
Theoretische Biophysikalische Chemie -...
20
0 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen DFG-CENTRUM FÜR FUNKTIONELLE NANOSTRUKTUREN Theoretische Biophysikalische Chemie QM/MM und Einbettungsverfahren Christoph Jacob KIT – Universität des Landes Baden-Württemberg und nationales Forschungszentrum in der Helmholtz-Gemeinschaft www.kit.edu
Transcript of Theoretische Biophysikalische Chemie -...

0 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
DFG-CENTRUM FÜR FUNKTIONELLE NANOSTRUKTUREN
Theoretische Biophysikalische ChemieQM/MM und Einbettungsverfahren
Christoph Jacob
KIT – Universität des Landes Baden-Württemberg undnationales Forschungszentrum in der Helmholtz-Gemeinschaft www.kit.edu

Quantum Chemistry
1 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Electronic Energy: Eel = Eel(R1, . . . ,RN)
positions of nuclei R1, . . . ,RN
Quantum Mechanics
Eel =⟨Ψel|Hel|Ψel
⟩electronic wavefunction: Ψel = Ψel(r1, . . . , rN)
coordinates of electrons r1, . . . , rN
Quantum Chemistry
wave-function theory (WFT)density-functional theory (DFT)
Force Field Methods
→ neglect electrons and model Eel directly
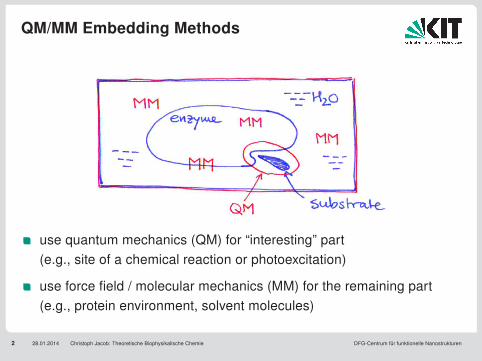
QM/MM Embedding Methods
2 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
use quantum mechanics (QM) for “interesting” part(e.g., site of a chemical reaction or photoexcitation)
use force field / molecular mechanics (MM) for the remaining part(e.g., protein environment, solvent molecules)

Force Field Approximation
3 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
“Standard” Force Field
Eel(R1, . . . ,RN) =12
bonds
∑i
ki (ri − r0i )
2 +12
angles
∑i
kθj (θj − θ0
j )2
+12
torsions
∑n
cos(nnωn − γn)
+ ∑I
∑J>I
14πε0
qIqJ
|RI −RJ |
+ ∑I
∑J>I
4εIJ
[(σIJ
|RI −RJ |
)12
−(
σIJ
|RI −RJ |
)6]
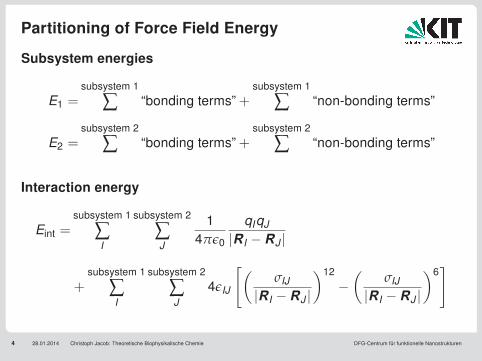
Partitioning of Force Field Energy
4 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Subsystem energies
E1 =subsystem 1
∑ “bonding terms” +subsystem 1
∑ “non-bonding terms”
E2 =subsystem 2
∑ “bonding terms” +subsystem 2
∑ “non-bonding terms”
Interaction energy
Eint =subsystem 1
∑I
subsystem 2
∑J
14πε0
qIqJ
|RI −RJ |
+subsystem 1
∑I
subsystem 2
∑J
4εIJ
[(σIJ
|RI −RJ |
)12
−(
σIJ
|RI −RJ |
)6]

QM/MM Embedding Methods
5 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Mechanical Embedding
Etot = EQM + EMM + Eint,MM
EQM is calculated for the isolated QM system→ embedding only affect energy expression
Electronic Embedding
Etot = E ′QM + EMM + Eint,MM
E ′QM is calculated for the embedded QM system
Polarizable Embedding
Etot = E ′QM + E ′MM + Eint,MM
MM charges are polarized by the QM subsystem

QM/MM: Covalent Bonds
6 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Interaction energy
Eint =subsystem 1
∑I
subsystem 2
∑J
14πε0
qIqJ
|RI −RJ |
+subsystem 1
∑I
subsystem 2
∑J
4εIJ
[(σIJ
|RI −RJ |
)12
−(
σIJ
|RI −RJ |
)6]
+ “bonding terms between subsystems”
Link Atoms
subtractive methods
Etot = E(QM)I+L − E(MM)
I+L + E(MM)tot
additive methods
Etot = E(QM)I+L + E(MM)
II + E(QM/MM)int

Quantum-Mechanical Subsystem Methods
7 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Subsystem methods are based on a partitioning of the energy:
Etot = EI + EII + Eint
QM/MM methods
partition force field (MM) energy:
EMMtot = EMM
I + EMMII + EMM
int
replace force-field EMMI by EMM
I from quantum chemistry:
EQM/MMtot = EQM
I + EMMII + EMM
int
Quantum-mechanical subsystem methods
partition quantum-mechanical (QM) energy:
EQMtot = EQM
I + EQMII + EQM
int
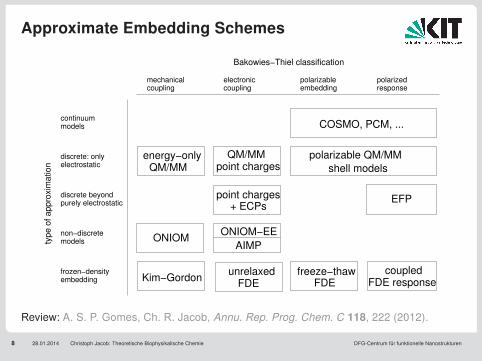
Approximate Embedding Schemes
8 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Kim−Gordon
electrostatic
discrete beyond purely electrostatic
non−discrete models
FDE responsecoupled
QM/MMpoint charges
point charges+ ECPs
couplingmechanical
couplingelectronic polarizable
embeddingpolarized response
unrelaxedFDE
ONIOM−EE
AIMP
polarizable QM/MM
shell models
EFP
frozen−density embedding
continuum models
typ
e o
f a
pp
roxim
atio
n
Bakowies−Thiel classification
energy−onlyQM/MM
COSMO, PCM, ...
ONIOM
freeze−thawFDE
discrete: only
Review: A. S. P. Gomes, Ch. R. Jacob, Annu. Rep. Prog. Chem. C 118, 222 (2012).

Density-Functional Theory (DFT) in a Nutshell
9 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Electronic Schrödinger equation
Hel Ψel(r1, . . . , rN) = Eel Ψel(r1, . . . , rN)
electronic Hamiltonian: Hel = T + VNe + Vee + ENN
electronic wavefunction: Ψel = Ψel(r1, . . . , rN)
coordinates of electrons r1, . . . , rN
→ problem depends on 3N electronic coordinates
Density-functional theory (DFT)
Idea: use electron density ρ(r) instead of the wavefunction
ρ(r) =∫
Ψel(r , r2, . . . , rN)d3r2 · · ·d3rN
→ electron density depends on only 3 coordinates

Density-Functional Theory (DFT) in a Nutshell
10 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Total energy functional
E [ρ] = Ts[ρ] + VNe[ρ] + J [ρ] + Exc[ρ] + ENN
electron–nuclear attraction:
VNe[ρ] =∫
ρ(r) vnuc(r) d3r
classical electron–electron Coulomb interaction:
J [ρ] =12
∫∫ρ(r)ρ(r ′)|r − r ′| d3rd3r ′
kinetic energy of noninteracting electrons with density ρ = ∑i |φi (r)|2
Ts[ρ] = − 12 ∑
i
∫φi ∗ (r)∆φi (r) d3r
everything else: exchange–correlation energy Exc[ρ]

Density-Functional Theory (DFT) in a Nutshell
11 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
⇒ derive equations for orbitals φi with ρ(r) = ∑i |φi (r)|2
Kohn–Sham equations[−∆
2+ vnuc(r) + vCoul[ρ](r) + vxc[ρ](r)
]φi (r) = εi φi (r)
classical Coulomb potential of the electrons:
vCoul[ρ](r) =12
∫ρ(r ′)|r − r ′| d3r ′
exchange–correlation potential
vxc[ρ](r) =δExc[ρ]
δρ(r)
exchange-correlation functional Exc[ρ] is not known
→ approximate functional have to be used
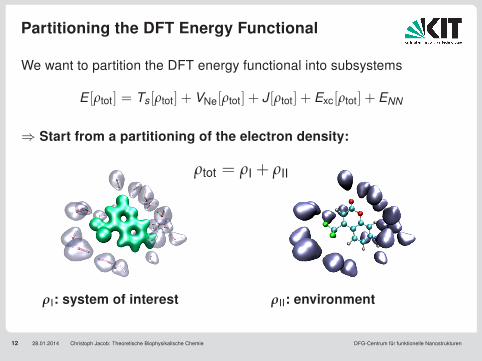
Partitioning the DFT Energy Functional
12 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
We want to partition the DFT energy functional into subsystems
E [ρtot] = Ts[ρtot] + VNe[ρtot] + J [ρtot] + Exc[ρtot] + ENN
⇒ Start from a partitioning of the electron density:
ρtot = ρI + ρII
ρI: system of interest ρII: environment

Partitioning the DFT Energy Functional
13 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
DFT total energy functional
E [ρtot] = Ts[ρtot] + VNe[ρtot] + J [ρtot] + Exc[ρtot] + ENN
Partitioned DFT total energy functional
E [ρtot] = E (I)NN + E (II)
NN + E (int)NN
+∫ (
ρI(r) + ρII(r))(
vnucI (r) + vnuc
II (r))
d3r
+12
∫ (ρI(r) + ρII(r)
)(ρI(r ′) + ρII(r ′)
)|r − r ′| d3rd3r ′
+ Exc[ρI] + Exc[ρII] + Enaddxc [ρI, ρII]
+ Ts[ρI] + Ts[ρII] + T nadds [ρI, ρII]
with the non-additive contributions
X nadd[ρI, ρII] = X [ρI + ρII]− X [ρI]− X [ρII]

Partitioning the DFT Energy Functional
14 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
DFT energy of subsystem I
E [ρI] = Ts[ρI] + VNe[ρI] + J [ρI] + Exc[ρI] + E (I)NN
DFT energy of subsystem II
E [ρII] = Ts[ρII] + VNe[ρII] + J [ρII] + Exc[ρII] + E (II)NN
DFT interaction energy
Eint[ρI, ρII] = E (int)NN +
∫ρI(r)v
nucII (r) d3r +
∫ρII(r)v
nucI (r) d3r
+∫
ρI(r)ρII(r ′)|r − r ′| d3rd3r ′ + Enadd
xc [ρI, ρII] + T nadds [ρI, ρII]
classical electrostatic interaction energynon-classical contributions from non-additive xc and kinetic energies
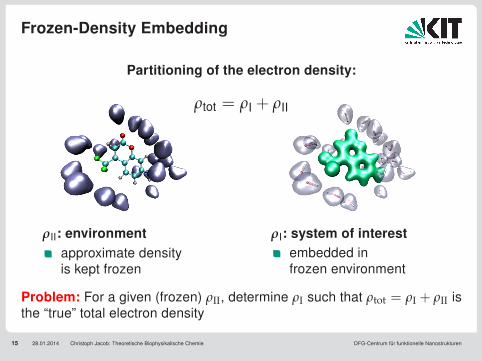
Frozen-Density Embedding
15 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Partitioning of the electron density:
ρtot = ρI + ρII
ρII: environmentapproximate densityis kept frozen
ρI: system of interestembedded infrozen environment
Problem: For a given (frozen) ρII, determine ρI such that ρtot = ρI + ρII isthe “true” total electron density

Frozen-Density Embedding
16 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
⇒ KS-like equations for orbitals φ(I)i of subsystem I[
−∇2
2+ vKS
eff [ρI](r) + vembeff [ρI, ρII](r)
]φi (r) = εi φi (r)
Embedding potential
vembeff [ρI, ρII](r) = vnuc
II (r) +∫
ρII(r ′)|r − r ′|dr ′
+δT nadd
s [ρI, ρII]
δρI+
δEnaddxc [ρI, ρII]
δρI
embedding potential is in principle exact
local potential that depends only on the electron densities
⇒ this is the potential that QM/MM methods should approximate!

Solvent Effects on Excitation Energies
17 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
large systems: many solvent molecules have to be considered
dynamics: many different structures (snapshots) required
⇒ construct approximate electron density for the solvent
⇒ calculate embedding potential with approximate functional for T nadds
Example: aminocoumarin C151 in water and n-hexane
solvent shell of 900 atoms
400 snapshots from classical MD simulations
excitation energies calculated withTD-DFT and FDE embedding potential
calculated shift: 0.17 eV (exp.: 0.22 eV)
J. Neugebauer, CRJ et al., J. Phys. Chem. A 109, 7805 (2005).
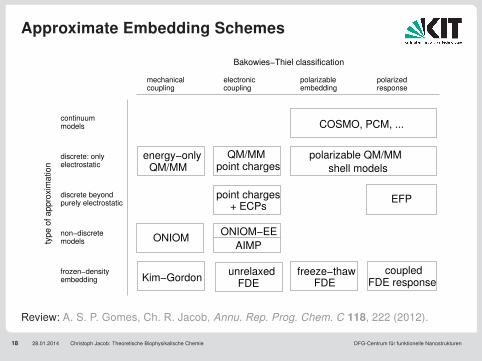
Approximate Embedding Schemes
18 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Kim−Gordon
electrostatic
discrete beyond purely electrostatic
non−discrete models
FDE responsecoupled
QM/MMpoint charges
point charges+ ECPs
couplingmechanical
couplingelectronic polarizable
embeddingpolarized response
unrelaxedFDE
ONIOM−EE
AIMP
polarizable QM/MM
shell models
EFP
frozen−density embedding
continuum models
typ
e o
f a
pp
roxim
atio
n
Bakowies−Thiel classification
energy−onlyQM/MM
COSMO, PCM, ...
ONIOM
freeze−thawFDE
discrete: only
Review: A. S. P. Gomes, Ch. R. Jacob, Annu. Rep. Prog. Chem. C 118, 222 (2012).

“Exact” Frozen-Density Embedding
19 28.01.2014 Christoph Jacob: Theoretische Biophysikalische Chemie DFG-Centrum für funktionelle Nanostrukturen
Embedding Wave-Function Theory (WFT) in DFT
Start from full DFT calculation of complex chemical system
Partition electron density as ρtot(r) = ρI(r) + ρII(r)
Reconstruct embedding potential for subsystem I
Use this embedding potential in accurate wave-function calculation
Approximate WFT-in-DFT embedding: Actinunyls in Cs2UO2Cl4
electronic spectrum of NpO2+2
and UO2+2 in Cs2UO2Cl4
accurate methods required→ relativistic Fock-space
coupled cluster (FSCC)A. S. P. Gomes, Ch. R. Jacob, L. Visscher, Phys. Chem. Chem. Phys. 10, 5353 (2008).
A. S. P. Gomes, Ch. R. Jacob, et al., Phys. Chem. Chem Phys. 15 15153 (2013).


















![140115 inklusives Gemeinwesen eichner [Kompatibilitätsmodus]€¦ · Sofie Eichner und Christian Jung 28.01.2014. Inklusion weltweit und vor Ort Inklusion ist eine lokale Verantwortung](https://static.fdokument.com/doc/165x107/606096a4397f1a4791274dcc/140115-inklusives-gemeinwesen-eichner-kompatibilittsmodus-sofie-eichner-und.jpg)