
Installation des Netvault6-GUI -...
132
Stable isotope and trace element composition of foraminiferal calcite - from incorporation to dissolution Dissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften - Dr. rer. nat. - im Fachbereich 2 (Biologie/Chemie) der Universität Bremen vorgelegt von Bärbel Hönisch Bremen, 2002
Transcript of Installation des Netvault6-GUI -...

Stable isotope and trace element composition of foraminiferal calcite -
from incorporation to dissolution
Dissertation
zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften
- Dr. rer. nat. -
im Fachbereich 2 (Biologie/Chemie)
der Universität Bremen
vorgelegt von
Bärbel Hönisch
Bremen, 2002

Tag des öffentlichen Kolloquiums:
14. November 2002
Gutachter der Dissertation:
Prof. Dieter Wolf-Gladrow
PD Dr. Ralph Schneider

Abstract
Long-term reconstruction of climate and environmental parameters from marine
sediments relies heavily on the reliability of proxy indicators from planktonic and benthic
foraminifera. Therefore, the aim of this dissertation is to improve our understanding and
confidence in planktonic foraminiferal proxies as indicators of seawater carbonate chemistry
and their stability in response to dissolution. Towards this goal laboratory experiments with
living specimens and empty shells collected from sediments were carried out.
In culture experiments with the living planktonic foraminifer Orbulina universa the
control of symbiont photosynthetic activity on the boron isotopic composition of shell calcite
was investigated (Publication I). Under low light (reduced photosynthetic rates) the boron
isotopic composition of the tests is 1.5‰ lower compared to shells grown under high light
(elevated photosynthetic rates). As boron isotope values trace seawater-pH, the lower δ11B
translates to a reduction in recorded pH of approximately 0.2 units. Data obtained for field-
grown, symbiont-barren Globigerina bulloides record significantly lighter δ11B than the
symbiont-bearing O. universa and therefore support the hypothesis that respiration and
photosynthesis are the key physiological parameters responsible for species-specific vital
effects. Although this experiment may indicate that symbiont-barren foraminifera reflect
ambient seawater chemistry more accurately than symbiont-bearing foraminifera, model
results suggest that photosynthesis- and respiration-driven offsets are constant over a wide
pH-range and do not compromise the reliability of δ11B in symbiont-bearing foraminifera as a
paleo-pH indicator.
The Ba/Ca ratio in foraminiferal shells has been shown to reflect seawater Ba
concentrations, which in turn are correlated to alkalinity. To investigate whether alkalinity
may directly influence the Ba incorporation into foraminiferal calcite and thereby compromise
the reliability of this alkalinity proxy, symbiont-bearing O. universa and symbiont-barren G.
bulloides were grown in seawater of constant Ba concentration at five different alkalinities
(Publication II). A weak negative correlation between the Ba uptake ratio in O. universa
shells and alkalinity was found under high light conditions. For an increase in alkalinity of
100 µmol kg-1 the distribution coefficient DBa (= Ba/Ca shell / Ba/Caseawater) decreased by
0.004. This change is well within the error of DBa determined to date and the weak influence
of alkalinity on Ba incorporation into foraminiferal shells is insignificant for
paleoreconstructions. Globigerina bulloides has not been calibrated for Ba before and the
experiments revealed that DBa in this species is the same as DBa in O. universa. In line with

the similar Ba/Ca uptake ratio of symbiont-bearing and symbiont-barren species, varying light
levels do not affect the Ba incorporation of O. universa.
To investigate the effect of undersaturated seawater on foraminiferal shell chemistry,
well preserved shells of the planktonic foraminifera species Globigerinoides sacculifer and
Neogloboquadrina pachyderma (sinistral coiling) were partially dissolved under controlled
conditions in the laboratory (Publication III). In addition to known dissolution effects on
Mg/Ca, δ18O and δ13C, significant effects on Sr/Ca and δ11B could be determined which are in
the same order of magnitude as observed for glacial/interglacial changes. Using previous
hypotheses to explain and discuss dissolution patterns, it becomes evident that the overall
process is not yet fully understood. While δ18O, δ13C, Mg/Ca and maybe Sr/Ca can be
explained by preferential dissolution of ontogenetic calcite and a shift of the bulk shell
chemistry to calcite secreted at greater depth (gametogenic and/or crust calcite), δ11B and
δ44Ca seem to be inconsistent with such an explanation and the dissolution patterns of these
elements need to be dominated by other processes. Reduced calcite stability due to higher
Mg/Ca was found to be insignificant to control the overall dissolution behavior in
foraminiferal shells and increasing Sr/Ca ratios demonstrate that crystal impurities are not
necessarily more prone to dissolution. The microstructural breakdown of shell surfaces, i.e.
the formation of fissures and crevices, indicates an increase in porosity upon shell corrosion
rather than the removal of outer calcite layers. The resulting increase in surface area leads to
the exposition of otherwise protected lattice areas and possibly allows certain elements to be
leached out. In summary, dissolution effects appear to be species-specific and depend on the
physico-chemical gradients encountered by vertically migrating foraminifera at different
locations.
The dissolution-driven weight loss of planktonic foraminifera shells in a defined narrow
size range has been proposed to reflect bottom water carbonate ion concentration ([CO32-]). A
number of recent studies used this relationship but disregarded a number of complications that
may limit the reconstructions. Publication IV combines experimental results on cultured
foraminifera and theoretical arguments to evaluate these complications: (1) The value chosen
for the pressure impact on the [CO32-] at calcite saturation is overestimated and should be 16
µmol kg-1 km-1 instead of 20 µmol kg-1 km-1. (2) The offset in [CO32-] between bottom and
pore water depends on the amount of organic matter that is being degraded within the
sediment and consequently the assumed constancy of the offset over time and space is highly
unlikely. (3) The initial weight of undissolved shells also changes between sites and over
geological timescales. Growth conditions such as [CO32-], light and temperature affect

respiration, symbiont photosynthesis and calcification processes and cause a significant
variability in initial shell weight. (4) As the dissolution susceptibility of foraminifera shells
varies between species, a single weight loss slope cannot be used for different species.
Correcting the published estimate of glacial bottom water [CO32-] for the various effects and
uncertainties discussed in Publication IV considerably reduces the published estimate of the
Atlantic glacial upper deep water [CO32-]-increase from +14 mol kg-1 to only +4 µmol kg-1.

Zusammenfassung
Die Rekonstruktion vergangener Klima- und Umweltbedingungen anhand von
marinen Sedimenten wird maßgeblich durch die Verläßlichkeit der Proxyindikatoren
bestimmt, die man aus Schalen planktischer und benthischer Foraminiferen gewinnen kann.
Das Ziel dieser Arbeit ist deshalb die Verbesserung des Verständnisses und Vertrauens in
Proxies aus planktischen Foraminiferen als Indikatoren mariner Karbonatchemie und der
Stabilität von Proxies in angelösten Schalen. Um mögliche Einflüsse zu untersuchen, wurden
Laborexperimente mit lebenden Foraminiferen und leeren Schalen aus Sedimenten
durchgeführt.
In Kulturexperimenten mit der lebenden planktischen Foraminifere Orbulina universa
wurde der Einfluß der Symbiontenaktivität auf die Borisotopenzusammensetzung der
Kalkschale untersucht (Publikation I). Im Vergleich zu Individuen, die unter hohem
Lichtangebot gewachsen sind (hohe Photosyntheseraten), ist die
Borisotopenzusammensetzung von Schalen, die unter geringem Lichtangebot (geringe
Photosyntheseraten) gebildet wurden, etwa 1.5‰ leichter. Da die Borisotopie ein Anzeiger
für den marinen pH-Wert ist, entspricht der geringere δ11B-Wert einer Verminderung des pH-
Werts um etwa 0,2 Einheiten. Die Borisotopenzusammensetzung der Symbionten-freien
Foraminifere Globigerina bulloides ist deutlich leichter als die der Symbionten-tragenden O.
universa und unterstützt damit die Hypothese, daß Respiration und Photosynthese die
physiologischen Schlüsselparameter für artspezifische Vitaleffekte darstellen. Obwohl dieses
Experiment nahelegen könnte, daß Symbionten-freie Foraminiferen die Meerwasserchemie
genauer aufzeichnen als Symbionten-tragende Arten, deuten Modellergebnisse daraufhin, daß
die durch Photosynthese und Respiration bedingten Unterschiede über weite pH-Bereiche
konstant sind und die Zuverlässigkeit von δ11B aus Schalen Symbionten-tragender
Foraminiferen als paläo-pH Indikator nicht beeinträchtigen.
Das Ba/Ca Verhältnis in Foraminiferenschalen spiegelt die Ba-Konzentration des
Meerwassers wider. Da die Ba-Konzentration des Meerwassers mit der Alkalität korreliert,
findet das Ba/Ca Verhältnis in Foraminiferenschalen Anwendung als Proxy für die Alkalität.
Um zu untersuchen, ob die Alkalität auch einen direkten Einfluß auf den Einbau von Ba2+ in
Foraminiferenkalk haben und damit die Zuverlässigkeit dieses Proxies beeinträchtigen
könnte, wurden die Symbionten-tragende Foraminifere O. universa und die Symbionten-freie
G. bulloides bei konstanter Ba Konzentration unter fünf unterschiedlichen Alkalitäten
gehältert. (Publikation II). Unter hohem Lichtangebot konnte dabei eine schwach negative

Korrelation zwischen Ba-Aufnahme in Schalen von O. universa und der Alkalität festgestellt
werden. Eine Zunahme der Alkalität in Höhe von 100 µmol kg-1 führt zu einer Abnahme von
0,004 im Verteilungskoeffizienten DBa (=Ba/CaForaminifere / Ba/CaMeerwasser). Dieser Unterschied
liegt innerhalb der natürlichen Varianz mit der planktische Foraminiferen Ba2+ einbauen und
ist damit unbedeutend für Paläorekonstruktionen. Über mögliche Vitaleffekte beim Einbau
von Ba/Ca in Schalen von Globigerina bulloides gab es vor dieser Studie keine Daten und die
Experimente konnten nun zeigen, daß diese Art Ba2+ im gleichen Verhältnis zur
Meerwasserkonzentration einbaut wie O. universa. In Übereinstimmung mit dem gleichen
Ba/Ca Aufnahmeverhältnis von Symbionten-tragenden und Symbionten-freien Arten zeigt
variierendes Lichtangebot keinen Einfluß auf den Ba2+-Einbau in O. universa.
Um den Einfluß von untersättigtem Meerwasser auf die Schalenchemie von
Foraminiferen zu untersuchen, wurden sehr gut erhaltene Schalen der planktischen
Foraminiferen Globigerinoides sacculifer und Neogloboquadrina pachyderma (links-
drehender Morphotyp) unter kontrollierten Bedingungen im Labor angelöst (Publikation III).
Zusätzlich zu den bereits bekannten Lösungseffekten auf Mg/Ca, δ18O und δ13C konnten
signifikante Effekte auf Sr/Ca und δ11B beobachtet werden, die in der Größenordnung von
Änderungen zwischen Warm- und Kaltzeiten liegen. Die Diskussion der gefundenen
Lösungsmuster anhand von früheren Erklärungshypothesen zeigt deutlich, daß man den
zugrundeliegenden Prozeß noch nicht vollständig versteht. δ18O, δ13C, Mg/Ca und eventuell
auch Sr/Ca können durch die bevorzugte Lösung von ontogenetischem Kalzit hinreichend gut
erklärt werden. Dabei wird das Gesamtsignal zur Chemie der äußeren Schale verschoben, die
in größeren Wassertiefen gebildet wird (gametogenetischer Kalzit und/oder Kruste). δ11B und
δ44Ca hingegen können nicht durch denselben Prozeß erklärt werden und die Lösungsmuster
dieser Elemente müssen deshalb durch andere Faktoren dominiert werden. Verstärkte
Lösungsanfälligkeit aufgrund von höheren Mg/Ca-Verhältnissen reicht nicht aus, um das
gesamte Lösungsverhalten von Foraminiferenschalen zu kontrollieren und zunehmende Sr/Ca
Verhältnisse zeigen, daß Verunreinigungen des Kristallgitters nicht notwendigerweise stärker
lösungsanfällig sind. Die Veränderung der Mikrostruktur einer Schalenoberfläche, d.h. die
Entstehung von Fissuren und kleinen Spalten, weist daraufhin, daß Lösung die Porosität
erhöht und die Schale nicht einfach schichtweise von außen nach innen abträgt. Das Resultat
ist eine Zunahme der Schalenoberfläche und damit die Exposition von Kristallgitterbereichen,
die in ungelösten Schalen vom aggressiven Meerwasser abgeschirmt sind. Damit besteht die
Möglichkeit, daß bestimmte Elemente bevorzugt herausgelöst werden können.
Zusammenfassend kann man sagen, daß Lösungseffekte artspezifisch sind und von den

physikochemischen Gradienten abhängen, die vertikal migrierende Foraminiferen an
unterschiedlichen Orten erfahren haben.
Der lösungsbedingte Gewichtsverlust planktischer Foraminiferenschalen einer definierten
Größenklasse korreliert mit der Karbonationenkonzentration ([CO32-]) des Wassers am
Meeresboden. Einige Studien haben diese Beziehung kürzlich angewendet, dabei aber eine
Reihe von Komplikationen nicht berücksichtigt, die die Aussagekraft der Rekonstruktionen
begrenzen. Publikation IV nutzt experimentelle Ergebnisse und theoretische Argumente, um
diese Komplikationen zu bestimmen und zu quantifizieren: (1) Der Wert für die
Druckabhängigkeit der [CO32-] bei Kalzitsättigung wurde zu groß gewählt und sollte 16 µmol
kg-1km-1 anstatt 20 µmol kg-1km-1 betragen. (2) Der Abbau organischen Materials im
Sediment verändert die Karbonationenkonzentration und bestimmt daher den [CO32-]-
Unterschied zwischen Boden- und Porenwasser. Räumlich und zeitlich gesehen ist es daher
höchst unwahrscheinlich, daß dieser Unterschied konstant ist. (3) Das Gewicht ungelöster
Schalen variiert zwischen verschiedenen Orten und über geologische Zeitskalen.
Wachstumsbedingungen wie [CO32-], Lichtangebot und Temperatur beeinflussen
physiologische Prozesse wie Respiration, Photosynthese der Symbionten und Kalzifizierung
und bedingen maßgebliche Unterschiede im Schalengewicht. (4) Da die Lösungsanfälligkeit
von Schalen unterschiedlicher Foraminiferenarten variiert, kann eine einzelne Beziehung
nicht universell für alle Arten angewendet werden. Korrigiert man Abschätzungen der [CO32-
]-Zunahme des glazialen Bodenwassers für die in Publikation IV diskutierten verschiedenen
Effekte, so reduziert sich die Zunahme für das obere Tiefenwasser des glazialen Atlantiks von
+14 mol kg-1 auf nur +4 µmol kg-1.

Danksagung
Das Gelingen dieser Arbeit verdanke ich der wissenschaftlichen Betreuung durch und Freundschaft mit Jelle Bijma, seinen Ideen, seinem Optimismus, Enthusiasmus und Überlebenswillen in Bor-schweren Zeiten.
Dieter Wolf-Gladrow danke ich für die Begutachtung und Betreuung der Arbeit, sowie für die Schaffung einer unvergleichlichen Arbeitsatmosphäre. Ebenso möchte ich mich bei Ralph Schneider für die Begutachtung bedanken.
I am especially grateful to Howie Spero, Ann Russell, David Lea, Dirk Nürnberg, Geert-Jan Brummer, Neven Loncaric, Abhijit Sanyal, Gary Hemming, Douglas Adams, Uli Groß, Michel Stoll and Frank Peeters. They have all contributed their time and considerable expertise to my work.
Nikolaus Gussone, Toni Eisenhauer, Silke Vetter, Folkmar Hauff und Anette Deyhle haben endlose Geduld mit immer wiederkehrenden Problemen am TIMS bewiesen und massgeblich zu den erfolgreichen Messungen beigetragen.
Similarly, this work would not have been possible without the advice and/or laboratory help and work of Pam Martin, Georges Paradis, Dotti Pak, Dave Winter and Sylvia Duncan.
I would like to thank Laurie Juranek, Megan Thomas and Heidi Iverson for their field help and a great summer on Catalina Island.
André Wischmeyer möchte ich für gute Nachbarschaft, Versorgung mit Kuchen, Schokolade und Musik danken. Ohne seine aufopfernde Unterstützung bei mathematischen und Computerfragen würde ich jetzt noch an den Problemen verzweifeln und hätte das Rennen nie gewonnen.
Richard Zeebe, Gert-Jan Reichart, Heiko Jansen und Christoph Völker danke ich für hilfreiche Diskussionen über Isotope und Spurenelemente, Lösungskinetik und Statistik.
Anja Terbrüggen gebührt besonderer Dank für die Organisation des C-Labors und seelische Aufbauarbeit bei der schlimmsten aller Laborkrankheiten: Coulometerfrust. Ebenso danke ich Frau Schwarz und Friedel Hinz für gute Zusammenarbeit.
Jan Helmke und Jürgen Pätzold danke ich für die Bereitstellung von Foraminiferen und Sedimentmaterial.
Das Überleben der Doktorarbeit bedeutet nicht nur wissenschaftliche Zusammenarbeit und Diskussion, sondern auch freundschaftliches Miteinander in der Arbeitsgruppe. Neben vorher genannten Gruppenmitgliedern möchte ich mich hier insbesondere bei Björn Rost, Albert Benthien, Ingrid Zondervan, Uta Schneider, Gerald Langer, Peter Köhler, Kai Schulz, Frank Gervais, Irini Mataliotaki, Ulf Riebesell, Uta Passow, Claudia Sprengel, Markus Geisen, Anja Engel, Markus Schartau, Ignacio Tebas, Silke Thoms und Christel Heemann bedanken.
Zuletzt, aber in mancherlei Hinsicht mehr als allen anderen, danke ich Hubertus Fischer, Uli Holzwarth und all denjenigen, die mich mit ihrer Freundschaft und ihrem Interesse in den vergangenen Jahren unterstützt haben.

Structure
This thesis is subdivided into 5 parts. Part 1 refers to the main context of this study.
Part 2 presents 4 manuscripts dealing with the main topic submitted or in preparation to be
submitted to reviewed scientific journals. To reduce repetitions, the references were excluded
from the manuscripts and combined in a separate chapter. Part 3 contains the conclusions of
this thesis and provides implications for future research. The appendix (part 4) presents the
report of working group 3 of the ESF Explanatory Workshop on "The ocean carbon cycle and
climate change", Delmenhorst, September 1-4, 2001, which deals with currently available
carbonate proxies and their major limitations.

Table of contents
1. INTRODUCTION AND MOTIVATION 1
1.1 The oceanic carbon cycle 1 1.1.1 The marine carbonate system 1 1.1.2 Carbonate chemistry in the light of biological activity 3 1.1.3 Glacial to interglacial changes in CO2 and future scenarios 5 1.2 The use of proxies in paleoceanography 6 1.3 Proxies and their limitations 11
2. PUBLICATIONS 15
2.1 Focus and outline of this study 15 2.2 Publication I: The influence of symbiont photosynthesis on the boron isotopic composition of foraminiferal shells 18 2.3 Publication II: Assessing the reliability of Ba/Ca as a tracer for alkalinity 31 2.4 Publication III: Post-depositional effects on trace metals and stable isotopes in foraminiferal calcite – Evidence from dissolution experiments 42 2.5 Publication IV: Comment to Broecker and Clark "Carbonate ion concentration in glacial-age deep waters of the Caribbean Sea" 71
3. SUMMARY AND OUTLOOK 80
3.1 Effects of symbiont photosynthesis and respiration on the stable boron isotopic composition of foraminiferal shells 80 3.2 The effect of alkalinity on planktonic foraminiferal Ba/Ca 81 3.3 Changes in planktonic foraminiferal shell chemistry after incubation in undersaturated seawater 82 3.4 Foraminifera collected from sediment cores - identifying their preservation state 83 3.5 Perspectives for future research 84
4. APPENDIX 87
Reconstructing and modeling past ocean carbonate chemistry – Working Group 3 report of the ESF Explanatory Workshop on "The ocean carbon cycle and climate change", Delmenhorst, September 1-4, 2001
5. REFERENCES 106

List of figures
page 1 Typical vertical seawater profiles of carbonate parameters....................................................4
2 Changes in surface ocean carbonate chemistry in response to increasing atmospheric
CO2.........................................................................................................................................6
3 Four species of planktonic foraminifera................................................................................8
4 The oxygen isotopic composition of marine calcites as a function of temperature and
seawater
δ18O..........................................................................................................................9
5 Boron speciation and isotope partitioning between B(OH)4- and B(OH)3 as a function of
seawater pH..........................................................................................................................10
6 Reconstructing past ocean alkalinity from foraminiferal Ba/Ca..........................................10
7 Comparison of the boron isotopic composition in shells of O. universa cultured under
HL and LL............................................................................................................................24
8 Comparison of the boron isotopic composition of the symbiont-bearing foraminifera O.
universa and the symbiont-barren G. bulloides taken from plankton tows and inorganic
carbonates.............................................................................................................................26
9 DBa in the subtropical, spinose, symbiont-bearing foraminifera Orbulina universa vs.
alkalinity...............................................................................................................................37
10 DBa in the subpolar, spinose planktonic foraminifera Globigerina bulloides compared
to alkalinity......................................... ................................................................................37
11 Comparison of Sr/Ca and Ba/Ca versus seawater pH in O. universa................................40
12 Microstructural breakdown of G. sacculifer shell surfaces monitored by scanning
electron microscopy............................................................................................................50
13 The effect of partial dissolution on Mg/Ca in G. sacculifer and N. pachyderma (sin.).......51
14 Averages of Mg/Ca in inner and outer calcite of 10 G. sacculifer shells as determined
by microprobe analysis of wall profiles...............................................................................53
15 The effect of partial dissolution on Sr/Ca in G. sacculifer and N. pachyderma (sin.).........54
16 The effect of partial dissolution on δ18O in G. sacculifer and N. pachyderma
(sin.)...........55
17 The effect of partial dissolution on δ13C in G. sacculifer and N. pachyderma (sin.)...........55
18 The effect of partial dissolution on δ44Ca in G. sacculifer and N. pachyderma (sin.).........56

19 The effect of partial dissolution on δ11B in G.
sacculifer.....................................................56
20 Schematic presentation of the life cycle of G. sacculifer: vertical migration and
varying calcification depths.................................................................................................59
21 Comparison of different dissolution rates of ontogenetic and gametogenic calcite and
the respective effect on changes of a heterogeneously distributed element in
foraminiferal calcite.............................................................................................................63
22 The effect of [CO3=] on planktonic foraminiferal shell weight............................................73
23 Foraminiferal shell weights versus pressure corrected [CO3=]...........................................77
A1 Present state of the δ11B proxy calibration..........................................................................91
A2 Increased foraminiferal shell weight under higher [CO32-] during shell growth
.................96
A3 Calcium carbonate content and calcite saturation in a modern sediment profile ...............99
List of tables
page 1 Dissolution effects on foraminiferal shell chemistry as observed in sediment
studies and laboratory dissolution experiments....................................................................13
2 Boron isotopic composition of cultured O. universa and modified seawater.......................22
3 Boron isotopic composition of plankton tow O. universa and G. bulloides........................ 22
4 Experimental Ba/Ca data for cultured shells.........................................................................36
5 Average weights of undissolved and dissolved foraminifera shells, dissolution estimates
and calcite saturation of experimental seawater....................................................................48
6 Dissolution experimental data: Minor and trace elements....................................................52
7 Numerical experiment on the dissolution susceptibility of foraminiferal Mg-calcite..........62
A1 Reconstructing past ocean carbonate chemistry: proxies, limitations and estimates..........90

A2 In situ investigation of sedimentary carbonate dissolution................................................95

General introduction 1
1. Introduction and motivation
Knowledge of the origin and amplitude of natural fluctuations in past climate systems
can be used to assess the stability of modern terrestrial and marine subsystems and their
potential range of variations in the future. Changes in the cycling of organic and inorganic
carbon in the ocean have been proposed (see for an overview Falkowski et al., 2000; Raven
and Falkowski, 1999; Sigman and Boyle, 2000) as mechanisms leading to the glacial-
interglacial changes in atmospheric carbon dioxide measured in ice-cores (Fischer et al.,
1999; Petit et al., 1999). In spite of the ocean's acknowledged importance in controlling
atmospheric carbon dioxide concentrations on glacial-interglacial timescales, the roles of
chemical and physical processes governing carbon transfers between the ocean and
atmosphere are still poorly understood. The chemical reactions determining the exchange of
CO2 between atmosphere and ocean are very complex and before we can go into the theory of
paleoceanographic reconstructions, the exchange reactions between ocean, atmosphere and
marine biosphere shall be introduced briefly.
1.1 The oceanic carbon cycle
1.1.1 The marine carbonate system The marine carbonate system encompasses the different dissolved inorganic carbon
species (CO2, H2CO3, HCO3- and CO3
2-), H+- and OH-- ions. These species are interrelated by
chemical reactions which determine their relative abundances in seawater. Following Henry's
law, gaseous CO2 dissolves into surface water directly proportional to the atmospheric partial
CO2 pressure (pCO2):
[CO2] aquatic = K0 (T, S) * pCO2, (1)
where K0 is the solubility coefficient of CO2 in seawater at a given temperature (T)
and salinity (S). The dissolved CO2 hydrates immediately with water to carbonic acid
(H2CO3), which itself dissociates to bicarbonate (HCO3-), carbonate (CO3
2-) and H+-ions:
CO2 (aq.) + H2O ⇔ H2CO3 ⇔ HCO3
- + H+ ⇔ CO32- + 2 H+ (2)
CO2 is therefore not only dissolved physically but dissociates to ionic species which
do not contribute to the aquatic partial pressure of CO2 (PCO2). This is the reason why

General introduction 2
significantly more CO2 dissolves in seawater than any other inert gas such as nitrogen or
oxygen. Because the concentration of H2CO3 is very small, it is usually combined with CO2
(aq.) to [CO2]. For the description of the carbonate system in seawater, stoichiometric
equilibrium constants, K1 and K2, are used which are related to the ion concentrations and
depend on temperature, pressure (P) and salinity:
K1(T,S, P) =H +[ ]HCO3
−[ ]CO2[ ] (3)
K2 (T,S, P) =H +[ ]CO3
2−[ ]HCO3
−[ ] (4)
Decreasing T and S and increasing P result in a shift of the relative ion concentrations
to the left-hand side of equation (2), i.e. especially [CO32-] will decrease and [CO2] (aq.)
increase in colder, deeper and less saline waters. The sum of the dissolved inorganic carbon
species is abbreviated as ΣCO2 or DIC and defined as follows:
DIC = [CO2] + [HCO3-] + [CO3
2-] (5)
In seawater, about 90% of the DIC is present as bicarbonate, approximately 9% as
carbonate and about 1% as dissolved CO2.
Another essential quantity for the description of the carbonate system is alkalinity,
which is closely related to the electrical charge balance in the ocean. The concept of alkalinity
is anything but trivial and has been regarded and defined in many different ways (Dickson,
1981). In general, alkalinity depends on a small charge excess of conservative cations ([Na+]
+ 2[Mg2+] + 2[Ca2+] + [K+]) over anions ([Cl-] + 2[SO42-]) which is mainly compensated for
by the anions of carbonic and boric acid ([HCO3-] + 2[CO3
2-] + [B(OH)4-]). As a very good
practical approximation, total alkalinity (TA) can also be described as the sum of the charges
of the major weak acids in seawater plus the charge of OH- and minus the charge of H+.
TA ≈ [HCO3-] + 2 [CO3
2-] + [B(OH)4-] + [OH-] - [H+] ± minor constituents (6)
Analytically, total alkalinity is regarded in terms of buffer capacity, i.e. the ability to
neutralize strong acids. This property is used to quantitatively determine alkalinity by titration
with HCl.

General introduction 3
Next to alkalinity, only DIC, pH and PCO2 can be determined analytically (for details
see DOE, 1994). As none of the carbonate system parameters varies independent from the
others, the interrelated dependency enables the oceanographer to calculate the entire
carbonate system (i.e. alkalinity, DIC, pH, PCO2, [HCO3-] and [CO3
2-]) with the knowledge
of no more than two of the constituents.
This was a very brief summary of the carbonate equilibria in the ocean. A detailed
description can be found in Zeebe and Wolf-Gladrow (2001). In the next section, we will see
how biological activity interacts with the thermodynamic equilibria just described.
1.1.2 Carbonate chemistry in the light of biological activity
One of the critical processes controlling the ocean-atmosphere CO2 exchange is
primary production in the surface ocean, and regeneration and cycling of biogenic materials in
the sea (e.g. Longhurst, 1991). Oceanic primary production takes place in the euphotic zone,
i.e. the upper layer of the ocean where sufficient light is available for photosynthesis. The
export of biogenic material from the surface to the deep ocean is called the biological carbon
pump, as it transfers inorganic carbon assimilated in the surface waters against the gradient to
the deep sea. Two biological carbon pumps can be distinguished, the organic carbon and the
inorganic calcium carbonate pump. The two pumps have opposite effects on the CO2
partitioning between ocean and atmosphere. While photosynthetically active organisms
sequester CO2 for the purpose of biomass production, the secretion of calcitic and aragonitic
skeletons by foraminifera, corals, pteropods and coccolithophores primarily increases surface
PCO2 (e.g. Frankignoulle and Canon, 1994; Wollast, 1994):
photosynthetic carbon fixation: 6 CO2 + 12 H2O → C6H12O6 + 6 O2 + 6 H2O (7)
carbonate precipitation: Ca2+ + 2 HCO3- → CaCO3 + CO2 + H2O (8)
Respiration processes in the deep ocean invert reaction (7) and release CO2 which
lowers the pH in the deep ocean and leads, in addition to the effects of higher pressure and
lower temperature, to the dissolution of calcium carbonates (reverse of reaction 8). An
example of the effects of photosynthesis, calcification, respiration and CaCO3 dissolution on
the distribution of the main dissolved constituents in seawater is displayed in Figure 1.
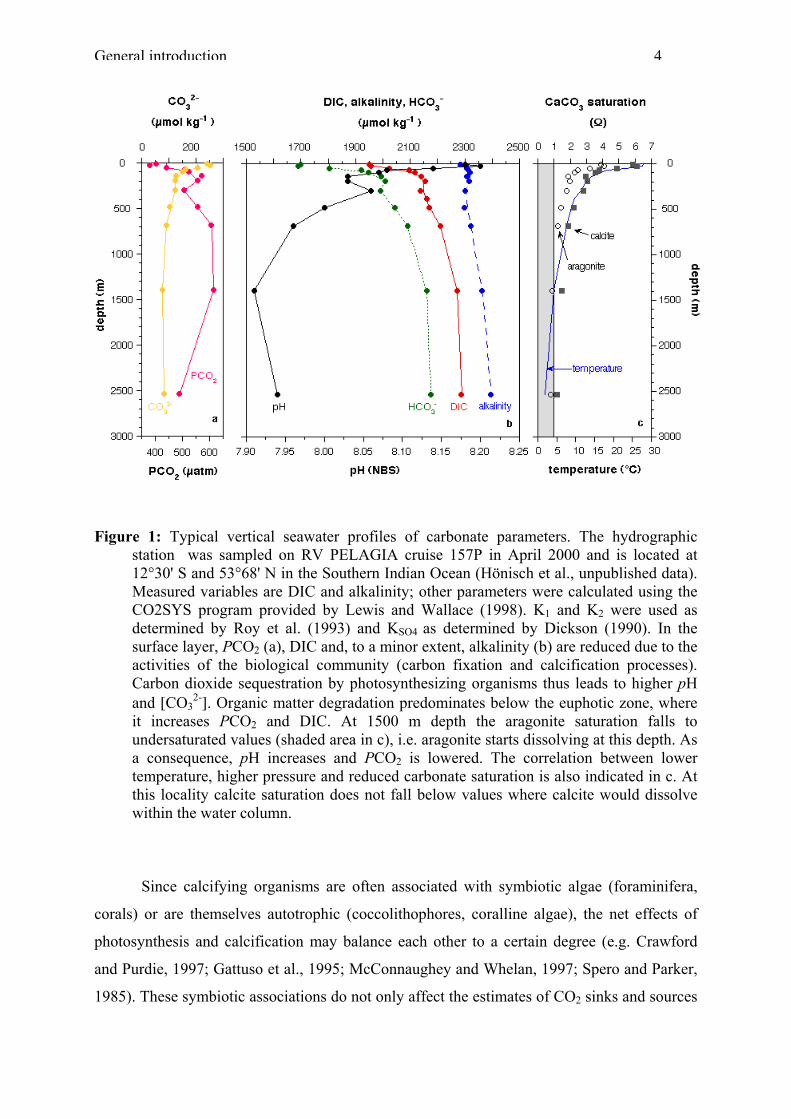
General introduction 4
Figure 1: Typical vertical seawater profiles of carbonate parameters. The hydrographic station was sampled on RV PELAGIA cruise 157P in April 2000 and is located at 12°30' S and 53°68' N in the Southern Indian Ocean (Hönisch et al., unpublished data). Measured variables are DIC and alkalinity; other parameters were calculated using the CO2SYS program provided by Lewis and Wallace (1998). K1 and K2 were used as determined by Roy et al. (1993) and KSO4 as determined by Dickson (1990). In the surface layer, PCO2 (a), DIC and, to a minor extent, alkalinity (b) are reduced due to the activities of the biological community (carbon fixation and calcification processes). Carbon dioxide sequestration by photosynthesizing organisms thus leads to higher pH and [CO3
2-]. Organic matter degradation predominates below the euphotic zone, where it increases PCO2 and DIC. At 1500 m depth the aragonite saturation falls to undersaturated values (shaded area in c), i.e. aragonite starts dissolving at this depth. As a consequence, pH increases and PCO2 is lowered. The correlation between lower temperature, higher pressure and reduced carbonate saturation is also indicated in c. At this locality calcite saturation does not fall below values where calcite would dissolve within the water column.
Since calcifying organisms are often associated with symbiotic algae (foraminifera,
corals) or are themselves autotrophic (coccolithophores, coralline algae), the net effects of
photosynthesis and calcification may balance each other to a certain degree (e.g. Crawford
and Purdie, 1997; Gattuso et al., 1995; McConnaughey and Whelan, 1997; Spero and Parker,
1985). These symbiotic associations do not only affect the estimates of CO2 sinks and sources

General introduction 5
for the ocean-atmosphere-interaction, but, as we will see later on, may also affect the
reliability of chemical recorders of past ocean conditions.
1.1.3 Glacial to interglacial changes in atmospheric CO2 and future scenarios
Observations from glacier icecores have shown that cyclic changes in atmospheric
CO2 levels occurred over the last 420,000 years with glacial periods displaying about 80
ppmv lower values compared to interglacials (~ 280 ppmv) (Fischer et al., 1999; Petit et al.,
1999). Isotope paleothermometry on the Vostok ice core revealed significant covariation
between air temperature and pCO2 of the past glacial cycles (Cuffey and Vimeux, 2001),
suggesting that CO2 may be an important forcing factor for climate. In contrast, Fischer et al.
(1999) observed that the pCO2 increase lags the warming of the last three deglaciations by
600 ± 400 years, rather arguing for an important feedback mechanism than a real climate
forcing function. However, the cyclicity between glacial and interglacial pCO2 cannot be
simply explained by higher oceanic CO2 solubility due to lower temperatures because the
concomitant sealevel decrease and salinity increase (e.g. Fairbanks, 1989) largely compensate
the pCO2 decrease due to cooling. Although many approaches have been made to determine
the major processes that control the state of the glacial ocean (e.g. Archer and Maier-Reimer,
1994; Boyle, 1988b; Broecker, 1997; Broecker and Clark, 2001b; Martin, 1990),
contradictions between theories and observations could not yet be excluded so that the
interactions between glacial-interglacial shifts in atmospheric CO2 and oceanic carbon
sequestration remain elusive (e.g. Anderson and Archer, 2002; Elderfield, 2002; Maher and
Dennis, 2001).
Understanding the origin of natural fluctuations in the past is crucial for predictions of
future variations (e.g. Stott and Kettleborough, 2002). Crowley (2000) estimated that only
about 25% of the 20th-century temperature increase can be attributed to natural variability.
Instead, most of the 20th-century warming is consistent with that predicted from green house
gas increases. Greenhouse gases absorb longwave (infra-red) radiation emitted from the earth
surface and thereby prevent the loss of solar energy to space. Concomitantly the global heat
budget increases. Atmospheric CO2 is one of those greenhouse gases. Since the industrial
revolution in the 19th-century, the atmospheric CO2 concentration has increased by >30%
from the average interglacial value of ~280 ppmv to 368 ppmv in 2000. The predictions for
the future exceed 900 ppmv by the year 2100 (Cox et al., 2000) if we do not manage to reduce
the current magnitude of CO2 emissions. The rise in atmospheric CO2 leads to changes in the
ocean carbonate chemistry (Figure 2) which could have strong impacts on the marine biota
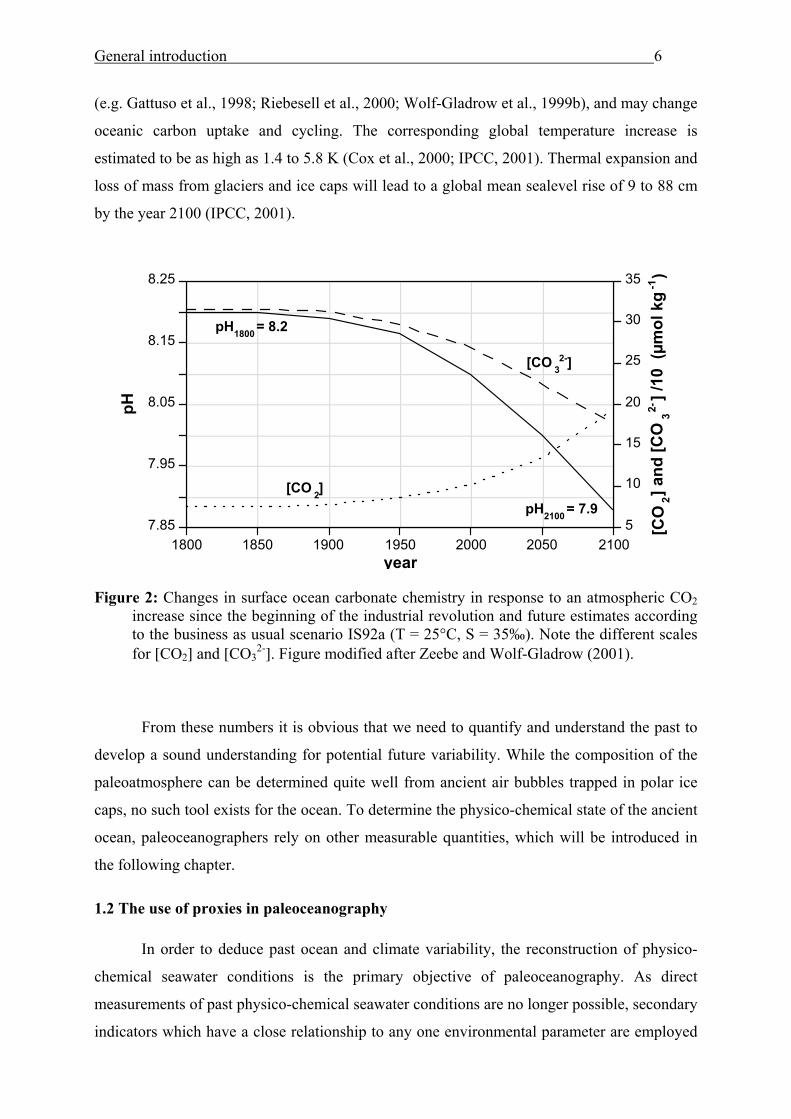
General introduction 6
(e.g. Gattuso et al., 1998; Riebesell et al., 2000; Wolf-Gladrow et al., 1999b), and may change
oceanic carbon uptake and cycling. The corresponding global temperature increase is
estimated to be as high as 1.4 to 5.8 K (Cox et al., 2000; IPCC, 2001). Thermal expansion and
loss of mass from glaciers and ice caps will lead to a global mean sealevel rise of 9 to 88 cm
by the year 2100 (IPCC, 2001).
7.85
7.95
8.05
8.15
8.25
5
10
15
20
25
30
35
1800 1850 1900 1950 2000 2050 2100
pH
[CO
2] and
[CO
32-] /
10 (
µmol
kg
-1)
year
pH1800 = 8.2
[CO 32-]
[CO 2]pH2100 = 7.9
Figure 2: Changes in surface ocean carbonate chemistry in response to an atmospheric CO2
increase since the beginning of the industrial revolution and future estimates according to the business as usual scenario IS92a (T = 25°C, S = 35‰). Note the different scales for [CO2] and [CO3
2-]. Figure modified after Zeebe and Wolf-Gladrow (2001).
From these numbers it is obvious that we need to quantify and understand the past to
develop a sound understanding for potential future variability. While the composition of the
paleoatmosphere can be determined quite well from ancient air bubbles trapped in polar ice
caps, no such tool exists for the ocean. To determine the physico-chemical state of the ancient
ocean, paleoceanographers rely on other measurable quantities, which will be introduced in
the following chapter.
1.2 The use of proxies in paleoceanography
In order to deduce past ocean and climate variability, the reconstruction of physico-
chemical seawater conditions is the primary objective of paleoceanography. As direct
measurements of past physico-chemical seawater conditions are no longer possible, secondary
indicators which have a close relationship to any one environmental parameter are employed

General introduction 7
for this task. These measurable descriptors for desired (but unobservable) variables are called
"proxies" (Wefer et al., 1999).
According to Lea (1999a), proxies can be divided into three classes: biotic
components (i.e. morphologic or taxonomic climate responses such as stomata density, floral
and faunal assemblage compositions etc.), chemical tracers and physical and mineralogical
sediment properties. This study is focussed on chemical tracers which comprise the largest
proxy group. They can be organized in three sub-categories: proxies of physical seawater
properties, such as temperature; proxies of seawater composition, such as nutrient
concentration and carbonate chemistry; and proxies of sediment particle flux, such as
productivity (see also Wefer et al., 1999). Empirical relationships between proxies and their
respective environmental parameters have been established in either laboratory studies or field
calibrations. Many of these chemical proxy relationships are based on foraminifera, a group
of unicellular organisms which secrete multi-chambered calcareous shells1. Foraminifera
occur in all ocean basins and may dwell in surface waters (planktonic species) as well as on
the seafloor (benthic species). Widespread as they occur, they have the potential to record
oceanwide seawater properties. The morphologic and geometric features of their skeleton, i.e.
the arrangement of their successive chambers, enable the micropaleontologist to identify the
different species (e.g. Kemle-von Mücke and Hemleben, 1999). Each of these species favors
different environmental conditions (e.g. Bijma et al., 1990; Darling et al., 1999; Rutherford et
al., 1999) and the knowledge of these habitat preferences allows to focus paleoreconstructions
on specific locations and timescales.
This study is focussed on planktonic foraminifera (Figure 3), whose individual life
spans are on the order of 2-4 weeks (Bijma et al., 1990; Spindler et al., 1979). High
abundances in the world ocean in addition to a short reproductive cycle make foraminifera an
important contributor of biogenic calcite to open ocean marine sediments (Bé et al., 1977) and
a valuable tool for the reconstruction of past ocean conditions. Planktonic foraminifera shells
are composed of extremely pure calcite, typically about 99% by weight. The remaining 1% is
comprised of minor and trace elements such as Mg, Sr, Ba and U. Since trace elements and
different isotopes of major and minor elements are incorporated directly from seawater during
shell precipitation, shell composition reflects both seawater composition and the physical and
biological conditions encountered during precipitation. We will now see how these shell
constituents can help to elucidate past ocean carbonate chemistry.
1 Some benthic species do not actively secrete shells but collect sediment material to construct exoskeletons. For obvious reasons these species are not used as chemical recorders in paleoceanography.
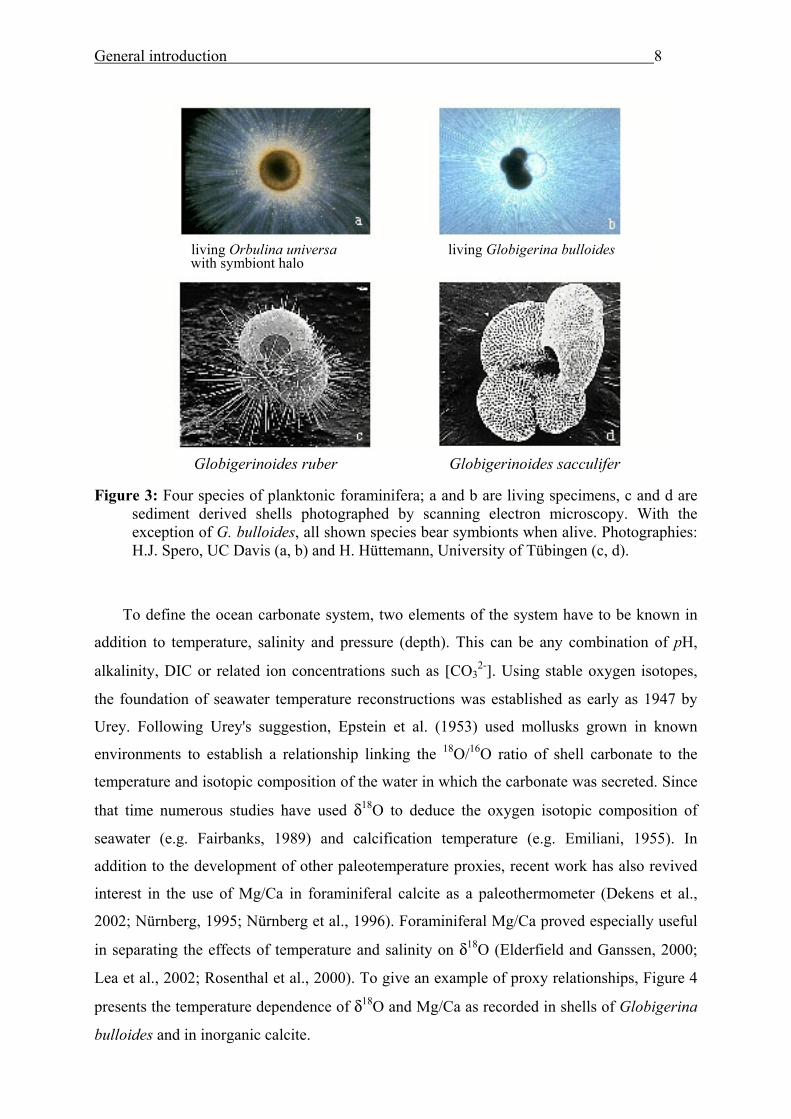
General introduction 8
living Orbulina universa living Globigerina bulloides with symbiont halo
Globigerinoides ruber Globigerinoides sacculifer
Figure 3: Four species of planktonic foraminifera; a and b are living specimens, c and d are sediment derived shells photographed by scanning electron microscopy. With the exception of G. bulloides, all shown species bear symbionts when alive. Photographies: H.J. Spero, UC Davis (a, b) and H. Hüttemann, University of Tübingen (c, d).
To define the ocean carbonate system, two elements of the system have to be known in
addition to temperature, salinity and pressure (depth). This can be any combination of pH,
alkalinity, DIC or related ion concentrations such as [CO32-]. Using stable oxygen isotopes,
the foundation of seawater temperature reconstructions was established as early as 1947 by
Urey. Following Urey's suggestion, Epstein et al. (1953) used mollusks grown in known
environments to establish a relationship linking the 18O/16O ratio of shell carbonate to the
temperature and isotopic composition of the water in which the carbonate was secreted. Since
that time numerous studies have used δ18O to deduce the oxygen isotopic composition of
seawater (e.g. Fairbanks, 1989) and calcification temperature (e.g. Emiliani, 1955). In
addition to the development of other paleotemperature proxies, recent work has also revived
interest in the use of Mg/Ca in foraminiferal calcite as a paleothermometer (Dekens et al.,
2002; Nürnberg, 1995; Nürnberg et al., 1996). Foraminiferal Mg/Ca proved especially useful
in separating the effects of temperature and salinity on δ18O (Elderfield and Ganssen, 2000;
Lea et al., 2002; Rosenthal et al., 2000). To give an example of proxy relationships, Figure 4
presents the temperature dependence of δ18O and Mg/Ca as recorded in shells of Globigerina
bulloides and in inorganic calcite.
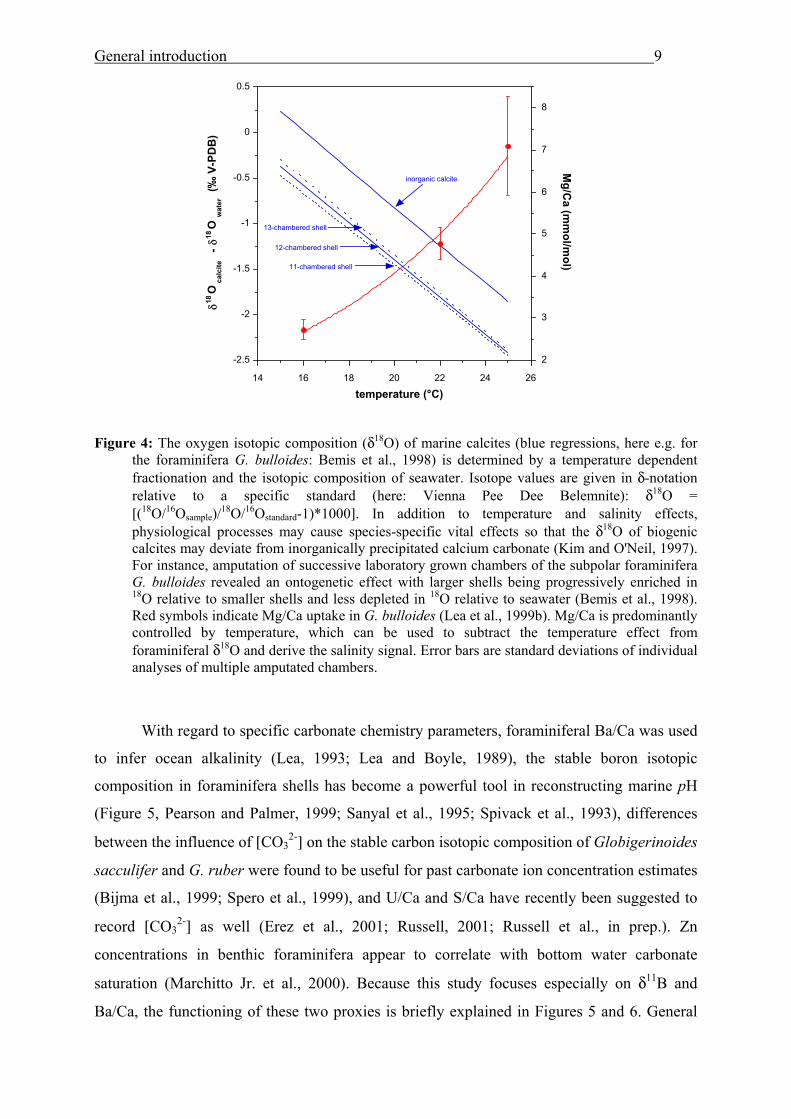
General introduction
-2.5
-2
-1.5
-1
-0.5
0
0.5
2
3
4
5
6
7
8
14 16 18 20 22 24 26
δ18O
cal
cite
- δ18
O w
ater
(‰ V
-PD
B)
Mg/C
a (mm
ol/mol)
temperature (°C)
12-chambered shell
11-chambered shell
13-chambered shell
inorganic calcite
9
Figure 4: The oxygen isotopic composition (δ18O) of marine calcites (blue regressions, here e.g. for
the foraminifera G. bulloides: Bemis et al., 1998) is determined by a temperature dependent fractionation and the isotopic composition of seawater. Isotope values are given in δ-notation relative to a specific standard (here: Vienna Pee Dee Belemnite): δ18O = [(18O/16Osample)/18O/16Ostandard-1)*1000]. In addition to temperature and salinity effects, physiological processes may cause species-specific vital effects so that the δ18O of biogenic calcites may deviate from inorganically precipitated calcium carbonate (Kim and O'Neil, 1997). For instance, amputation of successive laboratory grown chambers of the subpolar foraminifera G. bulloides revealed an ontogenetic effect with larger shells being progressively enriched in 18O relative to smaller shells and less depleted in 18O relative to seawater (Bemis et al., 1998). Red symbols indicate Mg/Ca uptake in G. bulloides (Lea et al., 1999b). Mg/Ca is predominantly controlled by temperature, which can be used to subtract the temperature effect from foraminiferal δ18O and derive the salinity signal. Error bars are standard deviations of individual analyses of multiple amputated chambers.
With regard to specific carbonate chemistry parameters, foraminiferal Ba/Ca was used
to infer ocean alkalinity (Lea, 1993; Lea and Boyle, 1989), the stable boron isotopic
composition in foraminifera shells has become a powerful tool in reconstructing marine pH
(Figure 5, Pearson and Palmer, 1999; Sanyal et al., 1995; Spivack et al., 1993), differences
between the influence of [CO32-] on the stable carbon isotopic composition of Globigerinoides
sacculifer and G. ruber were found to be useful for past carbonate ion concentration estimates
(Bijma et al., 1999; Spero et al., 1999), and U/Ca and S/Ca have recently been suggested to
record [CO32-] as well (Erez et al., 2001; Russell, 2001; Russell et al., in prep.). Zn
concentrations in benthic foraminifera appear to correlate with bottom water carbonate
saturation (Marchitto Jr. et al., 2000). Because this study focuses especially on δ11B and
Ba/Ca, the functioning of these two proxies is briefly explained in Figures 5 and 6. General
General introduction 10
descriptions of carbonate chemistry proxies and a discussion of their specific limitations can
also be found in the Appendix (Working Group 3 report of the ESF Explanatory Workshop on
"The ocean carbon cycle and climate change", Delmenhorst, September 1-4, 2001).
10
20
30
40
50
60
70
7 7.5 8 8.5 9 9.5 10
δ11B
(‰)
pH
seawater
modern marine carbonates
B(OH)3
B(OH)4-
b
0
100
200
300
400
7 7.5 8 8.5 9 9.5 10
conc
entr
atio
n (µ
mol
kg
-1)
pH
B(OH)4-
B(OH)3
a
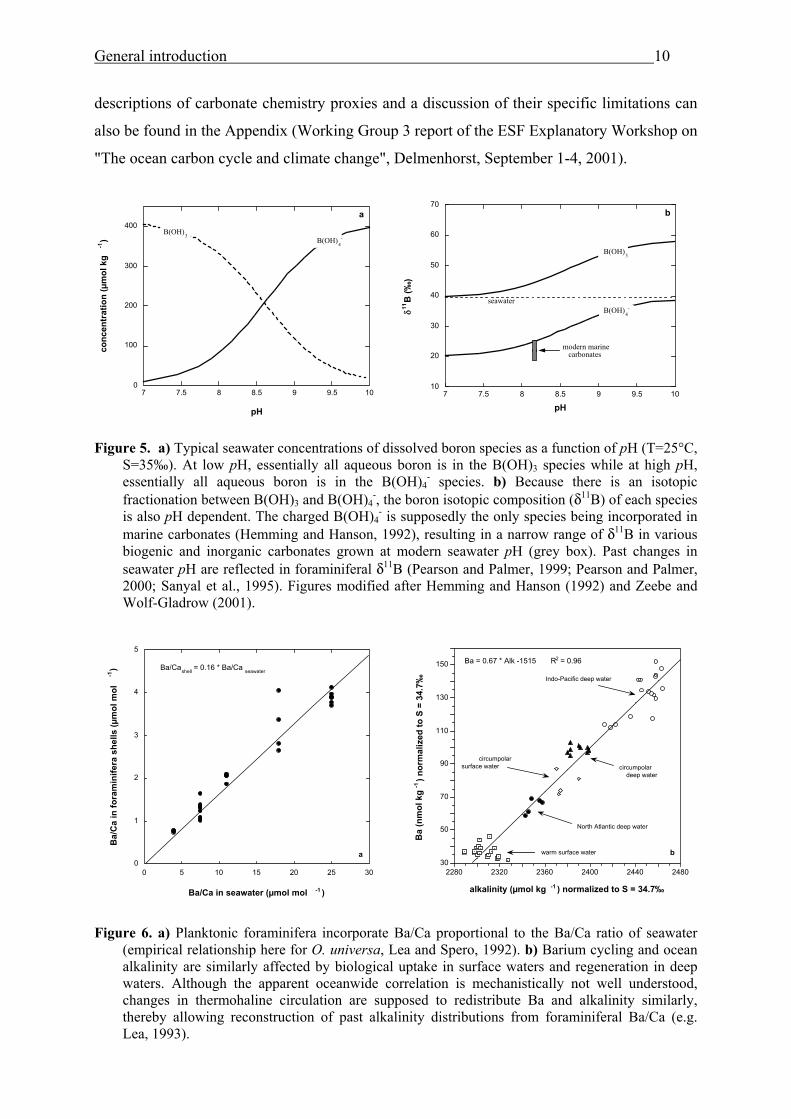
Figure 5. a) Typical seawater concentrations of dissolved boron species as a function of pH (T=25°C,
S=35‰). At low pH, essentially all aqueous boron is in the B(OH)3 species while at high pH, essentially all aqueous boron is in the B(OH)4
- species. b) Because there is an isotopic fractionation between B(OH)3 and B(OH)4
-, the boron isotopic composition (δ11B) of each species is also pH dependent. The charged B(OH)4
- is supposedly the only species being incorporated in marine carbonates (Hemming and Hanson, 1992), resulting in a narrow range of δ11B in various biogenic and inorganic carbonates grown at modern seawater pH (grey box). Past changes in seawater pH are reflected in foraminiferal δ11B (Pearson and Palmer, 1999; Pearson and Palmer, 2000; Sanyal et al., 1995). Figures modified after Hemming and Hanson (1992) and Zeebe and Wolf-Gladrow (2001).
0
1
2
3
4
5
0 5 10 15 20 25 30
Ba/
Ca
in fo
ram
inife
ra s
hells
(µm
ol m
ol-1
)
Ba/Ca in seawater (µmol mol -1 )
Ba/Cashell = 0.16 * Ba/Ca seawater
a30
50
70
90
110
130
150
2280 2320 2360 2400 2440 2480
Ba
(nm
ol k
g-1
) nor
mal
ized
to S
= 3
4.7‰
alkalinity (µmol kg -1 ) normalized to S = 34.7‰
Indo-Pacific deep water
circumpolar deep water
circumpolar surface water
North Atlantic deep water
warm surface water
Ba = 0.67 * Alk -1515 R2 = 0.96
b
Figure 6. a) Planktonic foraminifera incorporate Ba/Ca proportional to the Ba/Ca ratio of seawater
(empirical relationship here for O. universa, Lea and Spero, 1992). b) Barium cycling and ocean alkalinity are similarly affected by biological uptake in surface waters and regeneration in deep waters. Although the apparent oceanwide correlation is mechanistically not well understood, changes in thermohaline circulation are supposed to redistribute Ba and alkalinity similarly, thereby allowing reconstruction of past alkalinity distributions from foraminiferal Ba/Ca (e.g. Lea, 1993).

General introduction 11
In addition to chemical proxies of the seawater carbonate system, the preservation
state of carbonates can be used to estimate bottom water undersaturation for calcite and
aragonite. Relating the preservation state of carbonates in the sediment to the saturation state
of bottom water yields information on [CO32-]in situ. For example, the dissolution driven shell-
thinning of planktonic foraminifera (Broecker and Clark, 2001a; Lohmann, 1995) and the
CaCO3 size fraction index (Broecker and Clark, 1999) were found to approximate bottom
water [CO32-].
1.3 Proxies and their limitations The above mentioned proxies are but a small selection of the already available ones. A
remarkable range of available proxies (see, for an overview, Wefer et al., 1999) suggests that
the tools needed to do a comprehensive survey of past ocean-climate variability have already
been identified. However, many proxies bear uncertainties that complicate their interpretation.
There is abundant evidence that the incorporation of trace elements in foraminiferal calcite
does not take place according to thermodynamic equilibrium. Foraminifera, as living
organisms, actively precipitate their shells, thereby affecting both the structure and chemistry
of shell calcite. Active precipitation argues for significant biological and kinetic controls of
trace element substitution and isotope incorporation. As paleoceanographic reconstructions
can be no better than the proxies themselves, the principle task remaining is to refine and
validate these tools and ascertain which ones yield consistently reliable information.
Approaches to develop, calibrate and validate proxies are based on either field observations
(i.e. coretop sediments, sediment traps and plankton tows) or laboratory culture experiments.
Because environmental conditions often change in unison, using field data to quantify the
influence of variations in any single parameter is the more difficult approach. On the other
hand, laboratory culture experiments are limited by the lack of physico-chemical gradients
usually encountered by the vertically migrating foraminifera. However, the focus on selected
conditions is a major advantage of laboratory cultures. Culture data therefore provide an
important means by which sediment observations can be interpreted.
Beyond the uncertainties involved in specific element incorporation, one of the basic
assumptions in the use of proxies is that the primary signal remains unaltered after burial in
the geological record. However, sediment observations revealed significant variability in the
shell chemistry of planktonic foraminifera that could not be explained by oceanographic or
climatologic changes in the former habitat (Table 1). For instance, Savin and Douglas (1973)

General introduction 12
and Bender et al. (1975) first demonstrated that planktonic foraminiferal Mg/Ca decreases
with water depth and attributed this change to partial dissolution. Subsequently, Brown and
Elderfield (1996), Rosenthal et al. (2000) and Dekens et al. (2002) have attempted to explore
how this dissolution effect varies between species and within different ocean basins.
Similarly, stable oxygen isotope compositions were demonstrated to increase in deeper
sediment cores (Erez, 1979b; Rosenthal et al., 2000; Savin and Douglas, 1973). Ignoring post-
depositional alterations, for instance with regard to Mg/Ca and δ18O, would lead to
underestimates of the real habitat temperatures. Although it seems a reasonable assumption
that enhanced porewater acidity causes the observed variability, sediment observations and
the use of dilute acid in laboratory experiments bear uncertainties which limit data
interpretation. Furthermore, many chemical proxies have not yet been regarded in the light of
selective dissolution, and quantification of the amount of foraminiferal shell corrosion - using
indices of size fraction (Berger et al., 1982; Broecker and Clark, 1999), weight loss estimates
(Lohmann, 1995; Lohmann et al., 1999), reflectance (Helmke and Bauch, 2002) and
microstructural breakdown (Bé et al., 1974) of foraminiferal shells - underlies a number of
assumptions and restrictions (e.g. Publication IV of this study). All these uncertainties limit
the quality of the proxy data base.
Much effort has already been spent on refining available proxies. However, although
remarkable progress has been made on this field, the expansion of our knowledge also raises
new questions. This study aims to contribute to the understanding of the functioning and
reliability of planktonic foraminiferal proxies.

Tab
le 1
: Dis
solu
tion
effe
cts o
n fo
ram
inife
ral s
hell
chem
istry
as o
bser
ved
in se
dim
ent s
tudi
es a
nd la
bora
tory
dis
solu
tion
expe
rimen
ts.
Ref
eren
ce
Spec
ies
Cor
e lo
catio
nδ18
Oδ13
CM
g/C
a
Sr/C
aU
/Ca
Ba/
Ca
Cd/
Ca
Wat
er∆ C
O32-
‰
/km
‰/k
m%
/km
%
/km
%/k
m%
/km
%/k
mde
pth
µmol
kg-1
SE
DIM
EN
T C
OR
ET
OP
OB
SER
VA
TIO
NS:
Pl
ankt
onic
fora
min
ifera
R
usse
ll et
al.
(199
4)G
. sac
culif
er
Cea
ra R
ise
-20
-15
3000
-470
0m39
to
-3
Rus
sell
et a
l. (1
994)
G. s
accu
lifer
O
nton
g Ja
va P
late
au0
0 ?
1600
-450
0m20
to
-15
Bro
wn
& E
lder
field
(199
6)G
. sac
culif
er
Ont
ong
Java
Pla
teau
0 0
1600
-440
0m20
to
-15
Erez
(197
9)
G. s
accu
lifer
N
orth
Atla
ntic
0.5
45
00-4
950m
-11
to-2
2 H
astin
gs e
t al.
(199
8)G
. sac
culif
er
Equa
toria
l Atla
ntic
/Car
ibbe
an0
2540
-364
5m27
to
2 Lo
rens
et a
l. (1
977)
G. s
accu
lifer
Ea
st P
acifi
c R
ise,
cen
tral P
acifi
c-1
4 -4
580-
4000
m47
to
-19
Dek
ens e
t al.
(200
2)G
. sac
culif
er
Ont
ong
Java
Pla
teau
0.2
-5
1600
-450
0m20
to
-15
Dek
ens e
t al.
(200
2)G
. sac
culif
er
Cea
ra R
ise
0.3
-7
2800
-460
0m44
to
-3
Dek
ens e
t al.
(200
2)G
. sac
culif
er
Sier
ra L
eone
Ris
e
0 ?
-14
3100
-510
0m32
to
-7
Ros
enth
al e
t al.
(200
0)G
. sac
culif
er
Ont
ong
Java
Pla
teau
0.2
-12
1600
-340
0m20
to
-3
Ros
enth
al e
t al.
(200
0)G
. sac
culif
er
Cea
ra R
ise
0.2
-6
2800
-420
0m44
to
21
Ros
enth
al e
t al.
(200
0)G
. sac
culif
er
Sier
ra L
eone
Ris
e0
-0.1
0 29
00-5
100m
32 t
o -7
Le
aet
al.(
2000
)G
.rub
erO
nton
gJa
vaPl
atea
u-1
216
00-2
500m
20to
9Er
ez (1
979)
G
. rub
er
Nor
th A
tlant
ic0.
6
4500
-495
0m-1
1 to
-22
Lore
ns e
t al.
(197
7)G
. rub
er
East
Pac
ific
Ris
e, c
entra
l Pac
ific
-8
058
0-38
00m
47 t
o -2
1 D
eken
s et a
l. (2
002)
G. r
uber
O
nton
g Ja
va P
late
au0.
2-1
4 16
00-4
500m
20 t
o -1
5 D
eken
s et a
l. (2
002)
G. r
uber
C
eara
Ris
e0.
3-5
28
00-4
600m
44 t
o -3
D
eken
s et a
l. (2
002)
G. r
uber
Si
erra
Leo
ne R
ise
0
?-7
31
00-5
100m
32 t
o -7
Lo
rens
etal
.(19
77)
N.d
uter
trei
East
Paci
ficR
ise,
cent
ralP
acifi
c-2
10
1900
-470
0m20
to-4
5D
eken
s et a
l. (2
002)
N. d
uter
trei
O
nton
g Ja
va P
late
au0.
3-2
0 16
00-4
500m
20 t
o -1
5 D
eken
s et a
l. (2
002)
N. d
uter
trei
C
eara
Ris
e0.
2-2
1 28
00-4
600m
44 t
o -3
D
eken
s et a
l. (2
002)
N. d
uter
trei
Si
erra
Leo
ne R
ise
-0.1
-16
3100
-510
0m32
to
-7
Rus
sell
etal
.(19
94)
G.t
umid
aC
eara
Ris
e-3
6-4
430
00-4
700m
39to
-3R
usse
ll et
al.
(199
4)G
. tum
ida
Ont
ong
Java
Pla
teau
-16
-20
1600
-450
0m20
to
-15
Bro
wn
& E
lder
field
(199
6)G
. tum
ida
Ont
ong
Java
Pla
teau
-10
-5.3
1600
-440
0m20
to
15
Lore
ns e
t al.
(197
7)G
. tum
ida
East
Pac
ific
Ris
e, c
entra
l Pac
ific
-16
019
00-4
700m
20 t
o -4
5 Er
ez(1
979)
G.t
runc
atul
inoi
des
Nor
thA
tlant
ic0.
945
00-4
950m
-11
to-2
2Er
ez(1
979)
G.i
nfla
taN
orth
Atla
ntic
0.6
4500
-495
0m-1
1to
-22
Erez
(197
9)G
.hir
suta
Nor
thA
tlant
ic0.
745
00-4
950m
-11
to-2
2Er
ez(1
979)
G.c
ongl
obat
usN
orth
Atla
ntic
0.7
4500
-495
0m-1
1to
-22
Lore
ns e
t al.
(197
7)G
. con
glob
atus
Ea
st P
acifi
c R
ise,
cen
tral P
acifi
c0
058
0-38
00m
20 t
o -4
5 Er
ez(1
979)
G.s
ipho
nife
raN
orth
Atla
ntic
0.5
4500
-495
0m-1
1to
-22
Erez
(197
9)O
.uni
vers
aN
orth
Atla
ntic
0.1
4500
-495
0m-1
1to
-22
Erez
(197
9)P.
obliq
uilo
cula
taN
orth
Atla
ntic
-0.4
4500
-495
0m-1
1to
-22
Ben
thic
fora
min
ifera
R
usse
ll et
al.
(199
4)C
. wue
llers
torf
fi C
eara
Ris
e-2
9 -1
330
00-4
700m
39 t
o -3
M
cCor
kle
et a
l. (1
995)
C. w
uelle
rsto
rffi
Ont
ong
Java
Pla
teau
-0.2
6
-8-1
2-2
515
00-4
500m
20 t
o -1
5

Tab
le 1
con
tinue
d: D
isso
lutio
n ef
fect
s on
fora
min
ifera
l she
ll ch
emis
try a
s obs
erve
d in
sedi
men
t stu
dies
and
labo
rato
ry d
isso
lutio
n
ex
perim
ents
. R
efer
ence
Sp
ecie
s C
ore
loca
tion
δ18O
δ13C
Mg/
Ca
Sr/C
aU
/Ca
Ba/
Ca
Cd/
Ca
Wat
er d
epth
‰‰
%%
%%
%
LA
BO
RA
TO
RY
DIS
SOL
UT
ION
EX
PER
IME
NT
S:
Plan
kton
ic fo
ram
inife
ra
H
aley
& K
linkh
amm
er (2
002)
G. s
accu
lifer
C
arib
bean
-7
1125
40m
Ben
der e
t al.
(197
5)
G. s
accu
lifer
4.
4H
önis
ch e
t al.,
this
stud
y G
. sac
culif
er
Gul
f of A
qaba
00
01.
60
00
300m
Lea
& B
o yle
(199
1)G
. tru
ncat
ulin
oide
s0
Le
a &
Boy
le (1
991)
G. c
ongl
obat
us
Ber
mud
a R
ise
045
00m
H
aley
& K
linkh
amm
er (2
002)
O. u
nive
rsa
Car
ibbe
an-5
4-5
525
40m
H
önis
ch e
t al.,
this
stud
yN
. pac
hyde
rma
(sin
.)N
orw
egia
n Se
a0.
20.
2-2
53.
9-4
2 ?
043
?18
00m
Ben
thic
fora
min
ifera
Le
a &
Boy
le (1
993)
C
. wue
llers
torf
fi N
orw
egia
n Se
a
-1
3 ?
Lea
& B
oyle
(199
3)U
vige
rina
spp.
N
orth
wes
tern
Atla
ntic
-12
3430
m
Boy
le (1
988)
U
vige
rina
spp.
N
orth
wes
tern
Atla
ntic
2734
30m
Le
a &
Boy
le (1
993)
Ori
dors
alis
spp.
Ea
ster
n eq
uato
rial P
acifi
c
0 ?
3210
m
Cha
nges
in e
lem
ent/C
a ra
tios o
f cor
etop
sedi
men
t sam
ples
are
cal
cula
ted
as p
erce
ntag
e ch
ange
per
km
wat
er d
epth
, rel
ativ
e to
the
diss
olut
ion
onse
t in
a w
ater
dep
th p
rofil
e. S
imila
rly, i
soto
pe v
alue
s ar
e gi
ven
in a
bsol
ute
chan
ge p
er k
m. V
alue
s fo
r ∆C
O32-
(= [
CO
32-] in
situ
- [
CO
32-
] satu
ratio
n) i
ndic
ate
the
∆CO
32--r
ange
ove
r the
resp
ectiv
e de
pth
prof
ile a
nd w
ere
calc
ulat
ed u
sing
hyd
rogr
aphi
c da
ta fr
om n
earb
y W
OC
E an
d G
EOSE
CS
stat
ions
. Mar
ked
chan
ges
in s
hell
chem
istry
occ
ur w
here
∆C
O32-
falls
bel
ow 2
0 µm
ol k
g-1 (e
.g. D
eken
s et
al.,
200
2). O
bser
ved
diss
olut
ion
trend
s var
y in
tra- a
nd in
ters
peci
fical
ly.
Labo
rato
ry d
isso
lutio
n ex
perim
ents
hav
e be
en c
arrie
d ou
t und
er a
tmos
pher
ic p
ress
ure
usin
g m
odifi
ed s
eaw
ater
(Hön
isch
et a
l., th
is s
tudy
) or
dilu
te a
cid
(all
othe
r st
udie
s). W
ater
dep
th d
enot
es th
e de
pth
from
whi
ch th
e st
udy
mat
eria
l was
col
lect
ed. I
ndic
ated
val
ues
are
max
imum
ch
ange
s be
twee
n un
diss
olve
d an
d m
ost
diss
olve
d sh
ells
. N
ote
that
diff
eren
ces
in d
isso
lutio
n st
ate
exis
t be
twee
n la
bora
tory
stu
dies
. A
mbi
guou
s dis
solu
tion
trend
s are
den
oted
by
ques
tion
mar
ks.

Publications 15
2. Publications
2.1 Focus and outline of this study This dissertation reports of research on the use of planktonic foraminiferal proxies as
indicators of changes in seawater carbonate chemistry. In a first experimental series, living
planktonic foraminifera were investigated with regard to the incorporation of Ba/Ca and δ11B
as a function of physiological processes and seawater carbonate chemistry. A second set of
experiments concentrates on the preservation of foraminiferal shells and their chemical
composition after burial in the sediment.
I. The influence of symbiont photosynthesis on the boron isotopic composition of
foraminiferal shells Hönisch, B., J. Bijma, A.D. Russell, H.J. Spero, M.R. Palmer, A. Eisenhauer: The influence of symbiont photosynthesis on the boron isotopic composition of foraminiferal shells, Marine Micropaleontology, submitted 2002.
This part of the thesis investigates the reliability of δ11B as a proxy for paleo-pH and
the influence of symbiont photosynthetic activity. As microsensor studies have shown that pH
within the spine environment of planktonic foraminifera shows large variations due to
respiration and photosynthesis (Rink et al., 1998), it was investigated whether the known
boron isotopic fractionation between seawater and foraminiferal shells may be altered by
these physiological processes. The manuscript is based on laboratory experiments with living
Orbulina universa. Results of culture experiments are compared with field-grown O. universa
and Globigerina bulloides collected in plankton tows.
II. Assessing the reliability of Ba/Ca as a tracer for alkalinity Hönisch, B., A.D. Russell, J. Bijma, D.W. Lea, H.J. Spero: Assessing the reliability of Ba/Ca as a tracer for alkalinity; in preparation.
Culture experiments with the planktonic foraminifera Orbulina universa and
Globigerina bulloides have been carried out in order to investigate whether Ba- incorporation
during shell secretion is affected by seawater alkalinity. Inorganic precipitation experiments
predict such a linkage via increased in precipitation rates at higher alkalinities. As Ba2+ and
alkalinity vary proportionately in the ocean and the Ba/Ca ratio in foraminiferal shells is
assumed to reflect the seawater Ba2+ concentration, an influence of alkalinity on the Ba
incorporation could compromise the use of this proxy.

Publications 16
III. Post-depositional effects on trace metals and stable isotopes in foraminiferal calcite – Evidence from dissolution experiments
Hönisch, B., J. Bijma, N. Gussone, H.J. Spero, D. Nürnberg, D.W. Lea, A. Eisenhauer: Post-depositional effects on trace metals and stable isotopes in foraminiferal calcite – Evidence from dissolution experiments; in preparation. One of the basic assumptions in the use of proxies for paleoceanographic
reconstructions is that the primary signal remains unaltered after burial in the geological
archive. However, observations on sediment cores revealed significant variability in the shell
chemistry of planktonic foraminifera that could not be explained by oceanographic or
climatologic changes in the former habitat. Although it seems a reasonable assumption that
partial shell dissolution causes the observed variability, a number of uncertainties still remain.
Using extraordinarily well preserved shells of the tropical Globigerinoides sacculifer and the
polar Neogloboquadrina pachyderma (sin.), dissolution experiments under simulated natural
conditions have been carried out in the laboratory. Partially dissolved shells have been
analyzed with respect to minor and trace element to calcium ratios (Mg/Ca, Sr/Ca, Ba/Ca,
U/Ca, Cd/Ca) and stable isotopic compositions (δ18O, δ13C, δ44Ca, δ11B). The combination of
controlled laboratory conditions and the investigation of numerous proxies allows a detailed
discussion of the results with regard to the underlying dissolution mechanisms.
IV. The impact of the ocean carbonate chemistry on living foraminiferal shell weight: A comment to Broecker and Clark’s „Carbonate ion concentration in glacial-age deep waters of the Caribbean Sea“ Bijma, J., B. Hönisch, R.E. Zeebe: The impact of the ocean carbonate chemistry on living foraminiferal shell weight: A comment to Broecker and Clark’s „Carbonate ion concentration in glacial-age deep waters of the Caribbean Sea“. Geochemistry Geophysics Geosystems; in press.
Using the size normalized weight of planktonic foraminifera to determine their
preservation state and estimate the carbonate ion content of oceanic deep waters may be
compromised by a number of physico-chemical parameters. The assumptions made by
Broecker and Clark (2002) disregard existing evidence from culture experiments which
predict differences in shell wall thickness of planktonic foraminifera upon different growth
conditions. In addition to estimating the impact of uncertainties in the corrosivity of bottom
and pore waters, we quantify the effect of carbonate chemistry on shell growth of planktonic
foraminifera using combined data of various culture experiments.

Publications 17
Erklärung über den von mir geleisteten Anteil an den Publikationen Publikation I
Die Laborexperimente wurden von J. Bijma und mir geplant und in Zusammenarbeit mit A.
Russell und H. Spero durchgeführt. Ich habe die Proben gemessen, die Daten ausgewertet und
das Manuskript verfaßt.
Publikation II
Die Laborexperimente wurden von J. Bijma und mir geplant und in Zusammenarbeit mit D.
Lea, A. Russell und H. Spero durchgeführt. Ich habe die Daten ausgewertet und das
Manuskript verfaßt.
Publikation III
Die Laborexperimente habe ich in Zusammenarbeit mit Jelle Bijma geplant. Ich habe die
Experimente durchgeführt, ausgewertet und die Borisotope gemessen. Ich habe das
Manuskript verfaßt.
Publikation IV
Das Manuskript wurde in Zusammenarbeit mit J. Bijma und R. Zeebe geplant und verfaßt. Ich
habe die Kulturdaten zusammengestellt und ausgewertet.

Publication I 18
Publication I
The influence of symbiont photosynthesis on the boron isotopic composition of foraminiferal shells
Bärbel Hönisch, Jelle Bijma, Ann D. Russell, Howard J. Spero, Martin R. Palmer and Anton Eisenhauer
Marine Micropaleontology (submitted 2002)
.......................................................................................................................................................
Abstract Culture experiments were carried out with the planktonic foraminifer Orbulina
universa under high and low light levels in order to determine the influence of symbiont
photosynthetic activity on the boron isotopic composition of shell calcite. Under low light
(reduced photosynthetic rates) the boron isotopic composition of the tests is 1.5‰ lower
compared to shells grown under high light (elevated photosynthetic rates). In terms of inferred
pH, the lower boron isotope values correspond to a reduction in pH of approximately 0.2
units. The boron isotopic composition of Orbulina universa from plankton tows is similar to
shells grown under low light conditions in the laboratory. These data are consistent with
reduced symbiont concentrations in recently secreted shells. In addition to laboratory and
field grown O. universa, we present the first data for a symbiont-barren foraminifer,
Globigerina bulloides. Data obtained for G. bulloides fall ~1.4‰ below the field grown O.
universa. Although the plankton tow results are preliminary, they support the hypothesis that
respiration and photosynthesis are the key physiological parameters responsible for species-
specific vital effects. Model results have predicted that photosynthesis- and respiration-driven
offsets as presented here are constant over a wide pH range and thus do not reduce the
reliability of δ11B as a paleo-pH indicator.

Publication I 19
Introduction
Data from experiments with living foraminifera have confirmed the hypothesis that
seawater pH is the dominant environmental control on the 11B/10B content (δ11B) of
planktonic foraminifera shells (Hemming and Hanson, 1992; Sanyal et al., 2001; Sanyal et al.,
1996; Sanyal et al., 2000; Spivack et al., 1993). Although measurements of foraminiferal δ11B
are not yet a routine tool in paleoceanography, several studies have published paleo-pH
reconstructions across different geological timescales with encouraging results (Palmer et al.,
1998; Pearson and Palmer, 2000; Sanyal and Bijma, 1999; Sanyal et al., 1997; Sanyal et al.,
1995; Spivack et al., 1993).
Whereas pH is the primary environmental control on shell δ11B, several physiological
processes can modify the pH of the calcifying microenvironment, potentially complicating
straightforward interpretation of δ11B data. For instance, microelectrode studies have revealed
that pH in the calcifying microenvironment of symbiont-bearing foraminifera varies with light
levels (Jørgensen et al., 1985; Rink et al., 1998). Although symbionts remove CO2 during
photosynthesis, thereby increasing pH in the foraminiferal microenvironment, respiration
releases CO2 and decreases pH. Results from diffusion-reaction model simulations support
these microsensor studies (Wolf-Gladrow et al., 1999a), showing that respiration and
symbiont-photosynthesis, along with diffusion and chemical reactions, control the availability
of CO32- and HCO3
- for the calcification process. The carbonate ion effect on shell δ13C and
δ18O of planktonic foraminifera (Bijma et al., 1998; Spero et al., 1997) can also be partly
explained by the influence of these physiological processes (Zeebe, 1999; Zeebe et al., 1999).
Comparison of empirical δ11B versus pH-relationships has revealed significant offsets
between inorganic and biogenic calcification as well as between foraminifera species (Sanyal
et al., 2001). It was speculated that species-specificity could be due to differences in
microenvironment pH and/or due to differences in the relative proportion of calcite
precipitated during day and night (Sanyal et al., 2001). Similarly, Hemming et al. (1998)
attributed heavier boron isotopic compositions recorded in a coral during periods of high
productivity to enhanced symbiont photosynthetic activity and a therefore higher pH. This
study investigates the influence of symbiont photosynthetic activity on the boron isotopic
composition of O. universa grown in the laboratory. In order to estimate the effects on
naturally grown foraminifera, we compare experimental data with plankton tow samples of O.
universa and the symbiont-barren G. bulloides.

Publication I 20
2. Methods
2.1 Foraminifera collection and culturing
Foraminifera were cultured using previously established methods (Lea and Spero,
1992; Mashiotta et al., 1997; Spero et al., 1997). Juvenile (presphere) O. universa were hand
collected by scuba divers in July and August 2000 from surface waters of the San Pedro
Basin, approximately 2 km NNE of the Wrigley Institute for Environmental Studies, Santa
Catalina Island, California. Surface seawater for culturing was collected at the dive site,
filtered through a 0.8 µm membrane filter and its carbonate chemistry was subsequently
modified using the method of Sanyal et al. (2001). To reduce the number of shells required
for isotope analysis, the boron concentration in the culture solution was increased tenfold by
adding 0.27 g of boric acid (H3BO3) per L seawater. The pH was readjusted to ambient pH of
8.16 by titration with NaOH. Samples of the culture solution were taken at the beginning and
end of the experiment, acidified with ultrapure HCl and archived for later determination of the
isotopic value.
After collection, individual foraminifera were examined under an inverted light
microscope for identification of species and general condition and then transferred to 115-ml
glass jars containing the experimental filtered seawater. All culture jars were maintained at a
constant temperature in a 22 ± 0.3°C water bath, the approximate summer SST at the
collection site. For each experiment, seventy individuals were grown in the laboratory.
Foraminifera were grown under the following conditions: 1) a 12-hr high light:12-hr dark
cycle where light levels were adjusted to above Pmax (315-326 µmol photons m-2 s-1), and 2) a
12-hr low light (18-20 µmol photons m-2 s-1):12-hr dark cycle. Both experiments utilized high
output, cool white, fluorescent bulbs. The former light levels exceed the saturating irradiances
for symbionts in O. universa, whereas the latter are lower than the light compensation point
(Rink et al., 1998). During the 6-to 15-day culture period, O. universa secretes and calcifies a
spherical chamber. The foraminifera were fed a 1-day old Artemia sp. nauplius (brine shrimp)
every third day. Upon termination of the experiment following foraminiferal gametogenesis,
the empty shells were rinsed in ultrapure water and archived for later analysis.
Alkalinity was determined by Gran-titration at the start and termination of the
experiment. At the same time, dissolved inorganic carbon (DIC) samples were collected,
poisoned with saturated HgCl2 solution and measured coulometrically at the Alfred Wegener
Institute in Bremerhaven, Germany. Seawater pH values were determined potentiometrically
and are given on the NBS scale. Carbonate chemistry analyses were calibrated against

Publication I 21
certified reference material supplied by Dr. A.G. Dickson, University of California, San
Diego.
Plankton tow samples were collected at the dive site in order to determine the boron
isotopic composition of naturally grown O. universa and the symbiont-barren Globigerina
bulloides. Nets with a mesh size of 153 µm were towed at 0-20 m depth. Selected
foraminifera shells were rinsed in distilled water, dried and archived. The samples were
treated in a low temperature asher to remove organic matter and to better distinguish between
juvenile O. universa and G. bulloides. Approximately 300 shells of each species were
collected. Most O. universa had built their spherical chambers shortly before collection.
Shells were very thin and none of the collected specimens from the two species showed signs
of gametogenic calcification. Total sample weight before cleaning was no more than 1 mg for
O. universa and 0.6 mg for G. bulloides.
2.2 Analytical techniques
With the exception of the plankton tow samples, only gametogenic individuals from
the culture experiments were used for analysis. All specimens were rinsed in distilled water to
remove sea salts, dried and weighed. The shells of each experiment were pooled, crushed and
bleached with 4-6% sodium hypochlorite to remove organic matter and then rinsed,
ultrasonicated and centrifuged repeatedly with distilled water to remove soluble salt and
eventually adsorbed B. In a laminar flow bench, the cleaned carbonate was dissolved in 2N
quartz distilled HCl. The dissolved sample, containing approximately 5 ng of B, was loaded
on a rhenium zone refined filament, and 1 µl of boron-free seawater was added to enhance
ionization and suppress fractionation (Hemming and Hanson, 1994). Samples were dried at an
initial ion current of 0.8 A, followed by a 1 minute period at 1.2 A. Loaded filaments were
kept under an infrared lamp until mounted into the mass spectrometer. Isotope data were
collected on a Finnigan MAT 262 RPQ+ Thermal Ionization Mass Spectrometer (TIMS) at
GEOMAR in Kiel, Germany. The BO2- ion method was used following previously published
procedures (1997; Sanyal et al., 1996). For the culture experiments each sample was run at
least 4 times. Cultured foraminifera samples were measured at a filament temperature of 915
± 10°C. While we seldom observed time-dependent fractionation in these boron enriched
samples, the small plankton tow samples started fractionating after 20-30 minutes of
acquisition. We could therefore only complete two acceptable runs for O. universa and a
single acquisition for G. bulloides. However, initial values of the fractionating runs were
consistent with the results of acceptable analyses.

Publication I 22
Table 2. Boron isotopic composition of cultured O. universa and modified seawater
light
(µmol photons m-2 s-1)
pH
(culture water)
Seawater
δ11BMS (‰)
n
O. universa
δ11BC (‰)
n
δ11BNC
(‰)
321 ± 8 8.16 ± 0.02 -8.9 ± 0.1 5 -25.6 ± 0.6 4 22.0 ± 0.6
19 ± 2 8.15 ± 0.03 -9.1 ± 0.4 6 -27.2 ± 0.3 4 20.5 ± 0.3
Results are based on 70 shells per sample. Errors are expressed as 2σmean for multiple sample runs. δ11B (‰) = (Rs/Rstd-1)*1000, Rs = 11B/10B of sample, Rstd = 11B/10B of NBS 951 boric acid standard. Seawater standard = 39.5 ± 0.34‰. n = number of replicate analyses. δ11BNC is the δ11BC after conversion to the natural seawater scale (δ11BNS = 39.5‰), see text and Eq. 9 for details.
Table 3. Boron isotopic composition of plankton tow O. universa and G. bulloides
species ambient pH δ11B
(‰)
n
O. universa 8.12 ± 0.02 20.5 ± 0.5 2
G. bulloides 8.12 ± 0.02 19.0 ± 0.9 2*
Results are based on approximately 300 shells per sample. Errors are expressed as 2σmean for multiple sample runs. δ11B (‰) = (Rs/Rstd-1)*1000, Rs = 11B/10B of sample, Rstd = 11B/10B of NBS 951 boric acid standard. Seawater standard = 39.5±0.34‰. n = number of replicate analyses. * = runs incomplete according to criteria for acceptable runs, see text for details.
To rule out isobaric interferences on mass 42 with organic contamination (12C14N16O-
ions), mass 26 (12C14N-ions) was monitored during each measurement. No interferences were
detected. The 11B/10B ratio was corrected for isotopic interferences on mass 43 (10B16O17O-
ions) by subtraction of 0.00078 from the 43/42 ratio (Spivack and Edmond, 1986).
The fractionation ε between natural seawater (NS) and calcite (C) is usually calculated
as: ε(NS-C) = δ11BNS-δ11BC. This equation gives a good approximation when the isotopic
composition of NS and modified seawater (MS) are the same. Because the modified seawater
used in the culture experiments had a significantly different isotopic composition from natural
seawater, all analyses were corrected for this difference in order to allow comparisons to

Publication I 23
previously published data. To convert our data to the natural seawater scale we applied the
following equation (Zeebe and Wolf-Gladrow, 2001):
δ11BNC = αNS-MS * δ11BC + (αNS-MS – 1) *1000 (9)
where αNS-MS is a factor expressing the isotope difference between modified and natural
seawater. δ11BNC is the approximate value of the calcite if it had been grown in natural
seawater.
The boron isotopic compositions are listed in Table 2 and Table 3. Errors are
expressed as 2σmean. Repeated analyses of natural seawater used as a laboratory standard
resulted in an average value of 39.58 ± 0.34‰ (n = 9; filament temperature: 900 ± 10°C).
For laboratory intercomparison, additional analyses of the culture samples were
performed on a Micromass VG Sector 54 TIMS at the Southampton Oceanography Centre
(SOC), Southampton, UK. Analyses followed the method outlined in Palmer et al. (1998).
Samples and NBS 951 boric acid standard were measured at a filament temperature of 925 ±
10°C.
3. Results and Discussion
In the following section we present the data obtained from our experiments. The data
set is internally consistent and the results are reasonable with regard to theoretical
considerations. However, we found systematic offsets from previously published calibration
curves. Although the offsets do not affect the conclusions of this and most previous studies,
the underlying problem will be discussed in more detail at the end of the following section.
3.1 Laboratory experiments
The results of our experiments clearly show the influence of symbiont photosynthetic
activity on the boron isotopic composition of the shell. At equal culture water pH the δ11B of
low light O. universa shells is 1.5‰ lower than specimens grown under high light (Table 2,
Figure 7). If we shift the theoretical curve for δ11B of B(OH)4- (Kakihana et al., 1977) so it
passes through our HL data, the δ11B for the LL group would imply a decrease in pH of ~0.2
units.
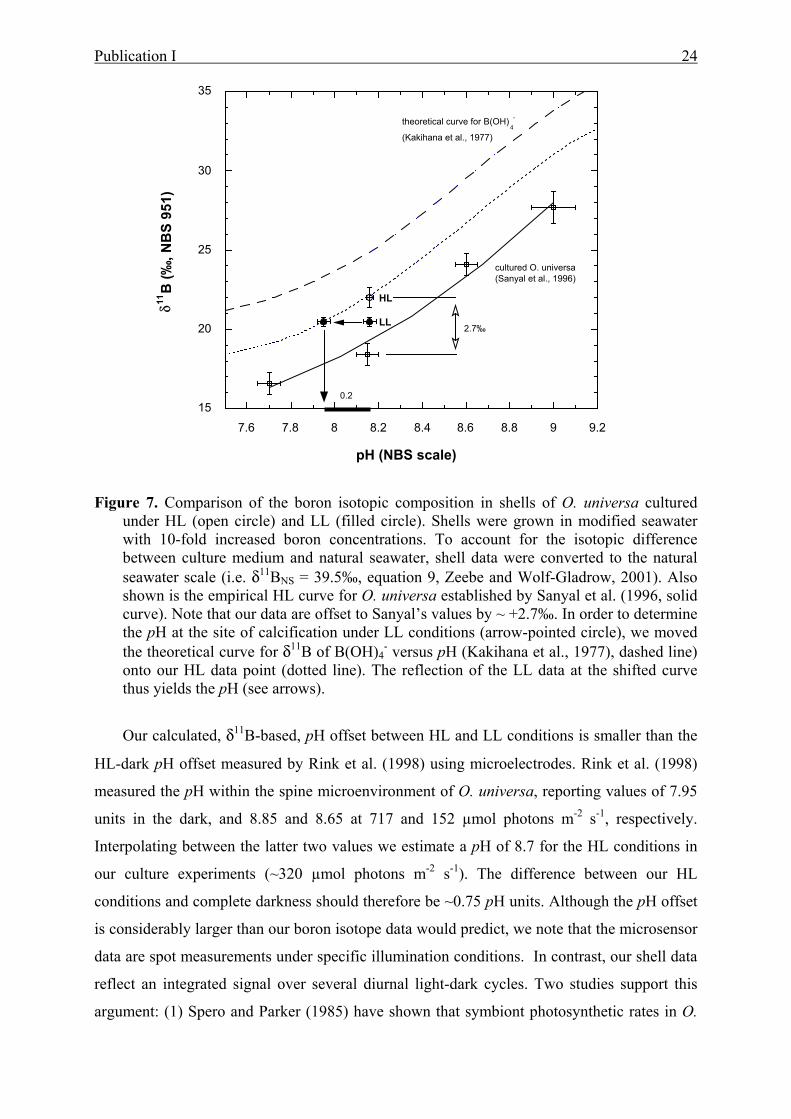
Publication I 24
15
20
25
30
35
7.6 7.8 8 8.2 8.4 8.6 8.8 9 9.2
δ11B
(‰, N
BS
951)
pH (NBS scale)
theoretical curve for B(OH)4-
(Kakihana et al., 1977)
HL
LL
cultured O. universa (Sanyal et al., 1996)
2.7‰
0.2
Figure 7. Comparison of the boron isotopic composition in shells of O. universa cultured under HL (open circle) and LL (filled circle). Shells were grown in modified seawater with 10-fold increased boron concentrations. To account for the isotopic difference between culture medium and natural seawater, shell data were converted to the natural seawater scale (i.e. δ11BNS = 39.5‰, equation 9, Zeebe and Wolf-Gladrow, 2001). Also shown is the empirical HL curve for O. universa established by Sanyal et al. (1996, solid curve). Note that our data are offset to Sanyal’s values by ~ +2.7‰. In order to determine the pH at the site of calcification under LL conditions (arrow-pointed circle), we moved the theoretical curve for δ11B of B(OH)4
- versus pH (Kakihana et al., 1977), dashed line) onto our HL data point (dotted line). The reflection of the LL data at the shifted curve thus yields the pH (see arrows).
Our calculated, δ11B-based, pH offset between HL and LL conditions is smaller than the
HL-dark pH offset measured by Rink et al. (1998) using microelectrodes. Rink et al. (1998)
measured the pH within the spine microenvironment of O. universa, reporting values of 7.95
units in the dark, and 8.85 and 8.65 at 717 and 152 µmol photons m-2 s-1, respectively.
Interpolating between the latter two values we estimate a pH of 8.7 for the HL conditions in
our culture experiments (~320 µmol photons m-2 s-1). The difference between our HL
conditions and complete darkness should therefore be ~0.75 pH units. Although the pH offset
is considerably larger than our boron isotope data would predict, we note that the microsensor
data are spot measurements under specific illumination conditions. In contrast, our shell data
reflect an integrated signal over several diurnal light-dark cycles. Two studies support this
argument: (1) Spero and Parker (1985) have shown that symbiont photosynthetic rates in O.

Publication I 25
universa display a daily periodicity. For any given 12-hour illumination period, symbionts
only photosynthesize at a maximum rate for 4-6 hours with lower rates during the remaining
illuminated period. Based on symbiont density and photosynthetic rates provided in that
study, the integrated photosynthetic rate for one light period is calculated at ~59 nmol C d-1
instead of ~87 nmol C d-1 which would be calculated if the maximum photosynthetic rate had
been maintained for the full 12 hour illuminated period. Therefore, the integrated symbiont
photosynthetic effect is only 68% of the spot pH measurements made by Rink et al. (1998).
With regard to pH, the computed integrated value for a full light period is therefore only 8.46
instead of 8.70. (2) Culture experiments by Lea et al. (1995) further showed that calcification
in O. universa varies among specimens and is not strictly limited to the daylight hours. They
calculated that on average, 33% of the spherical shell is precipitated during the night. Using a
simple mass balance, the influence of combining calcite secreted during the night (@ pH =
7.95) and during the day (@ pH = 8.46) would yield a weighted, time integrated pH of 8.29
for the HL group. The pH difference predicted for foraminifera grown under a HL-dark cycle
compared to shells grown in complete darkness is therefore reduced to ~0.34 instead of ~0.75
units.
Finally, it should be kept in mind that we did not keep the LL-foraminifera in the dark
but at ~19 µmol photons m-2 s-1. Although this is below the physiological compensation point
for the O. universa symbiotic association (association respiration rate = symbiont
photosynthetic rate) (Rink et al., 1998), symbiont photosynthesis still removes CO2.
Therefore the actual microenvironment pH under LL conditions should be higher than that in
shells grown in the dark. Using this line of argument, the calculated HL-dark pH difference
of ~0.34 units should be smaller for LL grown specimens. Our experimental result of a ~0.2
pH difference between LL and HL grown specimens agrees well with these calculations.
We also compared our results to those of a diffusion-reaction model (Zeebe et al.,
2001; subm.) which predicts a δ11B offset of approximately 3‰ between an integrated diurnal
record and shells grown under complete darkness. This value is considerably larger than our
experimental result (~1.5‰) due to yet unknown reasons. Additional model runs need to be
performed to explain the difference. Irrespective of the magnitude, the most important model
prediction is the constancy of this offset over a wide seawater pH range (7.9-8.5). The use of
δ11B as a paleo-pH proxy is therefore not compromised through physiological processes in the
spine environment.
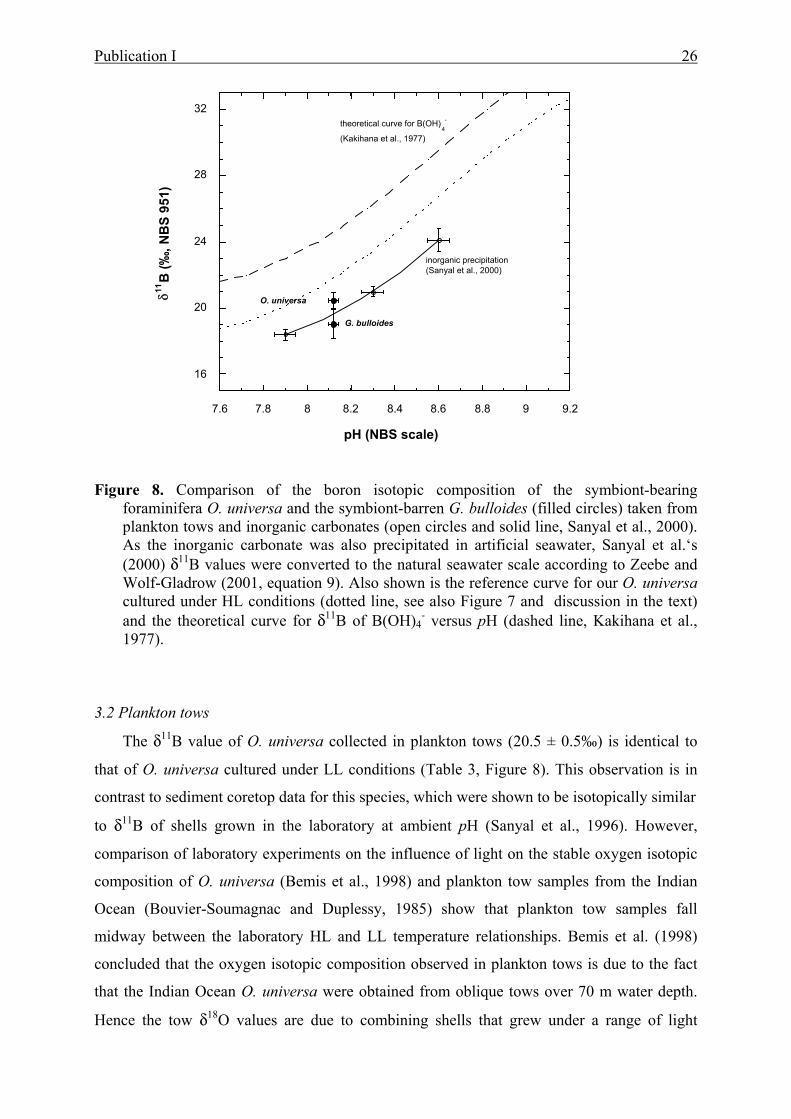
Publication I 26
16
20
24
28
32
7.6 7.8 8 8.2 8.4 8.6 8.8 9 9.2
δ11B
(‰, N
BS
951)
pH (NBS scale)
theoretical curve for B(OH)4-
(Kakihana et al., 1977)
inorganic precipitation(Sanyal et al., 2000)
O. universa
G. bulloides
Figure 8. Comparison of the boron isotopic composition of the symbiont-bearing
foraminifera O. universa and the symbiont-barren G. bulloides (filled circles) taken from plankton tows and inorganic carbonates (open circles and solid line, Sanyal et al., 2000). As the inorganic carbonate was also precipitated in artificial seawater, Sanyal et al.‘s (2000) δ11B values were converted to the natural seawater scale according to Zeebe and Wolf-Gladrow (2001, equation 9). Also shown is the reference curve for our O. universa cultured under HL conditions (dotted line, see also Figure 7 and discussion in the text) and the theoretical curve for δ11B of B(OH)4
- versus pH (dashed line, Kakihana et al., 1977).
3.2 Plankton tows
The δ11B value of O. universa collected in plankton tows (20.5 ± 0.5‰) is identical to
that of O. universa cultured under LL conditions (Table 3, Figure 8). This observation is in
contrast to sediment coretop data for this species, which were shown to be isotopically similar
to δ11B of shells grown in the laboratory at ambient pH (Sanyal et al., 1996). However,
comparison of laboratory experiments on the influence of light on the stable oxygen isotopic
composition of O. universa (Bemis et al., 1998) and plankton tow samples from the Indian
Ocean (Bouvier-Soumagnac and Duplessy, 1985) show that plankton tow samples fall
midway between the laboratory HL and LL temperature relationships. Bemis et al. (1998)
concluded that the oxygen isotopic composition observed in plankton tows is due to the fact
that the Indian Ocean O. universa were obtained from oblique tows over 70 m water depth.
Hence the tow δ18O values are due to combining shells that grew under a range of light

Publication I 27
conditions. Our plankton tow foraminifera were collected at depths down to 20 m. Light level
measurements made at the collection site in August 1987 yielded irradiance levels between
2188 µmol photons m-2 s-1 at the surface and 361 to 123 µmol photons m-2 s-1 at 18 to 27 m
water depth (Spero, unpublished data). These irradiances would suggest all the tow-collected
shells were exposed to light levels that were higher than the HL levels in the laboratory. To
explain the low isotopic value in the tows, we hypothesize that the thinly calcified specimens
collected in plankton tows are not fully calcified and may not contain the density of
symbionts expected from a similar sized sphere as it approaches gametogenesis. Thin-walled
shells could also reflect a disproportionate amount of dark (night) calcification relative to
gametogenic shells. The photosynthetic impact on the boron isotopic composition is therefore
assumed to be reduced at such an early stage suggesting plankton tow samples are not the
optimum source of O. universa material for testing the boron isotope calibration.
The δ11B of symbiont-barren G. bulloides (19.0 ± 0.9‰) was 1.4‰ lower than the O.
universa shells collected from the same plankton tows. Because this is the first δ11B datum
ever measured on a symbiont-barren species, it cannot be compared to literature data.
However, the dominant physiological process that affects the carbonate chemistry of G.
bulloides at the site of calcification is respiration. Although pH measurements have never
been conducted on this species, it is well known that the addition of respiratory CO2 decreases
pH in symbiont-bearing foraminifera by up to 0.3 units (Jørgensen et al., 1985; Rink et al.,
1998; Wolf-Gladrow et al., 1999a) and therefore would be expected to influence G. bulloides
similarly. Comparison of this datum point with data from inorganic precipitation experiments
(Sanyal et al., 2000) demonstrates that G. bulloides falls slightly below the inorganic
precipitation curve (Figure 8). Given the uncertainty of absolute differences between studies
and samples and the single datum presented here, the similarity between G. bulloides and the
inorganic precipitation experiments is promising. The lower δ11B compared to O. universa
and the inorganic precipitation results is reasonable under the assumption of a lower pH at the
site of calcification due to respiration.
3.3 Analytical offset
Our HL data are offset from Sanyal et al‘s (1996) empirical equation based on cultured
O. universa by approximately +2.7 ‰ (Figure 7). At this stage we cannot explain the offset.
Although part of the explanation could be due to lower light intensities in Sanyal’s
experiments (no special illumination was provided apart from the normal laboratory ceiling
lighting), it is unlikely that irradiances were lower than the LL levels studied in our

Publication I 28
experiments. Besides probable differences in the light regime, major differences between the
two experimental set-ups are the use of boron-enriched seawater and the fact that specimens
in our experiments underwent gametogenesis. While Sanyal et al. (2001) ruled out the
possibility that higher boron alkalinity in artificial seawater affects experimental δ11B values,
comparison between pregametogenic experimental individuals and postgametogenic shells
derived from sediments (Sanyal et al., 1996) confirmed that gametogenesis does not influence
the boron isotopic fractionation significantly. Since the experimental methods were equal
apart from these differences, there is no explanation for the offset to be expected from the
experimental point of view.
The only remaining difference is the laboratory and mass spectrometer on which the
samples were analyzed. Data for the previously published empirical relationships on
foraminifera and inorganic calcite were all established in the same laboratory (2001; Sanyal et
al., 1996; 2000). However, offsets between laboratories have already been reported in the
literature. For instance, Hemming et al. (1998) compared marine coral boron isotope data
studied by Vengosh et al. (1991), Hemming and Hanson (1992) and Gaillardet and Allègre
(1995). They found offsets up to 3‰ between studies, although measurements were
conducted on the same modern coral species. Analyses on the coral Porites (Hönisch and
Bijma, unpublished data), are similar to data published by Hemming and Hanson (1992) and
Gaillardet and Allègre (1995), indicating our analytical techniques are sound and comparable
between laboratories. Furthermore, data acquired on G. sacculifer at SOC (M.R. Palmer) are
offset by ~+2‰ to similar samples analyzed by Sanyal et al. (2001). Our own repeated
analyses on different samples of G. sacculifer revealed a much closer similarity in δ11B
between this species and O. universa than the one reported by Sanyal et al. (2001).
We suggest that the origin for the observed differences must be within the analytical
procedure. The offsets may be laboratory specific, maybe even specific for different
(biogenic) carbonates. Two possible causes of interlaboratory offsets include procedural
differences such as the temperature at which the analysis is performed, and differences in
standardization. For instance, the temperature at which the analysis is performed is species-
specific and is adjusted to the amount of boron present in the carbonate. Furthermore, many
laboratories use internal seawater standards to calibrate their data instead of the NBS 951
boric acid standard. Neither standard is a carbonate, and matrix differences may be more
important than assumed to date. The difference between the 43/42 ratio of biogenic
carbonates and seawater on the one hand, and the boric acid standard on the other, may be too

Publication I 29
large to make any of these non-carbonates a reasonable standard. There is a clear need to
define an international carbonate standard for boron isotopic analyses.
Despite the possibility of specific laboratory offsets, relative differences between
samples of the same species seem to be constant. Repeated analyses of our cultured samples
at SOC revealed a difference of ~2.2‰ between shells grown under HL (δ11B=23.9‰, n=2)
and LL (δ11B=21.7‰, n=1). Although the δ11B O. universa was measured ~2‰ heavier at
SOC compared to GEOMAR, the relative difference between the two cultured shell samples
obtained in both laboratories is the same within error. Relative comparisons with samples of
known δ11B-pH relationship are therefore feasible. However, comparison of absolute values
raised in different laboratories seems to be inappropriate until identification of the underlying
problem.
4. Conclusions
The results presented here suggest a dependence of O. universa δ11B on symbiont
photosynthetic activity similar to Hemming et al.'s (1998) observation on corals in high
productivity periods. Although the effect is significant, model results by Zeebe et al. (2001;
subm.) demonstrate that the δ11B offset is constant over a wide range of seawater pH (7.9-
8.5). Assuming that respiration and photosynthesis rates do not change with varying seawater
pH, the use of δ11B as a proxy for pH is not compromised through the vital effect presented
here.
If photosynthesis and respiration are the major parameters affecting deviations of the
shell isotopic signature from seawater pH, our results suggest that symbiont-bearing
foraminifera like O. universa and G. sacculifer should generally record higher δ11B values
and symbiont-barren species such as G. bulloides lower values compared to inorganic
calcites. Culture and field data presented here are consistent with this hypothesis but deviate
from earlier published data. In order to better understand the controls over δ11B in
foraminifera and to compare results from different laboratories, it is essential to resolve the
interlaboratory analytical offsets discussed here. Nevertheless, as long as modern samples of a
certain species are available, they can be used as a reference for ancient samples of the same
species. Using the shape of the theoretical relationship between pH and δ11B by Kakihana et
al. (1977), the differences in pH can be estimated.

Publication I 30
Acknowledgements
We thank the staff of the Catalina Marine Science Center for providing facilities to
make this work possible. We gratefully acknowledge field help by Laurie Juranek, M.
Thomas, H. Iverson and the Catalina dive crew. Invaluable was A. Sanyal’s introduction into
the secrets of δ11B analyses and boron enriched culture experiments. Of great benefit were
suggestions and comments by R. Zeebe, D. Lea, D. Wolf-Gladrow and H. Fischer and
technical discussions with A. Deyhle and N. Gussone. A review by N.G. Hemming was very
encouraging and the helpful suggestions are gratefully acknowledged. This research was
supported by DAAD grant D/00 20292 (BH), NEBROC (BH, JB), NSF grants OCE-9907044
(ADR), and OCE-9903632 (HJS). Analyses at SOC were funded by a European Commission
geochemical analytical facilities grant (BH, MRP).

Publication II 31
Publication II
Assessing the reliability of Ba/Ca as a tracer for alkalinity
Bärbel Hönisch, Ann D. Russell, Jelle Bijma, David W. Lea and Howard J. Spero
in preparation
.......................................................................................................................................................
Abstract
Here we report results of laboratory culture studies showing that seawater alkalinity
has a minor effect on the incorporation of Ba into the calcite shells of planktonic foraminifera
(Orbulina universa and Globigerina bulloides). The Ba/Ca ratio in foraminiferal shells has
been proposed as a proxy for alkalinity. If alkalinity itself has a significant influence on the
Ba incorporation into foraminiferal calcite, it would be impossible to use foraminiferal Ba/Ca
to differentiate between a change in seawater Ba2+ and a coincidental change in alkalinity.
Specimens of the symbiont-bearing species Orbulina universa and the symbiont-barren
Globigerina bulloides were grown in seawater of constant Ba2+ concentration at five different
alkalinities. The experimental alkalinity range comprised levels below, within and above the
range presently found in the ocean. We found a weak negative correlation between DBa and
alkalinity in O. universa shells under high light conditions: DBa = 0.27 (±0.04) – 4.39 (±1.58)*
10-5 * AT. For an increase in alkalinity of 100 µmol kg-1 DBa therefore decreases by 0.004.
This change is well within the error of DBa determined to date. The weak influence of
alkalinity on Ba incorporation into foraminiferal shells should not bias paleoreconstructions
based on foraminiferal Ba/Ca. Globigerina bulloides has not been calibrated for Ba before
and the experiments revealed that DBa in this species is the same as DBa in O. universa. In line
with the similar Ba/Ca uptake ratio of symbiont-bearing and symbiont-barren species, varying
light levels do not affect the Ba incorporation of O. universa.

Publication II 32
Introduction
One of the most intriguing scientific challenges today is to understand the interaction
between the atmospheric CO2 budget and the oceanic carbon cycle. Since both reservoirs are
tightly linked, information about one of them provides insight about the other. The values of
at least two carbonate system parameters are required to calculate the entire oceanic carbonate
chemistry. This can be any combination of dissolved inorganic carbon (DIC), alkalinity, pH
or related ion concentrations. Lea (1993) suggested that changes in the thermohaline
circulation should redistribute Ba2+ and alkalinity similarly, thereby allowing reconstruction
of past alkalinity distributions from benthic foraminiferal Ba/Ca. Using the modern oceanic
relationship between Ba2+ and alkalinity, Lea (1993) proposed an increase in alkalinity of
approximately 20-25 µmol kg-1 during the last glacial maximum when compared to the
Holocene. This could explain a significant amount of the glacial drop in pCO2.
Although this result is promising, recent research has shown that a number of
geochemical proxies are affected by the oceanic carbonate system. For instance, Lea et al.
(1999b) found that seawater pH influences shell Mg/Ca and Sr/Ca in the planktonic
foraminifera O. universa and G. bulloides. They attributed increased Sr/Ca under higher pH to
higher CO32- concentrations and thereby enhanced calcification rates. Similarly, shell δ13C
and δ18O of planktonic foraminifera vary inversely with the carbonate ion concentration of
seawater under conditions of constant δ13CDIC and δ18O of seawater (Bijma et al., 1998; Spero
et al., 1997). Vital as well as kinetic effects were found to explain the underlying mechanism
(Wolf-Gladrow et al., 1999a; Zeebe, 1999; Zeebe et al., 1999). More recently, Russell et al.
(2001, Russell, in prep. #548) observed a correlation between carbonate ion concentration and
U/Ca in foraminifera shells. If Ba incorporation into shell calcite is similarly affected, then it
would negate the use of this proxy for estimating alkalinity in paleoceans.
The specific motivation for this investigation is based on similarities in Ba2+ and Sr2+
incorporation into inorganic and biogenic calciumcarbonates. Ba and Sr are metals with ionic
radii greater than Ca and Mg (Ba2+: 1.47 Å, Sr2+: 1.31 Å, Ca2+: 1.00 Å, Mg2+: 0.72 Å,
according to Shannon (1976). Like Mg2+, Ba2+ and Sr2+ substitute for Ca2+ in biogenic calcite
(Lea and Boyle, 1991; Mackenzie et al., 1983; Speer, 1983). A general tendency has been
observed according to which partition coefficients of metals with ionic radii smaller than Ca
decrease with increasing precipitation rate, whereas those with an ionic radius larger than
Ca2+ increase with precipitation rate (Lorens, 1981). Inorganic precipitation experiments have
shown that the partition coefficients of Sr2+ and also Ba2+ depend strongly on precipitation
rate (Lorens, 1981; Morse and Bender, 1990; Tesoriero and Pankow, 1996) and observations

Publication II 33
on Sr2+ in planktonic foraminifera (Lea et al., 1999b), coccolithophorids (Stoll et al., 2001)
and apparently also in benthic foraminifera (Elderfield et al., 1996) are in agreement with
these inorganic experiments. Especially in the case of planktonic foraminifera Lea et al.
(1999b) showed that Sr/Ca increases in shells grown under higher pH. Under inorganic
conditions the adsorption of Ba and Sr (when both equally concentrated in the aqueous
solution) onto solid calcite is low but comparable (Zachara et al., 1991). Despite large
differences in seawater ion concentration, the relative uptake of Sr and Ba into planktonic
foraminiferal calcite (as reflected in the values of DSr and DBa) is similar (Lea, 1999b).
These similarities in the geochemical behavior of Ba and Sr led us to question whether
the incorporation of Ba2+ into biogenic calcite is affected by seawater carbonate chemistry.
Using laboratory experiments, we explore the influence of alkalinity on Ba/Ca in living
planktonic foraminifera.
Experimental Methods Collection and culturing of foraminifera
Foraminifera were cultured using previously established methods (Lea and Spero,
1992; Mashiotta et al., 1997; Spero et al., 1997). Juvenile (presphere) Orbulina universa and
small Globigerina bulloides were hand collected by scuba divers in July and August 2000
from surface waters of the San Pedro Basin, approximately 2 km NNE of the Wrigley
Institute for Environmental Studies, Santa Catalina Island, California. Surface seawater for
culturing was collected at the foraminifera collection site and filtered in the laboratory using
acid-cleaned 0.4 µm polycarbonate membrane filters and an acid-leached polysulfone filter
holder. After collection, individual foraminifera were examined under an inverted light
microscope for identification and inspection of general condition and then transferred to 120
ml glass jars containing the filtered culturing solutions. To avoid contamination of culture
water during transfer and feeding of specimens, sample handling was done wearing powder
free gloves and using acid-leached glass pipettes for feeding and transferring foraminifera. All
culture jars were maintained at a constant temperature in a 22 ± 0.1°C water bath, the
approximate summer SST at the collection site.
Foraminifera were grown under the following conditions: 1) a 12-hr high light
(HL):12-hr dark cycle where light levels were adjusted to above Pmax (299-365 µmol photons
m-2 s-1), and 2) a 12-hr low light (LL):12hr-dark cycle (23 µmol photons m-2 s-1). These light
The Geology Department
In the paragraph comparing the geochemistry of Ba and Sr you don’t specifically mention the influence of pH or CO3 on Sr/Ca incorporation \(aside from the precipitation rate issue\). I’d stick that in \(see Lea, Mashiotta, and Spero, 1999\)

Publication II 34
levels either exceed the saturating irradiances for symbionts in O. universa or fall below the
compensation point for photosynthetic activity (Rink et al., 1998).
During a 7- to 10-day culture period O. universa secretes and calcifies a spherical
chamber, whereas G. bulloides forms between two and four new chambers. Globigerina
bulloides and O. universa were fed a 1-day old Artemia sp. nauplius (brine shrimp) every
other or every third day respectively. After the foraminifera underwent gametogenesis, empty
shells were rinsed in ultrapure water and archived for later analysis. In addition, a sample of
the culture solution was acidified and archived to verify that the Ba2+ concentration remained
constant over the course of the experiment. Because the amount of calcite precipitated by
foraminifera does not require more than 0.1% of the initial Ca2+ present in the culture
seawater, Ca2+ concentrations were constant over the duration of the experiments.
Total alkalinity (AT) was modified by the addition of ultrapure HCl to lower AT or the
addition of ultrapure NaOH to increase AT. Initial and final alkalinity was determined by
Gran-titration with a Metrohm 785 titrino auto-titrator. Samples for DIC analysis were taken
at the beginning and the end of the experiment, poisoned with a few drops from a saturated
HgCl2 solution and measured coulometrically at the Alfred Wegener Institute.
Sample preparation
Only gametogenic individuals were used for analysis. All specimens were rinsed in
distilled water to remove sea salts, dried and weighed. Chambers secreted in the laboratory,
i.e. under controlled conditions, were separated from the field grown shell. Separating and
cleaning procedures followed methods established by Mashiotta et al. (1997). Briefly, shells
of O. universa were cracked open with a disposable scalpel and the juvenile test (if present)
was removed with a small brush. The fragments were then transferred to 0.5 ml
polypropylene centrifuge vials. Individual shells of O. universa shells were analysed where
possible, but for the smallest individuals, two or three shells were pooled to obtain at least 40
µg of uncleaned calcite. For G. bulloides, chambers grown in the laboratory were identified
by comparing the size of the specimen at collection with the size of the postgametogenic
shell. The laboratory-grown chambers were removed with a scalpel, pooled (25-35 chambers
per sample) and loaded into 0.5-ml polypropylene centrifuge vials. Samples were then subject
to a series of physical and chemical treatments including: oxidation in hot H2O2 – NaOH
solution to remove organic matter, 2-3 weak acid leaches (0.001N HNO3) and repeated rinses
in ultrapure water.

Publication II 35
Analytical Methodology
Sample analysis followed the multi-element inductively coupled plasma mass
spectrometry (ICP-MS) method previously described by Lea and Martin (1996). After
cleaning, 20-30 µg of purified foraminifera shells were dissolved in 0.5 ml of a 0.1 N HNO3
solution containing calibrated concentrations of 135Ba and 45Sc. The solutions were aspirated
into a Finnigan Element 2 high resolution magnetic sector ICP-MS. The 135Ba/138Ba and 45Sc/48Ca ratios are determined by pulse and analog acquisition modes, respectively. The
concentrations of Ba2+ and Ca2+ are then determined by isotope dilution and internal standard
calculation, respectively. Na/Ca was determined to be certain that the hydrogen peroxide –
sodium hydroxide solution used in the sample preparation was completely rinsed out. Several
analyses of a consistency standard with Ba2+ and Ca2+ concentrations similar to the
foraminiferal samples had a standard deviation of 0.4% for Ba2+, 1% for Ca2+, and 1.25% for
the Ba/Ca ratio. Analyses were performed at the University of California, Santa Barbara.
An average of 2-4 replicates was determined on O. universa. Due to the small sample
yield in G. bulloides merely one Ba/Ca analysis per alkalinity could be realised for this
species.
Results
Fifteen water samples were randomly selected from 184 total culture experiments to
verify the constancy of the trace and minor element concentrations over the course of the
experiment. These samples yielded ambient seawater concentrations of 37.8 ± 0.35 nmol kg-1
and 10 ± 0.09 mmol kg-1, for Ba2+ and Ca2+ respectively. The amount of Ba2+ incorporated
into the foraminiferal shell is negligible compared to the total Ba2+ present in the culturing
solutions (Lea and Spero, 1992). However, problems could arise from barium contamination
(e.g. during feeding) or adsorption onto the container walls. Although one of the water
samples showed a substantially higher Ba2+ concentration by 11%, the average change in the
seawater Ba2+ concentration was only 0.9% indicating that barium adsorption was negligible
and contamination was unlikely. Nevertheless, we cannot rule out the possibility that some of
the foraminifera experienced Ba2+ concentrations that may have differed slightly from the
average of 37.8 nmol kg-1.
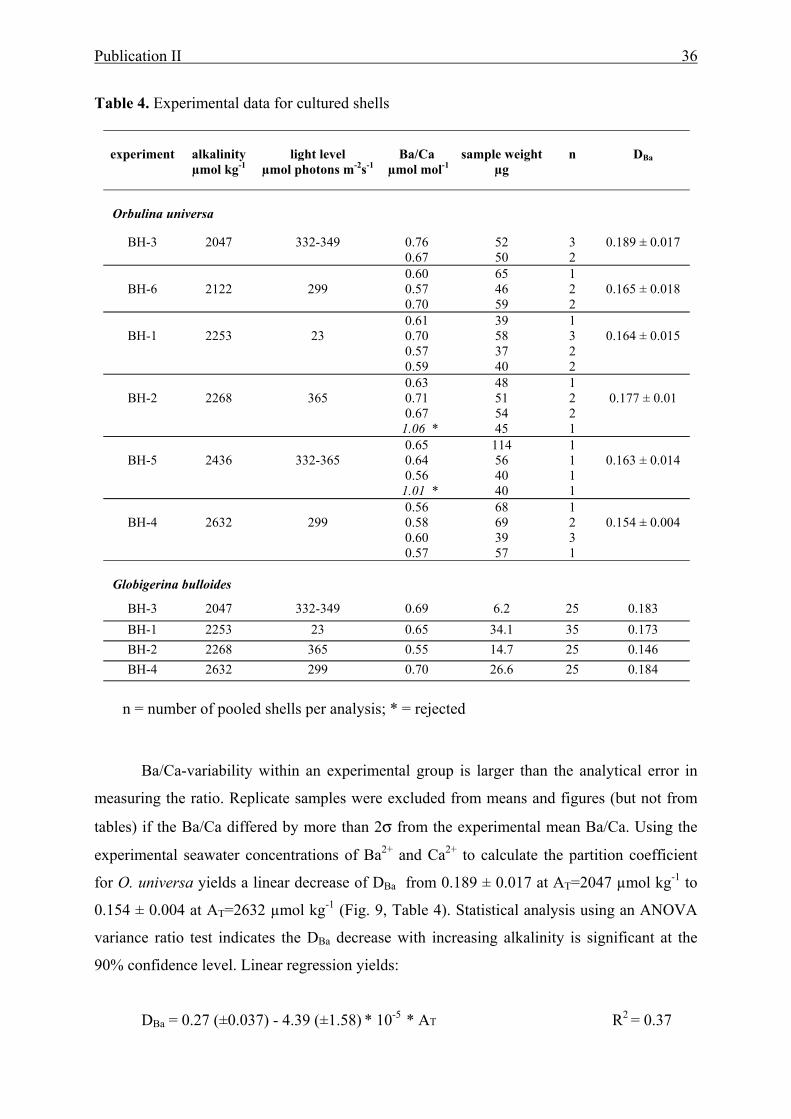
Publication II 36
Table 4. Experimental data for cultured shells
experiment
alkalinity µmol kg-1
light level
µmol photons m-2s-1
Ba/Ca
µmol mol-1
sample weight
µg
n
DBa
Orbulina universa
BH-3 2047 332-349 0.76 52 3 0.189 ± 0.017 0.67 50 2 0.60 65 1
BH-6 2122 299 0.57 46 2 0.165 ± 0.018 0.70 59 2 0.61 39 1
BH-1 2253 23 0.70 58 3 0.164 ± 0.015 0.57 37 2 0.59 40 2 0.63 48 1
BH-2 2268 365 0.71 51 2 0.177 ± 0.01 0.67 54 2 1.06 * 45 1 0.65 114 1
BH-5 2436 332-365 0.64 56 1 0.163 ± 0.014 0.56 40 1 1.01 * 40 1 0.56 68 1
BH-4 2632 299 0.58 69 2 0.154 ± 0.004 0.60 39 3 0.57 57 1
Globigerina bulloides
BH-3 2047 332-349 0.69 6.2 25 0.183 BH-1 2253 23 0.65 34.1 35 0.173 BH-2 2268 365 0.55 14.7 25 0.146 BH-4 2632 299 0.70 26.6 25 0.184
n = number of pooled shells per analysis; * = rejected
Ba/Ca-variability within an experimental group is larger than the analytical error in
measuring the ratio. Replicate samples were excluded from means and figures (but not from
tables) if the Ba/Ca differed by more than 2σ from the experimental mean Ba/Ca. Using the
experimental seawater concentrations of Ba2+ and Ca2+ to calculate the partition coefficient
for O. universa yields a linear decrease of DBa from 0.189 ± 0.017 at AT=2047 µmol kg-1 to
0.154 ± 0.004 at AT=2632 µmol kg-1 (Fig. 9, Table 4). Statistical analysis using an ANOVA
variance ratio test indicates the DBa decrease with increasing alkalinity is significant at the
90% confidence level. Linear regression yields:
DBa = 0.27 (±0.037) - 4.39 (±1.58) * 10-5 * AT R2 = 0.37
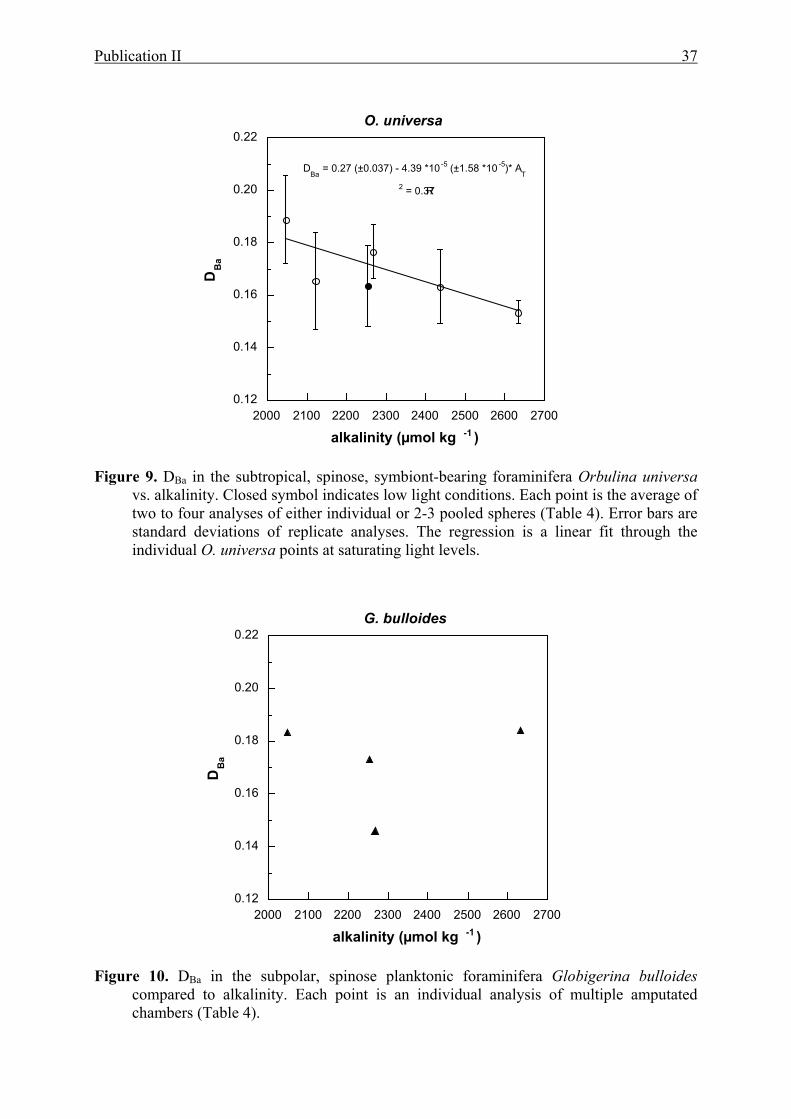
Publication II 37
0.12
0.14
0.16
0.18
0.20
0.22
2000 2100 2200 2300 2400 2500 2600 2700
O. universa
DB
a
alkalinity (µmol kg -1 )
DBa
= 0.27 (±0.037) - 4.39 *10-5 (±1.58 *10 -5)* AT
R2 = 0.37
Figure 9. DBa in the subtropical, spinose, symbiont-bearing foraminifera Orbulina universa vs. alkalinity. Closed symbol indicates low light conditions. Each point is the average of two to four analyses of either individual or 2-3 pooled spheres (Table 4). Error bars are standard deviations of replicate analyses. The regression is a linear fit through the individual O. universa points at saturating light levels.
0.12
0.14
0.16
0.18
0.20
0.22
2000 2100 2200 2300 2400 2500 2600 2700
G. bulloides
DB
a
alkalinity (µmol kg -1 )
Figure 10. DBa in the subpolar, spinose planktonic foraminifera Globigerina bulloides compared to alkalinity. Each point is an individual analysis of multiple amputated chambers (Table 4).

Publication II 38
DBa in G. bulloides ranges between 0.146 and 0.184 (Fig. 10). These partition
coefficients are comparable to the range observed in O. universa shells over the same
alkalinity range. However, given the small number of data points and non-linear distribution
of these data, it is not possible to determine whether or not a relationship between alkalinity
and DBa exists in G. bulloides.
In a final experimental series, we investigated the influence of symbiont
photosynthetic rate (as it varied with light levels) on O. universa DBa in ambient seawater (AT
= 2257 µmol kg-1). Under LL conditions DBa= 0.164 ± 0.015 was slightly lower than under
HL where DBa= 0.177 ± 0.01 (Fig. 9, Table 4). However, these partition coefficients are not
significantly different from each other.
Discussion
Recordability of Ba/Ca in planktonic foraminiferal shells
The average partition coefficients for Ba/Ca in O. universa and G. bulloides found in
this study are DBa= 0.166 ± 0.01 and DBa= 0.171 ± 0.02, respectively. Lea and Spero (1994)
calculated a value of DBa= 0.16 ± 0.01 (regression forced through zero) for cultured O.
universa. The combined partition coefficients of core-top fossil planktonic foraminifera yield
DBa= 0.19 ± 0.05 (Lea and Boyle, 1991). Hence, our values are in good agreement with the
literature.
Globigerina bulloides has not been calibrated for Ba before. Although this species is
symbiont-barren, the DBa is similar to that of the symbiont-bearing O. universa and G.
sacculifer. This finding is in contrast to other elements which are incorporated to different
degrees by symbiont-bearing and symbiont-barren species. For instance, Mashiotta et al.
(1997) observed strong contrasts between O. universa and G. bulloides in the uptake of Cd2+.
They suggested that symbiotic dinoflagellates either influence foraminiferal incorporation of
Cd2+ by sequestering Cd2+ from the calcifying microenvironment of O. universa, or by
photosynthetically enhancing calcification rate leading to Cd2+ exclusion. In contrast, Ba has
no reported biochemical function and in line with the similarity of HL and LL results for O.
universa, the uniformity of our results suggests that symbionts have no influence on the Ba
uptake.
In order to examine DBa in relation to calcification rate we refer to Mashiotta et al.
(1997) who estimated higher calcification rates for O. universa (~3000 µmol m-2 h-1, Lea et

Publication II 39
al., 1995) than for G. bulloides under identical culture conditions (~1700-2400 µmol m-2 h-1).
This difference is not reflected in the uptake of Ba during shell formation and the similarity in
DBa between O. universa and G. bulloides supports the generally held assumption that
species-specific differences are low for planktonic foraminifera and vital effects are not
important for the Ba uptake.
The observed negative correlation between Ba/Ca and alkalinity was not expected
from theoretical arguments which would have predicted a kinetic effect towards higher Ba/Ca
with increasing precipitation rate. To better compare the magnitude of the observed effect
with results on Sr/Ca, we also plotted Ba/Ca versus pH. Total CO2 was constant in our
experiments so that pH and [CO32-] varied almost linearly with the experimental alkalinity
range. While the kinetic effect documented for the pH-dependency of Sr/Ca is ~+1% per 0.1
pH unit (Fig. 4, Lea et al., 1999b), Ba/Ca decreases by ~2% per 0.1 pH unit (Fig. 11b). The
observed effect is thus similar in magnitude but opposite in direction.
The geochemical similarities between Sr and Ba discussed earlier do not extend to the
influence of alkalinity (or pH). This may be due to dissimilarities in sorptive behavior because
of differences in aqueous complexation (e.g. (van Cappellen et al., 1993). Alternatively, since
the mineral growth in foraminifera appears to follow a pattern which is predetermined by the
molecular organisation of the organic template (Langer, 1992), it may be that the organic
matrix controls the selection of certain divalent cations and discriminates against others.
Unfortunately the process of chamber formation and calcification in foraminifera (e.g. Bé et
al., 1979; Hemleben et al., 1977; Spero, 1988) is not very well understood at this level so that
this consideration remains rather speculative.
Paleoceanographical significance
The negative slope determined for O. universa under HL conditions implies that the
error yields a change in DBa of 0.004 (~ 0.017 µmol mol-1 in Ba/Ca) if alkalinity were to
change by 100 µmol kg-1. With such a large alkalinity change, ∆DBa would still be well within
the analytical error of the partition coefficient. For comparison, Lea (1993) found an increase
of 1.8 µmol mol-1 (~50 %) in the Ba/Ca of Circumpolar Deep Water during the last glacial
period. The alkalinity-dependency of Ba/Ca (<1 % per 10 µmol kg-1) would be negligible for
the proposed changes in alkalinity on glacial/interglacial time scales. Even under glacial
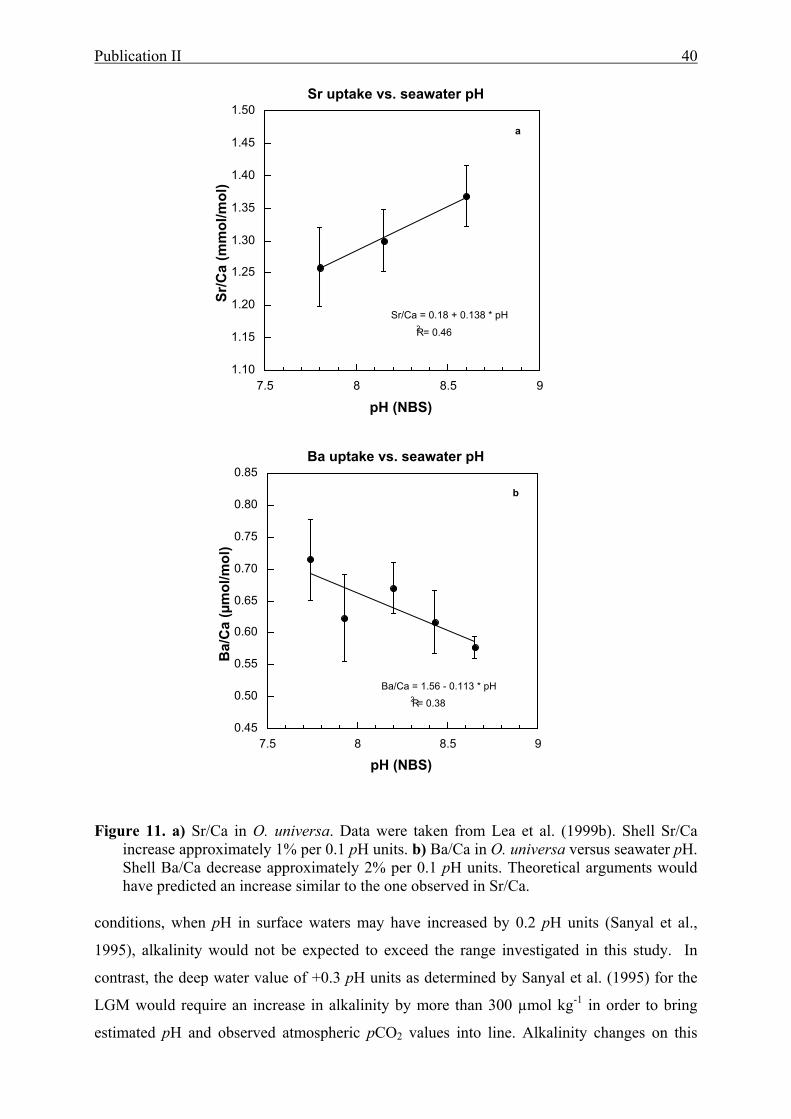
Publication II
1.10
1.15
1.20
1.25
1.30
1.35
1.40
1.45
1.50
7.5 8 8.5 9
Sr uptake vs. seawater pH
Sr/C
a (m
mol
/mol
)
pH (NBS)
Sr/Ca = 0.18 + 0.138 * pH R2 = 0.46
a
40
0.45
0.50
0.55
0.60
0.65
0.70
0.75
0.80
0.85
7.5 8 8.5 9
Ba uptake vs. seawater pH
Ba/
Ca
(µm
ol/m
ol)
pH (NBS)
Ba/Ca = 1.56 - 0.113 * pH
R2 = 0.38
b
Figure 11. a) Sr/Ca in O. universa. Data were taken from Lea et al. (1999b). Shell Sr/Ca increase approximately 1% per 0.1 pH units. b) Ba/Ca in O. universa versus seawater pH. Shell Ba/Ca decrease approximately 2% per 0.1 pH units. Theoretical arguments would have predicted an increase similar to the one observed in Sr/Ca.
conditions, when pH in surface waters may have increased by 0.2 pH units (Sanyal et al.,
1995), alkalinity would not be expected to exceed the range investigated in this study. In
contrast, the deep water value of +0.3 pH units as determined by Sanyal et al. (1995) for the
LGM would require an increase in alkalinity by more than 300 µmol kg-1 in order to bring
estimated pH and observed atmospheric pCO2 values into line. Alkalinity changes on this

Publication II 41
order could lead to a potentially large Ba/Ca change. However, the deep water pH estimate is
based on a sample of mixed benthic foraminifera species and as such not very reliable. In fact,
recent data by Anderson and Archer (2002) argue against such a strong increase in pH.
Conclusions
Our results demonstrate that species variability and symbiont effects are not important
for planktonic foraminifera. Orbulina universa and G. bulloides incorporate Ba in a similar
ratio to seawater concentration. Water column alkalinity and Ba/Ca are at most weakly linked
for these species, and the effect is too small to be significant for paleoceanographic
reconstructions. Alkalinity, the closest related component to the oceanic barium cycle, can
therefore be extensively ignored as a potential factor directly affecting the incorporation of
Ba2+ into planktonic foraminiferal calcite.
Acknowledgements
We thank the staff of the Catalina Marine Science Center for providing facilities to
make this work possible. We gratefully acknowledge field help by Laurie Juranek, Megan
Thomas, Heidi Iverson and the Catalina dive crew. The experiments could not have been
successfully completed without the laboratory work of Georges Paradis and Dotti Pak.
Support and comments from Christoph Völker and Hubertus Fischer are greatly appreciated.
This research was supported by DAAD grant D/00 20292 (BH), NEBROC (BH), NSF grant
OCE-9907044 (ADR), OCE-9906821 (DWL), OCE-9729203 and OCE-9903632 (HJS).

Publication III 42
Publication III
Post-depositional effects on trace metals and stable isotopes in foraminiferal calcite – Evidence from dissolution experiments
Bärbel Hönisch, Jelle Bijma, Nikolaus Gussone, Howard J. Spero, Dirk Nürnberg, David W. Lea and A. Eisenhauer
in preparation
.......................................................................................................................................................
Abstract
Sediment observations and laboratory dissolution experiments have increased
paleoceanographers' interest in post-depositional changes of chemical proxies recorded in
foraminifera shells. While previous studies are limited by a number of uncertainties, young
proxies like δ11B and δ44Ca have not yet been regarded in the light of dissolution. We used well
preserved planktonic foraminifera species Globigerinoides sacculifer and Neogloboquadrina
pachyderma (sinistral coiling) to partially dissolve them under controlled conditions in the
laboratory, simulating the natural processes at the seafloor. In addition to known dissolution
effects on Mg/Ca, δ18O and δ13C, we observe significant effects on Sr/Ca and δ11B on the order
of glacial/interglacial changes. Discussing previously hypothesized explanations for partial
dissolution, it will be shown that the overall process is not yet fully understood. While δ18O,
δ13C, Mg/Ca and maybe Sr/Ca can be explained by preferential dissolution of ontogenetic calcite
and a shift of the bulk shell chemistry to calcite secreted at greater depth, δ11B and δ44Ca seem to
be inconsistent with such a process and other explanations have to be investigated instead. Mg
impurities were found to be insufficient to reduce the overall stability of certain foraminiferal
shell components and increasing Sr/Ca ratios demonstrate that crystal impurities are not
necessarily more prone to dissolution. The microstructural breakdown of shell surfaces is
systematic and may be useful to estimate the preservation state of shells deposited in sediments.
Dissolution drives deep fissures into the shell wall, indicating that corrosion does not only
remove outer layers of a shell but also increases shell porosity. The resulting exposition of

Publication III 43
otherwise protected lattice areas possibly allows certain elements to be leached out. Dissolution
effects appear to be species-specific and depend on the physico-chemical gradients encountered
by vertically migrating foraminifera at different locations.
Introduction
The chemical composition of biogenic carbonates derived from marine sediments is
routinely used for paleoceanographic and paleoclimatic reconstructions. One of the basic
assumptions is that the primary signal remains unaltered after burial in the geological archive.
However, observations on sediment cores reveal significant variability in the shell chemistry of
planktonic foraminifera that can not be linked to oceanographic or climatologic changes in the
organism’s habitat. Part of the observed variability is assumed to be caused by dissolution. In
order to test this assumption, dissolution studies have focussed primarily on two experimental
approaches: (1) empirical observations on carbonate samples deposited in sediments at various
depths (e.g. Berger, 1967; Brown and Elderfield, 1996; Honjo and Erez, 1978; McCorkle et al.,
1995; Zachos et al., 1994); and (2) the determination of CaCO3 dissolution kinetics and changing
chemical composition in the laboratory (e.g. Bé et al., 1974; Bender et al., 1975; Boyle, 1988a;
Caron et al., 1990; Lea and Boyle, 1991; Lorens et al., 1977). Based on these and other
experiments, several hypotheses have been proposed:
1. Shell Mg increases the susceptibility to dissolution (Brown and Elderfield, 1996; Lorens et al.,
1977; Savin and Douglas, 1973).
2. Dissolution selectively removes the more soluble individuals and thus reduces their
contribution to the chemical bulk signal (Erez, 1979b).
3. Elements that are distributed homogeneously throughout a shell are not affected by dissolution
(Bender et al., 1975; Lea and Boyle, 1991; McCorkle et al., 1995).
4. Selective dissolution of ontogenetic calcite shifts bulk Mg/Ca and δ18O toward the chemistry
of the secondary crust (Brown and Elderfield, 1996; Dekens et al., 2002; Lohmann, 1995;
Rosenthal et al., 2000; Russell et al., 1994).
5. Although dissolution alters Mg/Ca and δ18O, the initial relationship remains constant such
that paired analyses of δ18O and Mg/Ca can be used to reconstruct the δ18O of seawater
(δ18Ow) (Rosenthal et al., 2000).

Publication III 44
Although it seems a reasonable assumption that dissolution causes the observed
variability, the impact of dissolution has not yet been quantified. Whereas interpretation of
results from sediment studies contains uncertainties about the real dissolution state,
recrystallization processes and mixing of shells from different time intervals, the use of dilute
acid in laboratory dissolution experiments results in a much more aggressive dissolution than at
the seafloor. We have carried out dissolution experiments under controlled laboratory conditions
in natural seawater whose carbonate chemistry has been manipulated. Demonstrably well
preserved shells of the two planktonic species Globigerinoides sacculifer and Neogloboquadrina
pachyderma (sinistral) have been partially dissolved. From these shells we present data on
Mg/Ca and Sr/Ca as well as stable isotope values such as δ18O, δ13C, δ11B and δ44Ca. In addition
to the bulk shell composition determined by ICP-MS (inductively coupled plasma mass
spectrometry), an electron microprobe was used to measure Mg/Ca in different parts of the
calcite of individual tests.
Methodology
Samples
We selected two sediment cores with unusually well preserved foraminifera shells: The
site of sediment core M 23352 (70°0‘N, 12°25‘W, 1819 m) is thought to have been situated
above the lysocline during the past 5 glacial-interglacial cycles. The small sieve size fraction
(279-309 µm) of the polar planktonic species N. pachyderma (sin.) was hand-picked from the 57
cm sediment horizon, which corresponds to marine isotope stage (MIS) 2. Scanning electron
microscopy (SEM) examination, as well as weight and reflectance analyses revealed that these
glacial foraminifera shells are perfectly preserved (Helmke and Bauch, 2002).
Box core GeoB 5815 (29°30,64‘N, 34°58,98‘E) was collected from a water depth of 326
m. At this locality, water column and pore waters are strongly supersaturated with respect to
calcite as reflected in the abundant occurrence of aragonitic pteropod shells within the sediment.
Because the sedimentation rate in this region is 20-30 cm ka-1 [pers. comm. J. Pätzold,
University of Bremen], one can infer that the samples are mostly Holocene and contamination
with glacial individuals is rare. Globigerinoides sacculifer was hand-picked from the 10-20 cm
sediment horizon. The shells were taken from the 315-450 µm and 450-500 µm sieve size
fraction. To remove clay and other adhering particles, the shells were cleaned ultrasonically in
artificial seawater (pH 8) and subsequently rinsed in ultrapure water (pH >8) to avoid the
crystallization of salts.

Publication III 45
Experimental Methods
The dissolution experiment presented here was designed to simulate natural conditions at
the seafloor. Adjusting the saturation state under laboratory conditions (15°C, atmospheric
pressure) was accomplished by reducing alkalinity and pH at natural concentrations of dissolved
inorganic carbon (DIC): Ultrapure 9.5 N HCl was added to 0.45 µm filtered North Sea water at
2137 µmol kg-1 DIC, and 2348 µmol kg-1 alkalinity. Four saturation states were prepared: 120,
90, 70 and 50% with respect to calcite (Table 5). Carbonate chemistry was monitored by
measuring DIC and alkalinity for the initial water batches and finally for each bottle separately.
Measurements were done coulometrically and by Gran titration, respectively. Saturation state
calculations are based on the CO2SYS program provided by Lewis and Wallace (1998), using K1
and K2 as determined by Roy et al. (1993) and KSO4 as determined by Dickson (1990). The two
size fractions of G. sacculifer and the shells of N. pachyderma (sin.) were incubated in individual
experiments. Due to dissolution in the closed system, alkalinity and calcite saturation increased
during the experiment.
Whole specimens were selected from the ultrasonically cleaned foraminifera shells for
the dissolution experiments. For G. sacculifer, five subsamples of similar weight and a fixed
number of shells (i.e. 35 shells of the large fraction and 100 shells of the small fraction) were
prepared for each saturation state. For the determination of the boron isotopic composition two
larger samples with 500 shells each were selected from the small G. sacculifer fraction and
incubated at 70 and 50% saturation. For N. pachyderma (sin.), no more than 600 shells were
available. These were split into groups of 100 shells and incubated at 70% saturation. Shells
were dried for 4 hours at 45°C and left in a desiccator overnight to cool. The shells were
weighed on an SARTORIUS ultramicro balance with a precision of ±1 µg.
The subsamples were incubated individually in acid leached 1-L PE-bottles. To avoid the
accumulation of shells and the formation of microzones with slightly increased saturations
(“artificial sediment“, Morse et al., 1979), the bottles were fit on a plankton wheel at lowest
spinning velocity (~1-1.5 turns/min). The incubation of all subsamples started at the same time,
while completion of each experiment depended on the different saturation states. Total
incubation time for individual samples thus lasted between 5 and 77 days (Table 5). Upon
termination of the experiment, the foraminifera were rinsed in ultrapure water, dried as described
above, weighed and counted to determine the loss of calcite per shell.

Publication III 46
Sample Preparation and Analytical Techniques
Stable oxygen and carbon isotopes
Stable isotope analyses of the calcite samples at the University of California, Davis,
required ≥15 µg of sample for precise measurements. Single shell analyses on G. sacculifer
resulted in an isotopic range that was much larger than the analytical uncertainty. Due to this
large natural variability we pooled 50 shells, crushed them and analyzed smaller subsamples.
Care was taken to select shells within 50 µm size increments for pooled shell analyses to
minimize ontogenetic factors that affect carbon and oxygen isotopes in foraminifera shells (e.g.
Berger et al., 1978). Duplicate analyses of three such samples of undissolved shells gave
reproducible results within the analytical uncertainty.
Depending on the dissolution state of N. pachyderma (sin.), 2-4 shells were pooled to
yield sufficient carbonate. Between 2 and 6 replicate analyses were conducted for each
dissolution state.
Samples were roasted at 375°C in vacuo for 30 minutes and analyzed with a Fisons
Optima isotope ratio mass spectrometer using an Isocarb common acid bath autocarbonate
system at 90°C. Results are given in delta notation (δ13C = [(13C/12Csample/13C/12Cstd–1)*1000]
and δ18O = [(18O/16Osample/18O/16Ostd–1)*1000]) relative to the Vienna Pee Dee belemnite
standard (V-PDB). The external precision of the measurements is ±0.06‰ and ±0.05‰ for δ18O
and δ13C respectively, based on repeat analyses of NBS 19 standard.
Minor elements
Preparation of foraminifera for trace metal analyses followed the rigorous multistep
cleaning method established by Boyle (1981) and Boyle and Keigwin (1985/1986). As the initial
design of the experiment included Ba/Ca, U/Ca and Cd/Ca, treatment with an alkaline chelating
solution was added according to the procedure by Lea and Boyle (1989) and Lea (1993). Briefly,
50 shells per sample were gently crushed and split in two. Half of each sample was cleaned using
the above named process. In order to test the corrosivity of the cleaning method, the other half
was repeatedly ultrasonicated in methanol and ultrapure water and then transferred to acid
leached microcentrifuge vials. Further cleaning steps were omitted for these splits.
Sample analysis followed the multi-element ICP-MS method previously described by Lea
and Martin (1996). After cleaning, 20-250 µg of purified foraminifera shells were dissolved in
dilute HNO3 solution containing calibrated concentrations of 25Mg, 45Sc, and 89Y. The solutions

Publication III 47
were aspirated into a Finnigan Element 2 high resolution magnetic sector ICP-MS and Mg, Ca
and Sr acquired by analog detector mode. The metal to Ca2+ ratios were estimated by isotope
dilution (25Mg/24Mg) and internal standard calculation (45Sc/48Ca and 89Y/88Sr). Na/Ca was
determined to confirm that the hydrogen peroxide – sodium hydroxide solution used in sample
preparation was completely rinsed out. Several analyses of a consistency standard with trace
metal and Ca2+ concentrations similar to the foraminiferal samples had a standard deviation of
0.83% for Mg/Ca and 0.7% for Sr/Ca (pers. comm. G. Paradis, UC Santa Barbara). Analyses
were performed at the University of California, Santa Barbara.
Additional Mg/Ca analyses were carried out using a wavelength-dispersive, automated
four-spectrometer CAMECA electronmicroprobe. Measurements were done according to the
method described by Nürnberg (1995). Briefly, shells of G. sacculifer (315-450 µm size fraction)
were rinsed in distilled water, embedded in resin, and ground and polished in order to reveal
fresh, relief-free calcite surfaces of chamber walls. The electron beam was focussed on spots
approximately 2-4 µm in diameter. Element concentrations in the ppm-range were detected
quantitatively, calculated stoichiometrically in oxide form, and reported as element/Ca ratios.
Mg-Periclase (synthetic) and Ca-Wollastonite (natural) served as standards. Between 26 and 54
spot analyses have been carried out on each of 10 individual shells. Measurements were done at
GEOMAR in Kiel, Germany.
Stable boron isotopes
Shells were crushed and bleached with 4-6% sodium hypochlorite to remove organic
matter. After at least 24 hours in bleach, the samples were rinsed 10 times in ultrapure water,
ultrasonicated and centrifuged to rinse out the bleach and remove soluble salts and adsorbed
boron. The cleaned carbonate was dissolved in 2N quartz distilled HCl, loaded on a rhenium
zone refined filament, and 1 µl of boron-free seawater was added to enhance ionization and
suppress fractionation (Hemming and Hanson, 1994). Isotope data were collected on a Finnigan
MAT 262 RPQ+ Thermal Ionization Mass Spectrometer (TIMS) at GEOMAR in Kiel, Germany.
The BO2- ion method followed the procedure outlined in Sanyal et al. (1996) and Sanyal et al.
(1997). Each sample was run at least 4 times. Analysis temperature was constant at 940 ± 10°C.
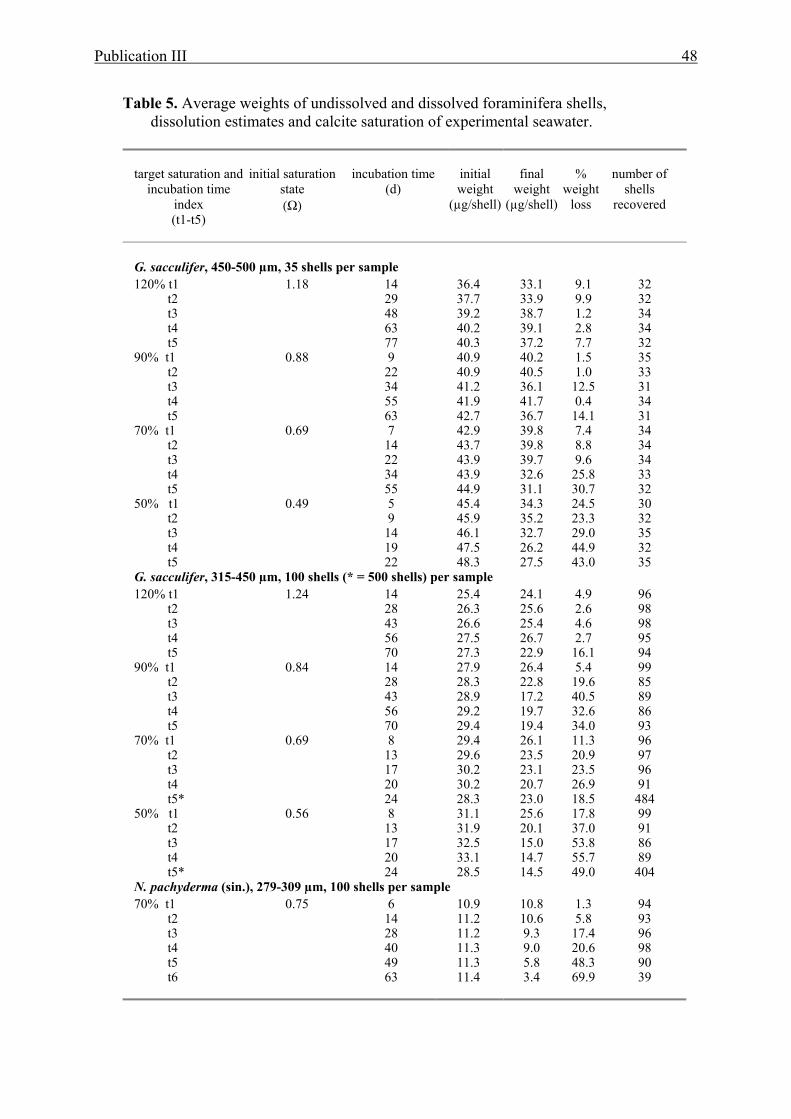
Publication III 48
Table 5. Average weights of undissolved and dissolved foraminifera shells, dissolution estimates and calcite saturation of experimental seawater.
target saturation and incubation time
index
initial saturation
state incubation time initial
weight (µg/shell)
final weight
(µg/shell)
% weight
loss
number of shells
recovered (d)
(Ω) (t1-t5)
G. sacculifer, 450-500 µm, 35 shells per sample 120% t1 1.18 14 36.4 33.1 9.1 32 t2 29 37.7 33.9 9.9 32 t3 48 39.2 38.7 1.2 34 t4 63 40.2 39.1 2.8 34
t5 77 40.3 37.2 7.7 32 90% t1 0.88 9 40.9 40.2 1.5 35 t2 22 40.9 40.5 1.0 33 t3 34 41.2 36.1 12.5 31 t4 55 41.9 41.7 0.4 34 t5 63 42.7 36.7 14.1 31 70% t1 0.69 7 42.9 39.8 7.4 34 t2 14 43.7 39.8 8.8 34 t3 22 43.9 39.7 9.6 34 t4 34 43.9 32.6 25.8 33 t5 55 44.9 31.1 30.7 32 50% t1 0.49 5 45.4 34.3 24.5 30 t2 9 45.9 35.2 23.3 32 t3 14 46.1 32.7 29.0 35 t4 19 47.5 26.2 44.9 32 t5 22 48.3 27.5 43.0 35 G. sacculifer, 315-450 µm, 100 shells (* = 500 shells) per sample 120% t1 1.24 14 25.4 24.1 4.9 96 t2 28 26.3 25.6 2.6 98 t3 43 26.6 25.4 4.6 98 t4 56 27.5 26.7 2.7 95 t5 70 27.3 22.9 16.1 94 90% t1 0.84 14 27.9 26.4 5.4 99 t2 28 28.3 22.8 19.6 85 t3 43 28.9 17.2 40.5 89 t4 56 29.2 19.7 32.6 86 t5 70 29.4 19.4 34.0 93 70% t1 0.69 8 29.4 26.1 11.3 96 t2 13 29.6 23.5 20.9 97 t3 17 30.2 23.1 23.5 96 t4 20 30.2 20.7 26.9 91 t5* 24 28.3 23.0 18.5 484 50% t1 0.56 8 31.1 25.6 17.8 99 t2 13 31.9 20.1 37.0 91 t3 17 32.5 15.0 53.8 86 t4 20 33.1 14.7 55.7 89 t5* 24 28.5 14.5 49.0 404 N. pachyderma (sin.), 279-309 µm, 100 shells per sample 70% t1 0.75 6 10.9 10.8 1.3 94 t2 14 11.2 10.6 5.8 93 t3 28 11.2 9.3 17.4 96 t4 40 11.3 9.0 20.6 98 t5 49 11.3 5.8 48.3 90 t6 63 11.4 3.4 69.9 39

Publication III 49
To rule out isobaric interferences on mass 42 due to residual organic matter
(12C14N16O-ions), we checked for mass 26 (12C14N-ions) during each measurement. No
interferences were detected. The 11B/10B ratio was corrected for isotopic interferences on mass
43 (10B16O17O-ions) by subtraction of 0.00078 from the 43/42 ratio (Spivack and Edmond,
1986). Results are reported in delta notation (δ11B=[(11B/10Bsample/11B/10Bstd–1) * 1000]
relative to the NBS 951 boric acid standard. Repeated analyses of natural seawater used as a
laboratory standard resulted in an average value of 39.5 ± 0.3‰.
Determination of δ44Ca
For N. pachyderma (sin.), 2-4 shells per dissolution state were selected and pooled.
Due to the strong seasonal signal in δ18O, the number of individuals of G. sacculifer (450-500
µm size fraction) was raised to 15. Three samples of each, undissolved and dissolved G.
sacculifer shells, were prepared for isotope analysis. Sample analysis followed the method
established by Nägler et al. (2000). Briefly, samples were dissolved in 2N ultrapure HCl and
spiked with a 43Ca-48Ca double spike. Samples were loaded on rhenium filaments and
measured on the Finnigan MAT 262 RPQ+ TIMS at GEOMAR. Each sample was run 1-3
times. Data are presented in delta notation relative to a natural CaF2 standard:
(δ44Ca=[(44Ca/40Casample/44Ca/40Ca std–1)*1000]. External reproducibility is 0.1‰.
Results Dissolution state and solubility
The respective dissolution state of each sample is reported in terms of weight loss [%]
per shell. Weight loss data and saturation states of the experimental seawater are shown in
Table 5. The calcite dissolution rates obtained in this study are in good agreement with
previous research and show both, intra- and interspecific variability in solution susceptibility.
The larger size fraction of G. sacculifer proved more solution resistant than the smaller size
fraction and G. sacculifer was generally more prone to dissolution than N. pachyderma (sin.).
Similar results have been obtained by Berger (1967; 1970) in studies of differential
dissolution on foraminiferal assemblages within the natural environment.
Visual inspection of the shells under the light microscope at 66-fold magnification
does not allow the detection and quantification of partial dissolution. Already at 5-6% weight
loss shells loose their shiny appearance and get opaque. Until disintegration starts and the
texture of severely dissolved shells gets almost sponge-like, wall microstructural changes
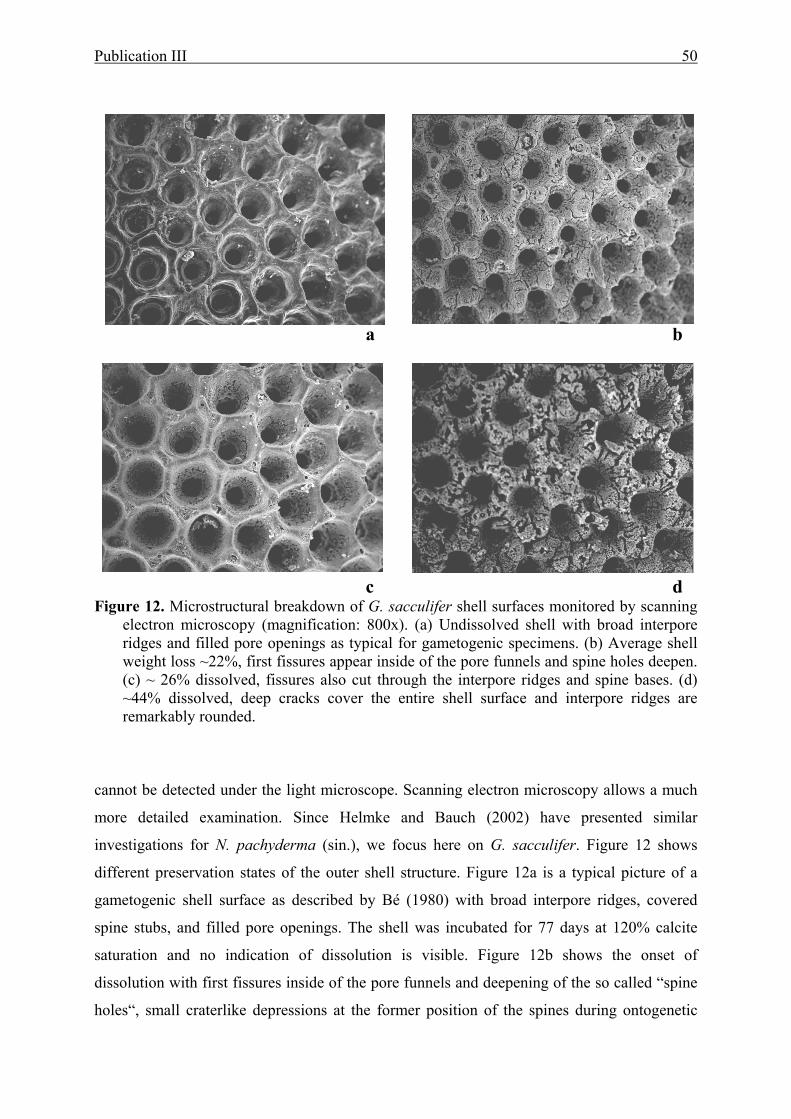
Publication III 50
a b
c d Figure 12. Microstructural breakdown of G. sacculifer shell surfaces monitored by scanning
electron microscopy (magnification: 800x). (a) Undissolved shell with broad interpore ridges and filled pore openings as typical for gametogenic specimens. (b) Average shell weight loss ~22%, first fissures appear inside of the pore funnels and spine holes deepen. (c) ~ 26% dissolved, fissures also cut through the interpore ridges and spine bases. (d) ~44% dissolved, deep cracks cover the entire shell surface and interpore ridges are remarkably rounded.
cannot be detected under the light microscope. Scanning electron microscopy allows a much
more detailed examination. Since Helmke and Bauch (2002) have presented similar
investigations for N. pachyderma (sin.), we focus here on G. sacculifer. Figure 12 shows
different preservation states of the outer shell structure. Figure 12a is a typical picture of a
gametogenic shell surface as described by Bé (1980) with broad interpore ridges, covered
spine stubs, and filled pore openings. The shell was incubated for 77 days at 120% calcite
saturation and no indication of dissolution is visible. Figure 12b shows the onset of
dissolution with first fissures inside of the pore funnels and deepening of the so called “spine
holes“, small craterlike depressions at the former position of the spines during ontogenetic
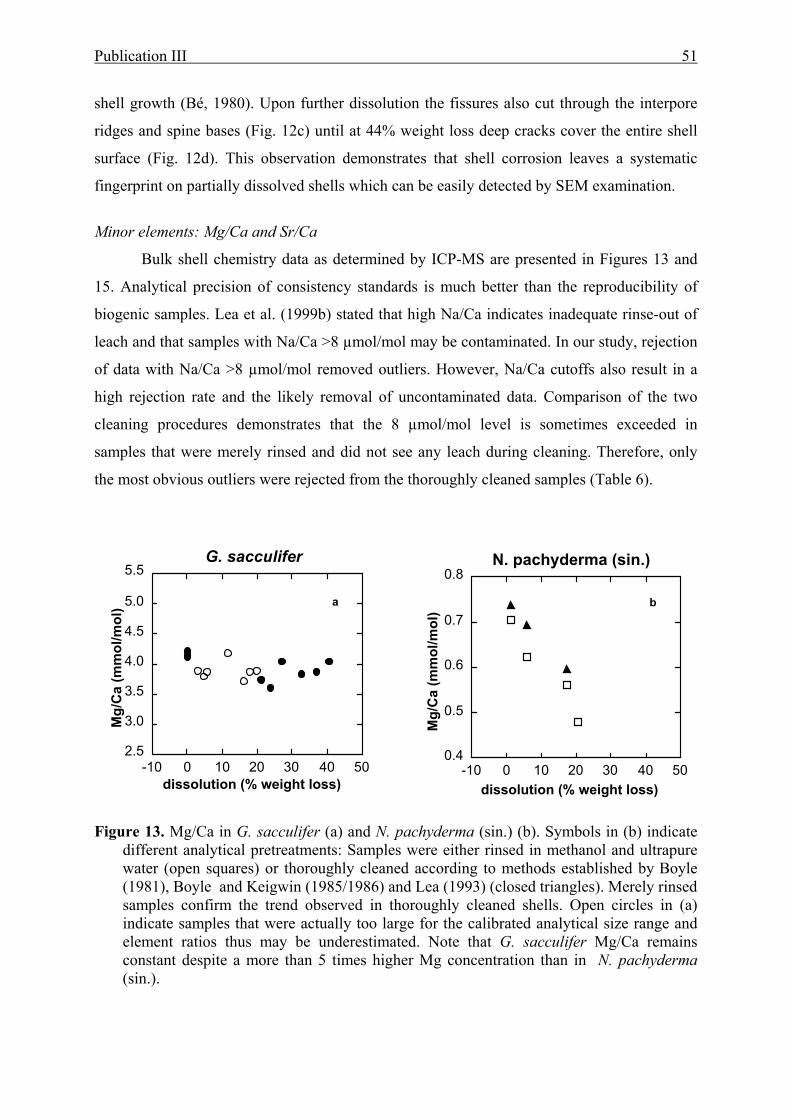
Publication III 51
shell growth (Bé, 1980). Upon further dissolution the fissures also cut through the interpore
ridges and spine bases (Fig. 12c) until at 44% weight loss deep cracks cover the entire shell
surface (Fig. 12d). This observation demonstrates that shell corrosion leaves a systematic
fingerprint on partially dissolved shells which can be easily detected by SEM examination.
Minor elements: Mg/Ca and Sr/Ca
Bulk shell chemistry data as determined by ICP-MS are presented in Figures 13 and
15. Analytical precision of consistency standards is much better than the reproducibility of
biogenic samples. Lea et al. (1999b) stated that high Na/Ca indicates inadequate rinse-out of
leach and that samples with Na/Ca >8 µmol/mol may be contaminated. In our study, rejection
of data with Na/Ca >8 µmol/mol removed outliers. However, Na/Ca cutoffs also result in a
high rejection rate and the likely removal of uncontaminated data. Comparison of the two
cleaning procedures demonstrates that the 8 µmol/mol level is sometimes exceeded in
samples that were merely rinsed and did not see any leach during cleaning. Therefore, only
the most obvious outliers were rejected from the thoroughly cleaned samples (Table 6).
2.5
3.0
3.5
4.0
4.5
5.0
5.5
-10 0 10 20 30 40 5
G. sacculifer
Mg/
Ca
(mm
ol/m
ol)
dissolution (% weight loss)
a
00.4
0.5
0.6
0.7
0.8
-10 0 10 20 30 40 50
N. pachyderma (sin.)
Mg/
Ca
(mm
ol/m
ol)
dissolution (% weight loss)
b
Figure 13. Mg/Ca in G. sacculifer (a) and N. pachyderma (sin.) (b). Symbols in (b) indicate
different analytical pretreatments: Samples were either rinsed in methanol and ultrapure water (open squares) or thoroughly cleaned according to methods established by Boyle (1981), Boyle and Keigwin (1985/1986) and Lea (1993) (closed triangles). Merely rinsed samples confirm the trend observed in thoroughly cleaned shells. Open circles in (a) indicate samples that were actually too large for the calibrated analytical size range and element ratios thus may be underestimated. Note that G. sacculifer Mg/Ca remains constant despite a more than 5 times higher Mg concentration than in N. pachyderma (sin.).
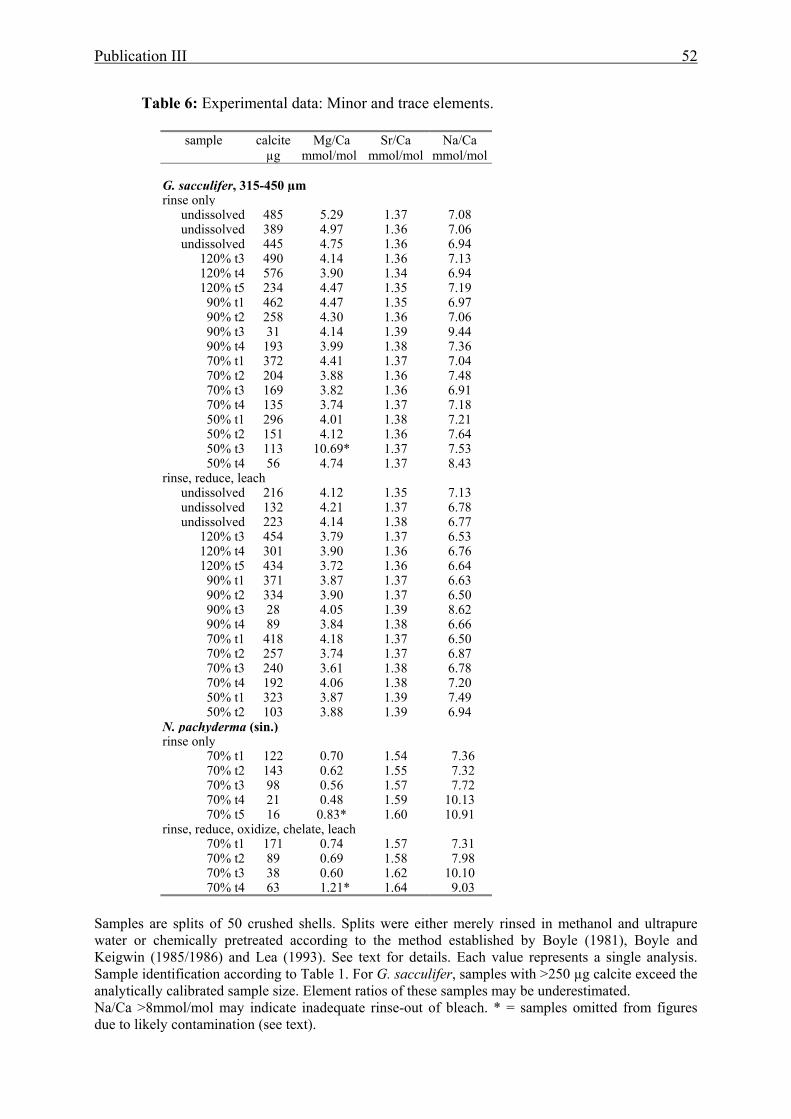
Publication III 52
Table 6: Experimental data: Minor and trace elements.
sample calcite µg
Mg/Ca mmol/mol
Sr/Ca mmol/mol
Na/Ca mmol/mol
G. sacculifer, 315-450 µm rinse only
undissolved 485 5.29 1.37 7.08undissolved 389 4.97 1.36 7.06undissolved 445 4.75 1.36 6.94
120% t3 490 4.14 1.36 7.13120% t4 576 3.90 1.34 6.94120% t5 234 4.47 1.35 7.19
90% t1 462 4.47 1.35 6.9790% t2 258 4.30 1.36 7.0690% t3 31 4.14 1.39 9.4490% t4 193 3.99 1.38 7.3670% t1 372 4.41 1.37 7.0470% t2 204 3.88 1.36 7.4870% t3 169 3.82 1.36 6.9170% t4 135 3.74 1.37 7.1850% t1 296 4.01 1.38 7.2150% t2 151 4.12 1.36 7.6450% t3 113 10.69* 1.37 7.5350% t4 56 4.74 1.37 8.43
rinse, reduce, leach undissolved 216 4.12 1.35 7.13undissolved 132 4.21 1.37 6.78undissolved 223 4.14 1.38 6.77
120% t3 454 3.79 1.37 6.53120% t4 301 3.90 1.36 6.76120% t5 434 3.72 1.36 6.64
90% t1 371 3.87 1.37 6.6390% t2 334 3.90 1.37 6.5090% t3 28 4.05 1.39 8.6290% t4 89 3.84 1.38 6.6670% t1 418 4.18 1.37 6.5070% t2 257 3.74 1.37 6.8770% t3 240 3.61 1.38 6.7870% t4 192 4.06 1.38 7.2050% t1 323 3.87 1.39 7.4950% t2 103 3.88 1.39 6.94
N. pachyderma (sin.) rinse only
70% t1 122 0.70 1.54 7.3670% t2 143 0.62 1.55 7.3270% t3 98 0.56 1.57 7.7270% t4 21 0.48 1.59 10.1370% t5 16 0.83* 1.60 10.91
rinse, reduce, oxidize, chelate, leach 70% t1 171 0.74 1.57 7.3170% t2 89 0.69 1.58 7.9870% t3 38 0.60 1.62 10.1070% t4 63 1.21* 1.64 9.03
Samples are splits of 50 crushed shells. Splits were either merely rinsed in methanol and ultrapure water or chemically pretreated according to the method established by Boyle (1981), Boyle and Keigwin (1985/1986) and Lea (1993). See text for details. Each value represents a single analysis. Sample identification according to Table 1. For G. sacculifer, samples with >250 µg calcite exceed the analytically calibrated sample size. Element ratios of these samples may be underestimated. Na/Ca >8mmol/mol may indicate inadequate rinse-out of bleach. * = samples omitted from figures due to likely contamination (see text).

Publication III 53
The Mg/Ca ratio in shells of N. pachyderma (sin.) is clearly reduced upon dissolution
(Fig. 13b). Applying the sediment core-top calibration of Nürnberg (1995) for left coiling N.
pachyderma (T°C = ln (Mg/Ca / 0.55) / 0.099), a temperature estimate of 3°C for least
dissolved shells and 0.9°C for shells that lost 17% of their initial weight can be calculated.
The temperature estimate is thus lowered by ~2 K.
Some samples were accidentally heavier than the weight range for which the method
was calibrated. The element ratios of these samples may be underestimated and we highlight
them in all plots as open symbols. However, a trend towards underestimated values was not
observed for any of the elements investigated. Data on N. pachyderma (sin.) are not affected
by this problem since all samples were within the calibrated weight range.
For G. sacculifer, no significant dissolution effect on Mg/Ca can be observed (Fig.
13a). The average Mg/Ca is 3.93 ± 0.18 mmol/mol. For comparison, microprobe analyses on
10 G. sacculifer shells yield clearly heterogeneous Mg/Ca distributions in individual shells.
Ratios for outer shell calcite average 4.61 ± 0.47 mmol/mol and 3.18 ± 0.54 mmol/mol for
inner calcite (Fig. 14). Since microprobe analyses are carried out without elaborate
preparation and cleaning procedures, it could be argued that the enrichment is due to
contaminating phases. A simple mass balance calculation based on the assumption that G.
sacculifer adds ~28% (Bé, 1980) of its shell weight during gametogenesis, yields a bulk shell
Mg/Ca ratio of 3.58 ± 0.52 mmol/mol. Given the large seasonal temperature variability at the
core site and the low number of shells investigated using microprobe compared to ICP-MS
measurements, the similarity between the two approaches is promising.
0 1 2 3 4 5 6 7
123456789
10G. sacculifer
Mg/Ca (mmol/mol)
shel
l No.
Figure 14. Averages of Mg/Ca in inner (grey bars) and outer calcite (black bars) of 10 G. sacculifer shells as determined by microprobe analysis of wall profiles. Each value is the average of 15-28 spot analyses. Approximately 20% higher Mg concentrations are encountered in outer compared to inner calcite.
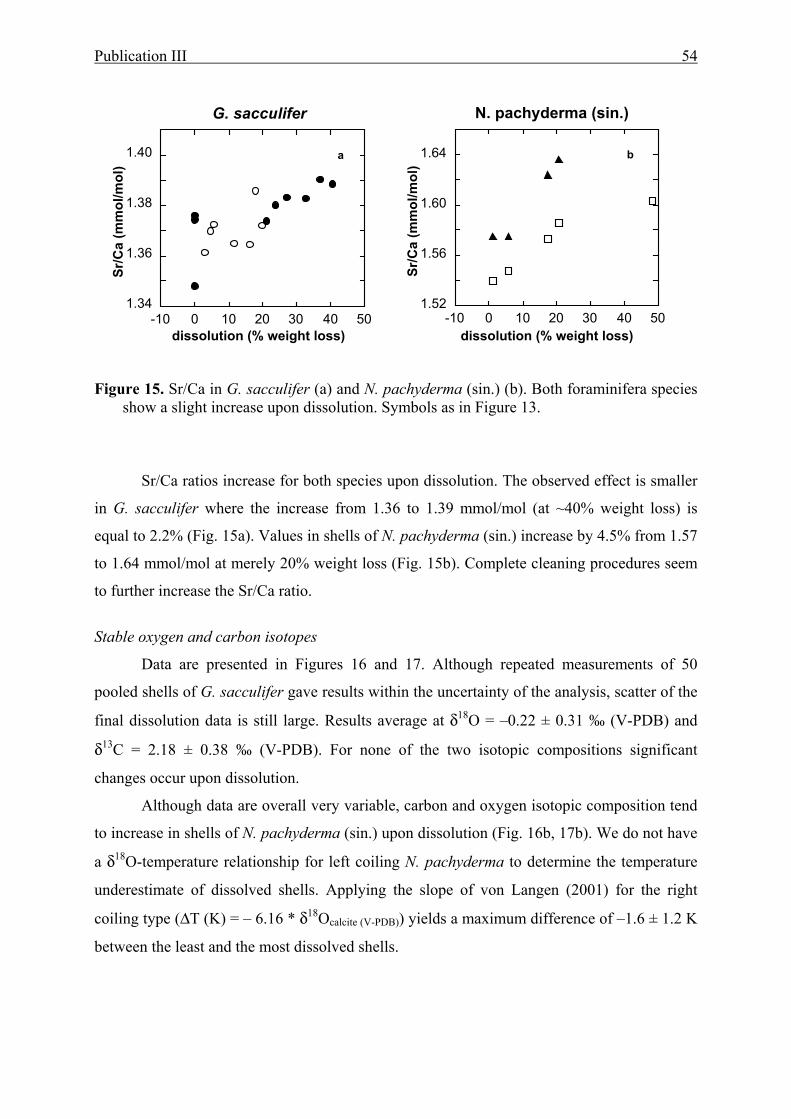
Publication III 54
1.52
1.56
1.60
1.64
-10 0 10 20 30 40 50
N. pachyderma (sin.)
Sr/C
a (m
mol
/mol
)
dissolution (% weight loss)
b
1.34
1.36
1.38
1.40
-10 0 10 20 30 40 50
G. sacculiferSr
/Ca
(mm
ol/m
ol)
dissolution (% weight loss)
a
Figure 15. Sr/Ca in G. sacculifer (a) and N. pachyderma (sin.) (b). Both foraminifera species
show a slight increase upon dissolution. Symbols as in Figure 13.
Sr/Ca ratios increase for both species upon dissolution. The observed effect is smaller
in G. sacculifer where the increase from 1.36 to 1.39 mmol/mol (at ~40% weight loss) is
equal to 2.2% (Fig. 15a). Values in shells of N. pachyderma (sin.) increase by 4.5% from 1.57
to 1.64 mmol/mol at merely 20% weight loss (Fig. 15b). Complete cleaning procedures seem
to further increase the Sr/Ca ratio.
Stable oxygen and carbon isotopes
Data are presented in Figures 16 and 17. Although repeated measurements of 50
pooled shells of G. sacculifer gave results within the uncertainty of the analysis, scatter of the
final dissolution data is still large. Results average at δ18O = –0.22 ± 0.31 ‰ (V-PDB) and
δ13C = 2.18 ± 0.38 ‰ (V-PDB). For none of the two isotopic compositions significant
changes occur upon dissolution.
Although data are overall very variable, carbon and oxygen isotopic composition tend
to increase in shells of N. pachyderma (sin.) upon dissolution (Fig. 16b, 17b). We do not have
a δ18O-temperature relationship for left coiling N. pachyderma to determine the temperature
underestimate of dissolved shells. Applying the slope of von Langen (2001) for the right
coiling type (∆T (K) = – 6.16 * δ18Ocalcite (V-PDB)) yields a maximum difference of –1.6 ± 1.2 K
between the least and the most dissolved shells.
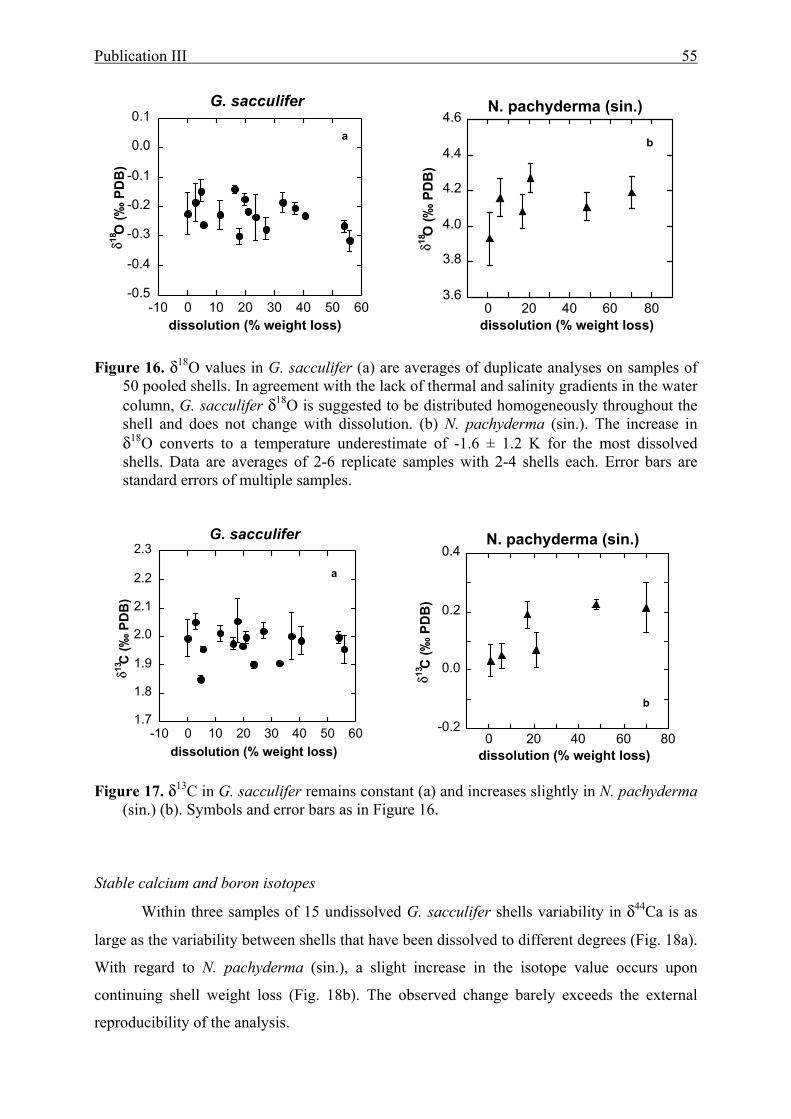
Publication III 55
-0.5
-0.4
-0.3
-0.2
-0.1
0.0
0.1
-10 0 10 20 30 40 50 60
G. sacculiferδ18
O (‰
PD
B)
dissolution (% weight loss)
a
3.6
3.8
4.0
4.2
4.4
4.6
0 20 40 60 80
N. pachyderma (sin.)
δ18O
(‰ P
DB
)
dissolution (% weight loss)
b
Figure 16. δ18O values in G. sacculifer (a) are averages of duplicate analyses on samples of 50 pooled shells. In agreement with the lack of thermal and salinity gradients in the water column, G. sacculifer δ18O is suggested to be distributed homogeneously throughout the shell and does not change with dissolution. (b) N. pachyderma (sin.). The increase in δ18O converts to a temperature underestimate of -1.6 ± 1.2 K for the most dissolved shells. Data are averages of 2-6 replicate samples with 2-4 shells each. Error bars are standard errors of multiple samples.
1.7
1.8
1.9
2.0
2.1
2.2
2.3
-10 0 10 20 30 40 50 60
G. sacculifer
δ13C
(‰ P
DB
)
dissolution (% weight loss)
a
-0.2
0.0
0.2
0.4
0 20 40 60 80
N. pachyderma (sin.)
δ13C
(‰ P
DB
)
dissolution (% weight loss)
b
Figure 17. δ13C in G. sacculifer remains constant (a) and increases slightly in N. pachyderma (sin.) (b). Symbols and error bars as in Figure 16.
Stable calcium and boron isotopes
Within three samples of 15 undissolved G. sacculifer shells variability in δ44Ca is as
large as the variability between shells that have been dissolved to different degrees (Fig. 18a).
With regard to N. pachyderma (sin.), a slight increase in the isotope value occurs upon
continuing shell weight loss (Fig. 18b). The observed change barely exceeds the external
reproducibility of the analysis.
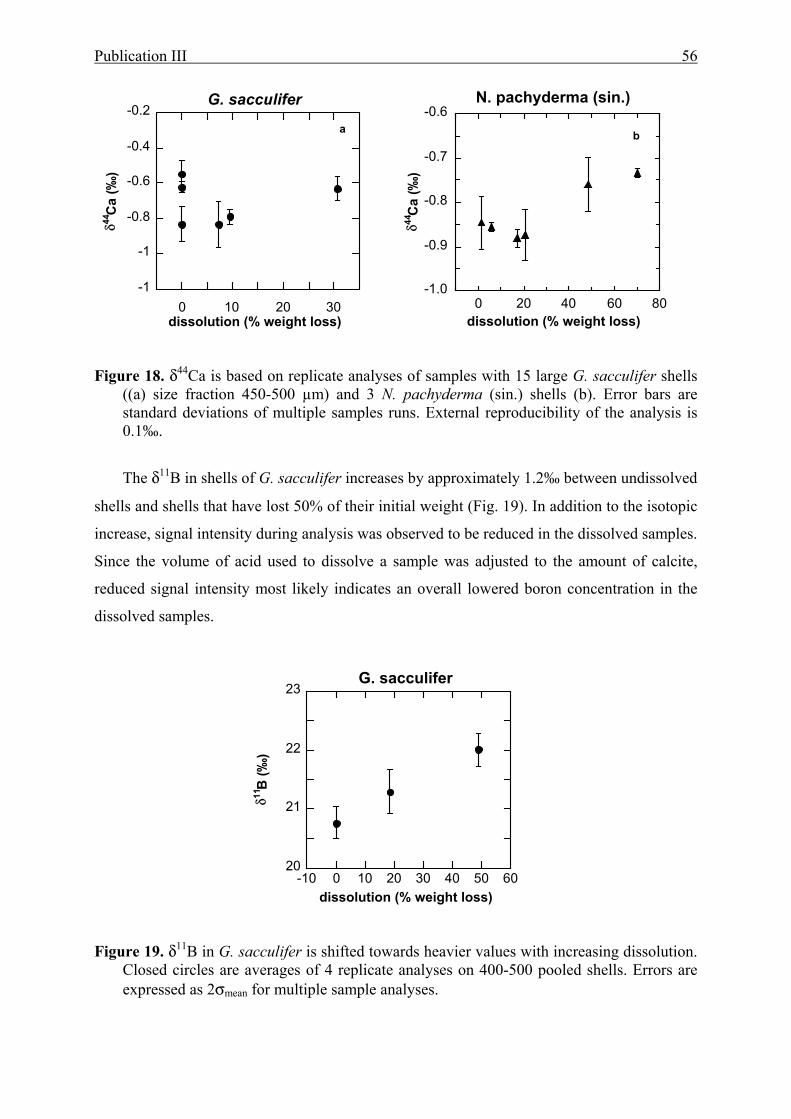
Publication III 56
-1
-1
-0.8
-0.6
-0.4
-0.2
0 10 20 30
G. sacculiferδ44
Ca
(‰)
dissolution (% weight loss)
a
-1.0
-0.9
-0.8
-0.7
-0.6
0 20 40 60 80
N. pachyderma (sin.)
dissolution (% weight loss)
δ44C
a (‰
)
b
Figure 18. δ44Ca is based on replicate analyses of samples with 15 large G. sacculifer shells
((a) size fraction 450-500 µm) and 3 N. pachyderma (sin.) shells (b). Error bars are standard deviations of multiple samples runs. External reproducibility of the analysis is 0.1‰.
The δ11B in shells of G. sacculifer increases by approximately 1.2‰ between undissolved
shells and shells that have lost 50% of their initial weight (Fig. 19). In addition to the isotopic
increase, signal intensity during analysis was observed to be reduced in the dissolved samples.
Since the volume of acid used to dissolve a sample was adjusted to the amount of calcite,
reduced signal intensity most likely indicates an overall lowered boron concentration in the
dissolved samples.
20
21
22
23
-10 0 10 20 30 40 50 60
G. sacculifer
δ11B
(‰)
dissolution (% weight loss)
Figure 19. δ11B in G. sacculifer is shifted towards heavier values with increasing dissolution.
Closed circles are averages of 4 replicate analyses on 400-500 pooled shells. Errors are expressed as 2σmean for multiple sample analyses.

Publication III 57
Discussion A conceptual model
One of the hypotheses that have been proposed to explain changes in foraminiferal
shell chemistry is the preferential dissolution of thinshelled individuals which are supposed to
have a different shell chemistry than thicker shelled individuals (Boyle and Rosenthal, 1996;
Erez, 1979a). Although we cannot rule out that non-encrusted individuals may be present in
sediments at other locations, all 25 shells examined by SEM for this study clearly showed
indices of encrustation (see also Figure 12). Furthermore, shell recovery in our experiments
was high (Table 5) and the few lost shells are due to random loss during sample handling and
fragmentation due to mechanical damage rather than selective dissolution. The chemical
changes observed in this study therefore have to be attributed to chemical changes within
individual shells.
Previous studies have already pointed out that foraminifera secrete different structural
types of calcite during their lifecycle and that chemical and isotopic differences exist between
these calcites. In particular, some species are known to deposit a thick crust during late
ontogeny which is usually associated with gametogenesis and often occurs in deeper waters of
different physico-chemical conditions (e.g Bé, 1980; Caron et al., 1990; Duplessy et al.,
1981). In G. sacculifer, gametogenic calcite is deposited over a period of only 16 hours (Bé,
1980). Considering that ontogenetic chamber formation in G. sacculifer takes ~4 weeks
(Bijma et al., 1994) and contributes ~2/3 to the final shell weight, the precipitation rate for
gametogenic calcification is very high and leads to structural differences between inner and
outer calcite (Bé, 1980).
In the literature, naming of the two calcite types is very confusing. The proximal type
may be described as inner, primary or ontogenetic calcite and the distal as outer, secondary,
gametogenic or crust calcite. With regard to partial shell dissolution in the sediment it was
suggested that ontogenetic calcite dissolves faster than the gametogenic crust which would
lead to a systematic shift of the initial bulk shell chemistry to the chemistry of the outer
calcite (Brown and Elderfield, 1996; Dekens et al., 2002; Lohmann, 1995; Rosenthal et al.,
2000; Russell et al., 1994). We use this concept to examine the dissolution behavior of the
shells studied herein.
Figure 20 illustrates the simplified two component system of a G. sacculifer that
secretes ~2/3 of its shell close to the sea surface and adds ~28% gametogenic calcite (Bé,
1980) at depth. The chemical and isotope values assigned to inner and outer calcite are based
on the bulk chemistry measured on our G. sacculifer shells (Figure 20). Mg/Ca for inner and

Publication III 58
outer calcite is known from microprobe analyses but the other values need to be assumed
based on theoretical considerations. Hydrographic data collected at the core site report that
vertical temperature and salinity differences are smaller than 0.2 K and 0.1‰ over the entire
depth range (Pätzold and participants, 2000). Upon migration to deeper waters in late
ontogeny of G. sacculifer, individuals are therefore supposed to have encountered
homogeneous temperatures and salinities but lower pH (derived from the nearby GEOSECS
station 405). While Erez et al. (1991) argue that gamete release of G. sacculifer in the Gulf of
Elat may occur at a depth of 200 m or even below, Bijma and Hemleben (1994) suggest that
G. sacculifer reproduces at ~80 m water depth in the central Red Sea. Because we do not want
to underestimate possible chemical differences between inner and outer calcite, we assume the
physico-chemical conditions at 250 m water depth for estimating the chemistry of the
gametogenic calcite. Based on these theoretical reflections we would expect lower δ11B in the
gametogenic calcite but similar δ18O, δ44Ca and Sr/Ca in inner and outer calcite (Sr
distribution in the water column is conservative). Mg/Ca is the only parameter which relies
directly on measured data (cf. Figure 14). δ13C is excluded from this model because in
addition to the "carbonate ion (or pH) effect" (Bijma et al., 1999; Spero et al., 1997) the signal
recorded in foraminiferal shells also depends on vertical changes in primary productivity
(changes in the seawater isotopic composition). Due to the lack of seawater isotope values we
cannot quantify these effects for this locality.
The higher Mg/Ca measured in outer compared to inner calcite is counterintuitive as
temperature is known to be the dominant parameter affecting Mg incorporation in
foraminiferal shells (Lea et al., 1999b). Hence, under constant water temperature conditions
we would expect similar Mg/Ca in inner and outer calcite. However, Nürnberg et al. (1996)
observed a similar pattern in laboratory grown G. sacculifer. They speculated that some sort
of a vital effect was responsible for the observed Mg variation and that this vital effect would
be usually hidden by the dominant temperature influence experienced by naturally migrating
foraminifera. Recently, Elderfield et al. (2002) observed that Mg/Ca increases with increasing
shell size. Because δ18O in the same shells does not follow this pattern Elderfield et al. (2002)
argued that temperature could not explain the increase but that Mg uptake must be a
nonequilibrium process which is partly decoupled from the dominant temperature effect. Our
shells are similar to Nürnberg et al.'s laboratory grown individuals in that they did not
experience temperature changes.

Publication III 59
ontogenetic (inner) calcite Mg/Ca = 3.18 mmol/mol Sr/Ca - 1.36 mmol/mol δ18 O - -0.28 ‰ (V-PDB) δ11 B - 20.9 ‰ at pH=8.25 δ44 Ca - -0.62 ‰
gametogenic (outer) calcite Mg/Ca = 4.61 mmol/mol Sr/Ca - 1.36 mmol/mol δ18 O - -0.28 ‰ (V-PDB) δ11 B - 19.9 ‰ at pH=8.12 δ44 Ca - -0.62 ‰
measured bulk shell chemistry of undissolved shells Mg/Ca = 4.16 ± 0.05 mmol/mol Sr/Ca - 1.36 ± 0.02 mmol/mol δ18 O - -0.28 ± 0.1 ‰ (V-PDB) δ13 C - 1.99 ± 0.06 ‰ (V-PDB) δ11 B - 20.77 ± 0.28 ‰ δ44 Ca - -0.62 ± 0.12 ‰
sedimentation
ontogenetic migration
gametogenic calcification at ~250m
ontogenetic calcification in surface water
Figure 20. Schematic presentation of vertical migration and varying calcification depths. The model G. sacculifer is assumed to secrete ~2/3 of the bulk calcite at the surface and ~28% as a gametogenic crust (Bé, 1980) at ~250 m depth (Erez et al., 1991). The model assumptions do not account for the juveniles' ascend from and subadults' descend to the reproduction depth. Chemical composition of ontogenetic and gametogenic calcite were either directly measured (Mg/Ca, cf. Figure 14) or estimated according to theoretical reflections on the physico-chemical gradients encountered at the core site (δ18O, δ11B, δ44Ca, Sr/Ca). A mass balance calculation was applied to bring theoretical reflections and measured bulk shell chemistry data into line.
Based on previous suggestions that shell chemistry is determined by a faster
dissolution rate of ontogenetic relative to gametogenic calcite, the following dissolution
pattern would be expected: no change in δ18O, δ44Ca and Sr/Ca, an increase in Mg/Ca and a
decrease in δ11B. With regard to Mg/Ca the story is a little bit more complicated: The Mg
content of biogenic calcite has been proposed to promote dissolution (Brown and Elderfield,
1996). Because higher Mg-incorporation usually predominates during early ontogeny in
warmer surface waters, the theory of higher solution susceptibility due to higher Mg content

Publication III 60
usually coincides with dissolution of early ontogenetic calcite. In this special case (higher
Mg/Ca in outer calcite) Mg/Ca should increase if the inner calcite dissolved faster than the
outer calcite or decrease if the higher Mg content of the outer calcite was sufficient to increase
the dissolution susceptibility of the outer calcite. On this background we will now discuss the
results obtained in this study.
Minor elements
The observed Mg/Ca decrease in shells of N. pachyderma (sin.) on the order of 20-
30% at only 17% weight loss is sizable and translates to a temperature underestimate of at
least 2 K in these shells. Neogloboquadrina pachyderma (sin.), migrates from subsurface
waters to 50-200 m water depth and this change of habitat is reflected in increasing δ18O of
this species (Kohfeld et al., 1996). Due to the encountered change in temperature it is likely
that Mg/Ca in N. pachyderma (sin.) displays a similar pattern to δ18O so that the observed
decrease in Mg/Ca is in line with the suggestion that ontogenetic calcite is preferentially
dissolved due to its higher magnesium content (Brown and Elderfield, 1996; Lorens et al.,
1977; Savin and Douglas, 1973).
For G. sacculifer a dissolution effect similar to the one in N. pachyderma (sin.) was
not observed despite the heterogeneous Mg distribution. This result is neither consistent with
a higher dissolution susceptibility of ontogenetic calcite (which would predict increasing
Mg/Ca for our shells) nor with Brown and Elderfields model that calcite with a higher Mg
content is less stable (which would predict decreasing Mg/Ca). As the difference in Mg/Ca
between outer (4.61 mmol/mol) and inner calcite (3.18 mmol/mol) is not as large as the one
determined for Globorotalia tumida (2.1 and 5.1 mmol/mol, respectively, Brown and
Elderfield, 1996), it may well be that it is too small to result in a measurable dissolution
effect. To test this hypothesis, we included the ion activity product (IAP) equation established
by Brown and Elderfield (1996) into a mass balance calculation and estimated the Mg/Ca of
shells that have lost 50% of their initial weight:
IAP = 4.17 * 10-9 + 1.97 * 10-11 * Mg/Ca (10) dissolution rate = κ * (1-Ω)η (11)
Ω = a
Ca 2+ * aCO32−
IAP (12)
where κ (d-1) denotes the dissolution rate constant, η (dimensionless) the dissolution reaction
order and Ω the saturation state with respect to Mg-calcite. As the rate constant κ is different

Publication III 61
for different biogenic carbonates and the reaction order η is not very well constrained, we
used several values cited in the literature (Hales and Emerson, 1997b; Keir, 1980; Morse,
1978; Walter and Morse, 1985). aCa2+ and aCO32- are the activities of Ca2+ and CO32- in
seawater which can be calculated using the seawater concentrations of [Ca2+] = 10.3 mmol kg-
1 and [CO32-] = 35 µmol kg-1 (value chosen in accordance to our experimental seawater
carbonate chemistry at 90% saturation, 15°C and atmospheric pressure) and the total activity
coefficients of these ions in seawater as provided by Millero and Pierrot (1998) (γtCa2+ = 0.203
and γtCO32- = 0.039). The amount of crust calcite secreted by G. sacculifer and G. tumida is
assumed to account for ~28% (Bé, 1980), respectively ~70% (Brown and Elderfield, 1996) of
the undissolved shell weight. Due to the heterogeneous Mg distribution, the relative
proportions of inner and outer calcite are supposed to change systematically with increasing
dissolution and so should the bulk Mg/Ca.
The results of our calculations are shown in Table 7. For any κ and η chosen, no
significant change in Mg/Ca of G. sacculifer can be determined. Similarly, 50% weight loss
in G. tumida does not reproduce the Mg/Ca variation (i.e. from 2.65 to 1.25 mmol/mol)
observed by Brown and Elderfield (1996). We also tested a much larger heterogeneity
between inner and outer calcite (10 and 2 mmol/mol, respectively). Only such a large contrast
between inner and outer calcite gives rise to a significant Mg/Ca reduction from 7.76 to 7.27
mmol/mol. However, using calibration equations for the temperature dependency of Mg/Ca in
G. sacculifer (Dekens et al., 2002; Nürnberg et al., 1996), such a heterogeneity would require
temperature differences of >17 K between surface habitat and encrustation depth.
We thus have to conclude that the difference in the Mg content between inner and
outer calcite is too small to cause a detectable dissolution effect. In addition, the strong Mg
dissolution effect observed in N. pachyderma (sin.) is striking with regard to the combination
of low Mg-content (~0.7 mmol/mol) and lower dissolution susceptibility of these shells. All in
all, these calculated dissolution rates argue against the suggestion that higher Mg
concentrations significantly increase the dissolution susceptibility of particular shell parts. We
conclude that Mg is either not the only dissolution-driving factor or that the Mg content is
coupled to some other parameter which is responsible for the overall dissolution behavior. An
alternative (or additional) explanation for different dissolution susceptibilities of inner and
outer calcite could be differences in surface area and/or crystal structure. A recent review
article by Morse and Arvidson (2002) summarizes that complex microstructure and reactvity
of the surface area of biogenic magnesian calcites strongly control the solubility. With regard
to G. sacculifer, the slowly secreted ontogenetic calcite shows well-developed polygonal
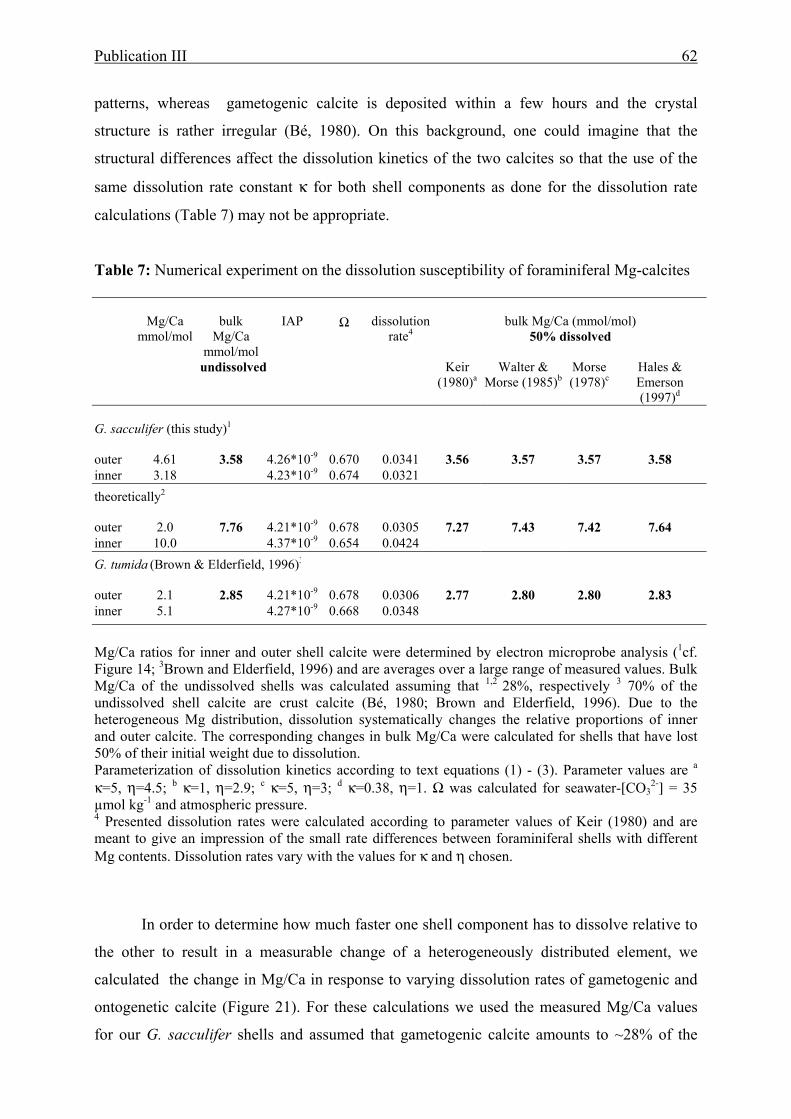
Publication III 62
patterns, whereas gametogenic calcite is deposited within a few hours and the crystal
structure is rather irregular (Bé, 1980). On this background, one could imagine that the
structural differences affect the dissolution kinetics of the two calcites so that the use of the
same dissolution rate constant κ for both shell components as done for the dissolution rate
calculations (Table 7) may not be appropriate.
Table 7: Numerical experiment on the dissolution susceptibility of foraminiferal Mg-calcites
Mg/Ca
mmol/mol
bulk
Mg/Ca mmol/mol
IAP
Ω
dissolution
rate4
bulk Mg/Ca (mmol/mol)
50% dissolved
undissolved Keir (1980)a
Walter & Morse (1985)b
Morse (1978)c
Hales & Emerson (1997)d
G. sacculifer (this study)1
outer 4.61 3.58 4.26*10-9 0.670 0.0341 3.56 3.57 3.57 3.58 inner 3.18 4.23*10-9 0.674 0.0321 theoretically2
outer 2.0 7.76 4.21*10-9 0.678 0.0305 7.27 7.43 7.42 7.64 inner 10.0 4.37*10-9 0.654 0.0424 G. tumida (Brown & Elderfield, 1996)3
outer 2.1 2.85 4.21*10-9 0.678 0.0306 2.77 2.80 2.80 2.83 inner 5.1 4.27*10-9 0.668 0.0348
Mg/Ca ratios for inner and outer shell calcite were determined by electron microprobe analysis (1cf. Figure 14; 3Brown and Elderfield, 1996) and are averages over a large range of measured values. Bulk Mg/Ca of the undissolved shells was calculated assuming that 1,2 28%, respectively 3 70% of the undissolved shell calcite are crust calcite (Bé, 1980; Brown and Elderfield, 1996). Due to the heterogeneous Mg distribution, dissolution systematically changes the relative proportions of inner and outer calcite. The corresponding changes in bulk Mg/Ca were calculated for shells that have lost 50% of their initial weight due to dissolution. Parameterization of dissolution kinetics according to text equations (1) - (3). Parameter values are a κ=5, η=4.5; b κ=1, η=2.9; c κ=5, η=3; d κ=0.38, η=1. Ω was calculated for seawater-[CO3
2-] = 35 µmol kg-1 and atmospheric pressure. 4 Presented dissolution rates were calculated according to parameter values of Keir (1980) and are meant to give an impression of the small rate differences between foraminiferal shells with different Mg contents. Dissolution rates vary with the values for κ and η chosen.
In order to determine how much faster one shell component has to dissolve relative to
the other to result in a measurable change of a heterogeneously distributed element, we
calculated the change in Mg/Ca in response to varying dissolution rates of gametogenic and
ontogenetic calcite (Figure 21). For these calculations we used the measured Mg/Ca values
for our G. sacculifer shells and assumed that gametogenic calcite amounts to ~28% of the
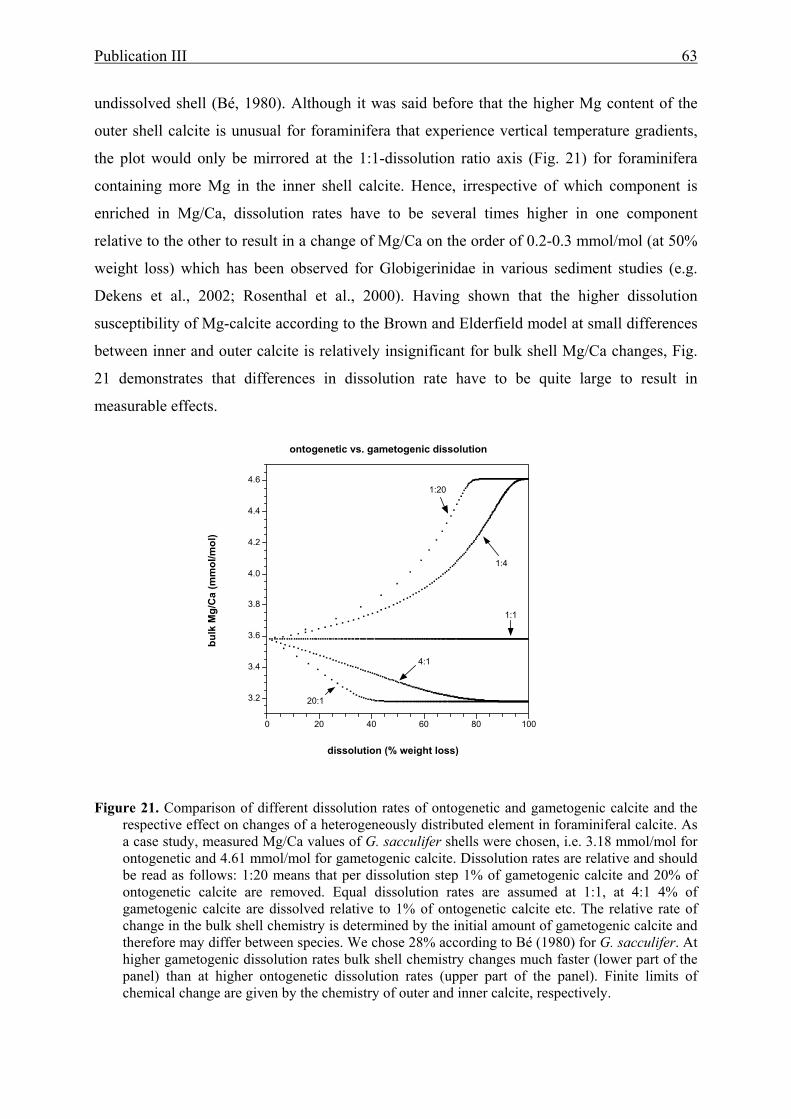
Publication III 63
undissolved shell (Bé, 1980). Although it was said before that the higher Mg content of the
outer shell calcite is unusual for foraminifera that experience vertical temperature gradients,
the plot would only be mirrored at the 1:1-dissolution ratio axis (Fig. 21) for foraminifera
containing more Mg in the inner shell calcite. Hence, irrespective of which component is
enriched in Mg/Ca, dissolution rates have to be several times higher in one component
relative to the other to result in a change of Mg/Ca on the order of 0.2-0.3 mmol/mol (at 50%
weight loss) which has been observed for Globigerinidae in various sediment studies (e.g.
Dekens et al., 2002; Rosenthal et al., 2000). Having shown that the higher dissolution
susceptibility of Mg-calcite according to the Brown and Elderfield model at small differences
between inner and outer calcite is relatively insignificant for bulk shell Mg/Ca changes, Fig.
21 demonstrates that differences in dissolution rate have to be quite large to result in
measurable effects.
3.2
3.4
3.6
3.8
4.0
4.2
4.4
4.6
0 20 40 60 80 10
ontogenetic vs. gametogenic dissolution
bulk
Mg/
Ca
(mm
ol/m
ol)
dissolution (% weight loss)
1:20
1:4
1:1
4:1
20:1
0
Figure 21. Comparison of different dissolution rates of ontogenetic and gametogenic calcite and the
respective effect on changes of a heterogeneously distributed element in foraminiferal calcite. As a case study, measured Mg/Ca values of G. sacculifer shells were chosen, i.e. 3.18 mmol/mol for ontogenetic and 4.61 mmol/mol for gametogenic calcite. Dissolution rates are relative and should be read as follows: 1:20 means that per dissolution step 1% of gametogenic calcite and 20% of ontogenetic calcite are removed. Equal dissolution rates are assumed at 1:1, at 4:1 4% of gametogenic calcite are dissolved relative to 1% of ontogenetic calcite etc. The relative rate of change in the bulk shell chemistry is determined by the initial amount of gametogenic calcite and therefore may differ between species. We chose 28% according to Bé (1980) for G. sacculifer. At higher gametogenic dissolution rates bulk shell chemistry changes much faster (lower part of the panel) than at higher ontogenetic dissolution rates (upper part of the panel). Finite limits of chemical change are given by the chemistry of outer and inner calcite, respectively.

Publication III 64
Although a few studies have shown that G. sacculifer Mg/Ca may be somewhat less
sensitive to dissolution than other species from the same location (Brown and Elderfield,
1996; Dekens et al., 2002; Hastings et al., 1998), varying degrees of Mg dissolution in G.
sacculifer have been observed in other studies (Dekens et al., 2002; Rosenthal and Lohmann,
2002, in press; Rosenthal et al., 2000). Hence, one might speculate whether the extent of G.
sacculifer's ontogenetic vertical migration changes from site to site so that this species
encounters different physico-chemical gradients at different locations. The G. sacculifer shells
investigated in this study are rather exceptional with their higher Mg/Ca in outer calcite.
While Mg is usually more concentrated in the inner calcite, combination of this pattern with a
lower structural stability of ontogenetic calcite may explain why dissolution drives Mg/Ca to
lower values in shells of foraminifera that encounter vertical temperature gradients during
their life cycle. This could explain why we observe lower Mg/Ca in partially dissolved shells
of N. pachyderma (sin.) whereas in G. sacculifer the structural and Mg-effects may be
balanced and Mg/Ca therefore remains constant.
In situ sediment studies showed that the Sr/Ca ratio of the planktonic G. tumida and
the benthic Cibicidoides wuellerstorffi is lowered by partial dissolution (Brown and
Elderfield, 1996; McCorkle et al., 1995). Brown and Elderfield (1996) concluded that
dissolution initially removes the most impure calcite before dissolving calcite with lower Mg
and Sr concentrations. Similarly, Haley and Klinkhammer (2002) studied the effect of
cleaning and dissolution on Orbulina universa in a flow-through system and observed that Sr
and Mg were removed preferentially. In contrast, shells of G. sacculifer either revealed a
slight increase in Sr/Ca upon dissolution (Bender et al., 1975; Haley and Klinkhammer, 2002)
or no systematic effect at all (Brown and Elderfield, 1996; Lorens et al., 1977). Our
increasing Sr/Ca data on G. sacculifer and N. pachyderma (sin.) line up with the results of
Bender et al. (1975) and Haley and Klinkhammer (2002). The magnitude of change found
herein for G. sacculifer is comparable with changes reported in the literature. For N.
pachyderma (sin.) no dissolution data have been reported before. Our experimental results
seem to be inconsistent with the assumption that Sr would be distributed homogeneously
throughout the shell. Although Sr incorporation primarily depends on seawater concentration
and seawater concentration is conservative, some indication exists that physiological
processes may affect Sr incorporation. In a recent study, Elderfield et al. (2002) observed
decreasing Sr/Ca with increasing foraminiferal shell size and attributed this pattern to lower
calcification rates in larger shells. Similarly, Lea et al. (1999b) attributed incrasing Sr/Ca at

Publication III 65
higher temperatures and higher pH to higher calcite precipitation rates under these conditions.
Given that gametogenic calcite in G. sacculifer is deposited at a very high precipitation rate
(Bé, 1980), Sr/Ca may be enriched in outer calcite. Using this line of argument, the observed
increase in Sr/Ca upon dissolution may be due to a shift of the bulk shell chemistry to the
outer calcite. However, as long as we do not have Sr/Ca microprobe data for these shells, we
cannot generalize this hypothesis because other studies have shown that Sr/Ca may strongly
decrease in shells of crust-depositing foraminifera such as G. tumida (Brown and Elderfield,
1996) and O. universa (Haley and Klinkhammer, 2001). The different dissolution effects
observed in different foraminifera species are especially surprising with regard to the
relatively uniform Sr-concentrations in planktonic foraminifera (1.2-1.6 mmol/mol; e.g. Lea,
1999b). Although at this stage we cannot describe the dissolution behavior of Sr/Ca in various
foraminifera species from different locations by a single explanation, it is evident that Sr in
calcite is not necessarily less stable than Ca.
Martin et al. (1999) studied Sr/Ca over Quaternary glacial cycles and measured
variations in shells of N. pachyderma (sin.) and other planktonic foraminifera species that are
of the same order (~ +0.07 mmol/mol during glacials) as the subtle changes determined in our
study for dissolution. Dissolution effects thus can cause significant bias in reconstructions of
glacial/interglacial variations.
Stable isotopes
The variability in stable oxygen and carbon isotopes between samples is quite large.
However, the observed dissolution trends appear reasonable with regard to theoretical
considerations: As mentioned above, previous studies suggested that dissolution shifts bulk
shell δ18O and δ13C towards the isotope chemistry of the outer calcite (Lohmann, 1995;
Rosenthal et al., 2000). Due to the addition of gametogenic calcite in deeper water sections
where temperatures are colder, δ18O was observed to increase in foraminifera that were
subject to dissolution (Dekens et al., 2002; Erez, 1979b; Lohmann, 1995; Rosenthal et al.,
2000). Based on sediment material, Rosenthal et al. (2000) suggested that dissolution affects
δ18O and Mg/Ca similarly. In contrast, a recent study by Dekens et al. (2002) found that the
effect is not always the same for both proxies.
As expected from the lack of thermal and salinity gradients at the core site and the
constancy of the Mg/Ca data, G. sacculifer does not show a dissolution effect in δ18O.
However, the lower Mg/Ca in N. pachyderma (sin.) seems also to be reflected in increased
δ18O. The temperature underestimate due to dissolution changes in both parameters is similar

Publication III 66
in magnitude (~-2 K and -1.6 ± 1.2 K, respectively), but due to the large variability in δ18O it
is not possible to confirm or disprove any of the arguments suggested by Rosenthal et al.
(2000) and Dekens et al. (2002).
Available estimates on the impact of dissolution on δ13C in planktonic foraminifera are
based on the symbiont bearing G. sacculifer (Lohmann, 1995; Rosenthal et al., 2000) and the
symbiont-barren Globorotalia truncatulinoides (Lohmann, 1995). Both studies attributed
lowered δ13C values in the symbiont-bearing G. sacculifer to reduced symbiont
photosynthetic activity at depth. Our G. sacculifer data may indicate a subtle trend towards
lower values but given the large variability, only comparison with literature data allows to
conclude that δ13C of the inner calcite of symbiont-bearing foraminifera must be enriched so
that preferential dissolution of the inner calcite shifts δ13C to the lower values of the outer
calcite. However, in line with the results obtained by Lohmann (1995) on G. truncatulinoides,
δ13C in N. pachyderma increases upon dissolution. Lohmann (1995) already pointed to the
size related change in G. truncatulinoides' δ13C, with δ13C increasing in larger shells due to
some "vital effect". Recent laboratory culture experiments now allow to attribute part of the
ontogenetic isotope enrichment to seawater carbonate chemistry: Spero et al. (1997) and
Bijma et al. (1998) found δ13C and δ18O to increase in foraminiferal calcite if secreted under
lower [CO32-] (or pH). The effect was especially strong for the symbiont-barren G. bulloides
(Spero et al., 1997). Peeters et al. (2002) confirmed the laboratory experiments in a field study
and found that due to calcification at depth where [CO32-] (pH) is lower, δ13C in foraminiferal
shells may be higher although primary productivity and therefore the isotopic composition of
the seawater is lower. Similarly, the higher δ13C in stronger dissolved shells of N.
pachyderma (sin.) (and G. truncatulinoides) may be due to a shift of the bulk shell signal to
the chemistry of the outer calcite which is supposed to have been secreted under lower [CO32-
] (pH) deeper in the watercolumn (Kohfeld et al., 2000; Kohfeld et al., 1996). Unfortunately,
no carbonate chemistry data were found to calculate the [CO32-] (pH) over the calcification
depth range of N. pachyderma (sin.) so that we cannot quantify the theoretical considerations.
Considering the empirical δ44Ca-temperature equation by Nägler et al. (2000) for G.
sacculifer and similar results on the temperature dependency of δ44Ca in N. pachyderma (Zhu
and MacDougall, 1998), a lower isotopic composition would be predicted for partially
dissolved shells if the signal were shifted towards the chemistry of the outer calcite (reflecting
lower temperatures deeper in the watercolumn). Assuming that the temperature record would

Publication III 67
change similar to δ18O and Mg/Ca (-1.6 to -2 K) and that the δ44Ca - temperature slope for N.
pachyderma (sin.) is similar to the one determined for G. sacculifer (Gussone and Eisenhauer,
unpublished data), δ44Ca should be lowered by ~0.4-0.5‰ upon dissolution. In contrast, we
observe slightly increased values in N. pachyderma (sin.). Instead of selectively dissolving the
ontogenetic calcite, some process must lead to the selective removal of 40Ca. However, δ44Ca
appears constant in G. sacculifer. On the one hand, this result is in line with the constant
Mg/Ca and δ18O and seems consistent with the lack of thermal gradients at the core site. On
the other hand, if selective removal of the light isotope is active in N. pachyderma (sin.), the
number of 15 shells per analysis for G. sacculifer may be too small to yield a representative
average so that the actual effect could remain hidden behind the natural variability.
Whichever process is responsible, the observed variation in N. pachyderma (sin.) is within the
analytical uncertainty and negligible for the paleoceanographic record.
Because gametogenic calcite in G. sacculifer is added at depth where pH is lower,
lower δ11B values would be expected if dissolution shifted the signal to the chemistry of the
outer calcite. The observed increase in δ11B (from 20.8 to 22‰) is not consistent with the
expectation. Because the δ11B value of the most dissolved sample exceeds the estimates for
both, ontogenetic and gametogenic calcite (20.9 and 19.9‰, respectively; Fig. 20), selective
removal of the light isotope seems to occur similar to the observations on δ44Ca in N.
pachyderma (sin.). We also found reduced signal intensity during analysis of the dissolved
shells. This could be indicative for an overall leach-out of boron from the shell calcite during
dissolution. Although we do not have any boron concentration data to corroborate this
hypothesis, a previous study on boron preservation in oyster shells presented evidence for
preferential removal of lattice bound boron due to sub-aerial weathering (Cook, 1977).
Considering the microstructural breakdown presented in Figure 12, dissolution does not seem
to be restricted to the shell surface but an increasing number of fissures cut through the entire
shell and expose lattice areas in the interior of the shell wall. Due to the larger surface area
preferential removal of impurities is therefore possible even though they do not need to be
concentrated in particular inner or outer shell areas.
The observed change in δ11B (~+1.2‰ at 50% weight loss) is almost as large as the
glacial-interglacial (G-I) increase determined by Sanyal et al. (1995) for planktonic
foraminifera (1.8 ± 1‰). Although 50% weight loss would be a rather poor preservation for
sediment material, smaller dissolution mediated changes have the potential to affect δ11B.

Publication III 68
Conclusions
We analyzed the elemental and isotopic composition (Mg/Ca, Sr/Ca, δ18O, δ13C, δ11B,
δ44Ca) in partially dissolved shells of two species of planktonic foraminifera, G. sacculifer
and N. pachyderma (sin.) from well preserved sediments located in the Red Sea and the
Norwegian Sea, respectively. In summary, the chemical composition of the G. sacculifer
shells selected for this study reacts rather inert on dissolution. Except for Sr/Ca and δ11B no
significant chemical changes could be observed. While the lack of thermal and salinity
gradients at the core site could explain the constancy of temperature recorders such as δ18O,
Mg/Ca and δ44Ca, comparison with N. pachyderma (sin.) and previous dissolution studies
revealed that dissolution patterns are much more complex than so far assumed.
With regard to Mg/Ca, our calculated dissolution rates do not support the suggestion
that higher Mg concentrations significantly increase the dissolution susceptibility. No
preferential removal of Mg could be observed in G. sacculifer despite the fact that the element
is enriched in the outer calcite and overall more than 5 times more concentrated than in N.
pachyderma (sin.) which in turn shows a strong dissolution effect. Calculations of dissolution
rates lead us to conclude that the low Mg content in foraminiferal shells is not a major factor
controlling the overall dissolution behavior. Other processes have to be investigated instead.
For example, differences in the crystal structure and surface area in addition to variations in
the chemical shell composition could be responsible for inter- and intraspecifically variable
dissolution effects.
Although the Mg-content appears relatively insignificant to preferentially dissolve
Mg-rich calcite, Mg/Ca, δ18O and δ13C in N. pachyderma (sin.) are generally consistent with
a shift of the signal to the chemistry of the outer calcite. Supported by literature data
(Lohmann, 1995) we can conclude that δ13C of symbiont-barren species (N. pachyderma, G.
truncatulinoides) increases upon partial shell dissolution while δ13C of symbiont-bearing
foraminifera (G. sacculifer) decreases. With regard to δ18O, the temperature effect of colder
water masses predominates over carbonate chemistry and symbiont effects so that dissolution
increases δ18O independent of whether a foraminifera bears symbionts or not.
Given that the physico-chemical gradients at the Red Sea location are negligible and
that the measured Mg/Ca difference between inner and outer calcite of G. sacculifer is rather
small, our δ18O, δ13C and Mg/Ca results seem consistent with the hypothesis that ontogenetic
calcite is more prone to dissolution than gametogenic calcite (Brown and Elderfield, 1996;
Lohmann, 1995; Rosenthal et al., 2000; Russell et al., 1994). However, the dissolution

Publication III 69
mediated increase in δ11B and apparently also in δ44Ca is not consistent with a shift of the
signal to a deeper shell secretion environment. The observed change in δ44Ca is too small to
be significant for paleoreconstructions. In contrast, the increase in δ11B by ~1.2‰ is more
than half as large as the G-I increase determined by Sanyal et al. (1995) for planktonic
foraminifera. Although this implies a potentially strong dissolution-bias for δ11B, the number
of data presented herein is very small and needs to be confirmed in extensive sediment studies
and for other foraminifera species as well.
Sr/Ca was shown to increase in both species with progressing dissolution. The subtle
increase was found to be consistent with previous results on G. sacculifer (Bender et al.,
1975; Haley and Klinkhammer, 2002) but disagrees when compared to other foraminifera
species (Brown and Elderfield, 1996; Elderfield et al., 2000; Haley and Klinkhammer, 2002;
McCorkle et al., 1995). With the information currently available we have no explanation for
this result. However, with regard to N. pachyderma (sin.) the observed dissolution effect
could be significant for paleoreconstructions. All in all, the hypothesis that the most impure
calcite is more prone to dissolution (e.g. Brown and Elderfield, 1996) is not supported by our
Sr/Ca results.
Although we started off with the aim to quantify the impact of dissolution, effects
seem to vary among foraminifera species and the sites from where they were collected, i.e. the
physico-chemical gradients under which the shells were secreted. Consequently, the
prediction of chemical trends in progressively dissolving foraminifera shell assemblages is
not a straight-forward task. A single process - such as the assumption that ontogenetic calcite
dissolves faster than gametogenic calcite - is insufficient to explain the various dissolution
effects. Partial dissolution does not seem to be a simple shell thinning process but rather
increases the porosity of foraminiferal shells, exposing lattice areas which are protected in
undissolved shells. Increasing the shell porosity opens a pathway for the leach-out of certain
elements or isotopes in addition to the potentially higher solubility of inner shell calcite.
Individual dissolution patterns may also be complicated by trace metal interactions at crystal
surfaces, which may depend on relative concentrations in the calcite lattice and thus be
location-specific. In order to circumvent the complexity of post-depositional effects on
foraminiferal shell chemistry, examination of additional dissolution indicators is required to
identify the preservation state and minimize uncertainties. The microstructural breakdown of
G. sacculifer allows a first estimate of shell corrosion.

Publication III 70
Acknowledgements
Jan Helmke (GEOMAR, Kiel) and Jürgen Pätzold (GeoB, University of Bremen)
kindly provided foraminifera and core material. The experiments could not have been
successfully completed without the laboratory work of Georges Paradis and Dave Winter.
Pamela Martin shared her knowledge about trace element cleaning techniques and Michael
Kriews offered his laboratory facilities. We gratefully acknowledge laboratory help by Anja
Terbrüggen, Friedel Hinz, Carmen Hartmann, Silke Vetter and Folkmar Hauff. This research
was supported by NEBROC.

Publication IV 71
Publication IV
The impact of the ocean carbonate chemistry on living foraminiferal shell weight: A comment to Broecker and Clark’s „Carbonate ion concentration in glacial-age deep
waters of the Caribbean Sea“
Jelle Bijma, Bärbel Hönisch and Richard E. Zeebe
Geochemistry Geophysics Geosystems (in press, 2002)
.......................................................................................................................................................
Broecker and Clark (2002) use the “size normalized weight” of planktic foraminifera to
estimate the carbonate ion concentration ([CO3=]) of Atlantic glacial upper deep water. This
method was introduced by Lohmann (1995) and is based on the fact that, within a defined size
fraction, dissolution decreases shell weight in proportion to the degree of undersaturation with
respect to CaCO3. Based on the following assumptions,
1) the thickness of the foraminiferal shell wall does not depend on growth conditions,
2) the saturation [CO3=] for calcite, [CO3
=]sat., increases by 20 µmol kg-1 km-1 (Broecker
and Clark, 2001a),
3) the weight loss slope is universal and ca. 0.3 µg (µmol kg-1)-1 (Broecker and Clark,
2001a),
4) the offset between the bottom water and pore water [CO3=] was the same during
glacial time as during the Holocene,
Broecker and Clark (2002) calculate that the [CO3
=] of Atlantic glacial upper deep water was
14 µmol kg-1 higher than during the Holocene. Although they recognise that some of their
assumptions are not strictly valid, they do not assess the impact of those assumptions on their
[CO3=] estimate. Here we comment on several of those assumptions and attempt to quantify
their impact on Broecker and Clark's (2002) calculations.

Publication IV 72
Assumption 1
Broecker and Clark (2002) provide evidence that this assumption is not valid. At the
same pressure-normalised [CO3=], P. obliquiloculata from the Pacific Ocean is consistently
10 µg heavier than those from the Indian Ocean. (Spero and Lea, 1993) have shown that G.
sacculifer cultured under high light intensities grows bigger and is more massive than under
lower light conditions. Hemleben et al. (1987) have demonstrated that G. sacculifer cultured
at higher temperatures, grows larger. Unfortunately they did not measure shell weight.
Chamber number/size/weight relationships for the symbiont barren species G. bulloides differ
from location to location (Spero and Lea, 1996). For instance, comparable ontogenetic stages
from the Chatham Rise are bigger and heavier than those from the San Pedro Basin.
Apparently, besides potential genetic differences (e.g. Darling et al., 1999; Darling et al.,
2000; Huber et al., 1997), growth conditions affect the size normalised weight.
Broecker and Clark (2002) point out that temperature and [CO3=] are likely candidates.
Elderfield (personal communication) showed that shell wall thickness is closely related to
growth temperature. On the other hand, Bijma et al. (1999) demonstrated that shell weight of
O. universa is primarily a function of the [CO3=] of the ambient water (Figure 22a)2. At the
same [CO3=], O. universa shells grow heavier under high light than under low light
conditions. Apparently, the [CO3=] at the site of calcification (SOC), reflects the ambient
[CO3=] but is modified by physiological processes (respiration, calcification and
photosynthesis) of the foraminifer and its symbionts (Wolf-Gladrow et al., 1999a). Broecker
and Clark (2002) point out that in today’s ocean, a very tight correlation exists between
surface water [CO3=] and temperature and that core-top shell weights can thus not be used to
distinguish between a temperature or a carbonate ion dependence (in equilibrium with today’s
atmosphere, the temperature impact on [CO3=] varies roughly between 5 to 6 µmol kg-1 K-1
for surface alkalinities between 2100 to 2400 µmol kg-1, respectively).
2 It can be demonstrated that [CO3=] primarily affects shell thickness and that shell weight is a derived parameter of that relationship. For reasons of comparability and simplicity we use the [CO3=]-weight relationship here.
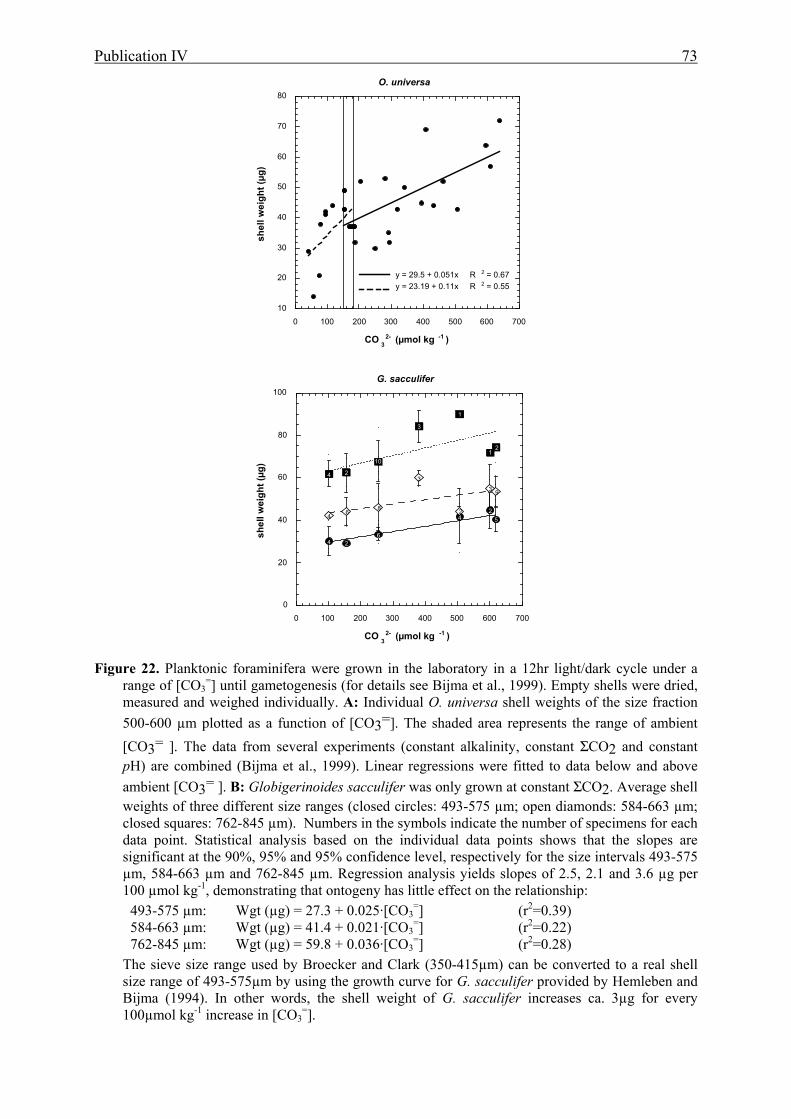
Publication IV
10
20
30
40
50
60
70
80
0 100 200 300 400 500 600 700
O. universa
shel
l wei
ght (
µg)
CO 32- (µmol kg -1 )
y = 29.5 + 0.051x R 2 = 0.67y = 23.19 + 0.11x R 2 = 0.55
73
0
20
40
60
80
100
0 100 200 300 400 500 600 700
G. sacculifer
shel
l wei
ght (
µg)
CO 32- (µmol kg -1 )
4 2
10
3
1
12
4 6 8
3
2
3 9
4 26
42
5
Figure 22. Planktonic foraminifera were grown in the laboratory in a 12hr light/dark cycle under a range of [CO3
=] until gametogenesis (for details see Bijma et al., 1999). Empty shells were dried, measured and weighed individually. A: Individual O. universa shell weights of the size fraction 500-600 µm plotted as a function of [CO3=]. The shaded area represents the range of ambient [CO3= ]. The data from several experiments (constant alkalinity, constant ΣCO2 and constant pH) are combined (Bijma et al., 1999). Linear regressions were fitted to data below and above ambient [CO3= ]. B: Globigerinoides sacculifer was only grown at constant ΣCO2. Average shell weights of three different size ranges (closed circles: 493-575 µm; open diamonds: 584-663 µm; closed squares: 762-845 µm). Numbers in the symbols indicate the number of specimens for each data point. Statistical analysis based on the individual data points shows that the slopes are significant at the 90%, 95% and 95% confidence level, respectively for the size intervals 493-575 µm, 584-663 µm and 762-845 µm. Regression analysis yields slopes of 2.5, 2.1 and 3.6 µg per 100 µmol kg-1, demonstrating that ontogeny has little effect on the relationship:
493-575 µm: Wgt (µg) = 27.3 + 0.025·[CO3=] (r2=0.39)
584-663 µm: Wgt (µg) = 41.4 + 0.021·[CO3=] (r2=0.22)
762-845 µm: Wgt (µg) = 59.8 + 0.036·[CO3=] (r2=0.28)
The sieve size range used by Broecker and Clark (350-415µm) can be converted to a real shell size range of 493-575µm by using the growth curve for G. sacculifer provided by Hemleben and Bijma (1994). In other words, the shell weight of G. sacculifer increases ca. 3µg for every 100µmol kg-1 increase in [CO3
=].

Publication IV 74
Barker and Elderfield (2002) have adopted an approach where they follow the evolution
of shell weight through time and compare this to predictions made from carbon system
modelling. They demonstrate that shell weight of G. bulloides decreases from the last
Termination towards the Holocene. This trend suggests that [CO3=] controls weight rather
than temperature since [CO3=] decreases while temperature increases during the deglaciation.
This finding is corroborated by laboratory experiments. We have evidence, albeit less well
constrained than for O. universa, that shell weight of G. sacculifer depends on the carbonate
chemistry of the ambient water (Figure 22b) and there is a priori no reason to assume that this
phenomenon is restricted to these two species or to foraminifera in general. A similar impact
of the carbonate chemistry has been demonstrated for corals (Gattuso and Buddemeier, 2000;
Gattuso et al., 1998; Kleypas et al., 1999) and for coccolithophorids (Riebesell et al., 2000;
Zondervan et al., 2001). The experimental results suggest that shell weight of individual G.
sacculifer increases by 3 µg for every 100 µmol kg-1 increase in [CO3=]. It should be noted
that only a part of the life cycle has been spent under controlled laboratory conditions and
hence that the real slope may be steeper. The point of the matter is that growth differences
have to be considered, not only between species and in space, but in time as well.
If we accept Sanyal et al.'s (1995) glacial surface water pH reconstruction, which is in
agreement with the ice-core pCO2 measurements and assume that the sites investigated by
Broecker and Clark (2002) were in equilibrium with the atmosphere, the glacial [CO3=] must
have been significantly higher. Depending on which scenario is followed to achieve glacial
pCO2 values, CO2 extraction or CaCO3 addition, the tropical glacial surface ocean [CO3=] was
50 to 120 µmol kg-1 higher, respectively, compared to the Holocene (Lea et al., 1999a).
Hence, before G. sacculifer settled to the ocean floor, glacial specimens must have been
heavier in weight than their Holocene counterparts from the same site. Using our empirical
relationship, 1.5 to 3.6 µg of the glacial weight increase in G. sacculifer was due to the fact
that shells grew heavier3. Hence, the effective glacial-Holocene weight difference decreases.
It is reasonable to assume that the change in the glacial surface [CO3=] was brought about by a
combination of both scenarios. Assuming that the average weight of glacial G. sacculifer was
2.6 µg heavier, the glacial interglacial weight difference for this species reduces from 4.9 to
2.3 µg. Consequently, using the 0.3 µg (µmol kg-1)-1 calibration of Broecker and Clark
(2002), the glacial [CO3=] increase reduces from 14 µmol kg-1 to 8 µmol kg-1.
3 Based on a glacial-interglacial temperature difference for tropical surface waters of 3 ºC, an average slope of [CO3
=] versus temperature of 5.5 µmol kg-1 K-1 and an experimental slope of 3µg weight increase for every 100µmol kg-1 increase in [CO3
=], the impact on shell weight of temperature alone is less than 0.5 µg.

Publication IV 75
Assumption 2 Although the true relationship between pressure and [CO3
=]sat. is exponential, a linear
approximation for the depth range between 3 to 4 km water depth is quite acceptable.
However, the coefficient of 20 µmol kg-1 km-1 (Ingle, 1975) used by Broecker and Clark
(2002) is the largest among a range of values. For instance, based on the relationship
[CO3=]sat. = 90·e(0.16·(z-4)) (Broecker and Takahashi, 1978), the change in the saturation [CO3
=]
between 3 to 4 km depth equals 13 µmol kg-1 km-1. Using the parameterisation of Millero
(1995), the saturation [CO3=] increases by 16 µmol kg-1 between 3 to 4 km depth. Although
Broecker and Clark (2002) acknowledge that the true slope may be in the range of 15 ± 2
µmol kg-1 km-1, they choose to use 20 µmol kg-1 km-1. Jansen et al. (2002) fitted the (more
convenient) equation of Broecker and Takahashi (1978) to the critical [CO3=] as calculated by
Millero (1995). Using this approximation ([CO3=]sat. = 88.7·e(0.189·(z-3.82))), the slope for the
depth range between 3 to 4 km is 16 µmol kg-1 km-1. A smaller pressure impact tends to
increase the weight loss slope and hence reduces the glacial [CO3=] estimate even more.
Assumption 3
Berger (1968) and Parker and Berger (1971) and many others after that, demonstrated
that planktic foraminifera are differently susceptible to dissolution. Hence, one could argue
that the critical [CO3=] is slightly different for each species and that therefore the weight loss
slopes must be species dependent and not universal. The fact that G. ruber does not show the
glacial-Holocene weight difference as the other species studied by Broecker and Clark (2002)
bolsters this contention. To verify the assumption that weight loss slopes are species
dependent we have replotted shell weights from Table 1 in Broecker and Clark (2001a)
against the pressure corrected [CO3=] (Figure 23). Based on the 20 µmol kg-1 km-1 change in
[CO3=]sat. used by Broecker and Clark (2001a), the slopes of the linear regressions are 0.46,
0.57 and 0.68 µg (µmol kg-1)-1 for G. sacculifer, P. obliquiloculata and N. dutertrei,
respectively. Using the smaller pressure effect on [CO3=]sat. of Jansen et al. (2002), the slopes
amend to 0.45, 0.66 and 0.65 µg (µmol kg-1)-1, respectively (Figure 23)44. Assuming an
average slope of 0.5 µg (µmol kg-1)-1, the estimated [CO3=] increase of glacial Atlantic upper
deep water, as calculated by Broecker and Clark (2001a), decreases by 43% from 14 to 8
4 Note that the inconsistency between the weight loss slopes and the ranking to dissolution provided by Parker and Berger Parker, F.L. and Berger, W.H., 1971. Faunal and solution patterns of planktonic foraminifera in

Publication IV 76
µmol kg-1. However, as argued above the slopes are species specific and ∆[CO3=] should
therefore be calculated on a per species basis. For the Caribbean cores the glacial-Holocene
increase in [CO3=] is then estimated to be 11 and 5 µmol kg-1 on the basis of G. sacculifer and
N. dutertrei, respectively. Apparently, the critical [CO3=] for G. sacculifer is higher than that
for N. dutertrei (i.e. at the same water depth, the Holocene-glacial weight difference for G.
sacculifer is larger than for N. dutertrei). The average of the two species is, of course, 8 µmol
kg-1 but the question arises which of the two species provides the best estimate?
Combining the impact of a steeper weight loss slope for G. sacculifer with that of
higher glacial surface water [CO3=] on G. sacculifer shell weight, reduces the glacial [CO3=]
increase estimate to 5 µmol kg-1.
Assumption 4
In addition to the assumption that the difference between the bottom and pore water
[CO3=] was the same during glacial time as during the Holocene, Broecker and Clark (2001a)
assume that the offset between bottom and pore water [CO3=] is constant with depth and
between different sites. However, because the rain ratio (carbonate carbon/organic carbon)
changes with depth and differs from location to location, these assumptions may not be valid.
In fact, Broecker and Clark (2001a) note that the observed correlation between the weights of
G. sacculifer, P. obliquiloculata and N. dutertrei for the core top samples from the Ceara Rise
demonstrates the variability of the [CO3=] offset between bottom and pore water.
Due to non-linear dissolution kinetics, the offset between bottom and pore water
[CO3=] changes drastically from above to below the saturation horizon (SH). Above the SH,
pore water is less saturated with respect to calcite than bottom water (due to respiration driven
pore water dissolution). Bottom water [CO3=] reaches the critical value at the SH; “interface”
dissolution starts and progresses exponentially towards greater depths. Because the
dissolution kinetics are not infinitely fast, an offset is created between the SH and the
lysocline. In this depth interval, called the transition zone, the saturation state of the pore
water increases from less saturated to more saturated than the bottom water5. This
demonstrates that the offset between bottom and pore water is not constant with depth and
surface sediments of the south Pacific. Deep-Sea Research, 18(1): 73-107. is most likely due to the combined effect of susceptability to dissolution and wall thickness (i.e. the initial shell weight). 5 Note that there is a depth where the [CO3
=] of bottom and pore water converge.
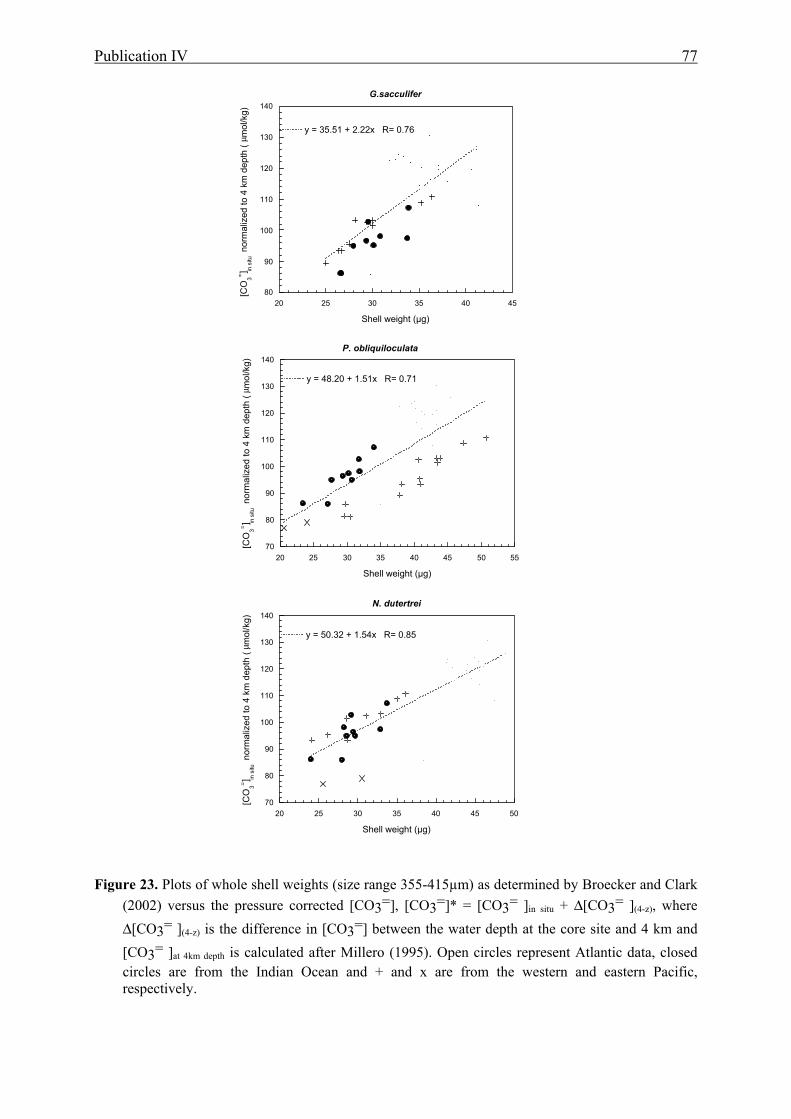
Publication IV
80
90
100
110
120
130
140
20 25 30 35 40 45
G.sacculifer
y = 35.51 + 2.22x R= 0.76
[CO
3= ] in s
itu n
orm
aliz
ed to
4 k
m d
epth
(µ m
ol/k
g)
Shell weight (µg)
77
70
80
90
100
110
120
130
140
20 25 30 35 40 45 50 55
P. obliquiloculata
y = 48.20 + 1.51x R= 0.71
[CO
3= ] in s
itu n
orm
aliz
ed to
4 k
m d
epth
(µ m
ol/k
g)
Shell weight (µg)
70
80
90
100
110
120
130
140
20 25 30 35 40 45 50
N. dutertrei
y = 50.32 + 1.54x R= 0.85
[CO
3= ] in s
itu n
orm
aliz
ed to
4 k
m d
epth
(µ m
ol/k
g)
Shell weight (µg)
Figure 23. Plots of whole shell weights (size range 355-415µm) as determined by Broecker and Clark (2002) versus the pressure corrected [CO3=], [CO3=]* = [CO3= ]in situ + ∆[CO3= ](4-z), where ∆[CO3= ](4-z) is the difference in [CO3=] between the water depth at the core site and 4 km and [CO3= ]at 4km depth is calculated after Millero (1995). Open circles represent Atlantic data, closed circles are from the Indian Ocean and + and x are from the western and eastern Pacific, respectively.

Publication IV 78
hence, that the slope of the weight-loss per unit change in [CO3=] changes below the SH.
Consequently, the size-normalised weight method should probably be restricted to cores that
have never seen in situ bottom water [CO3=] below the critical [CO3=].
One could argue that the variability in the [CO3
=] offset between bottom and pore
water is a fatal blow for Lohmann’s method. However, we should keep in mind that we are
dealing with a proxy and that such complications are to be expected. They basically set the
limit for the accuracy of the method. A better understanding of the [CO3=] variations between
bottom and pore water is needed to improve the robustness of the method.
Above the saturation horizon, the range of pH offsets between bottom and pore water,
for instance at the Ontong-Java Plateau, is somewhere between 0.02-0.04 pH units (Hales and
Emerson, 1996). This translates roughly to a [CO3=] offset between 5 to 10 µmol kg-1. Using
the average weight loss slope of 0.5 µg (µmol kg-1)-1, this implies that weight differences
between 2.5 to 5 µg are within the uncertainty for reconstructing bottom water [CO3=].
The restriction to cores that have never bathed in waters below the critical [CO3
=] has
also been noted by Broecker and Clark (2002). In addition, they argue that data from shallow
cores bathing in water with a [CO3=] higher than 120 µmol kg-1 should be omitted (because
the reduction in [CO3=] resulting from the release of respiration CO2 in the pore water is more
than compensated by the excess of bottom water [CO3=] over calcite saturation).
Some of the cores used by Broecker and Clark (2001a) to determine the weight loss
slopes are from below the SH (based on Jansen et al., 2002) or from in situ [CO3=] higher than
120 µmol kg-1. Limiting their data to [CO3=]sat. < [CO3
=]in situ < 120, the weight loss slopes for
G. sacculifer and N. dutertrei become 0.62 and 0.93 µg (µmol kg-1)-1, respectively. The
estimated glacial increase in [CO3=] for the Caribbean cores now decreases to 8 and 4 µmol
kg-1 on the basis of G. sacculifer and N. dutertrei, respectively.
If we combine the new estimate of the weight loss slope for G. sacculifer with the
impact of higher glacial surface water [CO3=] on this species, the ∆[CO3=] estimate based on
G. sacculifer reduces to 4 µmol kg-1. This brings the predicted average increase in [CO3=] of
Atlantic glacial upper deep water based on G. sacculifer (including the impact of higher
glacial surface water [CO3=] on initial shell weight) close to the prediction of 4 µmol kg-1
based on N. dutertrei (without an impact of higher glacial surface water [CO3=] on initial shell
weight). This could suggest that during growth the shell weight of N. dutertrei does not

Publication IV 79
respond strongly to the [CO3=], if at all. However, the predicted increase in [CO3=] of 4 µmol
kg-1 for Atlantic glacial upper deep water is clearly within the range of uncertainty related to
the variability in the [CO3=] offset between bottom and pore water leaving the question open
for further debate.
Acknowledgements
We would like to thank Howard J. Spero and David W. Lea for comments on earlier
versions of the manuscript and for many years of intensive collaboration in the laboratory in
which the shell weight measurements were generated.

Summary and outlook 80
3. Summary and outlook
To predict consequences of future climate variations especially with regard to
anthropogenic interference we have to understand the natural mechanisms that control the
climate system. The view into the past offers a rich source of information. However, the
quality of paleoceanographic reconstructions can be no better than the data base itself. This
thesis therefore deals primarily with the reliability of chemical proxies in planktonic
foraminifera shells as tracers of past seawater carbonate chemistry.
In the following section the main results are summarized and discussed in the light of
proxy applications in paleoceanography and paleoclimatology. Finally, perspectives for future
research are given.
3.1 Effects of symbiont photosynthesis and respiration on the stable boron isotopic
composition of foraminiferal shells
Culture experiments with living O. universa in combination with field-grown G.
bulloides and O. universa have shown that the pH in the microenvironment of planktonic
foraminifera is substantially affected by the photosynthetic sequestration of CO2, respectively
the release of CO2 during respiration processes. The observed difference in recorded pH
between shells grown under high light and low light conditions (∆pH ≈ 0.2) is only slightly
smaller than theoretical predictions based on microsensor pH-measurements (∆pH ≈ 0.34,
Rink et al., 1998) but significantly smaller than the model predictions cited in Publication I
(∆pH ≈ 0.4-0.76, Zeebe et al., subm.). New model runs have now shown that the difference
between model and culture data may be due to the 10-fold increased seawater boron
concentration used in the experiments (Zeebe et al., subm.). Taking into account that
increased boron concentrations modify the buffer capacity of the culture water, Zeebe et al.
(subm.) calculated different pH-gradients in the microenvironment compared to shells grown
in natural seawater. The smaller gradients calculated for shells grown in the modified
seawater are supposed to be responsible for the observed smaller differences between shells
grown under high light and low light. Upon correction of the boron concentration effect, the
model results are consistent with the culture data. This is a very promising result as it
demonstrates that the theoretical background for the use of δ11B as a paleoacidity-indicator is
well understood. The most important model prediction, however, is the constancy of the effect

Summary and outlook 81
over a wide seawater-pH range. This model outcome suggests that the reliability of δ11B is not
compromised by the physiological processes investigated in the culture experiments.
3.2 The effect of alkalinity on planktonic foraminiferal Ba/Ca
Theoretical arguments would have predicted higher Ba/Ca with increasing
precipitation rate. The arguments are based on experimentally determined similarities between
Sr2+ and Ba2+ incorporation into inorganic and biogenic calciumcarbonates (Lorens, 1981;
Morse and Bender, 1990; Tesoriero and Pankow, 1996; Zachara et al., 1991) and the finding
that Sr2+ uptake in planktonic foraminiferal calcite increases with pH, respectively
precipitation rate (Lea et al., 1999b). Similarly, Elderfield et al. (1996) developed a
biomineralization model for benthic foraminifera which argues for a calcification rate control
on trace element incorporation. However, the culture experiments carried out for Publication
II revealed slightly decreasing Ba2+ uptake in shells grown at higher alkalinities. Although the
observed effect is too small to significantly affect paleoceanographic reconstructions, it is
inconsistent with the theoretical predictions. A crucial point of this experiment, however, is
the use of planktonic and not benthic foraminifera which have so far been used for
reconstructions of glacial-interglacial variability (Lea, 1993; Lea and Boyle, 1990). Even
though planktonic foraminifera apparently react rather inert to changes in water column
alkalinity, benthic species may secrete their shells in environments which are actually
undersaturated with respect to calcite. It cannot be excluded that the incorporation of trace
elements under such conditions is different to the incorporation behavior of planktonic
foraminifera. For instance, (Marchitto Jr. et al., 2000) discovered that DZn and DCd in benthic
foraminifera may be affected by bottom water [CO32-] and argued that the supposed
dissolution effect on Ba/Ca in benthic foraminifera (Table 1, McCorkle et al., 1995) could be
equally due to an incorporation effect in undersaturated bottom water. In fact, preliminary
data on the benthic foraminifer Hoeglundia elegans indicate an anticorrelation between
bottom water alkalinity (or some associated parameter) and the Ba/Ca uptake ratio (G.J.
Reichart, unpublished data). This result is basically similar to the outcome of Publication II
and just as inconsistent with the theoretical predictions. However, it has to be pointed out that
H. elegans secretes an aragonitic shell and the result is thus not necessarily comparable to the
calcitic benthic foraminifera usually used for paleo-alkalinity estimates.
Laboratory experiments with benthic foraminifera are long-lasting and generally more
difficult than culture experiments with planktonic foraminifera. However, if additional data

Summary and outlook 82
prove the results described in Publication II and benthic coretop calibrations (G.J. Reichart,
unpublished data) to be true, alkalinity itself (or some associated parameter) may compromise
the use of benthic Ba/Ca as a tracer for past Ba2+ concentrations and paleo-alkalinity.
3.3 Changes in planktonic foraminiferal shell chemistry after partial dissolution in
undersaturated seawater
Previous research on the stability of chemical proxies in foraminifera is limited by a
number of uncertainties such as the ignorance of the actual dissolution state in sediments or
possible recrystallization processes. Simulated seafloor dissolution experiments carried out
for Publication III under controlled conditions in the laboratory and in combination with the
numerous proxies investigated allowed a detailed insight into the potential of partial shell
dissolution to change chemical proxies recorded in foraminiferal shells. Previous studies
focussed primarily on δ18O and Mg/Ca (Table 1). Although measured changes in the oxygen
isotopic composition of the shells studied herein were not significant, the observed variability
in δ18O and Mg/Ca is in line with known dissolution effects. However, the results presented in
Publication III should not be regarded as the final answer to all dissolution problems as
dissolution effects are very variable and have to be seen in the light of earlier research.
On the background of previous dissolution studies the results presented in Publication III
can be used to test different hypotheses previously put forward to explain observed effects.
Among these hypotheses the higher dissolution susceptibility of ontogenetic relative to
gametogenic calcite has the potential to explain the dissolution behavior of δ18O, δ13C, Mg/Ca
and maybe also Sr/Ca in shells of vertically migrating foraminifera that encounter physico-
chemical gradients during their ontogeny. However, δ44Ca and δ11B measured for this study
appear to increase upon dissolution and are therefore inconsistent with this model. δ44Ca and
δ11B have not been investigated in the light of partial dissolution before and additional studies
are required to confirm the observed trends. If dissolution shifts bulk shell chemistry to the
chemistry of calcite secreted at greater depth, δ44Ca and δ11B need to be significantly
dominated by other parameters which finally determine the relative stability of these elements
and isotopes in foraminiferal shell calcite.
Because it could be excluded that the higher Mg-content of certain shell parts may be
responsible for their lower stability, other parameters have to be considered to explain why
certain shell parts may be more soluble than others. For instance, gametogenic calcite is
precipitated much faster than ontogenetic calcite and different solubilities could be due to

Summary and outlook 83
differences in crystal structure or surface area. However, SEM examination of G. sacculifer
shells clearly showed that dissolution drives deep fissures into the shell surface,
demonstrating that dissolution also affects outer (gametogenic) calcite significantly and
exposes lattice areas to seawater which are protected in uncorroded shells. Based on these
results, partial dissolution does not seem to be a simple shell thinning process which would
argue for the outmost calcite layers to be removed first but increases the porosity, allowing
certain elements to be leached out of a shell.
The Sr/Ca data indicate that crystal impurities are not necessarily more prone to
dissolution. Although it was previously observed that Sr/Ca significantly decreases in
foraminifera species such as Orbulina universa (Haley and Klinkhammer, 2002), G. tumida
(Brown and Elderfield, 1996) and Globigerinoides ruber (D.W. Lea, personal
communication), other studies have shown subtle increases in Sr/Ca of G. sacculifer (Bender
et al., 1975; Haley and Klinkhammer, 2002). The different behavior found for different
foraminifera species can not be reconciled with the lack of pressure in laboratory dissolution
experiments and therefore has to be related to species-specific effects. Although higher
precipitation rates during gametogenic calcification could potentially explain why outer
calcite should be enriched in Sr and Sr/Ca therefore increases upon dissolution, comparison of
the herein observed dissolution pattern with previous studies remains puzzling. It could be
argued that in dependence of the overall elemental composition trace metals may interact at
mineral surfaces and thereby strengthen or weaken the stability of certain elements in the
crystal lattice. Considering the extent of vertical migrations, the encountered physico-
chemical vertical gradients and the overall specific seawater chemistry at different study sites,
dissolution effects may not only be species- but also location-specific (see also Table 1). In
summary, it has to be concluded that the process of partial dissolution is not yet very well
understood and that the prediction of dissolution effects is anything but a straight-forward
task. Without a fundamental understanding of the respective dissolution processes it is not yet
possible to quantify the extent to which certain proxies change upon dissolution and to correct
proxies measured in apparently corroded shells.
3.4 Foraminifera collected from sediment cores - identifying their preservation state Publication III has demonstrated that corrosion of planktonic foraminifera is reflected
in their shell microstructure and can easily be detected by SEM examination. This is an
important finding as it opens a simple way to evaluate the preservation state of shells used for
chemical analyses or bottom water acidity estimates. As discussed in Publication IV,

Summary and outlook 84
planktonic foraminiferal shell thickness or density is not only determined by post-depositional
dissolution but also varies with changing calcite saturation in surface waters. As demonstrated
herein, visual inspection of the shells allows to differentiate between real corrosion and less
favorable growth conditions. Comparable microstructural dissolution patterns have already
been observed by Volbers and Henrich (2002) for G. bulloides and Helmke and Bauch (2002)
for N. pachyderma (sin.). Compared to labor-intensive estimates of weight loss or break-up of
shells, SEM examination can be done on single shells and thus offers a relatively fast method
to judge whether the primary chemical signal of numerous downcore samples may have been
altered post-depositionally or not.
3.5 Perspectives for future research
The present study investigated the chemical composition of foraminifera shells in
response to changing seawater carbonate chemistry. Some open questions were answered,
however, as it is always in investigations of this kind, a wide field of new questions has been
opened as well. The boron isotopic composition of cultured and field-grown foraminifera
presented in Publication I was found to be significantly offset to previously established
empirical calibration curves. The reason for the observed offsets between studies and species
is most probably due to differences in the analytical methodology. Although it was assumed
that the differences between samples of the same species that are measured in the same
laboratory reflect the conditions under which the foraminifera were grown, differences and
uncertainties between laboratories and species are unsatisfactory with regard to
paleoceanographic reconstructions. The systematic reassessment of the analytical
methodology is therefore planned in the near future. In addition, we intend to produce and
establish an analytical standard with a boron isotopic composition and carbonate matrix
similar to the microfossils studied for paleoceanographic reconstructions. Foraminifera will
also be investigated in the light of changing salinity and temperature, parameters that have
recently been suggested to affect the boron isotopic fractionation during foraminiferal shell
secretion. This work will not be restricted to planktonic foraminifera alone, as there is a
pronounced need to investigate benthic foraminifera as well. Empirical calibration curves for
single benthic species are required which should be established preferentially in laboratory
culture experiments.
Publication II and III have shown that the mechanisms operating during trace element
incorporation and selective removal upon partial dissolution are still poorly understood.
Modeling studies in combination with microscale measurements of single foraminifera shell

Summary and outlook 85
walls are planned or partly underway in order to better understand the data obtained in this
and other studies. Cooperations with mineralogists and chemists are intended in order to
determine shell surface areas, yield a better understanding of how crystal structure could
affect the calcite stability and to find out which elements have the potential to interact and
thereby vary dissolution susceptibility.
The dissolution effect on δ11B clearly needs to be confirmed. As boron in calcite
occurs in trigonal and tetrahedral coordination states (Sen et al., 1994), and the isotopic
composition of each coordination state may be analog to the isotopic composition of trigonal
and tetrahedral boron in solution (personal communication N.G. Hemming), preferential
removal of one of the coordinations could possibly explain a change in isotopic composition
upon dissolution. Depending on which coordination is less stable, such a process would
remove isotopically light boron (if tetrahedral coordination is less stable) and leave the
residual with a heavier δ11B or vice versa (if trigonal coordination is less stable). Following
this assumption and given that boron in aragonite (corals) only occurs in the tetrahedral
coordination (Sen et al., 1994), no dissolution effect would be expected for aragonite. This
prediction could be easily tested in an experiment. However, this consideration clearly
depends on the assumption that only B(OH)4- (and no B(OH)3) adsorbed at the surface of a
foraminifera shell gets incorporated into the lattice (otherwise corals and foraminifera would
have a very different isotopic composition than shown e.g. in Hemming and Hanson, 1992)
and that a fractionation accompanies the structural transformation which isotopically enriches
the trigonal coordination state relative to the tetrahedral without changing the overall shell
isotope value (Sen et al., 1994). Because boron is assumed to substitute at the anion site in
CaCO3 and the CO32--site in calcite is larger than in aragonite (Sen et al., 1994), the
occurrence of the smaller trigonal coordination state in calcite could indicate that it is better
embedded in the calcite lattice than the larger tetrahedral coordination. Using this line of
argumentation, the observed increase in δ11B (Publication III) could be due to the higher
stability of the isotopically heavier trigonal coordination state in foraminiferal calcite.
However, this process would have to dominate over the preferential removal of inner
compared to outer calcite (as suggested for δ18O, δ13C and Mg/Ca) and the expected lower
δ11B incorporated at depth. As a first start, shells of Globigerinella equilateralis, picked and
partially dissolved in addition to the G. sacculifer shells from the Gulf of Aqaba core site
(Publication III), are available to challenge the G. sacculifer results. Furthermore, sediment
studies are planned with shells from different water depths.

Summary and outlook 86
Finally, we are also interested in the expansion of our knowledge to other marine
calcifiers. A unique set of hermatypic corals cultured under variable conditions of seawater
carbonate chemistry has already been analyzed for their boron isotopic composition. The
response to pH is significant, although different to the fractionation observed in foraminifera.
To complete this data set, the samples are already in preparation for minor and trace element
analyses such as Sr/Ca, Mg/Ca, U/Ca and Ba/Ca. Although the incorporation of these
elements into coral aragonite has not yet been calibrated with respect to changes in seawater
carbonate chemistry, evidence already exists that the chemical composition of coral skeletons
may respond in a manner similar to planktonic foraminifera (e.g. Cardinal et al., 2001; Cohen
et al., 2002; Kühl et al., 1995; Min et al., 1995; Shen and Dunbar, 1995).

Appendix 87
4. Appendix
Reconstructing and modeling past ocean carbonate chemistry –
Working Group 3 report of the ESF Explanatory Workshop on "The ocean carbon
cycle and climate change", Delmenhorst, September 1-4, 2001
Bärbel Hönisch (rapporteur), Jonathan Erez (discussion leader), Christina Crone, Heiko
Jansen, George P. Lohmann, Guy Munhoven, Martin R. Palmer, Ann D. Russell, Dieter A.
Wolf-Gladrow, Richard E. Zeebe, Patrizia Ziveri
.......................................................................................................................................................
Abstract
The chemical composition and preservation state of biogenic calcium carbonates
derived from marine sediments are routinely used for paleoceanographic and
paleoclimatologic reconstructions despite the fact that their calibration to environmental
parameters is often a matter of debate. Significant differences are observed between
laboratory culture experiments, field calibrations (as obtained from different locations and
depths in the water column) and core top-based analyses. The reasons for these discrepancies
are not well understood and call for further evaluation. Working group 3 reconsidered the
proxies currently available for reconstructions of the oceanic carbonate system. In addition to
the prevalent recorders of past ocean chemistry, foraminifera, we also discussed
coccolithophorids. Previous carbonate system reconstructions and limitations of several
proxies are summarized and suggestions for future research are proposed.

Appendix 88
Introduction
On glacial-interglacial time scales oceanic carbonate chemistry determines
atmospheric pCO2. However, the underlying mechanisms and possible feedbacks with climate
change are still not well understood. Knowledge of the nature and amplitude of natural
fluctuations in the past can be used to assess the stability of modern subsystems and their
potential range of variations in the future. Understanding the climate system therefore
requires the reconstruction of physical, chemical and biological parameters that characterize
the ocean carbonate system over glacial and interglacial time scales as well as the transitions
between them. Over the past decade a number of proxy relationships based predominantly on
foraminifera have been established on the basis of laboratory and field experiments. Among
others, the stable boron isotopic composition in foraminiferal shells was found to record
marine pH (Spivack et al., 1993), Ba/Ca was used to infer alkalinity (Lea and Boyle, 1989)
and differences between the influence of [CO32-] on the stable oxygen and carbon isotopic
composition of Globigerinoides sacculifer and G. ruber was found to be useful for past
carbonate ion concentration estimates (Bijma et al., 1999; Spero et al., 1999). More recently,
some new proxies such as foraminiferal U/Ca (Russell, 2001), S/Ca (Erez et al., 2001) and the
CaCO3 size fraction index (Broecker and Clark, 1999) have been found to record [CO32-].
In addition to these chemical proxies of the seawater carbonate system, the
preservation state of carbonates can be used to estimate bottom water undersaturation for
aragonite and calcite. One approach is to examine the depth of their lysoclines, which can be
defined as the levels of maximum solution rate increase in the deep sea (Berger, 1968).
Relating the preservation state of carbonates in the sediment to the saturation state of bottom
water yields information on [CO32-]in situ. For example, the dissolution-driven shell-thinning of
planktonic foraminifera was found to give a good approximation of bottom water [CO32-]
(Broecker and Clark, 2001a; Lohmann et al., 1999).
Unfortunately, the interpretation of these proxies is often hampered by their
dependency on additional variables. Species specificity, vital effects of the organisms, and
even the susceptibility to diagenesis complicate data interpretation. Hence, proxy
relationships are not as simple as we would like them to be. For example, the interpretation of
geochemical proxies in surface-dwelling planktonic foraminifera is complicated by the
presence of a significant fraction of calcite added at depth. Thus, since most calibrations are
empirical, a certain discrepancy exists between laboratory experiments and the real situation
at the seafloor. Combination of these proxy development approaches with better

Appendix 89
understanding of the calcification mechanisms and numerical models should yield higher
reliability of the proxies. Eventually, such new data and its proper modeling would render
further insight into the role and the impact of the carbon cycle on climate oscillations and in
particular resolve the mechanisms that control the operation of the oceanic carbon cycle.
Working group 3 discussed the carbonate system proxies (Table A1) and their
limitations. In order to obtain high-quality paleoreconstructions, future research needs to
focus on the removal of these uncertainties. Recommendations point towards field
investigations and culture experiments, as well as towards numerical models and the
improvement and further development of existing and new analytical techniques.
2. Proxies for ocean carbonate chemistry and their limitations 2.1 Carbonate chemistry 2.1.1 Ba/Ca to infer alkalinity
Ba is a nutrient-like tracer similar to Cd and δ13C, because biological activity extracts
these elements from surface waters and gravitation transfers them toward the seafloor in
sinking particles. On its way from the North Atlantic to the deep North Pacific, deep water is
progressively enriched in Ba. The close correlation between Ba and alkalinity in seawater
(Chan et al., 1977; Lea and Boyle, 1989) is mechanistically not well understood (Bishop,
1988; Chan et al., 1977; Chow and Goldberg, 1960; Lea, 1993), but is thought to be related to
the simultaneous release of alkalinity through CaCO3 dissolution and regeneration of Ba at
the seafloor. However, Lea (1993) suggested that changes in the thermohaline circulation
redistribute Ba and alkalinity similarly, thereby allowing reconstruction of past alkalinity
distributions from benthic foraminiferal Ba/Ca.
The main limitation of Ba as a paleoproxy is due to the short oceanic residence time
on the order of 10,000 years (Broecker and Peng, 1982; Chan et al., 1977). The Ba-alkalinity
correlation is not perfectly applicable on a time scale longer than this period. However, the
fact that Ba is incorporated into foraminifera shells in direct proportion to the seawater
concentration (Lea and Boyle, 1989; Lea and Spero, 1992; Lea and Spero, 1994) allows us to
estimate paleo-Ba concentrations from foraminifera deposited in sediments. Independent
estimates of seawater carbonate chemistry would offer an opportunity to verify whether the
present-day slope of the Ba-alkalinity relationship is applicable to the past as well. A
multiproxy approach would provide the best means of calculating alkalinity for various time
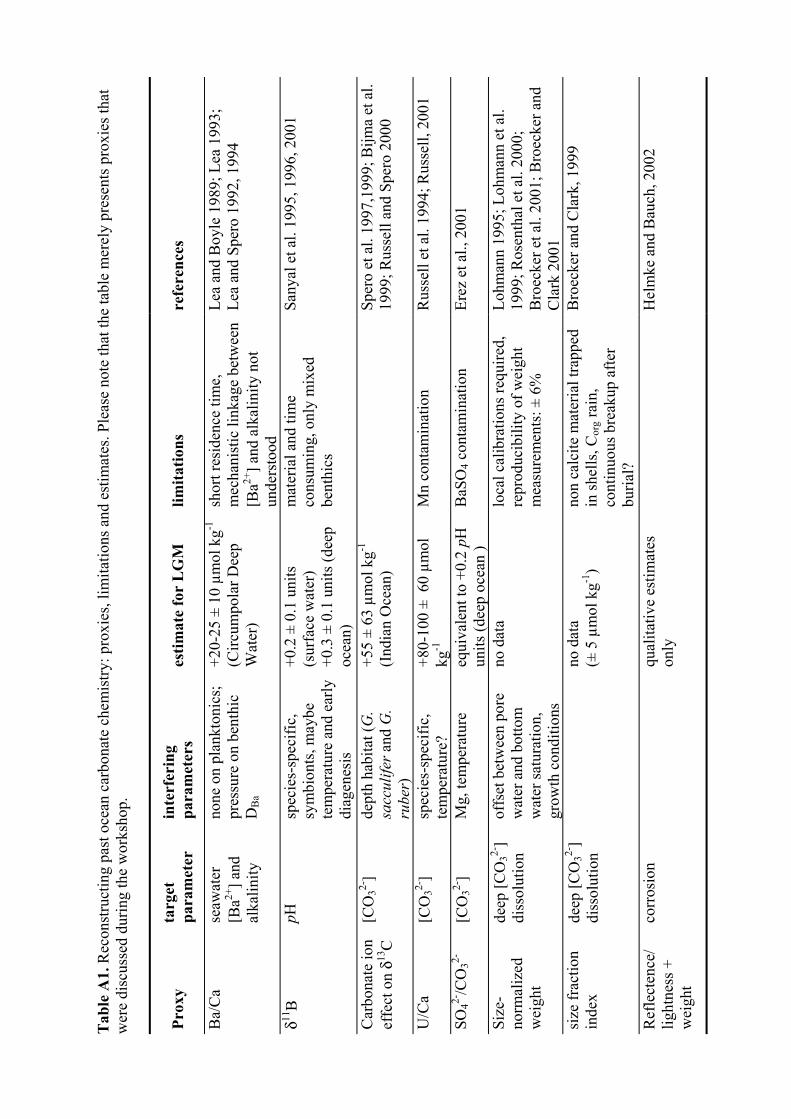
Tab
le A
1. R
econ
stru
ctin
g pa
st o
cean
car
bona
te c
hem
istry
: pro
xies
, lim
itatio
ns a
nd e
stim
ates
. Ple
ase
note
that
the
tabl
e m
erel
y pr
esen
ts p
roxi
es th
at
wer
e di
scus
sed
durin
g th
e w
orks
hop.
Pr
oxy
targ
et
para
met
er
inte
rfer
ing
para
met
ers
estim
ate
for
LG
M
limita
tions
re
fere
nces
Ba/
Ca
se
awat
er[B
a2+] a
nd
alka
linity
none
on
plan
kton
ics;
pr
essu
re o
n be
nthi
c D
Ba
+20-
25 ±
10
µmol
kg-1
(C
ircum
pola
r Dee
p W
ater
)
shor
t res
iden
ce ti
me,
m
echa
nist
ic li
nkag
e be
twee
n [B
a2+] a
nd a
lkal
inity
not
un
ders
tood
Lea
and
Boy
le 1
989;
Lea
199
3;
Lea
and
Sper
o 19
92, 1
994
δ11B
pH
spec
ies-
spec
ific,
sym
bion
ts, m
aybe
te
mpe
ratu
re a
nd e
arly
di
agen
esis
+0.2
± 0
.1 u
nits
(s
urfa
ce w
ater
) +0
.3 ±
0.1
uni
ts (d
eep
ocea
n)
mat
eria
l and
tim
e co
nsum
ing,
onl
y m
ixed
be
nthi
cs
Sany
al e
t al.
1995
, 199
6, 2
001
Car
bona
te io
n ef
fect
on
δ13C
[C
O32-
] de
pth
habi
tat (
G.
sacc
ulife
r and
G.
rube
r)
+55
± 63
µm
ol k
g-1
(Ind
ian
Oce
an)
Sp
ero
et a
l. 19
97,1
999;
Bijm
a et
al.
1999
; Rus
sell
and
Sper
o 20
00
U/C
a [C
O32-
] sp
ecie
s-sp
ecifi
c,
tem
pera
ture
? +8
0-10
0 ±
60
µmol
kg
-1
Mn
cont
amin
atio
n R
usse
ll et
al.
1994
; Rus
sell,
200
1
SO42-
/CO
32-
[CO
32-]
Mg,
tem
pera
ture
eq
uiva
lent
to +
0.2
pH
units
(dee
p oc
ean
) B
aSO
4 con
tam
inat
ion
Erez
et a
l., 2
001
Size
-no
rmal
ized
w
eigh
t
deep
[CO
32-]
diss
olut
ion
offs
et b
etw
een
pore
w
ater
and
bot
tom
w
ater
satu
ratio
n,
grow
th c
ondi
tions
no d
ata
loca
l cal
ibra
tions
requ
ired,
re
prod
ucib
ility
of w
eigh
t m
easu
rem
ents
: ± 6
%
Lohm
ann
1995
; Loh
man
n et
al.
1999
; Ros
enth
al e
t al.
2000
; B
roec
ker e
t al.
2001
; Bro
ecke
r and
C
lark
200
1 si
ze fr
actio
n in
dex
deep
[CO
32-]
diss
olut
ion
no
dat
a
(± 5
µm
ol k
g-1)
non
calc
ite m
ater
ial t
rapp
ed
in sh
ells
, Cor
g rai
n,
cont
inuo
us b
reak
up a
fter
buria
l?
Bro
ecke
r and
Cla
rk, 1
999
R
efle
cten
ce/
light
ness
+
wei
ght
corr
osio
n
qu
alita
tive
estim
ates
only
Hel
mke
and
Bau
ch, 2
002
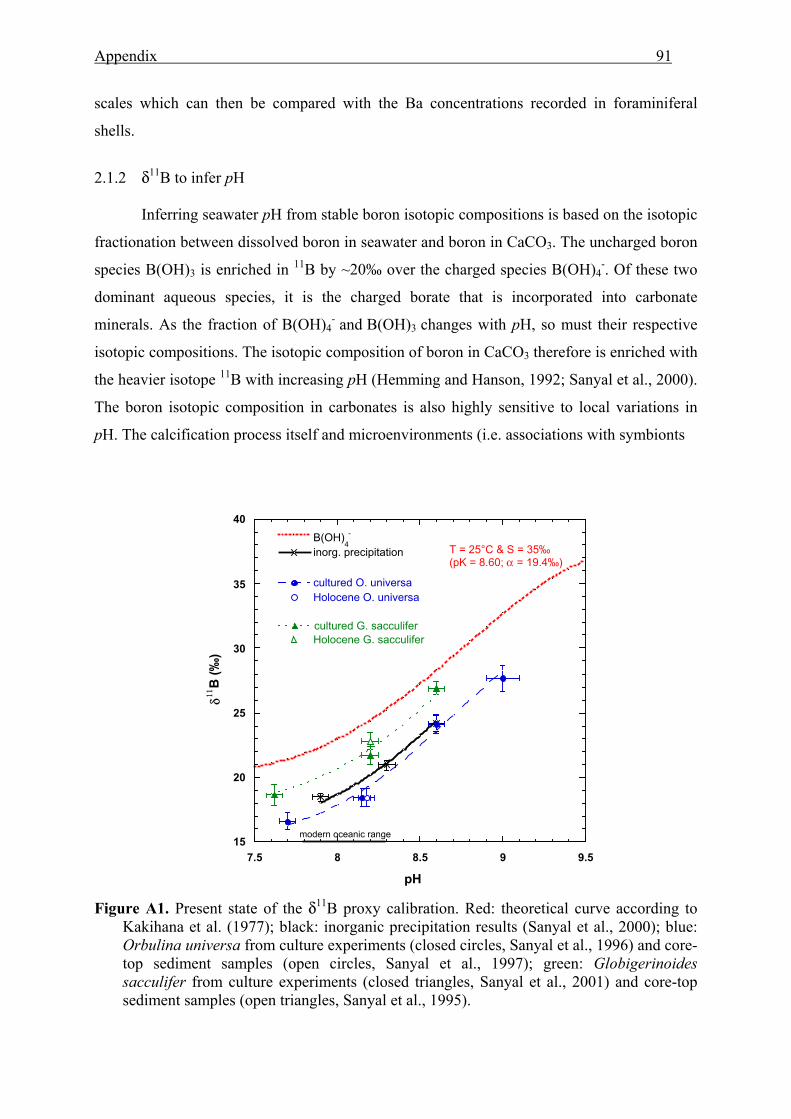
Appendix 91
scales which can then be compared with the Ba concentrations recorded in foraminiferal
shells.
2.1.2 δ11B to infer pH
Inferring seawater pH from stable boron isotopic compositions is based on the isotopic
fractionation between dissolved boron in seawater and boron in CaCO3. The uncharged boron
species B(OH)3 is enriched in 11B by ~20‰ over the charged species B(OH)4-. Of these two
dominant aqueous species, it is the charged borate that is incorporated into carbonate
minerals. As the fraction of B(OH)4- and B(OH)3 changes with pH, so must their respective
isotopic compositions. The isotopic composition of boron in CaCO3 therefore is enriched with
the heavier isotope 11B with increasing pH (Hemming and Hanson, 1992; Sanyal et al., 2000).
The boron isotopic composition in carbonates is also highly sensitive to local variations in
pH. The calcification process itself and microenvironments (i.e. associations with symbionts
15
20
25
30
35
40
7.5 8 8.5 9 9.5
inorg. precipitation
cultured O. universa
B(OH)4-
Holocene G. sacculifer
Holocene O. universa
cultured G. sacculifer
δ11B
(‰)
pH
modern oceanic range
T = 25°C & S = 35‰(pK = 8.60; α = 19.4‰)
Figure A1. Present state of the δ11B proxy calibration. Red: theoretical curve according to Kakihana et al. (1977); black: inorganic precipitation results (Sanyal et al., 2000); blue: Orbulina universa from culture experiments (closed circles, Sanyal et al., 1996) and core-top sediment samples (open circles, Sanyal et al., 1997); green: Globigerinoides sacculifer from culture experiments (closed triangles, Sanyal et al., 2001) and core-top sediment samples (open triangles, Sanyal et al., 1995).

Appendix 92
or precipitation within extrapallial fluids) must therefore be considered. While the planktonic
foraminifer G. sacculifer and benthic foraminifera appear to incorporate δ11B with little or no
fractionation compared to the theoretical curve, O. universa shows an offset from those
foraminifera by ~3.3 ‰ (Sanyal et al., 2001). The offset was suggested to be due to a vital
effect, although its nature could not be explained: both planktonic species are spinose and
symbiont-bearing and should therefore react similarly.
The effect of symbiont photosynthesis has recently been investigated in a diffusion-
reaction model by Zeebe at al. (subm.). They calculated a constant but significant offset
between δ11B in planktonic foraminiferal calcite and the isotopic signature of B(OH)4- in the
seawater medium. Recent laboratory culture data (Hönisch et al., subm.) are in good
agreement with the model results.
Stable boron isotopic analyses, using negative thermal ionisation mass spectrometry
(nTIMS), have several complications. First of all, the technique requires several hours of
permanent operator assistance and numerous replicate analyses until accurate values can be
obtained. Second, to achieve a reproducible result, approximately 4 ng boron are required per
analysis. Since foraminifera contain 5-15 ppm B (Hemming et al., 1998), up to 10 mg
foraminiferal calcite are needed per sample (when considering weight loss during cleaning
and multiple replicate analyses). Especially for the investigation of deep water chemistry the
second point is crucial, as the abundance of benthic foraminifera is too low to routinely allow
single-species analyses. Sanyal et al. (1995) therefore combined several species for their
deepwater record despite possible differences in habitat (epifaunal/infaunal) characterized by
a range of pH conditions, and species-specific offsets like the ones found for planktonic
foraminifera (Sanyal et al., 2001). These factors (Sanyal et al., 1997; Sanyal et al., 1996) may
have biased the obtained value, which suggested a 0.3 pH units increase for last glacial
deepwater (Sanyal et al., 1995).
In order to solve the problems named above, it is desirable to reduce the amount of
material required for analyses, to speed up measurements and to generally expedite the
analytical procedure so that the investigation of past ocean acidity can be realised extensively
in the future.

Appendix 93
2.1.3 Deconvolution of the carbonate ion effect to infer [CO32-]
This approach is based on the deconvolution of foraminiferal δ13C records to calculate
the change in surface [CO32-] and δ13CΣCO2 through time: The stable carbon and oxygen
isotopic compositions of planktonic foraminifera decrease with increasing carbonate ion
concentration (Bijma et al., 1999; Spero et al., 1999). Among the investigated planktonic
foraminifera, G. sacculifer and G. ruber share the same habitat but the slope in δ13C vs.
[CO32-] is twice as large in G. ruber as in G. sacculifer. This species-specific difference is
used to distinguish between the effect of [CO32-] and a simultaneous change in δ13CΣCO2.
Application to the sediment record leads to the estimate of +55 ± 63 µmol kg-1 [CO32-] for the
Indian Ocean during the last glacial (Spero et al., 1999). Unfortunately, this method is
restricted to tropical surface waters, where G. sacculifer and G. ruber occur.
2.1.4 U/Ca to infer [CO3
2-]
Laboratory experiments revealed that U/Ca in planktonic foraminifera shells is
inversely related to [CO32-] (Russell, 2001). The symbiont-barren G. bulloides incorporates
approximately twice U/Ca than the symbiont-bearing O. universa at the same [CO32-]. No
consistent temperature effect on the record has been found above 19°C. Application of the
U/Ca relationship to Caribbean cores suggested that glacial [CO32-] was 80-100 ± 60 µmol kg-
1 higher than during the Holocene.
Although the approach is generally promising, the study of several sediment cores
revealed that contamination by Mn-carbonates places a significant diagenetic overprint on the
incorporated U/Ca which may limit the general applicability of this proxy to sediments above
the redox front.
2.1.5 SO4
2-/ CO32- to infer [CO3
2-]
In laboratory culture experiments, Erez et al. (2001) observed a constant distribution
coefficient between SO42-/CO3
2- in the shells of benthic and planktonic foraminifera and SO42-
/CO32- ratio in seawater. Since the seawater SO4
2- inventory is not expected to have changed
on glacial-interglacial time scales, [CO32-] can be reconstructed. In situ calibrations of this
proxy in the Gulf of Eilat gave similar results to those of the laboratory experiments.
However, in the Little Bahama Bank a temperature effect was revealed which may have been
caused by changes in the Mg ion content, apparently affecting the SO42- content of
foraminiferal shells. Correction of this temperature effect leads to the empirical negative

Appendix 94
correlation between seawater [CO32-] and foraminiferal SO4
2-/CO32- as found in laboratory
culture experiments. Preliminary comparisons of SO42-/CO3
2- from Holocene and glacial
benthic foraminifera show variability in pH similar in magnitude to that estimated
independently from δ11B (approximately 0.2 pH units increase in the glacial deep Pacific).
The advantage of this proxy is the very small sample size required for routine
measurements using a Magnetic Sector ICP-MS. Hence it is practical to be used for benthic
foraminifera from deep sea sediments. However, the proxy is still under development and not
much is known about its limitations. One possible interference may be the contamination with
extraneous phases like barite (BaSO4).
2.2 Carbonate preservation 2.2.1 Size-normalized shell weight
The average mass of planktonic foraminifera is primarily determined by their size, but
there is a measurable secondary relationship of shell mass to water depth (Lohmann, 1995;
Lohmann et al., 1999; Rosenthal et al., 2000). Due to dissolution, the size-normalized mass of
nearly all species is lower in deeper water than it is in shallow water, and the decrease is
continuous over a wide range of carbonate saturation states, even well above the calcite
lysocline. Based on shells of the three species G. sacculifer, Pulleniatina obliquiloculata and
Neogloboquadrina dutertrei, Broecker and Clark (2001a) determined an average weight-loss
slope of 0.3 ± 0.05 µg (µmol kg-1)-1 decrease in pressure-corrected deep sea carbonate ion
concentration. This relationship allows estimates of changes in seawater carbonate content
from the size-normalized mass of planktonic foraminifera.
To use the relationship as a paleocarbonate ion proxy, this method requires that the
offset between pore and bottom water saturation is constant. However, numerous
investigations (Archer et al., 1989; Berelson et al., 1990; Berelson et al., 1994; Hales and
Emerson, 1996; Hales and Emerson, 1997a; Jahnke et al., 1994; Jahnke et al., 1997) have
applied microelectrodes and benthic flux chambers to validate the theory of respiration-driven
dissolution in-situ (Table A2, see also section 2.3.1). They conclude that 40-60 % of the
calcite dissolution above the saturation horizon can be attributed to metabolic processes. The
amount of organic matter reaching the seafloor varies between sites and depends on depth.
Assuming increased productivity on glacial time scales, the magnitude of this effect might
have been even stronger. Application of this proxy should therefore be restricted to locations
where strong changes in paleoproductivity are not expected.
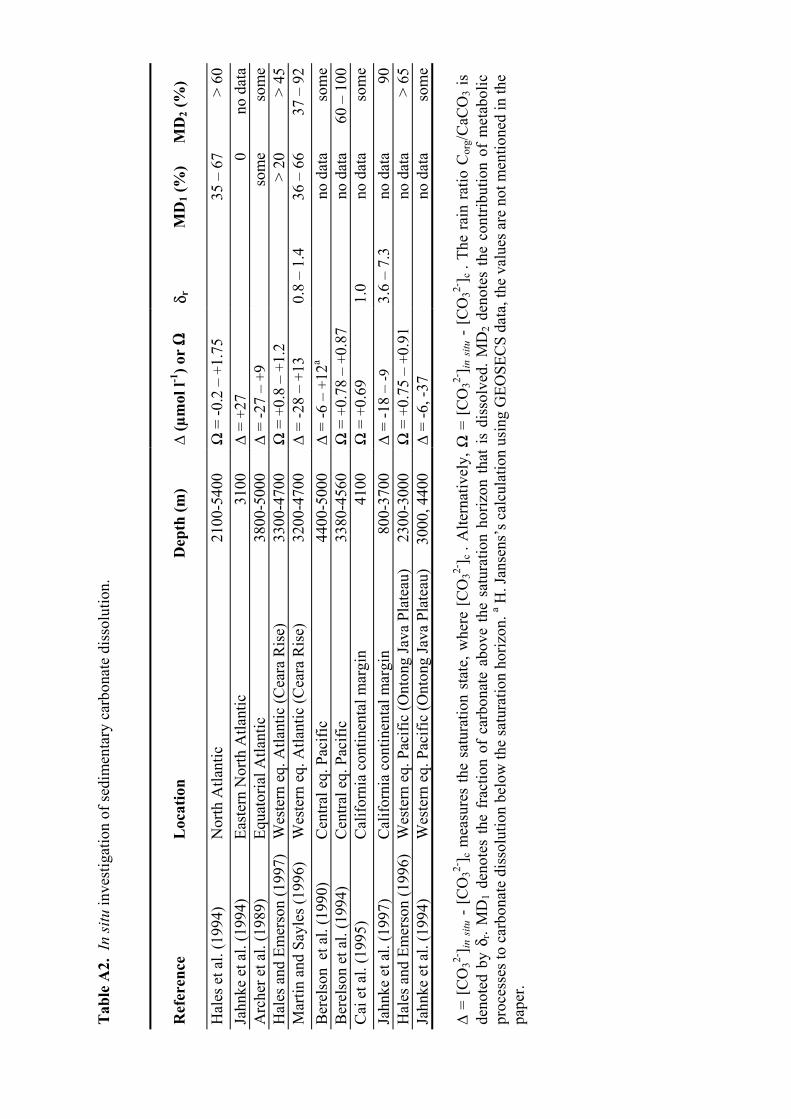
Tab
le A
2. I
n si
tu in
vest
igat
ion
of se
dim
enta
ry c
arbo
nate
dis
solu
tion.
R
efer
ence
L
ocat
ion
Dep
th (m
)
∆ (µ
mol
l-1) o
r Ω
δ r
M
D1 (
%)
MD
2 (%
)
Hal
es e
t al.
(199
4)
Nor
th A
tlant
ic
2100
-540
0
Ω =
-0.2
– +
1.75
35 –
67
> 60
Ja
hnke
et a
l. (1
994)
Ea
ster
n N
orth
Atla
ntic
31
00
∆ =
+27
0
noda
taA
rche
r et a
l. (1
989)
Eq
uato
rial A
tlant
ic
3800
-500
0
∆ =
-27
– +9
so
me
som
eH
ales
and
Em
erso
n (1
997)
W
este
rn e
q. A
tlant
ic (C
eara
Ris
e)
3300
-470
0
Ω =
+0.
8 –
+1.2
> 20
> 45
M
artin
and
Say
les (
1996
) W
este
rn e
q. A
tlant
ic (C
eara
Ris
e)
3200
-470
0
∆ =
-28
– +1
3 0.
8 –
1.4
36 –
66
37 –
92
Ber
elso
n e
t al.
(199
0)
Cen
tral e
q. P
acifi
c 44
00-5
000
∆
= -6
– +
12a
no
dat
aso
me
B
erel
son
et a
l. (1
994)
C
entra
l eq.
Pac
ific
3380
-456
0
Ω =
+0.
78 –
+0.
87
no
dat
a60
– 1
00
Cai
et a
l. (1
995)
C
alifo
rnia
con
tinen
tal m
argi
n 41
00
Ω =
+0.
69
1.0
no d
ata
som
e
Jahn
ke e
t al.
(199
7)
Cal
iforn
ia c
ontin
enta
l mar
gin
800-
3700
∆
= -1
8 –
-9
3.6
– 7.
3 no
dat
a90
H
ales
and
Em
erso
n (1
996)
W
este
rn e
q. P
acifi
c (O
nton
g Ja
va P
late
au)
2300
-300
0
Ω =
+0.
75 –
+0.
91
no
dat
a>
65
Jahn
ke e
t al.
(199
4)
Wes
tern
eq.
Pac
ific
(Ont
ong
Java
Pla
teau
)30
00, 4
400
∆
= -6
, -37
no d
ata
som
e
∆ =
[CO
32-] in
situ
- [
CO
32-] c
mea
sure
s th
e sa
tura
tion
stat
e, w
here
[C
O32-
] c . A
ltern
ativ
ely,
Ω =
[C
O32-
] in s
itu -
[C
O32-
] c . The
rai
n ra
tio C
org/C
aCO
3 is
de
note
d by
δr.
MD
1 de
note
s th
e fr
actio
n of
car
bona
te a
bove
the
satu
ratio
n ho
rizon
that
is d
isso
lved
. MD
2 de
note
s th
e co
ntrib
utio
n of
met
abol
ic
proc
esse
s to
carb
onat
e di
ssol
utio
n be
low
the
satu
ratio
n ho
rizon
. a H. J
anse
ns’s
cal
cula
tion
usin
g G
EOSE
CS
data
, the
val
ues a
re n
ot m
entio
ned
in th
e pa
per.
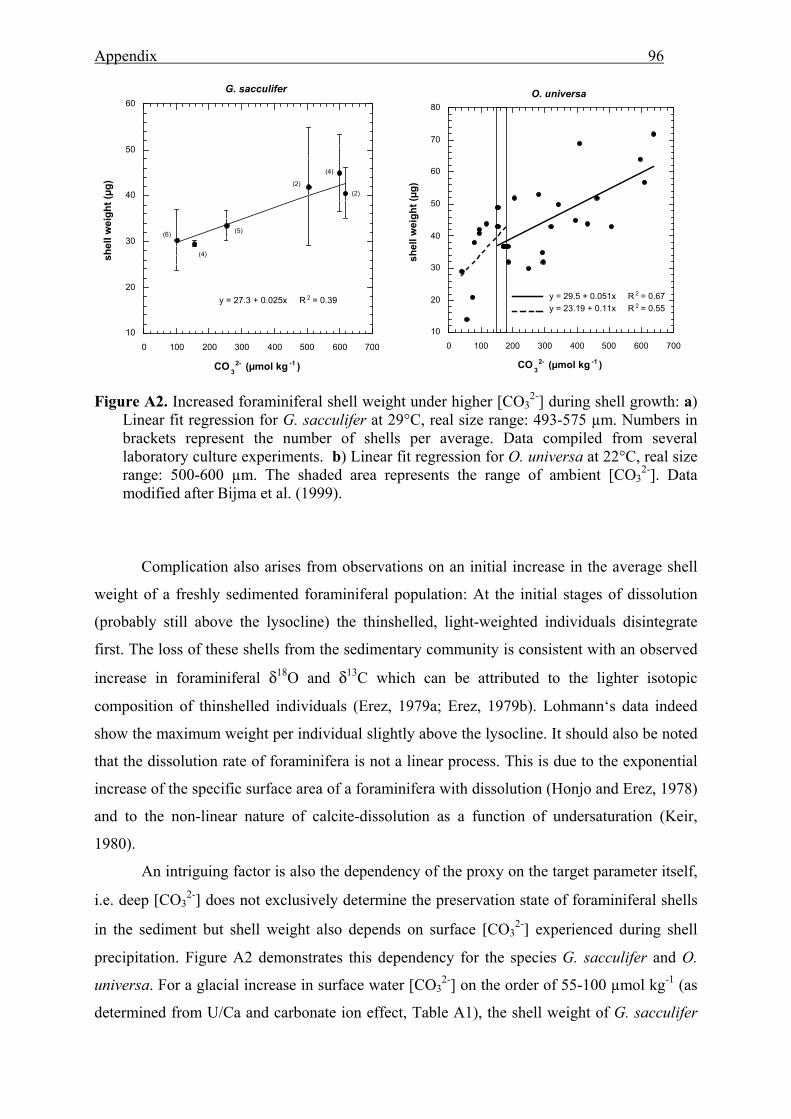
Appendix
10
20
30
40
50
60
0 100 200 300 400 500 600 700
G. sacculifersh
ell w
eigh
t (µg
)
CO 32- (µmol kg -1 )
(6)
(4)
(5)
(2)
(4)
(2)
y = 27.3 + 0.025x R 2 = 0.39
96
10
20
30
40
50
60
70
80
0 100 200 300 400 500 600 700
O. universa
shel
l wei
ght (
µg)
CO 32- (µmol kg -1 )
y = 29.5 + 0.051x R 2 = 0.67y = 23.19 + 0.11x R 2 = 0.55
Figure A2. Increased foraminiferal shell weight under higher [CO32-] during shell growth: a)
Linear fit regression for G. sacculifer at 29°C, real size range: 493-575 µm. Numbers in brackets represent the number of shells per average. Data compiled from several laboratory culture experiments. b) Linear fit regression for O. universa at 22°C, real size range: 500-600 µm. The shaded area represents the range of ambient [CO3
2-]. Data modified after Bijma et al. (1999).
Complication also arises from observations on an initial increase in the average shell
weight of a freshly sedimented foraminiferal population: At the initial stages of dissolution
(probably still above the lysocline) the thinshelled, light-weighted individuals disintegrate
first. The loss of these shells from the sedimentary community is consistent with an observed
increase in foraminiferal δ18O and δ13C which can be attributed to the lighter isotopic
composition of thinshelled individuals (Erez, 1979a; Erez, 1979b). Lohmann‘s data indeed
show the maximum weight per individual slightly above the lysocline. It should also be noted
that the dissolution rate of foraminifera is not a linear process. This is due to the exponential
increase of the specific surface area of a foraminifera with dissolution (Honjo and Erez, 1978)
and to the non-linear nature of calcite-dissolution as a function of undersaturation (Keir,
1980).
An intriguing factor is also the dependency of the proxy on the target parameter itself,
i.e. deep [CO32-] does not exclusively determine the preservation state of foraminiferal shells
in the sediment but shell weight also depends on surface [CO32-] experienced during shell
precipitation. Figure A2 demonstrates this dependency for the species G. sacculifer and O.
universa. For a glacial increase in surface water [CO32-] on the order of 55-100 µmol kg-1 (as
determined from U/Ca and carbonate ion effect, Table A1), the shell weight of G. sacculifer

Appendix 97
thus increases by approximately 1.6–2.9 µg – independent of concomitant changes in deep
water saturation. A detailed examination of available culture and sediment trap data is
required to better estimate the magnitude of the observed growth effects and to determine how
temperature affects the carbonate dependent growth variability at a certain locality over
glacial/interglacial time scales.
2.2.2 Reflectance/ lightness of foraminiferal shells
A qualitative estimate of carbonate corrosion prior to foraminiferal test fragmentation
is the combination of weight and light reflectance measurements of planktonic foraminiferal
tests of the polar species Neogloboquadrina pachyderma (sin.). The method was developed
by Helmke and Bauch (2002) and is restricted to regions and time intervals where carbonate
preservation is generally good. Carbonate corrosion leads to changes in the surface structure
of the calcite crystals and has a profound influence on the reflectivity of foraminiferal tests.
An inverse relationship between light reflectence and weight was found. Application to
Nordic Sea sediments revealed better preservation during glacial periods, which is consistent
with higher deep sea [CO32-] for this time scale. However, the method is yet far from being
used for quantitative estimates.
2.3 Estimating coccolithophorid paleoproductivity
Coccolithophorids are major contributors to the biogenic carbonate content in deep-sea
sediments (Archer et al., 2000; Milliman, 1993; Westbroek et al., 1993). Recently, there has
been increased interest in utilizing the elemental and isotopic chemistry of coccoliths. The
chemistry of coccolith carbonate may record different information than that of foraminiferal
carbonate because coccolithophorids, unlike foraminifera, are primary producers. Knowledge
about their paleoproductivity is of major importance for e.g. rain ratio estimates and
δ13Calkenone-based paleobarometer reconstructions (for review: Laws et al., 2001).
One limitation in the use of coccolith carbonate for geochemical studies has been their very
small size and therefore the inability to seperate monospecific coccolith assemblages for
analysis. New techniques now permit separation of fractions whose carbonate is highly
dominated (>70 % and often >90 %) by a single coccolith species (Stoll and Ziveri, in press).
As with foraminifera, calcite produced by different species of coccolithophorids has different
minor element partitioning and oxygen and carbon isotope fractionations. Specific
examination now opens a new field for paleoceanography.

Appendix 98
2.3.1 Coccolith Sr/Ca and stable carbon and oxygen isotopes to infer growth rate and cell size
The Sr/Ca ratio of coccoliths has been recently proposed as a potential indicator of
past coccolithophorid growth rates. The hypothesis is based on correlations between Sr/Ca in
polyspecific coccolith samples and primary productivity, alkenone-estimated growth rates,
and CaCO3 rain rates in deep sediment traps (e.g. Stoll and Schrag, 2000) across the
Equatorial Pacific upwelling region. Subsequently, a number of culture studies have
investigated controls over Sr/Ca ratios in coccoliths of several species. For identical
temperature and media composition, Sr partitioning is linearly related to rates of calcite
production/cell (Stoll et al., 2001). Higher calcification per cell at higher growth rates
observed in light-limited cultures of Gephyrocapsa oceanica, Calcidiscus leptoporus and
Emiliania huxleyi cultures (Paasche, in press; Stoll et al., in press) may suggest that active
uptake and calcification become increasingly important at higher growth rates. If coccolith
Sr/Ca is a reliable indicator of coccolithophorid productivity, it provides an index of past
productivity directly recorded by a primary producer. Furthermore, productivity estimates
from coccolith Sr/Ca do not rely on conventional determinations of sediment accumulation
rates which are often imprecise.
With regard to oxygen and carbon isotope fractionations, culture studies indicate
different nonequilibrium effects for different species of coccolithophorids (Dudley et al.,
1986; Ziveri et al., 2000; Ziveri et al., in prep.). These nonequilibrium effects appear to reflect
changing ecological and physiological responses of the organisms. In light- and nutrient-
replete cultures, the non-equilibrium effects in δ18O correlate highly with cell division rates
across a range of species. At similar calcification temperature and media composition, the
growth rates of the most common living species, E. huxleyi and G. oceanica, are strong and
δ18O is 3 ‰ offset with respect to equilibrium composition. In contrast, species with low
growth rates such as Umbilicosphaera sibogae var. foliosa have a δ18O fractionation effect of
~-2 ‰. Systematic relationships were also found between the carbon and oxygen isotopic
composition of the coccolith calcite for each species and the surface area/volume ratio of the
cells, which determines the diffusive flux of CO2 available to the cell (Ziveri et al., in prep.).
Clearly, more work is needed to test the validity of this proxy in constraining
coccolithophorid growth rates. Nevertheless, we are encouraged that qualitative or
quantitative determination of past variations in species-specific algal growth rates may be
possible.
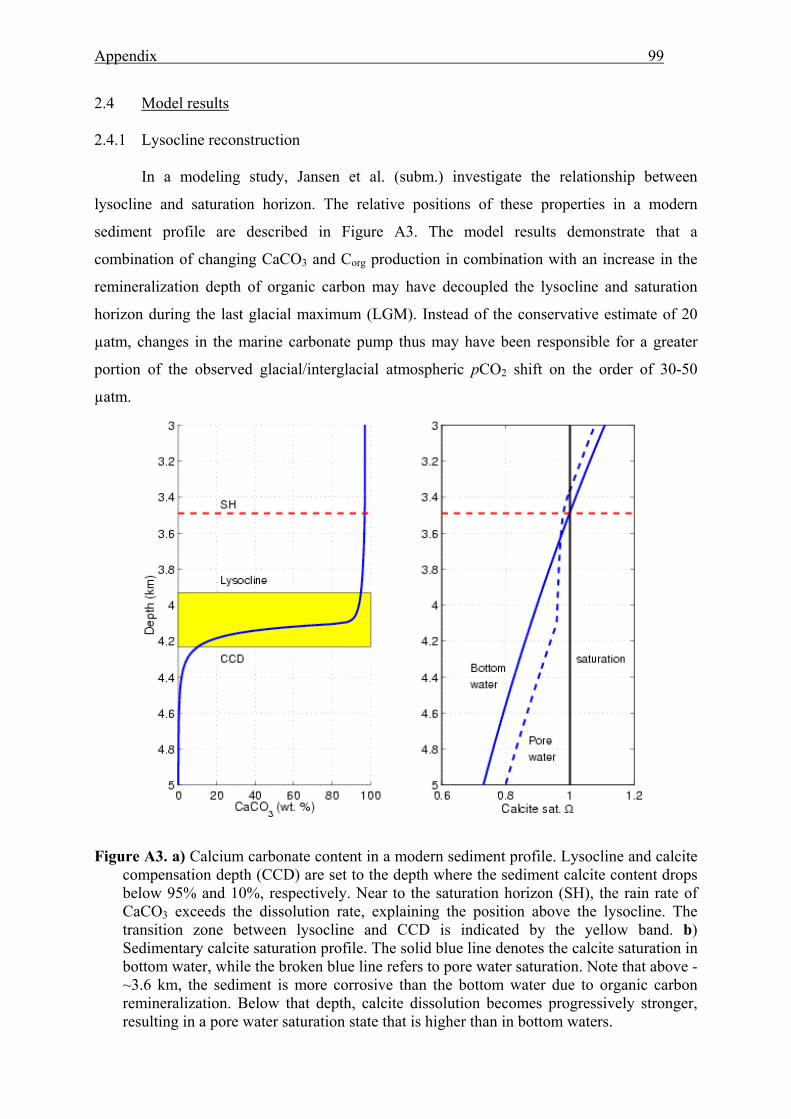
Appendix 99
2.4 Model results 2.4.1 Lysocline reconstruction
In a modeling study, Jansen et al. (subm.) investigate the relationship between
lysocline and saturation horizon. The relative positions of these properties in a modern
sediment profile are described in Figure A3. The model results demonstrate that a
combination of changing CaCO3 and Corg production in combination with an increase in the
remineralization depth of organic carbon may have decoupled the lysocline and saturation
horizon during the last glacial maximum (LGM). Instead of the conservative estimate of 20
µatm, changes in the marine carbonate pump thus may have been responsible for a greater
portion of the observed glacial/interglacial atmospheric pCO2 shift on the order of 30-50
µatm.
Figure A3. a) Calcium carbonate content in a modern sediment profile. Lysocline and calcite
compensation depth (CCD) are set to the depth where the sediment calcite content drops below 95% and 10%, respectively. Near to the saturation horizon (SH), the rain rate of CaCO3 exceeds the dissolution rate, explaining the position above the lysocline. The transition zone between lysocline and CCD is indicated by the yellow band. b) Sedimentary calcite saturation profile. The solid blue line denotes the calcite saturation in bottom water, while the broken blue line refers to pore water saturation. Note that above -~3.6 km, the sediment is more corrosive than the bottom water due to organic carbon remineralization. Below that depth, calcite dissolution becomes progressively stronger, resulting in a pore water saturation state that is higher than in bottom waters.

Appendix 100
Geological records suggest that during the last glacial, the Atlantic lysocline was 0.3-1
km shallower (Crowley, 1983; Curry and Lohmann, 1986) than today, while it was about 0.8
km deeper in the Pacific Ocean (Farrell and Prell, 1989). Assuming that the lysocline has not
changed its position relative to the saturation horizon, these changes roughly correspond to a
decrease in atmospheric pCO2 by approximately 20 µatm (Broecker et al., 2001). However,
ice core observations suggest a glacial/interglacial shift in atmospheric pCO2 of 80 µatm
(Neftel et al., 1982; Petit et al., 1999). To bring the two records into line, additional reduction
in atmospheric pCO2 can be brought about by decoupling the lysocline from the saturation
horizon, due to respiration-driven carbonate dissolution in the upper 10 cm of sediments, as
observed e.g. by Hales and Emerson (1996). By changing the amount of organic carbon
arriving at the seafloor, the amount of carbonate dissolution above the saturation horizon can
change dramatically, shoaling the lysocline relative to the saturation horizon.
In contrast to the model used by Archer and Maier-Reimer (1994), Jansen et al.
(subm.) do not consider the dissolution of CaCO3 in the water column, as until now, the
underlying mechanism of this proposed feature has not been found (Jansen and Wolf-
Gladrow, 2001; Jansen et al., 2002; Milliman et al., 1999). Although this might result in an
overestimation of the decoupling, it does not affect glacial/interglacial changes in the whole
ocean carbonate inventory. Archer and Maier-Reimer (1994) tested scenarios for a glacial
ocean where Corg production was three times as high and CaCO3 production 60 % lower than
at present. They concluded that such a variation, operated by a shift from calcareous to
siliceous organisms during glacial times, might have driven atmospheric pCO2 to glacial
values. More recently, Sigman et al. (1998) argued that an increase in respiration-driven
calcite dissolution has no significant effect on the decoupling of lysocline and saturation
horizon as increased shallow water dissolution of carbonates would deepen the lysocline due
to mass balance considerations. However, it is questionable whether carbonate production and
dissolution are balanced at all (Milliman, 1993).
In contrast to Sigman et al. (1998), Jansen et al. (subm.) demonstrate that a decoupling
of lysocline and saturation horizon is possible. However, respiration-driven dissolution can
only significantly influence the atmospheric pCO2 when the export ratio depends on primary
production. Otherwise, the amount of organic carbon reaching deep-sea sediments would be
too small. With fixed export ratios, variability in CaCO3 production has a greater influence on
atmospheric pCO2 than variability in organic carbon production has.
Organic carbon productivity has been assumed to have increased during the LGM in
the range of up to +100 % relative to today (Berger et al., 1989; Kumar et al., 1995; Paytan et

Appendix 101
al., 1996), while CaCO3 productivity ranged between –60 % and +60 % relative to modern
times (Archer et al., 2000; Broecker and Henderson, 1998; Kumar et al., 1995). These
estimates yield Corg: CaCO3 rain ratios that are comparable to modern rain ratios in high
productivity areas. Thus, glacial pCO2 levels of ~ 230-250 µatm pCO2 are achieved within the
assumed rain ratio ranges. The result of Archer and Maier-Reimer (1994), who found that the
glacial to interglacial shift in atmospheric pCO2 is completely explainable by a decoupling
mechanism could not be reproduced by Jansen et al. (subm.). Rather, their model indicates
that ~40-60 % of the glacial pCO2 reduction may be attributed to changes in the marine
carbonate pump.
3. Recommendations
The proxies and model results discussed above demonstrate that the ideal proxy-
relationship does not exist. The record of a target parameter may be compromised in several
ways. However, the influence of many of the interfering parameters could be corrected for, if
the nature of the interferences were known and other proxies applied to estimate the
magnitude of the specific error. To uncover possible interferences, further laboratory culture
experiments on recorder organisms such as foraminifera, coccolithophorids and corals are
required. The magnitude of these effects needs to be quantified through additional field data
and process modeling. Of equal importance is a better understanding of the biomineralization
mechanisms of the major groups that provide proxies (i.e. foraminifera, corals,
coccolithophorids and diatoms). Only with such an understanding it will be possible to
explain the deviations of proxy relationships from thermodynamic predictions and to verify
whether such deviations are constant or require to be corrected for. Finally, the effects of
dissolution and other diagenetic changes need to be studied in laboratory experiments and at
depth.
3.1 Multi-proxy approach
The general advantage of preservational proxies (except examination with scanning
electron microscopy) is their non-destructive character. The descriptive analysis of weight,
light reflectance and general intactness leaves foraminiferal samples untouched and allows
further chemical investigation. Apart from providing additional information on bottom water
carbonate saturation, knowledge about the preservation state can furthermore help to elucidate
possible interferences with dissolution effects on other chemical proxies like Mg/Ca. Adding
chemical analyses to the interpretation of preservational proxies may reduce their own

Appendix 102
uncertainties. This could be for instance the application of U/Ca and SO42-/CO3
2- to estimate
surface water carbonate chemistry and its possible influence on weight and size of planktonic
foraminifera.
Probably the most detailed knowledge on interferences exists in the field of chemical
proxies in biogenic carbonates. Several culture studies on living foraminifera led to the
establishment of empirical proxy-relationships and the discovery of some of their limitations
(Bemis et al., 2000; Bijma et al., 1999; Erez and Luz, 1982; Erez et al. 2001; Lea et al. 1995,
1999; Lea and Spero 1992, 1994; Mashiotta et al. 1997; Rink et al., 1998; Russell, 2001;
Sanyal et al., 2001; Sanyal et al., 1996; Spero et al., 1997; Zeebe, 1999). Supplementary
information to many of these proxies could be obtained by application of multi-element ICP-
MS analysis on the same sample (Lea and Martin, 1996). However, for many parameters a
reliable proxy does not even exist.
A crucial requirement to minimize uncertainties in the background information is
therefore the search for new proxies for parameters such as salinity, productivity or deep sea
carbonate chemistry. A first approach into this direction is given by the application of time-
of-flight secondary ion mass spectrometry (TOF-SIMS, see section 3.3) to foraminiferal shell
analysis. Furthermore, new recorders and proxies need to be found to reconstruct the
conditions below the calcite compensation depth, where carbonates are already dissolved and
no longer available. All in all, there is a clear need to combine available chemical and
preservational proxies in order to improve the quality of paleoceanographic reconstructions.
3.2 Laboratory and field verification
As evident from previous sections, the establishment of empirical relationships in
laboratory and field experiments and calcification studies are important steps towards the
mechanistic understanding of a proxy and its paleoceanographic robustness. Especially for the
recently developed proxies it would be desirable to investigate these in more detail. Important
aspects would be the U/Ca-calibration of the warm water species G. sacculifer and G. ruber
and to obtain better information on the temperature dependency of the already existing
relationships. The boron isotopic composition of planktonic foraminifera has recently been
suggested to be biased by temperature and partial dissolution (Wara et al., 2001). To question
this finding, a set of planktonic foraminifera from a dissolution experiment is currently being
analyzed. However, temperature effects will better be investigated in culture experiments,
where other parameters can be kept constant. The effects of temperature and Mg on SO42-
/CO32- are currently being studied.

Appendix 103
Although they are the most important recorders of deep water (carbonate) chemistry,
benthic foraminifera are poorly represented in terms of experimentally calibrated proxy-
relationships. Culture experiments are therefore required to expand our knowledge on already
established and new proxies like U/Ca and SO42-/CO3
2-. It would be interesting to investigate
whether the carbonate ion effect on δ13C and δ18O is applicable to benthic foraminifera as
well.
Culture experiments are an important tool but reveal a rather restricted picture of a
proxy. Under natural conditions, planktonic foraminifera migrate vertically during their
ontogeny and finally sink towards the seafloor, thereby experiencing variable water column
conditions that modify their shell chemistry (Erez and Honjo, 1981; Lohmann, 1995). To
further open this black box, plankton tow samples, sediment trap material, and surface
sediment samples are needed to quantify the effects of this modification. Furthermore,
dissolution experiments under simulated natural conditions are suggested to elucidate if part
of the CaCO3 is dissolved preferentially, which proxies are affected by dissolution and to
what extent.
With regard to possible contaminating phases, the SO42-/CO3
2- proxy needs to be
investigated in the light of special cleaning efforts. For foraminiferal Ba/Ca analyses, barite
contamination was found to be removeable by cleaning with an alkaline DTPA
(diethylenetriamine-pentaacetic acid) treatment (Lea and Boyle, 1989). Cleaning experiments
may help to determine the robustness of this new proxy.
3.3 Recorders for deep water carbonate chemistry
As indicated in section 2.4.1, an increase of the marine carbonate pump under glacial
conditions may have led to increased respiration-driven carbonate dissolution in the sediment.
When arguing with sediment dissolution, the related diffusion of [CO32-] out of the sediment
into the overlying bottom water needs to be considered to possibly having biased the glacial
record, favouring locally more alkaline conditions than actually present in the deep water. At
least for the Holocene this limitation can be tested. For instance the difference in δ11B
between G. sacculifer and benthic foraminifera appears to be similar in magnitude to the
expected difference in pH between surface and deep water (Sanyal et al., 1995). However,
how can we be sure that benthic foraminifera, that are living right at the sediment-water
interface, always recorded deep water carbonate chemistry and not an anomalous local
environment? In fact, recent data by Anderson and Archer (2002) argue against the strong pH
increase suggested by Sanyal et al. (1995). To solve this problem, epibenthic deep water

Appendix 104
recorders are needed. For instance the chemical composition of the (less abundant) deep sea
corals, ostracod shells or fish otoliths has hardly been investigated but these skeletal parts
may possibly offer useful information with regard to deep and midwater (carbonate)
chemistry.
3.4 Development of new methods
One possibility to reduce the amount of carbonate required for boron isotopic analyses
may be the application of TOF-SIMS. This technique uses a focussed, energetic ion beam that
detaches particles and ions from a sample surface. Furthermore, high resolution images can be
obtained by scanning the surface of a sample with the beam of a liquid metal ion gun. The
combination of surface imaging with depth profile capabilities allows the visualization of the
three-dimensional distribution of elements in a foraminiferal shell. The overall advantage of
this method is the simultaneous detection of all elements and masses on very small samples
(single foraminiferal shells or less). The development of this method is still in progress. First
results were published by Crone et al. (2000; subm.) and Vering et al. (2001). In addition to a
substantial reduction of the amount of carbonate required for boron isotopic analyses, this
analytical technique could simplify the establishment of new proxies. However, each analysis
takes many hours to establish acceptable levels of precision, so that it is still far from
becoming a routine analytical tool.
3.5 Models 3.5.1 Thermodynamics of uranium uptake
In order to better understand the interaction between vital effects of a foraminifer and
sea water chemistry, D.A. Wolf-Gladrow and A.D. Russell intend to develop a diffusion-
reaction model comparable to Wolf-Gladrow et al. (1997) and Zeebe et al. (1999).
3.5.2 Lysocline reconstruction
The existing box model of Jansen et al. (subm.) is a first approach to describe the
processes that are important at certain sediment depth intervals. However, lysocline and
saturation horizon shifted in opposite directions in the Atlantic relative to the Indopacific
under glacial conditions (see section 2.4.1). In order to better estimate the global validity, the
model is intended to be coupled to a global circulation model.

Appendix 105
3.5.3 Boron isotopic fractionation in seawater
R.E. Zeebe is currently recalculating the stable boron isotope fractionation factor
between B(OH)4- and B(OH)3. This value has never been measured and is fundamental to the
boron isotope paleo-pH recorder.
Acknowledgements
The authors thank J. Bijma and G. Ganssen for organization and invitations to the
workshop. Financial support was provided by the European Science Foundation and the
Hanse Wissenschafts Kolleg.

References 106
5. References Anderson, D.M. and Archer, D., 2002. Glacial-interglacial stability of ocean pH inferred from
foraminifer dissolution rates. Nature, 416: 70-72. Archer, D., Emerson, S. and Reimers, C., 1989. Dissolution of calcite in deep-sea sediments:
pH and O2 microelectrode results. Geochimica et Cosmochimica Acta, 53: 2831-2845. Archer, D. and Maier-Reimer, E., 1994. Effect of deep-sea sedimentary calcite preservation
on atmospheric CO2 concentration, Nature, pp. 260-263. Archer, D.A., Winguth, A., Lea, D. and Mahowald, N., 2000. What caused the
glacial/interglacial atmospheric pCO2 cycles? Reviews of Geophysics, 38(2): 159-189. Barker, S. and Elderfield, H., 2002. Foraminiferal calcification response tp glacial-interglacial
changes in atmospheric CO2. Science, 297: 833-836. Bé, A.W.H., 1980. Gametogenic calcification in a spinose planktonic foraminifer,
Globigerinoides sacculifer (BRADY). Mar. Micropal., 5: 283-310. Bé, A.W.H., Hemleben, C., Anderson, O.R. and Spindler, M., 1979. Chamber formation in
planktonic foraminifera. Micropaleontology, 25(3): 294-307. Bé, A.W.H. et al., 1977. Laboratory and field observations of living planktonic foraminifera.
micropleontology, 23(2): 155-179. Bé, A.W.H., Morse, J.W. and Harrison, S.M., 1974. Progressive dissolution and
ultrastructural breakdown of planktonic foraminifera. Dissolution of deep-sea carbonates: 27-55.
Bemis, B.E., Spero, H.J., Bijma, J. and Lea, D.W., 1998. Reevaluation of the oxygen isotopic composition of planktonic foraminifera: Experimental results and revised paleotemperature equations. Paleoceanography, 13(2): 150-160.
Bemis, B.E., Spero, H.J., Lea, D.W. and Bijma, J., 2000. Temperature influence on the carbon isotopic composition of Globigerina bulloides and Orbulina universa (planktonic foraminifera). Marine Micropaleontology, 38: 213-228.
Bender, M.L., Lorens, R.B. and Williams, D.F., 1975. Sodium, magnesium and strontium in the tests of planktonic foraminifera, Micropaleontology, pp. 448-459.
Berelson, W.M., Hammond, D.E. and Cutter, G.A., 1990. In situ measurements of calcium carbonate dissolution rates in deep-sea sediments. Geochimica et Cosmochimica Acta, 54: 3013-3020.
Berelson, W.M., Hammond, D.E., McManus, J. and Kilgore, T.E., 1994. Dissolution kinetics of calcium carbonate in equatorial Pacific sediments. Global Biogeochemical Cycles, 8(2): 219-235.
Berger, W.H., 1967. Foraminiferal ooze: solution at depth. Science, 156: 383-385. Berger, W.H., 1968. Planktonic foraminifera: selective solution and paleoclimatic
interpretation. Deep-Sea Research, 15: 31-43. Berger, W.H., 1970. Planktonic foraminifera: selective solution and the lysocline, Marine
Geology, pp. 111-138. Berger, W.H., Bonneau, M.-C. and Parker, F.L., 1982. Foraminifera on the deep-sea floor:
lysocline and dissolution rate. Oceanologica Acta, 5(2): 249-258. Berger, W.H., Smetacek, V. and Wefer, G., 1989. Ocean productivity and paleoproductivity -
an overview. In: W.H. Berger, V. Smetacek and G. Wefer (Editors), Production of the Ocean: Present and Past. John Wiley & Sons, pp. 1-34.
Bijma, J., Faber Jr., W.W. and Hemleben, C., 1990. Temperature and salinity limits for growth and survival of some planktonic foraminifers in laboratory cultures. Journal of Foraminiferal Research, 20(2): 95-116.

References 107
Bijma, J. and Hemleben, C., 1994. Population dynamics of the planktic foraminifer Globigerinoides sacculifer (Brady) from the central Red Sea, Deep-Sea Research I, pp. 485-510.
Bijma, J., Hemleben, C. and Wellnitz, K., 1994. Lunar-influenced carbonate flux of the planktic foraminifer Globigerinoides sacculifer (Brady) from the central Red Sea, Deep-Sea Research I, pp. 511-530.
Bijma, J., Spero, H.J. and Lea, D.W., 1998. Oceanic carbonate chemistry and foraminiferal isotopes: new laboratory results, Sixth International Conference on Paleoceanography.
Bijma, J., Spero, H.J. and Lea, D.W., 1999. Reassessing foraminiferal stable isotope geochemistry: Impact of the oceanic carbonate system (experimental results). In: G. Fischer and G. Wefer (Editors), Use of proxies in paleoceanography: Examples from the South Atlantic. Springer-Verlag, Berlin, Heidelberg, pp. 489-512.
Bishop, J.K.B., 1988. The barite-opal-organic carbon association in oceanic particulate matter. Nature, 332: 341-343.
Bouvier-Soumagnac, Y. and Duplessy, J.-C., 1985. Carbon and oxygen isotopic composition of planktonic foraminifera from laboratory culture, plankton tows and recent sediment: Implications for the reconstruction of paleoclimatic conditions and of the global carbon cycle. Journal of Foraminiferal Research, 15(4): 302-320.
Boyle, E. and Rosenthal, Y., 1996. Chemical hydrography of the South Atlantic during the last glacial maximum: Cd vs. δ13C. In: G. Wefer, W.H. Berger, G. Siedler and D.J. Webb (Editors), The South Atlantic: Present and past circulation. Springer-Verlag, Berlin Heidelberg, pp. 423-443.
Boyle, E.A., 1981. Cadmium, zinc, copper, and barium in foraminifera tests, Earth and Planetary Science Letters, pp. 11-35.
Boyle, E.A., 1988a. Cadmium: Chemical tracer of deepwater paleoceanography, Paleoceanography, pp. 471-489.
Boyle, E.A., 1988b. The role of vertical chemical fractionation in controlling late Quaternary atmospheric carbon dioxide, Journal of Geophysical Research, pp. 15701-15714.
Boyle, E.A. and Keigwin, L.D., 1985/1986. Comparison of Atlantic and Pacific paleochemical records for the last 215,000 years: changes in deep ocean circulation and chemical inventories. Earth and Planetary Science Letters, 76: 135-150.
Broecker, W. and Clark, E., 2001a. An evaluation of Lohmann's foraminifera weight dissolution index. Paleoceanography, 16(5): 531-534.
Broecker, W. and Takahashi, T., 1978. The relationship between lysocline depth and in situ carbonate ion concentration. Deep-Sea Research, 25: 65-95.
Broecker, W.S., 1997. Thermohaline circulation, the achilles heel of our climate system: Will man-made CO2 upset the current balance? Science, 278: 1582-1588.
Broecker, W.S. and Clark, E., 1999. CaCO3 distribution: A paleocarbonate ion proxy? Paleoceanography, 14(5): 596-604.
Broecker, W.S. and Clark, E., 2001b. Glacial-to Holocene redistribution of carbonate ion in the deep sea. Science, 294: 2152-2155.
Broecker, W.S. and Clark, E., 2002. Carbonate ion concentration in glacial age deep waters of the Caribbean Sea. Geochemistry Geophysics Geosystems.
Broecker, W.S. and Henderson, G.M., 1998. The sequence of events surrounding Termination II and their implications for the cause of glacial-interglacial CO2 changes. Paleoceanography, 13(4): 352-364.
Broecker, W.S., Lynch-Stieglitz, J., Clark, E., Hajdas, I. and Bonani, G., 2001. What caused the atmosphere's CO2 content to rise during the last 8000 years? Geochemistry Geophysics Geosystems, 2: paper # 2001GC000177.
Broecker, W.S. and Peng, T.-H., 1982. Tracers in the Sea. Lamont Doherty Earth Observatory, Columbia University, Palisades, New York, 689 pp.

References 108
Brown, S. and Elderfield, H., 1996. Variations in Mg/Ca and Sr/Ca ratios of planktonic foraminifera caused by postdepositional dissolution: Evidence of shallow Mg-dependent dissolution. Paleoceanography, 11(5): 543-551.
Cai, W.-J., Reimers, C.E. and Shaw, T., 1995. Microelectrode studies of organic carbon degradation and calcite dissolution at a California Continental rise site. Geochim Cosmochim. Acta, 59(3): 497-511.
Cardinal, D., Hamelin, B., Bard, E. and Pätzold, J., 2001. Sr/Ca, U/Ca and δ18O records in recent massive corals from Bermuda: relationships with sea surface temperature. Chemical Geology, 176: 213-233.
Caron, D.A., Anderson, O.R., Lindsey, J.L., Faber Jr., W.W. and Lin Lim, E., 1990. Effects of gametogenesis on test structure and dissolution of some spinose planktonic foraminifera and implications for test preservation, Marine Micropaleontology, pp. 93-116.
Carpenter, S.J. and Lohmann, K.C., 1992. Sr/Mg ratios of modern marine calcite: Empirical indicators of ocean chemistry and precipitation rate, Geochimica et Cosmochimica Acta, pp. 1837-1849.
Chan, L.H., Drummond, D., Edmond, J.M. and Grant, B., 1977. On the barium data from the Atlantic GEOSECS expedition. Deep-Sea Research, 24: 613-649.
Chow, T.J. and Goldberg, E.D., 1960. On the marine geochemistry of barium. Geochimica et Cosmochimica Acta, 20: 192-198.
Cohen, A.L., Owens, K.E., Layne, G.D. and Shimizu, N., 2002. The effect of algal symbionts on the accuracy of Sr/Ca paleotemperatures from coral. Science, 296: 331-333.
Cook, P.J., 1977. Loss of boron from shells during weathering and possible implications for the determination of paleosalinity. Nature, 268: 426-427.
Cox, P.M., Betts, R.A., Jones, C.D., Spall, S.A. and Totterdell, I.J., 2000. Acceleration of global warming due to carbon-cycle feedbacks in a coupled climate model. Nature, 408: 184-187.
Crawford, D.W. and Purdie, D.A., 1997. Increase of PCO2 during blooms of Emiliania huxleyi: Theoretical considerations on the asymmetry between acquisition of HCO3
- and respiration of free CO2. Limnol. Oceanogr., 42(2): 365-372.
Crone, C. et al., 2000. Investigation of planktonic foraminifera with TOF-SIMS and Laser-SNMS.
Crone, C., Vering, G., Hönisch, B., Bijma, J. and Arlinghaus, H.F., subm. Investigation of planktonic foraminifera with TOF-SIMS, 9th European Conference of Application of Surface and Interface Analysis
Crowley, T.J., 1983. Calcium carbonate preservation patterns in the central north Atlantic during the last 150,000 years. Marine Geology, 51: 1-14.
Crowley, T.J., 2000. Causes of climate change over the past 1000 years. Science, 289: 270-277.
Cuffey, K.M. and Vimeux, F., 2001. Covariation of carbon dioxide and temperature from the Vostok ice core after deuterium-excess correction. Nature, 412: 523-527.
Curry, W.B. and Lohmann, G.P., 1986. Late quaternary carbonate sedimentation at the Sierra Leone Rise (eastern equatorial Atlantic Ocean). Marine Geology, 70: 223-250.
Darling, K.F., Wade, C.M., Kroon, D., Leigh Brown, A.J. and Bijma, J., 1999. The diversity and distribution of modern planktic foraminiferal small subunit ribosomal RNA genotypes and their potential as tracers of present and past ocean circulations, Paleoceanography, pp. 3-12.
Darling, K.F. et al., 2000. Molecular evidence for genetic mixing of Arctic and Antarctic subpolar populations of planktonic foraminifers. Nature, 405: 43-47.

References 109
Dekens, P.S., Lea, D.W., Pak, D.K. and Spero, H.J., 2002. Core top calibration of Mg/Ca in tropical foraminifera: Refining paleotemperature estimation. Geochemistry Geophysics Geosystems, 3(4): 29 pp.
Dickson, A.G., 1981. An exact definition of total alkalinity and a procedure for the estimation of alkalinity and total inorganic carbon from titration data. Deep-Sea Research, 28 A: 609-623.
Dickson, A.G., 1990. Standard potential of the reaction: AgCl(s)+1/2H2(g)=Ag(s)+HCl(aq), and the standard acidity constant of the ion HSO4
- in synthetic seawater from 273.15 to 318.15K. Journal of Chemical Thermodynamics, 22: 113-127.
DOE, 1994. Handbook of methods for the analysis of the various parameters of the carbon dioxide system in seawater. ORNL/CDIAC-74, version 2.
Dudley, W.C., Blackwelder, P., Brand, L. and Duplessy, J.C., 1986. Stable isotopic composition of coccoliths. Marine Micropaleontology, 10: 1-8.
Duplessy, J.-C., Blanc, P.-L. and Bé, A., 1981. Oxygen-18 enrichment of planktonic foraminifera due to gametogenic calcification below the euphotic zone. Science, 213: 1247-1250.
Elderfield, H., 2002. Carbonate mysteries. Science, 296: 1618-1621. Elderfield, H., Bertram, C.J. and Erez, J., 1996. A biomineralization model for the
incorporation of trace elements into foraminiferal calcium carbonate, Earth and Planetary Science Letters, pp. 409-423.
Elderfield, H., Cooper, M. and Ganssen, G., 2000. Sr/Ca in multiple species of planktonic foraminifera: Implications for reconstructions of seawater Sr/Ca. Geochemistry Geophysics Geosystems 1: paper number 1999GC000031.
Elderfield, H. and Ganssen, G., 2000. Past temperature and δ18O of surface ocean waters inferred from foraminiferal Mg/Ca ratios. Nature, 405: 442-445.
Elderfield, H., Vautravers, M. and Cooper, M., 2002. The relationship between shell size and Mg/Ca, Sr/Ca, δ18O, and δ13C of species of planktonic foraminifera. Geochemistry Geophysics Geosystems, 3(8): 13pp.
Emiliani, C., 1955. Pleistocene temperatures. J. Geol. , 63: 538-578. Epstein, S., Buchsbaum, R., Lowenstam, H.A. and Urey, H.C., 1953. Revised carbonate-
water temperature scale. Bull. Geol. Soc. Am., 64: 1315-1326. Erez, J., 1979a. Influence of differential production and dissolution on the stable isotope
composition of planktonic foraminifera, Woods Hole Oceanographic Institution - Massachusetts Institute of Technology, 118p. pp.
Erez, J., 1979b. Modification of the oxygen isotope record in deep sea cores by Pleistocene dissolution cycles. Nature, 281: 535-538.
Erez, J., Almogi-Labin, A. and Avraham, S., 1991. On the life history of planktonic foraminifera: Linear reproduction cycle in Globigerinoides sacculifer (BRADY). Paleoceanography, 6(3): 295-306.
Erez, J. and Honjo, S., 1981. Comparison of isotopic composition of planktonic foraminifera in plankton tows, sediment traps and sediments. Paleog. Paleoclimat. Paleoecol., 33: 129-156.
Erez, J. and Luz, B., 1982. Temperature control of oxygen-isotope fractionation of cultured planktonic foraminifera. Nature, 297: 220-222.
Erez, J., Tishler, C., Berry, J. and Boyle, E.A., 2001. Sulfur in foraminifera shells, a new paleoceanographic proxy for the carbonate ion in seawater, 7th International Conference on Paleoceanography.
Fairbanks, R.G., 1989. A 17,000-year glacio-eustatic sea level record: influence of glacial melting rates at the Younger Dryas event and deep-ocean circulation. Nature, 342: 637-642.

References 110
Falkowski, P. et al., 2000. The global carbon cycle: A test of our knowledge of earth as a system. Science, 290: 291-296.
Farrell, J.W. and Prell, W.L., 1989. Climatic change and CaCO3 preservation: An 800,000 year bathymetric reconstruction from the central equatorial Pacific Ocean. Paleoceanography, 4(4): 447-466.
Fischer, H., Wahlen, M., Smith, J., Mastroianni, D. and Deck, B., 1999. Ice core records of atmospheric CO2 around the last three glacial terminations. Science, 283: 1712-1714.
Frankignoulle, M. and Canon, C., 1994. Marine calcification as a source of carbon dioxide: Positive feedback on increasing atmospheric CO2, Limnology and Oceanography, pp. 458-462.
Gaillardet, J. and Allègre, C.J., 1995. Boron isotopic compositions of corals: Seawater or diagenesis record? Earth Planet. Sci. Lett., 136: 665-676.
Gattuso, J.-P. and Buddemeier, R.W., 2000. Calcification and CO2. Nature, 407: 311-313. Gattuso, J.-P., Frankignoulle, M., Bourge, I., Romaine, S. and Buddemeier, R.W., 1998.
Effect of calcium carbonate saturation of seawater on coral calcification. Global and Planetary Change, 18(1-2): 37-46.
Gattuso, J.-P., Pichon, M. and Frankignoulle, M., 1995. Biological control of air-sea CO2 fluxes: effect of photosynthetic and calcifying marine organisms and ecosystems. Marine Ecology Progress series, 129: 307-312.
Hales, B. and Emerson, S., 1996. Calcite dissolution in sediments of the Ontong-Java Plateau: In situ measurements of porewater O2 and pH. Global Biogeochemical Cycles, 10(3): 527-541.
Hales, B. and Emerson, S., 1997a. Calcite dissolution in sediments of the Ceara Rise: In situ measurements of porewater O2, pH, and CO2 (aq.). Geochimica et Cosmochimica Acta, 61(3): 501-514.
Hales, B. and Emerson, S., 1997b. Evidence in support of first-order dissolution kinetics of calcite in seawater. Earth and Planetary Science Letters, 148: 317-327.
Hales, B., Emerson, S. and Archer, D., 1994. Respiration and dissolution in the sediments of the western North Atlantic: estimates from models of in situ microelectrode measurements of porewater oxygen and pH. Deep-Sea Research I, 41(4): 695-719.
Haley, B.A. and Klinkhammer, G.P., 2002. Development of a flow-through system for cleaning and dissolving foraminiferal tests. Chemical Geology, 185: 51-69.
Hastings, D.W., Russell, A.D. and Emerson, S.R., 1998. Foraminiferal magnesium in Globigerinoides sacculifer as a paleotemperature proxy. Paleoceanography, 13(2): 161-169.
Helmke, J.P. and Bauch, H.A., 2002. Glacial-interglacial carbonate preservation records in the Nordic Seas. Global and Planetary Change.
Hemleben, C., Bé, A.W.H., Anderson, O.R. and Tuntivate, S., 1977. Test morphology, organic layers and chamber formation of the planktonic foraminifer Globorotalia menardii (d'Orbigny). J. Foram. Res., 7(1): 1-25.
Hemleben, C. and Bijma, J., 1994. Foraminiferal population dynamics and stable carbon isotopes. In: R.e.a. Zahn (Editor), Carbon Cycling in the Glacial Ocean: Constraints on the Ocean's Role in Climate Change. NATO ASI Series. Springer-Verlag, Berlin Heidelberg, pp. 145-166.
Hemleben, C., Spindler, M., Breitinger, I. and Ott, R., 1987. Morphological and physiological responses of Globigerinoides sacculifer (Brady) under varying laboratory conditions. Marine Micropaelontology, 12(4): 305-324.
Hemming, N.G., Guilderson, T.P. and Fairbanks, R.G., 1998. Seasonal variations in the boron isotopic composition of a coral: A productivity signal? Global Biogeochemical Cycles, 12(4): 581-586.

References 111
Hemming, N.G. and Hanson, G.N., 1992. Boron isotopic composition and concentration in modern marine carbonates, Geochimica et Cosmochimica Acta, pp. 537-543.
Hemming, N.G. and Hanson, G.N., 1994. A procedure for the isotopic analysis of boron by negative thermal ionization mass spectrometry. Chemical Geology, 114: 147-156.
Hönisch, B., Bijma, J., Russell, A.D., Spero, H.J., Palmer, M.R. and Eisenhauer, A., subm. The influence of symbiont photosynthesis on the boron isotopic composition of foraminifera shells. Marine Micropaleontology.
Honjo, S. and Erez, J., 1978. Dissolution rates of calcium carbonate in the deep ocean; an in situ experiment in the North Atlantic Ocean. Earth and Planetary Science Letters, 40(2): 287-300.
Huber, B.T., Bijma, J. and Darling, K., 1997. Cryptic speciation in the living planktonic foraminifer Globigerinella siphonifera (d'Orbigny). Paleobiology, 23(1): 33-62.
Ingle, S.E., 1975. Solubility of calcite in the ocean. Marine Chemistry, 3: 301-319. IPCC, 2001. Climate change 2001. Cambridge University Press, 2002. Jahnke, R.A., Craven, D.B. and Gaillard, J., 1994. The influence of organic matter diagenesis
on CaCO3 dissolution at the deep-sea floor. Geochimica et Cosmochimica Acta, 58(13): 2799-2809.
Jahnke, R.A., Craven, D.B., McCorkle, D.C. and Reimers, C.E., 1997. CaCO3 dissolution in California continental margin sediments: The influence of organic matter remineralization. Geochimica et Cosmochimica Acta, 61(17): 3587-3604.
Jansen, H., Sanyal, A., Bijma, J. and Wolf-Gladrow, D.A., subm. Decoupling the calcium carbonate saturation horizon and lysocline depth: Implications for glacial-interglacial variations in atmospheric pCO2. Paleoceanography.
Jansen, H. and Wolf-Gladrow, D.A., 2001. Carbonate dissolution in copepod guts: a numerical model. Mar. Ecol. Prog. Ser., 221: 199-207.
Jansen, H., Zeebe, R.E. and Wolf-Gladrow, D.A., 2002. Modeling the dissolution of settling CaCO3 in the ocean. Global Biogeochemical Cycles, 16(2).
Jørgensen, B.B., Erez, J., Revsbech, N.P. and Cohen, Y., 1985. Symbiotic photosynthesis in a planktonic foraminiferan, Globigerinoides sacculifer (Brady), studied with microelectrodes. Limnol. Oceanogr., 30(6): 1253-1267.
Kakihana, H., Kotaka, M., Satoh, S., Nomura, M. and Okamoto, M., 1977. Fundamental studies on the ion-exchange of boron isotopes. Bull. Chem. Soc. Japan, 50: 158-163.
Keir, R.S., 1980. The dissolution kinetics of biogenic calcium carbonate in seawater. Geochimica et Cosmochimica Acta, 44: 241-252.
Kemle-von Mücke, S. and Hemleben, C., 1999. Foraminifera. In: D. Boltovskoy (Editor), South Atlantic Zooplankton. Backhuys Publishers, Leiden, pp. 43-73.
Kim, S.-T. and O'Neil, J.R., 1997. Equilibrium and nonequilibrium oxygen isotope effects in synthetic carbonates. Geochim. Cosmochim. Acta, 61(16): 3461-3475.
Kleypas, J.A. et al., 1999. Geochemical consequences of increased atmospheric carbon dioxide on coral reefs. Science, 284: 118-120.
Kohfeld, K.E., Anderson, R.F. and Lynch-Stieglitz, J., 2000. Carbon isotopic disequilibrium in polar planktonic foraminifera and its impact on modern and Last Glacial Maximum reconstructions. Paleoceanography, 15(1): 53-64.
Kohfeld, K.E., Fairbanks, R.G., Smith, S.L. and Walsh, I.D., 1996. Neogloboquadrina pachyderma (sinistral coiling) as paleoceanographic tracers in polar oceans: Evidence from Northeast Water Polynya plankton tows, sediment traps, amd surface sediments. Paleoceanography, 11(6): 679-699.
Kühl, M., Cohen, Y., Dalsgaard, T., Jørgensen, B.B. and Revsbech, N.P., 1995. Microenvironment and photosynthesis of zooxanthellae in scleractinian corals studied with microsensors for O2, pH and light. Marine Ecology Progress Series.

References 112
Kumar, N. et al., 1995. Increased biological productivity and export production in the glacial Southern Ocean. Nature, 373: 675-680.
Langer, M.R., 1992. Biosynthesis of glycosaminglycans in foraminifera: A review. Marine Micropaleontology, 19: 245-255.
Laws, E.A. et al., 2001. Controls on the molecular distribution and carbon isotopic composition of alkenones in certain haptophyte algae. Geochemistry Geophysics Geosystems, 2: paper number 2000GC000057.
Lea, D.W., 1993. Constraints on the alkalinity and circulation of glacial circumpolar deep water from benthic foraminiferal barium. Global Biogeochemical Cycles, 7(3): 695-710.
Lea, D.W., 1999a. Innovations in monitoring ocean history, An introduction to paleoceanographic proxies. In: Abrantes and Mix (Editors), Reconstructing Ocean History: A Window into the Future. Kluwer Academic//Plenum Publishers, New York, pp. 321-327.
Lea, D.W., 1999b. Trace elements in foraminiferal calcite. In: B.K. Sen Gupta (Editor), Modern Foraminifera. Kluwer Academic Publishers, Great Britain, pp. 259-277.
Lea, D.W., Bijma, J., Spero, H.J. and Archer, D., 1999a. Implications of a carbonate ion effect on shell carbon and oxygen isotopes for glacial ocean conditions. In: G. Fischer and G. Wefer (Editors), Use of proxies in paleoceanography: Examples from the South Atlantic. Springer-Verlag, Berlin Heidelberg, pp. 513-522.
Lea, D.W. and Boyle, E.A., 1989. Barium content of benthic foraminifera controlled by bottom-water composition. Nature, 338: 751-753.
Lea, D.W. and Boyle, E.A., 1990. Foraminiferal reconstruction of barium distributions in water masses of the glacial oceans. Paleoceanography, 5(5): 719-742.
Lea, D.W. and Boyle, E.A., 1991. Barium in planktonic foraminifera. Geochimica et Cosmochimica Acta, 55: 3321-3331.
Lea, D.W. and Boyle, E.A., 1993. Determination of carbonate-bound barium in foraminifera and corals by isotope dilution plasma-mass spectrometry, Chemical Geology, pp. 73-84.
Lea, D.W. and Martin, P.A., 1996. A rapid mass spectrometric method for the simultaneous analysis of barium, cadmium, and strontium in foraminifera shells. Geochimica et Cosmochimica Acta, 60(16): 3143-3149.
Lea, D.W., Martin, P.A., Chan, D.A. and Spero, H.J., 1995. Calcium uptake and calcification rate in the planktonic foraminifer Orbulina universa. Journal of Foraminiferal Research, 25(1): 14-23.
Lea, D.W., Martin, P.A., Pak, D.K. and Spero, H.J., 2002. Reconstructing a 350 ky history of sea level using planktonic Mg/Ca and oxygen isotope records from a Cocos Ridge core. Quaternary Science Reviews, 21: 283-293.
Lea, D.W., Mashiotta, T.A. and Spero, H.J., 1999b. Controls on magnesium and strontium uptake in planktonic foraminifera determined by live culturing. Geochimica et Cosmochimica Acta, 63(16): 2369-2379.
Lea, D.W., Pak, D.K. and Spero, H.J., 2000. Climate impact of late Quaternary equatorial Pacific sea surface temperature variations. Science, 289: 1719-1724.
Lea, D.W. and Spero, H.J., 1992. Experimental determination of barium uptake in shells of the planktonic foraminifera Orbulina universa at 22°C. Geochimica et Cosmochimica Acta, 56: 2673-2680.
Lea, D.W. and Spero, H.J., 1994. Assessing the reliability of paleochemical tracers: Barium uptake in the shells of planktonic foraminifera. Paleoceanography, 9(3): 445-452.
Lewis, E. and Wallace, D.W.R., 1998. Program developed for CO2 system calculations.

References 113
Lohmann, G.P., 1995. A model for variation in the chemistry of planktonic foraminifera due to secondary calcification and selective dissolution. Paleoceanography, 10(3): 445-457.
Lohmann, G.P., Rosenthal, Y. and McCorkle, D., 1999. Evidence for changes in carbonate ion concentration and foraminiferal calcite solubility during the last glaciation. Eos, Trans.
Longhurst, A.R., 1991. Role of the marine biosphere in the global carbon cycle. Limnol. Oceanogr., 36(8): 1507-1526.
Lorens, R.B., 1981. Sr, Cd, Mn and Co distribution coefficients as a function of calcite precipitation rate, Geochimica et Cosmochimica Acta, pp. 553-561.
Lorens, R.B., Williams, D.F. and Bender, M.L., 1977. The early nonstructural diagenesis of foraminiferal calcite, Journal of Sedimentary Petrology, pp. 1602-1609.
Mackenzie, F.T. et al., 1983. Magnesian calcites: Low temperature occurrence, solubility and solid solution behavior. In: R.J. Reeder (Editor), Carbonates: Mineralogy and Chemistry. Reviews in Mineralogy, Mineralogical Society of America, pp. 97-144.
Maher, B.A. and Dennis, P.F., 2001. Evidence against dust-mediated control of glacial-interglacial changes in atmospheric CO2. Nature, 411: 176-180.
Marchitto Jr., T.M., Curry, W.B. and Oppo, D.W., 2000. Zinc concentrations in benthic foraminifera reflect seawater carbonate chemistry. Paleoceanography, 15(3): 299-306.
Martin, J.H., 1990. Glacial-interglacial CO2 change; the iron hypothesis. Paleoceanography, 5: 1-13.
Martin, P.A., Lea, D.W., Mashiotta, T.A., Papenfuss, T. and Sarnthein, M., 1999. Variation of foraminiferal Sr/Ca over Quaternary glacial-interglacial cycles: Evidence for changes in mean ocean Sr/Ca? Geochemistry Geophysics Geosystems, 1: Paper No.: 1999GC000006.
Martin, W.R. and Sayles, F.L., 1996. CaCO3 dissolution in sediments of the Ceara Rise, western equatorial Atlantic. Geochim. Cosmochim. Acta, 60(2): 243-263.
Mashiotta, T.A., Lea, D.W. and Spero, H.J., 1997. Experimental determination of Cd uptake in shells of the planktonic foraminifera Orbulina universa and Globigerina bulloides: Implications for surface water paleoreconstructions. Geochimica et Cosmochimica Acta, 61: 4053-4065.
McConnaughey, T.A. and Whelan, J.F., 1997. Calcification generates protons for nutrient and bicarbonate uptake Earth-Science Reviews, 42: 95-117.
McCorkle, D.C., Martin, P.A., Lea, D.W. and Klinkhammer, G.P., 1995. Evidence of a dissolution effect on benthic foraminiferal shell chemistry: δ13C, Cd/Ca, Ba/Ca, and Sr/Ca results from the Ontong Java Plateau. Paleoceanography, 10(4): 699-714.
Millero, F.J., 1995. Thermodynamics of the carbon dioxide system in the oceans. Geochimica Cosmochimica Acta, 59(4): 661-667.
Millero, F.J. and Pierrot, D., 1998. A chemical equilibrium model for natural waters. Aquatic Geochemistry, 4: 153-199.
Milliman, J.D., 1993. Production and accumulation of calcium carbonate in the ocean: budget of a nonsteady state. Global Biogeochemical Cycles, 7(4): 927-957.
Milliman, J.D. et al., 1999. Biologically mediated dissolution of calcium carbonate above the chemical lysocline? Deep-Sea Research I, 46: 1653-1669.
Min, G.R. et al., 1995. Annual cycles of U/Ca in coral skeletons and U/Ca thermometry. Geochim. Cosmochim. Acta, 59(10): 2025-2042.
Morse, J.W., 1978. Dissolution kinetics of calcium carbonate in sea water: VI: The near-equilibrium dissolution kinetics of calcium carbonate-rich deep sea sediments, American Journal of Science, pp. 344-353.
Morse, J.W. and Arvidson, R.S., 2002. The dissolution kinetics of major sedimentary carbonate minerals. Earth-Science Reviews, 58.

References 114
Morse, J.W. and Bender, M.L., 1990. Partition coefficients in calcite: Examination of factors influencing the validity of experimental results and their application to natural systems, Chemical Geology, pp. 265-277.
Morse, J.W., De Kanel, J. and Harris, K., 1979. Dissolution kinetics of calcium carbonate in seawater: VII. The dissolution kinetics of synthetic aragonite and pteropod tests. American Journal of Science, 279: 488-502.
Nägler, T.F., Eisenhauer, A., Müller, A., Hemleben, C. and Kramers, J., 2000. The δ44 Ca-temperature calibration on fossil and cultured Globigerinoides sacculifer: New tool for reconstruction of past sea surface temperatures. Geochemistry Geophysics Geosystems, 1: paper number 2000GC000091.
Neftel, A., Oeschger, H., Schwander, J., Stauffer, B. and Zumbrunn, R., 1982. Ice core sample measurements give atmospheric CO2 content during the past 40,000 yr. Nature, 295: 220-223.
Nürnberg, D., 1995. Magnesium in tests of Neogloboquadrina pachyderma sinistral from high northern and southern latitudes. J. Foram. Res., 25(4): 350-368.
Nürnberg, D., Bijma, J. and Hemleben, C., 1996. Assessing the reliability of magnesium in foraminiferal calcite as a proxy for water mass temperatures, Geochimica et Cosmochimica Acta, pp. 803-814.
Paasche, E., in press. A review of the coccolithophorid Emiliania huxleyi (Prymnesiophyceae), with particular reference to growth, coccolith formation, and calcification-photosynthesis interactions. Phycology, 40.
Palmer, M.R., Pearson, P.N. and Cobb, S.J., 1998. Reconstructing past ocean pH-depth profiles. Science, 282: 1468-1471.
Parker, F.L. and Berger, W.H., 1971. Faunal and solution patterns of planktonic foraminifera in surface sediments of the south Pacific. Deep-Sea Research, 18(1): 73-107.
Pätzold, J. and cruise participants, 2000. Report and preliminary results of METEOR cruise M44/3, Aqaba (Jordan)-Safaga (Egypt)-Dubá (Saudi Arabia)-Suez (Egypt)-Haifa (Israel), 12.3.-26.3.-2.4.-4.4.1999, Fachbereich Geowissenschaften, Universität Bremen, Bremen.
Paytan, A., Kastner, M. and Chavez, F.P., 1996. Glacial to interglacial fluctuations in productivity in the Equatorial Pacific as indicated by marine barite. Science, 274: 1355-1357.
Pearson, P.N. and Palmer, M.R., 1999. Middle Eocene seawater pH and atmospheric carbon dioxide concentrations. Science, 284: 1824-1826.
Pearson, P.N. and Palmer, M.R., 2000. Atmospheric carbon dioxide concentrations over the past 60 million years. Nature, 406: 695-699.
Peeters, F.J.C., Brummer, G.-J.A. and Ganssen, G., 2002. The effect of upwelling on the distribution and stable isotope composition of Globigerina bulloides and Globigerinoides ruber (planktic foraminifera) in modern surface waters of the NW Arabian Sea. Global and Planetary Change. In press.
Petit, J.R. et al., 1999. Climate and atmospheric history of the past 420,000 years from the Vostok ice core, Antarctica. Nature, 399: 429-436.
Raven, J.A. and Falkowski, P.G., 1999. Oceanic sinks for atmospheric CO2. Plant, Cell and Environment, 22: 741-755.
Riebesell, U. et al., 2000. Reduced calcification of marine plankton in response to increased atmospheric CO2. Nature, 407: 364-367.
Rink, S., Kühl, M., Bijma, J. and Spero, H.J., 1998. Microsensor studies of photosynthesis and respiration in the symbiotic foraminifer Orbulina universa. Mar. Biol., 131(4): 583-595.

References 115
Rosenthal, Y. and Lohmann, G.P., 2002, in press. Accurate estimation of sea surface temperature using dissolution-corrected calibrations for Mg/Ca paleothermometry. Paleoceanography.
Rosenthal, Y., Lohmann, G.P., Lohmann, K.C. and Sherell, R.M., 2000. Incorporation and preservation of Mg in Globigerinoides sacculifer: Implications for reconstructing the temperature and 18O/16O of seawater. Paleoceanography, 15(1): 135-145.
Roy, R.N. et al., 1993. The dissociation constants of carbonic acid in seawater at salinities 5 to 45 and temperatures 0 to 45 deg. C. Mar. Chem., 44: 249-267.
Russell, A.D., 2001. New insights on environmental controls over the incorporation of uranium into foraminifera, 7th International Conference on Paleoceanography.
Russell, A.D., Emerson, S., Nelson, B.K., Erez, J. and Lea, D.W., 1994. Uranium in foraminiferal calcite as a recorder of seawater uranium concentrations. Geochimica et Cosmochimica Acta, 58(2): 671-681.
Russell, A.D., Hönisch, B., Spero, H.J. and Lea, D.W., in prep. Effects of changes in seawater carbonate ion concentration and temperature on the incorporation of U, Sr, and Mg into planktonic foraminiferal calcite, ms in preparation. to be submitted to Geochim. Cosmochim. Acta.
Rutherford, S., D'Hondt, S. and Prell, W., 1999. Environmental controls on the geographic distribution of zooplankton diversity. Nature, 400(400): 749-752.
Sanyal, A. and Bijma, J., 1999. A comparative study of northwest Africa and eastern equatorial Pacific upwelling zones as sources of CO2 during glacial periods based on boron isotope paleo-pH estimation, Paleoceanography, pp. 753-759.
Sanyal, A., Bijma, J., Spero, H.J. and Lea, D.W., 2001. Empirical relationship between pH and the boron isotopic composition of G. sacculifer: Implications for the boron isotope paleo-pH proxy. Paleoceanography, 16(5): 515-519.
Sanyal, A., Hemming, N.G. and Broecker, W.S., 1997. Changes in pH in the eastern equatorial Pacific across stage 5-6 boundary based on boron isotopes in foraminifera. Global Biogeochemical Cycles, 11(1): 125-133.
Sanyal, A. et al., 1996. Oceanic pH control on the boron isotopic composition of foraminifera: Evidence from culture experiments. Paleoceanography, 11(5): 513-517.
Sanyal, A., Hemming, N.G., Hanson, G.N. and Broecker, W.S., 1995. Evidence for a higher pH in the glacial ocean from boron isotopes in foraminifera. Nature, 373: 234-236.
Sanyal, A., Nugent, M., Reeder, R.J. and Bijma, J., 2000. Seawater pH control on the boron isotopic composition of calcite: Evidence from inorganic calcite precipitation experiments. Geochim. Cosmochim. Acta, 64(9): 1551-1555.
Savin, S.M. and Douglas, R.G., 1973. Stable isotope and magnesium geochemistry of recent planktonic foraminifera from the South Pacific. Geol. Soc. Am. Bull., 84: 2327-2342.
Sen, S., Stebbins, J.F., Hemming, N.G. and Ghosh, B., 1994. Coordination environments of B impurities in calcite and aragonite polymorphs: A 11B MAS NMR study. American Mineralogist, 79: 819-825.
Shannon, 1976. Revised effective ionic radii... Acta crystallog., A32: 751-767. Shen, G.T. and Dunbar, R.B., 1995. Environmental controls on uranium in reef corals.
Geochim. Cosmochim. Acta, 59(10): 2009-2024. Sigman, D.M. and Boyle, E.A., 2000. Glacial/interglacial variations in atmospheric carbon
dioxide. Nature, 407: 859-869. Sigman, D.M., McCorkle, D.C. and Martin, W.R., 1998. The calcite lysocline as a constraint
on glacial/interglacial low-latitude production changes. Global Biogeochemical Cycles, 12(3): 409-427.
Speer, J.A., 1983. Crystal chemistry and phase relations of orthorhombic carbonates. In: R.J. Reeder (Editor), Carbonates: Mineralogy and Chemistry. Reviews in Mineralogy. Mineralogical Society of America, pp. 145-190.

References 116
Spero, H.J., 1988. Ultrastructural examination of chamber morphogenesis and biomineralization in the planktonic foraminifer Orbulina universa. Marine Biology, 99: 9-20.
Spero, H.J., Bijma, J., Lea, D.W. and Bemis, B.E., 1997. Effect of seawater carbonate concentration on foraminiferal carbon and oxygen isotopes. Nature, 390: 497-500.
Spero, H.J., Bijma, J., Lea, D.W. and Russell, A.D., 1999. Deconvolving glacial ocean carbonate chemistry from the planktonic foraminifera carbon isotope record. In: Abrantes and Mix (Editors), Reconstructing Ocean History. A Window into the Future. Kluwer Academic/ Plenum Publishers, New York, pp. 329-342.
Spero, H.J. and Lea, D.W., 1993. Intraspecific stable isotope variability in the planktic foraminifera Globigerinoides sacculifer: Results from laboratory experiments. Mar. Micropal., 22: 221-234.
Spero, H.J. and Lea, D.W., 1996. Experimental determination of stable isotope variability in Globigerina bulloides: implications for paleoceanographic reconstructions, Marine Micropaleontology, pp. 231-246.
Spero, H.J. and Parker, S.L., 1985. Photosynthesis in the symbiotic planktonic foraminifer Orbulina universa, and its potential contribution to oceanic primary productivity. Journal of Foraminiferal Research, 15(4): 273-281.
Spindler, M., Hemleben, C., Bayer, U., Bé, A.W.H. and Anderson, O.R., 1979. Lunar periodicity of reproduction in the planktonic foraminifer Hastigerina pelagica. Mar. Ecol. Prog. Ser., 1: 61-64.
Spivack, A.J. and Edmond, J.M., 1986. Determination of boron isotope ratios by Thermal Ionization Mass Spectrometry of the Dicesium Metaborate Cation. Anal. Chem., 58: 31-35.
Spivack, A.J., You, C.-F. and Smith, H.J., 1993. Foraminiferal boron isotope ratios as a proxy for surface ocean pH over the past 21 Myr. Nature, 363: 149-151.
Stoll, H.M., Klaas, C.M., Probert, C.M., Ruiz Encinar, J. and García Alonso, J.I., 2001. Calcification rate and temperature effects on Sr partitioning in coccoliths of multiple species of coccolithophorids in culture. Global and Planetary Change, Special volume: From process studies to reconstruction of the paleoenvironment: advances in paleoceanography and -climatology.
Stoll, H.M., Rosenthal, Y. and Falkowski, P., in press. Climate proxies from Sr/Ca of coccolith calcite: calibrations from continuous culture of Emiliania huxleyi. Geochim. Cocmochim. Acta.
Stoll, H.M. and Schrag, D.P., 2000. Coccolith Sr/Ca as a new indicator of coccolithophorid calcification and growth rate. Geochemistry Geophysics Geosystems.
Stoll, H.M. and Ziveri, P., in press. Methods for separation of monospecific coccolith samples from sediments. Marine Micropaleontology.
Stott, P.A. and Kettleborough, J.A., 2002. Origins and estimates of uncertainty in predictions of twenty-first century temperature rise. Nature, 416: 723-726.
Tesoriero, A.J. and Pankow, J.F., 1996. Solid solution partitioning of Sr2+, Ba2+, and Cd2+ to calcite. Geochimica et Cosmochimica Acta, 60(6): 1053-1063.
Urey, H.C., 1947. The thermodynamic properties of isotopic substances. J. Chem. Soc.: 562-581.
van Cappellen, P., Charlet, L., Stumm, W. and Wersin, P., 1993. A surface complexation model of the carbonate mineral-aqueous solution interface. Geochim. Cosmochim. Acta, 57: 3505-3518.
Vengosh, A., Kolodny, Y., Starinsky, A., Chivas, A.R. and McCulloch, M.T., 1991. Coprecipitation and isotopic fractionation of boron in modern biogenic carbonates. Geochimica et Cosmochimica Acta, 55: 2901-2910.

References 117
Vering, G., Crone, C., Bijma, J. and Arlinghaus, H.F., 2001. TOF-SIMS characterization of planktonic foraminifera, 13th International Conference on Secondary Ion Mass Spectrometry and Related Topics.
Volbers, A.N.A. and Henrich, R., 2002. Present water mass calcium carbonate corrosiveness in the eastern South Atlantic inferred from ultrastructural breakdown of Globigerina bulloides in surface sediments. Chemical Geology, 186: 471-486.
von Langen, P.J., 2001. Non spinose planktonic foraminifera (Neogloboquadrina pachyderma) cultured for geochemical and paleoceanographic applications, University of California, Santa Barbara, 216 pp. pp.
Walter, L.M. and Morse, J.W., 1985. The dissolution kinetics of shallow marine carbonates in seawater: A laboratory study, Geochimica et Cosmochimica Acta, pp. 1503-1513.
Wara, M.W., Delaney, M.L., Bullen, T.D. and Ravello, A.C., 2001. Plio-Pleistocene records of boron in planktonic and benthic foraminifera: The roles of pH, temperature, and shell thinning, 7th International Conference on Paleoceanography.
Wefer, G., Berger, W.H., Bijma, J. and Fischer, G., 1999. Clues to ocean history: a brief overview of proxies. In: G. Fischer and G. Wefer (Editors), Use of proxies in paleoceanography: Examples from the South Atlantic. Springer-Verlag, Berlin Heidelberg, pp. 1-68.
Westbroek, P. et al., 1993. A model system approach to biological climate forcing: the example of Emiliania huxleyi. Global and Planetary Change, 8: 27-46.
Wolf-Gladrow, D., Bijma, J. and Zeebe, R.E., 1999a. Model simulation of the carbonate chemistry in the microenvironment of symbiont bearing foraminifera, Marine Chemistry, pp. 181-198.
Wolf-Gladrow, D. and Riebesell, U., 1997. Diffusion and reactions in the vicinity of plankton: A refined model for inorganic carbon transport. Marine Chemistry, 59: 17-34.
Wolf-Gladrow, D., Riebesell, U., Burkhardt, S. and Bijma, J., 1999b. Direct effects of CO2 concentration on growth and isotopic composition of marine plankton. Tellus, B 51: 461-476.
Wollast, R., 1994. The relative importance of biomineralization and dissolution of CaCO3 in the global carbon cycle, Bulletin de l'Institut océanographique, Monaco, pp. 13-35.
Zachara, J.M., Cowan, C.E. and Resch, C.T., 1991. Sorption of divalent metals on calcite. Geochimica et Cosmochimica Acta, 55: 1549-1562.
Zachos, J.C., Stott, L.D. and Lohmann, K.C., 1994. Evolution of early Cenozoic marine temperatures. Paleoceanography, 9(2): 353-387.
Zeebe, R.E., 1999. An explanation of the effect of seawater carbonate concentration on foraminiferal oxygen isotopes. Geochimica et Cosmochimica Acta, 63(13/14): 2001-2007.
Zeebe, R.E., Bijma, J. and Wolf-Gladrow, D., 1999. A diffusion-reaction model of carbon isotope fractionation in foraminifera. Marine Chemistry, 64: 199-227.
Zeebe, R.E., Bijma, J. and Wolf-Gladrow, D.A., 2001. Modeling pH profiles and stable boron isotopes in planktonic foraminifera, 7th International Conference on Paleoceanography.
Zeebe, R.E. and Wolf-Gladrow, D.A., 2001. CO in seawater: Equilibrium, kinetics, isotopes. Elsevier Oceanography Series, 65. Elsevier, 346 pp. pp.
2
Zeebe, R.E., Wolf-Gladrow, D.A. and Bijma, J., subm. Vital effects in foraminifera do not compromise the use of δ B as a paleo-pH indicator: Evidence from modeling. Paleoceanography.
11
Zhu, P. and MacDougall, J.D., 1998. Calcium isotopes in the marine environment and the oceanic calcium cycle. Geochim. Cocmochim. Acta, 62(10): 1691-1698.

References 118
Ziveri, P. et al., 2000. Species-specific stable isotope composition of coccolith calcite as a paleoceanographic proxy. EOS, Transactions of the American Geophysical Union (AGU), San Francisco, USA.
Ziveri, P., Probert, I., Stoll, H.M., Mackensen, A. and Keller, P., in prep. Origin of species-specific stable isotope "vital effects" in coccolith calcite.
Zondervan, I., Zeebe, R.E., Rost, B. and Riebesell, U., 2001. Decreasing marine biogenic calcification: A negative feedback on rising atmospheric pCO . Global Biogeochemical Cycles, 15(2): 507-516.
2




![DISSERTATION - elib.suub.uni-bremen.deelib.suub.uni-bremen.de/diss/docs/00010510.pdf · geringen Polarisierbarkeit ist es ein „hartes Anion“ (Superhalogenid)[6]. Aus diesen physikalischen](https://static.fdokument.com/doc/165x107/5e15cd1081ba745cb54d6ef2/dissertation-elibsuubuni-geringen-polarisierbarkeit-ist-es-ein-ahartes-anionaoe.jpg)











