
Molekularbiologische Untersuchungen zum Nachweis ... · 4.5 Quantifizierung mit der TaqMan®...
134
Lehrstuhl für Zellbiologie der Technischen Universität München Wissenschaftszentrum Weihenstephan Univ.-Prof. Dr. Bertold Hock Molekularbiologische Untersuchungen zum Nachweis arbuskulärer Mykorrhizapilze bei Wildpflanzenpopulationen landwirtschaftlicher Nutzflächen Holger Geue Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. J. Schnyder Prüfer der Dissertation: 1. Univ.-Prof. Dr. B. Hock 2. Priv.-Doz. Dr. P. Schröder Die Dissertation wurde am 2. Juli 2002 bei der Technischen Universität München eingereicht und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung und Umwelt am 11. September 2002 angenommen.
Transcript of Molekularbiologische Untersuchungen zum Nachweis ... · 4.5 Quantifizierung mit der TaqMan®...

Lehrstuhl für Zellbiologie der Technischen Universität München
Wissenschaftszentrum Weihenstephan
Univ.-Prof. Dr. Bertold Hock
Molekularbiologische Untersuchungen
zum Nachweis arbuskulärer Mykorrhizapilze
bei Wildpflanzenpopulationen landwirtschaftlicher Nutzflächen
Holger Geue
Vollständiger Abdruck der von der Fakultät Wissenschaftszentrum Weihenstephan für
Ernährung, Landnutzung und Umwelt der Technischen Universität München zur Erlangung
des akademischen Grades eines
Doktors der Naturwissenschaften (Dr. rer. nat.)
genehmigten Dissertation.
Vorsitzender: Univ.-Prof. Dr. J. Schnyder
Prüfer der Dissertation: 1. Univ.-Prof. Dr. B. Hock
2. Priv.-Doz. Dr. P. Schröder
Die Dissertation wurde am 2. Juli 2002 bei der Technischen Universität München eingereicht
und durch die Fakultät Wissenschaftszentrum Weihenstephan für Ernährung, Landnutzung
und Umwelt am 11. September 2002 angenommen.

II
„Die Wissenschaft fängt eigentlich erst dann an
interessant zu werden, wo sie aufhört.“
Justus von Liebig (1803-1873), deutscher Chemiker,
Begründer der Mineraldüngung in der Landwirtschaft

III
Meinem Lehrer
Karl Aldinger, *15. Juni 1952, U8. Mai 1996

IV
Inhaltsverzeichnis
1 Einleitung......................................................................................................................... 1
1.1 Arbuskuläre Mykorrhiza............................................................................................... 1
1.2 Identifizierung der arbuskulären Mykorrhizapilze ....................................................... 5
1.3 Molekularbiologische Untersuchungen bei arbuskulären Mykorrhizapilzen ............... 5
1.4 Zielsetzung.................................................................................................................... 6
2 Material und Methoden.................................................................................................. 7
2.1 Material ......................................................................................................................... 7
2.1.1 Versuchsflächen ..................................................................................................... 7
2.1.2 Probennahme im Freiland ...................................................................................... 9
2.1.3 Pflanzen................................................................................................................ 10
2.1.4 Arbuskuläre Mykorrhizapilze und sonstige Pilze ................................................ 11
2.1.5 Oligonukleotidprimer ........................................................................................... 12
2.1.6 Enzyme................................................................................................................. 12
2.1.7 Reagenzien ........................................................................................................... 13
2.1.8 Kits ....................................................................................................................... 14
2.1.9 Verbrauchsmaterial .............................................................................................. 15
2.1.10 Geräte ................................................................................................................... 16
2.1.11 Puffer und Färbelösungen .................................................................................... 17
2.2 Methoden .................................................................................................................... 21
2.2.1 Aufarbeitung der Proben ...................................................................................... 21
2.2.2 Produktion von Referenzkulturen ........................................................................ 22
2.2.3 Optimierung der DNA-Extraktion ....................................................................... 23
2.2.4 Bestimmung der DNA-Konzentration.................................................................. 27
2.2.5 Oligonukleotidprimer ........................................................................................... 28
2.2.6 Polymerase-Kettenreaktion .................................................................................. 30
2.2.6.1 Qualitative PCR-Analyse mit universellen Primern ......................................... 31
2.2.6.2 PCR-Analyse mit spezifischen Primern............................................................ 33
2.2.7 Gelelektrophorese................................................................................................. 34
2.2.8 Restriktionsfragment-Längenpolymorphismus-(RFLP-)Analyse........................ 35
2.2.9 Sequenzierung und Alignment ............................................................................. 36
2.2.10 Quantitativer Nachweis ........................................................................................ 36
2.2.11 Bestimmung der AMF anhand der Sporenmorphologie ...................................... 39
2.2.12 Färbung der intraradikalen Pilzstrukturen............................................................ 40

V
2.2.13 Bestimmung des Mykorrhizierungsgrades........................................................... 41
2.2.14 Phosphatbestimmung ........................................................................................... 42
3 Ergebnisse...................................................................................................................... 44
3.1 Optimierung der DNA-Extraktion aus Wurzelmaterial .............................................. 44
3.2 Untersuchung der Oligonukleotidprimer .................................................................... 45
3.2.1 Kontrolle der Amplifizierbarkeit.......................................................................... 49
3.2.2 Qualitativer Nachweis von A. longula mit spezifischen Primern ........................ 50
3.2.3 Qualitativer Nachweis von G. mosseae in Pflanzenwurzeln................................ 51
3.2.4 RFLP-Analyse der G. mosseae-Gruppe ............................................................... 52
3.2.5 Sequenzanalysen .................................................................................................. 54
3.3 Quantitativer Nachweis der AMF............................................................................... 59
3.3.1 Quantifizierung von A. longula mit der TaqMan® PCR ...................................... 59
3.3.2 Quantifizierung von G. mosseae mit der TaqMan® PCR..................................... 62
3.4 Zusammenfassung der molekularbiologischen und morphologischen Ergebnisse..... 65
3.5 Bestimmung der Mykorrhizapilze anhand der Sporenmorphologie ........................... 68
3.6 Bestimmung des Mykorrhizierungsgrades in Freilandproben .................................... 68
3.7 Phosphatgehalte .......................................................................................................... 74
3.8 Zusammenhang zwischen Mykorrhizierungsgrad und Phosphatgehalt...................... 79
4 Diskussion ...................................................................................................................... 82
4.1 DNA-Extraktion aus Wurzelmaterial.......................................................................... 82
4.2 Amplifizierbarkeit....................................................................................................... 83
4.3 Spezifische Primer ...................................................................................................... 84
4.4 Sequenzanalysen und RFLP........................................................................................ 86
4.5 Quantifizierung mit der TaqMan® real-time PCR ...................................................... 86
4.6 Identifikation der Mykorrhizapilze anhand der Sporenmorpholgie............................ 88
4.7 Mykorrhizierung und Phosphatgehalte ....................................................................... 89
4.8 Biodiversität: Arten und Individuen ........................................................................... 91
4.9 Fortpflanzung und Variabilität der AMF.................................................................... 93
4.10 Phylogenetische und Systematische Neuordnung der Morphospezies ....................... 95
4.11 Evolution und Artenzahl der AMF ............................................................................. 98
5 Zusammenfassung ...................................................................................................... 101
6 Literatur....................................................................................................................... 102
7 Anhang......................................................................................................................... 115

VI
Abbildungsverzeichnis
Abb. 1: Stammbaum der Glomales nach morphologischen Kriterien........................................ 2
Abb. 2: Typische morphologische Merkmale einer Glomus-Art in Pflanzenwurzeln ............... 2
Abb. 3: Wiese W02 mit Transsekt und den Positionen I, II und III. ................................... 7
Abb. 4: Weide W21 mit Transsekt und den Positionen 1 bis 5. ................................................ 8
Abb. 5: Übersicht der F.A.M.-Versuchsstation Klostergut Scheyern ........................................ 8
Abb. 6: Plantago lanceolata. ................................................................................................... 11
Abb. 7: Trifolium repens.... ...................................................................................................... 11
Abb. 8: Holcus lanatus............................................................................................................. 11
Abb. 9: Schematische Darstellung der Probenaufarbeitung..................................................... 21
Abb. 10: Anordnung der rDNA-Einheiten ............................................................................... 29
Abb. 11: Hybridisierungspositionen der Oligonukleotidprimer............................................... 30
Abb. 12: DNA-Längenmarker GeneRuler 100 bp DNA Ladder Plus...................................... 35
Abb. 13: Schematische Darstellung des Prinzip der TaqMan® real time PCR........................ 37
Abb. 14: DNA-Extrakte mit verschiedenen Zelllyse-Techniken ............................................. 45
Abb. 15: Amplifikate der PCR mit dem Primerpaar A2R-B2L (1) ......................................... 50
Abb. 16: Amplifikate der PCR mit dem Primerpaar A2R-B2L (2) ......................................... 50
Abb. 17: Amplifikate der PCR mit dem Primerpaar Aclong87For-Aclong400Rev ................ 51
Abb. 18: Amplifikate der PCR mit dem Primerpaar Glmoss85For-Glmoss387Rev (1).......... 52
Abb. 19: Amplifikate der PCR mit dem Primerpaar Glmoss85For-Glmoss387Rev (2).......... 52
Abb. 20: RFLP-Analyse von zehn PCR-Fragmenten der G. mosseae-Gruppe........................ 54
Abb. 21: Ergebnis der Sequenzanalyse im Bereich der Domäne D2....................................... 56
Abb. 22: Phylogenetischer Stammbaum der G. mosseae-Gruppe ........................................... 58
Abb. 23: Amplification-Plot von A. longula ............................................................................ 60
Abb. 24: Zusammenhang zwischen Kopienzahl und Ct-Wert der Kalibriergerade bei A.
longula. ...................................................................................................................... 60
Abb. 25: Kontrolle der Bindung des Primers P1 an die ermittelten Sequenzabschnitte. ......... 62
Abb. 26: Kontrolle der Bindung der Sonde F1 an die ermittelten Sequenzabschnitte............. 63
Abb. 27: Amplification-Plot von G. mosseae-Standards ......................................................... 63
Abb. 28: Zusammenhang zwischen den ermittelten Ct-Werten und der eingesetzten
Kopienzahl bei G. mosseae ........................................................................................ 64
Abb. 29: Sporenquetschpräparat von G. mosseae.................................................................... 69
Abb. 30: P. lanceolata-Wurzel nach Trypanblaufärbung ........................................................ 69
Abb. 31: Mykorrhizierungsraten nach Position (W02). ........................................................... 72

VII
Abb. 32: Mykorrhizierungsraten nach Pflanzen (W02) ........................................................... 72
Abb. 33: Mykorrhizierungsraten nach Probennahmeterminen (W02)..................................... 72
Abb. 34: Mykorrhizierungsraten nach Position (W21). ........................................................... 73
Abb. 35: Mykorrhizierungsraten nach Pflanzen (W21) ........................................................... 73
Abb. 36: Mykorrhizierungsraten nach Probenahmeterminen (W21)....................................... 73
Abb. 37: Kalibriergerade der Phosphatmessung. ..................................................................... 74
Abb. 38: Durchschnittlicher Phosphatgehalt nach Position (W02).......................................... 76
Abb. 39: Durchschnittlicher Phosphatgehalt nach Pflanze (W02)........................................... 76
Abb. 40: Durchschnittlicher Phosphatgehalt nach Probennahmeterminen (W02)................... 76
Abb. 41: Durchschnittlicher Phosphatgehalt nach Position (W21).......................................... 78
Abb. 42: Durchschnittlicher Phosphatgehalt nach Pflanze (W21)........................................... 78
Abb. 43: Durchschnittlicher Phosphatgehalt nach Probenahmeterminen (W21)..................... 78
Abb. 44: Hyphenkolonisierung in Abhängigkeit vom Phosphatgehalt des Bodens (W02) ..... 80
Abb. 45: Vesikelkolonisierung in Abhängigkeit vom Phosphatgehalt des Bodens (W02)...... 80
Abb. 46: Hyphenkolonisierung in Abhängigkeit vom Phosphatgehalt des Bodens (W21) ..... 81
Abb. 47: Vesikelkolonisierung in Abhängigkeit vom Phosphatgehalt des Bodens (W21)...... 81
Abb. 48: Stammbaum der Glomales nach MORTON & REDECKER (2001)............................... 96
Abb. 49: Stammbaum der Glomeromycota nach SCHWARZOTT et al. (2001). ........................ 98
Abb. 50: Zahl der neu beschriebenen AMF-Arten seit 1974. ................................................ 120

VIII
Tabellenverzeichnis
Tab. 1: Höhenlage der Beprobungspunkte. ................................................................................ 9
Tab. 2: Übersicht der Beprobungstermine auf der Wiese W02 und Weide W21. ..................... 9
Tab. 3: Schlag, Position und Pflanze der Ackerproben............................................................ 10
Tab. 4: Stämme und Herkunft der als Referenz verwendeten AMF. ....................................... 11
Tab. 5: Übersicht über die verwendeten Primer....................................................................... 12
Tab. 6: DNA-Verdünnungsreihe für den PicoGreen®-Assay................................................... 27
Tab. 7: Theoretische Bindungsspezifität von Oligonukleotiden. ............................................. 28
Tab. 8: Standard PCR-Reaktionsgemisch ................................................................................ 31
Tab. 9: Zusammensetzung des Master Mix A.......................................................................... 32
Tab. 10: Temperaturprogramm 1 für die PCR mit den Primern A2R-B2L. ............................ 32
Tab. 11: Größe der unterschiedlichen PCR-Produkte .............................................................. 33
Tab. 12: Zusammensetzung des Master Mix B........................................................................ 33
Tab. 13: Temperaturprogramm 2 für die PCR mit spezifischen Primern. ............................... 34
Tab. 14: Zusammensetzung eines 1%-igen Agarose-Geles. .................................................... 34
Tab. 15: Endonukleasen und ihre Erkennungssequenz. Pfeile geben die Schnittstelle an....... 36
Tab. 16: Master Mix C für die TaqMan® PCR-Analyse. ......................................................... 37
Tab. 17: Plasmid-Anzahl in der verwendeten Standardreihe. .................................................. 38
Tab. 18: Temperaturprogramm 3 für die TaqMan® PCR......................................................... 39
Tab. 19: Pipettierschema der Kalibrierreihe zur Phosphatbestimmung. .................................. 43
Tab. 20: Vergleich der Sequenzen spezifischer Primer mit Datenbanksequenzen .................. 46
Tab. 21: Ergebnisse der „virtuellen“ PCR ............................................................................... 49
Tab. 22: Abkürzungen der untersuchten Sequenzen ................................................................ 53
Tab. 23: Übersicht über die Schnittstellen des Enzyms AluI ................................................... 54
Tab. 24: Sequenzhomologie mit nächstverwandten Arten....................................................... 55
Tab. 25: Zusammenstellung der Ct-Werte der eingesetzten Standard-Verdünnungsreihe bei
A. longula. .................................................................................................................. 61
Tab. 26: Anzahl der ermittelten Kopienzahlen bei A. longula ................................................. 61
Tab. 27: Zusammenstellung der Ct-Werte der eingesetzten Standard-Verdünnungsreihe bei
G. mosseae. ................................................................................................................ 63
Tab. 28: Anzahl der ermittelten Kopienzahlen bei G. mosseae. .............................................. 64
Tab. 29: Ergebnisübersicht der molekularbiologischen Untersuchungen................................ 66
Tab. 30: Ergebnis der morphologischen Sporenuntersuchung................................................. 68
Tab. 31: Mykorrhizierung der Pflanzen auf der Wiese (W02)................................................. 70

IX
Tab. 32: Mykorrhizierung der Pflanzen auf der Weide (W21) ................................................ 71
Tab. 33: Phosphatgehalte der Wiese W02................................................................................ 75
Tab. 34: Phosphatgehalte der Weide W21 ............................................................................... 77

X
Abkürzungen
A Adenin
Abb. Abbildung
AC Arbuskel-Kolonisierungsrate nach MCGONIGLE et al. (1990)
AGE Agarose-Gelelektrophorese
A. longula A. longula BEG 8
AM arbuskuläre Mykorrhiza
AMF arbuskuläre Mykorrhizapilze (arbuscular mycorrhizal fungi)
Aqua dest. destilliertes Wasser
BEG La Banque Européene des Glomales (European Bank of Glomales)
bp Basenpaar(e) (base pairs)
BSA Rinderserumalbumin (Bovine Serum Albumin)
C Cytosin
DMSO Dimethylsulfoxid
DNA Desoxyribonukleinsäure (Desoxyribonuleic acid)
dNTP Desoxynucleosidtriphosphat
dTTP Desoxythymidintriphosphat
dUTP Desoxyuridintriphosphat
DTT Dithiothreitol
EDTA Ethylendiamintetraessigsäure
EtOH Ethanol
FAA Formaldehyd-Aceto-Alkohol
F.A.M. Forschungsverbund Agrarökosysteme München
FAM 6-Carboxyfluorescein
FeEDTA Eisenethylendiamintetraacetat
FG Frischgewicht
G Guanin
h Stunde(n)
HC Hyphen-Kolonisierungsrate nach MCGONIGLE et al. (1990)
INVAM International Culture Collection of Arbuscular & Vesicular-
Arbuscular Mycorrhizal Fungi
LSU große Untereinheit (large subunit)
M molar, Molarität (mol/L)
mg Milligramm

XI
mL Milliliter
mM millimolar
min Minute(n)
mm Millimeter
µg Mikrogramm
µL Mikroliter
µM mikromolar
µm Mikrometer
N (bei Lösungen) normal, Normalität
N (in Sequenzen) Nukleosid (Adenin, Cytosin, Guanin und Thymin)
NaOH Natriumhydroxid
NCBI National Center for Biotechnology Information
nm Nanometer
PCR Polymerase-Kettenreaktion (Polymerase Chain Reaction)
PVLG Polyvinyl-Lacto-Glycerin
PVP Polyvinylpyrrolidon
rDNA ribosomale DNA
RNA Ribonucleic acid, Ribonukleinsäure
RNase Ribonuklease
rpm Umdrehungen pro Minute (rounds per minute)
RT Raumtemperatur
s Sekunde(n)
sp. Spezies
SSU kleine Untereinheit (small subunit)
T Thymin
Tab. Tabelle
TAMRA 6-Carboxyltetramethylrhodamin
Taq Thermus aquaticus
TBE Tris-Borat-EDTA
TE Tris-EDTA
Tris Tris(hydroxymethyl)aminomethan
U Unit(s)
v/v Volumen pro Volumen
VAM vesikulär-arbuskuläre Mykorrhiza

XII
VC Vesikel-Kolonisierungsrate nach MCGONIGLE et al. (1990)
w/v Masse pro Volumen (weight per volume)

1 Einleitung 1
1 Einleitung
1.1 Arbuskuläre Mykorrhiza
Geschichtliches
Im Jahr 1845 entdeckten TULASNE & TULASNE Sporen der arbuskulären Mykorrhizapilze
(AMF) und beschrieben die ersten beiden Arten der neu eingeführten Gattung Glomus:
G. microcarpum und G. macrocarpum. 1885 definierte FRANK eine „auf Wurzelsymbiose
beruhende Ernährung gewisser Bäume durch unterirdische Pilze“ als „Mykorrhiza“. Der
Name setzt sich aus den beiden altgriechischen Worten ìýêçò (Pilz) und ñßæá (Wurzel)
zusammen, also „Pilzwurzel“. Es handelte sich bei dieser Form um die sogenannte
Ektomykorrhiza, die bei Bäumen vorkommt. Daneben kennt man heute noch sechs weitere
Formen solcher symbiotischer Zusammenschlüsse zwischen Pilzen und Pflanzenwurzeln
(PETERSON & FARQUHAR, 1994). Die arbuskuläre Mykorrhiza (AM) ist der häufigste Typ, der
auch als vesikulär-arbuskuläre Mykorrhiza (VAM) bezeichnet wird. Erst seit den 70er Jahren
des 20. Jahrhunderts wird die AM intensiv erforscht. Seitdem erkannte man die Bedeutung
der AMF für viele Pflanzen und damit den potentiellen Nutzen, vor allem für die
Landwirtschaft.
Vorkommen
Sowohl Sporen dieser Pilze als auch mykorrhizierte Pflanzen wurden in allen Klimazonen
und Kontinenten unseres Planeten vorgefunden. Glomus antarcticum kommt sogar in der
Antarktis vor (CABELLO et al., 1994). Etwa 80% aller Landpflanzen sind zu dieser Symbiose
befähigt (SMITH & GIANINAZZI-PEARSON, 1988; BONFANTE & PEROTTO, 1995; SMITH &
READ, 1997). Neben Kräutern und Gräsern können auch Bäume (z.B. Fraxinus excelsior,
Acer pseudoplatanus, Betula und Salix; FIEDLER, 2001), Farne (PETERSON et al., 1981), Bär-
lappgewächse (SCHMID & OBERWINKLER, 1993) und alle Klassen der Moose (STAHL, 1949;
PARKE & LINDERMAN, 1980) mit den AMF vergesellschaftet sein. Dieser enormen Zahl an
Phytobionten steht eine nur geringe Zahl an Mykobionten gegenüber: bisher wurden etwa 160
verschiedene AMF-Arten anhand ihrer Sporenmorphologie beschrieben. Die zunächst der
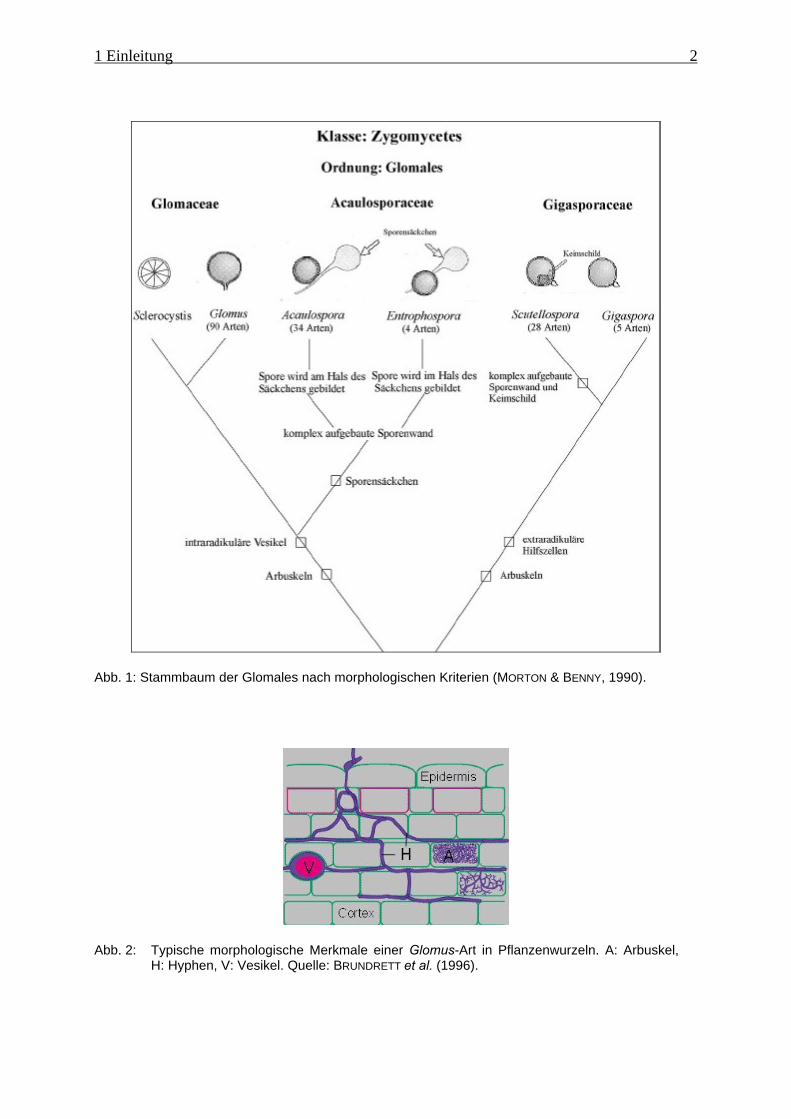
Ordnung Endogonales zugerechneten AMF wurden 1990 von MORTON & BENNY in der
neuen Ordnung Glomales zusammengefasst (Abb. 1). Die modernen Methoden der
Molekularbiologie, die in den letzten Jahren bahnbrechende Erkenntnisse in vielen
naturwissenschaftlichen Disziplinen bescherten, wurden ebenso erfolgreich auf die AMF
angewandt. Die daraus gewonnenen Erkenntnisse sind in Kapitel 4.9 zusammengefasst.
1 Einleitung 2
Abb. 1: Stammbaum der Glomales nach morphologischen Kriterien (MORTON & BENNY, 1990).
Abb. 2: Typische morphologische Merkmale einer Glomus-Art in Pflanzenwurzeln. A: Arbuskel,H: Hyphen, V: Vesikel. Quelle: BRUNDRETT et al. (1996).

1 Einleitung 3
Physiologie
Die Chlamydosporen dieser Pilze werden fast immer im Boden, selten auch in der Wurzel,
asexuell gebildet. Sie beinhalten meist sichtbare Lipidtröpfchen und andere Speicherstoffe.
Aus den Sporen keimen Hyphen, die, wenn sie nicht innerhalb weniger Tage auf die Wurzel
eines potentiellen Pflanzenpartners stoßen, wieder absterben. Ist Kontakt mit einem Phyto-
bionten hergestellt, so verdickt sich die Spitze der sogenannten runner hyphae und bildet an
der Wurzeloberfläche ein Appressorium aus (BONFANTE-FASOLO, 1984). Wie beim Eindrin-
gen eines pathogenen Pilzes reagiert die Pflanze zunächst mit Abwehrmaßnahmen (DUMAS-
GAUDOT et al., 1992; FRANKEN & GNADINGER, 1994; HARRISON & DIXON, 1994). Die ge-
nauen Abläufe der komplizierten Partnerfindung sind noch nicht bekannt (GIOVANETTI et al.,
1996). Offenbar erkennt die Pflanze jedoch sehr bald „ihren“ Pilz, so dass dieser ungehindert
eindringen kann. Die AMF bilden außerhalb der Wurzel ein feines weit verzweigtes extrara-
dikales Myzel aus, das zur Aufnahme von Wasser und mineralischen Nährstoffen dient. Da-
neben bilden sie ein intraradikales Myzel im Bereich von Wurzelkortex und Epidermis aus
(BONFANTE-FASOLO, 1984; Abb. 2). Der Zentralzylinder sowie das Meristem werden jedoch
nie kolonisiert (GIANINAZZI, 1991).
Neben diesem interzellulären Wachstum penetrieren die Hyphen die Zellwand der
Pflanzenzellen und bilden intrazellulär bäumchenförmig verzweigte Strukturen, die
sogenannten Arbuskel. Diese allen Glomales-Arten gemeinsame Struktur war auch für den
Mykorrhiza-Typ namensgebend. Die Plasmamembran der Pflanzenzelle bleibt bei der
Ausbildung des Arbuskels unbeschädigt und legt sich über die fein verzweigten Hyphen,
wobei ein neues apoplasmatisches Kompartiment entsteht, das von BONFANTE & PEROTTO
(1995) als Interface-Kompartiment beschrieben wurde. Dies ist der Ort des Stoffaustausches:
der Pilz stellt der Pflanze Phosphat und andere Mineralstoffe zur Verfügung, während die
Pflanze ihn mit Kohlenhydraten versorgt. Die genauen Transportmechanismen sind noch
nicht aufgeklärt (SMITH & READ, 1997). Der Pilz baut die erhaltenen Assimilate in Form der
Glucose zunächst in das Disaccharid Trehalose (O-á-D-Glucopyranosyl-(1�1)-á-D-
Glucopyranosid) oder Glycogen um, so dass die intrazelluläre Glucose-Konzentration
abgepuffert wird (SHACHAR-HILL et al., 1995). Aus den Kohlenhydraten werden anschließend
Lipide synthetisiert und diese zum extraradikalen Myzel transportiert (PFEFFER et al., 1999).
In den Wurzeln von den Vertretern der Unterordnung Glomineae können neben den
Arbuskeln auch Vesikel gebildet werden, bläschenförmige Speicherorgane, in denen Lipide
gespeichert werden (AMIJEE, 1989). Die Gigasporineae-Arten bilden im Unterschied zu den
Glomineae extraradikale Hilfszellen aus (MORTON & BENNY, 1990). Die extraradikalen

1 Einleitung 4
Hyphen bilden ein verzweigtes dreidimensionales Netzwerk, mit dem die AMF ein weit grö-
ßeres Volumen schneller und dichter durchdringen können, als dies der Pflanzenwurzel mög-
lich ist. Zusätzlich verfügt der Pilz über ein abweichendes Enzymsystem, so dass er Mineral-
stoffe, insbesondere Phosphat mit Hilfe einer alkalischen Phosphatase (ALLEN et al., 1981),
sehr viel besser aufschließen, und diese der Pflanze zuführen kann (JAKOBSEN et al., 1994).
Nutzen für die Landwirtschaft
Aufgrund der verbesserten Nährstoffversorgung der Pflanze wurde den AMF in den letzten
Jahren immer größere Bedeutung für eine nachhaltige Landwirtschaft beigemessen (BAREA &
JEFFRIES, 1995). Neben den in zahlreichen Gewächshausversuchen demonstrierten positiven
Effekten auf Wachstum, Qualität und Gesundheit der mykorrhizierten Pflanzen durch die ef-
fizientere Nährstoffausnutzung (POWELL & DANIEL, 1978) konnten auch eine erhöhte
Schwermetalltoleranz (BERRECK & HASELWANDTER, 2001), Trockenresistenz (DAVIES et al.,
1993), eine erhöhte Resistenz gegen Phytopathogene (HOOKER et al., 1994) sowie ein Ein-
fluss auf die Zusammensetzung von Pflanzengesellschaften (VAN DER HEIJDEN, 1998a,
1998b) beobachtet werden. BETHLENFALVAY & SCHÜEPP (1994) zeigten positive Auswirkun-
gen auf die Stabilität von landwirtschaftlich genutzten Flächen. Der gezielte Einsatz dieser
Pilze in Landwirtschaft und Gartenbau liegt daher nahe, sei es zur Verringerung der Dünge-
gaben oder zur Erhöhung der Gesundheit der Pflanzen. Weitere Einsatzgebiete sind die Be-
grünung von ariden Gebieten, schwermetallbelasteten Flächen oder Abraumhalden, um nur
einige Beispiele zu nennen (DODD, International Institute of Biotechnology, University of
Kent, mündliche Mitteilung). Mittlerweile wurden einige Firmen gegründet, die Pilzinokulum
kommerziell vermarkten.
Diversität und Funktionalität
Sowohl das Ausmaß als auch die Effektivität der Symbiose sind vom Genotyp der beteiligten
Symbionten abhängig (JAKOBSEN 1995). Während man den AMF häufig ein unspezifisches
Kolonisierungspotential nachgesagt hat (BONFANTE-FASOLO, 1984), konnten signifikante
Unterschiede zwischen einzelnen Arten hinsichtlich ihrer Funktionalität aufzeigt werden
(STREITWOLF-ENGEL et al., 1997). Auf der anderen Seite scheinen die Pflanzen eine gewisse
Auswahl treffen zu können. Während die überwiegende Mehrzahl der Gräser und Kräuter und
auch einige Bäume eine AM ausbilden, wurde bei den Vertretern der Brassicaceae, Chenopo-
diaceae und Cyperaceae bisher keine AM nachgewiesen (WEISSENHORN & FELDMANN, 1999).

1 Einleitung 5
Untersuchung der AMF im Feld
Gewächshausversuche liefern einen wichtigen Beitrag zur Erforschung der unterschiedlichen
AMF-Arten oder -Stämme. Häufig ist jedoch ihre Anwendbarkeit auf Freilandsysteme frag-
lich, da sie niemals die natürlichen Umstände einer komplexen Pflanzen- (und Pilz-) gemein-
schaft erklären können. Auf der anderen Seite ist es natürlich schwierig, aus dem Zusammen-
spiel vieler verschiedener Arten und Individuen eine Funktionalität zu erkennen, die noch
dazu von vielen weiteren Faktoren, wie Bodenbeschaffenheit, Klima oder Pathogenvorkom-
men beeinflusst wird. Dennoch ist es wichtig, das Zusammenspiel dieser Ökosysteme zu er-
fassen, um daraus entscheidende Rückschlüsse ziehen zu können. Für eine Bestandsaufnahme
der AMF sind eindeutige Unterscheidungs- und Identifizierungsmerkmale grundlegend.
1.2 Identifizierung der arbuskulären Mykorrhizapilze
Die Bestimmung der Pilzarten anhand ihrer Sporenmorphologie ist bei Freilandproben
überaus schwierig und meist nur auf Gattungsebene festzustellen. Grundsätzlich muss jede
Einzelspore individuell betrachtet werden, besondere Entwicklungsstadien und weitere
wichtige Merkmale der Sporen können fehlen. Zudem sollte zur zweifelsfreien Bestimmung
Vergleichsmaterial zur Verfügung stehen. Eine Identifizierung der AMF in planta ist ebenso
wenig möglich (SCHÜßLER et al., 2001a).
1.3 Molekularbiologische Untersuchungen bei arbuskulären
Mykorrhizapilzen
Um das Problem der AMF-Identifizierung zu lösen, wurden molekularbiologische Methoden
entwickelt. So beschrieben ROSENDAHL & SEN (1992) eine Isozym-Analyse. HAHN et al.
(1993) entwickelten spezifische Antikörper gegen Oberflächenproteine der Sporen. Die von
MULLIS et al. (1986) entwickelte Polymerase-Kettenreaktion (PCR), eine in vitro-Methode,
die die enzymatische Amplifikation von DNA-Sequenzabschnitten ermöglicht, wurde bald
auch von zahlreichen Wissenschaftlern für die Erforschung der AMF genutzt. Ein aus dem
Bakterium Thermus aquaticus stammendes, thermostabiles Enzym, die Taq DNA
Polymerase, bindet dabei monomere Desoxynukleosidtriphosphate (dNTP) komplementär zu
einer einzelsträngigen DNA-Matrix. Als Startpunkt fungieren an den aufgetrennten
Einzelsträngen Oligonukleotidprimer, die in 5‘�3‘-Richtung verlängert werden. Die
Reaktionen werden in einem Thermo-Cycler durchgeführt, wobei die Proben während 30-40

1 Einleitung 6
Zyklen bei jeweils drei verschiedenen Temperaturen inkubiert werden (Saiki, 1990). Die
PCR-Technik schaffte die Grundlage für zahlreiche Analysemethoden, die auch auf die AMF
übertragen wurden. LANFRANCO et al. (1995) publizierten eine RAPD-PCR und LONGATO &
BONFANTE (1997) untersuchten die Mikrosatellitenregionen mithilfe der PCR. Die
überwiegende Mehrheit konzentrierte sich jedoch auf die Entwicklung von Isolat- oder
Gruppen-spezifischen Primern für die PCR, und kombinierten diese Methode zum Teil mit
anderen Techniken wie der Restriktionsfragment-Längenpolymorphismus-(RFLP-)Analyse.
1.4 Zielsetzung
Ziel dieser Arbeit war es, die neu gewonnenen molekularbiologischen Erkenntnisse „im Feld“
anzuwenden. Mithilfe von PCR-Techniken sollten die ansonsten nicht unterscheidbaren AMF
direkt in der Wurzel nachgewiesen und zugeordnet werden. Trotz der Schwierigkeiten bei der
Probenaufarbeitung und bei der Wahl geeigneter DNA-Sequenzen sollte es möglich sein, auf-
bauend auf der Arbeit von BÖHM (2000), mit spezifischen Oligonukleotiden einzelne Arten
und Gruppen dieser Symbionten zu identifizieren. Im Mittelpunkt dieser Studie steht die
Pilzart Acaulospora longula sowie eine Gruppe nahe verwandter Arten um Glomus mosseae.
Neben dem Nachweis dieser Arten wurden auch molekularbiologische und histologische
Quantifizierungsmethoden angewandt. Daneben wurde die Phosphatversorgung der Pflanzen
berücksichtigt.
Die Untersuchungen fanden im Rahmen des Teilprojekts DI 4 (Mykorrhiza) des For-
schungsverbundes Agrarökosysteme München (F.A.M.) statt. Der F.A.M. verfügt mit dem
ehemaligen Klostergut in Scheyern über eine einzigartige Versuchsfläche. Der Betrieb wurde
1990 übernommen und auf zwei unterschiedliche Bewirtschaftungsformen umgestellt: Inte-
grierter Pflanzenbau und ökologischer Landbau. In Abstimmung mit anderen Teilprojekten
wurden eine Wiese und eine Weidefläche für eine intensive Beprobung ausgewählt. Daneben
fanden auch Beprobungen im Ackerland statt. Da es sich bei den arbuskulären Mykorrhiza-
pilzen um obligate Symbionten handelt (AZCON-AGUILAR & BAREA, 1995; DELP et al.,
1998), konnte eine Vermehrung der Referenzstämme nur mithilfe geeigneter Wirtspflanzen
stattfinden.

2 Material und Methoden 7
2 Material und Methoden
2.1 Material
2.1.1 Versuchsflächen
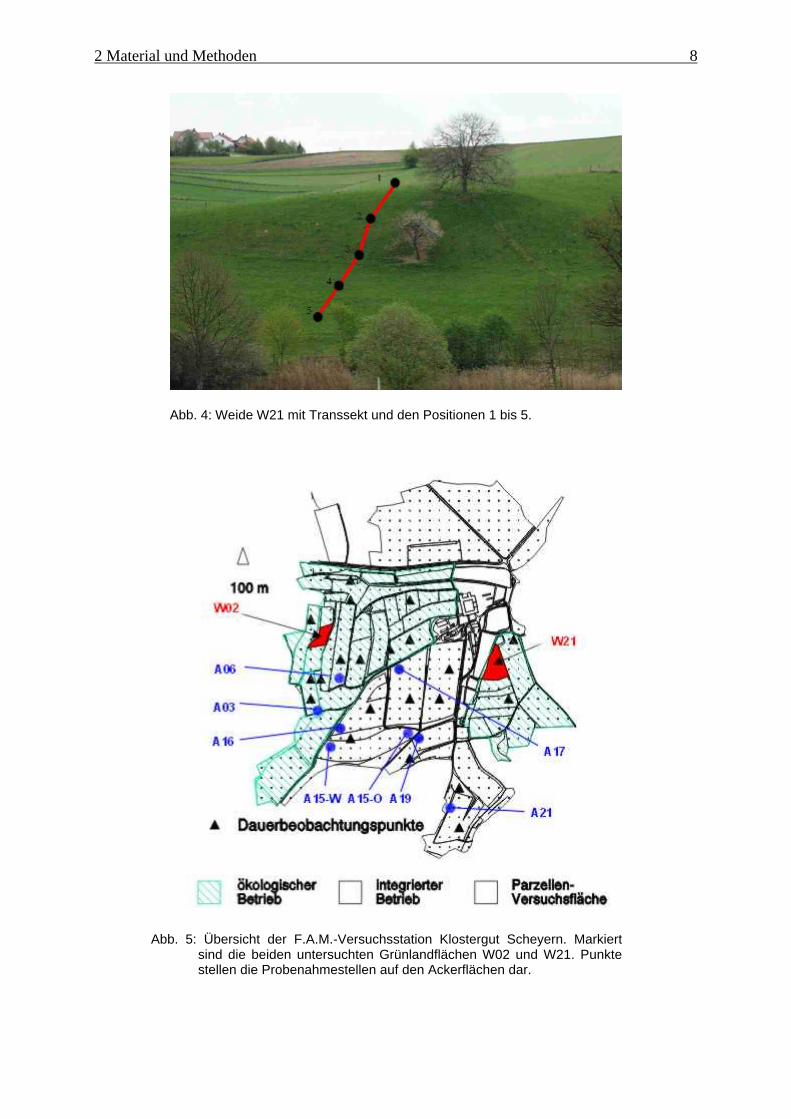
Beim Schlag W02 handelt es sich um eine Wiese mit südöstlicher Hanglage (Abb. 3). Der
Schlag W21 dient als Weide für Rinder und weist eine westliche Hanglage auf (Abb. 4). Auf-
grund der Hanglagen war eine möglichst hohe Phosphatvarianz zu erwarten. Die Pflanzenge-
sellschaften wurden nach SCHUBERT et al. (2001) bestimmt. Es handelt sich bei W02 um eine
Glatthafer-Wiese (Arrhenatherion elatioris BRAUN), bei W21 um eine Weidelgras-Weide
(Cynosurion cristati-Lolietum perennis BRAUN-BLANQUET & DE LEEUW) mit hohem Anteil an
Alopecurus pratensis und Poa trivialis. In Abb. 5 sind die untersuchten Flächen und Bepro-
bungspunkte in einer Übersichtskarte des Versuchsguts in Scheyern dargestellt.
Abb. 3: Wiese W02 mit Transsekt und den Positionen I, II und III.
2 Material und Methoden 8
Abb. 4: Weide W21 mit Transsekt und den Positionen 1 bis 5.
Abb. 5: Übersicht der F.A.M.-Versuchsstation Klostergut Scheyern. Markiertsind die beiden untersuchten Grünlandflächen W02 und W21. Punktestellen die Probenahmestellen auf den Ackerflächen dar.

2 Material und Methoden 9
2.1.2 Probennahme im Freiland
Entlang der Hänge von W02 und W21 wurden Transsekte gelegt und auf diesen 3 bzw. 5 Be-
probungspunkte mit gleichmäßigem Abstand definiert (Tab. 1).
Tab. 1: Höhenlage der Beprobungspunkte.
Position Wiese W02 Weide W21
oben 480 m 468 m
Mitte oben 464 m
Mitte 475 m 459 m
Mitte unten 456 m
unten 470 m 453 m
An den Beprobungspunkten wurden potenzielle Wirtspflanzen mit einem Spaten ausgesto-
chen. Dabei wurde die Pflanze samt einem würfelförmigen Wurzelballen von etwa 15 cm
Kantenlänge entnommen. Die Beprobungen wurden zu verschiedenen Zeitpunkten durchge-
führt. Jede Fläche wurde an fünf Terminen beprobt (Tab. 2).
Tab. 2: Übersicht der Beprobungstermine auf der Wiese W02 und Weide W21.
Termin Beprobung W02 Beprobung W21
05.05.1999 X
10.04.2000 X X
26.06.2000 X
01.08.2000 X
02.10.2000 X X
11.06.2001 X
22.08.2001 X X
Neben diesen beiden Grünlandflächen wurden am 27.08.2001 verschiedene Ackerflä-
chen stichprobenhaft beprobt um einen Vergleich zwischen Grünland und Ackerland treffen
zu können (Tab. 3). Die Lage der einzelnen Beprobungspunkte sind in Abb. 4 dargestellt.

2 Material und Methoden 10
Tab. 3: Schlag, Position und Pflanze der Ackerproben.
Schlag – Position Pflanze
A06 Südost Helianthus annuus
A15 West Zea mays
A15 Ost Zea mays
A16 West Triticum aestivum
A17 Nordwest Solanum tuberosum
A19 Nordwest Solanum tuberosum
A21 West Triticum aestivum
2.1.3 Pflanzen
Die AMF-Referenzkulturen wurden im Rahmen dieser Arbeit mit dem Spitzwegerich (Plan-
tago lanceolata, Blauetikett-Bornträger, Offstein) kultiviert. Dieser diente auch als Referenz
bei molekularbiologischen Untersuchungen: P. lanceolata lässt sich leicht mit einem Großteil
der Mykorrhizapilze kolonisieren und zeichnet sich durch seinen geringen Lichtbedarf und
seinen niederen Wuchs aus (WALKER und VESTBERG 1994). Außerdem ist er wenig anfällig
gegenüber pathogenen Pilzen. Um Inokulum lagerfähig zu machen, ist es sinnvoll, die AMF
durch „Trockenfallenlassen“ zur Sporulation anzuregen und dann die Mischung aus Substrat,
Sporen, Wurzel und Hyphen trocken und kühl aufzubewahren. Dazu wurde die Wasserzufuhr
eingestellt, das trockene oberirdische Pflanzenmaterial abgeschnitten und das Substrat für
6 bis 8 Wochen luftgetrocknet.
Bei den Probennahmen im Freiland wurde neben Spitzwegerich (Plantago lanceolata
L., Abb. 6) auch Weißklee (Trifolium repens L., Abb. 7) und Wolliges Honiggras (Holcus
lanatus L., Abb. 8) untersucht. Ein wichtiges Kriterium hierbei war das Vorkommen der
Wirtspflanze an allen Beprobungspunkten. Das Wollige Honiggras war auf W02 recht weit
verbreitet und aufgrund seiner Behaarung in jedem Wachstumsstadium leicht zu identifizie-
ren. Auf der Weide war die Verteilung der Grasarten sehr heterogen, so dass hier keine
Monokotyle beprobt wurde. Als stickstofffixierende Pflanze wurde Weißklee ausgewählt, der
ebenfalls weit verbreitet war. Allerdings musste bei der Entnahme darauf geachtet werden, die
Wurzeln zu treffen, da aufgrund von Stolonenbildung weiträumig Ausläufer entwickelt
wurden.

2 Material und Methoden 11
Abb. 6: Plantago lanceolata. Abb. 7: Trifolium repens. Abb. 8: Holcus lanatus.
2.1.4 Arbuskuläre Mykorrhizapilze und sonstige Pilze
Als Referenzmaterial wurden im Gewächshaus Kulturen verschiedener AMF herangezogen.
Für die Inokulation der Pflanzen wurden von Herrn Dr. Böhm in Tab. 4 aufgeführten AMF-
Stämme übernommen. Pilzkulturen mit BEG-Nummern sind in der Banque Européene des
Glomales (Dijon, Frankreich) registriert.
Tab. 4: Stämme und Herkunft der als Referenz verwendeten AMF.
Mykorrhizapilz Herkunft
Acaulospora longula SPAIN & SCHENCK (BEG 8) Walker, England
Entrophospora colombiana SPAIN & SCHENCK (BEG 39) Walker, England
Gigaspora margarita BECKER & HALL (BEG 34) Gianinazzi-Pearson, Frankreich
Gigaspora rosea NICOLSON & SCHENCK (BEG 9) Gianinazzi-Pearson, Frankreich
Glomus intraradices SCHENCK & SMITH (H3) von Alten, Deutschland
Glomus microaggregatum KOSKE, GEMMA & OLEXIA (BEG 56) Dodd, England
Glomus mosseae (NICOL. & GERD.) GERD. & TRAPPE (BEG 68) von Alten, Deutschland
Zur Untersuchung der Spezifität von Primern für die PCR fanden der Zuchtchampignon (Aga-
ricus bisporus (LANGE) IMBACH) und die Bäckerhefe (Saccharomyces cerevisiae MEYEN EX
E.C. HANSEN) aus dem Lebensmittelhandel Verwendung.

2 Material und Methoden 12
2.1.5 Oligonukleotidprimer
Zur Bestimmung der intraradikalen AM standen die unter Tab. 5 angegebenen Primer zur
Verfügung. Sämtliche Oligonukleotidprimer wurden von der Firma MWG-Biotech syntheti-
siert. Die Sonden wurden jeweils am 5'-Ende mit dem Reporterfarbstoff FAM (6-Carboxy-
Fluorescein) kovalent gebunden. Das 3'-Ende wurde mit dem Quencherfarbstoff TAMRA
(6-Carboxyl-Tetramethyl-Rhodamin) verknüpft und zusätzlich phosphoryliert um eine
Elongation durch die Polymerase zu verhindern.
Tab. 5: Übersicht über die verwendeten Primer.
Primer Sequenz Referenz
VANS1 5'-GTCTAGTATAATCGTTATACAGG-3' SIMON et al., 1992
VAACAU 5'-TGATTCACCAATGGGAAACCCC-3' SIMON et al., 1993a
VAGIGA 5'-TCACCAAGGGAAACCCGAAGG-3' SIMON et al., 1993a
VAGLO 5'-CAAGGGAATCGGTTGCCCGAT-3' SIMON et al., 1993a
VALETC 5'-ATCACCAAGGTTTAGTTGGTTGC-3' SIMON et al., 1993a
A2R 5'-AGCGAACAAGTACCGTGAGGG-3' BÖHM, 2000
B2L 5'-TAGACTCCTTGGTCCGTGTTTC-3' BÖHM, 2000
Aclong87For 5'-AATCAACTTGATAGTATGCGGG-3' BÖHM, 2000
Aclong400Rev 5'-TATGATCACTCCAACATCTAGATA-3' BÖHM, 2000
Glmoss85For 5'-TTGAAGTCAGTCATACCAACGG-3' BÖHM, 2000
Glmoss387Rev 5'-ACCGTAATACCCTAACATCATAAGC-3' BÖHM, 2000
Aclong251For 5'-ACCATCAGAAGCCAGGTGACTAG-3' BÖHM, 2000
Aclong338Rev 5'-TGGCTTTTCTTCGGAAAAGTGT-3' BÖHM, 2000
AclongSonde FAM-5'-CAATCCTCAATCCCGGTCACCACATTT-3'-TAMRA BÖHM, 2000
P1 5'-TGCAACGGATACCCTTCAGG-3' BÖHM et al., 1999
P2 5'-AATTCGTTCTAACCTTTTGACCGTAAT-3' BÖHM et al., 1999
F1 FAM-5'-AGCAAGCTTTCAACATCAAGGCAACGTATCA-3'-TAMRA BÖHM et al., 1999
2.1.6 Enzyme
AluI Restriktionsendonuklease MBI Fermentas, ER0011
Chitinase, von Streptomyces griseus, 717 U/g Sigma, C-1525
HinfI Restriktionsendonuklease MBI Fermentas, ER0801
HpaII Restriktionsendonuklease MBI Fermentas, ER0511
Protease (Proteinase K), 18,5 U/mg Sigma, P-0390
RNase A QIAGEN, 19101
RsaI Restriktionsendonuklease MBI Fermentas, ER1121
Taq DNA Polymerase, nativ MBI Fermentas, EP0071

2 Material und Methoden 13
2.1.7 Reagenzien
Agarose NEEO Roth, 2267.2
Ammoniummolybdat-Tetrahydrat Sigma, A-7302
L-Ascorbinsäure, Natriumsalz Sigma, A-7631
Benzylchlorid Fluka, 13270
Borsäure Sigma, B-6768
Bromphenolblau Sigma-Aldrich, B-5525
Calciumacetat-Hydrat Merck, 1.09325
Calciumchlorid Fluka, 21101
Calciumlactat Fluka, 21175
Calciumnitrat Merck, 1.02121
Desoxynukleotide (dNTP Mix), je 10 mM MBI Fermentas, R0192
Dimethylsulfoxid (DMSO) Merck, 1.02952
di-Natriumhydrogenphosphat Merck, 1.59323
1,4-Dithiothreitol (DTT) Sigma-Aldrich, D0632
DNA-Erase ICN, 821805
DNA-Längenmarker 20 bp DNA Ladder Biozym, 850320
DNA-Längenmarker GeneRuler™ 100bp DNA Ladder Plus MBI Fermentas, SM0321
Eisenethylendiamintetraacetat (FeEDTA) Riedel-de Haën, 03650
Essigsäure, 99-100%, reinst Sigma-Aldrich, 27221
Ethanol, absolut Sigma-Aldrich, 32205
Ethylendiamintetraessigsäure (EDTA) Sigma, E-5134
Ethidiumbromid Roth, 2218
Formaldehyd, 37%, ACS, säurefrei Merck, 1.04003
Glycerin, 100% Merck, 1.04092
Glyceringelatine Merck, 1.09242
Hexadecyltrimethylammoniumbromid
(Cetyltrimethylammoniumbromid, CTAB) Sigma-Aldrich, H6269
Iod Merck, 1.59192
Kaliumchlorid Merck, 4936
Kaliumdihydrogencitrat Fluka, 60215
Kaliumdihydrogenphosphat Merck, 1.04873
Kaliumhydroxid Sigma-Aldrich, 60370
Kaliumiodid Merck, 1.05051

2 Material und Methoden 14
Kaliumnitrat Merck, 1.05063
Kupfersulfat-Pentahydrat Merck, 1.59153
Ladungspuffer Loading Dye Solution MBI Fermentas, R0611
Magnesiumsulfat Merck, 1.05886
Mangan(II)chlorid Merck, 1.05927
Methanol, ACS, absolut, acetonfrei Sigma-Aldrich, 65543
Milchsäure Amresco, 0349
Molybdän(VI)oxid Merck, 1.00403
Natriumchlorid Merck, 1.06404
Natriumethylendiamintetraacetat (Na2EDTA) Sigma-Aldrich, 27285
Nucleon PhytoPure-Harz (in Kit enthalten) Amersham, RPN 8510
Nukleosidtriphosphate (dNTP) MBI Fermentas, R0191
Polyvinylalkohol Sigma-Aldrich, 81384
2-Propanol Sigma-Aldrich, 59304
Reagent DX Qiagen, 1011008
Rinderserumalbumin (BSA) MBI Fermentas, B14
Salzsäure, 32% Merck, 1.00319
Sorbinsäure Fluka, 85510
Tris(hydroxymethyl)aminomethan (Tris) J. T. Baker, 1414
Trypanblau Sigma, T-6146
Wasserstoffperoxid, 30% Merck, 1.08597
Zinksulfat Merck, 1.08883
2.1.8 Kits
DNeasy Plant Mini Kit QIAGEN, 69104
PicoGreen® dsDNA Quantitation Kit Molecular Probes, P11496
QIAquick Gel Extraction Kit QIAGEN, 28704
TaqMan® Universal PCR Master Kit ABI, 4304447

2 Material und Methoden 15
2.1.9 Verbrauchsmaterial
Deckgläser 22x22 mm Roth, H874
Deckgläser 24x60 mm Roth, H878
Duran-Flaschen, 100 mL, autoklavierbar Fisher, 60762001
Faltenfilter 150 mm Roth, 4799.1
Filterpipettenspitzen Safeseal-Tips, 10µL, steril Biozym, 692150
Filterpipettenspitzen Safeseal-Tips, 10µL, steril Biozym, 693010
Filterpipettenspitzen Safeseal-Tips, 100µL, steril Biozym, 692066
Filterpipettenspitzen Safeseal-Tips, 1000µL, steril Biozym, 692079
Floranid NK BASF
Gartenvlies Dehner, 241257
Hyperphos LS Pflanzenernährung
Kunststofftöpfe (9 cm) Firma Meyer 750908
Lichtdurchlässige Plastikkulturbeutel Sun Bag, transparent,
44x20 cm, 24 mm, 0,02 µm Filter Sigma, B-7026
Kunstlichtfilme KODAK EPY 64T Dinkel, 3 7260
Küvetten ½-Mikro Heiland, 370 490
Mikropistillen Merck, 2112100
Mikrotiterplatten (F-Form) Greiner, 655201
Objektträger, 76x26 mm Peske, 611-710
PCR-Reaktionsgefäße Micro PCR-Tubes, flat cap, 0,2 mL Hybaid, HB-TC-6202N
PCR-Reaktionsplatte OmniPlate 96 Hybaid, HB-TR3-MT
PCR-Reaktionsplattenabdeckung OmniPlate Lid PCR Hybaid, HB-TR3-MTL
Probenbeutel Rotilabo, 20x30 cm Roth, P283.1
Quarzsand Dorsilit, 0,6-1,2 mm, Nr. 7 BayWa, 430882000/1
Radigen Terraflor
Reaktionsgefäße 1,5 mL, PP, Safe-Lock Peske, 120-086
Seramis® Masterfoods
Stericlin-Papierautoklavierbeutel Medipha, 42651210
Sterilisationsklebeband Heiland, 326 202
TaqMan® 96 well reaction plate ABI, N801-0560
TaqMan® optical caps ABI, N801-0935
Wolframcarbid-Kugeln, 3 mm QIAGEN, 69997

2 Material und Methoden 16
2.1.10 Geräte
Analysesiebe 38, 160, 300, 500 µm Haan
Autoklav, S-ECZ Keller Apparatebau
Autoklav, 3870ELV Tuttenauer
Electrophoresis Power Supply ECPS 3000/150 Pharmacia
Elektrophoresekammern GNA 100 und GNA 200 Pharmacia
Fluoreszenzphotometer Reader SPECTRAFluor Plus Tecan
Gefriertrocknungsanlage Hetosicc Pfeiffer
Inkubationsschüttler New Brunswick Scientific
Kamera Camag Reprostar Polaroid
Kamera Nikon F-801s Nikon
Mikroskop Zeiss Axioplan Zeiss
Mikroprozessor pH-Meter pH 535, MultiCal WTW
PCR-Cycler Primer Express Hybaid
PCR-Cycler PCR Sprint Hybaid
Photometer Ultrospec 4000 Pharmacia
Pipette Eppendorf Reference, 0,5-10 µL Eppendorf
Pipette Eppendorf Reference, 10-100 µL Eppendorf
Pipette Eppendorf Reference, 100-1000 µL Eppendorf
Quarzglas-Präzisionsküvette SUPRASIL®, 5 mm Schichtdicke Hellma
Schüttler, Vortex VF 2 Janke und Kunkel
Schwingmühle MM 2 Retsch
Schwingmühlen-Adapter für 5 Reaktionsgefäße Retsch
Stereomikroskop Zeiss
Sterilbank Clean Air CA/RE 4 Haan
Vakuumexsikator Fisher, 60462038
Wasserbad Thermostat 2761 Eppendorf
Zentrifuge Biofuge Heraeus
Zentrifuge 5403 Eppendorf

2 Material und Methoden 17
2.1.11 Puffer und Färbelösungen
• Ammoniummolybdatlösung
Ammoniumheptamolybdat (NH4)6Mo7O24 · 4 H2O 2,50 g
Aqua dest. 40 mL
Bei ca. 50 °C lösen; nach dem Erkalten:
Aqua dest. ad 50 mL
Die Lösung ist mehrere Wochen haltbar.
• CAL-Vorratslösung (5x)
Calciumlactat 77 g
Calciumacetat 39,5 g
in 600 mL heißem Wasser gelöst; nach Abkühlen:
Essigsäure (ñ=1,06 g/mL) 89,5 mL
Aqua dest. ad 1000 mL
• Chitinase-Lösung (10 U/mL)
Kaliumdihydrogenphosphat 272 mg
Calciumchlorid 2,2 mg
Chitinase (717 U/g) 14 mg
Aqua dest. ad 1000 µL
mit KOH auf pH 6,0 einstellen.
• CTAB-Puffer (2x)
Tris-HCl 1,2 g
Natriumchlorid 8,2 g
EDTA 744 mg
Hexadecyltrimethylammoniumbromid (CTAB) 2 g
DTT 200 mg
Aqua dest. ad 100 mL
mit NaOH auf pH 8,0 einstellen.
• 0,5 M EDTA (pH 8,0)
EDTA 186,1 g
Aqua dest. ad 1000 mL
mit NaOH auf pH 8,0 einstellen.

2 Material und Methoden 18
• Extraktionspuffer (nach BAHNWEG et al., 1998)
CTAB-Puffer (2x) 100 mL
PVP 2 g
• FAA (Formaldehyd-Aceto-Alkohol) (KORMANIK & MCGRAW, 1984)
Formaldehyd (37%) 900 mL
Eisessig 50 mL
Ethanol (50%) 50 mL
• Hoagland-Nährlösung für die Pflanzenanzucht
(modifiziert nach SYLVIA & HUBELL, 1986)
Ca(NO3)2 · 4 H2O 246 mg
KNO3 152 mg
MgSO4 · 7 H2O 147 mg
FeEDTA 11 mg
NaCl 3 mg
H3BO3 0,86 mg
MnCl2 · 2 H2O 0,54 mg
ZnSO4 · 7 H2O 0,07 mg
CuSO4 · 5 H2O 0,02 mg
H2MoO4 · H2O 0,006 mg
Aqua dest. ad 1000 mL
mit H2SO4 auf pH 6,5 einstellen.
• Melzer's Reagenz für die Sporendiagnostik (KREISEL & SCHAUER, 1987)
Iod 1,5 g
Kaliumiodid 5 g
Aqua dest. ad 100 mL
• PCR Puffer (10x) von MBI Fermentas
100 mM Tris-HCl (pH 8,8)
500 mM KCl
0,8% Nonidet P40

2 Material und Methoden 19
• Phosphat-Standardlösung
Kaliumdihydrogenphosphat 383 mg
Aqua dest. ad 1000 mL
Ein Milliliter entspricht 0,2 mg P4O10.
• Proteinase K-Stammlösung (10 U/mL)
Proteinase K (18,5 U/mg) 0,54 mg
Aqua dest. 1000 µL
• PVLG-Einbettungsmedium für Sporen (MORTON et al., 1993)
Milchsäure (90%) 10 mL
Glycerin 1 mL
Polyvinylalkohol 1,66 g
Aqua dest. 10 mL
KOH pH 7,5
Aqua dest., Milchsäure und Glycerin in einer dunklen Flasche mischen,
Polyvinylalkohol zugeben und im Wasserbad bei 70 - 80 °C lösen.
• Reduktionslösung für die Phosphatbestimmung
Ascorbinsäure 125 mg
Salzsäure (c = 10 mol/L) 5 mL
Aqua dest. ad 10 mL
Die Lösung ist täglich frisch herzustellen.
• Resuspensionspuffer für die CTAB-Chloroform-Extraktion
Ammoniumacetat 77 mg
EDTA 9,3 mg
Aqua dest. ad 100 mL
• TBE-Puffer (5x) für die Gelelektrophorese (SAMBROOK et al., 1989)
Tris 54 g
Borsäure 27,5 g
0,5 M EDTA (pH 8,0) 20 mL
Aqua dest. ad 1000 mL

2 Material und Methoden 20
• TE-Puffer (20x)
10 mM Tris-HCl
1 mM EDTA
pH 7,5
• TE-Elutionspuffer (1x)
Tris 10,8 g
0,5 M EDTA (pH 8,0) 2 mL
Aqua dest. ad 1000 mL
• Trypanblau-Färbelösung (0,05 %, w/v)
(modifiziert nach KORMANIK & MCGRAW, 1984)
Trypanblau 0,5 g
Milchsäure 875 mL
Glycerin 63 mL
Aqua dest. ad 1000 mL
• Waschpuffer für die CTAB-Chloroform-Extraktion
Ammoniumacetat 77 mg
EtOH 76 mL
Aqua dest. ad 100 mL
2 Material und Methoden 21
2.2 Methoden
2.2.1 Aufarbeitung der Proben
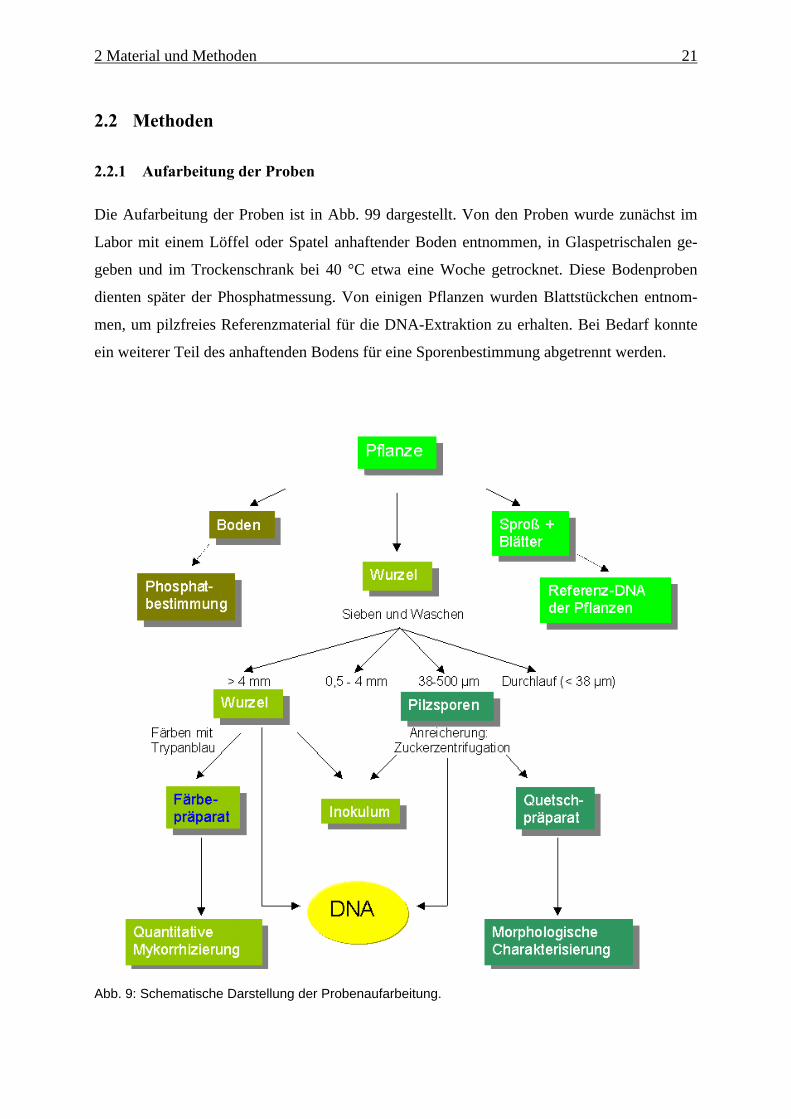
Die Aufarbeitung der Proben ist in Abb. 99 dargestellt. Von den Proben wurde zunächst im
Labor mit einem Löffel oder Spatel anhaftender Boden entnommen, in Glaspetrischalen ge-
geben und im Trockenschrank bei 40 °C etwa eine Woche getrocknet. Diese Bodenproben
dienten später der Phosphatmessung. Von einigen Pflanzen wurden Blattstückchen entnom-
men, um pilzfreies Referenzmaterial für die DNA-Extraktion zu erhalten. Bei Bedarf konnte
ein weiterer Teil des anhaftenden Bodens für eine Sporenbestimmung abgetrennt werden.
Abb. 9: Schematische Darstellung der Probenaufarbeitung.

2 Material und Methoden 22
Der Wurzelballen wurde in Bechergläsern mit Leitungswasser versetzt und die ineinan-
der verwachsenen Wurzeln der Versuchspflanze vorsichtig von anderen Wurzeln und anhaf-
tender Erde getrennt. Anschließend wurde mehrmals mit Leitungswasser gewaschen, bis
keine Trübung des Wassers mehr eintrat. Zuletzt wurden die Wurzeln in einem Becherglas
mit Aqua dest. im Ultraschallbad zweimal gereinigt, um eventuell noch anhaftende Humin-
stoffe zu entfernen, die bei der PCR stören würden. Die Wurzeln wurden anschließend mit
einem Zellstofftuch trockengetupft. Von den gewaschenen Wurzeln wurden mehrere kleine,
zufällig ausgewählte, ca. 1 cm lange Stücke abgeschnitten, mit flüssigem Stickstoff übergos-
sen und anschließend gefriergetrocknet. Aus ihnen wurde später DNA isoliert. Wie sich im
Lauf dieser Arbeit zeigte, musste die Aufarbeitung rasch erfolgen, da die DNA bereits nach
30 min durch Oxidationsprozesse degradiert sein konnte. Die restlichen Wurzeln wurden mit
Trypanblau gefärbt, um die Pilzstrukturen unter dem Mikroskop quantitativ zu bestimmen.
2.2.2 Produktion von Referenzkulturen
Zur methodischen Entwicklung und als Referenzen wurden verschiedene Stämme von AMF
kultiviert. Die Inokula wurden von Herrn Dr. Böhm zur Verfügung gestellt. Als Substrat
diente ein Gemisch aus Quarzsand (Körnung 0,6-1,2 mm, Dorsilit Nr. 7) und Seramis® (1:1,
v/v). Durch die Verwendung von Quarzsand lassen sich bei der Probenentnahme Wurzeln
und Pilzsporen leicht vom Substrat trennen; Seramis® erhöht als Tongranulat mit hohem
Feinporenanteil die Feuchtigkeit im Substrat und erleichtert dadurch die Kultur. 6,4 kg Sand
und 2 kg Seramis® wurden mit etwas Aqua dest. versetzt und gemeinsam bei 121 °C und 1,3
bar für 20 min autoklaviert, um Kontaminationen zu vermeiden. Je Liter Substrat wurden 760
mg Floranid NK, 110 mg Radigen und 1,5 g Gesteinsphosphat (Hyperphos) als Grunddün-
gung zugefügt und gut durchmischt. Kunststofftöpfe (MR9) wurden mit Gartenvlies ausgelegt
und zu etwa ¾ mit sterilem Substrat gefüllt. Darauf wurde 30 mL Inokulum (Substrat mit
AMF-Sporen und mykorrhizierten Wurzeln) gegeben und abschließend mit ca. 1 cm Quarz-
sand überschichtet. Samen von P. lanceolata wurden zur Oberflächensterilisation 20 min in
5%-ige Wasserstoffperoxidlösung gelegt und anschließend mehrmals mit Aqua dest. gewa-
schen. Zur Keimung wurden die Samen auf feuchte Filterpapierstücke in Petrischalen gelegt.
Nach etwa einer Woche wurden jeweils drei Keimlinge in die einzelnen Töpfe pikiert. Die
Töpfe wurden in Untersetzer gestellt und anschließend in Sunbags überführt und im Ge-
wächshaus bei ca. 22 °C kultiviert (WALKER & VESTBERG, 1994). Bei den Sunbags handelt es
sich um Kunststoffbeutel, die Kreuzkontaminationen verhindern, mittels eines integrierten

2 Material und Methoden 23
Mikrofilters Gasaustausch ermöglichen und die Luftfeuchtigkeit erhöhen. Die Pflanzen wur-
den zwei bis drei mal wöchentlich mit Aqua dest. gegossen. Zusätzlich wurde alle vier Wo-
chen mit einer modifizierten Hoagland-Nährlösung gedüngt, die bis auf Phosphat alle Makro-
und Mikronährstoffe enthält (SYLVIA & HUBBELL, 1986). Nach drei bis sechs Monaten
konnten die mykorrhizierten Wurzeln geerntet werden.
Zur Kontrolle der Spezifität von Primern wurden zur Gewinnung von DNA aus Blättern
und nicht-mykorrhiziertem Wurzelmaterial von Pflanzen deren oberflächen-sterilisierte Sa-
men in Petrischalen ausgesät, mit Aqua dest. feucht gehalten und nach etwa 5 Tagen aus den
entsprechenden Organen DNA extrahiert.
2.2.3 Optimierung der DNA-Extraktion
Um die DNA-Extraktion zu optimieren, wurden verschiedene Methoden und Protokolle gete-
stet. Mit den folgenden Schritten sollte die genomische DNA in möglichst reiner Form und in
hohen Ausbeuten aus der Zelle isoliert werden. Entscheidend war dabei die Abtrennung von
Polysacchariden, Polyphenolen und (bei Wurzeln und Bodenproben) Huminsäuren, die alle-
samt die folgende PCR stören konnten, ohne dabei die DNA-Ausbeute zu beeinträchtigen
(DEMEKE et al., 1992). Neben den gängigen Methoden zur DNA-Aufreinigung bei Tieren und
Mikroorganismen standen einige Modifikationen für Pflanzenmaterial sowie ein Kit der
Firma QIAGEN zur Isolierung von Pflanzen-DNA (aus Blättern, jedoch nicht aus Wurzeln)
zur Verfügung. BAHNWEG et al. (1998) und DOYLE & DOYLE (1987) beschäftigten sich spe-
ziell mit der Problematik der DNA-Extraktion aus Pflanzen.
Die vorgereinigten Wurzelstücke wurden in ein 2 mL-Reaktionsgefäß überführt, 1 mL
Aqua dest. hinzugefügt und etwa 1 min in ein Ultraschallbad gestellt, um möglicherweise
noch an der Oberfläche anhaftende Huminsäuren oder andere Stoffe zu entfernen. Anschlie-
ßend wurden die Wurzeln in ein weiteres Reaktionsgefäß überführt und der Vorgang so lange
wiederholt, bis keine Trübung mehr auftrat. Daraufhin wurden die Wurzeln auf Zellstoff ge-
trocknet und abgewogen. Es wurden jeweils zwischen 10 und 100 mg Frischgewicht bzw.
1 bis 20 mg Trockengewicht verwendet. Als Referenzen wurden auch Blattstücke von
Pflanzen oder Myzel von Pilzkulturen verwendet.

2 Material und Methoden 24
Der erste variable Schritt war der Aufschluss der Zellen, hierfür wurden verschiedene
Methoden angewandt:
• sofortiges Einfrieren in flüssigem Stickstoff und anschließende Gefriertrocknung –
Zellaufschluss als Lyophilisat
• sofortiges Einfrieren in flüssigem Stickstoff – Zellaufschluss in gefrorenem Zustand
• sofortiger Zellaufschluss in möglichst frischem Zustand
Das eigentliche Zerkleinern zu einem feinen Pulver erfolgte in 1,5 mL-Reaktionsgefäßen:
• Zermörsern der auf wenige Millimeter vorgeschnittenen Wurzelstückchen mittels
eines Mikropistills
• Zermahlen unter Zugabe von 2-4 Wolframcarbid-Kügelchen und Verwendung von
zwei Adaptoren für jeweils 5 Gefäße in einer Kugelschwingmühle MM2 für ca. 5 min
• Zelllyse mit einem Ultraschallstab
• Zellaufschluß mittels Ultra-Turrax
Um Verunreinigungen zu vermeiden, wurden alle Arbeitsflächen und Geräte mit 70%
Ethanol desinfiziert und autoklavierte Gefäße verwendet. Die Mikropistille wurden zunächst
2-3 h in 1 N Salzsäure auf 90 °C erhitzt und mit Aqua dest. gewaschen. Schließlich wurden
sie wie Scheren und Pinzetten mit DNA-Erase behandelt, welches sowohl DNA- als auch
RNA-Moleküle hydrolysiert. Anschließend wurde gründlich mit Aqua dest. gewaschen und
autoklaviert. Die Wolframcarbid-Kugeln besitzen aufgrund ihrer extremen Härte eine hohe
Durchschlagskraft. Außerdem konnten sie aufgrund ihrer magnetischen Eigenschaften leicht
aus den Ansätzen entfernt werden. Ihre Reinigung erfolgte durch fünfminütige Inkubation in
1 N HCl auf einem Magnetrührer und anschließendem Waschen in einem Sieb mit Aqua dest.
Zur Vermeidung von Aerosolen wurden ausschließlich Filterpipettenspitzen verwendet.
Extraktion mit dem DNeasy Plant Mini Kit von QIAGEN
Dieses kommerzielle System wurde entwickelt, um DNA aus oberirdischen Pflanzenteilen
oder Gewebekulturen zu isolieren. Garantiert wird dabei die sichere Abtrennung der meisten
PCR-Inhibitoren und die Gewinnung von qualitativ hochwertiger DNA. Das vom Hersteller
QIAGEN beigefügte Protokoll wurde mit geringen Modifikationen übernommen:
Zunächst wurden dem zerkleinerten Probenmaterial 400 µL Puffer AP1 und 4 µL
RNase beigefügt, mit einem Vortexer gemischt und 10 min im Wasserbad bei 65 °C inkubiert.
Bei der Verwendung von frischem Probenmaterial konnten alternativ Puffer AP1 und RNase

2 Material und Methoden 25
zusammen mit 1 µL Reagent DX (verhindert starkes Schäumen) bereits vor dem Mahlen mit
der Kugelschwingmühle zugefügt werden (Frau Dr. Boronowsky, Fa. QIAGEN, mündliche
Mitteilung). Der Puffer AP1 enthält EDTA, SDS, NaCl und Polyvinylpyrrolidon (PVP) in
wässriger Lösung. EDTA bindet Magnesiumionen und andere zweiwertige Kationen, die für
einige DNasen als Co-Faktoren fungieren (KOKILERA, 1995), und verhindert dadurch einen
enzymatischen DNA-Abbau. SDS denaturiert Proteine und trennt somit DNA-Protein-Kom-
plexe. NaCl hält SDS in Lösung. PVP begünstigt die Adsorption von Polyphenolen, die sonst
von Phenoloxidasen zu komplexbildenden Verbindungen oxidiert würden (SAUNDERS &
PARKES, 1999). Durch die RNase wird RNA verdaut, die in der PCR stören könnte. Um den
Zellaufschluss und damit die gewonnene DNA-Menge noch weiter zu steigern, wurden alter-
nativ zusätzlich jeweils 10 µL Chitinase- und Proteinase K-Lösung zugesetzt und vor der In-
kubation bei 65 °C zusätzlich 5 min bei RT bzw. 37 °C inkubiert.
Nach der Inkubation wurden 130 µl Puffer AP2 hinzugegeben und 5 min auf Eis inku-
biert. Der Puffer AP2 enthält Kaliumacetat und Essigsäure in wässriger Lösung. Durch die
Erhöhung der Ionenkonzentration präzipitieren die Proteine.
Anschließend wurde die extrahierte DNA auf eine QIAshredder spin column aufgetra-
gen und durch Zentrifugieren (2 min bei 15.000 rpm) filtriert. Größere Zellbestandteile blie-
ben dabei in dem Filter zurück, Präzipitate sammelten sich als Rückstand am Grund des Re-
aktionsgefäßes. Der Überstand (ca. 450 µL) wurde in ein neues 1,5 ml Reaktionsgefäß über-
führt.
Dem Überstand wurden 225 µL Puffer AP3 (enthält Guanidinium-Hydrochlorid) und
450 µL reiner Ethanol zugesetzt. Durch Zugabe eines monovalenten Salzes und Alkohol fällt
DNA spontan bei Raumtemperatur aus. Bei sehr kleinen DNA-Mengen unter 250 ng/µL (z.B.
Extraktion aus Sporen) wurde der Ansatz 30 min bis 24 h auf -20 °C in den Gefrierschrank
gestellt und zu Beginn der Extraktion 100 ng/µL tRNA (Poly-A) als Carrier hinzugegeben
(MÜLHARDT, 2000). Die Suspension wurde auf eine DNeasy mini spin column aufgetragen,
wobei die DNA an die Membran binden konnte und die restliche Flüssigkeit durch Zentrifu-
gieren (1 min bei 8.000 rpm) entfernt wurden.
Durch zweimaliges Waschen mit dem Puffer AW konnten störende Stoffe wie Polyphe-
nole oder noch vorhandene Salze des Puffer AP3 abgetrennt werden. Hierfür wurden 500 µL
Puffer AW auf die Säule aufgetragen und zunächst 1 min bei 8.000 rpm zentrifugiert. Beim
zweiten Waschschritt wurde die Membran durch 2 min Zentrifugieren bei 15.000 rpm ge-
trocknet. Der Puffer AW enthält NaCl, Tris-HCl und Ethanol.

2 Material und Methoden 26
Abschließend wurde die gereinigte DNA mit 100 µL eines auf 65 °C vorgewärmten
Elutionspuffer AE (enthält Tris und EDTA) eluiert. Zur Kontrolle der verwendeten Lösungen
auf DNA-Verunreinigungen wurde bei jeder Extraktion eine Extraktionskontrolle mit Aqua
dest. verwendet und später in einer PCR mit Universalprimern überprüft. Der DNA-Extrakt
wurde bei –20 °C gelagert.
CTAB-Chloroform-Extraktion (modifiziert nach DOYLE & DOYLE 1987)
100 mg pulverisiertes Material wurde 30 min bei 60 °C mit 800 µL 2x CTAB Puffer inkubiert
und anschließend mit Chloroform-Isoamylalkohol (24:1, v/v) extrahiert. Nach 10-minütiger
Zentrifugation bei 10.000 rpm wurde die wässrige Phase in ein neues Gefäß überführt, 500 µL
eisgekühlter 2-Propanol zugesetzt und vorsichtig gemischt. Die präzipitierte DNA wurde ab-
zentrifugiert und zweimal 10 min mit 1 mL Waschpuffer gereinigt. Anschließend wurde die
DNA abzentrifugiert, der Rückstand luftgetrocknet und danach in 100 µL
Resuspensionspuffer gelöst. Der gelösten DNA wurde 1 µL RNase A zugesetzt und 30 min
bei 37 °C inkubiert. Anschließend wurde die DNA mit 100 µL Aqua dest. verdünnt, 100 µL
7,5 M-Ammoniumacetat zugegeben und mit 750 µL kaltem Ethanol gefällt. Nach 5-minütiger
Inkubation auf Eis wurde bei 13.000 rpm zentrifugiert, das DNA-Präzipitat luftgetrocknet und
schließlich mit 100 µL TE-Puffer resuspendiert.
Protokoll der DNA-Extraktion (modifiziert nach BAHNWEG 1998)
100 mg frisches Material wurde in flüssigem Stickstoff tiefgefroren und gemahlen. Hierzu
wurden 500 µL Methanol (50% v/v), 25 µL Calciumchlorid (20%, w/v) und 1 % (w/v) DTT
auf Eis pipettiert. Der Ansatz wurde 10 min bei 4 °C inkubiert und anschließend 10 min bei
4 °C und 13.000 rpm zentrifugiert. Nach der Zentrifugation wurde der Überstand verworfen
und die Methanol-Calciumchlorid-Extraktion wiederholt. Danach wurde ein drittes Mal
extrahiert, diesmal mit 50% (v/v) Methanol und 1% DTT (ohne CaCl2). Zum Rückstand
wurden auf Eis (unter dem Abzug) 200 µL Benzylchlorid und 250 µL Extraktionspuffer
zugegeben. Der Ansatz wurde 10 min bei 4 °C geschüttelt. Anschließend wurden 150 µL
gekühltes Chloroform und 50 µL Nucleon PhytoPure-Harz hinzugefügt, weitere 5 min bei
4 °C geschüttelt und schließlich 10 min bei 4 °C und 2.000 rpm zentrifugiert. Der Überstand
wurde in ein neues Gefäß überführt und hierzu 150 µL Chloroform und 50 µL Nucleon
PhytoPure-Harz pipettiert. Nach weiteren 5 min Inkubation bei 4 °C wurde abermals wie
zuvor zentrifugiert. Von dem Überstand wurden vorsichtig 200 µL in ein neues Gefäß auf Eis
überführt und 100 µL 2-Propanol addiert, durch vorsichtiges Drehen gemischt und weitere
2 Material und Methoden 27
10 min auf Eis inkubiert. Die DNA wurde 10 min bei 4 °C und 13.000 rpm abzentrifugiert
und der Überstand verworfen. Anschließend folgten verschiedene Waschschritte bei RT mit
1) 500 µL 70%-igem Ethanol/0,1 M Natriumacetat, 2) 500 µL 80%-igem Ethanol, 3) 500 µL
100%-igem Ethanol und 4) 500 µL Chloroform. Abschließend wurde die DNA 30 min bei RT
getrocknet und in 100 µL TE-Puffer gelöst.
2.2.4 Bestimmung der DNA-Konzentration
Um das Ergebnis der Extraktionen quantitativ zu überprüfen, wurde ein Kit der Firma Mole-
cular Probes verwendet, mit dem es möglich war, doppelsträngige DNA sehr exakt zu quanti-
fizieren. PicoGreen® ist ein Reagenz, das hochselektiv an doppelsträngige DNA bindet. Wird
es in dieser Bindung mit Licht der Wellenlänge 502 nm angeregt, so fluoresziert es mit einer
Emissionswellenlänge von 523 nm. Da Fluoreszenz im Gegensatz zur Absorption sehr viel
genauer gemessen werden kann, entfallen bei dieser Methode störende Faktoren wie Konta-
minationen mit RNA oder Proteinen (MÜLHARDT, 2000).
Zunächst wurde die PicoGreen®-Stammlösung 1:200 mit 1x TE-Puffer verdünnt. Für
die Erstellung einer Kalibrationskurve wurde von der mitgelieferten dsDNA mit TE-Puffer
eine Stammlösung hergestellt (2 µg/mL). Mit dieser Stammlösung wurde eine DNA-Verdün-
nungsreihe hergestellt (Tab. 6). Die Lösungen wurden in Kavitäten einer 96er-Platte pipet-
tiert, 5 min bei Raumtemperatur im Dunkeln inkubiert und anschließend mit einem Fluores-
zenzphotometer gemessen. Die DNA-Standards wurden jeweils dreifach gemessen, die DNA-
Proben doppelt bestimmt.
Die zu messenden DNA-Proben wurden mit 1x TE-Puffer auf 100 µL verdünnt. Hierzu
wurde jeweils 100 µL PicoGreen®-Lösung gegeben und weiter wie bei den Standards verfah-
ren.
Tab. 6: DNA-Verdünnungsreihe für den PicoGreen®-Assay.
Stammlösung 1x TE-Puffer PicoGreen®-Lösung DNA-Konzentration
100 µL 0 µL 100 µL 1000 ng/mL
10 µL 90 µL 100 µL 100 ng/mL
1 µL 99 µL 100 µL 10 ng/mL
0,1 µL 99,9 µL 100 µL 1 ng/mL
0 µL 100 µL 100 µL 0 ng/mL

2 Material und Methoden 28
2.2.5 Oligonukleotidprimer
Für eine PCR werden Oligonukleotide benötigt, die den „Rahmen“ des zu amplifizierenden
Genabschnitts bilden. Jedes dieser Oligonukleotide oder „Primer“ besteht aus etwa 20 Nu-
kleotiden und bildet den Startpunkt für das Enzym DNA Polymerase. Da die DNA doppel-
strängig ist, verwendet man einen Forward-Primer, dessen Basensequenz der des codogenen
Stranges entspricht, und einen Reverse-Primer, dessen Basensequenz komplementär zum co-
dogenen Strang ist. Je nach Sequenz kann man zwischen universellen Primern unterscheiden,
die für ein bestimmtes Zielgen bei möglichst vielen Organismen passen, und spezifischen
Primern, die diese Zielregionen nur bei einer bestimmten Gruppe von Organismen erkennt.
Da in jedem Genom konservierte und variable Bereiche vorkommen, kann man die unter-
schiedlichen Primer entsprechend modellieren. Grundsätzlich gilt: je länger der Primer, desto
spezifischer kann er sein (Tab. 7). Eine besondere Art von Primern sind die DNA-Sonden, die
bis zu 30 Basen lang sein können und nicht als Startpunkt in einer PCR zum Einsatz kommen,
sondern hochspezifisch eine ganz bestimmte Zielregion erkennen sollen. Diese Sonden sind
meist mit einem Fluoreszenzfarbstoff markiert und können in einer quantitativen PCR einge-
setzt werden.
Tab. 7: Theoretische Bindungsspezifität von Oligonukleotiden.
Oligonukleotid TypischeBasenzahl
Theoretische Spezifität
Primer 20 1 : 420 = 1 : 1,1 ·1012
Sonde 25 1 : 425 = 1 : 1,1 ·1015
Universelle Primer dienten in dieser Arbeit zum einen zur Kontrolle der Amplifizier-
barkeit der extrahierten DNA (Vermeidung von falsch-negativen Ergebnissen) und zum ande-
ren in sogenannten nested PCRs (s. Kap. 2.2.6.2). Optimal für diese Arbeit wäre ein spezifi-
sches Primerpaar gewesen, das alle Arten der Ordnung Glomales und ausschließlich diese
amplifiziert. Ein solches Primerpaar war allerdings nicht gegeben. Des weiteren wurden
Primerkombinationen verwendet, die nur bestimmte Arten oder Gruppen der Glomales erken-
nen konnten. Weiterhin fanden spezifische DNA-Sonden Verwendung, die hochspezifisch
eingesetzt werden konnten, um die Verteilung einzelner Arten zu untersuchen.
Da von einigen Autoren bei der Entwicklung ihrer Primer nur sehr wenig Sequenzdaten zur
Verfügung standen, wurde die Qualität einzelner oder kombinierter Primer hinsichtlich ihrer
Spezifität mit den seither neu ermittelten Sequenzen untersucht:

2 Material und Methoden 29
• Mit dem Basic Local Alignment Search Tool (BLAST) des National Center for Bio-
technology Information (NCBI, http://www.ncbi.nlm.nih.gov/) wurden einzelne
Primersequenzen eingegeben und von dem Programm mit sämtlichen verfügbaren Se-
quenzdaten der Datenbank verglichen.
• Mit den Sequenzen eines Primerpaares sowie der zu erwartenden Fragmentlänge (bis
1000 bp) konnte mit dem Programm FastM der Fa. Genomatix (www.genomatix.de)
eine „virtuelle PCR“ durchgeführt werden. Auch hier verglich der Computer die vor-
handenen Sequenzen der Datenbank mit den eingegebenen Daten und führte eine PCR
in silico durch. Die Ergebnisse konnten später auch dazu genutzt werden, um anhand
der Fragmentlänge die PCR-Produkte zu überprüfen.
Zielregion der verwendeten Primer war die ribosomale DNA (rDNA). Diese kodiert für
die RNA der Ribosomen (rRNA): die große Unterheinheit (large subunit, LSU, bei Pilzen
meist als 25S rRNA angegeben), die kleine Untereinheit (small subunit, SSU oder 18S rRNA)
sowie die 5.8S rRNA. Diese drei kodierenden Genabschnitte sind durch hochvariable Introns,
die internal transcribed spacer (ITS1 und ITS2) voneinander getrennt. In Abb. 10 sind die
konservierten Bereiche der ribosomalen DNA mit den variablen Spacer-Regionen (ITS1 und
ITS2) dargestellt. Eine Übersicht der Bindungsorte einzelner Primer findet sich in Abb. 11.
Die rDNA wird sehr häufig für phylogenetische Studien verwendet (WHITE et al., 1990), da
sie einige Vorteile besitzt. An den Ribosomen findet die Proteinbiosynthese statt, deshalb
muss die rDNA in allen Lebewesen vorhanden sein. Außerdem handelt es sich bei der rDNA
um ein high copy gene; die einzelnen identischen rDNA-Einheiten sind zu Hunderten tandem-
förmig aneinandergereiht (ARNHEIM, 1983). Dies ist vor allem dann von Vorteil, wenn nur
sehr wenig DNA des Zielorganismus für die Analyse vorhanden ist.
Abb. 10: Anordnung der rDNA-Einheiten. IGS: Intergenic spacer, ITS1: Internal transcribed spacer 1;ITS2: Internal transcribed spacer 2; SSU: small subunit; LSU: large subunit; 5.8S: 5.8S-Untereinheit.

2 Material und Methoden 30
Abb. 11: Hybridisierungspositionen der Oligonukleotidprimer. IGS: Intergenic spacer; ITS1: Internaltranscribed spacer 1; ITS2: Internal transcribed Spacer 2; LSU: large subunit; SSU: smallsubunit.
2.2.6 Polymerase-Kettenreaktion
Zur Untersuchung der AMF wurden in einer PCR DNA-Fragmente der rDNA amplifiziert.
Eine PCR besteht aus folgenden Einzelschritten:
Denaturierung (95 °C)
Die doppelsträngige DNA wird dabei zunächst durch eine Erwärmung auf 95 °C aufge-
trennt.
Annealing (ca. 50 bis 60 °C)
Nach einer Temperaturabsenkung hybridisieren die Primer mit den komplementären
Matrixsträngen. Die Annealing-Temperatur hängt dabei von der Länge und dem GC-
Gehalt der Oligonukleotide ab (je kürzer die Primer und je niedriger ihr GC-Gehalt, de-
sto niedriger ist die optimale Hybridisierungstemperatur). Eine Absenkung der Tempe-
ratur kann zu einer erhöhten Produktausbeute führen, da die Primer leichter binden, al-
lerdings können dabei durch unspezifische Bindungen unerwünschte Nebenprodukte
entstehen.
Elongation (72 °C)
Bei 72 °C synthetisiert die DNA Polymerase den DNA-Doppelstrang. Danach beginnt
der Zyklus von neuem.
2 Material und Methoden 31
Die entstehenden Fragmente werden exponentiell vermehrt: aus einer einzigen Startko-
pie könnten nach 30 Zyklen rein rechnerisch 230, also über eine Milliarde identische Kopien
entstehen. In der Regel ebbt die Reaktion jedoch z.B. wegen Substratmangels ab (MÜLHARDT,
2000).
2.2.6.1 Qualitative PCR-Analyse mit universellen Primern
Optimierung der Reaktionsbedingungen
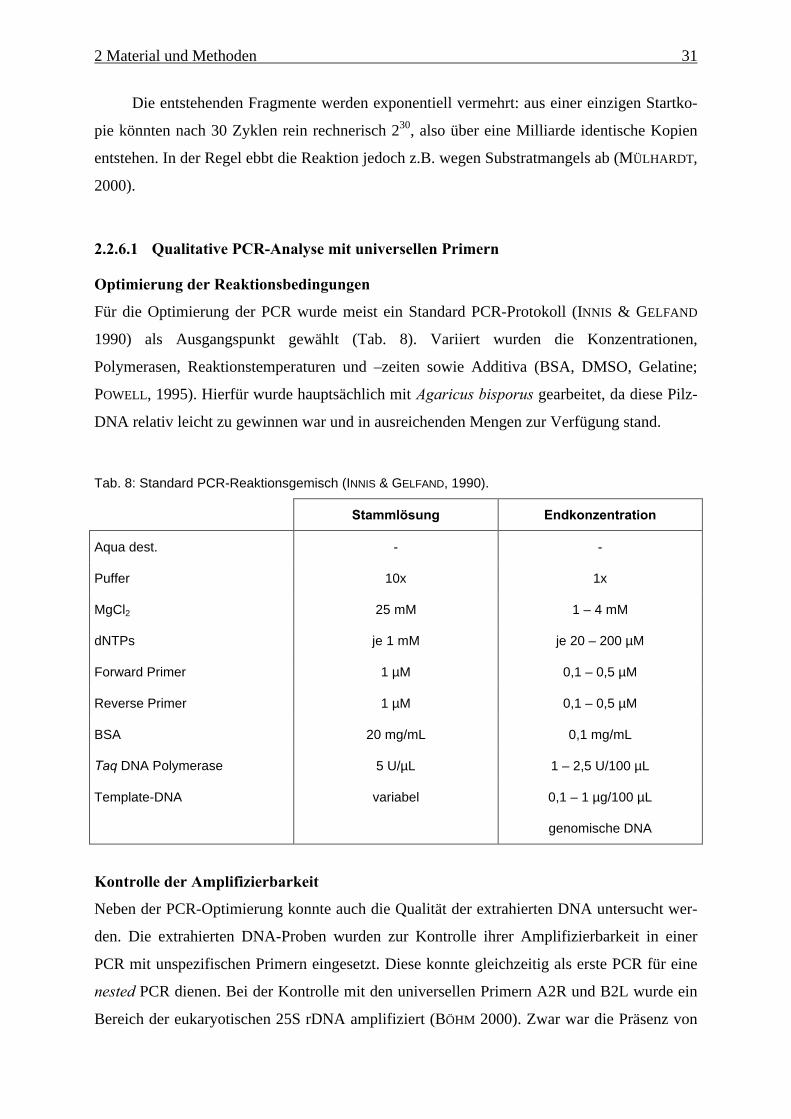
Für die Optimierung der PCR wurde meist ein Standard PCR-Protokoll (INNIS & GELFAND
1990) als Ausgangspunkt gewählt (Tab. 8). Variiert wurden die Konzentrationen,
Polymerasen, Reaktionstemperaturen und –zeiten sowie Additiva (BSA, DMSO, Gelatine;
POWELL, 1995). Hierfür wurde hauptsächlich mit Agaricus bisporus gearbeitet, da diese Pilz-
DNA relativ leicht zu gewinnen war und in ausreichenden Mengen zur Verfügung stand.
Tab. 8: Standard PCR-Reaktionsgemisch (INNIS & GELFAND, 1990).
Stammlösung Endkonzentration
Aqua dest. - -
Puffer 10x 1x
MgCl2 25 mM 1 – 4 mM
dNTPs je 1 mM je 20 – 200 µM
Forward Primer 1 µM 0,1 – 0,5 µM
Reverse Primer 1 µM 0,1 – 0,5 µM
BSA 20 mg/mL 0,1 mg/mL
Taq DNA Polymerase 5 U/µL 1 – 2,5 U/100 µL
Template-DNA variabel 0,1 – 1 µg/100 µL
genomische DNA
Kontrolle der Amplifizierbarkeit
Neben der PCR-Optimierung konnte auch die Qualität der extrahierten DNA untersucht wer-
den. Die extrahierten DNA-Proben wurden zur Kontrolle ihrer Amplifizierbarkeit in einer
PCR mit unspezifischen Primern eingesetzt. Diese konnte gleichzeitig als erste PCR für eine
nested PCR dienen. Bei der Kontrolle mit den universellen Primern A2R und B2L wurde ein
Bereich der eukaryotischen 25S rDNA amplifiziert (BÖHM 2000). Zwar war die Präsenz von
2 Material und Methoden 32
Pilz-DNA in diesem Stadium noch ungewiss, doch musste die sicher vorhandene Pflanzen-
DNA ein positives Ergebnis liefern. War kein Produkt erkennbar, so wurde die extrahierte
DNA als nicht amplifizierbar gewertet. Umgekehrt wurde die Probe bei Erscheinen einer
Pflanzen-Bande in weiteren PCRs eingesetzt.
Die Reaktionen wurden mit jeweils 1 µL Matrizen-DNA (ca. 10 ng) in einem Reak-
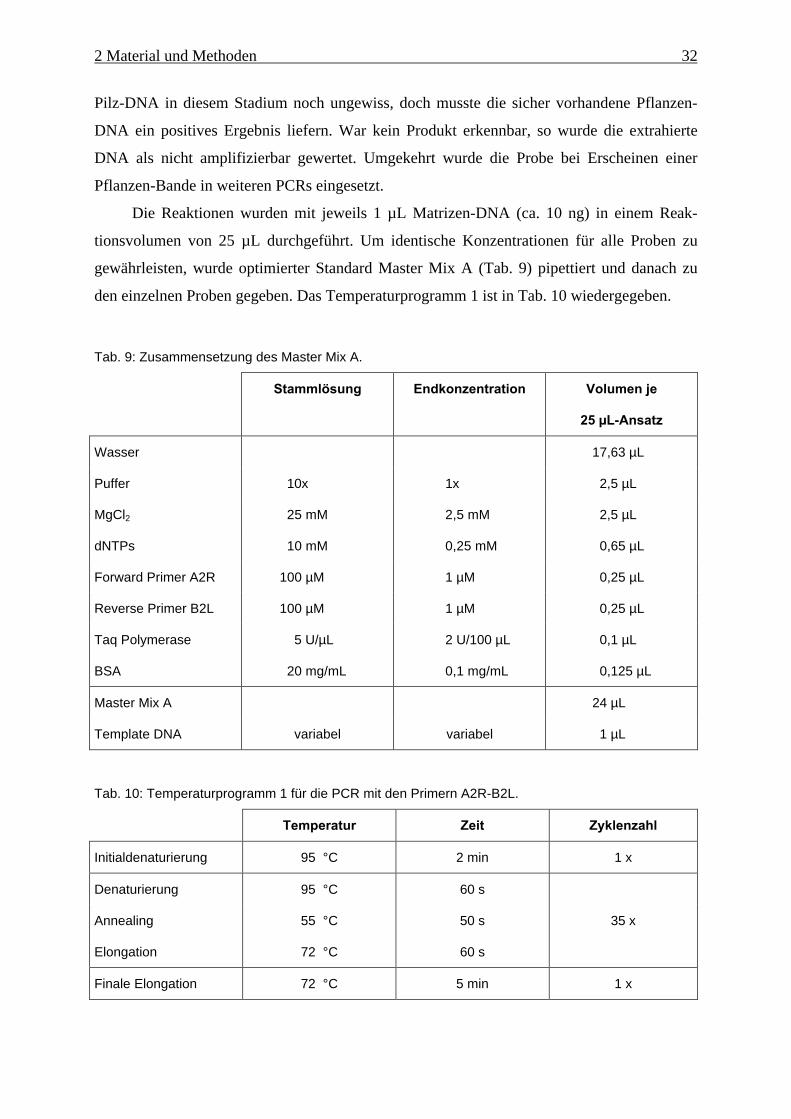
tionsvolumen von 25 µL durchgeführt. Um identische Konzentrationen für alle Proben zu
gewährleisten, wurde optimierter Standard Master Mix A (Tab. 9) pipettiert und danach zu
den einzelnen Proben gegeben. Das Temperaturprogramm 1 ist in Tab. 10 wiedergegeben.
Tab. 9: Zusammensetzung des Master Mix A.
Stammlösung Endkonzentration Volumen je
25 µL-Ansatz
Wasser 17,63 µL
Puffer 10x 1x 2,5 µL
MgCl2 25 mM 2,5 mM 2,5 µL
dNTPs 10 mM 0,25 mM 0,65 µL
Forward Primer A2R 100 µM 1 µM 0,25 µL
Reverse Primer B2L 100 µM 1 µM 0,25 µL
Taq Polymerase 5 U/µL 2 U/100 µL 0,1 µL
BSA 20 mg/mL 0,1 mg/mL 0,125 µL
Master Mix A 24 µL
Template DNA variabel variabel 1 µL
Tab. 10: Temperaturprogramm 1 für die PCR mit den Primern A2R-B2L.
Temperatur Zeit Zyklenzahl
Initialdenaturierung 95 °C 2 min 1 x
Denaturierung 95 °C 60 s
Annealing 55 °C 50 s
Elongation 72 °C 60 s
35 x
Finale Elongation 72 °C 5 min 1 x
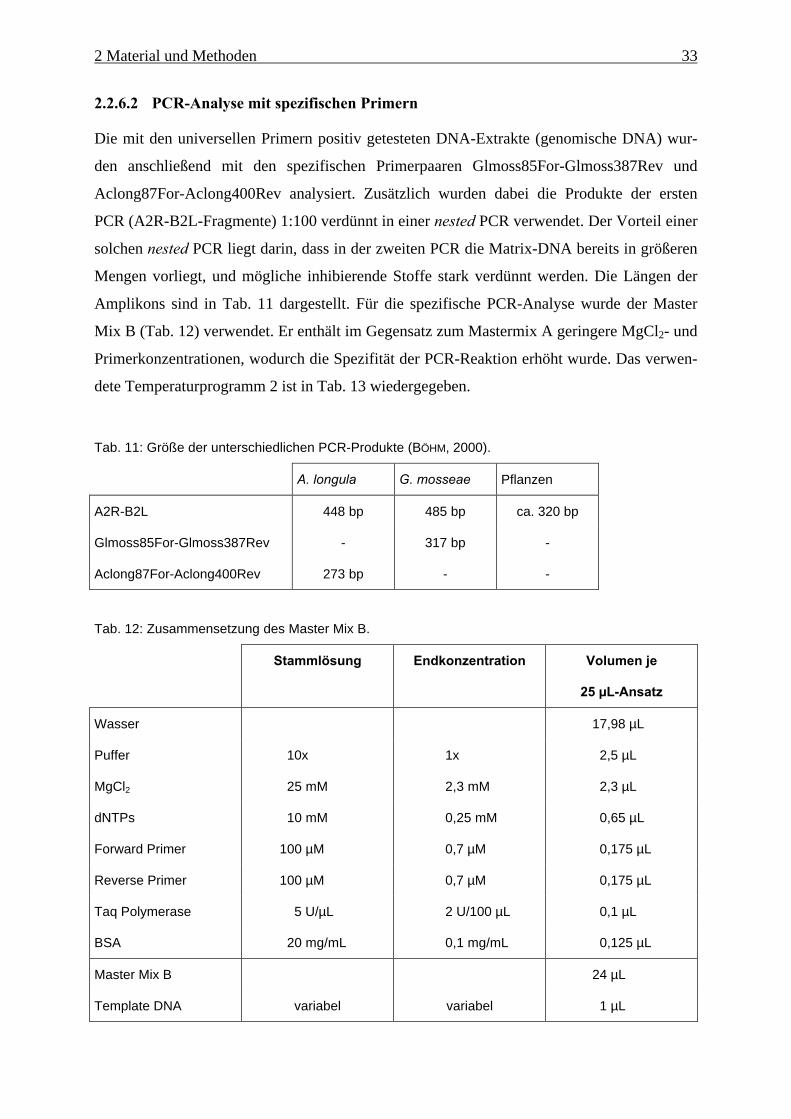
2 Material und Methoden 33
2.2.6.2 PCR-Analyse mit spezifischen Primern
Die mit den universellen Primern positiv getesteten DNA-Extrakte (genomische DNA) wur-
den anschließend mit den spezifischen Primerpaaren Glmoss85For-Glmoss387Rev und
Aclong87For-Aclong400Rev analysiert. Zusätzlich wurden dabei die Produkte der ersten
PCR (A2R-B2L-Fragmente) 1:100 verdünnt in einer nested PCR verwendet. Der Vorteil einer
solchen nested PCR liegt darin, dass in der zweiten PCR die Matrix-DNA bereits in größeren
Mengen vorliegt, und mögliche inhibierende Stoffe stark verdünnt werden. Die Längen der
Amplikons sind in Tab. 11 dargestellt. Für die spezifische PCR-Analyse wurde der Master
Mix B (Tab. 12) verwendet. Er enthält im Gegensatz zum Mastermix A geringere MgCl2- und
Primerkonzentrationen, wodurch die Spezifität der PCR-Reaktion erhöht wurde. Das verwen-
dete Temperaturprogramm 2 ist in Tab. 13 wiedergegeben.
Tab. 11: Größe der unterschiedlichen PCR-Produkte (BÖHM, 2000).
A. longula G. mosseae Pflanzen
A2R-B2L 448 bp 485 bp ca. 320 bp
Glmoss85For-Glmoss387Rev - 317 bp -
Aclong87For-Aclong400Rev 273 bp - -
Tab. 12: Zusammensetzung des Master Mix B.
Stammlösung Endkonzentration Volumen je
25 µL-Ansatz
Wasser 17,98 µL
Puffer 10x 1x 2,5 µL
MgCl2 25 mM 2,3 mM 2,3 µL
dNTPs 10 mM 0,25 mM 0,65 µL
Forward Primer 100 µM 0,7 µM 0,175 µL
Reverse Primer 100 µM 0,7 µM 0,175 µL
Taq Polymerase 5 U/µL 2 U/100 µL 0,1 µL
BSA 20 mg/mL 0,1 mg/mL 0,125 µL
Master Mix B 24 µL
Template DNA variabel variabel 1 µL
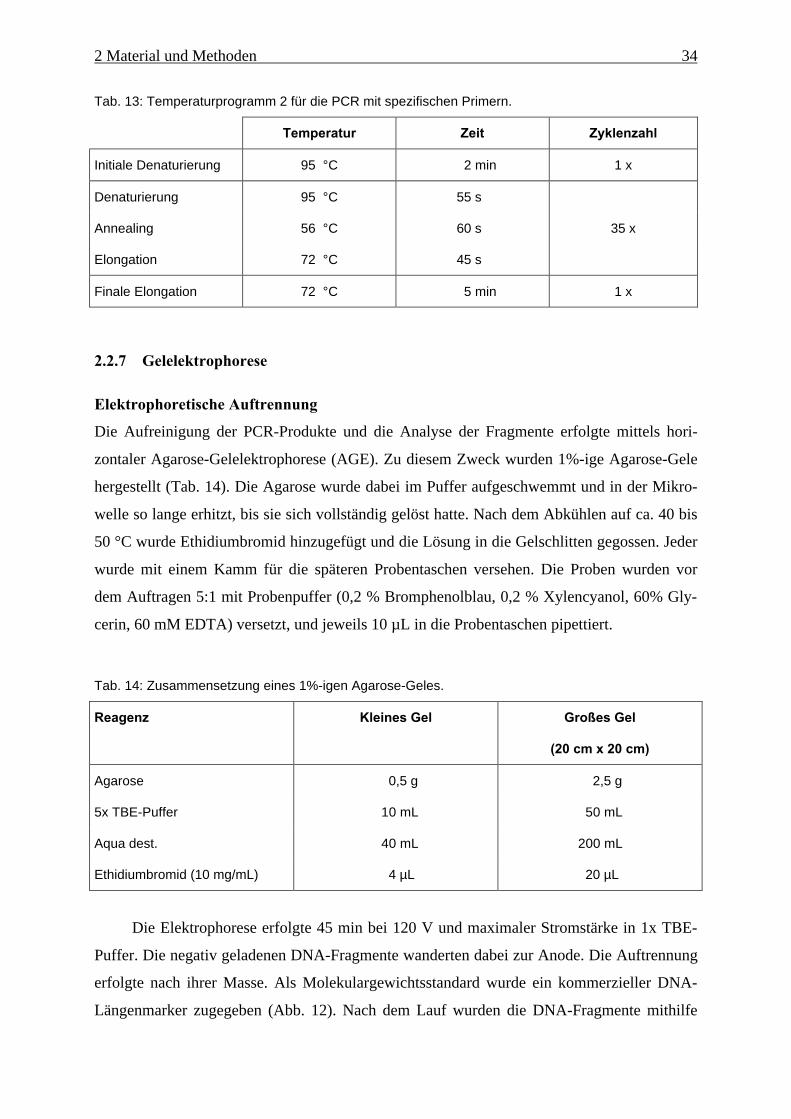
2 Material und Methoden 34
Tab. 13: Temperaturprogramm 2 für die PCR mit spezifischen Primern.
Temperatur Zeit Zyklenzahl
Initiale Denaturierung 95 °C 2 min 1 x
Denaturierung 95 °C 55 s
Annealing 56 °C 60 s
Elongation 72 °C 45 s
35 x
Finale Elongation 72 °C 5 min 1 x
2.2.7 Gelelektrophorese
Elektrophoretische Auftrennung
Die Aufreinigung der PCR-Produkte und die Analyse der Fragmente erfolgte mittels hori-
zontaler Agarose-Gelelektrophorese (AGE). Zu diesem Zweck wurden 1%-ige Agarose-Gele
hergestellt (Tab. 14). Die Agarose wurde dabei im Puffer aufgeschwemmt und in der Mikro-
welle so lange erhitzt, bis sie sich vollständig gelöst hatte. Nach dem Abkühlen auf ca. 40 bis
50 °C wurde Ethidiumbromid hinzugefügt und die Lösung in die Gelschlitten gegossen. Jeder
wurde mit einem Kamm für die späteren Probentaschen versehen. Die Proben wurden vor
dem Auftragen 5:1 mit Probenpuffer (0,2 % Bromphenolblau, 0,2 % Xylencyanol, 60% Gly-
cerin, 60 mM EDTA) versetzt, und jeweils 10 µL in die Probentaschen pipettiert.
Tab. 14: Zusammensetzung eines 1%-igen Agarose-Geles.
Reagenz Kleines Gel Großes Gel
(20 cm x 20 cm)
Agarose 0,5 g 2,5 g
5x TBE-Puffer 10 mL 50 mL
Aqua dest. 40 mL 200 mL
Ethidiumbromid (10 mg/mL) 4 µL 20 µL
Die Elektrophorese erfolgte 45 min bei 120 V und maximaler Stromstärke in 1x TBE-
Puffer. Die negativ geladenen DNA-Fragmente wanderten dabei zur Anode. Die Auftrennung
erfolgte nach ihrer Masse. Als Molekulargewichtsstandard wurde ein kommerzieller DNA-
Längenmarker zugegeben (Abb. 12). Nach dem Lauf wurden die DNA-Fragmente mithilfe

2 Material und Methoden 35
des Ethidiumkations detektiert, welches zwischen zwei benachbarte Basenpaare interkaliert
(KNIPPERS, 2001) und in diesem Zustand unter Anregung mit UV-Licht (366 nm) fluoresziert.
Die sichtbaren Banden wurden mit einer Polaroid-Kamera dokumentiert.
Abb. 12: DNA-Längenmarker GeneRuler 100 bp DNA Ladder Plus (MBI Fermentas).
Isolierung aufgereinigter DNA-Fragmente
Die aufgereinigten DNA-Banden wurden mit einem Skalpell aus dem Gel ausgeschnitten, und
mithilfe des QIAquick Gel Extraction Kits isoliert. Hierzu wurden die abgewogenen Gel-
stückchen mit dem dreifachen Volumen Puffer QG versetzt und die Gelstruktur durch 10 mi-
nütige Inkubation bei 50 °C aufgelöst. Anschließend wurden die herausgelösten DNA-Frag-
mente durch Zugabe von 2-Propanol gefällt, in eine QIAquick spin column überführt und bei
13.000 rpm 1 min zentrifugiert. Die DNA band dabei an die Säule und wurde mit 750 µL
Puffer PE gewaschen, der ebenfalls abzentrifugiert wurde. Anschließend wurden die DNA-
Fragmente mit 50 µL Puffer EB (10 mM Tris·Cl, pH 8,5) eluiert.
2.2.8 Restriktionsfragment-Längenpolymorphismus-(RFLP-)Analyse
Da das Primerpaar Glmoss85For-Glomoss387Rev nicht G. mosseae-spezifisch ist, sondern
theoretisch auch nahe verwandte Arten amplifizieren könnte, wurden die mit diesem Primer-
paar erhaltenen PCR-Produkte mit verschiedenen Restriktionsenzymen (Endonukleasen) un-
tersucht (Tab. 15). Dazu wurden zu jeweils 10 µL PCR-Produkt 2 µL Puffer Y+/Tango
(wurde mit dem Enzym geliefert), 1µL Enzym (1:10 verdünnt) und 7 µL Aqua dest. gegeben,

2 Material und Methoden 36
1 h inkubiert und anschließend über eine AGE analysiert (s. Kap. 2.2.7). Je nach Vorhanden-
sein einer bestimmten Schnittstelle ergeben sich daraus spezifische Bandenmuster.
Tab. 15: Endonukleasen und ihre Erkennungssequenz.Pfeile geben die Schnittstelle an.
Restriktionsenzym Erkennungsstelle
AluI AG�CT
Hin6I G�CGC
HpaII C�CGG
RsaI GT�AC
2.2.9 Sequenzierung und Alignment
Die Sequenzanalysen der DNA-Fragmente wurden von der Firma MWG Biotech (Ebersberg)
durchgeführt. Die erhaltenen Sequenzdaten wurden mit verschiedenen bekannten Sequenzen
einer Datenbank (NCBI) mithilfe des Programms BioEdit verglichen und ihre phylogeneti-
sche Verwandtschaft analysiert.
2.2.10 Quantitativer Nachweis
Die real-time PCR (HEID et al., 1996) dient der quantitativen Analyse bestimmter Genab-
schnitte, die auf dem Prinzip der 5’-Nuklease-Aktivität (HOLLAND et al., 1991) einiger DNA
Polymerasen beruht. Bei der TaqMan® PCR wird neben den beiden Primern eine spezifische
Sonde eingesetzt, bestehend aus einem Oligonukleotid (gewöhnlich aus 15 bis 30 Basen),
einem fluorogenen Reporter-Farbstoff (Fluorescein-Derivat) am 5’-Ende und einem Quen-
cher-Farbstoff (Rhodamin-Derivat) am 3’-Ende, das zusätzlich durch einen Phosphatrest
blockiert ist (LEE et al., 1993). Während der Reaktion wird bei jedem Zyklus die Fluoreszenz
durch Anregungen mit Licht der Wellenlänge von 488 nm gemessen. Bei der intakten Sonde
wird die Fluoreszenz des Reporter-Farbstoffes durch einen Energie-Transfer zum räumlich
nahegelegenen Quencher-Farbstoff verhindert (Abb. 13). Ist ein amplifizierbares Target vor-
handen, so hybridisiert die Sonde mit dem komplementären Matrizenstrang und wird während
der Extensionsphase durch die 5’�3’-Ex onukleaseaktivität der DNA Polymerase abgebaut.
Da durch diese Hydrolyse Reporter und Quencher räumlich voneinander getrennt werden,
steigt die Fluoreszenz entsprechend an und wird von einer CCD-Kamera in ein digitales
Signal umgesetzt.
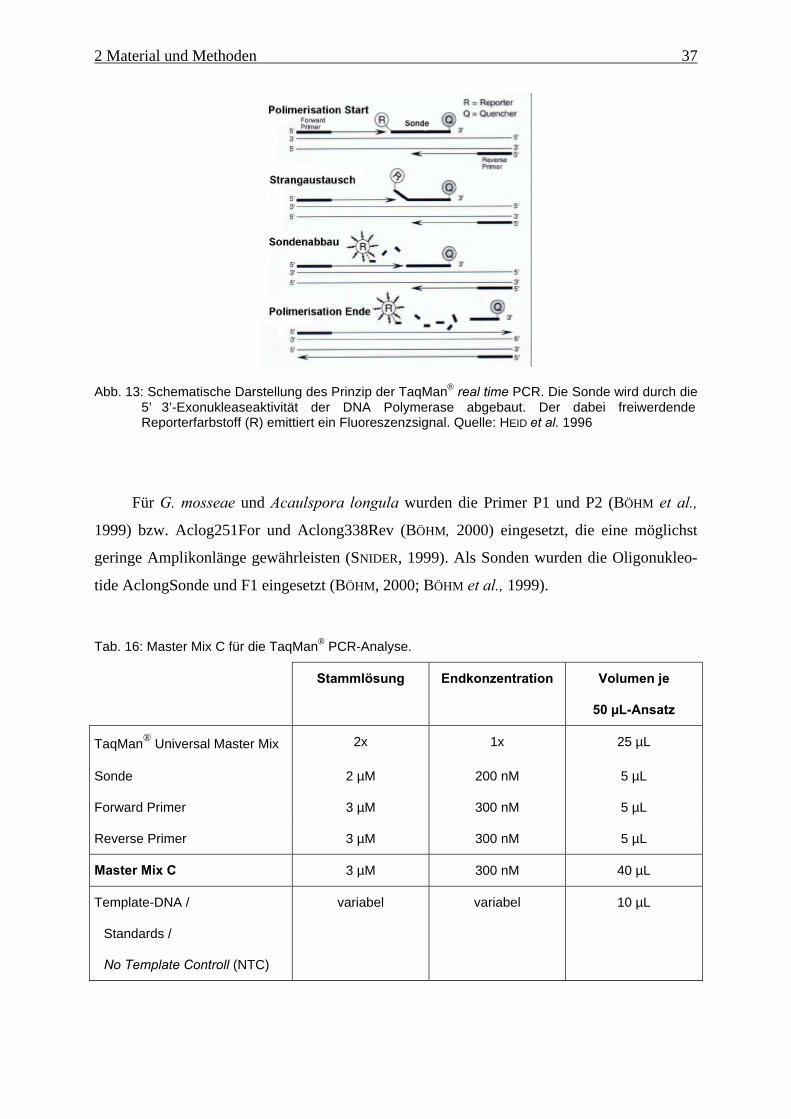
2 Material und Methoden 37
Abb. 13: Schematische Darstellung des Prinzip der TaqMan® real time PCR. Die Sonde wird durch die5’�3’-Exonukleaseaktivität der DNA Polymerase abgebaut. Der dabei freiwerdendeReporterfarbstoff (R) emittiert ein Fluoreszenzsignal. Quelle: HEID et al. 1996
Für G. mosseae und Acaulspora longula wurden die Primer P1 und P2 (BÖHM et al.,
1999) bzw. Aclog251For und Aclong338Rev (BÖHM, 2000) eingesetzt, die eine möglichst
geringe Amplikonlänge gewährleisten (SNIDER, 1999). Als Sonden wurden die Oligonukleo-
tide AclongSonde und F1 eingesetzt (BÖHM, 2000; BÖHM et al., 1999).
Tab. 16: Master Mix C für die TaqMan® PCR-Analyse.
Stammlösung Endkonzentration Volumen je
50 µL-Ansatz
TaqMan® Universal Master Mix 2x 1x 25 µL
Sonde 2 µM 200 nM 5 µL
Forward Primer 3 µM 300 nM 5 µL
Reverse Primer 3 µM 300 nM 5 µL
Master Mix C 3 µM 300 nM 40 µL
Template-DNA /
Standards /
No Template Controll (NTC)
variabel variabel 10 µL
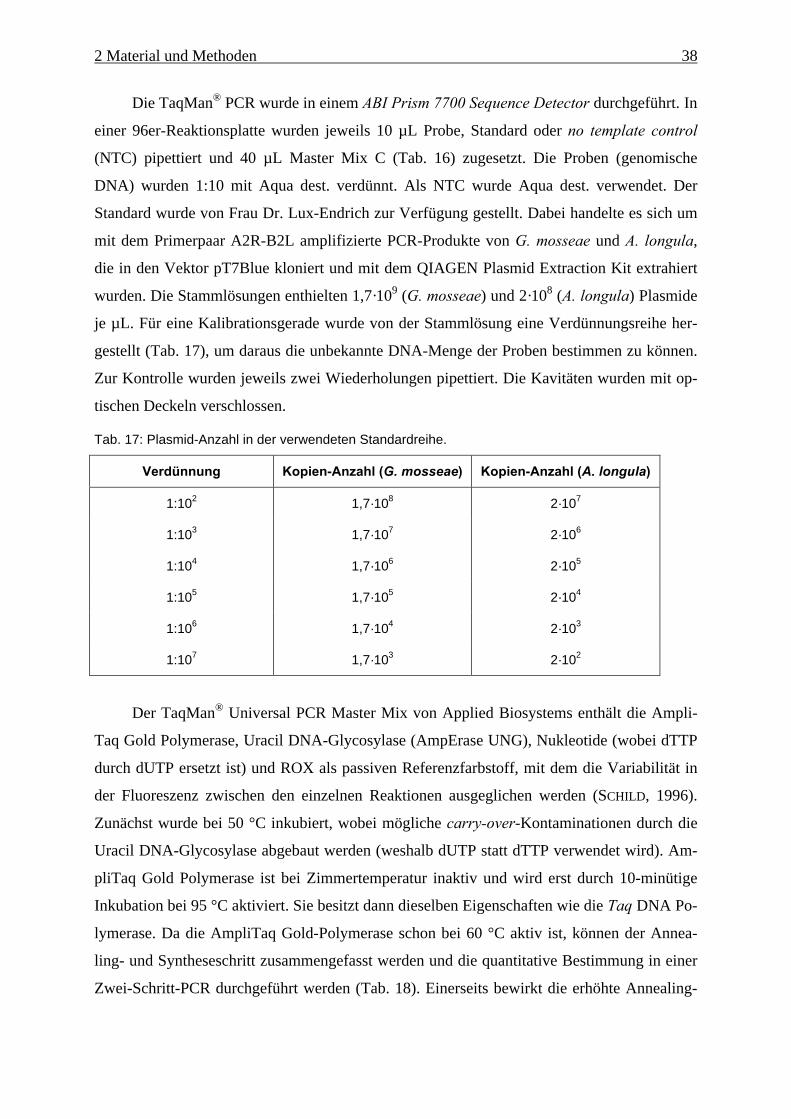
2 Material und Methoden 38
Die TaqMan® PCR wurde in einem ABI Prism 7700 Sequence Detector durchgeführt. In
einer 96er-Reaktionsplatte wurden jeweils 10 µL Probe, Standard oder no template control
(NTC) pipettiert und 40 µL Master Mix C (Tab. 16) zugesetzt. Die Proben (genomische
DNA) wurden 1:10 mit Aqua dest. verdünnt. Als NTC wurde Aqua dest. verwendet. Der
Standard wurde von Frau Dr. Lux-Endrich zur Verfügung gestellt. Dabei handelte es sich um
mit dem Primerpaar A2R-B2L amplifizierte PCR-Produkte von G. mosseae und A. longula,
die in den Vektor pT7Blue kloniert und mit dem QIAGEN Plasmid Extraction Kit extrahiert
wurden. Die Stammlösungen enthielten 1,7·109 (G. mosseae) und 2·108 (A. longula) Plasmide
je µL. Für eine Kalibrationsgerade wurde von der Stammlösung eine Verdünnungsreihe her-
gestellt (Tab. 17), um daraus die unbekannte DNA-Menge der Proben bestimmen zu können.
Zur Kontrolle wurden jeweils zwei Wiederholungen pipettiert. Die Kavitäten wurden mit op-
tischen Deckeln verschlossen.
Tab. 17: Plasmid-Anzahl in der verwendeten Standardreihe.
Verdünnung Kopien-Anzahl (G. mosseae) Kopien-Anzahl (A. longula)
1:102 1,7·108 2·107
1:103 1,7·107 2·106
1:104 1,7·106 2·105
1:105 1,7·105 2·104
1:106 1,7·104 2·103
1:107 1,7·103 2·102
Der TaqMan® Universal PCR Master Mix von Applied Biosystems enthält die Ampli-
Taq Gold Polymerase, Uracil DNA-Glycosylase (AmpErase UNG), Nukleotide (wobei dTTP
durch dUTP ersetzt ist) und ROX als passiven Referenzfarbstoff, mit dem die Variabilität in
der Fluoreszenz zwischen den einzelnen Reaktionen ausgeglichen werden (SCHILD, 1996).
Zunächst wurde bei 50 °C inkubiert, wobei mögliche carry-over-Kontaminationen durch die
Uracil DNA-Glycosylase abgebaut werden (weshalb dUTP statt dTTP verwendet wird). Am-
pliTaq Gold Polymerase ist bei Zimmertemperatur inaktiv und wird erst durch 10-minütige
Inkubation bei 95 °C aktiviert. Sie besitzt dann dieselben Eigenschaften wie die Taq DNA Po-
lymerase. Da die AmpliTaq Gold-Polymerase schon bei 60 °C aktiv ist, können der Annea-
ling- und Syntheseschritt zusammengefasst werden und die quantitative Bestimmung in einer
Zwei-Schritt-PCR durchgeführt werden (Tab. 18). Einerseits bewirkt die erhöhte Annealing-
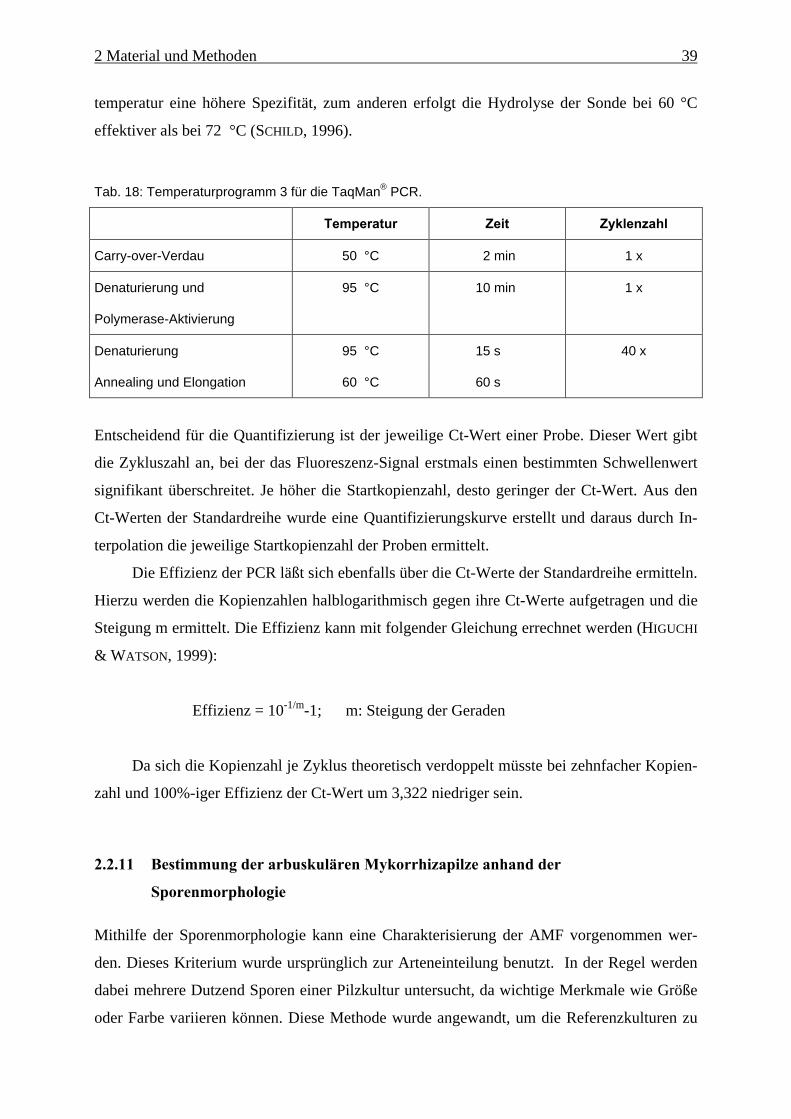
2 Material und Methoden 39
temperatur eine höhere Spezifität, zum anderen erfolgt die Hydrolyse der Sonde bei 60 °C
effektiver als bei 72 °C (SCHILD, 1996).
Tab. 18: Temperaturprogramm 3 für die TaqMan® PCR.
Temperatur Zeit Zyklenzahl
Carry-over-Verdau 50 °C 2 min 1 x
Denaturierung und
Polymerase-Aktivierung
95 °C 10 min 1 x
Denaturierung 95 °C 15 s
Annealing und Elongation 60 °C 60 s
40 x
Entscheidend für die Quantifizierung ist der jeweilige Ct-Wert einer Probe. Dieser Wert gibt
die Zykluszahl an, bei der das Fluoreszenz-Signal erstmals einen bestimmten Schwellenwert
signifikant überschreitet. Je höher die Startkopienzahl, desto geringer der Ct-Wert. Aus den
Ct-Werten der Standardreihe wurde eine Quantifizierungskurve erstellt und daraus durch In-
terpolation die jeweilige Startkopienzahl der Proben ermittelt.
Die Effizienz der PCR läßt sich ebenfalls über die Ct-Werte der Standardreihe ermitteln.
Hierzu werden die Kopienzahlen halblogarithmisch gegen ihre Ct-Werte aufgetragen und die
Steigung m ermittelt. Die Effizienz kann mit folgender Gleichung errechnet werden (HIGUCHI
& WATSON, 1999):
Effizienz = 10-1/m-1; m: Steigung der Geraden
Da sich die Kopienzahl je Zyklus theoretisch verdoppelt müsste bei zehnfacher Kopien-
zahl und 100%-iger Effizienz der Ct-Wert um 3,322 niedriger sein.
2.2.11 Bestimmung der arbuskulären Mykorrhizapilze anhand der
Sporenmorphologie
Mithilfe der Sporenmorphologie kann eine Charakterisierung der AMF vorgenommen wer-
den. Dieses Kriterium wurde ursprünglich zur Arteneinteilung benutzt. In der Regel werden
dabei mehrere Dutzend Sporen einer Pilzkultur untersucht, da wichtige Merkmale wie Größe
oder Farbe variieren können. Diese Methode wurde angewandt, um die Referenzkulturen zu

2 Material und Methoden 40
kontrollieren. Außerdem wurden in einem Vorversuch Bodenproben auf Sporen untersucht,
wobei jede Spore individuell bewertet wurde.
Für die Isolierung der Sporen wurden 2-3 Teelöffel Substrat bzw. Bodenprobe entnom-
men und in einem Becherglas mit Aqua dest. aufgeschwemmt. Nach der Nass-Siebmethode
(GERDEMANN & NICOLSON, 1963) wurden die Proben dann unter einem Wasserstrahl über
Siebe mit unterschiedlichen Porengrößen (5 mm, 500 µm und 38 µm) in einzelne Fraktionen
aufgeteilt. Die Sporen befanden sich zusammen mit Wurzelbruchstücken, Steinchen und an-
derem Material im Sieb mit der geringsten Maschenweite. Um die stark mit Fremdmaterial
durchmischten Sporen der Bodenproben weiter aufzureinigen wurden diese mit Aqua dest.
zentrifugiert (10 min, 2.000 rpm), der Überstand verworfen, der Rückstand mit 45% (w/v)
Saccharoselösung aufgeschlemmt und nochmals 2 min bei 1500 rpm zentrifugiert (DANIELS &
SKIPPER, 1984). Die Sporen blieben dabei aufgrund ihrer Dichte in der Saccharoselösung.
Diese wurde in das 38 µm-Sieb geschüttet und mit Aqua dest. nachgespült. Anschließend
wurden die Sporen mit einem kleinen Volumen Aqua dest. in eine Glas-Petrischale gespült
und unter einem Stereomikroskop aussortiert. Mit einer Pipette wurden die Sporen auf einen
Objektträger überführt und mit einem Deckglas fixiert. Zur Identifizierung wurden sie in Po-
lyvinylalkohol-Lacto-Glycerin (PVLG) und PVLG/Melzer’s Reagenz (1:1, v/v) eingebettet
(KREISEL & SCHAUER, 1987; MORTON, 1995). Durch leichten Druck auf das Deckglas wur-
den die Sporen aufgebrochen, um die Sporenwanddicke und den Sporenwandaufbau im Mi-
kroskop untersuchen zu können. Weiterhin wurde ihre Form, Größe, Farbe und Färbung
durch Melzer’s Reagenz bonitiert und mit Bestimmungshilfen (SCHENCK & PÉREZ, 1990;
INVAM) eine Kontrolle bzw. Artbestimmung durchgeführt.
2.2.12 Färbung der intraradikalen Pilzstrukturen
Um den Umfang der Kolonisierung der Pflanzenwurzeln durch AMF festzustellen, wurden
die intraradikalen Pilzstrukturen in den gereinigten Wurzeln mit Trypanblau angefärbt (modi-
fiziert nach KORMANIK & MCGRAW, 1984). Da die Wurzeln der drei untersuchten Pflanzen
unterschiedlich empfindlich gegenüber Kochen und Bleichen reagierten, musste das Protokoll
je nach Pflanze geändert werden.
Die Wurzeln wurden zunächst zur leichteren Handhabung in ein Vlies eingewickelt und
an den beiden Enden mit Büroklammern zugeheftet. Die Proben wurden einzeln in Flaschen
mit 10%iger Kaliumhydroxid-Lösung überführt und in einem Tischautoklaven auf 121 °C
erhitzt (H. lanatus nur auf 80 °C, da sich sonst die Cortexzellen ablösen). Durch das Kochen

2 Material und Methoden 41
in KOH wurde das Cytoplasma entfärbt. Nach Erreichen der Temperatur wurde langsam ab-
gekühlt und anschließend die gelblich gefärbte Lauge zweimal durch jeweils einminütiges
Waschen mit Aqua dest. entfernt.
Um eine bessere Färbung zu erzielen, wurde 3 min in einer 1%igen HCl-Lösung ange-
säuert. Anschließend wurden die Probenpäckchen in Trypanblau-Färbelösung überführt und
erneut für 5 min auf 121 °C erhitzt (H. lanatus 5 min auf 80 °C). Nach dem Abkühlen wurden
die Probenpäckchen mehrmals mit Leitungswasser gewaschen, bis keine Blaufärbung mehr
auftrat. Danach wurden die Klammern an den Seiten abgeschnitten und die Wurzeln aus dem
Vlies befreit. Die Wurzeln wurden dann auf Objektträger aufgezogen, in Kaisers Glyceringe-
latine eingebettet und mit einem Deckglas unter leichtem Druck fixiert.
2.2.13 Bestimmung des Mykorrhizierungsgrades
Die Auswertung wurde mit einem Lichtmikroskop (Zeiss Axioplan) bei 400-facher Vergröße-
rung durchgeführt. Die Dokumentation erfolgte über einen Fotoaufsatz mit einer Spiegelre-
flexkamera (Nikon F-801s). Für die Analyse der Mykorrhizierung wurden verschiedenste
Protokolle entwickelt. In dieser Arbeit wurde die Magnified Intersection-Methode angewandt
(MCGONIGLE et al., 1990).
Hierbei wurden die einzelnen Objektträger bei 200-facher Vergrößerung schlangenlini-
enförmig bewegt: zunächst an einem Ende beginnend vertikal; war das Ende erreicht, so
konnten die Objektträger anhand der Skala auf dem Objektträgertisch um 0,5 cm horizontal
verschoben werden und vertikal in entgegengesetzter Richtung abgesucht werden. War das
Ende abermals erreicht, so wurde wieder um 0,5 cm horizontal verschoben und vertikal abge-
sucht, so lange bis das horizontale Ende erreicht war. Wurde dabei mit dem Fadenkreuz des
Okulars ein Wurzelabschnitt getroffen, so wurde notiert, ob dieser Hyphen, Vesikel oder Ar-
buskel enthielt. Pro Stichprobe wurden durchschnittlich etwa fünfzig solcher Schnittpunkte
bewertet. Wurzeln ohne Cortex wurden nicht bonitiert. Durch Bildung der Summe von
Schnittpunkten mit Arbuskeln, Vesikeln, Hyphen und nicht-mykorrhizierten Wurzelab-
schnitten konnte die Hyphen-Kolonisierungsrate HC, Kolonisierungsrate mit Arbuskeln (AC)
und Koloniesierungsrate mit Vesikeln (VC) berechnet werden:

2 Material und Methoden 42
∑∑=
hnitteWurzelabsc
ArbuskelAC
∑∑=
hnitteWurzelabsc
VesikelVC
∑∑=
hnitteWurzelabsc
HyphenHC
Allerdings wiesen bereits MCGONIGLE et al. (1990) darauf hin, dass durch Trypanblau
auch Hyphen phytopathogener Pilze gefärbt sein können. Ein wichtiges Unterscheidungs-
merkmal sind hierbei die unseptierten Hyphen der arbuskulären Mykorrhizapilze.
2.2.14 Phosphatbestimmung
Für die Bestimmung des Phosphatgehalts in Bodenproben existieren zahlreiche Protokolle.
Hier wurde die CAL-Methode verwendet, bei der ein saurer, auf pH 4,1 gepufferter Calcium-
Acetat-Lactat (CAL)-Auszug hergestellt und anschließend photometrisch bestimmt wird. Die
CAL-Methode wird als Maß für die „pflanzenverfügbare Phosphatmenge“ definiert. Von den
im Boden zahlreichen Phosphatverbindungen werden hierbei diejenigen berücksichtigt, die
entweder leicht wasserlöslich sind, oder durch die Enzyme der Pflanzen zusätzlich aufge-
schlossen werden können. Im Unterschied zum Gesamtphosphatgehalt werden die – auch für
Pflanzen - schwerlöslichen Phosphate nicht berücksichtigt (Hoffmann, 1991). Das Protokoll
wurde auf Mikromaßstab reduziert, sowie die gemessene Wellenlänge auf das Absorptions-
maximum festgelegt.
Die getrockneten Bodenproben wurden mit einem Löffel durch ein feines Metallsieb
(Maschenweite 2 mm) gedrückt und eventuell beigemischtes Pflanzenmaterial entfernt. Von
dem homogenen Pulver wurde ca. 1000 mg genau eingewogen, in einem 100 mL Erlenmeyer-
Kolben mit 20 mL CAL-Gebrauchslösung versetzt, und 90 min auf einem Horizontalschüttler
mit 140 rpm geschüttelt. Anschließend wurde filtriert und dabei die ersten 1-2 mL des Filtrats
verworfen.
Für die Messung wurden ein UV/VIS-Photometer sowie 10 mm-Kunststoffküvetten
verwendet. Als Reagenzienblindwert wurden 500 µL CAL-Gebrauchslösung, 750 µL Wasser,
50 µL Molybdatreagenz und 50 µL Reduktionslösung in eine Küvette pipettiert und der
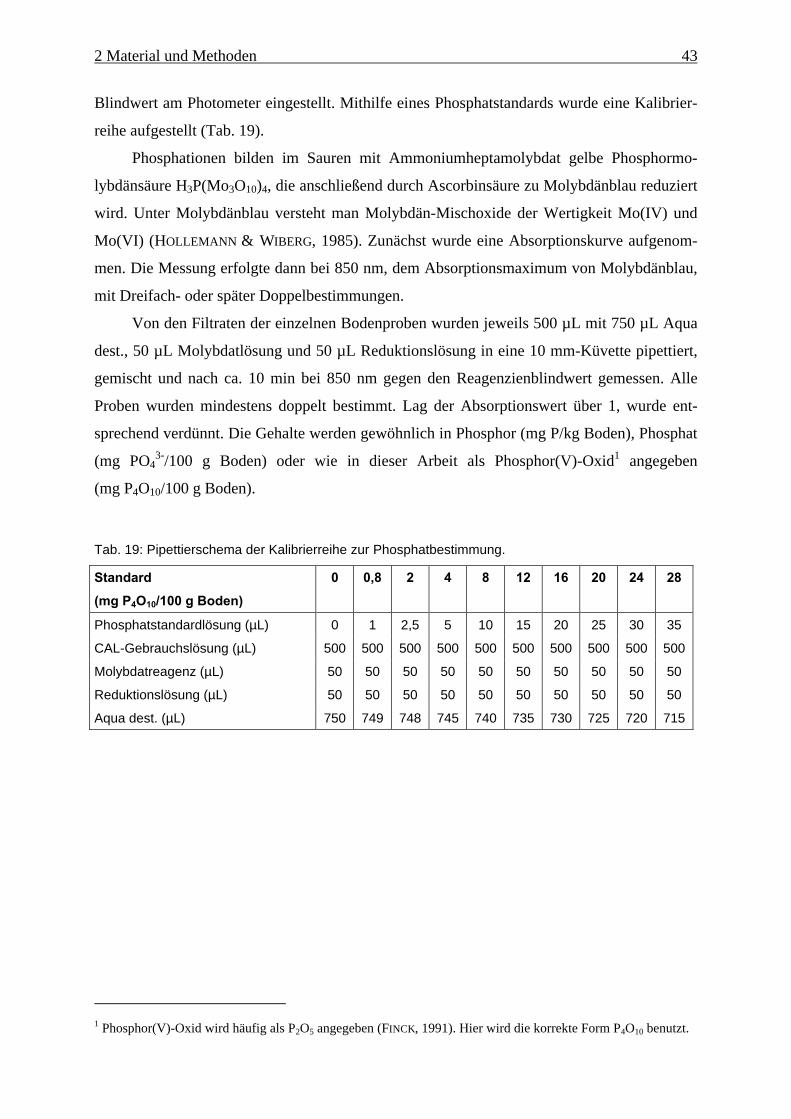
2 Material und Methoden 43
Blindwert am Photometer eingestellt. Mithilfe eines Phosphatstandards wurde eine Kalibrier-
reihe aufgestellt (Tab. 19).
Phosphationen bilden im Sauren mit Ammoniumheptamolybdat gelbe Phosphormo-
lybdänsäure H3P(Mo3O10)4, die anschließend durch Ascorbinsäure zu Molybdänblau reduziert
wird. Unter Molybdänblau versteht man Molybdän-Mischoxide der Wertigkeit Mo(IV) und
Mo(VI) (HOLLEMANN & WIBERG, 1985). Zunächst wurde eine Absorptionskurve aufgenom-
men. Die Messung erfolgte dann bei 850 nm, dem Absorptionsmaximum von Molybdänblau,
mit Dreifach- oder später Doppelbestimmungen.
Von den Filtraten der einzelnen Bodenproben wurden jeweils 500 µL mit 750 µL Aqua
dest., 50 µL Molybdatlösung und 50 µL Reduktionslösung in eine 10 mm-Küvette pipettiert,
gemischt und nach ca. 10 min bei 850 nm gegen den Reagenzienblindwert gemessen. Alle
Proben wurden mindestens doppelt bestimmt. Lag der Absorptionswert über 1, wurde ent-
sprechend verdünnt. Die Gehalte werden gewöhnlich in Phosphor (mg P/kg Boden), Phosphat
(mg PO43-/100 g Boden) oder wie in dieser Arbeit als Phosphor(V)-Oxid1 angegeben
(mg P4O10/100 g Boden).
Tab. 19: Pipettierschema der Kalibrierreihe zur Phosphatbestimmung.
Standard
(mg P4O10/100 g Boden)
0 0,8 2 4 8 12 16 20 24 28
Phosphatstandardlösung (µL) 0 1 2,5 5 10 15 20 25 30 35
CAL-Gebrauchslösung (µL) 500 500 500 500 500 500 500 500 500 500
Molybdatreagenz (µL) 50 50 50 50 50 50 50 50 50 50
Reduktionslösung (µL) 50 50 50 50 50 50 50 50 50 50
Aqua dest. (µL) 750 749 748 745 740 735 730 725 720 715
1 Phosphor(V)-Oxid wird häufig als P2O5 angegeben (FINCK, 1991). Hier wird die korrekte Form P4O10 benutzt.

3 Ergebnisse 44
3 Ergebnisse
Die Bonitierung der AMF in radix mit molekularbiologischen und „klassischen“
Experimenten hatte zum Ziel, die Verteilung und Häufigkeit einzelner AMF-Arten bei
bestimmten Pflanzen im Freiland zu bestimmen. Neben dem qualitativen Nachweis einzelner
Arten wurden zusätzliche quantifizierende Analysen durchgeführt, die Aufschluss über die
Rolle der AMF bei der Versorgung der Pflanzen mit Nährstoffen geben sollten.
3.1 Optimierung der DNA-Extraktion aus Wurzelmaterial
Zunächst war es nicht möglich gewesen, trotz scheinbar ausreichender Mengen an DNA und
diverser Optimierungsversuche, reproduzierbare PCR-Ergebnisse zu erhalten. Die Vermutung
lag nahe, dass das Problem an der DNA-Extraktion liegen könnte. Verdünnungsreihen, die
zum Ziel hatten, inhibierende Substanzen wie Polysaccharide soweit zu verdünnen, dass sie
keine negativen Effekte mehr auf das Ergebnis gehabt hätten, scheiterten ebenfalls. Polyphe-
nole können bei einer Zerstörung der Zelle, zum Beispiel durch Austrocknung, in kürzester
Zeit durch Phenoloxidasen und Peroxidasen oxidiert werden, dabei Radikale bilden und im
schlimmsten Fall an die DNA binden (ABU AL-SOUD & RADSTRÖM, 1988). Diese wäre dann
in der PCR von der Polymerase nicht mehr „lesbar“. Das Ziel der Extraktionsoptimierung war
daher, die DNA so schnell wie möglich zu isolieren, bevor sie durch Radikale angegriffen
wurde. Hierzu wurden drei verschiedene Methoden getestet. Die Extraktion nach DOYLE &
DOYLE (1987) war für DNA aus Pflanzenmaterial entwickelt worden. BAHNWEG (1998) hatte
eine Methode für schwieriges Material, wie Wurzeln entwickelt. Daneben stand der DNeasy
Plant Mini Kit zur Verfügung, der ebenfalls für Pflanzenmaterial entwickelt worden war.
Darüber hinaus wurden verschiedene Möglichkeiten der Zelllyse variiert (Abb. 14). Die
Extrakte wurden mithilfe eines Agarose-Gels analysiert und ihr DNA-Gehalt mit dem Pico-
Green® Assay bestimmt. Dabei zeigte sich, dass bei der Methode mit dem Ultraschallstab
zwar relativ viel DNA gewonnen werden konnte (über 60 µg/mL, Abb. 14, Spuren 1 und 2),
die DNA jedoch durch die starken Scherkräfte fragmentiert wurde. Das Zermösern mit einem
Micropistill brachte sehr geringe Ausbeuten (8 µg/mL, Spur 6), ebenso der Aufschluss mit
einem Ultra-Turrax (6 µg/mL, Spur 7). Die Extraktion mit dem QIAGEN-Kit in Kombination
mit der Schwingmühle brachte die besten Ergebnisse: relativ hohe Ausbeuten (40 µg/mL,
Spur 3) bei geringer Degradation. Noch höhere Ausbeuten ließen sich durch Inkubation mit
Protease und Chitinase bei 25 °C bzw. 37 °C erzielen (62 und 71 µg/mL, Spuren 4 und 5).
Allerdings wurde die DNA durch die erhöhte Temperatur etwas stärker degradiert.

3 Ergebnisse 45
Abb. 14: DNA-Extrakte mit verschiedenen Zelllyse-Techniken in 1%-Agarose-Gel.Zelllyse mit: 1 - Ultraschallstab, 2 - Ultraschallstab und Poly-A (als Träger-DNA), 3 - Schwingmühle, 4 - Schwingmühle und Inkubation mit Proteaseund Chitinase bei 25 °C, 5 - Schwingmühle und Inkubation mit Protease undChitinase bei 37 °C, 6 - Micropistill, 7 - Ultra-Turrax, 8 - Negativkontrolle(Aqua dest.) mit Schwingmühle; M: 100bp DNA Ladder Plus, m: 20 bp DNALadder. Darunter zum Vergleich die mit PicoGreen® ermittelten DNA-Kon-zentrationen der Extrakte.
3.2 Untersuchung der Oligonukleotidprimer
Um die Spezifität verschiedener Primer zu testen, wurden ihre Sequenzen mithilfe der
BLAST-Software analysiert (ALTSCHUL et al. 1997). Die eingesandten Sequenzen wurden mit
der Datenbank auf Übereinstimmungen überprüft. Die erhaltenen Ergebnisse wurden in zwei
Richtungen untersucht: 1) passte die spezifische Primersequenz zu allen Stämmen einer
Art/Gruppe und 2) schloss sie alle anderen Arten aus? Die Ergebnisse dieser Spezifitäts-Un-
tersuchung sind in (Tab. 20) dargestellt. Die Zahlen drücken die Basenhomologie aus.
Der von SIMON et al. (1992) beschriebene „Glomales-spezifische“ Primer VANS1 passt
nur bei einigen AM-Pilzarten 100%-ig. Zahlreiche Sequenzen hingegen, z.B. von Glomus
etunicatum, Acaulospora- und Scutellospora-Arten stimmten zum Teil nur mit 16 von 23 Ba-
sen mit der Primersequenz überein und wiesen mehrere Abweichungen auf.
3 Ergebnisse 46
Tab. 20: Vergleich der Sequenzen spezifischer Primer mit Datenbanksequenzen. Die Zahlen in derSpalte Basenhomologie geben die übereinstimmenden Basen an. Zahlen in Klammern ge-ben die Anzahl der Sequenzen einer Art an. Die Autoren sind in Tab. 1 aufgeführt.A.: Acaulospora, Ar.: Archaeospora, E.: Entrophospora, G.: Glomus, Gi.: Gigaspora, P.:Paraglomus, S.: Scutellospora.
Primer Länge
(Basen)
Spezifität
laut Autor(en)
Basenhomologie
VANS1 23 Glomales 23: G. intraradices, Gi. rosea, Gi. margarita,“Endophyte”
22: G. etunicatum
21: S. dipapillosa, S. pellucida, Gi. albida,G. vesiculiferum
19: G. etunicatum
17: A. rugosa
16: A. spinosa
ITS1F 22 Pilze 22: zahlreiche Glomales-Arten und andere Pilze,Chaetosphaeridium globosum (Grünalge)
ACAU1660 17 Acaulosporaceae 17: A. scrobiculata, A. longula, A. laevis, A. rugosa,A. morrowiae, E. colombiana
14: A. spinosa, Ar. leptoticha
ARCH1311 19 Paraglomaceae 19: P. occultum, P. brasilianum; 8 Pilzsequenzen(Rhynchostoma minutum, Acrospermum spp.,…)
GLOM1310 20 Glomaceae 20: G. caledonium, G. fasciculatum, G. intraradices,G. manihotis, G. sinuosum, G. vesiculiferum
19: G. caledonium, G. clarum, G. coremioides,G. coronatum, G. fragilistratum, G. mosseae,G. proliferum, G. verruculosum
LETC1670 18 G. etunicatum-Gruppe
18: G. claroideum, G. etunicatum, G. lamellosum,G. luteum, G. sp. S329, G. sp. W3234
17: Bensingtonia musae & ingoldii
GIGA5.8R 19 Gigasporaceae 19: Gigaspora (5 Arten), Scutellospora (4 Arten)
GLOM5.8R 19 Glomaceae 19: G. caledonium, G. clarum, G. dimorphicum,G. geosporum, G. intraradices, G. monosporum,G. mosseae
Aclong87For 22 A. longula 22: A. longula
Aclong251For 23 A. longula 23: A. longula
AclongSonde 27 A. longula 27: A. longula
3 Ergebnisse 47
Tab. 20 (Fortsetzung)
Primer Länge
(Basen)
Spezifität
laut Autor(en)
Annealing
Aclong338Rev 22 A. longula 22: A. longula
21: Blastobotrys capitolata
Aclong400Rev 24 A. longula 24: A. longula
19: A. tuberculata, A. spinosa
Glmoss85For 22 G. mosseae 22: G. mosseae, G. constrictum, G. coronatum,G. geosporum, E. infrequens
Glmoss387Rev 25 G. mosseae 25: G. mosseae, G. caledonium, G. constrictum,G. coronatum, G. geosporum, E. infrequens
P1 20 G. mosseae 20: G. mosseae
18: G. coronatum, E. infrequens
F1 31 G. mosseae 31: G. mosseae (18)
30: G. mosseae (6), G. constrictum, G. coronatum,E. infrequens
P2 27 G. mosseae 27: G. mosseae (19)
25: G. mosseae (1)
23: G. mosseae (5), G. geosporum, G. constrictum,G. coronatum
Der Primer ITS1F wurde als pilzspezifisch beschrieben (GARDES & BRUNS, 1993). Tat-
sächlich wurden zahlreiche Glomales-Arten mit übereinstimmender Sequenz gefunden. Aller-
dings „erkennt“ der Primer auch Chaetosphaeridium globosum, eine Grünalge.
Der von REDECKER (2000) publizierte Primer ACAU1660 ist für einige Arten der
Acaulosporaceae spezifisch. Allerdings diskriminiert er Acaulospora spinosa. ARCH1311
passt zu den beiden Paraglomus-Arten. Es wurden jedoch noch mindestens 8 weitere Pilzar-
ten gefunden, die 100%-ige Homologie besitzen. Bei dem Glomaceae-spezifischen Primer
GLOM1310 besaßen 7 Glomus-Arten eine Übereinstimmung in allen 20 Basen, 8 weitere
Glomus-Arten eine Übereinstimmung in 19 der 20 Basen. LETC1670 wurde als spezifisch für
die „Glomus etunicatum-Gruppe“ beschrieben. Es konnte völlige Übereinstimmung mit
Glomus etunicatum und fünf weiteren Glomus-Arten gefunden werden. Allerdings traf die
Sequenz auch mit 17 der 18 Basen auf zwei Bensingtonia-Arten zu. Bei den Primern

3 Ergebnisse 48
GIGA5.8R und GLOM5.8R wurden zahlreiche Arten der Gigasporaceae bzw. Glomaceae mit
völliger Übereinstimmung gefunden.
Die von BÖHM (2000) entwickelten Primer für A. longula (Aclong87For,
Aclong251For, AclongSonde, Aclong338Rev und Aclong400Rev) stimmten alle mit den in
der Datenbank vorhandenen Sequenzdaten von A. longula überein. Bei Aclong338Rev ergab
sich eine hohe Übereinstimmung von 21 der 22 Basen mit Blastobotrys capitolata. Die
G. mosseae-spezifischen Primer (Glmoss85For und Glmoss387Rev) stimmten zu 100% mit
Sequenzen von G. mosseae, G. caledonium, G. constrictum, G. coronatum, G. geosporum und
Entrophospora infrequens überein. P1 war identisch mit der G. mosseae-Sequenz. Allerdings
passten 18 von 20 Basen bei G. coronatum und E. infrequens. Bei P2 waren 19 von insgesamt
25 Datenbanksequenzen von G. mosseae mit dem Primer identisch. Die restlichen sechs Se-
quenzen differierten um bis zu vier Basen.
Im zweiten Schritt wurden die Primern in einer „virtuellen PCR“ getestet. Hierbei wur-
den die Primerpaare via Internet mithilfe der Software der Fa. Genomatix
(www.genomatix.de) mit Datenbanksequenzen verglichen. Zusätzlich zur Primersequenz
konnte die erwartete Produktlänge in Basenpaaren angegeben werden. Die Software ermittelte
solche Sequenzen, bei denen sowohl Fragmentlänge als auch die Bindungsstellen der beiden
Primer übereintrafen (Tab. 21).
Das Primerpaar VANS1-NS21 (SIMON et al. 1992) erbrachte ganze drei Sequenzen.
Dieses Ergebnis bestätigte die Tatsache, dass viele Glomales-Arten von VANS1 nicht erfasst
werden. Mit dem Primerpaar A2R-B2L wurden zahlreiche Sequenzen von Pilzen, Pflanzen
und Invertebraten gefunden. Damit war die universelle Bindungseigenschaft der beiden Oli-
gonukleotide bestätigt. Aclong87For und Aclong400Rev passten nur auf drei Sequenzen von
A. longula. Das Primerpaar ist also – bezogen auf bisher bekannte Sequenzen – spezifisch für
diese Art. Dem Ergebnis der in silico PCR mit den Primern Glmoss85For und Glmoss387Rev
zufolge, wurden neben G. mosseae mehrere, offenbar nah verwandte Arten, darunter
G. coronatum, G. constrictum, G. geosporum, G. caledonium, G. fragilistratum amplifiziert.
Die Spezifität dieses Primerpaares muß also auf eine G. mosseae-Gruppe ausgeweitet werden.
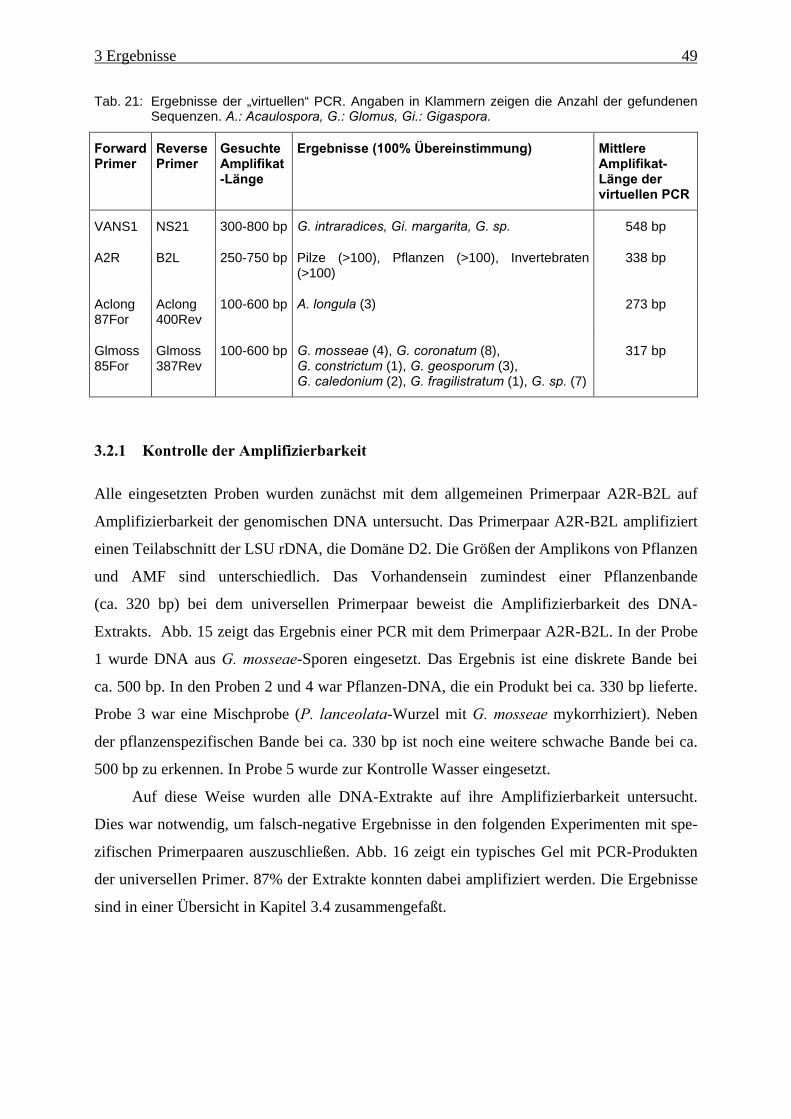
3 Ergebnisse 49
Tab. 21: Ergebnisse der „virtuellen“ PCR. Angaben in Klammern zeigen die Anzahl der gefundenenSequenzen. A.: Acaulospora, G.: Glomus, Gi.: Gigaspora.
ForwardPrimer
ReversePrimer
GesuchteAmplifikat-Länge
Ergebnisse (100% Übereinstimmung) MittlereAmplifikat-Länge dervirtuellen PCR
VANS1 NS21 300-800 bp G. intraradices, Gi. margarita, G. sp. 548 bp
A2R B2L 250-750 bp Pilze (>100), Pflanzen (>100), Invertebraten(>100)
338 bp
Aclong87For
Aclong400Rev
100-600 bp A. longula (3) 273 bp
Glmoss85For
Glmoss387Rev
100-600 bp G. mosseae (4), G. coronatum (8),G. constrictum (1), G. geosporum (3),G. caledonium (2), G. fragilistratum (1), G. sp. (7)
317 bp
3.2.1 Kontrolle der Amplifizierbarkeit
Alle eingesetzten Proben wurden zunächst mit dem allgemeinen Primerpaar A2R-B2L auf
Amplifizierbarkeit der genomischen DNA untersucht. Das Primerpaar A2R-B2L amplifiziert
einen Teilabschnitt der LSU rDNA, die Domäne D2. Die Größen der Amplikons von Pflanzen
und AMF sind unterschiedlich. Das Vorhandensein zumindest einer Pflanzenbande
(ca. 320 bp) bei dem universellen Primerpaar beweist die Amplifizierbarkeit des DNA-
Extrakts. Abb. 15 zeigt das Ergebnis einer PCR mit dem Primerpaar A2R-B2L. In der Probe
1 wurde DNA aus G. mosseae-Sporen eingesetzt. Das Ergebnis ist eine diskrete Bande bei
ca. 500 bp. In den Proben 2 und 4 war Pflanzen-DNA, die ein Produkt bei ca. 330 bp lieferte.
Probe 3 war eine Mischprobe (P. lanceolata-Wurzel mit G. mosseae mykorrhiziert). Neben
der pflanzenspezifischen Bande bei ca. 330 bp ist noch eine weitere schwache Bande bei ca.
500 bp zu erkennen. In Probe 5 wurde zur Kontrolle Wasser eingesetzt.
Auf diese Weise wurden alle DNA-Extrakte auf ihre Amplifizierbarkeit untersucht.
Dies war notwendig, um falsch-negative Ergebnisse in den folgenden Experimenten mit spe-
zifischen Primerpaaren auszuschließen. Abb. 16 zeigt ein typisches Gel mit PCR-Produkten
der universellen Primer. 87% der Extrakte konnten dabei amplifiziert werden. Die Ergebnisse
sind in einer Übersicht in Kapitel 3.4 zusammengefaßt.
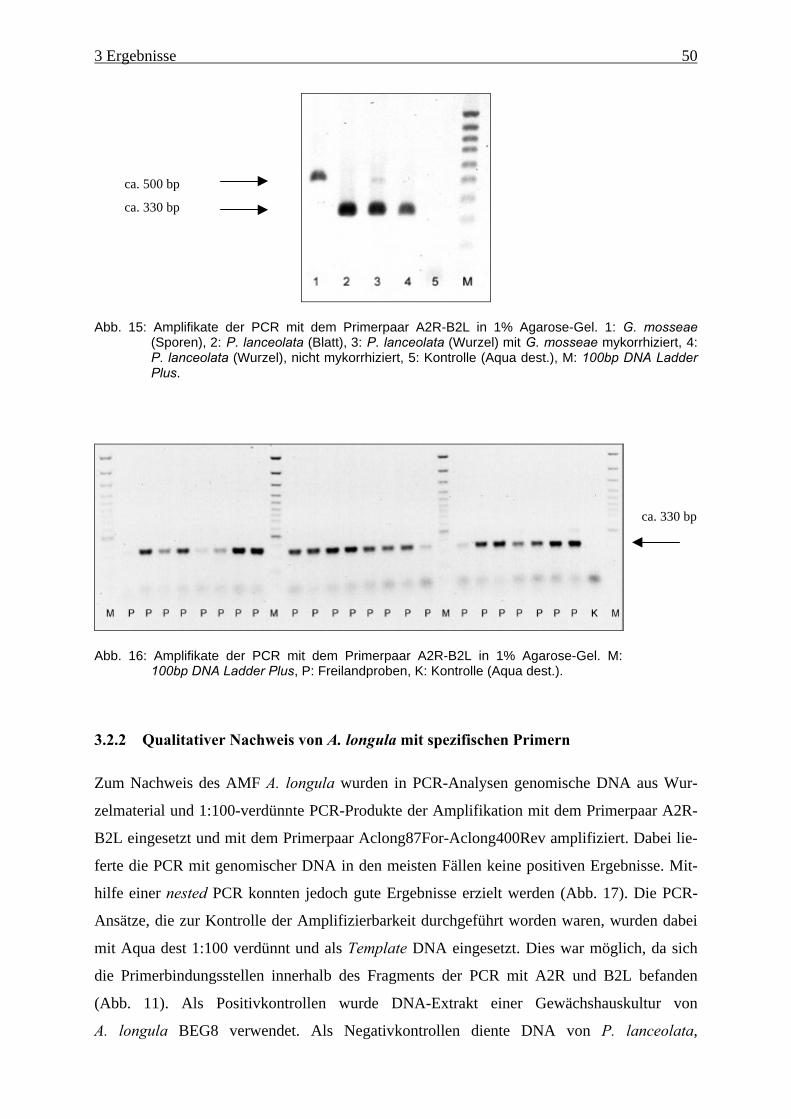
3 Ergebnisse 50
Abb. 15: Amplifikate der PCR mit dem Primerpaar A2R-B2L in 1% Agarose-Gel. 1: G. mosseae(Sporen), 2: P. lanceolata (Blatt), 3: P. lanceolata (Wurzel) mit G. mosseae mykorrhiziert, 4:P. lanceolata (Wurzel), nicht mykorrhiziert, 5: Kontrolle (Aqua dest.), M: 100bp DNA LadderPlus.
Abb. 16: Amplifikate der PCR mit dem Primerpaar A2R-B2L in 1% Agarose-Gel. M:100bp DNA Ladder Plus, P: Freilandproben, K: Kontrolle (Aqua dest.).
3.2.2 Qualitativer Nachweis von A. longula mit spezifischen Primern
Zum Nachweis des AMF A. longula wurden in PCR-Analysen genomische DNA aus Wur-
zelmaterial und 1:100-verdünnte PCR-Produkte der Amplifikation mit dem Primerpaar A2R-
B2L eingesetzt und mit dem Primerpaar Aclong87For-Aclong400Rev amplifiziert. Dabei lie-
ferte die PCR mit genomischer DNA in den meisten Fällen keine positiven Ergebnisse. Mit-
hilfe einer nested PCR konnten jedoch gute Ergebnisse erzielt werden (Abb. 17). Die PCR-
Ansätze, die zur Kontrolle der Amplifizierbarkeit durchgeführt worden waren, wurden dabei
mit Aqua dest 1:100 verdünnt und als Template DNA eingesetzt. Dies war möglich, da sich
die Primerbindungsstellen innerhalb des Fragments der PCR mit A2R und B2L befanden
(Abb. 11). Als Positivkontrollen wurde DNA-Extrakt einer Gewächshauskultur von
A. longula BEG8 verwendet. Als Negativkontrollen diente DNA von P. lanceolata,
ca. 330 bp
ca. 500 bp
ca. 330 bp
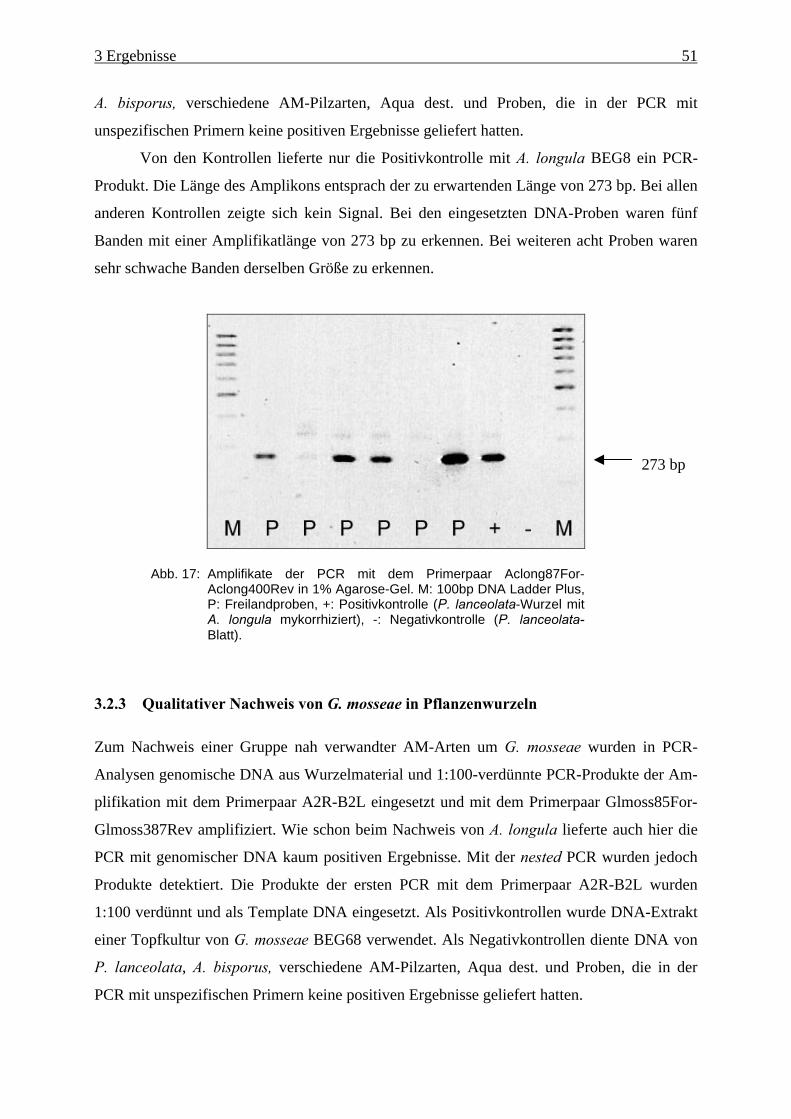
3 Ergebnisse 51
A. bisporus, verschiedene AM-Pilzarten, Aqua dest. und Proben, die in der PCR mit
unspezifischen Primern keine positiven Ergebnisse geliefert hatten.
Von den Kontrollen lieferte nur die Positivkontrolle mit A. longula BEG8 ein PCR-
Produkt. Die Länge des Amplikons entsprach der zu erwartenden Länge von 273 bp. Bei allen
anderen Kontrollen zeigte sich kein Signal. Bei den eingesetzten DNA-Proben waren fünf
Banden mit einer Amplifikatlänge von 273 bp zu erkennen. Bei weiteren acht Proben waren
sehr schwache Banden derselben Größe zu erkennen.
Abb. 17: Amplifikate der PCR mit dem Primerpaar Aclong87For-Aclong400Rev in 1% Agarose-Gel. M: 100bp DNA Ladder Plus,P: Freilandproben, +: Positivkontrolle (P. lanceolata-Wurzel mitA. longula mykorrhiziert), -: Negativkontrolle (P. lanceolata-Blatt).
3.2.3 Qualitativer Nachweis von G. mosseae in Pflanzenwurzeln
Zum Nachweis einer Gruppe nah verwandter AM-Arten um G. mosseae wurden in PCR-
Analysen genomische DNA aus Wurzelmaterial und 1:100-verdünnte PCR-Produkte der Am-
plifikation mit dem Primerpaar A2R-B2L eingesetzt und mit dem Primerpaar Glmoss85For-
Glmoss387Rev amplifiziert. Wie schon beim Nachweis von A. longula lieferte auch hier die
PCR mit genomischer DNA kaum positiven Ergebnisse. Mit der nested PCR wurden jedoch
Produkte detektiert. Die Produkte der ersten PCR mit dem Primerpaar A2R-B2L wurden
1:100 verdünnt und als Template DNA eingesetzt. Als Positivkontrollen wurde DNA-Extrakt
einer Topfkultur von G. mosseae BEG68 verwendet. Als Negativkontrollen diente DNA von
P. lanceolata, A. bisporus, verschiedene AM-Pilzarten, Aqua dest. und Proben, die in der
PCR mit unspezifischen Primern keine positiven Ergebnisse geliefert hatten.
273 bp
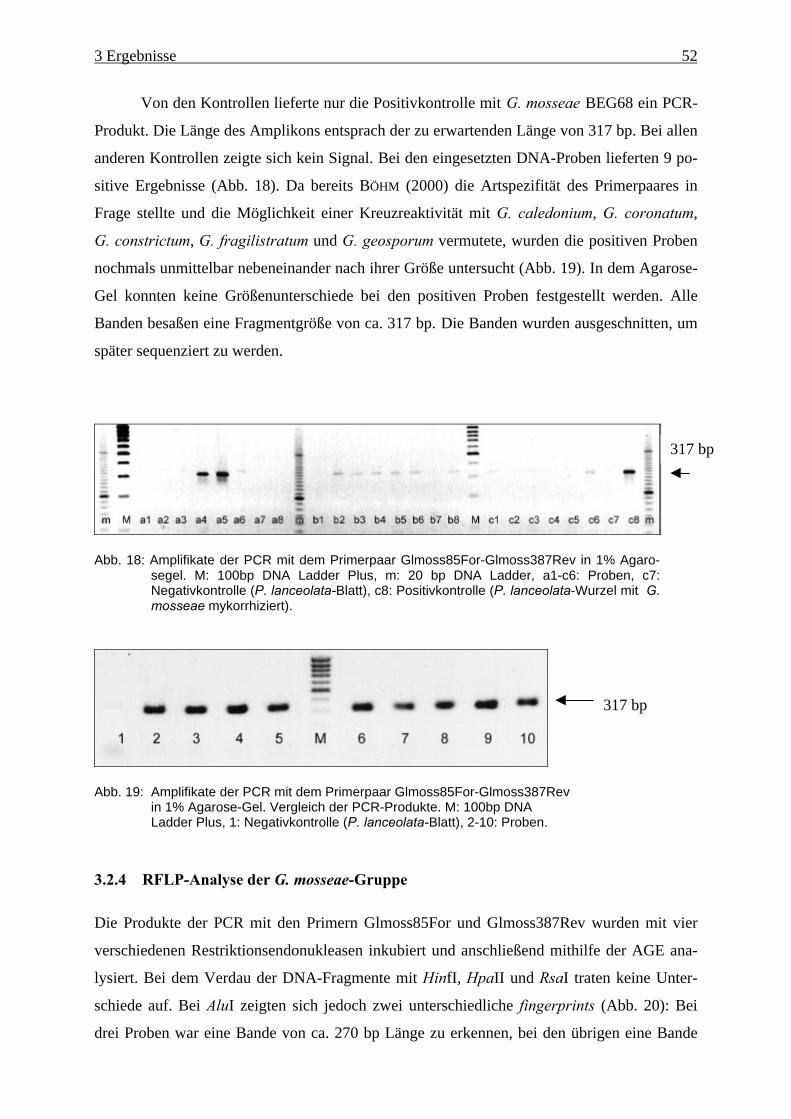
3 Ergebnisse 52
Von den Kontrollen lieferte nur die Positivkontrolle mit G. mosseae BEG68 ein PCR-
Produkt. Die Länge des Amplikons entsprach der zu erwartenden Länge von 317 bp. Bei allen
anderen Kontrollen zeigte sich kein Signal. Bei den eingesetzten DNA-Proben lieferten 9 po-
sitive Ergebnisse (Abb. 18). Da bereits BÖHM (2000) die Artspezifität des Primerpaares in
Frage stellte und die Möglichkeit einer Kreuzreaktivität mit G. caledonium, G. coronatum,
G. constrictum, G. fragilistratum und G. geosporum vermutete, wurden die positiven Proben
nochmals unmittelbar nebeneinander nach ihrer Größe untersucht (Abb. 19). In dem Agarose-
Gel konnten keine Größenunterschiede bei den positiven Proben festgestellt werden. Alle
Banden besaßen eine Fragmentgröße von ca. 317 bp. Die Banden wurden ausgeschnitten, um
später sequenziert zu werden.
Abb. 18: Amplifikate der PCR mit dem Primerpaar Glmoss85For-Glmoss387Rev in 1% Agaro-segel. M: 100bp DNA Ladder Plus, m: 20 bp DNA Ladder, a1-c6: Proben, c7:Negativkontrolle (P. lanceolata-Blatt), c8: Positivkontrolle (P. lanceolata-Wurzel mit G.mosseae mykorrhiziert).
Abb. 19: Amplifikate der PCR mit dem Primerpaar Glmoss85For-Glmoss387Revin 1% Agarose-Gel. Vergleich der PCR-Produkte. M: 100bp DNALadder Plus, 1: Negativkontrolle (P. lanceolata-Blatt), 2-10: Proben.
3.2.4 RFLP-Analyse der G. mosseae-Gruppe
Die Produkte der PCR mit den Primern Glmoss85For und Glmoss387Rev wurden mit vier
verschiedenen Restriktionsendonukleasen inkubiert und anschließend mithilfe der AGE ana-
lysiert. Bei dem Verdau der DNA-Fragmente mit HinfI, HpaII und RsaI traten keine Unter-
schiede auf. Bei AluI zeigten sich jedoch zwei unterschiedliche fingerprints (Abb. 20): Bei
drei Proben war eine Bande von ca. 270 bp Länge zu erkennen, bei den übrigen eine Bande
317 bp
317 bp

3 Ergebnisse 53
von ca. 130 bp. Anhand der Sequenzinformation (Kapitel 3.2.5) konnten mögliche Schnitt-
stellen des Enzyms AluI lokalisiert werden (Tab. 23). Bei den drei ungeschnittenen Fragmen-
ten (Pla5-004, HolIII-008 und ZeaA15) konnten auch in den Sequenzdaten keine Schnittstel-
len gefunden werden. Bei den restlichen Proben waren zwei Schnittstellen vorhanden, woraus
sich drei Fragmente mit 17, 121 und 128 bp ergaben. Diese Fragmentlängen stimmen mit de-
nen des Gels überein, unter der Voraussetzung, dass ein 17 bp-Fragment unter den gegebenen
Bedingungen im Gel nicht nachweisbar war und die beiden größeren Fragmente sich nicht
voneinander trennen ließen, also als eine Bande erschienen. Bei TraA21 fehlte die erste
Schnittstelle, so dass das zweite, größere Fragment eine Länge von 149 bp statt 121 bp besaß.
Bei HelA06 konnte über die erste Schnittstelle keine Aussage getroffen werden, da entspre-
chende Sequenzdaten fehlten. Hier waren auch im Gel keine eindeutigen Banden zu erkennen.
Tab. 22: Abkürzungen der untersuchten Sequenzen. Auf der linken Seite sind die Abkürzungen fürdie Sequenzen der Freiland-Isolate zusammengefaßt; auf der rechten Seite die verwendetenDatenbanksequenzen.
Ermittelte Sequenzen Datenbanksequenzen
Abkürzung Sequenz Abkürzung Sequenz
Glmoss68W G. mosseae BEG68 (Kontrolle)
Hel-A06 G. mosseae von H. annuus, A06,Juni 2000
HolI-108 G. mosseae von H. lanatus, W02-oben, August 2001
HolIII-008 G. caledonium von H. lanatus,W02-unten, August 2000
PlaIII-004 G. mosseae von P. lanceolata,W02-unten, April 2000
PlaIII-108 G. mosseae von P. lanceolata,W02-unten, August 2001
Pla4-004 G. mosseae von P. lanceolata,W21-unten Mitte, April 2000
Pla5-004 G. geosporum von P. lanceolata,W21-unten, April 2000
TraA21 G. mosseae von T. aestivum, A21,August 2001
ZeaA15 G. caledonium von Z. mays, A15,August 2001
EninfrMS05
GlcaleRMC
GlcaleRWZ
Glcoro28
Glcoro28K
Glfrag05
Glgeos11
Glgeos90
Glgeos106
Glmoss25
Glmoss68/2
Glmoss68/4
Glmoss68/6
Glmoss68/7
Entrophospora infrequens MS05
Glomus caledonium RMC658
Glomus caledonium RWZ658
Glomus coronatum BEG28
Glomus coronatum BEG28K
Glomus fragilistratum BEG05
Glomus geosporum BEG11
Glomus geosporum BEG90
Glomus geosporum BEG106
G. mosseae BEG25
G. mosseae BEG68 Klon 2
G. mosseae BEG68 Klon 4
G. mosseae BEG68 Klon 6
G. mosseae BEG68 Klon7

3 Ergebnisse 54
Abb. 20: RFLP-Analyse von zehn PCR-Fragmenten der G. mosseae-Gruppe mit dem Enzym AluI in1% Agarose-Gel. M: 100bp DNA Ladder Plus, 1: Gmoss68W, 2: Pla5-004, 3: Pla4-004, 4:PlaIII-004, 5: HelA06, 6: HolIII-008, 7: ZeaA15, 8: HolI-108, 9: TraA21, 10: PlaIII-108.Abkürzungen siehe Tab. 22.
Tab. 23: Übersicht über die Schnittstellen des Enzyms AluI. Abweichende Basen sind unterlegt.Schnittstellen sind mit Pfeilen (�) markiert, nicht geschnittene Positionen durch Striche (-),ungewisse Positionen durch ein Fragezeichen (?). Den Basen sind die Größen der Frag-mente angegeben. Abkürzungen siehe Tab. 22.
Position: 16-19 150-153 Fragmentlängen
AluI AG�CT AG�CT || || || ||Gmoss68W TAG�CTC GAG�CTT 17 – 128 - 121PlaIII-004 TAG�CTC GAG�CTT 17 – 128 - 121Pla4-004 TAG�CTC GAG�CTT 17 – 128 - 121HelA06 NNN?NNN GAG�CTT 17?- 128?- 121HolI-108 TAG�CTC GAG�CTT 17 – 128 - 121PlaIII-108 TAG�CTC GAG�CTT 17 – 129 - 121TraA21 TAG-TTC GAG�CTT 146 - 121HolIII-008 TAG-TTT GGG-CTT 267ZeaA15 TAG-TTT GGG-CTT 267Pla5-004 TAG-TTT GGG-CTT 267
3.2.5 Sequenzanalysen
Von der PCR mit den Primern Glmoss85For und Glmoss387Rev wurden zehn Amplikons
aufgereinigt und sequenziert, darunter neun Freilandproben und ein mit P. lanceolata kulti-
vierter Stamm von G. mosseae BEG68 (Glmoss68W) als Kontrolle. Von den erhaltenen Se-
quenzdaten wurden zunächst in der NCBI-Datenbank mithilfe des Programmes BLAST die
Sequenzen mit der größten Homologie ermittelt (Tab. 24). Mit diesen Datenbanksequenzen
und den sequenzierten Fragmenten von Glmoss85For und Glmoss387Rev wurde ein Annea-
ling durchgeführt. Zur Untersuchung der Sequenzhomologie wurde das Programm BioEdit
verwendet (HALL, 1999). Die Länge der sequenzierten Fragmente lag zwischen 265 und 270
Basenpaaren (Abb. 21). Die ersten 44 Nukleotide des Isolats HelA06 konnten bei der Sequen-
zierung nicht bestimmt werden. Sechs der neun sequenzierten Freiland-Stämme sowie die
Kontrolle G. mosseae BEG68 waren identisch oder unterschieden sich in nur einer Base
(0,4%) von G. mosseae-Stämmen aus der Datenbank. Diese sechs Isolate konnten folglich als
ca. 270 bpca. 130 bp
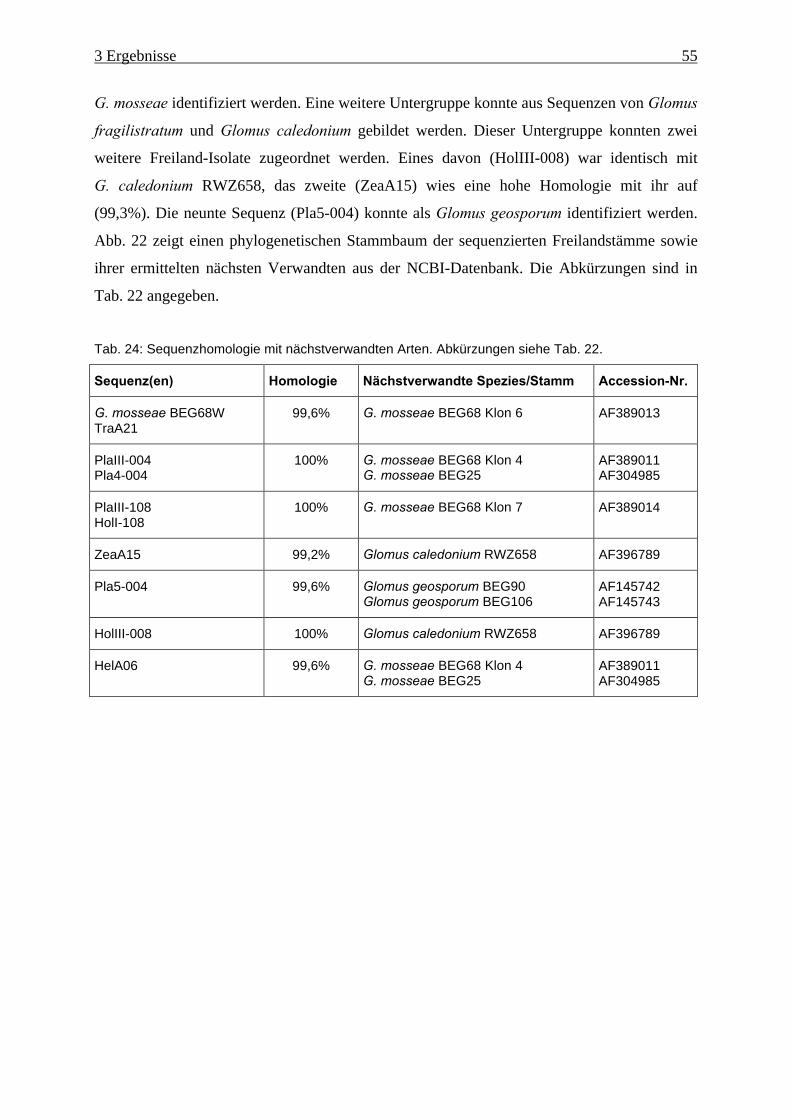
3 Ergebnisse 55
G. mosseae identifiziert werden. Eine weitere Untergruppe konnte aus Sequenzen von Glomus
fragilistratum und Glomus caledonium gebildet werden. Dieser Untergruppe konnten zwei
weitere Freiland-Isolate zugeordnet werden. Eines davon (HolIII-008) war identisch mit
G. caledonium RWZ658, das zweite (ZeaA15) wies eine hohe Homologie mit ihr auf
(99,3%). Die neunte Sequenz (Pla5-004) konnte als Glomus geosporum identifiziert werden.
Abb. 22 zeigt einen phylogenetischen Stammbaum der sequenzierten Freilandstämme sowie
ihrer ermittelten nächsten Verwandten aus der NCBI-Datenbank. Die Abkürzungen sind in
Tab. 22 angegeben.
Tab. 24: Sequenzhomologie mit nächstverwandten Arten. Abkürzungen siehe Tab. 22.
Sequenz(en) Homologie Nächstverwandte Spezies/Stamm Accession-Nr.
G. mosseae BEG68WTraA21
99,6% G. mosseae BEG68 Klon 6 AF389013
PlaIII-004Pla4-004
100% G. mosseae BEG68 Klon 4G. mosseae BEG25
AF389011AF304985
PlaIII-108HolI-108
100% G. mosseae BEG68 Klon 7 AF389014
ZeaA15 99,2% Glomus caledonium RWZ658 AF396789
Pla5-004 99,6% Glomus geosporum BEG90Glomus geosporum BEG106
AF145742AF145743
HolIII-008 100% Glomus caledonium RWZ658 AF396789
HelA06 99,6% G. mosseae BEG68 Klon 4G. mosseae BEG25
AF389011AF304985
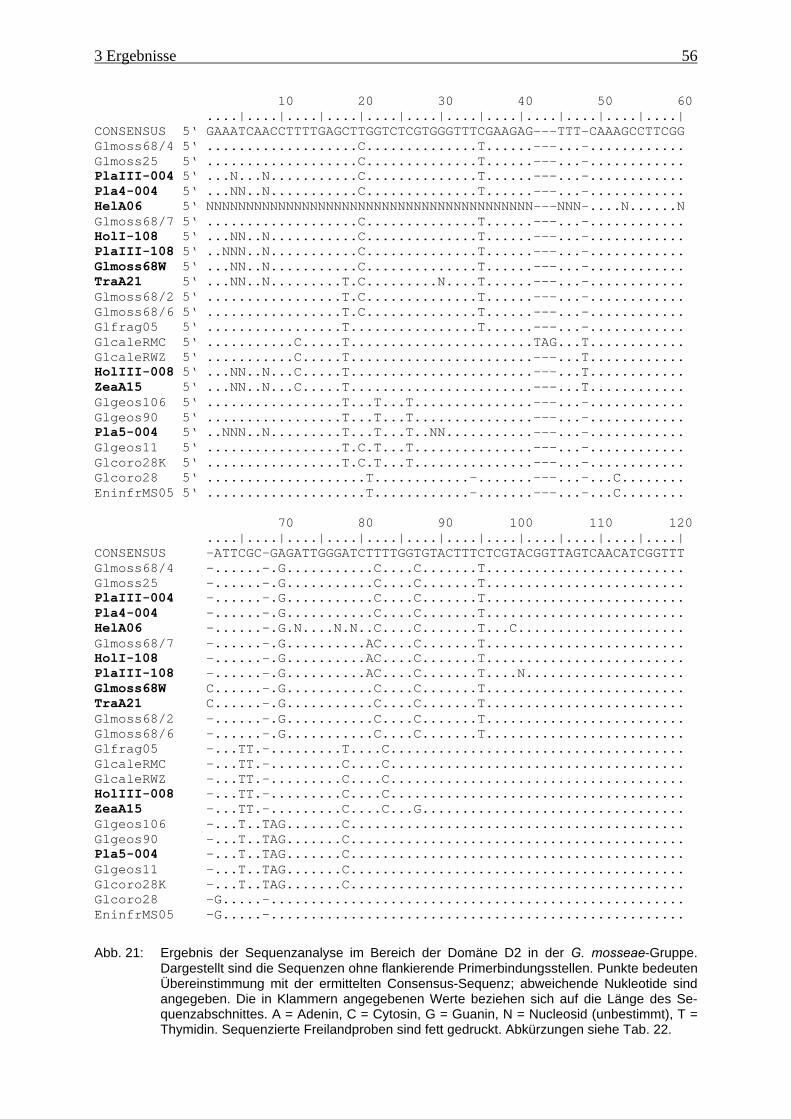
3 Ergebnisse 56
10 20 30 40 50 60 ....|....|....|....|....|....|....|....|....|....|....|....|CONSENSUS 5‘ GAAATCAACCTTTTGAGCTTGGTCTCGTGGGTTTCGAAGAG---TTT-CAAAGCCTTCGGGlmoss68/4 5‘ ...................C..............T......---...-............Glmoss25 5‘ ...................C..............T......---...-............PlaIII-004 5‘ ...N...N...........C..............T......---...-............Pla4-004 5‘ ...NN..N...........C..............T......---...-............HelA06 5‘ NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN---NNN-....N......NGlmoss68/7 5‘ ...................C..............T......---...-............HolI-108 5‘ ...NN..N...........C..............T......---...-............PlaIII-108 5‘ ..NNN..N...........C..............T......---...-............Glmoss68W 5‘ ...NN..N...........C..............T......---...-............TraA21 5‘ ...NN..N.........T.C.........N....T......---...-............Glmoss68/2 5‘ .................T.C..............T......---...-............Glmoss68/6 5‘ .................T.C..............T......---...-............Glfrag05 5‘ .................T................T......---...-............GlcaleRMC 5‘ ...........C.....T.......................TAG...T............GlcaleRWZ 5‘ ...........C.....T.......................---...T............HolIII-008 5‘ ...NN..N...C.....T.......................---...T............ZeaA15 5‘ ...NN..N...C.....T.......................---...T............Glgeos106 5‘ .................T...T...T...............---...-............Glgeos90 5‘ .................T...T...T...............---...-............Pla5-004 5‘ ..NNN..N.........T...T...T..NN...........---...-............Glgeos11 5‘ .................T.C.T...T...............---...-............Glcoro28K 5‘ .................T.C.T...T...............---...-............Glcoro28 5‘ ....................T............-.......---...-...C........EninfrMS05 5‘ ....................T............-.......---...-...C........
70 80 90 100 110 120 ....|....|....|....|....|....|....|....|....|....|....|....|CONSENSUS -ATTCGC-GAGATTGGGATCTTTTGGTGTACTTTCTCGTACGGTTAGTCAACATCGGTTTGlmoss68/4 -......-.G...........C....C.......T.........................Glmoss25 -......-.G...........C....C.......T.........................PlaIII-004 -......-.G...........C....C.......T.........................Pla4-004 -......-.G...........C....C.......T.........................HelA06 -......-.G.N....N.N..C....C.......T...C.....................Glmoss68/7 -......-.G..........AC....C.......T.........................HolI-108 -......-.G..........AC....C.......T.........................PlaIII-108 -......-.G..........AC....C.......T....N....................Glmoss68W C......-.G...........C....C.......T.........................TraA21 C......-.G...........C....C.......T.........................Glmoss68/2 -......-.G...........C....C.......T.........................Glmoss68/6 -......-.G...........C....C.......T.........................Glfrag05 -...TT.-.........T....C.....................................GlcaleRMC -...TT.-.........C....C.....................................GlcaleRWZ -...TT.-.........C....C.....................................HolIII-008 -...TT.-.........C....C.....................................ZeaA15 -...TT.-.........C....C...G.................................Glgeos106 -...T..TAG.......C..........................................Glgeos90 -...T..TAG.......C..........................................Pla5-004 -...T..TAG.......C..........................................Glgeos11 -...T..TAG.......C..........................................Glcoro28K -...T..TAG.......C..........................................Glcoro28 -G.....-....................................................EninfrMS05 -G.....-....................................................
Abb. 21: Ergebnis der Sequenzanalyse im Bereich der Domäne D2 in der G. mosseae-Gruppe.Dargestellt sind die Sequenzen ohne flankierende Primerbindungsstellen. Punkte bedeutenÜbereinstimmung mit der ermittelten Consensus-Sequenz; abweichende Nukleotide sindangegeben. Die in Klammern angegebenen Werte beziehen sich auf die Länge des Se-quenzabschnittes. A = Adenin, C = Cytosin, G = Guanin, N = Nucleosid (unbestimmt), T =Thymidin. Sequenzierte Freilandproben sind fett gedruckt. Abkürzungen siehe Tab. 22.
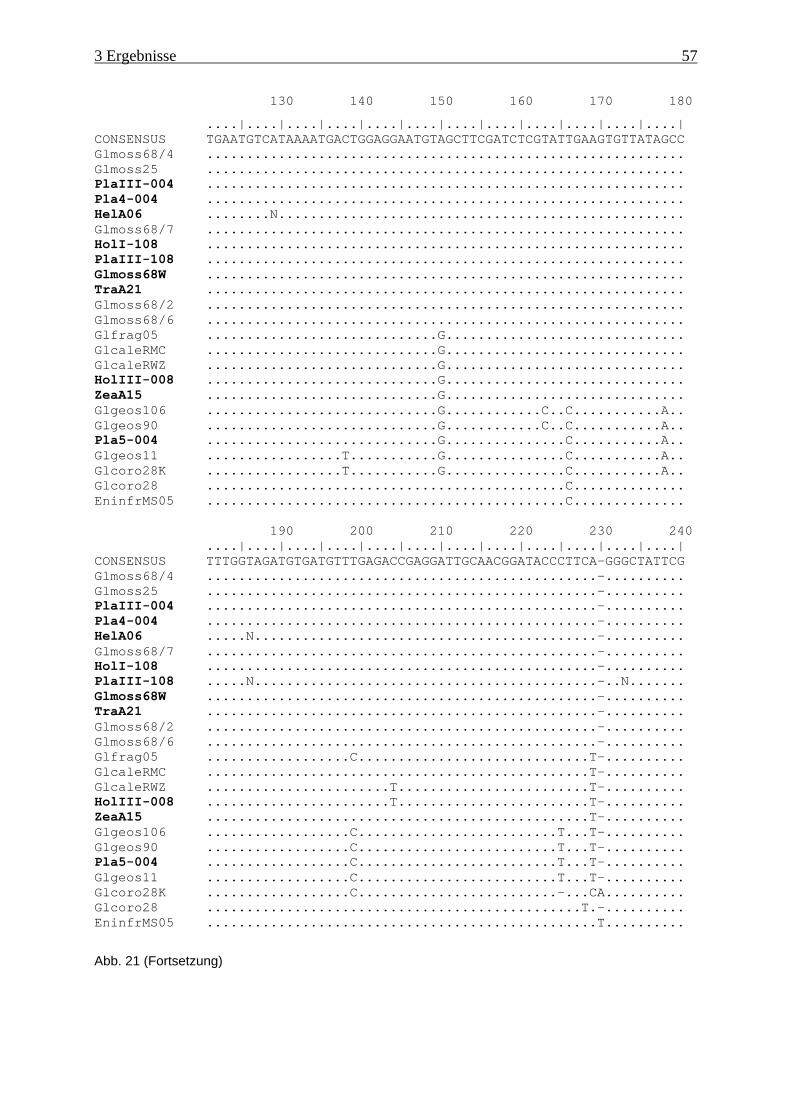
3 Ergebnisse 57
130 140 150 160 170 180
....|....|....|....|....|....|....|....|....|....|....|....|CONSENSUS TGAATGTCATAAAATGACTGGAGGAATGTAGCTTCGATCTCGTATTGAAGTGTTATAGCCGlmoss68/4 ............................................................Glmoss25 ............................................................PlaIII-004 ............................................................Pla4-004 ............................................................HelA06 ........N...................................................Glmoss68/7 ............................................................HolI-108 ............................................................PlaIII-108 ............................................................Glmoss68W ............................................................TraA21 ............................................................Glmoss68/2 ............................................................Glmoss68/6 ............................................................Glfrag05 .............................G..............................GlcaleRMC .............................G..............................GlcaleRWZ .............................G..............................HolIII-008 .............................G..............................ZeaA15 .............................G..............................Glgeos106 .............................G............C..C...........A..Glgeos90 .............................G............C..C...........A..Pla5-004 .............................G...............C...........A..Glgeos11 .................T...........G...............C...........A..Glcoro28K .................T...........G...............C...........A..Glcoro28 .............................................C..............EninfrMS05 .............................................C..............
190 200 210 220 230 240 ....|....|....|....|....|....|....|....|....|....|....|....|CONSENSUS TTTGGTAGATGTGATGTTTGAGACCGAGGATTGCAACGGATACCCTTCA-GGGCTATTCGGlmoss68/4 .................................................-..........Glmoss25 .................................................-..........PlaIII-004 .................................................-..........Pla4-004 .................................................-..........HelA06 .....N...........................................-..........Glmoss68/7 .................................................-..........HolI-108 .................................................-..........PlaIII-108 .....N...........................................-..N.......Glmoss68W .................................................-..........TraA21 .................................................-..........Glmoss68/2 .................................................-..........Glmoss68/6 .................................................-..........Glfrag05 ..................C.............................T-..........GlcaleRMC ................................................T-..........GlcaleRWZ .......................T........................T-..........HolIII-008 .......................T........................T-..........ZeaA15 ................................................T-..........Glgeos106 ..................C.........................T...T-..........Glgeos90 ..................C.........................T...T-..........Pla5-004 ..................C.........................T...T-..........Glgeos11 ..................C.........................T...T-..........Glcoro28K ..................C.........................-...CA..........Glcoro28 ...............................................T.-..........EninfrMS05 .................................................T..........
Abb. 21 (Fortsetzung)

3 Ergebnisse 58
250 260 270 ....|....|....|....|....|....|...CONSENSUS TCTGATCTCTGATACGTTGCCTTGATGTTGAAA 3‘ (266 bp)Glmoss68/4 ................................. 3‘ (266 bp)Glmoss25 ................................. 3‘ (266 bp)PlaIII-004 ................................. 3‘ (266 bp)Pla4-004 ................................. 3‘ (266 bp)HelA06 ................................. 3‘ (266 bp)Glmoss68/7 ................................. 3‘ (266 bp)HolI-108 ................................. 3‘ (266 bp)PlaIII-108 .............................N... 3‘ (266 bp)Glmoss68W ................................. 3‘ (267 bp)TraA21 ................................. 3‘ (267 bp)Glmoss68/2 ................................. 3‘ (266 bp)Glmoss68/6 ................................. 3‘ (266 bp)Glfrag05 ............A...............C.... 3‘ (266 bp)GlcaleRMC ............A...............C.... 3‘ (270 bp)GlcaleRWZ ............A...............C.... 3‘ (267 bp)HolIII-008 ............A...............C.... 3‘ (267 bp)ZeaA15 ............A...............C.... 3‘ (267 bp)Glgeos106 ............................C.... 3‘ (267 bp)Glgeos90 ............................C.... 3‘ (267 bp)Pla5-004 ............................C.... 3‘ (267 bp)Glgeos11 ............................C.... 3‘ (267 bp)Glcoro28K ............................C.... 3‘ (266 bp)Glcoro28 ................................. 3‘ (265 bp)EninfrMS05 ................................. 3‘ (266 bp)
Abb. 21: (Fortsetzung)
� Glomus coronatum BEG28 ������ � � Entrophospora infrequens MS05 � � � G. mosseae BEG68/2 + BEG68/6 � �� TraA21 � �� G. mosseae BEG68W � � G. mosseae BEG68/7, HolI-108, PlaIII-108 �������� G. mosseae BEG25+BEG68/4,PlaIII-004,Pla4-004, �� � HelA06��� � �� Glomus geosporum BEG90 + BEG106 � ������������ Pla5-004 � � ���� Glomus geosporum BEG11 ������� ���� Glomus coronatum BEG28K � ���� Glomus fragilistratum BEG05 ����� ���� Glomus caledonium RMC658 ������ ZeaA15 �� �� Glomus caledonium RWZ658, HolIII-008
Abb. 22: Phylogenetischer Stammbaum der G. mosseae-Gruppe. Grundlage bildete der Vergleich dersequenzierten Amplikons (fett abgebildet) mit Datenbanksequenzen. (�) entspricht einerSubstitution bei einer Gesamtlänge der Fragments von ca. 266 Nukleotiden. Abkürzungensiehe Tab. 22.

3 Ergebnisse 59
3.3 Quantitativer Nachweis der AMF
Für die Bestimmung der Kopienzahl wurde bei jedem Experiment eine Kalibrierkurve ange-
legt. Hierfür wurden Plasmide verwendet, die das mit dem Primerpaar A2R-B2L amplifizierte
Fragment der LSU rDNA von A. longula bzw. G. mosseae enthielten. Von den Plasmiden
wurden Verdünnungsreihen mit Aqua dest. hergestellt, die zur Berechnung einer Kalibrierge-
raden dienten. Die Plasmidverdünnungen, Proben und Kontrollen wurden in einer TaqMan®
96 well reaction plate vorgelegt. Hierzu wurde der Mastermix C pipettiert und die Kavitäten
mit TaqMan® optical caps verschlossen. Anschließend wurden in einem Abi Prism 7700
sequence detector 40 PCR-Zyklen durchgeführt und die dabei emittierte Fluoreszenz gemes-
sen. Die Software von Applied Biosystems errechnete automatisch für jeden Standard den Ct-
Wert, also diejenige Zykluszahl, bei dem das Fluoreszenz-Signal des Reporterfarbstoff erst-
malig über ein Hintergrundrauschen hinausragte. Die ermittelten Ct-Werte wurden
halblogarithmisch gegen die Kopienzahl der Standards aufgetragen. Die Stärke des Fluores-
zenz-Signals und damit der Ct-Wert waren abhängig von der eingesetzten Kopienzahl der
Ziel-DNA. Die in den Proben vorhandenen Kopienzahlen wurden durch Interpolation berech-
net. Da die Kopienzahl exponentiell zum Ct-Wert abnimmt, wurde das geometrische Mittel
der beiden Doppelbestimmungen ermittelt. Hieraus wurde auf die Kopienzahl je mg Pflan-
zenwurzel zurückgerechnet.
3.3.1 Quantifizierung von A. longula mit der TaqMan® PCR
Für die quantitative spezifische Bestimmung der Kopienzahl von A. longula-rDNA wurden
die beiden Primer Aclong251For und Aclong338Rev sowie die Sonde AclongSonde einge-
setzt. Die Länge des Amplikons betrug 92 bp. Für die Standardreihe wurde eine Verdün-
nungsreihe von klonierter A. longula-DNA hergestellt und mithilfe der ermittelten Ct-Werte
eine Standardkurve angefertigt. Abb. 23 zeigt den Verlauf der TaqMan® PCR mit den einge-
setzten A. longula-Standards. Der Rn-Wert entspricht dem Quotienten aus Emissionsintensität
des Reporterfarbstoffs und Emissionsintensität des passiven Referenzfarbstoffs. ÄRn
enstpricht dem Rn-Wert abzüglich dem Hintergrundsignal der ersten PCR-Zyklen. Die erhal-
tenen Werte sind in Tab. 25 zusammengefaßt und in Abb. 24 aufgetragen. Aus der Standard-
kurve ist der lineare Zusammenhang zwischen eingesetzter Kopienzahl und Signalstärke (Ct-
Wert) klar erkennbar. Mit der Steigung m der Korrelationsgeraden von -3,710 ergab sich eine
Effizienz der TaqMan® PCR von 86%. Der Korrelationskoeffizient der Standardkurve betrug
0,973.
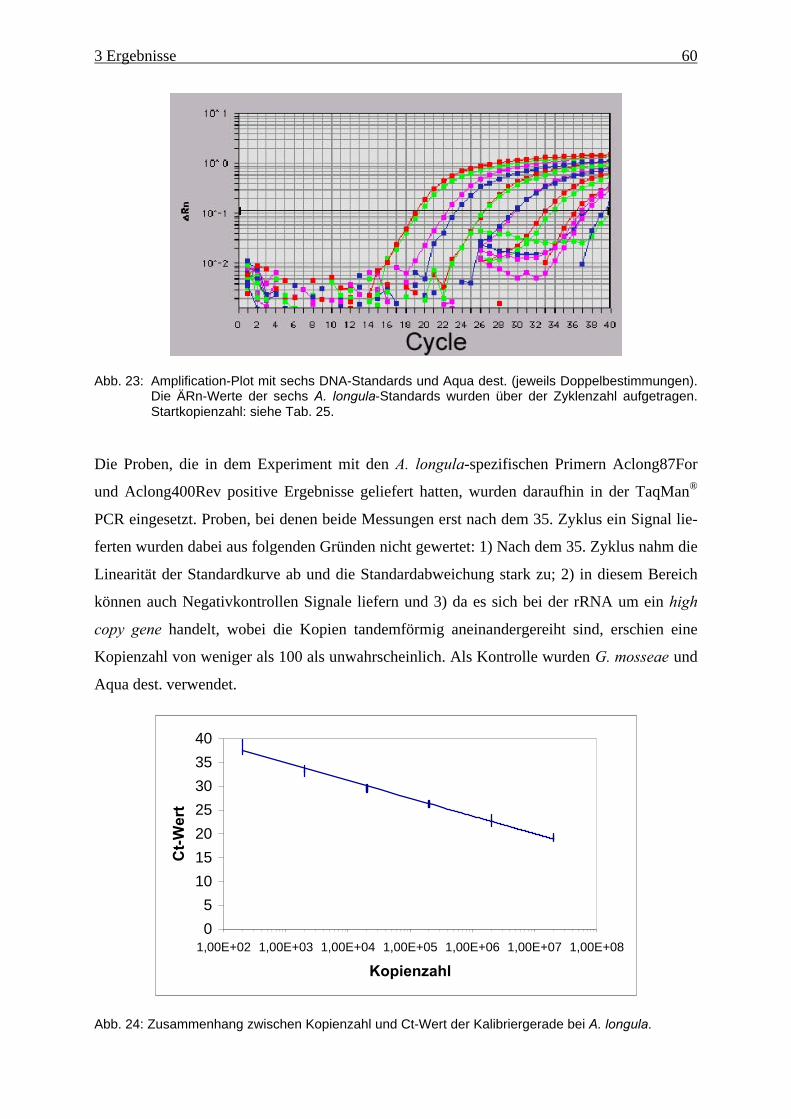
3 Ergebnisse 60
Abb. 23: Amplification-Plot mit sechs DNA-Standards und Aqua dest. (jeweils Doppelbestimmungen).Die ÄRn-Werte der sechs A. longula-Standards wurden über der Zyklenzahl aufgetragen.Startkopienzahl: siehe Tab. 25.
Die Proben, die in dem Experiment mit den A. longula-spezifischen Primern Aclong87For
und Aclong400Rev positive Ergebnisse geliefert hatten, wurden daraufhin in der TaqMan®
PCR eingesetzt. Proben, bei denen beide Messungen erst nach dem 35. Zyklus ein Signal lie-
ferten wurden dabei aus folgenden Gründen nicht gewertet: 1) Nach dem 35. Zyklus nahm die
Linearität der Standardkurve ab und die Standardabweichung stark zu; 2) in diesem Bereich
können auch Negativkontrollen Signale liefern und 3) da es sich bei der rRNA um ein high
copy gene handelt, wobei die Kopien tandemförmig aneinandergereiht sind, erschien eine
Kopienzahl von weniger als 100 als unwahrscheinlich. Als Kontrolle wurden G. mosseae und
Aqua dest. verwendet.
0
5
10
15
20
25
30
35
40
1,00E+02 1,00E+03 1,00E+04 1,00E+05 1,00E+06 1,00E+07 1,00E+08
Kopienzahl
Ct-
Wer
t
Abb. 24: Zusammenhang zwischen Kopienzahl und Ct-Wert der Kalibriergerade bei A. longula.
3 Ergebnisse 61
Die Ergebnisse sind in Tab. 26 zusammengefaßt. Bei A. longula BEG8 und sieben Proben
wurden Werte ermittelt, die eine Quantifizierung zuließen. Demnach befanden sich in der
Positivkontrolle 1889 Kopien je µL, die Proben enthielten zwischen 266 und 5926 Ko-
pien/µL.
Tab. 25: Zusammenstellung der Ct-Werte der eingesetzten Standard-Verdünnungsreihe bei A. longula.
Kopienzahl Ct-Wert 1 Ct-Wert 2 Mittelwert (n=2)
2 · 107 19,01 19,37 19,19
2 · 106 22,30 23,29 22,80
2 · 105 26,29 26,17 26,23
2 · 104 29,64 29,43 29,54
2 · 103 32,82 33,59 33,20
2 · 102 37,35 39,15 38,25
Tab. 26: Anzahl der ermittelten Kopienzahlen bei A. longula, geometrische Mittelwerte (n = 2) je µLDNA-Extrakt und Kopienzahl je mg Wurzel (FG).
Probe Kopienzahl A Kopienzahl B Mittelwert
je µL DNA-Ext.
Kopienzahl
je mg Wurzel
A. longula BEG8 1569 2274 1889 94450
PlaI-008 0 0 0 0
Pla3-004 2899 1283 1929 3445
Pla4-004 0 0 0 0
Pla5-004 775 790 782 3910
TriII-004 583 339 445 2967
TriII-008 0 0 0 0
Tri3-004 309 229 266 13300
Tri4-004 771 676 721 5150
Tri5-004 676 1074 852 5680
Tri5-106 0 0 0 0
HolIII-106 9221 3808 5926 4489
HelA06 0 0 0 0
SolA17 0 0 0 0
Aqua dest. 0 0 0

3 Ergebnisse 62
3.3.2 Quantifizierung von G. mosseae mit der TaqMan® PCR
Für die quantitative spezifische Bestimmung der Kopienzahl von G. mosseae-rDNA wurden
die beiden Primer P1 und P2 sowie die Sonde F1 verwendet. Die Länge des Amplikons betrug
109 bp. Da mit dem Primerpaar Glmoss85For und Glmoss387Rev nicht nur G. mosseae, son-
dern auch nah verwandte Arten amplifiziert wurden, musste zunächst die Bindung der
TaqMan®-Primer und –Sonde überprüft werden. Hierzu wurden die entsprechenden Sequenz-
abschnitte herangezogen und mit dem Primer P1 (Abb. 25) und der Sonde F1 (Abb. 26) ver-
glichen. Der Primer P2 lag außerhalb des sequenzierten Bereichs, so dass über seine Bin-
dungskapazität bei den unterschiedlichen Proben keine Aussage getroffen werden kann. Bei
denjenigen Proben mit AMF-Stämmen, die als G. caledonium (HolIII-008, ZeaA15) bzw.
G. geosporum (Pla5-004) identifiziert wurden, war aufgrund der Abweichungen in der Basen-
paarung eine verminderte Bindung der Primer und Sonden zu erwarten. Dennoch ist eine
Kreuzreaktivität nicht ausgeschlossen. Jedoch dürfte die PCR bei diesen Proben mit stark ver-
ringerter Effizienz abgelaufen sein. Somit kann die ermittelte Kopienzahl nicht als tatsächli-
che G. mosseae-Menge gewertet werden, sondern als Bruchteil der G. geosporum- bzw.
G. caledonium-Kopienzahl bezogen auf G. mosseae.
P1 3‘ ACGTTGCCTATGGGAAGTCC 5‘ ||||||||||||||||||||Gmoss68W 5‘ TGCAACGGATACCCTTCAGG 3‘PlaIII-004 5‘ TGCAACGGATACCCTTCAGG 3‘Pla4-004 5‘ TGCAACGGATACCCTTCAGG 3‘HelA06 5‘ TGCAACGGATACCCTTCAGG 3‘HolI-108 5‘ TGCAACGGATACCCTTCAGG 3‘PlaIII-108 5‘ TGCAACGGATACCCTTCAGG 3‘TraA21 5‘ TGCAACGGATACCCTTCAGG 3‘HolIII-008 5‘ TGCAACGGATACCCTTCTGG 3‘ZeaA15 5‘ TGCAACGGATACCCTTCTGG 3‘Pla5-004 5‘ TGCAACGGATACCTTTCTGG 3‘
Abb. 25: Kontrolle der Bindung des Primers P1 an die ermittelten Sequenz-abschnitte. Abweichende Basen sind unterlegt.
Für die Standardreihe wurde eine Verdünnungsreihe von klonierter G. mosseae-DNA herge-
stellt und mithilfe der ermittelten Ct-Werte eine Standardkurve angefertigt. Abb. 27 zeigt den
Verlauf der TaqMan® PCR mit den eingesetzten Standards. Die erhaltenen Werte sind in Tab.
27 zusammengefaßt und in Abb. 28 aufgetragen. Mit der Steigung m der Korrelationsgeraden
von -3,513 ergab sich eine Effizienz der TaqMan® PCR von 93%. Der Korrelationskoeffizient
der Standardkurve betrug 0,979.
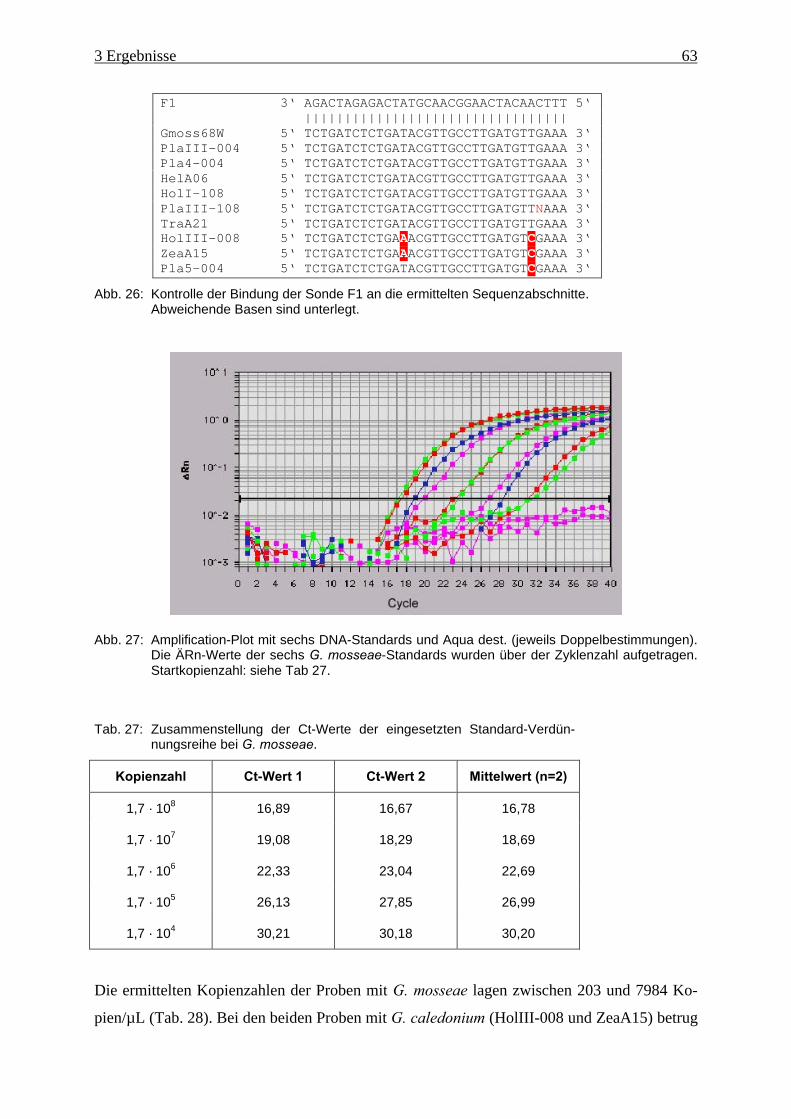
3 Ergebnisse 63
F1 3‘ AGACTAGAGACTATGCAACGGAACTACAACTTT 5‘ |||||||||||||||||||||||||||||||||Gmoss68W 5‘ TCTGATCTCTGATACGTTGCCTTGATGTTGAAA 3‘PlaIII-004 5‘ TCTGATCTCTGATACGTTGCCTTGATGTTGAAA 3‘Pla4-004 5‘ TCTGATCTCTGATACGTTGCCTTGATGTTGAAA 3‘HelA06 5‘ TCTGATCTCTGATACGTTGCCTTGATGTTGAAA 3‘HolI-108 5‘ TCTGATCTCTGATACGTTGCCTTGATGTTGAAA 3‘PlaIII-108 5‘ TCTGATCTCTGATACGTTGCCTTGATGTTNAAA 3‘TraA21 5‘ TCTGATCTCTGATACGTTGCCTTGATGTTGAAA 3‘HolIII-008 5‘ TCTGATCTCTGAAACGTTGCCTTGATGTCGAAA 3‘ZeaA15 5‘ TCTGATCTCTGAAACGTTGCCTTGATGTCGAAA 3‘Pla5-004 5‘ TCTGATCTCTGATACGTTGCCTTGATGTCGAAA 3‘
Abb. 26: Kontrolle der Bindung der Sonde F1 an die ermittelten Sequenzabschnitte.Abweichende Basen sind unterlegt.
Abb. 27: Amplification-Plot mit sechs DNA-Standards und Aqua dest. (jeweils Doppelbestimmungen).Die ÄRn-Werte der sechs G. mosseae-Standards wurden über der Zyklenzahl aufgetragen.Startkopienzahl: siehe Tab 27.
Tab. 27: Zusammenstellung der Ct-Werte der eingesetzten Standard-Verdün-nungsreihe bei G. mosseae.
Kopienzahl Ct-Wert 1 Ct-Wert 2 Mittelwert (n=2)
1,7 · 108 16,89 16,67 16,78
1,7 · 107 19,08 18,29 18,69
1,7 · 106 22,33 23,04 22,69
1,7 · 105 26,13 27,85 26,99
1,7 · 104 30,21 30,18 30,20
Die ermittelten Kopienzahlen der Proben mit G. mosseae lagen zwischen 203 und 7984 Ko-
pien/µL (Tab. 28). Bei den beiden Proben mit G. caledonium (HolIII-008 und ZeaA15) betrug
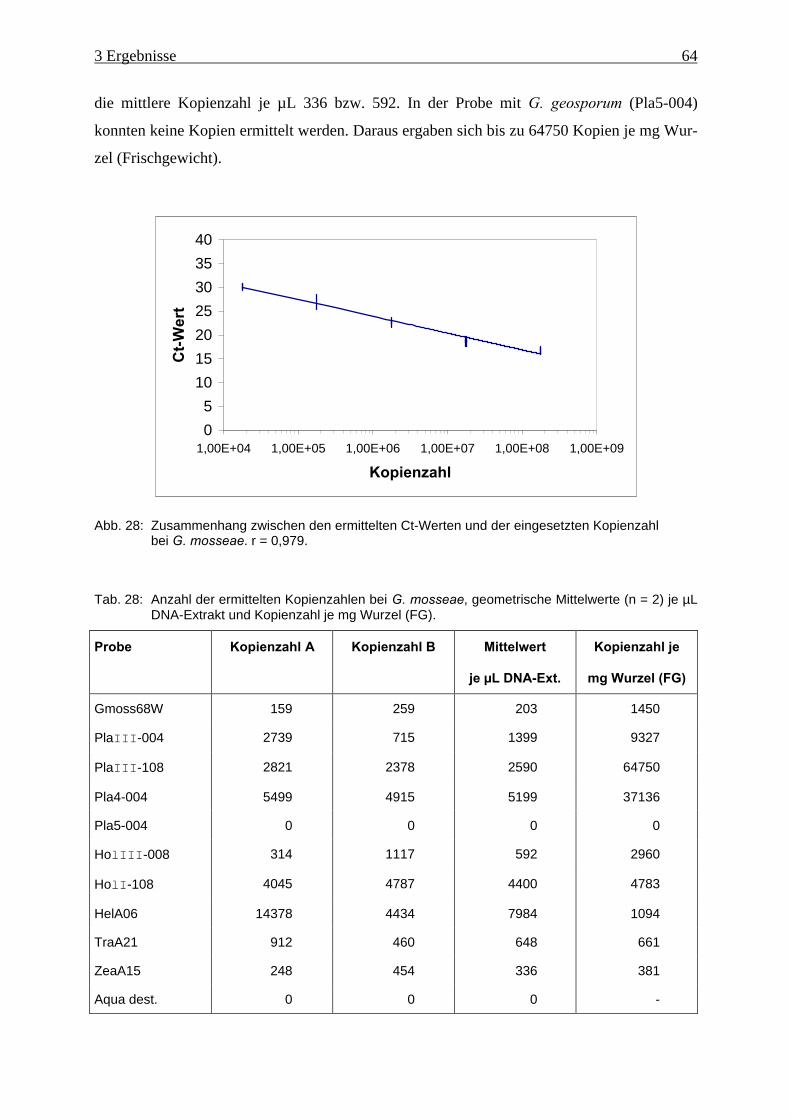
3 Ergebnisse 64
die mittlere Kopienzahl je µL 336 bzw. 592. In der Probe mit G. geosporum (Pla5-004)
konnten keine Kopien ermittelt werden. Daraus ergaben sich bis zu 64750 Kopien je mg Wur-
zel (Frischgewicht).
0
5
10
15
20
25
30
35
40
1,00E+04 1,00E+05 1,00E+06 1,00E+07 1,00E+08 1,00E+09
Kopienzahl
Ct-
Wer
t
Abb. 28: Zusammenhang zwischen den ermittelten Ct-Werten und der eingesetzten Kopienzahlbei G. mosseae. r = 0,979.
Tab. 28: Anzahl der ermittelten Kopienzahlen bei G. mosseae, geometrische Mittelwerte (n = 2) je µLDNA-Extrakt und Kopienzahl je mg Wurzel (FG).
Probe Kopienzahl A Kopienzahl B Mittelwert
je µL DNA-Ext.
Kopienzahl je
mg Wurzel (FG)
Gmoss68W 159 259 203 1450
PlaIII-004 2739 715 1399 9327
PlaIII-108 2821 2378 2590 64750
Pla4-004 5499 4915 5199 37136
Pla5-004 0 0 0 0
HolIII-008 314 1117 592 2960
HolI-108 4045 4787 4400 4783
HelA06 14378 4434 7984 1094
TraA21 912 460 648 661
ZeaA15 248 454 336 381
Aqua dest. 0 0 0 -

3 Ergebnisse 65
3.4 Zusammenfassung der molekularbiologischen und morphologischen
Ergebnisse
Aus 65 Proben von Pflanzenwurzeln wurde DNA extrahiert (Tab. 29). Von diesen DNA-Ex-
trakten konnten 56 in einer PCR mit unspezifischen Primern amplifiziert werden. In einer
PCR mit A. longula-spezifischen Primern ergaben fünf dieser positiv getesteten Proben (9%)
eine starke Bande, bei weiteren acht Proben (14%) waren schwache Banden erkennbar. Eine
quantitative PCR ergab zwischen 3910 und 13300 Kopien je mg Wurzelmasse bei den starken
Signalen und bis zu 5680 Kopien je mg bei den schwächeren Signalen. Allerdings konnten bei
sechs der acht Proben mit schwachen Signalen keine Kopien nachgewiesen werden.
In neun (16%) der Proben konnte mit den spezifischen Primern Glmoss85For und
Glmoss387Rev ein Produkt erhalten werden. Diese Produkte wurden sequenziert und durch
den Vergleich mit der Datenbank konnten sechs als G. mosseae, zwei als G. caledonium und
eines als G. geosporum identifiziert werden. Mit einer quantitativen PCR wurde die Kopien-
zahl je mg Wurzelmasse bestimmt. Bei den G. mosseae-Proben wurden zwischen 661 und
64750 Kopien/mg nachgewiesen, bei den beiden G. caledonium-Proben zwischen 381 und
2960 (G. mosseae-Äquivalente). Bei der G. caledonium-Probe konnten keine Kopien gemes-
sen werden.
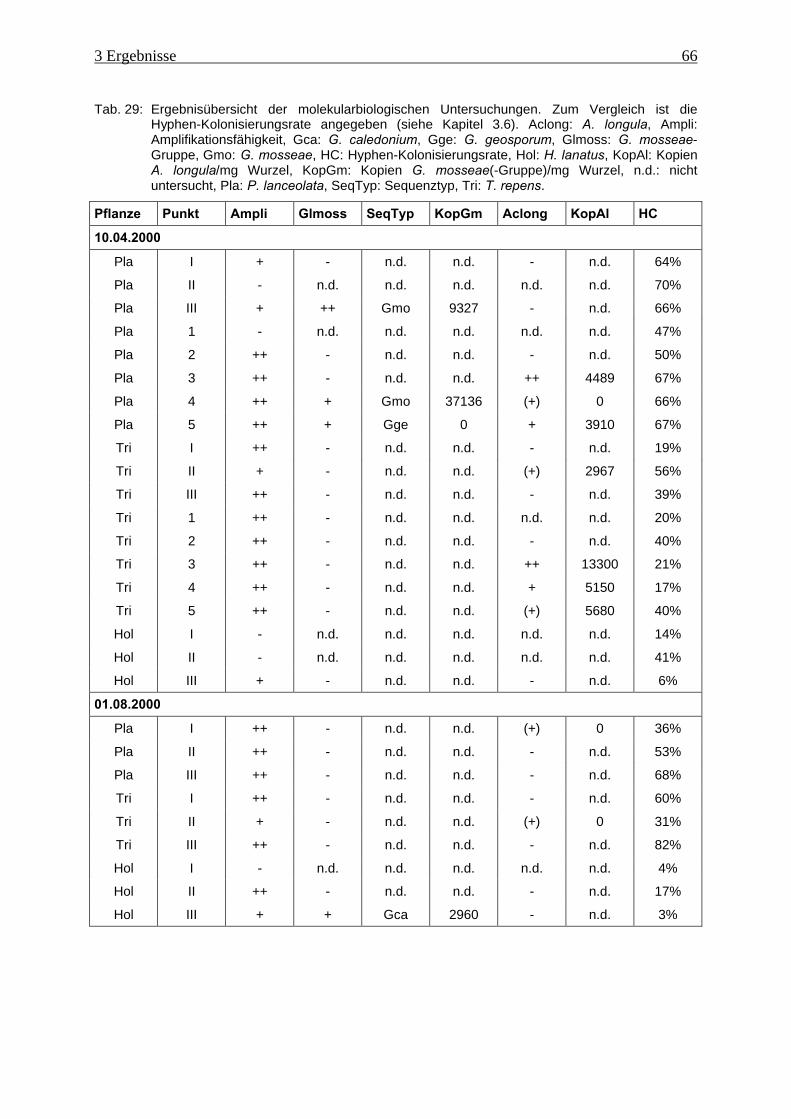
3 Ergebnisse 66
Tab. 29: Ergebnisübersicht der molekularbiologischen Untersuchungen. Zum Vergleich ist dieHyphen-Kolonisierungsrate angegeben (siehe Kapitel 3.6). Aclong: A. longula, Ampli:Amplifikationsfähigkeit, Gca: G. caledonium, Gge: G. geosporum, Glmoss: G. mosseae-Gruppe, Gmo: G. mosseae, HC: Hyphen-Kolonisierungsrate, Hol: H. lanatus, KopAl: KopienA. longula/mg Wurzel, KopGm: Kopien G. mosseae(-Gruppe)/mg Wurzel, n.d.: nichtuntersucht, Pla: P. lanceolata, SeqTyp: Sequenztyp, Tri: T. repens.
Pflanze Punkt Ampli Glmoss SeqTyp KopGm Aclong KopAl HC
10.04.2000
Pla I + - n.d. n.d. - n.d. 64%
Pla II - n.d. n.d. n.d. n.d. n.d. 70%
Pla III + ++ Gmo 9327 - n.d. 66%
Pla 1 - n.d. n.d. n.d. n.d. n.d. 47%
Pla 2 ++ - n.d. n.d. - n.d. 50%
Pla 3 ++ - n.d. n.d. ++ 4489 67%
Pla 4 ++ + Gmo 37136 (+) 0 66%
Pla 5 ++ + Gge 0 + 3910 67%
Tri I ++ - n.d. n.d. - n.d. 19%
Tri II + - n.d. n.d. (+) 2967 56%
Tri III ++ - n.d. n.d. - n.d. 39%
Tri 1 ++ - n.d. n.d. n.d. n.d. 20%
Tri 2 ++ - n.d. n.d. - n.d. 40%
Tri 3 ++ - n.d. n.d. ++ 13300 21%
Tri 4 ++ - n.d. n.d. + 5150 17%
Tri 5 ++ - n.d. n.d. (+) 5680 40%
Hol I - n.d. n.d. n.d. n.d. n.d. 14%
Hol II - n.d. n.d. n.d. n.d. n.d. 41%
Hol III + - n.d. n.d. - n.d. 6%
01.08.2000
Pla I ++ - n.d. n.d. (+) 0 36%
Pla II ++ - n.d. n.d. - n.d. 53%
Pla III ++ - n.d. n.d. - n.d. 68%
Tri I ++ - n.d. n.d. - n.d. 60%
Tri II + - n.d. n.d. (+) 0 31%
Tri III ++ - n.d. n.d. - n.d. 82%
Hol I - n.d. n.d. n.d. n.d. n.d. 4%
Hol II ++ - n.d. n.d. - n.d. 17%
Hol III + + Gca 2960 - n.d. 3%

3 Ergebnisse 67
Tab. 29: (Fortsetzung)
11.06.2001
Pla I + - n.d. n.d. - n.d. 33%
Pla II + - n.d. n.d. - n.d. 11%
Pla III - n.d. n.d. n.d. n.d. n.d. 6%
Tri I + - n.d. n.d. - n.d. 29%
Tri II - - n.d. n.d. - n.d. 10%
Tri III ++ - n.d. n.d. - n.d. 36%
Hol I + - n.d. n.d. - n.d. 18%
Hol II + - n.d. n.d. - n.d. 11%
Hol III + - n.d. n.d. ++ 4489 20%
22.08.2001
Pla I + - n.d. n.d. - n.d. 32%
Pla II + n.d. n.d. n.d. - n.d. 37%
Pla III + ++ Gmo 64750 - n.d. 56%
Pla 1 + - n.d. n.d. - n.d. 5%
Pla 2 ++ - n.d. n.d. - n.d. 17%
Pla 3 + - n.d. n.d. - n.d. 24%
Pla 4 + - n.d. n.d. - n.d. 38%
Pla 5 ++ - n.d. n.d. - n.d. 26%
Tri I ++ - n.d. n.d. - n.d. 27%
Tri II ++ - n.d. n.d. - n.d. 19%
Tri III + - n.d. n.d. - n.d. 17%
Tri 1 ++ - n.d. n.d. - n.d. 23%
Tri 2 + - n.d. n.d. - n.d. 26%
Tri 3 ++ - n.d. n.d. - n.d. 13%
Tri 4 ++ - n.d. n.d. - n.d. 27%
Tri 5 ++ - n.d. n.d. (+) 0 23%
Hol I + + Gmo 4783 - n.d. 0%
Hol II + - n.d. n.d. - n.d. 7%
Hol III ++ - n.d. n.d. - n.d. 16%
27.08.2001
Zea A15 + ++ Gca 381 - n.d. 4%
Sol A19 ++ - n.d. n.d. - n.d. 9%
Sol A17 ++ - n.d. n.d. (+) 0 13%
Hel A06 ++ ++ Gmo 1094 - n.d. 31%
Hel A06 ++ - n.d. n.d. (+) 0 12%
Tra A21 - + Gmo 661 n.d. n.d. 50%
Tra A16 (+) - n.d. n.d. n.d. n.d. 17%
Zea A15 (+) - n.d. n.d. n.d. n.d. 6%

3 Ergebnisse 68
3.5 Bestimmung der Mykorrhizapilze anhand der Sporenmorphologie
Neben der Überprüfung der Referenzkulturen aufgrund der Sporenbildung wurden auch Frei-
landproben von W02 und W21 stichprobenhaft auf vorhandene Sporen untersucht. Die iso-
lierten Sporen wurden zunächst nach ihrer Färbung in helle, bräunliche und dunkle Sporen
getrennt. Anschließend erfolgte die Untersuchung der Sporenwände. Die Ergebnisse der mor-
phologischen Bestimmung sind in Tab. 30 wiedergegeben. Demnach konnten 6 verschiedene
Sporentypen nachgewiesen werden. Eine exakte Artbestimmung war jedoch nicht möglich
(s. Kapitel 2.2.11). Die überwiegende Mehrzahl der gefundenen Sporen (ca. 70 %) gehörten
zu G. mosseae (Abb. 29) oder waren dieser Art sehr ähnlich. Gigaspora-Sporen konnten nicht
direkt nachgewiesen werden, jedoch fanden sich mehrere Hilfszellen.
Tab. 30: Ergebnis der morphologischen Sporenuntersuchung.
Häufigkeit Sporentyp
ca. 70 % Glomus mosseae-ähnlich
ca. 10 % Glomus geosporum-ähnlich
ca. 5 % Glomus pubescens- bzw. G. arborense-ähnlich
ca. 5 % Archaeospora leptoticha (Glomus leptotichum)-ähnlich
ca. 5 % Acaulospora delicata-ähnlich
ca. 5 % Gigaspora sp. (kein Sporennachweis aber Vorkommen von Hilfszellen)
3.6 Bestimmung des Mykorrhizierungsgrades in Freilandproben
Abb. 30 zeigt mit Trypanblau gefärbte Pilzhyphen, mehrere Arbuskel sowie ein Vesikel. Im
Hintergrund sind ungefärbte Pflanzenzellen zu erkennen. Die Bonitierung erfolgte je Probe an
ca. 30 bis 100 Stellen (n) für die Flächen W02 (Tab. 31) und W21 (Tab. 32). Während Pilz-
hyphen und Vesikel leicht zu erkennen waren, kann die Erfassung der Arbuskel unter Um-
ständen unvollständig gewesen sein, da ihre Identifizierung vor allem in dickeren Wurzelbe-
reichen, wo sie häufig nur als diffuse „Wolke“ zu erkennen waren, Probleme bereitete. Insge-
samt ist die mikroskopische Bonitierung der Mykorrhizierung gefärbter Präparate subjektiv
und nur bedingt aussagekräftig, was sich auch in den hohen Standardabweichungen wider-
spiegelt. Aus diesem Grund wurde auf eine statistische Auswertung verzichtet.
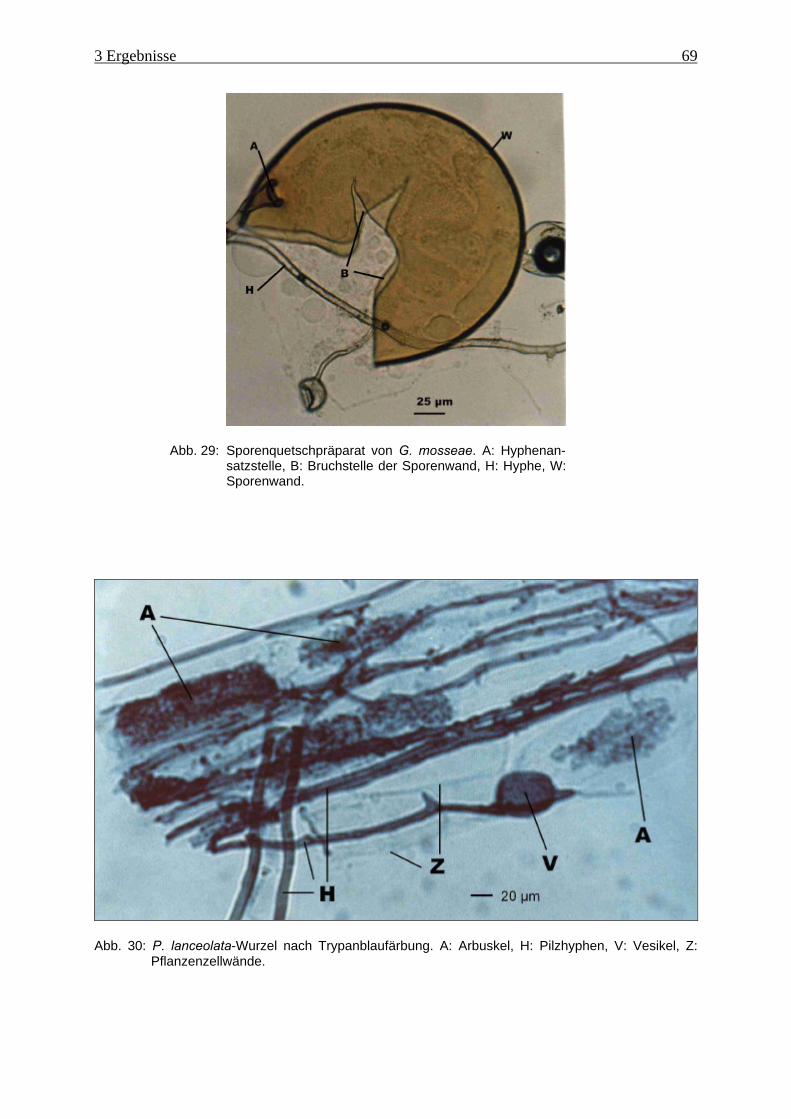
3 Ergebnisse 69
Abb. 29: Sporenquetschpräparat von G. mosseae. A: Hyphenan-satzstelle, B: Bruchstelle der Sporenwand, H: Hyphe, W:Sporenwand.
Abb. 30: P. lanceolata-Wurzel nach Trypanblaufärbung. A: Arbuskel, H: Pilzhyphen, V: Vesikel, Z:Pflanzenzellwände.
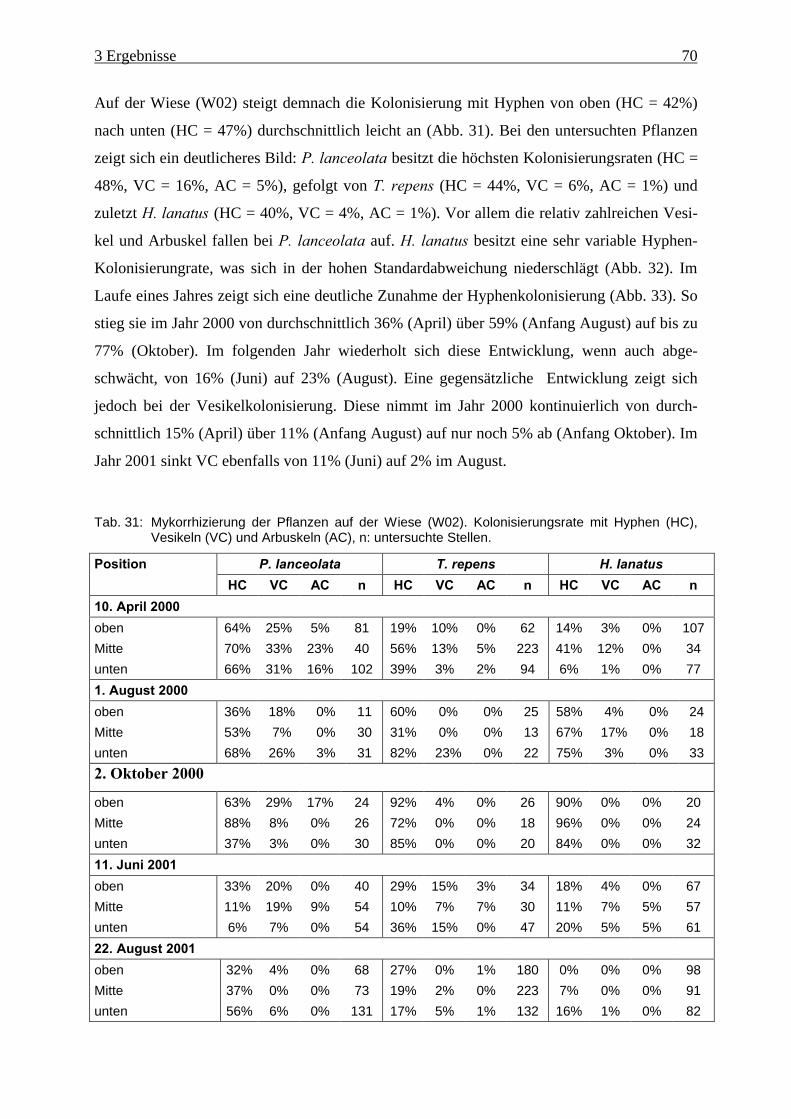
3 Ergebnisse 70
Auf der Wiese (W02) steigt demnach die Kolonisierung mit Hyphen von oben (HC = 42%)
nach unten (HC = 47%) durchschnittlich leicht an (Abb. 31). Bei den untersuchten Pflanzen
zeigt sich ein deutlicheres Bild: P. lanceolata besitzt die höchsten Kolonisierungsraten (HC =
48%, VC = 16%, AC = 5%), gefolgt von T. repens (HC = 44%, VC = 6%, AC = 1%) und
zuletzt H. lanatus (HC = 40%, VC = 4%, AC = 1%). Vor allem die relativ zahlreichen Vesi-
kel und Arbuskel fallen bei P. lanceolata auf. H. lanatus besitzt eine sehr variable Hyphen-
Kolonisierungrate, was sich in der hohen Standardabweichung niederschlägt (Abb. 32). Im
Laufe eines Jahres zeigt sich eine deutliche Zunahme der Hyphenkolonisierung (Abb. 33). So
stieg sie im Jahr 2000 von durchschnittlich 36% (April) über 59% (Anfang August) auf bis zu
77% (Oktober). Im folgenden Jahr wiederholt sich diese Entwicklung, wenn auch abge-
schwächt, von 16% (Juni) auf 23% (August). Eine gegensätzliche Entwicklung zeigt sich
jedoch bei der Vesikelkolonisierung. Diese nimmt im Jahr 2000 kontinuierlich von durch-
schnittlich 15% (April) über 11% (Anfang August) auf nur noch 5% ab (Anfang Oktober). Im
Jahr 2001 sinkt VC ebenfalls von 11% (Juni) auf 2% im August.
Tab. 31: Mykorrhizierung der Pflanzen auf der Wiese (W02). Kolonisierungsrate mit Hyphen (HC),Vesikeln (VC) und Arbuskeln (AC), n: untersuchte Stellen.
P. lanceolata T. repens H. lanatusPosition
HC VC AC n HC VC AC n HC VC AC n
10. April 2000
oben
Mitte
unten
64%
70%
66%
25%
33%
31%
5%
23%
16%
81
40
102
19%
56%
39%
10%
13%
3%
0%
5%
2%
62
223
94
14%
41%
6%
3%
12%
1%
0%
0%
0%
107
34
77
1. August 2000
oben
Mitte
unten
36%
53%
68%
18%
7%
26%
0%
0%
3%
11
30
31
60%
31%
82%
0%
0%
23%
0%
0%
0%
25
13
22
58%
67%
75%
4%
17%
3%
0%
0%
0%
24
18
33
2. Oktober 2000
oben
Mitte
unten
63%
88%
37%
29%
8%
3%
17%
0%
0%
24
26
30
92%
72%
85%
4%
0%
0%
0%
0%
0%
26
18
20
90%
96%
84%
0%
0%
0%
0%
0%
0%
20
24
32
11. Juni 2001
oben
Mitte
unten
33%
11%
6%
20%
19%
7%
0%
9%
0%
40
54
54
29%
10%
36%
15%
7%
15%
3%
7%
0%
34
30
47
18%
11%
20%
4%
7%
5%
0%
5%
5%
67
57
61
22. August 2001
oben
Mitte
unten
32%
37%
56%
4%
0%
6%
0%
0%
0%
68
73
131
27%
19%
17%
0%
2%
5%
1%
0%
1%
180
223
132
0%
7%
16%
0%
0%
1%
0%
0%
0%
98
91
82
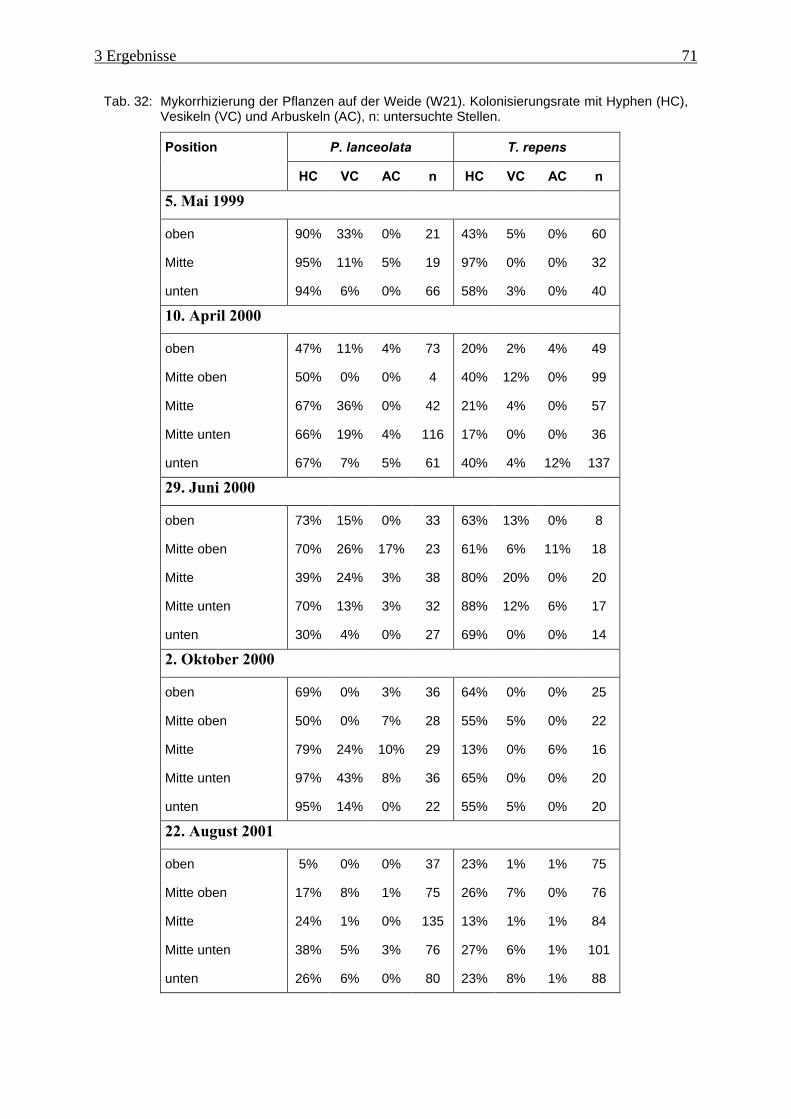
3 Ergebnisse 71
Tab. 32: Mykorrhizierung der Pflanzen auf der Weide (W21). Kolonisierungsrate mit Hyphen (HC),Vesikeln (VC) und Arbuskeln (AC), n: untersuchte Stellen.
P. lanceolata T. repensPosition
HC VC AC n HC VC AC n
5. Mai 1999
oben
Mitte
unten
90%
95%
94%
33%
11%
6%
0%
5%
0%
21
19
66
43%
97%
58%
5%
0%
3%
0%
0%
0%
60
32
40
10. April 2000
oben
Mitte oben
Mitte
Mitte unten
unten
47%
50%
67%
66%
67%
11%
0%
36%
19%
7%
4%
0%
0%
4%
5%
73
4
42
116
61
20%
40%
21%
17%
40%
2%
12%
4%
0%
4%
4%
0%
0%
0%
12%
49
99
57
36
137
29. Juni 2000
oben
Mitte oben
Mitte
Mitte unten
unten
73%
70%
39%
70%
30%
15%
26%
24%
13%
4%
0%
17%
3%
3%
0%
33
23
38
32
27
63%
61%
80%
88%
69%
13%
6%
20%
12%
0%
0%
11%
0%
6%
0%
8
18
20
17
14
2. Oktober 2000
oben
Mitte oben
Mitte
Mitte unten
unten
69%
50%
79%
97%
95%
0%
0%
24%
43%
14%
3%
7%
10%
8%
0%
36
28
29
36
22
64%
55%
13%
65%
55%
0%
5%
0%
0%
5%
0%
0%
6%
0%
0%
25
22
16
20
20
22. August 2001
oben
Mitte oben
Mitte
Mitte unten
unten
5%
17%
24%
38%
26%
0%
8%
1%
5%
6%
0%
1%
0%
3%
0%
37
75
135
76
80
23%
26%
13%
27%
23%
1%
7%
1%
6%
8%
1%
0%
1%
1%
1%
75
76
84
101
88
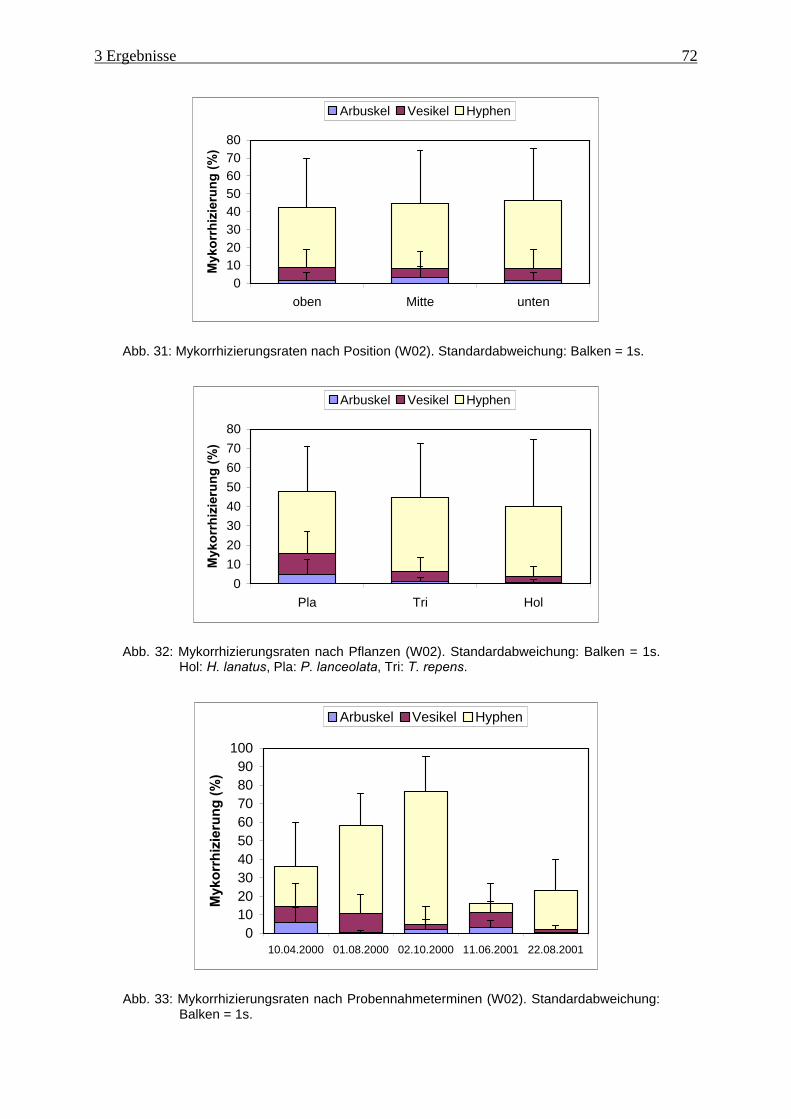
3 Ergebnisse 72
0
1020
3040506070
80
oben Mitte unten
Myk
orr
hiz
ieru
ng
(%
)
Arbuskel Vesikel Hyphen
Abb. 31: Mykorrhizierungsraten nach Position (W02). Standardabweichung: Balken = 1s.
0
10
20
30
40
50
60
70
80
Pla Tri Hol
Myk
orr
hiz
ieru
ng
(%
)
Arbuskel Vesikel Hyphen
Abb. 32: Mykorrhizierungsraten nach Pflanzen (W02). Standardabweichung: Balken = 1s.Hol: H. lanatus, Pla: P. lanceolata, Tri: T. repens.
0102030405060708090
100
10.04.2000 01.08.2000 02.10.2000 11.06.2001 22.08.2001
Myk
orr
hiz
ieru
ng
(%
)
Arbuskel Vesikel Hyphen
Abb. 33: Mykorrhizierungsraten nach Probennahmeterminen (W02). Standardabweichung:Balken = 1s.
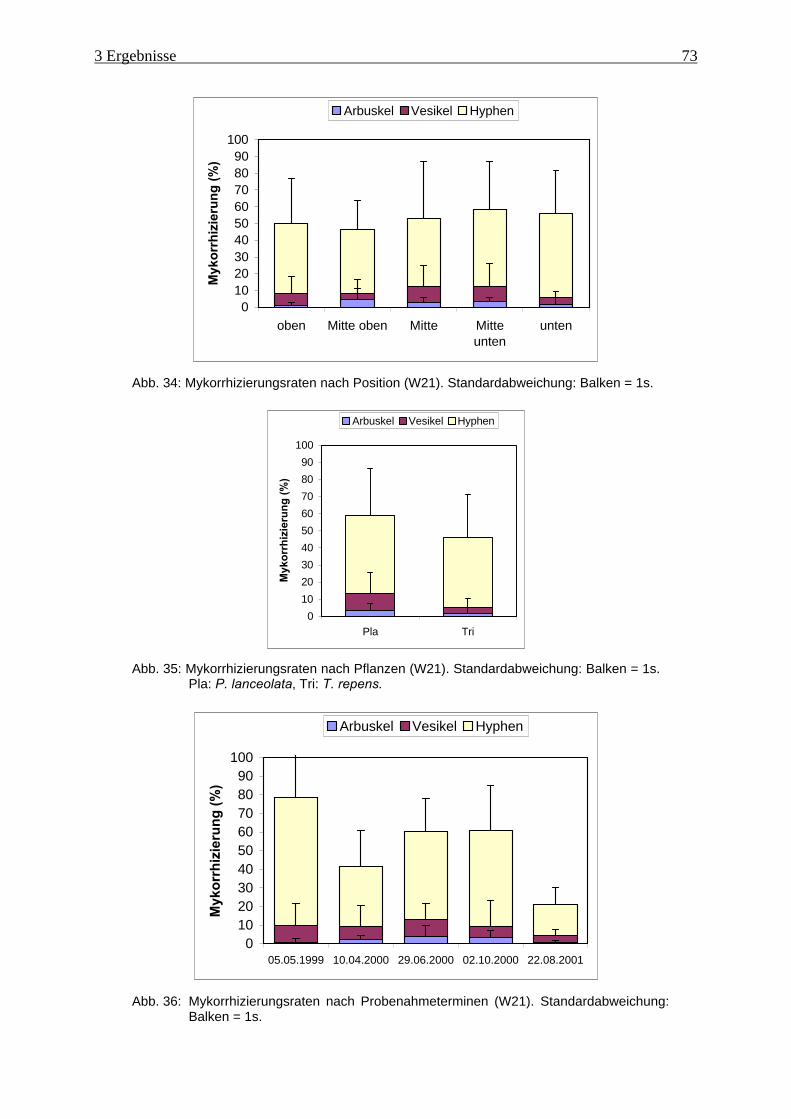
3 Ergebnisse 73
0102030405060708090
100
oben Mitte oben Mitte Mitteunten
unten
Myk
orr
hiz
ieru
ng
(%
)
Arbuskel Vesikel Hyphen
Abb. 34: Mykorrhizierungsraten nach Position (W21). Standardabweichung: Balken = 1s.
0
10
20
30
40
50
60
70
80
90
100
Pla Tri
Myk
orr
hiz
ieru
ng
(%
)
Arbuskel Vesikel Hyphen
Abb. 35: Mykorrhizierungsraten nach Pflanzen (W21). Standardabweichung: Balken = 1s.Pla: P. lanceolata, Tri: T. repens.
0102030405060708090
100
05.05.1999 10.04.2000 29.06.2000 02.10.2000 22.08.2001
Myk
orr
hiz
ieru
ng
(%
)
Arbuskel Vesikel Hyphen
Abb. 36: Mykorrhizierungsraten nach Probenahmeterminen (W21). Standardabweichung:Balken = 1s.
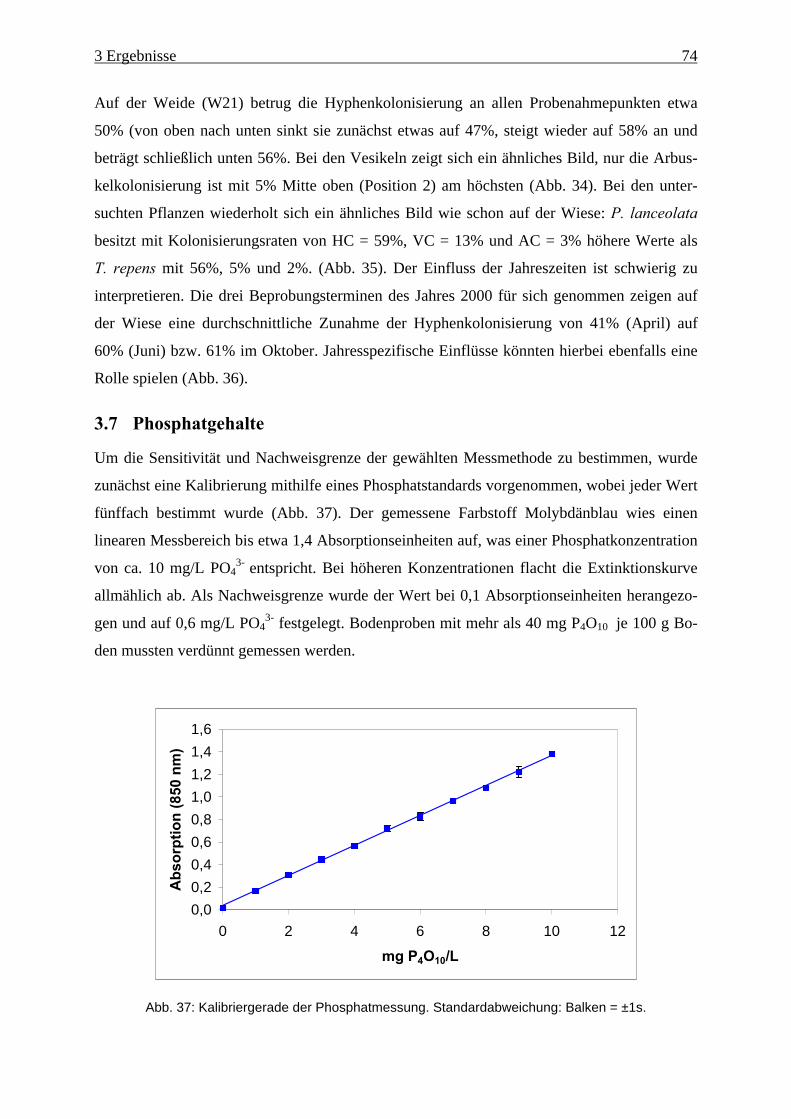
3 Ergebnisse 74
Auf der Weide (W21) betrug die Hyphenkolonisierung an allen Probenahmepunkten etwa
50% (von oben nach unten sinkt sie zunächst etwas auf 47%, steigt wieder auf 58% an und
beträgt schließlich unten 56%. Bei den Vesikeln zeigt sich ein ähnliches Bild, nur die Arbus-
kelkolonisierung ist mit 5% Mitte oben (Position 2) am höchsten (Abb. 34). Bei den unter-
suchten Pflanzen wiederholt sich ein ähnliches Bild wie schon auf der Wiese: P. lanceolata
besitzt mit Kolonisierungsraten von HC = 59%, VC = 13% und AC = 3% höhere Werte als
T. repens mit 56%, 5% und 2%. (Abb. 35). Der Einfluss der Jahreszeiten ist schwierig zu
interpretieren. Die drei Beprobungsterminen des Jahres 2000 für sich genommen zeigen auf
der Wiese eine durchschnittliche Zunahme der Hyphenkolonisierung von 41% (April) auf
60% (Juni) bzw. 61% im Oktober. Jahresspezifische Einflüsse könnten hierbei ebenfalls eine
Rolle spielen (Abb. 36).
3.7 Phosphatgehalte
Um die Sensitivität und Nachweisgrenze der gewählten Messmethode zu bestimmen, wurde
zunächst eine Kalibrierung mithilfe eines Phosphatstandards vorgenommen, wobei jeder Wert
fünffach bestimmt wurde (Abb. 37). Der gemessene Farbstoff Molybdänblau wies einen
linearen Messbereich bis etwa 1,4 Absorptionseinheiten auf, was einer Phosphatkonzentration
von ca. 10 mg/L PO43- entspricht. Bei höheren Konzentrationen flacht die Extinktionskurve
allmählich ab. Als Nachweisgrenze wurde der Wert bei 0,1 Absorptionseinheiten herangezo-
gen und auf 0,6 mg/L PO43- festgelegt. Bodenproben mit mehr als 40 mg P4O10 je 100 g Bo-
den mussten verdünnt gemessen werden.
0,0
0,2
0,4
0,6
0,8
1,0
1,2
1,4
1,6
0 2 4 6 8 10 12
mg P4O10/L
Ab
sorp
tio
n (
850
nm
)
Abb. 37: Kalibriergerade der Phosphatmessung. Standardabweichung: Balken = ±1s.
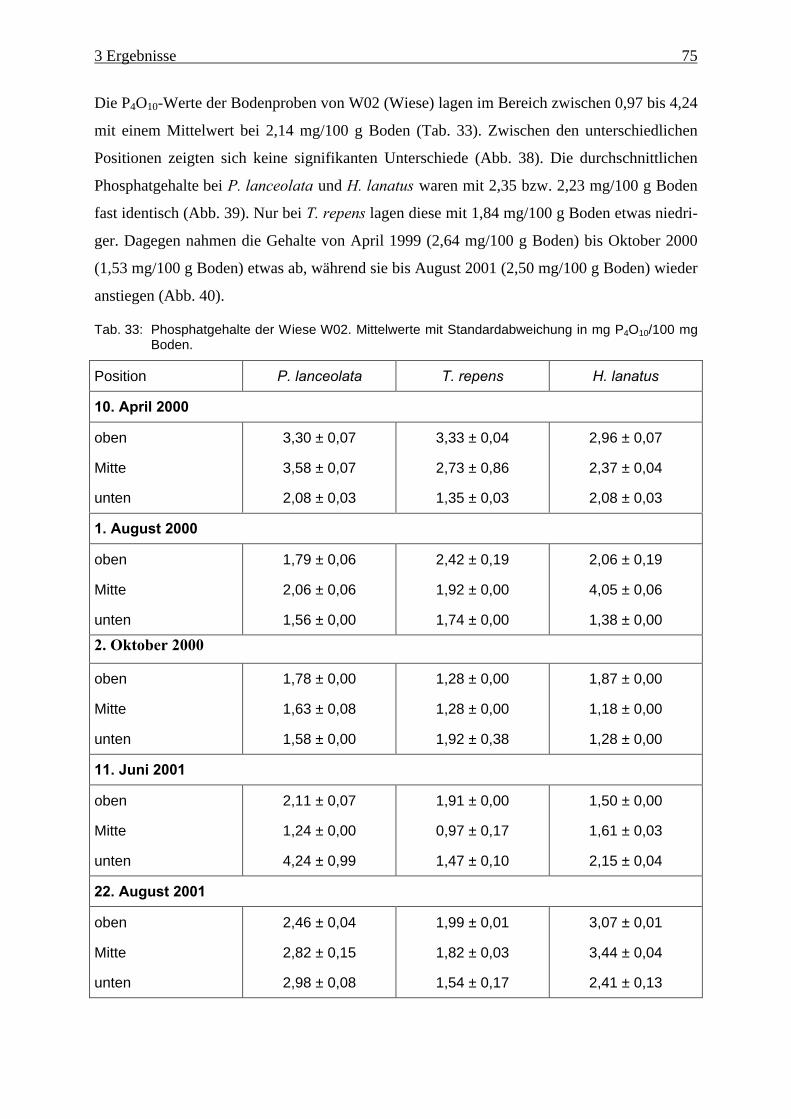
3 Ergebnisse 75
Die P4O10-Werte der Bodenproben von W02 (Wiese) lagen im Bereich zwischen 0,97 bis 4,24
mit einem Mittelwert bei 2,14 mg/100 g Boden (Tab. 33). Zwischen den unterschiedlichen
Positionen zeigten sich keine signifikanten Unterschiede (Abb. 38). Die durchschnittlichen
Phosphatgehalte bei P. lanceolata und H. lanatus waren mit 2,35 bzw. 2,23 mg/100 g Boden
fast identisch (Abb. 39). Nur bei T. repens lagen diese mit 1,84 mg/100 g Boden etwas niedri-
ger. Dagegen nahmen die Gehalte von April 1999 (2,64 mg/100 g Boden) bis Oktober 2000
(1,53 mg/100 g Boden) etwas ab, während sie bis August 2001 (2,50 mg/100 g Boden) wieder
anstiegen (Abb. 40).
Tab. 33: Phosphatgehalte der Wiese W02. Mittelwerte mit Standardabweichung in mg P4O10/100 mgBoden.
Position P. lanceolata T. repens H. lanatus
10. April 2000
oben
Mitte
unten
3,30 ± 0,07
3,58 ± 0,07
2,08 ± 0,03
3,33 ± 0,04
2,73 ± 0,86
1,35 ± 0,03
2,96 ± 0,07
2,37 ± 0,04
2,08 ± 0,03
1. August 2000
oben
Mitte
unten
1,79 ± 0,06
2,06 ± 0,06
1,56 ± 0,00
2,42 ± 0,19
1,92 ± 0,00
1,74 ± 0,00
2,06 ± 0,19
4,05 ± 0,06
1,38 ± 0,00
2. Oktober 2000
oben
Mitte
unten
1,78 ± 0,00
1,63 ± 0,08
1,58 ± 0,00
1,28 ± 0,00
1,28 ± 0,00
1,92 ± 0,38
1,87 ± 0,00
1,18 ± 0,00
1,28 ± 0,00
11. Juni 2001
oben
Mitte
unten
2,11 ± 0,07
1,24 ± 0,00
4,24 ± 0,99
1,91 ± 0,00
0,97 ± 0,17
1,47 ± 0,10
1,50 ± 0,00
1,61 ± 0,03
2,15 ± 0,04
22. August 2001
oben
Mitte
unten
2,46 ± 0,04
2,82 ± 0,15
2,98 ± 0,08
1,99 ± 0,01
1,82 ± 0,03
1,54 ± 0,17
3,07 ± 0,01
3,44 ± 0,04
2,41 ± 0,13
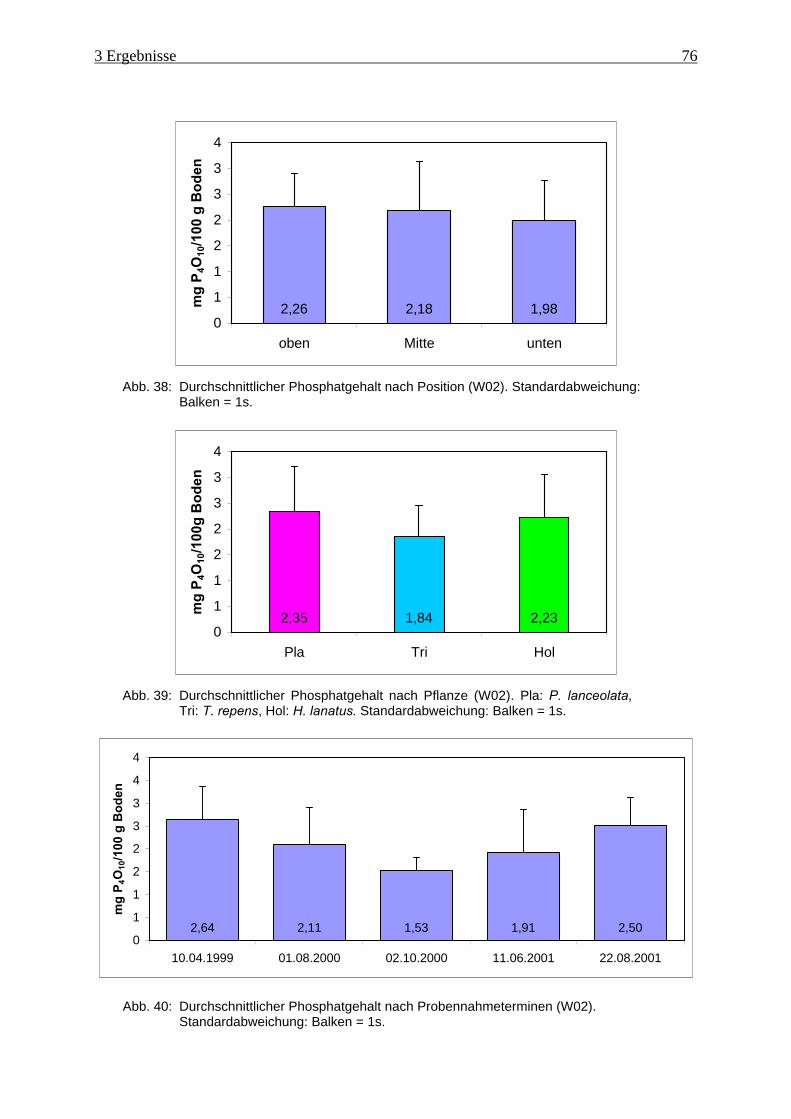
3 Ergebnisse 76
2,26 2,18 1,980
1
1
2
2
3
3
4
oben Mitte unten
mg
P4O
10/1
00 g
Bo
den
Abb. 38: Durchschnittlicher Phosphatgehalt nach Position (W02). Standardabweichung:Balken = 1s.
2,35 1,84 2,230
1
1
2
2
3
3
4
Pla Tri Hol
mg
P4O
10/1
00g
Bo
den
Abb. 39: Durchschnittlicher Phosphatgehalt nach Pflanze (W02). Pla: P. lanceolata,Tri: T. repens, Hol: H. lanatus. Standardabweichung: Balken = 1s.
2,64 2,11 1,53 1,91 2,500
1
1
2
2
3
3
4
4
10.04.1999 01.08.2000 02.10.2000 11.06.2001 22.08.2001
mg
P4O
10/1
00 g
Bo
den
Abb. 40: Durchschnittlicher Phosphatgehalt nach Probennahmeterminen (W02).Standardabweichung: Balken = 1s.
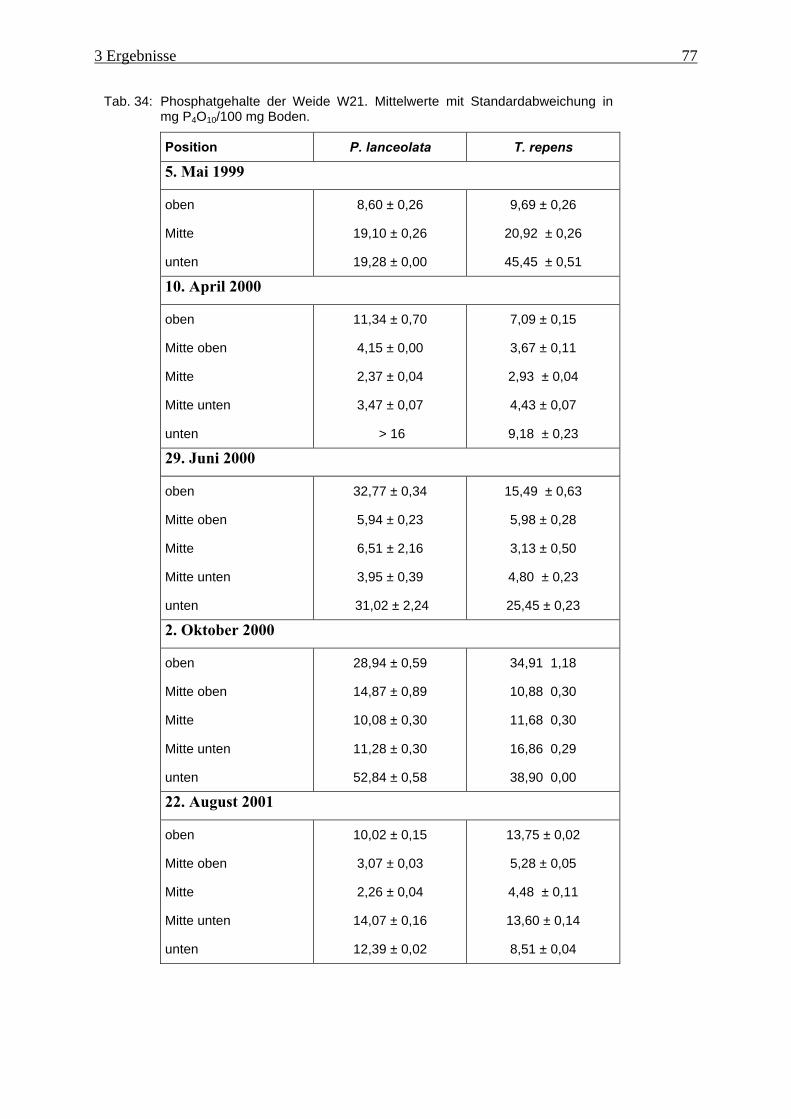
3 Ergebnisse 77
Tab. 34: Phosphatgehalte der Weide W21. Mittelwerte mit Standardabweichung inmg P4O10/100 mg Boden.
Position P. lanceolata T. repens
5. Mai 1999
oben
Mitte
unten
8,60 ± 0,26
19,10 ± 0,26
19,28 ± 0,00
9,69 ± 0,26
20,92 ± 0,26
45,45 ± 0,51
10. April 2000
oben
Mitte oben
Mitte
Mitte unten
unten
11,34 ± 0,70
4,15 ± 0,00
2,37 ± 0,04
3,47 ± 0,07
> 16
7,09 ± 0,15
3,67 ± 0,11
2,93 ± 0,04
4,43 ± 0,07
9,18 ± 0,23
29. Juni 2000
oben
Mitte oben
Mitte
Mitte unten
unten
32,77 ± 0,34
5,94 ± 0,23
6,51 ± 2,16
3,95 ± 0,39
31,02 ± 2,24
15,49 ± 0,63
5,98 ± 0,28
3,13 ± 0,50
4,80 ± 0,23
25,45 ± 0,23
2. Oktober 2000
oben
Mitte oben
Mitte
Mitte unten
unten
28,94 ± 0,59
14,87 ± 0,89
10,08 ± 0,30
11,28 ± 0,30
52,84 ± 0,58
34,91 1,18
10,88 0,30
11,68 0,30
16,86 0,29
38,90 0,00
22. August 2001
oben
Mitte oben
Mitte
Mitte unten
unten
10,02 ± 0,15
3,07 ± 0,03
2,26 ± 0,04
14,07 ± 0,16
12,39 ± 0,02
13,75 ± 0,02
5,28 ± 0,05
4,48 ± 0,11
13,60 ± 0,14
8,51 ± 0,04
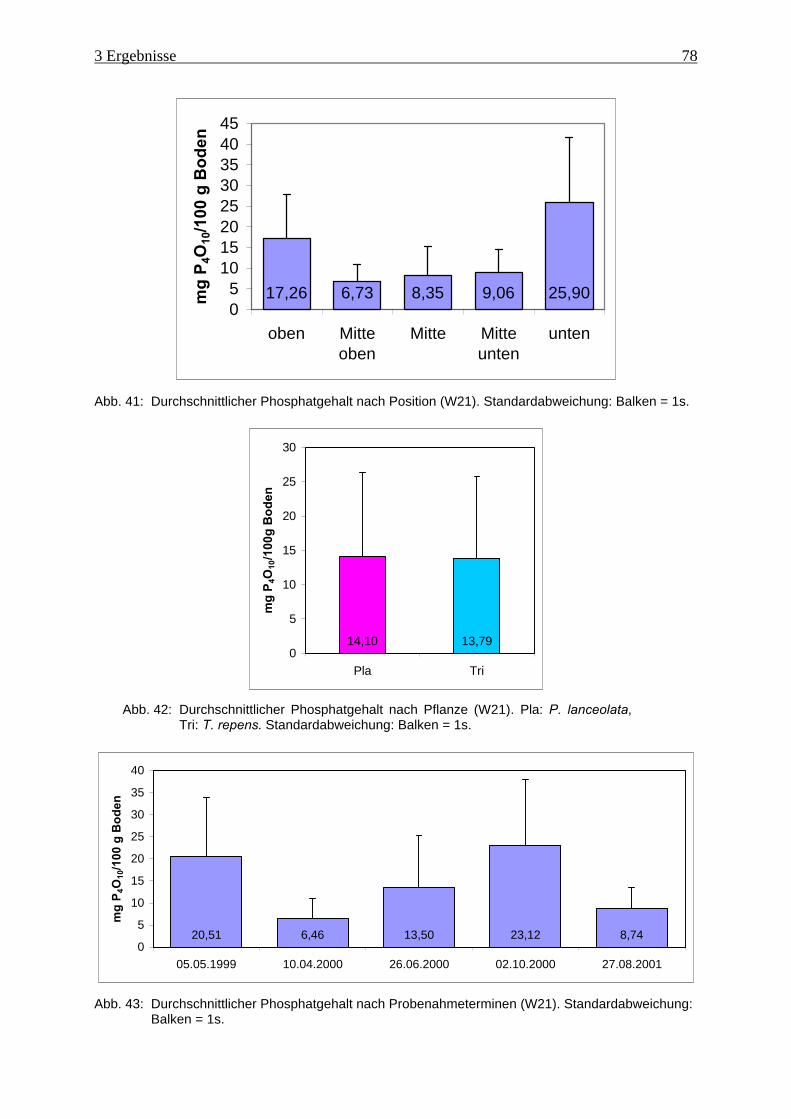
3 Ergebnisse 78
17,26 6,73 8,35 9,06 25,9005
1015202530354045
oben Mitteoben
Mitte Mitteunten
unten
mg
P4O
10/1
00 g
Bo
den
Abb. 41: Durchschnittlicher Phosphatgehalt nach Position (W21). Standardabweichung: Balken = 1s.
14,10 13,790
5
10
15
20
25
30
Pla Tri
mg
P4O
10/1
00g
Bo
den
Abb. 42: Durchschnittlicher Phosphatgehalt nach Pflanze (W21). Pla: P. lanceolata,Tri: T. repens. Standardabweichung: Balken = 1s.
20,51 6,46 13,50 23,12 8,740
5
10
15
20
25
30
35
40
05.05.1999 10.04.2000 26.06.2000 02.10.2000 27.08.2001
mg
P4O
10/1
00 g
Bo
den
Abb. 43: Durchschnittlicher Phosphatgehalt nach Probenahmeterminen (W21). Standardabweichung:Balken = 1s.

3 Ergebnisse 79
Die P4O10-Werte der Bodenproben von W21 (Weide) lagen breit gestreut im Bereich zwi-
schen 2,26 und 52,84 mg/100 g Boden mit einem Mittelwert von 13,94 mg/100 g Boden
(Tab. 34). Bei den unterschiedlichen Positionen zeigte sich, dass die flacheren Standorte
(„oben“ mit durchschnittlich 17,26 und „unten“ mit 25,90 mg/100 g Boden deutlich höhere
Werte besaßen, als die steileren „mittleren“ Standorte mit Werten zwischen 6,73 und
9,06 mg/100 g Boden (Abb. 41). Die durchschnittlichen Phosphatgehalte bei P. lanceolata
und T. repens waren mit 14,10 bzw. 13,79 mg/100 g Boden nahezu identisch (Abb. 42). Im
jahreszeitlichen Ablauf zeigte sich jeweils eine Abnahme über den Winter, sowie eine
deutliche Zunahme von April (6,46 mg/100 g Boden) über August (13,50 mg/100 g Boden)
bis Oktober (23,12 mg/100 g Boden; Abb. 43).
3.8 Zusammenhang zwischen Mykorrhizierungsgrad und Phosphatgehalt
Die einzelnen Mykorrhizierungswerte wurden für jede Pflanze mit den entsprechenden
Phosphatwerten korreliert. Die Hyphen- (HC) und Vesikel-Kolonisierungsraten (VC) wurden
getrennt dargestellt. Die Arbuskel-Kolonisierung (AC) wurde nicht berücksichtigt, da der
Nachweis von Arbuskeln relativ schwierig ist und nur geringe Kolonisierungsraten gefunden
wurden.
Auf der Wiese W02 zeigt sich bei P. lanceolata und T. repens praktisch kein direkter
Zusammenhang zwischen Hyphenkolonisierung HC und dem verfügbaren Phosphatgehalt des
anhaftenden Bodens (r = -0,139 bzw. –0,091; Abb. 44). Bei H. lanatus hingegen läßt sich bei
steigendem Phosphatgehalt eine Abnahme der Hyphenkolonisierung erkennen (r = - 0,389).
Auch bei der Vesikelkolonisierungsrate läßt sich bei P. lanceolata und T. repens kein direkter
Zusammenhang mit dem Phosphatgehalt feststellen (r = -0,066 bzw. 0,128). Bei H. lanatus
wiederum deutet sich ein Vesikel-Zunahme bei steigendem Phosphatgehalt an (r = 0,414;
Abb. 45).
Auch auf der Weide W21 ist keine direkte Korrelation zwischen Mykorrhizierung und
Phosphatgehalt bei P. lanceolata und T. repens erkennbar (Abb. 46 und Abb. 47). Selbst ver-
gleichsweise sehr hohe Phosphatwerte konnten mit starker Hyphenkolonisierung einhergehen.
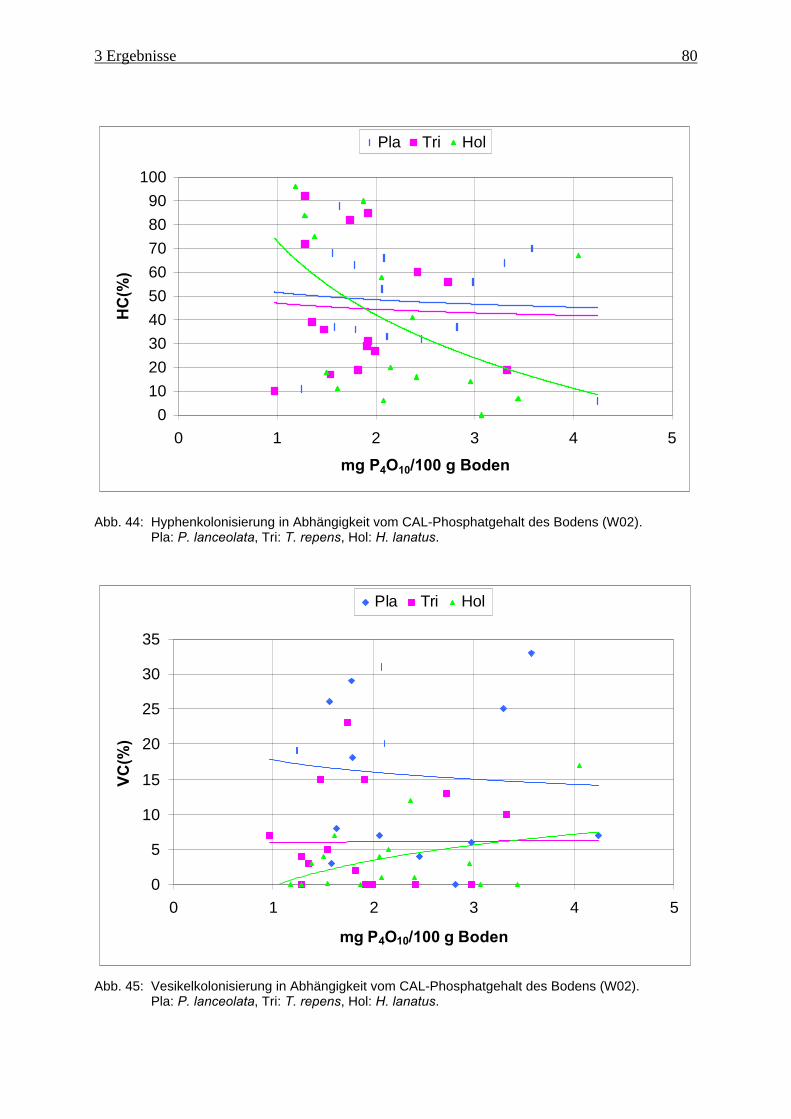
3 Ergebnisse 80
0
10
20
30
40
50
60
70
80
90
100
0 1 2 3 4 5
mg P4O10/100 g Boden
HC
(%)
Pla Tri Hol
Abb. 44: Hyphenkolonisierung in Abhängigkeit vom CAL-Phosphatgehalt des Bodens (W02).Pla: P. lanceolata, Tri: T. repens, Hol: H. lanatus.
Abb. 45: Vesikelkolonisierung in Abhängigkeit vom CAL-Phosphatgehalt des Bodens (W02).Pla: P. lanceolata, Tri: T. repens, Hol: H. lanatus.
0
5
10
15
20
25
30
35
0 1 2 3 4 5
mg P4O10/100 g Boden
VC
(%)
Pla Tri Hol
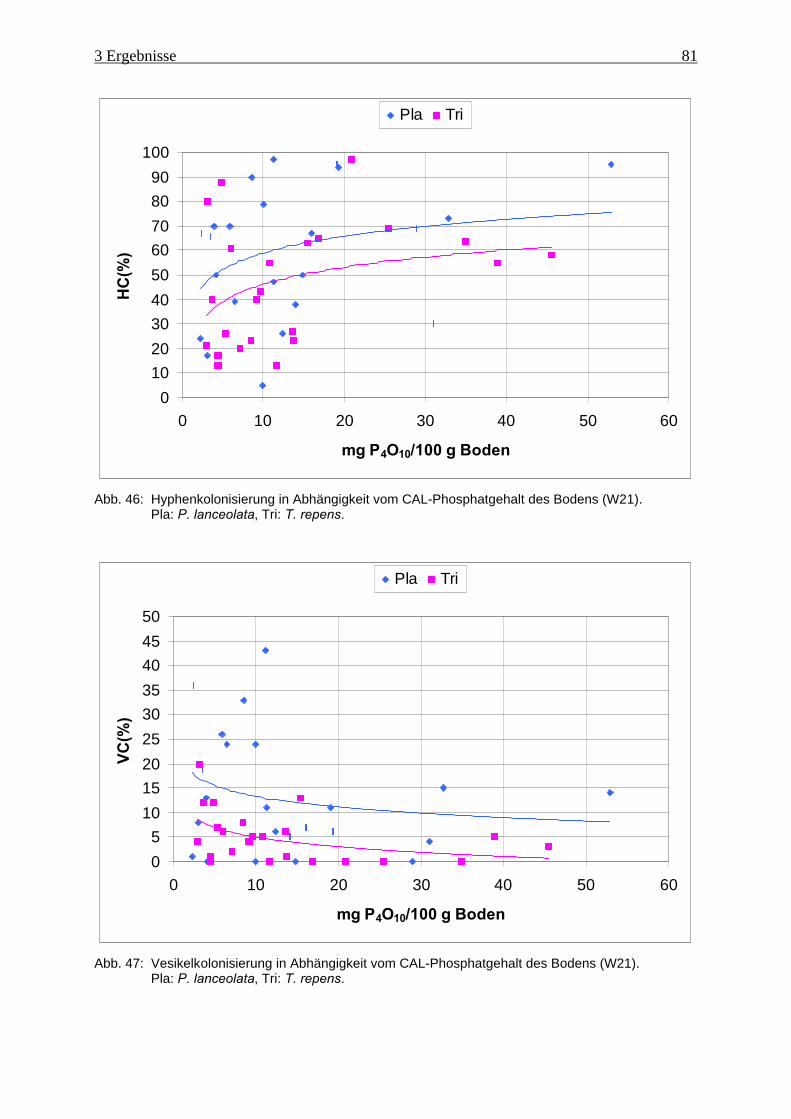
3 Ergebnisse 81
Abb. 46: Hyphenkolonisierung in Abhängigkeit vom CAL-Phosphatgehalt des Bodens (W21).Pla: P. lanceolata, Tri: T. repens.
Abb. 47: Vesikelkolonisierung in Abhängigkeit vom CAL-Phosphatgehalt des Bodens (W21).Pla: P. lanceolata, Tri: T. repens.
0
10
20
30
40
50
60
70
80
90
100
0 10 20 30 40 50 60
mg P4O10/100 g Boden
HC
(%)
Pla Tri
0
5
10
15
20
25
30
35
40
45
50
0 10 20 30 40 50 60
mg P4O10/100 g Boden
VC
(%)
Pla Tri

4 Diskussion 82
4 Diskussion
Parallel zum wachsenden Interesse an der arbusukären Mykorrhiza fand in den letzten Jahren
ein Umdenken in der Landwirtschaft hin zu einer nachhaltigen Landnutzung statt. Die For-
schungsergebnisse, die aus Gewächshausversuchen gewonnen werden konnten, versprechen
vielfache Anwendungsmöglichkeiten der AMF, so bei der Reduktion von Dünger, dem
Schutz vor Pathogenen und der Begrünung erodierter oder desertifizierter Flächen. Dennoch
ist bislang relativ wenig über sie bekannt und vor allem ihre biologische Funktion im Freiland
bietet noch zahlreiche Forschungsoptionen, z.B. die Einflüsse unterschiedlichen Arten auf die
Nährstoffversorgung der Pflanzen. Der artspezifische Nachweis einzelner AMF-Arten in ihrer
natürlichen Umgebung ist das zentrale Thema dieser Arbeit. Hierzu mußten die vorhandenen
Erkenntnisse der AMF-Forschung für eine Anwendung bei Freilandproben optimiert werden.
4.1 DNA-Extraktion aus Wurzelmaterial
Die Methode nach Doyle & Doyle (1987) brachte in der vorliegenden Arbeit nur wenig am-
plifizierbares Material. Die Methode nach BAHNWEG et al. (1998) führte zu guten Ergebnis-
sen. Allerdings nicht mit „frischestem Material“ (BAHNWEG, Institut für Biochemische Pflan-
zenpathologie, GSF-Forschungszentrum, mündliche Mitteilung), sondern mit gefriergetrock-
neten Proben. Der Vorteil dieser Extraktion ist, dass die Mengen beliebig variiert werden
können. Bei Benzylchlorid handelt es sich jedoch um ein starkes Reizgas, das ebenso wie
Chloroform und 2-Mercaptoethanol gesundheitlich bedenklich ist, so dass unter einem Abzug
gearbeitet werden muss. Die besten Ergebnisse wurden mit dem modifizierten QIAGEN
DNeasy Plant Mini Kit erzielt, wobei entscheidend war, dass die frischen Wurzeln
unverzüglich mit flüssigem Stickstoff schockgefroren wurden. Die Zelllyse der gefrierge-
trockneten Proben fand mit einer Kugelmühle in der Kältekammer statt. Die DNA-Extraktion
aus Wurzeln mit kochender Natronlauge und anschließender salzsaurer Neutralisation nach
REDECKER et al. (1997) konnte nicht reproduziert werden.
Auf den ersten Blick erscheint die Analyse von Sporen der beste und einfachste Weg,
um die Diversität und Verbreitung von AMF zu untersuchen. So entwickelten z.B. REDECKER
et al. (1997) eine PCR-RFLP-Methode zur Identifizierung einzelner Glomales-Arten. Die aus
Sporen gewonnene DNA ist jedoch keinesfalls frei von Fremd-DNA (s. Kap. 4.6). Auch spie-
gelt die Verteilung von Sporen nicht die tatsächliche Mykorrhizierung in einer Wurzel wider.
Sporen können ein Reservoir potentieller Mykobionten darstellen. Um die tatsächliche
Situation in den kolonisierten Wurzeln zu beschreiben, muß jedoch die Wurzel-DNA unter-

4 Diskussion 83
sucht werden. Die extrahierte DNA lieferte zunächst keine positiven Ergebnisse in der PCR,
deshalb wurde nach Gründen für die schlechte Amplifizierbarkeit gesucht. Es war bereits be-
kannt, dass sowohl Pflanzenmaterial, als auch Bodenproben viele Substanzen enthalten, wel-
che die DNA degradieren oder anderweitig die Polymerase-Kettenreaktion inhibieren können
(ABU AL-SOUD & RADSTRÖM, 1988). Zu diesen unerwünschten Reagenzien gehören neben
Polyphenolen und Polysacchariden in der Pflanze auch Huminstoffe im Boden. Polysaccha-
ride und Tannine stellen ein großes Problem bei der PCR dar. Sie sind bei DNA-Extraktion
nur schwer von der DNA abzutrennen und können DNA Polymerasen und Nukleasen inhibie-
ren oder zu unspezifischen Produkten führen. Polyphenolische Substanzen können durch
Phenoloxidasen und Peroxidasen, die bei Verletzung Wurzel oder deren Austrocknung freige-
setzt werden, zu freien Radikalen oxidiert werden. In dieser reaktiven Form können sie irre-
versibel an die DNA binden (ROWLAND & NGUYEN, 1993), die dadurch für die DNA Polyme-
rase unlesbar wird. Auch Huminsäuren besitzen eine inhibierende Wirkung auf die DNA Po-
lymerasen (TEBBE & VAJHEN, 1993). Allerdings haften diese in der Regel außen an der Wur-
zel und können durch eine gründliche Reinigung entfernt werden. Zunächst mußte also nach
einer geeigneten DNA-Extraktionsmethode gesucht werden.
Mithilfe von CTAB kann jedoch die DNA präzipitiert werden, während die Polysaccha-
ride in Lösung bleiben (MURRAY & THOMPSON, 1980). GEHRIG et al. (1998) extrahierten
DNA aus Blättern von Kalanchoe-Arten mittels der CTAB-Methode (DOYLE & DOYLE,
1987). Bei 40% der Extrakte konnte in einer PCR kein Amplifikat erhalten werden. Die glei-
che Extraktion mit sechs verschiedenen kommerziellen Kits führte zu noch geringeren Er-
folgsquoten. BOITEUX et al. (1999) untersuchten sieben verschiedene Extraktionsmethoden an
Daucus carota-Wurzeln und erzielten ebenfalls mit der CTAB-Methode die „besten Ergeb-
nisse“. BOBOWSKI et al. (1999) verwendeten ebendiese Methode zur Extraktion von DNA aus
Pflanzenblättern und –wurzeln. Dabei erhielten sie allerdings in nur 25% (Blätter) bzw. 3%
(Wurzeln) der Fälle amplifizierbares Material. LINDER et al. (2000) stellen schließlich sehr
treffend fest „that extraction and purification of DNA from roots can be challenging“.
4.2 Amplifizierbarkeit
Nach anfänglichen Schwierigkeiten konnten durch die optimierte DNA-Extraktion mit den
universellen Primern A2R und B2L die meisten DNA-Proben (etwa 90%) amplifiziert wer-
den. Das Zielgen der PCR, die rDNA, besitzt den Vorteil, dass es als high copy gene mit meh-
reren tausend Kopien innerhalb eines Genoms vorkommt (BUCHANAN et al., 2000). ROGERS

4 Diskussion 84
& BENDICH (1987) geben 200 bis 22.000 rDNA-Kopien je Pflanzengenom an. Da das rRNA-
Gen zudem in allen Organismen vorkommt, wird es gerne für phylogenetische Studien heran-
gezogen. Bei den AMF kann die geringe DNA-Menge und der hohe Anteil an Pflanzen-DNA
durch die hohe Kopienzahl der rDNA relativiert werden. TOTH et al. (1991) schätzten die
Pilz-Biomasse bei Allium cepa-Wurzeln, die zu 76% kolonisiert waren, auf nur 4,3%.
MALDONADO-MENDOZA et al. (2002) berechneten den Anteil von AMF-RNA auf 5-12% in
den am meisten kolonisierten Wurzeln, so dass wohl optimistisch betrachtet von einem Anteil
an Pilz-DNA von maximal 5% ausgegangen werden kann.
4.3 Spezifische Primer
Obwohl die SSU intensiver erforscht ist und zahlreiche Sequenzdaten zur Verfügung stehen,
birgt die LSU rDNA vielleicht die bessere Information. Die variable Domäne D2 bietet Ziel-
sequenzen für spezifische Primer und Endonukleasen (BÖHM, 2000). Außerdem konnte bisher
keine Polymorphie innerhalb einzelner Sporen wie bei der ITS-Region nachgewiesen werden
(Kap. 4.9). Mit den Primern Aclong87For und Aclong400Rev wurde bei fünf der amplifizier-
baren Proben (9%) ein positives Signal erhalten. Bei weiteren acht Extrakten (15%) waren
schwache Banden zu erkennen. Dabei könnte es sich um nahe Verwandte von A. longula han-
deln, bei denen die PCR weniger effizient ablief, oder um Proben mit sehr wenig A. longula-
DNA. Mit dem Primerpaar Glmoss85For und Glmoss387Rev wurden neun positive Signale
erhalten (17%), die entweder G. mosseae oder einer nah verwandten Art zuzuordnen sind.
Eine „Pilzspezifität“ der Primer reicht zum Nachweis der AMF in Wurzeln nicht aus,
denn neben Pflanzen- und AMF-DNA können die Wurzelproben durch Fremd-DNA konta-
miniert sein. So mussten bei fast allen Proben von H. lanatus und anderen Gräsern bei mikro-
skopischen Untersuchungen ein Befall mit Polymyxa graminis festgestellt werden.
VANDENKOORNHUYSE et al. (2002) untersuchten Wurzeln von Arrhenatherum elatius und
amplifizierten dabei Abschnitte der SSU rDNA mit pilzspezifischen Primern. Von den daraus
erhaltenen 49 Sequenztypen ließen sich nur drei den AMF zuordnen. Die restlichen 46 ge-
hörten zu den Basidiomycota, Ascomycota oder Zygomycota. Neben zahlreichen phytopatho-
genen Pilzen, die in Wurzeln vorkommen, können auch die AMF-Sporen selbst von Parasiten
befallen sein. So konnten REDECKER et al. (1999) nachweisen, dass einige Sequenzen aus
Scutellospora castanea-Sporen von Ascomyceten stammten. Daneben belegten WALLEY &
GERMIDA (1996) Sporenwand-assoziierte Bakterien.

4 Diskussion 85
Da es sich bei den AMF um obligate Symbionten handelt, ist die Kultur nicht kontami-
nierter Stämme unmöglich (Redecker et al., 1999). Versuche, axenische AMF-Kulturen zu
etablieren, sind bislang gescheitert. Zwar ist es möglich, Sporen zum Keimen zu bringen,
doch die Hyphen sterben spätestens nach zwei Wochen ab, wenn sie auf keine Wirtswurzel
stoßen und alle möglichen untersuchten Kombinationen von Kohlenstoff-Substraten konnten
den Pilz bisher nicht zur Vollendung seines Lebenszyklus anregen (Bago, 2000).
SIMON et al. (1992) veröffentlichten den ersten „Glomales-spezifischen“ Primer, nach-
dem sie die SSU rDNA-Sequenzen dreier Glomales-Arten miteinander verglichen hatten, die
damals zur Verfügung standen. Zahlreiche Autoren haben darauf hingewiesen, dass VANS1
„Probleme bei der PCR“ verursachte (CLAPP et al., 1995 und 1999; REDECKER, 1999) oder
nur „schwache Amplifikationsprodukte“ lieferte (GEHRIG et al. 1996). CLAPP et al. (1999)
zeigten, dass die Annealing site von VANS1 innerhalb der Glomales nicht konserviert ist.
Einige Arten wurden von diesem Primer überhaupt nicht erkannt (REDECKER, 2000).
SCHÜßLER et al. (2001a) bestätigten, dass VANS1 weder für die AMF im allgemeinen, noch
für eine ihrer taxonomischen oder phylogenetischen Gruppen spezifisch ist. Die Autoren
konnten auch belegen, dass die „familienspezifischen“ Primer VAACAU, VAGLO, VALETC
und VAGIGA (SIMON, 1993a) keine Spezifität für die Familien der Acaulosporaceae,
Glomaceae und Gigasporaceae besitzen.
Ein ähnliches Resultat fand sich bei dem pilzspezifischen Primer AM1 (HELGASON et
al., 1998), der zwar erfolgreich Pflanzen diskriminiert (bei Zea mays differieren 8 von 20 Ba-
sen), jedoch ebenso Geosiphon pyriforme, Archaeospora gerdemannii, Archaeospora trappei
(3/20) und Paraglomus occultum (4/20). Der Versuch, alle Vertreter der arbuskulären
Mykorrhizapilze durch geeignete Primer zu erfassen, muß bis heute als gescheitert betrachtet
werden, da nach wie vor im besten Fall Untergruppen amplifiziert werden können (SCHÜßLER
et al., 2001a).
REDECKER (2000) veröffentlichte Primer mit spezifischen Bindungseigenschaften für
eine G. mosseae/intraradices- (GLOM1310, GLOM5.8R), G. etunicatum/claroideum-
(LETC1670) und A. gerdemanni/trappei/G. occultum/brasilianum-Gruppe (ARCH1311) so-
wie für Acaulosporaceae (ACAU1660) und Gigasporaceae (GIGA5.8R). All diese Primer
amplifizieren jedoch die ITS1-Region, die durch ihre Hypervariabilität (s. Kap. 4.9) eine er-
höhte Diversität vortäuschen kann und ihre Anwendung bei Freilandproben deshalb in Frage
gestellt werden muß. Außerdem können auch mit diesen Primern nicht alle AMF-Arten (wie
z.B. G. versiforme) erfasst werden. Redeckers Analyse der Amplifikate mittels RFLP brachte
zwar „weitgehend übereinstimmende“, aber keine eindeutigen Ergebnisse. So konnten die

4 Diskussion 86
Gigaspora-Arten überhaupt nicht voneinander unterschieden werden. Eine vorgeschlagene
Sequenzierung der DNA-Fragmente ist bei hoher Probenzahl aufwändig und teuer.
4.4 Sequenzanalysen und RFLP
Die Sequenzanalysen im Bereich des mit dem Primerpaar Glmoss85For und Glmoss387Rev
amplifizierten Teilstückes zeigte eine Sequenzhomologie von insgesamt 87%. Alle Sequenzen
waren bis auf wenige Positionen innerhalb der ersten zehn Basen eindeutig. Nur bei HelA06
konnten die ersten 44 Nukleotide nicht ermittelt werden. Alle Sequenzen konnten im Ver-
gleich mit Datenbanksequenzen einer Art zugeordnet. Aus den erhaltenen Ergebnissen wurde
phylogenetischer Stammbaum erstellt. Es handelte sich dabei um sechs G. mosseae-, eine
G. geosporum- und zwei G. caledonium-Sequenzen. Außerdem konnten für die einzelnen
Arten charakteristische Substitutionsmuster identifiziert werden: Für G. mosseae 20>C,
70>G, 82>C, 87>C, 95>T, G. caledonium 12>C, 48>T, G. fragilistratum 78>T und
G. geosporum 225>T.
4.5 Quantifizierung mit der TaqMan® real-time PCR
BÖHM et al. (1999) entwickelten die real-time PCR für G. mosseae. Dabei wurden die Start-
kopien des G. mosseae templates (Bereich der LSU rDNA) von 11 bzw. 100 Sporen quantifi-
ziert, und zwischen 100.000 bis 350.000 Kopien je Spore gefunden. Das entspricht bei einer
angenommenen Kopienzahl von etwa 100 rDNA-Einheiten je Genom ca. 1000 bis 3500
Nuklei je Spore. Die real-time PCR hat gegenüber der kompetitiven PCR mit internen Stan-
dards den Vorteil, dass über einen großen DNA-Konzentrationsbereich gemessen werden
kann. Die Messung fand in 96er-Platten statt, dabei konnten maximal 40 Proben bestimmt
werden, da jede Probe mindestens doppelt bestimmt wurde und Standards und Kontrollen
mindestens 16 Kavitäten beanspruchten. Der Nachteil dieser Analysemethode sind die relativ
hohen Kosten.
Bei der Quantifizierung von A. longula betrug die Effizienz der PCR 86%. Der Korre-
lationskoeffizient betrug 0,973. Die Standardwerte lagen also bis zu 97,3% auf der Korrela-
tionsgeraden. Durchschnittlich ergeben sich zwischen 4489 und 94450 Kopien je mg Wurzel.
Dabei konnten bei sechs der acht schwachen Banden aus der PCR mit den A. longula-spezifi-
schen Primern keine Kopien ermittelt werden. Womöglich handelte es sich dabei um
A. longula-Varianten oder nah verwandte Arten, die an den entsprechenden Positionen der

4 Diskussion 87
Primer- und Sondenbindungsstellen Basensubstitutionen aufwiesen. Als Richtwert für eine
gewöhnliche PCR geben INNIS & GELFAND (1990) 3·105 Startkopien an.
Bei der Quantifizierung von G. mosseae betrug die Effizienz der PCR 93%. Der Korre-
lationskoeffizient betrug 0,979. Die Standardwerte lagen also bis zu 97,9% auf der Korrela-
tionsgeraden. Die Mittelwerte der gemessenen Kopienzahlen lagen zwischen 381 und 64750
Kopien je mg Wurzel. Allerdings ist zu beachten, dass es sich hierbei um „G. mosseae-Äqui-
valente“ handelt. Ein Vergleich der Probensequenzen mit denen des Primers P1 und der
Sonde F1 hatte gezeigt, dass bei den ermittelten G. caledonium und G. geosporum-Proben
jeweils eine bzw. zwei Basen abwichen. Dies könnte der Grund für eine verringerte PCR-
Effizienz gewesen sein und würde erklären, weshalb bei der G. geosporum-Probe von
P. lanceolata Pla5-004 keine Kopien ermittelt wurden. Weiterhin ist zu beachten, dass es sich
bei der rDNA um ein high copy gene handelt, so dass etwa 100 Startkopien je Genom vorla-
gen. Da diese fest miteinander auf einem Strang verbunden waren, wurden sie nur „paket-
weise“ pipettiert. Die Konsequenz ist eine umso höhere Standardabweichung bei geringerer
Kopienzahl.
Aus dem Ergebnis dieser Analyse lässt sich somit auf die Zahl der Genome oder Kerne
je mg Wurzel schließen. Da die Hyphen der AMF nicht septiert sind, ist eine Gleichsetzung
der Kernzahl mit der Zellzahl nicht möglich und eine Massenbestimmung schwierig. Über die
Zahl der Kerne in Sporen wurden einige Untersuchungen durchgeführt, die jedoch sehr
unterschiedlich ausgefallen sind, so untersuchten BIANCIOTTO & BONFANTE (1992)
cytofluorometrisch die Genomgröße zweier AMF-Arten. Sie errechneten dabei durch-
schnittlich 0,26 pg je Nukleus für G. versiforme und ca. 0,75 pg je Nukleus bei G. margarita.
Die Anzahl der Nuklei je Spore geben sie mit 9.000 für G. caledonium, 20.000 bei
G. margarita und 35.000 bei G. decipiens an. Allerdings beschreiben HOSNY et al. (1998)
lediglich 800 Nuklei und 0,88 pg DNA für S. castanea-Sporen und 1167 Nuklei bei
G. margarita. BURGGRAAF & BERINGER (1989) hingegen errechneten 20.000 Kerne pro
G. margarita-Spore und BÉCARD & PFEFFER (1993) geben 2400 Kerne an. BAHNWEG et al.
(1998) gehen bei Pilzen von einer haploiden Genomgröße von 2·107 bp und einer Kopienzahl
von 300 rDNA-Einheiten pro Genom und 9,1·108 bp je pg DNA aus. HOSNY et al. (1999) un-
tersuchten die rDNA von S. castanea und fanden eine Kopienzahl von 75 (40 bis 400) je
haploidem Genom. Sie stellten außerdem eine Größe des Genoms von 108 bis 109 bp fest,
verglichen sie mit anderen Pilzarten (1,4 bis 5·107 bp). Bei angenommenen 100 rDNA-Ein-
heiten je Genom und 0,5 pg je Nukleus ergibt dies ungefähr 45 bis 945 Kerne (22,5 bis 472 pg

4 Diskussion 88
AMF-DNA) je mg Wurzel bei A. longula und zwischen 4 und 648 Kerne (2 bis 324 pg AMF-
DNA) je mg Wurzel bei G. mosseae.
4.6 Identifikation der Mykorrhizapilze anhand der Sporenmorpholgie
Bei der Identifizierung der AMF mithilfe der Sporenmorphologie konnten sechs verschiede-
nen Typen unterschieden werden. Sporen vom Typ G. mosseae stellten dabei mit einem An-
teil von etwa 70% den größten Anteil. Dieses Ergebnis stimmt recht gut mit den Angaben
anderer Autoren überein (Kap. 4.8).
Die klassischen Studien zur Identifizierung der AMF-Arten beruhen auf der
Untersuchung der Sporenmorphologie. SCHENK & PÉREZ (1990) veröffentlichten hierzu einen
Bestimmungsschlüssel. Dieser basiert auf den Veröffentlichungen neu beschriebener Arten
und ist daher sehr uneinheitlich, so dass einzelne Beschreibungen nicht ohne weiteres
miteinander verglichen werden können. Während die Identifizierung auf Gattungsebene noch
relativ einfach ist, können bei der Bestimmung einzelner Arten vielerlei Probleme auftreten.
Charakteristika wie Größe oder Färbung sind abhängig vom Alter und Zustand der Sporen
sowie ihrer Umgebung (Substrat). Außerdem kann die sichere Identifizierung der Sporen
durch Parasitenbefall unmöglich werden. Bei der Bestimmung von Freiland-Sporen müssen
die meist einzeln isolierten Sporen individuell analysiert werden. Dadurch können einige
wichtige Unterscheidungsmerkmale nur bedingt berücksichtigt werden. Untersucht man die
Sporengröße an hunderten Sporen einer Einzelsporkultur, so erkennt man, dass diese einer
Gauß’schen Normalverteilungskurve entspricht. Außerdem sind junge Sporen in der Regel
kleiner als ältere. Die phänotypische Größe einer isolierten Freilandspore unbekannten Alters
ist daher kein sicheres Merkmal. Auch die Farbe einer Spore ist von ihrem Alter abhängig, so
sind jüngere heller. Außerdem können sie je nach Boden unterschiedlich getönt sein, so dass
Freilandsporen meist dunkler sind als Sporen aus Topfkulturen (KRAMADIBRATA et al. 2000).
Desgleichen kann die Sporen-Ontogenese aus entwickelten Sporen nicht mehr als Unterschei-
dungsmerkmal dienen. Mit der klassischen Methode werden „Morphotypen“ (MORTON &
BENNY, 1990; WALKER, 1992) beschrieben, wobei minimale Differenzen im Aufbau der
Sporenwand zur Unterscheidung von Arten ausreichen. Dass diese „Morphotypen“ jedoch
nicht synonym mit dem Begriff „Arten“ behandelt werden dürfen zeigt folgendes Beispiel:
A. gerdemannii und G. leptotichum wurden aufgrund ihres Sporenphänotyps beschrieben.
Nachdem SAWAKI et al. (1998) zeigen konnten, dass sie genotypisch identisch sind, wurden
beide als Archaeospora leptoticha zusammengefasst (s. Kap. 4.11).

4 Diskussion 89
Die Identifikation von Sporen wurde in dieser Arbeit bewusst nur dazu angewandt, um
einen ersten Überblick über das Vorhandensein bestimmter Typen zu erhalten. Sie sagt weder
etwas über die tatsächlichen Mykorrhizierungsverhältnisse in den Pflanzen aus, noch über das
Mykorrhizerungspotential, da viele AMF nur wenige, unregelmäßige oder gar keine Sporen
produzieren, oder diese sich in den Pflanzenwurzeln (z.B. G. intraradices) statt im Boden
befinden (SCHÜßLER et al. 2001a). Letztlich stellt die Sporenanalyse ein „Sporenbildungs-
potential“ fest (FITTER, Department of Biology, University of York, persönliche Mitteilung).
Auf eine intensivere Untersuchung der Sporen wurde deshalb zugunsten der
molekularbiologischen Methoden verzichtet.
4.7 Mykorrhizierung und Phosphatgehalte
Die angewandte Magnified Intersections-Methode nach MCGONIGLE et al. (1990) besitzt ge-
genüber der häufig verwendeten Grid-line Intersect-Methode (GIOVANETTI & MOSSE, 1980,
TROUVELOT et al., 1986) den Vorteil, dass einzelne kleine Bereiche keine höhere Kolonisie-
rungsrate vortäuschen wie kompakte, dichte Bereiche. GANGE et al. (1999) fanden heraus,
dass verschiedene Färbemethoden die Pilzhyphen unterschiedliche stark färben konnten. Die
Mykorrhizierung der einzelnen Pflanzen wies insgesamt starke Schwankungen auf, so dass
eine eindeutige Interpretation schwierig war. Bei der Hyphen-Kolonisierungsrate konnte
keine Abhängigkeit vom Standort oder der Pflanze festgestellt werden. Es sind lediglich Indi-
zien für eine jahreszeitliche Zunahme vorhanden. SANDERS & FITTER (1992) untersuchten die
Mykorrhizierung bei sechs verschiedenen Grasland-Pflanzenarten. Sie stellten bei
P. lanceolata und Rumex acetosa eine starke jahreszeitliche Schwankung fest. Bei Festuca
rubra, H. lanatus, Lathyrus pratensis und Trifolium pratense verlief die Kolonisierung
gleichmäßiger. Bei der Vesikel-Kolonisierungsrate zeigt sich ein differenzierteres Bild. So
deuten die Ergebnisse darauf hin, dass die Vesikeldichte zum Herbst hin abnimmt. Dies
könnte dergestalt interpretiert werden, dass die Assimilate zunächst in der Pflanzenwurzel,
also den Vesikeln gespeichert und im späteren Verlauf des Jahres während der Bildung von
Sporen in diese verlagert werden. FITTER et al. (1998) stellten eine Korrelation fest zwischen
dem C-Transfer und der Vesikelzahl in der korrespondierenden Pflanzenwurzel fest. Es ist
daher anzunehmen, dass der Anstieg der Vesikel-Kolonisierungsrate mit der hohen Assimi-
latproduktion der Pflanzen im Sommer zusammenfällt. Im Herbst dagegen nimmt die Assi-
milatproduktion wieder ab. Aus diesem Grund und in Kombination mit dem Temperaturabfall
und/oder zunehmender Trockenheit (bei Gewächshauskulturen wird der Pilz durch Einstellen

4 Diskussion 90
des Gießens zur Sporulation angeregt) könnten die AMF ihre Reservestoffe von den intrara-
dikalen Vesikeln in die neu gebildeten Sporen verlagern.
Die Phosphatgehalte fielen auf der Wiese W02 mit durchschnittlich 2 mg P4O10 je 100 g
Boden sehr gering aus. Auf der Weide lagen die Werte generell höher und erreichten in den
flacheren oberen und unteren Lagen Spitzenwerte im Gegensatz zu den steileren Mittellagen.
Dies deckt sich mit der Beobachtung, dass die Rinder die flachen Stellen bevorzugt zum Wie-
derkäuen aussuchten, was im Phosphateintrag durch Kot deutlich wird. Die jahreszeitliche
Zunahme auf der Weide könnte ebenfalls auf die Beweidung durch die Rinder und den damit
verbundenen P-Eintrag zurückzuführen sein. Den Winter über wurden sie im Stall gehalten,
so dass der P-Gehalt wieder abgenommen hat. Von dem geringen natürlichen, aus der Ver-
witterung von Mineralien entstandenen, Phosphatgehalt von 0,01-0,1% (davon ca. 60% in
anorganischer, ca. 40% in organischer Form) stehen den Pflanzen nur etwa 0,02-0,1 mg P/kg
zur Verfügung (BLUME, 1992). Düngerphosphate werden durch Umsetzungsprozesse langsam
an Eisenoxide absorbiert oder zu schwer löslichen Calciumphosphaten umgesetzt. Dadurch
steht es den Pflanzen nicht mehr zur Verfügung, so dass von einem Ausnutzunggrad von 10
bis 25% im ersten Jahr ausgegangen wird, allerdings tritt ein Depoteffekt ein. Bei
Flüssigdünger (Gülle) wird ein Großteil des Phosphats in tiefere Schichten gespült und kann
unter Umständen, wenn es in Gewässer gelangt zu deren Eutrophierung beitragen. Wenn hohe
Phosphatdüngegaben zu einer Hemmung der Mykorrhiza führen (BLUME, 1992), so kann eine
verringerte Düngung die Mykorrhiza fördern und womöglich die Phosphatausbeute (mit
gezielter Beimpfung durch geeignete Stämme) erhöhen.
Ein Zusammenhang zwischen Phosphatverfügbarkeit und Mykorrhizierung lässt sich
nur bei der Hyphenkonzentration von H. lanatus auf der Wiese feststellen. In Gewächshaus-
versuchen wurde bei Erhöhung des Nährstoffangebots für die Pflanze durch Düngung ein
Rückgang der Mykorrhiza beobachtet (FIEDLER, 2001). Diese Erkenntnis impliziert, dass im
umgekehrten Fall bei einer Verringerung der Düngemenge die Mykorrhizierung ansteigen
würde und dadurch ein Nährstoffmangel ausgeglichen werden könnte. Durch gezielte
Inokulierung (bei genauer Kenntnis der Funktionalität der AMF) landwirtschaftlicher
Nutzflächen könnte ein Wandel von einer high-input- in eine low-input-Landwirtschaft
durchaus denkbar sein (GALVEZ et al. 1995). Im Gegensatz zu den Gewächshausversuchen
mit kontrollierbaren Parametern können im Freiland sehr viele Faktoren eine Rolle spielen.
Da Grünflächen keine Monokulturen darstellen, stehen die einzelnen Pflanzen in Konkurrenz
und Phytopathogene können zusätzlich eine Mykorrhizierung begünstigen. Es mag
widersprüchlich erscheinen, dass eine Pflanze in einer mineralien- und wasserreichen

4 Diskussion 91
Umgebung auf die AMF angewiesen ist. Doch ein hohes Ressourcenangebot führt zu starkem
Pflanzenwachstum und –produktivität und damit zu einem Verdrängungswettbewerb
zwischen den Pflanzen (ALLEN & ALLEN, 1990). EOM et al. (2001) untersuchten den Einfluß
von Huftieren auf die Zusammensetzung von AMF-Arten (in diesem Fall Sporen-
Morphotypen) im Weideland. Sie fanden heraus, dass durch die Beweidung sowohl die
Wurzelkolonisierung mit AMF-Hyphen als auch die Diversität der AMF-Arten zunahm. Un-
einigkeit besteht über Möglichkeit des Assimilattransports zwischen verschiedenen Pflanzen
über ein AMF-Netz. WATKINS et al. (1996) berichteten vom Transport von bis zu 10% 13C-
markierter Verbindungen von einer anderen Pflanzen in eine AM-Wurzel. FITTER et al.
(1998) konnten jedoch belegen, dass die Assimilate ausschließlich in der Wurzel, nicht aber
in der übrigen „Empfänger-Pflanze“ zu finden waren. Die Autoren vermuten, dass die
Kohlenstoff-Verbindungen zwar über das Myzel verteilt werden und womöglich in Vesikeln
einer benachbarten Pflanzenwurzel deponiert werden, schließen aber einen Rücktransfer Pilz-
Pflanze aus.
4.8 Biodiversität: Arten und Individuen
Die Biodiversität setzt sich aus der Artenzahl und Artenverteilung zusammen. Bei den AMF
kann weder der Begriff Art genau definiert, noch eine Individuenzahl angegeben werden.
Eine klassische Definition des Artbegriffes setzt die fruchtbare Nachkommenschaft voraus
(ROSENDAHL, 1996). Bei den AMF handelt es sich jedoch um Lebewesen, die sich vegetativ
fortpflanzen. Eine Einteilung nach Sporenmerkmalen liefert bestenfalls „Morphotypen“.
Genetische Einteilungen liefern keine schlüssigeren Kriterien, da immer wieder von Polymor-
phien berichtet wurde (s. Kap. 4.9). Selbst wenn man sich auf „Karyotypen“ beschränkt, ist
eine Artabgrenzung problematisch. Auch die Suche nach einem „AMF-Individuum“ ist
schwierig, da nicht einmal einzelne Zellen innerhalb der unseptierten Hyphen zu definieren
sind. Die Sporen sind heteropolykaryotisch, so dass auch in diesem Fall die einzelnen Kerne
als kleinste (individuelle) Einheit anzusehen sind. Im Rahmen dieser Arbeit können deshalb
nur bedingt Aussagen über die Biodiversität der AMF getroffen werden. Einerseits konnten
mit den verfügbaren Mitteln nur einzelne Gruppen der AMF nachgewiesen, andererseits ist
eine Arteinteilung bei Haplonten naturgemäß problematisch. Eine Häufigkeit in Form von
Individuenzahlen kann ebensowenig angegeben werden. Die ermittelten Kopienzahlen stellen
in dieser Hinsicht einen Näherungswert dar, wobei jedoch bisher die Zusammenhänge
zwischen Kopien-, Kern- und Individuenzahl nur unzureichend erforscht sind.

4 Diskussion 92
MERRYWEATHER & FITTER (1998) untersuchten die Morphologie von AMF in Wurzeln
von Hyacinthoides non-scripta und ihre Sporen in der Wurzelzone. Sie fanden dabei in den
Wurzeln sechs Morphotypen (1 Scutellospora, 1 Acaulospora, 3 Glomus, 1 „Endophyt“) und
acht Sporentypen: hauptsächlich S. dipurpurascens und Acaulospora koskei neben Glomus
rubiforme und fünf Acaulospora-Arten. HELGASON et al. (1998) verglichen die AMF-Vertei-
lung in Wald und Ackerland. Sie sequenzierten einen Bereich der SSU rDNA von mykorrhi-
zierten Wurzeln, die mit den Primern AM1 und NS31 amplifiziert worden waren und stellten
signifikante Unterschiede zwischen Wald und Ackerland fest. Von 100 Acker-Sequenzen
ließen sich 92 G. mosseae zuordnen. Daneben fanden sie sechs weiter Glomus-, eine
Acaulospora- und eine Scutellospora-Sequenz. Diese Dominanz von G. mosseae und nah
verwandten Arten wurde in der Folgezeit durch verschiedene Autoren bestätigt. KJØLLER &
ROSENDAHL (2001) untersuchten dänische Ackerflächen, auf denen Erbsen angebaut waren.
Anhand der Sporenmorphologie konnten sie neben G. mosseae, G. geosporum, G. caledonium
und G. claroideum eine Scutellospora-Art feststellen. Die Sequenzierung von 39 Amplifika-
ten einer nested PCR erbrachte zwölf G. intrardices, sieben G. geosporum, achtzehn
G. caledonium und zwei G. fragilistratum-ähnliche Isolate. In drei Fällen konnte jeweils
G. intraradices und G. caledonium gemeinsam in einer Wurzelprobe festgestellt werden. Die
Abwesenheit von G. mosseae-Sequenzen erklären die Autoren mit einer Kolonisierung durch
diesen Pilz zu einem früheren Zeitpunkt. VANDENKOORNHUYSE et al. (2002) fanden bei der
Untersuchung von Arrhenatherum elatius-Wurzeln mit pilzspezifischen Primern (SSU rDNA)
drei AMF-Sequenztypen. Einer davon ließ sich der Gattung Glomus zuordnen, die beiden
anderen wiesen keine nähere Verwandschaft mit irgendeiner bekannten AMF-Art auf.
Aufgrund dieser Erkenntnisse lässt sich eine Artenzahl von bis zu 10 AMF in einem
Ökosystem vorhersagen, wobei auf landwirtschaftlichen Nutzflächen G. mosseae-verwandte
Arten dominant auftreten können. Die vorliegende Arbeit bestätigt diesen Befund: die Anzahl
von sechs verschiedenen Morphotypen stimmt sehr gut mit den Angaben der anderen Autoren
überein. Die Dominanz von G. mosseae im Grünland findet sich ebenfalls bestätigt: etwa 70%
der Sporen konnten als G. mosseae identifiziert werden, weitere 10% wurden der nah
verwandten Art G. geosporum zugeordnet. Auch die molekulargenetischen Untersuchen
ergaben ein verbreitetes Vorkommen von G. mosseae und verwandten Arten bei 17% der
untersuchten Pflanzen.

4 Diskussion 93
4.9 Fortpflanzung und Variabilität der AMF
Bei der langen Entwicklungszeit von über 400 Millionen Jahren und ihrer weltweiten Ver-
breitung hätten sich bei einer rein vegetativen Fortpflanzung weit mehr als die bekannten
166 AMF-Arten entwickeln können, da die divergierenden Genotypen keine Möglichkeit der
Rekombination besitzen. Die hohe genetische Diversität innerhalb einzelner AMF-Sporen ist
seit längerer Zeit bekannt und durch zahlreiche Publikationen belegt. Die spannende Frage, ob
sich die variablen rDNA Sequenzen innerhalb eines Kerns oder unter verschiedenen Nuklei
einer Spore befinden, bleibt ein offenes Rätsel (Redecker et al., 1999).
LLOYD-MACGILP et al. (1996) analysierten verschiedene Klone der ITS-Region aus
Sporen von G. mosseae, G. coronatum, G. fasciculatum und G. dimorphicum. Dabei konnten
sie Unterschiede von bis zu 6% innerhalb eines Isolats oder gar einer Spore feststellen.
Interessanterweise war die Variabilität zwischen Isolaten einer geographischen Region nicht
kleiner als zwischen Isolaten, die von verschiedenen Kontinenten stammten. HIJRI et al.
(1999) sequenzierten Klone von PCR-Produkten der ITS-Region einzelner Sporen. Sie kon-
struierten einen Stammbaum und konnten eine hohe Divergenz dieser Sequenzen feststellen.
REDECKER et al. (1999) konnten zwar anhand von Untersuchungen der 5.8S rDNA nachwei-
sen, dass einige Sequenzen von S. castanea, die sich nicht innerhalb der Glomales einordnen
ließen, zu den Ascomyceten gerechnet werden müssen (Phoma und Leptosphaeria). Sie
schlossen daraus, dass Ascomyceten entweder an der Sporenoberfläche leben, innerhalb der
Sporen als Parasiten vorkommen oder diese Sequenzen tatsächlich Teil des Genoms der AMF
sind. Allerdings wurde die Polymorphie unter den AMF nicht in Frage gestellt. ANTONIOLLI
et al. (2000) analysierten die Variabilität der ITS-Region bei Glomus mosseae und Gigaspora
margarita von einer Weidefläche in Australien. Dabei zeigte sich, dass aus einer einzelnen
Spore bis zu zwölf verschiedene Sequenzen mit einer mittleren Abweichung von 5,6 Substi-
tutionen je 100 Nukleotide (G. mosseae) bzw. neun verschiedenen Sequenzen mit 3,6 Substi-
tutionen (G. margarita) erhalten werden konnten. Datenbankvergleiche der einzelnen
Sequenzen erbrachten hohe Identitätsraten mit Isolaten rund um den Globus (u.a. Finnland,
Indonesien, Venezuela).
Bei den in dieser Arbeit untersuchten Sequenzen konnte keine nennenswerte
Variabilität innerhalb einzelner Isolate festgestellt werden. Obwohl die PCR-Produkte nicht
kloniert worden waren, verliefen die Sequenzierungen ohne Probleme. Nur einzelne Basen
konnten nicht eindeutig identifiziert werden. Diese unbestimmten Basen lagen jedoch nicht in
Bereichen, in denen sich einzelne Stämme voneinander unterschieden, so dass eine mögliche
Heterokaryose für diesen analysierten Bereich der LSU rDNA ausgeschlossen werden kann.

4 Diskussion 94
Die verwendeten spezifischen Primer sind aufgrund des hohen Konservierungsgrades der
LSU rDNA gegenüber der hochvariablen ITS-Region für diese Art der Untersuchung von
Vorteil. Einzelne Isolate von G. mosseae unterschieden sich in einigen wenigen Basen, so
dass unterschiedliche Stämme in allen untersuchten Flächen vorgefunden werden konnten.
VANDENKOORNHUYSE et al. (2001) untersuchten die genetische Diversität zweier
Glomus-Arten mit Hilfe von Inter Simple Sequence Repeat (ISSR) fingerprint. Dabei wurden
Elektrophoresetypen von einzelnen Sporen erstellt und diese miteinander verglichen. Die
hohe Zahl unterschiedlicher Elektrophorese-Profile und polymorpher Loci innerhalb der
beiden Arten ließ sie schlussfolgern, dass Mutationen alleine durch homologe Re-
kombinationen oder ein Kernaustausch durch Anastomose stattgefunden haben muß. Bereits
SANDERS et al. (1996) vermuteten als Grund für die heterokaryotische Natur der AMF den
Austausch von Nuklei durch die Fusion von Hyphen. Giovannetti et al. (1999) konnten tat-
sächlich eine Anastomose bei G. mosseae, G. caledonium und G. intraradices nachweisen.
Dabei fanden Fusionen von Hyphenwänden sowie ein Cytoplasma-Austausch zwischen
Hyphen statt, die aus einer einzigen Spore und mehreren Sporen eines Isolats gewachsen wa-
ren. Es konnten jedoch keine intergenerischen oder interspezifischen Fusionen beobachtet
werden. Bei den ebenfalls untersuchten Arten G. rosea und S. castanea konnte keine
Anastomose dokumentiert werden.
Anastomose ermöglicht den Austausch von genetischem Material. Diese als
Parasexualität bezeichnete Form der Rekombination ohne Meiose und Gameten
(PONTECORVO et al., 1953) würde einerseits die hohe Diversität innerhalb einzelner Sporen
und das Vorhandensein identischer Genotypen in verschiedenen Stämmen erklären, es könnte
aber auch der Grund für die geringe Artenzahl sein. Die sexuelle Fortpflanzung diploider
Organismen kann als „Motor der Evolution“ bezeichnet werden kann, da durch Meiose,
Crossing over und Rekombination immer neue Formen entstehen. Hingegen könnte die
Parasexualität als „Bremse der Evolution“, im Sinne einer geringen Artenzahl, fungiert haben.
Durch die Rekombination einzelner Kerne kann zwar eine hohe Anpassungsfähigkeit erzielt
werden, es entstehen jedoch dadurch keine neuen „Arten“, sondern der mögliche
Formenreichtum wird immer wieder durch eine Durchmischung von Kernen relativiert. Die
phänotypischen Eigenschaften der heteropolykaryotischen haploiden AMF würden demnach
durch Anzahl und Verhältnis der einzelnen Kerntypen beeinflusst. Bei der „normalen“
Sexualität diploider Organismen kann es dagegen vermutlich leichter und schneller zu
Fortpflanzungsbarrieren und damit zur Bildung neuer Arten kommen. Dies könnte auch die
geringe Wirtsspezifität der AMF erklären. Wenn ein Myzel mit einem heterogenen

4 Diskussion 95
Kernrepertoir neue Sporen ausbildet, werden diese zufällig mit Kernen unterschiedlichen
Genotyps ausgestattet. Da die Sporen tausende von Kernen enthalten können, besitzen
wahrscheinlich keine zwei Sporen eine exakt identische Kernzusammensetzung. Entspre-
chend ihrer Umweltbedingungen würden einzelne Sporen einen Vorteil erhalten und be-
stimmte Kerntypen begünstigt werden. Ändern sich die Umweltbedingungen, so könnte durch
Anastomose ein Genfluß stattfinden und die Karyotypen-Population mit „frischem Blut“
(= Kerne) aufgestockt werden.
Eine interessante These stellen RODRIGUEZ et al. (2001) auf. Auch sie hatten unter-
schiedliche Sequenzen bei Einzelsporen von Entrophospora infrequens gefunden, welche sich
zum Teil verschiedenen Glomus-Arten zuordnen ließen. E. infrequens gilt neben
G. globiferum, E. baltica, S. spinosissima und S. projecturata als nicht kultivierbar,
d.h. Einzelsporkulturen schlugen bisher stets fehl. Die Autoren vermuten deshalb, es könnte
sich hierbei um eine Hybrid-Form handeln, bei der genetisches Material (Nuklei)
verschiedener Arten durch Anastomose vermischt wurde. Bisher konnten jedoch nur
Anastomosen zwischen Isolaten einzelner Glomus-Arten dokumentiert werden.
4.10 Phylogenetische und Systematische Neuordnung der Morphospezies
Die ursprüngliche Zuordnung der AMF zur Ordnung Endogonales innerhalb der
Zygomyceten, war aufgrund der Sporokarpienbildung bei Endogone (sexuell) und Glomus
(asexuell) erfolgt. MORTON & BENNY (1990) fassten alle AMF in einer neuen Ordnung
Glomales zusammen, der sechs Gattungen angehörten. ALMEIDA & SCHENCK (1990) nahmen
aufgrund der Sporenposition in den Sporokarpien eine Neuordnung der 13 Sclerocystis-Arten
vor. Sie überführten fünf Arten in die Gattung Glomus und fassten die restlichen zur Art
S. coremioides zusammen. SIMON (1996) fand bei der Untersuchung der SSU rDNA eine
starke Abweichung zwischen Sequenzen von G. etunicatum einerseits, sowie G. mosseae,
G. intraradices, G. vesiculiferum und G. manihotis andererseits. REDECKER et al. (2000a)
kamen zu dem Schluß, dass die letzte verbliebene Sclerocystis-Art, S. coremioides, der
Gattung Glomus zuzuordnen sei. MORTON & REDECKER (2001) gliederten einige Arten der
Acaulosporaceae und Glomaceae aufgrund ihrer genetischen Abweichungen aus und grup-
pierten sie in zwei neu beschriebenen Familien (Archaeosporaceae und Paraglomaceae) mit
jeweils einer neuen Gattung (Archaeospora und Paraglomus; Abb. 48).

4 Diskussion 96
Abb. 48: Stammbaum der Glomales nach MORTON & REDECKER (2001).
SCHWARZOTT et al. (2001) stellten anhand von Untersuchungen der SSU rDNA von
30 Isolaten fest, dass die Gattung Glomus nicht monophyletisch ist. Neben den Familien
Archaesporaceae und Paraglomaceae konnten sie die restlichen Glomus-Vertreter in drei
Gruppen einteilen. Ihre Glomus mosseae-Gruppe (GlGrA) umfaßt in einer Untergruppe
(GlGrAa) u.a. die Arten G. caledonium, G. coronatum, G. fragilistratum, G. geosporum,
G. mosseae und G. verruculosum. Dies entspricht denjenigen Arten, die mit den Primern
Glom85For und Glom387Rev amplifiziert werden konnten. Eine weitere Untergruppe
(GlGrAb) besteht aus den Arten G. clarum, G. coremioides, G. fasciculatum, G. intraradices,
G. manihotis, G. proliferum, G. sinuosum und G. vesiculiferum und die Untergruppe GlGrAc
enthält das Isolat G. sp. W3347. Ihrer Glomus etunicatum-Gruppe (GlGrB) ordnen die Auto-
ren die Arten G. claroideum, G. etunicatum, G. lamellosum, G. luteum, G. manihotis
(clarum), G. microaggregatum und G. viscosum zu. Eine weitere Gruppe (GlGrC), bestehend
aus G. versiforme, G. spurcum und G. etunicatum, steht interessanterweise den
Acaulosporaceae näher als den beiden anderen Glomus-Gruppen. Ungeklärt bleibt dabei die
Stellung von G. manihotis und G. clarum, die sich sehr ähneln und sowohl in GlGrAa als
auch in GlGrB auftauchen. Ebenso finden sich Stämme von G. etunicatum in den Gruppen
GlGrC und GlGrB. Die Autoren schlagen für ihre Glomus-Gruppe C einen neuen Familien-

4 Diskussion 97
namen Diversisporaceae vor. Außerdem konnte nachgewiesen werden, dass Geosiphon
pyriforme (Kap. 4.11) innerhalb der Archaeosporaceae näher mit A. leptotichum verwandt ist
als mit A. trappei. Die Paraglomaceae weisen die größten genetischen Abweichungen im
Vergleich zu allen anderen Familien auf und repräsentieren vermutlich die ursprünglichsten
Vertreter der AMF.
Aus den phylogenetischen Befunden lassen sich verschiedene Schlussfolgerungen zie-
hen: Jedes Isolat, weist es auch eine noch so große morphologische Ähnlichkeit zu anderen
Arten auf, ist primär unbedingt als eigener Stamm bzw. Isolat zu behandeln. Die beiden
Unterordnungen Glominaeae und Gigasporineae sind in dieser Form nicht mehr haltbar. Die
erwogene Paraphylie der Glomales ist durch die neuesten phylogenetischen Erkenntnisse
widerlegt (SCHWARZOTT et al. 2001), so dass nunmehr alle AMF-Arten (inklusive
G. pyriforme) sicher einem gemeinsamen Vorfahren entstammen. Durch die Gruppierung von
G. pyriforme innerhalb der (dadurch paraphyletischen) Archaeosporaceae ergeben sich drei
Möglichkeiten: die Umbenennung G. pyriforme zu Archaeospora pyriforme, eine Zwei-
Familien-Lösung mit einer gemeinsamen Familie (oder Gattung) aus G. pyriforme und
A. leptotichum oder eine Drei-Familien-Lösung mit G. pyriforme, A. leptotichum und
A. trappei in drei Familien (oder Gattungen). Insgesamt sind die molekularbiologischen
Analysemethoden noch nicht erschöpft, so dass umfassende systematische und taxonomische
Änderungen zu erwarten sind. SCHÜßLER et al. (2001b) korrigierten den Ordnungsnamen
Glomales nach den Regeln des International Code of Botanical Nomenclature (GREUTER et
al., 2000) in Glomerales (Glomaceae in Glomeraceae, Paraglomaceae in Paraglomeraceae).
Außerdem erhoben sie die AMF zum neuen Phylum Glomeramycota (mit drei neuen
Ordnungen), das näher mit den Ascomycota und Basidiomycota verwandt ist als mit den
verbleibenden Gruppen der Zygomycota (Abb. 49). Die Diversisporales umfassen die
Gigasporaceae, Acaulosporaceae sowie die vorgeschlagene Familie Diversisporaceae
(Glomus-Gruppe C). Die Archaeosporaceae und Geosiphonaceae bilden die Archaeosporales
und die verbleibenden Paraglomaceae die Paraglomerales. Demnach enthält die Ordnung
Glomerales nur noch die beiden Glomus-Gruppen A und B, die in noch zu benennende
Familien einzuteilen sind. Die Neuordnung der verbliebenen Glomus-Arten in
möglicherweise zwei Familien (GrGlA und GrGlB) mit drei neuen Gattungen (GrGlA-
Untergruppen a bis c) scheitert bisher noch an der Tatsache, dass Untersuchungen der
Typusart G. microcarpum fehlen.

4 Diskussion 98
Abb. 49: Stammbaum der Glomeromycota nach SCHWARZOTT et al. (2001).
4.11 Evolution und Artenzahl der AMF
Die frühesten Fossilfunde der AMF werden auf 400 Millionen Jahre datiert. In fossilen
Aglaophyton major-Wurzeln hatten TAYLOR et al. (1995) Pilzstrukturen gefunden, die iden-
tisch mit heutigen Arbuskeln und Hyphen waren. Die Pilzart wurde als Glomites rhyniensis
beschrieben. REDECKER et al. (2000b) fanden Fossilien von unseptierten Hyphen und Sporen,
die denen der AMF sehr ähnlich waren, mit einem Alter von etwa 460 Millionen Jahren.
Simon et al. (1993b) bezifferten das Alter der Glomaceae, Gigasporaceae und
Acaulosporaceae mit ca. 355 Mio. Jahren. Die Autoren führten jedoch ein sehr viel früheres
Alter für die AMF an, gestützt auf phylogenetische Untersuchungen, das mit dem Erscheinen
der ersten Landpflanzen vor 475 Millionen Jahren zusammenfällt. Während dieser langen
Entwicklungszeit scheinen es manche Pflanzengruppen „vorgezogen“ zu haben, ihren Sym-
biose-Partner „aufzukündigen“ und eine Nährstoffversorgung ohne Mykobionten zu gewähr-
leisten (Trappe, 1987). Nachdem MORTON & BENNY (1990) die AMF in der Ordnung
Glomales zusammenfaßten bürgerte sich allmählich ein, die beiden Begriffe synonym zu
verwenden. GEHRIG et al. (1996) konnten jedoch zeigen, dass Geosiphon pyriforme zu dieser
Ordnung zählt. Dies ist die einzige bekannte Art, die Cyanobakterien (der Gattung Nostoc) als
Endosymbionten besitzt. Sie erinnert damit eher an Pflanzen, deren Chloroplasten ja ur-
sprünglich auch Cyanobakterien waren, oder Flechten als welche sie ursprünglich beschrieben

4 Diskussion 99
wurde. Ob es sich bei G. pyriforme um die ursprüngliche Symbioseform handelt, woraus sich
dann durch Koevolution arbuskuläre Mykorrhizen zwischen AMF und höheren Pflanzen mit
den typischen Arbuskeln entwickeln konnten, oder ob die Symbiose mit Cyanobakterien sich
erst später entwickelte, ist noch nicht geklärt. Da es sich bei AMF um obligate Symbionten
handelt, wäre eine Entwicklung von saprophytischen oder parasitischen Vorläufern über die
Zwischenform der Symbiose mit Cyanobakterien durchaus plausibel. Damit wäre
G. pyriforme der einzige noch existierende Vertreter dieser speziellen Form der Symbiose.
PIROZYNSKI & MALLOCH (1975) sehen das Zusammenspiel einer Alge und eines Oomyzeten
als Vorläufer der AM. Sporen vom Typ Glomus scheinen die ursprüngliche Form darzustel-
len, von der sich im Lauf der Evolution mehrere Seitenzweige mit modifizierter Morpologie
abgezweigt haben. Allerdings müsste dann der Acaulospora-Sporentyp mindestens zweimal
durch Konvergenz entstanden sein, da er sich bei Acaulospora und Archaeospora findet. Eine
andere Möglichkeit wäre, dass ursprünglich acauloide und glomoide Sporen gebildet wurden
– wie heute noch bei Archaospora leptoticha als „lebendem Fossil“ - und der eine oder andere
Sporentyp bei den anderen Arten später verloren ging (Redecker et al. 2000c).
Wie viele AMF-Arten konnten sich im Lauf dieser mehr als 400 Millionen Jahren ent-
wickeln? HAWKSWORTH (2001) schätzt die Zahl der Endogonales- und Glomales-Arten welt-
weit auf 1000. Wenn man diese Zahl entsprechend der derzeit beschriebenen Arten auf die
beiden Gruppen aufteilt, so kann man mit 800 bis 1000 AMF-Arten rechnen. Vor 1974 waren
acht Arten von arbuskuläre Mykorrhizapilzen beschrieben worden. Seitdem kamen ca. 158
hinzu (Anhang B), so dass heute etwa 167 Arten zu den Glomeromycota gezählt werden kön-
nen (inklusive Geosiphon pyriforme). Eine Übersicht der derzeit allgemein anerkannten Arten
findet sich in Anhang A. Während zwischen 1974 und 1989 die Zahl der Arten noch stark
zunahm, sind die Neubeschreibungen in den letzten zehn Jahren auf durchschnittlich drei pro
Jahr gesunken. Neben der Beschreibung neuer Spezies aus dem Freiland, führt auch die
Überprüfung kultivierter Arten zu Neufunden. So wurden z. B. die Arten G. eburneum und
G. luteum von G. spurcum ausgegliedert (KENNEDY et al., 1999). Andererseits wurden einige
Arten als Synonyme entlarvt. SCHWARZOTT et al. (2001) stellten fest, dass sich G. manihotis
und G. clarum genetisch kaum unterscheiden und wohl als eine Art anzusehen sind.
MERRYWEATHER & FITTER (1998) stellten eine hohe genetische Verwandschaft zwischen
S. dipurpurascens und S. calospora fest. Eine enge Verwandtschaft von A. rugosa, A. longula
und A. morrowiae wird ebenfalls diskutiert (INVAM). Die Artenzahlen wurden häufig auf-
grund von Pilz/Pflanzen-Raten und der Wirtsspezifität hochgerechnet. Da die AMF-Arten als
wirtsunspezifisch gelten (SANDERS, 1993) und viele Arten weltweit verbreitet sind (WALKER,

4 Diskussion 100
1992), läßt sich dieses Rechenmodell wohl nicht auf sie übertragen. Ob tatsächlich 1000 Ar-
ten existieren oder jemals beschrieben werden können bleibt ungewiß. Vielleicht wird aber
auch eines Tages der Artbegriff bei den AMF, Prokaryoten und anderen Haplonten
vollständig aufgegeben, zugunsten einer Klassifizierung nach Stämmen. Die hohen
Erwartungen jedenfalls, die in die molekularbiologischen Untersuchungsmethoden zur
Definition von Arten gesetzt wurden, konnten bisher ebensowenig erfüllt werden, wie bei der
klassischen Einteilung anhand der Sporenmorphologie.

5 Zusammenfassung 101
5 Zusammenfassung
In der vorliegenden Arbeit wurden arbuskuläre Mykorrhizapilze (AMF) in
landwirtschaftlichen Nutzflächen bei Plantago lanceolata, Trifolium repens, Holcus lanatus
und anderen Pflanzen mit spezifischen Primer in einer nested PCR nachgewiesen. Neben
Acaulospora longula wurde eine Gruppe nah verwandter Arten um Glomus mosseae erfasst,
die als dominant auf landwirtschaftlichen Flächen betrachtet werden kann. Die Isolate dieser
Gruppe konnten a posteriori durch Sequenzvergleiche als G. mosseae, G. caledonium und
G. geosporum identifiziert werden. Als Zielregion für die PCR diente die große Untereinheit
der ribosomalen DNA (LSU rDNA). Um die Pilze in planta nachweisen zu können, mussten
bestehende DNA-Extraktionsmethoden weiterentwickelt werden. 56 DNA-Extrakte aus
Wurzeln wurden mit universellen Primern als amplifizierbar getestet. A. longula konnte in
mindestens fünf Wurzelproben bei allen drei Grünland-Pflanzen nachgewiesen werden. Außer
in diesen Pflanzen wurde G. mosseae auch in Helianthus annuus und Triticum aestivum
gefunden. Die G. caledonium-Sequenzen stammten aus H. lanatus und Zea mays sowie G.
geosporum aus P. lanceolata. Aufgrund dieser Verbreitung konnte keine Wirtsspezifität der
untersuchten AMF festgestellt werden. Die PCR-Produkte der G. mosseae-Gruppe wurden
mit Endonukleasen behandelt, um Restriktionsmuster zu erhalten. Mit dem Enzym AluI
konnte G. mosseae von den übrigen Arten unterschieden werden. Von den gewonnenen
Sequenzdaten konnte unter Einbeziehung von Datenbank-Sequenzen ein phylogenetischer
Stammbaum erstellt werden. Mithilfe der quantitativen Taqman® real-time PCR wurde die
Kopienzahl der Mykorrhizapilze bestimmt. Von A. longula konnten bis zu 13.300 Kopien und
von G. mosseae bis zu 64.750 Kopien je mg Wurzel nachgewiesen werden.
Die Pilze wurden in der Wurzel mit Trypanblau gefärbt, die Kolonisierungsraten
mikroskopisch bonitiert und mit den pflanzenverfügbaren Phosphatgehalten der Rhizosphäre
korreliert. Aufgrund der hohen Standardabweichungen konnte kein signifikanter
Zusammenhang zwischen Mykorrhizierung und Standort, Jahreszeit, Pflanze oder
Phosphatverfügbarkeit festgestellt werden. Die Ergebnisse deuten jedoch darauf hin, dass die
Kolonisierung mit Hyphen im Jahresverlauf zunimmt, während die Vesikelzahl abzunehmen
scheint. Vermutlich werden die Speicherstoffe der Vesikel gegen Ende des Jahres zur Bildung
von Sporen extraradikal verlagert. P. lanceolata wies generell die höchste Hyphen-
Kolonisierungsrate auf und bei H. lanatus scheint es einen indirekten Zusammenhang
zwischen Hyphendichte und Phosphatkonzentration zu geben. Außerdem wurde eine
Sporenbestimmung vorgenommen, bei der sechs verschiedene Sporentypen unterschieden
werden konnten. Der häufigste Sporentyp ähnelte dabei G. mosseae.

6 Literatur 102
6 Literatur
ABU AL-SOUD W, RADSTRÖM P (1988) Capacity of Nine Thermostable DNA Polymerases To
Mediate DNA Amplification in the Presence of PCR-Inhibiting Samples. Appl Environ
Microbiol 64: 3748-3753
ALLEN MF, SEXTON JC, MOORE TS, CHRISTENSEN M (1981) Influence of phosphate source
on vesicular-arbuscular mycorrhizae of Bouteloua gracilis (H.B.K.) Lag ex Steud. New
Phytol 88: 683-693
ALLEN EB, ALLEN MF (1990) The Mediation of Competition by Mycorrhizae in Successional
and Patchy Environments. In: GRACE B, TILMAN D (eds.) Perspectives on Plant
Competition. Academic Press, San Diego, pp. 376-389
ALMEIDA RT, SCHENCK NC (1990) A Revision of the Genus Sclerocystis (Glomaceae,
Glomales). Mycologia 82: 703-714
ALTSCHUL SF, MADDEN TL, SCHÄFFER AA, ZHANG J, ZHANG Z, MILLER W, LIPMAN DJ
(1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search
programs. Nucleic Acids Res 25: 3389-3402
AMIJEE F (1989) Vesicular-arbuscular mycorrhizas: an ubiquitous symbiosis between fungi
and roots of vascular plants. Mycologist 3: 176-180
ANTONIOLLI ZI, SCHACHTMAN DP, OPHEL-KELLER K, SMITH SE (2000) Variation in rDNA
ITS sequences in Glomus mosseae and Gigaspora margarita spores from a permanent
pasture. Mycol Res 104: 708-715
ARNHEIM N (1983) Concerted evolution of multigene families. In: NEI M, KOEHN RK (eds.)
Evolution of Genes and Proteins. Sinauer, Sunderland, Mass., pp. 38-61
AZCON-AGUILAR C, BAREA JM (1995) Saprophytic growth of arbuscular mycorrhizal fungi.
In: VARMA A, HOCK B (eds.) Mycorrhiza. Springer, Berlin, pp. 391-407
BAGO B (2000) Putative sites for nutrient uptake in arbuscular mycorrhizal fungi. Plant and
Soil 226: 263-274
BAHNWEG G, SCHULZE S, MÖLLER EM (1998) DNA isolation from recalcitrant materials
such as trees, roots, bark and forest soil for the detection of fungal pathogens by
polymerase chain reaction. Analytical Biochemistry 262: 79-82

6 Literatur 103
BAREA JM, JEFFRIES P (1995) Arbuscular mycorrhizas in sustainable soil-plant systems. In:
VARMA A, HOCK B (eds.) Mycorrhiza. Springer, Berlin, pp. 521-560
BÉCARD G, PFEFFER PE (1993) Status of nuclear division in arbuscular mycorrhizal fungi
during in vitro development. Protoplasma 174: 62-68
BETHLENVALVAY GJ, SCHÜEPP H (1994) Arbuscular mycorrhizas and agrosystem stability. In:
GIANINAZZI S, SCHÜEPP H (eds.) Impact of arbuscular mycorrhizas on sustainable
agriculture and natural ecosystems. Birkhäuser, Basel, pp. 117-131
BERRECK M, HASELWANDTER K (2001) Effect of the arbuscular mycorrhizal symbiosis upon
uptake of cesium and other cations by plants. Mycorrhiza 10: 275-280
BIANCIOTTO V, BONFANTE P (1992) Quantification of the nuclear DNA content of two
aruscular mycorrhizal fungi. Mycol Res 96: 1071-1076
BLUME HP (1992) Handbuch des Bodenschutzes Bodenökologie und –belastung –
Vorbeugende und abwehrende Schutzmaßnahmen. Ecomed, Landsberg
BOBOWSKI BR, HOLE D, WOLF PG (1999) Identification of roots of woody species using
polymerase chain reaction (PCR) and restriction fragment length polymorphism (RFLP)
analysis. Mol Ecol 8: 485-491
BÖHM J (2000) Artspezifischer Nachweis arbuskulärer Mykorrhizapilze mit Hilfe von
Sequenzanalysen der ribosomalen DNA. Dissertation, Technische Universität München in
Weihenstephan, Lehrstuhl für Botanik
BÖHM J, HAHN A, SCHUBERT R, BAHNWEG G, ADLER N, NECHWATAL J, OEHLMANN R,
OßWALD W (1999) Real-time quantitative PCR: DNA Determination in Isolated Spores of
the Mycorrhizal Fungus G. mosseae and Monitoring of Phytophtora infestans and
Phytophtora citricola in their Respective Host Plants. J Phytopathology 147: 409-416
BOITEUX LS, FONSECA MEN, SIMON PW (1999) Effects of plant tissue and DNA purification
method on randomly amplified polymorphic DNA-based genetic fingerprinting analysis in
carrot. J Amer Soc Hort Sci 124: 32-38
BONFANTE P, PEROTTO S (1995) Strategies of arbuscular mycorrhizal fungi when infecting
host plants. New Phytol 130:3-21
BONFANTE–FASOLO P (1984) Anatomy and morphology of VA mycorrhizae. In: POWELL CL,
BAGYARAJ DJ (eds.) VA Mycorrhiza. CRC Press, Boca Raton, pp. 5-33

6 Literatur 104
BRUNDRETT M, BOUGHER N, DELL B, GROVE T, MALAJCZUK N (1996) Working with
Mycorrhizas in Forestry and Agriculture. ACIAR Monograph 32, Canberra
BUCHANAN BB, GRUISSEM W, JONES RC (2000) Biochemistry and Molecular Biology of
Plants. Courier Companies, p. 328
BURGGRAAF AJP, BERINGER JE (1989) Absence of nuclear DNA synthesis in vesicular-
arbuscular mycorrhizal fungi during in vitro development. Mycol Res 96: 25-33
CABELLO M, GASPAR L, POLLERO R (1994) Glomus antarcticum sp. nov., a vesicular-
arbuscular mycorrhizal fungus from Antarctica. Mycotaxon 51: 123-128
CLAPP JP, YOUNG JPW, MERRYWEATHER J W (1995) Diversity of fungal symbionts in
arbuscular mycorrhizas from a natural community. New Phytol 130: 259-265
CLAPP JP, FITTER H, YOUNG JPW (1999) Ribosomal small subunit sequence variation within
spores of an arbuscular mycorrhizal fungus, Scutellospora sp. Mol Ecol 8: 915-921
DANIELS BA, SKIPPER HD (1984) Methods for the recovery and quantitative estimation of
propagules from soil. In: SCHENCK NC (ed.) Methods and Principles of Mycorrhizal
Research. St. Paul, Minnesota, pp. 29-35
DAVIES FT, POTTER JR, LINDERMAN RG (1993) Drought Resistance of mycorrhizal pepper
plants independent of leaf P-concenctration – response in gas-exchange and water
relations. Physiologia Plantarum 87: 45-53
DELP G, ROSEWARNE GM, BARKER S (1998) The molecular route to understanding VAM
symbiosis. In: VARMA A, HOCK B (eds.) Mycorrhiza. Springer, Berlin, pp. 327-352
DEMEKE T, ADAMS RP (1992) The effect of plant polysaccharides and buffer aditives on
PCR. BioTechniques 12: 332-334
DOYLE JJ, DOYLE JL (1987) A rapid DNA isolation procedure for small quantities of fresh
leaf tissue. Phytochemical Bulletin 19: 11-15
DUMAT-GAUDOT E, FURLAN V, GRENIER J, ASSELIN A (1992) New acidic chitinase isoforms
induced in tobacco roots by vesicular-arbuscular mycorrhizal fungi. Mycorrhiza 1: 133-
136
ENARD W, KHAITOVICH P, KLOSE J, ZÖLLNER S, HEISSIG F, GIAVALISCO P, NIESELT-STRUWE
K, MUCHMORE E, VARKI A, RAVID R, DOXIADIS GM, BONTROP RE, PÄÄBO S (2002) Intra-
and Interspecific Variation in Primate Gene Expression Patterns. Science 296: 340-342

6 Literatur 105
EOM A-H, WILSON GWT, HARTNETT DC (2001) Effects of ungulate grazers on arbuscular
mycorrhizal symbiosis and fungal community structure iin tallgrass prairies. Mycologia
93: 233-242
FIEDLER HJ (2001) Böden und Bodenfunktionen in Ökosystemen, Landschaften und
Ballungsgebieten. Expert, Renningen-Malmsheim, Forum EIPOS, Band 7
FINCK A (1991) Düngung. Ulmer, Stuttgart
FITTER AH, GRAVES JD, WATKINS NK, ROBINSON D, SCRIMGEOUR C (1998) Carbon transfer
between plants and its control in networks of arbuscular mycorrhizas. Funct Ecol 12: 406-
412
FRANK B (1885) Über die auf Wurzelsymbiose beruhende Ernährung gewisser Bäume durch
unterirdische Pilze. Ber Deutsch Bot Ges 3: 128-145
FRANKEN P, GNADINGER F (1994) Analysis of parsley arbuscular endomycorrhiza: infection
development and mRNA levels of defense related genes. Mol Plant Microbe Interact 7:
612-620
GALVEZ L, DOUDS DD, WAGONER P, LONGNECKER LR, DRINKWATER LE, JANKE RR (1995)
An overwintering cover crop increases inoculum of VAM fungi in agricultural soil.
American Journal of Alternative Agriculture 10: 152-156
GANGE AC, BOWER E, STAGG PG (1999) A comparison of visualization techniques for
recording arbuscular mycorrizal colonization. New Phytol 142: 123-132
GARDES M, BRUNS TD (1993) ITS primers with enhanced specifity for basidiomycetes
application to the identification of mycorrhizae and rusts. Mol Ecol 2: 113-118
GEHRIG H, SCHÜßLER A, KLUGE M (1996) Geosiphon pyriforme, a Fungus forming
endocytobiosis with Nostoc (Cyanobacteria), is an ancestral member of the Glomales:
Evidence by SSU rRNA analysis. J Mol Evol 43: 71-81
GEHRIG H, VOLZ F, HEUTE V (1998) DNA from difficult plant and fungal species using
DNeasy Plant Kits. QIAGENNews 2: 14-17
GERDEMANN JW, NICOLSON TH (1963) Spores of mycorrhizal Endogone extracted from soil
by wet sieving and decanting. Trans Br Myc Soc 46: 235-244
GIANINAZZI S (1991) Vesicular-arbuscular (endo-) mycorrhizas: cellular, biochemical and
genetic aspects. Agricult, Ecosys Environm 35: 105-119

6 Literatur 106
GIOVANNETTI M, MOSSE B (1980) An evaluation of techniques for measuring vesicular-
arbuscular mycorrhizal infection in roots. New Phytol 8: 489-500
GIOVANNETTI M, SBRANA C, CIERNESI AS (1996) Analysis of factors involved in fungal
recognition responses to host-derived signals by arbuscular mycorrhizal fungi. New Phytol
133: 65-71
GIOVANNETTI M, AZZOLINI D, CITERNESI AS (1999) Anastomosis Formation and Nuclear and
Protoplasmic Exchange in Arbuscular Mycorrhizal Fungi. Appl Environ Microbiol
65: 5571-5575
GREUTER W, MCNEILL J, BARRIE FR, BURDET H-M, DEMOULIN V, FILGUEIRAS TS,
NICOLSON DH, SILVA PC, SKOG JE, TREHANE P, TURLAND NJ, HAWKSWORTH DL (2001)
International Code of Botanical Nomenclature. Regnum Vegetabile 131. Koeltz Scientific
Books, Königstein
HAHN A, BONFANTE P, HORN K, PAUSCH F, HOCK B (1993) Production of monoclonal
antiodies against surface antigens of spores from arbuscular mycorrhizal fungi by an
improved immunization and screening procedure. Mycorrhiza 4: 69-78
HALL TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis
program for Windows 95/98/NT. Nucl Acids Symp Ser 41: 95-98
HARRISON MJ, DIXON RA (1994) Spatial patterns of expression of flavonoid/isoflavonoid
pathway genes during interactions between roots of Medicago truncatula and the
mycorrhizal fungus Glomus versiforme. Plant J 6: 9-20
HAWKSWORTH DL (2001) The magnitude of fungal diversity: the 1.5 million species estimate
revisited. Mycol Res 105: 1422-1432
HEID CA, STEVENS J, LIVAK KJ, WILLIAMS PM (1996) Real Time Quantitative PCR. Genome
Res 6: 986-994
HELGASON T, DANIELL TJ, HUSBAND R (1998) Ploughing up the wood-wide web? Nature
394: 431
HIGUCHI R, WATSON R (1999) Kinetic PCR analysis using a CCD camera and without using
oligonulceotide probes. In: INNIS MA, GELFAND DH, SNINSKY JJ (eds.) PCR Applications.
Protocols for functional Genomics. Academic Press, San Diego, pp. 263-284

6 Literatur 107
HIJRI M, HOSNY M, VAN TUINEN D (1999) Intraspecific ITS polymorphism in Scutellospora
castanea (Glomales, Zygomycota) is structured within mulinucleate spores. Fung Genet
Biol 26: 141-151
HOFFMANN G (1991) Methodenbuch I. Die Untersuchung von Böden. VDLUFA-Verlag
Darmstadt
HOLLAND PM, ABRAMSON RD, WATSON R, GELFAND DH (1991) Detection of specific
polymerase chain reaction product by utilizing the 5‘�3‘ exonuclease activity of Thermus
aquaticus DNA polymerase. Proc Natl Acad Sci USA 88: 7276-7280
HOLLEMANN AF, WIBERG N (1985) Lehrbuch der Anorganischen Chemie. 33. Edition, de
Gruyter, Berlin
HOOKER JE, JAIZME-VEGA M, ATKINSON D (1994) Biocontrol of plant pathogens using
arbuscular mycorrhizal fungi. In: GIANINAZZI S, SCHÜEPP H (eds.) Impact of Arbuscular
Mycorrhizas on Sustainable Agriculture and Natural ecosystems. Birkäuser, Basel, pp.
191-200
HOSNY M, GIANINAZZI-PEARSON V, DULIEU H (1998) Nuclear content of 11 fungal species in
Glomales. Genome 41: 422-428
HOSNY M, HIJRI M, PASSERIEUX E, DULIEU H (1999) rDNA units are highly polymorphic in
Scutellospora castanea (Glomales, Zygomycetes). Gene 226: 61-71
INNIS MA, GELFAND DH (1990) Optimization of PCRs. In: INNIS MA, GELFAND DH,
SNINSKY JJ (eds.) PCR protocols, a guide to methods and applications. Academic Press,
San Diego pp. 3-11
JAKOBSEN I (1995) Transport of phosphorus and carbon in VA mycorrhizas. In: VARMA A,
HOCK B (eds.) Mycorrhiza. Springer, Berlin, pp. 297-324
JAKOBSEN I, JONER EJ, LARSEN J (1994) Hyphal phosphorus transport, a keystone to
mycorrhizal enhancement of plant growth. In: GIANINAZZI S, SCHÜEPP H (eds.) Impact of
Arbuscular Mycorrhizas on Sustainable Agriculture and Natural ecosystems. Birkhäuser,
Basel, pp. 133-146
KENNEDY LJ, STUTZ JC, MORTON JB (1999) Glomus eburneum and G. luteum, two new
species of arbuscular mycorrhizal fungi, with emendation of G. spurcum. Mycologia 91:
1083-1093

6 Literatur 108
KJØLLER R, ROSENDAHL S (2001) Molecular diversity of glomalean (arbuscular mycorrhizal)
fungi determined as distinct Glomus specific DNA sequences from roots of field grown
peas. Mycol Res 105: 1027-1032
KNIPPERS R (2001) Molekulare Genetik. Thieme, Stuttgart
KOKILERA L (1995) Comparative study of induction of endogenous DNA degradation in rat
liver nuclei and intact thymocytes. Comp Biochem Physiol 111 B: 35-43
KORMANIK PP, MCGRAW AC (1984) Quantification of vesicular-arbuscular mycorrhizae in
plant roots In: SCHENCK NC (ed.) Methods and Principles of Mycorrhizal Research. The
American Phytopathological Society, St. Paul, Minnesota, pp. 37-45
KRAMADIBRATA K, WALKER C, SCHWARZOTT D (2000) A New Species of Scutellospora with
a Coiled Germination Shield. Annals of Botany 86: 21-27
KREISEL H, SCHAUER F (1987) Methoden des mykologischen Laboratoriums. Fischer,
Stuttgart
LANFRANCO L, WYSS P, MARZACHI C (1995) Generation of RAPD-PCR primers for the
identification of isolates of G. mosseae, an arbuscular mycorrhizal fungus. Mol Ecol 4: 61-
65
LEE LG, CONNELL CR, BLOCH W (1993) Allelic discrimination by nick-translation PCR with
fluorogenic probes. Nucleic Acids Res 21: 3761-3766
LINDER CR, MOORE LA, JACKSON RB (2000) A Universal Molecular Method for Identifying
Underground Plant Parts to Species. Mol Ecol 9: 1549-1559
LLOYD-MACGILP SA, CHAMBERS SM, DODD JC (1996) Diversity of the ribosomal internal
transcribed spacers within and among isolates of G. mosseae and related mycorrhizal
fungi. New Phytol 133: 103-111
LONGATO S, BONFANTE P (1997) Molecular identification of mycorrhizal fungi by direct
amplification of microsatellite regions. Mycol Res 133: 103-112
MALDONADO-MENDOZA I, DEWBRE GR, VAN BUUREN ML, VERSAW WK, HARRISON MJ
(2002) Methods to estimate the proportion of plant and fungal RNA in an arbuscular
mycorrhiza. Mycorrhiza 12: 67-74
MCGONIGLE TP, MILLER MH, EVANS DG, FAIRCHILD GL, SWAN JA (1990) A new method
which gives an objective measure of colonization of roots by vesicular-arbuscular
mycorrhizal fungi. New Phytol 115: 495-501

6 Literatur 109
MERRYWEATHER J, FITTER A (1998) The arbuscular mycorrhizal fungi of Hyacinthoides non-
scripta I. Diversity of fungal taxa. New Phytol 138: 117-129
MORTON JB (1995) Taxonomic and phylogenetic divergence among five Scutellospora
species based on comparative developmental sequences. Mycologia 87: 127-137
MORTON JB, BENNY GL (1990) Revised classification of arbuscular mycorrhizal fungi
(Zygomycetes): A new order, Glomales, two new suborders, Glomineae and
Gigasporineae, and two new families, Acaulosporaceae and Gigasporaceae, with an
emendation of Glomaceae. Mycotaxon, 37: 441-491
MORTON JB, REDECKER D (2001) Two new families of Glomales, Archaeosporaceae and
Paraglomaceae, with two new genera Archaeospora and Paraglomus, based on concordant
molecular and morphological characters. Mycologia 93: 181-195
MORTON JB, BENTIVENGA SP, WHEELER WW (1993) Germplasm in the International
Collection of Arbuscular and Vesicular-Arbuscular Mycorrhizal Fungi (INVAM) and
procedures for culture development, documentation and storage. Mycotaxon 48: 491-528
MÜLHARDT C (2000) Molekularbiologie. Spektrum, Heidelberg
MULLIS KB, FALOONA F, SCHAARF S, SAIKI R, HORN G, ERLICH H (1986) Specific enzymatic
amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp
Quant Biol 51: 263-273
MURRAY MG, THOMPSON WF (1980) Rapid isolation of high molecular weight plant DNA.
Nucleic Acid Research 8: 4321-4325
PARKE JL, LINDERMAN RG (1980) Association of vesicular-arbuscular mycorrhizal fungi with
the moss Funaria hygrometrica. Can J Bot 58: 1898-1904
PETERSON RL, FARQUHAR ML (1994) Mycorrhizas – integrated development between roots
and fungi. Mycologia 86: 311-326
PETERSON RL, HOWARTH ML, WHITTIER DP (1981) Interactions between a fungal endophyte
and gametophyte cells in Psilotum nudum. Canad J Bot 59: 711-720
PFEFFER PE, DOUDS JR. DD, BÉCARD G (1999) Carbon Uptake and the Metabolism and
Transport of Lipids in an Arbuscular Mycorrhiza. Plant Physiology 120: 587-598
PIROZYNSKI KA, MALLOCH DW (1975) The Origin of Land Plants: A Matter of
Mycotrophism. BioSystems 6: 153-164

6 Literatur 110
PONTECORVO G, ROPFER JA, HEMMONS LJ, MACDONALD KD, BUFTON AWJ (1953) The
genetics of Aspergillus nidulans. Adv Genet 5: 141-238
POWELL SJ (1995) Protocol optimization and reaction specifity. In: NEWTON CR (ed.) PCR
essential data. John Wiley & Sons, Chichester, pp. 72-86
POWELL CL, DANIEL J (1978) Mycorrhizal fungi stimulate uptake of soluble and insoluble
phosphate fertilizer from phosphate deficient soil. New Phytol 80: 351-358
REDECKER D (2000) Specific PCR primers to identify arbuscular mycorrhizal fungi within
colonized roots. Mycorrhiza 10: 73-80
REDECKER D, THIERFELDER H, WALKER C (1997) Restriction analysis of PCR-amplified
internal transcribed spacers of ribosomal DNA as a tool for species identification in
different genera of the order Glomales. Appl Environ Microbiol 63: 1756-1761
REDECKER D, HIJRI M, DULIEU H (1999) Phylogenetic Analysis of a Dataset of Fungal 5.8S
rDNA Sequences Shows That Highly Divergent Copies of Internal Transcribed Spacers
Reported from Scutellospora castanea Are of Ascomycete Origin. Fungal Genetics and
Biology 28: 238-244
REDECKER D, MORTON JB, BRUNS TD (2000a) Molecular phylogeny of the arbuscular
mycorrhizal fungi Glomus sinuosum and Sclerocystis coremioides. Mycologia 92: 282-285
REDECKER D, KODNER R, GRAHAM LE (2000b) Glomalean Fungi from the Ordovician.
Science 289: 1920-1921
REDECKER D, MORTON JB, BRUNS TD (2000c) Ancestral lineages of arbuscular mycorrhizal
fungi (Glomales). Mol Phylogenet Evol 14: 276-284
RODRIGUEZ A, DOUGALL T, DODD JC, CLAPP JP (2001) The large subunit ribosomal RNA
genes of Entrophospora infrequens comprise sequences related to two different glomalean
families. New Phytologist 152: 159-167
ROGERS S, BENDICH AJ (1987) Ribosomal RNA genes in plants: variability in copy number
and in the intergenic spacer. Plant Mol Biol 9: 509-520
ROSENDAHL S (1996) A practical Approach to the Species Concept in the Glomales. In:
Mycorrhizas in Integrated Systems From Genes to Plant Development – Proceedings of the
4th European Symposium on Mycorrhizas. Granada, pp. 15-18
ROSENDAHL S, SEN R (1992) Isozyme analysis of mycorrhizal fungi and their mycorrhiza.
Meth Microbiol 24: 169-194

6 Literatur 111
ROWLAND LJ, NGUYEN B (1993) Use of Polyethylene Glycol for Purification of DNA from
Leaf Tissue of Woody Plants. BioTechniques 14: 735-736
SAIKI RK (1990) Amplification of Genomic DNA In: INNIS MA, GELFAND DH, SNINSKY JJ
(eds.) PCR protocols. Academic Press, San Diego, pp. 13-20
SAMBROOK J, FRITSCH EF, MANIATIS T (1989) Molecular cloning. Cold Spring Harbour
Laboratory Press, New York
SANDERS IR, FITTER AH (1992) The ecology and functioning of vesicular-arbuscular
mycorrhizas in co-existing grassland spezies. New Phytol 120: 517-524
SANDERS IR, ALT M, GROPPE K (1995) Identification of ribosomal DNA polymorphisms
among and within spores of the Glomales: application to studies on the genetic diversity of
arbuscular mycorrhizal fungal communities. New Phytol 130: 419-427
SANDERS IR, CLAPP JP, WIEMKEN A (1996) The genetic diversity of arbuscular mycorrhizal
fungi in natural ecosystems - a key to understanding the ecology and functioning of
mycorrhizal symbiosis. New Phytol 133: 123-134
SAUNDERS GC, PARKES HC (1999) Analytical molecular biology. The Royal Society of
Chemistry, Cambridge
SAWAKI H, SUGAWARA K, SAITO M (1998) Phylogenetic position of an arbuscular
mycorrhizal fungus, Acaulospora gerdemannii, and its synanamorph, Glomus leptochilum,
based upon 18S rRNA gene sequence. Mycoscience 39: 477-480
SCHENCK NC, PÉREZ Y (1990) Manual for the identification of VA mycorrhizal fungi. 3rd ed.,
Synergistic Publications, Gainesville
SCHILD TA (1996) Einführung in die Real-Time TaqMan PCR-Technologie. 7700 SDS
Workshop, PE Applied Biosystems, Weiterstadt
SCHMID E, OBERWINKLER F (1993) Mycorrhiza-like interaction between the achlorophyllous
gametophyte of Lycopodium clavatum L. and its fungal endophyte studied by light and
electron mycroscopy. New Phytol 124: 69-81
SCHUBERT R, HILBIG W, KLOTZ S (2001) Bestimmungsbuch der Pflanzengesellschaften
Deutschlands. Spektrum, Heidelberg
SCHÜßLER A, GEHRIG H, SCHWARZOTT D (2001a) Analysis of partial Glomales SSU rRNA
gene sequences: implications for primer design and phylogeny. Mycol Res 105: 5-15

6 Literatur 112
SCHÜßLER A, SCHWARZOTT D, WALKER C (2001b) A new fungal phylum, the
Glomeromycota: phylogeny and evolution. Mycol Res 105: 1413-1421
SCHWARZOTT D, WALKER C, SCHÜßLER A (2001) Glomus, the Largest Genus of the
Arbuscular Mycorrhizal Fungi (Glomales), Is Nonmonophyletic. Mol Phylogenet Evol
21: 190-197
SHACHAR-HILL Y, PFEFFER PE, DOUDS D, OSMAN SF, DONER LW, RATCLIFFE RG (1995)
Partitioning of intermediate carbon metabolism in VAM colonized leek. Plant Physiol 108:
7-15
SIMON L (1996) Phylogeny of the Glomales: deciphering the past to understand the present.
New Phytol 133: 95-101
SIMON L, LALONDE M, BRUNS TD (1992) Specific Amplification of 18S Fungal Ribosomal
Genes from Vesicular-Arbuscular Endomycorrhizal Fungi Colonizing Roots. Appl
Environm Microbiol 58: 291-295
SIMON L, LÉVESQUE RC, LALONDE M (1993a) Identification of Endomycorrhizal Fungi
Colonizing Roots by Fluorescent Single-Strand Conformation Polymorphism-Polymerase
Chain Reaction. Appl Environ Microbiol 59: 4211-4215
SIMON L, BOUSQUET J, LÉVESQUE RC (1993b) Origin and diversification of endomycorrhizal
fungi and coincidence with vascular land plants. Nature 363: 67-69
SMITH SE, GIANINAZZI-PEARSON V (1988) Physiological interactions between symbionts in
vesicular-arbuscular mycorrhizal plants. Annu Rev Plant Physiol Plan Mol Biol 39: 221-
244
SMITH SE, READ DJ (1997) Mycorrhizal symbiosis. Academic Press, San Diego
SNIDER J (1999) Whole cell assays. In: INNIS MA, GELFAND DH, SNINSKY JJ (eds.) PCR
Protocols. Academic Press, San Diego, pp. 329-333
STAHL M (1949) Die Mykorrhiza der Lebermoose mit besonderer Berücksichtigung der
thallösen Formen. Planta 37: 103-148
STREITWOLF-ENGEL R, BOLLER T, WIEMKEN A (1997) Clonal traits of two Prunella species
are determined by co-occuring arbuscular mycorrhizal fungi from a calcareous grassland. J
Ecol 85: 181-191
SYLVIA DM, HUBBELL DH (1986) Growth and sporulation of vesicular-arbuscular
mycorrhizal fungi in aeroponic and membrane systems. Symbiosis 1: 259:267

6 Literatur 113
TAYLOR TN, REMY W, HASS H (1995) Fossil arbuscular mycorrhizae from the Early
Devonian. Mycologia 87: 560-573
TEBBE C, VAHJEN W (1993) Interference of Humic Acids and DNA Extracted Directly from
Soil in Detection and Transformation of Recombinant DNA from Bacteria and a Yeast.
Appl Environ Microbiol 59: 2657-2665
TOTH R, MILLER RM, JARSTFER A, ALEXANDER A, BENNET EL (1991) The calculation of
intraradical fungal biomass from percent colonisation in vesicular-arbuscular mycorrhizae.
Mycologia 83: 553-558
TRAPPE JM (1987) Phylogenetic and ecologic aspects of mycotrophy in the angiosperms from
an evolutionary standpoint. In: SAFIR GR (ed.) Ecophysiology of VA Mycorrhizal plants.
CRC Press, Boca Raton, pp. 5-25
TROUVELOT A, KOUGH JL, GIANINAZZI-PEARSON V (1986) Mesure du taux de mycorhization
VA d'un système radiculaire. Recherche de méthodes d'estimation ayant une signification
fonctionnelle. In: GIANINAZZI-PEARSON V, GIANINAZZI S (eds.): Physiological and genetic
aspects of mycorrhizae. Institut National de la Recherche Agronomique, Paris, pp. 217-221
TULASNE LR, TULASNE C (1845) Fungi nonnulli hipogaei, novi v. minus cognito act. Giorn
Bot Ital 2: 55-63
VAN DER HEIJDEN MGA, KLIRONOMOS JN, URSIC M (1998a) Mycorrhizal fungal diversity
determines plant biodiversity ecosystem variability and productivity. Nature 396: 69-72
VAN DER HEIJDEN MGA, BOLLER T, WIEMKEN A (1998b) Different arbuscular mycorrhizal
fungal species are potential determinants of plant community structure. Ecology 79: 2082-
2091
VAN TUINEN D, JACQUOT E, ZHAO B (1998) Characterization of root colonization profiles by
a microcosm community of arbuscular mycorrhizal fungi using 25S rDNA-targeted nested
PCR. Mol Ecol 7: 879-887
VANDENKOORNHUYSE P, LEYVAL C, BONNIN I (2001) High genetic diversity in arbuscular
mycorrhizal fungi: evidence for recombination events. Heredity 87: 243-253
VANDENKOORNHUYSE P, BALDAUF SL, LEYVAL C, STRACZEK J, YOUNG JPW (2002)
Extensive Fungal Diversity in Plant Roots. Science 295: 2051
WALKER C (1992) Systematics and taxonomy of the arbuscular endomycorrhizal fungi
(Glomales) - a possible way forward. Agronomie 12: 887-897

6 Literatur 114
WALKER C, TRAPPE JM (1993) Names and epithets in the Glomales and Endogonales. Mycol
Res 97: 339-344
WALKER C, VESTBERG M (1994) A simple and inexpensive method for producing and
maintaining closed pot cultures of aruscular mycorrhizal fungi Agric Sci Finland 3: 233-
240
WALLEY FL, GERMIDA JJ (1996) Failure to decontaminate Glomus clarum NT4 spores is due
to spore wall-associated bacteria. Mycorrhiza: 6: 43-49
WATKINS NK, FITTER AH, GRAVES JD, ROBINSON D (1996) Carbon transfer between C3 and
C4 plants linked by a common mycorrhizal network, quantified using stable carbon
isotopes. Soil Biol Biochem 28: 471-477
WEISSENHORN I, FELDMANN F (1999) Perspektiven der Nutzung der arbuskulären Mykorrhiza
im niederländischen Gartenbau unter Glas. Mitteilungen der Biologischen Bundesanstalt,
363
WHITE TJ, BRUNS T, LEE S (1990) Amplification and direct sequencing of fungal ribosomal
RNA genes for phylogenetics. In: INNIS MA, GELFAND DH, SNINSKY JJ (eds.) PCR
protocols, a guide to methods and applications. Academic Press, San Diego, pp. 315-322

7 Anhang 115
7 Anhang
A. Übersicht der AMF-Arten
In der folgenden Übersicht sind sämtliche AMF-Arten (inklusive Geosiphon pyriforme) und
noch gebräuchliche Synonyme mit Autoren und Jahr der Beschreibung aufgeführt. Die
Gliederung entspricht dem neuesten Stand (SCHÜßLER et al., 2001b). Die Unterteilung der
Glomus-Arten ist bereits – soweit möglich – berücksichtigt. Glomus-Arten, die aufgrund
mangelnder Sequenzdaten bisher keiner der drei Gruppen zugeteilt werden konnten, werden
weiterhin in der Gattung Glomus geführt. Die Bezeichnungen entsprechen den Korrekturen
von WALKER & TRAPPE (1993) und SCHÜßLER et al. (2001b). Arten, von denen genetische
Informationen in der NCBI-Datenbank vorliegt, sind mit einem § gekennzeichnet.
STAMM GLOMEROMYCOTA WALKER & SCHÜßLER 2001
KLASSE GLOMEROMYCETES CAVALIER-SMITH 1998
ORDNUNG GLOMERALES (MORTON & BENNY 1990) WALKER & SCHÜßLER 2001
Familie Glomeraceae PIROZYNSKI & DALPÉ 1989
Gattung Glomus TULASNE & TULASNE 1845
Glomus mosseae-Gruppe (GlGrA)
Untergruppe a (GlGrAa)
§ G. caledonium (NICOLSON & GERDEMANN) TRAPPE & GERDEMANN 1974§ G. coronatum GIOVANNETTI, AVIO & SALUTINI 1991§ G. fragilistratum SKOU & JAKOBSEN 1989§ G. geosporum (NICOLSON & GERDEMANN) WALKER 1982§ G. mosseae (NICOLSON & GERDEMANN) GERDEMANN & TRAPPE 1974§ G. verruculosum BLASZKOWSKI 1997
Untergruppe b (GlGrAb)
§ G. clarum NICOLSON & SCHENCK 1979§ G. coremioides (BERK. & BROOME) REDECKER & MORTON 2000
= Sc. coccogena (PATOUILLARD) VON HÖHNEL 1910= Sc. coremioides BERKELEY & BROOME 1873= Sc. dussii (PATOUILLARD) VON HÖHNEL 1910
§ G. fasciculatum (THAXTER) GERDEMANN & TRAPPE emend. WALKER & KOSKE 1987§ G. intraradices SCHENCK & SMITH 1982 (G. intraradix)§ G. manihotis HOWELER, SIEVERDING & SCHENCK 1984 (G. manihot) = G. clarum? (oder
GlGrB?)§ G. sinuosum (GERDEMANN & BAKSHI) ALMEIDA & SCHENCK 1990
= Sc. pakistanica IQBAL & BUSHRA 1980= Sc. sinuosa GERDEMANN & BAKSHI 1976
§ G. vesiculiferum (THAXTER) GERDEMANN & TRAPPE 1974

7 Anhang 116
Glomus etunicatum-Gruppe (GlGrB)
§ G. claroideum (SCHENCK & SMITH 1982) WALKER & VERSTBERG 1998 (G. claroides)§ G. etunicatum BECKER & GERDEMANN 1977 (oder Diversisporales?)§ G. lamellosum DALPÉ, KOSKE & TEWS 1992§ G. luteum Kennedy, STUTZ & MORTON 1999§ G. manihotis HOWELER, SIEVERDING & SCHENCK 1984 (G. manihot) = G. clarum? (oder
GlGrAb?)§ G. microaggregatum KOSKE, GEMMA & OLEXIA 1986 (GlGrB)§ G. viscosum WALKER et al. 1995
Noch nicht zugeordnete Arten
G. aggregatum (SCHENCK & SMITH) KOSKE 1985G. albidum WALKER & RHODES 1981G. ambisporum SMITH & SCHENCK 1985G. antarticum CABELLO 1994G. arborense MCGEE 1986G. arenarium BLASZKOWSKI, TADYCH & MADEJ 2001G. australe (BERKELEY) BERCH 1983G. boreale (THAXTER) TRAPPE & GERDEMANN 1922G. botryoides ROTHWELL & VICTOR 1984G. callosum SIEVERDING 1988G. canadense (THAXTER) TRAPPE & GERDEMANN 1922G. cerebriforme MCGEE 1986G. chimonobambusae WU & LIU 1995G. citricola TANG & ZANG 1984= G. fistulosum SKOU & JAKOBSEN 1989= G. maculosum MILLER & WALKER 1986= G. multisubstensum MUKERJI, BHATTACHARJEE & TEWARI 1983G. clavisporum (TRAPPE) ALMEIDA & SCHENCK 1990= Sc. clavispora TRAPPE 1977= Sc. microcarpa IQBAL & BUSHRA 1980
§ G. constrictum TRAPPE 1977G. convolutum GERDEMANN & TRAPPE 1974G. corymbiforme BLASZKOWSKI 1995G. delhiense MUKERJI, BHATTACHARJEE & TEWARI 1983G. deserticola TRAPPE, BLOSS & MENGE 1984G. diaphanum MORTON & WALKER 1984
§ G. dimorphicum BOYETCHKO & TEWARI 1986G. dominikii BLASZKOWSKI 1988G. eburneum KENNEDY, STUTZ & MORTON 1999G. flavisporum (LANGE & LUND) TRAPPE & GERDEMANN 1964G. formosanum WU & CHEN 1986G. fragile (BERK. & BROOME) TRAPPE & GERDEMANN 1974G. fuegianum (SPEGAZZINI) TRAPPE & GERDEMANN 1922G. fulvum (BERK. & BROOME) TRAPPE & GERDEMANN 1922G. gibbosum BLASZKOWSKI 1997G. globiferum KOSKE & WALKER 1986G. glomerulatum SIEVERDING 1987(G. hadleyi ined.)G. halonatum ROSE & TRAPPE 1980 (G. halon, G. halonatus)G. heterosporum SMITH & SCHENCK 1985

7 Anhang 117
§ G. hoi Berch & TRAPPE 1985(G. hyalinum ined.)G. invermaium HALL 1977 (G. invermayanum)G. laccatum BLASZKOWSKI
G. lacteum ROSE & TRAPPE 1980G. liquidambaris (WU & CHEN) ALMEIDA & SCHENCK 1990= Sc. cunninghamia HU 1988= Sc. liquidambaris WU & CHEN 1987G. macrocarpum TULASNE & TULASNE 1845G. magnicaule HALL 1977G. melanosporum GERDEMANN & TRAPPE 1974G. microcarpum TULASNE & TULASNE 1845 => TypusartG. minutum BLASZKOWSKI, TADYCH & MADAJ 2000(G. mirificum ined.)
§ G. monosporum GERDEMANN & TRAPPE 1974G. mortonii BENTIVENGA & HETRICK 1991G. multicaule GERDEMANN & BAKSHI 1976G. multiforum TADYCH & BLASZKOWSKI 1997G. nanolumen KOSKE & GEMMA 1989G. pallidum HALL 1977G. pansihalos BERCH & KOSKE 1986G. przelewicensis BLASZKOWSKI 1988 = G. albidum?G. pubescens (SACCARDO & ELLIS) TRAPPE & GERDEMANN 1974G. pulvinatum (HENNINGS) TRAPPE & GERDEMANN 1975G. pustulatum KOSKE, FRIESE, WALKER & DALPÉ 1986G. radiatum (THAXTER) TRAPPE & GERDEMANN 1974G. reticulatum BHATTACHARJEE & MUKERJI 1980G. rubiforme (GERDEMANN & TRAPPE) ALMEIDA & SCHENCK 1990= Sc. indica BHATTACHARJEE & MUKERJI 1980= Sc. pachycaulis WU & CHEN 1986= Sc. rubiformis GERDEMANN & TRAPPE 1974G. scintillans ROSE & TRAPPE 1980G. segmentatum TRAPPE, SPOONER & IVORY 1979G. sterilum MEHROTRA & BAIJAL
G. taiwananse (WU & CHEN) ALMEIDA & SCHENCK 1990= Sc. taiwanensis WU & CHEN 1987G. tenebrosum (THAXTER) BERCH 1983G. tenerum TANDY emend. MCGEE 1986G. tenue (GREENHALL) HALL 1977G. tortuosum SCHENCK & SMITH 1982G. trimurales KOSKE & HALVORSON 1989G. tubiforme TANDY 1975 (G. tubaeforme)G. warcupii MCGEE 1986
(Gattung Sclerocystis Berkeley & Broome 1873) s. Glomus

7 Anhang 118
ORDNUNG DIVERSISPORALES WALKER & SCHÜßLER 2001
Familie Acaulosporaceae MORTON & BENNY 1990
Gattung Acaulospora (GERDEMANN & TRAPPE) emend. BERCH 1985
A. bireticulata ROTHWELL & TRAPPE 1979A. capsicula BLASZKOWSKI 1991A. cavernata BLASZKOWSKI 1989
§ A. colossica SCHULTZ, BEVER & MORTON 1999A. delicata WALKER, PFEIFFER & BLOSS 1986
§ A. denticulata SIEVERDING & TORO 1987A. dilatata MORTON 1986A. elegans TRAPPE & GERDEMANN 1974A. excavata INGLEBY & WALKER 1994A. foveata TRAPPE & JANOS 1982A. gedanensis BLASZKOWSKI 1988 (A. gdanskensis)
§ A. koskei BLASZKOWSKI 1995§ A. lacunosa MORTON 1986§ A. laevis GERDEMANN & TRAPPE 1974§ A. longula SPAIN & SCHENCK 1984 = A. morrowiae?§ A. mellea SPAIN & SCHENCK 1984§ A. morrowiae SPAIN & SCHENCK 1984
A. myriocarpa SPAIN, SIEVERDING & SCHENCK 1986A. nicolsonii WALKER, REED & SANDERS 1984A. paulineae BLASZKOWSKI Jahr?A. polonica BLASZKOWSKI 1988A. rehmii SIEVERDING & TORO 1987
§ A. rugosa MORTON 1986 = A. morrowiae?§ A. scrobiculata TRAPPE 1977§ A. spinosa WALKER & TRAPPE 1981
A. splendida SIEVERDING, CHAVERRI & ROJAS 1988A. sporocarpia BERCH 1985 (A. sporocarpa)A. taiwania HU 1988A. thomii BLASZKOWSKI 1988
§ A. tuberculata JANOS & TRAPPE 1982A. undulata SIEVERDING 1988A. walkeri KRAMADIBRATA & HEDGER 1990
Gattung Entrophospora AMES & SCHNEIDER 1979
E. baltica BLASZKOWSKI & TADYCH 1998§ E. colombiana SPAIN & SCHENCK 1984§ (E. contigua ined.)§ E. infrequens (HALL) AMES & SCHNEIDER 1979
E. kentinensis WU & LIU 1995E. schenckii SIEVERDING & TORO 1987
Familie Gigasporaceae MORTON & BENNY 1990
Gattung Gigaspora (GERDEMANN & TRAPPE) WALKER & SANDERS 1986
§ Gi. albida SCHENCK & SMITH 1982

7 Anhang 119
§ Gi. decipiens HALL & ABBOTT 1984§ Gi. gigantea (NICOLSON & GERDEMANN) GERDEMANN & TRAPPE 1974§ Gi. margarita BECKER & HALL 1976
= Gi. ramisporophora SPAIN, SIEVERDING & SCHENCK 1989§ Gi. rosea NICOLSON & SCHENCK 1979
= Gi. candida BHATTACHARJEE, MUKERJI, TEWARII & SKOROPAD 1982
Gattung Scutellospora WALKER & SANDERS 1986
S. alborosea (FERRER & HERRERA) WALKER & SANDERS 1980S. arenicola KOSKE & HALVORSON 1989S. armeniaca BLASZKOWSKI 1992
§ S. aurigloba (HALL) WALKER & SANDERS 1977 (S. auriglobosa)S. biornata SPAIN, SIEVERDING & TORO 1989
§ S. calospora (NICOLSON & GERDEMANN) WALKER & SANDERS 1986§ S. castanea WALKER 1993 = S. persica?§ S. cerradensis SPAIN & MIRANDA 1997§ S. coralloidea (TRAPPE, GERDEM. & HO) WALKER & SANDERS 1985 (S. coralloides)
S. crenulata HERRERA-PERAZA, CUENCA & WALKER 2001§ S. dipapillosa (WALKER & KOSKE) WALKER & SANDERS 1985§ S. dipurpurascens MORTON & KOSKE 1988§ S. erythropa (KOSKE & WALKER) WALKER & SANDERS 1984§ S. fulgida KOSKE & WALKER 1986§ S. gilmorei (TRAPPE & HERD.) WALKER & SANDERS 1974
S. gregaria (SCHENCK & NICOLSON) WALKER & SANDERS 1985S. hawaiiensis KOSKE & GEMMA 1995
§ S. heterogama (NICOLSON & GERDEMANN) WALKER & SANDERS 1985S. minuta (FERRER & HERRERA) WALKER & SANDERS 1980S. nigra (REDHEAD) WALKER & SANDERS 1979
§ S. nodosa BLASZKOWSKI 1991§ S. pellucida (NICOLSON & SCHENCK) WALKER & SANDERS 1986§ S. persica (KOSKE & WALKER) WALKER & SANDERS 1985
= Gi. tuberculata NEERAJ, MUKERJI, SHARMA & VARMA 1993§ S. projecturata KRAMADIBRATA & WALKER 2000
S. reticulata (KOSKE, MILLER & WALKER) WALKER & SANDERS 1983S. rubra STÜRMER & MORTON 1999S. savannicola (FERRER & HERRERA) Walker & Sanders 1980S. scutata WALKER & DIEDERICHS 1989S. spinosissima WALKER, CUENCA & SANCHEZ 1998S. tricalypta (HERRERA & FERRER) WALKER & SANDERS 1980S. verrucosa (KOSKE & WALKER) WALKER & SANDERS 1985S. weresubiae KOSKE & WALKER 1986
Familie Diversisporaceae fam. ined.
§ G. etunicatum BECKER & GERDEMANN 1977 (oder Glomus GlGrB?)§ G. spurcum (PFEIFFER, WALKER & BLOSS 1996) KENNEDY, STUTZ & MORTON 1999§ G. versiforme (KARSTEN) BERCH 1983
= G. epigaeum DANIELS & TRAPPE 1979

7 Anhang 120
ORDNUNG ARCHAEOSPORALES WALKER & SCHÜßLER 2001
Familie Archaeosporaceae MORTON & REDECKER 2001
Gattung Archaeospora MORTON & REDECKER 2001
§ Ar. gerdemannii (ROSE, DANIELS & TRAPPE) MORTON & REDECKER 2001= G. gerdemannii ROSE, DANIELS & TRAPPE 1979
§ Ar. leptoticha (SCHENCK & SMITH) MORTON & REDECKER 2001= A. appendicula SPAIN, SIEVERDING & SCHENCK 1984= A. gerdemannii NICOLSON & SCHENCK 1979= G. fecundisporum SCHENCK & SMITH 1982= G. leptotichum SCHENCK & SMITH 1982
§ Ar. trappei (AMES & LINDERMAN) MORTON & REDECKER 2001= A. trappei AMES & LINDERMAN 1976
Familie Geosiphonaceae
Gattung Geosiphon (KÜTZ.) V. WETTSTEIN 1915
§ Geo. pyriforme (KÜTZ.) V. WETTSTEIN 1915
ORDNUNG PARAGLOMERALES WALKER & SCHÜßLER 2001
Familie Paraglomeraceae MORTON & REDECKER 2001
Gattung Paraglomus MORTON & REDECKER 2001
§ P. brasilianum (SPAIN & MIRANDA) MORTON & REDECKER 2001= G. brasilianum SPAIN & MIRANDA 1996
§ P. occultum (WALKER) MORTON & REDECKER 2001= G. occultum WALKER 1982
B. Neubeschreibung von Arten
0
2
4
6
8
10
12
14
16
18
20
19
74
19
75
19
76
19
77
19
78
19
79
19
80
19
81
19
82
19
83
19
84
19
85
19
86
19
87
19
88
19
89
19
90
19
91
19
92
19
93
19
94
19
95
19
96
19
97
19
98
19
99
20
00
20
01
Abb. 50: Zahl der neu beschriebenen AMF-Arten seit 1974.

121
Danksagung
Mein besonderer Dank gilt Herrn Prof. Dr. Hock für die Überlassung des Themas und die
fachliche Unterstützung während meiner Arbeit an seinem Lehrstuhl.
Herrn Dr. Böhm möchte ich besonders für die von ihm geleistete Vorarbeit auf diesem Ge-
biet, die Überlassung von Pilzmaterial und seine konstruktive Kritik danken.
Für die gute Zusammenarbeit am Lehrstuhl für Zellbiologie bedanke ich mich bei allen übri-
gen Mitarbeitern, insbesondere bei Frau Dr. Lux-Endrich (Mykorrhiza) und Frau Rothe
(Computer-Problemchen). Ganz besonderer Dank gilt auch Frau Scholz, die sich überaus
kompetent um „den Papierkram“ sorgte.
Im Rahmen des F.A.M. möchte ich mich für die Kooperation bei Herrn Prof. Dr. Munch,
Herrn PD Dr. Schröder (F.A.M.-Leitung), Herrn Prof. Schnyder, Frau Dr. Huber-Sannwald
(Mykorrhiza), Herrn Dr. Schloter (TGGE) und Herrn Dr. Lebuhn (Gaeumannomyces) bedan-
ken.
Für ihre Ideen und Ratschläge rund um das Problem der DNA-Extraktion danke ich Herrn PD
Dr. Weller und Frau Dr. Römmelt, sowie Frau Dr. Boronowski (QIAGEN), die mit mir ge-
duldig zahlreiche Varianten am Telefon besprochen hat.
Mein besonderer Dank gilt meiner Familie, die jederzeit zu mir gestanden hat und meinen
Freunden, die von mir vernachlässigt wurden.
Bei Steffi bedanke ich mich für ihre Unterstützung und ihre Geduld mit mir.
Bei Herrn Aldinger, der leider viel zu früh von uns gegangen ist, möchte mich dafür bedan-
ken, dass es ihm nicht gelungen ist, mir von meinem Studium abzuraten.
Ich danke dem Bundesministerium für Forschung und Bildung für die finanzielle Förderung
des Projekts.

122
Lebenslauf
Persönliche Daten:
Name: Holger Geue
Geburtsdatum: 24. Juli 1972
Geburtsort: Augsburg
Staatsangehörigkeit: deutsch
Schulausbildung:
1979 – 1983 Wittelsbacher Grundschule in Augsburg
1983 – 1992 Holbein-Gymnasium in Augsburg
(Abschluß: Allgemeine Hochschulreife)
Buchpreis des Fonds der Chemischen Industrie
Hochschulstudium:
Okt. 1993 – Jun. 1998 Studium der Lebensmittelchemie an der Ludwig-Maximilians-
Universität in München
(Abschluß: Lebensmittelchemiker, 1. Staatsexamen)
Praktische Tätigkeiten:
Jul. 1992 – Sep. 1993 Wehrdienst als Sanitäts- und Stabsdienstsoldat beim
SanLehrBtl. 851 in München
Nov. 1996 - Dez. 1996 Wissenschaftliche Hilfskraft am Lehrstuhl für Lebensmittel-
chemie der Ludwig-Maximilians-Universität in München
Jul. 1998 - Jan. 1999 Wissenschaftlicher Angestellter am Lehrstuhl für Pharmazeuti-
sche Biologie der Ludwig-Maximilians-Universität in München
seit Feb. 1999 Wissenschaftlicher Angestellter am Lehrstuhl für Zellbiologie
der TU München in Freising-Weihenstephan
Von Februar 1999 bis April 2002 wurde die vorliegende Dissertation angefertigt.


















