
Seminar P6 – Elektrolytische Leitfähigkeit: Kapitelübersicht P6 PC Ph... · Elektrolyt, HCl ist...
12
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL Physikalisch-Chemisches Grundpraktikum für Pharmazeuten Seminar P6 – Elektrolytische Leitfähigkeit: Kapitelübersicht 1. Allgemeine Feststellungen 1.1. Die Besonderheiten von Elektrolytlösungen 1.2. Die Klassifikation von Elektrolyten 2. Die DEBYE-HÜCKEL-Theorie 2.1. Einleitung 2.2. Zentralion und Ionenwolke 2.3. Das DEBYE-HÜCKEL-Grenzgesetz 2.4. Der mittlere HENRYsche Aktivitätskoeffizient 3. Die Migration von Ionen im elektrischen Feld 3.1. Kräftebilanz und Driftgeschwindigkeit 3.2. Von der Driftgeschwindigkeit abgeleitete Größen 3.3. Trends bei der Ionengrenzleitfähigkeit 3.4. Der GROTTHUß-Mechanismus 4. Die Leitfähigkeit von Elektrolytlösungen 4.1. Widerstand, Leitwert, spezifischer Widerstand und Leitfähigkeit 4.2. Molare Leitfähigkeit und Äquivalentleitfähigkeit 4.3. Das Gesetz der unabhängigen Ionenwanderung 4.4. Die Konzentrationsabhängigkeit der Leitfähigkeit starker Elektrolyte 4.5. Die Konzentrationsabhängigkeit der Leitfähigkeit schwacher Elektrolyte 4.6. Die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit starker Elektrolyte 4.7. Die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit schwacher Elektrolyte PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL Physikalisch-Chemisches Grundpraktikum für Pharmazeuten Seminar P6 – Elektrolytische Leitfähigkeit 1. Allgemeine Feststellungen 1.1. Die Besonderheiten von Elektrolytlösungen Die Lösungen von Elektrolyten, die also Ladungsträger (Ionen) enthalten, verdienen gegenüber anderen Lösungen in mehrfacher Hinsicht besondere Aufmerksamkeit: Zum einen, weil die COULOMBschen Wechselwirkungen zwischen den geladenen Teilchen besonders stark und langreichweitig gegenüber anderen Wechselwirkungen sind und daher deutlicher zur Strukturierung der Lösung beitragen und ihre Eigenschaften in Abhängigkeit von der Konzentration stärker beeinflussen, zum anderen, weil Elektrolytlösungen den elektrischen Strom transportieren können, wobei auch hier Abhängigkeiten wie von der Konzentration oder der Temperatur existieren. 1.2. Die Klassifikation von Elektrolyten Zunächst sollen zwei Systeme zur Klassifikation von Elektrolyten vorgestellt werden. Erstens wird zwischen echten und potentiellen Elektrolyten unterschieden: Im Falle echter Elektrolyte liegen die Ionen bereits im Reinstoff vor, bspw. bei Natriumchlorid (NaCl). Im Falle potentieller Elektrolyte entstehen die Ionen hingegen erst im Kontakt mit dem Lösungsmittel, bspw. bei Chlorwasserstoff in Wasser (HCl; Salzsäure). Zweitens wird zwischen starken und schwachen Elektrolyten unterschieden: Starke Elektrolyte liegen im gesamten möglichen Konzentrations- bereich vollständig dissoziiert vor, bspw. Kaliumbromid (KBr) in Wasser, wohin- gegen für schwache Elektrolyte der Dissoziationsgrad stark von der Konzentra- tion abhängt, bspw. im Fall wässriger Essigsäure-Lösungen (HOAc). Ein in wässriger Lösung starker Elektrolyt verhält sich in anderen Lösungsmitteln aber unter Umständen wie ein typischer schwacher Elektrolyt. Ein Elektrolyt kann nach beiden Systemen klassifiziert werden; so ist NaCl, ebenso wie KBr, ein echter und starker Elektrolyt, HCl ist ein potentieller und starker Elektrolyt, HOAc schließlich ein potentieller und schwacher Elektrolyt. 2. Die DEBYE-HÜCKEL-Theorie 2.1. Einleitung Die Nettoladung ungleich Null der Ionen führt zu interionischen elektrostatischen Wechselwirkungen (wobei sich Kationen gegenseitig abstoßen, ebenso Anionen untereinander, während sich Kationen und Anionen gegenseitig anziehen) und auch zu signifikanten Wechselwirkungen mit dipolaren Lösungsmittelteilchen (sodass sich
Transcript of Seminar P6 – Elektrolytische Leitfähigkeit: Kapitelübersicht P6 PC Ph... · Elektrolyt, HCl ist...

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
Physikalisch-Chemisches Grundpraktikum für Pharmazeuten
Seminar P6 – Elektrolytische Leitfähigkeit: Kapitelübersicht
1. Allgemeine Feststellungen
1.1. Die Besonderheiten von Elektrolytlösungen
1.2. Die Klassifikation von Elektrolyten
2. Die DEBYE-HÜCKEL-Theorie
2.1. Einleitung
2.2. Zentralion und Ionenwolke
2.3. Das DEBYE-HÜCKEL-Grenzgesetz
2.4. Der mittlere HENRYsche Aktivitätskoeffizient
3. Die Migration von Ionen im elektrischen Feld
3.1. Kräftebilanz und Driftgeschwindigkeit
3.2. Von der Driftgeschwindigkeit abgeleitete Größen
3.3. Trends bei der Ionengrenzleitfähigkeit
3.4. Der GROTTHUß-Mechanismus
4. Die Leitfähigkeit von Elektrolytlösungen
4.1. Widerstand, Leitwert, spezifischer Widerstand und Leitfähigkeit
4.2. Molare Leitfähigkeit und Äquivalentleitfähigkeit
4.3. Das Gesetz der unabhängigen Ionenwanderung
4.4. Die Konzentrationsabhängigkeit der Leitfähigkeit starker Elektrolyte
4.5. Die Konzentrationsabhängigkeit der Leitfähigkeit schwacher Elektrolyte
4.6. Die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit starker Elektrolyte
4.7. Die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit schwacher Elektrolyte
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
Physikalisch-Chemisches Grundpraktikum für Pharmazeuten
Seminar P6 – Elektrolytische Leitfähigkeit
1. Allgemeine Feststellungen
1.1. Die Besonderheiten von Elektrolytlösungen
Die Lösungen von Elektrolyten, die also Ladungsträger (Ionen) enthalten, verdienen gegenüber anderen Lösungen in mehrfacher Hinsicht besondere Aufmerksamkeit: Zum einen, weil die COULOMBschen Wechselwirkungen zwischen den geladenen Teilchen besonders stark und langreichweitig gegenüber anderen Wechselwirkungen sind und daher deutlicher zur Strukturierung der Lösung beitragen und ihre Eigenschaften in Abhängigkeit von der Konzentration stärker beeinflussen, zum anderen, weil Elektrolytlösungen den elektrischen Strom transportieren können, wobei auch hier Abhängigkeiten wie von der Konzentration oder der Temperatur existieren.
1.2. Die Klassifikation von Elektrolyten
Zunächst sollen zwei Systeme zur Klassifikation von Elektrolyten vorgestellt werden. Erstens wird zwischen echten und potentiellen Elektrolyten unterschieden: Im Falle echter Elektrolyte liegen die Ionen bereits im Reinstoff vor, bspw. bei Natriumchlorid (NaCl). Im Falle potentieller Elektrolyte entstehen die Ionen hingegen erst im Kontakt mit dem Lösungsmittel, bspw. bei Chlorwasserstoff in Wasser (HCl; Salzsäure). Zweitens wird zwischen starken und schwachen Elektrolyten unterschieden: Starke Elektrolyte liegen im gesamten möglichen Konzentrations-bereich vollständig dissoziiert vor, bspw. Kaliumbromid (KBr) in Wasser, wohin-gegen für schwache Elektrolyte der Dissoziationsgrad stark von der Konzentra-tion abhängt, bspw. im Fall wässriger Essigsäure-Lösungen (HOAc). Ein in wässriger Lösung starker Elektrolyt verhält sich in anderen Lösungsmitteln aber unter Umständen wie ein typischer schwacher Elektrolyt. Ein Elektrolyt kann nach beiden Systemen klassifiziert werden; so ist NaCl, ebenso wie KBr, ein echter und starker Elektrolyt, HCl ist ein potentieller und starker Elektrolyt, HOAc schließlich ein potentieller und schwacher Elektrolyt.
2. Die DEBYE-HÜCKEL-Theorie
2.1. Einleitung
Die Nettoladung ungleich Null der Ionen führt zu interionischen elektrostatischen Wechselwirkungen (wobei sich Kationen gegenseitig abstoßen, ebenso Anionen untereinander, während sich Kationen und Anionen gegenseitig anziehen) und auch zu signifikanten Wechselwirkungen mit dipolaren Lösungsmittelteilchen (sodass sich

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
die Teilchen der Solvathülle tendenziell mit ihren partiell positiv geladenen Bereichen zum Ion hinwenden, falls es ein Anion ist, und mit ihren partiell negativ geladenen Bereichen, falls es ein Kation ist). Im Rahmen der DEBYE-HÜCKEL-Theorie wird das Lösungsmittel allerdings als strukturloses, kontinuierliches Medium (Dielektrikum) betrachtet.
2.2. Zentralion und Ionenwolke
Aufgrund der Abstoßung gleichnamiger Ladungen und der Anziehung ungleich-namiger Ladungen ist jedes Ion im zeitlichen Mittel mit mehr entgegengesetzter Ladung umgeben als mit gleicher Ladung. Jedes Ion umgibt sich mit einer Ionenwolke, für welche es das Zentralion darstellt. In dieser Ionenwolke befinden sich sowohl Kationen als auch Anionen, aber unmittelbar um das Zentralion herum ist die Ladungsdichte der zum Zentralion entgegengesetzt geladenen Ionen im Mittel höher als die gleich geladener Ionen. Je weiter man sich vom Zentralion entfernt, desto geringer wird die (betragsmäßige) Ladungsdichte, positive und negative Ladungen kommen mit immer ähnlicherer Wahrscheinlichkeit vor. Da die Wechsel-wirkungen zwischen allen Ionen der Lösung auftreten, ist jedes Ion gleichzeitig Zentralion für seine eigene Ionenwolke und Teil der Ionenwolken aller anderen Ionen. Dieser Strukturierung der Lösung wirkt die thermische Bewegung der Teilchen entgegen, durch welche die Ionenwolken in sehr kurzen Zeiten aufgelöst werden, sodass sie sich wieder rasch nachbilden müssen, nur um sich erneut bald aufzulösen.
2.3. Das DEBYE-HÜCKEL-Grenzgesetz
Aufgrund der Stärke und langen Reichweite der COULOMB-Kräfte weichen Elektrolytlösungen schon bei deutlich kleineren Konzentrationen signifikant vom Modell der ideal verdünnten Lösung ab als Lösungen von Nichtelektrolyten. Die Berücksichtigung des HENRYschen Aktivitätskoeffizienten (siehe P4 und P5) für den gelösten Stoff (bspw. bei der Analyse der Löslichkeit) oder des osmotischen Koeffizienten bei der Analyse kolligativer Eigenschaften (siehe P3) ist also bereits bei deutlich höheren Verdünnungen notwendig. Aus dem Wechselspiel zwischen elektrostatischen Kräften und thermischer Bewegung kann auf komplizierte Art und Weise eine Gleichung zur Berechnung des HENRYschen Aktivitätskoeffizienten der Spezies mit der Ladungszahl in verdünnten Elektrolytlösungen hergeleitet werden, das DEBYE-HÜCKEL-Grenzgesetz:
lg ⋅ ⋅ ⊝⁄ . (1)
Dabei ist lg der dekadische Logarithmus von , 0 ein von der Temperatur und dem Lösungsmittel abhängiger Faktor (bspw. ist für Wasser bei 25°C 0,510;wird in Gl. (1) dagegen der natürliche Logarithmus zugrunde gelegt, ist 1,175), ⊝ 1molL die Standard-Stoffmengenkonzentration (zur Bereinigung der Einhei-
ten) und
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
≡12
alle Ionen
(2)
die sogenannte molare Ionenstärke. Sie ist ein verallgemeinertes Maß für die Konzentration, in welches aber alle Ionen in der Lösung eingehen, und zwar quadratisch mit ihrer Ladungszahl und linear mit ihrer Stoffmengenkonzentration
. In dieser Definition besitzt sie auch die Einheit einer Stoffmengenkonzentration (etwa molL ). Die Ionenstärke ist eine Eigenschaft der gesamten Lösung, nicht eines einzelnen Ions. In das DEBYE-HÜCKEL-Grenzgesetz geht neben der Ionenstärke der Lösung – wodurch alle Ionen der Lösung miteinbezogen werden, was sinnvoll ist, da auch alle Ionen wechselwirken und somit zur Abweichung von der idealen Verdünnung beitragen – nur die Ladungszahl desjenigen Ions ein, für welches der Aktivitätskoeffizient berechnet werden soll, nicht aber eine Information über seine chemische Natur.
Das Grenzgesetz kann analog auch mit der molalen Ionenstärke
≡12
alle Ionen
(3)
formuliert werden, wobei dann in der Wurzel durch ⊝ 1molkg geteilt wird. Für einen 1-1-wertigen Elektrolyten ( | | 1, 1) ist als Spezialfall einfach und .
Es ist ersichtlich, dass der HENRYsche Aktivitätskoeffizient für unendliche Verdünnung gegen Eins strebt und für höhere Konzentrationen abnimmt, allerdings nach dem Grenzgesetz stets zwischen Null und Eins liegt (bei viel höheren Konzentrationen wird aber der HENRYsche Aktivitätskoeffizient tatsächlich sogar größer als Eins).
Man spricht vom DEBYE-HÜCKEL-Grenzgesetz, da es streng nur für den Grenzfall unendlicher Verdünnung gilt. Genau genommen liefert das Grenzgesetz die korrekte Tangente an die experimentelle Kurve lg vs. ⊝⁄ bei unendlicher Verdünnung. In der Praxis ist es im Fall von 1-1-wertigen Elektrolyten bis zu Konzentrationen von etwa 0,01molL anwendbar; bei 2-2-wertigen Elektrolyten ist der Anwendungsbereich aber schon auf Konzentrationen kleiner als 0,0005molL beschränkt. Es existieren allerdings erweiterte Formen, die für höhere Konzentrationen brauchbar sind, bspw. bis 1molL .
2.4. Der mittlere HENRYsche Aktivitätskoeffizient
Es existiert zwar für jedes Ion ein eigener HENRYscher Aktivitätskoeffizient. Da aufgrund der Ladungsneutralität der Lösung aber Kationen und Anionen immer nebeneinander vorliegen, ist praktischen Messungen nur ein mittlerer Aktivitäts-koeffizient zugänglich. Allgemein gilt für einen Elektrolyten K A , dass der

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
mittlere Aktivitätskoeffizient für den Elektrolyten das (stöchiometrisch gewichtete) geometrische Mittel der Aktivitätskoeffizienten der beiden einzelnen Ionen ist,
. (4)
Für einen 1-1-wertigen Elektrolyten ist daher
. (5)
Der mittlere Aktivitätskoeffizient eines Elektrolyten kann auch mittels des DEBYE-HÜCKEL-Grenzgesetzes bestimmt werden. Es lautet (unter Berücksichtigung der Elektroneutralitätsbedingung)
lg ⋅ | | ⋅ ⊝⁄ . (6)
3. Die Migration von Ionen im elektrischen Feld
3.1. Kräftebilanz und Driftgeschwindigkeit
Wird in einer Elektrolytlösung ein elektrisches Feld der Feldstärke angelegt, werden die Kationen (inklusive ihrer Solvathüllen) zur Kathode hin beschleunigt, die Anionen (inklusive ihrer Solvathüllen) zur Anode hin. Die beschleunigende Kraft auf ein Teilchen der Ladungszahl (und damit der Ladung ) besitzt den Wert
B, | | . (7)
Durch diese Beschleunigung erhöht sich die Geschwindigkeit der Teilchen. Diese erfahren jedoch auch abbremsende Kräfte. Im Falle einer ideal verdünnten Lösung der (dynamischen) Viskosität ist die einzige solche Kraft die Reibungskraft, die sich für ein kugelförmiges Teilchen mit dem Radius gemäß dem STOKESschen Gesetz berechnet als
R, 6 ⋅ ⋅ ⋅ . (8)
Je höher die Geschwindigkeit ist, auf die die Teilchen vom elektrischen Feld beschleunigt werden, desto höher ist auch die Reibungskraft. Die Teilchen erreichen ihre maximale Geschwindigkeit, die sogenannte Driftgeschwindigkeit D, , wenn sich beschleunigende und abbremsende Kräfte genau kompensieren:
B,D,
R, . (9)
Für eine ideal verdünnte Lösung (Index „0“ wegen → 0) ergibt sich dann die Driftgeschwindigkeit D, , zu
D, ,| |6 . (10)
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
3.2. Von der Driftgeschwindigkeit abgeleitete Größen
Es ist ersichtlich, dass die Driftgeschwindigkeit von der Feldstärke abhängt, und zwar ist sie proportional zu ihr. Zur Einordnung der Angabe einer Drift-geschwindigkeit ist daher immer noch die Angabe der zugehörigen Feldstärke notwendig. Um eine möglichst nur für das Ion selbst charakteristische Größe zu erhalten, wird die Driftgeschwindigkeit durch die Feldstärke geteilt. Die so definierte Größe heißt Ionenbeweglichkeit (in m V s )
≡ D, (11)
und ist (für nicht zu große Feldstärken) von der Feldstärke unabhängig. Die Ionenbeweglichkeit hängt allerdings vom Lösungsmittel und von der Temperatur ab (siehe Gl. (10); die Viskosität ist temperaturabhängig). Für spätere Zwecke ist es nützlich, die Ionenbeweglichkeit mit der FARADAY-Konstante 96500Cmol (sie entspricht der Ladung von einem Mol einfach positiv geladener Ladungsträger) zu multiplizieren; die so gewonnene Größe heißt Ionenäquivalentleitfähigkeit (in Sm mol )
≡ D, . (12)
Die Ionenäquivalentleitfähigkeit berücksichtigt also gewissermaßen den Beitrag zum Leitvermögen, den Ionen leisten, die ein Ladungsäquivalent von 1 Mol Elementar-ladungen transportieren. Der Betrag der Ladung von einem Mol eines einwertigen Ions stimmt ja z. B. überein mit dem Betrag der Ladung von 0,5 Mol eines zweiwertigen Ions (und ist allgemein äquivalent mit 1 | |⁄ Mol eines Ions mit der Ladungszahl ). Der reale Beitrag von 1 Mol Ionen zum Leitvermögen wird direkt von der molaren Ionenleitfähigkeit m, (in Sm mol ) erfasst, die wie folgt definiert ist:
m, ≡ | | | | . (13)
Genaugenommen entspricht der Zahlenwert von m, dem Beitrag zum Leitwert, den 1 Mol der ionischen Spezies zwischen planparallelen Elektroden im Abstand von 1 m leisten. Sowohl als auch m, hängen in nichtidealen Lösungen aufgrund interionischer Wechselwirkungen von der Konzentration ab. Besonders bedeutsam und daher in der Regel tabelliert ist der Grenzwert für unendliche Verdünnung, die Ionengrenzleitfähigkeit , bzw. m, , .
Als Maß für die Driftgeschwindigkeit eines Ions kann bei gegebener elektrischer Feldstärke gleichberechtigt jede der Größen Ionenbeweglichkeit, Ionenäquivalentleit-fähigkeit und molare Ionenleitfähigkeit dienen. Für den effektiven Beitrag einer ionischen Spezies zum Leitwert ist aber neben der Geschwindigkeit und der Ladungs-zahl die Konzentration dieser Ionen entscheidend. Es ist daher üblich, zusätzlich die Ionenleitfähigkeit (in Sm ) als Produkt der molaren Ionenleitfähigkeit und der Stoffmengenkonzentration der Ionen zu definieren:
≡ , | | | | . (14)

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
Der Zahlenwert von entspricht dann formal dem Beitrag zum Leitwert, den die ionische Spezies i in 1 m3 Lösung zwischen planparallelen Elektroden im Abstand von 1 m leistet.
3.3. Trends bei der Ionengrenzleitfähigkeit
Betrachtet man bspw. die Alkansäurerestionen Acetat (CH3COO ), Propionat (C2H5COO ) und Butyrat (C3H7COO ) in wässriger Lösung, so stellt man fest, dass mit steigender Größe des Alkylrests die Ionengrenzleitfähigkeit (bei derselben Temperatur) abnimmt. Dies steht im Einklang mit Gl. (10), nach der die Drift-geschwindigkeit bei unendlicher Verdünnung und damit auch die Ionengrenzleit-fähigkeit mit zunehmendem Radius abnimmt.
Auf den ersten Blick scheint es mit dieser Information kontraintuitiv, dass die Grenz-leitfähigkeit der Alkalimetall-Ionen Li , Na und K in Wasser in der gegebenen Reihenfolge (also von oberen nach unteren Perioden) zunimmt, obwohl der (kristallographische) Ionenradius größer wird. Entscheidend ist hier, dass mit im STOKESschen Gesetz und damit in Gl. (10) der hydrodynamische Radius gemeint ist, also der Radius des Ions inklusive seiner Solvathülle, denn dieser ganze Komplex bewegt sich durch die Lösung und ruft damit Reibungskräfte hervor. Nun ist es so, dass gegenüber dem Natrium-Ion das Lithium-Ion, weil letzteres bei gleicher Gesamtladung einen deutlich kleineren Ionenradius besitzt, eine deutlich höhere Ladungsdichte aufweist. Dadurch bindet das Lithium-Ion die dipolaren Lösungs-mittelteilchen seiner Solvathülle stärker an sich. Die Solvathülle ist so groß, dass das Lithium-Ion mitsamt seiner Solvathülle einen größeren hydrodynamischen Radius besitzt als das Natrium-Ion und das Kalium-Ion. Daher ist die Grenzleitfähigkeit des Lithium-Ions die kleinste der dreien.
3.4. Der GROTTHUß-Mechanismus
Bemerkenswert ist die Grenzleitfähigkeit des Protons und des Hydroxid-Ions in Wasser, die bis zu einer Größenordnung höher als die der vorher erwähnten Ionen ist. Dies liegt an einem besonderen Leitungsmechanismus für die beiden Eigenionen des Lösungsmittels, dem GROTTHUß-Mechanismus. Bspw. muss ein Proton nicht die gesamte Lösung bis hin zur Kathode durchqueren, um die Ladung zu transportieren, so wie es bei anderen Kationen der Fall ist, sondern die Wasserstoffbrückenbindung eines benachbarten Wassermoleküls zu dem Proton wird zu einer chemischen Bindung verfestigt, während die Bindung zu einem anderen Wasserstoffatom dieses Moleküls zu einer Wasserstoffbrückenbindung abgeschwächt wird. Die Bindung wird sozusagen umgeklappt. Auf diese Weise wird die Ladung durch eine Abfolge von Bindungs-bildungen und -brüchen transportiert, was deutlich schneller ist als die übliche Wanderung (Migration). Analog verläuft der Transport der negativen Ladung eines Hydroxid-Ions zur Anode.
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
In beiden Fällen verläuft der Ladungstransport umso rascher, je günstiger die Wassermoleküle bereits orientiert sind. Da dies im Falle von festem Eis besser gegeben ist als in flüssigem Wasser, ist die Protonenleitfähigkeit in Eis sogar noch einmal um zwei Größenordnungen höher als in flüssigem Wasser.
4. Die Leitfähigkeit von Elektrolytlösungen
4.1. Widerstand, Leitwert, spezifischer Widerstand und Leitfähigkeit
Während bislang die Bewegung der einzelnen Ionen im elektrischen Feld im Vordergrund stand, soll nun die Leitfähigkeit ganzer Elektrolytlösungen betrachtet werden. Der elektrische Widerstand einer gewissen Menge einer Elektrolytlösung in einer konduktometrischen Messzelle, in der zwischen zwei Elektroden ein elektrisches Feld angelegt wird, ist
⋅ Z . (15)
Dabei ist Z die Zellkonstante (in m ), welche alle geometrischen Faktoren der Messzelle umfasst, und der spezifische Widerstand der Lösung (in Ωm), welcher nur noch eine Eigenschaft der Lösung ist. In einer idealisierten Messzelle mit sehr großflächigen Elektrodenblechen, die in sehr geringem Abstand planparallel zueinan-der angeordnet sind (sodass das elektrische Feld als völlig homogen aufgefasst werden kann), entspricht die Zellkonstante einfach dem Verhältnis zwischen dem Abstand der Elektroden und der Querschnittsfläche einer Elektrode.
Eine Lösung, die den elektrischen Strom gut leitet, besitzt einen niedrigen Widerstand. Um eine Größe zu erhalten, die stattdessen einen hohen Wert besitzt, wenn die Lösung den elektrischen Strom gut leitet, wird der Kehrwert des elektrischen Widerstandes eingeführt, der Leitwert der Lösung (in Siemens S Ω )
≡1 1
⋅1Z≡
Z . (16)
Auch der Leitwert kann aufgeteilt werden in einen messzellabhängigen Teil, Z , und einen lösungsspezifischen Teil, . Letzterer wird als spezifischer Leitwert oder als (elektrische) Leitfähigkeit (griechischer Kleinbuchstabe Kappa) bezeichnet (in Sm Ω m ):
≡1
. (17)
Der Zahlenwert der Leitfähigkeit entspricht formal dem Leitwert von 1 m3 Lösung
zwischen planparallelen Elektroden im Abstand von 1 m. Die Leitfähigkeit einer
beliebigen Elektrolytlösung ist stets gleich der Summe der Leitfähigkeiten aller in
der Lösung existierenden Ionen (siehe Gl. (14)):

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
,
| |
∙ | |
. (18)
4.2. Molare Leitfähigkeit und Äquivalentleitfähigkeit
Im Falle einer ideal verdünnten Elektrolytlösung ist die Leitfähigkeit proportional zur Zahl der in einem bestimmten Lösungsvolumen enthaltenen Ionen. Wenn wir unterstellen, dass der Elektrolyt in einer ideal verdünnten Lösung stets vollständig dissoziiert, ist die Leitfähigkeit auch proportional zur nominalen Stoffmengen-konzentration, d. h. ∝ . Daher wird für starke und schwache Elektrolyte als weitere Größe der Quotient aus der Leitfähigkeit und der (nominalen) Stoffmengenkonzen-tration eingeführt, die molare Leitfähigkeit (in Sm mol )
m ≡ . (19)
Der Zahlenwert der molaren Leitfähigkeit entspricht dem Leitwert von 1 mol Elektrolyt zwischen planparallelen Elektroden im Abstand von 1 m. Für eine ideal verdünnte Lösung ist nun die molare Leitfähigkeit von der Konzentration unabhängig und somit nur noch eine Eigenschaft des Elektrolyten (sowie des Lösungsmittels und der Temperatur). Dieser Grenzwert für unendliche Verdünnung erhält das Symbol m, .
Vergleicht man verschiedene Elektrolyte hinsichtlich ihrer Befähigung zum Strom-transport, so muss man schließlich noch beachten, dass verschiedenwertige Elektrolyte bei gleicher Konzentration unterschiedlich viele Ladungsträger unterschiedlicher Ladung durch Dissoziation zur Verfügung stellen. So stellt bspw. eine Formeleinheit NaCl (in Wasser) zwei Formeleinheiten Ionen zur Verfügung, die beide einfach geladen sind, während eine Formeleinheit Calciumchlorid, CaCl2, (in Wasser) drei Formeleinheiten Ionen zur Verfügung stellt, von denen eines zweifach geladen ist (Ca2 ) und zwei einfach geladen sind (Cl ). Calciumchlorid hat im Vergleich zum Natriumchlorid also bereits einen „Startvorteil“ hinsichtlich der Leitfähigkeit, ohne dass man etwas über die Leitfähigkeit der einzelnen Ionen wissen müsste. Um diesen Effekt zu quantifizieren, wird für jeden Elektrolyt K A seine elektrochemische Wertigkeit
≡ | | (20)
definiert; so ist bspw. NaCl 1 ⋅ 1 1 und CaCl2 1 ⋅ 2 2. (Man beachte, dass die Ladungszahl eines einzelnen Ions ist, hingegen die elektrochemische Wertigkeit eines ganzen Elektrolyten darstellt.) Die für den Vergleich zwischen verschiedenen Elektrolyten optimale Größe ist also der Quotient aus molarer Leit-fähigkeit m und elektrochemischer Wertigkeit , die sogenannte Äquivalentleit-fähigkeit (in Sm mol )
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
≡ m . (21)
wird auch als Äquivalentkonzentration bezeichnet. (Man beachte, dass (Kleinbuchstabe Lambda) die Äquivalentleitfähigkeit eines einzelnen Ions ist, hingegen (Großbuchstabe Lambda) die Äquivalentleitfähigkeit eines ganzen Elektrolyten darstellt.) Auch die Äquivalentleitfähigkeit sollte für ideal verdünnte Lösungen konzentrationsunabhängig sein, dieser Grenzwert für unendliche Verdünnung heißt kurz auch Grenzleitfähigkeit .
4.3. Das Gesetz der unabhängigen Ionenwanderung
Für diesen Grenzfall unendlicher Verdünnung gilt, sowohl für starke als auch für schwache Elektrolyte, eine besonders einfache Beziehung zwischen der Grenzleit-fähigkeit des Elektrolyten und den Grenzleitfähigkeiten der einzelnen Ionen , , das Gesetz der unabhängigen Ionenwanderung:
, , . (22)
Da sich im Falle unendlicher Verdünnung die Ionen gegenseitig in ihrer Migration im elektrischen Feld nicht beeinflussen, addieren sich die Ionengrenzleitfähigkeiten einfach.
(Man beachte, dass zwar nach Gl. (18) für eine Lösung eines einzelnen Elektrolyten
stets gilt, aber (ohne Index „0“!) nur für einen starken
Elektrolyten gültig ist.)
Falls molare Grenzleitfähigkeiten verwendet werden, muss die Stöchiometrie des Elektrolyten explizit berücksichtigt werden:
m, m, , m, , . (23)
4.4. Die Konzentrationsabhängigkeit der Leitfähigkeit starker Elektrolyte
Die Leitfähigkeit bemisst, wie leitfähig die Lösung insgesamt ist. Dies wird durch zwei Faktoren bestimmt: wie viele Ladungsträger vorliegen, die den Strom transportieren, und wie gut die einzelnen Ladungsträger den Strom transportieren können. Gl. (18) zeigt, dass die Leitfähigkeit einer Elektrolytlösung umso größer ist, je höher die Konzentration der Ionen in der Lösung ist, je höher die Beweglichkeiten der Ionen sind und je höher die Ladungszahlen der Ionen sind. Wären die Beweglichkeiten der Ionen völlig unabhängig von der Konzentration der Ionen, wie in einer ideal verdünnten Lösung, sollte die Leitfähigkeit proportional zur Konzentration der Ionen sein. Grundsätzlich nehmen die Ionenbeweglichkeiten aber mit steigender Ionenkonzentration ab, da die Ionen durch die interionische Wechselwirkung in ihrer Beweglichkeit behindert werden. Dieser gegenläufige Effekt der Abnahme der Beweglichkeit mit steigender Ionenkonzentration erklärt, warum die Leitfähigkeit

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
starker Elektrolyte mit steigender Elektrolytkonzentration erst zunimmt, dann (falls noch keine Sättigung eingetreten ist) im Bereich einiger molL ein Maximum durchschreitet und schließlich sogar fällt. Ein starker Elektrolyt dissoziiert vollständig, sodass die Zunahme der Elektrolytkonzentration einhergeht mit einer entsprechenden linearen Zunahme der Ionenkonzentration. Wenn die Konzentration erhöht wird, werden zwar die einzelnen Teilchen unbeweglicher, was ungünstig für die Leitfähig-keit ist, aber vor allem erhöht sich die Anzahl der Ladungsträger, was günstig für die Leitfähigkeit ist. Bei geringen und mittleren Konzentrationen ist der letztere Effekt der dominante, also die Erhöhung der Zahl der Ladungsträger durch Konzentrations-erhöhung, daher steigt die Leitfähigkeit mit der Konzentration. Weil aber die Ladungsträger immer unbeweglicher werden, flacht die Kurve immer weiter ab, bis sie ein Maximum durchschreitet. Danach fällt , weil die zunehmende Immobilität der dominierende Faktor geworden ist. Ein davon unabhängiger Neben-faktor ist, dass mit steigender Konzentration die Viskosität der Lösung zunimmt (sowohl für starke als auch für schwache Elektrolyte), was ebenfalls die Beweglichkeit der Ionen reduziert.
4.5. Die Konzentrationsabhängigkeit der Leitfähigkeit schwacher Elektrolyte
Für schwache Elektrolyte spielt die Abnahme der Beweglichkeit der Ionen mit zunehmender Konzentration nur eine untergeordnete Rolle. Dennoch kann die Kurve
wie bei starken Elektrolyten ein Maximum aufweisen, weil mit zunehmender Konzentration der Dissoziationsgrad abnimmt (siehe 4.7.). Die dominierenden gegen-läufigen Effekte sind hier also: Erhöhung der Konzentration und Abnahme des Anteils der Ladungsträger.
4.6. Die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit starker Elektrolyte
Im Fall starker Elektrolyte bemisst die Äquivalentleitfähigkeit , wie mobil seine Ionen in der Lösung sind . Offenbar sind sie umso beweglicher (und somit umso fähiger zum Stromtransport), je geringer die Konzentration ist. Am beweglichsten sind sie im Grenzfall unendlicher Verdünnung, die Äquivalentleit-fähigkeit ist dann maximal und besitzt den Wert der Grenzleitfähigkeit .
Bei starken Elektrolyten lässt sich die Abnahme der Äquivalentleitfähigkeit mit der Konzentration hauptsächlich durch zwei Effekte erklären. Der Relaxationseffekt beschreibt, dass sich durch die Bewegung eines Ions im elektrischen Feld dieses Ion aus dem Zentrum seiner Ionenwolke bewegt. Es muss sich also ständig eine neue Ionenwolke um das Ion herum bilden. Dies geschieht aber nur mit einer gewissen Verzögerung, sodass die (zum Zentralion im Mittel entgegengesetzt geladene) Ionenwolke ständig hinterherhinkt. Somit liegt eine asymmetrische Ladungsver-teilung vor, die das betrachtete Ion bremst. Der elektrophoretische Effekt beschreibt, dass die Ionenwolke mit den Solvathüllen ihrer Ionen in die andere Richtung als ihr Zentralion wandert, was eine Strömung des Lösungsmittels in die zur Bewegung des
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
Zentralions entgegengesetzte Richtung hervorruft, welche ebenfalls das Ion bremst. Beide Effekte werden stärker, wenn die Konzentration erhöht wird.
Will man diese Effekte quantifizieren, so kann man dies mit dem BJERRUMschen Leitfähigkeitskoeffizienten bewerkstelligen, welcher (für starke und schwache Elektrolyte) einfach die Abweichung von der Grenzleitfähigkeit beschreibt,
≡, ,
. (24)
Explizit wird die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit starker Elektrolyte aber (für nicht zu konzentrierte Lösungen) durch das KOHLRAUSCHsche Quadratwurzelgesetz beschrieben,
⋅ ⊝⁄ , (25)
wobei 0 ein von der elektrochemischen Wertigkeit des Elektrolyten sowie von Viskosität und Permittivität/Dielektrizitätskonstante des Lösungsmittels abhängi-ger Faktor ist. Da für einen 1-1-wertigen starken Elektrolyten die molare Ionenstärke
ist, kann in diesem Fall auch geschrieben werden
⋅ ⊝⁄ . (26)
Das Gesetz kann weiterhin analog mit molaren Leitfähigkeiten formuliert werden, wobei die Konstante dann (abhängig von der Stöchiometrie) einen anderen Wert annimmt.
Im Falle starker Elektrolyte sollte also die Auftragung der Äquivalentleitfähigkeit gegen die Quadratwurzel der molaren Ionenstärke (bzw. der Stoffmengenkonzen-tration) (evtl. geteilt durch ⊝ 1molL ) eine fallende Gerade ergeben, die einen Ordinatenabschnitt gleich der Grenzleitfähigkeit des betrachteten Elektrolyten besitzt.
(Man beachte: Die Leitfähigkeit geht hingegen für → 0 gegen Null, einfach weil bei 0 keinerlei Ladungsträger vorliegen und somit auch keinerlei Strom transpor-tiert werden kann.)
4.7. Die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit schwacher Elektrolyte
Schwache Elektrolyte liefern in der KOHLRAUSCHschen Auftragung keine Gerade, sondern eine Hyperbel. Bei ihnen trifft das KOHLRAUSCHsche Quadratwurzelgesetz also nicht zu, was daran liegt, dass der Hauptgrund für die Konzentrationsabhängigkeit der Äquivalentleitfähigkeit für schwache Elektrolyte ein anderer als für starke ist. Entscheidend ist hier, dass das Ausmaß der Dissoziation stark von der Konzentration abhängt. Es wird zweckmäßig durch den Dissoziationsgrad beschrieben, der für einen beliebigen Elektrolyten K A
(27)

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
ist, wobei und die Konzentration von Kation bzw. Anion ist und die Ausgangskonzentration (nominale Konzentration). Für einen 1-1-wertigen Elektrolyt vereinfacht sich dies zu
. (28)
Für → 0 ist in aller Regel → 1, die Dissoziation ist also bei unendlicher Verdünnung vollständig (Die Eigendissoziation des Wassers kann die Dissoziation eines schwachen Elektrolyten aber zurückdrängen, sodass dann selbst bei unendlicher Verdünnung der Dissoziationsgrad von 1 verschieden ist; signifikant ist dieser Effekt etwa bei schwachen Säuren wie Phenol oder schwachen Basen wie Harnstoff, während stärkere Säuren wie Essigsäure oder stärkere Basen wie Ammoniak bei unendlicher Verdünnung praktisch als vollständig dissoziiert bzw. protoniert angesehen werden dürfen). Mit steigender Konzentration wird die Dissoziation stark zurückgedrängt, sodass der Dissoziationsgrad sinkt. Demnach ist die Tendenz zur Freisetzung von Ionen bei höheren Konzentrationen verringert, da aber nur Ionen zum Ladungs-transport beitragen können und nicht die neutralen nichtdissoziierten Spezies, bedeutet dies eine Abnahme der Äquivalentleitfähigkeit.
Für schwache Elektrolyte (und für starke Elektrolyte mit 1 ebenfalls) gilt exakt (siehe auch Gl. (24)):
. (29)
Die Äquivalentleitfähigkeit schwacher Elektrolyte wird also grundsätzlich durch die Beweglichkeit der Ionen und den Dissoziationsgrad bestimmt. In Gl. (29) ist im Gegensatz zum Gesetz der unabhängigen Ionenwanderung (siehe Gl. (22)) explizit enthalten, weil in der Definitionsgleichung (21) für die Äquivalentleitfähigkeit durch die nominale Stoffmengenkonzentration geteilt wird, die unvollständige Dissoziation also gar nicht berücksichtigt ist. Für verdünnte Lösungen schwacher Elektrolyte wird häufig die ARRHENIUS-Näherung getroffen: Die interionischen Wechsel-wirkungen seien vernachlässigbar, also 1, sodass
. (30)
Die Abweichung von der Grenzleitfähigkeit (zu welcher 1 gehört) lasse sich also komplett durch die unvollständige Dissoziation erklären, und der Anteil an dissoziierten Teilchen sei genau der Anteil an Äquivalentleitfähigkeit, der gegenüber dem Maximalwert vorliegt, also für den Stromtransport „zur Verfügung steht“.
Damit sind über den Dissoziationsgrad sowohl die Konzentration (Gl. (27) bzw. (28)) als auch die Äquivalentleitfähigkeit (Gl. (30)) miteinander verknüpft. Daraus lässt sich also eine Gleichung für die Konzentrationsabhängigkeit der Äquivalent-leitfähigkeit schwacher Elektrolyte herleiten. Üblicherweise wird dieser Zusammen-hang unter Zuhilfenahme der Dissoziationskonstante formuliert.
Für einen 1-1-wertigen Elektrolyten lautet die thermodynamische Dissoziations-konstante
PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
⊝⊝
eq , (31)
wobei die Konzentration an nichtdissoziiertem Elektrolyt ist (nicht etwa eine gemittelte Größe wie , man beachte die leicht unterschiedliche Nomenklatur ) und wieder ⊝ 1molL die Standard-Stoffmengenkonzentration (zur Bereinigung der Einheiten). Der Index bei ⊝ zeigt die Verwendung von Stoffmengenkonzen-trationen an, der Index eq bedeutet Gleichgewicht („equilibrium“). Die thermodyna-mische Dissoziationskonstante besteht also aus dem Massenwirkungsgesetz inklusive der HENRYschen Aktivitätskoeffizienten.
Für eine verdünnte Lösung wird nun genähert, dass letztere den Wert Eins besitzen, also vernachlässigbar sind. Es ergibt sich dann die konventionelle Dissoziations-konstante (für einen 1-1-wertigen Elektrolyten)
eq , (32)
in der zusätzlich üblicherweise nicht mehr durch ⊝ 1molL geteilt wird, weshalb die Einheit einer Konzentration besitzt und auch gilt
p lg ⊝ . (33)
Die Konzentration der Kationen und der Anionen ist für einen 1-1-wertigen Elektrolyten gemäß Gl. (28) jeweils gleich der Ausgangskonzentration, multipliziert mit dem Dissoziationsgrad, . Da der Anteil dissoziierter Moleküle ist, beträgt der Anteil nichtdissoziierter Moleküle 1 ; demnach ist die Konzentration nichtdissoziierter Moleküle 1 . Folglich ist
⋅1 1 . (34)
Wird nun die ARRHENIUS-Näherung, Gl. (30), für eingesetzt, ergibt sich eine Näherung für die Dissoziationskonstante:
1 1 . (35)
Diese genäherte Dissoziationskonstante heißt scheinbare Dissoziationskonstante app („app“ für „apparent“) und das Gesetz, das sie definiert und gleichzeitig den
gesuchten Zusammenhang zwischen Äquivalentleitfähigkeit und Konzentration für schwache Elektrolyte beschreibt, OSTWALDsches Verdünnungsgesetz:
app
1 . (36)
Ausmultiplizieren mit dem Nenner führt zu der linearisierten Form

PCPharm Uni Bonn WS 2018/2019 Seminar P6 ALEXANDER RIEGEL
app , (37)
nach der die Auftragung eine Gerade mit der Grenzleitfähigkeit als Ordinatenabschnitt ergibt, aus der Steigung kann app bestimmt werden. Weitere Umformung liefert eine alternative Linearisierung,
1 1app . (38)
Demnach ergibt die Auftragung eine Gerade mit dem Kehrwert der
Grenzleitfähigkeit als Ordinatenabschnitt, aus der Steigung kann app bestimmt werden. In beiden Fällen ist die ermittelte Dissoziationskonstante nur eine scheinbare, welche sich von der thermodynamischen durch Vernachlässigung der Aktivitäts-koeffizienten und durch Aufnahme der ARRHENIUS-Näherung unterscheidet. Die Grenzleitfähigkeit eines schwachen Elektrolyten kann experimentell oft nur ungenau bestimmt werden, da der rapide Anstieg der Äquivalentleitfähigkeit bei → 0 eine Extrapolation stark erschwert. Es bietet sich stattdessen an, die Grenzleitfähigkeit starker Elektrolyte, welche die Ionen des betrachteten schwachen Elektrolyten enthalten, nach KOHLRAUSCH zu bestimmen und dann gemäß dem Gesetz der unabhängigen Ionenwanderung, Gl. (22) bzw. (23), geschickt zu kombinieren, um die Grenzleitfähigkeit des schwachen Elektrolyten zu erhalten. Für Essigsäure ergibt sich z. B. HOAc HCl NaOAc NaCl , wie aus den folgenden vier Gleichungen (für einen schwachen und drei starke Elektrolyte) unmittelbar ersichtlich ist:
HOAc , H , OAc
HCl , H , Cl
NaOAc , Na , OAc
NaCl , Na , Cl

PCPharm Uni Bonn SS 2018 Seminar P6: Abbildungen ALEXANDER RIEGEL
Physikalisch-Chemisches Grundpraktikum für Pharmazeuten
Seminar P6 – Elektrolytische Leitfähigkeit: Abbildungen
Abbildung 1. Schematische Skizze der Struktur einer Elektrolytlösung. Jedes Ion, bspw. das Kation mit dem blauen Kreis oder das Anion mit dem roten Kreis, ist gleichzeitig ein Zentralion für seine Ionenwolke, die im zeitlichen Mittel zum Zentralion entgegengesetzt geladen ist, und Teil der Ionenwolke aller anderen Ionen. Die unregelmäßige thermische Bewegung der Teilchen bedingt eine kurze Lebensdauer der Wolken. [1]
Abbildung 2. Ionen strukturieren die dipolaren Lösungsmittelteilchen in ihrer Umgebung, hier das Beispiel von Natriumchlorid in Wasser. Es bilden sich Solvathüllen aus, in denen im Falle von Kationen die partiell negativ geladene Seite zum Ion hin orientiert ist, im Falle von Anionen die partiell positiv geladene Seite. [2]

PCPharm Uni Bonn SS 2018 Seminar P6: Abbildungen ALEXANDER RIEGEL
Abbildung 3. Schematische Darstellung des GROTTHUß-Mechanismus des Ladungstransports von Protonen (oben) und Hydroxidionen (unten) in Wasser. Die Ionen durchqueren die Lösung nicht wandernd, sondern der Ladungstransport wird durch „Bindungsumklappen“ besorgt. [3]
Abbildung 4. Für den GROTTHUß-Mechanismus günstige Ausrichtung der Wassermoleküle, mit einem Hydroniumion unten. Siehe den Link [4] für eine animierte Version.

PCPharm Uni Bonn SS 2018 Seminar P6: Abbildungen ALEXANDER RIEGEL
Abbildung 5. Abhängigkeit der Leitfähigkeit einiger wässriger Elektrolytlösungen bei 291K (* bei 288K) von der Konzentration. Alle Elektrolyte außer Essigsäure (CH3COOH) sind starke. [3]
Abbildung 6. Abhängigkeit der Äquivalentleitfähigkeit einiger wässriger Elektrolytlösungen bei 298K (* bei 291K) von der Konzentration. Alle Elektrolyte außer Essigsäure (CH3COOH) sind starke. [3]
PCPharm Uni Bonn SS 2018 Seminar P6: Abbildungen ALEXANDER RIEGEL
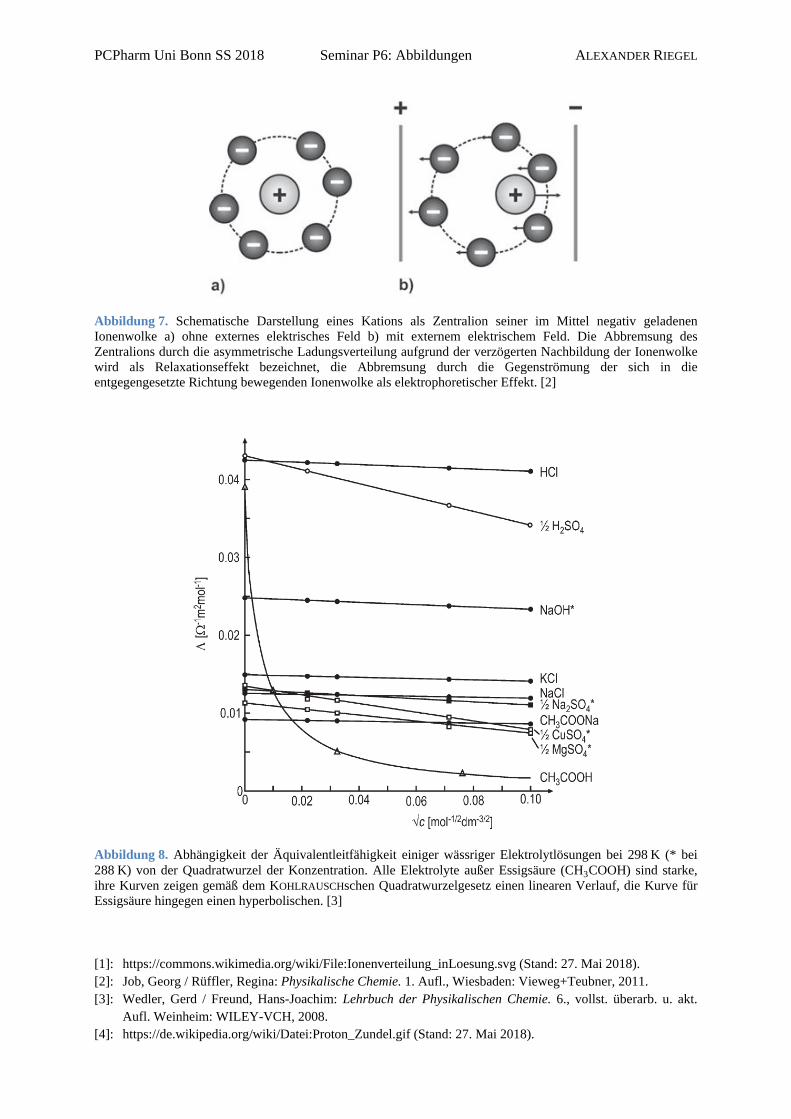
Abbildung 7. Schematische Darstellung eines Kations als Zentralion seiner im Mittel negativ geladenen Ionenwolke a) ohne externes elektrisches Feld b) mit externem elektrischem Feld. Die Abbremsung des Zentralions durch die asymmetrische Ladungsverteilung aufgrund der verzögerten Nachbildung der Ionenwolke wird als Relaxationseffekt bezeichnet, die Abbremsung durch die Gegenströmung der sich in die entgegengesetzte Richtung bewegenden Ionenwolke als elektrophoretischer Effekt. [2]
Abbildung 8. Abhängigkeit der Äquivalentleitfähigkeit einiger wässriger Elektrolytlösungen bei 298K (* bei 288K) von der Quadratwurzel der Konzentration. Alle Elektrolyte außer Essigsäure (CH3COOH) sind starke, ihre Kurven zeigen gemäß dem KOHLRAUSCHschen Quadratwurzelgesetz einen linearen Verlauf, die Kurve für Essigsäure hingegen einen hyperbolischen. [3]
[1]: https://commons.wikimedia.org/wiki/File:Ionenverteilung_inLoesung.svg (Stand: 27. Mai 2018). [2]: Job, Georg / Rüffler, Regina: Physikalische Chemie. 1. Aufl., Wiesbaden: Vieweg+Teubner, 2011. [3]: Wedler, Gerd / Freund, Hans-Joachim: Lehrbuch der Physikalischen Chemie. 6., vollst. überarb. u. akt.
Aufl. Weinheim: WILEY-VCH, 2008. [4]: https://de.wikipedia.org/wiki/Datei:Proton_Zundel.gif (Stand: 27. Mai 2018).


















