
Einfluss einer Gentherapie mit Thrombospondin 1 und 2 auf ... · 3.1 Bedeutung der Diabetischen...
86
Aus der Medizinischen Klinik 4 – Nephrologie und Hypertensiologie der Friedrich-Alexander-Universität Erlangen-Nürnberg Direktor: Prof. Dr. med. Kai-Uwe Eckardt Einfluss einer Gentherapie mit Thrombospondin 1 und 2 auf die Progression der Diabetischen Nephropathie in der Maus Inaugural-Dissertation zur Erlangung der Doktorwürde der Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg vorgelegt von Sebastian Graf aus Eichstätt
Transcript of Einfluss einer Gentherapie mit Thrombospondin 1 und 2 auf ... · 3.1 Bedeutung der Diabetischen...

Aus der Medizinischen Klinik 4 – Nephrologie und Hypertensiologie
der
Friedrich-Alexander-Universität Erlangen-Nürnberg
Direktor: Prof. Dr. med. Kai-Uwe Eckardt
Einfluss einer Gentherapie mit
Thrombospondin 1 und 2 auf die Progression
der Diabetischen Nephropathie
in der Maus
Inaugural-Dissertation
zur Erlangung der Doktorwürde
der Medizinischen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
vorgelegt von
Sebastian Graf
aus
Eichstätt

2
Gedruckt mit Erlaubnis der
Medizinischen Fakultät der Friedrich-Alexander-Universität Erlangen-Nürnberg
Dekan: Prof. Dr. med. Dr. h.c. Jürgen Schüttler Referent: PD Dr. rer. nat. Christoph Daniel Korreferentin: Prof. Dr. med. Kerstin Amann Tag der mündlichen Prüfung: 14.3.2012

3
Für meine Eltern

4
Inhaltsverzeichnis
1 Zusammenfassung ................................................................................. 7
2 Abstract ................................................................................................... 9
3 Einleitung .............................................................................................. 10
3.1 Bedeutung der Diabetischen Nephropathie ............................................ 10
3.2 Histologische Kennzeichen ..................................................................... 11
3.3 Pathomechanismen der Diabetischen Nephropathie .............................. 12
3.3.1 Biochemische Veränderungen durch Hyperglykämie ............................. 12
3.3.2 Hämodynamische Veränderungen ......................................................... 13
3.3.3 Beeinflussung durch das Renin-Angiotensin-System ............................. 14
3.3.4 Genetische Determinanten der Diabetischen Nephropathie ................... 14
3.3.5 Rolle der Zytokine ................................................................................... 14
3.4 TGF-β als Mediator bei der Diabetischen Nephropathie ......................... 15
3.4.1 TGF-β als verbindendes Element der Pathomechanismen .................... 15
3.4.2 Aktuelle Erkenntnisse über den Zusammenhang von TGF-β und der Diabetischen Nephropathie .............................................................. 16
3.4.3 Molekularstruktur und Signaltransduktionswege von TGF-β .................. 16
3.5 Aufbau und Funktion der Thrombospondine ........................................... 18
3.5.1 Familie der Thrombospondine ................................................................ 18
3.5.2 Biologische Rolle der Thrombospondine ................................................ 19
3.5.3 Molekulare Struktur der Thrombospondine 1 und 2 ................................ 20
3.5.4 Interaktion der Thrombospondine 1 und 2 mit TGF-β ............................. 21
3.6 Zielsetzung der Arbeit ............................................................................. 23
4 Material und Methoden ......................................................................... 24
4.1 Tierversuch ............................................................................................. 24
4.1.1 Versuchstiere .......................................................................................... 24
4.1.2 Experimentelles Design .......................................................................... 24
4.1.3 Vorgehensweise bei der Nephrektomie .................................................. 27
4.1.4 Töten der Tiere ....................................................................................... 27
4.2 Gewinnung & Analyse des Probenmaterials ........................................... 27
4.2.1 Glukosespiegel im Blut ........................................................................... 27
4.2.2 24 Stunden Sammelurin ......................................................................... 28
4.2.3 Gewichtskontrolle ................................................................................... 28
4.2.4 Serumgewinnung .................................................................................... 28
4.2.5 Bestimmung von Serumharnstoff und Serum- und Urinkreatinin ............ 28
4.2.6 Berechnung der Kreatinin-Clearance ...................................................... 29
4.2.7 Bestimmung der Proteinmenge im Urin .................................................. 29
4.2.8 Konservierung des Versuchstiergewebes ............................................... 30

5
4.3 Aufbereitung der Gewebe für die Immunhistochemie ............................. 31
4.3.1 Prozessieren des Gewebes .................................................................... 31
4.3.2 Anfertigen von Schnitten aus Gefriergewebe .......................................... 33
4.3.3 Anfertigen von Schnitten aus Paraffinblöcken ........................................ 33
4.4 (Immun)histochemische Färbungen ....................................................... 33
4.4.1 Periodsäure-Schiff-Färbung (PAS) ......................................................... 33
4.4.2 Immunfluoreszenzfärbung an Gefrierschnitten ....................................... 34
4.4.3 Immunfluoreszenzfärbung an Paraffinschnitten ...................................... 35
4.4.4 Immunperoxidasefärbung ....................................................................... 36
4.4.5 Primärantikörper ..................................................................................... 37
4.4.6 Sekundärantikörper und deren Substrate ............................................... 38
4.4.7 Mikroskopische Auswertung ................................................................... 39
4.5 Zymogramme .......................................................................................... 40
4.5.1 Proteinextraktion ..................................................................................... 40
4.5.2 Proteinbestimmung detergenzhaltiger Proben ........................................ 41
4.5.3 Anfertigen der nativen Polyacrylamid-Gele ............................................. 41
4.5.4 Beladen und Lauf der Gele ..................................................................... 41
4.5.5 Inkubation der Gele ................................................................................ 42
4.5.6 Blockieren der Matrixmetalloproteinasen ................................................ 42
4.5.7 Auswertung ............................................................................................. 42
4.5.8 Lösungen ................................................................................................ 43
4.6 Enzyme Linked Immunosorbent Assay ................................................... 44
4.6.1 Messung der Albuminurie ....................................................................... 44
4.6.2 Pufferlösungen ........................................................................................ 45
4.7 Real-Time PCR ....................................................................................... 45
4.7.1 RNA-Isolierung ....................................................................................... 45
4.7.2 Herstellung der cDNA ............................................................................. 46
4.7.3 Real-Time PCR ....................................................................................... 46
4.8 Statistische Auswertung.......................................................................... 47
4.9 Material ................................................................................................... 47
4.9.1 Chemikalien ............................................................................................ 47
4.9.2 Medikamente & Plasmide ....................................................................... 49
4.9.3 Geräte ..................................................................................................... 49
4.9.4 Verbrauchsmaterial ................................................................................. 51
4.9.5 Lösungen ................................................................................................ 51
5 Ergebnisse ............................................................................................ 52
5.1 Die Gentherapie hat keinen Einfluss auf die Blutzuckerwerte ................. 52
5.2 Die Gentherapie hat keinen Einfluss auf das Nierengewicht .................. 53
5.3 Thrombospondin 1 erhöht und Thrombospondin 2 reduziert das Level von aktivem TGF-β ........................................................................ 54

6
5.4 Modulation der Fibrose durch TSP1 und TSP2 ...................................... 56
5.5 Diabetische Tiere zeigen eine erhöhte MMP2 Aktivität ........................... 59
5.6 TSP2 schützt Podozyten vor einer Schädigung ...................................... 60
5.7 TSP2 hemmt die Infiltration von Entzündungszellen ............................... 61
5.8 Diabetes Mellitus Typ 1 führt zu einem renalen peritubulären Kapillarverlust ......................................................................................... 63
5.9 Die Behandlung mit TSP1 und TSP2 hat keinen Einfluss auf die Nierenfunktion ......................................................................................... 64
6 Diskussion ............................................................................................. 66
6.1 Aktuelle Datenlage .................................................................................. 66
6.2 Induktion eines Diabetes Mellitus und Entwicklung einer Diabetischen Nephropathie ..................................................................... 67
6.3 Aktivierung von TGF-β ............................................................................ 68
6.4 Modulation der Fibrose ........................................................................... 69
6.5 Infiltration durch Entzündungszellen ....................................................... 71
6.6 Schädigung der Podozyten ..................................................................... 71
6.7 Verlust der Kapillardichte ........................................................................ 72
6.8 Beeinflussung der Nierenfunktion ........................................................... 72
6.9 Zusammenfassung, Kritische Beurteilung und Ausblick ......................... 74
7 Abbildungsverzeichnis ......................................................................... 75
8 Literaturverzeichnis .............................................................................. 76
9 Abkürzungsverzeichnis ........................................................................ 84
10 Danksagung .......................................................................................... 86

7
1 Zusammenfassung
Hintergrund und Ziele: Diabetes Mellitus ist mittlerweile die häufigste
Ursache für eine fortgeschrittene Niereninsuffizienz und Dialysepflichtigkeit in
der westlichen Welt. Das Zytokin Transforming Growth Factor beta (TGF-β)
spielt eine zentrale Rolle bei der Krankheitsentstehung und Progression.
Damit TGF-β an seine Rezeptoren binden kann, muss es zuerst aktiviert
werden. Ein bereits bekannter Aktivator ist Thrombospondin 1 (TSP1). In
dieser Arbeit soll zum einen gezeigt werden, dass durch systemische Über-
expression von TSP1 die diabetische Nephropathie in vivo aggraviert wird.
Zum anderen soll demonstriert werden, dass die systemische Überexpres-
sion von TSP2 die Thrombospondin 1 vermittelte Aktivierung inhibiert und
damit die Progression der diabetischen Nephropathie reduziert.
Methodische Aspekte: Im Versuch wurde bei 27 Mäusen mittels Streptozoto-
cin (STZ) ein Diabetes Mellitus Typ 1 induziert. Bei einer Gruppe (n=9)
wurde mittels intramuskulärer Transfektion mit einem Plasmid TSP1 syste-
misch überexprimiert, bei einer weiteren Gruppe (n=9) wurde TSP2
systemisch überexprimiert. Bei einer dritten Gruppe (n=9) verwendeten wir
ein Kontrollplasmid (pUBlux). Als gesunde Kontrolle diente eine Gruppe von
8 Tieren ohne jegliche STZ-Injektion. Die diabetischen Tiere wurden 14
Wochen unter hyperglykämischen Bedingungen beobachtet. Mittels 24h-
Sammelurin wurde die Nierenfunktion überprüft. Das Level der Aktivierung
von TGF-β, der Grad der Fibrosierung, die Infiltration von Entzündungszel-
len, die Podozytenschädigung und der peritubuläre Kapillarverlust wurden
mit Hilfe von (immun)histochemischen Methoden in Nierenbiopsien überprüft.
Weiterhin gab eine Real-Time PCR Messung Auskunft über die Expression
von TGF-β1. Zymogramme zeigten die Aktivität der Matrix-Metalloproteinase
des Typs 2 (MMP2) an.
Ergebnisse: Es konnte gezeigt werden, dass die systemische
Überexpression von TSP1 das Level von aktivem TGF-β erhöht, und die
Überexpression von TSP2 das Level von aktivem TGF-β erniedrigt. Weiter-
hin führte die TSP2 Überexpression zu einer verringerten Deposition von
Kollagen IV, einer abgemilderten Infiltration durch Entzündungszellen
(Makrophagen, CD3+, CD4+, und CD8+ Zellen) und zu einem geringeren

8
Verlust der Podozytenanzahl. Die Aktivität von MMP2 und die Nierenfunktion
wurden durch die Überexpressionen nicht signifikant beeinflusst.
Praktische Schlussfolgerungen: Zusammenfassend konnten wir in dieser
Studie erstmalig in vivo nach dem Ursache-Wirkungs-Prinzip zeigen, dass
eine Überexpression von TSP1 die Aktivität von TGF-β bei der diabetischen
Nephropathie steigert und TSP2 ebendiese Aktivitätssteigerung verhindert.
Weiterhin reduzierte die TSP2 Überexpression die histologische Ausprägung
der diabetischen Nephropathie. Daher ist das Eingreifen in die
Thrombospondin-TGF-β-Achse eine vielversprechende Option bei der
Prävention und Therapie der diabetischen Nephropathie.

9
2 Abstract
Background: Diabetes is the most common cause of end-stage renal disease
in the western world. The cytokine Transforming Growth Factor beta (TGF-β)
plays a central role in the development and progression of diabetic
nephropathy. TGF-β needs to be activated to be able to bind to its receptors.
One known activator is Thrombospondin 1 (TSP1). This study aims to show,
that the systemic overexpression of TSP1 aggravates diabetic nephropathy
in vivo. Furthermore the study intends to demonstrate that the systemic
overexpression of TSP2 inhibits the TSP1 mediated activation of TGF-β and
therefore reduces the progression of diabetic nephropathy.
Methods: Type 1 diabetes was induced in wild-type mice (n=27) via the
injection of Streptocotocin. After intramuscular transfection of a plasmid one
group of 9 animals overexpressed TSP1, another group of 9 mice
overexpressed TSP2 and a control plasmid was used in a third group (n=9)
(pUBlux). We compared these three groups with one group of non-diabetic
mice (n=8). The animals lived 14 weeks under hyperglycemic conditions.
Renal function was evaluated with the help of 24h-urine samples. We
measured the level of activation of TGF-β, the degree of fibrosis/sclerosis,
the damage of podocytes and the loss of capillaries with
(immune)histochemical stainings of renal biopsies. With a real-time PCR, we
quantified the level of expression of TGF-β1. Furthermore zymographies
showed the activation of Matrix-Metalloproteinase 2 (MMP2).
Results: The systemic overexpression of TSP1 increased the level of active
TGF-β while TSP2 overexpression reduced it. Furthermore TSP2 overex-
pression reduced the deposition of Collagen Type IV and the influx of
inflammatory cells (macrophages, CD3+, CD4+ and CD8+ cells). Moreover,
TSP2 reduced the loss of podocytes. The activity of MMP2 and the renal
function was not significantly affected by the overexpressions.
Conclusions: This study shows for the first time in vivo that TSP1 increases
the level of active TGF-β in diabetic nephropathy, while TSP2 reduces it.
Furthermore, the overexpression of TSP2 ameliorated some histological
findings of diabetic nephropathy. Therefore, TSP1 blocking therapies may be
considered as a promising future treatment option for diabetic nephropathy.

10
3 Einleitung
Dieses Kapitel stellt einige Grundlagen der diabetischen Nephropathie dar:
Besonders berücksichtigt werden die verschiedenen Pathomechanismen, die
auch Möglichkeiten der Intervention, wie im Rahmen der vorgestellten
Studie, bieten.
3.1 Bedeutung der Diabetischen Nephropathie
Der Begriff „Diabetes Mellitus“ leitet sich vom altgriechischen διαβαίνειν
(hindurchfließen) und lateinischen mellitus (honigsüß) ab. Schon in der
Antike war das Krankheitsbild mit dem in Folge der Glucosurie honigsüß
schmeckenden Urin bekannt. Bis zum heutigen Tage hat sich der Diabetes
Mellitus mit einer Prävalenz von circa 10 % in Deutschland zu einer „Volks-
krankheit“ entwickelt, mit enormen gesundheitlichen Folgen für den Patienten
und immensen sozioökonomischen Auswirkungen auf die Gesellschaft.
Diabetes Mellitus ist jedoch nicht nur ein nationales Problem; weltweit wird
die Prävalenz weiter zunehmen [93]: Die Weltgesundheitsorganisation
rechnet mit einer Zunahme von aktuell 250 Millionen Erkrankten auf über 350
Millionen im Jahr 2030 [84].
Betroffene Patienten leiden dabei nicht so sehr an der Stoffwechselstörung
selbst, sondern an den Komplikationen, die ein langjähriger Diabetes mit sich
bringt: So kommt es in Folge einer Makroangiopathie zu einer erhöhten Rate
an kardiovaskulären Ereignissen wie Myokardinfarkten und Schlaganfällen.
Auf Seiten der kleinen Gefäße kommt es zu einer Retinopathie, Neuropathie,
Mikroangiopathie der kleinen Koronararterien und mehr. Ein weiteres betrof-
fenes Endorgan ist die Niere, deren durch die Stoffwechselstörung
verursachten Schäden unter dem Begriff der „Diabetischen Nephropathie“
subsummiert werden. Dabei kann die diabetische Nephropathie im fortge-
schrittenen Stadium bis zu einer chronischen Niereninsuffizienz und final zu
einer Dialysebedürftigkeit führen.
Mit der Prävalenzzunahme des Diabetes Mellitus kommt es auch zu einem
dramatischen Anstieg der Häufigkeit von niereninsuffizienten Patienten unter
den Diabetikern: Dies wird mittlerweile als „Katastrophe von globalem
Ausmaße“ angesehen [64]. So ist die diabetische Nephropathie mittlerweile

11
die häufigste Ursache für die fortgeschrittene Niereninsuffizienz in der
westlichen Welt [65]. In Deutschland ging man 2006 bei über 48.000 dialyse-
pflichtigen Patienten in 28 % der Fälle von Diabetes Mellitus als Verursacher
der Nierenfehlfunktion aus [70]. Neben den individuellen Auswirkungen auf
den Patienten ist die ökologische Dimension des Problems mit direkten
Behandlungskosten von 30.000 bis 50.000 Euro im Jahr beträchtlich [68].
Laut der Deutschen Diabetes Gesellschaft beruht die aktuelle Therapie der
diabetischen Nephropathie auf zwei Säulen [34]: Auf der einen Seite sollte
eine strikte Kontrolle des Blutzuckerspiegels beachtet werden. Zum anderen
sollten auch normotensive Patienten konsequent mit Blutdruckmedikamenten
behandelt werden. Hier werden vor allem ACE-Hemmer und Angiotensin-
Rezeptor-Blocker empfohlen, die auch in die Pathophysiologie und zugrun-
deliegenden Mechanismen eingreifen. Anzumerken ist, dass durch diese
Maßnahmen zwar die Progression der Krankheit verlangsamt, nicht jedoch
gestoppt wird [47].
Die Wahrscheinlichkeit, eine diabetische Nephropathie überhaupt erst zu
entwickeln, hängt stark von einer erfolgreichen Prävention und
Diabetesbehandlung ab: Angaben zum Risiko variieren daher je nach Studie:
Beispielsweise entwickeln 20 bis 40 % der Patienten mit DM Typ 1 eine Mik-
roalbuminurie – ein erstes Anzeichen einer Nierenschädigung [57]. Im Allge-
meinen wird das Risiko bei Typ 1 und 2 als gleichwertig eingeschätzt [63].
3.2 Histologische Kennzeichen
Die diabetische Nephropathie zeigt typische mikroskopische Veränderungen,
die partiell auch mit den klinischen Symptomen korrelieren [1, 28, 50]:
Die zuerst messbare und eine der zentralen Veränderungen ist die
Verdickung der glomerulären Basalmembran; zeitlich nahezu parallel findet
eine Verdickung der tubulären Basalmembran statt. Diesen Entwicklungen
folgt eine Expansion der mesangialen Matrix. Diese beruht vor allem auf
einer verstärkten Produktion von auch in der gesunden Niere vorhandenen
Komponenten der extrazellulären Matrix wie Kollagen Typ IV, Laminin und
Fibronektin. Es findet jedoch auch ein gestörter Umsatz und Abbau dieser
Moleküle statt. Die daraus folgende verstärkte Ablagerung zeigt sich als
diffuse Glomerulosklerose oder als noduläre Kimmelstiel-Wilson-Läsion.

12
Auch im Tubulointerstitium kommt es zu einer Fibrose. Diese beiden
Veränderungen korrelieren mit dem Abfall der Kreatinin-Clearance [59, 83].
Begleitend kommt es zu einer Hyalinose der glomerulären Gefäße und einer
Arteriolosklerose der afferenten und efferenten Arterien. Eine weitere renale
Zellpopulation nimmt ebenfalls Schaden: Es kommt zu Veränderungen und
zu einem Verlust von Podozyten und damit ihrer Filterfunktion [76].
Begleitend kommt es zu einer Infiltration durch Entzündungszellen, die auch
die Fibrose modulieren können [17-18, 29].
Das erste klinische Zeichen ist häufig die Mikroalbuminurie. Diese tritt jedoch
erst nach den ersten mikroskopischen Veränderungen auf. Sie kann sich
über das Stadium einer Makroalbuminurie bis zu einer chronischen Nierenin-
suffizienz entwickeln [53].
3.3 Pathomechanismen der Diabetischen Nephropathie
Die molekularen Mechanismen dieser Veränderungen sind sehr komplex.
Aber gerade in den letzten Jahren gelangte man zu einigen neuen Erkennt-
nissen über die einzelnen Faktoren. Neben den direkten biochemischen
Auswirkungen der Hyperglykämie und hämodynamischen Veränderungen
spielen das Renin-Angiotensin-System und genetische Ursachen eine Rolle.
Als ein verbindendes Element und gleichsam einer gemeinsamen
Endstrecke dienen Zytokine.
3.3.1 Biochemische Veränderungen durch Hyperglykämie
Eine hohe Glukosekonzentration per se führt bereits direkt zu Schäden an
der Niere. Beispielsweise konnte in vitro gezeigt werden, dass
Hyperglykämie die Apoptose von mesangialen Zellen fördert [51]. Dabei
kommt es vor allem auf die intrazelluläre Glukosekonzentration an, die durch
Glukosetranstporter wie GLUT1 und GLUT4 vermittelt wird [35].
Vier verschiedene Stoffwechselpfade sind hier von Bedeutung [13]:
1. Über den Polyolpfad wird die erhöhte Menge an zellulärer Glucose zu
Fructose umgebaut. Die entstehenden Intermediärmetabolite spielen
eine Rolle bei der Progression der diabetischen Nephropathie.

13
2. Der Hexosamin-Stoffwechselweg, in dem Aminozucker synthetisiert
werden, scheint ebenfalls für einige Manifestationen von diabetischen
Komplikationen verantwortlich zu sein.
3. Ein dritter untersuchter biochemischer Mechanismus der
hyperglykämischen Schädigung ist die Aktivierung von Proteinkinase
C, einem intrazellulären second messenger.
4. Als ein weiterer entscheidender biochemischer Prozess konnte die
nicht-enzymatische Glykosilierung von Proteinen identifiziert werden.
Diese Proteine werden als advanced glycation end products (AGE)
bezeichnet und können unter anderem über einen Rezeptor die
Genexpression von Zytokinen beeinflussen.
Weiterhin kommt es durch die Hyperglykämie zu einer Überproduktion von
reaktiven Sauerstoffradikalen im Rahmen der mitrochondrialen
Atmungskette. Diese Sauerstoffradikale scheinen ein übergeordnetes
Element zu sein, welches mit mindestens drei der genannten Mechanismen
interagiert [58].
3.3.2 Hämodynamische Veränderungen
Seit über zwei Dekaden ist bekannt, dass auch hämodynamische
Veränderungen bei der Entwicklung der diabetischen Nephropathie eine
Rolle spielen [31, 87]: Es kommt zu einer Hyperperfusion und Hyperfiltration
im Glomerulus, auch unabhängig vom systemischen Blutdruck. Die Verände-
rungen begünstigen auch den Verlust von Proteinen in den Primärharn. Der
Anstieg des glomerulären Plasmaflusses und der erhöhte transkapilläre
hydrostatische Druck resultieren aus einem Abfall des Widerstandes in den
efferenten und vor allem in den afferenten Arteriolen. In einer gesunden
Niere würde der gesteigerte Druck durch eine Autoregulation der Gefäße
kompensiert werden. Bei der diabetischen Nephropathie scheint die
Autoregulation gestört; viele Faktoren spielen hierbei eine Rolle (NO, ANP,
Angiotensin II, Zytokine und weitere). Der gesteigerte Druck und die
Proteinurie wiederum lösen strukturelle Veränderungen an der Niere aus.

14
3.3.3 Beeinflussung durch das Renin-Angiotensin-System
Ursprünglich wurde das Renin-Angiotensin-System (RAS) als systemischer
Regulator des Blutdruckes begriffen. Bei der diabetischen Nephropathie
spielt jedoch vielmehr das lokale RAS eine große Rolle und hat vielfältige
Effekte [66, 77, 86]:
So fördert das lokal aktivierte RAS sowohl über den Druck im Glomerulus als
auch über eine Schädigung der ultrafiltrierenden Membran die Proteinurie.
Angiotensin II selbst wirkt profibrotisch und proinflammatorisch.
Die Blockierung des Renin-Angiotensin-Systems über ACE-Hemmer oder
Angiotensinrezeptor-Blocker macht man sich in der Therapie der diabeti-
schen Nephropathie zu Nutze [47].
3.3.4 Genetische Determinanten der Diabetischen Nephropathie
Es gibt deutliche Hinweise, dass der genetische Hintergrund die Entwicklung
einer diabetischen Nephropathie beeinflusst [19, 49]. So entwickelt nur
ungefähr ein Drittel der Diabetiker einen Nierenschaden, jedoch teilweise
unabhängig von der Blutzuckerkontrolle. Außerdem lassen sich familiäre und
ethnische Häufungen feststellen.
Konsequenterweise wurden einige genetische Studien publiziert, in denen
man Polymorphismen in sog. Kandidatengenen identifizieren konnte.
Zurzeit werden genomweite Studien durchgeführt, bei denen man versucht,
sogenannte Einzelnukleotid-Polymorphismen (SNP) außerhalb der Kandi-
datengene zu lokalisieren.
3.3.5 Rolle der Zytokine
Zwischen den einzelnen Mechanismen als Mediatoren fungierend konnten
einige Zytokine identifiziert werden [26, 86]. Vascular endothelial growth
factor (VEGF) und Insulin-like growth factors (IGFs) wurden bereits intensiv
untersucht; die Ergebnisse sind jedoch nicht immer eindeutig und schlüssig.
Als wichtigstes Zytokin bei der Progression der diabetischen Nephropathie
wird der Wachstumsfaktor Transforming Growth Factor beta gesehen.

15
3.4 TGF-β als Mediator bei der Diabetischen Nephropathie
Transforming Growth Factor beta (TGF-β) gehört zu einer Gruppe von
Signalmolekülen, die an elementaren Steuerungsvorgängen im Körper
beteiligt sind und zu denen man unter anderem auch die Subfamilien TGF-α
und die bone morphogenitc proteins (BMPs) zählt [7]. TGF-β wird in nahezu
allen Gewebearten exprimiert und ist ein wichtiger Modulator von
Zellwachstum, Entzündung, Matrix-Synthese, Angiogenese und Apoptose.
Dementsprechend konnte seine Rolle bei der Kanzerogenese, bei der
Wundheilung, bei der Rheumatoiden Arthritis, in der embryonalen Entwick-
lung und bei fibrotischen Erkrankungen nachgewiesen werden [7, 62]. Zur
letzteren Gruppe zählt auch die diabetische Nephropathie, wo vor allem die
profibrotischen Effekte von TGF-β (vor allem der Isoform TGF-β1) auf die
extrazelluläre Matrix (ECM) gut untersucht sind: TGF-β stimuliert die
Synthese von Schlüsselproteinen der ECM wie Kollagen Typ I und IV,
Fibronektin und Laminin.
3.4.1 TGF-β als verbindendes Element der Pathomechanismen
Wie unter 3.3 beschrieben fungieren die Zytokine, allen voran TGF-β, als
Mediatoren bei den verschiedenen pathogenetischen Mechanismen:
1. Biochemische Prozesse: Hyperglykämie löst eine Erhöhung von AGE-
Proteinen, die Aktivierung von Protein Kinase C und einen Anstieg der
intrazellulären Glucosaminproduktion aus. Diese wiederum erhöhten
die TGF-β Expression in vivo und in vitro [62].
2. Hämodynamik: Anderseits spielen Zytokine auch eine Rolle bei der
Entwicklung der Hyperfiltration und Hyperperfusion [87].
3. Renin-Angiotensin-System: Weiterhin ist Angiotensin II in der Lage,
TGF-β in verschiedenen Nierenzellen zu induzieren [85] und darüber
die Synthese von Matrixmolekülen zu stimulieren [43].
4. Genetischer Hintergrund: Polymorphismen im TGF-β-Gen zeigen eine
Assoziation mit der Nephropathie beim Typ 1 Diabetiker [19].

16
3.4.2 Aktuelle Erkenntnisse über den Zusammenhang von TGF-β und
der Diabetischen Nephropathie
Die Zusammenhänge zwischen der diabetischen Nephropathie und TGF-β
(v.a. der Isoform TGF-β1) wurden bereits in einigen Studien dargestellt [95]:
So erhöht ein Medium mit erhöhter Glukosekonzentration in nahezu allen
renalen Zelllinien die Expression und die Bioaktivität von TGF-β, in einigen
Fällen werden auch die TGF-β Rezeptoren hochreguliert. Die
Antagonisierung mit Antisense-Oligonukleotiden führt konsequenterweise zu
einer reduzierten Expression von Matrixmolekülen in vitro [32].
Die Ergebnisse konnten in experimentellen Tiermodellen bestätigt werden
[30, 36, 42, 56, 73, 88]: Die Level von TGF-β1 auf mRNA- und auf
Proteinebene sind erhöht, sowohl im glomerulären als auch im tubulären
Kompartiment. Smad, ein Downstreammokelül von TGF-β, wird ebenfalls
signifikant aktiviert. In zwei diabetischen Tiermodellen konnten Ziyadeh et al.
zeigen, dass durch die Gabe von neutralisierenden monoklonalen anti-TGF-β
Antikörpern sowohl die frühen Zeichen einer diabetischen Nephropathie wie
die renale Hypertrophie als auch späte Merkmale wie die mesangiale Matrix-
akkumulation und der Serumkreatininanstieg verhindert oder abgemildert
werden konnten [74, 94]. Transgene Mäuse mit erhöhtem TGF-β1-Plasma-
Leveln entwickeln im Gegensatz dazu eine progressive renale Erkrankung,
die einer diabetischen Nephropathie sehr ähnlich ist ( dazu gehören unter
anderem eine mesangiale Matrixexpansion, verdickte Kapillarschlingen,
interstitielle Fibrose und eine tubuläre Atrophie) [44].
Auch Studien mit Patienten mit diabetischer Nephropathie untermauern die
Bedeutung von TGF-β [62]: So fand man in entnommenem Nierengewebe
ein erhöhtes Level von TGF-β auf Protein- und mRNA-Ebene.
3.4.3 Molekularstruktur und Signaltransduktionswege von TGF-β
Kenntnisse über den molekularen Aufbau von TGF-β helfen, seine
Funktionsweise zu verstehen [3, 7]:
Die drei Isoformen TGF-β1, TGF-β2 und TGF-β3 sind homodimerische
Polypeptide. TGF-β und das sogenannte latency assoziierte Protein (LAP)
werden zusammen als inaktives Proprotein synthetisiert (Molekulare Masse:
75 kDa), im Golgi-Apparat gespalten und extrazellulär sezerniert. Sie bleiben

17
jedoch weiter über nicht-kovalente Verbindungen assoziiert. Das LAP ist über
Disulfidbrücken mit dem latent-TGF-β-bindenden-Protein (LABP) verbunden.
Das LAB-Protein wiederum ist mit Proteinen der extrazellulären Matrix
verknüpft. Der Komplex aus TGF-β, LAP und LABP wird auch LLC (large
latent complex) genannt. Als Teil von LLC kann TGF-β nicht mit seinen
Rezeptoren interagieren und ist somit inaktiv. Verschiedene Moleküle, die
die Rezeptorbindungsstellen von TGF-β freisetzen und es damit aktivieren,
wurden bereits identifiziert: Matrix-Metalloproteinasen (MMP2 und MMP9),
Plasmin, Integrine, eine Behandlung mit leichten Säuren und TSP1 können
TGF-β aktivieren. Gemeinsam ist diesen Aktivatoren, dass sie jeweils
Indikatoren für eine Änderung bzw. Störung der extrazellulären Matrix sind.
Über diesen Schritt und weniger über seine absolute Konzentration wird
TGF-β reguliert. Verschiedenartige Variablen (unterschiedliche Aktivität,
gewebespezifische Verteilung, Verknüpfung mit der ECM und mehr) erklären
auch, dass die drei Isoformen in vitro, nicht jedoch in vivo nahezu identische
Effekte besitzen.
Abb. 3-1 Molekularstruktur von TGF-β
TGF-β ist über nicht-kovalente Bindungen mit dem latency assoziierten Protein (LAP) verbunden. Dieses steht über Disulfidbrücken in Verbindung mit dem latent-TGF-β-bindenden-Protein (LABP). Diese drei Moleküle bilden den large latent complex (LLC), welcher über Isopeptidbindungen mit der extrazellulären Matrix interagieren kann. Schema frei nach [3]
TGF-β Latency assoziiertes Protein (LAP)
Latent-TGF-β-bindendes-Protein (LABP)
Extrazelluläre Matrix
TGF-β
Latency assoziiertes Protein (LAP)
Latent-TGF-β-bindendes-Protein (LABP)
Extrazelluläre Matrix
Large Latent Complex (LLC)

18
Nach seiner Aktivierung bindet TGF-β an den drei Zelloberflächenrezeptoren
Typ I, II, und III. TGF-β bindet entweder am Typ III Rezeptor, der TGF-β dem
Typ II Rezeptor präsentiert, oder direkt an Typ II. Dieser Rezeptor rekrutiert,
bindet und transphosphoryliert den Typ I Rezeptor. Dadurch wird dessen
Proteinkinase aktiviert. Als intrazelluläre Signaltransduktoren fungieren dann
vor allem verschiedene Smad-Proteine, von denen man bis heute
mindestens zehn verschiedene kennt. Der aktivierte Typ I Rezeptor
phosporyliert Smad 2 oder 3, welche an Smad 4 binden. Der daraus resultie-
rende Komplex interagiert dann im Nukleus zellspezifisch mit Transkript-
ionsfaktoren, die die Genexpression steuern.
Zusammenfassend werden TGF-β und seine Effekte auf die Genexpression
vor allem über dessen Aktivierung reguliert. Als wichtiger Aktivator konnte ein
Thrombospondin identifiziert werden.
3.5 Aufbau und Funktion der Thrombospondine
3.5.1 Familie der Thrombospondine
Thrombospondine (TSP) sind eine kleine Familie von sezernierten
Glykoproteinen, die Wachstum, Adhäsion und Migration von Zellen
modulieren und außerdem die Angiogenese beeinflussen [10]: Die fünf
Mitglieder (TSP 1-5) werden aus funktionellen und strukturellen Gründen in
zwei Untergruppen eingeteilt: TSP1 und TSP2 sind Trimere mit drei
Untereinheiten (jeweils 180 kDA) und TSP 3-5 sind Pentamere, deren
Untereinheiten jeweils 105 kDa schwer sind. TSP1 wurde zuerst als
Thrombin-sensitives-Protein (TSP) identifiziert, welches als Antwort auf
Thrombin von den Thrombozyten sezerniert wird. TSP1 ist auch der am
besten untersuchte Vertreter aus dieser Familie.
Thrombospondine zählen zu den sog. matrizellulären Proteinen; das heißt,
sie interagieren sowohl mit der extrazellulären Matrix als auch mit Zellen an
sich. So ist TSP1 in der Lage an verschiedene Zelloberflächenrezeptoren wie
z.B. Integrinrezeptoren zu binden. Andererseits kann TSP1 auch mit
Strukturproteinen wie Kollagen, Fibronektin und Laminin interagieren und die
Aktivität von Proteasen, Zytokinen und Wachstumsfaktoren beeinflussen [8].

19
3.5.2 Biologische Rolle der Thrombospondine
Obwohl TSP1 und TSP2 in vielen embryonalen Geweben exprimiert werden,
haben sie einen offenkundigeren Einfluss beim Erwachsenen.
Entsprechende Knockout-Tiere haben einen relativ milden Phänotyp, der erst
bei exogenen Einflüssen stärker zu Tage tritt [45]. Dennoch konnten mit Hilfe
dieser Tiermodelle einige Funktionen genauer beschrieben werden [37].
Diese Funktionen unterscheiden sich partiell trotz der hohen Sequenzhomo-
logie (vgl. 3.5.3) zwischen TSP1 und 2, wohl aufgrund der unterschiedlichen
zeitlichen und räumlichen Expression:
1. TSP1 und 2 sind in der Lage, die Adhäsionsfähigkeit von Zellen und
der extrazellulären Matrix zu modulieren. Einige Mechanismen wurden
bis heute als Erklärung für diese Eigenschaft beschrieben: So kann
beispielsweise TSP1 die Struktur- und Adhäsionsmoleküle Kollagen,
Laminin und Fibronektin binden. TSP2 kann über eine Regulation der
Verfügbarkeit von MMP2 die Zelladhäsion von Fibroblasten
beeinflussen [9]. Wie bereits beschrieben, kann TSP1 auch über die
Aktivierung von TGF-β die Zusammensetzung der ECM beeinflussen.
2. Obwohl TSP1 und 2 unter bestimmten Umständen proangiogenetisch
wirken, sind sie doch im Allgemeinen sehr potente Inhibitoren der
Angiogenese [37]. Die dabei involvierten Mechanismen sind zum
Beispiel eine CD36-vermittelte Apoptose von Endothelzellen oder eine
Inhibition der Migration dieser Zellen. Diese Fähigkeiten machte man
sich in einer klinischen Phase II-Studie zu Nutze, bei der man ein
Fragment von TSP1 (ABT-510) als anti-angiogenetische
Krebstherapie einsetzte [4].
3. Die generellen Eigenschaften von Thrombospondinen bei Tumoren
sind dennoch wenig verstanden und widersprüchlich: So gibt es
Tumore, bei denen die Expression von TSP1 eine bessere Prognose
bedingen, bei anderen Entitäten jedoch eine schlechtere. Auch in vitro
und in vivo Ergebnisse zeigen sowohl eine Beschleunigung als auch
eine Verlangsamung der Tumorprogression [27].

20
4. Beim Prozess der Wundheilung sind die Ergebnisse klarer: Sowohl
TSP1 als auch TSP2 werden für eine „normale“ Wiederherstellung von
Haut und Bindegewebe benötigt. Nach einer Verletzung kommt es erst
zu einem Anstieg von TSP1, das von Entzündungszellen und
Thrombozyten sezerniert wird. In den späteren Phasen kommt es zu
einem Abfall von TSP1 und einem Anstieg von TSP2 durch
Fibroblasten. Bis heute konnten MMP2, MMP9 und TGF-β1 als
wichtige Mediatoren dieser Reaktion identifiziert werden [2, 11].
Die Vielfalt an Funktionen (nur die vier wichtigsten wurden aufgeführt) beruht
teilweise auf der komplexen Struktur der Thrombospondine.
3.5.3 Molekulare Struktur der Thrombospondine 1 und 2
Thrombospondin 1 und 2 bestehen aus drei identischen Polypeptiden, die
nahe am N-Terminus über Disulfidbrücken miteinander verbunden sind.
Jedes Polypeptid beinhaltet 6 verschiedene Domänen [14, 27]:
• die aminoterminale Domäne
• eine Region mit Homologien zu Prokollagen
• drei Typen von sich wiederholenden Domänen:
� Typ 1 ähnelt dabei Sequenzen in Komplementfaktoren wie
Properdin und interagiert mit TGF-β
� Typ 2 hat Homologien zum Epidermal Growth Factor
� Typ 3 hat Ähnlichkeiten zu Calmodulin
• ein carboxyterminales Ende
Geringere Gemeinsamkeiten zwischen TSP1 und TSP2 bestehen am
Aminoterminus [37] und höhere am carboxyterminalen Ende.

21
Abb. 3-2 Molekularstruktur von TSP1 und TSP2
Die Grafik zeigt schematisch den Aufbau von TSP1 und TSP2 mit den drei identischen Untereinheiten. Diese sind aus einem aminoterminalen Ende, einer Prokollagen ähnlichen Domäne, den Typ 1-3 Bereichen und dem carboxyterminalen Ende aufgebaut. Jeder Ab-schnitt kann mit verschiedenen Rezeptoren und Molekülen interagieren. Der Abschnitt, der mit TGF-β interagiert, liegt im Typ 1 Bereich. Die Sequenzhomologie zwischen TSP1 und TSP2 nimmt vom aminoterminalen zum carboxyterminalen Ende zu. Schema frei nach [27]
3.5.4 Interaktion der Thrombospondine 1 und 2 mit TGF-β
Die molekularen Mechanismen der Interaktion mit TGF-β konnten bereits
teilweise aufgeklärt werden. Wie unter Punkt 3.4.3 bereits erläutert wird TGF-
β als inaktives Zytokin in Verbindung mit dem latency assoziierten Protein
(LAP) (zusammen als small latent complex bezeichnet) sezerniert. Das
latent-TGF-β-bindende-Protein verknüpft das LAP mit der extrazellulären
Matrix. Der gesamte Komplex wird als large latent complex bezeichnet.
TSP1 ist in der Lage, sowohl den small latent complex als auch den large
latent complex von TGF-β zu binden und zu aktivieren [20, 71]. Es wurden
bisher zwei Sequenzen identifiziert, die für eine effiziente Aktivierung
notwendig sind [72]. Eines ist das WxxW Motiv vom Typ 1 Abschnitt im TSP1
Molekül. TSP1 bindet über dieses Motiv an das LAP und TGF-β selbst [92]
und wird dadurch in die korrekte Orientierung und Positionierung zu TGF-β
gebracht. Das zweite wichtige Motiv (K)RFK kann dann mit einer anderen
Bindungsstelle von TGF-β interagieren: Dies führt zu einer Konformationsän-
derung wahrscheinlich innerhalb des LAP. Weiterhin im Gesamtkomplex
inkorporiert kann TGF-β nun an seine Rezeptoren binden [40].
Sequenzhomologie
AminoterminalesEnde
Prokollagen homologe Domäne
Typ 1 Bereich
Typ 2 Bereich
Typ 3 Bereich
CarboxyterminalesEnde
22
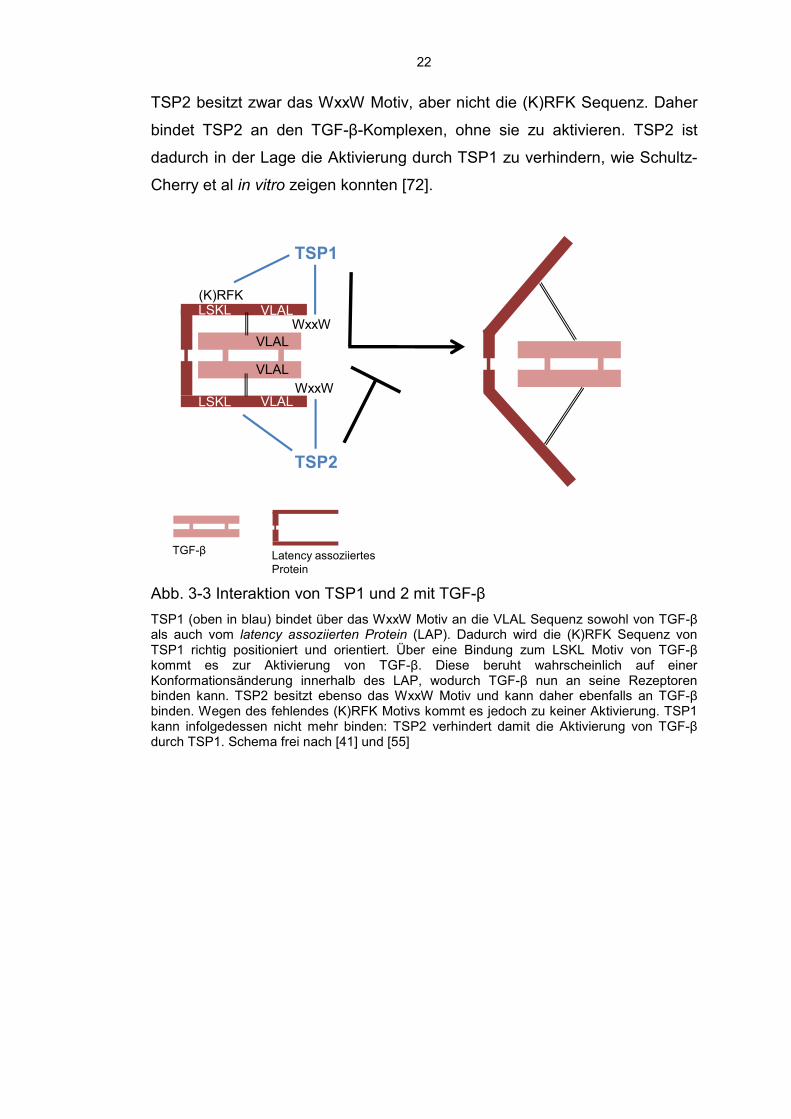
TSP2 besitzt zwar das WxxW Motiv, aber nicht die (K)RFK Sequenz. Daher
bindet TSP2 an den TGF-β-Komplexen, ohne sie zu aktivieren. TSP2 ist
dadurch in der Lage die Aktivierung durch TSP1 zu verhindern, wie Schultz-
Cherry et al in vitro zeigen konnten [72].
Abb. 3-3 Interaktion von TSP1 und 2 mit TGF-β
TSP1 (oben in blau) bindet über das WxxW Motiv an die VLAL Sequenz sowohl von TGF-β als auch vom latency assoziierten Protein (LAP). Dadurch wird die (K)RFK Sequenz von TSP1 richtig positioniert und orientiert. Über eine Bindung zum LSKL Motiv von TGF-β kommt es zur Aktivierung von TGF-β. Diese beruht wahrscheinlich auf einer Konformationsänderung innerhalb des LAP, wodurch TGF-β nun an seine Rezeptoren binden kann. TSP2 besitzt ebenso das WxxW Motiv und kann daher ebenfalls an TGF-β binden. Wegen des fehlendes (K)RFK Motivs kommt es jedoch zu keiner Aktivierung. TSP1 kann infolgedessen nicht mehr binden: TSP2 verhindert damit die Aktivierung von TGF-β durch TSP1. Schema frei nach [41] und [55]
TSP1
(K)RFK
WxxWVLAL
VLALLSKL
TSP2
WxxW
VLAL
LSKL
TGF-β
VLAL
Latency assoziiertesProtein

23
3.6 Zielsetzung der Arbeit
Viele Studien zeigen einen großen Nutzen in der Blockierung von TGF-β bei
fibrotischen Nierenerkankungen in Tiermodellen. Aufgrund der potentiellen
vielfältigen Nebenwirkungen entfällt diese Möglichkeit bei der Therapie am
Patienten. So zeigen TGF-β Knockout-Mäuse eine generalisierte
übersteigerte Immunantwort und sterben nach wenigen Wochen. Daher
bietet die lokale und spezifische Blockierung der TGF-ß Aktivierung eine
bessere Möglichkeit. In dieser Arbeit wird versucht, mittels systemischer
Überexpression von TSP2 die Thrombospondin 1 vermittelte TGF-ß-
Aktivierung zu blockieren und damit die Ausprägung einer experimentellen
diabetischen Nephropathie zu reduzieren. Zum anderen soll gezeigt werden,
dass mit Hilfe einer systemischen TSP1 Überexpression die diabetische
Nephropathie aggraviert werden kann.

24
4 Material und Methoden
4.1 Tierversuch
4.1.1 Versuchstiere
Für die Versuche wurden Mäuse aus dem Stamm C57/Bl6 verwendet. Die
Tiere wurden von Charles-River (Sulzfeld, Deutschland) bezogen oder
stammten aus eigener Zucht. Im Tierhaltungsraum wurde ein 12-stündiger
Wechsel von Tag und Nacht simuliert. Die Tiere wurden in Typ IV
Makrolonkäfigen gehalten und erhielten Standardfutter (Altromin 1324,
Spezialfutterwerke GmbH, Lage) und Trinkwasser ad libitum.
4.1.2 Experimentelles Design
Dieser Versuch wurde von der Nationalen Tieraufsichtsbehörde (Regierung
von Mittelfranken: 621-2531.31-17/05) genehmigt. Es wurden 55 männliche
Mäuse im Alter von 20 Wochen mit einem Gewicht zwischen 20 g und 30 g
verwendet. 39 Tiere waren den experimentellen Teilgruppen zugeordnet und
durchliefen den kompletten Versuchszeitraum von 18 Wochen. Als gesunde
Kontrollgruppe dienten 8 Tiere aus dem gleichen Stamm und mit vergleich-
barem Gewicht und Alter.
Den 39 Tieren aus der experimentellen Behandlungsgruppen wurde jeweils
die rechte Niere entfernt. Durch diese unilaterale Nephrektomie sollen die
durch die diabetische Stoffwechsellage verursachten Schäden an der
verbleibenden Niere potenziert werden [48].
Nach zwei Wochen Regenerationszeit wurde mit Hilfe einer intraperitonealen
Gabe von Streptozotocin (STZ) ein Diabetes Mellitus Typ 1 induziert. STZ
schädigt selektiv die Beta-Zellen im Pankreas und führt dadurch zu einem
Insulinmangel. Es erfolgten mehrere gewichtsadaptierte Applikationen an
verschiedenen Tagen über einen Zeitraum von drei Wochen mit 100 mg/kg
Körpergewicht (KG), 80 mg/kg KG oder 50 mg/kg (KG). Der Blutzuckerspie-
gel wurde mindestens jeden zweiten Tag gemessen. Über einen Zielwert von
mindestens 300 mg/dl Glukose im Blut wurde die Frequenz der STZ-
Applikation angepasst. Somit betrug die minimale Menge an STZ 300 mg/kg
KG an drei Tagen und die maximale Dosis 480 mg/kg KG an sechs Tagen.

25
Drei Wochen nach der ersten Injektion wurde mit der Insulintherapie
begonnen. Wir verwendeten ein langwirksames Insulin Glargin (Lantus® von
Sanofi-Aventis). Bis zum Ende des Experimentes erhielt jedes Tier jeden
zweiten Tag zwei Einheiten Insulin Glargin. Dadurch konnten wir die Tiere
vor einer diabetischen Ketoazidose schützen.
Nach einer weiteren Woche wurden die diabetischen Tiere randomisiert in
drei Gruppen eingeteilt und mit drei verschiedenen Plasmiden behandelt. Die
Plasmide dienten als Vektoren und führten zu einer systemischen und
dauerhaften Überexpression des jeweiligen Proteins. 10 µg der folgenden
Plasmide wurden jeweils in 15 µl PBS gelöst:
• pUBlux: Das Plasmid enthält einen Poly-Ubiquitin C Promoter und die
Sequenz für eine Firefly-Luciferase, in deren Anwesenheit Luciferin
mit Sauerstoff zu energiereichen Produkten reagiert. Bei deren Zerfall
kommt es zur Biolumineszenz. Dieses Plasmid wurde als
Kontrollplasmid verwendet.
• TSP1: Die multiple cloning site aus dem Plasmid pcDNA 3.1 His/C
wurde mit pUBlux ligiert und das Luciferasegen deletiert. Die Sequenz
von humanem TSP1 wurde mit Hilfe von Restriktionsenzymen (Kpnl
und Xbal) eingefügt. Nach der Deletion der schwachen
Startsequenzen wurde die starke Startsequenz ACCATGG mittels
Mutagenese-PCR eingefügt.
• TSP2: Bei der Herstellung des TSP2 Plasmides wurde ähnlich
verfahren: Statt einer TSP1 Sequenz wurde jedoch eine murine TSP2
Sequenz verwendet.
Zur Injektion der jeweiligen Lösungen wurden die Tiere sediert und der
rechte und linke Oberschenkel durch zwei einfache Hautschnitte freigelegt.
Es wurden in jedem Muskel an drei verschiedenen Injektionsstellen jeweils
10 µg Plasmid, die in 15 µl PBS gelöst waren, appliziert. Danach wurden die
Oberschenkelmuskel mit 6 Stromstößen mit einer Spannung von 50 V, einer
Dauer von 50 ms und einem Intervall von 950 ms elektroporiert. Die
Elektroporation (EPO) diente der Permeabilitätssteigerung der Muskelzellen,
um die Aufnahme der Vektoren in die Zellen (= Transfektion) zu erleichtern.
Die Tiere wurden weitere 10 Wochen beobachtet: Die Insulintherapie wurde
fortgeführt; es erfolgte mindestens einmal in der Woche eine Gewichts- und

26
Blutzuckerkontrolle. Am Ende des Experimentes wurden die Tiere unter
Narkose getötet und mehrere Proben und Gewebebiopsien entnommen.
Während des Versuchszeitraumes starben fünf Tiere, wahrscheinlich auf-
grund der diabetischen Stoffwechsellage. Ein durchschnittlicher Blutzucker-
wert von mindestens 300 mg/dl über 12-13 Wochen wurde als
Einschlusskriterium festgelegt. Sieben Tiere erfüllten dieses Kriterium nicht
und wurden aus der weiteren Wertung ausgeschlossen.
TSP1 Überexpression
n=9
pUBlux Überexpression
n=9
TSP2 Überexpression
n=9
Gesunde Kontrollgruppe
n=8
Abb. 4-1 Schema über Gruppenverteilung und Studiendesign
Am Tag Null wurden die Tiere unilateral nephrektomiert. Zwischen den Wochen 3 und 5 erfolgte die Induktion eines Diabetes Mellitus Typ 1 durch Applikationen von Streptozotocin. Nach einer einwöchigen Phase der Rekonvaleszenz begannen wir mit der Insulintherapie zur Prävention einer Ketoazidose. In der 9. Woche wurden die Tiere mit Plasmiden, die jeweils TSP1, TSP2 oder pUBlux enthielten, transfiziert. Nach insgesamt 18 Wochen und 14 Wochen unter Hyperglykämie wurden die Tiere getötet.
Unilaterale Nephrektomie
0
Applikationen von STZ
Transfektionvon TSP1 & 2und pUBlux
Töten der Tiere
Zeitachsein Wochen
3 6 9 18
Beginn der Insulintherapie

27
4.1.3 Vorgehensweise bei der Nephrektomie
Für die unilaterale Nephrektomie wurden die Tiere mit Isofluran (2 % bis 3 %
in Sauerstoff) narkotisiert. Danach wurde die rechte Flanke rasiert und
desinfiziert. Der retroperitoneale Raum wurde nach einem circa 1 cm großen
Schnitt in der Haut und der darunterliegenden Faszie dargestellt. Das
umliegende Fettgewebe wurde entfernt und die Niere dadurch freipräpariert.
Am Nierenhilus wurden die Nierenarterie und die Nierenvene mit Hilfe eines
Baumwollfadens und eines dreifachen chirurgischen Knotens abgebunden.
Anschließend wurde die rechte Niere unter minimalem Blutverlust entfernt.
Die Faszie wurde daraufhin mit einer fortlaufenden Naht und die Cutis mit
zwei bis drei Einzelknopfnähten verschlossen. Um eine Auskühlung zu
vermeiden, befanden die Tiere sich während der Operation auf einem Wär-
metisch und nach der Operation unter einer Wärmelampe.
4.1.4 Töten der Tiere
Unter Isoflurannarkose (siehe 4.1.3) wurde das Abdomen nach einem Längs-
und zwei Querbauchschnitten eröffnet. Die Präparation des retroperitonealen
Raumes diente zur Darstellung der Bauchaorta, der Vena cava inferior und
der verbliebenen Niere. Mit Hilfe einer Kanüle wurde aus der Vena cava
inferior jeweils circa 1 ml Blut entnommen. Nach einer Thorakotomie wurde
der linke Ventrikel punktiert und der Körperkreislauf mit 0,9 % NaCl-Lösung
gespült. Diese Maßnahme erhöhte die Qualität der später angefertigten
Gewebepräparate der Niere, indem die renalen Gefäße weitgehend frei von
Erythrozyten waren. Die noch verbliebene Niere wurde entnommen,
umliegendes Fett und die Kapsel wurden entfernt. Weiterhin wurde jedem
Tier ein Oberschenkelmuskel entnommen, um die lokale Plasmid-Expression
überprüfen zu können. Die Tiere starben nach Eröffnung der Bauchaorta.
4.2 Gewinnung & Analyse des Probenmaterials
4.2.1 Glukosespiegel im Blut
Um an Probenmaterial zu gelangen, wurde der Schwanz der Mäuse mit
einem Skalpell leicht verletzt und das austretende venöse Blut in einem
Blutglukosemessgerät analysiert. Die Messung erfolgte während der

28
Streptozotocinapplikationen jeden zweiten Tag, danach mindestens einmal
pro Woche.
4.2.2 24 Stunden Sammelurin
Die Tiere wurden einzeln für 24 Stunden in metabolische Käfige gesetzt. Die
gesammelte Urinmenge wurde daraufhin notiert und ein Teil des Urins bis
zur weiteren Analyse bei einer Temperatur von -20°C gelagert. Insgesamt
wurden dadurch an vier verschiedenen Zeitpunkten des Experimentes
Urinproben gewonnen (in den Wochen 1, 10, 14, 18). Im Urin bestimmten
wir die Albumin- und Kreatininkonzentrationen.
4.2.3 Gewichtskontrolle
Die Tiere wurden zu Beginn des Experimentes mehrmals pro Woche und im
Verlauf mindestens einmal pro Woche gewogen, um krankheitsbedingte
Gewichtsabnahmen rechtzeitig feststellen zu können.
4.2.4 Serumgewinnung
Serumproben wurden an zwei verschiedenen Zeitpunkten gewonnen. 4 Wo-
chen vor Versuchsende entnahmen wir den Tieren mit Hilfe einer Glaskapil-
lare retrobulbäres Blut.
Das Probenmaterial am Ende des Versuches stammte aus der Vena Cava
inferior. Anschließend wurden die zellulären Bestandteile für 10 Minuten bei
8000 rpm in einer Tischzentrifuge (Biofuge fresco, Heraeus) sedimentiert und
das sich im Überstand befindliche Serum abpippetiert. Wir lagerten die
Proben bis zur weiteren Analyse bei -20°C.
4.2.5 Bestimmung von Serumharnstoff und Serum- und Urinkreatinin
Kreatinin im Serum bzw. im Urin und Harnstoff im Serum wurden mit einem
automatischen Analysegerät (Beckmann Instruments GmbH, München) im
Klinischen Labor der Kinderklinik Erlangen gemessen.

29
4.2.6 Berechnung der Kreatinin-Clearance
Mit Hilfe folgender Formel erfolgte die Berechnung der Kreatinin-Clearance in
der Einheit µl pro Minute:
1440
24
×
×=
Serumim][Kreatinin
h/Urinmenge] im Urin[KreatininClearance
4.2.7 Bestimmung der Proteinmenge im Urin
Die Proteinkonzentration im Urin wurde nach dem Prinzip von Bradford
bestimmt. Hierbei entsteht durch Komplexierung von Coomassie Brilliant
Blau mit den Seitenketten von Proteinen ein Farbumschlag, der
photometrisch detektiert werden kann.
Die Quantifizierung erfolgte mittels des BioRad Protein Kits (BioRad,
München). Als Standard diente BSA in Konzentrationen von 10 µg/ml bis 500
µg/ml. Es wurden jeweils 10 µl vom Standard und den Proben als Duplikate
in eine Mikrotiterplatte mit 96 Vertiefungen (Sarstedt, Nümbrecht) pipettiert.
Die Proben wurden entweder unverdünnt oder in Verdünnungen bis 1:5
angesetzt, um Ergebnisse im linearen Messbereich der Standardkurve zu
erhalten. Nach dem Hinzufügen von jeweils 200 µl BioRad Reagenz wurde
die Absorption bei 595 nm an einem Mikrotiterplatten-Photometer (Tecan,
Crailsheim) gemessen.

30
4.2.8 Konservierung des Versuchstiergewebes
Nach Entnahme der verbliebenen Niere wurden Fett und Kapsel entfernt und
die Niere anschließend gewogen. Daraufhin wurde sie in mehrere Teile
geschnitten und unterschiedlich konserviert: (siehe auch 4.3)
Tabelle 1 Konservierung des Nierengewebes
Fixierung Anwendung Zusammensetzung/Hersteller
Tissue Tek Gefrierschnitte Sakura Finetek, Zoekterwomde, NL
MC Immunhistochemie Siehe 4.3.1
PAF Immunhistochemie Siehe 4.3.1
Zink Immunhistochemie Siehe 4.3.1
1% SDS Zymogramme 1 g SDS in 100 ml aqua dest.
Soerensen Puffer Semidünnschnitte 8,54 g Natriumhydrogenphosphat
1,985 h Kaliumhydrogenphosphat
3 % Glutaraldiaaldehyd
In 1 L aqua dest.
RLT Realtime-PCR Aus RNeasy MiniKit mit β-Mercaptoethanol
Ein Oberschenkelpräparat aus der TSP2 Gruppe wurde jeweils in PAF-
Lösung fixiert und weiterprozessiert, aus der TSP1 und pUBlux Gruppe
jeweils in MC-Lösung.

31
4.3 Aufbereitung der Gewebe für die Immunhistochemie
4.3.1 Prozessieren des Gewebes
Nach Entnahme der Niere wurde das Gewebe für die Dauer von mindestens
24h in Zinklösung, Paraformaldehyd oder Methyl Carnoy fixiert.
Zinklösung:
� 0,5 g Calciumacetat
� 5 g Zinkacetat
� 5 g Zinkchlorid
� in 1000 ml 0,1 M Trispuffer pH 7,4
Paraformaldehyd (PAF)
3 g Paraformaldehyd wurden in 100 ml PBS bei 60°C im Wasserbad gelöst und danach bei
4°C im Dunkeln gelagert.
Methyl Carnoy (MC)
� 60 % Methanol
� 30 % Chloroform
� 10 % Eisessig
Die Lösung wurde bei 4°C gelagert.
Das Gewebe wurde hierauf wie in der folgenden Tabelle dargestellt weiter
prozessiert.
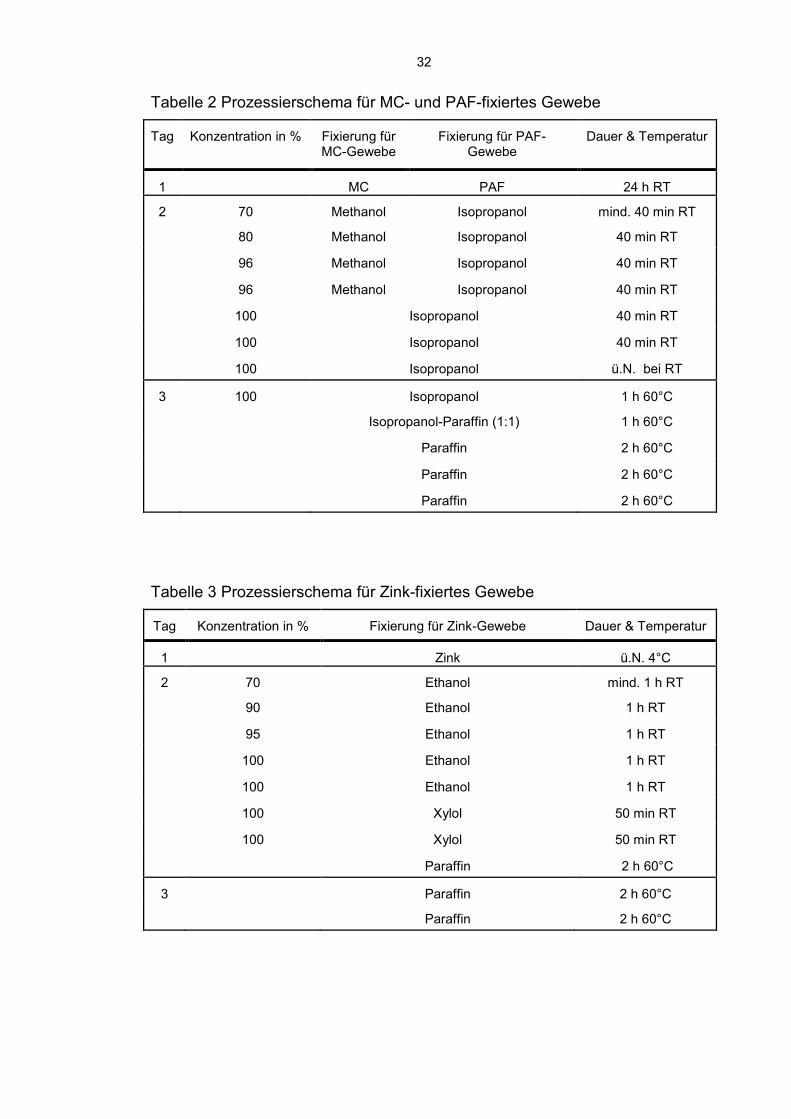
32
Tabelle 2 Prozessierschema für MC- und PAF-fixiertes Gewebe
Tag Konzentration in % Fixierung für MC-Gewebe
Fixierung für PAF-Gewebe
Dauer & Temperatur
1 MC PAF 24 h RT
2 70 Methanol Isopropanol mind. 40 min RT
80 Methanol Isopropanol 40 min RT
96 Methanol Isopropanol 40 min RT
96 Methanol Isopropanol 40 min RT
100 Isopropanol 40 min RT
100 Isopropanol 40 min RT
100 Isopropanol ü.N. bei RT
3 100 Isopropanol 1 h 60°C
Isopropanol-Paraffin (1:1) 1 h 60°C
Paraffin 2 h 60°C
Paraffin 2 h 60°C
Paraffin 2 h 60°C
Tabelle 3 Prozessierschema für Zink-fixiertes Gewebe
Tag Konzentration in % Fixierung für Zink-Gewebe Dauer & Temperatur
1 Zink ü.N. 4°C
2 70 Ethanol mind. 1 h RT
90 Ethanol 1 h RT
95 Ethanol 1 h RT
100 Ethanol 1 h RT
100 Ethanol 1 h RT
100 Xylol 50 min RT
100 Xylol 50 min RT
Paraffin 2 h 60°C
3 Paraffin 2 h 60°C
Paraffin 2 h 60°C

33
4.3.2 Anfertigen von Schnitten aus Gefriergewebe
Das Gewebe lagerte konserviert bei -70°C in Tissue Tek. Zur Anfertigung der
Schnitte wurden die Biopsien im Gefriermikrotom auf -20°C erwärmt. Die 5
µm dicken Schnitte wurden auf speziell für Gefriergewebe geeignete Super
Frost Objektträger gezogen und danach für 10 Minuten in -20°C kaltem
Aceton fixiert. In Alufolie verpackt wurden die Schnitte bis zum weiteren
Gebrauch in einem -70°C kalten Tiefkühlschrank gelagert.
4.3.3 Anfertigen von Schnitten aus Paraffinblöcken
Die bei Raumtemperatur gelagerten Gewebeblöcke wurden vor dem
Schneiden auf eine Kühlplatte mit -10°C überführt. Nach Erreichen der
gewünschten Temperatur wurden die Blöcke in das Mikrotom eingespannt.
Die 2 µm dicken Schnitte wurden dann auf der Oberfläche eines auf 45°C
erwärmten Wasserbad geglättet und danach die Objektträger aufgezogen.
Zur Entfernung des Paraffins und zur besseren Haftung wurden die Schnitte
in einem Ofen bei 60°C mindestens 20 min gebacken. Die fertigen Schnitte
wurden wiederum bei Raumtemperatur gelagert.
4.4 (Immun)histochemische Färbungen
Der Großteil der Schnitte wurde immunhistochemisch angefärbt. Das zu
untersuchende Protein fungiert als Antigen und geht mit dem hinzugefügten
Antikörper eine Bindung ein. Das Protein kann dann mit einem Chromogen
sichtbar gemacht werden.
4.4.1 Periodsäure-Schiff-Färbung (PAS)
Die PAS-Färbung beruht auf einer chemischen Reaktion. Es können dadurch
kohlenhydratreiche Strukturen wie Bestandteile der EZM angefärbt werden.
Periodsäure führt zu einer Oxidation von Glykolgruppen zu Aldehydgruppen.
Diese können danach durch das Schiffsche Reagenz sichtbar gemacht
werden. Das Schiffsche Reagenz ist eine farblose fuchsinschweflige Säure.
Bei der Reaktion wird Fuchsin wieder frei und bewirkt die typische rötliche

34
Färbung. Es wurde MC-fixiertes Gewebe verwendet und die Präparate zur
besseren Beurteilbarkeit mit Hämatoxylin gegengefärbt.
Tabelle 4 PAS-Färbung
Deparaffinieren 100 % Xylol 3 x 5 min
Rehydrieren 100 % Ethanol 3 x 3 min
95 % Ethanol 2 x 2 min
Aldehydgruppenbildung 2 % Periodsäure 15 min
Waschen Aqua dest. 2 x 3 min
Schiffsches Reagenz Schiffsches Reagenz 25 min
Waschen Aqua dest. 2 x 3 min
Gegenfärben Hämatoxylin 20 sec
Bläuen Laufendes Leitungswasser 5 min
Dehydrieren 70 % Ethanol 2 min
95 % Ethanol 2 x 2 min
100 % Ethanol 3 x 2 min
100 % Xylol 3 x 5 min
4.4.2 Immunfluoreszenzfärbung an Gefrierschnitten
Bei der Immunfluoreszenzfärbung wird der primäre Antikörper mit einem
fluoreszenzmarkierten sekundären Antikörper sichtbar gemacht.
Nach dem Auftauen der bei -70°C gelagerten Schnitte wurden diese in
Aceton überführt und dann luftgetrocknet. Die Präparate auf dem
Objektträger wurden mit einem wasserabweisenden Stift umkreist. Um
unspezifische Bindungen zu vermeiden, wurde jede Biopsie mit 50 µl von
einem 100% fetalem Kälberserum (FCS) inkubiert. Nach einem
Waschvorgang wurde der gewünschte primäre Antikörper (Anti CD4 in einer
Verdünnung von 1:200, Anti CD8 in einer Verdünnung von 1:400 in 1 % BSA
in TBS) über Nacht auf den Schnitten belassen. Am darauffolgenden Tag
wurden die Schnitte mit einem Sekundärantikörper in einer Verdünnung von
1:400 inkubiert. Eine Gegenfärbung der Kerne erfolgte mit 4′,6-Diamidin-2-
phenylindol (DAPI). Nach dem Eindecken mit Mowiol (Merck, Darmstadt)
wurden die Präparate möglichst kühl und im Dunklen gelagert, um die
Fluoreszenzintensität zu erhalten.

35
Tabelle 5 Immunfluoreszenzfärbung bei Gefrierschnitten
Auftauen der Schnitte 2-3 min bei RT
Fixierung Aceton 10 min bei 4°C
Trocknen 10 min bei RT
Waschen TBS 3 x 2 min
Blockieren 100 % FCS 30 min
Primärantikörper In TBS/BSA verdünnt ü.N. bei 4°C
Waschen TBS 3 x 2 min
Sekundärantikörper 1:400 verdünnt in TBS/BSA 2 h bei RT
Waschen TBS 3 x 2 min
Gegenfärben DAPI 10 sec
Waschen TBS 3 x 2 min
4.4.3 Immunfluoreszenzfärbung an Paraffinschnitten
Auch bei der Immunfluoreszenzfärbung an Paraffinschnitten wird der primäre
Antikörper mit einem sekundären Antikörper sichtbar gemacht. Wir setzten
einen Anti-CD31 Antikörper in einer Verdünnung von 1:50 ein.
Tabelle 6 Immunfluoreszenzfärbung an Paraffinschnitten
Deparaffinieren 100 % Xylol 3 x 5 min
Rehydrieren 100 % Ethanol 3 x 3 min
95 % Ethanol 2 x 2 min
Waschen TBS 3 x 2 min
Primärantikörper In TBS/BSA verdünnt ü.N. bei 4°C
Waschen TBS 3 x 2 min
Sekundärantikörper 1:250 verdünnt in TBS/BSA 2 h bei RT
Waschen TBS 3 x 2 min
Gegenfärben DAPI 10 sec
Waschen TBS 3 x 2 min

36
4.4.4 Immunperoxidasefärbung
Bei dieser Färbemethode wird ein biotinylierter Sekundärantikörper
eingesetzt, der von einer mit Avidin komplexierten Meerrettichperoxidase
detektiert werden kann. Nach Zugabe von Wasserstoffperoxid bewirken die
freiwerdenden Protonen durch ein Chromogen einen Farbumschlag. In
unserem Fall diente 3,3'-Diaminobenzidin (DAB) als Farbstoff.
Sämtliche Immunperoxidasefärbungen der Niere wurden an Zink-fixiertem
Gewebe angefertigt.
Tabelle 7 Immunperoxidasefärbung
Deparaffinieren 100 % Xylol 3 x 5 min
Rehydrieren 100 % Ethanol 3 x 3 min
95 % Ethanol 2 x 2 min
Blocken der endogenen
Peroxidase
10 % H2O2 in Methanol 20 min
Waschen TBS 3 x 3 min
Primärantikörper Verdünnt in 1 % BSA/TBS ü. N. bei 4°C
Waschen TBS 3 x 3 min
Sekundärantikörper 1:500 in 1 % BSA/TBS 30 min
Waschen TBS 3 x 3 min
Avidin-D Peroxidase 1:2000 in 1 % BSA/TBS
Waschen TBS 2 x 3 min
Chromogen DAB 10 min
Waschen Aqua dest. 5 min
Gegenfärbung Hämatoxylin 20 sec
Bläuen Laufendes Leitungswasser 3 x 2 min
Dehydrierung 70 % Ethanol 2 min
95 % Ethanol 2 x 2 min
100 % Ethanol 3 x 2 min
100 % Xylol 3 x 5 min
Eindecken Entellan

37
Bei ausgewählten Färbungen wurden die Präparate vor der Inkubation mit
dem Primärantikörper chemisch vorbehandelt, um die Antigenpräsentation zu
verstärken.
Tabelle 8 Verdünnung der Primärantikörper und Vorbehandlungen
Primärantikörper Verdünnung Vorbehandlung
CD3 1:100 Keine
F 4/80 1:25 10 mM Citronensäure pH 6 für 2 min bei 1000 Watt in
der Mikrowelle
Fibronektin 1:500 Keine
Kollagen I 1:30 0,1 % Pronase für 10 sec
1 % BSA/TBS für 30 mins
Kollagen IV 1:200 Keine
P-Smad 2/3 1:400 Keine
Wilms Tumor 1 1:250 Target Retrieval Solution für 2,5 min bei 1000 Watt in
der Mikrowelle
4.4.5 Primärantikörper
� Kollagen I, ein polyklonaler Kaninchen-Ak gegen Kollagen I in der
Ratte mit Kreuzreaktivität gegen Maus Kollagen I (AbD Serotec,
Düsseldorf)
� Kollagen IV, ein polyklonaler Ziegen-Ak gegen humanes Kollagen IV
mit Kreuzreaktivität gegen Maus Kollagen IV (Southern Biotechnology
Associates, Birmingham, UK)
� Fibronektin, ein polyklonaler Kaninchen-Ak gegen Maus Fibronektin
(Lab Vision Corporation, Fremont, CA, USA)
� CD3, ein monoklonaler Kaninchen-Ak gegen das humane CD3
Antigen mit Kreuzreaktivität gegen Maus CD3 (Thermo Scientific,
Fremont, CA, USA)
� Phospho-Smad 2/3, ein polyklonaler Kaninchen-Ak gegen die
phosphorylierte Form von humanem Smad 2/3 mit Kreuzreaktivität
gegen Maus P-Smad2/3 (sc-11769; Santa Cruz Biotechnology, Santa
Cruz, CA, USA)

38
� F 4/80, ein monoklonaler Ratten-Ak gegen ein auf Makrophagen
exprimiertes murines Glykoprotein (Caltag Laboratories, Burlingame,
CA, USA)
� CD8a, ein monoklonaler Ratten-Ak gegen CD8+ T-Lymphozyten in der
Maus (Caltag Laboratories, Burlingame, CA, USA)
� CD4, ein monoklonaler Ratten-Ak gegen CD4+ T-Lymphozyten in der
Maus (Caltag Laboratories, Burlingame, CA, USA)
� CD31, ein monoklonaler Ratten-Ak gegen das Oberflächenprotein
CD31 (PECAM-1) auf Maus-Endothelzellen (Cell Signalin, Boston,
MA, USA)
� Wilms Tumor 1, ein polyklonaler Ratten-Ak gegen Podozyten in der
Maus (Thermo Scientific, Fremont, CA, USA)
4.4.6 Sekundärantikörper und deren Substrate
� Biotinylierter Anti-Rabbit IgG Antikörper (Vector, Burlingame, USA)
� Biotinylierter Anti-Maus IgG Antikörper (Vector, Burlingame, CA, USA)
� Biotinylierter Anti-Ziegen IgG Antikörper (Vector, Burlingame, USA)
� Horseradish Peroxidase Avidin D (Vector, Burlingame, CA, USA)
� ImPACT DAB Kit (Vector, Burlingame, CA, USA)
� Alexa Fluor 488 Anti-Ratten Antikörper (Invitrogen, Eugene, USA)
� Cy 3-konjugierter Anti-Ratten Antikörper (Jackson Immuno Research,
Newmarket, UK)

39
4.4.7 Mikroskopische Auswertung
Die Quantifizierung der histologischen Färbungen erfolgte verblindet, d.h.
ohne Kenntnis der Gruppenzugehörigkeit des Präparates.
� Zur Evaluierung des glomerulären Schadens wurden 30 Querschnitte
pro Präparat bei 400-facher Vergrößerung auf PAS-Positivität
beurteilt. Dabei wurde ein von 0 bis 4 reichender Score für die
mesangiale Expansion angewandt: 0 = normal, 1 < 25 %, 2 < 50 %,
3 < 75 % und 4 > 75 %.
� Die Expression von Fibronektin wurde an 30 glomerulären
Querschnitten bei 400-facher Vergrößerung pro Biopsie bewertet.
Zum Einsatz kam der obengenannte semiquantitative Score.
� Die Einwanderung von Makrophagen wurde mit der F 4/80 Färbung
bestimmt. Es wurden positive Zellen bei 600-facher Vergrößerung in
25 Gesichtsfeldern im Cortex gezählt.
� Für die kortikale T-Lymphozyten Dichte wurden CD3+Zellen in 25
Gesichtsfeldern bei 600-facher Vergrößerung gezählt.
� Die Einwanderung von CD4+ und CD8+ T-Lymphozyten wurde in den
jeweiligen Färbungen bestimmt. Dazu wurden 20 Gesichtsfelder über
dem Cortex bei 400-facher Vergrößerung ausgezählt.
� Die tubulointerstitielle Deposition von Kollagen I wurde über 25
Gesichtsfelder bei 400-facher Vergrößerung anhand eines
semiquantitativen Scores ermittelt: unter 10 % des Gesichtsfeldes: 0
Punkte, unter 25 % 1 Punkt, 25 – 50 % 2 Punkte, 50 – 75 % 3 Punkte
und über 75 % 4 Punkte.
� Die Aktivierung von TGF-β wurde indirekt über den Anteil von
Phospho-Smad 2/3 positiven Zellkernen in 20 Glomeruli bei 600-
facher Vergrößerung analysiert.
� Die Anzahl an Podozyten wurde über WT 1 positive Zellkerne als
relativer Anteil an allen Zellkernen in 20 Glomeruli in 1000-facher
Vergrößerung gezählt.

40
� Die glomeruläre Deposition von Kollagen IV wurde an 20
Querschnitten pro Biopsie bei 600-facher Vergrößerung mit einem
semiquantitativen Score bewertet: unter 10 % Ausdehnung pro
Glomerulus: 0 Punkte, unter 25 % 1 Punkt, 25 – 50 % 2 Punkte, 50 –
75 % 3 Punkte und über 75 % 4 Punkte.
� Zur Evaluierung der Gefäßdichte wurde ein intraokulares Gitter mit 10
x 10 Quadraten verwendet. Jede Kammer, die keine CD31- Positivität
als Endothelzellmarker zeigte, wurde als negativ gewertet. Bei der
verwendeten Vergrößerung von 400 hatte ein Quadrat eine Größe von
0,0625 mm. Pro Biopsie wurden nach diesem Schema 13 mal 10 x 10
Quadrate ausgewertet.
4.5 Zymogramme
Bei Zymogrammen handelt es sich um eine Variante der SDS-Polyacrylamid-
Gelektrophorese, bei der man enzymatische Aktivitäten sichtbar machen
kann. In unserem Fall wurden Acrylamidgele mit Gelatine (hydrolisiertes
Kollagen) copolymerisiert, um die Aktivität von kollagendegradierenden
Proteasen zu demonstrieren.
4.5.1 Proteinextraktion
Das Nierengewebe für die Zymogramme wurde bei -70°C in einer 1-
prozentigen SDS-Lösung gelagert. Die Biopsien wurden langsam aufgetaut
und für das folgende Prozedere möglichst kühl gehalten. Die Proben wurden
mit einem Ultra-Turrax-Gerät (IKA, Staufen) mechanisch zerkleinert und
homogenisiert. Zum weiteren Aufschluss der Zellen wurde das Gewebe
zusätzlich einer Behandlung mit einem Ultraschallgerät (Bandelin, Berlin)
unterzogen. Dabei wurden sieben Zyklen mit einer maximalen Spannung
von 70 V und einer Amplitudeneinstellung von 50 % appliziert.
Nach dem Zentrifugieren mit 13000 rpm für 2 min wurden die Überstände
überführt.
41
4.5.2 Proteinbestimmung detergenzhaltiger Proben
Die Konzentrationsbestimmung in dem Protein-SDS-Gemisch erfolgte mittels
des BioRAD Dc Protein Assays. Die Proben wurden 1:5 oder 1:15 in PBS
verdünnt und jeweils 10 µl in Duplikaten in eine Mikrotiterplatte mit 96
Vertiefungen pipettiert. Als Standard diente BSA in Konzentrationen von
10µg/ml bis 500 µg/ml. Den Proben wurde 225 µl des BioRad Reagenzgemi-
sches hinzugefügt. Die Auslesung erfolgte an einem Mikrotiter-Photometer
bei einer Wellenlänge von 690 nm.
4.5.3 Anfertigen der nativen Polyacrylamid-Gele
Vier Gele mit einer Dicke von 0,75 mm wurden in einem Gelgießstand nach
folgendem Muster gegossen:
Tabelle 9 Zusammensetzung der Sammel- und Trenngele
Trenngel 8 % Sammelgel 5 %
Acrylamid 30 % 10,6 ml 2,64 ml
H2O Mit 0,2 % Gelatine: 13,3 ml 9,36 ml
Upper/Lower Tris Lower Tris: 8,0 ml Upper Tris: 4,0 ml
Temed 24 µl 24 µl
APS 200 µl 200 µl
Vor dem Auftragen des Sammelgels wurde das Trenngel 15 min getrocknet.
Pro Gel waren Taschen für jeweils 10 Proben vorhanden.
4.5.4 Beladen und Lauf der Gele
In jede Tasche wurden 30 µg Protein in einem Laufpuffer verdünnt pipettiert.
Die Menge des Laufpuffers orientierte sich am Volumen des Proteingemi-
sches und betrug mindestens 25 %.
Die Proteine wurden für 90 min bei 4°C und einer Spannung von 120 V
elektrophoretisch getrennt.

42
4.5.5 Inkubation der Gele
Um das SDS aus dem Gel und den aufgetrennten Proben zu entfernen,
wurden die Gele zunächst mit 2,5 % Triton X in 100 mM Tris-Lösung (pH 7,5)
für 2 mal 60 min behandelt. Im Folgenden wurden die Proben für 3 mal 20
Minuten mit einer Aktivationslösung auf einem Schüttler bei Raumtemperatur
gewaschen. Dieser Schritt diente zum Auswaschen von Triton X und zur
Aktivierung der Kollagenasen. Die Gele wurden anschließend bei 37°C für
36h in einem Brutschrank mit der Aktivationslösung inkubiert. Das Anfärben
der Kollagenstrukturen erfolgte mit einer 0,1 prozentigen Coomassielösung
für 2 bis 3 Stunden auf einem Schüttler. Um unspezifisches Anfärben zu
minimieren, wurden die Gele danach mit einer Entfärbungslösung für 5 bis 25
Minuten behandelt. Zur besseren Handhabung wurden die Gele in einen
Rahmen gespannt und zwischen zwei transparenten Folien getrocknet.
4.5.6 Blockieren der Matrixmetalloproteinasen
Um die Spezifität der Methode zu überprüfen, blockierten wir indirekt die
Aktivität der Metalloproteinasen durch Zugabe von Ethylendiamintetraacetat
(EDTA). Der Waschlösung wurde EDTA hinzugefügt und der Aktivationslö-
sung wurde EDTA im Austausch gegen die Elektrolyte beigegeben.
4.5.7 Auswertung
Nach dem Trocknen wurden die Gele mit einem handelsüblichen Scanner
(Hewlett Packard, Böblingen) in der Durchlichtmethode eingelesen.
Die weitere Auswertung erfolgte digital mit ImageJ (Open Source). Es
befanden sich aus jeder Gruppe jeweils zwei Proben auf vier verschiedenen
Gelen. Die Aktivität von MMP2 erschien als helle Bande, deren Intensität
gemessen wurde und von dem jeweils der Grauwert des Geles als
Hintergrundrauschen abgezogen wurde. Anschließend wurden die Werte
gegen die Kontrollgruppe normalisiert.
43
4.5.8 Lösungen
Laufpuffer Glycerin 50 %
Bromphenolblau 0,2 %
Waschlösung pH 7,5 Triton X 2,5 %
Aktivationslösung Tris 100 mM, pH 7,5
CaCl2 10 mM
NaCl 150 mM
ZnSO4 2 µM
Färbelösung Methanol 40 %
Essigsäure 10 %
Aqua dest. 50 %
Coomassie 0,1 %
Entfärbelösung Essigsäure 10 %
Methanol 40 %
Aqua dest. 50 %
Upper Tris pH 6,8 Aqua dest. 500 ml
Tris 30,3 g
SDS 20 % 10 ml
Lower Tris pH 8,8 Aqua dest. 1 l
Tris 181,7 g
SDS 20 % 20 ml
APS Ammoniumpersulfat 10 %

44
4.6 Enzyme Linked Immunosorbent Assay
Mittels eines Enzyme Linked Immunosorbent Assays (ELISA) können
Proteine in verschiedenen Proben wie Urin oder Serum nachgewiesen und
quantifiziert werden. Dabei bindet ein enzymmarkierter Antikörper an das
gewünschte Antigen und nach Zugabe eines Substrates kommt es dann zu
einer messbaren Farbreaktion.
4.6.1 Messung der Albuminurie
Um die Nierenschädigung abzuschätzen, bestimmten wir die Konzentration
von Albumin im Urin (Albuminurie). Wir benutzten ein Maus Albumin Elisa
Quantifizierungs-Kit von Bethyl Laboratories (Montgomery, TX, USA) und
beschichtete Mikrotiterplatten ELISA Nunc Maxisorp (Thermo Scientific,
Dänemark) mit 96 Vertiefungen. Der mitgelieferte Albumin-Ak wurde in dem
Coating Puffer nach Herstellerangaben verdünnt und die Mikrotiterplatte
damit ü.N. bei 4°C behandelt. Der mitgelieferte Standard wurde in der
Probenlösung in einer absteigenden Reihe verdünnt (500 ng/ml bis
7,81 ng/ml); die Proben je nach Vorkonzentration zwischen 1:500 bis 1:2000.
Proben und Standard wurden in Duplikaten auf die Mikrotiterplatte
aufgetragen und bei RT für 60 Minuten inkubiert. Für weitere 60 Minuten
wurde die Mikrotiterplatte mit einem Meerrettichperoxidase-Antikörper des
Kits behandelt. Zwischen den einzelnen Schritten erfolgten automatisierte 3-
fache Waschvorgänge mit der Waschlösung. Als Substrat dienten je 100 µl
ABTS Farbreagenz (1 ABTS Tablette in 50 ml ABTS-Puffer, Roche Applied
Science, Mannheim). Der Farbumschlag wurde nach 30 Minuten in einem
Mikrotiterplatte-Photometer bei 405 nm bestimmt.

45
4.6.2 Pufferlösungen
Coating Puffer 0,05 M Natriumcarbonat pH 9,6
Waschlösung � 50 mM Tris
� 0,14 M NaCl
� 0,05 % Tween 20 bei pH 8,0
Blockierungslösung � 50 mM Tris
� 0,14 NaCl
� 1% BSA bei pH 8,0
Probenlösung � 50 mM Tris
� 0,14 M NaCl
� 1 % BSA
� 0,05 % Tween 20 bei pH 8,0
4.7 Real-Time PCR
Die Real-Time PCR ist eine Vervielfältigungsmethode für Nukleinsäuren, die
auf dem Prinzip der Polymerase-Kettenreaktion (PCR) beruht. Zusätzlich zur
konventionellen PCR bietet die Real-Time PCR die Möglichkeit der
Quantifizierung der eingesetzten Nukleinsäuren durch Messung eines
Fluoreszenzsignals nach jedem PCR-Zyklus.
4.7.1 RNA-Isolierung
Sämtliche Schritte erfolgten mit RNAse-freien Pipettenspitzen, Reaktionsge-
fäßen und RNAse-freiem Wasser.
Das für die Real-Time PCR bestimmte Nierengewebe wurde bei -70°C in
RLT-Puffer gelagert. Dem Puffer wurde β-Mercaptoethanol zugesetzt, um
mögliche RNAsen zu inaktivieren und so die Qualität der Proben zu erhöhen.
Die eigentliche Isolierung der RNA erfolgte nach Auftauen der Proben mittels
des RNeasy Mini Kits (Quiagen, Hilden) den Herstellerangaben folgend. Die
jeweilige Konzentration wurde photometrisch (BioPhotometer, Eppendorf,
Hamburg) bei 260 nm bestimmt.

46
4.7.2 Herstellung der cDNA
Das Umschreiben von RNA in cDNA erfolgte mittels des VERSO cDNA Kits
(Abgene, UK) gemäß der Herstellerangaben. Als Vorlage diente RNA in einer
Konzentration von 15 ng/µl.
4.7.3 Real-Time PCR
Als Fluoreszenzsignal nutzten wir SYBR-Green. Die Signalintensität nimmt
dabei mit jedem Vervielfältigungsschritt zu und korreliert mit der ursprüngli-
chen RNA-Menge. Wir setzten 3 µl Probe und 17 µl Reaktionsgemisch ein.
Die Endkonzentrationen für Vorwärts- und Rückwärtsprimer betrugen jeweils
100 nM. Die Signalstärke wurde in der exponentiellen Phase der PCR
bestimmt und dann mit der delta-delta-CT Methode ausgewertet. Die
Ergebnisse wurden gegen die universell exprimierte 18S-RNA normalisiert.
Folgende Primer wurden eingesetzt:
18S
Forward: 5‘ TTGATTAAGTCCCTGCCCTTTGT 3‘
Reverse: 5‘ CGATCCGAGGGCCTCACTA 3‘
TGF-β1
Forward: 5‘ TGACGTCACTGGAGTTGTACGG 3‘
Reverse: 5‘ GGTTCATGTCATGGATGGTGC 3‘

47
4.8 Statistische Auswertung
Zur statistischen Auswertung nutzten wir GraphPad Prism 5.00 für Windows
(GraphPad Software, San Diego, CA). Die Evaluierung der Blutzuckerkurve
erfolgte mit einer 2-seitigen ANOVA-Analyse mit anschließendem Bonferroni-
Test. Für die statistische Auswertung der Überlebenskurve verwendeten wir
den Mantel-Cox Log-rank Test. Bei dem restlichen Großteil der Ergebnisse
wurden die Daten in einem ersten Schritt mittels des Kolmogorov-Smirnov-
Tests auf Normalverteilung überprüft. Es wurde daraufhin eine ANOVA-
Analyse mit Tukey’s Multiple Comparision Test durchgeführt. Alle Werte
werden als Mittelwerte ± SEM angegeben und das ermittelte
Signifikanzniveau wird folgendermaßen angegeben:
Tabelle 10 Signifikanzniveau
P Wert Signifikanzniveau Symbol
< 0,001 Hoch signifikant ***
0,001 bis 0,01 Sehr signifikant **
0,01 bis 0,05 signifikant *
> 0,05 Nicht signifikant Kein Symbol
4.9 Material
4.9.1 Chemikalien
beta-Mercaptoethanol Sigma, Taufkirchen
ABTS Roche, Mannheim
Aceton Chem solute, Renningen
Acrylamid AppliChem, Darmstadt
Ammoniumpersulfat AppliChem, Darmstadt
BSA (Rinderserumalbumin) Merck, Darmstadt
BioRad Protein Kit BioRad, München
BioRAD Dc Protein Assay BioRad, München
Bromphenolblau Merck, Darmstadt

48
Calciumacetat Merck, Darmstadt
Calciumchlorid Merck, Darmstadt
Chloroform Geyer GmbH, Renningen
Coomassie Merck, Darmstadt
DAB Vector Laboratories
Entellan Merck, Darmstadt
EDTA Merck, Darmstadt
Ethanol Merck, Darmstadt
Essigsäure Roth, Karlsruhe
Forene (Isofluran) Abbott, Wiesbaden
Fötales Kälberserum PAA, Pasching
Gelatine Serva, Heidelberg
Glycerin Merck, Darmstadt
Hämatoxylin III nach Gill Merck, Darmstadt
Isopropanol Merck, Darmstadt
Methanol Merck, Darmstadt
Mowiol Merck, Darmstadt
Natriumcarbonat Merck, Darmstadt
Natriumdihydrogenphosphat Merck, Darmstadt
Natriumchlorid Merck, Darmstadt
Ortho-phenylendiamin Merck, Darmstadt
Paraffin Merck, Darmstadt
Paraformaldehyd Merck, Damrstadt
PBS Dulbecco Biochrom AG, Berlin
Periodsäure VWR, Darmstadt
Pronase Qiagen, Düsseldorf
RNeasy Mini Kit Qiagen, Düsseldorf
Salzsäure 25% Merck, Darmstadt
SDS Roth, Karlsruhe
Schiff-Reagenz Merck, Darmstadt
Schwefelsäure Merck, Darmstadt
SYBR Green Invitrogen, Darmstadt
Target Retrieval Solution Dako, Glostrup, DK
Temed AppliChem, Darmstadt
Tris Merck, Damstadt
Triton X 100 BioRad, München

49
Tween 20 Merck, Darmstadt
Verso cDNA Kit Abgene, UK
Wasserstoffperoxid 30 % Chem solute, Renningen
Xylol Merck, Darmstadt
Zinkacetat Merck, Darmstadt
Zinkchlorid Sigma, Taufkirchen
Zinksulfat Merck, Darmstadt
4.9.2 Medikamente & Plasmide
Streptozotocin SIGMA, St. Louis, Miss., USA
Insulin Glargin (Lantus®) als Opti Set Pen
Sanofi-Aventis, Paris, Frankfreich
Thrombospondin 1, pUBTSP1 humanes TSP1 Plasmid, das in einen
langexprimierenden Vektor mit Ubiquitin
Promoter umkloniert wurde
Thrombospondin 2, pUBTSP2 murines TSP2 Plasmid, das in einen
langexprimierenden Vektor mit Ubiquitin
Promoter umkloniert wurde
Luciferase, pUBlux Firefly Luciferase, die in ein lang-
exprimierendes Plasmid mit Ubiquitin
Promoter umkloniert wurde
4.9.3 Geräte
Blutzuckermessgerät
Ascensia Elite und Sensoren Elite Sensor
Bayer, Leverkusen
Deionosierungsanlage
Seralpur pro90C
Veolia Water Systems GmbH, Wien
Elektroporator
Electro Square Porator ECM 830
BTX, Holliston, USA
Laufstation
Mini Trans-Blot Cell 37S
Bio-Rad, München
Magnetrührer
Heidolph MR3001
Heildorf, Schwabach
Mikroskope

50
Nikon eclipse 80i
Zeiss OPMI 1-FC
Nikon, Düsseldorf
Carl Zeiss, Jena
Mikroskopkamera Visitron Systems GmbH, Purchheim
Mikrotiterplatten-Photometer: Sunrise Tecan, Crailsheim
Mikrotiterplaten-Washer
Tecan M12/4R
Tecan, Crailsheim
Mikroliter- und Mehrkanalpipetten Eppendorf, Hamburg
Narkoseanlage
ZUA-82-GME Gasmisch-Einheit
Föhr Medical Instruments GmbH, Seeheim-
Ober Berrbach
Operationsbesteck Fine Science Tools, Heidelberg
OP-Tisch für Kleintiere Hans Sachs Elektronik, March-Hugstetten
Paraffinausblockeinheit
Leica EG1120
Leica, Bensheim
Real-Time-PCR ABI Prism 7000 SDS Applied Biosystems, USA
pH-Meter CG812 Schott Geräte, Mainz
Photometer BioPhotometer Hamburg, Eppendorf
Rotationsmikrotom
Microm HM335E
Mikrom GmbH, Walldorf
Stechnadeln für Insulinpen: Micro Fine BD Medical, Heidelberg
Thermomixer compact Eppendorf, Hamburg
Vortexer Heidolph REAX top Heidolph, Schwabach
Ultraschallgerät: Sonopuls HD 70 Bandelin, Berlin
UItra-Turrax: T10 basic IKA, Staufen
Waagen
Feinwaage U4100S
Tierwaage FTC3K01
Sartorius Universal, Göttingen
Kern & Sohn, Balingen, Frommern
Wasserbad Gesellschaft für Labortechnik, Burgwendel
Zentrifugen
Biofuge fresco, Heraeus
5810R
Thermo Scientific, Dänemark
Eppendorf, Hamburg

51
4.9.4 Verbrauchsmaterial
Deckgläser 24 x 50 mm Menzel Gläser, Braunschweig
Einwegskalpelle No. 21 und 11 PFM, Köln
Glaskapillare zur Blutentnahme Terumo Corporation, Tokyo, Japan
Injektionskanülen Gr.16, 18 und 20 B. Braun, Melsungen
Liquid Blocker SCI Science Services, München
Mikrotiterplatte
für Proteinurie
ELISA Nunc Maxisorp F96
Sarstedt, Nümbrecht
Thermo Scientific, Dänemark
Mikrotommesser Leica, Bensheim
Objektträger R. Langenbrinck, Emmendingen
Parafilm M Pechiney, USA
Pipettenspitzen
10 µl
200 µl und 1000 µl
Biozym Scientific GmbH, Hess, Oldendorf
Sarstedt, Nümbrecht
Polypropylenröhrchen 15 und 20 ml Greiner Bio-One, Frickenhausen
Reaktionsgefäße 1,5 und 2 ml Eppendorf, Hamburg
Super Frost Objektträger R. Langenbrinck, Emmendingen
Spritzen B. Braun, Melsungen
Tissue Tek Sakura Finetek, Zoekterwomde, NL
4.9.5 Lösungen
TBS pH 7,6 6,055 g Tris
8,52 g NaCl
Eingestellt mit HCL und mit Aqua dest. auf 1 Liter aufgefüllt
TBS/BSA TBS mit 1 % BSA
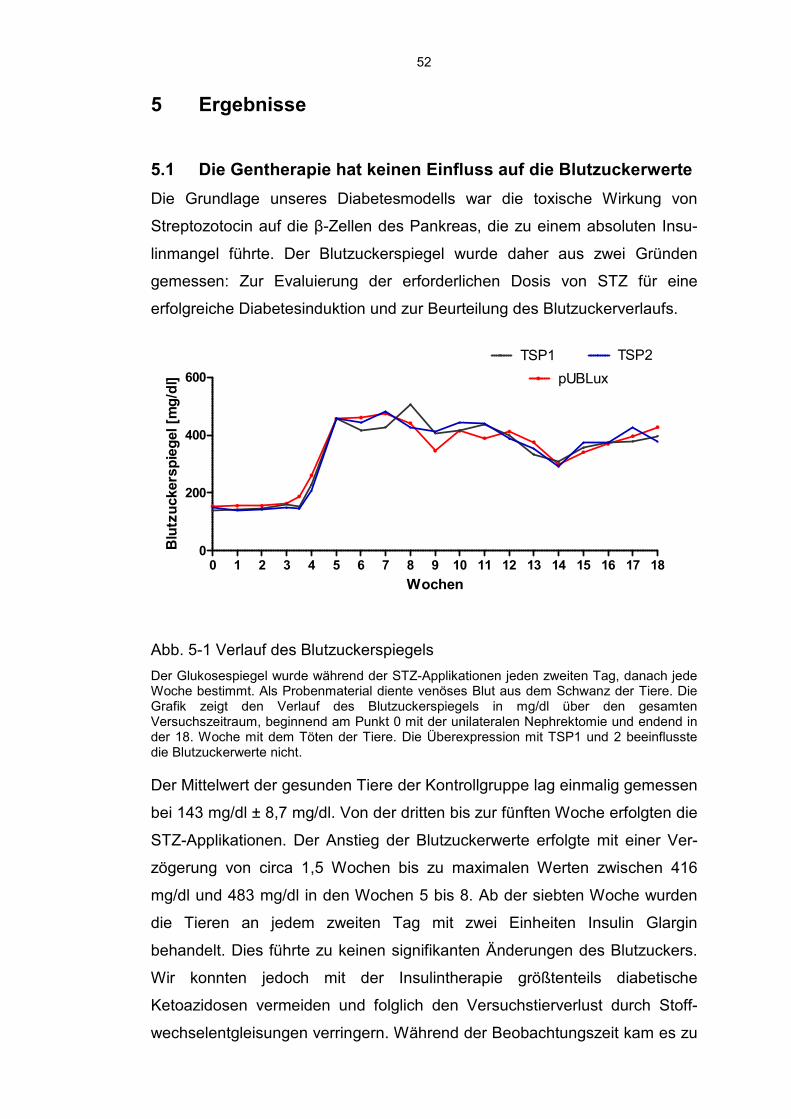
52
5 Ergebnisse
5.1 Die Gentherapie hat keinen Einfluss auf die Blutzuckerwerte
Die Grundlage unseres Diabetesmodells war die toxische Wirkung von
Streptozotocin auf die β-Zellen des Pankreas, die zu einem absoluten Insu-
linmangel führte. Der Blutzuckerspiegel wurde daher aus zwei Gründen
gemessen: Zur Evaluierung der erforderlichen Dosis von STZ für eine
erfolgreiche Diabetesinduktion und zur Beurteilung des Blutzuckerverlaufs.
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 180
200
400
600 pUBLux
TSP1 TSP2
Wochen
Blu
tzu
cker
spie
gel
[m
g/d
l]
Abb. 5-1 Verlauf des Blutzuckerspiegels
Der Glukosespiegel wurde während der STZ-Applikationen jeden zweiten Tag, danach jede Woche bestimmt. Als Probenmaterial diente venöses Blut aus dem Schwanz der Tiere. Die Grafik zeigt den Verlauf des Blutzuckerspiegels in mg/dl über den gesamten Versuchszeitraum, beginnend am Punkt 0 mit der unilateralen Nephrektomie und endend in der 18. Woche mit dem Töten der Tiere. Die Überexpression mit TSP1 und 2 beeinflusste die Blutzuckerwerte nicht.
Der Mittelwert der gesunden Tiere der Kontrollgruppe lag einmalig gemessen
bei 143 mg/dl ± 8,7 mg/dl. Von der dritten bis zur fünften Woche erfolgten die
STZ-Applikationen. Der Anstieg der Blutzuckerwerte erfolgte mit einer Ver-
zögerung von circa 1,5 Wochen bis zu maximalen Werten zwischen 416
mg/dl und 483 mg/dl in den Wochen 5 bis 8. Ab der siebten Woche wurden
die Tieren an jedem zweiten Tag mit zwei Einheiten Insulin Glargin
behandelt. Dies führte zu keinen signifikanten Änderungen des Blutzuckers.
Wir konnten jedoch mit der Insulintherapie größtenteils diabetische
Ketoazidosen vermeiden und folglich den Versuchstierverlust durch Stoff-
wechselentgleisungen verringern. Während der Beobachtungszeit kam es zu
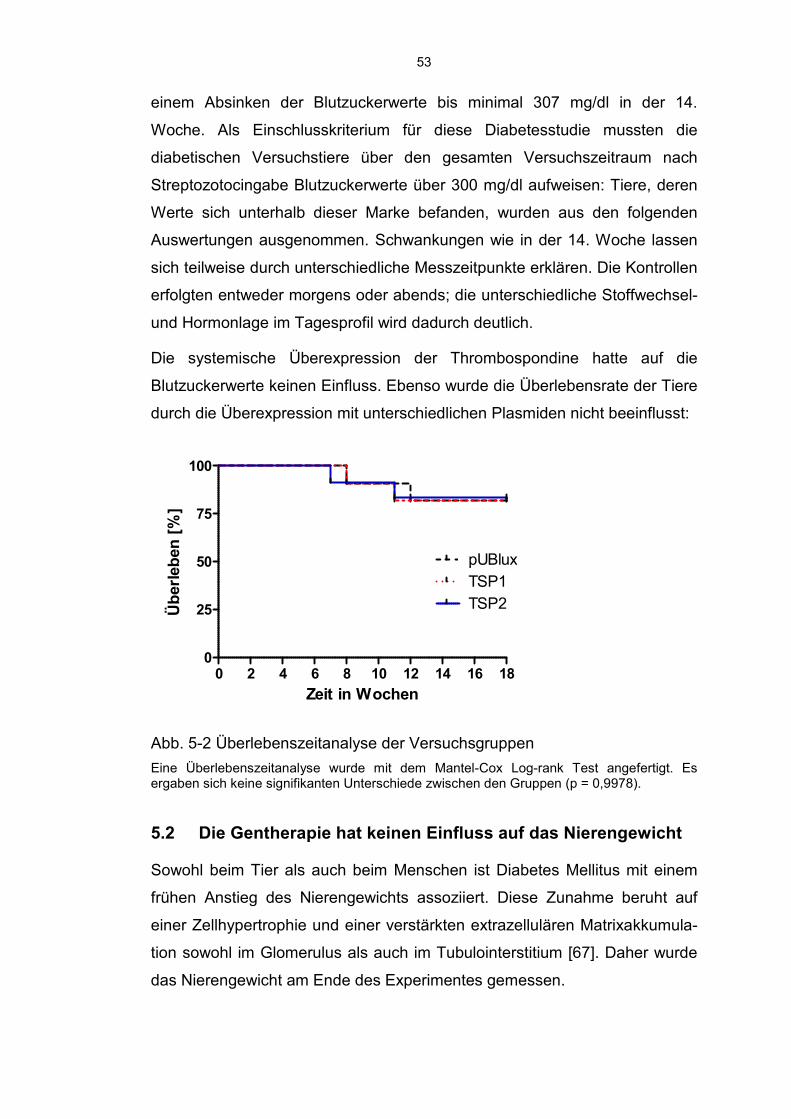
53
einem Absinken der Blutzuckerwerte bis minimal 307 mg/dl in der 14.
Woche. Als Einschlusskriterium für diese Diabetesstudie mussten die
diabetischen Versuchstiere über den gesamten Versuchszeitraum nach
Streptozotocingabe Blutzuckerwerte über 300 mg/dl aufweisen: Tiere, deren
Werte sich unterhalb dieser Marke befanden, wurden aus den folgenden
Auswertungen ausgenommen. Schwankungen wie in der 14. Woche lassen
sich teilweise durch unterschiedliche Messzeitpunkte erklären. Die Kontrollen
erfolgten entweder morgens oder abends; die unterschiedliche Stoffwechsel-
und Hormonlage im Tagesprofil wird dadurch deutlich.
Die systemische Überexpression der Thrombospondine hatte auf die
Blutzuckerwerte keinen Einfluss. Ebenso wurde die Überlebensrate der Tiere
durch die Überexpression mit unterschiedlichen Plasmiden nicht beeinflusst:
0 2 4 6 8 10 12 14 16 180
25
50
75
100
pUBlux
TSP1
TSP2
Zeit in Wochen
Üb
erle
ben
[%
]
Abb. 5-2 Überlebenszeitanalyse der Versuchsgruppen
Eine Überlebenszeitanalyse wurde mit dem Mantel-Cox Log-rank Test angefertigt. Es ergaben sich keine signifikanten Unterschiede zwischen den Gruppen (p = 0,9978).
5.2 Die Gentherapie hat keinen Einfluss auf das Nierengewicht
Sowohl beim Tier als auch beim Menschen ist Diabetes Mellitus mit einem
frühen Anstieg des Nierengewichts assoziiert. Diese Zunahme beruht auf
einer Zellhypertrophie und einer verstärkten extrazellulären Matrixakkumula-
tion sowohl im Glomerulus als auch im Tubulointerstitium [67]. Daher wurde
das Nierengewicht am Ende des Experimentes gemessen.

54
Kontrolle pUBlux TSP1 TSP20.0
2.5
5.0
7.5
10.0
12.5 ***
Nie
ren
ge
wic
ht
/ T
ierg
ew
ich
t [m
g/g
]
Abb. 5-3 Nierengewicht in Relation zum Tiergewicht
Die Nieren wurden am Ende des Experimentes gewogen und die gemessenen Werte in Relation gesetzt zum aktuellen Tiergewicht.
Bei allen diabetischen Tieren fanden wir eine Erhöhung des Nierengewichts
ungefähr um den Faktor 1,5. Zwischen den einzelnen Therapiegruppen
zeigten sich jedoch keine Unterschiede.
5.3 Thrombospondin 1 erhöht und Thrombospondin 2 reduziert
das Level von aktivem TGF-β
Ein wichtiger Mediator bei der Entwicklung der diabetischen Nephropathie ist
TGF-β. So führt z.B. dessen Aktivierung zu einer verstärkten Fibrose.
Thrombospondine können verschiedenartig mit TGF-β interagieren.
Thrombospondin 1 ist in der Lage, TGF-β zu aktivieren. Thrombospondin 2
blockiert Bindungsstellen so, dass TGF-β nicht mehr durch TSP1 aktiviert
werden kann. Um das Aktivitätsniveau von TGF-β zu beurteilen, untersuch-
ten wir eines seiner Signaltransduktionsmoleküle: Smad 2/3 liegt nach
Aktivierung von TGF-β vermehrt in phosporylierter Form vor. Daher
ermittelten wir den prozentualen Anteil von phosphoryliertem Smad 2/3
(P-Smad) pro Glomerulus mittels einer immunhistochemischen Färbung.
Weiter von Interesse war der Gesamtgehalt von TGF-β: Mit Hilfe einer Real-
Time PCR bestimmten wir die Expression von TGF-β1 und TGF-β2 (nicht
dargestellt) im kortikalen Anteil der Niere. Die eingesetzten Primer
unterscheiden nicht zwischen aktiver und passiver Form von TGF-β.
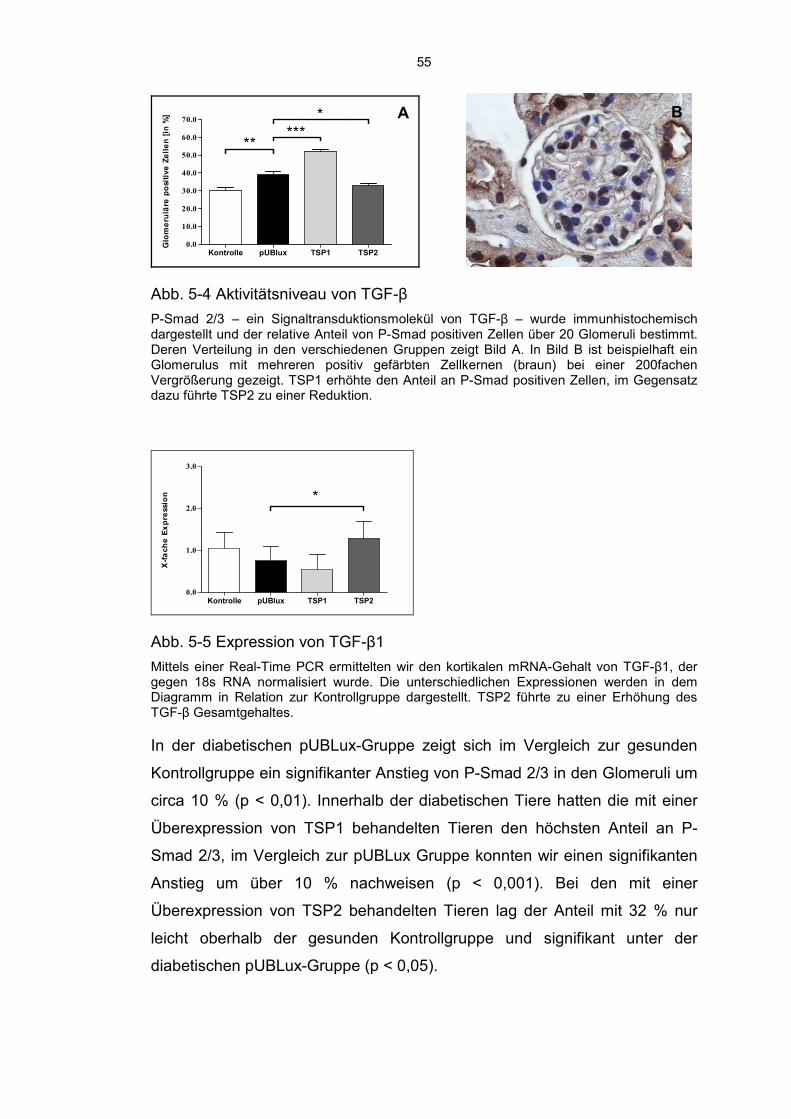
55
Kontrolle pUBlux TSP1 TSP20.0
10.0
20.0
30.0
40.0
50.0
60.0
70.0
*****
*
Glo
me
rulä
re p
osi
tive
Ze
lle
n [
in %
]
Abb. 5-4 Aktivitätsniveau von TGF-β
P-Smad 2/3 – ein Signaltransduktionsmolekül von TGF-β – wurde immunhistochemisch dargestellt und der relative Anteil von P-Smad positiven Zellen über 20 Glomeruli bestimmt. Deren Verteilung in den verschiedenen Gruppen zeigt Bild A. In Bild B ist beispielhaft ein Glomerulus mit mehreren positiv gefärbten Zellkernen (braun) bei einer 200fachen Vergrößerung gezeigt. TSP1 erhöhte den Anteil an P-Smad positiven Zellen, im Gegensatz dazu führte TSP2 zu einer Reduktion.
Kontrolle pUBlux TSP1 TSP20.0
1.0
2.0
3.0
*
X-f
ach
e E
xp
ress
ion
Abb. 5-5 Expression von TGF-β1
Mittels einer Real-Time PCR ermittelten wir den kortikalen mRNA-Gehalt von TGF-β1, der gegen 18s RNA normalisiert wurde. Die unterschiedlichen Expressionen werden in dem Diagramm in Relation zur Kontrollgruppe dargestellt. TSP2 führte zu einer Erhöhung des TGF-β Gesamtgehaltes.
In der diabetischen pUBLux-Gruppe zeigt sich im Vergleich zur gesunden
Kontrollgruppe ein signifikanter Anstieg von P-Smad 2/3 in den Glomeruli um
circa 10 % (p < 0,01). Innerhalb der diabetischen Tiere hatten die mit einer
Überexpression von TSP1 behandelten Tieren den höchsten Anteil an P-
Smad 2/3, im Vergleich zur pUBLux Gruppe konnten wir einen signifikanten
Anstieg um über 10 % nachweisen (p < 0,001). Bei den mit einer
Überexpression von TSP2 behandelten Tieren lag der Anteil mit 32 % nur
leicht oberhalb der gesunden Kontrollgruppe und signifikant unter der
diabetischen pUBLux-Gruppe (p < 0,05).
B A

Auf RNA-Ebene stellte sich ein anderes Expressionsmuster dar: Der kortikale
Gehalt von TGF-β war in der TSP1
Gruppe erhöht.
Zusammenfassend führte die TSP1
TGF-β; TSP2 verhindert
5.4 Modulation der Fibrose durch TSP1 und TSP2
Das prominenteste pathologische Merkmal der diabetischen Nephropathie ist
die Fibrosierung und Sklerosierung der Niere. Diese manifestieren sich
sowohl im Tubulointerstitium als auch in den Glomeruli.
Abb. 5-6 Histologische Veränderungen der diabetischen Nephropathie
Der Semidünnschnitt einer Niere aus der pUBluxhistopathologische Veränderungen der diabetigesunden Niere (Bild A):Basalmembran (GBM) und zuMesangium.
Einer der wichtigsten Mediatoren bei der Ent
Um den Einfluss auf dieses Zytokin und damit auf die Nierenfibrose durch
TSP1 und TSP2 zu beurteilen
küle. Über das Gesamtausmaß der Sklerose in den Glomeruli gab die PAS
Färbung Auskunft, in der kohlenhydratreiche Strukturen sichtbar wurden.
Drei spezifische Matrixproteine wurden mittels immunhistochemischer
Färbungen bewertet: Kollagen IV
im mesangialen interstitiellen Raum. Das zuletzt genan
untersuchten wir auf das Vorhandensein von
interstitiellen Fibrose nutzten wir eine Färbung gegen Kollagen I.
56
stellte sich ein anderes Expressionsmuster dar: Der kortikale
β war in der TSP1 Gruppe erniedrigt und in der TSP2
Zusammenfassend führte die TSP1 Überexpression zu einer Aktivierung von
β; TSP2 verhinderte eben diese.
Modulation der Fibrose durch TSP1 und TSP2
Das prominenteste pathologische Merkmal der diabetischen Nephropathie ist
die Fibrosierung und Sklerosierung der Niere. Diese manifestieren sich
sowohl im Tubulointerstitium als auch in den Glomeruli.
Histologische Veränderungen der diabetischen Nephropathie
Der Semidünnschnitt einer Niere aus der pUBlux-Gruppe (Bild B) verdeutlicht einige histopathologische Veränderungen der diabetischen Nephropathie im Vergleich zu einer gesunden Niere (Bild A): In diesen kommt es zur Verdickung der glomeruBasalmembran (GBM) und zu einer Akkumulation von extrazellulären Matrixproteinen im
Einer der wichtigsten Mediatoren bei der Entwicklung der Fibrose ist TGF
Um den Einfluss auf dieses Zytokin und damit auf die Nierenfibrose durch
TSP1 und TSP2 zu beurteilen, untersuchten wir verschiedene Matrixmole
küle. Über das Gesamtausmaß der Sklerose in den Glomeruli gab die PAS
kunft, in der kohlenhydratreiche Strukturen sichtbar wurden.
Drei spezifische Matrixproteine wurden mittels immunhistochemischer
Färbungen bewertet: Kollagen IV befindet sich sowohl in der GBM
ialen interstitiellen Raum. Das zuletzt genannte Kompart
untersuchten wir auf das Vorhandensein von Fibronektin. Zur Evaluation der
interstitiellen Fibrose nutzten wir eine Färbung gegen Kollagen I.
A
stellte sich ein anderes Expressionsmuster dar: Der kortikale
uppe erniedrigt und in der TSP2
Überexpression zu einer Aktivierung von
Modulation der Fibrose durch TSP1 und TSP2
Das prominenteste pathologische Merkmal der diabetischen Nephropathie ist
die Fibrosierung und Sklerosierung der Niere. Diese manifestieren sich
Histologische Veränderungen der diabetischen Nephropathie
Gruppe (Bild B) verdeutlicht einige schen Nephropathie im Vergleich zu einer
In diesen kommt es zur Verdickung der glomerulären einer Akkumulation von extrazellulären Matrixproteinen im
wicklung der Fibrose ist TGF-β.
Um den Einfluss auf dieses Zytokin und damit auf die Nierenfibrose durch
untersuchten wir verschiedene Matrixmole-
küle. Über das Gesamtausmaß der Sklerose in den Glomeruli gab die PAS-
kunft, in der kohlenhydratreiche Strukturen sichtbar wurden.
Drei spezifische Matrixproteine wurden mittels immunhistochemischer
befindet sich sowohl in der GBM als auch
nte Kompartiment
Fibronektin. Zur Evaluation der
interstitiellen Fibrose nutzten wir eine Färbung gegen Kollagen I.
B
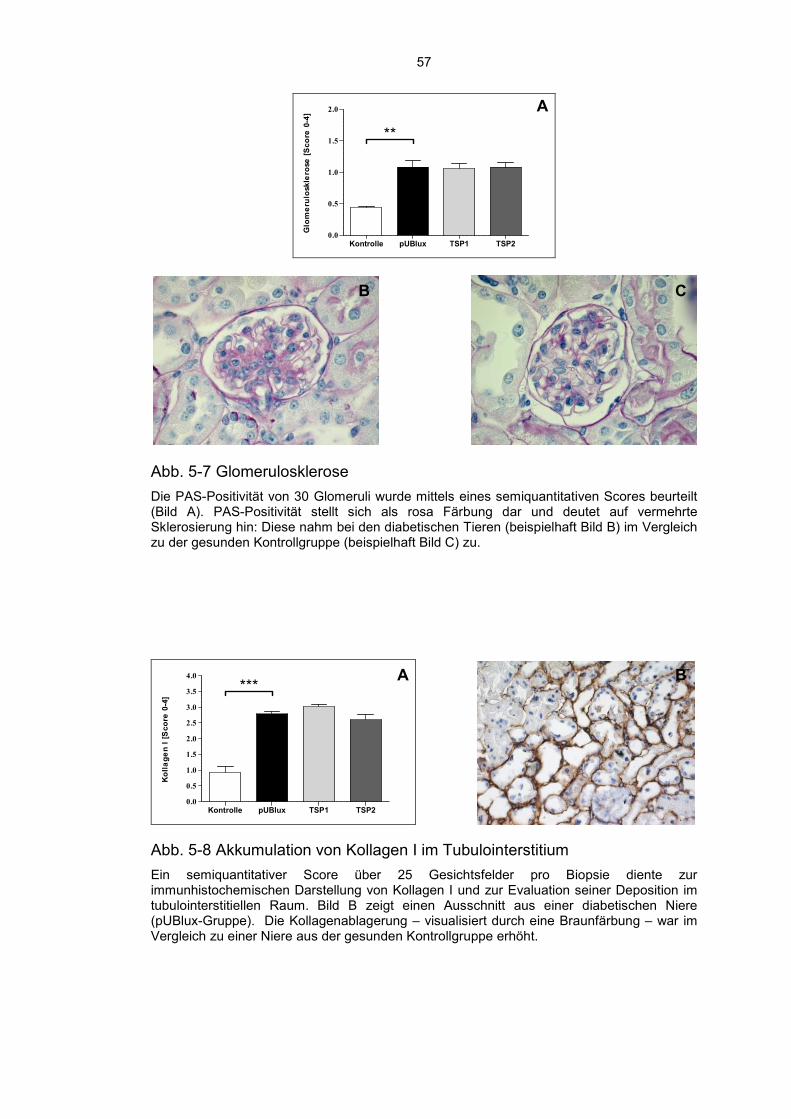
57
Kontrolle pUBlux TSP1 TSP20.0
0.5
1.0
1.5
2.0
**
Glo
me
rulo
skle
rose
[S
co
re 0
-4]
Abb. 5-7 Glomerulosklerose
Die PAS-Positivität von 30 Glomeruli wurde mittels eines semiquantitativen Scores beurteilt (Bild A). PAS-Positivität stellt sich als rosa Färbung dar und deutet auf vermehrte Sklerosierung hin: Diese nahm bei den diabetischen Tieren (beispielhaft Bild B) im Vergleich zu der gesunden Kontrollgruppe (beispielhaft Bild C) zu.
Kontrolle pUBlux TSP1 TSP20.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
***
Ko
lla
ge
n I
[S
co
re 0
-4]
Abb. 5-8 Akkumulation von Kollagen I im Tubulointerstitium
Ein semiquantitativer Score über 25 Gesichtsfelder pro Biopsie diente zur immunhistochemischen Darstellung von Kollagen I und zur Evaluation seiner Deposition im tubulointerstitiellen Raum. Bild B zeigt einen Ausschnitt aus einer diabetischen Niere (pUBlux-Gruppe). Die Kollagenablagerung – visualisiert durch eine Braunfärbung – war im Vergleich zu einer Niere aus der gesunden Kontrollgruppe erhöht.
A
B C
A B

58
Kontrolle pUBlux TSP1 TSP20.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5 *** ***
Ko
lla
ge
n I
V [
Sco
re 0
-4]
Kontrolle pUBlux TSP1 TSP20.0
0.5
1.0
1.5
2.0
2.5
3.0
***
Fib
ron
ek
tin
[S
core
0-4
]
Abb. 5-9 Akkumulation der Matrixmoleküle Kollagen IV und Fibronektin
Die entsprechenden Proteine wurden immunhistochemisch dargestellt. Über glomerulären Querschnitten wurde mittels eines semiquantitativen Scores die Deposition der Matrixmoleküle Kollagen IV (Bild A) und Fibronektin (Bild B) beurteilt. Die Therapie beeinflusste die Fibronektinakkumulation nicht, jedoch führte die TSP2 Überexpression zu einer signifikanten Verminderung der Kollagen IV Deposition. Bild D (TSP2) im Vergleich zu Bild C (pUBlux) verdeutlicht dies: Eine geringere Braunfärbung im Glomerulus spricht für eine geringere Kollagen IV Akkumulation.
Insgesamt waren alle vier untersuchten Fibrosemarker bei der diabetischen
pUBlux-Gruppe im Vergleich zur gesunden Kontrollgruppe signifikant erhöht
(mindestens p < 0,01), wenn auch nur auf relativ niedrigem absoluten
Niveau. Bezüglich der Glomerulosklerose, der Fibronektinakkumulation und
der Deposition von Kollagen I waren keine Unterschiede innerhalb der thera-
pierten Gruppen festzustellen. Jedoch führte die TSP2 Überexpression zu
einer signifikanten Reduktion von Kollagen IV im Glomerulus (p < 0,001).
C D
A B
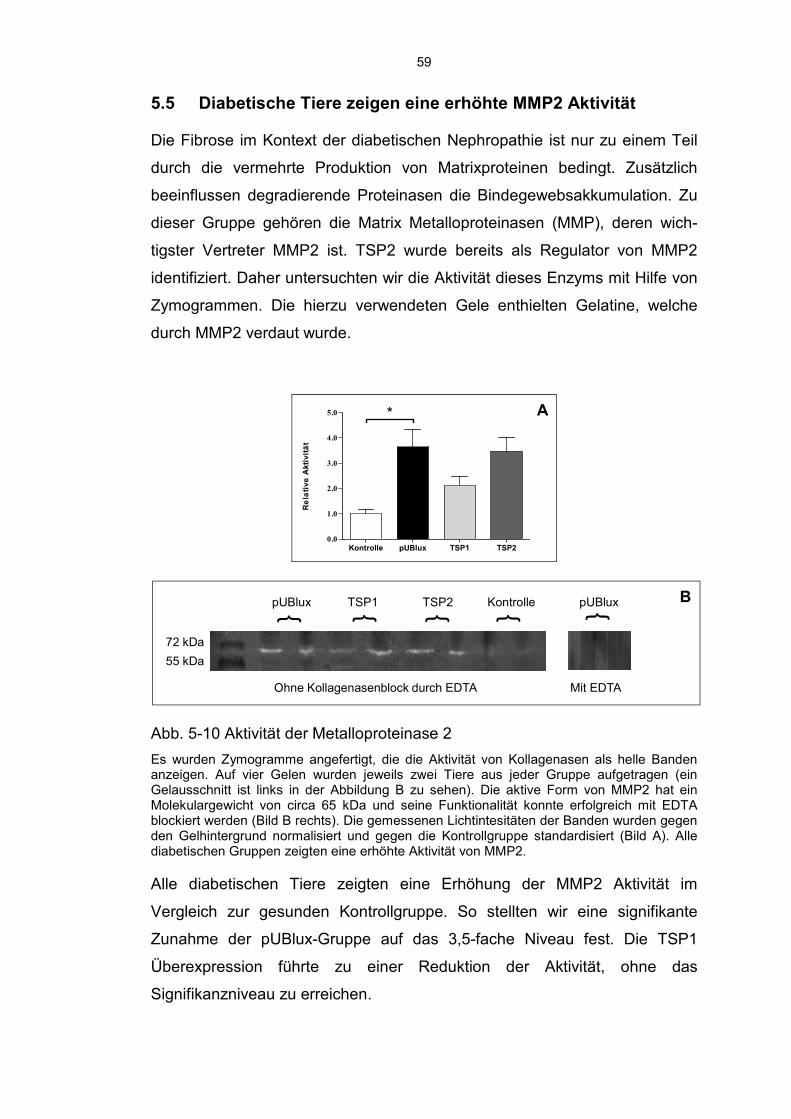
59
5.5 Diabetische Tiere zeigen eine erhöhte MMP2 Aktivität
Die Fibrose im Kontext der diabetischen Nephropathie ist nur zu einem Teil
durch die vermehrte Produktion von Matrixproteinen bedingt. Zusätzlich
beeinflussen degradierende Proteinasen die Bindegewebsakkumulation. Zu
dieser Gruppe gehören die Matrix Metalloproteinasen (MMP), deren wich-
tigster Vertreter MMP2 ist. TSP2 wurde bereits als Regulator von MMP2
identifiziert. Daher untersuchten wir die Aktivität dieses Enzyms mit Hilfe von
Zymogrammen. Die hierzu verwendeten Gele enthielten Gelatine, welche
durch MMP2 verdaut wurde.
Kontrolle pUBlux TSP1 TSP20.0
1.0
2.0
3.0
4.0
5.0 *
Re
lati
ve
Akti
vit
ät
Abb. 5-10 Aktivität der Metalloproteinase 2
Es wurden Zymogramme angefertigt, die die Aktivität von Kollagenasen als helle Banden anzeigen. Auf vier Gelen wurden jeweils zwei Tiere aus jeder Gruppe aufgetragen (ein Gelausschnitt ist links in der Abbildung B zu sehen). Die aktive Form von MMP2 hat ein Molekulargewicht von circa 65 kDa und seine Funktionalität konnte erfolgreich mit EDTA blockiert werden (Bild B rechts). Die gemessenen Lichtintesitäten der Banden wurden gegen den Gelhintergrund normalisiert und gegen die Kontrollgruppe standardisiert (Bild A). Alle diabetischen Gruppen zeigten eine erhöhte Aktivität von MMP2.
Alle diabetischen Tiere zeigten eine Erhöhung der MMP2 Aktivität im
Vergleich zur gesunden Kontrollgruppe. So stellten wir eine signifikante
Zunahme der pUBlux-Gruppe auf das 3,5-fache Niveau fest. Die TSP1
Überexpression führte zu einer Reduktion der Aktivität, ohne das
Signifikanzniveau zu erreichen.
72 kDa
55 kDa
} } } }
pUBlux KontrolleTSP2TSP1}
pUBlux
Ohne Kollagenasenblock durch EDTA Mit EDTA
A
B
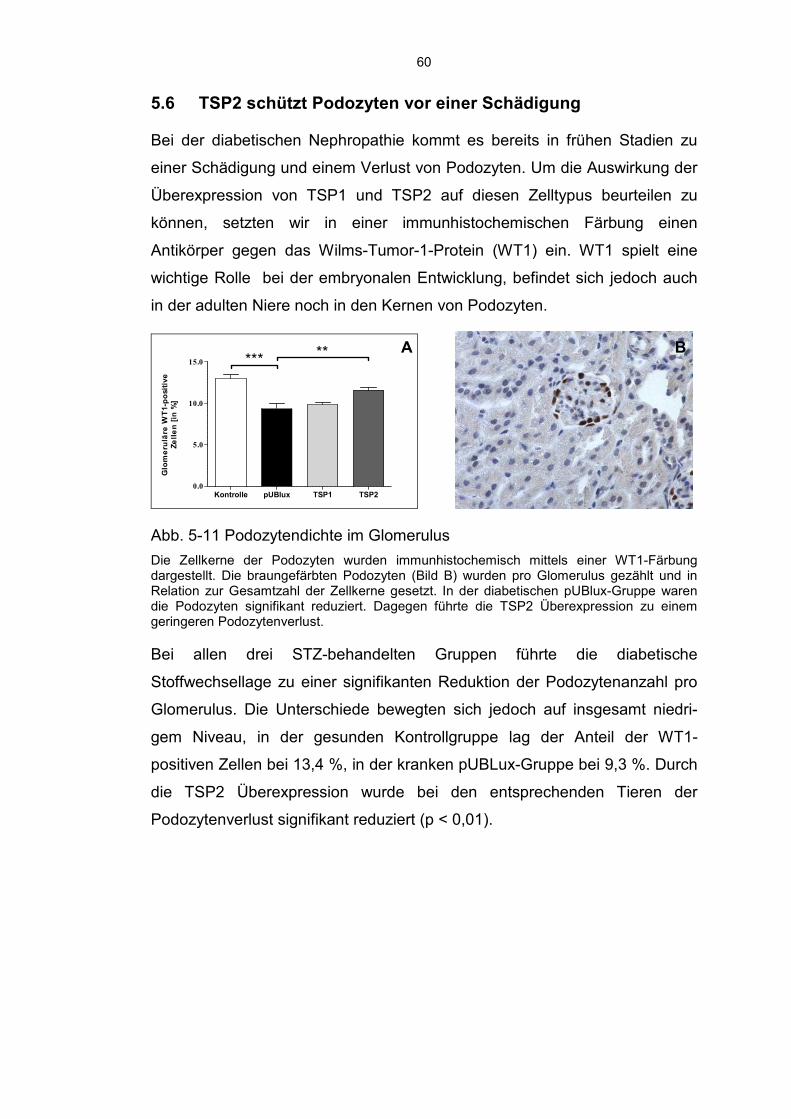
60
5.6 TSP2 schützt Podozyten vor einer Schädigung
Bei der diabetischen Nephropathie kommt es bereits in frühen Stadien zu
einer Schädigung und einem Verlust von Podozyten. Um die Auswirkung der
Überexpression von TSP1 und TSP2 auf diesen Zelltypus beurteilen zu
können, setzten wir in einer immunhistochemischen Färbung einen
Antikörper gegen das Wilms-Tumor-1-Protein (WT1) ein. WT1 spielt eine
wichtige Rolle bei der embryonalen Entwicklung, befindet sich jedoch auch
in der adulten Niere noch in den Kernen von Podozyten.
Kontrolle pUBlux TSP1 TSP20.0
5.0
10.0
15.0 *** **
Glo
me
rulä
re W
T1-p
osi
tive
Ze
lle
n [
in %
]
Abb. 5-11 Podozytendichte im Glomerulus
Die Zellkerne der Podozyten wurden immunhistochemisch mittels einer WT1-Färbung dargestellt. Die braungefärbten Podozyten (Bild B) wurden pro Glomerulus gezählt und in Relation zur Gesamtzahl der Zellkerne gesetzt. In der diabetischen pUBlux-Gruppe waren die Podozyten signifikant reduziert. Dagegen führte die TSP2 Überexpression zu einem geringeren Podozytenverlust.
Bei allen drei STZ-behandelten Gruppen führte die diabetische
Stoffwechsellage zu einer signifikanten Reduktion der Podozytenanzahl pro
Glomerulus. Die Unterschiede bewegten sich jedoch auf insgesamt niedri-
gem Niveau, in der gesunden Kontrollgruppe lag der Anteil der WT1-
positiven Zellen bei 13,4 %, in der kranken pUBLux-Gruppe bei 9,3 %. Durch
die TSP2 Überexpression wurde bei den entsprechenden Tieren der
Podozytenverlust signifikant reduziert (p < 0,01).
A B

61
5.7 TSP2 hemmt die Infiltration von Entzündungszellen
Einige aktuelle Studien betonen die Bedeutung der Entzündungsreaktion in
der Niere bei der Entstehung der diabetischen Nephropathie. Dabei spielen
verschiedene Typen von Entzündungszellen wie Maphrophagen und T-
Lymphozyten eine Rolle. Daher quantifizierten wir die Einwanderung von
Immunzellen mittels verschiedener immunhistochemischer Färbungen gegen
vier verschiedene Antigene: CD3+ Zellen entsprechen den T-Lymphozyten,
der Oberflächenmarker F 4/80 ist auf Makrophagen lokalisiert und die
Rezeptoren CD4 und CD8 befinden sich auf zwei verschiedenen Unterpo-
pulationen von T-Zellen.
Kontrolle pUBlux TSP1 TSP20.0
20.0
40.0
60.0 *** *
F4/8
0 p
osi
tive
Ze
lle
n p
ro G
esi
ch
tsfe
ld
Kontrolle pUBlux TSP1 TSP20.0
5.0
10.0
15.0
20.0
25.0 *** *
CD
3 p
osi
tiv
e Z
ell
en
pro
Ge
sic
hts
feld
Abb. 5-12 Infiltration von Makrophagen und T-Zellen
Wir inkubierten die Präparate mit Antikörpern gegen F 4/80 als Makrophagenmarker (Bild A) und gegen CD3 als T-Zellmarker (Bild B). Anschließend wurden die positiv gefärbten Zellen über 25 Gesichtsfelder im Kortexbereich bei 600-facher Vergrößerung gezählt. Die TSP2 Überexpression reduzierte signifikant die Anzahl der infiltrierenden Zellen, sowohl der Makrophagen als auch der T-Zellen. Bild C zeigt beispielhaft die Makrophageninfiltration, Bild D die T-Lymphozyteninfiltration.
C D
A B

62
Kontrolle pUBlux TSP1 TSP20.0
5.0
10.0
15.0
20.0
25.0 ****
CD
4+
Ze
lle
n p
ro G
esi
ch
tsfe
ld
Kontrolle pUBlux TSP1 TSP20.0
5.0
10.0
15.0
20.0
25.0
30.0
*****
CD
8+
Ze
lle
n p
ro G
esi
ch
tsfe
ld
Abb. 5-13 Differenzierung der T-Zellen in CD4+ und CD8+ Zellen
Zur genaueren Differenzierung der T-Zellen in CD4+ (Bild A) und CD8+ Zellen (Bild B) detektierten wir diese Zelltypen in einer Immunfluoreszenz und zählten positiv gefärbte Zellen über 20 Gesichtsfelder bei 400-facher Vergrößerung im Cortexbereich der Biopsien. Wir konnten eine signifikante Reduzierung durch TSP2 Überexpression der Infiltration sowohl von CD4+ Zellen, als auch von CD8+ Zellen feststellen. Bild C zeigt beispielhaft einen Ausschnitt einer Niere der pUBlux Gruppe.
Diabetes Mellitus führte bei allen drei STZ-behandelten Gruppen zu einer
signifikanten Steigerung der Infiltration der untersuchten Entzündungszellen.
Die Zellen befanden sich vor allem außerhalb der Glomeruli. Durch die TSP2
Überexpression wurde die Infiltration von CD3+ T-Zellen um 20 % und der
Influx von F 4/80 positiven Makrophagen um 45 % signifikant reduziert (bei
beiden p < 0,05). Der Effekt bei den T-Zellen wirkte sich nahezu in gleichem
Maße auf die Populationen der CD4+ und CD8+ Zellen aus (jeweils p < 0,05).
A B
C
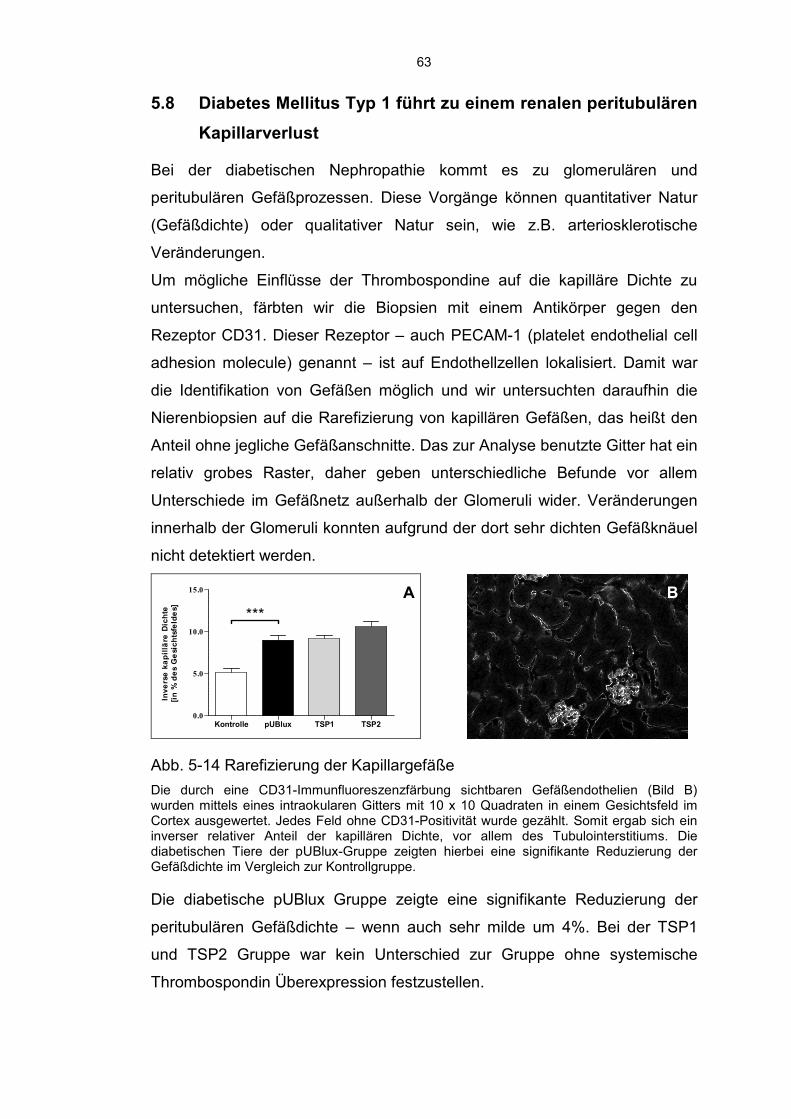
63
5.8 Diabetes Mellitus Typ 1 führt zu einem renalen peritubulären
Kapillarverlust
Bei der diabetischen Nephropathie kommt es zu glomerulären und
peritubulären Gefäßprozessen. Diese Vorgänge können quantitativer Natur
(Gefäßdichte) oder qualitativer Natur sein, wie z.B. arteriosklerotische
Veränderungen.
Um mögliche Einflüsse der Thrombospondine auf die kapilläre Dichte zu
untersuchen, färbten wir die Biopsien mit einem Antikörper gegen den
Rezeptor CD31. Dieser Rezeptor – auch PECAM-1 (platelet endothelial cell
adhesion molecule) genannt – ist auf Endothellzellen lokalisiert. Damit war
die Identifikation von Gefäßen möglich und wir untersuchten daraufhin die
Nierenbiopsien auf die Rarefizierung von kapillären Gefäßen, das heißt den
Anteil ohne jegliche Gefäßanschnitte. Das zur Analyse benutzte Gitter hat ein
relativ grobes Raster, daher geben unterschiedliche Befunde vor allem
Unterschiede im Gefäßnetz außerhalb der Glomeruli wider. Veränderungen
innerhalb der Glomeruli konnten aufgrund der dort sehr dichten Gefäßknäuel
nicht detektiert werden.
Kontrolle pUBlux TSP1 TSP20.0
5.0
10.0
15.0
***
Inv
ers
e k
ap
illä
re D
ich
te[i
n %
de
s G
esi
ch
tsfe
lde
s]
Abb. 5-14 Rarefizierung der Kapillargefäße
Die durch eine CD31-Immunfluoreszenzfärbung sichtbaren Gefäßendothelien (Bild B) wurden mittels eines intraokularen Gitters mit 10 x 10 Quadraten in einem Gesichtsfeld im Cortex ausgewertet. Jedes Feld ohne CD31-Positivität wurde gezählt. Somit ergab sich ein inverser relativer Anteil der kapillären Dichte, vor allem des Tubulointerstitiums. Die diabetischen Tiere der pUBlux-Gruppe zeigten hierbei eine signifikante Reduzierung der Gefäßdichte im Vergleich zur Kontrollgruppe.
Die diabetische pUBlux Gruppe zeigte eine signifikante Reduzierung der
peritubulären Gefäßdichte – wenn auch sehr milde um 4%. Bei der TSP1
und TSP2 Gruppe war kein Unterschied zur Gruppe ohne systemische
Thrombospondin Überexpression festzustellen.
A B

64
5.9 Die Behandlung mit TSP1 und TSP2 hat keinen Einfluss auf
die Nierenfunktion
Wir konnten einige Unterschiede auf histologischer und molekularer Ebene
durch die Überexpression feststellen. Um deren Effekt auf die Nierenleistung
zu beurteilen, untersuchten wir vier Nierenfunktionsparameter, unter
anderem die Ausscheidung von Albumin über den Urin – ein frühes
Kennzeichen der diabetischen Nephropathie. Kreatinin wurde im Serum und
Urin gemessen und daraus die Kreatinin-Clearance berechnet. Daneben
bestimmten wir die Serumharnstoffkonzentration als Retentionsparameter.
Die Konzentration von Albumin in den Mäuseurinen erhielten wir mittels
eines ELISA-Kits. Zusätzlich bestimmten wir die Proteinurie.
Kontrolle pUBlux TSP1 TSP20.0
20.0
40.0
60.0
80.0*
Se
rum
ha
rnst
off
[m
g/d
l]
Kontrolle pUBlux TSP1 TSP20.0
100.0
200.0
300.0
Kre
ati
nin
-Cle
ara
nc
e [
ul/
min
]
Abb. 5-15 Nierenfunktionsparameter am Endzeitpunkt
Aus dem Serum der Tiere wurde Harnstoff (Bild A) an einem automatischen Analysegerät bestimmt. Ebenso wurde die Kreatininkonzentration im Serum und im Urin bestimmt und daraus die Kreatinin-Clearance (Bild B) berechnet. Die Überexpression mit TSP1 und TSP2 hatte keinen Einfluss auf die Nierenfunktion.
Kontrolle pUBlux TSP1 TSP20.0
0.5
1.0
1.5
2.0
2.5**
Pro
tein
uri
e [
mg
/24
h]
Kontrolle pUBlux TSP1 TSP20.0
200.0
400.0
600.0
800.0
**
Alb
um
inu
rie
[µµ µµ
g/2
4h
]
Abb. 5-16 Protein- und Albuminurie
Die Proteinurie (Bild A) wurde mittels eines Bradford-Kits bestimmt, die Albuminurie (Bild B) mit Hilfe eines Elisa-Kits. Die diabetischen Tiere schieden signifikant höhere Konzentrationen an Protein und Albumin aus (pUBlux Gruppe versus Kontrolle). Die Überexpression zeigte keinen Einfluss auf diese beiden Parameter.
A B
A B

65
Die diabetischen Tiere zeigten signifikant erhöhte Werte an Serumharnstoff,
Proteinurie und Albuminurie. Der durch die STZ-Injektion ausgelöste
Diabetes Mellitus hatte also einen funktionseinschränkenden Effekt auf die
Niere. Die in Gefolge der Überexpression von TSP1 und TSP2 aufgetrete-
nen Veränderungen hatten jedoch keinen Einfluss auf die Nierenleistung.

66
6 Diskussion
6.1 Aktuelle Datenlage
Die diabetische Nephropathie ist mittlerweile die häufigste Ursache für ein
chronisches Nierenversagen mit Dialysepflichtigkeit in der westlichen Welt
[65]. Aufgrund der aktuellen Therapiemöglichkeiten ist man in der Lage, die
Progression der Erkrankung zu verlangsamen, nicht aber zu stoppen oder
umzukehren [47]. Daher ist die Erforschung der Mechanismen, die zur Ent-
wicklung der diabetischen Nephropathie beitragen, besonders wichtig.
Einer der wichtigsten Mediatoren bei der diabetischen Nephropathie ist
TGF-β, das als Prozytokin-Komplex sezerniert wird und extrazellulär aktiviert
werden muss [95]. Thrombospondin 1 und 2 spielen dabei eine Rolle:
In mehreren Studien konnte in vivo und in vitro gezeigt werden, dass ein
erhöhter Glukosespiegel TSP1 hochreguliert. So führt die Glukosestimulation
von Mesangiumzellen [61], proximalen Tubuluszellen [91], distalen Tubulus-
zellen [89], Endothelzellen, glatten Gefäßmuskelzellen und Fibroblasten [78]
zu einer Erhöhung der Expression von TSP1 und zu einer Aktivierung von
TGF-β in diesen Zellen. In Tierstudien konnte demonstriert werden, dass die
Produktion von TSP1, das in der gesunden Niere kaum exprimiert wird, stark
ansteigt [24]. In zwei Studien mit Nierenbiopsien von Patienten mit Diabetes
Mellitus Typ 1 [81] und Diabetes Mellitus Typ 2 [39] zeigte sich, dass die
Expression von TSP1 mit der Schwere der Erkrankung und der Aktivität von
TGF-β korreliert. Weiterhin demonstrierten Moczulski et al., dass TSP1
neben der Cyclooxygenase 1 als einziges Produkt von 198 Kandiatengenen
in peripheren mononukleären Blutzellen bei Patienten mit einer diabetischen
Nephropathie hochreguliert war [52]. Unsere Arbeitsgruppe konnte in einem
STZ-Diabetes-Modell mit TSP1 Knockout Mäusen zeigen, dass das Fehlen
von TSP1 protektiv bei der Entwicklung der diabetischen Nephropathie wirkt:
Der Grad der Matrixakkumulation, der TGF-β Aktivierung, des
Podozytenschadens, der Entzündungszelleninfiltration und der funktionellen
Schädigung waren in diabetische TSP1 defizienten Mäusen reduziert [24].

67
Auch in Tiermodellen anderer fibrotischer Nierenerkrankungen (Anti-GBM
Nephritis, Anti Thy 1 Modell) wirkte das Fehlen von TSP1 (durch Knockout
oder durch Blockierung der Aktivierung von TGF-β) protektiv [21-22, 38].
Zu Thrombospondin 2 existieren weitaus weniger Daten: In vitro konnten
Schultz-Cherry et al. zeigen, dass TSP2 in der Lage ist, die Aktivierung von
TGF-β durch TSP1 zu verhindern. Unsere Arbeitsgruppe konnte dies in zwei
Tierstudien bestätigen: In einem Experiment mit TSP2 Knockout Mäusen
waren einige Merkmale der anti-GBM Nephritis verstärkt [23]. Weiterhin
wirkte die Überexpression von TSP2 protektiv bei einem Rattenmodell der
mesangioproliferativen Glomerulonephritis (Anti Thy1 Modell) durch die
Inhibition der Zellproliferation, der Entzündungszellinfiltration und der TGF-β-
Aktivierung [25].
Ziel dieser Arbeit war es daher zu überprüfen, ob eine gentherapeutische
Überexpression von TSP2 vor der Entwicklung einer diabetischen
Nephropatie schützt bzw. die Überexpression von TSP1 diese aggraviert.
6.2 Induktion eines Diabetes Mellitus und Entwicklung einer
Diabetischen Nephropathie
Als Grundlage für unser Experiment diente uns ein Tiermodel, bei dem ein
Diabetes Mellitus Typ 1 durch Streptozotocinapplikation induziert wurde. STZ
– ursprünglich als Antibiotikum identifiziert – wirkt toxisch vor allem auf die
insulinproduzierenden Zellen im Pankreas. Dieses Tiermodel ist bereits seit
zwei bis drei Jahrzenten gut etabliert. STZ kann jedoch auch potentiell an-
dere Gewebe schädigen. Um diesem Nebeneffekt entgegenzuwirken, haben
wir uns für ein Low-Dose Regime (geringere Dosis auf mehrere Tage verteilt)
entschieden, wodurch das toxische Potential verringert werden kann [12]. Die
applizierte Menge richtete sich nach der erzielten Blutzuckersteigerung, so
dass wir den Tieren individuell zwischen 300 mg/kg KG an drei Tagen und
480 mg/kg KG an sechs Tagen verabreichten. Nichtsdestotrotz können
potentielle toxische Nebenwirkungen an der Niere nicht ausgeschlossen
werden.

68
Der Erfolg einer Diabetesinduktion hängt von der STZ-Aktivität und vom
Stamm der Tiere ab [79]: Wir setzten 300 mg/dl als minimalen
durchschnittlichen Blutzuckerwert über 12-13 Wochen fest; Tiere, die diesen
Wert nicht erreichten, wurden von der weiteren Auswertung ausgenommen.
Die drei wichtigsten Kennzeichen einer diabetischen Nephropathie sind eine
Verdickung der glomerulären Basalmembran, eine Expansion der
extrazellulären Matrix und als funktioneller Ausdruck der Schädigung eine
Proteinurie. Wir konnten immunhistologisch eine verstärkte Ablagerung der
wichtigsten Matrixmoleküle bei allen drei diabetischen Gruppen feststellen.
Diese drei Gruppen zeigten ebenso einen signifikanten Anstieg der
Albuminuriekonzentration zum Endzeitpunkt.
Zusammengefasst zeigten alle STZ-behandelten und ausgewerteten Tiere
eine milde bis moderate diabetische Nephropathie. Ungeachtet dessen
waren die Tiere physiognomisch gesund; die Überexpression hatte keine
makroskopisch erkennbaren Nebeneffekte.
6.3 Aktivierung von TGF-β
Einer der wichtigstes Mediatoren der diabetischen Nephropathie ist TGF-β.
Diese Studie zeigt zum ersten Mal an einem diabetischen Tiermodell, dass
TSP1 das Aktivitätsniveau von TGF-β erhöht und TSP2 dieses erniedrigt:
P-Smad 2/3 als Ausdruck der Aktivität von TGF-β war in der TSP1 Gruppe
erhöht und in der TSP2 Gruppe erniedrigt. Diese Änderungen komplettieren
Ergebnisse aus zwei früheren Studien unserer Arbeitsgruppe mit einem
Glomerulonephritismodell mit TSP2 Überexpression und einem
Diabetesmodell mit TSP1 Knockout-Mäusen.
Damit bestätigen diese Ergebnisse bisherige Beobachtungen, dass TGF-β
durch TSP1 aktiviert wird und diese Aktivierung durch TSP2 inhibiert wird.
Wir konnten dies jedoch zum ersten Mal in einem Tiermodel der diabetischen
Nephropathie nachweisen.
Die drei diabetischen Gruppen zeigten keine erhöhte Expression von TGF-β1
und TGF-β2 auf mRNA-Ebene. Bei den Messungen kann jedoch nicht
zwischen aktiver und passiver Form unterschieden werden. Die Ergebnisse
stehen in Kontrast zum Befund der meisten Tierstudien, bei denen im Rah-
men der diabetischen Nephropathie TGF-β hochreguliert wird [95].

69
TGF-β wird jedoch in Folge der diabetischen Nierenschädigung im tubulären
Ultrafiltrat und im Urin gefunden [82]. Dieser Mechanismus eines Verlustes
von TGF-β erklärt jedoch nicht den fehlenden Anstieg auf mRNA-Ebene.
Allerdings sollte an dieser Stelle noch einmal betont werden, dass TGF-β
seine Funktion vor allem über sein Aktivitätsniveau ausübt, und nicht über
seine Expression [3]. Innerhalb der diabetischen Gruppen war bei der TSP2
Gruppe die höchste Expression von Gesamt TGF-β1 festzustellen. Dies
könnte man als kompensatorischen Anstieg infolge der gestörten Aktivierung
interpretieren.
6.4 Modulation der Fibrose
Eines der zentralen Elemente der diabetischen Nephropathie ist die
glomeruläre und interstitielle Fibrose. Beim Menschen treten diese
Veränderungen 5-7 Jahre nach Erstmanifestation eines Diabetes Mellitus auf
[28]; bei unseren Tieren waren sie am Ende des Experiments nach 14
Wochen unter Hyperglykämie ebenfalls zu sehen. Insgesamt waren die
histopathologischen Veränderungen aufgrund der vergleichsweise kurzen
Krankheitsdauer eher milde und entsprachen denen einer beginnenden
diabetischen Nephropathie.
In vielen fibrotischen Erkrankungen, wie auch der diabetischen
Nephropathie, ist TGF-β bei der Entwicklung der Fibrose involviert, indem es
die Produktion von EZM-Molekülen wie Fibronektin, Laminin und Kollagenen
anregt [16]. Dementsprechend führen Anti-TGF-β-Therapien zu einer
geringeren Matrixakkumulation [40, 94]. In unserer Studie zeigte die TSP2
Gruppe einen signifikant geringeren Gehalt an Kollagen IV im Glomerulus,
wahrscheinlich durch eine Reduktion der TGF-β Aktivität. Der Gehalt zweier
weiterer Matrixmoleküle (Kollagen I im tubulointerstitiellen Raum und Fibro-
nektin im Glomerulus) war jedoch nicht signifikant verändert, was auf einen
zusätzlichen TGF-β-unabhängigen Mechanismus hindeuten könnte.
Nach den Ergebnissen einer Studie mit diabetischen TSP1 Knockout Mäu-
sen, bei der unsere Arbeitsgruppe eine Erniedrigung der Glomerulosklerose
feststellte [24], erwarteten wir eine Verschlechterung dieser histologischen
Veränderung bei den Tieren mit TSP1 Überexpression. Bei der TSP1 Gruppe
war jedoch trotz der gesteigerten TGF-β Aktivierung die Deposition keines

70
der untersuchten Matrixmoleküle signifikant verändert: Möglicherweise
befand sich das TGF-β-System und seine nachgeschalteten Systeme bereits
durch den Diabetes Mellitus auf einem so hohen Aktivitätsniveau, dass eine
weitere Aktivierung keine weiteren Konsequenzen im Bezug auf die
Matrixmolekülproduktion hatte.
Die Matrixakkumulation bei der diabetischen Nephropathie beruht jedoch
nicht nur auf der gesteigerten Produktion von Proteinen der EZM, sondern
auch auf dem gestörten Umsatz dieser Matrixmoleküle. Dabei sind unter
anderem sogenannte Matrix-Metalloproteinasen (MMP) beteiligt. Ein
Vertreter dieser Familie – MMP2 – kann bekanntermaßen mit TSP2 inter-
agieren. TSP2 kann einen Komplex mit MMP2 bilden und dann über einen
Rezeptor in Fibroblasten internalisiert werden, wie Yang et al. in vitro
demonstrierten [90]. Cheng et al. konnten außerdem zeigen, dass MMP2
eine epitheliale mesenchymale Transition (EMT) induzieren kann [15] und
über diesen Mechanismus an der Entstehung der diabetischen Nierenfibrose
beteiligt sein könnte: So reichte die transgene Überexpression von MMP2 in
renalen Tubulusepithelzellen aus, um das Spektrum einer fibrotischen
Nierenerkrankung zu generieren [15]. Die in vivo-Daten im Bezug auf die
diabetische Nephropathie geben ein uneinheitliches Bild ab [80]: So fanden
die meisten Autoren eine verminderte Aktivität bzw. ein reduziertes Level von
MMP2 in Tierexperimenten; in humanen Studien war deren Niveau oft
erhöht. In unserer Studie fand sich bei allen drei diabetischen Tieren eine
erhöhte Aktivität von MMP2.
Die TSP2 Gruppe in unserem Experiment zeigte eine unveränderte Aktivität,
in der TSP1 Gruppe war jedoch eine verringerte Aktivität zu sehen; der
Unterschied erreichte jedoch nicht das Signifikanzniveau von p < 0,05.
In vitro-Daten zeigen, dass TSP1 die MMP2-Aktivität vermindern kann [5].

71
6.5 Infiltration durch Entzündungszellen
Einige aktuelle Studien untermauern die Bedeutung von Entzündungszellen
bei der Progression der diabetischen Nephropathie: Chow et al. konnten in
einem diabetischen Mausmodell zeigen, dass die Makrophageninfiltration mit
der Schwere des Nierenschadens zunimmt [17]. Darüber hinaus kommt es
auch zu einer Leukozyteninfiltration [6, 54].
In unserem Experiment erniedrigte TSP2 die Anzahl sowohl der
infiltrierenden Makrophagen als auch der CD3+-Lymphozyten; dabei hatten
CD4+- und CD8+-Zellen jeweils einen nahezu gleichen Anteil. Die TSP1
Gruppe zeigte keinen Unterschied zur Gruppe ohne Thrombospondin-Über-
expression. Dies könnte für einen von TGF-β-unabhängigen zusätzlichen
Mechanismus sprechen, der ein weiteres Voranschreiten der diabetischen
Nephropathie verhindert. Die Rolle von TGF-β im Bezug auf Entzündungsre-
aktionen zeigt darüber hinaus ein kontroverses und uneindeutiges Bild: So
sterben Mäuse mit einem TGF-β1-Knockout an einer multifokalen Entzün-
dung [75]. Andererseits induziert TGF-β1 in mesangialen Zellen das
monocyte chemoattractant protein 1 [82], das Makrophagen stimuliert.
Stattdessen könnte TSP2 seine antiinflammatorische Komponente über die
Regulation von Entzündungsmediatoren ausüben. In einem Rheumatoiden-
Arthritis-Modell demonstrierten Park et al., wie eine Überexpression von
TSP2 zu einer verringerten Produktion von Interferon-γ, Tumor-Nekrose-
Faktor-α und einer Reduktion von gewebeständigen T-Lymphozyten führte
[60]. Die antiinflammatorische Wirkung von TSP2 konnte durch unsere
Arbeitsgruppe in zwei Tiermodellen einer Glomerulonephritis bestätigt
werden [23, 25].
6.6 Schädigung der Podozyten
Neben den mesangialen Zellen rücken die Podozyten immer mehr ins
Interesse bei der Erforschung der Entwicklung einer diabetischen
Nephropathie. Der Verlust von Podozyten schon am Beginn der Erkrankung
wurde bereits an Tiermodellen und menschlichen Nierenproben beschrieben
[76]. Auch die diabetischen Tiere dieser Studie zeigten eine erniedrigte
Podozytenanzahl pro Glomerulus. Die TSP2 Überexpression führte zu einem

72
geringeren Verlust an Podozyten. Der Mechanismus, wie TSP2 auf die
Podozyten wirken könnte, ist noch nicht vollständig geklärt. Es könnte sich
wiederum um einen TGF-β-abhängigen Signalweg handeln: So induzierte die
Behandlung mit einem hohen Level an TGF-β1 in vitro die Apoptose von Po-
dozyten [69]. Unsere Arbeitsgruppe stellte in einem TSP1-Knockout Modell
einer diabetischen Nephropathie fest, dass der Schädigungsgrad der Podo-
zyten vermindert war [24]. Anderseits könnten Podozyten sogar eine Quelle
für sezernierte Thrombospondine sein: So fanden Han et al. bei mit Glucose
stimulierten Podozyten eine erhöhte Expression von TSP1 [33].
6.7 Verlust der Kapillardichte
Die Gefäßveränderungen, die bei der diabetischen Nephropathie entstehen,
sind vor allem qualitativer Natur, wie eine Arterio- und Arteriolosklerose in
den Nierengefäßen. Dennoch untersuchten wir die Nierenbiopsien in dieser
Studie auf quantitative Unterschiede in der Kapillardichte. TSP1 und vor
allem TSP2 gelten nämlich als potente Angiogeneseinhibitoren [46]. In unse-
rer Studie konnten wir einen leichten Verlust der kapillaren Dichte in allen
diabetischen Tieren feststellen. Die Überexpression mit TSP1 bzw. 2 hatte
darauf jedoch keinen Einfluss. In einem Glomerulonephritis-Modell mit TSP2-
Knockout-Mäusen konnte unsere Arbeitsgruppe den angiogenese-
inhibitorischen Effekt von endogenem TSP2 nachweisen [23]; bei diesem
Modell war jedoch der Kapillarverlust insgesamt bei den kranken Tieren
wesentlich ausgeprägter.
6.8 Beeinflussung der Nierenfunktion
Die bisher diskutierten Unterschiede zwischen den Gruppen hatten keinen
Einfluss auf die untersuchten Nierenfunktionsparameter: Serumharnstoff,
Proteinurie und Albuminurie war bei allen diabetischen Gruppen erhöht. Die
paradox anmutende Erhöhung der Kreatinin-Clearance in den diabetischen
Gruppen, die ja für eine verbesserte Nierenleistung spräche, beruht wohl auf
einer Hyperfiltration, die zu Beginn einer diabetischen Nephropathie auftritt.
Es fanden sich bei keiner der 4 Nierenfunktionsparameter Unterschiede
innerhalb der Therapiegruppen. Ein längerer Beobachtungszeitraum, in dem

73
die diabetischen Veränderungen weiter vorangeschritten wären, hätte
Unterschiede eventuell besser zum Vorschein gebracht: In einer ähnlichen
Studie unserer Arbeitsgruppe an TSP1-Knockout Mäusen mit STZ-
induzierter diabetischer Nephropathie zeigten die Tiere nach 9,5 Wochen
eine diesem Experiment vergleichbare Proteinurie [24]. Nach 20 Wochen
verdreifachte sich diese und in der TSP1 Gruppe war erstmals im Verlauf ein
signifikanter Unterschied zur Wildtyp Gruppe zu sehen.

74
6.9 Zusammenfassung, Kritische Beurteilung und Ausblick
Zusammenfassend konnten wir in dieser Studie erstmalig in vivo nach dem
Ursache-Wirkungs-Prinzip zeigen, dass eine Überexpression von TSP1 die
Aktivität von TGF-β bei der diabetischen Nephropathie steigert und TSP2
ebendiese Aktivitätssteigerung verhindert. Weiterhin führte die TSP2
Überexpression zu einer verringerten Deposition von Kollagen IV, einer ab-
gemilderten Infiltration der Niere durch Entzündungszellen und zu einem
Erhalt der Podozytenanzahl.
Nichtsdestotrotz müssen die Einschränkungen dieser Studie diskutiert
werden. Ein zentraler Aspekt der Arbeit beinhaltet die Aktivierung von TGF-β
im Rahmen der diabetischen Nephropathie. Die Beteiligung anderer Faktoren
kann jedoch nicht ausgeschlossen werden. Und nicht immer kann zwischen
TGF-β vermittelten Effekten und direkt durch die Thrombospondine
vermittelten Effekten differenziert werden. Weiterhin müssen auch potentielle
Interaktionen des Insulins und toxische Nebenwirkungen von Streptozotocin
berücksichtigt werden. Ein Einfluss der Thrombospondine auf den
Blutzuckerspiegel selbst erscheint unwahrscheinlich, da es keinen
signifikanten Unterschied in den Serumglukoseverläufen der Gruppen gab.
Bei einer Übertragung der Ergebnisse auf die humane diabetische
Nephropathie muss man berücksichtigen, dass wir nach 14 Wochen unter
Hyperglykämie nur milde histopathologische Veränderungen beobachteten.
Beim Menschen tritt als erste histologische Veränderung nach 1-2 Jahren
eine Verdickung der GBM auf [28]; die Erkrankung kann dann über mehrere
Dekaden bis zu einer Niereninsuffizienz fortschreiten. Unser Modell spiegelte
also den Beginn einer progredienten diabetischen Nephropathie wider.
Trotz dieser Einschränkungen ist das Eingreifen in die Thrombospondin-
TGF-β-Achse, wie in unserer Studie durch die Überexpression von TSP2,
eine vielversprechende Option bei der Prävention und Therapie der diabeti-
schen Nephropathie. Weitere Studien z.B. über einen längeren Zeitraum
könnten weitere Aufschlüsse über die Wirkmechanismen und potentielle
Nebenwirkungen von Thrombospondin 2 hervorbringen.

75
7 Abbildungsverzeichnis
Abb. 3-1 Molekularstruktur von TGF-β .............................................................. 17
Abb. 3-2 Molekularstruktur von TSP1 und TSP2 .............................................. 21
Abb. 3-3 Interaktion von TSP1 und 2 mit TGF-β ............................................... 22
Abb. 4-1 Schema über Gruppenverteilung und Studiendesign ......................... 26
Abb. 5-1 Verlauf des Blutzuckerspiegels .......................................................... 52
Abb. 5-2 Überlebenszeitanalyse der Versuchsgruppen .................................... 53
Abb. 5-3 Nierengewicht in Relation zum Tiergewicht ........................................ 54
Abb. 5-4 Aktivitätsniveau von TGF-β ................................................................ 55
Abb. 5-5 Expression von TGF-β1...................................................................... 55
Abb. 5-6 Histologische Veränderungen der diabetischen Nephropathie ........... 56
Abb. 5-7 Glomerulosklerose.............................................................................. 57
Abb. 5-8 Akkumulation von Kollagen I im Tubulointerstitium ............................ 57
Abb. 5-9 Akkumulation der Matrixmoleküle Kollagen IV und Fibronektin .......... 58
Abb. 5-10 Aktivität der Metalloproteinase 2 ...................................................... 59
Abb. 5-11 Podozytendichte im Glomerulus ....................................................... 60
Abb. 5-12 Infiltration von Makrophagen und T-Zellen ....................................... 61
Abb. 5-13 Differenzierung der T-Zellen in CD4+ und CD8+ Zellen .................... 62
Abb. 5-14 Rarefizierung der Kapillargefäße ...................................................... 63
Abb. 5-15 Nierenfunktionsparameter am Endzeitpunkt ..................................... 64
Abb. 5-16 Protein- und Albuminurie .................................................................. 64

76
8 Literaturverzeichnis
1. Adler, S., Diabetic nephropathy: Linking histology, cell biology, and genetics. Kidney Int, 2004. 66(5): p. 2095-106.
2. Agah, A., T.R. Kyriakides, J. Lawler, and P. Bornstein, The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am J Pathol, 2002. 161(3): p. 831-9.
3. Annes, J.P., J.S. Munger, and D.B. Rifkin, Making sense of latent TGFbeta activation. J Cell Sci, 2003. 116(Pt 2): p. 217-24.
4. Baker, L.H., E.K. Rowinsky, D. Mendelson, R.A. Humerickhouse, R.A. Knight, J. Qian, R.A. Carr, G.B. Gordon, and G.D. Demetri, Randomized, phase II study of the thrombospondin-1-mimetic angiogenesis inhibitor ABT-510 in patients with advanced soft tissue sarcoma. J Clin Oncol, 2008. 26(34): p. 5583-8.
5. Bein, K. and M. Simons, Thrombospondin type 1 repeats interact with matrix metalloproteinase 2. Regulation of metalloproteinase activity. J Biol Chem, 2000. 275(41): p. 32167-73.
6. Bending, J.J., A. Lobo-Yeo, D. Vergani, and G.C. Viberti, Proteinuria and activated T-lymphocytes in diabetic nephropathy. Diabetes, 1988. 37(5): p. 507-11.
7. Blobe, G.C., W.P. Schiemann, and H.F. Lodish, Role of transforming growth factor beta in human disease. N Engl J Med, 2000. 342(18): p. 1350-8.
8. Bornstein, P., Diversity of function is inherent in matricellular proteins: an appraisal of thrombospondin 1. J Cell Biol, 1995. 130(3): p. 503-6.
9. Bornstein, P., L.C. Armstrong, K.D. Hankenson, T.R. Kyriakides, and Z. Yang, Thrombospondin 2, a matricellular protein with diverse functions. Matrix Biol, 2000. 19(7): p. 557-68.
10. Bornstein, P., Thrombospondins as matricellular modulators of cell function. J Clin Invest, 2001. 107(8): p. 929-34.
11. Bornstein, P., A. Agah, and T.R. Kyriakides, The role of thrombospondins 1 and 2 in the regulation of cell-matrix interactions, collagen fibril formation, and the response to injury. Int J Biochem Cell Biol, 2004. 36(6): p. 1115-25.
12. Breyer, M.D., E. Bottinger, F.C. Brosius, 3rd, T.M. Coffman, R.C. Harris, C.W. Heilig, and K. Sharma, Mouse models of diabetic nephropathy. J Am Soc Nephrol, 2005. 16(1): p. 27-45.
13. Brownlee, M., Biochemistry and molecular cell biology of diabetic complications. Nature, 2001. 414(6865): p. 813-20.
14. Carlson, C.B., J. Lawler, and D.F. Mosher, Structures of thrombospondins. Cell Mol Life Sci, 2008. 65(5): p. 672-86.
15. Cheng, S., A.S. Pollock, R. Mahimkar, J.L. Olson, and D.H. Lovett, Matrix metalloproteinase 2 and basement membrane integrity: a

77
unifying mechanism for progressive renal injury. FASEB J, 2006. 20(11): p. 1898-900.
16. Chiarelli, F., S. Gaspari, and M.L. Marcovecchio, Role of growth factors in diabetic kidney disease. Horm Metab Res, 2009. 41(8): p. 585-93.
17. Chow, F., E. Ozols, D.J. Nikolic-Paterson, R.C. Atkins, and G.H. Tesch, Macrophages in mouse type 2 diabetic nephropathy: correlation with diabetic state and progressive renal injury. Kidney Int, 2004. 65(1): p. 116-28.
18. Chow, F.Y., D.J. Nikolic-Paterson, R.C. Atkins, and G.H. Tesch, Macrophages in streptozotocin-induced diabetic nephropathy: potential role in renal fibrosis. Nephrol Dial Transplant, 2004. 19(12): p. 2987-96.
19. Conway, B.R. and A.P. Maxwell, Genetics of diabetic nephropathy: are there clues to the understanding of common kidney diseases? Nephron Clin Pract, 2009. 112(4): p. c213-21.
20. Crawford, S.E., V. Stellmach, J.E. Murphy-Ullrich, S.M. Ribeiro, J. Lawler, R.O. Hynes, G.P. Boivin, and N. Bouck, Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell, 1998. 93(7): p. 1159-70.
21. Daniel, C., Y. Takabatake, M. Mizui, Y. Isaka, H. Kawashi, H. Rupprecht, E. Imai, and C. Hugo, Antisense oligonucleotides against thrombospondin-1 inhibit activation of tgf-beta in fibrotic renal disease in the rat in vivo. Am J Pathol, 2003. 163(3): p. 1185-92.
22. Daniel, C., J. Wiede, H.C. Krutzsch, S.M. Ribeiro, D.D. Roberts, J.E. Murphy-Ullrich, and C. Hugo, Thrombospondin-1 is a major activator of TGF-beta in fibrotic renal disease in the rat in vivo. Kidney Int, 2004. 65(2): p. 459-68.
23. Daniel, C., K. Amann, B. Hohenstein, P. Bornstein, and C. Hugo, Thrombospondin 2 functions as an endogenous regulator of angiogenesis and inflammation in experimental glomerulonephritis in mice. J Am Soc Nephrol, 2007. 18(3): p. 788-98.
24. Daniel, C., K. Schaub, K. Amann, J. Lawler, and C. Hugo, Thrombospondin-1 is an endogenous activator of TGF-beta in experimental diabetic nephropathy in vivo. Diabetes, 2007. 56(12): p. 2982-9.
25. Daniel, C., A. Wagner, B. Hohenstein, and C. Hugo, Thrombospondin-2 therapy ameliorates experimental glomerulonephritis via inhibition of cell proliferation, inflammation, and TGF-beta activation. Am J Physiol Renal Physiol, 2009. 297(5): p. F1299-309.
26. Dronavalli, S., I. Duka, and G.L. Bakris, The pathogenesis of diabetic nephropathy. Nat Clin Pract Endocrinol Metab, 2008. 4(8): p. 444-52.
27. Esemuede, N., T. Lee, D. Pierre-Paul, B.E. Sumpio, and V. Gahtan, The role of thrombospondin-1 in human disease. J Surg Res, 2004. 122(1): p. 135-42.
28. Fioretto, P. and M. Mauer, Histopathology of diabetic nephropathy. Semin Nephrol, 2007. 27(2): p. 195-207.

78
29. Galkina, E. and K. Ley, Leukocyte recruitment and vascular injury in
diabetic nephropathy. J Am Soc Nephrol, 2006. 17(2): p. 368-77.
30. Gilbert, R.E., A. Cox, L.L. Wu, T.J. Allen, U.L. Hulthen, G. Jerums, and M.E. Cooper, Expression of transforming growth factor-beta1 and type IV collagen in the renal tubulointerstitium in experimental diabetes: effects of ACE inhibition. Diabetes, 1998. 47(3): p. 414-22.
31. Giunti, S., D. Barit, and M.E. Cooper, Mechanisms of diabetic nephropathy: role of hypertension. Hypertension, 2006. 48(4): p. 519-26.
32. Han, D.C., B.B. Hoffman, S.W. Hong, J. Guo, and F.N. Ziyadeh, Therapy with antisense TGF-beta1 oligodeoxynucleotides reduces kidney weight and matrix mRNAs in diabetic mice. Am J Physiol Renal Physiol, 2000. 278(4): p. F628-34.
33. Han, S.H., S. Yang, D.S. Jung, J.J. Li, J.J. Kim, S.J. Kwak, D.K. Kim, S.J. Moon, J.E. Lee, D.S. Han, and S.W. Kang, Gene expression patterns in glucose-stimulated podocytes. Biochem Biophys Res Commun, 2008. 370(3): p. 514-8.
34. Haslbeck , L., Neundörfer et al., Diabetische Neuropathie. Praxis-Leitlinien der Deutschen Diabetes-Gesellschaft (DDG). Diabetologie und Stoffwechsel 2008, 2008(3 Supp. 2): p. 134-140.
35. Heilig, C.W., L.A. Concepcion, B.L. Riser, S.O. Freytag, M. Zhu, and P. Cortes, Overexpression of glucose transporters in rat mesangial cells cultured in a normal glucose milieu mimics the diabetic phenotype. J Clin Invest, 1995. 96(4): p. 1802-14.
36. Hill, C., A. Flyvbjerg, H. Gronbaek, J. Petrik, D.J. Hill, C.R. Thomas, M.C. Sheppard, and A. Logan, The renal expression of transforming growth factor-beta isoforms and their receptors in acute and chronic experimental diabetes in rats. Endocrinology, 2000. 141(3): p. 1196-208.
37. Hiscott, P., L. Paraoan, A. Choudhary, J.L. Ordonez, A. Al-Khaier, and D.J. Armstrong, Thrombospondin 1, thrombospondin 2 and the eye. Prog Retin Eye Res, 2006. 25(1): p. 1-18.
38. Hochegger, K., S. Knight, C. Hugo, G. Mayer, J. Lawler, T.N. Mayadas, and A.R. Rosenkranz, Role of thrombospondin-1 in the autologous phase of an accelerated model of anti-glomerular basement membrane glomerulonephritis. Nephron Exp Nephrol, 2004. 96(2): p. e31-8.
39. Hohenstein, B., C. Daniel, B. Hausknecht, K. Boehmer, R. Riess, K.U. Amann, and C.P. Hugo, Correlation of enhanced thrombospondin-1 expression, TGF-beta signalling and proteinuria in human type-2 diabetic nephropathy. Nephrol Dial Transplant, 2008. 23(12): p. 3880-7.
40. Hugo, C., The thrombospondin 1-TGF-beta axis in fibrotic renal disease. Nephrol Dial Transplant, 2003. 18(7): p. 1241-5.
41. Hugo, C. and C. Daniel, Thrombospondin in renal disease. Nephron Exp Nephrol, 2009. 111(3): p. e61-6.

79
42. Isono, M., S. Chen, S.W. Hong, M.C. Iglesias-de la Cruz, and F.N. Ziyadeh, Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-beta-induced fibronectin in mesangial cells. Biochem Biophys Res Commun, 2002. 296(5): p. 1356-65.
43. Kagami, S., W.A. Border, D.E. Miller, and N.A. Noble, Angiotensin II stimulates extracellular matrix protein synthesis through induction of transforming growth factor-beta expression in rat glomerular mesangial cells. J Clin Invest, 1994. 93(6): p. 2431-7.
44. Kopp, J.B., V.M. Factor, M. Mozes, P. Nagy, N. Sanderson, E.P. Bottinger, P.E. Klotman, and S.S. Thorgeirsson, Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest, 1996. 74(6): p. 991-1003.
45. Kyriakides, T.R., Y.H. Zhu, L.T. Smith, S.D. Bain, Z. Yang, M.T. Lin, K.G. Danielson, R.V. Iozzo, M. LaMarca, C.E. McKinney, E.I. Ginns, and P. Bornstein, Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol, 1998. 140(2): p. 419-30.
46. Kyriakides, T.R. and S. Maclauchlan, The role of thrombospondins in wound healing, ischemia, and the foreign body reaction. J Cell Commun Signal, 2009.
47. Lewis, E.J., L.G. Hunsicker, R.P. Bain, and R.D. Rohde, The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med, 1993. 329(20): p. 1456-62.
48. Lopes, G.S., C.C. Lemos, C.A. Mandarim-De-Lacerda, and R. Bregman, Effect of unilateral nephrectomy on renal function of diabetic rats. Histol Histopathol, 2004. 19(4): p. 1085-8.
49. Maeda, S., Genetics of diabetic nephropathy. Ther Adv Cardiovasc Dis, 2008. 2(5): p. 363-71.
50. Mason, R.M. and N.A. Wahab, Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol, 2003. 14(5): p. 1358-73.
51. Mishra, R., S.N. Emancipator, T. Kern, and M.S. Simonson, High glucose evokes an intrinsic proapoptotic signaling pathway in mesangial cells. Kidney Int, 2005. 67(1): p. 82-93.
52. Moczulski, D.K., H. Fojcik, A. Wielgorecki, W. Trautsolt, B. Gawlik, S. Kosiorz-Gorczynska, M. Oczko-Wojciechowska, M. Wiench, K. Strojek, E. Zukowska-Szczechowska, and W. Grzeszczak, Expression pattern of genes in peripheral blood mononuclear cells in diabetic nephropathy. Diabet Med, 2007. 24(3): p. 266-71.
53. Mogensen, C.E., C.K. Christensen, and E. Vittinghus, The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes, 1983. 32 Suppl 2: p. 64-78.
54. Moriya, R., J.C. Manivel, and M. Mauer, Juxtaglomerular apparatus T-cell infiltration affects glomerular structure in Type 1 diabetic patients. Diabetologia, 2004. 47(1): p. 82-8.

80
55. Murphy-Ullrich, J.E. and M. Poczatek, Activation of latent TGF-beta by
thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev, 2000. 11(1-2): p. 59-69.
56. Nakamura, T., M. Fukui, I. Ebihara, S. Osada, I. Nagaoka, Y. Tomino, and H. Koide, mRNA expression of growth factors in glomeruli from diabetic rats. Diabetes, 1993. 42(3): p. 450-6.
57. Newman, D.J., M.B. Mattock, A.B. Dawnay, S. Kerry, A. McGuire, M. Yaqoob, G.A. Hitman, and C. Hawke, Systematic review on urine albumin testing for early detection of diabetic complications. Health Technol Assess, 2005. 9(30): p. iii-vi, xiii-163.
58. Nishikawa, T., D. Edelstein, X.L. Du, S. Yamagishi, T. Matsumura, Y. Kaneda, M.A. Yorek, D. Beebe, P.J. Oates, H.P. Hammes, I. Giardino, and M. Brownlee, Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature, 2000. 404(6779): p. 787-90.
59. Osterby, R., A. Hartmann, J.R. Nyengaard, and H.J. Bangstad, Development of renal structural lesions in type-1 diabetic patients with microalbuminuria. Observations by light microscopy in 8-year follow-up biopsies. Virchows Arch, 2002. 440(1): p. 94-101.
60. Park, Y.W., Y.M. Kang, J. Butterfield, M. Detmar, J.J. Goronzy, and C.M. Weyand, Thrombospondin 2 functions as an endogenous regulator of angiogenesis and inflammation in rheumatoid arthritis. Am J Pathol, 2004. 165(6): p. 2087-98.
61. Poczatek, M.H., C. Hugo, V. Darley-Usmar, and J.E. Murphy-Ullrich, Glucose stimulation of transforming growth factor-beta bioactivity in mesangial cells is mediated by thrombospondin-1. Am J Pathol, 2000. 157(4): p. 1353-63.
62. Pohlers, D., J. Brenmoehl, I. Loffler, C.K. Muller, C. Leipner, S. Schultze-Mosgau, A. Stallmach, R.W. Kinne, and G. Wolf, TGF-beta and fibrosis in different organs - molecular pathway imprints. Biochim Biophys Acta, 2009. 1792(8): p. 746-56.
63. Ritz, E. and S.R. Orth, Nephropathy in patients with type 2 diabetes mellitus. N Engl J Med, 1999. 341(15): p. 1127-33.
64. Ritz, E., I. Rychlik, F. Locatelli, and S. Halimi, End-stage renal failure in type 2 diabetes: A medical catastrophe of worldwide dimensions. Am J Kidney Dis, 1999. 34(5): p. 795-808.
65. Russell, T.A., Diabetic nephropathy in patients with type 1 diabetes mellitus. Nephrol Nurs J, 2006. 33(1): p. 15-28; quiz 29-30.
66. Ruster, C. and G. Wolf, Renin-angiotensin-aldosterone system and progression of renal disease. J Am Soc Nephrol, 2006. 17(11): p. 2985-91.
67. Schaefer, L., R.M. Schaefer, H. Ling, M. Teschner, and A. Heidland, Renal proteinases and kidney hypertrophy in experimental diabetes. Diabetologia, 1994. 37(6): p. 567-71.
68. Scherbaum and R. Eberhard, Prävention und Therapie der diabetischen Nephropathie. Deutsches Ärzteblatt, 2005. 3: p. 137-143.

81
69. Schiffer, M., P. Mundel, A.S. Shaw, and E.P. Bottinger, A novel role
for the adaptor molecule CD2-associated protein in transforming growth factor-beta-induced apoptosis. J Biol Chem, 2004. 279(35): p. 37004-12.
70. Schober-Hastenberg, H., Jahresbericht 2006/2007 QuaSi-Niere. http://www.bundesverband-niere.de/files/QuaSi-Niere-Bericht_2006-2007.pdf, 2007.
71. Schultz-Cherry, S., S. Ribeiro, L. Gentry, and J.E. Murphy-Ullrich, Thrombospondin binds and activates the small and large forms of latent transforming growth factor-beta in a chemically defined system. J Biol Chem, 1994. 269(43): p. 26775-82.
72. Schultz-Cherry, S., H. Chen, D.F. Mosher, T.M. Misenheimer, H.C. Krutzsch, D.D. Roberts, and J.E. Murphy-Ullrich, Regulation of transforming growth factor-beta activation by discrete sequences of thrombospondin 1. J Biol Chem, 1995. 270(13): p. 7304-10.
73. Sharma, K. and F.N. Ziyadeh, Renal hypertrophy is associated with upregulation of TGF-beta 1 gene expression in diabetic BB rat and NOD mouse. Am J Physiol, 1994. 267(6 Pt 2): p. F1094-01.
74. Sharma, K., Y. Jin, J. Guo, and F.N. Ziyadeh, Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes, 1996. 45(4): p. 522-30.
75. Shull, M.M., I. Ormsby, A.B. Kier, S. Pawlowski, R.J. Diebold, M. Yin, R. Allen, C. Sidman, G. Proetzel, D. Calvin, and et al., Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature, 1992. 359(6397): p. 693-9.
76. Siu, B., J. Saha, W.E. Smoyer, K.A. Sullivan, and F.C. Brosius, 3rd, Reduction in podocyte density as a pathologic feature in early diabetic nephropathy in rodents: prevention by lipoic acid treatment. BMC Nephrol, 2006. 7: p. 6.
77. Steckelings, U.M., F. Rompe, E. Kaschina, and T. Unger, The evolving story of the RAAS in hypertension, diabetes and CV disease: moving from macrovascular to microvascular targets. Fundam Clin Pharmacol, 2009. 23(6): p. 693-703.
78. Stenina, O.I., I. Krukovets, K. Wang, Z. Zhou, F. Forudi, M.S. Penn, E.J. Topol, and E.F. Plow, Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation, 2003. 107(25): p. 3209-15.
79. Tesch, G.H. and D.J. Nikolic-Paterson, Recent Insights into Experimental Mouse Models of Diabetic Nephropathy. Nephron Experimental Nephrology, 2006. 104(2): p. e57-e62.
80. Thrailkill, K.M., R. Clay Bunn, and J.L. Fowlkes, Matrix metalloproteinases: their potential role in the pathogenesis of diabetic nephropathy. Endocrine, 2009. 35(1): p. 1-10.
81. Wahab, N.A., L. Schaefer, B.S. Weston, O. Yiannikouris, A. Wright, A. Babelova, R. Schaefer, and R.M. Mason, Glomerular expression of

82
thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia, 2005. 48(12): p. 2650-60.
82. Wang, S.N., J. LaPage, and R. Hirschberg, Role of glomerular ultrafiltration of growth factors in progressive interstitial fibrosis in diabetic nephropathy. Kidney Int, 2000. 57(3): p. 1002-14.
83. White, K.E. and R.W. Bilous, Type 2 diabetic patients with nephropathy show structural-functional relationships that are similar to type 1 disease. J Am Soc Nephrol, 2000. 11(9): p. 1667-73.
84. Wild, S., G. Roglic, A. Green, R. Sicree, and H. King, Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care, 2004. 27(5): p. 1047-53.
85. Wolf, G., Link between angiotensin II and TGF-beta in the kidney. Miner Electrolyte Metab, 1998. 24(2-3): p. 174-80.
86. Wolf, G., New insights into the pathophysiology of diabetic nephropathy: from haemodynamics to molecular pathology. Eur J Clin Invest, 2004. 34(12): p. 785-96.
87. Wolf, G. and F.N. Ziyadeh, Cellular and molecular mechanisms of proteinuria in diabetic nephropathy. Nephron Physiol, 2007. 106(2): p. p26-31.
88. Yang, C.W., M. Hattori, H. Vlassara, C.J. He, M.A. Carome, E. Yamato, S. Elliot, G.E. Striker, and L.J. Striker, Overexpression of transforming growth factor-beta 1 mRNA is associated with up-regulation of glomerular tenascin and laminin gene expression in nonobese diabetic mice. J Am Soc Nephrol, 1995. 5(8): p. 1610-7.
89. Yang, Y.L., L.Y. Chuang, J.Y. Guh, S.F. Liu, M.Y. Hung, T.N. Liao, and Y.L. Huang, Thrombospondin-1 mediates distal tubule hypertrophy induced by glycated albumin. Biochem J, 2004. 379(Pt 1): p. 89-97.
90. Yang, Z., D.K. Strickland, and P. Bornstein, Extracellular matrix metalloproteinase 2 levels are regulated by the low density lipoprotein-related scavenger receptor and thrombospondin 2. J Biol Chem, 2001. 276(11): p. 8403-8.
91. Yevdokimova, N., N.A. Wahab, and R.M. Mason, Thrombospondin-1 is the key activator of TGF-beta1 in human mesangial cells exposed to high glucose. J Am Soc Nephrol, 2001. 12(4): p. 703-12.
92. Young, G.D. and J.E. Murphy-Ullrich, The tryptophan-rich motifs of the thrombospondin type 1 repeats bind VLAL motifs in the latent transforming growth factor-beta complex. J Biol Chem, 2004. 279(46): p. 47633-42.
93. Zimmet, P., K.G. Alberti, and J. Shaw, Global and societal implications of the diabetes epidemic. Nature, 2001. 414(6865): p. 782-7.
94. Ziyadeh, F.N., B.B. Hoffman, D.C. Han, M.C. Iglesias-De La Cruz, S.W. Hong, M. Isono, S. Chen, T.A. McGowan, and K. Sharma, Long-term prevention of renal insufficiency, excess matrix gene expression,

83
and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci U S A, 2000. 97(14): p. 8015-20.
95. Ziyadeh, F.N., Mediators of diabetic renal disease: the case for tgf-Beta as the major mediator. J Am Soc Nephrol, 2004. 15 Suppl 1: p. S55-7.

84
9 Abkürzungsverzeichnis
Abb. Abbildung ABTS 2,2'-Azino-di-(3-ethylbenzthiazolin)-6-sulfonsäure ACE Angiotensin-Converting-Encyme Ak Antikörper ANP Atriales natriuretisches Peptid APS Ammoniumperoxodisulfat Aqua dest. Destilliertes Wasser BSA Rinderserumalbumin C Celsius CD Cluster of differentiation cDNA Komplemtäre DNA d Tag DAB 3,3'-Diaminobenzidine DAPI 4′,6-Diamidin-2-phenylindol DM Diabetes Mellitus DNA Desoxyribonukleinsäure EDTA Ethylendiamintetraacetat ELISA Enzyme Linked Immunosorbent Assay EPO Elektroporation ECM/EZM Extrazelluläre Matrix FCS Fetales Kälberserum GBM glomeruläre Basalmembran GFR glomeruläre Filtrationsrate GLUT Glukosetransporter h Stunde kDA Kilodalton (atomare Masse) kg Kilogramm KG Körpergewicht LABP Latent-TGF-β-bindendes Protein LAP Latency assoziiertes Protein LLC Large latent complex MC Methyl Carnoy mg Milligramm min Minute MMP Matrix Metalloproteinasen ms Millisekunde mRNA Messenger Ribonukleinsäure µg Mikrogramm µl Mikroliter NaCl Natriumchlorid ODP Ortho-phenylendiamin P-Smad Phospholiertes Smad 2/3 PAF Paraformaldehyd PAS Periodsäure-Schiff-Färbung PBS Phosphate buffered saline PCR Polymerase-Kettenreaktion RAS Renin-Angiotensin-System

85
rpm Rounds per minute RT Raumtemperatur SDS sodium dodecylsulfate sec Sekunde STZ Streptozotocin SWK Stoffwechselkäfig T Temperatur TBS Tris buffered saline Temed Tetramethylethylendiamin TGF-β Transforming Growth Factor beta Tris Tris(hydroxymethyl)-aminomethan TRS Target Retrieval Solution TSP 1 Thrombospondin 1 TSP 2 Thrombospondin 2 ü.N. über Nacht V Volt WT 1 Wilms Tumor 1 ZN Zink

86
10 Danksagung
Ich danke Herrn Prof. Dr. med. Christian Hugo, der mittlerweile die
nephrologische Abteilung des Universitätsklinikums Dresden leitet, für die
Überlassung des Themas.
Weiterhin bedanke ich mich bei Frau Prof. Dr. med. Kerstin Amann aus der
nephropathologischen Abteilung in Erlangen für die Übernahme des
Zweitgutachtens.
Mein besonderer Dank gilt Herrn PD Dr. rer. nat. Christoph Daniel für die
erstklassige Betreuung. Seine immerwährende Unterstützung und seine
fachlichen Anregungen waren mir eine sehr große Hilfe.
Weiterhin möchte ich mich bei den Mitarbeitern der ehemaligen
Arbeitsgruppe Hugo bedanken, allen voran Tina, Tanja, Andrea und Susann.
Sie standen mir mit großer Geduld beim experimentellen Teil und bei der
Auswertung der Arbeit zur Seite.
Ein großes Dankeschön geht an meine Laborkollegin Regina für die große
wissenschaftliche und moralische Unterstützung.
Ein herzliches „Vergelt’s Gott“ geht an meine Freunde Benjamin und Christof,
die zeitgleich mit mir in der nephrologischen Abteilung promovierten.
Im Rahmen des SFB 423 der Deutschen Forschungsgemeinschaft und durch
die Dr. Ernst & Anita Bauer Stiftung erhielt ich eine finanzielle Förderung.
Vielen Dank.
Mein besonderer Dank gilt meinen Eltern und meinem Bruder Andreas.


















