
Journal of Materials Chemistry B - Polyactiva...c The Monash Institute of Pharmaceutical Science,...
6
This journal is © The Royal Society of Chemistry 2017 J. Mater. Chem. B Cite this: DOI: 10.1039/c7tb01518f Precise control of drug loading and release of an NSAID–polymer conjugate for long term osteoarthritis intra-articular drug delivery† Adrian Sulistio, ab Felisa Reyes-Ortega, ab Asha M. D’Souza, bc Sarah M. Y. Ng, bc David Valade, bc John F. Quinn, a Andrew C. Donohue, bc Friederike Mansfeld, a Anton Blencowe, d Greg Qiao, e Richard Prankerd, c Stephen Quirk, f Michael R. Whittaker, a Thomas P. Davis * ag and Russell J. Tait* bc A facile synthesis method of polymer diclofenac conjugates (PDCs) based on biocompatible polyurethane chemistry that provides a high drug loading and offers a high degree of control over diclofenac (DCF) release kinetics is described. DCF incorporating monomer was reacted with ethyl-L-lysine diisocyanate (ELDI) and different amounts of polyethylene glycol (PEG) in a one-step synthesis to yield polymers with pendent diclofenac distributed along the backbone. By adjusting the co-monomers feed ratio, the drug loading could be tailored accordingly to give DCF loading of up to 38 w/w%. The release rate could also be controlled easily by changing the amount of PEG in the backbone. Above 10 w/w% of PEG, the in vitro DCF release studies in physiological conditions showed an apparent zero-order profile with- out an initial burst effect for up to 120 days. The PDCs described may be suitable for long-term intra-articular (IA) delivery for the treatment of osteoarthritis (OA). A sustained course of non-steroidal anti-inflammatory drugs (NSAIDs) is often employed in the management of osteoarthritis (OA). 1 However, the long term use of NSAIDs can lead to severe systemic side effects such as gastrointestinal ulceration or hemorrhage, increased risk of heart attack, renal failure and depression. 2 Due to the localized nature of OA, a site specific intra-articular (IA) drug injection has become an attractive approach to administer the therapeutic agents directly into the affected area as it allows local therapeutic concentrations to be achieved at a much lower administered dose and minimizes the overall systemic exposure to the drug. 3–5 Keeping the injection frequency to a minimum presents a major challenge in IA drug delivery as the clearance of drug from the synovial fluid typically occurs rapidly. 1,4–7 As a result, various delivery vehicles which aim to prolong the residence time of the drug in the joint have been investigated, including liposomes, solid–lipid nanoparticles, polymeric nano- and micro-spheres and injectable hydrogels. 3,5 However, only a limited number of these delivery vehicles have made it to clinical trials. Larsen et al. 5 were able to show that the majority of these drug delivery vehicles were only able to prolong the residence time of the relevant drug in the knee for several days and only a few gave clinically relevant anti-inflammatory effects that lasted more than 7 days (up to 21 days). 5 Most of the above-mentioned materials rely on physical encapsulation through a self-assembly process as a means to load the NSAID into the vehicle. 8 While this process can provide a high drug load, the resulting dynamic structures have limited application in the sink conditions of a true physiological environment and typically suffer from a lack of control over the rate of drug release due to an inability to maintain the self- assembly. In the worst case, the extent of dilution which accompanies injection can lead to disassembly and immediate release of the encapsulated therapeutic. 8,9 In contrast, covalent conjugation of the drug to a polymer scaffold or matrix via tuneable biodegradable linkers provides a kinetic constraint to the rate of drug release independent of the assembled state of the polymer. 10,11 Polyurethanes have been used in a plethora of biomedical devices and drug delivery applications 11–27 due to demonstrated biocompatibility and, depending on the building blocks, biodegradability. 28,29 Herein, we introduce polymer-diclofenac conjugates (PDCs) based on polyurethane chemistry which could address the current limitations of burst release and low drug loading of more conventional drug delivery systems for drug delivery applications that require sustained drug release over a a ARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Monash University, 381 Royal Pde Parkville, VIC, 3052, Australia. E-mail: [email protected] b PolyActiva, Pty. Ltd, Level 9, 278 Collins Street Melbourne, VIC 3000, Australia. E-mail: [email protected] c The Monash Institute of Pharmaceutical Science, Parkville, VIC 3052, Australia d School of Pharmacy and Medical Sciences, University of South Australia, Mawson Lake, SA, 5095, Australia e Department of Chemical and Biomolecular Engineering, University of Melbourne, Parkville, VIC 3010, Australia f Kimberly-Clark Corporation, Roswell, 30022, Georgia, USA g Department of Chemistry, University of Warwick, Coventry, ULCV4 7AL, UK † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb01518f Received 6th June 2017, Accepted 24th July 2017 DOI: 10.1039/c7tb01518f rsc.li/materials-b Journal of Materials Chemistry B COMMUNICATION Published on 24 July 2017. Downloaded by Monash University on 03/08/2017 03:09:01. View Article Online View Journal
Transcript of Journal of Materials Chemistry B - Polyactiva...c The Monash Institute of Pharmaceutical Science,...

This journal is©The Royal Society of Chemistry 2017 J. Mater. Chem. B
Cite this:DOI: 10.1039/c7tb01518f
Precise control of drug loading and releaseof an NSAID–polymer conjugate for long termosteoarthritis intra-articular drug delivery†
Adrian Sulistio,ab Felisa Reyes-Ortega,ab Asha M. D’Souza,bc Sarah M. Y. Ng,bc
David Valade,bc John F. Quinn, a Andrew C. Donohue,bc Friederike Mansfeld,a
Anton Blencowe,d Greg Qiao, e Richard Prankerd,c Stephen Quirk, f
Michael R. Whittaker, a Thomas P. Davis *ag and Russell J. Tait*bc
A facile synthesis method of polymer diclofenac conjugates (PDCs)
based on biocompatible polyurethane chemistry that provides a
high drug loading and offers a high degree of control over diclofenac
(DCF) release kinetics is described. DCF incorporating monomer was
reacted with ethyl-L-lysine diisocyanate (ELDI) and different amounts
of polyethylene glycol (PEG) in a one-step synthesis to yield polymers
with pendent diclofenac distributed along the backbone. By adjusting
the co-monomers feed ratio, the drug loading could be tailored
accordingly to give DCF loading of up to 38 w/w%. The release rate
could also be controlled easily by changing the amount of PEG in the
backbone. Above 10 w/w% of PEG, the in vitro DCF release studies in
physiological conditions showed an apparent zero-order profile with-
out an initial burst effect for up to 120 days. The PDCs described may
be suitable for long-term intra-articular (IA) delivery for the treatment
of osteoarthritis (OA).
A sustained course of non-steroidal anti-inflammatory drugs(NSAIDs) is often employed in the management of osteoarthritis(OA).1 However, the long term use of NSAIDs can lead to severesystemic side effects such as gastrointestinal ulceration orhemorrhage, increased risk of heart attack, renal failure anddepression.2 Due to the localized nature of OA, a site specificintra-articular (IA) drug injection has become an attractiveapproach to administer the therapeutic agents directly intothe affected area as it allows local therapeutic concentrations
to be achieved at a much lower administered dose and minimizesthe overall systemic exposure to the drug.3–5
Keeping the injection frequency to a minimum presents amajor challenge in IA drug delivery as the clearance of drugfrom the synovial fluid typically occurs rapidly.1,4–7 As a result,various delivery vehicles which aim to prolong the residencetime of the drug in the joint have been investigated, includingliposomes, solid–lipid nanoparticles, polymeric nano- andmicro-spheres and injectable hydrogels.3,5 However, only a limitednumber of these delivery vehicles have made it to clinical trials.Larsen et al.5 were able to show that the majority of these drugdelivery vehicles were only able to prolong the residence time ofthe relevant drug in the knee for several days and only a few gaveclinically relevant anti-inflammatory effects that lasted more than7 days (up to 21 days).5
Most of the above-mentioned materials rely on physicalencapsulation through a self-assembly process as a means toload the NSAID into the vehicle.8 While this process can providea high drug load, the resulting dynamic structures have limitedapplication in the sink conditions of a true physiologicalenvironment and typically suffer from a lack of control overthe rate of drug release due to an inability to maintain the self-assembly. In the worst case, the extent of dilution whichaccompanies injection can lead to disassembly and immediaterelease of the encapsulated therapeutic.8,9 In contrast, covalentconjugation of the drug to a polymer scaffold or matrix viatuneable biodegradable linkers provides a kinetic constraint tothe rate of drug release independent of the assembled state ofthe polymer.10,11
Polyurethanes have been used in a plethora of biomedicaldevices and drug delivery applications11–27 due to demonstratedbiocompatibility and, depending on the building blocks,biodegradability.28,29 Herein, we introduce polymer-diclofenacconjugates (PDCs) based on polyurethane chemistry which couldaddress the current limitations of burst release and low drugloading of more conventional drug delivery systems for drugdelivery applications that require sustained drug release over a
a ARC Centre of Excellence in Convergent Bio-Nano Science and Technology,
Monash University, 381 Royal Pde Parkville, VIC, 3052, Australia.
E-mail: [email protected] PolyActiva, Pty. Ltd, Level 9, 278 Collins Street Melbourne, VIC 3000, Australia.
E-mail: [email protected] The Monash Institute of Pharmaceutical Science, Parkville, VIC 3052, Australiad School of Pharmacy and Medical Sciences, University of South Australia,
Mawson Lake, SA, 5095, Australiae Department of Chemical and Biomolecular Engineering, University of Melbourne,
Parkville, VIC 3010, Australiaf Kimberly-Clark Corporation, Roswell, 30022, Georgia, USAg Department of Chemistry, University of Warwick, Coventry, ULCV4 7AL, UK
† Electronic supplementary information (ESI) available. See DOI: 10.1039/c7tb01518f
Received 6th June 2017,Accepted 24th July 2017
DOI: 10.1039/c7tb01518f
rsc.li/materials-b
Journal ofMaterials Chemistry B
COMMUNICATION
Publ
ishe
d on
24
July
201
7. D
ownl
oade
d by
Mon
ash
Uni
vers
ity o
n 03
/08/
2017
03:
09:0
1.
View Article OnlineView Journal

J. Mater. Chem. B This journal is©The Royal Society of Chemistry 2017
prolonged period (e.g., for OA treatment). The synthesis methodemployed involves a step-growth polymerization in which a newdiclofenac-incorporating diol monomer is used with a corres-ponding diisocyanate co-monomer in the polyurethane synthesis.This method allows for facile control of the drug loading anddrug release kinetics through simple adjustment of the ratio of thediclofenac-incorporating diol monomer to other diol co-monomers(e.g. polyethylene glycol, PEG).
Wang et al.30 have extensively studied the hydrolysis of twodiclofenac incorporating monomers, diclofenac-( p-hydroxy-benzoate)-2-monoglyceride (DCF-PHB-MG) (2) and diclofenac-monoglycerides (DCF-MG) to show that under physiologicalconditions, the aryl ester construct (DCF-PHB-MG (2)) had amore rapid rate of hydrolysis compared to the alkyl ester, DCF-MG.They also showed two products arose directly from the hydrolysisof DCF-PHB-MG and DCF-MG and identified them as diclofenacand its cyclic lactam (an unwanted side-product). In view of theresults of Wang, et al.30 initial polymerizations were carried out totest the likelihood of releasing DCF from PDCs produced withthese DCF monomers. In contrast however, the PDCs obtainedfrom DCF-PHB-MG as the incorporating monomer successfullyreleased DCF under physiological conditions with almost nolactam observed. Furthermore, once incorporated into thepolymers, our unpublished studies also showed that little orno diclofenac was released from the polymer with the alkylester linkage (DCF-MG) as the incorporating monomer. Thisresult suggests that attaching the prodrug onto a polymericdelivery vehicle is able to protect the chemical fidelity of thetherapeutic cargo. Based on this finding, DCF-PHB-MG (2) waschosen as the preferred diclofenac incorporating monomer forthe subsequent polymerizations.
In the reported example, diclofenac (DCF, 1) was convertedinto a diclofenac-(p-hydroxybenzoate)-2-monoglyceride (DCF-PHB-MG, 2) as a polymerizable diol with DCF connected via a labilearyl ester linker (Scheme 1).31 The DCF–polymer conjugateswere synthesized in a one-pot synthesis by reacting ethyl-L-lysinediisocyanate (ELDI) with different ratios of the diol co-monomers,polyethylene glycol (PEG) and DCF-PHB-MG (2) in the presence ofa catalyst, dibutyltin dilaurate (DBTDL) to give linear polyurethanewith pendent drugs distributed along the backbone (Scheme 2).These monomers were chosen because of their known biocompat-ibility and their ability to biodegrade into non-toxic by-products.28
The stoichiometric ratio was adjusted such that there was anequimolar ratio of isocyanates to the total hydroxyl groups of the
diol monomers; PEG and DCF-PHB-MG (2) to enable the highestmolecular weight possible to be achieved within the thermo-dynamic and kinetic constraints of polyurethane synthesis.32,33
In order to investigate the versatility and tuneability of thedrug loading and release rate of the PDC, a series of DCFpolyurethanes with various DCF and PEG content have beensynthesized. Three different PEG lengths were chosen as theco-monomers: PEG200, PEG400 and PEG1000. PEG MWshigher than 1 kDa were not considered here as it would lowerthe polymer drug loading dramatically, making this polymerunsuitable for drug delivery applications. The different feed ratios,the molecular weight distribution and stoichiometric ratio of thefinal polymer composition are presented in Table S1, ESI.†
For each PEG length, several stoichiometric ratios weresystematically used in order to study the effect of the PEGcontent on the drug release rate. In addition, the stoichiometricratio was adjusted to give polymers with a similar range oftheoretical PEG content (w/w% weight of PEG per weight ofpolymer) regardless of the PEG length used. For example DCFP3 would have similar PEG w/w% content (11.8 w/w% PEG200)as P9 (10.3 w/w% PEG400) and P14 (10.4 w/w% PEG1000). Thiswas done to determine whether there was a direct relationshipbetween the PEG content (w/w%) and the release rate irrespectiveof PEG molecular weight. DCF polyurethanes without PEG wasalso synthesized to observe the effect on the release profile whenno PEG is present in the backbone. The GPC analysis of thepolymers showed they had average molecular weights within the
Scheme 1 Synthesis of diclofenac incorporating diol monomer (DCF-PHB-MG, 2).
Scheme 2 Synthesis of linear polyurethane diclofenac (DCF) polymerconjugate via step growth approach in a one pot system.
Communication Journal of Materials Chemistry B
Publ
ishe
d on
24
July
201
7. D
ownl
oade
d by
Mon
ash
Uni
vers
ity o
n 03
/08/
2017
03:
09:0
1.
View Article Online
This journal is©The Royal Society of Chemistry 2017 J. Mater. Chem. B
range Mn = 5.05–13.5 kDa and polydispersity indexes fromÐ = 1.35–3.56, common for any step-growth polymerization system(Table S1, ESI†).34,35
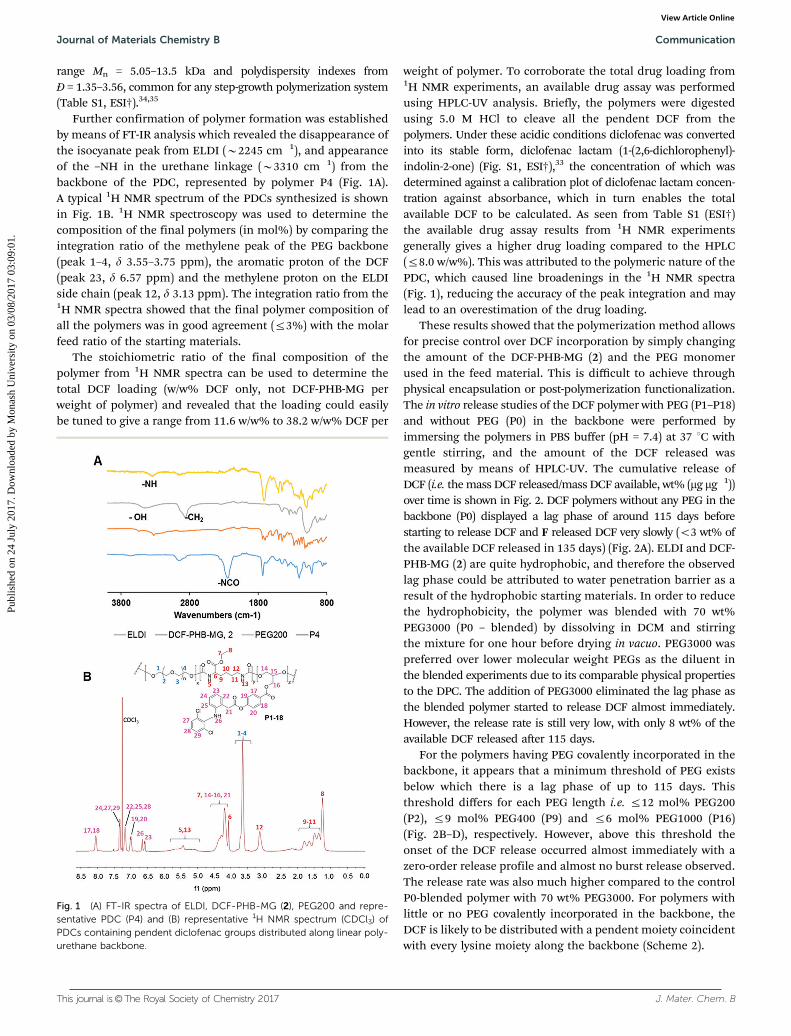
Further confirmation of polymer formation was establishedby means of FT-IR analysis which revealed the disappearance ofthe isocyanate peak from ELDI (B2245 cm�1), and appearanceof the –NH in the urethane linkage (B3310 cm�1) from thebackbone of the PDC, represented by polymer P4 (Fig. 1A).A typical 1H NMR spectrum of the PDCs synthesized is shownin Fig. 1B. 1H NMR spectroscopy was used to determine thecomposition of the final polymers (in mol%) by comparing theintegration ratio of the methylene peak of the PEG backbone(peak 1–4, d 3.55–3.75 ppm), the aromatic proton of the DCF(peak 23, d 6.57 ppm) and the methylene proton on the ELDIside chain (peak 12, d 3.13 ppm). The integration ratio from the1H NMR spectra showed that the final polymer composition ofall the polymers was in good agreement (r3%) with the molarfeed ratio of the starting materials.
The stoichiometric ratio of the final composition of thepolymer from 1H NMR spectra can be used to determine thetotal DCF loading (w/w% DCF only, not DCF-PHB-MG perweight of polymer) and revealed that the loading could easilybe tuned to give a range from 11.6 w/w% to 38.2 w/w% DCF per
weight of polymer. To corroborate the total drug loading from1H NMR experiments, an available drug assay was performedusing HPLC-UV analysis. Briefly, the polymers were digestedusing 5.0 M HCl to cleave all the pendent DCF from thepolymers. Under these acidic conditions diclofenac was convertedinto its stable form, diclofenac lactam (1-(2,6-dichlorophenyl)-indolin-2-one) (Fig. S1, ESI†),33 the concentration of which wasdetermined against a calibration plot of diclofenac lactam concen-tration against absorbance, which in turn enables the totalavailable DCF to be calculated. As seen from Table S1 (ESI†)the available drug assay results from 1H NMR experimentsgenerally gives a higher drug loading compared to the HPLC(r8.0 w/w%). This was attributed to the polymeric nature of thePDC, which caused line broadenings in the 1H NMR spectra(Fig. 1), reducing the accuracy of the peak integration and maylead to an overestimation of the drug loading.
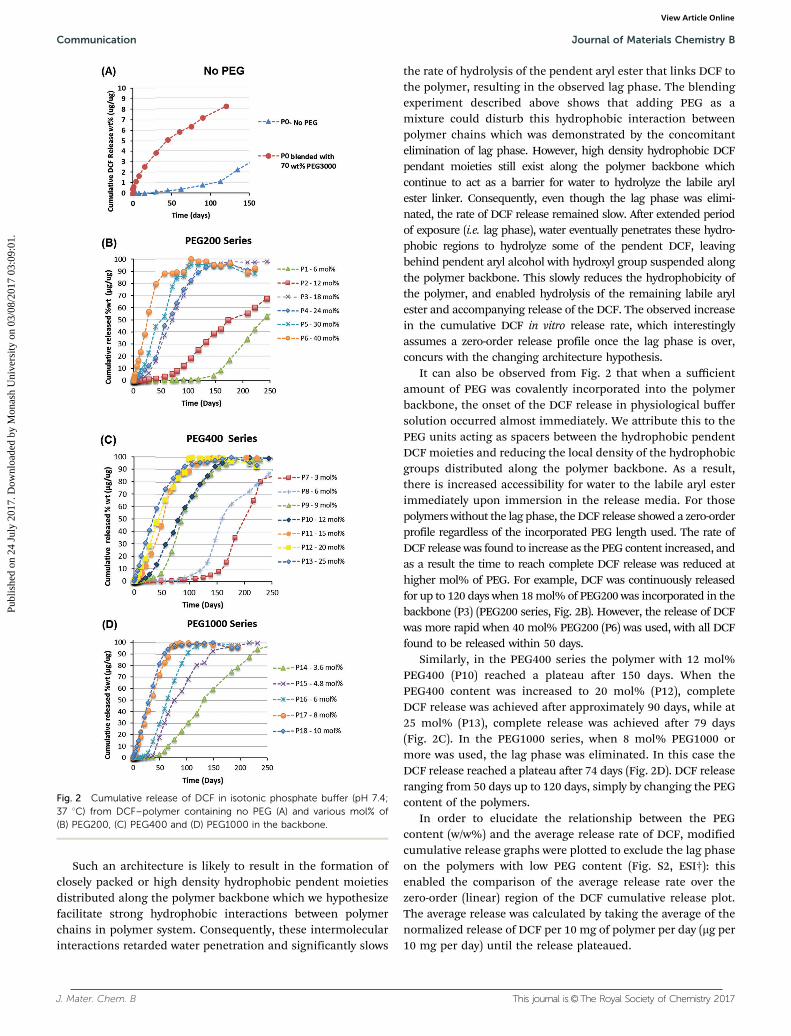
These results showed that the polymerization method allowsfor precise control over DCF incorporation by simply changingthe amount of the DCF-PHB-MG (2) and the PEG monomerused in the feed material. This is difficult to achieve throughphysical encapsulation or post-polymerization functionalization.The in vitro release studies of the DCF polymer with PEG (P1–P18)and without PEG (P0) in the backbone were performed byimmersing the polymers in PBS buffer (pH = 7.4) at 37 1C withgentle stirring, and the amount of the DCF released wasmeasured by means of HPLC-UV. The cumulative release ofDCF (i.e. the mass DCF released/mass DCF available, wt% (mg mg�1))over time is shown in Fig. 2. DCF polymers without any PEG in thebackbone (P0) displayed a lag phase of around 115 days beforestarting to release DCF and F released DCF very slowly (o3 wt% ofthe available DCF released in 135 days) (Fig. 2A). ELDI and DCF-PHB-MG (2) are quite hydrophobic, and therefore the observedlag phase could be attributed to water penetration barrier as aresult of the hydrophobic starting materials. In order to reducethe hydrophobicity, the polymer was blended with 70 wt%PEG3000 (P0 – blended) by dissolving in DCM and stirringthe mixture for one hour before drying in vacuo. PEG3000 waspreferred over lower molecular weight PEGs as the diluent inthe blended experiments due to its comparable physical propertiesto the DPC. The addition of PEG3000 eliminated the lag phase asthe blended polymer started to release DCF almost immediately.However, the release rate is still very low, with only 8 wt% of theavailable DCF released after 115 days.
For the polymers having PEG covalently incorporated in thebackbone, it appears that a minimum threshold of PEG existsbelow which there is a lag phase of up to 115 days. Thisthreshold differs for each PEG length i.e. r12 mol% PEG200(P2), r9 mol% PEG400 (P9) and r6 mol% PEG1000 (P16)(Fig. 2B–D), respectively. However, above this threshold theonset of the DCF release occurred almost immediately with azero-order release profile and almost no burst release observed.The release rate was also much higher compared to the controlP0-blended polymer with 70 wt% PEG3000. For polymers withlittle or no PEG covalently incorporated in the backbone, theDCF is likely to be distributed with a pendent moiety coincidentwith every lysine moiety along the backbone (Scheme 2).
Fig. 1 (A) FT-IR spectra of ELDI, DCF-PHB-MG (2), PEG200 and repre-sentative PDC (P4) and (B) representative 1H NMR spectrum (CDCl3) ofPDCs containing pendent diclofenac groups distributed along linear poly-urethane backbone.
Journal of Materials Chemistry B Communication
Publ
ishe
d on
24
July
201
7. D
ownl
oade
d by
Mon
ash
Uni
vers
ity o
n 03
/08/
2017
03:
09:0
1.
View Article Online
J. Mater. Chem. B This journal is©The Royal Society of Chemistry 2017
Such an architecture is likely to result in the formation ofclosely packed or high density hydrophobic pendent moietiesdistributed along the polymer backbone which we hypothesizefacilitate strong hydrophobic interactions between polymerchains in polymer system. Consequently, these intermolecularinteractions retarded water penetration and significantly slows
the rate of hydrolysis of the pendent aryl ester that links DCF tothe polymer, resulting in the observed lag phase. The blendingexperiment described above shows that adding PEG as amixture could disturb this hydrophobic interaction betweenpolymer chains which was demonstrated by the concomitantelimination of lag phase. However, high density hydrophobic DCFpendant moieties still exist along the polymer backbone whichcontinue to act as a barrier for water to hydrolyze the labile arylester linker. Consequently, even though the lag phase was elimi-nated, the rate of DCF release remained slow. After extended periodof exposure (i.e. lag phase), water eventually penetrates these hydro-phobic regions to hydrolyze some of the pendent DCF, leavingbehind pendent aryl alcohol with hydroxyl group suspended alongthe polymer backbone. This slowly reduces the hydrophobicity ofthe polymer, and enabled hydrolysis of the remaining labile arylester and accompanying release of the DCF. The observed increasein the cumulative DCF in vitro release rate, which interestinglyassumes a zero-order release profile once the lag phase is over,concurs with the changing architecture hypothesis.
It can also be observed from Fig. 2 that when a sufficientamount of PEG was covalently incorporated into the polymerbackbone, the onset of the DCF release in physiological buffersolution occurred almost immediately. We attribute this to thePEG units acting as spacers between the hydrophobic pendentDCF moieties and reducing the local density of the hydrophobicgroups distributed along the polymer backbone. As a result,there is increased accessibility for water to the labile aryl esterimmediately upon immersion in the release media. For thosepolymers without the lag phase, the DCF release showed a zero-orderprofile regardless of the incorporated PEG length used. The rate ofDCF release was found to increase as the PEG content increased, andas a result the time to reach complete DCF release was reduced athigher mol% of PEG. For example, DCF was continuously releasedfor up to 120 days when 18 mol% of PEG200 was incorporated in thebackbone (P3) (PEG200 series, Fig. 2B). However, the release of DCFwas more rapid when 40 mol% PEG200 (P6) was used, with all DCFfound to be released within 50 days.
Similarly, in the PEG400 series the polymer with 12 mol%PEG400 (P10) reached a plateau after 150 days. When thePEG400 content was increased to 20 mol% (P12), completeDCF release was achieved after approximately 90 days, while at25 mol% (P13), complete release was achieved after 79 days(Fig. 2C). In the PEG1000 series, when 8 mol% PEG1000 ormore was used, the lag phase was eliminated. In this case theDCF release reached a plateau after 74 days (Fig. 2D). DCF releaseranging from 50 days up to 120 days, simply by changing the PEGcontent of the polymers.
In order to elucidate the relationship between the PEGcontent (w/w%) and the average release rate of DCF, modifiedcumulative release graphs were plotted to exclude the lag phaseon the polymers with low PEG content (Fig. S2, ESI†): thisenabled the comparison of the average release rate over thezero-order (linear) region of the DCF cumulative release plot.The average release was calculated by taking the average of thenormalized release of DCF per 10 mg of polymer per day (mg per10 mg per day) until the release plateaued.
Fig. 2 Cumulative release of DCF in isotonic phosphate buffer (pH 7.4;37 1C) from DCF–polymer containing no PEG (A) and various mol% of(B) PEG200, (C) PEG400 and (D) PEG1000 in the backbone.
Communication Journal of Materials Chemistry B
Publ
ishe
d on
24
July
201
7. D
ownl
oade
d by
Mon
ash
Uni
vers
ity o
n 03
/08/
2017
03:
09:0
1.
View Article Online

This journal is©The Royal Society of Chemistry 2017 J. Mater. Chem. B
Fig. 3 shows the plot of average DCF release rate (mg per10 mg per day) of the zero-order region against the PEG content(w/w%, weight of PEG per weight of polymer). Fig. 3 reveals thatwhen the PEG content is lower than 10 w/w%, the average DCFrelease rate is similar (B10 mg per 10 mg per day) regardless ofwhat molecular weight of PEG was incorporated. However,above this level some differences were observed between thethree PEG series, indicating some molecular weight dependencein the release rate at higher PEG contents.
The PEG200 series showed a gradual increase in the averageDCF release rate from 10–15 mg per 10 mg per day as the level ofPEG content increases. The PEG400 series also showed similaraverage DCF release range (10–15 mg per 10 mg per day),however the observed increase in rate is greater as the incorporatedPEG content increases. The PEG1000 series showed, overall, a muchhigher average DCF release for the same PEG content as the PEG200and PEG400 series. The average DCF release increased sharply whenthe PEG content increases from 15 w/w% to 22 w/w%, however asthe PEG content increases further to 26 w/w%, no further increasein DCF average release rate was observed.
Conclusions
In conclusion, we have demonstrated an effective method forthe synthesis of polymer-diclofenac conjugates (PDCs) via poly-urethane chemistry that provides both a sustained release ofdiclofenac with no burst effect and high drug loading. Moreover,the method allows for facile control of the drug loading and releasekinetics simply by changing the feed ratio of the co-monomers.Application of this method allows the drug loading to be easilytailored by changing the feed ratio of the co-monomers, and givePDCs with up to 38 w/w% DCF loading. We have also demonstratedthat the drug release kinetics could easily be altered by changing theamount of PEG co-monomers to release all the pendent DCF in acontrolled manner from 50 days up to 120 days, depending onthe amount of PEG used and the desired application/treatmentperiod. The PDCs described may be suitable for long term intra-articular (IA) delivery for the treatment osteoarthritis (OA).Currently, some works are underway in testing the efficacy ofthe polymers in an in vivo validated animal model.
Acknowledgements
This collaborative research was conducted by PolyActiva, Pty.Ltd, University of Melbourne (ARC Linkage grant LP12020010 &LP140100284) and Monash University through the AustralianResearch Council Centre of Excellence in Convergent Bio-NanoScience and Technology (project number CE140100036). TPDwishes to acknowledge the award of an Australian LaureateFellowship.
References
1 P. Sarzi-Puttini, M. A. Cimmino, R. Scarpa, R. Caporali,F. Parazzini, A. Zaninelli, F. Atzeni and B. Canesi, Semin.Arthritis Rheum., 2005, 35, 1–10.
2 Coxib and traditional NSAID Trialists’ (CNT) Collaboration,Lancet, 2013, 382, 769–779.
3 N. Gerwin, C. Hops and A. Lucke, Adv. Drug Delivery Rev.,2006, 58, 226–242.
4 S. H. R. Edwards, Vet. J., 2011, 190, 15–21.5 C. Larsen, J. Østergaard, S. W. Larsen, H. Jensen, S. Jacobsen,
C. Lindegaard and P. H. Andersen, J. Pharm. Sci., 2008, 97,4622–4654.
6 S. G. Owen, H. W. Francis and M. S. Roberts, Br. J. Clin.Pharmacol., 1994, 38, 349–355.
7 H. R. Schumacher, Arthritis Rheum., 2003, 49, 413–420.8 H. M. Burt, A. Tsallas, S. Gilchrist and L. S. Liang, Expert
Opin. Drug Delivery, 2009, 6, 17–26.9 A. Wahab, M. E. Favretto, N. D. Onyeagor, G. M. Khan,
D. Douroumis, M. A. Casely-Hayford and P. Kallinteri,J. Microencapsulation, 2012, 29, 497–504.
10 S. S. Banerjee, N. Aher, R. Patil and J. Khandare, J. DrugDelivery, 2012, 103973, DOI: 10.1155/2012/103973.
11 J. Y. Cherng, T. Y. Hou, M. F. Shih, H. Talsma and W. E. Hennink,Int. J. Pharm., 2013, 450, 145–162.
12 S. A. Guelcher, A. Srinivasan, J. E. Dumas, J. E. Didier, S. McBrideand J. O. Hollinger, Biomaterials, 2008, 29, 1762–1775.
13 J. J. Loh, K. B. C. Sng and J. Li, Biomaterials, 2008, 29,3185–3194.
14 M. Ding, X. He, Z. Wang, J. Li, H. Tan, H. Deng, Q. Fu andQ. Gu, Biomaterials, 2011, 32, 9515–9524.
15 A. K. Mahanta, V. Mittal, N. Singh, D. Dash, S. Malik, M. Kumarand P. Maiti, Macromolecules, 2015, 48, 2654–2666.
16 H. Chen, Y. Li, Y. Liu, T. Gong, L. Wang and S. Zhou, Polym.Chem., 2014, 5, 5168–5174.
17 I. Omrani, N. Babanejad, H. K. Shendi and M. R. Nabid,Mater. Sci. Eng., C, 2017, 70, 607–616.
18 M. Ding, N. Song, X. He, J. Li, L. Zhou, H. Tan, Q. Fu andQ. Gu, ACS Nano, 2013, 7, 1918–1928.
19 G. Morral-Ruız, P. Melgar-Lesmes, C. Solans andM. J. Garcıa-Celma, J. Controlled Release, 2013, 171,163–171.
20 A. Basu, S. Farah, K. R. Kunduru, S. Doppalapudi, W. Khanand A. J. Domb, 8 – Polyurethanes for controlled drugdelivery, Advances in Polyurethane Biomaterials, WoodheadPublishing, 2016, pp. 217–246.
Fig. 3 Plot of average DCF release rate (mg per 10 mg per day) of thezero-order region against the PEG content (w/w%) in a physiologicalcondition. The dotted lines represent the polymer with lag phase.
Journal of Materials Chemistry B Communication
Publ
ishe
d on
24
July
201
7. D
ownl
oade
d by
Mon
ash
Uni
vers
ity o
n 03
/08/
2017
03:
09:0
1.
View Article Online

J. Mater. Chem. B This journal is©The Royal Society of Chemistry 2017
21 M. Boffito, E. Gioffredi, V. Chiono, S. Calzone, E. Ranzato,S. Martinotti and G. Ciardelli, Polym. Int., 2016, 65, 756–769.
22 C. Akduman, I. Ozguney and E. P. A. Kumbasar, Mater. Sci.Eng., C, 2016, 64, 383–390.
23 Z. Pan, Y. Ren, N. Song, Y. Song, J. Li, X. He, F. Luo, H. Tanand Q. Fu, Biomacromolecules, 2016, 17, 2148–2159.
24 Y. Yao, D. Xu, C. Liu, Y. Guan, J. Zhang, Y. Su, L. Zhao,F. Meng and J. Luo, RSC Adv., 2016, 6, 97684–97693.
25 Z. Pan, L. Yu, N. Song, L. Zhou, J. Li, M. Ding, H. Tan andQ. Fu, Polym. Chem., 2014, 5, 2901–2910.
26 C. Ferris, M. V. De Paz, A. Aguilar-De-Leyva, I. Caraballo andJ. A. Galbis, Polym. Chem., 2014, 5, 2370–2381.
27 X. He, M. Ding, J. Li, H. Tan, Q. Fu and L. Li, RSC Adv., 2014,4, 24736–24746.
28 J. P. Santerre, K. Woodhouse, G. Laroche and R. S. Labow,Biomaterials, 2005, 26, 7457–7470.
29 A. E. Hafeman, K. J. Zienkiewicz, A. L. Zachman, H. J. Sung,L. B. Nanney, J. M. Davidson and S. A. Guelcher, Biomaterials,2001, 32, 419–429.
30 F. Wang, J. Finnin, C. Tait, S. Quirk, I. Chekhtman,A. C. Donohue, S. M. Y. Ng, A. M. D’Souza, R. J. Tait andR. Prankerd, J. Pharm. Sci., 2016, 105, 773–785.
31 A. M. D’Souza, A. C. Donohue, R. J. Tait, F. H. Graichen,S. M. Y. Ng and A. Sulistio, Polymer–NSAID conjugate,WO2014000033 A1, 2014.
32 H. R. Kricheldorf, Macromolecules, 2003, 36, 2302–2308.33 B. Fernandez d’Arlas, L. Rueda, P. M. Stefani, K. de la Caba,
I. Mondragon and A. Eceiza, Thermochim. Acta, 2007, 459,94–103.
34 W. H. Stockmayer, J. Polym. Sci., Part A: Polym. Chem., 1952,9, 69–71.
35 J. K. Stille, J. Chem. Educ., 1981, 58, 862–866.
Communication Journal of Materials Chemistry B
Publ
ishe
d on
24
July
201
7. D
ownl
oade
d by
Mon
ash
Uni
vers
ity o
n 03
/08/
2017
03:
09:0
1.
View Article Online


















