
Quantenchemische Charateresierung von Cu(I)-Komplexen ... · Durch strahlungslose...
79
Bachelorarbeit " Quantenchemische Charakterisierung von Cu(I)-Komplexen ge- mischter P,N-Liganden auf [2.2]Paracyclophanbasis: Variation der Diarylphosphineinheit" "Quantum Chemical Characterization of Cu(I) Complexes with [2,2]Paracyclophane-Based P,N-Chelating Ligands: Variation of the Diarylphosphine Unit" Zur Erlangung des akademischen Grades Bachelor of Science (B.Sc.) im Studienfach Wirtschaftschemie Institut für Theoretische Chemie und Computerchemie der Heinrich-Heine-Universität Düsseldorf vorgelegt von Sandra Gladt Matrikelnummer: 2098832 Erstgutachterin: Frau Prof. Dr. Christel M. Marian Zweitgutachter: Herr Prof. Dr. Christian Ganter Düsseldorf Dienstag, 6. März 2018
Transcript of Quantenchemische Charateresierung von Cu(I)-Komplexen ... · Durch strahlungslose...

Bachelorarbeit
" Quantenchemische Charakterisierung von Cu(I)-Komplexen ge-
mischter P,N-Liganden auf [2.2]Paracyclophanbasis: Variation der
Diarylphosphineinheit"
"Quantum Chemical Characterization of Cu(I) Complexes with
[2,2]Paracyclophane-Based P,N-Chelating Ligands: Variation of
the Diarylphosphine Unit"
Zur Erlangung des akademischen Grades
Bachelor of Science (B.Sc.)
im Studienfach Wirtschaftschemie
Institut für Theoretische Chemie und Computerchemie
der Heinrich-Heine-Universität
Düsseldorf
vorgelegt von
Sandra Gladt
Matrikelnummer: 2098832
Erstgutachterin: Frau Prof. Dr. Christel M. Marian
Zweitgutachter: Herr Prof. Dr. Christian Ganter
Düsseldorf Dienstag, 6. März 2018


Abkürzungsverzeichnis I
Inhalt
Abkürzungsverzeichnis II
Tabellenverzeichnis III
Abbildungsverzeichnis V
1 Einleitung 1
2 Theorie 3
2.1 Photophysikalische Prozesse 3 2.2 TADF 4 2.3 Franck-Condon-Prinzip 4
2.4 Fluoreszenzratenkonstante 6 2.5 DFT 6
2.5.1 Hohenberg- Kohn-Theorem 7 2.5.2 Kohn-Sham-Formalismus 8
2.6 Hybrid-Funktionale 9
2.6.1 PBE0 9 2.6.2 BH-LYP 9
2.7 Basissätze 10 2.8 DFT/MRCI 10
2.9 ECP 11 3 Methodik 13
4 Ergebnisse und Auswertung 14
4.1 Rp-[κ2-N,P-4-(2′-Pyridyl)-1,2-diphenylphosphinyl[2.2]paracyclophan]chlorokupfer(I) 14
4.1.1 S0 Geometrie 14 4.1.2 DFT/MRCI 17
4.1.2.1 DFT/MRCI tight 17 4.1.2.2 Auswertung DFT/MRCI 20
4.2 Fluor-Substitution 25
4.2.1 Geometrie 25 4.2.2 DFT/MRCI 26
4.3 Methyl-Substitution 28 4.3.1 Geometrie 29
4.3.2 DFT/MRCI 30
4.4 T-Butyl-Substitution 33 4.4.1 Geometrie 33 4.4.2 DFT/MRCI 34
4.5 Stickstoff-Austausch 36 4.5.1 Geometrie 37 4.5.2 DFT/MRCI 39
4.6 Vergleich 42 5 Fazit 46
Literaturverzeichnis 48
Anhang 50
Eidesstattliche Erklärung 72
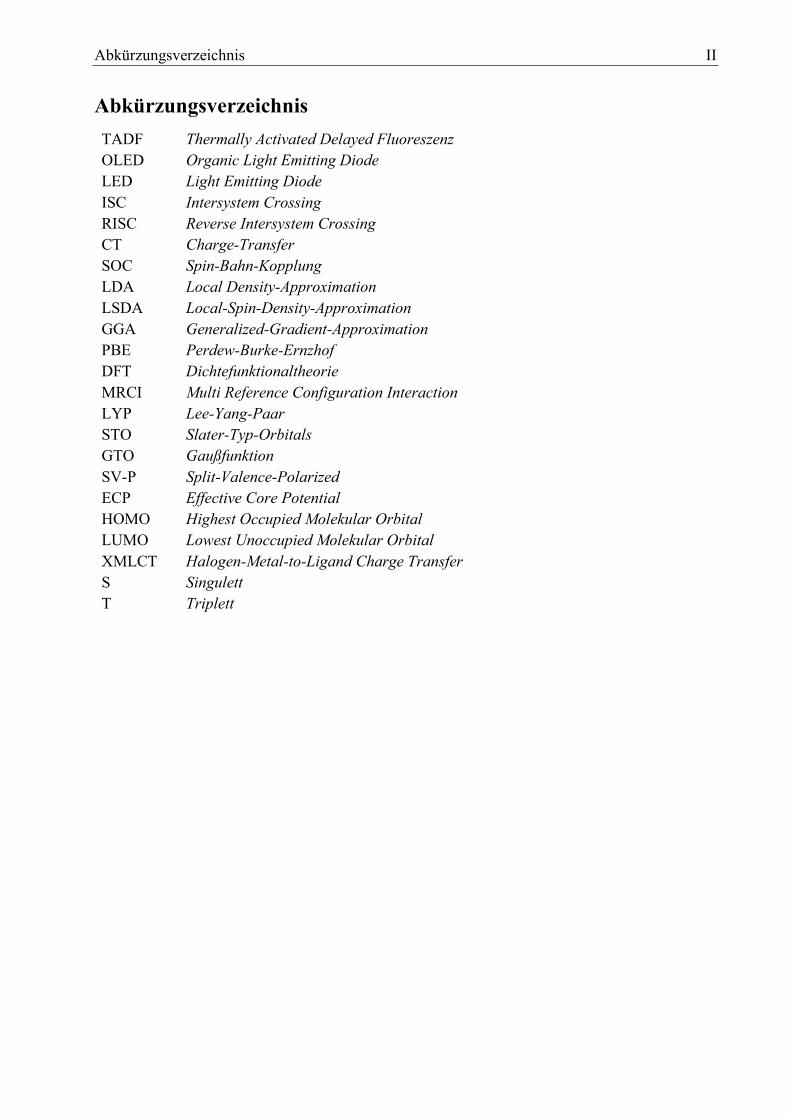
Abkürzungsverzeichnis II
Abkürzungsverzeichnis
TADF Thermally Activated Delayed Fluoreszenz
OLED Organic Light Emitting Diode
LED Light Emitting Diode
ISC Intersystem Crossing
RISC Reverse Intersystem Crossing
CT Charge-Transfer
SOC Spin-Bahn-Kopplung
LDA Local Density-Approximation
LSDA Local-Spin-Density-Approximation
GGA Generalized-Gradient-Approximation
PBE Perdew-Burke-Ernzhof
DFT Dichtefunktionaltheorie
MRCI Multi Reference Configuration Interaction
LYP Lee-Yang-Paar
STO Slater-Typ-Orbitals
GTO Gaußfunktion
SV-P Split-Valence-Polarized
ECP Effective Core Potential
HOMO Highest Occupied Molekular Orbital
LUMO Lowest Unoccupied Molekular Orbital
XMLCT Halogen-Metal-to-Ligand Charge Transfer
S Singulett
T Triplett

Tabellenverzeichnis III
Tabellenverzeichnis
Tabelle 1: Bindungslängen Vergleich mit Literaturwerten 15
Tabelle 2: Bindungswinkel Vergleich mit Literaturwerten 15
Tabelle 3: Bindungslängen des Moleküls 1 15
Tabelle 4: Bindungswinkel des Moleküls 1 16
Tabelle 5: Diederwinkel des Moleküls 1 16
Tabelle 6: relative Zustandsenergien im Vergleich MRCI -MRCI-tight 17
Tabelle 7: Vergleich Standard- und tight-Parameter: Zusammensetzung der Zustände 18
Tabelle 8: Standard- und tight-Parameter: Vergleich Oszillatorstärken 19
Tabelle 9: Anregungen Molekül 1 20
Tabelle 10: Molekül 1: Art der Übergänge 21
Tabelle 11:Molekül 1: Dipolmomente 22
Tabelle 11: Bindungslängen Molekül 2 25
Tabelle 12: Bindungswinkel Molekül 2 25
Tabelle 13: Diederwinkel Molekül 2 26
Tabelle 14: Charakterisierung der Zustände des Moleküls 2 26
Tabelle 15: Dipolmomente Molekül 2 27
Tabelle 16: Bindungslängen Molekül 3 29
Tabelle 17: Bindungswinkel Molekül 3 29
Tabelle 18: Diederwinkel des Moleküls 3 30
Tabelle 19: Charakterisierung der Zustände des Moleküls 3 30
Tabelle 20: Dipolmomente Molekül 3 32
Tabelle 21: Bindungslängen Molekül 4 33
Tabelle 22: Bindungswinkel Molekül 4 33
Tabelle 23: Diederwinkel Molekül 4 34
Tabelle 24: Charakterisierung der Zustände des Moleküls 4 34
Tabelle 25: Dipolmomente Molekül 4 35
Tabelle 27: Bindungslängen an der S0-Geometrie im Vergleich Molekül 1 und Molekül 5 37
Tabelle 28:Bindungslängen an der T1-Geometrie im Vergleich Molekül 1 und Molekül 5 37
Tabelle 29: Bindungswinkel an der S0-Geometrie im Vergleich Molekül 1 und Molekül 5 38
Tabelle 30:Bindungswinkel an der T1-Geometrie im Vergleich Molekül 1 und Molekül 5 38
Tabelle 31: Diederwinkel an der S0-Geometrie im Vergleich Molekül1 und Molekül 5 38
Tabelle 31: Diederwinkel an der T1-Geometrie im Vergleich Molekül 1 und Molekül 5 38

Tabellenverzeichnis IV
Tabelle 31: Bindungslängen Molekül 5 39
Tabelle 32: Bindungswinkel Molekül 5 39
Tabelle 33: Diederwinkel Molekül 5 39
Tabelle 34: Zustände des Moleküls 5 40
Tabelle 37: Dipolmomente Molekül 5 41
Tabelle 38: Energielücke HOMO-LUMO im Vergleich 42
Tabelle 39: Oszillatorstärke S1 im Vergleich 43
Tabelle 40: Vertikale Anregungsenergien im Vergleich 43
Tabelle 41: adiabatische Energien und Energielücke S1-T1 im Vergleich 44
Tabelle 42: Emissionswellenlängen im Vergleich 44
Tabelle 43: Fluoreszenzratenkonstante im Vergleich 44
Tabelle 30: Dipolmomente im Vergleich 45

Abbildungsverzeichnis V
Abbildungsverzeichnis
Abbildungen
Abbildung 1: Jablonski Diagramm der möglichen Übergänge: 3
Abbildung 2: Struktur Molekül 1 S0-Geometrie 14
Abbildung 3: Struktur Molekül 1 T1-Geometrie 14
Abbildung 4: Kolorierung und Nummerierung des Moleküls 1 15
Abbildung 5:HOMO des Moleküls 1 16
Abbildung 6: LUMO des Moleküls 1 16
Abbildung 7: Differenzdichte zw. S1 und S0 an der S0 Geometrie des Moleküls 1 21
Abbildung 8: Differenzdichte zw. T1 und S0 an der S0 Geometrie des Moleküls 1 21
Abbildung 9: Differenzdichte zw. T2 und S0 an der S0 Geometrie des Moleküls 1 21
Abbildung 10: Differenzdichte zw. S1 und S0 an der T1 Geometrie des Moleküls 1 21
Abbildung 11: Differenzdichte zw. T1 und S0 an der S0 Geometrie des Moleküls 1 21
Abbildung 12: Differenzdichte zw. T2 und S0 an der S0 Geometrie des Moleküls 1 21
Abbildung 13: Molekül 1: Dipolmomente 23
Abbildung 14: Struktur S0-Geometrie des Moleküls 2 25
Abbildung 15: Dipolmomente Molekül 2 27
Abbildung 16: Molekül 3 29
Abbildung 17: Dipolmomente Molekül 3 32
Abbildung 18: Struktur des Moleküls 4 an der S0-Geometrie 33
Abbildung 19: Dipolmomente Molekül 4 35
Abbildung 20: Aufbau Molekül 5 37
Abbildung 21: HOMO des Moleküls 5 39
Abbildung 22: LUMO des Moleküls 5 39
Abbildung 21: Dipolmomente Molekül 5 41
Diagramme
Diagramm 1: Vergleich der Zustandsenergien an der S0- und der T1-Geometrie 24
Diagramm 2: energetische Lage der Zustände des Moleküls 2 28
Diagramm 3: energetische Lage der Zustände des Moleküls 3 32
Diagramm 4: energetische Lage der Zustände des Moleküls 4 36
Diagramm 5: energetischer Lage der Zustände des Moleküls 5 42

Einleitung 1
1 Einleitung
Im Rahmen dieser Bachelorarbeit wird das Molekül Rp-[κ2-N,P-4-(2′-Pyridyl)-1,2-diphenylphos-
phinyl[2.2]paracyclophan]chlorokupfer(I) quantenchemisch untersucht. Das Enantiomer dieses
Moleküls wurde in der Arbeitsgruppe von Prof. Dr. Stefan Bräse am Karlsruher Institut für Tech-
nologie im Rahmen der Doktorarbeit von Carolin Braun synthetisiert und chemisch untersucht.
Dieser Art von Molekülen werden gute Katalysator-Eigenschaften, sowie die Fähigkeit zur ther-
misch aktivierte verzögerten Fluoreszenz (TADF) nachgesagt. Die Verwendbarkeit dieses Mole-
küls als OLED-Emitter (organic light emitting diode) ist im Wesentlichen davon abhängig, ob und
gegeben falls in welcher Größenordnung es in der Lage ist, TADF zu emittieren. Die TADF-Ei-
genschaften dieses Moleküls werden in dieser Bachelorarbeit genauer betrachtet und diskutiert.
OLEDs sind aktueller Forschungsbestandteil und werden für Handydisplays, Fernseher, Tablets
oder auch für Raumbeleuchtungen verwendet. Sie können wesentlich dünner aufgetragen werden
und sind zusätzlich stromsparender im Betrieb, verglichen mit den aktuell verwendeten LEDs.[1]
OLEDs lassen sich in drei Generationen unterteilen. Die erste Generation enthält stark fluoreszie-
rende Emitter, welche eine langsame ISC-Rate (intersystem crossing rate) aufweisen und eine sehr
geringe Phosphoreszenzratenkonstante besitzen. Aufgrund der Spinstatistik werden exitonische
Triplett-Zustände und Singulett-Zustände in einem Verhältnis 1:3 gebildet. Der Übergang S1-S0
ist ein spinerlaubter Übergang, wohingegen der T1-S0 ein spinverbotener Übergang ist. Bei dieser
Generation von OLEDs können also nur 25% der Exitonen geerntet werden, die restlichen 75%
relaxieren strahlungslos in den Grundzustand zurück[2]. Um mehr Quanteneffizienz zu erreichen,
wurde nach der Möglichkeit geforscht, auch die Triplett-Exitonen ernten zu können.
Zu den Emittern der OLEDs der zweiten Generation gehören metallorganische Komplexe, welche
5d-Metalle wie z.B. Platin oder Iridium enthalten. Diese Generation sind reine Phosphoreszenz-
Emitter. Werden diese Komplexe angeregt, relaxiert das Exiton schnell von den angeregten Sin-
gulett-Zuständen in den niedrigsten Triplett-Zustand. Dieser Vorgang, bei dem der Spin des
Exitons geändert wird, nennt man intersystem crossing (ISC). Aufgrund dieser Begebenheiten ist
es diesen Molekülen möglich, sowohl Singulett- als auch Triplett- Exitonen zu ernten und somit
eine Quanteneffizienz von 100% zu erreichen[3]. Wegen ihrer geringen Strahlungsratenkonstanten
sind diese Moleküle jedoch anfällig für z.B. Quenching-Prozesse und Bleaching-Reaktionen[4].
Auf Grund der Seltenheit von Platin und Iridium und ihrem hohen Preis auf dem Weltmarkt wurde
nach einer kostengünstigeren Alternative gesucht. Die dritte Generation von OLEDs enthält Emit-
ter aus organischen Donor-Akzeptor-Systemen und Übergangsmetallkomplexen mit kleiner

Einleitung 2
Singulett-Triplett-Aufspaltung (ΔES-T). Durch diese geringe Aufspaltung ΔES-T von ist ein schnel-
les ISC und ein reverse intersystem crossing (RISC) möglich. Es ist bei diesen Komplexen also
möglich, sowohl die Exitonen, welche der direkten Fluoreszenz entsprechen, als auch die Exitonen
der thermisch aktivierten verzögerten Fluoreszenz (TADF) zu ernten. Somit ist es mit OLEDs der
dritten Generation auch möglich, eine interne Quanteneffizienz von 100 Prozent zu erreichen[2].
Ebenso wie bei den OLEDS der zweiten Generation ist die Strahlungsratenkonstante bei den
OLEDs der dritten Generation sehr gering, wodurch die Moleküle anfällig für Konkurrenz-Reak-
tionen sind.

Theorie 3
2 Theorie
2.1 Photophysikalische Prozesse
Bei photophysikalischen Prozessen wird ein Molekül durch Absorption von einem Photon in einen
angeregten Zustand überführt. Um in den Grundzustand zurückzugelangen, muss das Molekül re-
laxieren. Dies kann entweder durch Strahlung, wie Fluoreszenz oder Phosphoreszenz, oder durch
strahlungslose Prozesse geschehen. Das Jablonski Diagramm führt einige Möglichkeiten zur Des-
aktivierung auf.
Abbildung 1: Jablonski Diagramm der möglichen Übergänge: 1-Absorption; 2 und 6 interne Konversion; 3: intersystem
crossing; 4 reverse intersystem crossing; 5: Fluoreszenz; 6: Phosophoreszenz [5]
Der Grundzustand eines Moleküls liegt meist mit einer Spinmultiplizität von S0 vor. Die Anregung
in die Singulett-Zustände sind erlaubte Übergänge, da es hierbei zu keiner Spinumkehr kommt.
Der Wechsel zwischen Singulett- und Triplett-Zuständen hingegen ist ein spinverbotener Über-
gang, da hier eine Spinumkehr stattfindet. Die erlaubten Übergänge finden sehr schnell statt, wo-
hingegen die spinverbotenen Übergänge wesentlich länger dauern und somit auch ein höheres Ri-
siko für Konkurrenz-Reaktionen besteht. Bei der Anregung in einen energetisch höheren Zustand
wird meist vertikal angeregt, wobei es auch zu 0-0-Übergänge kommen kann. Dies bedeutet, dass
nicht immer in den niedrigsten, sondern auch in höhere Schwingungszustände angeregt wird.
Durch strahlungslose Schwingungsrelaxationen kann das Molekül in den untersten Schwingungs-
zustand übergehen und von dort aus relaxieren. Es ist jedoch nicht zwingend nötigt, dass das Mo-
lekül in den untersten Schwingungszustand übergeht, denn es kann auch aus höheren Zuständen
relaxieren z.B. durch ISC und RISC. Sowohl die Fluoreszenz als auch die Phosphoreszenz können
unter Beachtung der Kasha-Regel[6] hauptsächlich aus den untersten angeregten Zuständen ent-
stammen. Die Fluoreszenz findet zwischen S1 & S0 und die Phosphoreszenz zwischen T1 & S0
statt. Obwohl die Phosphoreszenz einen verbotenen Übergang darstellt, ist sie durch die Spin-

Theorie 4
Bahn-Kopplung möglich. Die Emission von TADF-Emittern ist durch den Zwischenschritt von
ISC und RISC deutlich verzögert und meist schwächer im Vergleich zur normalen Fluoreszenz.
2.2 TADF
TADF-Emitter können sich aufgrund ihrer Quanteneffizienz als effektive OLEDs beweisen. Ihr
Einsatzgebiet könnten im wesentlichen Displays und Beleuchtungssysteme sein. Der Erste TADF-
Emitter wurde 1929 von Perrin entdeckt[7].Danach folgten einige andere Wissenschaftler, wie
1941 Lewis[8],und anschließend 1961 Parke und Hatchard, die TADF in Eosin und Benzyl beo-
bachteten[9]. Im Jahr 1968 fanden Wilkinson und Horrocks[10] heraus, dass es sich bei diese Emis-
sionen um verzögerte Fluoreszenz handeln musste. Adachi stellte aber als Erster den Zusammen-
hang zwischen der verzögerten Fluoreszenz und einer Quanteneffizienz von 100% auf[3].
Damit ein Komplex TADF aufweist, muss zunächst eine Anregung des Grundzustandes durch
elektrische Energie stattfinden. Hierbei wird das Molekül vertikal in energetisch höhere Zustände
angeregt, mit anschließender Relaxation in den S1-Zustand. Damit das RISC möglich wird, wel-
ches für die TADF benötigt wird, gibt es einige Voraussetzungen. Zum einen ist eine geringe
Energielücke (ΔES-T) notwendig, damit sie noch thermisch überwunden und das RISC stattfinden
kann. Als Voraussetzung für eine geringe Energielücke ist zum anderen die Austauschwechsel-
wirkung eines Systems zu betrachten. Die Austauschwechselwirkung ist gering, wenn die Dichte-
verteilung der Orbitale, welche an der Anregung beteiligt sind, nur geringfügig überlappen. Dies
ist der Fall bei einem Charge-Transfer-Übergang (CT). Bei dieser Anregung sind die ungepaarten
Elektronen weit voneinander entfernt, wodurch es zu einer geringfüge Überlappung kommt und
zu einem kleinen ΔES-T. Die Energielücke (ΔES-T) ist zwar ein wichtige Voraussetzung dafür, dass
ein Molekül TADF fähig ist, jedoch nicht allein ausschlaggebend. Ein weiteres wichtiges Krite-
rium ist die kurzreichweitige Spin-Bahn-Kopplung (SOC). Grund dafür ist, dass sich bei Komple-
xen deren Singulett und Triplett-Zustände dieselbe Besetzung der Raumorbitale aufweisen, die
Spin-Bahn-Kopplung aus Symmetriegründen auslöscht. Als Voraussetzung für die TADF-Fähig-
keit eines Moleküls ist es also wichtig, dass die Ratenkonstanten von Fluoreszenz und RISC groß
sind als die des strahlungslosen Zerfalls. CT-Übergänge weisen üblicherweise eine geringe Oszil-
latorstärke auf, weshalb es zu geringen Strahlungsraten kommt.
2.3 Franck-Condon-Prinzip
Das Franck-Condon-Prinzip gibt eine Auskunft über die Wahrscheinlichkeit von Übergängen zwi-
schen verschiedenen Schwingungszuständen eines Moleküls bei elektronischer Anregung. Grund-
lage dieses Prinzipes ist die Born-Oppenheimer-Näherung, welche besagt, dass sich Elektronen

Theorie 5
wesentlich schneller bewegen als die Kerne. Die Schrödingergleichung kann so in einen Elektro-
nen- und Kern- abhängigen Teil unterteilt werden. Diese Annahme gilt auch bei einer elektroni-
schen Anregung, weshalb die Kerne als feste Punkte im Koordinatensystem angesehen werden.
Bei einer elektronischen Anregung wird ein Elektron in einen höheren Schwingungszustand an-
gehoben. In welchen Vibrationszustand das Elektron angehoben wird, ist abhängig von den Vib-
rationswellenfunktionen der beiden beteiligten Zustände. Der wahrscheinlichste Übergang findet
in den Zustand statt, indem sich die Vibrationswellenfunktion am geringsten verändert. Die Inten-
sität dieser Anregung ist im Quadrat abhängig von dem Franck-Condon-Integral. Dieses Integral
wird von der Überlappung der Zustände beeinflusst, welche wiederum abhängig ist von ΔES-T. Mit
welcher Energie das Elektron angeregt werden muss bzw. mit welcher Emissionswellenlänge es
zurück in den Grundzustand fällt, ist abhängig von der Verschiebung des angeregten Zustandes
relativ zum Grundzustand.[11]
Der Zusammenhang zwischen dem Überlappungsintegral und dem Franck-Condon-Faktor wird
über das Übergangsdipolmoment hergestellt. Die Wahrscheinlichkeit eines elektronischen Über-
gangs kann wie folgt dargestellt werden:
𝜇𝐸𝐴 = ⟨𝛹𝜀𝐸𝛹𝜈𝐸|�̂�|𝛹𝜀𝐴𝛹𝜈𝐴⟩ (2-1)
�̂� ist der Operator des Dipolmoments.
�̂� = −𝑒 ∑ 𝑟𝑖 + 𝑒 ∑ 𝑍𝑖𝑅𝑖
𝑖𝑖
(2-2)
Die Integralform von 𝜇𝐸𝐴 lässt sich dann schreiben als:
𝜇𝐸𝐴 = −𝑒 ∑ ∫ 𝛹𝜀𝐸∗ 𝑟𝑖𝛹𝜀𝐴𝑑𝜏𝜀 ∫ 𝛹𝜈𝐸
∗ 𝛹𝜈𝐴𝑑𝜏𝜈 + 𝑒 ∑ 𝑍𝑙 ∫ 𝛹𝜀𝐸∗ 𝛹𝜀𝐴𝑑𝜏𝜀 ∫ 𝛹𝜈𝐸
∗ 𝑅𝑙𝛹𝜈𝐴𝑑𝜏𝜈
𝑙𝑖
(2-3)
Aufgrund der Orthogonalität der zwei elektronischen Zustände verschwindet der zweite Term der
oberen Gleichung. Es ergibt sich Folgende Gleichung:
𝜇𝐸𝐴 = −𝑒 ∑ ∫ 𝛹𝜀𝐸∗ 𝑟𝑖𝛹𝜀𝐴𝑑𝜏𝜀 ∫ 𝛹𝜈𝐸
∗ 𝛹𝜈𝐴𝑑𝜏𝜈 = �̂�𝜀𝐸 ,𝜀𝐴 𝑆(𝜈𝐸 ,
𝑖
𝜈𝐴) (2-4)
�̂�𝜀𝐸 ,𝜀𝐴 steht für das elektrische Übergangsdipolmoment und S(vE,vA) steht für das Überlappungs-
integral.
�̂�𝜀𝐸 ,𝜀𝐴 = −𝑒 ∑ ∫ 𝛹𝜀𝐸∗ 𝑟𝑖𝛹𝜀𝐴𝑑𝜏𝜀
𝑖
(2-5)

Theorie 6
S(νEνA) = ∫ ΨνE∗ ΨνAdτν (2-6)
Das Quadrat des Überlappungsintegral ist definiert als der Franck-Condon- Faktor |𝑆(𝜈𝐸𝜈𝐴)|2.
Somit ist das Übergangsdipolmoment proportional zum Franck-Condon-Faktor.[12]
2.4 Fluoreszenzratenkonstante
Die Fluoreszenzratenkonstante gibt Auskunft über die Geschwindigkeit der Fluoreszenzemission.
Eine Ratenkonstante alleine ist hierbei meist nicht aussagekräftigt genug. Besonders hinsichtlich
der TADF-Eigenschaften eines Moleküls müssten auch die Konkurrenz-Rektionen, wie z.B. Phos-
phoreszenz oder auch strahlungslose Übergänge, betrachtet werden. Die Fluoreszenzratenkon-
stante wird mit „Fermis Goldener Regel“ berechnet, welche sich auf die Störungstheorie stützt.
𝑘𝑟𝑎𝑑 =4𝑒2
3𝑐3ℏ4(𝐸𝑖 − 𝐸𝑓)
3|⟨𝑓|𝑟|𝑖⟩ |2 (2-7)
Ei steht für die Energie des Initialzustandes also des S1- Zustandes- Ef entspricht der Energie des
Endzustands. Die Differenz der Energien muss in cm-1 eingesetzt werden. |⟨𝑓|𝑟|𝑖⟩ | stellt das Über-
gangsdipolmoment des S1-Zustandes zum S0 -Zustande dar und wird in atomaren Einheiten ein-
gesetzt. Der vordere Teil der Rechnung lässt sich dann zu einem Vorfaktor von 2,0261·10-6 zu-
sammenfassen.[13]
2.5 DFT
Bei der Dichtefunktionaltheorie wird keine Wellenfunktion zur Berechnung der Elektronenstruk-
tur verwendet, da diese zu viele Informationen enthalten, welche bei der Energieberechnung nicht
von Relevanz sind. Wellenbasierte Methoden sind abhängig von 3N Koordinaten, wohingegen die
DFT nur noch von drei Koordinaten abhängig ist. Diese Reduktion ist durch den Zusammenhang
zwischen Gesamtenergie und Dichte möglich. Die elektronische Energie des Grundzustands ist
demnach nur von der Einelektronendichte abhängig. Das eingeführte Dichtefunktional (𝐸[𝜌]), von
Hohenberg und Kohn, ist die Verbindung zwischen elektronischer Grundzustandsenergie und Ei-
nelektronendichte. Laut Hohenberg und Kohn existiert für 𝐸[𝜌] ein Variationsprinzip. Es gibt eine
Einteilung in eine exakte Dichte 𝜌 und eine positive definite Elektronendichte 𝜌′.Die 𝜌′ besitzt die
Eigenschaft, dass sie bei Integration über den gesamten Raum die Gesamtelektronenzahl wieder-
gibt.
𝜌′(𝑟) = 𝑁 ∫ 𝛹 · 𝛹 𝑑𝑟2⃗⃗⃗⃗ … 𝑑𝑟𝑛⃗⃗⃗⃗ (2-8)

Theorie 7
𝑁 = ∫ 𝜌′(𝑟)𝑑𝑟 (2-9)
Für das exakte Dichtefunktional gilt so die folgende Gleichung:
𝐸0[𝜌0′ ] ≥ 𝐸0[𝜌0 ] (2-10)
Es gibt jedoch aktuell noch keine Konstruktionsvorschriften für die Einelektronendichte oder das
Dichtefunktional.
2.5.1 Hohenberg- Kohn-Theorem
Hohenberg und Kohn konnten den Beweis erbringen, dass alle externen Potentiale genau einer
Dichte zugeordnet werden können[14]. Da der Hamilton-Operator vollständig bekannt ist, lässt sich
die Mehrelektronenwellenfunktion bestimmen und daraus lässt sich schlussfolgern, dass alle Ob-
servablen als Funktion der Dichte dargestellt werden können. Versucht man nun das Dichtefunk-
tional ähnlich aufzuteilen wie die Schrödingergleichung, so ergibt sich folgende Gleichung für die
Gesamtenergie:
𝐸[𝜌] = 𝑇[𝜌] + 𝐸𝑛𝑒[𝜌] + 𝐽[𝜌] + 𝐾[𝜌] (2-11)
Hierbei steht T[𝜌] für die kinetische Energie, Ene[𝜌] für die Coulomb-Wechselwirkung zwischen
Kernen und Elektronen, J[𝜌] für die Coulomb-Abstoßung der Elektronen untereinander und K[𝜌]
für die Austauschwechselwirkungen. Durch das Variationsprinzip lässt sich so die Energie des
Grundzustandes bestimmen. Das Dichtefunktional stellt eine obere Grenze für die Energie dar. Für
J[𝜌] und Ene[𝜌] lassen sich klassische Ausdrücke formulieren.
𝐸𝑛𝑒[𝜌] = ∑ ∫𝑍𝛼 𝜌(𝑟)
𝑅𝛼⃗⃗⃗⃗⃗⃗ − 𝑟′⃗⃗⃗⃗
𝑑𝑟
𝛼
(2-12)
𝐽[𝜌] =1
2∫ ∫
𝜌(𝑟)𝜌(𝑟′⃗⃗⃗⃗ )
|𝑟 − 𝑟′⃗⃗⃗⃗ |𝑑𝑟𝑑𝑟′⃗⃗⃗⃗ (2-13)
Nach Gleichung (2-12) Zα die Kernkoordinate, Rα die Kernkoordinate und r steht für die Ortsva-
riable des Elektrons. Anders als für J[𝜌] und Ene[𝜌] lassen sich für T[𝜌] und K[𝜌] keine klassi-
schen Ausdrücke formulieren. Wären T[𝜌] und K[𝜌] bestimmbar würde man ein exaktes Ergebnis
berechnen können. So müssen für diese beiden Terme Näherungen zur Lösung hinzugezogen wer-
den.

Theorie 8
2.5.2 Kohn-Sham-Formalismus
Bei dem Kohn-Sham-Formalismus wird in einen exakt lösbaren Teil und einen Korrekturterm
aufgeteilt. Hierfür wird ein fiktives System mit nicht wechselwirkenden Teilchen angenommen,
welches jedoch dieselbe Einelektronendichte aufweist, wie das wechselwirkende System. Durch
diese Annahme lassen sich Einelektronenterme separieren. Da der Kohn-Sham-Formalismus von
einem Hartree-Fock-ähnlichen Ansatz ausgeht, lässt sich folgende Gleichung aufstellen:
�̂� = �̂� + �̂�(𝜆) + 𝜆�̂�𝑒𝑒 (2-14)
𝜆 kann Werte zwischen 0 und 1 annehmen, wobei 0 für ein nicht miteinander wechselwirkendes
System steht und 1 für ein nicht separables vollwechselwirkendes System steht. �̂�(𝜆) ist das ex-
terne Potential. Als Hilfsgrössen werden Kohn-Sham-Orbitale Φ und ihr antisymmetrisches Pro-
dukt eingeführt. Die Einelektronendichte ergibt sich dann als:
𝜌(𝑟) = ∑|𝛷𝑖(𝑟)|2
𝑛
𝑖
(2-15)
Nun lässt sich das kinetische Eigenfunktional T[𝜌] in eine Formel für ein nicht wechselwirkendes
System und in ein wechselwirkendes System einteilen.
𝑇𝑠[𝜌] = ∑ ⟨𝛷𝑖|−12 𝛻2|𝛷𝑖⟩
𝑛
𝑖
(2-16)
Dieser Term ist auch als Korrekturterm ΔT[𝜌] bekannt. Zur Ermittlung der Kohn-Sham-Orbitale
werden die entsprechenden Kohn-Sham-Gleichungen verwendet folg. Gleichung (2 17).
ℎ̂𝐾𝑆|𝛷𝑖(𝑟)2 (2-17)
Der effektive Einteilchenoperator nimmt die folgende Form an:
ℎ𝐾𝑆 =1
2𝛻2 + �⃗⃗�𝑒𝑓𝑓 (2-18)
�⃗⃗�𝑒𝑓𝑓 steht für das Kohn-Sham-Potential und wird wie folgt definiert:
�⃗⃗�𝑒𝑓𝑓(𝑟) = ∑𝑍𝛼
|𝑅𝛼⃗⃗⃗⃗⃗⃗ − 𝑟′⃗⃗⃗⃗ |
+ ∫𝜌(𝑟′⃗⃗⃗⃗ )
|𝑟 − 𝑟′⃗⃗⃗⃗ |𝑑𝑟′⃗⃗⃗⃗ + �̂�𝑋𝐶(𝑟)
𝛼
(2-19)
Der erste Term auf der rechten Seite der Gleichung (2-19) gibt die attraktiven Wechselwirkungen
zwischen Elektronen und Kernen wieder, der zweite Term stellt die Coulomb-Wechselwirkung
der Elektronen untereinander dar und der letzte Term steht für den Korrekturterm und das

Theorie 9
Austauschwechselwirkungspotential. Bei weiterer Zusammenfassung ergibt sich das Kohn-Sham-
Dichtefunktional zu:
𝐸𝐷𝐹𝑇[𝜌] = 𝐸𝑛𝑒[𝜌] + 𝐸𝑋𝐶[𝜌] (2-20)
mit der Austausch- Korrelationsfunktion.
𝐸𝑋𝐶[𝜌] = (𝑇[𝜌] − 𝑇𝑠[𝜌]) + (𝐸𝑒𝑒[𝜌] − 𝐽[𝜌]) (2-21)
Das Austausch-Korrelations-Funktional wird noch ein weiteres Mal in Austausch-Funktional
Ex[𝜌] und Korrelations-Funktional EC[𝜌] eingeteilt. Von diesen beiden trägt das Austausch-Funk-
tional mehr zum EXC[𝜌] bei. [15]
Für das Austauschkorrelationsfunktional gibt es verschiedene Lösungsansätze. Zum einen existiert
der lokale-Dichte-Ansatz (LDA-Ansatz). Dieser Ansatz ist der Einfachste, denn er geht von einem
nicht wechselwirkenden Elektronengas aus. Bei der lokalen-Spin-Dichte-Ansatz (LSDA) wird er-
weiternd zum LDA noch der Spin berücksichtig. Wesentlich genauer ist die generalisierte Gradi-
entennäherung. Hier wird ein gradientenkorrigiertes Dichtefunktional verwendet. [15,14]
2.6 Hybrid-Funktionale
Die in dieser Arbeit verwendeten Hybridfunktionale sind zum einen, das PBE0-Funktional bei den
Berechnungen der Geometrien und zum anderen das BHLYP-Funktional bei den DFT/MRCI-
Rechnungen.
2.6.1 PBE0
Das PBE0-Funktional kombiniert das auf die generalisierte-Gradienten-Näherung (GGA) beru-
hende PBE-Funktional (Perdew–Burke-Ernzerhof) mit einem exakten Austauschterm. Der dritte
Term ist ein weiteres Austausch-Korrelations-Funktional.[16] Zu 25 Prozent wird das Ergebnis aus
dem exakten Hartree-Fock-Austausch gebildet und zu 75 Prozent aus der DFT-Austauschenergie.
𝐸𝑥𝑐𝑃𝐵𝐸0 = 𝐸𝑥𝑐
𝑃𝐵𝐸 +1
4 (𝐸𝑥
𝐻𝐹 − 𝐸𝑥𝑃𝐵𝐸) (2-22)
2.6.2 BH-LYP
Das „Becke Half and Half“-Funktional (BHLYP-Funktional) wird hier verwendet, weil es das
einzige implementierte Funktional bei DFT/MRCI ist. Es besteht zur einen Hälfte aus dem Hart-
ree-Fock-Austauschintegral und zur anderen Hälfte aus dem B88-Autauschterm, dem LDA-Aus-
tauschterm (lokal-density approximation) und einem LYP-Korrelationsterm (Lee Yang Paar).[17]

Theorie 10
ExcBHLYP=
1
2 Ex
HF+1
2(Ex
B88[ρ]+ExLDA[ρ])+Ec
LYP[ρ] (2-23)
2.7 Basissätze
Eine gute Auswahl an Basissätzen ist wichtig für exakte Ergebnisse der Rechnungen. Bei einfa-
chen zweiatomigen Molekülen können Basissätze, wie slater-typ-orbtials (STO) verwendet wer-
den. Hier müssen nur 1- und 2-Zentren-Integrale berechnet werden. Bei komplexeren Molekülen
wird die Berechnung von STOs jedoch zu aufwendig, deshalb werden komprimierte Gaußfunkti-
onen (GTOs) verwendet. Außer bei sehr nahen oder sehr weiten Entfernungen von dem Atomkern,
beschreiben die Gaußfunktionen die attraktiven und repulsiven Wechselwirkungen zwischen Kern
und Elektron sehr gut. Je mehr primitive Gaußfunktionen für eine komprimierte Gaußfunktion
verwendet werden, desto genauer wird das Ergebnis. In dieser Arbeit werden außer für das Kup-
feratom SV-P (split-valence polarized) Basissätze verwendet. Dies bedeutet, dass nur eine STO
für die inneren Schalen verwendet werden jedoch mehrere STOs für den Valenzbereich. Die Po-
larisation bedeutet, dass zusätzliche p-Funktionen für Wasserstoffatome hinzugefügt werden, um
eine Polarisation zum Beispiel wechselnde Dipolmomente bei einer Anregung besser zu beschrei-
ben.[15]
Der andere verwendete Basissatz ist der cc-pVDZ-PP-Basissatz von Dunning. Dies ist ein Basis-
satz mit korrelationskonsistenter Basis. Das c-p steht für „correlation-consistent polarized“ also
korrelationskonsistent polarisiert. VDZ steht für Valenz double Zeta. Das PP am Schluss steht für
zusätzliche Pseudopotentiale.[18]
2.8 DFT/MRCI
Die DFT-Methode eignet sich nicht zur Berechnung von angeregten Zuständen. Dies ist der
Grund, warum eine Kombination aus DFT/MRCI herangezogen wird. Die multi reference confi-
guration interaction (MRCI) stellt die Korrelationsenergie als Linearkombination verschiedener
Konfigurationen dar, um zu berücksichtigen, dass die angeregten Zustände aus mehreren Elektro-
nenkonfigurationen bestehen. Sie gehört zu den Konfigurations-Wechselwirkungs-Methoden.
Durch die Einführung von Koeffizienten können die betrachteten Anregungen unterschiedlich ge-
wichtet werden. Die Koeffizienten werden variiert und folglich bestimmt.
𝛷0 = 𝑐0𝛹0 + ∑ 𝑐𝑎𝑟 𝛹𝑎𝑟 + ∑ 𝑐𝑎𝑟𝑏𝑠𝛹𝑎𝑟𝑏𝑠+…
𝑎<𝑏,𝑟<𝑠𝑎𝑟
(2-24)
𝛹0 stellt die Referenzdeterminante dar. Der zweite Term gibt die Einfachanregungen von einem
besetzten in ein unbesetztes Orbital wieder und der dritte Term die Doppelanregung usw. Werden

Theorie 11
alle Anregungen berücksichtig, spricht man von einer Full CI und man erlangt eine exakte Korre-
lationsenergie. Dies führt jedoch unwirtschaftlichen Rechenaufwand für größere Moleküle, wes-
halb bei dieser Methode bei großen Molekülen mit einem aktiven Raum gearbeitet wird. Kern-
nahe Orbitale und energetisch sehr hoch liegende Orbitale werden eingefroren. Anregungen finden
nur im aktiven Raum statt. Somit lässt sich der Rechenaufwand deutlich reduzieren. Die MRCI
beschränkt sich nicht auf eine Referenzdeterminante, wie man anhand der vorherigen Gleichung
erkennen kann, sondern es werden die Konfigurationen der berechneten Anregungen für die fol-
genden Terme verwendet.
Die Kombination von DFT und MRCI, welche von Grimme und Waletzke entdeckt wurde[19],
ermöglicht es, die Vorteile beider Methoden zu nutzen. Die DFT-Methode wird für die dynami-
sche Elektronenkorrelation verwendet, wohingegen die MRCI für die statische Elektronenkorre-
lation verwendet wird. Um eine Doppelzählung der dynamischen Korrelation bei der MRCI-Rech-
nung zu verhindern, wird die Hamilton-Matrix parametrisiert.[19,4]
Die Parametrisierung die bei dieser Arbeit angewendet wird ist die Lyskov-Parametrisierung.[20]
Diese verbessert die Beschreibung von Doppelanregungen mit vier offenen Schalen. Zur Anwen-
dung bei größeren Molekülen sind diese Parameter auch für eine engere Auswahlgrenze bei einer
Energiedifferenz zwischen den Konfigurationen von 0,8 Eh optimiert, welches zu einer verringer-
ten Rechenzeit führt.[20]
2.9 ECP
ECP steht für effektives Kernpotential. Dieses wird eingesetzt für schwere Elemente, bei denen
viele Gaußfunktionen für nicht-Valenzelektronen berechnet werden müssten. Da bei schweren
Elementen die relativistischen Effekte nicht vernachlässigt werden können, werden hier Pseudo-
potentiale eingeführt. Dabei werden die chemisch weniger relevanten inneren Elektronen durch
ein effektives Potential angenähert, welches mit den Valenzelektronen wechselwirkt. Pseudopo-
tentiale wurden schon 1934 durch Hans Heilmann eingeführt und verringern die Anzahl an zu
berechnenden Gaußfunktionen deutlich und somit die Rechenzeit. Es werden einige Näherungen
bei den Pseudopotentialen angewendet. Zum einen das Ein-Elektronenbild eingeführt, um Elekt-
ronen in Kern- und Valenzelektronen einteilen zu können. Die zweite Annahme ist, dass die Ein-
Elektronenzustände im Atomrumpf eingefroren sind und nicht polarisierbar sind. Diese Näherung
nennt man Frozen-Core-Approximation. Die dritte Näherung nimmt an, dass kein signifikanter
Überlapp zwischen Kern- und Valenzzuständen vorliegt, damit ein Wechsel der Elektronen zwi-
schen diesen Zuständen unwahrscheinlich ist. Häufig sind die Valenzfunktionen orthogonal zu den
Kern-Zuständen bei den Pseudopotentialen.[21] Der Vorteil von ECPs ist, dass auch

Theorie 12
skalarrelativistische Effekte mit eingebracht werden, welche besonders für schwere Elemente von
Bedeutung sind. Durch die hohe Anzahl an Kernladungen steigt die elektrostatische Anziehungs-
kraft des Kerns auf die Elektronen. Die kernnahen Elektronen werden dadurch so schnell beschleu-
nigt, dass sich ihre Geschwindigkeit, der der Lichtgeschwindigkeit nähert. Dadurch gilt die relati-
vistische Formel für die kinetische Energie nicht mehr. Dies führt zu einer Kontraktion der s-
Orbitale und einiger p-Orbitale. Die inneren Elektronen schirmen durch die Kontraktion die Kern-
ladung besser ab, infolgedessen werden die übrigen Orbitale energetisch angehoben.

Methodik 13
3 Methodik
Zu Beginn der Bachelorarbeit wurde das Molekül mit Hilfe der Software AVOGADRO konstru-
iert.[22] Die erhaltenen Koordinaten wurden in das Programm TURBOMOLE Version 7.0 [23]über-
führt und eine entsprechende Geometrieoptimierung durchgeführt. Die verwendeten Basissätze
sind SV(P) und cc-pVDZ-PP, wobei nur das Kupferatom ein ECP (Stuttgart-Koeln MCDHF RSC
ECP) und den Basissatz cc-pVDZ-PP erhalten hat. Das verwendete Funktional ist das PBE0-Hyb-
ridfunktional. Nach erfolgreicher Konvergenz bei der Geometrieoptimierung wurde mit dem Pro-
gramm AOFORCE (Komponente von TURBOMOLE) die Schwingungsfrequenzen berechnet.
Die Frequenzen wurden mit GMOLDEN[24] analysiert. Lag eine imaginäre Frequenz vor, wurde
die Struktur des Moleküls geändert. Hierfür wurde mit dem Programm VIBRATION die Struktur
um die vorhandene Schwingung variiert. Anschließend wurde erneut eine Geometrieoptimierung
mit den veränderten Koordinaten durchgeführt. Dies führte bei allen untersuchten Molekülen zu
einem positiven Ergebnis. Nur das Molekül mit der Methyl-Substitution gelangte so nicht zu ei-
nem absoluten Minimum. Auch nach der Erhöhung des Grids auf m5 konnte kein absolutes Mini-
mum gefunden werden. Die weiteren Rechnungen wurden mit der Energie und den Koordinaten
des Sattelpunktes weitergeführt, da die Torsionsschwingung der Methylgruppen nicht zu unter-
binden war. Nach der Geometrieoptimierung wurden DFT/MRCI-Rechnungen durchgeführt. Hier
wurde das Funktional von PBE0 auf BH-LYP geändert, da dies das einzige implementierten Funk-
tional in dem DFT/MRCI-Programm dar stellt. Mit diesem Programmen wurden jeweils die 5
niedrigsten Singulett- und Triplett-Zustände berechnet. Bei diesen Rechnungen wurde der imple-
mentieren Lyskov-Parameter[20] zur Berechnung verwendet. Bei dem unsubstituierten Molekül
wurde getestet, ob tight-Parameter mit esel=0.8 hätte verwendet werden können, oder ob doch
Standard-Parameter angewandt werden müssen. Um die Dipolmomente und die Übergangsdichten
zu bestimmen wurde anschließend die Differenzdichten erstellt. Hierfür wurde das Programm
GIMP[25] verwendet. Die Bindungslängen, Bindungswinkel und Diederwinkel wurden mit JMOL
bestimmt. Nachdem die geschlossenschaligen Berechnungen fertig waren, wurden weiterführende
Rechnungen mit unrestricted DFT (UDFT) gerechnet. Der Ablauf war exakt derselbe.

Ergebnisse und Auswertung 14
4 Ergebnisse und Auswertung
4.1 Rp-[κ2-N,P-4-(2′-Pyridyl)-1,2-diphenylphosphinyl[2.2]paracyclo-
phan]chlorokupfer(I)
Zu Beginn wurde sowohl eine Geometrieoptimierung des S0- als auch des T1–Zustands an dem in
der Überschrift benannten Molekül durchgeführt. Im Folgenden werden diese beiden Geometrien
miteinander verglichen und analysiert. Daraufhin wird im DFT/MRCI- Abschnitt auf die energe-
tische Lage der Zustände, die Oszillatorstärke etc. eingegangen, um davon ausgehend einen Zu-
sammenhang mit der Fähigkeit des Moleküls zur TADF herzustellen. Im weiteren Verlauf dieser
Arbeit wird dieses Molekül an den Phenyl-Einheiten substituiert und deshalb im Folgenden das
ursprüngliche Molekül als Molekül 1 bezeichnet.
4.1.1 S0 Geometrie
Die Geometrien des S0 und T1 Zustands weisen deutliche Abweichungen voneinander auf. Dies ist
darin begründet, dass im angeregten Zustand eine Bindung zwischen dem Kupferatom und einem
Wasserstoffatom am oberen Cyclophanring entsteht. Diese Wechselwirkung nennt man argos-
tisch. Ein elektrophiles Metallzentrum bildet hierbei schwache Wechselwirkungen zu einem Was-
serstoffatom aus. Es handelt sich um eine Drei Zentren-Zwei-Elektronen Bindung. Der Grund für
die Ausbildung dieser Brücke ist die Verschiebung der Elektronendichte zwischen Grundzustand
und angeregtem Zustand. Während das Molekül im Grundzustand eine hohe Elektronendichte an
dem Kupferatom aufweist, verschiebt sich die Elektronendichte im angeregtem Zustand zu dem
Pyridyl- und Cyclphan-Einheiten. Um diesen Mangel auszugleichen, bildet es eine Bindung zum
nächst gelegenen Wasserstoffatom aus. Die beiden folgenden Abbildungen zeigen die Strukturen
der zwei Optimierungen. wobei folgende Kolorierung und Nummerierung der Atome vorgenom-
men wurde (Abbildung 4).
Abbildung 2: Struktur Molekül 1 S0-Geometrie
Abbildung 3: Struktur Molekül 1 T1-Geometrie

Ergebnisse und Auswertung 15
Abbildung 4: Kolorierung und Nummerierung des Moleküls 1: Grün = Chlor; Kupfer = Kupfer; Orange = Phosphor; Blaue
= Stickstoff; Weiß = Wasserstoff; Grau = Kohlenstoff
Vergleicht man die berechneten Werte mit den experimentellen Werten der Doktorarbeit von
Carolin Braun (hier nur das Enantiomer betrachtet) wird deutlich, dass nur geringe Abweichungen
vorliegen.[26]
Die Werte von Carolin Braun entsprechen der Kristallstruktur, welche noch Reste von Lösungs-
mittelmolekülen enthalten, wodurch die geringen Abweichungen in Bindungslängen und Winkel
möglich sind.
Im Folgenden werden die Strukturen der unterschiedlichen Geometrien miteinander verglichen. In
Tabelle 3 sind die gemessenen Bindungslängen mit den stärksten Abweichungen (ab 5 pm) zwi-
schen den beiden Geometrien aufgeführt. Die weiteren Bindungslängen befinden sich in einer Ta-
belle im Anhang.
Tabelle 3: Bindungslängen des Moleküls 1
Wie man deutlich erkennen kann, liegen die größten Veränderungen rund um das metallische Kup-
feratom vor. Die Bindungslängen in der Pyridyl-Einheit, im para-Cyclophan-Einheit oder den
Tabelle 1: Bindungslängen Vergleich mit Literaturwer-
ten
Tabelle 2: Bindungswinkel Vergleich mit Literaturwer-
ten
Bindungs-
längen
[pm]
Berechneter
Wert
Literatur-
wert Differenz
Cu-N 207,4 203,1 4,3
Cu-P 219,1 216,8 2,3
Cu-Cl 219,6 222,5 -2,9
Bindungs-
winkel [°]
Berechneter
Wert
Literatur-
wert
Diffe-
renz
N-Cu-P 121,9 118,97 2,93
N-Cu-Cl 106,9 107,55 -0,65
P-Cu-Cl 128,2 132,57 -4,37
Bindungen [pm] S0 Geometrie T1 Geometrie S0- T1
P1-Cu2 219,1 233,6 -14,5
Cu2-Cl3 219,6 214,4 5,2
Cu2-N4 207,4 189,8 17,6
N4-C19 134,8 140,9 -6,1

Ergebnisse und Auswertung 16
Diarylphospin-Einheit ändern sich kaum. Die Tabellen 4 und 5 zeigen die Änderung der Bindungs-
winkel und der Diederwinkel, wobei nur noch jene betrachtet werden, welche Stickstoff-, Kupfer-
und das Phosphoratom beinhalten und Abweichungen von mehr als 5° aufweisen.
Tabelle 4: Bindungswinkel des Moleküls 1 Tabelle 5: Diederwinkel des Moleküls 1
Winkel [°] So T1 S0-T1
P1-Cu2-Cl3 128,2 104,5 23,7
P1-Cu2-N4 121,9 107,0 14,9
Cu2-P1-C5 108,7 115,0 -6,3
Cu2-P1-C36 115,9 106,9 9,0
Cl3-Cu2-N4 106,9 148,2 -41,3
C11-P1-C36 101,6 107,6 -6,0
Winkel [°] So T1 S0-T1
Cl3-Cu2-P1-C5 86,1 130,2 -44,1
Cl3-Cu2-P1-C11 -28,6 8,1 -36,7
N4-Cu2-P1-C5 -71,3 -45,4 -25,9
N4-Cu2-P1-C11 174,0 -167,4 341,4
Die größte Geometrieänderung entsteht durch die Bildung einer neuen Bindung zwischen dem
Kupfer- und einem Wasserstoffatom. Aus diesem Grund wird das Kupferatom näher zu der Cyc-
lophan-Einheit und somit auch näher zum Stickstoffatom gezogen. Die Bindungslänge verkürzt
sich um 17,6 pm. Dies ist die stärkste Veränderung der Bindungslänge an diesem Molekül. Durch
die Verkürzung der Kupfer-Stickstoffbindung muss der Chlor-Kupfer-Stickstoff-Winkel jedoch
größer werden. Aufgrund der Größe und der freien Elektronenpaare des Chloratoms ist es nicht
möglich, dass dieses näher an die Pyridyl-Einheit rückt. Um das Näherrücken des Chloratoms an
das Kupferatom auszugleichen, muss also der Winkel vergrößert werden. Der Abstand von Kup-
fer- zum Phosphoratom verlängert sich um 14.5 pm wohingegen sich der Chlor-Kupfer-Phosphor-
Bindungswinkel verkleinert. Dies ist darin begründet, dass die Diarylphosphin-Einheit sterisch
anspruchsvoll ist und somit eine Verkürzung der Phosphor-Kupfer-Bindung verhindert. Der Bin-
dungswinkel wird kleiner, weil das Chloratom der näher kommenden Pyridyl- Einheit ausweicht
und somit zwangsweise dem Phosphoratom nähern kommen muss. Anhand der Veränderung der
angegebenen Diederwinkel in Tabelle 5 kann man erkennen, dass die Phenylringe sich durch die
Geometrieveränderung auch anpassen, indem sie sich anders zueinander ausrichten. Die restlichen
Geometrieänderungen lassen sich anhand der HOMOLUMO-Anregung erklären, aus dem der
T1-Zustand hauptsächlich besteht.
Abbildung 5:HOMO des Moleküls 1
Abbildung 6: LUMO des Moleküls 1

Ergebnisse und Auswertung 17
Wie aus den Abbildungen 5 & 6 zu erkennen ist, handelt es sich bei dieser Anregung um einen
Charge-Transfer-Übergang, genauer gesagt um eine XMLCT. Im Abschnitt 4.1.2.2. wird auf die-
ses Thema genauer eingegangen. Eine XMLCT Anregung bedeutet, dass die Elektronendichte von
dem Kupfer-, Chlor- und dem Phosphoratom zu der Pyridyl-Einheit und dem damit verbundenen
Cyclophanring wandert. Anhand der HOMO- und der LUMO-Abbildung lassen sich einige der
kleineren Geometrieveränderung erklären. Zum Beispiel wird durch die überlappende Elektronen-
dichte zwischen dem C19- und dem C42-Atom die Bindung in der T1-Geometrie verkürzt. Im
Gegensatz dazu befindet sich im LUMO weniger Elektronendichte am Chloratom als im HOMO,
da diese zum Pyridyl- und zur der Cyclophan-Einheit wandert. Auf diesen Weg kann das Chlo-
ratom näher zum Kupferatom rücken. Die Bindungslänge zwischen Stickstoffatom und dem C19-
Atom wird aufgrund der abstoßenden Elektronendichten um 6,1 pm länger, wogegen die Bin-
dungswinkel aufgrund der HOMO-LUMO-Anregung kaum Änderung aufweisen.
4.1.2 DFT/MRCI
Nach der Geometrieoptimierung werden DFT/MRCI Einzelpunktrechnungen durchgeführt. Diese
können unter Umständen aufgrund ihres Rechenaufwandes sehr lange dauern. Deshalb wird an-
hand des Moleküls 1 getestet, ob diese Berechnungen auch mit tight-Parametern (esel = 0,8) durch-
zuführen sind, welche die Rechnung um einige Stunden verkürzen würden, oder ob doch Standard-
Parameter (esel =1) für exaktere Ergebnisse verwendet werden müssen.
4.1.2.1 DFT/MRCI tight
Um zu entscheiden, ob in den folgenden Rechnungen tight-Parameter zur Verkürzung der Rechen-
zeit verwendet werden können, werden anhand des Moleküls 1 einige Berechnungen zum Ver-
gleich durchgeführt. Sowohl für die S0- als auch die T1-Geometrie werden zwei Rechnungen, eine
mit tight-Parametern und eine mit Standard-Parametern, durchgeführt. Als erste Voraussetzung
für die Anwendbarkeit der tight-Parameter werden die Energien der Zustände betrachtet.
Tabelle 6: relative Zustandsenergien im Vergleich MRCI -MRCI-tight
MRCI
Esn-Es0 [eV]
MRCI-tight
Esn-Es0 [eV]
ΔEnergie (MRCI-
MRCI-tight) [eV]
S0-Geometrie
S0 0,00 0,00 0,00
S1 3,35 3,11 0,24
S2 3,64 3,33 0,31
S3 3,77 3,49 0,28
S4 3,86 3,66 0,20
S5 3,99 3,66 0,33
T1 3,11 2,76 0,35
T2 3,25 2,95 0,31
T3 3,32 3,01 0,31
T4 3,50 3,14 0,37
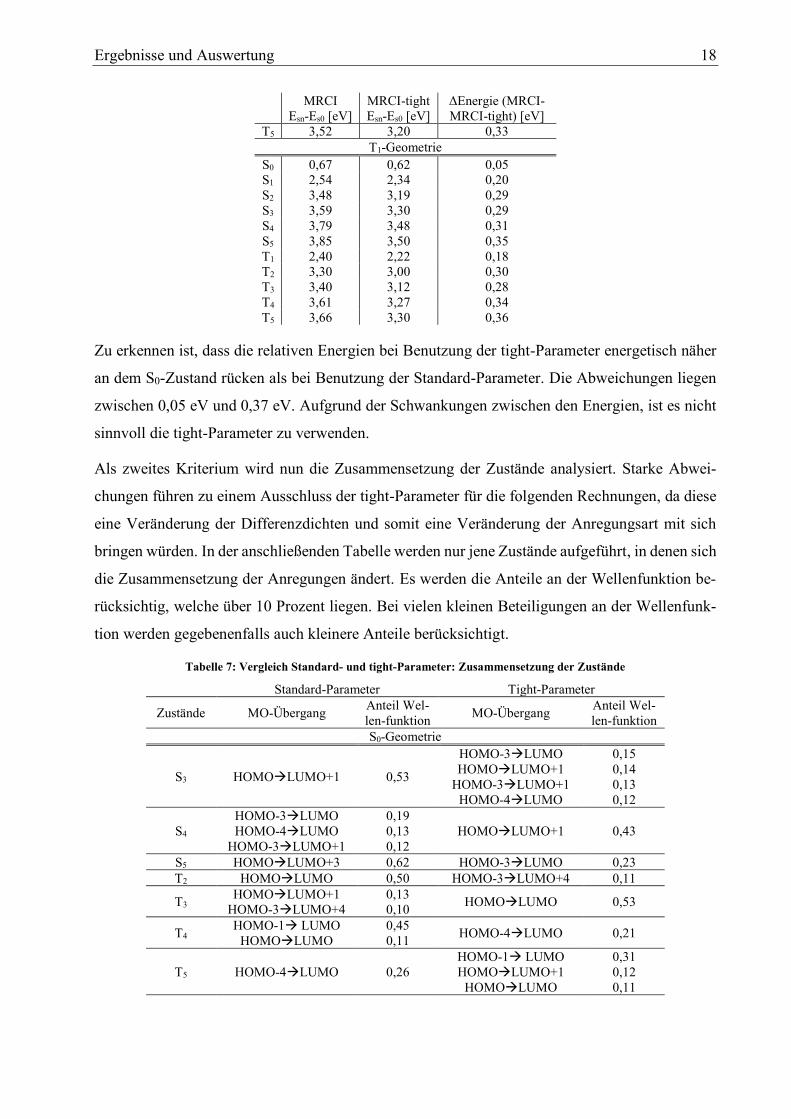
Ergebnisse und Auswertung 18
MRCI
Esn-Es0 [eV]
MRCI-tight
Esn-Es0 [eV]
ΔEnergie (MRCI-
MRCI-tight) [eV]
T5 3,52 3,20 0,33
T1-Geometrie
S0 0,67 0,62 0,05
S1 2,54 2,34 0,20
S2 3,48 3,19 0,29
S3 3,59 3,30 0,29
S4 3,79 3,48 0,31
S5 3,85 3,50 0,35
T1 2,40 2,22 0,18
T2 3,30 3,00 0,30
T3 3,40 3,12 0,28
T4 3,61 3,27 0,34
T5 3,66 3,30 0,36
Zu erkennen ist, dass die relativen Energien bei Benutzung der tight-Parameter energetisch näher
an dem S0-Zustand rücken als bei Benutzung der Standard-Parameter. Die Abweichungen liegen
zwischen 0,05 eV und 0,37 eV. Aufgrund der Schwankungen zwischen den Energien, ist es nicht
sinnvoll die tight-Parameter zu verwenden.
Als zweites Kriterium wird nun die Zusammensetzung der Zustände analysiert. Starke Abwei-
chungen führen zu einem Ausschluss der tight-Parameter für die folgenden Rechnungen, da diese
eine Veränderung der Differenzdichten und somit eine Veränderung der Anregungsart mit sich
bringen würden. In der anschließenden Tabelle werden nur jene Zustände aufgeführt, in denen sich
die Zusammensetzung der Anregungen ändert. Es werden die Anteile an der Wellenfunktion be-
rücksichtig, welche über 10 Prozent liegen. Bei vielen kleinen Beteiligungen an der Wellenfunk-
tion werden gegebenenfalls auch kleinere Anteile berücksichtigt.
Tabelle 7: Vergleich Standard- und tight-Parameter: Zusammensetzung der Zustände
Standard-Parameter Tight-Parameter
Zustände MO-Übergang Anteil Wel-
len-funktion MO-Übergang
Anteil Wel-
len-funktion
S0-Geometrie
S3 HOMOLUMO+1 0,53
HOMO-3LUMO
HOMOLUMO+1
HOMO-3LUMO+1
HOMO-4LUMO
0,15
0,14
0,13
0,12
S4
HOMO-3LUMO
HOMO-4LUMO
HOMO-3LUMO+1
0,19
0,13
0,12
HOMOLUMO+1 0,43
S5 HOMOLUMO+3 0,62 HOMO-3LUMO 0,23
T2 HOMOLUMO 0,50 HOMO-3LUMO+4 0,11
T3 HOMOLUMO+1
HOMO-3LUMO+4
0,13
0,10 HOMOLUMO 0,53
T4 HOMO-1 LUMO
HOMOLUMO
0,45
0,11 HOMO-4LUMO 0,21
T5 HOMO-4LUMO 0,26
HOMO-1 LUMO
HOMOLUMO+1
HOMOLUMO
0,31
0,12
0,11

Ergebnisse und Auswertung 19
Standard-Parameter Tight-Parameter
Zustände MO-Übergang Anteil Wel-
len-funktion MO-Übergang
Anteil Wel-
len-funktion
T1-Geometrie
S4
HOMO-2LUMO
HOMO-5LUMO
HOMO-8LUMO
0,31
0,16
0,11
HOMOLUMO+2
HOMO-2LUMO
0,44
0,20
Es sind Verdrehungen oder Änderungen der Zusammensetzung der Zustände zu erkennen. In der
S0-Geometrie sind die Zustände S3 und S4 bei Benutzung der tight-Parameter im Vergleich zu den
Standard-Parametern vertauscht. Die ΔES3-S4 liegt bei 0,09 eV in der Grundzustands-Geometrie.
Der S5-Zustand wird aus anderen Anregungen zusammengesetzt. Außerdem sind die Triplett-Zu-
stände T2 und T3 sowie T4 und T5 ebenfalls vertauscht (ΔET2-T3=0,07 eV; ΔET4-T5=0,02 eV). Im
Vergleich dazu scheinen die Zustände der T1-Geometrie geringer durch die Wahl der Parameter
beeinflusst zu werden. An dieser Stelle stimmt nur der S4-Zustand in beiden Berechnungen nicht
überein. Die Abweichungen führen dazu, dass die Benutzung der tight-Parameter eher ungünstig
ist. Alle folgenden Rechnungen werden also mit den Standard-Parametern durchgeführt. Um diese
Entscheidung noch einmal zu unterstützen, wird die Oszillatorstärke betrachtet, wobei nur diese
aufgeführt werden, welche eine Abweichung größer als 0,008 aufweisen:
Tabelle 8: Standard- und tight-Parameter: Vergleich Oszillatorstärken
Die zu erwartenden höheren Abweichungen der Oszillatorstärken bei Zuständen veränderter Zu-
sammensetzung lassen sich gemäß Tabelle 8 erkennen. Die Abweichung der Oszillatorstärke bei
der S0-Geometrie liegt am höchsten bei den Zuständen S3 & S4. Die Abweichung von dem Zustand
S5 von 0,00172 liegt trotz grundverschiedener Zusammensetzung des Zustandes nicht höher als
bei den Zuständen S3 & S4. In der T1-Geomtrie liegt nur die Oszillatorstärke des S4 Zustandes bei
höheren Abweichungen. Alles in Allem sind die Ergebnisse bei der Verwendung der tight-Para-
meter im Vergleich zu den Standard-Parametern zu stark. Sowohl die energetische Lage der Zu-
stände als auch teilweise ihre Zusammensetzung ändert sich und in Folge dessen ihre Oszillator-
starken. Letztere geben die Intensität des Übergangs wieder und sind besonders für die folgende
Auswertung von Bedeutung. Aufgrund der Ergebnisse in diesem Abschnitt werden im Folgenden
die DFT/MRCI-Rechnungen mit den Standard-Parametern durchgeführt.
Anregungen Standard-Parameter Tight-Parameter Differenz
S0-Geometrie
S0 S3 0,024 0,004 0,020
S0 S4 0,010 0,018 -0,008
T1-Geometrie
S0 S4 0,019 0,035 -0,015

Ergebnisse und Auswertung 20
4.1.2.2 Auswertung DFT/MRCI
Im vorherigen Abschnitt wurde die Zusammensetzung der Zustände genauer betrachtet. An dieser
Stelle ist bei den betrachteten Ergebnissen der Standard-Parametern aufgefallen, dass der T1-Zu-
stand an der S0-Geometrie nicht nur aus einer HOMO-LUMO Anregung besteht, sondern noch
eine weitere Anregung bei diesem Zustand einen Anteil an der Wellenfunktion aufweist. Die
HOMOLUMO-Anregung weist einen Anteil von 0,11 an der Wellenfunktion auf. Deutlich
wichtiger ist bei dem betrachtetem T1-Zustand die Anregung von HOMO-3 nach LUMO mit ei-
nem Anteil von 0,30 an der Wellenfunktion. Im Gegensatz zu dem T1-Zustand besteht der T2-
Zustand in der selben Geometrie hauptsächlich aus der HOMOLUMO-Anregung (0,50 Anteil
an der Wellenfunktion). Ausgehend von der Überlegung, dass sowohl die Anregung in den T1-
Zustand als auch in den S1-Zustand durch eine HOMOLUMO-Anregung stattfinden soll, wurde
von einer ähnlichen Geometrie beider Zuständen ausgegangen. Aus diesem Grund wurde in dieser
Arbeit nur die T1-Geomtrie optimiert. Die Zustände T1, T2 und T3 liegen alle unterhalb des S1-
Zustands und sind energetisch nahe beieinander. Bei der Berechnung der vertikalen Anregungs-
energien (ausgehend von der S0-Geometrie) kann es passieren, dass es zu einer Verdrehung der
Zustände kommt. Dies kann der Fall für die Zustände T1, T2, oder T3 sein. Betrachtet man nun die
berechneten Differenzdichten an der Triplett- und an der Grundzustands-Geometrie so fällt auf,
dass die Differenzdichte des T2-Zustands an der Grundzustands-Geometrie der Differenzdichte
des T1-Zustands an der Triplett-Geometrie stärker ähnelt. Die Differenzdichte des T1-Zustand an
der Grundzustands-Geometrie ähnelt keinem der anderen Differenzdichten an der Triplett-Geo-
metrie.
Tabelle 9: Anregungen Molekül 1
Grundzustands-Geometrie Triplett-Geometrie
Zustand MO-Übergang Anteil der Wel-
lenfunktion Zustand MO-Übergang
Anteil der Wel-
lenfunktion
T1 HOMO-3LUMO
HOMOLUMO
0,30
0,11 T1 HOMOLUMO 0,75
T2 HOMOLUMO 0,50 T2 HOMO-1 LUMO 0,52
T3 HOMOLUMO+1
HOMO-3LUMO+4
0,13
0,10 T3 HOMOLUMO+1 0,69
T4 HOMO-1LUMO
HOMOLUMO
0,45
0,11 T4
HOMO-3LUMO
HOMO-8LUMO
HOMO-2LUMO
0,20
0,16
0,15
T5 HOMO-4LUMO 0,26 T5 HOMOLUMO+2 0,72
Anhand Tabelle 9 kann man erkennen, dass der T1-Zustand an der S0-Geometrie dem T4-Zustand
an der T1-Geometrie am ehesten endspricht. Die Zuordnung fällt dann wie folgt aus: T1T4;
T2T1; T3T3; T4T2 und T5 wird komplett anders zusammengesetzt.

Ergebnisse und Auswertung 21
Aus diesem Grund wird bei den folgenden Berechnungen an der Grundzustands-Geometrie der
T2-Zustand für ΔES-T verwendet.
Abbildung 7: Differenzdichte zw.
S1 und S0 an der S0 Geometrie
des Moleküls 1
Abbildung 8: Differenzdichte zw.
T1 und S0 an der S0 Geometrie
des Moleküls 1
Abbildung 9: Differenzdichte zw.
T2 und S0 an der S0 Geometrie
des Moleküls 1
Abbildung 10: Differenzdichte
zw. S1 und S0 an der T1 Geomet-
rie des Moleküls 1
Abbildung 11: Differenzdichte
zw. T1 und S0 an der S0 Geomet-
rie des Moleküls 1
Abbildung 12: Differenzdichte
zw. T2 und S0 an der S0 Geomet-
rie des Moleküls 1
Die Restlichen Differenzdichten befinden sich im Anhang. Die folgende Tabelle 10 charakterisiert
der Übergänge anhand der Differenzdichten, um eine Einschätzung der Stärke der CT-Übergänge
geben zu können.
Tabelle 10: Molekül 1: Art der Übergänge
S0-Geometrie
S1 XMLCT; mit LC an Cyclophan und Phenyl
S2 XMLCT; mit LC an Cyclophan und Phenyl
S3 XMLCT; mit LC an Cyclophan und Phenyl; Elektronendichte
wandert vermehrt zu Pyridin und Cyclophan
S4 CT am Pyridin und Cyclophan; XMLCT Beitrag
S5 XMLCT
T1 LC am Pyridin und Cyclophan; XMLCT Beitrag
T2 XMLCT; mit LC an Cyclophan und Phenyl
T3 LC am untere Cyclophanring mit XMLCT-Beitrag
T4 XMLCT; mit LC an Cyclophan und Phenyl
T5 LC am Pyridin und Cyclophan; XMLCT Beitrag
T1-Geometrie
S1 XMLCT; mit LC an Cyclophan und Phenyl
S2 XMLCT; mit LC an Cyclophan und Phenyl

Ergebnisse und Auswertung 22
Anhand dieser Tabelle kann man erkennen, dass die zuvor getroffenen Zuordnung der Triplett-
Zustände mit Hilfe der enthaltenen Anregungen, sich nicht exakt in den Differenzdichten wieder
spiegelt. Es werden auch nur die Anregungen mit einer großen Beteiligung an der Wellenfunktion
betrachtet. Die anderen Anregungen beeinflussen den Zustand jedoch auch. Dies kann zu den be-
obachteten Abweichungen führen. Die vorhandenen Zustände weisen alle Charge-Transfer-Cha-
rakter auf. Generell sind sich die Zustände in ihren Differenzdichten sehr ähnlich. Es gibt nur
wenige Ausnahmen die deutliche Veränderungen aufweisen.
Charge-Transfer-Übergänge erfüllen die Bedingung für gute TADF-Eigenschaften. Deshalb ist es
von Vorteil, dass der Übergang S0S1 genau solch eine Eigenschaft aufweist. Im weiteren werden
die Dipolmomente genauer betrachtet. Diese geben Aufschluss, wie stark das Molekül polarisiert
ist und in welche Richtung. Die Raumrichtungen der Dipolmomente der Zustände S0, S1, T1, sowie
T2 der S0-Geometrie werden in der Tabelle 11 aufgeführt.
Tabelle 11:Molekül 1: Dipolmomente
Anhand der Werte in der Tabelle 11 kann man erkennen, dass der S1-Zustand das stärkste Dipol-
moment mit 10,38 D aufweist. Das Dipolmoment von T2 liegt mit 9,03 D dem Dipolmoment von
S1 am nächsten. Dieselbe Ausrichtung und Stärke des Dipolmoments ist ein wichtiges Kriterium
T1-Geometrie
S3 XMLCT; mit LC an Cyclophan und Phenyl
S4 XMLCT; mit LC an Cyclophan und Phenyl
S5 XMLCT; Übergang zum Cyclophan
T1 XMLCT; mit LC an Cyclophan und Phenyl
T2 XMLCT; mit LC an Cyclophan und Phenyl
T3 XMLCT; mit LC an Cyclophan und Phenyl
T4 XMLCT; mit LC an Cyclophan und Phenyl
T5 XMLCT; Übergang zum Cyclophan
Komponente
Zustand x y z Dipolmoment [D]
S0 -2,034 1,278 1,946 7,859
S1 -1,603 -3,415 1,561 10.378
T1 -2,278 0,649 1,888 7,698
T2 -1,648 -2,715 1,589 9,026

Ergebnisse und Auswertung 23
hinsichtlich der Überlappung des S1- und T1-Zustandes, welche das ISC und RISC beeinflusst. Um
diese Werte zu veranschaulichen, werden die Dipolmomente mit JMOL in das Molekül gelegt.
Die eingezeichneten Pfeile stellen die Dipolmomente der Zustände dar. Der T3-Zustand wurde
noch hinzugenommen, da auch er energetisch unterhalb des S1-Zustandes liegt. In Abbildung 13
wird erkennbar, dass die betrachteten Singulett-Zustände in verschiedene Richtungen innerhalb
des Moleküls weisen. Das Dipolmoment des S0-Zustandes sowie der des T1- und T3-Zustandes
weisen in Richtung des Chloratoms. Der S1- und der T2-Zustand orientieren sich jedoch in Rich-
tung der Pyridyl-Einheit. Würde man das Molekül solvatisieren, so könnte man bei den unter-
schiedlichen Dipolmomenten von S0 zu S1 und T2 mit sich ändernden Absorptions- und Emission-
spektren rechnen. Aufgrund der Verzerrung des Moleküls zwischen S0- und T1-Geometrie ist eine
Vorhersage jedoch schwer zu treffen. Das Diagramm 1 veranschaulicht die Lage der Zustände in
Abhängigkeit von dem S0-Zustand an der S0-Geometrie. Die energetische Lage der Zustände
wurde durch vertikale Anregungsenergien des S0-Zustandes an der S0-Geometrie berechnet. Die
adiabatische Energie kann nur zwischen den optimierten Zuständen berechnet werden und ist dem-
nach nur zwischen dem S0- und dem T1-Zustand möglich. Da jedoch besonders der ΔES1-T1 von
Bedeutung ist, wird die Energiedifferenz zwischen S0-Zustand an der S0-Geometrie und dem S1-
Zustand an der T1-Geometrie ermittelt und als Näherung für die adiabatische Energie des S1-Zu-
standes gewählt.
Abbildung 13: Molekül 1: Dipolmomente: blau = S0, rot = S1, lila = T1, orange = T2 und grün = T3
Ergebnisse und Auswertung 24
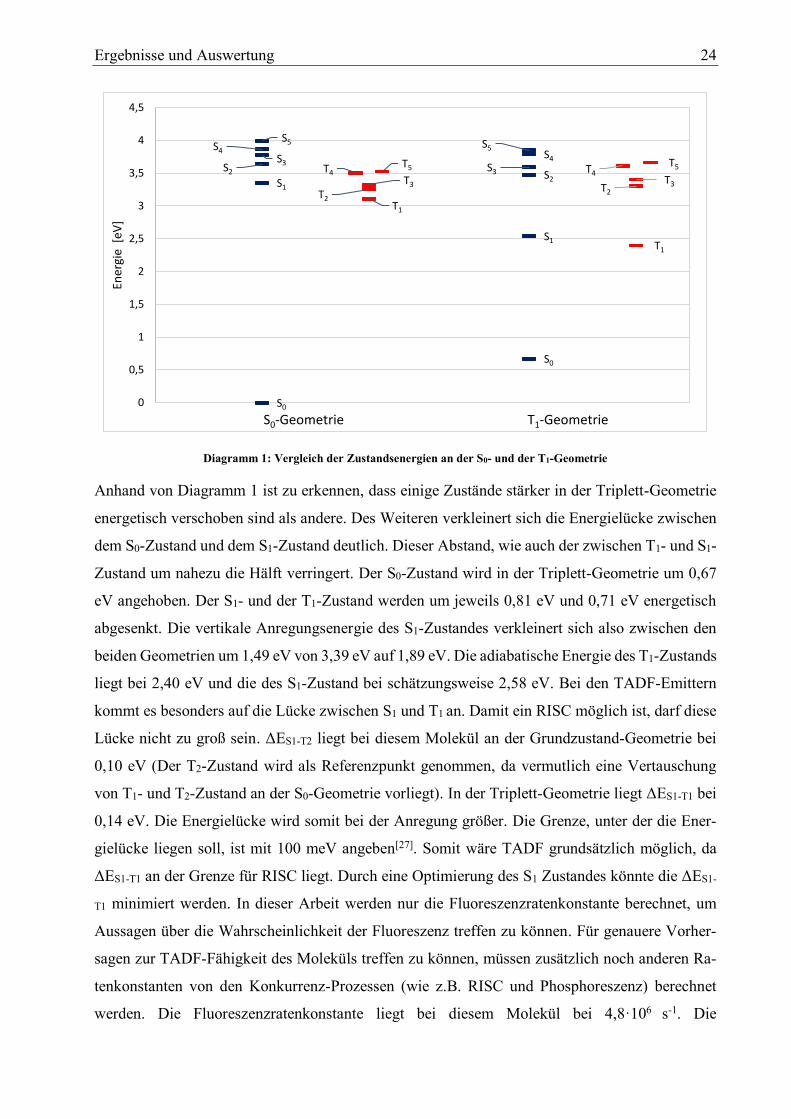
Diagramm 1: Vergleich der Zustandsenergien an der S0- und der T1-Geometrie
Anhand von Diagramm 1 ist zu erkennen, dass einige Zustände stärker in der Triplett-Geometrie
energetisch verschoben sind als andere. Des Weiteren verkleinert sich die Energielücke zwischen
dem S0-Zustand und dem S1-Zustand deutlich. Dieser Abstand, wie auch der zwischen T1- und S1-
Zustand um nahezu die Hälft verringert. Der S0-Zustand wird in der Triplett-Geometrie um 0,67
eV angehoben. Der S1- und der T1-Zustand werden um jeweils 0,81 eV und 0,71 eV energetisch
abgesenkt. Die vertikale Anregungsenergie des S1-Zustandes verkleinert sich also zwischen den
beiden Geometrien um 1,49 eV von 3,39 eV auf 1,89 eV. Die adiabatische Energie des T1-Zustands
liegt bei 2,40 eV und die des S1-Zustand bei schätzungsweise 2,58 eV. Bei den TADF-Emittern
kommt es besonders auf die Lücke zwischen S1 und T1 an. Damit ein RISC möglich ist, darf diese
Lücke nicht zu groß sein. ΔES1-T2 liegt bei diesem Molekül an der Grundzustand-Geometrie bei
0,10 eV (Der T2-Zustand wird als Referenzpunkt genommen, da vermutlich eine Vertauschung
von T1- und T2-Zustand an der S0-Geometrie vorliegt). In der Triplett-Geometrie liegt ΔES1-T1 bei
0,14 eV. Die Energielücke wird somit bei der Anregung größer. Die Grenze, unter der die Ener-
gielücke liegen soll, ist mit 100 meV angeben[27]. Somit wäre TADF grundsätzlich möglich, da
ΔES1-T1 an der Grenze für RISC liegt. Durch eine Optimierung des S1 Zustandes könnte die ΔES1-
T1 minimiert werden. In dieser Arbeit werden nur die Fluoreszenzratenkonstante berechnet, um
Aussagen über die Wahrscheinlichkeit der Fluoreszenz treffen zu können. Für genauere Vorher-
sagen zur TADF-Fähigkeit des Moleküls treffen zu können, müssen zusätzlich noch anderen Ra-
tenkonstanten von den Konkurrenz-Prozessen (wie z.B. RISC und Phosphoreszenz) berechnet
werden. Die Fluoreszenzratenkonstante liegt bei diesem Molekül bei 4,8·106 s-1. Die
S0
S1
S2
S3
S4
S5
T1
T2
T3
T4T5
S0
S1
S2
S3
S4
S5
T1
T2
T3
T4T5
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5En
ergi
e [
eV]
S0-Geometrie T1-Geometrie

Ergebnisse und Auswertung 25
Emissionswellenlänge liegt bei dem S1-Zustand bei 663 nm und bei dem T1-Zustand bei 717 nm.
Beide liegen im roten Lichtbereich an der Grenze zum infraroten Licht. Um Tendenzen vorhersa-
gen zu können werden im Folgenden Substitutionen an der Diarylphospin-Einheit vorgenommen.
Zum einen ist es interessant, ob sich durch die Substitution die ΔES1-T1 verändert, oder auch das
Dipolmoment oder die Differenzdichten. Die Gesamtheit dieser Faktoren könnte zur Verbesserung
der TADF-Eigenschaften führen. Außerdem kann gegebenenfalls die Emissionswellenlänge ver-
ändert werden und somit ein breiteres Spektrum an Farben gewonnen werden. Es werden Fluor-,
Methyl- und t-Butyl-Einheiten substituiert. Die Wirkung geht also von elektronenziehenden zu
elektronenschiebenden Substituenten.
4.2 Fluor-Substitution
An die beiden Phenylringe wird jeweils in para-Stellung ein Fluor substituiert.
Abbildung 14: Struktur S0-Geometrie des Moleküls 2
Die Nummerierung und die Kolorierung der Atome bleibt die selbe wie bei dem Standard Molekül,
wobei die Fluoratome blassgrün dargestellt werden und die Nummern 66 und 67 tragen. Im Wei-
teren wird dieses Molekül abgekürzt mit Molekül 2.
4.2.1 Geometrie
Die Geometrie im Grundzustand sowie in der T1-Geomtrie hat sich im Vergleich zu dem Molekül
1 durch die Substitution kaum verändert. Die Bindungslängen, die Bindungswinkel und die Die-
derwinkel, mit den größten Abweichungen zwischen den beiden berechneten Geometrien, werden
im Folgenden aufgeführt:
Tabelle 12: Bindungslängen Molekül 2 Tabelle 13: Bindungswinkel Molekül 2
Bindungen [pm] S0 T1 S0- T1
P1-Cu2 219,1 233,6 -14,5
Cu2-Cl3 219,9 214,5 5,4
Cu2-N4 206,7 189,8 16,9
N4-C19 134,8 140,9 -6,1
Winkel [°] So T1 S0-T1
P1-Cu2-Cl3 126,9 104,6 22,3
P1-Cu2-N4 122,2 106,3 15,9
Cu2-P1-C5 108,3 114,4 -6,1
Cu2-P1-C36 116,3 106,8 9,5
Cl3-Cu2-N4 107,6 148,7 -41,1
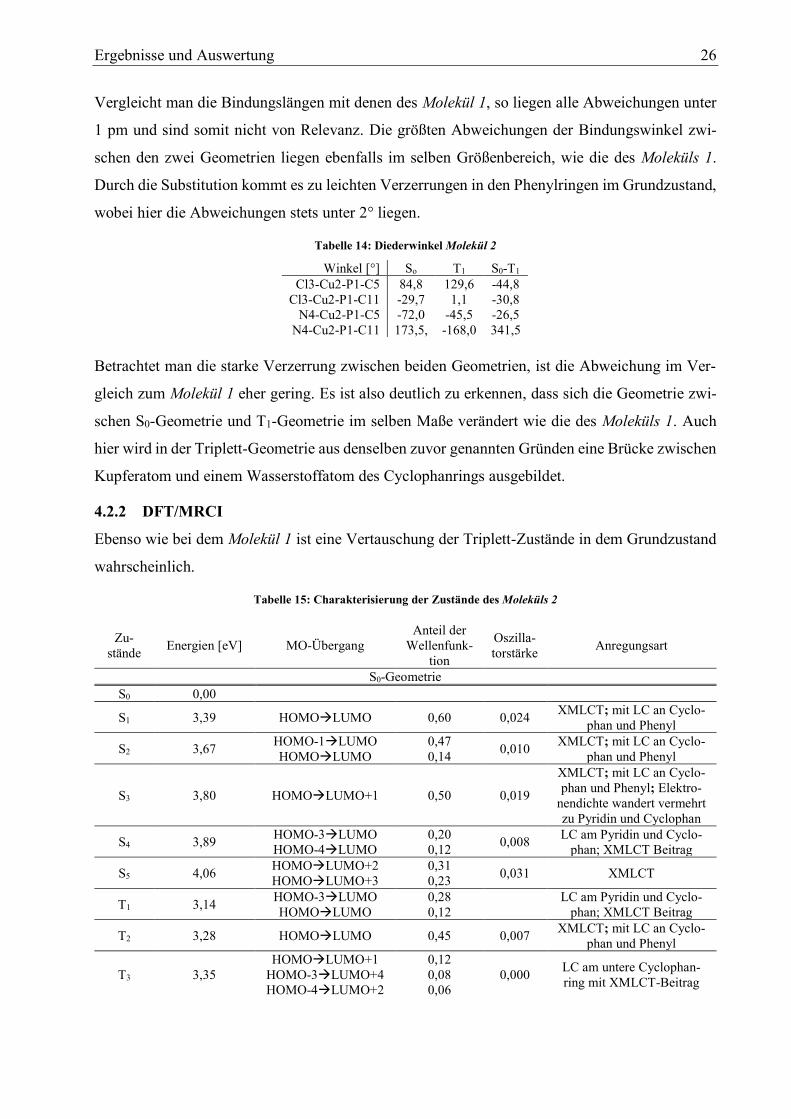
Ergebnisse und Auswertung 26
Vergleicht man die Bindungslängen mit denen des Molekül 1, so liegen alle Abweichungen unter
1 pm und sind somit nicht von Relevanz. Die größten Abweichungen der Bindungswinkel zwi-
schen den zwei Geometrien liegen ebenfalls im selben Größenbereich, wie die des Moleküls 1.
Durch die Substitution kommt es zu leichten Verzerrungen in den Phenylringen im Grundzustand,
wobei hier die Abweichungen stets unter 2° liegen.
Tabelle 14: Diederwinkel Molekül 2
Betrachtet man die starke Verzerrung zwischen beiden Geometrien, ist die Abweichung im Ver-
gleich zum Molekül 1 eher gering. Es ist also deutlich zu erkennen, dass sich die Geometrie zwi-
schen S0-Geometrie und T1-Geometrie im selben Maße verändert wie die des Moleküls 1. Auch
hier wird in der Triplett-Geometrie aus denselben zuvor genannten Gründen eine Brücke zwischen
Kupferatom und einem Wasserstoffatom des Cyclophanrings ausgebildet.
4.2.2 DFT/MRCI
Ebenso wie bei dem Molekül 1 ist eine Vertauschung der Triplett-Zustände in dem Grundzustand
wahrscheinlich.
Tabelle 15: Charakterisierung der Zustände des Moleküls 2
Zu-
stände Energien [eV] MO-Übergang
Anteil der
Wellenfunk-
tion
Oszilla-
torstärke Anregungsart
S0-Geometrie
S0 0,00
S1 3,39 HOMOLUMO 0,60 0,024 XMLCT; mit LC an Cyclo-
phan und Phenyl
S2 3,67 HOMO-1LUMO
HOMOLUMO
0,47
0,14 0,010
XMLCT; mit LC an Cyclo-
phan und Phenyl
S3 3,80 HOMOLUMO+1 0,50 0,019
XMLCT; mit LC an Cyclo-
phan und Phenyl; Elektro-
nendichte wandert vermehrt
zu Pyridin und Cyclophan
S4 3,89 HOMO-3LUMO
HOMO-4LUMO
0,20
0,12 0,008
LC am Pyridin und Cyclo-
phan; XMLCT Beitrag
S5 4,06 HOMOLUMO+2
HOMOLUMO+3
0,31
0,23 0,031 XMLCT
T1 3,14 HOMO-3LUMO
HOMOLUMO
0,28
0,12
LC am Pyridin und Cyclo-
phan; XMLCT Beitrag
T2 3,28 HOMOLUMO 0,45 0,007 XMLCT; mit LC an Cyclo-
phan und Phenyl
T3 3,35
HOMOLUMO+1
HOMO-3LUMO+4
HOMO-4LUMO+2
0,12
0,08
0,06
0,000 LC am untere Cyclophan-
ring mit XMLCT-Beitrag
Winkel [°] So T1 S0-T1
Cl3-Cu2-P1-C5 84,8 129,6 -44,8
Cl3-Cu2-P1-C11 -29,7 1,1 -30,8
N4-Cu2-P1-C5 -72,0 -45,5 -26,5
N4-Cu2-P1-C11 173,5, -168,0 341,5
Ergebnisse und Auswertung 27
Zu-
stände Energien [eV] MO-Übergang
Anteil der
Wellenfunk-
tion
Oszilla-
torstärke Anregungsart
T4 3,53 HOMO-1LUMO
HOMOLUMO
0,37
0,15 0,000
XMLCT; mit LC an Cyclo-
phan und Phenyl
T5 3,55 HOMO-4LUMO
HOMOLUMO+1
0,20
0,12 0,001
LC am Pyridin und Cyclo-
phan; XMLCT Beitrag
T1-Geometrie
S0 0,68
S1 1,89 HOMOLUMO 0,74 0,031 XMLCT; mit LC an Cyclo-
phan und Phenyl
S2 2,83 HOMOLUMO+1 0,67 0,006 XMLCT; mit LC an Cyclo-
phan und Phenyl
S3 2,96 HOMO-1LUMO 0,55 0,052 XMLCT; mit LC an Cyclo-
phan und Phenyl
S4 3,15
HOMO-2LUMO
HOMO-4LUMO
HOMO-6LUMO
0,40
0,11
0,10
0,020 XMLCT; mit LC an Cyclo-
phan und Phenyl
S5 3,20 HOMOLUMO+2 0,75 0,031 XMLCT; Übergang zum
Cyclophan
T1 1,75 HOMOLUMO 0,75 XMLCT; mit LC an Cyclo-
phan und Phenyl
T2 2,66 HOMO-1LUMO 0,52 0,004 XMLCT; mit LC an Cyclo-
phan und Phenyl
T3 2,75 HOMOLUMO+1 0,66 0,021 XMLCT; mit LC an Cyclo-
phan und Phenyl
T4 2,96
HOMO-4LUMO
HOMO-2LUMO
HOMO-7LUMO
0,22
0,17
0,13
0,009 XMLCT; mit LC an Cyclo-
phan und Phenyl
T5 3,01 HOMOLUMO+2 0,71 0,04 XMLCT; Übergang zum
Cyclophan
Die Differenzdichten sind fast identisch mit denen des Moleküls 1. Es ist also nicht mit einem
größeren Charge-Transfer-Übergang oder Dipolmoment zu rechnen.
Tabelle 16: Dipolmomente Molekül 2
Komponente
Zustand x y z Dipolmoment [D]
S0 -2,259 2,139 1,540 8,822
S1 -1,849 -2,503 1,211 8,488
T1 -2,533 1,468 1,475 8,334
T2 -1,945 -1,787 1,226 7,401
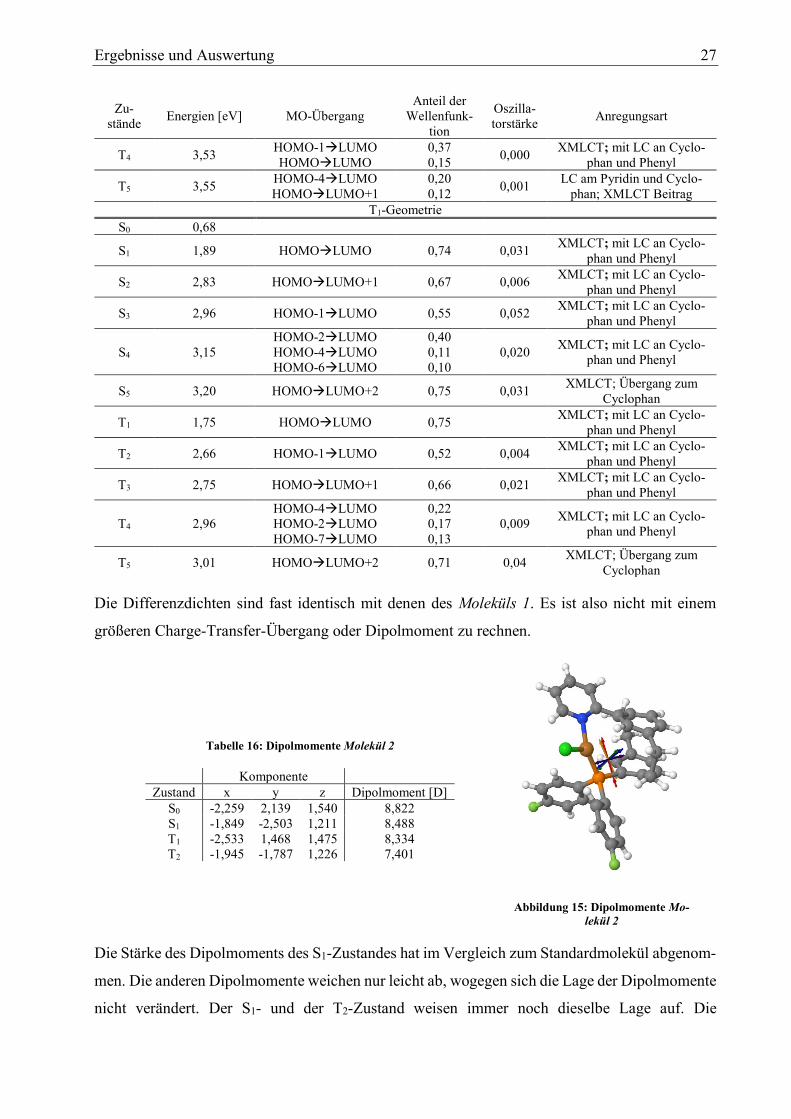
Abbildung 15: Dipolmomente Mo-
lekül 2
Die Stärke des Dipolmoments des S1-Zustandes hat im Vergleich zum Standardmolekül abgenom-
men. Die anderen Dipolmomente weichen nur leicht ab, wogegen sich die Lage der Dipolmomente
nicht verändert. Der S1- und der T2-Zustand weisen immer noch dieselbe Lage auf. Die
Ergebnisse und Auswertung 28
Differenzdichten sind auch hier fast identisch mit denen des Moleküls 1. Die Differenzdichten und
die Dipolmomente werden durch die Substitution kaum beeinflusst. Um die Auswirkung der Sub-
stitution einer stark elektronenziehenden Gruppe besser beurteilen zu können, wird im Diagramm
2 die energetische Lage der Zustände dargestellt.
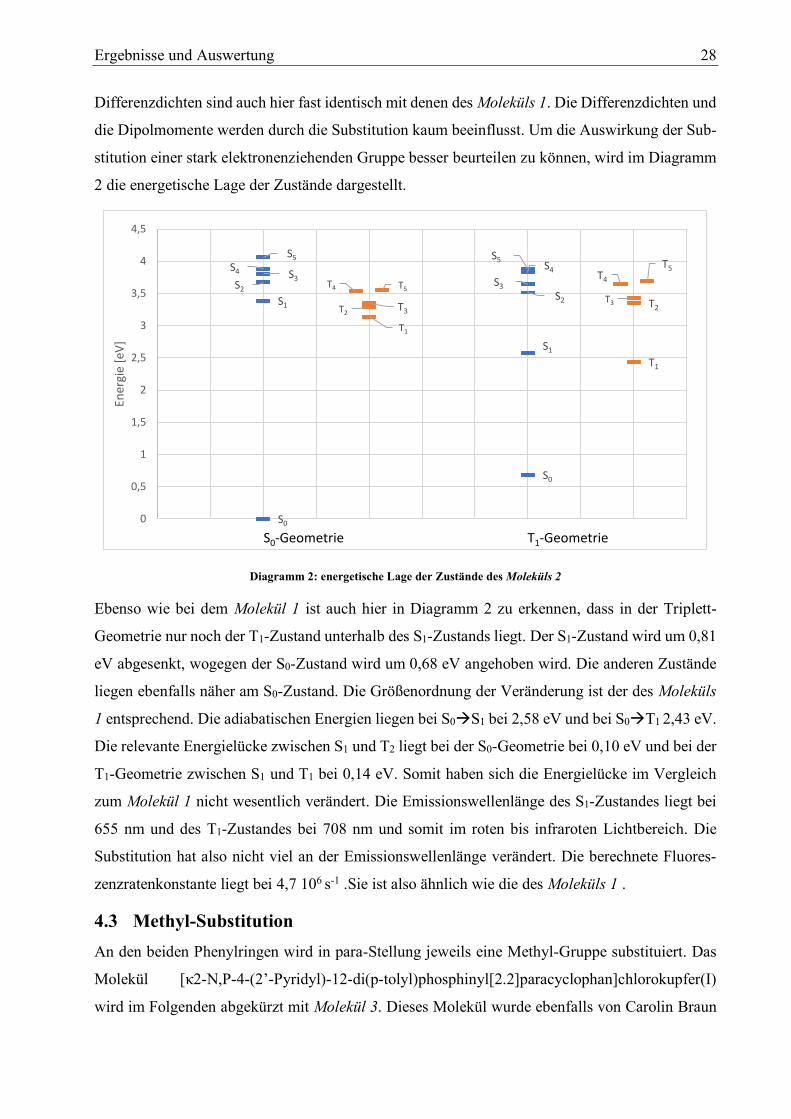
Diagramm 2: energetische Lage der Zustände des Moleküls 2
Ebenso wie bei dem Molekül 1 ist auch hier in Diagramm 2 zu erkennen, dass in der Triplett-
Geometrie nur noch der T1-Zustand unterhalb des S1-Zustands liegt. Der S1-Zustand wird um 0,81
eV abgesenkt, wogegen der S0-Zustand wird um 0,68 eV angehoben wird. Die anderen Zustände
liegen ebenfalls näher am S0-Zustand. Die Größenordnung der Veränderung ist der des Moleküls
1 entsprechend. Die adiabatischen Energien liegen bei S0S1 bei 2,58 eV und bei S0T1 2,43 eV.
Die relevante Energielücke zwischen S1 und T2 liegt bei der S0-Geometrie bei 0,10 eV und bei der
T1-Geometrie zwischen S1 und T1 bei 0,14 eV. Somit haben sich die Energielücke im Vergleich
zum Molekül 1 nicht wesentlich verändert. Die Emissionswellenlänge des S1-Zustandes liegt bei
655 nm und des T1-Zustandes bei 708 nm und somit im roten bis infraroten Lichtbereich. Die
Substitution hat also nicht viel an der Emissionswellenlänge verändert. Die berechnete Fluores-
zenzratenkonstante liegt bei 4,7 106 s-1 .Sie ist also ähnlich wie die des Moleküls 1 .
4.3 Methyl-Substitution
An den beiden Phenylringen wird in para-Stellung jeweils eine Methyl-Gruppe substituiert. Das
Molekül [κ2-N,P-4-(2’-Pyridyl)-12-di(p-tolyl)phosphinyl[2.2]paracyclophan]chlorokupfer(I)
wird im Folgenden abgekürzt mit Molekül 3. Dieses Molekül wurde ebenfalls von Carolin Braun
S0
S1
S2
S3S4
S5
T1
T2T3
T4 T5
S0
S1
S2
S3
S4
S5
T1
T2T3
T4
T5
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
Ener
gie
[eV
]
S0-Geometrie T1-Geometrie

Ergebnisse und Auswertung 29
während ihrer Doktorarbeit synthetisiert[26]. Es gibt jedoch keine Kristallstrukturen zu diesem Mo-
lekül. Die Nummerierung und Kolorierung der Atome bleibt dieselbe, wie bei dem Molekül 1,
wobei die zusätzlichen C-Atome die Nummern 68 und 64 erhalten. Bei diesem Molekül ergaben
sich einige Probleme bei der Berechnung des absoluten Minimums dieser Struktur. Die Schwin-
gung der Methylgruppen erschwerte die Suche. Um auf ein absolutes Minimum zukommen, wurde
mit dem bereits erwähnten TURBOMOLE Unterprogramm VIBRATION gearbeitet. Trotz des
Einsatzes dieses Programmes konnte kein Minimum gefunden werden. Anschließend wurde das
Grid von m3 auf m5 hochgesetzt, aber auch hier endete die Berechnung auf einen Sattelpunkt. Die
Schwingungsfrequenz der Berechnung mit einem Grid von m5 erreichte den Sattelpunkt mit der
kleinsten imaginären Frequenz. Die Torsionsschwingung der Methylgruppen stellten eine Heraus-
forderung dar. Deshalb wurde diese Geometrie mit der kleinsten imaginären Frequenz als Grund-
zustands-Geometrie verwendet. Bei der Triplett-Geometrie taucht dieses Problem nicht auf. Die
Rechnungen führten sofort in ein absolutes Minimum.
Abbildung 16: Molekül 3
4.3.1 Geometrie
Im Vergleich zum Molekül 1 hat sich durch diese Substitution nur geringfügige Änderungen an
der Geometrie ergeben. Die Bindungslängen und die Bindungswinkel, die sich am stärksten in den
beiden Geometrien verändern, bleiben dieselben wie beim Molekül 1. Die Abweichungen liegen
alle unterhalb von 1 pm bei den Bindungslängen und bei den Bindungswinkeln unterhalb von 3°,
wobei sich hauptsächlich die Bindungswinkel in den Phenylringen verändern.
Tabelle 17: Bindungslängen Molekül 3
Bindungen [pm] S0 T1 S0- T1
P1-Cu2 219,3 233,7 -14,4
Cu2-Cl3 219,6 214,3 5,3
Cu2-N4 207,8 190,7 17,1
N4-C19 134,8 140,9 -6,1
Tabelle 18: Bindungswinkel Molekül 3
Winkel [°] So T1 S0-T1
P1-Cu2-Cl3 128,6 106,3 22,3
P1-Cu2-N4 121,1 106,6 14,5
Cu2-P1-C36 116,1 107,3 8,8
Cl3-Cu2-N4 107,2 146,7 -39,5
Ebenso wie die Bindungslängen und die Bindungswinkel verändern sich auch die Diederwinkel
im Vergleich zum Molekül 1 kaum. Auch hier wird in der Triplett-Geometrie eine Bindung zwi-
schen dem Kupferatom und einem Wasserstoffatom des Cyclophanrings ausgebildet.
Ergebnisse und Auswertung 30
Tabelle 19: Diederwinkel des Moleküls 3
Die Abweichungen in den beiden Geometrien begründen sich somit in denselben Gründen, wie
die des Moleküls 1.
4.3.2 DFT/MRCI
Genau wie bei den beiden vorherigen Molekülen ist auch hier im Grundzustand die Reihenfolge
der Triplett-Zustände verwechselt. Der T2-Zustand an der S0-Geometrie ist wahrscheinlich der ei-
gentlich energetisch niedrigste Zustand. Es wird die Energielücke an der Grundzustands-Geomet-
rie zwischen S1- und T2-Zustand berechnet, um sie mit der Energielücke an der Triplett-Geomet-
rie vergleichen zu können. Im Folgenden sind die Zustände klassifiziert. Die relevanten Anregun-
gen von über zehn Prozent Anteil an der Wellenfunktion werden aufgeführt und deren Oszillator-
stärke angegeben. Außerdem wird die Anregungsart beschrieben.
Tabelle 20: Charakterisierung der Zustände des Moleküls 3
Zu-
stände Energie [eV] MO-Übergang
Anteil Wel-
lenfunktion
Os-
zilla-
tor-
stärke
Anregungsart
S0-Geometrie
S0 0,00
S1 3,30
HOMOLUMO 0,69 0,024 XMLCT; mit LC an Cyclophan
und Phenyl
S2 3,64 HOMO-1LUMO
HOMOLUMO
0,59
0,10 0,010
XMLCT; mit LC an Cyclophan
und Phenyl
S3
3,77
HOMOLUMO+1 0,58 0,018
XMLCT; mit LC an Cyclophan
und Phenyl; Elektronendichte
wandert vermehrt zu Pyridin und
Cyclophan
S4
3,87 HOMO-3LUMO
HOMO-4LUMO
HOMO-
3LUMO+1
0,20
0,13
0,12
0,007 LC am Pyridin und Cyclophan;
XMLCT Beitrag
S5 3,98 HOMOLUMO+2
HOMOLUMO+3
0,32
0,31 0,035 XMLCT
T1 3,11 HOMO-3LUMO
HOMOLUMO
0,27
0,21
LC am Pyridin und Cyclophan;
XMLCT Beitrag
T2 3,22
HOMOLUMO 0,40 0,009 XMLCT; mit LC an Cyclophan
und Phenyl
T3
3,35 HOMOLUMO+1
HOMO-
3LUMO+4
0,12
0,11 0,001
LC am untere Cyclophanring mit
XMLCT-Beitrag
T4 3,51 HOMO-1LUMO
HOMOLUMO
0,46
0,12 0,001
XMLCT; mit LC an Cyclophan
und Phenyl
Winkel [°] So T1 S0-T1
Cl3-Cu2-P1-C5 87,2 126,6 -39,4
Cl3-Cu2-P1-C11 -28,0 4,2 -32,2
N4-Cu2-P1-C5 -70,0 -48,1 -21,9
N4-Cu2-P1-C11 174,7, -170,6 345,3
Ergebnisse und Auswertung 31
Zu-
stände Energie [eV] MO-Übergang
Anteil Wel-
lenfunktion
Os-
zilla-
tor-
stärke
Anregungsart
T5
3,54 HOMO-4LUMO
HOMO-
3LUMO+2
0,205
0,11 0,001
LC am Pyridin und Cyclophan;
XMLCT Beitrag
T1-Geometrie
S0 0,66
S1 2,53
HOMOLUMO 0,73 0,033 XMLCT; mit LC an Cyclophan
und Phenyl
S2 3,47
HOMOLUMO+1 0,67 0,006 XMLCT; mit LC an Cyclophan
und Phenyl
S3 3,60
HOMO-1LUMO 0,56 0,056 XMLCT; mit LC an Cyclophan
und Phenyl
S4 3,78 HOMO-2LUMO
HOMO-5LUMO
0,29
0,13 0,018
XMLCT; mit LC an Cyclophan
und Phenyl
S5 3,86
HOMOLUMO+2 0,74 0,033 XMLCT; Übergang zum Cyclo-
phan
T1 2,39
HOMOLUMO 0,74 XMLCT; mit LC an Cyclophan
und Phenyl
T2 3,32
HOMO-1LUMO 0,53 0,004 XMLCT; mit LC an Cyclophan
und Phenyl
T3 3,39
HOMOLUMO+1 0,67 0,021 XMLCT; mit LC an Cyclophan
und Phenyl
T4
3,61 HOMO-4LUMO
HOMO-2LUMO
HOMO-9LUMO
0,19
0,16
0,12
0,009 XMLCT; mit LC an Cyclophan
und Phenyl
T5 3,68
HOMOLUMO+2 0,71 0,003 XMLCT; Übergang zum Cyclo-
phan
An der S0-Geometrie bestehen die Zustände aus denselben relevanten Anregungen wie bei dem
Molekül 2 und dem Molekül 1. Die Anteile an der Wellenfunktion weichen jedoch ab. In der
Triplett-Geometrie weichen die Zustände S4 und T4 leicht von der Zusammensetzung des Moleküls
1 ab, wobei die stärksten Anteile an der Wellenfunktion gleichbleiben. Die erstellten Diffe-
renzdichten weisen nur minimale Veränderungen auf. Auch hier wurde die energetische Lage der
Zustände nicht stark durch die Substitution beeinflusst. Betrachtet man die Dipolmomente der Zu-
stände, fällt auf, dass das Dipolmoment des S1-Zustandes größenbedingt wieder näher an dem des
Moleküls 1 liegt.
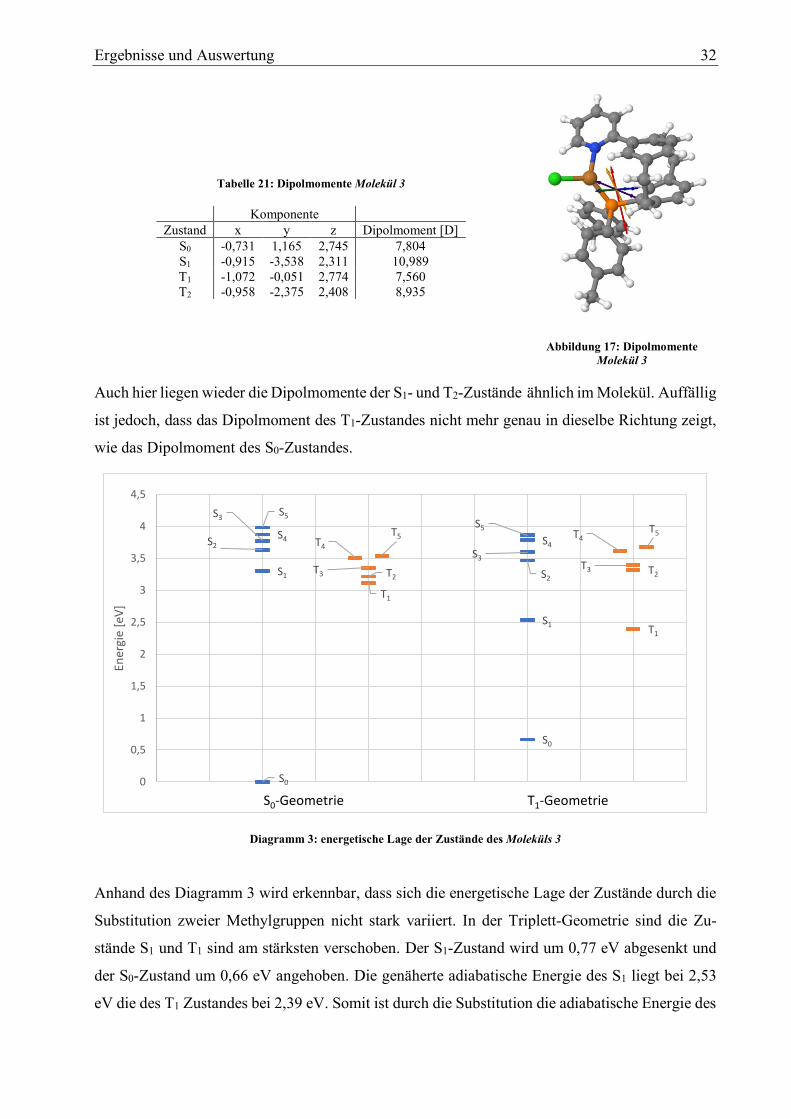
Ergebnisse und Auswertung 32
Tabelle 21: Dipolmomente Molekül 3
Komponente
Zustand x y z Dipolmoment [D]
S0 -0,731 1,165 2,745 7,804
S1 -0,915 -3,538 2,311 10,989
T1 -1,072 -0,051 2,774 7,560
T2 -0,958 -2,375 2,408 8,935
Abbildung 17: Dipolmomente
Molekül 3
Auch hier liegen wieder die Dipolmomente der S1- und T2-Zustände ähnlich im Molekül. Auffällig
ist jedoch, dass das Dipolmoment des T1-Zustandes nicht mehr genau in dieselbe Richtung zeigt,
wie das Dipolmoment des S0-Zustandes.
Diagramm 3: energetische Lage der Zustände des Moleküls 3
Anhand des Diagramm 3 wird erkennbar, dass sich die energetische Lage der Zustände durch die
Substitution zweier Methylgruppen nicht stark variiert. In der Triplett-Geometrie sind die Zu-
stände S1 und T1 sind am stärksten verschoben. Der S1-Zustand wird um 0,77 eV abgesenkt und
der S0-Zustand um 0,66 eV angehoben. Die genäherte adiabatische Energie des S1 liegt bei 2,53
eV die des T1 Zustandes bei 2,39 eV. Somit ist durch die Substitution die adiabatische Energie des
S0
S1
S2
S3
S4
S5
T1
T2T3
T4
T5
S0
S1
S2
S3
S4
S5
T1
T2T3
T4T5
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
Ener
gie
[eV
]
S0-Geometrie T1-Geometrie

Ergebnisse und Auswertung 33
T1-Zustandes verringert, wobei die des S1-Zustandes konstant ist. Die Energielücke S1-T2 im
Grundzustand beträgt 0,08eV und die Energielücke S1-T1 in der T1-Geometrie 0,14 eV. Im Ver-
gleich zum Molekül 1 ist die Energielücke an der Grundzustands-Geometrie kleiner geworden. An
der Triplett-Geometrie bleibt sie jedoch exakt gleich groß. Die Emissionswellenlänge von S1 be-
trägt 664 nm und die von T1 beträgt 717 nm. Der Emissionswellenlängenbereich hat sich durch
die Substitution nicht verändert. Die Fluoreszenzratenkonstante beträgt bei diesem Molekül
5,0·106 s-1.
4.4 T-Butyl-Substitution
Anschließend wird eine elektronenschiebende Einheit substituiert, die auch sterisch anspruchsvol-
ler ist als die vorherigen Substitutions-Einheiten. Eine t-Butyl-Einheit wird an je einen Phenylring
in para-Stellung angebracht. Dieses Molekül wird mit Molekül 4 abgekürzt. Die Nummerierung
Kolorierung der Atome entsprechen denen des Moleküls 1. Die zusätzlichen C-Atome werden mit
den Nummern 64, 65, 69, 73, 77, 79, 82 und 86 beziffert.
Abbildung 18: Struktur des Moleküls 4 an der S0-Geometrie
4.4.1 Geometrie
Ebenso wie bei den zuvor beschriebenen Molekülen ist die Abweichung zwischen Grundzustands-
geometrie und Triplett-Geometrie auf Grund der zusätzlichen Bindung am Kupferatom groß. Die
Veränderung durch die Substitution ist im Vergleich zum Molekül 1 jedoch gering. In den folgen-
den Tabellen sind die Bindungslängen (Abweichung größer als 5 pm), die Bindungswinkel (Ab-
weichung größer als 5°) und die relevanten Diederwinkel aufgeführt:
Tabelle 22: Bindungslängen Molekül 4 Tabelle 23: Bindungswinkel Molekül 4
Bindungen [pm] S0 T1 S0- T1
P1-Cu2 219,0 233,6 -14,6
Cu2-Cl3 219,7 214,5 5,2
Cu2-N4 208,1 190,0 18,1
N4-C19 134,7 140,9 -6,2
Winkel [°] So T1 S0-T1
P1-Cu2-Cl3 128,8 105,2 23,6
P1-Cu2-N4 121,6 106,7 14,9
Cu2-P1-C5 108,7 114,6 -5,9
Cu2-P1-C36 116,1 106,8 9,3
Cl3-Cu2-N4 106,5 147,9 -41,4
Ergebnisse und Auswertung 34
Tabelle 24: Diederwinkel Molekül 4
Winkel [°] So T1 S0-T1
Cl3-Cu2-P1-C5 86,2 130,0 -43,8
Cl3-Cu2-P1-C11 -28,6 7,3 -35,9
N4-Cu2-P1-C5 -71,1 -45,7 -25,4
N4-Cu2-P1-C11 174,1 -168,4 342,5
4.4.2 DFT/MRCI
Im Vergleich zum Molekül 1 haben sich die Zustände S2 und S3 an der S0-Geometrie in ihrer
Zusammensetzung geändert (Tabelle 24). Ihre Differenzdichten weisen jedoch keine großen Ab-
weichungen auf. An der T1-Geomtrie weicht nur der S5-Zustand in der Zusammensetzung ab. Die
Anteile an der Wellenfunktion verändern sich leicht.
Tabelle 25: Charakterisierung der Zustände des Moleküls 4
Zu-
stände Energie [eV] MO-Übergang
Anteil Wel-
lenfunktion
Oszilla-
torstärke Anregungsart
S0-Geometrie
S0 0,00
S1 3,32 HOMOLUMO 0,63 0,024 XMLCT; mit LC an Cyclophan
und Phenyl
S2 3,64 HOMO-1LUMO
HOMOLUMO
0,54
0,12 0,009
XMLCT; mit LC an Cyclophan
und Phenyl
S3 3,79 HOMO-2LUMO 0,54 0,017
XMLCT; mit LC an Cyclophan
und Phenyl; Elektronendichte
wandert vermehrt zu Pyridin
und Cyclophan
S4 3,88
HOMO-3LUMO
HOMO-4LUMO
HOMO-3LUMO+1
0,18
0,13
0,12
0,008 LC am Pyridin und Cyclophan;
XMLCT Beitrag
S5 4,00 HOMOLUMO+2
HOMOLUMO+3
0,34
0,25 0,035 XMLCT
T1 3,12 HOMO-3LUMO
HOMOLUMO
0,29
0,17
LC am Pyridin und Cyclophan;
XMLCT Beitrag
T2 3,24 HOMOLUMO 0,44 0,008 XMLCT; mit LC an Cyclophan
und Phenyl
T3 3,35 HOMOLUMO+1
HOMO-3LUMO+4
0,12
0,10 0,001
LC am untere Cyclophanring
mit XMLCT-Beitrag
T4 3,52 HOMO-1LUMO
HOMOLUMO
0,46
0,12 0,001
XMLCT; mit LC an Cyclophan
und Phenyl
T5 3,55 HOMO-4LUMO
HOMO-3LUMO+2
0,25
0,11 0,001
LC am Pyridin und Cyclophan;
XMLCT Beitrag
T1-Geometrie
S0 0,69
S1 2,55 HOMOLUMO 0,74 0,033 XMLCT; mit LC an Cyclophan
und Phenyl
S2 3,49 HOMOLUMO+1 0,67 0,007 XMLCT; mit LC an Cyclophan
und Phenyl
S3 3,62 HOMO-1LUMO 0,55 0,053 XMLCT; mit LC an Cyclophan
und Phenyl
S4 3,80 HOMO-2LUMO
HOMO-5LUMO
0,29
0,11 0,024
XMLCT; mit LC an Cyclophan
und Phenyl
S5 3,87 HOMOLUMO+3 0,75 0,032 XMLCT; Übergang zum Cyclo-
phan

Ergebnisse und Auswertung 35
Zu-
stände Energie [eV] MO-Übergang
Anteil Wel-
lenfunktion
Oszilla-
torstärke Anregungsart
T1 2,41 HOMOLUMO 0,75 XMLCT; mit LC an Cyclophan
und Phenyl
T2 3,35 HOMO-1LUMO 0,51 0,004 XMLCT; mit LC an Cyclophan
und Phenyl
T3 3,42 HOMOLUMO+1 0,66 0,021 XMLCT; mit LC an Cyclophan
und Phenyl
T4 3,63
HOMO-4LUMO
HOMO-2LUMO
HOMO-9LUMO
0,21
0,17
0,12
0,010 XMLCT; mit LC an Cyclophan
und Phenyl
T5 3,69 HOMOLUMO+3 0,72 0,003 XMLCT; Übergang zum Cyclo-
phan
Auch die Dipolmomente des S1- und T2-Zustands sind ähnlich im Molekül ausgerichtet, wie man
anhand von Abbildung 19 erkennen kann. Durch die Substitution einer elektronenschiebenden
Gruppe, wird das Dipolmoment des S1-Zustandes anscheinend verstärkt (siehe Tabelle 25). Die
Ausrichtungen der Dipolmomente verändert sich durch diese Substitution jedoch nicht. Die ener-
getische Lage der Zustände ändert sich durch die Substitution im Vergleich zum Molekül 1 kaum.
Tabelle 26: Dipolmomente Molekül 4
Komponenten
Zustand x y z Dipolmoment [D]
S0 -1,915 1,034 2,081 7,655
S1 -1,362 -3,695 1,884 11,096
T1 -2,135 0,084 2,047 7,522
T2 -1,504 -2,815 1,882 9,417
Abbildung 19: Dipolmomente Molekül 4
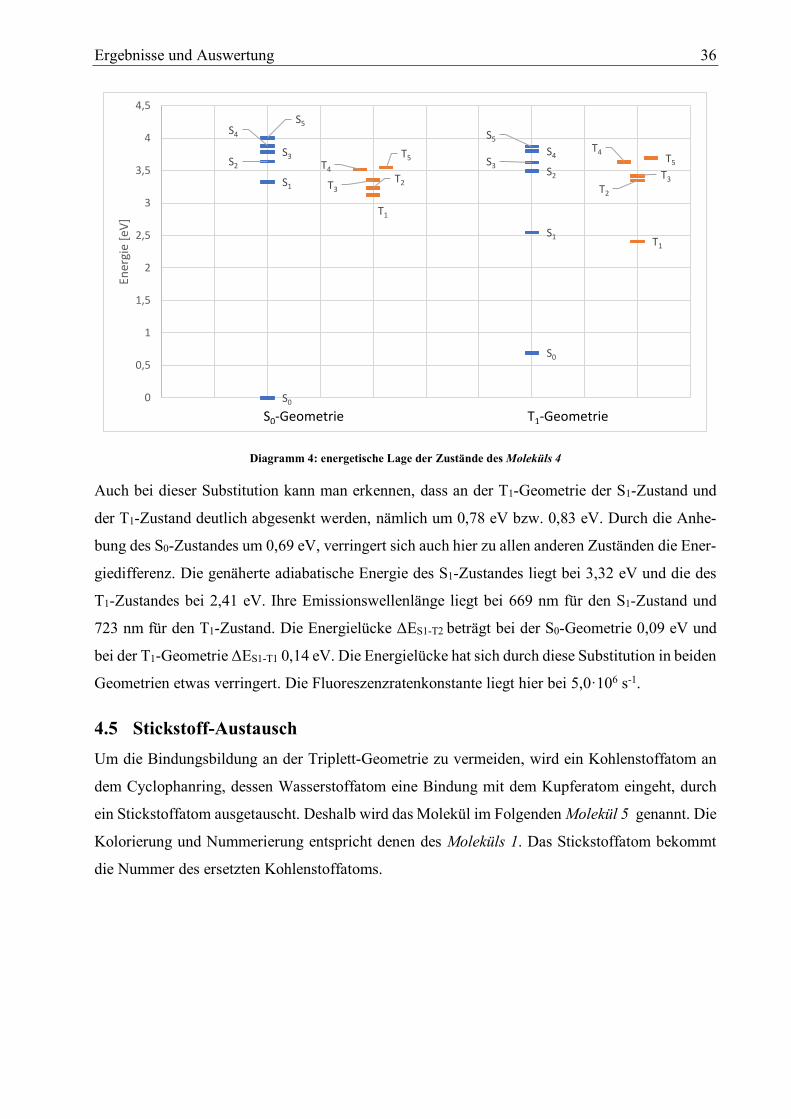
Ergebnisse und Auswertung 36
Diagramm 4: energetische Lage der Zustände des Moleküls 4
Auch bei dieser Substitution kann man erkennen, dass an der T1-Geometrie der S1-Zustand und
der T1-Zustand deutlich abgesenkt werden, nämlich um 0,78 eV bzw. 0,83 eV. Durch die Anhe-
bung des S0-Zustandes um 0,69 eV, verringert sich auch hier zu allen anderen Zuständen die Ener-
giedifferenz. Die genäherte adiabatische Energie des S1-Zustandes liegt bei 3,32 eV und die des
T1-Zustandes bei 2,41 eV. Ihre Emissionswellenlänge liegt bei 669 nm für den S1-Zustand und
723 nm für den T1-Zustand. Die Energielücke ΔES1-T2 beträgt bei der S0-Geometrie 0,09 eV und
bei der T1-Geometrie ΔES1-T1 0,14 eV. Die Energielücke hat sich durch diese Substitution in beiden
Geometrien etwas verringert. Die Fluoreszenzratenkonstante liegt hier bei 5,0·106 s-1.
4.5 Stickstoff-Austausch
Um die Bindungsbildung an der Triplett-Geometrie zu vermeiden, wird ein Kohlenstoffatom an
dem Cyclophanring, dessen Wasserstoffatom eine Bindung mit dem Kupferatom eingeht, durch
ein Stickstoffatom ausgetauscht. Deshalb wird das Molekül im Folgenden Molekül 5 genannt. Die
Kolorierung und Nummerierung entspricht denen des Moleküls 1. Das Stickstoffatom bekommt
die Nummer des ersetzten Kohlenstoffatoms.
S0
S1
S2
S3
S4
S5
T1
T2T3
T4
T5
S0
S1
S2
S3
S4
S5
T1
T2
T3
T4T5
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
Ener
gie
[eV
]
S0-Geometrie T1-Geometrie

Ergebnisse und Auswertung 37
Abbildung 20: Aufbau Molekül 5
4.5.1 Geometrie
Die Geometrie verändert sich stark im Vergleich zum Molekül 1. Durch den Austausch von einem
Kohlenstoffatom durch Stickstoffatom sollte eigentlich eine zusätzliche Bindung des Kupferatoms
unterdrückt werden. Im Gegensatz zu dieser Annahme bildet sich jedoch auch schon in der Grund-
zustands-Geometrie diese Bindung aus. Die Abweichungen an der Grundzustands-Geometrie im
Vergleich zum Molekül 1 sind relativ groß. Die folgenden Tabellen 27-31 vergleichen die Struk-
turen der beiden Moleküle sowohl in der T1- als auch in der S0-Geometrie. Die Bindungslängen
über 4 pm sind in Tabelle 27 & 28 aufgeführt. In beiden Geometrien kommt es zu leichten Abwei-
chungen bei den Bindungslängen.
Tabelle 27: Bindungslängen an der S0-Geometrie im Vergleich Molekül 1 und Molekül 5
Bindungen [pm] Molekül 5 Molekül 1 Differenz
P1-Cu2 224 219,1 -4,9
C42-N47 134,8 140,3 5,5
C46-N47 134 139,8 5,8
Tabelle 28:Bindungslängen an der T1-Geometrie im Vergleich Molekül 1 und Molekül 5
Bindungen [pm] Molekül 5 Molekül 1 Differenz
P1-Cu2 233,6 241,4 -7,8
P1-C5 214,4 220,3 -5,9
P1-C11 189,8 197,8 -8
P1-C36 139,7 134,1 5,6
Ebenso wie bei den Bindungslängen sind auch bei den Bindungswinkeln (Winkel über 4°) in bei-
den Geometrien Abweichungen zu erkennen. Die Bindungswinkel des Kupferatoms scheinen sich
im Vergleich zu Molekül 1 deutlich zu ändern. Der Winkel Cl3-Cu2-N4 an der T1-Geometrie ver-
ändert sich stark. Es kommt zu deutlichen Verzerrungen in beiden Geometrien.
Ergebnisse und Auswertung 38
Tabelle 29: Bindungswinkel an der S0-Geometrie im Vergleich Molekül 1 und Molekül 5
Winkel [°] Molekül 5 Molekül 1 Differenz
C19-C42-C47 114,1 118,3 4,20
P1-Cu2-Cl3 113,3 128,2 14,90
P1-Cu2-N4 112,9 121,9 9,00
Cu2-P1-C11 121,8 117,7 -4,10
Cu2-P1-C36 106,4 115,9 9,50
Cu2-N4-C17 124,1 112,4 -11,70
Cu2-N4-C19 114,6 127,1 12,50
C11-P1-C36 108,2 101,6 -6,60
Tabelle 30:Bindungswinkel an der T1-Geometrie im Vergleich Molekül 1 und Molekül 5
Winkel [°] Molekül 5 Molekül 1 Differenz
C19-C42-C47 119,4 114,6 4,8
P1-Cu2-Cl3 104,5 100 4,5
P1-Cu2-N4 107 122,6 -15,6
Cu2-P1-C5 115 110,7 4,3
Cu2-P1-C11 117 125,3 -8,3
Cu2-P1-C36 106,9 102 4,9
Cu2-N4-C17 117,1 126,5 -9,4
Cu2-N4-C19 124 111,8 12,2
Cl3-Cu2-N4 148,2 103,9 44,3
Die Diederwinkel ändern sich an der T1-Geometrie stärker als an der S0-Geometrie.
Tabelle 31: Diederwinkel an der S0-Geometrie im Vergleich Molekül1 und Molekül 5
Tabelle 32: Diederwinkel an der T1-Geometrie im Vergleich Molekül 1 und Molekül 5
Die Para-Cyclophan-Einheit ist durch die Änderung des Moleküls nicht mehr so stark verzerrt.
Die Pyridin-Einheit liegt jetzt stärker in einer Ebene mit dem oberen Cyclophanring. Die Phenyl-
Einheiten weisen eine leichte Drehung auf, sodass einer der beiden Ringe mehr mit dem unteren
Cyclophanring auf einer Ebene liegt
Im Weiteren wird die Geometrieverzerrung des Moleküls 5 an den beiden berechneten Geometrien
betrachtet. Dabei werden die Bindungslängen zwischen S0-Geometrie und T1-Geometrie mit mehr
als 5pm abweichen untenstehend aufgeführt,
Winkel [°] Molekül 5 Molekül 1 Differenz
Cl3-Cu2-P1-C5 76,7 86,1 -9,4
Cl3-Cu2-P1-C11 -43,3 -28,6 -14,7
N4-Cu2-P1-C5 -45,8 -71,3 25,5
N4-Cu2-P1-C11 -165,8 174,0 -8,2
Winkel [°] So Molekül 1 S0-T1
Cl3-Cu2-P1-C5 72,7 130,2 -57,5
Cl3-Cu2-P1-C11 -50,8 8,1 -58,9
N4-Cu2-P1-C5 -41,1 -45,4 4,3
N4-Cu2-P1-C11 -164,6 -167,4 2,8

Ergebnisse und Auswertung 39
Tabelle 33: Bindungslängen Molekül 5
Bindungen [pm] S0 T1 S0- T1
P1-Cu2 224,0 241,4 -17,4
Cu2-N4 210,1 197,8 12,3
C19-C42 148,0 142,7 5,3
Tabelle 34: Bindungswinkel Molekül 5
Winkel [°] So T1 S0-T1
P1-Cu2-N4 112,9 122,6 -9,7
Cl3-Cu-N47 141,6 157,4 -15,8
Die Bindungslängen P1-Cu2 und Cu2-N4 werden trotz der Geometrieveränderung an der Grund-
zustands-Geometrie, immer noch ,wenn auch nicht mehr so stark, in der Triplett-Geometrie
verzerrt. Die Cu2-Cl3-Bindung verändert sich nicht mehr signifikant dahingegen scheint nun einer
Verzerrung bei den Atomen C19-C42 zu entstehen. Bei den Bindungswinkeln liegt einer Verzer-
rung nur noch zwischen den Atomen P1-Cu-2-N4 vor. Die Bindung zwischen Kupferatom und
eingesetztem Stickstoffatom scheint bei einer Anregung in den T1-Zustand auch eine starke Ver-
zerrungen zu durchlaufen.
Tabelle 35: Diederwinkel Molekül 5
Die Differenzen bei den Diederwinkeln werden zwischen den beiden Geometrien kleiner. Das
bedeutet die Verzerrung ist nicht mehr so stark, wie bei dem Molekül 1. Die Änderungen der Ge-
ometrie, die jetzt zwischen den beiden Geometrien hervorgerufen wird, ist abhängig von der
HOMO-LUMO Anregungen. Diese Orbitale ähneln denen des Moleküls 1 stark.
Abbildung 21: HOMO des Moleküls 5
Abbildung 22: LUMO des Moleküls 5
4.5.2 DFT/MRCI
Im Vergleich zum Molekül 1 kommt es zu Abweichungen der Zusammensetzung der Zustände.
Im Allgemeinen ist zu beobachten, dass die Elektronendichte hauptsächlich zwischen dem Kup-
feratom, Phosphoratom, Chloratom, dem Pyridyl-Ring sowie dem oberen Cyclophan-Ring
Winkel [°] So T1 S0-T1
Cl3-Cu2-P1-C5 76,7 72,7 4
Cl3-Cu2-P1-C11 -43,3 -50,8 7,5
N4-Cu2-P1-C5 -45,8 -41,1 -4,7
N4-Cu2-P1-C11 -165,8 -164,6 -1,2
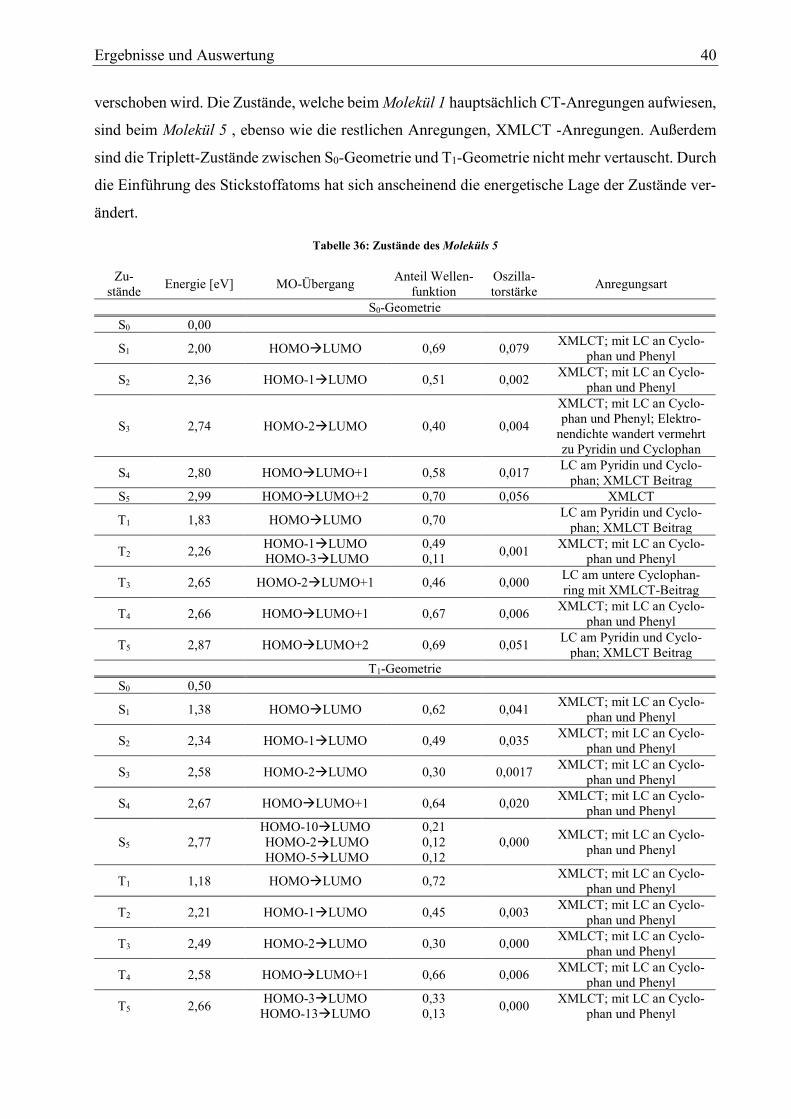
Ergebnisse und Auswertung 40
verschoben wird. Die Zustände, welche beim Molekül 1 hauptsächlich CT-Anregungen aufwiesen,
sind beim Molekül 5 , ebenso wie die restlichen Anregungen, XMLCT -Anregungen. Außerdem
sind die Triplett-Zustände zwischen S0-Geometrie und T1-Geometrie nicht mehr vertauscht. Durch
die Einführung des Stickstoffatoms hat sich anscheinend die energetische Lage der Zustände ver-
ändert.
Tabelle 36: Zustände des Moleküls 5
Zu-
stände Energie [eV] MO-Übergang
Anteil Wellen-
funktion
Oszilla-
torstärke Anregungsart
S0-Geometrie
S0 0,00
S1 2,00 HOMOLUMO 0,69 0,079 XMLCT; mit LC an Cyclo-
phan und Phenyl
S2 2,36 HOMO-1LUMO 0,51 0,002 XMLCT; mit LC an Cyclo-
phan und Phenyl
S3 2,74 HOMO-2LUMO 0,40 0,004
XMLCT; mit LC an Cyclo-
phan und Phenyl; Elektro-
nendichte wandert vermehrt
zu Pyridin und Cyclophan
S4 2,80 HOMOLUMO+1 0,58 0,017 LC am Pyridin und Cyclo-
phan; XMLCT Beitrag
S5 2,99 HOMOLUMO+2 0,70 0,056 XMLCT
T1 1,83 HOMOLUMO 0,70 LC am Pyridin und Cyclo-
phan; XMLCT Beitrag
T2 2,26 HOMO-1LUMO
HOMO-3LUMO
0,49
0,11 0,001
XMLCT; mit LC an Cyclo-
phan und Phenyl
T3 2,65 HOMO-2LUMO+1 0,46 0,000 LC am untere Cyclophan-
ring mit XMLCT-Beitrag
T4 2,66 HOMOLUMO+1 0,67 0,006 XMLCT; mit LC an Cyclo-
phan und Phenyl
T5 2,87 HOMOLUMO+2 0,69 0,051 LC am Pyridin und Cyclo-
phan; XMLCT Beitrag
T1-Geometrie
S0 0,50
S1 1,38 HOMOLUMO 0,62 0,041 XMLCT; mit LC an Cyclo-
phan und Phenyl
S2 2,34 HOMO-1LUMO 0,49 0,035 XMLCT; mit LC an Cyclo-
phan und Phenyl
S3 2,58 HOMO-2LUMO 0,30 0,0017 XMLCT; mit LC an Cyclo-
phan und Phenyl
S4 2,67 HOMOLUMO+1 0,64 0,020 XMLCT; mit LC an Cyclo-
phan und Phenyl
S5 2,77
HOMO-10LUMO
HOMO-2LUMO
HOMO-5LUMO
0,21
0,12
0,12
0,000 XMLCT; mit LC an Cyclo-
phan und Phenyl
T1 1,18 HOMOLUMO 0,72 XMLCT; mit LC an Cyclo-
phan und Phenyl
T2 2,21 HOMO-1LUMO 0,45 0,003 XMLCT; mit LC an Cyclo-
phan und Phenyl
T3 2,49 HOMO-2LUMO 0,30 0,000 XMLCT; mit LC an Cyclo-
phan und Phenyl
T4 2,58 HOMOLUMO+1 0,66 0,006 XMLCT; mit LC an Cyclo-
phan und Phenyl
T5 2,66 HOMO-3LUMO
HOMO-13LUMO
0,33
0,13 0,000
XMLCT; mit LC an Cyclo-
phan und Phenyl

Ergebnisse und Auswertung 41
Die Differenzdichten sind denen des Moleküls 1 sehr ähnlich. Es kommt zu leichten Vertauschun-
gen der Zustände, die Anregungen bleiben jedoch im Großen und Ganzen dieselben. S4 & S5 setz-
ten sich anders zusammen, als bei Molekül 1 die Differenzdichten sehen jedoch sehr ähnlich aus.
HOMO &LUMO von Molekül 5 entsprechen denen des Moleküls 1.
Ebenso wie die Zustände haben sich auch die Dipolmomente verändert und sind nicht mehr so
stark wie bei dem Molekül 1. Außerdem sind alle Dipolmomente – mit Ausnahme des Dipolmo-
ments von S0- in der selben Richtung.
Tabelle 37: Dipolmomente Molekül 5
Komponenten
Zustand x y z Dipolmoment [D]
S0 -2,26 1,71 1,89 8,64
S1 -1,55 -1,33 1,43 6,34
T1 -1,38 -1,57 1,44 6,45
T2 -1,51 -1,54 1,37 6,50
Abbildung 23: Dipolmomente Molekül 5
Die Veränderung des Moleküls hat auch die Anordnung der Zustände sichtlich geändert. Bei un-
serem Molekül 1 liegen bei der S0-Geometrie drei Triplett-Zustände unterhalb des S1-Zustandes.
Bei diesem ist es nur noch der T1-Zustand. Die Zustände T3 & T4 sind entartet und nicht mehr die
Zustände T4 & T5, wie bei den anderen Molekülen. In der Triplett-Geometrie ist keiner der Zu-
stände mehr entartet. Auch bei diesem Molekül kommt es zur Absenkung des S1- und T1-Zustan-
des, wobei der S0-Zustand in der Triplett-Geometrie angehoben wird. Der S0-Zustand wird um
0,50 eV angehoben und ist damit um rund 0,1 eV geringer als bei den vorherigen Molekülen. Der
S1-Zustand wird um 0,62 eV und der T1-Zustand um 0,64 eV abgesenkt. Diese beiden Zustände
werden also weniger stark abgesenkt, als bei den anderen betrachten Molekülen. Die geschätzte
adiabatische Anregungsenergie des S1-Zustandes liegt bei 1,38 eV und die des T1-Zustandes bei
1,18 eV. Somit ist die adiabatische Anregungsenergie um mindestens 1 eV geringer, als bei den
zuvor betrachteten Molekülen. ΔES1T1 liegt bei der S0-Geometrie bei 178 meV und in der Triplett-
Geometrie bei 198 meV. Die Energielücke liegt leicht oberhalb der Grenze, an der ISC und RISC
möglich sind. Durch die Substitution des Stickstoffatoms ist das Molekül ein ungeeigneter TADF-
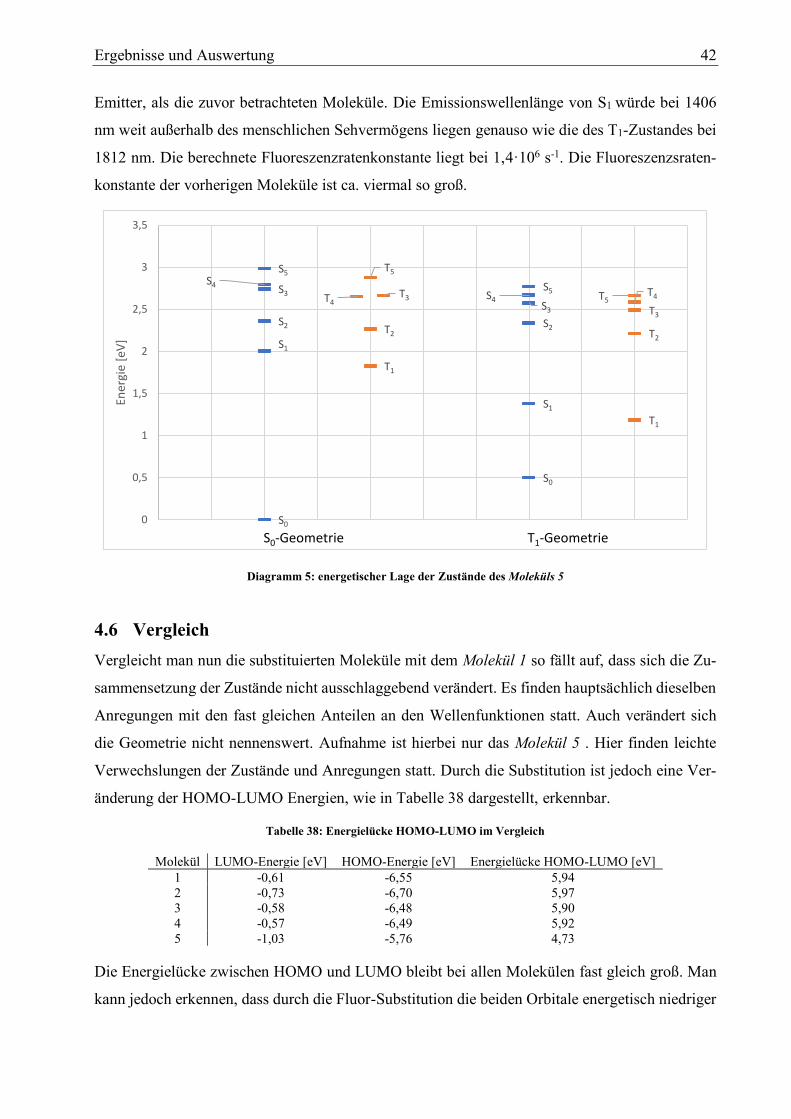
Ergebnisse und Auswertung 42
Emitter, als die zuvor betrachteten Moleküle. Die Emissionswellenlänge von S1 würde bei 1406
nm weit außerhalb des menschlichen Sehvermögens liegen genauso wie die des T1-Zustandes bei
1812 nm. Die berechnete Fluoreszenzratenkonstante liegt bei 1,4·106 s-1. Die Fluoreszenzsraten-
konstante der vorherigen Moleküle ist ca. viermal so groß.
Diagramm 5: energetischer Lage der Zustände des Moleküls 5
4.6 Vergleich
Vergleicht man nun die substituierten Moleküle mit dem Molekül 1 so fällt auf, dass sich die Zu-
sammensetzung der Zustände nicht ausschlaggebend verändert. Es finden hauptsächlich dieselben
Anregungen mit den fast gleichen Anteilen an den Wellenfunktionen statt. Auch verändert sich
die Geometrie nicht nennenswert. Aufnahme ist hierbei nur das Molekül 5 . Hier finden leichte
Verwechslungen der Zustände und Anregungen statt. Durch die Substitution ist jedoch eine Ver-
änderung der HOMO-LUMO Energien, wie in Tabelle 38 dargestellt, erkennbar.
Tabelle 38: Energielücke HOMO-LUMO im Vergleich
Molekül LUMO-Energie [eV] HOMO-Energie [eV] Energielücke HOMO-LUMO [eV]
1 -0,61 -6,55 5,94
2 -0,73 -6,70 5,97
3 -0,58 -6,48 5,90
4 -0,57 -6,49 5,92
5 -1,03 -5,76 4,73
Die Energielücke zwischen HOMO und LUMO bleibt bei allen Molekülen fast gleich groß. Man
kann jedoch erkennen, dass durch die Fluor-Substitution die beiden Orbitale energetisch niedriger
S0
S1
S2
S3
S4
S5
T1
T2
T4T3
T5
S0
S1
S2
S3
S4
S5
T1
T2
T3
T4T5
0
0,5
1
1,5
2
2,5
3
3,5
Ener
gie
[eV
]
S0-Geometrie T1-Geometrie

Ergebnisse und Auswertung 43
liegen. Durch die Substitution von Methyl- und t-Butyl-Einheiten werden das HOMO und das
LUMO im selben Maße energetisch angehoben. Bei Molekül 5 wird das LUMO deutlich abge-
senkt und das HOMO um ca. 1 eV im Vergleich zu den anderen Molekülen angehoben. Die
HOMO-LUMO-Lücke ist um 1 eV kleiner, als die der anderen. Es ist ein leichter Trend der ener-
getischen Lage der Grenzorbitale zu erkennen. Diese nimmt von elektronenziehender zu elektro-
nenschiebender Gruppe ab. Betrachtet man in Tabelle 39 die Oszillatorstärke des S1-Zustandes ist
festzustellen, dass die Stärke des Übergangs zwischen S0-Zustand und S1-Zustand im Grundzu-
stand durch die Substitutionen abnimmt. In der Triplett-Geometrie ist hingegen die Änderung der
Oszillatorstärke sehr gering. Bei Molekül 5 ist die Oszillatorstärke in beiden Geometrien höher.
Der Übergang ist bei diesem Molekül also intensiver.
Tabelle 39: Oszillatorstärke S1-Zustand im Vergleich
Die vertikalen Anregungsenergien, dargestellt in Tabelle 40, weisen nur sehr geringe Schwankun-
gen auf. Grundsätzlich ist zu erkennen, dass mit der Fluor- und t-Butyl-Substitution die Energien
angehoben werden. Durch die Substitution der Methyl-Einheit ändert sich die Energien am we-
nigsten. Die Vertikalen Anregungsenergie ändern sich bei Molekül 5 um ca. 1 eV. Alle Zustände
werden deutlich abgesenkt. Es ist jedoch kein Trend erkennbar.
Tabelle 40: Vertikale Anregungsenergien im Vergleich
Die Lage der S1- und T1-Zustände ändert sich kaum. Ihre adiabatische Energie schwankt zwischen
den Substitutionen ein wenig. Die größte Differenz ist zwischen dem Molekül 2 und dem Molekül
5 zu finden. Das Molekül 3 weist die geringsten adiabatischen Energien von den untersuchten
Molekülen auf, das Molekül 2 die höchsten. Die Energielücke zwischen S1- und T1-Zustand bleibt
bei der Triplett-Geometrie durch die Substitutionen unverändert bei 0,14 eV. Bei der S0-Geometrie
S1-S0-Geometrie S1-T1-Geometrie
Molekül 1 0,030 0,032
Molekül 2 0,024 0,031
Molekül 3 0,024 0,033
Molekül 4 0,024 0,033
Molekül 5 0,079 0,041
Vertikale
Anregungsenergien [eV] Molekül 1 Molekül 2 Molekül 3 Molekül 4 Molekül 5
S0 0 0 0 0 0,00
S1 3,35 3,39 3,30 3,32 2,00
S2 3,64 3,67 3,64 3,64 2,36
S3 3,77 3,80 3,77 3,79 2,74
S4 3,87 3,89 3,87 3,88 2,80
S5 4,00 4,06 3,98 4,00 2,99
T1 3,11 3,14 3,11 3,12 1,83
T2 3,25 3,29 3,22 3,24 2,26
T3 3,32 3,35 3,35 3,35 2,65
T4 3,50 3,53 3,51 3,51 2,66
T5 3,52 3,55 3,54 3,55 2,87

Ergebnisse und Auswertung 44
liegt ΔES1-T1 ungefähr 0,10 eV für Molekül 1 & Molekül 2 und bei ungefähr 0,09 eV für die Mole-
küle 3&4. Die elektronenschiebenden Moleküle verkleinern anscheinend die Energielücke zwi-
schen S1- und T1-Zustand, um diese Annahme zu stützen, müssten jedoch weitere Substitutionen
vorgenommen werden. Abweichend von diesem generellen Trend ist das Molekül 5 zu betrachten.
Es weist eine doppelt so große Energielücke zwischen S1- und T1-Zustand an der S0-Geometrie
auf. Auch ΔES1-T1 an der T1-Geometrie ist deutlich größer, als bei den anderen Molekülen. Die
adiabatischen Energien sind nur halb so groß im Vergleich zu den anderen Ergebnissen.
Tabelle 41: adiabatische Energien und Energielücke S1-T1 im Vergleich
Da sich die energetische Lage der Zustände kaum verändert, variiert auch ihre Emissionswellen-
länge kaum. Sie liegt bei allen Substitutionen an der Grenze zum infraroten Lichtbereich. Wobei
das Molekül 2 die kürzesten Wellenlängen aufweist und das Molekül 5 die längsten. Die Wellen-
länge des Molekül 3s ist fast identisch mit denen des Moleküls 1. Ein leichter Trend ist erkennbar.
Je stärker elektronenziehend die Gruppe ist, desto kürzer werden die Wellenlängen.
Tabelle 42: Emissionswellenlängen im Vergleich
Tabelle 43: Fluoreszenzratenkonstante im Vergleich
Die Fluoreszenzratenkonstanten liegen durchweg sehr nah beieinander. Es ist keine signifikante
Beeinflussung durch die Substitutionen erkennbar. Vergleicht man die Werte mit anderen Wer-
ten[28] von untersuchten TADF Molekülen, so fällt auf, dass die hier vorliegenden Werte höher
Energielücke S1-T1 an
der S0 -Geometrie [eV]
Energielücke S1-T1 an der
T1-Geometrie
Adiabatische
Energie S1[eV]
Adiabatische
Energie T1 [eV]
Molekül 1 0,10 0,14 2,58 2,40
Molekül 2 0,10 0,14 2,58 2,43
Molekül 3 0,08 0,14 2,53 2,39
Molekül 4 0,09 0,14 2,55 2,41
Molekül 5 0,18 0,20 1,38 1,18
Wellenlänge S1[nm] Wellenlänge T1[nm]
Molekül 1 663,0 717,3
Molekül 2 655,2 708,4
Molekül 3 663,6 717,4
Molekül 4 668,8 723,1
Molekül 5 1406,0 1812,0
Fluoreszenzratenkonstante [s-1]
Molekül 1 4,8·106
Molekül 2 4,7·106
Molekül 3 5,0·106
Molekül 4 5,0·106
Molekül 5 1,4·106

Ergebnisse und Auswertung 45
sind. Dies bedeutet, dass weniger Konkurrenz-Reaktionen in den untersuchten Molekül stattfin-
den.
Tabelle 44: Dipolmomente [D] im Vergleich
Da das Dipolmoment des S1-Zustands nicht bei ungefähr 10 D liegt, sondern nur noch bei ca. 8 D,
zeigt das Molekül 2 die größten Abweichungen hinsichtlich des Dipolmoments Außerdem liegt
das Dipolmoment des T2-Zustandes unterhalb der Dipolmomente der anderen Substitutionen.
Durch die Substitution der Butyl-Einheit wird im Gegensatz zur Fluor-Substitution das Dipolmo-
ment des S1-Zustandes um ca. 1 D erhöht. Das Molekül 2 zeigt im Vergleich zum Molekül 1 die
stärksten Beeinflussungen bei den Dipolmomenten. Das Molekül 5 zeigt außer bei dem Dipolmo-
ment von S0 generell kleinere Dipolmomente, als die anderen Moleküle.
Alles in Allem sind, wie in diesem Abschnitt besprochen, keine deutlichen Trends durch die Sub-
stitutionen zu erkennen. Dennoch führen sowohl die Substitution von Fluor oder auch t-Butyl zu
stärken Veränderung, als die Substitution von Methyl. Dementsprechend verändert sich durch die
Methyl-Substitution am wenigsten. Höchstwahrscheinlich ist der Einfluss dieser Einheit am ge-
ringsten. Sowohl bei der Fluor-, als auch bei der t-Butyl Substitution, kommt es weitestgehend zu
denselben Effekten (ausgenommen das Dipolmoment und den Wellenlängen). Die Fluor-Substi-
tution zeigt jedoch noch leicht stärkere Veränderungen als die t-Butyl-Substitution. Durch die Sub-
stitutionen an den Diarylphosphin-Einheiten, lassen sich anscheinend keine starken Veränderun-
gen hervorrufen.
S0 S1 T1 T2
Molekül 1 7,86 10,37 7,66 9,03
Molekül 2 8,82 8,49 8,33 7,40
Molekül 3 7,80 11,00 7,56 8,94
Molekül 4 7,66 11,10 7,52 9,42
Molekül 5 8,64 6,34 6,45 6,50

Fazit 46
5 Fazit
Ziel dieser Arbeit war es, das Molekül Rp-[κ2-N,P-4-(2′-Pyridyl)-1,2-diphenylphosphinyl[2.2]pa-
racyclophan]chlorokupfer(I) hinsichtlich seiner TADF-Eigenschaften zu untersuchen und mögli-
che Trends festzustellen, die sich aus der Substitution der Diarylphospineinheit ergeben.
Hierfür wurde sowohl die Grundzustands-geometrie, als auch im T1-Geometrie betrachtet. Die
Substitution ließ -wie erwartet- die Geometrie des Moleküls weitgehend unverändert. Die Bindung
zwischen dem Kupferatom und einem Wasserstoffatom des Cyclophanrings sollte durch Substitu-
tion eines Stickstoffatoms unterbunden werden. Dies war jedoch nicht der Fall. Das Molekül bil-
dete nun diese Brücke schon im Grundzustand aus.
Das HOMO und das LUMO sehen bei allen Molekülen sehr ähnlich aus. Es ist kein Trend hin-
sichtlich der energetischen Lage der Orbitale zu erkennen. Jedoch ist festzustellen, dass die Dia-
rylphosphineinheit sowohl bei dem HOMO also auch beim LUMO nicht relevant ist. Ebenso wie
die Orbitale ähneln sich auch die Differenzdichten sehr stark. Es ist kein deutlicher Unterschied
zu erkennen. Die meisten Orbitale weisen großen XMLCT-Charakter auf. Dieser erfüllt die An-
forderung an die Separation der Elektronendichte. Das Molekül 5 weist jedoch einen stärken
XMLCT-Charakter auf, besonders bei den Zuständen, die bei den anderen Molekülen nur einen
kleinen XMLCT-Beitrag hatten. Ein Unterschied zu der energetischen Lage der Zustände ist nur
beim Molekül 5 zu erwarten.
Die Dipolmomente sowie auch die Oszillatorstärken ändern sich zwischen den Molekülen kaum.
Ausnahme ist nur hier das Molekül 5, da die Zusammensetzung der Zustände leicht abweicht.
Die energetische Lage der Zustände ist bei allen Molekülen fast gleich. Es liegt immer eine Ver-
tauschung der Triplett-Zustände im Grundzustand vor und ΔEST ist ebenso wie die adiabatischen
und vertikalen Anregungsenergien fast immer gleich groß. Da bei allen Molekülen ΔEST fast iden-
tisch ist, sind auch alle Moleküle eher weniger als TADF-Emitter zu gebrauchen. Die Energie-
Lücke darf nicht größer als 100 meV sein und alle untersuchten Moleküle liegen leicht oberhalb
dieses Referenzwertes. Das RISC ist somit eher unwahrscheinlich. Ausnahme stellt hier nur das
Molekül 5 da. Die Triplett-Zustände sind an der Grundzustands-Geometrie nicht mehr vertauscht.
Generell sind die Zustände weiter aufgespalten. Dia adiabatischen Energien von S1 und T1 verrin-
gern sich deutlich, wohin gegen ΔEST deutlich zunimmt, sodass RISC bei diesem Molekül wahr-
scheinlich nicht stattfinden kann.

Fazit 47
Die Wellenlänge aller Moleküle liegt im roten bis infrarot Bereich auch hier ist kein Trend hin-
sichtlich rot oder blau Verschiebung zu erkennen. Das Molekül 5 hat eine Emissionswellenlänge
weit außerhalb des menschlichen Sehvermögens. IR Emitter
Die große ΔEST lässt den Schluss zu, dass diese Moleküle nicht gut als TADF-Emitter geeignet
sind. Das RISC ist eher unwahrscheinlich.
Gegeben falls kann das Molekül durch Substitutionen an der Pyridyl-Einheilt oder an der Cyclo-
phan-Einheit stärke beeinflusst werden. Dort findet hauptsächlich die Verschiebung der Elektro-
nendichte statt.
In der Bachelorarbeit von Andrea Garnica[29] wurden die gleichen Substitutionen nur an der
Pyrdin-Einheit des Moleküls 1 vorgenommen. Hierbei sind deutlichere Ergebnisse hinsichtlich
vorhandener Tendenzen erkennbar. Zum einen steigt die Anregungsenergien der Zustände bei
Substitution einer Gruppe mit +I-Effekt. Dieser Effekt ist bei meinen Untersuchungen nicht fest
zu stellen. Im Gegenteil die Fluor-Substitution führt zu leicht erhöhten Anregungsenergien. Des
Weiteren nimmt Δ ES1-T1 an beiden Geometrien mit der Fluor-Substitution bei der Untersuchung
von Frau Garnica zu. Bei meinen Ergebnissen ist dies nur bei der Triplett-Geometrie der Fall.
Auffälliger ist es eher, dass bei den anderen Substitutionen die Energielücke kleiner wird. Bei den
Emissionswellen länge ist eben Falls bei diesen beiden Arbeiten ein gegenläufiger Trend erkennen
bar.
Generell lässt sich sagen, dass durch die Substitutionen an diesem Molekül, keine wünschenswer-
ten Tendenzen erkennbar sind. Durch die gr0ße Energielücke Δ ES1-T1, welche an der Grenze zum
möglichen RISC liegt, werden die untersuchten Moleküle eher ungeeignet als TADF-Emitter sein.

Literaturverzeichnis 48
Literaturverzeichnis
[1] "Direct observation of intersystem crossing in a thermally activated delayed fluorescence
copper complex in the solid state", zu finden unter http://advances.sciencemag.org/con-
tent/2/1/e1500889.full, 2016.
[2] C. Adachi, M. A. Baldo, M. E. Thompson, S. R. Forrest, Journal of Applied Physics 2001,
90, 5048.
[3] Hartmut Yersin (Hrsg.) Highly Efficient OLEDs. Intersystem crossing processes in TADF
emitters, WILEY-VCH, 2018.
[4] Francis Perrin, La fluorescence des solutions. Induction moléculaire – Polarisation et
durée d'émission – Photochimi, 1929, Ann. Phys., Paris, S. 169–275.
[5] Gilbert N. Lewis, David Lipkin, and Theodore T. Magel, Am. Chem. Soc Am. Chem. Soc,
3005.
[6] C. A. Parker, C. G. Hatchard, Trans. Faraday Soc. 1961, 57, 1894.
[7] Wilkinson F and Horrocks A R 1968.
[8] Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi, M. P. Aldred, Chem.
Soc. Rev. 46 2017, 915.
[9] M. Kasha, Discuss. Faraday Soc. 1950, 9, 14.
[10] P. Klán, J. Wirz, Photochemistry of organic compounds. From concepts to practice, Wiley,
Chichester, U.K, 2009.
[11] S. Salzmann, V. Martinez-Junza, B. Zorn, S. E. Braslavsky, M. Mansurova, C. M. Marian,
W. Gärtner, The journal of physical chemistry. A 2009, 113, 9365.
[12] I. N. Levine, Quantum chemistry, 7. Aufl., Pearson, Boston, Mass., 2014.
[13] P. Hohenberg, W. Kohn, Phys. Rev. 1964, 136, B864-B871.
[14] C. Adamo, V. Barone, The Journal of Chemical Physics 1999, 110, 6158.
[15] A. D. Becke, Phys. Rev. A 1988, 38, 3098.
[16] T. H. Dunning, The Journal of Chemical Physics 1989, 90, 1007.
[17] S. Grimme, M. Waletzke, The Journal of Chemical Physics 1999, 111, 5645.
[18] M. Dolg, "Institute for Theoretical Chemistry", zu finden unter http://www.tc.uni-
koeln.de/cgi-bin/pp.pl?language=en,job=getreadme.
[19] "Avogadro - Free cross-platform molecular editor", zu finden unter https://avogadro.cc/,
2017.
[20] "Overview of the TURBOMOLE Program", zu finden unter http://www.turbomole-
gmbh.com/program-overview.html.

Literaturverzeichnis 49
[21] "MOLDEN a visualization program of molecular and electronic structure", zu finden unter
http://www.cmbi.ru.nl/molden/, 2017.
[22] I. Lyskov, M. Kleinschmidt, C. M. Marian, The Journal of Chemical Physics 2016, 144,
34104.
[23] "GIMP", zu finden unter https://www.gimp.org/.
[24] C. Braun 2017, 69.
[25] M. Y. Wong, E. Zysman-Colman, Advanced materials (Deerfield Beach, Fla.) 2017, 29.
[26] J. Föller, M. Kleinschmidt, C. M. Marian, Inorganic chemistry 2016, 55, 7508.
[27] Andrea Garnica, „Quantenchemische Charakterisierung von Cu(I)-Komplexen gemischter
P,N-Liganden auf [2.2]Paracyclophanbasis: Variation der Pyridyleinheit", Heinrich-Heine-
Universität, 2018

Anhang 50
Anhang
Koordinaten
Molekül 1 S0-Geometrie
S0-Geometrie T1-Geometrie
p 0,152384811 -2,38824865 0,24596679 0,16384699 2,40378734 0,10891358
cu 2,858664265 -0,05292461 -1,8450472 2,3467454 0,09609475 -2,80587018
cl 6,128935936 -1,29340019 -4,08001592 2,5550015 -2,18873687 -6,14536113
n 3,449093713 3,72067885 -0,9642925 3,349464 3,07961575 -1,08241599
c 1,43372421 -3,01243713 3,42045859 1,66811255 -2,68035836 3,21582898
c 4,055317881 -2,82025582 3,73106359 3,30416267 -0,77917135 4,06788798
c 5,169280707 -3,38368201 6,05386997 4,44128749 -0,96916081 6,43777113
c 3,672781531 -4,13175292 8,08774787 3,96574111 -3,05696116 7,9720146
c 1,05787827 -4,32636671 7,79265202 2,34543278 -4,96006784 7,13058839
c -0,056874061 -3,77925707 5,47140049 1,20345205 -4,78015193 4,76535797
c -0,436035718 -5,60450739 -0,91381542 -0,45985658 -5,67406129 -0,79117792
c -2,40366069 -7,12553011 -0,00213182 -2,7500644 -6,87116468 -0,21174553
c -2,700435593 -9,58080808 -0,90084868 -3,09744048 -9,41375109 -0,80977003
c -1,026863042 -10,5415013 -2,70222319 -1,16706741 -10,775675 -1,97824488
c 0,940105506 -9,03974172 -3,60286615 1,11377292 -9,58933626 -2,5586954
c 1,239595473 -6,57122907 -2,72033287 1,46742818 -7,04601666 -1,97901692
c 5,887382302 4,24363518 -0,54913721 5,84858738 3,30041253 -0,46736103
c 6,721561679 6,58274973 0,30828576 6,8542275 5,31180131 0,80023378
c 1,693416562 5,53487127 -0,62983096 1,68969497 5,06145183 -0,4447429
c 2,408037274 7,9558706 0,18731017 2,69474825 7,18234361 0,82523719
c 4,932705145 8,47602953 0,68872976 5,21183018 7,35353691 1,48052232
h 2,783661943 -5,38236248 -3,4333965 3,23801623 -6,11526051 -2,49391363
h -1,264209066 -12,4720213 -3,40588663 -1,4469935 -12,7694222 -2,45001784
h 2,256929634 -9,78427263 -5,01257643 2,62681079 -10,6430039 -3,49339444
h -3,72857866 -6,38130607 1,40175493 -4,27062319 -5,81233477 0,70503963
h -4,247259191 -10,7556958 -0,19013635 -4,89230769 -10,3355904 -0,35781409
h 5,23029734 -2,23719182 2,12639938 3,69421002 0,86431973 2,87940063
h 7,219877606 -3,23150494 6,26979159 5,71712458 0,52902276 7,07077881
h 4,543047812 -4,5663226 9,91295183 4,86539772 -3,20838599 9,82767711
h -0,124415199 -4,91443009 9,38438601 1,9686001 -6,60602008 8,3238778
h -2,108798942 -3,92938493 5,26497875 -0,05013105 -6,29149425 4,1220545
h 7,201636219 2,70060238 -0,98103699 7,05717617 1,72149267 -1,05386151
h 8,735271309 6,90909231 0,629585 8,87625561 5,36062972 1,21217136
h 5,503419227 10,3604215 1,32151817 5,9471133 9,04067297 2,41768133
h 0,973619775 9,43123169 0,35308095 1,42658384 8,77061851 1,19656702
c -2,934045408 -0,93879215 0,87390795 -2,8735242 -0,87623203 0,66498733
c -5,06221365 -1,16983702 -0,72446663 -4,88745724 -1,00101092 -1,09234067
c -7,315619451 -0,08401072 0,14618387 -7,18531812 0,03379346 -0,29211638

Anhang 51
S0-Geometrie T1-Geometrie
c -7,325566905 1,69557757 2,08598149 -7,29765945 1,69734472 1,74660996
c -5,054981032 2,43260962 3,22446096 -5,11102304 2,33870788 3,08037188
c -2,961461751 0,88764963 2,79342159 -3,00851555 0,78300672 2,72085864
c -0,96018677 5,00056703 -1,33437016 -0,89994297 4,95693811 -1,30698676
c -3,048880329 6,09836011 -0,06098478 -2,97223136 6,11940888 0,00753469
c -5,386615583 5,96638143 -1,28086841 -5,28747673 6,23553643 -1,23457977
c -5,79945114 4,30379998 -3,2768305 -5,83189651 4,73843194 -3,35090451
c -3,86763902 2,70579086 -4,1076437 -3,98954881 3,09974534 -4,28724355
c -1,416942676 3,31400609 -3,32823216 -1,51128819 3,49045223 -3,46487713
c -4,884941865 -2,02474556 -3,44569586 -4,55181432 -1,68013791 -3,8498074
c -4,475343686 0,23656627 -5,38360703 -4,69282423 0,70106154 -5,65367861
h -6,629440271 -3,03255188 -3,98217527 -6,00233006 -3,05225068 -4,45848488
h -3,333317291 -3,3838921 -3,69127093 -2,71458749 -2,61214946 -4,15482615
h -6,209856693 0,45571843 -6,51780022 -6,62682785 0,87642445 -6,4129474
h -2,953134985 -0,3039318 -6,69968362 -3,42866059 0,32480653 -7,26990712
c -4,724601525 5,02006257 4,38076254 -4,82232626 4,87609453 4,34666391
c -3,007520071 6,80583369 2,71673599 -2,93918363 6,61337594 2,83952528
h -3,873673648 4,91746808 6,28438122 -4,11966415 4,70578397 6,30671225
h -6,602751729 5,89254628 4,62028903 -6,6940154 5,78785298 4,46060558
h -1,057466235 6,69214326 3,43321726 -1,0267358 6,26579762 3,57619222
h -3,634606637 8,7734863 3,02989505 -3,3863505 8,60264353 3,29818232
h -7,011811384 6,9409604 -0,44748877 -6,85419833 7,23694045 -0,31883479
h -7,720361372 4,05500649 -4,0059642 -7,77525092 4,63029858 -4,05290147
h 0,200477249 2,36884746 -4,22058867 0,00091838 2,62588285 -4,60065651
h -9,058900558 -0,41208812 -0,92254714 -8,85769202 -0,19459555 -1,49051524
h -9,073687714 2,71942093 2,51279296 -9,05450051 2,7319851 2,1047988
h -1,20575039 1,2742829 3,82289167 -1,34321082 1,05824607 3,91542521
Koordinaten Molekül 2
S0-Geometrie T1-Geometrie
p 1,032080361 -2,473895461 -0,167677515 1,064868713 -2,486334581 -0,309799539
cu 3,766558165 -0,152252147 -2,235478629 3,26383561 0,012962378 -3,210027438
cl 7,07198563 -1,493450967 -4,365683783 3,509517999 -2,273297278 -6,548441134
n 4,356604675 3,615848878 -1,389785406 4,250301115 2,974235614 -1,442240012
c 2,306194333 -3,090476933 3,006460664 2,584800784 -2,760931346 2,784151114
c 4,930697476 -2,92158463 3,318934808 4,058326989 -0,763987999 3,719799283
c 6,054581611 -3,467784454 5,637147403 5,221054451 -0,936119388 6,07352743
c 4,525264234 -4,174277625 7,650057658 4,918449943 -3,130396712 7,485811161
c 1,913962719 -4,364018349 7,398028938 3,485473293 -5,148012533 6,600022209
c 0,81660664 -3,830974071 5,068533048 2,324019048 -4,955574473 4,249098392
c 0,456161405 -5,681270592 -1,341089039 0,391303506 -5,74521045 -1,195904192
c -1,518014066 -7,213261295 -0,461687485 -1,872930731 -6,926984423 -0,488957575
Anhang 52
S0-Geometrie T1-Geometrie
c -1,815028817 -9,662571093 -1,368068916 -2,295058254 -9,459689498 -1,061251715
c -0,106467968 -10,57364117 -3,150266411 -0,432377679 -10,80196588 -2,340907403
c 1,877318583 -9,103163126 -4,039171976 1,827856412 -9,675262056 -3,066329217
c 2,152688508 -6,645009435 -3,132350419 2,229334823 -7,139577837 -2,497175403
c 6,79544181 4,137527896 -0,972292823 6,742082416 3,174506362 -0,79027999
c 7,630054788 6,474914607 -0,110941305 7,742749184 5,174364618 0,499062589
c 2,601115948 5,43046299 -1,053456782 2,593372122 4,965000791 -0,82110277
c 3,317379516 7,849587482 -0,232841531 3,596212462 7,075524083 0,468993052
c 5,841712909 8,368263621 0,271377141 6,10466286 7,22566346 1,161471079
h 3,710046182 -5,458625405 -3,822446436 3,97419565 -6,229160638 -3,122675702
h 3,181835717 -9,887076632 -5,435177609 3,235867419 -10,78554847 -4,090388518
h -2,863481542 -6,483164971 0,929650344 -3,329421222 -5,86047246 0,51744157
h -3,348773914 -10,88334336 -0,717209236 -4,05050978 -10,40982506 -0,531566761
h 6,115603091 -2,366553958 1,711789063 4,313983434 0,952988935 2,600839466
h 8,098853785 -3,34609447 5,90261359 6,373354213 0,610191279 6,811843157
h 0,776662827 -4,928384442 9,027206265 3,298549169 -6,847193422 7,758752982
h -1,236931759 -3,969420128 4,872859134 1,208521917 -6,547947239 3,551998887
h 8,108685927 2,59313918 -1,402910902 7,947204412 1,588169089 -1,363463384
h 9,643635649 6,798379604 0,213107855 9,759020217 5,208427721 0,939720666
h 6,41153885 10,25165523 0,908030188 6,838614338 8,9042515 2,114826535
h 1,883849824 9,325432151 -0,065738915 2,336022262 8,673759262 0,823503035
c -2,055642034 -1,025443909 0,453069817 -1,971539131 -0,948255491 0,226718085
c -4,178761249 -1,247889357 -1,153593719 -3,959567017 -1,073245553 -1,560732092
c -6,431924699 -0,1551358 -0,290386976 -6,26973086 -0,041809701 -0,793492109
c -6,442626087 1,624014163 1,649510053 -6,412448637 1,621405737 1,243908701
c -4,173543223 2,353531924 2,79624845 -4,247190367 2,260367964 2,612766602
c -2,084618299 0,800615385 2,373781728 -2,140105857 0,702655888 2,285697186
c -0,054436922 4,903028087 -1,756143871 0,015243841 4,876597084 -1,720405899
c -2,136423789 6,012645974 -0,48099551 -2,074024299 6,041894468 -0,43498136
c -4,473730221 5,901645577 -1,702872063 -4,371889384 6,160087537 -1,709061116
c -4,896880259 4,246814329 -3,702901893 -4,889085586 4,663633599 -3,833054918
c -2,977833296 2,633576401 -4,533394862 -3,03494212 3,026686541 -4,746814236
c -0,522606958 3,220431736 -3,750583849 -0,567639969 3,417241208 -3,890800504
c -3,993591272 -2,096267827 -3,876579342 -3,585468468 -1,751562993 -4,313784
c -3,613078864 0,171732643 -5,810759953 -3,718259344 0,626914721 -6,121233798
h -5,723930885 -3,12641876 -4,416184223 -5,018606323 -3,133808291 -4,9401784
h -2,422951347 -3,432898354 -4,122614958 -1,738523616 -2,671389663 -4,595913561
h -5,368076788 0,402769614 -6,910641183 -5,645846754 0,795809276 -6,897855624
h -2,119461198 -0,370274591 -7,158406173 -2,437926725 0,253841241 -7,725680461
c -3,837284015 4,941590533 3,950329727 -3,976718751 4,797020876 3,883817677
c -2,092418673 6,71006158 2,299364337 -2,083186945 6,541656859 2,396092971
h -3,008212438 4,840489112 5,863709324 -3,292626661 4,62559936 5,850028334
h -5,711775184 5,826768273 4,167434481 -5,851226951 5,704341542 3,982906634
h -0,145708269 6,566259336 3,019437092 -0,17976519 6,211238517 3,162543853
Anhang 53
S0-Geometrie T1-Geometrie
h -2,692585013 8,684730039 2,620841404 -2,551847328 8,52890096 2,840387316
h -6,092349397 6,886801014 -0,869477781 -5,950598234 7,161676758 -0,814375248
h -6,81837132 4,01584297 -4,435903798 -6,823153301 4,556288689 -4,560656476
h 1,088621221 2,262337351 -4,639620421 0,960319981 2,563036325 -5,014059649
h -8,17240097 -0,475870471 -1,365851993 -7,923351402 -0,271004855 -2,017492695
h -8,188346198 2,654060395 2,070678553 -8,174154731 2,65679975 1,573553608
h -0,331216866 1,183651923 3,408298354 -0,499244036 0,972596691 3,513931799
f -0,392562624 -12,91261632 -4,024430336 -0,829285696 -13,21725841 -2,888967296
f 5,57630404 -4,675499894 9,877118608 6,02913277 -3,314509998 9,72683482
Koordinaten Molekül 3
S0-Geometrie T1-Geometrie
p 0,006786205 -1,914752029 0,154212464 0,00034262 -1,97691011 -0,03263527
cu 1,666155849 0,189470396 -3,007246572 1,04533061 0,42106454 -3,5915026
cl 3,465922095 -1,293430029 -6,439954944 -0,11760497 -1,74779151 -6,80792661
n 2,897756886 3,890788403 -2,555600143 2,83316331 3,29766012 -2,4002089
c 2,510300856 -2,617881696 2,458299233 2,6660416 -2,47329413 2,11255657
c 5,018056208 -2,491711728 1,634252437 4,55274812 -0,62563698 2,33654838
c 6,999038091 -3,115493392 3,25914267 6,58856132 -0,98856846 3,96198733
c 6,537828277 -3,873402009 5,746451945 6,82361534 -3,19603687 5,40121117
c 4,016473532 -3,993319991 6,560785148 4,94474858 -5,04152896 5,14531841
c 2,0326366 -3,385989074 4,949545708 2,89624729 -4,69772045 3,5306108
c -1,270000155 -5,06388244 -0,519932856 -1,30085059 -5,13032752 -0,51979803
c -2,757802643 -6,430539159 1,192750553 -3,10209492 -6,14042125 1,13842426
c -3,585660141 -8,852001765 0,592717821 -3,99301613 -8,59336687 0,79679777
c -2,947824946 -9,987639594 -1,716482204 -3,11875823 -10,107235 -1,18920851
c -1,466125276 -8,604174259 -3,412569669 -1,32065239 -9,07562078 -2,83528586
c -0,632527777 -6,170296155 -2,837114599 -0,41961494 -6,62520231 -2,51809555
c 5,305847135 4,191182131 -3,262949452 5,37406437 3,31857479 -2,86803147
c 6,64204085 6,420495217 -2,87740107 6,9615238 5,22664339 -2,15980969
c 1,639820446 5,835865687 -1,496721529 1,73687558 5,38805289 -1,16795541
c 2,858339013 8,158907219 -1,09676255 3,33424854 7,40419007 -0,45681487
c 5,383154752 8,444728945 -1,760205049 5,90345813 7,37488164 -0,8968543
h 0,5194011 -5,114037793 -4,201749143 0,92878904 -5,85013093 -3,87778717
h -0,955404912 -9,437565005 -5,23662143 -0,62691181 -10,2038429 -4,42493432
h -3,303914236 -5,588048026 3,001626936 -3,82885273 -5,00083773 2,70244633
h -4,75931798 -9,889159456 1,946431852 -5,41136966 -9,34410569 2,10334289
h 5,421604561 -1,912306715 -0,313590259 4,44553079 1,11227559 1,22582657
h 8,946652858 -3,005613722 2,569500856 8,03918132 0,48137268 4,09084448
h 3,601569319 -4,580485161 8,502172586 5,08776167 -6,79866425 6,22907477
h 0,089852375 -3,490417977 5,650168201 1,47362718 -6,18690164 3,36866876
h 6,150092925 2,556393215 -4,217359157 6,11232092 1,65974394 -3,8688255
Anhang 54
S0-Geometrie T1-Geometrie
h 8,61263011 6,563014625 -3,478191849 8,97131578 5,11415629 -2,61669329
h 6,343310628 10,2506517 -1,453596573 7,08404611 8,98633672 -0,37250705
h 1,784399304 9,745152692 -0,327716762 2,45531477 9,0784242 0,3760597
c -2,371143364 -0,223387221 2,029412779 -2,37147681 -0,20688416 1,73699445
c -4,991669645 -0,255348723 1,51058311 -4,93684613 -0,13378589 0,97617568
c -6,554888966 0,995353826 3,24407813 -6,60897799 1,0394385 2,65323553
c -5,583580353 2,74079525 4,959478647 -5,73543102 2,64604855 4,54846127
c -2,992736401 3,270707678 4,993229905 -3,15005722 3,08489115 4,85405389
c -1,420931037 1,564767928 3,737648332 -1,51268978 1,39053156 3,66052263
c -1,089905502 5,550981101 -0,957394101 -0,97858479 5,50405668 -0,88630969
c -2,306833617 6,804548133 1,077151948 -2,22240459 6,79379834 1,15413971
c -4,943731592 6,904761859 1,003114998 -4,82654971 7,12271989 0,97858965
c -6,331532611 5,321473281 -0,573292472 -6,31455769 5,73035993 -0,71247255
c -5,105494076 3,573052313 -2,127408613 -5,16092449 3,97729154 -2,31170332
c -2,518344989 3,948472235 -2,512702207 -2,54198411 4,14339885 -2,58264362
c -6,067941083 -1,078439185 -1,005503221 -5,82886126 -0,75881243 -1,66915532
c -6,424218975 1,187842612 -2,94538956 -6,55594919 1,67374366 -3,24335942
h -7,922520836 -1,98842007 -0,723114962 -7,48335031 -2,03080411 -1,61803337
h -4,849211682 -2,511037836 -1,887849431 -4,34779014 -1,78560836 -2,71223443
h -8,462778504 1,568752715 -3,152552533 -8,61027602 2,00066489 -3,10009565
h -5,731910797 0,559785084 -4,808095422 -6,13941537 1,26368886 -5,24660265
c -1,988187437 5,807375679 5,829745913 -2,17165289 5,5578307 5,87741234
c -0,99664161 7,448963582 3,544961017 -0,98290393 7,21572544 3,71296294
h -0,421657364 5,613421122 7,196408504 -0,71968877 5,28287521 7,35370346
h -3,507085364 6,839775059 6,816681678 -3,74776406 6,58625939 6,77497505
h 1,049168503 7,126658891 3,347444176 1,03551398 6,74039906 3,58837554
h -1,229273436 9,462216297 4,051637866 -1,07435883 9,21686376 4,30874726
h -5,948122776 8,005240488 2,440635237 -5,7884844 8,2197176 2,45101698
h -8,391360997 5,256451511 -0,383807316 -8,37650862 5,79274498 -0,55128888
h -1,544121496 2,879382104 -4,000803566 -1,69925251 3,15467645 -4,20564689
h -8,608919995 0,827525283 3,038210775 -8,63980598 0,96842067 2,25792756
h -6,888058912 3,900108255 6,073493754 -7,09882424 3,79557513 5,6002414
h 0,631831468 1,788169519 3,901971269 0,51177285 1,5001377 4,06445421
c -3,796432773 -12,62943408 -2,320653564 -4,05430227 -12,7649474 -1,53244694
h -2,501585163 -14,0383735 -1,478965062 -2,69763071 -14,1400035 -0,7343572
h -3,831298773 -12,96964747 -4,375836444 -4,28994459 -13,2350081 -3,5494916
h -5,700703177 -13,01505547 -1,564304833 -5,88105619 -13,0640716 -0,57680694
c 8,658029315 -4,555530097 7,507918759 9,00032558 -3,55098013 7,18706517
h 10,51716837 -4,242950078 6,622861242 10,7398867 -2,66569611 6,45635549
h 8,55105494 -6,563862685 8,070949042 9,3893378 -5,56852684 7,53114461
h 8,582154034 -3,424589852 9,260858834 8,59001209 -2,67095399 9,03834622

Anhang 55
Koordinaten Molekül 4
S0-Geometrie T1-Geometrie
p 0,254094596 0,146159791 -1,178189623 0,334874789 -0,03811117 -1,335916847
cu 2,846851261 2,444812897 -3,448613042 2,480937663 2,46120595 -4,273100565
cl 6,103629747 1,222764377 -5,713508887 2,740160064 0,182032183 -7,615418895
n 3,350265619 6,2815139 -2,749750551 3,431209191 5,472633991 -2,565112206
c 1,619840232 -0,277102372 1,987714196 1,865881821 -0,25949258 1,753235053
c 4,20606736 0,147659113 2,295251895 3,370127863 1,722743626 2,644919573
c 5,380148846 -0,271536555 4,621608429 4,524069003 1,576576279 5,011059631
c 4,017295238 -1,120497802 6,720523351 4,236143461 -0,542976791 6,566365435
c 1,414440087 -1,53789042 6,385880085 2,731713043 -2,524255845 5,63832645
c 0,234470721 -1,135880662 4,075896378 1,570042439 -2,401524151 3,285895986
c -0,269966041 -3,133412534 -2,161026946 -0,283381653 -3,307001059 -2,220947839
c -2,20753388 -4,660575661 -1,196439364 -2,552599424 -4,525353828 -1,60246353
c -2,409808222 -7,171888765 -1,93451975 -2,922155393 -7,046261576 -2,24544946
c -0,690197161 -8,270483004 -3,637340073 -1,062234284 -8,454806107 -3,511285136
c 1,23033991 -6,719862327 -4,58343759 1,200332119 -7,214282336 -4,095146068
c 1,44340573 -4,186334708 -3,872129731 1,588086693 -4,683023805 -3,47653542
c 5,786040716 6,88345675 -2,441756589 5,926176528 5,725618895 -1,949281133
c 6,584514328 9,269548171 -1,684604829 6,911736269 7,762831706 -0,706998444
c 1,557426103 8,057084736 -2,407342958 1,750432537 7,44579619 -1,954667982
c 2,231996494 10,51918554 -1,684276938 2,733779943 9,592372662 -0,710380114
c 4,75714542 11,12256248 -1,2900201 5,24830042 9,796897031 -0,054305792
h 2,966350781 -3,017542601 -4,659222481 3,355027856 -3,769429738 -4,032842252
h 2,615108794 -7,4677015 -5,917772558 2,71003882 -8,21003977 -5,087132205
h -3,58965304 -3,878250756 0,129971509 -4,051847454 -3,493265505 -0,622720667
h -3,952726571 -8,30429192 -1,15034968 -4,725058334 -7,926636152 -1,746266528
h 5,330503535 0,800007644 0,681712324 3,655353272 3,411847404 1,491990022
h 7,408052363 0,080516094 4,763322629 5,681003828 3,17436456 5,614913505
h 0,265504037 -2,20578445 7,970480234 2,449851857 -4,226530434 6,777113275
h -1,797129399 -1,481872505 3,906392196 0,426922219 -3,995736841 2,636135697
h 7,125500332 5,363124359 -2,878314976 7,149636533 4,149515856 -2,511826457
h 8,59869482 9,660747361 -1,450548255 8,932868547 7,836054924 -0,29409607
h 5,295347767 13,03990423 -0,732057074 5,965600203 11,50304012 0,862480095
h 0,764200986 11,96010115 -1,510145226 1,450209297 11,17357943 -0,362633051
c -2,856473559 1,540810622 -0,536978826 -2,718260863 1,467169259 -0,791070922
c -5,006154864 1,202905216 -2,08662292 -4,72666948 1,302155617 -2,551626027
c -7,27077608 2,262410886 -1,21367386 -7,03963877 2,314125195 -1,767143019
c -7,289710741 4,110116959 0,661357514 -7,178376552 3,994908569 0,255600017
c -5,018776571 4,938240461 1,734048989 -5,004131638 4,673201058 1,590820974
c -2,897040433 3,426218894 1,323896866 -2,881918906 3,139047137 1,251750198
c -1,100902217 7,435328753 -3,015932849 -0,837850446 7,306920478 -2,81873597
c -3,181140884 8,53379079 -1,728416358 -2,924704297 8,460800351 -1,519357341
c -5,543014213 8,315399527 -2,886933865 -5,23987123 8,538963443 -2,765902596
Anhang 56
S0-Geometrie T1-Geometrie
c -5,968007181 6,577601681 -4,815603741 -5,764992764 7,01480633 -4,867569953
c -4,024773229 4,988227759 -5,636439341 -3,903123764 5,386693571 -5,783999255
c -1,569368857 5,671064725 -4,938125237 -1,431673339 5,812377042 -4,961413104
c -4,850506952 0,25604507 -4,778961912 -4,378488074 0,60405055 -5,303005649
c -4,614243893 2,461658533 -6,80525605 -4,575690575 2,964827107 -7,125990294
h -6,54696931 -0,86644827 -5,235909712 -5,796303887 -0,807739845 -5,897755641
h -3,229771761 -1,02261261 -5,010574066 -2,520502575 -0,288037424 -5,600463685
h -6,41024859 2,593859224 -7,854251571 -6,518705113 3,101828053 -7,870191615
h -3,145699591 1,921807421 -8,181512816 -3,318671747 2,596812703 -8,749912211
c -4,730193429 7,574687312 2,78639149 -4,752122735 7,2258842 2,834159664
c -3,087844356 9,334805414 1,022897237 -2,902110708 8,98319944 1,307553399
h -3,840765172 7,56452953 4,67516828 -4,041277021 7,083526713 4,793450659
h -6,623944694 8,412413136 3,029234829 -6,637889536 8,108553606 2,947051873
h -1,119459115 9,27940351 1,694807299 -0,984001532 8,680875799 2,04856706
h -3,744654847 11,30037246 1,286524136 -3,38894915 10,96790198 1,745113101
h -7,167681032 9,286088304 -2,047879672 -6,819016193 9,532170992 -1,862454138
h -7,900327296 6,26616769 -5,488262087 -7,705880496 6,876828105 -5,571278949
h 0,046751537 4,724661182 -5,831228086 0,090441047 4,941670017 -6,078144735
h -9,023628607 1,856499805 -2,239047776 -8,704131942 2,05502714 -2,9700337
h -9,053105591 5,109957097 1,082570033 -8,948082967 5,013001352 0,597077409
h -1,135268552 3,885613862 2,311833331 -1,226412261 3,440951767 2,453094759
c -0,978437659 -11,0523851 -4,381440154 -1,573970881 -11,20970539 -4,230611425
c 1,101366021 -11,92306599 -6,194812832 0,70190333 -12,44613484 -5,520921857
h 1,074663947 -10,85065236 -7,983255104 1,207772237 -11,48388611 -7,301043869
h 0,823291958 -13,93658986 -6,66678965 0,249665863 -14,42772952 -5,990566635
h 2,997804107 -11,73281709 -5,347335368 2,382321456 -12,46113068 -4,284921228
c -3,546620857 -11,43358355 -5,686581248 -3,824516884 -11,30075652 -6,067624322
h -3,683543963 -10,27245429 -7,414472619 -3,414895624 -10,21530212 -7,801922698
h -5,136061154 -10,91238442 -4,441313911 -5,556254505 -10,51047039 -5,216639901
h -3,795228555 -13,43504581 -6,229370012 -4,229848142 -13,27378794 -6,618439255
c -0,848897489 -12,70410193 -1,993671961 -2,206056048 -12,73584568 -1,841043155
h 0,977168907 -12,45730642 -1,014553478 -0,615957449 -12,70307676 -0,489998305
h -1,047251821 -14,72105434 -2,498047238 -2,590598226 -14,72732686 -2,337097099
h -2,364494586 -12,23856955 -0,640388065 -3,891382943 -11,99597373 -0,861644958
c 5,234393864 -1,605824677 9,300378641 5,479264106 -0,766673515 9,168720114
c 3,955404419 0,072416723 11,29844497 3,415006872 -1,10141342 11,18630189
h 4,180069446 2,093440076 10,82973056 2,128072088 0,540520277 11,21497159
h 4,809896183 -0,255820935 13,17555541 4,277663929 -1,280522263 13,07906318
h 1,913955584 -0,321266004 11,45673576 2,264700549 -2,807265822 10,85108723
c 8,067760591 -1,02000238 9,303988203 7,035897202 1,577262288 9,844068442
h 8,449260823 0,979684467 8,848600173 5,862248021 3,300773378 9,902134973
h 9,109176556 -2,209254157 7,943473451 8,593485781 1,893103675 8,493310133
h 8,860842833 -1,394645189 11,19755123 7,884753672 1,33386595 11,73458443
c 4,881769881 -4,401788977 10,0094867 7,246864188 -3,07265609 9,191617398
Anhang 57
S0-Geometrie T1-Geometrie
h 2,867350406 -4,925866903 10,12528336 6,22249914 -4,846114875 8,801383155
h 5,746633346 -4,794121888 11,86948263 8,156709308 -3,265310986 11,06092519
h 5,783205782 -5,646804437 8,598815296 8,75042074 -2,875325501 7,758208604
Koordinaten Molekül 5
S0-Geometrie T1-Geometrie
p 0,822166523 -2,463957178 -0,324108033 0,823605346 -2,544209274 -0,35048915
cu 2,842888467 0,368020245 -2,736865179 2,828961789 0,73458596 -2,809132266
cl 6,192392 -1,220291474 -4,706319465 6,211345364 -1,307698071 -4,120256934
n 4,055894316 3,550061246 -0,697434817 4,200580961 3,851014163 -1,269152563
c 2,520747479 -3,04178488 2,642376748 2,546356397 -3,085298967 2,605947486
c 5,166646438 -3,027490459 2,495075358 5,189148193 -2,94084553 2,552665988
c 6,612923349 -3,574659079 4,629995655 6,582906299 -3,460933786 4,730112214
c 5,435827297 -4,11276333 6,925670942 5,354554205 -4,115497927 6,965728666
c 2,802391464 -4,126695211 7,081100418 2,720526054 -4,263748646 7,025451328
c 1,347440885 -3,607439961 4,947380875 1,318837866 -3,758846077 4,855837872
c 0,210897157 -5,688075197 -1,423707673 0,190282041 -5,757479522 -1,450501391
c -1,633617324 -7,262782756 -0,358808186 -1,720963445 -7,264786857 -0,405739056
c -1,944074288 -9,720141374 -1,248825931 -2,067531774 -9,730039322 -1,258695355
c -0,401919842 -10,62762949 -3,190123946 -0,502178731 -10,71061513 -3,144423231
c 1,448184252 -9,073076503 -4,238431801 1,411074834 -9,221718129 -4,174226107
c 1,758870053 -6,601715092 -3,368138459 1,762647421 -6,745605541 -3,336447033
c 6,434373127 3,938574236 0,02767483 6,604921488 4,278990709 -0,63604955
c 7,270028899 6,255539576 0,948532325 7,360443364 6,381061165 0,724604495
c 2,371924357 5,444959336 -0,592969759 2,309924298 5,509933622 -0,598297079
c 3,085761995 7,858576657 0,231100755 2,991706463 7,711897478 0,784409699
c 5,556623607 8,257031223 1,031843912 5,474860091 8,132075118 1,436003326
h 3,22058864 -5,370940212 -4,179672965 3,276750218 -5,573958226 -4,128385811
h -0,646549648 -12,56095654 -3,883625478 -0,778998512 -12,64877998 -3,810752931
h 2,664141012 -9,780603174 -5,754189142 2,64455215 -9,986216469 -5,647134607
h -2,846455705 -6,556503706 1,162130577 -2,960925935 -6,497436381 1,062345492
h -3,393888398 -10,93991397 -0,418955006 -3,568580018 -10,89684974 -0,444909699
h 6,079914073 -2,614536833 0,679177141 6,154761637 -2,443533826 0,789372687
h 8,67654152 -3,574350049 4,494633654 8,646321946 -3,345547956 4,67154213
h 6,57286958 -4,529051925 8,602470749 6,450929824 -4,513547085 8,673543161
h 1,871848102 -4,557226562 8,877524507 1,74913601 -4,780272053 8,776521089
h -0,716366038 -3,632069309 5,084822704 -0,744966941 -3,872307901 4,921297558
h 7,711391693 2,324453546 -0,193152173 7,952584053 2,849260434 -1,288812999
h 9,234398328 6,492066187 1,541345729 9,349807899 6,669453522 1,189001791
h 6,153612255 10,13053859 1,671461561 5,984199742 9,844571436 2,477927598
h 1,741254103 9,422161842 0,161206635 1,553946143 9,10525777 1,26496469
c -2,190381706 -1,031919944 0,542889701 -2,150963009 -1,049128241 0,550489325

Anhang 58
S0-Geometrie T1-Geometrie
c -4,334417472 -1,144082158 -1,048330247 -4,314821005 -1,142162815 -1,001372269
c -6,488849724 0,182130995 -0,257982747 -6,418286257 0,261663373 -0,205946406
c -6,377210424 2,028428794 1,613606321 -6,222633406 2,1193774 1,644893697
c -4,059490761 2,587638425 2,759338268 -3,880345608 2,627928041 2,765390634
c -2,091285545 0,870882674 2,394739864 -1,966093997 0,853305702 2,396308777
c -0,17020003 4,856384878 -1,600712957 -0,149021779 4,899282122 -1,520436315
c -2,422692063 6,093782274 -0,860647377 -2,49629273 6,160307986 -0,955491487
c -4,461564011 5,771247265 -2,508939555 -4,425131638 5,797073776 -2,674875888
c -4,408350908 3,938690474 -4,392441866 -4,28917292 3,937779368 -4,586204923
c -2,347834689 2,295503423 -4,464775439 -2,291130285 2,238809092 -4,4584727
n -0,206113024 2,936595969 -3,273607002 -0,241164747 2,786910962 -3,074157299
c -4,234990708 -2,087375524 -3,749375704 -4,274197249 -2,101195977 -3,694698303
c -2,569867028 -0,34502766 -5,500400561 -2,559990734 -0,419825748 -5,44829312
h -6,182955266 -2,112799286 -4,489903086 -6,224972104 -2,04550346 -4,423909317
h -3,513627614 -4,033659201 -3,907935394 -3,637248508 -4,076977515 -3,855250044
h -3,421642655 -0,325901307 -7,403924377 -3,397920853 -0,396963673 -7,356657125
h -0,647986606 -1,132444045 -5,691681762 -0,673202583 -1,289759839 -5,647904097
c -3,486424743 5,156155468 3,839165385 -3,235508309 5,210730974 3,77747352
c -2,933976499 7,2094868 1,724525713 -3,165315285 7,246212575 1,607560761
h -1,818037392 5,017084175 5,079382801 -1,371705949 5,13020008 4,707187913
h -5,058059104 5,85956253 5,016027395 -4,604660672 5,858876022 5,214252681
h -1,388956997 8,415975976 2,411495401 -1,923452454 8,794257525 2,241402149
h -4,604768765 8,442724344 1,552699586 -5,065177141 8,087110259 1,433768639
h -6,208217517 6,829939424 -2,171813185 -6,227212612 6,77390784 -2,373222408
h -6,065035874 3,592199044 -5,577961513 -5,912016605 3,546937988 -5,801077469
h -8,239784285 -0,037136935 -1,342015362 -8,18408163 0,094115406 -1,273215965
h -8,037270304 3,208474725 1,98350659 -7,837917172 3,362411859 2,005853587
h -0,309678012 1,180129984 3,40487405 -0,161889499 1,131989942 3,372731945
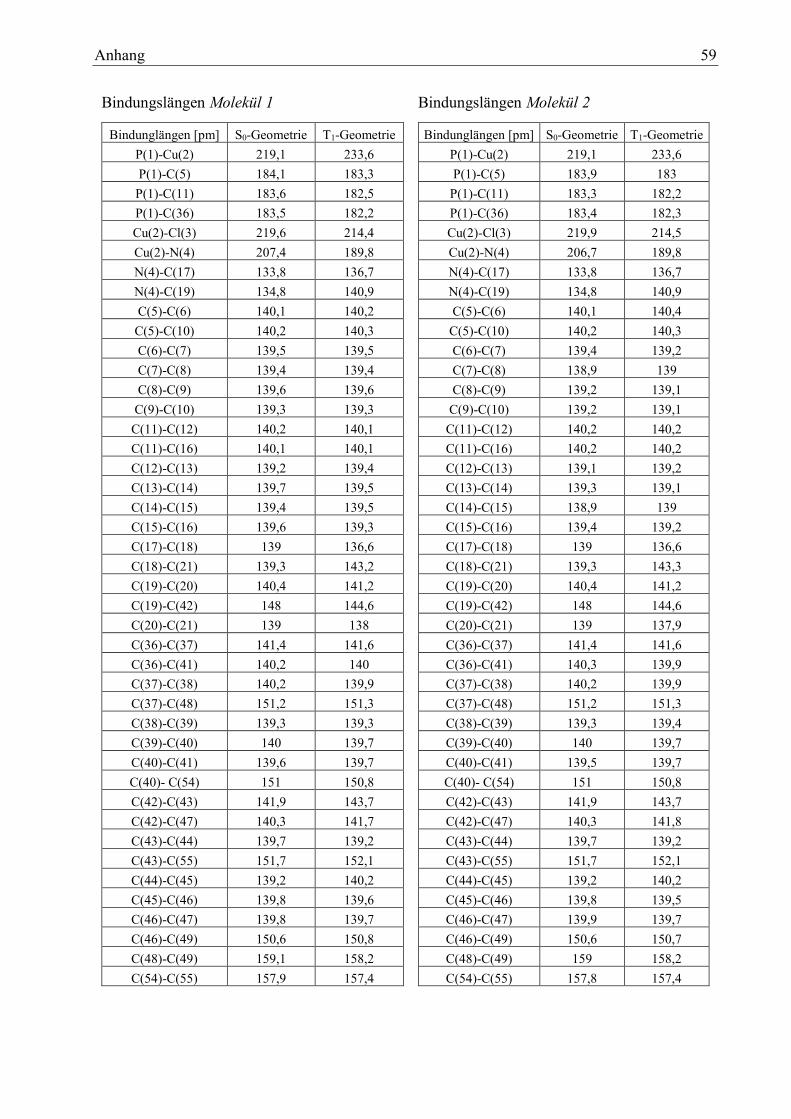
Anhang 59
Bindungslängen Molekül 1 Bindungslängen Molekül 2
Bindunglängen [pm] S0-Geometrie T1-Geometrie
P(1)-Cu(2) 219,1 233,6
P(1)-C(5) 184,1 183,3
P(1)-C(11) 183,6 182,5
P(1)-C(36) 183,5 182,2
Cu(2)-Cl(3) 219,6 214,4
Cu(2)-N(4) 207,4 189,8
N(4)-C(17) 133,8 136,7
N(4)-C(19) 134,8 140,9
C(5)-C(6) 140,1 140,2
C(5)-C(10) 140,2 140,3
C(6)-C(7) 139,5 139,5
C(7)-C(8) 139,4 139,4
C(8)-C(9) 139,6 139,6
C(9)-C(10) 139,3 139,3
C(11)-C(12) 140,2 140,1
C(11)-C(16) 140,1 140,1
C(12)-C(13) 139,2 139,4
C(13)-C(14) 139,7 139,5
C(14)-C(15) 139,4 139,5
C(15)-C(16) 139,6 139,3
C(17)-C(18) 139 136,6
C(18)-C(21) 139,3 143,2
C(19)-C(20) 140,4 141,2
C(19)-C(42) 148 144,6
C(20)-C(21) 139 138
C(36)-C(37) 141,4 141,6
C(36)-C(41) 140,2 140
C(37)-C(38) 140,2 139,9
C(37)-C(48) 151,2 151,3
C(38)-C(39) 139,3 139,3
C(39)-C(40) 140 139,7
C(40)-C(41) 139,6 139,7
C(40)- C(54) 151 150,8
C(42)-C(43) 141,9 143,7
C(42)-C(47) 140,3 141,7
C(43)-C(44) 139,7 139,2
C(43)-C(55) 151,7 152,1
C(44)-C(45) 139,2 140,2
C(45)-C(46) 139,8 139,6
C(46)-C(47) 139,8 139,7
C(46)-C(49) 150,6 150,8
C(48)-C(49) 159,1 158,2
C(54)-C(55) 157,9 157,4
Bindunglängen [pm] S0-Geometrie T1-Geometrie
P(1)-Cu(2) 219,1 233,6
P(1)-C(5) 183,9 183
P(1)-C(11) 183,3 182,2
P(1)-C(36) 183,4 182,3
Cu(2)-Cl(3) 219,9 214,5
Cu(2)-N(4) 206,7 189,8
N(4)-C(17) 133,8 136,7
N(4)-C(19) 134,8 140,9
C(5)-C(6) 140,1 140,4
C(5)-C(10) 140,2 140,3
C(6)-C(7) 139,4 139,2
C(7)-C(8) 138,9 139
C(8)-C(9) 139,2 139,1
C(9)-C(10) 139,2 139,1
C(11)-C(12) 140,2 140,2
C(11)-C(16) 140,2 140,2
C(12)-C(13) 139,1 139,2
C(13)-C(14) 139,3 139,1
C(14)-C(15) 138,9 139
C(15)-C(16) 139,4 139,2
C(17)-C(18) 139 136,6
C(18)-C(21) 139,3 143,3
C(19)-C(20) 140,4 141,2
C(19)-C(42) 148 144,6
C(20)-C(21) 139 137,9
C(36)-C(37) 141,4 141,6
C(36)-C(41) 140,3 139,9
C(37)-C(38) 140,2 139,9
C(37)-C(48) 151,2 151,3
C(38)-C(39) 139,3 139,4
C(39)-C(40) 140 139,7
C(40)-C(41) 139,5 139,7
C(40)- C(54) 151 150,8
C(42)-C(43) 141,9 143,7
C(42)-C(47) 140,3 141,8
C(43)-C(44) 139,7 139,2
C(43)-C(55) 151,7 152,1
C(44)-C(45) 139,2 140,2
C(45)-C(46) 139,8 139,5
C(46)-C(47) 139,9 139,7
C(46)-C(49) 150,6 150,7
C(48)-C(49) 159 158,2
C(54)-C(55) 157,8 157,4
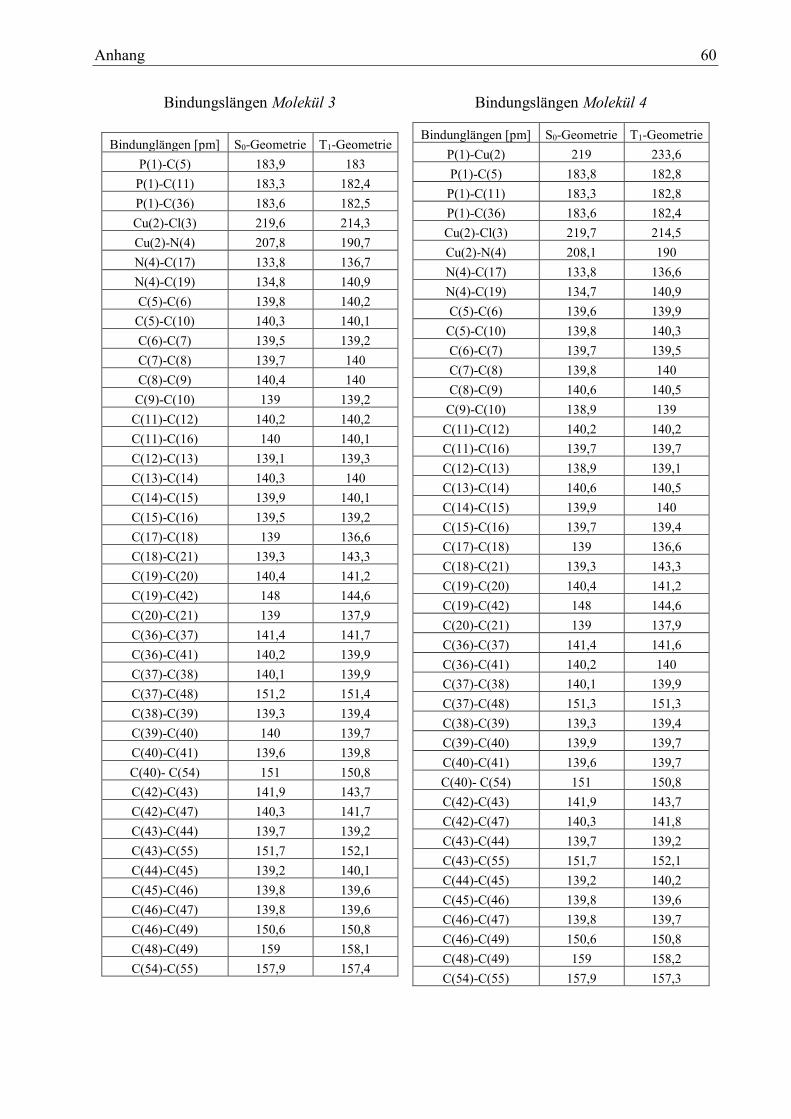
Anhang 60
Bindungslängen Molekül 3 Bindungslängen Molekül 4
Bindunglängen [pm] S0-Geometrie T1-Geometrie
P(1)-C(5) 183,9 183
P(1)-C(11) 183,3 182,4
P(1)-C(36) 183,6 182,5
Cu(2)-Cl(3) 219,6 214,3
Cu(2)-N(4) 207,8 190,7
N(4)-C(17) 133,8 136,7
N(4)-C(19) 134,8 140,9
C(5)-C(6) 139,8 140,2
C(5)-C(10) 140,3 140,1
C(6)-C(7) 139,5 139,2
C(7)-C(8) 139,7 140
C(8)-C(9) 140,4 140
C(9)-C(10) 139 139,2
C(11)-C(12) 140,2 140,2
C(11)-C(16) 140 140,1
C(12)-C(13) 139,1 139,3
C(13)-C(14) 140,3 140
C(14)-C(15) 139,9 140,1
C(15)-C(16) 139,5 139,2
C(17)-C(18) 139 136,6
C(18)-C(21) 139,3 143,3
C(19)-C(20) 140,4 141,2
C(19)-C(42) 148 144,6
C(20)-C(21) 139 137,9
C(36)-C(37) 141,4 141,7
C(36)-C(41) 140,2 139,9
C(37)-C(38) 140,1 139,9
C(37)-C(48) 151,2 151,4
C(38)-C(39) 139,3 139,4
C(39)-C(40) 140 139,7
C(40)-C(41) 139,6 139,8
C(40)- C(54) 151 150,8
C(42)-C(43) 141,9 143,7
C(42)-C(47) 140,3 141,7
C(43)-C(44) 139,7 139,2
C(43)-C(55) 151,7 152,1
C(44)-C(45) 139,2 140,1
C(45)-C(46) 139,8 139,6
C(46)-C(47) 139,8 139,6
C(46)-C(49) 150,6 150,8
C(48)-C(49) 159 158,1
C(54)-C(55) 157,9 157,4
Bindunglängen [pm] S0-Geometrie T1-Geometrie
P(1)-Cu(2) 219 233,6
P(1)-C(5) 183,8 182,8
P(1)-C(11) 183,3 182,8
P(1)-C(36) 183,6 182,4
Cu(2)-Cl(3) 219,7 214,5
Cu(2)-N(4) 208,1 190
N(4)-C(17) 133,8 136,6
N(4)-C(19) 134,7 140,9
C(5)-C(6) 139,6 139,9
C(5)-C(10) 139,8 140,3
C(6)-C(7) 139,7 139,5
C(7)-C(8) 139,8 140
C(8)-C(9) 140,6 140,5
C(9)-C(10) 138,9 139
C(11)-C(12) 140,2 140,2
C(11)-C(16) 139,7 139,7
C(12)-C(13) 138,9 139,1
C(13)-C(14) 140,6 140,5
C(14)-C(15) 139,9 140
C(15)-C(16) 139,7 139,4
C(17)-C(18) 139 136,6
C(18)-C(21) 139,3 143,3
C(19)-C(20) 140,4 141,2
C(19)-C(42) 148 144,6
C(20)-C(21) 139 137,9
C(36)-C(37) 141,4 141,6
C(36)-C(41) 140,2 140
C(37)-C(38) 140,1 139,9
C(37)-C(48) 151,3 151,3
C(38)-C(39) 139,3 139,4
C(39)-C(40) 139,9 139,7
C(40)-C(41) 139,6 139,7
C(40)- C(54) 151 150,8
C(42)-C(43) 141,9 143,7
C(42)-C(47) 140,3 141,8
C(43)-C(44) 139,7 139,2
C(43)-C(55) 151,7 152,1
C(44)-C(45) 139,2 140,2
C(45)-C(46) 139,8 139,6
C(46)-C(47) 139,8 139,7
C(46)-C(49) 150,6 150,8
C(48)-C(49) 159 158,2
C(54)-C(55) 157,9 157,3
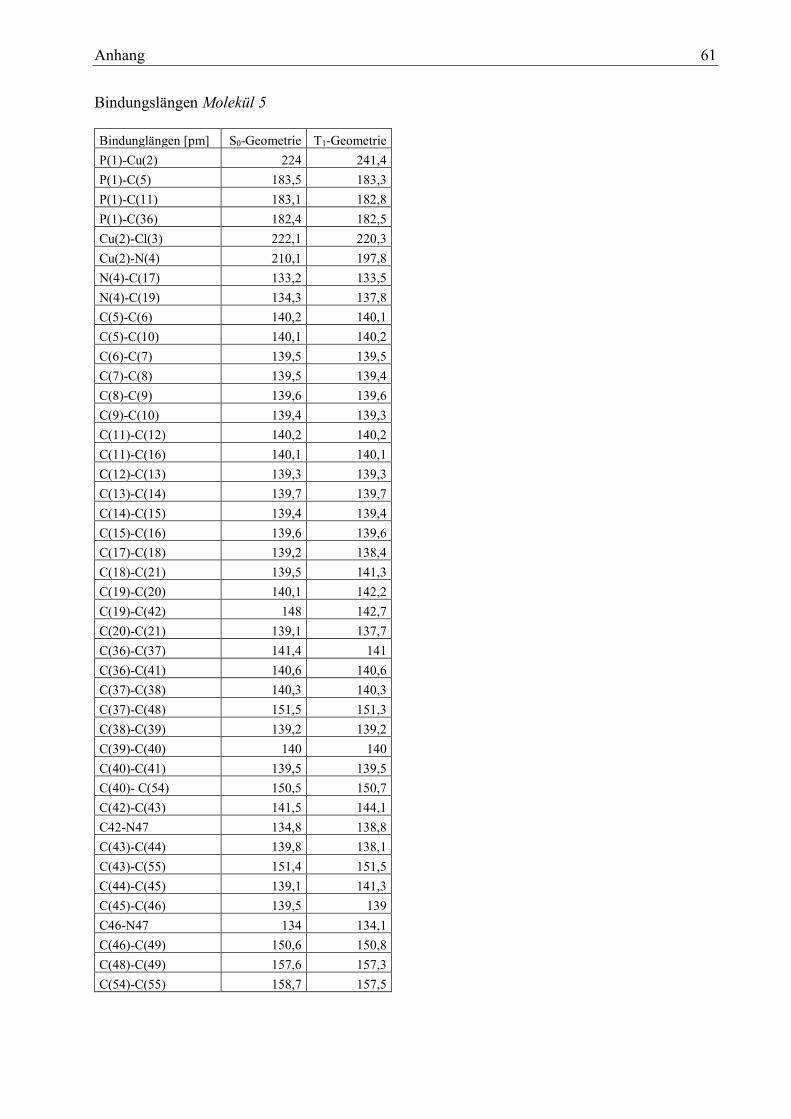
Anhang 61
Bindungslängen Molekül 5
Bindunglängen [pm] S0-Geometrie T1-Geometrie
P(1)-Cu(2) 224 241,4
P(1)-C(5) 183,5 183,3
P(1)-C(11) 183,1 182,8
P(1)-C(36) 182,4 182,5
Cu(2)-Cl(3) 222,1 220,3
Cu(2)-N(4) 210,1 197,8
N(4)-C(17) 133,2 133,5
N(4)-C(19) 134,3 137,8
C(5)-C(6) 140,2 140,1
C(5)-C(10) 140,1 140,2
C(6)-C(7) 139,5 139,5
C(7)-C(8) 139,5 139,4
C(8)-C(9) 139,6 139,6
C(9)-C(10) 139,4 139,3
C(11)-C(12) 140,2 140,2
C(11)-C(16) 140,1 140,1
C(12)-C(13) 139,3 139,3
C(13)-C(14) 139,7 139,7
C(14)-C(15) 139,4 139,4
C(15)-C(16) 139,6 139,6
C(17)-C(18) 139,2 138,4
C(18)-C(21) 139,5 141,3
C(19)-C(20) 140,1 142,2
C(19)-C(42) 148 142,7
C(20)-C(21) 139,1 137,7
C(36)-C(37) 141,4 141
C(36)-C(41) 140,6 140,6
C(37)-C(38) 140,3 140,3
C(37)-C(48) 151,5 151,3
C(38)-C(39) 139,2 139,2
C(39)-C(40) 140 140
C(40)-C(41) 139,5 139,5
C(40)- C(54) 150,5 150,7
C(42)-C(43) 141,5 144,1
C42-N47 134,8 138,8
C(43)-C(44) 139,8 138,1
C(43)-C(55) 151,4 151,5
C(44)-C(45) 139,1 141,3
C(45)-C(46) 139,5 139
C46-N47 134 134,1
C(46)-C(49) 150,6 150,8
C(48)-C(49) 157,6 157,3
C(54)-C(55) 158,7 157,5
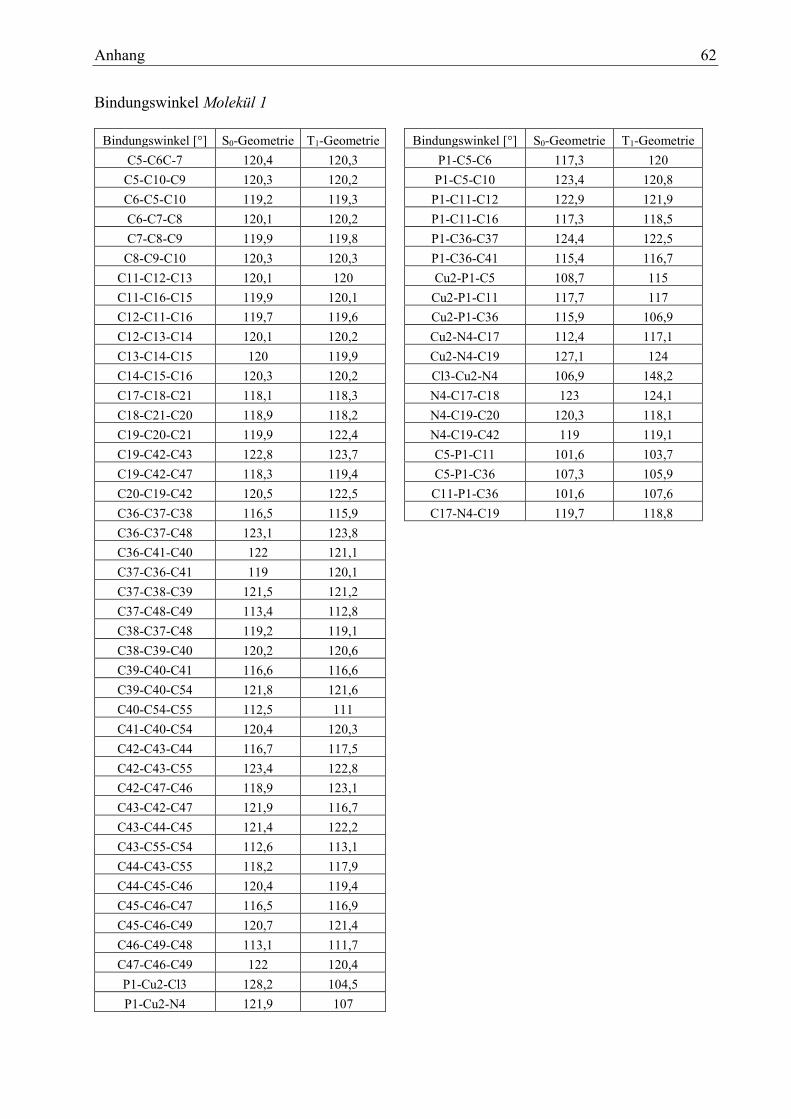
Anhang 62
Bindungswinkel Molekül 1
Bindungswinkel [°] S0-Geometrie T1-Geometrie Bindungswinkel [°] S0-Geometrie T1-Geometrie
C5-C6C-7 120,4 120,3 P1-C5-C6 117,3 120
C5-C10-C9 120,3 120,2 P1-C5-C10 123,4 120,8
C6-C5-C10 119,2 119,3 P1-C11-C12 122,9 121,9
C6-C7-C8 120,1 120,2 P1-C11-C16 117,3 118,5
C7-C8-C9 119,9 119,8 P1-C36-C37 124,4 122,5
C8-C9-C10 120,3 120,3 P1-C36-C41 115,4 116,7
C11-C12-C13 120,1 120 Cu2-P1-C5 108,7 115
C11-C16-C15 119,9 120,1 Cu2-P1-C11 117,7 117
C12-C11-C16 119,7 119,6 Cu2-P1-C36 115,9 106,9
C12-C13-C14 120,1 120,2 Cu2-N4-C17 112,4 117,1
C13-C14-C15 120 119,9 Cu2-N4-C19 127,1 124
C14-C15-C16 120,3 120,2 Cl3-Cu2-N4 106,9 148,2
C17-C18-C21 118,1 118,3 N4-C17-C18 123 124,1
C18-C21-C20 118,9 118,2 N4-C19-C20 120,3 118,1
C19-C20-C21 119,9 122,4 N4-C19-C42 119 119,1
C19-C42-C43 122,8 123,7 C5-P1-C11 101,6 103,7
C19-C42-C47 118,3 119,4 C5-P1-C36 107,3 105,9
C20-C19-C42 120,5 122,5 C11-P1-C36 101,6 107,6
C36-C37-C38 116,5 115,9 C17-N4-C19 119,7 118,8
C36-C37-C48 123,1 123,8
C36-C41-C40 122 121,1
C37-C36-C41 119 120,1
C37-C38-C39 121,5 121,2
C37-C48-C49 113,4 112,8
C38-C37-C48 119,2 119,1
C38-C39-C40 120,2 120,6
C39-C40-C41 116,6 116,6
C39-C40-C54 121,8 121,6
C40-C54-C55 112,5 111
C41-C40-C54 120,4 120,3
C42-C43-C44 116,7 117,5
C42-C43-C55 123,4 122,8
C42-C47-C46 118,9 123,1
C43-C42-C47 121,9 116,7
C43-C44-C45 121,4 122,2
C43-C55-C54 112,6 113,1
C44-C43-C55 118,2 117,9
C44-C45-C46 120,4 119,4
C45-C46-C47 116,5 116,9
C45-C46-C49 120,7 121,4
C46-C49-C48 113,1 111,7
C47-C46-C49 122 120,4
P1-Cu2-Cl3 128,2 104,5
P1-Cu2-N4 121,9 107
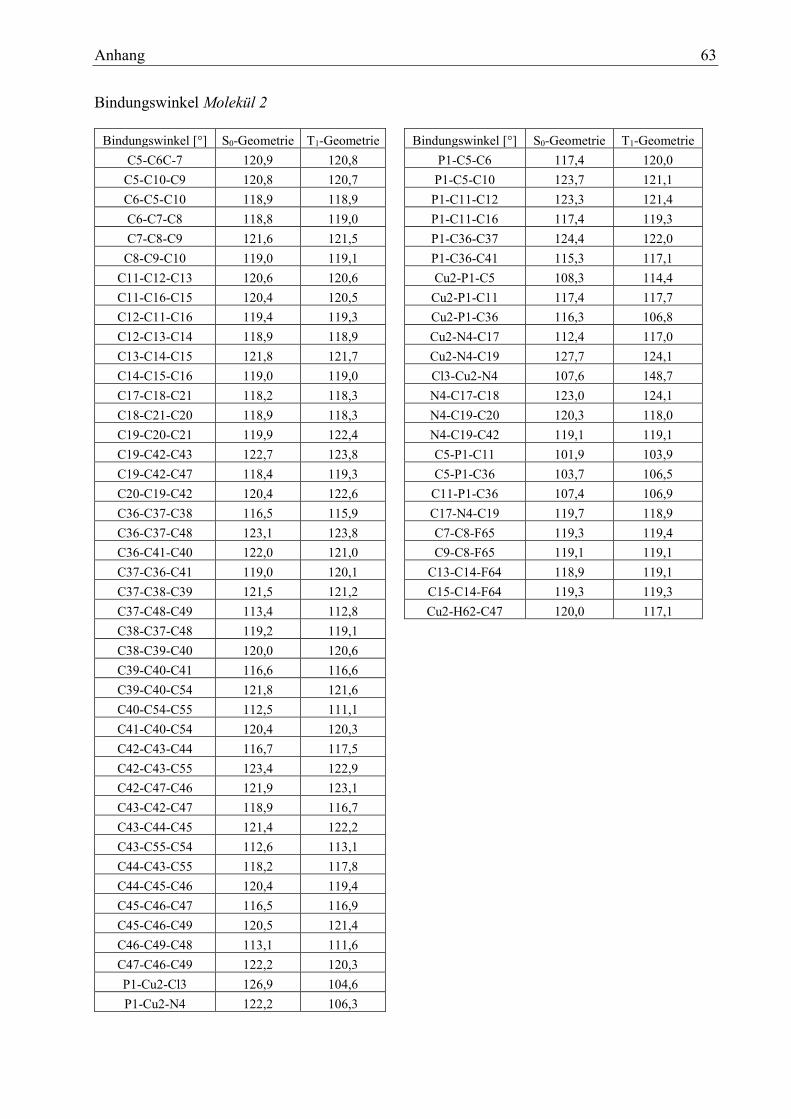
Anhang 63
Bindungswinkel Molekül 2
Bindungswinkel [°] S0-Geometrie T1-Geometrie Bindungswinkel [°] S0-Geometrie T1-Geometrie
C5-C6C-7 120,9 120,8 P1-C5-C6 117,4 120,0
C5-C10-C9 120,8 120,7 P1-C5-C10 123,7 121,1
C6-C5-C10 118,9 118,9 P1-C11-C12 123,3 121,4
C6-C7-C8 118,8 119,0 P1-C11-C16 117,4 119,3
C7-C8-C9 121,6 121,5 P1-C36-C37 124,4 122,0
C8-C9-C10 119,0 119,1 P1-C36-C41 115,3 117,1
C11-C12-C13 120,6 120,6 Cu2-P1-C5 108,3 114,4
C11-C16-C15 120,4 120,5 Cu2-P1-C11 117,4 117,7
C12-C11-C16 119,4 119,3 Cu2-P1-C36 116,3 106,8
C12-C13-C14 118,9 118,9 Cu2-N4-C17 112,4 117,0
C13-C14-C15 121,8 121,7 Cu2-N4-C19 127,7 124,1
C14-C15-C16 119,0 119,0 Cl3-Cu2-N4 107,6 148,7
C17-C18-C21 118,2 118,3 N4-C17-C18 123,0 124,1
C18-C21-C20 118,9 118,3 N4-C19-C20 120,3 118,0
C19-C20-C21 119,9 122,4 N4-C19-C42 119,1 119,1
C19-C42-C43 122,7 123,8 C5-P1-C11 101,9 103,9
C19-C42-C47 118,4 119,3 C5-P1-C36 103,7 106,5
C20-C19-C42 120,4 122,6 C11-P1-C36 107,4 106,9
C36-C37-C38 116,5 115,9 C17-N4-C19 119,7 118,9
C36-C37-C48 123,1 123,8 C7-C8-F65 119,3 119,4
C36-C41-C40 122,0 121,0 C9-C8-F65 119,1 119,1
C37-C36-C41 119,0 120,1 C13-C14-F64 118,9 119,1
C37-C38-C39 121,5 121,2 C15-C14-F64 119,3 119,3
C37-C48-C49 113,4 112,8 Cu2-H62-C47 120,0 117,1
C38-C37-C48 119,2 119,1
C38-C39-C40 120,0 120,6
C39-C40-C41 116,6 116,6
C39-C40-C54 121,8 121,6
C40-C54-C55 112,5 111,1
C41-C40-C54 120,4 120,3
C42-C43-C44 116,7 117,5
C42-C43-C55 123,4 122,9
C42-C47-C46 121,9 123,1
C43-C42-C47 118,9 116,7
C43-C44-C45 121,4 122,2
C43-C55-C54 112,6 113,1
C44-C43-C55 118,2 117,8
C44-C45-C46 120,4 119,4
C45-C46-C47 116,5 116,9
C45-C46-C49 120,5 121,4
C46-C49-C48 113,1 111,6
C47-C46-C49 122,2 120,3
P1-Cu2-Cl3 126,9 104,6
P1-Cu2-N4 122,2 106,3
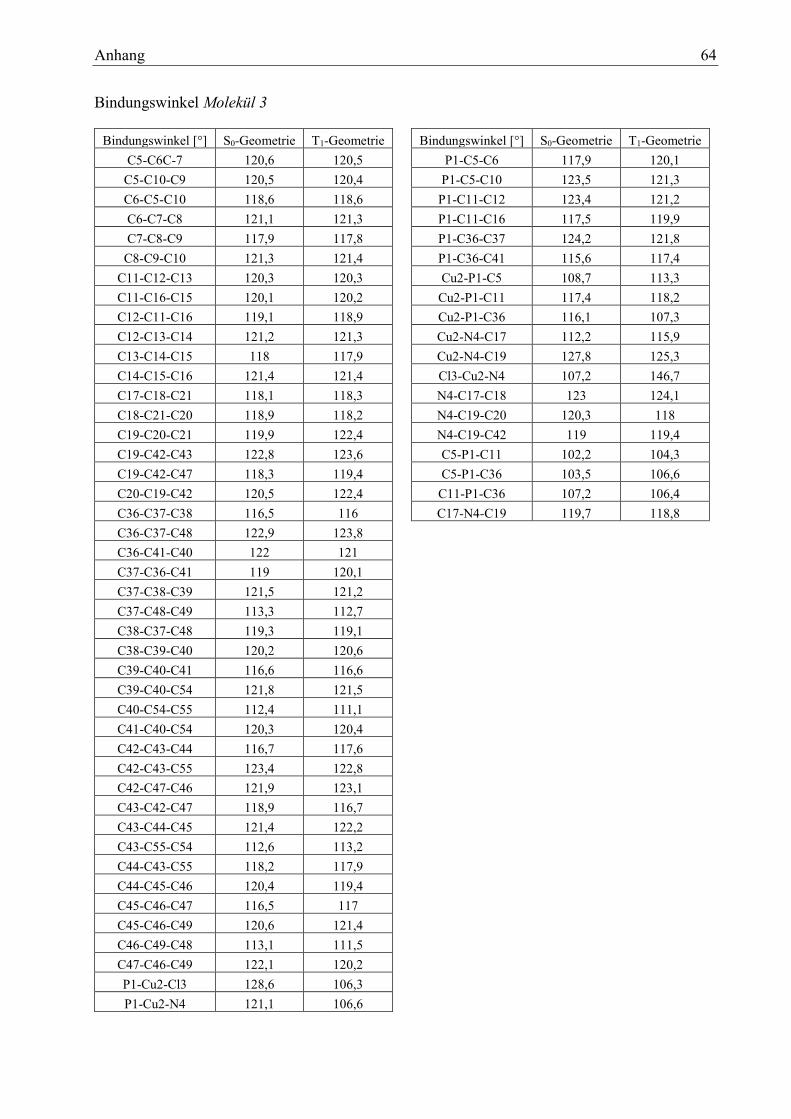
Anhang 64
Bindungswinkel Molekül 3
Bindungswinkel [°] S0-Geometrie T1-Geometrie Bindungswinkel [°] S0-Geometrie T1-Geometrie
C5-C6C-7 120,6 120,5 P1-C5-C6 117,9 120,1
C5-C10-C9 120,5 120,4 P1-C5-C10 123,5 121,3
C6-C5-C10 118,6 118,6 P1-C11-C12 123,4 121,2
C6-C7-C8 121,1 121,3 P1-C11-C16 117,5 119,9
C7-C8-C9 117,9 117,8 P1-C36-C37 124,2 121,8
C8-C9-C10 121,3 121,4 P1-C36-C41 115,6 117,4
C11-C12-C13 120,3 120,3 Cu2-P1-C5 108,7 113,3
C11-C16-C15 120,1 120,2 Cu2-P1-C11 117,4 118,2
C12-C11-C16 119,1 118,9 Cu2-P1-C36 116,1 107,3
C12-C13-C14 121,2 121,3 Cu2-N4-C17 112,2 115,9
C13-C14-C15 118 117,9 Cu2-N4-C19 127,8 125,3
C14-C15-C16 121,4 121,4 Cl3-Cu2-N4 107,2 146,7
C17-C18-C21 118,1 118,3 N4-C17-C18 123 124,1
C18-C21-C20 118,9 118,2 N4-C19-C20 120,3 118
C19-C20-C21 119,9 122,4 N4-C19-C42 119 119,4
C19-C42-C43 122,8 123,6 C5-P1-C11 102,2 104,3
C19-C42-C47 118,3 119,4 C5-P1-C36 103,5 106,6
C20-C19-C42 120,5 122,4 C11-P1-C36 107,2 106,4
C36-C37-C38 116,5 116 C17-N4-C19 119,7 118,8
C36-C37-C48 122,9 123,8
C36-C41-C40 122 121
C37-C36-C41 119 120,1
C37-C38-C39 121,5 121,2
C37-C48-C49 113,3 112,7
C38-C37-C48 119,3 119,1
C38-C39-C40 120,2 120,6
C39-C40-C41 116,6 116,6
C39-C40-C54 121,8 121,5
C40-C54-C55 112,4 111,1
C41-C40-C54 120,3 120,4
C42-C43-C44 116,7 117,6
C42-C43-C55 123,4 122,8
C42-C47-C46 121,9 123,1
C43-C42-C47 118,9 116,7
C43-C44-C45 121,4 122,2
C43-C55-C54 112,6 113,2
C44-C43-C55 118,2 117,9
C44-C45-C46 120,4 119,4
C45-C46-C47 116,5 117
C45-C46-C49 120,6 121,4
C46-C49-C48 113,1 111,5
C47-C46-C49 122,1 120,2
P1-Cu2-Cl3 128,6 106,3
P1-Cu2-N4 121,1 106,6
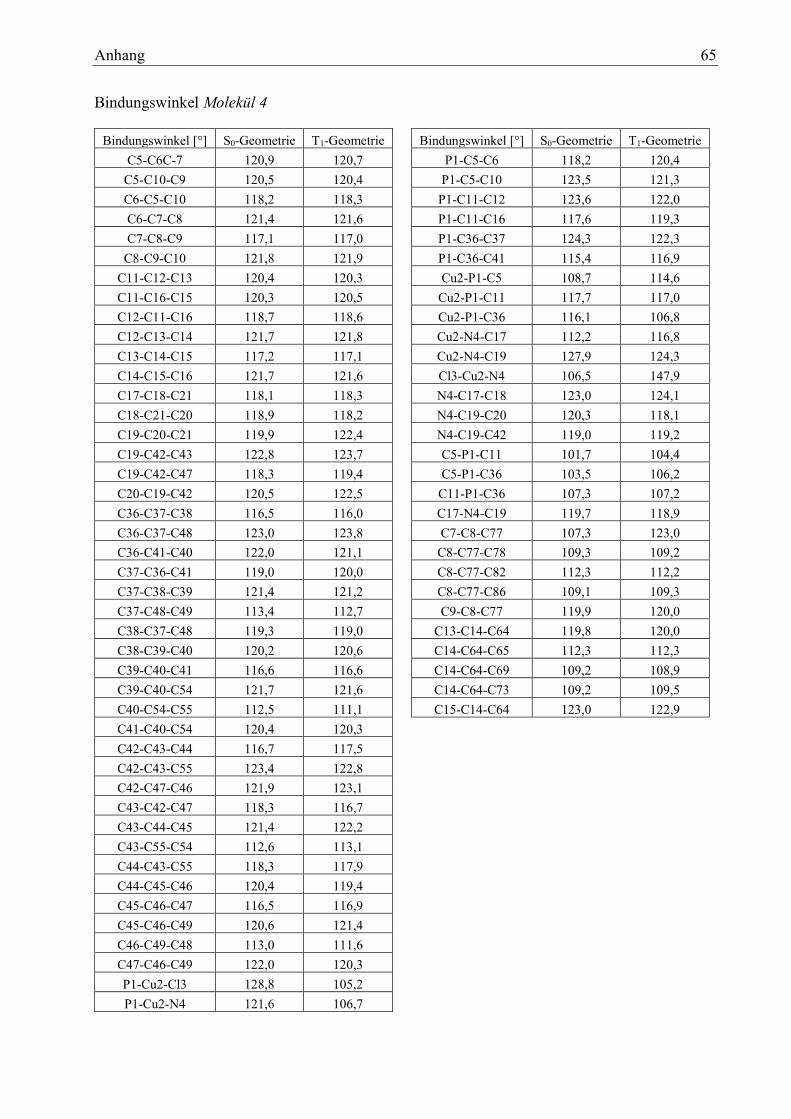
Anhang 65
Bindungswinkel Molekül 4
Bindungswinkel [°] S0-Geometrie T1-Geometrie Bindungswinkel [°] S0-Geometrie T1-Geometrie
C5-C6C-7 120,9 120,7 P1-C5-C6 118,2 120,4
C5-C10-C9 120,5 120,4 P1-C5-C10 123,5 121,3
C6-C5-C10 118,2 118,3 P1-C11-C12 123,6 122,0
C6-C7-C8 121,4 121,6 P1-C11-C16 117,6 119,3
C7-C8-C9 117,1 117,0 P1-C36-C37 124,3 122,3
C8-C9-C10 121,8 121,9 P1-C36-C41 115,4 116,9
C11-C12-C13 120,4 120,3 Cu2-P1-C5 108,7 114,6
C11-C16-C15 120,3 120,5 Cu2-P1-C11 117,7 117,0
C12-C11-C16 118,7 118,6 Cu2-P1-C36 116,1 106,8
C12-C13-C14 121,7 121,8 Cu2-N4-C17 112,2 116,8
C13-C14-C15 117,2 117,1 Cu2-N4-C19 127,9 124,3
C14-C15-C16 121,7 121,6 Cl3-Cu2-N4 106,5 147,9
C17-C18-C21 118,1 118,3 N4-C17-C18 123,0 124,1
C18-C21-C20 118,9 118,2 N4-C19-C20 120,3 118,1
C19-C20-C21 119,9 122,4 N4-C19-C42 119,0 119,2
C19-C42-C43 122,8 123,7 C5-P1-C11 101,7 104,4
C19-C42-C47 118,3 119,4 C5-P1-C36 103,5 106,2
C20-C19-C42 120,5 122,5 C11-P1-C36 107,3 107,2
C36-C37-C38 116,5 116,0 C17-N4-C19 119,7 118,9
C36-C37-C48 123,0 123,8 C7-C8-C77 107,3 123,0
C36-C41-C40 122,0 121,1 C8-C77-C78 109,3 109,2
C37-C36-C41 119,0 120,0 C8-C77-C82 112,3 112,2
C37-C38-C39 121,4 121,2 C8-C77-C86 109,1 109,3
C37-C48-C49 113,4 112,7 C9-C8-C77 119,9 120,0
C38-C37-C48 119,3 119,0 C13-C14-C64 119,8 120,0
C38-C39-C40 120,2 120,6 C14-C64-C65 112,3 112,3
C39-C40-C41 116,6 116,6 C14-C64-C69 109,2 108,9
C39-C40-C54 121,7 121,6 C14-C64-C73 109,2 109,5
C40-C54-C55 112,5 111,1 C15-C14-C64 123,0 122,9
C41-C40-C54 120,4 120,3
C42-C43-C44 116,7 117,5
C42-C43-C55 123,4 122,8
C42-C47-C46 121,9 123,1
C43-C42-C47 118,3 116,7
C43-C44-C45 121,4 122,2
C43-C55-C54 112,6 113,1
C44-C43-C55 118,3 117,9
C44-C45-C46 120,4 119,4
C45-C46-C47 116,5 116,9
C45-C46-C49 120,6 121,4
C46-C49-C48 113,0 111,6
C47-C46-C49 122,0 120,3
P1-Cu2-Cl3 128,8 105,2
P1-Cu2-N4 121,6 106,7
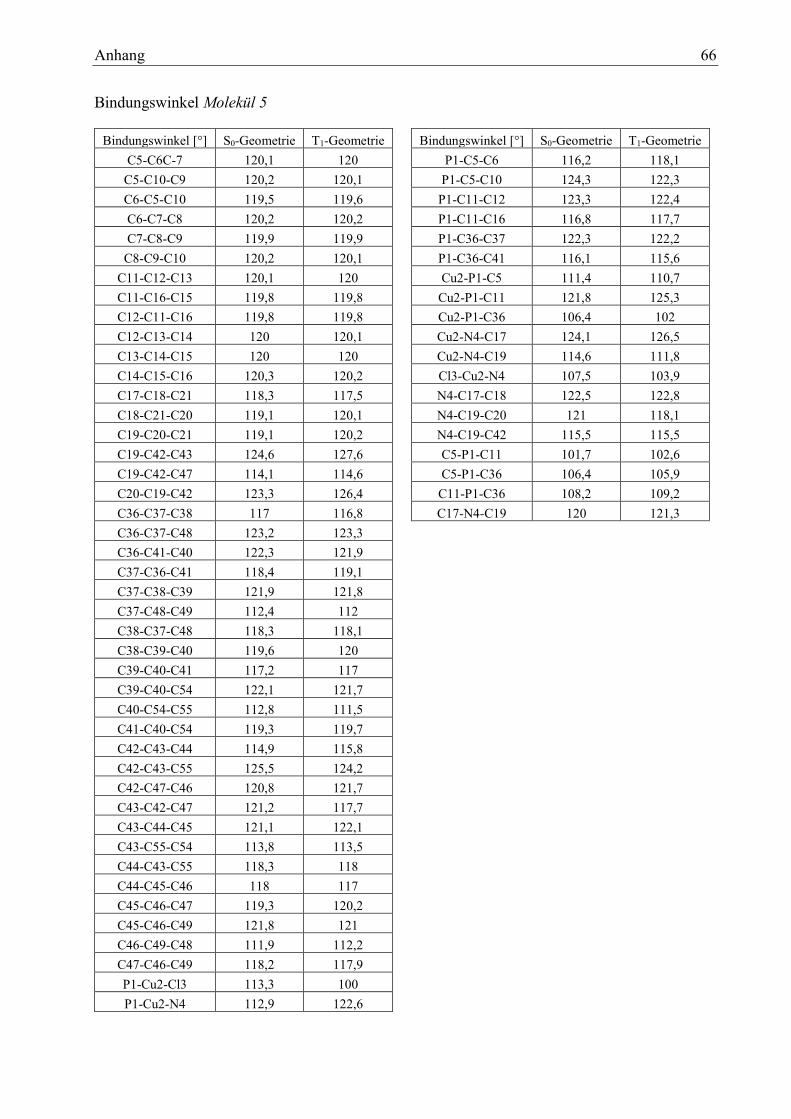
Anhang 66
Bindungswinkel Molekül 5
Bindungswinkel [°] S0-Geometrie T1-Geometrie Bindungswinkel [°] S0-Geometrie T1-Geometrie
C5-C6C-7 120,1 120 P1-C5-C6 116,2 118,1
C5-C10-C9 120,2 120,1 P1-C5-C10 124,3 122,3
C6-C5-C10 119,5 119,6 P1-C11-C12 123,3 122,4
C6-C7-C8 120,2 120,2 P1-C11-C16 116,8 117,7
C7-C8-C9 119,9 119,9 P1-C36-C37 122,3 122,2
C8-C9-C10 120,2 120,1 P1-C36-C41 116,1 115,6
C11-C12-C13 120,1 120 Cu2-P1-C5 111,4 110,7
C11-C16-C15 119,8 119,8 Cu2-P1-C11 121,8 125,3
C12-C11-C16 119,8 119,8 Cu2-P1-C36 106,4 102
C12-C13-C14 120 120,1 Cu2-N4-C17 124,1 126,5
C13-C14-C15 120 120 Cu2-N4-C19 114,6 111,8
C14-C15-C16 120,3 120,2 Cl3-Cu2-N4 107,5 103,9
C17-C18-C21 118,3 117,5 N4-C17-C18 122,5 122,8
C18-C21-C20 119,1 120,1 N4-C19-C20 121 118,1
C19-C20-C21 119,1 120,2 N4-C19-C42 115,5 115,5
C19-C42-C43 124,6 127,6 C5-P1-C11 101,7 102,6
C19-C42-C47 114,1 114,6 C5-P1-C36 106,4 105,9
C20-C19-C42 123,3 126,4 C11-P1-C36 108,2 109,2
C36-C37-C38 117 116,8 C17-N4-C19 120 121,3
C36-C37-C48 123,2 123,3
C36-C41-C40 122,3 121,9
C37-C36-C41 118,4 119,1
C37-C38-C39 121,9 121,8
C37-C48-C49 112,4 112
C38-C37-C48 118,3 118,1
C38-C39-C40 119,6 120
C39-C40-C41 117,2 117
C39-C40-C54 122,1 121,7
C40-C54-C55 112,8 111,5
C41-C40-C54 119,3 119,7
C42-C43-C44 114,9 115,8
C42-C43-C55 125,5 124,2
C42-C47-C46 120,8 121,7
C43-C42-C47 121,2 117,7
C43-C44-C45 121,1 122,1
C43-C55-C54 113,8 113,5
C44-C43-C55 118,3 118
C44-C45-C46 118 117
C45-C46-C47 119,3 120,2
C45-C46-C49 121,8 121
C46-C49-C48 111,9 112,2
C47-C46-C49 118,2 117,9
P1-Cu2-Cl3 113,3 100
P1-Cu2-N4 112,9 122,6
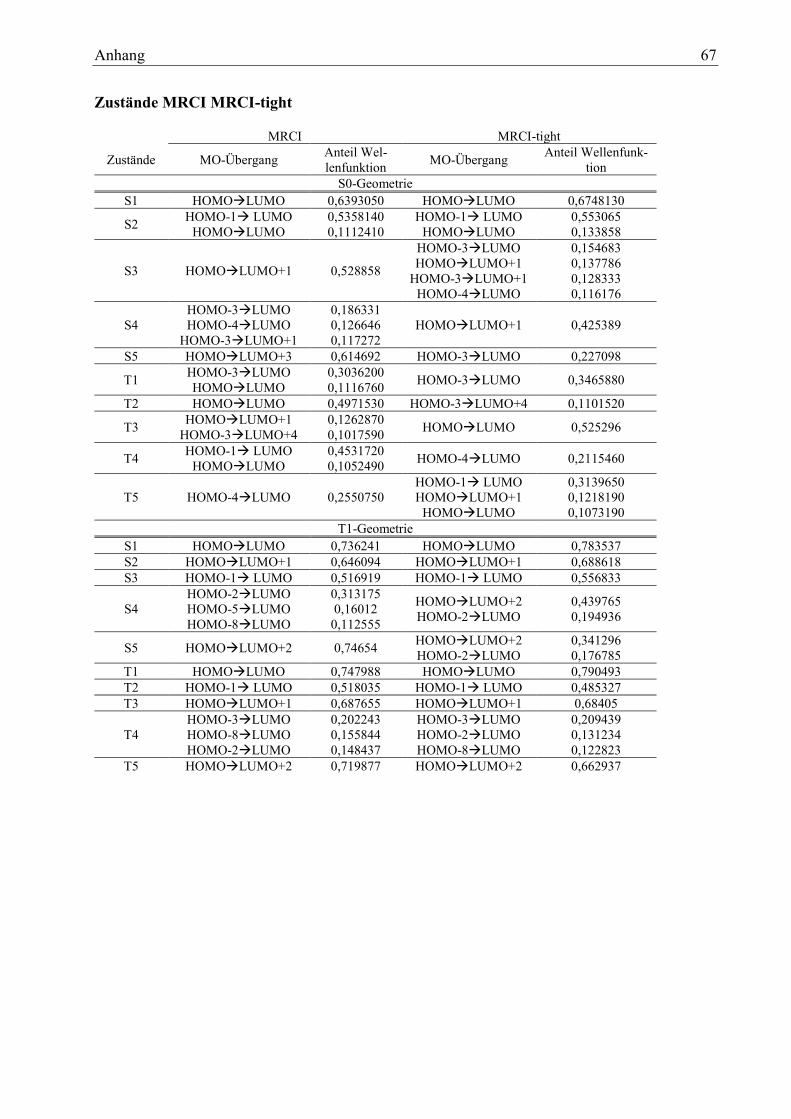
Anhang 67
Zustände MRCI MRCI-tight
MRCI MRCI-tight
Zustände MO-Übergang Anteil Wel-
lenfunktion MO-Übergang
Anteil Wellenfunk-
tion
S0-Geometrie
S1 HOMOLUMO 0,6393050 HOMOLUMO 0,6748130
S2 HOMO-1 LUMO
HOMOLUMO
0,5358140
0,1112410
HOMO-1 LUMO
HOMOLUMO
0,553065
0,133858
S3 HOMOLUMO+1 0,528858
HOMO-3LUMO
HOMOLUMO+1
HOMO-3LUMO+1
HOMO-4LUMO
0,154683
0,137786
0,128333
0,116176
S4
HOMO-3LUMO
HOMO-4LUMO
HOMO-3LUMO+1
0,186331
0,126646
0,117272
HOMOLUMO+1 0,425389
S5 HOMOLUMO+3 0,614692 HOMO-3LUMO 0,227098
T1 HOMO-3LUMO
HOMOLUMO
0,3036200
0,1116760 HOMO-3LUMO 0,3465880
T2 HOMOLUMO 0,4971530 HOMO-3LUMO+4 0,1101520
T3 HOMOLUMO+1
HOMO-3LUMO+4
0,1262870
0,1017590 HOMOLUMO 0,525296
T4 HOMO-1 LUMO
HOMOLUMO
0,4531720
0,1052490 HOMO-4LUMO 0,2115460
T5 HOMO-4LUMO 0,2550750
HOMO-1 LUMO
HOMOLUMO+1
HOMOLUMO
0,3139650
0,1218190
0,1073190
T1-Geometrie
S1 HOMOLUMO 0,736241 HOMOLUMO 0,783537
S2 HOMOLUMO+1 0,646094 HOMOLUMO+1 0,688618
S3 HOMO-1 LUMO 0,516919 HOMO-1 LUMO 0,556833
S4
HOMO-2LUMO
HOMO-5LUMO
HOMO-8LUMO
0,313175
0,16012
0,112555
HOMOLUMO+2
HOMO-2LUMO
0,439765
0,194936
S5 HOMOLUMO+2 0,74654 HOMOLUMO+2
HOMO-2LUMO
0,341296
0,176785
T1 HOMOLUMO 0,747988 HOMOLUMO 0,790493
T2 HOMO-1 LUMO 0,518035 HOMO-1 LUMO 0,485327
T3 HOMOLUMO+1 0,687655 HOMOLUMO+1 0,68405
T4
HOMO-3LUMO
HOMO-8LUMO
HOMO-2LUMO
0,202243
0,155844
0,148437
HOMO-3LUMO
HOMO-2LUMO
HOMO-8LUMO
0,209439
0,131234
0,122823
T5 HOMOLUMO+2 0,719877 HOMOLUMO+2 0,662937
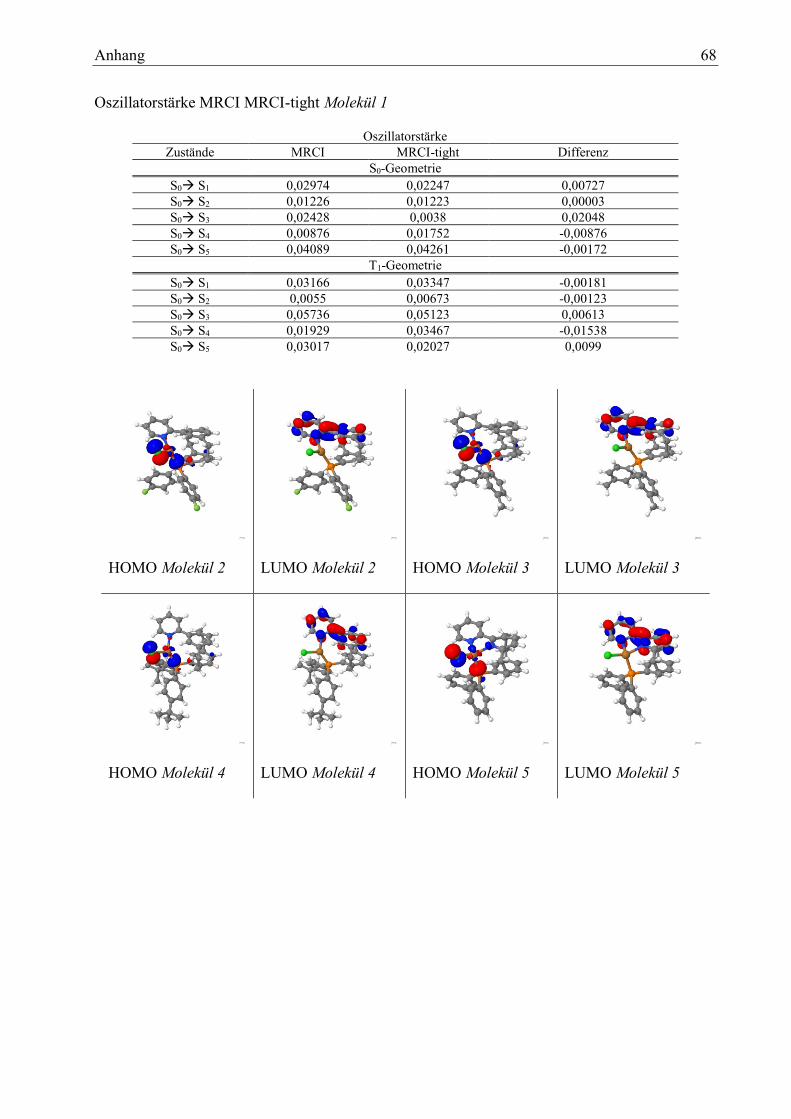
Anhang 68
Oszillatorstärke MRCI MRCI-tight Molekül 1
Oszillatorstärke
Zustände MRCI MRCI-tight Differenz
S0-Geometrie
S0 S1 0,02974 0,02247 0,00727
S0 S2 0,01226 0,01223 0,00003
S0 S3 0,02428 0,0038 0,02048
S0 S4 0,00876 0,01752 -0,00876
S0 S5 0,04089 0,04261 -0,00172
T1-Geometrie
S0 S1 0,03166 0,03347 -0,00181
S0 S2 0,0055 0,00673 -0,00123
S0 S3 0,05736 0,05123 0,00613
S0 S4 0,01929 0,03467 -0,01538
S0 S5 0,03017 0,02027 0,0099
HOMO Molekül 2
LUMO Molekül 2
HOMO Molekül 3
LUMO Molekül 3
HOMO Molekül 4
LUMO Molekül 4
HOMO Molekül 5
LUMO Molekül 5
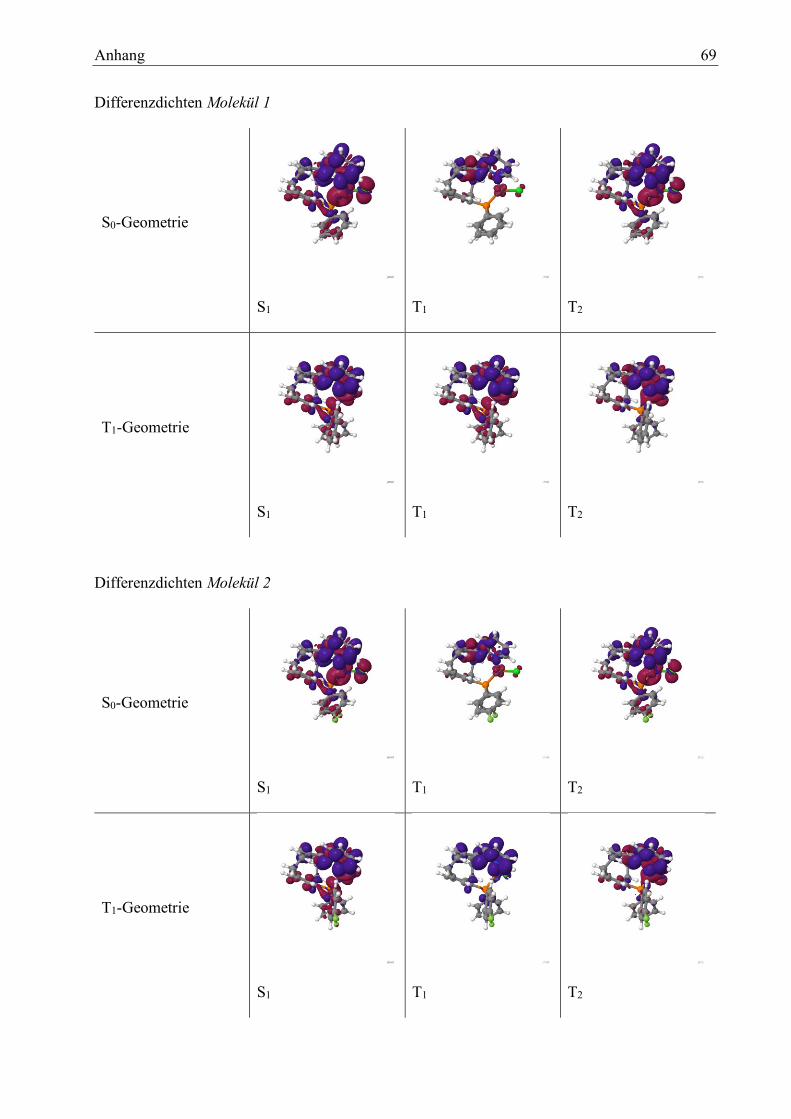
Anhang 69
Differenzdichten Molekül 1
S0-Geometrie
S1
T1
T2
T1-Geometrie
S1
T1
T2
Differenzdichten Molekül 2
S0-Geometrie
S1
T1
T2
T1-Geometrie
S1
T1
T2
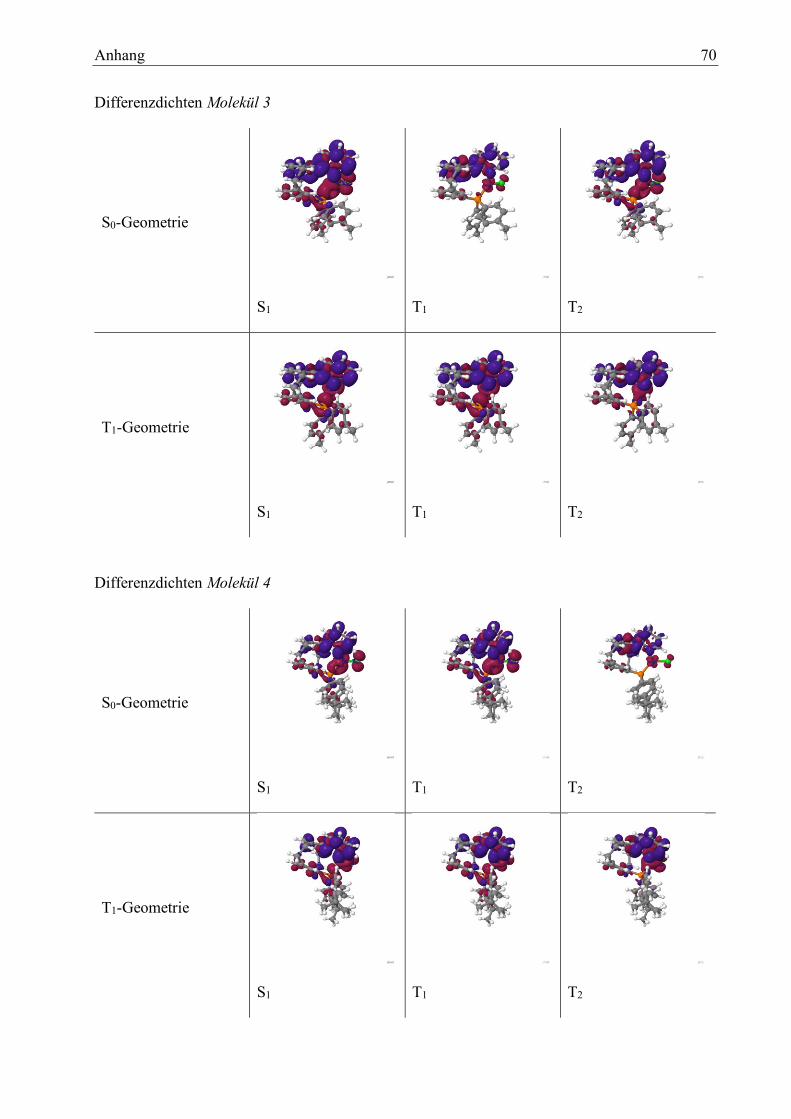
Anhang 70
Differenzdichten Molekül 3
S0-Geometrie
S1
T1
T2
T1-Geometrie
S1
T1
T2
Differenzdichten Molekül 4
S0-Geometrie
S1
T1
T2
T1-Geometrie
S1
T1
T2
Anhang 71
Differenzdichten Molekül 5
S0-Geometrie
S1
T1
T2
T1-Geometrie
S1
T1
T2
Molekül Übergangsdipolmoment S1 an der Triplett-
Geometrie [au]
Standrad-Molekül 0.831554
Molekül 2 0.8120436
Molekül 3 0.8466464
Molekül 4 0.8582698
Molekül 5 1.3756515

Eidesstattliche Erklärung 72
Eidesstattliche Erklärung
Ich,
Sandra Gladt, Wörthstraße 10, 46045 Oberhausen
versichere an Eides Statt durch meine Unterschrift, dass ich die vorstehende Arbeit selbstständig
und ohne fremde Hilfe angefertigt und alle Stellen, die ich wörtlich oder annähernd wörtlich aus
Veröffentlichungen entnommen habe, als solche eindeutig und unmissverständlich kenntlich ge-
macht habe, mich auch keiner anderen als der angegebenen Literatur oder sonstiger Hilfsmittel
bedient habe.
Ich versichere an Eides Statt, dass ich die vorgenannten Angaben nach bestem Wissen und Gewis-
sen getätigt habe und dass die Angaben der Wahrheit entsprechen und ich nichts verschwiegen
habe.
Die Strafbarkeit einer falschen eidesstattlichen Versicherung ist mir bekannt, namentlich die Straf-
androhung gemäß § 156 StGB bis zu drei Jahren Freiheitsstrafe oder Geldstrafe bei vorsätzlicher
Begehung der Tat bzw. gemäß § 161 Abs.1 StGB bis zu einem Jahr Freiheitsstrafe oder Geldstrafe
bei fahrlässiger Begehung.
Essen den 6. März 2018 _______________________________















![Wichtige Giftpflanzen des Grünlandes€¦ · Alkaloide [8] Toxine bleiben in Heu und Silage erhalten; können in Milch übergehen erwachsende Pferde und Rinder meiden i. d. R. die](https://static.fdokument.com/doc/165x107/605f32376d47bb07cb489bc7/wichtige-giftpflanzen-des-gr-alkaloide-8-toxine-bleiben-in-heu-und-silage-erhalten.jpg)


