
Veränderung der Mikrozirkulation nach experimenteller ... · Die SAB kann spontaner oder...
146
Aus dem Institut für Chirurgische Forschung der Ludwig-Maximilians-Universität München Vorstand: Prof. Dr. med. Ulrich Pohl Dissertation "Veränderung der Mikrozirkulation nach experimenteller Subarachnoidalblutung (SAB): Kinetik, Bedeutung und Identifikation von Mechanismen" Zum Erwerb des Doktorgrades der Humanbiologie an der Medizinischen Fakultät der Ludwig-Maximilians-Universität zu München Vorgelegt von Diplom Biochemiker Herrn Ari Christian da Fonseca Dienel aus Portimão/Portugal Jahr 2016
Transcript of Veränderung der Mikrozirkulation nach experimenteller ... · Die SAB kann spontaner oder...

Aus dem Institut für Chirurgische Forschung der Ludwig-Maximilians-Universität
München
Vorstand: Prof. Dr. med. Ulrich Pohl
Dissertation
"Veränderung der Mikrozirkulation nach experimentel ler
Subarachnoidalblutung (SAB): Kinetik, Bedeutung und
Identifikation von Mechanismen"
Zum Erwerb des Doktorgrades der Humanbiologie an der Medizinischen Fakultät der
Ludwig-Maximilians-Universität zu München
Vorgelegt von
Diplom Biochemiker Herrn Ari Christian da Fonseca Dienel
aus
Portimão/Portugal
Jahr
2016

Mit Genehmigung der Medizinischen Fakultät
der Universität München
Berichterstatter: Prof. Dr. med. Nikolaus Plesnila
Mitberichterstatter:
Prof. Dr. med. Andrej Khandoga Prof. Dr. med. Frank Christ
Mitbetreuung durch den promovierten Mitarbeiter:
PD. Dr. med. Karsten Schöller Dr. med. Nicole Terpolilli
Dekan: Tag der mündlichen Prüfung:
Prof. Dr. med. dent. Reinhard Hickel 13.09.2016

III
Inhaltsverzeichnis
Inhalt
Inhaltsverzeichnis ................................ ................................................................... III
Abkürzungsverzeichnis ............................. ............................................................ VII
1. Einleitung ..................................... ....................................................................... 10
1.1 Epidemiologie und Ätiologie ................................................................................................... 11
1.1.1 Aneurysmata ..................................................................................................................... 13
1.2 Klinische Beurteilung der SAB ............................................................................................... 15
1.2.1 Klinische Symptomatik ..................................................................................................... 15
1.2.2 Skalen zur Schweregradeinteilung ................................................................................ 15
1.2.3 Diagnose der Subarachnoidalblutung ........................................................................... 18
1.2.4 Therapie der Subarachnoidalblutung ............................................................................ 19
1.3 Pathophysiologie des Hirnschadens nach Subarachnoidalblutung ................................. 20
1.3.1 Frühe- und verzögerte Hirnschädigung ......................................................................... 22
1.3.1.1 Verzögerter Vasospasmus ........................................................................................... 25
1.3.1.2 Vasospasmen in der zerebralen Mikrozirkulation..................................................... 26
1.4 Stickstoffmonoxid (NO) ........................................................................................................... 27
1.4.1 Stickstoffmonoxidsynthase (NOS) ................................................................................. 29
1.5 NO im Gehirn ............................................................................................................................ 31
1.5.1 NO in der frühen Hirnschädigung nach SAB ................................................................ 32
1.5.2 NO-Donatoren ................................................................................................................... 33
1.5.3 Inhaliertes NO ................................................................................................................... 34
1.5.4 Rolle des NOs in der zerebralen Autoregulation ......................................................... 35
1.6 Rolle von Endothelin und Endothelin-Rezeptoren nach SAB ........................................... 36
1.7 Ziel der Arbeit ........................................................................................................................... 39
2. Material & Methoden............................. .............................................................. 40
2.1 Material ...................................................................................................................................... 40
2.1.1 Geräte ................................................................................................................................. 40
2.1.2 Verbrauchsmaterialien ..................................................................................................... 42
2.1.3 Inhalationsgase ................................................................................................................. 43
2.1.4 Chemikalien ....................................................................................................................... 43

IV
2.1.5 Intravitalmikroskop-Ausstattung ..................................................................................... 44
2.1.6 Software ............................................................................................................................. 45
2.1.7 Versuchstiere..................................................................................................................... 46
2.2 Methoden .................................................................................................................................. 48
2.2.1 Anästhesie und Antagonisierung.................................................................................... 48
2.2.2 Mechanische Beatmung .................................................................................................. 49
2.2.3 Regelung der Körpertemperatur ..................................................................................... 50
2.2.4 Messung des intrakraniellen Drucks (ICP) ................................................................... 50
2.2.5 Messung der zerebralen Durchblutung (CBF) ............................................................. 50
2.2.6 Induktion einer Subarachnoidalblutung (SAB) ............................................................. 51
2.2.7 Jugularis-Katheter ............................................................................................................. 52
2.2.8 Quantifizierung der Mikrozirkulation .............................................................................. 52
2.2.8.1 Analyse des zerebralen Gefäßbaums anhand des Strahler-Schemas ................. 52
2.2.8.2 Quantifizierung von Gefäßdurchmesser und Mikrovasospasmus ......................... 53
2.2.9 Intravitalmikroskopie ......................................................................................................... 54
2.2.9.1 Venöse und arterielle Katheterisierung der Maus .................................................... 54
2.2.9.2 Kranielles Fenster ......................................................................................................... 55
2.2.9.3 Intravitalmikroskopie zerebraler Mikrogefäße ........................................................... 56
2.2.10 Inhalation ......................................................................................................................... 56
2.2.10.1 Stickstoffmonoxid-Inhalation (iNO) ........................................................................... 56
2.2.10.2 Kohlendioxid-Inhalation .............................................................................................. 56
2.2.11 Anwendung von Clazosentan ....................................................................................... 56
2.2.12 Versuchsprotokolle ......................................................................................................... 58
2.2.12.1 Stickstoffmonoxid-Inhalation (iNO) ........................................................................... 58
2.2.12.2 Hemmung von Endothelin-1-Rezeptoren mit Clazosentan .................................. 59
2.2.12.2.1 Dosisfindung von Clazosentan .............................................................................. 59
2.2.12.2.2 Gabe von Clazosentan nach SAB ......................................................................... 60
2.2.12.3 Untersuchung der Rolle der NOS-Isoformen bei Vermittlung der CO2-Reaktivität zerebraler Gefäße....................................................................................................................... 61
3. Ergebnisse ..................................... ..................................................................... 62
3.1 Effekt von inhaliertem NO (iNO) auf die zerebrale Mikrozirkulation nach experimenteller Subarachnoidalblutung ...................................................................................... 62
3.1.1 Induktion der SAB ............................................................................................................. 62

V
3.1.2 Einfluss des inhalierten Stickstoffmonoxids auf die zerebrale Mikrozirkulation nach SAB ............................................................................................................................................... 63
3.1.2.1 Systemischer arterieller Blutdruck .............................................................................. 63
3.1.2.2 Physiologische Parameter ........................................................................................... 64
3.1.3 Einfluss von NO auf die arteriellen Gefäßdurchmesser ............................................. 64
3.1.4 Einfluss der NO-Inhalation auf die zerebralen Mikrovasospasmen .......................... 66
3.1.5 Räumliche Verteilung der Mikrovasospasmen ............................................................. 68
3.1.6 Effekt der NO-Inhalation auf den Grad der Mikrovasospasmen ............................... 70
3.1.7 Einfluss der NO-Inhalation auf Durchmesser zerebraler Venolen ............................ 72
3.2 Effekt der Endothelin-Rezeptor-Blockade auf die zerebrale Mikrozirkulation nach experimenteller Subarachnoidalblutung ...................................................................................... 72
3.2.1 Dosisfindung Clazosentan ............................................................................................... 72
3.2.2 Einfluss der ET1-Rezeptor-Blockade auf die zerebrale Mikrozirkulation ................. 74
3.2.2.1 MAP ................................................................................................................................. 74
3.2.2.2 Blut-Gas-Analyse ........................................................................................................... 74
3.2.2.3 Einfluss von 10 mg/kg KG/h Clazosentan auf den Durchmesser von zerebralen Arteriolen (9,5-55 µm) ................................................................................................................ 75
3.2.2.4 Effekt der ET1-Rezeptorblockade auf die posthämorrhagischen Mikro-vasospasmen .............................................................................................................................. 76
3.2.2.5 Effekt der ET1-Rezeptorblockade auf die Anzahl und den Konstriktionsgrad der Mikrovasospasmen..................................................................................................................... 78
3.3 Rolle der NO-Synthasen bei der CO2-induzierten Vasodilatation zerebraler Gefäße ... 80
3.3.1 Physiologische Parameter ............................................................................................... 80
3.3.2 Einfluss der NOS-Isoformen auf die CO2-Gefäßreaktivität ........................................ 80
3.3.3 Vasodilatation unter induzierter Hyperkapnie - Wildtyp-Mäusen .............................. 82
3.3.4 Vasodilatation unter induzierter Hyperkapnie – iNOS-defiziente Mäuse ................. 83
3.3.5 Vasodilatation unter induzierter Hyperkapnie – nNOS-defiziente Mäuse ................ 84
3.3.6 Vasodilatation unter induzierter Hyperkapnie – eNOS-defiziente Mäuse ................ 85
3.3.7 Vasodilatation unter induzierter Hyperkapnie der NOS-Isoformen in kleinen Arteriolen ...................................................................................................................................... 86
3.3.8 Mögliche Rolle der NOS bei der Formation von Mikrovasospasmen ....................... 86
4. Diskussion ..................................... ..................................................................... 89
4.1 Diskussion der Methoden ....................................................................................................... 89
4.1.1 Auswahl der Versuchstiere ............................................................................................. 89
4.1.2 Anästhesie ......................................................................................................................... 90

VI
4.1.3 Beatmung der Versuchstiere .......................................................................................... 91
4.1.4 Messung der zerebralen Durchblutung ......................................................................... 91
4.1.5 Wahl des SAB-Modells .................................................................................................... 92
4.1.6 Intravitalmikroskopie ......................................................................................................... 93
4.2. Diskussion der Ergebnisse .................................................................................................... 94
4.2.1 Hämodynamische Veränderungen nach SAB .............................................................. 94
4.2.2 Effekt von iNO und den Endothelin-Rezeptor-Blocker auf den systemischen Blutdruck nach SAB ................................................................................................................... 95
4.2.3 Veränderung der zerebralen Mikrozirkulation nach SAB ........................................... 96
4.2.3.1 Mikrovasospasmen ....................................................................................................... 96
4.2.3.2 Effekt von iNO ................................................................................................................ 97
4.2.3.3 Effekt der ET-1-Rezeptorblockade ............................................................................. 98
4.2.4 Rolle der NOS-Isoformen in der zerebralen Mikrozirkulation nach SAB ................. 99
5. Zusammenfassung ................................ ........................................................... 101
6. Literaturverzeichnis ........................... .............................................................. 102
A. Danksagung ..................................... ................................................................ 142
B. Lebenslauf ..................................... ................................................................... 143

VII
Abkürzungsverzeichnis
AA
ADMA
ATP
aSAB
AU
BH4
BBB
CA
CBF
CCT
CONSCIOUS
CT
cGMP
CI
CO2
CPP
CSF
CTA
CVS
CWp
DCI
DBI
DIND
DSA
EBI
EDRF
Arachidonsäure
Asymmetrisches Dimethylarginin
Adenosintriphosphat
Aneurysmatische Subarachnoidalblutung
Arbitrary Units, Perfusionseinheiten
Tetrahydrobiopterin
Blood Brain Barrier, Blut-Hirn-Schranke
Cerebral Autoregulation, Zerebrale Autoregulation
Cerebral Blood Flow, Zerebrale Durchblutung
Craniale Computertomographie
Clazosentan to Overcome Neurological Ischemia and Infarct
Occurring after Subarachnoid Hemorrhage
Computertomographie
Cyclic Guanosin Monophosphat, Zyklisches Guanosin-
Monophosphat
Confidence Interval, Konfidenzintervall
Kohlenstoffdioxid
Cerebral Perfusion Pressure, Zerebraler Perfusionsdruck
Cerebral Spinal Fluid, Liquor Zerebrospinalis
Computertomographische Angiographie
Cerebral Vasospasms, Zerebrale Vasospasmen
Circle Wilisii Perforation
Delayed Cerebral Ischämie, Verzögerte Zerebrale Ischämie
Delayed Brain Injury, Verzögerte Hirnverletzung
Delayed Ischemic Neurological Deficit, Verzögert Auftretendes
Ischämisches Neurologisches Defizit
Digitale Subtraktionsangiographie
Early Brain Injury, Frühe Hirnverletzung
Endothelium Derived Relaxing Factor

VIII
EEG
eNOS
ET
ETA
EtCO2
FITC
GCS
GMP
GSNO
GTN
GTP
ICP
iNO
iNOS
IVM
ISAT
LDF
LP
MAP
MCA
MMF
MMP
MRT
MVS
MW
n
NADPH
NAPSAH
7-NI
nNOS
NO
NOD
Elektroenzephalographie
Endotheliale NO-Synthase
Endothelin
Endothelin-Rezeptor-Antagonist
Endexpiratorisches (end tidal-) Kohlendioxid
Fluoresceinisothiocyanate
Gasgow Coma Scale
Guanosinmonophosphat
S-Nitrosoglutathion
Glyceryl Trinitrate (Nitroglycerin), Nitroglyzerin
Guanosintriphosphat
Intra Cranial Pressure, Intrakranieller Druck
Inhaliertes Stickstoffmonoxid
Induzierbare NO-Synthase
Intravitalmikroskopie
International Subarachnoid Aneurysma Trial
Laser-Doppler-Fluxmetrie
Lumbalpunktion
Middle Arterial Pressure, Mittlerer Arterieller Druck
Middle Cerebral Arterie, Arteria Cerebri Media
Medetomidin-Midazolam-Fentanyl
Matrix-Matalloprotease
Magnetische Resonanz Tomographie
Mikrovasospasmen
Mittelwert
Anzahl
Reduzierte Form des Nicotinamidadenindinukleotidphosphat
Non Aneurysmal Perimensephalic SAH, Nicht Aneurysmatische
Perimensephalische SAB
7-Nitroindazol
neuronale NO-Synthase
Stickstoffmonoxid
NO-Donator

IX
NOS
ONOO-
O2
O2-
p
PAASH
PARP
PDE
pETCO2
pH
PKC
ppm
ROI
ROS
SAB
SEM
sGC
SMTC
SNP
TCD
VEGF
WFNS
Stickstoff-Monoxid-Synthase
Peroxinitrit
Sauerstoff
Superoxid-Anion
Value of Probability, Signifikanzwert
Prognosis on Admission of Aneurysmal Subarachnoid Hemorrhage
Scale
Poly-(ADP-Ribose)-Polymerase
Phosphodiesterase
Endtidaler Kohlenstoffdioxid-Partialldruck
pH-Wert
Protein-Kinase-C
part per million
Region of Interest
Reactive Oxygen Species, Reaktive-Sauerstoff-Spezies
Subarachnoidalbutung
Standardfehler des Mittelwerts
Soluble Guanylat Cyclase, Löslische Guanylatcyklase
S-Methyl-L-Thiocitrullin
Sodium Nitroprusside, Natriumnitroprussid
Transkranielle Dopplersonographie
Vascular Endothelial Growth Factor ,Vaskulärer Endothelialer
Wachstumsfaktor
World Federation Neurological Surgeons

1. Einleitung
1. Einleitung
Die World Health Organisation (WHO) definiert den Schlaganfall als ein
Krankheitsbild, bei dem sich die klinischen Zeichen einer fokalen oder globalen
Störung zerebraler Funktionen rasch bemerkbar machen, mindestens 24 Stunden
anhalten oder zum Tode führen und offensichtlich nicht auf andere als vaskuläre
Ursachen zurückgeführt werden können.1 Demnach setzen akute fokale (oder globale)
neurologische Defizite ein aufgrund einer umschriebenen (oder globalen) Durchblu-
tungsstörung im Gehirn. Eine Durchblutungsstörung kann sowohl durch einen
Durchblutungsmangel oder eine Blutung hervorgerufen werden. Globale Hirndurchblu-
tungsstörung treten z. B. im Rahmen eines Herzstillstandes oder eines „Beinahe-
Ertrinkens“ auf. Generell unterscheidet man zwischen ischämischen Schlaganfällen,
spontanen intrazerebralen Hämatomen, Subarachnoidalblutungen sowie Hirnvenen-
und Sinusvenenthrombosen.2 Die Subarachnoidalblutung (SAB) ist ein relativ seltener
Subtyp des Schlaganfalls, der dadurch gekennzeichnet ist, dass Blut in den mit Hirn-
flüssigkeit (Liquor cerebrospinalis) gefüllten Subarachnoidalraum gelangt. Diese
Blutung wird häufig durch eine Aneurysmaruptur hervorgerufen.3,4 Der Subarach-
noidalraum wird begrenzt durch die Spinnenhaut (Arachnoidea mater) und die innere
Hirnhaut (Pia Mater), die das Hirnparenchym direkt bedeckt.
Abbildung 1: Schematische Darstellung eines Schädel s mit den entsprechenden Regionen die
nach SAB beeinträchtigt sind.

1. Einleitung
11
Die SAB kann spontaner oder traumatischer Natur sein.3,5 Die spontan auftretende
SAB macht etwa 5 % aller Schlaganfälle aus und wird zu 80-85% durch die Ruptur
eines intrakraniellen Aneurysmas hervorgerufen.3,5,6,6-8 Aufgrund deutlicher Verbesse-
rungen der mikrochirurgischen- und radiologisch-interventionellen Therapie-Verfahren
in den letzten Jahrzehnten, und der spezialisierten neurointensiv-medizinischen
Behandlung der aneurysmatischen Subarachnoidalblutung (aSAB), ist die Mortalitäts-
rate nach SAB zwar aktuell rückläufig,9-11 das Krankheitsbild hat jedoch noch immer
eine hohe Morbidität und Mortalität.4
1.1 Epidemiologie und Ätiologie
In den Vereinigten Staaten von Amerika sind Schlaganfälle die dritthäufigsten
Todesursachen. Jährlich erleiden circa 795.000 Menschen einen Schlaganfall, davon
610.000 erstmalig.12 In circa 5% der Fälle handelt es sich um eine Subarachnoidal-
blutung.3 Diese weist die schlechteste Prognose aller Schlaganfälle auf, sowohl was
das Überleben als auch die neurologischen Funktionsausfälle betrifft. Im europäischen
Raum ist eine ähnliche Inzidenz (7 bis 10 Fälle pro 100.000 Personen) wie in
Nordamerika zu finden.3,13-15 Die höchste Inzidenz wird jedoch für Finnland und Japan
dokumentiert, 20-30 Fälle pro 100.000 Personen jährlich.16-19 Auch Alter und
Geschlecht beeinflussen die Inzidenz der SAB. In der Altersgruppe von 25-45 Jahren
weisen vor allem Männer eine erhöhte SAB-Inzidenz auf, während in der Altersgruppe
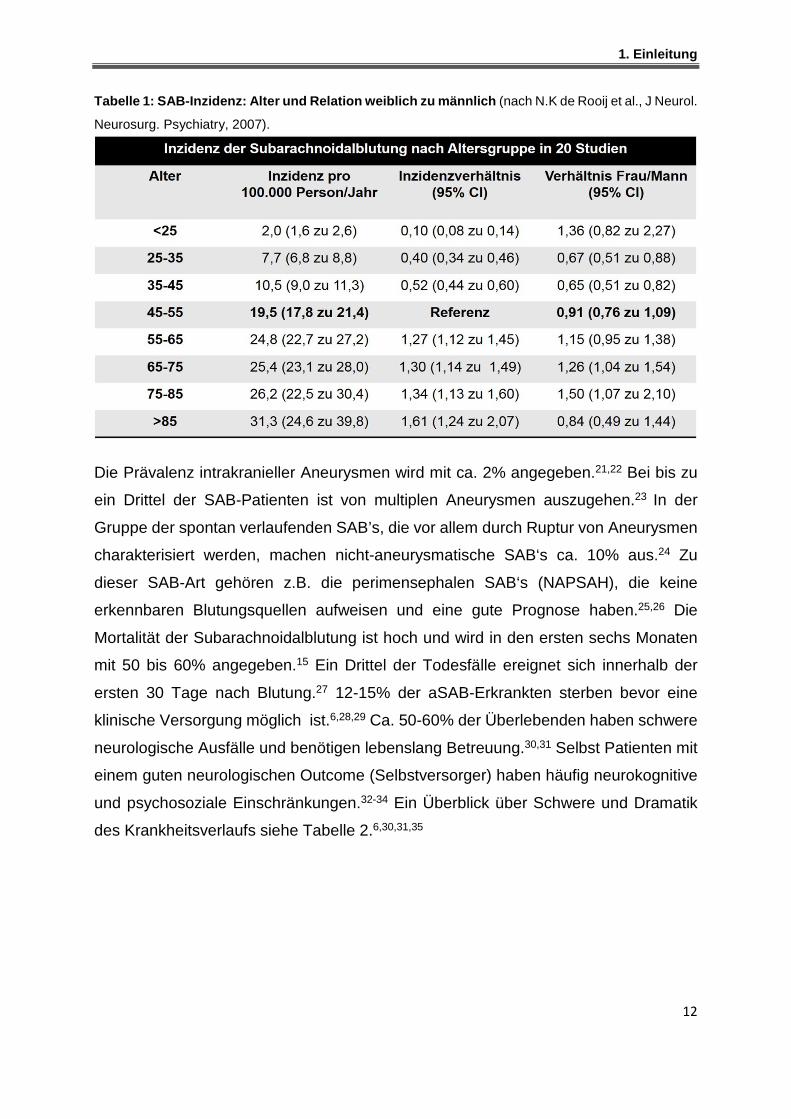
von 55 bis 85 Jahren, die SAB-Inzidenz bei Frauen deutlich höher ist.3,15,20
1. Einleitung
12
Tabelle 1: SAB-Inzidenz: Alter und Relation weiblic h zu männlich (nach N.K de Rooij et al., J Neurol.
Neurosurg. Psychiatry, 2007).
Die Prävalenz intrakranieller Aneurysmen wird mit ca. 2% angegeben.21,22 Bei bis zu
ein Drittel der SAB-Patienten ist von multiplen Aneurysmen auszugehen.23 In der
Gruppe der spontan verlaufenden SAB’s, die vor allem durch Ruptur von Aneurysmen
charakterisiert werden, machen nicht-aneurysmatische SAB‘s ca. 10% aus.24 Zu
dieser SAB-Art gehören z.B. die perimensephalen SAB‘s (NAPSAH), die keine
erkennbaren Blutungsquellen aufweisen und eine gute Prognose haben.25,26 Die
Mortalität der Subarachnoidalblutung ist hoch und wird in den ersten sechs Monaten
mit 50 bis 60% angegeben.15 Ein Drittel der Todesfälle ereignet sich innerhalb der
ersten 30 Tage nach Blutung.27 12-15% der aSAB-Erkrankten sterben bevor eine
klinische Versorgung möglich ist.6,28,29 Ca. 50-60% der Überlebenden haben schwere
neurologische Ausfälle und benötigen lebenslang Betreuung.30,31 Selbst Patienten mit
einem guten neurologischen Outcome (Selbstversorger) haben häufig neurokognitive
und psychosoziale Einschränkungen.32-34 Ein Überblick über Schwere und Dramatik
des Krankheitsverlaufs siehe Tabelle 2.6,30,31,35

1. Einleitung
13
Tabelle 2: Mortalität und Morbidität der SAB im Spo ntanverlauf (nach Bederson et al., Stroke,
2000; Hop et al., Stroke, 1998; Huang et al., Neurosurgery, 2002 und van Gijn et al., Lancet, 2007 )
Risikofaktoren die zu einer aneurysmatischen Subarachnoidalblutung beitragen
können sind arterielle Hypertonie, Hyperlipidämie, Zigarettenkonsum (Nikotinabusus),
Kokain-Missbrauch, exzessiver Alkoholkonsum und einige angeborene Bindegewebe-
störungen, wie z.B. das Ehlers-Danlos-Syndrom, das Marfan-Syndrom, Osteogenesis
imperfecta und Pseudoxanthoma elasticum.36-40 Einige Patienten weisen eine familiäre
Prädisposition für Aneurysmata und Subarachnoidalblutungen auf,41 dabei sind
Frauen vier Mal häufiger betroffen als Männer.42-44 Die Familienanamnese der
polyzystischen Nierenerkrankung22 und Hypertension36,45 scheinen u.a. das Risiko
einer SAB zu erhöhen. 5-20% der SAB-Patienten weisen eine positive Familien-
anamnese auf.46
1.1.1 Aneurysmata
Bei Aneurysmata handelt es sich um Aussackungen von Gefäßabschnitten, meist von
Arterien; sie sind spindel- oder sackförmig und entstehen infolge angeborener oder
erworbener Gefäßwandveränderungen. Aneurysmata sind wahrscheinlich das Ergeb-
nis einer Kombination von genetischen, hämodynamischen, oder durch Nikotin- oder

1. Einleitung
14
Alkoholmissbrauch induzierten Strukturfehler, die zu einer Abnahme der mittleren
Muskelschicht und der Tunica media, der Arterienwand, führen. Außerdem können
chronische hämodynamische induzierte intravaskuläre Schubspannungen für die
Bildung von aneurysmatischen Ausstülpungen in den Subarachnoidalraum
verantwortlich sein.37,47,48 Auch die Rolle von oxidativen Stress bei der Bildung und
Ruptur von Aneurysmata ist nicht zu unterschätzen.49 Aneurysmata entwickeln sich
meist in Gefäßgabelungen großer Gefäße, da turbulente Strömungen sich dort
bevorzugt entwickeln. Am häufigsten sind intrakranielle Aneurysmata an der A.
communicans anterior und der A. cerebri anterior lokalisiert (40%), gefolgt von der A.
carotis interna (30%, hauptsächlich am intraduralen supraklinoidal gelegenen Anteil).
In 20% der Fälle befinden sie sich an der A. cerebri media (MCA), d.h., dass sich die
überwiegende Mehrzahl von Aneurysmata (80-90%) im vorderen Teil der basalen
Hirnarterien befindet.21 Die restlichen 10-20% sind im hinteren Abschnitt des Circulus
arteriosus Willisii, bestehend aus A. basilaris und A. vertebralis, gelegen.50,51 Selten
werden intrazerebrale Aneurysmata durch Infektionen (sog. mykotische Aneurysmata)
oder traumatische Gefäßwandschädigung verursacht. Mit dem Anstieg der Größe des
Aneurysmas wird die Wandspannung erhöht, wodurch das Aneurysma zunehmend
anfällig für eine Ruptur wird. Nach dem Laplace’schen Gesetz verändert sich die
Wandspannung proportional zu der vierten Potenz des Aneurysmaradius, und dieser
ändert sich wiederum proportional zum arteriellen Druck. Deswegen erhöht sich das
Rupturrisiko durch Zunahme der Aneurysma-Größe drastisch.
Th = Pt x r i/h (N/m 2)
(Th = über die Gefäßwanddicke integrierte Wandspannung ; Pt = transmuraler Druck; r i =
Innenradius des Gefäßlumens; h = Wanddicke des Gefä ßes)
Als „kritischer Durchmesser“ gelten 4-10 mm.52-63 Zufällig diagnostizierte Aneurys-
mata über diese Größe sollten deswegen therapiert werden.63 Aneurysmata können
Durchmesser von mehr als 2,5 cm erreichen (Riesen- oder Giant- Aneurysma).64

1. Einleitung
15
1.2 Klinische Beurteilung der SAB
1.2.1 Klinische Symptomatik
Das Hauptsymptom der Subarachnoidalblutung ist der akute, schlagartig einsetzende
und sehr starke „Vernichtungs-Kopfschmerz“8,65 der bei ca. 80% der SAB-Patienten
beschrieben wird66 und der maximal innerhalb von einer Minute nach Blutungsbeginn
auftritt.67 Je nach Intensität und Lokalisation der Blutung können diese Kopfschmerzen
von Übelkeit, Erbrechen, Nackensteifigkeit und einer Bewusstseinseintrübung bis zum
Koma begleitet sein.6 Jeder zweite SAB-Patient leidet unter Hirnhautreizung (Menin-
gismus), in bestimmten Fällen aber ist der Meningismus um einige Stunden verzögert.
Die Meningen werden dann durch Blutabbauprodukte gereizt.48 In 25% der SAB-Fälle
treten subhyaloidale Glaskörperblutungen, das Terson-Syndrome, auf.68,69 Fokale
neurologische Defizite in der Initialphase oder epileptische Anfälle sprechen für ein
intrazerebrales Hämatom.70 Bei 10–30% der Patienten tritt als initiale Symptomatik
eine mildere Form des akuten Kopfschmerzes auf, die differentialdiagnostisch als
Migräne, akutes Zervikalsyndrom, hypertensive Krise oder Meningitis fehlinterpretiert
werden kann. Diese als „warning headache“ beschriebene Symptomatik kann bis zu
drei Wochen vor dem eigentlichen Blutungsereignis auftreten. Ursache ist meist eine
gedeckte Blutung aus dem später rupturierten Aneurysma.71-74
1.2.2 Skalen zur Schweregradeinteilung
Vigilanz/Grad der Bewusstlosigkeit, Alter und Blutvolumen in der initialen kranialen
Computertomographie (CCT) sind die Faktoren, die nach bisherigen Erkenntnissen
den größten Einfluss auf das Outcome von Patienten haben.75-77 Eine Vielzahl von
Skalen zur Graduierung der SAB wurden entwickelt.77 Im Jahr 1968 erschien die
Einteilung von Hunt und Hess, die eine modifizierte Form des 1956 entwickelten
Systems von Botterell darstellt.78 Ein Vorteil dieser Graduierung ist, dass sie weit
verbreitet und außerdem sehr einfach anzuwenden ist.79 Anhand der Hunt und Hess
Einteilung kann das operative Risiko in Bezug auf den Zustand des Patienten zum
Zeitpunkt der Operation abgeschätzt werden.80 Der Nachteil ist, dass sie eine
schwache Inter-Beobachter-Variabilität aufweist. Im Vergleich weisen Graduierungs-
skalen wie die WFNS (World Federation of Neurological Surgeons) und die PAASH

1. Einleitung
16
(Prognosis on Admission of Aneurysmal Subarachnoid Hemorrhage) eine starke Inter-
Beobachter-Variabilität auf.81 Im Jahr 1974 beschrieben Teasdale und Jennett die
Glasgow Coma Scale (GCS), ein Bewertungssystem zum Einschätzen einer
Bewusstseinsstörung, die ursprünglich bei traumatischen Kopfverletzungen zum
Einsatz kam und erst sekundär für die Beurteilung von Patienten nach Subarachnoidal-
blutung verwendet wurde.82 Im Gegensatz zu der Einteilung nach Hunt und Hess, wird
jede Rubrik einzeln graduiert. Es gibt drei verschiedene Rubriken, in die der Behandler
die Patienten einstuft, um dann am Ende die Punkte zu addieren.81 Es hat sich jedoch
als nachteilig erwiesen, dass die verbale Reaktion des Patienten so stark in das
Ergebnis einfließt, da Patienten mit Subarachnoidalblutung oft intubiert und so in ihrer
verbalen Kommunikation eingeschränkt sind.79 Im Jahr 1988 wurde unter dem Vorsitz
von Charles Drake die World Federation of Neurological Surgeons (WFNS) Scale
definiert. Es ist eine Fünf-Grad-Einteilung die an der Glasgow Coma Scale angelehnt
ist und beinhaltet zusätzlich das Vorhandensein eines fokalen neurologischen
Defizites.79,83 Die Prognosis on Admission of Aneurysmal Subarachnoid Hemorrhage
(PAASH) ist eine andere Fünf-Grad-Einteilugsskala die ausschließlich in Anlehnung
zur GCS entwickelt wurde.84 WFNS und PAASH verfügen über einen guten
prognostischen Wert im Hinblick auf Patienten Outcome, wirken übereinstimmend bei
der Beurteilung des überlappenden CI (Konfidenzintervall) und weisen eine ähnliche
Inter-Beobachter-Zuverlässigkeit auf. Trotzdem ist die PAASH-Skala leichter
anwendbar und zuverlässiger bei der Beurteilung eines schlechten Outcomes bei
SAB-Patienten als WFNS. Deswegen ist PAASH die geeignetste Skala für Beurteilung
eines klinischen Zustandes eines SAB-Patienten.81,85

1. Einleitung
17
Tabelle 3: Klinische Schweregradeinteilung nach HUN T und HESS, World Federation of
Neurological Surgeons (WFNS) und Prognosis on Admission of Aneurysmal Subarachnoid
Hemorrhage Scale (PAASH) (nach Degen et al., Stroke, 2011).
Wie in Tabelle 4 dargestellt, erlaubt die Einteilung nach Fisher (1980) eine
Klassifizierung in vier Gruppen anhand der computertomografischen Morphologie der
Subarachnoidalblutung.86 Die Graduierung erfolgt nach Menge an Blut im initialen
CCT. Diese Klassifizierung ist leicht anzuwenden und soll mit dem Vorkommen von
verzögertem Vasospasmus korrelieren.87,88
Tabelle 4: Klassifikation des Ausmaßes der Subarach noidalblutung in der Computertomo-
graphie (nach Fisher, Neurosurgery, 1980)

1. Einleitung
18
1.2.3 Diagnose der Subarachnoidalblutung
Bei klinischem Verdacht auf eine SAB wird meist eine kraniale Computertomografie
veranlasst, in der das subarachnoidale Blut hyperdens zur Darstellung kommt (Abbil-
dung 2) und die Blutung bewiesen werden kann.
Abbildung 2: Die native CCT zeigt eine ausgedehnte basal betonte SAB Fisher° III
Darüber hinaus lassen sich in der CCT eine intraparenchymatöse Blutungskom-
ponente oder ein Liquoraufstau erkennen. In vielen Fällen gibt bereits die Blut-
verteilung in der nativen CCT Hinweise auf die Lokalisation der Blutungsquelle. In der
akuten Phase ist die Magnetresonanztomografie (MRT) unter Verwendung geeigneter
Sequenzen zum Nachweis einer SAB genauso geeignet wie die CCT. Mit
zunehmender zeitlicher Latenz zum Blutungsereignis nimmt die Dichte der Blutung in
der CCT ab, dann ist die MRT der CCT zum Nachweis hämorrhagischer Residuen
sogar deutlich überlegen.89 Bei fehlendem Blutnachweis in der Bildgebung sollte etwa
sechs bis zwölf Stunden nach Einsetzen des Kopfschmerzes eine Lumbalpunktion
(LP) durchgeführt werden, bei der nicht der Nachweis des Blutes selbst, sondern ein
xanthochromer Überstand nach Zentrifugation als sicherster Nachweis einer SAB
gilt.6,90 Da das Timing der LP in Bezug auf den Beginn der SAB die Ergebnisse der
Analyse beeinflussen kann, werden ein paar Methoden zur Unterscheidung von
traumatischer und aneurysmatischer SAB vorgeschlagen wie z.B. den "drei
Röhrchentest", den Öffnungsdruck, und die visuelle Inspektion der Xanthochromie.91,92
Wird bildgebend, oder in der Liquordiagnostik eine Subarachnoidalblutung diagnosti-
ziert, muss die Blutungsquelle identifiziert werden; Mittel der Wahl bzw. Goldstandard

1. Einleitung
19
ist hierbei die digitale Subtraktionsangiografie (DSA). Sie liefert neben der
Aneurysmalokalisation wichtige Informationen bezüglich der Aneurysmakonfiguration
und der Lagebeziehung zu angrenzenden Gefäßen, welche die Auswahl des
therapeutischen Verfahrens zur Aneurysmaausschaltung entscheidend beeinflussen.
Die CT-Angiografie (CTA) ist mittlerweile in der Detektion von Blutungsquellen ähnlich
sensitiv und spezifisch wie die konventionelle Angiografie und aufgrund ihrer höheren
Verfügbarkeit sowie der nicht-invasiven und schnellen Anwendung sehr nützlich. Sie
ist jedoch bei der Darstellung kleiner Aneurysmen (< 3 mm) der DSA unterlegen.93,94
Lässt sich angiografisch keine Blutungsquelle nachweisen, sollte die Untersuchung ein
bis zwei Wochen nach dem Blutungsereignis wiederholt werden. Bei erneut fehlendem
Nachweis einer Blutungsquelle sollte dann eine MRT von Schädel und Halswirbelsäule
mit der Frage nach einem (teil-)thrombosiertem Aneurysma oder einer spinalen
Blutungsursache erfolgen.
1.2.4 Therapie der Subarachnoidalblutung
SAB-Patienten sollten in spezialisierten Zentren, die sowohl in Bezug auf die
intensivmedizinische Versorgung als auch auf die operative oder interventionelle
Ausschaltung der Blutungsquelle große Erfahrung haben, behandelt werden bzw.
verlegt werden, wenn der Allgemeinzustand es zulässt.15,66,95 Alle Patienten mit
gesicherter SAB müssen intensivmedizinisch betreut werden.15,96 Das Hauptrisiko in
der Frühphase der Subarachnoidalblutung ist die Nachblutung,15 die 15% der SAB-
Patienten erleiden97 und zu 70% tödlich enden.66 Deswegen sollte in der Akutphase
der systolische Blutdruck unter 140 mmHg gehalten werden. Es können hierfür
antihypertensive Substanzen wie Labetalol und Nicardipin intravenös verabreicht
werden.77,98,99 Eine Hyperglykämie, die die 10 mmol/l Grenze übersteigt, kommt bei
ein Drittel der SAB-Patienten vor und ist mit einer schlechten klinischen Prognose
assoziiert.100-103 Hyperthermie (Körpertemperatur über 38,3 °C) 100 ist nach
aneurysmatischen SAB ebenso mit einer schlechten Prognose assoziiert,104 ca. 50%
der SAB-Patienten entwickeln diesen Zustand,104 der medizinisch und physikalisch
behandelt werden sollte.15 Pharmakologisch kann eine SAB mit der Triple-"H"-
Therapie, Magnesium, Statinen, Fasudil, Erythropoietin, Nicardipine, Sildenafil,
Endothelin-Rezeptor-Antagonisten, NO-Donatoren (Nitroglyzerin, Nitroprusside) und

1. Einleitung
20
Nimodipine behandelt werden. Von den genannten Mitteln hat allerdings nur Nimodipin
einen durch klinische Studien nachgewiesenen positiven Effekt auf das Outcome nach
SAB.105-109 Zur Versorgung eines rupturierten Aneurysmas stehen die mikrochirur-
gische Versorgung mittels Clipping und die endovaskuläre Ausschaltung mit Coils
(Coiling) zur Verfügung. Die endovaskuläre Versorgung von Aneurysmata ist seit etwa
24 Jahren eine Alternative zur chirurgischen Versorgung.110 Coils sind Platinspiralen,
die mit Hilfe eines Katheters im Aneurysma platziert werden. Dort lösen sie eine lokale
Thrombose aus und trennen das Aneurysma vom Blutkreislauf. Der Zeitpunkt der
Aneurysmabehandlung hat Einfluss auf den weiteren Krankheitsverlauf. So kamen
mehrere klinische Studien zu dem Ergebnis, das eine frühe chirurgische Behandlung
des Aneurysmas die Reblutungsrate senkt.111 Auch erleichtert eine frühe Behandlung
des rupturierten Aneurysmas die Behandlung von Komplikationen wie zerebralen
Vasospasmen.112 Im International Subarachnoid Aneurysm Trial (ISAT) wurden
Patienten mit rupturierten Aneurysmata, deren Aneurysmata gleichermaßen für eine
neurochirurgische und endovaskuläre Versorgung geeignet waren, in zwei Gruppen
randomisiert.113,114 Die Aneurysmata der Patienten der einen Gruppe wurden mit Clips
und die Aneurysmata der Patienten der anderen Gruppe mit Coils versorgt. Für
Patienten, deren Aneurysma endovaskulär mit Coils versorgt wurde, ergab sich in
signifikant mehr Fällen eine bessere neurologische Erholung. Das Epilepsierisiko war
in dieser Gruppe ebenfalls niedriger. In der neurochirurgisch durch Clips behandelten
Patientengruppe war das Reblutungsrisiko geringer. Die Entscheidung für eine
chirurgische oder endovaskuläre Behandlung des Aneurysmas hängt aber von
mehreren Faktoren ab: Patientenalter, Aneurysmalokalisation und -morphologie, und
Allgemeinzustand des Patienten spielen ebenfalls eine Rolle.115
1.3 Pathophysiologie des Hirnschadens nach Subarach noidal-
blutung
Im Rahmen der Subarachnoidalblutung tritt Blut in den Subarachnoidalraum aus. Die
Menge ist abhängig von der Blutungsquelle und dem arteriellen Blutdruck. Das
zusätzliche Volumen führt im geschlossenen System des Neurokraniums zu einer
plötzlichen Erhöhung des intrakraniellen Drucks.116 Die Folgen einer solchen Druck-
erhöhung werden von der Monro-Kellie-Doktrin vorhergesagt, die feststellt, dass die

1. Einleitung
21
Summe der drei Komponenten in der Schädelkalotte, Gehirnparenchym, (arterielles
und venöses) Blut und Liquor cerebrospinalis stets gleich bleiben muss, um den
intrakraniellen Druck konstant zu halten (Abbildung 3). Kommt es zur Volumen-
zunahme im Subarachnoidalraum (wie z. B. bei einer SAB) können zunächst Blut und
Liquor verdrängt werden. Bis zu einem gewissen Grad kann so der intrakranielle Druck
im Normalbereich gehalten werden. Sind jedoch die Reservekapazitäten aufgebraucht
führt jede weitere Volumenzunahme zu einem linearen Anstieg des ICP.
Abbildung 3: Monro-Kellie-Doktrin
Die schlagartige Drucksteigerung durch das zusätzliche Blutvolumen bei der SAB
lässt den intrakraniellen Druck (Normalwert von 5-15 mmHg) auf deutlich erhöhte
Werte (28 mmHg bei kleinen Hämatomen, bis 80 mmHg bei großen Hämatomen)
ansteigen.117-120 Dies, sowie der Kontakt von Blut bzw. Blutbestandteilen mit der
schmerzempfindlichen Dura mater, sind die Ursachen des pathognomonischen,
schlagartig auftretenden starken Kopfschmerzes.8,65 Übersteigt der intrakranielle
Druck (auch nur kurzfristig) den systemischen Blutdruck, kann kein Blut nach
intrakraniell gelangen, was zur Minderdurchblutung bzw. zum Durchblutungsstillstand
(globale Ischämie) des Gehirnes führt. Aufgrund der kurzen Ischämietoleranz des
Gehirns118,121 kann in der Folge rasch ein Bewusstseinsverlust eintreten. Bleibt der ICP
dauerhaft erhöht, ist der Bewusstseinsverlust dauerhaft und führt in schneller Folge
r

1. Einleitung
22
zum irreversiblen Hirnschaden und zum Tod. Sinkt die intrakranielle Hypertension
nach einer initialen Druckspitze wieder ab und wird die zerebrale Durchblutung wieder
ermöglicht, kann das Bewusstsein schnell wiedererlangt werden. Eine weitere Folge
der initialen Blutung ist der Hydrozephalus. Entweder kann es durch Verschluss der
Liquorabflußwege durch Hämatome (v. a. im Bereich des Aquaeductus cerebri) zum
Hydrozephalus occlusivus kommen oder durch Hemmung der Resorption im Bereich
der Pacchionini Granulation50,122 zum Hydrozephalus aresorptivus. Eine weitere
Komplikation der SAB ist das Auftreten eines Hirnödems. Das Hirnödem wird
traditionell in zwei große Subtypen eingeteilt, in das zytotoxische und vasogene
Hirnödem. Von diesen weist vor allem das zytotoxische Hirnödem besondere
Resistenz gegenüber jegliche medizinische Behandlung auf.123 Durch immer weiter
steigendem Hirndruck wird die Perfusion des Gehirns weiter eingeschränkt, und es
kommt zur globalen Ischämie mit begleitenden disseminierten kortikalen und
hypothalamischen Läsionen.124-126 Die Blutung führt sowohl beim Menschen als auch
im experimentellen Modell zu einer sofortigen und andauernden Reduktion der
zerebralen Durchblutung.127-131 Es wird vermutet, dass die Menge und Lokalisation des
Blutes nach Subarachnoidalblutung verantwortlich für die Entstehung von zerebralen
Vasospasmen ist, und mit der Lokalisation und Ausprägung der Spasmen korreliert.50
1.3.1 Frühe- und verzögerte Hirnschädigung
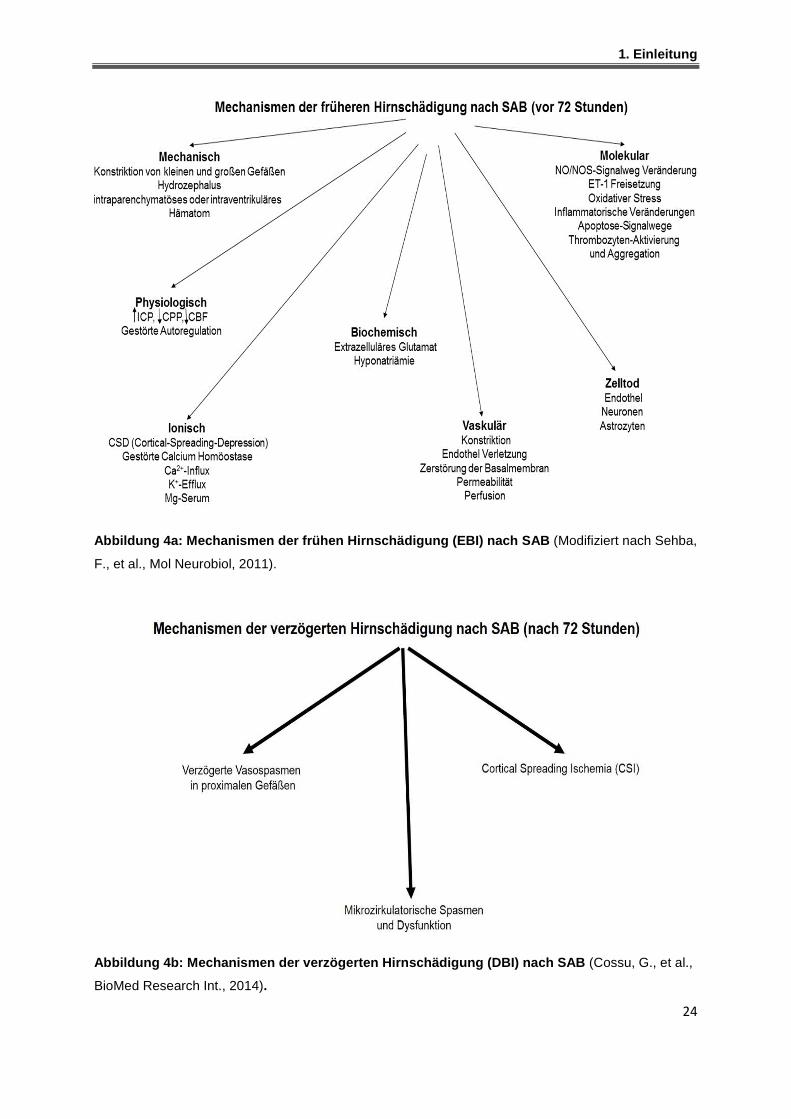
Der Begriff "Frühe Hirnschädigung" (Early Brain Injury, EBI) wird seit ca. 2004
verwendet. Er fasst die akuten pathophysiologischen Vorgänge, die innerhalb der
ersten 72 Stunden nach SAB-Blutung auftreten, zusammen.129,130 Eine schematische
Übersicht bietet Abbildung 4a. Zu den Ereignissen, die durch die Subarachnoidal-
blutung hervorgerufen werden, gehören die Beeinträchtigung der zerebralen
Autoregulation,132 die Störung der Blut-Hirn-Schranke,132 die Aktivierung von
Entzündungswegen und Apoptose,132 Exzitotoxizität und oxidativer Stress.132 Diese
Vorgänge werden zu gewissen Zeitpunkten durch Blut und Blutbestandteile und durch
transiente zerebrale Ischämie hervorgerufen.133-136 Bereits Sekunden nach der SAB
sind Veränderungen der zerebralen Physiologie (ICP-Anstieg, CPP- Verringerung,
CBF-Verringerung, Blutdruck-Anstieg), der Ionen-Homöostase und der elektrischen
Hirnaktivität (EEG-Amplitude) nachzuweisen.132 Ab der ersten Stunde nach SAB

1. Einleitung
23
treten folgende Ereignisse auf: Konstriktionen von kleinen und großen Gefäßen,137
Abbau von Kollagen-IV (größtes Protein der Basal Lamina),138 die Gefäßpermeabilität
erhöht sich während die Gefäßperfusion geringer wird,139 Zelltodsignalwege werden
aktiviert,132 Erhöhung der Glutamat-Konzentration in der zerebralen interstitiellen
Flüssigkeit,140 Anstieg des Hirnwassergehalt,141 der ICP stabilisiert sich auf einem
neuen Plateau, das über dem basalen Wert liegt,127,130 der CPP erholt sich,127 die
zerebrale Durchblutung und Autoregulation bleiben beeinträchtigt,142 und es entstehen
Veränderungen auf molekularer Ebene (starke Verringerung der NO-Konzentration,143
Thrombozyten-Aktivierung und intraluminale Aggregation,144 Anstieg der ET-1-
Konzentration,145 Anstieg des oxidativen Stresses,146,147 Erhöhung der Zytokin-
Expression).132 Der Begriff verzögerte Hirnverletzung (Delayed Brain Injury, DBI)
beschreibt pathophysiologische Mechanismen, die sich in der späteren Phase nach
SAB (3-21 Tage) ereignen und zu einer sekundären Schädigung des Hirngewebes
führen. Diese Vorgänge sind direkte Folge des EBI und können zu einer verzögert
auftretenden zerebralen Ischämie (Delayed Cerebral Ischemia, DCI) führen.148 DCI
wird in bis zu 30% der Fälle für schlechtes Outcome oder Tod von Patienten, deren
Aneurysma erfolgreich behandelt werden konnte, verantwortlich gemacht.149 Die
Ursachen sind noch nicht völlig geklärt, aber man vermutet, dass DCI u. a. durch
verzögert auftretende Vasospasmen, Thrombose, Dysfunktion der Mikrozirkulation
und Cortical Spreading Ischemia verursacht wird (Abbildung 4b). Diese Prozesse
werden wahrscheinlich durch Mechanismen der frühen Hirnschädigung ausgelöst bzw.
mitverursacht.133
1. Einleitung
24
Abbildung 4a: Mechanismen der frühen Hirnschädigung (EBI) nach SAB (Modifiziert nach Sehba,
F., et al., Mol Neurobiol, 2011).
Abbildung 4b: Mechanismen der verzögerten Hirnschäd igung (DBI) nach SAB (Cossu, G., et al.,
BioMed Research Int., 2014).

1. Einleitung
25
1.3.1.1 Verzögerter Vasospasmus
Zerebrale Vasospasmen, die erstmalig durch Robertson (1949), Ecker und Riemen-
schneider (1951) nach einer Subarachnoidalblutung beschrieben wurden,150,151 sind
Verengungen von zerebrale Arterien,122,152,153 die auch durch andere intrakraniellen
Blutungen hervorgerufen werden können.154,155,156,157 Verzögerte zerebrale Vasos-
pasmen treten frühestens drei Tage nach SAB auf. Sie erreichen den Höhepunkt ihrer
Inzidenz zwischen dem 6. und 11. Tag und können bis zu 3 Wochen nach der Blutung
vorherrschen.158 Im klinischen Alltag wird der Vasospasmus meist nicht per Angiogra-
phie, sondern mit Hilfe der transkraniellen Dopplersonographie (TCD) diagnostiziert.159
Mit diesem nicht invasiven Verfahren kann die Blutfließgeschwindigkeit in den
größeren Hirngefäßen bestimmt werden.159 Die zugrunde liegenden patho-
physiologischen Mechanismen sind bisher nicht völlig geklärt. Ein wichtiger Faktor für
die Entstehung eines verzögerten Vasospasmus scheint der Abbau von Erythrozyten
im Subarachnoidalraum zu sein. Dadurch werden Kalium- und vor allen Eisen-Ionen
freigesetzt, die zu einer erhöhten Expression von leistungsstarken Vasokonstriktoren
wie Thromboxan, Endothelin-1, Serotonin, Plättchen-Aktivierender-Faktoren, und 20-
Hydroxyeicosatetraenoid-Säure, die in der Zerebrospinalflüssigkeit (CSF) führen
können.160,161 Der intrazelluläre Calcium-Anstieg in den glatten Muskelzellen, und die
ionische Veränderung, wie z.B. der Anstieg des Kalium- und Verringerung des
Magnesium-Spiegels im Serum, sind weitere Faktoren die die Vasospasmenbildung
fördern.133 Ein großer Teil der Forschung wurde von der Annahme beeinflusst, dass
CVS für die verzögerte zerebrale Ischämie (DCI) verantwortlich ist. Heute ist es
allgemein anerkannt, dass nicht alle Patienten mit CVS DCI entwickeln. Umgekehrt
kann DCI oder DIND in Abwesenheit von CVS auftreten.133,135,162-166 Daher ist
anzunehmen, dass CVS nicht die einzige Ursache für die Entstehung von DCI
darstellt.167,168 Mechanismen die für die Pathogenese des DCI und für dessen
schlechte Prognose nach SAB beitragen, sind die frühe Hirnverletzung,132 Cortical
Spreading Depression,168,169 Entzündungen,170-173 Mikrothromben132,174 sowie CVS.175
Obwohl bei ca. 70% der SAB-Patienten angiographisch zerebrale Vasospasmen
nachgewiesen werden können, entwickeln nur 30% ein verzögertes neurologisches
Defizit (DIND).176 Weiterhin konnte gezeigt werden, dass das Outcome nach SAB
durch eine pharmakologische Hemmung der Vasospasmusentstehung nicht
verbessert werden kann.177,178 Diese Ergebnisse legen nahe, dass die Spasmen der

1. Einleitung
26
großen Hirngefäße nicht die einzige Ursache für die hohe Mortalität und
Morbiditätsrate der SAB beim Menschen sind.133 Dies legt die Vermutung nahe, dass
die zerebrale Mikrozirkulation eine entscheidende Rolle für die nach SAB
beobachteten Durchblutungsstörungen spielen könnte.179,180
1.3.1.2 Vasospasmen in der zerebralen Mikrozirkulat ion
Herz und Mitarbeiter konnten bereits 1975 zeigen, dass die Applikation von Blut auf
piale Mikrogefäße zu einer sofortigen Vasokonstriktion führt.181 Dass dieser Effekt
auch bei SAB-Patienten auftritt, wurde 2003 mittels orthogonal polarization spectral
imaging nachgewiesen: SAB-Patienten, die innerhalb von drei Tagen operiert wurden,
zeigten eine deutlich verringerte Kapillar-Dichte, außerdem wiesen kleine Arterien und
Arteriolen der Kortex-Oberfläche perlenschnurartige Einengungen, also Vasospas-
men, auf.182 Uhl et al. postulierten in ihrer Studie, dass diese Vasospasmen der
zerebralen Mikrozirkulation möglicherweise zur klinischen Symptomatik beitragen, und
diese den postoperativen Verlauf beeinflussen können.182 Pennings et al. zeigten
2004 bei SAB-Patienten mit ähnlicher Methodik nach Hyperventilation eine verstärkte
kontraktile Reaktion pialer Mikroarteriolen, sowie die Ausbildung von Spasmen dieser
Gefäße mit perlenschnurartigem Muster.183 Diese Veränderungen zeigten sich
lediglich bei Patienten, die früh nach SAB operiert wurden. Patienten die nach 48 Std.
operiert wurden, wiesen dieses Muster nicht auf.183 Um das Phänomen der früh
auftretenden Spasmen der zerebralen Mikrozirkulation genauer zu untersuchen,
wurde in unserer Arbeitsgruppe ein experimentelles Modell der SAB entwickelt, mit
dem die zerebrale Mikrozirkulation in vivo mittels Epifluoreszenz-Mikroskopie zu
unterschiedlichen Zeitpunkten nach SAB untersucht werden konnte. Mit diesem Modell
konnten gezeigt werden, dass drei und sechs Stunden nach SAB ca. 70% der Gefäße,
die von der MCA stammten, mikroarterioläre Kontraktionen aufwiesen, während nach
72 Stunden die Konstriktionshäufigkeit auf 56% gesunken war.184 Dieses Phänomen
wurde Mikrovasospasmus genannt und könnte für die frühe Störung der
Mikrozirkulation nach SAB mitverantwortlich sein. Die Ursachen bzw. Mechanismen,
die zum Mikrovasospasmus führen, sind noch unbekannt.167,180

1. Einleitung
27
1.4 Stickstoffmonoxid (NO)
Stickstoffmonoxid ist ein bioaktives Molekül, das mit anderen Molekülen sowohl
Redoxreaktionen als auch additive Reaktionen eingehen kann.185,186 Aufgrund seiner
geringen Größe kann es in kurzer Zeit biologische Membranen durchqueren und lokal
verschiedene Funktionen ausüben.187,188 Ende der 1970er Jahre bewiesen Furchgott
und Zawadzki et al., dass der sogenannte Endothelium derived relaxing factor (EDRF)
Stickstoffmonoxid ist.189-191 Ein Gas mit kurzer Halbwertszeit (t1/2 = 3-5 Sekunden),189
dass an der Regulation einer Vielzahl von biologischen und physiologischen
Vorgängen wie Modulation der Neurotransmission (z.B. Freisetzung von Dopamin im
Striatum192), am Immunsystem (z.B. MMP sezerniert durch Makrophagen193) und an
Lern- und Gedächtnis-Prozessen beteiligt ist.194-196,197 NO wird von der Isoenzym-
gruppe der NO-Synthasen (NOS) aus der semiessentiellen Aminosäure L-Arginin
synthetisiert.197,198 Im Hirngewebe gibt es alle drei Isoformen, die endotheliale
(eNOS),199-201 neuronale- (nNOS)202-204 und induzierbare NO-Synthase
(iNOS).201,205,206 Die NO-Synthasen katalysieren unter Mitwirkung von Tetrahydro-
biopterin (BH4), O2 und Kofaktoren wie NADPH die Umsetzung von L-Arginin zu L-
Citrullin und NO198 (Abbildung 5).
Abbildung 5: Vereinfachte Darstellung der NO-Synthe se.

1. Einleitung
28
Das endogen gebildete NO wirkt auf Gefäße stark vasodilatierend,189,190 indem es die
Aktivität der löslichen Guanylat-Cyclase (sGC) erhöht, was gleichzeitig zu einem
Anstieg des intrazellulären cGMP-Spiegels führt.207 In Abbildung 6 wird der NO-sGC-
cGMP-Signaltransduktionsweg erläutert: Stickstoffmonoxid (NO) wird enzymatisch
aus der Aminosäure L-Arginin durch drei Isoformen der NO-Synthase (eNOS, nNOS
und iNOS), gewonnen. Geringe Mengen endogen produziertes oder exogen
verabreichtes NO aktiviert die lösliche Guanylatzyklase (sGC), die GTP in zyklische
GMP konvertiert, was verschiedene physiologische und gewebeprotektive Effekte
vermittelt. Der Abbau von cGMP in GMP wird durch mehrere Phosphodiesterase
(PDE) Familien katalysiert. Exzessive Mengen von NO, gebildet unter pathologischen
Bedingungen und assoziiert mit erhöhtem Entzündungsgrad sowie oxydativem Stress
im Gewebe, können stark mit Superoxid-Anion (O2- •) reagieren, das zu Peroxynitrit
(ONOO-) wird. Peroxynitrit, gemeinsam mit anderen Oxidationsmitteln, induziert
Zellschäden durch Lipidperoxidation und ist verantwortlich für die Inaktivierung von
Enzymen und anderen Proteinen durch Oxidation und Nitrierung. Es aktiviert die
Matrix-Metalloproteasen (MMP) und das nukleare Enzym Poly-(ADP-Ribose)-
Polymerase (PARP), das letztendlich zu zelluläre Dysfunktion und Tod führt. Der NO-
sGC-cGMP-Signalweg kann durch Verringerung der Bioverfügbarkeit von NO (z.B.
durch chemische Wechselwirkung von NO mit O2•) oder durch Veränderung des
Redox-Zustandes der sGC selbst beeinträchtigt werden (beispielsweise durch
oxydativen Stress oder die Wirkung durch Peroxynitrit), wodurch es nicht mehr auf
endogenes NO oder NO-freisetzende Arzneimittel reagiert.208

1. Einleitung
29
Abbildung 6. Der NO-sGC-cGMP-Signalübertragungsweg (Modifiziert nach Evgenov et al., Nature Reviews, 2006).
1.4.1 Stickstoffmonoxidsynthase (NOS)
Die NOS ist ein Isoenzym, das in drei Formen vorliegt: endotheliale NOS (eNOS oder
NOS3), neuronale NOS (nNOS oder NOS1) und induzierbare NOS (iNOS oder NOS2).
Diese Namen reflektieren den Gewebetyp, in dem die NOS-Enzyme zum ersten Mal
beschrieben worden sind. Mittlerweile ist jedoch bekannt, dass jede dieser Isoformen
auch in einer Vielzahl anderer Gewebe exprimiert werden kann.209 Die drei Isoformen
sind sich ähnlich in Bezug auf Struktur und katalytische Mechanismen.198 eNOS wird
durch ein Gen kodifiziert, das auf Chromosom 7 lokalisiert ist. Es ist ein 135 kDa
Protein, das aus 1294 Aminosäuren besteht. Es wurde das erste Mal in
Gefäßendothelzellen identifiziert210,211 und später in andere Zelltypen vorgefunden,

1. Einleitung
30
wie z.B. in humanen neuronalen Zellen,212 humanen und Ratten-Astrozyten,213,214
humanen T-Zellen,215 humanen Knochenmarkzellen, humanen Osteoblasten und
Osteoklasten,216 humanen dermalen Fibroblasten,217 Ratten-Kardiomyozyten,218
Ratten-Hepatozyten219 und Kaninchen-Dickdarmzellen.220 Es wird konstitutiv
exprimiert und existiert sowohl in einer membrangebundenen als auch in einer
zytosolischen Form.198 Obwohl eNOS im Wesentlichen ein durch Kalzium-Calmodulin-
Komplex aktiviertes Enzym ist, wird es zusätzlich an endotheliale Rezeptoren,221-223
wie z.B. den M3-Muskarinrezeptor oder B1-Brandykininrezeptor,221 durch
Hypoxie,222,223 "Shear stress"221,224,225 und Hormone wie z.B. Östrogen226 stimuliert.
Durch die Stimulierung der Rezeptoren kommt es zu einer Erhöhung des Kalzium-
Einstroms in die Endothelzellen. Dies bewirkt die Phosphorylierung, und damit die
Aktivierung von membrangebundener und zytosolischer eNOS. Das so im Endothel
produzierte NO diffundiert in die glatten Gefäßmuskelzellen und induziert dort die
Gefäßrelaxation. Außerdem entfaltet es eine aggregationshemmende Wirkung auf
Thrombozyten und beeinflusst so die Durchblutung sowie den Gefäßtonus.221,227 Die
durch eNOS verursachte Vasodilatation ist in Arterien deutlich größer als in Venen.228
Die physiologische Konzentration des von eNOS produziertem NO liegt im
picomolaren (< 1nM),239 oder sub nanomolaren Bereich (ca.1-30 nM),229 und wird zur
Signaltransduk-tion nur in kurzen Schüben hergestellt.230 Die neuronale NOS, ein 160
kDa Protein,231,232 welches auf Chromosom 12 kodiert wird,234 ist eine konstitutive NO-
Synthase, die ähnlich dem eNOS abhängig von Ca2+ ist.198 Immunhistochemische
Untersuchungen haben nNOS in bestimmten Hirnregionen nachgewiesen wie Bulbus
olfactorius, Lamina tecti, Gyrus dentatus des Hippocampus, Stria terminalis,
diagonales Band von Broca, Hypophysenvorder- und -hinterlappen, Retina,
Cerebellum und Hypothalamus, dort insbesondere in den Nuclei supraopticus und
Nuclei paraventricularis.233,234 Darüber hinaus enthalten auch das Nebennierenmark
und periphere Darmnerven nNOS. Letztere werden auch als „nitrerge Nerven“
bezeichnet.235 Es übernimmt auch die Funktion eines Neurotransmitters, der die
Aktivität des sympathischen Nervensystems reguliert.236,237 Es ist auch an der
Regulation von gastrointestinaler Motilität, Kontrolle glatter Muskulatur sowie
neuroendokriner Prozesse beteiligt.237 Im Hirnstamm hemmt NO den Sympathikotonus
und kann dadurch Blutdruck und Herzfrequenz senken.238,239

1. Einleitung
31
Das iNOS-Gen ist lokalisiert auf Chromosom 17.198 iNOS ist im Gegensatz zu den
konstitutiven Isoformen eNOS und nNOS nicht ständig in aktiver Form im Gewebe
vorhanden, sondern induzierbar.198,240 iNOS wurde erstmalig aus Makrophagen isoliert
und wird in zahlreichen Geweben und Zellen exprimiert, wie z.B. Lunge, Muskelzellen,
Epithelien und Leukozyten. Im Gehirn wird iNOS in Endothelzellen, Mikroglia und
Astrozyten synthetisiert.198,241-245 iNOS ist Kalzium unabhängig und besitzt Calmodulin
als prothetische Gruppe.200 Es ist permanent aktiviert und kann NO für längere
Zeiträume erzeugen.229 Es ist ein Teil der unspezifischen Immunabwehr.246 Hier
fungiert das von diesem Enzym produziertes NO als zytotoxischen Agens gegen
Tumorzellen, Bakterien, Pilze und Protozoen.229,247 Dabei sind unzählige Stickstoff-
und Sauerstoffverbindungen entscheidend für die Herstellung des zytotoxischen
Milieus. Die Superoxide, die von der NADPH-Oxidase des Phagozyten hergestellt
werden, reagieren mit NO unter Bildung von Peroxynitrit.229,248 Studien berichten von
der Beteiligung von iNOS an der Regulation der renalen Funktion und des arteriellen
Blutdrucks.249 Auch nach Verletzungen soll iNOS an der Gefäßerweiterung mit
konsekutiver Hyperperfusion beteiligt sein.250 Die wesentliche Rolle der iNOS wird bei
iNOS-Knockoutmäusen erkannt, die vor allem anfälliger für Infektionen sind.251 Eine
Hyperaktivität der iNOS kann bei verschiedenen Erkrankungen (z.B. Morbus Crohn)
beobachtet werden. Im septischen Schock ist es verantwortlich für den ausgeprägten
Blutdruckabfall aufgrund gesteigerter Vasodilatation und Multiorganversagen.252
Anders als nNOS und eNOS, ist iNOS ein Enzym, dass über einen längeren Zeitraum
NO in höheren Konzentrationen synthetisiert.229 Diese Konzentration befindet sich im
nanomolaren (> 100 nM) Bereich.229,240
1.5 NO im Gehirn
Das Gehirn verbraucht ca. 20 % der körpereigenen Energie, obwohl es nur 2% der
Gesamtmasse des Körpers ausmacht. Eins der wichtigsten Aufgaben der zerebralen
Zirkulation ist das Gehirn mit Sauerstoff und Glukose zu versorgen, denn die neuronale
Aktivierung erfordert große Mengen an Energie, um den Ionenstrom zu regulieren, der
durch Depolarisation entsteht. Daher ist eine streng regulierte Durchblutung
essentiell.253 Der NO-Signalweg spielt eine wichtige Rolle bei der Regulation der
zerebralen Durchblutung. Das Verständnis der Mechanismen, die diese Prozesse

1. Einleitung
32
steuern, können nützliche Einblicke in die CBF-Veränderung geben, die während einer
Hirnverletzung auftreten. Zwei große Mechanismen liegen der Regulation des CBF
zugrunde, die Autoregulation und die neurovaskuläre Kopplung.253 eNOS-NO hat eine
Schlüsselrolle inne bei der Autoregulation, wogegen das nNOS produzierte NO
entscheidend bei der neurovaskulären Kopplung ist.253 Nach Hirnschädigung sind
diese NOS-Isomere in ihrer Funktionalität beeinträchtigt.254 Im Gehirn wird eNOS im
vaskulären Endothel und Plexus choroideus exprimiert. Das NO, das vom eNOS
stammt (eNOS-NO), spielt eine Schlüsselrolle für die Aufrechterhaltung der
Mikrozirkulation,254 fördert die Vasodilatation, erhöht die zerebrale Durchblutung,
hemmt die Thrombozyten-Aggregation und -Adhäsion,132,254,264 hemmt die
Leukozytenadhäsion,255,256 verringert die Proliferation der glatten Muskulatur und hat
u.a. eine antioxidative Wirkung,254,263 Das neuronale NO (nNOS-NO) agiert als ein
wichtiger Neurotransmitter, der die neuronale Plastizität, Gedächtnisbildung, zerebrale
Nerven, Systemdurchblutung, Übertragung von Schmerzsignale und Neurotrans-
mitter-Freisetzung, Schlaf-Wach-Zyklus und Hormonausschüttung (z.B. Östrogen)
reguliert. 257,258,262,270 Anders als eNOS und nNOS, wird die Exprimierung von iNOS
durch Makrophagen, Gliazellen und Tumorzellen in Anwesenheit von Zytokinen,
Endotoxinen und anderen inflamatorischen Agenzien bestimmt.259 Durch iNOS-
Aktivierung werden große Mengen (100-1000 Fach höher) an NO, im Vergleich zu
eNOS und nNOS, synthetisiert.260 Das aktivierte iNOS produziert kontinuierlich NO,
bis das Enzym abgebaut ist.261 Die NO-Funktion von eNOS und nNOS wird durch den
NO-sGC-cGMP-Signalweg vermittelt.
1.5.1 NO in der frühen Hirnschädigung nach SAB
Pathologische Veränderung im Stickstoffmonoxid (NO)/Stickstoffmonoxid-Synthase
(NOS)-Weg treten früh nach SAB auf.140,143,262 Experimentelle Versuche deuten auf
ein Mangel an NO hin, der nicht durch eine beeinträchtigte NO-Synthese zu erklären
ist, da die gesamte NOS-Aktivität, während der ersten 90 Minuten nach SAB
unverändert bleibt.263 Daher wird vermutet, dass die frühe Verringerung der zerebralen
NO-Konzentration nach SAB deshalb zustande kommt, weil NO durch Oxyhämo-
globin,264-266 durch Reaktion mit freien Radikalen,267 und der Reduzierung zu Nitriten268
deaktiviert wird. Dieser Effekt ist mit der Verringerung der zerebralen Durchblutung

1. Einleitung
33
vergesellschaftet.269 Daher werden dringend neue Therapien benötigt, die das
zerebrale NO-Defizit und die dadurch bedingte Abnahme der zerebralen Durchblutung
nach SAB verringern.270
1.5.2 NO-Donatoren
Stickstoffmonoxid-Donatoren (NOD) sind eine heterogene Gruppe von Medikamenten,
deren gemeinsames Merkmal die Fähigkeit ist, NO oder eine NO-verwandte Spezies,
wie das Nitrosonium-Kation (NO+) oder Nitroxyl-Anion (NO-), in vitro oder in vivo,
freizusetzen.271 Die Wirkung folgender NODs wurde experimentell oder klinisch nach
SAB untersucht: Organische Nitrate (z.B. Nitroglycerin, Isosorbid-5-Mononitrat,
Nicorandil, Pentaerythrittetranitrat); S-Nitrosothiole (z.B. S-Nitroso-N-
acetylpenicillamin und S-Nitroso-Glutathion); Sydnonimine (z.B. Molsidomin, SIN-1);
NONOate (JS-K, SPERMIN-NONOat und Proli-NONOat), anorganische Nitrate
(Natriumnitroprussid) und NO-Gas.272,273 Klassische NO-Donatoren, wie
Natriumnitroprussid (SNP) und Nitroglycerin (GTN), können nicht als Routine-
Therapeutika eingesetzt werden, da sie schwerwiegende Nebenwirkungen
besitzen.272 Zwar verursachte SNP im Tierversuch eine effektive Vasodilatation
zerebraler Gefäße, diese Wirkung war jedoch mit Hypotension und Hirnödem
vergesellschaftet.274,275 Intrathekale Behandlung mit SNP ruft, in der Mehrzahl der
Fälle, neben der positiven Wirkung auf Gefäßdurchmesser, Übelkeit, Erbrechen,
Migräne, Kopfschmerzen, Herzrhythmusstörung, unkontrollierten Anstieg der
zerebralen Durchblutung (CBF) und Zyanid-Vergiftung als Nebenwirkung hervor.276
GTN ist ein NO-Donator, der häufig zur Behandlung von Angina pectoris verwendet
wird. Nach SAB bei Primaten führte GTN zur Auflösung von Makrovasospasmen.277
Transdermale Verabreichung von GTN bei Menschen erhöhte nach SAB die
transkranielle Doppler (TCD) Geschwindigkeit und den CBF.278 Auch das klinische
Outcome verbesserte sich, und das DIND reduzierte sich.279 In allen Studien trat
allerdings eine ausgeprägte systemische Hypotension als Nebenwirkung auf. Dies
schränkt den klinischen Einsatz dieser vielsprechenden Substanz erheblich ein.272 Die
intrathekale Verabreichung von GTN führte experimentell zu einer vasodilatativen
Wirkung, ohne dass dadurch eine systemische Hypotonie hervorgerufen wurde.280
Allerdings wurde die Sicherheit und Wirksamkeit intrathekaler Anwendung am

1. Einleitung
34
Menschen noch nicht getestet.272 Die genannten Eigenschaften und Mängel der
klassischen NO-Donatoren veranlassten die Suche nach neuen NO-Donatoren
(NONOate, S-Nitrosothiole, Natriumnitrite, Sydnoniminen, NO-Gas), die in experimen-
tellen und klinischen Studien hinsichtlich ihres Potentials zur Vasospasmus
Behandlung, nach SAB, getestet werden.272 S-Nitrosothiole (z.B. S-Nitrosogluthation)
weisen nach experimenteller SAB, eine vasodilatative Wirkung auf zerebrale Arterien
auf. Außerdem hat dieser NO-Donator keinen Effekt auf de ICP und CPP. Jedoch weist
es Nebenwirkungen bzgl. systemische Blutdruckverminderung auf.140 Auch NANOate
weist eine relaxierende Wirkung auf die verengten zerebralen Gefäße auf.281,282 Als
Nebenwirkung wird durch NANOate die systemische Hypotension,283 und die
Hepatokarzinogenität, durch N-Nitrosopyrrolidine, gefördert.284 Experimentelle Stu-
dien zeigen, dass Sydnonimine, wie z.B. Molsidomin, einen gefäßrelaxierenden und
einen antithrombotischen Effekt auf die Mikrozirkulation aufweist. Auch hier zeigte sich
allerdings eine ausgeprägte Hypotension (um ca. 18,5 mmHg).285 Natriumnitrit, dass
für die Phase-2a an aSAB-Patienten untersucht wird, ist ein Medikament, das
zerebrale Vasospasmen verbessert.286 Es weist jedoch in klinischen Studien ebenfalls
für NO-Donoren typische Nebenwirkungen auf.287
1.5.3 Inhaliertes NO
Inhaliertes NO (iNO) wird seit 1991 als selektiver pulmonaler Vasodilatator verwendet,
um pulmonale Hypertonie zu behandeln.288,289 Der Vorteil von iNO liegt in der
sofortigen relaxierenden Wirkung auf pulmonale Widerstandsgefäße, so dass es
keinen systemischen hypotonen Nebeneffekt hervorruft.290-293 iNO hat auch
extrapulmonale Effekte, z.B. verringert es den ischämischen Reperfusionsschaden im
mesenterialen und myokardialen Gewebe.294,295 In experimentellen296-298 und
klinischen299 Studien wurden keine Veränderungen der zerebralen Durchblutung
(CBF) und der zerebralen Perfusion (CPP) an gesunden Tieren und Menschen
dokumentiert.300 Jedoch verursacht iNO nach experimenteller zerebraler Ischämie,
eine selektive Erweiterung der zerebralen Gefäße der ischämischen Penumbra, ohne
dass dabei der systemische Blutdruck abfällt. Es steigert die zerebrale Durchblutung,
verbessert das neurologische Outcome, und verhindert den ischämischen Zelltod.270
Aufgrund des Wirkmechanismus von iNO, könnte es möglicherweise eine vielverspre-

1. Einleitung
35
chende Behandlungsstrategie zur Vorbeugung des frühen Hirnschadens nach SAB
darstellen.301
1.5.4 Rolle des NOs in der zerebralen Autoregulatio n
Die Zerebrale Autoregulation (CA) ist ein homöostatischer Prozess bei dem
Widerstandsgefäße erweitert oder verengt werden, um die zerebrale Durchblutung
(CBF) konstant zu halten.302,303 Es werden zwei Haupttypen von CA unterschieden: a)
Stoffwechselregulation, die durch Veränderungen der Hirnstoffwechselrate angetrie-
ben wird304 und b) Druckregulation, die durch eine Antwort des CBF auf arterielle
Blutdruck-Veränderungen charakterisiert wird.305-309
Bei gesunden Erwachsenen wirken sich Veränderungen des systemischen Blutdrucks
(MAP), zwischen 60-160 mmHg, geringfügig oder gar nicht auf den CBF aus.302,310,335
Sobald der MAP ansteigt, konstringieren die kleinen zerebralen Arterien und Arteriolen
und sorgen dadurch für eine Aufrechterhaltung des CBF.311 Umgekehrt, dilatieren die
zerebralen Widerstandsgefäße sobald der systemische Blutdruck abfällt. Auch
dadurch wird der CBF konstant gehalten. Jenseits der autoregulatorischen Grenzen,
hängt die CBF direkt von MAP/CPP ab; eine arterielle Hypotonie führt zur
Mangeldurchblutung des Gehirns und ggf. Bewußtseinsverlust, während eine
ausgeprägte arterielle Hypertonie zur Öffnung der Blut-Hirn-Schranke und zum
Hirnödem führen kann.311
Die metabolische Regulation der Hirndurchblutung erfolgt über den arteriellen
pCO2.312,313 Bei gesunden Erwachsenen steigt im Bereich zwischen 25-75 mmHg der
CBF linear um 2-4% pro mmHg PaCO2 an und die zerebralen Gefäße dilatieren.335,336
Veränderungen in der zerebralen Zirkulation treten bei gesunden Menschen unter den
genannten Bedingungen innerhalb weniger Sekunden ein.312
Nach SAB sind autoregulatorische Mechanismen der zerebralen Durchblutung häufig
beeinträchtigt. Die Druckregulation, die Fähigkeit des CBF auf den veränderten
systemischen Blutdruck zu reagieren, wie auch die CO2-Reaktivitätsfähigkeit der
zerebralen Gefäße bleiben aus.128,131,132,142,314,315 Experimentell tritt eine schwere
autoregulatorische Störung innerhalb von zwei bis drei Stunden nach SAB auf und hält
bis zu drei Monate an.126 Klinisch tritt diese Beeinträchtigung in den meisten Fällen
innerhalb der ersten 72 Stunden nach aneurysmatischer SAB auf und korreliert in ihrer

1. Einleitung
36
Ausprägung mit der Schwere der SAB.315 Die genauen Mechanismen der
aufgehobenen CO2 -Reaktivität nach SAB sind nicht bekannt. Diskutiert wird u.a. eine
Entkoppelung der eNOS.316-318 Die funktionelle Entkopplung des eNOS vom Kofaktor
Tetrahydrobiopterin (BH4) geschieht unter ischämischen Bedingungen und ist für die
Erzeugung von Superoxid, anstatt NO, verantwortlich.319,320 Das Superoxid reagiert mit
NO zu toxischem Formperoxynitrit. Durch diese NO-Depletion verschärft sich die
Verringerung der zerebralen Durchblutung nach SAB und der neuronale Schaden wird
verstärkt. Die Gabe von L-Arginin- oder Pravastatin haben einen positiven Effekt auf
die zerebrale Autoregulation nach SAB.321,322 Weitere Untersuchungen müssen
zeigen, wie die gestörte Autoregulation nach SAB erfolgreich behandelt werden kann.
1.6 Rolle von Endothelin und Endothelin-Rezeptoren nach SAB
Endothelin wurde 1988 von Yanagisawa und Mitarbeiter entdeckt.323 Es wurde von
Aortenendothelzellen vom Schwein Isoliert, charakterisiert und kloniert.323 Endotheline
sind Oligopeptide, die in drei Isoformen auftreten: ET-1, ET-2 und ET-3.324 Sie
bestehen aus 21 Aminosäuren und werden hauptsächlich in Endothelzellen
hergestellt. Endothelin-1 (ET-1) ist der stärkste bisher bekannte Vasokonstriktor.325 Die
ET-1-Synthese wird reguliert durch physikalisch-chemische Faktoren326 (z.B.
Hypoxie), durch Vasokonstriktoren,327,328 Wachstumsfaktoren (z.B. VEGF),329 pH,391
Zytokine330 und Adhäsionsmoleküle.331 Diese induzieren die Transkription von ET-1-
mRNA und damit die innerhalb von Minuten einsetzende Synthese und Sekretion von
ET-1. Die Halbwertszeit der ET-1-mRNA beträgt etwa 15-20 Minuten und die
Plasmahalbwertszeit für ET-1 beträgt 4-7 Minuten.332 Diese schnelle Regulation
erlaubt den Endothelzellen, die ET-1-Syntheserate rasch zu verändern und somit den
Gefäßtonus schnell zu regulieren. Endothelzellen sezernieren 75-90% ihrer ET-1-
Produktion in das abluminale Interstitium,333,334 wo es mit Endothelinrezeptoren auf
den Gefäßmuskelzellen und perivaskulären Nervenzellen interagiert. Nur 25% der ET-
1 Gesamtproduktion wird in die Blutbahn sezerniert. Die Plasma-Konzentration von
ET-1 liegt im picomolaren Bereich,335 während die Schwellen-Konzentration für die ET-
1 induzierten Vasokonstriktionen meist im nanomolaren Bereich liegt.336 Seine
Wirkung wird über drei Rezeptoren vermittelt: Der ETA-, ETB1- und ETB2-Rezeptor.337
Diese Rezeptoren sind an die Phopsholipase-C via GTP-Binding-Protein gekoppelt.338

1. Einleitung
37
Die Vasokonstriktion wird vor allem durch ETA-Rezeptoren vermittelt, die besonders
stark in glatten Gefäßmuskelzellen exprimiert werden, während ETB1-Rezeptoren auf
Endothelzellen exprimiert werden, und eine durch NO vermittelte Vasodilatation
bewirken. ETB2-Rezeptoren, die in einem sehr geringen Umfang in glatten Gefäß-
muskelzellen exprimiert werden, vermittelt ebenso wie die ETA-Rezeptoren eine
Vasokonstriktion (Abbildung 7).338,339,382,340
Big ET-1 = Big Endothelin-1; ECE = Endothelin Conve rting Enzym
Abbildung 7: Vaskuläre Wirkung von Endothelin-1 (ET -1) (Modifiziert nach Agapitov et al., Jraas,
2002).
Der Signaltransduktionsweg für die ETA-Rezeptoren führt über eine Aktivierung hetero-
trimerischer G-Proteine (Gq/11) und Phospholipase-C (PLC) zu einer Freisetzung von
Inositoltriphosphat (IP3) und Diacylglycerol (DAG). IP3 aktiviert IP3-Rezeptoren (IP3R),
die sich in den Membranen des sarkoplasmatischen Retikulums (SR) befinden. Wird
IP3 an diese Membranproteine gebunden, wirken sie wie Ca2+-Kanäle und Ca2+ fließt

1. Einleitung
38
seinem Konzentrationsgradienten folgend aus dem SR in das Zytosol. Der daraus
resultierende Anstieg der Ca2+-Konzentration im Zytosol aktiviert die Calmodulin
(CaM) abhängige Myosin-Leichteketten-Kinase (MLCK), die über eine
Phosphorylierung der regulatorischen leichten Ketten des Myosins (MLC20) die
Konstriktion einleitet.341,342 Für eine schematische Verfolgung des ETA-Rezeptor-
Transduktionsweges siehe Abbildung 8.
Abbildung 8: Postuliertes Modell der Signaltransduk tionswege von ET A-Rezeptoren an glatten
Gefäßmuskelzellen.
AA = Arachidonsäure; PLA2 = Phospholipase A2; BK = Ca2+
- aktivierte K+-Kanäle; DAG = Diacylglycerol;
IP3 = Inositol Triphosphat; IP3R = IP3-Rezeptor; L-type = L-type Ca2+
-Kanäle; PLC = Phospholipase C;
RyR = Ryanodine Rezeptor; SERCA = sarkoplasmatische Ca2+
-ATPase.
Eine Reihe von Untersuchungen, die in den letzten Jahren durchgeführt wurden
zeigen, dass Endothelin-1 (ET-1) maßgeblich an der Pathogenese von zerebralen
Vasospasmen nach aneurysmatischer Subarachnoidalblutung (aSAB) bei Patienten
beteiligt ist.343-347 Weitere Studien zeigen, dass die Expression von ET-1 vor allem in

1. Einleitung
39
den ersten 72 Stunden nach SAB, verstärkt stattfindet.343,348,349 Dieses Ereignis wird
durch Thrombin und Oxyhämoglobin gefördert.350,351 Auch ist ein erhöhter ET-1-
Spiegel möglicherweise für die Beeinträchtigung der NO-Produktion nach SAB
verantwortlich. Diese Beeinträchtigung wird über die isoformspezifischen Protein-
Kinase-C (PKC)-vermittelte Hemmung der eNOS-Expression vermittelt.352 Präklinis-
che Studien legen nahe, dass ETA-selektive Antagonisten, wie z.B. Clazosentan,
Makrovasospasmen nach SAB auflösen können. Dies konnte auch in einer
randomisierten Phase-II-Studie an aSAB-Patienten gezeigt werden,353,354,355 In den
darauffolgenden Multizenterstudien zeigte sich allerdings keine signifikante Reduktion
von DIND, sowie keine Verbesserung des neurologischen Outcomes.178,353,354,356-359
Die Ergebnisse legen nahe, dass der verzögert auftretenden Makrovasospasmus nach
SAB keine kausale Rolle für die post-hämorrhagische Ischämie spielt, sondern
möglicherweise andere Mechanismen, wie z.B. eine Störung der zerebralen
Mikrozirkulation hier eine weit wichtigere Rolle einnimmt. Ob ET-1 möglicherweise
auch an Störungen der zerebralen Mikrozirkulation nach SAB beteiligt ist, ist bisher
allerdings völlig unklar.
1.7 Ziel der Arbeit
Das Ziel der Arbeit ist die Untersuchung der Ätiologie der zerebralen Mikrozirkulations-
störung nach experimenteller Subarachnoidalblutung. Die von unserer Arbeitsgruppe
erstmalig systematisch untersuchten Mikrovasospasmen sind ein in der Frühphase
nach Blutung auftretendes Phänomen, das sowohl beim Patienten, als auch nach
experimenteller Blutung auftritt, und das wahrscheinlich entscheidend zur frühen
mikrozirkulatorischen Dysfunktion und somit zum posthämorrhagischen ischämischen
Hirnschaden nach SAB beiträgt. Wie oben erläutert, könnten der Stickstoff-Monoxid-
Stoffwechsel, sowie Endothelin eine Schlüsselrolle in der Entwicklung der mikro-
arteriellen Konstriktion spielen. Deswegen wurde im Rahmen der vorliegenden
Dissertation nach experimenteller Subarachnoidalblutung der Einfluss von inhaliertem
Stickstoffmonoxid, sowie die Endothelin-Rezeptor-Blockade auf die zerebrale Mikrozir-
kulation untersucht.

2. Material & Methoden
2. Material & Methoden
Die Experimente und Datenanalyse wurden von Januar 2011 bis März 2014 am
Walter-Brendel-Zentrum für Experimentelle Medizin durchgeführt. Alle tierexperimen-
ellen Protokolle wurden von der zuständigen Behörde genehmigt (Regierung von
Oberbayern, Tierversuchsantrag: 55.2-1-54-2531-66/08). Die Planung, Ausführung,
Auswertung und Dokumentation der Versuche erfolgte nach den ARRIVE-
Richtlinien.360 Die Randomisierung erfolgte mittels Los-Zug, alle Versuche wurden von
einem verblindeten Untersucher durchgeführt und ausgewertet.
2.1 Material
Die Hersteller aller verwendeten Geräte und Verbrauchsmaterialien in der vorliegen-
den Arbeit sind in der Folge aufgelistet.
2.1.1 Geräte
Tabelle 5: Geräte
Bezeichnung Hersteller
Gefäß-Kauterisierer
Blut-Gas-Analysator
Bohrer-Ansatz
Diamantbohrer zur Trepanation
OP-Mikroskop
Druckwandler
Fine Scientific Tool GmbH, Heidelberg, D
Rapilab 1265, Global Siemens Healthcare
Headquarters, Siemens AG, Erlangen, D
Aeskulap GD 8730 R, 0,5 x 5,1 mm, B.
Braun Melsungen AG, Melsungen, D
Rewatronik Products; Wald Michelbach, D
Carl Zeiss AG; Oberkochen, D
DTX-Plus DT-XX, Becton-Dickinson,
Medical, Heidelberg, D

2. Material & Methoden
41
Heizplatte
ICP-Monitor
ICP-Sonde (Codmann Microsensor)
Inkubator
IVM-Mikroskop
Kleintierrespirator
Laserdoppler
Mikrokapnometer
Mikrokapnometer-Sonden
(10 und 20 ml)
Multi Gas Monitor zur NO-Messung
Perfusor secura
Perfusor (Sp 100i, Syringe pump)
Sauerstoffgehaltmesser
Signal-Transducer-Gerät
Signalanschlussbox
Stereotaktische Halterung
Waage
FHC, Bowdoinham, ME, USA
Codmann, Boston, USA
Codman, Norderstedt, D
Modell 7510, Drägerwerk AG Lübeck,
Lübeck, D
Modell MST 49, Leica Microsystems CMS
GmbH, Wetzlar, D
Mini Vent Typ 845, Hugo Sachs Elektronik,
March, D
Perimed Instruments GmbH, S
CI 240, Columbus Instruments, Ohio, USA
CI 240, Columbus Instruments, Ohio, USA
Industrial Scientific Corporation, Dortmund, D
B. Braun Melsungen AG, Melsungen, D
Corcom, USA
Oxidig, Drägerwerk AG Lübeck, Lübeck, D
Hugo Sachs Elektronik, March, D
Bedo Elektronik GmbH, Krefeld, D
Mod. 51600, Stoeting Co., Wood Dale, IL,
USA
Ohaus Corporation, CH

2. Material & Methoden
42
2.1.2 Verbrauchsmaterialien
Tabelle 6: Verbrauchsmaterialien
Bezeichnung Hersteller
Nahtmaterial zur Hautnaht
Faden zur Perforation
Katheter (Außendurchmesser: 0,61
mm & Innendurchmesser:0,28 mm)
Kleberhärter (Insta-Set)
Knochenzement (Poly-Plus Bondex)
Mikrochirurgische Gefäßclips
OP-Besteck
Sekundenkleber (Maxi-Cure)
Skalpell (N°11 & N°20)
Hemodynamic Monitoring System
Tubus (zur Intubation)
Gewebekleber - 3 M Vetbond
(Tissue Adhesive)
Ethibond 5-0, Ethicon, Norderstedt, D
Prolene 5-0, Ethicon, Norderstedt, D
Smiths Medical, Keene, USA
Drechsel & Mehr, Weiden, D
Engmann, Viernheim, D
Dentsply Dentry, Konstanz, D
Peter Lazig GmbH, Tuttlingen, D
FST, Berlin, D
Drechsel & Mehr, Weiden, D
Feather Safety Razorco.LTD, Okasaka, J
Spectramed, Deutschland
InsyteR, Decton Dickinson, USA
3 M Animal Care Products, St. Paul, USA

2. Material & Methoden
43
2.1.3 Inhalationsgase
Tabelle 7: Inhalationsgase
Bezeichnung Hersteller
CO2 (10 %ig, 10,0 L), UN 1956 Linde AG, Pullach, D
CO2 (99%, 13,4 L) UN 1013 Linde AG, Pullach, D
NO in N2 (Prüfgas, 268 mg/m3
Stickstoffmonoxid (200 ppm), Rest
Stickstoff); 10,0 L, RAL 6018
Linde AG, Pullach, D
2.1.4 Chemikalien
Tabelle 8: Chemikalien
Bezeichnung Hersteller
Atipamezolhidrochlorid
(2,5 mg/kg KG)
Bepanthen (Augen & Nasensalbe)
Clazosentan
D-PBS (mit CaCl2 und MgCl2)
Fentanyl
FITC-Dextran (Mw: 150.000 Da)
Flumazenil
Heparin
Orion Pharma GmbH, Hamburg, D
Bayer Vital, Leverkusen, D
Actelion, Allschwil, CH
Gibco, Invitrogen Life Technologies, Grand
Island , NY, USA
Jan Seen Cilag, Neuss, D
Sigma Chemicals, Deisendorf, D
Hikma Pharma GmbH, Niederolm, D
Rathiopharm GmbH, Ulm, D

2. Material & Methoden
44
Isofluran
Knochenzement (Poly-Plus Bondex)
Knochenzement (Aqualox)
Medetomidin
Midazolam
NaCl – 0,9% Lösung –
Naloxon-HCl
Abbot GmbH & Co. KG; Wiesbaden, D
Engmann, Viernheim, D
Dentsply Dentry, Konstanz, D
Voco, Cuxhaven, D
Orion Pharma GmbH, Hamburg, D
Roche, Basel, CH
Braun AG; Melsungen, D
Baxter, Lessines, B
PharmaSelect, Hannover, D
2.1.5 Intravitalmikroskop-Ausstattung
Tabelle 9: Intravitalmikroskop-Ausstattung
Bezeichnung Hersteller
Filter: Anregungsfilter (Bandpassfilter:
450-490 nm) und Reflektionspassfilter
(510 nm)
Leitz, München, D
Bewegungsregulator (Mikroskoptisch)
- Phytron IX - C - T
Fluoreszenzepiluminatoren (L3,
Poemopak)
Hitzefilter (7 mm)
Kamera SIT (Restlichtkamera),
C2400-08
Phytron Elektron Elektrik GmbH,
Gröbenzell, D
Leica, München, D
Leitz, München, D
Hamamatsu Photonics, Heersching, D

2. Material & Methoden
45
Kreutztisch
Objektive (5 x, 20 x)
Sperrfilter
(Bandpassfilter: 520-525 nm)
Xenon-Lampe (75 W) – XBO 75 W/2
Leitz, München, D
Leitz, Wetzlar, D
Leitz, München, D
Leitz, München, D
2.1.6 Software
Tabelle 10: Software
Anwendungsgebiet Programm/Hersteller
Leica - Bildbearbeitung /- Analyse,
Video-Aufnahme an
Intravitalmikroskop
FlexPro - Auswertung von zerebraler
Durchblutung (CBF) und Intrakra-
niellem Druck (ICP), graphische
Auswertung
Graphische Darstellung, Statistik
Aufnahme von ICP, LDF und MAP
Bildbearbeitung
Statistische Auswertung, schriftliche
Bearbeitung, Präsentation
Leica Application Suite (LAS),
Leica Microsystems,Wetzlar, D
FlexPro, Weisang, D
SigmaPlot 12, Erkrath, D
DaisyLab 5.0, meas X, Mönchengladbach ,
D
Adobe Photoshop 7.0, Adobe Systems
GmbH, München, D
Excel, Word, PowerPoint (Microsoft Office
2007, 2010), Redmond, WA, USA

2. Material & Methoden
46
Feinbewegung des Mikroskoptisches,
1µm Intervall steuerbar
Messaufnahmesystem (LDF,
MAP,ICP)
Alpha com V3.4.2 (Phyntron Elektron),
Gröbenzell, D
DASYLab ,National Instruments, München, D
2.1.7 Versuchstiere
Für die Versuchsreihen zur Untersuchung von inhaliertem NO und dem Endothelin-
Rezeptor-Inhibitor Clazosentan wurden männliche C57 bl/6 Mäuse mit einem Gewicht
von 20-24g und einem Alter von 6-8 Wochen verwendet (Charles River Laboratories,
Kisslegg, D). Zur Untersuchung der Rolle der NOS wurden homozygot transgene
nNOS-/- (NOS-1, Stamm Nummer 2633, genaue Bezeichnung: B6; 129S4
Nos1tm1Plh/J), iNOS-/- (NOS2, Stamm Nummer 2609, genaue Bezeichnung:
B6.129P2-Nos2tm1Lau/J) und eNOS-/-- (NOS-3, Stammnummer 2684, genaue
Bezeichnung: B6.129P2-Nos3tm1Unc/J) Zuchtpaare gekauft (Jackson Laboratories,
Maine, USA). Alle Mutationen wurden in Sv129-Zellen induziert, die resultierenden
transgenen Tiere wurden 10 Generationen in C57 bl/6 Mäuse zurückgekreuzt. Eine
Übersicht über die genauen Mutationen sowie den resultierenden Phänotyp gibt
Tabelle 11. Die Zucht erfolgte homozygot. Die Tiere wurden nach dem vom Hersteller
empfohlenen Protokoll genotypisiert. Als Kontrolltiere wurden männliche C57 bl/6
Mäuse verwendet. Die Haltung der Tiere erfolgte im Walter-Brendel-Zentrum,
entsprechend den erforderlichen speziellen Hygienebedingungen (FELASA
erweitert).361 Die Mäuse wurden bei einer Raumtemperatur von 22 ± 2°C in Standard
Makrolon Käfigen (Gruppenhaltung bei Tieren mit > 20 g KGW = 5 Tiere / Käfig Typ II
Iong) mit freiem Zugang zu Wasser und Futter bei einem Tag-Nachtrhythmus von 12
Stunden gehalten. Eine Stunde vor dem geplanten Versuchsbeginn wurden die Tiere
in Einzelkäfigen nüchtern gestellt; die postoperative Haltung erfolgte in Einzelkäfigen.

2. Material & Methoden
47
Tabelle 11.: Übersicht über Mutation, Genotyp und P hänotyp der NOS-Knockout-Mäuse
Detailanalyse NOS-1 NOS-2 NOS-3Symbol Nos-1 Tm1Plh Nos-2tm1Lau Nos-3tm1Unc
NameStickstoffmonoxid-Synthase-1,
neuronale Zellen; gezielte Mutation 1, Paul L Huang
Stickstoffmonoxid-Synthase-2, induzierbare Zellen; gezielte Mutation 1, Victor E Laubach
Stickstoffmonoxid-Synthase-3, endotheliale Zellen; gezielte
Mutation 1, University of North Carolina
Synonyme alphaNOS1, Kn, N2KO, nNOS-,
nNOS-, nNOS KO, NOSdeltaiNOS-, iNOS KO, NOS2-,
Nos2tm1Lau, NOS2tm/LauecNOS-, eNOS-, eNOSKO,
NOS3-
Gene
Lokalisierung: Chr5:117781032-117958840 bp, +
Strang, genetische Position: Chr5, 57.29 cM,
cytoband F
Lokalisierung: Chr11:78920787-78960254 bp, +
Strang, genetische Position: Chr11, 46.74 cM
Lokalisierung: Chr5:24364810-24384474 bp, +
Strang, genetische Position: Chr5, 11.32 cM
Eltern-Zelllinie J1 (ES Cell) E14TG2a (ES Cell) E14TG2a (ES Cell)Herkunft der Belastung 129S4/SvJae 129P2/OlaHsd 129P2/OlaHsd
Genotyp Homozygot 2 (hm2) Homozygot 2 (hm2) Homozygot 2 (hm2)
Allelzusammensetzung Nos1tm1Plh/Nos1tm1Plh Nos2tm1Lau/Nos2tm1Lau Nos3tm1Unc/Nos3tm1Unc
Genetischer Hintergrund B6.129S4-Nos1tm1Plh/J B6.129P2-Nos2tm1Lau/J B6.129P2-Nos3tm1Unc/J
Herz-Kreislauf-System
verringerte vaskuläre endotheliale Zellzahl, anomaler systemischer
arterieller Blutdruck, Verringerung der Herzmuskelkontraktilität, , anormale Angiogenesis, u.a.
Zellular
erhöhte fetale Herzmuskelzell-Apoptose, erhöhte Nieren-
Apoptose, verringerte Nierenzellproliferation, abnorme
RedoxaktivitätEndokrine / Exokrinen
Drüsenabnorme Eierstock-Morphologie,
kleinen EierstockWachstum / Größe /
KörperphänotypAbnahme des Körpergewichts fetale Wachstumsverzögerung
Hämatopoetischen Systems
abnorme Osteoklasten Physiologie
Homöostase / Stoffwechsel
glomeruläre Kapillarthrombose, Anstieg des
Serumharnstoffspiegels, erhöhter zirkulierender
Kreatinkinasespiegel, Zyanose, u.a.
Immunsystem verminderte entzündliche Reaktionanormale Entzündungsreaktion,
verminderte entzündliche Reaktion
abnorme Osteoklasten- Physiologie, erhöhte Entzündungsreaktion,
Nierenentzündung
Sterblichkeit / Altern partielle neonatale Letalität, teilweise postnatale Letalität
Muskel
vermindertes Skelettmuskelgewicht,
beeinträchtigte Kontraktilität der Skelettmuskulatur, Muskelermüdung
verringerte Herzmuskelkontraktionsfähigkeit,
erhöhte Herzkammer- Muskelkontraktilität
Nervensystem anormales Gehirnwellenmuster
Nieren / Harnwege
erhöhte Nierentubuli Apoptose, Albuminurie,
Nierenmikroaneurysma, Mesangiolysis, abnorme
glomeruläre Kapillarendothel- Morphologie, u.a.
Fortpflanzungsorgane verminderte Befruchtungsfrequenz
Atmungssystem anormale Tensid-Physiologie
Lungenblutungen , abnormalen Lungengefäß-Morphologie,
Lungengefäßstaus, abnorme Tensidsekretion, u.a.
Verhalten/Neurologisch anormaler Tagesrhythmus, anormales Schlafmuster
Skelett
abnorme Osteoklastenphysiologie,
verringerte Knochendichte, erhöhte Knochendichte, erhöhte
kompakte Knochendicke, abnormale Osteoblasten-
Physiologie
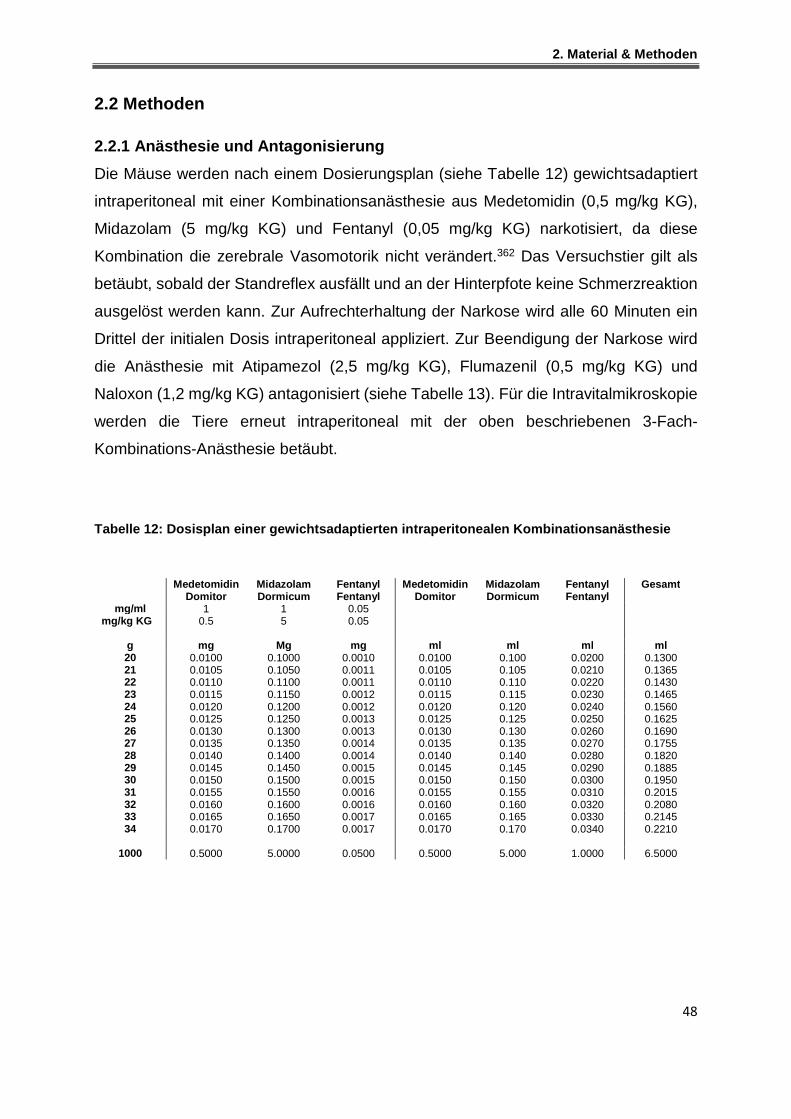
2. Material & Methoden
48
2.2 Methoden
2.2.1 Anästhesie und Antagonisierung
Die Mäuse werden nach einem Dosierungsplan (siehe Tabelle 12) gewichtsadaptiert
intraperitoneal mit einer Kombinationsanästhesie aus Medetomidin (0,5 mg/kg KG),
Midazolam (5 mg/kg KG) und Fentanyl (0,05 mg/kg KG) narkotisiert, da diese
Kombination die zerebrale Vasomotorik nicht verändert.362 Das Versuchstier gilt als
betäubt, sobald der Standreflex ausfällt und an der Hinterpfote keine Schmerzreaktion
ausgelöst werden kann. Zur Aufrechterhaltung der Narkose wird alle 60 Minuten ein
Drittel der initialen Dosis intraperitoneal appliziert. Zur Beendigung der Narkose wird
die Anästhesie mit Atipamezol (2,5 mg/kg KG), Flumazenil (0,5 mg/kg KG) und
Naloxon (1,2 mg/kg KG) antagonisiert (siehe Tabelle 13). Für die Intravitalmikroskopie
werden die Tiere erneut intraperitoneal mit der oben beschriebenen 3-Fach-
Kombinations-Anästhesie betäubt.
Tabelle 12: Dosisplan einer gewichtsadaptierten int raperitonealen Kombinationsanästhesie
Medetomidin Domitor
Midazolam Dormicum
Fentanyl Fentanyl
Medetomidin Domitor
Midazolam Dormicum
Fentanyl Fentanyl
Gesamt
mg/ml 1 1 0.05 mg/kg KG
0.5 5 0.05
g mg Mg mg ml ml ml ml 20 0.0100 0.1000 0.0010 0.0100 0.100 0.0200 0.1300 21 0.0105 0.1050 0.0011 0.0105 0.105 0.0210 0.1365 22 0.0110 0.1100 0.0011 0.0110 0.110 0.0220 0.1430 23 0.0115 0.1150 0.0012 0.0115 0.115 0.0230 0.1465 24 0.0120 0.1200 0.0012 0.0120 0.120 0.0240 0.1560 25 0.0125 0.1250 0.0013 0.0125 0.125 0.0250 0.1625 26 0.0130 0.1300 0.0013 0.0130 0.130 0.0260 0.1690 27 0.0135 0.1350 0.0014 0.0135 0.135 0.0270 0.1755 28 0.0140 0.1400 0.0014 0.0140 0.140 0.0280 0.1820 29 0.0145 0.1450 0.0015 0.0145 0.145 0.0290 0.1885 30 0.0150 0.1500 0.0015 0.0150 0.150 0.0300 0.1950 31 0.0155 0.1550 0.0016 0.0155 0.155 0.0310 0.2015 32 0.0160 0.1600 0.0016 0.0160 0.160 0.0320 0.2080 33 0.0165 0.1650 0.0017 0.0165 0.165 0.0330 0.2145 34
0.0170 0.1700 0.0017 0.0170 0.170 0.0340 0.2210
1000 0.5000 5.0000 0.0500 0.5000 5.000 1.0000 6.5000

2. Material & Methoden
49
Tabelle 13: Dosisplan einer gewichtsadaptierten int raperitonealen Antagonisierung
2.2.2 Mechanische Beatmung
Zur Aufrechterhaltung physiologischer Bedingungen erfolgt nach der Anästhesie die
Intubation mit einer speziell angefertigten Kanüle (20G) mit Silikonmanschette zur
Abdichtung der Stimmritze. Die Intubation erfolgt unter direkter visueller Kontrolle mit
einem Mikroskop. Die korrekte Tubuslage wird mittels Mikrokapnometrie verifiziert.
Nach erfolgreicher Intubation wird das Tier mit einem Kleintier-Respirator verbunden
und mit einem Gasgemisch aus 30% O2 und 70% Stickstoff kontrolliert beatmet
(Atemfrequenz: 140-200/min, Atemzugvolumen: 200-225 µl). Die Sauerstoffkonzen-
tration, die Beatmungsparameter und der endexspiratorische CO2-Partialdruck
(pETCO2) werden kontinuierlich überwacht, die Atemparameter ggf. angepasst.
Angestrebt wird ein pETCO2-Wert zwischen 35-45 mmHg. In einer vorherigen
Publikation unserer Arbeitsgruppe konnte gezeigt werden, dass die pETCO2-Werte
sehr gut mit dem im Blut gemessenen pCO2-Werten übereinstimmen, sodass dass
über die gesamte Versuchsdauer ein physiologischer pCO2 vorherrschte.362
Atipamezol Antisedan
Flumazenil Anexate
Naloxon Naloselect
Atipamezol Antisedan
Flumazenil Anexate
Naloxon Naloselect
Gesamt
mg/ml 5 0.1 0.4 mg/kg KG
2.5 0.5 1.2
g mg mg mg ml ml ml ml 20 0.0500 0.0100 0.0240 0.0100 0.1000 0.0600 0.1700 21 0.0525 0.0105 0.0252 0.0105 0.1050 0.0630 0.1785 22 0.0550 0.0110 0.0264 0.0110 0.1100 0.0660 0.1870 23 0.0575 0.0115 0.0276 0.0115 0.1150 0.0690 0.1955 24 0.0600 0.0120 0.0288 0.0120 0.1200 0.0720 0.2040 25 0.0625 0.0125 0.0300 0.0125 0.1250 0.0750 0.2125 26 0.0650 0.0130 0.0312 0.0130 0.1300 0.0780 0.2210 27 0.0675 0.0135 0.0324 0.0135 0.1350 0.0810 0.2295 28 0.0700 0.0140 0.0336 0.0140 0.1400 0.0840 0.2380 29 0.0725 0.0145 0.0348 0.0145 0.1450 0.0870 0.2465 30 0.0750 0.0150 0.0360 0.0150 0.1500 0.0900 0.2550 31 0.0775 0.0155 0.0372 0.0155 0.1550 0.0930 0.2635 32 0.0800 0.0160 0.0384 0.0160 0.1600 0.0960 0.2720 33 0.0825 0.0165 0.0396 0.0165 0.1650 0.0990 0.2805 34
0.0850 0.0170 0.0408 0.0170 0.1700 0.1020 0.2890
1000 2.5000 0.5000 1.2000 0.5000 5.0000 3.0000 8.5000

2. Material & Methoden
50
2.2.3 Regelung der Körpertemperatur
Die Körpertemperatur der Maus wird mit einer rektalen Temperatur-Sonde gemessen
und mit Hilfe einer rückgekoppelten Heizmatte konstant auf 37°C gehalten. Bei
chronischen Versuchen werden die Tiere nach Versuchsende bis zu 24 Stunden in
einem auf 34°C aufgewärmten Inkubator gehalten, um eine Hypothermie zu vermei-
den.
2.2.4 Messung des intrakraniellen Drucks (ICP)
Die ICP-Sonde wird zunächst in physiologischer Kochsalzlösung geeicht. Nach
Induktion der Narkose wird das Versuchstier in Bauchlage positioniert und die
Kopfhaut medial ca. 1 cm längs inzidiert. Nach Entfernen der Galea aponeurotica wird
der rechte Musculus temporalis dargestellt, inzidiert und stumpf nach lateral gedrängt.
Orientierend wird die durchschimmernde Arterie cerebri media aufgesucht. Caudal
davon erfolgt eine Bohrlochtrepanation mit einem Präzisions-Diamantenbohrer.
Anschließend wird die ICP-Sonde epidural eingebracht. Die Sonde wird mit Knochen-
zement fixiert und es erfolgt die Aufzeichnung einer Baseline über 10 bis 30 Minuten,
bevor der Versuch fortgesetzt wird.
2.2.5 Messung der zerebralen Durchblutung (CBF)
Die regional zerebrale Durchblutung (regionale cerebral blood flow, rCBF), wird nicht-
invasiv durch die intakte Schädeldecke mit Laser-Doppler-Fluxmetrie gemessen.363,364
Dazu wird das Versuchstier in Bauchlage positioniert und der linke Musculus
temporalis freigelegt. Dieser wird im Bereich der Ansatzsehne inzidiert und stumpf
nach lateral gedrängt. Anschließend wird die LDF-Sonde in einem 90° Winkel zur
Kalotte über dem Versorgungsgebiet der A. cerebri media angesetzt und mit
Gewebekleber befestigt. Die Laser-Doppler-Fluxmetrie misst über den Dopplershift die
Geschwindigkeit der zerebralen Durchblutung. Durch die Absorption des
eingestrahlten Laserlichtes wird die Erythrozytenkonzentration im jeweiligen
Messvolumen bestimmt.365 Das Produkt aus beiden Werten wird in Perfusions-
einheiten (Arbitrary Units, UA) angegeben. Es wurde zunächst unter Ruhebedin-
gungen zehn Minuten eine Baseline aufgezeichnet. Diese wurde mit 100%

2. Material & Methoden
51
gleichgesetzt; die weiteren CBF-Werte wurden dann in Relation zu dieser
Ruhedurchblutung als Prozent des Ausgangswertes dargestellt.315,316,365
2.2.6 Induktion einer Subarachnoidalblutung (SAB)
Die SAB wird durch Fadenperforation der A. cerebri media induziert. Es handelt sich
hierbei um ein modifiziertes Modell welches 1995 zum ersten Mal von Bederson für
die Ratte beschrieben und in unserer Arbeitsgruppe reproduzierbar auf die Maus
übertragen wurde.127 Hierfür wird auf der linken Seite in der Medioclavikularlinie ein
Hautschnitt ab dem oberen Teil des Sternums bis zur Mandibula ausgeführt. Im
Hautschnittbereich ist die Glandula sublingualis zu erkennen, diese wird stumpf
abpräpariert, um die A. carotis communis darzustellen. Nach Darstellung der Carotis-
Bifurkation wird zunächst die A. thyroidea ligiert. Anschließend wird die Arteria carotis
externa ca. 0,3 cm distal der Bifurkation ligiert. Dann wird ein Ligaturfaden ca. 0,1 cm
distal der Bifurkation vorgelegt. Nach temporären Clipping der Arteria carotis
communis wird die A. carotis externa mit einer Mikroschere zwischen den beiden
Ligaturen geöffnet und ein 3,0 cm monofiler Faden eingeführt. Anschließend wird
dieser Perforationsfaden mit der vorgelegten Ligatur fixiert und das Gefäß damit
abgedichtet. Nach Entfernung des Clips wird der Perforationsfaden über die Arteria
carotis externa in die Arteria carotis interna vorgeschoben. Unter kontinuierlicher
Kontrolle der zerebralen Durchblutung und des intrakraniellen Drucks wird der Faden
innerhalb der A. carotis interna nach intrakraniell vorgeschoben, bis er an der Media-
Bifurkation das Gefäß perforiert. Die Auslösung der Blutung wird bestätigt durch einen
abrupten massiven ICP-Anstieg (> 30 mmHg) und eine gleichzeitige drastische
Verringerung der zerebralen Durchblutung (CBF) auf weniger als 30% des
Ausgangswert. Nur wenn beide Kriterien erfüllt sind gilt die Blutung als erfolgreich
ausgelöst.366 Hiernach wird der Perforationsfaden bis in die Arteria carotis externa
zurückgezogen, und dieses Gefäß mit Hilfe der vorgelegten Ligatur anschließend
verschlossen. Nach der SAB-Induktion erfolgte eine Beobachtungsphase von 15
Minuten, in denen der intrakranielle Druck und die zerebrale Durchblutung aufge-
zeichnet wurden. Dann erfolgt der Wundverschluss mit monofilem Faden in
Einzelknopfnaht-Technik. Bei der Sham-Operation werden alle Schritte außer der
Perforation der A. cerebri media durchgeführt. Die Narkose wird antagonisiert, das Tier

2. Material & Methoden
52
nach Wiedererlangen der Reflexe extubiert und zur Weiterführung des Versuchs im
Inkubator bei 34°C und 25% Luftfeuchtigkeit gehalte n.
2.2.7 Jugularis-Katheter
Nach Positionierung des Versuchstiers auf dem Rücken wird mit der Pinzette auf der
linken Seite in der Medioclavicular-Linie ein ca. 1 cm langer Hautschnitt durchgeführt.
Die V. jugularis interna wird freipräpariert und möglichst weit cranial mit einem
Seidenfaden ligiert. Die Zuflüsse zur Vene werden ligiert bzw. kauterisiert. Ein
Haltefaden wird vorgelegt und das Gefäß nah am Übergang zur Vena subclavia
temporär geklippt. Das Gefäß wird nun zwischen Clip und Ligatur inzidiert. Ein mit
steriler Kochsalzlösung oder Therapiesubstanz vorgefüllter Katheter wird in das
Lumen platziert und mit der Halteligatur fixiert. Nach Öffnen des Clips wird einmalig
aspiriert, um die intravasale Lage des Katheters zu beweisen. Dann wird der Katheter
mit einer weiteren Ligatur und mit Fibrinkleber fixiert. Der Katheter wird nun mittels
subkutanter Tunnelung dorsal im Bereich des Nackens ausgeleitet, die Hautwunde mit
monofilem Faden in Einzelknopfnaht-Technik verschlossen.
2.2.8 Quantifizierung der Mikrozirkulation
2.2.8.1 Analyse des zerebralen Gefäßbaums anhand de s Strahler-Schemas
Das Strahler-System wurde ursprünglich zur Quantifizierung der Geomorphologie von
Tälern und Flusssystemen entwickelt.367 Heute wird es auch in den Biowissenschaften
angewendet, um z. B. neuronale Netzwerke, Gefäßsysteme oder auch Bronchial-
verzweigungen zu kategorisieren,368-372 also, um komplexe Systeme in vergleichbare
Kategorien einzuteilen. Dieses System wurde an unsere Fragestellung angepasst,184
um eine statistische Auswertung der Häufigkeit bzw. Anzahl von Mikrovasospasmen
an Gefäßen zu ermöglichen. Die Gefäße, die sich nicht weiter aufteilen, werden als
Kapillaren/(Gefäß-)Kategorie 0 bezeichnet, was einen Durchmesser von 5,0 bis 9,0
µm entspricht. Die Arteriolen (Gefäßdurchmesser 9,5-15 µm), aus welchen Kapillaren
entstammen, werden als Kategorie 1 bezeichnet usw. Eine Übersicht sowie ein
Echtbild zeigt Abbildung 9. Auf diese Weise werden für die vorliegende Studie nach
dem Strahler-Schema alle Gefäße am Anfang der Versuchsreihe kategorisiert. Nach

2. Material & Methoden
53
Applikation des Fluoreszenzfarbstoffs und der Identifizierung der arteriellen Gefäße
mittels der Flussrichtung werden zunächst die Kapillaren identifiziert. Danach werden
diese retrograd bis hin zu den Gefäßen der Kategorie 6 (Gefäßdurchmesser: 55-65
µm) verfolgt. Die Buchstaben sollen die Gefäßabschnitte und die tiefgestellten Zahlen
die Anzahl der Gefäßabschnitte einer einzelnen Kategorie definieren. Der gesamte
Gefäßbaum im freipräparierten Fenster wird digital aufgezeichnet. Die weitere
Auswertung erfolgte offline mit der Leica Application Suite (LAS) Software.
Strahler-Schema Zerebrale Mi krozirkulation in vivo
Abbildung 9: (a) zeigt eine schematische Darstellun g eines Gefäß-Baums nach Strahler-
Kategorien. (b) zeigt eine IVM-Übersichts-Aufnahme der zerebralen Mikrozirkulation. Die gelben
Zahlen bezeichnen die Gefäßkategorien, die Buchstab en die Gefäßabschnitte und die
tiefgestellten Zahlen die Anzahl der Gefäßabschnitt e einer einzelnen Kategorie.
2.2.8.2 Quantifizierung von Gefäßdurchmesser und Mi krovasospasmus
Nach der Quantifizierung der einzelnen Gefäßabschnitte nach dem Strahler-Schema,
folgt die Quantifizierung des Gefäßdurchmessers (Leica Application Suite (LAS), Leica
Microsystems). In Bildern mit 20x Vergrößerung wird ein Gefäßabschnitt in
a b
5x

2. Material & Methoden
54
ganzer Länge gescannt, um kontrahierte und nicht kontrahierte Bereiche zu
identifizieren. Abschnitte mit minimalem und maximalem Durchmesser werden dann
markiert und im Abstand von 1 µm dreimalig gemessen. Aus diesen Messungen wird
der Mittelwert gebildet. Der Grad der Konstriktion wird dann als Prozentwert der nicht-
kontrahierten Gefäßabschnitte angegeben (Abbildung 10).
Arteriole
Abbildung 10: Schematische Darstellung der Bestimmung eines Vasos pasmus an einer Arterio-
le. Die vertikalen Linien repräsentieren die Messpunkte zur Bestimmung des Gefäßdurchmessers im
nicht kontrahierten und im kontrahierten Bereich. Als Konstriktionsgrad wird das Verhältnis beider
Durchmesser bezeichnet.
2.2.9 Intravitalmikroskopie
2.2.9.1 Venöse und arterielle Katheterisierung der Maus
Die intravitalmikroskopische Untersuchung der zerebralen Zirkulation erfolgt drei
Stunden nach Induktion der SAB. Vor Beginn der Intravitalmikroskopie wird zur
kontinuierlichen Messung des arteriellen Blutdrucks die A. femoralis am linken Bein
mit einem heparinisierten Kunststoffkatheter (Außendurchmesser: 0,61 mm;
Innendurchmesser: 0,28 mm) kannuliert, der an einem Drucktransducer angesch-
lossen wird. Dieser wird vor Versuchsbeginn mit einem Quecksilber-Manometer
geeicht. Der Katheter wird kontinuierlich mit 0,9% NaCl-Kochsalzlösung (0,4 ml/h)
gespült, die Daten kontinuierlich aufgezeichnet. Am Ende eines jeden Versuchs wird
eine Blut-Gas-Analyse durchgeführt. Zur intravenösen Applikation des Fluoreszenz-
farbstoffs Fluoresceinisothiocyanat–Dextran (FITC-Dextran) und oder anderer Medi-
kamente wird die linke V. femoralis in gleicher Art und Weise katheterisiert.
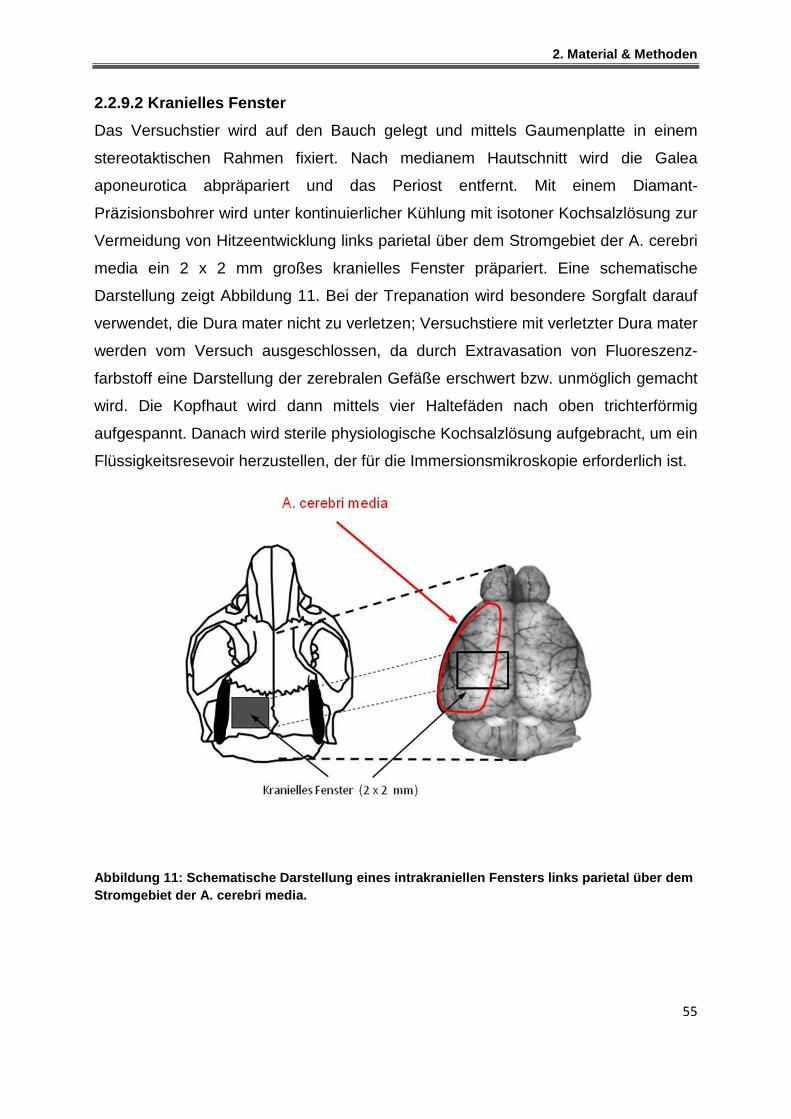
2. Material & Methoden
55
2.2.9.2 Kranielles Fenster
Das Versuchstier wird auf den Bauch gelegt und mittels Gaumenplatte in einem
stereotaktischen Rahmen fixiert. Nach medianem Hautschnitt wird die Galea
aponeurotica abpräpariert und das Periost entfernt. Mit einem Diamant-
Präzisionsbohrer wird unter kontinuierlicher Kühlung mit isotoner Kochsalzlösung zur
Vermeidung von Hitzeentwicklung links parietal über dem Stromgebiet der A. cerebri
media ein 2 x 2 mm großes kranielles Fenster präpariert. Eine schematische
Darstellung zeigt Abbildung 11. Bei der Trepanation wird besondere Sorgfalt darauf
verwendet, die Dura mater nicht zu verletzen; Versuchstiere mit verletzter Dura mater
werden vom Versuch ausgeschlossen, da durch Extravasation von Fluoreszenz-
farbstoff eine Darstellung der zerebralen Gefäße erschwert bzw. unmöglich gemacht
wird. Die Kopfhaut wird dann mittels vier Haltefäden nach oben trichterförmig
aufgespannt. Danach wird sterile physiologische Kochsalzlösung aufgebracht, um ein
Flüssigkeitsresevoir herzustellen, der für die Immersionsmikroskopie erforderlich ist.
Abbildung 11: Schematische Darstellung eines intrak raniellen Fensters links parietal über dem Stromgebiet der A. cerebri media.

2. Material & Methoden
56
2.2.9.3 Intravitalmikroskopie zerebraler Mikrogefäß e
Nach der Positionierung unter dem Mikroskop wird zunächst unter 5x Vergrößerung
eine Übersichtsaufnahme durchgeführt. Es folgt die Einteilung der Gefäße nach dem
Strahler-Schema. Untersucht werden piale Mikrogefäße (Arteriolen zwischen 9,5 und
65 µm und Venolen). Hiernach werden in höherer Vergrößerung (20x) mehrere
Regions of Interest (ROI) identifiziert und aufgezeichnet. Die Positionen werden
anhand der Tischkoordinaten und anhand der Anatomie markiert. Die fluoreszenz-
mikroskopischen Aufnahmen werden digital aufgezeichnet und für die weitere
Auswertung gespeichert. Die Gefäße werden mit der Gabe von 150-200 µl 0,5%ige
FITC-Dextran-Lösung (FITC-Dextran gelöst in 0,9%igen NaCl) visualisiert. Im Falle
eines Verlustes der Anfärbeintensität des Farbstoffes während der IVM, wird 1/3 des
initialen FITC-Dextran-Lösungsvolumen hinzugefügt.
2.2.10 Inhalation
2.2.10.1 Stickstoffmonoxid-Inhalation (iNO)
Stickstoffmonoxid (268 mg/m3 in Stickstoff) wird mit Atemgas (30% O2 und 70%
Stickstoff) gemischt, so dass das Versuchstier mit einer Endkonzentration von 50 ppm
beatmet werden kann. Die NO-Konzentration wird vor Beatmung des Versuchstieres
mit einem elektrochemischen Gasmonitor (Multi Gas Monitor zur NO-Messung,
Industrial Scientific Corporation, Dortmund, D) geprüft.
2.2.10.2 Kohlendioxid-Inhalation
Kohlenstoffdioxid (10% in Stickstoff) wird mit Atemgas (30% O2 und 70% Stickstoff)
gemischt, so dass das Versuchstier mit einer 7,5%igen Kohlenstoffdioxid-
Endkonzentration beatmet werden kann. Die CO2-Konzentration im Atemgas wird mit
Hilfe eines Sauerstoffgehaltmessers (Oxidig, Drägerwerk AG Lübeck, Lübeck, D)
kontinuierlich verfolgt.
2.2.11 Anwendung von Clazosentan
Clazosentan, (Ro 61-1790 / AXV-034343A / VML 588; Chemischer Name: [5-Methyl-
Pyridin-2-Sulfonsäure 6- (2-Hydroxy-Ethoxy) -5- (2-Methoxy-Phenoxy)-2-(2-1H-

2. Material & Methoden
57
Tetrazol-5-yl-Pyridin-4-yl)Pyrimidin-4-ylamid Dinatrium-Salz]; Molekulargewicht:
621,53 g/mol; Molekulare Formel: C25H21N9Na2O6S), ist ein gelb-weißes Pulver. Es
wird trocken gelagert und verdünnte Lösungen sollen vor Licht geschützt werden. Es
ist leicht wasserlöslich bis zu 4,8 mg/100 ml beim pH = 5,9 und 25 g/100 ml bei einem
pH-Wert über 7. Es ist eine schwache Disäure mit pKa-Werten von 3,3 und 4,5. Die
aktive Dosierung liegt in vitro bei 10-10 - 10-6 M und in vivo bei 3-10 mg/kg i.v. als Bolus
oder 3-10 mg/kg/h i.v. als Infusion. Es hat eine Plasma-Halbwertszeit von weniger als
eine Stunde in Ratten und Hunden. Clazosentan hat einen teratogenen Einfluss in
höheren Dosen und sollte nicht von schwangeren Frauen eingenommen werden.
Abbildung 12 stellt die Strukturformel von Clazosentans da.373
Abbildung 12: Chemische Struktur von Clazosentan
Clazosentan ist ein selektiver Endothelin-A (ETA) Rezeptorantagonist, der zu einer
Klasse von trifunktionellen Heteroarylsulfonamido Pyrimidinen gehört.373-375 Es wurde
durch Weiterentwicklung von Bosentans, dem ersten in klinischen Studien
verwendeten nichtpeptid ET-Rezeptor-Antagonisten,376 bindungsaffiner und wasser-
löslicher gemacht.373 Clazosentan fehlt jegliche Agonisten-Aktivität und ist 1000-fach
selektiver für die Hemmung des ETA- als für ETB- vermittelte Kontraktion.373 Aufgrund
der hohen Wasserlöslichkeit von Clazosentans eignet es sich besonders gut zur
parenteralen Anwendung.375 Studien zeigen, dass Clazosentan Vasospasmen in

2. Material & Methoden
58
großen zerebralen Gefäßen, nach experimenteller und klinischer Subarachnoidal-
blutung, umkehrt.373,377 Die Versuchsmäuse werden randomisiert und einer
Behandlung mit Clazosentan oder Vehikel (0,9%igen NaCl) unterzogen. Die
Behandlung erfolgt intravenös.
2.2.12 Versuchsprotokolle
2.2.12.1 Stickstoffmonoxid-Inhalation (iNO)
Bei männlichen C57 bl/6 Mäusen (n = 5 pro Gruppe) wird wie oben beschrieben eine
SAB induziert. Drei Stunden nach Subarachnoidalblutung wird die zerebrale
Mikrozirkulation mittels IVM untersucht, die Messungen erfolgen im Abstand von 10
Minuten. Nach intravenöser Bolusinjektion von FITC Dextran erfolgt eine
Quantifizierung des Gefäßbaums nach dem Strahler-Schema. Dann wird 30 Minuten
lang die Gefäßdurchmesser sowie Anzahl und Schwere der Mikrovasospasmen in
allen Gefäßabschnitten dokumentiert. Anschließend wird die iNO-Gruppe 40 min lang
mit 50 ppm NO zusätzlich zum normalen Atemgas behandelt. Die Zuschaltung des
Gases erfolgt randomisiert und die Kontrollgruppe wird weiter mit dem normalen
Atemgas (30% O2, 70% Stickstoff) beatmet. Alle 10 Minuten wird die zerebrale
Mikrozirkulation erneut quantifiziert. 30 Minuten nach Beendigung der NO-Inhalation
erfolgt die letzte Messung. Eine schematische Darstellung des Versuchsablaufs gibt
Abbildung 13. Während des gesamten Versuchs wird kontinuierlich der mittlere
arterielle Blutdruck aufgezeichnet. Am Ende des Versuchs wurde allen Versuchstieren
150 µl Blut zur Blut-Gas-Analyse entnommen.

2. Material & Methoden
59
Abbildung 13: Versuchsprotokoll für die NO-Inhalation-Versuchsrei he. (a) Kontrollgruppe und (b) iNO-Gruppe.
2.2.12.2 Hemmung von Endothelin-1-Rezeptoren mit Cl azosentan
2.2.12.2.1 Dosisfindung von Clazosentan
Da Clazosentan in der Literatur bisher nur bei Ratten378-380 verwendet wurde, wurde in
einer ersten Versuchsreihe im Rahmen einer Dosisfindung die wirksame Clazosentan-
Dosis ermittelt. Hierfür wird der Effekt von unterschiedlichen Clazosentan-
Konzentrationen (1 mg/kg KG/h, 3 mg/kg KG/h, 10 mg/kg KG/h, 20 mg/kg KG/h) auf
den systemischen Blutdruck und die zerebrale Durchblutung unter physiologischen
Bedingungen untersucht. Dafür wird gesunden C57 bl/6 Mäusen (n = 3 pro Gruppe)
nach Aufzeichnung einer zehn-minütigen Baseline-Messung die jeweilige Dosis
Clazosentan als i.v.-Dauerinfusion über 180 Minuten verabreicht. MAP und CBF
werden kontinuierlich über 180 Minuten aufgezeichnet. Als Dosis für die weiteren
Versuche wird die höchste Clazosentan-Dosis, die keinen Einfluss auf die gemesse-
nen Parameter zeigt, gewählt.
a. Kontrollgruppe
b. iNO-Gruppe
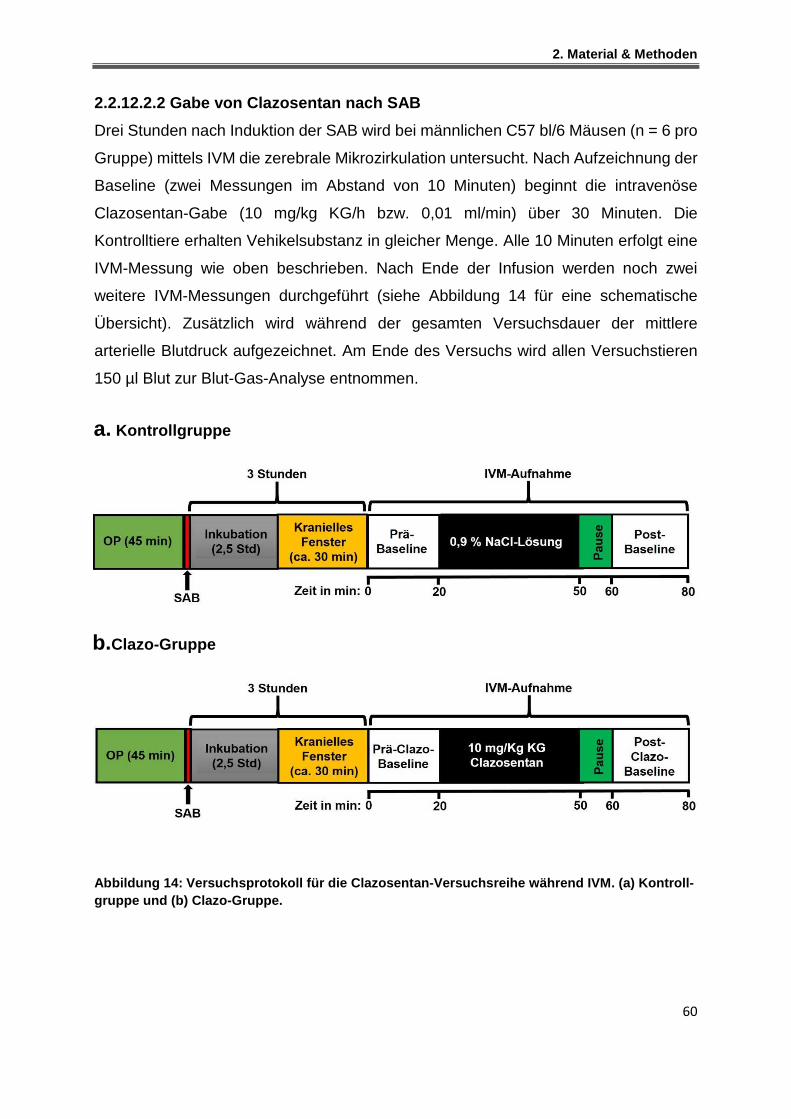
2. Material & Methoden
60
2.2.12.2.2 Gabe von Clazosentan nach SAB
Drei Stunden nach Induktion der SAB wird bei männlichen C57 bl/6 Mäusen (n = 6 pro
Gruppe) mittels IVM die zerebrale Mikrozirkulation untersucht. Nach Aufzeichnung der
Baseline (zwei Messungen im Abstand von 10 Minuten) beginnt die intravenöse
Clazosentan-Gabe (10 mg/kg KG/h bzw. 0,01 ml/min) über 30 Minuten. Die
Kontrolltiere erhalten Vehikelsubstanz in gleicher Menge. Alle 10 Minuten erfolgt eine
IVM-Messung wie oben beschrieben. Nach Ende der Infusion werden noch zwei
weitere IVM-Messungen durchgeführt (siehe Abbildung 14 für eine schematische
Übersicht). Zusätzlich wird während der gesamten Versuchsdauer der mittlere
arterielle Blutdruck aufgezeichnet. Am Ende des Versuchs wird allen Versuchstieren
150 µl Blut zur Blut-Gas-Analyse entnommen.
Abbildung 14: Versuchsprotokoll für die Clazosentan-Versuchsreihe während IVM. (a) Kontroll-gruppe und (b) Clazo-Gruppe.
a. Kontrollgruppe
b.Clazo-Gruppe

2. Material & Methoden
61
2.2.12.3 Untersuchung der Rolle der NOS-Isoformen b ei Vermittlung der CO 2-
Reaktivität zerebraler Gefäße
Männliche C57 bl/6 Mäuse (n = 15), eNOS-/- (n = 8), nNOS-/- (n = 8), iNOS-/- (n = 8),
werden narkotisiert und für die Intravitalmikroskopie vorbereitet. MAP und CBF werden
nach Messung einer Baseline kontinuierlich aufgezeichnet. Alle 10 min wird die
zerebrale Mikrozirkulation wie oben beschrieben aufgezeichnet. Vor jeder IVM-Phase
wird erneut ein Bolus FITC-Dextran i.v. appliziert. Die Untersuchung der CO2-
Inhalationsphase beginnt ab der 30. Minute 20 Minuten lang. 10 Minuten später erfolgt
eine abschließende Messung. Eine schematische Darstellung des Versuchsablaufs
gibt Abbildung 15. Am Ende des Versuchs wird allen Versuchstieren 150 µl Blut zur
Blut-Gas-Analyse entnommen.
Abbildung 15: Versuchsprotokoll für die CO 2-Inhalations-Versuchsreihe während IVM. (a) Kon-trollgruppe und (b) iNOS -/--, nNOS-/--, eNOS-/--Gruppe.
a. Kontrollgruppe
b. iNOS-/--, nNOS-/--, eNOS-/--Gruppe

3. Ergebnisse
3. Ergebnisse
3.1 Effekt von inhaliertem NO (iNO) auf die zerebra le Mikrozirkula-tion nach experimenteller Subarachnoidalblutung
3.1.1 Induktion der SAB
Vor SAB wiesen die Mäuse einen normalen intrakraniellen Druck zwischen 3 und 7
mmHg auf. Nach Induktion der Blutung stieg der ICP abrupt auf Werte von 92,3 ± 8,3
mmHg an. Während der nächsten Minuten sank der ICP-Wert und blieb jedoch zum
Zeitpunkt 15 Minuten (27,3 ± 3,5 mmHg) signifikant erhöht (Abbildung 16 a).
Gleichzeitig zeigte sich eine drastische Reduktion der regionalen zerebralen
Durchblutung auf 16,8% ± 4,2 % des Ausgangswertes, die sich ebenfalls während der
nächsten Minuten langsam zurückbildete. Am Ende der Beobachtungs-zeit, 15
Minuten nach SAB, erholte sich die zerebrale Durchblutung auf ca. 79,7% ± 10,1%
des Ausgangswertes (Abbildung 16 b). Dies entspricht den Werten, die in
vorangegangenen Studien der Arbeitsgruppe erhoben wurden.127 Aufgrund der gerin-
gen Abweichung kann in allen Gruppen von einer vergleichbar starken Blutung
ausgegangen werden.
Abbildung 16: (a) Intrazerebraler Druck (ICP) und (b) zerebrale D urchblutung (rCBF), vor,
während und nach der SAB Induktion.
Zeit [min]-10 -5 0 5 10 15 20
ICP
[mm
Hg
]
0
20
40
60
80
100
120 ICP (n=10) +/- SEM*p<0,05
SAB
vor SAB nach SAB
*
Zeit [min]-10 -5 0 5 10 15 20
rCB
F [%
]
0
20
40
60
80
100
120
rCBF (n=10) +/- SEM*p<0,05
SAB
vor SAB nach SAB
*

3. Ergebnisse
63
3.1.2 Einfluss des inhalierten Stickstoffmonoxids a uf die zerebrale Mikrozir-
kulation nach SAB
3.1.2.1 Systemischer arterieller Blutdruck
Der mittlere arterielle Blutdruck wurde kontinuierlich aufgezeichnet. Die NO-Inhalation
hatte keinen signifikanten Einfluß auf den MAP weder im Vergleich zu den Werten vor
NO-Gabe (vor iNO: 72,6 ± 10,5 mmHg und nach 40 min iNO: 72,0 ± 7,5 mmHg) noch
im Vergleich zur Kontrollgruppe (30. Min: 74,7 ± 4,7 mmHg und 70. Min: 61,6 ± 3,09
mmHg) (Abbildung 17).
Abbildung 17: Die Stickstoffmonoxid-Inhalation hat keinen Einfluss auf den systemischen
Blutdruck (gemessen in der A. femoralis).
Zeit [min]
0 10 20 30 40 50 60 70 80 90 100
MA
P [m
mH
g]
0
20
40
60
80
100
120 Kontrolle (SAB)SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p<0,05 vs 30 min
iNO (50 ppm)

3. Ergebnisse
64
3.1.2.2 Physiologische Parameter
Während des gesamten Versuchs wurde der arterielle pCO2 durch entsprechende
Anpassung der Beatmungsparameter im physiologischen Bereich gehalten. Am Ende
des Versuchs zeigte sich in Bezug auf pO2, pCO2 kein Unterschied zwischen den
Versuchsgruppen (Tabelle 14).
Tabelle 14: Tabellarische Darstellungen des pCO 2 und pO 2 nach 100 min Versuch.
MW ± SEM [SAB (n = 5); SAB + iNO (n = 5)]
3.1.3 Einfluss von NO auf die arteriellen Gefäßdurc hmesser
Direkt nach Beginn der NO-Inhalation (ab der 30. min) kam es in den kleinen und
kleinsten Arteriolen (Gefäßdurchmesser 9,5 bis 25 µm) zu einer signifikanten Dilatation
(Abbildung 18 b und c, in Kategorie 1: nach 30 min iNO: 126% ± 3,03%, nach 40 min
iNO: 126% ± 4,11% und 30 min nach iNO-Stopp: 134% ± 5,3%. In Kategorie 2 nach
40 min iNO: 117% ± 1,52% und 30 min nach iNO-Stopp: 122% ± 1,45%; p<0,05).
Dieser gefäßerweiternde Effekt blieb auch nach Beendigung der NO-Inhalation
erhalten und war bis zum Ende des Untersuchungszeitraums nachweisbar. In
Kapillaren (Durchmesser 5,0-9,0 µm, Kategorie 0, Abbildung 18 a) zeigte sich
trendmäßig die gleiche Veränderung. In den größeren Gefäßen (Kategorie 4-5,
Abbildung 18 e und f) zeigte sich kein Effekt der NO-Inhalation auf die Gefäßweite.

3. Ergebnisse
65
Abbildung 18: Einfluss der NO-Inhalation auf den Du rchmesser zerebraler Kapillaren (5,0-9,5 µm)
und Arteriolen (9,5-65 µm).
Zeit [min]
0 30 40 50 60 70 80 90 100
Kap
illar
en D
urch
mes
ser
[%]
0
80
100
120
140
160 Kontrolle (SAB)SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
Gefäßdurchmesser 5,0 - 9,5 µm
iNO (50 ppm)
Zeit [min]
0 30 40 50 60 70 80 90 100
Art
erio
len
Durc
hmes
ser
[%]
0
80
100
120
140
160Kontrolle (SAB) SAB + iNO
*
iNO (50 ppm)
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
Gefäßdurchmesser 9,5 - 15 µm
* *
Zeit [min]
0 30 40 50 60 70 80 90100
Art
erio
len
Dur
chm
esse
r [%
]
0
80
100
120
140
160Kontrolle (SAB)SAB + iNO
**
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
Gefäßdurchmesser 15 - 25 µm
iNO (50 ppm)
Zeit [min]0 30 40 50 60 70 80 90 100
Art
erio
len
Dur
chm
esse
r [%
]
0
80
100
120
140
160Kontrolle (SAB) SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
Gefäßdurchmesser 25 - 35 µm
iNO (50 ppm)
Zeit [min]0 30 40 50 60 70 80 90 100
Art
erio
len
Dur
chm
esse
r [%
]
0
80
100
120
140
160Kontrolle (SAB)SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
iNO (50 ppm)
Gefäßdurchmesser 35 - 45 µm
Zeit [min]0 30 40 50 60 70 80 90 100
Art
erio
len
Dur
chm
ess
er
[%]
0
80
100
120
140
160Kontrolle (SAB) SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
iNO (50 ppm)
Gefäßdurchmesser 45 - 55 µm

3. Ergebnisse
66
3.1.4 Einfluss der NO-Inhalation auf die zerebralen Mikrovasospasmen
Drei Stunden nach SAB zeigte sich in der zerebralen Mikrozirkulation eine signifikante
Anzahl von mikrovaskulären Kontraktionen (siehe Beispielbild in Abbildung 19).
Anzahl, Intensität und Verteilung entsprachen den Befunden in vorangegangenen
Studien.184
Abbildung 19: Mikrovaskuläre Kontraktionen von Arte riolen der Kategorie 1 und 2 drei Stunden
nach SAB.
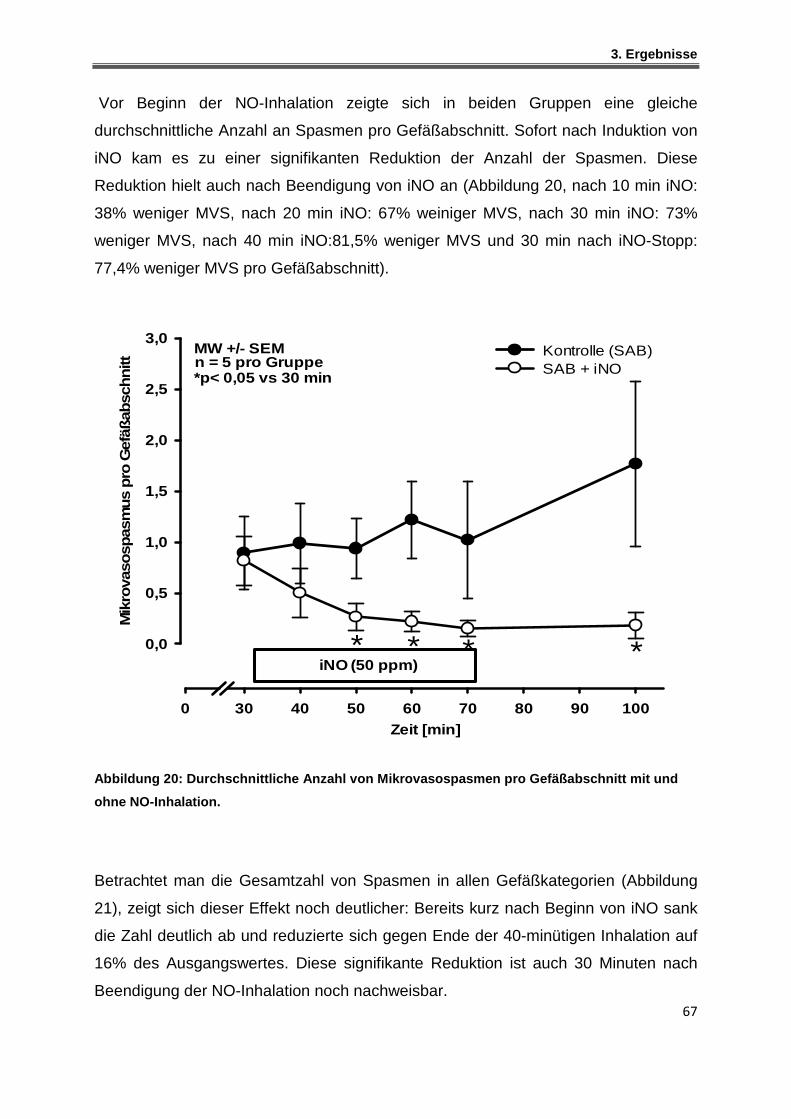
3. Ergebnisse
67
Vor Beginn der NO-Inhalation zeigte sich in beiden Gruppen eine gleiche
durchschnittliche Anzahl an Spasmen pro Gefäßabschnitt. Sofort nach Induktion von
iNO kam es zu einer signifikanten Reduktion der Anzahl der Spasmen. Diese
Reduktion hielt auch nach Beendigung von iNO an (Abbildung 20, nach 10 min iNO:
38% weniger MVS, nach 20 min iNO: 67% weiniger MVS, nach 30 min iNO: 73%
weniger MVS, nach 40 min iNO:81,5% weniger MVS und 30 min nach iNO-Stopp:
77,4% weniger MVS pro Gefäßabschnitt).
Abbildung 20: Durchschnittliche Anzahl von Mikrovas ospasmen pro Gefäßabschnitt mit und
ohne NO-Inhalation.
Betrachtet man die Gesamtzahl von Spasmen in allen Gefäßkategorien (Abbildung
21), zeigt sich dieser Effekt noch deutlicher: Bereits kurz nach Beginn von iNO sank
die Zahl deutlich ab und reduzierte sich gegen Ende der 40-minütigen Inhalation auf
16% des Ausgangswertes. Diese signifikante Reduktion ist auch 30 Minuten nach
Beendigung der NO-Inhalation noch nachweisbar.
Zeit [min]0 30 40 50 60 70 80 90 100
Mik
rova
sosp
asm
us p
ro G
efäß
absc
hnitt
0,0
0,5
1,0
1,5
2,0
2,5
3,0Kontrolle (SAB)SAB + iNO
* * * *iNO (50 ppm)
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min

3. Ergebnisse
68
Abbildung 21: Gesamtanzahl von Mikrovasospasmen in allen Gefäßkategorien mit und ohne
NO-Inhalation.
3.1.5 Räumliche Verteilung der Mikrovasospasmen
Vasospasmen zeigten sich in der vorliegenden Versuchsserie vor allem in Arteriolen
mit einem Kaliber von 9,5 bis 25 µm (Abbildung 22 b und 22 c). Hier zeigte sich
wiederum die signifikante Reduktion der Zahl der Spasmen durch die NO-Inhalation
(Abbildung 22 b und c, Kategorie 1: nach 10 min iNO: 50% weniger MVS, nach 20
min iNO: 78% weiniger MVS, nach 30 min iNO: 77% weniger MVS, nach 40 min
iNO:86,5% weniger MVS und 30 min nach iNO-Stopp: 85,6% weniger MVS. In
Kategorie 2 nach 10 min iNO: 41% weniger MVS, 20 min nach iNO: 68,5% weniger
MVS, 30 min nach iNO: 73,5% weniger MVS, 40 min nach iNO: 89% weniger MVS
und 30 min nach iNO-Stopp: 67% weniger MVS; p<0,05). In Gefäßen höherer
Kategorie (Durchmesser 25-45 µm, Abbildung 22 d, e) zeigten sich sehr wenige bis
keine Spasmen, auch hier hat iNO keinen signifikanten Effekt.
Zeit [min]
0 30 40 50 60 70 80 90 100
Mik
rova
sosp
asm
en [%
]
0
20
40
60
80
100
120
SAB + iNO (n = 5)Kontrolle (SAB; n = 5)
n=112 n=114n=108
n=116
n=126
n=113
n=86
n=58
n=30n=24
n=14n=20
n = Gesamt Mikrovasospasmen
iNO (50 ppm)

3. Ergebnisse
69
Abbildung 22: Räumliche Verteilung der Mikrovasospa smen (pro Gefäßabschnitt).
Gefäßdurchmesser 9,5-15 µm
Zeit [min]
0 30 40 50 60 70 80 90100
Mik
rova
sosp
asm
us p
ro G
efä
ßab
sch
nitt
[n]
0
1
2
3
4
5
6 Kontrolle (SAB) SAB + iNO
* * **
MW +/- SEMn = 5 pro Gruppe*p<0.05 vs 30 min
iNO (50 ppm)
Gefäßdurchmesser 15-25 µm
Zeit [min]
0 30 40 50 60 70 80 90100
Mik
rova
sosp
amus
pro
Gef
äßa
bsch
nitt
[n]
0
1
2
3
4
5
6Kontrolle (SAB) SAB + iNO
* * * **
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
iNO (50 ppm)
Gefäßdurchmesser 35-45 µm
Zeit [min]
0 30 40 50 60 70 80 90 100
Mik
rova
sosp
asm
us p
ro G
efäß
absc
hnitt
[n]
0
1
2
3
4
5
6Kontrolle (SAB) SAB + iNO
iNO (50 ppm)
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
Gefäßdurchmesser 25-35 µm
Zeit [min]
0 30 40 50 60 70 80 90100
Mik
rova
sosp
asm
en p
ro G
efäß
absc
hnitt
[n]
0
1
2
3
4
5
6Kontrolle (SAB) SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min
iNO (50 ppm)
Gefäßdurchmesser 5,0-9,0 µm
Zeit [min]
0 30 40 50 60 70 80 90100
Mik
rova
sosp
asm
en
pro
Gefä
ßab
schn
itt [n
]
0
1
2
3
4
5
6 Kontrolle (SAB) SAB + iNO
*
iNO (50 ppm)
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min

3. Ergebnisse
70
3.1.6 Effekt der NO-Inhalation auf den Grad der Mik rovasospasmen
Die überwiegende Mehrzahl der Spasmen ist leicht- bis mittelgradig, d. h. die Gefäße
wiesen einen Konstriktionsgrad zwischen 10 und 40% der Baseline auf. Höhergradige
Konstriktionen waren selten (Abbildung 23 a). Die NO-Inhalation führt zu einer raschen
Erweiterung der verengten Arteriolen, insbesondere der am häufigsten auftretenden
Spasmen mit einen Konstriktionsgrad zwischen 10-20% (Abbildung 23 c: p = 0,032, d:
p = 0,016 und Abbildung 23 e: p = 0,008). Auch hier lässt sich 30 Minuten nach Ende
der Therapie weiterhin ein signifikanter Effekt nachweisen (Abbildung 23 f).

3. Ergebnisse
Abbildung 23: Effekt der NO-Inhalation auf dem Mikr ovasospasmus im Hinblick auf den
Konstriktionsgrad. Die NO-Inhalation vermindert bzw. verhindert v. a. leicht- bis mittelgradige
Spasmen (10-30%), die jedoch die Mehrzahl aller Spasmen ausmachen.
Baseline - Vor NO-Inhalation (0-30 min)
Konstriktion [%]
10-2021-30
31-4041-50
51-60
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12 Kontrolle (SAB)SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p<0,05 vs SAB
Nach 10 min NO-Inhalation
Konstriktion [%]
10-2021-30
31-4041-50
51-60
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12 Kontrolle (SAB) SAB + iNO
MW +/- SEMn = 5 pro Gruppe*p<0,05 vs SAB
*
Nach 20 min NO-Inhalation
Konstriktion [%]
10-2021-30
31-4041-50
51-60
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12Kontrolle (SAB) SAB + iNO
**
* *
MW +/- SEMn = 5 pro Gruppe*p<0,05 vs SAB
Nach 30 min NO-Inhalation
Konstriktion [%]
10-2021-30
31-4041-50
51-60
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12Kontrolle (SAB) SAB + iNO
*
*
*
MW +/- SEMn = 5 pro Gruppe*p<0,05 vs SAB
Nach 40 min NO-Inhalation
Konstriktion [%]
10-2021-30
31-4041-50
51-60
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12Kontrolle (SAB) SAB + iNO
* *
MW +/- SEMn = 5 pro Gruppe*p<0,05 vs SAB
Baseline - 30 min nach NO-Inhalation
Konstriktion [%]
10-2021-30
31-4041-50
51-60
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12 Kontrolle (SAB) SAB + iNO
**
MW +/- SEMn = 5 pro Gruppe*p<0,05 vs SAB
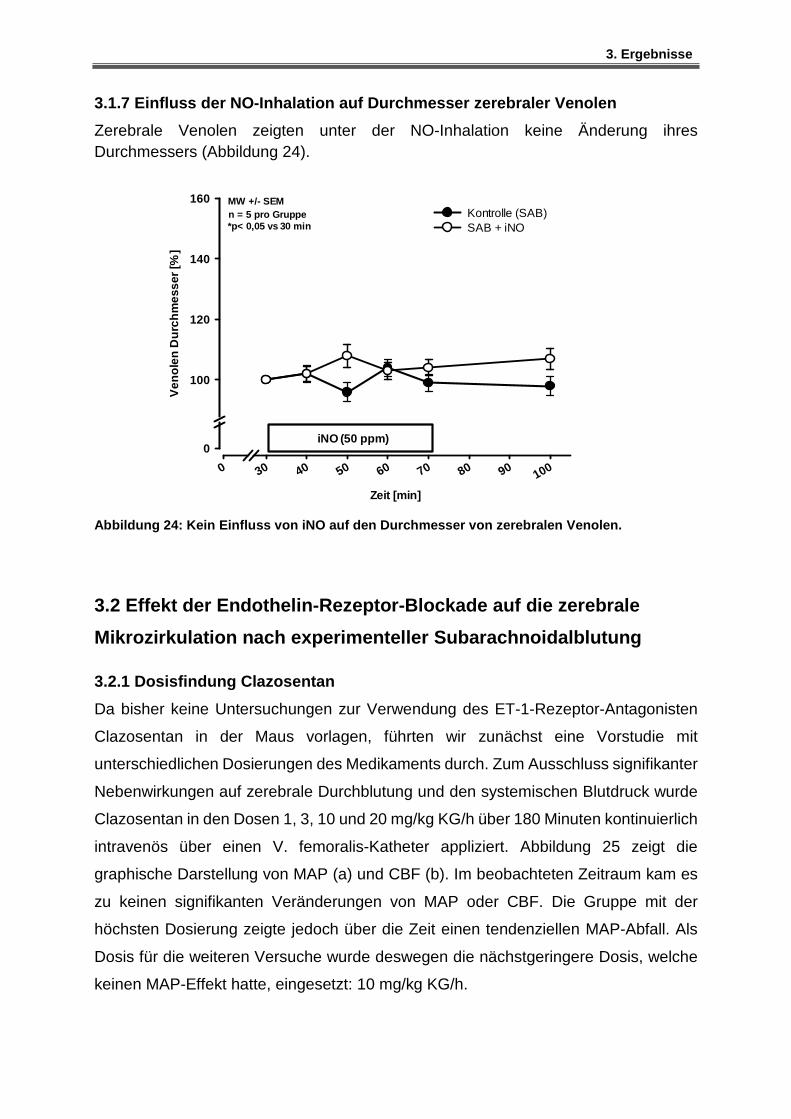
3. Ergebnisse
3.1.7 Einfluss der NO-Inhalation auf Durchmesser ze rebraler Venolen
Zerebrale Venolen zeigten unter der NO-Inhalation keine Änderung ihres Durchmessers (Abbildung 24).
Abbildung 24: Kein Einfluss von iNO auf den Durchme sser von zerebralen Venolen.
3.2 Effekt der Endothelin-Rezeptor-Blockade auf die zerebrale
Mikrozirkulation nach experimenteller Subarachnoida lblutung
3.2.1 Dosisfindung Clazosentan
Da bisher keine Untersuchungen zur Verwendung des ET-1-Rezeptor-Antagonisten
Clazosentan in der Maus vorlagen, führten wir zunächst eine Vorstudie mit
unterschiedlichen Dosierungen des Medikaments durch. Zum Ausschluss signifikanter
Nebenwirkungen auf zerebrale Durchblutung und den systemischen Blutdruck wurde
Clazosentan in den Dosen 1, 3, 10 und 20 mg/kg KG/h über 180 Minuten kontinuierlich
intravenös über einen V. femoralis-Katheter appliziert. Abbildung 25 zeigt die
graphische Darstellung von MAP (a) und CBF (b). Im beobachteten Zeitraum kam es
zu keinen signifikanten Veränderungen von MAP oder CBF. Die Gruppe mit der
höchsten Dosierung zeigte jedoch über die Zeit einen tendenziellen MAP-Abfall. Als
Dosis für die weiteren Versuche wurde deswegen die nächstgeringere Dosis, welche
keinen MAP-Effekt hatte, eingesetzt: 10 mg/kg KG/h.
Zeit [min]
0 30 40 50 60 70 80 90 100
Ven
olen
Dur
chm
esse
r [%
]
0
100
120
140
160Kontrolle (SAB) SAB + iNO
iNO (50 ppm)
MW +/- SEMn = 5 pro Gruppe*p< 0,05 vs 30 min

3. Ergebnisse
73
Abbildung 25: Clazosentan-Dosisfindung: Effekt unte rschiedlicher Dosen von Clazosentan auf
den systemischen arteriellen Blutdruck (MAP, a) und die regionale zerebrale Durchblutung
(rCBF, b). Die untersuchten Dosierungen 1 mg/Kg, 3 mg/Kg, 10 mg/Kg und 20 mg/Kg hatten keinen
signifikanten Effekt auf die untersuchten Parameter.
Clazosentan Dosisfindung CBF-Vergleichskurve
Zeit [min]
-50 0 50 100 150 200
rCB
F [%
]
0
20
40
60
80
100
120
140Clazosentan 1 mg/kgClazosentan 3 mg/kgClazosentan 10 mg/kgClazosentan 20 mg/kg
MW +/- SEMn = 3 pro Gruppe*p< 0,05
Clazosentan Dosisfindung MAP-Vergleichskurve
Zeit [min]
-50 0 50 100 150 200
MA
P [m
mH
g]
0
20
40
60
80
100
120
140
160 Clazosentan 1mg/kgClazosentan 3mg/ kgClazosentan 10 mg/kgClazosentan 20 mg/kg
MW +/- SEMn = 3 pro Gruppe*p< 0,05
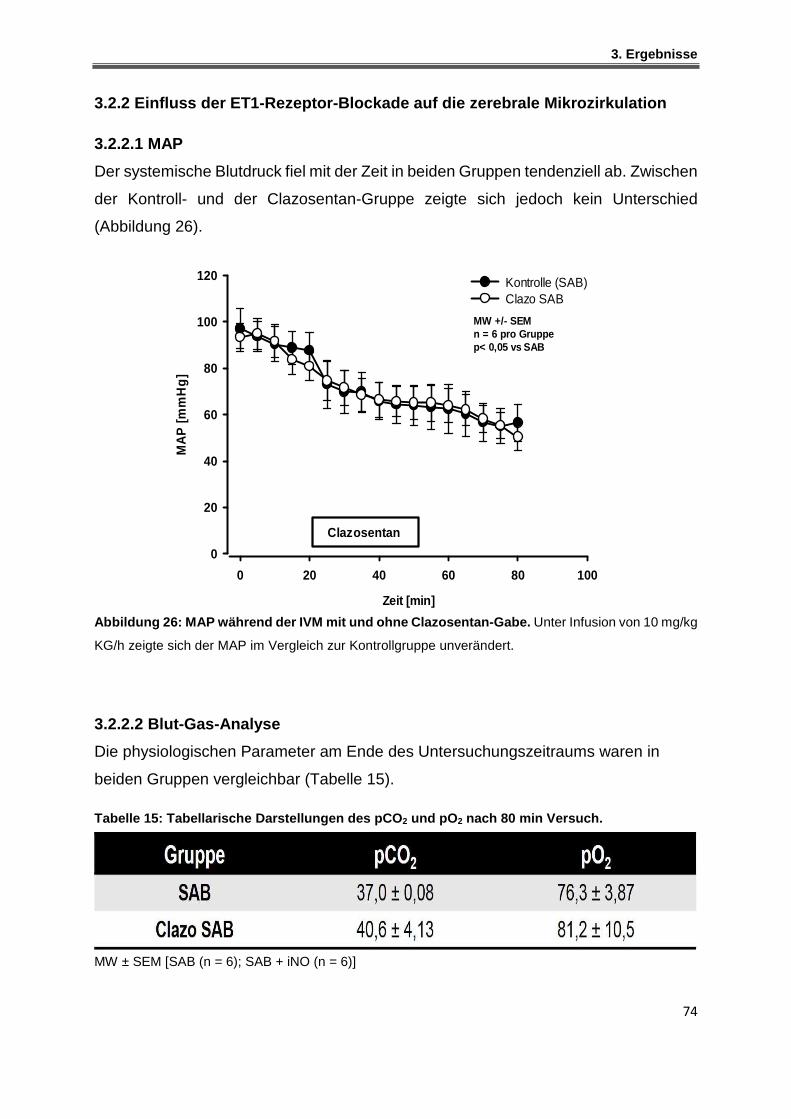
3. Ergebnisse
74
3.2.2 Einfluss der ET1-Rezeptor-Blockade auf die ze rebrale Mikrozirkulation
3.2.2.1 MAP
Der systemische Blutdruck fiel mit der Zeit in beiden Gruppen tendenziell ab. Zwischen
der Kontroll- und der Clazosentan-Gruppe zeigte sich jedoch kein Unterschied
(Abbildung 26).
Abbildung 26: MAP während der IVM mit und ohne Claz osentan-Gabe. Unter Infusion von 10 mg/kg
KG/h zeigte sich der MAP im Vergleich zur Kontrollgruppe unverändert.
3.2.2.2 Blut-Gas-Analyse
Die physiologischen Parameter am Ende des Untersuchungszeitraums waren in
beiden Gruppen vergleichbar (Tabelle 15).
Tabelle 15: Tabellarische Darstellungen des pCO 2 und pO 2 nach 80 min Versuch.
MW ± SEM [SAB (n = 6); SAB + iNO (n = 6)]
Zeit [min]
0 20 40 60 80 100
MA
P [m
mH
g]
0
20
40
60
80
100
120 Kontrolle (SAB)Clazo SAB
Clazosentan
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB

3. Ergebnisse
75
3.2.2.3 Einfluss von 10 mg/kg KG/h Clazosentan auf den Durchmesser von
zerebralen Arteriolen (9,5-55 µm)
Unter Clazosentan-Infusion zeigte sich in den Arteriolen von 9,5 bis 35 µm (Abbildung
27 a-c) keine signifikante Vasodilatation, die ET1-Rezeptor-Blockade führte lediglich
zu geringen Schwankungen des Gefäßdurchmessers.
Abbildung 27.: ET-Rezeptorblockade durch Clazosenta n führt in kleineren pialen Arteriolen zu
keiner signifikanten Vasodilatation. Die Werte der Clazosentan-Gruppe wurden auf die
Kontrollgruppe normalisiert (Durchmesser vor Beginn der Infusion entspricht 100%).
Zeit [min]
0 20 40 60 80 100
Du
rchm
esse
r vo
n A
rter
iole
n [%
]
0
80
100
120
140
Clazo SAB / SAB
Clazosentan
Gefäßdurchmesser 15 - 25 µm
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB
Zeit [min]
0 20 40 60 80 100
Dur
chm
esse
r vo
n A
rter
iole
n [%
]
0
80
100
120
140
Clazo SAB / SAB
Clazosentan
Gefäßdurchmesser 9,5 - 15 µm
MW +/- SEMn = 6 pro Gruppe*p< 0,05 vs SAB
Zeit [min]
0 20 40 60 80 100
Dur
chm
esse
r vo
n A
rter
iole
n [%
]
0
80
100
120
140
Clazo SAB / SAB
Clazosentan
Gefäßdurchmesser 25 - 35 µm
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB

3. Ergebnisse
76
Größere Gefäße bzw. Arteriolen mit einem Durchmesser von 35-45 µm reagierten auf
10 mg/kg KG/h Clazosentan mit einer signifikanten Vasodilatation in der 50. Minute
(126,6% ± 2,0%, p = 0,015). Diese ist jedoch rasch reversibel nach Ende der
Medikamentengabe (Abbildung 28 a). Arteriolen mit einem Durchmesser von 45-55
µm weisen in der 30. Minute nach Clazosentan-Infusion eine signifikante Erweiterung
der Gefäße von 121,01% ± 2,2% bei p = 0,03. Anschließend ist eine Verengung der
Gefäße zu beobachten (Abbildung 28 b).
Abbildung 28: ET-1-Rezeptorblockade mit Clazosentan (10 mg/kg KG/ h) auf größere zerebrale
Arteriolen. Unter Clazosentan-Infusion kommt es zu einer kurzfristigen signifikanten Vasodilatation bei
Arteriolen mit einem Durchmesser von 35-45 µm (a) und 45-55 µm (b).
3.2.2.4 Effekt der ET1-Rezeptorblockade auf die pos thämorrhagischen Mikro-
vasospasmen
Die Clazosentan-Infusion führte ab der 20. Min zur einen tendenziellen Verringerung
von Vasospasmenzahl bis zur 70. Min in Arteriolen der Kategorie 1 (Abbildung 29 a).
Arteriolen der Kategorie 2 (Abbildung 29 b) zeigten eine signifikante Verringerung der
MVS-Zahl gegenüber der Kontrolle in der 50. Min (die MVS-Zahl verringerte sich um
50% ± 3,5%, p = 0,041). In der Kategorie 3 (Abbildung 29 c) sind signifikante
Verringerungen bzgl. der MVZ-Zahl in der 50. Min (MVS-Zahl verringerte sich um 30%
Zeit [min]
0 20 40 60 80 100
Du
rchm
esse
r vo
n A
rter
iole
n [%
]
0
80
100
120
140
Clazo SAB / SAB
Clazosentan
Gefäßdurchmesser 35 - 45 µm
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB*
**
Zeit [min]
0 20 40 60 80 100
Du
rchm
esse
r vo
n A
rter
iole
ns
[%]
0
80
100
120
140
Clazo SAB / SAB
Clazosentan
Gefäßdurchmesser 45 - 55 µm
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB*
*

3. Ergebnisse
77
± 8,0%, p = 0,008), 70. Min (MVS-Zahl verringerte sich um 75% ± 0%, p = 0,015) und
80. Min (MVS-Zahl verringerte sich um 46,05% ± 0%, p = 0,015) zu verifizieren.
Abbildung 29: Effekt der ET1-Rezeptorblockade auf d ie Anzahl an Mikrovasospasmen. Unter
Clazosentan-Infusion kommt es zu einer signifikanten Mikrovasospasmenzahl Reduzierung bei
Arteriolen mit einem Durchmesser von 15-25 µm(b) und 25-35 µm (c).
Gefäßdurchmesser 15 -25 µm
Zeit [min]
0 20 40 60 80 100
Mik
orv
asos
pas
me
n p
ro G
efäß
absc
hn
itt [%
]
0
20
40
60
80
100
120
140
160
180
200
SABClazo SAB
Clazosentan
*
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB*
Gefäßdurchmesser 25 -35 µm
Zeit [min]
0 20 40 60 80 100
Mik
rova
sosp
asm
en
pro
Ge
fäß
absc
hnitt
[%]
0
50
100
150
200SABClazo SAB
Clazosentan
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB*
** *
Gefäßdurchmesser 9,5 -15 µm
Zeit [min]
0 20 40 60 80 100
Mik
orva
sosp
asm
en
pro
Gef
äß
abs
chn
itt [%
]
0
20
40
60
80
100
120
140
160
180
200SABClazo SAB
Clazosentan
MW +/- SEMn = 6 pro Gruppe*p< 0,05 vs SAB

3. Ergebnisse
78
3.2.2.5 Effekt der ET1-Rezeptorblockade auf die Anz ahl und den Konstrik-
tionsgrad der Mikrovasospasmen
Die überwiegende Mehrzahl der Spasmen ist mittelgradig ausgeprägt. Die Gefäße
wiesen vor allem MVS mit einen Konstriktionsgrad zwischen 10 und 30% der Baseline
auf. Höhergradige Konstriktion der Gefäße sind selten (Abbildung 30 a). Die Clazosen-
tan-Infusion (10 mg/kg KG/h) führt zu einer Erweiterung der Spasmen die einen
Konstriktionsgrad zwischen 10-30% aufweisen. 30 min nach Therapie bleiben MVS,
die einen Konstriktionsgrad zwischen 21-30% aufweisen, signifikant verringert
(Abbildung 30 f).

3. Ergebnisse
Nach 30 min Clazosentan-Infusion
Konstriktion [%]
10-2021-30
31-4041-50
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12
14
16SABClazo SAB
MW +/- SEMn = 6 pro Grupppe*p< 0,05 vs SAB
* *
Baseline - 20 min nach Clazosentans-Infusion
Konstriktion [%]
10-2021-30
31-4041-50
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12
14
16SABClazo SAB
MW +/- SEMn = 6 pro Gruppe*p< 0,05 vs SAB
* *
Baseline - 30 min nach Clazosentans-Infusion
Konstriktion [%]
10-2021-30
31-4041-50
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12
14
16SABClazo SAB
MW +/- SEMn = 6 pro Gruppep< 0,05 vs SAB
*
Abbildung 30: Effekt der ET-1-Rezeptorblockade auf die Anzahl und Konstriktionsgrad von
Mikrovasospasmen. Die Clazosentan-Infusion vermindert bzw. verhindert v. a. leicht- bis mittelgradige
Spasmen (10-30%), die jedoch die Mehrzahl aller Spasmen ausmachen
Baseline - Vor Clazosentan-Infusion (10-20 min)
Konstriktion [%]
10-2021-30
31-4041-50
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12
14
16SABClazo SAB
MW +/- SEMn = 6 pro Gruppe*p< 0,05 vs SAB*
Nach 10 min Clazosentan-Infusion
Konstriktion [%]
10-2021-30
31-4041-50
Mic
rova
sosp
asm
s / M
ouse
(n)
0
2
4
6
8
10
12
14
16SABClazo SAB
MW +/- SEMn = 6 pro Gruppe*p< 0,05 vs SAB
*
*
Nach 20 min Clazosentan-Infusion
Konstriktion [%]
10-2021-30
31-4041-50
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10
12
14
16SABClazo SAB
MW +/- SEMn = 6 pro Gruppe*p< 0,05 vs SAB
**
d
e f
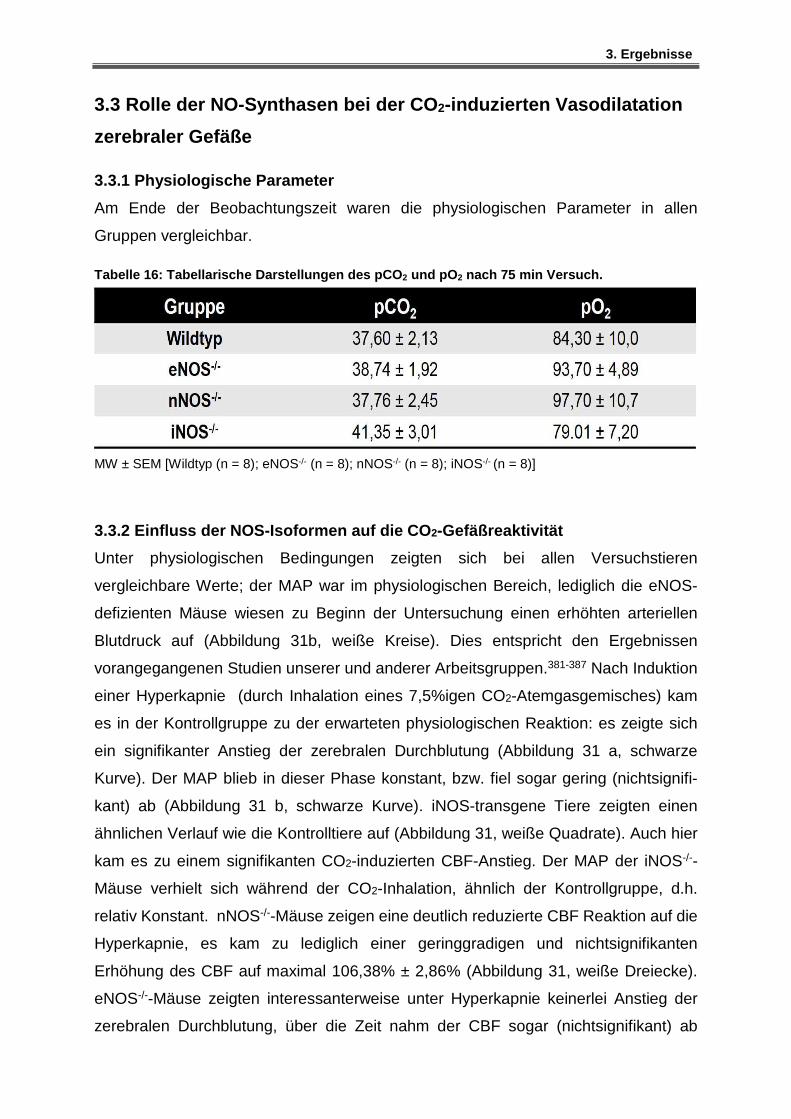
3. Ergebnisse
3.3 Rolle der NO-Synthasen bei der CO 2-induzierten Vasodilatation
zerebraler Gefäße
3.3.1 Physiologische Parameter
Am Ende der Beobachtungszeit waren die physiologischen Parameter in allen
Gruppen vergleichbar.
Tabelle 16: Tabellarische Darstellungen des pCO 2 und pO 2 nach 75 min Versuch.
MW ± SEM, Wildtyp (n = 8); eNOS-/- (n = 8); nNOS-/- (n = 8); iNOS-/- (n = 8)
MW ± SEM [Wildtyp (n = 8); eNOS-/- (n = 8); nNOS-/- (n = 8); iNOS-/- (n = 8)]
3.3.2 Einfluss der NOS-Isoformen auf die CO 2-Gefäßreaktivität
Unter physiologischen Bedingungen zeigten sich bei allen Versuchstieren
vergleichbare Werte; der MAP war im physiologischen Bereich, lediglich die eNOS-
defizienten Mäuse wiesen zu Beginn der Untersuchung einen erhöhten arteriellen
Blutdruck auf (Abbildung 31b, weiße Kreise). Dies entspricht den Ergebnissen
vorangegangenen Studien unserer und anderer Arbeitsgruppen.381-387 Nach Induktion
einer Hyperkapnie (durch Inhalation eines 7,5%igen CO2-Atemgasgemisches) kam
es in der Kontrollgruppe zu der erwarteten physiologischen Reaktion: es zeigte sich
ein signifikanter Anstieg der zerebralen Durchblutung (Abbildung 31 a, schwarze
Kurve). Der MAP blieb in dieser Phase konstant, bzw. fiel sogar gering (nichtsignifi-
kant) ab (Abbildung 31 b, schwarze Kurve). iNOS-transgene Tiere zeigten einen
ähnlichen Verlauf wie die Kontrolltiere auf (Abbildung 31, weiße Quadrate). Auch hier
kam es zu einem signifikanten CO2-induzierten CBF-Anstieg. Der MAP der iNOS-/--
Mäuse verhielt sich während der CO2-Inhalation, ähnlich der Kontrollgruppe, d.h.
relativ Konstant. nNOS-/--Mäuse zeigen eine deutlich reduzierte CBF Reaktion auf die
Hyperkapnie, es kam zu lediglich einer geringgradigen und nichtsignifikanten
Erhöhung des CBF auf maximal 106,38% ± 2,86% (Abbildung 31, weiße Dreiecke).
eNOS-/--Mäuse zeigten interessanterweise unter Hyperkapnie keinerlei Anstieg der
zerebralen Durchblutung, über die Zeit nahm der CBF sogar (nichtsignifikant) ab
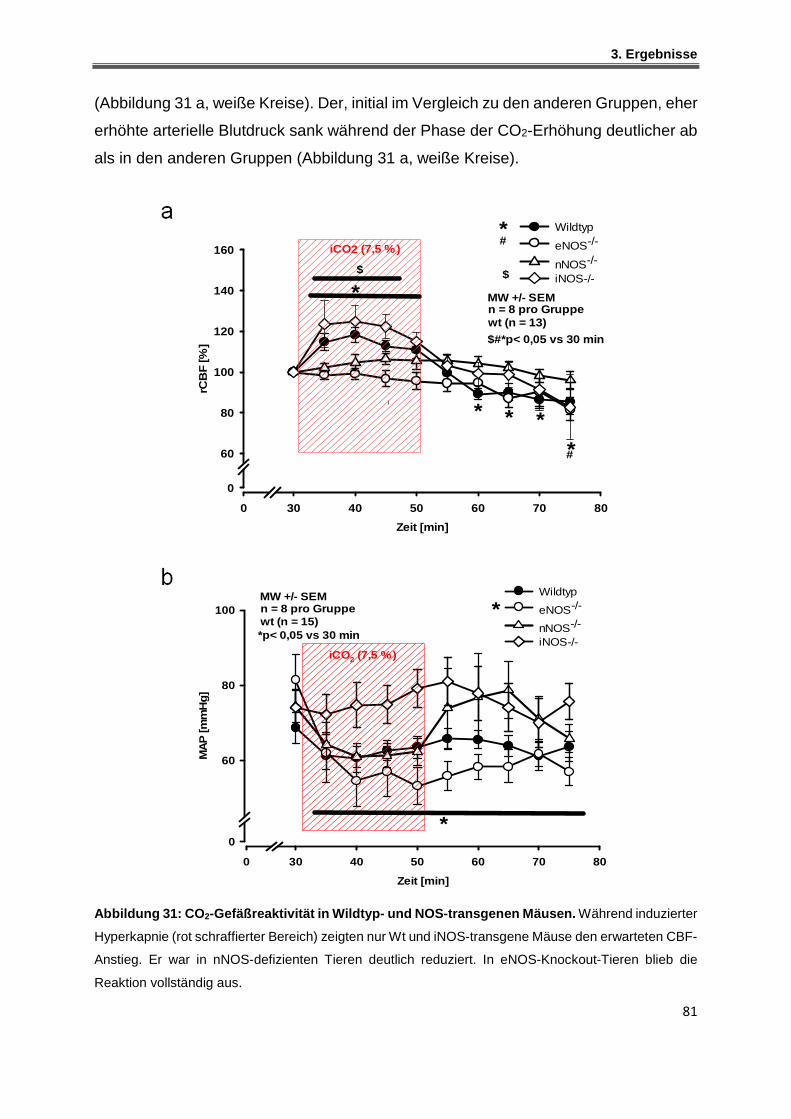
3. Ergebnisse
81
(Abbildung 31 a, weiße Kreise). Der, initial im Vergleich zu den anderen Gruppen, eher
erhöhte arterielle Blutdruck sank während der Phase der CO2-Erhöhung deutlicher ab
als in den anderen Gruppen (Abbildung 31 a, weiße Kreise).
Abbildung 31: CO 2-Gefäßreaktivität in Wildtyp- und NOS-transgenen Mä usen. Während induzierter
Hyperkapnie (rot schraffierter Bereich) zeigten nur Wt und iNOS-transgene Mäuse den erwarteten CBF-
Anstieg. Er war in nNOS-defizienten Tieren deutlich reduziert. In eNOS-Knockout-Tieren blieb die
Reaktion vollständig aus.
Zeit [min]
0 30 40 50 60 70 80
rCB
F [%
]
0
60
80
100
120
140
160
Wildtyp
eNOS-/-
nNOS-/-
iNOS-/-
iCO2 (7,5 %)
#
*#
MW +/- SEMn = 8 pro Gruppewt (n = 13)
$#*p< 0,05 vs 30 min
$ $
**
*
* *
Zeit [min]
0 30 40 50 60 70 80
MA
P [m
mH
g]
0
60
80
100
Wildtyp
eNOS-/-
nNOS-/-
iNOS-/-iCO2 (7,5 %)
MW +/- SEMn = 8 pro Gruppewt (n = 15)*p< 0,05 vs 30 min
*
*
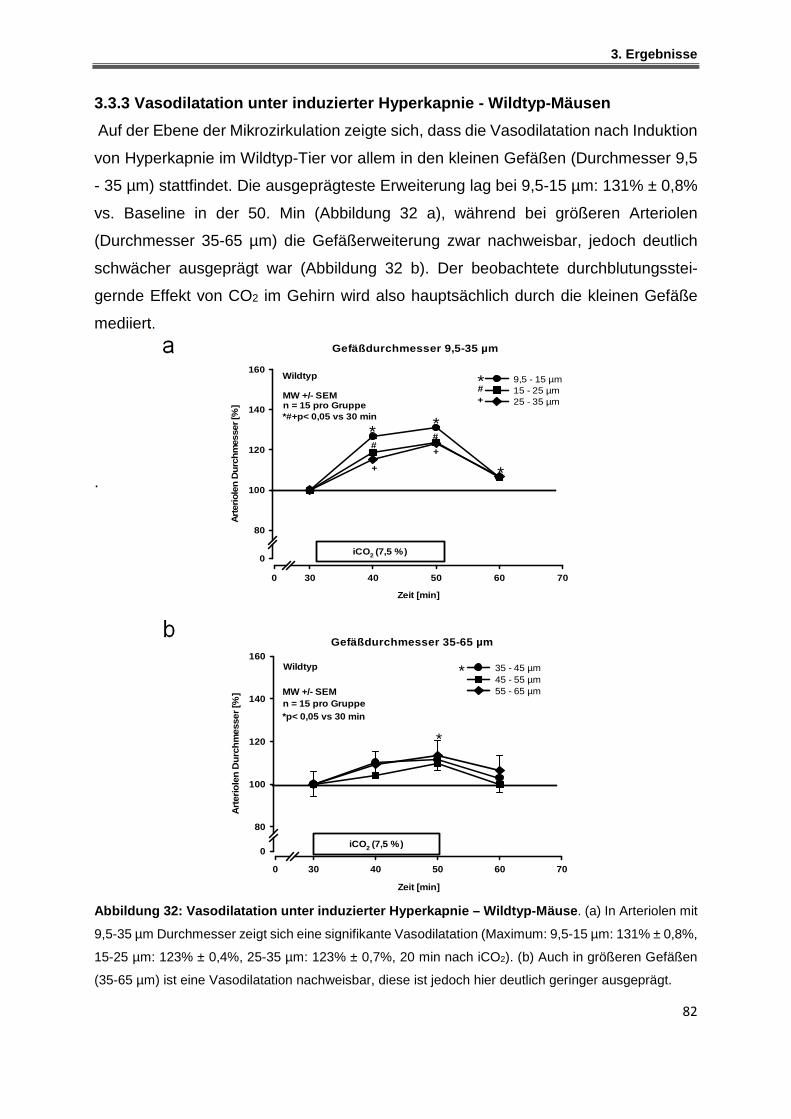
3. Ergebnisse
82
3.3.3 Vasodilatation unter induzierter Hyperkapnie - Wildtyp-Mäusen
Auf der Ebene der Mikrozirkulation zeigte sich, dass die Vasodilatation nach Induktion
von Hyperkapnie im Wildtyp-Tier vor allem in den kleinen Gefäßen (Durchmesser 9,5
- 35 µm) stattfindet. Die ausgeprägteste Erweiterung lag bei 9,5-15 µm: 131% ± 0,8%
vs. Baseline in der 50. Min (Abbildung 32 a), während bei größeren Arteriolen
(Durchmesser 35-65 µm) die Gefäßerweiterung zwar nachweisbar, jedoch deutlich
schwächer ausgeprägt war (Abbildung 32 b). Der beobachtete durchblutungsstei-
gernde Effekt von CO2 im Gehirn wird also hauptsächlich durch die kleinen Gefäße
mediiert.
.
Abbildung 32: Vasodilatation unter induzierter Hype rkapnie – Wildtyp-Mäuse . (a) In Arteriolen mit
9,5-35 µm Durchmesser zeigt sich eine signifikante Vasodilatation (Maximum: 9,5-15 µm: 131% ± 0,8%,
15-25 µm: 123% ± 0,4%, 25-35 µm: 123% ± 0,7%, 20 min nach iCO2). (b) Auch in größeren Gefäßen
(35-65 µm) ist eine Vasodilatation nachweisbar, diese ist jedoch hier deutlich geringer ausgeprägt.
Gefäßdurchmesser 35-65 µm
Zeit [min]
0 30 40 50 60 70
Arte
riole
n D
urch
mes
ser [
%]
0
80
100
120
140
16035 - 45 µm45 - 55 µm55 - 65 µm
Wildtyp *MW +/- SEMn = 15 pro Gruppe*p< 0,05 vs 30 min
iCO2 (7,5 %)
*
Gefäßdurchmesser 9,5-35 µm
Zeit [min]
0 30 40 50 60 70
Arte
riole
n D
urch
mes
ser [
%]
0
80
100
120
140
1609,5 - 15 µm15 - 25 µm25 - 35 µm
Wildtyp
MW +/- SEMn = 15 pro Gruppe*#+p< 0,05 vs 30 min
iCO2 (7,5 %)
* *
*
##
#
+
+
*
+
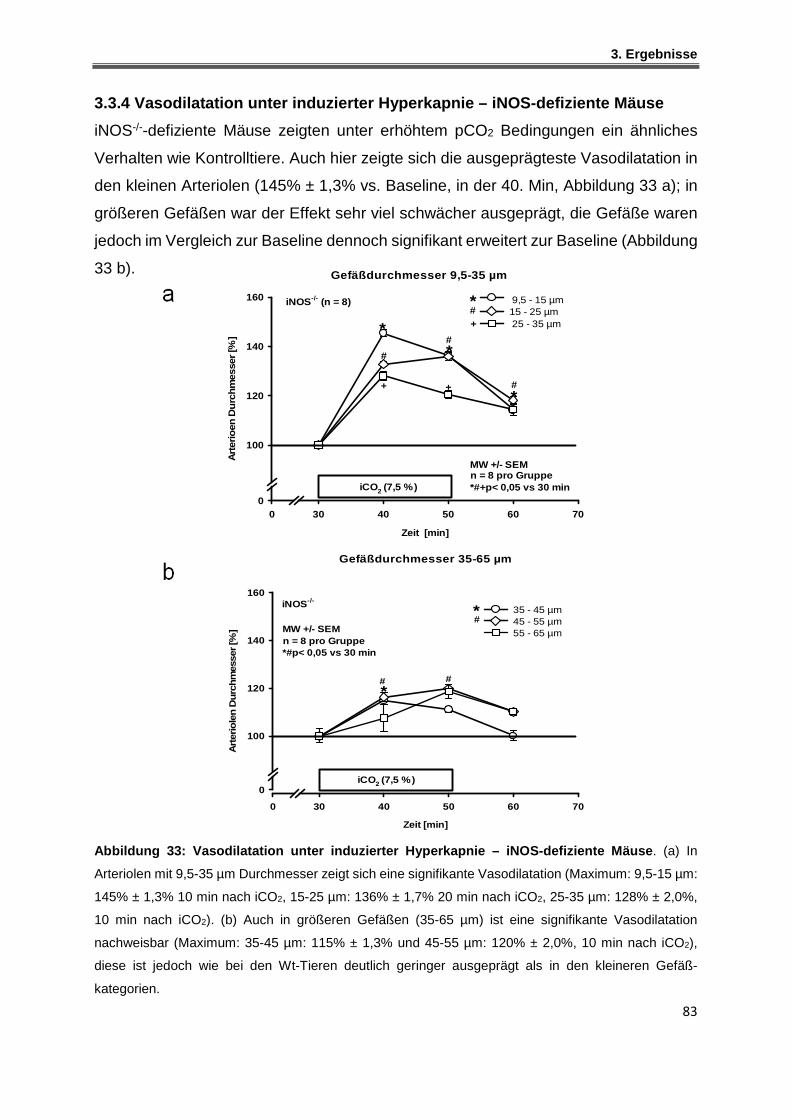
3. Ergebnisse
83
3.3.4 Vasodilatation unter induzierter Hyperkapnie – iNOS-defiziente Mäuse
iNOS-/--defiziente Mäuse zeigten unter erhöhtem pCO2 Bedingungen ein ähnliches
Verhalten wie Kontrolltiere. Auch hier zeigte sich die ausgeprägteste Vasodilatation in
den kleinen Arteriolen (145% ± 1,3% vs. Baseline, in der 40. Min, Abbildung 33 a); in
größeren Gefäßen war der Effekt sehr viel schwächer ausgeprägt, die Gefäße waren
jedoch im Vergleich zur Baseline dennoch signifikant erweitert zur Baseline (Abbildung
33 b).
Abbildung 33: Vasodilatation unter induzierter Hype rkapnie – iNOS-defiziente Mäuse . (a) In
Arteriolen mit 9,5-35 µm Durchmesser zeigt sich eine signifikante Vasodilatation (Maximum: 9,5-15 µm:
145% ± 1,3% 10 min nach iCO2, 15-25 µm: 136% ± 1,7% 20 min nach iCO2, 25-35 µm: 128% ± 2,0%,
10 min nach iCO2). (b) Auch in größeren Gefäßen (35-65 µm) ist eine signifikante Vasodilatation
nachweisbar (Maximum: 35-45 µm: 115% ± 1,3% und 45-55 µm: 120% ± 2,0%, 10 min nach iCO2),
diese ist jedoch wie bei den Wt-Tieren deutlich geringer ausgeprägt als in den kleineren Gefäß-
kategorien.
Gefäßdurchmesser 9,5-35 µm
Zeit [min]
0 30 40 50 60 70
Arte
rioen
Dur
chm
esse
r [%
]
0
100
120
140
160 9,5 - 15 µm15 - 25 µm 25 - 35 µm
MW +/- SEMn = 8 pro Gruppe*#+p< 0,05 vs 30 min
iNOS -/- (n = 8)
iCO2 (7,5 %)
*
*
#
#
#
++
+
*
*#
Gefäßdurchmesser 35-65 µm
Zeit [min]
0 30 40 50 60 70
Arte
riole
n D
urch
mes
ser [
%]
0
100
120
140
160
35 - 45 µm45 - 55 µm55 - 65 µmMW +/- SEM
n = 8 pro Gruppe*#p< 0,05 vs 30 min
iNOS -/-
iCO2 (7,5 %)
*
*# #
#

3. Ergebnisse
84
3.3.5 Vasodilatation unter induzierter Hyperkapnie – nNOS-defiziente Mäuse
Die nNOS-/--Mäuse wiesen unter erhöhtem pCO2 (7,5 % iCO2) eine im Vergleich zur
Kontrollgruppe deutlich verminderte CO2-induzierte Vasodilatation auf (15-25 µm: p =
0,006 nach 10 min iCO2, p = 0,049 nach 20 min iCO2: und p = 0,002 10 min nach iCO2,
25-35 µm: p = 0,022 nach 10 min iCO2). Im Vergleich zu den iNOS-/-- Mäusen war
ebenso eine verminderte CO2-induzierte Vasodilatation zu erkennen (15-25 µm: p =
0,033 10 min nach iCO2). Nur die kleineren Arteriolen mit einem Durchmesser von 9,5-
25 µm wiesen eine geringe, jedoch im Vergleich zur Baseline signifikante
Vasodilatation auf (116% ± 1,0% bei Kategorie 1 und 108% ± 1,1% bei Kategorie 2,
Abbildung 34 a). Bei größeren Arteriolen (von 35-65 µm) war der vasodilatative Effekt
nahezu nicht nachweisbar (Abbildung 34 b). Der Anteil von nNOS an der CO2-
induzierten Vasodilatation erscheint also vor allem in kleineren Gefäßen wichtig.
Abbildung 34: Vasodilatation unter induzierter Hype rkapnie – nNOS-defiziente Mäuse . Die CO2-
bedingte zerebrale Vasodilatation war in nNOS-Knockout-Mäusen signifikant erhöht und deutlich
geringer ausgeprägt als in der Kontrollgruppe, vor allem im Kaliber 15-25 µm. (a) In Arteriolen mit 9,5-
25 µm Durchmesser zeigt sich eine geringe Vasodilatation (Maximum: 9,5-15 µm: 116% ± 2% 20 min
nach iCO2 und 15-25 µm: 108% ± 1,0% 10 min nach iCO2), während in größeren Gefäßen (b) eine
Vasodilatation nahezu komplett ausblieb.
Gefäßdurchmesser 9,5-35 µm
Zeit [min]
0 30 40 50 60 70
Arte
riole
n D
urch
mes
ser [
%]
0
80
100
120
140
1609,5 - 15 µm15 - 25 µm25 - 35 µm
nNOS -/-
iCO2 (7,5 %)
MW +/- SEMn = 8 pro Gruppe*#p< 0,05 vs 30 min
*
*
##
#
Gefäßdurchmesser 35-65 µm
Zeit [min]
0 30 40 50 60 70
Arte
riole
n D
urch
mes
ser [
%]
0
80
100
120
140
160 35 - 45 µm45 - 55 µm55 - 65 µm
nNOS -/-
MW +/- SEMn = 8 pro Gruppep< 0,05 vs 30 min
iCO2 (7,5 %)

3. Ergebnisse
85
3.3.6 Vasodilatation unter induzierter Hyperkapnie – eNOS-defiziente Mäuse
Die auffälligsten Veränderungen im Vergleich zur Kontrollgruppe zeigten sich, wie in
der CBF-Messung, auch auf der Ebene der Mikrozirkulation bei den eNOS-/--Mäusen.
Vor allem war die Reaktion kleiner Arteriolen der eNOS-/--Mäuse im Vergleich zur
Kontrollgruppe besonders verringert (15-25 µm: p = 0,005 nach 10 min iCO2, p = 0,049
nach 20 min iCO2: und p = 0,022 10 min nach iCO2). Auch im Vergleich zu iNOS-/--
Mäusen war der Gefäßdurchmesser deutlich geringer (9,5-15 µm: p = 0,05 nach 10
min iCO2; 15-25 µm: p = 0,001 nach 10 min iCO2, p = 0,038 nach 20 min iCO2; 25-35
µm: p = 0,028 nach 10 min iCO2). Bei größeren Arteriolen (von 35-65 µm) war der
vasodilatative Effekt bei eNOS-/--Mäusen auch verringert (35-45 µm: p = 0,026 nach
20 min iCO2 im Verglich zur Kontrolle und p = 0,003 nach 10 min iCO2 im Vergleich
zu iNOS-/--Mäusen). In den Arteriolen mit 25-35 µm Durchmesser kam es zu einer
geringfügigen Vasodilatation (117% ± 1,0%, Abbildung 35 a), die auch nur zu einem
Zeitpunkt (50 min) im Vergleich zur Baseline statistisch signifikant war. Der Anteil von
eNOS an der CO2-induzierten Vasodilatation erscheint also vor allem in kleineren
Gefäßen wichtig.
Abbildung 35: Vasodilatation unter induzierter Hype rkapnie – eNOS-defiziente Mäuse . Die CO2-
bedingte zerebrale Vasodilatation war deutlich geringer ausgeprägt als bei Wildtyp-Mäusen. Lediglich
in Gefäßen der Kategorie 3 kam es zu einer geringen Vasodilatation (a), In größeren Arteriolen (b) hat
iCO2 nur eine ganz schwache Wirkung auf die Gefäße.
Gefäßdurchmesser 35-65 µm
Zeit [min]
0 30 40 50 60 70
Art
erio
len
Dur
chm
esse
r [%
]
0
80
100
120
140
16035 - 45 µm 45 - 55 µm 55 - 65 µm
eNOS-/-
iCO2 (7,5 %)
MW +/- SEMn = 8 pro Gruppep< 0,05 vs 30 min
Gefäßdurchmesser 9,5-35 µm
Zeit [min]
0 30 40 50 60 70
Art
erio
len
Dur
chm
esse
r [%
]
0
80
100
120
140
1609,5 - 15 µm15 - 25 µm25 - 35 µm
eNOS-/-
iCO2 (7,5 %)
MW +/- SEMn = 8 pro Gruppe*p< 0,05 vs 30 min
*
*

3. Ergebnisse
86
3.3.7 Vasodilatation unter induzierter Hyperkapnie der NOS-Isoformen in klei-
nen Arteriolen
Abbildung 36 zeigt für jeden Genotyp die Gefäßkategorie mit der ausgeprägtesten
vasodilatatorischen Reaktion, d. h. Arteriolen mit einem Durchmesser zwischen 9,5 -
15 µm. Es ist deutlich erkennbar, dass die Arteriolen der Wildtyp- und iNOS-/--Gruppe
im Vergleich zu nNOS- und vor allem eNOS-transgenen Tieren eine deutlich
ausgeprägtere CO2-Reaktivität zeigen.
Abbildung 36: Einfluss der NOS- auf die CO2-induzie rte Vasodilatation, Gefäße mit einem
Durchmesser von 9,5-15 µm.
3.3.8 Mögliche Rolle der NOS bei der Formation von Mikrovasospasmen
Bei der Untersuchung der zerebralen Mikrozirkulation im Rahmen der Untersuchung
der CO2-Reaktivität fielen bereits unter physiologischen Bedingungen charakteris-
tische Gefäßveränderungen bei den NOS-transgenen Mäusen auf: die Arteriolen
wiesen segmentale Konstriktionen auf, die an die Mikrovasospasmen, wie sie nach
Gefäßdurchmesser 9,5-15 µm
Zeit [min]
0 30 40 50 60 70
Arte
riole
n D
urch
mes
ser [
%]
0
100
120
140
160 Wildtyp
eNOS-/-
nNOS-/-
iNOS-/-
iCO2 (7,5 %)
* *
#
#
MW +/- SEMn = 8 pro Gruppewt (n = 15)*+#p< 0,05 vs 30 min
#
*
#
*
+
+

3. Ergebnisse
87
Subarachnoidalblutung auftreten, erinnerten. Beispielbilder zeigt Abbildung 37. Neben
den Versuchsgruppen ist in Abbildung 37 f beispielhaft, wie der Befund drei Stunden
nach SAB zeigt. Daraufhin wurde das IVM-Protokoll angepasst und die zerebrale
Mikrozirkulation der NOS-defizienten Tiere basierend auf den Versuchen nach SAB
untersucht und ausgewertet. Für die Analyse wurden lediglich die ersten 10 min von
der Baseline (insgesamt sind es 30 min Baseline) herangezogen.
Abbildung 37: Beginnend mit einer Gesamtübersicht des zerebralen Mikrogefäßbaumes durch IVM,
konnte mit einer 20 fachen Vergrößerung (Immersionsobjektiv) Arteriolen, 9,5-35 µm, besichtigt und
analysiert werden. Die Häufigkeit von MVS ergab sich wie folgt: SAB > eNOS -/- > nNOS-/- > iNOS-/-. Die
Wildtyp-Mäuse dienten als Kontrolle. Die weißen Pfeile weisen auf MVS hin.
a b
c d
e f

3. Ergebnisse
eNOS-/--Mäuse wiesen insbesondere eine signifikante Anzahl von MVS in Arteriolen
auf, mit Durchmessern von 15-25 µm. Dies gilt auch nNOS-/--Mäuse. Im Vergleich zu
Wt-Tieren, die dieses Phänomen nur sehr vereinzelt zeigen, handelt es sich um einen
signifikanten Unterschied (Abbildung 38 a). Die Ausprägung erinnert an das
Vorkommen von Spasmen nach SAB, ist jedoch etwas geringer ausgeprägt. Die
vorkommenden mikroarteriolären Konstriktionen sind – ebenfalls wie beim posthä-
morrhagischen MVS – hauptsächlich leicht- bis mittelgradig (10-40%).
Abbildung 38: Vorkommen und Ausprägung von Mikrovasospasmen bei N OS-defizienten
Mäusen. ( a) eNOS-/-- und weniger stark ausgeprägt die nNOS-defizienten Mäuse zeigen eine signifi-
kante Anzahl von MVS v.a. in den kleinen Gefäßarteriolen (ähnlich wie bei SAB). (b) Die Spasmen sind,
ebenfalls wie nach SAB, vor allem gering bis mittelgradig (10-40%).
Konstriktion [%]
10-20 21-30 31-40 41-50
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
1
2
3
4
5
6
7Wildtyp
eNOS-/-
nNOS-/-
iNOS-/-
*
*
**
*
MW +/- SEMn = 8 pro Gruppewt (n = 15)*p< 0,05 vs Wildtyp
Arteriolen Durchmesser [µm]
9,5-15 15-25 25-35 35-45
Mik
rova
sosp
asm
en p
ro M
aus
[n]
0
2
4
6
8
10Wildtyp
eNOS-/-
nNOS-/-
iNOS-/-
*
* *
MW +/- SEMn = 8 pro Gruppewt (n = 15)*p< 0,05 vs Wildtyp
*

4. Diskussion
4. Diskussion
4.1 Diskussion der Methoden
4.1.1 Auswahl der Versuchstiere
Nagetiere, insbesondere Mäuse, sind die am häufigsten verwendete Spezies in der
Schlaganfallforschung, da sie aufgrund ihrer schnellen Reproduktionsfähigkeit, sowie
ihrer kleinen Körpergröße kostengünstig und schnell gezüchtet werden können. Sie
haben ein robustes Herz-Kreislauf-System, das die komplikationslose Durchführung
von Langzeit-Narkosen ermöglicht. Ein Nachteil bei der Verwendung von Mäusen ist
die Schwierigkeit der Durchführung von operativen Eingriffen. Das Problem kann
jedoch durch die Anwendung mikrochirurgischer Techniken sowie die Verwendung
entsprechend dimensionierter Instrumente (Mikroskop, Mikrosonden) behoben
werden. Ein weiterer Nachteil ist, dass Mäuse kein gyrencephales Gehirn besitzen,
was die Übertragbarkeit mancher Ergebnisse auf den Menschen nicht sicher
gewährleistet. Ein großer Vorteil der Maus ist jedoch die Möglichkeit zur genetischen
Manipulation durch die Verwendung von transgenen Mausmodellen, z.B. die
Züchtung von Mäusen mit spezifischen Gendeletionen (Knock-Out-Maus). Wichtige
Vorteile der Verwendung von Knockoutmäusen sind, dass das Deaktivieren eines
Gens eine umfassendere und präzisere Ausschaltung ermöglicht als eine
medikamentöse Blockade, zudem ist eine genetische Manipulationen oft der einzige
Weg zur Bestimmung der genauen Funktion eines Gens.388 NOS-Inhibitoren weisen
eine geringe Selektivität bzgl. der verschiedenen NOS-Isoformen auf, aufgrund der
schlechten Interaktionsfähigkeit der Inhibitoren mit der Arginin-Bindungsstelle.389 So
ist die inhibitorische Potenz von 1400 W, NAP und L-VNIO für nNOS und eNOS
schwach ausgeprägt.389-393 Lediglich für iNOS gibt es genügend selektive Inhibitoren,
wie z.B. 1400 W, Aminoguanidin, Isothiourea. Ein weiterer Grund für die Verwendung
eines Mausmodells in der vorliegenden Studie ist, dass die dünne Dura Mater und der
schmale Subarachnoidalraum der Maus Epifluoreszenzmikroskopie bei intakter Dura
erlaubt.127 Dies ist von Vorteil, da hierdurch operationsbedingte Artefakte minimiert
werden. Bei größeren Versuchstieren wie z.B. Ratten ist dies nicht möglich.394 Hier
muss die Dura Mater entfernt werden und das Gehirn während der Messungen
konstant mit einem künstlichem Liquor gespült werden.394 Dies ist sehr störanfällig,
außerdem besteht die Gefahr der Entwicklung eines Hirnödems.

4. Diskussion
90
4.1.2 Anästhesie
Um Aussagen über die zerebrale Mikrozirkulation und insbesondere über dessen
Veränderungen machen zu können, muss sichergestellt werden, dass die
Untersuchungen unter physiologischen Bedingungen stattfinden. Veränderungen von
systemischem Blutdruck, Körpertemperatur, pCO2 und pO2 können zu einer
Beeinflussung der zerebralen Durchblutung führen. Deswegen ist wichtig, dass diese
Parameter im Laufe eines Versuchs engmaschig überwacht und kontrolliert werden.
In der vorliegenden Studie wurden die Versuchstiere mechanisch beatmet, die
Temperatur konstant gehalten, die Atemparameter wurden kontinuierlich und
nichtinvasiv mittels Mikrokapnometrie überwacht und angepasst. In einer vorange-
gangenen Studie362 konnte gezeigt werden, dass mit dem in der Studie verwendeten
Setup auch über eine längere Narkosedauer physiologische Parameter eingehalten
werden können. Mehrfache Blutentnahmen zur Blutgasanalyse, welche aufgrund des
geringen Blutvolumens der Maus schnell zur Hypotension führen können, werden so
vermieden. Auch die Narkoseform kann die Versuchsergebnisse signifikant
beeinflussen. Isofluran, das am häufigsten tierexperimentell meist verwendete volatile
Betäubungsmittel395,396 entfaltet in vielerlei Hinsicht eine ähnliche Wirkung wie
Halothan,397 das vasodilatativ wirkt.398 Isofluran führt dosisabhängig zu einer globalen
CBF-Erhöhung sowie zum systemischen Blutdruckabfall und es beeinträchtigt die
zerebrale Autoregulation.399 Isofluran kann auch neuroprotektiv wirken400,401 indem es
nach zerebraler Ischämie die Infarktgröße,402 die neuronale Apoptose (durch
Hochregulierung von antiapoptotischen Faktoren wie MAP-Kinase),403-405 die
Exzitotoxizität,29,183 das Hirnödem,406 und das neurologische Defizit vermindert406
sowie die Öffnung der Blut-Hirn-Schranke verringert.405 Ähnliche neuroprotektive
Effekte lassen sich auch nach SAB nachweisen.407,408 Da auch andere volatile
Anästhetika wie Sevofluran, Desfluran und Enfluran ähnliche Wirkungen wie Isofluran
ausüben,395 wurde in der vorliegenden Studie für die intravitalmikroskopischen
Untersuchungen, kein volatiles Anästhetikum, sondern eine Injektionsanästhesie aus
Medetomidin, Fentanyl und Midazolam (MMF) verwendet. Die intraperitoneale
Applikation dieser Kombination bei Nagern ist ein weitverbreitetes Verfahren in der
Veterinärmedizin, das gut steuerbar ist und eine große therapeutische Breite aufweist.
Entscheidend für diese Arbeit war allerdings, dass dieses Narkoseprotokoll keinen,
bzw. nur einen geringen Einfluss auf die zerebrale Mikrozirkulation hat.362 Auch für die

4. Diskussion
91
MMF-Narkose gibt es einige wenige Hinweise auf eine neuroprotektive Wirkung; so
wurde gezeigt, dass es nach Retinaischämie unter MMF zu einer Zunahme der Zahl
überlebender Ganglienzellen um etwa 40% des Ausgangswertes kommt,409 zudem
wurde berichtet, dass die Narkose mit Medetomidin und alpha-2-Adrenorezeptor-
Agonisten die Größe der Nekrose nach fokaler zerebraler Ischämie verringern
kann,410,411 Midazolam scheint aber eine geringe neuroprotektive Wirkung zu haben.412
Die dargestellten Nebenwirkungen von MMF sind jedoch im Vergleich zu volatilen
Anästhetika und anderen Injektionsnarkotika wie Propofol (fördert Blutdruckabfall und
Atemdepression) und Ketamin (Blutdruck- und Herzfrequenzsteigernd) als gering
einzuschätzen.
4.1.3 Beatmung der Versuchstiere
Das in der aktuellen Studie verwendete Narkoseprotokoll beinhaltet ein Opiat. Da
Opiate atemdepressiv wirken, mussten die Versuchstiere intubiert und kontrolliert
beatmet werden.413 Die kontrollierte Beatmung hat Auswirkungen auf den Säure-
Basen-Haushalt und die Blutgase des Versuchstieres. Die Beatmung muss daher so
gesteuert werden, dass die Zielgrößen sich im physiologischen Bereich befinden, da
Abweichungen vom Normbereich Auswirkungen auf die zerebrale Mikrozirkulation
haben würden. Zielgrößen, die während des Versuchs besonders beachtet werden
müssen sind Sauerstoffpartialdruck (pO2), und Kohlendioxidpartialdruck (pCO2), der
mittels Mikrokapnometrie gemessen wird.362,414,415 Das Hauptziel der mechanischen
Beatmung ist, den Erhalt des physiologischen paCO2 im Blut, sowie eine ausreichende
Gewebeoxygenierung für das Versuchstier zu garantieren.
4.1.4 Messung der zerebralen Durchblutung
Die Laser-Doppler-Flowmetrie ist eine nicht-invasive Methode zur Messung der
lokalen zerebralen Durchblutung. Die Technik basiert auf dem Prinzip, dass
monochromatisches Licht seine Wellenlänge ändert, wenn es von bewegten Objekten
(wie z.B. Erythrozyten) reflektiert wird (Doppler-Effekt). Die Menge und die
Frequenzänderung des reflektierten Lichts sind abhängig von der Anzahl und der
Geschwindigkeit der im Gewebe zirkulierenden Blutzellen. So kann mit der LDF

4. Diskussion
92
zuverlässig jede dynamische Veränderung der zerebralen Durchblutung detektiert
werden.416-421 Insgesamt ist die LDF für die Studie angemessen, weil sie a) nicht-
invasiv ist b) eine zusätzliche Applikation von Tracern (z.B. Radionuklide) nicht
erforderlich ist und c) ICP- und IVM-Messungen gleichzeitig zur CBF-Messung
durchgeführt werden können. Zudem erfordern die Untersuchungen der vorliegenden
Studie, dass schnelle kortikale Durchblutungsveränderungen verfolgt werden können;
hierbei sind vor allem Änderungen im Vergleich zur Baseline von Interesse. Absolute
Werte sind daher erforderlich. Nachteile der LDF-Methode liegen in der begrenzten
Eindringtiefe. Andere Techniken, die für die Messung der zerebralen Durchblutung
verwendet werden können, sind die Autoradiographie, die absolute CBF-Werte
ermöglicht. Diese Methode ist allerdings technisch sehr aufwendig und erlaubt nur die
Messung einzelner Messzeitpunkte da das Versuchstier für die Messung getötet
werden muss.422 Alle anderen Methoden zur Bestimmung der zerebralen Durch-
blutung, wie z.B. Perfusions-CT, Magnetresonanztomographie, oder Thermodilution,
können wegen mangelnder Auflösung oder Größe der Sonden nicht bei Mäusen
eingesetzt werden.
4.1.5 Wahl des SAB-Modells
Die Subarachnoidalblutung kann im Mausmodell entweder durch ein- oder mehrmalige
Injektion von Blut in die Cisterna Magna,423-425 Perforation einer intracisternalen
Vene426 oder Faden-Perforation der A. cerebri media127,427,428 vorgenommen werden.
Das Cisterna-Magna-Injektions-Modell und das Venen-Dissektions-Modell können
schnell durchgeführt werden, sind gut reproduzierbar und führen zu einem akuten
Anstieg des ICP sowie zu einer Abnahme der CBF.423-426 Diese Veränderungen sind
jedoch im Gegensatz zu den Vorgängen nach SAB im Patienten innerhalb kurzer Zeit
nach Injektion reversibel. Negativ für die vorliegende Studie ist die bei beiden Modellen
notwendige Verletzung der Dura mater, die die Intravitalmikroskopie behindern oder
verfälschen kann. Beiden Modellen fehlt jedoch der plötzliche, heftige Blutungsbeginn
und der endotheliale Schaden, die charakteristisch für die aneurysmatische SAB
sind.267 Daher ist das MCA-Perforations-Modell aktuell das geeigneste Modell zur
Erforschung der Pathophysiologie der SAB,427 da es die Folgen einer aneurys-
matischen Subarachnoidalblutung (verzögerte zerebralen Vasospasmen,429-434 frühe

4. Diskussion
93
Vasospasmen in der Mikrozirkulation,133,184,435 neurologische Dysfunktionen,429,431-
433,436,437 Hirnödem, 436,437 Hydrocephalus,438,439 eine klinisch relevante Mortalität von
ungefähr 30%,127,429,436,437 ICP-Anstieg127,184 und CBF-Abfall127,184) am besten
reproduziert und eine gute Standardisierbarkeit ermöglicht.427 Durch eine gleichzeitige
und kontinuierliche Messung von ICP und CBF am MCA-Perforations-Modell wird
kontrolliert, dass die Subarachnoidalblutung standardisiert ausgelöst werden kann.127
Außerdem ist für unsere Studie die Tatsache von Vorteil, dass keine Öffnung der Dura
Mater nötig ist und daher die Intravitalmikroskopie gut durchgeführt werden kann.
Nachteile dieses Modells sind das nicht direkt kontrollierbare austretende Blutungsvo-
lumen und die komplexe OP, die viel Erfahrung verlangt.
4.1.6 Intravitalmikroskopie
Die Visualisierung des Plasmas und somit der zerebralen pialen Mikrogefäße erfolgte
durch repetitive i.v. Injektionen von Fluoresceinisothiocyanat-Dextran (FITC-
Dextran).363,440 Ein Vorteil der i.v. Farbstoff-Injektion ist, dass eine Verletzung der Blut-
Hirn-Schranke durch Extravasation des Farbstoffes sofort offensichtlich ist.
Versuchstiere, bei denen dies der Fall war, wurden von der Analyse ausgeschlossen,
da bei traumatischer Präparation eine artifizielle Beeinflussung der Versuchs-
ergebnisse nicht auszuschließen bzw. wahrscheinlich ist. Eine Alternative zur
Verwendung eines i.v. Farbstoffs wäre eine Maus (tie2GFP-Maus), das Green-
Fluorescence-Protein (GFP) in Endothelzellen exprimiert.441,442 In so einer Maus
könnte eine Hämodilution durch Injektion des Farbstoffs sowie mögliche systemische
oder lokale Effekte des Farbstoffs vermieden werden. Der Nachteil einer tie2GFP-
Maus in einem SAB-Model könnte in der zusätzlichen genetischen Manipulation
liegen, die den natürlichen Zustand der Mikrogefäße des Versuchstieres beeinflussen
könnte sowie deren im Vergleich zum Farbstoff geringere Verfügbarkeit.
Die Eindringtiefe des Intravitalmikroskops liegt bei 50 µm. Damit können piale Gefäße,
die für unsere Studie relevant waren, mit hoher optischer und zeitlicher Auflösung
untersucht werden. Nichtinvasive, bildgebende Verfahren, wie die Computertomo-
graphie oder Magnetresonanztomographie, die in der klinischen Anwendung weit
verbreitet sind, und auch in Tierversuchen zum Einsatz kommen, bieten zwar eine
zunehmend bessere räumliche Auflösung, jedoch erlaubt nur die Intravitalmikroskopie

4. Diskussion
94
eine hochauflösende Analyse der Veränderung der zerebralen Mikrozirkulation in
Echtzeit.
4.2. Diskussion der Ergebnisse
Da viele Patienten nach SAB in der frühen und nur wenige in der verzögerten
Hirnschädigungsphase, in der Spasmen der großen Hirngefäße auftreten, sterben, ist
das Ziel der vorliegenden Arbeit zu untersuchen inwiefern die zerebrale
Mikrozirkulation an der Frühmortalität nach SAB beteiligt ist und welche Mechanismen
dieser Schädigung zugrundeliegen. Fast zwei Drittel der Todesfälle (ca. 61%) treten
in den ersten 48 Stunden nach SAB auf.443 25% der SAB Patienten443 erreichen nicht
das Krankenhaus während 33-36% der SAB-Patienten behandelt werden aber
dennoch an den Folgen der frühen Hirnschädigung nach SAB sterben.443,444 Der
pathologische Zustand in den ersten 72 Stunden nach SAB wird durch Beeinträch-
tigung der zerebralen Autoregulation,132 Störung der Blut-Hirn-Schranke,132 Aktivie-
rung von Entzündungswegen und Apoptose,132 Exzitotoxizität und oxidativer Stress,
gestörte Kalzium Homöostase, Vasospasmen132 und durch Dysfunktion der zerebralen
Mikrozirkulation182-184 charakterisiert. Diese Vorgänge werden zu gewissen Zeitpunk-
ten in der frühen Hirnschädigungsphase durch Blut, und dessen Bestandteile, durch
Aktivierung von Thrombozyten und durch transiente zerebrale Ischämie hervorgeru-
fen.133-136,144
4.2.1 Hämodynamische Veränderungen nach SAB
Nach Auslösung der SAB mittels dem MCA-Perforations-Modell zeigte sich ein
massiver intrakranieller Druckanstieg (von 3 auf 100 mmHg) mit einem gleichzeitigen
starken Abfall der regionalen zerebralen Durchblutung um ca. 85 % im Vergleich zum
Ausgangswert.127 Der intrakranielle Druckanstieg stoppte als dieser den systemi-
schen-arteriellen Blutdruck erreichte.127,130 Durch die nun beginnende Verteilung des
Blutes im gesamten Subarachnoidalraum fiel der ICP innerhalb von 5-8 min bis auf ca.
20 mmHg, während die zerebrale Durchblutung sich auf bis zu 80% des Ausgangs-
wertes erholte. Auf diesem Niveau hielten sich der ICP und die CBF auch danach noch
Konstant. Ähnliche Werte werden auch bei SAB-Patienten die eine Re-Blutung erlitten

4. Diskussion
95
beobachtet. Dies legt nahe dass es bei der Maus und beim Menschen zu einem
vergleichbaren Zeit- und Druckverlauf kommt.445
4.2.2 Effekt von iNO und den Endothelin-Rezeptor-Bl ocker auf den systemi-
schen Blutdruck nach SAB
Eine arterielle Hypotension ist mit einer deutlichen Verschlechterung der Prognose von
SAB-Patienten vergesellschaftet.446 Daher der MAP in dem Bereich zwischen 70 und
110 mmHg gehalten werden sollte.447 Die Verabreichung der NO-Vorstufe L-
Arginin448,449 oder des NO-Donators S-Nitrosoglutathion (GSNO),140 rufen eine
deutliche Reduktion des Hirnschadens nach experimenteller SAB hervor. Allerdings
verursacht GSNO einen drastischen, wenn auch vorübergehenden, Abfall des
systemischen Blutdrucks.140 Dieses Phänomen tritt auch nach der Anwendung von L-
Arginine auf.450,451 Dagegen hat die NO-Inhalation, ein zugelassenes Verfahren zur
Behandlung von pulmonaler Hypertonie, weder am Versuchstier noch beim Menschen
eine nachteilige Wirkung auf den systemischen Blutdruck.452-454,270,455,456,457 Hohe iNO-
Dosen wirken jedoch Blutdrucksenkend,458,459 vor allem ab Konzentrationen von 300
ppm.459 In unserer Studie, in der maximal 50 ppm inhaliertes NO appliziert wurden,
konnte kein Effekt auf den systemischen Blutdruck nachgewiesen werden. Vielmehr
tendiert der MAP bei den Kontrolltieren (mit induzierter SAB) um ca. 10 mmHg geringer
zu sein als bei der iNO-Gruppe.
Clazosentan ist ein selektiver Endothelin-Rezeptor-Blocker,373-375 der experimentell
und klinisch erfolgreich zur Behandlung von Spätvasospasmen nach SAB eingesetzt
wurde.373,377 Nach der CONSCIOUS-1-Studie waren die Nebenwirkungen, die bei der
Behandlung durch Clazosentan auftraten, u.a. Hypotonie.354 Die ET-1-Rezeptor-
Blockade durch Clazosentan soll experimentell jedoch eine kontinuierliche
Verringerung der zerebralen Durchblutung verhindern.379 In unsere Studie zeigte die
ET-Rezeptor-Blockade bei gesunden Wildtyp-Mäusen, innerhalb von drei Stunden,
keinen signifikanten Effekt auf den systemischen Blutdruck373 oder den CBF. Auch
unterscheidet sich nach SAB der MAP der mit Clazosentan behandelten Tieren nicht
von den Kontrolltieren.

4. Diskussion
96
4.2.3 Veränderung der zerebralen Mikrozirkulation n ach SAB
4.2.3.1 Mikrovasospasmen
Der Vasospasmus und die daraus resultierende zerebrale Ischämie, sind die meist
gefürchteten Komplikationen nach SAB. Frühere Untersuchungen waren vor allem auf
den verzögerten zerebralen Makrovasospasmus fokussiert. Jedoch ist seit einigen
Jahren bekannt, dass in den ersten 72 Stunden nach SAB auch die zerebrale
Mikrozirkulation beeinträchtig ist und damit zerebrale Mikrovasospasmen auftre-
ten.182-184 Aufgrund der technischen Möglichkeiten werden in der Klinik nur qualitative
Aussagen über den Durchmesser der großen Gefäße, wie der A. cerebri media und
der A. cerebri anterior, getroffen. Eine Reduktion des Gefäßdurchmessers in einem
dieser Gefäße kann jedoch durch eine ausreichende Kolateralisierung kompensiert
werden. Die Frage, ob nicht auch die kleinen Widerstandsgefäße in Form von
Arteriolen von Mikrovasospasmen betroffen sind, stand immer zur Debatte.460-462
Gestützt wurde dies durch die Tatsache, dass der diagnostizierte Vasospasmus der
großen Gefäße keinen positiven prädiktiven Wert bzgl. der neurologischen Defizite
nach SAB aufweist. 2003 konnten von Uhl et al. erstmals der Nachweis erbracht
werden, dass Mikrovasospasmen auch beim Menschen auftreten.182 Am Patienten war
es jedoch nicht möglich, zu verschiedenen Zeitpunkten und an denselben Stellen
qualitative und quantitative Aussagen über die entstandenen Mikrovasospasmen zu
treffen. Somit ergab sich die Aufgabe, für die nähere Erforschung der Mikrovaso-
spasmen ein Tiermodell zu etablieren. So konnten wir 2012 das erste Mal die Existenz
von Mikrovasospasmen nach experimenteller SAB nachweisen.184 Diese Befunde
konnten in der aktuellen Arbeit reproduziert werden: 3-5 Stunden nach SAB treten
akute Vasokonstriktionen überwiegend in Arteriolen geringen Durchmessers (9,5-25
µm) auf. Kapillaren hingegen wiesen fast keine Mikrovasospasmen auf, während
Venolen überhaupt keine Vasospasmen aufwiesen. Die überwiegende Mehrzahl der
arteriolären Mikrovasospasmen ist mittelgradig, zwischen 10-40%. Freies Hämoglobin
ist eine mögliche Ursache für diese Mikrovasokonstriktion.463 Abbildung 39 stellt
schematisch die Ursachen der Mikrovasospasmenbildung dar.

4. Diskussion
97
Abbildung 39: Mögliche Ursachen zur Mikrovasospasme n-Bildung
4.2.3.2 Effekt von iNO
Stickstoffmonoxid (NO) ist ein endogener Vasodilator der direkt auf die glatten
Gefäßmuskelzellen wirkt und zur Gefäßrelaxierung führt.282 Damit es zur
Gefäßrelaxierung kommt, wird unter physiologischen Bedingungen der NO-sGC-
cGMP-Signaltrans-duktionsweg aktiviert.208 Nach SAB ist der NO/NOS-Weg
gestört137,140,261 und dies könnte für die Genese und Entwicklung der frühen
Hirnschädigung entscheidend sein.464-471 Blutabbauprodukte, vor allem das
Hämoglobin, sind für die Depletion von NO aus dem zerebrovaskulären System
verantwortlich. Die NO-Depletion verursachte eine Verringerung der Expression von
sGC,472 sowie ein Anstieg der Hydrolyse-Rate von cGMP.473 Diese Beobachtungen
stehen im Einklang mit der Beeinträchtigung der NO vermittelten Vasodilatation nach
SAB.473 In der vorliegenden Studie soll der durch SAB verursachte NO-Mangel durch
iNO wieder hergestellt werden. Drei bis fünf Stunden nach SAB, weist iNO einen
vasodilatativen Effekt auf spastische Arteriolen (9,5-25 µm) auf. Der vasodilatatorische

4. Diskussion
98
Effekt findet sofort nach Beginn der iNO statt. Nach 40 Minuten NO-Inhalation waren
die MVS fast komplett verschwunden und 30 Minuten nach iNO-Stopp war die Zahl
der Vasospasmen praktisch gleich gering geblieben. Während iNO verschwinden vor
allem MVS, die einen Konstriktionsgrad zwischen 10-40% aufweisen. 30 Minuten nach
iNO-Stopp blieb die Anzahl der leichten und mittelstarken MVS konstant gering. Durch
unsere Studie erkennt man, dass die Beeinträchtigung der zerebralen Mikrozirkulation,
mit Hilfe des inhalierten NO (50 ppm) aufgehoben werden kann. Das Inhalieren von
50 ppm NO hat keine negative Auswirkung auf den systemischen Blutdruck.270 Frühere
Untersuchungen haben außerdem gezeigt, dass die NO-Inhalation nach SAB die
Mortalität und das Hirnödem senkt.127,270 Die vorliegende Studie deutet darauf hin,
dass diese Neuroprotektion durch eine Verbesserung der zerebralen Mikrozirkulation
durch Reduktion des Mikrovasospasmus verursacht wird. Deswegen ist die NO-
Inhalation eine vielverspre-chende Therapieoption in der frühen Phase nach
Subarachnoidalblutung weil es die Homöostase der zerebralen Mikrozirkulation
wiederherstellt.
4.2.3.3 Effekt der ET-1-Rezeptorblockade
ET-1 ist eines der stärksten Vasokonstriktoren in der zerebralen Zirkulation. Es wirkt
hauptsächlich durch Andocken an den ETA-Rezeptor.435,540 Clazosentan, ein selektiver
ETA-Rezeptor-Antagonist reduziert die Entwicklung von zerebralen Makrovasos-
pasmen nach SAB.474,475 In der aktuellen Studie zeigte die ET1-Rezeptorblockade
durch Clazosentan (10 mg/kg KG) einen präventiven Effekt auf die zerebrale
Mikrozirkulation. Die ET-1-Rezeptor-Blockade führte zur Erweiterung von zerebralen
Mikrogefäßen, vor allem von Arteriolen mit einem Kaliber zwischen 35-55 µm. Die
kleineren Arteriolen (9,5-35 µm) wiesen keine signifikante Gefäßrelaxierung auf. Eine
deutliche Abnahme von MVS findet sich vor allem in größere Arteriolen. 30 min nach
Beendigung der Clazosentan-Gabe bleiben die Anzahl der MVS mit einem
Konstriktionsgrad zwischen 21-30% deutlich reduziert. Auch eine Behandlung mit
einem Endothelin-Rezeptor-Antagonisten scheint die Homöostase der zerebralen
Mikrozirkulation wieder herstellen zu können. Der Effekt beschränkt sich im Vergleich
zu iNO allerdings auf die größeren Arteriolen.

4. Diskussion
99
4.2.4 Rolle der NOS-Isoformen in der zerebralen Mik rozirkulation nach SAB
Die Regulation der Durchblutungsverteilung zwischen verschiedenen Hirnarealen ist
entscheidend für die Kopplung von Gewebedurchblutung und Stoffwechsel. Auf der
lokalen Ebene ist bekannt, dass Stickstoffmonoxid eine Schlüsselrolle bei der
Modulation des mikrovaskulären Tonus und somit der lokalen Durchblutung des
Gehirns spielt.228,476 Stickstoffmonoxid tritt mit anderen lokalen vasoaktiven
Mediatoren (z.B. Prostaglandin und Endothelin) und mit dem neuronalen Netzwerk in
Wechselwirkung, die für die Festlegung des Gefäßtonus und Durchblutung verant-
wortlich sind. 246,534 Obwohl es drei verschiedene Isoformen der NO-Synthase (eNOS,
nNOS, iNOS) gibt, ist allgemein akzeptiert, dass die lokale Regulation des Gefäßtonus
hauptsächlich durch die Freisetzung von NO durch eNOS vermittelt wird. 534,477 In
dieser Arbeit sollte geklärt werden, welche NOS-Isoformen (endotheliale, neuronale
oder induzierbare), insbesondere in der zerebralen mikrovaskulären Regulation
beteiligt sind, und welche nach SAB beeinträchtigt wird.
Es ist bekannt, dass ein hyperkapnisches Milieu die NO Freisetzung aktiviert, und so
die selektive Relaxation zerebraler Gefäße bei gesunden Tieren und Menschen
induziert.313,478,479 Nach SAB reagierten die zerebralen Arteriolen jedoch nicht auf CO2.
Dieser Mangel an CO2-Reaktionsfähigkeit wurde durch einen vollständigen Verlust von
CO2-induzierter Hyperämie begleitet. Darüber hinaus zeigen Daten aus unserer
Arbeitsgruppe, dass eine Erhöhung des arteriellen CO2-Partialdrucks nicht in der Lage
ist, spastische Mikrogefäße zu dilatieren, und die zerebrale Duchblutung nach SAB zu
erhöhen.480 Ähnliche Befunde wurden auch bei SAB-Patienten erhoben.182,183
Auch in der aktuellen Arbeit konnte durch Inhalation von 7,5% CO2 die Hirndurch-
blutung bei Wildtyp-Mäusen signifikant erhöht werden. Auch iNOS-defiziente-Mäuse
zeigten im hyperkapnischen Milieu, deutlich erhöhte CBF-Werte. bei eNOS-/-- und
nNOS-/--Mäusen zeigten nach Inhalation von CO2 allerdings keine wesentliche
Erhöhung der zerebralen Durchblutung.
Während der CO2-Inhalation sind die Arteriolen mit einem Kaliber zwischen 9,5 und 45
µm vor allem bei Wildtyp-, wie auch bei iNOS-/--Mäusen deutlich erweitert. eNOS- und
nNOS-defiziente Mäuse zeigten dagegen im hyperkapnischen Milieu (7,5% iCO2) eine
schwache CO2-Reaktivität der Arteriolen.

4. Diskussion
100
Interessanterweise zeigten NOS-defiziente-Mäuse bereits unter Kontrollbedingungen
Mikrovasospasmen. Diese traten vor allem in eNOS-/--defizienten Mäusen auf, gefolgt
von nNOS-/--Mäusen und zuletzt iNOS-/--Mäusen. Ein ähnliches Bild ist auch bei SAB-
Mäusen zu finden. In den drei erwähnten Knockoutmäusegruppen waren die MVS
leicht oder mittelgradig (10-40%). Die Ähnlichkeit zwischen den bei eNOS und nNOS
defizienten Tieren und nach SAB beobachteten Mikrovasospasmen läßt darauf
schließen, dass NO möglicherweise an der Bildung von Mikrovasospasmen nach SAB
beteiligt ist. Diese Annahme wird dadurch unterstützt, dass inhaliertes NO zu einer
massiven Verminderung der Mikrovasospasmen nach SAB führt.
Der Grund für die erwähnten pathologischen Veränderungen der zerebralen
Mikrozirkulation nach SAB liegen daher aller Wahrscheinlichkeit nach in der gestörten
Funktionalität von endothelialen NOS und neuronalen NOS. Ein möglicher molekularer
Mechanismus ist die funktionelle Entkopplung der eNOS vom ihrem Co-Faktor
Tetrahydrobiopterin (BH4). Diese geschieht unter ischämischen Bedingungen, und hat
zur Folge, dass die eNOS statt NO Superoxidanion produziert.319,320,200,481,482 Ausser-
dem könnte das asymmetrische Dimethylarginin (ADMA), ein endogener eNOS-
Inhibitor, der in der zerebrospinalen Flüssigkeit von SAB-Patienten vorkommt,468 auch
für die Entstehung von Mikrovasospasmen verantwortlich sein. Ein erhöhter ET-1
Spiegel im Gehirn, nach SAB, ist auch verantwortlich für die Beeinträchtigung der NO-
Produktion. Diese Beeinträchtigung wird verursacht über die isoformspezifische
Protein-Kinase-C-vermittelte Hemmung der eNOS-Expression.352
Bemerkenswert ist, dass nach Applikation von S-Methyl-L-Thiocitrullin (SMTC) —
einem nNOS-Inhibitor —, die Durchblutung des menschlichen Unterarms abnahm.483
Dadurch zeigen Capettini et al., dass nicht nur die eNOS, sondern auch die nNOS eine
wichtige Rolle in der Vasorelaxation spielt.484 Da die nNOS, der eNOS in Aufbau,
Struktur und Regulation sehr ähnlich ist, könnte ein ähnliches Entkopplungs-
phänomen wie bei eNOS auch bei der nNOS stattfinden und zu einer dysfunktionellen
zerebralen Mikrogefäßregulation führen. Diese Annahme wird unterstützt durch die
Beobachtung, dass die pharmakologische Hemmung der nNOS durch den selektiven
Inhibitor 7-Nitroindazol (7-NI), eine starke Entkopplung der hämodynamischen von der
neuronalen Aktivität zur Folge hat.485

5. Zusammenfassung
5. Zusammenfassung
Die frühe Beeinträchtigung der zerebralen Mikrozirkulation ist entscheidend für die
Pathophysiologie der Subarachnoidalblutung (SAB). Das Vorkommen von Vasospas-
men in zerebralen Mikrogefäßen ist wahrscheinlich einer der Mechanismen, der für
posthämorrhagische Ischämie bei SAB-Patienten verantwortlich ist.182 Auch in dieser
Arbeit konnte gezeigt werden, dass Vasospasmen ab der vierten Stunde nach SAB
vor allem in zerebralen Arteriolen vermehrt vorkommen. Die Gabe von inhaliertem
Stickstoffmonoxid (iNO) konnte die Anzahl von zerebralen Mikrovasospasmen nach
SAB um 85% reduzieren. Damit konnte erstmals gezeigt werden, dass Spasmen der
zerebralen Mikrozirkulation maßgeblich an der Perfusionsstörung nach SAB beteiligt
sind. Damit wurde auch die Hypothese bestätigt, dass post-hämorrhagische
Mikrovasospasmen auf dem Boden einer lokalen NO-Depletion entstehen. Dies wurde
durch Untersuchungen an eNOS defizienten Tieren bestätigt, die bereits unter
Kontrollbedingungen ausgeprägte Mikrovasospasmen aufzeigen. Auf Grund seiner
ausgesprochen positiven Wirkung auf die zerebralen Mikrovasospasmen, seines gut
definierten und niedrigen Nebenwirkungsprofils und seiner einfachen Anwendbarkeit,
könnte die Inhalation von NO ein potentes Mittel zur Behandlung der post-
hämorrhagischen Ischämie sein. Die Endothelin-Rezeptor-Blockade durch Clazosen-
tan, die bisher Anwendung in der verzögerten Phase bei SAB-Patienten fand, um
Arterien zu relaxieren und Vasospasmen zu reduzieren, zeigt auch in der Frühphase
nach SAB, eine gefäßrelaxierende Wirkung, vor allem in Arteriolen mit einem Kaliber
zwischen 35-55 µm. Eine deutliche Reduzierung von MVS kann man vor allem in den
größeren Arteriolen beobachten. Diese Befunde bestätigen die nicht signifikante
Wirkung von Clazosentan auf das Outcome von SAB-Patienten und könnten darauf
hinweisen, dass iNO oder die Kombination von iNO und Clazosentan eine
vielversprechendere Therapieform der SAB darstellen könnte.

6. Literaturverzeichnis
6. Literaturverzeichnis
1. Hatano S. Experience from a multicentre stroke register: a preliminary report. Bulletin of the World Health Organization. 1976;54(5):541-553.
2. Eschenfelder Ch C, Zeller JA, Stingele R. [Stroke: causes and classification].
Hamostaseologie. 2006;26(4):298-308. 3. de Rooij NK, Linn FH, van der Plas JA, Algra A, Rinkel GJ. Incidence of
subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender and time trends. Journal of Neurology, Neurosurgery, and Psychiatry. 2007;78(12):1365-1372.
4. Odom MJ, Zuckerman SL, Mocco J. The role of magnesium in the management
of cerebral vasospasm. Neurology Research International. 2013;2013:943914. 5. Konczalla J, Platz J, Schuss P, Vatter H, Seifert V, Guresir E. Non-aneurysmal
non-traumatic subarachnoid hemorrhage: patient characteristics, clinical outcome and prognostic factors based on a single-center experience in 125 patients. BMC Neurology. 2014;14:140.
6. van Gijn J, Kerr RS, Rinkel GJ. Subarachnoid haemorrhage. Lancet.
2007;369(9558):306-318. 7. Rodriguez-Rodriguez A, Egea-Guerrero JJ, Ruiz de Azua-Lopez Z, Murillo-
Cabezas F. Biomarkers of vasospasm development and outcome in aneurysmal subarachnoid hemorrhage. Journal of the Neurological Sciences. 2014;341(1-2):119-127.
8. van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and
management. Brain . Journal of Neurology. 2001;124(Pt 2):249-278. 9. Juvela S. Aspirin and delayed cerebral ischemia after aneurysmal subarachnoid
hemorrhage. J Neurosurg. 1995;82(6):945-952. 10. Lovelock CE, Rinkel GJ, Rothwell PM. Time trends in outcome of subarachnoid
hemorrhage: Population-based study and systematic review. Neurology. 2010;74(19):1494-1501.
11. Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a
review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. The Lancet. Neurology. 2003;2(1):43-53.
12. Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics--
2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18-e209.

6. Literaturverzeichnis
103
13. Ingall T, Asplund K, Mahonen M, Bonita R. A multinational comparison of subarachnoid hemorrhage epidemiology in the WHO MONICA stroke study. Stroke. 2000;31(5):1054-1061.
14. Linn FH, Rinkel GJ, Algra A, van Gijn J. Incidence of subarachnoid hemorrhage:
role of region, year, and rate of computed tomography: a meta-analysis. Stroke. 1996;27(4):625-629.
15. Steiner T, Juvela S, Unterberg A, et al. European Stroke Organization
guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovascular Diseases. 2013;35(2):93-112.
16. Fujinaka T, Yoshimine T, Mashimo T. [Management of aneurysmal
subarachnoid hemorrhage]. Masui. The Japanese Journal of Anesthesiology. 2012;61(9):962-970.
17. Inagawa T. Trends in incidence and case fatality rates of aneurysmal
subarachnoid hemorrhage in Izumo City, Japan, between 1980-1989 and 1990-1998. Stroke. 2001;32(7):1499-1507.
18. Ohkuma H, Fujita S, Suzuki S. Incidence of aneurysmal subarachnoid
hemorrhage in Shimokita, Japan, from 1989 to 1998. Stroke. 2002;33(1):195-199.
19. Sarti C, Tuomilehto J, Salomaa V, et al. Epidemiology of subarachnoid
hemorrhage in Finland from 1983 to 1985. Stroke. 1991;22(7):848-853. 20. Magnetic Resonance Angiography in Relatives of Patients with Subarachnoid
Hemorrhage Study G. Risks and benefits of screening for intracranial aneurysms in first-degree relatives of patients with sporadic subarachnoid hemorrhage. The New England Journal of Medicine. 1999;341(18):1344-1350.
21. Rinkel GJ. Natural history, epidemiology and screening of unruptured
intracranial aneurysms. Revue Neurologique. 2008;164(10):781-786. 22. Rinkel GJ, Djibuti M, Algra A, van Gijn J. Prevalence and risk of rupture of
intracranial aneurysms: a systematic review. Stroke. 1998;29(1):251-256. 23. Baumann F, Khan N, Yonekawa Y. Patient and aneurysm characteristics in
multiple intracranial aneurysms. Acta Neurochirurgica. Supplement. 2008;103:19-28.
24. Venti M. Subarachnoid and intraventricular hemorrhage. Frontiers of Neurology
and Neuroscience. 2012;30:149-153. 25. Samaniego EA, Dabus G, Fuentes K, Linfante I. Endovascular treatment of
severe vasospasm in nonaneurysmal perimesencephalic subarachnoid hemorrhage. Neurocritical Care. 2011;15(3):537-541.

6. Literaturverzeichnis
104
26. Kapadia A, Schweizer TA, Spears J, Cusimano M, Macdonald RL. Nonaneurysmal perimesencephalic subarachnoid hemorrhage: diagnosis, pathophysiology, clinical characteristics and long-term outcome. World Neurosurgery. 2014;82(6):1131-1143
27. Hop JW, Rinkel GJ, Algra A, van Gijn J. Case-fatality rates and functional
outcome after subarachnoid hemorrhage: a systematic review. Stroke. 1997;28(3):660-664.
28. Schievink WI, Wijdicks EF, Parisi JE, Piepgras DG, Whisnant JP. Sudden death
from aneurysmal subarachnoid hemorrhage. Neurology. 1995;45(5):871-874. 29. Truelsen T, Bonita R, Duncan J, Anderson NE, Mee E. Changes in
subarachnoid hemorrhage mortality, incidence, and case fatality in New Zealand between 1981-1983 and 1991-1993. Stroke. 1998;29(11):2298-2303.
30. Bederson JB, Awad IA, Wiebers DO, et al. Recommendations for the
management of patients with unruptured intracranial aneurysms: A Statement for healthcare professionals from the Stroke Council of the American Heart Association. Journal of Cerebral Circulation. 2000;31(11):2742-2750.
31. Hop JW, Rinkel GJ, Algra A, van Gijn J. Quality of life in patients and partners
after aneurysmal subarachnoid hemorrhage. Stroke. 1998;29(4):798-804. 32. Mayer SA, Kreiter KT, Copeland D, et al. Global and domain-specific cognitive
impairment and outcome after subarachnoid hemorrhage. Neurology. 2002;59(11):1750-1758.
33. Powell J, Kitchen N, Heslin J, Greenwood R. Psychosocial outcomes at three
and nine months after good neurological recovery from aneurysmal subarachnoid haemorrhage: predictors and prognosis. Journal of Neurology, Neurosurgery, and Psychiatry. 2002;72(6):772-781.
34. Hackett ML, Anderson CS. Health outcomes 1 year after subarachnoid
hemorrhage: An international population-based study. The Australian Cooperative Research on Subarachnoid Hemorrhage Study Group. Neurology. 2000;55(5):658-662.
35. Huang J, van Gelder JM. The probability of sudden death from rupture of
intracranial aneurysms: a meta-analysis. Neurosurgery. 2002;51(5):1101-1105. 36. Broderick JP, Viscoli CM, Brott T, et al. Major risk factors for aneurysmal
subarachnoid hemorrhage in the young are modifiable. Stroke. 2003;34(6):1375-1381.
37. Feigin VL, Rinkel GJ, Lawes CM, et al. Risk factors for subarachnoid
hemorrhage: an updated systematic review of epidemiological studies. Stroke. 2005;36(12):2773-2780.

6. Literaturverzeichnis
105
38. Teunissen LL, Rinkel GJ, Algra A, van Gijn J. Risk factors for subarachnoid hemorrhage: a systematic review. Stroke. 1996;27(3):544-549.
39. Schievink WI, Michels VV, Piepgras DG. Neurovascular manifestations of
heritable connective tissue disorders. A review. Stroke. 1994;25(4):889-903. 40. Qureshi AI, Suri MF, Yahia AM, et al. Risk factors for subarachnoid hemorrhage.
Neurosurgery. 2001;49(3):607-612. 41. Okamoto K, Horisawa R, Kawamura T, et al. Family history and risk of
subarachnoid hemorrhage: a case-control study in Nagoya, Japan. Stroke. 2003;34(2):422-426.
42. Lozano AM, Leblanc R. Familial intracranial aneurysms. Journal of
Neurosurgery. 1987;66(4):522-528. 43. Leblanc R. Familial cerebral aneurysms. A bias for women. Stroke.
1996;27(6):1050-1054. 44. Ronkainen A, Hernesniemi J, Puranen M, et al. Familial intracranial aneurysms.
Lancet. 1997;349(9049):380-384. 45. Bromberg JE, Rinkel GJ, Algra A, van den Berg UA, Tjin ATML, van Gijn J.
Hypertension, stroke, and coronary heart disease in relatives of patients with subarachnoid hemorrhage. Stroke. 1996;27(1):7-9.
46. Schievink WI, Torres VE, Piepgras DG, Wiebers DO. Saccular intracranial
aneurysms in autosomal dominant polycystic kidney disease. Journal of the American Society of Nephrology : JASN. 1992;3(1):88-95.
47. Brisman JL, Song JK, Niimi Y, Berenstein A. Treatment options for wide-necked
intracranial aneurysms using a self-expandable hydrophilic coil and a self-expandable stent combination. AJNR. American Journal of Neuroradiology. 2005;26(5):1237-1240.
48. Schievink WI. Intracranial aneurysms. The New England Journal of Medicine.
1997;336(1):28-40. 49. Starke RM, Chalouhi N, Ali MS, et al. The role of oxidative stress in cerebral
aneurysm formation and rupture. Current Neurovascular Research. 2013;10(3):247-255.
50. Priebe HJ. Aneurysmal subarachnoid haemorrhage and the anaesthetist.
British Journal of Anaesthesia. 2007;99(1):102-118. 51. Poeck K. [Heinz Ganshirt --forerunner of research on cerebrovascular diseases
in Germany]. Fortschritte der Neurologie-Psychiatrie. 2001;69 Suppl 1:S45-47.

6. Literaturverzeichnis
106
52. Broderick JP, Adams HP, Jr., Barsan W, et al. Guidelines for the management of spontaneous intracerebral hemorrhage: A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 1999;30(4):905-915.
53. Ebina K, Shimizu T, Sohma M, Iwabuchi T. Clinico-statistical study on
morphological risk factors of middle cerebral artery aneurysms. Acta Neurochirurgica. 1990;106(3-4):153-159.
54. Ferguson GG. Physical factors in the initiation, growth, and rupture of human
intracranial saccular aneurysms. J Neurosurg. 1972;37(6):666-677. 55. Kassell NF, Torner JC. Size of intracranial aneurysms. Neurosurgery.
1983;12(3):291-297. 56. Orz Y, Kobayashi S, Osawa M, Tanaka Y. Aneurysm size: a prognostic factor
for rupture. British Journal of Neurosurgery. 1997;11(2):144-149. 57. Rosenorn J, Eskesen V. Does a safe size-limit exist for unruptured intracranial
aneurysms? Acta Neurochirurgica. 1993;121(3-4):113-118. 58. Suzuki J, Ohara H. Clinicopathological study of cerebral aneurysms. Origin,
rupture, repair, and growth. J Neurosurg. 1978;48(4):505-514. 59. Wiebers DO, Whisnant JP, Sundt TM, Jr., O'Fallon WM. The significance of
unruptured intracranial saccular aneurysms. J Neurosurg. 1987;66(1):23-29. 60. Wiebers DO, Whisnant JP, Huston J, 3rd, et al. Unruptured intracranial
aneurysms: natural history, clinical outcome, and risks of surgical and endovascular treatment. Lancet. 2003;362(9378):103-110.
61. Yoshimoto T, Mizoi K. Importance of management of unruptured cerebral
aneurysms. Surgical Neurology. 1997;47(6):522-525. 62. Zacks DJ, Russell DB, Miller JD. Fortuitously discovered intracranial
aneurysms. Archives of Neurology. 1980;37(1):39-41. 63. Jeong YG, Jung YT, Kim MS, Eun CK, Jang SH. Size and location of ruptured
intracranial aneurysms. Journal of Korean Neurosurgical Society. 2009;45(1):11-15.
64. Sughrue ME, Saloner D, Rayz VL, Lawton MT. Giant intracranial aneurysms:
evolution of management in a contemporary surgical series. Neurosurgery. 2011;69(6):1261-1270.
65. Grooters GS, Sluzewski M, Tijssen CC. How often is thunderclap headache
caused by the reversible cerebral vasoconstriction syndrome? Headache. 2014;54(4):732-735.

6. Literaturverzeichnis
107
66. Bederson JB, Connolly ES, Jr., Batjer HH, et al. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 2009;40(3):994-1025.
67. Ducros A, Bousser MG. Thunderclap headache. Bmj. 2013;346:e8557. 68. McCarron MO, Alberts MJ, McCarron P. A systematic review of Terson's
syndrome: frequency and prognosis after subarachnoid haemorrhage. Journal of Neurology, Neurosurgery, and Psychiatry. 2004;75(3):491-493.
69. Rheinboldt M, Francis K, Parrish D, Harper D, Blase J. Terson syndrome in
conjunction with ruptured intracranial aneurysm and penetrating intracranial injury: a review of two cases. Emergency Radiology. 2014;21(2):215-218.
70. Silverman IE, Restrepo L, Mathews GC. Poststroke seizures. Archives of
Neurology. 2002;59(2):195-201. 71. Hijdra A, Braakman R, van Gijn J, Vermeulen M, van Crevel H. Aneurysmal
subarachnoid hemorrhage. Complications and outcome in a hospital population. Stroke. 1987;18(6):1061-1067.
72. Jakobsson KE, Saveland H, Hillman J, et al. Warning leak and management
outcome in aneurysmal subarachnoid hemorrhage. J Neurosurg. 1996;85(6):995-999.
73. Linn FH, Wijdicks EF, van der Graaf Y, Weerdesteyn-van Vliet FA, Bartelds AI,
van Gijn J. Prospective study of sentinel headache in aneurysmal subarachnoid haemorrhage. Lancet. 1994;344(8922):590-593.
74. Tolias CM, Choksey MS. Will increased awareness among physicians of the
significance of sudden agonizing headache affect the outcome of subarachnoid hemorrhage? Coventry and Warwickshire Study: audit of subarachnoid hemorrhage (establishing historical controls), hypothesis, campaign layout, and cost estimation. Stroke. 1996;27(5):807-812.
75. Hijdra A, van Gijn J, Nagelkerke NJ, Vermeulen M, van Crevel H. Prediction of
delayed cerebral ischemia, rebleeding, and outcome after aneurysmal subarachnoid hemorrhage. Stroke. 1988;19(10):1250-1256.
76. Rosengart AJ, Schultheiss KE, Tolentino J, Macdonald RL. Prognostic factors
for outcome in patients with aneurysmal subarachnoid hemorrhage. Stroke. 2007;38(8):2315-2321.
77. Suarez JI, Tarr RW, Selman WR. Aneurysmal subarachnoid hemorrhage. The
New England Journal of Medicine. 2006;354(4):387-396. 78. Hunt WE, Hess RM. Surgical risk as related to time of intervention in the repair
of intracranial aneurysms. J Neurosurg. 1968;28(1):14-20.

6. Literaturverzeichnis
108
79. Rosen DS, Macdonald RL. Subarachnoid hemorrhage grading scales: a
systematic review. Neurocritical Care. 2005;2(2):110-118. 80. Wood MJ, Nowitzke AM. Epidemiological aspects of spontaneous subarachnoid
haemorrhage in Queensland, Australia. Journal of Clinical Neuroscience 2005;12(7):770-774.
81. Degen LA, Dorhout Mees SM, Algra A, Rinkel GJ. Interobserver variability of
grading scales for aneurysmal subarachnoid hemorrhage. Stroke. 2011;42(6):1546-1549.
82. Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A
practical scale. Lancet. 1974;2(7872):81-84. 83. Report of World Federation of Neurological Surgeons Committee on a Universal
Subarachnoid Hemorrhage Grading Scale. Journal of Neurosurgery. 1988;68(6):985-986.
84. Takagi K, Tamura A, Nakagomi T, et al. How should a subarachnoid
hemorrhage grading scale be determined? A combinatorial approach based solely on the Glasgow Coma Scale. Journal of Neurosurgery. 1999;90(4):680-687.
85. van Heuven AW, Dorhout Mees SM, Algra A, Rinkel GJ. Validation of a
prognostic subarachnoid hemorrhage grading scale derived directly from the Glasgow Coma Scale. Stroke. 2008;39(4):1347-1348.
86. Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to
subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6(1):1-9.
87. Klimo P, Jr., Schmidt RH. Computed tomography grading schemes used to
predict cerebral vasospasm after aneurysmal subarachnoid hemorrhage: a historical review. Neurosurgical Focus. 2006;21(3):E5.
88. Frontera JA, Claassen J, Schmidt JM, et al. Prediction of symptomatic
vasospasm after subarachnoid hemorrhage: the modified fisher scale. Neurosurgery. 2006;59(1):21-27.
89. Fiebach JB, Schellinger PD, Gass A, et al. Stroke magnetic resonance imaging
is accurate in hyperacute intracerebral hemorrhage: a multicenter study on the validity of stroke imaging. Stroke. 2004;35(2):502-506.
90. van Gijn J. Slip-ups in diagnosis of subarachnoid haemorrhage. Lancet.
1997;349(9064):1492.

6. Literaturverzeichnis
109
91. Czuczman AD, Thomas LE, Boulanger AB, et al. Interpreting red blood cells in lumbar puncture: distinguishing true subarachnoid hemorrhage from traumatic tap. Journal of Emergency Medicine. 2013;20(3):247-256.
92. Shah KH, Edlow JA. Distinguishing traumatic lumbar puncture from true
subarachnoid hemorrhage. The Journal of Emergency Medicine. 2002;23(1):67-74.
93. Lubicz B, Levivier M, Francois O, et al. Sixty-four-row multisection CT
angiography for detection and evaluation of ruptured intracranial aneurysms: interobserver and intertechnique reproducibility. AJNR. American Journal of Neuroradiology. 2007;28(10):1949-1955.
94. Romijn M, Gratama van Andel HA, van Walderveen MA, et al. Diagnostic
accuracy of CT angiography with matched mask bone elimination for detection of intracranial aneurysms: comparison with digital subtraction angiography and 3D rotational angiography. AJNR. American Journal of Neuroradiology. 2008;29(1):134-139.
95. Boogaarts HD, van Amerongen MJ, de Vries J, et al. Caseload as a factor for
outcome in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. Journal of Neurosurgery. 2014;120(3):605-611.
96. Green DM, Burns JD, DeFusco CM. ICU management of aneurysmal
subarachnoid hemorrhage. Journal of Intensive Care Medicine. 2013;28(6):341-354.
97. Ohkuma H, Tsurutani H, Suzuki S. Incidence and significance of early
aneurysmal rebleeding before neurosurgical or neurological management. Stroke. 2001;32(5):1176-1180.
98. Kim SY, Kim SM, Park MS, Kim HK, Park KS, Chung SY. Effectiveness of
nicardipine for blood pressure control in patients with subarachnoid hemorrhage. Journal of Cerebrovascular and Endovascular Neurosurgery. 2012;14(2):84-89.
99. Cannon CM, Levy P, Baumann BM, et al. Intravenous nicardipine and labetalol
use in hypertensive patients with signs or symptoms suggestive of end-organ damage in the emergency department: a subgroup analysis of the CLUE trial. BMJ Open. 2013;3(3).
100. Wartenberg KE, Schmidt JM, Claassen J, et al. Impact of medical complications
on outcome after subarachnoid hemorrhage. Critical Care Medicine. 2006;34(3):617-623.
101. Dorhout Mees SM, van Dijk GW, Algra A, Kempink DR, Rinkel GJ. Glucose
levels and outcome after subarachnoid hemorrhage. Neurology. 2003;61(8):1132-1133.

6. Literaturverzeichnis
110
102. Lanzino G, Kassell NF, Germanson T, Truskowski L, Alves W. Plasma glucose levels and outcome after aneurysmal subarachnoid hemorrhage. Journal of Neurosurgery. 1993;79(6):885-891.
103. Juvela S, Siironen J, Kuhmonen J. Hyperglycemia, excess weight, and history
of hypertension as risk factors for poor outcome and cerebral infarction after aneurysmal subarachnoid hemorrhage. Journal of Neurosurgery. 2005;102(6):998-1003.
104. Dorhout Mees SM, Luitse MJ, van den Bergh WM, Rinkel GJ. Fever after
aneurysmal subarachnoid hemorrhage: relation with extent of hydrocephalus and amount of extravasated blood. Stroke. 2008;39(7):2141-2143.
105. Siasios I, Kapsalaki EZ, Fountas KN. Cerebral vasospasm pharmacological
treatment: an update. Neurology Research International. 2013;2013:571328. 106. Rinkel GJ, Feigin VL, Algra A, van den Bergh WM, Vermeulen M, van Gijn J.
Calcium antagonists for aneurysmal subarachnoid haemorrhage. The Cochrane Database of Systematic Reviews. 2005(1):CD000277.
107. Dorhout Mees SM, Rinkel GJ, Feigin VL, et al. Calcium antagonists for
aneurysmal subarachnoid haemorrhage. The Cochrane Database of Systematic Reviews. 2007(3):CD000277.
108. Ott S, Jedlicka S, Wolf S, et al. Continuous selective intra-arterial application of
nimodipine in refractory cerebral vasospasm due to aneurysmal subarachnoid hemorrhage. BioMed Research International. 2014;2014:970741.
109. Reddy D, Fallah A, Petropoulos JA, Farrokhyar F, Macdonald RL, Jichici D.
Prophylactic Magnesium Sulfate for Aneurysmal Subarachnoid Hemorrhage: A Systematic Review and Meta-analysis. Neurocritical Care. 2014;21(2):356-364
110. Guglielmi G, Vinuela F, Dion J, Duckwiler G. Electrothrombosis of saccular
aneurysms via endovascular approach. Part 2: Preliminary clinical experience. J Neurosurg. 1991;75(1):8-14.
111. Whitfield PC, Kirkpatrick PJ. Timing of surgery for aneurysmal subarachnoid
haemorrhage. The Cochrane database of systematic reviews. 2001(2):CD001697.
112. Mayberg MR, Batjer HH, Dacey R, et al. Guidelines for the management of
aneurysmal subarachnoid hemorrhage. A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Stroke. 1994;25(11):2315-2328.
113. Molyneux A, Kerr R, Stratton I, et al. International Subarachnoid Aneurysm Trial
(ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomised trial. Lancet. 2002;360(9342):1267-1274.

6. Literaturverzeichnis
111
114. Molyneux AJ, Kerr RS, Yu LM, et al. International subarachnoid aneurysm trial
(ISAT) of neurosurgical clipping versus endovascular coiling in 2143 patients with ruptured intracranial aneurysms: a randomised comparison of effects on survival, dependency, seizures, rebleeding, subgroups, and aneurysm occlusion. Lancet. 2005;366(9488):809-817.
115. Britz GW. ISAT trial: coiling or clipping for intracranial aneurysms? Lancet.
2005;366(9488):783-785. 116. Kim DJ, Czosnyka Z, Kasprowicz M, et al. Continuous monitoring of the Monro-
Kellie doctrine: is it possible? Journal of Neurotrauma. 2012;29(7):1354-1363. 117. Bailes JE, Spetzler RF, Hadley MN, Baldwin HZ. Management morbidity and
mortality of poor-grade aneurysm patients. J Neurosurg. 1990;72(4):559-566. 118. Jakobsen M, Enevoldsen E, Bjerre P. Cerebral blood flow and metabolism
following subarachnoid haemorrhage: cerebral oxygen uptake and global blood flow during the acute period in patients with SAH. Acta Neurologica Scandinavica. 1990;82(3):174-182.
119. Nornes H. The role of intracranial pressure in the arrest of hemorrhage in
patients with ruptured intracranial aneurysm. J Neurosurg. 1973;39(2):226-234. 120. Nornes H. Cerebral arterial flow dynamics during aneurysm haemorrhage. Acta
Neurochirurgica. 1978;41(1-3):39-48. 121. Grote E, Hassler W. The critical first minutes after subarachnoid hemorrhage.
Neurosurgery. 1988;22(4):654-661. 122. Kassell NF, Torner JC, Haley EC, Jr., Jane JA, Adams HP, Kongable GL. The
International Cooperative Study on the Timing of Aneurysm Surgery. Part 1: Overall management results. Journal of Neurosurgery. 1990;73(1):18-36.
123. Raslan A, Bhardwaj A. Medical management of cerebral edema. Neurosurgical
Focus. 2007;22(5):E12. 124. Crompton MR. The Pathogenesis of Cerebral Infarction Following the Rupture
of Cerebral Berry Aneurysms. Brain. Journal of Neurology. 1964;87:491-510. 125. Wijdicks EF, Vermeulen M, Murray GD, Hijdra A, van Gijn J. The effects of
treating hypertension following aneurysmal subarachnoid hemorrhage. Clinical Neurology and Neurosurgery. 1990;92(2):111-117.
126. Neil-Dwyer G, Lang DA, Doshi B, Gerber CJ, Smith PW. Delayed cerebral
ischaemia: the pathological substrate. Acta Neurochirurgica. 1994;131(1-2):137-145.

6. Literaturverzeichnis
112
127. Feiler S, Friedrich B, Scholler K, Thal SC, Plesnila N. Standardized induction of subarachnoid hemorrhage in mice by intracranial pressure monitoring. Journal of Neuroscience Methods. 2010;190(2):164-170.
128. Jakubowski J, Bell BA, Symon L, Zawirski MB, Francis DM. A primate model of
subarachnoid hemorrhage: change in regional cerebral blood flow, autoregulation carbon dioxide reactivity, and central conduction time. Stroke. 1982;13(5):601-611.
129. Piepgras A, Thome C, Schmiedek P. Characterization of an anterior circulation
rat subarachnoid hemorrhage model. Stroke. 1995;26(12):2347-2352. 130. Bederson JB, Germano IM, Guarino L. Cortical blood flow and cerebral
perfusion pressure in a new noncraniotomy model of subarachnoid hemorrhage in the rat. Stroke. 1995;26(6):1086-1091.
131. Rasmussen G, Hauerberg J, Waldemar G, Gjerris F, Juhler M. Cerebral blood
flow autoregulation in experimental subarachnoid haemorrhage in rat. Acta Neurochirurgica. 1992;119(1-4):128-133.
132. Sehba FA, Pluta RM, Zhang JH. Metamorphosis of subarachnoid hemorrhage
research: from delayed vasospasm to early brain injury. Molecular Neurobiology. 2011;43(1):27-40.
133. Cossu G, Messerer M, Oddo M, Daniel RT. To look beyond vasospasm in
aneurysmal subarachnoid haemorrhage. BioMed Research International. 2014;2014:628597.
134. Friedrich V, Flores R, Muller A, Sehba FA. Escape of intraluminal platelets into
brain parenchyma after subarachnoid hemorrhage. Neuroscience. 2010;165(3):968-975.
135. Stein SC, Browne KD, Chen XH, Smith DH, Graham DI. Thromboembolism and
delayed cerebral ischemia after subarachnoid hemorrhage: an autopsy study. Neurosurgery. 2006;59(4):781-787
136. Nau R, Haase S, Bunkowski S, Bruck W. Neuronal apoptosis in the dentate
gyrus in humans with subarachnoid hemorrhage and cerebral hypoxia. Brain Pathology. 2002;12(3):329-336.
137. Sehba FA, Friedrich V, Jr., Makonnen G, Bederson JB. Acute cerebral vascular
injury after subarachnoid hemorrhage and its prevention by administration of a nitric oxide donor. Journal of Neurosurgery. 2007;106(2):321-329.
138. Sehba FA, Mostafa G, Knopman J, Friedrich V, Jr., Bederson JB. Acute
alterations in microvascular basal lamina after subarachnoid hemorrhage. Journal of Neurosurgery. 2004;101(4):633-640.

6. Literaturverzeichnis
113
139. Friedrich V, Flores R, Muller A, Sehba FA. Luminal platelet aggregates in functional deficits in parenchymal vessels after subarachnoid hemorrhage. Brain Research. 2010;1354:179-187.
140. Sehba FA, Ding WH, Chereshnev I, Bederson JB. Effects of S-
nitrosoglutathione on acute vasoconstriction and glutamate release after subarachnoid hemorrhage. Stroke.1999;30(9):1955-1961.
141. Milhorat TH. Acute hydrocephalus after aneurysmal subarachnoid hemorrhage.
Neurosurgery. 1987;20(1):15-20. 142. Kamiya K, Kuyama H, Symon L. An experimental study of the acute stage of
subarachnoid hemorrhage. Journal of Neurosurgery. 1983;59(6):917-924. 143. Sehba FA, Schwartz AY, Chereshnev I, Bederson JB. Acute decrease in
cerebral nitric oxide levels after subarachnoid hemorrhage. Journal Cerebral Blood Flow and Metabolism. 2000;20(3):604-611.
144. Sehba FA, Mostafa G, Friedrich V, Jr., Bederson JB. Acute microvascular
platelet aggregation after subarachnoid hemorrhage. Journal of Neurosurgery. 2005;102(6):1094-1100.
145. Josko J, Hendryk S, Jedrzejowska-Szypula H, Gwozdz B, Herman ZS, Gawlik
R. Influence endothelin ETA receptor antagonist--BQ-123--on changes of endothelin-1 level in plasma of rats with acute vasospasm following subarachnoid hemorrhage. Journal of physiology and pharmacology. Journal of the Polish Physiological Society. 1998;49(3):367-375.
146. Gaetani P, Marzatico F, Rodriguez y Baena R, et al. Arachidonic acid
metabolism and pathophysiologic aspects of subarachnoid hemorrhage in rats. Stroke. 1990;21(2):328-332.
147. Marzatico F, Gaetani P, Cafe C, Spanu G, Rodriguez y Baena R. Antioxidant
enzymatic activities after experimental subarachnoid hemorrhage in rats. Acta Neurologica Scandinavica. 1993;87(1):62-66.
148. Macdonald RL. Delayed neurological deterioration after subarachnoid
haemorrhage. Nature reviews. Neurology. 2014;10(1):44-58. 149. Dorsch NW. A review of cerebral vasospasm in aneurysmal subarachnoid
haemorrhage Part II: Management. Journal of clinical neuroscience. Journal Neurosurgical Society of Australasia. 1994;1(2):78-92.
150. Robertson EG. Cerebral lesions due to intracranial aneurysms. Brain : Journal
of Neurology. 1949;72(Pt. 2):150-185. 151. Ecker A, Riemenschneider PA. Arteriographic demonstration of spasm of the
intracranial arteries, with special reference to saccular arterial aneurysms. J Neurosurg. 1951;8(6):660-667.

6. Literaturverzeichnis
114
152. Kassell NF, Sasaki T, Colohan AR, Nazar G. Cerebral vasospasm following
aneurysmal subarachnoid hemorrhage. Stroke.1985;16(4):562-572. 153. Weir B. The pathophysiology of cerebral vasospasm. British Journal of
Neurosurgery. 1995;9(3):375-390. 154. Maeda K, Kurita H, Nakamura T, et al. Occurrence of severe vasospasm
following intraventricular hemorrhage from an arteriovenous malformation. Report of two cases. Journal of Neurosurgery. 1997;87(3):436-439.
155. Carvi y Nievas MN, Archavlis E. Atypical causes of nontraumatic intracranial
subarachnoid hemorrhage. Clinical Neurology and Neurosurgery. 2009;111(4):354-358.
156. Martin NA, Doberstein C, Zane C, Caron MJ, Thomas K, Becker DP.
Posttraumatic cerebral arterial spasm: transcranial Doppler ultrasound, cerebral blood flow, and angiographic findings. Journal of Neurosurgery. 1992;77(4):575-583.
157. Raps EC, Galetta SL, Broderick M, Atlas SW. Delayed peripartum
vasculopathy: cerebral eclampsia revisited. Annals of Neurology. 1993;33(2):222-225.
158. Wilkins RH. Cerebral vasospasm. Critical Reviews in Neurobiology.
1990;6(1):51-77. 159. Aaslid R, Markwalder TM, Nornes H. Noninvasive transcranial Doppler
ultrasound recording of flow velocity in basal cerebral arteries. J Neurosurg. 1982;57(6):769-774.
160. Takeuchi K, Miyata N, Renic M, Harder DR, Roman RJ. Hemoglobin, NO, and
20-HETE interactions in mediating cerebral vasoconstriction following SAH. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2006;290(1):R84-89.
161. Zimmermann M, Seifert V. Endothelin and subarachnoid hemorrhage: an
overview. Neurosurgery. 1998;43(4):863-875. 162. Vergouwen MD, Ilodigwe D, Macdonald RL. Cerebral infarction after
subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke. 2011;42(4):924-929.
163. Treggiari-Venzi MM, Suter PM, Romand JA. Review of medical prevention of
vasospasm after aneurysmal subarachnoid hemorrhage: a problem of neurointensive care. Neurosurgery. 2001;48(2):249-261.
164. Fergusen S, Macdonald RL. Predictors of cerebral infarction in patients with
aneurysmal subarachnoid hemorrhage. Neurosurgery. 2007;60(4):658-667.

6. Literaturverzeichnis
115
165. Jakobsen M, Enevoldsen E, Dalager T. Spasm index in subarachnoid
haemorrhage: consequences of vasospasm upon cerebral blood flow and oxygen extraction. Acta Neurologica Scandinavica. 1990;82(5):311-320.
166. Weir B, Grace M, Hansen J, Rothberg C. Time course of vasospasm in man. J
Neurosurg. 1978;48(2):173-178. 167. Castanares-Zapatero D, Hantson P. Pharmacological treatment of delayed
cerebral ischemia and vasospasm in subarachnoid hemorrhage. Annals of Intensive Care. 2011;1(1):12.
168. Leng LZ, Fink ME, Iadecola C. Spreading depolarization: a possible new culprit
in the delayed cerebral ischemia of subarachnoid hemorrhage. Archives of Neurology. 2011;68(1):31-36.
169. Dreier JP. The role of spreading depression, spreading depolarization and
spreading ischemia in neurological disease. Nature Medicine. 2011;17(4):439-447.
170. Edvinsson L, Povlsen GK. Late cerebral ischaemia after subarachnoid
haemorrhage: is cerebrovascular receptor upregulation the mechanism behind? Acta Physiologica. 2011;203(1):209-224.
171. Muroi C, Mink S, Seule M, Bellut D, Fandino J, Keller E. Monitoring of the
inflammatory response after aneurysmal subarachnoid haemorrhage in the clinical setting: review of literature and report of preliminary clinical experience. Acta Neurochirurgica. Supplement. 2011;110(Pt 1):191-196.
172. Nakura T, Osuka K, Inukai T, Takagi T, Takayasu M. Soluble gp130 regulatess
interleukin-6 in cerebrospinal fluid after subarachnoid haemorrhage. Journal of Neurology, Neurosurgery, and Psychiatry. 2011;82(9):952-954.
173. Sarrafzadeh A, Schlenk F, Gericke C, Vajkoczy P. Relevance of cerebral
interleukin-6 after aneurysmal subarachnoid hemorrhage. Neurocritical Care. 2010;13(3):339-346.
174. Rabinstein AA, Lanzino G, Wijdicks EF. Multidisciplinary management and
emerging therapeutic strategies in aneurysmal subarachnoid haemorrhage. The Lancet. Neurology. 2010;9(5):504-519.
175. Crowley RW, Medel R, Dumont AS, et al. Angiographic vasospasm is strongly
correlated with cerebral infarction after subarachnoid hemorrhage. Stroke. 2011;42(4):919-923.
176. Dorsch NW. Cerebral arterial spasm--a clinical review. British Journal of
Neurosurgery. 1995;9(3):403-412.

6. Literaturverzeichnis
116
177. Macdonald RL, Higashida RT, Keller E, et al. Randomised trial of clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid hemorrhage undergoing surgical clipping (CONSCIOUS-2). Acta Neurochirurgica. Supplement. 2013;115:27-31.
178. Macdonald RL, Higashida RT, Keller E, et al. Clazosentan, an endothelin
receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). The Lancet. Neurology. 2011;10(7):618-625.
179. Schubert GA, Seiz M, Hegewald AA, Manville J, Thome C. Acute hypoperfusion
immediately after subarachnoid hemorrhage: a xenon contrast-enhanced CT study. Journal of Neurotrauma. 2009;26(12):2225-2231.
180. Schubert GA, Seiz M, Hegewald AA, Manville J, Thome C. Hypoperfusion in the
acute phase of subarachnoid hemorrhage. Acta Neurochirurgica. Supplement. 2011;110(Pt 1):35-38.
181. Herz DA, Baez S, Shulman K. Pial microcirculation in subarachnoid
hemorrhage. Stroke. 1975;6(4):417-424. 182. Uhl E, Lehmberg J, Steiger HJ, Messmer K. Intraoperative detection of early
microvasospasm in patients with subarachnoid hemorrhage by using orthogonal polarization spectral imaging. Neurosurgery. 2003;52(6):1307-1315.
183. Pennings FA, Bouma GJ, Ince C. Direct observation of the human cerebral
microcirculation during aneurysm surgery reveals increased arteriolar contractility. Stroke. 2004;35(6):1284-1288.
184. Friedrich B, Muller F, Feiler S, Scholler K, Plesnila N. Experimental
subarachnoid hemorrhage causes early and long-lasting microarterial constriction and microthrombosis: an in-vivo microscopy study. Journal Cerebral Blood Flow and Metabolism. 2012;32(3):447-455.
185. Stamler JS. Redox signaling: nitrosylation and related target interactions of nitric
oxide. Cell. 1994;78(6):931-936. 186. Beligni MV, Lamattina L. Nitric oxide: a non-traditional regulator of plant growth.
Trends in plant Science. 2001;6(11):508-509. 187. Ignarro LJ. Biosynthesis and metabolism of endothelium-derived nitric oxide.
Annual Review of Pharmacology and Toxicology. 1990;30:535-560. 188. Anbar M. Nitric oxide: a synchronizing chemical messenger. Experientia.
1995;51(6):545-550. 189. Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived
relaxing factor produced and released from artery and vein is nitric oxide.

6. Literaturverzeichnis
117
Proceedings of the National Academy of Sciences of the United States of America. 1987;84(24):9265-9269.
190. Palmer RM, Ferrige AG, Moncada S. Nitric oxide release accounts for the
biological activity of endothelium-derived relaxing factor. Nature. 1987;327(6122):524-526.
191. Furchgott RF, Zawadzki JV. The obligatory role of endothelial cells in the
relaxation of arterial smooth muscle by acetylcholine. Nature. 1980;288(5789):373-376.
192. Hanbauer I, Wink D, Osawa Y, Edelman GM, Gally JA. Role of nitric oxide in
NMDA-evoked release of [3H]-dopamine from striatal slices. Neuroreport. 1992;3(5):409-412.
193. Ridnour LA, Windhausen AN, Isenberg JS, et al. Nitric oxide regulates matrix
metalloproteinase-9 activity by guanylyl-cyclase-dependent and -independent pathways. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(43):16898-16903.
194. Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annual
Review of Biochemistry. 1994;63:175-195. 195. Vincent SR. Nitric oxide neurons and neurotransmission. Progress in
Neurobiology. 2010;90(2):246-255. 196. Paul V, Ekambaram P. Involvement of nitric oxide in learning & memory
processes. The Indian Journal of Medical Research. 2011;133:471-478. 197. Buerk DG, Barbee KA, Jaron D. Nitric oxide signaling in the microcirculation.
Critical Reviews in Biomedical Engineering. 2011;39(5):397-433. 198. Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure,
function and inhibition. The Biochemical Journal. 2001;357(Pt 3):593-615. 199. McCarron RM, Chen Y, Tomori T, et al. Endothelial-mediated regulation of
cerebral microcirculation. Journal of physiology and pharmacology. Journal of the Polish Physiological Society. 2006;57 Suppl 11:133-144.
200. Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease:
from marvel to menace. Circulation. 2006;113(13):1708-1714. 201. Yamakawa H, Jezova M, Ando H, Saavedra JM. Normalization of endothelial
and inducible nitric oxide synthase expression in brain microvessels of spontaneously hypertensive rats by angiotensin II AT1 receptor inhibition. Journal Cerebral Blood Flow and Metabolism. 2003;23(3):371-380.

6. Literaturverzeichnis
118
202. Bult H, Boeckxstaens GE, Pelckmans PA, Jordaens FH, Van Maercke YM, Herman AG. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature. 1990;345(6273):346-347.
203. Estrada C, Mengual E, Gonzalez C. Local NADPH-diaphorase neurons
innervate pial arteries and lie close or project to intracerebral blood vessels: a possible role for nitric oxide in the regulation of cerebral blood flow. Journal Cerebral Blood Flow and Metabolism. 1993;13(6):978-984.
204. Iadecola C, Beitz AJ, Renno W, Xu X, Mayer B, Zhang F. Nitric oxide synthase-
containing neural processes on large cerebral arteries and cerebral microvessels. Brain Research. 1993;606(1):148-155.
205. Iadecola C, Zhang F, Casey R, Clark HB, Ross ME. Inducible nitric oxide
synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke. 1996;27(8):1373-1380.
206. Forster C, Clark HB, Ross ME, Iadecola C. Inducible nitric oxide synthase
expression in human cerebral infarcts. Acta Neuropathologica. 1999;97(3):215-220.
207. Arnold WP, Mittal CK, Katsuki S, Murad F. Nitric oxide activates guanylate
cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proceedings of the National Academy of Sciences of the United States of America. 1977;74(8):3203-3207.
208. Evgenov OV, Pacher P, Schmidt PM, Hasko G, Schmidt HH, Stasch JP. NO-
independent stimulators and activators of soluble guanylate cyclase: discovery and therapeutic potential. Nature Reviews. Drug Discovery. 2006;5(9):755-768.
209. Fischmann TO, Hruza A, Niu XD, et al. Structural characterization of nitric oxide
synthase isoforms reveals striking active-site conservation. Nature Structural Biology. 1999;6(3):233-242.
210. Marsden PA, Heng HH, Scherer SW, et al. Structure and chromosomal
localization of the human constitutive endothelial nitric oxide synthase gene. The Journal of Biological Chemistry. 1993;268(23):17478-17488.
211. Forstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-
dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(5):1788-1792.
212. Abe K, Pan LH, Watanabe M, Konno H, Kato T, Itoyama Y. Upregulation of
protein-tyrosine nitration in the anterior horn cells of amyotrophic lateral sclerosis. Neurological Research. 1997;19(2):124-128.

6. Literaturverzeichnis
119
213. Colasanti M, Persichini T, Fabrizi C, et al. Expression of a NOS-III-like protein in human astroglial cell culture. Biochemical and Biophysical Research Communications. 1998;252(3):552-555.
214. Iwase K, Miyanaka K, Shimizu A, et al. Induction of endothelial nitric-oxide
synthase in rat brain astrocytes by systemic lipopolysaccharide treatment. The Journal of Biological Chemistry. 2000;275(16):11929-11933.
215. Reiling N, Kroncke R, Ulmer AJ, Gerdes J, Flad HD, Hauschildt S. Nitric oxide
synthase: expression of the endothelial, Ca2+/calmodulin-dependent isoform in human B and T lymphocytes. European Journal of Immunology. 1996;26(3):511-516.
216. Helfrich MH, Evans DE, Grabowski PS, Pollock JS, Ohshima H, Ralston SH.
Expression of nitric oxide synthase isoforms in bone and bone cell cultures. Journal of bone and mineral research. Journal of the American Society for Bone and Mineral Research. 1997;12(7):1108-1115.
217. Wang R, Ghahary A, Shen YJ, Scott PG, Tredget EE. Human dermal fibroblasts
produce nitric oxide and express both constitutive and inducible nitric oxide synthase isoforms. The Journal of Investigative Dermatology. 1996;106(3):419-427.
218. Balligand JL, Kobzik L, Han X, et al. Nitric oxide-dependent parasympathetic
signaling is due to activation of constitutive endothelial (type III) nitric oxide synthase in cardiac myocytes. The Journal of Biological Chemistry. 1995;270(24):14582-14586.
219. Zimmermann H, Kurzen P, Klossner W, Renner EL, Marti U. Decreased
constitutive hepatic nitric oxide synthase expression in secondary biliary fibrosis and its changes after Roux-en-Y choledocho-jejunostomy in the rat. Journal of Hepatology. 1996;25(4):567-573.
220. Xue C, Pollock J, Schmidt HH, Ward SM, Sanders KM. Expression of nitric oxide
synthase immunoreactivity by interstitial cells of the canine proximal colon. Journal of the Autonomic Nervous System. 1994;49(1):1-14.
221. Church JE, Fulton D. Differences in eNOS activity because of subcellular
localization are dictated by phosphorylation state rather than the local calcium environment. Journal of Biological Chemistry. 2006;281(3):1477-1488.
222. Hoffmann A, Gloe T, Pohl U. Hypoxia-induced upregulation of eNOS gene
expression is redox-sensitive: a comparison between hypoxia and inhibitors of cell metabolism. Journal of Cellular Physiology. 2001;188(1):33-44.
223. Le Cras TD, Xue C, Rengasamy A, Johns RA. Chronic hypoxia upregulates
endothelial and inducible NO synthase gene and protein expression in rat lung. The American Journal of Physiology. 1996;270(1 Pt 1):L164-170.

6. Literaturverzeichnis
120
224. Uematsu M, Ohara Y, Navas JP, et al. Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. The American Journal of Physiology. 1995;269(6 Pt 1):C1371-1378.
225. Malek AM, Izumo S, Alper SL. Modulation by pathophysiological stimuli of the
shear stress-induced up-regulation of endothelial nitric oxide synthase expression in endothelial cells. Neurosurgery. 1999;45(2):334-344.
226. Ruehlmann DO, Mann GE. Rapid non-genomic vasodilator actions of
oestrogens and sex steroids. Current Medicinal Chemistry. 2000;7(5):533-541. 227. Loscalzo J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis.
Circulation Research. 2001;88(8):756-762. 228. Moncada S, Higgs A. The L-arginine-nitric oxide pathway. The New England
Journal of Medicine. 1993;329(27):2002-2012. 229. Thomas DD, Ridnour LA, Isenberg JS, et al. The chemical biology of nitric oxide:
implications in cellular signaling. Free Radical Biology & Medicine. 2008;45(1):18-31.
230. Anggard E. Nitric oxide: mediator, murderer, and medicine. Lancet.
1994;343(8907):1199-1206. 231. Hall AV, Antoniou H, Wang Y, et al. Structural organization of the human
neuronal nitric oxide synthase gene (NOS1). The Journal of Biological Chemistry. 1994;269(52):33082-33090.
232. Ohkuma H, Itoh K, Shibata S, Suzuki S. Morphological changes of
intraparenchymal arterioles after experimental subarachnoid hemorrhage in dogs. Neurosurgery. 1997;41(1):230-235.
233. Vincent SR, Kimura H. Histochemical mapping of nitric oxide synthase in the rat
brain. Neuroscience. 1992;46(4):755-784. 234. Reuss S, Decker K, Rosseler L, Layes E, Schollmayer A, Spessert R. Nitric
oxide synthase in the hypothalamic suprachiasmatic nucleus of rat: evidence from histochemistry, immunohistochemistry and western blot; and colocalization with VIP. Brain Research. 1995;695(2):257-262.
235. Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase
indicating a neural role for nitric oxide. Nature. 1990;347(6295):768-770. 236. Cabrera C, Bohr D. The role of nitric oxide in the central control of blood
pressure. Biochemical and Biophysical Research Communications. 1995;206(1):77-81.
237. Yun HY, Dawson VL, Dawson TM. Nitric oxide in health and disease of the
nervous system. Molecular Psychiatry. 1997;2(4):300-310.

6. Literaturverzeichnis
121
238. Kagiyama S, Tsuchihashi T, Abe I, Fujishima M. Enhanced depressor response
to nitric oxide in the rostral ventrolateral medulla of spontaneously hypertensive rats. Hypertension. 1998;31(4):1030-1034.
239. Spyer KM. Annual review prize lecture. Central nervous mechanisms
contributing to cardiovascular control. The Journal of Physiology. 1994;474(1):1-19.
240. Tedeschi E, Menegazzi M, Margotto D, Suzuki H, Forstermann U, Kleinert H.
Anti-inflammatory actions of St. John's wort: inhibition of human inducible nitric-oxide synthase expression by down-regulating signal transducer and activator of transcription-1alpha (STAT-1alpha) activation. The Journal of Pharmacology and Experimental Therapeutics. 2003;307(1):254-261.
241. Cho HJ, Xie QW, Calaycay J, et al. Calmodulin is a subunit of nitric oxide
synthase from macrophages. The Journal of Experimental Medicine. 1992;176(2):599-604.
242. Guo FH, De Raeve HR, Rice TW, Stuehr DJ, Thunnissen FB, Erzurum SC.
Continuous nitric oxide synthesis by inducible nitric oxide synthase in normal human airway epithelium in vivo. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(17):7809-7813.
243. Bogdan C. Nitric oxide and the immune response. Nature Immunology.
2001;2(10):907-916. 244. Lyons CR, Orloff GJ, Cunningham JM. Molecular cloning and functional
expression of an inducible nitric oxide synthase from a murine macrophage cell line. The Journal of Biological Chemistry. 1992;267(9):6370-6374.
245. Xie QW, Cho HJ, Calaycay J, et al. Cloning and characterization of inducible
nitric oxide synthase from mouse macrophages. Science. 1992;256(5054):225-228.
246. Moncada S. Nitric oxide gas: mediator, modulator, and pathophysiologic entity.
The Journal of Laboratory and Clinical Medicine. 1992;120(2):187-191. 247. Hibbs JB, Jr., Taintor RR, Vavrin Z, Rachlin EM. Nitric oxide: a cytotoxic
activated macrophage effector molecule. Biochemical and Biophysical Research Communications. 1988;157(1):87-94.
248. Fang FC. Perspectives series: host/pathogen interactions. Mechanisms of nitric
oxide-related antimicrobial activity. The Journal of Clinical Investigation. 1997;99(12):2818-2825.
249. Mattson DL, Maeda CY, Bachman TD, Cowley AW, Jr. Inducible nitric oxide
synthase and blood pressure. Hypertension. 1998;31(1):15-20.

6. Literaturverzeichnis
122
250. Rubinstein I, Abassi Z, Coleman R, Milman F, Winaver J, Better OS. Involvement of nitric oxide system in experimental muscle crush injury. The Journal of Clinical Investigation. 1998;101(6):1325-1333.
251. Mashimo H, Goyal RK. Lessons from genetically engineered animal models. IV.
Nitric oxide synthase gene knockout mice. The American Journal of Physiology. 1999;277(4 Pt 1):G745-750.
252. Kolb H, Kolb-Bachofen V. Nitric oxide in autoimmune disease: cytotoxic or
regulatory mediator? Immunology Today. 1998;19(12):556-561. 253. Garry PS, Ezra M, Rowland MJ, Westbrook J, Pattinson KT. The role of the
nitric oxide pathway in brain injury and its treatment--from bench to bedside. Experimental Neurology. 2015;263:235-243.
254. Toda N, Ayajiki K, Okamura T. Cerebral blood flow regulation by nitric oxide in
neurological disorders. Canadian Journal of Physiology and Pharmacology. 2009;87(8):581-594.
255. Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology,
and pharmacology. Pharmacological Reviews. 1991;43(2):109-142. 256. Broos K, Feys HB, De Meyer SF, Vanhoorelbeke K, Deckmyn H. Platelets at
work in primary hemostasis. Blood Reviews. 2011;25(4):155-167. 257. Schuman EM, Madison DV. Nitric oxide and synaptic function. Annual Review
of Neuroscience. 1994;17:153-183. 258. Toda N, Okamura T. The pharmacology of nitric oxide in the peripheral nervous
system of blood vessels. Pharmacological Reviews. 2003;55(2):271-324. 259. Marletta MA. Mammalian synthesis of nitrite, nitrate, nitric oxide, and N-
nitrosating agents. Chemical Research in Toxicology. 1988;1(5):249-257. 260. Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the
expression of inducible nitric oxide synthase. Nitric oxide : biology and chemistry / Journal of the Nitric Oxide Society. 2010;23(2):75-93.
261. MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function.
Annual Review of Immunology. 1997;15:323-350. 262. Schwartz AY, Sehba FA, Bederson JB. Decreased nitric oxide availability
contributes to acute cerebral ischemia after subarachnoid hemorrhage. Neurosurgery. 2000;47(1):208-214.
263. Sehba FA, Chereshnev I, Maayani S, Friedrich V, Jr., Bederson JB. Nitric oxide
synthase in acute alteration of nitric oxide levels after subarachnoid hemorrhage. Neurosurgery. 2004;55(3):671-677

6. Literaturverzeichnis
123
264. Afshar JK, Pluta RM, Boock RJ, Thompson BG, Oldfield EH. Effect of intracarotid nitric oxide on primate cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 1995;83(1):118-122.
265. Kajita Y, Suzuki Y, Oyama H, et al. Combined effect of L-arginine and
superoxide dismutase on the spastic basilar artery after subarachnoid hemorrhage in dogs. J Neurosurg. 1994;80(3):476-483.
266. Watkins LD. Nitric oxide and cerebral blood flow: an update. Cerebrovascular
and Brain Metabolism Reviews. 1995;7(4):324-337. 267. Sobey CG, Faraci FM. Subarachnoid haemorrhage: what happens to the
cerebral arteries? Clinical and Experimental Pharmacology & Physiology. 1998;25(11):867-876.
268. Pluta RM. Delayed cerebral vasospasm and nitric oxide: review, new
hypothesis, and proposed treatment. Pharmacology & Therapeutics. 2005;105(1):23-56.
269. Hlatky R, Goodman JC, Valadka AB, Robertson CS. Role of nitric oxide in
cerebral blood flow abnormalities after traumatic brain injury. Journal Cerebral Blood Flow and Metabolism. 2003;23(5):582-588.
270. Terpolilli NA, Kim SW, Thal SC, et al. Inhalation of nitric oxide prevents ischemic
brain damage in experimental stroke by selective dilatation of collateral arterioles. Circulation Research. 2012;110(5):727-738.
271. Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. British
Journal of Pharmacology. 2007;151(3):305-321. 272. Fathi AR, Bakhtian KD, Pluta RM. The role of nitric oxide donors in treating
cerebral vasospasm after subarachnoid hemorrhage. Acta Neurochirurgica. Supplement. 2011;110(Pt 1):93-97.
273. Scatena R, Bottoni P, Pontoglio A, Giardina B. Pharmacological modulation of
nitric oxide release: new pharmacological perspectives, potential benefits and risks. Current Medicinal Chemistry. 2010;17(1):61-73.
274. Allen GS, Gross CJ. Cerebral arterial spasm. Part 7: In vitro effects of alpha
adrenergic agents on canine arteries from six anatomical sites and six blocking agents on serotonin-induced contractions of the canine basilar artery. Surgical Neurology. 1976;6(2):63-70.
275. Allen GS. Cerebral arterial spasm. Part 8: The treatment of delayed cerebral
arterial spasm in human beings. Surgical Neurology. 1976;6(2):71-80. 276. Ram Z, Spiegelman R, Findler G, Hadani M. Delayed postoperative
neurological deterioration from prolonged sodium nitroprusside administration. Case report. Journal of Neurosurgery. 1989;71(4):605-607.

6. Literaturverzeichnis
124
277. Nakao K, Murata H, Kanamaru K, Waga S. Effects of nitroglycerin on
vasospasm and cyclic nucleotides in a primate model of subarachnoid hemorrhage. Stroke. 1996;27(10):1882-1887.
278. Reinert M, Wiest R, Barth L, Andres R, Ozdoba C, Seiler R. Transdermal
nitroglycerin in patients with subarachnoid hemorrhage. Neurological Research. 2004;26(4):435-439.
279. Tanaka Y, Masuzawa T, Saito M, et al. Combined administration of Fasudil
hydrochloride and nitroglycerin for treatment of cerebral vasospasm. Acta Neurochirurgica. Supplement. 2001;77:205-207.
280. Marbacher S, Neuschmelting V, Graupner T, Jakob SM, Fandino J. Prevention
of delayed cerebral vasospasm by continuous intrathecal infusion of glyceroltrinitrate and nimodipine in the rabbit model in vivo. Intensive Care Medicine. 2008;34(5):932-938.
281. Pluta RM, Oldfield EH, Boock RJ. Reversal and prevention of cerebral
vasospasm by intracarotid infusions of nitric oxide donors in a primate model of subarachnoid hemorrhage. Journal of Neurosurgery. 1997;87(5):746-751.
282. Clatterbuck RE, Gailloud P, Tierney T, Clatterbuck VM, Murphy KJ, Tamargo
RJ. Controlled release of a nitric oxide donor for the prevention of delayed cerebral vasospasm following experimental subarachnoid hemorrhage in nonhuman primates. Journal of Neurosurgery. 2005;103(4):745-751.
283. Aihara Y, Jahromi BS, Yassari R, Sayama T, Macdonald RL. Effects of a nitric
oxide donor on and correlation of changes in cyclic nucleotide levels with experimental vasospasm. Neurosurgery. 2003;52(3):661-667.
284. Keefer LK. Progress toward clinical application of the nitric oxide-releasing
diazeniumdiolates. Annual Review of Pharmacology and Toxicology. 2003;43:585-607.
285. Giedrojc J, Weichert W, Breddin HK. [Antithrombotic effects of molsidomine
alone and in combination with other antithrombotic agents in a thrombosis model of the microcirculation of the rat]. Zeitschrift fur Kardiologie. 1991;80 Suppl 5:35-39.
286. Oldfield EH, Loomba JJ, Monteith SJ, et al. Safety and pharmacokinetics of
sodium nitrite in patients with subarachnoid hemorrhage: a phase IIa study. Journal of Neurosurgery. 2013;119(3):634-641.
287. Pluta RM, Oldfield EH, Bakhtian KD, et al. Safety and feasibility of long-term
intravenous sodium nitrite infusion in healthy volunteers. PloS One. 2011;6(1):e14504.

6. Literaturverzeichnis
125
288. Frostell C, Fratacci MD, Wain JC, Jones R, Zapol WM. Inhaled nitric oxide. A selective pulmonary vasodilator reversing hypoxic pulmonary vasoconstriction. Circulation. 1991;83(6):2038-2047.
289. Pepke-Zaba J, Higenbottam TW, Dinh-Xuan AT, Stone D, Wallwork J. Inhaled
nitric oxide as a cause of selective pulmonary vasodilatation in pulmonary hypertension. Lancet. 1991;338(8776):1173-1174.
290. Ichinose F, Roberts JD, Jr., Zapol WM. Inhaled nitric oxide: a selective
pulmonary vasodilator: current uses and therapeutic potential. Circulation. 2004;109(25):3106-3111.
291. Peliowski A, Canadian Paediatric Society F, Newborn C. Inhaled nitric oxide use
in newborns. Paediatrics & Child Health. 2012;17(2):95-100. 292. Finer NN, Barrington KJ. Nitric oxide for respiratory failure in infants born at or
near term. The Cochrane Database of Systematic Reviews. 2006(4):CD000399.
293. Bloch KD, Ichinose F, Roberts JD, Jr., Zapol WM. Inhaled NO as a therapeutic
agent. Cardiovascular Research. 2007;75(2):339-348. 294. Fox-Robichaud A, Payne D, Hasan SU, et al. Inhaled NO as a viable
antiadhesive therapy for ischemia/reperfusion injury of distal microvascular beds. The Journal of Clinical Investigation. 1998;101(11):2497-2505.
295. Nagasaka Y, Fernandez BO, Garcia-Saura MF, et al. Brief periods of nitric oxide
inhalation protect against myocardial ischemia-reperfusion injury. Anesthesiology. 2008;109(4):675-682.
296. Rosenberg AA, Kinsella JP, Abman SH. Cerebral hemodynamics and
distribution of left ventricular output during inhalation of nitric oxide. Critical Care Medicine. 1995;23(8):1391-1397.
297. Lopes Cardozo RH, de Beaufort AJ, Gesink BJ, et al. Inhalation of nitric oxide:
effect on cerebral hemodynamics and activity, and antioxidant status in the newborn lamb. Biology of the Neonate. 1996;69(4):284-292.
298. Kusuda S, Shishida N, Miyagi N, Hirabayashi M, Kim TJ. Cerebral blood flow
during treatment for pulmonary hypertension. Archives of Disease in Childhood. Fetal and Neonatal Edition. 1999;80(1):F30-33.
299. Vavilala MS, Roberts JS, Moore AE, Newell DW, Lam AM. The influence of
inhaled nitric oxide on cerebral blood flow and metabolism in a child with traumatic brain injury. Anesthesia and Analgesia. 2001;93(2):351-353 , 353rd contents page.

6. Literaturverzeichnis
126
300. Kuebler WM, Kisch-Wedel H, Kemming GI, et al. Inhaled nitric oxide induces cerebrovascular effects in anesthetized pigs. Neuroscience Letters. 2003;348(2):85-88.
301. Terpolilli NA, Moskowitz MA, Plesnila N. Nitric oxide: considerations for the
treatment of ischemic stroke. Journal Cerebral Blood Flow and Metabolism. 2012;32(7):1332-1346.
302. Paulson OB, Strandgaard S, Edvinsson L. Cerebral autoregulation.
Cerebrovascular and Brain Metabolism Reviews. 1990;2(2):161-192. 303. Ruland S, Aiyagari V. Cerebral autoregulation and blood pressure lowering.
Hypertension. 2007;49(5):977-978. 304. Voldby B, Enevoldsen EM, Jensen FT. Cerebrovascular reactivity in patients
with ruptured intracranial aneurysms. Journal of Neurosurgery. 1985;62(1):59-67.
305. Giller CA. A bedside test for cerebral autoregulation using transcranial Doppler
ultrasound. Acta Neurochirurgica. 1991;108(1-2):7-14. 306. Steinmeier R, Bauhuf C, Hubner U, et al. Slow rhythmic oscillations of blood
pressure, intracranial pressure, microcirculation, and cerebral oxygenation. Dynamic interrelation and time course in humans. Stroke. 1996;27(12):2236-2243.
307. Rosner MJ. Introduction to cerebral perfusion pressure management.
Neurosurgery Clinics of North America. 1995;6(4):761-773. 308. Lang EW, Chesnut RM. A bedside method for investigating the integrity and
critical thresholds of cerebral pressure autoregulation in severe traumatic brain injury patients. British Journal of Neurosurgery. 2000;14(2):117-126.
309. Czosnyka M, Richards H, Kirkpatrick P, Pickard J. Assessment of cerebral
autoregulation with ultrasound and laser Doppler wave forms--an experimental study in anesthetized rabbits. Neurosurgery. 1994;35(2):287-292.
310. Lassen NA. Cerebral blood flow and oxygen consumption in man. Physiological
Reviews. 1959;39(2):183-238. 311. Udomphorn Y, Armstead WM, Vavilala MS. Cerebral blood flow and
autoregulation after pediatric traumatic brain injury. Pediatric Neurology. 2008;38(4):225-234.
312. Severinghaus JW, Lassen N. Step hypocapnia to separate arterial from tissue
PCO2 in the regulation of cerebral blood flow. Circulation Research. 1967;20(2):272-278.

6. Literaturverzeichnis
127
313. Lavi S, Egbarya R, Lavi R, Jacob G. Role of nitric oxide in the regulation of cerebral blood flow in humans: chemoregulation versus mechanoregulation. Circulation. 2003;107(14):1901-1905.
314. Ebel H, Rust DS, Leschinger A, et al. Vasomotion, regional cerebral blood flow
and intracranial pressure after induced subarachnoid haemorrhage in rats. Zentralblatt fur Neurochirurgie. 1996;57(3):150-155.
315. Schmieder K, Moller F, Engelhardt M, et al. Dynamic cerebral autoregulation in
patients with ruptured and unruptured aneurysms after induction of general anesthesia. Zentralblatt fur Neurochirurgie. 2006;67(2):81-87.
316. Lam JM, Smielewski P, Czosnyka M, Pickard JD, Kirkpatrick PJ. Predicting
delayed ischemic deficits after aneurysmal subarachnoid hemorrhage using a transient hyperemic response test of cerebral autoregulation. Neurosurgery. 2000;47(4):819-825.
317. Lang EW, Diehl RR, Mehdorn HM. Cerebral autoregulation testing after
aneurysmal subarachnoid hemorrhage: the phase relationship between arterial blood pressure and cerebral blood flow velocity. Critical Care Medicine. 2001;29(1):158-163.
318. Ratsep T, Asser T. Cerebral hemodynamic impairment after aneurysmal
subarachnoid hemorrhage as evaluated using transcranial doppler ultrasonography: relationship to delayed cerebral ischemia and clinical outcome. Journal of Neurosurgery. 2001;95(3):393-401.
319. Vasquez-Vivar J, Kalyanaraman B, Martasek P, et al. Superoxide generation by
endothelial nitric oxide synthase: the influence of cofactors. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9220-9225.
320. Stuehr D, Pou S, Rosen GM. Oxygen reduction by nitric-oxide synthases. The
Journal of Biological Chemistry. 2001;276(18):14533-14536. 321. Zimmermann C, Haberl RL. L-arginine improves diminished cerebral CO2
reactivity in patients. Stroke. 2003;34(3):643-647. 322. Tseng MY, Czosnyka M, Richards H, Pickard JD, Kirkpatrick PJ. Effects of acute
treatment with pravastatin on cerebral vasospasm, autoregulation, and delayed ischemic deficits after aneurysmal subarachnoid hemorrhage: a phase II randomized placebo-controlled trial. Stroke. 2005;36(8):1627-1632.
323. Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor
peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411-415.
324. Inoue A, Yanagisawa M, Kimura S, et al. The human endothelin family: three
structurally and pharmacologically distinct isopeptides predicted by three

6. Literaturverzeichnis
128
separate genes. Proceedings of the National Academy of Sciences of the United States of America. 1989;86(8):2863-2867.
325. Hocher B, Schwarz A, Fagan KA, et al. Pulmonary fibrosis and chronic lung
inflammation in ET-1 transgenic mice. Am J Respir Cell Mol Biol. 2000;23(1):19-26.
326. Malek A, Izumo S. Physiological fluid shear stress causes downregulation of
endothelin-1 mRNA in bovine aortic endothelium. The American Journal of Physiology. 1992;263(2 Pt 1):C389-396.
327. Barton M, Shaw S, d'Uscio LV, Moreau P, Luscher TF. Angiotensin II increases
vascular and renal endothelin-1 and functional endothelin converting enzyme activity in vivo: role of ETA receptors for endothelin regulation. Biochemical and Biophysical Research Communications. 1997;238(3):861-865.
328. Ito H, Hirata Y, Adachi S, et al. Endothelin-1 is an autocrine/paracrine factor in
the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. The Journal of Clinical Investigation. 1993;92(1):398-403.
329. Matsuura A, Yamochi W, Hirata K, Kawashima S, Yokoyama M. Stimulatory
interaction between vascular endothelial growth factor and endothelin-1 on each gene expression. Hypertension. 1998;32(1):89-95.
330. Corder R, Carrier M, Khan N, Klemm P, Vane JR. Cytokine regulation of
endothelin-1 release from bovine aortic endothelial cells. Journal of Cardiovascular Pharmacology. 1995;26 Suppl 3:S56-58.
331. Schwarting A, Schlaak J, Lotz J, Pfers I, Meyer zum Buschenfelde KH, Mayet
WJ. Endothelin-1 modulates the expression of adhesion molecules on fibroblast-like synovial cells (FLS). Scand J Rheumatol. 1996;25(4):246-256.
332. Yanagisawa M, Inoue A, Takuwa Y, Mitsui Y, Kobayashi M, Masaki T. The
human preproendothelin-1 gene: possible regulation by endothelial phosphoinositide turnover signaling. Journal of Cardiovascular Pharmacology. 1989;13 Suppl 5:S13-17.
333. Yoshimoto S, Ishizaki Y, Sasaki T, Murota S. Effect of carbon dioxide and
oxygen on endothelin production by cultured porcine cerebral endothelial cells. Stroke. 1991;22(3):378-383.
334. Unoki H, Fan J, Watanabe T. Low-density lipoproteins modulate endothelial
cells to secrete endothelin-1 in a polarized pattern: a study using a culture model system simulating arterial intima. Cell and Tissue Research. 1999;295(1):89-99.
335. Lam HC, Takahashi K, Ghatei MA, Warrens AN, Rees AJ, Bloom SR.
Immunoreactive endothelin in human plasma, urine, milk, and saliva. Journal of Cardiovascular Pharmacology. 1991;17 Suppl 7:S390-393.

6. Literaturverzeichnis
129
336. Rubanyi GM, Polokoff MA. Endothelins: molecular biology, biochemistry,
pharmacology, physiology, and pathophysiology. Pharmacological Reviews. 1994;46(3):325-415.
337. Rothoerl RD, Ringel F. Molecular mechanisms of cerebral vasospasm following
aneurysmal SAH. Neurological Research. 2007;29(7):636-642. 338. Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure
and salt homeostasis by endothelin. Physiological Reviews. 2011;91(1):1-77. 339. Levin ER. Endothelins. The New England Journal of Medicine.
1995;333(6):356-363. 340. Seo B, Oemar BS, Siebenmann R, von Segesser L, Luscher TF. Both ETA and
ETB receptors mediate contraction to endothelin-1 in human blood vessels. Circulation. 1994;89(3):1203-1208.
341. Somlyo AV, Bond M, Somlyo AP, Scarpa A. Inositol trisphosphate-induced
calcium release and contraction in vascular smooth muscle. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(15):5231-5235.
342. Simonson MS, Dunn MJ. Cellular signaling by peptides of the endothelin gene
family. FASEB J. 1990;4(12):2989-3000. 343. Thampatty BP, Sherwood PR, Gallek MJ, et al. Role of endothelin-1 in human
aneurysmal subarachnoid hemorrhage: associations with vasospasm and delayed cerebral ischemia. Neurocritical Care. 2011;15(1):19-27.
344. Fassbender K, Hodapp B, Rossol S, et al. Endothelin-1 in subarachnoid
hemorrhage: An acute-phase reactant produced by cerebrospinal fluid leukocytes. Stroke. 2000;31(12):2971-2975.
345. Suzuki K, Meguro K, Sakurai T, Saitoh Y, Takeuchi S, Nose T. Endothelin-1
concentration increases in the cerebrospinal fluid in cerebral vasospasm caused by subarachnoid hemorrhage. Surgical Neurology. 2000;53(2):131-135.
346. Kastner S, Oertel MF, Scharbrodt W, Krause M, Boker DK, Deinsberger W.
Endothelin-1 in plasma, cisternal CSF and microdialysate following aneurysmal SAH. Acta Neurochirurgica. 2005;147(12):1271-1279.
347. Kessler IM, Pacheco YG, Lozzi SP, de Araujo AS, Jr., Onishi FJ, de Mello PA.
Endothelin-1 levels in plasma and cerebrospinal fluid of patients with cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Surgical Neurology. 2005;64 Suppl 1:S1:2-5.
348. Hansen-Schwartz J, Hoel NL, Zhou M, Xu CB, Svendgaard NA, Edvinsson L.
Subarachnoid hemorrhage enhances endothelin receptor expression and

6. Literaturverzeichnis
130
function in rat cerebral arteries. Neurosurgery. 2003;52(5):1188-1194; 1194-1185.
349. Vikman P, Beg S, Khurana TS, Hansen-Schwartz J, Edvinsson L. Gene
expression and molecular changes in cerebral arteries following subarachnoid hemorrhage in the rat. J Neurosurg. 2006;105(3):438-444.
350. Kasuya H, Weir BK, White DM, Stefansson K. Mechanism of oxyhemoglobin-
induced release of endothelin-1 from cultured vascular endothelial cells and smooth-muscle cells. Journal of Neurosurgery. 1993;79(6):892-898.
351. Peltonen S, Juvela S, Kaste M, Lassila R. Hemostasis and fibrinolysis activation
after subarachnoid hemorrhage. Journal of Neurosurgery. 1997;87(2):207-214. 352. Ramzy D, Rao V, Tumiati LC, et al. Elevated endothelin-1 levels impair nitric
oxide homeostasis through a PKC-dependent pathway. Circulation. 2006;114(1 Suppl):I319-326.
353. Vajkoczy P, Meyer B, Weidauer S, et al. Clazosentan (AXV-034343), a selective
endothelin A receptor antagonist, in the prevention of cerebral vasospasm following severe aneurysmal subarachnoid hemorrhage: results of a randomized, double-blind, placebo-controlled, multicenter phase IIa study. Journal of Neurosurgery. 2005;103(1):9-17.
354. Macdonald RL, Kassell NF, Mayer S, et al. Clazosentan to overcome
neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008;39(11):3015-3021.
355. Macdonald RL, Higashida RT, Keller E, et al. Randomized trial of clazosentan
in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling. Stroke. 2012;43(6):1463-1469.
356. Kramer A, Fletcher J. Do endothelin-receptor antagonists prevent delayed
neurological deficits and poor outcomes after aneurysmal subarachnoid hemorrhage?: a meta-analysis. Stroke. 2009;40(10):3403-3406.
357. Nogueira RG, Bodock MJ, Koroshetz WJ, et al. High-dose bosentan in the
prevention and treatment of subarachnoid hemorrhage-induced cerebral vasospasm: an open-label feasibility study. Neurocritical Care. 2007;7(3):194-202.
358. Shaw MD, Vermeulen M, Murray GD, Pickard JD, Bell BA, Teasdale GM.
Efficacy and safety of the endothelin, receptor antagonist TAK-044 in treating subarachnoid hemorrhage: a report by the Steering Committee on behalf of the UK/Netherlands/Eire TAK-044 Subarachnoid Haemorrhage Study Group. J Neurosurg. 2000;93(6):992-997.

6. Literaturverzeichnis
131
359. Vergouwen MD. Effect of endothelin-receptor antagonists on delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage remains unclear. Stroke. 2009;40(12):e714; author reply e715-716.
360. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, Group NCRRGW.
Animal research: reporting in vivo experiments: the ARRIVE guidelines. British Journal of Pharmacology. 2010;160(7):1577-1579.
361. Guillen J. FELASA guidelines and recommendations. Journal of the American
Association for Laboratory Animal Science : JAALAS. 2012;51(3):311-321. 362. Thal SC, Plesnila N. Non-invasive intraoperative monitoring of blood pressure
and arterial pCO2 during surgical anesthesia in mice. Journal of Neuroscience Methods. 2007;159(2):261-267.
363. Kataoka H, Kim SW, Plesnila N. Leukocyte-endothelium interactions during
permanent focal cerebral ischemia in mice. Journal Cerebral Blood Flow and Metabolism. 2004;24(6):668-676.
364. Plesnila N, Putz C, Rinecker M, et al. Measurement of absolute values of
hemoglobin oxygenation in the brain of small rodents by near infrared reflection spectrophotometry. Journal of Neuroscience Methods. 2002;114(2):107-117.
365. Frerichs KU, Feuerstein GZ. Laser-Doppler flowmetry. A review of its
application for measuring cerebral and spinal cord blood flow. Molecular and chemical neuropathology / sponsored by the International Society for Neurochemistry and the World Federation of Neurology and Research Groups on Neurochemistry and Cerebrospinal Fluid. 1990;12(1):55-70.
366. Schmid-Elsaesser R, Zausinger S, Hungerhuber E, Baethmann A, Reulen HJ.
A critical reevaluation of the intraluminal thread model of focal cerebral ischemia: evidence of inadvertent premature reperfusion and subarachnoid hemorrhage in rats by laser-Doppler flowmetry. Stroke. 1998;29(10):2162-2170.
367. Strahler AN. Geomorphic Significance of Valleys and Parks of the Kaibab and
Coconino Plateaus, Arizona. Science. 1944;100(2593):219-220. 368. Jiang ZL, Kassab GS, Fung YC. Diameter-defined Strahler system and
connectivity matrix of the pulmonary arterial tree. Journal of Applied Physiology. 1994;76(2):882-892.
369. Lapi D, Marchiafava PL, Colantuoni A. Pial microvascular responses to transient
bilateral common carotid artery occlusion: effects of hypertonic glycerol. Journal of Vascular Research. 2008;45(2):89-102.
370. Nordsletten DA, Blackett S, Bentley MD, Ritman EL, Smith NP. Structural
morphology of renal vasculature. American journal of physiology. Heart and Circulatory Physiology. 2006;291(1):H296-309.

6. Literaturverzeichnis
132
371. Toroczkai Z. Topological classification of binary trees using the Horton-Strahler
index. Physical review. E, Statistical, Nonlinear, and Soft Matter Physics. 2002;65(1 Pt 2):016130.
372. Uylings HB, Smit GJ, Veltman WA. Ordering methods in quantitative analysis of
branching structures of dendritic trees. Advances in Neurology. 1975;12:347-354.
373. Roux S, Breu V, Giller T, et al. Ro 61-1790, a new hydrosoluble endothelin
antagonist: general pharmacology and effects on experimental cerebral vasospasm. The Journal of Pharmacology and Experimental Therapeutics. 1997;283(3):1110-1118.
374. Breu V, Clozel M, Burri K, Hirth G, Neidhart W, Ramuz H. In vitro
characterisation of Ro 46-8443, the first non-peptide antagonist selective for the endothelin ETB receptor. FEBS Letters. 1996;383(1-2):37-41.
375. Bruderer S, Detishin V, Tsvitbaum N, Dingemanse J. Influence of different
degrees of liver impairment on the pharmacokinetics of clazosentan. British Journal of Clinical Pharmacology. 2011;71(1):52-60.
376. Clozel M, Breu V, Gray GA, et al. Pharmacological characterization of bosentan,
a new potent orally active nonpeptide endothelin receptor antagonist. The Journal of Pharmacology and Experimental Therapeutics. 1994;270(1):228-235.
377. Sabri M, Ai J, Macdonald RL. Dissociation of vasospasm and secondary effects
of experimental subarachnoid hemorrhage by clazosentan. Stroke. 2011;42(5):1454-1460.
378. Chen G, Tariq A, Ai J, et al. Different effects of clazosentan on consequences
of subarachnoid hemorrhage in rats. Brain Research. 2011;1392:132-139. 379. Schubert GA, Schilling L, Thome C. Clazosentan, an endothelin receptor
antagonist, prevents early hypoperfusion during the acute phase of massive experimental subarachnoid hemorrhage: a laser Doppler flowmetry study in rats. Journal of Neurosurgery. 2008;109(6):1134-1140.
380. Vatter H, Konczalla J, Weidauer S, et al. Characterization of the endothelin-B
receptor expression and vasomotor function during experimental cerebral vasospasm. Neurosurgery. 2007;60(6):1100-1108.
381. Kleinbongard P, Dejam A, Lauer T, et al. Plasma nitrite reflects constitutive nitric
oxide synthase activity in mammals. Free Radical Biology & Medicine. 2003;35(7):790-796.
382. Godecke A, Decking UK, Ding Z, et al. Coronary hemodynamics in endothelial
NO synthase knockout mice. Circulation Research. 1998;82(2):186-194.

6. Literaturverzeichnis
133
383. Gregg AR, Schauer A, Shi O, Liu Z, Lee CG, O'Brien WE. Limb reduction
defects in endothelial nitric oxide synthase-deficient mice. The American Journal of Physiology. 1998;275(6 Pt 2):H2319-2324.
384. Shesely EG, Maeda N, Kim HS, et al. Elevated blood pressures in mice lacking
endothelial nitric oxide synthase. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(23):13176-13181.
385. Huang PL, Huang Z, Mashimo H, et al. Hypertension in mice lacking the gene
for endothelial nitric oxide synthase. Nature. 1995;377(6546):239-242. 386. Wood KC, Cortese-Krott MM, Kovacic JC, et al. Circulating blood endothelial
nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2013;33(8):1861-1871.
387. Schwarzmaier SM, Terpolilli NA, Dienel A, et al. Endothelial nitric oxide
synthase mediates arteriolar vasodilatation after traumatic brain injury in mice. Journal of Neurotrauma. 2015;32(10)731-738.
388. Nelson RJ. The use of genetic "knockout" mice in behavioral endocrinology
research. Hormones and Behavior. 1997;31(3):188-196. 389. Boer R, Ulrich WR, Klein T, Mirau B, Haas S, Baur I. The inhibitory potency and
selectivity of arginine substrate site nitric-oxide synthase inhibitors is solely determined by their affinity toward the different isoenzymes. Molecular Pharmacology. 2000;58(5):1026-1034.
390. Garvey EP, Oplinger JA, Furfine ES, et al. 1400W is a slow, tight binding, and
highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. The Journal of Biological Chemistry. 1997;272(8):4959-4963.
391. Zhang HQ, Fast W, Marletta MA, Martasek P, Silverman RB. Potent and
selective inhibition of neuronal nitric oxide synthase by N omega-propyl-L-arginine. Journal of Medicinal Chemistry. 1997;40(24):3869-3870.
392. Babu BR, Griffith OW. N5-(1-Imino-3-butenyl)-L-ornithine. A neuronal isoform
selective mechanism-based inactivator of nitric oxide synthase. The Journal of Biological Chemistry. 1998;273(15):8882-8889.
393. Pigott B, Bartus K, Garthwaite J. On the selectivity of neuronal NOS inhibitors.
British Journal of Pharmacology. 2013;168(5):1255-1265. 394. Ellis EF, Wei EP, Cockrell CS, Choi S, Kontos HA. The effect of PGF2 alpha on
in vivo cerebral arteriolar diameter in cats and rats. Prostaglandins. 1983;26(6):917-923.

6. Literaturverzeichnis
134
395. Deng J, Lei C, Chen Y, et al. Neuroprotective gases--fantasy or reality for clinical use? Progress in Neurobiology. 2014;115:210-245.
396. Gargiulo S, Greco A, Gramanzini M, et al. Mice anesthesia, analgesia, and care,
Part I: anesthetic considerations in preclinical research. ILAR Journal / National Research Council, Institute of Laboratory Animal Resources. 2012;53(1):E55-69.
397. Hoffman WE, Thomas C, Albrecht RF. The effect of halothane and isoflurane
on neurologic outcome following incomplete cerebral ischemia in the rat. Anesthesia and Analgesia. 1993;76(2):279-283.
398. Inoue S, Kawaguchi M, Kurehara K, et al. Mild hypothermia can enhance pial
arteriolar vasodilation induced by isoflurane and sevoflurane in cats. Critical Care Medicine. 2002;30(8):1863-1869.
399. Li CX, Patel S, Auerbach EJ, Zhang X. Dose-dependent effect of isoflurane on
regional cerebral blood flow in anesthetized macaque monkeys. Neuroscience Letters. 2013;541:58-62.
400. Burchell SR, Dixon BJ, Tang J, Zhang JH. Isoflurane provides neuroprotection
in neonatal hypoxic ischemic brain injury. Journal of Investigative Medicine : the official publication of the American Federation for Clinical Research. 2013;61(7):1078-1083.
401. Zhao P, Zuo Z. Isoflurane preconditioning induces neuroprotection that is
inducible nitric oxide synthase-dependent in neonatal rats. Anesthesiology. 2004;101(3):695-703.
402. Xiong L, Zheng Y, Wu M, et al. Preconditioning with isoflurane produces dose-
dependent neuroprotection via activation of adenosine triphosphate-regulated potassium channels after focal cerebral ischemia in rats. Anesthesia and Analgesia. 2003;96(1):233-237.
403. Matchett GA, Allard MW, Martin RD, Zhang JH. Neuroprotective effect of volatile
anesthetic agents: molecular mechanisms. Neurological Research. 2009;31(2):128-134.
404. Zhang HP, Yuan LB, Zhao RN, et al. Isoflurane preconditioning induces
neuroprotection by attenuating ubiquitin-conjugated protein aggregation in a mouse model of transient global cerebral ischemia. Anesthesia and Analgesia. 2010;111(2):506-514.
405. Altay O, Suzuki H, Hasegawa Y, et al. Isoflurane attenuates blood-brain barrier
disruption in ipsilateral hemisphere after subarachnoid hemorrhage in mice. Stroke. 2012;43(9):2513-2516.

6. Literaturverzeichnis
135
406. Khatibi NH, Ma Q, Rolland W, et al. Isoflurane posttreatment reduces brain injury after an intracerebral hemorrhagic stroke in mice. Anesthesia and Analgesia. 2011;113(2):343-348.
407. Li H, Yin J, Li L, Deng J, Feng C, Zuo Z. Isoflurane postconditioning reduces
ischemia-induced nuclear factor-kappaB activation and interleukin 1beta production to provide neuroprotection in rats and mice. Neurobiology of Disease. 2013;54:216-224.
408. Altay O, Suzuki H, Hasegawa Y, Ostrowski RP, Tang J, Zhang JH. Isoflurane
on brain inflammation. Neurobiology of Disease. 2014;62:365-371. 409. Ozden S, Isenmann S. Neuroprotective properties of different anesthetics on
axotomized rat retinal ganglion cells in vivo. Journal of Neurotrauma. 2004;21(1):73-82.
410. Jolkkonen J, Puurunen K, Koistinaho J, et al. Neuroprotection by the alpha2-
adrenoceptor agonist, dexmedetomidine, in rat focal cerebral ischemia. European Journal of Pharmacology. 1999;372(1):31-36.
411. Maier C, Steinberg GK, Sun GH, Zhi GT, Maze M. Neuroprotection by the alpha
2-adrenoreceptor agonist dexmedetomidine in a focal model of cerebral ischemia. Anesthesiology. 1993;79(2):306-312.
412. Ito H, Watanabe Y, Isshiki A, Uchino H. Neuroprotective properties of propofol
and midazolam, but not pentobarbital, on neuronal damage induced by forebrain ischemia, based on the GABAA receptors. Acta Anaesthesiologica Scandinavica. 1999;43(2):153-162.
413. Dalkara T, Irikura K, Huang Z, Panahian N, Moskowitz MA. Cerebrovascular
responses under controlled and monitored physiological conditions in the anesthetized mouse. Journal Cerebral Blood Flow and Metabolism. 1995;15(4):631-638.
414. Scheid P, Lofaso F, Isabey D, Harf A. Respiratory response to inhaled CO2
during positive inspiratory pressure in humans. Journal of Applied Physiology. 1994;77(2):876-882.
415. Larsen R. [Multiple organ failure--light at the end of the tunnel?]. Der
Anaesthesist. 2001;50(11):817-818. 416. Rajan V, Varghese B, van Leeuwen TG, Steenbergen W. Review of
methodological developments in laser Doppler flowmetry. Lasers in Medical Science. 2009;24(2):269-283.
417. Eyre JA, Essex TJ, Flecknell PA, Bartholomew PH, Sinclair JI. A comparison of
measurements of cerebral blood flow in the rabbit using laser Doppler spectroscopy and radionuclide labelled microspheres. Clinical physics and physiological measurement : an official journal of the Hospital Physicists'

6. Literaturverzeichnis
136
Association, Deutsche Gesellschaft fur Medizinische Physik and the European Federation of Organisations for Medical Physics. 1988;9(1):65-74.
418. Kocher M. Metabolic and hemodynamic activation of postischemic rat brain by
cortical spreading depression. Journal Cerebral Blood Flow and Metabolism. 1990;10(4):564-571.
419. Carter LP. Surface monitoring of cerebral cortical blood flow. Cerebrovascular
and Brain Metabolism Reviews. 1991;3(3):246-261. 420. Piper RD, Lambert GA, Duckworth JW. Cortical blood flow changes during
spreading depression in cats. The American Journal of Physiology. 1991;261(1 Pt 2):H96-102.
421. Morita-Tsuzuki Y, Bouskela E, Hardebo JE. Vasomotion in the rat cerebral
microcirculation recorded by laser-Doppler flowmetry. Acta Physiologica Scandinavica. 1992;146(4):431-439.
422. Flohr H, Hoppe A. Autoradiographic technique to assess distribution of blood
flow within organs. Pflugers Archiv : European Journal of Physiology. 1969;310(1):16-21.
423. Lin CL, Calisaneller T, Ukita N, Dumont AS, Kassell NF, Lee KS. A murine
model of subarachnoid hemorrhage-induced cerebral vasospasm. Journal of Neuroscience Methods. 2003;123(1):89-97.
424. Solomon RA, Antunes JL, Chen RY, Bland L, Chien S. Decrease in cerebral
blood flow in rats after experimental subarachnoid hemorrhage: a new animal model. Stroke. 1985;16(1):58-64.
425. Delgado TJ, Brismar J, Svendgaard NA. Subarachnoid haemorrhage in the rat:
angiography and fluorescence microscopy of the major cerebral arteries. Stroke. 1985;16(4):595-602.
426. Altay T, Smithason S, Volokh N, Rasmussen PA, Ransohoff RM, Provencio JJ.
A novel method for subarachnoid hemorrhage to induce vasospasm in mice. Journal of Neuroscience Methods. 2009;183(2):136-140.
427. Buhler D, Schuller K, Plesnila N. Protocol for the induction of subarachnoid
hemorrhage in mice by perforation of the Circle of Willis with an endovascular filament. Translational Stroke Research. 2014;5(6):653-659.
428. Titova E, Ostrowski RP, Zhang JH, Tang J. Experimental models of
subarachnoid hemorrhage for studies of cerebral vasospasm. Neurological Research. 2009;31(6):568-581.
429. Gao J, Wang H, Sheng H, et al. A novel apoE-derived therapeutic reduces
vasospasm and improves outcome in a murine model of subarachnoid hemorrhage. Neurocritical Care. 2006;4(1):25-31.

6. Literaturverzeichnis
137
430. Kamii H, Kato I, Kinouchi H, et al. Amelioration of vasospasm after
subarachnoid hemorrhage in transgenic mice overexpressing CuZn-superoxide dismutase. Stroke. 1999;30(4):867-871.
431. McGirt MJ, Parra A, Sheng H, et al. Attenuation of cerebral vasospasm after
subarachnoid hemorrhage in mice overexpressing extracellular superoxide dismutase. Stroke. 2002;33(9):2317-2323.
432. McGirt MJ, Lynch JR, Parra A, et al. Simvastatin increases endothelial nitric
oxide synthase and ameliorates cerebral vasospasm resulting from subarachnoid hemorrhage. Stroke. 2002;33(12):2950-2956.
433. Parra A, McGirt MJ, Sheng H, Laskowitz DT, Pearlstein RD, Warner DS. Mouse
model of subarachnoid hemorrhage associated cerebral vasospasm: methodological analysis. Neurological Research. 2002;24(5):510-516.
434. Saito A, Kamii H, Kato I, et al. Transgenic CuZn-superoxide dismutase inhibits
NO synthase induction in experimental subarachnoid hemorrhage. Stroke. 2001;32(7):1652-1657.
435. Sehba FA, Friedrich V. Early micro vascular changes after subarachnoid
hemorrhage. Acta Neurochirurgica. Supplement. 2011;110(Pt 1):49-55. 436. Liu S, Tang J, Ostrowski RP, et al. Oxidative stress after subarachnoid
hemorrhage in gp91phox knockout mice. The Canadian Journal of Neurological Sciences. Le Journal Canadien des Sciences Neurologiques. 2007;34(3):356-361.
437. Sozen T, Tsuchiyama R, Hasegawa Y, et al. Role of interleukin-1beta in early
brain injury after subarachnoid hemorrhage in mice. Stroke. 2009;40(7):2519-2525.
438. Okubo S, Strahle J, Keep RF, Hua Y, Xi G. Subarachnoid hemorrhage-induced
hydrocephalus in rats. Stroke. 2013;44(2):547-550. 439. Lackner P, Vahmjanin A, Hu Q, Krafft PR, Rolland W, Zhang JH. Chronic
hydrocephalus after experimental subarachnoid hemorrhage. PloS One. 2013;8(7):e69571.
440. Rovainen CM, Woolsey TA, Blocher NC, Wang DB, Robinson OF. Blood flow
in single surface arterioles and venules on the mouse somatosensory cortex measured with videomicroscopy, fluorescent dextrans, nonoccluding fluorescent beads, and computer-assisted image analysis. Journal Cerebral Blood Flow and Metabolism. 1993;13(3):359-371.
441. Hillen F, Kaijzel EL, Castermans K, oude Egbrink MG, Lowik CW, Griffioen AW.
A transgenic Tie2-GFP athymic mouse model; a tool for vascular biology in

6. Literaturverzeichnis
138
xenograft tumors. Biochemical and Biophysical Research Communications. 2008;368(2):364-367.
442. Dunphy MP, Entenberg D, Toledo-Crow R, Larson SM. In vivo
microcartography and subcellular imaging of tumor angiogenesis: a novel platform for translational angiogenesis research. Microvascular Research. 2009;78(1):51-56.
443. Broderick JP, Brott TG, Duldner JE, Tomsick T, Leach A. Initial and recurrent
bleeding are the major causes of death following subarachnoid hemorrhage. Stroke. 1994;25(7):1342-1347.
444. Cross DT, 3rd, Tirschwell DL, Clark MA, et al. Mortality rates after subarachnoid
hemorrhage: variations according to hospital case volume in 18 states. Journal of Neurosurgery. 2003;99(5):810-817.
445. Nornes H, Magnaes B. Recurrent haemorrhage and haemostasis in patients
with ruptured intracranial saccular aneurysms. Acta Neurologica Scandinavica. Supplementum. 1972;51:473-476.
446. Kataoka K, Taneda M. Reversible arterial hypotension after acute aneurysmal
subarachnoid hemorrhage. Surgical Neurology. 1985;23(2):157-161. 447. Magder SA. The highs and lows of blood pressure: toward meaningful clinical
targets in patients with shock. Critical Care Medicine. 2014;42(5):1241-1251. 448. Yang MF, Sun BL, Xia ZL, Zhu LZ, Qiu PM, Zhang SM. Alleviation of brain
edema by L-arginine following experimental subarachnoid hemorrhage in a rat model. Clinical Hemorheology and Microcirculation. 2003;29(3-4):437-443.
449. Sun BL, Zhang SM, Xia ZL, et al. L-arginine improves cerebral blood perfusion
and vasomotion of microvessels following subarachnoid hemorrhage in rats. Clinical Hemorheology and Microcirculation. 2003;29(3-4):391-400.
450. Mehta S, Stewart DJ, Levy RD. The hypotensive effect of L-arginine is
associated with increased expired nitric oxide in humans. Chest. 1996;109(6):1550-1555.
451. Bath PM, Willmot M, Leonardi-Bee J, Bath FJ. Nitric oxide donors (nitrates), L-
arginine, or nitric oxide synthase inhibitors for acute stroke. The Cochrane Database of Systematic Reviews. 2002(4):CD000398.
452. Albert J, Norman M, Wallen NH, Frostell C, Hjemdahl P. Inhaled nitric oxide
does not influence bleeding time or platelet function in healthy volunteers. European Journal of Clinical Investigation. 1999;29(11):953-959.
453. Krejcy K, Schmetterer L, Kastner J, et al. Role of nitric oxide in hemostatic
system activation in vivo in humans. Arteriosclerosis, Thrombosis, and Vascular Biology. 1995;15(11):2063-2067.

6. Literaturverzeichnis
139
454. Samama CM, Diaby M, Fellahi JL, et al. Inhibition of platelet aggregation by
inhaled nitric oxide in patients with acute respiratory distress syndrome. Anesthesiology. 1995;83(1):56-65.
455. Terpolilli NA, Kim SW, Thal SC, Kuebler WM, Plesnila N. Inhaled nitric oxide
reduces secondary brain damage after traumatic brain injury in mice. Journal of Cerebral Blood Flow and Metabolism. 2013;33(2):311-318.
456. Inglessis I, Shin JT, Lepore JJ, et al. Hemodynamic effects of inhaled nitric oxide
in right ventricular myocardial infarction and cardiogenic shock. Journal of the American College of Cardiology. 2004;44(4):793-798.
457. Frostell CG, Blomqvist H, Hedenstierna G, Lundberg J, Zapol WM. Inhaled nitric
oxide selectively reverses human hypoxic pulmonary vasoconstriction without causing systemic vasodilation. Anesthesiology. 1993;78(3):427-435.
458. Day RW, Lynch JM, White KS, Ward RM. Acute response to inhaled nitric oxide
in newborns with respiratory failure and pulmonary hypertension. Pediatrics. 1996;98(4 Pt 1):698-705.
459. Hogman M, Frostell C, Arnberg H, Sandhagen B, Hedenstierna G. Prolonged
bleeding time during nitric oxide inhalation in the rabbit. Acta Physiologica Scandinavica. 1994;151(1):125-129.
460. Hansen-Schwartz J. Cerebral vasospasm: a consideration of the various
cellular mechanisms involved in the pathophysiology. Neurocritical Care. 2004;1(2):235-246.
461. Janjua N, Mayer SA. Cerebral vasospasm after subarachnoid hemorrhage.
Current Opinion in Critical Care. 2003;9(2):113-119. 462. Stein SC, Levine JM, Nagpal S, LeRoux PD. Vasospasm as the sole cause of
cerebral ischemia: how strong is the evidence? Neurosurgical Focus. 2006;21(3):E2.
463. Sun BL, Zheng CB, Yang MF, Yuan H, Zhang SM, Wang LX. Dynamic
alterations of cerebral pial microcirculation during experimental subarachnoid hemorrhage. Cellular and Molecular Neurobiology. 2009;29(2):235-241.
464. Durmaz R, Ozkara E, Kanbak G, et al. Nitric oxide level and adenosine
deaminase activity in cerebrospinal fluid of patients with subarachnoid hemorrhage. Turkish Neurosurgery. 2008;18(2):157-164.
465. Khaldi A, Zauner A, Reinert M, Woodward JJ, Bullock MR. Measurement of
nitric oxide and brain tissue oxygen tension in patients after severe subarachnoid hemorrhage. Neurosurgery. 2001;49(1):33-38.

6. Literaturverzeichnis
140
466. Ng WH, Moochhala S, Yeo TT, Ong PL, Ng PY. Nitric oxide and subarachnoid hemorrhage: elevated level in cerebrospinal fluid and their implications. Neurosurgery. 2001;49(3):622-626.
467. Woszczyk A, Deinsberger W, Boker DK. Nitric oxide metabolites in cisternal
CSF correlate with cerebral vasospasm in patients with a subarachnoid haemorrhage. Acta Neurochirurgica. 2003;145(4):257-263.
468. Jung CS, Oldfield EH, Harvey-White J, et al. Association of an endogenous
inhibitor of nitric oxide synthase with cerebral vasospasm in patients with aneurysmal subarachnoid hemorrhage. Journal of Neurosurgery. 2007;107(5):945-950.
469. Medele RJ, Stummer W, Reulen HJ, Steiger HJ. Evidence for peroxidative
damage by nitric oxide in experimental chronic cerebral vasospasm. Neurological Research. 1996;18(3):277-280.
470. Sayama T, Suzuki S, Fukui M. Role of inducible nitric oxide synthase in the
cerebral vasospasm after subarachnoid hemorrhage in rats. Neurological Research. 1999;21(3):293-298.
471. Yamamoto S, Nishizawa S, Yokoyama T, Ryu H, Uemura K. Subarachnoid
hemorrhage impairs cerebral blood flow response to nitric oxide but not to cyclic GMP in large cerebral arteries. Brain Research. 1997;757(1):1-9.
472. Kasuya H, Weir BK, Nakane M, et al. Nitric oxide synthase and guanylate
cyclase levels in canine basilar artery after subarachnoid hemorrhage. Journal of Neurosurgery. 1995;82(2):250-255.
473. Sobey CG, Quan L. Impaired cerebral vasodilator responses to NO and PDE V
inhibition after subarachnoid hemorrhage. The American Journal of Physiology. 1999;277(5 Pt 2):H1718-1724.
474. van Giersbergen PL, Dingemanse J. Tolerability, pharmacokinetics, and
pharmacodynamics of clazosentan, a parenteral endothelin receptor antagonist. European Journal of Clinical Pharmacology. 2007;63(2):151-158.
475. van Giersbergen PL, Gunawardena KA, Dingemanse J. Influence of ethnic
origin and sex on the pharmacokinetics of clazosentan. Journal of Clinical Pharmacology. 2007;47(11):1374-1380.
476. Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp
Pharmacol. 2006(176 Pt 1):213-254. 477. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction:
testing and clinical relevance. Circulation. 2007;115(10):1285-1295.

6. Literaturverzeichnis
141
478. Lavi S, Gaitini D, Milloul V, Jacob G. Impaired cerebral CO2 vasoreactivity: association with endothelial dysfunction. American journal of physiology. Heart and Circulatory Physiology. 2006;291(4):H1856-1861.
479. Fathi AR, Yang C, Bakhtian KD, Qi M, Lonser RR, Pluta RM. Carbon dioxide
influence on nitric oxide production in endothelial cells and astrocytes: cellular mechanisms. Brain Research. 2011;1386:50-57.
480. Friedrich B, Michalik R, Oniszczuk A, Abubaker K, Kozniewska E, Plesnila N.
CO2 has no therapeutic effect on early microvasospasm after experimental subarachnoid hemorrhage. Journal Cerebral Blood Flow and Metabolism. 2014;34(8):e1-6.
481. Sabri M, Ai J, Knight B, et al. Uncoupling of endothelial nitric oxide synthase
after experimental subarachnoid hemorrhage. Journal of Cerebral Blood Flow and Metabolism. 2011;31(1):190-199.
482. Luo S, Lei H, Qin H, Xia Y. Molecular mechanisms of endothelial NO synthase
uncoupling. Curr Pharm Des. 2014;20(22):3548-3553. 483. Forstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur
Heart J. 2012;33(7):829-837. 484. Capettini LS, Cortes SF, Gomes MA, et al. Neuronal nitric oxide synthase-
derived hydrogen peroxide is a major endothelium-dependent relaxing factor. American Journal of Physiology. Heart and Circulatory Physiology. 2008;295(6):H2503-2511.
485. Stefanovic B, Schwindt W, Hoehn M, Silva AC. Functional uncoupling of
hemodynamic from neuronal response by inhibition of neuronal nitric oxide synthase. Journal of Cerebral Blood Flow and Metabolism. 2007;27(4):741-754.

A. Danksagung
A. Danksagung
Für die Aufnahme in das Walter-Brendel-Zentrum für experimentelle Medizin möchte
ich mich bei Prof. Dr. med. U. Pohl herzlich bedanken.
Mein außerordentlicher Dank gilt meinem Doktorvater und Betreuer Prof. Dr. med.
Nikolaus Plesnila für das Dissertationsthema und die sehr gute Betreuung. Mit seiner
fachlichen Kompetenz und seiner persönlichen Zuverlässigkeit hat er mich stets
unterstützt und motiviert.
Von Herzen danke ich auch meiner Mitbetreuerin, Frau Dr. med. Nicole Terpolilli, die
mich in meiner Dissertation von Anfang an begleitet hat, mir jederzeit geduldig zur
Seite stand und mich durch ihre Leidenschaft am Forschen und Heilen immer wieder
motiviert hat. Von Ihr habe ich viel gelernt.
Ganz herzlich bedanke ich mich auch bei meinem Mitbetreuer PD. Dr. med. Karsten
Schöller, der mich als Doktorand engagiert hat. Er hat mich in seiner Zeit in München
stets unterstützt und aktiv begleitet.
Für die tatkräftige und freundschaftliche Unterstützung bei der Durchführung der
chirurgischen Experimente und Beschaffung der erforderlichen Materialien danke ich
in besonderer Weise Nicole Heumos.
Dr. biol. hum. Jürgen Peters danke ich herzlich für seine Hilfestellung und Offenheit in
Fragen zur Statistik und Computertechnik. Gerne erinnere ich mich an unsere
weltanschaulichen und philosophischen Gespräche.
Zum Schluss denke ich auch dankbar an meinen Eltern und meine Geschwister die
mich immer motivierten.
Laudate Dominum

B. Lebenslauf
Persönliche Daten
Name: Ari Christian da Fonseca Dienel
Geburtsdatum: 04.07.1979
Geburtsort: Portimão/ Portugal
Nationalität: Deutsch/Portugiesisch
Aktuelle Beschäftigung
01.2011 – 04.2015 Doktorand am Walter-Brendel-Zentrum (AG Plesnila), Institut
für Chirurgische Forschung, Ludwig-Maximilians-Universität zu
München: Thema der Dissertation “ Veränderung der Mikrozir-
kulation nach experimenteller Subarachnoidalblutung (SAB):
Kinetik, Bedeutung und Identifikation von Mechanismen”.
Wissenschaftliche Projekte
10.2010 – 11.2010 Molekularbiologische Projektarbeit am Paul-Flechsig-Institut, in
Leipzig. Thema: „Untersuchung von Zellzyklus Inhibitoren, P16
(Tumorsupressor Protein/Gen), im Alzheimer-Hirn“
08.2010 – 10.2010 Projektarbeit am Paul-Flechsig-Institut, Hirnforschungszentrum,
in Leipzig. Thema „Analyse des Vorhandenseins und der
potentiellen Sekretion von Matrix-relevanten Molekülen in
verschiedenen Zelllinien (MDCK, N2A, COS, SY5Y, GT1-7)“

B. Lebenslauf
144
03.2008 – 09.2008 Diplomarbeit am Paul-Flechsig-Institut in Leipzig (Alzheimer-
Forschungszentrum) Thema: “Einfluss der Pseudophospho-
rylierung auf die Tau-Phosphorylierungsmuster, Zytoskelett-
stabilität und Zellvitalität in N2a Neuroblastomazellen”
02.2006 – 04.2006 Projektarbeit am Paul-Flechsig-Institut, in Leipzig (Alzheimer-
Forschungszentrum).Thema: „Einfluss der Dimerisierung auf
die Phosphorylierung von Tau-Proteinen in murinen Neuroblas-
tomazellkulturen (N2a)“.
07.2004 – 10.2004 Projektarbeit am Biotechnologie-Zentrum in Leipzig. Thema:“
Kultivierung und Charakterisierung von embryonalen Kardio-
myozyten aus dem Huhn, für Anwendung auf Micro-Elektrode-
Arrays (MEAs)“
Bildung
10.2000 – 09.2008 Diplom in Biochemie an der Universität Leipzig
10.1999 – 03.2000 Sprachkurs "Deutsch als Fremdsprache (DAF)" an der
Universität Trier.
09.1995 – 07.1998 Sekundarschule in Silves / Portugal; Abschluss: Abitur

B. Lebenslauf
145
Wehrdienst/Bundeswehr
11.1998 – 09.1999 Wehrdienst als Sanitätssoldat in Rennerod und Idar-Oberstein
(Dienstgrad: OG)
Andere Aktivitäten
06.08.2013 Simultanübersetzung aus dem Brasilianischen ins Deutsche für einen Dokumentarfilm „Der verlorene Vater“ im Bayerischer Rundfunk
05.2010 – 06.2010 Verkauf von Gesundheits- und ökologische Gartenanbau Lite-ratur als selbstständiger Verkäufer in Schweden.
05.2000 – 09.2000
02.2001 – 03.2001 Elektrikerhelfer in der Firma Elektro-Schmitz, in Trier
11.2008 – 04.2010
07.2001-09.2001 Sitzfertigung bei Mercedes-Benz für die E-Klasse,
07.2002-09.2002 in Sindelfingen

Eidestattliche Versicherung
Ich, Ari Christian da Fonseca Dienel, erkläre hiermit an Eides statt,
dass ich die vorliegende Dissertation mit dem Thema
„Veränderung der Mikrozirkulation nach experimenteller Subarachnoidal-
blutung (SAB): Kinetik, Bedeutung und Identifikation von Mechanismen“
selbständig verfasst, mich außer der angegebenen keiner weiteren Hilfsmittel bedient
und alle Erkenntnisse, die aus dem Schrifttum ganz oder annähernd übernommen
sind, als solche kenntlich gemacht und nach ihrer Herkunft unter Bezeichnung der
Fundstelle einzeln nachgewiesen habe.
Ich erkläre des Weiteren, dass die hier vorgelegte Dissertation nicht in gleicher oder
in ähnlicher Form bei einer anderen Stelle zur Erlangung eines akademischen
Grades eingereicht wurde.
München, 13.09.2016


















