Beeinflussung der Geschwindigkeit ionischer … · - M. Dakkouri, W. Bodenmüller, J. Chem. Educ....
15
V5 Beeinflussung der Geschwindigkeit ionischer Reaktionen durch Fremdsalzzusatz Aktualisiertes Versuchsskript: 13. Mai 2016
Transcript of Beeinflussung der Geschwindigkeit ionischer … · - M. Dakkouri, W. Bodenmüller, J. Chem. Educ....

V5
Beeinflussung der Geschwindigkeit
ionischer Reaktionen durch
Fremdsalzzusatz
Aktualisiertes Versuchsskript: 13. Mai 2016

2
Physikalisch-Chemisches Praktikum für Fortgeschrittene
Änderung der Reaktionsgeschwindigkeit bei Fremdsalzzusatz (Salzeffekt)
Versuchsziel
Man führt die alkalische Entfärbung von Phenolphthalein mit NaOH bei verschiedenen
Ionenstärken durch und prüft, ob die gezeigten Zusammenhänge durch das Experiment
bestätigt werden. Die Ionenstärken werden durch unterschiedliche Konzentrationen
von KCl oder Na2SO4 eingestellt.
Fragen zur Selbstkontrolle
(1) Was sind die grundsätzlichen Unterschiede zwischen idealem Verhalten von Gasen,
Mischungen und Elektrolyt-Lösungen?
(2) Zeigen Sie, dass beim Herstellen einer idealen Mischung die Mischungswärme Null ist
und der freiwillige Ablauf des Mischungsvorgangs allein auf die Zunahme der Entropie
zurückzuführen ist.
(3) Welche Methoden zur experimentellen Bestimmung von Aktivitätskoeffizienten gibt
es?
(4) Worin liegen die grundsätzlichen Fehler der Debye-Hückel-Theorie?
(5) Wie groß ist der Debye-Radius eines 1-1, eines 1-2 und eines 2-4 Elektrolyten in
Wasser, bei einer Konzentration. von 10-3
M und einer Temperatur von 18°C?
(6) Wie lassen sich die Aktivitätskoeffizienten von konzentrierteren Lösungen berechnen?
(7) Geben Sie Beispiele für Ionenreaktionen an, die mit einer Bindungsbildung
einhergehen und bei denen
(a) zA und zB das gleiche Vorzeichen haben
(b) zA und zB verschiedene Vorzeichen haben
(c) zA und zB gleich Null ist
Bitte Kolloquiums-Themenliste beachten!!!!!

3
3. Literatur
Pflichtliteratur:
Debye-Hückel-Onsager Theorie:
- J.O’M. Bockris, A.K.N. Reddy, Modern Electrochemistry 1, Kapitel 3.3
Zum Versuch:
- L. Nicholson, J. Chem. Educ. 66(9) (1989) 725.
- M. Dakkouri, W. Bodenmüller, J. Chem. Educ. 74(5) (1997) 556
Zusatzliteratur:
- L. Hile, J. Chem. Educ. 68(5) (1991) 446
- M.D. Barnes, V.K. La Mer, J. Am. Chem. Soc. 64 (1942) 2312
- M. Dakkouri, W. Bodenmüller, J. Chem. Educ. 74(5) (1997) 556
- R. Winans, C.A. Brown, J. Chem. Educ. 52(8) (1975) 526

4
4.Theorie
4.1 Die Aktivität aus elektrochemischer Sicht
Ein Ion bildet in Lösung eine „Wolke“ von Ionen bevorzugt entgegengesetzten
Ladungsvorzeichens um sich aus. Diese Ionenwolke stabilisiert ihr Zentralion und vermindert
etwas dessen Reaktivität bezüglich chemischer Reaktionen. Die dem Ion verbleibende
„wirksame“ Konzentration, die sogenannte Aktivität ai ist somit nur ein Bruchteil fi
der
wirklichen Konzentration ci:
i i ia f c (1)
Der Aktivitätskoeffizient fi beschreibt die Abweichungen vom idealen Verhalten.
In unendlich verdünnten Lösungen verschwinden die interionischen Wechselwirkungen und
es gilt:
0lim 1iI
f
(2)
Hierbei ist I die sogenannte Ionenstärke1
:
21
2i i
i
I z c bzw. 21
2m i i
i
I z m (mit Molalität mi) (3)
Da die Einzelionenaktivitäten ai experimentell nicht zugänglich sind, definiert man den
sogenannten mittleren Aktivitätskoeffizienten f , der durch das geometrische Mittel der
Einzelionenaktivitätskoeffizienten gegeben ist. Für einen Elektrolyten, der in x Kationen und
y Anionen zerfällt, gilt:
x y x yf f f (4)
4.2 Die Debye-Hückel-Theorie
Siehe Zusatzliteratur (!!!):
J.O’M. Bockris, A.K.N. Reddy, Modern Electrochemistry 1, Kapitel 3.3
Debye-Hückel’sches Gesetz zur Berechnung von Aktivitätskoeffizienten von geladenen
Komponenten bzw. Grenzgesetz für verdünnte Elektrolytlösungen:
2
log1
i mi
m
A z If
Ba I
bzw. 2log i i mf A z I (5)
_______________________________________________________________________ 1
Der Begriff der Ionenstärke geht auf Lewis und Randall zurück, die schon vor der theoretischen Berechnung der
Aktivitätskoeffizienten durch Debye und Hückel empirisch fanden, dass der Logarithmus des mittleren
Aktivitätskoeffizienten eines Elektrolyten proportional der Summe der Produkte zi²∙ci sämtlicher Ionen in der Lösung ist.
Hieraus ist ersichtlich, dass in genügend verdünnten Lösungen die chemische Natur der Ionen ohne Bedeutung ist, und nur
elektrostatische Kräfte für das Abweichen vom Idealverhalten verantwortlich sind.

5
4.3 Die Brønsted'sche Gleichung
Chemische Reaktionen in Lösungen, die unter Beteiligung von Ionen ablaufen, zeigen
oftmals große Abweichungen von den klassischen Geschwindigkeitsgleichungen. So
beobachtet man eine Abhängigkeit der Geschwindigkeitskonstante von der Ionenstärke der
Lösung, was bedeutet, dass auch Salze, die nicht an der eigentlichen Reaktion teilnehmen,
sogenannte Neutral- oder Fremdsalze, einen Einfluss auf die Kinetik der Reaktion ausüben.
Brønsted ging von der allgemein gebräuchlichen Vorstellung aus, dass die reagierenden
Stoffe instabile Zwischenzustände durchlaufen, bevor sie in den Endzustand übergehen. Eine
bimolekulare Reaktion verläuft danach in der Weise, dass zunächst ein aus den
Reaktionspartnern A und B gebildeter, aktivierter Komplex X‡
entsteht, der sich mit den
Ausgangsstoffen im Gleichgewicht befindet, und der seinerseits in die Endprodukte zerfällt:
Die Geschwindigkeit der Reaktion wird durch die Zustandssummen der Edukte und des
aktivierten Komplexes bestimmt.
Brønsted gelangte empirisch zu folgender Formel:
A A BA BX
X
dc f f=const c k c c
dt f
(6)
Die von Brønsted gefundene Gleichung (6) lässt sich auch ableiten, indem man die
Konzentrationen in der Geschwindigkeitsgleichung durch Aktivitäten ersetzt. Nimmt man die
Bildung des aktivierten Komplexes X‡ als geschwindigkeitsbestimmend an, so erhält man,
wiederum für eine bimolekulare Reaktion:
X
BABA
AX
BBAAXX
BAX
f
ffcck
dt
dc
dt
dc
cfcfkdt
dcf
aakdt
da
(7)
Für verdünnte Lösungen lassen sich die Aktivitätskoeffizienten berechnen und somit
quantitative Aussagen über die Reaktionsgeschwindigkeit machen.
4.4 Der primäre Salzeffekt
Die Änderung der Geschwindigkeitskonstante k mit der Ionenstärke Im nennt man den
primären kinetischen Salzeffekt.
Nach Brønsted gilt:
0
0log log log
A B
X
A B
X
f fk k
f
f fk k
f
(8)
6
wobei k0
die Geschwindigkeitskonstante bei unendlicher Verdünnung darstellt. Mit Gleichung
(5), unter der Annahme, dass zX = zA + zB, ergibt sich:
0
2 2 20
22 20
2 2 2 20
log log log log log
log
log
log 2
A B X
A m B m X m
A m B m A B m
m A B A A B B
k k f f f
k Az I Az I Az I
k Az I Az I A z z I
k A I z z z z z z
0log log 2 A B mk k A z z I (9)
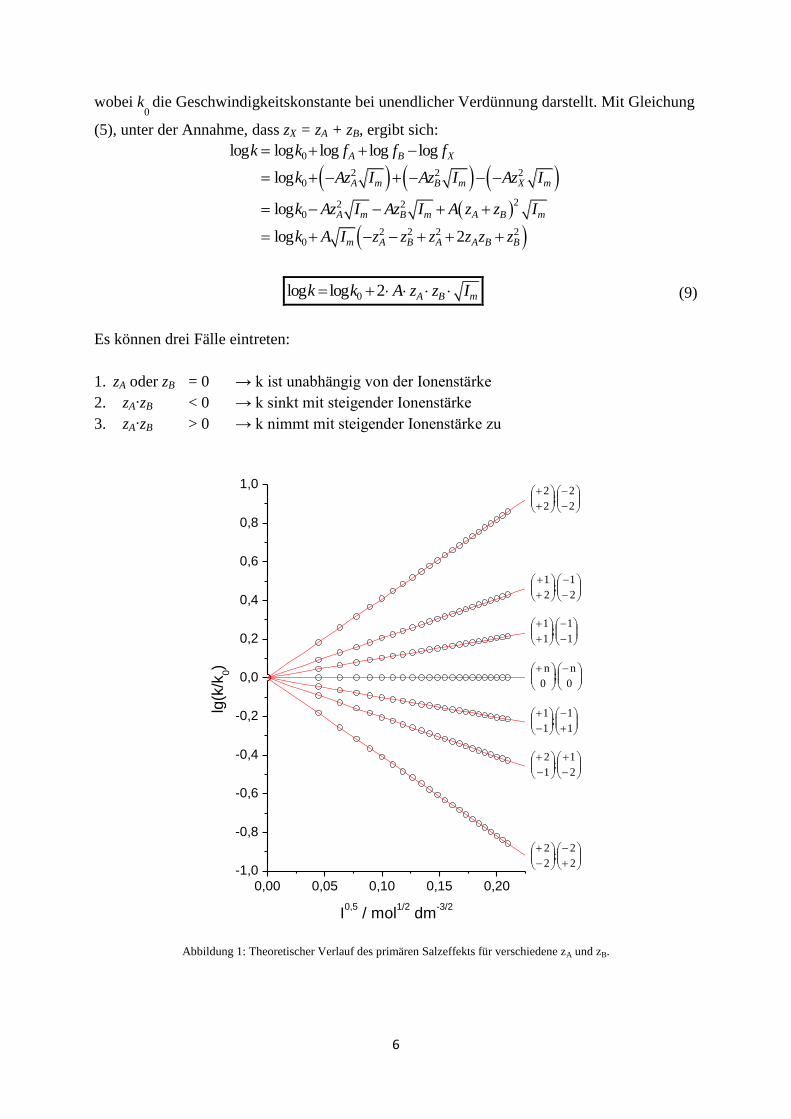
Es können drei Fälle eintreten:
1. zA oder zB = 0 → k ist unabhängig von der Ionenstärke
2. zA∙zB < 0 → k sinkt mit steigender Ionenstärke
3. zA∙zB > 0 → k nimmt mit steigender Ionenstärke zu
Abbildung 1: Theoretischer Verlauf des primären Salzeffekts für verschiedene zA und zB.
0,00 0,05 0,10 0,15 0,20
-1,0
-0,8
-0,6
-0,4
-0,2
0,0
0,2
0,4
0,6
0,8
1,0
lg(k
/k0)
I0,5
/ mol1/2
dm-3/2
2
2;
2
2
2
1;
2
1
1
1;
1
1
0
n;
0
n
1
1;
1
1
2
1;
1
2
2
2;
2
2

7
5.Theorie zum Versuch
Abbildung 2: Phenolphthalein: (a) neutrale Form, (b) gefärbtes Dianion, (c) Carbinol-Base.
Phenolphthalein (a) ist ein farbloses Lacton, das im pH-Bereich von 8,5 bis 10,0 unter
Öffnung des Lacton-Ringes in das rosa gefärbte Dianion (b) übergeht. In stärker alkalischem
Medium ist ein langsames Abblassen der rosa Farbe zu beobachten. Es stellt sich eine
Gleichgewichtsfärbung ein, die von der OH–-Konzentration abhängig ist. Diese Entfärbung
ist auf die Bildung der dreifach negativ geladenen Carbinol-Base (c) zurückzuführen und
kann folgendermaßen formuliert werden:
P2-
+ OH- [X]
‡ POH
3- (10)
Nimmt man für die Hinreaktion einen bimolekularen und für die Rückreaktion einen
monomolekularen Mechanismus an, so liefert die Anwendung der Brønsted'schen Formel
(3) folgende Geschwindigkeitsgleichungen:
2
1 1 23
3
2 2 33
P OH
P OHX
POH
POHX
f fv k c c
f
fv k c
f
(11)
Nach Debye-Hückel (7/9) erhält man, unter der Voraussetzung, dass sich die Ladung des
kritischen Komplexes X additiv aus den Ladungen der Edukte zusammensetzt:
21 1 2
01 1
10
log log 2
Az z IA B m
P OH
A B m
v k c c
k k Az z I
(12)
und
2 2 3
02 2log log
POHv k c
k k
(13)
Der Theorie nach zeigt also die Hinreaktion einen primären Salzeffekt, während die
Rückreaktion unabhängig von der Ionenstärke der Lösung ist.
Reaktion (10) ist eine umkehrbare Reaktion; die Differentialgleichung hierzu lautet:
3
1 2 2 3POH
P OH POH
dck c c k c
dt
(14)

8
Bei OH--Überschuss erhält man eine Reaktion quasi-erster Ordnung, so dass man cOH- in die
Geschwindigkeitskonstante einbeziehen kann:
3
1 2 2 3
01 2 3 2 3
'
'
POH
P POH
P POH POH
dck c k c
dt
k c c k c
(15)
Im Gleichgewicht (g) ist
30POH
dc
dt
und somit
01 2 23 3' g g
P POH POHk c c k c
oder
02 3
2 1
3
'
g
P POH
g
POH
c ck k
c
(16)
Gleichung (16) in (15) eingesetzt ergibt:
03 2 33 0
1 2 3 1
3
03 1 2
33
3
' '
'
g
POH P POHPOHgP POH
POH
gPOH Pg POHPOH
POH
c c cdck c c k
dt c
dc k cc c
dt c
03 1 2
333
01 2
33
3
'
'ln
POH PggPOHPOHPOH
g PgPOHPOH
POH
dc k cdt
cc c
k cc c t C
c
Bei t = 0 ist cPOH3- = 0; es ist also
3ln g
POHc C
und
03 1 2
333
'ln
g
POH PggPOHPOHPOH
c k ct
cc c
(17)
Mit (16) ergibt sich
0 01 2 3 1 2
1 2 1
3 3
' '' '
g
P POH Pg g
POH POH
k c c k ck k k
c c
(18)
(18) in (17) eingesetzt liefert schließlich:
3
1 2
33
1' ln
g
POHg
POHPOH
ck k
t c c
(19)

9
Die Konzentration an Farbstoff ist nach dem Lambert-Beer‘schen Gesetz proportional zum
Logarithmus der Transmission T:
2
03 2 2
0
1 2
log
1 log log' ln
log log
P
POH P P
g
t g
c const T
c c c
T Tk k
t T T
(20)
Auflösen nach Tt ergibt:
'0 1 2
'1 2
log log logexp
k k tg gt
k k t
T T e TT
e
(21)
Trägt man die gemessene Transmission Tt gegen t auf, so kann man mit Hilfe von Gl. (21)
eine Kurve an den Verlauf anfitten und so die Parameter T0, T
g
(Transmission zum Zeitpunkt
0 bzw. Transmission im Gleichgewicht) und 1 2'k k bestimmen.
Für die Gleichgewichtskonstante K ergibt sich:
03
2
log log
log
gg
POHg g
OH OHP
c T TK
c c T c
(22)
Mit
1
2
1 1'OH
kK
k
k k c
lassen sich die Geschwindigkeitskonstanten k1 und k2 berechnen zu
1 21
2 1 2 1
'
1
'
OH
OH
k kk
cK
k k k k c
(23)
Wird durch Fremdsalzzusatz die Ionenstärke Im variiert, erhält man verschiedene Werte für
k1, k2 und K. Trägt man nun log k1 gegen mI auf, so sollte sich bei starker Verdünnung eine
Grenzgerade mit der Steigung 4A ergeben (s. Abbildung 1 und Beispielgraphen im Anhang).

10
6. Versuchsdurchführung
Herstellung der Salz-Lösungen:
Zur Herstellung der KCl-Stammlösung werden ca. 1,2 g KCl in ein 100 ml Becherglas
eingewogen; hierbei ist die genaue Masse auf 0,1mg genau zu notieren, um später die genaue
Ionenkonzentration der einzelnen Proben bestimmen zu können. Das KCl wird in etwas
demin. Wasser gelöst, in einem 100 ml Messkolben gegeben und aufgefüllt. Für Na2SO4
verfährt man in gleicher Weise, wobei man hier mit einer Einwaage von ca. 0,7 g arbeitet.
Vorbereitung der Proben:
Die Proben werden jeweils in einem 100 ml Messkolben hergestellt; hierzu werden zunächst
50 ml 0,1 M NaOH vorgelegt und anschließend die jeweilige Menge Salz, hineinpipettiert und
ggf. mit demin. Wasser aufgefüllt.
Probe V(NaOH) / ml V(KCl) / mL V(Na2SO4) / mL V(H2O) / mL
1 50 0 0 50
2 50 5 0 45
3 50 10 0 40
4 50 25 0 25
5 50 50 0 0
6 50 0 5 45
7 50 0 10 40
8 50 0 25 25
9 50 0 50 0
Vorbereitung der Spektrometer:
Vor Beginn der Messung werden alle drei Spektrometer auf die Wellenlänge 560 nm
eingestellt und anschließend kalibriert. Dies geschieht, indem bei wassergefüllter Küvette,
zunächst das eingebaute Messinstrument mit Hilfe der Feineinstellung (Grobbereich: 5) auf
100% Transmission eingestellt wird und der Set-Button für den Maximalwert betätigt wird
(siehe Abbildung); ab diesem Zeitpunkt dürfen keine Änderungen an den elektronischen
Einstellungen des Spektrometer mehr erfolgen! Im nächsten Schritt wird am Küvettenwahlrad
die Position 3 gewählt (hier befindet sich eine lichtundurchlässige Schicht) und der Software
der Wert für 0% Transmission, durch Drücken auf den Set-Button für den Minimalwert,
mitgeteilt.
Messung:
Zu Beginn der Messung werden 1000 μl der 2 mM Phenolphtaleinlösung (LM: 0,5 EtOH / 0,5
H2O) zum jeweiligen Kolben mit einer Eppendorfpipette gegeben; ab hier startet die
Zeitmessung (Start-Button). Dann wird die Küvette zweimal mit der Lösung gespült um
anschließend die Probe zu messen (5ml Einwegspritze mit Kanüle verwenden). Nach jeder
Messung wird die Probe entfernt und die Küvette mit demin. Wasser gespült; nach
Versuchende werden die Küvetten gespült und bleiben anschließend mit demin. Wasser
gefüllt.
Jede Probe durchläuft eine Messzeit von 3600 s.

11
Abbildung 3: Bedienoberfläche des Messprogramms VISIONlite.
Auswertung:
Die Software speichert die Messwerte in einer eigenen Datei. Jede Messreihe wird in eine
eigene Datei geschrieben und muss als .csv Datei exportiert werden um daraufhin mit Origin
geöffnet werden zu können. Hierbei wird jeder Messpunkt in eine eigene Zeile geschrieben.
Die erste Spalte enthält die Zeit in Minuten, die zweite die Transmission in Prozent.

12
7. Auswertung
Zeitlicher Verlauf der Transmission Tt (21):
'0 1 2
'1 2
log log logexp
k k tg gt
k k t
T T e TT
e
Fit mit Origin zur Bestimmung von T0, T
g, k‘1+ k2 (s. Zusatzmaterialien):
1. Tt gegen t auftragen
2. Im Diagramm in der Menüleiste Analysis → Non-linear Curve Fit auswählen
3. Category New: Speicherort für die neue Funktion festlegen
4. Function New
a. Als Funktion eingeben:
y=10^(((log(T0)-log(Tg))+log(Tg)*exp(k*x))/exp(k*x))
(y entspricht Tt, x entspricht t und k entspricht k‘1 + k2
b. User defined Parameter Names: T0, Tg, k
c. "save as "y-Skript
d. Action → Dataset (Spalten x und y zuordnen: assign)
e. Action → Fit (Anfangs-Parameter vorgeben: Tg und T0 aus Kurvenverlauf
abschätzen, k ca. 0,0001), auf 1 Iter. klicken (Kurve wird eingezeichnet), auf
100 Iter. klicken bis die berechnete Kurve mit dem gemessenen Verlauf gut
übereinstimmt und sich die Parameter nicht mehr ändern
• Die Gleichgewichtskonstante K berechnet sich nach Gleichung (22).
• k1 und k2 sind nach Gleichung (23) zu bestimmen.
Ergebnisse in übersichtlicher, tabellarischer Form präsentieren!!!

13
Anhang:
Formel für die Debye-Hückel-Konstanten A und B
Das Debye-Hückel-Gesetz wurde hergeleitet, so dass sich folgende Form ergab:
32 20
0
00
24
ln1 2
1
A LM mr Bm
m A LMm
r B
eN I
k TA If z z z z
Ba I Ne a I
k T
Mit der inversen Debye-Länge
20
0
2 A m
B
e N I
k Tergibt sich
20
08ln
1
LMr B
LM
e
k Tf z z
a
Für stark verdünnte Lösungen ist 1 LMa , so dass das Grenzgesetz gilt:
20
0
ln8 r B
z z ef
k T
Für A gilt
2 20 0
0 04 2
A LM
B B
e e NA
k T k T bzw.
32 20
0
24 A LM
B
eA N
k T
Für B gilt
0
0
2 A LM
r B
NB e
k T
Für T 25°C, r 78,38 und LM 997,05 kg m-3
ergeben sich die Werte
lnA 1,1744 (kg mol-1
)1/2
und lnB 3,285.10
9 (kg mol
-1)1/2
m-1
.
A und B beziehen sich hier auf den natürlichen Logarithmus des mittleren
Aktivitätskoeffizienten. Um A bezüglich des dekadischen Logarithmus anzugeben, wird
durch ln(10) geteilt, da ln log ln 10a a . Damit gilt für die Konstanten A und B
bezüglich des dekadischen Logarithmus logf des mittleren Aktivitätskoeffizienten:
32 20
log0
12
ln(10) 4 A LMB
eA N
k T0,510 (kg mol
-1)1/2
0
log0
2
ln(10)A LM
r B
e NB
k T1,427
.10
9 (kg mol
-1)1/2
m-1
.
Typische Werte für den mittleren Ionendurchmesser a liegen zwischen 3 und 9 Å für
anorganische Salze. Durch Analyse der Messergebnisse lässt sich mithilfe der Debye-Hückel-
Theorie ein mittlerer Ionendurchmesser a für das (größere) Phenolphthalein-Dianion und
das Hydroxid-Ion abschätzen.
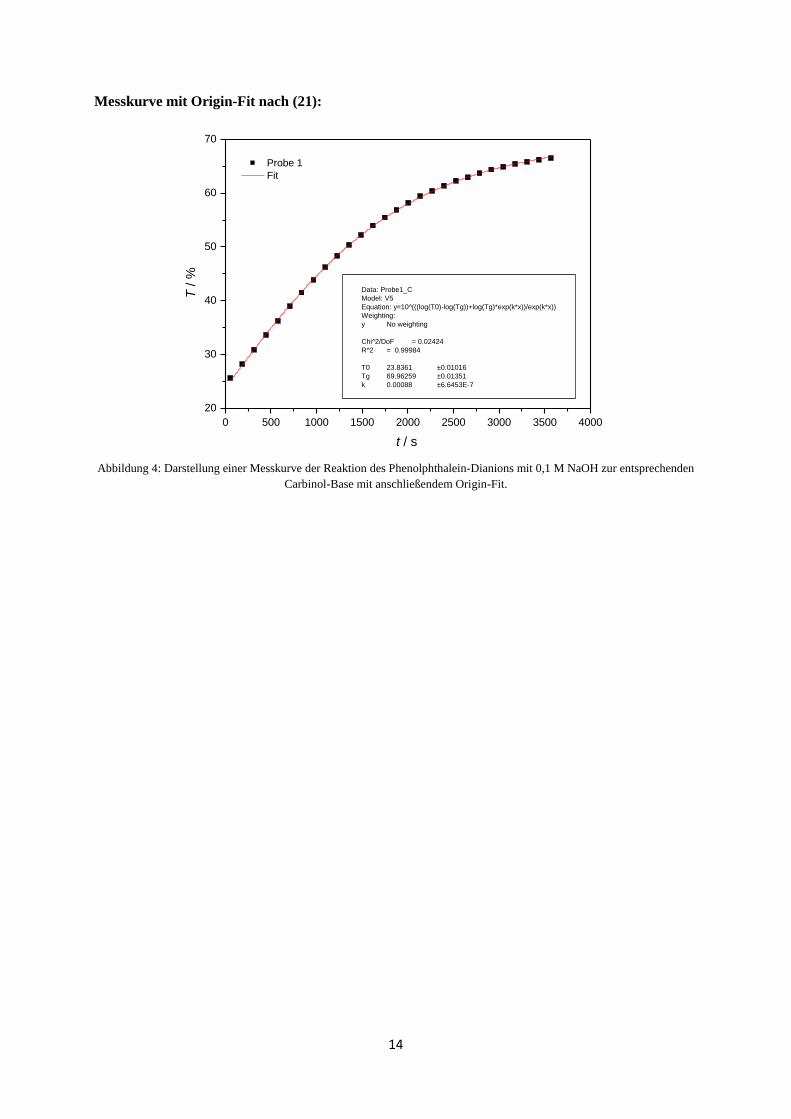
14
Messkurve mit Origin-Fit nach (21):
0 500 1000 1500 2000 2500 3000 3500 4000
20
30
40
50
60
70
Data: Probe1_C
Model: V5
Equation: y=10^(((log(T0)-log(Tg))+log(Tg)*exp(k*x))/exp(k*x))
Weighting:
y No weighting
Chi^2/DoF = 0.02424
R^2 = 0.99984
T0 23.8361 ±0.01016
Tg 69.96259 ±0.01351
k 0.00088 ±6.6453E-7
Probe 1
Fit
T / %
t / s
Abbildung 4: Darstellung einer Messkurve der Reaktion des Phenolphthalein-Dianions mit 0,1 M NaOH zur entsprechenden
Carbinol-Base mit anschließendem Origin-Fit.

15
Auswertung (Origin o. Excel) des primären Salzeffekts (9):
y = 0.4893x - 2.0365
R² = 0.5607y = 0.89x - 2.1316R² = 0.8313
-2.4
-2.3
-2.2
-2.1
-2.0
-1.9
-1.8
-1.7
-1.6
-1.5
-1.4
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35
lg (k1
/ m
ol-1
s-1L)
Im1/2 / mol1/2 kg-1/2
Fremdsalzeffekt
Debye-Hückel Grenzgesetz
Debye-Hückel GesetzNa2SO4-Zusatz
KCl-Zusatz
Abbildung 5: Darstellung des primären Salzeffekts bei Zugabe von KCl und Na2SO4 bei der Reaktion des Phenolphthalein-
Dianions mit 0,1 M NaOH zur entsprechenden Carbinol-Base; theoretisch bestimmtes Atheo. = 0,510 kg1/2 mol-1/2.
0.0
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
1.0
0.00 0.05 0.10 0.15 0.20 0.25 0.30 0.35 0.40 0.45 0.50
f ±
Im1/2 / mol1/2 kg-1/2
Fremdsalzeffekt
Debye-Hückel Grenzgesetz
Debye-Hückel Gesetz
Abbildung 6: Mittlerer Aktivitätskoeffizient als Funktion der Wurzel der (molalen) Ionenstärke für das Debye-Hückel-Gesetz
und das Grenzgesetz für starke Verdünnung.














![[Chem] Schoops - Analytische Chemie](https://static.fdokument.com/doc/165x107/5571f82249795991698cb7d7/chem-schoops-analytische-chemie.jpg)