
Regenwasser Analyse WS 12 13 Els - haw-hamburg.de · Laborprojekt WS2012/13: Meza, Schuster, Arndt,...
29
1 Regenwasser Analyse Wintersemester 2012/2013 Laborprojekt VerfasserInnen: Adriana Meza Artmann Yasmin Arndt Ulrike Schuster Jens Heseding Betreuender Professor: Prof. Dr. Olaf Elsholz Hochschule für Angewandte Wissenschaften Hamburg Campus Bergedorf Fakultät Life Science Lohbrügger Kirchstraße 65 21033 Hamburg
Transcript of Regenwasser Analyse WS 12 13 Els - haw-hamburg.de · Laborprojekt WS2012/13: Meza, Schuster, Arndt,...

1
Regenwasser Analyse Wintersemester 2012/2013
Laborprojekt VerfasserInnen: Adriana Meza Artmann Yasmin Arndt Ulrike Schuster Jens Heseding
Betreuender Professor: Prof. Dr. Olaf Elsholz Hochschule für Angewandte Wissenschaften Hamburg Campus Bergedorf Fakultät Life Science Lohbrügger Kirchstraße 65 21033 Hamburg

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Hinweis:
Dieser Bericht bezieht sich auf den Bericht unserer Vorgängergruppen, vor allem der des SS 2011
und dient als Ergänzung. Zur besseren Lesbarkeit wurden große Teile des Vorgängerberichtes
übernommen. Auch die Abbildungen stammen, falls nicht anders angegeben, aus oben
genanntem Bericht.

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Inhaltsverzeichnis
1. Vorbereitung ....................................................................................................................................... 4
1.1. Beschreibung der Probenahmestellen ......................................................................................... 4
1.2. Kurzanleitung: Vorgehen im Labor ............................................................................................... 7
1.3. Wiegen der Probenmenge ........................................................................................................... 7
1.4. Vorgehen bei geringem Probenvolumen ..................................................................................... 8
2. Benennung der Proben ....................................................................................................................... 9
3. Beschreibung der Analyseverfahren ................................................................................................... 9
3.1. Leitfähigkeit .................................................................................................................................. 9
3.1.1. Grundlagen ............................................................................................................................ 9
3.1.2. Messung .............................................................................................................................. 10
3.2. pH ............................................................................................................................................... 11
3.2.1. Grundlagen .......................................................................................................................... 11
3.2.2. Messung .............................................................................................................................. 12
3.3. Photometrie ............................................................................................................................... 12
3.3.1. Prinzip .................................................................................................................................. 12
3.3.2. Photometer3 ........................................................................................................................ 14
3.3.3. Messung .............................................................................................................................. 14
3.4. Ionenchromatographie (IC) ........................................................................................................ 15
3.4.1. Grundlagen .......................................................................................................................... 16
3.4.2. Vorbereitung der Analyse .................................................................................................... 18
3.4.3. Beispiel eines Chromatogramms ......................................................................................... 19
3.5. Fließinjektionsanalyse (FIA) ........................................................................................................ 20
3.5.1. Grundlagen .......................................................................................................................... 20
3.5.2. Mechanismus ...................................................................................................................... 21
3.5.3. Vorbereitung der Analyse .................................................................................................... 21
3.5.4. Messung .............................................................................................................................. 22
4. Messergebnisse ................................................................................................................................. 23
5. Fazit ................................................................................................................................................... 26

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
1. Vorbereitung
1.1. Beschreibung der Probenahmestellen
Es sind zwei Probenahmestellen für die Regenwasseranalyse auf dem Gelände der Hochschule für Angewandte Wissenschaften eingerichtet. Die eine Probenahmestelle befindet sich auf dem Dach des Südflügels der Mensa, die andere unten im Südosten des Grundstückes, in der Nähe des Lieferanteneinganges. Näheres zeigen folgende Abbildungen:
Abbildung 1: Übersicht der Probenahmestellen

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Abbildung 2: Probenahmestelle Dach
Abbildung 3: Probenahmestelle unten

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Abbildung 4: Probenahmestelle unten, Nahansicht
Abbildung 5: Skizze zur Probenahmestelle Dach
Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
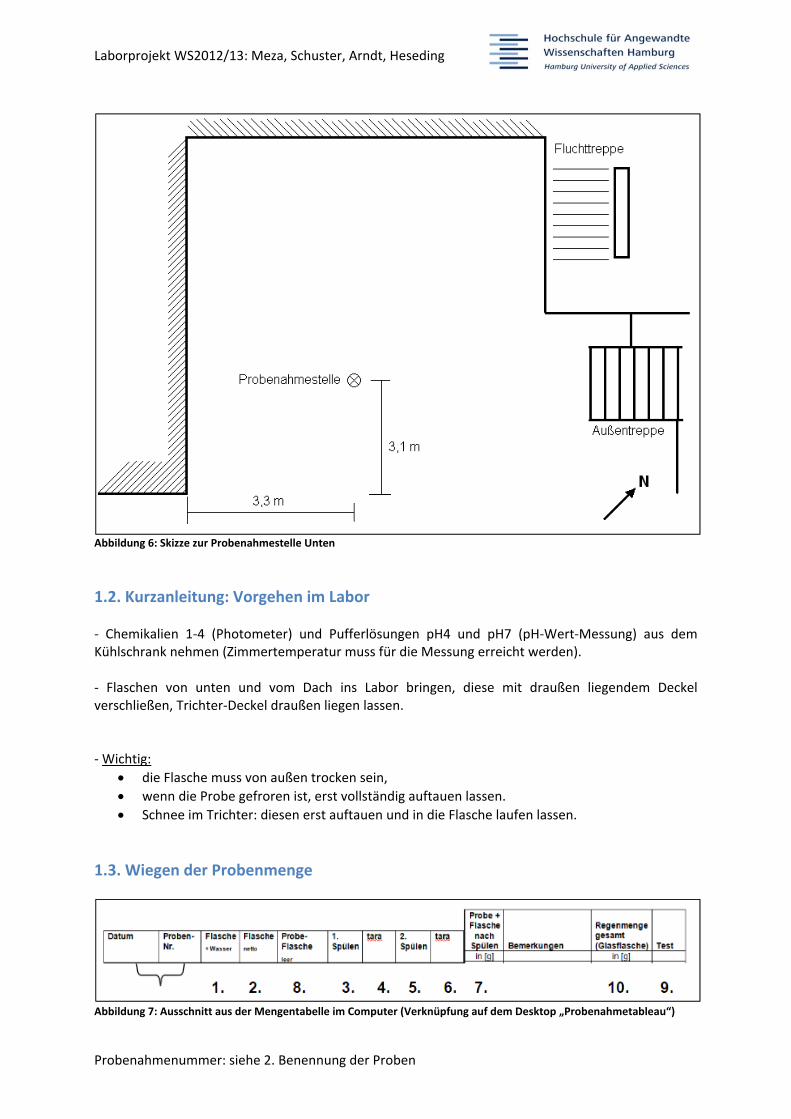
Abbildung 6: Skizze zur Probenahmestelle Unten
1.2. Kurzanleitung: Vorgehen im Labor
‐ Chemikalien 1‐4 (Photometer) und Pufferlösungen pH4 und pH7 (pH‐Wert‐Messung) aus dem Kühlschrank nehmen (Zimmertemperatur muss für die Messung erreicht werden). ‐ Flaschen von unten und vom Dach ins Labor bringen, diese mit draußen liegendem Deckel verschließen, Trichter‐Deckel draußen liegen lassen. ‐ Wichtig:
die Flasche muss von außen trocken sein,
wenn die Probe gefroren ist, erst vollständig auftauen lassen.
Schnee im Trichter: diesen erst auftauen und in die Flasche laufen lassen.
1.3. Wiegen der Probenmenge
Abbildung 7: Ausschnitt aus der Mengentabelle im Computer (Verknüpfung auf dem Desktop „Probenahmetableau“)
Probenahmenummer: siehe 2. Benennung der Proben

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
1. Flasche1 von unten/vom Dach inkl. Niederschlag messen (mit Deckel) 2. Probenflasche2 messen (ohne Probe, mit Deckel) 3. Spülen: ganz kleinen Schluck Niederschlag in Probenflasche füllen und diese mit Inhalt und Deckel messen 4. Probeflasche mit kleinem Schluck Niederschlag (aus 3.) schütteln und konditionieren ausschütten (in separates Becherglas für Abfall) und erneut messen 5. zweites Spülen, siehe 3. 6. zweites Tara, siehe 4. 7. restliche Probe in Probenflasche kippen und Gesamtgewicht messen 8. Flasche von unten/vom Dach ohne Probe messen 9. hier nichts eintragen, dort wird mittels eines Computerprogramms überprüft, ob man sich vermessen hat. Nichts manipulieren! 10. Wert errechnet sich automatisch
Abbildung 8: Ausschnitt aus der Tabelle Konzentration
1. Den Wert von der Tabelle(Menge) von 10. Übernehmen 2. Hier wird die Regenmenge automatisch berechnet, anhand der Trichtergröße, etc.
1.4. Vorgehen bei geringem Probenvolumen
Auf den beiden Probeflaschen ist mittels grüner Markierungen vorgegeben, bis zu welchem Probevolumen die folgenden Arbeitsschritte vonnöten sind. Ziel ist es, so wenig Probe wie möglich zum Spülen zu verbrauchen. Es wird eine geringe Menge der Probe in das entsprechende Reagenzglas (Dach/Unten) gegeben und dieses damit gespült. Nun wird mit einer Spritze mit Luer‐Lock‐Verbinder, die noch vorhandene Spülprobe im Reagenzglas aufgezogen und die Spritze damit gespült. Der Luer‐Lock‐Verbinder wird nun durch einen Filter ersetzt und die Spülprobe langsam tröpfchenweise in die Probeflasche (klein) gegeben. Nach der Spülung der Probeflasche wird das Spülwasser verworfen. Die Probeflasche wird im gespülten Zustand mit Deckel gewogen und danach mit einem Aufkleber versehen und beschriftet (siehe 2. Benennung der Proben). Das Reagenzglas wird mit der Probe gefüllt und mit der Spritze + Filter tröpfchenweise in die Probenflasche gegeben. Wichtig: Im Reagenzglas müssen am Ende mindestens 5 ml (nicht filtriert!) verbleiben.
1 Mit Flaschen sind die Glasflaschen gemeint, in denen die Proben die Woche über gesammelt werden 2 Mit Probenflaschen sind die PET‐Flaschen gemeint, die oben im Wandschrank mit der Aufschrift Laborprojekt im Raum S.2.18 stehen

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Abbildung 9: Luer‐Lock‐Verbinder (Foto: Yasmin Arndt)
Diese Arbeitsschritte müssen mit der 2. Probe (Probe Dach bzw. Probe Unten) wiederholt werden.
2. Benennung der Proben
Am Beispiel: P12017 Das P steht für das Laborprojekt und die nachfolgende 12 für das Jahr 2012. Die dreistellige‐Zahl zum Schluss gibt die tatsächliche fortlaufende Probennummer an, wobei zwischen ungeraden und geraden Zahlen zu unterscheiden ist: ungerade = Probenbehälter steht oben auf dem Dach gerade = Probenbehälter steht unten, am Boden Während des Ausmessens, für die Probemenge, werden die Proben jeweils in PET Flaschen gefüllt. Diese müssen beschriftet werden und später im Kühlschrank aufbewahrt werden. Wenn alle Messungen vorbei sind, werden die Proben nochmal mit einem HPLC‐Filter filtriert, damit keine Stoffe mehr von Mikroorganismen umgewandelt werden können und somit die Messergebnisse verfälschen.
3. Beschreibung der Analyseverfahren
3.1. Leitfähigkeit
3.1.1. Grundlagen
Die spezifische Leitfähigkeit bzw. Leitwert ist ein Maß für die Fähigkeit eines Stoffes den elektrischen Strom zu leiten:
1
R: Ohm [Ω] G: Siemens[ S]
elektrischer Leitwert: ∗ ∗
ä

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
ɣ: Leitfähigkeit A: Querschnittsfläche l: Länge ρ: spezifischer Widerstand Eine zwei‐Elektroden‐Messzelle, bestehend aus zwei gegenüberliegenden, gleichgroßen Festkörperelektroden mit der Fläche A, sowie dem Abstand d zueinander:
Abbildung 10: zwei‐elektroden‐Messzelle
Mit angelegter Wechselspannung fangen die Ionen in der Elektrolytlösung an sich zwischen den Festkörpern hin‐ und her zu bewegen. Kalibriert wird das Leitfähigkeitsmessgerät durch eine Lösung mit bekannter Leitfähigkeit, um eventuelle Abweichungen aufzuzeigen. Die Messung ist stark Temperaturabhängig, weshalb immer bei einer Referenztemperatur von 25 °C gemessen werden sollte. Um dem Effekt entgegen zu wirken, wird eine Temperaturkompensation eingeführt. Die Temperaturabhängigkeit der Leitfähigkeit beträgt zwischen 1‐5% pro Grad Celsius je nach gelöstem Stoff. Das Ergebnis wird in mS/cm oder μS/cm mit der entsprechenden Temperatur angegeben.
3.1.2. Messung
Zuerst muss die Probe auf Zimmertemperatur gebracht werden. Da die Leitfähigkeit stark temperaturabhängig ist, sollte die Probe immer bei Zimmertemperatur gemessen werden, um die Werte untereinander vergleichen zu können. Das Leitfähigkeitsmessgerät IA 82 sowie den Rührer einschalten und die Leitfähigkeit des Reinstwassers messen. Liegt diese im Normalbereich von ca. 0,3 bis 2 μS/cm kann dieses Gerät so verwendet werden. Sonst muss das Reinstwasser ausgewechselt und nochmals gemessen werden, bis dieser Wert erreicht wird. Der Messzylinder (10 ml) muss nun vorbereitet werden. Es muss ein Plastikröhrchen, welches in etwa die gleiche Größe hat wie der Zylinder eingesetzt werden. In das Röhrchen kommt ein kleiner Rührfisch. Nun die Probe (ca. 5 ml) in den Messzylinder überführen. Die Probe soll während der Messung gerührt werden, damit ein stetiges durchströmen der Sonde gewährleistet ist. Dann wird die Leitfähigkeitssonde in die Probe eingetaucht. Aufpassen, dass das Rührfischchen nicht gegen die Sonde stößt und diese beschädigt. Den ersten Wert aufschreiben, da die Leitfähigkeit nicht konstant ist. Diese verändert sich eventuell auf Grund der CO2 –Konzentration in der Luft.

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Abbildung 11: Veränderung der Leitfähigkeit durch CO2
3.2. pH
Die Angabe des pH‐Wertes ist dimensionslos und beschreibt den negativen dekadischen Logarithmus der Wasserstoffionen‐Konzentration.
3.2.1. Grundlagen
Die pH‐Elektrode ist als Einstabmesskette aufgebaut und besteht aus einer Kombination aus Arbeitselektrode und Referenzelektrode in Form einer Silber‐Silberchlorid‐Elektrode 2. Art. Als Elektrolytlösung wird 3 molare KCl‐Lösung verwendet, dadurch ist ein minimaler Ionenfluss gewährleistet, der ausreicht, um die Redox‐Gleichgewichte an der Elektrodenoberfläche einzustellen.
Abbildung 12: pH‐Elektrode
Arbeitselektrode: Der Aufbau einer Arbeitselektrode ist im nachstehenden Bild dargestellt. Ihre wesentlichen Bestandteile sind: die Glasmembran, die Innenpufferflüssigkeit, das Ableitelement mit dem daran angeschlossenen Kabel und eine innere Abschirmung. Das Herzstück der Elektrode ist jedoch die Glasmembran. Die elektrische Ladung, die an der Membraninnenseite entsteht, wird über Ableitelement, Ableitung und angeschlossenes Kabel zum Messverstärker geführt. Als Ableitelement wird meistens ein Silberdraht, ummantelt mit Silberchlorid, eingesetzt, der den Kontakt zum Innenpuffer herstellt. Die elektrische Abschirmung umhüllt innerhalb des Glasrohres das Messsystem und ist an den Außenmantel des Elektrodenkabels (Koaxkabel) angeschlossen.

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Wichtig: 1. Sollte nur sehr wenig Probe (hier Regenwasser) vorhanden sein, wird für die Leitfähigkeitsmessung und die pH‐Messung eine Probe genommen. Allerdings muss immer ZUERST die Leitfähigkeit und erst dann der pH‐Wert gemessen werden, da an der pH‐Sonde sich KCl‐Lösung befindet, die den Messwert bei der Leitfähigkeitsmessung stark beeinträchtigen würde. 2. Die pH‐Kalibrierlösung immer als erstes aus dem Kühlschrank nehmen, wenn das Labor betreten wird, da die pH‐Messung temperaturabhängig ist (Zimmertemperatur erforderlich). 3. Wenn man mit der pH‐Sonde einen Wert über und unter den Werten von 5‐6 misst, muss die Probe nochmals mit einem pH‐Papier überprüft werden (gegebenenfalls auch Standardlösung überprüfen).
3.2.2. Messung
Das Gerät IA 108 einschalten und die Kappe abnehmen, in welcher sich KCl‐Lösung befindet (falls diese mal erneuert werden muss, steht im Überschrank neue. Die Sonde sollte immer mit dieser Lösung benetzt sein, da sie sonst austrocknet!). Die Sonde sollte mit Reinstwasser gespült und anschließend kalibriert werden. Zum Kalibrieren die Sonde erst in die Kalibrierlösung mit dem pH‐Wert 4 halten, liegt der gemessene pH‐Wert bei 4 ± 0,2 so ist diese Kalibrierung in Ordnung. Die Sonde erneut spülen und nun in die Kalibrierlösung mit dem pH‐Wert 7 halten, liegt der gemessene pH‐Wert bei 7 ± 0,1 so ist auch diese Kalibrierung in Ordnung. Falls die Werte abweichen, muss neu kalibriert werden, dafür bitte die Anleitung an der Wand verwenden. (Dieses Gerät wird allerdings sehr häufig von den Praktikanten kalibriert, so dass dies meist überflüssig ist). Nun den pH‐Wert der Probe messen, wobei ca. 1 Minute auf einen stabilen Wert gewartet werden sollte. Auch hierfür sollten, genau wie bei der Leitfähigkeitsmessung, ca. 5 ml in einen Messzylinder(10 ml) überführt werden.
3.3. Photometrie
3.3.1. Prinzip3
Die photometrische Analyse ist ein Messverfahren, bei dem man die Konzentration einer Probe bestimmt, indem man die Schwächung eines Lichtstrahls nach Durchgang durch die in Lösung befindliche absorbierende Substanz mit der Konzentration korreliert.
Abbildung 133: Lichtdurchgang durch eine Probe
3 Skript Praktikum Allgemeine und Anorganische Chemie (HAW – Hamburg)

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
ɸ0 : Lichtstrom vor Absorption ɸ: Lichtstrom nach Absorption d: Schichtdicke der Küvette Werden sich im Grundzustand ( Egrund) befindende Moleküle mit Licht bestrahlt, absorbieren diese, Quanten der Energie h*ν aus dem eingestrahlten Licht und werden somit in einen angeregten Zustand ( Eang)gebracht:
Egrund ‐ Eang = h*ν Die absorbierten Quanten werden meist nicht in Form von Licht sondern z.B. als Wärme wieder abgegeben. Als Maß für die Lichtschwächung werden die Messgrößen Absorption (Abs), Transmission (T) und Extinktion (E) gebraucht:
ɸ ɸɸ
ɸɸ
lgɸɸ
lg
Grundlage der Photometrie ist das Lambert‐Beer´sche Gesetz, das die Proportionalität zwischen Konzentration und Extinktion angibt:
ɛ ∗ ∗ ɛ: stoffspezifischer molarer dekadischer Extinktionskoeffizient c: Konzentration d: Schichtdicke der durchstrahlten Probe

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
3.3.2. Photometer3
Auf der nachstehenden Abbildung wird der schematische Aufbau eines Einstrahlphotometers
gezeigt:
Abbildung 144: schematischer Aufbau eines Einstrahlsphotometers
Von einer Lichtquelle wird ein Lichtstrahl gesendet. Diese wird durch eine Linse gebündelt und im Monochromator spektral zerlegt. Durch Drehen des Monochromators, wird die geeignete Wellenlänge eingestellt. Danach gelangt das Lichtbündel durch die Spalte und durchstrahlt die Küvette, in der sich die Probe befindet. Das nicht absorbierte Licht fällt auf eine Photozelle und verursacht einen Kurzschlussstrom, der ein Maß für die Lichtintensität ist und somit auch für die Konzentration.
3.3.3. Messung
Ansetzen der Chemikalien: Pufferlösung: 16 g tri – Natriumcitrat – Dihydrat in Reinstwasser lösen und mit Reinstwasser auf 100 ml auffüllen Natriumsalicylat – Lösung (NaSal): 16 g Natriumsalicylat in Reinstwasser lösen und mit Reinstwasser auf 100 ml auffüllen Dichlorisocyanurat – Lösung (DClC): 2,5 g Natriumhydroxid in 25 ml Reinstwasser lösen, anschließend 0,2 g Dichlorisocyanurat‐Dihydrat darin lösen und mit Reinstwasser auf 100 ml auffüllen Katalysator – Lösung: 0,12 g Natriumnitroprussid‐Dihydrat in Reinstwasser lösen und mit Reinstwasser auf 100 ml auffüllen Ammonium – Stammlösung 100 mg/l: 1 ml NH4
+ ‐ Merck – Stammlösung (1000mg/l) mit Reinstwasser auf 10 ml auffüllen

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
NH4+ ‐ Standardlösung 1 mg/l:
500 µl der NH4
+‐ Stammlösung (100 mg/l) in Reinstwasser lösen und mit Reinstwasser auf 50 ml auffüllen NH4
+ ‐ Standardlösung 5 mg/l: 2,5 ml der NH4
+‐ Stammlösung (100 mg/l) in Reinstwasser lösen und mit Reinstwasser auf 50 ml auffüllen Materialien: 5 Einwegküvetten 1 Küvettenständer 1 ml – Festpipette 500 µl – Festpipette 5 Parafilmstücke (2 x 2 cm) Stoppuhr Durchführung: Es wird vom Reinstwasser, von den Standardlösungen und von jeder Regenwasserprobe jeweils 1 ml in eine Einwegküvette pipettiert. Anschließend wird in die erste Küvette jeweils 500 µl Pufferlösung, NaSal, DClC und Katalysator pipettiert4. Die Stoppuhr wird gestartet, die Einwegküvette wird mit Parafilm abgedeckt und leicht geschwenkt. Dieser Vorgang wird bei jeder Einwegküvette wiederholt. Es ist wichtig, dass alle Küvetten nach der gleichen Reaktionszeit gemessen werden. Deswegen muss man den Katalysator in festen Zeitabständen, z.B. alle 2 Minuten (d.h. jeweils in der 0., 2., 4., 6. und 8. Minute), pipettieren. Die Küvetten sollen hierbei nicht am Anfang vorbereitet sondern nacheinander angesetzt werden. Im Photometer werden die Parameter 1 und 5 eingestellt: λ = 656 nm. Als Referenzmessung dient destilliertes Wasser (eine Einwegküvette mit dest. Wasser in die Referenzposition des Photometers stellen). In unserem Versuch erfolgte die photometrische Messung genau 20 Minuten nach pipettieren des Katalysators. Das bedeutet, dass jeweils in der 20, 22., 24., 26. und 28. Minute gemessen wurde. Die Berechnung erfolgt mittels Microsoft‐Excel. Anhand des destillierten Wasser und der Standardlösung wird eine Kalibriergerade erstellt und mithilfe deren Funktionsgleichung die Ammoniumkonzentration in den Regenwasserproben ermittelt.
3.4. Ionenchromatographie (IC)
Für unsere Messungen haben wir dasselbe Gerät wie unsere Vorgängergruppe verwendet: „732 Basic IC“ von der Firma Metrohm.
4 Es empfiehlt sich, im Küvettenständer 4 Einwegküvetten als Ablage für die Pipettenspitzen einzusetzen, um für die jeweiligen Chemikalien dieselben Spitzen während des Versuches verwenden zu können.

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Abbildung 155: Innenansicht des verwendeten IC‐Geräts
3.4.1. Grundlagen
Die Trennsäule: Im IA‐Labor wird die Säule „Metrosepp A Supp 4“ von der Firma Metrohm (siehe Abbildung 15) verwendet. Die Säule besteht aus einem PEEK‐Mantel (PEEK: Polyetheretherketon) und einer Trennphase aus Polyvinylalkohol‐Partikeln, die mit Ammoniumgruppen belegt sind. Der Eluent: Hier handelt es sich um eine gemischte Natrium‐Hydrogen‐Carbonat (NaHCO3) und Natrium‐Carbonat (Na2CO3) ‐Lösung. Die Stoffanteile sind: n (NaHCO3) = 1,7 mmol/l n (Na2CO3) = 1,8 mmol/l Achtung: Der Eluent muss unbedingt vor der Benutzung entgast werden. Wenn Sauerstoff und andere Gase in die IC‐Anlage eindringen, können diese die Messung stören und das System beschädigen. Im IA‐Labor verwendeten wir für die Entgasung eine Vakuumfiltration. Das „Six‐Port‐Ventil“:

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Das „Six‐Port‐Ventil“ ist eine gängige Methode zur Einspritzung der Probe in chromatographischen Verfahren. Den sechs Aus‐ bzw. Eingängen verdankt es seinen Namen. Zunächst muss die Probe in die Probeschleife gesaugt werden. Das Probenvolumen ist sehr gering, es beträgt nur 20 µl. Um sicherzustellen, dass die Probeschleife komplett gefüllt ist, werden aber mindestens 3 ml Probe in das System gegeben, da auch der Zulaufschlauch zunächst gefüllt und mit Probe gespült werden muss. Sobald in der Software am PC auf „Inject“ gedrückt wird, schaltet das Ventil um und der Eluent befördert die Probe auf die Trennsäule.
Abbildung 166: Six‐Port‐Ventil mit Probeschleife
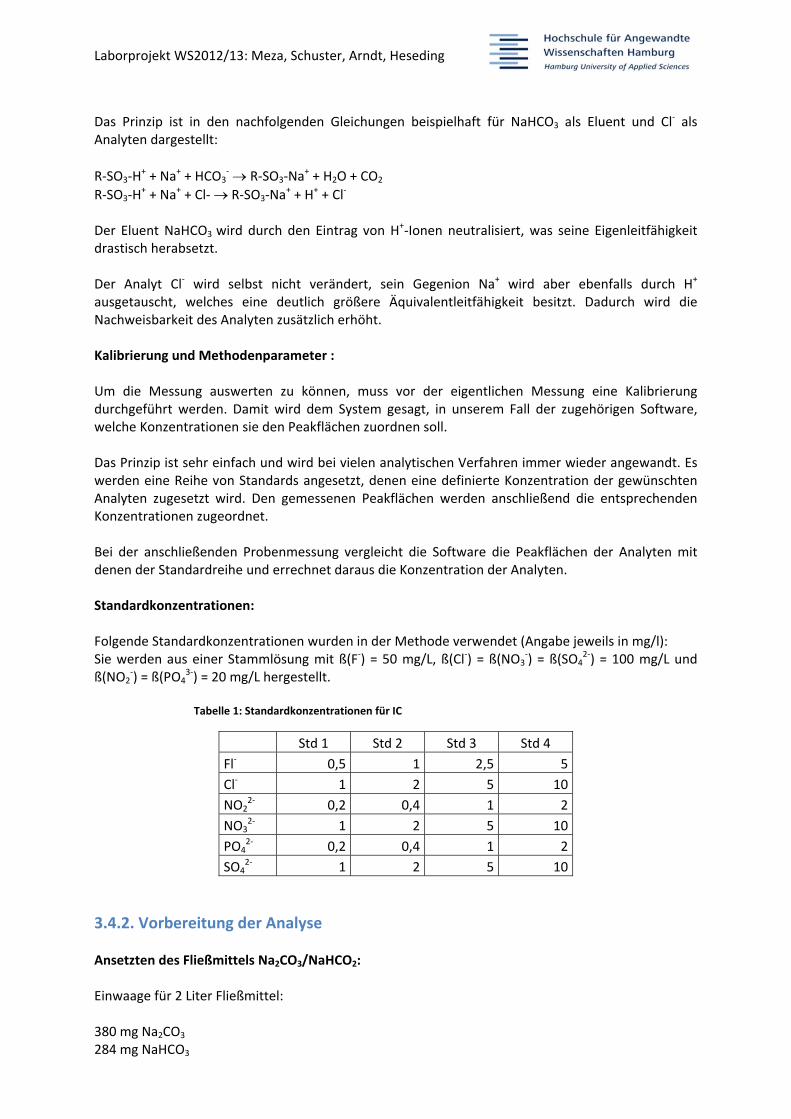
Der Detektor: Grundsätzlich kann zwischen elektrochemischen (Leitfähigkeit, potentiometrische (Detektion) und spektrometrischen (Photometrie, Fluoreszenz) Detektoren unterschieden werden. An der IC in dem IA‐Labor wird ein Leitfähigkeitsdetektor (auch konduktometrischer Detektor genannt) verwendet. Über eine Elektrodenanordnung wird elektrochemisch die Leitfähigkeit der einzelnen Analyten gemessen. Man erhält im Anschluss ein Diagramm mit mehreren Peaks. Zu jeder Zeit, zu der eine erhöhte Leitfähigkeit vorhanden ist, erscheint ein Peak. Da jeder Analyt eine charakteristische Retentionszeit (Verzögerung) aufweist, können die Peaks den verschiedenen Analyten zugeordnet werden. Die Peakfläche fällt umso höher aus, je mehr Anionen vorhanden sind, d.h. je höher die Konzentration eines Analyten ist. Die chemische Suppression: Der Eluent weist in der Regel eine Eigenleitfähigkeit auf. Unter Umständen ist die Veränderung der Leitfähigkeit einzelner Analyten relativ zu dieser Eigenleitfähigkeit gering. Das hat zur Folge, dass die Nachweisgrenze für den Analyten sehr hoch ausfällt und geringere Konzentrationen nicht erfasst werden. Deshalb wurde die Methode der chemischen Suppression entwickelt. Bei dieser Methode wird der Eluent mit den Analyten nach der Trennsäule nicht direkt zu dem Detektor, sondern zunächst über einen Suppressor geleitet. Dieser Suppressor hat die Aufgabe, die Eigenleitfähigkeit des Eluenten und dadurch auch die Nachweisgrenzen für die Anionen drastisch zu senken. Das funktioniert mit einem stark sauren Kationenaustauscher, der sowohl in den Eluenten wie in den Analyten H+‐Ionen einführt.
Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Das Prinzip ist in den nachfolgenden Gleichungen beispielhaft für NaHCO3 als Eluent und Cl‐ als
Analyten dargestellt:
R‐SO3‐H+ + Na+ + HCO3
‐ R‐SO3‐Na+ + H2O + CO2
R‐SO3‐H+ + Na+ + Cl‐ R‐SO3‐Na
+ + H+ + Cl‐ Der Eluent NaHCO3
wird durch den Eintrag von H+‐Ionen neutralisiert, was seine Eigenleitfähigkeit drastisch herabsetzt. Der Analyt Cl‐ wird selbst nicht verändert, sein Gegenion Na+ wird aber ebenfalls durch H+
ausgetauscht, welches eine deutlich größere Äquivalentleitfähigkeit besitzt. Dadurch wird die Nachweisbarkeit des Analyten zusätzlich erhöht. Kalibrierung und Methodenparameter : Um die Messung auswerten zu können, muss vor der eigentlichen Messung eine Kalibrierung durchgeführt werden. Damit wird dem System gesagt, in unserem Fall der zugehörigen Software, welche Konzentrationen sie den Peakflächen zuordnen soll. Das Prinzip ist sehr einfach und wird bei vielen analytischen Verfahren immer wieder angewandt. Es werden eine Reihe von Standards angesetzt, denen eine definierte Konzentration der gewünschten Analyten zugesetzt wird. Den gemessenen Peakflächen werden anschließend die entsprechenden Konzentrationen zugeordnet. Bei der anschließenden Probenmessung vergleicht die Software die Peakflächen der Analyten mit denen der Standardreihe und errechnet daraus die Konzentration der Analyten. Standardkonzentrationen: Folgende Standardkonzentrationen wurden in der Methode verwendet (Angabe jeweils in mg/l): Sie werden aus einer Stammlösung mit ß(F‐) = 50 mg/L, ß(Cl‐) = ß(NO3
‐) = ß(SO42‐) = 100 mg/L und
ß(NO2‐) = ß(PO4
3‐) = 20 mg/L hergestellt.
Tabelle 1: Standardkonzentrationen für IC
Std 1 Std 2 Std 3 Std 4
Fl‐ 0,5 1 2,5 5
Cl‐ 1 2 5 10
NO22‐ 0,2 0,4 1 2
NO32‐ 1 2 5 10
PO42‐ 0,2 0,4 1 2
SO42‐ 1 2 5 10
3.4.2. Vorbereitung der Analyse
Ansetzten des Fließmittels Na2CO3/NaHCO2: Einwaage für 2 Liter Fließmittel: 380 mg Na2CO3 284 mg NaHCO3

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Ansetzen der Standards: Folgende Fertiglösungen wurden verwendet:
Tabelle 2: Eingesetzte Fertiglösungen
Konzentration ß= mg/l Bestellnummer
Chlorid (Cl‐) 1000 ± 2
Nitrat (NO32‐) 995 ± 5
Sulfat (SO42‐) 1001 ±2
Fluorid (Fl‐) 1003 ± 2
Nitrit (NO2‐) 1001 ± 5
Phosphat (PO42‐) 999 ± 2
Bei den Standardlösungen, die blau hinterlegt sind handelt es sich um einen Mischstandard in dem drei Anionen bereits miteinander kombiniert vorliegen. Zwischenstandard mit Wasser ansetzen: 1000 µl Cl‐ ; NO3
‐ ; SO42‐ ‐ Lösung, 500 µl F‐ ‐ Lösung, 200 µl
NO2‐ ‐ Lösung und 200 µl PO4
3‐ auf 10 ml Aus dem Zwischenstandard (Z_Std) die vier Kalibrierstandards ansetzen. Als Lösemittel Eluat Na2CO3/NaHCO2 verwenden. Std 1: 1mL Z_Std auf 100 mL ; Std 2: 1mL Z_Std auf 50 mL Std 3: 1mL Z_Std auf 20 mL ; Std 4: 1mL Z_Std auf 10 mL Methodenparameter: Wie bereits eingangs erwähnt, wird für die Analyse der Regenwasserproben eine spezielle Methode verwendet. Die Parameter der Methode sind hier nochmal zusammengestellt: Verwendetes Gerät: 732 Basic IC von Metrohm Methodenname: Regenwasser2010.mtw Probenvolumen: 20 µl Säule: Metrosepp A Supp 4, l = 250 mm, Ø = 4 mm Eluent: 1,7 mmol/L NaHCO3; 1,8 mmol/L Na2CO3 Fließgeschwindigkeit: 1 ml/min Temperatur: 20 °C Methodendauer: 15 min Kalibrierung: Externe Standards, Auswertung über Fläche
3.4.3. Beispiel eines Chromatogramms
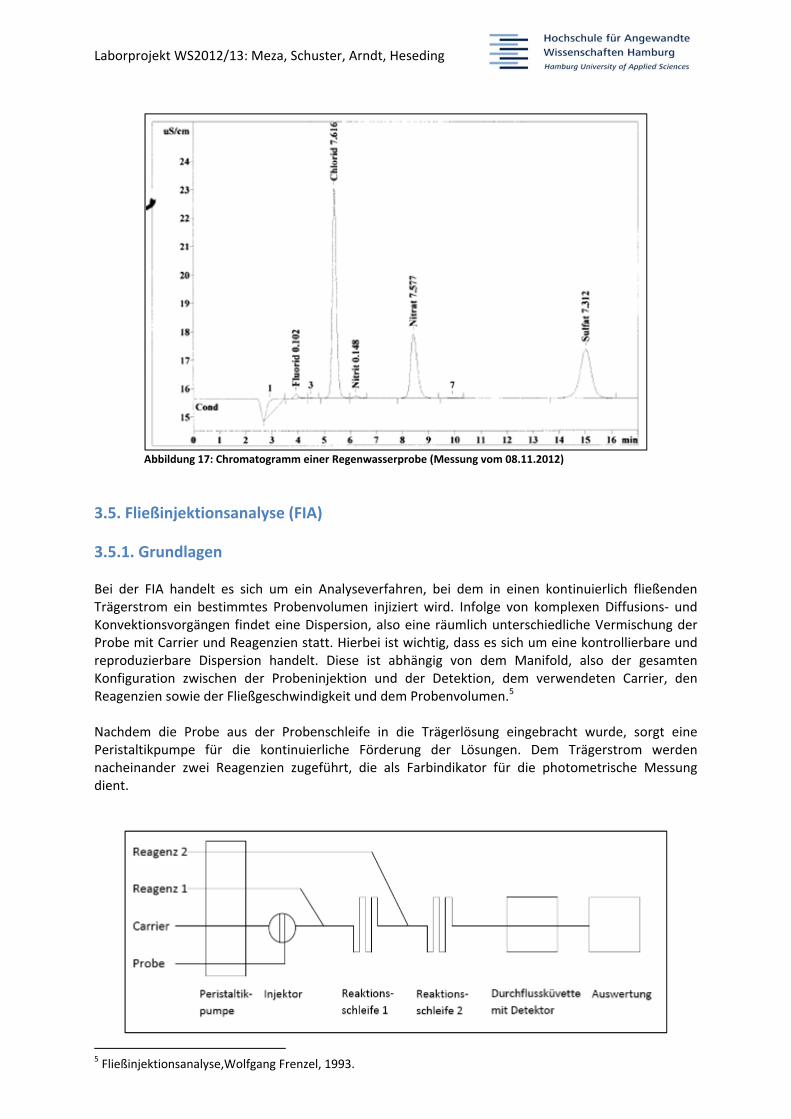
Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Abbildung 17: Chromatogramm einer Regenwasserprobe (Messung vom 08.11.2012)
3.5. Fließinjektionsanalyse (FIA)
3.5.1. Grundlagen
Bei der FIA handelt es sich um ein Analyseverfahren, bei dem in einen kontinuierlich fließenden Trägerstrom ein bestimmtes Probenvolumen injiziert wird. Infolge von komplexen Diffusions‐ und Konvektionsvorgängen findet eine Dispersion, also eine räumlich unterschiedliche Vermischung der Probe mit Carrier und Reagenzien statt. Hierbei ist wichtig, dass es sich um eine kontrollierbare und reproduzierbare Dispersion handelt. Diese ist abhängig von dem Manifold, also der gesamten Konfiguration zwischen der Probeninjektion und der Detektion, dem verwendeten Carrier, den Reagenzien sowie der Fließgeschwindigkeit und dem Probenvolumen.5 Nachdem die Probe aus der Probenschleife in die Trägerlösung eingebracht wurde, sorgt eine Peristaltikpumpe für die kontinuierliche Förderung der Lösungen. Dem Trägerstrom werden nacheinander zwei Reagenzien zugeführt, die als Farbindikator für die photometrische Messung dient.
5 Fließinjektionsanalyse,Wolfgang Frenzel, 1993.

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Abbildung 18: Fließschema FIA6
Das Besondere an dem Verfahren der FIA ist die Tatsache, dass sich kein Gleichgewicht im Sinne eines Endzustandes einstellt, sondern dass es sich um ein dynamisches Gleichgewicht handelt. Wird das Manifold und die exakte zeitliche Kontrolle der Abläufe des FIA‐Systems eingehalten, ist das Ergebnis reproduzierbar und es kann ein definierter Zustand an jedem beliebigen Ort im FIA System sowie zu jedem Zeitpunkt erfasst werden. Daraus ergeben sich verschiedene Vorteile dieses Analyseverfahrens wie beispielsweise kurze Analysezeiten, hoher Probendurchsatz und geringer Probenvolumen‐ und Reagenzverbrauch. Für die Messungen für dieses Laborprojekt wurde die FIAstar 5000 der Fa. Foss/Tecator verwendet.
3.5.2. Mechanismus
Bei der Reaktion für den quantitativen Nachweis von Ammonium in den Regenwasserproben handelt es sich um die bekannte Berthelot‐Reaktion, die wie in DIN 38406/5.beschrieben modifiziert ist. Es wird statt Hypochlorit nach DIN Dichlorisocyanursäure und statt Phenol Natriumsalicylat verwendet. Im ersten Schritt wird die Ammoniumlösung auf einen alkalischen pH‐Wert gebracht. Das ist wichtig, damit sich das Gleichgewicht der Reaktion auf die Seite von Ammoniak verlagert also vollständig als NH3 vorliegt. Im zweiten Schritt reagiert das Ammoniak mit der Dichlorisocyanursäure zu Aminchlorid. Im dritten Schritt findet eine Reaktion mit Natriumsalicyat statt, wobei diese Reaktion vom Natriumnitroprussid Dihydrat katalysiert wird. In einer mehrstufigen Reaktion erfolgt dann die Bildung eines Indophenolblau ähnlichen Farbstoffs. Die Lösung hat dann eine bläulich ‐ grünliche Färbung und kann photometrisch detektiert werden.
3.5.3. Vorbereitung der Analyse
Ansetzen der Chemikalien: Carrier (Citratpuffer): 10 g tri‐Natriumcitrat‐Dihydrat 0,25 ml Polyethylenglycolodecylether w=3% Reagenz 1 (Natriumsalicylat‐Lösung): 8,5 g Natriumsalicylat 0,2 g Natriumnitroprussid Dihydrat 0,25 ml Polyethylenglycolodecylether w=3% Reagenz 2 (Dichlorisocyanursäure‐Lösung): 5 g Natriumhydroxid 0,4 g Dichlorisocyanursäure Alle Lösungen werden mit entgastem Reinstwasser jeweils auf 250 ml aufgefüllt. Das Entgasen ist wichtig, um Luftblasen im FIA System vorzubeugen. Ansetzen der Standards7:
6 Studienprojekt_Regen. pdf, Sarah Flashaar, 2009. 7 nach der Arbeitsvorschrift der HAW für die FIA Ammoniumbestimmung, September 2008.

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
Aus der Ammonium‐Stammlösung β(NH4) = 1.000 mg/l werden vier Standards zu jeweils 100 ml hergestellt. Tabelle 3: Massenkonzentration der Standards
Lösung Massenkonzentration (mg/l) Pipettiervorlagen [µl]
Standard 1 0,2 20
Standard 2 0,5 50
Standard 3 1,0 100
Standard 4 2,0 200
Alle Lösungen werden mit Reinstwasser jeweils auf 100 ml aufgefüllt und täglich neu angesetzt. Einstellung bzw. Überprüfung der Geräteparameter6 : Einsetzen der Injektionsschleife Vi = 200 μL Einsetzen der entsprechenden Filter in den Filterhalter des Detektors Referenzfilter = 880 nm, Messwellenlängenfilter = 660 nm Einsetzen des passenden Manifolds Einstellen der Fließgeschwindigkeiten: Tabelle 4: Pumprate der Lösungen
Lösung Pumprate (ml/min) Schlauchcode
Reagenz 1 0,6 Gelb/Orange
Reagenz 2 0,6 Gelb/Orange
Carrier 0,6 Gelb/Orange
Probe 2,8 Rot/ Rot
Vorbereiten der Regenwasserproben: durch membranfiltrieren (0,45 µl Einmal‐Filter).
3.5.4. Messung
Nachdem das Reinstwasser, der Carrier und die Reagenzien angeschlossen wurden und sichergestellt wurde, dass die Luft aus dem System entfernt ist, wird die Methode geladen (in diesem Fall die laborinterne Methode NH4_02 bis 2 bei einer Temperatur von 40º C). Am Anfang jeder Probenmessung steht die Kalibrierung mit den vier Standards aus der Ammonium‐Stammlösung. Nach Beendigung der Kalibrierung der Methode, wird der Korrelationskoeffizient r2 geprüft. Die Bezugsfunktion wird akzeptiert, wenn dieser mindestens 0,9990 beträgt. Anschließend wird die Probenliste erstellt und die Proben gemessen, wobei nach wie vor sichergestellt sein muss, dass sich keine Luftblasen in den Schläuchen oder der Durchfluss‐Küvette für die photometrische Messung befinden. Bei der folgenden Auswertung der Ergebnisse ist zu beachten, ob die Proben direkt nach der Probennahme gemessen wurden, da sich sonst in der Zwischenzeit ein Teil des Ammoniums abgebaut hat und ggf. sich keine richtige Aussage über den Ammoniumgehalt der jeweiligen Probe machen lässt bzw. es keine Vergleichbarkeit gibt, wenn die Proben zu unterschiedlichen Zeitpunkten nach der Probenahme gemessen wurden.

23
4. Messergebnisse
Tabelle 5: Regenwassermenge (Wochenmischproben)
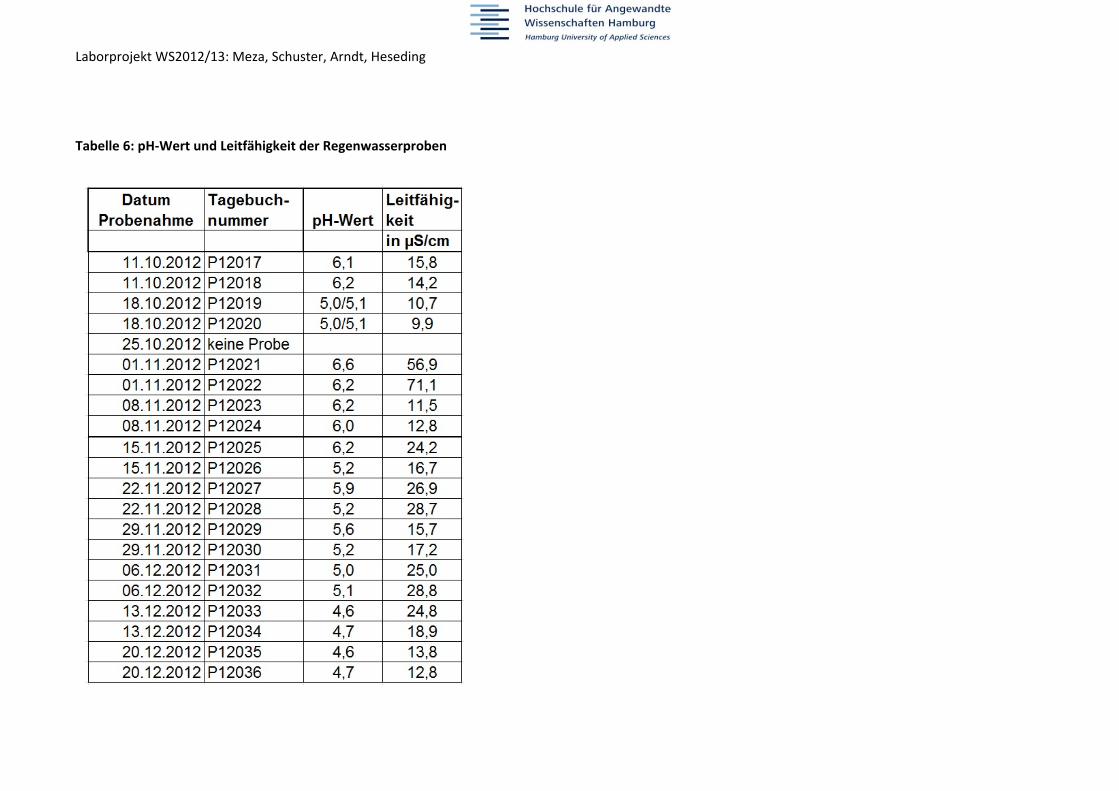
Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding Tabelle 6: pH‐Wert und Leitfähigkeit der Regenwasserproben
25
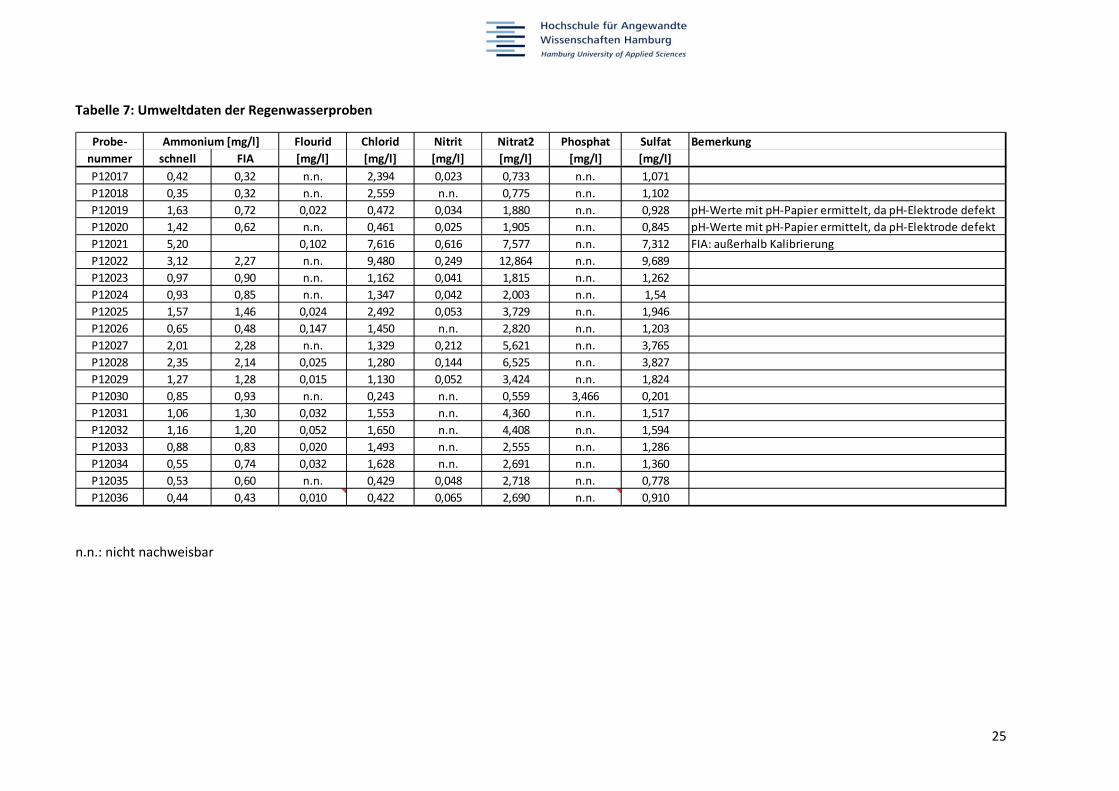
Tabelle 7: Umweltdaten der Regenwasserproben
n.n.: nicht nachweisbar
Probe‐ Flourid Chlorid Nitrit Nitrat2 Phosphat Sulfat Bemerkung
nummer schnell FIA [mg/l] [mg/l] [mg/l] [mg/l] [mg/l] [mg/l]
P12017 0,42 0,32 n.n. 2,394 0,023 0,733 n.n. 1,071
P12018 0,35 0,32 n.n. 2,559 n.n. 0,775 n.n. 1,102
P12019 1,63 0,72 0,022 0,472 0,034 1,880 n.n. 0,928 pH‐Werte mit pH‐Papier ermittelt, da pH‐Elektrode defekt
P12020 1,42 0,62 n.n. 0,461 0,025 1,905 n.n. 0,845 pH‐Werte mit pH‐Papier ermittelt, da pH‐Elektrode defekt
P12021 5,20 0,102 7,616 0,616 7,577 n.n. 7,312 FIA: außerhalb Kalibrierung
P12022 3,12 2,27 n.n. 9,480 0,249 12,864 n.n. 9,689
P12023 0,97 0,90 n.n. 1,162 0,041 1,815 n.n. 1,262
P12024 0,93 0,85 n.n. 1,347 0,042 2,003 n.n. 1,54
P12025 1,57 1,46 0,024 2,492 0,053 3,729 n.n. 1,946
P12026 0,65 0,48 0,147 1,450 n.n. 2,820 n.n. 1,203
P12027 2,01 2,28 n.n. 1,329 0,212 5,621 n.n. 3,765
P12028 2,35 2,14 0,025 1,280 0,144 6,525 n.n. 3,827
P12029 1,27 1,28 0,015 1,130 0,052 3,424 n.n. 1,824
P12030 0,85 0,93 n.n. 0,243 n.n. 0,559 3,466 0,201
P12031 1,06 1,30 0,032 1,553 n.n. 4,360 n.n. 1,517
P12032 1,16 1,20 0,052 1,650 n.n. 4,408 n.n. 1,594
P12033 0,88 0,83 0,020 1,493 n.n. 2,555 n.n. 1,286
P12034 0,55 0,74 0,032 1,628 n.n. 2,691 n.n. 1,360
P12035 0,53 0,60 n.n. 0,429 0,048 2,718 n.n. 0,778
P12036 0,44 0,43 0,010 0,422 0,065 2,690 n.n. 0,910
Ammonium [mg/l]
Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
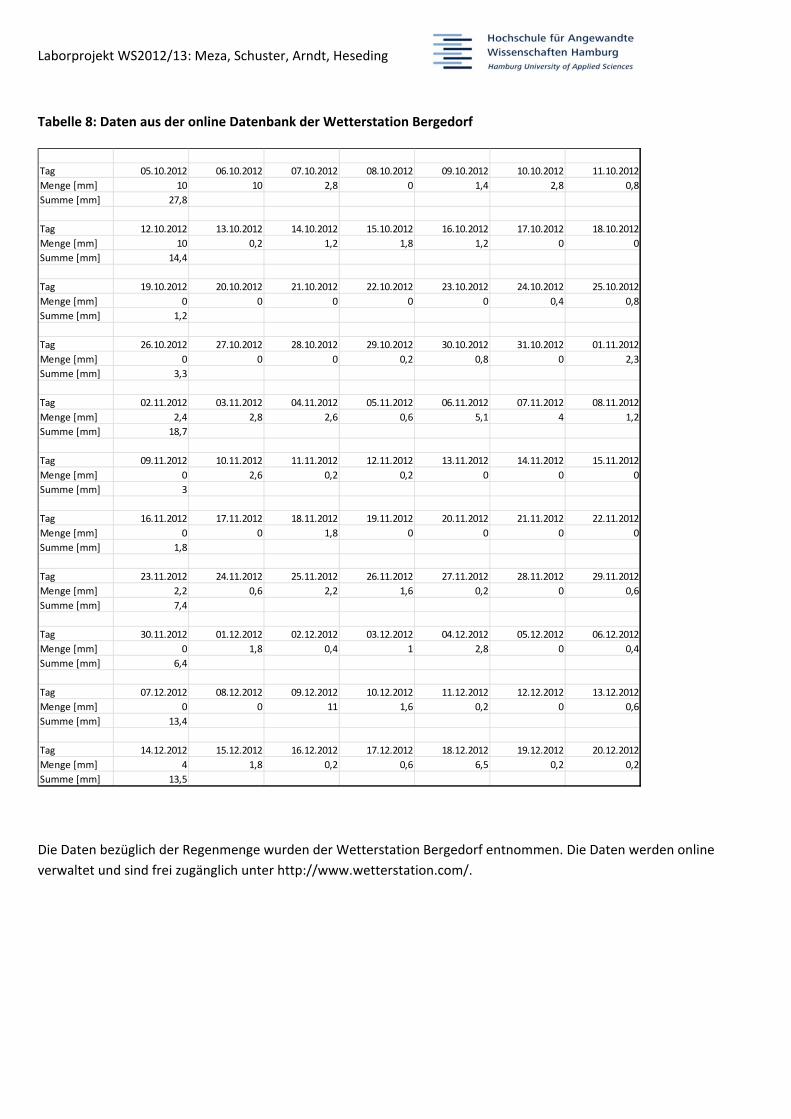
Tabelle 8: Daten aus der online Datenbank der Wetterstation Bergedorf
Die Daten bezüglich der Regenmenge wurden der Wetterstation Bergedorf entnommen. Die Daten werden online
verwaltet und sind frei zugänglich unter http://www.wetterstation.com/.
Tag 05.10.2012 06.10.2012 07.10.2012 08.10.2012 09.10.2012 10.10.2012 11.10.2012
Menge [mm] 10 10 2,8 0 1,4 2,8 0,8
Summe [mm] 27,8
Tag 12.10.2012 13.10.2012 14.10.2012 15.10.2012 16.10.2012 17.10.2012 18.10.2012
Menge [mm] 10 0,2 1,2 1,8 1,2 0 0
Summe [mm] 14,4
Tag 19.10.2012 20.10.2012 21.10.2012 22.10.2012 23.10.2012 24.10.2012 25.10.2012
Menge [mm] 0 0 0 0 0 0,4 0,8
Summe [mm] 1,2
Tag 26.10.2012 27.10.2012 28.10.2012 29.10.2012 30.10.2012 31.10.2012 01.11.2012
Menge [mm] 0 0 0 0,2 0,8 0 2,3
Summe [mm] 3,3
Tag 02.11.2012 03.11.2012 04.11.2012 05.11.2012 06.11.2012 07.11.2012 08.11.2012
Menge [mm] 2,4 2,8 2,6 0,6 5,1 4 1,2
Summe [mm] 18,7
Tag 09.11.2012 10.11.2012 11.11.2012 12.11.2012 13.11.2012 14.11.2012 15.11.2012
Menge [mm] 0 2,6 0,2 0,2 0 0 0
Summe [mm] 3
Tag 16.11.2012 17.11.2012 18.11.2012 19.11.2012 20.11.2012 21.11.2012 22.11.2012
Menge [mm] 0 0 1,8 0 0 0 0
Summe [mm] 1,8
Tag 23.11.2012 24.11.2012 25.11.2012 26.11.2012 27.11.2012 28.11.2012 29.11.2012
Menge [mm] 2,2 0,6 2,2 1,6 0,2 0 0,6
Summe [mm] 7,4
Tag 30.11.2012 01.12.2012 02.12.2012 03.12.2012 04.12.2012 05.12.2012 06.12.2012
Menge [mm] 0 1,8 0,4 1 2,8 0 0,4
Summe [mm] 6,4
Tag 07.12.2012 08.12.2012 09.12.2012 10.12.2012 11.12.2012 12.12.2012 13.12.2012
Menge [mm] 0 0 11 1,6 0,2 0 0,6
Summe [mm] 13,4
Tag 14.12.2012 15.12.2012 16.12.2012 17.12.2012 18.12.2012 19.12.2012 20.12.2012
Menge [mm] 4 1,8 0,2 0,6 6,5 0,2 0,2
Summe [mm] 13,5
Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
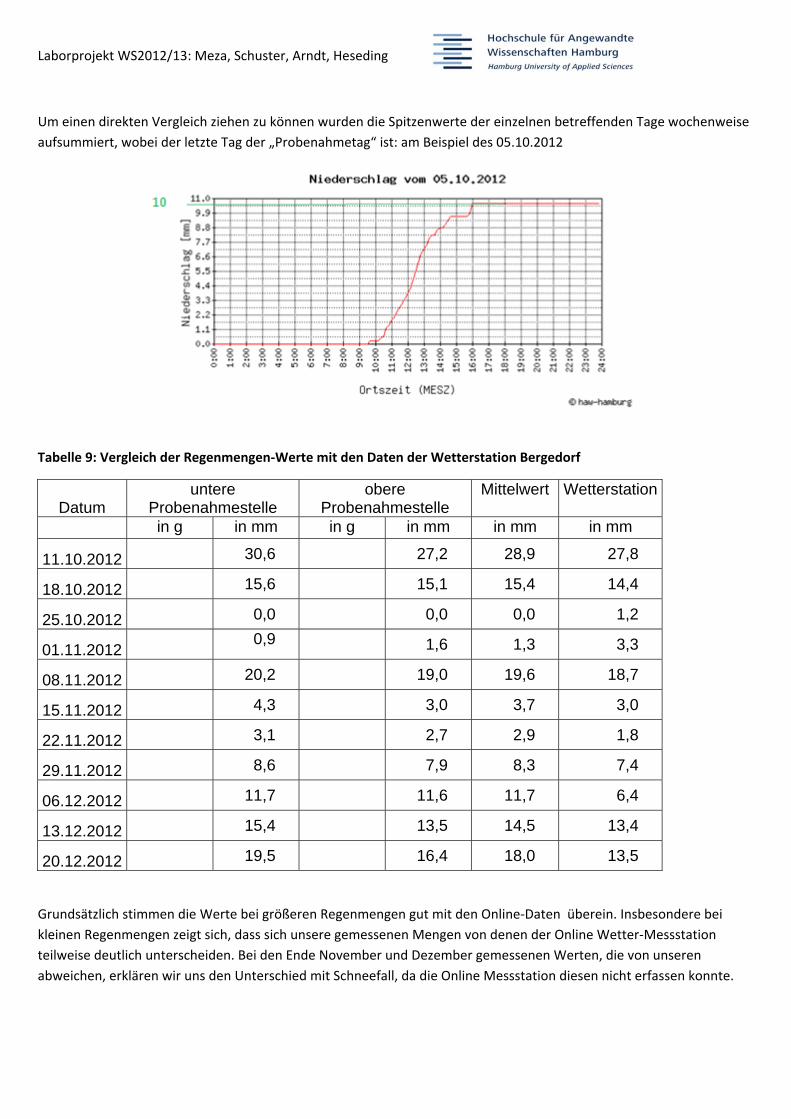
Um einen direkten Vergleich ziehen zu können wurden die Spitzenwerte der einzelnen betreffenden Tage wochenweise
aufsummiert, wobei der letzte Tag der „Probenahmetag“ ist: am Beispiel des 05.10.2012
Tabelle 9: Vergleich der Regenmengen‐Werte mit den Daten der Wetterstation Bergedorf
Datum untere
Probenahmestelle obere
Probenahmestelle Mittelwert Wetterstation
in g in mm in g in mm in mm in mm
11.10.2012 30,6 27,2 28,9 27,8
18.10.2012 15,6 15,1 15,4 14,4
25.10.2012 0,0 0,0 0,0 1,2
01.11.2012 0,9 1,6 1,3 3,3
08.11.2012 20,2 19,0 19,6 18,7
15.11.2012 4,3 3,0 3,7 3,0
22.11.2012 3,1 2,7 2,9 1,8
29.11.2012 8,6 7,9 8,3 7,4
06.12.2012 11,7 11,6 11,7 6,4
13.12.2012 15,4 13,5 14,5 13,4
20.12.2012 19,5 16,4 18,0 13,5
Grundsätzlich stimmen die Werte bei größeren Regenmengen gut mit den Online‐Daten überein. Insbesondere bei
kleinen Regenmengen zeigt sich, dass sich unsere gemessenen Mengen von denen der Online Wetter‐Messstation
teilweise deutlich unterscheiden. Bei den Ende November und Dezember gemessenen Werten, die von unseren
abweichen, erklären wir uns den Unterschied mit Schneefall, da die Online Messstation diesen nicht erfassen konnte.

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
5. Fazit
Der Niederschlag, der ursprünglich der chemischen Zusammensetzung von verdampften Meerwasser entspricht, wird
während des Transports in der Atmosphäre zusätzlich mit Inhaltsstoffen angereichert, die sowohl natürlicher als auch
anthropogener Natur sein können. Ein wesentlicher Prozess dabei ist die Oxidation von Stickoxiden und Schwefeldioxid,
die vor allem durch die Verbrennung fossiler Brennstoffe in die Atmosphäre eingetragen werden. Es entstehen (z.T. weit
von der Emissionsquelle entfernt) Salpeter‐ und Schwefelsäure. Aus der Landwirtschaft werden durch Evaporation von
Düngern vor allem NH4OH und NH3 freigesetzt. Teile der durch industrielle Emissionen eingebrachten Säuren HNO3 und
H2SO4 werden durch CaCO3 von kontinentalem Staub und dem NH4OH aus der Dünnung neutralisiert. Der nicht
neutralisierte Rest ist im Regenwasser in Form von freien Protonen präsent und bestimmt den pH‐Wert des
Regenwassers (APPELO & POSTMA 1999). Die Zusammensetzung des Regenwassers wird nach SIGG & STUMM (1996)
hauptsächlich durch die Konzentrationen der starken Säuren H2SO4, HNO3 und HCl sowie der basischen Komponenten
NH3 und Carbonat bestimmt. Das natürliche Gleichgewicht mit CO2 in der Atmosphäre und die Anwesenheit kleiner
Konzentrationen anderer Säuren (z. B. organischer Säuren) tragen ebenfalls zum Säure‐Base‐Gleichgewicht des
Niederschlagswassers bei. Die wichtigsten gelösten Bestandteile des Niederschlags sind demnach Cl‐, SO42‐,SO3
2‐, NO3‐,
NO2‐, HCO3
‐, Na+, K+, Ca2+, Mg2+und NH4+, das mit Luftsauerstoff unter Abgabe von Protonen zu NO3
‐oxidiert werden
kann. Mit Werten zwischen 0,01 und 1 mg/l ist Ammonium das wichtigste Kation im Niederschlag (MATTHEß 1994). Die
von uns analysierten Werte liegen zwischen 0,3 mg/l und ca. 5 mg/l; der Median liegt bei 0,85 mg/L, das 10%‐Percentil
bei ß(NH4+) = 0,4 mg/L und das 90%‐Perzentil bei ß(NH4
+) = 2,2 mg/L . Der Grenzwert für Trinkwasser liegt bei 0,5mg pro
L8. Diese Erhöhungen sind für Menschen als unbedenklich anzusehen; zwar ist Ammonium ein Nervengift, für eine Letale
Dosis Sind allerdings stark erhöhte Werte (> 30mg/L) über einen längeren Zeitraum erforderlich. In den Niederschlags‐
Analysen wurden pH‐Werte von 4,6 bis 6,6 gemessen. Die Leitfähigkeit liegt im Bereich zwischen 10 und 30 μS/cm,
wobei Spitzenwerte bei ca. 70 µS/cm beobachtet wurden. Die Aufenthaltszeit des Probenwassers im Gelände kann den
Eintrag von organischer Substanz und eine Veränderung der Sauerstoffsättigung zur Folge haben. Da der Nitratwert ein
redox‐sensibler Parameter ist, machen sich diese Einflüsse hier besonders bemerkbar. Bei erhöhter Sauerstoffsättigung
durch Aufnahme von Luftsauerstoff wird Ammonium zu Nitrat oxidiert, bei Sauerstoffverbrauch durch eingebrachte
organische Substanz wird das Nitrat reduziert (VORNEHM et al. 2003).9 Im Vergleich mit der Online Wetter‐Messstation
zeigen sich teilweise stärkere Unterschiede mit den von uns ermittelten Volumina. Einflussgrößen könnten hier u.a. die
Sonneneinstrahlung und damit verbundene Verdampfungsprozesse über den Tag, Windeinflüsse o.ä. sein. In wie fern
diese allerdings die Differenzen erklären können, müsste gesondert mit einer Analyse des Aufbaus der Wetterstation
erfolgen. Bei den von Ende November bis Dezember gemessenen Werten die von unseren teilweise stark abweichen,
erklären wir uns den Unterschied mit Schneefall, da die Online Messstation diesen nicht erfassen konnte; siehe
ebenfalls Berichte zur Regenwasseranalyse vorangegangener Gruppen. Die Spanne des pH‐Wertes variiert im Laufe der
Messphase zwischen einem pH‐Wert von 4,6 bis 6,6. Bei der von uns durchgeführten Analyse ist auffällig, das die am
Boden aufgefangenen Proben immer einen Ammoniumgehalt leicht unterhalb der Proben auf dem Dach aufweisen. In
wie weit hier die photodynamischen Einflüsse eine Rolle spielen, kann evtl. durch einen variierten Probenaufbau in
folgenden Versuchen ermittelt werden.
Ein kurzer allgemeiner Kommentar zu weiterführender Literatur: Die ersten Schritte zu einem verantwortungsvollen
Umgang mit Wasser im Haushalt sind wassersparendes Verhalten und der Einsatz wassersparender Armaturen. In
Großstädten wird die Nutzung von Regenwasser in Haushalten eine eher untergeordnete Rolle spielen. Denkbar wäre
8 http://www.dvgw.de/wasser/recht‐trinkwasserverordnung/trinkwasserverordnung/anlage‐3, 17.03.2013 9 http://edoc.ub.uni‐muenchen.de/4041/1/Vornehm_Christine.pdf: Hydro‐geochemische Untersuchungen zum System
Niederschlag – Boden – Grundwasser im Grundgebirge des Bayerischen Waldes; 17.03.2013

Laborprojekt WS2012/13: Meza, Schuster, Arndt, Heseding
hier die Regenwassernutzung in Einrichtungen wie Flughäfen, Gewerbebetrieben und Fußballstadien. Dort wird
Trinkwasser in sehr großem Umfang nur für Bewässerung und Toilettenspülung gebraucht. Das Niederschlagswasser des
Stadiondaches im Berliner Olympiastadion etwa wird seit Fertigstellung im Sommer 2004 vollständig auf dem Gelände
bewirtschaftet. Die Hälfte der Regenmenge ist für die Bewässerung des Spielfeldes vorgesehen, der Rest versickert. Eine
Nutzung für die Toilettenspülung erfolgt hingegen nicht. Zu erwähnen ist, dass bei der Planung des neuen
Dienstgebäudes für das Umweltbundesamt in Dessau die Nutzung von Regenwasser geprüft, auf die Realisierung jedoch
aus ökologischen wie ökonomischen Gründen verzichtet wurde. Hingegen gibt es im gewerblichen und industriellen
Bereich verschiedene zweckmäßige Einsatzmöglichkeiten für Regenwasser. Beispielsweise zur Reinigung von Tierställen
in der Landwirtschaft, für große Klimaanlagen mit Kühltürmen, Autowaschanlagen und als Prozesswasser in der
Industrie. In Haushalten, in denen die Sparmöglichkeiten – außer der Regenwassernutzung – ausgeschöpft wurden,
beträgt der durchschnittliche Verbrauch circa 95 Liter pro Person und Tag. Durch die zusätzliche Verwendung von
Regenwasser in Garten und Haushalt können zusätzlich ca. 35 bis 40 Liter Trinkwasser pro Person und Tag durch
Regenwasser ersetzt werden, vorausgesetzt der Regenwasserertrag ist entsprechend hoch. Bezüglich der Nutzung von
Regenwasser zum Wäschewaschen wird in anderen Kapiteln des zitierten Berichtes hingewiesen.10
10 http://www.umweltdaten.de/publikationen/fpdf‐l/2973.pdf: Versickerung und Nutzung von Regenwasser; 17.03.2013










