
Bachelor-Thesis Zur Lipase-katalysierten Darstellung von...
49
Fachbereich Agrarwirtschaft und Lebensmittelwissenschaften Studiengang Bioprodukttechnologie Bachelor-Thesis Zur Lipase-katalysierten Darstellung von Peroxycarbonsäuren mit Harnstoff- Wasserstoffperoxid Vorgelegt von: Jan-Niklas Franzen Am: 23. Januar 2012 Betreuender Professor: Herr Prof. Dr. Mark Rüsch gen. Klaas Korreferent: Frau Prof. Dr. Christine Wittmann URN: urn:nbn:de:gbv:519-thesis 2012-0020-5
Transcript of Bachelor-Thesis Zur Lipase-katalysierten Darstellung von...

Fachbereich Agrarwirtschaft und Lebensmittelwissenschaften Studiengang Bioprodukttechnologie
Bachelor-Thesis
Zur Lipase-katalysierten Darstellung von Peroxycarbonsäuren mit Harnstoff-Wasserstoffperoxid
Vorgelegt von: Jan-Niklas Franzen
Am: 23. Januar 2012
Betreuender Professor: Herr Prof. Dr. Mark Rüsch gen. Klaas
Korreferent: Frau Prof. Dr. Christine Wittmann
URN: urn:nbn:de:gbv:519-thesis 2012-0020-5

2
Abstract
The chemo-enzymatic synthesis of peroxy acids has been optimized using Novozym 435, the
immobilized form of Candida antacrtica lipase B, and the complex urea-hydrogen peroxide.
Urea-hydrogen peroxide, an anhydrous form of hydrogen peroxide, has the potential of releasing
hydrogen peroxide in a controlled manner and thus avoids the need to add the aqueous hydrogen
peroxide slowly to the reaction micture. As a Solvent and reactant diethyl carbonate and ethyl
acetate were used, yields up to 6,34 mmol Peroxyacetic Acid and 1,23 mmol Peroxycarbonic
acid were reached. Further conversion-dominated factors were investigated for future prospects
in industrial or laboratory applications.

3
Inhaltsverzeichnis
Abstract…………………………………………………………………………………………………………... 21. Einleitung……………………………………………………………………………………………………… 42. Stand der Wissenschaft und Technik…………………………………………………………………………... 5
2.1 Peroxycarbonsäuren………………………………………………………………………………………… 52.2 Novozym® 435 / Lipasen……………………………………………………………………….................... 72.3 Wasserstoffperoxid / Harnstoff-Wassertoffperoxid………………………………………………………… 8
3. Ziel der Arbeit………………………………………………………………………………………………….. 124. Material und Methoden………………………………………………………………………………………… 13
4.1 Chemikalien/Reagenzien…………………………………………………………………………………… 134.2 Methoden………………………………………………………………………………………………….... 13
4.2.1 Bestimmung der Konzentration des Harnstoff-Wasserstoffperoxid-Adduktes………………………… 134.2.2 Enzymatisch katalysierte Synthese von Peroxycarbonsäuren………………………………………….. 144.2.3 Variationen der enzymatisch katalysierten Synthese von Peroxycarbonsäuren………………………... 144.2.4 Cerimetrische Bestimmung der Wasserstoffperoxidkonzentration und
iodometrische Bestimmung der Peroxycarbonsäurenkonzentration………………………………………….. …. 155. Versuchsergebnisse…………………………………………………………………………………………….. 16
5.1 Bestimmung der Konzentration des Harnstoff-Wasserstoffperoxid-Adduktes…………………………….. 165.2 Enzymatisch katalysierte Synthese von Peroxycarbonsäuren; Reaktionskriterien nach Ernst (2011) 18
5.2.1 Synthese 1: Oxidation von Essigsäureethylester zu Peroxyessigsäure…………………………………. 185.2.1 Synthese 2: Oxidation von Diethylcarbonat zu Peroxykohlensäure……………………………………. 18
5.3 Variationen der enzymatisch katalysierten Synthese von Peroxycarbonsäuren……………………………. 195.3.1 Synthese 3: Reduktion der Reaktionszeit auf 2 und 4 Stunden………………………………………… 195.3.2 Synthese 4: Unterlassung der Filtration des Reaktionsmediums nach Abschluss der Reaktion……….. 205.3.3 Synthese 5: Erhöhung der Reaktionstemperatur auf 50°C……………………………………………... 225.3.4 Synthese 6: Hinzugabe von Harnstoff-Wasserstoffperoxid in Intervallen……………………………... 22
6. Diskussion…………………………………………………………………………………………………….... 236.1 Synthese 1: Oxidation von Essigsäureethylester zu Peroxyessigsäure……………………………………... 236.2 Synthese 2: Oxidation von Diethylcarbonat zu Peroxykohlensäure………………………………………... 26
6.3 Variationen der enzymatisch katalysierten Synthese von Peroxycarbonsäuren………………………............ 306.3.1 Synthese 3: Reduktion der Reaktionszeit auf 2 und 4 Stunden…………………………………………... 306.3.2 Synthese 4: Unterlassung der Filtration des Reaktionsmediums nach Abschluss der Reaktion…............. 316.3.3 Synthese 5: Erhöhung der Reaktionstemperatur auf 50°C………………………………………….......... 346.3.4 Synthese 6: Hinzugabe von Harnstoff-Wasserstoffperoxid in Intervallen………………………….......... 36
7. Schlussfolgerung und Ausblick………………………………………………………………………………... 388. Zusammenfassung……………………………………………………………………………………………... 399. Literatur………………………………………………………………………………………………………... 4110. Verwendete Abkürzungen……………………………………………………………………………………. 4311. Abbildungsverzeichnis………………………………………………………………………………………... 4412. Tabellenverzeichnis…………………………………………………………………………………………... 4613. Selbständigkeitserklärung…………………………………………………………………………………...... 4814. Anhang………………………………………………………………………………………………………... 49

4
1. Einleitung
Chemo-enzymatische Verfahren zur Herstellung pharmazeutischer, chemischer und
lebensmitteltechnischer Produkte haben in den letzten Jahrzenten kontinuierlich an Bedeutung
gewonnen. Durch rapide Fortschritte in der Molekularbiologie werden Biotransformationen
zunehmend konkurrenzfähig zu konventionell-chemischen Verfahren. Dies ist vor allem auf die
verhältnismäßig kostengünstige Bereitstellung leistungsfähiger Enzyme zurückzuführen,
verbunden mit einem wachsenden Know-How bezüglich ihrer Einsatzgebiete abseits ihrer
biochemischen Rolle im Organismus. Während bis zu Beginn der 80er Jahre aus Kostengründen
größtenteils nur sehr komplexe Produkte (z.B. Antibiotika, Insulin) durch fermentative
Methoden hergestellt wurden, werden heutzutage auch vergleichsweise einfache Verbindungen
unter Zuhilfenahme isolierter Biokatalysatoren produziert. Da die meisten der kommerziell
erhältlichen Lipasenpräparationen im Vergleich zu anderen Enzymen immer noch
verhältnismäßig teuer angeboten werden, sind Lipasen-katalysierte Reaktionen hauptsächlich
dann industriell interessant, wenn sie zur Herstellung von Produkten guten kommerziellen
Wertes dienen (Bornscheuer, 1999).
Peroxycarbonsäuren als Produkt erfüllen dieses Kriterium. Neben ihrer industriellen Anwendung
als Bleichmittel und Desinfektionsmittel sind Peroxycarbonsäuren wichtige Oxidationsmittel in
der chemischen Industrie sowie in der organischen Laborsynthese. Die industrielle
Standardmethode für die Synthese von Peroxycarbonsäuren ist die Reaktion von Carbonsäuren
mit Wasserstoffperoxid, katalysiert durch eine starke Säure. Björking et al. (1990) entwickelte
als erstes eine chemo-eynzymatische Synthesemethode für Peroxycarbonsäuren, katalysiert
durch Novozym® 435, eine immobilisierte Lipase B von Candida antarctica. Ein störender
Aspekt bezüglich Effektivität und Ökonomität dieser Methode ist die Verwendung von
wässrigem Wasserstoffperoxid. Einerseits bergen die Lagerung und der Umgang gewisse
Risiken, was gerade bei industrieller Umsetzung mit Kosten verbunden ist. Andererseits
bewirken zu hohe Konzentrationen von Wasserstoffperoxid im Reaktionsmedium eine komplette
Denaturierung der Lipase B des Novozym® 435, weshalb eine kontinuierliche Zugabe von
Wasserstoffperoxid in Intervallen notwendig ist (Ankudey; Olivio; Peeples, 2007). In dieser
Arbeit wird deshalb auf den Einsatz von wässrigem Wasserstoffperoxid verzichtet, und
stattdessen Harnstoff-Wasserstoffperoxid, ein unstabiles nicht kovalentes Addukt aus Harnstoff
und Wasserstoffperoxid, verwendet. Harnstoff-Wasserstoffperoxid hat das Potential,
Wasserstoffperoxid in einer kontrollierbaren Art und Weise freizusetzen.
Das Ziel dieser Studie ist die Durchführung einer chemo-enzymatischen Synthese von
Peroxycarbonsäuren, katalysiert durch Novozym® 435, unter Verwendung von Harnstoff-

5
Wassertstoffperoxid als Oxidationsmittel. Als Edukte dienen Essigsäureethylester und
Diethylcarbonat, folglich werden als Produkte Peroxyessigsäure und
Monoperoxykohlensäuremonoethylester (Abk. Peroxykohlensäure) erwartet.
2. Stand der Wissenschaft und Technik
2.1 Peroxycarbonsäuren
Peroxycarbonsäuren (veraltet: Persäuren) sind Carbonsäuren, die eine Peroxogruppe (-O-O-)
besitzen. Sie können als Derivate des Wasserstoffperoxids angesehen werden und besitzen ein
starkes Oxidationsvermögen. Industrielle Anwendung finden sie hauptsächlich als Bleichmittel
und Desinfektionsmittel/Antiseptikum. Eines der meist verwendeten Desinfektionsmittel ist die
Peroxyessigsäure (Abb. 1), welche bereits 1902 untersucht wurde (www.kesla.de, 2011).
CH3
O
O
O
H
Abbildung 1: Peroxyessigsäure
Es hat sich herausgestellt, dass sie als Desinfektionsmittel keine Wirkungslücken oder Bereiche
abgeschwächter Wirkung zeigt, zusätzlich wird eine Resistenzentwicklung durch eine
unspezifische Wirkweise, eine Art „kaltes Verbrennen“ der Keime, vermieden. Sie besitzt die
schnellste Wirkungsgeschwindigkeit aller bekannten Mikrobizide und ist dazu umweltfreundlich,
da sie nach Anwendung in Sauerstoff und Essigsäure zerfällt (Patett, 2001).
In der organischen Laborsynthese finden Peroxycarbonsäuren ein breites Anwendungsspektrum
als Oxidationsmittel. Beispiele sind die C=C-Epoxidierung, Bayer-Villinger-Oxidation,
Hydroxilierung von aromatischen Ringen und die Oxidation von Aminen (Rüsch gen. Klaas;
Warwel, 2000). Der Epoxidierung kommt hier die größte Bedeutung zu, Björking et al. (1990)
beschrieb als erstes die chemo-enzymatische Epoxidation von Alkenen durch
Peroxycarbonsäuren.
Die Standardmethode für die Synthese von Peroxycarbonsäuren ist die Reaktion von
Carboxylsäuren mit Wasserstoffperoxid, katalysiert durch eine starke Säure (Abb. 2) (Rüsch gen.
Klaas; Warwel, 2000).

6
O
R OH R
O
OOH+ H2O2
[H+]
+ H2O
Abbildung 2: Säurekatalytische Herstellung einer Peroxycarbonsäure (Rüsch gen. Klaas; Warwel, 2000)
Die Lage des Gleichgewichtes und die Geschwindigkeit der Gleichgewichtsstellung wird
beeinflusst durch die Größe des Restes „R“ der verwendeten Carbonsäure, demnach müssen
Reaktionsbedingungen wie Temperatur, Menge und Acidität des Katalysators entsprechend
angepasst werden. Da strukturempfindliche oder langkettige Substrate und vor allem die
entstehenden Peroxycarbonsäuren unter den dann erforderlichen Bedingungen meist nicht stabil
bleiben, ist diese Methode für Substrate dieser Art nicht geeignet (Patett, 2001). Andere
Verfahren für die Synthese von einzelnen Peroxycarbonsäuren (Folli, 1968) sind meist zu teuer
und komplex für einen weitfassenden Gebrauch, es besteht also ein Bedarf für eine neue, simple
und vielseitig anwendbare Methode. Eine diesen Ansprüchen entsprechende Synthesemethode
wurde als erstes von Björkling, Kirk und Kollegenschaft entwickelt, einer Gruppe, angestellt
beim weltweit führenden dänischen Enzymhersteller Novo Nordisk A/S. Sie publizierten, dass
mittelkettige Fettsäuren mit Wasserstoffperoxid zu den entsprechenden Peroxycarbonsäuren mit
Umsätzen von 33-54% oxidiert werden können, sofern die Reaktion durch die immobilisierte
Lipase-B des Mikroorganismus Candida antarctica (Novozym® 435) katalysiert wird (Rüsch
gen. Klaas; Warwel, 2000). Am „Institut für Biochemie und Technologie der Fette“ in Münster
wurde diese Methode in diverse Richtungen weiterentwickelt, wodurch viele neue
Peroxycarbonsäuren durch mildere und schonendere Reaktionsbedingungen synthetisiert wurden
konnten (Patett, 2001). Durch die Verwendung eines Esters als Lösungsmittel ließen sich mit
dieser Methode die besten Ergebnisse erzielen, eine allgemeine Reaktionsgleichung der
enzymatisch katalysierten Herstellung von Peroxycarbonsäuren ist in Abbildung 3 dargestellt:
R1
O
OR2
+ H2O2 [Novozym ® 435]R1
O
OOH
in Ester
R2OH+
Abbildung 3: Allgemeine Reaktionsgleichung enzymatisch katalysierter Synthese von Peroxycarbonsäuren (Rüsch gen. Klaas; Warwel, 2000)
Rüsch gen. Klaas und Warwel (2000) publizierten, dass die enzymatische Peroxycarbonsäuren-
Synthese aus Essigsäureethylester und Essigsäuremethylester unter gleichen

7
Reaktionsbedingungen effektiver ist als die enzymatische Peroxycarbonsäuren-Synthese aus
Essigsäure. Unter anderem aus diesem Grund werden in dieser Arbeit Ester als Edukte
verwendet.
2.2 Novozym® 435 / Lipasen
Das von Björking et al. (1990) bei der chemo-enzymatischen Peroxycarbonsäurensynthese
verwendete Novozym® 435 (EC: 3.1.1.3) ist eine immobilisierte Lipase-B des Mikroorganismus
Candida antarctica. Zur Herstellung wird das Gen von Candida antarctica, welches Lipase-B
codiert, in einen Wirt-Mikroorganismus, aspergillus oryzae geklont. Das exprimierte Enzym
wird anschließend auf einem makroporösem Polyacrylharz immobilisiert (Ankudey; Olivio;
Peeples, 2006). Lipasen (systematischer Name: Triacylglycerol-Acylhydrolasen) können neben
Mikroorganismen auch aus Pflanzen, Pilzen, und Säugetierorganismen isoliert werden.
Natürliche Substrate der Lipasen sind Triacylglyceride und abgeleitete Verbindungen wie Di-
und Monoacylglyceride, wie alle Hydrolasen benötigen sie keine Coenzyme. Lipase-B von
Candida antarctica hat gemeinsam mit einer Cutinase aus Fusarium solani pisi als einzige
bekannte Lipasen keine bewegliche Oligopeptideinheit („Deckel“) über dem aktivem Zentrum
aufzuweisen. Diese Eigenschaft und ihre Fähigkeit zur Konformationsänderung ermöglicht es
ihr, ein sehr breites Substratspektrum, auch abseits der Tri-, Di- und Monoacylglyceride, zu
akzeptieren (Bornscheuer, 1998).
Hauptsächliche Einsatzgebiete von Novozym® 435 in Industrie und Forschung sind die
Veresterung, die Umesterung und die Hydrolyse (Bornscheuer et al., 1998). Amkudey, Olivio
und Peeples (2006) stellten fest, dass Novozym® 435 die Perhydrolysis von Octansäure
effektiver als eine Menge anderer getesteter Enzyme katalysiert. Rüsch gen. Klaas und Warwel
(2000) kamen ebenfalls zu der Erkenntnis, dass Novozym® 435, neben 30 anderen getesteten
Lipasen, für die Peroxysäurensynthese aus Fettsäuren mit Wasserstoffperoxid am besten
geeignet ist.
Auch zur enzymatischen Synthese von Peroxyessigsäure aus Essigsäureethylester hat sich
Novozym® 435 gegenüber Novonymes NS-40054 und Novonymes NS-40079 als bester
Katalysator herausgestellt (Ernst, 2011). Aus diesem Grund wird es in dieser Studie eingesetzt.
Lipase-B von Candida antarctica ist wie alle Lipasen eine Serin-Esterase. Katalysiert werden
Reaktionen durch die drei Aminosäuren Serin, Histidin und Asparaginsäure („katalytische
Triade“). In Abbildung 4 sind die katalysierenden Aminosäuren der Lipase-B aus Candida
antarctica dargestellt.

8
Abbildung 4: Katalytische Triade mit der Aminosäuren-Nummerierung des aktiven Zentrums der Lipase-B aus Candida antarctica (Rüsch gen. Klaas; Warwel, 2000)
Bei der enzymatischen Synthese einer Peroxycarbonsäure aus einem Ester findet als erster
Schritt eine Hydrolyse statt. Bei einer Hydrolyse eines Esters durch Lipase-B Candida antarctica
wird der Substratester vom Serin im aktiven Zentrum unter Ausbildung eines tetrahedralen
Intermediates nukleophil angegriffen. Im Anschluss entsteht unter Abspaltung eines Alkohols
ein Acyl-Enzym-Komplex, unterstützt durch das Carboxylatanion des Asparaginsäureesters und
durch den Imidazolrest des Histidins, welcher das freiwerdende Proton des Serinrestes
übernimmt. Das negativ geladene Intermediat wird durch mehrere Aminosäurereste in der
Oxyaniontasche stablisiert. Das Acyl-Enzym wird schließlich von einem Nukleophil angegriffen
und ein zweites tetrahedrales Intermediat wird gebildet. Dieses Intermediat zerfällt anschließend
wieder in das freie Enzym und das zu erwartende Produkt.
Dient Wasser im Reaktionsmedium als Nukleophil, so entsteht als Produkt eine Säure, bei
Wasserstoffperoxid entsprechend eine Peroxysäure. Wird ein Alkohol als Nukleophil verwendet,
entsteht ein neuer Ester, auch Umesterung genannt (Bornscheuer, 1998).
2.2 Wasserstoffperoxid / Harnstoff-Wassertoffperoxid
Wie bereits erwähnt, ist ein störender Aspekt hinsichtlich Effektivität und Ökonomität der
enzymatisch katalysierten Synthese von Peroxycarbonsäuren die Verwendung von wässrigem
Wasserstoffperoxid als Oxidationsmittel. Zwar ist der Umgang mit Wasserstoffperoxid immer
noch wesentlich sicherer als zum Beispiel der Umgang mit flüssigem Sauerstoff, dennoch
kommt es immer wieder zu Unfällen beim Einsatz sowie bei der Lagerung (Jones, 1999).
Wasserstoffperoxid in allen Formen ist thermodynamisch instabil und zersetzt sich
kontinuierlich zu Wasser und Sauerstoff. Obwohl 20 Tonnen wässriges 70 % Wasserstoffperoxid
durchschnittlich nur 0,3 % des aktiven Sauerstoffs pro Jahr verlieren, entwickeln sich dadurch 13
dm3 Sauerstoff pro Tag. Dies ist genug, um bei der Lagerung beschichtetes Material gefährlich
HO- NH
O
N
N
O NH-
H O
O
O-HN
Aspartat 105
Histidin 224
Serin 1050.

9
unter Druck zu setzen oder für eine Sauerstoffanreicherung im oberen Gasraum des
Lagercontainers zu sorgen. Dies kann die Explosionsgefahr drastisch steigern.
Des Weiteren kann Wasserstoffperoxid in Konzentrationen über 40 % explosive Gemische mit
organischen Komponenten bilden. Solche Mixturen können konventionellen Sprengstoffen in
ihrer Sprengkraft gleichen und sind zusätzlich wesentlich sensitiver bezüglich der Auslösung
einer Detonation (Tabelle 1) (Jones, 1999).
Tabelle 1: Vergleich der Explosivkraft und Sensitivität konventioneller Sprengstoffe mit Wasserstoffperoxid Gemischen(Jones, 1999)
Stoff Sprengkraft SensitivitätNitroglycerin 52 2-585 % mol/mol Wasserstoffperoxid/Glycerin 46 10-1570 % mol/mol Wasserstoffperoxid/ Polyethylen 30 22Pikrinsäure 32 75Trinitrotoluol (TNT) 30 15097 % mol/mol Wasserstoffperoxid/Wasser 17 nicht sensitiv
Besondere Vorsicht ist beim Erhitzen von Wasserstoffperoxid geboten. Die Zersetzungsrate
erhöht sich bei Temperaturanstieg etwa um den Faktor 2,3 per 10°C, zusätzlich ist
Wasserstoffperoxiddampf in Konzentrationen über 39 % schon unter Atmosphärendruck
explosiv (Jones, 1999; Hudlický, 1990).
Der wohl störendste Aspekt hinsichtlich der Verwendung von Wasserstoffperoxid in der
enzymatisch katalysierten Synthese von Peroxycarbonsäuren ist allerdings nicht die unter
Umständen gefährliche Lagerung oder der mit Gefahren verbundene Einsatz. Bereits Björking
(1990) berichtete, dass zu hohe Konzentrationen von wässrigem Wasserstoffperoxid zu einer
kompletten Denaturierung der Lipase-B von Candida antarctica führen. Dies ist auf die Tatsache
zurückzuführen, dass durch Oxidationsreaktionen, ausgelöst durch Wasserstoffperoxid, die
Tertiärstruktur des Enzyms irreparabel geschädigt wird. Für eine nachhaltige und effiziente
Synthese ist es aus diesem Grund zwingend notwendig, das wässrige Wasserstoffperoxid der
Reaktion in angepassten Mengen und Zeitintervallen hinzuzugeben (Ankudey et al, 2006). Das
Ziel ist hierbei, die Konzentration von frei verfügbarem Wasserstoffperoxid möglichst konstant
auf einem Level zu halten. Einerseits soll so eine funktionierende Synthese sichergestellt
werden, andererseits eine Denaturierung der Lipase verhindert werden. Übertragen auf die
industrielle Anwendung ist diese Tatsache alles andere als optimal. Die kontinuierliche Zugabe
ist einerseits mit entsprechenden Kosten verbunden, andererseits birgt ein kontinuierlicher
Batch-Reaktor, der dem Reaktionsmedium selbstständig wässriges Wasserstoffperoxid
hinzugibt, seinerseits neue Sicherheitsrisiken.

10
In dieser Studie wird, um diese Problemstellung zu umgehen, Wasserstoffperoxid durch
Harnstoff-Wasserstoffperoxid (Abb.5) als Oxidationsmittel ersetzt.
Abbildung 5: Harnstoff-Wasserstoffperoxid (auch Carbamidperoxid; Percarbamid)
Harnstoff-Wasserstoffperoxid (auch Carbamidperoxid oder Percarbamid) ist eine nicht
kovalente, wasserfreie Additionsverbindung zwischen Harnstoff und Wasserstoffperoxid. Die
Summenformel lautet CO(NH2)2.H2O2 (1:1). Man spricht auch von " festem
Wasserstoffperoxid", da es in kristalliner Form vorliegt. Der Schmelzpunkt, welcher in diesem
Fall zur Zersetzung in Harnstoff und Wasserstoffperoxid führt, beträgt 80-90°C
(www.chemicalland21.com).
Harnstoff-Wasserstoffperoxid hat das Potential, Wasserstoffperoxid in einer kontrollierbaren Art
und Weise freizusetzen. Hierdurch fällt die Notwendigkeit weg, das wässrige Wassertoffperoxid
dem Reaktionsmedium der enzymatisch katalysierten Synthese von Peroxycarbonsäuren in
Intervallen hinzuzugeben. Die Abwesenheit von Wasser im Reaktionsmedium ist auch von
Vorteil, da es unerwünschte Reaktionen der Edukte und Produkte minimiert.
Auch eine im Vergleich zu wässrigem Wasserstoffperoxid verhältnismäßig sichere Lagerung
und Handhabung ist gegeben. Harnstoff-Wasserstoffperoxid ist zwar als starkes Oxidationsmittel
feuergefährlich, bei Vermischung mit organischen Substanzen, z.B. Ether oder Aceton, können
durch Oxidationsvorgänge von freigesetztem Wasserstoffperoxid weiterhin explosive Lösungen
entstehen. Wird jedoch bei der Lagerung und Handhabung darauf geachtet, dass das Addukt
nicht mit Feuchtigkeit in Kontakt kommt und eine kühle Lagertemperatur von 2-8 °C eingehalten
wird, verläuft die Zersetzung in Harnstoff und Wasserstoffperoxid sehr langsam. Dies stellt eine
deutliche Risikominimierung im Vergleich von wässrigem Wasserstoffeproxid dar. Zusätze von
Natrium- oder Ammoniumdihydrogenphosphat bzw. Zinksulfat verbessern die Wärmestabilität,
Harnstoff-Wasserstoffperoxid kommt auch mit Stärke tablettiert in den Handel
(www.chemicalland21.com). Industriell wird es unter einer Vielzahl von Produktbezeichnungen
unter anderem zur Desinfektion bei der Wundbehandlung, zum Haarefärben/Blondieren, als
Bleichmittel für Zähne und als Entwickler für Blaupausen eingesetzt (Römpp, 1995). Prinzipiell
O
NH2 NH2
HO
OH

11
findet es überall dort Anwendung, wo eine kontrollierbare, automatische Freisetzung von
Wasserstoffperoxid von Nöten ist, bzw. gegenüber der manuellen Zugabe in Intervallen von
Vorteil ist.
Ankudey, Olivo und Peeples (2006) untersuchten erstmals erfolgreich die Lipasen-katalysierte
Epoxidation von diversen Alkenen unter Verwendung von Harnstoff-Wasserstoffperoxid als
Oxidationsmittel. Sie stellten fest, dass sich die besten Umsätze mit Essigsäureethylester als
Lösungsmittel und Edukt erreichen ließen. Hierzu wurde Peroxyessigsäure aus
Essigsäureethylester synthetisiert, katalysiert durch Novozym® 435. Wasserfreies Harnstoff-
Wasserstoffperoxid wurde genutzt als Oxidator, welcher einmalig in angepasster Menge am
Anfang der Reaktion hinzugegeben wurde. Die entstehende Peroxyessigsäure wurde in situ als
Oxidationsmittel genutzt, um im Reaktionsmedium enthaltene Alkene zu den entsprechenden
Epoxiden zu oxidieren.
Ríos, Salazar und Olivo (2007) führten in Folge unter Zuhilfenahme von Harnstoff-
Wasserstoffperoxid eine Bayer-Villinger-Oxidation von substituierten Cyclohexanonen durch.
Wieder wurde hierzu, enzymatisch katalysiert, Peroxyessigsäure aus Essigsäureethylester in situ
produziert, um im Reaktionsmedium enthaltene Cyclohexanone zu den jeweiligen Lactonen zu
oxidieren. Wasserfreies Harnstoff-Wasserstoffperoxid wurde einmalig in angepasster Menge am
Anfang der Reaktion hinzugegeben. Sowohl die Gruppe um Ankudey (2006) als auch die
Gruppe um Ríos (2007) waren erfolgreich mit ihrem Vorhaben und setzten Harnstoff-
Wasserstoffperoxid effektiv als Oxidant in der in situ Synthese von Peroxyessigsäure aus
Essigsäureethylester ein.
Um den Effekt der eventuellen Denaturierung des Novozym® 435 durch Harnstoff-
Wasserstoffperoxid zu untersuchen, recycelten Ankudey, Olivo und Peeples (2006) das Enzym
nach jeder Epoxidations-Reaktion durch Filtration und verwendeten es erneut. Es stellte sich
heraus, dass das Enzym seine Aktivität bei bis zu 6 Runden von Alken Epoxidationen beibehielt,
wobei weiterhin Umsätze von 75-100 % erreicht wurden. Dies unterstreicht die Nachhaltigkeit
und das Potential des Harnstoff-Wasserstoffperoxid-Addukts, Wasserstoffperoxid in für
enzymatisch katalysierte Reaktionen geeigneten Konzentrationen und Zeitintervallen
freizusetzen.

12
3. Ziel der Arbeit
Unter Berücksichtigung und vergleichend mit der Studie der enzymatisch katalysierten
Herstellung von Peroxyessigsäure mit wässrigem Wasserstoffperoxid (Ernst, 2011), ist das Ziel
der Arbeit die Untersuchung der Durchführung einer enzymatischen Synthese von
Peroxycarbonsäuren, katalysiert durch Novozym® 435, unter Verwendung von Harnstoff-
Wassertstoffperoxid als Oxidationsmittel.
Als Lösungsmittel und gleichzeitig als Edukte dienen Essigsäureethylester oder Diethylcarbonat,
erwartet wird die Synthese von Peroxyessigsäure bzw. Peroxykohlensäure. Die Reaktions-
gleichung der zu erwartenden Synthese der Peroxyessigsäure ist in Abbildung 6 dargestellt:
Abbildung 6: Reaktionsgleichung der enzymatisch katalysierten Synthese von Peroxyessigsäure aus Essigsäureethylester, unter Verwendung von Harnstoff-Wasserstoffperoxid als Oxidationsmittel
Die Reaktionsgleichung der durchzuführenden Synthese von Peroxykohlensäure aus Diethylcarbonat
setzt sich wie folgt zusammen (Abb. 7):
Abbildung 7: Reaktionsgleichung der enzymatisch katalysierten Synthese von Peroxykohlensäure aus Diethylcarbonat, unter Verwendung von Harnstoff-Wasserstoffperoxid als Oxidationsmittel
Der Schwerpunkt dieser Studie liegt auf der Realisierbarkeit der Durchführung dieser beiden
Synthesen. Zusätzlich werden umsatzbestimmende Kriterien wie Reaktionsdauer, Abfiltration
des Enzyms nach Abschluss der Reaktion, Reaktionstemperatur und die die schrittweise
Hinzugabe von Harnstoff-Wasserstoffperoxid, im Vergleich zur anfänglichen Gesamtzugabe,
untersucht.
CH3
O
O OHCH3
O
O CH3+ CO(NH2)2 [Novozym® 435]
+ C2H6O + CO(NH2)2. H2O2
+ CO(NH2)2 [Novozym® 435]+ C2H6O + CO(NH2)2. H2O2
O
O
O
C2H5
C2H5
O
O
O
OH
C2H5

13
4. Material und Methoden
4.1 Chemikalien/Reagenzien
Für alle enzymatisch katalysierten Peroxysäurensynthesen wird das Harnstoff-
Wasserstoffperoxid-Addukt des Herstellers Alfa Aesar GmbH & Co (CAS: 124-43-6; Charge:
10159973) verwendet. Als Edukte dienen Essigsäureethylester ( %) des Herstellers Carl
Roth GmbH & Co (CAS: 141-78-6; Charge: 30360285), sowie Diethylcarbonat des Herstellers
Merck Schuchardt OHG (CAS: 105-58-8; Charge: 56210898). als Katalysator wird in allen
Synthesen Novozym® 435 des Herstellers Codexis Inc. (CAS: 9001-62-1; Lot: LC200219)
genutzt.
Für die cerimetrische Bestimmung der Wasserstoffperoxidkonzentration nach Abschluss der
Reaktion wird Ferroinlösung des Herstellers Merck Schuchardt OHG (Charge: 0CC602981),
Schwefelsäure (10 %) des Herstellers Carl Roth GmbH & Co (CAS: 7664-93-9; Charge:
46575509), sowie Cer(IV)-sulfatlösung (0,1 M) des Herstellers Carl Roth GmbH & Co (CAS:
13590-82-4 [Wasserfrei]; Charge: 1111695) verwendet.
Für die obligatorisch im Anschluss folgende iodometrische Bestimmung der
Peroxysäurenkonzentration wird Iod-Kaliumiodid-Lösung (30 %) des Herstellers Merck
Schuchardt OHG (CAS: 7553-56-2; Charge: 58310992), Stärkelösung (1 %) (Lösliche Stärke
nach Zulkowsky) des Herstellers Merck Schuchardt OHG (Lot: 823E940857) sowie
Natriumthiosulfatlösung (0,1 M) des Herstellers Sigma-Aldrich Co. LLC (CAS: 7772-98-7
[Wasserfrei]; Charge: SZE92950) verwendet.
4.2 Methoden
4.2.1 Bestimmung der Konzentration des Harnstoff-Wasserstoffperoxid-Adduktes
Das in dieser Studie angewendete Harnstoff-Wasserstoffperoxid-Addukt wird vom Hersteller
Alfa Aesar GmbH & Co mit einer Konzentration von 97 % angegeben. Es wird empfohlen, das
Addukt trocken und kühl (2-8°C) zu lagern. Aufgrund von unangemessener Lagerung
(Raumtemperatur; ø 21°C) bis zum Beginn des Projektes, wird vermutet, dass die vom
Hersteller angegebene Konzentration nicht mehr gegeben ist. Zur Kontrolle wird die
Wasserstoffperoxidkonzentration des Adduktes cerimetrisch bestimmt (siehe Kap. 4.2.4). Da es
sich um ein 1:1 Addukt handelt, ist diese Konzentration äquivalent zur Harnstoff-
Wasserstoffperoxid-Konzentration im Produkt.

14
4.2.2 Enzymatisch katalysierte Synthese von Peroxycarbonsäuren
Alle in dieser Thesis durchgeführten enzymatisch katalysierten Synthesen von
Peroxycarbonsäuren orientieren sich vom Versuchsaufbau an der Studie der enzymatisch
katalysierten Darstellung von Peroxyessigsäure mit wässrigem Wasserstoffperoxid von Ernst
(2011). Bis auf wenige Abweichungen werden sie nach identischem Schema durchgeführt. Als
Reaktionsgefäß wird ein 100 ml Erlenmeyerkolben verwendet. In diesem erfolgt, entsprechend
der zu synthetisierenden Peroxycarbonsäure, die Zugabe von 20 ml Essigsäureethylester (
%), oder 20 ml Diethylcarbonat. Im Anschluss erfolgt die Zugabe von 0,40 g Novozym® 435
und 1,9924 g Harnstoff-Wasserstoffperoxid. Der Erlenmeyerkolben wird mit einem Stopfen
luftdicht verschlossen. Unter Anwendung eines Magnetrührers wird die Probe bei einer
Umdrehung von 7,5/s exakt 6 Stunden bei Raumtemperatur gerührt. Der Versuchsaufbau ist in
Abbildung 8 wiedergegeben.
Abbildung 8: Versuchsaufbau; Peroxysäuresynthese Abbildung 9: Versuchsaufbau; Abfiltration des Enzyms
Im Anschluss wird das Novozym® 435 mit Hilfe eines Glastrichters mit Filterpapier des
Herstellers Macherey-Nagel (MN 615. ø 70mm) abfiltriert und die Lösung in einen zweiten 100
ml Erlenmeyerkolben überführt (Abb. 9).
Die Wasserstoffperoxidkonzentration der filtrierten Lösung wird cerimetrisch bestimmt, die
abschließende Bestimmung der Peroxysäurenkonzentration erfolgt iodometrisch (siehe Kap.
4.2.4).
4.2.3 Variationen der enzymatisch katalysierten Synthese von Peroxycarbonsäuren
In dieser Studie werden u.a. Variationen des Versuchsaufbaus der enzymatisch katalysierten
Synthese von Peroxycarbonsäuren vorgenommen, um umsatzbestimmende Kriterien der
Reaktion zu untersuchen. Untersucht werden eine Veränderung der Reaktionszeit, das
100 ml Erlenmeyerkolben
Glastrichter
1
2
3
45 6
7
8
9
1 10
2
3
45 6
7
8
9
11
100 ml Erlenmeyerkolben
Stopfen
Magnetrührer mit Heizfunktion
Magnetrührkern

15
Unterlassen der Abfiltration des Enzyms nach Abschluss der Reaktion, eine Erhöhung der
Reaktionstemperatur und die schrittweise Hinzugabe von Harnstoff-Wasserstoffperoxid zum
Reaktionsmedium, im Vergleich zur anfänglichen Gesamtzugabe. Eine Veränderung der
Reaktionszeit wird vorgenommen, um Erkenntnisse über die Mikrokinetik der Reaktion zu
gewinnen. Der Versuchsaufbau ist identisch mit dem des Kap. 4.2.2, als einzige Änderung wird
die Reaktionszeit von 6 Stunden auf 4, sowie auf 2 Stunden reduziert. Das Unterlassen der
Abfiltration des Enzyms nach Abschluss der Reaktion dient dazu, zu untersuchen, ob neben dem
Enzym auch andere Stoffe, wie z.B. Wasserstoffperoxid oder Peroxysäuren, durch das
Abfiltrieren teilweise nicht in der Lösung verbleiben. Der Versuchsaufbau ist identisch mit dem
des Kap. 4.2.2, mit dem Unterschied, dass die cerimetrische und iodometrische Bestimmung der
Wasserstoffperoxid- und Peroxysäurenkonzentration ohne vorherige Filtration des
Reaktionsmediums stattfindet. Eine Erhöhung der Reaktionstemperatur wird angewendet, um
mögliche Folgeeffekte, wie eine schnellere Zersetzung des Harnstoff-Wasserstoffperoxid-
Adduktes, eine verbesserte Enzymaktivität oder eine zu vermeidende Zersetzung der
entstandenen Peroxysäuren zu untersuchen. Der Versuchsaufbau ist analog zu dem des Kap.
4.2.2, allerdings wird der als Reaktionsgefäß dienende Erlenmeyerkolben von dem Magnetrührer
mit einem Heizfeld von 50°C erhitzt. Die schrittweise Hinzugabe von Harnstoff-
Wasserstoffperoxid zum Reaktionsmedium wird durchgeführt, um zu untersuchen, ob die
dadurch veränderte Konzentration von freiem Wasserstoffperoxid einen Einfluss auf den Umsatz
an Peroxycarbonsäuren hat. Der Versuchsaufbau ist identisch mit dem des Kap. 4.2.2, allerdings
werden die benötigten 1,9924 g Harnstoff-Wasserstoffperoxid in 5 äquivalente Teilmengen von
0,3985 g separiert. Eine Teilmenge wird dem Reaktionsmedium zu Anfang hinzugegeben, die
Zugabe der anderen Teilmengen erfolgt stündlich über den Zeitraum von 4 Stunden.
4.2.4 Cerimetrische Bestimmung der Wasserstoffperoxidkonzentration und iodometrische
Bestimmung der Peroxycarbonsäurenkonzentration
Die Bestimmung der Wasserstoffperoxidkonzentration wird in dieser Studie in allen Synthesen
cerimetrisch durchgeführt, die Bestimmung der Peroxycarbonsäurenkonzentration erfolgt
iodometrisch. Für die cerimetrische Bestimmung der Wasserstoffperoxidkonzentration wird die
zu bestimmende Probe zur Verdünnung in einen 100 ml Erlenmeyerkolben eingewogen, welcher
mit 50 ml dest. Wasser aufgefüllt wird. Die Menge der eingewogenen Probe und der
Verdünnungsfaktor variieren nach zu erwartender Wasserstoffperoxid- und

16
Peroxysäurenkonzentration. Es erfolgt die Zugabe von 3 Tropfen Ferroinlösung und 5 ml
Schwefelsäure (10 %).
Die Lösungen werden über eine Bürette mit Cer(IV)-sulfatlösung (0.1 M) unter Anwendung
eines Magnetrührers bis zum Farbumschlag titriert. Die molare Wasserstoffperoxidkonzentration
der Probe kann nun kalkulatorisch bestimmt werden (siehe Formel, Anhang). Der
Versuchsaufbau der Titration via Bürette ist in Abbildung 10 dargestellt.
Abbildung 10: Versuchsaufbau; Titration via Bürette
Mit der austitrierten Lösung kann nun eine iodometrische Bestimmung der
Peroxysäurenkonzentration durchgeführt werden. Hierzu wird der Lösung zusätzlich 1,5 ml Iod-
Kaliumiodid-Lösung (30 %) mittels Dispenser zugesetzt, anschließend werden 2,5 ml
Stärkelösung (1 %) in den Erlenmeyerkolben pipettiert.
Die Lösung wird über eine Bürette unter Anwendung eines Magnetrührers mit
Natriumthiosulfatlösung (0,1 M) bis zum Farbumschlag titriert (Abb. 10). Die molare
Peroxysäurenkonzentration der Probe kann nun kalkulatorisch bestimmt werden (siehe Formel,
Anhang).
5. Versuchsergebnisse
5.1 Bestimmungen der Konzentration des Harnstoff-Wasserstoffperoxid-Adduktes
Zur Bestimmung der Konzentration des Harnstoff-Wasserstoffperoxid-Adduktes wurde die
Wasserstoffperoxidkonzentration des Adduktes cerimetrisch nach Kap. 4.2.4 bestimmt. Da es
50
40
30
20
10
0
1234 5 6 7
89
1 10
234 5 6 7
8911
Titrationslösung
verdünnte Probenlösung
Bürette
17
sich um ein 1:1 Addukt handelt, ist diese molare Konzentration äquivalent zur molaren
Harnstoff-Wasserstoffperoxid-Konzentration im Produkt.
Zur Verdünnung wurde 1 g des Adduktes in 10 ml dest. Wasser gelöst. Für eine
Dreifachbestimmung wurde von 3 Äquivalenten von dieser Lösung von 0,5 ml, gelöst in 50 ml
dest. Wasser, cerimetrisch die Wasserstoffperoxidkonzentration bestimmt. Die Messergebnisse
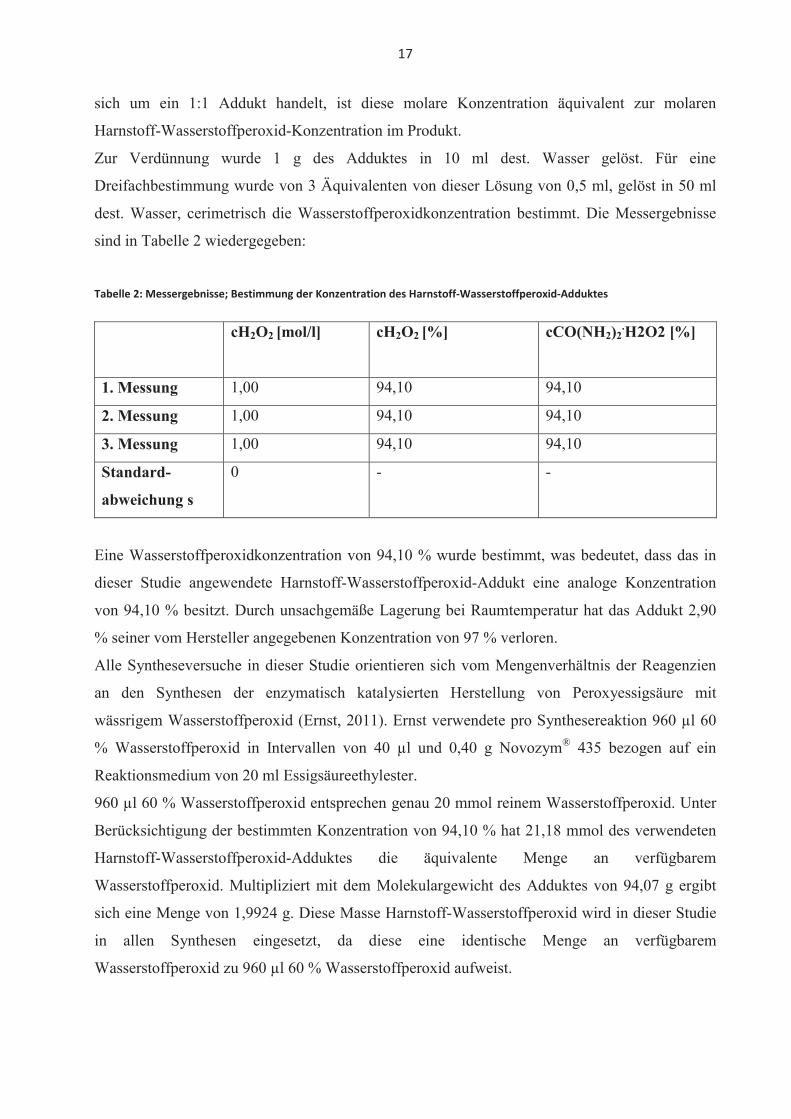
sind in Tabelle 2 wiedergegeben:
Tabelle 2: Messergebnisse; Bestimmung der Konzentration des Harnstoff-Wasserstoffperoxid-Adduktes
cH2O2 [mol/l] cH2O2 [%] cCO(NH2)2.H2O2 [%]
1. Messung 1,00 94,10 94,10
2. Messung 1,00 94,10 94,10
3. Messung 1,00 94,10 94,10
Standard-
abweichung s
0 - -
Eine Wasserstoffperoxidkonzentration von 94,10 % wurde bestimmt, was bedeutet, dass das in
dieser Studie angewendete Harnstoff-Wasserstoffperoxid-Addukt eine analoge Konzentration
von 94,10 % besitzt. Durch unsachgemäße Lagerung bei Raumtemperatur hat das Addukt 2,90
% seiner vom Hersteller angegebenen Konzentration von 97 % verloren.
Alle Syntheseversuche in dieser Studie orientieren sich vom Mengenverhältnis der Reagenzien
an den Synthesen der enzymatisch katalysierten Herstellung von Peroxyessigsäure mit
wässrigem Wasserstoffperoxid (Ernst, 2011). Ernst verwendete pro Synthesereaktion 960 μl 60
% Wasserstoffperoxid in Intervallen von 40 μl und 0,40 g Novozym® 435 bezogen auf ein
Reaktionsmedium von 20 ml Essigsäureethylester.
960 μl 60 % Wasserstoffperoxid entsprechen genau 20 mmol reinem Wasserstoffperoxid. Unter
Berücksichtigung der bestimmten Konzentration von 94,10 % hat 21,18 mmol des verwendeten
Harnstoff-Wasserstoffperoxid-Adduktes die äquivalente Menge an verfügbarem
Wasserstoffperoxid. Multipliziert mit dem Molekulargewicht des Adduktes von 94,07 g ergibt
sich eine Menge von 1,9924 g. Diese Masse Harnstoff-Wasserstoffperoxid wird in dieser Studie
in allen Synthesen eingesetzt, da diese eine identische Menge an verfügbarem
Wasserstoffperoxid zu 960 μl 60 % Wasserstoffperoxid aufweist.

18
5.2 Enzymatisch katalysierte Synthese von Peroxycarbonsäuren; Reaktionskriterien nach
Ernst (2011)
5.2.1 Synthese 1: Oxidation von Essigsäureethylester zu Peroxyessigsäure
Für einen Direkten Vergleich der Effizienz der Peroxyessigsäuresynthese mit Harnstoff-
Wasserstoffperoxid im Vergleich zu der Peroxyessigsäuresynthese mit konventionellem
wässrigem Wasserstoffperoxid, wurde in Synthese 1 Essigsäureethylester zu Peroxyessigsäure
nach Kapitel 4.2.2 oxidiert.
Nach einer dreifachen cerimetrischen Bestimmung der Wasserstoffperoxidkonzentration sowie
dreifacher iodometrischer Bestimmung der Peroxyessigsäurekonzentration, ergab der Mittelwert
der molaren Wasserstoffperoxidkonzentration 0,1550 [mol/l], der Mittelwert der molaren
Peroxyessigsäurekonzentration 0,3170 [mol/l]. Die entsprechenden Messergebnisse und
kalkulierten Stoffmengen sind in Tabelle 3 wiedergegeben:
Tabelle 3: Synthese 1; Messergebnisse; Oxidation von Essigsäureethylester zu Peroxyessigsäure
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxyessigsäure
[mol/l]
nPeroxyessigsäure
[mmol]
1. Messung 0,1600 3,20 0,3100 6,20
2. Messung 0,1550 3,10 0,3200 6,40
3. Messung 0,1500 3,00 0,3210 6,42
Mittelwert x̄ 0,1550 3,10 0,3170 6,34
Standard-
abweichung s
4,08*10-3 - 4,97*10-3 -
5.2.2 Synthese 2: Oxidation von Diethylcarbonat zu Peroxykohlensäure
Da die in Synthese 1 angewendete Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid
zufriedenstellend verlief, wurde in Synthese 2 nach gleichem Versuchsablauf Diethylcarbonat zu
Peroxykohlensäure oxidiert.
Nach Abschluss der Reaktion wurde die Wasserstoffperoxidkonzentration dreifach cerimetrisch
und die Peroxysäurekonzentration dreifach iodometrisch bestimmt. Der Mittelwert der molaren
Wasserstoffperoxidkonzentration ergab 0,0493 [mol/l], der Mittelwert der molaren

19
Peroxykohlensäurekonzentration 0,0393 [mol/l]. Die entsprechenden Messergebnisse und
kalkulierten Stoffmengen sind in Tabelle 4 wiedergegeben:
Tabelle 4: Synthese 2; Messergebnisse; Oxidation von Diethylcarbonat zu Peroxykohlensäure
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxykohlensäure
[mol/l]
nPeroxykohlensäure
[mmol]
1. Messung 0,0500 1,00 0,0400 0,80
2. Messung 0,0460 0,92 0,0380 0,76
3. Messung 0,0520 1,04 0,0400 0,80
Mittelwert x̄ 0,0493 0,99 0,0393 0,79
Standard-
abweichung s
2,49*10-3 - 0,943*10-3 -
5.3 Variationen der enzymatisch katalysierten Synthese von Peroxycarbonsäuren
5.3.1 Synthese 3: Reduktion der Reaktionszeit auf 2 und 4 Stunden
Um Erkenntnisse über den Reaktionsablauf zu gewinnen, wurde in Synthese 3 Diethylcarbonat,
in 2 externen Reaktionen, nach Kapitel 4.2.2 zu Peroxykohlensäure oxidiert. Die erste Reaktion
wurde nach 2 Stunden durch Abfiltration des Enzyms beendet, die zweite Reaktion
gleichermaßen nach 4 Stunden.
Der Mittelwert der molaren Wasserstoffperoxidkonzentration der Reaktion mit 2 Stunden
Reaktionsdauer ergab nach dreifacher cerimetrischer Bestimmung 0,0433 [mol/l], der Mittelwert
der Reaktion mit 4 Stunden 0,0483 [mol/l]. Der Mittelwert der molaren
Peroxykohlensäurekonzentration der Reaktion mit 2 Stunden ergab nach dreifacher
iodometrischer Bestimmung 0,0100 [mol/l], der Mittelwert der Reaktion mit 4 Stunden 0,0160
[mol/l]. Die entsprechenden Messergebnisse und kalkulierten Stoffmengen der Reaktion mit 2
Stunden Reaktionszeit sind in Tabelle 5 wiedergegeben, entsprechende Werte der Reaktion mit 4
Stunden Reaktionszeit sind in Tabelle 6 dargestellt.

20
Tabelle 5: Synthese 3; Messergebnisse; Oxidation von Diethylcarbonat zu Peroxykohlensäure; Reduktion der Reaktionszeit auf 2 Stunden
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxykohlensäure
[mol/l]
nPeroxykohlensäure
[mmol]
1. Messung 0,0400 0,80 0,0100 0,20
2. Messung 0,0450 0,90 0,0100 0,20
3. Messung 0,0450 0,90 0,0100 0,20
Mittelwert x̄ 0,0433 0,87 0,0100 0,20
Standard-
abweichung s
2,36*10-3 - 0 -
Tabelle 6: Synthese 3; Messergebnisse; Oxidation von Diethylcarbonat zu Peroxykohlensäure; Reduktion der Reaktionszeit auf 4 Stunden
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxykohlensäure
[mol/l]
nPeroxykohlensäure
[mmol]
1. Messung 0,0450 0,90 0,0180 0,36
2. Messung 0,0500 1,00 0,0150 0,30
3. Messung 0,0500 1,00 0,0150 0,30
Mittelwert x̄ 0,0483 0,97 0,0160 0,32
Standard-
abweichung s
2,36*10-3 - 1,41*10-3 -
Aufgrund der bei dieser Synthese entstandenen geringen Peroxysäurenkonzentrationen und den
damit verbundenen zur Titration benötigten geringen Mengen Natriumthiosulfatlösung, ist das
Ergebnis dieser Synthese nur von bedingter Aussagekraft. Dennoch ist ein Trend der
Reaktionskinetik zu erkennen.
5.3.2 Synthese 4: Unterlassung der Filtration des Reaktionsmediums nach Abschluss der
Reaktion
Zur Überprüfung ob neben dem Enzym auch andere Stoffe, wie z.B. Wasserstoffperoxid oder
Peroxysäuren, durch das Abfiltrieren teilweise nicht in der Lösung verbleiben, wurde in
Synthese 4 Essigsäureethylester und Diethylcarbonat nach Kapitel 4.2.2 zu Peroxyessigsäure
bzw. Peroxykohlensäure oxidiert. Nach Abschluss der Reaktion wurde die Wasserstoffperoxid-

21
sowie Peroxysäurekonzentration direkt dreifach cerimetrisch und iodometrisch, ohne die
obligatorische Filtration, bestimmt.
Der Mittelwert der molaren Wasserstoffperoxidkonzentration der Reaktion mit
Essigsäureethylester ergab 0,2483 [mol/l], der Mittelwert der molaren
Peroxyessigsäurekonzentration 0,3116 [mol/l]. Die Messergebnisse und kalkulierten
Stoffmengen der Reaktion mit Essigsäureethylester sind in Tabelle 7 wiedergegeben:
Tabelle 7: Synthese 4; Messergebnisse; Oxidation von Essigsäureethylester zu Peroxyessigsäure; Unterlassung der Filtration des Reaktionsmediums nach Abschluss der Reaktion
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxyessigsäure
[mol/l]
nPeroxyessigsäure
[mmol]
1. Messung 0,2450 4,90 0,3100 6,20
2. Messung 0,2500 5,00 0,3100 6,20
3. Messung 0,2500 5,00 0,3150 6,30
Mittelwert x̄ 0,2483 4,97 0,3116 6,23
Standard-
abweichung s
2,36*10-3 - 2,36*10-3 -
Der Mittelwert der molaren Wasserstoffperoxidkonzentration der Reaktion mit Diethylcarbonat
ergab 0,0683 [mol/l], der Mittelwert der molaren Peroxykohlensäurekonzentration 0,0353
[mol/l]. Die Messergebnisse und kalkulierten Stoffmengen der Reaktion mit Diethylcarbonat
sind in Tabelle 8 dargestellt:
Tabelle 8: Synthese 4; Messergebnisse; Oxidation von Diethylcarbonat zu Peroxykohlensäure; Unterlassung der Filtration des Reaktionsmediums nach Abschluss der Reaktion
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxykohlensäure
[mol/l]
nPeroxykohlensäure
[mmol]
1. Messung 0,0700 1,40 0,0360 0,72
2. Messung 0,0650 1,30 0,0350 0,70
3. Messung 0,0700 1,40 0,0350 0,70
Mittelwert x̄ 0,0683 1,37 0,0353 0,71
Standard-
abweichung s
2,36*10-3 - 0,47*10-3 -

22
5.3.3 Synthese 5: Erhöhung der Reaktionstemperatur auf 50°C
Um temperaturabhängige Reaktionskriterien wie eine schnellere Zersetzung des Harnstoff-
Wasserstoffperoxid-Adduktes, eine verbesserte Enzymaktivität oder eine zu vermeidende
Zersetzung der zu synthetisierenden Peroxycarbonsäuren zu untersuchen, wurde in Synthese 5
die Reaktionstemperatur auf 50° C erhöht. Als Edukt diente Diethylcarbonat, welches, unter
angesprochener Temperaturerhöhung, nach Kapitel 4.2.2 zu Peroxykohlensäure oxidiert wurde.
Nach dreifacher cerimetrischer Bestimmung der Wasserstoffperoxidkonzentration ergab der
Mittelwert 0,0667 [mol/l], der Mittelwert der molaren Peroxykohlensäurekonzentration ergab
nach dreifacher iodometrischer Bestimmung 0,0616 [mol/l]. Die entsprechenden
Messergebnisse und kalkulierten Stoffmengen sind in Tabelle 9 wiedergegeben:
Tabelle 9: Synthese 5; Messergebnisse; Oxidation von Diethylcarbonat zu Peroxykohlensäure; 50°C
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxykohlensäure
[mol/l]
nPeroxykohlensäure
[mmol]
1. Messung 0,0700 1,40 0,0650 1,30
2. Messung 0,0650 1,30 0,0600 1,20
3. Messung 0,0650 1,30 0,0600 1,20
Mittelwert x̄ 0,0667 1,33 0,0616 1,23
Standard-
abweichung s
2,36*10-3 - 2,36*10-3 -
5.3.4 Synthese 6: Hinzugabe von Harnstoff-Wasserstoffperoxid in Intervallen
Um Kenntnisse über die Zersetzungsgeschwindigkeit des Harnstoff-Wasserstoffperoxids und
damit verbundene Änderungen der Ausbeute zu gewinnen, wurde in Synthese 6 das Addukt dem
Reaktionsmedium in 5 Intervallen zugegeben. Durchgeführt wurde der Synthese sowohl mit
Essigsäureethylester als auch mit Diethylcarbonat als Edukt, bis auf die Zugabe in Intervallen
war der Versuchsaufbau äquivalent zu Kapitel 4.2.2.
In der Reaktion mit Essigsäureethylester ergab der Mittelwert der molaren
Wasserstoffperoxidkonzentration 0,2667 [mol/l], der Mittelwert der molaren
Peroxyessigsäurekonzentration ergab 0,2930 [mol/l]. Die entsprechenden Messergebnisse und
kalkulierten Stoffmengen sind in Tabelle 10 wiedergegeben:

23
Tabelle 10: Synthese 6; Messergebnisse; Oxidation von Essigsäureethylester zu Peroxyessigsäure; Hinzugabe von Harnstoff-Wasserstoffperoxid in Intervallen
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxyessigsäure
[mol/l]
nPeroxyessigsäure
[mmol]
1. Messung 0,2600 5,20 0,3000 6,00
2. Messung 0,2760 5,52 0,2900 5,80
3. Messung 0,2700 5,40 0,2900 5,80
Mittelwert x̄ 0,2667 5,33 0,2930 5,86
Standard-
abweichung s
6,89*10-3 - 4,73*10-3 -
Der Mittelwert der molaren Wasserstoffperoxidkonzentration der Reaktion mit Diethylcarbonat
ergab 0,0617 [mol/l], der Mittelwert der molaren Peroxykohlensäurekonzentration 0,0253
[mol/l]. Die entsprechenden Messergebnisse und kalkulierten Stoffmengen sind in Tabelle 11
dargestellt:
Tabelle 11: Synthese 6; Messergebnisse; Oxidation von Diethylcarbonat zu Peroxykohlensäure; Hinzugabe von Harnstoff-Wasserstoffperoxid in Intervallen
cH2O2
[mol/l]
nH2O2
[mmol]
cPeroxykohlensäure
[mol/l]
nPeroxykohlensäure
[mmol]
1. Messung 0,0650 1,30 0,0260 0,52
2. Messung 0,0600 1,20 0,0250 0,50
3. Messung 0,0600 1,20 0,0250 0,50
Mittelwert x̄ 0,0617 1,23 0,0253 0,51
Standard-
abweichung s
2,36*10-3 - 4,73*10-4 -
6. Diskussion
6.1 Synthese 1: Oxidation von Essigsäureethylester zu Peroxyessigsäure
Die Auswertung von Synthese 1 bestätigt die Durchführbarkeit der enzymatischen Synthese von
Peroxyessigsäure mit Harnstoff-Wasserstoffperoxid als Oxidationsmittel. Der gemessene
Mittelwert der synthetisierten Stoffmenge Peroxyessigsäure von 6,34 mmol zeugt von einem den
Erwartungen entsprechenden Reaktionsablauf.

24
Es zeigt sich, dass das Harnstoff-Wasserstoffperoxid Addukt sich in für diese Reaktion
geeignetem Maße zersetzt. Der Zerfall von Harnstoff-Wasserstoffperoxid in freien Harnstoff und
Wasserstoffperoxid erfolgt in Lösung in Essigsäureethylester einem chemischen Gleichgewicht.
Während der Zerfall des Adduktes in kristalliner, „fester“ Form unter geeigneten
Lagerbedingungen nur sehr langsam voranschreitet, scheint in Lösung in Essigsäureethylester
das chemische Gleichgewicht von freiem Harnstoff und Wasserstoffperoxid, zu dem nicht
kovalent gebundenem Addukt, für die enzymatische Synthese von Peroxyessigsäure geeignet zu
sein. Dies bedeutet, dass einerseits genug freies Wasserstoffperoxid in Lösung ist um mit Acyl-
Enzym-Komplexen zu reagieren um Peroxyessigsäure entstehen zu lassen, andererseits die
Konzentration niedrig genug ist um eine zu hohe Denaturierung des Novozym® 435 zu
verhindern.
Eine solche Gleichgewichtsstellung war zu erwarten. Bereits Ankudey, Olivio und Peeples
(2006), welche Epoxidationen in Essigsäureethylester unter Verwendung von Harnstoff-
Wasserstoffperoxid durch eine enzymatische in situ Synthese von Peroxyessigsäure
durchführten, stellten bei anschließendem Recylcling des Enzyms Novozym® 435 fest, dass es
seine Aktivität bei bis zu 6 aufeinanderfolgenden Epoxidationsreaktionen nahezu unverändert
beibehielt. Dies lässt darauf schließen, dass eine Denaturierung einer kleinen Konzentration der
Lipase stattfindet. Allerdings ist diese, im technischen Maßstab, verglichen mit dem
Mehraufwand der kontinuierlichen Zugabe von wässrigem Wasserstoffperoxid bei der
konventionellen enzymatischen Peroxysäurensynthese, zu vernachlässigen. Nicht zuletzt lässt
sich gerade bei der kontinuierlichen Zugabe von wässrigem Wasserstoffperoxid eine
Denaturierung einer großen Menge des Enzyms nicht verhindern. Schließlich ist es im
technischen Maßstab nahezu unmöglich, dem Reaktionsmedium jeweils exakt die Konzentration
an Wasserstoffperoxid hinzuzugeben, welche der jeweiligen Konzentration der verfügbaren
Acyl-Enzym-Komplexe entspricht.
Ein Vergleich der in Synthese 1 synthetisierten Stoffmenge an Peroxyessigsäure und der
Stoffmenge Wasserstoffperoxid zu Ende der Reaktion, bezogen zu den selben Werten der
enzymatischen Peroxyessigsäuresynthese mit wässrigem Wasserstoffperoxid nach Ernst (2011),
ist in Tabelle 12 wiedergegeben. Bis auf die kontinuierliche Zugabe von wässrigem
Wasserstoffperoxid statt der anfänglichen Zugabe von Harnstoff-Wasserstoffperoxid, wurde die
Synthese von Ernst ebenfalls nach Kapitel 4.2.2 durchgeführt.

25
Tabelle 12: Vergleich von Peroxyessigsäuresynthese aus Essigsäureethylester mit Harnstoff-Wasserstoffperoxid zu Peroxyessigsäuresynthese aus Essigsäureethylester mit wässrigem Wasserstoffperoxid (60%) (Ernst, 2011)
Peroxyessigsäuresynthese mit
wässrigem Wasserstoffperoxid
(60%) (Ernst, 2011)
Synthese 1:
Peroxyessigsäuresynthese mit
Harnstoff-Wasserstoffperoxid
nWasserstoffperoxid [mmol] 13,92 3,10nPeroxyessigsäure [mmol] 9,17 6,34
Ernst bestimmte nach Abschluss der Reaktion eine Stoffmenge Wasserstoffperoxid von 13,92
mmol. Diese erscheint im Verhältnis zu der in der Synthese mit Harnstoff-Wasserstoffperoxid
bestimmten Stoffmenge von 3,10 mmol sehr hoch. Die verhältnismäßig hohe Konzentration lässt
vermuten, dass die von Ernst alle 15 Minuten zugefügten Mengen von 40 µl 60 %
Wasserstoffperoxid sehr hoch dosiert waren. Geht man davon aus, dass während der gesamten
Reaktion ein ähnlicher Überschuss von 13,92 mmol bzw. eine Konzentration von 0,6960 [mol/l]
Wasserstoffperoxid im Medium vorherrschte, ist diese Methode aufgrund der entsprechenden
Denaturierungsrate des Novozyms® 435 wirtschaftlich suboptimal.
In der Synthese mit Harnstoff-Wasserstoffperoxid hingegen wurde nach Abschluss der Reaktion
eine Stoffmenge Wasserstoffperoxid von 3,10 mmol bestimmt. Dies lässt keinen direkten
Rückschluss auf die Stoffmenge an freiem Wasserstoffperoxid zu, da bei der cerimetrischen
Titration auch das gebundene Harnstoff-Wassertoffperoxid mit einbezogen wird (siehe Kap.
5.1). Allerdings lässt sich hieraus ableiten, dass von den 21,18 mmol Harnstoff-
Wasserstoffperoxid, welche zu Anfang der Reaktion hinzugegeben wurden, nach Abschluss der
Reaktion weniger als 3,10 mmol in seiner gebundenen Form vorliegt. Der hohe Verbrauch des
Adduktes lässt auf einen hohen Umsatz schließen. Dies muss allerdings relativiert werden,
vergleicht man die Peroxyessigsäureausbeute von Ernst (9,17 mmol) mit der Ausbeute aus der
Synthese mit Harnstoff-Wasserstoffperoxid (6,34 mmol). Zu erklären ist dies aller
Wahrscheinlichkeit nach durch Eigenzerfall des Wasserstoffperoxids. Dieser muss teilweise
nach Trennung des Adduktes stattgefunden haben, sodass dem Enzym nur ein Teil des
anfänglich im Addukt gebundenen Wasserstoffperoxids zur Verfügung stand.
Zur Visualisierung ist die in Synthese 1 synthetisierte Stoffmenge an Peroxyessigsäure und die
Wasserstoffperoxidmenge zu Ende der Reaktion, verglichen mit den selben Messwerten der
enzymatischen Peroxyessigsäuresynthese mit wässrigem Wasserstoffperoxid von Ernst, in
Abbildung 11 dargestellt:

26
Abbildung 11: Vergleich von Peroxyessigsäuresynthese aus Essigsäureethylester mit Harnstoff-Wasserstoffperoxid zu Peroxyessigsäuresynthese aus Essigsäureethylester mit wässrigem Wasserstoffperoxid (60%) (Ernst, 2011)
Eine niedrigere Ausbeute an Peroxyessigsäure bezogen auf die Synthese von Ernst, war zu
erwarten. Auch Ankudey, Olivio und Peeples (2006) publizierten, dass bei der Lipase-
katalysierten Epoxidation von diversen Alkenen durch in situ Synthese von Peroxyessigsäure
unter Verwendung von Harnstoff-Wasserstoffperoxid als Oxidationsmittel, generell niedrigere
Umsätze zustande kamen, wie bei äquivalenten Epoxidationen mit wässrigem
Wasserstoffperoxid. Folglich scheint das Enzym bei Verwendung von wässrigem
Wasserstoffperoxid größeren Konzentrationen von freiem Wasserstoffperoxid ausgesetzt zu sein.
Dies führte einerseits dazu, dass die Reaktion aufgrund der besseren Verfügbarkeit schneller
voranschreitet, andererseits steigt die Denaturierungsrate des Enzyms entsprechend. Unter
Berücksichtigung der Recycelbarkeit des Novozym® 435 ist deshalb die schonendere
Synthesemethode unter Verwendung von Harnstoff-Wasserstoffperoxid der konventionellen
enzymatischen Synthesemethode durchaus vorzuziehen.
6.2 Synthese 2: Oxidation von Diethylcarbonat zu Peroxykohlensäure
Die Auswertung von Synthese 2 bestätigt, dass auch die Durchführung der enzymatischen
Synthese von Peroxykohlensäure mit Harnstoff-Wasserstoffperoxid als Oxidationsmittel möglich
ist. Der gemessene Mittelwert der synthetisierten Stoffmenge Peroxykohlensäure von 0,79 mmol
ist zwar im Vergleich zu der in Synthese 1 erzeugten Stoffmenge von 6,34 mmol
Peroxyessigsäure relativ gering, zeugt aber von einem funktionierenden Reaktionsablauf. Eine
0123456789
101112131415
Peroxyessigsäuresynthese aus Essigsäureethylester mit
wässrigem Wasserstoffperoxid (60%) (Ernst, 2011)
Synthese 1: Peroxyessigsäuresynthese aus
Essigsäureethylester mit Harnstoff-Wasserstoffperoxid
nWasserstoffperoxid [mmol]
nPeroxyessigsäure [mmol]

27
Umsatzeinbuße bei Verwendung eines anderen Lösungsmittel und Eduktes als
Essigsäureethylester war zu erwarten. Schließlich publizierten Ankudey, Olivo und Peeples
(2006), dass bei der Alken-Epoxidation mit Harnstoff-Wasserstoffperoxid, Essigsäureethylester
als Lösungsmittel und Edukt für die in situ Peroxysäuresynthese mit Abstand die besten Umsätze
hervorbrachte. Auch Ríos, Salazar und Olivo (2007) wählten bei der Oxidation von
substituierten Cyclohexanonen durch enzymatisch katalysierte in situ Peroxysäuresynthese
Essigsäureethylester als am besten geeignetes Lösungsmittel und Edukt.
Durch den Überschuss an Diethylcarbonat im Reaktionsmedium entsteht in Synthese 2
ausschließlich Monoperoxykohlensäuremonoethylester (siehe Abb. 7). Erst im theoretischen
Fall, dass alle Diethylcarbonat Moleküle zu Monoperoxykohlensäuremonoethylester (hier:
Peroxykohlensäure) oxidiert worden wären, würde Novozym® 435 die Oxidation zu der
Diperoxykohlensäure katalysieren. Ein Problem bei der Peroxysäurensynthese aus
Diethylcarbonat ist die Unstabilität der Peroxykohlensäure. Der bezogen auf die
Peroxyessigsäuresynthese verhältnismäßig geringe Umsatz könnte deswegen darin begründet
sein, dass Peroxykohlensäure zwar in größeren Stoffmengen entsteht, ein großer Teil aber nach 6
Stunden Reaktionszeit bereits wieder in Ethanol, Wasser, Kohlenstoffdioxid und Sauerstoff
zerfallen ist. Eine andere Möglichkeit könnte eine schlechtere Substratspezifität des Novozym®
435 bezogen auf Diethylcarbonat sein. So könnte es für die katalytischen Triaden der Lipase
energetisch und/oder räumlich aufwändiger sein, dass im Gegensatz zu Essigsäureethylester 2
Esterverbindungen aufweisende Diethylcarbonatmolekül, vom Serin im aktivem Zentrum unter
Ausbildung eines tetrahedralen Intermediates nukleophil anzugreifen (Kap 2.2).
Nach Abschluss der Reaktion wurde eine Stoffmenge Wasserstoffperoxid von 0,986 mmol
bestimmt, was nur ein Drittel der bestimmten Stoffmenge Wasserstoffperoxid von 3,10 mmol
aus Synthese 1 entspricht. Generell sind in allen enzymatischen Peroxykohlensäurensynthesen
dieser Studie deutlich geringere Wasserstoffperoxidkonzentrationen gemessen worden als bei
entsprechenden Peroxyessigsäuresynthesen. Der Grund hierfür könnte in dem teilweisen Zerfall
der Peroxykohlensäure liegen. Zersetzt sich diese bereits vor der cerimetrischen Titration in
Ethanol, Wasser, Kohlenstoffdioxid und Sauerstoff, so geht in der Summe Wasserstoffperoxid
im Reaktionsmedium verloren.
Zum Vergleich der enzymatischen Synthese von Peroxykohlensäure mit Harnstoff-
Wasserstoffperoxid, zu der enzymatischen Synthese von Peroxykohlensäure mit wässrigem
Wasserstoffperoxid, sind in Tabelle 13 die Messwerte von Synthese 2 mit den Messwerten einer
Peroxykohlensäuresynthese, durchgeführt durch Trojan (2010), dargestellt. Trojan verwendete
ebenfalls 20 ml Diethylcarbonat und 0,40 g Novozym® 435, setzte aber statt Harnstoff-

28
Wasserstoffperoxid äquivalent zu Ernst 960 μl 60 % Wasserstoffperoxid ein. Über eine
Reaktionszeit von 24 h fügte er dem Reaktionsmedium per Dosierpumpe im Abstand von 48 min
jeweils eine Menge von 0,04 μl der wässrigen Wasserstoffperoxidlösung zu.
Tabelle 13: Vergleich von Peroxykohlensäuresynthese aus Diethylcarbonat mit Harnstoff-Wasserstoffperoxid [6h ] zu Peroxykohlensäuresynthese aus Diethylcarbonat mit wässrigem Wasserstoffperoxid (60%) [24h] (Trojan, 2011)
Peroxykohlensäuresynthese mit
wässrigem Wasserstoffperoxid (60%)
[24h] (Trojan, 2010)
Synthese 2: Peroxykohlensäuresynthese mit
Harnstoff-Wasserstoffperoxid
[6h ]
nWasserstoffperoxid
[mmol]23,48 0,99
nPeroxykohlensäure
[mmol]1,10 0,79
Trojan (2010) bestimmte nach Abschluss der Reaktion eine Stoffmenge Wasserstoffperoxid von
23,48 mmol. Diese erscheint selbst im Verhältnis zu der in der Peroxyessigsäuresynthese mit
wässrigem Wasserstoffperoxid von Ernst (2011) gemessenen Stoffmenge von 13,92 mmol
erstaunlich hoch. Trojan wendete im Vergleich zu Ernst eine 6-fache Reaktionszeit an, erhöhte
die Anzahl der Zugabeintervalle des wässrigen Wasserstoffperoxids und reduzierte die
Intervallvolumen um das Zehnfache. Der hohe Wasserstoffperoxid Wert zu Ende der Reaktion
ist also höchst wahrscheinlich in einer fehlerhaften Dosierung oder fehlerhaften Titration
begründet, da die gemessene Stoffmenge von 23,48 mmol höher ist als die 21 mmol, welche dem
Reaktionsmedium ursprünglich insgesamt zugefügt werden sollten. Gleichwohl ist auch diese
von Trojan angewendete Methode der Peroxykohlensäuresynthese, aufgrund der entsprechend zu
Ernst noch höheren Denaturierungsrate des Novozyms® 435, wirtschaftlich suboptimal.
Wie bereits erwähnt, wurde in Synthese 2 mit Harnstoff-Wasserstoffperoxid nach Abschluss der
Reaktion eine Stoffmenge Wasserstoffperoxid von 0,99 mmol bestimmt. Diese lässt, wie auch
bei Synthese 1, keinen direkten Rückschluss auf die Stoffmenge an freiem Wasserstoffperoxid
zu. Allerdings ist davon auszugehen, dass der im Verhältnis zur Synthese von Trojan sehr
geringe Wert, einen nachhaltigen Umgang mit dem Enzym erleichtert. Trojan erzielte mit einer
synthetisierten Stoffmenge von 1,10 mmol Peroxykohlensäure, bezogen auf Synthese 2, eine
höhere Ausbeute von 39,24 %. Dennoch ist unter Berücksichtigung der Reaktionszeit von 24 h
und einer zu Synthese 2 äquivalenten Menge von 0,4 g Novozym® 435, nach Abschluss der
Reaktion wahrscheinlich ein großer Teil der Lipase denaturiert.
Zur Visualisierung ist die in Synthese 2 synthetisierte Stoffmenge Peroxykohlensäure und die
Wasserstoffperoxidmenge zu Ende der Reaktion, verglichen mit den selben Messwerten der

29
enzymatischen Peroxykohlensäuresynthese mit wässrigem Wasserstoffperoxid von Trojan, in
Abbildung 11 dargestellt:
Abbildung 12: Vergleich von Peroxykohlensäuresynthese aus Diethylcarbonat mit Harnstoff-Wasserstoffperoxid zu Peroxykohlensäuresynthese aus Diethylcarbonat mit wässrigem Wasserstoffperoxid (60%) (Trojan, 2011)
Ein Hindernis trat bei Synthese 2 bei der Durchführung der cerimetrischen Bestimmung der
Wasserstoffperoxidkonzentration, sowie bei der iodometrischen Bestimmung der
Peroxykohlensäurekonzentration nach Kapitel 4.2.4 auf. Als entsprechende Proben zur
Verdünnung in einen 100 ml Erlenmeyerkolben mit dest. Wasser aufgefüllt wurden, war ein 2-
Phasen-System klar ersichtlich. Während bei der Peroxyessigsäuresynthese in Synthese 1 alle
verwendeten Chemikalien gut bis sehr gut in Wasser löslich sind, ist Diethylcarbonat als
„praktisch unlöslich in Wasser“ angegeben (Römmp, 1995). Da alle anderen verwendeten wie
entstandenen Chemikalien in Synthese 2 wasserlöslich sind, ist davon auszugehen, dass das 2-
Phasen-System ausschließlich durch das Diethylcarbonat verursacht wurde. Um trotzdem eine
möglichst genaue iodometrische wie cerimetrische Bestimmung sicherzustellen, wurde das 2-
Phasen-System mit Hilfe einer Pipette gründlich gerührt, bevor die zu bestimmende Probe
genommen wurde.
02468
1012141618202224
Peroxykohlensäuresynthese aus Diethylcarbonat mit wässrigem
Wasserstoffperoxid (60%) [Reaktionszeit 24h] (Trojan, 2011)
Synthese 2: Peroxykohlensäuresynthese aus Diethylcarbonat mit Harnstoff-
Wasserstoffperoxid
nWasserstoffperoxid [mmol]
nPeroxykohlensäure [mmol]

30
6.3 Variationen der enzymatisch katalysierten Synthese von Peroxycarbonsäuren
6.3.1 Synthese 3: Reduktion der Reaktionszeit auf 2 und 4 Stunden
Die Auswertung von Synthese 3 ermöglicht eine grobe Einsicht in den Reaktionsablauf der in
Synthese 2 durchgeführten Peroxykohlensäuresynthese aus Diethylcarbonat. Zum Vergleich sind
die kalkulierten Stoffmengen der in Synthese 3 durchgeführten Peroxykohlensäuresynthese mit 2
und 4 Stunden Reaktionszeit, sowie die Stoffmengen aus Synthese 2 mit 6 Stunden Reaktionszeit
in Tabelle 14 wiedergegeben. Für eine Übersicht des Reaktionsablaufes sind die entsprechenden
Stoffmengen in Abbildung 13 in einem Liniendiagramm aufgeführt.
Tabelle 14: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid mit 2, 4 und 6 Stunden Reaktionszeit
Synthese 3:
Peroxykohlensäuresynthese
mit Harnstoff-
Wasserstoffperoxid [2 h]
Synthese 3:
Peroxykohlensäuresynthese
mit Harnstoff-
Wasserstoffperoxid [4 h]
Synthese 2:
Peroxykohlensäuresynthese
mit Harnstoff-
Wasserstoffperoxid [6 h]
nWasserstoffperoxid
[mmol]0,87 0,97 0,99
nPeroxykohlensäure
[mmol]0,20 0,32 0,79
Abbildung 13: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid mit 2, 4 und 6 Stunden Reaktionszeit
0
0,2
0,4
0,6
0,8
1
1,2
0 2 4 6 8
Stof
fmen
ge [m
mol
]
Zeit [h]
Wasserstoffperoxid
Peroxykohlensäure
Linear (Wasserstoffperoxid)
Expon. (Peroxykohlensäure)

31
Wie schon erwähnt, ist aufgrund der bei dieser Synthese entstandenen geringen
Peroxykohlensäurekonzentrationen und den damit verbundenen zur Titration benötigten
geringen Mengen Natriumthiosulfatlösung, das Ergebnis dieser Reaktion nur von bedingter
Aussagekraft. Dennoch lässt sich über den Zeitraum von 4 h ein exponentieller Anstieg der
Stoffmenge Peroxykohlensäure im Reaktionsmedium beobachten. Ausgehend von der
gemessenen Stoffmenge von 0,20 mmol nach 2 Stunden Reaktionszeit, steigert sich die
Stoffmenge nach 6 Stunden Reaktionszeit um 395 % auf 0,79 mmol. Hieraus lässt sich ableiten,
dass sich die Peroxykohlensäureausbeute, bei Verlängerung der Reaktionszeit, unter Umständen
weiter exponentiell ausweiten ließe.
Die Stoffmenge Wasserstoffperoxid steigt über den Zeitraum von 4 h linear von 0,87 mmol auf
0,99 mmol, was einem Anstieg von 13,79 % entspricht. Theoretisch ist es nicht möglich, dass die
Konzentration bei einer anfänglichen Gesamtzugabe des Adduktes während der Reaktion steigt.
Der Anstieg könnte deshalb darin begründet sein, dass zu Anfang nicht die komplette Zugabe im
Reaktionsmedium gelöst war. Zu beobachten war dies an leichten Rückstanden am
Reaktionsgefäß. Erst durch das kontinuierliche Rühren des Magnetrührers gingen diese
Rückstände nach und nach in Lösung, was den gemessenen Anstieg zur Folge gehabt haben
könnte. Dennoch unterstreicht die verhältnismäßig niedrige Konzentration über den gesamten
Reaktionszeitraum die Nachhaltigkeit der Synthesemethode, bezogen auf eine mögliche
Denaturierung bzw. Wiederverwendbarkeit des Enzyms.
6.3.2 Synthese 4: Unterlassung der Filtration des Reaktionsmediums nach Abschluss der
Reaktion
Bis auf Synthese 4 wurden alle Peroxysäurensynthesen nach Kapitel 4.2.2 nach
Reaktionsabschluss filtriert, um das Enzym vom Reaktionsmedium zu trennen und die Reaktion
zu beenden. Verwendet wurde stets Filterpapier des Herstellers Macherey-Nagel (MN 615. ø 70
mm). Die Auswertung von Synthese 4 zeigt geringe Auswirkungen auf die Stoffmengen von
Wasserstoffperoxid und Peroxysäuren zu Ende der Reaktion, wenn die cerimetrische und
iodometrische Titration ohne vorherige Filtration erfolgen. Hierfür wurde das Reaktionsgefäß
vor einer Probennahme ca. 5 min stehen gelassen, um eine Sedimentation des Enzyms zu
erreichen. Für eine genaue Titration ist es wichtig, dass möglichst wenig bis gar kein Enzym in
der Probe vorhanden ist. Wenn dies der Fall wäre, würde nach Verdünnung der Probe der hohe
Wassergehalt zu unerwünschten Enzymreaktionen führen.
32
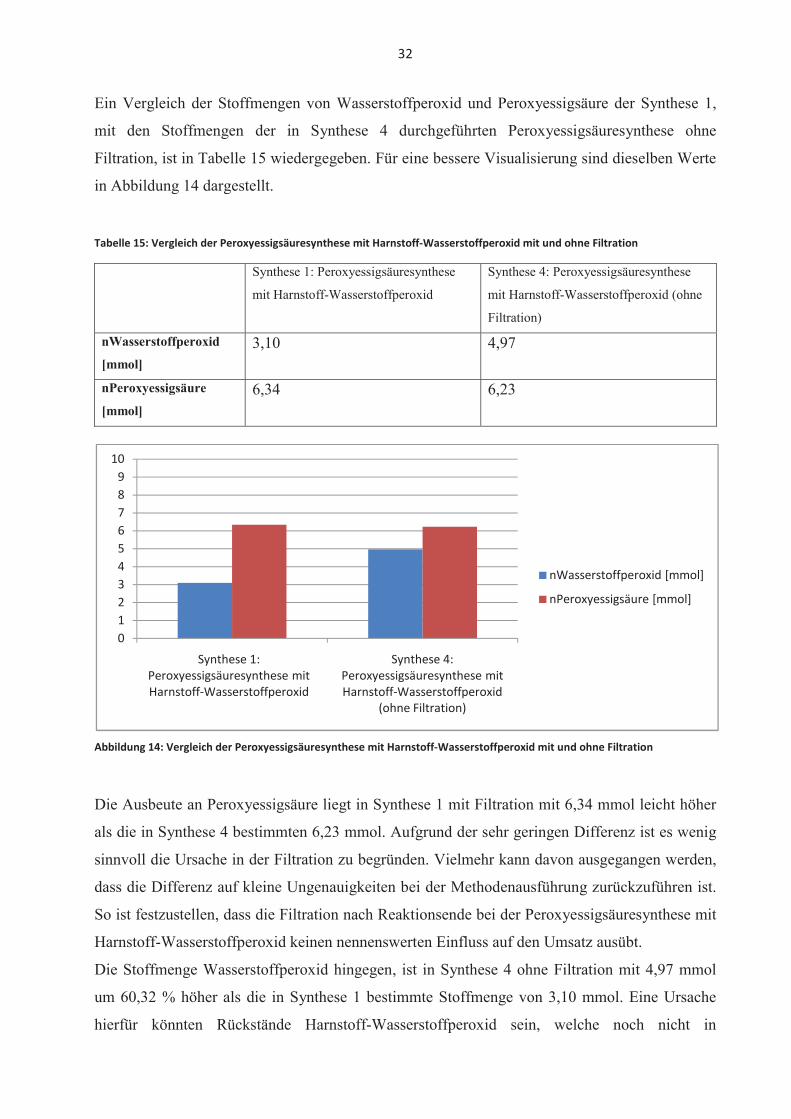
Ein Vergleich der Stoffmengen von Wasserstoffperoxid und Peroxyessigsäure der Synthese 1,
mit den Stoffmengen der in Synthese 4 durchgeführten Peroxyessigsäuresynthese ohne
Filtration, ist in Tabelle 15 wiedergegeben. Für eine bessere Visualisierung sind dieselben Werte
in Abbildung 14 dargestellt.
Tabelle 15: Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit und ohne Filtration
Synthese 1: Peroxyessigsäuresynthese
mit Harnstoff-Wasserstoffperoxid
Synthese 4: Peroxyessigsäuresynthese
mit Harnstoff-Wasserstoffperoxid (ohne
Filtration)
nWasserstoffperoxid
[mmol]3,10 4,97
nPeroxyessigsäure
[mmol]6,34 6,23
Abbildung 14: Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit und ohne Filtration
Die Ausbeute an Peroxyessigsäure liegt in Synthese 1 mit Filtration mit 6,34 mmol leicht höher
als die in Synthese 4 bestimmten 6,23 mmol. Aufgrund der sehr geringen Differenz ist es wenig
sinnvoll die Ursache in der Filtration zu begründen. Vielmehr kann davon ausgegangen werden,
dass die Differenz auf kleine Ungenauigkeiten bei der Methodenausführung zurückzuführen ist.
So ist festzustellen, dass die Filtration nach Reaktionsende bei der Peroxyessigsäuresynthese mit
Harnstoff-Wasserstoffperoxid keinen nennenswerten Einfluss auf den Umsatz ausübt.
Die Stoffmenge Wasserstoffperoxid hingegen, ist in Synthese 4 ohne Filtration mit 4,97 mmol
um 60,32 % höher als die in Synthese 1 bestimmte Stoffmenge von 3,10 mmol. Eine Ursache
hierfür könnten Rückstände Harnstoff-Wasserstoffperoxid sein, welche noch nicht in
0123456789
10
Synthese 1: Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid
Synthese 4: Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid
(ohne Filtration)
nWasserstoffperoxid [mmol]
nPeroxyessigsäure [mmol]
33
Essigsäureethylester gelöst waren. Wurden diese normalerweise nach Abschluss der Reaktion
per Filtration entfernt, so verblieben sie in Synthese 4 in der zu titrierenden Probe und wurden
zur Verdünnung mit dest. Wasser gemischt.
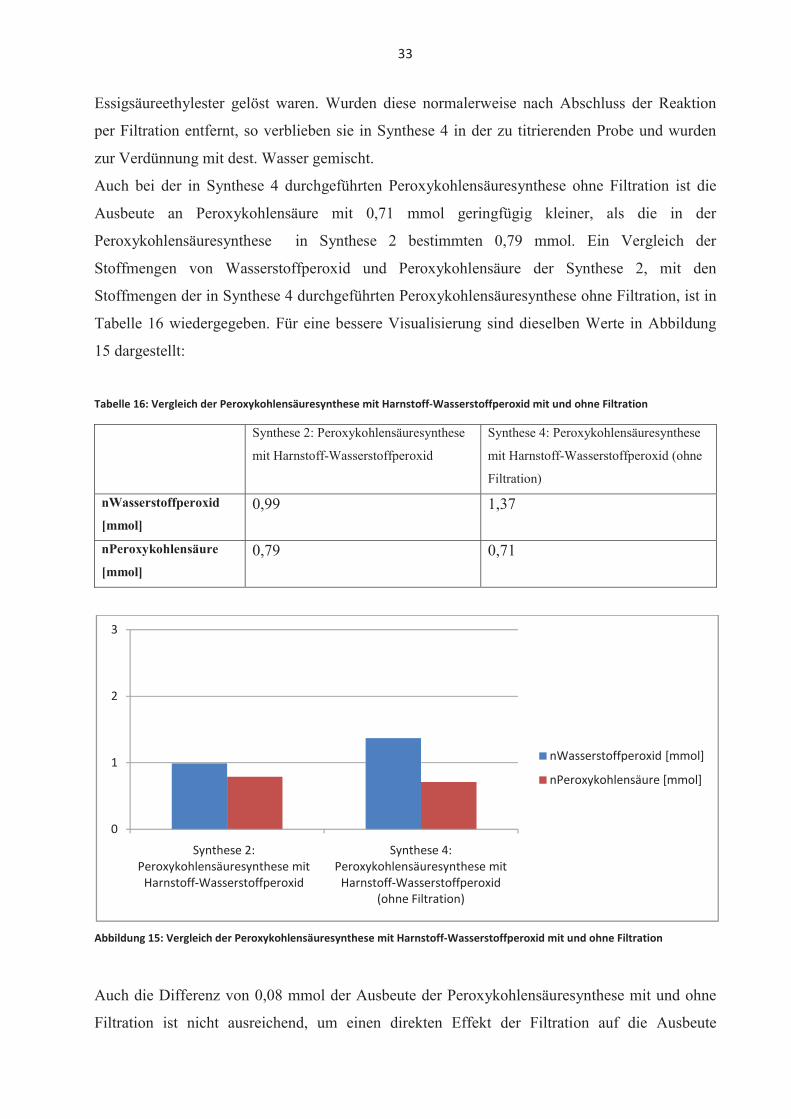
Auch bei der in Synthese 4 durchgeführten Peroxykohlensäuresynthese ohne Filtration ist die
Ausbeute an Peroxykohlensäure mit 0,71 mmol geringfügig kleiner, als die in der
Peroxykohlensäuresynthese in Synthese 2 bestimmten 0,79 mmol. Ein Vergleich der
Stoffmengen von Wasserstoffperoxid und Peroxykohlensäure der Synthese 2, mit den
Stoffmengen der in Synthese 4 durchgeführten Peroxykohlensäuresynthese ohne Filtration, ist in
Tabelle 16 wiedergegeben. Für eine bessere Visualisierung sind dieselben Werte in Abbildung
15 dargestellt:
Tabelle 16: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid mit und ohne Filtration
Synthese 2: Peroxykohlensäuresynthese
mit Harnstoff-Wasserstoffperoxid
Synthese 4: Peroxykohlensäuresynthese
mit Harnstoff-Wasserstoffperoxid (ohne
Filtration)
nWasserstoffperoxid
[mmol]0,99 1,37
nPeroxykohlensäure
[mmol]0,79 0,71
Abbildung 15: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid mit und ohne Filtration
Auch die Differenz von 0,08 mmol der Ausbeute der Peroxykohlensäuresynthese mit und ohne
Filtration ist nicht ausreichend, um einen direkten Effekt der Filtration auf die Ausbeute
0
1
2
3
Synthese 2: Peroxykohlensäuresynthese mit
Harnstoff-Wasserstoffperoxid
Synthese 4: Peroxykohlensäuresynthese mit
Harnstoff-Wasserstoffperoxid (ohne Filtration)
nWasserstoffperoxid [mmol]
nPeroxykohlensäure [mmol]

34
nachzuweisen. Allerdings wirkt sich, wie auch bei der Peroxyessigsäuresynthese, die
Unterlassung der Filtration entscheidend auf die molare Stoffmenge Wasserstoffperoxid zu Ende
der Reaktion aus. Im Vergleich zu Synthese 2, bei der eine Stoffmenge von 0,99 mmol gemessen
wurde, beträgt der Anstieg der Stoffmenge in Synthese 4 mit 1,37 mmol rund 38,38 %. Als
Ursache werden auch hier Rückstände Harnstoff-Wasserstoffperoxid vermutet, welche noch
nicht in Diethylcarbonat gelöst waren und durch Unterlassung der Filtration mit in die zu
titrierende Probe gelangten.
Zusammenfassend ist festzustellen, dass Synthese 4 gezeigt hat, dass sowohl bei der
Peroxyessigsäuresynthese als auch bei der Peroxykohlensäuresynthese, die abschließende
Filtration durch Filterpapier des Herstellers Macherey-Nagel (MN 615. ø 70mm) keinen
relevanten Einfluss auf die Peroxysäureausbeute ausübt.
6.3.3 Synthese 5: Erhöhung der Reaktionstemperatur auf 50°C
Synthese 5 zeigt eine beachtliche Steigerung der Peroxykohlensäureausbeute bei Erhöhung der
Reaktionstemperatur von 21°C (Synthese 2) auf 50°C. Die bestimmten Stoffmengen
Wasserstoffperoxid und Peroxykohlensäure zu Ende der Reaktion von Synthese 5, sind,
verglichen mit den äquivalenten Werten aus Synthese 2, in Tabelle 17 angegeben. In Abbildung
16 sind die Stoffmengen diagrammatisch dargestellt.
Tabelle 17: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid bei 21°C und 50°C Reaktionstemperatur
Synthese 2: Peroxykohlensäuresynthese
mit Harnstoff-Wasserstoffperoxid
(21°C)
Synthese 5: Peroxykohlensäuresynthese
mit Harnstoff-Wasserstoffperoxid
(50 °C)
nWasserstoffperoxid
[mmol]0,99 1,33
nPeroxykohlensäure
[mmol]0,79 1,23

35
Abbildung 16: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid bei 21°C und 50°C Reaktionstemperatur
Die Peroxykohlensäureausbeute ließ sich, im Vergleich zu Synthese 2, durch eine Erhöhung um
29°C von 0,79 mmol auf 1,23 mmol um insgesamt 55,70 % steigern. Eine Ursache könnte eine
Annährung an das Temperaturoptimum der Lipase für diese Reaktion sein. Novozym® 435 wird,
u.a. aufgrund seiner Immobilisierung auf makroporösem Acrylharz, als relativ thermostabil
angegeben (www.sigmaaldrich.com). Ankudey et al (2007) nutzten bei der in situ
Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid stets eine Reaktionstemperatur von
27°C.
Eine andere oder auch ergänzende Ursache könnte darin begründet sein, dass der Zerfall des
Harnstoff-Wasserstoffperoxid Adduktes temperaturbedingt schneller und in größerem Maße
erfolgt. Dies hätte zur Folge, dass das Enzym verhältnismäßig größeren Mengen freien
Wasserstoffperoxids ausgesetzt ist. Einerseits würde hierdurch die Synthese beschleunigt,
andererseits würde es einen nachhaltigen Umgang mit dem Enzym erschweren. Generell ist eine
Reaktionstemperatur von 50°C in dieser Reaktion kritisch, da unerwünschte Reaktionen des
freien Wasserstoffperoxids mit dem Enzym, welche zur Denaturierung führen, begünstigt oder
beschleunigt werden können. Es würde sich deshalb anbieten, Synthese 2 und Synthese 5
mehrmals zu wiederholen, und dabei das Enzym zu recyceln und wiederzuverwenden. Unter
Berücksichtigung des Gesamtumsatzes und der Recyclingraten könnte so die Nachhaltigkeit der
beiden angewendeten Reaktionstemperaturen der Peroxykohlensäuresynthese mit Harnstoff-
Wasserstoffperoxid genauer untersucht werden.
0
1
2
3
Synthese 2: Peroxykohlensäuresynthese mit
Harnstoff-Wasserstoffperoxid
Synthese 5: Peroxykohlensäuresynthese mit
Harnstoff-Wasserstoffperoxid (50°C)
nWasserstoffperoxid [mmol]
nPeroxykohlensäure [mmol]

36
6.3.4 Synthese 6: Hinzugabe von Harnstoff-Wasserstoffperoxid in Intervallen
Die Auswertung von Synthese 6 zeigt einen Umsatzverlust bei der Peroxyessigsäure- und
Peroxykohlensäuresynthese, wenn Harnstoff-Wasserstoffperoxid, statt in einer anfänglichen
Gesamtzugabe, dem Reaktionsmedium in Intervallen zugegeben wird.
Ein Vergleich der Stoffmengen von Wasserstoffperoxid und Peroxykohlensäure der Synthese 2,
mit den Stoffmengen der in Synthese 6 durchgeführten Peroxykohlensäuresynthese mit
Intervallzugabe von Harnstoff-Wasserstoffperoxid, ist in Tabelle 18 und Abbildung 17
wiedergegeben:
Tabelle 18: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid mit anfänglicher Gesamtzugabe zuZugabe in Intervallen
Synthese 2: Peroxykohlensäuresynthese
mit Harnstoff-Wasserstoffperoxid
(anfängliche Gesamtzugabe)
Synthese 6: Peroxykohlensäuresynthese
mit Zugabe von Harnstoff-
Wasserstoffperoxid in Intervallen
nWasserstoffperoxid
[mmol]0,99 1,23
nPeroxykohlensäure
[mmol]0,79 0,51
Abbildung 17: Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid mit anfänglicher Gesamtzugabe zu Zugabe in Intervallen
Die Peroxykohlensäureausbeute verringerte sich in Synthese 6 mit 0,51 mmol gegenüber der
Ausbeute von 0,79 mmol von Synthese 2 um 35,44 %. Dies lässt darauf schließen, dass die 5
0
1
2
3
Synthese 2: Peroxykohlensäuresynthese mit
Harnstoff-Wasserstoffperoxid (anfängliche Gesamtzugabe)
Synthese 6: Peroxykohlensäuresynthese mit
Zugabe von Harnstoff-Wasserstoffperoxid in Intervallen
nWasserstoffperoxid [mmol]
nPeroxykohlensäure [mmol]

37
Teilmengen von 0,3985 g Harnstoff-Wasserstoffperoxid, welche stündlich zugegeben wurden,
die Konzentration an freiem Wasserstoffperoxid im Reaktionsmedium im Vergleich zu Synthese
2 eher niedrig hielten. Die Konzentration an freiem Wasserstoffperoxid stieg stündlich mit jeder
Zugabe aufgrund des sich neu einstellenden Gleichgewichtes von Harnstoff-Wasserstoffperoxid
zu freiem Harnstoff und Wasserstoffperoxid. Die so erzeugte, über den Reaktionszeitraum von 4
Stunden ansteigende Konzentration, scheint für die enzymatische Peroxykohlensäuresynthese
schlechter geeignet zu sein als die durch anfängliche Gesamtzugabe des Adduktes erzeugte
Konzentration. Dies unterstreicht zum einen die deutlich niedrigere Ausbeute, als auch die nach
Reaktionsende gemessene leicht höhere Stoffmenge Wasserstoffperoxid von 1,23 mmol.
Ein ähnlicher Rückschluss kann aus der in Synthese 6 durchgeführten Peroxyessigsäuresynthese
mit Zugabe von Harnstoff-Wasserstoffperoxid in Intervallen gezogen werden. Ein Vergleich der
Stoffmengen von Wasserstoffperoxid und Peroxyessigsäure der Synthese 1, mit den
Stoffmengen der Peroxykohlensäuresynthese mit Intervallzugabe, ist in Tabelle 19 und
Abbildung 18 wiedergegeben:
Tabelle 19: Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit anfänglicher Gesamtzugabe zuZugabe in Intervallen
Synthese 1: Peroxyessigsäuresynthese
mit Harnstoff-Wasserstoffperoxid
(anfängliche Gesamtzugabe)
Synthese 6: Peroxyessigsäuresynthese
mit Zugabe von Harnstoff-
Wasserstoffperoxid in Intervallen
nWasserstoffperoxid
[mmol]3,10 5,33
nPeroessigsäure [mmol] 6,34 5,86
Abbildung 18: Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit anfänglicher Gesamtzugabe zuZugabe in Intervallen
0123456789
10
Synthese 1: Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid (anfängliche Gesamtzugabe)
Synthese 6: Peroxyessigsäuresynthese mit
Zugabe von Harnstoff-Wasserstoffperoxid in Intervallen
nWasserstoffperoxid [mmol]
nPeroxyessigsäure [mmol]

38
Die Peroxyessigsäureausbeute verringerte sich in Synthese 6 mit 5,86 mmol gegenüber der
Ausbeute von 6,34 mmol von Synthese 2 um 7,57 %. Dies ist zwar deutlich weniger als der
Ausbeuteverlust der Peroxykohlensäuresynthese mit Intervallzugabe von 35,44 %, dennoch wird
ersichtlich, dass auch bei der Peroxykohlensäuresynthese die anfängliche Gesamtzugabe von
Vorteil ist.
7. Schlussfolgerung und Ausblick
Die Ausführungen dieser Studie haben gezeigt, dass 20 ml Essigsäureethylester oder
Diethylcarbonat, unter Anwendung von 0,40 g Novozym® 435 und 1,9924 g Harnstoff-
Wasserstoffperoxid, mit Ausbeuten bis zu 6,34 mmol zu ihren entsprechenden Peroxysäuren
oxidiert werden können. Aus Gründen der Effizienz, Kosteneinsparung und Nachhaltigkeit wäre
es interessant zu untersuchen, wie sich eine Reduzierung der Enzymmenge im Reaktionsmedium
auf die Umsatzgeschwindigkeit und Denaturierungsrate auswirken würde. Ankudey, Olivio und
Peeples (2006) fanden heraus, dass eine Reduzierung der Enzymmenge bei der Epoxidation
durch in situ Synthesen von Peroxyessigsäure aus Essigsäureethylester mit Harnstoff-
Wasserstoffperoxid, geringfügige bis keine Änderungen beim Umsatz zur Folge hatte.
Epoxidationen führten sie in 3 ml Essigsäureethylester mit 1 mmol des zu epoxidierenden
Alkens, 1,10 mmol Harnstoff-Wasserstoffperoxid und 50 mg Novozym® 435 durch. Bei einer
Reduzierung der Enzymmenge bis zu 5 mg konnten keine Umsatzverluste festgestellt werden
(Tabelle 20) (Ankudey; Olivio; Peeples, 2006).
Tabelle 20: Einfluss der Reduzierung der Enzymmenge auf Umsatz bei der Epoxidation durch in situ Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid (Ankudey; Olivio; Peeples, 2006)
Enzym [mg] Reaktionszeit [h] Umsatz [%]
50 5 82
40 5 86
25 5 86
15 5 85
10 5 85
5 5 86
2 5 81
1 5 61

39
Aufgrund der ähnlichen Reaktionskriterien wie bei Synthesen dieser Studie, ist es
wahrscheinlich, dass auch diese mit wesentlich kleineren Enzymmengen zu ähnlichen Ausbeuten
führen würden. Zu beachten ist allerdings, dass durch kleinere Enzymmengen, welche dadurch
verhältnismäßig größeren Mengen freiem Wasserstoffperoxid ausgesetzt wären, die
Denaturierungsrate steigen könnte. Um dies zu überprüfen würde sich anbieten, in dieser Studie
durchgeführte Synthesen mit anschließendem Enzymrecycling zu wiederholen. Äquivalente
Synthesen mit kleineren Enzymmengen, welche ebenfalls mit anschließendem Enzymrecycling
wiederholt werden würden, könnten durch Vergleiche der möglichen Wiederholungen ohne
Umsatzverluste die Annahme bestätigen oder widerlegen.
Ebenfalls aus Gründen der Effizienz, Kosteneinsparung und Nachhaltigkeit wäre es lohnenswert,
den Einfluss der Reduzierung der verwendeten Menge Harnstoff-Wasserstoffperoxid auf die
Ausbeute zu untersuchen. Die in Synthesen dieser Studie verwendete Menge ist meist nie
komplett in Lösung gegangen, was an Rückständen im Reaktionsgefäß und den Messungen der
Wasserstoffperoxidkonzentrationen in Synthese 3 und 4 gut ersichtlich war. Aus diesem Grund
ist es wahrscheinlich, dass auch hier eine Reduzierung nicht zwanghaft zu Ausbeuteverlusten
führt.
8. Zusammenfassung
Unter Berücksichtigung und vergleichend mit der Studie der enzymatisch katalysierten Synthese
von Peroxyessigsäure mit wässrigem Wasserstoffperoxid (Ernst, 2011), zeigt diese Arbeit unter
gleichen Reaktionsbedingungen eine funktionierende Synthesemethode zur enzymatisch
katalysierten Darstellung von Peroxysäuren mit Harnstoff-Wasserstoffperoxid. Bei der
enzymatischen Peroxysäurensynthese mit wässrigem Wasserstoffperoxid ist es notwendig, das
wässrige Wasserstoffperoxid dem Reaktionsmedium in Intervallen zuzugeben, um eine
Denaturierung des Enzyms zu verhindern. Diese Studie zeigt durch den Einsatz des Harnstoff-
Wasserstoffperoxiod-Adduktes eine Alternative auf, welche dem Reaktionsmedium durch eine
anfängliche Gesamtzugabe zugesetzt werden kann. Dies ist möglich, da Harnstoff-
Wasserstoffperoxid das Potential besitzt, Wasserstoffperoxid in einer kontrollierbaren Art und
Weise freizusetzen. Einerseits erleichtert dies die Durchführung, andererseits wird eine
niedrigere Denaturierungsrate vermutet. Unter Verwendung von 20 ml Essigsäureethylester oder
Diethylcarbonat als Lösungsmittel wie Edukt, werden mit 1,9924 g Harnstoff-
Wasserstoffperoxid, katalysiert durch 0,40 g Novozym® 435, Ausbeuten bis zu 6,34 mmol
Peroxyessigsäure und 1,23 mmol Peroxykohlensäure erreicht. Aufgrund der verringerten

40
Konzentration an freiem Wasserstoffperoxid im Reaktionsmedium fallen die Ausbeuten
geringfügig kleiner aus als bei äquivalenten Synthesen mit wässrigem Wasserstoffperoxid.
Allerdings haben die vergleichsweise geringen Mengen Wasserstoffperoxid im Medium eine
reduzierte Denaturierungsrate zur Folge, was die Synthese mit Harnstoff-Wasserstoffperoxid zu
einer ernsthaften Alternative werden lässt. Die anfängliche Gesamtzugabe des Harnstoff-
Wasserstoffperoxids stellt sich, hinsichtlich der maximalen Ausbeute, sowohl bei der
Peroxyessigsäure- als auch bei der Peroxykohlensäuresynthese, gegenüber der Zugabe des
Adduktes in Intervallen als vorteilhaft heraus. Weitere Untersuchungen zeigen, dass die in dieser
Studie angewendete Filtration nach Abschluss der Reaktion, keinen Einfluss auf den Verbleib an
Peroxysäuren in der filtrierten Lösung hat. Um den Umsatz der enzymatischen
Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid zu steigern, würde eine
Verlängerung der in dieser Studie angewendeten Reaktionszeit von 6 Stunden in Erwägung
kommen. Dies unterlegen Messungen der Peroxykohlensäurekonzentration nach 2, 4 und 6
Stunden, welche einen exponentiellen Anstieg zeigen. Die Peroxykohlensäureausbeute lässt sich,
in ansonsten äquivalenten Synthesen, durch eine Erhöhung der Reaktionstemperatur von 21°C
auf 50°C um 55,70 % steigern. Diese Reaktionstemperatur ist allerdings insofern kritisch, da
unerwünschte Reaktionen des freien Wasserstoffperoxids mit dem Enzym, welche zur
Denaturierung führen, begünstigt oder beschleunigt werden können.

41
9. Literatur
Monografien
- Bornscheuer, U.: Neue Strategien zum Einsatz von Lipasen und Esterasen in der organischen
Synthese. Aachen: Shaker, 1999
- Bersin, T.H.: Kurzes Lehrbuch der Enzymologie. 3. Auflage. St. Gallen: VAG, 1950
- Hudlický, H.: Oxidations in organic chemistry. Third printing. York: ACS Monograph, 1997
- Jones, C.W.: Application of Hydrogen Peroxide and Derivates. Cambridge: The Royal Society
of Chemistry, 1999
- Römpp, H.: Römpp Chemie-Lexikon. 9. Auflage. Version 1.0. Stuttgart/New York: Thieme,
1995
Periodika
- Ankudey, E.G., Olivio, H. F., Peeples, T.L. (2006), Lipase-mediated epoxidation utilizing urea-
hydrogen peroxide in ethyl acetate. Green Chemistry. DOI 10.1039/b604984b
- Folli, U., Iarossi, D., Montanari, F., Torre, G. (1968), Asymmetric induction and
configurational correlations in oxidation at sulphur. Part III. Oxidations of aryl alkyl sulphides to
sulphoxides by optically active peroxy-acids. WO 94/02367
- Godtfredsen, S.E., Kirk, O., Björking, F., Christensen, L.B. (1990), A process for preparing
peroxy carboxylic acids. WO 91/06574
- Ríos, M.Y., Salazar, E., Olivo, H.F. (2007), Baeyer-Villinger oxidation of substituded
cyclohexanones via lipase-mediated perhydrolysis utilizing urea-hydrogen peroxide in ethyl
acetate. Green Chemistry. DOI 10.1039/b618175a

42
- Rüsch gen. Klaas, M., Warwel, S. (2000), Lipase-Catalyzed Peroxy Fatty Acid generation and
Lipid Oxidation. Bornscheuer, T.: Enzymes in Lipid Modification.Weinheim: Wiley-VCH, 2000
- Rüsch gen. Klaas, M., Warwel, S. (2000), New Percarboxylic Acids by Lipase Catalysis:
Preparation and Oxidation Properties. Adam, W.: Peroxide Chemistry – Mechanistic and
Preparative Aspects of Oxygen Transfer. Weinheim: Wiley-VCH, 2000
Internet
http://chemicalland21.com/industrialchem/inorganic/UREA%20HYDROGEN%20PEROXIDE.h
tm 15.12.2011
http://www.kesla.de/fobst.htm. 02.12.2011
http://www.sigmaaldrich.com/catalog/ProductDetail.do?N4=L4777|SIGMA&N5=SEARCH_
CONCAT_PNO|BRAND_KEY&F=SPEC. 04.01.2012
Sonstige Quellen
- Ernst, R.: Enzymatische Herstellung von Percarbonsäuren – Versuche mit verschiedenen
Enzymen. Hausarbeit. Hochschule Neubrandenburg: nicht veröffentlicht, 2011
- Patett, N.: Untersuchungen zur Inaktivierung von Bakteriosporen mit neu synthetisierten
Persäuren. Diplomarbeit. Hochschule Neubrandenburg, 2001
- Trojan, P.: Die mikrobizide Wirkung von Percarbonsäuren. Studienarbeit. Hochschule
Neubrandenburg: nicht veröffentlicht, 2010

43
10. Verwendete Abkürzungen
Dest. Wasser – Destilliertes Wasser
Peroxykohlensäure - Monoperoxykohlensäuremonoethylester

44
11. Abbildungsverzeichnis
Abb. 1 : Peroxyessigsäure………………………………………………………………… 5
Abb. 2 : Säurekatalytische Herstellung einer Peroxycarbonsäure ………………….......... 6
Abb. 3 : Reaktionsgleichung enzymatisch katalysierter Synthese von
Peroxycarbonsäuren……………………………………………………………... 6
Abb. 4 : Katalytische Triade mit der Aminosäuren-Nummerierung des aktiven Zentrums
der Lipase-B aus Candida antarctica…………………………….......................... 8
Abb. 5 : Harnstoff-Wasserstoffperoxid (auch Carbamidperoxid; Percarbamid)…………. 10
Abb. 6 : Reaktionsgleichung der enzymatisch katalysierten Synthese von
Peroxyessigsäure aus Essigsäureethylester, unter Verwendung von Harnstoff-
Wasserstoffperoxid als Oxidationsmittel………………………........................... 12
Abb. 7 : Reaktionsgleichung der enzymatisch katalysierten Synthese von
Peroxykohlensäure aus Diethylcarbonat, unter Verwendung von Harnstoff-
Wasserstoffperoxid als Oxidationsmittel………………………………………... 12
Abb. 8 : Versuchsaufbau; Peroxysäurensynthese………………………………………… 14
Abb. 9 : Versuchsaufbau; Abfiltration des Enzyms………………………………………. 14
Abb. 10 : Versuchsaufbau; Titration via Bürette…………………………………………... 16
Abb. 11 : Vergleich von Peroxyessigsäuresynthese aus Essigsäureethylester mit
Harnstoff-Wasserstoffperoxid zu Peroxyessigsäuresynthese aus
Essigsäureethylester mit wässrigem Wasserstoffperoxid (60%)………….......... 26
Abb. 12 : Vergleich von Peroxykohlensäuresynthese aus Diethylcarbonat mit Harnstoff-
Wasserstoffperoxid zu Peroxykohlensäuresynthese aus Diethylcarbonat mit
wässrigem Wasserstoffperoxid (60%)…………………………………………... 29
Abb. 13 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
mit 2, 4 und 6 Stunden Reaktionszeit…………………………………………… 30
Abb. 14 : Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit
und ohne Filtration………………………………................................................. 32
Abb. 15 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
mit und ohne Filtration………………………………........................................... 33
Abb. 16 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
bei 21°C und 50°C Reaktionstemperatur………………………………………... 35

45
Abb. 17 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
mit anfänglicher Gesamtzugabe zu Zugabe in
Intervallen………………………………………………………………….......... 36
Abb. 18 : Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit
anfänglicher Gesamtzugabe zu Zugabe in
Intervallen………………………………………………………………….......... 37

46
12. Tabellenverzeichnis
Tab. 1 : Vergleich der Explosivkraft und Sensitivität konventioneller Sprengstoffe mit
Wasserstoffperoxid Gemischen………………………………………………... 9
Tab. 2 : Messergebnisse; Bestimmung der Konzentration des Harnstoff-
Wasserstoffperoxid-Adduktes…………………………………………………. 17
Tab. 3 : Synthese 1; Messergebnisse; Oxidation von Essigsäureethylester zu
Peroxyessigsäure……………………………………………………………….. 18
Tab. 4 : Synthese 2; Messergebnisse; Oxidation von Diethylcarbonat zu
Peroxykohlensäure……………………………………………………………... 19
Tab. 5 : Synthese 3; Messergebnisse; Oxidation von Diethylcarbonat zu
Peroxykohlensäure; Reduktion der Reaktionszeit auf 2 Stunden……………… 20
Tab. 6 : Synthese 3; Messergebnisse; Oxidation von Diethylcarbonat zu
Peroxykohlensäure; Reduktion der Reaktionszeit auf 4 Stunden……………… 20
Tab. 7 : Synthese 4; Messergebnisse; Oxidation von Essigsäureethylester zu
Peroxyessigsäure; Unterlassung der Filtration des Reaktionsmediums nach
Abschluss der Reaktion………………………………………………………… 21
Tab. 8 : Synthese 4; Messergebnisse; Oxidation von Diethylcarbonat zu
Peroxykohlensäure; Unterlassung der Filtration des Reaktionsmediums nach
Abschluss der Reaktion………………………………………………………… 21
Tab. 9 : Synthese 5; Messergebnisse; Oxidation von Diethylcarbonat zu
Peroxykohlensäure; 50°C………………………………………………………. 22
Tab. 10 : Synthese 6; Messergebnisse; Oxidation von Essigsäureethylester zu
Peroxyessigsäure; Hinzugabe von Harnstoff-Wasserstoffperoxid in Intervallen 23
Tab. 11 : Synthese 6; Messergebnisse; Oxidation von Diethylcarbonat zu
Peroxykohlensäure; Hinzugabe von Harnstoff-Wasserstoffperoxid in
Intervallen………………………………………………………………………. 23
Tab. 12 : Vergleich von Peroxyessigsäuresynthese aus Essigsäureethylester mit
Harnstoff-Wasserstoffperoxid zu Peroxyessigsäuresynthese aus Essigsäure-
ethylester mit wässrigem Wasserstoffperoxid (60%)………………………….. 25
Tab. 13 : Vergleich von Peroxykohlensäuresynthese aus Diethylcarbonat mit Harnstoff-
Wasserstoffperoxid [6h ] zu Peroxykohlensäuresynthese aus Diethylcarbonat
mit wässrigem Wasserstoffperoxid (60%) [24h]……………………………… 28

47
Tab. 14 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
mit 2, 4 und 6 Stunden Reaktionszeit…………………………………………... 30
Tab. 15 : Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit
und ohne Filtration…………………………………………………………. 32
Tab. 16 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
mit und ohne Filtration…………………………………………………………. 33
Tab. 17 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
bei 21°C und 50°C Reaktionstemperatur………………………………………. 34
Tab. 18 : Vergleich der Peroxykohlensäuresynthese mit Harnstoff-Wasserstoffperoxid
mit anfänglicher Gesamtzugabe zu Zugabe in Intervallen……………………... 36
Tab. 19 : Vergleich der Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid mit
anfänglicher Gesamtzugabe zu Zugabe in Intervallen……………………... 37
Tab. 20 : Einfluss der Reduzierung der Enzymmenge auf Umsatz bei der Epoxidation
durch in situ Peroxyessigsäuresynthese mit Harnstoff-Wasserstoffperoxid…… 38

48
Selbstständigkeitserklärung
Hiermit versichere ich, dass ich die vorliegende Arbeit selbstständig angefertigt habe und keine anderen als die angegebenen Quellen und Hilfsmittel benutzt habe.
_____________________________ _____________________________ Ort, Datum Unterschrift

49
Anhang
Formel zu Bestimmung der Wasserstoffperoxidkonzentration bei der cerimetrischen
Titration:
cH202 [mol/l] = Verbrauch Cer(IV)-Sulfatlösung [ml] * 0,5 * 0,1[mol/l] * VerdünnungsfaktorVprobe[ml]
Formel zu Bestimmung der Peroxysäurenkonzentration bei der iodometrischen Titration:
cPersäure [mol/l] = Verbrauch Na2S2O3[ml] * 0,5 * 0,1[mol/l] * VerdünnungsfaktorVprobe[ml]




![Polysulfonylamine, XII [1] N-Acyl-dimesylamine (N.N ...zfn.mpdl.mpg.de/data/Reihe_B/43/ZNB-1988-43b-1495.pdf · 1497 A. Blaschette et al. N-Acyl-dimesylamine Tab. II. Spektroskopische](https://static.fdokument.com/doc/165x107/5eccdbe82a00c011850e8902/polysulfonylamine-xii-1-n-acyl-dimesylamine-nn-zfnmpdlmpgdedatareiheb43znb-1988-43b-1495pdf.jpg)


![Kombinatorische Untersuchungen zur heterogen katalysierten ... · Da Biogas bisweilen hohe Konzentrationen schwefel- haltiger Verunreinigungen enthalten kann [11], würde der Einsatz](https://static.fdokument.com/doc/165x107/5d4cdcd388c993821c8b521e/kombinatorische-untersuchungen-zur-heterogen-katalysierten-da-biogas-bisweilen.jpg)
![Acyl- und Alkylidenphosphane, XV [1] 2.2 ...zfn.mpdl.mpg.de/data/Reihe_B/36/ZNB-1981-36b-0016.pdf · G. Becker et al. Acyl- und Alkylidenphosphane 17 [11, s. auch 12] ergibt für](https://static.fdokument.com/doc/165x107/5e117fb1d56d93523e1f3b17/acyl-und-alkylidenphosphane-xv-1-22-zfnmpdlmpgdedatareiheb36znb-1981-36b-0016pdf.jpg)



![[XLS] · Web viewDie Ballonfahrt Lankers, Katrin Elfenblick Franzen, Jonathan (Die Korrekturen) ausgeschieden Amann, Peter Kalabrien Marvi Hämmer (Lehrprogramm) McPartlin, Anna Weil](https://static.fdokument.com/doc/165x107/5ba0212509d3f242318c4844/xls-web-viewdie-ballonfahrt-lankers-katrin-elfenblick-franzen-jonathan-die.jpg)




