
Grundlagen der anorganischen Chemie Vorlesung WS 2011/2012 · 3.1.1 Lewis-Formeln . . . . . . . . ....
85
-
Upload
hoangxuyen -
Category
Documents
-
view
228 -
download
0
Transcript of Grundlagen der anorganischen Chemie Vorlesung WS 2011/2012 · 3.1.1 Lewis-Formeln . . . . . . . . ....

Grundlagen der anorganischen Chemie
Vorlesung WS 2011/2012
Rudolf Schimassek und Micha Wildermuth
Dozenten: Prof. Powell und Dr. Kleist
Dieses Dokument erhebt keinen Anspruch auf Vollständigkeit und Fehlerfreiheit.
Wer einen Fehler �ndet kann es uns mitteilen oder ihn behalten.

2
• Klausur: Mittwoch 14.März.12 (14:00 � 16:00 Uhr)
• Internetadresse für Folien, . . . :www.itcp.kit.edu/grunwaldt/kleist
→ Studium und Lehre Passwort: kit2012
Quellen:
• Vorlesung und alle damit verbundenen Quellen
• http://de.wikipedia.org/ (Bilder)
• andere Webseiten über Google-Bildersuche

Inhaltsverzeichnis
1 Atombau 7
1.1 Rutherford (1911) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.1.1 Atombausteine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
1.2 Bohr'sches Atommodell (1913) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71.2.1 Bohr'sches Postulat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
1.3 Atomorbitaltheorie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.3.1 Schrödinger-Gleichung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81.3.2 Quantenzahlen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91.3.3 Elektronenkon�guration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91.3.4 Besetzungsregeln . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101.3.5 Grundzustand . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2 Periodensystem der Elemente (PSE) 11
2.0.6 Begri�e . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 112.1 Bezeichnung der Elemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11
2.1.1 Atommasse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.2 Eigenschaften der Elemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
2.2.1 Atomradien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.2.2 Ionisierungsenergie I . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122.2.3 Elektronena�nität EA . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
3 Chemische Bindungen 13
3.1 Kovalente Bindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133.1.1 Lewis-Formeln . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
3.2 Formalladungen (Elektronenbuchhaltung) . . . . . . . . . . . . . . . . . . . . . . . . . . . 143.3 Mesomerie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
4 Reaktionen 15
4.1 Aktivierungsenergie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154.2 Entropie (Unordnung) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154.3 Standardbildungenthalpie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 154.4 Elektronennegativität . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 164.5 Oxidationszahlen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174.6 Polare Bindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174.7 Ionengitter . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184.8 Gitterenergie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
4.8.1 Beispiel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184.8.2 Born�Haber�Kreisprozess . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184.8.3 Born�Haber�Kreisprozess für NaCl . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
5 Metalle 21
5.1 typische Eigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215.2 Die Metallbindung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
5.2.1 Bsp. Na�Metall . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215.3 Metallstrukturen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
3

4 INHALTSVERZEICHNIS
6 Lösungen 256.1 Inhalt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 256.2 Konzentrationsmaÿe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 256.3 Struktur von H2O . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 256.4 Wasser als Lösungsmittel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
6.4.1 Eigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 266.5 Elektrolyt� und nicht Elektrolyt�Lösungen . . . . . . . . . . . . . . . . . . . . . . . . . . 266.6 Lösungsenthalpie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
6.6.1 z.B.: Au�ösung von KCl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
7 Energetik 297.1 Entropie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
7.1.1 Beispiel: Entropieänderung für die Transformation Wasser → Eis . . . . . . . . . . 297.2 Freie Standardbildugsenthalpie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 307.3 Beein�ussung der Löslichkeit durch Temperatur und Druck . . . . . . . . . . . . . . . . . 307.4 Das Massenwirkungsgesetz . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 307.5 Verschiebung von Gleichgewichtslagen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 327.6 Löslichkeit und Löslichkeitsprodukt . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
7.6.1 Fall 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 327.6.2 Fall 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 327.6.3 Fall 3 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
7.7 Verschiebung von Gleichgewichtslagen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337.7.1 Druckänderungen: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337.7.2 Temperaturänderungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
7.8 Metastabile Systeme / Katalysatoren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337.9 Energie-Diagramm für eine chemische Reaktion . . . . . . . . . . . . . . . . . . . . . . . . 34
8 Säuren und Basen 358.1 Theorien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
8.1.1 Das Arrhenius�Konzept . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358.1.2 Das Brønsted�(Lowry)�Konzept . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358.1.3 Das Lewis�Konzept . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 36
8.2 pH�Wert, Ionenprodukt des Wassers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 368.3 Säurestärke . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
8.3.1 Starke Säuren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 378.3.2 Schwache Säuren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 378.3.3 Protolysegrad . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 378.3.4 Mehrprotonige Säuren: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
8.4 Metallsalze . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 388.4.1 Schwach basische Anionen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
8.5 Pu�erlösungen: . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 388.6 Pu�ersysteme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 398.7 Redox-Systeme . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 39
8.7.1 Summengleichung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 398.8 Konzentrationsabhängigkeit von Potentialen . . . . . . . . . . . . . . . . . . . . . . . . . . 40
8.8.1 Bestimmung von Gleichgewichtskonstanten . . . . . . . . . . . . . . . . . . . . . . 408.8.2 pH-Abhängigkeit des Potentials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
8.9 Elektrolyse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 418.9.1 Beispiel: (PE) Lithium�Batterie . . . . . . . . . . . . . . . . . . . . . . . . . . . . 428.9.2 Beispiel: (SE) Blei�Akku . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 428.9.3 Beispiel: Brennsto�zellen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
8.10 Korrosion und Lokalelemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 428.10.1 Lokalelement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 428.10.2 Korrosion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
9 Hauptgruppenelemente 439.1 Wassersto� . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
9.1.1 Eigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 439.2 Edelgase: Gruppe 18 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
9.2.1 Eigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 449.2.2 Vorkommen, Darstellung, Verwendung . . . . . . . . . . . . . . . . . . . . . . . . . 449.2.3 Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45

INHALTSVERZEICHNIS 5
9.3 Halogene: Gruppe 17 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 469.3.1 Eigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 469.3.2 Vorkommen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 469.3.3 chemische Eigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 479.3.4 Oxidationswirkung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 479.3.5 Darstellung und Verwendung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 479.3.6 Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
9.4 Chalkogene: Gruppe 16 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 519.4.1 allgemeine Eigenschaften . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 529.4.2 Vorkommen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 529.4.3 Eigenschaften der Elemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 529.4.4 Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
9.5 Sticksto�gruppe: Gruppe 15 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 589.5.1 Vorkommen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 589.5.2 Die Elemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 589.5.3 Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 609.5.4 Oxide des Sticksto�s . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 619.5.5 Sauersto��Säuren des Sticksto� . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 639.5.6 Oxide des Phosphors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 649.5.7 Sauersto�säuren des Phosphors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 649.5.8 Arsenverbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
9.6 Kohlensto�: Gruppe 14 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 659.6.1 Vorkommen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 659.6.2 Elemente . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 669.6.3 Darstellung und Verwendung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 679.6.4 C�Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 689.6.5 H�Verbindungen von Si . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 719.6.6 O�Verbindungen von Si . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
9.7 Alkalimetalle: Gruppe 1 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 739.7.1 Vorkommen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 739.7.2 Darstellung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 749.7.3 Verwendung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 749.7.4 Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 759.7.5 O�Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 759.7.6 Salze von Oxosäuren . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
9.8 Erdalkalimetalle: Gruppe 2 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 779.8.1 Vorkommen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 789.8.2 Darstellung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 789.8.3 Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
9.9 Borgruppe: Gruppe 13 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 809.9.1 Vorkommen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 819.9.2 Modi�kationen und Darstellung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 819.9.3 Verbindungen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 83
10 Anmerkungen 85

6 INHALTSVERZEICHNIS

Kapitel 1
Atombau
1g Kohlensto� + 1, 333g Sauersto� −→ Kohlenmonooxid
1g Kohlensto� + 2 ∗ 1, 333g Sauersto� −→ Kohlendioxid
1.1 Rutherford (1911)
02He
2+ Id−→ | 99%Id−→Goldfolie
Ergebnisse:⇒ Masse und positive Ladung im Kern⇒ Elektronen machen fast das gesamte Volumen aus
1.1.1 Atombausteine
Elektron Proton Neutron0−1e
11p
10n
Masse 9, 11 ∗ 10−31kg = 5, 5 ∗ 10−4u 1, 673 ∗ 10−27kg = 1, 007 ∗ 10−4u 1, 675 ∗ 10−27kg = 1, 009 ∗ 10−4uLadung −1, 6 ∗ 10−19C = −e −1, 6 ∗ 10−19C = +e −1, 6 ∗ 10−19C = +e︸ ︷︷ ︸
Kernbausteine = Nukleonen
1.2 Bohr'sches Atommodell (1913)
nur für Wassersto�
−FC = FZ
FC = − e2
4 ∗ π ∗ ε0 ∗ r2
FZ =m ∗ v2
r
⇒ m ∗ v2
r=
e2
4 ∗ π ∗ ε0 ∗ r2
Energie des Elekrtons
E = Ekin + Epot
=1
2m ∗ v2 − e2
4 ∗ π ∗ ε0 ∗ r
7

8 KAPITEL 1. ATOMBAU
=e2
8 ∗ π ∗ ε0 ∗ r− e2
4 ∗ π ∗ ε0 ∗ r
=e2
4 ∗ π ∗ ε0 ∗ r
⇒ Elektronen sind strahlungsfrei auf bestimmten Bahnen⇒ Energieniveaus (= Schalen) → K,L,M, ... (n = 1, 2, 3, ...)
Grundzustand: niedrigste Energieangeregte Zustände: durch Energiezufuhr → kehrt in Grundzustand zurück → Photon
1.2.1 Bohr'sches Postulat
Qunatelung der Energie
Bahndrehimpuls
m ∗ v ∗ r = n ∗ h
2 ∗ πh = 6, 626 ∗ 10−34Js
⇒ v =n ∗ h
2 ∗ π ∗m ∗ r
⇒ r =h2 ∗ ε0
π ∗m ∗ e2∗ n2 = 0, 053nm ∗ n2
v =1
n∗ e2
2 ∗ h ∗ ε0=
1
n∗ 2, 18 ∗ 106m
s
E = − m ∗ e4
8 ∗ h2 ∗ ε20∗ 1
n
Problem:
• keine Mehrelektronensysteme
• Widerspruch zur Heisenberg'schen Unschärferelation ∆x ∗∆(m ∗ v) > h∆v ≈ 104m
s → ∆x ≈ 70nm⇒ Elektron auch als Welle mit Aufenthaltswahrscheinlichkeiten
1.3 Atomorbitaltheorie
Wellencharakter des Elektrons
λ =h
m ∗ v
⇒ stehende Welle (Kreisbahnen): n ∗ λ = 2 ∗ π ∗ r
1.3.1 Schrödinger-Gleichung
⇒ Ergebnis:
• begrenzte Anzahl an Schwingungszuständen, die erlaubt sind
• räumliche Ladungs- und Energieverteilung
⇒ Atomorbitale beschrieben durch 4 Quantenzahlen

1.3. ATOMORBITALTHEORIE 9
1.3.2 Quantenzahlen
1. Hauptquantenzahl n:
• bestimmt Energieniveaus (Schalen)
• n = 1, 2, 3, ... (K,L,M, ...)
2. Nebenquantenzahl l:
• bestimmt Atomorbital-�Sorte�/-From
• l ≤ n− 1
3. magnetische Quantenzahl ml:
• bestimmt Anzahl
• −l ≤ ml ≤ +l
• Beispiele:
� l = 0→ s-Orbital 1 mal s� l = 1→ p-Orbital ml : −1, 0,+1 : 3 mal p-Orbitale
� l = 2 :→ d-Orbital ml : −2,−1, 0,+1,+2 : 5 mal d-Orbitale
4. Spinquantenzahl ms
• Besetzung eines Atomorbitals mit 2e− unterschiedlicher Spinquantenzahl
• ms = + 12 ,−
12
• Beispiele:
� 1s-OrbitalQuantenzahl: n = 1l l = 0 ms = + 1
2 2e− → ms = − 12
1.3.3 Elektronenkon�guration
Mehrelektronenatome
• Schalen, Orbitale wie bei Wassersto�
• Gestalt und Richtung maximaler Elektronendichte

10 KAPITEL 1. ATOMBAU
1.3.4 Besetzungsregeln
1. Pauli-Prizip:
• keine 2 Elektronen eines Atoms dürfen in allen Quantenzahlen übereinstimmen⇒ nur 2 Elektronen je Atomorbital
• maximale Zahl von Elektronen pro SchaleSchale e−-Zahl Orbitalen = 1 ⇒ 2e− 3sn = 2 ⇒ 8e− 3s+ 3pn = 3 ⇒ 18e− 3s+ 3p+ 3dn = 4 ⇒ 32e− 3s+ 3p+ 3d+ 3f
2. Hund'sche Regel:
Besetzung der Unterschalen, sodass die Zahl der Elektronen mit gleicher Spinrichtung ineiner Unterschale maximal wird→ Au�üllen der Unterschalen in energetischer Reihenfolge
1.3.5 Grundzustand
• erst s einfach, dann doppelt
• dann px, py, pz einfach, dann doppelt, etc.
• Beispiel:
� Li: 1s2 2s1 ∧= [He] 2s1
� N: 1s2 2s2 2p3 ∧= [He] 2s2 2p3
� Cl: 1s2 2s2 2p6 3s2 3p5 ∧= [Ne] 3s2 3p5
• Achtung: Au�üllend von d-Orbitalen erst nach den s-Orbitalen der Folgenden Perioden
• Nebengruppenelemente / Hauptgruppenelemente

Kapitel 2
Periodensystem der Elemente (PSE)
Elemente mit analoger Elektronenko�guration der äuÿeren Schale haben ähnliche Eigenschaften⇒ Gruppeneinteilung im PSE
Beispiel:
• 1. Gruppe: Alkalimetalle (Li, Na, K, Rb, Cs)
� Valenzelktronenkon�guration ns1
� Eigenschaften:
∗ sehr reaktiv∗ +I-wertige Ionen∗ weiche Leichtmetalle∗ niedrige Schmelztemperatur
• 7. Gruppe: Halogene (F, Cl, Br, I)
� Valenzelktronenkon�guration ns2np5
� Eigenschaften:
∗ sehr reaktionsfähig∗ -I-wertige Ionen∗ typische Nichtmetalle
Peridische Wiederholung von Elementeigenschaften (nach der Hauptquantenzahl)
2.0.6 Begri�e
• Hauptgruppen (d- und f-Orbitale sind vollständig oder gar nicht besetzt) (Gruppen 1, 2, 13 - 18)
• Nebengruppen (Besetzung der noch freien d-Orbitale) Übergangsmetalle (Gruppen 3 - 11)
• Lanthanoide (4f)
• Actinoide (5f)
2.1 Bezeichnung der ElementeNukleonenzahl→Ordnungszahl→
147N ← Elementsymbol
Ordnungszahl = Kernladungszahl = Protonenzahl = Elektronenzahl des neutralen Elements
Nuklid: Atomsorte einer festen Protonenzahl, aber unterschiedlicher Neutronenzahl
Beispiel: Nuklide des Wassersto�s 11H
21H
31H (Fehler in der Vorlesung!)
Isotope: Isotope gehören zu einer Elementsorte, ist also synonym zu Nuklid
Beispiel: Isotopedes Kohlensto�s 126 C
136 C
Isobare: Isobare gehören nicht zu einer Elementsorte und sind veschiedene Elenmente gleicher Masse
Beispiel: 146 C
146 N
11

12 KAPITEL 2. PERIODENSYSTEM DER ELEMENTE (PSE)
2.1.1 Atommasse
m =∑ni=1mi ∗ hi n: Zahl der Isotope
mi : Masse des jeweiligen Isotopshi : Häu�gkeit
Bezeichnung Charakteristikum Beispiele BemerkungIsotope gleiche Protonenzahl 12
6 C136 C
Isotone gleiche Neutronenzahl 136 C
147 N
Isobare gleiche Massenzahl 177 N
178 O
179 F wenn sehr schwer
⇒ β - ZerfallSpiegelkerne Neutronen- und Protonen- 3
1H32He siehe Isobare
zahl vertauschtIsodiaohare gleicher Neutronenüberschuss 12
6 C147 N
Isomere unterschiedliche innere Zustände 9943Tc
99m43 Tc
2.2 Eigenschaften der Elemente
Die Eigenschaften der Elemente ändern sich in Abhängigkeit von ihrer Stellung im PSE.
2.2.1 Atomradien
In einer Periode nehmen die Radien der Atome ab, weil die immer gröÿer werdernde Kernladung dieElektronenhülle immer stärker zusammenzieht. (siehe Zeff (e�ektive Kernladung) - Slaters Regeln). Ineiner Gruppe nehmen die Radien der Atome zu, da weitere Schalen hinzukommen und diese Volumen-vergröÿerung nicht völlig von der Kontraktion durch die wachsende Kernladungszahl ausgeglichen wird.
2.2.2 Ionisierungsenergie I
Die 1. Ionisierungsenergie ist die Mindestenergie, die benötigt wird, um ein Elektron vollständig aus demAtom zu entfernen. (X + I → X+ + e−).Die Ionisierungsenergie ist ein Maÿfür die Festigkeit, mit der das e− am Atom gebunden ist.Innerhalb einer Periode nimmt die Ionisierungsenergie aufgrund der zunehmenden Kernladung zu. BeiEdelgasen hat die Ionisierungsenergie jeweils ein Maximum (Edelgaskon�guration). Bei Alkalimetallensinkt die Ionisierungsenergie und weiÿt ein Minimum auf.
2.2.3 Elektronena�nität EA
Die Elektronena�nität ist die Energie, die frei wird (negative EA-Werte) oder benötigt wird (postiveEA-Werte), wenn an einem Atom ein Elektron unter Bildung eines negativ geladenen Ions angelagertwird. (Y + e− → Y − + EA).Achtung: Nicht mit Elektronennegativität zu verwechseln.

Kapitel 3
Chemische Bindungen
3.1 Kovalente Bindungen
Eine kovalente Einfachbindung besteht aus einem Paar von Elektronen, das zwei Atome gemeinsam an-gehört.
Beispiel: H2
Jedes H-Atom hat ein Elektron, das symmetrisch um den Atomkern verteilt ist. Die zwei H-Atome kom-men zusammen, die Atomorbitale (1s) überlagern sich, sodass die Elektronenwolke im Bereich zwischenden Atomkernen dichter wird.Skizze
H⊙⊙
H → H2 = (H −H)
Obwohl die Elektronen dem Molkül als ganzem gehören, ist jedes H-Atom an zwei elektronen beteiligtund hat damit eine Elktronenkon�guration, die derjenigen des nächsten Edelgases entspricht.→ Edelgasregel: Die Lewis-Theorie betont das Erreichen der Edelgaskon�guration als Ziel für jedes Atom.
3.1.1 Lewis-Formeln
Molkülstrukturen können als Strichformeln gezeichnet werden, Jeder Bindestrich zwischen zwei Atomensymbolisiert ein gemeinsames Elektronenpaas. Die übrigen Valenzelektronen werden als Punkte oderStriche angegeben.
(8-N)-Regel
Die Zahl der kovalenten Bindungen ergibt sich oft aus der Zahl der Elektronen, die noch fehlen, um diekon�guration des nächsten Edelgases zu erreichen. Da bei den Nichtmetallen die Zahl der Valenzelektronengleich der Hauptgruppennummer ist (alte Nomenklatur) werden 8-N Elektronen benötigt.
Beispiele
• F2:Fluor ist in der 7. Hauptruppe.Durch Bildung einer kovalenten Bindung von zwei Fluoratomen kommt jedes zu einem Elektrone-noktett
(Elektronenkon�guration von Ne)
• N2:Mehrfachbindungen können entstehen, wenn mehr als zwei Elektronen zu einer Elektronenpaarbin-dung zusammentreten. Ein N-Atom besitzt drei ungepaarte Valenzelektronen
(N 1s22s22p3)
13

14 KAPITEL 3. CHEMISCHE BINDUNGEN
• NO: ·_
N ≡ O| ⇒ Radikal (ein ungepaartes Elektron)
Um Lewis-Formeln im Einklang mit der Oktett-Regel richtig zu formulieren, muss die Gesamtzahl derValenzelektronen so auf bindende und einsame Elektronen aufgeteilt werden, dass jedes Atom 8 Elektro-nen (4 Paare) und jedes H-Atom von zwei Elektronen umgeben ist. Die Anzahl der Elektronen, die anBindungen beteiligt sind, ergibt sich folgendermaÿen:
Anzahl der Bindungselektronen= 2 * (Anzahl der H-Atome) + 8 * (Anzahl der übrigen Atome) - (Gesamtzahl der Valenzelektronen)
Beispiel: H2CO Bindungeselektronen:
2 ∗ 2+ 8 ∗ 2− 12
8
3.2 Formalladungen (Elektronenbuchhaltung)
Ein Atom, an dem der Lewis-Formal so viele Bindungsstriche zusammenkommen, wie nach der (8-N)-Ragel zu erwarten sind, hat keine Formalladung. Wenn nicht, muss dies gekennzeichnet werden.→ Formalladung
Beispiele
• CO-MolekülValenzelektronen: 4 + 6 = 10Nach den Regeln muss eine Dreifachbindung formuliert werden. Durch die gleichmäÿige Aufteilungder Bindungselektronen erhält sowohl das C-Atom, als auch das O-Atom 5 Elektronen. Also hatdas C-Atom ein e− zu viel und das O-Atom ein e− zu wenig.
|C ≡ O|⊕
• HNO3-MolekülDas N-Atom ist von 3 O-Aoomen ungeben. An einem O-Atom ist das H-AtomValenzelektronen: 1(H) + 5(N) + (3 ∗ 6)(O) = 24Bindungselektronen: (2 ∗ 1) + (8 ∗ 4)− 24 = 10 (5 Paare)
Mesomerie: Beide Formen sind gleichwertig → Grenzformen
3.3 Mesomerie
Wenn mehrere gleichwertige Formeln angegeben werden können, gelten diese als Grenzformeln (Mesome-rie)
Bedingungen
1. Die räumliche Anordnung der Kerne muss erhalten bleiben.
2. Zwei aneinander gebundene Atome sollen keine Formalladungen mit gleichem Vorzeichen haben.
3. Die Wichtigsten Grenzformeln sind die, mit der kleinsten Anzahl der Formalladungen.
4. Das elektronennegativste Atom sollte keine positive Formalladung erhalten (siehe aber CO)

Kapitel 4
Reaktionen
Bei einer Chemischen Reation �ndet eine Umverteilung von Atomen statt. Dabei erfolgt eine sto�icheVeränderung und auch ein Energieumsatz.
• Wenn Energie benötigt wird, läuft die Reaktion endothermisch ab.Die Reaktionswärme (in Joule)ist mit positivem Vorzeichen als +∆H angegeben.
Beispiel: Ba(OH)2 ∗ 8H2O +NH4SCN −→ Ba2+ + 2SCN− + 2NH3 + 10H2O + ∆H
• Wenn Energie freigesetzt wird, läuft die Reaktion exothermisch ab. Die Reaktionswärme (in Joule)ist mit nagativem Vorzeichen als −∆H angegeben.
Beispiel: CaCl2 + 6H2O −→ CaCl2 ∗ 6H2O −∆H
Die Reaktion von Eisen mit Luftsauersto�:
3Fe+ 2O2 −→ Fe3O4
3 ∗ 56u+ 4 ∗ 16u −→ 3 ∗ 56u ∗ 4 ∗ 16u
168u+ 64u −→ 168u+ 64u
⇒ Masse bleibt erhalten
4.1 Aktivierungsenergie
Exotherme Reationen, die aber gehemmt sind, beötigen eine Aktivierung.Beispiel:
2H2 +O2 −→ 2H2O −∆H
wobeiE1 die AktivierungsenergieE2 die Reaktionsenthalpie ∆H
ist.
4.2 Entropie (Unordnung)
Beispiel:Ba(OH)2 ∗ 8H2O +NH4SCN −→ Ba2+
(aq) + 2SCN−(aq) + 2NH3(g) + 10H2O
⇒ Entropie hat zugenommenDie Enthalpie ∆H ist positiv, aber die freie Enthalpie ∆G ist negativ. Gibbs-Helmholtz-Gleichung:
∆G = ∆H − T ∗∆S ∆G∧= freie Enthalpie T
∧= Temperatur [K] ∆S
∧= Entropie
4.3 Standardbildungenthalpie
Die Standardbildungsenthalpie ist die Enthalpie, die bei der Bildung einer Verbindung aus den Elementenunter Standardbedingungen frei oder benötigt wird.
15

16 KAPITEL 4. REAKTIONEN
Beispiel
C +O2 −→ CO2∆H0f = −393, 8
kJ
mol
Für Elemente in ihrem bei Standardbedingungen stabilen Zustand setzt man die Standardbildungsent-halpien gleich Null. Z.B. ∆H0
f = 0 für Wassersto� als H2, Sticksto� als N2, Kohlensto� als Graphit (nichtals Diamant)
Allgemein
∆H0 =∑
∆H0f (Produkte)−
∑∆H0
f (Edukte)
Beispiele
1. Bildung von Synthesegas aus Kohlensto� und Wasser:
C(s) +H2O(g) −→ CO(g) +H2(g)
∆H0f (H2O(g)) = −241, 8 kJ
mol ∆H0f = (CO(g)) = −110, 6 kJ
mol
∆H0 =∑
∆H0f (Produkte)−
∑∆H0
f (Edukte)
= (−110, 6 + 0)− (0 + (−241, 8)) kJmol = 131, 2 kJmol (endotherm)
2. Reduktion von Eisen(III)-oxid durch Kohlenmonoxid:
Fe2O3(s) + 3CO(g) −→ 2Fe(s) + 3CO2(g)
∆H0f (Fe2O3(s)) = −824, 8 kJ
mol ∆H0f = (CO(g)) = −110, 6 kJ
mol ∆H0f = (CO2(g)) = −398, 8 kJ
mol
∆H0 =∑
∆H0f (Produkte)−
∑∆H0
f (Edukte)
= (2 ∗ 0 + 3 ∗ (−398, 8))− (−824, 8 + 3 ∗ (−110, 6)) kJmol = −24, 8 kJmol (exotherm)
4.4 Elektronennegativität
Unter der Elektronegativität χ versteht man die Fähigkeit eines Atoms, die Elektronen einer Atom-bindung zu sich herüber zu ziehen. Elektronennegativität kann in verschiedenen Weisen errechnet undtabelliert werden. Die Pauli-Zahlenwerte werden am häu�gsten verwendet. Je gröÿer der Zahlenwert fürdie Elektronegativität ist, um so stärker zieht das jeweilige Atom Bindungselektronen an.
Beispiel:H2O,χ(H) = 2, 20 χ(O) = 3, 44
Die bindenden Elektronenpaare sind zu Sauersto� hin verschoben. Das Molekül ist wegen seines gewinkel-ten Aufbaus ein Dipol mit negativer Ladung bei O und positiver Ladung bei H. χ zeigt im Periodensystemeinen systematischen Gang. Die Elektronegativität nimmt innerhalb einer Hauptgruppe von oben nachunten ab, in einer Periode nimmt sie von links nach rechts zu. Die Polarität einer Bindung läÿt sich ausder Di�erenz Dc der beteiligten Atome abschätzen. Ist ∆χ hinreichend groÿ, dann kann das bindendeElektronenpaar ganz dem elektronegativeren Bindungspartner zugeordnet werden. Dann ergibt sich eineIonenbindung. Ionenbindung liegt in der Regel in Verbindungen der elektropositiven (χ� 1). Metalle derersten und zweiten Hauptgruppe vor, die durch Paarung von Valenzelektronen keine Edelgaskon�gurationerreichen würden.
Beispiel:NaCl : χ(Na) = 0.93 χ(Cl) = 3, 16∆χ = 2, 23
Es ist nicht Na-Cl zu formulieren, sondern das bindende Elektronenpaar ist dem Cl-Atom zugeordnet, essind Ionen entstanden: Na+Cl−.
Schema der Werte von χ im Periodensystem:

4.5. OXIDATIONSZAHLEN 17
4.5 Oxidationszahlen
Im Fall der Ionenverbindung NaCl ist die Ladung der Ionen gleich der Oxidationszahl der Atome. Oxi-dationszahlen können auch bei Verbindungen mit Atombindungen (kovalente Verbindungen) verwendetwerden. Oxidationszahlen sind Ladungen oder �ktive Ladungen, die den Atomen einer Verbindung nach-bestimmten Regeln zugewiesen werden. Um Verwechslungen in Fällen zu vermeiden, in denen die Oxi-dationszahl nicht einer tatsächlichen Ionenladung entspricht, verwendet man römische Zahlen für dieOxidationszahlen.
Die Regeln
1. Bei einatomigen Ionen ist die Oxidationszahl gleich der Ladung.
2. Bindungselektronen zwischen gleichartigen Atomen werden zu gleichen Teilen zwischen diesen ver-teilt. Elemente erhalten die Oxidationszahl 0.
3. Die Summe der Oxidationszahlen aller Atome eines mehratomigen Ions ist gleich der Ladung diesesIons. Bei neutralen Verbindungen ist sie 0.
4. Fluor besitzt als elektronegativstes Element in allen Verbindungen die Oxidationszahl -I.
5. Sauersto� hat in den meisten Verbindungen die Oxidationszahl -II. Ausnahmen sind Peroxide (-I);Hyperoxide (− 1
2 ) und in OF2 (+II nach Regel 4).
6. Wassersto� besitzt in den meisten Fällen die Oxidationszahl +I. In Metallhydriden wie NaH hatWassersto� die Oxidationszahl -I.
7. In Verbindungen der Nichtmetalle ist die Oxidationszahl des elektronegativeren Elements negativund entspricht der Ionenladung, die für Ionenverbindungen dieses Elements gilt.
Beispiel
SO2−4 χ(O) > χ(S)
+I
H2
+V I
S−IIO4 [
+V I
S−IIO4 ]2−
4.6 Polare Bindungen
Wenn sich im Periodensystem weit links stehende Elemente mit weit rechts stehenden Elementen verbin-den, entstehen Ionenverbindungen (Wenn ∆χ >2)
Beispiel
2Na(s) + Cl2(g) −→ Na+Cl−(s)
Hiermit erreichen Kationen und Anionen Edelmetallkon�guration:
Na(1s22s22p63s1) −→ Na(1s22s22p6) + e−∧= [Ne]
Cl(1s22s22p63s23p5) −→ Cl(1s22s22p63s23p6)∧= [Ar]
Zwischen den entstandenen Ionen sind elektrostatische Kräfte wirksam:
Coulombsche KraftFC ∼
q1 ∗ q2
r2
←Ladung←Abstand der Ionen
Die elektrostatischen Kräfte sind ungerichtet und das von den Ionen ausgehende elektrische Feld ist inallen Raumrichtungen gleich. Ein Kation wie Na+ wird mehr als nur ein Cl− binden und auch umgekehrtwird ein Anion wie Cl− mehr als nur ein Na+ binden. Es entsteht ein Ionengitter.Im Fall von NaCl sind die Na+-Ionen von jeweils sechs Cl−-Ionen in Form eines Oktaeders umgeben,ebenso sind die Cl−-Ionen von sechs Na+-Ionen koordiniert. Die jeweils sechs Abstände zum nächstenNachbarn sind in der NaCl-Struktur gleich. Es bestehen gleich groÿe Bindungskräfte zu jedem Nachbarn.Die Zahl der nächsten Nachbarn um eine Atomsorte heiÿt die Koordinationszahl KZ. Sie ist eine cha-rakteristische Gröÿe für eine Kristallstruktur. Für eine (binäre) Verbindung der Zusammensetzung ABn(n = 1, 2, 3) wird KZ für beide Atomsorten angegeben.Für NaCl ergibt sich KZ 6 : 6. 10.11.11

18 KAPITEL 4. REAKTIONEN
4.7 Ionengitter
Im Fall von NaCl sind die Na+ �Ionen von jeweils 6 Cl− �Ionen in Form eines Oktaeders umgeben.Ebenso sind Cl− �Ionen von 6 Na+ �Ionen koordiniert.
Die jeweils 6 Abstände zum nächsten Nachbarn sind in der NaCl�Struktur gleich. Es bestehen gleichgroÿe Bindungskräfte zu jedem Nachbarn. Die Zahl der nächsten Nachbarn heiÿt Koordinationszahl(KZ) Für NaCl: KZ 6:6
Für eine binäre Verbindung der Zusammensetzung ABn (n=1,2,3) wird KZ für beide Atomsortenangegeben.
4.8 Gitterenergie
• beim Zusammenfügen von unendlich entfernten, im Gaszustand be�ndlichen positiv und negativgeladenen Ionen zu einem Kristall wird die Gitterenergie frei.
4.8.1 Beispiel
Na+(g) + Cl−(g) → NaCl(s) ∆HGitter = −788
kJ
mol
4.8.2 Born�Haber�Kreisprozess
Zur Bestimmung der Gitterenergie bedient man sich des Born�Haber�Kreisprozesses, der von Max Bornund Fritz Haber unabhängig voneinander 1916 entwickelt wurde.
• Basiert auf dem Satz von Hess: Die Reaktionsenthalpie einer chemischen Reaktion hat einen festenBetrag, unabhängig davon, in wie vielen (und welchen) Schritten die Reaktion abläuft
4.8.3 Born�Haber�Kreisprozess für NaCl
Na(s) + 1/2Cl2(g) → NaCl(s) ∆H0f (NaCl) < 0
∆H0f = ∆HSub + I.P.+ 1/2∆HDiss + E.A.+ ∆HGitter
∆HGitter = ∆Hf −∆HSub − I.P.− 1/2∆HDiss − E.A.
∆HGitter = −411− 108− 496− 122− (−349) = −788kJ
mol
Abkürzungen:∆HSub Sublimationsenergie I.P. Ionisierungspotential∆HDiss Dissoziationsenergie
Der erste Schritt beim Aufbau eines Ionnkristalls ist die Bildung eines Ionenpaars Na+Cl− Bei derAnnäherung von Na+ und Cl− �Ionen:
1. sinkt das Coulomb�Potential ( q2
r →e2
r )
2. steigt ein abstoÿendes Potential schnell an

4.8. GITTERENERGIE 19
Um in ähnlicher Weise den Coulombanteil (Uc) an der Gitterenergie berechnen zu können, ist dieUmgebung der Ionen zu beachten Für ein Na+ �Ion in NaCl besteht diese Umgebung aus:
• 6Cl− im Abstand d
• 12Na+ im Abstand√
2d
• 8Cl− im Abstand√
3d
• 6Na+ im Abstand√
4d
• 24Cl− im Abstand√
5d
•...
Die Coulomb-Energie des NaCl-Gitters ergibt sich als Summe der Einzelbeiträge:
z+z−r
wobei die Häu�gkeit der Abstände zu beachten ist. Für �r�werden die entsprechenden Abstände alsVielfache von d eingesetzt. z+ , z− sind die Ladungen (hier: z+ = z− = 1)
Anziehende Energiebeiträge positiv, abstoÿende negativ anzugeben.
U = −z+z−e2
d((6− 12√
2) + (
8√3
)− (6√4
) + (24√
5) . . . )
Der Klammerausdruck, der Grenzwert für unendlich viele Summanden, ist die Madelung-Konstante,die nur von der Gittergeometrie abhängt. Für den NaCl�Strukturtyp ist der Zahlenwert 1,748
auf 1 Mol NaCl bezogen:
Uc = −NA ∗Mh+z−e
2
dM → Madelung-Konstante
Uc ist näherungsweise gleich der UGitterWerden verschiedene Verbindungen gleicher Struktur betrachtet, so ergibt sich aus der Betrachtung
ders Coulomb�Anteils eine Vergröÿerung der Gitterenergie bei:
1. eine Erhöhung der Ladung der Ionen (vgl. NaCl und BaO)NaCl (d=238 pm), z+ = z− = 1; UGitter = −788 kJ
mol
BaO (d=276pm), z+ = z− = 2; UGitter = −3128 kJmol
2. eine Verkleinerung des Abstands der Ionen (vgl. BaO und MgO)BaO (d=276pm), z+ = z− = 2; UGitter = −3128 kJ
mol
MgO (d=212pm), z+ = z− = 2; UGitter = −3936 kJmol
Gitterenergien sind ein Maÿ für der Särke der Bindungen im Ionenkristall.UGitter[
kJmol ] Smp. [◦C] Ritzhörte (Mohs)
NaCl −788 80 2, 5BaO −3128 1925 3, 3MgO −3936 2642 6, 0

20 KAPITEL 4. REAKTIONEN

Kapitel 5
Metalle
15.11.1175% der Elemente des Periodensystems sind Metalle
5.1 typische Eigenschaften
• Die Absorption für sichtbares Licht ist hoch. Hieraus folgt das groÿe Spiegelre�exionsvermögen. Dasist die Ursache für den metallischen Glanz und Undurchsichtigkeit.
• gute elektrische Leitfähigkeit
• gute Wärmeleitfähigkeit
• Metalle sind leicht duktil (verformbar).
Unterschied zu Sto�en mit ionischer Bindung bzw. kovalenter Bindung: Die Halbmetalle zeigen Metall-glanz aber unterscheiden sich von den Metallen durch die Gröÿe und die Temperaturabhängigkeit derelektrischen Leitfähigkeit. (Elemente: B, Si, Ge, As, Sb, Te) Der metallische Charakterin einer Gruppedes PSE nimmt von oben nach unten zu.
5.2 Die Metallbindung
5.2.1 Bsp. Na�Metall
Na hat 1 Valenzelektron. Für die Bindung zwischen Na�Atomen scheint weder die Ionenbindung nochdie Atombindung eine befriedigende Lösung zu sein. Die Ionenbindung benötigt Na+ und Na−�Ionen(ungünstige Elektronena�nität von Na) Die Atombindung ergibt keine keine Edelgasschale(Na � Na) Die Bindung zwischen den Na�Atomen kann man anhand eines Ëlektronengasmodells �verstehen.Jedes Na�Atom trägt sein Valenzelektron zu diesem Elektronengas bei, in das Na+�Ionen eingebettetsind. Die Bindung ergibt sich als elektrostatische Anziehung zwische den Na+�Ionen und der nega-tiven Ladungsdichte zwischen den Atomrümpfen. Bei dieser Betrachtungsweise entspricht das Prinzipder Metallbindung dem der Atombindung. Der Unterschied besteht in der räumlichen Ausrichtung derBindungselektronen bei der Atombindung, während bei der Metallbindung die wenigen zur Verfügung ste-henden Elektronen mit mehreren Atomrümpfen wechselwirken können. Die hohe elektrische Leitfähigkeitzeigt die Existenz von freibeweglichen Ladungsträgern (Elektronen).
5.3 Metallstrukturen
Die wichtigsten Metallstrukturen ergeben sich durch die dichtest-mögliche Packung gleichartiger Kugeln.
Draufsicht: Seitenansicht:
In der Ebene lassen sich um eine Kugel (Atom) maximal 6 gleich groÿe Kugeln (Atome) herumle-gen.Werden nun 2 Schichten möglichst raumsparend übereinandergelegt, so berühren in der �auf�Lücke�Stellung� weitere 3 Kugeln aus der 2. Schicht die zuerst betrachtete Kugel. Eine auf der anderen Seiteebenfalls auf Lücke angelegte 3. Schicht vergröÿert die Koordinationszahl der betrachteten Kugel auf denWert 12 nächste Nachbarn. 2 Möglichkeiten:
21

22 KAPITEL 5. METALLE
kubisch dichteste Packung (kdp) hexagonal dichteste Packung (hdp)
1. hdp, A3-Typ, Magnesium-Typ (⇒ ABAB-Stepelung)
2. kdp, A1-Typ, Kupfer-Typ (⇒ ABCABC-Stapelung)
3. A2-Typ, Wolfram-Typ, kubisch-raumzentrierte Struktur. Die KZ eines Atoms ist 8+6 (8 nächsteund 6 übernächste Nachbarn, die etwa 15 % weiter entfernt sind; diese sind selbst wieder Zentrenvon solchen Kuben):
• A1�Typ, kdp � Elementarzelle � Flächenzentrierter WürfelDie Stapelrichtung ist die Raumdiagonale. Bei kdP und hdP ist die Raumfüllung dieselbe und läÿtsich leicht für die kdP herleiten:
Volumen der Elementarzelle:V = a3
Kantenlänge
Zu der Elementarzelle zählen 8 Atome (in den Ecken), die nur zu einem Achtel innerhalbdieser liegen und 6 Atome (auf den Flächen), die zur Hälfte innerhalb der ElementarzelleliegenDaraus ergibt sich eine e�ektive Zahl von Atomen, die innerhalb der Zelle liegen:
Zeff =8
8+
6
2= 4
Das Volumen der Atome ist also:
VAtome = 4 ∗ 4
3πr3 =
16
3πr3
Mit r = a∗√
24 ergibt sich dann:
16
3πr3 = f ∗ a3
r =a
2∗ 3
√3 ∗ f2 ∗ π
f =16 ∗ π ∗ r3
3 ∗ a3= 74%
⇒ r =a
2∗ 3
√3 ∗ f2 ∗ π
≈ a ∗√
2
4
17.11.1117.11.11
A1�Typ: • kubisch dichteste Packung (kdP)
• Raumfüllung: 74 %
A2�Typ: • Innenzentrierte kubische Elementarzelle (→ Wolframtyp)
• Raumfüllung: 68%
A3�Typ: • hexagonal dichteste Packung (hdP)
• Raumfüllung 74 %

5.3. METALLSTRUKTUREN 23
⇒ 80 % der metallischen Elemente kristallisieren in den Strukturen A1, A2, A3. Viele Metalle sindpolymorph. In Abhängigkeit von der Temperatur kristallisieren sie in mehr als einer Struktur.
z.B. α−Fe(A2) 906◦C←→ γ−Fe(A2) 1401◦C←→ δ−Fe(A2) 1536◦C←→ Fe(l)
Eisen ist in der Form γ�Fe duktiler und damit leichter bearbeitbar als in der α�Form
Mit der Vorstellung der dichtest gepacktesten, gleichartigen Kugeln, die durch ungerichtete Kräftemiteinander verbunden sind, läÿt sich die Duktilität der Metalle verstehen. Eine Verschiebung von Atom-rümpfen ist möglich, ohne dass vermehrte elektrostatische Abstoÿung auftritt und ihne dass gerichteteBindungen aufgehoben werden. Feste Verbindungen, deren Kristallstruktur durch Atombindungen zu-sammengehalten werden (z.B. Diamant, Silicium) sind spröde, da es bei einer Scherung einer Atomreihezum Bruch von Elektronenpaarbindungen kommt.
Ionenkristalle sind spröde, da beim gleichen Vorgang bereits nach der Verschiebung um nur einenAtomdurchmesser gleichnamige Ladungen in Kontakt gebracht werden können.
Hohe Duktilität wird begünstigt, wenn in den Kristallitin (kleine Kirstallbausteine) der polykristal-linen Metallstücke zum einen die Gleitebenen möglichst glatt sind. Dies ist gleichbedeutend mit einermöglichst dichten Packung auf der Ebene und wenn zum andern glatten Ebenen in möglichst unter-schiedlichen Orientierungen auftreten.
Im A1�Typ sind beide Forderungen am besten erfüllt:
• dichtest gepackte Schichten
• kubische Elementarzelle mit der Packung senkrecht zu den 4 Raumdiagonalen
A3�Typ:
• dichtest gepackte Schichten
• aber nur in einer Orientierung
A2�Typ:
• kubisch aber keine dichteste Packung

24 KAPITEL 5. METALLE

Kapitel 6
Lösungen
6.1 Inhalt
• Solvatation, Hydratation
• Thermodynamik
• Beein�ussung der Löslichkeit durch Temperatur und Druck
• Lösungen sind homogene Gemische
• Die Komponente mit dem gröÿten Mengenanteil wird Lösungsmittel genannt
• Die Menge eines gelösten Sto�es in einer gegebenen Menge Lösungsmittel nennt man Konzentra-tion
• Eine Lösung, in der die maximal au�ösbare Menge eines Sto�es enthalten ist, heiÿt gesättigteLösung
• Lösungen mit geringerer Konzentration sind ungesättigte Lösungen
6.2 Konzentrationsmaÿe
Molarität:Anzahl Mole eines Sto�es, die in einem Volumen von 1 Liter vorhanden sind. z.B.: 1 l wässrige HCl-Lösungenthält 0,2 mol HCl. c(HCl) = 0,2 molar = 0,2 M für eine 0,2 M HCl-Lösung muss man (Konzentration)
(M(H) +M(Cl)) ∗ cl = 36, 461g
mol∗ 0, 2mol
6.3 Struktur von H2O
O�Atom: 6 Valenzelektronen → 2s22p4
Hybridisierung der 2s und 2p�Orbitale: Hybridisierung → 4 ∗ sp3�Orbitale (Abb.3) → tetraedrischerAufbau wegen maximalem Abstand zwischen den einzelnen sp3�Orbitalen. Der ideale Tetraeder-Winkelist 109◦. Aber die Freien Elektronenpaars brauchen mehr Platz und drücken damit die H-Atome zusam-men (→ 105◦)
Durch zusätzliche Bindungen (H�Brücken) ist Wasser �üssig:
Bei Eis gibt es 12 unterschiedliche Modi�kationen. 22.11.11
25

26 KAPITEL 6. LÖSUNGEN
6.4 Wasser als Lösungsmittel
Wasser ist bei Raumtemperatur �üssig Die Existenz der Wassersto�brücken ist die Ursache mancherBesonderheiten.
Molekül Masse [u] Schmelzpunkt [◦C] Siedepunnkt [◦C]H2O 18, 02 0 +100H2S 34, 08 −85 −60H2Se 80, 98 −60 −41H2Te 129, 62 −49 −2
H2O O ist sp3�Hybridisiert → H�BrückenWinkel zwischen den beiden H�Atomen: 105◦ (H2O) → bei gröÿeren Atomen (S, Se, Te) wird der
Winkel kleiner (90◦C)Grund für unterschiede: Energieniveau�Unterschiede zwischen s� und p�Orbitalen (bei Sauersto�
geringerer Unterschied) → sp3�Hybridisierung bei weiteren Atomen nicht möglich.
6.4.1 Eigenschaften
Wasser ist auch ein sehr wichtiges Lösungsmittel für ionisch aufgebaute und polare Sto�e, indem sich diepolaren Wassermoleküle um die geladenen Teilchen des gelösten Sto�es herumlagern:Hydratation
Beispiel:NaCl→ Na+
(aq) + Cl−(aq)
Na+(aq) stehtfür ein Ion des Typs [Na(H2O)6]+, bei dem 6 Wassermoleküle in Form eines Oktaeders
mit ihren negativen geladenen O�Atomen an des Na+�Ion koordiniert sind.Wieviel Mol Wasser sind in einem Liter Wasser?
→ n =1000g
18 gmol
= 55, 4939 mol
6.5 Elektrolyt� und nicht Elektrolyt�Lösungen
Wenn eine wässrige Lösung freibewegliche Ionen enthält, sprechen wir von einer Elektrolyt�Lösung.Die Ionen folgen dem elektrischen Feld, wenn eine Spannung an in die Lösung tauchende Elektrodenangelegt wird.
Die positiv geladenen Ionen, die Kationen, wandern zu rKathode. Die negativ geladenen Ionen, dieAnionen wandern zur Anode Die Ionen sind die Träger der elektrischen Ladung.
Nichtelektrolytlösungen kommen zustande, wenn nichtionische Sto�e, die zur Ausbildung von Was-sersto�brückenbindungen befähigt sind, in Wasser aufgelöst werden. In der sind diese Verbindungen, dieOH�Gruppen enthaltenwie Alkohol, Ethylenglykol, Zucker und auch Säuren wie Schwefelsäure. Beispielefür Verbindungen mit anderen polaren Gruppen, die auch in Wasser löslich sind, sind Harnsto�, Acetonund Formaldehyd.
Unpolare Verbindungen sind in der Regel nicht mit Wasser mischbar. Z.B. Öl, halogenierte Kohlen-wassersto�moleküle wie CCl4, CHCl3
6.6 Lösungsenthalpie
Wenn eine Substanz in einem Lösungsmittel gelöst wird, wird Energie freigesetzt (−∆H) oder aufgenom-men (+∆H). Bei konstantem Druck und o�enem Gefäÿ entspricht die se Energie der Lösungsenthalpie.
6.6.1 z.B.: Au�ösung von KCl
1. Die Energie, die gebraucht wird, um die Kristallstruktur des KCls unter Bildung gasförmiger Ionenaufzubrechen. ( = Gitterenergie)
KCl(s) → K+(g) + Cl−(g)∆H = 701, 2
kJ
mol
2. Die freigesetzte Hydratationsenthalpie bei der Bildung von hydratisierten, gelösten Ionen aus dengasförmigen Ionen
K+(g) + Cl−(g) → K+
(aq) + Cl−(aq)∆H = −684, 1kJ
mol

6.6. LÖSUNGSENTHALPIE 27
Gesamt:
KCl(s) → K+(aq) + Cl−(aq)∆H = +17, 1
kJ
mol
→ Lösungsenthalpie
zu 2.: Die Hydratationsenthalpie ist genau genommen die Summe von 3 Energiewerten:
1. Die noptwendige Energie, um einige Wassersto�brücken im Wasser zu lösen
2. die freigesetzte Energie bei der Hydratation der Kalium�Ionen
3. die freigesetzte Energie bei der Hydratation der Chlorid�Ionen
Lösung enthält: [Cu(NH3)4]2+ (blau)CrO2−
4 (gelb)

28 KAPITEL 6. LÖSUNGEN

Kapitel 7
Energetik
24.11.117.1 Entropie
Die Näherung exotherme Reaktionen laufen (spätestens nach Aktivierung) freiwillig ab, endotherme Re-aktionen müssen durch Energiezufuhr erzwungen werden gilt nur für Reaktionen mit groÿer ∆H.
Bisher haben wir nur den 1. Hauptsatz der Thermodynamik berücksichtigt.
Energie kann umgewandelt, aber weder erzeugt noch vernichtet werden.
2.Hauptsatz der Themodynamik
Eine Aussage über, ob ein Vorgang freiwillig ablaufen wird. Hier ist die Entropie S von zentraler BedeutungBei einer spontanen Zustandsänderung vergröÿert sich die Entropie. Freiwillig stellt sich somit immer nurein Zustand mit geringer Ordnung ein.
∆Sges = ∆SSys + ∆Sumg
Alle Entropiee�ekte müssen berücksichtigt werden. Die Gesamtänderung der Entropie (∆Sges) ist dieSumme aus der Entropieänderung des Systems (∆SSys) und der Umgebung (∆SUmg)
7.1.1 Beispiel: Entropieänderung für die Transformation Wasser → Eis
(bei normalem Durck) ∆S in Jmol∗K
Temp. [◦C] ∆SSys ∆SUmg ∆Sges+1 -22,13 +22,05 −0, 08← ∆Sges < 0
→ Wasser gefriert nicht0 -21,99 +21,99 0,00 ← ∆Sges = 0
→ weder Schmelzen noch Gefrieren sind begünstigt → Gleichgewicht-1 -21,85 +21,93 +0, 08← ∆Sges > 0
→ Wasser gefriert spontan
Die Zunahme der Gesamtentropie kann als Kriterium für das freiwillige Ablaufen eines Vorgangsdienen. Die Entropie des Universums nimmt ständig zu. Nach Rudolf Clausius können die ersten beidenHauptsätze zusammengefasst werden:
• Die Energie des Universums ist konstant. Die Entropie des Universums strebt einem Maximum zu.Gibbs-Helmholtz-Gleichung
∆G = ∆H − T ∗∆S
∆G: freie Reaktionsenthalpie∆G < 0 läuft die Reaktion freiwillig ab. ∆G == ist das System im Gleichgewicht ∆G > 0 läuft die
Reaktioon nicht freiwillig ab.Jetzt können wir verstehen, warum sich HCl in Wasser au�öst:
Sto� ∆H T ∗∆S ∆G[ kJmol ]KCl +17,1 +24,1 -7,0AgF -20,5 -5,8 -14,7
29

30 KAPITEL 7. ENERGETIK
7.2 Freie Standardbildugsenthalpie
Die freie Standardbildungsenthalpie ∆G0f kann man berechnen, als Summe der freien Standardbildungs-
enthalpien der einzelnen Reaktanden. Die erhaltenen ∆G0-Werte gelten für Standardbedingungen (Druckund Temperatur) aber auch in Bezug auf die Aktivitäten (näherungsweise Konzentrationen) der Reak-tanden.
Für eine allgemeine Reaktion:
aA+ bB → xX + yY
Für everdünnte Lösung:
∆G = ∆G0 +RT.ln[x]x ∗ [Y ]y
[A]a ∗ [B]b
Na+ + Cl− → NaCl
∆G = ∆G0 +RTln[NaCl]
[Na+][Cl−]; [Na+] = konz.
7.3 Beein�ussung der Löslichkeit durch Temperatur und Druck
Wie sich eine Temperatur- bzw Druckänderung auf die Löslichkeit einer Substanz auswirkt hängt davonab, ob beim Herstellen einer gesättigten Lösung Energie freigesetzt oder aufgenommen wird.
Beispiele
Wir haben eine Lösung, die sich im Gleichgewicht mit ungelösten Bodenkörper be�ndet (gesättigte Lö-sung). Zur Herstellung der Lösung sei die Zufuhr von Energie notwendig. Bei einer Erhöhung der Tem-peratur wird Wärme aufgenommen, wenn ein Teil des Bodenkörpers in Lösung geht.
Na+(aq) + Cl−(aq) ⇔ NaCl(s)
Aa(aq) +Bb(aq) ⇔ AB(s); ∆G < 0
Bei Temperatur-Erhöhung wird das Gleichgewicht nach links verschoben
Aa(aq) +Bb(aq) ⇔ AB(s); ∆G > 0
Bei Temperatur-Erhöhung wird das Gleichgewicht nach rechts verschoben Das Prinzip des kleinstenZwangs �Le Châtelier�'s Prinzip
Bei Temperaturerniedrigung weicht das System aus, indem ein Vorgang mit Energieabgabe verläuftund gelöster Sti� scheidet sich ab.
29.11.1129.11.11
7.4 Das Massenwirkungsgesetz
Wenn Substanzen miteinander eine reversible chem. Reaktion eingehen, so stellt sich ein dynamischerGleichgewichtszustand ein. Die Reaktionsgeschwindigkeiten der Hinreaktion und der Rückreaktionsind gleich. Die Konzentrationen aller beteiligten Substanzen bleiben konstant. Die Konzentrationenstehen zueinander in einem Verhältnis, welches durch das Massenwirkungsgesetz (MWG) erfasst wird.Für die allgemeine Reaktion
aA+ bB xX + yY
lautet das MWG:
K =[X]x ∗ [Y ]y
[A]a ∗ [B]b
Gleichgewichtskonstante K ist temperaturabhängigSie ist unabhängig von:
• den anwesenden Sto�mengen
• Druck
• An- oder Abwesenheit eines Katalysators

7.4. DAS MASSENWIRKUNGSGESETZ 31
Verhalten in Abhängigkeit von K:
• Wenn K groÿ ist, liegt das Gleichgewicht auf der rechten Seite (es läuft weitgehend die Hintreaktionab).
• Wenn K klein ist, liegt das Gleichgewicht auf der linken Seite.
Der Reaktionsquotient Q entspricht dem Ausdruck für K, wenn beliebige Konzentrationen vorliegen.
• Wenn Q = K, liegt ein Gleichgewichtszustand vor.
• Wenn Q < K, läuft die Reaktion von links nach rechts
• Wenn Q > K, läuft die Reaktion von rechts nach links
Aber
∆G = ∆G0 +RT ∗ lnK R ist die Gaskonstante
⇒ 0 = ∆G0 +RT ∗ lnK
∆G0 = −RT ∗ lnK
Bei negativen ∆G0 -Werten ist K>1, die Produkte sind bevorzugt.
Beispiel
2H2(g) +O2(g) 2H2O(g)∆G0f = −228, 6
kJ
mol
∆G0 = −R ∗ T ∗ lnK
2 ∗ (−228, 6) = (8, 3145 ∗ 10−3) ∗ 298 ∗ lnK
K = e184,34 = 1080
Bei positiven ∆G0 -Werten sind die Reaktanden bevorzugt
Beispiel
2(g) +O2(g) 2NO(g)∆G0f (NO) = +86, 7
kJ
mol
∆G0 = 2 ∗ (86, 7) = −R ∗ T ∗ lnK
K = e−70 = 10−31
Bei ∆G0 = 0 gilt:
[A]a ∗ [B]b = [X]x ∗ [Y ]y
→ Gleichgewicht
Beispiel
H2 + I2 2HI
Bei einer Temperatur von 490 ◦C in diene Volumen von 1 l reagieren 1 mmol H2 und 1 mmol I2 zu einemGeisch aus 1,544 mmol HI, 0,228 mmol H2 und 0,228 mol I2
Umgekehrt, werden 2 mmol HI auf die gleiche Temperatur erhitzt, �ndet die Reaktion 2HI H2 +I2statt. Aus 2 mmol HI bildet sich ein Gemisch aus 1,544 mol HI, 0,228 mmol H2 und 0,228 mmol I2

32 KAPITEL 7. ENERGETIK
7.5 Verschiebung von Gleichgewichtslagen
Das Prinzip des kleinsten Zwangs, formuliert von Le Châtelier (1884), lautet:
Ein im Gleichgewicht be�ndliches System weicht eiem Zwang aus und es stellt sich ein neuesGleichgewicht ein.
Jede Änderung der Bedingungen ist ein solcher Zwang.
• Konzentrationsänderungen
• Druckänderungen
• Temperaturänderungen
7.6 Löslichkeit und Löslichkeitsprodukt
Eine gesättigte Lösung steht mit festen Bodenkörper des gelösten Sto�es im Gleichgewicht.
AB(s) AB(aq) A+(aq) +B−(aq)
Auf das Gleichgewicht, das in homogener Phase besteht, kann das MWG angewendet werden:
K =[A+] ∗ [B−]
[AB]
[AB] ist die Konzentration an gelöstem, undissoziiertem AB und bei den meisten Elektrolyten ist sieunmessbar klein. Diese Konzentration darf als konstant angesehen werden, solange Bodenkörper vor-handen ist, der mit der Konzentration über das linke Gleichgewicht verbunden ist. [AB] wird mit derGleichgewichtskonstante zu einer neuen Konstante kombiniert, dem Löslichkeitsprodukt: Ksp, KL oderLLP
KL = [A+] ∗ [B−]
pKL = −log10KL
p-Werte: für X ist pX als −log10X de�niert
Beispiel
X = 10−10; pX = −log10(10−10) = 10
Beispiel
AgCl mit pKL = 10
Beispiel
KL = [Ag+] ∗ [Cl−] = 10−10mol2
l2
7.6.1 Fall 1
[Ag+][Cl−] = KL die Lösung ist gesättigt. Wegen:
[Ag+] = [Cl−] ⇒ [Ag+][Cl−] = 10−10mol2
l2
[Ag+]2 = 10−10mol2
l2
[Ag+] = 10−5moll
7.6.2 Fall 2
[Ag+][Cl−] > KL werden der Lösung Ag+ oder Cl− zugefügt, so ist diese übersättigt. Als Folge wird soviel AgCl ausfallen, bis wieder gilt:
[Ag+][Cl−] = 10−10mol2
l2

7.7. VERSCHIEBUNG VON GLEICHGEWICHTSLAGEN 33
Beispiel:
Zu einer ges. Lösung wird Cl− zugegeben, bis [Cl−] = 10−2moll . Dann fällt so viel AgCl aus bis [Ag+] =
10−8moll ⇒ 10−2 ∗ 10−8 = 10−10mol
l 01.12.11
7.6.3 Fall 3
[Ag+] ∗ [Cl−] < KL → die Lösung ist ungesättigt un kann weiteren Bodensatz au�ösen
Eine Verringerung von [Ag+] oder [Cl−] ergibt sich beim Verändern der Lösung oder durch chemischeReaktionen, in denen diese Ionen Aisgangssto�e sind: vor vor allem Komplexbildungsreaktionen von Ag+
durch Ammoniak:Ag+ + 2NH3 [Ag(NH3)2]+
→ stabil � Gleichgewicht liegt rechts
7.7 Verschiebung von Gleichgewichtslagen
7.7.1 Druckänderungen:
Der Druck wirkt sich vor allem bei Reaktionen von Gasen aus sofern ∆n! = 0
Beispiel
2SO2(g) +O2(g) 2SO3(g)
Edukte: 3 mol; Produkte: 2 mol
Wenn die Reaktion in einem geschlossenen Gefäÿ abläuft, verringert sich der Druck Richtung Pro-dukte. Wird der Druck erhöht, weicht es aus, indem sich das Gleichgewichtin Richtung der Produkteverschiebt. Bei Erniedrugung des Drucks verschiebt sich das Gleichgewicht in Richtung der Edukte.
⇒ Bei Druckerhöhung verschiebt sich das Gleichgewicht auf die Seite, aus der weniger Teilchensind und umgekehrt bei niedrigerem Druck zur Seite mit mehr Teilchen.
7.7.2 Temperaturänderungen
Die Ammoniak-Synthese ist eine exotherme Reaktion.
N2(g) + 3H2(g) 2NH3(g)∆H = −92, 4kJ
mol
Wird die Temperatur erhöht, so versucht das System der Temperaturerhöhung durch Wärmeverbrauchauszuweichen. Die endotherme Rückreaktion läuft ab und das Gleichgewicht verlagert sich auf die Seiteder Edukte. Hohe Ausbeute an NH3 werden somit erreicht, wenn die Temperatur möglichst niedrig ist.Bei niedrigen Temperaturen läuft die Reaktion extrem langsam ab, bis zur Einstellung des Gleichgewichtswären Jahrmillionen notwendig.
⇒ Temperaturerhöhung bewirkt eine Verschiebung auf die endotherme Seite, Temperaturer-niedrigung zu einer Verschiebung zur exothermen Seite.
7.8 Metastabile Systeme / Katalysatoren
Die Gleichgewichtskonstante sagt nur etwas über Konzentrationen im Gleichgewichtszustand aus (∆G =0), n icht aber, wie schnell dieser Zustand erreicht wird.
• Eine Reaktion mit ∆G > 0 wird sicher nicht freiwillig ablaufen
• Eine Reaktion mit ∆G < 0 kann durchaus eine Aktivierung benötigen, ehe es zur Produktbildungkommt.
2H2 +O2 2H2O; ∆G0f = −228, 6
kJ
mol
Die Reaktionsgeschwindigkeit ist äuÿerst klein, das System ist metastabil.

34 KAPITEL 7. ENERGETIK
7.9 Energie-Diagramm für eine chemische Reaktion
Exotherme Reaktion Endotherme Reaktion
wobei E1 die Aktivie-rungsenergie und E2 dieReaktionsenthalpie ist
Metastabile Systeme sind kinetisch gehemmt. Diese Hemmung kann durch Zuführung von Energie(Aktivierungsenergie) überwunden werden. Diese Aktivierungsenergie kann durch Zugabe eines Kataly-sators herabgesetzt werden. Katalysatoren sind Sto�e, die den Reaktionsmechanismus verändern, abersie treten in der Brutto-Reaktion nicht auf, da sie unverändert aus der Reaktion hervorgehen (was einezwischenzeitliche Veränderung nicht ausschlieÿt). Die Lage des Gleichgewichts wird durch den Katalysa-tor nocht verändert, nur die Einstellung des Gleichgewichts wird beschleunigt. Die Katalysierte Reaktionbesitzt eine kleinere Aktivierungsenergie als die unkatalysierte: die Geschwindigkeit der katalysierten Re-aktion ist gröÿer.
2 Möglichkeiten:
• Homogene Katalyse:
die reagierenden Sto�e und der Katalysator liegen in der selben Phase vor.
• Heterogene Katalyse:
verschieden Phasen: Bsp: NH3-Snthese nach dem Haber-Bosch-Verfahrenwird durch einen Eisen-Oxid-Basiert-Katalysator beschleunigt. Dieser ist nurüber 400◦C wirksam. Deshalb wird die Reaktion bei hohem Durck (günstig)und hohen Temperaturen (ungünstig) durchgeführt.
06.12.11

Kapitel 8
Säuren und Basen
8.1 Theorien
8.1.1 Das Arrhenius�Konzept
• Eine Säure bildet H+�Ionen in wässriger Lösung:
HCl −→ H+(aq) + Cl−(aq)
• Eine Base bildet OH−�Ionen in wässriger Lösung:
NaOH −→ Na+(aq) +OH−(aq)
• Die Reaktion zwischen eine Säure und einer Base heiÿt Neutralisation. Dabei entstehen Wasserund ein Salz:
Säure + Base −→ Salz +H2O
HCl +NaOH −→ NaCl +H2O (Na+ + Cl−)(aq)
8.1.2 Das Brønsted�(Lowry)�Konzept
• Säuren sind Protonendonatoren (→ Moleküle, die H+�Ionen abspalten können)Bei der Abgabe von Protonen wird aus der Säure ihre konjugierte Base
• Basen sind Protonenakzeptoren (→ Moleküle, die H+�Ionen aufnehmen können)Beim Aufnehmen von Protonen wird aus der Base ihre konjugierte Säure
• Die Säure�Base�Reaktion spielt sich zwischen zwei konjugierten Säure�Base�Paaren ab:
Säure 1 + Base 2 Base 1 + Säure 2
Diese Gleichung beschreibt die Summe zweier Teilgleichungen:Säure 1 H+ + Base 1 Bsp: HCl H+ + Cl−
Base 2 +H+ Säure 2 Bsp: H2O +H+ H3O+
Insgesamt: HClSäure 1
+ H2OBase 2
H3O+
Säure 2+ Cl−
Base 1
• Besonderheit bei Wasser:
NaOH +H2O Na+ +OH− +H2O
Das OH−�Ion wirkt als Base und das Wasser wirkt als Säure, das heiÿt, dass obwohl sich praktischnichts ändert, handelt es sich hier um ein Gleichgewicht
• Neutralisation
H3O+(aq) +OH−(aq) H2O +H2O
35

36 KAPITEL 8. SÄUREN UND BASEN
8.1.3 Das Lewis�Konzept
Es kommt auf die Bildung einer kovalenten Bindung (Koordinationsbindung) zwischen Basen� und Säure�Teilchen an. Eine Lewis�Base stellt ein Elektronenpaar zur Verfügung. Eine Lewis�Säure wirkt alsElektronenpaar�Akzeptor.
• Lewis�Base: NH3
• Lewis�Säure: BF3
BF3 +NH3 −→
• NH3 ist sowohl eine Lewis� als auch eine Brønsted�Base: NH3 +H+ −→ NH+4
• Komplexe entstehen aus Lewis�Säure/Base�Paaren:
Cu2+
Lewis�Säure+ 4NH3
Lewis�Base−→ [Cu(NH3)4]2+
8.2 pH�Wert, Ionenprodukt des Wassers
Der Säuregrad einer wässrigen Lösung ist durch die Konzentration anH+ (Arrhenius) bzw.H3O+ (Brøns-
ted) gegeben:pH = − log10[H+] bzw. pH = − log10[H3O
+]
In reinem Wasser liegt das Gleichgewicht (Autoprotolyse) nahezu vollständig auf der linken Seite:
2H2O H3O+ +OH−
Massenwirkungsgesetz:
K =[H3O
+] ∗ [OH−]
[H2O]2
Da nur wenige H2O�Moleküle Ionen bilden, ist die Konzentration des nicht protolysierten Wassers prak-tisch gleich der Gesamtkonzentration an Wasser:
[H2O] =1000 gl18 g
mol
= 55, 5mol
l
Daher kann man die Konzentration an H2O zusammen mit K als eine neue Konstante KW (das Ionen-produkt des Wassers) de�nieren:
K ∗ [H2O]2 = [H3O+] ∗ [OH−] =: KW
Bei 25◦C:
KW = 10−14mol
l⇒ pKW = − log10 10−14 = 14
Das bedeutet auch, dass 10−14moll = [H3O
+] ∗ [OH−] = 10−7moll ∗ 10−7mol
lDamit ergibt sich für pH� und pOH�Wert:
pH = − log10[H3O+] = 7 pOH = − log10[OH−] = 7
⇒ pH + pOH = 14
Demnach gilt:[H3O
+] > [OH−]⇒ pH < 7
[H3O+] = [OH−]⇒ pH = 7
[H3O+] < [OH−]⇒ pH > 7

8.3. SÄURESTÄRKE 37
8.3 Säurestärke08.12.11
HA+H2O H3O+ +A−
K =[H3O
+] ∗ [A−]
[HA] ∗ [H2O]
K ∗ [H2O] = KS =[H3O
+] ∗ [A+]
[HA]
pKS = −log10KS
Bei Säure�Base�Paaren heiÿen die zusammengehörigen Säuren und Basen:konjugierte Säure / Base → konjugiertes Säure�Base�Paar
• Ein�uss der Elektronegativität:
NH3 H2O HF
(→) Säurestärke nimmt von links nach rechts zu
• Ein�uss der Atomgröÿe:
H2O H2S H2Se
(→) Säurestärke nimmt von links nach rechts zu
HF UCl HBr HIHOCl HOBr HOI
(←) Säurestärke nimmt von rechts nach links zu
HOCl HClO2 HClO3 HClO4
(→) Säurestärke nimmt von links nach rechts zu
8.3.1 Starke Säuren
HCl +H2O −→← H3O+ + Cl−
pH = − log10 c0
für Konzentrationen kleiner 10−7 muss die Autoprotolyse des Wassers berücksichtigt werden.→+10−7moll
8.3.2 Schwache Säuren
HAc+H2O H3O+ +Ac−
Ks =[H3O
+] ∗ [Ac−]
[HAc]⇒ Ks =
[H3O+]2
[HAc]
Näherung: [HAc] ≈ c0 ⇒ [H3O+] =
√Ks ∗ c0
⇒ pH = 12 (pKs − log10 c0)
genau: [HAc] = c0 − [H3O+] ⇒ quadratische Gleichung
Beispiel
c0 = 0, 1moll ⇒ pH ≈ 2, 88
c0 = 0, 001moll ⇒ pH ≈ 3, 9
8.3.3 Protolysegrad
α =c0 − [HA]
c0⇒ 0 ≤ α ≤ 1
(α gegen 1 → starke Säure)

38 KAPITEL 8. SÄUREN UND BASEN
Oswaldscher Verdünnungssatz
zunehmende Verdünnung → α wird gröÿer
8.3.4 Mehrprotonige Säuren:
H2SO4 +H2O HSO−4 +H3O+
HSO4 +H2O SO2−4 +H3O
+
pH-Werte von Hydroxiden:NaOH Na+ +OH−
Ca(OH)2 Ca2+ + 2OH−
pOH = −log10c0
Beispiel: c0 = 0, 1moll ⇒ pOH = 1⇒ pH = 14− pOH = 13
8.4 Metallsalze
Reaktion Säure� / Base�Wirkung pKs pH(fast keine Rückreaktion) c0 = 0, 1moll
AlCl3 Al3+ + 3Cl− [Al(H2O)6]3+H2O [Al(H2O)5(OH)]2+ +H3O+ 4,9 3,0
FeCl3 Fe3+ + 3Cl− [Fe(H2O)6]3+ +H2O [Fe(H2O)5(OH)]2+ +H3O+ 2,2 1,6
NH4Cl NH+4 + Cl− NH+
4 +H2O NH3 +H3O+ 9,2 5,1
KHSO4 K+ +HSO−4 HSO−4 +H2O SO2−
4 +H3O+ 1,9 1,5
8.4.1 Schwach basische Anionen
NaAc: Ac− +H2O HAC +OH− KB = [HAC]∗[OH−][AC−] ; pKB = −log10KB
Na2CO3: CO2−3 +H2O HCO−3 +OH− KB =
[HCO−3 ]∗[OH−]
[CO2−3
;
pKB(CO2−3 ) = 14− pKS(CO2−
3 ) = 14− 10, 37 = 3, 63
KS =[H3O
+] ∗ [A−]
[HA]
KB =[HA] ∗ [OH−]
[A−]
KS ∗KB = [H3O+] ∗ [OH−] = KW
pKS + pKB = pKW = 14
8.5 Pu�erlösungen:
Gemisch aus schwacher Säure und deren konjugierten Baseim menschlichen Körper wichtige Pu�erlösungen:
• HAc/Ac−
• H2PO−4 /HPO
2−4
Reaktion mit Säure:H3O
+ +A− H2O +HA
Reaktion mit Base:OH− +HA H2O +A−
KS =[H3O
+] ∗ [A−]
[HA]
beste Pu�erwirkung: [A−] = [HA] ⇒ pH = pKS[HA][A−] ändert sich bei Säure oder Basezugabe.13.12.11

8.6. PUFFERSYSTEME 39
8.6 Pu�ersysteme
Beispiel
Sto� pH pH mit 0,01 mol HCl ∆pH1 l Wasser 7,0 2,0 5,01 l HAc/Ac− mit c = 0, 011moll 4,7 4, 7− log( 0,021
0,0001 ) = 3, 4 1,31 l HAc/Ac− mit c = 0, 11moll 4,7 4, 7− log( 0,12
0,10 ) = 4, 6 0,1
Indiokatoren: farbige schwache Säuren, die in deprotonierter Form eine andere Farbe haben.
8.7 Redox-Systeme
Oxidationszahlen:0
Cl2,+I
Na+,+II
Mg−IF2,
+ 83
Fe3
−IIO4 = FeO ∗ Fe2O3
Oxidation: Abgabe von ElektronenErhöhung der Oxidationsstufe: Zn→ Zn2+ + 2e−
Oxidationsmittel: Reagenz zur Oxidation, wird dabei selbst reduziert.
Reduktion: Aufnahme von Elektronen
Erniedrigung der Oxidationsstufe:0
Cl2 + 2e− → 2−ICl−
Reduktionsmittel: Reagenz zur Reduktion, wird dabei selbst oxidiert.
Redox-System: Kopplung beider Reaktionstypen.Beispiel:Red.: M2+ + 2e− →MOx.: H2 → 2H+ + 2e−
Redox: M2+ +H2 →M + 2H+
8.7.1 Summengleichung
• Auswahl des Redoxpaars
• Elektronenblianz
• Ladungsbilanz
• Massenbilanz
Beispiel
Cr+V IO−II4
2−+ 3e− + 8H3O
+ → Cr3+3++ 12H2O
unterschiedliche Redoxpaare haben unterschiedliche Reduktions� und Oxidationsvermögen.→ Reduktions�bzw. Oxidationsvermögen muss im Verhältnis zum Reaktionspartner (= 2. Redoxpaar) gesehenwerden.
Da nur Potentialdi�erenzen mesbar sind, muss ein Bezugepunkt gewählt werden. Dieser wird durch dieNormalwassersto�elektrode de�niert.
Normalwassersto�elektrode: eine platinierte Platin-Elektrode über die Wassersto� mit einem Druckvon 1,013 bar geleitet wird.
⇒ Bestimmung von Potentialdi�erenzen möglich.⇒ elektrochemische Spannungsreihe⇒ Aussage über das Ablaufen von Reaktionen möglich

40 KAPITEL 8. SÄUREN UND BASEN
8.8 Konzentrationsabhängigkeit von Potentialen
Normalpotentials bezogen auf Aktivitäten von a = 1.Änderung der Konzentration → Potentialänderung⇒ Nernstsche Gleichung für Halbzellenpotentiale
E = E0 +R ∗ Tz ∗ F
∗ lncOxcRed
mitT = TemperaturR = 8, 134 J
mol∗KLF = 96487 C
molz = Anzahl der übertragenen e− pro Atom
⇒ E = E0 + 0,059z ∗ log10
cOx
cRed
Beispiel
+V II
MnO−4 + 5e− + 8H3O+ →
+II
Mn2+ + 12H2O
E = 1, 51 +0, 059
5∗ log c(MnO−4 ) ∗ c8(H3O
+)
c(Mn2+)
• Konstante Konzentrationen (Feststo�e, H2O) nicht in Nernstsche Gleichung
• bei Gasen: Partialdruck
2−ICl− →
0
Cl2 + 2e−
E = E0 +0, 059
2∗ log10
PCl2c2Cl−
8.8.1 Bestimmung von Gleichgewichtskonstanten
Beispiel
Cr2+ + Fe3+ Cr3+ + Fe2+
Cr2+ → Cr3+ + e− E0 = −0, 41V
Fe3+ + e− → Fe2+ E0 = +0, 77V
E1 = E01 +
0, 059
1∗ log10
c(Cr3+)
c(Cr2+)
E2 = E02 +
0, 059
1∗ log10
c(Fe3+)
c(Fe2+)
∆E = E2 − E1 = ∆E0 + 0, 059 ∗ log10
c(Cr2+) ∗ c(Fe3+)
c(Fe2+) ∗ c(Cr3+)
bei 50 % Umsatz:
c(Cr2+) = c(Cr3+) c(Fe2+) = c(Fe3+) ⇒ ∆E = ∆E0
Reaktion endet, wenn ∆E = 0⇒ E1 = E2
E02 + 0, 059 ∗ log10
c(Fe3+)
c(Fe2+)= E0
1 + 0, 059 ∗ log10
c(Cr3+)
c(Cr2+)
∆E0 = E02 − E0
1 = 0, 059 ∗ log10
c(Cr3+) ∗ c(Fe2+)
c(Fe3+) ∗ c(Cr2+)
log10Kc =∆E0
0, 059∗ z

8.9. ELEKTROLYSE 41
8.8.2 pH-Abhängigkeit des Potentials
H2 + 2H2O → 2H3O+ + 2e−
E = E0 +0, 059
2∗ log10 c
2(h3O+)
E = 0, 059 ∗ log10 c(H3O+)
E = −0, 059pH
in neutraler Lösung: pH = 7 ⇒ E = −0, 413Vnur Redoxpaare mit E < −0, 413V können in neutraler Lösung H2 entwickeln
saurer:+III
MnO−2 + 5e− + 8H3O+ →
+II
Mn2+ + 12H2O
leicht basisch:+V II
MnO−4 + 3e− + 2H2O →MnO2 + 4OH−
stark basisch:MnO4 + e− →MnO2−
4 15.12.11
Pasivierung: einige Metalle mit E0 < 0 lösen sich nicht in Säure unter Wassersto�entwicklung, weil sieeine unlösliche Oxidschicht (z.B. Al, Cr) ausbilden.
Konzentrationskette: 2 gleiche Redoxpaare mit einer Potentialdi�erenz durch einen Konzentrations-unterschied. ∆E! = 0, bis c1 = c2
Elektronen zweiter Art :
Kalomelelektrode:
Hg-Elektrode, ummantelt mit Hg2Cl2, Pt�Draht als elektrische Zuleitung.Elektrolyt: KCl�Lösung gesättigt mit Hg2Cl2
20
Hg + 2Cl− →+I
Hg2Cl + 2e−
E0 = +0, 24V
AgCl�Elektrode:
Ag�Draht beschichtet mit AgClElektrolyt: KCl�Lösung gesättigt mit AgCl
Ag + Cl− → AgCl + e−
8.9 Elektrolyse
Umkehrung von galvanischen Elementen durch anlegen einer extrenen Zersetzungsspannung.
∆Ezers = ∆EEMK(+∆EOhm) + ∆EU
∆EU Überspannung
Überspannung vor allem bei Reaktionen mit Gasentwicklung: kinetische Hemmung an bestimmten Me-tallen.In Gegenwart mehrerer Redoxpaare:
• Reduktion der Kationen mit dem gröÿten Potential
• Oxidation des Redoxpaars mit dem kleinsten Potential
Umkehrung des Daniell�Elements :hohe Überspannung von H2 an Zn ⇒ nur deswegen Entladung von Zn2+ und nicht von H3O
+
Elektrochemische Stromquellen :
Primärelemente: Redoxprozesse setzen Energie frei, die als elektrische Energie genutzt werdenkann.
Sekundärelemente (Akkumulatoren): wie Primärelemente, aber reversibel (�wiederau�adbar�).
Brennsto�zellen: kontinuierliche Zuführung des Brennsto�es.

42 KAPITEL 8. SÄUREN UND BASEN
Beispiel
PE: Leclanché�Element:Kohle�Elektroden mit Mantel aus Elektrolyt: NH4Cl
Ox.:0
Zn→+II
Zn2+ + 2e−
Red.: 2+IV
MnO2 + 2e− + 2H2O → 2+III
MnO(OH) + 2OH−
2MnO2 + Zn+ 2NH4Cl→ 2MnO(OH) + Zn(NH3)2Cl2 ∆E0 ≈ 1, 5V
8.9.1 Beispiel: (PE) Lithium�Batterie
Ox.: Li→ Li+ + e−
2+IV
S OCl2 + 4e− →0
S ++IV
S O4 + 4Cl− ∆E = 3, 6V
8.9.2 Beispiel: (SE) Blei�Akku
in 20 % H2SO4
Entladung:
Ox.:0
Pb+ SO2−4 →
+II
PbSO4 + 2e−
Red.:+IV
Pb O2 + 2e− + SO2−4 + 4H3O
+ →+II
PbSO4 + 6H2OPb+ PbO2 + 2H2SO4 → 2PbSO4 + 2H2O ∆E ≈ 2V
Entladung ⇒ Verdünnung der H2SO4 ⇒ Änderung der Dichte (→ Messung des Ladungszustands)Au�aden durch Elektrolyse
8.9.3 Beispiel: Brennsto�zellen
Ox.:0
H2 →+I
H2O + 2e−
Red.: 1/2O2 + 2e− → O2−
H2 + 1/2O2 → H2O
8.10 Korrosion und Lokalelemente
8.10.1 Lokalelement
2H3O+ + 2e− → H2 + 2H2O
→ kinetisch gehemmt an Zn, aber nicht an Cu⇒ Verunreinigung der Zn.Ober�äche mit Cu fördert das Au�ösen des Zn.
8.10.2 Korrosion
Al, Cr PassivierungFe: �Rosten�⇒ Schutzschichten auf Fe aus unedleren Metallen

Kapitel 9
Hauptgruppenelemente
häu�gste Elemete in der Erdkruste:Element Massenanteil Element Massenanteil
[in %] [in %]O 45,50 P 0,112Si 27,2 Mn 0,106Al 8,3 F 0,054Fe 6,2 Ba 0,039Ca 4,66 Sr 0,038Mg 2,76 S 0,034Na 2,27 C 0,018K 1,84 Zr 0,016Ti 0,63 V 0,014H 0,15 Cl 0,013Summe: 99,51 Summe: 0,444
9.1 Wassersto�
9.1.1 Eigenschaften
• Ordnungszahl: 1
• Elektronenkon�guration: 1 s1
Abgabe von e− Protonen H+/H3O+
Aufnahme von e−: Hydratation H−
nur mit stark elektropositiven Metallen
• maximal eine kovalente Bindung
• Ionisierungsenergie [eV]: 13,6
• Elektronegativität: 2,2
• Schmelzpunkt: −259◦C
• Siedepunkt: −253◦C
• typisches Nichtmetall
• Ausbildung von Wassersto�brücken (z.B. mit HF, H2O)
• Isotopene�ekt: Eigenschaft H2 D2
Schmelzpunkt [in K] 14, 0 18, 7Siedepunkt [in K] 20, 4 23, 7Verdampfungsenthalpie [kJ/mol] 0, 117 0, 197Dissoziationenergie bei 25◦C [kJ/mol] 436 444
10.01.12
43

44 KAPITEL 9. HAUPTGRUPPENELEMENTE
9.2 Edelgase: Gruppe 18
9.2.1 Eigenschaften
He Ne Ar Kr Xe RnOrdnungszahl 2 10 18 36 54 86e−-Kon�guration 1s1 [He]2s22p6 [Ne]3s23p6 [Ar]3d104s24p6 [Kr]4d105s25p6 [Xe]4f145d106s26p6
IE1 [eV] 24, 6 21, 6 15, 8 14, 0 12, 1 10, 7Smp [◦C] −272 −249 −189 −157 −112 −71Sdp [◦C] −269 −229 −168 −153 −108 −62vdW-Radium [pm] 120 160 190 200 220 −Farbe des Lichts2 gelb rot rot gelbgrün violett weiÿ
1 Ionisierungsenergie2 bei Gasentladungsröhren
• farblose, geruchslose Gase
• ungiftig, nicht brennbar
• in festem Zustand nehmen alle Edelgase Kristallstruktur in kdP an (He auch hdP)
• kovalente Bindungen möglich
� für Kr, Xe
� nur mit stark elektronegativen Elementen (z.B. F, O, Cl)
• thermodynamisch stabil nur Xe�Flouride
9.2.2 Vorkommen, Darstellung, Verwendung
• Edelgase sind Bestandteil der Luft (ca. 1 % (hauptsächlich Ar: ca. 0,93 %))
• 4019K + 0
−1 e→4018 Ar
• He in Erdgas (bis zu 8 %)
• technische Gewinnung:
fraktionierende Destillation (Linde�Verfahren)Prinzip: Joule�Thomson�E�ekt: komprimiertes Gas kühlt bei Expansion ab.
• Labormaÿstab:
Luft = 4N2 +O2 + EG
Luft+ 2Cu −→ 2CuO
Luft+ 3Mg −→Mg3N2
• Verwendung:
� Schutzgas (Ar)
� Füllgase von Glühlampen, Halogenlampen (v.a. Kr wegen niedrigen Wärmeleitfähigkeit)
� Gasentladungsröhren
� Hochdrucklampen (Xe): Flutlicht, Leuchttürme
� Ballonfüllungen (He)
� Tauchgas (10 % O2 in He)
9.2. EDELGASE: GRUPPE 18 45
9.2.3 Verbindungen
He, Ne, Ar bilden nur ClathrateKr, Xe Verbindungen mit F, O, Cl → direkte Reation nur mit F2 nur nach Aktivierung
• Halogenide:
400◦C Mikrowellen: Xe+ F2 � XeF2
400◦C, 6bar: XeF2 + F2 � XeF4
300◦C, 60bar: XeF4 + F2 �+V I
XeF6
→ wirken oxidierend:+IV
Xe−IF4 + 4
−IJ− −→
0
Xe+ 20
J2 + 4−IF−
XeF4 + 2SF4 −→ Xe+ 2SF6
XeF6 + 6HCl −→ Xe+ 3Cl2 + 6HF
→ Zersetzung in Wasser:XeF2 +H2O −→ Xe+ 2HF + 1/2O2
KrF2:
Kr + F2elektrischeEntladung−−−−−−−−−−−−−−→ KrF2
stärkstes bekanntes Oxidationmittel
5KrF2 + 20
Au −→ 2+V
AuF5 + 5Kr
KrF2 + 2+I
AgF −→ 2+II
AgF2 +KrKrF2 +H2O −→ Kr + 2HF + 1/2O2
[Auch: XeCl2, XeCl4, XeBr2]
• Oxide, Oxi�ouride:
XeO3: 3+IV
XeF4 + 6H2O −→0
Xe+ 12HF + 3+V I
XeO3
XeF6 + 3H2O −→ XeO3 + 6HFXeO3 → explosiv: XeO3 −→ Xe+ 1, 5O2
beständig in H2O: XeO3 +H2O � H2XeO4
Xe-Säure (schwach sauer, stark oxidierend)+V III
Xe O4: Ba2XeO6 + 2H2SO4 −→ XeO4 + 2BaSO4 + 2H2O
[Auch: XeOF2, XeOF4, XeO2F2]
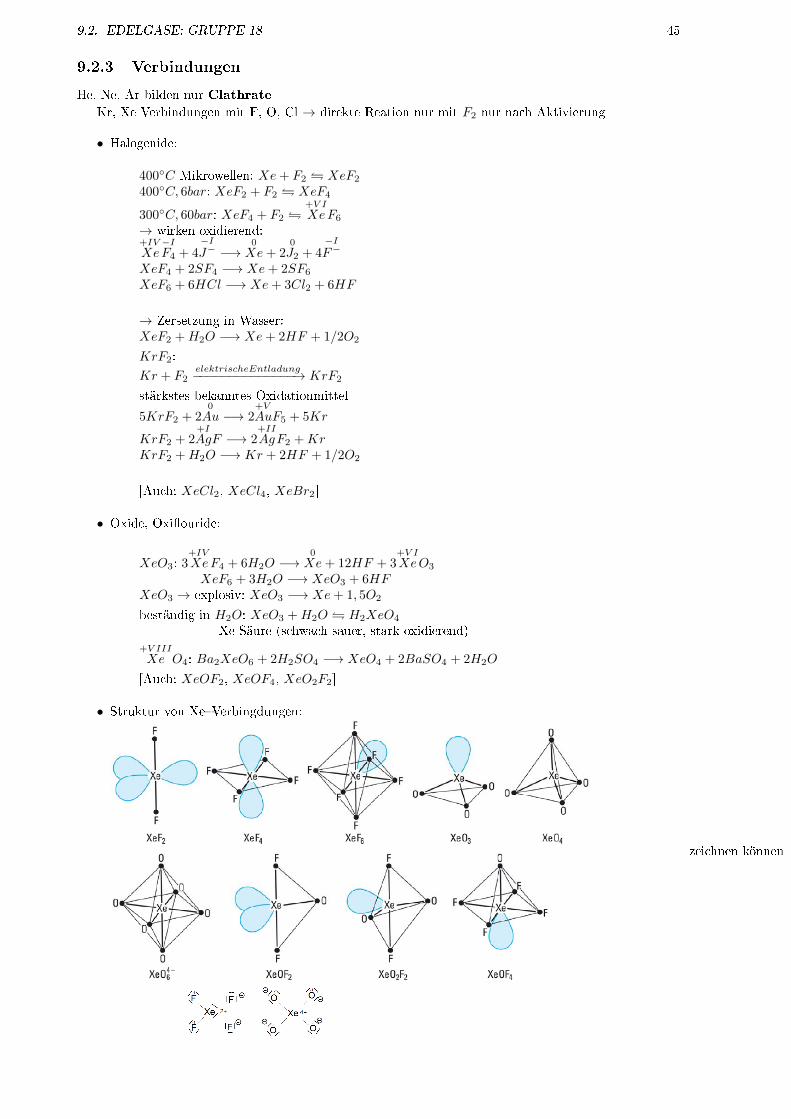
• Struktur von Xe�Verbingdungen:
zeichnen können

46 KAPITEL 9. HAUPTGRUPPENELEMENTE
9.3 Halogene: Gruppe 17
9.3.1 Eigenschaften
F Cl Br IOrdnungszahl 9 17 35 53e−-Kon�guration [He]2s22p5 [Ne]3s23p5 [Ar]3d104s24p5 [Kr]4d105s25p5
Elektrongnegativität 4, 1 2, 98 2, 7 2, 2Elektronena�nität [eV] −3, 4 −3, 6 −3, 4 −3, 1Ionisierungsenergie [eV] 17, 5 13, 0 11, 8 10, 4Smp [◦C] −220 −101 −7 114Sdp [◦C] −188 −34 59 185Dissoziationsenergie [kJ/mol] 159 243 193 151Standardpotential (2X−/X2) [V] +2, 87 +1, 36 +1, 07 +0, 54Bindungslänge (X�X) [pm] 142 199 228 267
• � Salzbildner�
• typische Nichtmetalle
• sehr elektrisch negativ Reaktionfreudig
• Valenzelektronenkon�guration: s2p5 ⇒ e−�Aufnahme exotherm
• X− in Ionenverbindungen
• 1 kovalente Bindung möglich
• F in allem Verbindungen: Oxidationszahl: −I
• Cl, Br, J: auch: +I, +III, +V , +V IIin Verbindungen mit F, O, Cl
•+I
Br−IF ,
+III
J−ICl3,
+V II
Cl−IIO4
−
, BrF5, JF7
• F: Wechselwirkung zwischen den beiden Atomen in einem Elementmolekül sind sehr niedrig (Van�der�Waals�Kräfte)
9.3.2 Vorkommen
nicht elementar, nur gebunden
• F:
� Flussspat: CuF2
� Apatite: Cu5(PO4)3(OH2F )
� Kryolith: Na3AlF612.01.11
• Cl, Br
� gelöst im Meerwasser (ca. 2 % Cl−, 0,01 % Br−)
� Salzlagerstätten: Steinsalz (NaCl), Sylin (KCl)
• I
� in Meerwasser
� zusätzlich als (IO−3 ) im Chilesalpeter (NaNO3)

9.3. HALOGENE: GRUPPE 17 47
9.3.3 chemische Eigenschaften
• F:
� reagiert mit alle Elementen, auÿer mit He, Ne, Ar, N2 direkt
� andere Elemente in höchsten Oxidationsstufen (+V II
J F7,+II
AgF2,+V
AuF5)
� Passivierung von Cu, Ni, Stahl, Monel (Cu�Ni�Legierung)
� Reaktion mit Glas:
Bei Spuren von H2O oder HF:
2F2 + 2H2O → 4HF +O2
4HF + SiO2 → SiF42H2O
• Cl:
� reagiert mit allem Elementen auÿer allen Edelgasen, O2, N2
mit 1. und 2. Hauptgruppe: Salzemit Nichtmetallen: kovalente Chloride (PCl3, PCl5, SCl2, SCl4)
� Cl2 gut löslich in H2O0
Cl2 +H2O→←− H
−ICl +HO
−ICl
� HOCl starkes Oxidatiions� und Desinfektionsmittel
• Br: ähnlichg wie Cl, weniger heftig
• I:
� Reaktion mit S, P, Fe, Al, . . .
9.3.4 Oxidationswirkung
F2 + 2X− → X2 + 2F−
X= Cl, Br, ICl2 + 2X− → X2 + 2Cl−
X= Br, IBr2 + 2I− → J2 + 2Br−
9.3.5 Darstellung und Verwendung
• F
� nicht durch chem. Oxidation herstellbar → Schmelz�usselektrolyse (wasserfrei, sonst O2)Elektrolyt: KF ∗ xHF
�K2MnF6 + 2SbF5 → 2KSbF6 +′′MnF ′′4 → 2KSbF6 +MnF3 + 1/2F2
• Cl:
� technische Elektrolyse: Diaphragma bzw. Membranverfahren oder Amalgamverfahren
2Na+ + 2Cl− + 2H2O → 2Na+ + 2OH− +H2 + Cl2
früher: Deacon�Verfahren:
2HCl + 1/2O2430◦C−−−−→CuCl2
H2O + Cl2
48 KAPITEL 9. HAUPTGRUPPENELEMENTE
Labor: Weldon�Verfahren:
4HCl +MnO4 → 2H2O +MnCl2 + Cl2
16HCl + 2KMnO4 → 8H2O + 2KCl + 2MnCl2 + 5Cl2
� Verwendung:
∗ organische Chemie, Industrie (80 %)
∗ HCl, Br2, TiCl4: Darstellung
∗ Bleichen, Desinfektion
� ökologische Probleme:
∗ FCKWs (Ozonschicht)
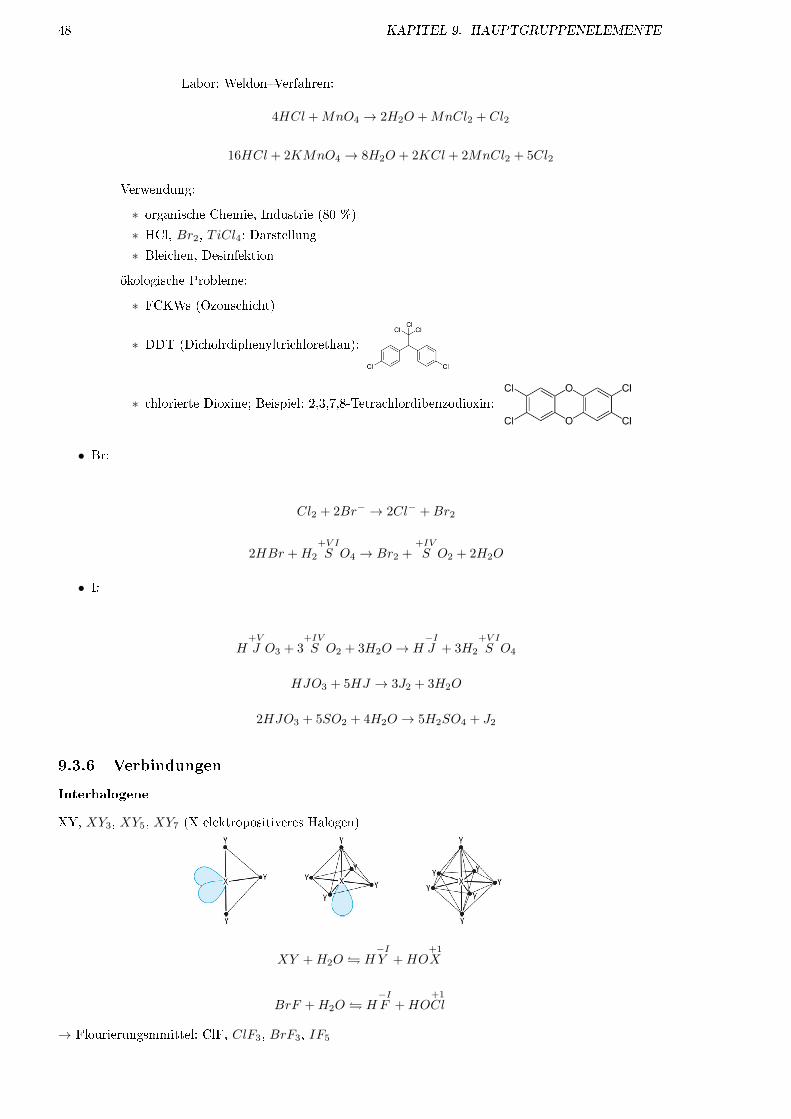
∗ DDT (Dicholrdiphenyltrichlorethan):
∗ chlorierte Dioxine; Beispiel: 2,3,7,8-Tetrachlordibenzodioxin:
• Br:
Cl2 + 2Br− → 2Cl− +Br2
2HBr +H2
+V I
S O4 → Br2 ++IV
S O2 + 2H2O
• I:
H+V
J O3 + 3+IV
S O2 + 3H2O → H−IJ + 3H2
+V I
S O4
HJO3 + 5HJ → 3J2 + 3H2O
2HJO3 + 5SO2 + 4H2O → 5H2SO4 + J2
9.3.6 Verbindungen
Interhalogene
XY, XY3, XY5, XY7 (X elektropositiveres Halogen)
XY +H2O � H−IY +HO
+1
X
BrF +H2O � H−IF +HO
+1
Cl
→ Flourierungsmmittel: ClF, ClF3, BrF3, IF5

9.3. HALOGENE: GRUPPE 17 49
ClFfarbloses GasDE: 256 kJ/mol
↓
Smp.Sdp.ReaktionsfähigkeitDisproportionierung←−
BrFhellrotes GasDE: 280 kJ/molBE: -94 kJ/mol
BrCldunkelrote Flüssig-keitDE: 218 kJ/molBE: +15 kJ/mol
IFbraunes PulverDE: 271 kJ/molBE: -96 kJ/molDisproportionierungbei T > −14◦C
IClrote KristalleDE: 211 kJ/molBE: +18 kJ/mol
IBrrotbraune KristalleDE: 179 kJ/molBE: +41 kJ/mol
DE:Dissoziationsenergiebei T = 298KBE:Bildungsenthalpie
ClF3
farbloses GasBE: -165 kJ/mol
ClF5
farbloses GasBE: -255 kJ/mol
BrF3
farblose FlüssigkeitBE: -256 kJ/mol
BrF5
farblose FlüssigkeitBE: -429 kJ/mol
IF3
gelbes PulverBE: -486 kJ/molDisproportionierungbei T > −28◦C
IF5
farblose FlüssigkeitBE: -841 kJ/mol
IF7
farbloses GasBE: -962 kJ/mol
(ICl3)2
gelbe KristalleBE: -90 kJ/mol
Polyhalogenide
I2 schlecht lösich in H2O aber gut in KI�Lösung I− + I2 � I−3
Halogenwassersto�e
• HF, HCl, HBr, HI
• polare Bindungen:δ+
H�
δ−�
X�|
• gut löslich in H2O:HX +H2O −→← H3O
+ +X−
• Bildung aus den Elementen:H2 +X2 → 2HX
F und Cl: Explosion; Br: 200◦C und Pt�Katalysator; I: über 500◦C
• technische Darstellung: HF: CaF2 +H2SO4 → CuSO4 + 2HFin Glas: 2CuF2 + 2H2SO4 + SiO2 → 2CuSO4 + 2H2O + SiF4
SiF4 + 2HF → H2SiF6
• Verwendung:
� AlF3; Kryolith�Herstellung, FCKW
� Glasindustrie: Ätzen, Polieren
• wässrige Lösung: Flusssäure (40 %�ig), mittelstarke Säure
• HCl:

50 KAPITEL 9. HAUPTGRUPPENELEMENTE
� technische Darstellung: über Chlorid�H2SO4�Verfahren:
NaCl +H2SO420◦C→ NaHSO4 +HCl
NaCl +NaHSO480◦C→ Na2SO4 +HCl
� sowie Nebenprodukt organischen Chlorierungen
R3 − CH + Cl2 → R3 − C − Cl +HCl
� wässrige Lösung: Salzsäure (37 � 38 %�ig), starke, nichtoxidierende Säure → löst nur unedleMetalle auf17.01.12
� NaCl +H2SO4 −→ NaHSO4 +HCl
• HBr, HJ:
� nicht aus Salzen mit H2SO4 wegen der teilweisen Oxidation zu I2, Br2
→ Hydrolyse von PX3 (PBr3, PI3)
PX3 + 3H2O −→ 3HX +H3PO3
� HI sehr starke Säure, aber oxidationsemp�ndlich: 4HI +O2 −→ 2I2 + 2H2O
Halogenide
• X−�Anionen
• 1. & 2. Hauptgruppe: typische Salze
• Nichtmetalle: kovalente HalogenideGase: BF3, SiF4, PF5, CF4
Flüssigkeiten: SCl2, PCl3, CCl4, SiBr4
Sauersto�säuren der Halogene
von links nach rechts mehr mesomete Grenzstrukturen⇒ stabileres Anion, leichteres Abspalten von H+
→ Tendenz zur Disproportionierung:
3+I
ClO− −→+V
ClO−3 + 2−ICl−
alle Halogensauersto�säuren: starke Oxidationsmittel (besonders HOCl)
HOCl: (oder auch HClO)H2O + Cl2 HCl +HOCl
wasserfrei: HOCl nicht bekannt,Cl2O→ Anhydrid
2HOCl Cl2O +H2O
Oxidationsstufe Cl Br I+1 HClO HBrO4 HIO+3 HClO2
+5 HClO3 HBrO3 HIO3
+7 HClO4 HBrO4 HIO4, H5IO6, H7I3O14
Säurestärke Säure Name Anion Name↓ HClO hypochlorige Säure ClO− Hypochlorit↓ HClO2 chlorige Säure ClO−2 Chlorit↓ HClO3 Chlorsäure ClO−3 Chlorat↓ HClO4 Perchlorsäure ClO−4 Perchlorat

9.4. CHALKOGENE: GRUPPE 16 51
Hypochlorite
Cl2 in alkalische LösungCl2 + 2NaOH NaCl +NaOCl +H2O
• HClO2: schnelle Zersetzung:
H+III
Cl O2 −→ 4+IV
Cl O2 +HCl + 2H2O
• Chlorite:2ClO2 + 2NaOH +H2O2 −→ 2NaClO2 +O2 + 2HO
• HClO3:Ba(ClO3)2 +H2SO4 −→ 2HClO3 +BaSO4
• HClO4: beständigste HClOx�Verbindung
anodische Oxidation von ClO−3 :
+V
ClO−3 +H2O −→+V II
Cl O−4 + 2e− + 2H+
KClO4 +H2SO4 −→ HCLO4 +KHSO4
4+V
ClO−3 −→ 3+V II
Cl O−4 +−ICl−
Oxide der Halogene
Cl, Br, J: in positiver Oxidationsstufeinstabile Verbindungstarke OxidationsmittelBsp.: Cl2O, ClO2
Sauersto��ouride
+II
O−IF2:
2F2 + 2OH− → 2F− +OF2 +H2OOxidationsmittel, Flourierungsmittel→ räumlicher Bau wie H2O
+I
O2
−IF2:
O2 + F2elektrischeEntladung−−−−−−−−−−−−−−→ O2F2
→ räumlicher Bau wie H2O2
HOF :
HOF +H2O → HF +H2O2
Pseudohalogene:
Cn−, SCN−, OCN−, N−3ähneln sehr stark dem Verhalten der Halogene (Element�Moleküle)
9.4 Chalkogene: Gruppe 16
siehe Folie [Nr]O S Se Te Po
Ordnungszahl 8 16 34 52 84e−-Kon�guration [He]2s22p4 [Ne]3s23p4 [Ar]3d104s24p4 [Kr]4d105s25p4 [Xe]4f145d106s26p4
Elektronegativität 3, 5 2, 4 2, 5 2, 0 1, 8IE [eV] 13, 6 10, 4 9, 8 9, 0 8, 4Smp [◦C] −219 120 220 450Sdp [◦C] −183 445 685 1390Diss.-Energie [kJ/mol] 498 423 333 258
IE: Ionisierungsenergie; Diss.-Energie: Dissoziationsenergie

52 KAPITEL 9. HAUPTGRUPPENELEMENTE
9.4.1 allgemeine Eigenschaften
• �Erzbildner�
• Sonderstellung von Sauersto�
• Valenzelektronenkon�guration: s2p4
Oxidationsstufen: −2, −1 (für O)−2, +4, +6 (für S, Se, Te)
Stabilität der Oxidationsstufel +6 nimmt mit zunehmender Ordnungszahl ab
H2
+V I
Se O4 stärkeres Oxidationsmittel als H2
+V I
S O4+IV
S O2 stärkeres Reduktionsmittel als+IV
Se O2
sauere Charakter der Oxide: SO2 > SeO2 > TeO2
SO3 > SeO3 > TeO3
→ Schwefelsäure: starke Säure und Tellursäure: schwache Säure
9.4.2 Vorkommen
• O:
� häu�gstes Element in der Erdkruste
� elementar (O2) in Luft (21%)
� gebunden in H2O und Verbindungen (→ Oxide, Silikate, Carbonate)
• S:
� elementar in Lagerstätten
� gebunden in Sul�den: ErzlagerstättenSchwermetallsul�de:
∗ Pyrit (FeS2)
∗ Zinkblende (ZnS)
∗ Bleiglanz (PbS)
∗ Kupferkies (CuFeS2)
∗ Zinnober (HgS)
∗ Gips (CaSO4 ∗ 2H2O)
∗ Anhydrit (CaSO4)
9.4.3 Eigenschaften der Elemente
• O:
� Disauersto� O2:
∗ farbloses, geruchsloses Gas
∗ �üssig: hellblau
∗ Lewis�Formel
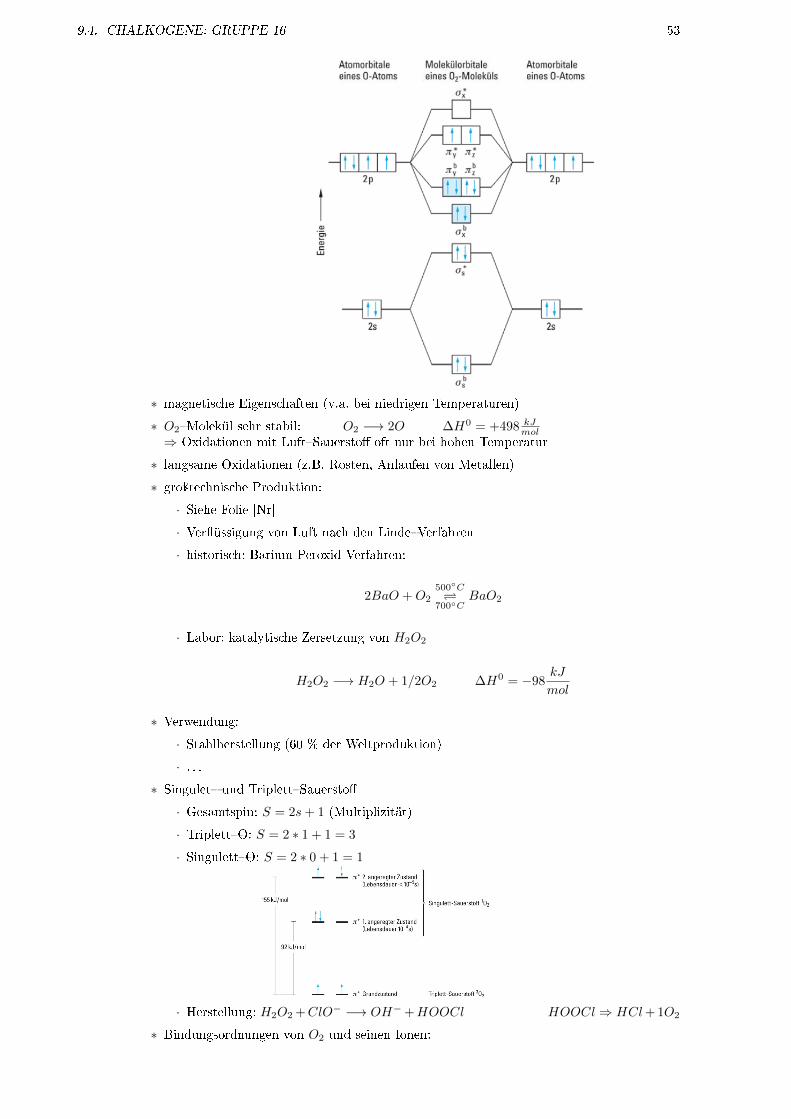
9.4. CHALKOGENE: GRUPPE 16 53
∗ magnetische Eigenschaften (v.a. bei niedrigen Temperaturen)
∗ O2�Molekül sehr stabil: O2 −→ 2O ∆H0 = +498 kJmol
⇒ Oxidationen mit Luft�Sauersto� oft nur bei hohen Temperatur
∗ langsame Oxidationen (z.B. Rosten, Anlaufen von Metallen)
∗ groÿtechnische Produktion:
· Siehe Folie [Nr]· Ver�üssigung von Luft nach den Linde�Verfahren
· historisch: Barium-Peroxid-Verfahren:
2BaO +O2500◦C
700◦CBaO2
· Labor: katalytische Zersetzung von H2O2
H2O2 −→ H2O + 1/2O2 ∆H0 = −98kJ
mol
∗ Verwendung:
· Stahlherstellung (60 % der Weltproduktion)
· . . .∗ Singulet� und Triplett�Sauersto�
· Gesamtspin: S = 2s+ 1 (Multiplizität)
· Triplett�O: S = 2 ∗ 1 + 1 = 3
· Singulett�O: S = 2 ∗ 0 + 1 = 1
· Herstellung: H2O2 +ClO− −→ OH−+HOOCl HOOCl⇒ HCl+ 1O2
∗ Bindungsordnungen von O2 und seinen Ionen:
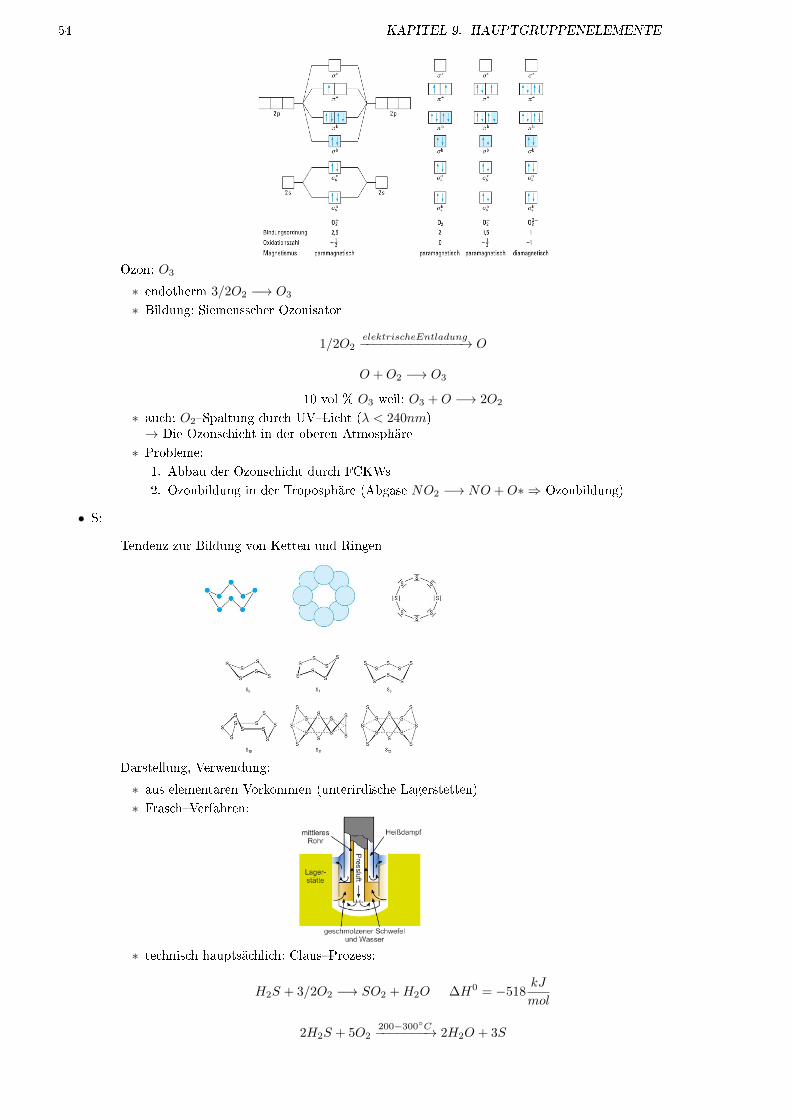
54 KAPITEL 9. HAUPTGRUPPENELEMENTE
� Ozon: O319.01.12
∗ endotherm 3/2O2 −→ O3
∗ Bildung: Siemensscher Ozonisator
1/2O2elektrischeEntladung−−−−−−−−−−−−−−→ O
O +O2 −→ O3
10 vol % O3 weil: O3 +O −→ 2O2
∗ auch: O2�Spaltung durch UV�Licht (λ < 240nm)→ Die Ozonschicht in der oberen Atmosphäre
∗ Probleme:
1. Abbau der Ozonschicht durch FCKWs
2. Ozonbildung in der Troposphäre (Abgase NO2 −→ NO +O∗ ⇒ Ozonbildung)
• S:
� Tendenz zur Bildung von Ketten und Ringen
� Darstellung, Verwendung:
∗ aus elementaren Vorkommen (unterirdische Lagerstetten)
∗ Frasch�Verfahren:
∗ technisch hauptsächlich: Claus�Prozess:
H2S + 3/2O2 −→ SO2 +H2O ∆H0 = −518kJ
mol
2H2S + 5O2200−300◦C−−−−−−−→ 2H2O + 3S
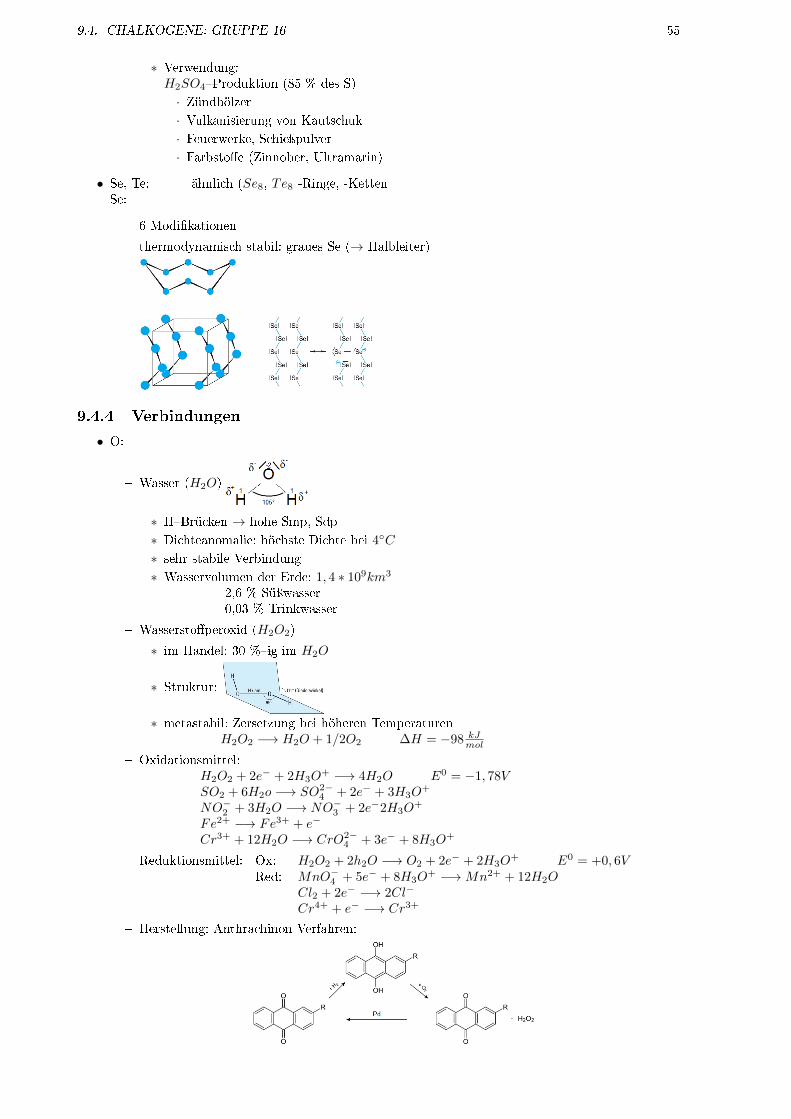
9.4. CHALKOGENE: GRUPPE 16 55
∗ Verwendung:H2SO4�Produktion (85 % des S)· Zündhölzer· Vulkanisierung von Kautschuk· Feuerwerke, Schieÿpulver· Farbsto�e (Zinnober, Ultramarin)
• Se, Te: ähnlich (Se8, Te8 -Ringe, -KettenSe:
� 6 Modi�kationen
� thermodynamisch stabil: graues Se (→ Halbleiter)
9.4.4 Verbindungen
• O:
� Wasser (H2O)
∗ H�Brücken → hohe Smp, Sdp∗ Dichteanomalie: höchste Dichte bei 4◦C
∗ sehr stabile Verbindung∗ Wasservolumen der Erde: 1, 4 ∗ 109km3
2,6 % Süÿwasser0,03 % Trinkwasser
� Wassersto�peroxid (H2O2)
∗ im Handel: 30 %�ig im H2O
∗ Struktur:
∗ metastabil: Zersetzung bei höheren TemperaturenH2O2 −→ H2O + 1/2O2 ∆H = −98 kJ
mol
� Oxidationsmittel:H2O2 + 2e− + 2H3O
+ −→ 4H2O E0 = −1, 78VSO2 + 6H2o −→ SO2−
4 + 2e− + 3H3O+
NO−2 + 3H2O −→ NO−3 + 2e−2H3O+
Fe2+ −→ Fe3+ + e−
Cr3+ + 12H2O −→ CrO2−4 + 3e− + 8H3O
+
� Reduktionsmittel: Ox: H2O2 + 2h2O −→ O2 + 2e− + 2H3O+ E0 = +0, 6V
Red: MnO−4 + 5e− + 8H3O+ −→Mn2+ + 12H2O
Cl2 + 2e− −→ 2Cl−
Cr4+ + e− −→ Cr3+
� Herstellung: Anthrachinon-Verfahren:

56 KAPITEL 9. HAUPTGRUPPENELEMENTE
� Peroxide: Salze von H2O2:
∗ O2−2 �Anoinen
∗ Bekannt: Peroxide von 1. HG, Ca, Sr, Ba
� Peroxide +H2O:gekühlt: Na2O2 + 2H2O −→ H2O2 +NaOHohne Kühlung: Na2O2 +H2O −→ 2NaOH + 1/2O2
2Na+O2 −→ Na2O2 →BleichmittelBaO + 1/2O2 −→ BaO2
� Hyperoxide: O−2 �Anion
M−1/2
O2 (M = K, Rb, Cs) starke Oxidationsmittel
� Dioxygenyle:O2 −→ O+
2 + e− ∆H0 = +1168 kJmol
O2 + PtF6 −→ O+2 (PtF6)−
• Wassersto��Verbindungen von S, Se, Te
� H2 + S600◦C−−−−→ H2S
� H2 + Se300−400◦C−−−−−−−→ H2Se
� Al2Te3 + 6HCl −→ 3H2Te+ 2AlCl3 stark endotherm
� Labor: FeS + 2HCl −→ FeCl2 +H2S
� H2X: farblose, sehr giftige Gase:Sto�: Winkel Sto�: WinkelH2O 104, 4◦ H2Se 91◦
H2S 92, 3◦ H2Te 89, 5◦
� reduzierende Wirkung:H2S + Cl2 −→ 2HCl + SH2S +H2SO4 −→ SO−2 + S + 2H2O
� Säure: H2X +H2O H3O+ +HX−
HX− +H2O H3O+ +X2−
H2S, H2Se, H2Te Säurestärke nimmt von links nach rechts zu
� Sul�de:S2−�Anionen Sul�deHS−�Anionen Hydrogensul�de→ Metallsul�de schwer löslich⇒ Kationentrennungsgangim sauren (pH = 0) kleine S2−�Konzentration
Ar2S3, S . . .im basischen:höhere S2−�konz (NH4)2S�Gruppe: (NH4)2S, . . .24.01.12
� Polysul�de, Polysulfane:
S2−n �Ketten: |
−S−− Sn −
−S−|
Na2Sn +HCl −→ 2NaCl +H2Sn
� Schwefeloxide:
SnO, S7O, S7O2
∗ SO2: farbloses, stechend riechendes Gas, gut lösich in H2O
H2O + SO2 H2SO3

9.4. CHALKOGENE: GRUPPE 16 57
S +O2 −→ SO2 ∆H0B = −297kJ/mol
4FeS2 + 11O2 −→ 2Fe2O3 + 8SO2
∗ SO3: 3SO3 S3O3
SO3 starkes Oxidationsmittel
• Sauersto�säuren des Schwefels:
� H2SO4
∗ Darstellung nach Kontaktverfahren:
SO2 + 1/2O2 −→ SO3 ∆H0 = −99kJ/mol
→ Kat: V2O5/SiO2; Reaktion bei 420− 440◦C
V2O5 + SO2 −→ V2O4 + SO3; V2O4 + 1/2O2 −→ V2O5
SO3 +H2SO4 −→ H2S2O7 (SO3 schlecht löslich inH2O)
H2S2O7 +H2O −→ 2H2SO4
∗ ölige farblose Flüssigkeit (98%)∗ Oleum: rauchende H2SO4 (SO3�Überschuss)∗ Trocknungsmittel∗ Verkohlung von organischen Sto�en∗ Oxidationsmittel: 2H2SO4 + Cu −→ CuSO4 + SO2 + 2H2O
statt Cu auch Ag, Hg möglich; nicht mit Au, Pt; passiviert Fe∗ starke zweibasige Säure:· Salze:
HSO−4 : HydrogensulfatSO2−
4 : Sulfate
Struktur:
· Halogenderivate (Ersetzen von OH durch Cl)
SO3 +HCl −→ HSO3Cl (Sulfonierungsmittel)
· H2SO5: Caro'sche Säure: nur ein H+ wird abgespalten:
· H2S2O8:
· H2SO3 (Schwe�ige Säure)
· H2S2O3 (Thioschwefelsäure)
SO3 +H2S−80◦C−−−−→ H2S2O3
S8 + 8Na2SO3 −→ 8Na2S2O3 (Fixiersalz: Photographie)
⇒ Reduktionsmittel (Bleichindustrie)
Na2S2O3 + 4Cl2 + 5H2O −→ Na2SO4 +H2SO4 + 8HCl

58 KAPITEL 9. HAUPTGRUPPENELEMENTE
· wichtige Halogenverbindungen:SF6: inaktiv, SchutzgasSOCl2: Thionylchlorid
SO2 + PCl5 −→ SOCl2 + POCl3 (Chlorierungsmittel)SO2Cl2: Sulferylchlorid
SO2 + Cl2 −→ SO2Cl2 (Chlorierungsmittel)
9.5 Sticksto�gruppe: Gruppe 15
N P As Sb BiOrdnungszahl 7 15 33 51 83e−-Kon�guration [He]2s22p3 [Ne]3s23p3 [Ar]3d104s24p3 [Kr]4d105s25p3 [Xe]4f145d106s26p3
Elektronegativität 3, 0 2, 1 2, 2 1, 8 1, 7IE [eV] 14, 5 11, 0 9, 8 8, 6 7, 3Smp [◦C] −210 441 8172 630 271Sdp [◦C] −196 2801 6163 1635 1580
1 weiÿer Phosphor2 graues Arsen (unter Luftabschluss, 27 bar)3 graues Arsen sublimiert bei Normaldruck
• Valenzelektronenkon�guration: s2p3 ⇒ Oxidationsstufen: -3, +3, -5 (wird mit zunehmender Ord-nungszahl instabil)
9.5.1 Vorkommen
• N:
� Hauotbestandteil der Luft (78% N2)
� gebunden:
∗ Salpeter NaNO3
∗ Eiweiÿe
• P:
� sehr reaktiv → nur gebunden: Apatit Ca5(PO4)3, (OH, F, Cl) (Knochen von Wirbeltierenbestehen aus Apatit)
• As:
� sehr selten
� häu�ger Arsenide: FeAsS, CoAsS, NiAsS
� positiv polarisiertes As: As4S4 (Realgar)
• Sb:
� Sul�de, Metallantimonide
� Sb2S3: Grauspieÿglanz
• B:
� Bismutglanz: Bi2S3
� Bismutocker: Bi2O3
9.5.2 Die Elemente
• N:
� gasörmig
� |N ≡ N | → isoelektrisch: |C ≡ O|⊕, |N ≡ O|⊕, |C ≡ N |� N2 2N ∆H0 = +945kJ/mol sehr stabil: Inertgas
� technische Herstellung: Linde�Verfahren
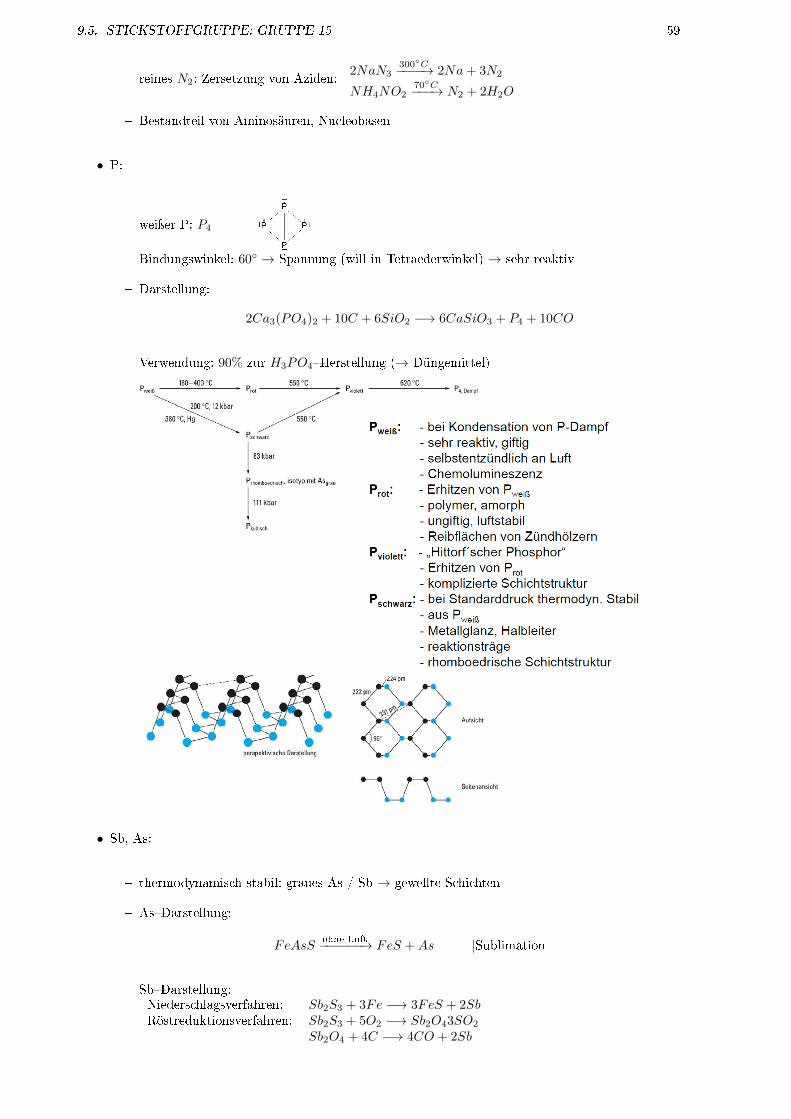
9.5. STICKSTOFFGRUPPE: GRUPPE 15 59
� reines N2: Zersetzung von Aziden:2NaN3
300◦C−−−−→ 2Na+ 3N2
NH4NO270◦C−−−→ N2 + 2H2O
� Bestandteil von Aminosäuren, Nucleobasen
• P:
� weiÿer P: P4
Bindungswinkel: 60◦ → Spannung (will in Tetraederwinkel) → sehr reaktiv
� Darstellung:
2Ca3(PO4)2 + 10C + 6SiO2 −→ 6CaSiO3 + P4 + 10CO
� Verwendung: 90% zur H3PO4�Herstellung (→ Düngemittel)
• Sb, As:
� thermodynamisch stabil: graues As / Sb → gewellte Schichten
� As�Darstellung:
FeAsSohne Luft−−−−−−→ FeS +As |Sublimation
� Sb�Darstellung:Niederschlagsverfahren: Sb2S3 + 3Fe −→ 3FeS + 2SbRöstreduktionsverfahren: Sb2S3 + 5O2 −→ Sb2O43SO2
Sb2O4 + 4C −→ 4CO + 2Sb

60 KAPITEL 9. HAUPTGRUPPENELEMENTE
• Bi:
� nur 1 Modi�kation, analog Asgrau → Metall
9.5.3 Verbindungen
• H�Verbindungen des N: NH3, N2H4, HN3, Nh2OH
� NH3:
∗ farbloses Gas, leicht zu ver�üssigen
∗ Struktur: Winkel: 107◦; Pyramide
∗ Autoprotolyse: 2NH3 NH+4 +NH−2
∗ Bildung solvatisierter Elektronen: Na+NH3 Na+am + e−am
∗ schwach basisch: NH3 +H2O NH+4 +OH− pkB = 4, 75
NH3 +HCL −→ NH4Cl
∗ Struktur NH+4 :
∗ NH+4 : ähnlich zu Alkalimetallen M+
∗ Groÿtechnische Darstellung: Haber-Bosch-Verfahren
∗ Verwendung: Düngemittel, HNO3�Produktion∗ Salze:· NH+
4 (Ammonium)· NH−2 (Amide)· NH2− (Imide)· N3− (Nitride)
∗ Hydrazin:
· N2H4

9.5. STICKSTOFFGRUPPE: GRUPPE 15 61
∗ Disproportionierung:3N2H4 −→ 4NH3 +N2
∗ wässrige Lösung reduzierend, basisch∗ Reaktion mit O2:
N2H4 +O2 −→ N2 + +2H2O ∆H0 = −625kJ/mol
∗ technische Herstellung:· Rasching�Synthese:
NH3 +NaOCl −→ NaOH + NH2ClChloramin
NaOH +NH2Cl +NH3 −→ NaCl +H2O +N2H4
2NH3 +NaOCl −→ N2H4 +NaCl +H2O
∗ Verwendung:· Herbizide· pharmazeutische Industrie· Polymerisation (Starter)
� Sticksto�wassersto�säure HN3:
∗ wasserfrei explosiv: 2HN3 −→ 3N2 +H2 ∆H0 = −538kJ/mol
∗ bis 20 %�ig in Wasser: schwach sauer: HN3 +H2O −→ H3O+ + N−3
Azid
∗ Azide von 1. und 2. Hauptgruppe: kontrollierte Zersetzung: 2NaN3300◦C−−−−→ 2Na+ 3N2
∗ N−3 : linear, symmetrisch
∗ HN3: 2 unterschiedliche N�N�Bindungen
NaNH2 +N2O190◦C−−−−→ NaN3 +H2O
• Hydride von P, As, Sb, Bi←−−−−−−−−−−Stabilität nimmt zu
(Sb, B: thermisch instabil)
Mg3P2 + 6HCl −→ 2PH3 + 3MgCl2
AsCl3 + 6H −→ AsH3 + 3HCl
4AsCl3 + 3LiAlH4 −→ 3LiCl + 3AlCl3 + 4AsH3
� PH3: Phosphan:
∗ farbloses, giftiges Gas∗ Geruch: Knoblauch
� P2H4: Diphosphan:
∗ farblose Flüssigkeit∗ selbstentzündlich∗ Disproportionierung zu PH3 und höheren Phosphanen
� AsH3: Arsan (Arsenhydrid)
∗ farbloses, giftiges Gas∗ thermische Zersetzung: As�Spiegel (Marsh'sche Probe)
9.5.4 Oxide des Sticksto�s
• N2O Lachgas:
� farblos
� reaktionsträge
� Zerfall erst bei Temperaturen über 600◦C
� Verwendung: Narkosegas

62 KAPITEL 9. HAUPTGRUPPENELEMENTE
� Herstellung: thermische Zersetzung:
NH4NO3200◦C−−−−→ N2O + 2H2O ∆H0 = −124kJ/mol
bei 300◦C : NH4NO3 −→ 2H2O +N2 + 1/2O2 → Exploison
� Struktur:
� Treibhausgas, zerstört Ozon
• NO, N2O2:
� NO farbloses, giftiges Gas 1/2N2 + 1/2O2→←− NO ∆H0 = +90kJ/mol
� technische Darstellung: Ostwald�Verfahren:
4NH3 + 5O2800−900◦C−−−−−−−→Pt−Kat
4NO + 6H2O
∗ Kontaktzeit am Pt�Netz ca 1 ms → Abschreckung → kein Zerfall in Elemente
� Labor:8H3O
+ + 2NO−3 + 3Ci −→ 3Cu2+ + 2NO + 12H2O
∗ |.
N = O||· paramagnetisch, Bindungsordnung: 2,5· nur mäÿig reaktiv, obwohl Radikal
∗ ||O =_
N −_
N = O|| schwache Bindung (Gleichgewicht: 2NO→←− N2O2)
∗ spontane Oxidation zu NO2: 2NO +O2 2NO2 ∆H0 = −114kJ/mol
• N2O3:
� blaue Flüssigkeit
� NO +NO2(Abkühlen)
N2O3Zerfall bei T>−10◦C
(Anhydrid des salpetrigen Säure)
� Anhydrid von HNO2: N2O3 + 2OH− −→ 2NO−2Nitrit
+H2O
� Abb.18 ↔ [NO+][NO−2 ] planar
• NO2, N2O4:
� NO2 braunes, giftiges Gas
2NO2braun
N2O4farblos
∆H0 = −57kJ/mol
� startke Oxidationsmittel
� NO2: gemischtes Anhydrid von HNO2 und HNO3
N2O4 + 2OH− −→ NO−3Nitrate
+NO−2 +H2O
� NO2�Reduktion: Nitrit NO−2
� NO2�Oxidation: Nitryl NO+2
• N2O5:
� Anhydrid der Salpetersäure
� 2HNO3 → H2O +N2O5 Zersetzung bei Raumtemperatur zu NO2, O2
� ↔ [NO+2 ][NO−3 ]

9.5. STICKSTOFFGRUPPE: GRUPPE 15 63
• NH2OH:
� Hydroxylamin (OH�Derivat von NH3):
∗ starkes Reduktionsmittel∗ Disproportionierung:
sauer: 4NH2OH −→ 2NH3 +N2O + 3H2Obasisch: 3NH2OH −→ NH3 +N2 + 3H2O
9.5.5 Sauersto��Säuren des Sticksto�
• HNO3 (Salpetersäure)
� Herstellung:N2O4 +H2O + 1/2O2 −→ 2HNO3
Teilschritte:1. N2O4 +H2O −→ HNO3 +HNO3
2. 3HNO2 −→ HNO3 + 2NO +H2O3. 2NO +O2 −→ N2O4
� Verwendung: Düngemittel (NH4NO3), Sprengsto�
� wasserfrei:
∗ farblose Flüssigkeit∗ teilweise Zersetzung beim Sieden: 4HNNO3 −→ 4NO2 + 2H2O + O2 (auch bei Raum-temperatur mit Licht)
� konzentrierte HNO3: 65%�ig
� rauchend: gelöstes NO2
� starkes Oxidationsmittel: (aber nicht mit Au, Pt)
NO−3 + 3e− + 4H3O+ −→ NO + 6H2O E0 = +0, 96V
Cu −→ Cu2+ + 2e−
Ag −→ Ag+ + e−
� Passivierung: Cr, Al, Fe
� Königswasser: löst auch Au, Pt (HCl : HNO3 = 3 : 1)
HNO3 + 3HCl −→ NOCl + 2H2O + 2Cl �aktives Chlor�
� Nitrate: NO−3
� Zersetzung:
∗ 1. Hauptgruppe: KNO3 −→ KNO2 + 1/2O2
∗ Schwermetalle: Hg(NO3)2 −→ HGO + 2NO2 + 1/2O2
∗ Verwendung: Oxdationsmittel, Düngemittel, Schwarzpulver 31.01.12
• HNO2 (salpetrige Säure)
� im Reinzustand nicht bekanntnur in verdünnter Lösung
� Disproportionierung (HNO3�Darstellung)
� Reduktionsmittel:+III
N O−2 �+V
N O3 + 2e− + 2H3O+
� Oxidationsmittel:+III
N O−2 + e− + 2H3O+
+II
N O + 3H2O E0 = +0, 996V
� Abb.1
� Darstellung:
KNO3∆T−−→ KNO2 + 1/2O2
NO +NO2 + 2NaOH −→ 2NaNO2 +H2O

64 KAPITEL 9. HAUPTGRUPPENELEMENTE
9.5.6 Oxide des Phosphors
P4O6, P4O10
• P4O6:P4 + 3O2
stöchiom. Menge−→ P4O6 ∆H0
B = −1641kJ/mol
� Struktur: abgeleitet von P4, P�O�P�Bindungen statt P�P�Bindungen
� giftig
� beständig an Luft bei Raumtemperatur, aber bei 70◦C Reaktion zu P4O10 → Anhydrid derPhosphon�Säure H3PO3(H2PHO3)
6H2O + P4O6 −→ 4H3PO3
• P4O10:P4 + 5O2 −→ P4O10 ∆H0
B = −2986kJ/mol
� Trocknungsmittel
� sehr heftige Reaktion mit H2O:
P4O10 + 6H2O −→ 4H3PO4ortho�Phosphorsäure
∆H0 = −378kJ/mol
� kein Oxidationsmittel
9.5.7 Sauersto�säuren des Phosphors
Durch Kondensation könnendann beispielsweise
Trimetaphosphorsäure(Metaphosphorsäuren:(HPO3)n, Ringe) oder
Triphosphorsäure(Polyphosphorsäuren:Hn+2PnO3n+1, Ketten)entstehen.
Besonderheit:
• Wenn ein freies EP am Phosphor vorhande wäre ⇒ Isomerisierung (siehe Abb.2)
• Kondensation (H2O�Abspaltung):

9.6. KOHLENSTOFF: GRUPPE 14 65
� Polyphosphorsäuren Hn+2PnO3n+1
� Metaphosphorsäure: Ringe (HPO3)n
� Monophosphoräuren: H3POn
� Diphosphorsäuren: H4P2On
• Orthophosphorsäure: H3PO4
� farblose Kristalle
� gut lösich in H2O
� 85 %�ig im Handel
� mittelstarke, 3�basige Säure
� Salze:
∗ Dihydrogenphosphate: H2PO−4
∗ Hydrogenphosphate: HPO2−4
∗ Phosphate: PO3−4
� Bedeutung:
∗ Phosphatpu�er∗ Zusatz in Cola
� Korrosionsschutz von Stahl (Zink�Phosphorisierung)
� Düngemittel
� Backpulver
� Herstellung:
∗ nasser Aufschluss:
Ca3(PO4)2 + 3H2SO4 −→ 3CaSO4 + 2H3PO4
∗ trockener Aufschluss:
P4 + 5O2 −→ P4O10Hydrolyse−−−−−−−→ H3PO4
9.5.8 Arsenverbindungen
Struktur von Arsensul�den
9.6 Kohlensto�: Gruppe 14
C Si Ge Sn PbOrdnungszahl 6 14 32 50 82e−-Kon�guration [He]2s22p2 [Ne]3s23p2 [Ar]3d104s24p2 [Kr]4d105s25p2 [Xe]4f145d106s26p2
Elektronegativität 2, 5 1, 7 2, 0 1, 7 1, 6IE [eV] 11, 3 8, 1 7, 9 7, 3 7, 4Smp [◦C] 38001 1410 947 232 327
1 Graphit bei 0, 2bar
9.6.1 Vorkommen
• C:
� elemenar als Graphit, Diamant
� chem. gebunden: CarbonateCaCO3: Kalkstein, Kreide, MarmorCaCO3 ∗MgCO3: DolomitMgCO3: Magnesit

66 KAPITEL 9. HAUPTGRUPPENELEMENTE
� Bestandteil von Lebewesen
� Kohle, Erdöl, Erdgas
� CO2 in Luft (0,038 %)
• Si:
� zweithäu�gstes Element der Erdkruste
� nicht elementar, überwiegend als SiO2 (→ Sand) und in Silikaten
• Ge:
� in seltenen sul�dischen Mineralien
• Sn:
� selten elementar, sonst in Erzen (SnO2, Zinnstein, Cu2FeSnS4 (Stannin))
• Pb:
� PBS Bleiglanz
� PbCO3: Weiÿbleierz
� PbCrO4: Rotbleierz
� PbMnO4: Gelbbleierz
� Blei(II)�Verbindungen stabiler als Blei(IV)�Verbindungen
9.6.2 Elemente
• C:Diamant Graphit
Hybridisierung: sp3 sp2
Struktur: 3D SchichtstrukturHärte: sehr hart Verschiebbarkeit der Schichten
→ Schleifmittel, Bohrer, Glasschneider → SchmiermittelElektronen: alle e− lokalisiert delokalisierte (p�p)�π�Bindungen
über gesamte SchichtEigenschaften: → farblos, nichtleitend schwarz, elektrisch leitend,
Anwendung als ElektrodenmaterialVerbrennung: zu CO2: 800◦C zu CO2: 700◦C
� CDiamant −→ CGraphit ∆H = −1, 9kJ/mol
� Phasendiagramm Kohlensto�:
� Diamanten:
∗ thermodynamisch stabiler bei hohen Drücken
∗ Umwandlung von Graphit in Diamant: für ausreichende Geschwindigkeit: 1500◦C, 60 kbar(in Gegenwart von Fe, Co, Ni, Pt katalysierte Reaktion) → synthetische Diamanten
∗ Diamantenherstellung: neuerer Methode CWD
∗ natürliche Diamanten: entstanden unter Druck
� Graphit:
∗ künstliches Graphit durch thermische Zersetzung von Kohle, Erdöl, Erdgas

9.6. KOHLENSTOFF: GRUPPE 14 67
∗ Struktur:
∗ spezielle Kon�gurationen: Koks, Ruÿ, Holzkohle, . . .Bezeichnung Eigenschaften und VerwendungRuÿ Reifenindustrie, Pigment für FarbenAktivkohle groÿes Adsorbtionsvermögen → KatalyseFaserkohlensto� leichtes, stabiles Material (→ Sport)Pyrographit hohe Anisotropie (Temperaturbeständigkeit)
→ Hitzeschilde, RöntgenmonochromatorenGlaskohlensto� leicht, spröde, hart → MedizintechnikGraphitfolien Dichtungen
� Fullerene:
∗ neuere Modi�kation des Kohlensto�s
• Si, Ge, Sn (graues Sn): 02.02.12
� Diamantgitter
� Eigenhalbleiter (kleiner Teil der e− delokalisiert)
� durch Dotierung mit As, Ga → n�, p�dotierte Halbleiter
α− Sngraues Sn
130◦C β − Sn
weiÿes SnH0 = +2kJ/mol
• Pb:
� typ. Metallgitter kdp
� läuft wie Alkalimetalle an Luft an
• Si, Ge, Sn, Pb:
� keine Graphit�Struktur
9.6.3 Darstellung und Verwendung
• Si:
� technische Darstellung:
SiO2 + 2C1800◦C−−−−−→ Si+ 2CO ∆H0 = +690kJ/mol
� für Halbleitertechnik braucht man hochreines Si:
Si+ 3HCl300◦C
1100◦CHSiCl3 +H2
� weitere Aufreinigung:
∗ Einkristalle (nicht viele kleine Kristalle zusammen, sondern nur ein groÿer):1. Zonenschmelzen2. Czochralski�Verfahren
• Sn:SnO2 + 2C −→ Sn+ 2CO ∆H0 = +360kJ/mol

68 KAPITEL 9. HAUPTGRUPPENELEMENTE
� Recycling von Weiÿblech (verzinntes Fe�Blech → elektrolytische Abscheidung)
� beständig an Luft, Wasser, aber Reaktion mit:
∗ Säuren:Sn+ 2HCl −→ SnCl2 +H2
∗ Basen:
Sn+ 4H2O + 2OH− −→ [+IV
Sn (OH)6]2− + 2H2
• Pb:PbS + 1, 5O2 −→ PbO + SO2
PbO + CO −→ Pb+ CO2
� Passivierung mit: H2SO4, HCl, HF
� lösich in HNO3 und heiÿen Laugen
� wird von Wasser an Luft angegri�en: Pb+ 1/2O2 +H2O −→ Pb(OH)2
� Verwendung:∗ Pb�Rohre
∗ Geschosse
∗ Akku�Platten
∗ Legierungen
9.6.4 C�Verbindungen
• Carbide:
� Verbindungen mit Metallen, Si, B (C mit gröÿeren EN)
� Kovalent: SiC
∗ sehr hart
∗ chemisch resistent
∗ Schleifmittel
∗ Bohrer
∗ hochtemperaturbeständige Bauteile
∗ Darstellung: SiO2 + 3C2200◦
−−−→ SiC + 2CO ∆H0 = +625kJ/mol
� Salzartig: CaC2
∗ → Ca2+ + C2−2
∗ Struktur:
∗ Darstellung: CaO + 3C2200◦C−−−−−→ CaC2 + CO ∆H0 = +465kJ/mol
∗ Herstellung von Acetylen: CaC2 + 2H2O −→ Ca(OH)2 +HC ≡ CH∗ Herstellung vo Kalksticksto�: CaC2 +N2 −→ CaCN2
Düngemittel+ C
CaCN2 + 3H2O −→ CaCO3 + 2NH3
• Oxide von C:
� CO:
∗ farbloses, geruchsloses Gas
∗ sehr giftig
∗ isoelektrisch zu Sticksto�: |C ≡ O|⊕C + 1/2O2 −→ CO ∆H0
B = −111kJ/mol
∗ technische Erzeugung: → H2�Darstellung Synthesegas
∗ Labor: HCOOHH2SO4−−−−→ H2O + CO
∗ Verbrennung an Luft: CO + 1/2O2 → CO2 ∆H0 = −283kJ/mol

9.6. KOHLENSTOFF: GRUPPE 14 69
∗ Reduktionsmittel: z.B. Fe�Oxide bei höherer Temp. → Hochofenprozess
Gichtgas:· 55 % Stichsto�· 30 % CO· 15 % CO2
· guter Brennwert∗ mit Metallen Carbonlykomplexe:· Beispiel: Reinigung von Ni (Mond�Verfahren)
Ni+ 4CO80◦C
180◦CNi(CO)4
� CO2:
∗ farbloses, geruchsloses Gas∗ nicht brennbar, unterhält Verbrennung nicht (⇒ Feuerlöscher)∗ schwerer als Luft → Erstickungsgefahr∗ leite Ver�üssigung unter Druck∗ kristallin in festem Zustand → Trockeneis → Sublimiert bei −78◦C
∗ gut löslich in H2O: → CO2�haltige Getränke ||O = C = O||∗ Darstellung:
C +O2 −→ CO2 ∆H0B = −394kJ/mol
· Kalkbrennen:CaCO3
1000◦C−−−−−→ CaC + CO2
· Labor:CaCO3 + 2HCl −→ CaCl2 + h2O + CO2
· Boudouard�Gleichgewicht:
CO2 + C(s) 2CO ∆H0 = +173kJ/mol
· Wassergas�Gleichgewicht:
CO2 +H2 Co+H2O ∆H0 = +41kJ/mol
∗ Bedeutung: Tiere / P�anzen: Atmung, Photosynthese∗ 0,038 % CO2 in der Atmosphäre→ CO2 absorbiert Wärmestrahlung der Erdober�äche → Treibhause�ekt
� Kohlensäure, Carbonate:
CO2 +H2O H2CO3 pK = 2, 6
H2CO3 +H2O H3O+ + HCO−3
HydrogencarbonatpKS = 3, 8
HCO−3 +H2O H3O+ + CO2−
3 pKS = 10, 3

70 KAPITEL 9. HAUPTGRUPPENELEMENTE
∗ H2CO3 nicht isolierbar aus wässriger Lösung
∗ Bedeutung: Wasserhärte:
· temporäre Härte (Carbonat�Härte)
CaCO3 +H2O + CO2 Ca2+ + 2HCO−3
Erwärmen verringert sich die Ca2+�Konzentration (le Châtelier)
· permanente Härte (Sulfat�Härte)CaSO4
∗ Derivate des H2CO3:
· Harnsto�: Düngemittel
CO2 + 2NH360bar−−−→ CONH2ONH4
−H2O−−−−→ CO(NH2)2
· Phosgen: Giftgas CO + Cl2 −→ COCl2
• N�Verbindungen des C:
� HCN: Blausäure
∗ farblose, giftige Flüssigkeit
∗ Geruch: Bittermandel
∗ sehr schwache Säure
∗ 2 Tautomere: |C ≡ N −H07.02.12
∗ Darstellung:
CH4 +NH31200◦C−−−−−→Kat
HCN + 3H2
∗ Salze:
· Cyanide CN− ⇒ |C ≡ N |· Reaktion mit Säure: 2KCN +H2O + CO2 −→ K2CO3 + 2HCN
· Cyanide auch durch Einleiten von HCN in Lauge → Cyanidlaugerei:Lösen von Ag, Au aus Erzen: 4Ag+ 8CN−+ 2H2O+O2 −→ 4[Ag(CN)2]−+ 4OH−
· Dicyan (CN)2 |N ≡ C − C ≡ N | → Pseudohalogen
· Cyansäure: HOCN 2 Tautomere: H −O − C ≡ N | O = C = N −HSalze stabil: |O − C ≡ N | ↔ O = C = N
· Thiocyanate: |S − C ≡ N |
• Halogen� und Schwefelverbindungen:
� CCl4:
∗ Lösungsmittel
∗ Feuerlöschmittel
� CF4:
∗ Polytetra�ourethylen → Te�on
� CS2:
∗ farblose, giftige Flüssigkeit
∗ Lösungsmittel: Fette, Öle, Phosphor, Schwefel

9.6. KOHLENSTOFF: GRUPPE 14 71
9.6.5 H�Verbindungen von Si
• Kettenförmige Silane: SinH2n+2 n <= 15
• endotherme Verbindungen, beständig bei RT (ohne O2, H2O)
• bei Erhitzen:SiH4 −→ So+ 2H2 ∆H0 = −34kJ/mol
• Selbstentzündung an Luft: SiO2 +H2O
•δ−C −
δ+
Hδ+
Si−δ−H
9.6.6 O�Verbindungen von Si
• SiO2:
� polymerer harter Feststo�
� Struktur:
� hohe Bindungesenergie durch π�Bindungsanteile
� Modi�kationen:
� rasches Abkühlen von SiO2�Schmelzen: Gläser → nicht�kristallin
� Vorkommen in der Natur:
∗ Kristalle:
· Bergkristall (farblos)(Quarz: Mineral mit chemischer Formel SiO2)
· Amethyst (lila)(mit Metallen wie Al, Fe, Ca, Mg, Li, Na verunreinigter Quarz)
· Rosenquarz (rosarot)(Varietät des Minerals Quarz mit Einschlüssen von Dumortierit, ein Borsilikat, daswiederrum Einschlüsse von Eisen und Titan enthält, die ihm die Farbe verleihen)
∗ mikrokristallin
· Achat (alle Farben auÿer kobaltblau, magenta, lila und pink, immer gestreift)(mikrokristalline Varietät des Minerals Quarz)
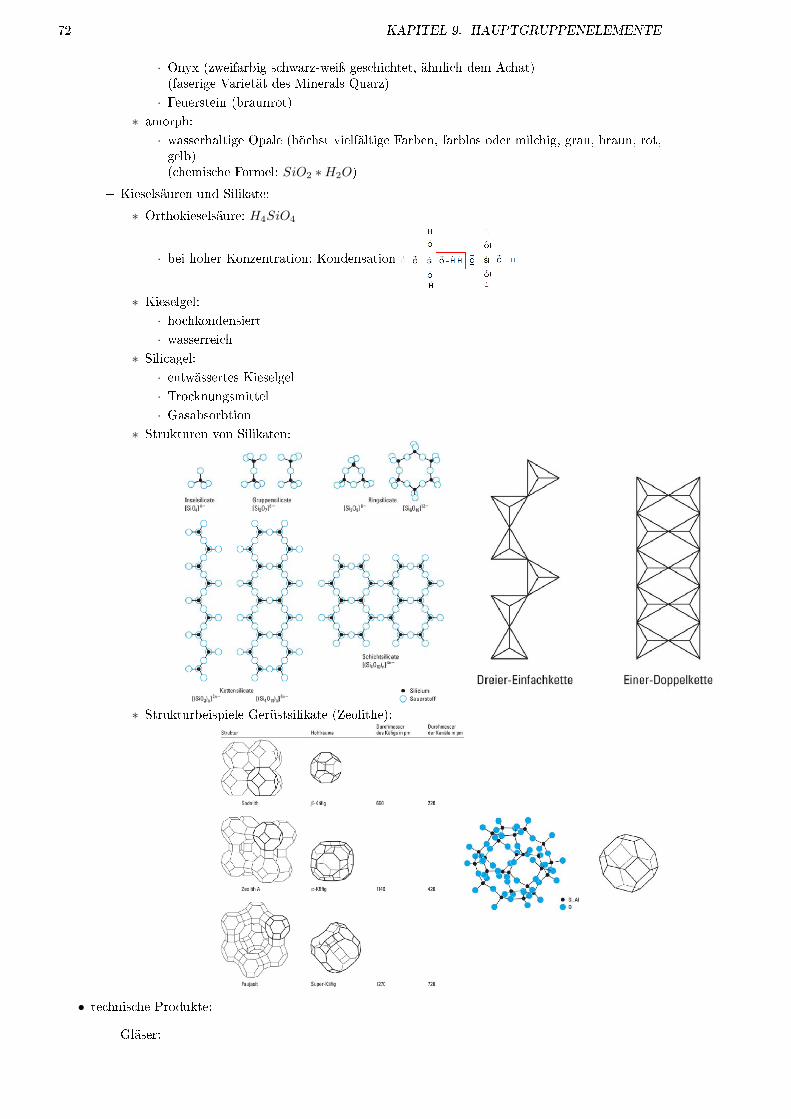
72 KAPITEL 9. HAUPTGRUPPENELEMENTE
· Onyx (zweifarbig schwarz-weiÿ geschichtet, ähnlich dem Achat)(faserige Varietät des Minerals Quarz)
· Feuerstein (braunrot)
∗ amorph:
· wasserhaltige Opale (höchst vielfältige Farben, farblos oder milchig, grau, braun, rot,gelb)(chemische Formel: SiO2 ∗H2O)
� Kieselsäuren und Silikate:
∗ Orthokieselsäure: H4SiO4
· bei hoher Konzentration: Kondensation
∗ Kieselgel:
· hochkondensiert· wasserreich
∗ Silicagel:
· entwässertes Kieselgel· Trocknungsmittel· Gasabsorbtion
∗ Strukturen von Silikaten:
∗ Strukturbeispiele Gerüstsilikate (Zeolithe):
• technische Produkte:
� Gläser:

9.7. ALKALIMETALLE: GRUPPE 1 73
∗ nur Nahordnung
∗ metastabil
∗ Netzwerkbildner: SiO2, B2O3, P2O5, . . .
∗ Netzwerkwandler: Na2O, K2O, CaO, . . .
� Keramiken:
∗ Glaskeramik:
· durch teilweise Entglasung
∗ Tonkeramik:
· durch Brennen von Tonen (Schichtsilikate)
∗ Wasserglas:Si2O + 2Na2CO3 −→ Na4SiO4 + 2CO2
∗ Silikone:
· Struktur:
· Silikonherstellung: R = CH3, Ph
6RCl + 3Si300−400◦C−−−−−−−→ RSiCl3 +R2SiCl2 +R3 + SiCl
Hydrolyse:
RSi(OH)3Verzweigungsstelle
+R2Si(OH)2Kettenglied
+R3Si(OH)Kettenende
−→ Silikone
Anwendung: Schmier� und Isolationsmittel, Dichtungen, Släuche, Kabel . . .
9.7 Alkalimetalle: Gruppe 1
Li Na K Rb CsOrdnungszahl 3 11 19 37 55e−-Kon�guration [He]2s1 [Ne]3s1 [Ar]4s1 [Kr]5s1 [Xe]6s1
Elektronegativität 1, 0 1, 0 0, 9 0, 9 0, 9IE [eV] 5, 4 5, 1 4, 3 4, 2 3, 9Smp [◦C] 181 98 64 39 28Sdp [◦C] 1347 881 754 688 705Dichte (20◦C) [g/cm3] 0, 53 0, 97 0, 86 1, 53 1, 90Ionenradius1 [pm] 76 102 138 152 167HE von M+ [kJ/mol] −519 −406 −322 −293 −264Standardpotential [V] −3, 04 −2, 71 −2, 92 −2, 92 −2, 92Flammenfärbung kaminrot gelb violett violett blau
IE: Ionisierungsenergie HE: Hyratationsenthalpie 1 für M+ bei KZ 6
Lithium � Magnesium: Schrägbeziehung (ähnliche Eigenschaften)
9.7.1 Vorkommen
• sehr reaktiv → nur gebunden
• Li: Spodumen LiAl[Si2P6]
• Na: Tektosilikate:
� Naturfeldspat Na[AlSi3O8]
� Steinsalz NaCl (auch 3 % in Meerwasser)
� Soda Na2Co3 ∗ 10H2O
� Kryolith Na3AlF6
• K: wie Na im Salzlaugen

74 KAPITEL 9. HAUPTGRUPPENELEMENTE
� Sylvin KCl
� Carrnallit KCl ∗MgCl2 ∗ 6H2O
� Kalifeldspat K[AlSi3O8]
• Rb, Cs: zusammen mit anderen
9.7.2 Darstellung
• chem. Reduktion aus Verbindungen schwierig wegen E0
→ häu�g elektrochem. Reaktionwichtig: H2O-frei → Schmelzen
• Na:
� elektrolytisch aus:
∗ NaCl�Schmelze → Downs�VerfahrenSmp. von NaCl (808◦C) wird durch 60 % CaCl2 auf ca. 600◦C herabgesetztKath: 2Na+ + 2e− −→ 2NaAmode: 2Cl −→ Cl2 + 2e−
∗ (NaOH�Schmelze → Castner�Verfahren) technisch nicht bedeutend
• Li:
� Schmelzelektrolyse von LiCl/KCl�Mischung
• K:
� Reduktion von KCl�Schmelze mit Na ⇒ Na�K�Legierung ⇒ reines K durch Destillation
• Rb, Cs:
Cs2Cr2O7 + 2Zr500◦C−−−−→V ac.
2Cs+ 2ZrO2 + Cr2O3
9.7.3 Verwendung
• Li:
� Batterien
� Legierungen:
∗ Härten von Pb, Mg, Al
∗ Flugzeugbau
� Li2CO3:
∗ Al�Herstellung
∗ Glas�, Keramikindustrie
� 6Li2H: Kernfusion
� Li+: Medikamente gegen Depressionen
• Na:
� Synthese von Na2O2, NaNH4, NaH, NaCN
� früher bedeutend: Na�Pb�Legierung→ Tetraethylblei: Pb(C2H5)4 (Antiklopfmittel in Benzin)
� Ti�Darstellung:

9.7. ALKALIMETALLE: GRUPPE 1 75
∗ Hunter�Verfahren:
TiCl4 + 4Na −→ Ti+ 4NaCl ∆H0 = −869kJ/mol
� Beleuchtungstechnik: Na�Damp�ampen
� Reduktionsmittel im Labor
� Trocknung org. Lösungsmittel
• Cs:
� 137Cs: Strahlenquelle in Medizin
9.7.4 Verbindungen
• Hydride: 2M +H2 −→ 2 MHNaCl�Struktur
• LiH:
Li+ 0, 5H2600◦C−−−−→ LiH ∆H0
B = −91kJ/mol
LiH +H2O −→ LiOh+H2
4LiH +AlCl3Ether−−−−→ LiAlH4
Redunktionsmittel+ 3LiCl
• NaH:
Na+ 0, 5H2300◦C−−−−→ NaH ∆H0 = −57kJ/mol
BF3 + 4NaH125◦C−−−−→Ether
NaBH4Reduktionsmittel
+ 3NaF
9.7.5 O�Verbindungen
Alkalimetalle bei Erhitzen an Luft:4Li+O2 → 2Li2O Lithiumoxid[6Li+N2 −→ 2Li3N ]2Na+O2 −→ Na2O2 NatriumperoxidM +O2 −→MO2 M�Hyperoxid (M = K, Rb, Cs)
• Oxide: M2O:
� Li2O
∗ Glasindustrie
∗ therm. Zersetzung von LiOH, Li2CO3, LiNO3
� Na2O:
∗ hygroskopisch:Na2O2 + 2Na −→ 22Na2O
NaNO3 + 5NaN3 −→ 3Na2O + 8N2
• Peroxide: M2O2:
� Na2O2:
∗ stabil bis 500◦C
∗ starkes Oxidationsmittel → Papier�, Textilbleiche
M2O2 +H2SO4 −→M2SO4 +H2O2
M2O2 + CO2 −→M2CO3 + 1/2O2
• Hyperoxide: MO2:
� nur stabil mit K, Rb, Cs
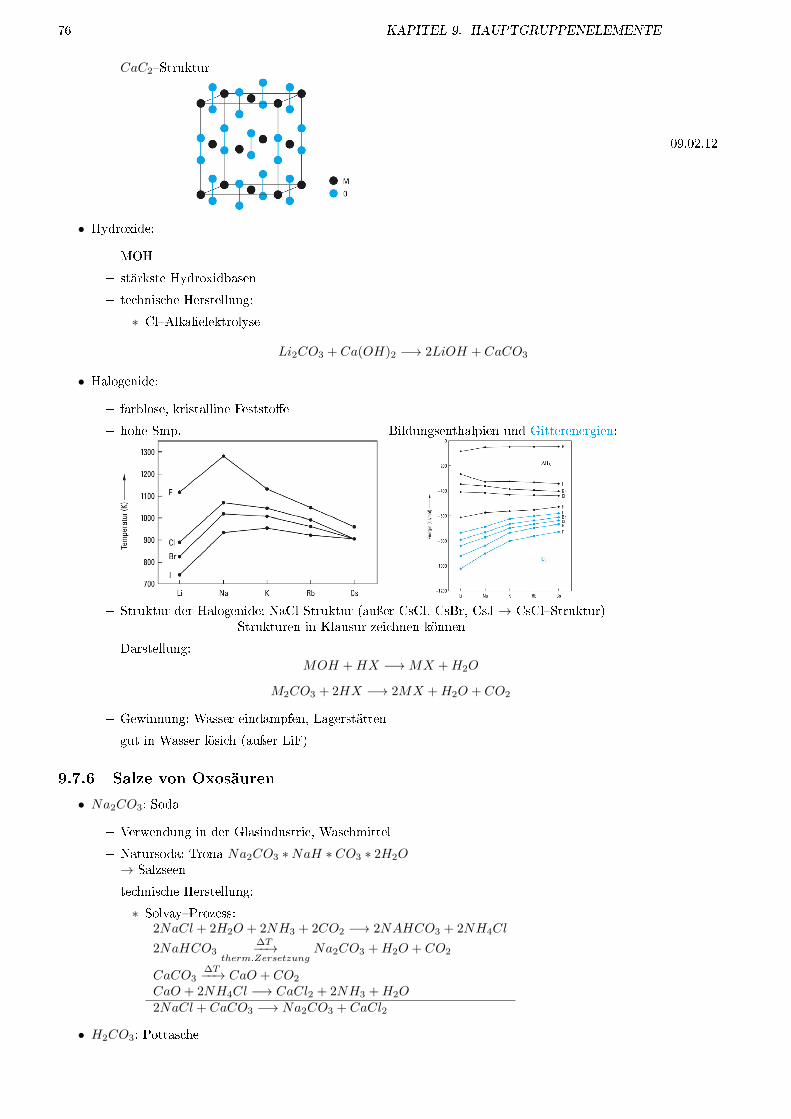
76 KAPITEL 9. HAUPTGRUPPENELEMENTE
� CaC2�Struktur
09.02.12
• Hydroxide:
� MOH
� stärkste Hydroxidbasen
� technische Herstellung:
∗ Cl�Alkalielektrolyse
Li2CO3 + Ca(OH)2 −→ 2LiOH + CaCO3
• Halogenide:
� farblose, kristalline Feststo�e
� hohe Smp. Bildungsenthalpien und Gitterenergien:
� Struktur der Halogenide: NaCl-Struktur (auÿer CsCl, CsBr, CsJ → CsCl�Struktur)Strukturen in Klausur zeichnen können
� Darstellung:MOH +HX −→MX +H2O
M2CO3 + 2HX −→ 2MX +H2O + CO2
� Gewinnung: Wasser eindampfen, Lagerstätten
� gut in Wasser lösich (auÿer LiF)
9.7.6 Salze von Oxosäuren
• Na2CO3: Soda
� Verwendung in der Glasindustrie, Waschmittel
� Natursoda: Trona Na2CO3 ∗NaH ∗ CO3 ∗ 2H2O→ Salzseen
� technische Herstellung:
∗ Solvay�Prozess:2NaCl + 2H2O + 2NH3 + 2CO2 −→ 2NAHCO3 + 2NH4Cl
2NaHCO3∆T−−→
therm.ZersetzungNa2CO3 +H2O + CO2
CaCO3∆T−−→ CaO + CO2
CaO + 2NH4Cl −→ CaCl2 + 2NH3 +H2O2NaCl + CaCO3 −→ Na2CO3 + CaCl2
• H2CO3: Pottasche

9.8. ERDALKALIMETALLE: GRUPPE 2 77
� nicht analog zum Solvay�Prozess
2KOH + CO2 −→ K2CO3 +H2O
• Na2SO4:2NaCl +MgSO4 −→MgCl2 +Na2SO4
2NaCl +H2SO4 −→ 2HCl +Na2SO4
� Verwendung in der Glas�, Textil� und Papierindustrie
• NaNO3: Chilesalpeter
� Verwendung:
∗ KNO3�Herstellung∗ Düngemittel
Na2CO3 + 2HNO3 −→ 2NaNO3 +H2O + CO2
• KNO3: Kalisalpeter
� Verwendung:
∗ Pyrotechnik∗ Schwarzpulverbestandteil∗ Düngemittel
K2CO3 + 2HNO3 −→ 2KNO3 +H2O + CO2
NaNO3 +KCl KNO3 +NaCl
• Löslichkeit von MNO3 und MCl (M = Na, K):
9.8 Erdalkalimetalle: Gruppe 2
Be Mg Ca Sr BaOrdnungszahl 4 12 20 38 56e−-Kon�guration [He]2s2 [Ne]3s2 [Ar]4s2 [Kr]5s2 [Xe]6s2
Elektronegativität 1, 5 1, 2 1, 0 1, 0 0, 91.IE [eV] 9, 3 7, 6 6, 1 5, 7 5, 22.IE [eV] 18, 2 15, 0 11, 9 11, 0 10, 0Smp [◦C] 1285 650 845 771 726Sdp [◦C] 2477 1105 1483 1385 1696Dichte (20◦C) [g/cm3] 1, 85 1, 74 1, 54 2, 63 3, 62Ionenradius1 [pm] 45 72 100 118 135HE von M2+ [kJ/mol] −2494 −1921 −1577 −1443 −1305Standardpotential [V] −1, 85 −2, 36 −2, 87 −2, 89 −2, 90
IE: Ionisierungsenergie HE: Hydratisierungsenthalpie 1: für M2+ bei KZ 6
• Häu�g in Oxidationsstufe +2
• Darstellung über Schmelzelektrische Verfahren
• teilweise char. Flammenfärbungen
• reagieren auch mit Luftsticksto� zu Erdalkalinitriden
• Mg dritthäu�gste Ionenkonzentration in Meerwasser

78 KAPITEL 9. HAUPTGRUPPENELEMENTE
9.8.1 Vorkommen
• Be:
� relativ selten, z.B. Beryll Be3Al2[Si6O18]
� Cr-haltig Smaragd (grün)
� Fe-haltig Aquamarin (hellblau)
• Mg:
� Dolomit CaMg(CO3)2
� Magnesit MgCO3
� Silikate:
∗ Olivin: (Mg,Fe)2SiO4
∗ Talk Mg3[Si4O10](OH)2
� Spinell MgAl2O4
� 0, 13% Mg im Meerwasser
• Ca:
� gesteinsbildende Mineralien
� Feldspate, z.B. Ca[Al2Si2O8]
� Kalkstein, Kreide, Marmor CaCO3
� Gips CaSO4 ∗ 2H2O, Anhydrit CaSO4
� Dolomit, Apatit Ca5(PO4)3(OH,Cl, F )
� Flussspat CaF2
• Sr:
� Strontianit SrCO3
� Cölestin SrSO4
• Ba:
� Whiterit BaCO3
� Schwerspat BaSO4
9.8.2 Darstellung
• Schmelzelektrolyseverfahren:
� Be: BeCl2/NaCl (Gemisch)
� Mg: MgCl2
� Ca: CaCl2/CaF2 (Gemisch)Eisenkathoden in Schmelze → langsamen herausziehen aus Schmelze und Ca erstarrt in Formvon langen Stangen
� Sr: entsprechend Ca
� Ba: BaO aus Zersetzung von BaCO3
3BaO + 2Al −→ 3Ba+Al2O3
3BaO + Si −→ 2Ba+BaSiO3

9.8. ERDALKALIMETALLE: GRUPPE 2 79
9.8.3 Verbindungen
• Be:
� Schrägbeziehung zu Al
� Be�Bindungen sehr eher kovalent als bei anderen Erdalkalimetallen
� höhere Ionisierungsenergie
� höhere Elektronegativität
� Be2+ sehr klein→ Be�Verbindungen kovalenter
� lineare BeX2�Moleküle nut sp�Hybridisierung ⇒ e−�Mangelverbindungen
� Erhöhung der KZ auf 4 durch:
1. Dreizentrenbildungen: (BeH2)n (Abb.1) ⇒ Be�H�Be�Dreizentren�2e−�Bingungen2. koordinative Bindungen: I(BeCl2)n (Abb.2)
• Mg:
� Oxide entstehen bei Verbrennung an Luft
� Magnesiumhydrid MgH2:
∗ Mg +H2 −→MgH2 ∆H0B = −74 kJ
mol
∗ aus Diethylmagnesium: Mg(C2H5)2 −→MgH2 + 2C2H4
� Magnesiumoxid MgO:
∗ Verbrennung an Luft: Mg + 1/2O2 −→MgO ∆H0B = −602 kJ
mol
∗ technisch: thermische Zersetzung von MgCO3 −→MgO + CO2
� Magnesiumhydroxid Mg(OH)2:
MgCl2Ca(OH)2 −→Mg(OH)2 + CaCl2
� Magnesiumcarbonat MgCO3:
∗ Magnesit, wichtigstes Mg-Mineral, groÿe Lagerstätten∗ Herstellung von MgO
� Grignard-Verbindungen RMgX:
∗ RX +Mg −→ RMgX (R = org. Reste, Lösungsmittel: Ether)∗ organische Chemie: Alkylierungen, Arylierungen
• Ca:
� Calciumhydrid CaH2:
∗ Ca+H2 −→ CaH2 ∆H0B = −186 kJ
mol
∗ H2-Entwicklung in Wasser: CaH2 + 2H2O −→ Ca(OH)2 + 2H2
� Calciumoxid CaO (Ätzkalk, gebrannter Kalk):
∗ Kalkbrennen: CaCO31000−1200◦C−−−−−−−−−→ CaO + CO2
∗ Bedeutung bei Stahl-Produktion
� Calciumhydroxid Ca(OH)2 (gelöschter Kalk):
∗ H2O + CaO −→ Ca(OH)2 ∆H0 = −65 kJmol
∗ Luftmörtel (wasseranfällig)
∗ billigste Base ⇒ technishce Verwendung (Soda-, Chlorkalk-Synthese)
� Calciumchlorid CaCl2:
∗ Abfallprodukt bei Sodaherstellung∗ Trocknungsmittel
� Calciumcarbonat CaCO3:
∗ Modi�kationen:· Calcit (Kalkspat): Kalkstein, Marmor, Kreide

80 KAPITEL 9. HAUPTGRUPPENELEMENTE
· Aragonit: Perlen· Vaterit (Mineral aus der Mineralklasse Carbonate)
� Calciumsulfat CaSO4:
∗ Gips: CaSO4 ∗ 2H2O
∗ gebrannter Gips: CaSO4 ∗ 0, 5H2O
∗ Stuckgips: CaSO4 ∗ (0, 18− 0, 48)H2O
∗ Anhydrit: CaSO4
� Calciumphosphate (z.B. Ca(H2PO4)2):
∗ Hauptbestandteil von Phosphatdüngemitteln
∗ Backpulver
∗ Zahncremes
� Calciumcarbid CaC2 und Kalksticksto� CaCN2: siehe Kohlensto�
� Mörtel:
∗ Luftmörtel:
· Kalkmörtel: besteht aus gelöschtem Kalk und Sand, schwindet beim ErhärtenErhärtung: Ca(OH)2 + CO2 −→ CaCO3 +H2O
· Gipsmörtel: dehnt sich beim Erhärten aus
∗ Wassermörtel: Zement
· Durch Brennen von Kalkstein/Ton-Mischungen bei 1450◦C
· Erhärtet auch unter Wasser
• Ba:
� Bariumsulfat BaSO4:
∗ nicht löslich in Wasser, chemisch sehr beständig
∗ Malerfarbe (�Permanentweiÿ�)
∗ Füllsto� in Papier- und Gummiindustrie
� Bariumoxid BaO:
∗ NaCl-Struktur∗ Technisch: BaCO3 + C −→ BaO + 2CO
� Bariumperoxid BaO2: siehe Sauersto� (Seite 56)
� Wasserlösliche Ba�Verbindungen giftig
� Ba2O2 einizeges stabiles Peroxid der Erdalkalimetalle
9.9 Borgruppe: Gruppe 13
B Al Ga In TlOrdnungszahl 5 13 31 49 81e−-Kon�guration [He]2s22p1 [Ne]3s23p1 [Ar]3d104s24p1 [Kr]4d105s25p1 [Xe]4f145d106s26p1
Elektronegativität 2, 0 1, 5 1, 8 1, 5 1, 41. IE [eV] 8, 3 6, 0 6, 0 5, 8 6, 12. IE [eV] 25, 1 18, 8 20, 5 18, 9 20, 43. IE [eV] 37, 9 28, 4 30, 7 28, 0 29, 8Smp [◦C] 21801 660 30 156 302Sdp [◦C] 3660 2467 2400 2080 1457Dichte [g/cm3] 2, 462 2, 70 5, 91 7, 31 11, 85SE [kJ/mol] 5701 327 277 243 182Standardpotential4 [V] −0, 873 −1, 68 −0, 53 −0, 34 +0, 72
IE: Ionisierungsenergie SE: Sublimationsenthalpie 1β-rhomboedrisches Bor 2α-rhomboedrisches Bor3 B/B(OH)3
4 für M/M3+

9.9. BORGRUPPE: GRUPPE 13 81
9.9.1 Vorkommen
• B:
� Borate (z.B. Borax Na2B4O7 ∗ 10H2O)
� Borsäure H3BO3 in heiÿen Quellen
• Al:
� häu�gstes Metall der Erdrinde, dritthäu�gstes Element
� Feldspate (Albit Na[AlSi3O8]), Glimmer (Muskovit KAl2[AlSi3O10](OH)2), Schichtsilikate(Kaolinit Al4[SiO10](OH)8)
� Al2O3: Korund; Rubin (rot), Saphir (blau)
� Kryolith Na3AlF6 (weitgehend abgebaut)
� Bauxit: Gemenge aus Böhmit/Diaspor AlO(OH) und Hydrargillit Al(OH)3
(Bauxit enthält auch Fe-Verbindungen)
• Ga, In:
� Begleiter von Zn in Zinkblende
• Tl:
� Begleiter von Zn in Zinkblende und von Fe in Pyrit
9.9.2 Modi�kationen und Darstellung
• Bor:
� kovalente Bindungen wegen hoher IE und groÿer EN (alle anderen Elemente mit weniger alsvier Valenzelektronen: Metallgitter)
� einziges Element der Nichtmetalle, das weniger als 4 Valenzelektronen hat
� komplizierte Strukturen (Grund: vier Valenzorbitale, aber nur drei Elektronen ⇒ Elektronen-mangel, Raumnetzstrukturen nur durch Mehrzentrenbindungen)
� Mehrzentrendindung bei B:
� B12�Ikosaeder: Ikosaedrische Struktur (20�Flächner)
� mehrere bekannte Modi�kationen, alle enthalten B12-Ikosaeder, wobei alle B-Atome äquivalentsind und 5 Nachbarn haben
� Darstellung:
∗ Darstellung in hoher Reinheit schwierig (B korrosiv, Bildung von Metallboriden, Reduktionvon CO2 und SiO2)
∗ Reduktion von Borhalogeniden mit Wassersto�:
2BCl3 + 3H21000−1400◦C−−−−−−−−−→ 2B + 6HCl ∆H0 = +262
kJ
mol

82 KAPITEL 9. HAUPTGRUPPENELEMENTE
∗ thermische Zersetzung von BI3 an Wolframdrähten:
2BI3800−1000◦C−−−−−−−−→ 2B + 3I2 ∆H0 = −71
kJ
mol
∗ Schmelz�usselektrolyse von KBF4/KCl/B2O3-Mischungen
� Verwendung:
∗ 10B: Kerntechnik (hoher Neutroneneinfangquerschnitt)
∗ Borfasern für Verbundwerksto�e (hohe Festigkeit und Temperaturbeständigkeit)
� elementares Bor ist sehr reaktiv (würde CO2 reduzieren)
� Dimerisierung: Beispiel�Reaktion siehe Folie 21 → Diboran (weitere: Folie 22 / 23)
• Aluminium:
� Silberweiÿes Leichtmetall, kubisch-�ächenzentrierte Packung
� sehr dehnbar ⇒ feine Drähte, dünne Folien
� beständig an Luft, in Wasser und oxidierenden Säuren (Passivierung durch dünne Oxid- bzw.Hydroxidschicht)
∗ Eloxal-Verfahren: elektrochemische Oxidation zur Erzeugung einer harten, dickeren Oxid-schicht (0, 01− 0, 02mm) zum Schutz
∗ Eloxiertes Al beständig gegen Meerwasser, Säuren, Laugen
� Wassersto�entwicklung mit verdünnten Säuren:
Al + 3H3O+ −→ Al3+ + 1, 5H2 + 3H2O E0 = −1, 68V
� Amphoterer Charakter von Al(OH)3:
Al(OH)3 + 3H3O+ −→ [Al(H2O)6]3+
Al(OH)3 +OH− −→ [Al(OH)4]−
⇒ keine Schutzschicht in start saurer oder alkalischer Lösung
� bei ausreichend groÿem oder kleinem pH verliert Al die Schutzschicht
� Verbrennung an Luft: 2Al + 1, 5O2 −→ Al2O3 ∆H0 = −1677kJ/mol
� Aluminothermisches Verfahren:
∗ Reduktion von Metalloxiden mit Al
∗ möglich für alle Metalloxide mit kleinerem ∆H0B
∗ Bsp: ∆H0B(Cr2O3) = −1130kJ/mol
Cr2O3 + 2Al −→ Al2O3 + 2Cr ∆H0 = −547kJ/mol
∗ Thermitschweiÿen:
3Fe3O4 + 8Al −→ 4Al2O3 + 9Fe ∆H0 = −3341kJ/mol
⇒ Schweiÿnaht durch Bildung von �üssigem Fe (T bis zu 2400◦C)
� wichtigstes Gebrauchsmetall nach Eisen
� Weltproduktion (2008): 39, 3 ∗ 106t
� Elektrolyse aus wässriger Lösung nicht möglich (negatives Potential)
� Ausgangsmaterial: Bauxit (AlO(OH) mit Fe2O3-Verunreinigung)
∗ wichtig: Abtrennung des Fe2O3 vor der Schmelzelektrolyse (sonst Fe-Abscheidung)
∗ technisches Verfahren: Bayer-Verfahren:
Bauxit+NaOH170◦C,Druck−−−−−−−−→ Na[Al(OH)4]
löslich+Fe2O3unlöslich
Filtration, Impfen−−−−−−−−−−−→ Al(OH)3+NaOH1200◦C−−−−−→ Al2O3+H2O

9.9. BORGRUPPE: GRUPPE 13 83
∗ Schmelzpunkterniedrigung (Smp. (Al2O3) = 2050◦C) durch Kryolith-Zugabe (Eutekti-kum: 10, 5%Al2O3, 89, 5% Kryolith; Smp. = 960◦C)
· Al sammelt sich �üssig am Boden desElektrolyseofens
· Schmelze schützt Al vor Oxidation· Reinheit: 99, 8− 99, 9% Al (Rest: Fe, Si)
� Verwendung:
∗ Elektrotechnik (gute Leitfähigkeit)
∗ chemischer Apparatebau (chemische Widerstandsfähigkeit)
∗ Fahrzeug-, Flugzeug-, Schi�-, Hausbau
∗ Haushaltsgegenstände
∗ Al-Pulver für Anstrichmittel
∗ Aluminothermische Verfahren
∗ Al-Folien als Verpackungsmaterial
9.9.3 Verbindungen
• B:
� einzigartiges chemisches Verhalten
� nicht als B3+-Kation
� kovalente Bindungen mit sp2-hybridisierten B-Atomen (trigonal planar)
� BX3-Verbindungen sind Elektronenmangelverbindungen
∗ Ausbildunge von π-Bindungen (Bsp: BF3)
∗ Mehrzentrenbindungen (Bsp: B2H6)
� Mesomerie von BF3:
� Dimerisierung: (BH3 nicht beständig) −→
� Strukturen von Boranen: beteiligte Zwei- und Dreizentrenbindungen
∗ Zweizentren-BH-Bindung: B −H∗ Zweizentren-BB-Bindung: B −B
∗ Dreizentren-BHB-Bindung:
∗ o�ene Dreizentren-BBB-Bindung:
∗ geschlossene Dreizentren-BBB-Bindung:
� Strukturen von Boranen:

84 KAPITEL 9. HAUPTGRUPPENELEMENTE
unbesetzte Bezeichnung BeispielEcken
1 nido-Borane BnHn+4
(nidus = Nest)2 arachno-Borane BnHn+6
(arachne = Spinne)3 hypho-Borane BnHn+8
(hypho = Netz)
� wichtige Bor-Verbindungen:
∗ Borcarbid B13C2: sehr hart ⇒ Schleifmittel, Panzerplatten
∗ Natriumborhydrid NaBH4: Hydrierungsmittel
∗ ortho-Borsäure H3BO3:
· kristalliner Feststo� (Smp. 171◦C), planare Schichtstruktur
· schlecht löslich in Wasser
· wirkt als OH−�Akzeptor statt als H+�Donator
H3BO3oder:B(OH)3
+ 2H2O [B(OH)4]− +H3O+
∗ Bornitrid BN :
· thermisch sehr beständig, relativ inert ⇒ feuerfeste Auskleidungen
· mehrere Modi�kationen, z.B. hexagonales BN (Graphitstruktur)
• Al:
� keine (p-p)π-Bindungen ⇒ Dimerisierung von AlX3
� Lithiumalanat LiAlH4: Hyfrierungsmittel
� Aluminiumhydroxid Al(OH)3: amphoter, löslich in Säuren und Laugen
� Aluminiumhydroxid AlO(OH): 2 Modi�kationen: Diaspor, Böhmit
� Aluminiumoxid Al2O3:
∗ γ−Al2O3: hygroskopisch, Trägermaterial für Katalysatoren, durch Entwässern von Böhmit
∗ α − Al2O3: Korund, durch Glühen von γ − Al2O3 bei 1000◦C nicht hygrpskopisch, sehrhart, technische Herstellung aus Bauxit, Bedeutung in Al-Darstellung

Kapitel 10
Anmerkungen
• σ∗ ist das antibindende Orbital zu σ⇒ � X* � ist Anti�Orbital zum Orbital � X � ohne �* �
• Luft siedet bei −194◦C
• Bindungsabstand wird kleiner bei höherer Bindungsordnung
• Der Name von Ionen kommt nicht von der Ladung, sondern von dem Pol Bei der Elektrolyse, zuder sie sich hin bewegen: Kationen (pos. geladen) Kathode (−)
Anionen (neg. geladen) Anode (+)
• Impfen: (Impf-)Kristall in Lösung einbringen
85

![Synthesis of microporous polymeric BINOL-derived ...€¦ · Lewis acids Organocatalysis ] and McMillan [12 , 13] in 2000. After that, es and Brønsted acids. These with Lewis acid](https://static.fdokument.com/doc/165x107/5ffd4cd44dee5333805d1f85/synthesis-of-microporous-polymeric-binol-derived-lewis-acids-organocatalysis.jpg)
















