Elektrochemische Messtechniken zur globalen und …€¦ · A. Schulte , S. Belger, M ... Wien (A)...
167
Elektrochemische Messtechniken zur globalen und lokalen Korrosionsuntersuchung an Nickel-Titan-Formgedächtnislegierungen Dissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften der Fakultät für Chemie der Ruhr-Universität Bochum vorgelegt von Sascha Belger Bochum, November 2005
Transcript of Elektrochemische Messtechniken zur globalen und …€¦ · A. Schulte , S. Belger, M ... Wien (A)...

Elektrochemische Messtechniken
zur globalen und lokalen Korrosionsuntersuchung
an Nickel-Titan-Formgedächtnislegierungen
Dissertation
zur Erlangung des Grades eines
Doktors der Naturwissenschaften
der Fakultät für Chemie der
Ruhr-Universität Bochum
vorgelegt von
Sascha Belger
Bochum, November 2005

Diese Arbeit wurde in der Zeit von Juni 2001 bis November 2005 am Lehrstuhl für
analytische Chemie, AG Elektroanalytik & Sensorik unter der Leitung von
Prof. Dr. W. Schuhmann angefertigt
Eingereicht am 15.11.2005
Tag der mündlichen Prüfung 09.12.2005
Referent Prof. Dr. W. Schuhmann
Koreferent Prof. W. S. Sheldrick PhD
Prüfer Prof. Dr. Ch. Wöll

Veröffentlichungen
Teile dieser Arbeit sind Veröffentlicht in:
Publikationen
Mikrochimica Acta (eingereicht)
S. Belger, A. Schulte und W. Schuhmann
„Sequential injection stripping voltammetry for routine on-line nickel dissolution
studies on biomedical NiTi shape memory alloys”
Materials Science & Engineering A: Structural Materials:
Properties Microstructure and Processing, 378 (2004), S. 523-526
A. Schulte, S. Belger, M. Etienne und W. Schuhmann
„Imaging localised corrosion of NiTi shape memory alloys by means of alternating
current scanning electrochemical microscopy (AC-SECM)”
Materialwissenschaft und Werkstofftechnik, 35 (2004), S. 276-279
S. Belger, A. Schulte, C. Hessing, M. Pohl und W. Schuhmann
„Alternating current scanning electrochemical microscopy (AC-SECM) studies on the
surface of electrochemically polished NiTi shape memory alloys”
Shape Memory Materials and its Applications - Materials Science Forum,
394 (2001), S. 145-148
A. Schulte, S. Belger und W. Schuhmann
„Corrosion of NiTi shape-memory alloys: Visualization by means of potentiometric
"constant-distance" scanning electrochemical Microscopy”
Vorträge und Poster
European Syposium on Martensitic Transformation and Shape Memory,
Cirencester (UK) 2003
A. Schulte, S. Belger, M. Etienne und W. Schuhmann
Vortrag „Imaging localized corrosion of NiTi shape memory alloys by means of
alternating current scanning electrochemical microscopy (AC-SECM)”

Veröffentlichungen
6. Tagung AK Elektrochemische Analysenmethoden GDCh-GÖCh – ELACH,
Wien (A) 2003
A. Schulte, S. Belger, M. Etienne, B. Ballesteros Katemann und W. Schuhmann
Vortrag „Elektrochemische Rastermikroskopie im Wechselstrom Modus –
Untersuchung lokaler Korrosionsphänomene“
6. Tagung AK Elektrochemische Analysenmethoden GDCh-GÖCh – ELACH,
Wien (A) 2003
S. Belger, T. Erichsen, A. Schulte, J. Choi, M. Epple und W. Schuhmann
Poster „Sequential-Injection stripping analysis of Ni-release from corroding NiTi
shape memory alloys“
ISE, Düsseldorf 2002
S. Belger, A. Schulte, B. Ballesteros Katemann und W. Schuhmann
Poster „Visualisation of loclized corrosion of NiTi shape-memory alloys by means of
AC-SECM”
European Conference on Shape Memory and Superelastic Technologies
(SMST), Kunming (China) 2001
A. Schulte, S. Belger und W. Schuhmann
Vortrag „Corrosion of NiTi Shape Memory Alloys: Visualisation by Means of
Potentiometric “Constant-Distance” Scanning Electrochemical Microscopy”

Danksagung
Mein herzlicher Dank gilt Allen, die zum Gelingen dieser Arbeit beigetragen haben.
Besonderer Dank gilt:
Herrn Prof. Dr. Schuhmann, für die interessante Aufgabenstellung, die
Diskussionsbereitschaft und seine guten Ratschläge.
Albert Schulte, der immer zu ausgiebigen Gesprächen bereit war und mir hilfsbereit
zur Seite Stand.
Thomas Erichsen, der mir in apparativen sowie Fragen zur Softwareprogrammierung
bereitwillig Unterstützung zukommen ließ.
Christian Hessing und Jan Frenzel, die mir in materialwissenschaftlichen Fragen gern
Auskunft erteilten.
Allen Mitarbeitern der AG Elektroanalytik & Sensorik für die ständige Diskussions-
bereitschaft, das sehr gute Arbeitsklima, der Durchsicht dieser Arbeit und einer
unvergesslichen Zeit an der Ruhr-Universität.
Den Werkstätten der Fakultät für Chemie, für die hervorragende und schnelle
Umsetzung neuer Geräteteile.
Meinen Eltern für die jahrelange Unterstützung, und ganz besonders meiner Frau
und meinem Sohn, die so manches Mal auf meine Anwesenheit verzichten mussten,
aber mit trotzdem immer zur Seite standen. Mit dem Rückhalt den ich durch euch
genießen durfte, wurde diese Arbeit erst möglich.

Inhaltsverzeichnis I
Inhaltsverzeichnis
1 Einleitung.............................................................................................................1
2 Stand der Forschung..........................................................................................3
2.1 Formgedächtnislegierungen (FGLs) ..............................................................3
2.1.1 Der Formgedächtniseffekt und die martensitische Umwandlung ...........3
2.1.2 Korrosion und Biokompatibilität der NiTi-FGLs ......................................4
2.2 Elektrochemische Rastermikroskopie (SECM)..............................................7
2.2.1 Aufbau eines SECM ...............................................................................9
2.2.2 Funktionsweise eines SECM................................................................12
2.2.2.1 Annäherung der Ultramikroelektrode an nicht leitende Oberflächen 13
2.2.2.2 Annäherung der Ultramikroelektrode an leitende Oberflächen......... 15
2.2.2.3 Oberflächenabbildung mit dem SECM.............................................. 16
2.2.2.3.1 Der „constant-height“ Modus......................................................17
2.2.2.3.2 Der „constant-distance” Modus..................................................19
2.2.3 Funktionsweise der optischen Höhenkontrolle .....................................20
2.3 Ionenselektive Rastermikroskopie ...............................................................23
2.4 SECM im Wechselstrom Modus (AC-SECM)..............................................27
2.5 Sequentielle Injektionsanalyse (SIA) ...........................................................31
2.5.1 Aufbau und Funktionsweise eines Fließ-Injektions-Analysen-Systems31
2.5.2 Aufbau und Funktionsweise eines SIA.................................................32
2.5.3 Der Online-General-Analyser (OLGA)..................................................33
2.6 Voltammetrische Strippinganalyse ..............................................................35
2.6.1 Anodische Strippinganalyse (ASV).......................................................35
2.6.2 Adsorptive Strippinganalyse (AdSV) ....................................................36
2.6.3 Elektrodentypen für die voltammetrische Strippinganalyse..................37
2.7 Voltammetrische Strippinganalyse im SIA...................................................38
3 Problemstellung................................................................................................40
4 Ergebnisse eigener Arbeit ...............................................................................42
4.1 Probenvorbereitung .....................................................................................42
4.1.1 Probengeometrie ..................................................................................42
4.1.2 Weiterführende Probenbearbeitung .....................................................42
4.1.2.1 Polieren der NiTi-Proben .................................................................. 43
4.1.2.2 Proben für die SECM........................................................................ 44

Inhaltsverzeichnis II
4.1.3 Probencharakterisierung mittels XPS-Analyse.....................................44
4.2 Sequentielle Injektions-Strippinganalyse (SISA) .........................................48
4.2.1 Vorversuche im stationären System.....................................................48
4.2.1.1 Herstellung und Test von Quecksilberfilmelektroden (MFE) ............ 49
4.2.1.1.1 MFE auf Basis einer Iridium-Scheibenelektrode........................49
4.2.1.1.2 MFE auf Basis einer Carbonfasermikroelektrode (CFME).........51
4.2.2 Adaption der AdSV an das OLGA-System...........................................52
4.2.2.1 Integration eines Potentiostaten ....................................................... 54
4.2.2.2 Softwareentwicklung ......................................................................... 55
4.2.2.2.1 DPV-Software ............................................................................55
4.2.2.2.2 OLGA-Software..........................................................................55
4.2.2.3 Wahl der Messzelle........................................................................... 61
4.2.2.4 Erster Test des Systems................................................................... 62
4.2.2.5 Adaption eines PalmSens-Potentiostaten an das SISA-System ...... 63
4.2.2.5.1 Entwicklung einer Software für den PalmSens-Potentiostaten..63
4.2.2.5.2 Programmierung eines PlugIn-Systems ....................................66
4.2.2.6 Zweiter Test des Systems................................................................. 68
4.2.2.6.1 Überarbeitung des Designs der Durchflussmesszelle ...............69
4.2.2.7 Konzeption alternativer Elektrodentypen .......................................... 71
4.2.2.7.1 Quecksilberfilmelektroden auf Basis einer Gold- ...........................
Scheibenelektrode .....................................................................71
4.2.2.7.2 Wismutfilmelektroden (BiFE)......................................................72
4.2.2.8 Evaluierung der BiFE im SISA-System............................................. 78
4.2.2.8.1 Erweiterung des SISA-Systems.................................................78
4.2.2.8.2 Test der BiFE mittels AdSV im SISA-System ............................79
4.2.2.8.3 Die miniaturisierte Rührmesszelle..............................................81
4.2.2.8.4 Die Walljet-Durchflussmesszelle (neues Design) ......................82
4.2.2.9 Optimierung der Wismutfilmstruktur.................................................. 84
4.2.2.10 Abschließende Evaluierung des SISA-Systems ........................... 87
4.2.3 Online Bestimmung des Nickelaustritts aus korrodierendem NiTi .......90
4.3 Hochaufgelöste lokale Korrosionsuntersuchung mittels SECM ..................95
4.3.1 Potentiometrische SECM mit Mikro-pH-Sonden im „constant-distance“
Modus...................................................................................................95
4.3.2 AC-SECM.............................................................................................97

Inhaltsverzeichnis III
4.3.2.1 Entwicklung einer Tiltwinkel-Kontrolle für das SECM ....................... 98
4.3.2.2 Hochaufgelöste AC-SECM an NiTi-FGLs mit Pt-Ultramikroelektroden
im „constant-height“ Modus ............................................................. 102
4.3.2.2.1 Abbildung von Mikroeinschlüssen in einer NiTi-Oberfläche.....107
4.3.2.2.2 Abbildung einer Mikrostrukturierten NiTi-Oberfläche ...............108
4.3.2.2.3 Abbildung einer geätzten NiTi-Oberfläche ...............................111
4.3.2.3 Hochaufgelöste AC-SECM im „constant-distance“ Modus ............. 114
4.3.2.3.1 Evaluierung von Pt-Scheibenelektroden im „constant-distance“
AC-SECM.................................................................................114
4.3.2.3.2 Hochaufgelöste AC-SECM im „constant-distance“ Modus an
geätzten Nitinol-Oberflächen....................................................117
5 Zusammenfassung und Ausblick..................................................................120
6 Experimenteller Teil........................................................................................123
6.1 Chemikalien, Geräte und Materialien ........................................................123
6.1.1 Chemikalienliste .................................................................................123
6.1.2 Geräteliste ..........................................................................................124
6.1.3 Materialien ..........................................................................................125
6.1.4 Aufbau des SISA-Systems .................................................................126
6.1.5 Aufbau des SECM..............................................................................128
6.2 Elektrodenherstellung ................................................................................129
6.2.1 Herstellung von Glaskohlenstoff-, Platinmikro- und..................................
Iridiummakroelektroden ......................................................................129
6.2.2 Herstellung von schwingungsfähigen Carbonfaserelektroden ...........131
6.2.3 Herstellung von schwingungsfähigen Platinnanoelektroden ..............132
6.2.4 Herstellung der miniaturisierten Referenzelektrode ...........................133
6.3 Beschichtung der Elektroden.....................................................................133
6.3.1 Quecksilberfilmelektroden ..................................................................133
6.3.2 Wismutfilmelektroden .........................................................................134
6.3.3 Mikro-pH-Elektroden auf Basis IrO2-beschichteter CFMEs................134
6.4 Aktivitätstest der pH-Sonden .....................................................................135
6.5 Probenvorbereitung ...................................................................................136
6.5.1 Material...............................................................................................136
6.5.2 Sägen und Einbetten ..........................................................................136
6.5.3 Polieren ..............................................................................................137

Inhaltsverzeichnis IV
6.5.3.1 Mechanisches Polieren................................................................... 137
6.5.3.2 Elektrolytisches Polieren................................................................. 138
6.6 Beschreibung der Experimente .................................................................139
6.6.1 Typisches SISA-Experiment...............................................................139
6.6.2 Typisches SECM-Experiment.............................................................139
7 Anhang.............................................................................................................142
7.1 Auszug aus dem Code der SISA-Software................................................142
8 Literaturverzeichnis........................................................................................146

Einleitung 1
1 Einleitung
Für industrielle Zwecke und medizinische Anwendungen wird stets nach neuen,
innovativen Materialien gesucht, um die physikalischen sowie chemischen
Eigenschaften von mechanischen Bauteilen oder medizinischen Geräten sowie
Implantaten zu verbessern. Dies soll eine längere Haltbarkeit, aufgabenorientierte,
verbesserte Eigenschaften oder auch gesteigerte Biokompatibilität ermöglichen.
Zudem wird versucht neue Funktionalitäten zu entwickeln, die Prozesse vereinfachen
können oder neue ermöglichen.
Eine bemerkenswerte Kategorie unter den metallischen Werkstoffen bilden die Form-
Gedächtnis Legierungen (FGLs). Sie können durch Erwärmen oder Abkühlen über
bzw. unter ihre Umwandlungstemperatur die Form verändern und bieten somit ein
breit gefächertes Spektrum an Anwendungsmöglichkeiten, wie z.B. als Aktoren,
Haltevorrichtungen, etc.
Seit der Entdeckung der Form-Gedächtnis Eigenschaft der nahezu gleichatomigen
Legierung aus Nickel und Titan, auch Nitinol genannt, im Jahre 1963, steht ein
vergleichsweise kostengünstiges Material mit Form-Gedächtnis-Effekt zur Verfügung.
Zudem lässt es, je nach Verhältnis der beiden Metalle, eine Umwandlung bei
Raumtemperatur zu. Ferner bringt das Material, wie andere FGLs auch, aufgrund
seiner Superelastizität hervorragende Dämpfungseigenschaften mit sich, die es für
industrielle Anwendungen noch attraktiver machen.
Ein wichtiger Aspekt bei metallischen Werkstoffen ist das Korrosionsverhalten. Es
bestimmt die möglichen Einsatzgebiete und trägt zur Haltbarkeit und Biokompatibilität
bei. Somit ist es wichtig, neue Materialien auf ihr Korrosionsverhalten hin zu prüfen
und Einflüsse auf die Korrosion resultierend aus verschiedenen Herstellungsver-
fahren, Bearbeitungsmethoden und Beschichtungen zu erfassen.
Die Korrosion von Metallen ist ein chemischer/elektrochemischer Prozess, bei dem
die Oberfläche des Materials an Schwachstellen angegriffen und teilweise aufgelöst
wird. Das aufgelöste Metall geht ionisch in Lösung und hinterlässt im Falle der
Lochfraßkorrosion eine sich ausbreitende Vertiefung. Dies hat mehrere lokale und
globale, also am Korrosionsort und z.B. in der umgebenden Lösung, messbare
Effekte zur Folge. Zum einen verändert sich aufgrund der austretenden Ionen die
lokale Leitfähigkeit der Lösung und evtl. durch z.B. Hydroxidbildung (z.B. Me + 2H2O
→ Me(OH)2 + 2H+) auch der pH-Wert der Lösung. Zum anderen verändert sich

Einleitung 2
aufgrund des Materialabtrags die Topographie des Materials. Außerdem können die
herausgelösten Metallionen selbst nachgewiesen werden. Daraus ergeben sich
mehrere Möglichkeiten der Korrosionsuntersuchung mittels lokaler und globaler
Analysenmethoden.
Die lokale Betrachtung von Oberflächen ist mit optischen sowie verschiedenen
Rastermikroskopiearten möglich. Doch erhält man bei den meisten dieser Verfahren
lediglich ein Abbild der Oberfläche ohne jegliche weiterführenden Informationen.
Lediglich die konfokale Lasermikroskopie (CLSM) und die Raster-Kraft-Mikroskopie
(AFM) geben Aufschluss über die tatsächliche Topographie einer Oberfläche.
Dennoch wird durch diese Techniken eine für die Korrosion ganz wesentliche
Information, die elektrochemische Aktivität der Oberfläche, nicht erfasst.
Ein globaler Effekt der Korrosion ist die Erhöhung der Konzentration von Metallionen
im Korrosionsmedium. Um diese zu bestimmen, stehen der Analytik verschiedene
Methoden zur Metallspurenanalyse zur Verfügung. Die Kosten jedoch sind häufig
sehr hoch und die Verfahren apparativ sehr aufwändig wie z.B. eine massen-
spektrometrische Bestimmung von Metallionen mit induktiv gekoppeltem Plasma
(ICP-MS). Eine kostengünstige Analyse mittels Strippingvoltammetrie (SV) wäre eine
Alternative, jedoch ist die Korrosion ein längerer Prozess, so dass Langzeit-
experimente nötig wären.
In der vorliegenden Arbeit soll ein neuer Weg eröffnet werden, Metalloberflächen
mittels elektrochemischer Rastermikroskopie im Wechselstrommodus (AC-SECM)
sowie mittels potentiometrischer SECM zu visualisieren. Anhand der Ergebnisse
dieser Arbeit soll aufzeigt werden, dass topographische sowie elektrochemische
Informationen über Metalloberflächen vor, während und nach der Lochfraßkorrosion
mit hoher lateraler Auflösung erhalten werden können.
Darüber hinaus wird in dieser Arbeit das Ausbluten von Metallionen aus
korrodierenden Nitinol-Werkstücken durch eine automatisierte, adsorptive Stripping-
voltammetrie (AdSV) untersucht. Um diese Messungen kostengünstig und mit
geringem Aufwand durchzuführen, wird in der vorliegenden Arbeit ein System
entwickelt, welches automatisierte Langzeitexperimente an korrodierenden Metall-
proben ermöglicht.

Stand der Forschung 3
2 Stand der Forschung
2.1 Formgedächtnislegierungen (FGLs)
Die Formgedächtnislegierungen zeichnen sich durch ihren Formgedächtniseffekt und
ihre Superelastizität aus, die bei Standardlegierungen nicht beobachtet werden
können. Erstmals entdeckt wurde dieses Verhalten 1951 bei einer Au-47,5 at% Cd
Legierung und 1963 bei NiTi-Legierungen[1]. Nitinol hat sich als wichtigste und am
meisten verwendete Formgedächtnislegierung etabliert.
Formgedächtnislegierungen gehören zu der Gruppe der Funktionswerkstoffe, die
sich durch ihre außergewöhnlichen physikalischen Eigenschaften von den durch ihre
mechanischen Eigenschaften gekennzeichneten Strukturwerkstoffen abgrenzen[2].
Sie werden dementsprechend eingesetzt, um eine bestimmte funktionelle Aufgabe
zu erfüllen.
Die Einsatzgebiete von z.B. NiTi-FGLs liegen nicht nur in typisch industriellen
Anwendungen, wie z.B. Kupplungen, Aktoren oder Ventilen[3], sondern auch im
medizinischen Bereich. Hier ist mittlerweile ein breites Spektrum an Verwendungs-
zwecken für Nitinol etabliert, wie Geräte für minimalinvasive Eingriffe, Drähte für
Zahnspangen, Stents, Klammern und Knochenimplantate[4, 5].
2.1.1 Der Formgedächtniseffekt und die martensitische
Umwandlung
Der Formgedächtniseffekt der FGLs sowie die Superelastizität beruhen auf einer
strukturellen Phasenumwandlung der Hochtemperaturphase Austenit in die
Niedertemperaturphase Martensit, welche martensitische Umwandlung (MT) genannt
wird[6, 7]. Die Phasenumwandlung ist in diesem Fall reversibel und kann thermisch
oder mechanisch induziert sein. Die thermische Umwandlung beginnt bei der
Martensit-Start-Temperatur (Ms), bei der sich erste Martensitkristalle im Austenit-
gefüge formen, und ist bei der Martensit-Finish-Temperatur (Mf) abgeschlossen.
Durch eine anschließende Erwärmung des Materials kann die austenitische Phase
wieder hergestellt werden. Die Umwandlung beginnt in diesem Fall bei der Austenit-
Start-Temperatur (As) und ist bei der Austenit-Finish-Temperatur (Af) beendet.
Kennzeichnend für einen solchen Zyklus ist eine thermische Hysterese, so dass sich
die Umwandlungstemperaturen unterscheiden.

Stand der Forschung 4
Eine sehr vereinfachte Erklärung der Vorgänge im Material soll hier die Grundlagen
in Kürze näher bringen. Die Phasenumwandlung erfolgt diffusionslos, d.h. die Atome
behalten ihre Nachbaratome und es findet lediglich eine Umstrukturierung des
Kristallgitters, im Allgemeinen zu einer niedrigeren Symmetrie, beim Übergang der
Nieder- in die Hochtemperaturphase statt. Diese Strukturänderung verläuft nach
einem scherungsähnlichen Mechanismus (siehe Abbildung 2-1). Für diese Scherung
gibt es zwei mögliche Orientierungen, wie sie in Abbildung 2-1 A und B angedeutet
sind, die Struktur des Gitters ist jedoch die gleiche[8].
Dieses allgemein gültige Verhalten trifft auch auf die NiTi-FGLs zu, wobei hier eine
Umwandlung vom kubisch raumzentrierten Austenit zu einer monoklinen Kristall-
struktur im Martensit stattfindet. Eine detailliertere Beschreibung des gesamten
Phänomens ist in der Literatur[9, 10] zu finden.
2.1.2 Korrosion und Biokompatibilität der NiTi-FGLs
Das Korrosionsverhalten und die damit verbundene Funktions- bzw. Lebensdauer ist
ein wichtiger Aspekt für jede Art von Werkstücken. Im Bereich der medizinischen
Anwendung ist die Korrosion zusätzlich ein essentieller Faktor hinsichtlich der
Austenit AustenitMartensit Martensit
A
B
Austenit AustenitMartensit Martensit
A
B
Austenit AustenitMartensit Martensit
A
B
Abbildung 2-1: vereinfachte schematische Darstellung der martensitischen Umwandlung
Stand der Forschung 5
Biokompatibilität[4, 11, 12, 13]. Das bei der Korrosion aufgelöste Material gelangt un-
weigerlich meist über einen langen Zeitraum in den menschlichen Körper und
entfaltet dort seine spezifische Wirkung.
In der Medizin stieß Nitinol trotz seiner hervorragenden Materialeigenschaften wegen
des hohen Nickelgehalts von ca. 50at% lange Zeit auf Ablehnung. Die Folgen von
einer zu hohen Nickelkonzentration im menschlichen Körper oder einer dauerhaften
Exposition von Gewebe an nickelhaltige Oberflächen sind schon seit längerem
bekannt. Die Auswirkungen reichen von zytotoxischen über allergieauslösende bis
hin zu krebserregenden Effekten[14, 15, 16]. Jedoch ist NiTi mit einer passivierenden
TiO2-Schicht bedeckt, die die Legierung vor Korrosion schützt. Bei der Abwägung für
oder gegen solch ein neuartiges Material wurde oft nicht berücksichtigt, dass die
Nickelfreisetzung aus Nitinol mit einer glatten und fehlerfreien Passivschicht so
niedrig ist, dass sie unter der durchschnittlichen täglichen Nickelaufnahme eines
Menschen über Nahrungsmittel und Luft liegt[4].
Frühe Experimente zur Korrosionsbeständigkeit bescheinigen NiTi eine weitaus
bessere Korrosionsstabilität als dem meist in der Medizin verwendeten chirurgischen
Edelstahl[17]. Dazu wurde die allgemein anerkannte Methode der anodischen
dynamischen Polarisation (ADP) zur Untersuchung verwendet, bei der eine
Potentialrampe beginnend beim freien Korrosionspotential in Richtung zunehmender
Spannung gefahren wird und die abrupte Zunahme der Stromdichte als Anzeichen
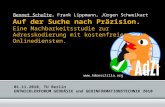
für beginnende Korrosion angenommen wird (siehe Abbildung 2-2). Aus dem Plateau
in der Strom-Spannungskennlinie bis zum Anstieg des Stroms kann zusätzlich
U
I
Plateaustrom
Durchbruchspotential
U
I
Plateaustrom
Durchbruchspotential
Abbildung 2-2: schematische Darstellung einer typischen anodischen dynamischen Polarisationskurve

Stand der Forschung 6
entnommen werden, dass die Oberfläche des Materials mit einer Passivschicht
bedeckt ist. Diese wird bei beginnender Korrosion durchbrochen. Aus diesem Grund
spricht man auch an dieser Stelle von der Durchbruchsspannung (Eb).
Weitere anerkannte Testmethoden, die der ADP sehr ähnlich sind, sind z.B. die
zyklische dynamische Polarisation (CDP) und der Kratztest (scratch test, ST). Die
CDP beginnt äquivalent zur ADP, jedoch wird nach erreichen einer genügend hohen
Stromdichte das Potential wieder zum Ausgangswert zurück geführt. Dies ermöglicht
zusätzlich zur Bestimmung der Durchbruchsspannung, das Repassivierungspotential
(Ep) zu ermitteln. Ebenfalls zur Bestimmung des Ep wird der ST benutzt. Hier wird die
Passivschicht mit einer feinen Diamantnadel angeritzt und danach ein angelegtes
Potential schrittweise abgesenkt. Sinkt auch die Stromdichte, beginnt die
Repassivierung.
Viele Veröffentlichungen präsentieren Ergebnisse aus solchen Messreihen, wobei
die Ergebnisse von 100 bis 1800 mV für Eb streuen[18, 19, 20, 21, 22, 23, 24]. Daher haben
diese Studien wenig zur Aufklärung des tatsächlichen Eb beigetragen und
ermöglichen so keine Einstufung der Korrosionsbeständigkeit des Nitinol. Es wird
sogar davon berichtet, dass die Korrosionsbeständigkeit von Nitinol besser ist als bei
einem 316L Edelstahl, der auch für die Fertigung chirurgischer Instrumente und
Implantate benutzt wird[23]. Dieser Standpunkt wird kontrovers diskutiert, so
behaupten z. B. Kim et al, dass sich die Korrosionsbeständigkeit der beiden
Materialien genau gegensätzlich verhält[20].
Der Austritt von Ni2+ durch Lagerung von NiTi in einer physiologischen Salzlösung
wurde schon früh über längere Zeiträume bestimmt[25]. Bei dieser Untersuchung
wurde allerdings die Lösung nach einer bestimmten Zeit ausgewechselt und off-line
analysiert, meist mit AAS. Die Ergebnisse zeigten einen geringeren Austritt an Ni2+
aus der NiTi-Legierung als aus Edelstählen wie SUS316 oder SUS304, welche auch
für medizinische Applikationen verwendet werden, trotz eines wesentlich höheren
Nickelgehalts im Nitinol. Es wurde auch davon berichtet, dass der Austritt von Nickel
nach einigen Tagen abnimmt oder sogar unter die Nachweisgrenze absinkt[18].
Es wurden zudem auch Zytotoxizitätstests durchgeführt[26], um die direkte Reaktion
des menschlichen Körpers auf die Legierung beurteilen zu können. Auch hier zeigten
sich starke Schwankungen, wahrscheinlich aufgrund der unterschiedlichen
Testmethoden. Die Beurteilung der schädlichen Wirkung von Nitinol reicht von nicht
nachweisbarer Zytotoxizität[27, 28] über eine geringe Zytotoxizität vergleichbar mit

Stand der Forschung 7
anderen Materialien für Implantate[29] bis hin zu sehr viel höherer Zytotoxizität als z.B.
Edelstahl[26].
Ein häufiger Ansatz, um die Korrosionsstabilität und somit auch die Biokompatibilität
zu verbessern, ist die Veränderung der Oberfläche des Materials. Dies findet
entweder durch Glätten der Oberfläche mittels Polieren statt, da eine glatte
Oberfläche weniger Angriffsmöglichkeiten für Korrosion bietet, oder durch
Modifizieren der Passivschicht. Zu diesem Zweck wird die Oxidschicht aufgelöst und
durch z. B. thermische Behandlung, elektrolytisches Polieren oder chemische
Oxidation erneuert[30, 24]. Durch diese Verfahren wird die Passivschicht homogener
und teilweise dicker als eine natürlich aufgewachsene Oxidschicht.
Die Vielfalt der zuvor erwähnten Vorbehandlungen der FGLs ist einer der Gründe für
die enorme Streuung der vorher beschriebenen Testreihen. Deren Ergebnisse
wurden teilweise an FGLs erhalten, die unterschiedlich vorbehandelt und in
verschiedenen Lösungen untersucht wurden. Schon die Herstellung der Legierung
hat einen großen Einfluss auf deren Korrosionsbeständigkeit[20]. Shabalovskaya et al
hat diese Zusammenhänge vor Kurzem in einem umfassenden Übersichtsartikel
aufgezeigt[13].
Eine detaillierte Untersuchung der Effekte, die für die Korrosion bei NiTi-FGLs
verantwortlich sind, im Besonderen auf lokaler Ebene stehen weiterhin aus.
2.2 Elektrochemische Rastermikroskopie (SECM)
Das elektrochemische Rastermikroskop wurde Ende der achtziger Jahre von Bard et
al.[31] und Engstrom[32] et al. entwickelt. Im prinzipiellen Aufbau und Funktionsweise
ähnelt es anderen Rastermikroskopen, wie z. B. dem Rastertunnelmikroskop (STM)
und dem Rasterkraftmikroskop (AFM). Grundlage eines jeden Rastermikroskops ist
das Rastern einer Oberfläche mit einer Sonde, die eine auf die Sondenspitze
begrenzte Wechselwirkung mit der zu untersuchenden Oberfläche eingeht. Dies ist
z. B. bei der STM der Tunnelstrom, bei der AFM Anziehungs- bzw. Abstoßungskräfte
zwischen Sondenspitze und Oberfläche. Die Gemeinsamkeit des SECM, AFM und
STM liegt darin, dass mit der Rastersonde eine Oberfläche in Linien mit
vorgegebenem Abstand abgefahren wird und aus den durch Wechselwirkung der
Sondenspitze mit der Probenoberfläche erhaltenen Informationen ein Abbild der
Oberflächentopographie erstellt wird. Das SECM kann zusätzlich zu der Oberflächen-

Stand der Forschung 8
topographie weitere Informationen über die elektrochemischen Eigenschaften der
Oberfläche erfassen.
Der Nachteil des SECM gegenüber anderen Rastermikroskopietechniken liegt in der
geringeren Auflösung. Während es mit AFM und STM möglich ist, Auflösungen bis in
den atomaren Bereich zu erhalten, ist das Auflösungsvermögen des SECM durch die
Dimensionen der Messsonde, dem Abstand zur Probe[33] und durch die
Positionierbarkeit begrenzt.
SECM-Messsonden sind im allgemeinen in Form von scheibenförmigen Ultramikro-
elektroden (UME) realisiert, die meist in Durchmessern bis in den unteren Mikro-
meterbereich verfügbar sind[34, 35, 36]. In den letzten Jahren konnten die SECM-
Messsonden jedoch bis in den Nanometerbereich verkleinert werden[37, 38, 39].
Das Messsignal selbst besteht bei einem typischen SECM-Experiment aus einem
Faraday’schen Strom, welcher fließt, wenn die Messsonde in eine Elektrolytlösung
eintaucht, die eine reversibel oxidierbare oder reduzierbare Spezies (einen Redox-
mediator) enthält. Dabei muss an der SECM-Sonde ein ausreichendes Potential
anliegen, um den Mediator elektrochemisch umzusetzen. Bei der einfachsten Form
dieses Experiments wird die UME rasterförmig in konstanter Höhe über eine Probe
geführt. Diese Form des SECM-Versuchs wird „constant-z“ Modus bzw. „constant-
height“ Modus genannt (siehe Abschnitt 2.2.2.3.1). Das aufgezeichnete Stromsignal
enthält dabei Informationen über die Topographie[40], lokale Leitfähigkeit[41],
Konzentrationsprofile[42] und chemisch aktive Bereiche der Probenoberfläche („hot
spots“)[43].
Zudem besteht die Möglichkeit, Oberflächen mit dem SECM nicht nur passiv
abzubilden, sondern sie auch aktiv zu modifizieren. So lassen sich mit dem SECM
Mikrostrukturen in z. B. Metall-[44, 45] und Halbleiteroberflächen[46, 47] ätzen sowie
Metalle[48] und Polymere[49] gezielt unterhalb der Sondenspitze abscheiden.
In jüngerer Zeit wird das SECM auch zur Charakterisierung biologischer Proben
genutzt. So sind Untersuchungen an Enzymen[33, 50, 51], an Antikörpern[52],
Antigenen[53] und an DNA[54] durchgeführt und Ionentransporte durch Hautporen[55]
beobachtet worden. Neuere Entwicklungen im Bereich der SECM gehen in die
Richtung der Beobachtung der Aktivität einzelner lebender Zellen. So wurde z.B.
jüngst die Freisetzung chemischer Botenstoffe aus einzelnen sekretorischen Zellen
mittels des SECM beschrieben[56, 57].

Stand der Forschung 9
Eine wichtige Neuerung gelang mit der Integration eines Höhenkontroll-Modus
(„constant-distance“ Modus, siehe Abschnitt 2.2.2.3.2) in den Aufbau des SECM[58].
Dadurch wurde es möglich, die Messsonde nicht in konstanter Höhe über eine
Oberfläche zu führen, sondern in einem konstanten Abstand. Die enormen Vorteile
des „constant-distance“ Modus liegen in der Separation des Topographiesignals vom
elektrochemischen Signal. Im „constant-height“ Modus hingegen sind beide Signale
,wie oben erwähnt, überlagert. Der „constant-distance“ Modus erlaubt es folglich drei-
dimensionale Strukturen mit einem hohen Aspektverhältnis abzubilden. Zudem ergibt
sich auch eine Verbesserung bei Anwendungen zur Oberflächenmodifizierung, da
man mit dieser Technik nun auch dreidimensionale Strukturen abscheiden kann.
Dies ist z. B. in Form von freistehenden Polypyrroltürmen auf Goldoberflächen[59]
gelungen. Dazu wurde während der Abscheidung die UME von der wachsenden
Polymerschicht zurückgezogen und so immer ein konstanter Abstand zu dieser
beibehalten.
Bisher wurden nur amperometrische Methoden vorgestellt, jedoch sind prinzipiell
auch potentiometrische Sonden in das SECM integrierbar. Es wurden z. B. verschie-
dene Mikro-pH-Sonden entwickelt und im SECM zur Abbildung von pH-aktiven Ober-
flächen genutzt[60]. Ebenso konnten Ionenselektive Ca2+-Sensoren zur Abbildung der
Auflösung von kalziumhaltigen Oberflächen verwendet werden[61].
In den letzten Jahren sind durch Adaption von Impedanztechniken an das SECM
auch Experimente im Wechselstrommodus möglich[62] geworden. Bei dieser Form
der SECM wird eine Wechselspannung fester Frequenz und Amplitude an die Sonde
angelegt und der Widerstand des Lösungsmittels dient als Messsignal. Wie bei der
amperometrischen SECM enthält das Signal Informationen über die Topographie, die
lokale Leitfähigkeit und über Konzentrationsprofile, jedoch ohne einen Redox-
mediator einsetzen zu müssen.
2.2.1 Aufbau eines SECM
Der typische Aufbau eines SECM für einen Betrieb im „constant-height“ Modus
besteht aus drei Schrittmotoren für die Bewegung der Probe und/oder der SECM-
Sonde in alle drei Raumrichtungen, einem Computer zur Steuerung des Geräts und
zur Messwertaufnahme, einem Potentiostaten zum Betrieb der elektrochemischen
Messzelle und zur Messung des Sondenstroms, und der Messzelle selbst, die mit
dem Elektrolyten gefüllt ist. In der Messzelle befinden sich in einer Dreielektroden-
Stand der Forschung 10
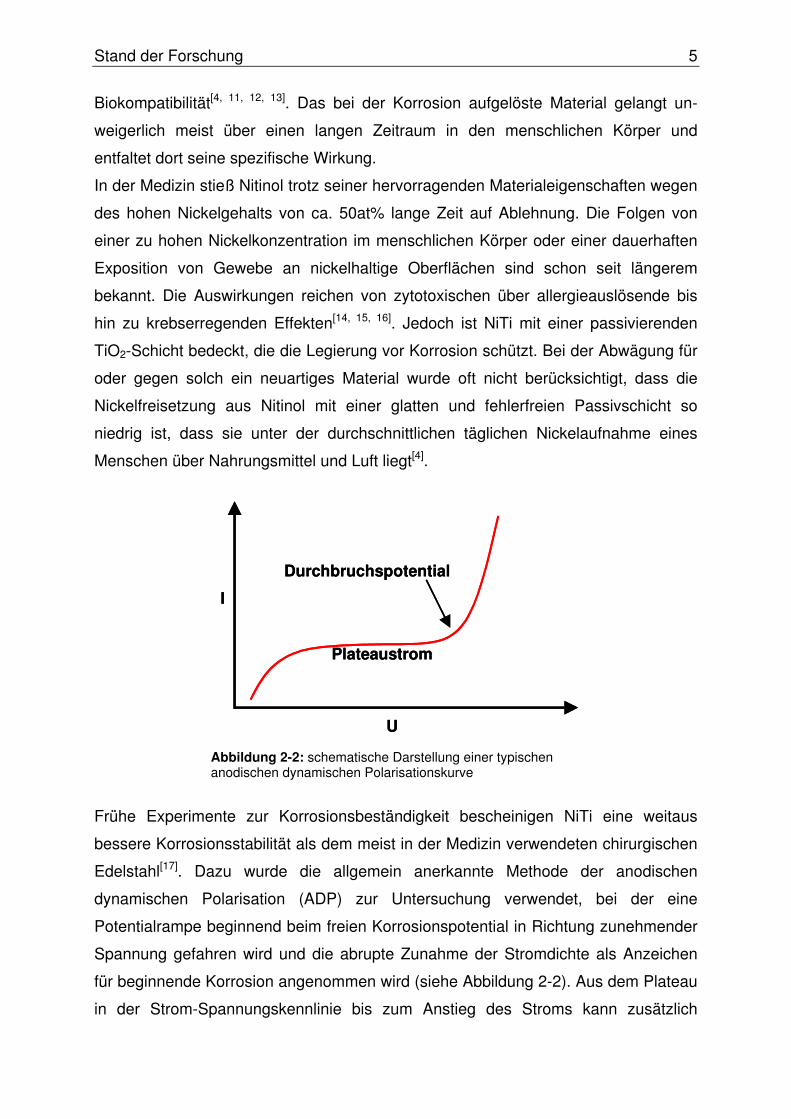
anordnung die scheibenförmige UME als Arbeitselektrode (SECM-Sonde), eine
geeignete Referenzelektrode und eine Platin-Gegenelektrode. Eine schematische
Darstellung eines SECM ist in der Abbildung 2-3 zu sehen.
Die Konzeption soll im Folgenden etwas genauer beschrieben werden. Die
Positioniereinheit besteht aus drei Schrittmotoren, die die am Probentisch fixierte
Messzelle in x-, y- und z-Richtung verschieben können. Die Mikroelektrode nimmt
eine fixierte z-Position ein und die Messzelle wird relativ zur ortsfesten SECM-Sonde
in die drei Raumrichtungen bewegt. Die Schrittmotoren werden über eine Motorkarte
angesprochen, die wiederum die Steuerbefehle für die Positionierung vom PC
entgegen nimmt. Da die Auflösung des SECM stark von einer genauen
Positionierung abhängt, werden hochpräzise Schrittmotoren verwendet, die möglichst
einen Mikroschrittmodus erlauben. Die Genauigkeit der Positionierung kann noch
erhöht werden, indem zusätzlich zu den Schrittmotoren Piezoaktoren eingesetzt
y
z
RE
CE
schwingungsgedämpfter Tisch
CE
REWE
PotentiostatMotorsteuerkarten
Iout Ein
PC für die Hardwaresteuerung und Datenaufnahme
ProbeElektrolyt
elektrochemischeMesszelle
x
AD/DA seriell
Mikroelektrode
y
z
RE
CE
schwingungsgedämpfter Tisch
CE
REWE
PotentiostatMotorsteuerkarten
Iout Ein
PC für die Hardwaresteuerung und Datenaufnahme
ProbeElektrolyt
elektrochemischeMesszelle
x
AD/DA seriell
Mikroelektrode
Abbildung 2-3: schematische Darstellung eines SECM für den „constant-height“ Modus
Stand der Forschung 11
werden. Diese schränken jedoch auf der anderen Seite aufgrund ihrer geringen
Verfahrwege von maximal hundert Mikrometern den Abbildungsbereich stark ein.
Ein Potentiostat wird benötigt, um der UME je nach Art des verwendeten Redoxme-
diators ein anodisches oder kathodisches Potential aufzuprägen, das ausreicht, die
Redoxspezies in der Lösung elektrochemisch umzusetzen. Gleichzeitig misst der
Potentiostat den über die SECM-Sonde fließenden Strom und gibt dieses Signal an
den Steuerrechner weiter. Oft wird ein Bipotentiostat verwendet, um bei Bedarf eben-
falls ein Potential an die zu untersuchende Probe anlegen zu können.
Die Anforderungen an den Computer sind nach heutigen Maßstäben sehr gering. Es
reicht im Normalfall ein PC mit 80486 Prozessor, auf dem die oft selbst-
programmierte Steuerungssoftware des SECM läuft. Die Kommunikation mit der
Schrittmotorkarte ist im allgemeinen über den seriellen Anschluß hergestellt,
während die Ansteuerung des Potentiostaten und die Aufnahme des SECM-
Sondenstroms über eine AD/DA-Wandlerkarte erfolgt.
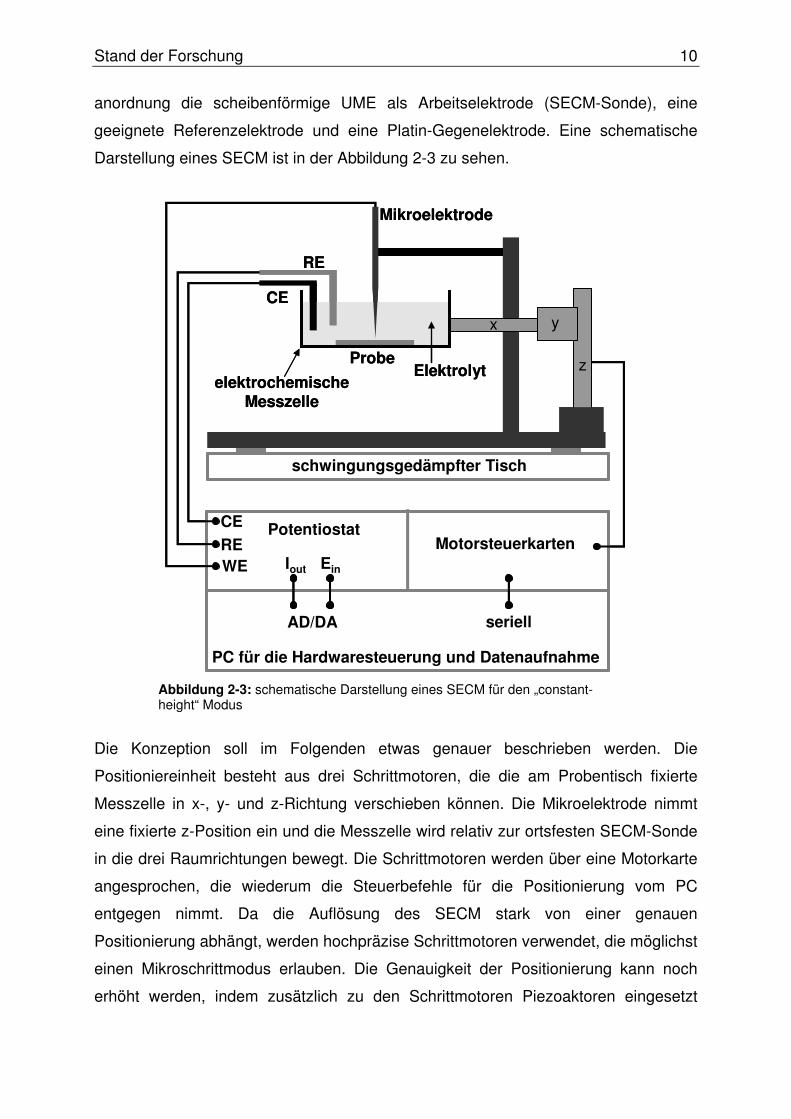
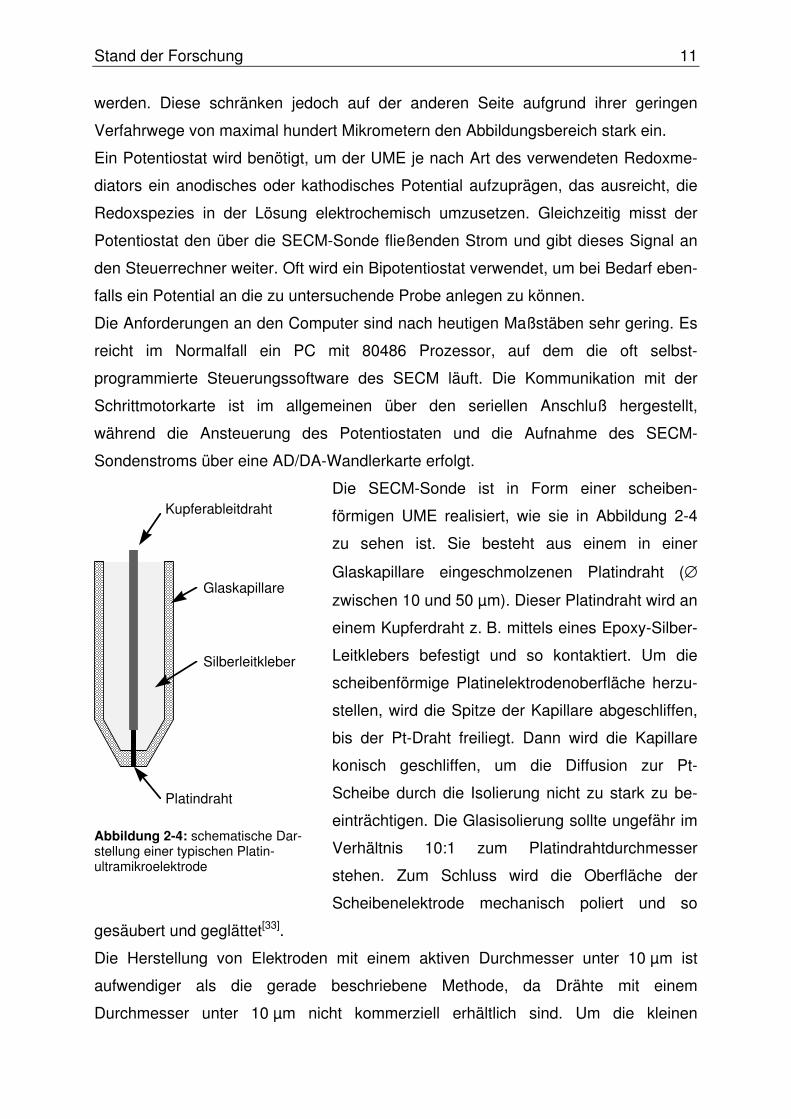
Die SECM-Sonde ist in Form einer scheiben-
förmigen UME realisiert, wie sie in Abbildung 2-4
zu sehen ist. Sie besteht aus einem in einer
Glaskapillare eingeschmolzenen Platindraht (∅
zwischen 10 und 50 µm). Dieser Platindraht wird an
einem Kupferdraht z. B. mittels eines Epoxy-Silber-
Leitklebers befestigt und so kontaktiert. Um die
scheibenförmige Platinelektrodenoberfläche herzu-
stellen, wird die Spitze der Kapillare abgeschliffen,
bis der Pt-Draht freiliegt. Dann wird die Kapillare
konisch geschliffen, um die Diffusion zur Pt-
Scheibe durch die Isolierung nicht zu stark zu be-
einträchtigen. Die Glasisolierung sollte ungefähr im
Verhältnis 10:1 zum Platindrahtdurchmesser
stehen. Zum Schluss wird die Oberfläche der
Scheibenelektrode mechanisch poliert und so
gesäubert und geglättet[33].
Die Herstellung von Elektroden mit einem aktiven Durchmesser unter 10 µm ist
aufwendiger als die gerade beschriebene Methode, da Drähte mit einem
Durchmesser unter 10 µm nicht kommerziell erhältlich sind. Um die kleinen
Kupferableitdraht
Glaskapillare
Silberleitkleber
Platindraht
Abbildung 2-4: schematische Dar-stellung einer typischen Platin-ultramikroelektrode

Stand der Forschung 12
Dimensionen zu erreichen, werden meist größere Platindrähte geätzt und z. B. mit
einer Polymerschicht isoliert[39]. Eine einfachere Methode erlaubt es, die Elektroden
mittels eines Laserpullers gleichzeitig mit der isolierenden Glaskapillare auszuziehen
und so den eingebetteten Platindraht zu verjüngen[37].
2.2.2 Funktionsweise eines SECM
Zu Beginn einer Messung muss die Sonde an die Probenoberfläche angenähert
werden. Dies wird mit dem z-Motor bewerkstelligt, wobei der Abstand zur Probe
ständig kontrolliert werden muss, um ein Aufsetzen auf der Probe bzw. Abbrechen
der Elektrode zu vermeiden. Die Kontrolle des Annäherungsvorgangs kann optisch
mit Hilfe eines integrierten Mikroskops[63, 64], mit einer automatisierten Höhenkon-
trolle, wie sie in Abschnitt 2.2.3 näher erläutert wird, oder durch Verfolgen des elek-
trochemischen Signals während der Annäherung geschehen.
Um die letzte Methode und die eigentliche Messung besser verständlich zu machen,
ist es notwendig auf die Eigenschaften von UME näher einzugehen. Ausführliche
Diskussionen über dieses Thema finden sich in[34, 65, 66] und zur Funktionsweise des
SECM in[31, 67].
Die UME befindet sich zusammen mit der
Referenz- und der Gegenelektrode in der
mit Elektrolytlösung gefüllten Messzelle.
Die Lösung enthält eine z.B. oxidierbare
Spezies die bei Anlegen eines genügend
hohen anodischen Potentials gemäß R →
O + e- umgesetzt wird. Als Folge dieser
Umsetzung fließt über die UME ein mess-
barer Strom. Dieser Faraday’sche Strom
geht bei UME sehr schnell in einen
konstanten Diffusionsgrenzstrom über.
Dies liegt daran, dass sich an einer UME
nahezu unmittelbar ein charakteristisches
hemisphärisches Diffusionsfeld ausbildet, welches für einen erhöhten
Massentransport zur Elektrodenoberfläche im Vergleich zu einer Makroelektrode
verantwortlich ist. Der Diffusionsgrenzstrom einer scheibenförmigen UME lässt sich
durch folgende Gleichung beschreiben[67]:
R O
Abbildung 2-5: Darstellung des hemi-
sphärischen Diffusionsfelds bei UME und der
oxidativen Umsetzung eines Mediators

Stand der Forschung 13
It,∞∞∞∞ = 4nFDca
n = Zahl der umgesetzten Elektronen F = Faraday-Konstante c = Konzentration von R D = Diffusionskoeffizient von R a = Elektrodenradius
Der gemessene Strom kann also durch Veränderung (Blockade) des Diffusionsfeldes
oder durch Variation der Konzentration des Redoxmediators steigen oder fallen.
Dieser Effekt wird bei der SECM-Messung im sogenannten Feedbackmodus
ausgenutzt.
2.2.2.1 Annäherung der Ultramikroelektrode an nicht leitende
Oberflächen
Bei Annäherung der UME an eine nicht leitende Oberfläche wird bei der Auf-
zeichnung der Annäherungskurve (I/t) ein schneller Rückgang des Stromsignals ver-
zeichnet.
Dies liegt darin begründet, dass das
Diffusionsfeld der UME bei einem
kritischen Abstand zur Oberfläche der
Probe eingeschränkt wird (siehe Abbildung
2-6). Es gelangen weniger Mediatormole-
küle pro Zeiteinheit zur Elektrodenober-
fläche und werden dort umgesetzt, was zu
einem Absinken des Faraday’schen
Stromes führt. Dieses Phänomen wird als
negativer Feedback bezeichnet und oft bei
der Annäherung der Sonde an eine
Oberfläche als Abbruchkriterium genutzt. Das Stromminimum ist bei Aufsetzen der
Elektrode auf der Probenoberfläche erreicht und besteht nur noch aus einem
schwachen Reststrom. Der Strom ist nicht gleich Null, da sich nie ein perfekter
Abschluss zwischen Elektrodenfläche und Probe erreichen lässt, so dass die
Diffusion zur Elektrode niemals vollständig unterbunden wird.
Der Effekt des negativen Feedbacks bei isolierend ummantelten Scheibenelektroden
ist theoretisch behandelt worden[68]. Es sind folgende Kriterien zur Berechnung
festgelegt worden:
• Die Probenoberfläche ist planar, unbegrenzt ausgedehnt und planparallel zur
Mikroelektrode ausgerichtet.
nicht leitend
Abbildung 2-6: Darstellung des negativen Feedbacks

Stand der Forschung 14
• Der Radius der Isolierung der Scheibenelektrode ist um ein Vielfaches größer als
der Radius der aktiven Elektrodenoberfläche.
• Die Umsetzung des Redoxmediators an der Mikroelektrode verläuft diffusions-
kontrolliert. Im Falle leitender Oberflächen verläuft die Umsetzung des Redox-
mediators an diesen ebenfalls unter Diffusionskontrolle (siehe Abschnitt 2.2.2.2)
Durch Interpolation der einzelnen Werte, die ausführliche Herleitung kann in der
Literatur[69] nachgelesen werden, erhält man für den negativen Feedback eine
Näherungslösung.
Diese Näherung hat eine Abweichung von max. 1.2% von den durch Simulation be-
rechneten Werten und besitzt ihre Gültigkeit für Werte von d/r im Bereich von 0.05
bis 20.
Abbildung 2-7 zeigt die graphische Darstellung dieser Näherungslösung. Es ist
erkennbar, dass die Abnahme des Faraday’schen Stromes bei einem Abstand zur
nicht leitenden Oberfläche einsetzt, der etwa dem fünffachen des aktiven
Elektrodenradius entspricht. Der Einfluss der Isolierungsdicke auf den Strom ist in
der Formel nicht berücksichtigt. Hier wurde eine Isolierung, die dem 10-100-fachen
des Elektrodenradius entspricht, angenommen. Die Dicke der Isolierungsschicht trägt
zu der oben erwähnten Abweichung der Werte bei.
0
0.2
0.4
0.6
0.8
1
1.2
0 2 4 6 8 10 12 14 16
d / r
I t / I
t,o
o
Abbildung 2-7: theoretisch berechnete Annäherungskurve für den negativen Feedback

Stand der Forschung 15
2.2.2.2 Annäherung der Ultramikroelektrode an leitende Oberflächen
Bei der Annäherung der UME an eine
leitende Oberfläche wird im Gegensatz
zur nicht leitenden Oberfläche ein starker
Stromanstieg beobachtet. Die Begrün-
dung liegt hier nicht in der geometrischen
Veränderung des Diffusionsfeldes,
sondern in einer an der leitenden
Oberfläche stattfindenden Rückreaktion
des Redoxmediators gemäß O + e- → R,
sobald sich die Elektrode nahe genug an
der Oberfläche befindet (siehe Abbildung
2-8). Diese Rückreaktion kann statt-
finden, da sich an der leitenden Proben-
oberfläche ein Potential in Abhängigkeit von dem Konzentrationsverhältnis der
Redoxspezies in seiner oxidierten und reduzierten Form gemäß Nernst ausbildet.
[[[[ ]]]][[[[ ]]]]RO
logn059.0
EE 0⋅⋅⋅⋅++++====
E0 = Normalpotential n = Zahl der ausgetauschten Elektronen [O] = Konzentration der oxidierten Form [R] = Konzentration der reduzierten Form Unter der Annahme, dass die Lösung eine hohe Konzentration an einer reduzier-
baren Spezies enthält, wird der logarithmische Ausdruck in der Gleichung negativ
und das Potential an der Oberfläche kleiner als das Standardpotential der Reaktion
R → O + e-, so dass die Rückreaktion einsetzt. Dies führt zu einem erhöhten Strom-
fluss an der UME, da diese nicht mehr nur durch die Diffusion der reduzierten Form
des Mediators aus dem weiter entfernten Lösungsvolumen angewiesen ist, sondern
zusätzlich auch eine Erhöhung der Konzentration von R im eigenen Diffusionsfeld
vorfindet. Weil die UME die oxidierte Form liefert, die die leitende Probe wieder
reduziert und umgekehrt, wird dieser Reaktionsablauf auch Redoxrecycling genannt.
Das Potential der Oberfläche bleibt dabei trotz Konzentrationsänderung konstant,
weil es sich aus dem Konzentrationsverhältnis des Redoxpaares über die gesamte
Oberfläche ergibt und diese im Vergleich zur Reaktionsfläche sehr groß ist.
leitend
R O
Abbildung 2-8: Darstellung des positiven Feedbacks
Stand der Forschung 16
Der Effekt des ansteigenden Stromes bei der Annäherung der UME an eine leitende
Oberfläche wird in Analogie zur Annäherung an eine nicht leitende Oberfläche positi-
ver Feedback genannt. Jedoch liegt hier nicht nur eine Abhängigkeit des Stromes
vom aktiven Elektrodenradius und dem Abstand zur Probe vor, sondern ebenso eine
Abhängigkeit von der Art der Probe und eventuell vom heterogenen Ladungstransfer
zwischen O und der leitenden Oberfläche. Ist der Ladungstransfer langsam im
Vergleich zur Diffusion der reduzierten Form von der Probenoberfläche zur UME, ist
der Strom nicht mehr diffusionskontrolliert. Dieser Effekt wurde zur Untersuchung von
Reaktionskinetiken verwendet[67, 70].
Auch das Phänomen des positiven Feedbacks wurde theoretisch behandelt[68, 69]. Die
Näherung weicht um max. 0,7 % von der Simulation ab und hat wiederum einen
Gültigkeitsbereich für d/r von 0,05 bis 20, wobei hier aber die Dicke der Isolierung nur
einen sehr geringen Einfluss hat.
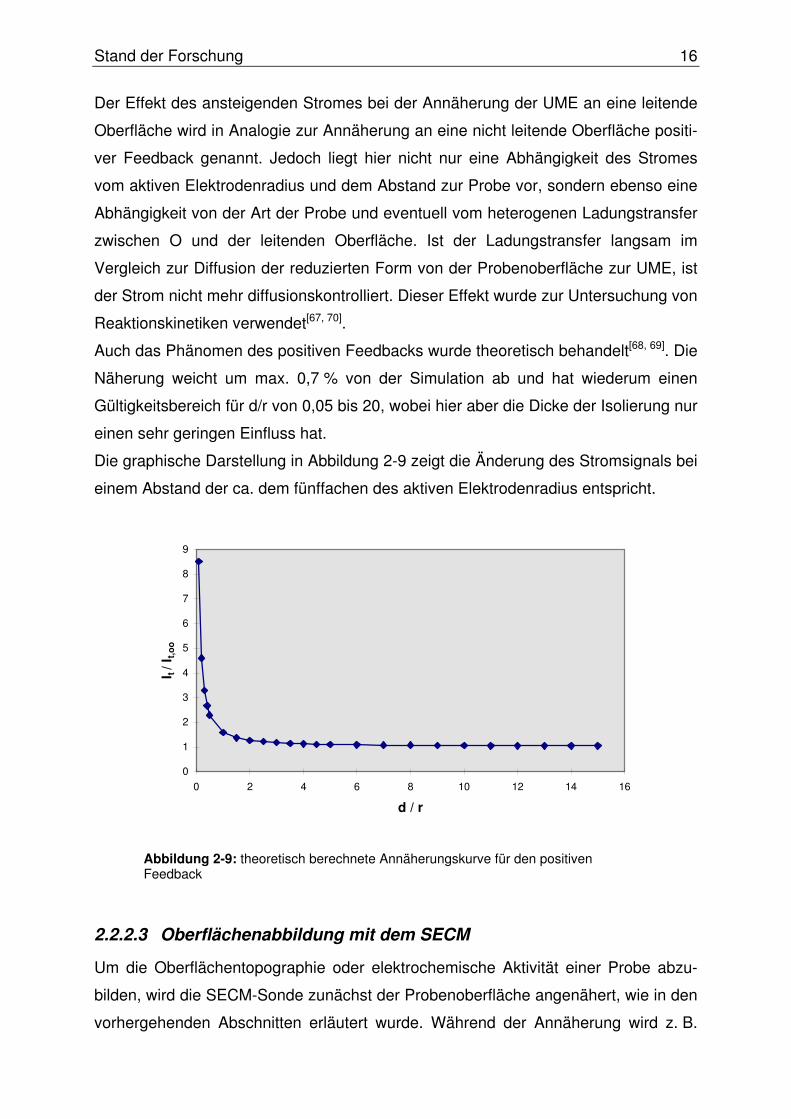
Die graphische Darstellung in Abbildung 2-9 zeigt die Änderung des Stromsignals bei
einem Abstand der ca. dem fünffachen des aktiven Elektrodenradius entspricht.
2.2.2.3 Oberflächenabbildung mit dem SECM
Um die Oberflächentopographie oder elektrochemische Aktivität einer Probe abzu-
bilden, wird die SECM-Sonde zunächst der Probenoberfläche angenähert, wie in den
vorhergehenden Abschnitten erläutert wurde. Während der Annäherung wird z. B.
0
1
2
3
4
5
6
7
8
9
0 2 4 6 8 10 12 14 16
d / r
I t / I
t,o
o
Abbildung 2-9: theoretisch berechnete Annäherungskurve für den positiven Feedback

Stand der Forschung 17
das Stromsignal der UME beobachtet (computergestützte Aufzeichnung einer An-
näherungskurve) und der Schrittmotor gestoppt, sobald eine Änderung des Stromes
verzeichnet wird. Dabei kann je nach Oberfläche (leitend/nichtleitend) ein positiver
oder negativer Feedback auftreten. Genau diese beiden Effekte sind letztlich auch
die Grundlage für die ortsaufgelöste Untersuchung der elektrochemischen Aktivität
und der Topographie der Probenoberflächen.
Nach der Positionierung der Elektrode über der Probe und Festlegung des Arbeitsab-
standes wird in der Steuerungssoftware eine Strecke x gewählt, die der abzufahren-
den Linienlänge entspricht, sowie die Anzahl der lateral abzufahrenden Linien und
eine Strecke ∆y, die den Abstand zwischen den Linien wiedergibt. Die Messung kann
je nach SECM im „constant-height“ Modus oder im „constant-distance“ Modus durch-
geführt werden. In Abhängigkeit des gewählten Modus wird während des Abbildens
entweder nur das elektrochemische Signal oder zusätzlich noch ein Höhensignal auf-
gezeichnet. Nach vollständigem Abrastern werden die Daten über eine Auswertungs-
software in ein dreidimensionales Bild umgesetzt. In diesen Darstellungen werden
zum einen der fließende Strom an der UME gegen die x-y-Länge („constant-height“
Modus), zum anderen zusätzlich die Höheninformation gegen die x-y-Koordinaten
(„constant-distance“ Modus) aufgetragen.
Einen genaueren Einblick in den Ablauf und die Funktionsweise der beiden
Abbildungsmodi sollen in den nächsten Abschnitten gegeben werden.
2.2.2.3.1 Der „constant-height“ Modus
Der UME wird über den Potentiostaten ein Potential aufgeprägt, welches ausreicht,
um die in Lösung befindliche Redoxspezies diffusionskontrolliert umzusetzen. Die
Annäherung erfolgt langsam (v < 1 µm/s) über den z-Schrittmotor oder gegebenen-
falls über den z-Piezo bis eine Veränderung des Stroms eintritt. Die Änderung kann,
wie in den vorherigen Abschnitten beschrieben wurde, einer Zunahme oder Ab-
nahme je nach Art der Oberfläche und des Redoxmediators entsprechen. Die Mess-
elektrode wird so über der Probe positioniert, dass die Stromänderung ca. 20 bis
30% der maximalen Variation bei vollkommener Annäherung an die Oberfläche
entspricht. Für das weitere Experiment wird die z-Position (der Arbeitsabstand) nicht
mehr verändert, sondern nur noch laterale Bewegungen über die Fläche ausgeführt
(siehe Abbildung 2-10)

Stand der Forschung 18
Die Anzahl der Linien sowie deren Länge und Abstand zueinander werden nach der
Annäherung festgelegt und abgefahren. Bei dieser Bewegung wird das Stromsignal
aufgezeichnet, welches durch die Topographie und Leitfähigkeit der Oberfläche
örtlich variieren kann.
Befindet sich eine Erhöhung auf einer nicht leitfähigen Probe, so nimmt der Strom-
fluss an der UME ab, da sich der Abstand zwischen der Probenoberfläche und der
UME verringert und somit das Diffusionsfeld stärker eingeschränkt wird. Dies ent-
spricht dem erwähnten negativen Feedback. Ist hingegen eine Vertiefung in der
Oberfläche vorhanden, so steigt der Strom an, weil sich das Diffusionsfeld der Elek-
trode weiter ausdehnen kann und ein höherer Massentransport zur Elektrodenober-
fläche stattfindet. Der negative Feedback-Effekt nimmt also ab. Bei einer elektro-
chemisch aktiven Oberfläche verhält es sich umgekehrt, da ein größerer Abstand den
positiven Feedback abschwächt und ein geringerer ihn verstärkt. Im Anschluß an die
Messung kann der Strom mit Hilfe der Annäherungskurve in eine Höheninformation
umgerechnet werden.
Der Einfluss der unterschiedlichen Leitfähigkeiten der Probe auf den Strom ist in den
Auswirkungen dem der Topographie recht ähnlich. Erreicht man mit der Sonde eine
elektrochemisch aktive Zone auf der Probe, so steigt der Strom in Folge des
Redoxrecyclings an. Im Gegensatz dazu fällt der Strom beim Übergang von einer
leitfähigen Region zu einer nicht leitfähigen ab, da das Redoxrecycling abbricht.
Wie an den obigen Ausführungen zu erkennen ist, stößt man hier auf eine
Limitierung des „constant-height“ Modus. Wenn die zu untersuchende Probe nämlich
Abbildung 2-10: Darstellung der Elektrodenbewegung im „constant-height“ Modus

Stand der Forschung 19
beide Oberflächenbeschaffenheiten in sich vereint, also uneben ist und leitende und
nicht leitende Regionen besitzt, überlagern sich die elektrochemischen und topo-
graphischen Informationen. Dies kann leicht zu Fehlinterpretationen der Mess-
ergebnisse führen, da die ansteigenden und abfallenden Stromsignale nicht mehr
eindeutig der Topographie oder der Reaktivität zugeordnet werden können.
Bei zu großen Höhenunterschieden der Probe wird ein weiterer Nachteil des
„constant-height“ Modus sichtbar. Ist eine Struktur auf der Probenoberfläche höher
als der vorher eingestellte Arbeitsabstand zwischen Sondenspitze und Probe, wird
die Messsonde bei der lateralen Bewegung über die Oberfläche in diese hineinfahren
und kann dabei die Probe oder sich selbst beschädigen. Das Problem besteht vor
allem bei sehr kleinen UME, da der Arbeitsabstand nur etwa dem dreifachen des
Elektrodendurchmessers entspricht, infolge dessen also nicht viel Spielraum für
Topographieänderungen bleibt.
2.2.2.3.2 Der „constant-distance” Modus
Betrachtet man die im letzten Abschnitt erläuterten Nachteile, ist eine aktive Höhen-
kontrolle, welche den Abstand zwischen Probe und Sonde konstant hält und somit
die Separation der Topographie und der elektrochemischen Information gewähr-
leistet, eine logische Weiterentwicklung[33, 71].
Diese Weiterentwicklung wurde mit dem „constant-distance“ Modus realisiert, der
aufgrund eines vom Feedback-Strom unabhängigen Höhensignals über einen
Regelkreislauf den Abstand der UME zur Oberfläche bei Änderung dieses Signals
korrigiert (siehe Abbildung 2-11).
Abbildung 2-11: Darstellung der Elektrodenbewegung im „constant-distance“ Modus

Stand der Forschung 20
2.2.3 Funktionsweise der optischen Höhenkontrolle
Um eine Höhenkontrolle für das SECM zu verwirklichen, gibt es im wesentlichen drei
bisher etablierte Ansätze. Der erste setzt auf ein elektrochemisches Signal, welches
konstant gehalten wird. Wie in den vorhergehenden Kapiteln ausgeführt, hängt das
amperometrische Signal der UME im SECM vom Abstand zur Oberfläche der Probe
ab. Wird die Messsonde im Feedbackbereich platziert, kann das Stromsignal zur
Kontrolle der Höhe verwendet werden. Ebenso ist dies durch Konstanthalten der
Impedanz beim Anlegen einer Wechselspannung möglich[72]. Diese Methode
funktioniert allerdings nur zuverlässig, wenn davon ausgegangen werden kann, dass
die Probe an jeder Stelle der Oberfläche die gleiche elektrochemische Aktivität
besitzt.
Der zweite Ansatz ist stromunabhängig und ist aus einer Kombination von AFM mit
SECM entstanden. In diesem Fall werden modifizierte AFM-Kantilever als SECM-
Sonde benutzt und die Höhe mit der bewährten AFM-Technik kontrolliert[73, 74].
Der dritte Ansatz ist ebenfalls stromunabhängig und nutzt die Dämpfung der
horizontalen Resonanzschwingung der UME durch hydrodynamische Scherkräfte.
Die Methoden, um die UME in Schwingung zu versetzten und die Dämpfung zu
registrieren, variieren. Vorgestellt wurden Techniken zur Höhenkontrolle, welche auf
eine Art Stimmgabel zurückgreifen[75], oder einen Piezo zur Anregung der
Schwingung nutzen[71]. Das Auslesen der Dämpfung kann optisch (optische Höhen-
kontrolle) oder über einen zweiten „Empfänger“-Piezo (nichtoptische Höhenkontrolle)
geschehen[76].
Die Detektion der Änderung der Schwingungsamplitude und Phase erfolgt bei der
optischen Höhenkontrolle über die Variation der Oszillation des Fresnel’schen
Beugungsmusters der UME, das bei der Fokussierung eines Lasers auf die
Elektrodenspitze entsteht und mit einer geteilten Photodiode ausgelesen wird.
Dieses Prinzip erlaubt nicht nur den „constant-distance“ Modus mit amperome-
trischen SECM-Sonden, sondern auch eine Annäherung von Messsonden, die kein
Stromsignal und somit weder einen positiven noch einen negativen Feedback liefern,
wie z.B. potentiometrische Sonden.
Für das SECM im „constant-distance“ Modus sind schwingungsfähige UME
notwendig, die bei Fokussierung eines Laserstrahls auf die Elektrodenspitze ein
hinreichend starkes Beugungsmuster auf der Photodiode erzeugen[33]. Solche UME
(siehe Abbildung 2-12) lassen sich herstellen, indem Glaskapillaren, wie sie auch für
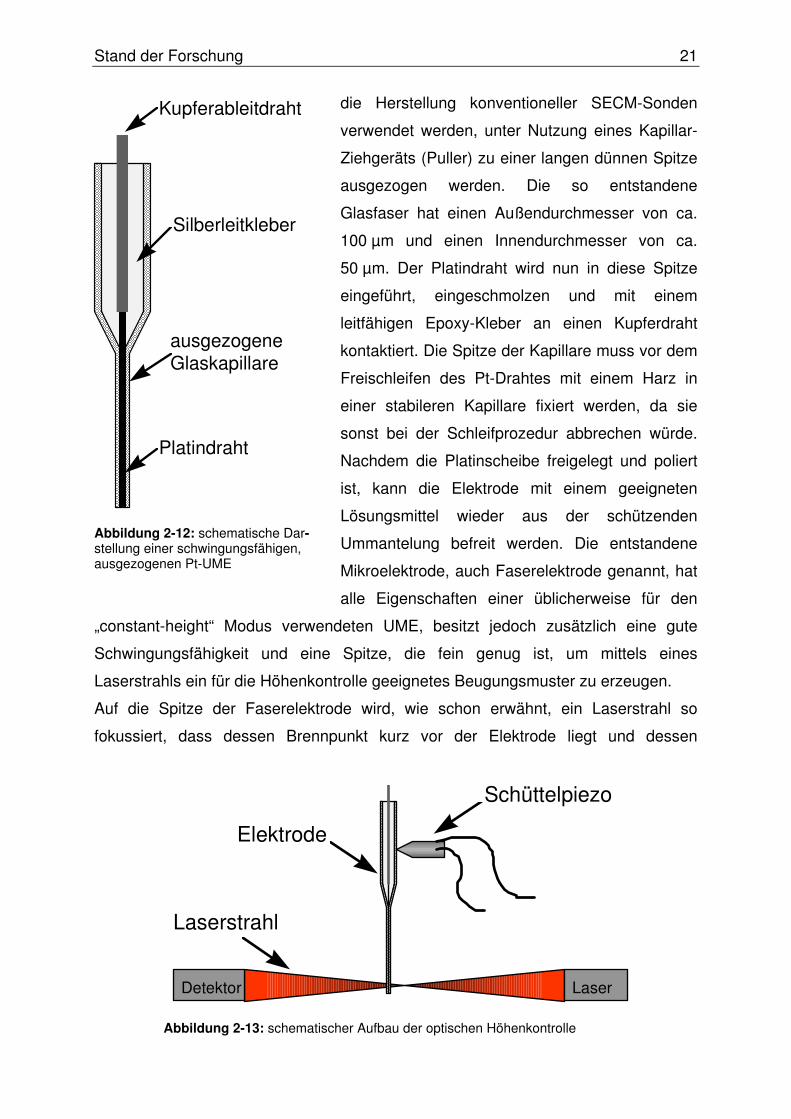
Stand der Forschung 21
die Herstellung konventioneller SECM-Sonden
verwendet werden, unter Nutzung eines Kapillar-
Ziehgeräts (Puller) zu einer langen dünnen Spitze
ausgezogen werden. Die so entstandene
Glasfaser hat einen Außendurchmesser von ca.
100 µm und einen Innendurchmesser von ca.
50 µm. Der Platindraht wird nun in diese Spitze
eingeführt, eingeschmolzen und mit einem
leitfähigen Epoxy-Kleber an einen Kupferdraht
kontaktiert. Die Spitze der Kapillare muss vor dem
Freischleifen des Pt-Drahtes mit einem Harz in
einer stabileren Kapillare fixiert werden, da sie
sonst bei der Schleifprozedur abbrechen würde.
Nachdem die Platinscheibe freigelegt und poliert
ist, kann die Elektrode mit einem geeigneten
Lösungsmittel wieder aus der schützenden
Ummantelung befreit werden. Die entstandene
Mikroelektrode, auch Faserelektrode genannt, hat
alle Eigenschaften einer üblicherweise für den
„constant-height“ Modus verwendeten UME, besitzt jedoch zusätzlich eine gute
Schwingungsfähigkeit und eine Spitze, die fein genug ist, um mittels eines
Laserstrahls ein für die Höhenkontrolle geeignetes Beugungsmuster zu erzeugen.
Auf die Spitze der Faserelektrode wird, wie schon erwähnt, ein Laserstrahl so
fokussiert, dass dessen Brennpunkt kurz vor der Elektrode liegt und dessen
Kupferableitdraht
ausgezogeneGlaskapillare
Silberleitkleber
Platindraht
Abbildung 2-12: schematische Dar-stellung einer schwingungsfähigen, ausgezogenen Pt-UME
Laserstrahl
Schüttelpiezo
Elektrode
LaserDetektor
Abbildung 2-13: schematischer Aufbau der optischen Höhenkontrolle

Stand der Forschung 22
Fresnel’sches Beugungsmuster auf eine geteilte Photodiode projiziert wird, so wie es
in der Abbildung 2-13 zu erkennen ist.
Die Elektrode wird mit einem Schüttelpiezo, welcher eine sinusförmige Spannung
von einem Funktionsgenerator erhält, zur Schwingung in Resonanz angeregt. Die am
Funktionsgenerator einzustellende Frequenz und Amplitude zur Erzeugung der Re-
sonanzschwingung sind individuell bei jeder Elektrode unterschiedlich. Die geteilte
Photodiode ist so geschaltet, dass sie ein Differenzsignal der beiden Diodenhälften,
auf die das oszillierende Beugungsbild der Elektrode projiziert wird, an einen Lock-in-
Verstärker weitergibt. Der Lock-in-Verstärker vergleicht das Anregungs- mit dem
Schwingungssignal, verstärkt es phasenselektiv und liefert einen Messwert der bei
eingestellter Resonanzfrequenz maximal ist.
Die in Resonanz schwingende Elektrode wird in Analogie zu den oben beschriebe-
nen Annäherungsarten mit Hilfe des z-Schrittmotors an die Probenoberfläche heran-
geführt. Bei genügend geringem Abstand zu dieser, wird die Frequenz, die Amplitude
und die Phase der Resonanzschwingung durch den Einfluss von hydrodynamischen
Scherkräften gedämpft. Diese Dämpfung führt zu einem Absinken des Signals am
Lock-in-Verstärker und kann als Abbruchkriterium der Annäherung genutzt werden.
Die Stärke der Scherkräfte ist von der Viskosität der Lösung abhängig und hat im All-
gemeinen eine Reichweite von unter 1 µm in die Lösung hinein. Der Effekt ist noch
nicht genauer untersucht worden[33]. Allgemein kann jedoch davon ausgegangen
werden, dass die Viskosität der Lösung in der Nähe der Oberfläche eine Kraft auf die
Spitze der Elektrode ausübt, die eine Abstandsabhängigkeit aufweist.
Die Annäherung im SECM erfolgt automatisiert. Das Anfangssignal des Lock-in-
Verstärkers bei großem Abstand der Sonde zur Probe wird wiederum über eine AD-
Karte in den SECM-Steuerrechner eingelesen. Der Steuersoftware wird dann ein
prozentualer, auf den Anfangswert des Lock-in-Signals bezogener Wert vorgegeben,
bei dem der Annäherungsvorgang gestoppt werden soll. Die Auftragung des Lock-in-
Signals gegen die zurückgelegte Strecke bildet die Annäherungskurve, die typischer-
weise erst in einem konstanten Abstand, parallel zur x-Achse (Zeitachse) verläuft und
dann bei Einsetzen der Scherkräfte linear abfällt. Aus der berechneten Steigung der
abfallenden Geraden erhält der rückgekoppelte Regelkreislauf für den „constant-
distance“ Modus die Parameter zur Konstanthaltung der Höhe.
Die Regelsoftware versucht bei der lateralen Bewegung der Sonde über die Ober-
fläche den bei der Annäherung festgelegten prozentualen Anteil des Ausgangs-Lock-

Stand der Forschung 23
in-Signals konstant zu halten. Dies geschieht, indem sie den z-Motor (bzw. z-Piezo)
in die entsprechende Richtung steuert und somit die Höhenunterschiede der Ober-
fläche ausgleicht. Ein wählbarer Dämpfungsfaktor soll dabei ein Überschwingen der
Höhenregelung vermeiden, die z. B. aufgrund nicht-scherkraftabhängiger Schwan-
kungen (z.B. Rauschen der Photodiode, Trittschall, etc.) im Lock-in-Signal erfolgen
würde.
Die Auftragung der z-Motorposition gegen die x-y-Koordinaten ergibt die von dem
elektrochemischen Signal entkoppelte Topographieinformation. Die Topographie
einer Probe wird allerdings nicht richtig dimensioniert wiedergegeben. Wird z. B. eine
25 µm breite Bandstruktur abgebildet, so wird in der höhenkontrollierten Topogra-
phieabbildung eine 2 mal um die Elektrodendicke verbreiterte Strukturdarstellung
erhalten. Dies liegt darin begründet, dass der Rand der Isolierung der Elektrode beim
Auftreffen zuerst und beim Verlassen der Bandstruktur bis zuletzt den Scherkräften
ausgesetzt ist. Somit ist die Auflösung des SECM nicht mehr allein durch die
elektrochemisch aktive Fläche der Elektrode, wie es für den „constant-height“ Modus
der Fall ist, sondern durch den Gesamtradius der Sonde begrenzt.
2.3 Ionenselektive Rastermikroskopie
Das elektrochemische Rastermikroskop ist im Abschnitt 2.2 ausführlich diskutiert
worden. Die hohe Auflösung, die Möglichkeit verschiedene Abbildungsmodi
(„constant-height“ oder „constant-distance“) einzusetzen und lokal chemische
Aktivitäten zu detektieren, machen es zu einem hervorragenden Werkzeug, um
chemische Prozesse zu verfolgen und zu visualisieren. So können nicht nur mit Hilfe
des positiven und negativen Feedbacks (Abschnitt 2.2.2) elektrochemisch aktive
Stellen auf einer Probe detektiert werden, sondern wie schon erwähnt, auch lokal
Enzym- und Zellaktivitäten beobachtet und Elektronentransferkinetiken gemessen
werden. Solche Messungen werden mit amperometrischen SECM-Sonden
durchgeführt, die aber nicht geeignet sind, um alle interessierenden chemischen
Systeme zu untersuchen. In der medizinischen Diagnostik sind zum Beispiel Alkali-
und Erdalkaliionenkonzentrationen von großer Bedeutung und der pH-Wert einer
Lösung ist für viele Reaktionen ein entscheidender Parameter. Die
amperometrischen Messverfahren sind für derlei Analysen oft nicht ausreichend
selektiv oder ungeeignet.

Stand der Forschung 24
Eine Alternative bietet der Einsatz potentiometrischer Elektroden, die selektiv auf be-
stimmte Ionen ansprechen und mittlerweile für viele verschiedene Ionenarten verfüg-
bar sind. Eine allgemeine theoretische Abhandlung über ionenselektive Elektroden
ist in[77] zu finden. Die Anwendung potentiometrischer ionenselektiver Mikrosensoren
ist in der Elektrophysiologie schon seit langem erfolgreich zur lokalen Bestimmung
von Ionenkonzentrationen etabliert worden[78].
Naheliegend ist also eine Verknüpfung der Technik des SECM mit potentiome-
trischen Mikrosensoren als Messsonden. Vorraussetzung ist aber, dass an den zu
untersuchenden Oberflächen lokal ausreichend große Aktivitätsgradienten des
Analyten vorhanden sein müssen, da sonst nur ein nicht aussagekräftiges kon-
stantes Potential gemessen würde. Ändert sich das Konzentrationsprofil einer Ionen-
spezies lokal, kann mit einer SECM-Aufnahme ein ionenselektives Abbild der aktiven
Zentren der Entstehung dieser Spezies erhalten werden.
In den letzten fünfzehn Jahren haben mehrere Arbeitsgruppen ihre Forschungen in
diese Richtung vorangetrieben. Eine der ersten Veröffentlichungen[79] beschreibt die
Untersuchung des Austritts von Chloridionen aus Poren einer Polyanilin-beschich-
teten Elektrode mittels einer für Chloridionen-selektiven Ag/AgCl-Elektrode. Ein
anderes Beispiel ist die Verwendung einer Referenzelektrode als SECM-Sonde, um
Potentialschwankungen an einer korrodierenden Metalloberfläche zu verfolgen[80, 81].
Sehr interessant sind neuere Entwicklungen in Richtung der sogenannten pH-Mikros-
kopie. Nachdem erste pH-Abbildungen von wasserzersetzenden Metallelektroden
und korrodierenden Oberflächen mit Hilfe der Fluoreszenzmikroskopie und einem
pH-abhängigen Fluoreszenzfarbstoffs gemacht wurden[82], ist diese Methode weiter-
entwickelt worden, indem ein Fluoreszenzfarbstoff auf einer optischen Faser
immobilisiert und mit einem AFM über die Probe gerastert wurde[83].
Parallel dazu entwickelte sich die potentiometrische pH-Mikroskopie unter der
Nutzung des SECM. Vorreiter auf diesem Gebiet waren Bard et al., die 1993 eine
Arbeit über potentiometrische pH-Rastermikroskopie veröffentlichten[60]. Die pH-
Sonde bestand in diesem Fall aus einer Sb-Elektrode von meist 40 µm aktivem
Durchmesser. Hergestellt wurde sie durch Einspritzen von geschmolzenem Antimon
in eine Glaskapillare und anschließendem Ausziehen derselben zu einer dünnen
Spitze. Das verwendete SECM entsprach prinzipiell der Bauweise, die in Abschnitt
2.2.1 erläutert wurde, wobei die Messanordnung um ein hochohmiges Voltmeter zur
Bestimmung des Sondenpotentials gegen eine Ag/AgCl-Referenz erweitert wurde.

Stand der Forschung 25
Ein Problem beim Einsatz potentiometrischer SECM-Sonden ist die Annäherung der
Sonde an die Probenoberfläche, da für potentiometrische Elektroden ein positiver
bzw. negativer Feedback nicht auftritt und so nicht für die Positionierung verwendet
werden kann. In Abschnitt 2.2.2.1 und 2.2.2.2 wurden ausführlich die bestehenden
Möglichkeiten der Elektrodenannäherung beschrieben, wenn keine zusätzliche
Höhenkontrolle vorhanden ist. Bard et al. umgingen das Problem des fehlenden
Feedbacks, indem sie die Antimonscheibenelektrode während der Annäherung als
amperometrische und erst nach der Positionierung als potentiometrische Sonde ein-
setzten[60]. Für Arbeiten in wässriger Lösung bietet sich dabei an, an die SECM-
Sonde ein Potential von etwa -0,8 V anzulegen, was ausreicht, um Sauerstoff zu
reduzieren, so dass kein zusätzlicher Redoxmediator benötigt wird. Dies ist von
Vorteil, da bestimmte Redoxmediatoren bei der anschließenden pH-Bestimmung
stören würden[84].
Die Antimonelektroden zeigten eine für diese Art Elektroden typische Empfindlichkeit
von 40-50 mV/pH, die im Vergleich zu anderen pH-Sonden eher gering ist[77, 84] und
eine Ansprechzeit im Millisekundenbereich. Zur Validierung des Messprinzips wurden
mit dem SECM im „constant-height“ Modus verschiedene Testsysteme untersucht.
Diese Messungen wurden analog der in Abschnitt 2.2.2.3.1 beschriebenen Weise
durchgeführt, lediglich das Stromsignal war durch das Potentialsignal der pH-Elektro-
de ersetzt worden. Als Testobjekte dienten eine 25 µm Pt-Scheibenelektrode, an der
elektrolytisch Wasser zersetzt wurde, eine korrodierende Silberjodidscheibe und in
einer Kapillare immobilisierte Urease bzw. Hefezellen. Eines dieser Experimente soll
im folgenden genauer erläutert werden.
Die glasummantelte Pt-Scheibenelektrode (Generatorelektrode) wurde in eine Mess-
zelle eingesetzt und diese mit einem Phosphatpuffer (pH 7) gefüllt. An die Mikroelek-
trode wurde ein kathodisches Potential (-2 V vs. Pt-Gegenelektrode) angelegt, dass
ausreicht, um die Wasserzersetzung zu bewirken, aber niedrig genug ist, um eine
störende Gasentwicklung zu vermeiden.
H2O + e- →→→→ ½ H2 + OH-
Die pH-Änderung in der Nähe der Generatorelektrode konnte detektiert werden und
zu einem Abbild ihrer pH-Aktivität umgerechnet werden. Der Einfluss der Pufferstärke
auf die Diffusion der OH--Ionen konnte anhand des Radius der pH-Abbildung nach-
vollzogen werden. Zudem wurde dieses Experiment dazu genutzt, die Abhängigkeit

Stand der Forschung 26
des Potentials der pH-Elektrode vom Abstand zur Probenoberfläche durch lang-
sames Zurückziehen (z-Scan) in das Lösungsvolumen hinein und Auftragung des
Potentials gegen den Abstand zu visualisieren.
Zwei Jahre später wiederum veröffentlichten Bard et al. eine Studie über poten-
tiometrische Rastermikroskopie, basierend auf ionenselektiven Zweikanal-
Mikropipetten-Elektroden als SECM-Sonden[85]. Die Technik zur Herstellung solcher
Elektroden ist in der Elektrophysiologie entwickelt worden[86, 87] und basiert auf
ausgezogenen Glaskapillaren, die Öffnungen im unteren Mikrometerbereich
aufweisen. Diese werden mit einer ionenselektiven Polymermembran versehen, mit
einem Elektrolyten befüllt und mit einer Ag/AgCl-Ableitelektrode ausgestattet. Bard
nutzte eine der Öffnungen zur Bestimmung der Ionenkonzentrationen (u.a. H+) und
die andere zur Annäherung der Sonde an die Oberfläche der Probe. Zur Positionie-
rung potentiometrischer Sonden in einem kontrollierten Abstand zur Probenober-
fläche ist auch die Zuhilfenahme eines optischen Mikroskops beschrieben worden[63,
64]. Klusmann und Schultze benutzten eine Mikropipetten-pH-Sonde mit Flüssigmem-
bran für Untersuchungen von pH-Diffusionsfeldern an Pt-Mikroelektroden[64], an
denen Wasser elektrolytisch zersetzt wurde. Die Mikropipette hatte einen aktiven
Durchmesser von ca. 1 µm, was eine hohe Auflösung zur Folge hatte. Die
Empfindlichkeit entsprach etwa den nach Nernst erwarteten 59 mV/pH und die An-
sprechzeit lag unter einer Sekunde. Die Sonde wurde mit Hilfe des optischen Mikros-
kops auf definierte Abstände zur Oberfläche der Generatorelektrode angenähert und
der Einfluss des Abstandes auf das Sondenpotential ermittelt. Aufgrund der nicht
absolut planaren Fläche der Probe konnte allerdings während der Messung kein
konstanter Abstand eingehalten werden, was quantitative Aussagen erschwerte.
Wipf et al.[63] benutzten hingegen eine AEIROF-Elektrode (Anodic Electrodeposited
IRidium Oxide Film electrodes) auf der Basis von CFME für eine pH-Mikroskopie. Die
CFME wurden in Anlehnung an das in Abschnitt 2.2.1 für die Herstellung der
typischen UME beschriebene Verfahren hergestellt. Der aktive Durchmesser ist
durch den Durchmesser der verwendeten Carbonfaser auf 5 µm festgelegt. Ihr
Gesamtdurchmesser inklusive der Glasisolierung lag bei ungefähr 40 µm. Die Be-
schichtung mit Iridiumoxid erfolgte mittels literaturbekannter Methoden[88, 89].
Abbildungen des pH-Profils einer wasserzersetzenden 10 µm Pt-Scheibenelektrode
wurden bei verschiedener Pufferkonzentration und an immobilisierter Glukose-

Stand der Forschung 27
oxidase mit Ferrocenniumionen als Redoxmediator im „constant-height“ Modus
durchgeführt.
Aufbauend auf diesem Experiment sind im Rahmen meiner Diplomarbeit CFME
basierende AEIROF-Elektroden zur Visualisierung des pH-Profils an wasserzer-
setzenden UMEs eingesetzt worden. Die Messungen wurden im „constant-distance“
Modus unter zu Hilfenahme der optischen Höhenkontrolle (siehe Abschnitt 2.2.2.3.2
und 2.2.3) durchgeführt[162]. Somit gelang eine einfachere Positionierung der
Elektrode an der Probenoberfläche und eine genauere Abbildung des pH-Profils
unter Berücksichtigung dessen Abstandsabhängigkeit.
2.4 SECM im Wechselstrom Modus (AC-SECM)
Im letzten Abschnitt wurde eine Alternative zur klassischen, amperometrischen
SECM beschrieben. Die potentiometrische SECM deckt einen großen Bereich vorher
nicht erfassbarer Oberflächenphänomene ab und ist dabei selektiv für bestimmte
Ionen. Ein in den letzten Jahren in der Arbeitsgruppe entwickelter Wechselstrom-
Modus für das SECM stellt eine weitere Alternative zu den beiden bisher
vorgestellten dar.
Die AC-SECM ist einer Verschmelzung der elektrochemischen Impedanzspektros-
kopie (EIS) bzw. der lokalen elektrochemischen Impedanzspektroskopie (LEIS) mit
der SECM. Bei der elektrochemischen Impedanzspektroskopie wird eine Wechsel-
spannung mit konstanter Amplitude an ein System angelegt und unter Variation der
Frequenz die Stromantwort sowie die Phasenverschiebung aufgezeichnet. Es wird
versucht, das so erhaltene Impedanzspektrum mittels eines Ersatzschaltbildes zu
erklären, welches im Allgemeinen aus einer Anordnung von ohmschen Widerständen
und Kondensatoren besteht. Die einzelnen Elemente des Ersatzschaltbildes werden
dann auf die elektrochemischen Prozesse im System übertragen.
Wird z. B. eine Redoxreaktion mittels EIS untersucht, so würde das zu einem Ersatz-
schaltbild, wie in Abbildung 2-14 dargestellt, führen. An der Phasengrenze Arbeits-
elektrode/Lösung bildet sich zuerst die elektrochemische Doppelschicht aus, welche
mit einem Plattenkondensator Cdl vergleichbar ist. Zudem kann die zur Elektronen-
übertragung zwischen Elektrode und Redoxspezies erforderliche Aktivierungsenergie
als Durchtrittswiderstand (charge-transfer-restistor) Rct angesehen werden. Die
Warburg-Impedanz Zw repräsentiert die Diffusion, welche für den Elektronenübertrag
ebenfalls eine wichtige Rolle spielt. Die Warburg-Impedanz selbst entspricht einer

Stand der Forschung 28
Reihenschaltung eines Verlustkondensator und eines frequenzabhängigen Wider-
standes. Zuletzt muss noch der Lösungsmittelwiderstand einbezogen werden, der
hier als ohmscher Widerstand RΩΩΩΩ dargestellt ist.
Die mittels EIS, welche hauptsächlich in der Korrosionsforschung eingesetzt wird, be-
stimmten Informationen beziehen sich jedoch auf eine gesamte Probe und spiegeln
aufgrund des integralen Charakters nicht die lokalen Gegebenheiten an der Ober-
fläche wieder. Dabei ist die Frequenzabhängigkeit des Stroms z. B. an einer korro-
dierenden Stelle einer Metalloberfläche eine ganz andere als die an einer passivier-
ten[90].
CE WE
Cdl
RΩΩΩΩ
ZW Rct
CE WE
Cdl
RΩΩΩΩ
ZW Rct
Abbildung 2-14: Ersatzschaltskizze für eine Redoxreaktion, siehe Text zur Erklärung der Elemente
passiv aktiv
Äquipotentialflächen
Messgerät
Elektroden
passiv aktiv
Äquipotentialflächen
Messgerät
Elektroden
Abbildung 2-15: schematische Darstellung der Versuchsanordnung zur lokalen elektrochemischen Impedanz Spektroskopie

Stand der Forschung 29
Einen Ansatz lokalisierte Informationen an der Probenoberfläche zu ermitteln, stellte
Lillard et al 1992 in Form der LEIS vor[91]. Hier werden zwei Elektroden im konstanten
vertikalen Abstand zueinander an die Probenoberfläche geführt und das Potential
zwischen ihnen gemessen (siehe Abbildung 2-15). Bei dem Elektrodenpaar kann es
sich entweder um zwei Pt-Elektroden[92, 93, 94] oder um zwei Referenzelektroden[95]
handeln. Insgesamt kann ein solches System aus bis zu 6 Elektroden bestehen. An
einer korrodierenden Stelle kann aufgrund eines sich in die Lösung ausbreitenden
Potentialgradienten ein Potentialunterschied zwischen diesen beiden Elektroden
verzeichnet und mit Hilfe des bekannten Lösungswiderstandes und der angelegten
Wechselspannung die Impedanz berechnet werden. Auf diese Weise ist es möglich
lokale Impedanzspektren aufzunehmen.
Eine Weiterentwicklung dieser Methode ist das LEIM (local electrochemical
impedance mapping). Bei diesem Verfahren werden ebenfalls zwei Elektroden wie in
Abbildung 2-15 an der Probenoberfläche positioniert, jedoch kein Frequenzspektrum,
sondern eine Wechselspannung mit fixer Frequenz und Amplitude angelegt, während
das Elektrodenpaar lateral über die Oberfläche geführt wird[95]. Eine Auftragung der
berechneten Impedanz gegen die x,y-Position ergibt ein Impedanzabbild der Ober-
fläche.
Die laterale Auflösung dieser Messmethode ist jedoch sehr gering, da die Elektroden
meist einen Durchmesser von über 100 µm haben und noch der Abstand zwischen
den Elektroden mit eingeht.
Kürzlich wurde die AC-SECM vorgestellt[62, 96], die aus der Kombination der LEIS
bzw. der LEIM und dem SECM entstanden ist. Die AC-SECM kommt im Gegensatz
zur LEIM mit einem Dreielektroden-System aus und benötigt im Vergleich zur
amperometrischen SECM einen zusätzlichen Lock-in-Verstärker. Der Lock-in-
Verstärker wird mit dem Potentiostaten gekoppelt und gibt diesem eine Wechsel-
spannung von fester Frequenz und Amplitude vor. Diese Wechselspannung wird der
SECM-Sonde aufgeprägt, welche als Arbeitselektrode geschaltet ist. Die Strom-
antwort wird in den Lock-in-Verstärker eingespeist, mit der Anregungsspannung
verglichen, phasensensitiv verstärkt und das Signal in Form der Impedanz-Magnitude
und Phasenverschiebung ausgegeben.
Die Messung ist nur unter Einhaltung bestimmter Rahmenparameter möglich, wie
anhand der Skizzen in Abbildung 2-14 und Abbildung 2-16 erläutert werden kann.

Stand der Forschung 30
Das Schaltbild in Abbildung 2-14 reduziert sich auf eine Reihenschaltung aus dem
Lösungswiderstand RΩΩΩΩ und der Doppelschichtkapazität Cdl, wie in Abbildung 2-16
dargestellt, wenn der verwendete Elektrolyt keine Redoxspezies enthält, die bei der
angelegten Wechselspannung umgesetzt werden kann. Durch diese Vereinfachung
des elektrochemischen Systems werden Zw und Rct eliminiert, die den Impedanz-
anteil des Elektronenübertrags der Redoxreaktion repräsentieren (siehe Abbildung
2-14).
Die Doppelschichtkapazität ist eine störende Größe, da sich die AC-SECM-Messung
auf den Widerstand des Lösungsmittels bezieht. Durch die Wahl des Elektrolyten und
der Messfrequenz kann dieses Schaltbild effektiv auf RΩΩΩΩ reduziert werden. Es
werden im Allgemeinen Lösungen von sehr geringer Leitfähigkeit wie 0,1 M NaCl
eingesetzt und Frequenzen ab 1 kHz verwendet. Bei solch hohen Frequenzen hat
die Doppelschichtkapazität keinen Einfluss auf das Messsignal mehr[97].
Eine Annäherung der SECM-Sonde an eine Oberfläche führt zu einer Annäherungs-
kurve, die der unter Abschnitt 2.2.2.1 beschriebenen für amperometrische Sonden im
Verlauf ähnelt. Der Grund für Abnahme des beobachteten Signals ist noch nicht
gänzlich verstanden[97], doch wird möglicherweise durch die teilweise Hinderung der
Ionenbeweglichkeit zwischen Arbeits- und Gegenelektrode durch die Proben-
oberfläche im Nahfeldwechselwirkungsbereich hervorgerufen. Dies würde zu einer
erhöhten Impedanz führen. Handelt es sich um eine leitende Oberfläche, kann ein
Teil des durch die Wechselspannung hervorgerufenen Stroms über den elektrischen
Leiter abgeführt werden, wodurch die Impedanz weniger stark zunimmt. Ein
Vergleich zweier Annäherungskurven auf eine Glas- und auf eine Goldoberfläche ist
in Abschnitt 4.3.2.2 dargestellt.
CE WE
CdlRΩΩΩΩ
CE WE
CdlCdlRΩΩΩΩ
Abbildung 2-16: Ersatzschaltbild für die Impedanz bei der AC-SECM

Stand der Forschung 31
2.5 Sequentielle Injektionsanalyse (SIA)
Analysensysteme wie die Fließinjektionsanalyse (FIA) oder die daraus entwickelte
Sequentielle-Injektionsanalyse (SIA) ermöglichen automatisierte Prozesskontrolle in
Echtzeit über einen rückgekoppelten Regelkreislauf. Die heutigen Geräte zeichnen
sich durch hervorragende Zuverlässigkeit, Reproduzierbarkeit, Flexibilität und
Rentabilität im Vergleich zu manuell durchgeführten Analysen aus. Sie sind
mittlerweile in verschiedenen Anwendungsgebieten etabliert, wie der medizinischen
Diagnostik[98, 99, 100], der Lebensmittelanalytik[101, 102, 103], der Biotechnologie[104, 105, 106]
und kürzlich auch der Schwermetallspurenanalyse[107, 108, 109, 110].
2.5.1 Aufbau und Funktionsweise eines Fließ-Injektions-Analysen-
Systems
Wie erwähnt, ist das SIA eine Weiterentwicklung des FIA-Systems. Das Funktions-
prinzip der Fließinjektionsanalyse basiert auf einem kontinuierlich fließenden Strom
eines Trägerelektrolyten, der mittels einer Schlauchpumpe aufrechterhalten wird. In
diesen Trägerstrom kann ein Probensegment injizierte werden, welches mit dem
Strom zu einem Detektorsystem getragen wird[111], wo das Messsignal aufgezeichnet
wird (siehe Abbildung 2-17). Als Detektoren kommen meist Photometer, aber auch
Potentiostaten[112] zum Einsatz.
Probe
Träger-lösung
Detektor
Abfall
Pumpen
Injektionsventil
Probe
Träger-lösung
Detektor
Abfall
Pumpen
Injektionsventil
Abbildung 2-17: schematischer Aufbau eines FIA-Gerätes

Stand der Forschung 32
Die in den Trägerstrom injizierte Probe vermischt sich auf dem Weg zum Detektor
zum Teil mit dem Trägerelektrolyt. Dieser Vorgang wird Dispersion genannt und
kommt bei laminarer Strömung im wesentlichen durch Diffusion zustande. Das
Ausmaß der Dispersion, repräsentiert durch den Dispersionskoeffizienten D, ist von
mehreren Parametern abhängig: z. B. der Größe des Probensegments, dem
Schlauchdurchmesser oder der Pumpgeschwindigkeit[113, 114].
max
o
c
cD =
Die Durchmischung des Probensegments mit den Nachbarsegmenten kann gezielt
dazu genutzt werden, Verdünnungen durchzuführen oder die Vermischung mit einem
Reagenz zu gewährleisten. Das Reagenz muss dazu zusätzlich in den Trägerstrom,
angrenzend an das Probensegment, injiziert werden. Die Strecke zwischen Ventil
und Detektor dient dabei als Dispersions- und somit Reaktionsstrecke.
Aufgrund des kontinuierlichen Pumpens macht sich der hohe Verbrauch an Träger-
lösung nachteilig bei den FIA-Systemen bemerkbar. Zudem kann es durch dieses
Verfahren zu einem hohen Verbrauch an Probenlösung kommen. Beides ist aus
wirtschaftlichen Gründen unerwünscht.
2.5.2 Aufbau und Funktionsweise eines SIA
Basierend auf den Erkenntnissen der FIA wurde die Sequentielle-Injektions-Analyse
(SIA) entwickelt, um die zum Ende des letzten Kapitels aufgeführten Nachteile zu
beheben.
Bei diesem System, das erstmals 1990 von Ruzicka und Marshall vorgestellt
wurde[115], wird auf einen kontinuierlichen Trägerstrom verzichtet. Die Pumpeinheit
besteht häufig aus einer Spritzenpumpe mit Zweiwegeventil[109, 116, 117], aufgrund des
unzureichenden Start/Stop-Verhaltens kommen selten Schlauchpumpen[117, 118, 119]
zum Einsatz. Anstatt manueller Injektionsventile werden computergesteuerte
Multipositionierventile verwendet. Diese verfügen über mehrere Anschlüsse, an die
Gefäße mit unterschiedlichen Reagenzien und Probe angeschlossen werden können
(siehe Abbildung 2-18).
Die Probe und Reagenzlösungen werden zunächst in das Schlauchsegment
zwischen dem Ventil und der Pumpe angesaugt und anschließend im laminaren
Fluss zum Detektor gepumpt. Die wesentlichen Vorgänge während der Vermischung
(Dispersion, etc.) entsprechen denen der FIA. Aufgrund des sequentiellen Pumpvor-

Stand der Forschung 33
gangs ist der Verbrauch an Trägerelektrolyt, Probenlösung und Reagenzien wesent-
lich geringer als bei einem FIA-System.
SIA-Systeme weisen folgende Limitierungen auf: Ein laminarer Fluss durch den
Detektorbereich des Gerätes lässt sich nur durch einen Kolbenhub der
Spritzenpumpe bewerkstelligen. Daher bestimmt der Kolbenhub, und damit die
gewählte Größe der Spritzenpumpe, das maximale Schlauchvolumen zwischen
Ventil und Detektor. Da dieses die Reaktionsstrecke darstellt, ist das Volumen der
Reaktionsstrecke auf das Volumen der Spritzenpumpe beschränkt. Als Kostenfaktor
macht sich der hohe Preis der Multipositionierventile bemerkbar, da er mit der Anzahl
der verfügbaren Anschlüsse steigt.
2.5.3 Der Online-General-Analyser (OLGA)
Auf der Basis der zuvor beschriebenen Nachteile des SIA-Systems wurde das
Gesamtkonzept überarbeitet und der Online-General-Analyser (OLGA) entwickelt[120].
Bei dem OLGA-System wurden die typischen einschränkenden Elemente eines SIA-
Systems ersetzt. Eine Kolbenhubpumpe ist an die Stelle der Spritzenpumpe getreten
und erlaubt aufgrund der geringen Pulsation einen laminaren Strom ohne
Volumenbegrenzung. Die hohe Präzision der Pumpe wird durch ihren Schrittmotor-
Probe
Träger-lösung
Reagenz
Detektor
Abfall
Multipositionierventil
Spritzenpumpe
Probe
Träger-lösung
Reagenz
Detektor
Abfall
Multipositionierventil
Spritzenpumpe
Abbildung 2-18: schematischer Aufbau eines SIA-Gerätes

Stand der Forschung 34
antrieb gewährleistet, der es erlaubt, eine Kolbenumdrehung in 360 Einzelschritte zu
unterteilen. Das Kolbenvolumen beträgt 10 µl und ermöglicht die ausreichend
genaue Dosierung von Volumina bis zu 2 µl. Die maximale Pumpgeschwindigkeit von
50 µl/s liegt unter der von Spritzenpumpen, ist jedoch für die vorgesehene
Anwendung ausreichend.
Wie in der Abbildung 2-19 zu sehen ist, wurde das teure Multipositionsventil gegen
einen Manifold aus Acrylglas ausgetauscht. Um diesen Verteiler herum sind
standardmäßig acht kostengünstige Magnetquetschventile angeordnet. Es können
zusätzlich externe Ventile zugeschaltet werden.
Das Gerät verfügt über einen integrierten Potentiostaten, einen Controller für die
Kolbenhubpumpe und eine zusätzliche Schlauchpumpe. Die Ansteuerung erfolgt
durch eine in Visual Basic 6 konzipierte Software. Eine serielle Schnittstelle des PCs
dient zur Kommunikation mit der Kolbenhubpume und eine parallele Schnittstelle zur
Kommunikation mit dem Potentiostaten und zum Schalten der Ventile.
Abbildung 2-19: fotografische Abbildung des OLGA-Systems

Stand der Forschung 35
2.6 Voltammetrische Strippinganalyse
Die Strippingvoltammetrie (SV) hat sich zu einem hervorragenden Werkzeug der
Spurenanalyse von Schwermetallen und organischen Verbindungen entwickelt[121].
Neben der Atomabsorptionsspektroskopie (AAS) und der induktiv gekoppelten
Plasma-Massenspektrometrie (ICP-MS) ist sie die sensitivste Methode, um Schwer-
metalle bis in den subnanomolaren Konzentrationsbereich zu detektieren[121, 122, 123,
124]. Die SV zeichnet sich gegenüber anderen Methoden durch einen einfacheren und
kostengünstigeren aparativen Aufbau aus, da lediglich ein Potentiostat mit
zugehöriger Messzelle benötigt wird. Die hohe Sensitivität, welche von der
Anreicherungszeit abhängt, wird erreicht, indem der Analyt vor der voltammetrischen
Bestimmung bei einem konstanten Potential an der Elektrode angereichert wird. Dies
geschieht unter konvektiven Bedingungen. Der anschließende Bestimmungsschritt,
bei dem der angereicherte Analyt wieder von der Elektrode entfernt wird (stripping),
kann mit jeder voltammetrischen Methode in ruhender Lösung durchgeführt werden.
Der zu verwendende Elektrolyt, sowie dessen pH-Wert, der zu benutzende Modus
(siehe folgende Kapitel) als auch das erreichbare Detektionslimit hängen dabei von
dem Analyten ab[121, 125].
2.6.1 Anodische Strippinganalyse (ASV)
Die ASV ist die älteste Strippingmethode und wird im Allgemeinen zur Bestimmung
von Schwermetallen an einer Quecksilberelektrode durchgeführt (siehe Abbildung
2-20). Während der kathodischen Anreicherung an der Elektrode bildet der Analyt mit
dem Hg ein Amalgam und wird so Bestandteil der Elektrode.
Die anschließende Bestimmung des Analyten erfolgt durch anodisches Herauslösen
des Analyten aus dem Hg. Hier kann jedes beliebige voltammetrische Verfahren
benutzt werden. Jedoch sind Methoden wie die Differenz-Puls-Voltammetrie (DPV)
und Square-Wave-Voltammetrie (SWV) bevorzugt, da sie aufgrund der Kompen-
sation des kapazitiven Ladestroms an sich eine höhere Nachweisgrenze garantieren
als einfache Methoden wie Linear-Sweep-Voltammetrie (LSV) oder cyclische
Voltammetrie (CV).

Stand der Forschung 36
2.6.2 Adsorptive Strippinganalyse (AdSV)
Für den Nachweis von Metallen, die kein Amalgam bilden, sowie für nichtmetallische
Analyten kann die AdSV, auch kathodische Strippinganalyse genannt, verwendet
werden. Dazu wird für den Nachweis von Metallionen wie z. B. Ni2+ ein
Komplexbildner hinzugegeben und das komplexierte Ion unter Anlegen eines
geeigneten Potentials an der Elektrode adsorbiert. Da der adsorbierte Analyt immer
noch oxidiert, jedoch im Komplex gebunden vorliegt, erfolgt der Stripping-Schritt im
Gegensatz zur ASV kathodisch.
Die Bedingungen, wie die Anreicherung in gerührter Lösung und die Auswahl der
voltammetrischen Methode, sind äquivalent zu denen der ASV. Lediglich die Auswahl
eines geeigneten Puffers und eines passenden Komplexbildners müssen auf den
jeweiligen Analyten abgestimmt werden (siehe Abbildung 2-21).
Für einige Analyten sind bereits eine große Anzahl an verschiedenen Reagenzien als
Komplexbildner entdeckt worden. So wird z. B. das Dimethylglyoxim (DMG), das
ursprünglich in der Gravimetrie verwendet wurde, auch zur Komplexierung von Ni2+
in der AdSV eingesetzt[126]. Bemerkenswerterweise wird DMG heutzutage immer
E- 1.0 V
t
I
Ep
Ip
kath
odis
chan
odis
ch
Ed
StrippingMen+ + Hg + ne-M(Hg)Men+ + Hg + ne- M(Hg)
Metall-Anreicherung
gerührt Nicht gerührt Nicht gerührt
E- 1.0 V
t
I
Ep
Ip
kath
odis
chan
odis
ch
Ed
StrippingMen+ + Hg + ne-M(Hg)Men+ + Hg + ne- M(Hg)
Metall-Anreicherung
gerührt Nicht gerührt Nicht gerührt
Abbildung 2-20: Skizze und schematischer Strom-Spannungs-Verlauf bei einer anodischen Strippinganalyse

Stand der Forschung 37
noch häufig benutzt[118, 127], obwohl die Verwendung von Komplexbildnern wie z. B.
Cyclohexan-1,2-dion (Nioxim) zu einer Verdoppelung der Sensitivität führt[128].
2.6.3 Elektrodentypen für die voltammetrische Strippinganalyse
Im Laufe der Jahre sind viele verschiedene Elektrodentypen wie Kohlepaste-
Elektroden, die den Komplexbildner für die AdSV schon enthalten[129, 130], poly-
kristalline Goldfilmelektroden[131] und Dentalamalgamelektroden[132, 133] entwickelt
worden. Durchgesetzt haben sich jedoch nur drei Elektrodentypen, die im Folgenden
detaillierter beschrieben werden sollen.
Die typischen Arbeitselektroden für die Strippinganalyse sind Quecksilberelektroden.
Basierend auf den Erfahrungen aus der Polarographie wurden zunächst Quecksilber-
tropfenelektroden[126] (HMDE, hanging mercury drop electrode) in der ASV
verwendet. HMDEs bestehen aus einer Glaskapillare mit einem Quecksilberreservoir
an deren Ende ein Hg-Tropfen hängt, der als erneuerbare Elektrode dient.
Quecksilber besitzt eine hohe Überspannung gegen Wasserstoff und bietet ein
breites für die ASV nutzbares Potentialfenster. Der entscheidende Vorteil bei der
HMDE liegt darin, dass durch Abschlagen des Hg-Tropfens nach einer Messung,
durch Bildung eines neuen Tropfens an der Kapillare für jede Messung eine frische,
unverschmutzte Elektrodenoberfläche vorliegt.
Trotz der offensichtlichen Vorteile dieses Elektrodentyps musste über neue
Elektrodendesigns aufgrund gesetzlicher Vorgaben in Bezug auf die Giftigkeit von
Quecksilber nachgedacht werden. Zudem war es für einige Messanordnungen (siehe
Abschnitt 2.7) unpraktisch eine HMDE zu benutzen. Für einige Anwendungen war
Britton-Robinson-Puffer (pH 6,5 – 7,2)
2,5-Dimercapto-1,3,4-thiadiazolAl (III)
HEPES Puffer (pH 7,7)8-HydroxyquinolinCu (II)
Acetat-EDTA-Puffer (pH 4,6)4-(2-Pyridylazo) ResorcinFe (II)
Ammoniakpuffer (pH 9,2)DimethylglyoximNi (II)
Ammoniakpuffer (pH 9,2)DimethylglyoximCo (II)
ElektrolytReagenzElement
Britton-Robinson-Puffer (pH 6,5 – 7,2)
2,5-Dimercapto-1,3,4-thiadiazolAl (III)
HEPES Puffer (pH 7,7)8-HydroxyquinolinCu (II)
Acetat-EDTA-Puffer (pH 4,6)4-(2-Pyridylazo) ResorcinFe (II)
Ammoniakpuffer (pH 9,2)DimethylglyoximNi (II)
Ammoniakpuffer (pH 9,2)DimethylglyoximCo (II)
ElektrolytReagenzElement
Abbildung 2-21: Auszug aus einer Tabelle[121] mit unterschiedlichen Analyten, den für die AdSV geeigneten Komplexbildnern und Puffersystemen

Stand der Forschung 38
eine höhere mechanische Stabilität der Elektrode wie bei Feststoffelektroden
erwünscht[134]. Daher wurden zunächst Quecksilberfilmelektroden (MFE, mercury film
electrode) entwickelt worden, um nicht auf die Vorteile des Quecksilbers verzichten
zu müssen. Sie basieren auf einer Substratelektrode, die kein Amalgam bildet, wie
Iridium[135, 136, 137], Glaskohlenstoff[134, 138] oder Carbonfasern[139], auf der das Hg
elektrochemisch abgeschieden werden kann. Dies ermöglicht eine neue Methode in
der ASV, bei der das Hg zusammen mit dem Analyten während der Anreicherung,
also in situ, abgeschieden und anschließend beim Stripping-Prozess wieder
aufgelöst wird.
Andere Ansätze verfolgten aufgrund der Toxizität des Hg den vollkommenen Verzicht
auf Quecksilber, ohne dabei zu große Einbußen in der Funktionalität hinnehmen zu
müssen. Eine ernsthafte Konkurrenz zur Quecksilberfilmelektrode stellt die
Wismutfilmelektrode (BiFE, bismuth film electrode) dar. Dieser Elektrodentyp wurde
erstmals im Jahr 2000 von Wang et al[140] für die ASV verwendet. BiFEs bieten
annähernd die gleiche Wasserstoffüberspannung und nur ein geringfügig kleineres
nutzbares Potentialfenster als die quecksilberbasierenden Elektroden. Darüber
hinaus ist sie mechanisch stabiler.
Sie weisen eine vergleichbare Sensitivität, Reproduzierbarkeit und Peakseparation
sowie einen ebenso geringen Grundstrom wie MFEs auf. Die Elektrodenherstellung
gestaltet sich ähnlich einfach. Bei den BiFEs wird Wismut elektrolytisch auf der
Arbeitselektrode abgeschieden[140, 141]. Die Eignung von BiFEs für die AdSV wurde
durch die Arbeiten von Bobrowski et al[141, 142] und der Gruppe von Economou[143]
bestätigt.
2.7 Voltammetrische Strippinganalyse im SIA
Das Einsatzgebiet der in den letzten Kapiteln ausführlich diskutierten Stripping-
analyse umfasst aufgrund ihrer Flexibilität die Bestimmung von Schwermetallen in
Bodenproben[127, 144, 145], in Blutproben[139], Leitungs- [123, 145], See-[128, 146] und
Industrieabwässern[147], von Fungiziden[148], von pharmazeutischen Produkten und
anderen Biomolekülen[121]. Viele dieser Anwendungsgebiete erfordern Routine-
analysen z. B. zur Überwachung der Abwässer oder zur Qualitätssicherung von
Produkten in Fertigungsabläufen.
Im Kapitel 2.5.2 wurde die gute Eignung des SIA-Systems für die automatisierte
Routineanalyse vorgestellt. Eine Kopplung der FIA oder SIA mit der SV war in den

Stand der Forschung 39
letzten Jahren ein Forschungsziel mehrer Arbeitsgruppen und führte zu einem
kostengünstigen, sehr flexiblen, automatisierten System zur Spurenanalyse, haupt-
sächlich im Bereich der Schwermetalle.
Im Jahr 2000 wurde von unserer AG bereits eine Arbeit über die Kopplung der ASV
mit einem SIA-System, des sogenannten SISA-Systems (Sequential-Injection-
Stripping-Analysis), veröffentlicht[149]. Die Studie basierte auf dem OLGA-System
(Kapitel 2.5.3) und einer chemisch modifizierten GC-Elektrode, welche selektiv für
Hg2+ ist. Die zur Detektion genutzte DPV wurde von einem externen Potentiostaten
ausgeführt, der durch ein von der OLGA gesendetes Triggersignal die Messung
startete. Aufgrund des Elektrodendesigns ist dieses System jedoch auf einen
Analyten limitiert.
Kürzlich veröffentlichten Mikkelsen et al[147] eine Studie über eine Art FIA-System,
welches die DPASV (differential pulse anodic stripping voltammetry) als Detektions-
methode für unterschiedliche Schwermetalle in Abwässern der Nickelverhüttung
nutzt. Die Stabilität der quecksilberfreien Silber-Arbeitselektroden erreichte mehrere
Tage und die Signalqualität stand der einer HMDE in nichts nach.
Auch eine Adaption der ASV an SIA-Systeme gelang in den letzten Jahren
Suteerapataranon et al[116]. In der Veröffentlichung wird ein SIA-System zur
Bestimmung mehrerer Schwermetalle, welches jedoch auf eine MFE als Arbeits-
elektrode zurückgreift, vorgestellt. Die Technik der SIA erlaubte in diesem Fall sogar
eine automatisierte Regenerierung des Quecksilberfilms. Durch die Durchflusszelle
konnte eine Hg2+-Lösung gepumpt werden, aus der eine elektrochemische
Beschichtung der Kohlenstoffelektrode möglich war. Jedoch birgt diese Methode
wieder die teure Entsorgung der Abwässer in sich, da das Hg2+ stark toxisch ist.
Eine wichtige Komponente in der Kopplung der SV an die FIA- und SIA-Systeme ist
das Design einer geeigneten Messzelle. Es wurden neben Dünnschicht-
Durchflusszellen auch Wall-Jet-Zellen[119, 150] verwendet. Ihr Vorteil liegt in der
Sensitivitätserhöhung der Messung durch einen verbesserten hydrodynamischen
Transport des Analyten zur Elektrode[151, 152].
Theoretische Hintergründe zu diesem Phänomen können der Literatur[153, 154] ent-
nommen werden.

Problemstellung 40
3 Problemstellung
NiTi-FGLs sind aufgrund ihrer Eigenschaften, hauptsächlich wegen des
Formgedächtniseffektes und der Superelastizität, ein besonders interessantes
Material für die Industrie und die Medizin. Für beide Einsatzgebiete ist eine genaue
Kenntnis über das Korrosionsverhalten dieser Legierung von großer Bedeutung.
Vor der Verwendung von NiTi unter mechanischer und/oder thermischer Belastung
ist eine Untersuchung des Korrosionsverhaltens unerlässlich, um eine Aussage über
die Zuverlässigkeit und Lebenserwartung der Bauteile machen zu können. Zudem
können so Herstellungsverfahren und evtl. Schutzschichten entwickelt und optimiert
werden, welche die Korrosionsstabilität erhöhen und somit ein qualitativ
hochwertiges Werkstück garantieren, welches eine hohe Funktionsdauer aufweist.
Für medizinische Anwendung ist es wichtig sicherzustellen, dass so wenig Ni wie
möglich in den menschlichen Körper gelangt. Ni ist als allergieauslösend und krebs-
erregend bekannt und deshalb im menschlichen Körper unerwünscht[155, 156]. Folglich
ist eine Methode gefragt, mit der Ni-Ausblutungen aus z.B. Knochenimplantaten oder
Stents, die aus Nickel-haltigen Legierungen, wie Nitinol, gefertigt werden, verfolgt
und quantifiziert werden können.
An die Analysenmethoden werden stark unterschiedliche Anforderungen gestellt.
Zum einen interessiert die globale Korrosion, mit der die gesamte Nickelfreisetzung
einer Probe bestimmt wird. Hierüber lässt sich die potentielle Gefährlichkeit der
Probe für den menschlichen Organismus abschätzen und die Güte einer eventuell
aufgebrachten Schutzschicht, welche die Nickelausblutung verhindern soll,
bestimmen. Darüber hinaus ist eine Lokalisierung der Korrosionsvorgänge gefordert,
um den Beginn und die Ausbreitung von Lochfraßkorrosion zu untersuchen und so
gegebenenfalls die Auslöser der Korrosion durch optimierte Herstellung oder
Bearbeitung zu unterdrücken.
Für das Untersuchen lokaler Phänomene ist das SECM prädestiniert. Wie schon
erwähnt ist es mit dieser Art der Rastermikroskopie möglich, chemische Prozesse
und Aktivitäten an Oberflächen zu visualisieren. Dies ist auf unterschiedliche Arten
möglich. Amperometrische Sonden können verwendet und somit der Einfluss der
Oberfläche auf den Mediator als Messsignal genutzt werden oder ionenselektive

Problemstellung 41
potentiometrische Sonden, die bei der Korrosion entstehende Ionenkonzentrations-
gradienten visualisieren können. Es ist ebenso möglich, die noch sehr neue Methode
des AC-SECM einzusetzen, da sie elektrochemische Unterschiede an der
Oberfläche aufzeigen kann und auf Leitfähigkeitsänderungen des Elektrolyten,
welche durch austretende Ionen hervorgerufen werden können, reagiert. Zudem
lässt sich diese Analysenmethode ohne einen zugesetzten Redoxmediator
durchführen, so dass die chemischen Vorgänge an der Oberfläche von der
Messmethode unbeeinflusst bleiben.
Um eine Aussage über die Biokompatibilität eines Nitinol-Werkstückes machen zu
können, muss man es einem Elektrolyten aussetzen und diesen über einen längeren
Zeitraum in regelmäßigen Zeitintervallen auf seinen Nickelgehalt hin prüfen. Die
AdSV hat sich, wie vorher schon ausgeführt, als ein sehr sensitives und vom Aufbau
her einfaches und kostengünstiges Werkzeug zur Nickelspurenanalyse heraus-
gestellt. Da eine Langzeituntersuchung über mehrere Stunden oder Tage eine
Vielzahl von Messungen verursacht, ist hier zusätzlich eine Automatisierung von
Nöten. Die sequentielle Injektionsanalytik bringt alle Vorrausetzungen mit, um eine
automatische Probenahme mit der AdSV zu verknüpfen. Es ist möglich mit einem
angepassten SIA-System nicht nur die Probe zu nehmen, sondern diese auch direkt
mit Puffern und Reagenzien zu mischen und zur elektrochemischen Messzelle zu
pumpen, in der die AdSV durchgeführt werden kann.
Im Rahmen dieser Arbeit soll die Adaption der SECM-Technik an die Korrosions-
problematik sowie die Möglichkeiten des SECM bei hochaufgelösten, lokalen
Korrosionsuntersuchung an NiTi-FGLs gezeigt werden. Zudem soll die Entwicklung
eines gekoppelten AdSV-SIA-Systems beschrieben und dessen Leistungsfähigkeit
zur automatisierten Langzeitdetektion von Nickelausblutungen aus Nitinol-
Werkstücken gezeigt werden.

Ergebnisse eigener Arbeit 42
4 Ergebnisse eigener Arbeit
4.1 Probenvorbereitung
Essentiell für analytische Experimente ist die Probennahme und –aufbereitung. Die
Optimierung dieser Prozedur für die in dieser Arbeit benutzten Proben ist im
folgenden detailliert dargelegt.
Die für diese Arbeit verwendeten Proben wurden nahezu ausschließlich aus dem
vom SFB-459 zur Verfügung gestellten Material hergestellt. Einige Ausnahmen, auf
die im Text gesondert hingewiesen wird, stammen von Projektpartnern aus diesem
SFB.
4.1.1 Probengeometrie
Die Form des vorhandenen Materials (Abschnitt 6.5.1) war für die geplanten
Experimente im SECM und im SIA-System nicht geeignet, da sie für die Proben-
tische bzw. Messzellen zu groß waren. Zudem sind meist definierte Oberflächen
erforderlich, um vergleichende Aussagen über die ermittelten Korrosionsraten treffen
zu können oder den Ort der potentiellen Korrosion einzugrenzen. Zu diesem Zweck
wurde das Material mittels einer Trennscheibe auf die passende Größe
zurechtgeschnitten.
Die Vorgabe der Dimension wird durch den nächsten Schritt der Probenvorbereitung
festgelegt, da die so zugeschnittenen Proben in einem Epoxidharz eingebettet
werden. Die für den Einbettungsprozess (siehe hierzu Kapitel 6.5.2) benutzte
Maschine kann Material mit einem Durchmesser unter 20 mm einbetten.
Vor der Verwendung der Proben muss der Übergang der NiTi-Fläche zum
Einbettungsmaterial mit Zweikomponentenkleber oder Sekundenkleber abgedichtet
werden, da der Mikrospalt ausreicht, um an dieser Stelle Spaltkorrosion beginnen zu
lassen. Diese setzt deutlich schneller ein als die hier interessierende Lochfraß-
korrosion auf der Fläche der Nitinolscheibe und würde die Ergebnisse verfälschen.
4.1.2 Weiterführende Probenbearbeitung
Bevor die Proben poliert werden, müssen sie noch von der Rückseite elektrisch
kontaktiert werden. Dies hat zweierlei Gründe, zum einen muss für das elektro-
lytische Polieren (siehe Abschnitt 6.5.3.2) ein Potential an die Probe angelegt werden
können und zum anderen ist es für die geplanten Experimente nötig, die Probe an

Ergebnisse eigener Arbeit 43
einen Potentiostaten anschließen zu können. Eine praktikable Lösung, ist die Probe
von der Rückseite zu kontaktieren und den Draht zur Seite hin wegzuführen, wie es
in Kapitel 6.5.2 beschrieben ist. So wird wieder eine für die SECM-Experimente
wichtige, plane Auflagefläche der Probe erhalten.
4.1.2.1 Polieren der NiTi-Proben
Das Polieren der eingebetteten Proben ist aus zwei Gründen notwendig. Zum einen
wird für eine SECM-Messung eine möglichst plane Oberfläche benötigt und zum
anderen resultiert daraus eine definierte Fläche, die für alle Experimente die gleichen
Vorraussetzungen mitbringt. Dies ist wichtig, da so die erhaltenen Ergebnisse
vergleichbar werden.
Es wurden für diese Arbeit zwei unterschiedliche Poliermethoden benutzt. Es handelt
sich dabei um das mechanische und elektrolytische Polieren.
Das mechanische Polieren wird mit Hilfe einer Poliermaschine auf einem rotierenden
Polierfilz durchgeführt (siehe dazu Abschnitt 6.5.3.1). Hierbei ist darauf zu achten,
dass die Polierscheiben nicht für andere, etwa eisenhaltige Materialien benutzt
wurde, da es sonst zu einem Materialeintrag in die NiTi-Oberfläche kommen kann.
Dieser würde das Korrosionsverhalten beeinflussen.
Ein beobachteter Nebeneffekt bei der mechanischen Polierprozedur ist das Heraus-
treten einzelner Körner des Materials. Dies deutet darauf hin, dass die Körner eine
unterschiedliche Härte aufweisen und daraus ein ungleichmäßiger Materialabtrag
Abbildung 4-1: AFM-Abbildung einer mechanisch polierten NiTi-Oberfläche

Ergebnisse eigener Arbeit 44
resultiert. Besonders deutlich treten die Korngrenzen hervor, die mittels AFM unter-
sucht wurden (Abbildung 4-1) und eine Tiefe von ca. 21 nm aufwiesen.
Das elektrolytische Polieren (vgl. Abschnitt 6.5.3.2) basiert auf dem elektro-
chemischen Auflösen des Materials, wobei Elektrolyt, Potential und Dauer des
Auflösungsprozesses darauf optimiert sind, die aus der Fläche des Metalls
herausragenden Spitzen aufzulösen und so die Oberfläche zu glätten.
Die resultierende Restrauhigkeit der Oberfläche ist größer als bei dem mechanischen
Poliervorgang, jedoch bringt diese Methode den Vorteil des geringeren Zeitaufwands
mit sich. Zudem führt das elektrolytische Polieren zu einer besseren Passivierung der
Oberfläche (vgl. Kapitel 2.1.2). Möglicherweise aktiviert die Polierprozedur die Ober-
fläche, so dass eine umfangreichere Oxidation stattfinden kann.
4.1.2.2 Proben für die SECM
Wie aus Abschnitt 4.3 hervorgeht, findet ein
SECM-Experiment in einer kleinen elektro-
chemischen Messzelle statt. Die Proben-
geometrie lässt nicht zu, diese in eine vor-
handene Messzelle einzusetzen. Dieses
Problem lässt sich leicht lösen, indem die Probe
selbst Teil der Messzelle wird. Zu diesem Zweck
wird ein ca. 1 cm langes Stück einer Photo-
meterküvette abgesägt und auf die Probe
geklebt. Die entstandene Messzelle (siehe
Abbildung 4-2) bietet genügend Volumen für ausreichend Elektrolyt und die drei
erforderlichen Elektroden.
4.1.3 Probencharakterisierung mittels XPS-Analyse
Die Reinheit sowie die Zusammensetzung der hergestellten Proben wurden mittels
XPS analysiert.
Um aus den erhaltenen Messdaten die Spektren darstellen zu können, muss die
gemessene Energie noch um die Anregungsenergie reduziert werden, da nur die
Bindungsenergie von Interesse ist. Der Zusammenhang zwischen kinetischer und
Bindungsenergie wird durch folgende Gleichung wiedergegeben:
EB = h*νννν - Ekin - ΦΦΦΦS
Abbildung 4-2: Fotografische Ab-bildung der aufgeklebten SECM-Messzelle.
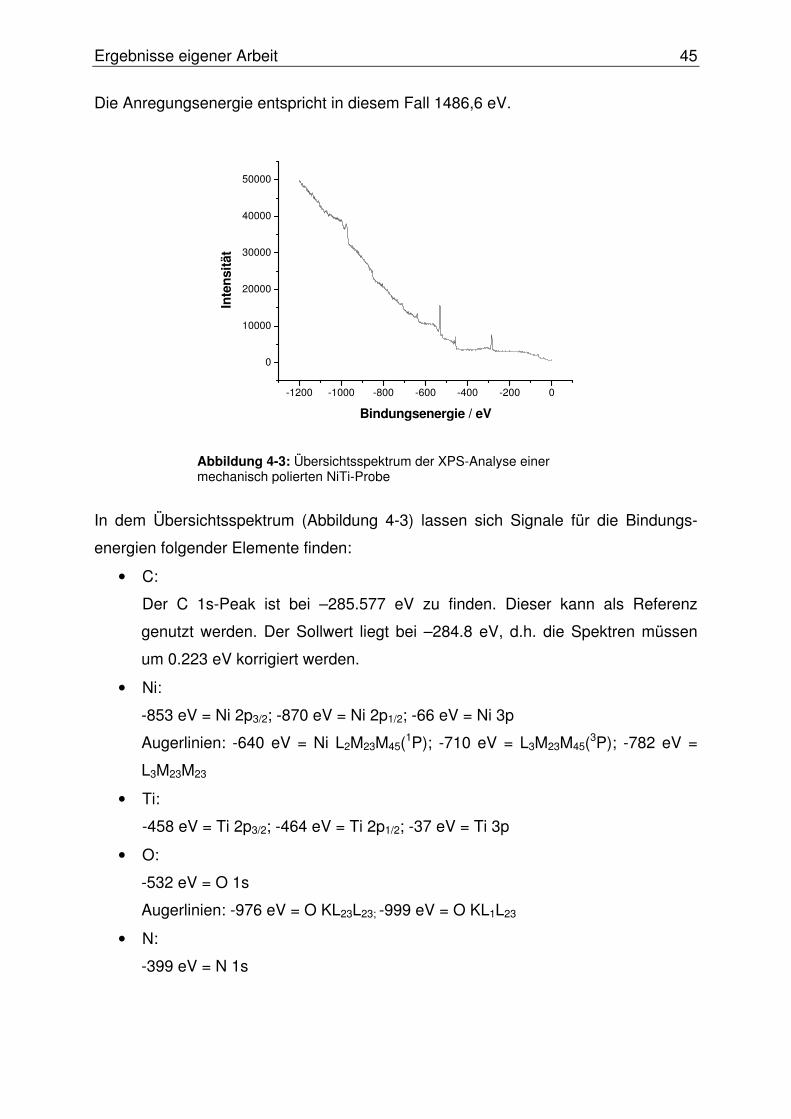
Ergebnisse eigener Arbeit 45
Die Anregungsenergie entspricht in diesem Fall 1486,6 eV.
In dem Übersichtsspektrum (Abbildung 4-3) lassen sich Signale für die Bindungs-
energien folgender Elemente finden:
• C:
Der C 1s-Peak ist bei –285.577 eV zu finden. Dieser kann als Referenz
genutzt werden. Der Sollwert liegt bei –284.8 eV, d.h. die Spektren müssen
um 0.223 eV korrigiert werden.
• Ni:
-853 eV = Ni 2p3/2; -870 eV = Ni 2p1/2; -66 eV = Ni 3p
Augerlinien: -640 eV = Ni L2M23M45(1P); -710 eV = L3M23M45(
3P); -782 eV =
L3M23M23
• Ti:
-458 eV = Ti 2p3/2; -464 eV = Ti 2p1/2; -37 eV = Ti 3p
• O:
-532 eV = O 1s
Augerlinien: -976 eV = O KL23L23; -999 eV = O KL1L23
• N:
-399 eV = N 1s
-1200 -1000 -800 -600 -400 -200 0
0
10000
20000
30000
40000
50000
Inte
nsi
tät
Bindungsenergie / eV
Abbildung 4-3: Übersichtsspektrum der XPS-Analyse einer mechanisch polierten NiTi-Probe

Ergebnisse eigener Arbeit 46
Nachdem die Elemente identifiziert worden sind, wurden einzelne Spektren in der für
die jeweiligen Elemente wichtigen Energieregionen mit höherer Auflösung aufgenom-
men.
-285.6 eV = C 1s
-289 eV = C 1s (in z.B.
Carboxyl oder Carbonat)
A (C 1s) = 3837.49953
Der Kohlenstoff ist im XPS-
Spektrum immer vorhanden
und kann auch hier als Ad-
sorbat angesehen werden.
Ein Hinweis auf Carbide (-
281 bis –283 eV) ist hier
nicht zu finden.
-454 eV = Ti 2p3/2 (element.
Ti)
-459 eV = Ti 2p3/2 (in TiO2)
-465 eV = Ti 2p1/2 (in TiO2)
A (Ti 2p) = 12857.7681
Da Titan an Luft innerhalb
kürzester Zeit oxidiert, liegt
hier fast ausschließlich TiO2
an der Oberfläche vor. Dies
ist an den, vom Element-
peak aus, um ca. 5 eV
nach links verschobenen
Bindungsenergien erkennbar.
-298 -296 -294 -292 -290 -288 -286 -284 -282 -280 -278 -276400
600
800
1000
1200
1400
1600
1800
2000
2200
Inte
nsi
tät
Bindungsenergie / eV
Abbildung 4-4: XPS-Spektrum für C 1s
-472 -470 -468 -466 -464 -462 -460 -458 -456 -454 -452 -450
3000
4000
5000
6000
7000
8000
Inte
nsi
tät
Bindungsenergie / eV
Abbildung 4-5: XPS-Spektrum für Ti 2p
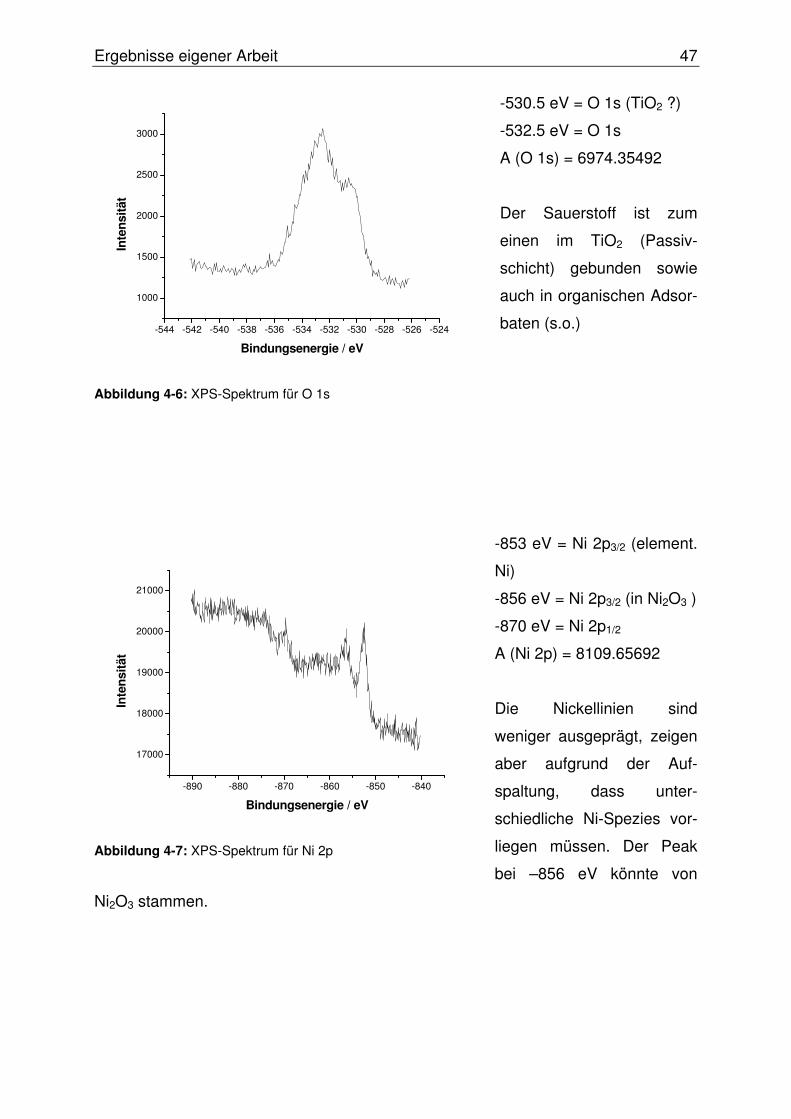
Ergebnisse eigener Arbeit 47
-530.5 eV = O 1s (TiO2 ?)
-532.5 eV = O 1s
A (O 1s) = 6974.35492
Der Sauerstoff ist zum
einen im TiO2 (Passiv-
schicht) gebunden sowie
auch in organischen Adsor-
baten (s.o.)
-853 eV = Ni 2p3/2 (element.
Ni)
-856 eV = Ni 2p3/2 (in Ni2O3 )
-870 eV = Ni 2p1/2
A (Ni 2p) = 8109.65692
Die Nickellinien sind
weniger ausgeprägt, zeigen
aber aufgrund der Auf-
spaltung, dass unter-
schiedliche Ni-Spezies vor-
liegen müssen. Der Peak
bei –856 eV könnte von
Ni2O3 stammen.
-544 -542 -540 -538 -536 -534 -532 -530 -528 -526 -524
1000
1500
2000
2500
3000In
ten
sitä
t
Bindungsenergie / eV
Abbildung 4-6: XPS-Spektrum für O 1s
-890 -880 -870 -860 -850 -840
17000
18000
19000
20000
21000
Inte
nsi
tät
Bindungsenergie / eV
Abbildung 4-7: XPS-Spektrum für Ni 2p
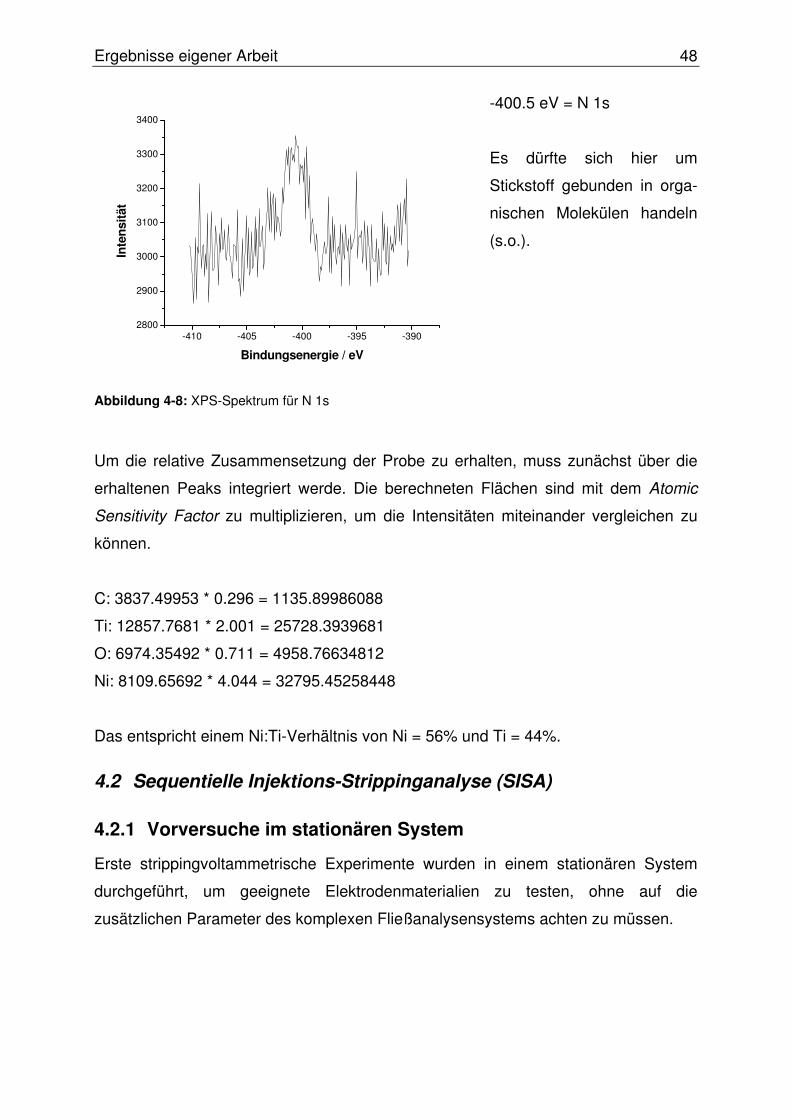
Ergebnisse eigener Arbeit 48
-400.5 eV = N 1s
Es dürfte sich hier um
Stickstoff gebunden in orga-
nischen Molekülen handeln
(s.o.).
Um die relative Zusammensetzung der Probe zu erhalten, muss zunächst über die
erhaltenen Peaks integriert werde. Die berechneten Flächen sind mit dem Atomic
Sensitivity Factor zu multiplizieren, um die Intensitäten miteinander vergleichen zu
können.
C: 3837.49953 * 0.296 = 1135.89986088
Ti: 12857.7681 * 2.001 = 25728.3939681
O: 6974.35492 * 0.711 = 4958.76634812
Ni: 8109.65692 * 4.044 = 32795.45258448
Das entspricht einem Ni:Ti-Verhältnis von Ni = 56% und Ti = 44%.
4.2 Sequentielle Injektions-Strippinganalyse (SISA)
4.2.1 Vorversuche im stationären System
Erste strippingvoltammetrische Experimente wurden in einem stationären System
durchgeführt, um geeignete Elektrodenmaterialien zu testen, ohne auf die
zusätzlichen Parameter des komplexen Fließanalysensystems achten zu müssen.
-410 -405 -400 -395 -3902800
2900
3000
3100
3200
3300
3400In
ten
sitä
t
Bindungsenergie / eV
Abbildung 4-8: XPS-Spektrum für N 1s

Ergebnisse eigener Arbeit 49
4.2.1.1 Herstellung und Test von Quecksilberfilmelektroden (MFE)
Wie in Abschnitt 2.6.3 erwähnt, gibt es verschiedene Elektrodentypen, die für die
Strippinganalyse geeignet sind. Jedoch ist nicht jede dieser Elektroden auch für den
Einsatz in einem SIA-System zu favorisieren.
Der klassische Elektrodentyp für die Strippinganalyse ist die HMDE (siehe Abschnitt
2.6.3). Sie bringt den Vorteil mit sich, dass für jede Messung ein frischer
Quecksilbertropfen und somit eine saubere Elektrodenoberfläche zur Verfügung
steht. Da jedoch nach jeder Messung das benutzte Quecksilber in die Lösung
abgegeben wird, wäre es schwierig, den entstehenden Quecksilberabfall
automatisiert aus einem SIA-System abzuführen.
Am besten geeignet für ein fließendes System scheinen dementsprechend Film- oder
Feststoffelektroden zu sein. Einige davon wurden im Stand der Forschung schon
angesprochen. Sie bieten zwar nicht für jede Messung eine frische Oberfläche, dafür
entfällt das Problem der Quecksilberabführung. Zudem wäre es zumindest für die
Filmelektroden möglich, sie in situ, also bei der Anreicherung des Analyten[140], oder
vor einem Messzyklus zu erstellen.
Die MFE ist nach der HMDE eine der am häufigsten genutzten Elektroden zur
Strippinganalyse. Sie bietet den gleichen Vorteil des großen nutzbaren Potential-
fensters und der hohen Wasserstoffüberspannung, kann aber nicht die leicht
erneuerbare Elektrodenoberfläche der HMDE bieten.
Die Beschichtung der Elektroden erfolgt gemäß einer Veröffentlichung von Tercier et
al[157]. Die polierte und gesäuberte Elektrode wird durch Anlegen eines konstanten
Potentials von –400 mV für eine der Elektrodenfläche angepasste Zeit (siehe
folgende Abschnitte) mit Quecksilber beschichtet. Die Lösung, aus der das Hg
abgeschieden wird, besteht aus einer 5 mM HgAc-Lösung in 0,1 M HClO4, welche
zuvor 5 Minuten lang mit Argon gespült werden muss, um störenden Sauerstoff aus
der Lösung zu entfernen.
4.2.1.1.1 MFE auf Basis einer Iridium-Scheibenelektrode
Iridiumscheibenelektroden sind aufgrund ihrer vernachlässigbaren Amalgambildung
ein ideales Substrat für eine MFE[157, 135]. Hier wurde eine nach der Anleitung in
Abschnitt 6.2.1 hergestellte Ir-Elektrode mit 125 µm Durchmesser verwendet.
Die Abscheidung des Quecksilbers erfolgt nach der oben erwähnten Methode für
eine Dauer von 600 s. In Abbildung 4-9 ist der Verlauf einer Abscheidung mittels
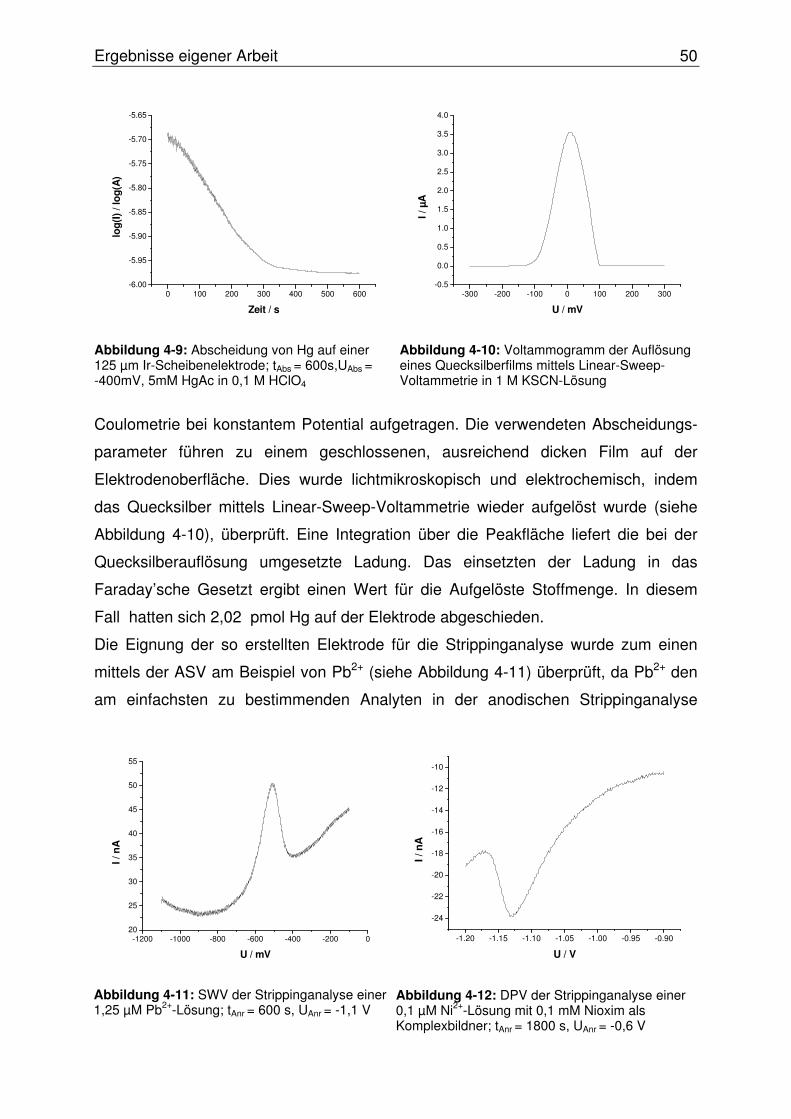
Ergebnisse eigener Arbeit 50
Coulometrie bei konstantem Potential aufgetragen. Die verwendeten Abscheidungs-
parameter führen zu einem geschlossenen, ausreichend dicken Film auf der
Elektrodenoberfläche. Dies wurde lichtmikroskopisch und elektrochemisch, indem
das Quecksilber mittels Linear-Sweep-Voltammetrie wieder aufgelöst wurde (siehe
Abbildung 4-10), überprüft. Eine Integration über die Peakfläche liefert die bei der
Quecksilberauflösung umgesetzte Ladung. Das einsetzten der Ladung in das
Faraday’sche Gesetzt ergibt einen Wert für die Aufgelöste Stoffmenge. In diesem
Fall hatten sich 2,02 pmol Hg auf der Elektrode abgeschieden.
Die Eignung der so erstellten Elektrode für die Strippinganalyse wurde zum einen
mittels der ASV am Beispiel von Pb2+ (siehe Abbildung 4-11) überprüft, da Pb2+ den
am einfachsten zu bestimmenden Analyten in der anodischen Strippinganalyse
0 100 200 300 400 500 600-6.00
-5.95
-5.90
-5.85
-5.80
-5.75
-5.70
-5.65lo
g(I
) / l
og
(A)
Zeit / s
Abbildung 4-9: Abscheidung von Hg auf einer 125 µm Ir-Scheibenelektrode; tAbs = 600s,UAbs = -400mV, 5mM HgAc in 0,1 M HClO4
-300 -200 -100 0 100 200 300-0.5
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
4.0
I / µ
A
U / mV
Abbildung 4-10: Voltammogramm der Auflösung eines Quecksilberfilms mittels Linear-Sweep-Voltammetrie in 1 M KSCN-Lösung
-1200 -1000 -800 -600 -400 -200 020
25
30
35
40
45
50
55
I / n
A
U / mV
Abbildung 4-11: SWV der Strippinganalyse einer 1,25 µM Pb2+-Lösung; tAnr = 600 s, UAnr = -1,1 V
-1.20 -1.15 -1.10 -1.05 -1.00 -0.95 -0.90
-24
-22
-20
-18
-16
-14
-12
-10
I / n
A
U / V
Abbildung 4-12: DPV der Strippinganalyse einer 0,1 µM Ni2+-Lösung mit 0,1 mM Nioxim als Komplexbildner; tAnr = 1800 s, UAnr = -0,6 V
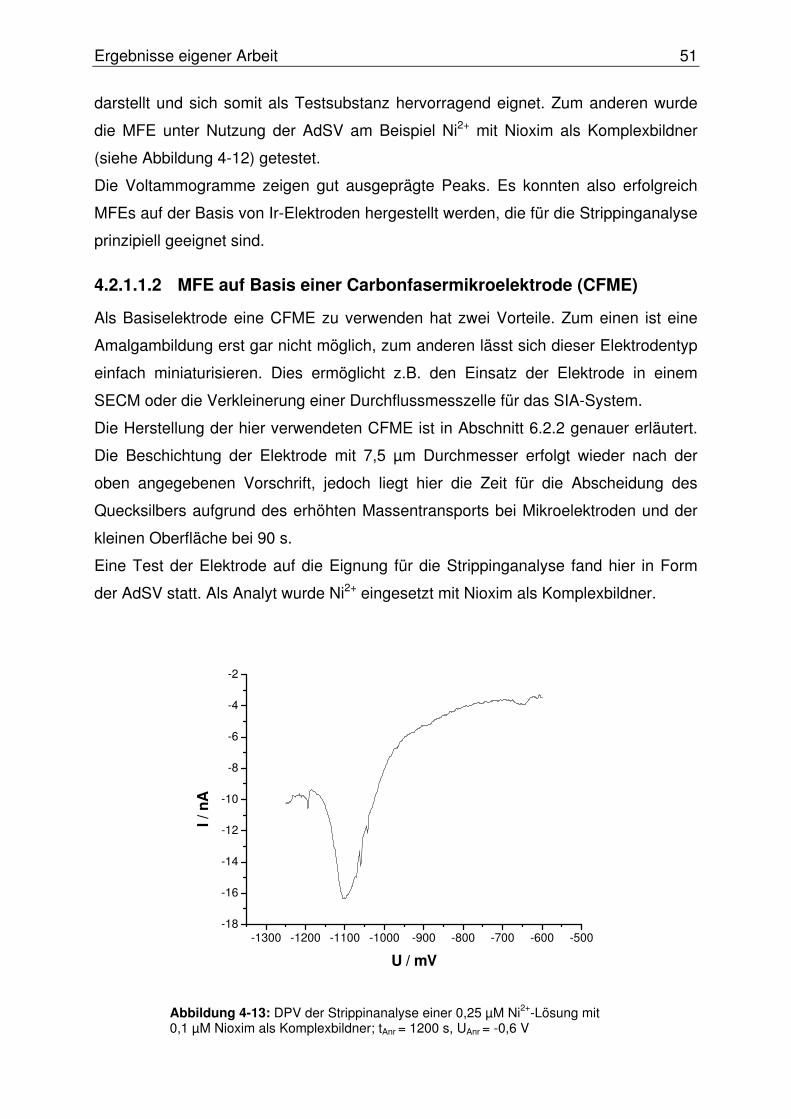
Ergebnisse eigener Arbeit 51
darstellt und sich somit als Testsubstanz hervorragend eignet. Zum anderen wurde
die MFE unter Nutzung der AdSV am Beispiel Ni2+ mit Nioxim als Komplexbildner
(siehe Abbildung 4-12) getestet.
Die Voltammogramme zeigen gut ausgeprägte Peaks. Es konnten also erfolgreich
MFEs auf der Basis von Ir-Elektroden hergestellt werden, die für die Strippinganalyse
prinzipiell geeignet sind.
4.2.1.1.2 MFE auf Basis einer Carbonfasermikroelektrode (CFME)
Als Basiselektrode eine CFME zu verwenden hat zwei Vorteile. Zum einen ist eine
Amalgambildung erst gar nicht möglich, zum anderen lässt sich dieser Elektrodentyp
einfach miniaturisieren. Dies ermöglicht z.B. den Einsatz der Elektrode in einem
SECM oder die Verkleinerung einer Durchflussmesszelle für das SIA-System.
Die Herstellung der hier verwendeten CFME ist in Abschnitt 6.2.2 genauer erläutert.
Die Beschichtung der Elektrode mit 7,5 µm Durchmesser erfolgt wieder nach der
oben angegebenen Vorschrift, jedoch liegt hier die Zeit für die Abscheidung des
Quecksilbers aufgrund des erhöhten Massentransports bei Mikroelektroden und der
kleinen Oberfläche bei 90 s.
Eine Test der Elektrode auf die Eignung für die Strippinganalyse fand hier in Form
der AdSV statt. Als Analyt wurde Ni2+ eingesetzt mit Nioxim als Komplexbildner.
-1300 -1200 -1100 -1000 -900 -800 -700 -600 -500-18
-16
-14
-12
-10
-8
-6
-4
-2
I / n
A
U / mV
Abbildung 4-13: DPV der Strippinanalyse einer 0,25 µM Ni2+-Lösung mit 0,1 µM Nioxim als Komplexbildner; tAnr = 1200 s, UAnr = -0,6 V

Ergebnisse eigener Arbeit 52
Das Voltammogramm in Abbildung 4-13 zeigt einen deutlichen Ni2+-Peak bei –1,1 V.
Somit ist dieser Elektrodentyp für die Strippinganalyse prinzipiell verwendbar.
4.2.2 Adaption der AdSV an das OLGA-System
Wie in Abschnitt 3 erwähnt, ist ein Ziel dieser Arbeit ein automatisiertes,
elektrochemisches Messsystem zu entwickeln, welches erlaubt Nickelkonzentra-
tionen kontinuierlich und mit hohem Probendurchsatz zu bestimmen. Die Aufzeich-
nung der Konzentration soll während der Korrosion eines NiTi-Werkstücks in einem
Langzeitexperiment in Echtzeit erfolgen.
Zu diesem Zweck soll auf Basis eines in der Arbeitsgruppe entwickelten SIA-Geräts
namens OLGA (online general analyzer), welches bereits erfolgreich für die
Bestimmung von Hg2+ mittels ASV eingesetzt werden konnte[149], eine Kopplung der
adsorptiven Strippinganalyse mit der sequentiellen Injektionsanalyse stattfinden.
Für die erfolgreiche Durchführung der Strippinganalyse wird eine elektrochemische
Puffer
Isopropanol
Pumpe
Abfall
Standard
Mischzelle
Probe
Durchflusszelle
PC für die Ansteuerung der Hardware und Datenerfassung
Funktionsgenerator+ Potentiostat
Magnetquetschventile Manifold
Puffer
Isopropanol
Pumpe
Abfall
Standard
Mischzelle
Probe
Durchflusszelle
PC für die Ansteuerung der Hardware und Datenerfassung
Funktionsgenerator+ Potentiostat
Magnetquetschventile Manifold
Abbildung 4-14: schematische Abbildung des OLGA-Systems für die Pb2+-Bestimmung mittels ASV

Ergebnisse eigener Arbeit 53
Messzelle, Puffer, in dem die Messung durchgeführt werden kann und ggf. einen
Komplexbildner, der mit dem Analyt einen adsorbierbaren Komplex bildet, benötigt.
Apparativ sind ein Potentiostat sowie eine Rührvorrichtung, meist Magnetrührer,
notwendig, da die Anreicherung des Analyten an der Elektrode unter Konvektion
stattfinden soll.
Der flexible Aufbau des OLGA-Systems ermöglicht die Durchführung der Stripping-
analyse prinzipiell nach Modifikationen und Erweiterungen am Gerät und an der
Steuersoftware. Die über einen Verteiler und mit Magnetquetschventilen
angeschlossenen Gefäße (siehe Abbildung 4-14) können den nötigen Puffer,
Standardlösung, etc. aufnehmen.
Somit kann automatisch eine Probe genommen und zur Ver-
dünnung und pH-Einstellung mit Puffer in die Mischkammer
gepumpt werden. Die Mischzelle verfügt, wie in Abbildung 4-15
schematisch dargestellt, über einen Ein- bzw. Auslass, der von
der Oberseite mittels eines Schlauchs zum konisch zulaufenden
Boden der Mischkammer führt. Dies gewährleistet eine gute
Durchmischung der Lösung allein aufgrund der durch den
Pumpvorgang entstehenden Verwirbelungen innerhalb der
Mischkammer. Dies wurde durch Ersetzen der Probenlösung
durch einen Farbstoff innerhalb eines Testzyklus optisch
überprüft. Aus der Mischzelle kann die Lösung zur Messzelle
(siehe dazu die folgenden Kapitel) gepumpt werden, in welcher dann die
Anreicherung in bewegter Lösung, wie in der Strippingvoltammetrie üblich, stattfindet.
Im Anschluß kann nach Beenden des Pumpvorgangs in ruhender Lösung die
Strippingvoltammetrie erfolgen.
Zur Bestimmung der Konzentration eines Analyten ist eine Kalibrierung des Systems
unerlässlich. Meist erfolgt diese mit einer Standardlösung bekannter Konzentration.
In dem hier entwickelten System entfällt eine vorhergehende Kalibrierung, da mittels
des Standardadditionsverfahrens eine interne Referenz gegeben ist. Zu diesem
Zweck wird das Gefäß „Standardlösung“ (siehe Abbildung 4-14) benötigt. Die
Standardaddition kann prinzipiell aus beliebig vielen Additionsschritten bestehen
(siehe dazu auch Kapitel 4.2.2.2.2). Es besteht jedoch eine Limitierung des
Gesamtvolumens der Mischung aus Standardlösung, Probe und Puffer durch das
Volumen der Mischkammer, das somit auch die Anzahl der Additionen einschränkt.
Ein/Auslass Ein/Auslass
Abbildung 4-15: Skizze der im OLGA-System ver-wendeten Misch-zelle

Ergebnisse eigener Arbeit 54
Die Standardaddition bietet in diesem Fall noch einen zusätzlichen Vorteil. Da das
Gerät auf Messungen über mehrere Stunden oder auch Tage ausgelegt ist, die
Elektrode aber über diesen Zeitraum möglicherweise an Aktivität verliert, wird dieser
systematische Fehler durch die interne Kalibrierung automatisch berücksichtigt und
eliminiert.
4.2.2.1 Integration eines Potentiostaten
Der im OLGA-System integrierte Potentiostat lässt nur amperometrische Messungen
zu und verfügt über keine weiteren Schnittstellen, um die Funktionalität zu erweitern.
Aufgrund dieser Tatsache musste das SIA-Gerät durch eine externe Lösung um die
erforderlichen voltammetrischen Messmethoden, wie DPV, ergänzt werden. Die
Kopplung eines EG&G PAR Model 264a Potentiostaten mit dem OLGA-System, wie
es für die frühere Hg2+-Bestimmung benutzt wurde, ließ keine elektronische Daten-
aufnahme zu. Die Voltammogramme wurden auf einem Schreiber ausgegeben.
Diese Technik ist für Langzeitexperimente mit hohem Datenaufkommen unzu-
reichend, da sie mit einer langwierigen manuellen Auswertung verknüpft ist.
Die erste apparative Lösung bestand aus der Integration eines Potentiostaten der
Firma Jaissle, welcher mit einem HP-Funktionsgenerator gekoppelt wurde, um das
komplexe Potential-Zeit-Profil aufzuprägen. Eine DPV-Potentialrampe ist in der
Abbildung 4-16 schematisch dargestellt. Es handelt sich dabei um eine lineare
Strommessung
t
E
StrommessungStrommessung
t
E
Abbildung 4-16: schematische Darstellung einer DPV-Potentialrampe

Ergebnisse eigener Arbeit 55
Potentialrampe, die mit einer Rechteckspannung überlagert wird. Die Strommessung
erfolgt jeweils direkt vor einem Potentialpuls und am Ende eines Pulses. Die
Differenz der beiden Stromwerte ergibt einen Messpunkt. Die anzulegende Potential-
rampe wurde demzufolge durch den programmierbaren Funktionsgenerator über den
externen Anschluss des Potentiostaten übermittelt. Die Berechnung des Voltammo-
gramms wurde von der selbst programmierten Software übernommen.
Die Verbindung zwischen dem Steuer-PC und dem Funktionsgenerator wurde über
eine RS232 Schnittstelle hergestellt. Zudem wurde eine zweite Verbindung zum
OLGA-System benötigt, über die ein Start der Messung per Triggersignal ermöglicht
wurde. Die vom Potentiostaten gelieferten Stromwerte konnten über eine AD-
Wandlerkarte eingelesen werden.
4.2.2.2 Softwareentwicklung
4.2.2.2.1 DPV-Software
Die entwickelte DPV-Software besteht aus zwei Hauptprogrammteilen. Die Software
erlaubt die Programmierung des Funktionsgenerators mit der entsprechenden
Rampenfunktion, sowie die Berechnung des Voltammogramms aus den vom
Potentiostaten gelieferten Stromwerten.
Die Parameter der DPV-Rampenfunktion, wie Pulshöhe, Pulsbreite, Pulsfrequenz,
etc. können über eine Eingabemaske an das Programm übergeben werden. Die
Software berechnet daraus einzelne Datenpunkte die den Eckpunkten der Rampe
(vgl. Abbildung 2-1) entsprechen und übermittelt diese an den Funktionsgenerator.
Die vom Potentiostaten gelieferten Stromwerte werden von der Software unter
Berücksichtigung der Zeitskala aufgezeichnet und auf Stufen überprüft, die durch die
Potentialpulse hervorgerufen worden sind. Nach Erkennung der Stufen erfolgt die
Differenzbildung. Die Zeitskala kann mit Hilfe der Spannungsvorschubsgeschwindig-
keit in das zugehörige Potential umgerechnet werden und erlaubt somit die
Zuordnung der berechneten Stromdifferenzen zur zugehörigen Spannung.
4.2.2.2.2 OLGA-Software
Die Pumpe des OLGA-Systems wird über ein RS232 Interface von einem PC
angesteuert. Die Ventilsteuerung des SIA-Gerätes benötigt zusätzlich einen
Parallelanschluss.

Ergebnisse eigener Arbeit 56
Für das Gerät stand eine Steuersoftware, geschrieben in Visual Basic 6, zur Ver-
fügung. Diese stammte aus früheren Anwendungen in der Fermentationsanalyse und
musste für die automatisierte Strippinganalyse geändert, bzw. erweitert werden.
Hier sollen nur die wichtigsten Aspekte der Softwareentwicklung aufgegriffen werden,
da die Erläuterung des gesamten Programms an dieser Stelle zu umfassend wäre
und nicht im Rahmen dieser Arbeit konzipiert wurde[158].
Die Kommunikation zwischen PC und OLGA beschränkt sich darauf, dass der PC die
auszuführenden Befehle an die OLGA überträgt und das SIA-Gerät auf Anfrage
Statusmeldungen zurückgibt.
In dem in Abbildung 4-17 zu sehenden Hauptfenster können vor der Messung über
die Menüs Einstellungen vorgenommen und über die Schaltflächen in der oberen
Leiste unter anderem die Messung gestartet bzw. abgebrochen werden. Am unteren
Rand des Fensters werden während der Messung Statusmeldungen eingeblendet,
so dass immer Informationen über den aktuellen Stand der Messung, sowie über die
Dauer der aktuellen Aktion und des gesamten Experiments verfügbar sind. Zudem
handelt es sich bei dem Hauptfenster um eine sogenannte MDI-Form. Innerhalb
Abbildung 4-17: Screenshot des Hauptfensters der OLGA-Software

Ergebnisse eigener Arbeit 57
dieses Fensters lassen sich untergeordnete Fenster aufrufen, die MDI-Child genannt
werden. Diese können nur innerhalb des Hauptfensters bewegt werden und sind mit
diesem verknüpft.
Die auszuführenden Befehle werden vor dem Start der Messung über eine
Eingabemaske an die Software übergeben. Das Eingabefenster für die Parameter
der Messung ist in 6 Bereiche, aufgeteilt (siehe Abbildung 4-18). Hierbei bestehen
Pumpparameter immer aus einem Satz von Geschwindigkeit und Dauer. Daraus
ergibt sich das jeweilige Volumen und lässt zusätzlich eine hohe Flexibilität für die
Pumpgeschwindigkeiten zu, so dass für jeden Pumpvorgang eine optimal
angepasste Geschwindigkeit vorgegeben werden kann. Dies ist gerade für den
Anreicherungsvorgang in der Strippinganalyse interessant, da hier eher niedrige
Geschwindigkeiten gewählt werden, während für das Befüllen der Mischkammer
höhere Geschwindigkeiten ab 15 µl/s aufwärts verwendet werden. Dies führt zu
stärkeren Verwirbelungen der Lösung in der Mischzelle und somit zu einer besseren
Durchmischung des Puffers mit dem Analyten und dem Komplexbildner.
Das Segment „System“ beinhaltet die Eingabefelder für die allgemeinen
Pumpvorgänge. Die wichtigsten Einstellungen in dieser Gruppe sollen hier näher
erläutert werden. Beim Parameter „Vorspülen“ werden Geschwindigkeit und Zeit so
eingestellt, dass die Probe vor der Messung exakt bis zur Messzelle gepumpt wird.
Damit wird das Totvolumen des Verbindungsschlauchs zwischen Verteiler und
Messzelle überbrückt, so dass für die Anreicherung Probenlösung direkt an der
Messzelle anliegt. Der Parametersatz „Injektion“ bestimmt das Volumen der
Abbildung 4-18: Screenshot der Parametereingabemaske des SISA-Systems

Ergebnisse eigener Arbeit 58
Probenahme. Unter „Zelle nachspülen“ kann festgelegt werden, mit welcher
Puffermenge nach einer Messung die Messzelle gereinigt werden soll. Da die
Möglichkeit besteht, die Pumpe mit Isopropanol (oder anderen Lösungen) zu spülen,
sind die beiden Einträge „Isopropanol“ und „Nachspülen“ aufgeführt. Hier wird
festgelegt, wie viel Isopropanol durch die Pumpe gespült und mit welcher Puffer-
menge dieses wieder aus dem System gepumpt werden soll. Das Isopropanol soll
dazu dienen, evtl. vorhandene Luftbläschen aus der Pumpe zu entfernen. Diese
können sich sonst festsetzten, so dass Pumpvorgänge kaum noch möglich sind, bzw.
die Volumina und Geschwindigkeiten von den Sollwerten erheblich abweichen. In
den allgemeinen Einstellungen ist der Punkt „Zyklen bis Isopropanol“ zu finden. Die
hier eingetragene Zahl gibt an, nach wie vielen Messzyklen die Pumpe mit
Isopropanol gespült wird. Ist an dieser Stelle eine Null eingetragen, wird der
Spülschritt nie ausgeführt.
Im „Probe“-Segment finden sich Zusatzparameter zur Probenahme wieder. Da der
Schlauch für die Probennahme ebenfalls ein Eigenvolumen hat, muss vor dem
Ansaugen der Probe erst die alte Lösung aus dem Schlauch entfernt werden, damit
frische Probe am Verteiler anliegt. Der Parametersatz unter „Ansaugen“ muss folglich
mindestens das Volumen des Probenschlauchs ergeben. Die alte Probelösung wird
anschließend mit Puffer in den Abfallbehälter gespült. Die Menge des dabei
eingesetzten Puffers wird unter „Ausspülen“ festgelegt.
Der Abschnitt „Kalibrieren“ enthält alle Parameter, die für die automatische
Standardaddition erforderlich sind. Die beiden Punkte „Ansaugen“ und „Ausspülen“
sind hier wieder für das Durchspülen des Verbindungsschlauchs zwischen Verteiler
und Vorratsgefäß verantwortlich, damit frische Standardlösung am Verteiler anliegt.
„Injizieren/Addition“ gibt an, wie viel Standardlösung zur Addition eingesetzt wird.
Hier ist darauf zu achten, dass abhängig von der Konzentration der Standardlösung
und dem Volumen der Mischkammer Werte eingetragen werden, die eine sinnvolle
Standardaddition ergeben. Der Parameter „Anzahl der Additionen“ ist selbst-
erklärend. Ergibt die Anzahl der Additionen multipliziert mit dem Volumen der
verwendeten Standardlösung und unter Berücksichtigung des Probevolumens einen
Wert, der größer als das Volumen der Mischzelle ist, dann erscheint hier eine
Fehlermeldung. Wird eine Null eingetragen, wird keine Standardaddition
durchgeführt.

Ergebnisse eigener Arbeit 59
Das Segment „Reagenz“ ist für die adsorptive Variante der Strippinganalyse wichtig,
da hier festgelegt wird, wie viel Komplexbildner zugegeben werden soll. Natürlich ist
diese Option auch nutzbar, um evtl. Reagenzien zuzusetzen, die die Sensitivität der
ASV verbessern sollen, wie es in der Literatur beschrieben wird[159].
Im Abschnitt „Stripping“ werden Einstellungen für die Anreicherung des Analyten
festgelegt. Die „Durchflusszyklen“ bestimmen wie oft eine Probe durch die Messzelle
gepumpt werden soll. Dies stellt eine Erweiterung der Standardanreicherungs-
prozedur dar, bei der die Probe nur einmal durch die Durchflusszelle gepumpt wird.
Hier ist nun die Möglichkeit gegeben, die Probe mehrmals vor und zurück zu
pumpen, um somit die Menge an angereichertem Analyten an der Elektroden-
oberfläche zu erhöhen. Die Idee basiert auf der Tatsache, dass bei einer
Anreicherung in der Strippinganalyse nie die gesamte Menge an Analyt aus der
Lösung durch Diffusion an die Elektrodenoberfläche gelangt, so dass bei einem
erneuten Durchfluss immer noch genügend Analyt vorhanden ist, um an der
Elektrode eine Erhöhung der angereicherten Analytkonzentration zu erzielen. Die
„Anlagerungszeit“ gibt an, wie lang die Anreicherung stattfinden soll und muss mit
dem Potentiostaten abgestimmt sein. Die „Pumpgeschwindigkeit“ kann in einem
Intervall angegeben werden. Dies hat folgende Bewandtnis: Alle drei in der Gruppe
„Stripping“ zusammengefassten Parameter sind von einander abhängig. Es können
also Parameter vorgeben werden und durch betätigen der „Neuberechnung“-
Schaltfläche wird eine Fittingroutine gestartet, die versucht eine Lösung für die
gewünschten Parameter zu finden, die möglichst nah an den Vorgaben liegt.
Die drei allgemeinen Einstellungen „Isopropanol“, „Reaktorvolumen“ und „Wartezeit“
befinden sich in keiner speziellen Kategorie und sind am unteren Rand des Fensters
zu finden. Die Einstellung „Isopropanol“ wurde oben schon erklärt, das
„Reaktorvolumen“ bezieht sich auf das Volumen der Mischkammer. Da diese
auswechselbar ist, muss hier das Volumen der Mischzelle eingetragen werden, weil
die Software diesen Wert für Berechnungen von Pumpvolumina benötigt. Die
„Wartezeit“ ist eine Funktion, die für Langzeitmessungen eingeführt wurde. Wird hier
eine Null eingetragen, dann läuft die OLGA kontinuierlich. Das bedeutet, nach einem
abgeschlossenen Messzyklus beginnt das Gerät sofort wieder mit der nächsten
Probenahme und startet somit einen neuen Zyklus. Das kann je nach Einstellung
bereits nach 10 Minuten der Fall sein. Jedoch sind viele Reaktionen nicht so schnell
und die Bestrebungen gehen immer in die Richtung der Kostenminimierung für die
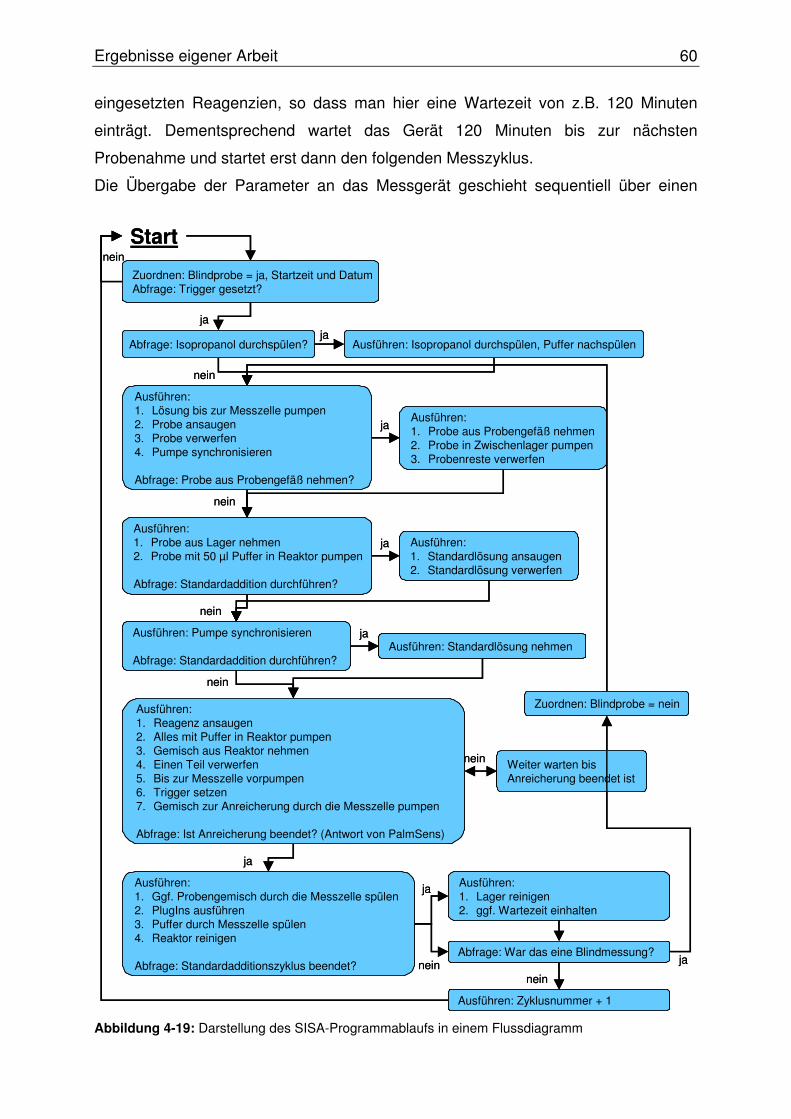
Ergebnisse eigener Arbeit 60
eingesetzten Reagenzien, so dass man hier eine Wartezeit von z.B. 120 Minuten
einträgt. Dementsprechend wartet das Gerät 120 Minuten bis zur nächsten
Probenahme und startet erst dann den folgenden Messzyklus.
Die Übergabe der Parameter an das Messgerät geschieht sequentiell über einen
Zuordnen: Blindprobe = ja, Startzeit und DatumAbfrage: Trigger gesetzt?
Abfrage: Isopropanol durchspülen? Ausführen: Isopropanol durchspülen, Puffer nachspülen
Ausführen:1. Lösung bis zur Messzelle pumpen2. Probe ansaugen3. Probe verwerfen4. Pumpe synchronisieren
Abfrage: Probe aus Probengefäß nehmen?
Ausführen: Pumpe synchronisieren
Abfrage: Standardaddition durchführen?
Ausführen:1. Standardlösung ansaugen2. Standardlösung verwerfen
Ausführen:1. Probe aus Lager nehmen2. Probe mit 50 µl Puffer in Reaktor pumpen
Abfrage: Standardaddition durchführen?
Ausführen: Standardlösung nehmen
Ausführen:1. Reagenz ansaugen2. Alles mit Puffer in Reaktor pumpen3. Gemisch aus Reaktor nehmen4. Einen Teil verwerfen5. Bis zur Messzelle vorpumpen6. Trigger setzen7. Gemisch zur Anreicherung durch die Messzelle pumpen
Abfrage: Ist Anreicherung beendet? (Antwort von PalmSens)
Ausführen:1. Probe aus Probengefäß nehmen2. Probe in Zwischenlager pumpen3. Probenreste verwerfen
Abfrage: War das eine Blindmessung?
Startnein
jaja
nein
ja
nein
ja
nein
ja
Ausführen:1. Ggf. Probengemisch durch die Messzelle spülen2. PlugIns ausführen3. Puffer durch Messzelle spülen4. Reaktor reinigen
Abfrage: Standardadditionszyklus beendet?
Ausführen: 1. Lager reinigen 2. ggf. Wartezeit einhalten
Weiter warten bisAnreicherung beendet ist
nein
nein
ja
ja
nein ja
Ausführen: Zyklusnummer + 1
nein
Zuordnen: Blindprobe = nein
Zuordnen: Blindprobe = ja, Startzeit und DatumAbfrage: Trigger gesetzt?
Abfrage: Isopropanol durchspülen? Ausführen: Isopropanol durchspülen, Puffer nachspülen
Ausführen:1. Lösung bis zur Messzelle pumpen2. Probe ansaugen3. Probe verwerfen4. Pumpe synchronisieren
Abfrage: Probe aus Probengefäß nehmen?
Ausführen: Pumpe synchronisieren
Abfrage: Standardaddition durchführen?
Ausführen:1. Standardlösung ansaugen2. Standardlösung verwerfen
Ausführen:1. Probe aus Lager nehmen2. Probe mit 50 µl Puffer in Reaktor pumpen
Abfrage: Standardaddition durchführen?
Ausführen: Standardlösung nehmen
Ausführen:1. Reagenz ansaugen2. Alles mit Puffer in Reaktor pumpen3. Gemisch aus Reaktor nehmen4. Einen Teil verwerfen5. Bis zur Messzelle vorpumpen6. Trigger setzen7. Gemisch zur Anreicherung durch die Messzelle pumpen
Abfrage: Ist Anreicherung beendet? (Antwort von PalmSens)
Ausführen:1. Probe aus Probengefäß nehmen2. Probe in Zwischenlager pumpen3. Probenreste verwerfen
Abfrage: War das eine Blindmessung?
Startnein
jaja
nein
ja
nein
ja
nein
ja
Ausführen:1. Ggf. Probengemisch durch die Messzelle spülen2. PlugIns ausführen3. Puffer durch Messzelle spülen4. Reaktor reinigen
Abfrage: Standardadditionszyklus beendet?
Ausführen: 1. Lager reinigen 2. ggf. Wartezeit einhalten
Weiter warten bisAnreicherung beendet ist
nein
nein
ja
ja
nein ja
Ausführen: Zyklusnummer + 1
nein
Zuordnen: Blindprobe = nein
Abbildung 4-19: Darstellung des SISA-Programmablaufs in einem Flussdiagramm

Ergebnisse eigener Arbeit 61
Timer, welcher so den Ablauf der Messung koordiniert. Die gesamte Timerroutine ist
im Anhang 7.1 angefügt. Sie ist während der Experimente im Funktionsumfang
ständig weiterentwickelt worden und wurde im späteren Verlauf hauptsächlich um
Spül- und Reinigungsschritte erweitert. Diese konnten die Sensitivität und
Reproduzierbarkeit verbessern. Der komplexe Messablauf ist zur Veranschaulichung
in einem Flussdiagramm in Abbildung 4-19 zu sehen.
Fast die gesamte Timerroutine besteht aus einer einzigen „Select-Case“ Anweisung,
die folgendermaßen funktioniert: Eine Zählvariable erhält anfangs den Wert 0, was
dazu führt, dass beim ersten Durchlauf des Timers die Anweisungen unter „Case 0“
ausgeführt werden. Danach wird die Variable meist um 1 erhöht und die Routine wird
verlassen. Beim nächsten Timeraufruf wird dann der nächste „Case“ ausgeführt bzw.
der, der laut der Zählvariablen an der Reihe ist.
4.2.2.3 Wahl der Messzelle
Es bietet sich an, in einem SIA-System eine Durchflussmesszelle zu verwenden,
jedoch ist diese im Gegensatz zu einem FIA-System nicht zwingend erforderlich. Da
bei der SIA die Möglichkeit gegeben ist, die Lösungen vor- und zurückzupumpen,
kann auch eine angepasste konventionelle Messzelle benutzt werden. Die Anfor-
derungen an eine für die AdSV geeignete elektrochemische Messzelle lassen sich
leicht mit denen für das SIA-System vereinen. Im Allgemeinen wird eine
RE
CE
WE Dichtungen
RE
CE
WE Dichtungen
Abbildung 4-20: Skizze der Durchflussmesszellen (altes Design) mit großem Zellvolumen (ca. 290 µl)

Ergebnisse eigener Arbeit 62
elektrochemische Messzelle, die ein Zwei- oder Dreielektrodensystem aufnehmen
und mit einer Pumpvorrichtung befüllt werden kann, benötigt.
Erste Experimente wurden mit einer aus früheren Applikationen in der Fermen-
tationskontrolle vorhandenen Durchflussmesszelle (siehe Abbildung 4-20), die so
modifiziert wurde, dass sie die kleineren Elektroden für die Strippingvoltammetrie
aufnehmen kann, durchgeführt.
Die Messzelle ist aus Plexiglas gefertigt und erlaubt eine Dreielektrodenanordnung.
Sie verfügt über einen zylindrischen Innenraum mit einem Durchmesser von 9 mm
und einer Höhe von 4,5 mm, in dem sich die Arbeitselektrode und die miniaturisierte
Ag/AgCl-Referenzelektrode gegenüberliegen. Der Ein- und Auslass haben einen
Durchmesser von 1 mm, wobei in dem Einlass eine Edelstahlkanüle eingeklebt ist,
die als Gegenelektrode dient. Die Arbeitselektrode wird mit einem ebenfalls aus
Plexiglas bestehenden Schraubverschluss von der Unterseite der Zelle eingesetzt.
Der Übergang ist mit einem O-Ring abgedichtet.
4.2.2.4 Erster Test des Systems
Pb2+ wurde für die ersten Probemessungen als Analyt gewählt, da die Blei-
bestimmung mittels ASV unkomplizierter als die Nickelanalyse mittels AdSV ist und
somit für den ersten Test des Systems besser geeignet ist. Die verwendete Pb2+-
Konzentration wurde mit 165 mM extrem hoch gewählt, um eine zu niedrige
Analytkonzentration als Fehlerquelle ausschließen zu können. Als Arbeitselektrode
wurde eine MFE auf Basis einer Iridiumelektrode gewählt, wie sie in Abschnitt
4.2.1.1.1 beschrieben ist, jedoch mit 2 mm Durchmesser.
Der erste Test des Systems zeigte apparative sowie softwareseitige Fehler auf. Die
Differenzbildung, die von der selbst programmierten Software übernommen wurde,
wies eine große Fehleranfälligkeit in der Erkennung der Potentialpulse bzw. der
daraus resultierenden Strompulse auf. Diese wurden oft falsch ausgelesen und
erbrachten folglich bei der Berechnung keine korrekten Ergebnisse.
Zudem brachte die apparative Lösung aus Kombination des Funktionsgenerators mit
dem Potentiostaten noch weitere Nachteile mit sich. Die Übertragung der Rampen-
funktion vom PC zum Funktionsgenerator war aufgrund der Anbindung über eine
RS232-Schnittstelle und der großen Menge an zu übertragenen Datenpunkten sehr
langsam. So dauerte eine Änderung der Messparameter länger als 30 Min.
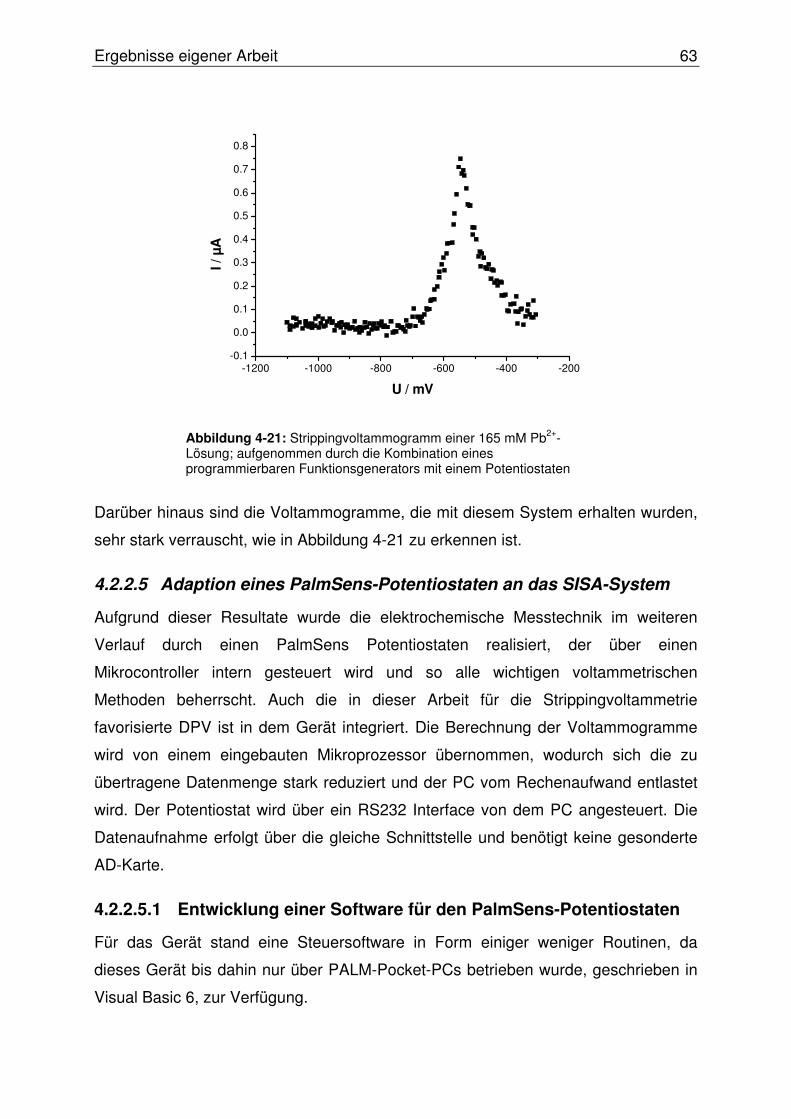
Ergebnisse eigener Arbeit 63
Darüber hinaus sind die Voltammogramme, die mit diesem System erhalten wurden,
sehr stark verrauscht, wie in Abbildung 4-21 zu erkennen ist.
4.2.2.5 Adaption eines PalmSens-Potentiostaten an das SISA-System
Aufgrund dieser Resultate wurde die elektrochemische Messtechnik im weiteren
Verlauf durch einen PalmSens Potentiostaten realisiert, der über einen
Mikrocontroller intern gesteuert wird und so alle wichtigen voltammetrischen
Methoden beherrscht. Auch die in dieser Arbeit für die Strippingvoltammetrie
favorisierte DPV ist in dem Gerät integriert. Die Berechnung der Voltammogramme
wird von einem eingebauten Mikroprozessor übernommen, wodurch sich die zu
übertragene Datenmenge stark reduziert und der PC vom Rechenaufwand entlastet
wird. Der Potentiostat wird über ein RS232 Interface von dem PC angesteuert. Die
Datenaufnahme erfolgt über die gleiche Schnittstelle und benötigt keine gesonderte
AD-Karte.
4.2.2.5.1 Entwicklung einer Software für den PalmSens-Potentiostaten
Für das Gerät stand eine Steuersoftware in Form einiger weniger Routinen, da
dieses Gerät bis dahin nur über PALM-Pocket-PCs betrieben wurde, geschrieben in
Visual Basic 6, zur Verfügung.
-1200 -1000 -800 -600 -400 -200-0.1
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
I / µ
A
U / mV
Abbildung 4-21: Strippingvoltammogramm einer 165 mM Pb2+-Lösung; aufgenommen durch die Kombination eines programmierbaren Funktionsgenerators mit einem Potentiostaten

Ergebnisse eigener Arbeit 64
Die Software zur Steuerung des Potentiostaten ist ein eigenständiges, von der des
OLGA-Systems unabhängiges Programm. Sie soll auch dazu dienen, den
Potentiostaten als Einzelgerät und nicht nur in Kombination mit der SIA-Einheit
nutzen zu können. Da die Programmierung der Kommunikation zwischen
Potentiostat und PC im wesentlichen komplexe, über die serielle Schnittstelle
gesendete Bytefolgen umfasst, möchte ich hier nur auf die Nutzeroberfläche und
einige nützliche Funktionen eingehen.
Die Software wurde mit einer Autoerkennung des Potentiostaten ausgestattet, die es
nicht mehr erfordert Kenntnis darüber zu haben, an welchem seriellen Port das Gerät
angeschlossen ist (siehe Abbildung 4-22).
Das Hauptfenster in Abbildung 4-23 zeigt alle verfügbaren Funktionen. Wichtig sind
hier nur der mittlere Bereich und der Abschnitt „Control“. Dieser Abschnitt ermöglicht
die manuelle Steuerung des Geräts, damit die Messzelle evtl. ein- und ausgeschaltet
oder ein Potential zur vorhergehenden Konditionierung aufgeprägt werden kann.
Der für die Strippinganalyse wichtigste Teil der Software verbirgt sich in dem
Segment „Method“. Hier kann die voltammetrische Methode ausgewählt werden, mit
der die Bestimmung des Schwermetalls bei der Strippinganalyse durchgeführt
werden soll. Zur Verfügung stehen bei dem aktuellen Entwicklungstand Linear-
Sweep-Voltammetrie, Differenz-Puls-Voltammetrie, Square-Wave-Voltammetrie,
Normal-Puls-Voltammetrie, Wechselstrom-Voltammetrie und cyclische Voltammetrie.
Zudem muss hier die Dauer und das Potential für die Anreicherung unter tdep und
Edep eingestellt werden. tequ kann genutzt werden, um zwischen Anreicherung und
Stripping eine Wartezeit einzurichten. Dies ist in der Strippinganalyse üblich, da die
Anreicherung unter konvektiven Bedingungen stattfindet und das Stripping in
ruhender Lösung. So können einige Sekunden ohne Pumpvorgänge eingerichtet
Abbildung 4-22: Screenshot des Startfensters der PalmSens-Software mit integrierter automatischer Anschlusserkennung

Ergebnisse eigener Arbeit 65
werden, damit sich die Lösung beruhigt. Es ist ebenso möglich zwischen den
Messungen auf das Ruhepotential umzustellen oder ein Potential aufzuprägen. Es
hat sich gezeigt, dass es besser ist ein Potential von –1,1 V, wie unter Est.by zu sehen
ist, anzulegen (siehe Abschnitt 4.2.2.8.2). Der Grund liegt in der Elektroden-
beschichtung, wie in den kommenden Kapiteln noch erläutert werden wird.
Wichtig ist zu erwähnen, dass die Schaltfläche „Start Measurement“ zwei
unterschiedliche Funktionsweisen besitzt. Im Standardmodus wird einfach die
Messung, welche gerade im Potentiostaten gespeichert ist, gestartet. Dies ist für die
Nutzung des Potentiostaten ohne Kopplung an ein anderes Gerät gedacht. Ist
allerdings über den Schalter „Remote Control“ der Fernsteuermodus aktiviert, dann
wartet das Programm, bis es von einer anderen Software den Befehl bekommt, zu
starten. In diesem Fall von der OLGA-Software, die eine temporäre Datei entfernen
muss, die zuvor von der PalmSens-Software erstellt wurde. Die Software des
Potentiostaten erkennt den Verlust der Datei, da sie während der Warteperiode
kontinuierlich deren Präsens überprüft, und startet die Messung. Nach Abschluss des
Abbildung 4-23: Screenshot des Hauptfensters des PalmSens-Steuerprogramms. Unter „Method“ werden die Parameter für die Strippinganalyse mittels DPV eingestellt.

Ergebnisse eigener Arbeit 66
Messung erstellt sie eine neue Datei. Die OLGA-Software erkennt, dass die Datei
wieder vorhanden ist und startet den nächsten Zyklus.
Das Teilfenster „Potential and Current“ zeigt alle aktuellen Mess- und Statuswerte
des Potentiostaten an. Die restlichen sichtbaren Elemente dienen der Überprüfung
und Entwicklung des Programms und werden in späteren Versionen nicht mehr zu
sehen sein.
Die Software bietet alle wichtigen Methoden, die für die Strippinganalyse interessant
sind. Die Bedienung ist einfach und die Kopplung mit der OLGA-Software
unkompliziert gelöst. Somit ist apparativ eine gute Vorraussetzung für die
automatisierte Strippinganalyse geschaffen.
4.2.2.5.2 Programmierung eines PlugIn-Systems
Um eine Datenvisualisierung während der laufenden Messung zu erhalten, wurde
das OLGA-Programm mit einem PlugIn-System ausgestattet, welches sich die in
Kapitel 4.2.2.2.2 beschriebene MDI-Eigenschaft des Hauptfensters zunutze macht.
PlugIns sind Zusatzprogrammteile, die in das Hauptprogramm eingefügt werden
können und dieses im Funktionsumfang erweitern, ohne das es selbst verändert
werden muss. Das System basiert auf PlugIns, die in Form von ActiveX-Dynamic-
Abbildung 4-24: Screenshot des Startfensters der OLGA-Software; im unteren Teilfenster werden angemeldete PlugIns mit Information angezeigt
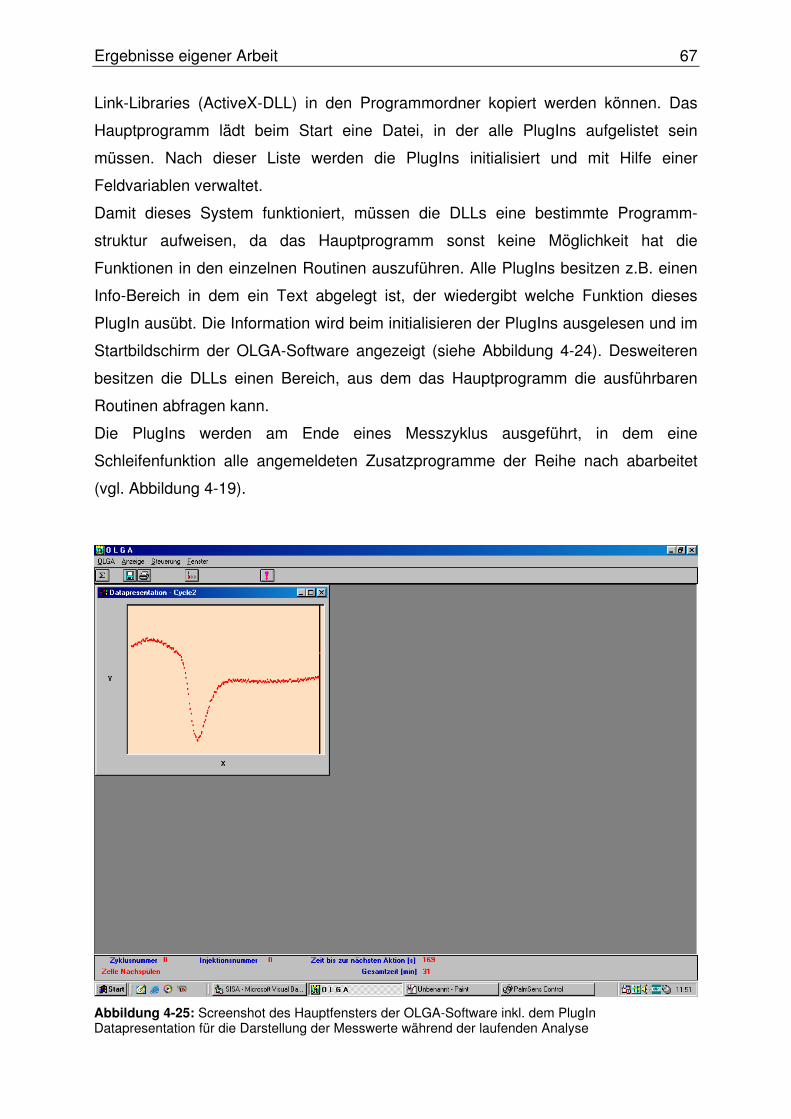
Ergebnisse eigener Arbeit 67
Link-Libraries (ActiveX-DLL) in den Programmordner kopiert werden können. Das
Hauptprogramm lädt beim Start eine Datei, in der alle PlugIns aufgelistet sein
müssen. Nach dieser Liste werden die PlugIns initialisiert und mit Hilfe einer
Feldvariablen verwaltet.
Damit dieses System funktioniert, müssen die DLLs eine bestimmte Programm-
struktur aufweisen, da das Hauptprogramm sonst keine Möglichkeit hat die
Funktionen in den einzelnen Routinen auszuführen. Alle PlugIns besitzen z.B. einen
Info-Bereich in dem ein Text abgelegt ist, der wiedergibt welche Funktion dieses
PlugIn ausübt. Die Information wird beim initialisieren der PlugIns ausgelesen und im
Startbildschirm der OLGA-Software angezeigt (siehe Abbildung 4-24). Desweiteren
besitzen die DLLs einen Bereich, aus dem das Hauptprogramm die ausführbaren
Routinen abfragen kann.
Die PlugIns werden am Ende eines Messzyklus ausgeführt, in dem eine
Schleifenfunktion alle angemeldeten Zusatzprogramme der Reihe nach abarbeitet
(vgl. Abbildung 4-19).
Abbildung 4-25: Screenshot des Hauptfensters der OLGA-Software inkl. dem PlugIn Datapresentation für die Darstellung der Messwerte während der laufenden Analyse
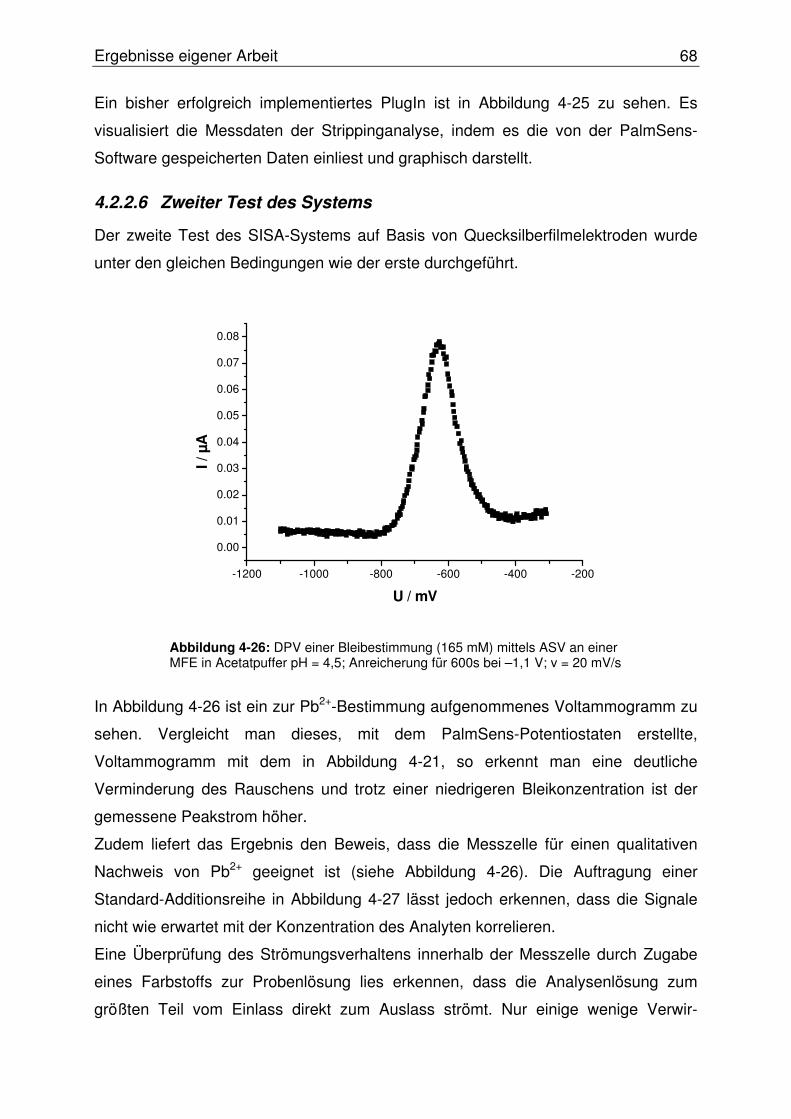
Ergebnisse eigener Arbeit 68
Ein bisher erfolgreich implementiertes PlugIn ist in Abbildung 4-25 zu sehen. Es
visualisiert die Messdaten der Strippinganalyse, indem es die von der PalmSens-
Software gespeicherten Daten einliest und graphisch darstellt.
4.2.2.6 Zweiter Test des Systems
Der zweite Test des SISA-Systems auf Basis von Quecksilberfilmelektroden wurde
unter den gleichen Bedingungen wie der erste durchgeführt.
In Abbildung 4-26 ist ein zur Pb2+-Bestimmung aufgenommenes Voltammogramm zu
sehen. Vergleicht man dieses, mit dem PalmSens-Potentiostaten erstellte,
Voltammogramm mit dem in Abbildung 4-21, so erkennt man eine deutliche
Verminderung des Rauschens und trotz einer niedrigeren Bleikonzentration ist der
gemessene Peakstrom höher.
Zudem liefert das Ergebnis den Beweis, dass die Messzelle für einen qualitativen
Nachweis von Pb2+ geeignet ist (siehe Abbildung 4-26). Die Auftragung einer
Standard-Additionsreihe in Abbildung 4-27 lässt jedoch erkennen, dass die Signale
nicht wie erwartet mit der Konzentration des Analyten korrelieren.
Eine Überprüfung des Strömungsverhaltens innerhalb der Messzelle durch Zugabe
eines Farbstoffs zur Probenlösung lies erkennen, dass die Analysenlösung zum
größten Teil vom Einlass direkt zum Auslass strömt. Nur einige wenige Verwir-
-1200 -1000 -800 -600 -400 -200
0.00
0.01
0.02
0.03
0.04
0.05
0.06
0.07
0.08
I / µ
A
U / mV
Abbildung 4-26: DPV einer Bleibestimmung (165 mM) mittels ASV an einer MFE in Acetatpuffer pH = 4,5; Anreicherung für 600s bei –1,1 V; v = 20 mV/s
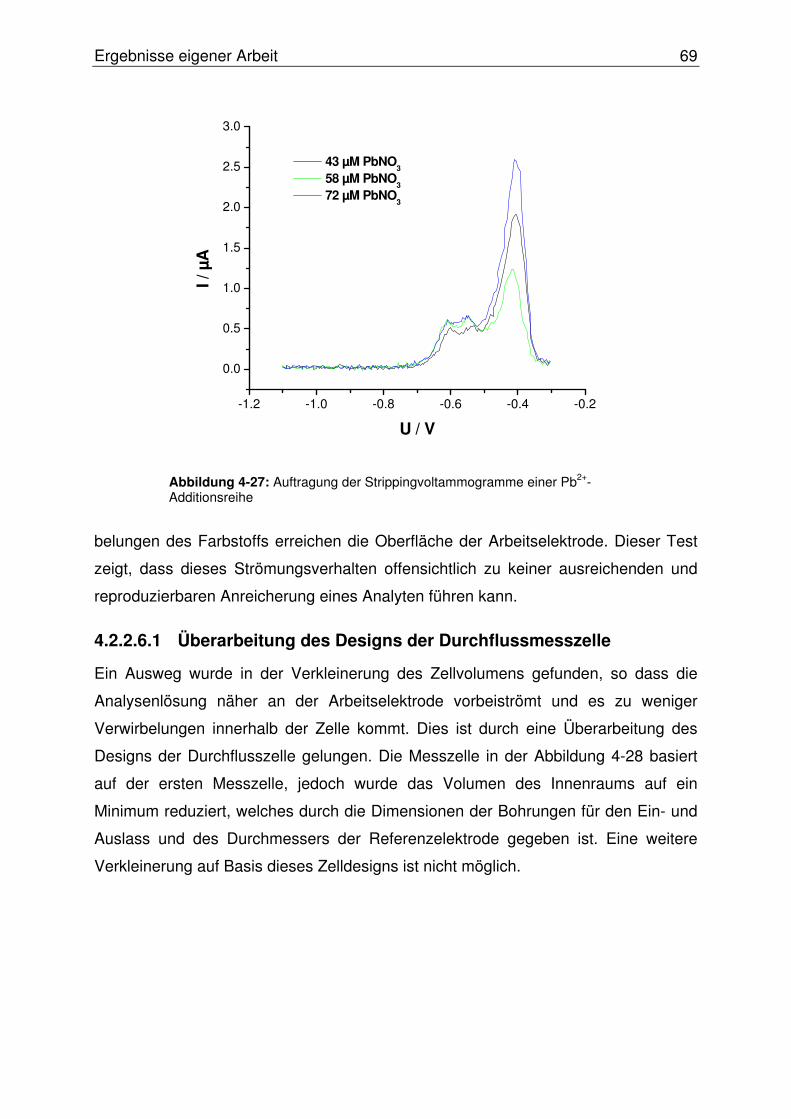
Ergebnisse eigener Arbeit 69
belungen des Farbstoffs erreichen die Oberfläche der Arbeitselektrode. Dieser Test
zeigt, dass dieses Strömungsverhalten offensichtlich zu keiner ausreichenden und
reproduzierbaren Anreicherung eines Analyten führen kann.
4.2.2.6.1 Überarbeitung des Designs der Durchflussmesszelle
Ein Ausweg wurde in der Verkleinerung des Zellvolumens gefunden, so dass die
Analysenlösung näher an der Arbeitselektrode vorbeiströmt und es zu weniger
Verwirbelungen innerhalb der Zelle kommt. Dies ist durch eine Überarbeitung des
Designs der Durchflusszelle gelungen. Die Messzelle in der Abbildung 4-28 basiert
auf der ersten Messzelle, jedoch wurde das Volumen des Innenraums auf ein
Minimum reduziert, welches durch die Dimensionen der Bohrungen für den Ein- und
Auslass und des Durchmessers der Referenzelektrode gegeben ist. Eine weitere
Verkleinerung auf Basis dieses Zelldesigns ist nicht möglich.
-1.2 -1.0 -0.8 -0.6 -0.4 -0.2
0.0
0.5
1.0
1.5
2.0
2.5
3.0
43 µM PbNO3
58 µM PbNO3
72 µM PbNO3
I / µ
A
U / V
Abbildung 4-27: Auftragung der Strippingvoltammogramme einer Pb2+-Additionsreihe

Ergebnisse eigener Arbeit 70
Eine Überprüfung des Strömungsverhaltens durch Zugabe des Farbstoffs bestätigte
eine Verbesserung. Die Lösung bildete weniger Verwirbelungen, jedoch konnten
diese nicht ganz unterdrückt werden, und die Arbeitselektrode liegt nun im Zentrum
des Stroms der Analysenlösung. Die Abbildung 4-29 zeigt ein Stripping-
voltammogramm einer Bleibestimmung mit der überarbeiteten Messzelle. Trotz einer
geringeren Konzentration an Pb2+ ist der Peakstrom bei gleichbleibenden
Messparametern höher als in Abbildung 4-26. Dies bestätigt, dass die Anreicherung
des Analyten nun effektiver erfolgt.
Ein weiteres Problem stellten die verwendeten MFEs dar. In folgenden
Langzeitexperimenten verschlechterte sich die Signalqualität der MFE schon nach
wenigen Stunden. Dies könnte mehrer Ursachen haben, wie z.B. die Oxidation durch
gelösten Sauerstoff oder auch die mechanische Instabilität des Hg-Films. Der
Sauerstoff wurde zwar aus allen Lösungen im OLGA-System mittels einer per-
manenten Argonzufuhr zu entfernen, jedoch werden in dem System PTFE-Schläuche
verwendet, da diese chemisch inert sind. Doch besitzen diese bekanntermaßen eine
hohe Sauerstoffpermeabilität, so dass über diesen Weg wieder Sauerstoff in das
System gelangen wird.
RE
CE
WE Dichtungen
RE
CE
WE Dichtungen
Abbildung 4-28: Skizze der überarbeiteten Durch-flussmesszelle mit geringerem Zellvolumen (ca. 75 µl)
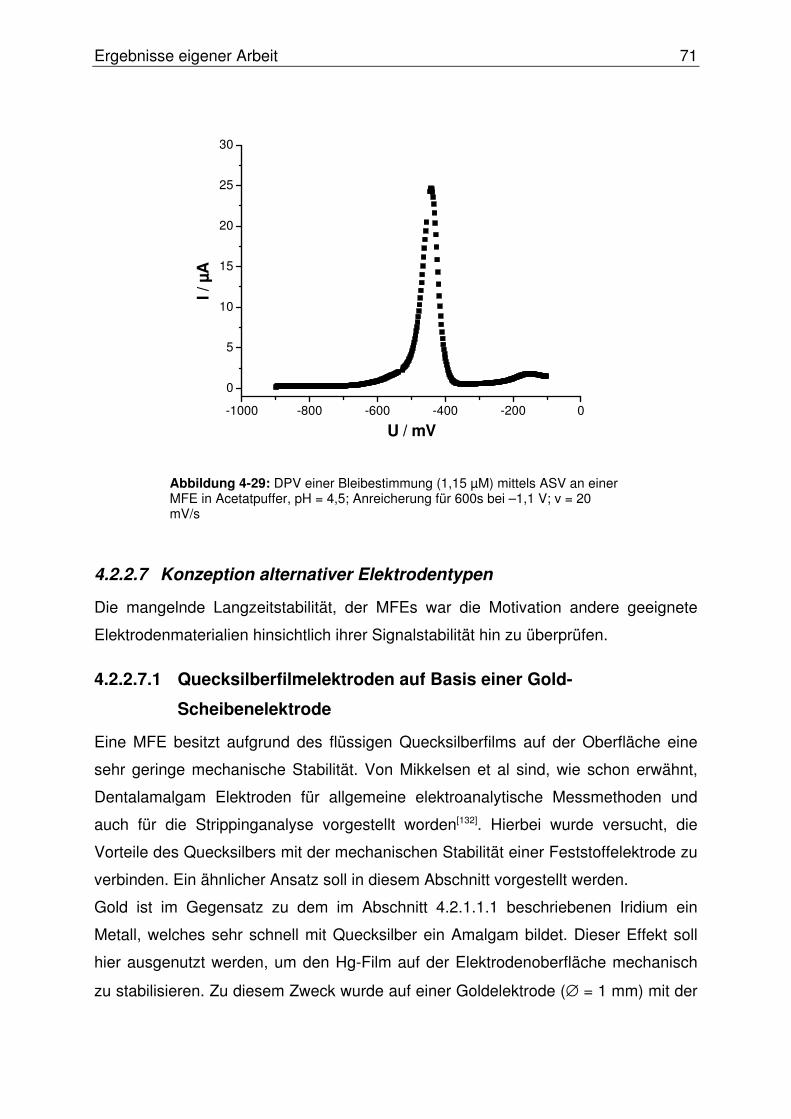
Ergebnisse eigener Arbeit 71
4.2.2.7 Konzeption alternativer Elektrodentypen
Die mangelnde Langzeitstabilität, der MFEs war die Motivation andere geeignete
Elektrodenmaterialien hinsichtlich ihrer Signalstabilität hin zu überprüfen.
4.2.2.7.1 Quecksilberfilmelektroden auf Basis einer Gold-
Scheibenelektrode
Eine MFE besitzt aufgrund des flüssigen Quecksilberfilms auf der Oberfläche eine
sehr geringe mechanische Stabilität. Von Mikkelsen et al sind, wie schon erwähnt,
Dentalamalgam Elektroden für allgemeine elektroanalytische Messmethoden und
auch für die Strippinganalyse vorgestellt worden[132]. Hierbei wurde versucht, die
Vorteile des Quecksilbers mit der mechanischen Stabilität einer Feststoffelektrode zu
verbinden. Ein ähnlicher Ansatz soll in diesem Abschnitt vorgestellt werden.
Gold ist im Gegensatz zu dem im Abschnitt 4.2.1.1.1 beschriebenen Iridium ein
Metall, welches sehr schnell mit Quecksilber ein Amalgam bildet. Dieser Effekt soll
hier ausgenutzt werden, um den Hg-Film auf der Elektrodenoberfläche mechanisch
zu stabilisieren. Zu diesem Zweck wurde auf einer Goldelektrode (∅ = 1 mm) mit der
-1000 -800 -600 -400 -200 0
0
5
10
15
20
25
30
I / µ
A
U / mV
Abbildung 4-29: DPV einer Bleibestimmung (1,15 µM) mittels ASV an einer MFE in Acetatpuffer, pH = 4,5; Anreicherung für 600s bei –1,1 V; v = 20 mV/s

Ergebnisse eigener Arbeit 72
oben beschriebenen Methode 100 s lang Quecksilber abgeschieden. Dies führt
umgehend zu einer Goldamalgambildung auf der Elektrodenoberfläche.
Auch dieser Elektrodentyp wurde auf die Tauglichkeit für die AdSV hin überprüft,
indem in einem stationären System Ni2+ mit Nioxim als Komplexbildner nach-
gewiesen wurde (siehe Abbildung 4-30).
Damit konnte gezeigt werden, dass diese Elektrode prinzipiell für die
Strippinganalyse geeignet ist. Weitere Tests machten jedoch deutlich, dass es sich
als schwierig erweist, eine Kalibrierung durchzuführen. Bei AdSV-Versuchen mit
verschiedenen Ni2+-Konzentrationen, zeigte sich keine Korrelation zwischen
eingesetzter Analytkonzentration und resultierendem Messsignal.
4.2.2.7.2 Wismutfilmelektroden (BiFE)
Die Suche nach einem quecksilberfreien Elektrodenmaterial hat die Entwicklung der
Wismutfilmelektroden in den letzten Jahren vorangetrieben. Sie haben sich in
aktuellen Publikationen als leistungsfähiger Ersatz für die quecksilberbasierenden
Elektroden in der Strippinganalyse erwiesen. Während Nachweisgrenze und
Sensitivität vergleichbar sind, ist der nutzbare Potentialbereich geringfügig kleiner, da
sich das Bi bereits bei einem Potential positiver als –200 mV aufzulösen beginnt und
die Höhe der Wasserstoffüberspannung des Quecksilbers nicht erreicht wird. Jedoch
-1300 -1200 -1100 -1000 -900 -800 -700 -600 -500-600
-550
-500
-450
-400
-350
-300I /
nA
U / mV
Abbildung 4-30: DPV der Strippinganalyse einer 2 nM Ni2+-Lösung mit 0,1 M Nioxim als Komplexbildner; tAnr = 600s, UAnr = -0,6V

Ergebnisse eigener Arbeit 73
ist dieses Potentialfenster für die hier beabsichtigten Messungen ausreichend. Die
BiFE bringt gegenüber der MFE aber auch zwei Vorteile mit sich. Bi ist nicht toxisch,
im Gegensatz zu Hg, was nicht nur die Handhabung erleichtert, sondern auch die
Entsorgung der Lösungen, und Bi ist mechanisch stabiler als Hg. Dies sollte bei den
Strömungsverhältnissen in einer Durchflussmesszelle, in welcher die Elektrode
nachher eingesetzt werden soll, eine längere Lebensdauer der Beschichtung
gewährleisten.
Als Basiselektrode wird eine glasummantelte, nach der Beschreibung in Abschnitt
6.2.1 angefertigte Glaskohlenstoffelektrode mit 1 mm Durchmesser verwendet. GC
ist aus der Literatur bereits als gutes Substrat für die Wismutfilmabscheidung
bekannt[125, 141, 143].
Eine verbreitete Methode der Wismutbeschichtung geht auf eine Arbeit von Wang et
al[140] zurück. Hier wird aus einer leicht sauren (pH = 4,5), acetatgepufferten BiIII-
Lösung das Bi bei –1,2 V abgeschieden. Dies führte bei ersten Experimenten zu
keinem zufriedenstellenden Ergebnis. Die bei der Abscheidung auftretende heftige
Gasentwicklung führte zu einem löchrigen, ungleichmäßigen, nicht die gesamte
Elektrodenfläche bedeckenden Wismutfilm.
Durch entgasen der Lösung mittels Argon, um den Sauerstoff auszutreiben und somit
evtl. auftretende Oxidbildung auszuschließen, und starkem Rühren während des
Abscheidungsprozesses, um entstehende Gasbläschen sofort von der Elektroden-
Abbildung 4-31: lichtmikroskopische Abbildung einer BiFE (∅ = 1mm). Die Beschichtung erfolgte nach einer von Wang et al veröffentlichten Methode[140], jedoch mit optimierten Parameter.

Ergebnisse eigener Arbeit 74
oberfläche zu entfernen, wurde versucht, die Filmqualität zu verbessern.
Bei lichtmikroskopischen Untersuchungen erschien der so entstandene Film
homogener, bedeckte die ganze Elektrodenoberfläche und zeigte nur noch vereinzelt
größere Poren, die einige Mikrometer Durchmesser haben (siehe Abbildung 4-31).
Ein Film ohne große Poren konnte erst durch Herabsetzen des Abscheidungs-
potentials erreicht werden. Das optimierte Abscheidungspotential kann mittels
cyclovoltammetrischer Analyse für jede Elektrode individuell bestimmt werden (siehe
-600 -400 -200 0 200 400
-4
-2
0
2
4
6
8
-350 mV
I / µ
A
U / mV
Abbildung 4-32: CycloVoltammogramm einer GC-Elektrode (∅ = 1mm) in einer 100 mg/ml Bi-Lösung in 5 M Acetat-Puffer. Das Abscheidungspotential wurde hier z.B. auf –350 mV für die folgende Bi-Beschichtung festgelegt.
Abbildung 4-33: Abbildung einer mit 0,3 µm Poliersuspension polierten GC-Elektrodenoberfläche mittels AFM

Ergebnisse eigener Arbeit 75
Abbildung 4-32), um so die Filmerstellung zu perfektionieren. Dabei wird das
Potential so gewählt, dass es sich genügend negativ vom Reduktionspeak befindet,
um eine schnelle Deposition zu gewährleisten, aber nicht zu einer Gasentwicklung
führen kann.
Der Vergleich der Topographie der GC-Elektrodenoberfläche vor (siehe Abbildung
4-33) und nach (siehe Abbildung 4-34) der Abscheidung des Wismuts lässt gut die
inselförmige Struktur des aufgewachsenen Films erkennen. Aus den AFM
Abbildungen kann die Porengröße auf ca. 400 nm mit einer Tiefe von ca. 250 nm
abgeschätzt werden. Dies entspricht einer deutlichen Verkleinerung der Porengröße
im Vergleich zur Abscheidung bei -1,2 V.
Die BiFE wurde im Anschluß, wie die im letzten Kapitel beschriebenen MFEs, in
einem stationären Testsystem anhand von Ni2+ und Nioxim als Komplexbildner
überprüft. Erste Messungen zeigten, dass diese Elektrode für AdSV-Messungen
zwar verwendbar ist, jedoch Schwächen in der Reproduzierbarkeit und
Signalstabilität aufweist, wie die Auftragung des Peakstroms bei unterschiedlichen
Ni2+-Konzentrationen in Abbildung 4-35 aufzeigt.
Hier ist eindeutig zu erkennen, dass der Peakstrom der DPVs bei der Steigerung der
Ni-Konzentration von 0 (Nioxim) auf 100 nM einen Sprung von ca 120 nA vollzieht,
wobei die Steigerung von 100 auf 200 nM keinen Effekt hat. Dies kann aber kein
Sättigungseffekt sein, da die Peakhöhe sogar bei gleichbleibender Konzentration,
Abbildung 4-34: Abbildung einer auf einer GC-Elektrode abgeschiedenen Bi-Schicht
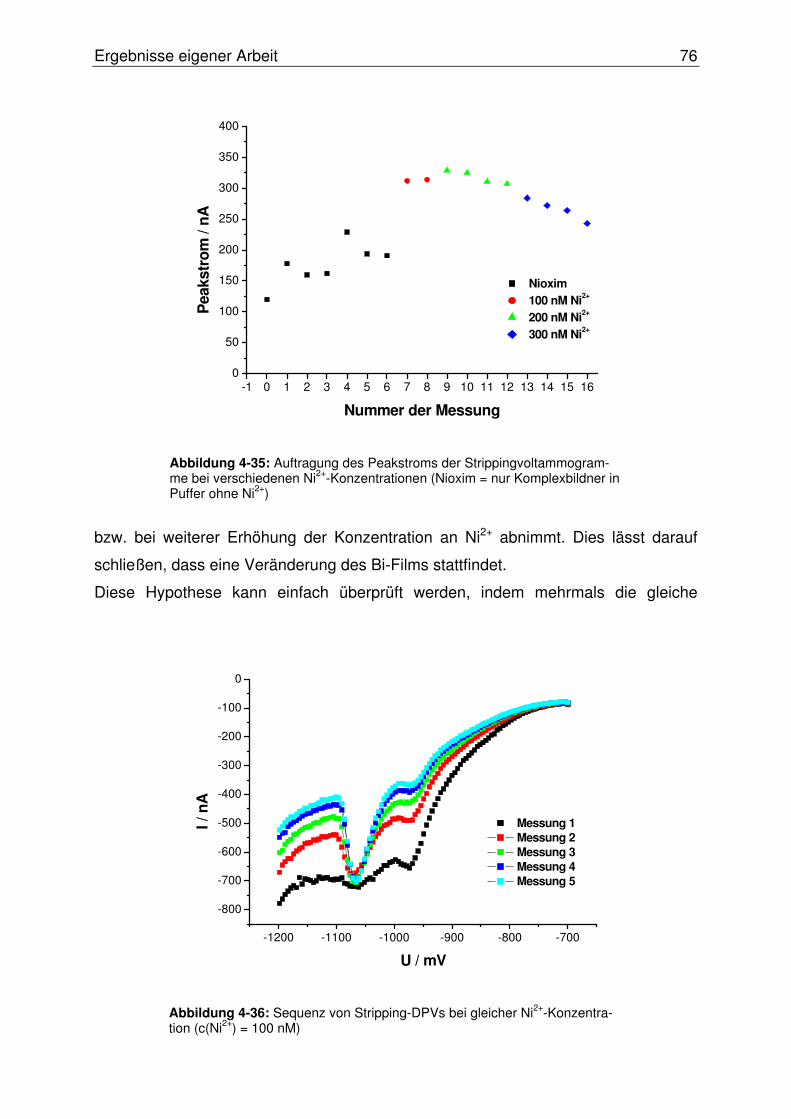
Ergebnisse eigener Arbeit 76
bzw. bei weiterer Erhöhung der Konzentration an Ni2+ abnimmt. Dies lässt darauf
schließen, dass eine Veränderung des Bi-Films stattfindet.
Diese Hypothese kann einfach überprüft werden, indem mehrmals die gleiche
-1 0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 160
50
100
150
200
250
300
350
400
Nioxim 100 nM Ni2+
200 nM Ni2+
300 nM Ni2+
Pea
kstr
om
/ n
A
Nummer der Messung
Abbildung 4-35: Auftragung des Peakstroms der Strippingvoltammogram-me bei verschiedenen Ni2+-Konzentrationen (Nioxim = nur Komplexbildner in Puffer ohne Ni2+)
-1200 -1100 -1000 -900 -800 -700
-800
-700
-600
-500
-400
-300
-200
-100
0
Messung 1 Messung 2 Messung 3 Messung 4 Messung 5
I / n
A
U / mV
Abbildung 4-36: Sequenz von Stripping-DPVs bei gleicher Ni2+-Konzentra-tion (c(Ni2+) = 100 nM)

Ergebnisse eigener Arbeit 77
Konzentration an Ni2+ hintereinander gemessen wird. In Abbildung 4-36 ist eine
Auftragung von 5 Stripping-DPVs dargestellt, die mit einer frisch beschichteten
Elektrode bei gleichbleibender Ni2+-Konzentration von 100 nM aufgenommen
wurden. Es ist deutlich zu erkennen, dass die Grundlinie mit jeder Messung in
Richtung des geringeren Reduktionsstroms verschoben wird. Der Effekt nimmt mit
Abnahme des Potentials zu. Zudem steigt die Höhe des Reduktionspeaks des
Nickelkomplexes (bei ca. – 1070 mV) mit steigender Zahl der Messzyklen an. Diese
Zunahme lässt auf eine Aktivierung der Oberfläche mit der Zahl der Messzyklen
schließen.
Die Aktivierung der Elektrodenoberfläche kann durch einen Konditionierungsschritt
vor der eigentlichen Ni-Messung im Vorfeld erreicht werden, so dass die Ni-
Bestimmung dadurch nicht mehr beeinflusst wird. Die Konditionierung besteht aus 10
CVs, die mit einem Startpotential von –0,7 V bis zum Endpotential von –1,2 V in
nickelfreier Pufferlösung aufgenommen werden. Der Grundstrom geht dabei auf
einen konstanten Wert zurück und ermöglicht so die nachfolgende störungsfreie Ni-
Bestimmung.
Eine Ni-Kalibrierung, die nach einer derartigen Konditionierung der Elektrode erstellt
wurde ist in Abbildung 4-37 dargestellt.
Die beschriebenen Optimierungen führten zu einer für die AdSV brauchbaren BiFE.
100 150 200 250 300 350 40080
100
120
140
160
180
200
R= 0.97223
Pea
kstr
om
/ n
A
c(Ni2+) / nM
Abbildung 4-37: Kalibrierfunktion einer Ni2+-Bestimmung. Medium: 10 ml 0,01 M Ammoniakpuffer pH 9,1; 0,1 mM Nioxim als Komplexbildner
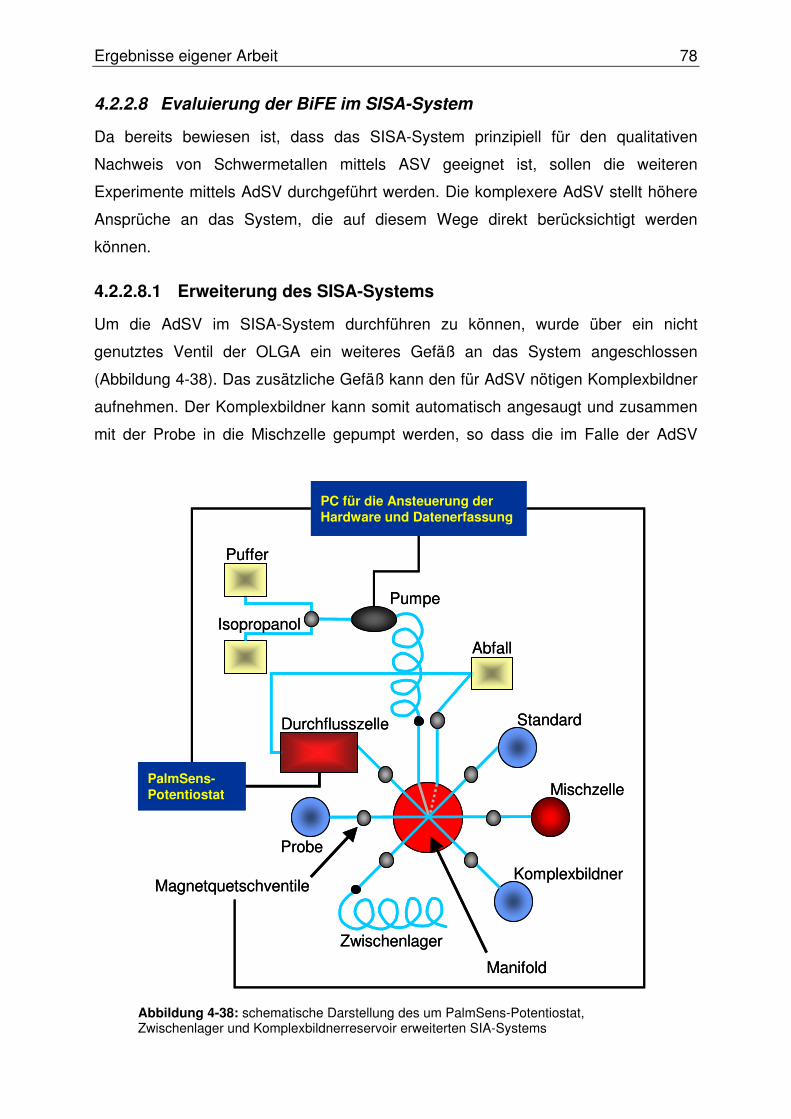
Ergebnisse eigener Arbeit 78
4.2.2.8 Evaluierung der BiFE im SISA-System
Da bereits bewiesen ist, dass das SISA-System prinzipiell für den qualitativen
Nachweis von Schwermetallen mittels ASV geeignet ist, sollen die weiteren
Experimente mittels AdSV durchgeführt werden. Die komplexere AdSV stellt höhere
Ansprüche an das System, die auf diesem Wege direkt berücksichtigt werden
können.
4.2.2.8.1 Erweiterung des SISA-Systems
Um die AdSV im SISA-System durchführen zu können, wurde über ein nicht
genutztes Ventil der OLGA ein weiteres Gefäß an das System angeschlossen
(Abbildung 4-38). Das zusätzliche Gefäß kann den für AdSV nötigen Komplexbildner
aufnehmen. Der Komplexbildner kann somit automatisch angesaugt und zusammen
mit der Probe in die Mischzelle gepumpt werden, so dass die im Falle der AdSV
Puffer
Isopropanol
Pumpe
Abfall
Standard
Mischzelle
Komplexbildner
Zwischenlager
Probe
Durchflusszelle
PC für die Ansteuerung der Hardware und Datenerfassung
PalmSens-Potentiostat
Magnetquetschventile
Manifold
Puffer
Isopropanol
Pumpe
Abfall
Standard
Mischzelle
Komplexbildner
Zwischenlager
Probe
Durchflusszelle
PC für die Ansteuerung der Hardware und Datenerfassung
PalmSens-Potentiostat
Magnetquetschventile
Manifold
Abbildung 4-38: schematische Darstellung des um PalmSens-Potentiostat, Zwischenlager und Komplexbildnerreservoir erweiterten SIA-Systems
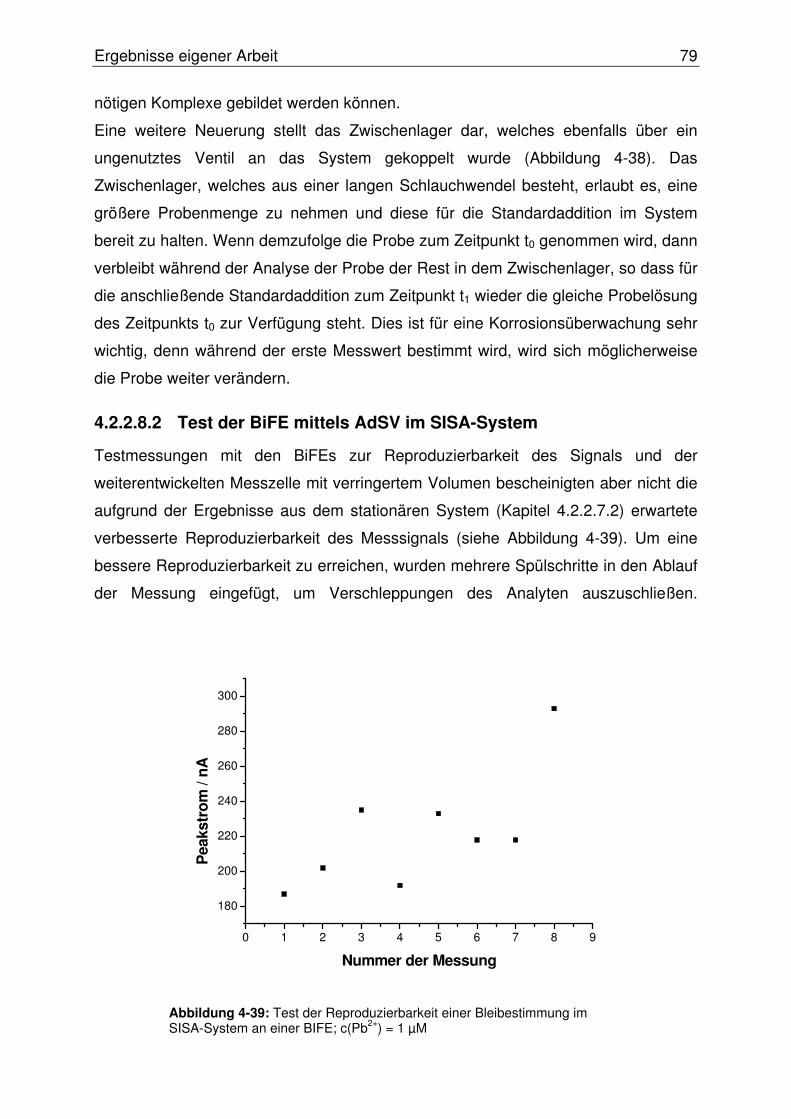
Ergebnisse eigener Arbeit 79
nötigen Komplexe gebildet werden können.
Eine weitere Neuerung stellt das Zwischenlager dar, welches ebenfalls über ein
ungenutztes Ventil an das System gekoppelt wurde (Abbildung 4-38). Das
Zwischenlager, welches aus einer langen Schlauchwendel besteht, erlaubt es, eine
größere Probenmenge zu nehmen und diese für die Standardaddition im System
bereit zu halten. Wenn demzufolge die Probe zum Zeitpunkt t0 genommen wird, dann
verbleibt während der Analyse der Probe der Rest in dem Zwischenlager, so dass für
die anschließende Standardaddition zum Zeitpunkt t1 wieder die gleiche Probelösung
des Zeitpunkts t0 zur Verfügung steht. Dies ist für eine Korrosionsüberwachung sehr
wichtig, denn während der erste Messwert bestimmt wird, wird sich möglicherweise
die Probe weiter verändern.
4.2.2.8.2 Test der BiFE mittels AdSV im SISA-System
Testmessungen mit den BiFEs zur Reproduzierbarkeit des Signals und der
weiterentwickelten Messzelle mit verringertem Volumen bescheinigten aber nicht die
aufgrund der Ergebnisse aus dem stationären System (Kapitel 4.2.2.7.2) erwartete
verbesserte Reproduzierbarkeit des Messsignals (siehe Abbildung 4-39). Um eine
bessere Reproduzierbarkeit zu erreichen, wurden mehrere Spülschritte in den Ablauf
der Messung eingefügt, um Verschleppungen des Analyten auszuschließen.
0 1 2 3 4 5 6 7 8 9
180
200
220
240
260
280
300
Pea
kstr
om
/ n
A
Nummer der Messung
Abbildung 4-39: Test der Reproduzierbarkeit einer Bleibestimmung im SISA-System an einer BIFE; c(Pb2+) = 1 µM
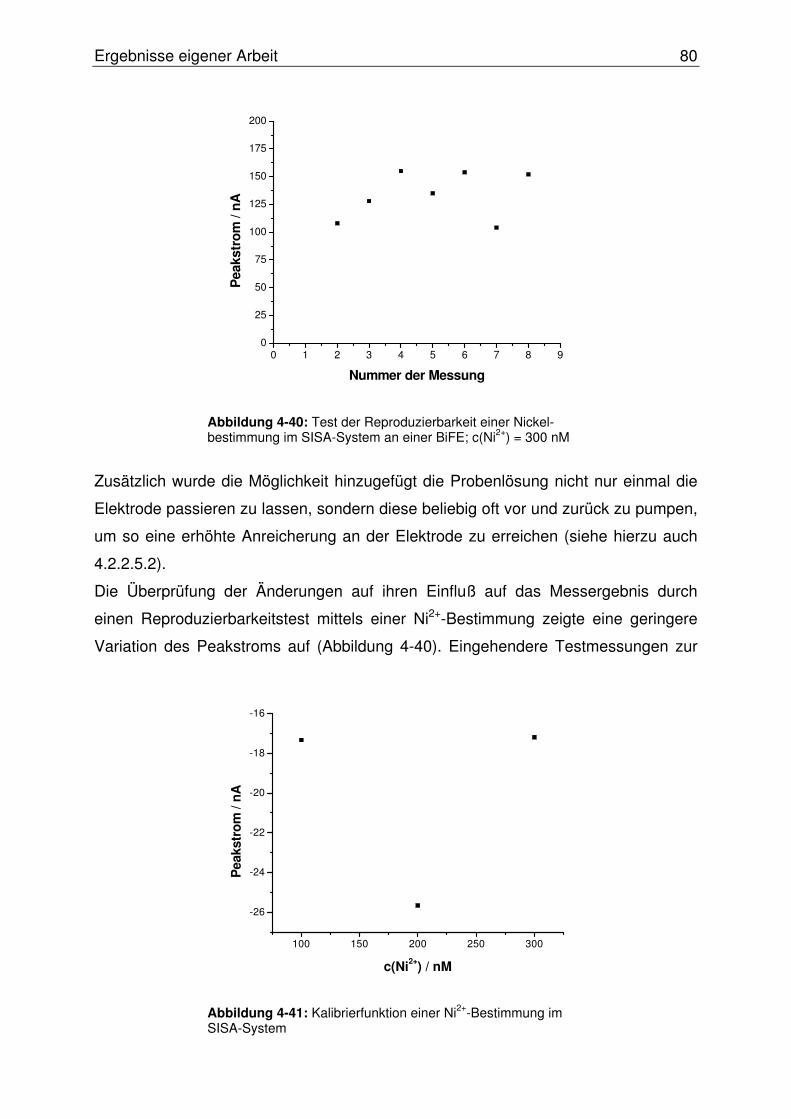
Ergebnisse eigener Arbeit 80
Zusätzlich wurde die Möglichkeit hinzugefügt die Probenlösung nicht nur einmal die
Elektrode passieren zu lassen, sondern diese beliebig oft vor und zurück zu pumpen,
um so eine erhöhte Anreicherung an der Elektrode zu erreichen (siehe hierzu auch
4.2.2.5.2).
Die Überprüfung der Änderungen auf ihren Einfluß auf das Messergebnis durch
einen Reproduzierbarkeitstest mittels einer Ni2+-Bestimmung zeigte eine geringere
Variation des Peakstroms auf (Abbildung 4-40). Eingehendere Testmessungen zur
0 1 2 3 4 5 6 7 8 90
25
50
75
100
125
150
175
200
Pea
kstr
om
/ n
A
Nummer der Messung
Abbildung 4-40: Test der Reproduzierbarkeit einer Nickel-bestimmung im SISA-System an einer BiFE; c(Ni2+) = 300 nM
100 150 200 250 300
-26
-24
-22
-20
-18
-16
Pea
kstr
om
/ n
A
c(Ni2+) / nM
Abbildung 4-41: Kalibrierfunktion einer Ni2+-Bestimmung im SISA-System

Ergebnisse eigener Arbeit 81
Kalibrierung des Systems zeigten jedoch, dass das Problem weiterhin bei der
Messzelle zu suchen ist, da eine Kalibrierung des Systems nicht möglich war (siehe
Abbildung 4-41).
Die offensichtlich ungleichmäßigen Verwirbelungen in der Messzelle reichten
anscheinend aus, um den quantitativen Nachweis des Schwermetalls weiterhin zu
stören. Aufgrund dieser Tatsache, soll im nächsten Kapitel eine Weiterentwicklung
einer Messzelle vorgestellt werden, die an die stationären Systeme angelehnt ist.
4.2.2.8.3 Die miniaturisierte Rührmesszelle
In einem stationären System zur Strippinganalyse wird eine elektrochemische
Messzelle verwendet, in der die konvektiven Bedingungen zur Anreicherung des
Analyten an der Arbeitselektrode mittels eines externen Rührers, meist Magnet-
rührer, erreicht werden. Es sollen damit einheitliche Bedingungen für jede Messung
und ein besserer Massentransport zur Elektrode gewährleistet werden. Dieses
Konzept lässt sich auch auf ein SIA-System übertragen.
Zu diesem Zweck wurde eine miniaturisierte
Rührmesszelle mit einem Innenvolumen
von 1 ml entwickelt, die sich, wie in
Abbildung 4-42 zu sehen ist, über einen
seitlichen Ein-/Auslass befüllen und nach
der Messung wieder entleeren lässt. Die
Konvektion während der Anreicherung wird
über einen Magnetrührer erreicht, der sich
über das OLGA-System ein- und
ausschalten lässt. Die drei Elektroden
werden hier durch Löcher im Deckel der
Messzelle hindurchgeführt. Als Gegen-
elektrode dient hier ein Platindraht und als
Referenz eine, miniaturisierte Ag/AgCl-
Elektrode (siehe Abschnitt 6.2.4). Die vorher verwendete einschraubbare
Referenzelektrode fand hier aufgrund ihres Durchmessers keinen Platz. Der Vorteil
dieser Messzelle liegt in der schon bekannten Technik aus der stationär
durchgeführten Strippinganalyse.
Die Experimente mit diesem Messzellentyp zeichneten sich durch eine verbesserte
Korrelation zwischen der Konzentration des Analyten und der Signalhöhe aus, wie in
CERE
WE
Magnetrührfisch
CERE
WE
Magnetrührfisch
Abbildung 4-42: schematische Darstellung einer miniaturisierte Rührmesszelle für das SISA-System; Zellvolumen = 1 ml
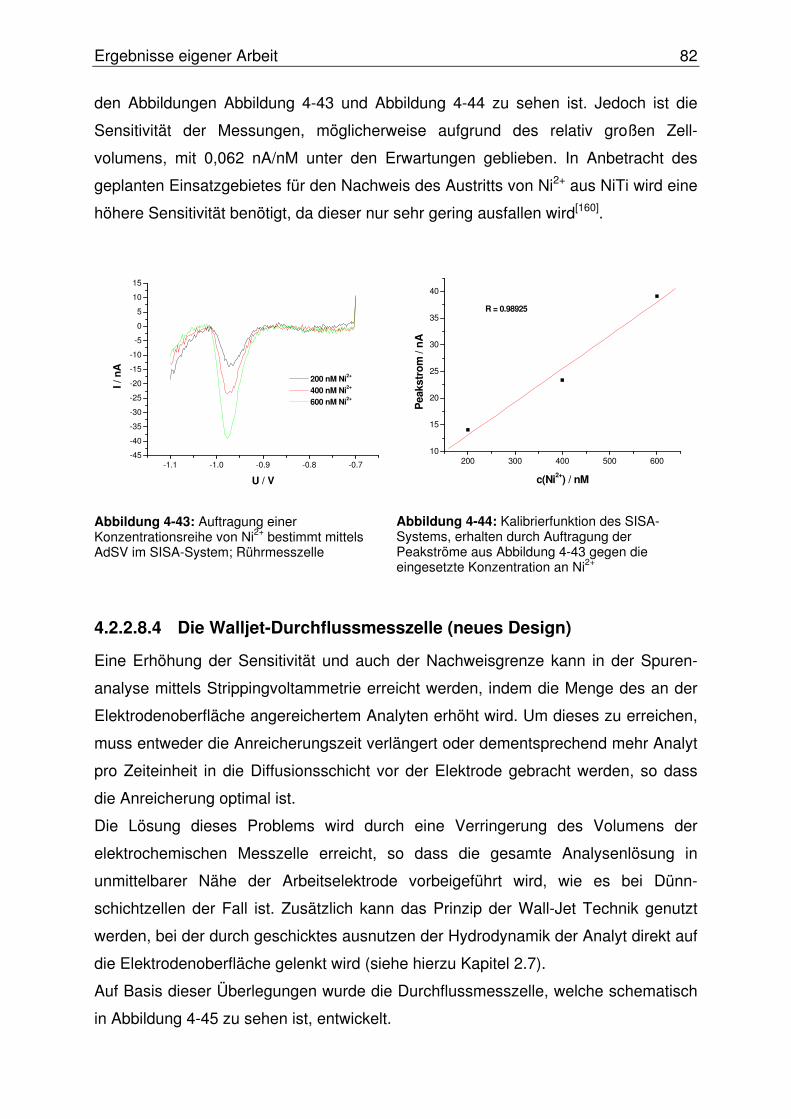
Ergebnisse eigener Arbeit 82
den Abbildungen Abbildung 4-43 und Abbildung 4-44 zu sehen ist. Jedoch ist die
Sensitivität der Messungen, möglicherweise aufgrund des relativ großen Zell-
volumens, mit 0,062 nA/nM unter den Erwartungen geblieben. In Anbetracht des
geplanten Einsatzgebietes für den Nachweis des Austritts von Ni2+ aus NiTi wird eine
höhere Sensitivität benötigt, da dieser nur sehr gering ausfallen wird[160].
4.2.2.8.4 Die Walljet-Durchflussmesszelle (neues Design)
Eine Erhöhung der Sensitivität und auch der Nachweisgrenze kann in der Spuren-
analyse mittels Strippingvoltammetrie erreicht werden, indem die Menge des an der
Elektrodenoberfläche angereichertem Analyten erhöht wird. Um dieses zu erreichen,
muss entweder die Anreicherungszeit verlängert oder dementsprechend mehr Analyt
pro Zeiteinheit in die Diffusionsschicht vor der Elektrode gebracht werden, so dass
die Anreicherung optimal ist.
Die Lösung dieses Problems wird durch eine Verringerung des Volumens der
elektrochemischen Messzelle erreicht, so dass die gesamte Analysenlösung in
unmittelbarer Nähe der Arbeitselektrode vorbeigeführt wird, wie es bei Dünn-
schichtzellen der Fall ist. Zusätzlich kann das Prinzip der Wall-Jet Technik genutzt
werden, bei der durch geschicktes ausnutzen der Hydrodynamik der Analyt direkt auf
die Elektrodenoberfläche gelenkt wird (siehe hierzu Kapitel 2.7).
Auf Basis dieser Überlegungen wurde die Durchflussmesszelle, welche schematisch
in Abbildung 4-45 zu sehen ist, entwickelt.
-1.1 -1.0 -0.9 -0.8 -0.7-45
-40
-35
-30
-25
-20
-15
-10
-5
0
5
10
15
200 nM Ni2+
400 nM Ni2+
600 nM Ni2+
I / n
A
U / V
Abbildung 4-43: Auftragung einer Konzentrationsreihe von Ni2+ bestimmt mittels AdSV im SISA-System; Rührmesszelle
200 300 400 500 60010
15
20
25
30
35
40
R = 0.98925
Pea
kstr
om
/ n
A
c(Ni2+) / nM
Abbildung 4-44: Kalibrierfunktion des SISA-Systems, erhalten durch Auftragung der Peakströme aus Abbildung 4-43 gegen die eingesetzte Konzentration an Ni2+

Ergebnisse eigener Arbeit 83
Die Messzelle besteht aus zwei Plexiglasteilen, die miteinander verschraubt werden.
Der untere Teil enthält eine Bohrung für die Arbeitselektrode, die gegenüber dem
Einlass angeordnet ist. Zudem befindet sich im unteren Teil der Zelle eine PTFE-
Dichtung, welche bei geschlossener Messzelle Ober- und Unterteil dicht miteinander
verbindet. Der obere Teil enthält drei Bohrungen, wobei die mittlere als Einlass und
die beiden äußeren als Auslass dienen. Die Auslässe sind symmetrisch zum Einlass
angeordnet, damit die eingepumpte Lösung gleichmäßig zu beiden Seiten abgeführt
werden kann und somit ein gleichförmiges Strömungsprofil an der Arbeitselektrode
herrscht. In der mittleren Bohrung ist zusätzlich eine Edelstahlkanüle eingeklebt, die
als Gegenelektrode dient. In den Boden des Oberteils ist ein Kanal gefräst, welcher
die Verbindung zwischen dem Ein- und den Auslässen darstellt. Er besitzt eine Breite
von 2 mm, dies entspricht dem Durchmesser der Arbeitselektrode, und eine Tiefe
von 0,7 mm. Von der Rückseite der Messzelle kann die schon in der ersten
Durchflussmesszelle (siehe Abschnitt 4.2.2.3) verwendete Ag/AgCl-Referenz
eingeschraubt werden, ohne in diesem Fall für das Zellvolumen bestimmend zu sein,
da sie über eine 1 mm Bohrung mit dem linken Auslasskanal verbunden ist.
CE
WE
RE
Dichtung
CE
WE
RE
Dichtung
Abbildung 4-45: schematische Abbildung der Wall-Jet Durchflussmesszelle für das SISA-System.
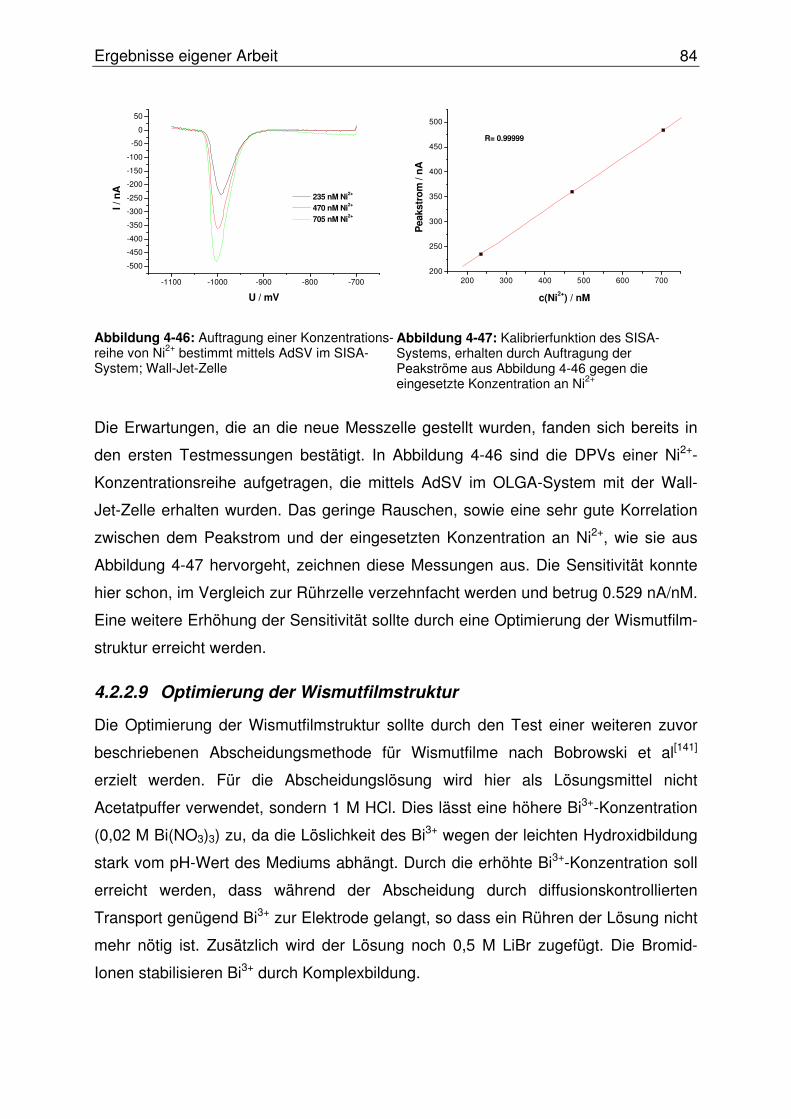
Ergebnisse eigener Arbeit 84
Die Erwartungen, die an die neue Messzelle gestellt wurden, fanden sich bereits in
den ersten Testmessungen bestätigt. In Abbildung 4-46 sind die DPVs einer Ni2+-
Konzentrationsreihe aufgetragen, die mittels AdSV im OLGA-System mit der Wall-
Jet-Zelle erhalten wurden. Das geringe Rauschen, sowie eine sehr gute Korrelation
zwischen dem Peakstrom und der eingesetzten Konzentration an Ni2+, wie sie aus
Abbildung 4-47 hervorgeht, zeichnen diese Messungen aus. Die Sensitivität konnte
hier schon, im Vergleich zur Rührzelle verzehnfacht werden und betrug 0.529 nA/nM.
Eine weitere Erhöhung der Sensitivität sollte durch eine Optimierung der Wismutfilm-
struktur erreicht werden.
4.2.2.9 Optimierung der Wismutfilmstruktur
Die Optimierung der Wismutfilmstruktur sollte durch den Test einer weiteren zuvor
beschriebenen Abscheidungsmethode für Wismutfilme nach Bobrowski et al[141]
erzielt werden. Für die Abscheidungslösung wird hier als Lösungsmittel nicht
Acetatpuffer verwendet, sondern 1 M HCl. Dies lässt eine höhere Bi3+-Konzentration
(0,02 M Bi(NO3)3) zu, da die Löslichkeit des Bi3+ wegen der leichten Hydroxidbildung
stark vom pH-Wert des Mediums abhängt. Durch die erhöhte Bi3+-Konzentration soll
erreicht werden, dass während der Abscheidung durch diffusionskontrollierten
Transport genügend Bi3+ zur Elektrode gelangt, so dass ein Rühren der Lösung nicht
mehr nötig ist. Zusätzlich wird der Lösung noch 0,5 M LiBr zugefügt. Die Bromid-
Ionen stabilisieren Bi3+ durch Komplexbildung.
-1100 -1000 -900 -800 -700
-500
-450
-400
-350
-300
-250
-200
-150
-100
-50
0
50
235 nM Ni2+
470 nM Ni2+
705 nM Ni2+
I / n
A
U / mV
Abbildung 4-46: Auftragung einer Konzentrations-reihe von Ni2+ bestimmt mittels AdSV im SISA-System; Wall-Jet-Zelle
200 300 400 500 600 700200
250
300
350
400
450
500
R= 0.99999
Pea
kstr
om
/ n
A
c(Ni2+) / nM
Abbildung 4-47: Kalibrierfunktion des SISA-Systems, erhalten durch Auftragung der Peakströme aus Abbildung 4-46 gegen die eingesetzte Konzentration an Ni2+
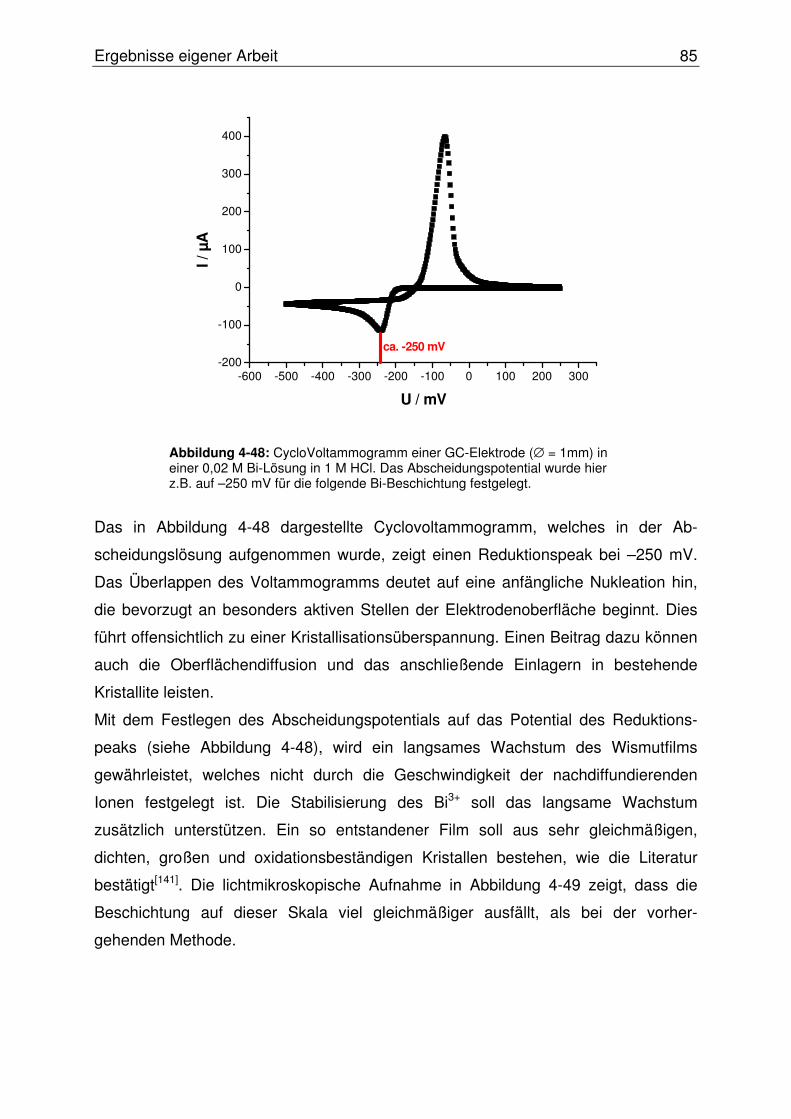
Ergebnisse eigener Arbeit 85
Das in Abbildung 4-48 dargestellte Cyclovoltammogramm, welches in der Ab-
scheidungslösung aufgenommen wurde, zeigt einen Reduktionspeak bei –250 mV.
Das Überlappen des Voltammogramms deutet auf eine anfängliche Nukleation hin,
die bevorzugt an besonders aktiven Stellen der Elektrodenoberfläche beginnt. Dies
führt offensichtlich zu einer Kristallisationsüberspannung. Einen Beitrag dazu können
auch die Oberflächendiffusion und das anschließende Einlagern in bestehende
Kristallite leisten.
Mit dem Festlegen des Abscheidungspotentials auf das Potential des Reduktions-
peaks (siehe Abbildung 4-48), wird ein langsames Wachstum des Wismutfilms
gewährleistet, welches nicht durch die Geschwindigkeit der nachdiffundierenden
Ionen festgelegt ist. Die Stabilisierung des Bi3+ soll das langsame Wachstum
zusätzlich unterstützen. Ein so entstandener Film soll aus sehr gleichmäßigen,
dichten, großen und oxidationsbeständigen Kristallen bestehen, wie die Literatur
bestätigt[141]. Die lichtmikroskopische Aufnahme in Abbildung 4-49 zeigt, dass die
Beschichtung auf dieser Skala viel gleichmäßiger ausfällt, als bei der vorher-
gehenden Methode.
-600 -500 -400 -300 -200 -100 0 100 200 300-200
-100
0
100
200
300
400
ca. -250 mV
I / µ
A
U / mV
Abbildung 4-48: CycloVoltammogramm einer GC-Elektrode (∅ = 1mm) in einer 0,02 M Bi-Lösung in 1 M HCl. Das Abscheidungspotential wurde hier z.B. auf –250 mV für die folgende Bi-Beschichtung festgelegt.

Ergebnisse eigener Arbeit 86
Damit jede Elektrode mit der gleichen Filmdicke beschichtet wird, wird die
Abscheidung bei einem bestimmten Ladungsumsatz abgebrochen. Der optimale
Ladungsumsatz und somit die beste Bi-Schicht wurden bestimmt. Zu diesem Zweck
wurde eine Reihe von AdSV-Experimenten mit unterschiedlicher Bi-Schichtdicke auf
der Arbeitselektrode durchgeführt. In Abbildung 4-50 ist die Auftragung des
Peakstroms einer Ni-Bestimmung gegen die umgesetzte Ladungsdichte während der
Bi-Abscheidung dargestellt. Es ist ein Maximum des Peakstroms bei einer Ladungs-
dichte von 1,9 mC/mm2 erkennbar. Mit dieser Schichtdicke sollte die beste
Sensitivität erreicht werden. Bei einer Ladungsdichte < 0,9 mC/mm2 ist der Bi-Film
nicht geschlossen und die Elektrode ist unbrauchbar. Bei einer Ladungsdichte ≥
2,6 mC wird die Beschichtung zu dick und haftet offensichtlich nicht mehr
ausreichend an der Oberfläche, so dass es häufig zu einem Verlust der Bi-Schicht
während der Messung kommt und das Experiment abgebrochen werden muss.
Die so optimierten BiFEs sollten für die geplante Applikation ideal sein. Ihr Verhalten
bei strippingvoltammetrischen Messungen im Durchflusssystem wird in den
folgenden Kapiteln über die AdSV-SIA-Kopplung noch weiter ausgeführt.
Abbildung 4-49: Lichtmikroskopische Abbildung einer BiFE (∅ = 1 mm), die nach einer Beschreibung von Bobrowski et al beschichtet wurde.
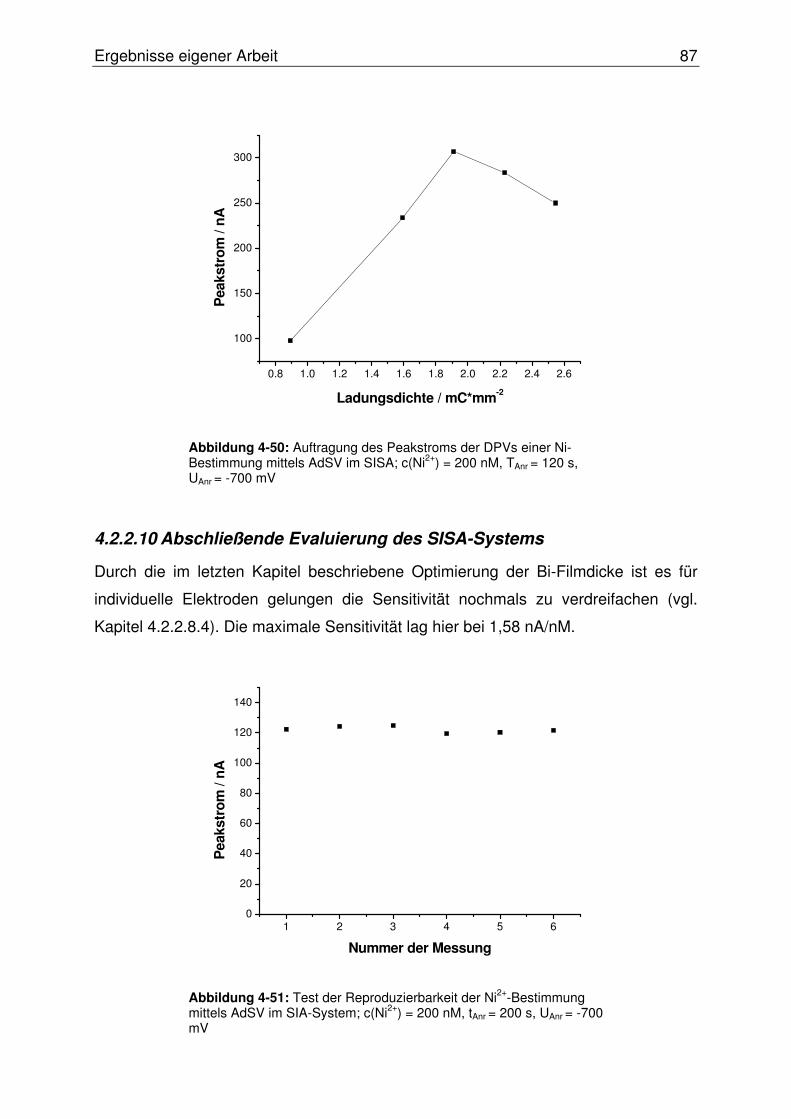
Ergebnisse eigener Arbeit 87
4.2.2.10 Abschließende Evaluierung des SISA-Systems
Durch die im letzten Kapitel beschriebene Optimierung der Bi-Filmdicke ist es für
individuelle Elektroden gelungen die Sensitivität nochmals zu verdreifachen (vgl.
Kapitel 4.2.2.8.4). Die maximale Sensitivität lag hier bei 1,58 nA/nM.
0.8 1.0 1.2 1.4 1.6 1.8 2.0 2.2 2.4 2.6
100
150
200
250
300
Pea
kstr
om
/ n
A
Ladungsdichte / mC*mm-2
Abbildung 4-50: Auftragung des Peakstroms der DPVs einer Ni-Bestimmung mittels AdSV im SISA; c(Ni2+) = 200 nM, TAnr = 120 s, UAnr = -700 mV
1 2 3 4 5 60
20
40
60
80
100
120
140
Pea
kstr
om
/ n
A
Nummer der Messung
Abbildung 4-51: Test der Reproduzierbarkeit der Ni2+-Bestimmung mittels AdSV im SIA-System; c(Ni2+) = 200 nM, tAnr = 200 s, UAnr = -700 mV

Ergebnisse eigener Arbeit 88
Auch die Reproduzierbarkeit der Messung konnte stark verbessert werden und ist in
Abbildung 4-51 beispielhaft an einer Ni2+-Bestimmung, bei der eine Nickelstandard-
lösung unter Zugabe des Komplexbildners DMG der AdSV im OLGA-System
unterzogen wurde, zu sehen. Die Nickelkonzentration betrug nach Verdünnung
200 nM. Die Peakhöhe erreichte einen durchschnittlichen Wert von 122 nA bei einer
Standardabweichung von nur 2,1 nA. Dieses Ergebnis bescheinigt der Kombination
aus Wall-Jet-Messzelle und der optimierten BIFE mit DMG als Komplexbildner eine
gute Signalstabilität und eine hervorragende Reproduzierbarkeit.
Die Messungen wurden bei einer Anreicherungszeit tAnr = 200 s und einem Potential
von UAnr = -700 mV mittels DPV erhalten. Für diese Parameter muss noch der lineare
Messbereich bestimmt werden. Der lineare Messbereich ist bei der Stripping-
voltammetrie von der Anreicherungszeit abhängig. Es ist somit möglich, indem für
niedrige Konzentrationen die Anreicherungszeit angehoben und für hohe
Konzentrationen dementsprechend herabgesetzt wird, den linearen Bereich an die
jeweilige Anforderung anzupassen. Für die geplanten Korrosionsuntersuchungen ist
der Parameter tAnr schwer abzuschätzen, da anfänglich im reinen Puffer kein Nickel
vorhanden ist und während der Korrosion die Konzentration unter Umständen sehr
stark ansteigen kann, sobald sich erste Korrosions-Pits gebildet haben. Aus diesem
0 200 400 600 800 10000
100
200
300
400
500
600
700
800
900
1000
Pea
kstr
om
/ n
A
c(Ni2+) / nM)
Abbildung 4-52: Bestimmung des linearen Messbereichs der Nickelquantifizierung mittels AdSV im SISA-System; c(DMG) = 0,13 mM, TAnr = 200s, UAnr = -700 mV
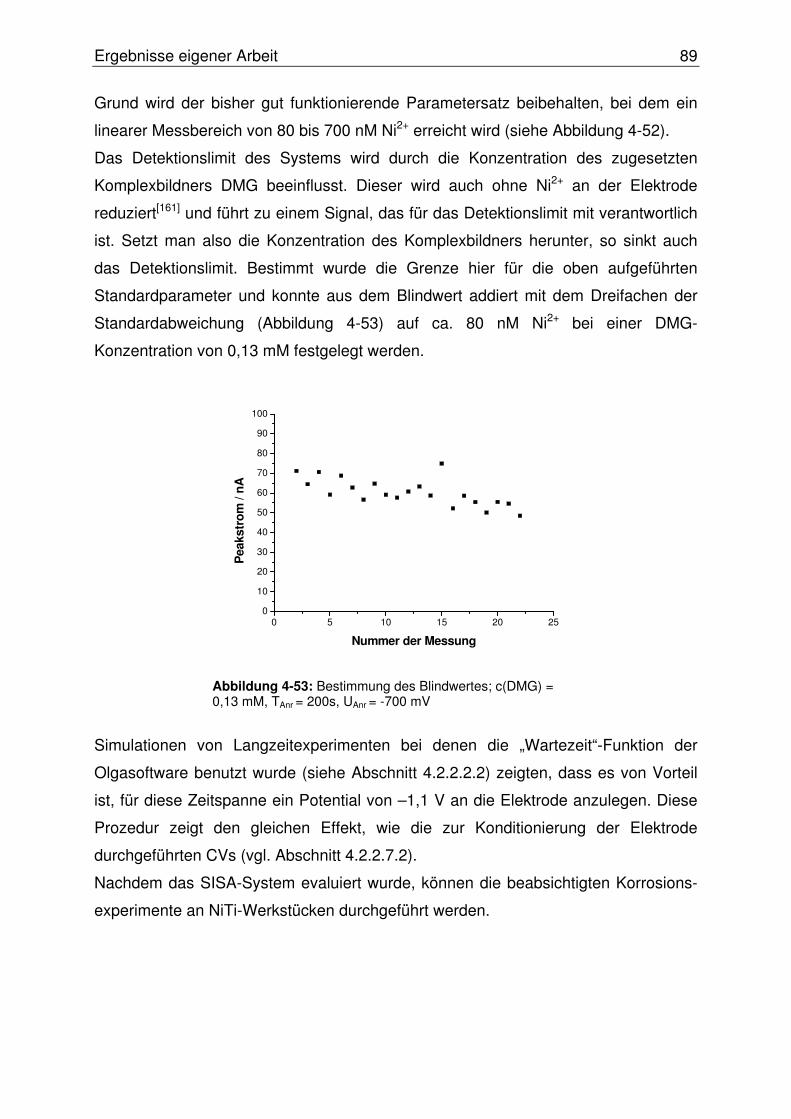
Ergebnisse eigener Arbeit 89
Grund wird der bisher gut funktionierende Parametersatz beibehalten, bei dem ein
linearer Messbereich von 80 bis 700 nM Ni2+ erreicht wird (siehe Abbildung 4-52).
Das Detektionslimit des Systems wird durch die Konzentration des zugesetzten
Komplexbildners DMG beeinflusst. Dieser wird auch ohne Ni2+ an der Elektrode
reduziert[161] und führt zu einem Signal, das für das Detektionslimit mit verantwortlich
ist. Setzt man also die Konzentration des Komplexbildners herunter, so sinkt auch
das Detektionslimit. Bestimmt wurde die Grenze hier für die oben aufgeführten
Standardparameter und konnte aus dem Blindwert addiert mit dem Dreifachen der
Standardabweichung (Abbildung 4-53) auf ca. 80 nM Ni2+ bei einer DMG-
Konzentration von 0,13 mM festgelegt werden.
Simulationen von Langzeitexperimenten bei denen die „Wartezeit“-Funktion der
Olgasoftware benutzt wurde (siehe Abschnitt 4.2.2.2.2) zeigten, dass es von Vorteil
ist, für diese Zeitspanne ein Potential von –1,1 V an die Elektrode anzulegen. Diese
Prozedur zeigt den gleichen Effekt, wie die zur Konditionierung der Elektrode
durchgeführten CVs (vgl. Abschnitt 4.2.2.7.2).
Nachdem das SISA-System evaluiert wurde, können die beabsichtigten Korrosions-
experimente an NiTi-Werkstücken durchgeführt werden.
0 5 10 15 20 250
10
20
30
40
50
60
70
80
90
100
Pea
kstr
om
/ n
A
Nummer der Messung
Abbildung 4-53: Bestimmung des Blindwertes; c(DMG) = 0,13 mM, TAnr = 200s, UAnr = -700 mV

Ergebnisse eigener Arbeit 90
4.2.3 Online Bestimmung des Nickelaustritts aus korrodierendem
NiTi
Die hier verwendeten Proben wurden von dem
Stangenmaterial mit ∅ = 12,5 mm und der
Zusammensetzung Ni = 55,73 Ti = 44,04 (wt%),
welches dem SFB 459 zur Verfügung gestellt wurde
(siehe Abschnitt 6.5.1), nach der Prozedur aus
Kapitel 6.5 hergestellt. Es wurde das mechanische
Polieren als Endbearbeitung verwendet und der
Rand mit einem Zweikomponentenkleber ab-
gedichtet.
Da NiTi, wie erwähnt, eine oxidische Passivschicht
besitzt, wurde zunächst das Durchbruchspotential
bestimmt. Das Durchbruchspotential ist die
Spannung bei der die Passivschicht aufbricht und
Korrosion beginnt. Dieses kann mit einem linearen
Spannungsvorschub mittels eines Potentiostaten
bestimmt werden.
Zu diesem Zweck wird die Probe, in eine elektrochemische Messzelle eingebaut und
als Arbeitselektrode geschaltet (Abbildung 4-54). Zunächst wird das Ruhepotential
der unbelasteten Probe bestimmt. Dies kann zwischen 30 Minuten und mehreren
Stunden aufgenommen werden. Für unsere Zwecke sind 30 Minuten ausreichend,
da sich das Potential im weiteren Verlauf nur noch vernachlässigbar verändert. Das
bestimmte Ruhepotential dient in der anschließenden Messung als Startpotential und
lag in diesem Fall bei –142 mV. Die Spannung wurde mit einer Geschwindigkeit von
0,167 mV/s auf 1,6 V erhöht. Die langsame Vorschubgeschwindigkeit ist nötig, um an
der Grenzfläche zwischen Probe und Elektrolyt ein Gleichgewicht aufrecht zu
erhalten.
Das Voltammogramm in Abbildung 4-55 zeigt die Strom/Spannungskurve der NiTi-
Probe. Der Beginn des starken Stromanstiegs zeigt den Durchbruch der Passiv-
schicht an, so dass das Durchbruchspotential auf 1,4 V bestimmt werden kann.
Dieses Potential wird im weiteren Verlauf der Experimente benötigt, um evtl.
Korrosion durch anlegen einer Spannung zu induzieren.
A
B
C
Ni2+
A
B
C
Ni2+
Abbildung 4-54: schematische Darstellung der Korrosionszelle; A = Platindrahtgegenelektrode, B = in Polymer eingebettetes NiTi als Arbeitelektrode, C = Ag/AgCl-Referenzelektrode
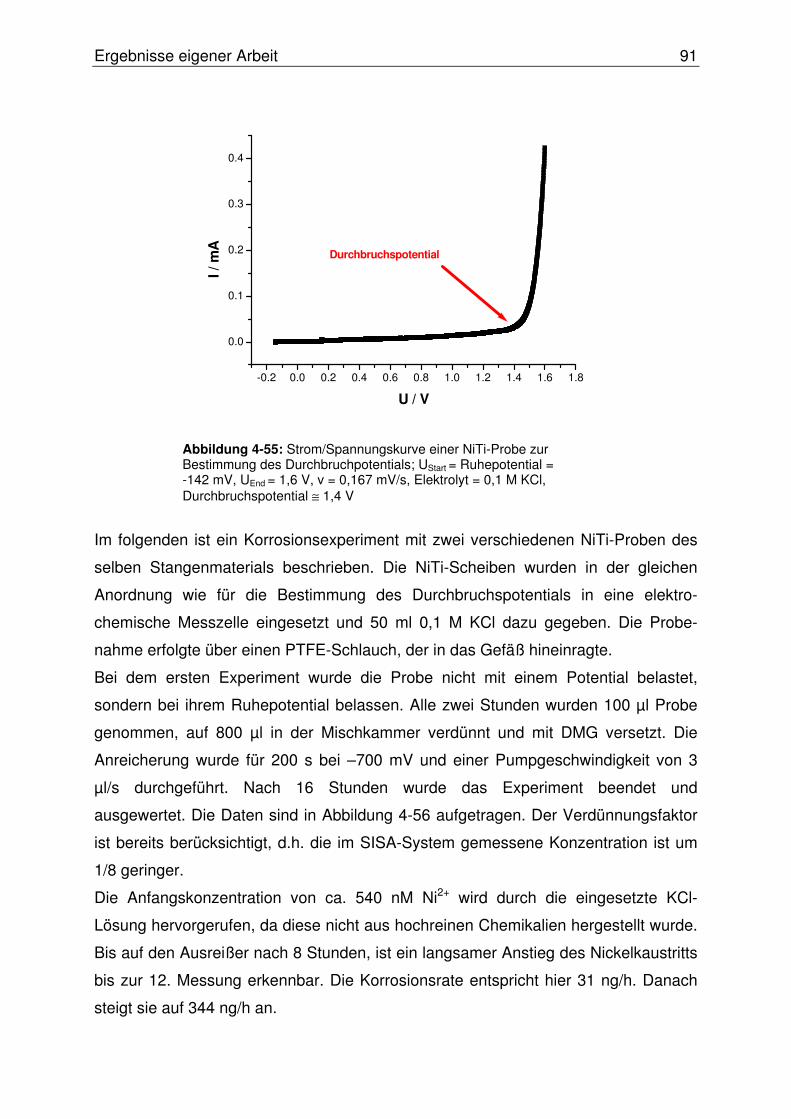
Ergebnisse eigener Arbeit 91
Im folgenden ist ein Korrosionsexperiment mit zwei verschiedenen NiTi-Proben des
selben Stangenmaterials beschrieben. Die NiTi-Scheiben wurden in der gleichen
Anordnung wie für die Bestimmung des Durchbruchspotentials in eine elektro-
chemische Messzelle eingesetzt und 50 ml 0,1 M KCl dazu gegeben. Die Probe-
nahme erfolgte über einen PTFE-Schlauch, der in das Gefäß hineinragte.
Bei dem ersten Experiment wurde die Probe nicht mit einem Potential belastet,
sondern bei ihrem Ruhepotential belassen. Alle zwei Stunden wurden 100 µl Probe
genommen, auf 800 µl in der Mischkammer verdünnt und mit DMG versetzt. Die
Anreicherung wurde für 200 s bei –700 mV und einer Pumpgeschwindigkeit von 3
µl/s durchgeführt. Nach 16 Stunden wurde das Experiment beendet und
ausgewertet. Die Daten sind in Abbildung 4-56 aufgetragen. Der Verdünnungsfaktor
ist bereits berücksichtigt, d.h. die im SISA-System gemessene Konzentration ist um
1/8 geringer.
Die Anfangskonzentration von ca. 540 nM Ni2+ wird durch die eingesetzte KCl-
Lösung hervorgerufen, da diese nicht aus hochreinen Chemikalien hergestellt wurde.
Bis auf den Ausreißer nach 8 Stunden, ist ein langsamer Anstieg des Nickelkaustritts
bis zur 12. Messung erkennbar. Die Korrosionsrate entspricht hier 31 ng/h. Danach
steigt sie auf 344 ng/h an.
-0.2 0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8
0.0
0.1
0.2
0.3
0.4
Durchbruchspotential
I / m
A
U / V
Abbildung 4-55: Strom/Spannungskurve einer NiTi-Probe zur Bestimmung des Durchbruchpotentials; UStart = Ruhepotential = -142 mV, UEnd = 1,6 V, v = 0,167 mV/s, Elektrolyt = 0,1 M KCl, Durchbruchspotential ≅ 1,4 V

Ergebnisse eigener Arbeit 92
Die Passivschicht konnte offensichtlich einen Austritt von Nickel in die Lösung von
Anfang an nicht unterbinden. Somit muss ein Defekt in der Oxidschicht angenommen
werden. Nach ca. 12 Stunden muss sich dieser Defekt soweit ausgeweitet haben
-2 0 2 4 6 8 10 12 14 16 181.0
1.5
2.0
2.5
3.0
3.5
m(N
i) /
µg
Zeit / h
Abbildung 4-56: Auftragung der durch Korrosion ausgetretenen Masse an Nickel gegen die Zeit der Exposition der NiTi-Probe ermittelt mit dem SISA-System; Probenfläche: A≈ 113 mm2, Elektrolyt: 50 ml 0,1 M KCl, Anreicherung: 200s –700 mV
0 5 10 15 20 250.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
m(N
i) /
µg
Zeit / h
Abbildung 4-57: Auftragung der durch Korrosion ausgetretenen Masse an Nickel gegen die Zeit der Exposition der NiTi-Probe ermittelt mit dem SISA-System; Probenfläche: A≈ 113 mm2, Elektrolyt: 50 ml 0,1 M KCl, Anreicherung: 200s –700 mV
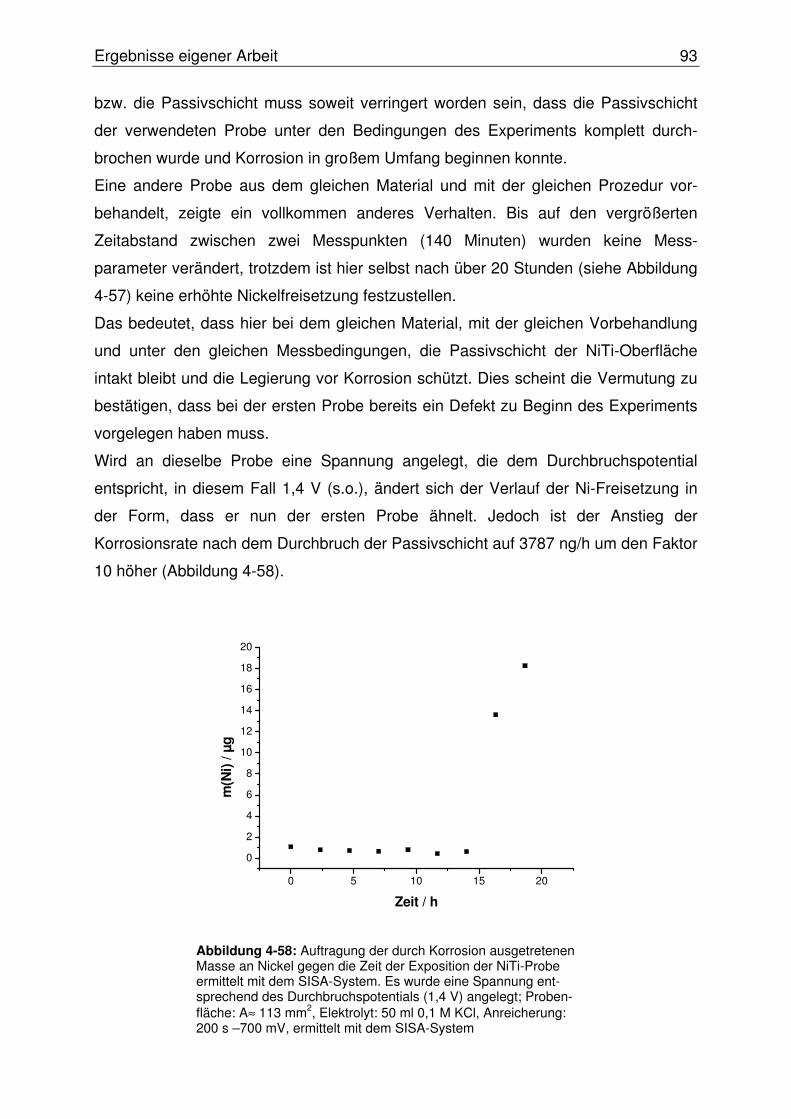
Ergebnisse eigener Arbeit 93
bzw. die Passivschicht muss soweit verringert worden sein, dass die Passivschicht
der verwendeten Probe unter den Bedingungen des Experiments komplett durch-
brochen wurde und Korrosion in großem Umfang beginnen konnte.
Eine andere Probe aus dem gleichen Material und mit der gleichen Prozedur vor-
behandelt, zeigte ein vollkommen anderes Verhalten. Bis auf den vergrößerten
Zeitabstand zwischen zwei Messpunkten (140 Minuten) wurden keine Mess-
parameter verändert, trotzdem ist hier selbst nach über 20 Stunden (siehe Abbildung
4-57) keine erhöhte Nickelfreisetzung festzustellen.
Das bedeutet, dass hier bei dem gleichen Material, mit der gleichen Vorbehandlung
und unter den gleichen Messbedingungen, die Passivschicht der NiTi-Oberfläche
intakt bleibt und die Legierung vor Korrosion schützt. Dies scheint die Vermutung zu
bestätigen, dass bei der ersten Probe bereits ein Defekt zu Beginn des Experiments
vorgelegen haben muss.
Wird an dieselbe Probe eine Spannung angelegt, die dem Durchbruchspotential
entspricht, in diesem Fall 1,4 V (s.o.), ändert sich der Verlauf der Ni-Freisetzung in
der Form, dass er nun der ersten Probe ähnelt. Jedoch ist der Anstieg der
Korrosionsrate nach dem Durchbruch der Passivschicht auf 3787 ng/h um den Faktor
10 höher (Abbildung 4-58).
0 5 10 15 20
0
2
4
6
8
10
12
14
16
18
20
m(N
i) /
µg
Zeit / h
Abbildung 4-58: Auftragung der durch Korrosion ausgetretenen Masse an Nickel gegen die Zeit der Exposition der NiTi-Probe ermittelt mit dem SISA-System. Es wurde eine Spannung ent-sprechend des Durchbruchspotentials (1,4 V) angelegt; Proben-fläche: A≈ 113 mm2, Elektrolyt: 50 ml 0,1 M KCl, Anreicherung: 200 s –700 mV, ermittelt mit dem SISA-System

Ergebnisse eigener Arbeit 94
Das bedeutet, dass die Oberfläche einer NiTi-Legierung durch ihre oxidische
Passivschicht sehr gut vor Korrosion geschützt ist, solange diese intakt ist. Bei der
zuerst gezeigten Probe muss bereits zu Beginn des Experiments ein Defekt in der
Oxidschicht vorgelegen haben, der für das anfängliche, langsame Ausbluten von Ni2+
und der nach 12 Stunden einsetzenden Lochfraßkorrosion verantwortlich ist. Denn
eine fehlerfreie Passivschicht hält, wie die zweite Messung zeigt, länger als 12
Stunden einer angelegten Spannung im Grenzbereich des Durchbruchspotentials
stand.

Ergebnisse eigener Arbeit 95
4.3 Hochaufgelöste lokale Korrosionsuntersuchung mittels SECM
SECM ist wie in Abschnitt 2.2.2 ausführlich erklärt, eine Methode zur Visualisierung
elektrochemischer Eigenschaften von Oberflächen. Dies kann in der Korrosions-
untersuchung ausgenutzt werden, um Schwachstellen in einer Passivschicht oder
Wachstum von Lochfraßkorrosion aufzufinden und zu verfolgen. Die Passivschicht
von NiTi-FGLs sollte wie ein Isolator wirken, so dass an Defekten eine höhere
elektrochemische Aktivität erwartet werden kann als an intakten Stellen.
Wenn Korrosion stattfindet, können mehrere Effekte zu einer Signaländerung im
SECM führen. Zum einen ist an dieser Stelle nicht geschütztes Metall frei zugänglich,
welches eine höhere Leitfähigkeit besitzt als eine passivierte Metalloberfläche. Zum
anderen herrscht durch die Auflösung des Metalls lokal eine erhöhte Ionen-
konzentration. Zusätzlich kann sich die Topographie aufgrund von Lochfraß ändern.
Die folgenden Kapitel werden aufzeigen, mit welchen SECM-Methoden die Korrosion
an NiTi-Legierungen am besten erfasst werden kann.
4.3.1 Potentiometrische SECM mit Mikro-pH-Sonden im „constant-
distance“ Modus
Wenn eine NiTi-Legierung korrodiert, dann löst sich Ni2+ aus dem Metall. Dieses
bildet in einem genügend basischen Medium Ni(OH)2 und entfernt somit Hydroxid-
ionen aus der Lösung. Das führt dementsprechend zu einer pH-Änderung in
NiTi FGL
lokale Korrosion
pH-Mikrosensor
H+H+
NiTi FGL
lokale Korrosion
pH-Mikrosensor
H+H+
Abbildung 4-59: schematische Darstellung einer lokal korrodierenden NiTi-FGL und der damit verbundenen pH-Änderung, die mittels pH-Sensor erfasst wird

Ergebnisse eigener Arbeit 96
Richtung niedrigerer pH-Werte. Mit einer, während meiner Diplomarbeit[162]
entwickelten, potentiometrischen SECM mittels Mikro-pH-Sonden kann ein solcher
Effekt erfasst und visualisiert werden. In Abbildung 4-59 ist der Versuchsablauf
schematisch dargestellt.
Um einen solchen Effekt abzubilden, wurde eine NiTi-Probe wie in Kapitel 4.1.1
beschrieben vorbereitet, kontaktiert und mit einer Küvette versehen. Im Anschluß
wurde die Oberfläche mit einer feinen Nadel angeritzt, um die Passivschicht lokal zu
verletzen. Die Probe wurde als Arbeitselektrode an einen Potentiostaten an-
Abbildung 4-60: videomikroskopische Aufnahme eines Ritzes in der Passivschicht einer NiTi-FGL unter Potentialbelastung; E = 1,6 V
Abbildung 4-61: dreidimensionale Darstellung des pH-Profils eines Kratzers in einer NiTi-Oxidschicht

Ergebnisse eigener Arbeit 97
geschlossen und ein Potential > 1,6 V, also oberhalb des Durchbruchspotentials,
angelegt. Wie in Abbildung 4-60 zu sehen ist, steigen Gasblässchen bevorzugt an
den Orten mit verletzter Passivschicht auf. Das deutet schon darauf hin, dass die
elektrochemische Aktivität an dieser Stelle höher ist.
Die Abbildung des Risses im „constant-distance“ Modus mittels einer Mikro-pH-
Sonde zeigt einen Signalanstieg, der auf eine pH-Erniedrigung hinweist, wenn die
pH-Sonde direkt über dem Riss in der Oxidschicht lokalisiert ist. Die dreidimensionale
Abbildung 4-61 gibt einen Ausschnitt des pH-Profils über dem Kratzer auf der
Oberfläche wieder.
Eine genauere Betrachtung einer einzelnen Linie aus dieser Rasteraufnahme
(Abbildung 4-62) zeigt eine in Hinblick auf zukünftige Experimente zu geringe Orts-
auflösung. Diese hängt zum einen von der Diffusion der H+-Ionen, die mit der Wahl
der Pufferstärke beeinflussbar ist, zum anderen von der Geometrie der Elektrode ab.
Die Elektrodengeometrie entspricht einer Kugelform mit einem ungefähren Durch-
messer von 10 µm, die durch die elektrochemische Abscheidung des pH-sensitiven
Iridiumoxids (vgl. Kapitel 6.3.3) hervorgerufen wird.
Somit ist das Verfahren der potentiometrischen SECM im „constant-distance“ Modus
mittels Mikro-pH-Sensoren prinzipiell geeignet, um Korrosion an NiTi-FGLs zu
visualisieren.
4.3.2 AC-SECM
Eine neue Methode, von der bereits in ersten Experimenten in der Arbeitsgruppe
vielversprechende Ergebnisse[163] erhalten wurden, die sie für die Korrosions-
forschung interessant erscheinen lassen, soll nun auf Nitinol übertragen werden.
535
540
545
550
555
560
565
570
575
0 100 200 300 400 500 600
x / µm
Po
ten
tial
/ m
V (
vs A
g/A
gC
l)
Abbildung 4-62: pH-Linienscan über einen Ritz in einer NiTi-Passivschicht

Ergebnisse eigener Arbeit 98
Die Funktionsweise der AC-SECM wurde im Abschnitt 2.4 bereits erklärt. Die
Möglichkeit mit dieser Methode elektrochemische Aktivitäten an Oberflächen zu
visualisieren, ohne einen Redoxmediator einsetzen zu müssen, macht sie zu einem
interessanten Werkzeug für die Korrosionsuntersuchung an NiTi-FGLs. Es sollte
möglich sein, Defekte in der Passivschicht und Lochfraßkorrosion aufgrund der lokal
höheren Leitfähigkeit im Vergleich zur passivierten Oberfläche der Legierung mit
hoher lateraler Auflösung sichtbar zu machen. Aufgrund der hohen Auflösung wäre
die AC-SECM eine gute Ergänzung zur potentiometrischen SECM mittels Mikro-pH-
Sensoren.
Die lokale Impedanz wird hier in Form der Magnitude R als Potential analog vom
Lock-In Verstärker ausgegeben. Der Wert der Magnitude kann über eine AD-Karte in
den PC eingelesen werden und hat somit die festgelegte Einheit mV. Ebenso ist es
möglich die Phasenverschiebung Φ aufzuzeichnen. Sie wird hier ebenfalls in mV,
also dem proportionalen Wert der Spannung am Ausgang des Lock-In Verstärkers
ausgegeben.
4.3.2.1 Entwicklung einer Tiltwinkel-Kontrolle für das SECM
Bei der SECM gibt es, wie bei allen anderen Rastermikroskopiearten, das Problem
der Abhängigkeit von Auflösung und Messdauer. Je höher die Auflösung einer
Messung, desto länger dauert diese, da mehr Linien, mehr Datenpunkte und evtl.
eine langsamere Scangeschwindigkeit nötig sind. Im Falle der SECM ist ins-
besondere die Messzeit im „constant-height“ Modus sehr viel kürzer als im „constant-
distance“ Modus (siehe Abschnitt 2.2.2.3). Die geringere Geschwindigkeit im
„constant-distance“ Modus liegt in der Überprüfung und ggf. Nachregelung des
Abstands zwischen Probe und Sondenspitze für jeden aufgenommenen Datenpunkt
begründet.
Oft ist es allerdings nicht möglich, eine flache Probe mit dem „constant-height“
Modus abzubilden. Obwohl die Probenoberfläche sehr plan sein kann, ist die Probe
häufig nicht exakt waagerecht auf dem Probenhalter platziert. Diese Abweichung, der
sogenannte Tiltwinkel, führt zu einer abstandsabhängigen Signaldrift in der
Abbildung. Im ungünstigsten Fall kann dieser Verkippungswinkel eine Kollision der
Sonde mit der Oberfläche während des Scannens verursachen. Dabei können
Sonde sowie die Probenoberfläche zerstört werden.

Ergebnisse eigener Arbeit 99
Um nun mit relativ hoher Messgeschwindigkeit solche Proben abzubilden, ist es
denkbar einen Modus einzuführen, der zwischen der Messung in konstanter Höhe
und der Messung mit scherkraftabhängiger Höhenkontrolle angesiedelt ist. Da die
Probenoberfläche selbst weitgehend plan ist, ist es nicht nötig für jeden Datenpunkt
den Abstand zu bestimmen und nachzuregeln, sondern es reicht aus, den Tiltwinkel
der Oberfläche zu bestimmen und diesen auszugleichen. Somit wird der Abstand
zwischen Sonde und Probe im Rahmen der Genauigkeit der Winkelbstimmung und
der Planarität der Oberfläche konstant gehalten, ohne den enormen Zeitaufwand der
scherkraftabhängigen Höhenkontrolle während der Messung. Dies ist gerade bei der
Beobachtung von Korrosionsphänomenen wichtig, da sich die Probe mit der Zeit
stark verändern kann.
Die Winkelkorrektur ist nach einem einfachen Prinzip umgesetzt worden. Die
Elektrode wird gerade soweit von der Oberfläche entfernt positioniert, dass sie sich
nicht im Nahfeld befindet. Danach werden drei Annäherungskurven an verschie-
denen Stellen auf der Probenoberfläche aufgenommen (siehe Abbildung 4-63) und
bei einer vorher festgelegten Änderung des Signals abgebrochen. Aus den
erhaltenen Koordinaten lässt sich eine Ebene aufspannen und der Winkel in x- und
y-Richtung berechnen.
X-TiltwinkelY-Tiltwinkel
1.
2.
3.
Elektrode
X-TiltwinkelY-Tiltwinkel
1.
2.
3.
Elektrode
Abbildung 4-63: schematische Darstellung der Bestimmung der Tiltwinkel einer Probe im SECM mittels der Annäherung an drei Punkten

Ergebnisse eigener Arbeit 100
Zu diesem Zweck wurde ein neues Modul für die SECM-Software programmiert und
in das bestehende Programm integriert. Das Modul erlaubt die Erfassung der Tilt-
winkel vollkommen automatisch. In Abbildung 4-64 ist die Benutzeroberfläche dieses
Moduls zu sehen. Auf der rechten Seite können die Parameter für die Annäherungen
eingestellt werden. Die Schrittweite ergibt sich aus dem Parameter „Steps/Timer“,
dabei entspricht 1 „Step“ 10 nm. Sie sollte nur wenige 100 nm betragen, da sonst die
Genauigkeit der Winkelbestimmung leidet. Multipliziert mit dem Parameter „Timer“,
der in Millisekunden angeben wird, ergibt sich die Annäherungsgeschwindigkeit.
Diese sollte nicht über 1 µm/s liegen, damit gute Ergebnisse erzielt werden können.
Der Abstand zwischen den Annäherungspunkten wird unter „(+)x-Dist.“ und „(+)y-
Dist.“ eingetragen und sollte den Ausmaßen des späteren Abbildungsgebietes
entsprechen. Damit wird der Winkel exakt für das interessierende Gebiet erfasst. Der
Parameter „z-Dist“ gibt wieder, wie weit die Elektrode nach einer erfolgten
Annäherung wieder von der Oberfläche entfernt werden soll. Hier sollte ein Wert
eingetragen werden, der gewährleistet, dass die Elektrode außerhalb des
Feedbackbereichs positioniert wird. Bei Betätigung der Schaltfläche „F“ öffnet sich
Abbildung 4-64: Screenshot der Benutzeroberfläche des Tiltwinkelkorrektur-Moduls
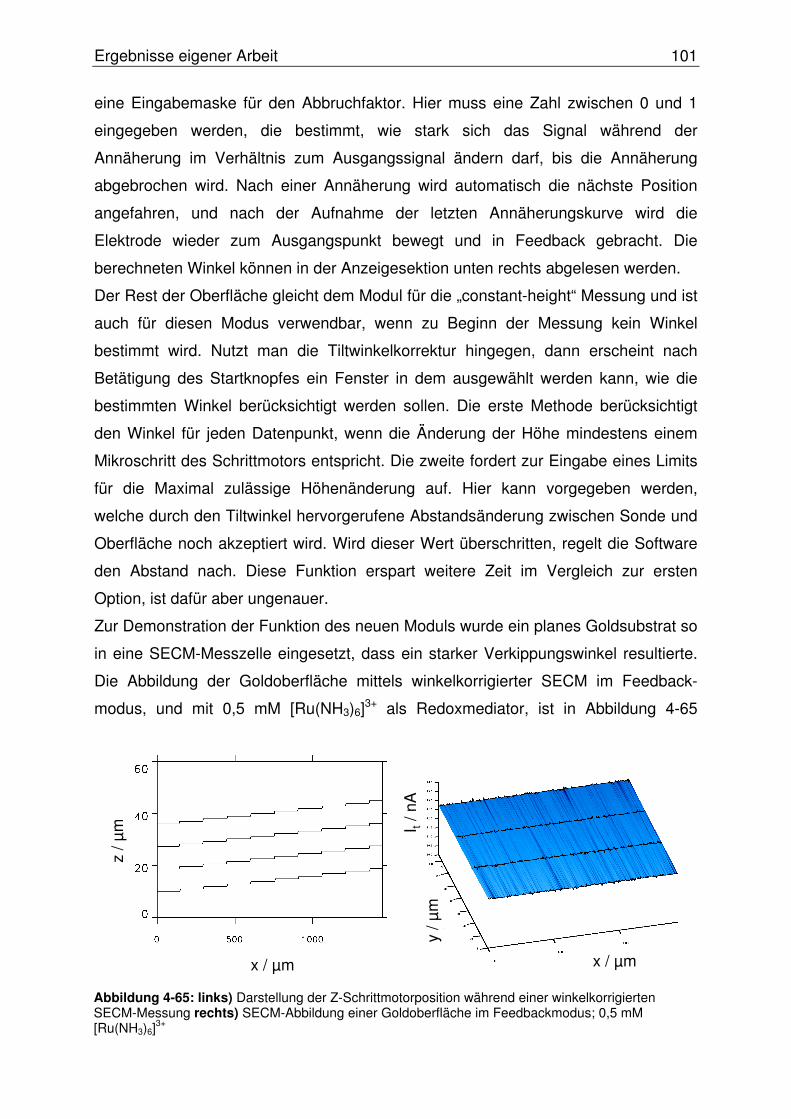
Ergebnisse eigener Arbeit 101
eine Eingabemaske für den Abbruchfaktor. Hier muss eine Zahl zwischen 0 und 1
eingegeben werden, die bestimmt, wie stark sich das Signal während der
Annäherung im Verhältnis zum Ausgangssignal ändern darf, bis die Annäherung
abgebrochen wird. Nach einer Annäherung wird automatisch die nächste Position
angefahren, und nach der Aufnahme der letzten Annäherungskurve wird die
Elektrode wieder zum Ausgangspunkt bewegt und in Feedback gebracht. Die
berechneten Winkel können in der Anzeigesektion unten rechts abgelesen werden.
Der Rest der Oberfläche gleicht dem Modul für die „constant-height“ Messung und ist
auch für diesen Modus verwendbar, wenn zu Beginn der Messung kein Winkel
bestimmt wird. Nutzt man die Tiltwinkelkorrektur hingegen, dann erscheint nach
Betätigung des Startknopfes ein Fenster in dem ausgewählt werden kann, wie die
bestimmten Winkel berücksichtigt werden sollen. Die erste Methode berücksichtigt
den Winkel für jeden Datenpunkt, wenn die Änderung der Höhe mindestens einem
Mikroschritt des Schrittmotors entspricht. Die zweite fordert zur Eingabe eines Limits
für die Maximal zulässige Höhenänderung auf. Hier kann vorgegeben werden,
welche durch den Tiltwinkel hervorgerufene Abstandsänderung zwischen Sonde und
Oberfläche noch akzeptiert wird. Wird dieser Wert überschritten, regelt die Software
den Abstand nach. Diese Funktion erspart weitere Zeit im Vergleich zur ersten
Option, ist dafür aber ungenauer.
Zur Demonstration der Funktion des neuen Moduls wurde ein planes Goldsubstrat so
in eine SECM-Messzelle eingesetzt, dass ein starker Verkippungswinkel resultierte.
Die Abbildung der Goldoberfläche mittels winkelkorrigierter SECM im Feedback-
modus, und mit 0,5 mM [Ru(NH3)6]3+ als Redoxmediator, ist in Abbildung 4-65
x / µm
z / µ
m I t/ n
Ay
/ µm
x / µmx / µm
z / µ
m I t/ n
Ay
/ µm
x / µm
Abbildung 4-65: links) Darstellung der Z-Schrittmotorposition während einer winkelkorrigierten SECM-Messung rechts) SECM-Abbildung einer Goldoberfläche im Feedbackmodus; 0,5 mM [Ru(NH3)6]
3+

Ergebnisse eigener Arbeit 102
(rechts) dargestellt. Der Sondenstrom der 10 µm Pt-Elektrode war über den
abgebildeten Bereich der Fläche konstant. Dies beweist, dass der Abstand zwischen
Sondenspitze und Probenoberfläche während der Messung ebenfalls konstant blieb.
Die Darstellung der Motorposition (Abbildung 4-65 links) zeigt, dass der
Verkippungswinkel der Probe während der Messung berücksichtigt wurde.
Die Messungen im folgenden Kapitel wurden, wenn erforderlich, unter Korrektur des
Tiltwinkels durchgeführt.
4.3.2.2 Hochaufgelöste AC-SECM an NiTi-FGLs mit Pt-
Ultramikroelektroden im „constant-height“ Modus
Als Sonde für die AC-SECM haben sich Pt-UMEs bewährt[62] und sind folglich für die
in diesem Kapitel vorgestellten Experimente verwendet worden. Elektroden mit
10 µm Durchmesser wurden nach dem in Abschnitt 6.2.1 beschriebenen Verfahren
hergestellt und erfolgreich an das bestehende SECM für den AC-Modus adaptiert.
Erste Annäherungskurven auf leitenden und nichtleitenden Oberflächen konnten
aufgenommen werden und sind in Abbildung 4-66 dargestellt. Auffällig ist, dass im
Gegensatz zur amperometrischen SECM kein positiver Feedback auf leitenden
Oberflächen beobachtet werden kann, sondern lediglich eine geringere negative
Wechselwirkung als auf einem Isolator. Dieses Ergebnis lässt sich mit den bekannten
-5 0 5 10 15 20 25 30 35 40 45 500.64
0.72
0.80
0.88
0.96
R/R
d / µm
Gold
Glas
∞∞ ∞∞
Arbeitsabstand
-5 0 5 10 15 20 25 30 35 40 45 500.64
0.72
0.80
0.88
0.96
R/R
d / µm
Gold
Glas
∞∞ ∞∞
Arbeitsabstand
Abbildung 4-66: SECM-Annäherungskurven einer 10 µm Pt-Scheiben-elektrode an eine Gold- bzw. Glasoberfläche; Elektrolyt = 1mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak
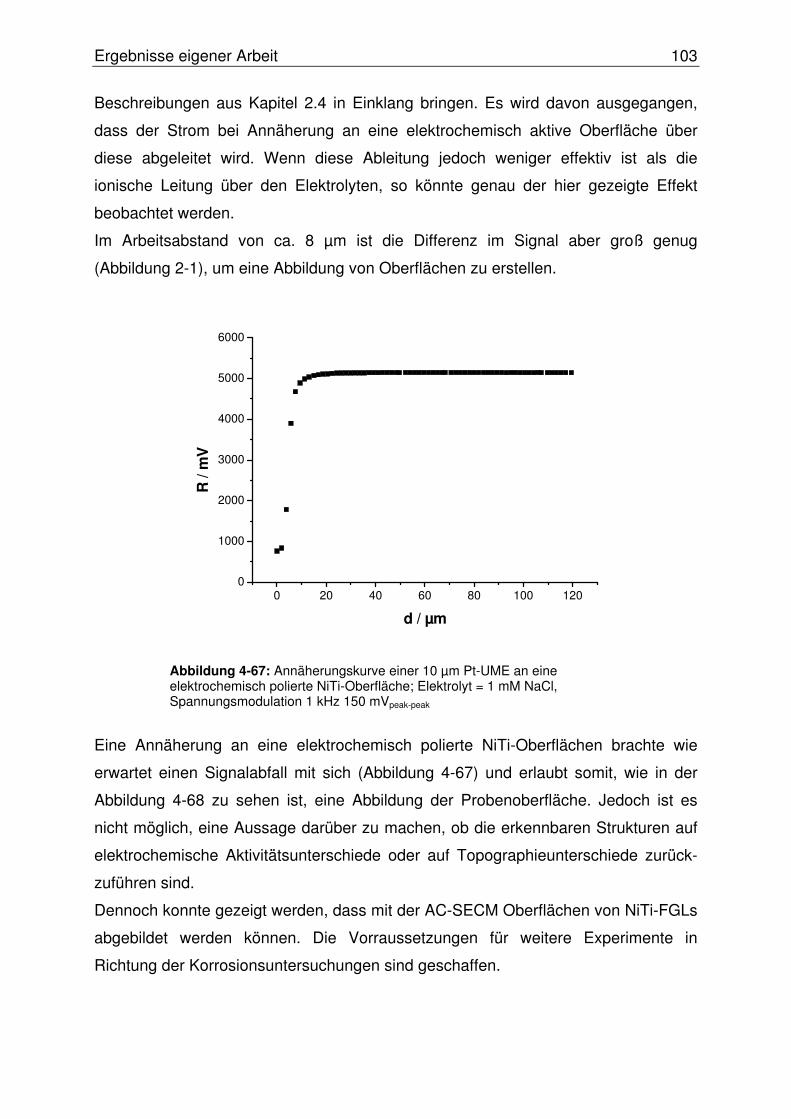
Ergebnisse eigener Arbeit 103
Beschreibungen aus Kapitel 2.4 in Einklang bringen. Es wird davon ausgegangen,
dass der Strom bei Annäherung an eine elektrochemisch aktive Oberfläche über
diese abgeleitet wird. Wenn diese Ableitung jedoch weniger effektiv ist als die
ionische Leitung über den Elektrolyten, so könnte genau der hier gezeigte Effekt
beobachtet werden.
Im Arbeitsabstand von ca. 8 µm ist die Differenz im Signal aber groß genug
(Abbildung 2-1), um eine Abbildung von Oberflächen zu erstellen.
Eine Annäherung an eine elektrochemisch polierte NiTi-Oberflächen brachte wie
erwartet einen Signalabfall mit sich (Abbildung 4-67) und erlaubt somit, wie in der
Abbildung 4-68 zu sehen ist, eine Abbildung der Probenoberfläche. Jedoch ist es
nicht möglich, eine Aussage darüber zu machen, ob die erkennbaren Strukturen auf
elektrochemische Aktivitätsunterschiede oder auf Topographieunterschiede zurück-
zuführen sind.
Dennoch konnte gezeigt werden, dass mit der AC-SECM Oberflächen von NiTi-FGLs
abgebildet werden können. Die Vorraussetzungen für weitere Experimente in
Richtung der Korrosionsuntersuchungen sind geschaffen.
0 20 40 60 80 100 1200
1000
2000
3000
4000
5000
6000
R /
mV
d / µm
Abbildung 4-67: Annäherungskurve einer 10 µm Pt-UME an eine elektrochemisch polierte NiTi-Oberfläche; Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak

Ergebnisse eigener Arbeit 104
Erste Experimente zur Visualisierung lokaler Korrosion von NiTi wurden unter
drastischen Bedingungen durchgeführt. Während der SECM-Messung wurde ein
Potential von 1,6 V an die NiTi-Probe angelegt. Dies liegt um einige 100 mV über
dem Durchbruchspotential (vgl. Abschnitt 4.2.3) und sollte eine starke Korrosion
garantieren.
In Abbildung 4-69 ist ein Ergebnis eines solchen Experiments zu sehen. Auf der
Abbildung 4-68: Abbildung der Oberfläche einer elektrochemisch polierten NiTi-FGL mittels AC-SECM im „constant-height“ Modus; Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak
600 µm
1000
µm
3046 mV
3524 mV
600 µm
1000
µm
3046 mV
3524 mV
Abbildung 4-69: rechts) Abbildung einer korrodieren NiTi-Oberfläche mittels AC-SECM; Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak links) lichtmikroskopische Abbildung der Korrodierten Probe nach dem Experiment

Ergebnisse eigener Arbeit 105
linken Seite ist eine lichtmikroskopische Abbildung der Nitinol-Oberfläche nach dem
Korrosionsexperiment dargestellt. Die Lochfraßkorrosion hat sich weit im Material
ausgebreitet. Der rotumrandete Ausschnitt wurde mit dem SECM im
Wechselstrommodus erfasst und zeigt in dem Bereich der Korrosion einen Anstieg
der Magnitude um bis zu 500 Einheiten. Dies kann auf den bereits bekannten zwei
Phänomenen beruhen. Zum einen wird ein Topographieeffekt erfasst, da das
Material, wie in der linken Abbildung zu sehen, stark abgetragen wurde. Zum
anderen treten während der Korrosion Metallionen aus der Oberfläche aus, die lokal
die Leitfähigkeit der Lösung erhöhen.
Nachdem gezeigt werden konnte, dass Korrosionsphänomene mit der AC-SECM
visualisierbar sind, wurden Experimente zur Beobachtung des Beginns der
Lochfraßkorrosion und zum Wachstum der Löcher durchgeführt. Dazu mussten erst
geeignete Areale auf der Oberfläche der Probe lokalisiert werden. Eine Möglichkeit,
um zu erreichen, ist die Beobachtung der Oberfläche durch ein Videomikroskop
während kurzzeitig eine Spannung oberhalb des Durchbruchpotentials an die Probe
angelegt wird. Dies führt umgehend zur Gasentwicklung an defekten oder dünnen
Stellen in der Passivschicht, neben denen dann die Elektrode positioniert werden
kann. Nach einer kurzen Repassivierungsphase kann der Bereich abgerastert
werden. Die drei folgenden Abbildungen sollen beispielhaft solche aktiven Bereiche
zeigen.
Abbildung 4-70: Visualisierungen von Inhomogenitäten in der elektrochemischen Aktivität von NiTi-Passivschichten mittels AC-SECM links) dreidimensionale Ansicht rechts) zweidimensionale Ansicht der gleichen Inhomogenität; die helleren Regionen entsprechen einer höheren Aktivität; Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak

Ergebnisse eigener Arbeit 106
Positive Signale werden an den defekten Stellen erhalten, die mit anderen
Mikroskopiearten nicht entdeckt werden können, da sie sich nur durch ihre
elektrochemische Aktivität von der umgebenden Fläche unterscheiden. Solche
Gebiete sollten besonders anfällig für Lochfraßkorrosion sein. Um diese Behauptung
zu beweisen, wurde eine Bilderserie (siehe Abbildung 4-72) mit dem SECM erstellt,
wobei zwischen den Messungen jeweils für 30 Minuten ein Potential von 1,8 V
angelegt wurde, um den Lochfraß zu forcieren. Während der SECM-Messung wurde
die Probe wieder auf Ruhepotential geschaltet.
Wie auf den Bildern zu erkennen ist, breitet sich der Defekt in der Passivschicht mit
der Messzeit weiter aus, was darauf schließen lässt, dass diese Stellen für die
Lochfraßkorrosion anfällig sind bzw. hier lokale Korrosion stattfindet. Es konnte
gezeigt werden, dass mit der AC-SECM Inhomogenitäten in der Oxidschicht von
NiTi-FGLs erfasst werden können und dass das Voranschreiten von Loch-
fraßkorrosion verfolgt werden kann. Die Visualisierung dieser Phänomene ist
aufgrund der Variation der Elektrolytimpedanz möglich, die wie schon erwähnt von
der lokalen elektrochemischen Aktivität der Oberfläche, der Topographie und der
lokalen Ionenkonzentration abhängt.
Abbildung 4-71: Visualisierung zweier angrenzender Inhomogenitäten in der elektrochemischen Aktivität von NiTi-Passivschichten mittels AC-SECM; Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak

Ergebnisse eigener Arbeit 107
4.3.2.2.1 Abbildung von Mikroeinschlüssen in einer NiTi-Oberfläche
Mit dem gleichen Verfahren wie im letzten Abschnitt beschrieben, sollte eine NiTi-
Oberfläche mit Mikroeinschlüssen abgebildet werden.
Die Einschlüsse sind beim elektrochemischen Polieren aus der Oberfläche
hervorgetreten, bzw. das umgebende Material wurde bei dem Polierverfahren
abgetragen und die Einschlüsse sind zurückgeblieben. Sie sind auf Verun-
reinigungen beim Herstellungsprozess der Legierung zurückzuführen. Das elektro-
chemische Verhalten dieser Teilchen war bisher nicht bekannt.
Wie in Abbildung 4-73 zu sehen ist, ist es möglich die Einschlüsse mit dem AC-
SECM abzubilden, obwohl der Teilchendurchmesser unter dem der SECM-Sonde
lag. Der Durchmesser der Teilchen lag im Bereich weniger Mikrometer, wie
Aufnahmen mit dem REM belegen. Da hier mit einer 10 µm Pt-Elektrode gemessen
A B
C
1,8 V
1,8 V
30 Min
30 Min
A B
C
1,8 V
1,8 V
30 Min
30 Min
Abbildung 4-72: Abbildung eines Defektes in der NiTi-Passivschicht mittels AC-SECM. Während der Abbildung war die Probe auf Ruhepotential geschaltet, zwischen den Messungen wurde für 30 Minuten 1,8 V an die Probe angelegt. Die helleren Bereiche entsprechen einer höheren Aktivität. Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak

Ergebnisse eigener Arbeit 108
wurde, war es nicht möglich die Einschlüsse mit einer höheren Auflösung zu
erfassen.
Die Abbildung 4-73 zeigt, dass die Verunreinigungen offensichtlich von unter-
schiedlicher chemischer Zusammensetzung sind, da sie zu positiven sowie negativen
Signalen im AC-SECM führen. Somit sind Teilchen mit höherer und geringerer
elektrochemischer Aktivität in dem Material vorhanden.
4.3.2.2.2 Abbildung einer Mikrostrukturierten NiTi-Oberfläche
Eine Aufgabe des SFB 459 ist es, Methoden zur Mikrostrukturierung und
Verbesserung der mechanischen und der Korrosionseigenschaften von NiTi-FGLs zu
entwickeln. Um Mikrostrukturen auf NiTi-Oberflächen zu erzeugen wurde das
Focused-Ion-Beam (FIB) Verfahren angewendet. Bei dem hier verwendeten Gerät
werden Galliumionen beschleunigt und auf die Oberfläche geschossen. Je nach
verwendeter Ionendosis, welche die Anzahl der Ionen pro cm2 angibt, werden die
Galliumionen in die Oberfläche implantiert, amorphisieren diese oder brechen diese
auf und tragen sie ab. Dieses Abtragen des Materials wird auch sputtern genannt.
Eine Versuchreihe sollte zeigen, welche Ionendosis benötigt wird, damit der Sputter-
prozess einsetzt und ob noch andere Nebeneffekte durch das Implantieren der
Galliumionen auftreten. Hierfür wurde eine NiTi-Oberfläche präpariert, die in unter-
schiedliche Bereiche aufgeteilt wurde. Die unterschiedlichen Bereiche wurden jeweils
mit einer anderen Dosis an Galliumionen beschossen.
Abbildung 4-73: Abbildung einer NiTi-Oberfläche mit Mikro-einschlüssen mittels AC-SECM; Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak
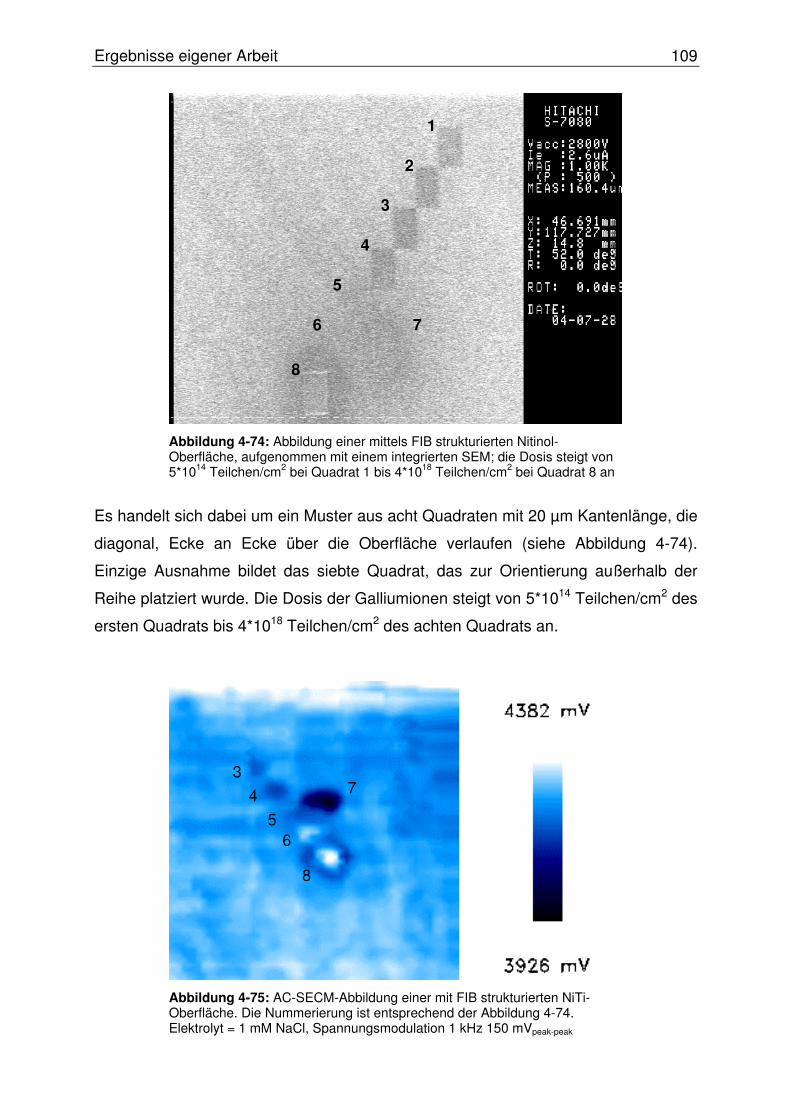
Ergebnisse eigener Arbeit 109
Es handelt sich dabei um ein Muster aus acht Quadraten mit 20 µm Kantenlänge, die
diagonal, Ecke an Ecke über die Oberfläche verlaufen (siehe Abbildung 4-74).
Einzige Ausnahme bildet das siebte Quadrat, das zur Orientierung außerhalb der
Reihe platziert wurde. Die Dosis der Galliumionen steigt von 5*1014 Teilchen/cm2 des
ersten Quadrats bis 4*1018 Teilchen/cm2 des achten Quadrats an.
1
2
3
4
5
6
8
7
1
2
3
4
5
6
8
7
Abbildung 4-74: Abbildung einer mittels FIB strukturierten Nitinol-Oberfläche, aufgenommen mit einem integrierten SEM; die Dosis steigt von 5*1014 Teilchen/cm2 bei Quadrat 1 bis 4*1018 Teilchen/cm2 bei Quadrat 8 an
8
65
374
8
65
374
Abbildung 4-75: AC-SECM-Abbildung einer mit FIB strukturierten NiTi-Oberfläche. Die Nummerierung ist entsprechend der Abbildung 4-74. Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak
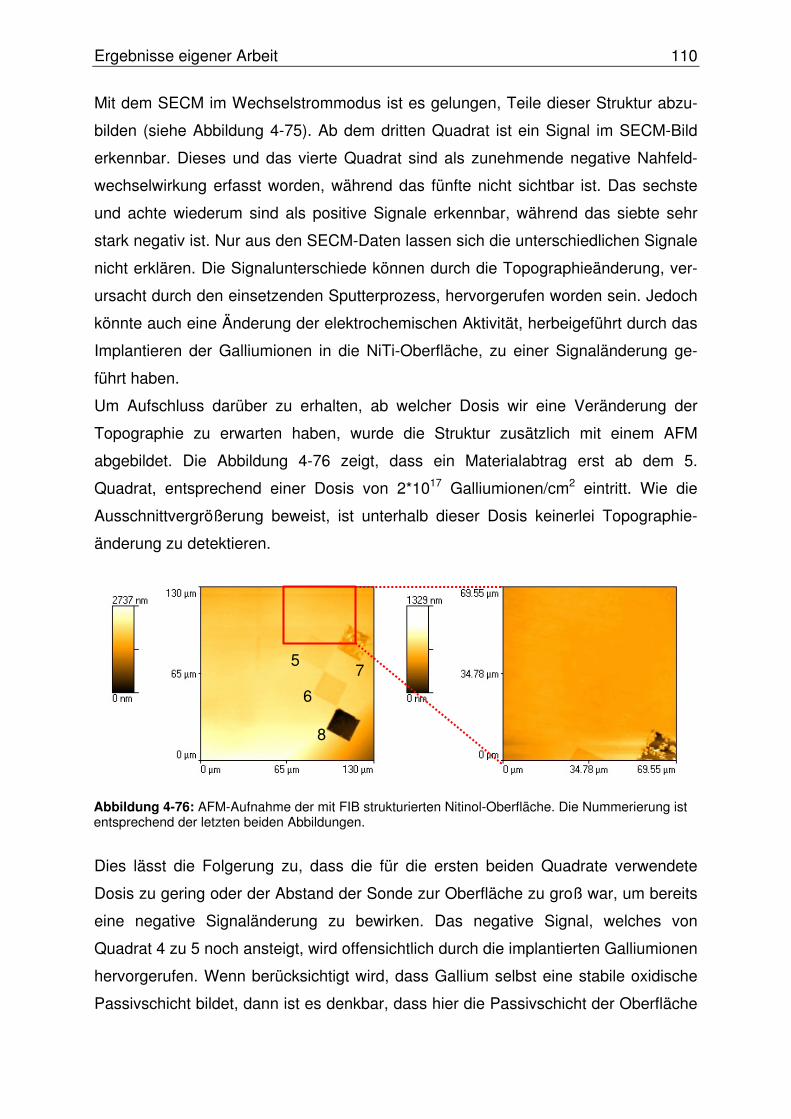
Ergebnisse eigener Arbeit 110
Mit dem SECM im Wechselstrommodus ist es gelungen, Teile dieser Struktur abzu-
bilden (siehe Abbildung 4-75). Ab dem dritten Quadrat ist ein Signal im SECM-Bild
erkennbar. Dieses und das vierte Quadrat sind als zunehmende negative Nahfeld-
wechselwirkung erfasst worden, während das fünfte nicht sichtbar ist. Das sechste
und achte wiederum sind als positive Signale erkennbar, während das siebte sehr
stark negativ ist. Nur aus den SECM-Daten lassen sich die unterschiedlichen Signale
nicht erklären. Die Signalunterschiede können durch die Topographieänderung, ver-
ursacht durch den einsetzenden Sputterprozess, hervorgerufen worden sein. Jedoch
könnte auch eine Änderung der elektrochemischen Aktivität, herbeigeführt durch das
Implantieren der Galliumionen in die NiTi-Oberfläche, zu einer Signaländerung ge-
führt haben.
Um Aufschluss darüber zu erhalten, ab welcher Dosis wir eine Veränderung der
Topographie zu erwarten haben, wurde die Struktur zusätzlich mit einem AFM
abgebildet. Die Abbildung 4-76 zeigt, dass ein Materialabtrag erst ab dem 5.
Quadrat, entsprechend einer Dosis von 2*1017 Galliumionen/cm2 eintritt. Wie die
Ausschnittvergrößerung beweist, ist unterhalb dieser Dosis keinerlei Topographie-
änderung zu detektieren.
Dies lässt die Folgerung zu, dass die für die ersten beiden Quadrate verwendete
Dosis zu gering oder der Abstand der Sonde zur Oberfläche zu groß war, um bereits
eine negative Signaländerung zu bewirken. Das negative Signal, welches von
Quadrat 4 zu 5 noch ansteigt, wird offensichtlich durch die implantierten Galliumionen
hervorgerufen. Wenn berücksichtigt wird, dass Gallium selbst eine stabile oxidische
Passivschicht bildet, dann ist es denkbar, dass hier die Passivschicht der Oberfläche
8
6
57
8
6
57
Abbildung 4-76: AFM-Aufnahme der mit FIB strukturierten Nitinol-Oberfläche. Die Nummerierung ist entsprechend der letzten beiden Abbildungen.

Ergebnisse eigener Arbeit 111
durch das Implantieren verstärkt wurde und somit eine geringere elektrochemische
Aktivität gemessen werden muss.
In den letzten Kapiteln wurde aufgezeigt, dass ein Zusammenhang zwischen
erhöhter elektrochemischer Aktivität und dem Beginn von Korrosion besteht. Auf-
grund der hier gezeigten Ergebnisse, gibt es Grund zu der Annahme, dass Gallium
ebenso als Korrosionsinhibitor fungieren kann, da es, implantiert in die NiTi-Ober-
fläche, dessen elektrochemische Aktivität herabsetzt.
Auch kann durch Kombination der SECM- und AFM-Ergebnisse leicht erklärt werden,
weshalb Quadrat 5 nicht im SECM-Bild erscheint. Hier löschen sich offensichtlich
zwei gegenläufige Effekte aus. Aufgrund der höheren Ionendosis wäre ein im
Vergleich zum 4. Quadrat stärker negatives Signal zu erwarten. Jedoch beginnt hier
bereits der Sputterprozess. Somit ist an dieser Stelle eine Vertiefung, aufgrund derer
das Signal ansteigen sollte.
Bei Quadrat 6 und 8 überwiegt hingegen der Topographieeffekt und sorgt somit für
einen Anstieg des Signals im SECM. Die Tiefe der Struktur beträgt hier 180 nm bzw.
1900 nm.
Eine Ausnahme bildet das Quadrat 7, welches wie erwähnt außerhalb der
diagonalen Reihe platziert wurde und zur Orientierung dient. Die Tiefe liegt mit 480
nm, wie erwartet, zwischen denen des sechsten und achten Quadrats. Jedoch ist
hier das SECM-Signal stark negativ. Aufklärung findet sich wiederum in der AFM-
Abbildung, die anzeigt, dass sich in diesem Quadrat viele kleine Körner befinden.
Diese dürften aus abgetragenem Material einer benachbarten Struktur bestehen, da
das Material durch den Sputterprozess seitlich aus dem Quadrat transportiert wird.
Dieses Material sollte eine hohe Konzentration an Gallium enthalten, das offen-
sichtlich für eine Signalabsenkung sorgt. Diese scheint den Topographieeinfluss
deutlich zu übertreffen.
Das Fazit dieses Experiments ist, dass FIB eine geeignete Methode zur
Mikrostrukturierung von NiTi-Oberflächen ist und ein Potential zur Verbesserung der
Korrosionsbeständigkeit durch Implantieren von Gallium besitzt.
4.3.2.2.3 Abbildung einer geätzten NiTi-Oberfläche
Für manche Anwendungen, z.B. dem Beschichten[164] oder Implantieren von Nitinol
ist eine raue Oberfläche anstatt einer polierten erwünscht. Eine Möglichkeit eine raue
Oberfläche herzustellen ist das elektrochemische Ätzen der Legierung. Interessant

Ergebnisse eigener Arbeit 112
ist in diesem Zusammenhang die elektrochemische Aktivität der geätzten Oberfläche
und somit die Anfälligkeit für Korrosion.
Es wurde ein Gebiet auf der Oberfläche einer geätzten Nitinol-Probe mittels Vickers-
Mikrohärteeindrücken markiert und mit einem REM abgebildet. Die Mikrohärteein-
drücke sollen ein Wiederfinden des mittels REM abgebildeten Gebiets im SECM
gewährleisten. Zudem liefert die REM-Aufnahme bereits eine Vorstellung der Ober-
flächenstruktur. In Abbildung 4-77 ist diese Aufnahme zu sehen. Bei den vier
pyramidalen Vertiefungen nahe der Ecken des Bildes handelt es sich um die
Mikrohärteeindrücke.
Die AC-SECM-Messung wurde mit den Standardparametern von 1 kHz Frequenz
und einer Spannung von 150 mVpeak-peak durchgeführt. Um den mit dem REM
abgebildeten Bereich besser wiederfinden zu können, wurde unter Zuhilfenahme
eines Lichtmikroskops und den Mikrohärteeindrücken mit einem wasserfesten
Folienstift ein Rahmen um das Gebiet gezogen. Das Ergebnis der SECM-Messung
ist in Abbildung 4-78 (links) zu sehen.
Die helleren Bereiche lassen sich mit den geätzten Löchern aus der REM-Aufnahme
der gleichen Fläche (Abbildung 4-77) in Einklang bringen. Zur Verdeutlichung zeigt
Abbildung 4-78 (rechts) eine Überlagerung der Abbildung 4-77 mit dem SECM-Bild.
Es ist eine eindeutige Korrelation zwischen den positiven Signalen des AC-SECM
Abbildung 4-77: Abbildung der Oberfläche einer elektrochemisch geätzten NiTi-Probe mittels REM
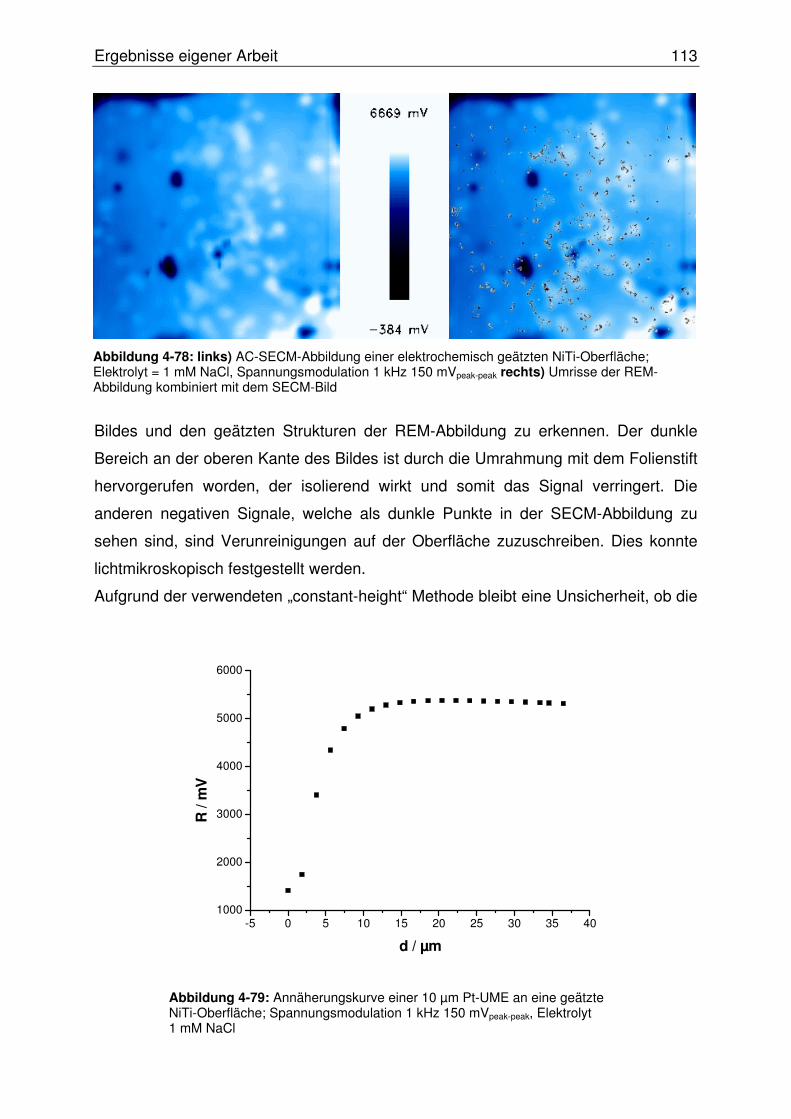
Ergebnisse eigener Arbeit 113
Bildes und den geätzten Strukturen der REM-Abbildung zu erkennen. Der dunkle
Bereich an der oberen Kante des Bildes ist durch die Umrahmung mit dem Folienstift
hervorgerufen worden, der isolierend wirkt und somit das Signal verringert. Die
anderen negativen Signale, welche als dunkle Punkte in der SECM-Abbildung zu
sehen sind, sind Verunreinigungen auf der Oberfläche zuzuschreiben. Dies konnte
lichtmikroskopisch festgestellt werden.
Aufgrund der verwendeten „constant-height“ Methode bleibt eine Unsicherheit, ob die
Abbildung 4-78: links) AC-SECM-Abbildung einer elektrochemisch geätzten NiTi-Oberfläche; Elektrolyt = 1 mM NaCl, Spannungsmodulation 1 kHz 150 mVpeak-peak rechts) Umrisse der REM-Abbildung kombiniert mit dem SECM-Bild
-5 0 5 10 15 20 25 30 35 401000
2000
3000
4000
5000
6000
R /
mV
d / µm
Abbildung 4-79: Annäherungskurve einer 10 µm Pt-UME an eine geätzte NiTi-Oberfläche; Spannungsmodulation 1 kHz 150 mVpeak-peak, Elektrolyt 1 mM NaCl

Ergebnisse eigener Arbeit 114
positiven Signale nur auf Topographie beruhen. Aus der Annäherungskurve in
Abbildung 4-79, die vor dem Scan aufgenommen wurde, lässt sich ein maximaler R-
Wert von 5400 mV entnehmen. In der Abbildung steigt die Magnitude jedoch im
Bereich der unteren linken Ecke bis auf einen Wert von über 6600 mV. Hier könnte
also eine durch Ionenaustritt aus der Oberfläche verstärkte positive Signaländerung
vorliegen. Eine Überprüfung dieser Messergebnisse erfolgte im „constant-distance“
Modus unter Verwendung einer optischen, scherkraftabhängigen Höhenkontrolle.
4.3.2.3 Hochaufgelöste AC-SECM im „constant-distance“ Modus
Wie in den letzten Kapiteln beschrieben wurde, konnte die AC-SECM für die
Visualisierung von NiTi-Oberflächen und Korrosionsphänomenen an diesen erfolg-
reich angewendet werden. Offensichtlich besteht aber aufgrund der oftmals nur
wenige Mikrometer großen Strukturen Bedarf an einer höheren Auflösung und, wie
im letzten Abschnitt erwähnt, auch an einer „constant-distance“ Variante der AC-
Messung.
Vor kurzem wurden in unserer Arbeitsgruppe Pt-Nanoelektroden entwickelt[37], die für
das SECM mit nichtoptischer, scherkraftabhängiger Höhenkontrolle evaluiert sind[38].
Die Herstellung der Elektroden unter Nutzung eines Laserpullers ist im Abschnitt
6.2.3 beschrieben.
4.3.2.3.1 Evaluierung von Pt-Scheibenelektroden im „constant-
distance“ AC-SECM
Pt-Elektroden mit einem Durchmesser < 10 µm konnten problemlos an das hier
verwendete SECM mit optischer Höhenkontrolle adaptiert werden, wie die folgenden
Abschnitte dieser Arbeit zeigen werden. Mit den kleineren Elektroden ist eine
Verbesserung der Auflösung bis zu einem Faktor von 1000 (bei einer 10 nm Elek-
trode) zu erreichen.
Die Durchmesser der verwendeten Elektroden liegen herstellungsbedingt (siehe
dazu Kapitel 6.2.3) zwischen ~10 nm und ~5 µm. Abbildung 4-80 zeigt beispielhaft
zwei Cyclovoltammogramme von Elektroden, die für die AC-SECM mit optischer,
scherkraftabhängiger Höhenkontrolle verwendet wurden. Die Durchmesser der
beiden Elektroden wurde auf 4 µm bzw. 300 nm bestimmt.
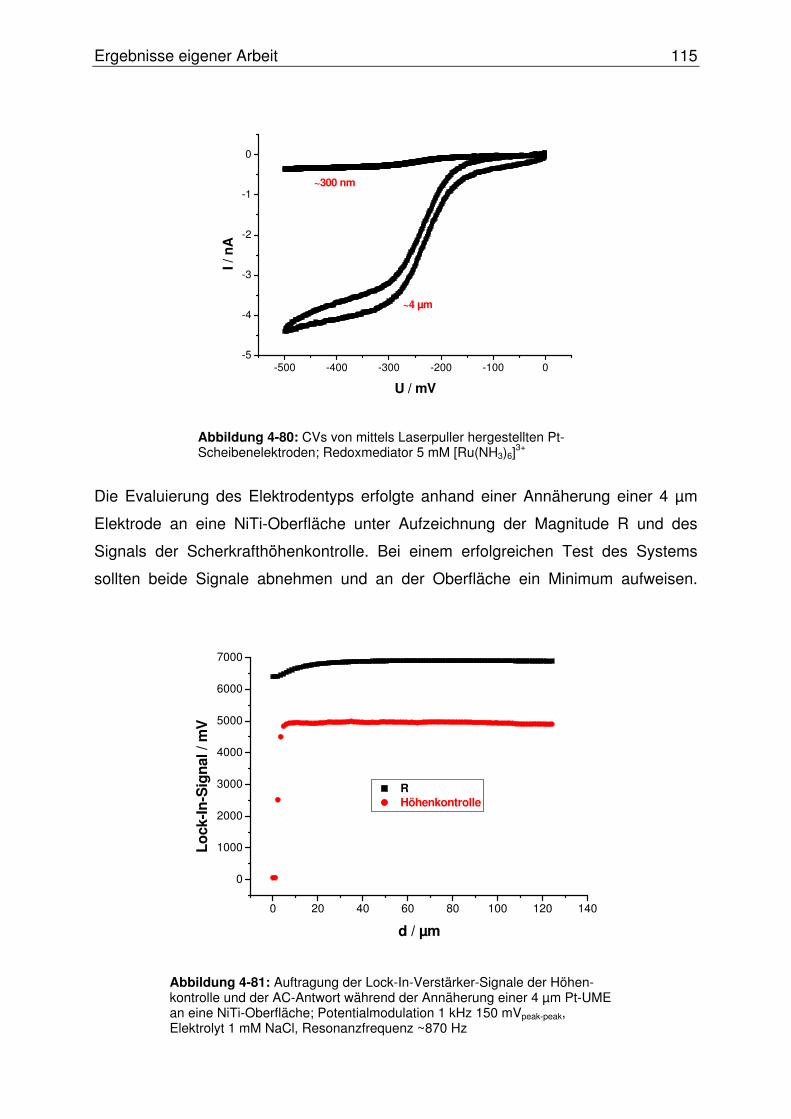
Ergebnisse eigener Arbeit 115
Die Evaluierung des Elektrodentyps erfolgte anhand einer Annäherung einer 4 µm
Elektrode an eine NiTi-Oberfläche unter Aufzeichnung der Magnitude R und des
Signals der Scherkrafthöhenkontrolle. Bei einem erfolgreichen Test des Systems
sollten beide Signale abnehmen und an der Oberfläche ein Minimum aufweisen.
-500 -400 -300 -200 -100 0-5
-4
-3
-2
-1
0
~4 µm
~300 nm
I / n
A
U / mV
Abbildung 4-80: CVs von mittels Laserpuller hergestellten Pt-Scheibenelektroden; Redoxmediator 5 mM [Ru(NH3)6]
3+
0 20 40 60 80 100 120 140
0
1000
2000
3000
4000
5000
6000
7000
R Höhenkontrolle
Lo
ck-I
n-S
ign
al /
mV
d / µm
Abbildung 4-81: Auftragung der Lock-In-Verstärker-Signale der Höhen-kontrolle und der AC-Antwort während der Annäherung einer 4 µm Pt-UME an eine NiTi-Oberfläche; Potentialmodulation 1 kHz 150 mVpeak-peak, Elektrolyt 1 mM NaCl, Resonanzfrequenz ~870 Hz

Ergebnisse eigener Arbeit 116
Dieser Effekt ist in Abbildung 4-81 zu sehen. Typischerweise beginnt das Signal der
Magnitude schon wesentlich früher abzusinken jedoch lediglich um 500 Einheiten auf
ca. 6400 mV, während das Signal der Scherkrafthöhenkontrolle erst in unmittelbarer
Nähe der Oberfläche bis auf 0 abfällt. Dies ist ein aus der höhenkontrollierten,
amperometrischen SECM bekannter Effekt und kann in Kapitel 2.2.2.3.2
nachgelesen werden.
Als Testprobe wurde die Oberfläche einer Nitinol-Probe mit einem Kratzer präpariert,
der ebenfalls unter Aufnahme beider Signale abgerastert wurde. Die daraus
entstandenen dreidimensionalen Abbildungen der Oberfläche sind in Abbildung 4-82
zu sehen. Die linke Abbildung gibt die Topographie der Teststruktur wieder. Die
Struktur des Kratzers wurde mittels des z-Piezos nachgeregelt und kann hoch-
aufgelöst dargestellt werden. Die rechte Abbildung gibt den R-Wert wieder, der von
der Impedanz des Elektrolyten abhängt.
Überraschender Weise wird im Bereich des Kratzers ein negatives Magnituden-
Signal registriert. Die Erklärung dafür könnte in der Funktionsweise des AC-SECM
begründet liegen. Maßgeblich verantwortlich für das Signal, welches bei der SECM
im Wechselstrommodus gemessen wird, ist der Widerstand der Lösung, gemessen
zwischen der Arbeits- und der Gegenelektrode. Der Widerstand nimmt bei der
Annäherung der SECM-Sonde an die Oberfläche zu, da der immer dünner werdende
Elektrolytfilm einen höheren Widerstand bildet (siehe dazu Abschnitt 2.4). Es ist leicht
vorstellbar, dass der Widerstand noch weiter zunimmt, wenn die Elektrode in eine
Abbildung 4-82: Abbildung eines repassivierten Kratzers in einer Nitinol-Probe mittels „constant-distance“ AC-SECM links) Auftragung des topographischen Signals der Höhenkontrolle rechts) Auftragung des R-Wertes; Potentialmodulation 1 kHz 150 mVpeak-peak, Elektrolyt 1 mM NaCl

Ergebnisse eigener Arbeit 117
Vertiefung abgesenkt wird und damit der Ladungstransport zwischen Arbeits- und
Gegenelektrode noch stärker behindert wird.
4.3.2.3.2 Hochaufgelöste AC-SECM im „constant-distance“ Modus an
geätzten Nitinol-Oberflächen
Die Analyse der geätzten NiTi-Probe kann nun mit den kleineren Elektroden und der
scherkraftabhängigen Höhenkontrolle fortgesetzt werden. Zu diesem Zweck wurde
eine Sonde mit 300 nm aktivem Durchmesser in dem mit Folienstift markierten
Bereich an die Oberfläche der Probe angenähert. Ein Ausschnitt von 125 µm x
145 µm wurde abgerastert (siehe Abbildung 4-83).
Die Abbildung verdeutlicht die Steigerung der Auflösung des SECM und lässt keine
Drift im Signal erkennen, die auf eine Änderung des Abstands zwischen SECM-
Sonde und Probenoberfläche schließen lässt.
Der Vergleich der REM-Aufnahme mit der AC-Abbildung der Oberfläche (siehe
Abbildung 4-84 A & C) zeigt eine noch deutlichere Deckung der positiven Signale mit
den geätzten Strukturen, als dies in Kapitel 4.3.2.2.3 der Fall ist. Die Löcher in der
Oberfläche werden als helle Flächen sichtbar, welche einer lokalen Erniedrigung der
Impedanz des Elektrolyten entsprechen. Aus dem Topographiebild (Abbildung 4-84
B) lässt sich entnehmen, dass die Topographieänderungen, hervorgerufen durch die
Abbildung 4-83: AC-SECM-Abbildung einer elektrochemisch geätzten NiTi-Oberfläche mit einer 300 nm Pt-Elektrode im „constant-distance“ Modus; Potentialmodulation 1,2 kHz 150 mVpeak-peak, Elektrolyt 1 mM NaCl

Ergebnisse eigener Arbeit 118
geätzten Löcher, nicht durch die Höhenkontrolle nachgeregelt werden. Die Be-
gründung liegt in der Elektrodengeometrie. Die aktive Oberfläche der Platinscheibe
hat zwar einen Durchmesser von nur 300 nm, jedoch vergrößert die Glasisolierung
den Gesamtquerschnitt auf 30 bis 40 µm. Dies liegt gerade im Bereich der
Dimensionen der größeren Vertiefungen und führt dazu, dass die Elektrode nicht in
die Löcher abgesenkt werden kann, sondern in quasi konstanter Höhe darüber hin-
weg bewegt wird.
Offensichtlich ist die Vermutung, dass die positiv verstärkten Signale im Bereich der
geätzten Strukturen auf einer erhöhten elektrochemischen Aktivität basieren (vgl.
Kapitel 4.3.2.2.3), falsch. Denn das Signal steigt bei der höhenkontrollierten SECM
maximal auf die Höhe des Ausgangssignals an, wie aus der Annäherungskurve (hier
nicht gezeigt) entnommen werden kann. Die Erhöhung des Signals über das
Ausgangsniveau hinaus, welches in der „constant-height“-Abbildung beobachtet
werden konnte, muss eine andere Ursache haben. Wie die Erfahrung zeigt, kann
vielmehr angenommen werden, dass die Oberfläche mit der Elektrode berührt wurde
A B
C
A B
C
Abbildung 4-84: Abbildung der Oberfläche einer geätzten NiTi-Scheibe A) Auftragung der Magnitude R der AC-SECM-Messung B) topographische Abbildung der Oberfläche mittels höhenkontrollierter AC-SECM C) REM-Aufnahme der Oberfläche, das rote Rechteck zeigt den Bereich der SECM-Messung an

Ergebnisse eigener Arbeit 119
und diese die Passivschicht lokal verletzt hat. Der Kontakt der Sonde mit dem
ungeschützten Metall könnte zu dem beobachteten Signalanstieg geführt haben.

Zusammenfassung und Ausblick 120
5 Zusammenfassung und Ausblick
Ziel dieser Arbeit war es, geeignete Werkzeuge zur Korrosionsuntersuchung an NiTi-
FGLs zu entwickeln, zu evaluieren und einzusetzen. Es sollten dabei zwei
unterschiedliche Wege beschritten werden. Zum einen wurde ein hochsensitives
Messverfahren für die Langzeitbestimmung der globalen Nickelfreisetzung aus
Nitinol-Werkstücken benötigt, zum anderen eine Methode zur Visualisierung lokaler
Korrosionsphänomene mit hoher lateraler Auflösung. Damit sollten Möglichkeiten
geschaffen werden, die Bildung von Lochfraßkorrosion und vor allem die
Nickelfreisetzung aus unterschiedlich hergestellten und bearbeiteten NiTi-
Werkstücken zu erfassen. Die erzielten Ergebnisse sollten dazu dienen optimierte
Verfahren zum Legieren von Nickel mit Titan und zur Bearbeitung dieser
Legierungen zu entwickeln. Zudem sollten sie ermöglichen, z.B. Beschichtungen zum
Korrosionsschutz auf ihre Wirksamkeit zu prüfen. Schließlich sollte dies zu Produkten
mit einer höheren Haltbarkeit und in der Medizin mit einem geringeren Risiko für den
Patienten führen.
Es konnten in Langzeitstabilität und Sensitivität optimierte Bi-Filmelektroden für die
automatisierte Bestimmung von Ni2+ mittels AdSV hergestellt werden. Die elektro-
chemische Beschichtung kann problemlos mit einem einfachen Verfahren auf einer
1 mm GC-Elektrode durchgeführt werden.
Die Entwicklung einer neuen Wall-Jet Durchflussmesszelle, trägt zu einer erhöhten
Sensitivität und verbesserten Nachweisgrenze bei. Diese neue Messzelle lies sich
erfolgreich in das bestehende OLGA-System integrieren und zeigte in ersten
Langzeitexperimenten zusammen mit der Bi-Filmelektrode ihr Potential für die
Online-Überwachung von Korrosionsvorgängen.
Die Software zur Steuerung der OLGA wurde erweitert und an die Problematik der
Strippingvoltammetrie angepasst. Sie gewährleistet eine hohe Flexibilität für die
Pumpparameter, um die Messung in unterschiedlichen Konzentrationsbereichen
verschiedener Analyten abzudecken. Sie erlaubt es, die Analysenlösung mehrfach
zur Anreicherung durch die Messzelle zu transportieren, beherrscht automatische
Standardaddition und kann über ein PlugIn-System erweitert werden. Ein erstes
PlugIn zur on-line-Visualisierung der vom Potentiostaten aufgenommenen
Messdaten wurde bereits erfolgreich integriert.

Zusammenfassung und Ausblick 121
Eine PC-Software zur Steuerung des PalmSens-Potentiostaten und zur Daten-
aufnahme wurde erstellt. Diese arbeitet über ein einfaches Dateiaustausch-System
mit der OLGA-Software zusammen und ermöglicht so die synchrone Zusammen-
arbeit der beiden Programme. Zudem ist es möglich, die Software unabhängig von
dem SIA-System nur für die Steuerung des Potentiostaten zu benutzen. Alle
wichtigen voltammetrischen Messtechniken sind bereits integriert.
Die Visualisierung von Korrosionsphänomenen ist mit der potentiometrischen SECM
unter Verwendung eines Mikro-pH-Sensors sowie mit der AC-SECM gelungen.
Die AC-SECM konnte erfolgreich für unterschiedliche Ansprüche optimiert werden.
Es besteht nun die Wahl zwischen einer geringeren Auflösung mit einer 10 µm Pt-
UME im „constant-height“ Modus, über die Möglichkeit bei dieser Messung den
Tiltwinkel der Probe korrigieren zu lassen, bis hin zur hochaufgelösten, mit Pt-
Nanoelektroden durchgeführten AC-SECM im „constant-distance“ Modus. Die Dauer
der Messung steigt mit den genannten Verbesserungen der Auflösung jedoch an, so
dass für die Beobachtung eines mit der Zeit wachsenden Korrosionslochs eher die
erste Methode gewählt werden sollte und zur Visualisierung von Strukturen im
unteren Mikrometerbereich die letztere. An mehreren Beispielen wurde die flexible
Einsatzmöglichkeit für unterschiedlich bearbeitete NiTi-Oberflächen demonstriert.
Neue Messreihen mit dem SISA-System, zum Vergleich verschiedener Proben, die
von den Projektpartnern des SFB 459 geliefert werden, sind bereits in Planung. Hier
sollen Auswirkungen auf die Nickelausblutung durch verschiedene Herstellungs-
prozesse, wie die schmelzmetallurgische oder pulvermetallurgische Herstellung,
aufgezeigt werden. Auch die Auswirkungen der Nachbearbeitung, wie das
Lösungsglühen, soll auf diesem Wege überprüft werden. Es soll herausgefunden
werden welchen Einfluss z.B. die spanende Bearbeitung oder das Aufbringen von
Verschleißschutzschichten auf den Nickelaustritt haben.
Es wäre möglich die Bi-Filmelektrode direkt im SISA-Gerät zu erstellen und eine
automatische Regenerierung des Films durchzuführen, indem die Wismutab-
scheidungslösung in die Messzelle gepumpt wird und dort die Deposition stattfindet.
Das SISA-System könnte zudem mit einem automatisierten Probengeber kombiniert
werden. So wäre ein korrosionsbezogenes Hochdurchsatz-Screening einfach und
kostengünstig zu realisieren.

Zusammenfassung und Ausblick 122
Denkbar wäre auch, den Nickelaustritt unter mechanischer oder thermischer
Belastung zu untersuchen. Elektrochemische Messzellen, die beheizt werden können
oder in denen cyclische Dehnungs- bzw. Torsionssversuche mit NiTi-Proben durch-
geführt werden können, müssten zu diesem Zweck entwickelt werden.
Auch ein Transfer der Strippingvoltammetrie in das SECM wäre denkbar. Somit
könnten lokale Ni2+-Konzentrationen an Nitinol-Oberflächen bestimmt werden.
Zukünftige AC-SECM-Experimente sollen weiteren Aufschluss über die Entstehungs-
herde von Lochfraßkorrosion bringen und ebenfalls wie schon bei der SISA den
Vergleich unterschiedlicher Proben ermöglichen. Hier allerdings auf lokaler Ebene.
Ein weiteres Ziel wäre die Reduzierung der isolierenden Glasummantelung der Pt-
Nanoelektroden. Somit könnte die Topographieauflösung der „constant-distance“ AC-
SECM erhöht werden.
Untersuchungen von NiTi-Proben mittels AC-SECM, die eine Phasenumwandlung
vollziehen, wären mit einer temperaturgeregelten Messzelle durchführbar. Die
Ergebnisse solcher Messungen könnten Aufschluss über das lokale Korrosions-
verhalten von NiTi im martensitischen Zustand im Vergleich zum austenitischen
Zustand liefern.
AC-SECM-Messungen an gedehnten NiTi-Proben könnten durch Entwicklung einer
Messzelle mit Spannvorrichtung durchgeführt werden und Aufschluss über das
Verhalten der Passivschicht unter mechanischer Belastung bringen. Auf dem
gleichen Weg könnten Schutzschichten auf ihre Haltbarkeit untersucht werden.

Experimenteller Teil 123
6 Experimenteller Teil
6.1 Chemikalien, Geräte und Materialien
6.1.1 Chemikalienliste
Chemikalien:
Die Reinheit aller verwendeten Chemikalien war p.a. oder von höchstem erhältlichem
Standard. Verwendetes Wasser wurde dreifach destilliert. Ausnahmen sind
gesondert gekennzeichnet.
Natriumhexachloroiridat(III)-hydrat Aldrich, Chem. Co.,
Milwaukee, Wisconsin (USA)
Rutheniumhexamintrichlorid Sigma, Deisenhofen
Salzsäure Baker, Deventer (NL)
Schwefelsäure Baker, Deventer (NL)
Natriumhydroxid Baker, Deventer (NL)
Aceton Baker, Deventer (NL)
Wasser, Ultrex II Baker, Deventer (NL)
Nickelnitratlösung in 2% HNO3,
Instra Analyzed, 1000 µg/ml Ni Baker, Deventer (NL)
1,2-Cyclohexandiondioxim Sigma-Aldrich Chemie GmbH,
Steinheim
Ammoniumchlorid, Suprapur Merck KGaA, Darmstadt
Kaliumchlorid Baker, Deventer (NL)
Ammoniaklösung 25%, Suprapur Merck KGaA, Darmstadt
Salzsäure 30%, Suprapur Merck KGaA, Darmstadt
Wismut(III)nitrat-Pentahydrat Sigma-Aldrich Chemie GmbH,
Steinheim
Lithiumbromid-Hydrat Sigma-Aldrich Chemie GmbH,
Steinheim
Dimethylglyoxim Sigma-Aldrich Chemie GmbH,
Steinheim
Ethanol absolut Merck KGaA, Darmstadt
Methanol, Lichrosolv Merck KGaA, Darmstadt

Experimenteller Teil 124
Isopropanol Merck KGaA, Darmstadt
6.1.2 Geräteliste
Komponenten des SECM mit Höhenkontrolle:
Profilschiene 90 mm Owis, Staufen
Profilschiene 65 mm Owis, Staufen
Messtisch, xy, MT 60 25 mm Owis, Staufen
2-Phasenschrittmotoren, Digimic 25 mm Owis, Staufen
3-Kanal Spannungsverstärker PS 500 Owis, Staufen
Verschiebetisch, xy, VT 45 25 mm Owis, Staufen
Messtisch, z, MT 75 25 mm, mit 200 µm
Piezoverstellung Owis, Staufen
Anregungspiezo, PSt 150/5/7 Pickelmann, München
Schrittmotorenkarte Tecuria, Chur (CH)
Messwertkarte 14 Bit AD/DA Kosiol, Frankfurt
I/O-Karte Kosiol, Frankfurt
Relais Outputkarte PCLD-785 Kosiol, Frankfurt
PC, Intel Pentium, 133MHz, 16 MB RAM Vobis AG, Aachen
Laserdiode LDM 145/670/1 mW Atos, Pfungstadt
Photodiode Spot 4D Laser 2000, Weßling
Lock-in Verstärker Signal Recovery 5210 AMETEK Advanced Measurement
Technology Inc., Oak Ridge (USA)
Lock-in Verstärker EG&G 7265 DSP AMETEK Advanced Measurement
Technology Inc., Oak Ridge (USA)
Potentiostat/Galvanostat Jaissle 1002 PC.T. Jaissle Elektronik GmbH, Waiblingen
Potentialmeter Wenking Model PPT 85 Bank Elektronik, Goettingen
Netzgerät PE 1542 Philips, Amsterdam (NL)
Netzgerät Conrad, Hirschau
Geräte des SISA-Systems:
OLGA Sensolytics GmbH, Bochum
Potentiostat PalmSens Palm Instruments BV, Houten (NL)
Potentiostat MP 75 Bank Elektronik, Goettingen
PC, AMD K6III, 400MHz, 320 MB RAM Silicon Computer, Bochum

Experimenteller Teil 125
Geräte zur Elektrodenherstellung:
Potentiostat EG&G PAR 263 A EG&G, Bad Wildbad
Potentiostat Autolab PGSTAT 12 Eco Chemie B.V., Utrecht (NL)
Kapillarpuller PP830 Narishige, London (UK)
Laserpuller Model P-2000 Sutter Instruments Co., Novato
(USA)
pH-Meter, pH 23 Knick, Berlin
Geräte zur Probenherstellung:
Poliergerät Planopol-2 Struers A/S, Ballerup (DK)
Schleifgerät Knuth-Rotor 2 Struers A/S, Ballerup (DK)
Trennmaschine Cuto 10 Jean-Wirtz GmbH & Co. KG,
Düsseldorf
6.1.3 Materialien
Platindraht 10 µm, 25 µm Goodfellow, Cambridge (UK)
Iridiumdraht 2 mm Goodfellow, Cambridge (UK)
Glaskohlenstoffstäbe SigradurG 1 mm HTW Hochtemperatur-Werkstoffe
GmbH, Tierhaupten
Borosilikat-Glaskapillaren Hilgenberg GmbH, Hilgenberg
Quarz-Glaskapillaren Hilgenberg GmbH, Hilgenberg
Aluminiumoxidpolierpasten
(1 µm; 0,3 µm) Leco GmbH, Kirchheim
Diamantsuspension 3 µm Leco GmbH, Kirchheim
Polierfilz Leco GmbH, Kirchheim
Silber-Leitlack Leitsilber L100 Pollin Electronic, Pförring
Zweikomponenten-Silber-Epoxy-Kleber
epo-tek H 20 E Polytek, Waldbronn
Carbonfasern 7,5 µm Grafil, Coventry (UK)
Carbon-Epoxy-Kleber, Electrodag 5513 Acheson Colloids, Scheemda (NL)
Zweikomponenten Silicongummi
Sylgard 184 Dow Corning Co., Midland,
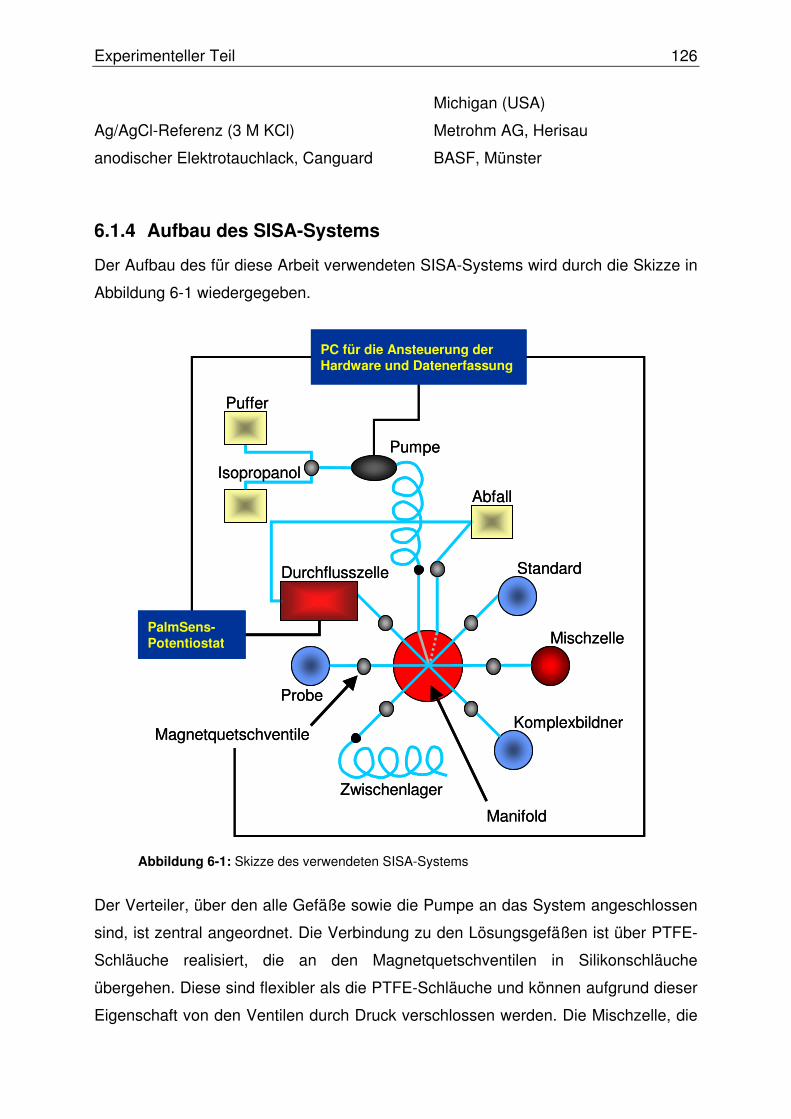
Experimenteller Teil 126
Michigan (USA)
Ag/AgCl-Referenz (3 M KCl) Metrohm AG, Herisau
anodischer Elektrotauchlack, Canguard BASF, Münster
6.1.4 Aufbau des SISA-Systems
Der Aufbau des für diese Arbeit verwendeten SISA-Systems wird durch die Skizze in
Abbildung 6-1 wiedergegeben.
Der Verteiler, über den alle Gefäße sowie die Pumpe an das System angeschlossen
sind, ist zentral angeordnet. Die Verbindung zu den Lösungsgefäßen ist über PTFE-
Schläuche realisiert, die an den Magnetquetschventilen in Silikonschläuche
übergehen. Diese sind flexibler als die PTFE-Schläuche und können aufgrund dieser
Eigenschaft von den Ventilen durch Druck verschlossen werden. Die Mischzelle, die
Puffer
Isopropanol
Pumpe
Abfall
Standard
Mischzelle
Komplexbildner
Zwischenlager
Probe
Durchflusszelle
PC für die Ansteuerung der Hardware und Datenerfassung
PalmSens-Potentiostat
Magnetquetschventile
Manifold
Puffer
Isopropanol
Pumpe
Abfall
Standard
Mischzelle
Komplexbildner
Zwischenlager
Probe
Durchflusszelle
PC für die Ansteuerung der Hardware und Datenerfassung
PalmSens-Potentiostat
Magnetquetschventile
Manifold
Abbildung 6-1: Skizze des verwendeten SISA-Systems

Experimenteller Teil 127
zur Vermischung des Analyten mit dem Puffer und dem Komplexbildner dient, und
die Durchflusszelle sind in den Kapiteln 4.2.2 und 4.2.2.8.4 genauer beschrieben.
Die Pumpvorgänge werden mittels einer Schrittmotor getriebenen Kolbenhubpumpe
mit 10 µl Kolbenvolumen durchgeführt, die von einem PC über den parallelen
Anschluss gesteuert wird. Die Ventile sowie der PalmSens-Potentiostat werden
ebenfalls von dem PC gesteuert, jedoch über jeweils eine separate serielle
Verbindung.
In der fotografischen Abbildung 6-2 ist das weiße Gehäuse des OLGA-Systems zu
sehen, welches die Pumpe (im Vordergrund) und den Verteiler mit den
Quetschventilen (im Hintergrund) beherbergt. Die Durchflussmesszelle befindet sich
an einer Stativstange befestigt vor der OLGA mit dem PalmSens-Potentiostaten im
Vordergrund. Die zweite Halterung an der Stativstange dient zur Befestigung einer
elektrochemischen Messzelle, in der die zu untersuchende Probe einem Elektrolyten
ausgesetzt werden kann. Der zusätzliche Potentiostat, welcher sich auf dem OLGA-
Gehäuse befindet, kann zum Anlegen einer Spannung an die Probe genutzt werden.
Der Puffer und der Abfallbehälter befinden sich rechts neben der Apparatur.
Abbildung 6-2: fotografische Abbildung des für die Korrosionsuntersuchungen verwendeten SISA-Systems

Experimenteller Teil 128
6.1.5 Aufbau des SECM
Der Aufbau des verwendeten SECM entspricht der in Abbildung 6-3 gezeigten Skizze
(der Laser und die Photodiode wurden zur besseren Übersichtlichkeit nicht
eingezeichnet, s.u.).
Die x-y-z-Positioniereinheit besteht aus drei Schrittmotoren mit 10 nm Verstellweg
pro Mikroschritt. Damit lässt sich die Messzelle relativ zur feststehenden SECM-
Sonde positionieren. Zur feineren und schnelleren Regulierung des Abstandes
zwischen Probe und Sonde, welches gerade für den „constant-distance“ Modus von
Bedeutung ist, befindet sich in z-Richtung zusätzlich zum Schrittmotor ein
Piezoverschiebetisch. Der Piezo wird über einen Hochspannungsverstärker von der
im PC befindlichen AD/DA-Karte angesteuert, anders als bei den Schrittmotoren, die
über eine gesonderte Motorensteuerkarte, welche über den seriellen Anschluß mit
dem Rechner kommuniziert, angesprochen werden. Die Signale der Messsonde
werden wahlweise, je nach Experiment von einem Potentiostaten, einem
Potentiometer oder einem Lock-in Verstärker registriert und vom PC über die AD/DA-
Karte ausgelesen.
Der Aufbau der Höhenkontrolle ist identisch mit der in Abbildung 2-13 dargestellten
Skizze. Der Strichlaser wird mit einem gesonderten Labornetzteil betrieben, ebenso
der Differenzverstärker der geteilten Photodiode. Zur Ansteuerung des Schüttel-
Vibrationsgedämpfter Tisch
Gegenelektrode
Referenzelektrode
MikroelektrodeSchüttel-Piezo
elektrochemische Zellemit Glasfenster
PotentiometerPotentiostatLock-in Verstärker
PC für die Ansteuerung der Hardware und Datenerfassung
Controllerkarten f. Positionier-einheit und AD/DA Karte
Z-Piezo
Y
Z
X
Vibrationsgedämpfter Tisch
Gegenelektrode
Referenzelektrode
MikroelektrodeSchüttel-Piezo
elektrochemische Zellemit Glasfenster
PotentiometerPotentiostatLock-in Verstärker
PC für die Ansteuerung der Hardware und Datenerfassung
Controllerkarten f. Positionier-einheit und AD/DA Karte
Z-Piezo
Y
Z
X
Abbildung 6-3: schematische Darstellung des für diese Arbeit verwendeten SECM

Experimenteller Teil 129
piezos wird der integrierte Frequenzgenerator des Lock-in Verstärkers genutzt, der
auch das Signal der Photodiode aufnimmt, mit der Anregungsfrequenz vergleicht und
phasenselektiv verstärkt. Das resultierende Signal wird über die AD/DA-Karte des PC
ausgelesen.
Das SECM befindet sich in einem Faraday-Käfig auf einem schwingungsgedämpften
Wägetisch, um Störungen durch elektromagnetische Strahlung und durch Er-
schütterungen zu vermeiden. Die Peripherie befindet sich auf und neben dem
Faraday-Käfig, wie in der fotografischen Abbildung 6-4 zu sehen ist.
6.2 Elektrodenherstellung
6.2.1 Herstellung von Glaskohlenstoff-, Platinmikro- und
Iridiummakroelektroden
Die Herstellung der GC-, Pt- und Ir-Elektroden ist nahezu identisch und entspricht der
in Kapitel 2.2.1 beschriebenen Vorgehensweise für Pt-Mikroelektroden.
Abbildung 6-4: fotografische Abbildung des verwendeten SECM

Experimenteller Teil 130
Als Isolierung dienen Borosilikat-Glaskapillaren unterschiedlichen Durchmessers (für
GC-Elektroden: ∅a = 1,5 mm, ∅i = 1,275 mm; Pt-Elektroden: ∅a = 1,5 mm, ∅i =
1,125 mm; Ir-Elektroden: ∅a = 2,0 mm, ∅i = 1,61 mm), die auf eine Länge von ca.
5 cm mit Hilfe eines Diamantglasritzers gekürzt werden. Ein Ende wird unter Drehen
der Kapillare mittels eines Bunsenbrenners zugeschmolzen. Nach Abkühlen der
Kapillare wird ein 1 cm langes Stück des 10 µm Platin- oder 1 mm Iridiumdrahtes,
bzw. des 1mm GC-Stabes in die Kapillare eingeführt und bis in die zugeschmolzene
Spitze nach vorn bewegt. Im Anschluß daran wird mit einer Wasserstrahlpumpe ein
Vakuum an die Kapillare angelegt und mit einer Heizwendel der Draht/Stab ungefähr
bis zur Hälfte in das Glas eingeschmolzen ohne Luftblasen mit einzuschließen. Das
Glas wird am zugeschmolzenen Ende mit einem Schleifpapier abgetragen, bis der
Draht/Stab freigelegt ist, der die scheibenförmige Elektrodenfläche bildet. Dann kann
die Elektrode mit Hilfe einer rotierenden Computerfestplatte, die mit einem feinen
Sandpapier beklebt ist, plan geschliffen werden. Bei den Pt-Mikroelektroden wird
danach das Kapillarende konisch geschliffen, bis das Verhältnis des Durchmessers
der aktiven Elektrodenfläche zur Isolierung ca. 1:10 beträgt. Dieser Schritt ist bei den
anderen Elektrodentypen nicht notwendig. Anschließend wird die Elektroden-
oberfläche mit Aluminiumoxidpaste, (3, 1 und 0,3 µm Körnung) mit Hilfe eines
Polierfilzes poliert. Die Kontaktierung erfolgt über einen Kupferdraht, der mit dem
abschleifeneinschmelzen+Platindraht
konischschleifen
+ polieren
kontaktieren
Abbildung 6-5: schematische Darstellung der Elektrodenherstellung am Beispiel einer Platinscheibenelektrode

Experimenteller Teil 131
Metalldraht über einen Silber-Epoxy-Leitkleber, bzw. der mit dem GC-Stab mittels
eines Silberleitlacks, verbunden ist. Der Kleber muss 20 Min, der Lack ca. 2 Std. bei
80° C in einem Ofen ausgehärtet werden. Die vollständige Herstellungsprozedur ist
zur Veranschaulichung in Abbildung 6-5 in Form eines Fließdiagramms dargestellt.
6.2.2 Herstellung von schwingungsfähigen Carbonfaserelektroden
Carbonfasern von ca. 7,5 µm Durchmesser und etwa 3,5 cm Länge werden zur
Entfernung von Fett und anderen Verunreinigungen in einer Soxlett-Apparatur eine
Stunde in Aceton zum Sieden erhitzt.
Einzelne, gereinigte Carbonfasern werden mit einem Carbon-Epoxy-Leitkleber an
einen Kupferdraht befestigt und kontaktiert. Nach dem Antrocknen des Klebers an
Luft wird der Kupferdraht in eine Glaskapillare eingefädelt und vorsichtig soweit
hindurch gezogen, bis sich die Klebestelle in der Mitte der Kapillare befindet. Im
Anschluß wird der Draht mit einem Zweikomponenten-Silicongummi in der Kapillare
fixiert, wobei das Aushärten der Siliconmischung in einem Ofen für 5-10 Min. bei
200° C erfolgt.
Die Kapillaren werden mit dem Kupferdraht nach unten zeigend in ein
Kapillarziehgerät so eingespannt, dass sich die Verbindungsstelle zwischen Carbon-
Silicongummi
Kupferableitdraht
Carbon-Epoxy-Leitkleber
CarbonfaserElektro-Depositionslack
Glaskapillare
Abbildung 6-6: schematische Darstellung einer schwingungs-fähigen CFME

Experimenteller Teil 132
faser und Draht direkt unter der Heizwendel befindet. Mit geringem Gewicht (ein
Metallgewicht) und Temperatur („heater level“ = 66) wird die Kapillare gezogen, so
dass sie eben durchtrennt wird. Das herausragende Ende der Faser wird durch die
Abscheidung eines anodischen Elektrotauchlacks (2 Min. bei +3,5 V) isoliert. Bei der
Abscheidung wird die Carbonfaser als Anode an eine Gleichspannungsquelle
angeschlossen, während eine Platindraht-Ringelektrode als Gegenelektrode dient.
Der Lack wird bei 200° C für 20 Min. in einem Ofen getrocknet. Im Anschluß kann die
scheibenförmige Elektrodenfläche durch einen Schnitt mit einem Skalpell quer zur
Faserachse freigelegt werden.
6.2.3 Herstellung von schwingungsfähigen Platinnanoelektroden
In eine Quarzglaskapillare von 10 cm Länge und einem Außendurchmesser von
0,9 mm sowie einem Innendurchmesser von 0,3 mm wird ein ca. 2 cm langes Stück
Platindraht von 25 µm Durchmesser eingefädelt. Der Draht wird mittig in der Kapillare
positioniert und diese mittels eines weichen Tuchs von Fett befreit bevor sie auf den
Schlitten des Laserpullers befestigt wird. Die beiden Schlitten des Pullers werden mit
einer Metallklammer arretiert, so dass sie keine Zugkraft auf die eingespannte
Kapillare ausüben können. Die beiden Enden der Kapillare werden mittels
Silikonschläuchen mit einer Wasserstrahlpumpe verbunden und ein Vakuum erzeugt.
Im Anschluß daran wird der Laserpuller für 40 s mit folgenden Parametern betrieben:
Heat: 775, Filament: 5, Velocity: 100, Delay: 120, Pull: 1.
Darauf folgt eine 20-sekündige Abkühlphase, um den Laser nicht zu überhitzen.
Diese Prozedur wird fünfmal wiederholt, damit der Platindraht optimal im Glas
eingeschmolzen ist. Nach der letzten Einschmelzphase wird innerhalb von 10 s die
Klammer entfernt, welche die Schlitten zusammenhält, und der Puller mit folgenden
Parametern gestartet:
Heat: 600, Filament: 2, Velocity: 130, Delay: 150, Pull: 220.
Nach wenigen Sekunden erfolgt der Hardpull des Laserpullers, aus dem zwei
Elektroden resultieren. Diese müssen zunächst lichtmikroskopisch auf Mängel, wie
z.B. Unterbrechungen im Platindraht, untersucht werden, bevor sie analog zu der
Beschreibung in Kapitel 6.2.1 kontaktiert werden.
Im Anschluss wird die Elektrode in eine Schleifapparatur eingesetzt, die sie in axiale
Rotation (ca. 2500 U/min) versetzt. Über eine Mikrometerschraube wird die Elektrode

Experimenteller Teil 133
vorsichtig auf ein feines Schleifpapier (Körnung≥ 500) abgesenkt und für ca. 5
Minuten poliert.
Die Überprüfung des aktiven Durchmessers der Elektrode erfolg mittels cyclischer
Voltammetrie in einer 0,5 mM [Ru(NH3)6]3+-Lösung.
6.2.4 Herstellung der miniaturisierten Referenzelektrode
Eine auf die gewünschte Länge gekürzte Pasteurpipette bildet die Hülle der
Referenzelektrode. Bei Bedarf kann der Auslauf der Pipette durch Erhitzen in einer
Bunsenbrennerflamme abgewinkelt werden, bevor ein Keramikdiaphragma in die
Spitze des Auslaufs eingeschmolzen wird. In den Glaskörper der Pipette wird in der
Nähe des oberen Randes ein Loch mittels eines Diamantbohrers gebohrt. Dieses
dient zum späteren nachfüllen des Innenelektrolyten und wird vorerst mit einem
schmalen Streifen Parafilm verschlossen. Abschließend wird die Pipette mit Hilfe
einer Spritze luftblasenfrei mit 3 M KCl-Lösung befüllt, so dass die Lösung bis wenige
mm unter den Rand der Pipette reicht.
Ein Silberdraht von ca. 5 cm Länge und einem Durchmesser von 0,5 mm wird bis zur
Hälfte zu einer Wendel gedreht. Die Drahtwendel wird in eine Mischung aus 1 M KCl
und 1 M HCl und für 10 Minuten chloridisiert. Zu diesem Zweck wird mittels eines
Labornetzteils eine Spannung von 10 V zwischen dem Silberdraht und einer
Platinringgegenelektrode angelegt, wobei der Silberdraht die Anode und der
Platinring die Kathode bildet. Im Anschluß wird der chloridisierte Draht so in die
vorher präparierte Pasteurpipette abgesenkt, dass sich die Wendel im inneren der
Pipette befindet und das Ende des Drahts mindestens 1 cm herausragt.
Abschließend wird das Ende der Pipette, aus dem der Draht herausragt, mit
Siegelwachs verschlossen.
6.3 Beschichtung der Elektroden
6.3.1 Quecksilberfilmelektroden
Zur Herstellung der Abscheidungslösung werden durch Zugabe von 4,24 ml 70
prozentige HClO4 zu dem entsprechenden Volumen an tridestillierten Wasser, 500 ml
0,1 M HClO4-Lösung vorbereitet. Im Anschluß werden 649,1 mg Quecksilberacetat
darin gelöst.
Aus der Depositionslösung muss vor der Verwendung der Sauerstoff durch
Einleitung von Argon ausgetrieben werden.

Experimenteller Teil 134
Eine Iridiumelektrode mit 2 mm Durchmesser wird zur Befreiung von Verunreinigung-
en mit einem Polierfilz und einer 0,3 µm Aluminiumoxidpaste poliert. Die Paste wird
im Ultraschallbad von der Elektrodenoberfläche entfernt und die Elektrode
zusammen mit einer Platingegen- und einer Ag/AgCl-Referenzelektrode in die
Depositionslösung getaucht.
Zur Abscheidung wird für 60 s ein Potential von –250 mV angelegt. Nach Abspülen
mit tridestilliertem Wasser kann die Elektrode für Messungen verwendet werden.
6.3.2 Wismutfilmelektroden
Die Depositionslösung wird gemäß der Literaturangabe[141] durch Lösen von
97,01 mg Bi(NO3) und 524,15 mg LiBr in 1 M HCl hergestellt.
Die zuvor mittels einer 0,3 µm Aluminiumoxidpaste und einem Polierfilz gereinigten
1mm GC-Elektrode wird unter zur Hilfenahme eines starken Wasserstrahls von den
Resten der Polierpaste befreit und als Arbeitselektrode in die Abscheidungslösung
getaucht. Eine Platingegen- und eine Ag/AgCl-Referenzelektrode komplettieren die
Dreielektrodenanordnung.
Zunächst wird ein zyklisches Voltammogramm mit einem Start/Endpotential von
250 mV und einem Umkehrpotential von –500 mV erstellt. Das Potential des
Reduktionspeaks wird für die folgende Abscheidung bei konstanter Spannung
verwendet.
Die Abscheidung des Wismutfilms erfolgt mittels Coulometrie bei konstantem
Potential, wobei das vorher ermittelte Potential eingesetzt wird. Der Abbruch der
Abscheidung erfolgt automatisch bei einem festgelegten Ladungsumsatz von
1,5 mC.
6.3.3 Mikro-pH-Elektroden auf Basis IrO2-beschichteter CFMEs
Zur Erstellung der Depositionslösung werden 0,13 g Na3IrCl6•xH2O in 10 ml 0,1 M
HCl gelöst und 2 h unter Rückfluss zum Sieden erhitzt[88]. Nach dem Abkühlen stellt
man unter Argonatmosphäre mit einer 3 M NaOH-Lösung einen pH-Wert von
ungefähr 12,5 ein. Die alkalische Lösung ist nicht an Luft stabil und muss deshalb
immer unter Argon gehalten werden.

Experimenteller Teil 135
Die Abscheidung des Iridiumoxids wird in einer abgewandelten Art nach einer
Publikation von Baur et al.[63] durchgeführt. Dazu wird in einer Dreielektroden-
anordnung mit der Carbonfaser als Arbeitselektrode, einer Platingegenelektrode und
einer Ag/AgCl-Referenzelektrode ein cyclisches Voltammogramm bestehend aus
zwei Zyklen im Potentialbereich von -300 bis +1400 mV aufgenommen. Aus dem
zweiten Zyklus wird das Abscheidungspotential für die folgende Deposition bei
konstantem Potential abgelesen. Es entspricht dem Potential des beginnenden
Sauerstoffpeaks, der meist bei ca. +1,2 V liegt. Die Abscheidung bei konstantem
Potential dauert typischerweise zwischen 60 und 120 s, je nach gewünschter
Oxidschichtdicke. Die pH-Sensoren sind nach kurzem Abspülen mit destilliertem
Wasser sofort nutzbar.
6.4 Aktivitätstest der pH-Sonden
Die pH-Sensoren werden durch Registrierung des Sondenpotentials gegen eine
Ag/AgCl-Elektrode mittels eines hochohmigen Potentiometers während der Titration
eines 25 mM Phosphatpuffers mit verdünnter HCl oder NaOH-Lösung überprüft. Der
pH-Wert der Lösung wird dabei mit einem pH-Meter und einer Glaselektrode verfolgt.
Die Potentialänderungen der pH-Mikrosonde werden über eine AD/DA-Karte in einen
PC eingelesen.
Die Beurteilung der Sonden erfolgt durch Bestimmung der Sensitivität mittels
Auftragung des Potentials gegen den pH-Wert und durch Abschätzen der
Ansprechzeit aus der aufgenommenen Kalibrierkurve.
elektrochemischeAbscheidung von IrOx
Generierung des pH-sensitiven Ir-Oxids
Carbonfaser Iridiumoxid-Film
Abbildung 6-7: schematischer Ablauf der Iridiumoxidabscheidung auf einer Carbonfaser

Experimenteller Teil 136
6.5 Probenvorbereitung
6.5.1 Material
Zu Beginn des SFB-459 wurde allen Projektpartnern NiTi-Standardmaterial für
Experimente zur Verfügung gestellt. Es handelt sich dabei um Material
unterschiedlicher Zusammensetzung und Form (Abbildung 6-8).
6.5.2 Sägen und Einbetten
Von Stangenmaterialien wurden dem-
entsprechend Scheiben zwischen 1 und 5
mm Dicke gesägt (siehe Abbildung 6-9 B)
und Bleche wurden in viereckige Stücke mit
einer Kantenlänge unter 1 cm geschnitten.
Dazu wird die Materialprobe mit dem
Polymergranulat in eine Form gegeben und
für 12 Minuten bei 170 °C mit 30 kN
gepresst. Der so entstandene Rohling
(Abbildung 6-9 A) wird entgratet und das
NiTi mittels eines groben Schleifpapiers
(80er Körnung) freigelegt. Im Anschluss
wird die so entstandene NiTi-Scheibe mit
Schmirgelpapier (320er, 500er und 1000er
Abbildung 6-8: fotografische Abbildung des vom SFB-459 zur Verfügung gestellten NiTi-Standardmaterials. oben) Bleche der Dicke 1,5 mm und der Zusammensetzung Ni: 55,73 Ti: 44,04 (wt%) unten links) Stangenmaterial mit ∅ = 12,5 mm, Zusammensetzung Ni: 55,73 Ti: 44,04 (wt%) unten rechts) Stangenmaterial mit ∅ = 15,25 mm, Zusammensetzung Ni: 55,26 Ti: 44,98 (wt%)
B
A
B
A
Abbildung 6-9: A) in Polymermatrix eingebettete und polierte NiTi-Scheibe B) NiTi-Scheibe von Stangen-material

Experimenteller Teil 137
Körnung) plan geschliffen und für das anschließende Polieren vorbereitet.
Zum Kontaktieren des NiTi wird an der Rückseite der eingebetteten Probe das
Polymer mittig aufgebohrt und eine Kupferlitze/draht an die Metallrückseite angelötet
(siehe Abbildung 6-10). Anschließend wird das Loch mit einem Zweikomponenten-
kleber verschlossen, so dass nur die Vorderseite des NiTi Kontakt zur Umgebung
hat. Die Rückseite wird nach Aushärten des Klebers wieder plangeschliffen.
6.5.3 Polieren
6.5.3.1 Mechanisches Polieren
Zum mechanischen Polieren steht das Gerät Planopol-2 zur Verfügung. Dieses ist
mit einer rotierenden Polierfilzscheibe ausgestattet.
Das Polieren findet in drei Stufen statt. In der ersten wird eine Diamantsuspension
auf das Filz aufgegeben, welche eine Partikelgröße von 6 µm aufweist. In der
zweiten und dritten Stufe wird eine Al2O3-Paste, mit einer Korngröße von 3 bzw.
1 µm auf das Filz aufgetragen. Es ist auf sauberes Arbeiten zu achten, da sonst die
Poliersuspension aus dem vorhergehenden Schritt verschleppt werden kann und für
die resultierende Rauhigkeit bestimmend ist.
Nach Beendigung des Poliervorgangs wird die Probe im Ultraschallbad mit Ethanol
gereinigt.
Abbildung 6-10: Schematischer Querschnitt durch eine eingebettete Niti-Probe, die von der Unterseite mit einem Kupferdraht elektrisch kontaktiert ist.

Experimenteller Teil 138
6.5.3.2 Elektrolytisches Polieren
Der Elektrolyt besteht aus einem Gemisch aus 21,4% Perchlorsäure und 78,6%
Essigsäure und wird während des Poliervorgangs mit Hilfe eines Magnetrührers
gerührt. Die Probe wird an dem rückwärtig angebrachten Draht als Arbeitselektrode
kontaktiert. Ein ringförmiges Platinblech dient als Gegenelektrode. Es wird für 7,5
Minuten ein Potential von 10 V angelegt. Nach der Auftragung in Abbildung 6-11 D
bewirkt dieser Parametersatz ein Minimum an Oberflächenrauhigkeit. Diese Methode
wurde von einem Projektpartner aus dem SFB 459 entwickelt und veröffentlicht[165].
Nach Abschluss der Polierprozedur wird die Probe mittels Ethanol gereinigt.
A B C D
A
B
C
D
Polierzeit [min]
Mit
tler
e ar
ith
met
isch
e R
auh
tief
e P
a[µ
m]
A B C D
A
B
C
D
Polierzeit [min]
Mit
tler
e ar
ith
met
isch
e R
auh
tief
e P
a[µ
m]
Abbildung 6-11: oben) Auftragung der mittleren arithmetischen Rauhtiefe gegen die Polierzeit, beim elektrochemischen Polieren. unten) REM-Aufnahmen der NiTi-Oberfläche in den oben gekennzeichneten Polierstadien.

Experimenteller Teil 139
6.6 Beschreibung der Experimente
6.6.1 Typisches SISA-Experiment
Nach Anschließen des Probengefäßes an das System wird durch alle Schläuche des
Systems jeweils frische Lösung aus den angeschlossenen Gefäßen gespült, um
Rückstände aus der Apparatur auszuwaschen.
Eine nach der Anleitung aus Kapitel 6.3.2 präparierte Elektrode wird umgehend in die
Bodenplatte der Durchflussmesszelle (siehe Kapitel 4.2.2.8.4) eingeschraubt, so
dass die Elektrodenoberfläche plan mit der Bodenplatte abschließt. Anschließend
wird das Oberteil der Messzelle mit der Platte fest verschraubt und die
Durchflusszelle in die Halterung gesetzt.
Nun wird frische Pufferlösung durch die Messzelle gepumpt, die sich durch die
Bohrung für die Referenzelektrode drückt. Erst wenn die Bohrung komplett mit Puffer
gefüllt ist, wird die Pumpe gestoppt und die Referenz eingesetzt. Diese
Vorgehensweise verhindert, dass Luftblasen in den Bohrungen der Messzelle
eingeschlossen werden und den leitenden Kontakt zur Referenzelektrode
unterbrechen.
Im Anschluß wird die Luft aus den Schläuchen zwischen Messzelle und
Abfallbehälter gepumpt und die Elektroden mit dem Potentiostat verbunden. Nach
dem Starten der Steuersoftware für das OLGA-System sowie für den Potentiostaten,
wird für die Potentiostatsoftware die Fernsteuerfunktion aktiviert. Entsprechen die
Einstellungen für die Messung den Vorstellungen, kann bereits der Startknopf
betätigt werden. In der OLGA-Software müssen die Parameter über das Menü
überprüft werden bevor die Messung gestartet wird.
Die Messung kann jeder Zeit nach belieben über die Stop-Schaltfläche beendet
werden.
6.6.2 Typisches SECM-Experiment
Nachdem die Messsonde im dafür vorgesehenen Halter des SECM (siehe Abbildung
6-3) befestigt wurde, kann die Messzelle mit der Probe an diese herangeführt und
manuell grob vorpositioniert werden.
Soll eine Messung im „constant-distance“ Modus erfolgen, werden im Folgenden die
für den Betrieb der scherkraftabhängigen Höhenkontrolle benötigten Einstellungen
vorgenommen. Der Laser wird auf die Spitze der Messsonde fokussiert und die

Experimenteller Teil 140
Photodiode an dem entstehenden Beugungsmuster ausgerichtet. Nun wird der
Anregungspiezo vorsichtig an die Elektrode herangeführt, bis er sie gerade berührt.
Dies kann am Schwinden des Beugungsmusters erkannt werden, da der Piezo die
Elektrode aus dem Strahlengang drückt. Der Laser wird dementsprechend
nachjustiert und der Lock-in Verstärker eingeschaltet. Im Anschluß wird die
Resonanzfrequenz der Elektrode bestimmt, indem man den zur Verfügung
stehenden Frequenzbereich durchstimmt und beim maximalen Signalausschlag am
Lock-in Verstärker einregelt. Bei den CFMEs liegt die Resonanzfrequenz typischer-
weise zwischen 150 und 250 Hz, bei den Pt-Nanoelektroden bei ca. 900 Hz.
Die Annäherung der Elektrode an die Oberfläche erfolgt nach grobem Abschätzen
des Abstandes zunächst mit dem z-Schrittmotor, wobei am Monitor des
Steuerrechners das Lock-in-Signal verfolgt wird. Die letzten ca. 200 µm werden mit
0,88 µm/s, der langsamsten verfügbaren Geschwindigkeit in diesem Programmteil
der Steuerungssoftware, zurückgelegt. Bei einem starken Abfall des Signals durch
die einsetzenden Scherkräfte wird der z-Motor sofort angehalten und soweit
zurückgesetzt, bis wieder ein maximales Lock-in-Signal registriert werden kann.
Die Feinpositionierung und die Festlegung der Parameter für die Höhenregelung
(computergesteuerte rückgekoppelte Regelschleife) werden aus einem anderen
Programmteil gestartet. Die Annäherung wird jetzt mit einem Piezoaktor
durchgeführt, dessen Annäherungsgeschwindigkeit auf 0,01 µm/s eingestellt wird. Im
Programm wird vor der Annäherung zuerst ein prozentualer Wert (meist 70%) des
maximalen Lock-in Signals festgelegt (Sollwert), bei dessen Unterschreitung die
Annäherung automatisch unterbrochen wird und das Programm zur Festlegung eines
Punktes auf der aufgezeichneten Annäherungskurve auffordert. Eine Gerade wird
zwischen diesem und dem Punkt des Abbruchs berechnet. Aus der Steigung der
Geraden erhält die Software die nötigen Parameter für die „constant-distance“
SECM-Messung.
Nach Festlegen eines Dämpfungsfaktors, der ein Überschwingen der Regelschleife
verhindern soll, werden die Abbildungsparameter, wie z.B. Länge, Abstand und
Anzahl der Linien, aber auch die Datenerfassungsrate und Scangeschwindigkeit der
Messung vorgegeben.
Nach dem Start der Messung wird die Sonde lateral in x-Richtung über die
Probenoberfläche geführt. Bei einer Veränderung des Lock-in-Signals stoppt diese
Bewegung und die Höhenkontrolle regelt den Abstand der Probe zur Elektrode mit

Experimenteller Teil 141
Hilfe des z-Piezo nach, bis das Signal wieder mit dem Sollwert übereinstimmt. Nach
Erreichen der vorgegebenen x-Länge wird die Messsonde höhenkontrolliert zum
Startpunkt der Linie zurückgeführt, bevor die Bewegung in y-Richtung erfolgt. Diese
Prozedur wiederholt sich, bis die Anzahl der eingestellten Linien erreicht ist. Der
abgespeicherte Datensatz des Experiments kann anschließend mit einer speziellen
Software dreidimensional dargestellt werden.

Anhang 142
7 Anhang
7.1 Auszug aus dem Code der SISA-Software
Im Folgenden ist die Timerroutine aus der OLGA-Software angefügt: Sub injektnorm() Dim i As Integer Dim FileNum As Integer Dim Filename, strInput As String Dim anlagerzeit As Integer, dauer As Integer Static intDepT, intCondT, intEqT, intMeasT As Integer Static StartZeit As Single Dim endzeit As Single, ZeitDif As Single Static Datum As Variant, lastDatum As Variant Static Blind As Boolean Filename = "c:\temp\Sisa.tmp" anlagerzeit = intDepT aktstandard% = 1 Select Case zähler% Case 0: Blind = True If FileExist(Filename) Then 'PalmControl/ Remotebefehlübergabe FileNum = FreeFile Open Filename For Input As #FileNum Input #FileNum, strInput intCondT = CInt(strInput) Input #FileNum, strInput Input #FileNum, strInput intDepT = CInt(strInput) Input #FileNum, strInput Input #FileNum, strInput intEqT = CInt(strInput) Input #FileNum, strInput Input #FileNum, strInput intMeasT = CInt(strInput) Close #FileNum StartZeit = Timer lastDatum = Date Else Exit Sub End If Case 1: 'Isopropan durchspülen If (isospülzahl > 0) Then If ((zyklus Mod isospülzahl) = 0) Then Call isopropansaug Else zähler = zähler + 2 End If Else zähler = zähler + 2: Exit Sub End If Case 2: Call isopropausspülen Case 3: 'Lösung bis zur Zelle pumpen Call vorspülen Case 4: 'Probe spülen Call probespülen Case 5: 'Probe ausspülen

Anhang 143
Call probeausspülen Case 6: 'Pumpe syncronisieren Call pumpsync Case 7: 'Probe nehmen If ((zyklus Mod (zwischenkalib_zyklen + 1)) = 0) And SampleInj = False Then Call injektprobe Else zähler = zähler + 1 End If Case 8: 'Probe für folgende Standardad. zwischenlagern Call probelagern SampleInj = True zähler = 99 Case 100: 'Reste verwerfen Call probeausspülen zähler = 4 Exit Sub Case 9: 'Probe aus Lager nehmen Call injektprobe Case 10: 'Probe mit 50 µl Puffer in den Reaktor spülen Call inreaktor("Probe") Case 11: 'Kalibrierlösung spülen If (zwischenkalib_zyklen > 0) And (zyklus Mod (zwischenkalib_zyklen + 1) <> 0) Then Call kalibrierspülen Else zähler = zähler + 1 End If Case 12: 'Kalibrierlösung verwerfen Call kalibrierausspülen Case 13: 'Pumpe synchronisieren Call pumpsync Case 14: 'Kalibrierlösung ansaugen Call injektstandard(zyklus Mod (zwischenkalib_zyklen + 1)) Case 15: 'Reagenz ansaugen Call ansaugnad Case 16: 'Puffer + ggf Standard + ggf Reagenz in Mischkammer spülen Call inreaktor("Standard") 'Nur für statische Meßzelle 'zähler = zähler + 2 Case 17: 'Gemisch aus Mischkammer saugen Call ausreaktor Case 18: 'Teil des Angesaugten in Abfall spülen Call reaktorverwerfen Case 19: 'Probe bis zur Meßzelle vorpumpen Call vorspülen Case 20:

Anhang 144
'Trigger setzen Call Kill(Filename) 'nur für statische Meßzelle 'Rührer starten 'Call Stirrer(anlagzeit) 'zähler = zähler + 1 Case 21: 'Probe, ggfs. mit Standard und Reagenz, durch Messzelle spülen Call vorspülzelle(ZyklusZeit%) Case 22: 'OLGA wartet, bis ein neuer File vom StrippingProgramm geschrieben wurde If (FileExist(Filename) = False) Then olga1Aktion$ = "Warte auf StrippingDPV" Exit Sub Else If Ungerade(durchflzykl%) = False Then dauer% = reaktorvolume% / CInt(stdSpeed$) Call Pumpe(Format$(dauer%), stdSpeed$, "+") End If End If Case 23: 'PlugIns ausführen i = -1 Do i = i + 2 Call PlugInObj(i).ExeMe Loop Until i >= UBound(PlugInObj) 'Wartezeit vor nächster Messreihe 'If zwischenkalib_zyklen = (zyklus Mod (zwischenkalib_zyklen + 1)) Then ' Do ' Datum = Date ' endzeit = Timer ' If Datum = lastDatum Then ' ZeitDif = endzeit - StartZeit ' Else ' ZeitDif = 86400 - StartZeit + endzeit ' End If ' DoEvents ' Loop Until ZeitDif >= DelayTime 'End If Call vorspülzelle 'Lösung aus der "Meßzelle" pumpen (nur für statische Meßzelle nötig) 'Call ausreaktor Case 24: 'Reaktor reinigen Call cleanReaktor Case 25: 'Lager reinigen If zwischenkalib_zyklen = (zyklus Mod (zwischenkalib_zyklen + 1)) Then Call cleanAd SampleInj = False End If Case 26: If OlgaKonT.pause.Checked Then Exit Sub Case 27: 'Wartezeit abwarten If zwischenkalib_zyklen = (zyklus Mod (zwischenkalib_zyklen + 1)) Then Do Datum = Date endzeit = Timer If Datum = lastDatum Then ZeitDif = endzeit - StartZeit Else ZeitDif = 86400 - StartZeit + endzeit End If DoEvents Loop Until ZeitDif >= DelayTime End If 'Zurücksetzen nach Blindmessung

Anhang 145
If (zyklus Mod (zwischenkalib_zyklen + 1)) = 0 And Blind = True Then Blind = False zähler = 3 Exit Sub End If zyklus = zyklus + 1 zähler = 0 Exit Sub End Select zähler% = zähler% + 1 End Sub

Literaturverzeichnis 146
8 Literaturverzeichnis
[1] W. J. Buehler, J. V. Gilfrich und R. C. Wiley. J Appl Phys (1963), 34, Seite
1475-7.
[2] E. Hornbogen und H. Warlimont in: Metallkunde, 1991, 2. Auflage. Auflage,
1-8. Springer-Verlag, Berlin
[3] L. M. Schetky. Materials Science Forum (2000), 327-3, Seite 9-16.
[4] M. Es-Souni, M. Es-Souni und H. Fischer-Brandies. Anal Bioanal Chem
(2005), 381 [3], Seite 557-567.
[5] S. Miyazaki. Shape Memory Materials (1998), Seite 267-281.
[6] E. Hornbogen. Advances in Science and Technology (Faenza, Italy) (1995),
4 [New Horizons for Materials], Seite 111-122.
[7] K. Shimizu, M. Irie und T. Tadaki in: Memory Effects in Materials.
Introduction to Shape Memory Materials, 1986, 168 pp.
[8] K. Otsuka und C. M. Wayman. Shape Memory Materials (1998), Seite 1-26.
[9] K. Otsuka und C. M. Wayman. Shape Memory Materials (1998), Seite 27-48.
[10] T. Saburi. Shape Memory Materials (1998), Seite 49-96.
[11] J. Liu, D. Yang, W. Wang, J. Chen und Y. Cai. Gongneng Cailiao (2000), 31
[6], Seite 584-586, 589.
[12] S. Zhu, X. Yang, Z. Cui und K. Yao. Gongneng Cailiao (1998), 29 [Suppl.],
Seite 683-688.
[13] S. A. Shabalovskaya. Bio-Med Mater Eng (2002), 12 [1], Seite 69-109.
[14] M. Lamberti, B. Perfetto, T. Costabile, N. Canozo, A. Baroni, F. Liotti, N.
Sannolo und M. Giuliano. Toxicol Appl Pharmacol (2004), 195 [3], Seite 321-
330.
[15] E. Denkhaus und K. Salnikow. Crit Rev Oncol/Hematol (2002), 42 [1], Seite
35-56.
[16] D. G. Barceloux. J Toxicol-Clin Toxicol (1999), 37 [2], Seite 239-258.
[17] K. M. Speck und A. C. Fraker. J Dent Res (1980), 59 [10], Seite 1590-1595.

Literaturverzeichnis 147
[18] D. J. Wever, A. G. Veldhuizen, J. De vries, H. J. Busscher, D. R. A. Uges und
J. R. Van horn. Biomaterials (1998), 19 [7-9], Seite 761-769.
[19] G. Rondelli und B. Vicentini. Biomaterials (1999), 20 [8], Seite 785-792.
[20] H. Kim und J. W. Johnson. Angle Orthod (1999), 69 [1], Seite 39-44.
[21] M. Es-souni, M. Es-souni und H. Fischer-brandies. Biomaterials (2002), 23
[14], Seite 2887-2894.
[22] C. Kuphasuk, Y. Oshida, C. J. Andres, S. T. Hovijitra, M. T. Barco und D. T.
Brown. J Prosthet Dent (2001), 85 [2], Seite 195-202.
[23] R. Venugopalan und C. Trepanier. Minim Invasive Ther Allied Technol
(2000), 9 [2], Seite 67-73.
[24] C. Trepanier, M. Tabrizian, L. Yahia, L. Bilodeau und D. L. Piron. J Biomed
Mater Res (1998), 43 [4], Seite 433-440.
[25] N. Fujita, Y. Sato, F. Uratani und M. Miyagi. Report of Osaka Prefectural
Industrial Technology Research Institute (1985), 86, Seite 32.
[26] M. Assad, S. Lombardi, S. Berneche, E. A. Desrosiers, L. H. Yahia und C. H.
Rivard. Ann Chir (1994), 48 [8], Seite 731-736.
[27] D. J. Wever, A. G. Veldhuizen, M. M. Sanders, J. M. Schakenraad und J. R.
Vanhorn. Biomaterials (1997), 18 [16], Seite 1115-1120.
[28] C. Trepanier, T. K. Leung, M. Tabrizian, L. Yahia, J. G. Bienvenu, J. F.
Tanguay, D. L. Piron und L. Bilodeau. J Biomed Mater Res (1999), 48 [2],
Seite 165-171.
[29] D. Bogdanski, M. Koller, D. Muller, G. Muhr, M. Bram, H. P. Buchkremer, D.
Stover, J. Choi und M. Epple. Biomaterials (2002), 23 [23], Seite 4549-4555.
[30] S. A. Shabalovskaya, J. Anderegg, F. Laab, P. A. Thiel und G. Rondelli. J
Biomed Mater Res Part B (2003), 65b [1], Seite 193-203.
[31] A. J. Bard, F. R. F. Fan, J. Kwak und O. Lev. Anal Chem (1989), 61 [2], Seite
132-8.
[32] R. C. Engstrom, M. Weber, D. J. Wunder, R. Burgess und S. Winquist. Anal
Chem (1986), 58 [4], Seite 844-8.

Literaturverzeichnis 148
[33] C. Kranz in:, Dissertation, 1996. TU München
[34] R. M. Wightman, M. R. Deakin, D. O. Wipf und P. Kovach. Proceedings -
Electrochem Soc (1987), 87-3 [Proc. Symp. Electrochem. Solid State Sci.
Educ. Grad. Undergrad. Level, 1987], Seite 25-36.
[35] F. R. F. Fan, M. V. Mirkin und A. J. Bard. J Phys Chem (1994), 98 [5], Seite
1475-81.
[36] C. Lee, C. J. Miller und A. J. Bard. Anal Chem (1991), 63 [1], Seite 78-83.
[37] B. B. Katemann und W. Schuhmann. Electroanalysis (2002), 14 [1], Seite 22-
28.
[38] B. B. Katemann, A. Schulte und W. Schuhmann. Electroanalysis (2004), 16
[1-2], Seite 60-65.
[39] P. Sun, Z. Q. Zhang, J. D. Guo und Y. H. Shao. Anal Chem (2001), 73 [21],
Seite 5346-5351.
[40] K. Borgwarth, D. Ebling und J. Heinze. Electrochim Acta (1995), 40 [10],
Seite 1455-60.
[41] J. Kwak und A. J. Bard. Anal Chem (1989), 61 [17], Seite 1794-9.
[42] C. Lee, J. Kwak und A. J. Bard. Proceedings of the National Academy of
Sciences of the United States of America (1990), 87 [5], Seite 1740-3.
[43] N. Casillas, S. Charlebois, W. H. Smyrl und H. S. White. J Electrochem Soc
(1994), 141 [3], Seite 636-642.
[44] D. Mandler und A. J. Bard. J Electrochem Soc (1989), 136 [10], Seite 3143-4.
[45] O. E. Husser, D. H. Craston und A. J. Bard. J Electrochem Soc (1989), 136
[11], Seite 3222-9.
[46] D. Mandler und A. J. Bard. J Electrochem Soc (1990), 137 [8], Seite 2468-72.
[47] S. Meltzer und D. Mandler. J Chem Soc, Faraday Trans (1995), 91 [6], Seite
1019-24.
[48] K. Borgwarth, C. Ricken, D. G. Ebling und J. Heinze. Ber Bunsen-Ges
(1995), 99 [11], Seite 1421-6.

Literaturverzeichnis 149
[49] C. Kranz, M. Ludwig, H. E. Gaub und W. Schuhmann. Adv Mater (1995), 7
[1], Seite 38-40.
[50] B. R. Horrocks, D. Schmidtke, A. Heller und A. J. Bard. Anal Chem (1993),
65 [24], Seite 3605-14.
[51] D. T. Pierce, P. R. Unwin und A. J. Bard. Anal Chem (1992), 64 [17], Seite
1795-804.
[52] G. Wittstock, K. J. Yu, H. B. Halsall, T. H. Ridgway und W. R. Heineman.
Anal Chem (1997), 67 [19], Seite 3578-82.
[53] H. Shiku, T. Matsue und I. Uchida. Anal Chem (1996), 68 [7], Seite 1276-8.
[54] F. Turcu, A. Schulte, G. Hartwich und W. Schuhmann. Angew Chem-Int Edit
(2004), 43 [26], Seite 3482-3485.
[55] E. R. Scott, A. I. Laplaza, H. S. White und J. B. Phipps. Pharm Res (1993),
10 [12], Seite 1699-1709.
[56] A. Hengstenberg, A. Blochl, I. D. Dietzel und W. Schuhmann. Angew Chem,
Int Edit (2001), 40 [5], Seite 905-908.
[57] S. Isik, M. Etienne, J. Oni, A. Blochl, S. Reiter und W. Schuhmann. Anal
Chem (2004), 76 [21], Seite 6389-6394.
[58] A. Hengstenberg, C. Kranz und W. Schuhmann. Chem-Eur J (2000), 6 [9],
Seite 1547-1554.
[59] C. Kranz, H. E. Gaub und W. Schuhmann. Adv Mater (1996), 8 [8], Seite
634-637.
[60] B. R. Horrocks, M. V. Mirkin, D. T. Pierce, A. J. Bard, G. Nagy und K. Toth.
Anal Chem (1993), 65 [9], Seite 1213-1224.
[61] M. Etienne, A. Schulte, S. Mann, G. Jordan, L. D. Dietzel und W.
Schuhmann. Anal Chem (2004), 76 [13], Seite 3682-3688.
[62] B. B. Katemann, A. Schulte, E. J. Calvo, M. Koudelka-Hep und W.
Schuhmann. Electrochem Commun (2002), 4 [2], Seite 134-138.
[63] D. O. Wipf, F. Y. Ge, T. W. Spaine und J. E. Baur. Anal Chem (2000), 72
[20], Seite 4921-4927.

Literaturverzeichnis 150
[64] E. Klusmann und J. W. Schultze. Electrochim Acta (1997), 42 [20-22], Seite
3123-3134.
[65] J. Heinze. Angew Chem (1993), 105 [9], Seite 1327-49.
[66] D. Shoup und A. Szabo. J Electroanal Chem Interfac (1984), 160 [1-2], Seite
27-31.
[67] A. J. Bard, F. R. F. Fan und M. V. Mirkin. Electroanal Chem (1994), 18, Seite
243-373.
[68] J. Kwak und A. J. Bard. Anal Chem (1989), 61 [11], Seite 1221-7.
[69] M. V. Mirkin, F. R. F. Fan und A. J. Bard. J Electroanal Chem (1992), 328 [1-
2], Seite 47-62.
[70] D. O. Wipf und A. J. Bard. J Electrochem Soc (1991), 138 [2], Seite 469-74.
[71] M. Ludwig, C. Kranz, W. Schuhmann und H. E. Gaub. Rev Sci Instr (1995),
66 [4], Seite 2857-60.
[72] R. T. Kurulugama, D. O. Wipf, S. A. Takacs, S. Pongmayteegul, P. A. Garris
und J. E. Baur. Anal Chem (2005), 77 [4], Seite 1111-1117.
[73] C. Kranz, G. Friedbacher und B. Mizaikoff. Anal Chem (2001), 73 [11], Seite
2491-+.
[74] J. V. Macpherson und P. R. Unwin. Anal Chem (2001), 73 [3], Seite 550-557.
[75] D. Oyamatsu, Y. Hirano, N. Kanaya, Y. Mase, M. Nishizawa und T. Matsue.
Bioelectrochemistry (2003), 60 [1-2], Seite 115-121.
[76] B. B. Katemann, A. Schulte und W. Schuhmann. Chemistry--A European
Journal (2003), 9 [9], Seite 2025-2033.
[77] E. Pungor. Anal Sci (1998), 14 [2], Seite 249-256.
[78] D. Ammann in: Ion-Selective Microelectrodes: Principles, Design and
Application, 1986, 346 pp.. Springer-Verlag, Berlin
[79] G. Denuault, M. H. T. Frank und L. M. Peter. Faraday Discuss (1992), 94,
Seite 23-35.
[80] H. S. Isaacs. Int Corros Conf Ser (1974), nace-3 [Localized Corros.], Seite
158-67.

Literaturverzeichnis 151
[81] R. J. O'Halloran, L. F. G. Williams und C. P. Lloyd. Corrosion (Houston, TX,
U S) (1984), 40 [7], Seite 344-9.
[82] R. C. Engstrom, S. Ghaffari und H. Qu. Anal Chem (1992), 64 [21], Seite
2525-9.
[83] A. A. Panova, P. Pantano und D. R. Walt. Anal Chem (1997), 69 [8], Seite
1635-1641.
[84] S. Glab, A. Hulanicki, G. Edwall und F. Ingman. Anal Chem (1989), 21 [1
1], Seite 29-47.
[85] K. Toth, G. Nagy, C. Wei und A. J. Bard. Electroanalysis (1995), 7 [9], Seite
801-810.
[86] R. C. Thomas in: Ion-Sensitive Intracellular Microelectrodes. How to Make
and Use Them, 1978, 110 pp.. Academic Press, London
[87] F. W. Orme. Glass Microelectrodes (1969), Seite 376-95.
[88] J. E. Baur und T. W. Spaine. J Electroanal Chem (1998), 443 [2], Seite 208-
216.
[89] S. A. M. Marzouk, S. Ufer, R. P. Buck, T. A. Johnson, L. A. Dunlap und W. E.
Cascio. Anal Chem (1998), 70 [23], Seite 5054-5061.
[90] S. R. Taylor. Prog Org Coat (2001), 43 [1-3], Seite 141-148.
[91] R. S. Lillard, P. J. Moran und H. S. Isaacs. J Electrochem Soc (1992), 139
[4], Seite 1007-12.
[92] S. J. Badger, S. B. Lyon und S. Turgoose. J Electrochem Soc (1998), 145
[12], Seite 4074-4081.
[93] I. Annergren, D. Thierry und F. Zou. J Electrochem Soc (1997), 144 [4], Seite
1208-1215.
[94] A. Safavi, N. Maleki, E. Shams und H. R. Shahbaazi. Electroanalysis (2002),
14 [13], Seite 929-934.
[95] A. M. Mierisch, J. Yuan, R. G. Kelly und S. R. Taylor. J Electrochem Soc
(1999), 146 [12], Seite 4449-4454.

Literaturverzeichnis 152
[96] A. S. Baranski und P. M. Diakowski. Journal of Solid State Electrochemistry
(2004), 8 [10], Seite 683-692.
[97] B. Ballesteros Katemann in: Elektrochemische Rastermikroskopie in
Nanomerterdimensionen und hochaufgelöste Charakterisierung von
Korrosionsphänomenen mittels lokaler Impedanz, Dissertation, 2001. Ruhr-
Universität Bochum
[98] J. M. Fernandez-Romero, M. D. Luque de Castro und M. Valcarcel. Anal
Chim Acta (1993), 276 [2], Seite 271-9.
[99] O. Elekes, D. Moscone, K. Venema und J. Korf. Clin Chim Acta (1995), 239
[2], Seite 153-65.
[100] M. J. McGrath, E. I. Iwuoha, D. Diamond und M. R. Smyth. Biosens
Bioelectron (1995), 10 [9/10], Seite 937-43.
[101] I. Ogbomo, R. Kittsteiner-Eberle, U. Englbrecht, U. Prinzing, J. Danzer und
H. L. Schmidt. Anal Chim Acta (1991), 249 [1], Seite 137-43.
[102] B. Baier, T. Joedicke, M. Lueck, O. Mueke, J. Harms und E. Krueger. Mschr
Brauwiss (1992), 45 [3], Seite 76-8.
[103] I. Ogbomo, T. Becker, W. Hummel, J. Danzer und H. L. Schmidt. Mschr
Brauwiss (1997), 50 [5/6], Seite 108-113.
[104] H. Luedi, M. B. Garn, P. Bataillard und H. M. Widmer. J Biotechnol (1990), 14
[1], Seite 71-9.
[105] U. Englbrecht und H. L. Schmidt. J Chem Technol Biotechnol (1992), 53 [4],
Seite 397-400.
[106] K. Schuegerl, L. Brandes, X. Wu, J. Bode, J. Il Ree, J. Brandt und B.
Hitzmann. Anal Chim Acta (1993), 279 [1], Seite 3-16.
[107] A. Economou und P. R. Fielden. Talanta (1998), 46 [5], Seite 1137-1146.
[108] J. F. Van Staden und R. E. Taljaard. Talanta (2004), 64 [5], Seite 1203-1212.
[109] C. L. Da silva und J. C. Masini. Fresenius J Anal Chem (2000), 367 [3], Seite
284-290.

Literaturverzeichnis 153
[110] R. E. Taljaard und J. F. Van Staden. Anal Chim Acta (1998), 366 [1-3], Seite
177-186.
[111] J. Ruzicka und A. K. Ryan in: Flow Injection Analysis - Principles, Tutorials
and Resources, 1. Auflage. Auflage. CD-ROM
[112] E. A. Hutton, B. Ogorevc und M. R. Smyth. Electroanalysis (2004), 16 [19],
Seite 1616-1621.
[113] J. Ruzicka und E. H. Hansen. Anal Chim Acta (1988), 214 [1-2], Seite 1-27.
[114] J. Ruzicka. Analyst (Cambridge, U K) (1994), 119 [9], Seite 1925-34.
[115] J. Ruzicka und G. D. Marshall. Anal Chim Acta (1990), 237 [2], Seite 329-43.
[116] S. Suteerapataranon, J. Jakmunee, Y. Vaneesorn und K. Grudpan. Talanta
(2002), 58 [6], Seite 1235-1242.
[117] W. W. Kubiak, R. M. Latonen und A. Ivaska. Talanta (2001), 53 [6], Seite
1211-1219.
[118] A. Economou und A. Voulgaropoulos. J Autom Methods Manag Chem
(2003), 25 [6], Seite 133-140.
[119] P. Richter, M. I. Toral und B. Abbott. Electroanalysis (2002), 14 [18], Seite
1288-1293.
[120] W. Schuhmann, H. Wohlschlaeger, J. Huber, H. L. Schmidt und H. Stadler.
Anal Chim Acta (1995), 315 [1-2], Seite 113-22.
[121] A. Z. Abu zuhri und W. Voelter. Fresenius J Anal Chem (1998), 360 [1], Seite
1-9.
[122] P. M. Bersier, J. Howell und C. Bruntlett. Analyst (1994), 119 [2], Seite 219-
232.
[123] N. Y. Stozhko und O. V. Inzhevatova. J Anal Chem (2004), 59 [9], Seite 854-
860.
[124] M. Korolczuk, A. Moroziewicz und M. Grabarczyk. Anal Bioanal Chem
(2005), 382 [7], Seite 1678-1682.
[125] J. Wang. Princeton Applied Research, Application Note (1985), A-7
[126] H. W. Nuernberg. Pure Appl Chem (1982), 54 [4], Seite 853-78.

Literaturverzeichnis 154
[127] J. Opydo. Mikrochim Acta (2001), 137 [3-4], Seite 157-162.
[128] Donat J. R. and Bruland K. W.. Anal Chem (1988), 60 [3], Seite 240-244.
[129] Z. Q. Zhang, H. Liu, H. Zhang und Y. F. Li. Anal Chim Acta (1996), 333 [1-2],
Seite 119-124.
[130] L. G. Shaidarova, N. A. Ulakhovich, I. L. Fedorova und Y. G. Galyametdinov.
Journal of Analytical Chemistry (Translation of Zhurnal Analiticheskoi Khimii)
(1996), 51 [7], Seite 687-692.
[131] M. H. Fonticelli, D. Posadas und R. I. Tucceri. J Electroanal Chem (2003),
553, Seite 157-161.
[132] O. Mikkelsen und K. H. Schroder. Electroanalysis (2003), 15 [8], Seite 679-
687.
[133] O. Mikkelsen, K. H. Schroder und T. A. Aarhaug. Coll Czech Chem Commun
(2001), 66 [3], Seite 465-472.
[134] A. Economou und P. R. Fielden. Trac-Trends Anal Chem (1997), 16 [5],
Seite 286-292.
[135] C. Belmont, M. L. Tercier, J. Buffle, G. C. Fiaccabrino und M. Koudelka-Hep.
Anal Chim Acta (1996), 329 [3], Seite 203-214.
[136] C. Belmont-Hebert, M. L. Tercier, J. Buffle, G. C. Fiaccabrino, N. F. de Rooij
und M. Koudelka-Hep. Anal Chem (1998), 70 [14], Seite 2949-2956.
[137] K. Fushimi, K. Azumi und M. Seo. ISIJ Int (1999), 39 [4], Seite 346-351.
[138] H. J. Diederich, S. Meyer und F. Scholz. Fresenius J Anal Chem (1994), 349
[8-9], Seite 670-675.
[139] B. J. Feldman, J. D. Osterloh, B. H. Hata und A. Dalessandro. Anal Chem
(1994), 66 [13], Seite 1983-1987.
[140] J. Wang, J. M. Lu, S. B. Hocevar, P. A. M. Farias und B. Ogorevc. Anal
Chem (2000), 72 [14], Seite 3218-3222.
[141] A. Krolicka und A. Bobrowski. Electrochem Commun (2004), 6 [2], Seite 99-
104.

Literaturverzeichnis 155
[142] A. Krolicka, A. Bobrowski, K. Kalcher, J. Mocak, I. Svancara und K. Vytras.
Electroanalysis (2003), 15 [23-24], Seite 1859-1863.
[143] M. Morfobos, A. Economou und A. Voulgaropoulos. Anal Chim Acta (2004),
519 [1], Seite 57-64.
[144] E. A. Hutton, J. T. Van elteren, B. I. Ogorevc und M. R. Smyth. Talanta
(2004), 63 [4], Seite 849-855.
[145] U. A. Kirgoz, S. Marin, M. Pumera, A. Merkoci und S. Alegret. Electroanalysis
(2005), 17 [10], Seite 881-886.
[146] A. Cobelo-Garcia, J. Santos-Echeandia, R. Prego und O. Nieto.
Electroanalysis (2005), 17 [10], Seite 906-911.
[147] O. Mikkelsen, S. M. Skogvold, K. H. Schroder, M. I. Gjerde und T. A.
Aarhaug. Anal Bioanal Chem (2003), 377 [2], Seite 322-326.
[148] M. J. G. :de la Huebra, P. Hernandez, O. Nieto, Y. Ballesteros und L.
Hernandez. Fresenius J Anal Chem (2000), 367 [5], Seite 474-478.
[149] I. Turyan, T. Erichsen, W. Schuhmann und D. Mandler. Electroanalysis
(2001), 13 [1], Seite 79-82.
[150] E. O. Martins und G. Johansson. Anal Chim Acta (1982), 140 [1], Seite 29-
37.
[151] Y. L. Huang, S. B. Khoo und M. G. S. Yap. Electroanalysis (1994), 6 [11-12],
Seite 1077-1082.
[152] B. Soucazeguillous und W. Kutner. Electroanalysis (1997), 9 [1], Seite 32-39.
[153] A. J. Dalhuijsen, T. H. Van der Meer, C. J. Hoogendoorn, J. C. Hoogvliet und
W. P. Van Bennekom. J Electroanal Chem Interfac (1985), 182 [2], Seite
295-313.
[154] H. B. Hanekamp und H. J. Van Nieuwkerk. Anal Chim Acta (1980), 121,
Seite 13-22.
[155] F. El feninat, G. Laroche, M. Fiset und D. Mantovani. Adv Eng Mater (2002),
4 [3], Seite 91-104.

Literaturverzeichnis 156
[156] S. A. Shabalovskaya. International Materials Reviews (2001), 46 [5], Seite
233-250.
[157] M. L. Tercier und N. a. B. J. Parthasarathy. Electroanalysis (1995), 7 [1],
Seite 55-63.
[158] Sensolytics GmbH, Bochum
[159] L. Lin, N. S. Lawrence, S. Thongngamdee, J. Wang und Y. H. Lin. Talanta
(2005), 65 [1], Seite 144-148.
[160] J. Ryhanen, M. Kallioinen, W. Serlo, P. Peramaki, J. Junila, P. Sandvik, E.
Niemela und J. Tuukkanen. Journal of Biomedical Materials Research
(1999), 47 [4], Seite 472-480.
[161] L. A. M. Baxter, A. Bobrowski, A. M. Bond, G. A. Heath, R. L. Paul, R. Mrzljak
und D. Zarebski. Anal Chem (1998), 70 [7], Seite 1312-1323.
[162] S. Belger in: Mikro-pH-Elektroden zur lateral aufgelösten Untersuchung von
lokalen pH-Modulationen mittels scherkraftkontrollierter elektrochemischer
Rastermikroskopie, Diplomarbeit, 2001. Ruhr-Universität Bochum
[163] B. B. Katemann, C. G. Inchauspe, P. A. Castro, A. Schulte, E. J. Calvo und
W. Schuhmann. Electrochim Acta (2003), 48 [9], Seite 1115-1121.
[164] J. Choi, D. Bogdanski, M. Koller, S. A. Esenwein, D. Muller, G. Muhr und M.
Epple. Biomaterials (2003), 24 [21], Seite 3689-3696.
[165] M. Pohl, C. Hessing und J. Frenzel. Materials Science & Engineering, A:
Structural Materials: Properties, Microstructure and Processing (2004), a378
[1-2], Seite 191-199.

Sascha Belger
Lebenslauf
Adresse Sascha Belger
Horneburger Str. 17
45711 Datteln
Tel.: 02363 – 569699
Email: [email protected]
Persönliche Informationen Geburt 03. März 1975 in Recklinghausen
Familienstand verheiratet seit 10. September 2004
Kinder einen Sohn, geboren am 25. Dezember 2004
Staatsangehörigkeit Deutsch
Schule 1981 - 1985 Westerbachschule, städt. Kath. Grundschule
Oer-Erkenschwick
1985 - 1994 Städt. Gymnasium Oer-Erkenschwick
04/1994 Allgemeine Hochschulreife
Zivildienst 1994-1995 Stadtsportverband Oer-Erkenschwick
Universität 1995 - 2001 Chemiestudium an der Ruhr-Universität Bochum
05/2001 Abschluss mit Diplom, Thema der Diplomarbeit:
„Mikro-pH-Elektroden zur lateral aufgelösten Unter-
suchung von lokalen pH-Modulationen mittels scherkraft-
kontrollierter elektrochemischer Rastermikroskopie“
2001 - 2005 Promotionsstudium an der Ruhr-Universität Bochum
„Elektrochemische Messtechniken zur globalen und
lokalen Korrosionsuntersuchung an Nickel-Titan-
Formgedächtnislegierungen“

Berufserfahrung seit 2001 wiss. Hilfskraft/Mitarbeiter in der AG Elektroanalytik &
Sensorik bei Prof. Dr. Schuhmann
Weiterbildung 07/1997 Prüfung der eingeschränkten Sachkenntnis nach § 5
Absatz 2 der Chemikalien – Verbotsverordnung
02/1998 Einführung in die EDV-gestützte Literatursuche
05/2002 TCP/IP veranschaulicht
07/2002 Windows (NT/2000/XP) Systemadministration I


















