert: der mit -1 multiplizierte Zehner-Logarithmus der Aktivität derOxoniumionen (H3O+)
• RDE: Rotierende Disk-Elektrode
• RR: Resonance Raman
• RPM: Rotations per minute
• SAM: Self-assembled monolayer (selbstorganisierende Monoschicht)
• SERS: surface enhanced raman spectroscopy
• SERRS: surface enhanced resonance raman spectroscopy
• SPR: surface plasmon resonance
• tBLM Tethered bilayer lipid membrane (verankerte Lipiddoppelschichtmem-branen)
• TR-SERRS: time-resolved surface enhanced resonance raman spectroscopy
• ø: Durchmesser

ABBILDUNGSVERZEICHNIS 158
Abbildungsverzeichnis1.1 Die Atmungskette. . . . . . . . . . . . . . . . . . . . . . . . . . . . 21.2 Die mitochondrale Atmungskette und ihre Lokalisation im Mitochon-
drium. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41.3 Die Röntgenstruktur des Cytochrom c (Rinderherz) mit Häm - Grup-
pe. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51.4 Die Röntgenstruktur des Cytochrom c (Rinderherz) mit Häm - Grup-
pe und Liganden. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61.5 Die Röntgenstruktur der Cytochrom-c-Oxidase von Paracoccus de-
nitrificans und ihre redox-aktiven Metallzentren. Das binukleare CuA-Zentrum befindet sich in Untereinheit II (gelb). Die RedoxzentrenHäm a, Häm a3 und CuB sind dagegen in Untereinheit I (grün). . . 8
1.6 Vergrößerung der Cytochrom-c-Oxidase-Redox-Zentren von Para-coccus denitrificans mit den entsprechenden Liganden. Die Seiten-ketten von Häm a enthalten unter anderem Vinyl - Gruppen (-CH=CH2), Formyl - Gruppen (-CH=O) und eine Farnesyl - Gruppe(-CHOH(-CH2CH2CH=CCH3)3-CH3). . . . . . . . . . . . . . . . . 9
1.7 Protonenwege in der Cytochrom-c-Oxidase von Paracoccus denitri-ficans. Der K-Weg der CcO führt über Lys-354 und Thr-351 zurHydroxyl-Gruppe von Tyr-280 am katalytischen Zentrum. Der D-Weg führt über Asp-124 über eine Reihe von Aminosäuren zu Glu-278 und von dort aus weiter ins katalytische Zentrum. Die Unterein-heiten III und IV wurden aus Gründen der Übersichtlichkeit wegge-lassen. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
1.8 Das Modell des konventionellen katalytischen Cytochrom-c-Oxidase-Zyklus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
1.9 Immobilisierung eines Proteins mit Hilfe der His-Tag-Technologie. . 131.10 Die zwei unterschiedlichen Konfigurationsmöglichkeiten der uns zur
Verfügung stehenden Cytochrom-c-Oxidase. Der His-Tag und dieVerankerung an die Metalloberfläche sind vergrößert dargestellt. Linksim Bild: Das Enzym mit His-Tag an Untereinheit II (orange). Rechtsim Bild: Das Enzym mit His-Tag an Untereinheit I (rot). Die Un-tereinheiten III und IV sind gelb bzw. schwarz dargestellt. . . . . . 14
2.1 Die Feldverteilung eines Oberflächenplasmons senkrecht zur Grenz-fläche. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.2 Das Verhältnis von Wellenvektoren eines aus dem Dielektrikum ein-fallenden Photons und eines Oberflächenplasmons. . . . . . . . . . 23
2.3 Die Prismenkopplung nach der Kretschmann-Konfiguration. . . . . 242.4 Links: Änderung der Dispersionsrelation des Oberflächenplasmons
durch Anlagerung einer dielektrischen Schicht. Rechts: ResultierendeVerschiebung des Anregungswinkels im Winkelspektrum. . . . . . . 25
2.5 Erläuterung der Kinetikmessung. . . . . . . . . . . . . . . . . . . . 26

ABBILDUNGSVERZEICHNIS 159
2.6 Die Impedanz in der komplexen Gaußschen Zahlenebene. . . . . . . 292.7 Strom- und Spannungs-Liniendiagramm für Widerstand und Kon-
densator. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 322.8 Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plot für einen
reinen Widerstand (rot: 50 kΩ und blau: 100 kΩ). . . . . . . . . . . 342.9 Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plot für einen
Kondensator (rot: 5µF und blau: 50µF). . . . . . . . . . . . . . . . 362.10 Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plot für eine
Parallelschaltung aus Kondensator und Widerstand. . . . . . . . . 382.11 Frequenzreduzierter Admittanz-, Nyquist- und Bode-Plot für eine
Reihenschaltung aus Kondensator und Widerstand. . . . . . . . . . 402.12 Frequenzreduzierter Admittanz-, Bode- und Nyquist-Plot eines idea-
len Monolayer-Ersatzschaltkreises. . . . . . . . . . . . . . . . . . . 412.13 Frequenzreduzierter Admittanz-, Bode- und Nyquist-Plot eines idea-
len ptBLM-Ersatzschaltkreises. . . . . . . . . . . . . . . . . . . . . 432.14 (a) „Dreiecksform“ des angelegten Potentials als Funktion der Zeit.
(b) Strom-/Zeit-Kennlinie einer Messlösung, die eine oxidierte undeine reduzierte Form einer Redox-Spezies enthält. (c) Cylovoltam-mogramm dieser Messlösung. Ea: anodisches Spitzenpotential; Ec:kathodisches Spitzenpotential; E1 und E2: Umkehrpotentiale; Ia undIc: anodischer bzw. kathodischer Spitzenstrom. . . . . . . . . . . . 45
2.15 Schematisches Cyclovoltammogramm einer ideal oberflächengebun-denen Redoxspezies. . . . . . . . . . . . . . . . . . . . . . . . . . . 52
2.16 Cyclovoltammogramm (a) ohne und (b) mit IR-Drop. . . . . . . . 532.17 Das Prinzip der zeitaufgelösten Messmethode. . . . . . . . . . . . . 552.18 Die Energieniveaus bei der elastischen und inelastischen Streuung. 572.19 Erläuterung des resonanten Ramaneffekts. . . . . . . . . . . . . . . 612.20 Porphyringerüst eines D4h - Metalloporphyrins mit den Substituen-
ten X und Y. Die zu unterscheidenden Kohlenstoffatome sind mit #,$ und m gekennzeichnet. M steht für das zentrale Metallatom undN für ein Stickstoffatom. . . . . . . . . . . . . . . . . . . . . . . . . 63
2.21 Absorptionspektrum von Cytochrom c mit der schematischen Dar-stellung des 4-Orbital-Modells. . . . . . . . . . . . . . . . . . . . . 64
2.22 Illustration der dominanten Moden für Metalloporphyrine im Marker-Banden-Bereich, bezogen auf Abbildung 2.20. . . . . . . . . . . . . 65
3.1 Die Herstellung einer TSG-Elektrode. . . . . . . . . . . . . . . . . . 673.2 Die Funktionalisierung der TSG-Elektrode. . . . . . . . . . . . . . 683.3 Die Herstellung einer ptBLM-Probe. In diesem Fall ist die Cytochrom-
c-Oxidase mit His-Tag an Untereinheit I (rot) verwendet worden.Die Untereinheiten II, III und IV sind in orange, gelb und schwarzdargestellt. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
3.4 Die Dialyse. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71

ABBILDUNGSVERZEICHNIS 160
3.5 Die Architektur einer ptBLM-Probe, bei der als Protein die Cyto-chrom-c-Oxidase mit His-Tag an Untereinheit I verwendet wordenist. Die horizontalen Längenangaben beziehen sich auf den Teil desEnzyms, der aus dem Bilayer in die Lösung weist. Die funktionali-sierte Elektrode ist vergrößert abgebildet. . . . . . . . . . . . . . . 72
3.6 Die zwei unterschiedlichen Konfigurationsmöglichkeiten der uns zurVerfügung stehenden Cytochrom-c-Oxidase. Links im Bild: Das En-zym mit His-Tag an Untereinheit II. Aktivierbar durch direktenElektronentransfer. Rechts im Bild: Das Enzym mit His-Tag an Un-tereinheit I. Aktivierbar durch Cytochrom c (grün). Die Unterein-heiten III und IV sind gelb bzw. schwarz dargestellt. . . . . . . . . 74
3.7 Die Cytochrom-c-Oxidase N139C. Dargestellt sind nur die Unterein-heiten I (rot) und II (orange). . . . . . . . . . . . . . . . . . . . . . 75
4.1 Der Messaufbau für SPR, EIS und CV. . . . . . . . . . . . . . . . . 764.2 Das Zelldesign der SPR-Zelle und der Einbau in den Reiter. . . . . 784.3 Komplette Messzelle mit Halterung (Reiter) und den Anschlüssen
für die Elektroden bzw. dem Ein- und Auslass für den Lösungsaus-tausch. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
5.1 Kinetikmessung zur Überprüfung des ptBLM-Aufbaus. Die dazuge-hörigen SPR-Spektren wurden an Luft (schwarz), vor (rot) und nach(blau) der Anbindung, sowie vor (orange) und nach (grün) dem PBS-Spülen aufgenommen. . . . . . . . . . . . . . . . . . . . . . . . . . 81
5.2 Überprüfung des ptBLM-Aufbaus mit Hilfe der EIS. Oben: Bode-Plot. Unten: frequenzreduzierter Admittanz- und Nyquist-Plot. DieMesspunkte sind in dieser Darstellung miteinander verbunden. . . . 83
5.3 Aktivierung der Cytochrom-c-Oxidase durch reduziertes Cytochromc. Links oben: Bode-Plot. Unten: Nyquist- und frequenzreduzierterAdmittanz-Plot. Die Messpunkte sind in dieser Darstellung mitein-ander verbunden. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
5.4 Widerstand des ptBLM-Systems aufgetragen gegen die Konzentra-tion des reduzierten Cytochrom c. . . . . . . . . . . . . . . . . . . . 87
5.5 Scan-Raten-Studie (schwarz: 10 mV/s, rot: 50 mV/s, orange: 100 mV/s,blau: 500mV/s, cyan: 900 mV/s) mit oxidiertem (links) und che-misch reduziertem (rechts) Cytochrom c in Kombination mit der pt-BLM immobilisierten Cytochrom-c-Oxidase in der up-Konfiguration. 88
5.6 Der Randles–Sevcik Plot für oxidiertes (grün, schwarz) und reduzier-tes (rot) Cytochrom c mit (rot, grün) und ohne (schwarz) Cytochrom-c-Oxidase in der up-Konfiguration. . . . . . . . . . . . . . . . . . . 89
5.7 Peakpotentiale als Funktion der Scan-Rate (Trumpet-Plot) des re-duzierten Cytochrom c in Kombination mit der up-Konfiguration. . 91

ABBILDUNGSVERZEICHNIS 161
5.8 50 mV/s Cyclovoltammogramm von oxidiertem (links) und redu-ziertem (rechts) Cytochrom c [1 mMol/l] in Kombination mit derCytochrom-c-Oxidase in der up-Konfiguration (mit und ohne Sau-erstoff). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
5.9 Ergebnisse der Potentialtitration für eine 1, 82 · 10!4 molare Cyto-chrom c-Lösung. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
5.10 Potentialtitration: Auftragung Widerstand gegen Konzentration er-gibt bei allen Potentialen den erwarteten exponentiellen Abfall. DieMesspunkte sind in dieser Darstellung miteinander verbunden. . . . 97
5.11 Potentialtitration: Auftragung Widerstand gegen Potential. Der mög-liche Kurvenverlauf ist in dieser Darstellung integriert. . . . . . . . 98
5.12 Cyclovoltammogramme (Scan-Rate von 10 mV/s) von der Cytochrom-c-Oxidase in der down-Konfiguration mit (rot) und ohne Sauerstoff(schwarz) und von der Cytochrom-c-Oxidase in der up-Konfiguration(blau). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 100
5.13 Cyclovoltammogramme der Mutante N139C und des Wild-Typ inGegenwart (links, Scan-Rate: 10 mV/s) und Abwesenheit (rechts,Scan-Rate: 100 mV/s) von Sauerstoff. . . . . . . . . . . . . . . . . . 101
6.1 Funktionalisierung der Silberelektrode durch einen SAM (Self As-sembling Monolayer). . . . . . . . . . . . . . . . . . . . . . . . . . . 105
6.2 Die Cytochrom-c-Oxidase in der down-Konfiguration, angebundenauf einer aufgerauhten Silberelektrode. . . . . . . . . . . . . . . . . 106
7.1 Der Messaufbau für potentiostatische SERRS-Messungen. . . . . . 1087.2 Der erweiterte Messaufbau für zeitaufgelöste SERRS-Messungen. . 1097.3 Das Zelldesign der alten Messzelle. . . . . . . . . . . . . . . . . . . 1117.4 Bild der alten Messzelle ohne Objektiv und Tefloneinsatz. . . . . . 1117.5 Das Zelldesign der neuen Messzelle. Der Deckel mit Objektiv, Refe-
renz- und Gegenelektrode ist nicht eingezeichnet. . . . . . . . . . . 1137.6 Querschnitt der neuen Messzelle. Der Deckel mit Objektiv, Referenz-
und Gegenelektrode ist nicht eingezeichnet. Der herausnehmbare Zy-linder (cyan) besteht aus Teflon. Der Einsatz besteht aus Teflon(weiß) und Plexiglas. . . . . . . . . . . . . . . . . . . . . . . . . . . 113
8.1 Elektrochemisches Aufrauhen der Silberelektroden. . . . . . . . . . 1148.2 AFM-Bilder der unterschiedlich aufgerauhten Silberelektroden. (a)
keine Aufrauhung, (b) Aufrauhung mit 20/5/5, (c) 70/20/20, (d)210/60/60. Die Höhenskala geht bei allen vier Bildern von 0 nm bis200 nm. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
8.3 Nanoscope-Bilder der unterschiedlich aufgerauhten Silberelektroden.(a) keine Aufrauhung, (b) Aufrauhung mit 20/5/5, (c) 70/20/20, (d)210/60/60. Die unterschiedlichen Höhenskalen sind in dem jeweiligenBild eingezeichnet. . . . . . . . . . . . . . . . . . . . . . . . . . . . 117

ABBILDUNGSVERZEICHNIS 162
8.4 SERRS-Spektren von oxidiertem (rot) und chemisch reduziertem(blau) Cytochrom c. Die Messungen werden mit einer DTP - modi-fizierten Silberoberfläche durchgeführt. . . . . . . . . . . . . . . . . 119
8.5 Potentialtitration des Cytochrom c ((a) 40mV, (b) 90 mV , (c) 140 mV,(d) 240 mV, (e) 350 mV). . . . . . . . . . . . . . . . . . . . . . . . . 120
8.6 Rücktitration des Cytochrom c ((a) 350mV, (b) 190 mV, (c) 90 mV,(d) -110 mV, (e) -210 mV, (f) -310 mV, (g) -410mV, (h) -510mV). . 121
8.7 Potentialtitration der Cytochrom-c-Oxidase ((a)-710mV, (b)-610 mV,(c)-510mV, (d)-410 mV, (e)-310mV, (f)-210mV, (g)-110mV). . . . 124
8.8 Die zu den Peaks gehörenden Moden für die oxidierten (rot: -110 mV)und reduzierten Spektren (blau: -710mV). . . . . . . . . . . . . . . 124
8.9 Wiederholte Oxidation (rot und schwarz: -110 mV) und Reduktion(blau: -710 mV) der Cytochrom-c-Oxidase. . . . . . . . . . . . . . . 125
8.10 Die Parameter der zeitaufgelösten SERRS-Messung. . . . . . . . . 1278.11 SERRS-Spektren der down-Konfiguration für die Parametereinstel-
lungen a (oxidiert, rot) und b (reduziert, blau). . . . . . . . . . . . 127A.1 Gemessene Winkelbereiche der SPR-Spektren. Links: an Luft. Rechts:
in Lösung. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 135A.2 Datensatz, der mit Hilfe des komplexen nichtlinearen Fitalgorithmus
der Software ZView gefittet wurde. Der verwendete Schaltkreis zumFitten symbolisiert ein ptBLM-System. Das Schaltkreissymbol !!+!steht für ein CPE-Element. . . . . . . . . . . . . . . . . . . . . . . 136
A.3 Beispiel einer Basislinien-Korrektur mit Hilfe der Gpes - Software. 138A.4 Bestimmung der Peakhöhen und der Lage der Peaks mit Hilfe der
Origin - Software. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139A.5 Die in dieser Diplomarbeit durchgeführte Vorbehandlung der Ra-
manspektren. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141A.6 Der Rotationsramaneffekt. . . . . . . . . . . . . . . . . . . . . . . . 148A.7 Konstruktionszeichnung der neuen SERRS-Zelle. Die Längenanga-
ben sind in Millimeter. . . . . . . . . . . . . . . . . . . . . . . . . . 149A.8 Messaufbau der rotierenden Scheiben-Elektrode. . . . . . . . . . . . 150A.9 Nanofokusbild: Unterschied zwischen einer handpolierten (links) und
einer maschinenpolierten Probe (rechts). . . . . . . . . . . . . . . . 151A.10 Nanofokusbild der polierten Silberelektrode. . . . . . . . . . . . . . 152A.11 AFM-Bild der polierten Silberelektrode. . . . . . . . . . . . . . . . 153A.12 AFM-Bild der aufgerauhten (20/5/5) Silberelektrode. . . . . . . . . 154A.13 AFM-Bild der aufgerauhten (70/20/20) Silberelektrode. . . . . . . 155A.14 AFM-Bild der aufgerauhten (210/60/60) Silberelektrode. . . . . . . 156

TABELLENVERZEICHNIS 163
Tabellenverzeichnis5.1 Fit-Ergebnisse der EIS-Messung zur Überprüfung des ptBLM - Auf-
baus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 835.2 Fit-Ergebnisse des linearen Fits für den Randles–Sevcik Plot. . . . 905.3 Fit-Ergebniss des Trumpet-Plots. . . . . . . . . . . . . . . . . . . . 906.1 Poliervorschrift für Ag-Elektroden . . . . . . . . . . . . . . . . . . 1036.2 Protokoll zum Aufrauhen der Ag-Elektroden mit (70/20/20). . . . 1048.1 Ergebnisse der AFM und Nanoscope-Daten. . . . . . . . . . . . . . 1158.2 Moden und Lagen der Banden sowohl für Spektren mit oxidiertem
und reduziertem Cytochrom c, als auch für Spektren mit Cytochromc, bei denen ein Potential von 40 mV bzw. 350mV an die Elektrodeangelegt wird. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122
8.3 Moden und Lagen der Banden der Cytochrom-c-Oxidase Spektren,bei denen ein Potential von -10 mV bzw. -710 mV an die Elektrodeangelegt wird. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 129
A.1 Lagen und Höhen der Peaks für das gewählte Beispiel . . . . . . . 137

LITERATUR 164
Literatur[1] J. M. Berg, J. L. Tymoczko, and L. Stryer, Biochemistry (Fifth Edition). W.
H. Freeman and Company, 2002.
[2] K. Ataka, F. Giess, W. Knoll, R. Naumann, S. Haber-Pohlmeier, B. Rich-ter, and J. Heberle, “Oriented attachment and membrane reconstitutionof his-tagged cytochrome-c-oxidase to a gold electrode: In situ monitoringby surface-enhanced infrared absorption spectroscopy,” J. Am. Chem. Soc.,vol. 126, pp. 16199–16206, Dec. 2004.
[3] R. A. Scott and A. G. Mauk, Cytochrome c, a multidisciplinary approach.University Science Books, 1996.
[4] R. E. Dickerson, T. Takano, D. Eisenberg, O. B. Kallai, L. Samson,A. Cooper, and E. Margoliash, “Ferricytochrome c. general features of thehorse and bonito proteins at 2.8 angström resolution,” J. Biol. Chem.,vol. 246, no. 5, pp. 1511–1535, 1971.
[5] T. Takano, O. B. Kallai, R. Swanson, and R. E. Dickerson, “The structureof ferrocytochrome c at 2.45 angström resolution,” J. Biol. Chem., vol. 248,pp. 5234–5255, Aug. 1973.
[6] G. W. Bushnell, G. V. Louie, and G. D. Brayer, “High-resolution three-dimensional structure of horse heart cytochrome c,” Journal of MolecularBiology, vol. 214, pp. 585–595, July 1990.
[7] G. T. Babcock and M. Wikstrom, “Oxygen activation and the conservationof energy in cell respiration,” Nature, vol. 356, pp. 301–309, Mar. 1992.
[8] S. Yoshikawa, K. Shinzawa-Itoh, and T. Tsukihara, “X-ray structure andthe reaction mechanism of bovine heart cytochrome c oxidase,” Journal ofInorganic Biochemistry, vol. 82, pp. 1–7, Nov. 2000.
[9] C. Ostermeier, A. Harrenga, U. Ermler, and H. Michel, “Structureat 2.7 angström resolution of the paracoccus denitrificans two-subunitcytochrome-c-oxidase complexed with an antibody fv fragment,” PNAS,vol. 94, pp. 10547–10553, Sept. 1997.
[10] H. Witt and B. Ludwig, “Isolation, analysis, and deletion of the gene codingfor subunit iv of cytochrome-c-oxidase in paracoccus denitrificans,” J. Biol.Chem., vol. 272, pp. 5514–5517, Feb. 1997.
[11] R. B. Gennis, “How does cytochrome oxidase pump protons?,” 1998,, vol. 95,pp. 12747–12749, 1998.
[12] H. Michel, “The mechanism of proton pumping by cytochrome c oxidase,”PNAS, vol. 95, pp. 12819–12824, Oct. 1998.

LITERATUR 165
[13] D. Proshlyakov, T. Ogura, K. Shinzawa-Itoh, S. Yoshikawa, and T. Kitagawa,“Resonance raman/absorption characterization of the oxo intermediates ofcytochrome c oxidase generated in its reaction with hydrogen peroxide: Phand h2o2 concentration dependence,” Biochemistry, vol. 35, pp. 8580–8586,July 1996.
[14] M. Fabian, W. W. Wong, R. B. Gennis, and G. Palmer, “Mass spectrome-tric determination of dioxygen bond splitting in the peroxy intermediate ofcytochrome-c-oxidase,” PNAS, vol. 96, pp. 13114–13117, Nov. 1999.
[15] E. Hochuli, W. Bannwarth, H. Dobeli, R. Gentz, and D. Stuber, “Geneticapproach to facilitate purification of recombinant proteins with a novel metalchelate adsorbent,” Nat Biotech, vol. 6, pp. 1321–1325, Nov. 1988.
[16] E. Hochuli, H. Dobeli, and A. Schacher, “New metal chelate adsorbent se-lective for proteins and peptides containing neighbouring histidine residues,”Journal of Chromatography A, vol. 411, pp. 177–184, 1987.
[17] M. Friedrich, J. Robertson, D. Walz, W. Knoll, and R. Naumann, “Electronicwiring of membrane proteins a kinetic study of cytochrome c oxidase,” ...nochnicht veröffentlicht.
[18] R. H. Ritchie, “Plasma losses by fast electrons in thin films,” Phys. Rev.,vol. 106, pp. 874–, June 1957.
[19] T. Turbadar, “Complete absorption of light by thin metal films,” Proceedingsof the Physical Society, vol. 73, no. 1, pp. 40–44, 1959.
[20] A. Otto, “Excitation of nonradiative surface plasma waves in silver by themethod of frustrated total reflection,” Zeitschrift für Physik A Hadrons andNuclei, vol. V216, pp. 398–410, Aug. 1968.
[21] E. Kretschmann and H. Raether, “Radiative decay of non radiative surfaceplasmons excited by light (surface plasma waves excitation by light and decayinto photons applied to nonradiative modes),” Zeitschrift für Naturforschung,vol. 23A, pp. 2135, 2136, 1968.
[22] W. Knoll, “Interfaces and thin films as seen by bound electromagnetic waves,”Annual Review of Physical Chemistry, vol. 49, no. 1, pp. 569–638, 1998.
[23] B. Rothenhausler, J. Rabe, P. Korpiun, and W. Knoll, “On the decay of plas-mon surface polaritons at smooth and rough ag-air interfaces: A reflectanceand photo-acoustic study,” Surface Science, vol. 137, pp. 373–383, Feb. 1984.
[24] T. Fließbach, Elektrodynamik (4.Auflage). Spektrum Akademischer Verlag,2005.
[25] C. Gerthsen, Gerthsen Physik. Springer, 2002.

LITERATUR 166
[26] J. Homola, “Present and future of surface plasmon resonance biosensors,”Analytical and Bioanalytical Chemistry, vol. 377, pp. 528–539, Oct. 2003.
[27] I. Pockrand, “Surface plasma oscillations at silver surfaces with thin trans-parent and absorbing coatings,” Surface Science, vol. 72, pp. 577–588, Apr.1978.
[28] J. F. Tassin, R. L. Siemens, W. T. Tang, G. Hadziioannou, J. D. Swalen, andB. A. Smith, “Kinetics of adsorption of block copolymers revealed by surfaceplasmons,” J. Phys. Chem., vol. 93, pp. 2106–2111, Mar. 1989.
[29] Macdonald, Impedance Spectroscopy. John Wiley & Sons, 1987.
[30] F. Kohlrausch, “Über die elektromotorische kraft sehr dünner gasschichtenauf metallplatten,” Annalen der Physik und Chemie , série 2 no. 148, vol. 224,p. 143, 1873.
[31] O. Heaviside, Electromagnetic Theory, The Complete and Unabridged Editionof Vols. I, II, and III. With a critical and historical introduction by ErnstWeber. Dover Publications, 1950.
[32] I. K’Owino and O. Sadik, “Impedance spectroscopy: A powerful tool for rapidbiomolecular screening and cell culture monitoring,” Electroanalysis, vol. 17,no. 23, pp. 2101–2113, 2005.
[33] I. N. Bronstein and K. A. Semendjajew, Taschenbuch der Mathematik. HarriDeutsch, 2000.
[34] H. W. Bode, “Variable equalizers,” Bell Syst. Tech. J, vol. 17, pp. 229–244,1938.
[35] J. H. Sluyters, “On the impedance of galvanic cells i,” Rec. Trav. Chim,vol. 79, pp. 1092–1100, 1960.
[36] J. H. Sluyters and J. J. C. Oomen, “On the impedance of galvanic cells ii,”Rec. Trav. Chim, vol. 79, pp. 1101–1110, 1960.
[37] J. E. Bauerle, “Study of solid electrolyte polarization by a complex admittan-ce method,” Journal of Physics and Chemistry of Solids, vol. 30, pp. 2657–2670, Dec. 1969.
[38] A. K. Jonscher, “The dielectric response: A review of data and their newinterpretation,” Physics of thin films, vol. 11, pp. 205–317, 1980.
[39] C. Whitehouse, R. O’Flanagan, B. Lindholm-Sethson, B. Movaghar, andA. Nelson, “Application of electrochemical impedance spectroscopy to thestudy of dioleoyl phosphatidylcholine monolayers on mercury,” Langmuir,vol. 20, pp. 136–144, Jan. 2004.

LITERATUR 167
[40] R. Naumann, S. Schiller, F. Giess, B. Grohe, K. Hartman, I. Karcher, I. Ko-per, J. Lubben, K. Vasilev, and W. Knoll, “Tethered lipid bilayers on ultraflatgold surfaces,” Langmuir, vol. 19, pp. 5435–5443, June 2003.
[41] R. Naumann, E. K. Schmidt, A. Jonczyk, K. Fendler, B. Kadenbach, T. Lie-bermann, A. Offenhausser, and W. Knoll, “The peptide-tethered lipid mem-brane as a biomimetic system to incorporate cytochrome c oxidase in a func-tionally active form,” Biosensors and Bioelectronics, vol. 14, pp. 651–662,Oct. 1999.
[42] F. Giess et al., “The protein-tethered lipid bilayer: A novel mimic of thebiological membrane,” Biophys. J., vol. 87, pp. 3213–3220, Nov. 2004.
[43] R. A. Marcus and N. Sutin, “Electron transfers in chemistry and biology,”Biochimica et Biophysica Acta (BBA) - Reviews on Bioenergetics, vol. 811,pp. 265–322, Aug. 1985.
[44] R. A. Marcus, “On the theory of electron-transfer reactions. vi. unified treat-ment for homogeneous and electrode reactions,” The Journal of ChemicalPhysics, vol. 43, pp. 679–701, 1965.
[45] J. E. B. Randles, “A cathode ray polarograph. part ii. the current-voltagecurves,” Trans. Faraday Soc, vol. 44, pp. 327 – 338, 1948.
[46] A. Sevcik, “Oscillographic polarography with triangular voltage,” Collect.Czech. Chem. Commun., vol. 13, pp. 349–377, 1948.
[47] D. Britz, Digital Simulation In Electrochemistry. Springer, 2005.
[48] A. J. Bard and L. Faulkner, Electrochemical Methods. John Wiley & Sons,2001.
[49] J. Heinze, “Cyclovoltammetrie - die spektroskopie des elektrochemikers,” An-gewandte Chemie, vol. 96, no. 11, pp. 823–840, 1984.
[50] V. Balzani, Electron Tranfer in Chemistry Vol. 1-5. Wiley-VCH Verlag, 2001.
[51] D. Pletcher, A first course in electrode processes. The Electrochemical Con-sultancy, 1991.
[52] B. Speiser, “Elektroanalytische methoden i: Elektrodenreaktionen und chro-noamperometrie,” Chemie in unserer Zeit, vol. 15, no. 1, pp. 21–26, 1981.
[53] B. Speiser, “Elektroanalytische methoden ii: Cyclische voltammetrie,” Che-mie in unserer Zeit, vol. 15, no. 2, pp. 62–67, 1981.
[54] P. He and L. R. Faulkner, “Intelligent, automatic compensation of solutionresistance,” Anal. Chem., vol. 58, no. 3, pp. 517–523, 1986.

LITERATUR 168
[55] H. Wackerbarth, K. Udo, G. Walter, and P. Hildebrandt, “Novel time-resolvedsurface-enhanced (resonance) raman spectroscopic technique for studying thedynamics of interfacial processes: Application to the electron transfer reactionof cytochrome c at a silver electrode,” Applied Spectroscopy, vol. Volume 53Number 3, pp. 283–291(9), 1999.
[56] A. Smekal, “Zur quantentheorie der dispersion,” Naturwissenschaften,vol. V11, pp. 873–875, Oct. 1923.
[57] C. V. Raman and K. S. Krishnan, “A new type of secondary radiation,”Nature, vol. 121, pp. 501–502, 1928.
[58] N. B. Colthup, Introduction to infrared and Raman spectroscopy. AcademicPress, 1990.
[59] R. J. H. Clark and T. J. Dines, “Resonance raman spectroscopy, and itsapplication to inorganic chemistry. new analytical methods,” AngewandteChemie International Edition in English, vol. 25, pp. 131–158, 1986.
[60] K. Kneipp, H. Kneipp, and M. Feld, Surface-enhanced Ramanscattering andbiophysics. Journal of Physics: Condensed Matter, 2002.
[61] V. L. Ginzburg, I. L. Fabelinsky, and M. D. Galanin, “Peter p. shorygin: Anappreciation on his 90th birthday,” Journal of Raman Spectroscopy, vol. 32,no. 6-7, pp. 407–411, 2001.
[62] A. C. Albrecht, “On the theory of raman intensities,” The Journal of ChemicalPhysics, vol. Volume 34, pp. 1476–1484, 1961.
[63] T. G. Spiro and T. C. Strekas, “Resonance raman spectra of hemoglobin andcytochrome c: Inverse polarization and vibronic scattering,” PNAS, vol. 69,pp. 2622–2626, Sept. 1972.
[64] D. A. Long, Raman Spectroscopy. McGraw-Hill International Book Company,1977.
[65] H. und Wolf, Molekülphysik und Quantenchemie. Springer-Verlag, 1998.
[66] M. Fleischmann, P. J. Hendra, and A. J. McQuillan, “Raman spectra ofpyridine adsorbed at a silver electrode,” Chemical Physics Letters, vol. 26,pp. 163–166, May 1974.
[67] A. Otto, Topics in applied physics, Volume 54, Light Scattering in Solids IV.Springer, 1984.
[68] A. Otto, “Surface-enhanced raman scattering of adsorbates,” Journal of Ra-man Spectroscopy, vol. 22, no. 12, pp. 743–752, 1991.

LITERATUR 169
[69] E. Koglin, “Oberflächenverstärkte raman-spektroskopie (sers) von biopoly-meren,” Fresenius’ Journal of Analytical Chemistry, vol. V321, pp. 638–639,Jan. 1985.
[70] M. Gouterman, The Porphyrins, Vol. III, Part A, pp 1-156. Academic Press,New York, 1978.
[71] Fran and Adar, The Porphyrins, Vol. III, Part A, pp: 167-223. AcademicPress, New York, 1978.
[72] T. G. Spiro, R. S. Czernuszewicz, and X.-Y. Li, “Metalloporphyrin structureand dynamics from resonance raman spectroscopy,” Coordination ChemistryReviews, vol. 100, pp. 541–571, Apr. 1990.
[73] S. Hu, K. Morris, J. Singh, K. Smith, and S. T., “Complete assignment ofcytochrome c resonance raman spectra via enzymatic reconstitution withisotopically labeled hemes,” J . Am. Chem. SOC., vol. 115, pp. 12446–12458,1993.
[74] R. Felton and N.-T. Yu, The Porphyrins, Vol. III, Part A, pp. 347-393.Academic Press, New York, 1978.
[75] T. Kitagawa and O. Y., Structure and Bonding 64 (Seite 71-114). Springer-Verlag, 1987.
[76] T. Kitagawa, M. Abe, and H. Ogoshi, “Resonance raman spectra ofoctaethylporphyrinato-ni(ii) and meso-deuterated and 15n substituted de-rivatives. i. observation and assignments of nonfundamental raman lines,”The Journal of Chemical Physics – November 15, 1978 – Volume 69, Issue10, pp. 4516-4525, vol. 69, pp. 4516–4525, 1978.
[77] M. Abe, T. Kitagawa, and K. Y., “Resonance raman spectra ofoctaethylporphyrinato-ni(ii) and meso-deuterated and 15n substituted de-rivatives. ii. a normal coordinate analysis,” The Journal of Chemical Physics– November 15, 1978 – Volume 69, Issue 10, pp. 4526-4534, vol. 69, pp. 4526–4534, 1978.
[78] M. Friedrich, F. Giess, R. Naumann, W. Knoll, K. Ataka, J. Heberle, J. Hra-bakova, D. Murgida, and P. Hildebrandt, “Active site structure and redoxprocesses of cytochrome c oxidase immobilised in a novel biomimetic lipidmembrane on an electrode,” Chemical Communications, no. 21, pp. 2376–2377, 2004.
[79] J. H. P. H. Jana Hrabakova, Kenichi Ataka and D. H. Murgida, “Long distanceelectron transfer in cytochrome c oxidase immobilised on electrodes. a surfaceenhanced resonance raman spectroscopic study,” Phys. Chem. Chem. Phys,vol. 8, p. 759–766, 2006.

LITERATUR 170
[80] S. W. Han, Y. C. Ching, S. L. Hammes, and D. L. Rousseau, “Vibrationalstructure of the formyl group on heme a. implications on the properties ofcytochrome c oxidase.,” Biophys J., vol. 60, p. 45–52, 1991.
[81] S. Iwata, C. Ostermeier, B. Ludwig, and H. Michel, “Structure at 2.8 a resolu-tion of cytochrome c oxidase from paracoccus denitrificans,” Nature, vol. 376,pp. 660–669, Aug. 1995.
[82] U. Pfitzner, A. Odenwald, T. Ostermann, L. Weingard, B. Ludwig, and O.-M. H. Richter, “Cytochrome c oxidase (heme aa3) from paracoccus denitrifi-cans: Analysis of mutations in putative proton channels of subunit i,” Journalof Bioenergetics and Biomembranes, vol. 30, pp. 89–97, Feb. 1998.
[83] J. Hosler, J. Fetter, M. Tecklenburg, M. Espe, C. Lerma, and S. Ferguson-Miller, “Cytochrome aa3 of rhodobacter sphaeroides as a model for mitochon-drial cytochrome c oxidase. purification, kinetics, proton pumping, and spec-tral analysis,” J. Biol. Chem., vol. 267, pp. 24264–24272, Dec. 1992.
[84] M. Svensson-Ek, J. Abramson, G. Larsson, S. Tornroth, P. Brzezinski, andS. Iwata, “The x-ray crystal structures of wild-type and eq(i-286) mutantcytochrome c oxidases from rhodobacter sphaeroides,” Journal of MolecularBiology, vol. 321, pp. 329–339, Aug. 2002.
[85] M. H. M. Olsson and A. Warshel, “Monte carlo simulations of proton pumps:On the working principles of the biological valve that controls proton pum-ping in cytochrome c oxidase,” PNAS, vol. 103, pp. 6500–6505, Apr. 2006.
[86] T. Baumgart, M. Kreiter, H. Lauer, R. Naumann, G. Jung, A. Jonczyk,A. Offenhausser, and W. Knoll, “Fusion of small unilamellar vesicles ontolaterally mixed self-assembled monolayers of thiolipopeptides,” Journal ofColloid and Interface Science, vol. 258, pp. 298–309, Feb. 2003.
[87] D. T. Sawyer, A. Sobkowiak, and J. L. Roberts, Electrochemistry for Che-mists. John Wiley & Sons, 1995.
[88] N. S. Knoll, Kenichi, “functional tethered bilayer lipid membranes,” Ultrathinelectrochemical chemo- and biosensors Springer, pp. 239–254, 2004.
[89] S. Sivasankar, S. Subramaniam, and D. Leckband, “Direct molecular levelmeasurements of the electrostatic properties of a protein surface,” Proc. Natl.Acad. Sci, vol. 95, p. 12961–12966, 1998.
[90] P. E. Smith, R. M. Brunne, A. E. Mark, and W. F. Van Gunsteren, “Dielec-tric properties of trypsin inhibitor and lysozyme calculated from moleculardynamics simulations,” J. Phys. Chem., vol. 97, pp. 2009–2014, Mar. 1993.

LITERATUR 171
[91] E. Laviron, “General expression of the linear potential sweep voltammogramin the case of diffusionless electrochemical systems,” Journal of Electroana-lytical Chemistry, vol. 101, pp. 19–28, July 1979.
[92] J. Hirst, J. Duff, G. Jameson, M. Kemper, B. Burgess, and F. Armstrong,“Kinetics and mechanism of redox-coupled, long-range proton transfer in aniron-sulfur protein. investigation by fast-scan protein-film voltammetry,” J.Am. Chem. Soc., vol. 120, pp. 7085–7094, July 1998.
[93] L. Jeuken, J. McEvoy, and F. Armstrong, “Insights into gated electron-transfer kinetics at the electrode-protein interface: A square wave voltam-metry study of the blue copper protein azurin,” J. Phys. Chem. B, vol. 106,pp. 2304–2313, Mar. 2002.
[94] A. Haas, D. Pilloud, K. Reddy, G. Babcock, C. Moser, J. Blasie, and P. Dut-ton, “Cytochrome c and cytochrome c oxidase: Monolayer assemblies andcatalysis,” J. Phys. Chem. B, vol. 105, pp. 11351–11362, Nov. 2001.
[95] M. C. R. James D. Burgess and F. M. Hawkridge, “Cytochrome c oxidaseimmobilized in stable supported lipid bilayer membranes,” Langmuir, vol. 14,pp. 2467–2475, 1998.
[96] J. K. Cullison, F. M. Hawkridge, N. Nakashima, and S. Yoshikawa, “A studyof cytochrome c oxidase in lipid bilayer membranes on electrode surfaces,”Langmuir, vol. 10, pp. 877–882, Mar. 1994.
[97] D. E. Reed and F. M. Hawkridge, “Direct electron transfer reactions of cyto-chrome c at silver electrodes,” Anal. Chem., vol. 59, no. 19, pp. 2334–2339,1987.
[98] D. Millo, A. Ranieri, W. Koot, C. Gooijer, and G. vanderZwan, “Towardscombined electrochemistry and surface-enhanced resonance raman of hemeproteins: Improvement of diffusion electrochemistry of cytochrome c at sil-ver electrodes chemically modified with 4-mercaptopyridine,” Anal. Chem.,vol. 78, no. 15, pp. 5622–5625, 2006.
[99] J. Burgess, M. Rhoten, and F. Hawkridge, “Observation of the resting andpulsed states of cytochrome c oxidase in electrode-supported lipid bilayermembranes,” J. Am. Chem. Soc., vol. 120, pp. 4488–4491, May 1998.
[100] Y. A. Ivashchuk-Kienbaum, “Monitoring of the membrane potential in pro-teoliposomes with incorporated cytochrome-c oxidase using the fluorescentdye indocyanine,” Journal of Membrane Biology, vol. 151, pp. 247–259, June1996.
[101] R. Naumann et al., “Simulation of active and passive ion transport acrosstethered bilayers,” ...noch nicht veröffentlicht.

LITERATUR 172
[102] L. Lü, J. Fuh, and J.-S. Wong, Laser induced materials and processes forrapid prototyping. Kluwer Academic Publishers.
[103] D. Millo, A. Bonifacio, A. Ranieri, M. Borsari, C. Gooijer, and G. vander-Zwan, “Voltammetric and surface-enhanced resonance raman spectroscopiccharacterization of cytochrome c adsorbed on a 4-mercaptopyridine mono-layer on silver electrodes,” Langmuir, vol. 23, pp. 4340–4345, Apr. 2007.
[104] P. Hildebrandt and M. Stockburger, “Cytochrome c at charged interfaces.1. conformational and redox equilibria at the electrode/electrolyte interfaceprobed by surface-enhanced resonance raman spectroscopy,” Biochemistry,vol. 28, pp. 6710–6721, 1989.
[105] Murgida, Hildebrandt, and Waldeck, “Surface-enhanced resonance ramanspectroscopic and electrochemical study of cytochrome c bound on electrodesthrough coordination with pyridinyl-terminated self-assembledmonolayers,”J.Phys.Chem.B, vol. 108, pp. 2261–2269, 2004.
[106] V. Ziswiler et al., Biologie. Springer, 1981.
[107] H.-m. Lee, T. Das, D. Rousseau, D. Mills, S. Ferguson-Miller, and R. Gen-nis, “Mutations in the putative h-channel in the cytochrome c oxidase fromrhodobacter sphaeroides show that this channel is not important for protonconduction but reveal modulation of the properties of heme a,” Biochemistry,vol. 39, pp. 2989–2996, Mar. 2000.
[108] P. Hildebrandt, F. Vanhecke, G. Buse, T. Soulimane, and A. G. Mauk, “Re-sonance raman study of the interactions between cytochrome c variants andcytochrome c oxidase,” Biochemistry, vol. 32, no. 40, pp. 10912–10922, 1993.
[109] P. Boulas, M. Gómez-Kaifer, and L. Echegoyen, “Supramolekulare elektro-chemie,” Angewandte Chemie, vol. 110, no. 3, pp. 226–258, 1998.
[110] J. M. Peloquin, E. J. Bylina, D. C. Youvan, and D. F. Bocian, “Resonanceraman studies of genetically modified reaction centers from rhodobacter cap-sulatus,” Biochemistry, vol. 29, no. 36, pp. 8417–8424, 1990.
[111] C. Le Moigne, B. Schoepp, S. Othman, A. Vermeglio, and A. Desbois, “Dis-tinct structures and environments for the three hemes of the cytochrome bc1complex from rhodospirillum rubrum. a resonance raman study using b-bandexcitations,” Biochemistry, vol. 38, no. 3, pp. 1066–1076, 1999.
[112] F. Gao, H. Qin, D. Knaff, L. Zhang, L. Yu, C. Yu, and M. Ondrias, “Q-band resonance raman spectra of oxidized and reduced mitochondrial bc1complexes,” Biochemistry, vol. 37, no. 27, pp. 9751–9758, 1998.

LITERATUR 173
[113] D. D. Hobbs, A. Kriauciunas, S. Guner, D. B. Knaff, and M. R. Ondrias,“Resonance raman spectroscopy of cytochrome bc1 complexes from rhodospi-rillum rubrum: Initial characterization and reductive titrations,” Biochimicaet Biophysica Acta (BBA) - Bioenergetics, vol. 1018, pp. 47–54, July 1990.
[114] K. R. Beebe, R. J. Pell, and M. B. Seasholtz, Chemometrics: A PracticalGuide. J. Wiley & Sons, 1998.
[115] M. Friedrich, V. Kirste, J. Zhu, R. Gennis, W. Knoll, and R. Naumann,“Activity of membrane proteins immobilized in a biomimetic architecture asa function of packing density,” ...noch nicht veröffentlicht.
[116] J. Vanderkooi, G. Maniara, T. Green, and D. Wilson, “An optical methodfor measurement of dioxygen concentration based upon quenching of phos-phorescence,” J. Biol. Chem., vol. 262, pp. 5476–5482, Apr. 1987.

DanksagungenAll denen, die zum Gelingen dieser Arbeit beigetragen haben, gilt mein Dank.
Für die freundliche Betreuung seitens des Max-Planck-Instituts für Polymerfor-schung bedanke ich mich besonders bei Frau Dr. Naumann. Über den Beistandbei Fragen und Problemen hinaus engagierte sie sich für meine Arbeit und meinePerson. Ich danke Herrn Prof. Kremer und Herrn Prof. Knoll für die Möglichkeitmeine Diplomarbeit am MPI-P zu absolvieren.
Dank geht an Marcel Friedrich für die Einführung in die hohe Kunst der TR-SERRS-Spektroskopie und in die Herstellung und Untersuchung von ptBLM-Sys-temen. Der Dank geht auch an Vinzenz Kirste, Slavoj Kresak und Asmorom Ki-brom für die Unterstützung im Labor und für die Hilfe bei den elektrochemischenMessungen.
Außerdem bedanke ich mich bei Hans-Jörg Menges für die vielen Ratschläge unddie Unterstützung im Raman-Labor.
Den Werkstätten am MPI-P (Feinmechanik und Elektronik) danke ich für die pro-fessionelle Realisierung meiner Konstruktionen und die schnelle Bearbeitung un-planmäßiger Aufträge. Hierbei gilt besonderer Dank Herrn Gerstenberg und HerrnRichter für die Unterstützung bei der Konstruktion der neuen SERRS-Zelle.
Für die Anfertigung der AFM- und Nanoscope-Aufnahmen bedanke ich mich beiHelma Burg und Andreas Best.
Ich bedanke mich bei der gesamten Knoll-Gruppe für die herzliche Aufnahme unddas angenehme, internationale Arbeitsklima.
Mein herzlichster Dank gilt meiner gesamten Familie und meiner Freundin fürdie aufbauende Unterstützung.


















