
Rutheniumkatalysierte Hydratisierung von terminalen...
144
Rutheniumkatalysierte Hydratisierung von terminalen Alkinen und deren Einsatz in Synthesestrategien Von der Fakultät für Mathematik, Informatik und Naturwissenschaften der RWTH Aachen University zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigte Dissertation vorgelegt von Diplom-Gymnasiallehrer Thomas Clemens Kribber aus Haselünne Berichter: Priv.-Doz. Dr. L. Hintermann Universitätsprofessor Dr. C. Bolm Tag der mündlichen Prüfung: 31. Juli 2009 Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.
Transcript of Rutheniumkatalysierte Hydratisierung von terminalen...

Rutheniumkatalysierte Hydratisierung von terminalen Alkinen und deren Einsatz in Synthesestrategien
Von der Fakultät für Mathematik, Informatik und
Naturwissenschaften der RWTH Aachen University zur Erlangung des
akademischen Grades eines Doktors der Naturwissenschaften
genehmigte Dissertation
vorgelegt von
Diplom-Gymnasiallehrer
Thomas Clemens Kribber
aus Haselünne
Berichter:
Priv.-Doz. Dr. L. Hintermann
Universitätsprofessor Dr. C. Bolm
Tag der mündlichen Prüfung: 31. Juli 2009
Diese Dissertation ist auf den Internetseiten der Hochschulbibliothek online verfügbar.

Die vorliegende Arbeit wurde unter Leitung von PD Dr. Lukas Hintermann in der Zeit von
Dezember 2005 bis Mai 2009 am Institut für Organische Chemie der RWTH Aachen
University durchgeführt.
Ich danke Herrn PD Dr. Lukas Hintermann für die Möglichkeit der Anfertigung dieser
Dissertation unter den guten Arbeitbedingungen in seiner Arbeitsgruppe und die vielen
hilfreichen Diskussionen.
Herrn Prof. Dr. Carsten Bolm danke ich für die dauerhafte Unterstützung während dieser
Zeit und die freundliche Übernahme des Korreferats.
Teile dieser Arbeit sind bereits veröffentlicht:
1. Highly Active in Situ Catalysts for Anti-Markovnikov Hydration of Terminal
Alkynes
A. Labonne, T. Kribber, L. Hintermann, Org. Lett. 2006, 8, 5853.
2. Iterative Synthesis of Oligo-1,4-Diols via Catalytic anti-Markovnikov
Hydration of Terminal Alkynes
T. Kribber, A. Labonne, L. Hintermann, Synthesis 2007, 2809.
3. The AZARYPHOS Family of Ligands for Ambifunctional Catalysis: Syntheses
and Use in Ruthenium-Catalyzed anti-Markovnikov Hydration of Terminal
Alkynes
L. Hintermann, T. T. Dang, L. Labonne, T. Kribber, L. Xiao, P. Naumov, Eur. J.
Chem. 2009, 15 (29), 7167.

Inhaltsverzeichnis
i
Inhaltsverzeichnis 1. Einleitung...................................................................................................................... 1
1.1. Hydratisierung von Alkinen ........................................................................................ 1 1.2. Metallkatalysierte Hydratisierung von Alkinen .......................................................... 2 1.3. Mechanismus der MARKOVNIKOV-selektiven Hydratisierung von Alkinen ............... 4 1.4. Hydratisierung von Alkinen mit anti-MARKOVNIKOV-Selektivität............................. 6 1.5. Mechanismus der rutheniumkatalysierten anti-MARKOVNIKOV-Hydratisierung ...... 12 1.6. Synthetische Anwendungen der anti-MARKOVNIKOV-Hydratisierung ..................... 18
2. Aufgabenstellung ..................................................................................................... 19 3. Hauptteil...................................................................................................................... 20
3.1. Optimierung der rutheniumkatalysierten anti-MARKOVNIKOV-Hydratisierung von
terminalen Alkinen .................................................................................................. 20 3.1.1. Methodenentwicklung und Modellreaktion....................................................... 20 3.1.2. Untersuchungen zur Katalysatorvariation ......................................................... 22 3.1.3. Effekt der Katalysatorbeladung und Konzentration .......................................... 24 3.1.4. Komplexe ohne bifunktionale Liganden ........................................................... 27 3.1.5. Der Lösungsmitteleffekt in der Hydratisierung................................................. 29 3.1.6. Der Einfluss der Wassermenge auf die Hydratisierung..................................... 31 3.1.7. Der Einfluss des Acetonitrils auf die Hydratisierung........................................ 32 3.1.8. Der Einfluss von Additiven auf die Katalyse .................................................... 34 3.1.9. Der Ligandeneffekt in Mischkomplexen........................................................... 35
3.1.9.1. Die Synthese der Ruthenium(II)-Mischkomplexe...................................... 36 3.1.9.2. Ruthenium(II)-Mischkomplexe mit Phosphinliganden in der Katalyse..... 38 3.1.9.3. Heteroleptische Ruthenium(II)-Mischkomplexe mit Phosphit- und anderen Liganden .................................................................................................................. 43 3.1.10. Austausch des Gegenions ............................................................................ 44 3.1.11. Die Optimierungen auf einen Blick............................................................. 47
3.2. Anwendung der anti-MARKOVNIKOV-Hydratisierung in der Synthese..................... 48 3.2.1. Hydratisierung von Propargylalkoholen............................................................ 48 3.2.2. Hydratisierung von Propargylalkoholen mit Ruthenium-Katalysatoren ........... 49 3.2.3. Herstellung von 5,6-Dihydropyran-2-onen durch iterative Synthese von 1,3-Diolen .......................................................................................................................... 53
3.2.3.1. Synthese von 5,6-Dihydropyran-2-onen mittels rutheniumkatalysierter anti-MARKOVNIKOV-Hydratisierung von terminalen Alkinen........................................ 53 3.2.3.2. Synthesestrategien für 5,6-Dihydropyran-2-one ........................................ 54 3.2.3.3. Einsatz der anti-MARKOVNIKOV-Hydratisierung in der Synthese.............. 54
3.2.4. Stereochemischer Verlauf der anti-MARKOVNIKOV-Hydratisierung von Propargylalkoholen...................................................................................................... 58 3.2.5. Iterative Synthese von Poly-1,4-diolen via rutheniumkatalysierter anti-MARKOVNIKOV-Hydratisierung................................................................................... 60
3.2.5.1. Iterative Synthesemethoden........................................................................ 60 3.2.5.2. Kettenabschlüsse und Ringschluss ............................................................. 64 3.2.5.3. Stereochemische Aspekte der Poly-1,4-diole............................................. 68

Inhaltsverzeichnis
ii
4. Zusammenfassung und Ausblick ....................................................................... 72 5. Experimenteller Teil ............................................................................................... 76
5.1. Allgemeines............................................................................................................... 76
5.1.1. Allgemeine Arbeitstechniken............................................................................ 76 5.1.2. Reinigung und Trocknung der verwendeten Lösungsmittel ............................. 76 5.1.3. Kommerzielle Chemikalien............................................................................... 76 5.1.4. Analytische Methoden....................................................................................... 77 5.1.5. Chromatographie............................................................................................... 78
5.2. Allgemeine Arbeitsvorschriften ................................................................................ 79 5.2.1. Durchführung der Reaktionskinetik-Messung mittels HPLC ........................... 79 5.2.2. Synthese von [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (L) (acetonitril)] hexafluorophosphat-Komplexen mit variablen Koliganden L (AAV1)................................................................................. 79 5.2.3. Iterative Synthese von Oligo-1,4-diolen via katalytischer anti-MARKOVNIKOV Hydratisierung von Terminalen Alkinen..................................................................... 80
5.2.3.1. anti-MARKOVNIKOV Hydratisierung von terminalen Alkinen (AAV2) ..... 80 5.2.3.2. Allenylzink-bromid (66) Lösung in THF................................................... 80 5.2.3.3. Propargylierung von Aldehyden (AAV3) .................................................. 80 5.2.3.4. Einführen der TBS Schutzgruppe (AAV4) ................................................ 81 5.2.3.5. WITTIG Reaktion (AAV5) .......................................................................... 81 5.2.3.6. Addition von Lithium Methylpropiolat an Aldehyde (AAV6) .................. 81
5.3. Synthese und Arbeitsvorschriften ............................................................................. 82 5.3.1. Synthese der η5-Cyclopentadienylruthenium(II)-Komplexe ............................ 82
5.3.1.1. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (acetonitril)] hexafluorophosphat (22)............................ 82 5.3.1.2. [η5-Cyclopentadienylruthenium(II) bis-(triphenylphosphin) (acetonitril)] hexafluorophosphat (28) ......................................................................................... 83 5.3.1.3. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (iso-butyronitril)] hexafluorophosphat (29).................... 83 5.3.1.4. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (benzonitril)] hexafluorophosphat (30)........................... 84 5.3.1.5. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat (31)...................... 85 5.3.1.6. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (triphenylphosphin) (acetonitril)] hexafluorophosphat (32)................................................................................................................................. 85 5.3.1.7. [η5-Cyclopentadienalruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (tris-ortho-tolylphosphin) (acetonitril)] hexafluorophosphat (34) ......................................................................................... 86 5.3.1.8. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (tri-1-naphthylphosphin) (acetonitril)] hexafluorophosphat (35) .......................................................................................................................... 87 5.3.1.9. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (tributylphosphin) (acetonitril)] hexafluorophosphat (36)................................................................................................................................. 87 5.3.1.10. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (tri-para-tolylphosphin) (acetonitril)] hexafluorophosphat (37) .......................................................................................................................... 90

Inhaltsverzeichnis
iii
5.3.1.11. [η5-Cyclopentadienylruthenium(II) (2-(diphenylphosphino)-6-(2,4,6-triphenylphenyl)pyridin) (tri-para-fluorophenylphosphin) (acetonitril)] hexafluorophosphat (38).......................................................................................... 90 5.3.1.12. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (tri-para-chlorophenylphosphin) (acetonitril)] hexafluorophosphat (39).......................................................................................... 91 5.3.1.13. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (tri-para-methoxyphenylphosphin) (acetonitril)] hexafluorophosphat (40).......................................................................................... 92 5.3.1.14. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (tri-4-trifluoromethylphenylphosphin) (acetonitril)] hexafluorophosphat (41).......................................................................................... 92 5.3.1.15. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (triethylphosphit) (acetonitril)] hexafluorophosphat (42)93 5.3.1.16. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (triphenylphosphit) (acetonitril)] hexafluorophosphat (43)................................................................................................................................. 94 5.3.1.17. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (acetonitril)] (rac)-TRISPHAT (44)................................... 94 5.3.1.18. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (triphenylphosphin) (acetonitril)] (rac)-TRISPHAT (45) .. 95
5.3.2. Synthese von 1,3-Diolen und deren Derivate.................................................... 96 5.3.2.1. 3-Benzylether-1-octin (46) ......................................................................... 96 5.3.2.2. 3-Tetrahydropyran-2-yloxy-1-octin (47).................................................... 96 5.3.2.3. 3-Tetrahydropyran-2-yloxy-1-octanal (47a) .............................................. 97 5.3.2.4. 3-tert-Butyldimethylsilyloxy-1-octin (48).................................................. 98 5.3.2.5. 3-Benzoyloxy-1-octin (49) ......................................................................... 99 5.3.2.6. 3-Methoxymethyloxy-1-octin (50) ........................................................... 100 5.3.2.7. 3-Methoxy-1-octin (51) ............................................................................ 100 5.3.2.8. 3-Methoxymethyloxyoctanal (52) ............................................................ 101 5.3.2.9. 5-Methoxymethyloxy-3-hydroxy-decansäure-tert-butylester (53)........... 102 5.3.2.10. 6-Pentyl-5,6-dihydro-2-pyron oder Massoialacton (54)......................... 103 5.3.2.11. 5-Methoxymethyloxy-3-hydroxy-1-decin (55) ...................................... 103 5.3.2.12. 3,5-Methylendioxy-1-decin (56) ............................................................ 104 5.3.2.13. 6,8-Methylendioxy-4-hydroxy-1-tridecen (57) ...................................... 105 5.3.2.14. 6,8-Methylendioxy-4-acryloyloxy-1-tridecen (58) ................................ 106 5.3.2.15. 5,6-Dihydro-6-(2,4-methylendioxy-nonyl)-pyran-2-on (59).................. 107 5.3.2.16. 1,3-Dibenzoyloxy-octan (61) ................................................................. 108
5.3.3. Synthese der Oligo-1,4-diole und deren Derivate ........................................... 111 5.3.3.1. 1-Phenylbut-3-in-1-ol (67) ....................................................................... 111 5.3.3.2. 1-Phenyl-3-butin-1-ylacetat (68) .............................................................. 112 5.3.3.3. 4-Acetoxy-4-phenylbutanal (69) .............................................................. 112 5.3.3.4. 4-Hydroxy-1-phenyl-6-heptin-1-ylacetat (70).......................................... 113 5.3.3.5. 4-tert-Butyldimethylsilyloxy-1-phenyl-6-heptin-1-ylacetat (71) ............. 114 5.3.3.6. 4-tert-Butyldimethylsilyloxy-7-hydroxy-1-phenyl-9-decin-1-ylacetat (72)............................................................................................................................... 114 5.3.3.7. 4,7-Bis(tert-butyldimethylsilyloxy)-1-phenyl-9-decin-1-ylacetat (73) .... 115 5.3.3.8. 4,7-Bis(tert-butyldimethylsilyloxy)-10-hydroxy-1-phenyl-12-tridecin-1-ylacetat (74) ........................................................................................................... 116 5.3.3.9. 4,7,10-Tris(tert-butyldimethylsilyloxy)-1-phenyl-12-tridecin-1-ylacetat (75)............................................................................................................................... 116

Inhaltsverzeichnis
iv
5.3.3.10. (E)-6-Acetoxy-6-phenyl-2-hexensäuremethylester [(E)-78] ................. 117 5.3.3.11. 7-Acetoxy-4-hydroxy-7-phenyl-2-heptinsäuremethylester (79) ............ 118 5.3.3.12. (E)-9-Acetoxy-6-(tert-butyldimethylsilyloxy)-9-phenyl-2-nonensäuremethylester [(E)-80]............................................................................ 118 5.3.3.13. 10-Acetoxy-7-(tert-butyldimethylsilyloxy)-4-hydroxy-10-phenyl-2-decinsäuremethylester (81) ................................................................................... 119 5.3.3.14. (E)-15-Acetoxy-6,9,12-tris(tert-butyldimethylsilyloxy)-15-phenyl-2-pentadecensäuremethylester [(E)-82] ................................................................... 120 5.3.3.15. 16-Acetoxy-7,10,13-tris(tert-butyldimethylsilyloxy)-4-hydroxy-16-phenyl-2-hexadecinsäuremethylester (83) ............................................................ 121 5.3.3.16. 16-Acetoxy-4,7,10,13-tetrakis(tert-butyldimethylsilyloxy)-16-phenyl-2-hexadecinsäuremethylester (84) ............................................................................ 121 5.3.3.17. 16-Hydroxy-4,7,10,13-tetrakis(tert-butyldimethylsilyloxy)-16-phenyl-2-hexadecinsäure (85)............................................................................................... 122 5.3.3.18. 17-Phenyl-5,8,11,14-tetrakis(tert-butyldimethylsilanyloxy)-1-oxa-zyklo-hepta-3-decin-2-on (86)......................................................................................... 123 5.3.3.19. 4-tert-Butyldimethylsilyloxy-4-phenyl-1-butin (87).............................. 124 5.3.3.20. 7-tert-Butyldimethylsilyloxy-4-hydroxy-7-phenyl-2-heptinsäuremethylester (88) .................................................................................. 125
A Literaturverzeichnis ...................................................................................... 126 B Abkürzungsverzeichnis ................................................................................ 133 C Danksagung ...................................................................................................... 136 D LEBENSLAUF................................................................................................ 138

1. Einleitung
1
1. Einleitung
1.1. Hydratisierung von Alkinen
Die Addition von Wasser an Alkine ist eine synthetische Methode um
Carbonylverbindungen herzustellen, die im Gegensatz zur energieintensiven Redox-
Chemie atomökonomisch ist.1 Im Allgemeinen verlaufen die Hydratisierungen von
Alkinen, wie auch die der Alkene, in ihrer Selektivität nach der MARKOVNIKOV-Regel
(1870), welche sich durch den Mechanismus der elektrophilen Addition erklären lässt.
Man erhält also durch die Addition von Wasser an terminale Alkine üblicherweise die
entsprechenden Methylketone (MARKOVNIKOV-Produkt) bzw. Acetaldehyd im Falle von
Acetylen (Schema 1.1a). Bei unsymmetrischen internen Alkinen können sich zwei
Regioisomere bilden (Schema 1.1b).
Schema 1.1: Allgemeine Darstellung der Hydratisierung von Alkinen.
Die Hydratiserung von Alkinen verbindet das Gebiet der Alkinchemie mit dem der
Carbonylverbindungen, was viele Möglichkeiten zur Anwendung bei organischen
Synthesen eröffnet.

1. Einleitung
2
1.2. Metallkatalysierte Hydratisierung von Alkinen
Im Jahre 1860 entdeckte BERTHELOT das Acetylen und beschrieb dessen Hydratisierung in
Schwefelsäure.2 Er nahm an, dass er den Vinylalkohol erhalten hatte und nicht, wie sich
später herausstellte, ein Gemisch aus Acetaldehyd und Crotonaldehyd.3 Danach beschrieb
1881 KUTSCHEROFF die Hydratisierung von Acetylen zu Acetaldehyd in Gegenwart von
Schwefelsäure und Quecksilbersalzen als erste Metallkatalysatoren in dieser Umsetzung.4
Bei der Reaktion von KUTSCHEROFF wird Acetylen durch eine Quecksilbernitrat- oder
Quechsilberchloridlösung geleitet; nach der Hydrolyse liegt Acetaldehyd liegt.5 Dabei
wurde durch Vergleiche herausgefunden, dass Quecksilbersulfat [Quecksilber(II)-Oxid
gelöst in Schwefelsäure] die höchste katalytische Aktivität unter allen Quecksilbersalzen
aufwies.
Die KUTSCHEROFFsche Reaktion und allgemein die Acetylenchemie hatten von 1910 bis
1950 eine immense Bedeutung für die Herstellung großtechnischer Zwischenprodukte, wie
Acetaldehyd, Vinylchlorid, Vinylacetat und chlorierter Lösemittel.6 Jedoch wurde
Acetylen als Ausgangsmaterial für Basischemikalien weitgehend durch Olefine aus der
Petrochemie abgelöst.7 Des Weiteren sind heutzutage die meisten Produkte, wie Ethanol,
Essigsäure, Ethylacetat usw., die über Acetaldehyde und Derivate davon hergestellt
werden könnten, auch einfacher großtechnisch aus Ethylen, Methanol und Kohlenmonoxid
zu synthetisieren. Bei letzteren Methoden wird auf den Einsatz der hochtoxischen
Quecksilbersalze verzichtet. Trotzdem lassen sich noch heute Anwendungen für die
Hydratisierung von Alkinen mit Quecksilbersalzen in Totalsynthesen finden, da die
Reaktion verschiedene Funktionalitäten tolleriert, selektiv ist und gute Ausbeuten erzielt.
Ein Beispiel hierfür ist die beschriebene Totalsynthese von Tetrodotoxin, in der ein Alkin
mittels Quecksilberoxid katalytisch hydratisiert wurde (Schema 1.2).8
Schema 1.2: Quecksilberkatalysierte Alkinhydratisierung in der Totalsynthese von Tetrodotoxin.8
Neben Quecksilbersulfat finden heute auch noch andere modifizierte Quecksilber-
Katalysatorsysteme Verwendung, wie z. B. das von NEWMAN [HgSO4/Dowex 50 – ein

1. Einleitung
3
Polystyrolsulfonat-Harz/H2O]9 oder NISHIZAWA und Mitarbeitern [Hg(OTf)2•(TMU)2, H2O,
CH3CN/CH2Cl2]10 (TMU = Tetramethylharnstoff).
Im Laufe der Zeit wurden neben Quecksilbersalzen weitere Katalysatoren für die
Hydratisierung von Alkinen entwickelt, die auf Gold(III) [NaAuCl4],11 Ruthenium(II),12
Ruthenium(III),13 Rhodium(III),14 Iridium(I),15 Iridium(III),16 Platin [PtCl4(CH2=CH2)17
oder PtCl418] und anderen Metallen19 basieren.20 Besonders geeignet für die praktische
Anwendung sind sowohl der Palladium(II)-Katalysator PdCl2(MeCN)2 von UTIMOTO für
die regioselektive Hydratisierungen unter Nachbargruppen-Beteiligung,21 als auch Gold(I)-
Katalysatoren basierend auf [Au(PPh3)]+.22 Diese kationischen Goldkomplexe können in
situ aus a) [AuX(PPh3)] (X = Cl, CF3CO2, MeSO3, NO3) und Bortrifluorid oder b)
[AuCl(PPh3)] und AgY (Y = nicht nukleophiles Gegenion) oder c) [AuMe(PPh3)] und
einer starken Säure (H2SO4, HBF4, MeSO3H) hergestellt werden.22a,b Nachdem TELES und
Mitarbeiter von der BASF berichtet hatten, dass Gold(I)-Spezies zusammen mit einem
sauren Cokatalysator die Addition von Methanol an Alkine katalysieren, entwickelten
HAYASHI und TANAKA mit ihren Mitarbeitern ein katalytisches Gold(I)-System mit Säure
in wässrigem Methanol für die Hydratisierung von Alkinen.22c Die höchste Aktivität bei
der Hydratisierung von 1-Octin konnte dabei mit [AuMe(PPh3)] (0.01 mol%) und
Schwefelsäure (50 mol%) in Gegenwart von Kohlenmonoxid (1 atm) (→ 99% 2-Octanon,
70 °C, 1 h), als Additiv, oder mit CF3SO3H anstelle der Schwefelsäure und ohne
Kohlenmonoxid erzielt werden (→ 99% 2-Octanon, 70 °C, 1 h). Unter den gleichen
Reaktionsbedingungen (70 °C, 1 h) war auch die Wahl des Lösungsmittels entscheidend,
wobei die Ausbeuten in iso-Propanol (→ 71%), Dioxan (→ 56%), Acetonitril (→ 53%)
und Tetrahydrofuran (→ 11%) sowie in Dichlormethan, Dimethylformamid oder Toluol
geringer waren als in Methanol. Darüber hinaus hängt die Effektivität des Katalysators von
der Art der Säure ab, die das Anion zum kationischen Gold(I)-Komplex stellt. Ohne
Additiv war die Ausbeute des H2SO4-Systems nur gering (35%), während sie mit
CF3SO3H (→ 99%), CH3SO3H (→ 77%) und H3PW12O40 (→ 80%) unter gleichen
Bedingungen wesentlich höher lag. Der neue Gold(I)-Katalysator wurde anschließend für
verschiedene Alkine unter standardisierten Reaktionsbedingungen (0.1–1 mol%
[AuMe(PPh3)], 50 mol% H2SO4, 1 mmol Alkin, 0.5 mL Wasser, 1–3 mL Methanol, 70 °C)
getestet. Interessanterweise reagierten die propargylischen Alkohole zu einem Gemisch aus
Methylketonen (MARKOVNIKOV-Addition) und α,β-ungesättigten Aldehyden (anti-
MARKOVNIKOV-Addition, gefolgt von einer Eliminierung bzw. MEYER-SCHUSTER-
Umlagerung), während alle anderen getesteten Alkine nur die MARKOVNIKOV-Produkte

1. Einleitung
4
ergaben. Dieses Phänomen in Verbindung mit dem Gold(I)-Komplex wurde von
MIZUSHIMA und seinen Mitarbeitern nicht weiter untersucht, sondern es wurde lediglich
auf die Arbeiten von WAKATSUKI und GROTJAHN verwiesen, welche sich speziell mit der
anti-MARKOVNIKOV-Hydratisierung von Alkinen beschäftigt hatten.
Neben diesem System wurde kürzlich ein Gold(I)-Phosphin-Komplex beschrieben, bei
dem durch die Wahl von SPhos23 als Ligand und NTf224 als weniger stark koordinierendes
Gegenion im Vergleich zu OTf - die Azidität am Metallzentrum ausreichend erhöht werden
und die Hydratisierung von substituierten Alkinen zu den entsprechenden Ketonen bei
Raumtemperatur und ohne weiteres Additiv durchgeführt werden konnte. Diese Neuerung
des Katalysatorsystems ermöglicht auch die Umsetzung von säurelabilen
Alkinverbindungen unter milden und selektiven Bedingungen.25 Ein weiteres modifiziertes
katalytisches System auf Basis eines Gold(I)-Komplexes wurde von NOLAN und
Mitarbeitern beschrieben. Durch den Einsatz von N-Heterozyklischen-Carbenen als
Liganden im Komplex konnten sie im Gegensatz zu HAYASHI und TANAKA die
Hydratisierung von verschiedenen Alkinen, sowohl mit interner als auch terminaler
Alkinfunktion, mit sehr hohen Turnovers, ohne Säure und mit einer Katalysatorbeladung
von 50–100 ppm in Dioxan/Wasser (2:1) bei 120 °C in 18 h durchführen. Allerdings ist
eine equimolare Menge an Silberhexafluoroantimonat, bezogen auf den Gold(I)-Komplex,
vonnöten.26
1.3. Mechanismus der MARKOVNIKOV-selektiven Hydratisierung von
Alkinen
Betrachtet man den Mechanismus der KUTSCHEROFF-Reaktion vom heutigen
Wissensstandpunkt aus, so scheint er relativ simpel. Allerdings sind einige Aspekte noch
nicht im Detail bekannt.27

1. Einleitung
5
R H
Hg2+
HO
R H
H
HO
R Hg+
H
HR
Hg2+
O
R
O
R Hg+
HH
H+
H2O
H+
1
2
3
4
5
Schema 1.3: Mechanismus der Hg2+-katalysierten Hydratisierung von Alkinen.
Das Alkin bindet an eine freie Koordinationsstelle des Quecksilberkations und bildet den
kurzlebigen π-Komplex 1 (Schema 1.3). Die Regioselektivität des Angriffs von Wasser an
die koordinierte Dreifachbindung wird mit sterischen und elektronischen Aspekten erklärt.
Die Stabilisierung der positiven Partialladung am internen Kohlenstoffatom der
Dreifachbindung ist größer und die sterische Abstoßung zwischen dem Quecksilberion und
dem Alkin-Substituenten stärker als mit dem terminalen Wasserstoff, weshalb der Angriff
des Nukleophiles an die Dreifachbindung eher am internen Kohlenstoffatom stattfinden
wird.28 Somit bildet sich nach der Anlagerung von Wasser vermutlich die β-
Hydroxyethinylquecksilber-Spezies 2, die bisher allerdings weder isoliert noch
spektroskopisch charakterisiert werden konnte. Danach kommt es zur säureinitiierten
Abspaltung des Quecksilbers (Proto-de-mercurierung) und zur Freisetzung des Ketons 5,
von dem angenommen wird, dass es aus dem vorher gebildeten Enol 4 durch
Tautomerisierung entsteht.
Die Mechanismen anderer Metallkatalysatoren verlaufen ähnlich wie mit Quecksilber. Sie
unterscheiden sich jedoch in der Stabilität der Zwischenstufen. Die Besetzung der d-
Elektronenschalen kann in einigen Fällen Einfluss auf die Addition von Wasser an das
bereits koordinativ gebundene Substrat haben und durch Säure beschleunigt werden. Des
Weiteren ist die Stereochemie der Wasseraddition (syn oder anti) an die Dreifachbindung
noch nicht bekannt. Zwar wurde ein Mechanismus zur Gold(I)-katalysierten Addition von
Methanol an Alkine im Stil einer syn-Addition vorgeschlagen,22a,b aber ob dieser auf die

1. Einleitung
6
Gold(I)-katalysierte Hydratisierung von Alkinen übertragbar ist bleibt unklar. Schließlich
ist die Addition von Wasser an Alkine sehr selektiv, selbst in der Gegenwart von Alkohol
oder Carbonsäuren, die die Reaktionen beschleunigen, was auf einen spezifischen
Reaktionsmechanismus hindeuten würde.29
1.4. Hydratisierung von Alkinen mit anti-MARKOVNIKOV-Selektivität
Die bisher genannten Methoden der Hydratisierung von Alkinen verlaufen bis auf wenige
Ausnahmen mit MARKOVNIKOV-Selektivität. Zur Herstellung der anti-MARKOVNIKOV-
Produkte kann anstelle der Hydratisierung auch der indirekte Weg über die
stöchiometrische Hydroborierung oder Hydrosilierung, gefolgt von einer Oxidation,
beschritten werden.30
Erst mehr als 100 Jahre nachdem KUTSCHEROFF die erste metallkatalysierte Hydratisierung
von Alkinen beschrieben hatte, wurde 1998 von TOKUNAGA und WAKATSUKI eine
Katalyse mit einem Ruthenium(II)-Komplex veröffentlicht, mit der das anti-
MARKOVNIKOV-Produkt aus terminalen Alkinen hergestellt werden kann. Die ersten Tests
von TOKUNAGA und WAKATSUKI ergaben für die Hydratisierung von 1-Octin in Ethanol
oder iso-Propanol in Gegenwart von 5 mol% RuCl2(C6H6)(PPh3)31 ein Aldehyd/Keton-
Verhältnis von 1:8.12b, 32 Nach Kombination des Arenruthenium(II)-Komplexes mit 20
verschiedenen Phosphinen als Liganden stellten sich die Phosphine PPh2(C6F5) und P(m-
C6H4SO3Na)3 als die Besten heraus. Durch Optimierung der Versuchsbedingungen konnte
mit dem Katalysatorsystem aus 10 mol% RuCl2(C6H6){PPh2(C6F5)} und 30 mol%
PPh2(C6F5) in wässrigem iso-Propanol bei 65–100 °C eine Ausbeute von 75% Octanal mit
nur 4.5% des regioisomeren Hexylmethylketons erzielt werden. Dieses System kann nicht
nur einfache aliphatische terminale Alkine hydratisieren, sondern auch Substrate mit
Phenyl-, Chloralkyl- oder Benzylether-Gruppen. Allerdings sind die Ausbeuten bei
Substraten wie tert-Butylacetylen oder Phenylacetylen sehr gering, was durch die sterische
Hinderung bzw. elektronische Deaktivierung begründet ist.12b Nach weiteren Studien
konnten WAKATSUKI und Mitarbeiter die Aktivität und die Selektivität mit
Katalysatorkomplexen des Typs CpRuX(PR3)233 wesentlich verbessern im Vergleich zu
RuCl2(C6H6){PPh2(C6F5)}.12c, 34 Das anfänglich verwendete CpRuCl(PPh3)2 gab bei der
Hydratisierung von 1-Octin nur 35% Ausbeute an Octanal bei einer Katalysatorbeladung
von 30 mol%. Weiterführende Experimente zeigten, dass zweizähnige Phosphanliganden

1. Einleitung
7
(dppm, dppe, dppp, dppb),35 aber überraschenderweise auch elektronenreiche einzähnige
Phosphanliganden (PMePh2, PMe2Ph, PMe3), gute Resultate lieferten. Die höchsten
Ausbeuten bei der Hydratisierung von 1-Hexin lieferten CpRuCl(dppm) (1 mol%
Katalysator in iso-Propanol/Wasser 3:1 bei 100 °C) und CpRuCl(PMePh2)2 (5 mol%) mit
jeweils >99% Ausbeute an Hexanal. Es wurden keine Nebenprodukte (Keton)
nachgewiesen. Um die Anwendungsbreite dieses katalystischen Systems festzustellen,
wurde CpRuCl(dppm) in der Hydratisierung weiterer terminaler Alkine eingesetzt, was zu
guten bis sehr guten Ergebnissen führte. Es konnten terminale Alkine mit Ether-, Ester-
oder Nitril-Gruppen hydratisiert werden und sogar das sterisch anspruchsvolle tert-
Butylacetylen und Phenylacetylen reagierten zu 3,3-Dimethylbutyraldehyd bzw.
Phenylacetaldehyd, welche nicht durch die Hydroformylierung von Olefinen erhalten
werden können. Die Beispiele belegen den großen Anwendungsbereich dieses
Katalysatorsystems.12c In einem zu diesen Entwicklungen begleitenden Patent wurde
gezeigt, dass [CpRu(L2)]X-Komplexe mit stickstoffhaltigen Chelatliganden (L2) in der
katalystischen Hydratisierung von 1-Octin aktiv sind. Die besten Liganden sind in diesem
Fall 2,2´-Bipyridin (52% Umsatz), Phenanthrolin (49% Umsatz) und PyBOX-iPr36 (92%
Umsatz). Allerdings konnte kein Unterschied zwischen den kationischen
[CpRu(L2)(Lösungsmittel)]PF6-Komplexen mit entweder Phosphin- oder stickstoffhaltigen
Liganden im Vergleich zu den entsprechenden neutralen Chloro-Komplexen festgestellt
werden.34
Im selben Jahr wie WAKATSUKI veröffentlichten GROTJAHN und Mitarbeiter ihre Arbeit
auf dem Gebiet der Katalysatorentwicklung für anti-MARKOVNIKOV-Hydratisierung von
Alkinen. Dabei postulierten sie als Struktur für den Katalysatorkomplex folgende (Abb.
1.1):
Abbildung 1.1: Allgemeine Komplexstruktur.
Die Anordnung von Metall und Liganden, die mit ihren funktionellen Gruppen als
Protonen- oder Wasserstoffbindungsakzeptor fungieren können, leiteten sie aus der
Metallenzymkatalyse ab. Ausgehend von einem [CpRu(PR3)2]+-Fragment und einem
sterisch anspruchsvollen Imidazolylphosphinliganden erhielten sie den Ruthenium(II)-
Komplex 6 (Abb. 1.2). Mittels Kristallstrukturanalyse konnten sie zeigen, dass dieser

1. Einleitung
8
Komplex als Aquakomplex mit zwei Wasserstoffbrückenbindungen zwischen den freien
Elektronenpaaren der Stickstoffatome aus den Imidazolylphosphinliganden und den
Wasserstoffatomen des Wassers, das an das Metallzentrum koordinierte, vorliegt.12e
Abbildung 1.2: Der in der katalytischen Hydratisierung von Alkinen als Katalysator verwendete
Ruthenium(II)-Imidazolylphosphan-Komplex von GROTJAHN.
Ein Vorteil des neuen Katalysators 6 ist neben der geringeren Beladung bei der
Hydratisierung von Hexin (2 mol%, Aceton, 5 Äquiv H2O, 70 °C, 21 h, 92% Umsatz), dass
es bei Reaktionen mit Nitrilen zu keiner Deaktivierung kommt und Reaktionen mit
säureempfindlichen Propargylacetaten problemlos vonstatten gehen. Außerdem ist der
Katalysator auf eine Vielzahl von Substraten anwendbar und erzielt hohe
Regioselektivitäten von ≥100:1 zugunsten der Aldehyde. Jedoch deutete trotz der
Kristallstruktur und der damit verbundenen Erkenntnis, dass hier das Prinzip der
kooperativen Bindung zum Wasser vorliegt, wegen der quantitativ ähnlichen katalytischen
Aktivität von 6 im Vergleich zu der von CpRuCl(dppm) (1 mol%, Isopropanol/Wasser,
100 °C, 12 h, 95% Ausbeute) nichts auf einen grundlegend anderen (also: einen
bifunktionellen) Reaktionsmechanismus hin. Dies änderte sich aber nachdem ein direkter
kinetischer Vergleich in Aceton bei 70 °C durchgeführt wurde und der Komplex 6 eine
90fach höhere Aktivität aufwies als CpRuCl(dppm).37 Darüber hinaus präsentierten
GROTJAHN und LEV im Jahre 2004 weitere Modifikationen des Komplexes, bei denen die
wesentliche Neuerung in der Veränderung der Liganden bestand. Als aktivsten Katalysator
beschrieben sie den [CpRu(PPh2-Py-tBu)2(MeCN)]PF6-Komplex 7 (Abb. 1.3), bei dem die
bisher verwendeten Imidazolylphosphinliganden durch 6-substituierte-2-
Diphenylphosphanylpyridine ersetzt wurden.

1. Einleitung
9
Abbildung 1.3: Der Rutheniumkatalysator für katalytische Hydratisierung von Alkinen mit
anti-MARKOVNIKOV-Selektivität nach GROTJAHN (2004).
Komplex 7 katalysiert sogar bei Raumtemperatur die Umsetzung von alkylsubstituierten
Alkinen zu Aldehyden und ist fast 1000fach schneller als der bis dato aktivste Katalysator
von WAKATSUKI. Dies scheint nicht mehr allein durch einfache sterische und elektronische
Ligandenvariationen erklärbar zu sein, sondern deutet viel mehr auf das Auftreten der
kooperativen Katalyse hin.20
Im Zuge der Arbeit von GROTJAHN und LEV wurden sowohl die Phosphine als auch der
anionische Ligand verändert und getestet. Die beste Kombination mit dem [CpRuII]-
Fragment bildete das 6-tert-Butyl-2-diphenylphosphino-pyridin (tBuPyPPh2), wobei die
tert-Βutylgruppe wegen ihres sterischen Anspruchs eine wichtige Rolle spielt (s. Kap. 1.5.).
Des Weiteren wurden Alternativen zum Cyclopentadienylliganden getestet, die
Unterschiede in ihrer Elektronendichte und sterischen Hinderung aufwiesen. Allerdings
führten diese Variationen (Tris-pyrazolyl-borat = Tp, η5-Indenyl, η5-
Pentamethylcyclopentadienyl = η5-Cp*) nicht zu aktiven Katalysatoren oder im Fall des
Tp´s nur zu geringen Aktivitäten. Als Ergebnis der Arbeit wurde festgehalten, dass
[CpRuII]+ das derzeit geeignetste Metallfragment in Katalysatoren für die Hydratisierung
von terminalen Alkinen darstellt. Zum einen würde ein elektronenreiches und sterisch
anspruchsvolleres Metallzentrum eine Abspaltung der Phosphine und eine irreversible
Umwandlung der als Katalysezwischenstufe postulierten Metallacylkomplexe zu
Carbonylkomplexen bewirken. Zum anderen würde ein elektronenärmeres und sterisch
weniger anspruchsvolles Metallzentrum nicht die Isomerisierung des Alkins zu einem
Vinyliden-Komplex vollziehen,37 der als wichtiger Schritt im anti-MARKOVNIKOV-
Mechanismus angesehen wird (s. Kap 1.4.).12d
Ergänzend zum Aspekt der kooperativen Katalyse mit P,N-Liganden sei auf die Arbeit von
BREIT und CHEVALLIER hingewiesen, die die Selbstanordnung von zweizähnigen Liganden
durch Wasserstoffbrückenbindungen für die rutheniumkatalysierte anti-MARKOVNIKOV-

1. Einleitung
10
Hydratisierung von terminalen Alkinen ausgehend von einem [CpRuII]+-Fragment
beschrieben haben.38 Durch Kombination des Katalysatorvorläufers [CpRu(MeCN)3]PF6
mit einer Mischung aus Phosphinoisochinolon und Phosphinoaminopyridin im Verhältnis
1:1 erhielten sie unter anderem Komplex 8 (Abb. 1.4), der in ihrem Fall der aktiviste und
selektivste Katalysator für die Hydratisierung war.
Ru PPh2Ph2P
N N
O
PF6
8
NCOtBu
H
H
IsochinolonAminopyridin
Abbildung 1.4: Selbstanordnung von zweizähnigen Liganden durch Wasserstoffbrückenbindungen.
Unter den Reaktionsbedingungen (2–10 mol% 8, wässriges Aceton, 120 °C, 26–96 h)
wurden Ausbeuten zwischen 65% und 91% erzielt und meist lag die Regioselektivität über
99:1. Es war auch möglich einen tertitären Propargylalkohol innerhalb eines Steroids zu
hydratisieren ohne durch Eliminierung ein Aldol zu erhalten.38
HINTERMANN und Mitarbeiter hatten und haben sich zum Ziel gemacht die anti-
MARKOVNIKOV-Hydratisierung in der organischen Synthese anzuwenden und zu etablieren.
Nachdem die wegweisende Arbeit der Gruppe um GROTJAHN zum Thema bifunktionale
Katalyse mit Katalysator 7 veröffentlicht worden war37 schien vor dem Hintergrund dieser
Ergebnisse das Problem der katalytischen Aktivität fürs erste gelöst zu sein. Allerdings
wurde die benötigte Menge des Katalysators für die Forschungsprojekte zu einem
Problem,20 da der im Komplex 7 verwendete Phosphinpyridinligand über sechs Stufen mit
einer niedrigen Gesamtausbeute von 6% synthetisiert werden muss39, 40 und der
Vorläuferkomplex [CpRu(MeCN)3]PF6 luftempfindlich und sehr teuer ist.41
Daher wurde zuerst die Synthese der Phosphinpyridinliganden optimiert. Letztendlich
konnte die Sequenz auf zwei Stufen verkürzt werden. In der ersten Stufe wird 2,6-
Dibrompyridin mit sterisch anspruchsvollen Aryl-GRIGNARD-Verbindungen in einer
KUMADA-Kupplung selektiv zum monoarylierten Brompyridin umgesetzt. Dieses konnte
im zweiten Schritt durch nukleophile Phosphinierung in das luftstabile 6-Aryl-2-

1. Einleitung
11
diphenylphosphinopyridin überführt werden. Man erhielt hohe Ausbeuten für
unterschiedlich substituierte Aryl-GRIGNARD-Verbindung (78–86% Ausbeute).12f, 42
Im Zusammenhang mit der Optimierung des Katalysatorsystems wurde entdeckt, dass zwei
Äquivalente der Phosphinpyridinliganden zusammen mit dem luftstabilen Komplex
[CpRu(η6-C10H8)]PF6 (9)43 (C10H8 = Naphthalen) zu einem in situ Katalysator reagieren,
der hohe Aktivitäten in der anti-MARKOVNIKOV-Hydratisierung von terminalen Alkinen
zeigt.
Abbildung 1.5: In situ Katalysator für die anti-MARKOVNIKOV-Hydratisierung.
Die in situ Katalysatoren wurden aus 9 und 10–14 durch Ligandenaustausch (Abb. 1.5) in
Acetonitrillösung bei 60 °C erhalten und mittels 31P-NMR-Spektroskopie und ESI-MS-
Spektrometrie wurde gezeigt, dass die Komplexe [CpRu(10–14)2(MeCN)]PF6 analog zu
dem von GROTJAHN beschriebenen Komplex 7 aufgebaut sind. Die katalytischen
Hydratisierungen wurden in Aceton (1–5 mol% [Ru], 45–65 °C, 1–20 h, 69–99% Ausbeute)
durchgeführt, wobei die Größe des Substituenten R des Phosphinopyridinliganden
entscheidend für die Aktivität des Katalysators war. Diese stieg mit folgender Reihenfolge
der Substitutenten R an: Ph ˂ Mes ˂ tBu ˂ 2,4,6-(iPr)3C6H2 ˂ 2,4,6-Ph3C6H2.12f Dieser
Verlauf korreliert ungefähr mit dem Raumanspruch der Liganden, was überraschte, da es
aufgrund der mechanistischen Modelle für die Reaktion nicht vorhergesagt werden
konnte.12c-d, 37 Der in situ gebildete Komplex aus 9 und 12 zeigte eine sehr ähnliche
Reaktivität wie der Komplex 7 von GROTJAHN. Der aktivste Komplex (aus 9 und 14)
dieser Serie war um den Faktor vier schneller als die bis dahin schnellste
Katalysatorvariation von GROTJAHN. Eine weitere Vereinfachung des Katalysatorsystems
wurde durch die Erkenntnis möglich, dass der Ligandenaustausch auch unter den
Reaktionsbedingungen stattfindet. Somit wurden die Katalysen durchgeführt, indem man 9

1. Einleitung
12
mit dem entsprechenden Liganden 10–14 in Aceton, das fünf Äquivalente Wasser enthielt,
mischte und bei 70 °C über Nacht rühren ließ.12g
Zusätzlich zu den Phosphinopyridinliganden 10–14 wurden noch eine Reihe weiterer Aza-
arylphosphinen synthetisiert, die zusammen mit Komplex 9 sehr aktive in situ
Katalysatoren ergaben.42
1.5. Mechanismus der rutheniumkatalysierten anti-MARKOVNIKOV-
Hydratisierung
Mechanistische Untersuchungen von BIANCHINI und Mitarbeitern brachten eine erste
Erklärung für die Desaktivierung von Ruthenium(II)-Katalysatoren.44 Sie beschrieben, dass
das Ruthenium(II) mit dem Alkin einen Vinylidenkomplex bildet, der nach Addition von
Wasser zu einer Ruthenium-Acyl-Spezies führt. Diese zersetzt sich dann zum inaktiven
Rutheniumcarbonylkomplex unter Bildung von Alkanen anstelle der Aldehyde
(Schema 1.4).
Schema 1.4: Mechanismus der Hydratisierung nach BIANCHINI.
Einen ersten Vorschlag für den Mechanismus der anti-MARKOVNIKOV-Hydratisierung von
Alkinen lieferten 1998 WAKATSUKI und Mitarbeiter. Analog zum Mechanismus von
BIANCHINI gingen sie von einer Addition von Wasser an ein
Rutheniumvinylidenintermediat aus, so dass sich eine Ruthenium-Acyl-Spezies bildet.
Diese würde dann nach Protonierung der Kohlenstoff-Metall-Bindung zur Abspaltung des
Aldehyds führen (Schema 1.5).12b

1. Einleitung
13
R H[Ru]
C [Ru]H
R [Ru]-
OHH
R
[Ru]
OR
H H
H2O
- H+
H+
[Ru]-
OR
H H HH+
oderRH
O
H H
Schema 1.5: Erster Vorschlag für den Mechanismus der anti-MARKOVNIKOV-Hydratisierung.
Zwar konnte dieser Mechanismus die Bildung von Aldehyden erklären, aber nicht korrekt
die Ergebnisse der Experimente mit deuteriummarkierten Substraten vorhersagen. Diese
Experimente zeigten, dass das Wasserstoffatom des terminalen Alkins am endständigen
Kohlenstoffatom verbleibt (Schema 1.6).12d
Schema 1.6: Experimente mit deuteriummarkierten Substraten.
Deshalb schlug WAKATSUKI 2001 einen neuen Mechanismus vor, der experimentell jedoch
kaum belegt war (Schema 1.7). Danach findet ausgehend von einem π-Komplex A, der
sich aus Ruthenium(II) und Alkin bildet, eine externe Protonierung statt, die zu einem
Vinyl-ruthenium(IV)-Intermediat B führt. Dieses wird durch eine 1,2-Verschiebung von
Wasserstoff (in Schema 1.7, Deuterium) stabilisiert zu C und durch Addition von OH-
gefolgt von einer Tautomerisierung in einen Ruthenium(IV)-Hydrido-Acyl-Komplex D
überführt, welcher reduktiv den Aldehyd eliminiert.

1. Einleitung
14
R D[RuII]
R
H [RuIV]
D
C [RuIV]H
R D[RuIV]
OHH
RD
[RuIV]
OR
D
H+
OH-
R D
D
OR
H H
H H
A
B
C
D
Schema 1.7: Postulierter Mechanismus von WAKATSUKI für die anti-MARKOVNIKOV-Hydratisierung.
Dieser Mechanismus kann nun die Experimente mit den deuteriummarkierten Substraten
erklären und wird durch DFT-Rechnungen gestützt. Offen bleibt jedoch, wieso es zu einer
externen Protonierung in einem nahezu neutralen Reaktionsmedium kommt, wo doch ein
π-Alkin-Vinyliden-Gleichgewicht bekannt ist, und wieso es beim Ruthenium(IV)-Hydrido-
Acyl-Komplex nicht zu einem Protonenaustausch mit dem Lösungsmittel und
anschließender Decarbonylierung kommt, so wie im bis dahin gebräuchlichen
Mechanismus postuliert ist (Schema 1.5).
Durch die Einführung der bifunktionalen Katalyse durch GROTJAHN und Mitarbeiter und
die von ihnen beschriebene Beschleunigung der Reaktionskinetik12e, 37 ergaben sich
wichtige Forderungen an einen zu postulierenden Mechanismus: dieser muss in der Lage
sein, die enorme Beschleunigung der Katalyse durch die Phosphinopyridinliganden zu
erklären. Es konnte nachgewiesen werden, dass sich eine Wasserstoffbrückenbindung
zwischen dem Wasserstoffatom am endständigen Kohlenstoffatom des terminalen Alkins
und dem Stickstoff des Pyridylphosphans ausbildet.37 Ein weiterer Hinweis war die
Erkenntnis, dass sterisch anspruchsvolle Substituenten R (Abb. 1.6 a) in C-6 Position am
Pyridylphosphan für die anti-MARKOVNIKOV-Hydratisierung vonnöten sind, da sterisch
weniger anspruchsvolle Substituenten R den Katalysator durch eine P,N,P-Koordination
blockieren (Abb 1.6 b) und einfache Pyridylphosphane (R = H) in einer irreversible
Addition mit den am Ruthenium koordinierten Alkinen reagieren.37b

1. Einleitung
15
Ru(S)
Ph2P PPh2
N
R
N
R
Ru PPh2
NPh2P
R
N
Ra) b)
(S) = Lösungsmittel Abbildung 1.6: Der Substituenteneffekt der Pyridylphosphinliganden.
In einer ersten Vermutung über den Mechanismus der bifunktional katalysierten
Hydratisierung von Alkinen nahm GROTJAHN an, dass das freie Elektronenpaar des
Stickstoffatoms im Pyridiylphosphanliganden als Säure-Base-Katalysator für die
Protonierung des Alkins wirkt. Dies würde zum Ruthenium(IV)-Vinyl-Intermediat führen,
wie im Mechanismus von WAKATSUKI vorgeschlagen.37b Weiterführende Arbeiten der
gleichen Gruppe konnten die Vermutung jedoch nicht bestätigen. Um den Mechanismus
mit all seinen Intermediaten aufzuklären folgte man der Annahme aller bisher
vorgeschlagenen Mechanismen, die von einem Ruthenium-Alkin-π-Komplex ausgingen.
Darüber hinaus unterstrichen DFT-Rechnungen die Bedeutung des Komplexes 18
(Schema 1.8) für den Reaktionsweg.45
1. Einleitung
16
H H
RuCl
R2P PR2
N N
RuR2P PR2
N
RuO
R2P PR2
N NHHN
RuR2P PR2
N N
RuR2P PR2
N NCCH
R1
RuR2P PR2
N HNO
R1
+ H2O
- H2O
H2O, 0 °C
HCCH-40 °C
> 0 °C- R1CH2CHO
DCM,KX,H2OKX
DCM
+ X- + X-
+ X- + X-
+ X-
> -40 °C,0-3h
15
17
18
19
20
16
R = Ph oder MeX = B(C6F5)4 oder B[3,5-(CF3)2C6H2]4
> 0 °C- R1CH2CHO
Ru
+ X-
PhMe2P PMe2PhHH
18a
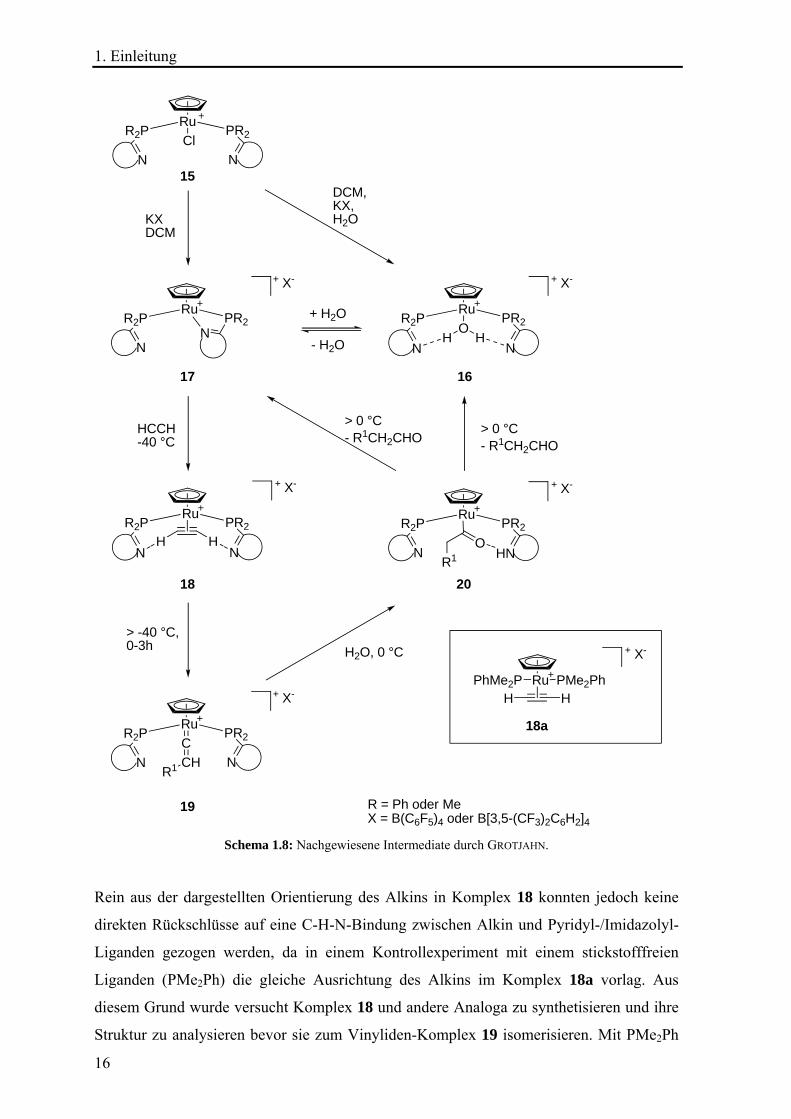
Schema 1.8: Nachgewiesene Intermediate durch GROTJAHN.
Rein aus der dargestellten Orientierung des Alkins in Komplex 18 konnten jedoch keine
direkten Rückschlüsse auf eine C-H-N-Bindung zwischen Alkin und Pyridyl-/Imidazolyl-
Liganden gezogen werden, da in einem Kontrollexperiment mit einem stickstofffreien
Liganden (PMe2Ph) die gleiche Ausrichtung des Alkins im Komplex 18a vorlag. Aus
diesem Grund wurde versucht Komplex 18 und andere Analoga zu synthetisieren und ihre
Struktur zu analysieren bevor sie zum Vinyliden-Komplex 19 isomerisieren. Mit PMe2Ph

1. Einleitung
17
als Ligand und CpRu(COD)Cl als Vorläuferkomplex gelang GROTJAHN und Mitarbeitern
die Umsetzung mit Acetylen zum entsprechenden Komplex 18a (X = Cl), während sie für
die Komplexe mit Pyridyl- und Imidazolyl-Liganden zunächst die Aqua-Komplexe 16
herstellen mussten. Diese bildeten gelagert unter Vakuum unerwartet durch
Wasserabspaltung den Chelatkomplex 17, der im Gleichgewicht mit Komplex 16 und
Wasser steht. Im Fall des Pyridyl-Liganden (tBuPyPMe2) konnte eine Umsetzung von 16
oder 17 mit Acetylen zu Komplex 19 bei –40 °C beobachtet werden. Beim Imidazolyl-
Liganden46 war dies nicht möglich, da der entsprechende Aqua-Komplex bis 10 °C inert
gegenüber der Reaktion mit Acetylen war und danach direkt über die Zwischenstufen 18
und 19 zu einem unsymmetrischen Acylprodukt reagierte.45 Diese unterschiedlichen
Verhalten in der Transformation der Komplexintermediate zeigen deutlich den Effekt der
kinetischen Beschleunigung, der durch den Liganden mit seinem basischen Stickstoffatom
hervorgerufen wird. GROTJAHN schließt durch einen Vergleich der Kopplungen (1JCH und 2JCH) aus dem NMR-Spektrum mit denen in einer Literaturstelle47 vorhergesagten
Kopplungen für eine HCCH–OH2 Bindung darauf, dass die Wasserstoffbrückenbindungen
in Komplex 18 mit (tBuPyPMe2) als Ligand vorhanden sind. Diese Schlussfolgerung
konnte durch die Beobachtung der 2hJCN-Kopplung (hier: 3 ± 0.5 Hz) bestättigt werden, da
diese ein direkter Nachweis für die Wasserstoffbrückenbindung ist. Wie schon erwähnt
isomerisieren die Komplexe vom Typ 18, die nicht mit Pyridyldiphenylphosphinliganden
(R = Ph), sondern nur mit Pyridyldimethylphosphinliganden (R = Me), nachgewiesen
werden konnten, zu Komplex 19. Diese Isomerisierung verläuft mit unterschiedlichen
Liganden unterschiedlich schnell und Komplex 19 reagiert bei 0 °C und in Gegenwart von
Wasser zum Acyl-Komplex 20. Es konnte durch NMR-Spektroskopie und
Markierungsexperimente nachgewiesen werden, dass die möglichen Tautomere von 20
nicht vorliegen. Somit konnte zum ersten Mal ein Acyl-Intermediat 20 für die anti-
MARKOVNIKOV-Hydratisierung von Alkinen nachgewiesen und gezeigt werden, dass das
für die Katalyse benötigte Proton am basischen Pyridinliganden lokalisiert ist und dieser
Ligand eine Wasserstoffbrückenbindung zum acylischen Sauerstoffatom bildet.48 In
weiteren Arbeiten will GROTJAHN den Einfluss der bifunktionalen Liganden auf die
Bildung von 20 und die Abspaltung des Aldehyds untersuchen.49
Die Studien von GROTJAHN und Mitarbeitern zeigen deutlich, dass der Mechanismus der
bifunktionalen Katalyse zumindest für das untersuchte System nicht mit dem von
WAKATSUKI vorgeschlagenen Mechanismus vereinbar ist, da keine Protonierung des

1. Einleitung
18
Metallzentrums beobachtet werden konnte, sondern die des Stickstoffs im bifunktionalen
Liganden.
1.6. Synthetische Anwendungen der anti-MARKOVNIKOV-Hydratisierung
Generell ist die Verwendung von Wasser als Reagenz in organischen Reaktionen aus Sicht
der Ökonomie, Sicherheit und Umwelt ein sehr interessanter Punkt.50 Bisher ist die anti-
MARKOVNIKOV-selektive Hydratisierung von terminalen Alkinen nur selten synthetisch
verwendet worden. Bekannt ist jedoch, dass diese Reaktion auf viele Substrate anwendbar
ist. In der Katalyse werden sowohl Sauerstoffverbindungen, wie Ketone, Ester, TBS-Ether
und THP-Acetale,12e, 37a, 12f als auch Stickstoffverbindungen wie Amide, Imide38 und
Sulfonamide37a toleriert.

2. Aufgabenstellung
19
2. Aufgabenstellung
Ein Ziel dieser Arbeit war es die katalytische anti-MARKOVNIKOV-Hydratisierung von
terminalen Alkinen weiterzuentwickeln und zu optimieren um die besten
Reaktionsbedingungen für die Anwendung in Synthesestrategien zu erhalten. In diesem
Zusammenhang sollten die Parameter Substratkonzentration, Lösungsmittel und
Katalysatorbeladung variiert werden, was erste Hinweise auf die Reaktivität der
Rutheniumkomplexe unter verschiedenen Bedingungen liefern sollte. Anschließend sollte
das katalytische Verhalten der Rutheniumkomplexe genauer in den Fokus gestellt werden
indem man neben den oben genannten Parametern unterschiedliche Wassermengen und
verschiedene Additive in der Katalyse testet. Hieraus sollte sich ergeben, ob die Katalyse
positiv oder negativ beeinflusst werden kann und in wie weit sie empfindlich gegenüber
Nebenprodukten oder Verunreinigungen ist. Des Weiteren sollte der Katalysator selbst
durch die Wahl der Liganden modifiziert werden um die Effizienz zu erhöhen und um
weitere Informationen über den Mechanismus bzw. den Ablauf der Reaktion zu erhalten.
Neben der Optimierung des Katalysators und der Reaktionsbedingungen sollten
erste Anwendungen für die katalytische Hydratisierung in Synthesestrategien entwickelt
werden. Ziel dabei war es das Potential und die Effizienz dieser atomökonomischen
Reaktion in der Synthese von Naturstoffen und naturstoffähnlichen Verbindungen
aufzuzeigen. Mit Hilfe der anti-MARKOVNIKOV-Hydratisierung sollten sowohl Poly-1,3-
diole also auch Poly-1,4-diole und deren Derivate hergestellt werden.

3. Hauptteil
20
3. Hauptteil
3.1. Optimierung der rutheniumkatalysierten anti-
MARKOVNIKOV-Hydratisierung von terminalen Alkinen
Aus den Arbeiten von WAKATSUKI, GROTJAHN und unserer Arbeitsgruppe wird deutlich,
dass die optimalen Bedingungen für die rutheniumkatalysierte Hydratisierung von
terminalen Alkinen bisher höchstens für Einzelfälle beschrieben sind und eine
systematische und umfassende Suche nach den besten Reaktionsparametern noch nicht
durchgeführt worden ist.
3.1.1. Methodenentwicklung und Modellreaktion51 Um den Reaktionsverlauf der rutheniumkatalysierten Hydratisierung möglichst zeitnah
verfolgen zu können und die Messzeit gering zu halten sollte anstelle der von LABONNE
verwendeten analytischen Methoden eine neue entwickelt werden.42 Denn jede Probe war
mittels einer GC-Messung analysiert worden, welche unter den verwendeten Bedingungen
eine Messzeit von einer Stunde aufwies. Stattdessen sollte nun die HPLC mit UV-Detektor
in Kombination mit einer Kieselgelsäule (Kromasil® 100 Si-5μ) genutzt werden um eine
Messzeit von unter 15 min zu erreichen, so dass die Proben nach der Entnahme aus der
Reaktionslösung bis zur Analyse, keine Lagerung erforderten, während derer sekundäre
Reaktionen auftreten könnten. Auf diese Weise lassen sich die Messpunkte für die
Dokumentation des Reaktionsverlaufes enger setzen, was eine erheblich genauere
Erfassung und Genauigkeit der Katalysen ermöglicht. Als Substrat für die Modellreaktion
fiel die Wahl auf 4-Phenyl-1-butin, da dieses Alkin bzw. dessen Hydratisierungsprodukt
UV-aktiv ist und somit vom UV-Detektor der HPLC leicht detektiert werden kann.
Darüber hinaus lässt es sich mit einer Retentionszeit von ca. 3 min (Kromasil® 100 Si-5μ,
Laufmittel: TBME/Hexan, 1:5; Flussgeschwindigkeit 1 mL/min) gut vom 4-Phenylbutanal,
dem Hydratisierungprodukt trennen, welches eine Retentionszeit von ca. 5 min hat. Zur
quantitativen Bestimmung des Reaktionsproduktes wurde als interner Standard
Pivaloylanilid (Tab. 3.1, Eintrag 12) gewählt, das mit in die Reaktionslösung gegeben
wurde. Diese Verbindung beeinflusst unter den gegebenen Reaktionsbedingungen nicht

3. Hauptteil
21
die Katalyse und hat, im Vergleich zu den anderen getesteten Substanzen (Tab. 3.1), die
geeignetste Retentionszeit.
Eintrag
Substanz Retentionszeit tR [min]a
1
5.5
2
3.5
3 3.6
4
3.3
5 O
5.3
6
16.0
7
4.8
8
4.5
9 O O
3.6
10
56.0
11
17.3
12
7.0
13
6.0
a) HPLC (Kromasil® 100 Si-5μ, 20 °C, λ = 284 nm, TBME/Hexan, 1:5, 1.0 mL/min).
Tabelle 3.1: Retentionszeiten verschiedener Substanzen als potentielle interne Standards.
Mit diesen Ergebnissen konnte die Modellreaktion (Schema 3.1) für die weiteren
kinetischen Untersuchungen aufgestellt werden:

3. Hauptteil
22
2 mol% Katalysator
Aceton, Wasser,Pivaloylanilid, 50 °C
O
Schema 3.1: Modellreaktion für die kinetischen Messungen.
Ein Standardchromatogramm, das es auszuwerten galt, ist in Abbildung 3.1 dargestellt.
Abbildung 3.1: HPLC-Chromatogramm einer gemessenen Probe bei λ = 213 nm.
3.1.2. Untersuchungen zur Katalysatorvariation Um den von GROTJAHN beschriebenen Effekt der bifunktionalen Katalyse12e, 37 sowie um
die Ergebnisse seiner Arbeit und der von LABONNE42, 12f zu reproduzieren, wurden die
Komplexe [CpRu(tBuPyPPh2)2(MeCN)]PF6 (7), [CpRu(tAmPyPPh2)2(MeCN)]PF6 (21)
und [CpRu{(2,4,6Ph3C6H2)PyPPh2}2(MeCN)]PF6 (22) (Abb. 3.2) synthetisiert. Die

3. Hauptteil
23
Komplexsynthese erfolgte durch Ligandenaustausch mit dem Vorläuferkomplex
[CpRu(η6-C10H8)]PF643 (9) (C10H8 = Naphthalen) und den Liganden 12, 14 und 2-
Diphenylphosphino-6-tert-pentylpyridin in Acetonitril (vgl. Abb. 1.5).
Abbildung 3.2: Ruthenium(II)-Komplexe.
Die Komplexe 7, 21 und 22 wurden in der Modellreaktion (vgl. Schema 3.1) mit einer
Katalysatorbeladung von 2 mol% eingesetzt. Die Hydratisierung des 4-Phenyl-1-butins zu
4-Phenylbutanal konnte mit der unter Kap. 3.1.1. beschriebenen HPLC-Methode verfolgt
und durch Quantifizierung über den internen Standard in einem Umsatz-Reaktionszeit-
Diagramm ausgewertet werden. Aus den Umsatzkurven der einzelnen Katalysen (Abb. 3.3)
ist der beschriebene Einfluss der Substituentengröße am Pyridylphosphin auf die
katalytische Hydratisierung deutlich erkennbar.
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
72122
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.3: Vergleich der Komplexe 7, 21 und 22 in der Katalyse.

3. Hauptteil
24
Mit steigendem sterischen Anspruch der Substituenten am Phosphanliganden nimmt die
Geschwindigkeit der Katalyse zu. Der Unterschied zwischen Komplex 7 mit tBuPyPPh2
als Liganden und 21 mit tAmPyPPh2 ist wegen der Ähnlichkeit ihrer Substituenten noch
gering, nimmt aber beim Komplex 22, der (2,4,6Ph3C6H2)PyPPh2 als Liganden trägt, stark
zu. Der Komplex 22 liefert nach 1 h ca. 50%igen und nach 6 h nahezu vollständigen
Umsatz, während die Komplexe 7 und 21 erst nach 3–3.5 h 50% und nach 9 h fast
vollständigen Umsatz erreichen. Dies bestätigt die bereits veröffentlichten Ergebnisse in
dem Sinne, dass die Aktivität der Katalysatorkomplexe vom sterischen Anspruch ihrer
Liganden abhängt.12e, 37, 12f-g, 42 Für die nachfolgenden Untersuchungen in dieser Arbeit
wird das Ergebnis, das der Komplexes 22 in der Katalyse ergeben hat, als Referenz
verwendet, da dieses das bisher beste Resultate in der rutheniumkatalysierten
Hydratisierung darstellt.
3.1.3. Effekt der Katalysatorbeladung und Konzentration Nachdem aus den obigen Untersuchungen hervor ging, dass sterisch anspruchsvolle
Liganden die Aktivität des Katalysators erhöhen stellte sich als nächstes die Frage welchen
Einfluss die Katalysatorbeladung auf die Reaktionsgeschwindigkeit hat und in wieweit die
Geschwindigtkeit von der Konzentration der Reaktionslösung abhängt. Aus diesem Grund
wurde eine Serie mit 1 mol%, 2 mol%, 3 mol% und 4 mol% Katalysatorbeladung sowohl
bei einer Substratkonzentration von 0.25 M als auch von 0.5 M durchgeführt.
Bei beiden Serien steigt der Umsatz pro Zeit mit der Katalysatorbeladung, wobei es eine
größere Differenz zwischen 1 mol% und 2 mol% gibt, während die Unterschiede zwischen
2 mol%, 3 mol% und 4 mol% geringer ausfallen. Nach einer Anfangsphase von ungefähr
30 min, in der die unterschiedlichen Katalysatorbeladungen sehr ähnliche Umsätze zeigen,
kann man bis zur dritten Stunde der Katalyse die größten Unterschiede im Verlauf
feststellen. Danach gleichen sich die Kurven im Zeitraum von der vierten Stunde bis zur
sechsten Stunde aneinander und asymptotisch an den vollständigen Umsatz an (Abb. 3.4).

3. Hauptteil
25
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
1 mol%2 mol%3 mol%4 mol%
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, X mol% [CpRu(14)2(MeCN)]PF6, 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
a)
Abbildung 3.4a: Versuchsreihe zur Katalysatorbeladung bei c = 0.25 M.
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
1 mol%2 mol%3 mol%4 mol%
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, X mol% [CpRu(14)2(MeCN)]PF6, 1 mL (c = 0.5 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
b)
Abbildung 3.4b: Versuchsreihe zur Katalysatorbeladung bei c = 0.5 M.
Dies trifft jedoch nicht für die Reaktionen mit 1 mol% Katalysatorbeladung zu. Bei der
Konzentration von 0.25 M ist diese Menge an Katalysator zwar ausreichend um eine
nahezu vollständige Umsetzung des Alkins zum Aldehyd zu erzielen, aber dies geschieht
mit einer Reaktionszeit von ca. 9 h wesentlich langsamer als bei höherer
Katalysatorbeladung (Abb. 3.4a). Erhöht man die Konzentration auf 0.5 M so bleibt der

3. Hauptteil
26
Umsatz bei 55-60% stehen (Abb. 3.4b), was eventuell durch experimentelle
Abweichungen, die bei solch geringen Katalysatormengen eine größere Auswirkung haben,
oder aber durch Desaktivierung des Katalysators hervorgerufen wurde. Durch mehrere
Wiederholungsexperimente konnte jedoch ausgeschlossen werden, dass es an einem
praktischen Fehler bei der Durchführung lag. Somit scheint es sich um eine Desaktivierung
des Katalysators zu handeln. Ein Indiz hierfür liefert das 31P-NMR-Spektrum der
Reaktionslösung, da nach der Katalyse ein Peak bei 48.7 ppm statt wie erwartet bei 44.3
ppm (aktiver Katalysatorkomplex 22) vorhanden war. Dies deutet auf eine neue
Komplexspezies hin, die während der Katalyse gebildet wurde und inaktiv zu sein scheint.
Durch ein Experiment, bei dem der Komplex 22 in Lösung vorgelegt und
Kohlenstoffmonooxid durch die Lösung geleitet wurde, konnte gezeigt werden, dass der
Peak bei 44.3 ppm im Laufe der Zeit in den Peak bei 48.7 ppm übergeht. Folglich handelt
es sich beim Peak bei 48.7 ppm um einen Rutheniumcarbonyl-Komplex, der keine
katalytische Aktivität in der Hydratisierung zeigt.
Ein direkter Vergleich der unterschiedlichen Katalysatorbeladungen bei den beiden
Konzentrationen zeigt, dass die Unterschiede in der Aktivität minimal sind (ausgenommen
1 mol%) (Abb. 3.5). Somit scheint der Effekt der Substratkonzentration gerade bei höheren
Mengen an Katalysator geringer zu sein.
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
1 mol% bei c = 0.25 M2 mol% bei c = 0.25 M3 mol% bei c = 0.25 M4 mol% bei c = 0.25 M1 mol% bei c = 0.5 M2 mol% bei c = 0.5 M3 mol% bei c = 0.5 M4 mol% bei c = 0.5 M
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, X mol% [CpRu(14)2(MeCN)]PF6, Y mL (1 mL bei c = 0.25 M; 2 mL bei c = 0.5 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.5: Direkter Vergleich der Katalysen bei unterschiedlicher Konzentration und
Katalysatorbeladung.

3. Hauptteil
27
Die kinetischen Messungen mit unterschiedlichen Katalysatorbeladungen haben gezeigt,
dass die Unterschiede in der Geschwindigkeit sich bezüglich der Endausbeute mit der Zeit
(nach 4–6 h) egalisieren. Die Veränderung der Substratkonzentration hat unter den
angegebenen Bedingungen nur eine geringe Auswirkung auf den Reaktionsverlauf und die
Geschwindigkeit. Auf Grund der ähnlichen Aktivität des Katalysators unter verschiedenen
Reaktionsbedingungen wurde für die weiteren Katalysen die Menge an Ruthenium(II)-
Komplex auf 2 mol% und die Substratkonzentration auf 0.25 M festgesetzt, da dies gut zu
händelende Bedingungen sind und die Katalysatorbeladung ausreichend erscheint.
3.1.4. Komplexe ohne bifunktionale Liganden Neben den Ruthenium(II)-Komplexen mit bifunktionalen Liganden wurden auch
Katalysatoren mit zweizähnigen Phosphinliganden getestet. Dieser Typ von Katalysatoren
war bereits von WAKATSUKI als aktiv für die Hydratisierung von Alkinen beschrieben
worden12c, 34 und es war gezeigt worden, dass es keinen Unterschied zwischen den
kationischen [CpRu(L2)(Lösungsmittel)]PF6-Komplexen (L = Ligand) und den
entsprechenden neutralen Chloro-Komplexen gibt.34 Deshalb wurden die kationischen
Komplexe 23–2652 unter den Bedingungen der Modellreaktion in der Katalyse eingesetzt.
Ebenso wurde mit dem kommerziell erhältlichen Komplex 27 und dem selbst hergestellten
Komplex 28 verfahren (Abb. 3.6).
Abbildung 3.6: Weitere Ruthenium(II)-Komplexe für die Hydratisierung.

3. Hauptteil
28
Wider Erwarten zeigte keiner der genannten Komplexe eine Aktivität in der
Hydratisierung, obwohl der Komplex 27 aufgrund der Angaben von WAKATSUKI einen
Umsatz hätte bewirken sollen.12c Da sich aber die von WAKATSUKI beschriebene Reaktion
in Temperatur, Lösungsmittel und umzusetzendem Substrat von der Modellreaktion
unterschied wurden die Bedingungen etwas angepasst und die Hydratisierung von 1-
Phenylbutin wurde nochmals in einem Lösungsmittelgemisch aus iso-Propanol/Wasser
(3:1) bei 100 °C und mit 2 mol% Katalysatorbeladung durchgeführt. Unter diesen
Bedingungen konnte mit Komplex 27 nach 9 h ein Umsatz von 64% erreicht werden,
jedoch stieg dieser nicht weiter an. Vergleicht man dies mit Komplex 22 unter den
gleichen Bedingungen, so wird deutlich, dass dessen Aktivität um ein vielfaches höher
liegt, da er bereits nach 10 min einen nahezu vollständigen Umsatz (93%) erzeugt
(Abb. 3.7).
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
2227
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 1.25 mL (c = 0.31 M) iso -Propanol, 0.375 mL (41.64 Äquiv) H2O, Pivaloylanilid als Interner Standard, 100 °C.
Abbildung 3.7: Vergleich von Komplex 22 und 27 in der Hydratisierung von 4-Phenyl-1-butin
unter veränderten Bedingungen.
Die Abweichung der Ergebnisse für Komplex 27 im Vergleich zur Literatur (>99%
Ausbeute bei der Hydratisierung von 1-Hexin)12c kann durch das verwendete Substrat
erklärt werden, da in einem Kontrollversuch mit dem aliphatischen Alkin 1-Octin ein
nahezu identischer Umsatz im Vergleich zur Literaturangabe, welcher durch GC-Analytik
bestimmt wurde, erhalten werden konnte. Aufschlussreich war jedoch, dass der Komplex
22 in iso-Propanol/Wasser bereits nach 10 min fast quantitative Umsätze lieferte. Dies

3. Hauptteil
29
stellt eine Erhöhung der Reaktionsgeschwindigkeit um ein Vielfaches dar. Die Begründung
hierfür dürfte nicht ein Konzentrationseffekt sein, da die Konzentrationen mit 0.31 M und
0.25 M (Modellreaktion) sehr ähnlich waren. Ebenso wenig dürfte die Wahl des
Lösungsmittels oder die Wassermenge entscheidend sein. Zwar war die Wassermenge mit
42 Äquivalenten in etwa achtmal so groß wie unter Modellreaktionsbedingungen, aber von
wesentlich entscheidender Bedeutung ist die Erhöhung der Temperatur [°C] um das
Doppelte. Zur allgemeinen Abklärung des Einflusses des Lösungsmittels und der
Wassermenge auf die rutheniumkatalysierte Hydratisierung von Alkinen wurden weitere
Versuchsreihen durchgeführt (s. Kap. 3.1.5 und 3.1.6). Der Einfluss der Temperatur [°C],
die beim Lösungsmittelgemisch iso-Propanol/Wasser doppelt so hoch war wie bei Aceton
und etwaige Druckeffekte wurden jedoch nicht genauer untersucht.
3.1.5. Der Lösungsmitteleffekt in der Hydratisierung Um den Einfluss des Lösungsmittels beurteilen zu können wurde eine Serie von Katalysen
mit unterschiedlichen Lösungsmitteln unter den standardisierten Bedingungen der
Modellreaktion durchgeführt. Insgesamt wurden neben Aceton sieben weitere
Lösungsmittel und ein Gemisch aus Aceton/Wasser (50:50) getestet.
Keines der Lösungsmittel, abgesehen von dem Lösungsmittelgemisch aus Aceton/Wasser,
lieferte bessere Ergebnisse als Aceton, was nicht auf ein Löslichkeitsproblem
zurückzuführen ist, da alle eine homogene Lösung ergaben (Abb. 3.8).

3. Hauptteil
30
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
Aceton–Wasser, 50:50AcetonDimethoxyethanTHFDimethoxymethanTrifluorethanolDiethylketonDioxanEthanol
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% [CpRu(14)2(MeCN)]PF6, 1 mL (c = 0.5 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.8: Unterschiedliche Lösungsmittel für die Hydratisierung.
Als schlechtestes Lösungsmittel für die Reaktion stellte sich Ethanol heraus. Dies dürfte
vermutlich daran liegen, dass es die durch Dissoziation von Acetonitril freiwerdende
Koordinationsstelle am Komplex 22 belegt indem es durch die freien Elektronenpaare des
Sauerstoffatoms an das Metallzentrum bindet, so wie das Wassermolekül in Komplex 16.
Eine anschließende Dissoziation des Ethanols scheint zwar stattzufinden, da die Katalyse
noch abläuft, aber nur verlangsamt. Ein Indiz für diese Vermutung liefert das 2,2,2-
Trifluorethanol, da die Katalyse mit diesem Lösungsmittel schneller als mit Ethanol abläuft,
was in der schnelleren Dissoziation des Lösungsmittels bedingt durch den elektronischen
Effekt der CF3-Gruppe auf die Hydroxygruppe begründet sein dürfte. Als ein alternatives
Lösungsmittel für Aceton dürfte sich am ehesten ein Ether und von denen das
1,2-Dimethoxyethan eignen, da dieses zwar in den ersten Stunden die Katalyse etwas
verlangsamt, aber nach 9 h in etwa zum gleichen Umsatz führt. Eine Erhöhung des
Umsatzes pro Zeit zeigt in der Anfangsphase nur das Gemisch aus Aceton und Wasser im
Vergleich zu Aceton, was auf eine Beschleunigung der Katalyse durch eine höhere
Wasserkonzentration im Reaktionsmedium hindeutet.

3. Hauptteil
31
3.1.6. Der Einfluss der Wassermenge auf die Hydratisierung Wie sich in den vorherigen Unterkapiteln angedeutet hat scheint Wasser einen großen
Einfluss auf die Geschwindigkeit der Katalyse zu haben. Um die Relation zwischen
Wassermenge (gemessen in Äquivalente zum Substrat) und Geschwindigkeit der
rutheniumkatalysierten Hydratisierung von Alkinen zu bestimmen wurden Katalysen mit
2.5, 5 (Standard), 10, 20, 36.65 und 55.5 Äquivalenten Wasser durchgeführt. Als Ergebnis
dieser Versuchsreihe kann festgehalten werden, dass die Reaktionsgeschwindigkeit mit
Zunahme der Wassermenge steigt, sich jedoch nach 6 h die Umsätze der Katalysen bis auf
die mit 2.5 Äquivalenten Wasser nahezu angeglichen haben (Abb. 3.9).
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
2.5 Äquiv5 Äquiv10 Äquiv20 Äquiv36.65 Äquiv55.5 Äquiv
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% [CpRu(14)2(MeCN)]PF6, 2 mL (c = 0.25 M) Aceton, X Äquiv H2O, Pivaloylanilid als Interner Standard, 50 °C.
Aceton:Wasser (ν/ν)87.9 : 143.4 : 121.1 : 110.1 : 1
5 : 13 : 1
Abbildung 3.9: Der Einfluss von Wasser auf die Katalyse.
Des Weiteren geht aus den Verlaufskurven hervor, dass es einen größeren Unterschied
zwischen dem Versuch mit 2.5 Äquivalenten Wasser und den drei Versuchen mit 5, 10 und
20 Äquivalenten Wasser gibt. Die letzteren drei zeigen eine ähnliche Geschwindigkeit.
Einen größeren Unterschied gibt es auch zwischen den Katalysen mit 20 und
36.65 Äquivalenten und zwischen den Katalysen mit 36.65 und 55.5 Äquivalenten.
Der Effekt des Wassers, das mit steigendem Anteil im Lösungsmittel die Hydratisierung
beschleunigt, könnte ausgehend von dem von GROTJAHN postulierten Mechanismus mit
den nachgewiesen Intermediaten45, 48, 49 (s. Kap 1.5) erklärt werden. Nach Dissoziation des
Acetonitrils aus Komplex 22 wird der in der Katalyse aktive Aqua-Komplex 16 schneller
gebildet. Dies ist vorteilhaft, da das Wasser nicht so stark wie Acetonitril an das

3. Hauptteil
32
Metallzentrum koordiniert, so dass dadurch die Anlagerung des Alkins und die Bildung
des π-Komplexes 18 erleichert wird. Außerdem kann die Addition von Wasser an den
Vinyliden-Komplex 19 zum Acyl-Komplex 20 ebenfalls schneller ablaufen. Ein anderer
Erklärungsansatz wäre, der vermutlich wahrscheinlicher ist, dass das
Hexafluorophosphatanion mit steigendern Wassermenge nicht mehr an das Komplexkation
koordiniert und somit die Anlagerung eines Alkins an die von Acetonitril freiwerdende
Koordinationsstelle erleichtert wird (siehe Kap. 3.1.10).
3.1.7. Der Einfluss des Acetonitrils auf die Hydratisierung In diesem Kapitel soll der Vermutung nachgegangen werden, dass Acetonitril die
rutheniumkatalysierte Hydratisierung von terminalen Alkinen mit Komplex 22 hemmt (s.
Kap. 3.1.3). Um diese Vermutung zu untermauern, wurden die Katalysen zusätzlich mit
Acetonitril versetzt. Die Menge an Acetonitril wurde dabei variiert (5, 10, 20 und 50 mol%)
und zur Auswertung mit einer Probe ohne zusätzliches Acetonitril verglichen (Abb. 3.10).
In letzterem Fall ist zu beachten, dass ein mol-Äquivalent Acetonitril schon im Katalysator
enthalten ist.
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
0%5%10%20%50%
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% [CpRu(14)2(MeCN)]PF6, 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C, X mol% MeCN.
Abbildung 3.10: Der Effekt von Acetonitril auf die Katalyse.
Aus den Verlaufskurven ist ersichtlich, dass das zugegebene Acetonitril die Katalyse
verlangsamt. Dabei besteht eine direkte Korrelation zwischen der Verlangsamung der

3. Hauptteil
33
Katalyse und der zugefügten Menge an Acetonitril. Die hemmende Wirkung, die durch
diese Experimente bestätigt wird, liegt wohl darin, dass das Acetonitril mit steigender
Konzentration in der Lösung die nach der Dissoziation aus Komplex 22 oder nach
Abspaltung des Aldeyds aus Acyl-Komplex 20 freigewordene Koordinationsstelle am
Metallzentrum schneller besetzt als Wasser oder das nächste Substartmolekül. Somit muss
sich aus dem neu entstandenen Komplex 22 erst wieder das Acetonitril abspalten, bevor
sich der aktive Komplex 18 bzw. 17 bilden und sich das Alkin koordinativ an das
Metallzentrum unter Bildung des π-Komplexes 18 binden kann um dann entsprechend dem
postulierten Mechanismus (s. Kap. 1.5) weiter zu reagieren.
Diese Ergebnisse machen deutlich, dass bei der Synthese der Komplexe entsprechend
sorgfältig gearbeitet werden muss, damit kein überschüssiges Acetonitril, das nicht am
Metall gebunden ist, nach der Komplexbildung als Verunreinigung zurückbleibt.
Alternativ kann zur Komplexbildung anstelle des Acetonitrils (s. Kap 3.1.2) auch iso-
Butyronitril oder Benzonitril verwendet werden, was zu den Komplexen 29 und 30 führt
(Abb. 3.11).
RuN
PPh2Ph2P
N
R
PF6-
N
R
RuN
PPh2Ph2P
N
RPh
Ph
Ph Ph
R =
PF6-
N
R
29
30
RuPF6
9
NR PPh2
+ 2
tBuCN50 °C, 25 h
PhCN50 °C, 138 h
14
Abbildung 3.11: Alternative Nitrile im Ruthenium(II)-Komplex für die Hydratisierung.
Der Einbau von iso-Butylnitril bzw. von Benzonitril könnte zu einer Aktivitätssteigerung
der Komplexe in der Hydratisierung führen, da sich iso-Butyronitril wegen seines sterisch
anspruchsvollen iso-Propylrest schneller vom Metallzentrum abspalten und langsamer
wieder anlagern sollte. Benzonitril könnte sich weniger wegen sterischen als viel mehr
wegen elektronischen Gründen leichter vom Metallzentrum abspalten als Acetonitril.

3. Hauptteil
34
Jedoch zeigen die kinetischen Auswertungen der Katalysen, dass beide Nitrile im
Vergleich zum Acetonitril die Reaktion verlangsamen (Abb. 3.12).
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
222930
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.12: Die iso-Butyro- und Benzonitril-Komplexe 29 und 30 in der Katalyse.
Der erwünschte Effekt der Steigerung der Aktivität durch die Variation des Nitrils im
Rutheniumkomplex ist nicht eingetreten. Es bleibt festzuhalten, dass Acetonitril das derzeit
am besten geeignete Nitril für die Komplexbildung ist und diese Komplexe die höchste
Aktivität aufweisen. Allerdings verlangsamt Acetonitril die Katalyse erheblich, sobald es
über das durch Abspaltung aus dem Rutheniumkomplex festgelegte Maß hinaus im
Reaktionsmedium vorliegt.
3.1.8. Der Einfluss von Additiven auf die Katalyse Die Verlangsamung der Katalyse durch freies Acetonitril (s. Kap. 3.1.7) deutet daraufhin,
dass es eine gewisse Empfindlichkeit der rutheniumkatalysierten Hydratisierung von
terminalen Alkinen gegenüber Additiven gibt. Um Abzuklären welche Additive einen
positiven oder negativen Effekt auf die Kinetik der Katalyse haben, wurden sieben
Substanzen in der Modellreaktion zu gesetzt und der Reaktionsverlauf kinetisch verfolgt
(Abb. 3.13).

3. Hauptteil
35
0
20
40
60
80
100
0,0 0,5 1,0 1,5 2,0 2,5 3,0 3,5 4,0 4,5
Reaktionszeit t [h]
Aus
beut
e A
ldes
hyd
[%]
kein Additiv
Benzoesäure
Tetrabutylammoniumhexafluorophosphat
Tetrabutylammonium-toluol-4-sulfonat
2-Amino-benzoesäure
Tetrabutylammoniumchlorid
Tetraethylammoniumacetat-tetrahydrat
Hünig-Base
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% [CpRu(14)2(MeCN)]PF6, 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C, 20 mol% Additiv.
Abbildung 3.13: Additive in der rutheniumkatalysierten Hydratisierung von Alkinen.
Die Katalyse wurde nicht negativ durch Benzoesäure, Tetrabutylammonium-toluol-4-
sulfonat und -hexafluorophosphat, dem Gegenion zum kationischen Ruthenium(II)-
Komplex (s. Kap. 3.10), beeinflusst. 2-Amino-benzoesäure bewirkt eine minimale
Verschlechterung des Umsatzes. Zu einem Stillstand der Katalyse führten
Tetrabutylammoniumchlorid, Tetraethylammoniumacetat-tetrahydrat, weil Acetat
vermutlich stark an die freiwerdende Koordinationsstelle des Rutheniums bindet, und die
Hünig-Base. Im Falle basischer Additive wird der Katalysator möglicherweise als
Acetylid-Komplex blockiert.
Diese Resultate belegen, dass die Katalyse gewisse Additive toleriert und ein etwaiges
Entstehen der Carbonsäure aus dem Produkt der Hydratisierung durch Oxidation nicht die
Aktivität des Komplexes beeinträchtigt, was durch Benzoesäure als inertes Additiv belegt
wird. Insgesamt konnte jedoch keine Verbesserung des katalytischen Systems durch die
Additive erzielt werden.
3.1.9. Der Ligandeneffekt in Mischkomplexen In den vorherigen Kapiteln wurde gezeigt, dass sich die Geschwindigkeit und die Aktivität
der Ruthenium(II)-Komplexe mit bi- und nicht bifunktionalen Liganden für die
Hydratisierung von terminalen Alkinen durch die Katalysatorbeladung, die

3. Hauptteil
36
Substratkonzentration, das Lösungsmittel und Additive beeinflussen lassen. Die Komplexe
mit bifunktionalen und sterisch anspruchsvollen Liganden stellten sich dabei als die
aktivsten heraus, wie es teilweise schon von GROTJAHN im Zusammemhang mit dem
Substituenteneffekt in C-6 Position am Pyridylphophanliganden beschrieben wurde.37b
Bisher wurden jedoch nur homoleptische Bis-Komplexe in der Katalyse eingesetzt.
Heteroleptische Bis-Komplexe vom Typ [CpRu(L1)(L2)(MeCN)]PF6 (L1,L2 = Liganden)
mit zwei unterschiedlichen Liganden wurden noch nicht verwendet. Der Vorteil solcher
Mischkomplexe wäre, dass man über den einen Liganden das basische Stickstoffatom, das
für die Koordinierung des Wassers und des Alkins und die spätere Protonierung zu
Komplex 20 im vorgeschlagenen Mechanismus45, 48, 49 essentiell vonnöten ist, einführt,
aber über den zweiten Liganden versucht werden kann, die elektronischen Eigenschaften
des Metallzentrums und die Raumumgebung um die Koordinationsstelle zu verändern.
Allerdings ist aufgrund des Mechanismus nicht vorhersagbar, ob solche Mischkomplexe in
der Katalyse aktiv sind, weil alle bisherigen bifunktionellen Katalysatoren für die Reaktion
jeweils zwei funktionalisierte Liganden enthielten. Die Annahme geht zwar dahin, dass
auch ein einzelner funktionalisierter Ligand ausreichend sein dürfte um den Vinyliden-
Komplex 19 und nach Wasseraddition und Protonierung den Acyl-Komplex 20 zu bilden,
aber bekannt ist dies nicht.
3.1.9.1. Die Synthese der Ruthenium(II)-Mischkomplexe
Bei der Synthese der Mischkomplexe kann in Analogie zu der der homoleptischen-bis-
Komplexe verfahren werden. Der Vorläuferkomplex [CpRu(η6-C10H8)]PF6Fehler!
Textmarke nicht definiert. (9) (C10H8 = Naphthalen) wird mit einem Äquivalent von
Ligand 14 in Acetonitril innerhalb von 20 h bei Raumtemperatur zum monosubstituierten
Komplex 31 umgesetzt, der isoliert wird (Schema 3.2). Es ist bei diesem, wie auch beim
nächsten Syntheseschritt darauf zu achten, dass die stöchiometrischen Verhältnisse aufs
genaueste eingehalten werden, da sich sonst die entsprechenden homoleptischen-bis-
Komplexe anstelle der Mischkomplexe bilden können.

3. Hauptteil
37
RuPF6
9
NPh2P
Ph
Ph Ph
MeCN
RT, 20 h
RuPh2P
N
PF6-
Ph
Ph
NN
PhMe
Me
14 31
RuPh2P
N
PF6-
Ph
Ph
NL
PhMe
1 Äquiv L,Aceton, RT, 3 h
Schema 3.2: Synthese der Mischkomplexe.
Der Komplex 31 kann in Acetone mit diversen Phosphinen und Phosphiten zu den
entsprechenden Mischkomplexen umgesetzt werden (Abb. 3.14). Die entsprechenden
Ruthenium(II)-Komplexe konnten mittels 31P-NMR-Spektroskopie und ESI-MS-
Spektrometrie eindeutig charakterisiert werden. Neben sterisch anspruchsvollen und
weniger anspruchsvollen Liganden konnten auch Liganden mit unterschiedlichen
elektronischen Eigenschaften zu den Mischkomplexen umgesetzt werden, wobei die
Synthese die Bildung hochreiner Verbindungen erlaubte.
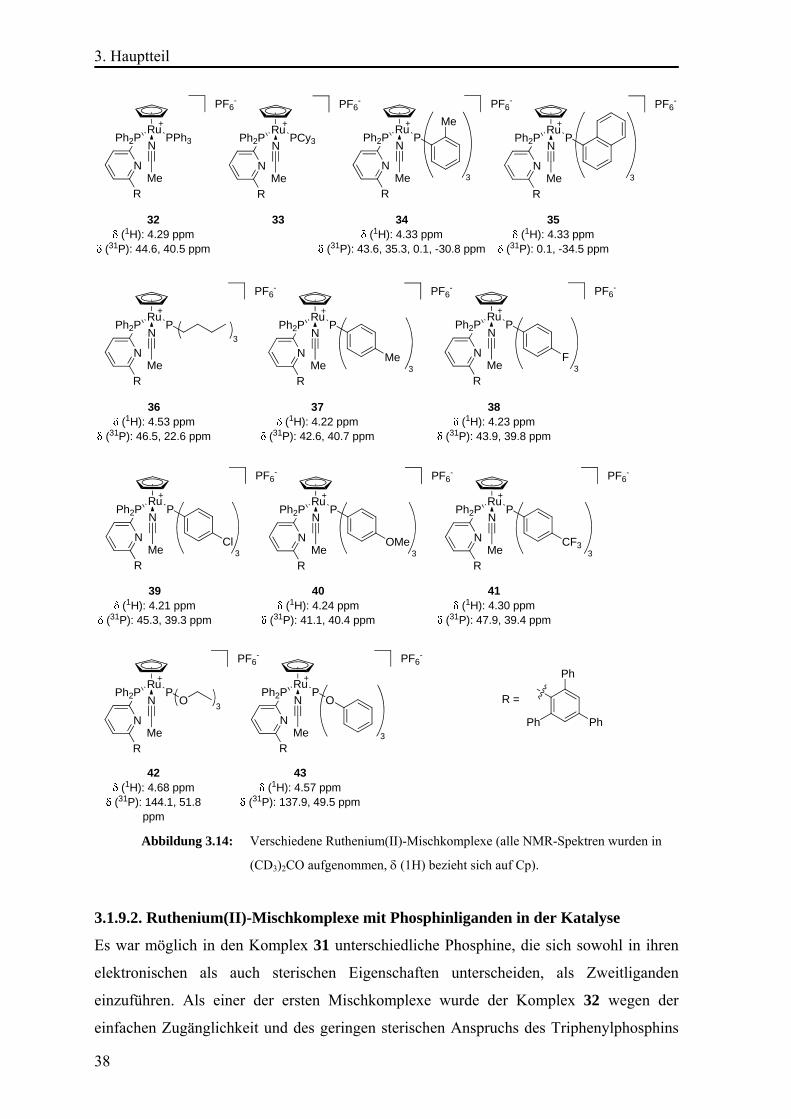
3. Hauptteil
38
RuN
PPh2P
N
RMe
PF6-
3
RuN
PCy3Ph2P
N
RMe
PF6-
RuN
PPh2P
N
RMe
PF6-
O 3
RuN
PPh2P
N
RMe
PF6-
O
3
RuN
PPh3Ph2P
N
RMe
PF6-
RuN
PPh2P
N
RMe
PF6-
F3
RuN
PPh2P
N
RMe
PF6-
Cl3
RuN
PPh2P
N
RMe
PF6-
Me
3
RuN
PPh2P
N
RMe
PF6-
3
RuN
PPh2P
N
RMe
PF6-
Me3
RuN
PPh2P
N
RMe
PF6-
OMe3
RuN
PPh2P
N
RMe
PF6-
CF33
Ph
Ph Ph
R =
32(1H): 4.29 ppm
(31P): 44.6, 40.5 ppm
33 34(1H): 4.33 ppm
(31P): 43.6, 35.3, 0.1, -30.8 ppm
35(1H): 4.33 ppm
(31P): 0.1, -34.5 ppm
36(1H): 4.53 ppm
(31P): 46.5, 22.6 ppm
37(1H): 4.22 ppm
(31P): 42.6, 40.7 ppm
38(1H): 4.23 ppm
(31P): 43.9, 39.8 ppm
39(1H): 4.21 ppm
(31P): 45.3, 39.3 ppm
40(1H): 4.24 ppm
(31P): 41.1, 40.4 ppm
41(1H): 4.30 ppm
(31P): 47.9, 39.4 ppm
42(1H): 4.68 ppm
(31P): 144.1, 51.8ppm
43(1H): 4.57 ppm
(31P): 137.9, 49.5 ppm
Abbildung 3.14: Verschiedene Ruthenium(II)-Mischkomplexe (alle NMR-Spektren wurden in
(CD3)2CO aufgenommen, δ (1H) bezieht sich auf Cp).
3.1.9.2. Ruthenium(II)-Mischkomplexe mit Phosphinliganden in der Katalyse
Es war möglich in den Komplex 31 unterschiedliche Phosphine, die sich sowohl in ihren
elektronischen als auch sterischen Eigenschaften unterscheiden, als Zweitliganden
einzuführen. Als einer der ersten Mischkomplexe wurde der Komplex 32 wegen der
einfachen Zugänglichkeit und des geringen sterischen Anspruchs des Triphenylphosphins
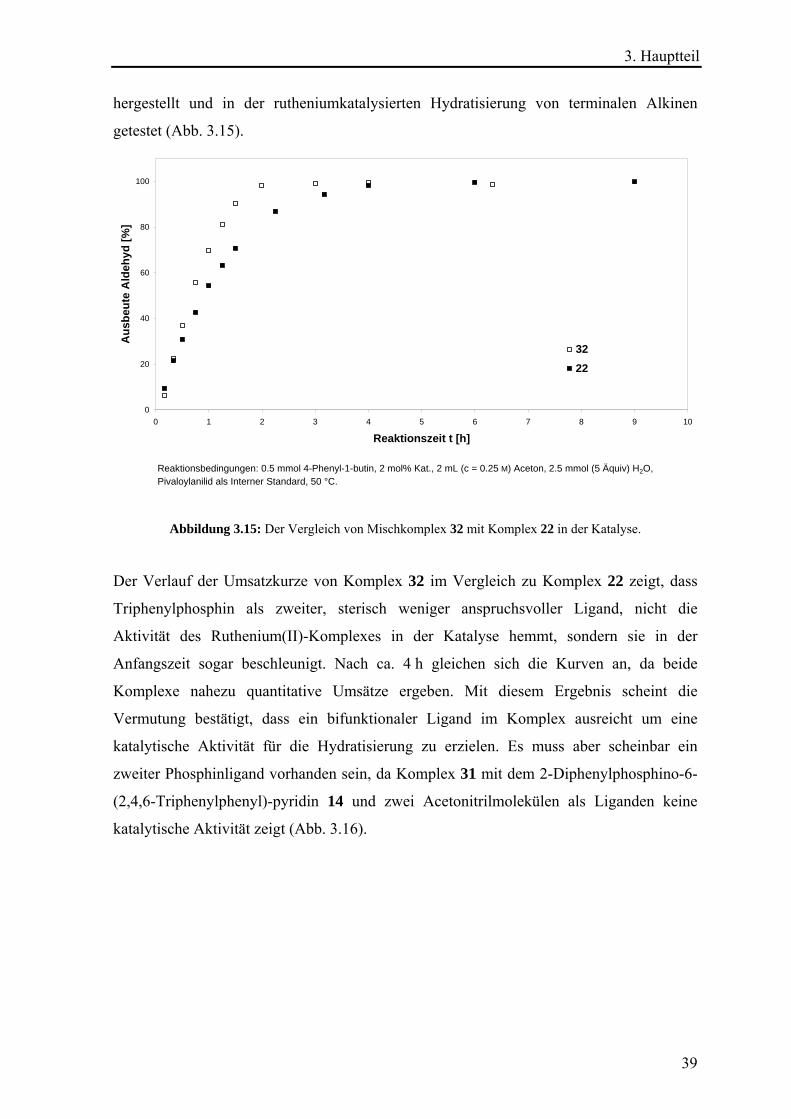
3. Hauptteil
39
hergestellt und in der rutheniumkatalysierten Hydratisierung von terminalen Alkinen
getestet (Abb. 3.15).
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
3222
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.15: Der Vergleich von Mischkomplex 32 mit Komplex 22 in der Katalyse.
Der Verlauf der Umsatzkurze von Komplex 32 im Vergleich zu Komplex 22 zeigt, dass
Triphenylphosphin als zweiter, sterisch weniger anspruchsvoller Ligand, nicht die
Aktivität des Ruthenium(II)-Komplexes in der Katalyse hemmt, sondern sie in der
Anfangszeit sogar beschleunigt. Nach ca. 4 h gleichen sich die Kurven an, da beide
Komplexe nahezu quantitative Umsätze ergeben. Mit diesem Ergebnis scheint die
Vermutung bestätigt, dass ein bifunktionaler Ligand im Komplex ausreicht um eine
katalytische Aktivität für die Hydratisierung zu erzielen. Es muss aber scheinbar ein
zweiter Phosphinligand vorhanden sein, da Komplex 31 mit dem 2-Diphenylphosphino-6-
(2,4,6-Triphenylphenyl)-pyridin 14 und zwei Acetonitrilmolekülen als Liganden keine
katalytische Aktivität zeigt (Abb. 3.16).

3. Hauptteil
40
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
2231
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O,Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.16: Komplex 31 in der Katalyse.
Zusätzlich wurden Phosphine wie Tributylphosphin, Tri(para-tolyl)phosphin, Tri(ortho-
tolyl)phosphin, Tricyclohexylphosphin, Tri(1-naphthyl)phosphin, Tri(para-
fluorphenyl)phosphin, Tri(para-chlorphenyl)phosphin, Tri(para-methoxyphenyl)phosphin
und Tri(para-trifluoromethylphenyl)phosphin als Liganden im Mischkomplex getestet um
die sterischen und elektronischen Verhältnisse am Metallzentrum zu verändern. All diese
Katalysatorvariationen hatten bis auf einen Fall negative Auswirkungen. Der Komplex mit
Tricyclohexylphosphin (33) als zweiten Liganden war nahezu inaktiv in der Katalyse (Abb.
3.17), was an der räumlichen Abschirmung der Koordinationsstelle durch die
Cyclohexylgruppen liegen dürfte, die eine Anlagerung des Alkins und die Bildung des π-
Komplexes 18 verhindert. Allerdings bildet sich der Komplex 33 trotz des sterisch
gehinderten Phosphinliganden, während dies bei den Komplexen 34 und 35 in Aceton
nicht der Fall war. Das 31P-NMR-Spektrum vom Versuch der Synthese von 34 mit
Tri(ortho-tolyl)phosphin als Ligand zeigt, dass sich der Komplex 22 (43.6 ppm) und
möglicherweise [CpRu(oTol3P)2(MeCN)]PF6 (35.3 ppm) gebildet haben und darüber
hinaus noch freies Phosphin (oTol3P = –30.8 ppm) vorliegt. Hierin scheint auch der Grund
für die Inaktivität des verwendeten Katalysatorgemisches zu liegen. Aufschlussreich ist an
dieser Stelle, dass nicht der heteroleptische Ruthenium(II)-Komplex gebildet werden kann.
Dies ist wohl darauf zurück zu führen, dass das ortho-Tolylphosphin durch die
Methylgruppe in sterisch ungünstiger Position den zuvor in den Komplex eingeführten

3. Hauptteil
41
2,4,6Ph3C6H2PyPPh2-Liganden verdrängt, was schlussendlich zu den homoleptischen
Komplexen führt. In diesem Zusammenhang wundert es nicht, dass sich Komplex 35 mit
Tri(1-naphthyl)phosphin als sterisch sehr anspruchsvollen Koliganden nicht gebildet hat.
Es konnte in diesem Fall im 31P-NMR-Spektrum nur der Mono-Chelat-Komplex mit
2,4,6Ph3C6H2PyPPh2 (0.1 ppm) und freies Tri(1-naphthyl)phosphin (–34.5 ppm)
nachgewiesen werden.
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
372236333435
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.17: Mischkomplexe mit sterisch anspruchsvollen Phosphinliganden in der Katalyse.
Erst der Komplex 36 mit Tributylphosphin als Koliganden brachte Umsätze, die aber weit
unter denen des Komplexes 22 blieben. Wir waren in der Lage von Komplex 36 einen
Kristall zu züchten und zum ersten Mal eine Kristallstruktur von einem Cyclopentadienyl-
Ruthenium(II)-Komplex mit einem 6-substituierten Pyridylphosphin zu erhalten. Aus der
Kristallstrukturanalyse (Abb. 3.18) wird ersichtlich, dass die Butylreste des koordinierten
Tributylphosphins die mit Acetonitril besetzte Koordinationsstelle so abschirmen, dass
eine Anlagerung und Reaktion am Metallzentrum erschwert wird. Auffällig ist auch die
Orientierung der Pyridylgruppe, die mit ihrem Stickstoff von der Koordinationsstelle des
Acetonitrils wegzeigt.

3. Hauptteil
42
Abbildung 3.18: Kristallstruktur von Komplex 35 [Darstellung: ORTEP (50% Wahrscheinlichkeit
des Ellipsoids, H-Atome in kalkulierter Position mit freigewähltem Durchmesser,
PF6-Gegenion weggelassen)].
Der Vergleich von Komplex 37 mit Tri(para-tolyl)phosphin als zweiten Liganden mit
Komplex 22 (Abb. 3.17) zeigt, dass beide eine ähnliche Aktivität in der Hydratisierung
zeigen. Bezieht man jetzt noch Komplex 32 mit in den Vergleich ein, so scheint es, dass
die Methylgruppe in para-Stellung am Phenylrest des Phosphinliganden zwar zu keiner
sterischen Hinderung führt, aber durch einen elektronischen Effekt auf das Phosphoratom
und somit das Metall die Aktivität in der Katalyse leicht beeinflusst. Die Variation des
Substituenten in para-Stellung um den Einfluss verschiedener elektronischer Effekte
untersuchen zu können führte mit den Komplexen 38–41 (Abb. 3.14) zu dem Ergebnis,
dass para-substituierte-Triphenylphosphine als Koligand unabhängig vom elektronischen
Effekt, den die Substituenten ausüben, alle sehr aktiv sind (Abb. 3.19). Die kleinen
Unterschiede lassen sich so interpretieren, dass σ-Akzeptorliganden (X=F, Cl) etwas
höhere Aktivitäten induzieren als σ-Donoren (X=Me), aber die Datenlage ist nicht ganz
eindeutig, da z.B. Komplex 41 (X=CF3) nicht in dieses Bild passt.

3. Hauptteil
43
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
323738394041
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
X = HX = MeX = FX = ClX = OMeX = CF3
Abbildung 3.19: Mischkomplexe mit para-substituierten-Triphenylphosphinliganden in der Katalyse.
3.1.9.3. Heteroleptische Ruthenium(II)-Mischkomplexe mit Phosphit- und anderen
Liganden
Neben den Phosphinen wurden als Liganden in den Mischkomplexen auch Phosphite
getestet (Abb. 3.14). Die Mischkomplexe 42 mit Triethylphosphit und 43 mit
Triphenylphosphit sind zwar aktive Katalysatoren für die Hydratisierung von terminalen
Alkinen, jedoch kann man aus dem Umsatz-Reaktionszeit-Diagramm ersehen, dass sie
wesentlich langsamer sind, als der als Standard angenommene Komplex 22 (Abb. 3.20).
Sie liegen mit ihrer Aktivität in etwa im Bereich des Mischkomplexes 36. Hier ist der
elektronische Effekt des Liganden vermutlich der ausschlaggebende Punkt für die
Verlangsamung der Katalyse.

3. Hauptteil
44
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
224243
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.20: Mischkomplexe mit Phosphitliganden in der Katalyse.
3.1.10. Austausch des Gegenions In der Arbeit von LABONNE wurde gezeigt, dass der Austausch des Gegenions
Hexafluorophosphat durch rac-TRISPHAT im Komplex 22 einen positiven Einfluss auf die
katalytische Aktivität hat.42 Leider liessen die angegebenen Daten nur eine grobe
Schätzung des Effekts zu, weshalb zum besseren Vergleich das Experiment mit Komplex
44, der durch einen Anionenaustausch mit (HNBu3)(rac)-TRISPHAT in DCM aus Komplex
22 hergestellt wird, wiederholt wurde (Abb. 3.21).
RuN
PPh2Ph2P
N
RMe
Ph
Ph Ph
R =
X-
N
R
RuN
PPh3Ph2P
N
RMe
X-
OPO
OOO O
ClCl
ClCl
ClCl
Cl
Cl
ClCl
Cl
Cl
X- =
44 45 Abbildung 3.21: Komplexe mit TRISPHAT als Gegenion für die Hydratisierung.
Vergleicht man die Umsatzkurve von 44 mit derjenigen von 22 unter den
Standardbedingungen so fällt auf, dass der Komplex mit TRISPHAT als Gegenion in den

3. Hauptteil
45
ersten 3 h der Katalyse schneller ist, aber nach ca. 4 h beide Komplexen den gleichen,
nahezu quantitativen Umsatz erzielen (Abb. 3.22). Dies stimmt von der Tendenz mit den
beschriebenen Ergebnissen überein. Da ein beschleunigender Effekt der Katalyse durch
den Austausch des Gegenions bewirkt wurde sollte überprüft werden, ob auch die Aktivität
des bis dato schnellsten Katalysators 32 durch den Anionenaustausch, der zu Komplex 45
(Abb. 3.21) führt, weiter gesteigert werden kann.
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionzeit t [h]
Aus
beut
e A
ldeh
yd [%
]
45322244
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% Kat., 2 mL (c = 0.25 M) Aceton, 2.5 mmol (5 Äquiv) H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.22: TRISPHAT-Komplexe in der Katalyse.
Vergleicht man die Umsatzkurven von Komplex 32 und 45 so fällt auf, dass sie einen fast
identischen Verlauf haben, was darauf schließen lässt, dass das Gegenion in diesem Fall
keinen signifikanten Einfluss mehr auf die Katalyse hat. Zur weiteren Erhöhung der
Geschwindigkeit der Katalyse mit Komplex 44 wurde dieser in Kombination mit 55.5
Äquivalenten Wasser in der Modellreaktion eingesetzt (Abb. 3.23).

3. Hauptteil
46
0
20
40
60
80
100
0 1 2 3 4 5 6 7 8 9 10
Reaktionszeit t [h]
Aus
beut
e A
ldeh
yd [%
]
55.5 Äquiv
5 Äquiv
Reaktionsbedingungen: 0.5 mmol 4-Phenyl-1-butin, 2 mol% [CpRu(14)2(MeCN)]Trisphat, 2 mL (c = 0.25 M) Aceton, X Äquiv H2O, Pivaloylanilid als Interner Standard, 50 °C.
Abbildung 3.23: Der Effekt von Gegenion und Wassser auf die Katalyse.
Das Ergebnis war mit einem ähnlichen Verlauf der Geschwindigkeitskurven überraschend
und weißt darauf hin, dass an dieser Stelle ein intrinsisches Optimum an Aktivität erreicht
zu sein scheint, weil eine Kombination der beiden einzelnen Effekte (Wassergehalt, Art
des Anions) keine Steigerung der Reaktionsgeschwindigkeit mehr mit sich bringt.
Die Erhöhung der Geschwindigkeit im Vergleich von Katalysator 22 zu 44 dürfte in der
Art des Gegenions begründet sein. Das Hexafluorophosphat-Anion assoziiert sich eng an
das Komplexkation und bildet ein Kontaktionenpaar, in dem es aus sterischen Gründen
schwieriger wird das Acetonitril im Komplex durch ein Substratmolekül zu ersetzen. Im
Gegensatz dazu zeigt TRISPHAT, das seine negative Ladung über viele Atome verteilen
kann und lipophil ist, eine schwächere Anion/Kation-Interaktion und liegt vollständig
dissoziiert vor. Dies führt wohl zu der höheren Geschwindigkeit im Vergleich von
Katalysator 44 zu 22, da die Reaktion an der mit Acetonitril besetzen Koordinationsstelle
nicht aus sterischen Gründen durch das Gegenion behindert wird. Die Erhöhung der
Aktivität durch Steigerung der Wassermenge (s. Kap. 3.1.6), wie es noch bei Katalysator
22 beobachtet werden konnte, ist bei Katalysator 44 nicht zu sehen, was daran liegen dürfte,
dass das TRISPHAT-Anion bereits vollständig dissoziiert vorliegt, während das
Hexafluorophosphat-Anion, welches in einem Gleichgewicht aus Komplex und Komplex-
Kation mit solvatisiertem Gegenion steht, noch assoziiert und erst durch das Wasser
vollständig dissoziiert wird.

3. Hauptteil
47
3.1.11. Die Optimierungen auf einen Blick Als Ergebnis dieser umfassenden kinetischen Studien lässt sich festhalten, dass das beste
Katalysatorsystem für die rutheniumkatalysierte anti-MARKOVNIKOV-Hydratisierung von
terminalen Alkinen aus Komplex 32 in Aceton mit 55.5 Äquivalenten Wasser bei 50 °C
bei einer Substratkonzentration von 0.25 M besteht. Diese Bedingungen stellen eine
erhebliche Verbesserung bezogen auf die Reaktionsgeschwindigkeit sowie die
Zugänglichkeit des Komplexes dar. Es erwies sich aufgrund dieser Erkenntnis als unnötig
zwei Äquivalente eines bifunktionalen P,N-Liganden in den Komplex
[CpRu(L1)(L2)(S)]+PF6- einzuführen, da ein einzelnes in Kombination mit einem
Äquivalent eines einfachen Phosphins wie PPh3 ausreicht. Es konnte gezeigt werden, dass
die Wahl des Lösungsmittels entscheidend für die Katalyse ist und dass Additive durch
Besetzung der freien Koordinationsstelle am Metallzentrum wenn überhaupt meist einen
hemmenden Einfluss auf die Reaktion haben. Im Bereich der Ligandenvariation konnte,
wie oben bereits erwähnt, ein bifunktionaler P,N-Ligand mit einem „Platzhalter“-Liganden
kombiniert werden, wobei die Testreihe ergab, dass als „Platzhalter“ ein elektonenarmes
und sterisch wenig anspruchsvolles Triarylphosphin am besten geeignet ist. Bei dem so
optimierten System war die Wahl des Gegenions, der noch beim Komplexpaar 22/44 einen
Einfluss hatte, nicht mehr von Belang für die Geschwindigkeit der Katalyse.
Im Bereich des Wassergehalts des Lösungsmittels besteht noch die Möglichkeit der
weiteren Optimierung des Systems, allerdings ist diese durch die Löslichkeit von Substrat
und Katalysator beschränkt, da hier mit 55.5 Äquivalenten Wasser die Grenze der
Löslichkeit erreicht ist. Eventuell ließe sich die Löslichkeit des Katalysators in Wasser
durch das Einführen z. B. einer oder mehrerer Sulfonat-Gruppen in der Liganden-
Peripherie erhöhen. Dies würde dann weitere Steigerungen des Wasseranteils im
Lösungsmittel erlauben, was vielleicht die Reaktionsgeschwindigkeit weiter beschleunigt,
aber auch zu einem zweiphasigen Katalysatorsystem führen könnte.

3. Hauptteil
48
3.2. Anwendung der anti-MARKOVNIKOV-Hydratisierung in der
Synthese
3.2.1. Hydratisierung von Propargylalkoholen Durch die Addition von Acetylen an Aldehyde erhält man Propargylalkohole,53 welche
durch die Entwicklungen der letzten Jahre auch in enantiomerenreiner Form zugänglich
geworden sind. Neben der wohl bekanntesten Reaktion, der asymmetrischen Addition von
Alkinen an Aldehyde mittels Zn(OTf)2/N-Methylephedrin, die von CARREIRA und
Mitarbeitern beschrieben wurde,54 führen auch die Transfer-Hydrierung55 oder die
Reduktion von Carbonylverbindungen mit chiralen Oxazaborolidin-Katalysatoren56 zu
enantiomerenreinen Propargylalkoholen. Eine Kombination dieser Alkinylierungen mit der
anti-MARKOVNIKOV-Hydratisierung sollte theoretisch zu einer neuen und komplett
stereoselektiven Synthese von Aldolen führen, die bei einer iterativen Verlängerung
stereoselektiv Poly-1,3-diole ergeben würde (Abb. 3.4).
Schema 3.3: Synthese von Poly-1,3-diolen via iterative Alkinylierung und
anti-MARKOVNIKOV-Hydratisierung.
Der Nutzen solch einer Synthesestrategie liegt klar auf der Hand, wenn man die Bedeutung
der Aldolderivate als Intermediate für die asymmetrische Synthese betrachtet. Außerdem
ist dieser Syntheseweg im Idealfall komplett atomökonomisch, da sowohl die
asymmetrische Addition des Acetylens, das hier als Nukleophil reagiert, an den Aldehyd
mit der Bildung des stereogenen Zentrums als auch die Hydratisierung des
Propargylalkohols zum Aldol katalytische Reaktionen sind, bei denen kein
„Abfall“ entsteht. Dies ist jedoch etwas geschönt, da durch den Begriff der Atomökonomie
nicht die benötigte Menge an Katalysator, die stark von dessen Effizienz in der Katalyse
abhängt, berücksichtigt wird. Um wirklich von Atomökonomie sprechen zu können müsste

3. Hauptteil
49
die Leistung des Katalysators, in diesem Fall vor allem des Rutheniumkatalysators für die
Hydratisierung, weiter optimiert werden und er müsste nach der Reaktion zurückgewonnen
und wieder eingesetzt werden können. Entwicklungen in diesem Bereich erscheinen aber
erst dann als sinnvoll, wenn gezeigt werden kann, dass das Prinzip der vorgestellten
Synthesestrategie im Kern realisierbar ist. Ein erstes Hindernis auf diesem Weg war die
Hydratisierung von ungeschützten Propargylalkoholen, da die Katalysatoren für die anti-
MARKOVNIKOV-Hydratisierung nicht in der Lage waren diese umzusetzen. Aus diesem
Grund müssen die Alkohole zuvor mit einer Schutzgruppe versehen werden, was sicherlich
nicht atomökonomisch ist, aber später den Vorteil hat, dass man selektiv weitere
Funktionalisierungen vornehmen kann.
3.2.2. Hydratisierung von Propargylalkoholen mit Ruthenium-
katalysatoren Aufgrund der Arbeiten von LABONNE sind die Liganden für die Komplexe vom Typ
[CpRu(L)2(MeCN)]PF6 (vgl. Kap. 1.4) wesentlich leichter zugänglich geworden,12f, 42 so
dass es möglich wurde sich intensiver dem Thema der Hydratisierung propargylischer
Alkohole zu widmen. Als Modelsubstrate für die (kinetischen) Studien wurden geschützte
Versionen des 1-Octin-3-ols gewählt, welches sowohl als racemische als auch als
enantiomerenangereicherte Verbindung kommerziell erhältlich ist.
Abbildung 3.24: Geschützte 1-Octin-3-ole.
Erste Versuche zur Hydratisierung der geschützten Alkine 46-49 (Abb. 3.25) in Aceton mit
5 Äquivalenten Wasser bei 50 oder 70 °C mit 3 mol% des Katalysators 21 ergaben, dass

3. Hauptteil
50
das THP-Acetal 47 mit 20% Umsatz (bestimmt mittels 1H-NMR-Spektroskopie) die besten
Resultate lieferte (Tab. 3.2).57
Eintrag Substrat Zeit
[h]
Temp.
[°C]
Produkte
[molare%]a
1 R = Bn (46) 24 50 0 0 2 “ 48 70 <5 10 3 R = THP (47) 24 50 <10 0 4 “ 48 70 20 <5 5 R = TBS (48) 48 70 0 0 6 R = Bz (49) 48 70 0 10
a) Die Produkte wurden mittels 1H-NMR-Spektroskopie analysiert und quantifiziert und als molare Zusammensetzung des Produktgemisches angegeben.
Tabelle 3.2: Erste Hydratisierungen von unterschiedlich geschützten Octinolen.
Neben dem Aldehyd des jeweils eingesetzten Propargylalkohols wurde als Nebenprodukt
(E)-2-Octenal in unterschiedlichen Mengen festgestellt. Zur Optimierung führte man
weitere Versuche mit 47 in unterschiedlichen Lösungsmitteln durch (Tab. 3.3). Am Ende
dieser Versuchsreihe erwies sich Aceton, wie schon in den Modellstudien zur
Reaktionsoptimierung, als das geeignetste Lösungsmittel. Unter den optimierten
Bedingungen aus Tab. 3.3 war es möglich in einem 5 mmol Ansatz mit 5 mol% 21 in
Aceton (c = 0.2 M) mit 5 Äquivalenten Wasser nach 30 h bei 40 °C eine Ausbeute von
44% (nach Säulenchromatographie) zu erhalten.57

3. Hauptteil
51
Eintrag Lösungsmittel Zeit
[h]
Temp.
[°C]
Produkte
[mol-%]a
1 Aceton 44 RT 19 2 2 iPrOH 44 RT 4 4 3 THF 44 RT 2 3 4 TBME 44 RT 4 1 5 tBuOH 44 RT 2 1 6 EtOH 3 45 4 4 7 MeOH 3 45 5 3 8 H2O 44 45 17 1
a) Die Produkte wurden mittels 1H-NMR-Spektroskopie analysiert und quantifiziert und als molare Zusammensetzung des Produktgemisches angegeben. Abweichungen zu 100% sind durch das Edukt und unbestimmte Nebenprodukte gegeben.
Tabelle 3.3: Der Lösungsmitteleinfluss bei der Hydratisierung von 47.
Das Ergebnis, dass das THP-Acetal 47 die besten Umsätze in der Katalyse lieferte,
überraschte etwas, da nicht ohne weiteres zu erklären ist, warum sowohl der Benzylether
46 als auch der TBS-Ether 48 kaum reagierten. Es wurde erst vermutet, dass in den beiden
letztgenannten Fällen Chloridverunreinigungen aus der Schützung des Alkohols die
Katalyse negativ beeinflusst haben könnten. Deshalb wurde eine neue Versuchsreihe mit
sorgfältig gereinigten Edukten 47-49 und zusätzlich den beiden Verbindungen 50 und 51,
die in ihren elektronischen Eigenschaften 47 ähneln, jedoch weniger sterisch anspruchsvoll
sind, durchgeführt (Tab. 3.4). Bei den Versuchen wurden die Komplexe 21 und 22 mit
unterschiedlichen Katalysatorbeladungen eingesetzt und die Reaktionsgemische mittels 1H-NMR-Spektroskopie analysiert.

3. Hauptteil
52
Eintrag Substrat Katalysator
[mol%]
Zeit
[h]
Produkte
[mol-%]a
21
22
1 R = MOM (50) 5 17 18 12 48 2 “ 5 17 43 3 29 3 “ 5 50 69 6 9 4 “ 10 24 67 6 1 5 “ 10 50 72 6 9 6 R = THP (47) 5 17 17 6 33 7 “ 5 17 48 3 36 8 “ 5 50 58 3 27 9 “ 10 24 66 3 12 10 “ 10 50 70 3 6 11 R = TBS (48) 5 50 n.d. n.d. 12 “ 5 24 12 1 58 13 “ 5 50 24 12 36 14 “ 10 24 18 8 70 15 “ 10 50 36 12 24 16 R = Me (51) 5 17 9 39 19 17 “ 5 50 11 54 3 18 “ 10 50 4 39 28 19 “ 5 17 38 24 3 20 “ 5 24 33 24 42 21 “ 5 66 33 27 35 22 “ 10 24 35 24 41 23 “ 10 66 34 24 30 24 R = Bn (46) 5 17 24 12 48 25 “ 10 17 n.d. n.d.
a) Die Produkte wurden mittels 1H-NMR-Spektroskopie analysiert und quantifiziert und als molare Zusammensetzung des Produktgemisches angegeben. Abweichungen zu 100% sind durch unbestimmte Nebenprodukte gegeben.
Tabelle 3.4: Hydratisierung von geschützten 1-Octin-3-ol-Derivaten zu Aldolprodukten.
Die Ergebnisse aus Tabelle 3.4 liefern insbesondere auch wegen der Erhöhung der
Katalysatorbeladung ein detaillierteres Bild. Die höhere Beladung war dabei wegen der
Decarbonylierung des Katalysators zwingend vonnöten (vgl. Kap. 3.1.3). Die positiven
Ergebnisse mit dem THP-Acetal 47 wurden bestätigt und das MOM-Acetal 50 erwies sich
mit gleichen (vgl. Eintrag 4 und 8) oder leicht besseren Ausbeuten (vgl. Eintrag 3 und 8)
als eine ebenfalls geeignete Ausgangsverbindung. Der Aldehyd des MOM-Ethers 50

3. Hauptteil
53
konnte in einem präparativen Ansatz nach säulenchromatographischer Reinigung mit 63%
Ausbeute isoliert werden. Dagegen konnte der TBS-Ether trotz verlängerter Reaktionszeit
nur zu einem geringen Teil umgesetzt werden (vgl. Eintrag 13 und 15) und in allen Fällen
entstand vermutlich durch Eliminierung aus dem Katalyseprodukt das Nebenprodukt (E)-
2-Octenal. Dieser α,β-ungesättigte Aldehyd entstand in maximalen Ausbeuten bei der
Hydratisierung von Methylether 51.
Schlussendlich kann festgehalten werden, dass man durch rutheniumkatalysierte anti-
MARKOVNIKOV-Hydratisierung von geschützten Propargylalkoholen die Aldolprodukte
erhalten kann und sich hierfür das THP-Acetal 47 und das MOM-Acetal 50 als Substrate
am besten eignen.
3.2.3. Herstellung von 5,6-Dihydropyran-2-onen durch iterative Synthese
von 1,3-Diolen
3.2.3.1. Synthese von 5,6-Dihydropyran-2-onen mittels rutheniumkatalysierter anti-
MARKOVNIKOV-Hydratisierung von terminalen Alkinen
Bereits während der Optimierungsarbeit wurde nach Anwendungsmöglichkeiten für die
rutheniumkatalysierte anti-MARKOVNIKOV-Hydratisierung von terminalen Alkinen gesucht,
mit dem Ziel diese atomökonomische Reaktion in der Synthese von Naturstoffen oder
naturstoffähnlichen Verbindungen einzusetzen. Als ein mögliches Syntheseziel wurden die
5,6-Dihydropyran-2-one ausgewählt (Abb. 3.25). Diese finden sich als strukturelles
Merkmal in vielen Naturstoffen finden.58 Generell sind natürlich vorkommende Lactone
auf vielen pharmakologischen Gebieten aktiv und das Strukturelement 5,6-Dihydropyran-
2-on ist in vielen dieser Substanzen vorhanden. Daraus resultiert ein gesteigertes
pharmakologisches sowie chemisches Interesse, da gezeigt werden konnte, dass sowohl
natürliche als auch synthetisierte 5,6-Dihydropyran-2-one zytotoxisch sind und die HIV
Protease hemmen, die Apoptose einleiten und gegen Brustkrebs wirken können. Ein Teil
dieser Aktivität beruht vermutlich auf der konjugierten Doppelbindung, die als Michael
Akzeptor reagieren kann.59
Abbildung 3.25: Allgemeine Struktur der 5,6-Dihydropyran-2-one.

3. Hauptteil
54
3.2.3.2. Synthesestrategien für 5,6-Dihydropyran-2-one
In der Literatur sind viele verschiedene Methoden zur Darstellung von 5,6-Dihydropyran-
2-on-Ringen beschrieben. Die am häufigsten zitierten und verwendeten Methoden sind die
Lactonisierung von substituierten δ-Hydroxysäure-Derivaten, die Oxidation von
substituierten Dihydropyranderivaten und die Ringschlussmetathese von Substanzen
Homoallyl-acrylaten. Neben diesen Methoden gibt es noch weitere, wie beispielsweise die
intramolekulare HORNER-WADSWORTH-EMMONS-Reaktion, die BAEYER-VILLIGER-
Oxidation oder die Zykloaddition, diese finden aber seltener Anwendung.59
Da die natürlich vorkommenden 5,6-Dihydropyran-2-one stereogene Zentren im Pyronring
an den Positionen C-5 und C-6, oder aber in einer der Seitenketten enthalten, werden
stereoselektive Reaktionen zu ihrer Synthese benötigt. Zur Erzeugung eines
Chiralitätszentrums in der Seitenkette lässt sich praktisch jede bekannte stereoselektive
Reaktion verwenden. Hingehen lassen sich die stereogenen Zentren in den Ringpositionen
C-5 und C-6 des Pyrons grob mit zwei Strategien aufbauen: entweder durch Verwendung
eines chiralen Vorläufers oder durch eine asymmetrische (enantioselektive) Reaktion.
Dabei kommen als chirale Verläufer Kohlenhydrate (vor allem Monosaccharide),
Hydroxysäuren und Epoxide in Frage. Eine Vielzahl der Pyrone wurde über
asymmetrische Reaktionen, wie die SHARPLESS Epoxidierung oder Dihydroxylierung,
aldolartige Reaktionen, Allylierungen oder Cycloadditionen hergestellt.59
3.2.3.3. Einsatz der anti-MARKOVNIKOV-Hydratisierung in der Synthese
Aus der Klasse der 5,6-Dihydropyran-2-one wurde als erstes Ziel für die Anwendung der
anti-MARKOVNIKOV-Hydratisierung von Propargyloxy-Derivaten zu Aldolprodukten das
Massoialacton (54) ausgewählt. Dieses α,β-ungesättigte-δ-Lacton wurde erstmals 1937
von ABE aus der Rinde des Massoi-Baumes (Cryptocarya massoia) isoliert.60 Bis heute
sind eine Reihe von Synthesen sowohl für das natürlich vorkommende (–)-Massoialacton
als auch für das nicht natürlich vorkommende Enantiomer [(+)-Massoialacton] bekannt.59
Nachdem durch die oben erwähnten Studien gezeigt werden konnte, dass aus dem MOM-
Acetal 50 der Aldehyd 52 durch rutheniumkatalysierte Hydratisierung hergestellt werden
kann, wurde dieser Aldehyd 52 mit dem durch LDA deprotonierten tert-Butylacetat in
einer nukleophilen Addition zum Hydroxyester 53 mit einer Ausbeute von 68% umgesetzt.
Anschließend konnte durch Rühren in Trifluoressigsäure gleichzeitig die Methoxymethyl-
Schutzgruppe und der tert-Butyl-Rest entfernt werden, worauf das nicht isolierte

3. Hauptteil
55
Zwischenprodukt direkt zum Massoialacton 54 zyklisierte, welches mit 71% Ausbeute
isoliert wurde (Schema 3.4).
Schema 3.4: Die Synthese vom Massoialacton.
Somit ist es durch den Einsatz der anti-MARKOVNIKOV-Hydratisierung und einfacher
Folgereaktionen gelungen das Massoialacton (±)-54 ausgehend von 3-Methoxymethyloxy-
1-octin 50 in nur drei Stufen in einer redoxneutralen Sequenz herzustellen, was eine
Verkürzung der Syntheseroute und das Einsparen von Oxidations- und
Reduktionsreaktionen im Vergleich zu einer kürzlich erschienen Arbeit bedeutet.61 Dieses
Beispiel belegt die Anwendbarkeit der rutheniumkatalysierten anti-MARKOVNIKOV-
Hydratisierung im Sinne einer atomökonomischen, redoxneutralen Synthese von
Aldolderivaten durch Hydratisierung von Propargylalkohol-Derivaten.
Neben der Synthese des Massoialactons sollte auch ein Derivat aus der Familie der
polyacetat-abgeleiteten Pyrone (Abb. 3.26) durch zwei weitere Iterationen zu einem 1,3-
Diolderivat bzw. einem 1,3,5-Triolderivat hergestellt werden.62 Mit dieser Synthese soll
gezeigt werden, dass Poly-1,3-diole, die zu Naturstoffen oder naturstoffähnlichen
Verbindungen führen, einfach und ökonomisch effizient zugänglich sind.
Abbildung 3.26: Cryptocarya Diacetat und ein davon abgeleitetes Pyron.

3. Hauptteil
56
Ausgehend vom Alkinylacetal 50 konnte in nur sieben Stufen das Pyron 59 erhalten
werden. Die erste Stufe bestand aus der Hydratisierung des Alkins 50 mit 10 mol% 22 in
Aceton mit 5 Äquivalenten Wasser bei 50 °C. Der Aldehyd 52 konnte durch
Säulenchromatographie vom Rückstand der Hydratisierung in guten Ausbeuten (63%)
erhalten werden. Es erwies sich in der Folge als einfacher den Aldehyd nicht zu isolieren,
sondern ihn direkt in der Folgereaktion einzusetzen. Somit konnte nach der Hydratisierung
und nach dem Ersetzten des Aceton/Wasser-Gemisches durch THF in einer nukleophilen
Addition mit 5 Äquivalenten Ethynylmagnesiumchlorid bei 0 °C der Aldehyd 52 zum
Hydroxyalkin 55, das als ein Gemisch aus zwei Diastereomeren erhalten wurde, umgesetzt
werden. Die rutheniumkatalysierte Hydratisierung des MOM-Acetals 50 konnte später
auch unter den optimierten Bedingungen (vgl. Kap. 3.1 und 3.1.6) mit 4 mol%
Katalysatorbeladung (22) in Aceton/Wasser (3:1) bei 50 °C in 5 statt 17.5 h durchgeführt
werden. Allerdings ist im Anschluss das Entfernen des Aceton/Wasser-Gemisches etwas
aufwendiger, da sich das Wasser nicht vollständig im Membranpumpenvakuum
abdestillieren lässt. Deshalb gibt man Dichlormethan zur Reaktionslösung um eine
Phasentrennung zwischen organischer und wässriger Phase zu erzielen. Die organische
Phase wird dann via Kanüle in einen anderen Kolben überführt, dort eingeengt und der
Rückstand mit THF und wenig Magnesiumsulfat versetzt, um mögliche mittransferierte
Wasserspuren zu binden. Mit dem so gelösten Aldehyd kann dann die GRINGARD-Addition
durchgeführt werden. Bevor eine weitere Hydratisierung vorgenommen werden kann,
muss erst die freie Hydroxygruppe des Alkins 55 geschützt werden. Ein Versuch, eine
weitere MOM-Gruppe einzuführen, scheiterte, weil das zyklische Acetal 56 unter den
Reaktionsbedingungen gebildet wurde. Deshalb wurde nun das Hydroxyalkin 55 in
Dimethoxymethan gelöst und mit Trifluormethansulfonsäure versetzt, so dass als
Diastereomerengemisch das zyklische Acetal 56 entsteht. Die nächsten beiden Stufen der
Synthese ähneln den ersten beiden: das Alkin 56 wird zum Aldehyd hydratisiert, welcher
mit Allylmagnesiumbromid in Ether bei 0 °C zum Hydroxyalken 57 reagiert. In einer
Acylierungsreaktion mit Acryloylchlorid in Gegenwart von Triethylamin und DMAP
entsteht aus Verbindung 57 der Acylester 58, welcher in einer Ringschlussmetathese mit
GRUBBS I Katalysator63 zum 5,6-Dihydro-6-(2,4-methylendioxynonyl)-pyran-2-on 59
zyklisiert (Schema 3.5).

3. Hauptteil
57
OH OMOM OMOM
O
MOMO OHO OO O OH
O O O
O
O O O
O
LiBr (25 mol%),pTsOH (8 mol%)
DMM,Toluol,RT, 48 h 50 (74%)
22 (10 mol%)
Aceton,H2O (5 Äquiv),50 °C, 17.5 h
(5 Äquiv),THF,0 °C, 40 min
MgCl
DMM
TfOH(20 mol%)
CH2CHCOCl(1.5 Äquiv),
Et3N (3 Äquiv),DCM,
RT, 10 min
GRUBBS I Kat (10 mol%)
DCM, 4 h
1) 22 (4 mol%),Aceton/H2O (3:1),50 °C, 5 h
2) MgBr
(5 Äquiv),Ether,0 °C, 1 h
52 (63%)
55 (74%)56 (89%)57 (73%)
58 (74%) 59 (85%)
Schema 3.5: Synthese vom 5,6-Dihydro-6-(2,4-methylendioxynonyl)-pyran-2-on.
Diese Synthesestrategie ist hoch effizient, was sowohl die Anzahl der Stufen als auch die
Möglichkeit zum Upscaling angeht. Vor allem fällt die Absenz von Redox-
Transformationen auf, die bei vergleichbaren Synthesen viel Raum einnehmen. Anstelle
von teuren und in stöchiometrischen Mengen benötigten Reagenzien wurden relativ
einfache Ausgangsmaterialien verwendet und anspruchsvolle Reagenzien nur in
katalytischen Mengen eingesetzt. Es sind bisher noch keine enantiomerenangereicherten
Verbindungen hergestellt worden. Dies sollte aber durch den Einsatz der entsprechenden
enantioselektiven Reaktionen (vgl. Kap. 3.2.1.) möglich sein, da die rutheniumkatalysierte
anti-MARKOVNIKOV-Hydratisierung von Propargylalkoholen unter Retention des
stereogenen Zentrums verläuft (vgl. Kap. 3.2.4.).

3. Hauptteil
58
3.2.4. Stereochemischer Verlauf der anti-MARKOVNIKOV-Hydratisierung
von Propargylalkoholen Durch die in Kapitel 3.2.2. beschriebene Methode ist es möglich verlässliche
Untersuchungen zum stereochemischen Verlauf anzustellen indem man enantiomerenreine
sekundäre Propargylalkohole als Substrate verwendet und sie in Produkte bekannter
Konfiguration überführt. In diesem Fall wurde für die Studien das THP-Acetal 47
verwendet, da die THP-Schutzgruppe einfach einzuführen ist, sich leicht wieder entfernen
lässt und das Acetal 47 mit guten Ausbeuten hydratisiert werden kann. Das Acetal wurde
mit 5 mol% 22 als Katalysator in Aceton mit 5 Äquivalenten Wasser bei 50 °C in 17.5 h
zum Aldehyd 60 umgesetzt, der nicht isoliert, sondern nach Entfernen des Acetons in
Methanol mit Natriumborhydrid bei 0 °C direkt reduziert und ohne Reinigung mit
Methanol/Salzsäure zum 1,3-Diol entschützt wurde. Das 1,3-Diol konnte mit
Benzoylchlorid in Gegenwart von Triethylamin und DMAP in das Dibenzoat 61 überführt
werden (Schema 3.6).
Schema 3.6: Synthese von 1,3-Dibenzoyloxy-octan aus 1-Octin-3-ol.
Die beiden Benzoatgruppen machen es möglich die Probe mit der chiralen HPLC mit UV-
Detektor zu analysieren. Neben dem racemischen 1-Octin-3-ol wurde auch das
enantiomerenangereicherten (R)-1-Octin-3-ol (98+% ee)64 und das (S)-1-Octin-3-ol (98+%
ee)65 in der gleichen Sequenz eingesetzt und gemäss der Syntheseroute in Schema 3.6 zu
den entsprechenden Dibenzoylderivaten umgesetzt. Die HPLC-Chromatogramme zeigen,
dass die Derivate der enantiomerenangereicherten Octine auch nach der anti-
MARKOVNIKOV-Hydratisierung ihren Enantiomerenüberschuß behalten (Abb. 3.27). Dies
bedeutet, dass die Hydratisierung unter Erhalt des Enatiomerenüberschusses verläuft.

3. Hauptteil
59
Abbildung 3.27: HPLC-Spektren der 1,3-Dibenzoyloxy-octane, gemessen auf einer chiralen OD Säule,
iPrOH/Hexan = 5:95, Fluss = 0.5 mL/min, λ = 230 nm; a) racemische Probe 61; b) (R)-
61; c) (S)-61; Peaks vor 10 min sind auf HLPC-Lösungsmittelunreinheiten
zurückzuführen.
Weil der genaue Mechanismus der katalytischen anti-MARKOVNIKOV-Hydratisierung noch
nicht bekannt ist, konnte nicht mit letzter Sicherheit ausgeschlossen werden, dass eine
Inversion der Konfiguration am stereogenen Zentrum stattgefunden hatte. Zur Klärung der
absoluten Konfiguration wurde deshalb ausgehend vom enantiomerenangereicherten (S)-
MOM-Acetal 50 durch Hydratisierung der (S)-Aldehyd 52 hergestellt, der durch Oxidation
mit Kaliumpermanganat in die Carbonsäure 62 überführt wurde. Nach Entschützen und
Veresterung erhielt man den Hydroxyester 63, dessen Drehwert mit dem Literaturwert66
für eine (S)-konfiguriete Probe übereinstimmte (Schema 3.7). Somit ist auch die
Möglichkeit einer stereospezifischen Inversion während der rutheniumkatalysierten anti-
MARKOVNIKOV-Hydratisierung ausgeschlossen.

3. Hauptteil
60
Schema 3.7: Synthese von (S)-3-Hydroxyoctansäuremethylester; [α]D
20: +22.9 (c = 1, CHCl3),
Literatur [α]D20: +24 (c = 1, CHCl3).
3.2.5. Iterative Synthese von Poly-1,4-diolen via rutheniumkatalysierter
anti-MARKOVNIKOV-Hydratisierung67 Das Ziel, eine katalytische sequentielle Synthese von Poly-1,3-diolen zu entwickeln,
konnte schließlich realisiert werden (s. Kap. 3.2.3.). Die anfänglichen Probleme mit der
rutheniumkatalysierten anti-MARKOVNIKOV-Hydratisierung von Propargyloxy-Derivaten
führte jedoch dazu, dass zuerst die Hydratisierung von Homo-propargyloxy-Derivaten zu
4-Oxybutanalen als die weniger Problematische untersucht wurde. Diese Substanzen ließen
sich wesentlich einfacher in der Hydratisierung umsetzen, selbst mit weniger aktiven
Katalysatoren. Aus diesem Grund wurde erst die iterative Synthese von Poly-1,4-
diolderivaten entwickelt, welche die Machbarkeit einer atomökonomischen Synthese von
komplexen Molekülen via C-C-Kupplungsreaktionen und katalytischen
Heterofunktionalisierungen aufzeigen sollte.
3.2.5.1. Iterative Synthesemethoden
Die katalytische Hydratisierung ist eine zukunftsträchtige Möglichkeit für nachhaltige
Synthesestrategien, da die Produkte durch eine „abfallfreie“ Addition von Wasser an die
Dreifachbindung des Alkins erhalten werden können. Dem gegenüber stehen
konventionelle Methoden aus energie- und materialintensiven Redoxreaktionen, die auch
noch Nebenprodukte ergeben.68 Deshalb ist die rutheniumkatalysierte anti-MARKOVNIKOV-
Hydratisierung eine gute Methode zur Herstellung von Aldehyden aus terminalen Alkinen
(Schema 3.8).

3. Hauptteil
61
Ru
PF6
9
NR PPh2
6414
R = tAmylR = 2,4,6-Ph3C6H2
R H2O+ RO
9 + 2 L (1-5 mol%)
Aceton, 60 °C, 3-15 h
L =
Schema 3.8: Rutheniumkatalysierte anti-MARKOVNIKOV-Hydratisierung von terminalen Alkinen.
Mit der Entwicklung des in situ Katalysatorsystems basierend auf dem Komplex 912g und
den Liganden 64 und 14, das die Vorteile der bereits bekannten Katalysatoren in den
Punkten Aktivität, Handhabung und Verfügbarkeit der Liganden und des
Vorläuferkomplexes vereint, konnte erstmalig eine Synthesestrategie in Form der iterativen
Synthese69 von Poly-1,4-diolderivaten durch C-C-Kupplungsreaktionen und anti-
MARKOVNIKOV-Hydratisierung in Angriff genommen werden (Schema 3.9). Ausgehend
von einem Aldehyd I wird durch Propargylierung der Homopropargylalkohol II erhalten,
welcher im nächsten Schritt geschützt und danach katalytisch und regioselektiv hydratisiert
wird, so dass man den Aldehyd III erhält. Weitere Iterationen mit dem Aldehyd III führen
zu beliebig langen Poly-1,4-diolketten.
O
RR
R
OSG
O
OH
I III
II
M
Addition
sequentielle
Synthese
1. Schützung2. Hydratisierung
Iterationen
Schema 3.9: Synthesestrategie zur Herstellung von Poly-1,4-diolen.
Neben dieser Methode sind Poly-1,4-diolfragmente nicht stereoselektiv aus
Furfuraldehyden oder -ketonen durch Hydrogenolyse hergestellt worden.70 Des Weiteren
ist eine iterative Synthese ausgehend von einer Allylierung eines Aldehyds gefolgt von
einer Hydroborierung/Oxidation zu einem 1,4-Diol bekannt, wobei eine SWERN-Oxidation
des terminalen Alkohols dann den Aldehyd für eine weitere Iteration liefert.71 Durch diese

3. Hauptteil
62
Reduktions- und Oxidationsschritte kann man mit hohem Material- und Ressourceneinsatz
ebenfalls die Diole erhalten.
Bei der Umsetzung der Synthesestrategie zur Herstellung von Poly-1,4-diolen ging man
von Benzaldehyd 65 aus, welcher durch Zugabe von GAUDEMAR´s Allenylzinkbromid 6672
zum Homoallylalkohol 67 (Gemisch aus Alkin 67 und Allen 67a im Verhältnis 92:8) in
hohen Ausbeuten reagierte (Schema 3.10).72b Zur Herstellung des
Allenylzinkbromidreagenzes 66 sei gesagt, dass es nach Literaturvorschrift aus Zinkstaub
und Propargylbromid in THF hergestellt wurde, aber die Temperatur der Reaktion bei 0 °C
anstelle von –10 °C gehalten wurde.72c Darüber hinaus musste das Reagenz 66 kurz vor
jeder Reaktion frisch hergestellt werden, da es sich nicht lagern ließ.72b Propargylierungen
von Aldehyden mit Zinkreagenzien sind sowohl mit vorher hergestellten Reagenzien72a-b, 73
als auch unter BARBIER-Bedingungen in DMF,74 THF75 oder Wasser bekannt.76 Jedoch
waren die BARBIER-Bedingungen bei größeren Ansätzen weniger praktikabel, da sich die
Temperaturkontrolle wesentlich schwieriger gestaltete und es schnell zu Überhitzungen
kam. Somit war der Gebrauch der frisch hergestellten Allenylzinkbromidverbindung 66
nach GAUDEMAR sicherer für Großansätze der Propargylierung. Ein Überschuss an
Reagenz 66 war nötig um die Bildung von Nebenprodukten wie die des 1,5-Diphenylbut-
2-in-1,5-diols durch zweifache Addition von Benzaldehyd 65 zu unterdrücken.
Schema 3.10: Erste Iteration.
Der Alkohol 67 wurde durch Acetylierung unter Standardbedingungen zum Acetat 68
umgesetzt, welches Allen 68a enthielt (68/68a = 10:1 bis 25:1, je nach Aufarbeitungs- und

3. Hauptteil
63
Reinigungsmethode). Alternativ wurde das Rohprodukt von 67 ohne Zwischenreinigung
direkt zu 68 acetyliert, was eine höhere Gesamtausbeute ergab. Das Allen 68a konnte
durch vorsichtige Destillation oder durch Ausfrieren vom Isomer 68 bei –20 °C abgetrennt
werden. Allerdings stellte sich heraus, dass dies nicht notwendig war, da das Allen 68a die
Hydratisierung des Esters 68 zum Aldehyd 69 durch den in situ Katalysator (9 + 2 • 64, vgl.
Schema 3.8) nicht störte und unter den Reaktionsbedingungen nicht reagierte. Stattdessen
ließ es sich einfach durch Säulenchromatographie vom Aldehyd 69 abtrennen. Es erwies
sich in der Folge als einfacher, den Aldehyd nicht zu isolieren, sondern ihn direkt in der
Folgereaktion einzusetzen. Aus diesem Grund wurde der Ester 68 hydratisiert und das
Rohprodukt nach Entfernen des Lösungsmittels und erneutem Lösen des Rückstandes in
THF mit Allenylzinkbromid 66 in hoher Gesamtausbeute zum Propargylalkohol 70
umgesetzt. Der Alkohol 70 konnte mit TBSCl in Gegenwart von Imidazol in DMF
geschützt und in den Silylether 71 überführt werden. Die katalytische anti-MARKOVNIKOV-
Hydratisierung dieses Alkins (Schema 3.11) verlief ohne weitere Probleme, so dass das
Rohprodukt direkt mit Allenylzinkbromid 66 zum Alkohol 72 umgesetzt werden konnte.
Dieser wurde wiederum mit einer TBS-Schutzgruppe versehen und gab das Alkin 73. Die
nächste Hydratisierung gefolgt von der nächsten Propargylierung von Alkin 73 zu Alkohol
74 konnte ebenfalls problemlos durchgeführt werden. Nach dem Schützen der
Hydroxygruppe erhielt man als Produkt von vier Propargylierungen und drei
Hydratisierungen ausgehend von Benzaldehyd 65 den Trisilylether 75 (Ausbeute über alle
Stufen, 54%).
Schema 3.11: Fortsetzung der Iteration.

3. Hauptteil
64
3.2.5.2. Kettenabschlüsse und Ringschluss
Nachdem durch iterative Synthese die Poly-1,4-diole 71, 73 und 75 erhalten worden waren,
sollten diese modifiziert und durch Kettenabschlüsse die Iteration beendet werden. In
einem ersten Ansatz wurde ein Abschluss der Kette zum einen durch eine WITTIG-
Reaktion mit dem Phosphoniumylid 76 und zum anderen durch Addition des
Lithiummethylpropiolat 77 versucht (Schema 3.12). Beide Reaktionen ergeben
Carbonsäurederivate, die später durch Entschützen und eine YAMAGUCHI-
Makrolactonisierung in Zyklen überführt werden können.
Ph
OAc
OPh
OAc
68 69
Ph
OAc
Ph
OAc
CO2Me
9 + 2 * 64(3 mol% [Ru])
Aceton, H2O70 °C, 14 h
OH
CO2Me
CO2MePh3P
76
THF, 50 °C, 2 h
Li CO2Me
77
THF, -78 °C, 10 min
78 (94%) 79 (87%) Schema 3.12: Modelversuche zum Kettenabschluss.
Bevor mit den längerkettigen Poly-1,4-diolen diese Abschlusssequenzen durchgeführt wurden,
testete man die beiden Reaktionen an dem einfacher herzustellenden Ester 68. Dieser wurde
katalytisch hydratisiert und ohne Reinigungsschritt direkt mit einem Überschuss an Ylid 76 in
den ungesättigten Ester 78 in hohen Ausbeuten und guter Stereoselektivität (E/Z = 25:1)
überführt. Ebenfalls gelang die Addition vom Lithiumester 77 (hergestellt aus LDA und
Methylpropiolat) an den Aldehyd 69, der durch Hydratisierung aus 68 erhalten wurde, zum γ-
Hydroxypropiolester 79 in guten Ausbeuten. Wichtig hierbei ist es die Reaktion bei –78 °C
abzubrechen, da sich beim Erwärmen das Produkt wegen der empfindlichen
Propiolatdreifachbindung zersetzt.
Nach diesen erfolgreichen Vorversuchen wurden die Reaktionen auch mit längerkettigen
Poly-1,4-diolen durchgeführt (Schema 3.13).
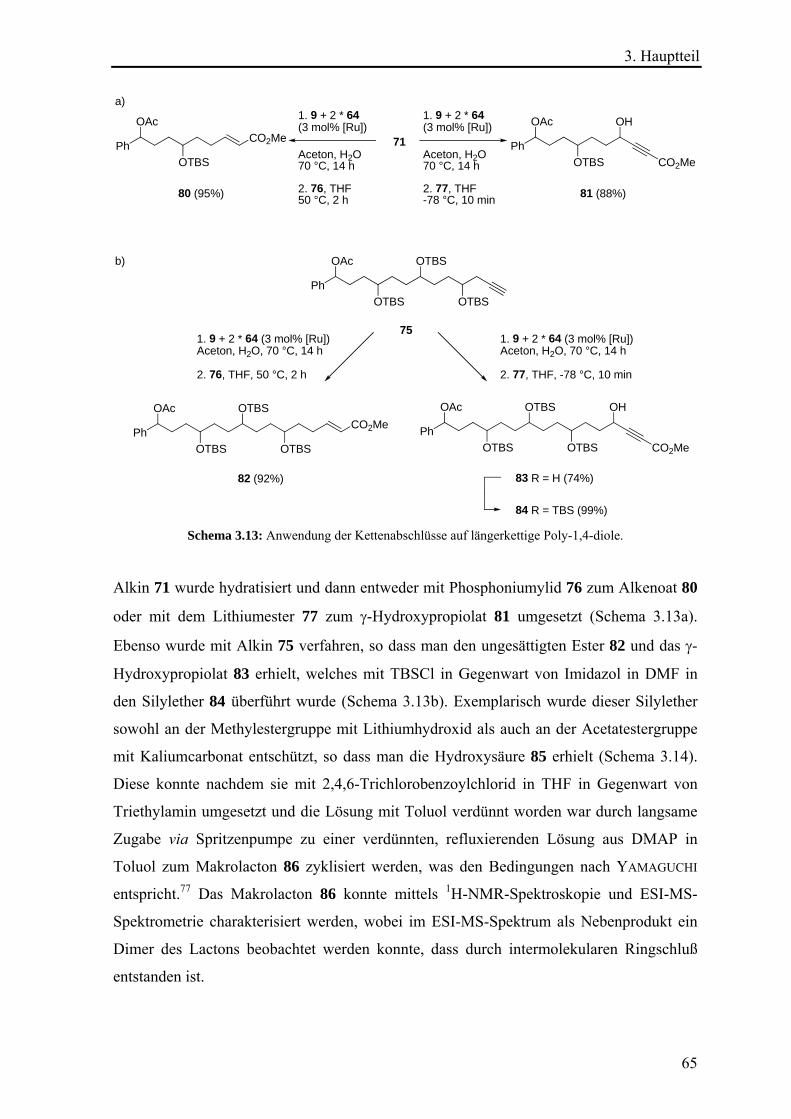
3. Hauptteil
65
Aceton, H2O70 °C, 14 h
2. 77, THF-78 °C, 10 min
1. 9 + 2 * 64(3 mol% [Ru])
71
Ph
OAc
OTBS
OTBS
OTBS
75
Aceton, H2O70 °C, 14 h
2. 76, THF50 °C, 2 h
1. 9 + 2 * 64(3 mol% [Ru])
Ph
OAc
OTBS
CO2MePh
OAc
OTBS
OH
CO2Me
80 (95%) 81 (88%)
1. 9 + 2 * 64 (3 mol% [Ru])Aceton, H2O, 70 °C, 14 h
2. 76, THF, 50 °C, 2 h
1. 9 + 2 * 64 (3 mol% [Ru])Aceton, H2O, 70 °C, 14 h
2. 77, THF, -78 °C, 10 min
Ph
OAc
OTBS
OTBS
OTBS
CO2Me Ph
OAc
OTBS
OTBS
OTBS
OH
CO2Me
b)
a)
82 (92%) 83 R = H (74%)
84 R = TBS (99%) Schema 3.13: Anwendung der Kettenabschlüsse auf längerkettige Poly-1,4-diole.
Alkin 71 wurde hydratisiert und dann entweder mit Phosphoniumylid 76 zum Alkenoat 80
oder mit dem Lithiumester 77 zum γ-Hydroxypropiolat 81 umgesetzt (Schema 3.13a).
Ebenso wurde mit Alkin 75 verfahren, so dass man den ungesättigten Ester 82 und das γ-
Hydroxypropiolat 83 erhielt, welches mit TBSCl in Gegenwart von Imidazol in DMF in
den Silylether 84 überführt wurde (Schema 3.13b). Exemplarisch wurde dieser Silylether
sowohl an der Methylestergruppe mit Lithiumhydroxid als auch an der Acetatestergruppe
mit Kaliumcarbonat entschützt, so dass man die Hydroxysäure 85 erhielt (Schema 3.14).
Diese konnte nachdem sie mit 2,4,6-Trichlorobenzoylchlorid in THF in Gegenwart von
Triethylamin umgesetzt und die Lösung mit Toluol verdünnt worden war durch langsame
Zugabe via Spritzenpumpe zu einer verdünnten, refluxierenden Lösung aus DMAP in
Toluol zum Makrolacton 86 zyklisiert werden, was den Bedingungen nach YAMAGUCHI
entspricht.77 Das Makrolacton 86 konnte mittels 1H-NMR-Spektroskopie und ESI-MS-
Spektrometrie charakterisiert werden, wobei im ESI-MS-Spektrum als Nebenprodukt ein
Dimer des Lactons beobachtet werden konnte, dass durch intermolekularen Ringschluß
entstanden ist.

3. Hauptteil
66
Schema 3.14: YAMAGUCHI-Zyklisierung zum Makrolacton.
Es konnte somit gezeigt werden, dass die rutheniumkatalysierte anti-MARKOVNIKOV-
Hydratisierung von terminalen Alkien eine nützliche Synthesemethode ist, die in
mehrstufigen Reaktionssequenzen eingesetzt werden kann. Durch die Entwicklung des in
situ Katalysatorsystems12f war es möglich die erste Anwendung für diese Reaktion in einer
iterativen Synthesesequenz von Poly-1,4-diolen und deren Derivaten aufzuzeigen, wobei
es gelang bis zu vier anti-MARKOVNIKOV-Hydratisierungen in Serie durchzuführen. Eine
wichtige Neuerung des in situ Katalysatorsystems konnte dabei etabliert werden. Es wurde
nämlich entdeckt, dass nicht, wie im Originalprotokoll, der vorher hergestellte Komplex
[CpRu(L)2(MeCN)]PF6 37a, 12f für die Katalyse vonnöten ist, sondern dass das Mischen von
Komplex 9 mit 2 Äquivalenten Ligand und Alkin in Aceton/Wasser und erhitzen auf 70 °C
ausreicht um die Reaktion zu starten. Dies vereinfacht die Prozedur, spart Zeit und liefert
vor allem vergleichbare Ergebnisse zum vorgebildeten Komplex.
Die grundlegende Idee hinter der Anwendung von katalytischen
Heterofunktionalisierungensreaktionen in der Synthese ist die Erhöhung der
Atomökonomie und der Effizienz der Syntheseroute durch Verringerung der energie- und
abfallintensiven Redoxreaktionen. Die Effizienz der oben dargestellten iterativen Synthese
lässt sich gut in Schema 3.15 erkennen, das zeigt, dass der Hexadecinsäuremethylester 84
mit seiner Strukturkomplexität aus einfachen Startmaterialien, welche aus
Basischemikalien hergestellt werden können, zugänglich ist und in wenigen Schritten zu
einem naturstoffähnlichen Makrolacton umgesetzt werden kann.
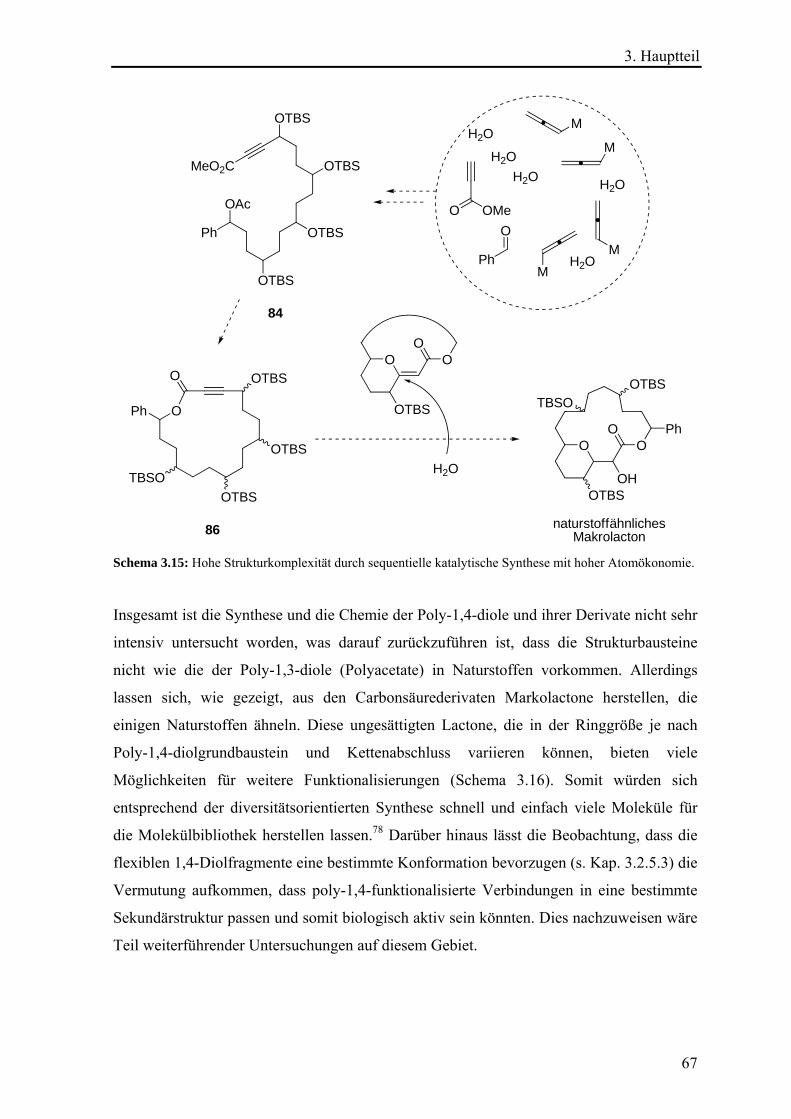
3. Hauptteil
67
86
84
OTBS
OAc
Ph OTBS
OTBS
OTBS
MeO2C
OMeO
Ph
O
MM
M
M
H2O
H2O
H2O
H2OH2O
OOO
OTBS
OOO Ph
OTBS
OTBS
TBSO
OHH2O
naturstoffähnlichesMakrolacton
OPh
TBSOOTBS
OTBS
O OTBS
Schema 3.15: Hohe Strukturkomplexität durch sequentielle katalytische Synthese mit hoher Atomökonomie.
Insgesamt ist die Synthese und die Chemie der Poly-1,4-diole und ihrer Derivate nicht sehr
intensiv untersucht worden, was darauf zurückzuführen ist, dass die Strukturbausteine
nicht wie die der Poly-1,3-diole (Polyacetate) in Naturstoffen vorkommen. Allerdings
lassen sich, wie gezeigt, aus den Carbonsäurederivaten Markolactone herstellen, die
einigen Naturstoffen ähneln. Diese ungesättigten Lactone, die in der Ringgröße je nach
Poly-1,4-diolgrundbaustein und Kettenabschluss variieren können, bieten viele
Möglichkeiten für weitere Funktionalisierungen (Schema 3.16). Somit würden sich
entsprechend der diversitätsorientierten Synthese schnell und einfach viele Moleküle für
die Molekülbibliothek herstellen lassen.78 Darüber hinaus lässt die Beobachtung, dass die
flexiblen 1,4-Diolfragmente eine bestimmte Konformation bevorzugen (s. Kap. 3.2.5.3) die
Vermutung aufkommen, dass poly-1,4-funktionalisierte Verbindungen in eine bestimmte
Sekundärstruktur passen und somit biologisch aktiv sein könnten. Dies nachzuweisen wäre
Teil weiterführender Untersuchungen auf diesem Gebiet.

3. Hauptteil
68
Schema 3.16: Funktionalisierungsmöglichkeiten des ungesättigten Lactons.
3.2.5.3. Stereochemische Aspekte der Poly-1,4-diole
Alle Propargylierungen in der Synthese der 1,4-Diole wurden mit dem achiralen
Zinkreagenz 66 durchgeführt, weshalb es sich beim Homopropargylalkohol 67 um ein
Racemat handelt, während die Iteration zu 70 ein 1:1 Gemisch aus zwei Diastereomeren
gab. Das Fehlen der Diastereoselektivität war für alle Reaktionen von 66 mit einem 4-
Acetoxy- oder 4-tert-Butyldimethylsilyloxy-aldehyd unter den angegebenen Bedingungen
charakteristisch. Deshalb müssen die Ausgangsverbindungen 72 und 73 als vier
diastereomere Enantiomerenpaare vorliegen und die Verbindungen 74 und 75 als
Gemische aus acht diasteromeren Enantiomerenpaare. Nach vier Iterationen müssten
folglich die Produkte 83 und 84 zu gleichen Anteilen aus 16 Diastereomeren bestehen.
Allerdings sollte es möglich sein mit asymmetrischen Reagenzien79 oder Katalysatoren80
die Propargylierung von Aldehyden in asymmetrischer Form durchzuführen, wobei die
Konfiguration eines jeden Stereozentrums durch die asymmetrischen Reagenzien oder
Katalysatoren bestimmt wäre. Die oben beschriebene nicht stereoselektive Synthese bietet
die Möglichkeit der Einbindung der 1,4-Diole aus der rutheniumkatalysierten anti-
MARKOVNIKOV-Hydratisierung von terminalen Alkinen in den Aufbau von
stereodivergenten Molekülbibliotheken im Sinne der diversitätsorientierten Synthese
(DOS).78 Eine Folge dieser nicht stereoselektiven Synthese ist es, dass die NMR-Spektren
der Produkte aus einer Überlagerung der Signale der Diastereomere bestehen. Durch diese
Überlagerung der Signale werden die spezifischen Unterschiede der Diastereomere, die

3. Hauptteil
69
sich aufgrund der jeweils bevorzugten Konformation ergeben, sofort deutlich. Dieser Punkt
wurde offensichtlicher bei der Auswertung der 1H-NMR-Spektren der Zwischenprodukte
mit einer freien Hydroxygruppe (70, 72, 74, 79, 81, 83). Obwohl das Spektrum aus der
Überlagerung von bis zu 16 Diastereomeren besteht kann es nahezu so ausgewertet werden,
als ob nur eine Verbindung vorläge, was der Auszug aus dem 1H-NMR-Spektrum von 74
illustriert (Abb. 3.28).
Abbildung 3.28: Das 1H-NMR Spektrum (400 MHz) von 74. a) Aliphatischer Bereich von δ = 1.2–
3.9 ppm, b) H-Signal bei δ = 5.7 ppm, c) OH-Signal bei δ = 2.79 ppm, d) OH-Signal
bei δ = 2.23 ppm.
Das Signal für H-1 (in α-Position zu Phenyl, Abb. 3.28b) erscheint als einziges pseudo-
Triplet und weist somit nicht auf das Gemisch aus acht Diastereomeren hin. Im Gegensatz
dazu stehen zwei eigenartige Multiplets (bei δ = 2.79 und 2.23 ppm) mit je einem Integral
von 0.5 H. Ein Protonenaustauschexperiment mit D2O lieferte den Nachweis dafür, dass es
sich bei diesen beiden Multiplets um die Signale des Wasserstoffs der Hydroxygruppe
handelt. Vergleicht man die Signale mit den Referenzspektren der kürzeren 1,4-Diole so
ergibt sich daraus, dass das gut strukturierte Multiplet bei δ = 2.79 ppm (Abb. 3.28c) eine

3. Hauptteil
70
zufällig regelmäßige Anordnung von vier Doubletts mit je einer Kopplungskonstante von 3JHH = 7.7 Hz darstellt. Das Multiplet bei δ = 2.23 ppm (Abb. 3.28d) ist zwar auch eine
Überlagerung von vier Doubletts (3JHH = 3.8–4.4 Hz) der vier Diastereomere, aber in einer
unregelmäßigen Anordnung. Allerdings sind es nur die Signale der Hydroxygruppe, die
auf die Anwesenheit von acht Diastereomeren in der Verbindung 74 hinweisen. Dies lässt
die Vermutung zu, dass es eine intramolkulare Wasserstoffbrückenbindung zwischen der
Hydroxygruppe (Donor) und dem Sauerstoff der Silylethergruppe (Akzeptor) gibt, die die
Diastereomere in einer bestimmten Konformation fixiert. Hierfür spricht auch, dass die
Signale der Hydroxygruppe in zwei klare Spektralbereiche getrennt sind, es eine
Hochfrequenzverschiebung der OH-Signale gibt und man gut definierte Signale für die 3JHOCH-Kopplung des Wasserstoffs der Hydroxygruppe erhält.
Bisher ist nur von einigen wenigen 1,4-Diolen, im wesentlichen von den TADOLLEn,81
bekannt, dass sie wasserstoffverbrückte Chelate bilden. Über einfachgeschützte 1,4-
Diolfragmente und ihre Konformation gibt es kaum Informationen. Eine Suche in der
Cambridge Strukturdatenbank ergab, dass es nur ein Beispiel für eine intramolekulare
Bindung zwischen einer Hydroxygruppe und einer tert-Butyldimethylsilylether-Gruppe in
einer 1,4-funktionalisierten Verbindung gibt.82 In den anderen Fällen wo dieses
Strukturmotiv mögliche wäre, werden stattdessen Wasserstoffbrückenbindungen zu
Akzeptoren wie Carbonylgruppen oder solche intermolekularer Art ausgebildet. Deshalb
wurde für weitere Untersuchungen eine einfache Modelverbindung hergestellt, die aus nur
zwei Diastereomeren besteht (Schema 3.17).
Schema 3.17: Synthese eines Testsubstrates für die Studien der intramolekularen
Wasserstoffbrückenbindungen in einem monogeschützten 1,4-Diol.
Dafür wurde Alkohol 67 mit TBSCl in Gegenwart von Imidazol in DMF geschützt und
anschließend in der anti-MARKOVNIKOV-Hydratisierung selektiv umgesetzt, so dass man
nach Addition des Lithiummethylpropiolats 77 den Alkohol 88 als ein Gemisch zweier
Diastereomere (88a/88b, 60:40) erhielt. Diese Verbindung hat sowohl einen
elektronenreichen Wasserstoffbrückenbindungs-Akzeptor als auch einen elektronenarmen
Wasserstoffbrückenbindungs-Donor und zeigt tatsächlich gut aufgelöste Signale für die

3. Hauptteil
71
Hydroxygruppe im 1H-NMR-Spektrum bei δ = 2.45 ppm (d, 3J = 5.4 Hz, 0.6 H) und bei δ
= 3.07 ppm (d, 3J = 7.2 Hz, 0.4 H) in CDCl3. Ausgehend von den angenommenen
Konformationen, die bei den beiden Diastereomeren vorherrschend sein dürften (Abb. 3.29)
und unter Einbeziehung der Größe der 3JHCOH-Kopplung83 sowie der Ergebnisse des 2D
NOESY-Spektrums deutet sich an, dass die Relativkonfigurationen der Diastereomere 88a
und 88b den angenommenen entsprechen. Allerdings muss diese
Konfigurationsbestimmung als vorläufig angesehen werden, da die unterschiedlichen
Signallagen für δ (OH) der Diastereomere möglicherweise auch die Folge des Effekts der
Entschirmung durch die Alkinfunktion84 sein könnte, welche das Wasserstoffatom der
Hydroxygruppe im Diastereomer 88b mehr beeinflusst als in 88a (und vermutlich ebenso
in den Alkinolen 70, 72, 74, 79, 81, 83).
Abbildung 3.29: Vorläufige Bestimmung der relativen Konfiguration der Diastereomere
88a und 88b auf Basis von NMR-Daten.

4. Zusammenfassung
72
4. Zusammenfassung und Ausblick
Im Rahmen dieser Arbeit wurde die rutheniumkatalysierte anti-MARKOVNIKOV-
Hydratisierung von terminalen Alkinen optimiert. Dazu wurde ein analytisches Verfahren
entwickelt um den Reaktionsverlauf der kinetischen Modellreaktion (Schema 4.1) mittels
HPLC-Chromatographie mit angeschlossenem UV-Dektor und internem Standard
verfolgen zu können.
Schema 4.1: Modellreaktion.
In der Katalyse wurden unterschiedliche Rutheniumkomplexe eingesetzt und getestet,
wobei neben den bis dato in der Literatur beschriebenen Homo-bis-
phosphanruthenium(II)-Komplexen auch Komplexe vom Typ [CpRu(L1)(L2)(MeCN)]PF6
mit einem P,N-Liganden (L1) und einem einfachen Triarylphosphinen (L2), wie PPh3 (32),
verwendet wurden. Letztere erwiesen sich als die erheblich aktivieren. Für das umfassende
Screening wurde der Homo-bis-phosphanruthenium(II)-Komplex 22 verwendet. Dieser
wurde in der Katalyse mit unterschiedlichen Lösungsmitteln, Substratkonzentrationen,
Katalysatorbeladungen, Wassermengen und Additiven getestet um die optimalen
Bedingungen und die Faktoren, die einen Einfluss auf die Katalyse haben, zu finden.
Neben der Variation der Reaktionsbedingungen stellte die Modifikation der Katalysatoren
durch Gegenion- und Ligandenaustausch hin zu den Mischkomplexen eine wesentliche
Neuerung dar. Als Liganden wurden neben den verschiedenen Azaarylphosphinen weitere
Phosphine als auch Phosphite verwendet, die sich in ihren sterischen und elektronischen
Eigenschaften deutlich unterschieden.
Abbildung 4.1: Ruthenium(II)-Komplexe.

4. Zusammenfassung
73
Als Ergebnis dieser umfassenden kinetischen Studien stellte sich heraus, dass der beste
Katalysator für die anti-MARKOVNIKOV-Hydratisierung von terminalen Alkinen der
Komplex 32 ist. Dieser zeigt in Aceton (c = 0.25 M) mit 55.5 Äquivalenten Wasser bei
50 °C die höchste Aktivität und Reaktionsgeschwindigkeit. Aufgrund dieser Aktivität
konnte gezeigt werden, dass es ausreicht einen bifunktionalen P,N-Liganden mit einem
sterisch wenig anspruchsvollen Triarylphosphin zu kombinieren um einen aktiven
Katalysator zu erhalten. Somit ist es nicht mehr vonnöten, wie bisher in der Literatur
beschrieben, zwei bifunktionale P,N-Liganden im Katalysator zu verwenden, was sowohl
wegen der aufwendigeren Synthese der bifunktionalen Liganden zu einer Material- und
Kosteneinsparung bei der Herstellung der Katalysatoren führt als auch einen Hinweis für
den postulierten Mechanismus gibt. Im Allgemeinen hatte die Wahl des Lösungsmittels
und die Zugabe von Additiven einen großen Effekt auf die Katalyse. Neben der Hemmung
der rutheniumkatalysierten Hydratisierung von terminalen Alkinen durch Acetonitril und
einige Basen konnte gezeigt werden, dass Säuren (Benzoesäure) den Reaktionsverlauf der
Katalyse nicht stören und die Steigerung der Wassermenge zu einer Erhöhung der
Reaktionsgeschwindigkeit führt. Das Maximum des Wasseranteils lag bei 55.5
Äquivalenten, da hier die Löslichkeit des Katalysators und des Alkins im Aceton/Wasser-
Gemisch noch gerade gegeben war. Dieses Löslichkeitsproblem des Ruthenium(II)-
Komplexes in Wasser könnte in weiterführenden Untersuchungen durch das Einführen z. B.
einer oder mehrerer Sulfonat-Gruppen in der Ligandenperipherie verringert werden und die
Löslichkeit steigern, was eine weitere Erhöhung des Wasseranteils erlauben würde und
somit vielleicht eine nochmalige Beschleunigung der Katalyse bewirken oder gar zu einem
zweiphasigen Katalysatorsystem führen würde. Ein heterogenes System würde natürlich
die Atomökonomie der Reaktion wesentlich verbessern, da so der Katalysator über die
wässrige Phase abgetrennt und rezyklisiert werden könnte unter der Voraussetzung, dass
die Decarbonylierung des Katalysators reduziert werden kann.
Neben der Optimierung der Katalyse konnte die anti-MARKOVNIKOV-Hydratisierung in
Synthesestrategien eingesetzt werden und führte dabei entweder zu Aldolprodukten und
Poly-1,3-diolen oder aber zu Poly-1,4-diolen. Die Poly-1,4-diole führten in einer iterativen
Synthesesequenz aus einfachen Grundchemikalien zu strukturkomplexen Poly-1,4-
diolderivaten und naturstoffähnlichen Verbindungen, welche sich aufgrund der nicht
stereoselektiven Syntheseweise bestens für den Aufbau von Molekülbibliotheken im Sinne
der diversitätsorientierten Synthese eignen. Ausgehend von den in dieser Arbeit

4. Zusammenfassung
74
beschriebenen Poly-1,4-diolderivaten sollte es möglich sein weitere Makrolactone
herzustellen, diese zu funktionalisieren und biologischen Aktivitätstest zu unterziehen.
Schema 4.2: Iterative Synthese von Poly-1,4-diolen.
Die Hydratisierung der Propargylalkohole zu den Aldolen bzw. 1,3-Diolen gestaltete sich
anfänglich schwieriger als die Hydratisierung zu den 1,4-Diolen, da es verstärkt zur
Deaktivierung des Katalysators durch Carbonylierung kam. Letzten Endes war es jedoch
möglich unter Anwendung der katalytischen anti-MARKOVNIKOV-Hydratisierung in einer
Synthesestrategie den Naturstoff 54 (Massoialacton) und das Derivat 59 zu erhalten, das
aus der Familie der Polyacetat-abgeleiteten Pyrone stammt.
Schema 4.3: Synthese von Naturstoffen aus Propargylalkoholen.

4. Zusammenfassung
75
Dieser Einsatz in den unterschiedlichen Synthesestrategien führte durch die Verringerung
der energie- und abfallintensiven Schritte zu einer Erhöhung der Atomökonomie und der
Effizienz der jeweiligen Syntheseroute. Außerdem sollte es möglich sein bei zukünftigen
Synthesen die Stereochemie mit bekannten stereoselektiven Reaktionen zu kontrollieren,
da gezeigt werden konnte, dass die Katalyse unter Retention des Stereozentrums verläuft.

5. Experimenteller Teil
76
5. Experimenteller Teil
5.1. Allgemeines
5.1.1. Allgemeine Arbeitstechniken Alle luft- und feuchtigkeitsempfindlichen Reaktionen wurden in mit Magnetrührkernen
versehenen Schlenkrohren/-kolben durchgeführt, die vor Verwendung im
Ölpumpenvakuum mit einem Heißluftfön ausgeheizt, mit Argon belüftet und mit einem
Septum oder Glasstopfen mit Kunststoffdichtung verschlossen wurden.85 Die nur
luftempfindlichen Reaktionen wurden unter einem permanenten, schwachen
Argonüberdruck durchgeführt. Lösungsmittel, flüssige und gelöste Reagenzien wurden mit
Hilfe von mit Schutzgas gespülten Polypropylenspritzen oder Hamilton Glas-Microliter-
Spritzen mit Einweg- oder V2A-Stahlkanülen zugegeben oder entnommen. Feststoffe
wurden im Argongegenstrom hinzugefügt.
5.1.2. Reinigung und Trocknung der verwendeten Lösungsmittel Die Trocknung und Reinigung des Lösungsmittels Tetrahydrofuran erfolgte nach
Standardvorschrift,86 was bedeutet, dass das Tetrahydrofuran mehrere Tage über
Kaliumhydroxid vorgetrocknet, danach über neutralem Aluminiumoxid filtriert,
anschließend unter Rückfluss über Natrium/Benzophenon erhitzt und unter Argon
destilliert wurde. Aus Sicherheitsgründen wurde diese Methode jedoch nach einiger Zeit
nicht mehr verwendet, so dass THF kommerziell in absoluter bzw. HPLC-Reinheit
bezogen wurde. Aceton und Acetonitril wurden von der Firma ROTH in der Qualität
≥99.8% Ph. Eur., reinst (Aceton) und HPLC-Reinheit (Acetonitril) bezogen. Die übrigen
Lösungsmittel, die nicht in der vorgenannten Reinheit benötigt wurden, wurden teils
vorgetrocknet (EtOAc über CaCl2) und destilliert.
5.1.3. Kommerzielle Chemikalien Alle in dieser Arbeit verwendeten Reagenzien wurden von den Firmen Acros, Alfa Aesar,
Fluka, Lancaster, Merck, Roth, Sigma-Aldrich und Strem Chemical Inc. bezogen. Das
RuCl3 wurde dankenswerterweise von Umicore zur Verfügung gestellt.

5. Experimenteller Teil
77
5.1.4. Analytische Methoden 1H-NMR-Spektroskopie: Die Spektren wurden mit den Geräten Gemini 300 (300 MHz),
Inova 400 (400 MHz) oder Unity 500 (500 MHz) der Firma Varian gemessen. Die
chemischen Verschiebungen δ wurden in parts per million (ppm) relativ zu
Tetramethylsilan (δ = 0.00 ppm) als internen Standard angegeben.
13C-NMR-Spektroskopie: Die Spektren wurden auf einem Gemini 300 (75MHz), einem
Inova 400 (100 MHz) oder einem Unity 500 (125 MHz) der Firma Varian
aufgenommen. Die Chemischen Verschiebungen δ wurden in ppm relativ zu
Tetramethylsilan (δ = 0.00 ppm) als internen Standard angegeben.
31P-NMR-Spektroskopie: Die Spektren wurden auf einem Varian Mercury 300 (121
MHz) oder Varian Inova 400 (162 MHz) gemessen.
Massenspektren: Für die Messung von Massenspektren wurden ein Varian MAT 212
oder ein MAT 95 der Firma Finnigan verwendet. Die Angaben erfolgten in der
atomaren Masseneinheiten m pro Elementarladung z. In Klammern wurden die
Intensitäten der Werte in Prozent des Basispeaks angegeben. Falls kein [M+]-
oder [M+ + H]-Signal vorlag, wurde die Massenzahl des größten Fragments
genannt.
Infrarotspektren: Zur Messung von IR-Spektren wurden die Geräte PE 1760 FT oder 100
FT-IR der Firma Perkin-Elmer verwendet. Die Messung der Spektren erfolgte als
Film oder in Lösung. Angegeben wurden nur die charakteristischen
Absorptionsbanden mit den Wellenzahlen in cm-1.
Elementaranalysen: Diese Analysen wurden mit dem Gerät CHN-Rapid der Firma
Heraeus durchgeführt. Die Angabe der Werte erfolgte in Massenprozent in der Art
und Weise, dass zuerst die gefundenen und dahinter in Klammern die theoretisch
berechneten Werte für Kohlenstoff und Wasserstoff aufgeführt wurden. Als rein
wurden die Substanzproben angesehen, wenn Δ(C,H,N) ≤ 0.3 galt.

5. Experimenteller Teil
78
Optische Drehung: Die Messung des Drehwertes erfolgte auf einem Perkin-Elmer 241
Polarimeter. Die Messungen wurden bei Raumtemperatur und einer Lichtfrequenz
von 589 nm in einer Küvette (d = 10 cm) durchgeführt und es wurden
Lösungsmittel mit HPLC-Reinheitsgrad verwendet.
Kristallographie: Die Kristalle wurden auf einem Bruker SMART mit CCD Area
Detektor vermessen.
5.1.5. Chromatographie Dünnschichtchromatographie: Die Dünnschichtchromatographie (DC) wurde mit
Kieselgel 60 Glasfertigplatten (Schichtdicke 0.25 mm) der Firma Merck
durchgeführt. Die Detektion erfolgte neben der Fluoreszenzlöschung bei λ = 254
nm durch Eintauchen in eine Entwicklerlösung aus (NH4)6[Mo7O24] • 4 H2O (10 g),
Ce(SO4)2 • 4 H2O (0.1 g), konz. Schwefelsäure (10 mL) und dest. Wasser (190 mL)
mit anschließendem Erwärmen mittels Heißluftfön.
Säulenchromatographie: Zur Aufreinigung der Verbindungen wurde Kieselgel 60 der
Firma Merck (Korngröße 43 μm bis 60 μm) als Säulenfüllmaterial (stationäre
Phase) verwendet.
HPLC: Die analytischen HPLC-Messungen wurden mit einem Gerät der Firma Knauer
(Smartline Autosampler 3800, Smartline Manager 5000, Smartline Pump 1000,
WellChrom DIODE ARRAY DETCTOR K-2800) mit UV-Detektion durchgeführt.
Als stationäre Phasen wurden eine Kromasil® 100 Si-5μ der Firma CS
Chromatographie Service und eine OD-Säule der Firma Chiral Technologies
(früher Daicel Chemical Industries Ltd.) eingesetzt (Länge: 25 cm, Durchmesser:
0.46 cm).

5. Experimenteller Teil
79
5.2. Allgemeine Arbeitsvorschriften
5.2.1. Durchführung der Reaktionskinetik-Messung mittels HPLC
In ein Schlenkrohr wurde unter Argon 4-Phenyl-1-butin (70 μL, 0.5 mmol),
Pivaloylanilid87 (74 mg, 0.42 mmol, w/w Pivaloylanilid:4-Phenylbutanal 1:1) und der
Ruthenium(II)-Komplex (2 mol%) trocken eingewogen. Zu diesem Gemisch wurde aus
einer frisch angesetzten und entgasten88 Aceton/Wasser-Stammlösung das Lösungs-
mittelgemisch (2 mL enthielten 45 mg, 2.5 mmol, 5 Äquiv H2O) gegeben, das Schlenkrohr
mit einem Septum verschlossen und die Reaktionslösung auf 50 °C erwärmt. Zur
Vorbereitung der HPLC-Probe wurden mit einer Hamilton-Spritze 20 μL aus der
Reaktionslösung entnommen und mit 5 mL des HPLC-Laufmittels (TBME/Hexan, 1:5)
verdünnt. Die verdünnte Lösung wurde dann in ein GC/MS-Gläschen gefüllt und mittels
HPLC-Spektroskopie mit UV-Detektor analysiert (Kromasil® 100 Si-5μ, 20 °C, λ =
213 nm, TBME/Hexan = 1:5, 1 mL/min). Durch Integration der Peakflächen von 4-
Phenylbutanal und Pivaloylanilid konnte man die Spektren auswerten und in Abhängigkeit
der Zeit ein Diagramm erstellen. Für die Auswertung der Integralverhältnisse wurde eine
Referenzierung mit gereinigtem 4-Phenylbutanal und Pivaloylanilid durchgeführt. Dazu
wurde jeweils eine Stammlösung aus 10 mg Substrat in 10 mL Laufmittel angesetzt und
mittels obenen beschriebener Methode analysiert. Der Vergleich der Integralflächen ergab
ein Verhältnis von Pivaloylanilid zu 4-Phenylbutanal von 0.62:1, woraus sich für den
Absorptionsintensitätsfaktor r ein Wert von 0.62 ergibt.
5.2.2. Synthese von [η5-Cyclopentadienylruthenium(II) (2-
diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) (L) (acetonitril)]
hexafluorophosphat-Komplexen mit variablen Koliganden L (AAV1)
In ein Schlenkrohr wurde unter Argon ein Gemisch aus dem Vorläuferkomplex [η5-
Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin) bis-
(acetonitril)] hexafluorophosphat 31 (1 Äquiv) und Ligand (1 Äquiv) eingewogen und mit
entgastem Aceton (50 mL/mmol) versetzt. Das Gemisch wurde für 3.5 h bei RT gerührt
bevor das Lösungsmittel am Membranpumpenvakuum entfernt wurde. Der ölige
Rückstand wurde in entgastem Petrolether (50 mL/mmol) aufgenommen, zu einem
feinkristallinen Pulver verkratz und für 10 min bei hoher Geschwindigkeit gerührt bevor

5. Experimenteller Teil
80
via Kanüle die Mutterlauge abfiltriert wurde. Das so erhaltene Pulver wurde zweimal mit
Petrolether gewaschen und nach Dekantieren der Waschlösung via Kanüle im
Hochvakuum getrocknet.
5.2.3. Iterative Synthese von Oligo-1,4-diolen via katalytischer anti-
MARKOVNIKOV Hydratisierung von Terminalen Alkinen 5.2.3.1. anti-MARKOVNIKOV Hydratisierung von terminalen Alkinen (AAV2)
Ein Schlenkrohr wurde mit [CpRu(η6-naphthalen)]PF6 9 (3 mol%),89 Ligand L (6 mol%),
Alkin (1 Äquiv), dest. Wasser (5 Äquiv) und Aceton (2 mL/mmol) befüllt. Der
Flüssigkeitspegel wurde auf dem Schlenkrohr markiert bevor weiteres Aceton (0.5
mL/mmol) zugegeben wurde. Anschließend wurde das zusätzlich zugegebene Aceton im
Membranpumpenvakuum wieder entfernt (dieser Schritt dient der Entfernung von O2). Die
erhaltene Lösung wurde unter Argon auf 70 °C erhitzt und im geschlossenen Rohr gerührt
(ACHTUNG: Druckentwicklung). Nach vollständiger Reaktion (12–18 h, überwacht
mittels DC), wurde die Lösung auf Raumtemperatur abgekühlt und das Lösungsmittel im
Membranpumpenvakuum entfernt. Der nicht gereinigte Aldehyd wurde in THF (als eine
0.5 M Lösung) gelöst und direkt für weitere Umsetzungen verwendet (Annahme: Aldehyd
= 1 Äquiv).
5.2.3.2. Allenylzink-bromid (66) Lösung in THF72c
Ein Schlenkrohr wurde unter Argon mit Zinkstaub (1.1 Äquiv), THF (2 mL/mmol) und
TMSCl (1–2 Tropfen/mmol Zinkstaub) befüllt. Die Lösung wurde für kurze Zeit zum
Refluxieren gebracht, so dass der Zinkstaub sinterte. Danach wurde die Lösung auf 0 °C
(Eisbad) abgekühlt und es wurde langsam Propargylbromid zu getropft. Nach vollständiger
Zugabe wurde die Lösung für 30 min bei 0 °C gerührt bevor sie verwendet wurde.
5.2.3.3. Propargylierung von Aldehyden (AAV3)
Eine Lösung aus Aldehyd (1 Äquiv) in THF (2 mL/mmol) wurde bei 0 °C zu einer
Allenylzink-Lösung 66 (1.5 Äquiv in THF) gegeben und für 20 min bei dieser Temperatur
gerührt. Anschließend wurde die Reaktion durch die Zugabe von ges. wässriger NH4Cl-
Lösung (4 M, 1.5 mL/mmol), wässriger HCl-Lösung (2.4 M, 0.1 mL/mmol) und TBME
(1.4 mL/mmol) beendet. Die wässrige Phase wurde mit TBME (1.5 mL/mmol) extrahiert
und die vereinigten organischen Phasen wurden mit dest. Wasser, ges. wässriger NaHCO3-

5. Experimenteller Teil
81
Lösung (0.9 M) und dest. Wasser (jeweils 1.5 mL/mmol) gewaschen. Danach wurde die
Lösung über MgSO4 getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels
Säulenchromatographie (TBME/Hexan, normalerweise 1:5→1:3) gereinigt.
5.2.3.4. Einfügen der TBS Schutzgruppe (AAV4)
Der sekundäre Alkohol (1 Äquiv), TBSCl (2 Äquiv) und Imidazol (3 Äquiv) wurden in
DMF (2 mL/mmol) bei Raumtemperatur gerührt. Nach Beendigung (DC-Kontrolle; 10–
48 h) wurde die Reaktion durch Zugabe von wässriger HCl-Lösung (2.4 M, 3 mL/mmol)
beendet. Die wässrige Phase wurde mit TBME (2 × 3 mL/mmol) extrahiert und die
vereinigten organischen Phasen wurden mit dest. Wasser, ges. wässriger NaHCO3-Lösung
(0.9 M) und dest. Wasser (jeweils 5 mL/mmol) gewaschen. Danach wurde die Lösung über
MgSO4 getrocknet, filtriert und eingeengt. Der Rückstand wurde mittels
Säulenchromatographie (TBME/Hexan, normalerweise 1:5→1:3) gereinigt.
5.2.3.5. WITTIG Reaktion (AAV5)
Zum in THF (0.5 M) gelösten Aldehyd (1 Äquiv), den man aus AAV2 erhalten hat, wurde
Methyl-triphenylphosphoranyliden-acetat (2.5 Äquiv) gegeben und die Lösung wurde für
2 h bei 50 °C gerührt. Die Reaktion wurde durch Zugabe von wässriger HCl-Lösung
(2.4 M, 2–3 mL/mmol) und TBME (3 mL/mmol) beendet. Die organische Phase wurde mit
dest. Wasser, ges. wässriger NaHCO3-Lösung (0.9 M) und dest. Wasser (jeweils 4–
5 mL/mmol) gewaschen und danach über MgSO4 getrocknet. Nach Filtration und
Einengen im Membranpumpenvakuum erhielt man das Rohprodukt, welches durch
Säulenchromatographie (TBME/Hexan, normalerweise 1:5→1:3) gereinigt wurde.
5.2.3.6. Addition von Lithium Methylpropiolat an Aldehyde (AAV6)
In einem Schlenkkolben wurde unter Argon n-Butyllithium (5 Äquiv) zu Diisopropylamin
(5 Äquiv) in THF (5 mL/mmol) gegeben und die Lösung für 15 min bei 0 °C (Eisbad)
gerührt und dann auf –78 °C abgekühlt. Anschließend tropfte man langsam Methyl
propiolat (5 Äquiv) dazu. Nach weiteren 20 min wurde der Aldehyd (1 Äquiv, in THF,
siehe AAV2) bei –78 °C tropfweise zur Reaktionslösung gegeben. Die Reaktion wurde
nach 5 min durch Zugabe von ges. wässriger NH4Cl-Lösung (4 M, 4 mL/mmol) und TBME
(5 mL/mmol) bei –78 °C beendet. Die wässrige Phase wurde mit TBME (5 mL/mmol)
extrahiert und die vereinigten organischen Phasen wurden mit ges. wässriger NaHCO3-
Lösung (0.9 M) und zweimal mit dest. Wasser (jeweils 5–6 mL/mmol) gewaschen. Die

5. Experimenteller Teil
82
organische Phase wurde danach über MgSO4 getrocknet, filtriert und eingeengt und der
erhaltene Rückstand durch Säulenchromatographie (TBME/Hexan; normalerweise
1:4→1:2) gereinigt.
5.3. Synthese und Arbeitsvorschriften
5.3.1. Synthese der η5-Cyclopentadienylruthenium(II)-Komplexe
5.3.1.1. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (acetonitril)] hexafluorophosphat (22)
In einem trockenen Schlenkrohr wurden unter Argon-Schutzgasatmosphäre der
Vorläuferkomplex [CpRu(η6-naphthalen)]PF6 9 (0.475 g, 1.08 mmol) mit dem Aza-
arylphosphan 2-Diphenylphosphino-6-(2,4,6-triphenylphenyl)pyridin 14 (1.23 g, 2.16
mmol) vorgelegt und mit entgastem Acetonitril (50 mL) versetzt. Man ließ die Lösung für
72 h bei 60 °C rühren bevor man das überschüssige Lösungsmittel erst im
Membranpumpen- und danach im Hochvakuum entfernte. Den öligen Rückstand nahm
man in entgastem Petrolether (25 mL) auf und rührte ihn für 1 h bei 50 °C. Anschließend
verkratzte man den Rückstand zu einem feinkristallinen Pulver und rührte für weitere
30 min bei 50 °C. Die Mutterlauge wurde via Kanüle abfiltriert und das so erhaltene Pulver
zweimal mit Petrolether (je 25 mL) gewaschen. Nach Filtration via Kanüle und Trocknen
im Hochvakuum erhielt man ein feines hellgelbes Pulver.
1H-NMR (400 MHz, CD3CN): δ = 1.49 (t, J = 1.3 Hz, 3 H, CH3), 3.92 (s, 5 H, Cp), 6.68–
6.77 (m, 6 H, Ar), 6.86–6.92 (m, 4 H, Ar), 7.01 (t, J = 7.4 Hz, 4 H, Ar), 7.08–7.17
(m, 26 H, Ar), 7.23–7.35 (m, 6 H, Ar), 7.38–7.43 (m, 2 H, Ar), 7.45–7.51 (m, 4 H,
Ar), 7.68–7.70 (m, 4 H, Ar), 7.76–7.81 (m, 4 H, Ar) ppm. 31P-NMR (160 MHz, CD3CN): δ = 44.4 (s), –144.8 (sept, J = 707 Hz, PF6) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.42

5. Experimenteller Teil
83
5.3.1.2. [η5-Cyclopentadienylruthenium(II) bis-(triphenylphosphin) (acetonitril)]
hexafluorophosphat (28)
In einem trockenen Schlenkrohr wurden unter Argon der Vorläuferkomplex [CpRu(η6-
naphthalen)]PF6 9 (87.9 mg, 0.2 mmol) mit Triphenylphosphin (105 mg, 0.4 mmol)
vorgelegt und mit entgastem Acetonitril (5 mL) versetzt. Die klare, gelbe Lösung ließ man
für 43 h bei 60 °C rühren bevor man das überschüssige Lösungsmittel erst im
Membranpumpen- und danach im Hochvakuum entfernte. Den öligen Rückstand nahm
man in entgastem Petrolether (5 mL) auf und rührte für 1 h bei 50 °C. Anschließend
verkratzte man den Rückstand zu einem feinkristallinen Pulver und rührte für weitere
30 min bei 50 °C gerührt. Die Mutterlauge filtrierte man via Kanüle ab und das so
erhaltene Pulver wusch man zweimal mit Petrolether (je 5 mL). Nach Filtration via Kanüle
und Trocknen im Hochvakuum erhielt man ein feines hellgelbes Pulver.
1H-NMR (400 MHz, (CD3)2CO): δ = 2.36 (t, J = 1.4 Hz, 3 H, CH3), 4.60 (s, 5 H, Cp),
7.18–7.23 (m, 12 H, Ar), 7.32–7.37 (m, 12 H, Ar), 7.43–7.48 (m, 6 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 41.9 (s), –144.4 (sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 731.3 (10) [M+], 691.1 (100).
5.3.1.3. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (iso-butyronitril)] hexafluorophosphat (29)
In einem trockenen Schlenkrohr wurden unter Argon der Vorläuferkomplex [CpRu(η6-
naphthalen)]PF6 9 (43.9 mg, 0.1 mmol) mit dem Aza-arylphosphan 2-Diphenylphosphino-
6-(2,4,6-triphenylphenyl)pyridin 14 (113.5 mg, 0.2 mmol) vorgelegt und mit entgastem
und durch neutrales Al2O3 filtriertes Isobutyronitril (2.5 mL) versetzt. Die Lösung ließ man
für 25 h bei 50 °C rühren bevor man das überschüssige Lösungsmittel im

5. Experimenteller Teil
84
Membranpumpenvakuum entfernte. Den Rückstand nahm man in entgastem Petrolether (5
mL) auf, verkratzte ihn zu einem feinkristallinen Pulver und rührte für weitere 30 min bei
50 °C. Die Mutterlauge filtrierte man via Kanüle ab und das erhaltene Pulver wusch man
zweimal mit Petrolether (je 5 mL). Nach Filtration via Kanüle und Trocknen im
Hochvakuum erhielt man ein feines hellgelbes Pulver.
1H-NMR (300 MHz, (CD3)2CO): δ = 0.98 (d, J = 7.0 Hz, 6 H, 2 x CH3), 1.42 (s, 1 H,
CH), 2.96 (sept x t, J = 7.0, 1.1 Hz, 1 H, CH), 4.06 (s, 5 H, Cp), 6.84–6.90 (m, 2 H,
Ar), 6.93–7.57 (m, 50 H, Ar), 7.74 (s, 4 H, Ar), 7.85–7.89 (m, 4 H, Ar) ppm. 31P-NMR (120 MHz, (CD3)2CO): δ = 42.9 (s), –144.3 (sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1301.07 [M+ – iPrCN], 734.53.
5.3.1.4. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (benzonitril)] hexafluorophosphat (30)
In einem trockenen Schlenkrohr wurden unter Argon der Vorläuferkomplex [CpRu(η6-
naphthalen)]PF6 9 (43.9 mg, 0.1 mmol) mit dem Aza-arylphosphan 2-Diphenylphosphino-
6-(2,4,6-triphenylphenyl)pyridin 14 (113.5 mg, 0.2 mmol) vorgelegt und mit entgastem
und durch neutrales Al2O3 filtriertes Benzonitril (2 mL) versetzt. Die Lösung ließ man 138
h bei 50 °C rühren bevor man das überschüssige Lösungsmittel im Hochvakuum entfernte.
Den Rückstand nahm man in entgastem Petrolether (5 mL) aufgenommen und verkratzte
ihn zu einem feinkristallinen Pulver. Die Mutterlauge filtrierte man via Kanüle ab und das
so erhaltene Pulver wusch man zweimal mit Petrolether (je 5 mL). Nach Filtration via
Kanüle und Trocknen im Hochvakuum wurde ein feines hellgelbes Pulver erhalten.
1H-NMR (400 MHz, (CD3)2CO): δ = 4.14 (s, 5 H, Cp), 6.87–6.92 (m, 2 H, Ar), 6.97–7.65
(m, 54 H, Ar), 7.71 (d, J = 1.9 Hz, 2 H, Ar), 7.77 (d, J = 1.9 Hz, 2 H, Ar), 7.78–
7.81 (m, 1 H, Ar), 7.86–7.90 (m, 4 H, Ar) ppm. 31P-NMR (162 MHz, (CD3)2CO): δ = 42.2 (s), –144.4 (sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1301.11 [M+ – PhCN], 734.55.

5. Experimenteller Teil
85
5.3.1.5. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat (31)
In einem trockenen Schlenkrohr wurden unter Argon-Schutzgasatmosphäre der
Vorläuferkomplex [CpRu(η6-naphthalen)]PF6 9 (1 Äquiv) mit dem Aza-arylphosphan 2-
Diphenylphosphino-6-(2,4,6-triphenylphenyl)pyridin 14 (1 Äquiv) vorgelegt und mit
entgastem Acetonitril (20 mL/mmol) versetzt. Man ließ die Lösung für 20 h bei RT rühren
bevor man das überschüssige Lösungsmittel im Membranpumpenvakuum entfernte. Den
öligen Rückstand nahm man in entgastem Petrolether (20 mL/mmol) auf, verkratzte ihn zu
einem feinkristallinen Pulver und filtrierte via Kanüle die Mutterlauge ab. Das so erhaltene
Pulver wurde zweimal mit Petrolether gewaschen und nach Filtration via Kanüle im
Hochvakuum getrocknet.
1H-NMR (400 MHz, (CD3)2CO): δ = 2.15 (d, J = 1.4 Hz, 6 H, CH3), 4.32 (s, 5 H, Cp),
7.08–7.98 (m, 30 H, Ar) ppm. 31P-NMR (162 MHz, (CD3)2CO): δ = 51.1 (s), –144.4 (sept, J = 708 Hz, PF6) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.42
5.3.1.6. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (triphenylphosphin) (acetonitril)] hexafluorophosphat (32)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (100 mg, 0.104
mmol) zusammen mit Triphenylphosphin (27.3 mg, 0.104 mmol) zu Komplex 32
umgesetzt.

5. Experimenteller Teil
86
1H-NMR (300 MHz, (CD3)2CO): δ = 2.23 (t, J = 1.4 Hz, 3 H, CH3), 4.29 (s, 5 H, Cp),
6.72–6.79 (m, 2 H, Ar), 6.99–7.07 (m, 6 H, Ar), 7.09–7.49 (m, 29 H, Ar), 7.51–
7.57 (m, 2 H, Ar), 7.75–7.78 (m, 2 H, Ar), 7.87–7.90 (m, 2 H, Ar) ppm. 31P-NMR (120 MHz, (CD3)2CO): δ = 44.6 (d, J = 35.8 Hz), 40.5 (d, J = 36.0 Hz),
–144.4 (sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1036.67 [M+], 996.27, 734.60.
5.3.1.7. [η5-Cyclopentadienalruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (tris-(ortho-tolyl)phosphin) (acetonitril)]
hexafluorophosphat (34)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tri(ortho-tolyl)phosphin (30.4 mg, 0.1 mmol) umgesetzt. Gemäss 31P-NMR ist der Komplex zumindest in Aceton nicht stabil.
1H-NMR (400 MHz, (CD3)2CO): δ = 1.67 (d, J = 1.9 Hz, 3 H, CH3), 2.37 (s, 9 H, 3 x
Me), 4.33 (s, 5 H, Cp), 6.68–6.73 (m, 2 H, Ar), 7.01–7.99 (m, 40 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 43.6 (s, [CpRu(2,4,6Ph3C6H2PyPPh2)2(MeCN)]PF6),
35.3 (s, [CpRu(2,4,6Ph3C6H2PyPPh2)(oTol3P)(MeCN)]PF6), 0.1 (s, [CpRu (η2-
2,4,6Ph3C6H2PyPPh2)(MeCN)]PF6), –30.8 (s, oTol3P)90, –144.4 (sept, J = 708 Hz,
PF6) ppm.
MS (ESI, CHCl3): m/z = 1037.78 [M+ – MeCN], 734.29.

5. Experimenteller Teil
87
5.3.1.8. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (tri-(1-naphthyl)phosphin) (acetonitril)]
hexafluorophosphat (35)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tri(1-naphthyl)phosphin (41.2 mg, 0.1 mmol) umgesetzt. In Aceton
liegt gemäss 31P-NMR freies Tri(1-naphthyl)phosphin und Mono-Chelat-Komplex mit
2,4,6Ph3C6H2PyPPh2 vor.
1H-NMR (400 MHz, (CD3)2CO): δ = 1.67 (d, J = 1.9 Hz, 3 H, CH3), 4.33 (s, 5 H, Cp),
6.94–6.99 (m, 3 H, Ar), 7.16–8.02 (m, 44 H, Ar), 8.50–8.56 (m, 4 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 0.1 (s, ), –34.5 (s, [P(Naphth)3])91, –144.4 (hept, J =
708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1144.91 [M+ – MeCN], 734.48.
5.3.1.9. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (tributylphosphin) (acetonitril)] hexafluorophosphat (36)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-(diphenylphosphino)-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tributylphosphin (25 μL, 0.1 mmol) zu Komplex 36 umgesetzt.

5. Experimenteller Teil
88
Kristallisationsversuch:
Nach Einengen der Reaktionslösung und Aufnahme des Rückstandes in Aceton wurde die
Lösung mit Petrolether überschichtet und bei 4 °C gelagert, wobei man inunter ca. 10
Tagen einen Kristalle erhielt.
Kristallographische Daten:
Kristallisiert aus Aceton/Petrolether
Summenformel C60H65F6N2P3Ru
Molekülgewicht [g mol−1] 1122.12
Kristallfarbe, -beschaffenheit gelb, Block
Kristalldimensionen [mm3] 0.34 × 0.12 × 0.08
Temperatur [K] 130 (2)
Kristallsystem Monoklin
Raumgruppe P21/n
Z 4
Reflexionen für Zellbestimmung 81939
2θ Bereich der Zellbestimmung [°] 2.29–21.48
Parameter der Zelleinheit
a [Å] 18.3756 (11)
b [Å] 14.6058 (9)
c [Å] 22.0886 (13)
α [°] 90

5. Experimenteller Teil
89
β [°] 111.7290 (11)
γ [°] 90
V [Å3] 5507.1 (6)
F(000) 2328
Dx [g cm−3] 1.353
μ(Mo Kα) [mm−1] 0.432
Scan Typ ω
2θmax [°] 21.48
Transmissionsfaktoren (min; max) 0.867; 0.9663
alle gemessenen Reflexionen 16265
Symmetry-independent reflections 9259
Rint 0.091
Reflexion mit I > 2σ (I) 16265
Reflexion bei der Verfeinerung 5982
verfeinerte Parameter 650
Final R(F) [I > 2σ (I) Reflexionen] 0.0532
ωR(F2) (alle Daten) 0.1060
Gewichte ω = [σ2(F02 ) + (0.0460P)2 + 0.0000P]−1
mit P = (F02 + 2Fc
2 ) / 3
Anpassung 0.884
Δρ (max; min) [e Å−3] 1.161; −0.588
1H-NMR (400 MHz, (CD3)2CO): δ = 0.80 (t, J = 7.2 Hz, 9 H, 3 x CH3), 1.13–1.40 (m, 15
H, CH2), 1.62–1.74 (m, 3 H, CH2), 2.43 (t, J = 1.3 Hz, 3 H, CH3), 4.53 (s, 5 H, Cp),
7.01–7.06 (m, 1 H, Ar), 7.13–7.59 (m, 25 H), 7.72 (d, J = 2.1 Hz, 1 H, Ar), 7.78 (d,
J = 2.0 Hz, 1 H, Ar), 7.84–7.89 (m, 2 H, Ar) ppm. 31P-NMR (162 MHz, (CD3)2CO): δ = 46.5 (d, J = 37.8 Hz), 22.6 (d, J = 37.8 Hz), –144.4
(sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 935.96 [M+ – MeCN], 734.23.

5. Experimenteller Teil
90
5.3.1.10. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (tri-(para-tolyl)phosphin) (acetonitril)] hexafluorophosphat
(37)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tri(para-tolyl)phosphin (30.4 mg, 0.1 mmol) zu Komplex 37
umgesetzt.
1H-NMR (400 MHz, (CD3)2CO): δ = 2.18 (t, J = 1.4 Hz, 3 H, CH3), 2.31 (br s, 9 H, 3 x
CH3), 4.22 (s, 5 H, Cp), 6.79–6.85 (m, 2 H, Ar), 6.91–6.97 (m, 4 H, Ar), 7.02–7.59
(m, 32 H, Ar), 7.79–7.82 (m, 2 H, Ar), 7.89–7.94 (m, 2 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 42.6 (d, J = 35.5 Hz), 40.7 (d, J = 35.5 Hz), –144.4
(sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1037.84 [M+ – MeCN], 775.06, 734.53.
5.3.1.11. [η5-Cyclopentadienylruthenium(II) (2-(diphenylphosphino)-6-(2,4,6-
triphenylphenyl)pyridin) (tri-para-fluorophenylphosphin) (acetonitril)]
hexafluorophosphat (38)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tri(para-fluorphenyl)phosphin (31.6 mg, 0.1 mmol) zu Komplex 38
umgesetzt.

5. Experimenteller Teil
91
1H-NMR (400 MHz, (CD3)2CO): δ = 2.25 (t, J = 1.3 Hz, 3 H, CH3), 4.23 (s, 5 H, Cp),
6.77–6.84 (m, 2 H, Ar), 6.92–6.98 (m, 6 H, Ar), 7.02–7.58 (m, 30 H, Ar), 7.76–
7.78 (m, 2 H, Ar), 7.84–7.89 (m, 2 H, Ar) ppm. 31P-NMR (162 MHz, (CD3)2CO): δ = 43.9 (dq, J = 35.5 Hz, J = 2.8 Hz), 39.8 (d, J = 35.5
Hz), –144.4 (sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1090.16 [M+], 1052.71, 735.27.
5.3.1.12. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (tri-para-chlorophenylphosphin) (acetonitril)]
hexafluorophosphat (39)
RuN
PPh2P
N
RMe
Ph
Ph Ph
R =
PF6-
Cl3
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tri(para-chlorphenyl)phosphin (36.6 mg, 0.1 mmol) zu Komplex 39
umgesetzt.
1H-NMR (400 MHz, (CD3)2CO): δ = 2.26 (br s, 3 H, CH3), 4.21 (s, 5 H, Cp), 6.83–6.90
(m, 2 H, Ar), 6.93–6.97 (m, 1 H, Ar), 7.06–7.62 (m, 35 H, Ar), 7.81–7.86 (m, 2 H,
Ar), 7.91–7.95 (m, 2 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 45.3 (d, J = 35.4 Hz), 39.3 (d, J = 35.4 Hz), –144.4
(sept, J = 708.5 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1099.70 [M+ – MeCN], 1052.71, 734.51.

5. Experimenteller Teil
92
5.3.1.13. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (tri-para-methoxyphenyl)phosphin) (acetonitril)]
hexafluorophosphat (40)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tri(para-methoxyphenyl)phosphin (35.2 mg, 0.1 mmol) zu
Komplex 40 umgesetzt.
1H-NMR (400 MHz, (CD3)2CO): δ = 2.24 (t, J = 1.3 Hz, 3 H, CH3), 3.78 (s, 9 H, 3 x
CH3), 4.24 (s, 5 H, Cp), 6.75–6.80 (m, 6 H, Ar), 6.81–6.90 (m, 4 H, Ar), 6.92–6.98
(m, 6 H, Ar), 7.09–7.57 (m, 22 H, Ar), 7.77 (d, J = 1.9 Hz, 1 H, Ar), 7.79 (d, J = 1.9
Hz, 1 H, Ar), 7.87–7.90 (m, 2 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 41.1 (d, J = 36.1 Hz), 40.4 (d, J = 36.1 Hz), –144.4
(sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1120.75 [M+], 1086.43, 871.57, 735.59.
5.3.1.14. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (tri-(4-trifluoromethylphenyl)phosphin) (acetonitril)]
hexafluorophosphat (41)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (96.1 mg, 0.1
mmol) zusammen mit Tri(para-trifluoromethylphenyl)phosphin (46.6 mg, 0.1 mmol) zu
Komplex 41 umgesetzt.

5. Experimenteller Teil
93
1H-NMR (400 MHz, (CD3)2CO): δ = 2.31 (t, J = 1.3 Hz, 3 H, CH3), 4.30 (s, 5 H, Cp),
6.88–6.94 (m, 2 H, Ar), 6.98 (ddd, J = 7.6, 4.4, 1.3 Hz, 1 H, Ar), 7.07–7.64 (m,
34 H, Ar), 7.78–7.82 (m, 1 H, Ar), 7.83 (s, 2 H, Ar), 7.90–7.92 (m, 2 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 47.9 (d, J = 35.2 Hz), 39.4 (d, J = 35.2 Hz), –144.4
(sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1200.07 [M+], 964.26, 801.89, 734.33.
5.3.1.15. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (triethylphosphit) (acetonitril)] hexafluorophosphat (42)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (89.7 mg, 0.0933
mmol) zusammen mit Triethylphosphit (16 μL, 0.0933 mmol) zu Komplex 42 umgesetzt.
1H-NMR (400 MHz, (CD3)2CO): δ = 1.10 (t, J = 7.0 Hz, 9 H, 3 x CH3), 2.35 (br s, 3 H,
CH3), 3.83–3.95 (m, 6 H, 3 x CH2), 4.68 (s, 5 H, Cp), 7.15–7.61 (m, 26 H, Ar), 7.69–7.73
(m, 2 H, Ar), 7.83–7.87 (m, 2 H, Ar) ppm. 31P-NMR (162 MHz, (CD3)2CO): δ = 144.1 (d, J = 65.3 Hz), 51.8 (d, J = 65.3 Hz), –
144.4 (sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 900.07 [M+ – MeCN], 734.53.

5. Experimenteller Teil
94
5.3.1.16. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (triphenylphosphit) (acetonitril)] hexafluorophosphat (43)
Gemäß AAV1 wurde das [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-
(2,4,6-triphenylphenyl)pyridin) bis-(acetonitril)] hexafluorophosphat 31 (95.3 mg, 0.099
mmol) zusammen mit Triphenylphosphit (26 μL, 0.099 mmol) zu Komplex 43 umgesetzt.
1H-NMR (400 MHz, (CD3)2CO): δ = 2.12 (br s, 3 H, CH3), 4.57 (s, 5 H, Cp), 6.95–7.99
(m, 45 H, Ar) ppm. 31P-NMR (160 MHz, (CD3)2CO): δ = 137.9 (d, J = 65.0 Hz), 49.5 (d, J = 65.0 Hz), –
144.4 (sept, J = 708 Hz, PF6) ppm.
MS (ESI, CHCl3): m/z = 1084.60 [M+], 1043.92 [M+ – MeCN – PF6], 964.27, 808.46,
734.33.
5.3.1.17. [η5-Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (acetonitril)] (rac)-TRISPHAT (44)
In einem trockenen Schlenkrohr wurden unter Argon der Vorläuferkomplex [η5-
Cyclopentadienylruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl) pyridin)
(acetonitril)] hexafluorophosphat 22 (298 mg, 0.2 mmol) zusammen mit (HNBu3)(rac)-
TRISPHAT (105 mg, 0.4 mmol) vorgelegt und mit entgastem DCM (10 mL) versetzt. Die
gelbe Lösung ließ man für 24 h bei RT rühren und reinigte das Produkt mittels
Säulenchromatographie (DCM/PE, 60:40→70:30→100:0). Die gelben Produktfraktionen
wurden vereinigt und eingeengt und der Rückstand in DCM (1 mL) und MeCN (1 mL)
aufgenommen und wieder eingeengt. Den Rückstand nahm man in Petrolether auf und
verkratzte ihn zu einem feinkristallinen Pulver, die Mutterlauge filtrierte man via Kanüle

5. Experimenteller Teil
95
ab und das so erhaltene Produkt trocknete man erst am Membranpumpen- und danach am
Hochvakuum.
1H-NMR (300 MHz, C6D6): δ = 1.40 (s, 3 H, CH3), 3.98 (s, 5 H, Cp), 6.60–7.29 (m, 52 H),
7.49 (dt, J = 6.9, 1.5 Hz, 4 H), 7.42 (d, J = 1.5 Hz, 4 H) ppm. 31P-NMR (120 MHz, C6D6): δ = 44.1 (s), –80.0 (s, PTRISPHAT) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.42
5.3.1.18. [η5-Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (triphenylphosphin) (acetonitril)] (rac)-TRISPHAT (45)
In einem trockenen Schlenkrohr wurden unter Argon der Vorläuferkomplex [η5-
Cyclopentadienylruthenium(II) (2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin)
(triphenylphosphin) (acetonitril)] hexafluorophosphat 32 (245.9 mg, 0.208 mmol)
zusammen mit (HNBu3)(rac)-TRISPHAT (198.6 mg, 0.208 mmol) vorgelegt und mit
entgastem DCM (5 mL) versetzt. Die Lösung ließ man für 24 h bei RT rühren und reinigte
das Produkt mittels Säulenchromatographie (DCM/PE, 60:40→70:30→100:0). Die gelben
Produktfraktionen wurden vereinigt und eingeengt und der Rückstand in DCM (1 mL) und
MeCN (1 mL) aufgenommen und wieder eingeengt. Den Rückstand nahm man in
Petrolether auf, verkratzte ihn zu einem feinkristallinen Pulver und filtrierte die
Mutterlauge via Kanüle ab. Das so erhaltene Produkt trocknete man erst am
Membranpumpen- und danach am Hochvakuum.
1H-NMR (400 MHz, (CD3)2CO): δ = 2.22 (s, 3 H, CH3), 4.60 (s, 5 H, Cp), 6.72–6.79 (m,
2 H, Ar), 6.99–7.56 (m, 39 H, Ar), 7.74–7.79 (m, 2 H, Ar), 7.85–7.92 (m, 2 H, Ar)
ppm. 31P-NMR (162 MHz, (CD3)2CO): δ = 44.6 (d, J = 35.7 Hz), 40.7 (d, J = 35.7 Hz),
–80.7 (s, PTRISPHAT) ppm.
MS (ESI, MeOH): m/z = 995.84 (100) [M+ – MeCN], 734.29 (52).

5. Experimenteller Teil
96
5.3.2. Synthese von 1,3-Diolen und deren Derivate 5.3.2.1. 3-Benzylether-1-octin (46)
1-Octin-3-ol (1.434 mL, 10 mmol) wurde zusammen mit Benzylbromid (1.914 mL, 16
mmol) und Tetrabutylammoniumchlorid (0.445 g, 1.6 mmol) in DCM (10 mL) gelöst.
Dann tropfte man unter Rühren langsam wässrige 50%ige Natriumhydroxidlösung (3 g,
37.5 mmol) zu und nach vollständiger Zugabe hielt man die Lösung für 24 h am
Refluxieren. Anschließend beendete man die Reaktion durch Zugabe von ges. wässriger
NaHCO3-Lösung (50 mL) und verdünnte mit TBME (100 mL). Die organische Phase
wurde nochmals mit ges. wässriger NaHCO3-Lösung (50 mL) und danach mit dest. Wasser
(zweimal je 50 mL) gewaschen. Die wässrige Phase wurde rückextrahiert und die
vereinigten organischen Phasen wurden über MgSO4 getrocknet, filtriert und eingeengt.
Nach Säulenchromatographie (TBME/Hexan, 1:30) erhielt man eine leicht gelbliche
Flüssigkeit (2.1 g, 0.97 mmol, 97%).
Rf = 0.60 (TBME/Hexan, 1:20). 1H-NMR (CDCl3, 300 MHz): δ = 0.85–0.91 (m, 3H, CH3), 1.21–1.37 (m, 4 H, 2 x
CH2), 1.41–1.54 (m, 2 H, CH2), 1.65–1.86 (m, 2 H, CH2), 2.46 (d, J = 2.0 Hz, 1 H,
CH), 4.08 (dt, J = 6.7, 1.9 Hz, 1H, CH), 4.51 (d, J = 11.6 Hz, 1 H, CH2), 4.81 (d, J
= 11.9 Hz, 1 H, CH2), 7.23–7.39 (m, 5 H, Ar) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.0 (CH3), 22.5 (CH2), 24.9 (CH2), 31.5 (CH2), 35.6
(CH2), 68.5 (CH), 70.5 (CH2), 73.7 (CH), 82.8 (C), 127.7 (CH), 128.0 (CH), 128.4
(CH), 137.9 (C) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.92
5.3.2.2. 3-Tetrahydropyran-2-yloxy-1-octin (47) OTHP
Die Verbindung wurde gemäß Literaturvorschrift,93 wie folgt, hergestellt: 1-Octin-3-ol
(2.92 mL, 20 mmol) wurde zusammen mit 3,4-Dihydro-2H-pyran (3.65 mL, 40 mmol) und
pTsOH (38 mg, 0.2 mmol, 1 mol%) in DCM (30 mL) gegeben und bei 0 °C (Eisbad) für
2 h gerührt. Die Reaktion beendete man durch die Zugabe von ges. wässriger NaHCO3-
Lösung (50 mL) und verdünnte mit TBME (100 mL). Die organische Phase wusch man

5. Experimenteller Teil
97
nochmals mit ges. wässriger NaHCO3-Lösung (50 mL) und danach mit dest. Wasser (100
mL). Die wässrige Phase wurde rückextrahiert und die vereinigten organischen Phasen
über Na2SO4 getrocknet, filtriert und eingeengt. Nach Säulenchromatographie
(TBME/Hexan, 1:20) erhielt man eine leicht gelbliche Flüssigkeit (4.04 g, 0.192 mmol,
96%, Mischung aus Diastereomeren A/B = 60:40).
Rf = 0.41 und 0.33 (TBME/Hexan, 1:20). 1H-NMR (CDCl3, 300 MHz): δ = 0.86–0.93 (m, 3 H, CH3), 1.28–1.36 (m, 4 H, 2 x CH2),
1.38–1.90 (m, 10 H, 5 x CH2), 2.37 (d, J = 2.0 Hz, 1 H, CH, A), 2.43 (d, J = 2.2 Hz,
1 H, CH, B), 3.49–3.58 (A), 3.76–3.86 (B), 3.97–4.07 (m, 2 H, CH2), 4.28 (dt, J =
6.7, 2.2 Hz, 1 H, CH, B), 4.41 (dt, J = 6.7, 2.0 Hz, 1 H, CH, A), 4.75 (m, 1 H, CH,
B), 4.98 (m, 1 H, CH, A) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.0 (CH3), 19.1 (B) / 19.3(A) (CH2), 22.5 (CH2), 24.7
(B) / 25.0 (A) (CH2), 25.4 (B) / 25.5 (A) (CH2), 30.5 (CH2), 31.5 (CH2), 35.6
(CH2), 62.3 (CH2), 64.8 (A) / 67.1 (B) (CH), 72.5 (B) / 73.1 (A) (CH), 83.0 (C),
95.6 (A) / 98.2 (B) (CH) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.94
5.3.2.3. 3-Tetrahydropyran-2-yloxy-1-octanal (47a)
3-(Tetrahydropyran-2-yloxy)-1-octin 47 (210 mg, 1 mmol) und und [η5-
Cyclopentadienalruthenium(II) bis-(2-(diphenylphosphino)-6-(2,4,6-triphenyl)pyridin)
(acetonitril)] hexafluorophosphat 22 (149 mg, 0.1 mmol, 10 mol%) wurden in einem
Gemisch aus Aceton (2 mL) und Wasser (90 mg, 5 mmol, 5 Äquiv) gelöst. Das
Lösungsmittelvolumen wurde markiert bevor weiteres Aceton (2 mL/mmol) zugeben und
zur Entgasung der Lösung bis zur Markierung im Vakuum wieder eingeengt wurde.
Danach wurde die Lösung bei 50 °C für 17.5 h unter Argonatmosphäre gerührt.
Anschließend engte man die Lösung ein und nach Säulenchromatographie (TBME/Hexan,
1:10) erhielt man den Aldehyd 47a (100.5 mg, 0.44 mmol, 44%) als ein Gemisch aus zwei
Diastereomeren.

5. Experimenteller Teil
98
Rf = 0.2 (TBME/Hexan, 1:10)
Diastereomer A 1H-NMR (CDCl3, 400 MHz): δ = 0.86–0.93 (m, 3 H, CH3), 1.27–1.35 (m, 4 H, 2 x CH2),
1.48–1.86 (m, 10 H, 5 x CH2), 2.50–2.72 (m, 2 H, CH2), 3.44–3.55 / 3.79–3.94 (m,
2 H, CH2), 4.15 (m, 1 H, CH), 4.68–4.72 (m, 1 H, CH), 9.80–9.85 (m, 1 H, CH)
ppm. 13C-NMR (CDCl3, 100 MHz): δ = 14.3 (CH3), 20.0 (CH2), 22.8 (CH2), 25.4 (CH2), 25.6
(CH2), 31.2 (CH2), 32.0 (CH2), 35.9 (CH2), 48.3 (CH2), 63.0 (CH2), 72.5 (CH), 97.9
(CH), 201.7 (CH) ppm.
Diastereomer B 1H-NMR (CDCl3, 400 MHz): δ = 0.86–0.93 (m, 3 H, CH3), 1.27–1.35 (m, 4 H, 2 x CH2),
1.48–1.86 (m, 10 H, 5 x CH2), 2.50–2.72 (m, 2 H, CH2), 3.44–3.55 / 3.79–3.94 (m,
2 H, CH2), 4.07 (m, 1 H, CH), 4.66 (m, 1 H, CH), 9.80–9.85 (m, 1 H, CH) ppm. 13C-NMR (CDCl3, 100 MHz): δ = 14.3 (CH3), 20.2 (CH2), 22.8 (CH2), 25.1 (CH2), 25.6
(CH2), 31.2 (CH2), 32.0 (CH2), 34.8 (CH2), 49.5 (CH2), 63.2 (CH2), 73.1 (CH), 98.9
(CH), 202.2 (CH) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.95
5.3.2.4. 3-tert-Butyldimethylsilyloxy-1-octin (48)
Gemäß AAV4 ausgehend von 1-Octin-3-ol (2.91 mL, 20 mmol) wurde der Silylether als
gelbe Flüssigkeit (4.32 g, 90% Ausbeute) erhalten.
Rf = 0.82 (TBME/Hexan, 1:30).
IR (film): ν = 3310, 2934, 2859, 1466, 1342, 1254, 1090, 838, 779, 655, 629 cm–1. 1H-NMR (400 MHz, CDCl3): δ = 0.12 (d, J = 9.34 Hz, 6 H, 2 x CH3), 0.90 (s, 12 H, 3 x
CH3), 1.19–1.36 (m, 4 H, 2 x CH2), 1.36–1.47 (m, 2 H, CH2), 1.63–1.70 (m, 2 H,
CH2), 2.37 (d, J = 1.92 Hz, 1 H, CH), 4.33 (dt, J = 6.59, 1.92 Hz, 1 H, CH) ppm. 13C-NMR (100 MHz, CDCl3): δ = –4.9 (CH3), –4.4 (CH3), 14.1 (CH3), 18.4 (C), 22.7

5. Experimenteller Teil
99
(CH3), 24.9 (CH3), 25.9 (3 x CH3), 31.5 (CH2), 38.6 (CH2), 62.8 (CH), 71.8 (CH),
85.8 (C) ppm.
MS (EI, 70 eV): m/z (%) = 239.1 (26) [M+], 215.1 (20), 173.0 (19), 127.0 (53), 113.0 (28),
83.0 (61), 75.1 (100).
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.96
5.3.2.5. 3-Benzoyloxy-1-octin (49) OBz
Die Verbindung wurde gemäß Literaturvorschrift,97 wie folgt, hergestellt: 1-Octin-3-ol
(1.43 mL, 10 mmol) wurde zusammen mit Triethylamin (2.5 mL, 18 mmol) und DMAP
(61 mg, 1 mmol) in DCM (10 mL) vorgelegt und mit Benzoylchlorid (1.6 mL, 14 mmol)
versetzt. Nach drei Stunden rühren bei RT wurde die Reaktion durch Zugabe von dest.
Wasser (20 mL) und TBME (60 mL) abgebrochen. Die organische Phase wurde zweimal
mit wässriger NaOH-Lösung (2 M, 20 mL) und danach mit dest. Wasser (20 mL)
gewaschen. Die wässrige Phase wurde rückextrahiert und die vereinigten organischen
Phasen wurden über MgSO4 getrocknet, filtriert und eingeengt. Nach
Säulenchromatographie (TBME/Hexan, 1:25) erhielt man ein gelbliches Öl (2.2 g,
9.6 mmol, 95%).
Rf = 0.29 (TBME/Hexan, 1:25). 1H-NMR (CDCl3, 300 MHz): δ = 0.85–0.91 (m, 3 H, CH3), 1.30–1.45 (m, 4 H, 2 x CH2),
1.48–1.61 (m, 2 H, CH2), 1.86–1.98 (m, 2 H, CH2), 2.48 (d, J = 2.2 Hz, 1 H, CH),
5.61 (dt, J = 6.4, 2.2 Hz, 1 H, CH), 7.44 (m, 2 H, 2 x CH), 7.57 (m, 1 H, CH), 8.07
(m, 2 H, 2 x CH) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.0 (CH3), 22.5 (CH2), 24.6 (CH2), 31.3 (CH2), 34.7
(CH2), 64.4 (CH), 73.6 (CH), 81.4 (C), 128.4 (CH), 129.8 (CH), 129.9 (C), 133.2
(C), 165.5 (C) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.98

5. Experimenteller Teil
100
5.3.2.6. 3-Methoxymethyloxy-1-octin (50) OMOM
Die Verbindung wurde gemäß Literaturvorschrift,99 wie folgt, hergestellt: 1-Octin-3-ol
(2.92 mL, 20 mmol) wurde zusammen mit Lithiumbromid (434 mg, 5 mmol) und pTsOH
(305 mg, 1.6 mmol) in Toluol (10 mL) gelöst und mit Dimethoxymethan (10 mL) versetzt.
Nach 48 h rühren bei RT beendete man die Reaktion durch Zugabe von 25%iger wässriger
Ammoniaklösung (5 mL), dest. Wasser (20 mL) und TBME (20 mL). Die organische
Phase wurde zweimal mit dest. Wasser (15 mL) gewaschen, die wässrige Phase
rückextrahiert und die vereinigten organischen Phasen über MgSO4 getrocknet, filtriert und
eingeengt. Nach Säulenchromatographie (TBME/Hexan, 1:10) des Rückstandes erhielt
man das Produkt als farblose Flüssigkeit (2.5 g, 14.7 mmol, 74%).
Rf = 0.61 (TBME/Hexan, 1:20). 1H-NMR (CDCl3, 300 MHz): δ = 0.87–0.93 (m, 3 H, CH3), 1.28–1.37 (m, 4 H, 2 x CH2),
1.42–1.54 (m, 2 H, CH2), 1.69–1.79 (m, 2 H, CH2), 2.40 (d, J = 2.22 Hz, 1 H, CH),
3.38 (s, 3 H, CH3), 4.32 (dt, J = 6.68, 2.23 Hz, 1 H, CH), 4.61 (d, J = 6.93 Hz, 1 H,
CH2), 4.93 (d, J = 6.68 Hz, 1 H, CH2) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.5 (CH3), 22.7 (CH2), 24.0 (CH2), 31.6 (CH2), 35.7
(CH2), 55.7 (CH3), 65.6 (CH), 73.5 (CH), 82.8 (C), 94.3 (CH2) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.100
5.3.2.7. 3-Methoxy-1-octin (51)
Die Verbindung wurde gemäß Literaturvorschrift,101 wie folgt, hergestellt: 1-Octin-3-ol
(0.146 mL, 1 mmol) wurde zusammen mit Methyliodid (0.125 mL, 2 mmol) und
Kaliumhydroxid (0.224 g, 4 mmol) in DMSO (2 mL) gelöst. Die Lösung ließ man für 1 h
bei RT rühren bevor man die Reaktion durch Zugabe von ges. wässriger NaHCO3-Lösung
(10 mL) beendete und mit TBME (20 mL) verdünnte. Die organische Phase wurde
nochmals mit ges. wässriger NaHCO3-Lösung (10 mL) und danach mit dest. Wasser (20
mL) gewaschen. Nach Rückextraktion der wässrigen Phase und trocknen der vereinigten
organischen Phasen über MgSO4, filtrieren und einengen konnte man durch

5. Experimenteller Teil
101
Säulenchromatographie (TBME/Hexan, 1:30) das Produkt als eine leicht gelbliche
Flüssigkeit (122.5 mg, 0.874 mmol, 87%) erhalten.
Rf = 0.65 (TBME/Hexan, 1:20). 1H-NMR (CDCl3, 400 MHz): δ = 0.90 (t, J = 6.9 Hz, 3H, CH3), 1.24–1.36 (m, 4 H, 2 x
CH2), 1.41–1.49 (m, 2 H, CH2), 1.63–1.79 (m, 2 H, CH2), 2.43 (d, J = 2.2 Hz, 1 H,
CH), 3.41 (s, 3 H, CH3), 3.93 (dt, J = 6.6, 1.9 Hz, 1H, CH) ppm. 13C-NMR (CDCl3, 100 MHz): δ = 14.1 (CH3), 22.7 (CH2), 24.9 (CH2), 31.6 (CH2), 35.6
(CH2), 56.5 (CH3), 71.2 (CH), 73.7 (CH), 82.8 (C) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.102
5.3.2.8. 3-Methoxymethyloxyoctanal (52)
3-Methoxymethyloxy-1-octin 50 (170.3 mg, 1 mmol) und [η5-
Cyclopentadienalruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenyl)pyridin)
(acetonitril)] hexafluorophosphat 22 (148.7 mg, 0.1 mmol, 10 mol%) wurden in einem
Gemisch aus Aceton (2 mL) und dest. Wasser (90 mg, 5 mmol, 5 Äquiv) gelöst. Das
Lösungsmittelvolumen wurde markiert bevor weiteres Aceton (2 mL/mmol) zugeben und
zur Entgasung der Lösung bis zur Markierung im Vakuum wieder eingeengt wurde.
Danach wurde die Lösung bei 50 °C für 17.5 h unter Argonatmosphäre gerührt und
anschließend eingeengt, so dass man nach Säulenchromatographie (TBME/Hexan, 1:10)
den Aldehyd als farbloses Öl (118.6 mg, 0.63 mmol, 63%) erhielt.
Rf = 0.17 (TBME–Hexan, 1:10). 1H-NMR (CDCl3, 400 MHz): δ = 0.87–0.93 (m, 3 H, CH3), 1.27–1.39 (m, 6 H, 3 x CH2),
1.49–1.66 (m, 2 H, CH2), 2.60 (ddd, J = 16.2, 6.0, 2.8 Hz, 2 H, CH2), 3.36 (s, 3 H,
CH3), 4.08 (dt, J = 6.3, 6.0 Hz, 1 H, CH), 4.67 (q, J = 6.9, 6.3 Hz, 1 H, CH2), 9.80–
9.82 (m, 1 H, CH) ppm. 13C-NMR (CDCl3, 100 MHz): δ = 14.2 (CH3), 22.8 (CH2), 25.1 (CH2), 31.9 (CH2), 35.1
(CH2), 48.9 (CH2) 55.8(CH3), 73.3 (CH), 95.9 (CH2), 201.4 (CH) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.103

5. Experimenteller Teil
102
5.3.2.9. 5-Methoxymethyloxy-3-hydroxy-decansäure-tert-butylester (53)
In einem Schlenkrohr wurden unter Argon 3-Methoxymethyloxy-1-octin 50 (85 mg, 0.5
mmol) und [η5-Cyclopentadienalruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) (acetonitril)] hexafluorophosphat 22 (37 mg, 0.025 mmol, 5
mol%) in einem Gemisch aus Aceton (1 mL) und Wasser (45 mg, 2.5 mmol, 5 Äquiv)
gelöst. Das Lösungsmittelvolumen wurde markiert bevor weiteres Aceton (2 mL/mmol)
zugeben und zur Entgasung der Lösung bis zur Markierung im Membranpumpenvakuum
wieder eingeengt wurde. Danach ließ man die Lösung bei 50 °C für 6 h unter
Argonatmosphäre rühren, engte sie ein und nahm den Rückstand in abs. THF (1 mL) auf.
Zu der Lösung tropfte man langsam folgende frisch hergestellte Lösung: Bei 0 °C legte
man unter Argon Diisopropylamin (0.354 mL, 2.5 mmol) in abs. THF (10 mL) vor und
versetzte es langsam mit n-Butyllithium (1.57 mL, 2.5 mmol). Danach wurde die Lösung
auf –78 °C abgekühlt, tert-Butylacetat (0.338 mL, 2.5 mmol) zu getropft und für 30 min
gerührt. Nach vollständigem Zutropfen der Reaktionslösung aus dem ersten Schritt
beendete man die Reaktion nach 10 min bei –78 °C durch Zugabe von ges. wässriger
NH4Cl-Lösung (5 mL), dest. Wasser (20 mL) und TBME (25 mL). Die organische Phase
wurde mit dest. Wasser (20 mL) gewaschen, die wässrige Phase rückextrahiert und die
vereinigten organischen Phasen über MgSO4 getrocknet, filtriert und eingeengt. Nach
Säulenchromatographie (TBME/Hexan, 1:5) erhielt man den Ester 53 (104 mg, 0.34 mmol,
68%).
Rf = 0.33 und 0.26 (TBME/Hexan, 1:3).
IR (film): ν = 3472, 2932, 2349, 1729, 1463, 1369, 1256, 1154, 954, 917, 844, 761, 608,
529 cm–1. 1H-NMR (CDCl3, 300 MHz): δ = 0.85–0.92 (m, 3 H, CH3), 1.24–1.36 (m, 6 H, 3 x CH2),
1.46 (s, 9 H, 3 x CH3) 1.49–1.82 (m, 4 H, 2 x CH2), 2.38 (d, J = 6.4 Hz, 2 H, CH2),
2.41–2.44 (m, 0.5 H, OH), 3.40 (d, J = 7.9, 3 H, CH3), 3.41–3.46 (m, 0.5 H, OH),
3.72–3.87 (m, 1 H, CH), 4.08–4.28 (m, 1 H, CH), 4.62–4.72 (m, 2 H, CH2) ppm. 13C NMR (CDCl3, 75 MHz): δ = 14.1 (CH3), 22.7 (CH2), 24.7/25.0 (CH2), 28.2 (3 x CH3),
32.1 (CH2), 34.3/35.0 (CH2), 40.9/41.2 (CH2), 42.8/43.1 (CH2), 55.9 (CH3),
65.0/66.9 (CH), 75.5/76.4 (CH), 81.1 (C), 95.3/96.4 (CH2), 172.0 (C) ppm.
MS (EI, 70 eV): m/z (%) = 231.4 (5) [M+ - OtBu], 217.3 (11), 203.3 (11), 177.2 (23),

5. Experimenteller Teil
103
145.2 (89), 127.2 (22), 115.2 (62), 57.3 (86), 45.3 (100).
Elementaranalyse (C16H32O5): Ber.: C 63.13, H 10.60; Gef.: C 62.91, H 10.57.
5.3.2.10. 6-Pentyl-5,6-dihydro-2-pyron oder Massoialacton (54)
Der 5-Methoxymethyloxy-3-hydroxy-decansäure-tert-butylester 53 (55 mg, 0.18 mmol)
wurde in 5 mL Triflouressigsäure bei RT für 7 d gerührt. Die Reaktion wurde durch die
Zugabe von 50%iger NaOH (3 g, 37.5 mmol) und TBME (20 mL) beendet. Die organische
Phase wurde mit dest. Wasser (20 mL) gewaschen, die wässrige Phase wurde
rückextrahiert und die vereinigten organischen Phasen wurden über MgSO4 getrocknet,
filtriert und eingeengt. Nach Säulenchromatographie (TBME/Hexan, 1:3) erhielt man das
Lacton 54 (23.7 mg, 0.127 mmol, 71% Ausbeute).
Rf = 0.24 (TBME/Hexan, 1:3). 1H-NMR (CDCl3, 300 MHz): δ = 0.90 (t, J = 6.9 Hz, 3 H, CH3), 1.19–1.81 (br m, 8 H, 4
x CH2), 2.29–2.36 (m, 2 H, CH2), 4.36–4.48 (m, 1 H, CH), 6.02 (dd, J = 9.5, 1.5 Hz,
1 H, CH), 6.83–6.92 (m, 1 H, CH) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.104
5.3.2.11. 5-Methoxymethyloxy-3-hydroxy-1-decin (55)
3-Methoxymethyloxy-1-octin 50 (170.2 mg, 1 mmol) und [η5-
Cyclopentadienalruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin)
(acetonitril)] hexafluorophosphat 22 (74.4 mg, 0.05 mmol, 5 mol%) wurden unter Argon in
einem entgasten Lösungsmittelgemisch aus Aceton (3 mL) und Wasser (1 mL, 55.5 mmol,
55.5 Äquiv) gelöst. Danach ließ man die Lösung bei 50 °C für 3.5 h rühren, bevor man sie
einengte und den Rückstand in abs. THF (4 mL) aufnahm. Zu der Lösung gab man bei
0 °C (Eisbad) Ethinylmagnesiumchlorid-Lösung (10 mL, 5 mmol, 5 Äquiv) und rührte für
40 min. Die Reaktion beendete man durch die Zugabe von ges. wässriger NH4Cl-Lösung
(25 mL) und verdünnte mit TBME (50 mL). Die organische Phase wurde mit ges.
wässriger NaHCO3-Lösung (25 mL) und zweimal mit dest. Wasser (je 25 mL) gewaschen.

5. Experimenteller Teil
104
Die wässrige Phase wurde rückextrahiert und die vereinigten organischen Phasen wurden
MgSO4 getrocknet, filtriert und eingeengt. Nach Säulenchromatographie (TBME/Hexan,
1:10→1:3→1:0) erhielt man das Alkin 55 als hellgelbes Öl (120.1 mg, 0.735 mmol, 74%).
Rf = 0.32 (TBME/Hexan, 1:3).
IR (film): ν = 3429, 3308, 2931, 2859, 2114, 1716, 1622, 1577, 1541, 1465, 1380, 1307,
1217, 1151, 1098, 1037, 918, 758, 664, 632, 561 cm–1. 1H-NMR (CDCl3, 300 MHz): δ = 0.86–0.93 (m, 3 H, CH3), 1.25–1.39 (m, 6 H, 3 x CH2),
1.47–1.65 (m, 2 H, CH2), 1.83–2.07 (m, 2 H, CH2), 2.49 (dd, J = 7.9, 2.2 Hz, 1 H,
CH), 2.90 (br s, 1 H, OH), 3.41 (d, J = 7.2 Hz, 3 H, CH3), 3.76– 3.99 (m, 1 H, CH),
4.54–4.73 (m, 3 H, CH, CH2) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.1 (CH3), 22.7 (CH2), 24.6/24.8 (CH2), 32.0 (CH2),
34.5/34.6 (CH2), 41.7/42.5 (CH2), 55.9/56.1 (CH3), 59.6/61.0 (CH), 72.7/73.1 (CH),
75.7/76.2 (CH), 84.7 (C), 95.6/96.3 (CH2) ppm.
MS (EI, 70 eV): m/z (%) = 183.3 (2) [M+ – CH3O], 169.3 (1), 145.3 (13), 111.2 (100),
99.2 (21), 83.2 (14), 55.3 (42), 45.3 (90).
Elementaranalyse (C12H22O3): Ber.: C 67.26, H 10.35; Gef.: C 67.24, H 10.34.
5.3.2.12. 3,5-Methylendioxy-1-decin (56)
5-Methoxymethyloxy-3-hydroxy-1-decin 55 (630 mg, 2.94 mmol) wurde in
Dimethoxymethan (10 mL) gelöst und mit TfOH (52 μL, 0.59 mmol) versetzt. Nach 5 min
rühren bei RT beendete man die Reaktion durch Zugabe von 25%iger wässriger NH3-
Lösung (10 mL), dest. Wasser (30 mL) und TBME (50 mL). Die organische Phase wurde
zweimal mit dest. Wasser (je 50 mL) gewaschen, die wässrige Phase rückextrahiert und die
vereinigten organischen Phasen über MgSO4 getrocknet, filtriert und eingeengt. Nach
Säulenchromatographie (TBME/Hexan, 1:10) des Rückstandes erhielt man das farblose
und flüssige Produkt 56 (474.6 mg, 2.60 mmol, 89%).
Rf = 0.59 und 0.5 (TBME/Hexan, 1:10).
IR (film): ν = 3307, 2930, 2858, 2770, 2127, 1587, 1542, 1465, 1402, 1379, 1362, 1257,
1235, 1215, 1183, 1138, 1111, 1032, 982, 945, 901, 843, 809, 758, 667, 633, 569,
517 cm–1.

5. Experimenteller Teil
105
1H-NMR (CDCl3, 300 MHz): δ = 0.86–0.93 (m, 3 H, CH3), 1.24–1.67 (m, 8 H, 4 x CH2),
1.75–2.02 (m, 2 H, CH2), 2.55 (dd, J = 16.6, 2.2 Hz, 1 H, CH), 3.48–3.59 (m, 0.5 H,
CH), 3.86–3.96 (m, 0.5 H, CH), 4.36 (dt, J = 7.2, 2.2 Hz, 0.5 H, CH), 4.70 (d, J =
6.4 Hz, 0.5 H, CH2), 4.82–4.87 (m, 0.5 H, CH), 4.89 (d, J = 6.4 Hz, 0.5 H, CH2),
5.08 (d, J = 6.4 Hz, 0.5 H, CH2), 5.23 (d, J = 6.4 Hz, 0.5 H, CH2) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.0 (CH3), 22.5 (CH2), 24.4/24.5 (CH2), 31.6/31.7
(CH2), 35.5/35.6 (CH2), 36.2/37.9 (CH2), 63.4/66.4 (CH), 72.5/76.2 (CH),
73.4/75.8 (CH), 81.0/81.6 (C), 88.7/93.4 (CH2) ppm.
MS (EI, 70 eV): m/z (%) = 181.4 (26) [M]+, 145.3 (25), 111.3 (44), 99.2 (21), 71.4 (14),
55.3 (43), 45.4 (100).
Elementaranalyse (C11H18O2): Ber.: C 72.49, H 9.95; Gef.: C 72.35, H 9.91.
5.3.2.13. 6,8-Methylendioxy-4-hydroxy-1-tridecen (57)
3,5-Methylendioxy-1-decin 56 (300 mg, 1.65 mmol) und [η5-
Cyclopentadienalruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin)
(acetonitril)] hexafluorophosphat 22 (97.9 mg, 0.066 mmol, 4 mol%) wurden unter Argon
in einem entgasten Lösungsmittelgemisch aus Aceton (4.94 mL) und Wasser (1.65 mL,
91.6 mmol, 55.5 Äquiv) gelöst. Danach ließ man die Lösung bei 50 °C für 5 h rühren,
bevor man entgastes DCM (15 mL) zugab um eine Phasentrennung zwischen organischer
und wässriger Phase einzustellen. Die organische Phase wurde via Kanüle in einen anderen
Schlenkkolben überführt, die wässrige Phase weitere zweimal mit DCM (5 mL) gewaschen
und die vereinigten organischen Phasen wurden eingeengt. Den Rückstand nahm man in
abs. Ether (6.6 mL) auf, versetzte ihn mit MgSO4 und kühlte auf 0 °C (Eisbad) ab. Zu der
Lösung gab man bei 0 °C Allylmagnesiumbromid (0.8 M in Ether, 11.2 mL, 8.9 mmol, 5
Äquiv), welches frisch nach Literaturvorschrift hergestellt wurde,105 und rührte für 60 min.
Die Reaktion beendete man durch die vorsichtige Zugabe von ges. wässriger NH4Cl-
Lösung (10 mL) und dest. Wasser (10 mL) und verdünnte mit TBME (30 mL). Die
organische Phase wurde mit ges. wässriger NaHCO3-Lösung (20 mL) und zweimal mit
dest. Wasser (je 25 mL) gewaschen, die wässrige Phase rückextrahiert und die vereinigten
organischen Phasen über MgSO4 getrocknet, filtriert und eingeengt. Nach
Säulenchromatographie (TBME/Hexan, 1:10) erhielt man den Allylalkohol 57 als farblose
Flüssigkeit (291.7 mg, 1.20 mmol, 73%).

5. Experimenteller Teil
106
Rf = 0.23 (TBME/Hexan, 1:10).
IR (film): ν = 3450, 3076, 2929, 2857, 2775, 2676, 1641, 1463, 1434, 1407, 1383, 1355,
1238, 1177, 1133, 1080, 1027, 914, 802 cm–1. 1H-NMR (CDCl3, 300 MHz): δ = 0.86–0.93 (m, 3 H, CH3), 1.23–1.97 (br m, 10 H, 5 x
CH2), 2.15–2.35 (m, 2 H, CH2), 2.90–3.10 (br s, 1 H, OH), 3.51–3.62 (m, 0.5 H,
CH), 3.78–4.01 (m, 2 H, 2 x CH), 4.09–4.26 (m, 0.5 H, CH), 4.69–4.75 (m, 1 H,
CH2), 4.85–4.94 (m, 1 H, CH), 5.06–5.18 (m, 2 H, CH2), 5.75–5.92 (m, 1 H, CH)
ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.0 (CH3), 22.6 (CH2), 24.5/24.6/25.2 (CH2),
31.7/32.2/32.3 (CH2), 34.9/35.0/35.8/35.9 (CH2), 37.6/37.8/39.7/40.0 (CH2),
41.8/41.90/41.95/42.16/42.2 (2 x CH2), 67.0/68.3 (CH), 70.3/70.5/71.8/72.1 (CH),
73.8 (CH), 86.9/87.0/93.2/93.4 (CH2), 117.6/117.7/118.0/118.2 (CH2), 134.7 (CH)
ppm.
MS (EI, 70 eV): m/z (%) = 241.4 (2) [M]+, 201.4 (62), 171.3 (82), 153.3 (79), 127.3 (94),
109.3 (100), 97.2 (33), 83.3 (69), 71.3 (78), 55.3 (84), 45.3 (17).
Elementaranalyse (C14H26O3): Ber.: C 69.38, H 10.81; Gef.: C 69.27, H 10.63.
5.3.2.14. 6,8-Methylendioxy-4-acryloyloxy-1-tridecen (58)
6,8-Methylendioxy-4-hydroxy-1-tridecen 57 (260 mg, 1.073 mmol) wurde in DCM (2 mL)
gelöst und mit Acryloylchlorid (0.131 mL, 1.61 mmol), Triethylamin (0.446 mL, 3.22
mmol) und DMAP (26 mg, 0.21 mmol) versetzt. Nach 10 min bei RT beendete man die
Reaktion durch Zugabe von ges. wässriger NaHCO3-Lösung (15 mL), dest. Wasser (10 mL)
und DCM (50 mL). Die organische Phase wurde mit dest. Wasser (20 mL) gewaschen, die
wässrige Phase rückextrahiert und die vereinigten organischen Phasen über MgSO4
getrocknet, filtriert und eingeengt. Nach Säulenchromatographie (TBME/Hexan, 1:5)
erhielt man das Dien 58 als farblose Flüssigkeit (234.2 mg, 0.79 mmol, 74%).
Rf = 0.8 und 0.68 (TBME/Hexan, 1:3).
IR (film): ν = 3433, 3078, 2930, 2857, 2774, 1724, 1639, 1463, 1435, 1406, 1295, 1271,
1194, 1136, 1033, 988, 917, 808 cm–1. 1H-NMR (CDCl3, 400 MHz): δ = 0.86–0.92 (m, 3 H, CH3), 1.21–1.34 (m, 8 H, 4 x

5. Experimenteller Teil
107
CH2), 1.37–1.84 (m, 3 H, 1.5 x CH2), 1.92–2.23 (m, 1 H, CH2), 2.31–2.48 (m, 1 H,
CH2), 3.47–3.68 (m, 2 H, 2 x CH), 3.79–4.05 (m, 1 H, CH), 4.61–4.66 (m, 1 H,
CH2), 4.81–4.92 (m, 1 H, CH2), 5.01–5.28 (m, 2 H, CH, CH2), 6.06–6.16 (m, 1 H,
CH), 6.36–6.43 (m, 1 H, CH2) ppm. 13C-NMR (CDCl3, 100 MHz): δ = 14.1 (CH3), 22.7 (CH2), 24.68/24.70/25.1/25.2 (CH2),
31.8–40.4 (5 x CH2), 68.2/68.7/70.4/70.6 (CH), 70.1/70.2/71.7/71.7 (CH),
73.0/73.6/76.41/76.45 (CH), 87.1/87.2/93.4/93.5 (CH2), 118.0/118.1 (CH2), 128.6
(CH), 130.55/130.61 (CH2), 133.2 (CH), 165.6 (C) ppm.
MS (EI, 70 eV): m/z (%) = 295.4 (3) [M+], 255.4 (13), 225.4 (40), 195.3 (20), 183.3 (32),
157.3 (56), 153.3 (81), 135.3 (44), 123.3 (53), 109.3 (75), 97.3 (44), 79.3 (49), 67.3
(58), 55.3 (100).
Elementaranalyse (C17H28O4): Ber.: C 68.89, H 9.52; Gef.: C 68.77, H 9.24.
5.3.2.15. 5,6-Dihydro-6-(2,4-methylendioxynonyl)-pyran-2-on (59)
Man legt das Acylester 58 (100 mg, 0.337 mmol) in entgastem DCM (50 mL) vor und
versetzt es mit GRUBBS I Katalysator63 (27.8 mg, 10 mol%). Nachdem die Lösung für 4 h
am Refluxieren gehalten worden war entfernte man das Lösungsmittel im
Membranpumpenvakuum und erhielt nach Säulenchromatographie (TBME/Hexan, 1:1) ein
Diastereomerengemisch des Pyranons als farblose Flüssigkeit [(A) 52.6 mg, (B) 24.6 mg,
0.288 mmol, 85%)].
Diastereomer A
Rf = 0.325 (TBME/Hexan, 1:1).
IR (film): ν = 3438, 2930, 2858, 2774, 1725, 1465, 1426, 1385, 1249, 1195, 1142, 1029,
958, 819, 731, 664, 553 cm–1. 1H-NMR (CDCl3, 300 MHz): δ = 0.89 (t, J = 6.7 Hz, 3 H, CH3), 1.23–1.50 (m, 7 H, 3.5
x CH2), 1.51–1.65 (m, 2 H, CH2), 1.71–1.94 (m, 2 H, CH2), 2.07–2.18 (m, 1 H,
CH2), 2.25–2.55 (m, 2 H, CH2), 3.51–3.62 (m, 1 H, CH), 3.83–4.02 (m, 1 H, CH),
4.58–4.77 (m, 2 H, CH, CH2), 5.03–5.06 (m, 1 H, CH2), 5.99–6.06 (m, 1 H, CH),
6.85–6.95 (m, 1 H, CH) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.0 (CH3), 22.5 (CH2), 24.5 (CH2), 29.2/29.9 (CH2)

5. Experimenteller Teil
108
31.7 (CH2), 35.8 (CH2), 37.2/37.9 (CH2), 40.3/41.6 (CH2), 71.7/72.2 (CH),
73.9/74.5 (CH), 76.4 (CH), 93.3 (CH2), 121.2/121.4 (CH), 145.2/145.3 (CH), 164.3
(C) ppm.
MS (EI, 70 eV): m/z (%) = 269.4 (20) [M+], 250.3 (3), 238.4 (3), 221.4 (4), 197.3 (67),
183.3 (25), 167.2 (95), 139.3 (82), 109.3 (27), 97.2 (100). 68.3 (62), 55.3 (68).
Elementaranalyse (C15H24O4): Ber.: C 67.14, H 9.01; Gef.: C 66.98, H 8.84.
Diastereomer B
Rf = 0.225 (TBME/Hexan, 1:1).
IR (film): ν = 3435, 2930, 2859, 2782, 1723, 1462, 1430, 1387, 1247, 1183, 1135, 1018,
952, 818, 755, 726, 663, 554 cm–1. 1H-NMR (CDCl3, 300 MHz): δ = 0.82–0.94 (m, 3 H, CH3), 1.21–1.62 (m, 8 H, 4 x CH2),
1.63–1.99 (m, 4 H, 2 x CH2), 2.29–2.49 (m, 2 H, CH2), 3.84–3.98 (m, 1 H, CH),
4.12–4.28 (m, 1 H, CH), 4.56–4.76 (m, 1 H, CH), 4.78–4.96 (m, 2 H, CH2), 5.98–
6.07 (m, 1 H, CH), 6.85–6.95 (m, 1 H, CH) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.0 (CH3), 22.7 (CH2), 25.1/25.3 (CH2), 28.9/29.9
(CH2), 31.6 (CH2), 32.5 (CH2), 34.5/34.9 (CH2), 37.9/40.1 (CH2), 67.1/67.4 (CH),
71.7/71.7 (CH), 74.1/74.8 (CH), 86.9/87.0 (CH2), 121.3/121.4 (CH), 145.1/145.2
(CH), 164.3 (C) ppm.
MS (EI, 70 eV): m/z (%) = 269.4 (26) [M+], 250.3 (2), 239.3 (12), 221.3 (5), 197.2 (73),
183.3 (27), 167.2 (96), 149.2 (40), 139.3 (79), 109.3 (48), 97.2 (100). 68.3 (71),
55.3 (81).
Elementaranalyse (C15H24O4): Ber.: C 67.14, H 9.01; Gef.: C 66.90, H 8.80.
5.3.2.16. 1,3-Dibenzoyloxy-octan (61)
Man löste unter einer Argon-Schutzgasatmosphäre in einem Schlenkrohr 3-
(Tetrahydropyran-2-yloxy)-1-octin 47 (100 mg, 0.475 mmol) und [η5-
Cyclopentadienalruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin)
(acetonitril)] hexafluorophosphat 22 (35.3 mg, 0.024 mmol, 5 mol%) in einem Gemisch
aus Aceton (0.95 mL) und Wasser (43 mg, 2.375 mmol, 5 Äquiv). Das
Lösungsmittelvolumen wurde markiert bevor man weiteres Aceton (2 mL/mmol) zugab
und zur Entgasung der Lösung bis zur Markierung im Vakuum wieder eingeengte. Danach

5. Experimenteller Teil
109
ließ man die Lösung bei 50 °C für 17.5 h unter Argonatmosphäre rühren. Anschließend
engte man die Lösung ein, nahm den Rückstand in Methanol (4.5 mL) auf und gab bei
0 °C (Eisbad) Natriumborhydrid (46.7 mg, 1.235 mmol, 2.6 Äquiv) zu. Nach 15 min
beendete man diese Reaktion durch die Zugabe von ges. wässriger NaCl-Lösung (30 mL)
und TBME (30 mL). Die wässrige Phase wurde rückextrahiert und die vereinigten
organischen Phasen über Na2SO4 getrocknet, filtriert und eingeengt.
Den Rückstand löste man in Methanol (5 mL) und wässriger HCl (2.4 M, 2.5 mL) und
rührte es für 20 min bei RT. Die Reaktionslösung wurde mit TBME (20 mL) in den
Scheidetrichter überführt und die wässrige Phase abgetrennt. Die organische Phase wurde
zweimal mit ges. wässriger NaHCO3-Lösung (je 20 mL) gewaschen, die wässrige Phase
rückextrahiert und die vereinigten organischen Phasen über MgSO4 getrocknet, filtriert und
eingeengt. Man versetzte den Rückstand zweimal mit Toluol (10 mL) und eingeengte ihn
wieder ein um das Methanol azeotropisch zu entfernen.
Nun wurde der Rückstand mit Dichlormethan (4.5 mL), Et3N (0.99 mL, 7.125 mmol,
15 Äquiv), DMAP (116.1 mg, 0.95 mmol, 2 Äquiv) und Benzoylchlorid (0.552 mL,
4.75 mmol, 10eq.) versetzt. Nach 70 min rühren bei RT beendete man die Reaktion durch
Zugabe von 25%iger wässriger NH3-Lösung (10 mL) und verdünnte mit TBME (30 mL).
Die organische Phase wurde mit wässriger HCl (2.4 M, 10 mL), ges. wässriger NaHCO3
(30 mL) und dest. Wasser (30 mL) gewaschen, über MgSO4 getrocknet, filtriert und
eingeengt. Nach Säulenchromatographie (TBME/Hexan, 1:3) des Rückstandes erhielt man
das geschützte Diol 61 als hell bräunliches Öl (41 mg, 0.116 mmol, 24 %).
Rf = 0.81 (TBME/Hexan, 1:3).
IR (film): ν = 2931, 2861, 1719, 1602, 1451, 1313, 1272, 1107, 1070, 1026, 711 cm-1. 1H-NMR (CDCl3, 300 MHz): δ = 0.83–0.93 (m, 3 H, CH3), 1.25–1.47 (m, 6 H, 3 x CH2),
1.65–1.97 (m, 2 H, CH2), 2.18 (q, J = 6.4 Hz, 2 H, CH2), 4.34–4.53 (m, 2 H, CH2),
5.36 (dt, J = 5.9 Hz, 1 H, CH), 7.35–7.48 (m, 4 H, Ar-H), 7.49–7.58 (m, 2 H, Ar-H),
7.97–8.09 (m, 4 H, Ar-H) ppm. 13C-NMR (CDCl3, 75 MHz): δ = 14.1 (CH3), 22.6 (CH2), 25.1 (CH2), 31.8 (CH2), 33.3
(CH2), 34.4 (CH2), 61.7 (CH2), 72.3 (CH), 128.5 (4 x CH), 129.7 (4 x CH), 130.3
(C), 130.6 (C), 133.0 (2 x CH), 166.3 (C), 166.7 (C) ppm.
HPLC (Chiralcel-OD, 20 °C, λ = 230 nm, Heptan/iPrOH = 95:5, 1.0 mL/min): tR =
10.4 min (S), 14.6 min (R).
MS (EI, 70 eV): m/z (%) = 249 (3) [M+], 232 (10), 127 (6), 110 (23), 105 (100), 107 (75),

5. Experimenteller Teil
110
91 (55), 77 (40).
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.106
Ausgehend von (R)-3-(Tetrahydropyran-2-yloxy)-1-octin erhielt man folgenden Wert:
HPLC (Chiralcel-OD, 20 °C, λ = 230 nm, Heptan/iPrOH = 95:5, 1.0 mL/min): tR =
14.55 min (R).
Ausgehend von (S)-3-Methoxymethyloxy-1-octin erhielt man folgenden Wert:
HPLC (Chiralcel-OD, 20 °C, λ = 230 nm, Heptan/iPrOH = 95:5, 1.0 mL/min): tR =
10.4 min (S).
5.3.2.17. (S)-3-Hydroxyoctansäuremethylester (63)
Man löste unter einer Argon-Schutzgasatmosphäre in einem Schlenkrohr (S)-3-
Methoxymethyloxy-1-octin (S)-50 (85.2 mg, 0.5 mmol) und [η5-
Cyclopentadienalruthenium(II) bis-(2-diphenylphosphin-6-(2,4,6-triphenylphenyl)pyridin)
(acetonitril)] hexafluorophosphat 22 (37.2 mg, 0.025 mmol, 5 mol%) in einem Gemisch
aus Aceton (0.95 mL) und Wasser (45 mg, 2.5 mmol, 5 Äquiv). Das
Lösungsmittelvolumen wurde markiert bevor man weiteres Aceton (2 mL/mmol) zugab
und zur Entgasung der Lösung bis zur Markierung im Vakuum wieder eingeengte. Danach
ließ man die Lösung bei 50 °C für 6 h unter Argonatmosphäre rühren. Anschließend engte
man die Lösung ein, nahm den Rückstand in tert-Butanol (3 mL) auf und gab wässrige
NaH2PO4-Lösung (1.25 M, 2 mL) und wässrige KMnO4-Lösung (1 M, 3 mL) zu. Nach 70
min beendete man diese Reaktion durch die Zugabe von ges. wässriger Na2SO3-Lösung
(6 mL) und stellte mit wässriger HCl (2.4 M) einen pH-Wert von 3 ein.107 Die Lösung
wurde TBME (30 mL) extrahiert und die organische Phase wurde mit ges. wässriger
NaHCO3-Lösung neutral gewaschen. Die Lösung wurde über Na2SO4 getrocknet, filtriert
und eingeengt. Den Rückstand löste man in Methanol (10 mL) und konz. H2SO4 (0.5 mL)
und rührte es für 45 min bei 75 °C. Die Reaktion wurde durch die Zugabe von ges.
wässriger NaHCO3-Lösung (30 mL) mit TBME (50 mL) beendet. Die organische Phase
wurde zweimal mit dest. Wasser (30 mL) gewaschen, die wässrige Phase rückextrahiert
und die vereinigten organischen Phasen über MgSO4 getrocknet, filtriert und eingeengt.
Nach Säulenchromatographie (TBME/Hexan, 1:3) des Rückstandes erhielt man den
Methylester 63 als farblose Flüssigkeit (40 mg, 0.23 mmol, 46 %).

5. Experimenteller Teil
111
Rf = 0.55 (TBME/Hexan, 1:1). 1H-NMR (CDCl3, 300 MHz): δ = 0.85–0.92 (m, 3 H, CH3), 1.20–1.57 (m, 8 H, 4 x CH2),
2.41 (dd, J = 16.5, 9.0 Hz, 1 H, CH2), 2.52 (dd, J = 16.5, 3.1 Hz, 1 H, CH2), 2.85 (d,
J = 4.0 Hz, 1 H, OH), 3.71 (s, 3 H, CH3), 3.96–4.04 (m, 1 H, CH) ppm.
Drehwert: [a]D20 = 22.9 (c = 1 g/L, CHCl3).66
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.108
5.3.3. Synthese der Oligo-1,4-diole und deren Derivate 5.3.3.1. 1-Phenylbut-3-in-1-ol (67)
Zum Allenylzink (66; 1 M in THF, 0.15 mol, 1.5 Äquiv) wurde langsam bei 0 °C (Eisbad)
Benzaldehyd (10.2 mL, 0.10 mol, 1 Äquiv) getropft. Nach vollständiger Zugabe wurde die
Reaktionsmischung für 30 min gerührt und dann durch Zugabe von ges. wässriger NH4Cl-
Lösung (4 M, 250 mL), wässriger HCl (2.4 M, 50 mL) und TBME (300 mL) beendet. Die
wässrige Phase wurde mit TBME (100 mL) extrahiert und die vereinigten organischen
Phasen wurden mit dest. Wasser, ges. wässriger NaHCO3-Lösung (0.9 M) und dest. Wasser
(jeweils 300 mL) gewaschen. Nach dem Trocknen über MgSO4, Filtrieren und Einengen
des Lösungsmittels wurde das Rohprodukt mittels Säulenchromatographie (TBME/Hexan,
1:3→1:1) gereinigt, so dass man ein farbloses Öl erhielt (10.8 g, 74%), welches Spuren
vom Allenisomer 67a (67/67a, 92:8) enthielt.
1H-NMR (400 MHz, CDCl3): δ = 2.06 (t, J = 2.7 Hz, 1 H, H-3), 2.56 (d, J = 3.1 Hz, 1 H,
OH), 2.62 (AB-System, pseudo-dd, J = 6.4, 2.6 Hz, 2 H, H-2), 4.84 (dt, J = 6.4, 2.9
Hz, 1 H, H-1), 7.25–7.41 (m, 5 H, Ar) ppm. Ausgewählte Signale für Allen 3a: δ =
2.35 (br d, J = 3.9 Hz, 1 H, OH), 4.87–4.96 (m, 2 H), 5.22–5.27 (m, 1 H), 5.43 (q, J
= 7.1 Hz, 1 H) ppm.
Die spektroskopischen Daten stimmen mit denen in der Literatur überein.109

5. Experimenteller Teil
112
5.3.3.2. 1-Phenyl-3-butin-1-ylacetat (68) OAc
DMAP (0.757 g, 6.2 mmol, 10 mol%) und Et3N (12.95 mL, 93 mmol, 1.5 Äquiv) wurden
bei 0 °C (Eisbad) zu einer Lösung von Alkohol 67 (9.086 g, 62 mmol) in TBME (0.5 M)
gegeben. Essigsäureanhydrid (5.9 mL, 62 mmol, 1 Äquiv) wurde unter Rühren langsam
zugetropft. Nach vollständiger Reaktion (ca. 30 min, DC-Kontrolle) beendete man die
Reaktion durch die Zugabe von dest. Wasser (150 mL). Die wässrige Phase wurde mit
TBME (25 mL) extrahiert und die vereinigten organischen Phasen mit dest. Wasser, ges.
wässriger NaHCO3-Lösung (0.9 M) und dest. Wasser (jeweils 100 mL) gewaschen.
Nach dem Trocknen über MgSO4, Filtrieren und Einengen des Lösungsmittels destillierte
man das erhaltene Öl bei 80 °C (0.2 mbar) und erhielt eine farblose Flüssigkeit (10.845 g,
93%).
Diese ist eine Mischung aus Produkt 68 und Allenisomer 68a im Verhältnis 92:8.
In einer alternativen Synthese, beginnend mit Benzaldehyd (Ansatzgröße: 0.1 Mol), wurde
der verunreinigte Alkohol 67 ohne Reinigung direkt acetyliert und destilliert, was die
Verbindung 68 (16.18 g, 86%) gab.
1H-NMR (300 MHz, CDCl3): δ = 1.97 (t, J = 2.7 Hz, 1 H, H-4), 2.10 (s, 3 H, CH3), 2.69
(ddd, J = 16.8, 6.4, 2.7 Hz, 1 H, H-2), 2.78 (ddd, J = 16.8, 6.9, 2.6 Hz, 1 H, H-2),
5.89 (t, J = 6.6 Hz, 1 H, H-1), 7.27–7.41 (m, 5 H, Ph). Ausgewählte Signale für
Allen 68a: d = 4.79–4.9 (m, 2 H), 5.42 (q, J = 6.7 Hz, 1 H), 6.29 (dt, J = 6.7, 2.3 Hz,
1 H) ppm. 13C-NMR (75 MHz, CDCl3): δ = 21.1 (CH3), 26.5 (CH2), 70.7 (CH), 73.5 (CH), 79.5 (C),
126.5 (CH), 128.4 (CH), 128.5 (CH), 139.0 (C), 170.0 (C) ppm.
5.3.3.3. 4-Acetoxy-4-phenylbutanal (69)
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) ausgehend von 68 (250
mg, 1.33 mmol) wurde der Aldehyd 69 als farbloses Öl (252 mg, 92%) erhalten.

5. Experimenteller Teil
113
Rf = 0.24 (TBME/Hexan, 1:3).
IR (film): ν = 3034, 2936, 2827, 2727, 1734, 1495, 1451, 1373, 1239, 1025, 763, 702, 546
cm–1. 1H-NMR (400 MHz, CDCl3): δ = 2.08 (s, 3 H, CH3), 2.10–2.28 (m, 2 H, H-2), 2.44–2.50
(m, 2 H, H-3), 5.77 (dd, J = 7.6, 5.9 Hz, 1 H, H-4), 7.26–7.38 (m, 5 H, Ar), 9.73 (t,
J = 1.3 Hz, 1 H, H-1) ppm. 13C-NMR (100 MHz, CDCl3): δ = 21.3 (CH3), 28.8 (CH2), 40.0 (CH2), 75.0 (CH), 126.3
(CH), 128.2 (CH), 128.6 (CH), 139.6 (C), 170.1 (C), 200.9 (CH) ppm.
MS (EI, 70 eV): m/z (%) = 206 (1) [M+], 188 (1), 163 (80), 146 (45), 120 (100), 107 (75),
91 (55), 77 (40).
Elementaranalyse (C12H14O3): Ber.: C 69.88, H 6.84; Gef.: C 69.62, H 6.91.
5.3.3.4. 4-Hydroxy-1-phenyl-6-heptin-1-ylacetat (70)
Gemäß AAV2 ([Ru] = 5 mol%; mit L = 2-Diphenylphosphin-6-(tert-butyl)pyridin) und
AAV3 wurde der Ester 70 ausgehend von 68 (1.035 g, 5.5 mmol) als farbloses Öl (1.1 g,
82%) erhalten.
Rf = 0.32 (TBME/Hexan, 1:3).
IR (film): ν = 3476, 3291, 3033, 2932, 2363, 1735 cm–1. 1H-NMR (400 MHz, CDCl3): δ = 1.41–1.66 (m, 2 H, CH2), 1.79–2.15 (m, 3 H, CH, CH2),
2.03–2.06 (m, 1 H), 2.07 (s, 3 H, CH3), 2.25–2.45 (m, 2 H, CH2), 3.72–3.81 (m, 1 H,
CH), 5.72–5.80 (m, 1 H, CH), 7.27–7.37 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = 21.4 (CH3), 27.6 (CH2), 32.0/32.2 (CH2), 32.5/32.6
(CH2), 69.4/69.6 (CH), 71.1/71.2 (CH), 75.7/76.0 (CH), 80.5 (C), 126.4 (CH),
127.9 (CH), 128.4 (CH), 140.2/140.3 (C), 170.3 (C) ppm.
MS (EI, 70 eV): m/z (%) = 246 (3) [M+], 203 (97), 186 (25), 185 (35), 147 (100), 129 (43),
120 (64), 107 (99), 91 (61), 79 (43).
Elementaranalyse (C15H18O3): Ber.: C 73.15, H 7.37; Gef.: C 73.61, H 7.27.

5. Experimenteller Teil
114
5.3.3.5. 4-tert-Butyldimethylsilyloxy-1-phenyl-6-heptin-1-ylacetat (71)
Gemäß AAV4 ausgehend von 70 (7.66 g, 31.1 mmol) wurde der Ester als farbloses Öl
(10.9 g, 98%) erhalten.
Rf = 0.7 (TBME/Hexan, 1:3).
IR (film): ν = 3303, 2943, 2363, 1740, 1463, 1369, 1242, 1094, 1034, 838, 779, 698, 635,
541 cm–1. 1H-NMR (400 MHz, CDCl3): δ = 0.02–0.06 (m, 6 H, 2 × CH3), 0.87/0.87 (s, 9 H, 2 ×
CH3), 1.40–1.77 (m, 2 H, CH2), 1.79–2.05 (m, 3 H, CH, CH2), 2.07 (s, 3 H, CH3),
2.21–2.47 (m, 2 H, CH2), 3.78–3.86 (m, 1 H, CH2), 5.70–5.77 (m, 1 H, CH2), 7.26–
7.38 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = –4.7 (CH3), –4.5 (CH3), 18.0 (C), 21.2 (CH3), 25.8
(CH3), 27.0/27.1 (CH2), 31.2/31.4 (CH2), 32.0/32.2 (CH2), 70.0 (CH), 70.1/70.3
(CH), 75.7/75.9 (CH), 81.1/81.2 (C), 126.3 (CH), 127.7 (CH), 128.2 (CH),
140.2/140.3 (C), 170.0 (C) ppm.
MS (ESI, CHCl3): m/z = 383.2 [M+ + Na].
Elementaranalyse (C21H32O3Si·0.2 H2O): Ber.: C 69.26, H 8.97; Gef.: C 69.19, H, 9.28.
5.3.3.6. 4-tert-Butyldimethylsilyloxy-7-hydroxy-1-phenyl-9-decin-1-ylacetat (72) OAc
OTBS
OH
Gemäß AAV2 ([Ru] = 2 mol%, mit L = 2-Diphenylphosphin-6-(2,4,6-
triphenylphenyl)pyridin) und AAV2 ausgehend von 71 (1.00 g, 2.77 mmol) wurde der
Ester als Öl (1.15 g, > 95%) erhalten.
Rf = 0.30 (TBME/Hexan, 1:3).
IR (film): ν = 3503, 3302, 2941, 2363, 1732, 1460, 1371, 1247, 1045, 838, 764, 699, 638,
541 cm–1. 1H-NMR (300 MHz, CDCl3): δ = –0.02–0.05 (m, 6 H, 2 × CH3), 0.87 (s, 9 H, 3 × CH3),

5. Experimenteller Teil
115
1.30–2.00 (m, 8 H, 4 x CH2), 2.00–2.05 (m, 1 H, CH), 2.06 (s, 3 H, CH3), 2.09 (d, J
= 4.7 Hz, 0.5 H, OH), 2.25–2.47 (m, 2 H, CH2), 2.48–2.54 (m, 0.5 H, OH), 3.64–
3.78 (m, 2 H, 2 x CH), 5.70 (t, J = ~7 Hz, 1 H, CH), 7.26–7.40 (m, 5 H, Ar) ppm. 13C-NMR (75 MHz, CDCl3): δ = –4.5 (CH3), –4.6 (CH3), 18.1 (C), 21.3 (CH3), 25.9
(CH3), 27.3/27.4 (CH2), 31.5–32.8 (4 × CH2), 69.9/70.2/70.7/70.8 (CH),
71.3/71.4/71.5/71.6 (CH), 76.0 (CH), 80.9 (C), 126.4/126.5 (CH), 127.9 (CH),
128.4 (CH), 140.5/140.6 (C), 170.3 (C) ppm.
MS (ESI, MeOH): m/z = 441.3 [M+ + Na].
Elementaranalyse (C24H38O4Si): C 68.86, H 9.15; Gef.: C 69.35, H 9.34.
5.3.3.7. 4,7-Bis(tert-butyldimethylsilyloxy)-1-phenyl-9-decin-1-ylacetat (73) OAc
OTBS
OTBS
Gemäß AAV4 ausgehend von 72 (2.26 g, 5.4 mmol) wurde der Ester als Öl (2.9 g, 99%)
erhalten.
Rf = 0.84 (TBME/Hexan, 1:3).
IR (film): ν = 3307, 2943, 1741, 1465, 1369, 1244, 1064, 838, 776, 699, 636, 546 cm–1. 1H-NMR (400 MHz, CDCl3): δ = –0.03–0.07 (m, 12 H, 4 × CH3), 0.86/0.87 (s, 9 H, 3 x
CH3), 0.88 (s, 9 H, 3 x CH3), 1.28–1.75 (m, 6 H, 3 x CH2), 1.77–1.99 (m, 2 H, CH2),
1.94–1.97 (m, 1 H, CH), 2.06 (s, 3 H, CH3), 2.22–2.38 (m, 2 H, CH2), 3.60–3.70 (m,
1 H, CH), 3.71–3.83 (m, 1 H, CH), 5.71 (t, J = ~7 Hz, 1 H, CH), 7.26–7.37 (m, 5 H,
Ar) ppm. 13C-NMR (75 MHz, CDCl3): δ = –4.5 bis –4.3 (4 × CH3), 18.2 (2 × C), 21.4 (CH3), 25.9
(CH3), 26.0 (CH3), 27.3/27.4 (CH2), 31.6–32.7 (4 × CH2), 70.0 (CH), 71.0/71.1
(CH), 71.5/71.7/71.8 (CH), 76.2 (CH), 81.6 (C), 126.4/126.5 (CH), 127.8 (CH),
128.4 (CH), 140.6 (C), 170.2 (C) ppm.
MS (ESI, CHCl3): m/z = 555.5 [M+ + Na].
Elementaranalyse (C30H52O4Si2·0.2 H2O): Ber.: C 67.16, H 9.84; Gef.: C 66.96, H 9.74.

5. Experimenteller Teil
116
5.3.3.8. 4,7-Bis(tert-butyldimethylsilyloxy)-10-hydroxy-1-phenyl-12-tridecin-1-ylacetat
(74)
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) und AAV3 ausgehend
von 73 (1 g, 1.88 mmol) wurde der Hydroxyester als Öl (0.9 g, 82%) erhalten.
Rf = 0.33 (TBME/Hexan, 1:3).
IR (film): ῦ = 3428, 3310, 2952, 2119, 1737, 1465, 1371, 1251, 1065, 836, 775, 701, 633,
544 cm–1. 1H-NMR (400 MHz, CDCl3): δ = –0.04–0.06 (m, 12 H, 4 × CH3), 0.86 (s, 9 H, 3 x CH3),
0.88 (s, 9 H, 3 x CH3), 1.24–1.74 (m, 10 H, 5 x CH2), 1.75–1.98 (m, 2 H, CH2),
1.99–2.04 (m, 1 H, CH), 2.06 (s, 3 H, CH3), 2.19–2.25 (m, 0.5 H, OH), 2.28–2.46
(m, 2 H, CH2), 2.73–2.83 (m, 0.5 H, OH), 3.57–3.79 (m, 3 H, 3 x CH), 5.70 (t, J =
~7 Hz, 1 H, CH), 7.25–7.37 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = –4.5 (4 × CH3), 18.1 (2 × C), 21.2 (CH3), 25.8 (2 ×
CH3), 26.9/27.3 (CH2), 31.3–32.9 (6 × CH2), 69.8/70.1 (CH), 70.4/70.7 (CH), 71.4–
72.2 (2 × CH), 76.0 (CH), 80.7/80.9 (C), 126.2/126.3 (CH), 127.7 (CH), 128.2
(CH), 140.3/140.4 (C), 170.1 (C) ppm.
MS (ESI, MeOH): m/z = 613.6 [M+ + Na].
Elementaranalyse (C33H58O5Si2): Ber.: C 67.07, H 9.89; Gef.: C 67.33, H 10.43.
5.3.3.9. 4,7,10-Tris(tert-butyldimethylsilyloxy)-1-phenyl-12-tridecin-1-ylacetat (75)
Gemäß AAV4 ausgehend von 74 (3 g, 5.076 mmol) wurde der Ester als Öl (3.6 g, 99%)
erhalten.
Rf = 0.9 (TBME/Hexan, 1:3).
IR (film): ν = 3306, 2944, 1740, 1465, 1369, 1247, 1070, 837, 771, 699, 636, 532 cm–1. 1H-NMR (400 MHz, CDCl3): δ = –0.03–0.07 (m, 18 H, 6 × CH3), 0.85–0.88 (m, 27 H, 9

5. Experimenteller Teil
117
x CH3), 1.27–2.02 (m, 12 H, 6 x CH2), 1.91–1.95 (m, 1 H, CH), 2.06 (s, 3 H, CH3),
2.23–2.38 (m, 2 H, CH2), 3.55–3.67 (m, 2 H, 2 x CH), 3.73–3.82 (m, 1 H, CH),
5.70 (t, J = ~7 Hz, 1 H, CH), 7.25–7.37 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = –4.6 bis –4.4 (6 × CH3), 18.0 (3 × C), 21.2 (CH3),
25.7–25.9 (3 × CH3), 27.3 (CH2), 31.4–32.8 (6 × CH2), 69.8 (CH), 70.9/71.0 (CH),
71.6/71.7/71.8 (CH), 72.1/72.2 (CH), 76.0 (CH), 81.5 (C), 126.3 (CH), 127.6 (CH),
128.2 (CH), 140.5 (C), 170.1 (C) ppm.
MS (ESI, CHCl3): m/z = 727.8 [M+ + Na].
Elementaranalyse (C39H72O5Si3): Ber.: C 66.42, H 10.29; Gef.: C 65.84, H 9.98.
5.3.3.10. (E)-6-Acetoxy-6-phenyl-2-hexensäuremethylester [(E)-78]
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) und AAV5 ausgehend
von 68 (160 mg, 0.88 mmol) wurden das (E)- und das (Z)-Isomer des Methylsäureesters als
Öl ((E): 217 mg, 94%; (Z): 8.5 mg, 4%; Verhältnis von E/Z = 25:1) erhalten.
Rf = 0.43 (TBME/Hexan, 1:3).
IR (film): ν = 3032, 2950, 1729, 1657, 1495, 1437, 1372, 1314, 1237, 1032, 852, 761, 702,
549 cm–1. 1H-NMR (300 MHz, CDCl3): δ = 1.85–2.32 (m, 4 H, 2 x CH2), 2.08 (s, 3 H, CH3), 3.72 (s,
3 H, OCH3), 5.74 (dd, J = 7.2, 5.9 Hz, 1 H, H-6), 5.82 (dt, J = 15.8, 1.4 Hz, 1 H, H-
2), 6.94 (dt, J = 15.7, 6.8 Hz, 1 H, H-3), 7.26–7.38 (m, 5 H, Ar) ppm. 13C-NMR (75 MHz, CDCl3): δ = 21.2 (CH3), 28.3 (CH2), 34.5 (CH2), 51.5 (CH3), 75.2
(CH), 121.5 (CH), 126.4 (CH), 128.1 (CH), 128.6 (CH), 140.0 (C), 147.9 (CH),
166.9 (C), 170.2 (C) ppm.
MS (EI, 70 eV): m/z = 263.2 (3) [M+], 219.1 (14), 202.1 (38), 163.0 (63), 143.1 (85),
121.1 (67), 107.1 (87), 100.1 (100), 91.1 (31).
Elementaranalyse (C15H18O4): Ber.: C 68.68, H 6.92; Gef.: C 68.41, H, 6.70.

5. Experimenteller Teil
118
(Z)-78 1H-NMR (300 MHz, CDCl3): δ = 1.85–2.17 (m, 2 H, CH2), 2.08 (s, 3 H, CH3), 2.64–2.77
(m, 2 H, CH2), 3.68 (s, 3 H, OCH3), 5.75 (dd, J = 8.1, 5.7 Hz, 1 H, H-6), 5.79 (dt, J
= 11.5, 1.7 Hz, 1 H, H-2), 6.20 (dt, J = 11.5, 7.5 Hz, 1 H, H-3), 7.27–7.35 (m, 5 H,
Ar) ppm.
5.3.3.11. 7-Acetoxy-4-hydroxy-7-phenyl-2-heptinsäuremethylester (79)
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) und AAV6 ausgehend
von 68 (0.1 g, 0.531 mmol) wurde der Methylsäureester als Öl (135 mg, 87%) erhalten.
Rf = 0.14 (TBME/Hexan, 1:3).
IR (film): ν = 3416, 3032, 2955, 2855, 2236, 1717, 1496, 1437, 1374, 1251, 1028, 957,
754, 702, 542 cm–1. 1H-NMR (400 MHz, CDCl3): δ = 1.64–2.15 (m, 4 H, 2 x CH2), 2.08 (s, 3 H, CH3),
3.02/3.04 (d, J = 5.7/5.5 Hz, 1 H, OH), 3.76 (s, 3 H, OCH3), 4.42–4.50 (m, 1 H, H-
4), 5.73–5.78 (m, 1 H, H-7), 7.27–7.37 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = 21.2 (CH3), 31.5/31.6 (CH2), 32.6 (CH2), 52.8 (CH3),
61.2/61.3 (CH), 75.3/75.4 (C), 76.1 (CH), 87.7 (C), 126.2 (CH), 127.9 (CH), 128.3
(CH), 139.8 (C), 153.5 (C), 170.3 (C) ppm.
MS (ESI, MeOH): m/z = 313.0 [M+ + Na].
Elementaranalyse (C16H18O5): Ber.: C 66.19, H 6.25; Gef.: C 66.13, H 6.62.
5.3.3.12. (E)-9-Acetoxy-6-(tert-butyldimethylsilyloxy)-9-phenyl-2-
nonensäuremethylester [(E)-80]
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) und AAV5 ausgehend
von 71 (166 mg, 0.46 mmol) wurden das (E)- und das (Z)-Isomer des Methylsäureesters als
Öl ((E): 190 mg, 95%; (Z): 7.6 mg, 4%; Verhältnis von E/Z = 25:1) erhalten.

5. Experimenteller Teil
119
Rf = 0.58 (TBME/Hexan, 1:3).
IR (film): ν = 3032, 2951, 1730, 1658, 1496, 1438, 1372, 1317, 1239, 1040, 837, 775, 702,
543 cm–1. 1H-NMR (300 MHz, CDCl3): δ = –0.03/0.00/0.07 (s, 6 H, 2 × CH3), 0.86/0.86 (s, 9 H, 3 x
CH3), 1.27–2.00 (m, 6 H, 3 x CH2), 2.07 (s, 3 H, CH3), 2.11–2.27 (m, 2 H, CH2),
3.63–3.72 (m, 1 H, CH), 3.72 (s, 3 H, OCH3), 5.7 (t, J = 7.1 Hz, 1 H, CH), 5.79 (dq,
J = 15.7, 1.6 Hz, 1 H, CH), 6.95 (dtd, J = 15.6, 7.1, 1.3 Hz, 1 H, CH), 7.25–7.38 (m,
5 H, Ar) ppm. 13C-NMR (75 MHz, CDCl3): δ = –4.4 (CH3), –4.3 (CH3), 18.2 (C), 21.4 (CH3), 26.0
(CH3), 28.1 (CH2), 31.6/31.8 (CH2), 32.7 (CH2), 35.0/35.1 (CH2), 51.6 (CH3),
70.9/71.0 (CH), 76.2 (CH), 121.0 (CH), 126.6 (CH), 128.1 (CH), 128.6 (CH),
140.6/140.7 (C), 149.5 (CH), 167.2 (C), 170.5 (C) ppm.
MS (ESI, MeOH): m/z = 457.1 [M+ + Na].
Elementaranalyse (C24H38O5Si): Ber.: C 66.32, H 8.81; Gef.: C 66.06, H 8.51.
(Z)-80 1H-NMR (300 MHz, CDCl3): δ = 0.02/0.04 (s, 6 H, 2 × CH3), 0.88/0.89 (s, 9 H, 3 x CH3),
1.32–2.04 (m, 6 H, 3 x CH2), 2.09 (s, 3 H, CH3), 2.55–2.75 (m, 2 H, CH2), 3.66–3.77 (m, 1
H, CH), 3.72 (s, 3 H, OCH3), 5.68–5.83 (m, 2 H, 2 x CH), 6.21 (dtd, J = 11.4, 7.3, 1.9 Hz,
1 H, CH) 7.25–7.41 (m, 5 H, Ar) ppm.
5.3.3.13. 10-Acetoxy-7-(tert-butyldimethylsilyloxy)-4-hydroxy-10-phenyl-2-
decinsäuremethylester (81)
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) und AAV6 ausgehend
von 71 (1 g, 2.76 mmol) wurde der Methylsäureester als Öl (1.13 g, 88%) erhalten.
Rf = 0.19 (TBME/Hexan, 1:3).
IR (film): ν = 3398, 3031, 2955, 2858, 2236, 1719, 1496, 1437, 1374, 1254, 1053, 949,
837, 756, 702, 544 cm–1. 1H-NMR (400 MHz, CDCl3): δ = –0.01–0.06 (br s, 6 H, 2 × CH3), 0.86/0.88 (s, 9 H,

5. Experimenteller Teil
120
3 x CH3), 1.32–1.99 (m, 8 H, 4 x CH2), 2.07 (s, 3 H, CH3), 2.63/2.66 (d, J = 5.2 Hz,
0.5 H, OH), 3.24/3.30 (d, J = 7.4 Hz, 0.5 H, CH), 3.69–3.81 (m, 1 H, CH), 3.77 (s,
3 H, OCH3), 4.42–4.54 (m, 1 H, CH), 5.70 (t, J = ~7 Hz, 1 H, CH), 7.24–7.39 (m, 5
H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = –4.4 bis –4.3 (2 × CH3), 18.3 (C), 21.5 (CH3), 26.1
(CH3), 31.2–32.7 (4 × CH2), 52.9/53.0 (CH3), 61.9/62.4 (CH), 71.1/71.3/71.4 (CH),
76.0/76.1 (CH), 76.3/76.5 (C), 88.2/88.3 (C), 126.5/126.6 (CH), 128.0/128.1 (CH),
128.5 (CH), 140.3/140.4/140.5/140.5 (C), 153.8 (C), 170.4 (C) ppm.
MS (ESI, MeOH): m/z = 485.1 [M+ + Na].
Elementaranalyse (C25H38O6Si): Ber.: C 64.90, H 8.28; Gef.: C 64.89, H 8.63.
5.3.3.14. (E)-15-Acetoxy-6,9,12-tris(tert-butyldimethylsilyloxy)-15-phenyl-2-
pentadecensäuremethylester [(E)-82]
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) und AAV5 ausgehend
von 75 (250 mg, 0.354 mmol) wurde das (E)-Isomer des Methylsäureesters als Öl (255 mg,
92%) erhalten.
Rf = 0.80 (TBME/Hexan, 1:3).
IR (film): ν = 2946, 1734, 1656, 1464, 1370, 1247, 1065, 837, 775, 702, 665, 538 cm–1. 1H-NMR (300 MHz, CDCl3): δ = –0.04–0.05 (m, 18 H, 6 x CH3), 0.86–0.87 (mehrere s,
27 H, 9 x CH3), 1.21–1.62 (m, 12 H, 6 x CH2), 1.73–2.00 (m, 2 H, CH2), 2.06 (s, 3
H, CH3), 2.15–2.32 (m, 2 H, CH2), 3.51–3.72 (m, 3 H, H-6, H-9, H-12), 3.73 (s, 3
H, OCH3), 5.70 (t, J = ~7 Hz, 1 H, H-15), 5.82 (d, J = 15.7 Hz, 1 H, H-2), 6.98 (dt,
J = 15.6, 6.9 Hz, 1 H, H-3), 7.25–7.37 (m, 5 H, Ar) ppm. 13C-NMR (75 MHz, CDCl3): δ = –4.6 bis –4.3 (6 × CH3), 18.1 (3 × C), 21.3 (CH3), 25.9
(3 × CH3), 27.9/28.0 (CH2), 31.4–32.9 (6 × CH2), 35.20 (CH2), 51.4 (CH3), 71.7–
72.4 (3 × CH), 76.2 (CH), 120.8 (CH), 126.5 (CH), 127.8 (CH), 128.4 (CH), 140.7
(C), 149.7 (CH), 167.1 (C), 170.3 (C) ppm.
MS (ESI, MeOH): m/z = 801.8 [M+ + Na].
Elementaranalyse (C42H78O7Si3): Ber.: C 64.73, H 10.09; Gef.: C 65.20, H 10.17.

5. Experimenteller Teil
121
5.3.3.15. 16-Acetoxy-7,10,13-tris(tert-butyldimethylsilyloxy)-4-hydroxy-16-phenyl-2-
hexadecinsäuremethylester (83)
Gemäß AAV2 ([Ru] = 2 mol%, mit L = 2-Diphenylphosphin-6-(2,4,6-tri-iso-
propylphenyl)pyridin) und AAV6 ausgehend von 75 (0.5 g, 0.71 mmol) wurde der
Methylsäureester als Öl (423 mg, 74%) erhalten.
Rf = 0.46 (TBME/Hexan, 1:3).
IR (film): ν = 3415, 3029, 2953, 2857, 2236, 1720, 1466, 1437, 1371, 1253, 1061, 938,
837, 770, 701, 666 cm–1. 1H-NMR (300 MHz, CDCl3): δ = –0.04–0.07 (mehrere s, 18 H, 6 x CH3), 0.86–0.90
(mehrere s, 27 H, 9 x CH3), 1.23–2.00 (m, 16 H, 8 x CH2, und 0.5 OH), 2.06 (s, 3 H,
CH3), 2.82–2.91 (m, 0.5 H, OH), 3.52–3.72 (m, 3 H, 3 x CH), 3.77 (s, 3 H, OCH3),
4.44–4.57 (m, 1 H, H-4), 5.70 (t, J = ~7 Hz, 1 H, H-16), 7.24–7.39 (m, 5 H, Ar)
ppm. 13C-NMR (75 MHz, CDCl3): δ = –4.5 bis –4.2 (6 × CH3), 18.2 (3 × C), 21.4 (CH3), 26.0
(3 × CH3), 31.1–33.1 (8 × CH2), 52.8/52.9 (CH3), 61.8/62.5 (CH), 71.7–72.6 (3 ×
CH), 76.2 (C), 76.3 (CH), 88.4/88.5 (C), 126.6/126.7 (CH), 128.0 (CH), 128.6
(CH), 140.8 (C), 154.0 (C), 170.5 (C) ppm.
MS (ESI, MeOH): m/z = 829.9 [M+ + Na].
Elementaranalyse (C43H78O8Si3): Ber.: C 63.97, H 9.74; Gef.: C 63.79, H 10.02.
5.3.3.16. 16-Acetoxy-4,7,10,13-tetrakis(tert-butyldimethylsilyloxy)-16-phenyl-2-
hexadecinsäuremethylester (84)
Gemäß AAV4 ausgehend von 83 (430 mg, 0.533 mmol) wurde der Methylsäureester als Öl
(487 mg, 99 % Ausbeute) erhalten.
Rf = 0.79 (TBME/Hexan, 1:3).
IR (film): ν = 2947, 2859, 2237, 1728, 1466, 1368, 1250, 1073, 938, 838, 777, 700, 667
cm–1.

5. Experimenteller Teil
122
1H-NMR (400 MHz, CDCl3): δ = –0.06–0.14 (mehrere s, 24 H, 8 x CH3), 0.82–0.94
(mehrere s, 36 H, 12 x CH3), 1.20–1.99 (m, 16 H, 8 x CH2), 2.06 (s, 3 H, CH3),
3.48–3.70 (m, 3 H, H-7, H-10, H-13), 3.76 (s, 3 H, OCH3), 4.43/4.47 (t, J = 6.2 Hz,
1 H, H-4), 5.70 (t, J = ~7 Hz, 1 H, H-16), 7.23–7.36 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz. CDCl3): δ = –4.4 bis –4.2 (8 × CH3), 18.2–18.3 (4 × C), 21.4 (CH3),
25.8–26.0 (4 × CH3), 31.5–33.8 (8 × CH2), 51.8/51.9/52.7 (CH3), 62.7/62.9 (CH),
71.7–72.7 (3 × CH), 75.8 (C), 76.2 (CH), 88.9 (C), 126.4/126.5 (CH), 127.8 (CH),
128.4 (CH), 140.6 (C), 153.8 (C), 170.2 (C) ppm.
MS (ESI, CHCl3/EtOH): m/z = 990.0 [M+ + EtOH + Na].
Elementaranalyse (C49H92O8Si4·1.25 H2O): Ber.: C 62.34, H 10.09; Gef.: C 62.34, H
10.01.
5.3.3.17. 16-Hydroxy-4,7,10,13-tetrakis(tert-butyldimethylsilyloxy)-16-phenyl-2-
hexadecinsäure (85)
Der Methylsäureester 84 (445 mg, 0.48 mmol) wurde zusammen mit Lithiumhydroxid
(101.3 mg, 2.41 mmol) in einem Gemisch aus THF und dest. Wasser (5:1, 42 mL) gelöst
und für 4 h bei 60 °C gerührt. Die Reaktion wurde durch Zugabe von ges. wässriger
NH4Cl-Lösung (20 mL) und TBME (50 mL) beeendet und die organische Phase wurde mit
dest. Wasser (25 mL) gewaschen. Nach Rückextraktion der wässrigen Phase wurden die
vereinigten organischen Phasen über MgSO4 getrocknet, filtriert und einengt. Den
Rückstand nahm man in Methanol (42 mL) auf, versetzte ihn mit K2CO3 (133.5 mg, 0.96
mmol) und rührte ihn für 16 h bei RT. Durch Zugabe von dest. Wasser (20 mL) und
TBME (100 mL) wurde die Reaktion beendete und eine Rückextraktion der wässrigen
Phase durchgeführt. Die vereinigten organischen Phasen wurden über MgSO4 getrocknet,
filtriert und eingeengt. Nach Säulenchromatographie (TBME/Hexan, 1:3) erhielt man die
Hydroxysäure 85 als geldliches Öl (197 mg, 47%).
Rf = 0.12 (TBME/Hexan, 1:3).
IR (film): ν = 3489, 2951, 2858, 1719, 1465, 1362, 1254, 1071, 937, 837, 771, 701, 665
cm–1. 1H-NMR (400 MHz, CD3OD): δ = –0.04–0.18 (m, 24 H, 8 x CH3), 0.69–1.05 (m, 36

5. Experimenteller Teil
123
H, 12 x CH3), 1.25–1.87 (m, 16 H, 8 x CH2), 3.43–3.51 (br s, 1 H, OH), 3.58–3.69
(m, 2 H, 2 x CH), 3.69–3.77 (m, 1 H, CH), 4.48–4.59 (m, 2 H, 2 x CH), 7.17–7.24
(m, 1 H, CH), 7.25–7.35 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz. CD3OD): δ = –4.7 bis –3.95 (8 × CH3), 19.0 (4 × C), 26.6 (12 ×
CH3), 33.2–35.8 (8 × CH2), 63.8/63.9 (CH), 73.0–73.6 (3 × CH), 75.3 (CH), 78.1
(C), 88.4 (C), 127.0 (CH), 128.1 (2 x CH), 129.1 (2 x CH), 146.1/146.2 (C), 156.2
(C) ppm.
MS (ESI, CHCl3/MeOH): m/z = 947.6 [M+ + 2•MeOH + Na].
Elementaranalyse (C46H88O4Si7): Ber.: C 63.83, H 10.25; Gef.: C 63.48, H 10.32.
5.3.3.18. 17-Phenyl-5,8,11,14-tetrakis(tert-butyldimethylsilanyloxy)-1-oxa-zyklo-
hepta-3-decin-2-on (86)
Die Carbonsäure 85 wurde unter Argon in einem ausgeheizten Schlenkrohr zusammen mit
Triethylamin (20.8 μL, 0.156 mmol) und THF (2.5 mL) für 15 min gerührt bevor 2,4,6-
Trichlorobenzoylchlorid (12.2 μL, 0.078 mL) zugegeben wurde. Die Lösung wurde für
3.5 h gerührt, anschließend mit Toluol (16.65 mL) verdünnt und mittels Spritzenpumpe
über einen Zeitraum von 3 h zu einer refluxierenden Lösung aus DMAP (88.9 mg, 0.728
mmol) in Toluol (40.5 mL) gegeben. Nach vollständiger Zugabe wurde die Lösung für 1 h
am Refluxieren gehalten und durch Zugabe von wässriger HCl (2.4 M, 15 mL) wurde die
Reaktion beendete. Die organische Phase wurde mit dest. Wasser (30 mL) und ges.
wässriger NaHCO3-Lösung gewaschen und die wässrige Phase rückextrahiert. Die
vereinigten organischen Phasen wurden über MgSO4 getrocknet, filtriert und eingeengt.
Nach Säulenchromatographie (EtOAc/Hexan, 1:2) erhielt man das Lacton 86 (36.3 mg,
83%) als farbloses Öl.
Rf = 0.21 (EtOAc/Hexan, 1:2).
IR (film): ν = 3079, 2960, 2927, 2855, 2236, 1948, 1817, 1719, 1577, 1547, 1495, 1461,
1411, 1369, 1260, 1207, 1084, 1011, 938, 822, 699, 644 cm–1. 1H-NMR (400 MHz, CD3OD): δ = –0.06–0.18 (m, 24 H), 0.77–0.96 (m, 36 H), 1.25–1.87

5. Experimenteller Teil
124
(m, 16 H, 8 x CH2), 3.58–3.91 (m, 3 H, 3 x CH), 4.48–4.59 (m, 2 H, 2 x CH), 7.25–
7.45 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz. CD3OD): δ = –4.6 bis –4.4 (8 × CH3), 18.2 (4 × C), 25.8–26.0 (12 ×
CH3), 29.5–33.6 (8 × CH2), 63.1 (CH), 67.3 (C), 70.1–72.8 (3 × CH), 74.8 (CH),
89.1 (C), 126.1 (CH), 128.1 (2 x CH), 129.9 (2 x CH), 137.7 (C), 157.7 (C) ppm.
MS (ESI, CHCl3/MeOH): m/z = 887.3 [M+ + K], 869.3 [M+ + Na].
Elementaranalyse (C46H86O6Si4): Ber.: C 65.19, H 10.23; Gef.: C 65.21, H 10.31.
5.3.3.19. 4-tert-Butyldimethylsilyloxy-4-phenyl-1-butin (87)
Ph
OTBS
Gemäß AAV4 ausgehend von 67 (2.00 g, 13.7 mmol) wurde das Silan als Öl (3.461 g,
97 % Ausbeute; enthält 5 % vom Allenisomer) erhalten.
Rf = 0.94 (TBME/Hexan, 1:3).
IR (film): ν = 3306, 3028, 2941, 2363, 1598, 1465, 1362, 1254, 1097, 937, 841, 778, 697,
636, 543 cm–1. 1H-NMR (400 MHz, CDCl3): δ = –0.08 (s, 3 H, CH3), 0.07 (s, 3 H, CH3), 0.89 (s, 9 H, 3 x
CH3), 1.95 (t, J = 2.7 Hz, 1 H, CH), 2.48 (ddd, J = 16.6, 5.8, 2.7 Hz, 1 H, CH2),
2.59 (ddd, J = 16.6, 7.1, 2.6 Hz, 1 H, CH2), 4.81 (dd, J = 6.9, 5.9 Hz, 1 H, CH),
7.21–7.38 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = –4.7 (CH3), –4.6 (CH3), 18.4 (C), 25.9 (CH3), 31.1
(CH2), 70.0 (CH), 73.8 (CH), 81.7 (C), 125.9 (CH), 127.4 (CH), 128.1 (CH), 143.9
(C) ppm.
MS (EI, 70 eV): m/z (%) = 245.0 (4) [M+ – CH3], 221.1 (31), 203.0 (100), 185.0 (4), 128.1
(9), 115.0 (3), 97.1 (22), 75.1 (35).
Elementaranalyse (C16H24OSi): Ber.: C 73.79, H 9.29; Gef.: C 73.47, H 8.81.

5. Experimenteller Teil
125
5.3.3.20. 7-tert-Butyldimethylsilyloxy-4-hydroxy-7-phenyl-2-heptinsäuremethylester
(88)
Ph
OTBS
OH
CO2Me
Gemäß AAV2 (mit L = 2-Diphenylphosphin-6-(tert-amyl)pyridin) und AAV6 ausgehend
von 87 (0.5 g, 1.92 mmol) wurde der Methylsäureester als Öl (432 mg, 62%, Mischung aus
Diastereomeren A/B = 60:40) erhalten.
Rf = 0.4 (TBME/Hexan, 1:3).
IR (film): ν = 3401, 3027, 2945, 2862, 2238, 1715, 1444, 1257, 1080, 843, 773, 700, 541
cm–1. 1H-NMR (400 MHz, CDCl3): δ = –0.13/–0.08 (s, 3 H, CH3), 0.04/0.07 (s, 3 H, CH3),
0.89/0.91 (s, 9 H, 3 x CH3), 1.72–2.08 (m, 4 H, 2 x CH2), 2.45 (d, J = 5.4 Hz, 0.6 H,
OH-A), 3.07 (d, J = 7.2 Hz, 0.4 H, OH-B), 3.77/3.78 (s, 3 H, OCH3), 4.43–4.53 (m,
1 H, H-4), 4.77 (t, J = 5.6 Hz, 0.6 H, H-7 A), 4.85 (dd, J = 5.7, 4.6 Hz, 0.4 H, H-7
B), 7.19–7.37 (m, 5 H, Ar) ppm. 13C-NMR (100 MHz, CDCl3): δ = –4.9 (B, CH3), –4.9 (A, CH3), –4.6 (B, CH3), –4.5 (A,
CH3), 18.3/18.4 (C), 26.0 (CH3), 31.9 (B, CH2), 32.6 (A, CH2), 35.4 (B, CH2), 36.0
(A, CH2), 52.8 (CH3), 61.8 (B, CH), 62.2 (A, CH), 74.2 (B, CH), 74.4 (A, CH),
76.2/76.4 (C), 88.1/88.2 (C), 125.8 (CH), 127.1 (CH), 128.1 (CH), 143.8 (B, C),
144.3 (A, C), 153.7 (C), 170.3 (C) ppm.
MS (ESI, CHCl3): m/z = 385.1 [M+ + Na].
Elementaranalyse (C20H30O4Si·0.2 H2O): Ber.: C 65.61, H 8.37; Gef.: C, 65.55; H, 8.27.

A Literaturverzeichnis
126
A Literaturverzeichnis
[1] B. M. Trost, Sciene 1991, 254, 1471.
[2] M. Berthelot, Ann. Chim. Phys. 1863, 67, 52.
[3] H. Lagermarck, A. Eltekoff, Chem. Ber. 1877, 10, 637.
[4] M. Kutscheroff, Chem. Ber. 1881, 14, 1540.
[5] W. Kirsten (Hrsg.), Organisch-präparative Chemie, Bd. 1 – Die Katalyse in der
organischen Chemie, B. N. Dolgow, VEB – Deutscher Verlag der Wissenschaft,
Berlin, 1963, 492.
[6] Acetaldehyde. In Ullmann’s Encyclopedia of Industrial Chemistry, 7. Aufl., Wiley-
VCH: Weinheim, 2006.
[7] K. Weissermel, H.-J. Arpe, Industrielle Organische Chemie – Bedeutende Vor- und
Zwischenprodukte, 3. Auflage, VCH, Weinheim – Basel – Cambridge – New York,
1988, S. 97.
[8] N. Ohyabu, T. Nishikawa, M. Isobe, J. Am. Chem. Soc. 2003, 125, 8798.
[9] M. S. Newman, J. Am. Chem. Soc. 1953, 75, 4740.
[10] M. Nishizawa, M. Skwarczynski, H. Imagawa, T. Sugihara, Chem. Lett. 2002, 12.
[11] a) Y. Fukuda, K. Utimoto, J. Org. Chem. 1991, 56, 3729; b) Y. Fukuda, K. Utimoto,
Bull. Chem. Soc. Jpn. 1991, 64, 2013.
[12] a) J. Halpern, B. R. James, A. L. W. Kemp, J. Am. Chem. Soc. 1966, 88, 5142; b) M.
Tokunaga, Y. Wakatsuki, Angew. Chem. Int. Ed. 1998, 37, 2867; Angew. Chem. 1998,
110, 3024; c) T. Suzuki, M. Tokunaga, Y. Wakatsuki, Org. Lett. 2001, 3, 735; d) M.
Tokunaga, T. Suzuki, N. Koga, T. Fukushima, A. Horiuchi, Y. Wakatsuki, J. Am.
Chem. Soc. 2001, 123, 11917; e) D. B. Grotjahn, C. D. Incarvito, A. L. Rheingold,
Angew. Chem. Int. Ed. 2001, 40, 3884; Angew. Chem. 2001, 113, 4002; f) A. Labonne,
T. Kribber, L. Hintermann, Org. Lett. 2006, 8, 5853; g) T. Kribber, A. Labonne, L.
Hintermann, Synthesis 2007, 2809.
[13] a) J. Halpern, B. R. James, A. L. W. Kemp, J. Am. Chem. Soc. 1961, 83, 4097; b) M.
M. Taqui Khan, S. B. Halligudi, S. Shukla, J. Mol. Catal. 1990, 58, 299; c) I. K.
Meier, J. A. Marsella, J. Mol. Catal. 1993, 78, 31.
[14] J. Blum, H. Huminer, H. Alper, J. Mol. Catal. 1992, 75, 153.

A Literaturverzeichnis
127
[15] a) C. S. Chin, W.-T. Chang, S. Yang, K.-S. Joo, Bull. Korean Chem. Soc. 1997, 18,
324; b) T. Hirabayashi, Y. Okimoto, A. Saito, M. Morita, S. Sakaguchi, Y. Ishii,
Tetrahedron Lett. 2006, 62, 2231.
[16] a) S. Y. Kim, C. S. Chin, M. S. Eum, J. Mol. Catal. A: Chem. 2006, 253, 245; b) S.
Ogo, K. Uehara, T. Abura, Y. Watanabe, S. Fukuzumi, J. Am. Chem. Soc. 2004, 126,
16520.
[17] a) W. C. Hiscox, P. W. Jennings, Organometallics 1990, 9, 1997; b) J. W. Hartman, W.
C. Hiscox, P. W. Jennings, J. Org. Chem. 1993, 58, 7613.
[18] W. Baidossi, M. Lahav, J. Blum, J. Org. Chem. 1997, 62, 669.
[19] a) I. K. Meier, J. A. Marsella, J. Mol. Catal. 1993, 78, 31; b) N. X. Hu, Y. Aso, T.
Otsubo, F. Ogura, Tetrahedron Lett. 1986, 27, 6099.
[20] Für weitere Informationen: L. Hintermann, A. Labonne, Synthesis 2007, 1121.
[21] K. Imi, K. Imai, K Utimoto, Tetrahedron Lett. 1987, 28, 3127.
[22] a) J. H. Teles, S. Brode, M. Chabanas, Angew. Chem. Int. Ed. 1998, 37, 1415; b) M.
Schulz, J. H. Teles, U.S. Patent 6037482, 2000; Chem. Abstr. 1997, 127, 121499; c) E.
Mizushima, K. Sato, T. Hayashi, M. Tanaka, Angew. Chem. Int. Ed. 2002, 41, 4563;
Angew. Chem. 2002, 114, 4745; d) R. Casado, M. Contel, M. Laguna, P. Romero, S.
Sanz, J. Am. Chem. Soc. 2003, 125, 11925.
[23] SPhos = 2-Dicyclohexylphosphino-2',6'-dimethoxybiphenyl.
[24] NTf2 = Bis(trifluoromethansulfonyl)imidat.
[25] A. Leyva, A. Corma, J. Org. Chem. 2009, 74, 2067.
[26] N. Marion, R. Ramon, S. Nolan, J. Am. Chem. Soc. 2009, 131, 448.
[27] a) S. Patai (Hrsg.), The Chemistry of the Carbon–Carbon Triple Bond, P. F. Hudrlik,
A. M. Hudrlik, John Wiley & Sons, Chichester, 1978, 240; b) W. L. Budde, R. E.
Dessy, J. Am. Chem. Soc. 1963, 85, 3964; c) M. Bassetti, B. Floris, Gazz. Chim. Ital.
1986, 116, 595; d) M. Bassetti, B. Floris, G. Illuminati, Organometallics 1985, 4, 617;
e) M. Bassetti, B. Floris, J. Org. Chem. 1986, 51, 4140; f) M. Bassetti, B. Floris, J.
Chem. Soc., Perkin Trans. 2 1988, 227.
[28] a) F. Alonso, I. P. Beletskaya, M. Yus, Chem. Rev. 2004, 104, 3079; b) M. Beller, J.
Seayad, A. Tillack, H. Jiao, Angew. Chem. Int. Ed. 2004, 43, 3368.
[29] P. W. Jennings, J. W. Hartman, W. C. Hiscox, Inorg. Chim. Acta 1994, 222, 317.
[30] a) J. March, Advanced Organic Chemistry, 4. Aufl., Wiley, New York, 1992, S. 787; b)
H. Sakurai, M. Ando, N. Kawada, K. Sato, A. Hosomi, Tetrahedron Lett. 1986, 27, 75.
[31] M. A. Bennett, A. K. Smith, J. Chem. Soc., Dalton Trans. 1974, 233.

A Literaturverzeichnis
128
[32] M. Tokunaga, Y. Wakatsuki, Jap. Patent 11319576 A2, 1999; Chem. Abstr. 1999, 131,
352828.
[33] Cp = η5-Cyclopentadienyl; R = Aryl, Alkyl, verbrückende Alkyle; X = Cl, nicht
nukleophile Gegenionen.
[34] T. Suzuki, Y. Wakatsuki, M. Tokunaga, Jap. Patent 2002114730 A2, 2002; Chem.
Abstr. 2002, 136, 309681.
[35] dppm = Bis(diphenylphosphino)methan; dppe = 1,2-Bis(diphenylphosphino)ethan;
dppp = 1,3-Bis(diphenylphosphino)propan; dppb = 1,4-Bis(diphenylphosphino)butan.
[36] Py-BOX-iPr = 2,6-Bis(4-iso-propyl-2-oxazolin-2-yl)pyridin.
[37] a) D. B. Grotjahn, D. A. Lev, J. Am. Chem. Soc. 2004, 126, 12232; b) D. B. Grotjahn,
Chem. Eur. J. 2005, 11, 7146.
[38] B. Breit, F. Chevallier, Angew. Chem. Int. Ed. 2006, 45, 1599; Angew. Chem. 2006,
118, 1629.
[39] J. Baur, H. Jacobsen, P. Burger, G. Artus, H. Berke, L. Dahlenburg, Eur. J. Inorg.
Chem. 2000, 7, 1411.
[40] Hinweis: Komplex 7 ist kommerziell erhältlich (Strem Chemicals Inc., 250mg = 122
€).
[41] Preis: 1g = 366 € (Strem Chemicals Inc.).
[42] A. Labonne, Dissertation, RWTH Aachen, 2007.
[43] E. P. Kündig, F. R. Monnier, Adv. Synth. Catal. 2004, 346, 901.
[44] C. Bianchini, J. A. Casares, M. Peruzzini, A. Romerosa, F. Zanobini, J. Am. Chem.
Soc. 1996, 118, 4585.
[45] D. B. Grotjahn, V. Miranda-Sato, E. J. Kragulj, D. A. Lev, G. Erdogan, X. Zeng, A. L.
Cooksy, J. Am. Chem. Soc. 2008, 130, 20.
[46] siehe Ligand in Komplex 6.
[47] M. Pecul, J. Leszczynski, J. Sadlej, J. Chem. Phys. 2000, 112, 7930.
[48] D. B. Grotjahn, E. J. Kragulj, C. D. Zeinalipour-Yazdi, V. Miranda-Sato, D. A. Lev,
A. L. Cooksy, J. Am. Chem. Soc. 2008, 130, 10860.
[49] D. B. Grotjahn, Dalt. Trans. 2008, 6497.
[50] M. Tokunaga, J. F. Larrow, F. Kakiuchi, E. N. Jacobsen, Science 1997, 277, 936.
[51] Teile dieser Ergebnisse wurden zusammen mit Christian Matzen im Rahmen seiner
Forschungsarbeit erstellt.
[52] Von Li Xiao synthetisiert, unveröffentlichte Ergebnisse.
[53] W. Reppe, Liebigs Ann. Chem. 1955, 596, 1.

A Literaturverzeichnis
129
[54] a) D. E. Frantz, R. Fässler, C. S. Tomooka, E. M. Carreira, Acc. Chem. Res. 2000, 33,
373; b) D. Boyall, D. E. Frantz, E. M. Carreira, Org. Lett. 2002, 4, 2605; c) H. Sasaki,
D. Boyall, E. M. Carreira, Helv. Chim. Acta 2001, 84, 964; d) N. K. Anand, E. M.
Carreira, J. Am. Chem. Soc. 2001, 123, 9687.
[55] K. Matsumura, S. Hashiguchi, T. Ikariya, R. Noyori, J. Am. Chem. Soc. 1997, 119,
8738.
[56] E. J. Corey, C. J. Helal, Angew. Chem. 1998, 110, 2092.
[57] Teile dieser Ergebnisse wurden zusammen mit Eva Paciok im Rahmen ihrer
Forschungsarbeit (01.03.-12.04.2006) erstellt.
[58] a) H. M. R. Hoffmann, J. Rabe, Angew. Chem. p Int. Ed. Engl. 1985, 24, 94; b) E.
Negishi, M. Kotora, Tetrahedron 1997, 53, 6707, c) I. J. Collins, Chem. Soc., Perkin
Trans. 1 1999, 1377; d) N. B. Carter, A. E. Nadany, J. B. Sweeney, J. Chem. Soc.,
Perkin Trans. 1 2002, 2324.
[59] J. Macro, M. Carda, J. Murga, E. Falomir, Tetrahedron 2007, 63, 2929.
[60] S. J. Abe, J. Chem. Soc. Jpn. 1937, 58, 246.
[61] G. Sabitha, P. Gopal, J. S. Yadav, Synth. Commun. 2007, 37, 1495.
[62] Polyacetat-Pyron Naturstoffe: a) S. E. Drewes, B. M. Sehlapelo, M. M. Horn, R. Scott-
Shaw, P. Sandor, Phytochem. 1995, 38, 1427; b) J. O. Andrianaivoravelona, S. Sahpaz,
C. Terreaux, K. Hostettmann, H. Stoeckli-Evans, J. Rasolondramanitra, Phytochem.
1999, 52, 265; c) S. Tosaki, Y. Horiuchi, T. Nemoto, T. Ohshima, M. Shibasaki, Chem.
Eur. J. 2004, 10, 1527.
[63] GRUBBS I Katalysator: Benzyliden-bis(tricyclohexylphosphin)-dichlororuthenium.
[64] Chemikalie über Alfa Aesar bezogen, Produktnummer L19045.
[65] Chemikalie über Alfa Aesar bezogen, Produktnummer L19046.
[66] P. Lemieux, Can. J. Chem. 1951, 29, 415.
[67] Basiert in Teilen auf der Publikation: T. Kribber, A. Labonne, H. Hintermann,
Synthesis 2007, 2809.
[68] a) M. B. Smith, J. March, Advanced Organic Chemistry, 5. Aufl., John Wiley & Sons:
New York, 2001, 1514; b) C. F. Cullis, A. Fish, in The Chemistry of the Carbonyl
Group, S. Patai, Hrsg., Interscience: London, 1966, 79.
[69] a) N. Feuerbacher, F. Vögtle, Top. Curr. Chem. 1998, 197, 1; b) T.-L. Ho, Tactics of
Organic Synthesis, John Wiley & Sons: New York, 1994, 15.
[70] C. R. Russell, K. Alexander, W. O. Erickson, L. S. Hafner, L. E. Schniepp, J. Am.
Chem. Soc. 1952, 74, 4543.

A Literaturverzeichnis
130
[71] a) V. Caprio, M. A. Brimble, D. P. Furkert, Tetrahedron 2001, 57, 4023; b) K. Meilert,
M. A. Brimble, Org. Biomol. Chem. 2006, 4, 2184.
[72] a) P. Knochel, in Science of Synthesis, Vol. 3; I. O’Neil, Hrsg., Thieme: Stuttgart,
2003, 5; b) J.-L. Moreau, in The Chemistry of Ketenes, Allenes and Related
Compounds, S. Patai, Hrsg., John Wiley: Chichester, 1980, 363; c) M. Gaudemar, in
Organometallic Syntheses, Vol. 3, R. B. King, J. J. Eisch, Hrsg., Elsevier: Amsterdam,
1986, 409; d) M. Gaudemar, Ann. Chim. (Paris) 1956, 1, 161.
[73] a) A. Yanagisawa, S. Habaue, H. Yamamoto, Tetrahedron 1992, 48, 1969; b) A.
Yanagisawa, S. Habaue, H. Yamamoto, J. Org. Chem. 1989, 54, 5198.
[74] W.-L. Wu, Z.-J. Yao, Y.-L. Li, J.-C. Li, Y. Xia, Y.-L. Wu, J. Org. Chem. 1995, 60,
3257.
[75] a) M. V. Papadopoulou, Chem. Ber. 1989, 122, 2017; b) H. B. Henbest, E. R. H.
Jones, I. M. S. Walls, J. Chem. Soc. 1949, 2696.
[76] a) L. W. Bieber, M. F. da Silva, R. C. da Costa, L. O. S. Silva, Tetrahedron Lett. 1998,
39, 3655; b) I. Yavari, F. Riazi-Kermani, Synth. Commun. 1995, 25, 2923.
[77] J. Inanaga, K. Hirata, H. Saeki, T. Katsuki, M. Yamaguchi, Bull. Chem. Soc. Jpn.
1979, 52, 1989.
[78] M. D. Burke, S. L. Schreiber, Angew. Chem. Int. Ed. 2004, 43, 46; Angew. Chem.
2004, 116, 48.
[79] a) C. Lai, J. A. Soderquist, Org. Lett. 2005, 7, 799; b) S. Konishi, H. Hanawa, K.
Maruoka, Tetrahedron: Asymmetry 2003, 14, 1603; c) S. E. Denmark, T. Wynn, J.
Am. Chem. Soc. 2001, 123, 6199; d) T.-P. Loh, M.-J. Lin, K.-L. Tan, Tetrahedron Lett.
2003, 44, 507; e) G. E. Keck, D. Kirshnamurthy, X. Chen, Tetrahedron Lett. 1994, 35,
8323; f) E. J. Corey, C.-M. Yu, D.-H. Lee, J. Am. Chem. Soc. 1990, 112, 878; g) G. P.
Boldrini, L. Lodi, E. Tagliavini, C. Tarasco, C. Trombini, A. Umani-Rochi, J. Org.
Chem. 1987, 52, 5447; h) N. Ikeda, I. Arai, H. Yamamoto, J. Am. Chem. Soc. 1986,
108, 483.
[80] a) M. Inoue, M. Nakada, Org. Lett. 2004, 6, 2977; und zitierte Literatur; b) M.
Bandini, P. G. Cozzi, P. Melchiorre, R. Tino, A. Umani-Rochi, Tetrahedron:
Asymmetry 2001, 12, 1063; c) C.-M. Yu, S.-K. Yoon, H.-S. Choi, K. Baek, Chem.
Commun. 1997, 763.
[81] D. Seebach, A. K. Beck, A. Heckel, Angew. Chem. Int. Ed. 2001, 40, 92; Angew.
Chem. 2001, 113, 96.

A Literaturverzeichnis
131
[82] a) R. S. Coleman, S. R. Gurrala, S. Mitra, A. Raao, J. Org. Chem. 2005, 70, 8932; b)
CSD-code: RAXGUT.
[83] B. Bernet, A. Vasella, Helv. Chim. Acta 2000, 83, 995.
[84] H. Günther, NMR-Spektroskopie, Thieme: Stuttgart, 1992.
[85] D. F. Shriver, M. D. Drezdzon, The Manipulation of Air-Sensitive Compounds, 2.
Aufl., Wiley, Chichester, 1986.
[86] H. Witte, W. Seelinger, Liebigs Ann. Chem. 1974, 996.
[87] Das Pivaloylanilid wurde nach Literaturvorschrift hergestellt: W. M. Seganish, P.
DeShong, J. Org. Chem. 2004, 69, 6790.
[88] Zum Entgasen des Lösungsmittels wurde für einige Minuten Argon durchgeleitet. [89] E. P. Kündig, F. R. Monnier, Adv. Synth. Catal. 2004, 346, 901.
[90] J. A. S. Howell, N. Fey, J. D. Lovatt, P. C. Yates, P. McArdle, D. Cunningham, E.
Sadeh, H. E. Gottlieb, Z. Goldschmidt, M. B. Hursthouse, M. E. Light, J. Chem. Soc.,
Dalton Trans. 1999, 3015.
[91] T. E. Müller, J. C. Green, D. M. P. Mingos, C. M. McPartlin, C. Whittingham, D. J.
Williams, T. M. Woodroffe, J. Organomet. Chem. 1998, 551, 313.
[92] M. Tiecco, L. Testaferri, A. Temperini, L. Bagnoli, F. Marini, C. Santi, R. Terlizzi,
Eur. J. Org. Chem. 2004, 16, 3447.
[93] K. F. Bernady, M. B. Floyd, J. F. Poletto, M. J. Weiss, J. Org. Chem. 1979, 44, 1438.
[94] R. C. Larock, K. Narayanan, Tetrahedron 1988, 44, 6995; D. S. Brown, S. V. Ley, S.
Vile, M. Thompson, Tetrahedron 1991, 47, 1329; D. Hickman, K. Hodgetts, P.
Mackman, T. Wallace, J. M. Wardleworth, Tetrahedron 1996, 52, 2235.
[95] C. Ensch, M. Hesse, Helv. Chim. Act. 2003, 86, 233.
[96] D. A. Evans, T. C. Crawford, R. C. Thomas, J. A. Walker, 1976, 41, 3947.
[97] M. S. Wolfe, Synth. Commun. 1997, 27, 2975.
[98] K. Nakamura, K. Takenaka, Tetrahedron: Asymmetry 2002, 13, 415.
[99] J.-L. Gras, Y.-Y. K. W. Chang, A. Guerin, Synthesis 1985, 74.
[100] A. Lee, Y. Hu, S. Chu, Tetrahedron 2001, 57, 2121.
[101] R. A. W. Johnstone, M. E. Rose, Tetrahedron 1979, 35, 2169.
[102] Y. Fukuda, K. Utimoto, Bull. Chem. Soc. Jpn. 1991, 64, 2013.
[103] Y. Suzuki, S.J. Danko, Y. Kono, J.M. Daly, S. Takeuchi, Agric Biol.Chem. 1985, 49,
149.
[104] T. Mino, S. Masuda, M. Nishio, M. Yamashita, J. Org. Chem. 1997, 62, 2633.

A Literaturverzeichnis
132
[105] E. A. Hill, W. A. Boyd, H. Desai, A. Darki, L. Bivens, J. Organomet. Chem. 1996,
514, 1.
[106] H. Acharya, Y. Kobayashi, Tetrahedron 2006, 62, 3329.
[107] A. Abiko, J. C. Roberts, T. Takemasa, S. Masmune, Tetrahedron Lett. 1986, 27, 4537.
[108] D. Blanc, J. Madec, F. Popowyck, T. Ayad, P. Phansavath, V. Ratovelomanana-Vidal,
J.-P. Genet, Adv. Synth. Catal. 2007, 349, 943.
[109] K. Behrens, B. O. Kneisel, M. Noltemeyer, R. Brückner, Liebigs Ann. 1995, 385.

B Abkürzungsverzeichnis
133
B Abkürzungsverzeichnis
λ Wellenlänge
ν Wellenzahl
AAV Allgemeine Arbeitsvorschrift
abs. absolut
Äquiv Äquivalent(e)
Ar Arylische Protonen (bei NMR-Analytik)
Bn Benzyl
br breit
Bz Benzoyl
CDCl3 deuteriertes Chloroform
CH2Cl2 oder DCM Dichlormethan
Cp η5-Cyclopentadienyl
Cy Cyclohexyl
d Dublett (bei NMR-Analytik)
DC Dünnschichtchromatographie
dest. Destilliert
DMAP 4-(Dimethylamino)-pyridin
DMF N,N-dimethylformamid
dppb 1,4-Bis(diphenylphosphino)butan
dppe 1,2-Bis(diphenylphosphino)ethan
dppm Bis(diphenylphosphino)methan
dppp 1,3-Bis(diphenylphosphino)propan
EI Elektronenstoßionisation
ESI Elektornengasionisation
Et Ethyl
EtOAc Ethylacetat
EtOH Ethanol
eV Elektronenvolt
GC/MS Gaschromatographie/Massenspektroskopie
ges. gesättigter
h Stunden

B Abkürzungsverzeichnis
134
HCl Salzsäure
HPLC High Performance Liquid Chromatographie
HV Hochvakuum
i iso
iPrOH Isopropanol
IR Infrarotspektroskopie
J Kopplungskonstante
konz. Konzentriert
L Ligand
m meta
Me Methyl
MeCN Acetonitril
MgSO4 Magnesiumsulfat
MOM Methoxymethyl
Naph oder Naphth Naphthalin
NaSO4 Natriumsulfat
NMR(-Spektroskopie) (engl.: nuclear magnetic resonance) kernmagnetische
Resonanzspektroskopie
p para
Ph Phenyl
ppm parts per million
q Quartett
R organischer Rest
Rf ratio to front
Rt Retentionszeit
rac racemisch
RT Raumtemperatur
s Singulett
t Triplett
t oder tert tertiär
TBME tert-Butylmethylether
TBS tert-Butyldimethylsilan
TFA Trifluoressigsäure
TfOH Trifluormethansulfonsäure

B Abkürzungsverzeichnis
135
THF Tetrahydrofuran
THP Tetrahydronpyran
TMS Trimethylsilyl
TMSCl Trimethylsilylchlorid
UV ultraviolett

C Danksagung
136
C Danksagung
Mein besonderer Dank gilt meinem Doktorvater, Herrn PD Dr. Lukas Hintermann, für die mir
gebotene Möglichkeit in seinem Arbeitskreis dieses Forschungsthema bearbeiten zu können
und für die hervorragenden Arbeitsbedingungen.
Herrn Prof. Dr. Carsten Bolm danke ich für die freundliche Übernahme des Korreferats und
die Unterstützung während der letzten Jahre bei der Erstellung der Arbeit.
Mein nächster Dank gebührt allen Mitarbeitern der analytischen Abteilungen, der Werkstatt
und der Chemikalienausgabe des Instituts für Organische Chemie der RWTH Aachen
University, ohne deren Hilfe die Anfertigung dieser Arbeit kaum möglich gewesen wäre.
Herrn Prof. Dr. Ulli Englert und seinen Mitarbeitern danke ich für die Hilfe bei der Lösung
der Kristallstruktur.
Ich danke allen derzeitigen und ehemaligen Mitgliedern des Arbeitskreises Hintermann und
Bolm für die schöne gemeinsame Zeit und die gute Arbeitsatmosphäre. Mein spezieller Dank
richtet sich dabei an meine Arbeitskreiskollegen Aurélie Labonne, die mir dieses
Forschungsthema schon während meiner Examensarbeit näher gebracht hat, und Marco
Schmitz für die zahlreichen Gespräche. Des Weiteren Dr. Jörg Sedelmeier für seine
Freundschaft und die vielen hilfreichen Diskussion, ebenso wie Agathe Mayer und Anke
Krebs mit ihren offenen Ohren und Anregungen. Herzlich danken möchte ich auch meiner
lieben Kollegin Christiane Metje für ihre Hilfe und ihre Freundschaft. Ohne sie wären die
Mittagspausen wesentlich langweiliger geworden und sie war stets für mich auch außerhalb
der Chemie in manch schwieriger Situation da.
Bei Anke Krebs, Agathe Mayer, und Dr. Jörg Sedelmeier möchte ich mich für das zügige und
gewissenhafte Korrekturlesen dieser Doktorarbeit bedanken.
Meinen Forschungsstudenten Eva Paciok und Christian Matzen danke ich für ihre engagierte
Mitarbeit in meinem Labor.

C Danksagung
137
Dem „Daumenfussball“- und AoE-Team, Dr. Jörg Sedelmeier, Marcus Frings, Dr. Toni
Rantanen, Dr. Lorenzo Zani, Hans-Bernd Mieskes und Florian Boeck gilt der Dank für
zahlreiche hart umkämpfte Partien in sportlich fairer und feucht fröhlicher Atmosphäre.
Ich danke auch meinen langjährigen Freunden aus der Schulzeit, die immer dann für
Abwechselung gesorgt haben, wenn man sie gebraucht hat und stets ein offenes Ohr hatten.
Mein größter Dank geht jedoch an meine Familie, die mich auf dem gesamten Weg immer
unterstützt hat und für mich da war. Sie hat erst all dies möglich gemacht.

D Lebenslauf
138
D LEBENSLAUF
Persönliche Daten
Name: Thomas Clemens Kribber
Geburtsdatum: 01. September 1981
Geburtsort: Haselünne
Staatsangehörigkeit: deutsch
Familienstand: ledig
Schulausbildung
1988–1992 Paulusschule in Haselünne
1992–1994 Orientierungsschule in Haselünne
1994–2001 Kreisgymnasium St. Ursula in Haselünne
Akademischer Werdegang
09/ 2001–11/2005 Studium der Fächer Chemie und kath. Religionslehre für die
Lehrämter für die Sekundarstufe II und für die Sekundarstufe I
an der RWTH Aachen University. Examensarbeit unter der
Leitung von Prof. Dr. Carsten Bolm an der RWTH Aachen
University.
12/2005–07/2009 Promotion in der Organischen Chemie unter der Leitung von
PD Dr. Lukas Hintermann am Institut für Organische Chemie
der RWTH Aachen University.


















