FGFR3Spektrum Genetik der Skelettdysplasien · Nachdem die Achondroplasie auf Chromosom 4p kartiert...
8
Zusammenfassung Das Spektrum der durch Mutationen des Fibroblastenwachstumsfaktorre zeptor3 ( FGFR3)Gens verursachten Skelettdysplasien umfasst inzwischen 8 verschiedene Krankheitsbilder. Durch Analyse der Funktion des FGFR3 bei Skelettentwicklung und Wachstum wird versucht, die Patho physiologie der durch FGFR3Muta tionen bedingten Störungen zu ver stehen und diese Erkenntnisse in eine GenotypPhänotypKorrelation einzu bringen. Damit werden auch erste Grundlagen für neue Therapieansätze der Achondroplasie geschaffen. Schlüsselwörter FGFR3, Skelettdysplasie, Wachstumsfuge, Signalkaskade, Achondroplasie Therapieansatz FGFR3Spectrum Abstract The spectrum of skeletal dysplasias caused by mutations of the fibroblast growth factor receptor 3 ( FGFR3) gene now comprises eight different conditions. The analysis of FGFR3 function during skeletal development and growth should provide insights to understand the pathophysiology of defects due to FGFR3 mutations to complement data on genotypepheno type correlations. These studies will set the stage to develop new thera peutic strategies to treat achondro plasia. Keywords FGFR3, skeletal dysplasia, growth plate, signaling pathway, achondroplasia therapeutic approach Einleitung In der großen heterogenen Gruppe der Osteochondrodysplasien gibt es eine umschriebene Anzahl von Ske letterkrankungen, die durch Defekte des Fibroblastenwachstumsfaktor Rezeptor 3( FGFR3)Gens bedingt sind. Dass die Krankheitsbilder Achondro plasie, Hypochondroplasie und Tha natophore Dysplasie eine gemeinsa me genetische Grundlage haben, war schon lange vor den Erfolgen der Mo lekulargenetik vermutet worden, als Spranger (Spranger, 1985) diese Ske lettdysplasien aufgrund von klini schen und röntgenologischen Befun den als sog. AchondroplasieFamilie zusammenfasste. Namensgeber war die Achondroplasie, die mit einer Fre quenz von ca. 1:16.000 häufigste Form der genetisch bedingten Ske lettentwicklungsstörungen. Inzwi schen ist diese Einschätzung durch die Forschung bestätigt und erweitert worden. Bei der letzten Internationa len Konferenz zum Thema Skelettdys plasien (Hall, 2002) wurden 33 Krank heitsgruppen abgegrenzt, wobei die AchondroplasieGruppe sechs Entitä ten umfasste, zu der inzwischen noch weitere Krankheitsbilder hinzugekom men sind (Abb.1). Die Erweiterung der Achondroplasie bzw. FGFR3SkelettdysplasieFamilie erfolgte über klinisch/röntgenologi sche Ansatzpunkte aber auch durch neue genetische, molekulargeneti sche bzw. zellbiologische Erkennt nisse. Die wesentlichen Informationen dazu sind aktuell verfügbar über Genetik der Skelettdysplasien 9 medgen 16 (2004) UniversitätsKinderklinik Mainz FGFR3Spektrum Bernhard Zabel
Transcript of FGFR3Spektrum Genetik der Skelettdysplasien · Nachdem die Achondroplasie auf Chromosom 4p kartiert...

ZusammenfassungDas Spektrum der durch Mutationendes Fibroblastenwachstumsfaktorre�zeptor�3 (FGFR3)�Gens verursachtenSkelettdysplasien umfasst inzwischen8 verschiedene Krankheitsbilder. Durch Analyse der Funktion desFGFR3 bei Skelettentwicklung undWachstum wird versucht, die Patho�physiologie der durch FGFR3�Muta�tionen bedingten Störungen zu ver�stehen und diese Erkenntnisse in eineGenotyp�Phänotyp�Korrelation einzu�bringen. Damit werden auch ersteGrundlagen für neue Therapieansätzeder Achondroplasie geschaffen.
SchlüsselwörterFGFR3, Skelettdysplasie,Wachstumsfuge, Signalkaskade,Achondroplasie Therapieansatz
FGFR3�SpectrumAbstractThe spectrum of skeletal dysplasiascaused by mutations of the fibroblastgrowth factor receptor 3 (FGFR3)gene now comprises eight differentconditions. The analysis of FGFR3function during skeletal developmentand growth should provide insights tounderstand the pathophysiology ofdefects due to FGFR3 mutations tocomplement data on genotype�pheno�type correlations. These studies willset the stage to develop new thera�peutic strategies to treat achondro�plasia.
KeywordsFGFR3, skeletal dysplasia, growth plate, signaling pathway,achondroplasia therapeuticapproach
EinleitungIn der großen heterogenen Gruppeder Osteochondrodysplasien gibt eseine umschriebene Anzahl von Ske letterkrankungen, die durch Defektedes Fibroblastenwachstumsfaktor Rezeptor 3 (FGFR3) Gens bedingtsind.
Dass die Krankheitsbilder Achondro plasie, Hypochondroplasie und Tha natophore Dysplasie eine gemeinsa me genetische Grundlage haben, warschon lange vor den Erfolgen der Mo lekulargenetik vermutet worden, alsSpranger (Spranger, 1985) diese Ske lettdysplasien aufgrund von klini schen und röntgenologischen Befun den als sog. Achondroplasie Familiezusammenfasste. Namensgeber wardie Achondroplasie, die mit einer Fre quenz von ca. 1:16.000 häufigsteForm der genetisch bedingten Ske lettentwicklungsstörungen. Inzwi schen ist diese Einschätzung durchdie Forschung bestätigt und erweitertworden. Bei der letzten Internationa len Konferenz zum Thema Skelettdys plasien (Hall, 2002) wurden 33 Krank heitsgruppen abgegrenzt, wobei dieAchondroplasie Gruppe sechs Entitä ten umfasste, zu der inzwischen nochweitere Krankheitsbilder hinzugekom men sind (Abb.1).
Die Erweiterung der Achondroplasie bzw. FGFR3 Skelettdysplasie Familieerfolgte über klinisch/röntgenologi sche Ansatzpunkte aber auch durchneue genetische, molekulargeneti sche bzw. zellbiologische Erkennt nisse. Die wesentlichen Informationendazu sind aktuell verfügbar über
Gen
etik
der
Ske
lett
dys
pla
sien
9medgen 16 (2004)
Universitäts Kinderklinik Mainz
FGFR3�Spektrum
Bernhard Zabel

OMIM (Online Mendelian Inheritancein Man – National Center for Biotech nology Information: http://www3.ncbi.nlm.nih.gov/Omim/).
Spektrum der mit FGFR3�Mutationen assoziiertenErkrankungen
Thanatopore DysplasieAchondroplasieHypochondroplasieDas klinische Spektrum reicht von derletalen Thanatophoren Dysplasie (TD)über die Achondroplasie (ACH) alsschwere Erscheinungsform bis hin zurHypochondroplasie (HCH) als leichte ste Erkrankungsvariante. Der Erbgangist in allen drei Fällen autosomal do minant. Es gibt gemeinsame Haupt symptome, die aber in TD, ACH bzw.HCH unterschiedlich stark ausge prägt sind. Im Vordergrund steht derdisproportionierte Kleinwuchs mit rhi zomel verkürzten Extremitäten sowierelativ langem Rumpf und lumbalerHyperlordose, außerdem der Makro zephalus mit eingesunkener Nasen wurzel bei allgemeiner Mittelgesichts hypoplasie. Radiologisch imponierenmetaphysäre Veränderungen an denverkürzten Röhrenknochen sowie eineingeengter Wirbelkanal. Die Extremedieses Spektrums sind zum einen beider TD ein schon vor Geburt ausge prägter schwerer, disproportionierterKleinwuchs mit Makrozephalus, (evtl.Kleeblattschädel) und sehr schmalemThorax mit stark verkürzten Rippen,der die respiratorische Insuffizienzbedingt, an der die Kinder meist un mittelbar nach der Geburt versterben.
Zum anderen ist es die HCH, die beider Diagnose häufig Schwierigkeitenbereitet, da beim Minderwuchs mit ei ner Erwachsenengröße von ca. 145cm eine Disproportion im Bereich derExtremitäten und eine verstärkte Len denlordose sowie charakteristischeradiologische Zeichen kaum in Er scheinung treten müssen und auchKopfform und Facies in den meistenFällen völlig unauffällig sind (Hilbert etal., 1998).
Nachdem die Achondroplasie aufChromosom 4p kartiert worden war,galt das in der terminalen Region deskurzen Arms von Chromosom 4(4p16.3) liegende Fibroblastenwachs tumsfaktorrezeptor 3 (FGFR3) Genals mögliches ACH Kandidatengen.1994 wurde dann erstmals eineFGFR3 Mutation als Ursache der Er krankung identifiziert (Shiang et al.,1994). In der Folge zeigte sich, dasspraktisch allen ACH Fällen eine ganzspezifische FGFR3 Mutation in derTransmembrandomäne zugrundeliegt. Sie betrifft das Nukleotid an Po sition 1138, wodurch eine Aminosäu rekonversion Gly380Arg resultiert(außerdem gibt es nur noch zweiACH Einzelfallbeschreibungen mit ei nem Gly→Arg Aminosäureaustauschin unmittelbarer Nachbarschaft anPosition 375). Wenn bei familiärenFällen beide Eltern von ACH betroffensind und beim zu erwartenden Kinddie ACH Mutation homozygot vor liegt, resultiert ein TD Phänotyp. Inden folgenden Jahren wurden auchbei der Thanatophoren Dysplasie mitden Unterformen Typ I bzw. Typ II(TD1, TD2) sowie bei der Hypochon
droplasie umschriebene FGFR3 Mu tationen gefunden (Abb.1). Da die TD Fälle röntgenologisch so eindeutigsind (auch die Abgrenzung zwischenTyp I und Typ II aufgrund vorhandeneroder fehlender „Telefonhörerform“ derFemora), ist der Nachweis einer derTD typischen FGFR3 Mutationen in zwischen in nahezu 100% der Fällemöglich. Dagegen liegt die Treffer quote bei Patienten mit HCH immernoch unter 70% der untersuchtenFälle. Dies ist teilweise durch einefehlerhafte Diagnosestellung zu erklä ren, da HCH aufgrund der Variabilitätdes klinischen Bildes, das vom ACH Phänotyp bis hin zum Minderwuchsmit uncharakteristischen Röntgenbe funden reicht, oft differentialdiagno stische Schwierigkeiten bereitet. Einweiterer Grund ist sicher auch, dasssich die aufwändige FGFR3 Diagno stik in der Regel auf die 3 HCH Muta tions hot spots (N540K/T/S undI538V, K650N/Q bzw. N328I) be schränkt. Zu berücksichtigen ist je doch darüber hinaus eine genetischeHeterogenität, da gezeigt werdenkonnte, dass es familiäre Fälle mit ty pischer HCH Symptomatik gibt, diekeine Kopplung zum FGFR3 Genlo kus aufweisen.
Auch bei klinisch/röntgenologischeindeutig erscheinenden ACH Befun den bleibt es wichtig, diese Diagno sen durch FGFR3 Analyse zu bestäti gen. Es können dabei unerwarteteBefunde auftreten, wie z.B. die Iden tifizierung von TD Mutationen beiACH ähnlichem Phänotyp, was bishernur teilweise durch den Nachweis ei nes Mosaikstatus für die Mutation er
Gen
etik
der
Ske
lett
dys
pla
sien
10 medgen 16 (2004)
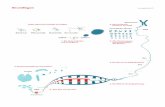
Abb 1 Schematische Darstellung desFGFR3�Moleküls mit Extra� und Intra�zellulärbereich sowie Transmembran�bereich TM
Spezifische Domänen sind: die Immun globulin ähnlichen Domänen Ig I, Ig II, IgIII (Ig III mit den Spleißvarianten IIIb bzw.IIIc), saure Domäne A sowie die zweige teilte Tyrosinkinase Domäne TK1 bzw.TK2.
Eingezeichnet ist die Lokalisation einerReihe der typischen FGFR3 Mutationenvon Skelettdysplasien des FGFR3 Spek trums (s. Text).

klärbar war. Solche Ergebnisse sindnicht nur von akademischen Interes se, sondern werfen auch konkreteFragen zur genetischen Beratung auf,insbesondere bei der Besprechungvon Pränatalbefunden.
Platyspondyle letale Skelett�dysplasie Typ San DiegoEine Erweiterung der FGFR3 Dyspla sien erfolgte durch Untersuchung derBezüge zu anderen Osteochondrody splasie Gruppen. Die platyspondylenletalen Skelettdysplasien (PLSD) sindeine heterogene Gruppe zu der auchdie TD gerechnet wird. Weitere Vertre ter, die als umschriebene Krankheits bilder abgegrenzt wurden, sind PLSDTyp San Diego, PLSD Typ Torranceund PLSD Typ Luton. Die Überprü fung dieser Erkrankungen auf FGFR3 Mutationen ergab, dass sich bei Fäl len mit PLSD Typ San Diego (PLSD SD) TD1 typische FGFR3 Mutationennachweisen lassen, wohingegen dieAnalyse der anderen PLSD Variantennegativ verlief. Eine Erklärung derPLSD SD spezifischen radiologischenund vor allem histologischen Verän derungen (Nachweis von Einschluss körperchen im Bereich des erweiter ten rauen endoplasmatischen Retiku lums) steht noch aus.
SADDAN�DysplasieDas klinische FGFR3 Spektrum wur de weiter ergänzt (Überblick z.B. Vajoet al., 2000), indem man Fälle identi fizierte, die vom Schweregrad zwi schen den TD Formen und der ACHlagen. Dies kommt auch im Namenfür dieses Krankheitsbild zum Aus druck: SADDAN für Severe Achon�droplasia with Developmental Delayand Acanthosis Nigricans. Die zu grunde liegende FGFR3 missense Mutation betrifft Aminosäureposition650, wobei je nach Substitution TD1bzw. TD2, SADDAN oder HCH resul tieren kann. Um dies zu erklären wirdspekuliert, dass es je nach Qualitätdes Aminosäure Austausches an Po sition Lys650 zu gravierenden Unter schieden hinsichtlich der FGFR3 Ty rosinkinase Aktivität kommt, wasunterschiedlich schwere Skelettdys plasien zur Folge haben kann.
Nichtsyndromatische koronareKraniosynostose/Muenke�Syndrom, Crouzon�Syndrom mitAcanthosis NigricansParallel zur Analyse von FGFR3 Kan didatenerkrankungen verlief die Su che nach Syndromen, die durchFGFR1 bzw. FGFR2 Mutationen be dingt sein können. Die Liste umfasstinzwischen Pfeiffer , Apert , Crouzon ,Jackson Weiss und Beare Steven son cutis gyrata Syndrom, allesKrankheitsbilder, die als syndromati sche Kraniosynostoseformen zu sammengefasst werden können. ImZuge der FGFR Analytik wurdenschließlich auch Patienten identifi ziert, deren Kraniosynostose Symp tomatik auf FGFR3 Mutationen zu rückzuführen war. Konkret betrifft dieseinerseits familiäre oder sporadischeFälle mit sog. nichtsyndromatischerkoronarer Kraniosynostose (CCS) mitder Mutation Pro250Arg, die auch alsMuenke Syndrom bezeichnet werden,sowie andererseits Fälle mit Crouzon Syndrom und Acanthosis Nigricans(CAN) mit dem Befund Ala391Glu. Fürden Pro250Arg Aminosäureaustauschist bemerkenswert, dass wenn erFGFR3 betrifft, die Konsequenz dasMuenke Syndrom ist, wenn diesemissense Mutation aber im FGFR1 bzw. im FGFR2 Gen vorliegt, jeweilsdas Pfeiffer bzw. das Apert Syndromresultieren.
Es ist zu konstatieren, dass das an fangs so umschriebene klinische Bildder FGFR3 Pro250Arg Mutation inzwi schen Überlappungen zu definiertenKraniosynostose Syndromen zeigenkann, die z.B. auch den normaler weise durch TWIST Genmutationenbedingten Saethre Chotzen SyndromPhänotyp einschließen können. Kli nisch kann das Krankheitsbild darü ber hinaus Symptome wie Brachy daktylie, Hautveränderungen, Innen ohrschwerhörigkeit und psychomoto rische Retardierung umfassen. Zur Er klärung der beobachteten inter undintrafamiliären Variabilität werden u.a.modifier Gene bzw. auch ge schlechtsspezifische Faktoren verant wortlich gemacht.
Die FGFR3 Mutation der Fälle mitCrouzon Syndrom und Hautverände rungen entspricht nicht den typischen
FGFR2 Crouzon Mutationen, die ex trazellulär im Bereich der Ig III Domä ne lokalisiert sind. Die CAN Mutation(Ala391Glu) liegt in der FGFR3 Trans membran Domäne in unmittelbarerNähe zur klassischen ACH Mutation.Die Patienten haben die typische Zei chen des Crouzon Syndrom Phäno typs aber höchstens nur subtile Merk male der Achondroplasie Skelettdy splasie Familie.
Bei einer Reihe von genetischen Er krankungen können Hautbefunde imSinne einer Acanthosis Nigricansbzw. einer epidermalen Hyperplasieauftreten. Dies betrifft sowohl einigeStörungen aus der Kraniosynostose wie auch aus der Skelettdysplasie Gruppe, die einerseits auf FGFR2 (Beare Stevenson Syndrom) oder an derseits auf FGFR3 Mutationen (CAN,CCS, SADDAN und TD) beruhen. Ins besondere für die Acanthosis Nigri cans wird vermutet, dass ihre Patho genese mit gestörten Wachstumsfak torrezeptor Tyrosinkinase Signalkas kaden zusammenhängt.
Interessanterweise zeigen die Patien ten der Kraniosynostosegruppe – imGegensatz zu den FGFR3 assoziier ten Skelettdysplasien – ein ungestör tes Längenwachstum. Somit führenMutationen im FGFR3 Gen nicht nurzu ähnlichen Erkrankungen mit gra duell unterschiedlichen Schweregra den, wie dies bei den Mitgliedern der“Achondroplasie Familie“ der Fall ist,sondern sie können auch Ursachephänotypisch sehr verschiedenerKrankheitsbilder sein. Erklärbar wirddies vielleicht, wenn deutlich wird,warum bestimmte Mutationen spezi fische Auswirkungen besonders inder Wachstumsfuge bei dem Vorgangder enchondralen Ossifikation haben,während andere zu Störungen vor wiegend im Bereich der Schädelnäh te führen.
CATSHL�PhänotypSchließlich ist in dieser Übersichtüber FGFR3 Phänotyp Genotyp Kor relationen als neuster Befund zumFGFR3 Spektrum zu erwähnen, dasskürzlich über eine Familie mit domi nant vererbter FGFR3 Mutation be richtet wurde, deren wesentliches kli nisches Merkmal Hochwuchs ist. Als
Gen
etik
der
Ske
lett
dys
pla
sien
11medgen 16 (2004)

weitere Symptome werden Innenohr schwerhörigkeit, Kamptodaktylie, Ge lenküberstreckbarkeit, thorakolumba le Skoliose, Mikrocephalie und Ent wicklungsverzögerung angegeben.Bei der Mutation handelt es sich umeine missense Mutation (Arg621His)in der katalytischen Schleife im Be reich des Beginns der Tyrosinkinase Domäne. Es wird vermutet, dass essich um eine loss of function Muta tion handelt, die aber nicht einerFGFR3 Haploinsuffizienz gleichzuset zen ist. Begründet wird dies mit derTatsache, dass die Deletion einesFGFR3 Allels wie sie beim Wolf Hirschhorn Syndrom mit größeren4p16 Deletionen beobachtet werden nicht zu vergleichbaren Skelettbe funden führt. Man nimmt eher an,dass von der R621H Mutation betrof fene FGFR3 Moleküle eine reduzierteTyrosinkinase Aktivität zeigen. Diesebisher nur als Kurzbericht veröffent lichte Mitteilung (Toydemir et al.,2003) nährt weitere Hoffnungen, dieman bereits aufgrund des Fgfr3�knock�out Mausmodells hatte, nach dem sich herausstelle, dass die Kö pergröße dieser Tiere die der Normal mäuse übertraf (Deng et al., 1996).Die Erwartungen richten sich nun dar auf, dass durch die genaue Aufklä rung der normalen und der pathologi schen FGFR3 Funktion die Grundlagegeschaffen werden kann, um die vonFGFR3 Mutationen betroffenen Er krankungen direkt therapeutisch zubeeinflussen.
Maligne ErkrankungenDer Vollständigkeit halber sollte indiesem Zusammenhang auch noch
kurz auf onkologisch relevanteFGFR3 Defekte eingegangen werden.Bei Multiplen Myelomen hat manchromosomale Translokationen ge funden, die die genomische FGFR3 Region betrafen und eine pathologi sche Regulation des FGFR3 Gens zurFolge hatten. Es sind außerdem so matische FGFR3 Mutationen vor al lem in Blasen Karzinomen aber auchbei Dickdarm Krebs identifiziert wor den, wobei es sich hauptsächlich umTD , aber auch um SADDAN undACH Mutationen handelt. Zusam menhänge mit den beschriebenenFGFR3 bedingten Skeletterkrankun gen konnte bisher nicht nachgewie sen werden, wobei für die FGFR3 Skelettdysplasien auch kein erhöhtesTumorrisiko bekannt ist.
Molekulare PhysiologieHauptwirkorte des FGFR3 sind Ske lett und ZNS sowie epitheliale Struk turen, wobei unterschiedliche Spleiß formen (IIIc bzw. IIIb) und Liganden(FGFs) involviert sind. Im Bereich desKnorpel/Knochen Systems ist dieFGFR3 Rolle in der Wachstumsfugeund bei der dort ablaufenden enchon dralen Ossifikation von besonderemInteresse. Diese epi /metaphysäreRegion ist es, in der fast ausschließ lich das gesamte Längenwachstumdes Röhrenknochens erfolgt. Es han delt sich um einen hoch organisiertenGewebsbereich, der durch verschie dene Zonen (Ruhezone, Prolifera tionszone, prähypertrophe und hyper trophe Zone) so strukturiert ist, dassChondrozyten eine Abfolge von Proli ferations und Differenzierungstufendurchlaufen. Dabei werden sie durch
eine Reihe von Faktoren beeinflusst,bevor sie das Endstadium der Diffe renzierung erreicht haben, Apoptosedurchlaufen und durch Knochen er setzt werden. Eine genaue Regulationder verschiedenen Schritte ist ent scheidend für den Ablauf des Wachs tums. Die Architektur der Wachs tumsfuge sichert dabei, dass Prolife ration und Reifung der Chondrozytengerichtet und kontrolliert verläuft. Ge ringe Abweichungen bei Richtung undGeschwindigkeit des Wachstumskönnen zu Disproportionen bestimm ter Skelettanteile führen, wie sie fürviele Skelettdysplasien charakteri stisch sind (Abb. 2).
Von wesentlicher Bedeutung für dieWachstumsgeschwindigkeit ist dieDauer, die ein Chondrozyt in der Pro liferationszone verbringt bzw. die Ba lance zwischen Proliferation und Dif ferenzierung. Eine Reihe von Wachs tumsfaktoren und ihre Rezeptoren so wie Signalpeptide und Transkriptions faktoren sind an diesen Prozessenbeteiligt. Hervorzuheben sind Genka skaden, die über Fibroblastenwachs tumsfaktoren (FGFs), und ihre Rezep toren laufen, darüber hinaus spielensog. bone morphogenetic proteins(BMPs) eine wesentliche Rolle sowieder parathyroid hormone related pep�tide (PTHrP)/PTHrP�Rezeptor/Indianhedgehog (IHH) Regelkreis (Ornitzund Marie, 2002).
Bei Letzterem bilden das Parathor mon verwandte Peptid PTHrP (dasvon der periartikulären Region ausabgegeben wird) und das Signalpep tid IHH (exprimiert im Übergang von
Gen
etik
der
Ske
lett
dys
pla
sien
12 medgen 16 (2004)
Abb 2 Schematische Darstellung der Wachstums�fuge
(A) mit ihren verschiedenen Zonen sowie ein Über�blick über wesentliche Regelkreise, die an derSteuerung der dort ablaufenden enchondralenOssifikation beteiligt sind (A: rechter Teil) sowie
(B) Schema des epi/metaphysären Anteils einesRöhrenknochens.
PTHrH/PTHrP R/IHH Regelkreis (blau): PTHrP (parathyroid hormone related peptide), PTHrP R(PTHrP Rezeptor), IHH (Indian hedgehog), PTC(patched = IHH Rezeptor) SMO (smoothened = assoziiertes Signalmolekül).
FGF FGFR Kaskade (schwarz, FGFR3: rot): Fibro blastenwachstumsfaktoren FGFs/FGF18, FGF Rezeptoren FGFR1, FGFR2, FGFR3.
BMP Kaskade (grün): BMPs (bone morphogeneticproteins).
(C) Darstellung der verschiedenen Wachstums�fugen�Zonen im Normalzustand (N) und beiAchondroplasie (ACH).
Vergleichende Übersicht über Histologie undFGFR Wirkorte (nach Aviezer et al., 2003).

der prähyhpertrophen zur hypertrohenZone) einen geschlossenen Regel kreis, der die Rate der Chondrozyten Differenzierung steuert. Das Systemfunktioniert autoregulatorisch auf derGrundlage einer IHH Hemmung überdie PTHrP Rezeptor Expression, derin unmittelbarer Nähe zu den IHH produzierenden Zellen aktiv ist. DieRückkopplung zur Induktion derPTHrP Produktion läuft über den IHH Rezeptor PTC und weitere Zwischen stationen wie SMO im Perichondrium.Das Endergebnis davon ist, dass im mer nur ein bestimmtes Chondrozy ten Kontingent den Schritt zur Diffe renzierung in hypertrophe Zellendurchläuft. Teilweise verknüpft mitdiesem Regelkreis sind die BMPs, diedie Chondrozyten Proliferation unter stützen, aber verzögernd auf ihre Dif ferenzierung und Reifung wirken.
Vor diesem Hintergrund ist dieFGFR3 Wirkung einzuordnen, die nor malerweise für einen Gleichgewichts zustand der in verschiedenen Zonender Wachstumsfuge lokalisierten pro liferierenden und dann sich differen zierenden Chondrozyten sorgt. Funk tionell ist dies ein hemmender Ein fluss, mit dem ein beschleunigtesAufbrauchen der Zellschichten verhin dert wird. Fällt der Regelkreis da durch weg, dass von den Chondrozy ten kein FGFR3 exprimiert wird,kommt es zu einem überschießendenWachstum. Dies zeigte sich an denFgfr3�knock�out Mäusen, bei denendas Fgfr3 Gen ausgeschaltet wurdeund die größer als vergleichbare Wild typ Tiere sind.
Der FGFR3 Effekt wird teilweise di rekt an den Chondrozyten bewirkt,wobei Fibroblasten Wachstumsfakto ren (FGFs) – und dabei insbesondereFGF18 – als Liganden eine wichtigeSteuerungsfunktion haben. FGF18koordiniert außerdem z.B. über ande re FGFRs die Osteogenese mit derChondrogenese (Liu et al., 2002). DieFGFR3 Wirkung geschieht aber auchteilweise indirekt über die Einwirkungauf die gerade angesprochenenIHH/PTHrP bzw. BMP Regelkreise(Minina et al., 2002) (Abb. 2).
Molekulare PathologieDie FGFR gehören zu der Gruppe derTyrosinkinase Rezeptoren. Diesespielen – wie bereits angesprochen –eine zentrale Rolle bei der Signal transduktion für Zellwachstum und differenzierung. Es sind bisher vierverschiedene Gene (FGFR1�4) be kannt, die für strukturell sehr ähnlichaufgebaute Rezeptoren kodieren. Siebestehen aus einer hydrophoben Sig nalpeptidsequenz, der sich drei gly kosylierte, Immunglobulin ähnlicheDomänen anschließen. Es folgt dieTransmembrandomäne und wesent lich für den intrazellulären Anteil isteine zweigeteilte Tyrosinkinasedomä ne (Abb.1).
Die normalen Abläufe auf zellulärerEbene bestehen in der Bindung derLiganden an die Immunglobulindomä nen II und III des Rezeptors sowie anHeparansulfat Proteoglycane. DieFolge ist eine FGFR3 Dimerisierung,was wiederum eine Autophosphory lierung der intrazellulären Tyrosinkina
se (TK) Domäne induziert. Dadurchwerden die FGFR Signaltransduk tionskaskaden aktiviert, die wiederumdie Aktivität von Transkriptionsfakto ren beeinflussen. Eine wichtige durchFGFR3 signalling aktivierte Signal transduktionskaskade ist der STAT(signal transducer and activator oftranscription) Signalweg, der über be stimmte Faktoren (u.a. p21) direkt in hibitorisch auf Zellzyklus und Zell wachstum wirkt (Sahni et al., 1999).Zum anderen wirkt der Liganden ge
Gen
etik
der
Ske
lett
dys
pla
sien
13medgen 16 (2004)
Abb 3 Schematische Darstellung der Aus�wirkungen von FGFR3�Mutationen auf zellu�lärer Ebene, Hauptelemente der FGFR3�Sig�nalkaskaden sowie mögliche Achondropla�sie�Therapieansatzpunkte (s. Text) (* =FGFR3�Mutation).
Einschub oben links (nach Webster und Donoghue, 1997):
Mutationsabhängige FGFR3 Aktivierung: (a) normale Situation; (b) sehr starke Aktivierung durch extrazelluläre
kovalente Disulfidbrückenbindungen beiTD1 Fall bzw. bei
(d) = intrazelluläre Mutation im Bereich der Tyrosinkinase Domäne bei TD2 Fall;
(c) geringere Aktivierung durch die weniger stabile Mutations bedingte Wasserstoff brückenbindung im Bereich der Transmembran Domäne bei ACH.
Zellschema (Chondrozyt)
Membranbereich und intrazellulär links(olivgrün)FGF Rezeptor Abbau über sog. Cbl Proteine(Ubiquitin Ligasen bzw. Adaptor Moleküle) undProteine (Ub), die die Ubiquitinilierung steuern.Bei mutierten Rezeptoren erfolgt ein vermehr tes Recycling (statt lysosomaler Abbau).
Intrazellulär Mitte links (hellblau)Aktivierung des STAT (signal transducer andactivator of transcription) Signalwegs, der sichüber bestimmte Faktoren (u.a. p21) direkt inhi bitorisch auf Zellzyklus und Zellwachstum aus wirkt.
Intrazellulär Mitte rechts (gelb)MAPK (mitogen activated protein kinase) Sig nalweg, der ausgehend von Wachstumsfakto ren u.a. über Ras und Raf bzw. MAPK Kinasen(MKKs) und ERKs (extracellular signal regula ted kinases) zu Zielgenen führt, die an derExtrazellulärmatrix Produktion beteiligt sind.
Therapieansätze(1) Verhinderung der Liganden bedingten Re
zeptoraktivierung z.B. durch Liganden Anta gonisten oder neutralisierende Antikörper;
(2) die Verhinderung der Rezeptor Dimerisie rung durch spezifische Antikörper;
(3) die Verhinderung der Rezeptor Phosphory lierung bzw. der Rezeptor Aktivierung durchRezeptor Tyrosin Kinase Inhibitoren;
(4) Einwirkung auf die FGFR3 Signaltransduk tion durch geeignete Inhibitoren oder durchModulation in Richtung alternativer path ways.
(5) Einwirkung auf die FGFR3 Genexpressionauf der Ebene der DNA bzw. mRNA z.B.durch RNAi Technologie.
(6) Blockierung des bei ACH aktivierten MAP Kinase Signalwegs durch die Wirkung desC Typ Natriuretischen Peptids (CNP) übereine Guanylyl Cyclase (GC B) durch die Be reitstellung von cyclischen GMP (cGMP) alsBotenstoff.

bundene Rezeptor auf den MAPK(mitogen activated protein kinase)Signalweg, der ausgehend von Wachs tumsfaktoren u.a. über Ras und Rafbzw. verschiedene MAPK Kinase(MKK) und MAPK Familienmitglieder(z.B. ERKs, extracellular signal�regu�lated kinases) zur Regulation von Ziel genen führt, die an der Extrazellulär matrix Produktion beteiligt sind. Überdie spezifischen Aktivierungsmusterdieser Kaskaden in Chondrozyten istjedoch noch wenig bekannt (Stantonet al., 2003) (Abb. 3).
Liegt ein durch eine FGFR3 Mutationveränderter Rezeptor vor, so führendie dadurch bedingten Konforma tionsänderungen zur konstitutiven,d.h. Liganden unabhängigen FGFR3 Aktivierung. Diese scheinen graduellunterschiedlich zu sein und mit demresultierenden klinischen Bild zu kor relieren (Webster und Donoghue,1997). So kann es bei bestimmtenTD1 Mutationen (R248C) zum Amino säureaustausch mit Einbau desschwefelhaltigen Cystein im Extrazel lulärbereich kommen, so dass zwi schen zwei mutierten FGFR3 aberran te Disulfidbrückenbindungen entste hen, die zur konstitutiven Rezeptor Dimerisierung führen [Abb. 3 (b)]. Da gegen scheint die TD2 Mutation(K650E) im Bereich der intrazellulärgelegenen Tyrosinkinase Domäne ineiner direkten Aktivierung mit über schießender TK Aktivität zu resultie ren [Abb. 3 (d)]. Auch bei der ACH Mutation im Transmembranbereich(Gly380Arg) kann es in Abwesenheitvon Liganden zur Dimerisierung undAutophosphorylierung kommen, waseine konstitutive Rezeptoraktivierungbedeutet. Basis dafür scheint eineWasserstoffbrückenbindung zwischender Arginin Seitenkette des mutiertenFGFR3 Moleküls und der gegenüber liegenden Carboxylgruppe des Wild typ Moleküls zu sein [Abb. 3 (c)]. Derleichtere Phänotyp der ACH gegen über der klinisch schwer betroffenenTD erklärt sich vermutlich durch diegeringere Stabilität der Wasserstoff brückenbindung im Vergleich zur ko valenten Disulfidbindung bei TD, dieeinen stärkeren Grad der FGFR3 Ak tivierung induziert. Abhängig von die ser Mutations spezifischen Aktivie rung wird die Proliferation der Chon
drozyten gehemmt und ihre terminaleDifferenzierung gestört. Diese Regu lationsprozesse laufen insbesondereüber die STAT Kaskade ab (s. Abb. 3).Beim Menschen und im Mausmodellder ACH bzw. der TD (Brodie undDeng, 2003) ist dies auch durch eineVerschmälerung der entsprechendenZonen in der Wachstumsfuge nach weisbar. Dies führt dann letztendlichzu einem reduzierten Längenwachs tum als wesentliches Kennzeichen ei ner Gruppe von FGFR3 Skelettdy splasien. Neue Befunde von Maus modellen haben die Bedeutung einerzweiten Signalkaskade in den Vorder grund des Interesses gerückt. Es han delt sich dabei um den ERK1/2 MAP Kinase Signalweg, der in Chondrozy ten bei überschießender Aktivierungdurch mutierten FGFR3 auf noch we nig verstandenem Weg die Bildungder extrazellulären Matrix beeinträch tigt. Dass dies entscheidend zumACH Phänotyp beiträgt, ergibt sichaus den Untersuchungen, die zeigen,dass eine Blockierung dieser Reak tionskette vermittelt durch CNP (C�type natriuretic peptide) die Sympto matik korrigieren kann (Yasoda et al.,2004) (s. Abb. 3).
Schließlich konnte nachgewiesenwerden, dass der physiologische Ab bau der FGF Rezeptoren über sog.Cbl Proteine abläuft, die als Ubiqui tin Ligasen bzw. multifunktionelleAdaptor Moleküle den Prozess desRezeptor Recyclings bzw. dessen (ly sosomalen) Abbau steuern. MutierteRezeptoren werden vor der selektivenUbiquitinylierung bewahrt und ver mehrt einem Recycling unterworfen,was zu einer ausgeprägten FGFR3 Regenerierung in den Chondrozytenführt. Auch dies kann wesentlich zueiner Verstärkung der Wirkung vonFGFR3 Mutationen beitragen (Cho etal., 2004) (Abb. 3).
Die sporadischen FGFR Mutationenhaben auch noch in einem anderenZusammenhang Interesse hervorge rufen. Es war bisher nicht schlüssigerklärbar, warum diese spezifischenMutationen besonders väterlichen Ur sprungs sind und warum sie mit fort schreitendem Alter des Vaters zuneh men. Neuste Daten lassen vermuten,dass die verstärkte Aktivierung der
Signaltransduktion, die die bekanntennegativen Auswirkungen auf den Ge samtorganismus hat, einen positivenSelektionseffekt für mutationstragen de Zellen in der Spermiogenese be wirkt (Goriely et al., 2003).
Therapeutische AspekteDie Übersicht über die Zellbiologieder ACH (Abb. 3) kann auch als Aus gangspunkt für mögliche ACH Thera pieansätze dienen. Die besondere Si tuation besteht darin, dass eine spe zifische Punktmutation, die ein be stimmtes Zelloberflächenmolekül be trifft, einen eindrucksvollen Phänotypbewirkt, der aber doch sehr gewebe spezifisch auf das Skelettsystem be schränkt ist. Damit konzentrieren sichdie Therapieansätze auf das FGFR3 Genprodukt und die davon ausgehen den Signal und Wirkkaskaden.
Extrazellulärer Ansatzpunkte sind (1) die Verhinderung der Liganden be
dingten Rezeptoraktivierung durchLiganden bzw. Heparin basierteAntagonisten oder neutralisieren de Antikörper, bzw.
(2) die Verhinderung der Rezeptor Di merisierung durch spezifischeAntikörper.
Intrazelluläre Ansatzpunkte sind ins besondere (3) die Verhinderung der Rezeptor
Phosphorylierung bzw. der Rezep tor Aktivierung durch Rezeptor Ty rosin Kinase Inhibitoren,
(4) die Einwirkung auf die FGFR3 Sig naltransduktion durch geeigneteInhibitoren oder über die Modula tion alternativer pathways oderschließlich
(5) die direkte Einwirkung auf dieFGFR3 regulierte Genexpressionauf der Ebene der DNA bzw.mRNA (z.B. durch RNA interferen�ce (RNAi) Technologie).
Erste Behandlungsversuche z.B. mitTK Inhibitoren sind bereits mit Organ kulturen von ACH Mäusen erfolgreichdurchgeführt worden (Aviezer et al.,2003). Weiterhin wurden inzwischenhumane Antikörper gegen FGFR3 ge neriert, die jetzt an ACH Tieren gete stet werden sollen (Rauchenberger etal., 2003). Sie würden auch dafür sor gen, dass die bei ACH vermehrt ge
Gen
etik
der
Ske
lett
dys
pla
sien
14 medgen 16 (2004)

nerierten und stabilisierten Rezepto ren neutralisiert würden. (6) Das neueste ACH Therapiekon
zept beruht auf der Wirkung desC Typ Natriuretischen Peptids(CNP) auf das Knochenwachstum.Dem liegt die Blockierung des beiACH aktivierten MAPKinase Sig nalwegs zugrunde, wobei CNPdies über die Aktivierung einerGuanylyl Cyclase (GC B) und dieresultierende Synthese von cGMPbewirkt (Yasoda et al., 2004).
Die in den letzten Jahren propagier ten, auf Wachstumshormongabe be ruhenden, ACH Behandlungskonzep te haben nicht überzeugt und auf wändige Verlängerungs Operationenerscheinen nur für eine umschriebeneZahl von Patienten ein Alternativkon zept. In dieser Situation scheint sichjetzt die intensive Grundlagenfor schung auszuzahlen und einen Aus blick auf gezielte Behandlungsmög lichkeiten der Achondroplasie zu ge ben. Man sollte sich aber im Klarendarüber sein, dass es wahrscheinlichnoch Jahre dauern wird, um ein effi zientes, möglichst nebenwirkungs freies Behandlungsverfahren zu eta blieren, das hoffentlich nicht nur dasLängenwachstum verbessert, son dern auch ACH Komplikationen wieSpinalkanalengen verhindern hilft.
In der Zwischenzeit arbeiten wir miteinem Gesundheitsvorsorge Konzeptfür Kinder mit Achondroplasie, dasals Gemeinschaftsinitiative desBundesverbandes KleinwüchsigeMenschen und ihre Familien (BKMF)(http://www.bkmf.de) und der Univer sitäts Kinderkliniken Magdeburg,Mainz und München entwickelt wor den ist. Diese Empfehlungen sind indem Band „Achondroplasie und Hy pochondroplasie. Diagnostik und Be treuung von Kindern mit spezifischenKleinwuchsformen“ zusammenge fasst (Mohnike et al. 2001). Die Pa tienten erfordern einen multidiszipli nären Ansatz, da je nach Lebensalterunterschiedliche Probleme und Kom plikationen auftreten können. Diesekönnen u.a. durch Engen im Bereichdes craniocervicalen Übergangs unddes Spinalkanals gegeben sein, wo durch die Betreuung durch Neuropä diater, Neuroradiologen und Neuro
Gen
etik
der
Ske
lett
dys
pla
sien
15medgen 16 (2004)
GfH�Jahrestagung 12.–15.6.2004in München
ReduzierterTeilnehmerbeitragfür GfH�Mitglieder bis zur Deadline 31.3.2004
GfH�Mitglieder�Tarif: 250 €
(Anmeldung über die www.eshg.orgunter Nennung der GfH Mitglieds zugehörigkeit und dem reduziertenTeilnehmertarif)
GfH�Studenten�Tarif: 60 €
(Studenten melden sich unter der Vorlage ihres Ausweise direkt bei der GfH Geschäftsstelle per Fax an:089 55027856)
Deutsche Gesellschaft
für Humangenetik
UnterbringungEurokongress GmbH Ms. Renate von Franckenstein Isartorplatz 3 80331 München Tel. +49 (0)89 210 986 0 Fax +49 (0)89 210 986 98 [email protected] www.eurokongress.de

chirurgen erforderlich werden kann.Die engen Verhältnisse im HNO Be reich können die Grundlage für Mittel ohr Hörstörungen sein und sekundärdadurch Sprachentwicklungsverzöge rungen bedingen, wobei die geistigeEntwicklung bei ACH generell in kei ner Weise beeinträchtigt ist. An sprechpartner wären u.a. HNO undKlinik für Kommunikationsstörungen.Die statomotorische Entwicklung wirdnur anfangs durch ACH typische Mu skelhypotonie verzögert. In dieserPhase hat die Physiotherapie einenwichtigen, wissenschaftlich freilichnoch nicht einwandfrei belegten, Stel lenwert bei der Betreuung der Betrof fenen. Schließlich ist die orthopädi sche Begleitung von der Kleinkindzeitbis zum Erwachsenenalter u.a. wegenmöglicher Beinfehlstellungen (meistO Beinstellung) und Wirbelsäulen Haltungsschäden sowie in Hinblickauf die Planung eines Eingriffs zur Ex tremitäten Verlängerung bzw. Ach senkorrektur von besonderer Bedeu tung. Letztere Option auf mögliche 15cm Längenzuwachs wird aber meisterst nach dem 15. Lebensjahr unddann auf ausdrücklichen Wunsch derBetroffenen in Betracht gezogen. Auf grund der vielen Fachdisziplinen, diebei der Betreuung der Patienten undihrer Familien gefragt sind, bietet sicheine schwerpunktmäßige, koordinier te Organisationsstruktur an, wie siez.B. in Mainz mit dem von der Univer sitäts Kinderklinik betreuten sog.Kleinwuchszentrum besteht. Bundes weit wird gleichzeitig eine Netzwerk struktur im Rahmen einer BMBF Initi ative zur Diagnostik, Betreuung undErforschung von Skelettdysplasien alsSKELNET Projekt etabliert. Ein ent sprechendes Netzwerk auf europäi scher Ebene, in das die Mainzer Kin derklinik eingebunden ist, hat seineArbeit bereits aufgenommen (Europe an Skeletal Dysplasia Network ESDNhttp://www. esdn.org).
LiteraturAviezer D, Golembo M, Yayon A (2003) Fibrob last growth factor receptor 3 as a therapeutictarget for achondroplasia genetic short limbeddwarfism. Curr Drug Targets 4:353 365.
Brodie SG, Deng C X (2003) Mouse models or thologous to FGFR3 related skeletal dysplasias.Pediatr Pathol Mol Med 22:87 103.
Cho JY, Guo C, Torello M, Lunstrum GP, Iwata T,Deng C, Horton WA (2004) Defective lysosomaltargeting of activated fibroblast growth factor re ceptor 3 in achondroplasia. Proc Natl Acad SciUSA 101:609 614.
Deng C, Wynshaw Boris A, Zhou F, Kuo A, Le der P (1996) Fibroblast growth factor receptor 3is a negative regulator of bone growth. Cell84:911 921.
Goriely A, McVean GAT, Röjmyr M, IngemarssonB, Wilkie AOM (2003) Evidence for selective ad vantage of pathogenic FGFR2 mutations in themale germ line. Science 301:643 646.
Hall CM (2002) International nosology and clas sification of constitutional disorders of bone(2001). Am J Med Genet 113:65 77
Hilbert M, Hilbert K, Spranger J, Wildhardt G,Winterpacht A, Wüchner C, Zabel B (1998)·Hy pochondroplasie, Achondroplasie und Thanato phore Dysplasie als Folge von Mutationen desFibroblastenwachstumsfaktorrezeptor 3 Gens(FGFR3). Monatsschr Kinderheilkd 146:687–691.
Liu Z, Xu J, Colvin JS, Ornitz DM (2002) Coordi nation of chondrogenesis and osteogenesis byfibroblast growth factor 18. Genes & Dev16:859 869.
Minina E, Kreschel C, Naski MC, Ornitz DM,Vortkamp A (2002) Interaction of FGF, Ihh/Pthlh,and BMP signalling integrates chondrocyte pro liferation and hypertrophic differentiation. DevCell 3:439 449.
Mohnike K, Klingebiel K H, Zabel B, Hrsg.Achondroplasie und Hypochondroplasie. Dia gnostik und Betreuung von Kindern mit spezifi schen Kleinwuchsformen. Palatium, Mannheim,2001.
Ornitz DM, Marie PJ (2002) FGF signaling path ways in endochondral and intramembranousbone development and human genetic diseaseGenes & Dev 16:1446 1465
Rauchenberger R, Borges E, Thomassen Wolf E,Rom E, Adar R, Yaniv Y, Malka M, Chumakov I,Kotzer S, Resnitzky D, Knappik A, Reiffert S,Prassler J, Jury K, Waldherr D, Bauer S, Kretz schmar T, Yayon A, Rothe C (2003) Human com binatorial Fab library yielding specific and func tional antibodies against the human fibroblastgrowth factor receptor 3. J Biol Chem 278:38194 38205.
Sahni M, Ambrosetti DC, Mansukhani A, GertnerR, Levy D, Basilico C (1999) FGF signaling inhi bits chondrocyte proliferation and regulatesbone development through the STAT 1 pathway.Genes & Dev 13:1361 1366
Shiang R, Thompson LM, Zhu Y Z, Church DM,Fiedler TJ, Bocian M, Winokur ST, Wasmuth JJ(1994) Mutations in the transmembrane domainof FGFR 3 causes the most common geneticform of dwarfism, achondroplasia. Cell (UnitedStates), Jul 29 1994, 78(2) p335 42.
Spranger J (1985) Pattern recognition in bonedysplasias. In: CJ Papadatos & CS BartsocasHrsg. Endocrine genetics and genetics ofgrowth. Liss, New York, pp315 342.
Stanton LA, Underhill TM, Beier F (2003) MAP ki nases in chondrocyte differentiation. Dev Biol263:165 175.
Toydemir R, Longo N, Brassington A, Bayrak Toydemir P, Krakowiak P, Jorde LB, Bamshad M(2003).A new syndrome caused by a novel loss of function mutation in FGFR3. Am J Hum Ge net 73(Suppl):171.
Vajo Z, Francomano C, Wikin DJ (2000) The mo lecular and genetic basis of fibroblast growthfactor receptor 3 disorders: The achondroplasiafamily of skeletal dysplasias, Muenke craniosy nostosis, and Crouzon syndrome with acantho sis nigricans. Endocrine Reviews 21: 23 39.
Webster MK, Donoghue DJ (1997) FGFR activa tion in skeletal disorders: too much of a goodthing. Trends Genet 13:178 182.
Yasoda A, Komatsu Y, Chusho H, Miyazawa T,Ozasa A, Miura M, Kurihara T, Rogi T, Tanaka S,Suda M, Tamura N, Ogawa Y, Nakao K (2004)Overexpression of CNP in chondrocytes rescuesachondroplasia through a MAPK dependentpathway. Nat Med 10:80 86.
KorrespondenzadresseProf. Dr. Bernhard ZabelUniv. Kinderklinik MainzLangenbeckstr. 155101 MainzTel. (06131) 17 6826Fax (06131) 17 [email protected] mainz.de
Gen
etik
der
Ske
lett
dys
pla
sien
16 medgen 16 (2004)