
Pharmakologie und Toxikologie für Biomediziner · Praktikum Pharmakologie und Toxikologie für...
58
Praktikum Pharmakologie und Toxikologie für Biomediziner Zentrum Pharmakologie und Toxikologie 16.04.2012 – 26.04.2012 Sommersemester 2012 Prof. Dr. Andreas Pich
Transcript of Pharmakologie und Toxikologie für Biomediziner · Praktikum Pharmakologie und Toxikologie für...

Praktikum Pharmakologie und Toxikologie für Biomediziner
Zentrum Pharmakologie und Toxikologie
16.04.2012 – 26.04.2012
Sommersemester 2012
Prof. Dr. Andreas Pich
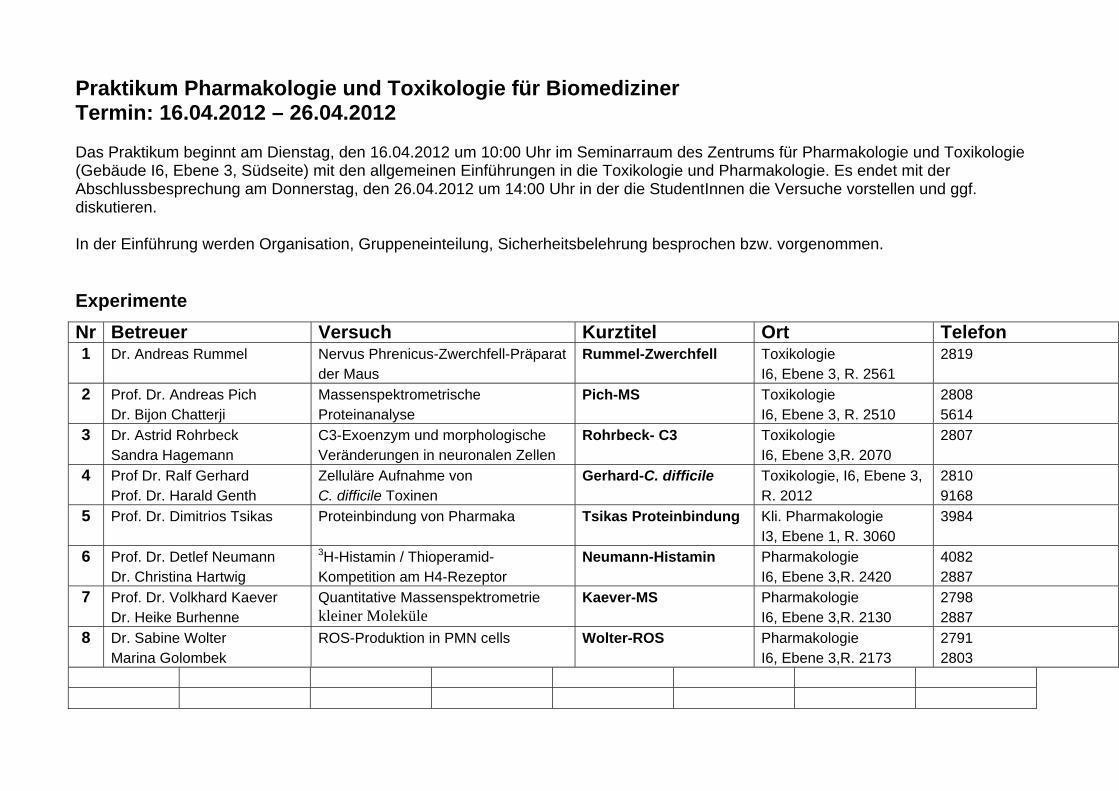
Praktikum Pharmakologie und Toxikologie für Biomediziner Termin: 16.04.2012 – 26.04.2012 Das Praktikum beginnt am Dienstag, den 16.04.2012 um 10:00 Uhr im Seminarraum des Zentrums für Pharmakologie und Toxikologie (Gebäude I6, Ebene 3, Südseite) mit den allgemeinen Einführungen in die Toxikologie und Pharmakologie. Es endet mit der Abschlussbesprechung am Donnerstag, den 26.04.2012 um 14:00 Uhr in der die StudentInnen die Versuche vorstellen und ggf. diskutieren. In der Einführung werden Organisation, Gruppeneinteilung, Sicherheitsbelehrung besprochen bzw. vorgenommen. Experimente Nr Betreuer Versuch Kurztitel Ort Telefon 1 Dr. Andreas Rummel Nervus Phrenicus-Zwerchfell-Präparat
der Maus Rummel-Zwerchfell Toxikologie
I6, Ebene 3, R. 2561 2819
2 Prof. Dr. Andreas Pich Dr. Bijon Chatterji
Massenspektrometrische Proteinanalyse
Pich-MS Toxikologie I6, Ebene 3, R. 2510
2808 5614
3 Dr. Astrid Rohrbeck Sandra Hagemann
C3-Exoenzym und morphologische Veränderungen in neuronalen Zellen
Rohrbeck- C3 Toxikologie I6, Ebene 3,R. 2070
2807
4 Prof Dr. Ralf Gerhard Prof. Dr. Harald Genth
Zelluläre Aufnahme von C. difficile Toxinen
Gerhard-C. difficile Toxikologie, I6, Ebene 3, R. 2012
2810 9168
5 Prof. Dr. Dimitrios Tsikas Proteinbindung von Pharmaka Tsikas Proteinbindung Kli. Pharmakologie I3, Ebene 1, R. 3060
3984
6 Prof. Dr. Detlef Neumann Dr. Christina Hartwig
3H-Histamin / Thioperamid-Kompetition am H4-Rezeptor
Neumann-Histamin Pharmakologie I6, Ebene 3,R. 2420
4082 2887
7 Prof. Dr. Volkhard Kaever Dr. Heike Burhenne
Quantitative Massenspektrometrie kleiner Moleküle
Kaever-MS Pharmakologie I6, Ebene 3,R. 2130
2798 2887
8 Dr. Sabine Wolter Marina Golombek
ROS-Produktion in PMN cells Wolter-ROS Pharmakologie I6, Ebene 3,R. 2173
2791 2803

Montag 16.04. 10:00 Uhr
Einführung Toxikologie
EinführungToxikologie
EinführungToxikologie
Einführung Toxikologie
EinführungToxikologie
EinführungToxikologie
Einführung Toxikologie
Montag 16.04. 14:00
Einführung Pharmakologie
EinführungPharmakologie
EinführungPharmakologie
Einführung Pharmakologie
EinführungPharmakologie
EinführungPharmakologie
Einführung Pharmakologie
Gruppe 1 Gruppe 2 Gruppe 3 Gruppe 4 Gruppe 5 Gruppe 6 Gruppe7 Dienstag 17.04.10:00
Rummel Zwerchfell
WolterROS
GerhardC. difficile
Rohrbeck C3
Dienstag 17.04.14:00
Kaever MS
WolterROS
GerhardC. difficile
Rohrbeck C3
RummelZwerchfell
Neumann Histamin
Pich MS
Mittwoch 18.04.10:00
Kaever MS
Tsikas‐Proteinbindung
WolterROS
Gerhard C. difficile
RohrbeckC3
Neumann Histamin
Pich MS
Mittwoch 18.04. 14:00
Pich MS
KaeverMS
WolterROS
Gerhard C. difficile
RohrbeckC3
RummelZwerchfell
Neumann Histamin
Donnerstag 19.04. 10:00
Pich MS
KaeverMS
Tsikas‐Proteinbindung
Wolter ROS
GerhardC. difficile
RohrbeckC3
Neumann Histamin
Donnerstag 19.04. 14:00
Neumann Histamin
PichMS
KaeverMS
Wolter ROS
GerhardC. difficile
RohrbeckC3
Rummel Zwerchfell
Freitag 20.04. 10:00
Neumann Histamin
PichMS
KaeverMS
Tsikas‐Proteinbindung
WolterROS
GerhardC. difficile
Rohrbeck C3
Freitag 20.04.14:00
Tsikas‐Proteinbindung
Neumann Histamin
PichMS
Kaever MS
WolterROS
GerhardC. difficile
Rohrbeck C3
Wochenende . Wochenende . Wochenende . Wochenende . Wochenende . Wochenende . Wochenende . Wochenende . Montag 23.04. 10:00
Rohrbeck C3
Neumann Histamin
PichMS
Kaever MS
Tsikas‐Proteinbindung
WolterROS
Gerhard C. difficile
Montag 23.04. 14:00
Rohrbeck C3
RummelZwerchfell
Neumann Histamin
Pich MS
KaeverMS
WolterROS
Gerhard C. difficile
Dienstag 24.04.10:00
Gerhard C. difficile
RohrbeckC3
Neumann Histamin
Pich MS
KaeverMS
Tsikas‐Proteinbindung
Wolter ROS
Dienstag 24.04.14:00
Gerhard C. difficile
RohrbeckC3
RummelZwerchfell
Neumann Histamin
PichMS
KaeverMS
Wolter ROS
Mittwoch 25.04. 10:00
Wolter ROS
GerhardC. difficile
RohrbeckC3
Neumann Histamin
PichMS
KaeverMS
Tsikas‐Proteinbindung
Mittwoch 25.04. 14:00
Wolter ROS
GerhardC. difficile
RohrbeckC3
Rummel Zwerchfell
Neumann Histamin
PichMS
Kaever MS
Donnerstag 26.05.10:00
Neumann Histamin
PichMS
Kaever MS
Donnerstag 26.04. 14:00
Abschluss‐ Seminar
Abschluss‐Seminar
Abschluss‐Seminar
Abschluss‐ Seminar
Abschluss‐Seminar
Abschluss‐Seminar
Abschluss‐ Seminar
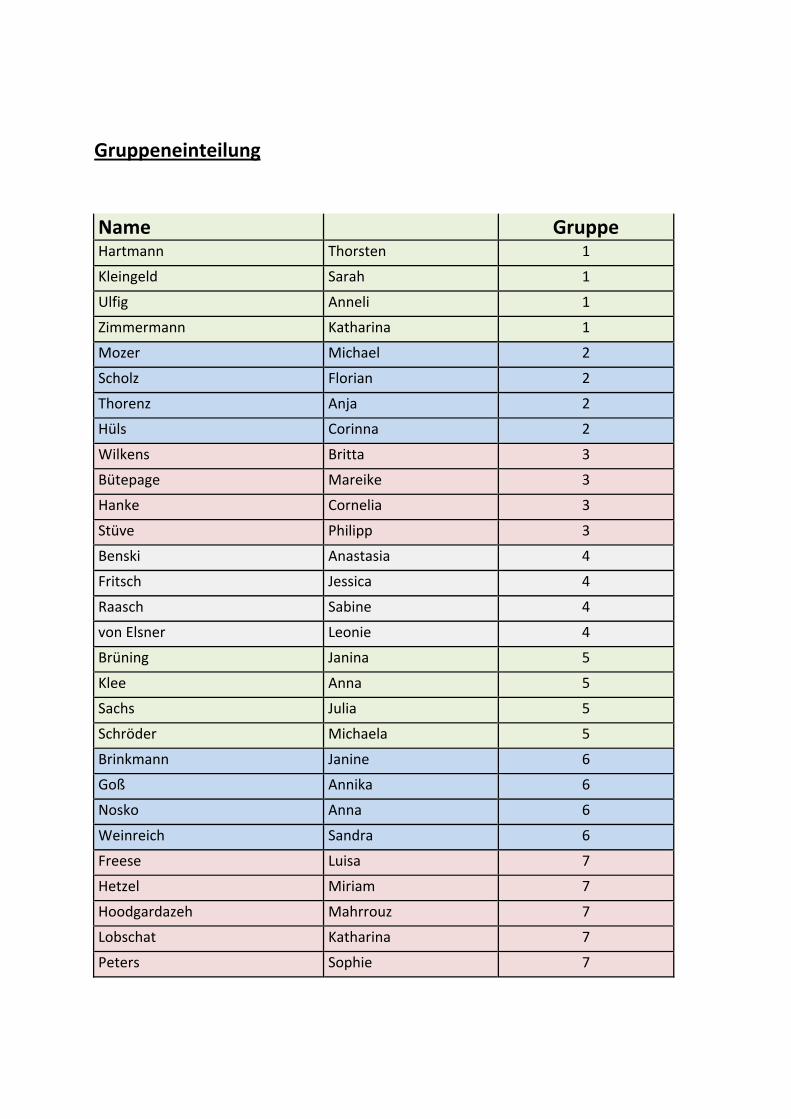
Gruppeneinteilung
Name Gruppe Hartmann Thorsten 1
Kleingeld Sarah 1
Ulfig Anneli 1
Zimmermann Katharina 1
Mozer Michael 2
Scholz Florian 2
Thorenz Anja 2
Hüls Corinna 2
Wilkens Britta 3
Bütepage Mareike 3
Hanke Cornelia 3
Stüve Philipp 3
Benski Anastasia 4
Fritsch Jessica 4
Raasch Sabine 4
von Elsner Leonie 4
Brüning Janina 5
Klee Anna 5
Sachs Julia 5
Schröder Michaela 5
Brinkmann Janine 6
Goß Annika 6
Nosko Anna 6
Weinreich Sandra 6
Freese Luisa 7
Hetzel Miriam 7
Hoodgardazeh Mahrrouz 7
Lobschat Katharina 7
Peters Sophie 7

20.03.2012
1
Sommersemester 2012
Termines. AblaufplanBeginn: 10:00 Uhr/14:00 Uhr oder nach Absprache
Abschlussbesprechung:• Dnnerstag, 26.04.2012, 14:00 Uhr Seminarraum
Pharmakologie/Toxikologie
Protokolle• Gruppenprotokolle • Abgabe bis 14.05.2012• Testat bis zum 11.06.2012
Biostoff VOGefahrstoff VOStr Schu VO
Mikroorganismen
Chemikalien / Proteine
Radioaktivität
GenT GesetzGenT SiVO
Selbst- und Fremdschutz
sauber arbeiten und keine Kontaminationen
an die Abteilung angepasste Informationen
Betriebsanweisung Aushang
Kittel Handschuhe Türklinke!Desinfektion dann waschenAerosolbildung verhindern (sterilfiltrieren/ schallen)
SchutzbrilleEssen, ∅ Trinken, ∅ SchminkenAbzug / Werkbank benutzen
• Sicherheitsausstattung
• Feuerlöscher, 1. Hilfe, Augendusche…
• gewissenhafter Umgang mit Geräten und Chemikalien• „Spätfolgen“
GHSGHS--SymboleSymbole
Global Harmonized System of Global Harmonized System of Classification and Labelling of ChemicalsClassification and Labelling of Chemicals

20.03.2012
2
GHSGHS--SymbolSymbol GHSGHS--SymbolSymbol
GHSGHS--SymbolSymbolGHSGHS SymbolSymbol
Berufskrankheit; 0; 0%Tätlichkeiten:Angriffe von Tieren, Patienten;
1; 1%
Sonstige Unfallereignisse:z.B. Strom, Verbrennungen,
Vergiften; 5; 5%
Sportunfall; 2; 2% Wegeunfall; 34; 30%
Unfälle 2008 an der MHH
Personenkreis: Mitarbeiter, Studenten, Schüler, Azubis, Praktikantenca. 10.000 Personen ca. 100 gemeldete Unfälle
Verheben, verstauchen:Transport von Gegenständen;
1; 1%
Sich stoßen, anstoßen:Ausführen einer Körperbewegung;
bücken, aufrichten, gehen usw.;
4; 4%Bewegte Teile:Umgang mit Gegenständen,Tür, Fenster, Werkzeug, MaschinenGetroffen, erfasst, gequetscht
werden; 13; 12%
Fallen aus der Höhe:Von erhöhtem Standplatz, Gleichgewicht verlieren;
5; 5%
Gehen und Laufen:Ausrutschen, stolpern,
hängenbleiben, umknicken; 24; 21%
Sich schneiden, stechen:Scharfe, spitze Gegenstände geraten außer Kontrolle, beim Umgang, beim Transport, beim
Reinigen usw.; 15; 14%
Heiße Stoffe / Teile; ätzende
Stoffe; 6; 5%
Sich lösende, herumfliegende Teile:Staub, Splitter usw.: Auge wird
getroffen; 0; 0%
Toxikologie
Arzneimitteltoxikologie Gewerbetoxikologieepidemiologische Toxikologie
i t ll T ik l i
FachrichtungenMethoden
MedizinBiochemie
Biologie experimentelle ToxikologieKosmetikatoxikologieLebensmitteltoxikologieBiozidtoxikologieRisikoabschätzungLebensmitteltoxikologieUmwelttoxikologie
klinische Toxikologie
BiologieChemie
MolekularbiologieGenetikPhysik
Statistik
Pharmakologie & Toxikologie
Toxikologie :E t l ti
Tierversuch g
Extrapolation Mensch
Pharmakologie : Tierversuch klinischer Versuch

20.03.2012
3
Tierversuch
Toxikologie
Extrapolation
hohe Dosen
niedrige Dosen
Mensch
Risikoabschätzung
Versuch: 10 Glasflaschen auf einen Steinfußboden fallen lassen und bestimmen, wieviel zerbrechen
Versuch 10 Flaschen aus 100 cm 9 zerbrechen
Fall-höhe
Ergebnis
Versuch 10 Flaschen aus 100 cm 9 zerbrechen
Risikoabschätzung
Versuch: 10 Glasflaschen auf einen Steinfußboden fallen lassen und bestimmen, wieviel zerbrechen
Versuch 10 Flaschen aus 100 cm 9 zerbrechen
Fall-höhe
Ergebnis
Risikoabschätzung
Versuch: 10 Glasflaschen auf einen Steinfußboden fallen lassen und bestimmen, wieviel zerbrechen
Versuch 10 Flaschen aus 100 cm 9 zerbrechen
Fall-höhe
Ergebnis
Versuch 10 Flaschen aus 100 cm 9 zerbrechen
Extrapolation
100 Flaschen aus ? 1000 Flaschen aus ?10.000 Flaschen aus ?
10 cm1 cm
0,1 cm
Versuch 10 Flaschen aus 100 cm 9 zerbrechen
Dioxin (TCDD)
LebertumoreRatten: bei 10 ng Dioxin pro kg bei 1 ng - -
+
”
Indol-Carbinol: 500 mg pro kg Broccoli
Bindung an den Arylhydrocarbonsäure-Rezeptorwie Dioxin
Indol-Carbinol (Broccoli)
Grenzwert-berechnung
Ames, BN, Magav, R, Gold LS (1987) Science 236, 271
1 mg Broccoli pro Tag
Ethylalkohol
Im Tierversuch schwach karzinogene Wirkung wie Dioxin
Ames, BN, Magav, R, Gold LS (1987) Science 236, 271
Grenzwert-berechnung I Bier in 50 Jahren

20.03.2012
4
Dioxin (TCDD)
Toxizität im Tier LD50g / kg μ
Meerschweinchen 1 Ratte 40Kaninchen 115 Beagle-Hund 200 Hamster 5000
Behandlung akuter Vergiftungen
Ingo JustInstitut für ToxikologieMedizinische HochschuleHannover
Rahmenbedingungen
Laien sind fast immer die ersten Personen am Auffindeort einer verunfallten oder vergifteten Person.
Der Laienhelfer soll am Auffindeort die Vitalfunktionen sichern und stabilisierendie Transportfähigkeit des Patienten herstellen bzw. sicherstellen
Besondere Risiken drohen dem Laienhelfer bei:
Anwesenheit von Giftgasen (z.B. CO, HCN, CO2, Phosgen)
Bestehende Brand- oder Explosionsgefahr
Ingestion von hochtoxischen Substanzen (z.B. Organophosphate)
Selbstgefährdung
Selbstschutz geht vor Rettung!
Kurze Orientierung: Brand? Chemikalien? Hinweisschilder?
Bei V. a. Giftgas: Rettung ohne Atemschutz nur über kürzeste Distanzen möglich!
Sicherheitsabstand einhalten, Rettungskräfte anfordern!
GHSGHS--SymbolSymbol
GHSGHS S b lS b lGHSGHS--SymbolSymbolGlobal Harmonized System of Global Harmonized System of
Classification and Labelling of ChemicalsClassification and Labelling of Chemicals
GHSGHS--SymbolSymbol
G SG S SSGHSGHS--SymbolSymbol

20.03.2012
5
Aufrechterhaltung und Überwachung der Vitalfunktionen Atmung und Kreislauf
Grundsätze für den Laienhelfer
Verhinderung zusätzlicher Komplikationen (Aspiration)
Verhinderung einer Selbstgefährdung
Gefahren:1. Selbstgefährdung und Kontamination der Rettungskette
2. Resorption lipophiler Giftstoffe mit verzögerter systemischer Wirkung
Kontamination der Haut
Gefahren:1. Selbstgefährdung und Kontamination der Rettungskette
2. Resorption lipophiler Giftstoffe mit verzögerter systemischer Wirkung
Kleidung aufschneiden und wegklappen (nicht ausziehen) !
Kontamination der Haut
Spülen der betroffenen Hautareale nur mit viel Wasser. Keine Seife oder Bürsten verwenden. Im Kopfbereich Haupthaar nicht vergessen!
Bei öligen und stark haftenden Kontaminationen Polytheylenglykol 400 (z.B. Lutrol®) statt Wasser.
Bei Kontamination mit einer stark giftigen Substanz ist eineanschließende stationäre Überwachung unabweisbar.
Kontamination des Auges
Das betroffene Auge wird mit Wasservon nasal nach temporal gespült.
Neutralisationsversuche sind verboten!
Kontamination des Auges
Bei Lidkrampf keinesfalls Gewalt zumÖffnen des Augenlides anwenden!
unverzügliche Vorstellung in eineraugenärztlichen Ambulanz.
Lokalanästhetika
Verteilung
Aufnahme(Absorption)
Behandlungsprinzipien akuter Intoxikationen
Sicherung der Vitalfunktionen
Gift tf
Ausscheidung(Exkretion)
g(Distribution)
Biotransformation(Metabolisierung)
Giftentfernung - Minderung der Giftresorption- Beschleunigung der Giftelimination
Antidotgabe

20.03.2012
6
Niemals Erbrechen auslösen !
Spezifische Maßnahmen bei Vergiftungsverdacht
Keine Flüssigkeiten einflössen !
Ausnahme: Verätzungen
mechanische Reizung des RachensGabe von Ipecacuanha Sirup
Methoden
induziertes Erbrechen
UAW bei Ipecacfortgesetztes Erbrechen
Emetin, CephalinWirkeintritt nach 20-30 min
Kontraindikationenätzende Substanzenschäumende Substanzen, Lösungsmittel, aliphatische KW(drohender) Bewusstseinsverlust / Krampfanfall
fortgesetztes Erbrechen
Methode
Intubation
Magenschlauch legenaspirierenLavage mit Portionen zu 200 - 300 ml
Magenspülung
WirksamkeitEntleerungsrate variabel
durchschnittlich werden nur 30% des ingestierten Materials entfernt
Risikenbeschleunigte Passage durch den PylorusWasserintoxikation bei KindernHerzrhythmusstörungenMagenperforation
hohe Adsorptionsleistung durch große Oberfläche1000 - 2000 qm pro g Aktivkohle
Gabe von Aktivkohle
keine Adsorption vonanorganischen SubstanzenSäuren und Laugenorganischen Lösungsmitteln
Ethylenglykol, Methanol, Ethanol
Einmalgabe 1g / kg KGtrinken oder über Magensonde
Verteilung(Distribution)
Aufnahme(Absorption)
Toxikokinetik
Niere (renal) - Ultrafiltration- tubuläre Sekretion
Galle (biliär)Lunge
Ausscheidung(Exkretion)
Phase I: Funktionalisierung, CYPPhase II: Konjugation
Biotransformation(Metabolisierung)
ParacetamolParacetamol
CYP
hepatische Detoxifizierung
HWZ 1-3 h
HWZ* >12 h
Acetylcystein

20.03.2012
7
Voraussetzungwasserlösliche Substanzgeringe Plasmaeiweißbindung überwiegende Verteilung
Hämodialyse
Vorteil Korrektur von Elektrolyt-und Säure/Basestörungen
überwiegende Verteilung im Körperwasser VD < 2 L/kg
ADHMethanol Form-
aldehydAmeisen-
säureALDH10x ↓
Methanol CH3-OH
C1-Stoff-wechsel
weitverbreitetes organ. LösungsmittelHaushaltsprodukte, Kühlmittel, Farbverdünner/-entfernerkeine ! Steuern
Sehstörungen ab 3. Tag (reversibel)Erblindung ab 5. Tag (irreversibel)
pulmonal 10%
langsame renale Elimination⇒ metabol. Azidose
am 2. bis 4. d
LD: 30-50 ml
HWZ 3 -30 hdosisabhängig
ADHMethanol Form-
aldehydAmeisen-
säureALDH
Methanol CH3-OH
pulmonal TherapieHemmung der Oxidation des Methanols durch
Fomepizol(Ethanol 1 ‰ für 3-5 d)
Hämodialyse
primäre Verfahren: Verminderung der Giftresorption
sekundäre Verfahren: beschleunigte Ausscheidung
Gabe von Aktivkohle
induziertes ErbrechenMagenspülungHautdekontamination
Augenspülung
intrakorporalerepetitive KohlegabeUrinalkalisierung(forcierte Diurese)Steigerung der hepat.Metabolisierung
extrakorporaleHämodialyseHämoperfusion
wichtige hämodialysierbareSubstanzen
nicht hämodialysierbareSubstanzen
Methanol
Hämodialyse
Benzodiazepine
Neuroleptika
Antidepressiva
MethanolEthylenglykol SalicylateLithiumMetformin

Einführungin die Pharmakologie (Pharmakodynamik)
undAsthma-Versuch
Roland Seifert
Institut für PharmakologieSommertertial 2012
PharmakodynamikAnalyse der Wirkungen von Wirkstoffen auf den OrganismusArzneistoff hat nützliche Wirkung; Gift hat schädliche Wirkung
PharmakokinetikWirkungen des Organismus auf den Wirkstoff
- Absorption (A)- Distribution (D)- Metabolismus (M)- Elimination (E)
ArzneimittelZubereitung eines Arzneistoffs zur Anwendung beim Menschen(z. B. Tabletten, Salben, Injektionslösungen)
1. Arzneimittelspezialität2. Generikum
International Non-Proprietary Name (INN) = internationaler Freiname
Was ist Pharmakologie?Pharmakologie analysiert die Interaktionen von Wirkstoffen mit dem lebendenOrganismus
Rezeptoren als pharmakologische Zielstrukturen
Parameter Ligand-gesteuerte G-Protein-gekoppelteIonenkanäle Rezeptoren
Lokalisation
Struktur
Signal-transduktion
Effektoren
Wirkungs-eintritt
Plasmamembran
oligomerisch;zentrale Ionenpore
direkt
Ionenkanäle
Millisekunden(sehr schnell)
Plasmamembran
7-Transmembran-domänen
G-Proteine
Ionenkanäle,Enzyme
Sekunden(schnell)
AgonistAllosterischerModulator
Antagonist
AgonistAnt-agonist
Rezeptoren als pharmakologische Zielstrukturen
Parameter Tyrosinkinase-gekoppelte NukleäreRezeptoren Rezeptoren
Lokalisation
Struktur
Signal-transduktion
Effektoren
Wirkungs-eintritt
AgonistAnt-agonist
Plasmamembran
1-Transmembran-Domäne
direkt
Proteinkinasen,Proteinphosphorylierung
Minuten - Stunden(langsam)
AgonistRezeptor
Kinase
ATP ADPInhibitor P
Zytosol→ Kern
Monomere
direkt
Gentranskription,Proteinbiosynthese
Stunden(sehr langsam)
DNA-Bindung
Ligand-Bindung
Rezeptoren als pharmakologische Zielstrukturen
Ligand-gesteuerte G-Protein-gekoppelteIonenkanäle Rezeptoren
Beispiele
Nikotinische Cholinozeptoren
GABAA-Rezeptor
5-HT3-Rezeptor
Glutamat (NMDA)-Rezeptoren
Adrenozeptoren
Muskarinische Cholinozeptoren
Dopamin-Rezeptoren
Serotonin-Rezeptoren
Tyrosinkinase-gekoppelte NukleäreRezeptoren Rezeptoren
Beispiele
Insulin-Rezeptor
Zytokin-Rezeptoren
Wachstumsfaktor-Rezeptoren
Schilddrüsenhormon-Rezeptor
Glucocorticoid-RezeptorMineralocorticoid-Rezeptor
Xenobiotika-Rezeptoren
Enzyme als pharmakologische Zielstrukturen
Enzym Inhibitor Indikation
Acetylcholinesterase Neostigmin Myasthenia gravis(AchE) E605 (irreversibel) Insektizid, Kampfstoff
Cyclooxygenase Ibuprofen Schmerzen(COX) ASS (irreversibel) Thrombozytenaggre-
gationshemmung
HMG-CoA-Reduktase Simvastatin Hypercholesterinämie
Angiotensin-conver- Ramipril Hypertonie, Herzin-ting enzyme (ACE) suffizienz, KHK
Phosphodiesterase 5 Sildenafil erektile Dysfunktion,(PDE) pulmonale Hypertonie
Dihydrofolat- Trimethoprim bakterielle InfektionenReduktase Methotrexat Autoimmunerkrankung

Transporter und Ionenkanäle als pharmakologische Zielstrukturen
Transporter Inhibitor Indikation
5-HT-Reuptake selektive Serotonin- DepressionWiederaufnahme-Inhibitoren
Protonenpumpe Omeprazol (irreversibel) Ulcus duodeni
Na+/K+/2Cl- Schleifendiuretika Lungenödem, hypertensiveKo-Transporter Krise, Herzinsuffizienz
Kanal Ligand Indikation
Na+-Kanäle Lidocain (Blocker) LokalanästhetikumTetrodotoxin (Aktivator) Delikatesse (Fugu)
Ca2+-KanäleL-Typ Dihydropyridine (Blocker) HypertonieN-Typ ω-Conotoxin (Blocker) Schwere Tumorschmerzen
K+-Kanäle Amiodaron (Blocker) ArrhythmienSulfonylharnstoffe (Blocker) Diabetes mellitus Typ II
TM I II III IV V VI VII
-Asp N -Amin Ser
Ser
HO
HO
extra-zellulär
intra-zellulär Gα
i1 i2 i3
C-Terminus
N-Terminus
Ionenpaarbindung
H-Brückenbindung
e1 e2 e3
HelixPlasma-membran
+
Glykosylierung
P PPhosphorylierung
Ser Ser
β γ
GPCR-Struktur
Gα GPCR Agonist
ternärer Komplex
γ β
GPCRAgonist
Gα
GDP
γ β
GDP
G-Protein-Zyklus
Gα
GTPγ β
γ β
GTP
GPCR
Agonist
Gα
GTP EffektorPLC ↑K+ ↑Ca2+ ↓MAPK ↑Effektor
AC ↑↓PLC ↑
Gα
Pi
GDP
PertussistoxinB. pertussis
Bakterielle ADP-Ribosyltransferasen
CholeratoxinV. cholerae
1
2
3
4
5
66
VERSUCH 6
βAR - Gs - AC - Weg: Pharmakologische Eingriffsmöglichkeiten
Adrenalin „first messenger“Katecholamin, H
Herzstillstandanaphylaktischer Schock
βAR-AntagonistenKHKHypertoniechronischeHerzinsuffizienz
AMPPDEs
CNG-Kanäle
Protein + ATP Protein ∼ + ADPP
Protein + Pi
Phosphatasen
CholeratoxinBakterielle AC-Toxine
(ExoY, CyaA, EF)
AC5-InhibitorenHerzinsuffizienz
β1AR
Gs
ATP PPi cAMP „second messenger“
ACs I - IX
PKAPDE-Inhibitoren
AsthmaVERSUCH 7
Veränderte Zellfunktion:→ positive Inotropie → Hemmung der → positive Chronotropie Neutrophilenaktivierung
VERSUCH 8
Desensitisierung
Reduzierte Antwort einer Zelle nach mehrmaliger Agonist-Stimulation eines Rezeptors
β2AR: Therapie des Asthma bronchiale
μOPR/MOPR: Therapie von Tumorschmerzen, Substitutionbei Heroinabhängigen
α1AR: Therapie des Schnupfens
D2R: Therapie des Morbus Parkinson
Wirkung
Zeit
1 2 3 4
Stimulation
Molekularer Mechanismus der Desensitisierung
C
A
G E
Rezeptor- und G-Proteinaktivierung
1.
C
A
G E
Rezeptor- und G-Proteinaktivierung
2.
GRK: GPCR-KinasePP P
ARRESTIN
Internalisierung
PP
P
4.
5. A
P PA
Downregulation =Verschwinden vonR von derPlasmamembran
Dephosphorylierung von R
Resensitisierung6.
A
G E
3.
ARRESTIN
PP P
R/G-EntkopplungDesensitisierung
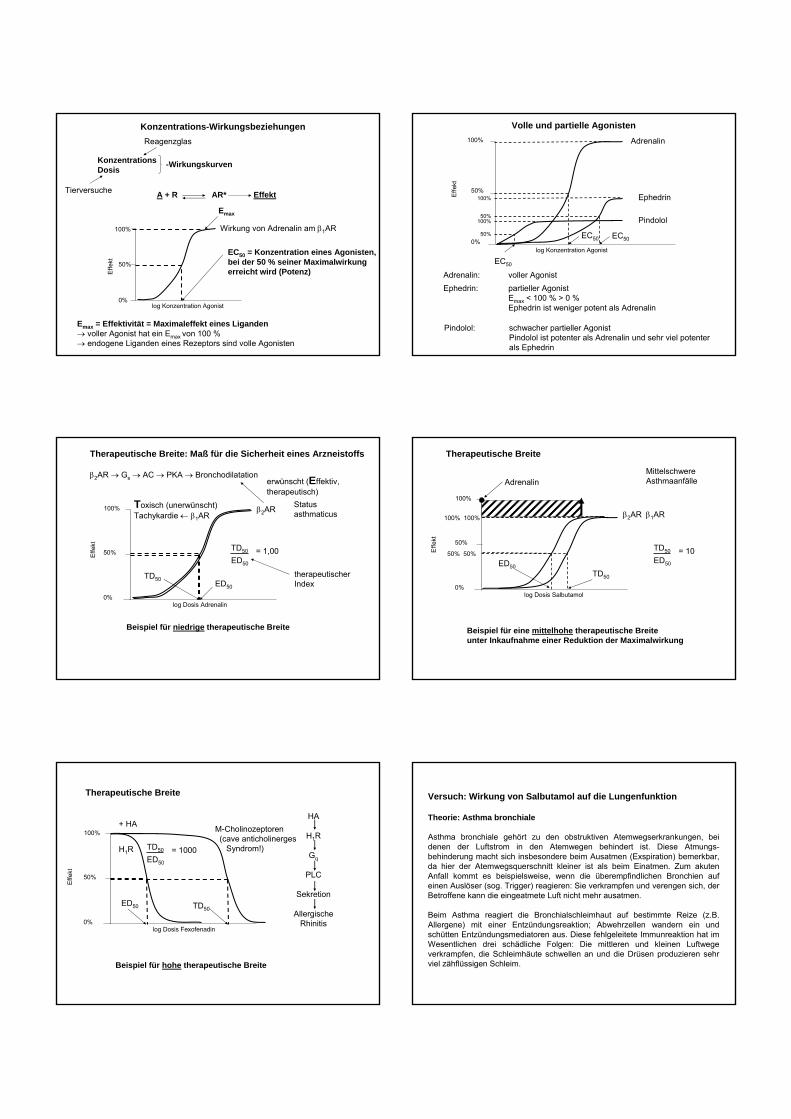
KonzentrationsDosis
Reagenzglas
Tierversuche
100%
50%
log Konzentration Agonist0%
EC50 = Konzentration eines Agonisten, bei der 50 % seiner Maximalwirkungerreicht wird (Potenz)
Wirkung von Adrenalin am β1AR
Emax = Effektivität = Maximaleffekt eines Liganden→ voller Agonist hat ein Emax von 100 %→ endogene Liganden eines Rezeptors sind volle Agonisten
Emax
Effe
kt
-Wirkungskurven
A + R AR* Effekt
Konzentrations-Wirkungsbeziehungen100%
50%
log Konzentration Agonist0%
Effe
kt
Adrenalin
EC50
Adrenalin: voller Agonist
Volle und partielle Agonisten
100%
50%
Ephedrin
EC50
Ephedrin: partieller AgonistEmax < 100 % > 0 %Ephedrin ist weniger potent als Adrenalin
Pindolol
EC50
100%
50%
Pindolol: schwacher partieller AgonistPindolol ist potenter als Adrenalin und sehr viel potenterals Ephedrin
Therapeutische Breite: Maß für die Sicherheit eines Arzneistoffs
β2AR → Gs → AC → PKA → Bronchodilatation
100%
50%
log Dosis Adrenalin0%
Effe
kt
β2ARToxisch (unerwünscht)Tachykardie ← β1AR
ED50
TD50
TD50
ED50
= 1,00
therapeutischerIndex
Statusasthmaticus
erwünscht (Effektiv, therapeutisch)
Beispiel für niedrige therapeutische Breite
Therapeutische Breite
100%
50%
log Dosis Salbutamol0%
Effe
kt
β2AR
ED50TD50
TD50
ED50
= 10
MittelschwereAsthmaanfälle
Beispiel für eine mittelhohe therapeutische Breiteunter Inkaufnahme einer Reduktion der Maximalwirkung
β1AR
50% 50%
100% 100%
Adrenalin
Therapeutische Breite
100%
50%
log Dosis Fexofenadin0%
Effe
kt
H1R
ED50 TD50
TD50
ED50
= 1000
Beispiel für hohe therapeutische Breite
M-Cholinozeptoren(cave anticholinerges
Syndrom!)
+ HAHA
H1R
Gq
PLC
Sekretion
AllergischeRhinitis
Versuch: Wirkung von Salbutamol auf die Lungenfunktion
Theorie: Asthma bronchiale
Asthma bronchiale gehört zu den obstruktiven Atemwegserkrankungen, bei denen der Luftstrom in den Atemwegen behindert ist. Diese Atmungs-behinderung macht sich insbesondere beim Ausatmen (Exspiration) bemerkbar, da hier der Atemwegsquerschnitt kleiner ist als beim Einatmen. Zum akuten Anfall kommt es beispielsweise, wenn die überempfindlichen Bronchien auf einen Auslöser (sog. Trigger) reagieren: Sie verkrampfen und verengen sich, der Betroffene kann die eingeatmete Luft nicht mehr ausatmen.
Beim Asthma reagiert die Bronchialschleimhaut auf bestimmte Reize (z.B. Allergene) mit einer Entzündungsreaktion; Abwehrzellen wandern ein und schütten Entzündungsmediatoren aus. Diese fehlgeleitete Immunreaktion hat im Wesentlichen drei schädliche Folgen: Die mittleren und kleinen Luftwege verkrampfen, die Schleimhäute schwellen an und die Drüsen produzieren sehr viel zähflüssigen Schleim.

Abb.1: Gesunder (links) und asthmatischer Bronchus (rechts)
Die wichtigsten bronchienerweiternden Medikamente (Bronchodilatatoren) sind β2-Sympathomimetika. Sie wirken entspannend auf die Muskulatur der Atemwege, so dass sich diese weiten. Zur Dauerbehandlung gehören auch entzündungshemmende Medikamente; in dieser Gruppe spielen die Glucocorticoide die Hauptrolle. Sie hemmen nachhaltig den Entzündungsprozess und vermindern die Überempfindlichkeit der Bronchien, so dass diese weniger zum Verkrampfen und zu akuten Anfällen neigen.Auch beim Gesunden haben β2-Sympathomimetika bronchodilatatorischeWirkungen. Diese werden beim Doping missbräuchlich ausgenutzt. Dies ist der Grund dafür, dass so viele Leistungssportler „Asthmatiker“ sind.Im Rahmen des Praktikums demonstrieren Hochschullehrer im Selbstversuch die Anwendung von β2-Sympathomimetika. Leider ist wegen juristischer Unsicherheiten der Selbstversuch durch Studenten nicht möglich.
Stoffklasse:
Wirkmechanismus:
Halbwertszeit:
Einzeldosis:
Indikationen:
Kontraindikationen:
Nebenwirkungen:
Interaktionen:
β2-Sympathomimetikum
bronchodilatierend; starker partieller Agonist an den β2-Adrenozeptoren (der Bronchien)
ca. 1 h
bei Bedarf 1-2 Hübe, bis zu 4 x täglich (1 Hub: 100 µg)
Asthma bronchiale, chronische obstruktive Bronchitits (COPD)
akuter Herzinfarkt, schwere Hyperthyreose, Phäochromozytom, schwere KHK, Leber- und Niereninsuffizienz, Tachyarrhythmien
Tachykardie, Unruhegefühl, Herzrhythmusstörungen, Angina-Pectoris-Anfälle, Tremor, Blutzuckeranstieg
Antidiabetika, β-Sympathomimetika, Anticholinergika, Theophyllin, β–Rezeptorenblocker, MAO-Hemmer, trizyklische Antidepressiva
Versuch: Salbutamol (Sultanol®)
Strukturformel:
Bedarf • Sultanol® Dosieraerosol, 100 µg• Peak-Flow-Messgeräte• Blutdruckmessgerät• Stoppuhr
• Als Basiswerte werden bei allen Probanden persönlicher Peak Expiratory Flow (PEF), Herzfrequenz (HF) und Blutdruck (BP) bestimmt
• Inhalation von 1-2 Hüben Sultanol®• Unmittelbar nach der Applikation des Arzneistoffs
sowie nach 3, 5 und 10 Minuten werden PEF, HF und BP bestimmt
Durchführung:
Zeitplan:
21
Größe (cm)Alter1)GeschlechtInitialen/NameNr.
1) Das Probandenalter wird als „Lebensjahr“ definiert. Ein 24jähriger befindet sich in seinem 25. Lebensjahr.Auf diese Vereinbarung hat man sich geeinigt, um auch Säuglinge unter 1 Jahr schlüssig einordnen zu können. Sie befinden sich im „1. Lebensjahr“.
Versuchsdurchführung:
PEFHFBP
2
PEFHFBP
1
10 min5 min3 minnachSalbutamol
vor Salbutamol
ParameterNr.
Messwerte
Peak Flow (l/min)
Eintrag Rote Liste®
Hauptgruppe: 28.B.1.1.2.2.1.1.1.Sultanol® Dosier-Aerosol FCKW-frei Druckgasinhalation, SuspensionRp Hersteller GlaxoSmithKline Zus.: 1 Sprühstoß (75 mg) enth.: Salbutamolsulfat 0,12 mg (entspr. 0,1 mg Salbutamol). Weit. Bestandteile: Norfluran (HFA 134a).
PZN 386391927,633 x 1 Dosier-Aerosol (N3) (3×mind. 200 Sprühstöße)
PZN 152873721,872 x 1 Dosier-Aerosol (N2) (2×mind. 200 Sprühstöße)
PZN 152872015,951 Dosier-Aerosol (N1) (mind. 200 Sprühstöße)
Anw.: Zur Verhütung u. Behandl. v. obstruktiven Atemwegserkrank. (z. B. Asthma bronchiale, chronische Bronchitis, Lungenemphysem).Gegenanz.: Gleichz. Behandl. mit β-Rezeptorenblockern.Anw.-beschränk.: S 2 a-g. Überempfindlichkeit gg. and. Sympathomimetika, Thyreotoxikose.Schwang./Stillz.: S 2Nebenw.: S 2. Gelegentl. (1-10%): Erweiterung der peripheren Gefäße, Änderungen des Geschmacksempfindens. Sehr selten (<0,1%): Herzrhythmusstör. (u. a. supraventrikuläre Tachykardie), zentralnervös stimulierende Wirk. nach Inhalation, die sich in Übererregbarkeit äußerten (überwiegend bei Kdrn. bis zu 12 J.), akute Überempfindlichkeitsreakt. (z. B. Bronchospasmus, Kollaps). Myokardiale Ischämie.Wechselw.: S 2. Gleichz. Gabe von Procarbazin kann hypertone Reakt. auslösen. Gleichz. Behandl. mit AM vom Typ Mutterkornalkaloide (z. B. Ergotamin) nur mit Vorsicht, da wechselseitige Beeinflussung d. Vasomotorik schwer voraussagbar ist u. diese ebenso zu vasokonstriktorischen wie -dilatator. Reakt. führen kann.Tox.: S 2 Warnhinw.: Dop.!Hinw.: Reaktionsvermögen! (V) Dies gilt in verstärktem Maß bei Behandlungsbeginn, bei höheren Dos. sowie bei Zusammenwirken m. Alkohol od. Beruhigungs- u. Schlafmitteln.Dos.: Erw. u. Kdr. (ab 12 J.): 1 Einzeldos. =1-2 Sprühstöße (entspr. 0,1-0,2 mg Salbutamol). Kdr. (<12 J.): 1 Einzeldos. =1 Sprühstoß (entspr. 0,1 mg Salbutamol). Akutbehandl. plötzlich auftretender Bronchialkrämpfe u. anfallsweise auftretender Atemnot: 1 Einzeldos. = Erw. u. Kdr. (ab 12 J.): 1-2 Sprühstöße; Kdr. (<12 J.): 1 Sprühstoß. Gezielte Vorbeugung bei Anstrengungsasthma od. vorhersehbarem Allergenkontakt: 1 Einzeldos. = Erw. u. Kdr. (ab 12 J.): 1-2 Sprühstöße; Kdr. (<12 J.): 1 Sprühstoß, 10-15 Min. vorher. Tagesgesamtdos.: 12 Sprühstöße bei Erw. u. Kdrn. (ab 12 J.), 6 Sprühstöße bei Kdrn. (<12 J.).Lag.: Lagerungshinweis! KP.: 10×1 Dosier-Aerosol (mind. 200 Sprühstöße)
visomat® handy IV (Auszug aus der Gebrauchsanweisung)

Nervus Phrenicus-Zwerchfell-Präparat der Maus
Versuchstier: 2 Mäuse, Stamm NMRI (Naval Marine Research Institute), ca. 30 g schwer Pharmaka Konzentration der Stammlösung⌠mg/ml⌡ . (+)-Atracurium 10 Physostigmin 0,4 Suxamethoniumchlorid 20 Botulinum Neurotoxin A (BoNT/A) 100 U (=MLD, 500 pg) . Nährlösung Krebs-Ringer-Lösung mit 11 mmol/l Glucose, Carbogen-durchspült (95 % O2, 5 % CO2) . Apparaturen und Instrumente Apparatur für isolierte Organe mit Umlaufthermostat, 5 ml Badvolumen, 37○C. 1 Dehnungsmessstreifen zur isometrischen Registrierung der Kontraktionen (DMS- Aufnehmer) 1 PC 1 elektrisches Reizgerät 1 Halterung mit Reizelektroden Operationsbrett, Petrischalen, Präparierbesteck Versuchsvorbereitung Die Maus wird in CO2-Narkose durch Entbluten getötet. Danach wird die Maus in Rückenlage auf dem Operationsbrett fixiert, das Fell über dem Thorax nach Medianschnitt bis zur Wirbelsäule abgelöst und das Sternum entfernt. Anschließend wird die Brustwand linksseitig etwa 0,5 cm oberhalb des Zwerchfells horizontal durchtrennt und weitgehend herausgeschnitten. Aus präparatorischen Gründen wird lediglich der linke N. phrenicus sowie das linke Zwerchfell verwandt. Der linke N. phrenicus wird möglichst weit kopfwärts mit einem Faden angeschlungen, oberhalb der Schlinge durchtrennt und bis zum Zwerchfell vorsichtig freipräpariert. Der freipräparierte, angeschlungene Nerv wird zum Schutz vor Beschädigung und Austrocknung bei der Fortsetzung der Präparation in den Costodialphragmalwinkel gelegt. Das linke Zwerchfell wird dorsal von der Wirbelsäule gelöst, medial von der rechten Zwerchfellhälfte abgetrennt und dann mit dem Rippenbogen zur weiteren Feinpräparation in eine mit Puffer gefüllte Petrischale gelegt. Aus dem Zwerchfell wird unter Schonung des Nerven ein ca. 0,7 x 1,5 cm großer Abschnitt (vom Rippenbogen zum Centrum tendineum verlaufend) herausgetrennt. Das Zwerchfellpräparat wird nunmehr in der Versuchsapparatur zwischen einem Haken (Rippenbogen) und einer Klemme (Centrum tendineum) fixiert, wobei letztere über einen Faden mit dem Dehnungsmessstreifen verbunden ist. Schließlich wird der N. phrenicus schonend durch zwei Ringelelektroden gezogen. Danach wird das Präparat in das mit Krebs-Ringer-Lösung (carbogengesättigt, 37○C) gefüllte Organbad vollständig eingetaucht.

Prinzip der Registrierung Am N. phrenicus-Zwerchfell-Präparat der Maus, welches von BÜLBRING (1946) eingeführt wurde, kann demonstriert werden, dass die Hemmung der neuromuskulären Übertragung durch Muskelrelaxantien spezifisch in der Endplatte lokalisiert ist. Die Kontraktionen des Zwerchfells werden „isometrisch“ über einen DMS-Aufnehmer auf dem PC registriert. Das Präparat wird indirekt über den Nerven gereizt. Die Reizung erfolgt mit Rechteckimpulsen bei einer Frequenz von 1 Hz und supramaximaler Intensität. Die Impulsdauer beträgt bei indirekter Reizung 0,1 ms. Durchführung des Versuchs Sofort nach Einhängen des Präparates in das Organbad wird mit der indirekten Reizung begonnen. Pharmakonhaltige Lösungen werden in der angegebenen Reihenfolge mit einer Pipette der Badflüssigkeit zugesetzt. Nach Eintritt des Effektes wird das Pharmakon durch mehrmaliges Wechseln der Badflüssigkeit ausgespült. Reihenfolge der Versuchsschritte Pharmakon Konzentration der
Stammlösung (mg/ml)
Erwünschte Endkonzentration in Badflüssigkeit (mg/ml) (bitte errechnen)
Volumen der Stammlösung für 4 ml Badvolumen (µl)
Maus 1 Zugabe von BoNT/A 500 pg ad 4 mL Maus 2 1. (+)-Atracurium nach Eintritt des Effektes spülen
10
3 (3x1uL)
wenn indirekter Reizerfolg wiederherstellt 2. (+)-Atracurium nach ca. 15 min Physostigmin wenn indirekter Reizerfolg wiederherstellt, spülen
10
0,4
3
2
nach ca. 15 min 3. Suxamethonium nach ca. 15 min Physostigmin nach ca. 10 min spülen
20
0,4
2
2

Erläuterung der Ergebnisse 1. BoNT/A
Die aus den Bakterium C. botulinum stammenden BoNT sind 150 KDa große Proteine, die nach einer Latenzzeit die Freisetzung des Neurotransmitters Acetylcholin (ACh) aus den präsynaptischen Nervenenden hemmen. Sie sind also periphere Muskelrelaxanzien, welche im Gegensatz zu den übrigen Pharmaka nicht den postsynap-tischen Rezeptor blockieren, sondern hochspezifisch am Motorneuron angreifen.
2. (+)-Atracurium
(+)-Atracurium hemmt die neuromuskuläre Übertragung am Skelettmuskel. Die direkte Reizung des Muskels wird durch Atracurium nicht behindert. Bei der Nervenreizung freigesetztes Acetylcholin führt zur Depolarisation der postsynaptischen Membran. Dadurch kommt es zum Entstehen eines Aktionspotentials, das über die gesamte Muskelfaser fortgeleitet wird und eine Kontraktion des Muskels zur Folge hat. Der Depolarisation liegen Permeabilitätsänderungen für kleine Ionen wie Na+, K+ und Ca2+ zugrunde. Atracurium zählt zu den nicht depolarisierenden Substanzen, da es am Acetylcholinrezeptor bindet, aber keine Permeabilitätsänderungen hervorruft. So kann die vorher wirksame indirekte elektrische Reizung bzw. das dabei freigesetzte Acetylcholin nicht mehr wirken. 3. Physostigmin
Atracurium und Acetylcholin konkurrieren um denselben Rezeptor an der motorischen Endplatte. Dabei handelt es sich um einen kompetitiven Antagonismus. Bei Atracurium-Dosierungen, die gerade eine Blockierung der neuromuskulären Überleitung hervorrufen, kann durch Vermehrung der Acetylcholin-Moleküle am Rezeptor der kompetitive Antagonismus zwischen Atracurium und Acetylcholin zugunsten des Acetylcholins entschieden und damit eine Überleitung wieder herstellt werden. Eine Hemmung des raschen Acetylcholinabbaues durch reversible Hemmung der spezifischen, in der Basalmembran lokalisierten Cholinesterase (hier durch Physostigmin, aber auch Pyridostigmin, Neostigmin, Edrophonium) bewirkt eine Anhäufung von Acetylcholin im Gebiet des Rezeptors sowie eine Verdrängung von Atracurium, wodurch die neuromuskuläre Impulsübertragung wieder hergestellt wird.

4. Suxamethonium
Suxamethonium wirkt auf die Membran der motorischen Endplatte ähnlich wie ACh, d.h. es depolarisiert die Membran. Da es aber im Gegensatz zu Acetylcholin nur sehr langsam durch Cholinesterasen inaktiviert wird, tritt in der Anfangsphase der Applikation eine Dauerpolarisierung ein. Obwohl Suxamethonium weiter auf die Endplatte einwirkt, kommt es im weiteren Verlauf zu einer Repolarisation der postsynaptischen Membran. Aufgrund einer Desensibilisierung der Acetylcholin-Rezeptoren ist die neuromuskuläre Übertragung weiterhin blockiert. Bei depolarisierenden Substanzen ist die Gabe von Cholinesterasehemmern nicht sinnvoll, da Acetylcholin und Suxamethonium beide depolarisierend wirken. Beziehungen zur praktischen Arzneitherapie Aus den vorliegenden Versuchen ergeben sich wichtige Anwendungsmöglichkeiten für die praktische Therapie. Die vorgestellten Substanzen haben vor allem in der Anästhesie einen großen Einsatzbereich gefunden, da zu vielen operativen Eingriffen eine zuverlässige Muskelrelaxation eine wesentliche Voraussetzung ist. Wie die Praktikumsversuche zeigen, ist eine Blockade der neuromuskulären Erregungsübertragung und damit eine Muskelrelaxation durch zwei Typen von Pharmaka möglich. Muskelrelaxantien vom Typ des (+)-Atracuriums besetzen die postsynaptischen Acetylcholinrezeptoren und verhindern eine Depolarisation der postsynaptischen Membran durch ACh, sie führen damit zu einer elektrischen Stabilisierung der postsynaptischen Membran (Stabilitätsblock). Wegen geringer Nebenwirkungen (Kreislaufreaktionen, Histaminfreisetzung, Bronchokonstriktion) werden heute in der Praxis an Stelle von (+)-Atracurium vielfach andere Pharmaka mit gleichem Wirkungsmechanismus verwandt (z.B. Gallamin, Pancuronium). Sie alle werden unter dem Begriff der stabilisierenden oder besser nicht polarisierenden Muskelrelaxantien zusammengefasst. Ihre Wirkung kann durch Cholinesterasehemmer wie Physostigmin (Erhöhung der Acetylcholinkonzentration und kompetitive Verdrängung des Muskelrelaxans) wieder aufgehoben werden, was z.B. bei vorzeitigem Operationsabbruch oder Überdosierung von Bedeutung ist. Daneben kommen auch Pharmaka vom Wirkungstyp des Suxamethoniums in der Anästhesie zur Anwendung, die durch Besetzung der Acetylcholinrezeptoren eine länger dauernde Depolarisierung der postsynaptischen Membran und damit eine Hemmung der neuromuskulären Erregungsübertragung bewirken (Depolarisationsblock). Substanzen dieses Wirktyps werden als depolarisierende Muskelrelaxantien bezeichnet. Außerhalb der operativen Fächer haben Pharmaka dieses Typs bei der Behandlung von Tetanuskrämpfen ihre Anwendung gefunden. Für den praktischen Einsatz ist von Belang, dass der Depolarisationsblock nicht durch Cholinesterasehemmer antagonisiert werden kann. Während bei den bisher genannten Beispielen das therapeutische Ziel in einer passageren Unterbrechung der neuromuskulären Überleitung besteht, muss bei der Myastenia gravis eine Verbesserung der Überleitung angestrebt werden. Diese Erkrankung beruht auf einem offenbar autoimmunologisch bedingten Mangel an Acetylcholinrezeptoren. Cholinesterasehemmer wie Neostigmin verlängern die Wirkdauer von ACh im synaptischen Spalt und führen damit zu einer Verbesserung der neuromuskulären Impulsübertragung. BoNT/A wird bei chronischen lokal umschriebenen Muskelverkrampfungen (Blepharospasmus, zervikale Dystonien) eingesetzt. Es wirkt über 3-4 Monate, indem es den verkrampften Muskel paralysiert.

Massenspektrometrische Proteinanalyse
Betreuer: Prof. Dr. Andreas Pich: Tel.: 0511-532-2808, E-mail: [email protected] Dr. Bijon Chatterji: Tel.: 0511-532-4256, E-Mail: [email protected] Praktikumsort: Institut für Toxikologie, Gebäude I6, Ebene 3, Raum 2510 / 2011 Vorbereitung: Zur Vorbereitung sollten Sie sich mit folgenden Themengebieten vertraut machen:
- Grundlagen der Massenspektrometrie - Ionisierungsverfahren (MALDI, ESI) - Massenanalysatoren (TOF, Ionenfallen, Orbitrap, Quadrupol, Hybridgeräte) - Tandem MS (MS/MS, a-,b-,c-Ionen / x-,y-,z-Ionen) - Kenntnis des Versuchablaufs
Einleitung Massenspektrometrische Techniken werden für die Identifizierung und Charakterisierung einzelner Proteine und komplexer Proteome genutzt und haben sich in der Zwischenzeit zur Standardmethode für die Bestimmung der Aminosäuresequenz und den Nachweis posttranslationaler Modifikationen entwickelt. In der Pharmakologie und Toxikologie werden Proteine als Zielstrukturen von Pharmaka und Toxinen mit massenspektrometrischen Techniken identifiziert und potentielle Modifikationen ermittelt, die z.B. einen Effekt auf die Signalweitergabe haben. Da immer mehr Proteine als Pharmaka Verwendung finden, werden auch diese zur Identifizierung und Absicherung der Primärstruktur im Massenspektrometer analysiert (Qualitätssicherung). Intakte Proteine lassen sich zwar im Massenspektrometer darstellen (top down Ansatz) aber eine Identifizierung oder Strukturanalyse ist nur möglich wenn das Protein in hohen Konzentrationen und sehr rein vorliegt. Daher bedient man sich eines Tricks und zerlegt Proteine mit Hilfe von spezifischen Endoproteinasen (Trypsin) in Peptide und analysiert diese anschließend im Massenspektrometer (bottom up Ansatz). Die Massen der nachweisbaren Peptide bilden den so genannten „peptide mass fingerprint“ (PMF) (Abb.1). Peptide werden unterschiedlich gut im Massenspektrometer abgebildet, was in erster Linie mit der Ionisierbarkeit aber auch anderen Faktoren zusammenhängt. Peptide mit intensiven Signalen werden als Vorläuferionen (precursor ions) im Massenspektrometer isoliert, fragmentiert und anschließend die Massen der Fragmente bestimmt (MS/MS-

Analyse, a-,b-,c-Ionen / x-,y-,z-Ionen) (Abb. 1). Da Peptide unter den gewählten Bedingungen nur einmal statistisch verteilt am Peptidrückrad zerbrechen, kann anhand der Abstände der Fragmentmassen die Aminosäuresequenz ermittelt werden. Dieses Verfahren ist allerdings sehr aufwendig und langsam. Daher setzt man heutzutage Datenbanken ein, in denen alle bekannten Proteinsequenzen enthalten sind. Durch die großen Fortschritte der Genomsequenzierungen stehen für viele Organismen die codierenden Bereiche und damit die Proteinsequenzen fest. In Datenbanken werden nun die Proteine in silico in tryptische Peptide und Peptidfragmente zerlegt. Geeignete Software-Tools (MASCOT, SEQUEST, PARAGON) durchsuchen mit den experimentell ermittelten Daten die Datenbank und finden das Protein mit den höchsten Übereinstimmungen heraus. Für die Bewertung der Ergebnisse erhält man einen bestimmten Wahrscheinlichkeitswert, z.B. Mascot Score, der die Qualität der Analyse widerspiegelt.
Abb. 1. Massenspektrometrische Proteinanalyse. Im oberen Spektrum wurde ein Protein im Massen-spektrometer gemessen. Eine Identifizierung ist nicht möglich. Strukturinformationen können nach Verdau mit Trypsin und durch Erzeugung von MS/MS-Spektren erhalten werden. Der Verdau wird im Labor ü.N. durchgeführt. Die Isolierung der Vorläufermassen, ihre Fragmentierung und die anschließende Analyse der Fragmentmassen erfolgt in Millisekunden im Massenspektrometer. Die Zuordnung der Massen erfolgt mit Hilfe von Datenbanken.

Praktikumsaufgabe Die Identifizierung von Proteinen mit MS-Techniken soll erlernt werden. Jede StudentIn erhält eine Coomassie brilliant blue gefärbte Proteinbande. Diese wird ausgeschnitten, tryptisch verdaut und der MS-Analyse zugeführt. Die Identität der Proteine ist den Praktikumsteilnehmern nicht bekannt und soll im Rahmen des Experiments ermittelt werden. Ablaufplan
Zeit MS-Funktionsanalyse Tag 1 14:00 Begrüßung, Einführung Tag 1 14:30 Auswertung des Gels, Ausschneiden der Banden Tag 1 15:00 Entfärben der Banden Tag 1 15:15 Kolloquium: MS- MS/MS-Techniken Tag 1 16:30 Trypsinzugabe
Tag 1 ca. 17:00 Ende Tag 1 Tag 2 10:00 Proben auf MALDI-Target auftragen Tag 2 10:30 MS- MS/MS-Messung im MALDI-TOF/TOF Tag 2 11:00 Auswertung der MS-Daten, Datenbankvergleiche Tag 2 12:00 Ende Tag 2
Massenspektrometrische Protein-Analyse Materialien Skalpell
Eppendorf-Reaktionsgefäße Pipetten und Spitzen
Lösungen 50 % Acetonitril Acetonitril 20 mM Ammoniumhydrogencarbonat, pH 8,5 Trypsinlösung (12,5 ng/μl), 20 mM Ammoniumhydrogenkarbonat, 10 % Acetonitril Matrix: α-Cyano-Hydroxy-Zimtsäure, 1 μg/μl in 50 % Acetonitril, 0,2 % TFA Peptidstandard
Tryptischer Verdau 1) Coomassie-gefärbte Bande genau ausschneiden und zerkleinern. 2) Bande in E-cup überführen und 2 x 1 min bei RT mit 250 μl
Ammoniumhydrogencarbonat waschen. 3) Bande mit 250 μl 50 % Acetonitril überschichten, 30 min bei 37 °C schütteln,
wiederholen. 4) Zugabe von 50 – 100 μl Acetonitril (je nach Bandengröße), 2 min Inkubation bei RT
bis die Gelstücke milchig weiß aussehen. 5) Das Acetonitril entfernen und Gelstücke im Vakuum trocknen (Speed-Vac) 6) 20 μl Trypsinlösung auf die trockenen Gelstücke geben und ca. 30-60 min auf Eis
inkubieren. 7) Probe bei 37 °C über Nacht inkubieren.

8) Probe kurz zentrifugieren, aus dem flüssigen Überstand ca. 1,0 μL abnehmen In der Regel enthält dieser Überstand genügend Peptide, um eine aussagekräftige MS-Analyse anzufertigen.
Massenspektrometrische Messung
1) Von jeder Probe werden 1,0 μL auf einen Spot eines MALDI-targets aufgetragen und sofort mit 0,8 μL Matrix-Lösung versetzt, durch vorsichtiges hin-und her-Pipettieren gemischt und zum Trocknen stehen gelassen (ca. 5 min „dried droplet-Methode).
2) Zusätzlich werden 0,5 μl eines Peptidstandards auf die entsprechenden Kalibrierungsspots gegeben, mit 0,5 μl Matrix-Lösung versetzt und getrocknet.
3) Das target wird in das MALDI-TOF/TOF-Massenspektrometer eingeschleust und die Peptidmassen werden gemessen.
4) Die Messung und Auswertung der MS-Daten wird unter Anleitung durchgeführt. Literatur
1 Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature (2003) 422, 198-207

Immunhistochemische Untersuchungen von C3bot induzierten morphologischen Veränderungen in kultivierten hippokampalen Zellen Praktikumsort Institut für Toxikologie (Direktor: Prof. Dr. med. Ingo Just) Gebäude I6, Ebene 3, Raum 2070 Ansprechpartner Dr. Astrid Rohrbeck [email protected] Sandra Hagemann [email protected] Tel. 532-2807/-2844 Allgemeine Hinweis Bitte bringen Sie Kittel mit! Vorbereitung: Zur Vorbereitung sollten Sie sich mit folgenden Themengebieten vertraut machen:
- Biochemie des Zytoskeletts (Aktin, Tubulin, Intermediärfilamente) - Signaltransduktionsvorgänge der kleinen GTPasen (Rho-Familie) - Grundlagen der Immunfluoreszenz - Kenntnis des Versuchablaufs
Einleitung Clostridium botulinum C3 Exoenzym
Das C3 Exoenzym von Clostridium botulinum ist namensgebend für die Familie der C3-ähnlichen ADP-Ribosyltransferasen. Das C3 Exoenzym hat ein einem Molekulargewicht von ca. 25 kDa und überträgt eine ADP-Ribose des Cosubstrates NAD auf die Aminosäure Asparagin 41 der Zielproteine RhoA, B und C. Durch die Mono-ADP-Ribosylierung der Rho-GTPasen werden diese funktionell inaktiviert und es kommt nachfolgend zur Inhibition der Aktin-Polymerisierung (Aktories, 2004). Dies führt nachfolgend zu einer Umverteilung der Aktinfilamente und zu einer Depolimerisation der Aktin-Stress-Fasern, was zu deutlichen morphologischen Veränderungen führt. Anhand der morphologischen Veränderungen (Abb.1) kann gezeigt werden, dass eine C3-Behandlung eine Zellteilung (Cytokinese) verhindert wobei die Kernteilung (Karyokinese) scheinbar unverändert bleibt, was in Folge zu einer Mehrkernbildung führt. Es konnte bereits gezeigt werden, dass eine Depolymerisation des Aktinzytoskeletts zu einer Mehrkernbildung führt (Gachet, 2001). Daher ist zu vermuten, dass durch die C3-Behandlung eine Rho-abhängige Reorganisation des Aktinzytoskeletts verursacht wird. Diese Modifikation des Aktinzytoskeletts wirkt sich negativ auf die Ausbildung des kontraktilen Rings aus und beeinflusst den Zellzyklus. Neben der Mehrkernbildung ist aufgrund der C3-Behandlung auch ein multipolarer Phänotyp zu erkennen.

Abbildung 1: Morphologische Veränderung nach C3 Inkubation im Fluoreszenzmikroskop. Die obere Reihe zeigt die unbehandelten Kontrollzellen. In der mittleren und unteren Reihe sind die mit C3 behandelten Zellen dargestellt. In der mittleren Reihe ist exemplarisch eine Multikernbildung und in der unteren Reihe der multipolare Phänotyp gezeigt. Ziel des Versuches Visualisierung der C3-induzierten morphologischen Veränderungen. Bewertung der C3-induzierten Multikernbildung und scheinbar bipolaren Zellen mittels Fluoreszenzmikroskopie. Ablaufplan 10.00 – 10.15 Uhr Begrüßung 10.15- ca.11.30 Uhr Waschen, Fixierung, Permeabilsierung und
Blockierung der Zellen ca.10.30 – 11.30 Uhr Kolloquium 11.30 – 12.00 Uhr Überführung der Deckgläschen 12.00 – 13.00 Uhr 1.AK Inkubation Mittagspause 13.00 – 13.30 Uhr Waschen und erneute Permeabilisierung 13.30 – 14.00 Uhr 2.AK Pause, Besprechung offener Fragen 14.00 – 15.30 Uhr Waschen, Antifade ab 15.30 Uhr Mikroskopische Auswertung und im
Anschluss Abschlussbesprechung (Die Zeiten können sich je nach experimentellen Gegebenheiten etwas nach hinten verschieben.) Durchführung
- aussäen der HT22 Zellen auf Deckgläschen - 24h später altes Medium entfernen und Zellen vergiften (500nM C3botWT) - 24h bzw. 48h Inkubation bei 37°C, 5%CO2

- nach Ablauf der C3-Inkubationsdauer altes Medium absaugen - Zellen mit kaltem PBS waschen - 20 Minuten mit 4% Paraformaldehyd in PBS bei Raumtemperatur fixieren - anschließend das Paraformaldehyd absaugen, die Zellen erneut dreimal mit kaltem
PBS waschen - um die Zytoskelettstrukturen für die Fluoreszenzstoffe zugänglich zu machen,
werden die Zellen für 5 Minuten mit 0,1-0,3% Triton X-100 in PBS permeabilisiert
- um unspezifische Bindungen der Fluoreszenzstoffe zu vermeiden, folgt eine Inkubation mit 5% BSA in PBS für 30min (Blockierung)
- 1.AK ansetzen (α-Tubulin /Ratte): Verdünnung: 1:500 in 5% BSA - im Anschluss daran werden die Deckgläschen in eine mit Parafilm ausgelegte
feuchte Kammer gelegt (Dabei ist darauf zu achten, dass die Oberseite der Deckgläschen mit den fixierten Zellen nach oben gelegt werden!!!)
- 80µl 1.AK auf jedes Deckgläschen pipettieren - 1h bei RT inkubieren - anschließend die Zellen zweimal mit PBS waschen - nochmalige Inkubation mit 0,1-0,3% Triton X-100 für 5 Minuten - 2.AK ansetzten in 5% BSA, alle zusammen (z.B. 1000µl Gesamtvolumen): -
Alexa 488 α-rat ( Tubulin) 1:1000 = 1µl (ZK, 4°C, blaue Box, gelbes Eppi) Rhodamin/Phalloidin (Actinfärbung) 1:1000 = 1µl (ZK, -20°C, AK’s, grüne Box)
Dapi (Kernfärbung) 1:100 = 10µl (4°C) - 80µl der 2.AK-Lösung auf die Deckgläschen geben - Deckgläschen für 30min, abgedeckt mit Alufolie, stehen lassen - AK-Lösung absaugen und zweimal für fünf Minuten mit kaltem PBS und dann
zweimal mit kalten 0,1 % TWEEN 20 in PBS waschen - zuletzt folgt noch ein Waschschritt mit Aqua dest. - um ein Ausbleichen der Fluoreszenz zu verhindern, werden 10µl des Antifade (Pro
Long® Antifade Kit) auf die Objektträger vorgelegt - Deckgläschen kurz abtropfen lassen und mit den Zellen nach unten auf das
Antifade bzw. Objektträger legen (ggf Luftblasen vorsichtig rausdrücken) - nach einer Trocknungszeit von einigen Minuten bis einer Stunde werden die
Ränder der Deckgläser mit Nagellack versiegelt (zur längeren Aufbewahrung) - die Objektträger bis zur Auswertung durch die Fluoreszenzmikroskopie dunkel
und bei 4°C lagern - mikroskopische Auswertung erfolgt am Zeiss Axiovert 200 M Mikroskop
Literatur
1 Aktories K, Wilde C, Vogelsgesang M, Rho-modifying C3-like ADP-ribosyltransferases Rev Physiol Biochem Pharmacol (2004) 152:1-222
2 Gachet Y, Tournier S, BA Miller J, Hyams JS, A MAP kinase-dependent actin checkpoint ensures proper spindle orientation in fission yeast; Nature (2001) 412, 352 - 355

Funktionelle Evaluation des Drei-Domänen-Modells „Großer clostridialer Zytotoxine“
Praktikumsort Institut für Toxikologie (Direktor: Prof. Dr. med. Ingo Just) Gebäude I 6, Ebene 3, R.2020. Ansprechpartner Dr. Ralf Gerhard [email protected] Dr. Harald Genth [email protected] Tel. 532-2810/-9168 Allgemeine Hinweise: Mitzubringen sind: Kittel Ihre Vorbereitung sollte umfassen:
GTP-bindende Proteine, GTPase Zyklus Regulation des Zytoskeletts der Zelle Rezeptor-vermittelte Endozytose Generierung von Antikörpern in Tieren, „Impfung“
1. Einleitung Rho-GTPasen sind zentrale Regulatorproteine der Zellmotilität, der Endo- und Exozytose sowie des Zellzyklus. Daher sind Rho-GTPasen bevorzugte Zielsubstrate bakterieller Toxine. Die Toxine dienen pathogenen Bakterien zu dem Zweck, die Signalweitergabe der Zielzelle zu manipulieren (z.B. um das Bakterium aufzunehmen) oder um Abwehrmechanismen der Zelle auszuschalten. Diese Strategie ist auch der Funktionsmechanismus der Toxine A und B aus C. difficile. Sie gehören zur Familie der „Grossen clostridialen Zytotoxine“, einer Gruppe hochmolarer Glycosyltransferasen, die Rho-GTPasen inaktivieren. Toxin A und Toxin B schädigen durch ihre Aktivität das Darmepithel und können eine Durchfallerkrankung oder eine „pseudomembranösen Kolitis“ (PMC) verursachen. Die Toxine zeigen eine Drei Domänen-Struktur (Abb. 1). Diese Domänen erlauben die Aufnahme des Toxins in die Zielzelle („Autotransporter“) sowie die enzymatische Aktivität (Abb. 2).
Abb. 1. Drei-Domänen-Struktur der „Grossen clostridialen Zytotoxine“ (Toxin B). Die Rezeptor-bindende Domäne setzt sich aus sich wiederholenden Oligopeptid-Elementen zusammen, die möglicherweise an Zuckerstrukturen des unbekannten Rezeptors binden. Diese multivalente Bindung löst eine Rezeptor-vermittelte Endozytose aus. Eine hydrophobe Region in der Mitte des Moleküls stellt eine Transmembran-Domäne dar, welche eine Pore oder einen Kanal formt, durch den die katalytische Domäne in das Zytoplasma gelangt. Die Transferase-Aktivität befindet sich in der N-terminalen Domäne.

Abb. 2. Aufnahme der Toxine in die Zielzelle. 1. Das Toxin bindet an einen unbekannten Rezeptor. 2. Die Bindung des Toxins an den Rezepetor löst die Endozytose aus. 3. Das endozytierte Vesikel wird zum Endosom prozessiert. 4. Eine vesikuläre ATPasae pumpt Protonen in das endosomale Lumen. Dabei sinkt der pH-Wert auf etwa 5,5. 5. Die pH-Absenkung führt zu einer Konformationsänderung des Toxins; die hydrophobe Transmembran-Domäne (TMD) bewegt sich zur Oberfläche des Moleküls, um mit der endosomalen Membran zu interagieren. Die Ausbildung der Pore wird durch Hemmstoffe der Protonen-ATPase blockiert. 6. Die katalytische Domäne wird durch eine Protease abgespalten und gelangt durch die Pore in das Zytoplasma. 7. Die katalytische Domäne mono-glucosyliert Rho-GTPasen im Zytoplasma. 2. Ziel des Versuches: Vertieftes Verständnis der Toxinaufnahme
Hemmung der Translokation aus dem Endosom durch Bafilomycin Hemmung der Toxinwirkung durch Antitoxine
3. Testsysteme
Depolymerisation des Aktinzytoskeletts - „Zytotoxizitäts-Testung“ a) Zellabrundung b) Aktinfärbung
Testsystem „Antitoxin – Toxin Interaktion“ („Dot-Blot“)

4. Zeitplan
Bafilomycin Toxin-Antitoxin-Testing („Dot-Blot“)
Zytotoxizitäts- Testung
9:00- 9:15 Tag 1 Begrüßung 9:15 – 10:00 Vergiftung Membran beladen,
Blocken Vorinkubation Vergiftung
10:00 – 10:45 Kolloquium 10:45 – 11:15 Zellen fixieren Waschen, 1.AK
Mittagspause 13:00 – 13:30 Waschen 13:30 – 14:15 2.AK 13.30 – 14:15 Mikroskopie Waschen 14:15 – 15:00 ECL/Quantifzierung Dokumentation
Zellen auszählen 15:00 Abschlussbesprechung
5. Praktikumsaufgabe 1 5.1 Hintergrund: Bafilomycin A1 ist ein Pilzgift aus Streptomyces griseus, das die Protonen-ATPase eukaryonter Zellen inhibiert. Dadurch wird die Ansäuerung endosomaler Vesikel unterbunden. Da die Senkung des intravesikulären pH-Wertes auf 5.0 notwendig für die Konformationsänderung der Toxine zur Einlagerung in die Vesikelmembran ist, kann somit die zytotoxizität (intrazelluläre Wirkung) verhindert werden. 5.2 Durchführung Material: Bafilomycin-Stock [0,3 mM] in DMSO Toxin B [200 ng/µl] 10 mM Tris-HCl pH 7,4, 50 mM NaCl, 0,1 mM MnCl2 Zellen: Swiss 3T3 Fibroblasten in 4er-wells PBS:
Fixierlösung: PBS + 3,7% (v/v) Formalin Es werden nach folgendem Schema die Substanzen in ihrer Reihenfolge zu den Zellen (in 500 µl Medium) zugegeben:
# 1 Kontrolle
# 2 Toxin B (1 µl)
# 3 1. Bafilomycin A1 (5 µl) 2. Toxin B (1 µl)
# 4 Bafilomycin A1 (5 µl)
Die morphologischen Änderungen werden nach 30 und 90 min mikroskopisch untersucht. Danach erfolgt die Fixierung der Zellen und Färbung des Aktinzytoskeletts mit Alexa Fluor 568-Phalloidin:
Absaugen des Kulturmediums Zugabe von 0,3 ml Fixierlösung pro well 10 min Inkubation bei RT 2 x waschen mit je 0,5 ml PBS Inkubation mit Alexa Fluor 568-Phalloidin (15 µl Stock in 1 ml PBS)
Je 0,25 ml pro well unter Lichtausschluss 2 x waschen mit je 0,5 ml PBS Montieren der Deckgläser auf Objektträger

6. Praktikumsaufgabe 2 – Hemmung der Toxinaufnahme durch Antitoxine 6.1 Hintergrund: Die katalytische Domäne (CDB1), die Transmembrandomäne (CDB2) sowie die Rezeptorbindedomäne (CDB3) des TcdB wurden kloniert und als rekombinante Proteine in E. coli exprimiert. Da die singulären Domänen untoxisch sind, eignen sie sich als Antigene zur Immunisierung von Kaninchen. Die gewonnen Antitoxine sollen in diesem Praktikum auf ihre Fähigkeit getestet werden:
1. Bindung an die clostridialen Toxine bzw. die funktionellen Domänen 2. Neutralisation der zytotoxischen Wirkung der Toxine
6.2 Definitionen:
Domäne/Funktion Aminosäuren Masse[kDa]
holo-TcdB Holo-Toxin 1-2366 270 CDB1 Katalytische 1-546 60 CDB2 Transmembran 901-1750 94 CDB3 Rezeptorbindung 1751-2366 68
6.3 Material
• Nitrozellulose-Blotmembranen, Schleicher & Schuell, 0,4 µm Porengröße • Toxinlösungen (200 µg/ml): TcdB • Lösungen der katalytischen Domänen (200 µg/ml): TcdB1-546, TcdB901-1750, TcdB1751-2366 • Antitoxine (0,5 mg/ml): anti-TcdB, anti-CDB1, anti-CDB2, anti-CDB3 • Zweitantikörper(1 mg/ml): anti-Rabbit IgG, Peroxidase gekoppelt • TBST (Tris-buffered Saline Tween): 50 mM TRIS pH 7,2; 150 mM NaCl; 0,05 % (w/v)
Tween20 • Milchpulver, fettfrei • ECL-ROTI Lumineszenz-Reagenz

6.4 Nachweis der Toxin – Antitoxin – Interaktion („Dot Blot“) 1. Die bereitgestellten Proteinlösungen werden mit ddH2O 1:10 verdünnt. 2.Jeder Teilnehmer erhält eine Nitrocellulose-Membran, auf die je 5 „Dots“ à 2µl vorsichtig aufgetragen werden (Schema). 3. Die beladenen Membranen werden 30’ getrocknet. 4. Das Blocken unspezifischer Protein-Bindungsstellen auf den Membranen erfolgt mit 5% Milchpulver in TBST für 60’. 5. Die Membranen werden gründlich mit ddH2O und TBST gespült. 6. Pro Membran wird ein Erstantikörper (Antitoxin) im Verhältnis 1:10.000 in TBST eingesetzt. Die Inkubationsdauer beträgt 60’.
Membran 1 2 3 4 Anti-Toxin Anti-holo-TcdB Anti-CDB1 Anti-CDB2 Anti-CDB3
7. Die Membranen werden 3x10’ mit TBST gewaschen. 8. Die Membranen werden mit dem Zweitantikörper im Verhältnis 1:2.000 in TBST für 60’ inkubiert. 9. Die Membranen werden erneut 3x10’ mit TBST gewaschen. 10. Die Entwicklung erfolgt mit ECL-ROTI. Das ROTI-Reagenz enthält Luminol, welches von der an den Zweitantikörper gekoppelten Peroxidase in Anwesenheit von H2O2 oxidiert wird. Das Reaktionsprodukt chemoluminesziert und kann daher unter Lichtausschluss detektiert werden. Zunächst werden die beiden Komponenten des Lumineszenz-Reagenzes (H2O2 und Luminol) im Verhältnis 1:1 gemischt. Die Mischung wird dann auf die Membranen (hintereinander) gegeben und für 1’ abgedunkelt (Alufolie) unter Schwenken inkubiert. Die Detektion der Lumineszenz erfolgt unter Lichtausschluss (Deckel zu) mit der Kodak Station. Die Schwärzung der Punkte wird semi-quantitativ erfasst. 6.5 Zytotoxizitäts-Testung 1. Die Toxinlösung wird im Verhältnis 1:1.000 mit ddH2O verdünnt. Diese Toxinlösung wird im Verhältnis 1:1 mit den unverdünnten Antitoxinen bzw. ddH2O als Kontrolle versetzt. Als weitere Kontrolle werden je 5 µl der unverdünnten Antitoxine mit 5µl ddH2O versetzt. Alle Ansätze werden 10’ auf Eis inkubiert. 2. Die Fibroblasten werden in einer 24-Ansatz-Platten kultiviert. Um Material zu sparen wird das Medium je Loch auf 200 µl reduziert. Die Zellen werden mit den Toxin- bzw. Antikörperlösungen
TcdB
CDB1
CDB3
CDB2
TcdB
CDB1
CDB3
CDB2
TcdB
CDB1
CDB3
CDB2
TcdB
CDB1
CDB3
CDB2
TcdB
CDB1
CDB3
CDB2

versetzt. Dafür werden je 2µl der Lösungen aus 2. zu je 200 µl Medium pro Ansatz gegeben. Ein Ansatz wird mit 2µl ddH2O versetzt. Tabelle zeigt das Schema. 3. Die Zellen werden für 4 Stunden bei 37°C und 5% CO2 inkubiert. 4. Nach der Inkubation erfolgt die Auswertung des Versuches mit dem Phasen-Kontrast-Mikroskop. Dazu wird von jedem Ansatz eine repräsentative Aufnahme mit der Digitalkamera des Mikroskops gemacht (Software: Kappa Image Base, Programm „Control“). Zur Quantifizierung werden die erhaltenen Bilder im Programm „Metreo“ geöffnet. Durch Erzeugen eines Rasters erhält man 6 Quadranten, die dann ausgezählt werden. Man zählt zunächst die komplett abgerundeten Zellen und dann alle weiteren. Pro Aufnahme werden sechs Quadranten ausgezählt. Das höchste und niedrigste Ergebnis werden gestrichen; die übrigen vier Werte werden ausgewertet (Mittelwertbildung, Standardabweichung). Die prozentuale Abrundung [R%] wird berechnet:
N (abgerundet) R % = ---------------------------------------------------- x100
N (abgerundet) + N (weitere Zellen) 6.6 Zusammenfassung der Ergebnisse
1 Toxin – Antitoxin – Interaktion („Dot-Blot“). Bitte tragen Sie die Interaktionen in semi-quantitativer Erfassung ein: ++ starke Kreuzreaktivität + Kreuzreaktivität besteht 0 keine Kreuzreaktivität
holo-TcdB CDB1 CDB2 CDB3
Anti-Holo-TcdB Anti-CDB1 Anti-CDB2 Anti-CDB3
2 Zytotoxische Wirkung auf Fibroblasten. Bitte tragen Sie die R %-Werte ein:
6.7 Diskussion:
1. Formulieren Sie Hypothesen, warum die Antitoxine anti-CDB1 und anti-CDB2 nur eine schwache Kreuzreaktivität mit dem holo-Toxin zeigen.
2. Das Antitoxin anti-CDB1 blockiert in vitro die Glucosyltransferase-Aktivität des holo-Toxins. Warum vermag er dennoch nicht die zytotoxische Aktivität im Zytotoxizitätsassay zu blockieren ?
3. Welche weiteren (theoretischen) Möglichkeiten könnten Sie sich vorstellen, um die zytotoxische Wirkung der Toxine zu blockieren ? Gehen Sie hierzu die einzelnen Schritte der Toxinaufnahme und –prozessierung anhand Abb. 2 im Detail durch !
4. Auf welchen Zelltypen könnte die Inkubation der Antitoxine allein eine Wirkung zeigen ?
Zusatz TcdB + Zusatz Zusatz allein ddH2O Anti-Holo-TcdB Anti-CDB1 Anti-CDB2 Anti-Holo-TcdB

1
KURS: BIOMEDIZIN
Thema: Pharmakokinetik und Pharmakodynamik:
Proteinbindung von Pharmaka
Dozent: Prof. Dimitrios Tsikas, Alexander Zörner
Betreuer: Bibiana Beckmann, Anke Böhmer
Ort: Institut für Klinische Pharmakologie der MHH
EINFÜHRUNG, LEHRINHALT UND ZIEL DES KURSES
Folgende kurze Definitionen können für die Begriffe PHARMAKOKINETIK und
PHARMAKODYNAMIK gemacht werden: Die Pharmakokinetik beschreibt den zeitlichen
Verlauf der Konzentration eines Arzneistoffes – Pharmakons – und seiner Metabolite in
Blut und Urin. Die Pharmakodynamik beschreibt den biologischen Effekt des
Pharmakons an seinem Wirkort.
Oral verabreichte Pharmaka werden im Darm resorbiert und in der Leber
metabolisiert (siehe „First Pass Effect“), bevor sie die systemische Zirkulation erreichen
(ABB. 1). Resorbierte Pharmaka werden über das Blut an ihren jeweiligen Wirkort
transportiert. Die Bioverfügbarkeit (F, fraction) beschreibt den prozentuellen Anteil des
applizierten Pharmakons, der seinen Wirkort erreicht. Intravenös verabreichte Pharmaka
umgehen zunächst die Leber und erreichen direkt das Blut. Die Bioverfügbarkeit
intravenös verabreichter Pharmaka ist definitionsgemäß 100% (F = 1).
Abb. 1. Resorption, Verteilung, Proteinbindung, Metabolismus
(Elimination) und Ausscheidung von Pharmaka über die Niere
(und die Galle).
Gelb (hell) = lipophil; Blau (dunkel) = hydrophil.

2
Die Metabolisierung von Pharmaka erfolgt in der Regel in zwei Stufen oder zwei
Phasen: Phase 1 und Phase 2. Phase 1-Reaktionen sind Oxidationsreaktionen und
werden durch das Cytochrom P450 (CYP)-System katalysiert. Die Phase 1-Metabolite sind
(in der Regel) Substrate für die Phase 2-Reaktionen – Konjugationsreaktionen. Ziel
der Metabolisierung ist die Inaktivierung und Elimination von Pharmaka. Eine gesteigerte
Ausscheidung über die Niere wird durch Steigerung der Hydrophilie des Pharmakon und
seiner Metabolite erreicht (ABB. 1).
In Protein-haltigen Flüssigkeiten und im Gewebe binden Pharmaka reversibel an
Proteine. Dabei ist nur das freie, d.h. ungebundene Pharmakon, in der Lage, Barrieren
(z.B. Zellmembranen) zu überwinden und an seinem Wirkort (z.B. Rezeptor) zu
gelangen, wo es schließlich seine pharmakologische Wirkung entfalten kann. Die Bindung
von Pharmaka an Plasmaproteine – Proteinbindung (PB) ist von besonderer Bedeutung
für die Pharmakokinetik und Pharmakodynamik. Die Verfügbarkeit, d.h. die Konzentration
oder Menge eines Pharmakon an seinem Wirkort, der Metabolismus des Pharmakon in
der Leber (und anderen Organen) sowie seine Ausscheidung über die Niere hängen
entscheidend vom Ausmaß seiner Bindung an Proteine ab: Die Fraktion der Protein-
gebundenen Pharmaka ist „wirkungslos“, und kann weder metabolisiert noch über die
Niere (ABB. 2) ausgeschieden werden.
Abb. 2. Zusammenhang zwischen prozentueller renaler Ausscheidung
in unveränderter Form und Plasma-Proteinbindung von
Pharmaka. Jedes Symbol repräsentiert ein Pharmakon.
R ist der Korrelationskoeffizient zwischen „Renale
Ausscheidung (%)“ und „Proteinbindung (%)“. Ein Wert von 1
für R würde einer 100%igen Korrelation entsprechen.
0 20 40 60 80 100
0
20
40
60
80
100
R = - 0.87
Ren
ale
Au
ssch
eid
un
g (
%)
Proteinbindung (%)

3
In diesem Kurs werden pharmakokinetische (PK) und pharmakodynamische (PD)
Grundlagen vermittelt. Am Beispiel von PARACETAMOL und SALICYLSÄURE, zwei Pharmaka
mit recht unterschiedlichen PK/PD-Eigenschaften einschließlich deutlich unterschiedlich
starker Proteinbindungsgraden (TABELLE 1, ABB. 3), wird das Ausmaß der Proteinbindung
an Albumin, eines der relevantesten Transportproteine im Blut, experimentell bestimmt.
Tabelle 1. Pharmakokinetische (PK) und pharmakodynamische (PD)
Daten von Paracetamol und Salicylsäure
PK/PD-Größe Paracetamol Salicylsäure *
_______________________________________________________________________
Anwendung Schmerz, Fieber Keratolyse (Haut)
Effektive Plasma-Konz. 60 – 120 µM 1100 – 2200 µM
Toxische Plasma-Konz. > 2000 µM > 1500 µM (Tinnitus)
Elimin.-Halbwertszeit 2.0 ± 0.4 h 2 – 19 h
Bioverfügbarkeit 88 ± 15% 100%
Verteilungsvolumen 0.95 L/kg 0.2 L/kg
Renale Ausscheidung 3 ± 1% 2 -30% (pH-abhängig)
Proteinbindung 20% 80 – 95% (Konz.-abhängig)
_______________________________________________________________________
* Hauptmetabolit von Acetylsalicylsäure (Aspirin)
NHC
O
CH3
OH
OHO
OH
Paracetamol, Acetaminophen(N-(4-hydroxyphenyl)acetamid)
Salicylsäure(2-Hydroxybenzoesäure)
Abb. 3. Chemische Struktur von Paracetamol und Salicylsäure

4
Die Proteinbindung von Pharmaka kann mit Hilfe folgender Techniken im Labor bestimmt
werden:
1) Equilibrium-Dialyse
2) Mikrodialyse
3) ULTRAFILTRATION
4) Ultrazentrifugation
5) Erythrocyten-Verteilungsmethode
In diesem Praktikum wird das Verfahren der Ultrafiltration, die meist verwendete
Methode, angewandt. Dabei wird eine Lösung des Pharmakons in Plasma oder wie im
vorliegenden Fall in einer gepufferten, wässrigen, Albumin-haltigen Lösung bei kleiner g-
Zahl unter Verwendung einer semipermeablen Membran mit einem definierten cut-off
(z.B. 10 kDa) zentrifugiert. Dabei wird ein Ultrafiltrat von kleinem Volumen gewonnen,
in welchem das freie Pharmakon aufgrund des herrschenden, d.h. nicht gestörten
Gleichgewichts, in der gleichen Konzentration vorliegt wie in der Proteinfraktion: CF. Die
Proteinbindung PB (in Prozent) wird dann mit Hilfe der Formel über das analytisch
erfassbare CF berechnet:
PB (%) = [ CG / CT ] × 100 = [ (CT – CF) / CT ] × 100 Glg. 1
wobei: CB die Konzentration des gebundenen Pharmakon ist
CT die bekannte (eingesetzte) Gesamtkonzentration des Pharmakon ist
Zu diesem Zweck wird die Konzentration des freien Paracetamol bzw. der freien
Salicylsäure im Ultrafiltrat CF mittels HPLC (High-Performance Liquid
Chromatography) mit UV-Detektion (HPLC-UV) bestimmt (Abbn. 4 und 5). Die
Konzentration von Albumin im Puffer ist konstant (30 g/L), während Paracetamol und
Salicylsäure bei unterschiedlichen Gesamt-Konzentrationen eingesetzt werden (CT = 0,
50, 100, 250 µM), um das Vorliegen einer Konzentrations-abhängigen PB dieser
Pharmaka zu untersuchen. Die Konzentration von Albumin, Paracetamol und Salicylsäure
liegt jeweils in physiologischen bzw. pharmakologisch relevanten Bereichen vor.

5
Abb. 4. HPLC-Chromatogramm eines Standardgemisches aus
Paracetamol und Salicylsäure in Phosphat-Puffer. Für weitere
Erläuterungen siehe den Text. Siehe auch Abb. 5.
Abb. 5. Kalibriergeraden für die HPLC-Analyse von Paracetamol und
Salicylsäure in Phosphat-Puffer. Für weitere Erläuterungen
siehe den Text. Vergleich auch Abb. 4.
0 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20
0,0
2,0
4,0
6,0
8
10
12
14 AU
Paracetamol PHT Salicylsäure
PHT
0 50 100 150 200 250
0
50
100
150
200
250
300 Paracetamol Salicylsäure
Pea
k he
ight
(23
0 nm
)
Konzentration (µM)

6
EXPERIMENTELLER TEIL
(s. Arbeitsprotokoll für Details)
1. Herstellung von Lösungen (je 0, 50, 100, 250 µM) von Paracetamol und
Salicylsäure in Albumin-freiem (Kalibrierlösungen zur Bestimmung von CT) und
Albumin-haltigem (30 g/L) Phosphatpuffer (100 mM, pH 7.4)
2. Durchführung der Ultrafiltration, Gewinnung des Ultrafiltrats
3. HPLC-Analyse des Ultrafiltrats und der Kalibrierlösungen – Paracetamol und
Salicylsäure werden gleichzeitig analysiert
HPLC-Bedingungen
Säule: Nucleodur C18 Gravity
(250 x 4 mm i.D., 5 µm Partikelgröße)
Mobile Phase: 45 mM (NH4)2SO4-Acetonitril, 90:10 (v/v)
(keine pH-Einstellung)
Flussrate: 1 mL/min
Detektion: UV (bei 230 nm)
Analysenzeit: 20 min
Retentionszeit: 6.2 min für Paracetamol
10.3 min für Salicylsäure
Auswertung und Diskussion der Ergebnisse
(s. Arbeitsprotokoll für Details)
1. Berechnung der Konzentration CF von Paracetamol und Salicylsäure (in µM) aus
HPLC-Analysen; möglich ist auch die Verwendung von Peakhöhen (PH)
2. Ermittlung der PB für die untersuchten Konzentrationen nach Glg. 1 oder Glg. 2
3. Graphische und tabellarische Präsentation und Beschreibung der Ergebnisse:
a) Vergleich zwischen Paracetamol und Salicylsäure
b) Überprüfung einer Konzentrationsabhängigkeit der PB für beide Pharmaka

7
ARBEITSPROTOKOLL ZUR PROTEINBINDUNG
Herstellung von Lösungen, Durchführung der Ultrafiltration, HPLC-Analyse,
PB-Berechnung
1) Es werden 10 mM-Lösungen von Paracetamol (MG=151.2) und
Salicyläure (MG=160.1 für das Na-Salz) in destilliertem Wasser
hergestellt
2) Es wird eine 30 g/L Albumin-Lösung in Phosphatbuffer (100 mM, pH
7.4) hergestellt. Als Albumin wird humanes Serum-Albumin (HSA)
verwendet
3) Es werden je 0, 50, 100 und 250 µM Gemische von Paracetamol und
Salicylsäure in HSA-freiem Phosphatpuffer hergestellt: je 0, 40, 80
und 200 µL Aliquots der 10 mM-Lösungen zu 8000, 7920, 7840 und
7600 µL des Phosphatpuffers ���� CT
4) Es werden je 0, 50, 100 und 250 µM Gemische von Paracetamol und
Salicylsäure in der HSA-Lösung hergestellt: siehe oben
���� CF
5) Je 2000 µL der HSA-Paracetamol-Salicylsäure-Lösungen werden mit
Hilfe von Ultrafiltrationskartuschen (Vivaspin, cut-off 10 kDa,
Sartorius) durch Zentrifugation bei ca. 300 ×g für 2 Minuten bei 10
°C ultrafiltriert ���� CF
Wichtig: das Ultrafiltrat-Volumen sollte 50 – 100 µL betragen, d.h.
maximal 2.5 – 5% des Gesamtvolumens, damit das Bindungs-
Gleichgewicht nicht gestört wird

8
6) Je 50 µL der Proben aus Schritt 3) ( ���� CT ) und der Ultrafiltrate
( ���� CF ) werden mit je 200 µL der mobilen Phase verdünnt und in
geeignete Gefäße für die HPLC überführt.
7) HPLC-Analyse der Proben mittels eines automatischen
Probenaufgebers
8) Auswertung der HPLC-Analysen, Bestimmung der Proteinbindung
(PB) nach Glg. 1
Bemerkung: Aufgrund der Gleichbehandlung aller Proben
einschließlich der Verdünnung mit Puffer vor der HPLC-Analyse ist
die Berechnung der Konzentrationen von Paracetamol und
Salicylsäure Cf und CT nicht unbedingt erforderlich. Anstelle von CF
und CT kann in Glg. 1 Höhe (PH) der HPLC-Peaks von Paracetamol
und Salicylsäure im Ultrafiltrat bzw. in Proben vom Schritt 3) ohne
Einheiten eingesetzt werden: CF = PHF, und CT = PHT (siehe Glg.
2).
PB (%) = [ (PHT– PHF) / PHT ] × 100 Glg. 2
Literatur
[1] Pharmakologie und Toxikologie, Heinz Lüllmann, Klaus Mohr, Thieme
Verlag
[2] Pharmakokinetik, H. Derendorf, T. Gramatté, H.G. Schäfer,
Wissenschaftliche Verlagsgesellschaft mbH Stuttgart, 2002

Kursteil ‚Histamin-Bindungsassay’
Christina Hartwig, Kerstin Beste
Histamin und Histamin-Rezeptoren
Histamin oder auch 2-(4-Imidazolyl)ethylamin ist ein biogenes Amin. Die Synthese von
Histamin erfolgt durch Decarboxilierung von L-Histidin. Es wirkt als Neurotransmitter und
lokaler Mediator, wobei der Metabolismus durch Methylierung und Oxidation von statten
geht. Bekannt ist, dass Histamin vor allem von Zellen des Immunsystems wie Mastzellen,
basophilen Granulozyten, DCs und T-Zellen aber auch von Neuronen und enterochromaffinen
Zellen produziert wird. Histamin wird von 4 verschiedenen (bisher bekannten) Rezeptoren
erkannt. Diese sind G-Protein gekoppelt und kommen auf vielen verschiedenen Zellen vor.
Über den H1-R wirkt Histamin unter anderem im ZNS auf den Wachheitsstatus des
Organismus. Histamin ist aber über den H1-R ebenso an der Vasodilatation und Ödembildung
beteiligt. Über den H2-R wirkt Histamin dagegen vor allem auf die H+-Sekretion der
Belegzellen im Magen, bei den Kardiomyocyten ist es für die positive Inotropie
verantwortlich. Der H3-R ist relativ spezifisch für präsynaptische Neuronen und das Histamin
sorgt hier für eine Suppression der Ausschüttung von Neurotransmittern. Der jüngste im
Bunde ist der H4-R, er wurde erst vor wenigen Jahren entdeckt und spielt eine wichtige Rolle
bei Entzündungsreaktionen. Die genaue Funktion von Histamin im Zusammenhang mit dem
H4-R wird noch diskutiert. Es wurden jedoch bereits sehr stark wirksame Antagonisten
identifiziert, die möglicherweise als Entzündungshemmer eingesetzt werden können.
SF9-Zellen
Sf-9 ist eine immortalisierte Insekten-Zelllinie aus Ovar-Zellen von Spodoptera frugiperda,
einer Mottenart (Nachtfalter), die zur Familie der Eulenfalter (Noctuidae) und zur Ordnung
der Schmetterlinge (Lepidoptera) zählt. Sie enthalten keine eigenen Histamin-Rezeptoren
oder G-Proteine und eignen sich aus diesem Grund hervorragend, um beispielsweise humane
oder murine Histaminrezeptoren via Plasmidtransfer einzubringen.
Prinzip und Durchführung des [3H] Histamin-Kompetitionsbindungsassay am Histamin
H4-Rezeptor
Für den Histamin-Bindungsassay werden ausschließlich radioaktiv markiertes [3H] Histamin
und ein H4-Rezeptor-Antagonist (Thioperamid) verwendet. Das radioaktiv markierte [3H]
Histamin bindet an den H4-Rezeptor, der in den Membranen von SF9-Zellen exprimiert ist.
Thioperamid ist ein Antagonist (genauer gesagt, ein inverser Agonist) für den H4-Rezeptor
und verdrängt das [3H] Histamin aus dieser Bindestelle am Rezeptor.
Versuchsprotokoll:
Bis zur Zugabe von [3H ]Histamin wird auf Eis gearbeitet! • SF9-Membran: Die Membranproben werden bei -80°C gelagert. Eine Membranprobe
(1 Eppi) wird aufgetaut und zuerst 5x mit einer Insulinspritze mit 22G-Kanüle (dunkelgrau), dann 15 mal mit einer 27G-Kanüle (hell-grau) homogenisiert. Die erforderliche Konzentration von 500 µg Protein/ml (siehe Berechnungen) wird mit 1x Bindungspuffer eingestellt. Dann wird nochmals gut gevortext. Der H4-Rezeptor ist sehr thermolabil. Deshalb darf die Membran erst direkt vor der Verwendung aufgetaut werden, da der Rezeptor sonst denaturiert. Ist die Membran nicht gut genug homogenisiert und sind noch “Klümpchen” vorhanden, bekommt man artifizielle Resultate.
• Thioperamid: Eine 50 mM Stocklösung in H2O-bidest steht zur Verfügung. Sie wird soweit mit H2O-Bidest verdünnt, bis die Konzentration 10-fach höher ist als die finale Konzentration (siehe Berechnungen).
• [3H]Histamin: Kenndaten: 14,2 Ci/mmol; 1 mCi/ml. Wird mit Bindungspuffer auf 100 nM verdünnt (siehe Berechnungen).
Nr. SF9-Membran Bindungspuffer/0,2% (m/v) BSA [3H]Histamin Kompetitor (50 µl)
1-3 50 µg in 100 µl
300 µl
10 nM final in 50 µl
H2O bidest
4-6 Thioperamid 30 nM final
7-9 Thioperamid 100 nM final
10-12 Thioperamid 300 nM final
13-15 Thioperamid 1 µM final
16-18 Thioperamid 3 µM final
19-21 Thioperamid 10 µM final
22-24 H2O bidest

• Zunächst werden 300 µl des Bindungspuffer/BSA-Gemisches in die Probenröhrchen pipettiert.
• Dann werden 50 µl der entsprechenden Thioperamidkonzentationen oder H2O-Bidest zu den Ansätzen gegeben.
• Nun werden je 100 µl der Membran in die Probenröhrchen pipettiert. Wichtig ist, dass die Membranen gut suspendiert wurden!
• Ab hier wird im Radioisotopenlabor weitergearbeitet: Die Probenröhrchen werden in einen im Radioisotopenlabor vorhandenen Ständer transferiert und dann wird die radioaktives Histamin dazugegeben.
• Die Probenröhrchen werden dann auf dem Innova-Horizontalschüttler bei RT und 250 rpm 60min lang geschüttelt. In dieser Zeit bindet das mit 3H radioaktiv markierte Histamin an den H4-Rezeptor in der Membran und es stellt sich ein Gleichgewicht zwischen freiem und gebundenem [3H]Histamin sowie dem Kompetitor Thioperamid ein.
• Kurz vor Ende der Inkubationszeit wird der Brandell-Harvester vorbereitet: Der Harvester wird ohne Matte mit kaltem 1x Bindungspuffer gespült, dann wird die Harvestermatte aufgelegt und mit 0,3%igem (m/v) Polyethylenenimin beschichtet. Ohne Äquilibrierung würde auch das ungebundene [3H]Histamin an der Filtermatte hängenbleiben.
• Nun wird der Ständer mit den 24 Teströhrchen und mit 24 Leerröhrchen („Dummys“) bestückt und nach Anleitung filtriert.
• Die vom Harvester ausgestanzten Filterkreise werden mit einer Pinzette abgenommen und in Szintillationsröhrchen gegeben. In jedes Röhrchen werden mit der Dispensette 4 ml Szintillator Rotiszint Eco Plus gefüllt.
• Außerdem wird in ein Röhrchen eine Radioaktivitätsvorlage pipettiert, d. h. 50 µl der Radioaktivität, die auch in die Proben pipettiert wurden.
• Zusätzlich werden noch Wischproben von den benutzten Pipetten, dem Mixer, dem Ständer und dem Arbeitsplatz gemacht.
• Die Proben werden 1 Stunde stehen gelassen, so dass sich die Radioaktivität aus den Filterplättchen herauslösen kann, dann wird kräftig geschüttelt und anschließend im Betaszintillationscounter 1 Minute mit Programm 3 gemessen.
• Die Auswertung erfolgt mit dem Graph Pad Prism Programm (siehe Anleitung). 10x Bindungspuffer (5 l): Tris 750 mM 454g MgCl2x6H2O 125 mM 127,5g EDTA 10 mM 14,61g pH=7,4; Lagerung bei 4°C
3H Kompetitionsbindungsassay-Berechnungen
• SF9 Membran: H4 hH4R + Gαi2 + β1γ2 2090 µg/ml. Davon 717 µl + 2283 µl Bindungspuffer: 500 µg/ml.
• Thioperamid: 50mM. Verdünnung 1:50: 490µl bidest + 10µl Thioperamid: 1mM. Für 2 Gruppen: 1.100 µM Verdünnung für final 10 µM : 40 µl 1mM Thioperamid + 360 µl Bidest (1:10) 2.30 µM Verdünnung für final 3 µM : 10 µl 1 mM Thioperamid + 323 µl Bidest (1:33,3)

3.10 µM Verdünnung für final 1 µM : 40 µl 100 µM Thioperamid + 360 µl Bidest (1:10). 4.3 µM Verdünnung für final 300 nM : 10 µl100 µM Thioperamid +323 µl Bidest (1:33,3) 5.1 µM Verdünnung für final 100 nM : 40 µl 10 µM Thioperamid + 360 µl Bidest (1:10) 6.300 nM Verdünnung für final 30 nM : 10 µl 10 µM Thioperamid + 323 µl Bidest (1:33,3)
• 1xBindungspuffer/0,33% (m/v) BSA: Verdünnung 1:10 aus 10 x BB, davon 9,67 ml + 330 µl 10% (m/v) BSA: 0,2% (m/v) BSA final.
• [3H]Histamin: 14,2 Ci/mmol,1 mCi/ml. 14,2 Ci ≈ 1 mmol. 1 Ci ≈ 70,42 µmol. 1 mCi ≈ 70,42 nmol. 70,42 nmol/ml Lösung→70,42µmol/l Lösung→70,42 µM ist die Stocklösung. Gebraucht wird eine Arbeitslösung von 100 nM, final ist sie dann 10nM. Verdünnung1: Pro Gruppe: 704,2→2 µl 70,42µM [3H]Histamin Stocklösung + 1406 µl 1 x BB→100 nM. Stocklösung vorher gut vortexen!!!!
• Bilanzierung der Radioaktivität: Pro Gruppe: 2µl ≈74MBq≈2µCi.Ca. 60 MBq sind wäßriger Abfall (Kanister); ca. 10MBq sind organischer Abfall (Kanister); ca. 4 MBq sind fester Abfall (gelber Sack). Die Filterplättchen enthalten keine Radioaktivität mehr (durch den Szintillator herausgelöst).

Quantitative MS-Analytik kleiner Moleküle Prof. Dr. Volkahard Kaever, Tel.: 0511-532-2798, [email protected]
Dr. Heike Burhenne. Tel.: 0511-532-2887, [email protected]
Phosphodiesterasen: Inhibition der PDE4B mit Rolipram
Zyklische Nukleotide und Phosphodiesterasen Zylische Nukleotide (cNMPs) wie cAMP und cGMP sind als wichtige second messenger an
einer Vielzahl zellulärer Prozesse beteiligt. Hierzu zählen neben Zellwachstum,
Herzkontraktion und der Relaxation glatter Muskelzellen u.a. auch Entzündungsreaktionen.
Ein Anstieg der cAMP-Konzentration bewirkt eine Aktivierung der Protein-Kinase A. Die
daraus resultierende Phosphorylierung verschiedener Proteine führt schließlich zu einer
Hemmung inflammatorischer und immunmodulierender Zellen.
Die Wirkung zyklischer Nukleotide wird durch Phosphodiesterasen (PDEs) terminiert. Diese
Enzyme katalysieren die Hydrolyse von cNMPs wie cAMP und cGMP zu den
entsprechenden Monophosphaten AMP bzw. GMP, und
spielen damit eine wichtige Rolle in der Regulation der cNMP-
Konzentration in den Zellen. Derzeit sind 11 verschiedene
PDE-Isoformen bekannt, von denen in Säugern u.a. die PDE4
vornehmlich cAMP hydrolysiert.
Selektive Inhibitoren der PDE4 wie das Rolipram oder
Rofluminast werden derzeit in der Behandlung von Asthma
und der chronisch obstruktiven Lungenerkrankung (COPD)
eingesetzt bzw. befinden sich in der Erprobung. Ziel des
Praktikumsversuches ist es, die Inhibition der PDE4 Rolipram

exemplarisch mit Rolipram zu untersuchen.

Bestimmung des gebildeten AMP Die Messung des Edukts cAMP sowie des Produkts AMP erfolgt in diesem Versuch mittels
Flüssigchromatographie-gekoppelter Massenspektrometrie (LC-MS). Um zufällige Fehler
seitens des Analysegerätes ausgleichen zu können, werden Proben und Kalibrierreihe mit
einem internen Standard (Tenofovir) versetzt. Sowohl AMP bzw. cAMP als auch der interne
Standard werden im Zuge der Chromatographie auf einer mit porösem Kohlenstoff gefüllten
Säule (PGC) von den anderen Bestandteilen der Lösung abgetrennt und in einem Single-
Massenspektrometer analysiert. Hier erfolgt zunächst eine Ionisierung der Substanzen in der
Ionenquelle des Massenspektrometers. Anschließend werden die gebildeten Ionen anhand
ihres Masse/Ladungsverhältnisses (m/z) selektiert und erreichen schließlich den
Ionendetektor. Durch Bestimmung der Anzahl der ankommenden Ionen, kann mit Hilfe der
Kalibrierreihe auf die gebildete AMP-Menge (bzw. cAMP) geschlossen werden.
Versuchsdurchführung: Vorhandene Lösungen: Reaktionspuffer (100 mM Tris/HCL, 16,6 mM MgCl2, 3,4 mM EDTA) cAMP-Stock 1 mM AMP-Stock 1 mM Rolipram-Stock 10 mM Interner Standard (Tenofovir) 100 µM PDE4B 11,25 µg/ml Millipore-Wasser Eluent A 10 mM Ammoniumacetat; pH 10 (eingestellt mit NH3) Erstellung einer Kalibrierreihe:
Zum Erstellen einer Kalibrierreihe erhalten Sie eine 1 mM-Stocklösung AMP und cAMP.
Erstellen Sie daraus eine Kalibrierreihe mit den im Schema aufgeführten Konzentrationen.
Dabei wird zunächst ein Mix aus den beiden Stocklösungen hergestellt. Aus diesem Mix wird
der erste Kalibrierpunkt hergestellt. Jede weitere Verdünnung wird anschließend aus der
vorangegangenen erstellt. Verwenden Sie zum Verdünnen eine 1:2-Verdünnung des
Reaktionspuffers.

Assay
Ziel dieses Versuchs ist es, eine Inhibitionskurve zu erstellen. Dabei wird durch Zugabe
steigender Inhibitorkonzentrationen eine Hemmung der Phosphodiesteraseaktivität
herbeigeführt. Aus der daraus resultierenden Inhibitionskurve lässt sich der IC50-Wert des
Inhibitors für eine bestimmte Phosphodiesterase ermitteln.
Der hier untersuchte Inhibitor ist das Rolipram. Von diesem sollen fünf unterschiedliche
Konzentrationen untersucht werden: 0 µM, 0,1 µM, 1 µM, 10 µM und 100 µM.
Rolipram liegt Ihnen als 10 mM-Stocklösung (gelöst in DMSO) vor. Stellen Sie aus dieser
zunächst die fünf Konzentrationen her. Es wird, wie bei der Kalibrierreihe, jede Rolipram-
Konzentration durch Verdünnen der vorherigen hergestellt. Beachten Sie dabei, dass für den
Assays das Rolipram um den Faktor 10 verdünnt wird! Verwenden Sie zum Verdünnen
Millipore-Wasser.
Um einen störenden Einfluss des DMSO auf die Reaktion auszuschließen, setzen Sie
zusätzlich eine sog. DMSO-Kontrolle an. Stellen Sie dafür eine 1:10-Verdünnung des DMSO
mit Millipore-Wasser her.

Die zu untersuchende Phosphodieesterase ist die PDE4B. Die Konzentration der
Stocklösung beträgt 11,25 µg/ml. Stellen Sie hieraus eine 1:100 Verdünnung her. Sie
benötigen für den Assay ca. 200 µl. Verwenden Sie zum Verdünnen Millipore-Wasser.
Ein Substrat der PDE4B ist das cAMP. Dieses liegt Ihnen in einer Stocklösung von 1 mM
vor. Stellen Sie hieraus 200 µL einer 100 µM-Lösung. Verwenden Sie zum Verdünnen
Millipore-Wasser.
Für die Auswertung der HPLC-MS -Analyse wird ein interner Standard (Tenofovir) benötigt.
Tenofovir liegt Ihnen in einer 100 µM-Lösung vor. Stellen Sie hiervon mindestens 1600 µL
einer 200 nM-Lösung her. Verwenden Sie zum Verdünnen Eluent A.
Pipettierschema:
Erstellen Sie zu jeder Inhibitorkonzentration 3fach-Werte. Beschriften Sie sowohl 1,5 mL-
Eppendorf-Tubes als auch Analysegefäße mit den jeweiligen Inhibitorkonzentrationen.
Beschriften Sie zusätzlich Analysegefäße mit den Kalibrierpunkten E1-E6. Arbeiten Sie
anschließend nach folgendem Fließschema:
finale Rolipram-Konzentration
anzusetzende Rolipram-Konzentration
VRolipram-Stock Vvorangegangene
Lösung
VWasser VLsg. gesamt
100 µM - - 100 µL 10 µM - 100 µL 1 µM - 100 µL 0,1 µM - 100 µL 0 µM (Milliporewasser) - - 100 µL
0 µM (1:10 DMSO`) - - - 100 µL
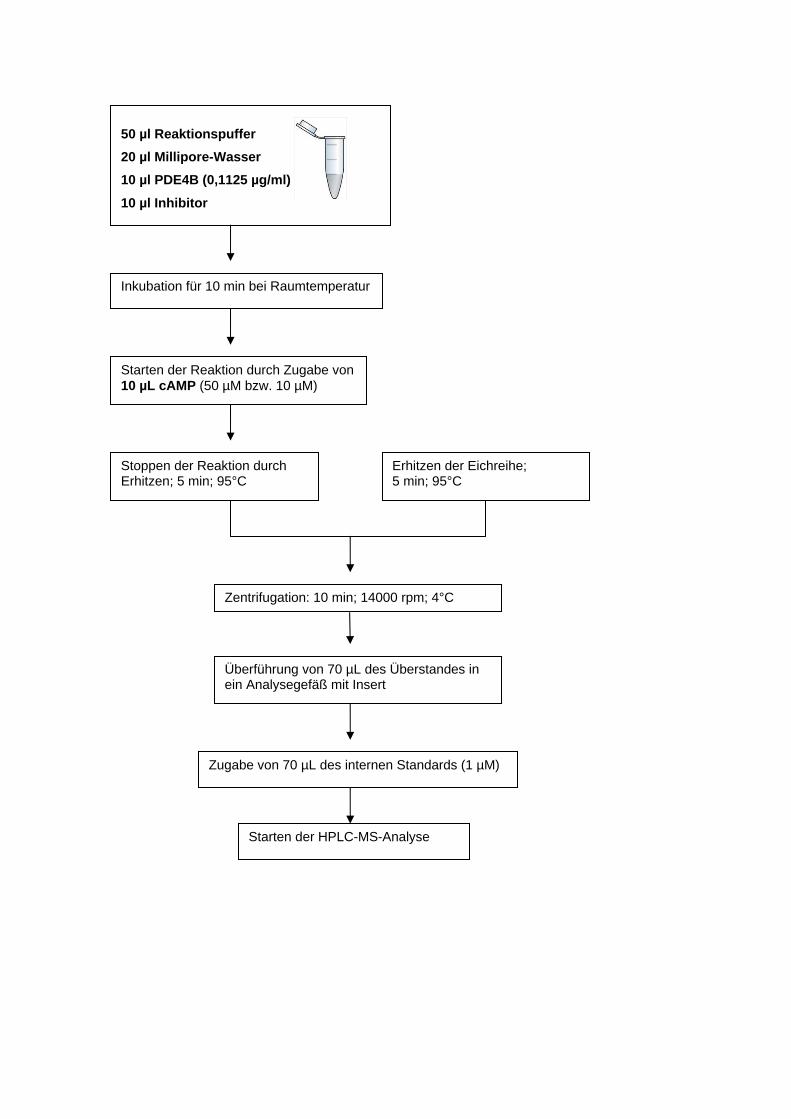
50 µl Reaktionspuffer 20 µl Millipore-Wasser 10 µl PDE4B (0,1125 µg/ml)
10 µl Inhibitor
Inkubation für 10 min bei Raumtemperatur
Starten der Reaktion durch Zugabe von 10 µL cAMP (50 µM bzw. 10 µM)
Stoppen der Reaktion durch Erhitzen; 5 min; 95°C
Erhitzen der Eichreihe; 5 min; 95°C
Zentrifugation: 10 min; 14000 rpm; 4°C
Überführung von 70 µL des Überstandes in ein Analysegefäß mit Insert
Zugabe von 70 µL des internen Standards (1 µM)
Starten der HPLC-MS-Analyse

Auswertung
Die Auswertung erfolgt über die Peakflächen der erhaltenen MS-Signale. Eine Integration
der Signale erfolgt in der Regel automatisch durch die Gerätesoftware, muss jedoch in
einigen Fällen manuell durchgeführt werden.
Um Messungenauigkeiten des Gerätes bei der Auswertung berücksichtigen zu können,
haben Sie Tenefovir als internen Standard mit vermessen. Auch seine Peakflächen werden
bestimmt.
Erstellen Sie in Excel eine Kalibriergerade, indem Sie das Verhältnis der Analytpeakfläche
(AMP bzw. cAMP) zu der Peakfläche des internen Standards in Abhängigkeit der
Analytkonzentration darstellen. Lassen Sie sich die Ausgleichsgerade, den Korrelationsfaktor
r2 sowie die dazugehörige Geradengleichung darstellen.
Die Auswertung der Proben erfolgt nun ebenfalls über das Peakflächenverhältnis von Analyt
zu internem Standard mittels der oben erhaltenen Geradengleichung. Bilden Sie aus den
3fach-Werten Mittelwerte, stellen Sie diese in Abhängigkeit von der Rolipram-Konzentration
dar und schätzen Sie den IC-50-Wert ab. Geben Sie diesen Wert in Ihrem Protokoll an.
Des Weiteren sollten Sie folgende Fragen beantworten:
1. Welche anderen analytischen Methoden hätten Sie zur Bestimmung der PDE-
Aktivität verwenden können?
2. Welche Vor- bzw. Nachteile bietet die LC-MS-Technik?
3. Welche spezifischen PDE-Inhibitoren kennen Sie?

Versuch 8:
Produktion reaktiver Sauerstoffspezies durch neutrophile Granulozyten – Hemmung durch β2-adrenerge Rezeptorliganden
Dr. Sabine Wolter. 0511-532-2791, [email protected]
Marina Golombek
Hintergrund
Bei der Pathophysiologie des Asthma Bronchiale spielen unterschiedliche Zelltypen eine Rolle. Dabei sind die Beiträge von Mastzellen, Lymphozyten und eosinophilen Granulozyten gut untersucht. Neben diesen Zellen des Immunsystems sind aber auch neutrophile Granulozyten (Neutrophile), insbesondere bei der Entstehung von schwereren Asthmaanfällen, involviert.
Ein Mediator den aktivierte Neutrophile produzieren ist das O2.—Molekül, eine
reaktive Sauerstoffspezies (ROS). Diese sind nützlich in der Abwehr von fungialen und bakteriellen Infektionen, können aber im Falle der aus dem Gleichgewicht geratenen Immunantwort beim Asthma zur Verschlimmerung des Krankheitsbildes führen.
Abb. 1: Signalschema zur Aktivierung der ROS Produktion in neutrophilen Granulozyten: Das Peptid fMLF (N-Formyl-L-methionyl-L-leucyl-L–phenylalanin) aktiviert über den Formylpeptid Rezeptor (FPR) und einen Giα-gekoppelten Mechanismus die NOX Oxidase, welche die Reduktion von Sauerstoff zur reaktiven Sauerstoffspezies (ROS) O2
.- katalysiert. Wird der β2-adrenerge Rezeptor (β2AR) aktiviert, so führt dies zu einem Anstieg des intrazellulären cAMP und darüber zu einer Hemmung der Oxidase. PIP2: Phosphatidylinositol-4,5-bisphosphat, PLC: Phospholipase C, DAG: Diacylglycerin, PKC: Protein-kinase C, AC 9: Adenylylzyklase 9
Zur Therapie von Asthma bronchiale werden Liganden am β2-adrenergen Rezeptor eingesetzt. Kurzfristig führt dies zur Dilatation der Gefäßmuskulatur und erleichtert das Atmen bei akuten Asthmaanfällen („Reliever“). Gleichzeitig können aber auch über vorhandene β2-adrenerge Rezeptoren inflammatorische

Prozesse der Immunzellen gehemmt werden. So beispielsweise die ROS-Produktion von neutrophilen Granulozyten (siehe Abb. 1).
Literatur
Monteseirín,J.Neutrophils and asthmaJ Investig Allergol Clin Immunol, 2009, 19, 340-354
Barnes, P. J. New molecular targets for the treatment of neutrophilic diseasesJ Allergy Clin Immunol, 2007, 119, 1055-1062
Methode
Isolation neutrophiler Granulozyten aus humanem Vollblut
• Für den Versuch werden 25 ml Vollblut benötigt, die mit PBS 1 + 1 verdünnt werden.
Freiwillige Spender aus der Gruppe der Praktikanten! • Das Blut wird vorsichtig in ein 50 ml Röhrchen pipettiert, in das 25 mL
Ficoll vorgelegt wurden. Dazu den Auslauf der Pipetierhilfe langsam stellen und das Röhrchen fast waagerecht halten. Wichtig ist, dass sich Blut und Ficoll nicht durchmischen, sondern eine saubere Trennlinie entsteht. • 20’ bei RT mit 800 g ohne Bremse zentrifugieren. (Sorvall RT 6000 = 2000rpm/Stufe 3,5) • Mit einer Pipette wird das Plasma abgenommen, die milchige
Leukozytenbande in der Interphase entfernt, dann das Ficoll bis knapp vor das Erythrozytensediment abgesaugt.
Es dürfen keine Leukozyten in die untere Granulo- bzw. Erythrozytenschicht gelangen.
Abb.2: Zellen nach Zentrifugation von Vollblut auf einem Ficoll-Gradienten

• Zu der Granulo- und Erythrozytenschicht werden 36 mL bidest H2O gegeben und sanft 1’ zur Lyse der Erythrozyten geschwenkt. Dann 4 mL 10x PBS zur Äquilibrierung dazugegeben.
Die Lyse darf nicht zu lange dauern, denn es sollen nur die Erythrozyten entfernt, nicht auch die Granulozyten geschädigt werden. • Die Lysate werden 10’ mit 400g ( 1400 rpm/Stufe 3) zentrifugiert. RT mit Bremse • Der Überstand wird verworfen und zum Pellet werden nochmals 18 mL
bidest H2O gegeben, um die restlichen Erythrozyten zu zerstören. 30’’ schwenken, dann 2 mL 10x PBS dazugegeben. • Nochmal zentrifugieren wie oben beschrieben, anschließend • die Granulozyten in 25 ml PBS waschen, 10’ 600 g = 1600 rpm
• Überstand gründlich entfernen. Reste des Puffers können mit einer blauen Eppendorfspitze abgenommen werden, und dabei kann auch noch vorhandenes Hämoglobin, das über dem weißen Zellpellet steht, abgenommen werden. • Die Zellen werden in 2 mL PBS aufgenommen und gezählt; dazu werden 5 µL der Zellen 1:10 vorverdünnt, und 50 µl Trypanblau-Gebrauchslsg. zugegeben.
Zellzählung siehe Beiblatt • Dann die Zellen auf die benötigte Zellzahl von 1x106 Zellen/mL einstellen
und bei 4°C auf Eis lagern. Ficoll: Biocoll Separating Solution von Biochrom Dichte 1,077 g/mL, Cat. No. L6115
PBS (10x): Dulbecco´s PBS without Ca&Mg PAA Laboratories GmbH, Cat.No. H15-011
Messung der ROS-Produktion von Neutrophilen
• Der Assay wird in einer 96 well-Platte mit 3 x Werten in einem
Gesamtvolumen von 200 µL durchgeführt. • Gemessen wird am Biotek Plattenleser Synergy 4 bei 37°C (Aufheizphase beachten). • Konzentrationsreihen der β2AR Agonisten ansetzten, jeweils das 20x der
finalen Konzentration. Pro Wert werden 3x10 µL Ligandenlösung benötigt (+ Volumen für die nächste Verdünnungsstufe)

(S)-Salbutamol 100 µl 10 mM Stammlsg. in H2O 12 µl 10 mM = 600 µM final 10 µl 6 mM = 300 µM 10 µl 2 mM = 100 µM 10 µl 200 µM = 10 µM 10 µl 20 µM = 1 µM 10 µl 2 µM = 100 nM

(R)-Salbutamol 50 µl 10 mM Stammlsg. in H2O 10 µl 6 mM = 300 µM final 10 µl 2 mM = 100 µM 10 µl 200 µM = 10 µM 10 µl 20 µM = 1 µM 10 µl 2 µM = 100 nM 10 µl 200 nM = 10 nM (R)-Isoproterenol 5 µl 10 mM Stammlsg. in H2O 10 µl 20 µM = 1 µM final 10 µl 2 µM = 100 nM 10 µl 200 nM = 10 nM 10 µl 20 nM = 1 nM 10 µl 2 nM = 100 pM 10 µl 200 pM = 10 pM Ligandenlösung lt. Pipetierplan in die Platte vorlegen. (Konzentration ansteigend!) Außerdem in Reihe A 1 – 9 je 20 µl PBS vorlegen = unstimulierte Kontrollen Reihe B 1 - 9 je 10 µl PBS vorlegen = stimulierte Kontrollen (fMLP) fMLP
20 µl fMLP Stammlg. 1 mM in Dimethylformamid, daraus entsprechende Menge 20 µM Lösung herstellen
Mastermix ansetzen, es werden 80 µl/200 µl eingesetzt = 2,5 x der finalen Konzentration Erst zuletzt unmittelbar vor Gebrauch frisch ansetzen. Benötigt werden 7 ml Lösung (72 Proben + Sicherheitsvolumen) 5,3 µl Cytochalasin B 1 mg/ml in DMSO (sehr giftig) = 0,75 µg/ml ( 1 : 1333) = 0,3 µg/ml final im Assay 875 µl CaCl2 20 mM in H2O = 2,5 mM (1 : 8) = 1 mM final 875 µl Cytochrom C 2 mM in PBS = 250 µM (1 : 8) = 100 µM final ad 7 ml mit PBS auffüllen Jeweils 80 µl zügig in die Platte geben.
Anschließend 100 µl der gezählten Zellen dazugeben (Multipette) = (1x105 Zellen/well final) Dann am vorgeheizten Platten-Reader: 3’ Vorinkubation (Programm Marina Praktikum) bei 37°C
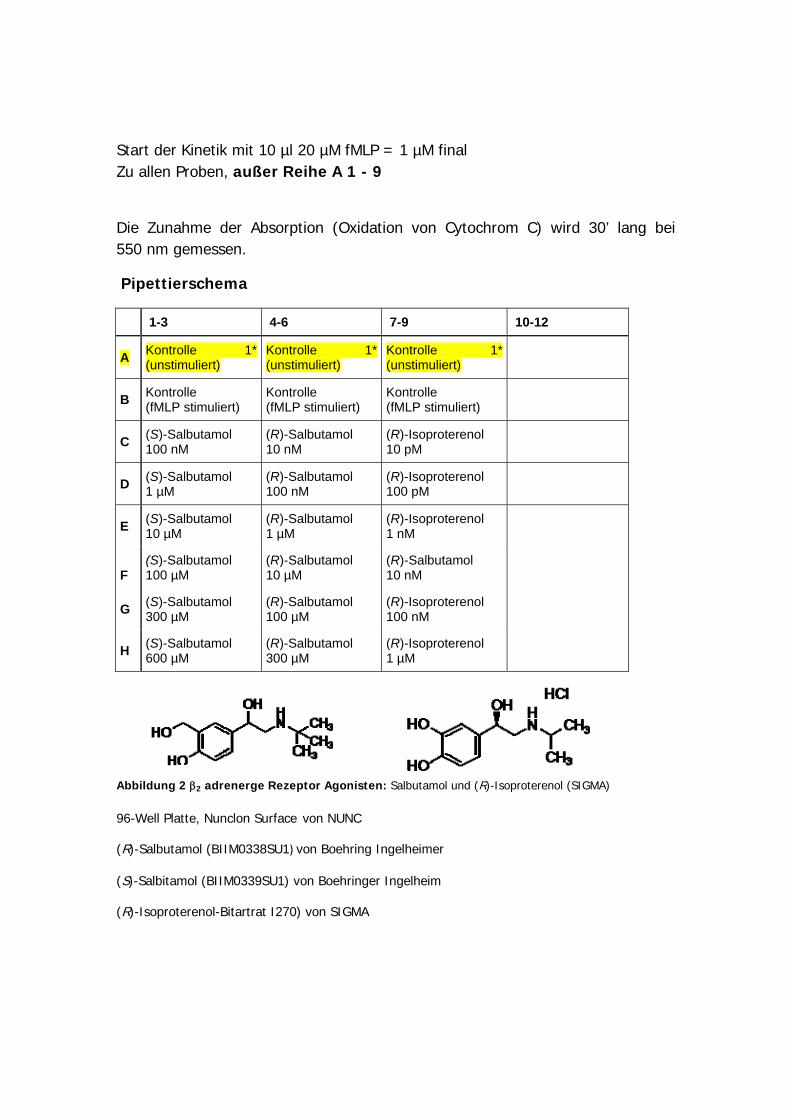
Start der Kinetik mit 10 µl 20 µM fMLP = 1 µM final Zu allen Proben, außer Reihe A 1 - 9
Die Zunahme der Absorption (Oxidation von Cytochrom C) wird 30’ lang bei 550 nm gemessen.
Pipettierschema
1-3 4-6 7-9 10-12
A Kontrolle 1* (unstimuliert)
Kontrolle 1* (unstimuliert)
Kontrolle 1* (unstimuliert)
B Kontrolle (fMLP stimuliert)
Kontrolle (fMLP stimuliert)
Kontrolle (fMLP stimuliert)
C (S)-Salbutamol 100 nM
(R)-Salbutamol 10 nM
(R)-Isoproterenol 10 pM
D (S)-Salbutamol 1 µM
(R)-Salbutamol 100 nM
(R)-Isoproterenol 100 pM
E (S)-Salbutamol 10 µM
(R)-Salbutamol 1 µM
(R)-Isoproterenol 1 nM
F
(S)-Salbutamol 100 µM
(R)-Salbutamol 10 µM
(R)-Salbutamol 10 nM
G (S)-Salbutamol 300 µM
(R)-Salbutamol 100 µM
(R)-Isoproterenol 100 nM
H (S)-Salbutamol 600 µM
(R)-Salbutamol 300 µM
(R)-Isoproterenol 1 µM
96-Well Platte, Nunclon Surface von NUNC
(R)-Salbutamol (BIIM0338SU1) von Boehring Ingelheimer
(S)-Salbitamol (BIIM0339SU1) von Boehringer Ingelheim
(R)-Isoproterenol-Bitartrat I270) von SIGMA
Abbildung 2 β2 adrenerge Rezeptor Agonisten: Salbutamol und (R)-Isoproterenol (SIGMA)

Auswertung
In GraphPad Prism kann mit den Daten der kompletten Kinetik gerechnet werden (inkl. Fehlerrechnung). Da letztlich die Werte zu Beginn (t=0) und des Plateaus (t=30) die gesamte Produktion von ROS widerspiegeln, kann ein Teil der Auswertung mit einem Tabellenkalkulationsprogramm durchgeführt werden.
Eine Quantifizierung der entstandenen ROS im 96-Well Format ist schwierig und findet nicht statt. Es wird normalisiert:
In GraphPad PRISM oder einem anderen Programm mit Kurvenanpassung (R, etc.) werden die normalisierten Werte den jeweiligen log(cLigand) zugeordnet und als Graph dargestellt. Über nicht-lineare Regression erhält man die Dosis-Wirkungskurven.
Ansprechpartner
Marina Golombek, Technische Assistentin, [email protected]
Michael Reinartz, cand. rer. nat, [email protected]
Dr. rer. nat. Sabine Wolter, wiss. Mitarbeiterin, [email protected]
Δ<A550,xn>= [<A550,xn>(t=30) - <A550,Kontrolle 1>(t=30)] - [<A550,xn>(t=0) - <A550,Kontrolle 1>(t=0)]
Formel 1 Berechnung von ΔA550: Nach Abzug des Hintergrundes werden die Differenzen der Absorptionen bei 550 nM zu den Zeitpunkten t=0 und t=30 gebildet.
Inhibition der ROS Produktion / % von 1 µM (R)-Iso = 100% -(Δ<A550,xn>-Δ<A550,H7-9>)/(Δ<A550,B1-9>-Δ<A550,H7-9>)*100%
Formel 2 Normalisierung auf (R)-Isoproterenol: Nach der Normalisierung auf den ΔA550-Wert von 1 µM (R)-Isoproterenol werden die Werte in Prozente der maximalen Inhibition transformiert.


















