
10. VorlesungModern Methods in Drug Discovery WS04/051 Cytochrom P450, Polymorphismus, Transporter.

Drug Metabolism Reviews, 39: 481–499, 2007Copyright © Informa Healthcare USA, Inc.ISSN: 0360-2532 print / 1097-9883 onlineDOI: 10.1080/03602530701498455
481
INHIBITION OF CYTOCHROMES P450: EXISTING AND NEW PROMISING THERAPEUTIC TARGETS
Inge SchusterUniversität Wien, Fakultät für Lebenswissenschaften, Institut für MedizinischeChemie, Wien, Austria
Rita BernhardtUniversität des Saarlandes, Institut für Biochemie, Saarbrücken, Germany
Mammalian cytochromes P450 have been shown to play highly important roles in themetabolism of drugs and xenobiotics as well as in the biosynthesis of a variety of endoge-nous compounds, many of them displaying hormonal function. The role of P450s as thera-peutic targets is still inadequately recognized although several P450 inhibitors becameefficient drugs that even reached blockbuster status. Here, we try to give a comprehensiveoverview on cytochromes P450s, which are already well-established targets – particularlyfocussing on the treatment of infectious diseases, metabolic disorders and cancer – and onthose, which have a high potential to become successful targets. In addition, the design ofinhibitors of cytochromes P450 will be discussed.
Key Words: P450; Drug target; P450 inhibition.
INTRODUCTION
It is not only a great honor, but also a great pleasure for us to contribute to this specialissue of Drug Metabolism Reviews paying tribute to the work of Prof. Dr. RonaldW. Estabrook in this area and, generally in the whole field of P450 research. Within the 50 yearsafter P450 discovery, mammalian cytochromes P450 were shown to be involved not only inthe biotransformation of drugs and xenobiotics but to play a crucial role also in the synthesisand metabolism of a variety of endogenous compounds such as steroids, fatty acids, prostag-landins, vitamins and bile acids. In many facets of this field, Ron’s investigations led togroundbreaking discoveries, contributing importantly to our present knowledge about mech-anisms and functions of P450’s. However, with his practical sense, Ron recognized also thepotential of P450’s for commercial applications, ranging from their use in chemical synthe-sis to their exploitation in pharmaceutical strategies. The present article deals with one ofthese latter aspects, namely the role of P450’s as targets for drug development, in particularfocusing on low molecular weight inhibitors of P450 function. In contrast to the overwhelm-ing amount of investigation related to P450 mechanisms and functions, in our opinion the
Address correspondence to Rita Bernhardt, Universität des Saarlandes, Institut für Biochemie, PF 15 11 50,D-66041 Saarbrücken, Germany; E-mail: [email protected]
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

482 I. SCHUSTER AND R. BERNHARDT
target function of P450’s has not received as much attention in the scientific community as itdeserves. With this review on present approaches to target P450’s and respective achieve-ments, we hope to stimulate the awareness of this promising research area.
While fungal P450s are long-established, successful targets in antimycotic therapy(for review see: Schuster, 1993), there is ample evidence that a variety, if not most ofhuman P450’s can serve as valuable therapeutic targets too. In general, targets are definedas biomolecules, which are underlying/associated with disease processes, can be modu-lated by pharmaceutical intervention, and thereby restore/improve health. Current strate-gies in the drug discovery process start with the identification and validation of biologicaltargets. As a prerequisite termed “druggability”, targets possess structural folds, whichenable interactions with (mostly) small drug-like compounds and, thereby, modulation offunction. Based on bioinformatic and chemoinformatic approaches, predictions of so-called “druggable” human targets have been made, resulting in total numbers between 2000and 3000 genes/gene products (Russ and Lampel, 2005). However, only a small fractionof these druggable targets has been successfully exploited for therapy until now. A recentanalysis of all classes of FDA-approved drugs (>20,000 products, December 2005) with aknown mode-of-action indicates that only 266 human-genome derived proteins are usedas targets so far (Overington et al., 2006), including several P450-forms (see Table 1).
DRUGS TARGETING CYTOCHROMES P450
Within the 10 most commonly drugged families, cytochromes P450 hold the tenthposition, sharing 1.9% of all FDA-approved drugs (Overington et al., 2006). In fact, thisshare is still far below the actual therapeutic potential of the cytochrome P450 superfam-ily, which displays an enormously wide range of activities from disposing xenobiotics tothe production of a broad variety of response modifiers that are essential in cell and organhomeostasis. Deficiencies/overexpression of individual forms of cytochrome P450 lead todisturbances in gene regulation, signal transduction, etc. and can cause/contribute todiverse pathological states, including cardiovascular diseases and cancer, thus makingP450 enzymes valuable targets for therapeutic intervention. For the conserved overallstructural fold with the preserved heme-binding core at the bottom of the substrate-bindingpocket, druggability appears a common characteristic of the families of human P450’s(Russ and Lampel, 2005).
Validated Targets in Endogenous Metabolism
Many products arising from cytochrome P450-dependent oxidation of endogenousmetabolites display hormone function, when—in complex with their cognate nuclearreceptors – they regulate the expression of various important genes. In case of unbalancedhigh hormone concentrations, signaling via the nuclear receptor can become massivelydisturbed and lead to pathologic conditions. Concerning sexual hormones, it is well-established that overproduction of estrogens can promote the development of breastcancer, while excessive androgens can promote the growth of prostate cancer. Relating tocorticoids, high cortisol levels are connected to the metabolic syndrome, increased aldos-terone levels were demonstrated to induce hypertension (for review see Hakki andBernhardt 2006). In order to block the undesired signaling in diseases caused by excessiveestrogens, androgens or aldosterone, the classical therapy aims at inhibition of the corre-sponding nuclear receptors by specific antihormones (Fig. 1A).
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

483
Tab
le 1
Inhi
bito
rs ta
rget
ing
P450
’s in
volv
ed in
end
ogen
ous
met
abol
ism
.
CY
PIn
hibi
tion
ofD
rugs
, dev
elop
men
t cpd
s.In
dica
tions
Ref
eren
ces
4A20
-HE
TE
syn
thes
isH
ET
0016
, DD
MS
Ang
ioge
nesi
s, e
ndot
hel.c
ell
prol
ifer
atio
nL
jubi
mov
& G
rant
, 200
5C
hen
et a
l., 2
005
5AT
hrom
boxa
ne s
ynth
ase
(in
com
bina
tion
wit
h T
X
rece
ptor
ant
agon
ist
Rid
ogre
l (Ja
nsse
n) O
zagr
el (C
aym
an P
harm
a)
Ter
bogr
el (
Boe
hrin
ger)
, Fur
egre
late
, D
azm
egre
l (Pf
izer
)
Thr
ombo
lysi
s af
ter
hear
t inf
arct
ion,
A
sthm
a,(I
nfla
mm
ator
y bo
wel
di
seas
e),(
glio
ma
inva
sive
ness
)
Dru
g B
ank
Dog
ne e
t al.,
200
2V
ande
n B
ossc
he e
t al.,
19
9211
A1,
11B
1, 1
1B2
Ster
oido
gene
sis(
chol
este
rol
side
cha
in c
leav
age,
co
rtic
oid
synt
hesi
s)
Am
inog
luth
etim
ide
Mit
otan
e (B
rist
ol-
Mye
rs-S
quib
b) to
xic.
irre
vers
ible
Adr
enoc
orti
cal c
ance
r, (
Cus
hing
sy
ndro
me)
Dru
g B
ank
Cai
et a
l., 1
997
17A
1A
ndro
gen
prod
ucti
onK
etoc
onaz
ole
(Jan
ssen
) A
bira
tero
ne P
I/I
I (B
oehr
inge
r) A
zoly
le s
tero
ids
Lia
rozo
le P
III
Pros
tate
can
cer
Atta
rd e
t al.2
006
O’D
onne
ll et
al.,
2004
19A
1A
rom
atas
e (E
stro
gen
prod
ucti
on)
Ana
stro
zole
(A
stra
zene
ca),
Let
rozo
le
(Nov
artis
), A
min
oglu
teth
imid
e,
Hes
pere
tin
(Fla
vono
id)
Exe
mes
tane
(P
fize
r) I
rrev
ersi
ble,
(ad
ione
der
ivat
.)
Tes
tola
cton
e (i
rrev
ersi
ble?
)
Est
roge
n de
pend
ent c
ance
r (B
reas
t ca
ncer
)B
rueg
gem
eier
et a
l., 2
005
Dru
g B
ank
24A
1C
alci
trio
l-24
-hyd
roxy
latio
n(i
nact
ivat
ion)
VID
400
(Nov
arti
s), C
TA
018,
CT
AP
101,
201,
C
TA
R39
8 (C
ytoc
hrom
a PI
/II)
(ir
reve
rsib
le)
Pso
rias
is, S
econ
dary
hyp
er-
para
thyr
oidi
smS
chus
ter
et a
l., 2
001
Kah
ram
an e
t al.,
200
426
A1
Ret
inoi
c ac
id o
xida
tion
(i
nact
ivat
ion)
Lia
rozo
le (
Jans
sen,
Bar
rier
The
rape
utic
s in
c.)
RA
MB
As
(ret
inoi
c ac
id m
etab
olis
m
bloc
king
age
nts)
: R11
6010
, Ram
bazo
le
(R11
5866
; PI)
Icht
hyos
is (
Orp
han
Dru
g S
tatu
s 20
04)(
Acn
e, P
sori
asis
)N
jar,
200
2Pa
tel e
t al.,
200
7
51A
1L
anos
tero
l 14-
a-de
met
hy-
lase
(st
erol
syn
thes
is)
Flu
cona
zole
, Itr
acon
azol
e, V
oric
onaz
ole
Syst
emic
myc
oses
,Top
ical
/Su
perf
icia
l myc
oses
De
Wit
h, 2
005
Not
e: M
arke
ted
drug
s: b
old,
com
poun
ds in
dev
elop
men
t: re
gula
r. F
or d
etai
ls s
ee te
xt.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

484 I. SCHUSTER AND R. BERNHARDT
A different, more straightforward strategy interferes directly with the cause of theunbalanced hormone levels. Concerning steroid hormones, a set of P450 enzymes playskey roles in their production and metabolism and, by this means exerts tight control on hor-mone levels. In case of too high levels of a hormone, inhibition of its synthesis (and/orstimulation of its breakdown) by selective inhibitors of the corresponding steroid hydroxy-lase has been successfully exploited and became a standard therapy e.g., in the case ofestrogen induced breast cancer (see below; Fig. 1B, Table 1). On the other hand, unbal-anced low hormone levels resulting from enhanced metabolism to inactive products candisable beneficial signaling, required e.g., for growth control or cell homeostasis. In thiscase, inhibition of hormone catabolism via respective P450 enzymes can lead to increasedand sustained hormone levels and function (Fig. 1C). The feasibility of this approach hasbeen successfully demonstrated by the approval of liarozole, an inhibitor of—the non-steroid—retinoic acid breakdown via CYP26A1, for the treatment of ichthyosis (see below;Table 1), and the clinical trials with inhibitors of vitamin D breakdown via CYP24A1, inthe indication of psoriasis, but also in different forms of cancer (see below; Table 1).
Starting with compounds that already became successful therapeutics (reachingeven blockbuster status), the following sections will give a synopsis of the present statusof P450 inhibitors in research and development (summary: Table 1).
Figure 1
FunctionHormone
Antagonist
Receptor
A
Function Inactive metabolite
Hormone Prehormone
P450
P450
Receptor
C
Function
Prehormone Hormone
Inactive metabolite
P450 P450
Receptor
B
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 485
CYP19A1: a target of blockbusters. Inhibition of aromatase, first describedby the group of Brodie (Schwarzel et al., 1973), was soon thereafter recognized as a pow-erful strategy to treat estrogen-dependent diseases, in particular breast cancer(Brueggemeier et al., 2005; Simpson and Dowsett, 2002; Jonat and Mundhenke, 2007;Jonat et al. 2007). Since estrogen dependency of tumour growth was shown in the major-ity of breast cancer patients (in 75% of postmenopausal and 60% of premenopausalwomen (McGurie, 1980), inhibition of estrogen production and/or estrogen function byantiestrogens appeared a highly promising approach.
After orimeten (aminogluthetimide), a relatively unselective inhibitor also of otherP450’s (see also below and Table 1), had proven efficacy in clinical trials (Santen et al.,1977), much effort was put into the design and synthesis of improved aromatase inhibi-tors, aiming at compounds with high affinity and selectivity towards aromatase. In fact,aromatase inhibitors of the second and third generation, became standard therapeutics, inparticular in postmenopausal breast cancer (Brueggemeier et al., 2005). In these patients,the ovaries are no longer the principle source of estrogen, but many other tissues are capa-ble of producing the hormone too and responding to its growth-stimulating activity in anautocrine/paracrine manner (Simpson et al., 2005; Simpson and Dowsett, 2002). Althoughthe total amounts of estrogens synthesized in the extragonadal tissues and distributed tothe circulation might be small, high local concentrations could be achieved, as demon-strated e.g., in breast tumors of postmenopausal women that contained at least 20-foldhigher estrogen levels than the surrounding plasma (Pasqualini et al., 1996).
Consequently, reduction of growth stimulation by locally produced estrogen with inhibi-tors of aromatase became a standard protocol in the treatment of estrogen dependent tumors(Howell et al., 2001; Jonat and Mundhenke 2007; Jonat et al., 2007). The recently introducedArimidex (anastrozole; accelerated launch by AstraZeneca in 2002) turns out to be the leadinghormonal breast cancer therapy in US, Japan and France with sales of $ 1,5 billion in 2006(AstraZeneca, annual report 2006). At the same time, Femara (letrozole), launched by Novartisin Europe and US by the end of 2004, is already listed under the 10 top products of the com-pany and may reach blockbuster status (> $ 1 billion) 2007 (Novartis, annual report 2006, quar-terly report 2007). Moreover, exemestane, an androstendione-derived irreversible inhibitorfrom Pfizer achieved sales of $ 320 million in 2006 (Pfizer, annual report).
CYP51A1: another blockbuster target. Although the successful applicationof aromatase inhibitors a) points to the validity and feasibility of targeting cytochromesP450 and b) the financial returns indicate a sound potential for blockbusters, still only fewdistinct P450-forms have been exploited for therapeutic strategies. A prominent exampleis lanosterol-14alpha-demethylase (CYP51A1), a P450 crucially involved in the biosyn-thesis of sterols and present in all biological kingdoms. A wide array of potent, clinicallyeffective inhibitors has been designed against the fungal form (for review see e.g.,Sheehan et al., 1999). Within this group of inhibitors, fluconazole, itraconazole, and vori-conazole have broadened therapeutic options and became major players in the rapidlyexpanding systemic antifungal market (amounting to $3.3 billion in 2003, de With et al.,2005). However, there is also a strong demand for the design of new inhibitors because ofa) a substantially rising resistance to established antifungals and b) predictions about thefuture incidence of severe systemic mycoses, which warn about a strong increase due tothe growing number of patients with reduced immune responses caused by older age,infections, and chemotherapy.
While inhibition of CYP51 is still restricted to antifungal therapy, the enzyme is consid-ered a valuable target also in other infections, with bacteria (e.g. with mycobacterium
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

486 I. SCHUSTER AND R. BERNHARDT
tuberculosis) as well as with lower eukaryotes (e.g. with trypanosoma; for review seeLepesheva and Waterman, 2007). In addition, there is increasing interest to use inhibitorsof human CYP51A1 to treat various human diseases. Blocking cholesterol synthesis atthis step is thought to counteract dyslipidemias and to inhibit growth of some forms ofcancer. The recent elucidation of the crystal structure of a microbial form (Podust et al.,2001) paved the way for a computational pharmacophore modeling of human CYP51 aim-ing at an improved design of potent inibitors (Ekins et al., 2007).
CYP5A1: renaissance of an old target. Thromboxane synthase (CYP5A1) andprostaglandin synthase (CYP8A1) play essential, however opposing roles in the hemody-namic status of the body. Thromboxane synthase converts the cyclooxygenase product,prostanglandin H2, into thromboxane A2 (TXA2), which is a powerful contractor of smoothmuscles and inducer of platelet aggregation. TXA2 is involved in a host of pathologicalstates, such as thromboembolic disorders, myocardial ischemia, pulmonary hypertension,asthma and preeclampsia. Direct, selective inhibition of CYP5A1 might have advantagesover inhibition of cyclooxygenase since the formation of the potent vasodilator and plateletaggregation antagonist prostacyclin is not impaired. As the TXA2-precursor prostaglandinH2 exerts agonistic actions on the TXA2/prostaglandin endoperoxide receptors, compoundswith a dual action, namely as inhibitors of CYP5A1 and antagonists of the receptors, havebeen designed since the 1980’s and several compounds were approved as drugs (for reviewsee Patscheke, 1990). Ridogrel (Van den Bossche et al., 1992) is used as an adjunctive ther-apy to induce thrombolysis in patients suffering acute myocardial infarction (DrugBank).Ozagrel (OKY-046) is available for the indication of asthma (Dogne et al., 2002).
In the last decade, new indications like tumor invasiveness (focusing on malignantglioma) or preeclampsia have added to the existing therapeutic interest, stimulating sev-eral groups from academia and companies to take up the design of new improved com-pounds and to perform mechanistic studies and clinical trials (e.g., Masereel, B, Rolin S.[FUNDP Namur]: Conception, synthèse et évaluation biologique d’antagonistes duthromboxane A2. Program 1995–2005).
CYP17A1: promising old target, but still unsatisfying results. CYP17 is akey enzyme in the androgen biosynthesis. Androgens not only play a critical role in the devel-opment and maintenance of sexual characteristics in human males, but are also growth factorsfor such severe and widespread diseases as benign prostatic hyperplasia (BPH) and prostaticcancer (Van den Bossche et al., 1992; Bostwick et al., 1992). Also here, classical therapeuticalconcepts like blocking the androgen receptor (e.g., by flutamide) or administering gonadorelinanalogs are controversial (Long et al., 2000). They block only testicular androgen formation(the same is true for orchiectomy and estrogen treatment), but not adrenal androgen formationwhich amounts up to 10–20% of the overall androgen production (Hartman et al., 2002).Therefore, new strategies to inhibit the production of testosterone and dihydrotestosteronedirectly by blocking CYP17 and/or the 5α-reductase have been supposed and lead to impres-sive results (De Coster et al., 1996., for review see: Hakki and Bernhardt, 2006).
Ketoconazole, introduced more than 20 years ago by Janssen and then a break-through in antifungal therapy, caused various side effects including gynecomastia in malepatients that was due to the powerful inhibition of androgen synthesis via CYP17A1(Trachtenberg et al., 1984; Williams et al., 1986). Focusing on androgen deprivation,ketoconazole was initially used to treat androgen responsive prostate cancer; later on, itwas found that high-dose ketoconazole (up to 3×400 mg/day) was effective even afterdevelopment of hormone resistant disease (Small et al., 1997). At these high doses, keto-conazole inhibits various other P450’s, including steroidogenic forms (e.g., CYP11B1,
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 487
CYP27B1 (Denner et al., 1995a,b, 1998)) and forms involved in the metabolism ofco-medicated anti-cancer agents (enhancing their efficacies), and has a direct killing effecton cancer cells. Although FDA has not approved this high-dose ketoconazole treatment,well-designed trials and active therapy have been undertaken, carefully following admin-istration guidelines and monitoring drug levels and bioavailability to reduce side effects asfar as possible (Lam, 2004). In the search for improved inhibitors with high specificity andselectivity for CYP17A1, several groups from academia (Angelastro et al., 1989; Ayubet al., 1987; Nakajin et al., 1988, 1989; Jarman et al., 1990; McCague et al., 1990; Barrieet al., 1989, 1994; Rowlands et al., 1995; Potter et al., 1996; Hartmann et al., 2002) andcompanies have designed a variety of steroidal and nonsteroidal compounds for this tar-get. At this time, only one compound, Arbiraterone acetate, a mechanism based steroidalinhibitor has entered clinical development (Attard et al., 2006, O’Donnell et al., 2004). Inaddition, the non-steroidal compound Liarozole exhibited good anti-tumor effects in aphase III clinical study (probably due to the concomitant block of CYP26A1; see below.Stearns et al., 1993).
CYP11A1/B1,B2: important targets, dirty drugs. Several drugs are in clini-cal use as inhibitors of steroidogenesis by members of the CYP11-family, but show littleselectivity for the distinct enzymes and cause polypharmacological side effects. This isparticularly the case with mitotane, an old compound (approved by FDA/CDER 1970)that has been extensively used in all forms of hypercortisolism and is the drug of choice inthe treatment of inoperable adrenocortical cancer. The cytotoxic compound acts in a mul-tifactorial way: it does not only inhibit steroid biosynthesis, but also causes adrenocortico-lytic effects and modifications of steroid peripheral metabolism (Cai et al., 1997). Otherinhibitors such as metyrapone—clinically used to block adrenal CYP11B1—and ketocon-azole interfere with reactions catalysed by a variety of other cytochromes P450 andextraadrenal effects are likely (Sonino and Boscaro, 1999). In order to substitute for these“dirty” drugs, efforts are undertaken by many groups to develop more specific and selec-tive inhibitors of distinct steroid hydroxylases (reviewed by Hakki and Bernhardt, 2006).
CYP11B2 has been under consideration of being a drug target for about a decade. Itis known for many years that aldosterone leads to hypertension as described for familialhyperaldosteronism (Stowasser et al., 1992). In addition, chronic aldosterone elevation inplasma has also been diagnosed in other diseases such as adenoma, idiopathic hyperaldos-teronism and in cases of insufficient renal flow. Although the use of aldosterone antago-nists shows clinical benefit in the treatment of these diseases, it also leads to severe sideeffects like gynaecomastia and endocrinal dysregulation. Therefore, trials to inhibit thesynthesis of aldosterone directly have been published. It turned out, however, thatCYP11B2 is not an easy drug target, since it shares 93% sequence identity on the proteinlevel with CYP11B1. This makes the development of selective inhibitors rather difficult.This target faced, however, a new challenge when it was discovered that aldosterone notonly causes hypertension but can also be involved in congestive heart failure. The RALESand EPHESUS trials clearly demonstrated that patients treated after severe heart failurewith a mineralocorticoid receptor antagonist in addition to conventional treatment,showed an up to 30% decreased mortality and morbidity (Pitt et al., 1999, 2001, 2002). Itwas demonstrated that elevated aldosterone levels lead to an increase in blood volume andmay stimulate cardiac fibroblasts resulting in cardiac hypertrophy, myocardial fibrosis,ventricular arrhythmia and other adverse effects (Brilla et al., 2000; Young and Funder,2000; Lijnen and Petrov, 2000; Ramires et al., 1998). Since the antagonists (spironolac-tone as well as eplerenone) used to treat patients in the above mentioned trials also caused
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

488 I. SCHUSTER AND R. BERNHARDT
severe side effects (Mantero and Lucarelli, 2000; MacFadyen et al., 1997; Sobermann andWeber, 2000; Pitt et al., 2001; Delyani, 2000)., it seems reasonable also in this case toinhibit aldosterone production directly.
Recent developments have convincingly demonstrated that a specific and selectiveinhibition of CYP11B2 is possible (Denner et al., 1995a,b; Ehmer et al., 2002, Bureiket al., 2004, Ulmschneider et al., 2005a,b). The data support previous results, which on thebasis of CYP11B1 and CYP11B2 modeling (Belkina et al., 2001) and site-directedmutagenesis (Böttner and Bernhardt, 1996; Böttner et al., 1996, 1998) postulated rathersignificant differences in the active sites of these very similar proteins. Several types ofchemical compounds show inhibitory effects on CYP11B2. Besides the well-known azolecompounds, which are used to treat fungal infections and thereby often exert a side effecton adrenal steroid hydroxylases, steroidal compounds as well as non-steroidal compoundswere found to specifically inhibit CYP11B2. Non- steroidal inhibitors may be advanta-geous, since it can be expected that they have fewer side effects on the endocrine systemthan steroidal compounds. Some of these compounds showed very high selectivity againstCYP11B2 which makes them interesting leads for further optimization as therapeuticagents (reviewed by Hakki and Bernhardt, 2006).
CYP26A1: new retinoid—sparing target with a huge market potential.
Focusing on the design of CYP17A1 inhibitors, clinical testing of a compoundnamed liarozole (Janssen) revealed side effects characteristic for vitamin A (all-trans ret-inoic acid—ATRA) hypervitaminosis, namely cheilitis, dry mouth, pruritus, and xerosis(Miller, 1998). In fact, increased ATRA-levels were found, caused by inhibition of ATRAcatabolism via a P450 enzyme, later identified as CYP26A1 (Van Wauve et al., 1992;White et al., 1997). ATRA is an endogenous hormone, which plays an essential role in thenormal proliferation and differentiation of epithelial cells, vision, and reproduction (Rooset al., 1998). Inside target cells, hormonal activity is terminated on hydroxylation byCYP26A1, which may be over-expressed in tumor cells and is further inducible by ATRA(White et al., 1997). Application of ATRA in pharmacological doses has resulted in bene-ficial therapeutic effects in various cancers and in hyperproliferative, inflammatory skindiseases (review: Miller, 1998; Roos et al., 1998). However, severe side effects with evenfatal outcome and development of resistance on prolonged treatment limited an otherwisesuccessful therapy. Inhibition of CYP26A1 is a new strategy to raise local hormone levelsand function by blocking a too rapid catabolism. Liarozole, the first example of retinoicacid metabolism blocking agents (“RAMBAs”), showed efficacy in various clinical trialsagainst metastatic prostate cancer and dermatological indications such as ichthyosis, acneand psoriasis. In 2004, Liarozole, has received orphan drug designation for the treatmentof congenital ichthyosis from the FDA and the European Commission. Aiming at succes-sors of liarozole with higher potency and selectivity for CYP26A1, several compoundsunderwent testing and showed promising profiles. Rambazole (R115866) has entered an oralclinical development program in the indication psoriasis and acne (Barrier Therapeutics,information February 2007). For a recent review on CYP26A1 inhibitors see Njar, 2002.
CYP24A1: promising new target, compounds still in development. Apromising group of new P450 targets are vitamin D-hydroxylases, which control synthesisand metabolism of hormonally active vitamin D (VD). VD-metabolites, in particular1α,25(OH)2D3, play a key role in regulating calcium metabolism, and moreover areinvolved in the regulation of various other physiological systems, such as the maintenanceof cell homeostasis, the control of immune responses, and the function of the central ner-vous system (Brown et al., 1999). VD deficiencies (common especially in elder subjects
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 489
with low outdoor activities) can cause/contribute to a variety of pathological states (fordetails see Zittermann, 2003), which can be improved by substitution of active VD metab-olites/analogues. Respective strategies for the treatment of type I rickets and osteoporosisare well-established, as well as for hyperproliferative dermatological diseases like psoria-sis. Beneficial effects against various forms of cancer were shown in many clinical trials;however, development of severe hypercalcemia still limits a broad use of the agents. Thesame holds true for indications like autoimmune disorders and neuronal aging (Nagpalet al., 2005).
In target cells, levels and function of active VD/analogs are short-lived due to self-induced, rapid inactivation by CYP24A1. High CYP24A1 expression might be also causedby different inducing agents (diverse endogenous compounds and xenobiotics), as well asby gene amplification (found in various tumors). Consequently, VD-deficiency may locallyarise and cause/reinforce disorders (reviewed by Schuster et al., 2006). Aiming at increased/sustained hormone levels, a project on azole-type inhibitors of CYP24A1 was started atNovartis in the early 1990’s. Selective compounds, which blocked metabolism of1α,25(OH)2D3, but not its endogenous synthesis by CYP27B1at low nanomolar concentra-tion, strongly increased and prolonged hormonal function in model systems – monitored bytranscription of D-responsive genes and growth reduction (Posner et al., 2004). A com-pound termed VID400 was chosen for development in the indication psoriasis (Schusteret al., 2001).
C-24 analogs of 1α,25(OH)2D licensed to Cytochroma Inc. (Markham, Ontario,Canada) and termed “vitamin D signal amplifiers” are both, potent inhibitors of CYP24A1and agonists of the vitamin D signaling pathway (Kahraman et al., 2004). The lead com-pound CTA018 is at the beginning of clinical Phase II in the indication: topical treatmentof mild to moderate psoriasis, (www.cytochroma.com).
Selective inhibitors of CYP24A1 offer a very wide field of therapeutic application(see above), used as single compounds to raise endogenous hormone levels or in combina-tion with low doses of exogenous vitamin D/analogs, thereby diminishing/abrogating theirhypercalcemic side effects.
CYP4 family: novel targets with great therapeutic potential. For theirfunction in the metabolism of fatty acids and synthesis of biologically active productsfrom arachidonic acid, members of the large CYP4 family are important players inorgan/organism homeostasis and pathology. Specific expression of distinct forms incertain cell types (e.g., CYP4X1 in neurons, CYP4F3 in polymorphonuclear leuko-cytes, CYP4F12 in small intestine, overexpression of CYP4Z1 in certain tumors, etc.)point to special roles of CYP4’s in these cells (Murray, 2004) and to a realistic poten-tial for modulation.
CYP4A plays a vital role in angiogenesis via production of the arachidonic acidproduct 20-HETE, which is a potent vasoconstrictor and inducer of growth factor stimu-lated angiogenesis. In model systems, addition of 20-HETE or transfection of CYP4A ledto endothelial cell growth and angiogenesis (reviewed in Ljubimov and Grant, 2005).Recent data show that inhibition of CYP4A with two mechanistically different com-pounds, N-hydroxy-N-(4-butyl-2-methylphenol) formamidine (HET0016) and dibromo-dodecenyl methylsulfonimide (DDMS), abolished the angiogenic response to growthfactors (VEGF, FGF-2, and EGF) (Chen et al., 2005).
Consequently, CYP4A appears a highly valuable target for designing selectiveinhibitors, which block neovascularization in pathological states, in particular in retinopa-thies and solid tumors.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

490 I. SCHUSTER AND R. BERNHARDT
Targeting Cytochromes P450 From the Families 1, 2, and 3
Of a total number of 57 functional human P450 enzymes, 40% are comprised in fam-ilies 1 to 3. Most of these forms are involved into the metabolism of exogenous compoundsand show broad overlapping substrate specificities, especially those five P450’s responsiblefor the metabolism of around 90% of all registered drugs (for review see: Guengerich et al.,2005). The promiscuity of these enzymes can be understood from their crystal structures,which have been already solved for 8 major P450s of families 1–3 (CYP1A2, CYP 2A6,CYP2B4, CYP2C5, CYP2C8, CYP2C9, CYP2D6, CYP3A4; structures and references inMMDB: Entrez’s 3D-structure database: http://www.ncbi.nlm.nih.gov/Database/ ) and theplasticity of their active sites. As an example, CYP3A4 the most abundant and promiscuousP450 in humans that accommodates especially large lipophilic ligands, can adopt multipleconformations, fitting simultaneously even two relatively large compounds (ketoconazole,erythromycin) in its active site (Ekroos and Sjögren, 2006).
For their high promiscuity, inhibition of drug metabolizing P450’s, has been consid-ered primarily in terms of unwanted drug-drug interactions causing poor metabolic andtoxicity profiles. In principle, each substrate of one of these P450’s can be regarded as amore or less strongly acting inhibitor on the metabolism of all the other substrates of theenzyme, thereby increasing their levels and lifetime. Importantly, oxidation of many sub-strates can lead to reactive products, which inactivate the particular enzyme and, conse-quently prevent metabolism of all its other substrates from exogenous or endogenoussources until fresh enzyme is generated by biosynthesis. Concerning CYP3A4, mecha-nism-based inactivation has been shown by numerous structurally unrelated drugs, e.g., byerythromycin, tamoxifen, fluoxetine, dihydralazine, ritonavir and many others (for reviewsee: Zhou et al., 2004).
Stabilisation of metabolically highly labile drugs and increasing their plasma levelscan be regarded also as an asset resulting from drug-drug interactions. Ritonavir, a syn-thetic peptidomimetic HIV protease inhibitor and (irreversible) inhibitor of CYP3A4 (seeabove) has been recently approved by the FDA to be co-administered in a low-dose withother protease inhibitors (lopinavir, darunavir: approved 2000 and 2006, respectively;http://www.fda.gov/). By this means, the resulting blood levels of Lopinavir or Darunavirare enhanced and, accordingly the effectiveness against HIV infection. Using pharmaco-genetic approaches and therapy-monitoring to avoid adverse effects as far as possible, thesuccessful principle of inhibiting drug inactivation might find broader application, also inother indications.
A different, new strategy deals with the therapeutic potential of P450’s in cancertherapy. By differential expression, a preferential localization of individual P450 forms(e.g., of CYP1B1, CYPP2J2, and CYP2W1) has been observed in tumors (Rodriguez-Antona and Ingelman-Sundberg, 2006). These tumor-associated P450’s might be directlyinvolved in cancerogenesis by their capacity to activate procarcinogens. Therefore, inhibi-tors of these forms could be exploited to reduce their carcinogenic potential. Inhibitorscould also diminish inactivation of chemotherapeutics by CYPs especially expressed atthe tumor site. On the other hand, these P450-forms could activate specifically designedprodrugs to cytotoxic compounds only at the tumor site. For its specific expression incolon and adrenal tumors, but not in other tissues from adults, the recently discoveredCYP2W1 appears a particularly suitable target for a respective cancer therapy (Karlgrenand Ingelman Sundberg, 2007). However, at this time most attention is paid to CYP1B1,which is overexpressed in a wide variety of human tumors and might crucially contribute
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 491
to tumor growth by converting 17ß-estradiol to 4-hydroxyestradiol. Based on CYP1B1 asa promising target, various groups have started anticancer strategies, which are still in pre-clinical development or in the early clinical phase (reviewed by McFadyen and Murray,2005).
DESIGNING INHIBITORS OF CYPS
The design of small-molecule inhibitors of P450 enzymes follows two mainapproaches, namely synthesis of substrate analogs or of compounds linked to the heme iron.Aiming at efficient and safe therapeutics, both routes offer advantages and disadvantages.
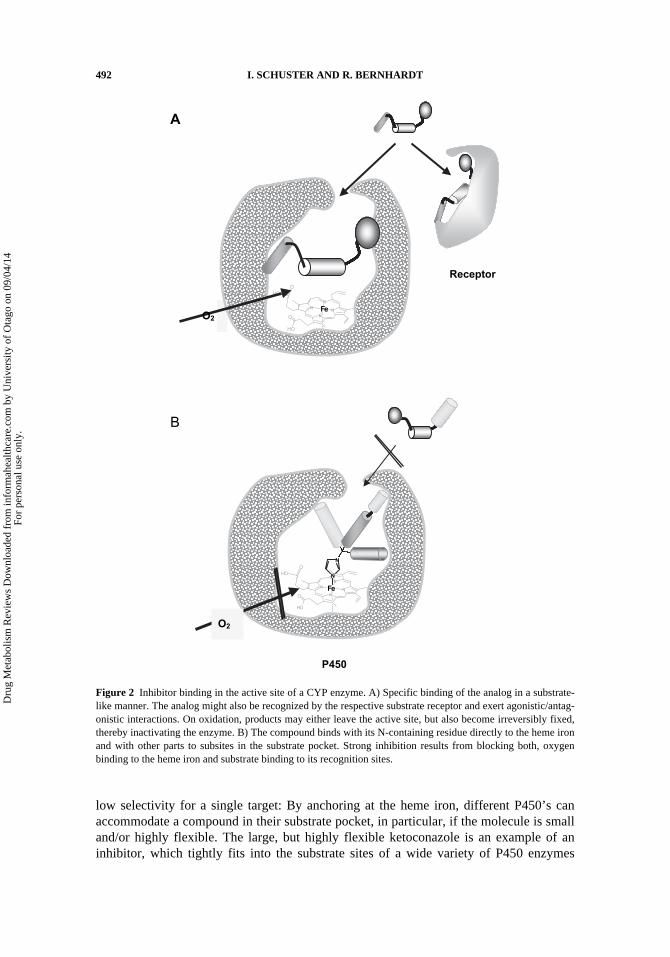
CYP’s involved in the synthesis and metabolism of endogenous compounds—e.g.,in the steroid hormone cascade show high specificities for a distinct substrate or to a classof closely related structures. Therefore, substrate analogs can be created with high speci-ficity (KD in nM-range) and selectivity for a single CYP (Fig. 2A). A drawback of thesecompounds is that they can fit well into the cognate receptors, too, resulting in undesiredagonistic/antagonistic activities. Nevertheless, some types of agonistic inhibitors, whichblock e.g the breakdown of hormones by CYP24A1 or CYP26A1, can reinforce thepotency of inhibition (see above). A weakness of substrate analogues may also be a lowmetabolic stability, needing high, possibly toxic doses to reach the target cell in sufficientconcentration.
Mechanism-based (suicide) inhibitors are even more specific than reversible sub-strate analogues (Kent et al., 2001). Binding to a target P450 in substrate-like manner isfollowed by conversion to a reactive intermediate that binds covalently to the enzyme (orother biomolecules in the neighborhood) and cause inactivation (see previous section). Inother words, a chemical knock-out of target P450’s takes place, which can be relieved bynew enzyme synthesis, in the absence of the inhibitors. Although this inhibitor profileappears most favorable, the covalently modified P450 can potentially be recognized as aneoantigen and elicit immune responses (Bonkovsky, 2007, Boelsterli, 2003). Autoanti-bodies directed against various P450 enzymes (e.g., CYP11A1, CYP17A1, CYP21A1;CYP2D6, CYP2A6, CYP2C9, CYP1A2, CYP2E1, CYP3A) have been detected in thesera of patients suffering from (drug-induced) hepatic and extrahepatic autoimmune dis-eases (Obermayer-Straub et al., 2000). For the relatively low incidence of these seriousside effects, they remain largely undetected during drug development.
A different strategy is based on compounds, which are anchored to the sixth ligationsite of the central heme iron with a lone electron pair, e.g. from a heterocyclic nitrogen.Substitution of the heterocycle with residues of appropriate length, shape, rigidity, andpolarity results in a specific, high affinity fit in the substrate pocket. These types of inhibi-tors – most of them comprising imidazole or triazole residues and termed “azoles”- func-tion by (at least) two mechanisms: i) They compete with substrate docking in the activesite and ii) block oxygen binding and activation at the heme iron (Fig. 2B). Moreover,inhibitor binding can induce conformational changes, which impede accessibility to thesubstrate pocket and/or the interactions between the components of the electron transferchain (Anderson and Chapman, 2005).
As shown in Table 1, azoles are the most successful type of P450 targeting drugs,applied in a wide range of indications and reaching blockbuster status in some of them(breast cancer and antifungal therapy). Azoles can be designed with appropriate lipophiliccharacter to enable rapid uptake into target cells and passage to their molecular targets,and to achieve optimized pharmacokinetic properties. However, azoles might exhibit a
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.
492 I. SCHUSTER AND R. BERNHARDT
low selectivity for a single target: By anchoring at the heme iron, different P450’s canaccommodate a compound in their substrate pocket, in particular, if the molecule is smalland/or highly flexible. The large, but highly flexible ketoconazole is an example of aninhibitor, which tightly fits into the substrate sites of a wide variety of P450 enzymes
Figure 2 Inhibitor binding in the active site of a CYP enzyme. A) Specific binding of the analog in a substrate-like manner. The analog might also be recognized by the respective substrate receptor and exert agonistic/antag-onistic interactions. On oxidation, products may either leave the active site, but also become irreversibly fixed,thereby inactivating the enzyme. B) The compound binds with its N-containing residue directly to the heme ironand with other parts to subsites in the substrate pocket. Strong inhibition results from blocking both, oxygenbinding to the heme iron and substrate binding to its recognition sites.
FeN
N
NN
OHO
O
HO
O2
Receptor
A
FeN
N
NN
OOH
O
OH
N
N
O2
P450
B
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 493
(Schuster, 1985). For the promiscuity of drug metabolizing enzymes, binding of a distinctdrug can result in adverse side effects on co-medication with drugs, which are substratesfor the particular enzyme (drug-drug interactions; see above). On the other hand,enhanced metabolic stability of the azole itself can result if it efficiently suppresses oxy-gen binding, which is essentially required for its oxidation. Table 2 lists advantages anddisadvantages associated with the different strategies of P450-inhibition.
In addition to these established and successful strategies, we would like to add a newapproach, namely modulation of P450 activities by interference with the protein-proteininteractions between the P450 and its electron-transfering protein. Considering potentialregulators of protein-protein complexes, we hypothesize that small, highly charged com-pounds may bind to protein interfaces and thereby alter protein functions.
OUTLOOK
During the past years it became obvious that cytochromes P450 not only play anexceptional role in drug and xenobiotics metabolism and in the biosynthesis of endoge-nous compounds, but also gain increasing importance as novel therapeutic targets for drugdevelopment. Because of their role in the generation of essential biological response mod-ifiers and in the maintenance of their levels, overexpression (e.g. in tumors) or deficien-cies (e.g. CYP27B1 in ricketts) of distinct functionally active P450s can substantiallycontribute or even cause a very broad range of pathological disorders. Normalizing P450function could, therefore be regarded as a causative treatment rather than a symptomaticone. Inhibition of P450s with lipophilic, small molecules has proven a feasible approach,already leading to the production of blockbuster drugs in the indications estrogen-inducedcancer and systemic mycoses. Moreover, advantages of using these types of inhibitorsover other methods to block P450-mediated signaling became observable, resulting fromtheir favourable pharmacokinetic properties.
It is obvious that many more P450s than recently exploited may serve as successfultherapeutic targets. In the post-genomic era, reliable expression systems are available formost of the P450s, which are prerequisites to develop efficient methods to test for theirfunction, but also to screen for specific and selective inhibitors of their function. For the
Table 2 Strategies of P450 inhibition.
Plus Minus
Substrate analogues Reversible inhibitors:High specificity and selectivity
can be achievedAgonistic/antagonistic activity at
the cognate receptorMechanism-based inhibitors:
Very high selectivity, knock-out of the target P450
Immune-mediated idiosyncratic reactions possible
Inhibitors of CYP oxidation (azoles)
High specificity Reduced selectivity: potential to bind to other CYP‘s
Devoid of agonistic/ antagonistic activity
Drug-drug interactions
Enhanced metabolic stabilityInhibitors of
electron transferModulation of Protein-protein interactions
Novel concept Outcome uncertainPhysiological relevance
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

494 I. SCHUSTER AND R. BERNHARDT
low homology of different P450s, there is a realistic opportunity to detect potent, selectiveinhibitors of individual forms, which do not substantially interfere with other forms thatwould cause detrimental side effects (as it is still shown with present inhibitors, e.g., of theCYP11-family). Optimism results also from studies on P450s exhibiting greater than 90%identity in their amino acid sequences (e.g., CYP11B1 and CYP11B2), for which effectiveand selective inhibitors could be found.
Recent screening methods using high throughput technologies, combinatorial strate-gies and large compound libraries will reveal a host of new lead structures not only foralready well-accepted drug targets such as steroidogenic P450, but also for the majority ofP450s, which have not yet been addressed as targets. Optimizing lead structures withrespect to specificity, selectivity, efficacy and absence of toxicity in appropriate modelsystems can then result in compounds, suitable to enter the pre-clinical and clinical drugdevelopment programs. We expect that at least some of the programs will provide us withnovel drugs with a great potential in various indications, particularly also to becomeblockbusters in contributing to the fight against the greatest threats of human beings: car-diovascular diseases and cancer.
ACKNOWLEDGMENT
The authors would like to thank Ron Estabrook for his wonderful friendship andfor fruitful discussions.
The help of Gabi Schon in preparing the manuscript is thankfully acknowledged.The work is supported by a grant from the Fonds der Chemischen Industrie to R.B.
REFERENCES
Anderson, J. L., Chapman, S. K. (2005). Ligand probes for heme proteins. Dalton Trans: 13–24.Angelastro, M. R., Laughlin, M. E., Schatzman, G. L., Bey, P., Blohm, T. R. (1989). 17,-(Cyclopro-
pylamino)-Androst-5-en-3,-ol, a Selective Mechanism-Based Inhibitor of CytochromeP45017alpha (Steroid 17alpha-hydroxylase/C17, 20Lyase). Biochem. Biophys. Res. Commun.162:1571–1577.
Attard, G., Sarker, D., Reid, A., Molife, R., Parker, C., de Bono, J. S. (2006). Improving the out-come of patients with castration-resistant prostate cancer through rational drug development.British Journal of Cancer 95:767–774.
Ayub, M., Level, M. (1987). Inhibition of Testicular 17α-hydroxylase and 17, 20-Lyase but not 3-Hydroxysteroid Dehydrogenase-Isomerase or 17-Hydroxysteroid Oxidoreductase by Keto-conazole and Other Imidazole drugs. J. Steroid Biochem. 28:521–531.
Barrie, S. E., Rowlands, M. G., Foster, A. B., Jarman, M. (1989). Inhibition of 17α-hydroxylase/C17, 20Lyase by bifluranol and its analogues. J. Steroid Biochem. 33:1191–1195.
Barrie, S. E., Potter, G. A., Goddard, P. M., Haynes, B. P., Dowsett, M., Jarman, M. (1994). Pharma-cology of Novel Steroidal Inhibitors of Cytochrome P45017alpha (17alpha-Hydroxylase/C17, 20 Lyase. J. Steroid Biochem. Mol. Biol. 50:267–273.
Belkina, N. V., Lisurek, M., Ivanov, A. S., Bernhardt, R. (2001). Modelling of three-dimensionalstructures of cytochromes P450 11B1 and 11B2. J. Inorg. Biochem. 87:197–207.
Boelsterli, U. A. (2003). Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity.Toxicol. Appl. Pharmacol. 192:307–322.
Böttner, B., Bernhardt, R. (1996a). Changed ratios of glucocorticoids/mineralocorticoids caused bypoint mutations in the putative I-helix regions of CYP11B1 and CYP11B2. Endocr. Res.4:455–461.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 495
Böttner, B., Schrauber, H., Bernhardt, R. (1996b). Engineering a mineralocorticoid- to a glucocorti-coid-synthesizing cytochrome P450. J. Biol. Chem. 271:8028–8033.
Böttner, B., Denner, K., Bernhardt, R. (1998). Conferring novel aldosterone synthesis to humanCYP11B1 by replacing key amino acid residues for CYP11B2 specific ones. Eur. J. Biochem.252:458–466.
Bonkovsky, H. L. (2007). AASLD-FDA-NiH-PhRMA Hepatotoxicity Meeting www.fda.gov/cder/livertox/presentations2007.htm
Bostwick, D. G., Cooner, W. H., Denis, L., Jones, G. W., Scardino, P. W., Murphy, G. P. (1992).The association of benign prostatic hyperplasia and cancer of the prostate. Cancer 70:291–301.
Brilla, C. G. (2000). Aldosterone and moyocardial fibrosis in heart failure. Herz 25:299–306.Brown, A. J., Ritter, C., Slatopolsky, E. (1999). Vitamin D. Am. J. Physiol. 277:157–175.Brueggemeier, R. W., Hackett, J. C., Diaz-Cruz, E. S. (2005). Aromatase inhibitors in the treatment
of breast cancer. Endocrine Reviews 26:331–345.Bureik, M., Hübel, K., Dragan, C.-A., Scher, J., Becker, H., Lenz, N., Bernhardt, R. (2004). Devel-
opment of test systems for the discovery of selective human aldosterone synthase (CYP11B2)and 11β-hydroxylase (CYP11B1) inhibitors. Discovery of a new lead compound for the ther-apy of congestive heart failure, myocardial fibrosis and hypertension. Molecular and CellularEndocrinology 217:249–254.
Cai, W., Counsell, R. E., Schteingart, D. E., Sinsheimer, J. E., Vaz, A. D. N., Wotring, L. L. (1997).Adrenal proteins bound by a reactive intermediate of mitotane. Cancer Chemother. Pharma-col. 39:537–540.
Chen, P., Guo, M., Wygle, D., Edwards, P. A., Falck, J. R., Roman, R. J., Scicli, A. G. (2005).Inhibitors of Cytochrome P450 4A Suppress Angiogenic Responses. Amer. J. Pathol.166:615–624.
De Coster, R., Wouters, W., Bruynseels, J. (1996). P-450 dependent enzymes as targets for prostatecancer therapy, J. Steroid Biochem. Mol. Biol. 56:133–143.
Delyani, J. A. (2000). Mineralocorticoid receptor antagonists: the evolution of utility and pharma-cology. Kidney Int. 57:1408–1411.
Denner, K., Doehmer, J., Bernhardt, R. (1995a). Cloning of CYP11B1 and CYP11B2 from normalhuman adrenals and their functional expression in COS- and V79 Chinese Hamster cells.Endocr. Res. 21:443–448.
Denner, K., Vogel, R., Schmalix, W., Doehmer, J., Bernhardt, R. (1995b). Cloning and stableexpression of the human mitochondrial cytochrome P45011B1 cDNA in V79 chinese hamstercells and their application for testing of potential inhibitors.” Pharmacogenetics 5:89–96.
Denner, K., Bernhardt, R. (1998). Inhibition studies of steroid conversions mediated by humanCYP11B1 and CYP11B2 expressed in cell cultures. In: Oxygen Homeostasis and its Dynam-ics. Tokyo, Berlin, Heidelberg, New York, Springer-Verlag, pp. 231–236.
Dogne, J. M., de Leval, X., Benoit, P., Rolin, S., Pirotte, B., Masereel, B. (2002). Therapeutic poten-tial of thromboxane inhibitors in asthma. Expert. Opin. Investig. Drugs 11:275–281.
De With, K., Steib-Bauert, M., Knoth, H., Dörje, F., Strehl, E., Rothe, U., Maier, L., Kern, W. V.(2005). Hospital use of systemic antifungal drugs. BMC Clin. Pharmacol. 5:1 http://www.biomedcentral.com/1472-6904/5/1.
Ehmer, P. B., Bureik, M., Bernhardt, R., Muller, U., Hartmann, R. W. (2002). Development of a testsystem for inhibitors of human aldosterone synthase (CYP11B2): screening in fission yeastand evaluation of selectivity in V79 cells. J. Steroid Biochem. Mol. Biol. 81:173–179.
Ekins, S., Mankowski, D. C., Hoover, D. J., Lawton, M. P., Treadway, J. L., Harwood, H. J. (2007).Three Dimensional-Quantitative Structure Activity Relationship Analysis of Human CYP51Inhibitors. Drug. Metab. Dispos. 35:493–500.
Ekroos, M., Sjögren, T. (2006). Structural basis for ligand promiscuity in cytochrome P450 3A4.Proc. Natl. Acad. Sci. U.S.A. 103:13682–13687.
Guengerich, F. P., Wu, Z. L., Bartelson, C. J. (2005). Function of human cytochrome P450s: charac-terization of the orphans. Biochem. Biophys. Res. Commun. 338:465–469.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

496 I. SCHUSTER AND R. BERNHARDT
Hakki, T., Bernhardt, R. (2006). CYP 17- and CYP11B-dependent steroid hydroxylases as drugdevelopment targets. Pharmacology & Therapeutics 111:27–52.
Hartmann, R. W., Ehmer, P. B., Haidar, S., Hector, M., Jose, J., Klein, C. D. P., Seidel, S. B.,Sergejew, T. F., Wachall, B. G., Wächter, G. A., Zhuang, Y. (2002). Inhibition of CYP17,a new strategy of the Treatment of prostate cancer. Arch. Pharm. Med. Chem. 4:119–128.
Howell, A., Howell S. J., Clarke, R., Anderson E. (2001). Where do selective estrogen receptormodulators (SERMs) and aromatase inhibitors (ALs) now fit into breast cancer treatmentalgorithms? J. Steroid Biochem. Mol. Biol. 79:227–237.
Jarman, M., Barrie, S. E., Deadman, J. J., Houghton, J., McCague, R. (1990). Hydroxyperfluoroa-zobenzenes: Novel Inhibitors of Enzymes of Androgen Biosynthesis. J. Med. Chem.33:2452–2455.
Jonat, W., Mundhenke, C. (2007a). The FACE Trial: Letrozole or Anastrozole as Initial AdjuvantTherapy? Cancer Invest. 25:14–18.
Jonat, W., Hilpert, F., Kaufmann, M. (2007b). Aromatase inhibitors: a safety comparison. ExpertOpinion on Drug Safety 6:165–174.
Kahraman, M., Sinishtaj, S., Dolan, P. M., Kensler, T. W., Peleg, S., Saha, U., Chuang, S. S.,Bernstein, G., Korczak, B., Posner, G. H. (2004). Potent, selective and low-calcemic inhibitorsof CYP24 hydroxylase: 24-sulfoximine analogues of the hormone 1 alpha,25-dihydroxyvitaminD(3). J. Med. Chem. 47:6854–6863.
Karlgren, M., Ingelman-Sundberg, M. (2007) Tumour-specific expression of CYP2W1: its potentialas a drug target in cancer therapy. Expert Opin. Ther. Targets 11:61–67.
Kent, U. M., Jushchyshyn M. I., Hollenberg, P. W. (2001). Mechanism-Based Inactivators as Probesof Cytochrome P450 Structure and Function. Curr. Drug Metab. 2:215–243.
Lam, R. (2004). High Dose Ketoconazole Plus Hydrocortisone (HDK+ HC). PCRI Insights News-letter 7(2) (http://www.prostate-cancer.org/education/andeprv/Lam_HDK.html)
Lepesheva, G. I., Waterman, M. R. (2007). Sterol 14alpha-demethylase cytochrome P450 (CYP51),a P450 in all biological kingdoms. Biochim. Biophys. Acta 1770:467–477.
Lijnen, P., Petrov, V. (2000). Induction of cardiac fibrosis by aldosterone. J. Mol. Cell. Cardiac.32:865–879.
Ljubimov, A. V., Grant, M. B. (2005). P450 in the angiogenesis affair. Am. J. Pathol. 166:341–344.Long, B., Grigoryev, D. N., Nnane, I. P., Liu, Y., Ling, Y. Z., Brodie, A. M. (2000). Antiandrogenic
effects of novel androgen synthesis inhibitors on hormone-dependent prostate cancer. CancerRes. 60:6630–6640.
MacFadyen, R. J., Barr, C. S., Struthers, A. D. (1997). Aldosterone blockade reduces vascular col-lagen turnover, improves heart rate variability and reduces early morning rise in heart rate inheart failure patients. Cardiovasc. Res. 35:30–34.
Mantero, F., Lucarelli, G. (2000). Aldosterone antagonism in hypertension and heart failure. Ann.Endocrinol. 61:52–60.
McCague, R., Rowlands, M. G., Barrie, S. E., Houghton, J. (1990). Inhibition of Enzymes ofEstrogen and Androgen Biosynthesis by Ester of 4-Pyridylacetic acid. J. Med. Chem.33:3050–3055.
McFadyen M. C. E., Murray, G. I. (2005). Cytochrome P450 1B1: a novel anticancer therapeutictarget. Future Oncol. 1:259–263.
McGurie, W. L. (1980). Steroid hormone receptors in breast cancer treatment strategy. Recent Prog.Horm. Res. 36:135–156.
Miller W. H. (1998). The emerging role of retinoids and retinoic acid metabolism blocking agents inthe treatment of cancer. Cancer 83:1471–1482.
Murray, M. (2004). Review of: Identification of a novel mammary-restricted cytochrome P450,CYP4Z1, with overexpression in breast carcinoma. Breast Cancer Online (www.bco.org):7(10).
Nagpal, S., Na, S., Rathnachalam, R. (2005). Noncalcemic Actions of Vitamin D Receptor Ligands.Endocrine Reviews 26:662–687.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 497
Nakajin, S., Takahashi, K., Shinoda, M. (1988). Inhibitory Effect and Spectral Changes on Pig Tes-ticular Cytochrome P-450 (17αhydroxylase/C17, 20-lyase) by 20,-Hydroxy-C21-steroids.Yakugaku Zasshi 108:1188–1195.
Nakajin, S., Takahashi, K., Shinoda, M. (1989). Inhibitory effect and Interaction of Stanozolol withPig Testicular Cytochrome P-450 (17α-hydroxylase/C17, 20-Lyase). Chem. Pharm. Bull.(Tokyo) 37:1855–1858.
Njar V. C. O. (2002). Cytochrome P450 retinoic acid 4-hydroxylase inhibitors: potential agents forcancer therapy. Mini Rev. Med. Chem 2:261–269.
Obermayer-Straub, P., Strassburg, C. P., Manns, M. P. (2000). Target proteins in human autoimmu-nity: cytochromes P450 and UDP-glucuronyltransferases. Can. J. Gastroenterol. 14:429–439.
O’Donnell, A., Judson, I., Dowsett, M., Raynaud, F., Dearnaley, D., Mason, M., Harland, S.,Robbins, A., Halbert, G., Nutley, B., Jarman, M. (2004). Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostatecancer. Br. J. Cancer 90:2317–2325.
Overington, J. P., Al-Lazikani, B., Hopkins, A. L. (2006) How many drug targets are there? NatureReviews Drug Discovery 5:993–996.
Pasqualini, J. R., Chetrite, G., Blacker, C., Feinstein M. C., Delalonde, L., Talbi, M. Maloche, C.(1996). Concentration of estrone, estradiol, and estrone sulfate and evaluation of sulfatase andaromatase activities in pre and postmenopausal breast cancer patients. J. Clin. Endocrinol.Metab. 81:1460–1464.
Patel, J., Khandelwal, A., Chopra, P., Handratta, V., Njar, V. (2007). Murine toxicology and phar-macokinetics of novel retinoic acid metabolism blocking agents. Cancer Chemother. Phar-macol. Mar 8: (Epub ahead of print).
Patscheke, H. (1990). Current concepts for a drug-induced inhibition of formation and action ofthromboxane A2. Blut 60:261–268.
Pitt, B., Zannad, F., Remme, W. J., Cody, R., Castaigne, A., Peerez, A., Palensky, J., Wittes, J.(1999). The effect of spironolactone on morbidity and mortality in patients with severeheart failure. Randomized Aldosterone Evaluation study Investigators. N. Engl. J. Med.341:709–717.
Pitt, B., Williams, G., Remme, W., Martinez, F., Lopez-Sendon, J., Zannad, F., Neaton, J., Roniker, B.,Hurley, S., Burns, D., Bittman, R., Kleiman, J. (2001). The EPHESUS trial: eplerenone inpatients with heart failure due to systolic dysfunction complicating acute myocardial infarc-tion. Eplerenone Post-AMI Heart Failure Efficacy and Survival Study. Cardiovasc. DrugsTher. 1:79–87.
Pitt, B. (2002). Do diuretics and aldosterone receptor antagonists improve ventricular remodeling? J.Card. Fail. 8:491–493.
Podust, L. M., Stojan, J., Poulos, T. L., Waterman, M. R. (2001). Substrate recognition sites in14alpha-sterol demethylase from comparative analysis of amino acid sequences and X-raystructure of Mycobacterium tuberculosis CYP51. J. Inorg. Biochem. 87:227–235.
Posner, G. H., Crawford, K. R., Yang, H. W., Kahraman, M., Jeon, H. B., Li, H., Lee, J. K., Suh, B. C.,Hatcher, M. A., Labonte, T., Usera, A., Dolan, P. M., Kensler, T. W.. Peleg, S., Jones, G.,Zhang, A., Korczak, B., Saha, U., Chuang, S. S. (2004). Potent, low-calcemic, selectiveinhibitors of CYP24 hydroxylase: 24-sulfone analogs of the hormone 1α,25-dihydroxyvita-min D3. J. Steroid Biochem. Mol. Biol. 89–90:5–12.
Potter, G. A., Barrie, S. E., Jarman, M., Rowlands, M. (1996). Novel Steroidal Inhibitors of HumanCytochrome P45017alpha (17α-Hydroxylase/C17–20 Lyase): Potential Agents for the Treat-ment of Prostatic Cancer. J. Med. Chem. 38:2463–2471.
Ramires, F. J., Sun, Y., Weber, K. T. (1998). Myocardial fibrosis associated with aldosterone orangiotensin II administration: attenuation by calcium channel blockade. J. Mol. Cell. Cardiol.30:475–483.
Rodriguez-Antona C., Ingelman-Sundberg, M. (2006). Cytochrome P450 pharmacogenetics andcancer. Oncogene 25:1679–1691.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

498 I. SCHUSTER AND R. BERNHARDT
Roos T. C., Jugert, F. K., Merk, H. F., Bickers, D. R. (1998). Retinoid metabolism in the skin. Phar-macol. Rev 50:315–333.
Rowlands, M. G., Barrie, S. E., Chan, F., Houghton, J., Jarman, M., McCague, R., Potter, G. A.(1995). Esters of 3-Pyridylacetic Acid that Combine Potent Inhibition of 17α-Hydroxylase/C17,20-Lyase (Cytochrome P45017R) with Resistance to Esterase hydrolysis. J. Med. Chem.38:4191–4197.
Russ, A. P., Lampel, S. (2005). The druggable genome: an update. DDT 10:1607–1610.Santen, R. J., Samojlik, E., Lipton, A., Harvey, H., Ruby, E. R., Wells, S. A., Kendall, J. (1977).
Kinetic, hormonal and clinical studies with aminoglutethimide in breast cancer. Cancer.39:2948–2958.
Schuster, I. (1993). Ergosterol Synthesis: The major Target in Antimycotic Therapy. In: Ruckpaul, K.,Rein, R. ed Frontiers in Biotransformation, Vol. 8, Berlin: Akademie Verlag, pp. 147–185.
Schuster, I. (1985). The interaction of representative members from two classes of antimycotics–theazoles and the allylamines–with cytochromes P-450 in steroidogenic tissues and liver. Xeno-biotica 15:529–546.
Schuster, I., Egger, H., Bikle, D., Herzig, G., Reddy, G. S., Stuetz, A., Stuetz, P., Vorisek, G. (2001).Selective inhibition of vitamin D hydroxylases. Steroids 66:409–422.
Schuster, I., Astecker, N., Egger, H., Herzig, G., Reddy, G. S., Schuessler, M., Vorisek, G.,Wachter, C. (2006). Inhibitors of vitamin D-hydroxylases: mechanistic tools and therapeuticaspects. In: Stolzt VD ed. New Topics in Vitamin D Research, Nova Science Publishers, NY:pp. 67–144.
Schwarzel, W. C., Kruggel, W. G., Brodie, H. J. (1973). Studies on the mechanism of estrogen bio-synthesis. 8. The development of inhibitors of the enzyme system in human placenta. Endo-crinology 92:866–880.
Sheehan, D. J., Hitchcock, C. A., Sibley, C. M. (1999). Current and Emerging Azole AntifungalAgents. Clin. Microbiol. Rev. 12:40–79.
Simpson, E. R., Misso, M., Hewit, K. N., Hill, R. A., Boon, W. C., Jones, M. E., Kovacic, A., Zhou, J.,Clyne, C. D. (2005). Estrogen – the Good, the Bad, and the Unexpected. Endocrine Reviews26:322–330.
Simpson, E. R., Dowsett, M. (2002). Aromatase and its inhibitors: significance for breast cancertherapy. Recent Prog. Horm. Res. 57:317–338.
Small, E. J., Baron, A., Bok, R. (1997). Simultaneous antiandrogen withdrawal and treatment withketoconazole and hydrocortisone in patients with “advanced” prostate carcinoma. Cancer80:1755–1759.
Soberman, J. E., Weber, K. T. (2000). Spironolactone in congestive heart failure. Curr. HypertensRep. 2:451–456.
Sonino, N., Boscaro, M. (1999). Medical therapy for Cushing’s disease. Endocrinal Metab. Clin.North Am. 28:211–222.
Stearns, M. E., Wang, M., Fudge, K. (1993). Liarozole and 13-cis-retinoic acid anti-prostatic tumoractivity. Cancer Res. 53:3073–3077. Erratum in: Cancer Res. 53:5831 (1993).
Stowasser, M., Gordon, R. D., Tunny, T. J., Klemm, S. A., Finn, W. L., Krek, A. L. (1992). Familialhyperaldosteronism type II: five families with a new variety of primary aldosteronism. Clin.Exp. Pharmacol. Physiol. 19:319–322.
Trachtenberg, J. (1984). Ketoconazole Therapy in Advanced Prostatic Cancer. J. Urol. 132:61–63.Ulmschneider, S., Muller-Vieira, U., Mitrenga, M., Hartmann, R. W., Oberwinkler-Marchais, S.,
Klein, C. D., Bureik, M., Bernhardt, R., Antes, I., Lengauer, T. (2005a). Synthesis and evalu-ation of imidazolylmethylenetetrahydronaphthalenes and imidazolylmethyleneindanes:potent inhibitors of aldosterone synthase. J. Med. Chem. 48:1796–1805.
Ulmschneider, S., Muller-Vieira, U., Klein, C. D., Antes, I., Lengauer, T., Hartmann, R. W. (2005b).Synthesis and evaluation of (pyridylmethylene)tetrahydronaphthalenes/-indanes and structur-ally modified derivatives: potent and selective inhibitors of aldosterone synthase. J. Med.Chem. 48:1563–1575.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

INHIBITION OF CYTOCHROMES P450 499
Van den Bossche, H., Willemsens, G., Bellens, D., Janssen, P. A. (1992a). Ridogrel: a selectiveinhibitor of the cytochrome P450-dependent thromboxane synthesis. Biochem. Pharmacol.43:739–744.
Van den Bossche, H. (1992b). Inhibitors of P450-dependend steroid biosynthesis: from research tomedical treatment. J. Steroid. Biochem. Mol. Biol. 43:1003–1021.
Van Wauwe J., Van Nyen, G., Coene, M. C., Stoppie, P., Cools, W., Goossens, J. et al. (1992).Liarozole, an inhibitor of retinoic acid metabolism, exerts retinoid-mimetic effects in vivo.J. Pharmacol. Exper. Ther. 261:773–779.
Williams, G., Kerle, D. J., Doble, A., Dunlop, H., Smith, C., Allen, J., Yeo, T., Bloom, S. R. (1986).Objective Responses to Ketoconazole Therapy in Patients with Relapsed Progressive ProstateCancer. Br J. Urol. 58:45–51.
White, J. A., Beckett-Jones, B., Guo, Y-D, Dilworth F. J., Bonasoro, J., Jones, G., Petkovich, M.(1997). cDNA Cloning of Human Retinoic Acid-metabolizing Enzyme (hP450RAI) Identi-fies a Novel Family of Cytochromes P450 (CYP26). J. Biol. Chem 272:18538–18541.
Young, M., Funder, J. W. (2000). Aldosterone and heart. Trends Endocr. Metab. 11:224–226.Zhou, S., Chan, E., Lim, L. Y., Boelsterli, U. A., Li, S. C., Wang, J., Zhang, Q., Xu, A. (2004).
Therapeutic drugs that behave as mechanism-based inhibitors of cytochrome P450 3A4.Curr. Drug. Metab. 5:415–442
Zittermann, A. (2003). Vitamin D in preventive medicine: are we ignoring the evidence? Brit.J. Nutr. 89:552–572.
Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

Dru
g M
etab
olis
m R
evie
ws
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Uni
vers
ity o
f O
tago
on
09/0
4/14
For
pers
onal
use
onl
y.

















