
Vorlesung SS 2002 FU-Berlin Professor M....
108
1 Stereochemie Vorlesung SS 2002 FU-Berlin Professor M. Kalesse
Transcript of Vorlesung SS 2002 FU-Berlin Professor M....

1
Stereochemie
Vorlesung SS 2002FU-Berlin
Professor M. Kalesse

2
Einführung in die Stereochemie1) Molekulare Konnektivität beschreibt, in welcher Reihenfolge die Atome einer Substanz miteinander verbunden sind.
2) Verbindungen mit identischer Summenformel aber unterschiedlicher Konnektivität nennt man Isomere.Z.B. für C2H6O: CH3CH2OH oder CH3OCH3
3) Verbindungen mit identischer Konnektivität aber unterschiedlicher Anordnung der Atome im Raum werdenStereoisomere genannt.
3
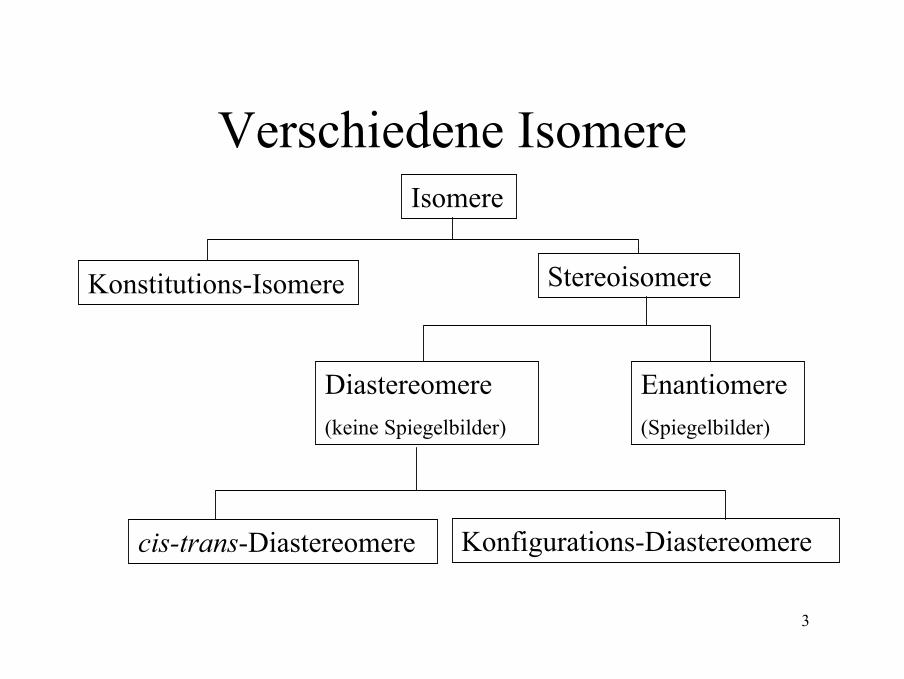
Verschiedene IsomereIsomere
Stereoisomere
Enantiomere(Spiegelbilder)
Diastereomere(keine Spiegelbilder)
Konfigurations-Diastereomerecis-trans-Diastereomere
Konstitutions-Isomere
4
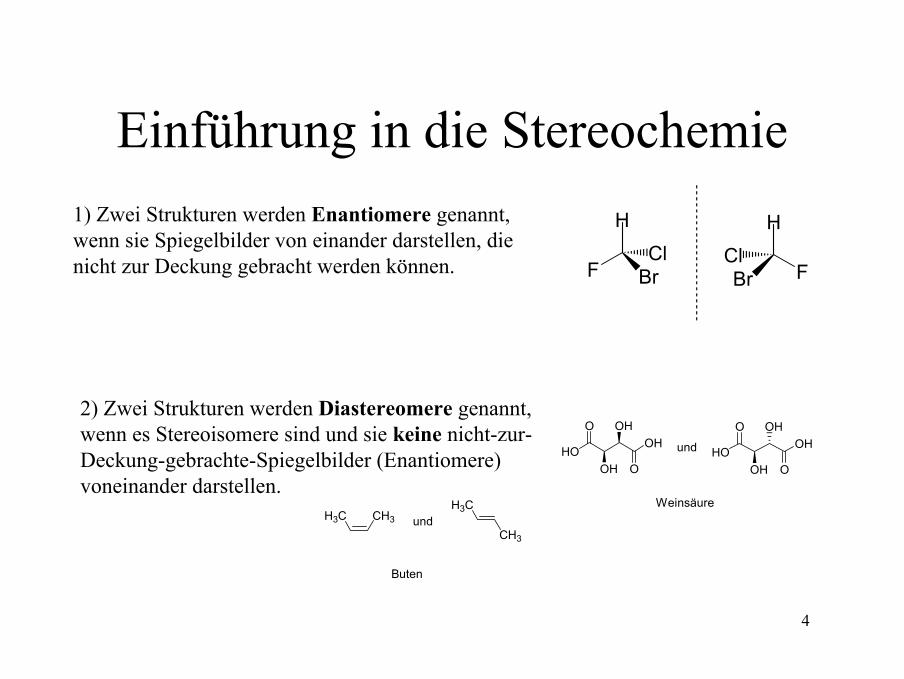
Einführung in die Stereochemie1) Zwei Strukturen werden Enantiomere genannt, wenn sie Spiegelbilder von einander darstellen, die nicht zur Deckung gebracht werden können.
H
F BrCl
H
FBrCl
2) Zwei Strukturen werden Diastereomere genannt, wenn es Stereoisomere sind und sie keine nicht-zur-Deckung-gebrachte-Spiegelbilder (Enantiomere) voneinander darstellen.
HOOH
O
O
OH
OHHO
OHO
O
OH
OH
Weinsäure
und
CH3H3CH3C
CH3
und
Buten
5
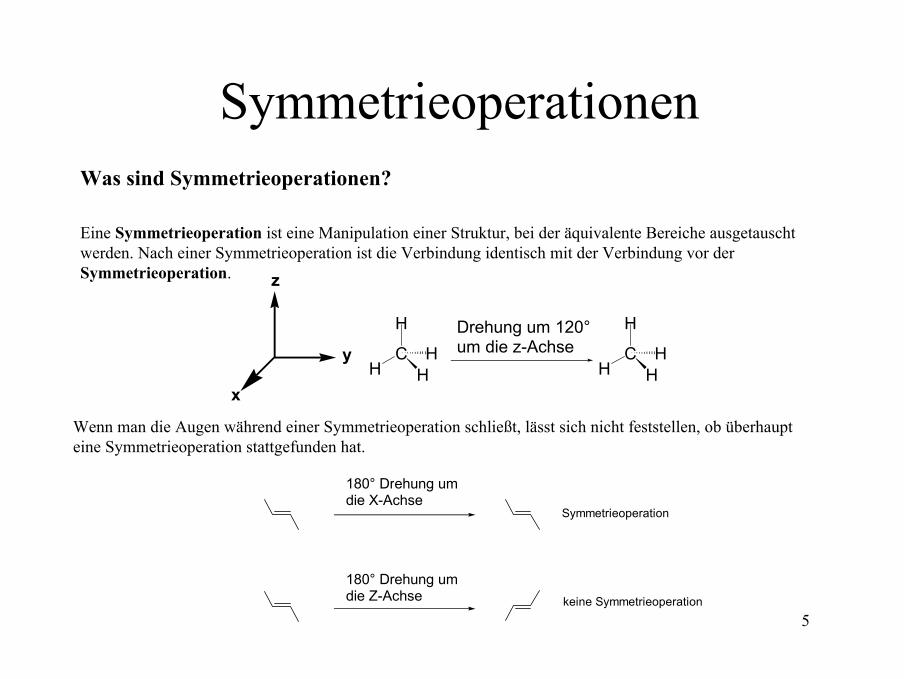
SymmetrieoperationenWas sind Symmetrieoperationen?
Eine Symmetrieoperation ist eine Manipulation einer Struktur, bei der äquivalente Bereiche ausgetauscht werden. Nach einer Symmetrieoperation ist die Verbindung identisch mit der Verbindung vor der Symmetrieoperation.
H
CH H
H
z
y
x
Drehung um 120°um die z-Achse
H
CH H
H
Wenn man die Augen während einer Symmetrieoperation schließt, lässt sich nicht feststellen, ob überhaupt eine Symmetrieoperation stattgefunden hat.
180° Drehung umdie X-Achse
180° Drehung um die Z-Achse
Symmetrieoperation
keine Symmetrieoperation

6
Symmetrieoperationen
H
CH H
H
H
CH H
H
Achse, um welche die Symmetrieoperation (Rotation um 120°)durchgeführt wurde.
1. Rotation: ≥1, Rotation um eine Achse (von 0 bis 360°)2. Reflektion: Reflektion in einer Ebene, die durch die Struktur verläuft3. Inversion aller Atome durch einen zentralen Punkt:Die Operation verändert alle Punkte (x, y, z) zu (-x, -y, -z)4. Rotation-Reflektion: ≥1Wiederholungen der folgenden Operationen:Rotation um eine Achse, dann Reflektion in einer Ebene senkrecht zur Rotationsachse
Anwendung dieser Operationen auf molekulare Strukturen
Ein Symmetrieelement ist eine geometrische Einheit (Punkt, Linie, Ebene),an welcher die Symmetrieoperation durchgeführt wird.

7
Symmetrieoperationen und Symmetrieelemente
Symmetrieelement Symmetrieoperation1. Drehachse = Cn n Drehungen von (360°/n)
2. Spiegelebene = σ Spiegelung an einer Ebene
3. Inversionszentrum = i Inversion
4. Dreh-Spiegel-Achse = Sn n Wiederholungen von: Drehungen um (360°/n) dann Spiegelung an einer Ebene senkrecht zur Achse
8
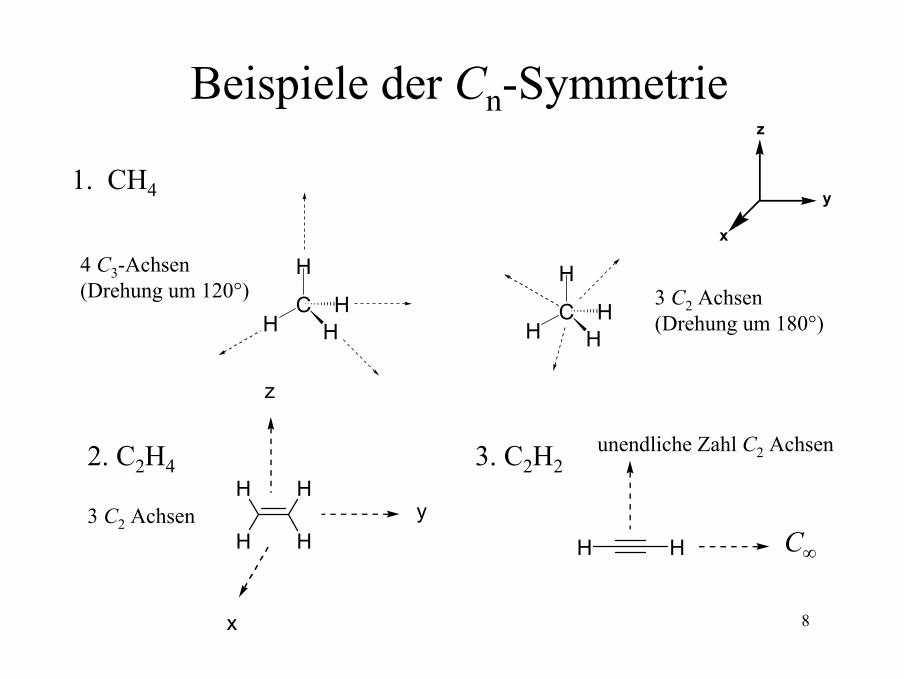
Beispiele der Cn-Symmetriez
y
x
H
CH H
HH
CH H
H
1. CH4
4 C3-Achsen(Drehung um 120°) 3 C2 Achsen
(Drehung um 180°)
H
HH
H
z
y
x
2. C2H4
3 C2 AchsenHH
3. C2H2unendliche Zahl C2 Achsen
C∞
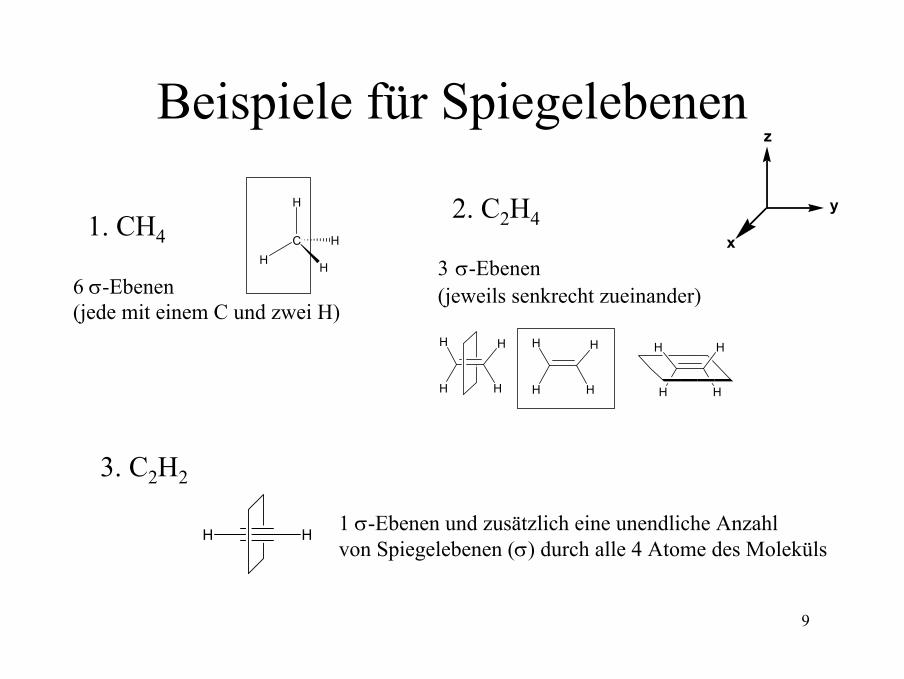
9
Beispiele für Spiegelebenen
1. CH4
6 σ-Ebenen (jede mit einem C und zwei H)
H
CH H
H
z
y
x
2. C2H4
3 σ-Ebenen (jeweils senkrecht zueinander)
H
HH
H H
HH
H HH
HH
HH
3. C2H2
1 σ-Ebenen und zusätzlich eine unendliche Anzahl von Spiegelebenen (σ) durch alle 4 Atome des Moleküls
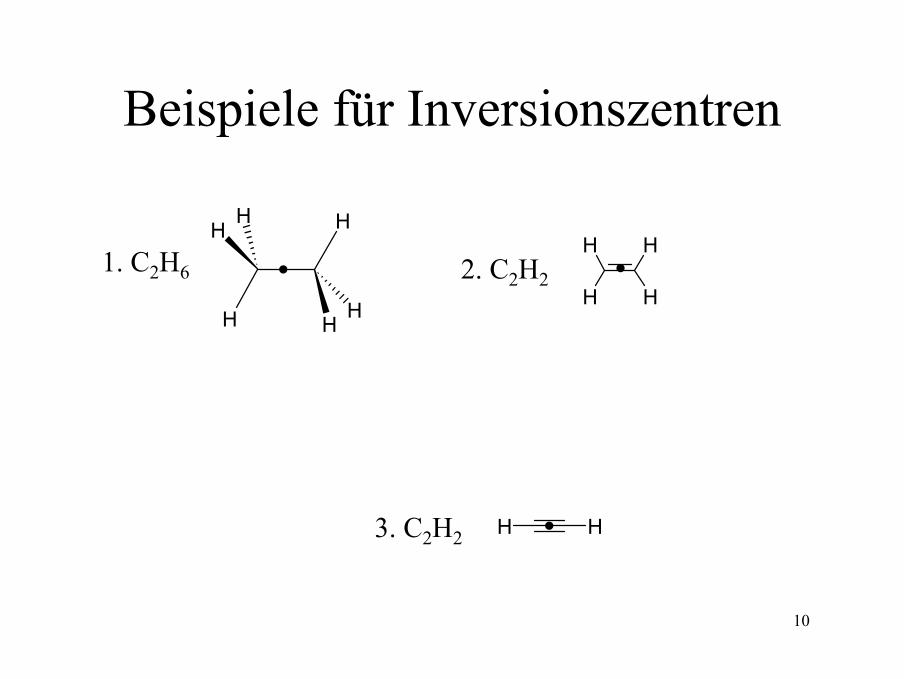
10
Beispiele für Inversionszentren
H
HH
HH
H
H
H
H
H
HH
1. C2H6 2. C2H2
3. C2H2
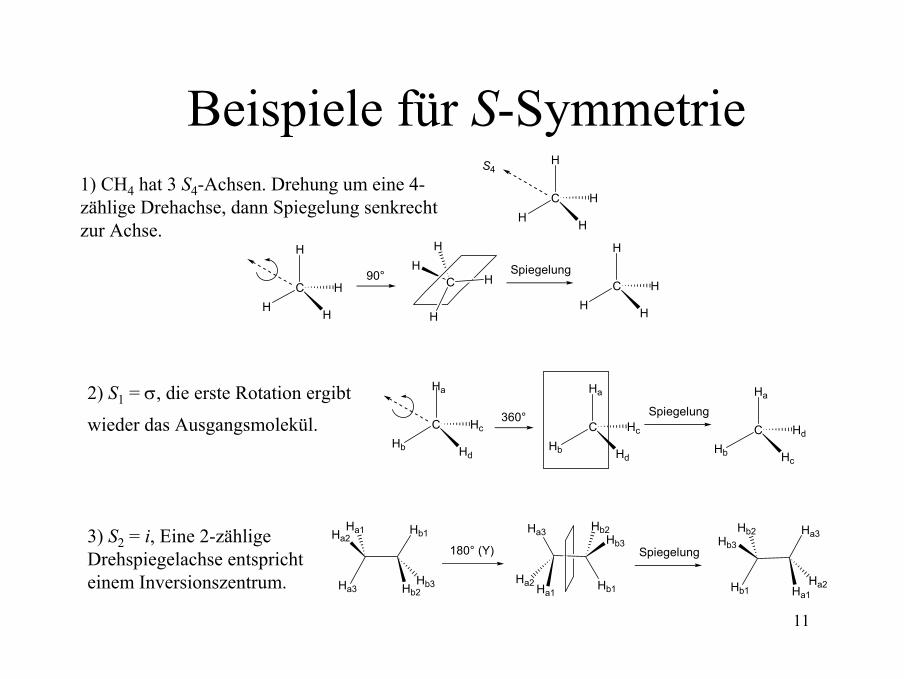
11
Beispiele für S-SymmetrieH
CH H
H
S4
H
CH H
H90°
H
C H
HH Spiegelung
H
CH H
H
360° Spiegelung
Ha
CHb Hd
Hc
Ha
CHb Hd
Hc
Ha
CHb Hc
Hd
Hb1
Hb2Hb3
Ha2Ha1
Ha3
180° (Y)
Hb1
Hb2Hb3
Ha2 Ha1
Ha3
Spiegelung
Hb1
Hb2Hb3
Ha2Ha1
Ha3
1) CH4 hat 3 S4-Achsen. Drehung um eine 4-zählige Drehachse, dann Spiegelung senkrecht zur Achse.
2) S1 = σ, die erste Rotation ergibt
wieder das Ausgangsmolekül.
3) S2 = i, Eine 2-zählige Drehspiegelachse entspricht einem Inversionszentrum.
12
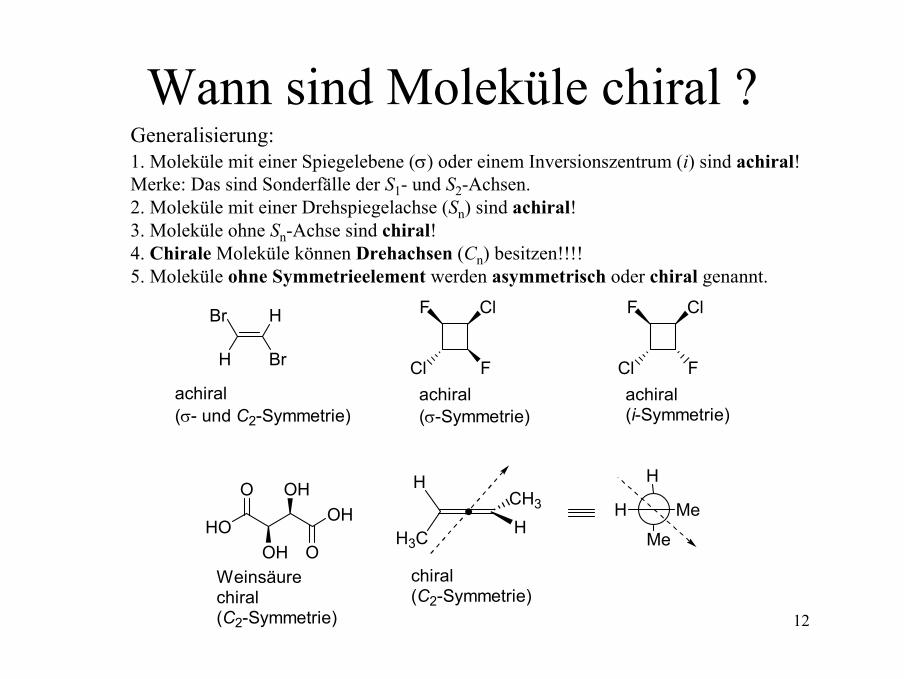
Wann sind Moleküle chiral ?Generalisierung:1. Moleküle mit einer Spiegelebene (σ) oder einem Inversionszentrum (i) sind achiral! Merke: Das sind Sonderfälle der S1- und S2-Achsen.2. Moleküle mit einer Drehspiegelachse (Sn) sind achiral!3. Moleküle ohne Sn-Achse sind chiral! 4. Chirale Moleküle können Drehachsen (Cn) besitzen!!!!5. Moleküle ohne Symmetrieelement werden asymmetrisch oder chiral genannt.
Br
H
H
Br
achiral(σ- und C2-Symmetrie)
ClF
FClachiral(σ-Symmetrie)
ClF
FClachiral(i-Symmetrie)
HOOH
O
O
OH
OHWeinsäurechiral(C2-Symmetrie)
H
H3C HCH3
chiral(C2-Symmetrie)
H Me
H
Me

13
Was sind chirale Moleküle?1) Moleküle mit einem chiralen Zentrum:Ein Chiralitätszentrum ist ein Atom (meistens ein C-Atom) mit vier verschiedenen Substituenten; auch oft stereogenes Zentrum genannt. Achtung: Jedes Chiralitätszentrum ist auch immer ein stereogenes Zentrum. Aber ein stereogenes Zentrummuss nicht unbedingt ein Chiralitätszentrum sein. Ein streogenes Zentrum ist dadurch definiert, dass der Positionstausch von zwei Substituenten ein Enantiomer oder Diastereomer erzeugt.Beispiele:Chirale Verbindung mit Chiralitätszentren (stereogenen Zentren)
HOOH
OH
OH
O
O
Weinsäure
meso-Weinsäure
21 3
4
HOOH
OH
OH
O
O2
1 34
C2 und C3 sind Chiralitätszentren oder auch stereogene Zentren. Das Molekül ist chiral.
C2 und C3 sind Chiralitätszentren oder auch stereogene Zentren. Aber: Das Molekül ist achiral.Meso-Verbindungen sind achirale-Verbindungen mit mehr als einem Chiralitätszentrum aufgrund der internen Symmetrie. Hier: Ein Beispiel für ein Inversionszentrum (i, bzw. S2) oder eine Spiegelebene nach Rotation.
HOOH
OH
OH
O
O
meso-Weinsäure
i = S2
Rotation
HO OHOH
OOHO
Spiegelebene (S1)

14
Stereogene Zentren
F
Cl
Cl
Stereogenes Zentrum
HF
Cl
ClH
Austausch von 2 Substituenten am stereogenen Zentrum
Ebenfalls keine chirale Verbindung
Achirale Verbindung (Spiegelebene); das stereogene Zentrum ist kein Chiralitätszentrum (zwei Substituenten sind identisch).
Die achirale Verbindung ist ein Diastereomer der ursprünglichenachiralen Verbindung.
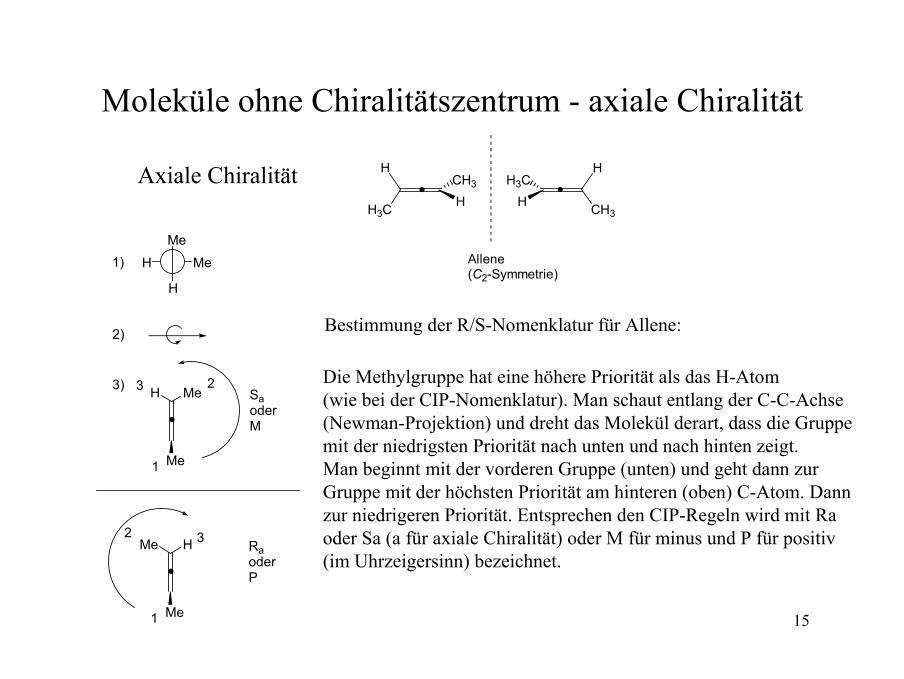
15
Moleküle ohne Chiralitätszentrum - axiale Chiralität
Axiale Chiralität H
H3C HCH3
Allene(C2-Symmetrie)
H
CH3H
H3C
Bestimmung der R/S-Nomenklatur für Allene:
H
Me
H Me
MeH
Me1
23
1)
2)
3)SaoderM
HMe
Me1
32RaoderP
Die Methylgruppe hat eine höhere Priorität als das H-Atom (wie bei der CIP-Nomenklatur). Man schaut entlang der C-C-Achse(Newman-Projektion) und dreht das Molekül derart, dass die Gruppe mit der niedrigsten Priorität nach unten und nach hinten zeigt. Man beginnt mit der vorderen Gruppe (unten) und geht dann zur Gruppe mit der höchsten Priorität am hinteren (oben) C-Atom. Dann zur niedrigeren Priorität. Entsprechen den CIP-Regeln wird mit Ra oder Sa (a für axiale Chiralität) oder M für minus und P für positiv (im Uhrzeigersinn) bezeichnet.
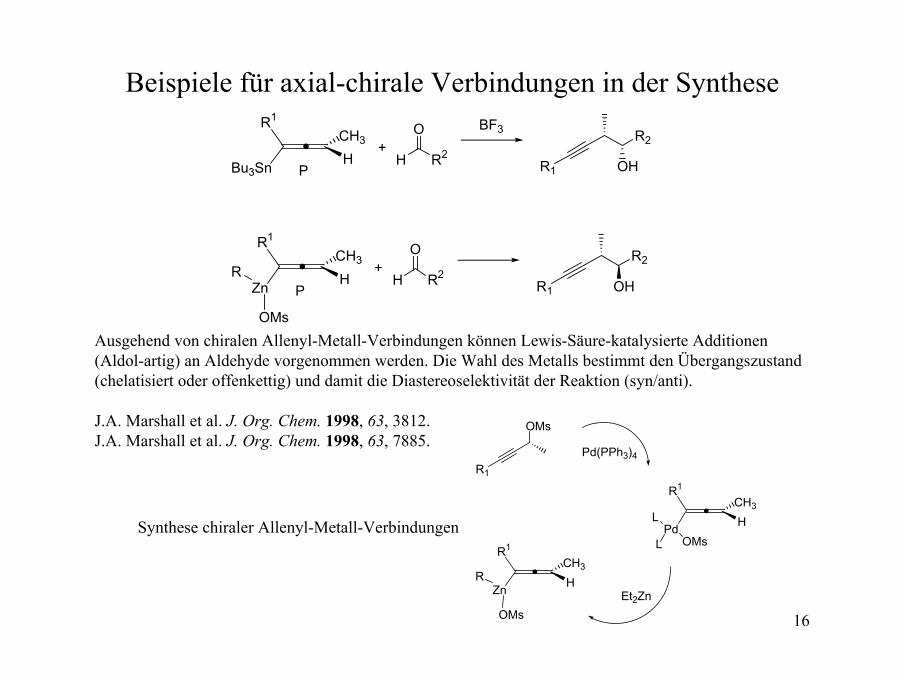
16
Beispiele für axial-chirale Verbindungen in der SyntheseR1
Bu3Sn HCH3
P+
O
H R2
BF3 R2
OHR1
R1
Zn HCH3
P+
O
H R2R2
OHR1R
OMs
Ausgehend von chiralen Allenyl-Metall-Verbindungen können Lewis-Säure-katalysierte Additionen(Aldol-artig) an Aldehyde vorgenommen werden. Die Wahl des Metalls bestimmt den Übergangszustand (chelatisiert oder offenkettig) und damit die Diastereoselektivität der Reaktion (syn/anti).
J.A. Marshall et al. J. Org. Chem. 1998, 63, 3812.J.A. Marshall et al. J. Org. Chem. 1998, 63, 7885.
R1
Pd HCH3
OMs
R1
R1
Zn HCH3
R
OMs
OMsL
L
Pd(PPh3)4
Et2Zn
Synthese chiraler Allenyl-Metall-Verbindungen
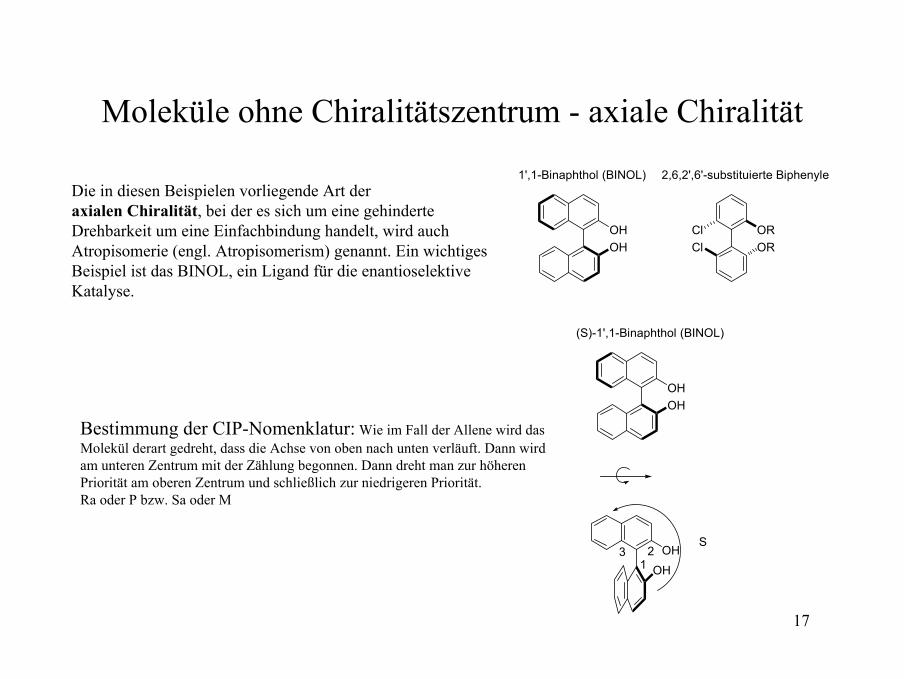
17
Moleküle ohne Chiralitätszentrum - axiale Chiralität
Die in diesen Beispielen vorliegende Art der axialen Chiralität, bei der es sich um eine gehinderteDrehbarkeit um eine Einfachbindung handelt, wird auchAtropisomerie (engl. Atropisomerism) genannt. Ein wichtiges Beispiel ist das BINOL, ein Ligand für die enantioselektiveKatalyse.
OHOH
Cl ORORCl
2,6,2',6'-substituierte Biphenyle1',1-Binaphthol (BINOL)
OHOH
(S)-1',1-Binaphthol (BINOL)
OHOH1
23S
Bestimmung der CIP-Nomenklatur: Wie im Fall der Allene wird das Molekül derart gedreht, dass die Achse von oben nach unten verläuft. Dann wird am unteren Zentrum mit der Zählung begonnen. Dann dreht man zur höheren Priorität am oberen Zentrum und schließlich zur niedrigeren Priorität. Ra oder P bzw. Sa oder M
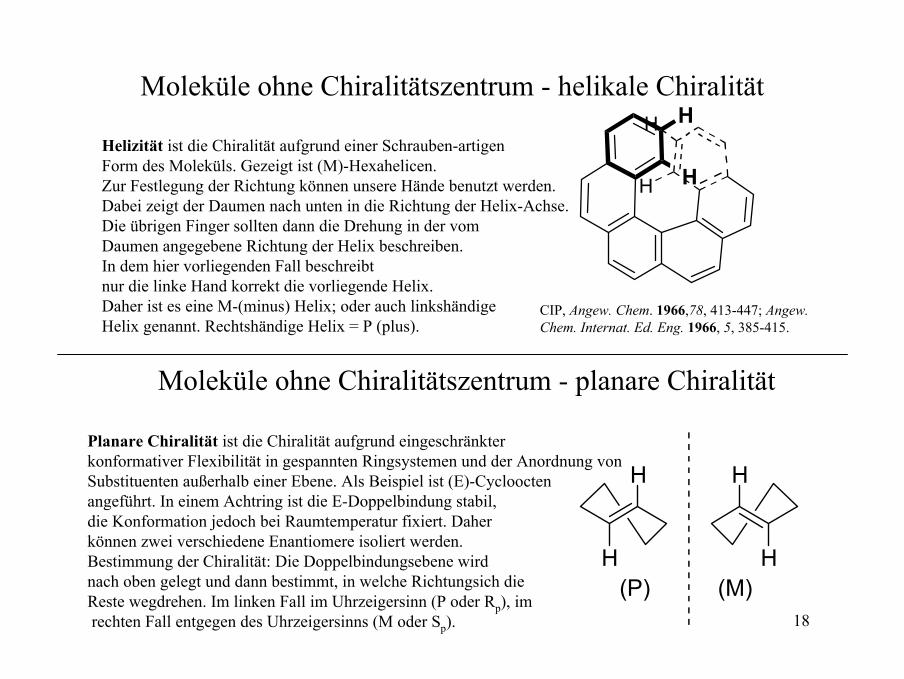
18
Moleküle ohne Chiralitätszentrum - helikale Chiralität
H
HH
H
Moleküle ohne Chiralitätszentrum - planare Chiralität
Helizität ist die Chiralität aufgrund einer Schrauben-artigenForm des Moleküls. Gezeigt ist (M)-Hexahelicen.Zur Festlegung der Richtung können unsere Hände benutzt werden.Dabei zeigt der Daumen nach unten in die Richtung der Helix-Achse. Die übrigen Finger sollten dann die Drehung in der vomDaumen angegebene Richtung der Helix beschreiben. In dem hier vorliegenden Fall beschreibt nur die linke Hand korrekt die vorliegende Helix.Daher ist es eine M-(minus) Helix; oder auch linkshändigeHelix genannt. Rechtshändige Helix = P (plus).
H
H
H
H(P) (M)
Planare Chiralität ist die Chiralität aufgrund eingeschränkter konformativer Flexibilität in gespannten Ringsystemen und der Anordnung vonSubstituenten außerhalb einer Ebene. Als Beispiel ist (E)-Cyclooctenangeführt. In einem Achtring ist die E-Doppelbindung stabil, die Konformation jedoch bei Raumtemperatur fixiert. Daher können zwei verschiedene Enantiomere isoliert werden. Bestimmung der Chiralität: Die Doppelbindungsebene wird nach oben gelegt und dann bestimmt, in welche Richtungsich die Reste wegdrehen. Im linken Fall im Uhrzeigersinn (P oder Rp), imrechten Fall entgegen des Uhrzeigersinns (M oder Sp).
CIP, Angew. Chem. 1966,78, 413-447; Angew. Chem. Internat. Ed. Eng. 1966, 5, 385-415.

19
Homotope Gruppen
HO OHHabH
2
Im 1,3-Propandiol betrachten wir die beiden H-Atome an C2.Werden beide H-Atome jeweils durch ein Deuterium ersetzt,entstehen zwei neue Moleküle, die jedoch beide achiral sind unddie ihre gegenseitigen Spiegelbilder darstellen. Man sagt, dieseAtome sind homotopisch (engl. homotopic).
HO OHHaD
HO OHDbH
identische Moleküle
Zwei Gruppen (hier H-Atome) sind homotopisch, wenn siedurch eine Cn-Drehachse ineinander überführbar sind. Hier überführt eine C2-Achse die beiden H-Atome ineinander.
20
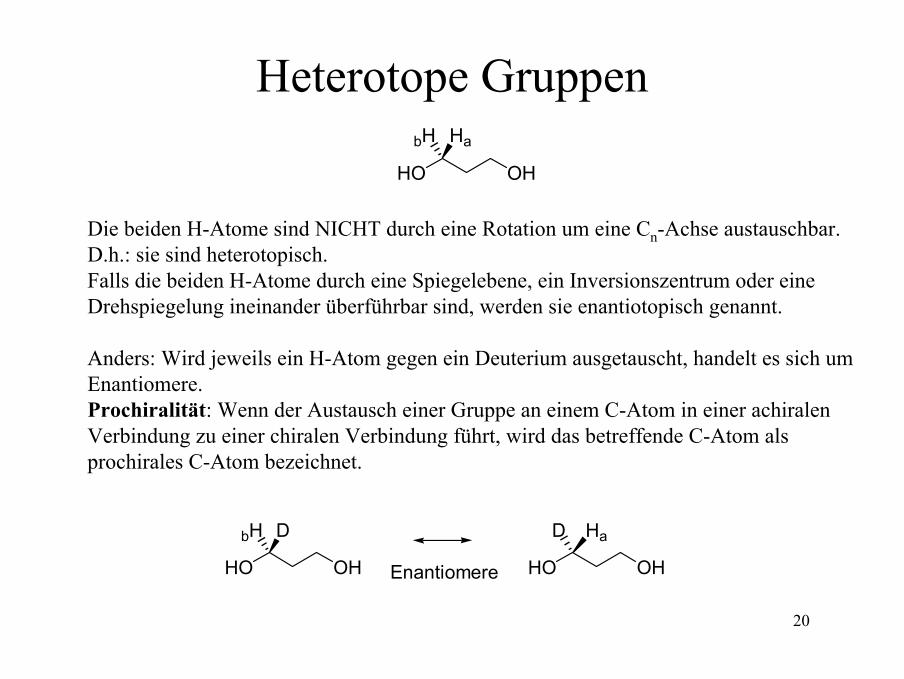
Heterotope Gruppen
HO
HabH
OH
Die beiden H-Atome sind NICHT durch eine Rotation um eine Cn-Achse austauschbar. D.h.: sie sind heterotopisch.Falls die beiden H-Atome durch eine Spiegelebene, ein Inversionszentrum oder eineDrehspiegelung ineinander überführbar sind, werden sie enantiotopisch genannt.
Anders: Wird jeweils ein H-Atom gegen ein Deuterium ausgetauscht, handelt es sich umEnantiomere.Prochiralität: Wenn der Austausch einer Gruppe an einem C-Atom in einer achiralenVerbindung zu einer chiralen Verbindung führt, wird das betreffende C-Atom alsprochirales C-Atom bezeichnet.
EnantiomereHO
DbH
OH HO
HaD
OH
21
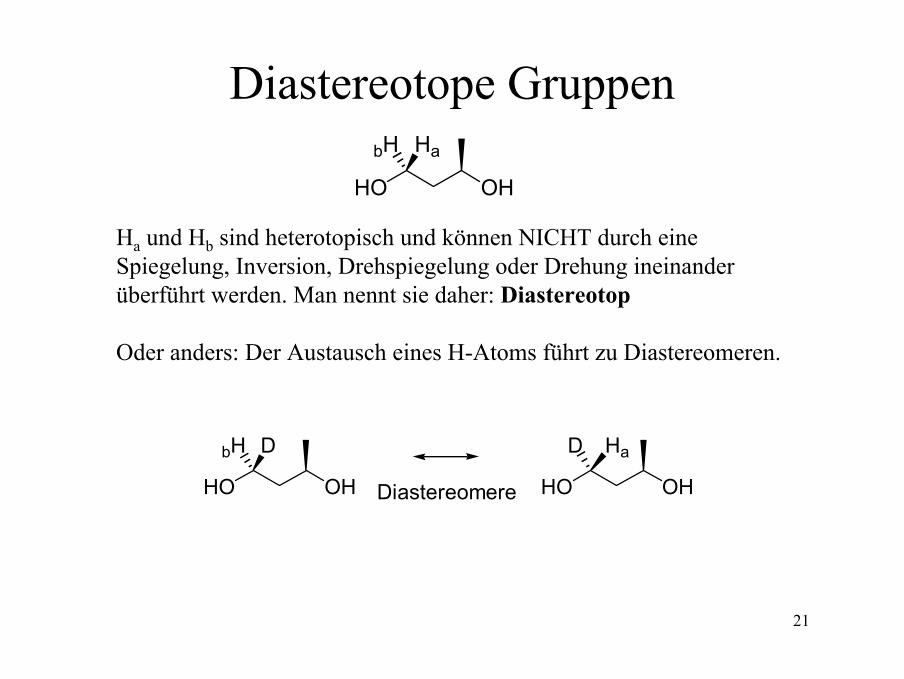
Diastereotope Gruppen
HO
HabH
OH
Ha und Hb sind heterotopisch und können NICHT durch eine Spiegelung, Inversion, Drehspiegelung oder Drehung ineinanderüberführt werden. Man nennt sie daher: Diastereotop
Oder anders: Der Austausch eines H-Atoms führt zu Diastereomeren.
DiastereomereHO
DbH
OH HO
HaD
OH
22
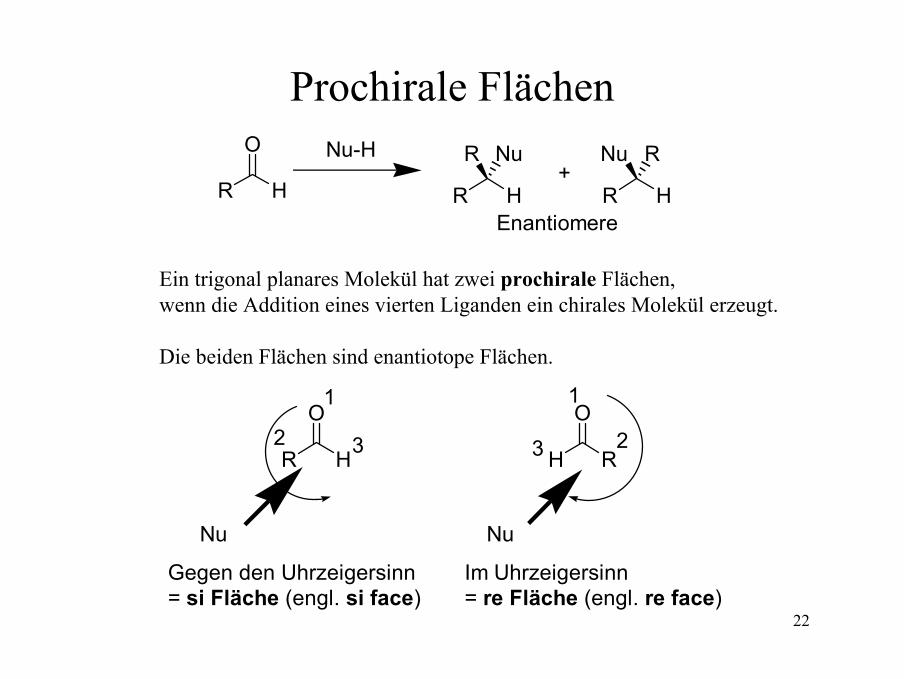
Prochirale FlächenO
R H
Nu-H
R H
R Nu
R H
Nu R+
Enantiomere
Ein trigonal planares Molekül hat zwei prochirale Flächen,wenn die Addition eines vierten Liganden ein chirales Molekül erzeugt.
Die beiden Flächen sind enantiotope Flächen.
O
R H
O
RH
NuNu
1
2 3
1
23
Gegen den Uhrzeigersinn= si Fläche (engl. si face)
Im Uhrzeigersinn= re Fläche (engl. re face)

23
Dynamische StereochemieMoleküle sind NICHT statisch. Sie unterliegen Rotationen und Streckungen. Eine Konformation beschreibt die exakte Anordnung der Atome im Raum.Verschiedene Konformationen können durch Drehungen um Einfachbindungen ineinander überführt werden. Unter Konformationsanalyse versteht man dieAbschätzung der relativen Energie-Werte verschiedener Konformationen einesMoleküls und deren Einfluss auf die chemischen Eigenschaften.
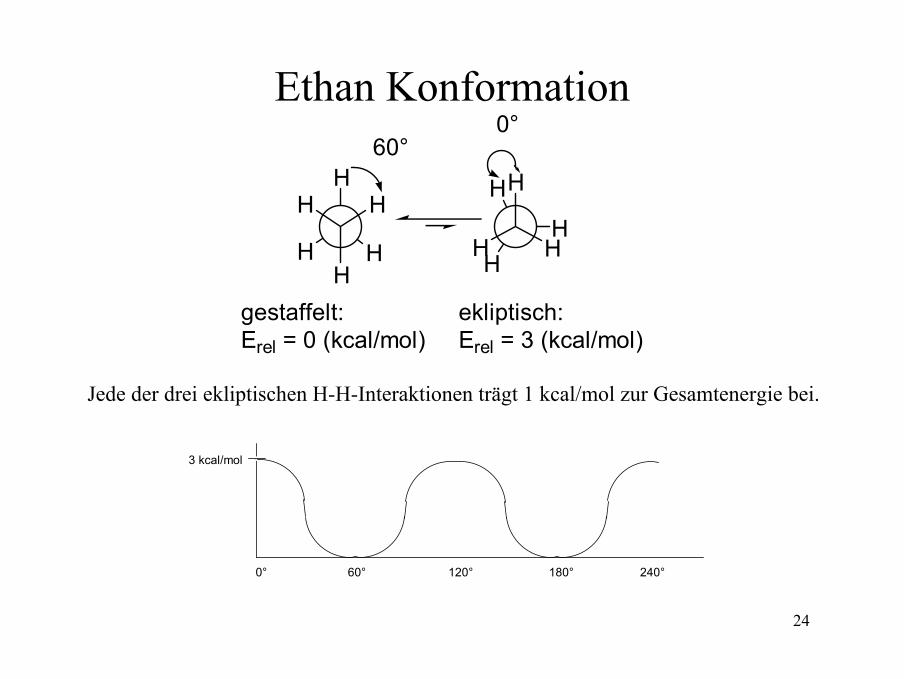
Konformationsanalyse von Ethan:Jede ekliptische-Anordnung (engl. eclipsed) zweier benachbarter H-Atome führt zu einem Anstieg der relativen Energie um 1 kcal/mol. Daher hat die ekliptische-Konformationeine um 3 kcal/mol höher Energie als die gestaffelte Konformation (engl. staggered).
H
H H
H
H
H
HH
H
H
H
H
Gestaffelte Konformation
Ekliptische Konformation
Erel = 0 (kcal/mol) Erel = 3 (kcal/mol)
24
Ethan Konformation
H
H H
H
H
H
HH
H
H
H
H
0°60°
gestaffelt: Erel = 0 (kcal/mol)
ekliptisch: Erel = 3 (kcal/mol)
Jede der drei ekliptischen H-H-Interaktionen trägt 1 kcal/mol zur Gesamtenergie bei.
3 kcal/mol
0° 60° 120° 180° 240°
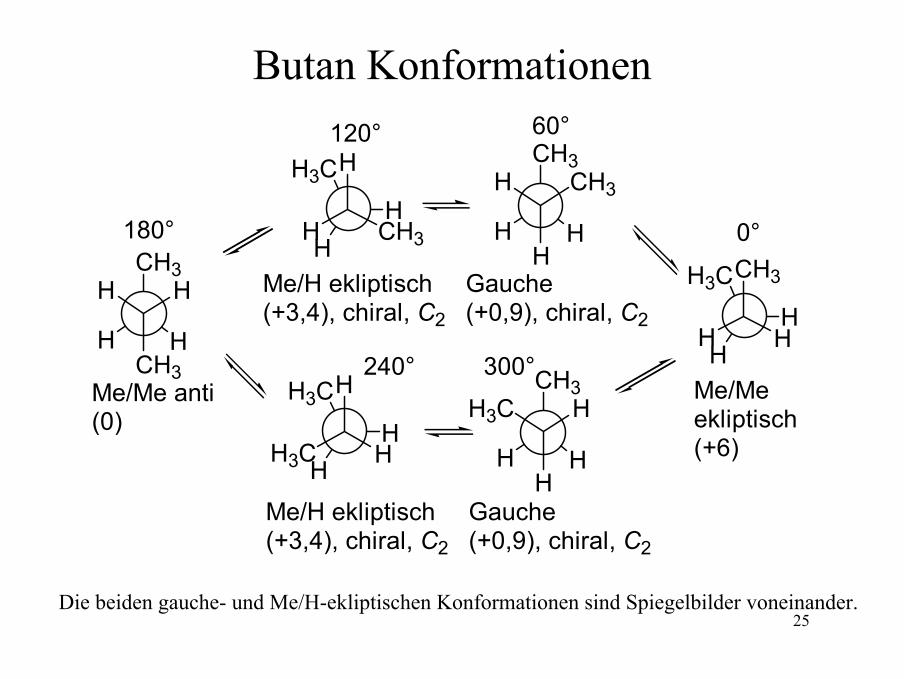
25
Butan Konformationen
CH3
H H
CH3
H
H
HH
H3C
H
H
CH3CH3
H H
H
CH3
H
CH3
H H
H
H
H3C
HH
H3C
H
CH3
H
H3CH
H3C
H
H
H
180°
Me/Me anti(0)
120°
Me/H ekliptisch(+3,4), chiral, C2
Gauche(+0,9), chiral, C2
Me/H ekliptisch(+3,4), chiral, C2
Gauche(+0,9), chiral, C2
Me/Me ekliptisch(+6)
60°
0°
300°240°
Die beiden gauche- und Me/H-ekliptischen Konformationen sind Spiegelbilder voneinander.
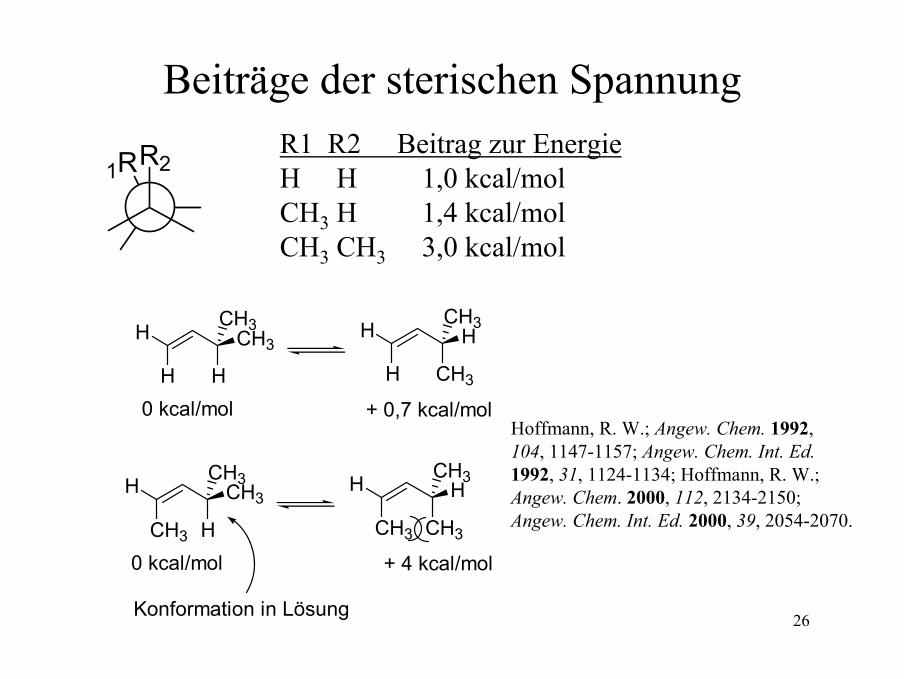
26
Beiträge der sterischen Spannung
1RR2R1 R2 Beitrag zur EnergieH H 1,0 kcal/molCH3 H 1,4 kcal/molCH3 CH3 3,0 kcal/mol
CH3
CH3
H
H
CH3 H
CH3
H
CH3
CH3
+ 4 kcal/mol 0 kcal/mol
CH3
H
H
H
CH3 H
H
H
CH3
CH3
+ 0,7 kcal/mol 0 kcal/mol
Konformation in Lösung
Hoffmann, R. W.; Angew. Chem. 1992, 104, 1147-1157; Angew. Chem. Int. Ed.1992, 31, 1124-1134; Hoffmann, R. W.; Angew. Chem. 2000, 112, 2134-2150; Angew. Chem. Int. Ed. 2000, 39, 2054-2070.
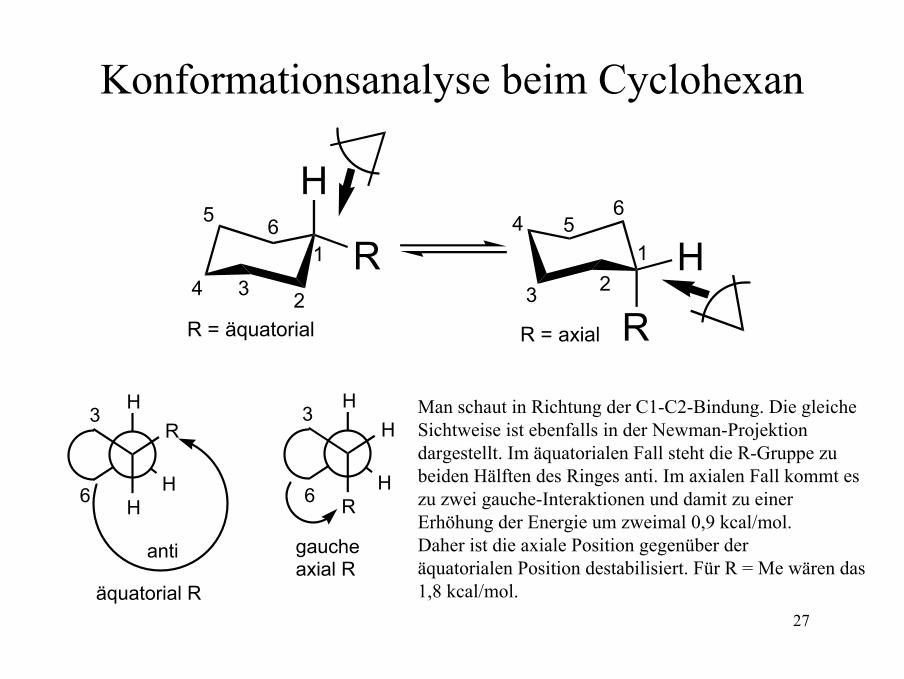
27
Konformationsanalyse beim Cyclohexan
H
H
R
H
H
H
H
R
3
6 6
3
anti
äquatorial R
gaucheaxial R
H
R
H
R
R = äquatorial R = axial
1
234
5 61
23
4 56
Man schaut in Richtung der C1-C2-Bindung. Die gleiche Sichtweise ist ebenfalls in der Newman-Projektiondargestellt. Im äquatorialen Fall steht die R-Gruppe zubeiden Hälften des Ringes anti. Im axialen Fall kommt eszu zwei gauche-Interaktionen und damit zu einer Erhöhung der Energie um zweimal 0,9 kcal/mol.Daher ist die axiale Position gegenüber der äquatorialen Position destabilisiert. Für R = Me wären das 1,8 kcal/mol.

28
Die A-Werte für Cyclohexane
H
R
H
R∆G°
Der A-Wert = - ∆G°
Für den Fall, dass R vorzugsweise äquatorial liegt, ist A positiv und somit ∆G° negativ.Je größer der Rest ist, um so größer ist der A-Wert.
R A-Werte (kcal/mol)-CCH 0,45-CH3 1,7-CH2CH3 1,8-CH(CH3)2 2,2-Si(CH3)3 2,5-C6H5 2,8-C(CH3)3 4,8

29
Der Zusammenhang zwischen Energie und Selektivität
B oder CABetrachtet wird die Transformation von A zu B oder C.
Thermodynamische Kontrolle:Selektivität = B/C = e (-∆∆G°/RT) R = Gaskonstante (1,99 cal/mol K)
T = Temperatur in Kelvin (Raumtemp. = 25°C, 298 K)
Ein ∆∆G+ von 1,4 kcal/mol ergibt ein Verhältnis von 10:1 für B/C.Ein ∆∆G+ von 2,8 kcal/mol ergibt ein Verhältnis von 100:1 für B/C.
Kinetische Kontrolle:Selektivität = B/C = e (-∆∆G+/RT) R = Gaskonstante (1,99 cal/mol K)
T = Temperatur in Kelvin (Raumtemp. = 25°C, 298 K)
Ein ∆∆G° von 1,4 kcal/mol ergibt ein Verhältnis von 10:1 für B/C.Ein ∆∆G° von 2,8 kcal/mol ergibt ein Verhältnis von 100:1 für B/C.
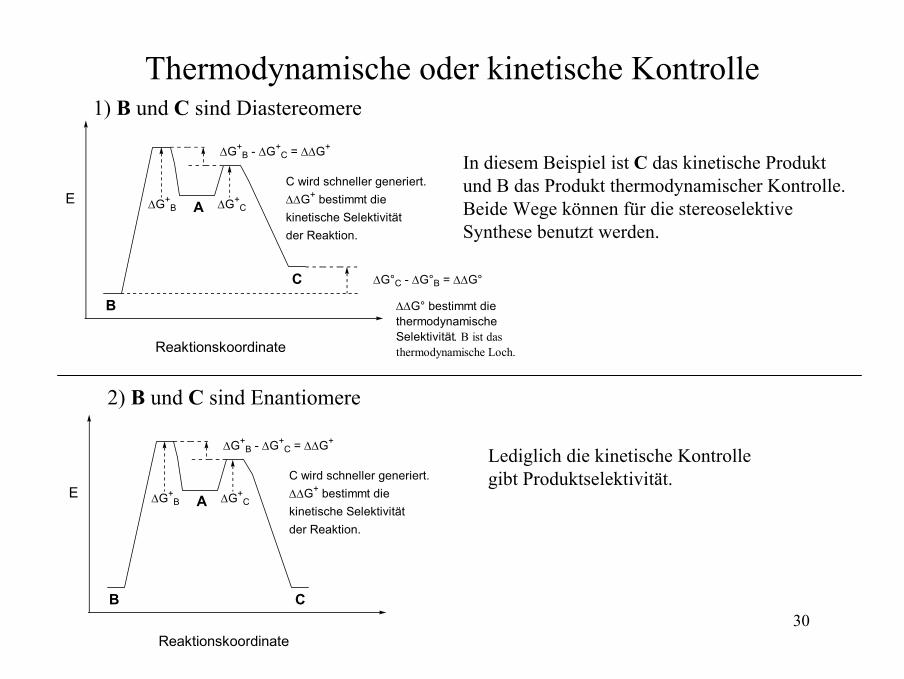
30
Thermodynamische oder kinetische Kontrolle 1) B und C sind Diastereomere
A
B
C
E
Reaktionskoordinate
∆G+B ∆G+
C
∆G+B - ∆G+
C = ∆∆G+
C wird schneller generiert.∆∆G+ bestimmt die kinetische Selektivität der Reaktion.
∆G°C - ∆G°B = ∆∆G°
∆∆G° bestimmt die thermodynamische Selektivität. Β ist dasthermodynamische Loch.
In diesem Beispiel ist C das kinetische Produkt und B das Produkt thermodynamischer Kontrolle.Beide Wege können für die stereoselektiveSynthese benutzt werden.
A
B C
E
Reaktionskoordinate
∆G+B ∆G+
C
∆G+B - ∆G+
C = ∆∆G+
C wird schneller generiert.∆∆G+ bestimmt die kinetische Selektivität der Reaktion.
2) B und C sind Enantiomere
Lediglich die kinetische Kontrollegibt Produktselektivität.

31
Thermodynamische Kontrolle oder kinetische Kontrolle
B oder CABetrachtet wird die Transformation von A zu B oder C.
Die Produktverteilung wird von einem der folgenden zwei Faktoren beeinflusst: 1. Thermodynamische Kontrolle: Die Reaktionszusammensetzung und Produktverteilung wird durchdie relative Energie der Produkte bestimmt.Um eine Reaktion unter thermodynamischer Kontrolle durchzuführen, müssen entwedera) die Produkte B und C reversibel A wiederherstellen können, oder b) B und C müssen in einem direkten Gleichgewicht stehen, welches nicht notwendiger Weise über A gehen muss.
2. Kinetische Kontrolle: Die Reaktions- und Produktverteilung wird von der Geschwindigkeit bestimmt, mit der B und C gebildet werden. Das Produkt, welches schneller gebildet wird, entstehtim Überschuß.
Merke:1) Diastereomere haben unterschiedliche Bildungsenthalpien. Wenn B und C Diastereomere sind, dann kann sowohl die kinetische Kontrolle als auch die thermodynamische Kontrolle zu Stereoselektivitäten führen.2) Enantiomere haben identische Bildungsenthalpien: Wenn B und C Enantiomere sind, dann kann lediglich die kinetische Kontrolle zur stereoselektiven Bildung eines Enantiomeren führen.
32
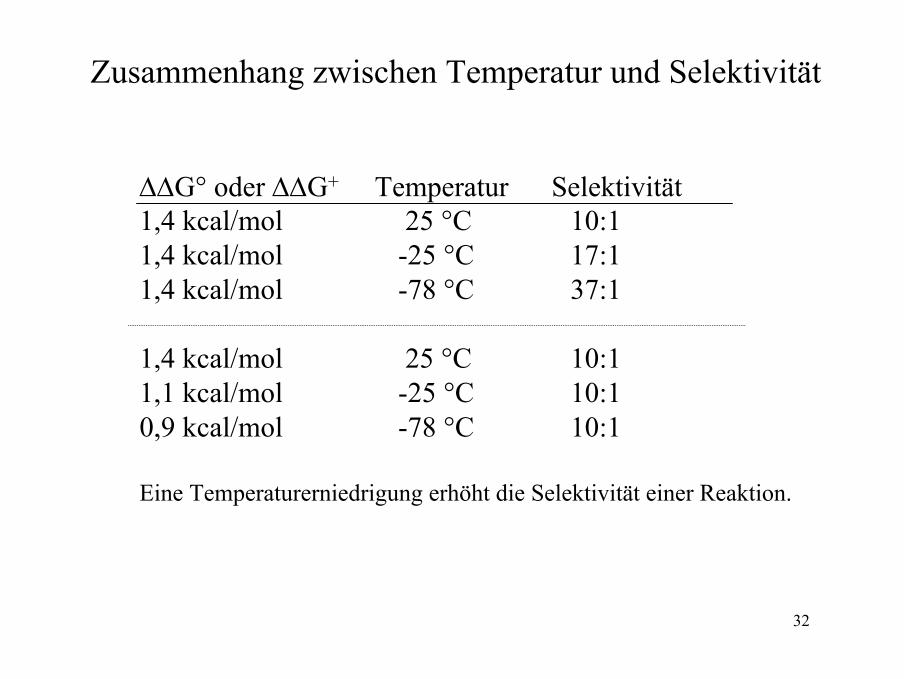
Zusammenhang zwischen Temperatur und Selektivität
∆∆G° oder ∆∆G+ Temperatur Selektivität1,4 kcal/mol 25 °C 10:11,4 kcal/mol -25 °C 17:11,4 kcal/mol -78 °C 37:1
1,4 kcal/mol 25 °C 10:11,1 kcal/mol -25 °C 10:10,9 kcal/mol -78 °C 10:1
Eine Temperaturerniedrigung erhöht die Selektivität einer Reaktion.

33
Es gibt verschiedene Formen, eine Reaktion zu beschreiben
B oder CABetrachtet wird die Transformation von A zu B oder C.
Wir nehmen an, dass B und C Diastereomere sind und in einem Verhältnis von 3: 1 für B:Centstehen.Die Selektivität kann dann wie folgt wiedergegeben werden.
a) Diastereomeren Verhältnis oder kurz er (engl. diastereomer ratio).Selektivität = B/C = 3:1 oder auch 75 : 25
b) Diastereoselektivität oder kurz ds (engl. diastereoselectivity)Hier würde man von 75% Diastereoselektivität sprechen.
c) % Diastereomerenüberschuss oder kurz %de (engl. diastereomeric excess)Hier wären es 50%de%de = %Hauptprodukt - %Unterschussisomer = 75% - 25% =50%
Für Enantiomere gelten die analogen Ausdrücke: Enantiomerenverhältnis, kurz er (engl.Enantiomeric ratio; Enantioselektivität, kurz es (engl. Enantioselectivity);Enantiomerenüberschuss oder %ee (engl. enantiomeric excess).
34
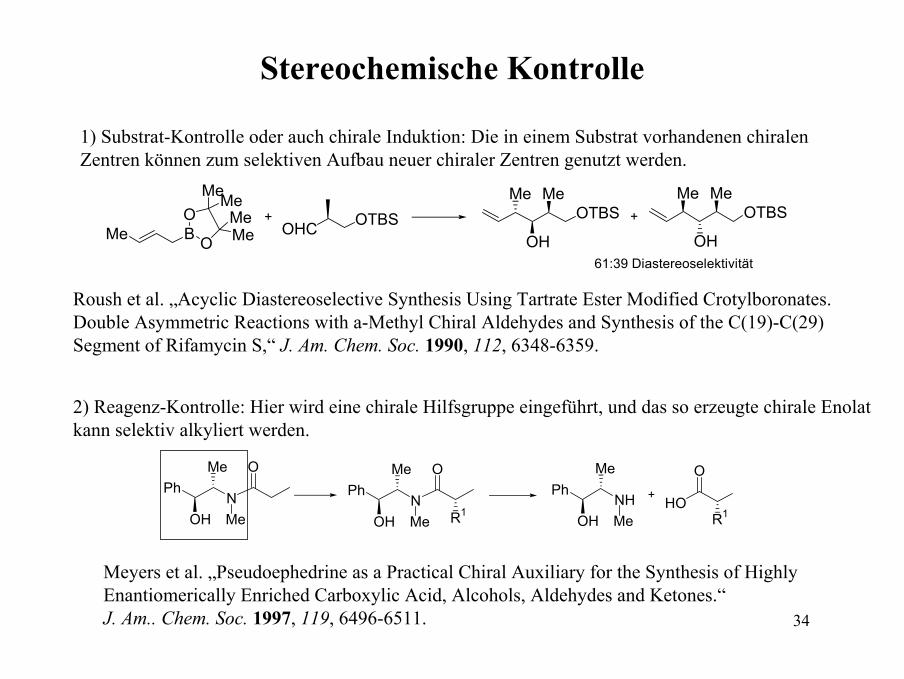
Stereochemische Kontrolle
1) Substrat-Kontrolle oder auch chirale Induktion: Die in einem Substrat vorhandenen chiralenZentren können zum selektiven Aufbau neuer chiraler Zentren genutzt werden.
Me B O
OMeMe
MeMe
OHC OTBS+ OTBSMe
OH
MeOTBS
Me
OH
Me+
61:39 Diastereoselektivität
Roush et al. „Acyclic Diastereoselective Synthesis Using Tartrate Ester Modified Crotylboronates.Double Asymmetric Reactions with a-Methyl Chiral Aldehydes and Synthesis of the C(19)-C(29) Segment of Rifamycin S,“ J. Am. Chem. Soc. 1990, 112, 6348-6359.
2) Reagenz-Kontrolle: Hier wird eine chirale Hilfsgruppe eingeführt, und das so erzeugte chirale Enolatkann selektiv alkyliert werden.
PhN
OH
Me O
Me
PhN
OH
Me O
Me R1
PhNH
OH
Me O
Me R1HO+
Meyers et al. „Pseudoephedrine as a Practical Chiral Auxiliary for the Synthesis of HighlyEnantiomerically Enriched Carboxylic Acid, Alcohols, Aldehydes and Ketones.“ J. Am.. Chem. Soc. 1997, 119, 6496-6511.
35
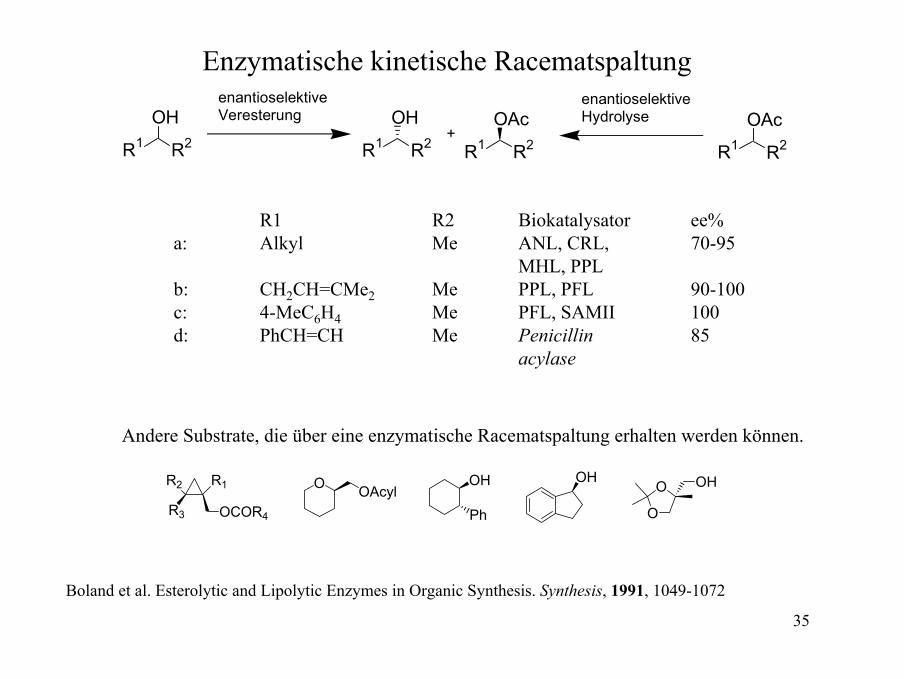
Enzymatische kinetische Racematspaltung
R1 R2
OHenantioselektive Veresterung
R1 R2
OH
R1 R2
OAc+
R1 R2
OAcenantioselektive Hydrolyse
R1 R2 Biokatalysator ee%a: Alkyl Me ANL, CRL, 70-95
MHL, PPLb: CH2CH=CMe2 Me PPL, PFL 90-100c: 4-MeC6H4 Me PFL, SAMII 100d: PhCH=CH Me Penicillin 85
acylase
Andere Substrate, die über eine enzymatische Racematspaltung erhalten werden können.
OCOR4R3
R2 R1 O OAcylOH
Ph
OH
O
O OH
Boland et al. Esterolytic and Lipolytic Enzymes in Organic Synthesis. Synthesis, 1991, 1049-1072

36
Kinetische Racematspaltung: Chemische Methoden
• Zwei Enantiomere, hier R+S genannt, werden mit einem chiralen Reagenz umgesetzt.
R + S
Chirales Reagenz R-D +(S-D)
S (+R)Trennung R-D
derivatisiertesProdukt
Sisolierte Ausgangsverbindung
+
Dabei werden die beiden Enantiomere mit unterschiedlichen Geschwindigkeiten umgesetzt.
R R-D S S-DkR
Reagenz
kS
Reagenz
Das Verhältnis der beiden Geschwindigkeitskonstanten wird S genannt: S = kR/kS
ln[(1-C)(1-ee)]
ln[(1-C)(1-ee)]S =
C = Konversionee = Enantiomeren-Überschußdes reisolierten Eduktes
Verhältnis zwischen S und C (%) für eine kinetische Razemattrennung mit 99% ee.S C S C1.5 99.999 5.0 86.6 1.9 99.86 7.0 79.22.4 98.9 10.0 72.13.0 96.44.0 91.3
37
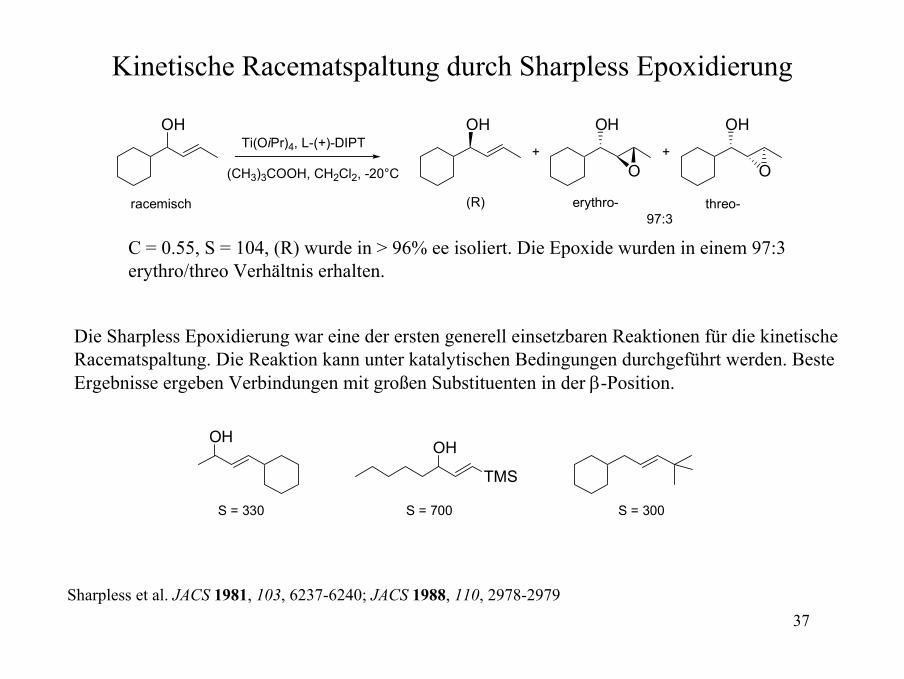
Kinetische Racematspaltung durch Sharpless Epoxidierung
OH OH
(R)
OH
O
OH
O
Ti(OiPr)4, L-(+)-DIPT
(CH3)3COOH, CH2Cl2, -20°C+
racemisch
+
erythro- threo-97:3
C = 0.55, S = 104, (R) wurde in > 96% ee isoliert. Die Epoxide wurden in einem 97:3 erythro/threo Verhältnis erhalten.
Die Sharpless Epoxidierung war eine der ersten generell einsetzbaren Reaktionen für die kinetische Racematspaltung. Die Reaktion kann unter katalytischen Bedingungen durchgeführt werden. Beste Ergebnisse ergeben Verbindungen mit großen Substituenten in der β-Position.
OHOH
TMS
S = 330 S = 700 S = 300
Sharpless et al. JACS 1981, 103, 6237-6240; JACS 1988, 110, 2978-2979
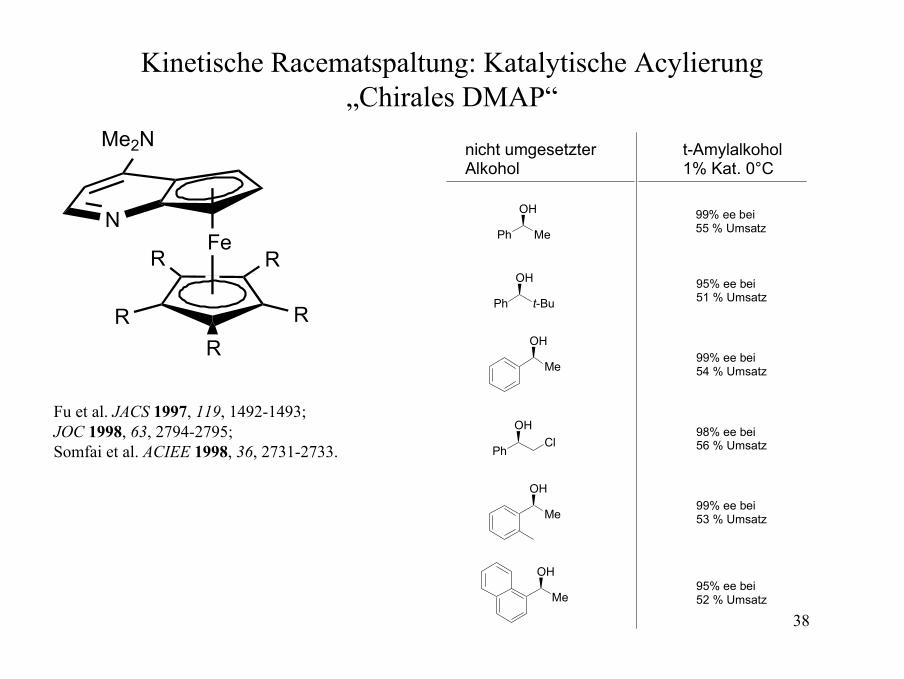
38
Kinetische Racematspaltung: Katalytische Acylierung„Chirales DMAP“
N
Me2N
RRR
R RFe Ph Me
OH
Ph t-Bu
OH
Me
OH
Ph
OHCl
Me
OH
Me
OH
nicht umgesetzter Alkohol
t-Amylalkohol1% Kat. 0°C
99% ee bei55 % Umsatz
95% ee bei51 % Umsatz
99% ee bei54 % Umsatz
98% ee bei56 % Umsatz
99% ee bei53 % Umsatz
95% ee bei52 % Umsatz
Fu et al. JACS 1997, 119, 1492-1493;JOC 1998, 63, 2794-2795;Somfai et al. ACIEE 1998, 36, 2731-2733.
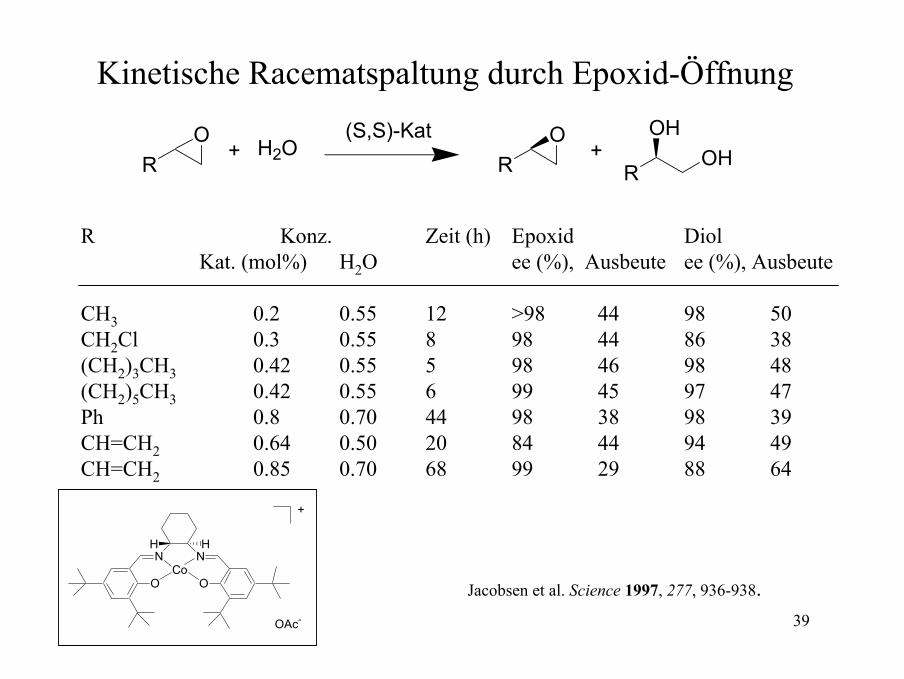
39
Kinetische Racematspaltung durch Epoxid-Öffnung
RO
+ H2O(S,S)-Kat
RO
+R
OHOH
NN
O O
HH
Co
OAc-
+
R Konz. Zeit (h) Epoxid DiolKat. (mol%) H2O ee (%), Ausbeute ee (%), Ausbeute
CH3 0.2 0.55 12 >98 44 98 50CH2Cl 0.3 0.55 8 98 44 86 38(CH2)3CH3 0.42 0.55 5 98 46 98 48(CH2)5CH3 0.42 0.55 6 99 45 97 47Ph 0.8 0.70 44 98 38 98 39CH=CH2 0.64 0.50 20 84 44 94 49CH=CH2 0.85 0.70 68 99 29 88 64
Jacobsen et al. Science 1997, 277, 936-938.
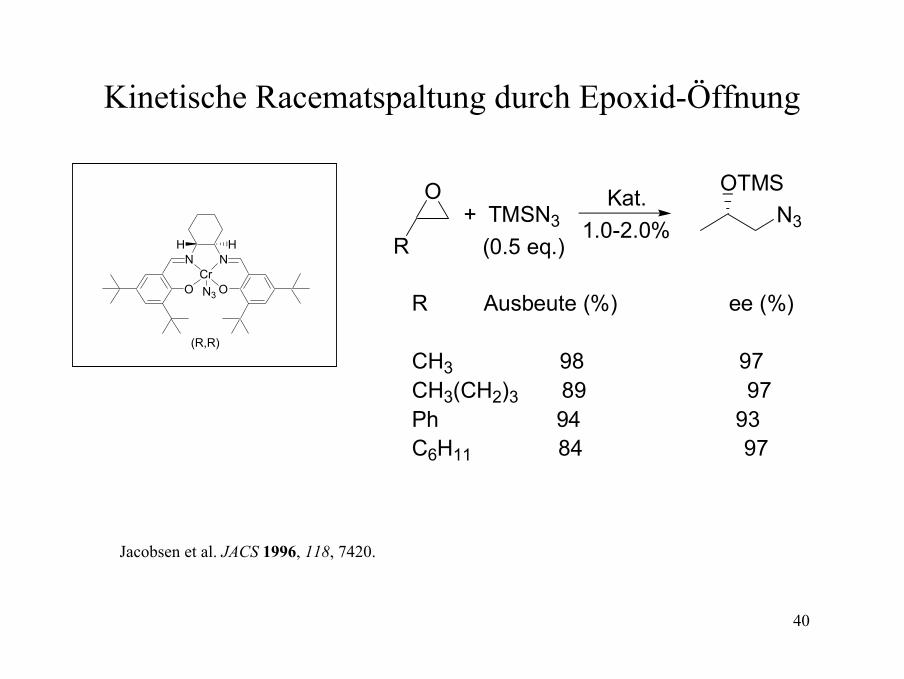
40
Kinetische Racematspaltung durch Epoxid-Öffnung
N N
O O
H H
CrN3
(R,R)
O
RTMSN3 N3
OTMS+
(0.5 eq.)
Kat.1.0-2.0%
R Ausbeute (%) ee (%)
CH3 98 97CH3(CH2)3 89 97Ph 94 93C6H11 84 97
Jacobsen et al. JACS 1996, 118, 7420.
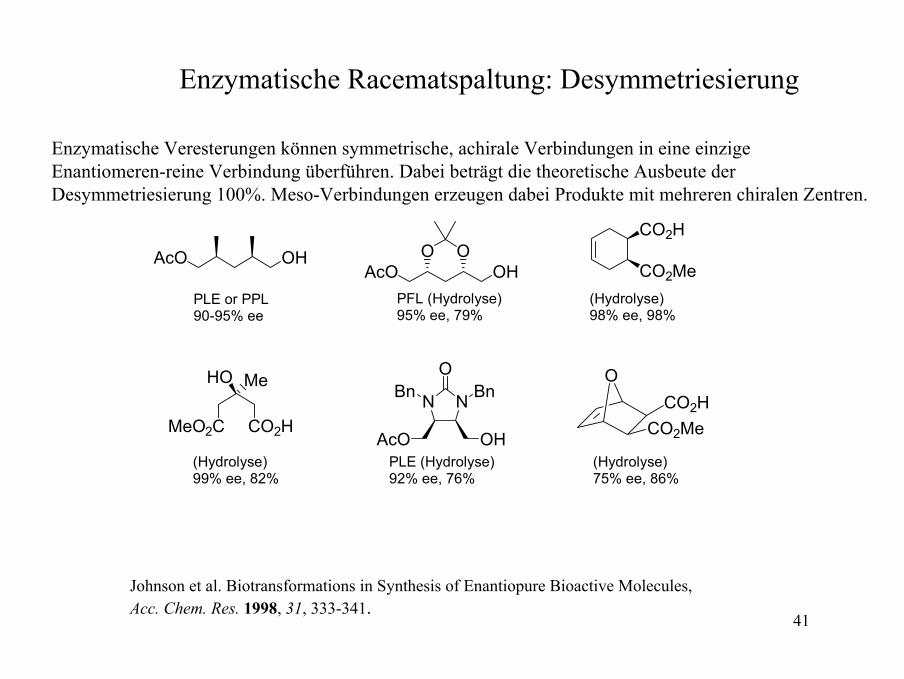
41
Enzymatische Racematspaltung: Desymmetriesierung
Enzymatische Veresterungen können symmetrische, achirale Verbindungen in eine einzige Enantiomeren-reine Verbindung überführen. Dabei beträgt die theoretische Ausbeute der Desymmetriesierung 100%. Meso-Verbindungen erzeugen dabei Produkte mit mehreren chiralen Zentren.
AcO OHAcO OH
O OCO2H
CO2Me
MeO2C CO2H
HO MeNN
OBnBn
OHAcO
OCO2H
CO2Me
PLE or PPL90-95% ee
PFL (Hydrolyse)95% ee, 79%
(Hydrolyse)99% ee, 82%
(Hydrolyse)98% ee, 98%
PLE (Hydrolyse)92% ee, 76%
(Hydrolyse)75% ee, 86%
Johnson et al. Biotransformations in Synthesis of Enantiopure Bioactive Molecules,Acc. Chem. Res. 1998, 31, 333-341.
42
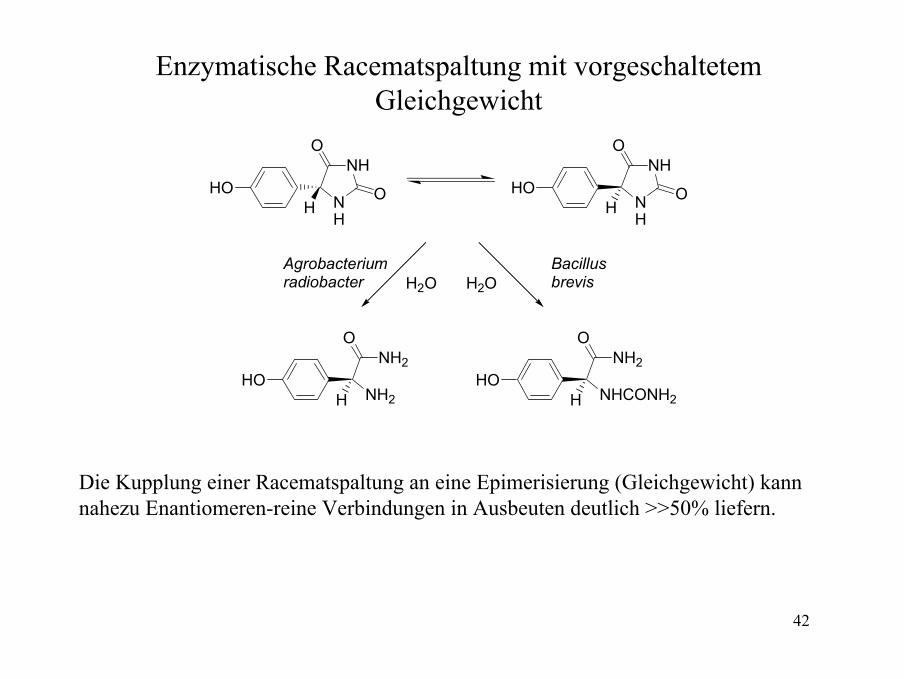
Enzymatische Racematspaltung mit vorgeschaltetemGleichgewicht
NH
NH
O
OH
HO
H2OH2O
NH
NH
O
OH
HO
NH2
NHCONH2
O
HHO
NH2
NH2
O
HHO
Bacillusbrevis
Agrobacteriumradiobacter
Die Kupplung einer Racematspaltung an eine Epimerisierung (Gleichgewicht) kannnahezu Enantiomeren-reine Verbindungen in Ausbeuten deutlich >>50% liefern.
43
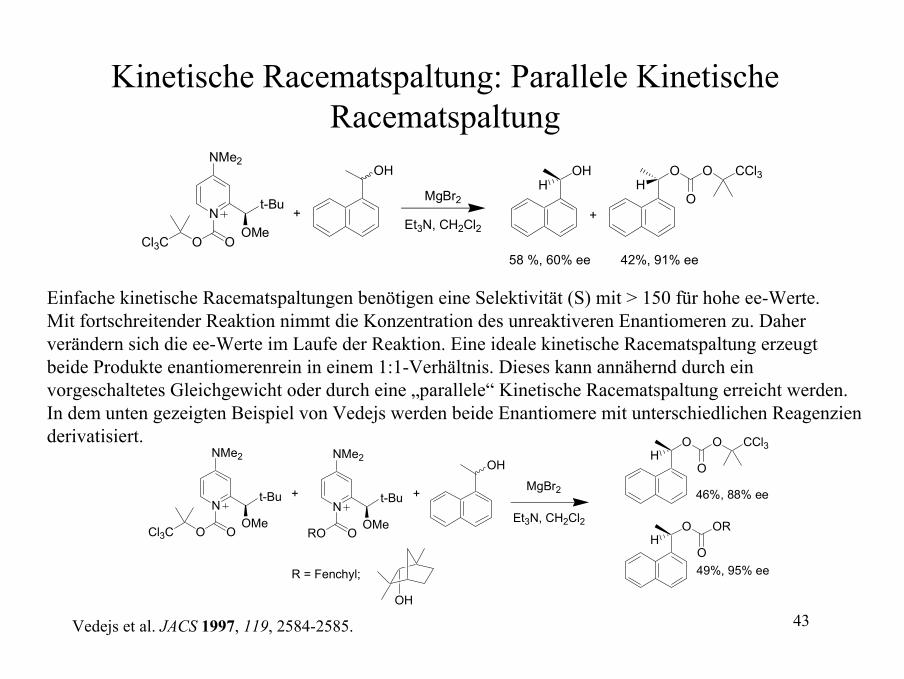
Kinetische Racematspaltung: Parallele Kinetische Racematspaltung
N
NMe2
OMe
t-Bu
OOCl3C
OH
MgBr2
OHH
OH
O CCl3
O
Et3N, CH2Cl2+
58 %, 60% ee 42%, 91% ee
+
Einfache kinetische Racematspaltungen benötigen eine Selektivität (S) mit > 150 für hohe ee-Werte. Mit fortschreitender Reaktion nimmt die Konzentration des unreaktiveren Enantiomeren zu. Daher verändern sich die ee-Werte im Laufe der Reaktion. Eine ideale kinetische Racematspaltung erzeugt beide Produkte enantiomerenrein in einem 1:1-Verhältnis. Dieses kann annähernd durch ein vorgeschaltetes Gleichgewicht oder durch eine „parallele“ Kinetische Racematspaltung erreicht werden. In dem unten gezeigten Beispiel von Vedejs werden beide Enantiomere mit unterschiedlichen Reagenzien derivatisiert.
N
NMe2
OMe
t-Bu
OOCl3C
N
NMe2
OMe
t-Bu
ORO
OH
MgBr2
OH
OH
OR
O
OH
O CCl3
O
+
R = Fenchyl;
+
Et3N, CH2Cl2
46%, 88% ee
49%, 95% ee
Vedejs et al. JACS 1997, 119, 2584-2585.
44
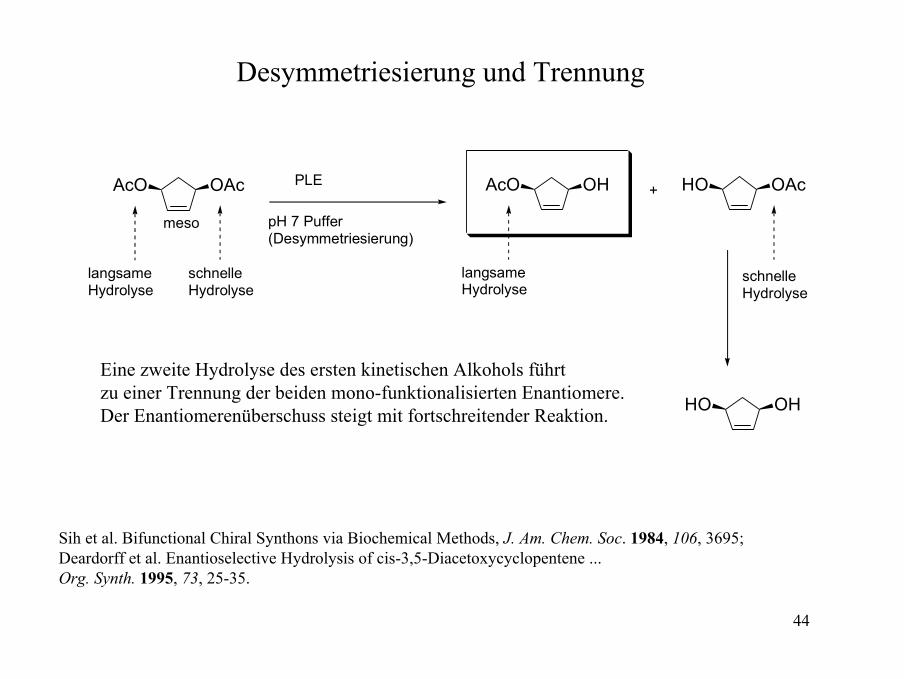
Desymmetriesierung und Trennung
OAcAcO
meso
langsame Hydrolyse
schnelle Hydrolyse
PLE
pH 7 Puffer(Desymmetriesierung)
OHAcO
langsame Hydrolyse
+ OAcHO
schnelle Hydrolyse
OHHO
Eine zweite Hydrolyse des ersten kinetischen Alkohols führtzu einer Trennung der beiden mono-funktionalisierten Enantiomere.Der Enantiomerenüberschuss steigt mit fortschreitender Reaktion.
Sih et al. Bifunctional Chiral Synthons via Biochemical Methods, J. Am. Chem. Soc. 1984, 106, 3695; Deardorff et al. Enantioselective Hydrolysis of cis-3,5-Diacetoxycyclopentene ... Org. Synth. 1995, 73, 25-35.
45
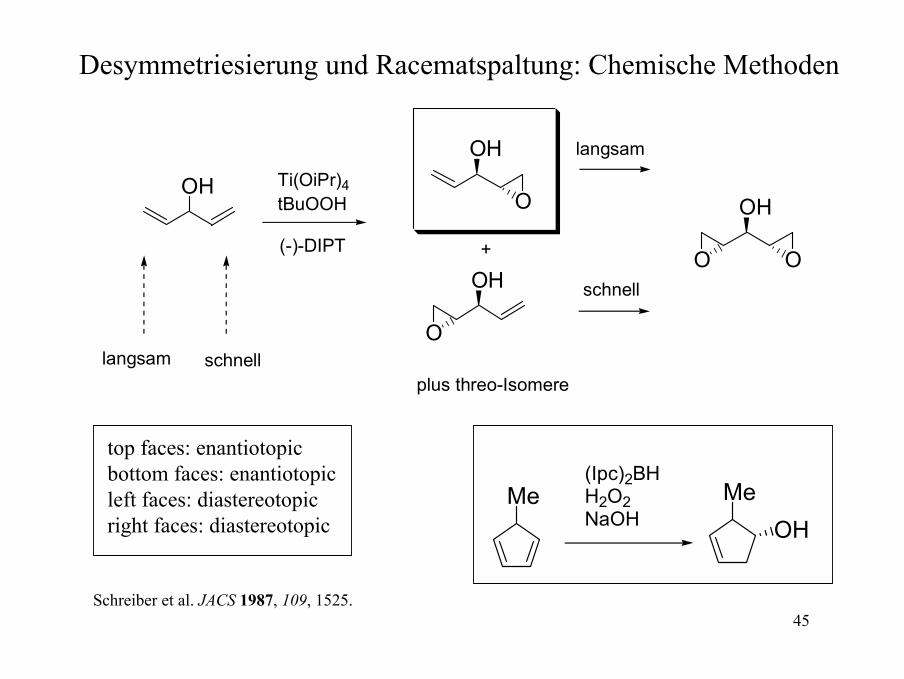
OH
schnelllangsam
Ti(OiPr)4tBuOOH
(-)-DIPT
OH
OH
O
O
plus threo-Isomere
+
OH
O O
langsam
schnell
Desymmetriesierung und Racematspaltung: Chemische Methoden
top faces: enantiotopicbottom faces: enantiotopicleft faces: diastereotopicright faces: diastereotopic
Me(Ipc)2BHH2O2NaOH
MeOH
Schreiber et al. JACS 1987, 109, 1525.
46
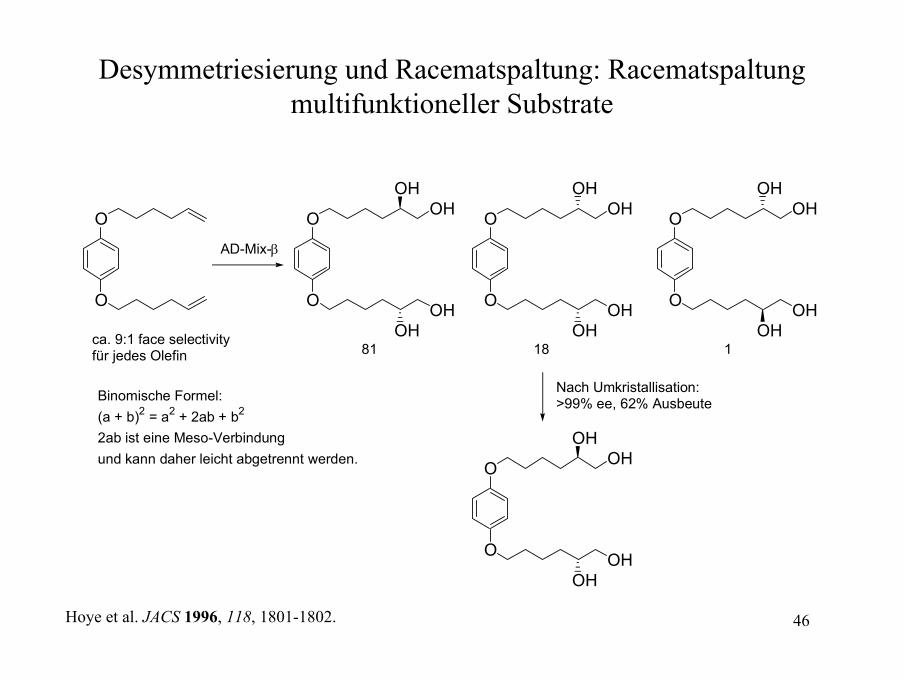
Desymmetriesierung und Racematspaltung: Racematspaltungmultifunktioneller Substrate
O
O
O
O
OH
OH
OH
OH
O
O
OH
OH
OH
OH
O
O
OH
OH
OH
OH
AD-Mix-β
ca. 9:1 face selectivityfür jedes Olefin 81 18 1
Binomische Formel:(a + b)2 = a2 + 2ab + b2
2ab ist eine Meso-Verbindungund kann daher leicht abgetrennt werden. O
O
OH
OH
OH
OH
Nach Umkristallisation: >99% ee, 62% Ausbeute
Hoye et al. JACS 1996, 118, 1801-1802.
47
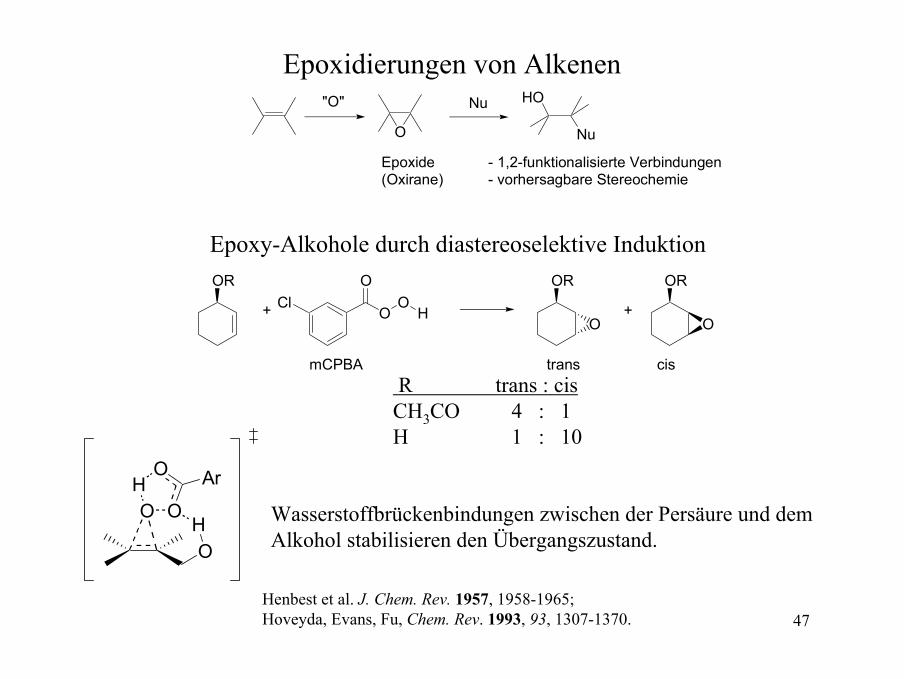
Epoxidierungen von Alkenen"O"
O
Nu HO
Nu
Epoxide(Oxirane)
- 1,2-funktionalisierte Verbindungen- vorhersagbare Stereochemie
ORCl
O
OO
H
OR
O
OR
O+
mCPBA trans cis
+
Epoxy-Alkohole durch diastereoselektive Induktion
R trans : cis CH3CO 4 : 1H 1 : 10
OH
OO
OH Ar
Wasserstoffbrückenbindungen zwischen der Persäure und dem Alkohol stabilisieren den Übergangszustand.
Henbest et al. J. Chem. Rev. 1957, 1958-1965; Hoveyda, Evans, Fu, Chem. Rev. 1993, 93, 1307-1370.
48
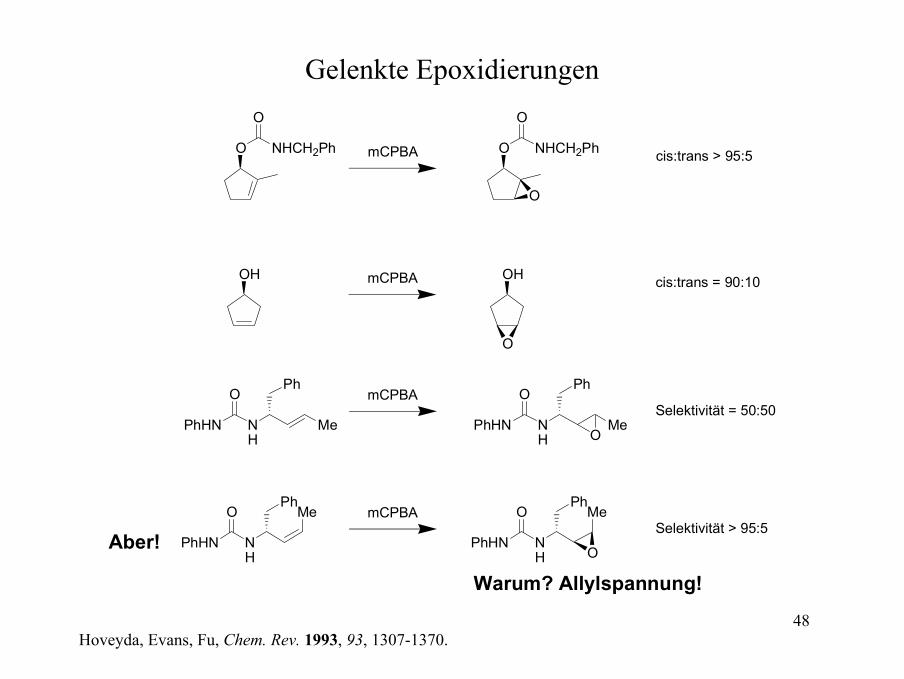
Gelenkte Epoxidierungen
OH
PhHN NH
OPh
Me
PhHN NH
Me
OPh
O NHCH2Ph
O
OH
O
PhHN NH
OPh
O
Me
O NHCH2Ph
O
O
PhHN NH
Me
OPh
O
mCPBA cis:trans > 95:5
mCPBA cis:trans = 90:10
mCPBASelektivität = 50:50
mCPBASelektivität > 95:5
Aber!
Warum? Allylspannung!
Hoveyda, Evans, Fu, Chem. Rev. 1993, 93, 1307-1370.

49
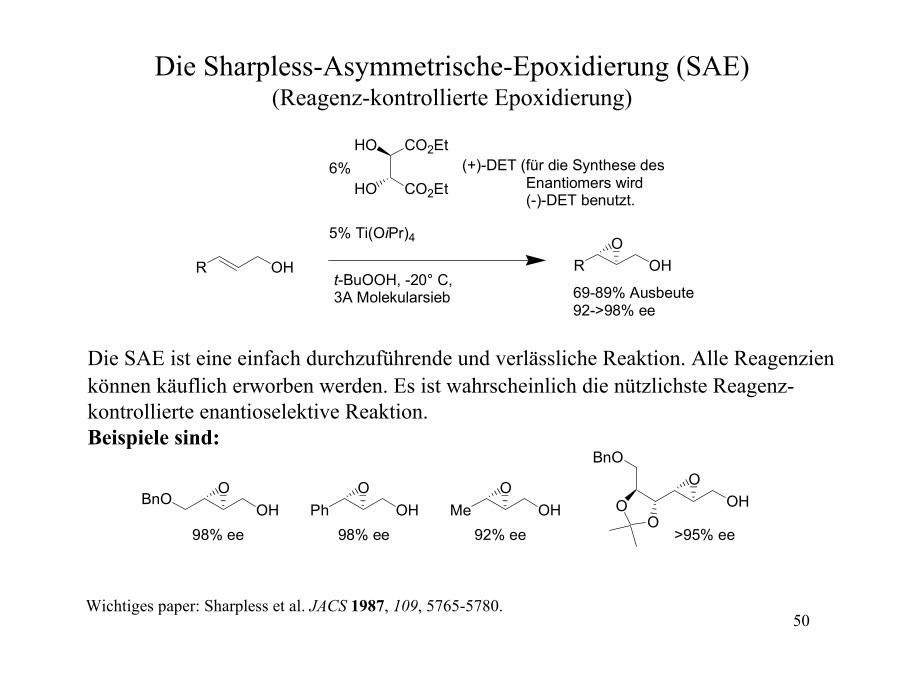
Gelenkte Epoxidierungen mit VO(acac)2
OH
OH
t-BuOOH
t-BuOOH
OH
O
OH
O
VO(acac)298:2 Diastereoselektivität
VO(acac)2
>99:1 Diastereoselektivität
VO(acac)2 katalysiert die Epoxidierung von Alkenen mit t-BuOOH.
Me
OH
Me
OH
Me
OH
t-BuOOH
t-BuOOH
t-BuOOH
Me
OH
O
Me
OH
O
Me
OH
OVO(acac)24:1 Diastereoselektivität
VO(acac)22.5:1 Diastereoselektivität
VO(acac)22.5:1 Diastereoselektivität
Acyclische Stereokontrolle ist sehr oft wenig selektiv
Hoveyda, Evans, Fu, Chem. Rev. 1993, 93, 1307-1370.
50
Die Sharpless-Asymmetrische-Epoxidierung (SAE)(Reagenz-kontrollierte Epoxidierung)
R OH
CO2Et
CO2EtHO
HO
t-BuOOH, -20° C, 3A Molekularsieb
6% (+)-DET (für die Synthese des Enantiomers wird (-)-DET benutzt.
5% Ti(OiPr)4
R OHO
69-89% Ausbeute92->98% ee
Die SAE ist eine einfach durchzuführende und verlässliche Reaktion. Alle Reagenzien können käuflich erworben werden. Es ist wahrscheinlich die nützlichste Reagenz-kontrollierte enantioselektive Reaktion.Beispiele sind:
OHBnO
OPh OH
OMe OH
OOH
OBnO
OO
98% ee 98% ee 92% ee >95% ee
Wichtiges paper: Sharpless et al. JACS 1987, 109, 5765-5780.
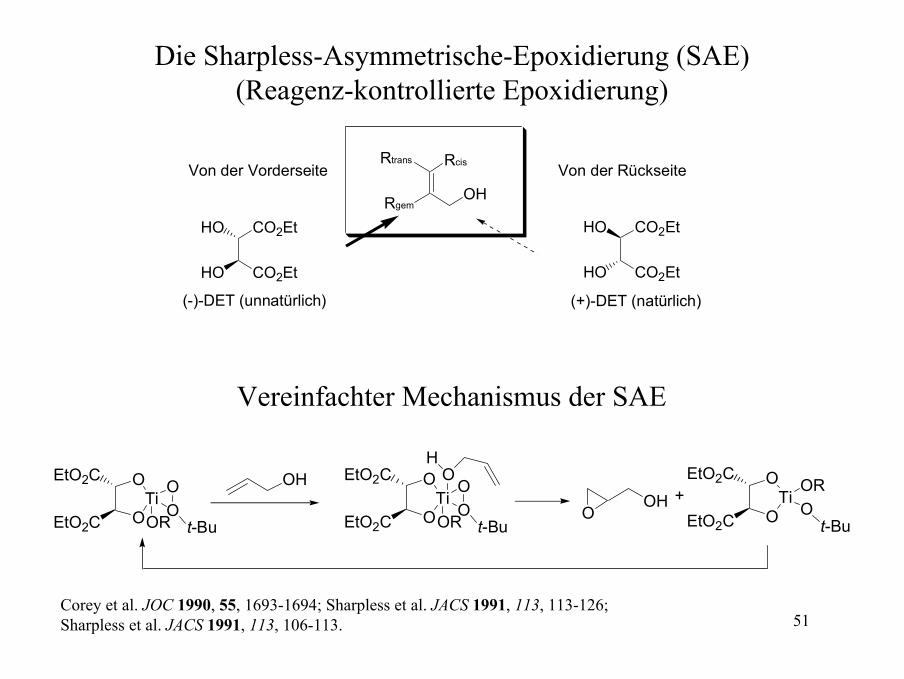
51
Die Sharpless-Asymmetrische-Epoxidierung (SAE)(Reagenz-kontrollierte Epoxidierung)
CO2Et
CO2EtHO
HO
OH
RcisRtrans
Rgem
CO2Et
CO2EtHO
HO
Von der Rückseite
(+)-DET (natürlich)
Von der Vorderseite
(-)-DET (unnatürlich)
Vereinfachter Mechanismus der SAE
OTi
O
OO
EtO2C
EtO2C OR t-Bu
OH
OTi
O
OO
EtO2C
EtO2C OR t-Bu
OH
OHO O
TiO
OOR
EtO2C
EtO2C t-Bu
+
Corey et al. JOC 1990, 55, 1693-1694; Sharpless et al. JACS 1991, 113, 113-126; Sharpless et al. JACS 1991, 113, 106-113.
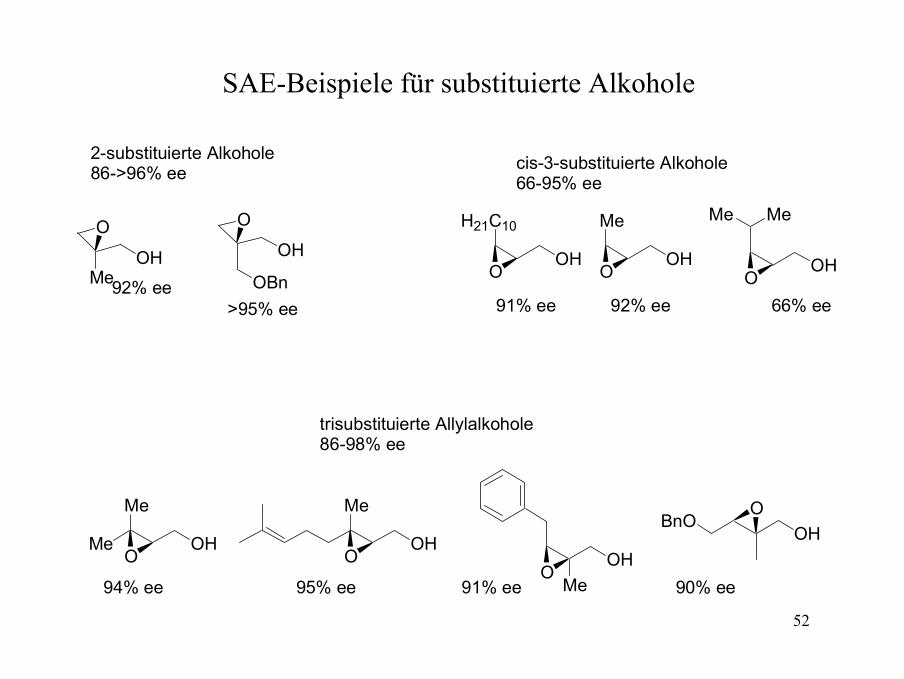
52
SAE-Beispiele für substituierte Alkohole
O
MeOH
O
OH
OBn
Me
OHO OH
O
Me MeH21C10
OHO
Me OHO
Me
OHO
Me
OHO
Me
BnOOH
O
2-substituierte Alkohole86->96% ee
92% ee>95% ee 92% ee
cis-3-substituierte Alkohole66-95% ee
91% ee 66% ee
trisubstituierte Allylalkohole86-98% ee
94% ee 95% ee 91% ee 90% ee
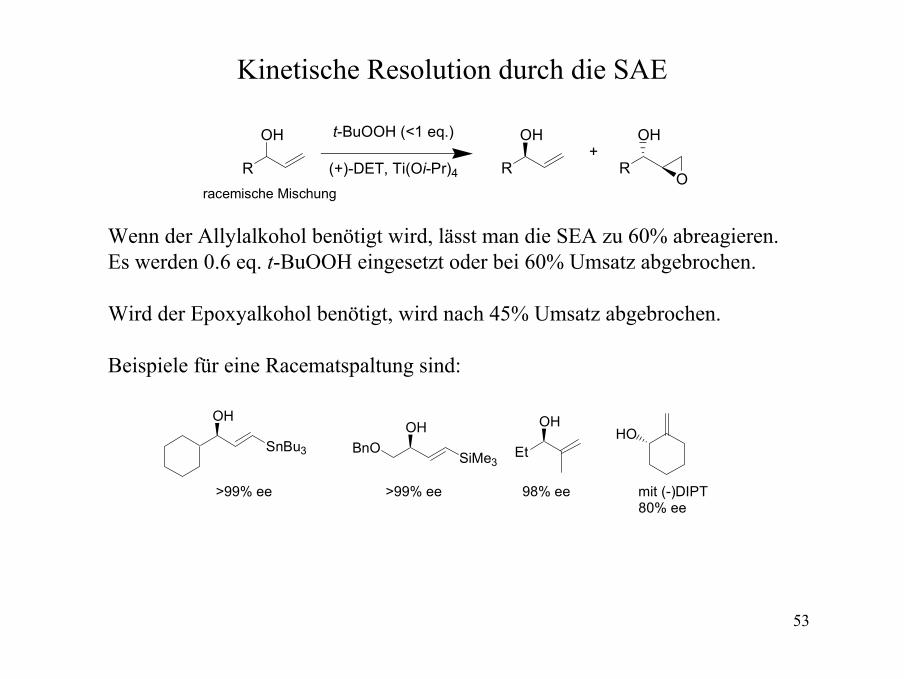
53
Kinetische Resolution durch die SAE
R
OH t-BuOOH (<1 eq.)
(+)-DET, Ti(Oi-Pr)4 R
OH
R
OH+
Oracemische Mischung
Wenn der Allylalkohol benötigt wird, lässt man die SEA zu 60% abreagieren.Es werden 0.6 eq. t-BuOOH eingesetzt oder bei 60% Umsatz abgebrochen.
Wird der Epoxyalkohol benötigt, wird nach 45% Umsatz abgebrochen.
Beispiele für eine Racematspaltung sind:
SnBu3
OH
BnOSiMe3
OH
Et
OHHO
>99% ee >99% ee 98% ee mit (-)DIPT80% ee
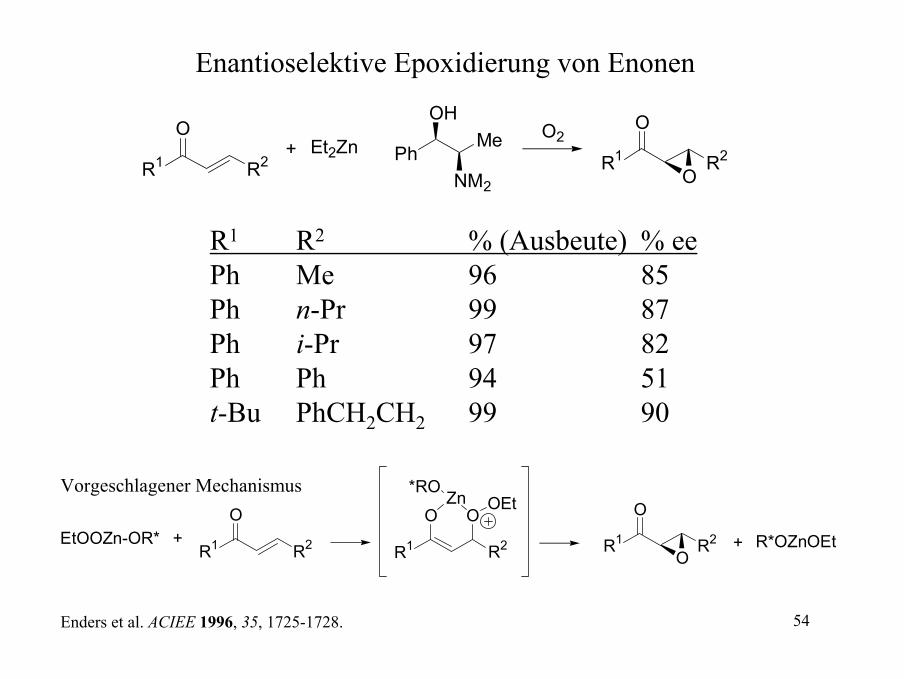
54
Enantioselektive Epoxidierung von Enonen
R1 R2
OEt2Zn Ph
MeOH
NM2
O2
R1 R2
O
O+
R1 R2 % (Ausbeute) % eePh Me 96 85Ph n-Pr 99 87Ph i-Pr 97 82Ph Ph 94 51t-Bu PhCH2CH2 99 90
R1 R2
O O OZn
R1 R2
OEt*RO
R1 R2
O
OEtOOZn-OR* + + R*OZnOEt
Vorgeschlagener Mechanismus
Enders et al. ACIEE 1996, 35, 1725-1728.
55
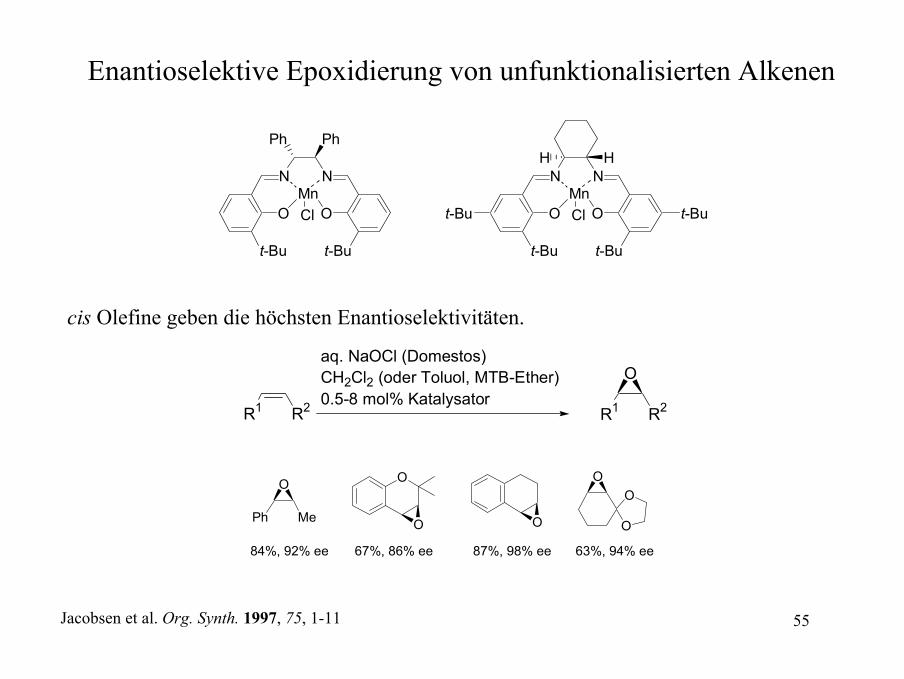
Enantioselektive Epoxidierung von unfunktionalisierten Alkenen
N N
OO
PhPh
t-But-Bu
MnCl
N N
OO
t-But-Bu
MnClt-Bu t-Bu
H H
cis Olefine geben die höchsten Enantioselektivitäten.
R1 R2
aq. NaOCl (Domestos)CH2Cl2 (oder Toluol, MTB-Ether)0.5-8 mol% Katalysator
R1 R2
O
Ph Me
O
O
O
O O
OO
84%, 92% ee 67%, 86% ee 87%, 98% ee 63%, 94% ee
Jacobsen et al. Org. Synth. 1997, 75, 1-11
56
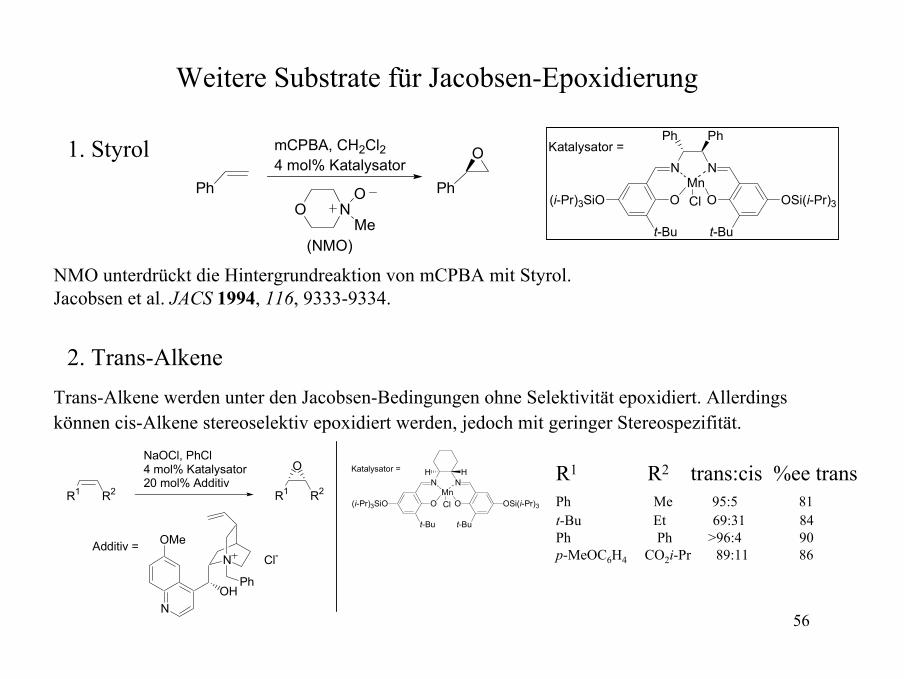
Weitere Substrate für Jacobsen-Epoxidierung
1. StyrolPh
NOO
Me(NMO)
Ph
OmCPBA, CH2Cl24 mol% Katalysator
NMO unterdrückt die Hintergrundreaktion von mCPBA mit Styrol.Jacobsen et al. JACS 1994, 116, 9333-9334.
N N
OO
PhPh
t-But-Bu
MnCl(i-Pr)3SiO OSi(i-Pr)3
Katalysator =
2. Trans-AlkeneTrans-Alkene werden unter den Jacobsen-Bedingungen ohne Selektivität epoxidiert. Allerdings können cis-Alkene stereoselektiv epoxidiert werden, jedoch mit geringer Stereospezifität.
R2R1
N
N
OMe
OHPh
Cl-
R2R1
ONaOCl, PhCl4 mol% Katalysator20 mol% Additiv
Additiv =
N N
OO
t-But-Bu
MnCl(i-Pr)3SiO OSi(i-Pr)3
Katalysator = H H R1 R2 trans:cis %ee transPh Me 95:5 81t-Bu Et 69:31 84Ph Ph >96:4 90p-MeOC6H4 CO2i-Pr 89:11 86

57
Enantioselektive Epoxidierung nicht-funktionalisierter Alkene
PhPh
Me
PhMe
Ph Ph
69%, 93% ee 87%, 88% ee 91%, 95% ee 61%, 86% ee
Jacobsen et al. JOC 1994, 59, 4378-4380.
3. Trisubstituierte Alkene mit ungesättigten Substituenten.
4. Tetrasubstituierte Alkene mit ungesättigten Substituenten werden mit ee-WertenZwischen 35-97% ee epoxidiert. Jacobsen et al. THL 1995, 36, 5123-5126.
Mn
O
R R
H H
Mn
O
R R
H
H
Mn
O
H R
R
H
ORR
H H
ORH
R H
cis trans
Mechanismus der Jacobsen Epoxidierung.T. Linker, ACIEE 1997, 36.

58
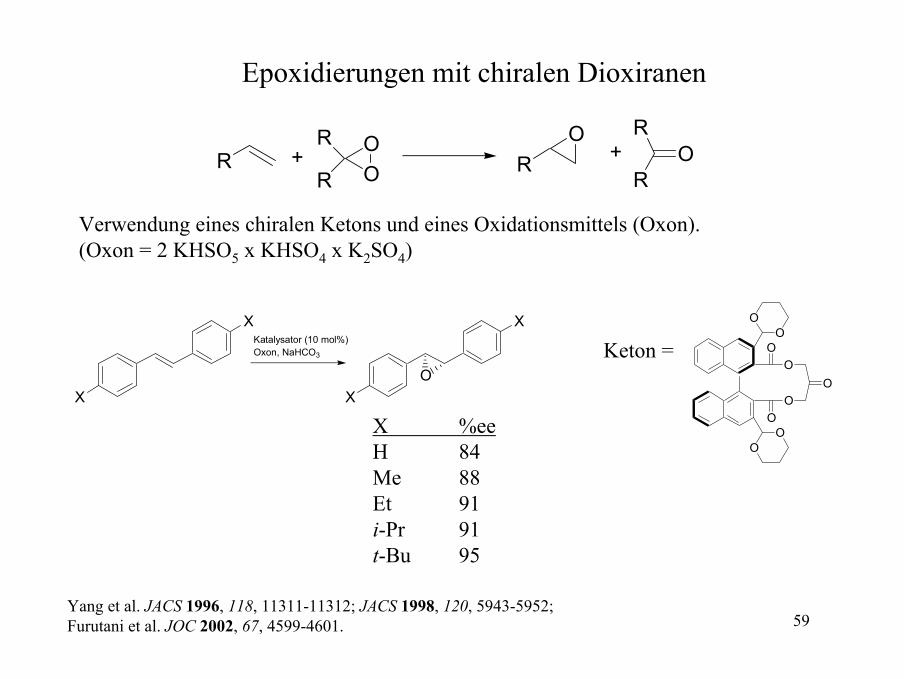
Chirale Dioxirane für die enantioselektive Epoxidierung
Katalysator (20-30 mol%), Oxon, K2CO3, +Na2B4O7xH2O + EDTA
O
OOO
OO
aus Fructose
Katalysator
Alken Ausbeute (%) ee(%)75 97
93 92
70 90
70 91
66 94
90 24
81 28
PhPh
Wang et al. JACS 1997, 119, 11224-11235.
PhMe
Ph OH
BuBu
PhC10H21
Ph
Ph
Ph
Me
59
Epoxidierungen mit chiralen Dioxiranen
R +OOR
RR
O+
R
RO
Verwendung eines chiralen Ketons und eines Oxidationsmittels (Oxon).(Oxon = 2 KHSO5 x KHSO4 x K2SO4)
X
XKatalysator (10 mol%)Oxon, NaHCO3
X
X
O
O
OO
OO
OO
OO
Keton =
X %eeH 84Me 88Et 91i-Pr 91t-Bu 95
Yang et al. JACS 1996, 118, 11311-11312; JACS 1998, 120, 5943-5952; Furutani et al. JOC 2002, 67, 4599-4601.

60
Enantioselektive Dihydroxylierung von Alkenen mit OsO4 – die Liganden
OsO4
H2O OHHO
NRO
N
MeO
H
NOR
N
OMe
H
Pseudo-Enantiomere
R = H, DHQD (Dihydrochinidin)(engl. dihydroquinidine)
R = H, DHQ (Dihydrochinin)(engl. dihydroquinine)
O
N
N
HOMe
O
N
N
HMeO
NNO
N
N
HMeO
O
N
N
HOMe
N N
(DHQD)2PHAL
Dihydrochinidin-Phthalazin
(DHQ)2PHAL
Dihydrochinin-Phthalazin
Kolb et al. Chem. Rev. 1994, 94, 2483-2547; Bolm et al. ACIIE 1997, 36, 741-743.
61
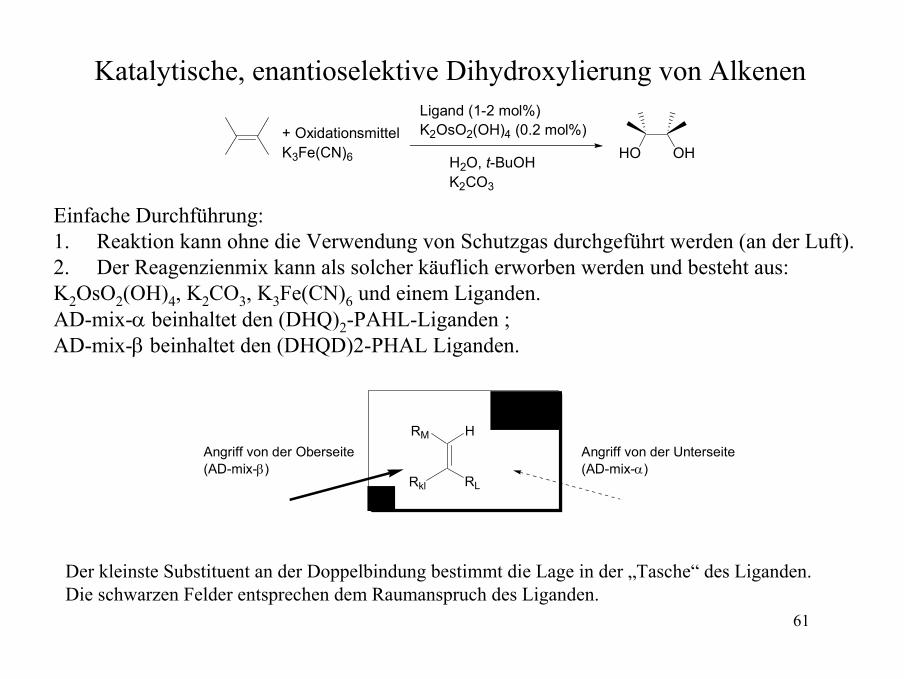
Katalytische, enantioselektive Dihydroxylierung von Alkenen
OHHO+ OxidationsmittelK3Fe(CN)6
Ligand (1-2 mol%)K2OsO2(OH)4 (0.2 mol%)
H2O, t-BuOHK2CO3
Einfache Durchführung:1. Reaktion kann ohne die Verwendung von Schutzgas durchgeführt werden (an der Luft).2. Der Reagenzienmix kann als solcher käuflich erworben werden und besteht aus: K2OsO2(OH)4, K2CO3, K3Fe(CN)6 und einem Liganden. AD-mix-α beinhaltet den (DHQ)2-PAHL-Liganden ; AD-mix-β beinhaltet den (DHQD)2-PHAL Liganden.
HRM
RLRkl
Angriff von der Unterseite(AD-mix-α)
Angriff von der Oberseite(AD-mix-β)
Der kleinste Substituent an der Doppelbindung bestimmt die Lage in der „Tasche“ des Liganden.Die schwarzen Felder entsprechen dem Raumanspruch des Liganden.

62
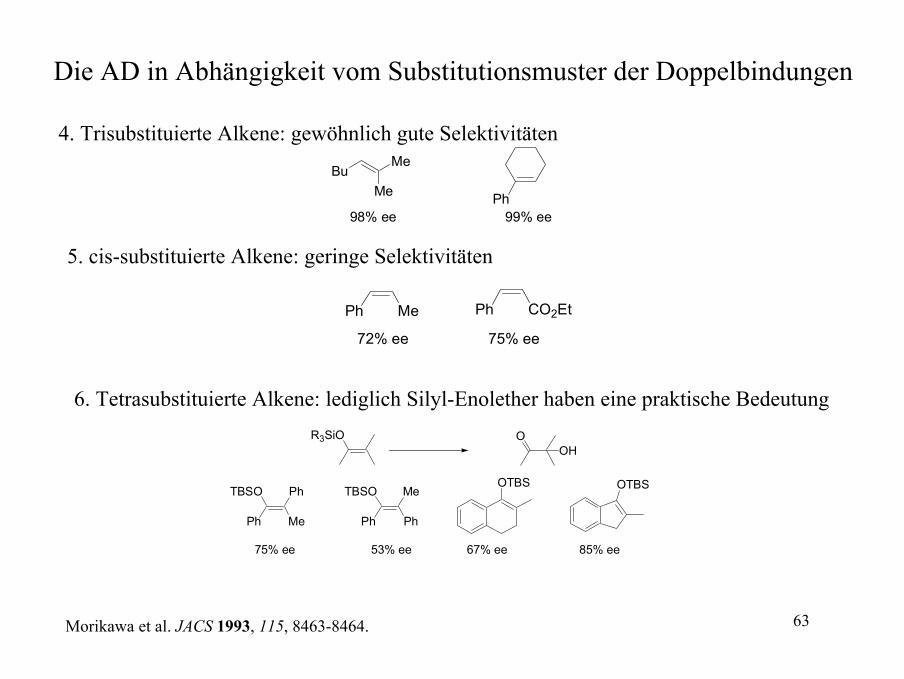
Die AD in Abhängigkeit vom Substitutionsmuster der Doppelbindungen
1. Monosubstituierte Alkene = gewöhnlich hohe Selektivitäten
C8H17 Ph PhOO
O
Ph
84% ee 97% ee 88% ee 77% ee 72% ee
2. trans-disubstituierte Alkene = ausgezeichnete Selektivitäten
BuBu
BuCO2Et
PhMe
CO2Et
C13H27Ph
Ph
97% ee 99% ee 97% ee >99% ee 93% ee
Bu
Me
Ph
MeMe
C13H2778% ee 94% ee 79% ee
3. 1,1-Disubstituierte Alkene
63
Die AD in Abhängigkeit vom Substitutionsmuster der Doppelbindungen
4. Trisubstituierte Alkene: gewöhnlich gute Selektivitäten
BuMe
Me
Ph98% ee 99% ee
5. cis-substituierte Alkene: geringe Selektivitäten
Ph Me Ph CO2Et
72% ee 75% ee
6. Tetrasubstituierte Alkene: lediglich Silyl-Enolether haben eine praktische BedeutungR3SiO O
OH
MePh
PhTBSO
PhPh
MeTBSOOTBS OTBS
75% ee 53% ee 67% ee 85% ee
Morikawa et al. JACS 1993, 115, 8463-8464.
64
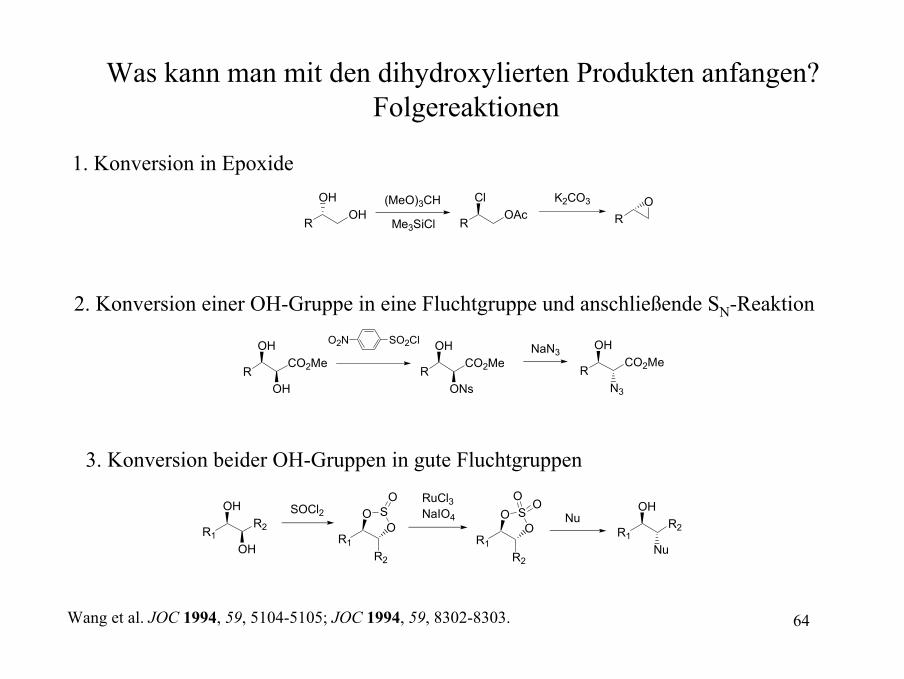
Was kann man mit den dihydroxylierten Produkten anfangen? Folgereaktionen
1. Konversion in Epoxide
ROH
OH (MeO)3CH
Me3SiCl ROAc
Cl K2CO3
RO
2. Konversion einer OH-Gruppe in eine Fluchtgruppe und anschließende SN-Reaktion
RCO2Me
OH
OH
O2N SO2Cl
RCO2Me
OH
ONs
NaN3
RCO2Me
OH
N3
3. Konversion beider OH-Gruppen in gute Fluchtgruppen
R1R2
OH
OH
SOCl2
R1R2
OSO
O RuCl3NaIO4
R1R2
OSO
OO
R1R2
OH
Nu
Nu
Wang et al. JOC 1994, 59, 5104-5105; JOC 1994, 59, 8302-8303.
65
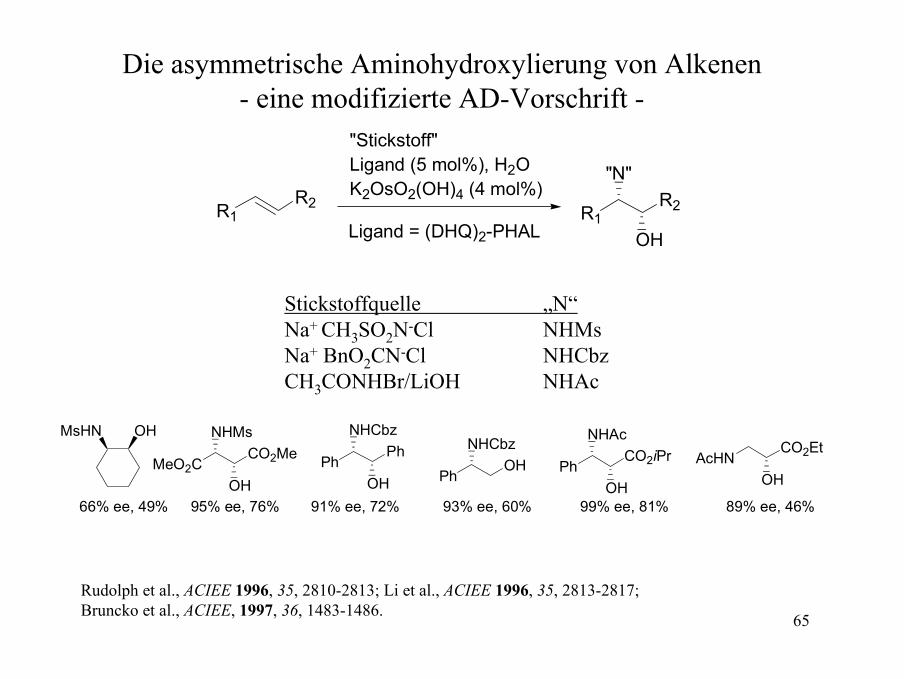
Die asymmetrische Aminohydroxylierung von Alkenen- eine modifizierte AD-Vorschrift -
R1R2
"Stickstoff"Ligand (5 mol%), H2OK2OsO2(OH)4 (4 mol%)
R1R2
OH
"N"
Ligand = (DHQ)2-PHAL
Stickstoffquelle „N“Na+ CH3SO2N-Cl NHMsNa+ BnO2CN-Cl NHCbzCH3CONHBr/LiOH NHAc
OHMsHN
MeO2CCO2Me
NHMs
OHPh
PhNHCbz
OH PhOH
NHCbzAcHN
CO2Et
OHPh
CO2iPrNHAc
OH66% ee, 49% 95% ee, 76% 91% ee, 72% 93% ee, 60% 99% ee, 81% 89% ee, 46%
Rudolph et al., ACIEE 1996, 35, 2810-2813; Li et al., ACIEE 1996, 35, 2813-2817; Bruncko et al., ACIEE, 1997, 36, 1483-1486.
66
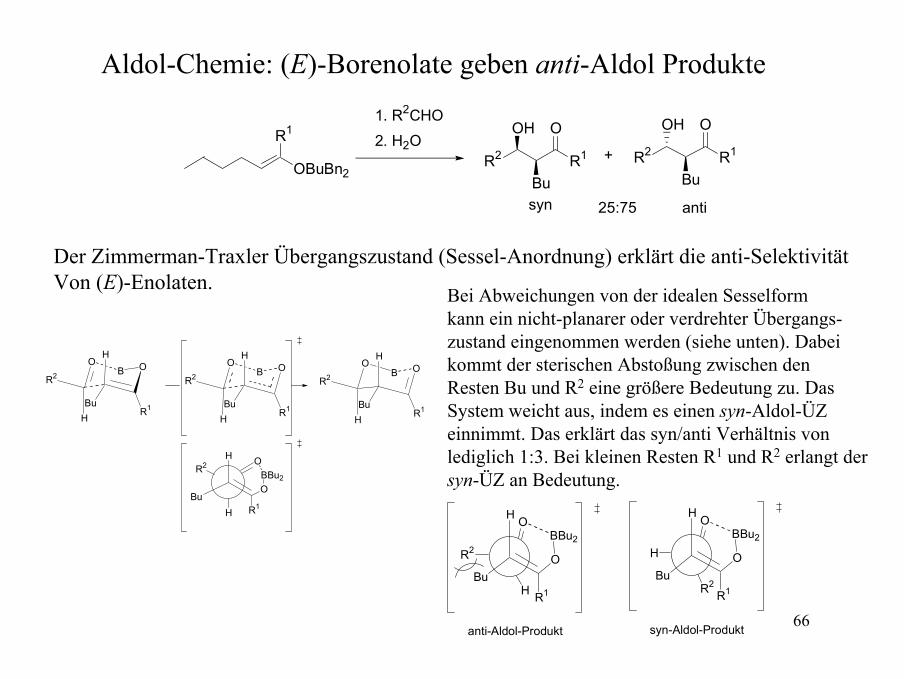
Aldol-Chemie: (E)-Borenolate geben anti-Aldol Produkte
OBuBn2
R1
R2 R1
OH
Bu
O
R2 R1
Bu
OH O1. R2CHO2. H2O
+
syn anti25:75
Der Zimmerman-Traxler Übergangszustand (Sessel-Anordnung) erklärt die anti-SelektivitätVon (E)-Enolaten.
H
Bu
R1
O
BBu2
O
R2
H
H
Bu
R1
O
BBu2
O
H
R2
anti-Aldol-Produkt syn-Aldol-Produkt
Bei Abweichungen von der idealen Sesselformkann ein nicht-planarer oder verdrehter Übergangs-zustand eingenommen werden (siehe unten). Dabeikommt der sterischen Abstoßung zwischen denResten Bu und R2 eine größere Bedeutung zu. DasSystem weicht aus, indem es einen syn-Aldol-ÜZeinnimmt. Das erklärt das syn/anti Verhältnis von lediglich 1:3. Bei kleinen Resten R1 und R2 erlangt der syn-ÜZ an Bedeutung.
OBO
HBu
H
R1
R2OB
O
HBu
H
R1
R2
OH
BuR1
OBBu2
R2
H
OBO
HBu
H
R1
R2
67
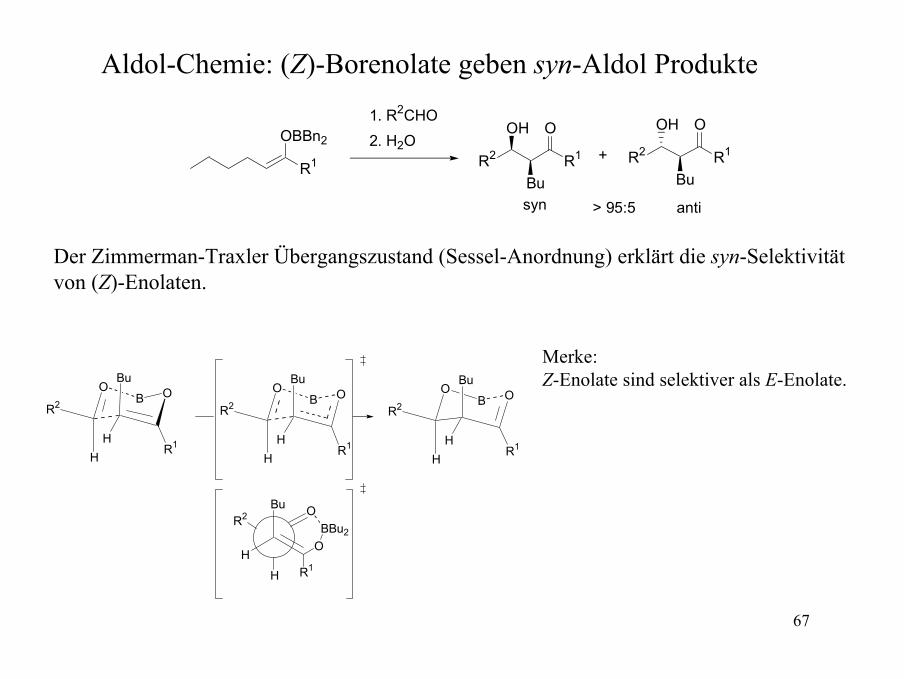
Aldol-Chemie: (Z)-Borenolate geben syn-Aldol Produkte
R1
OBBn2
R2 R1
OH
Bu
O
R2 R1
Bu
OH O1. R2CHO2. H2O
+
syn anti> 95:5
OBO
HH
Bu
R1
R2OB
O
HH
Bu
R1
R2
OBu
HR1
OBBu2
R2
H
OBO
HH
Bu
R1
R2
Der Zimmerman-Traxler Übergangszustand (Sessel-Anordnung) erklärt die syn-Selektivitätvon (Z)-Enolaten.
Merke:Z-Enolate sind selektiver als E-Enolate.
68
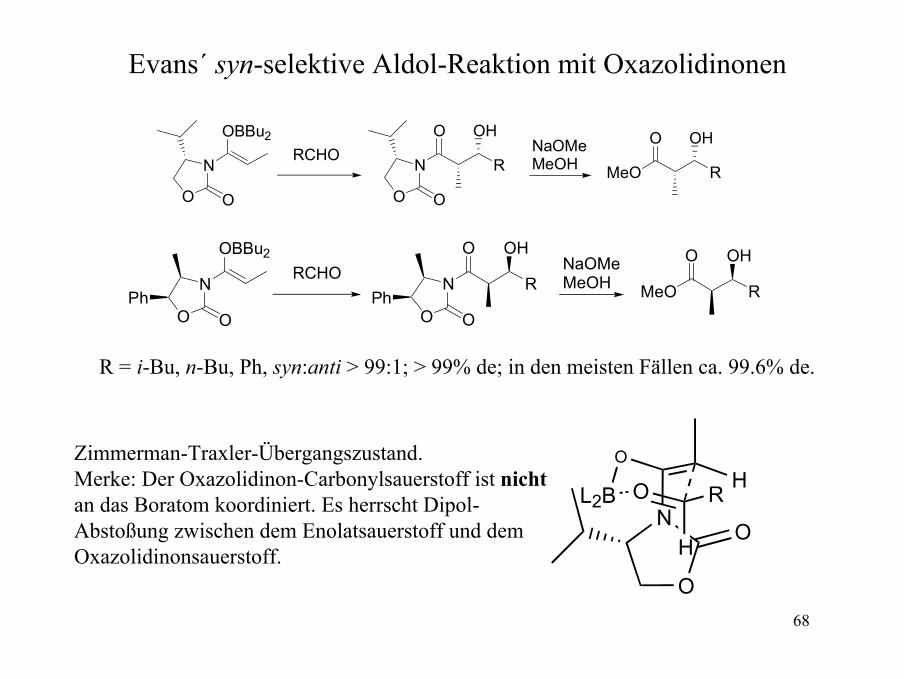
Evans´ syn-selektive Aldol-Reaktion mit Oxazolidinonen
N
O O
OBBu2RCHO N
O O
O
R
OHNaOMeMeOH MeO
O
R
OH
N
O O
OBBu2
PhRCHO N
O O
O
R
OH
Ph
NaOMeMeOH MeO
O
R
OH
R = i-Bu, n-Bu, Ph, syn:anti > 99:1; > 99% de; in den meisten Fällen ca. 99.6% de.
L2B O
O
RH
N
O
HO
Zimmerman-Traxler-Übergangszustand. Merke: Der Oxazolidinon-Carbonylsauerstoff ist nichtan das Boratom koordiniert. Es herrscht Dipol-Abstoßung zwischen dem Enolatsauerstoff und demOxazolidinonsauerstoff.
69
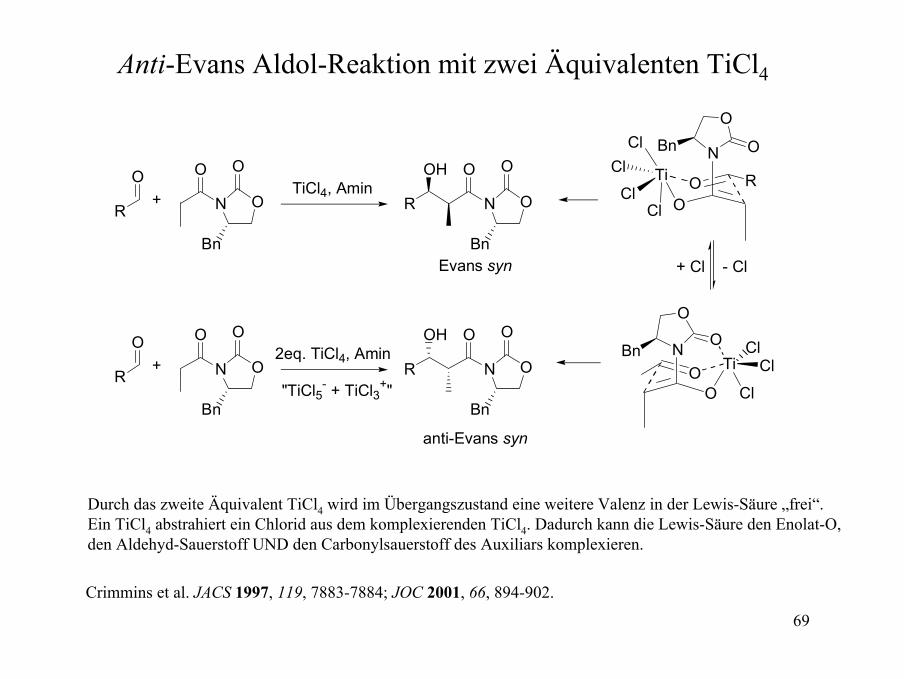
Anti-Evans Aldol-Reaktion mit zwei Äquivalenten TiCl4
R
OON
Bn
OO
R
OON
Bn
OO
ON
Bn
OO
R
OH
ON
Bn
OO
R
OH Ti
OO
N
O
Bn OCl
ClCl
ClR
O
TiON
O
BnO
Cl
ClCl
+TiCl4, Amin
+ 2eq. TiCl4, Amin
Evans syn
anti-Evans syn
- Cl+ Cl
"TiCl5- + TiCl3
+"
Durch das zweite Äquivalent TiCl4 wird im Übergangszustand eine weitere Valenz in der Lewis-Säure „frei“.Ein TiCl4 abstrahiert ein Chlorid aus dem komplexierenden TiCl4. Dadurch kann die Lewis-Säure den Enolat-O, den Aldehyd-Sauerstoff UND den Carbonylsauerstoff des Auxiliars komplexieren.
Crimmins et al. JACS 1997, 119, 7883-7884; JOC 2001, 66, 894-902.
70
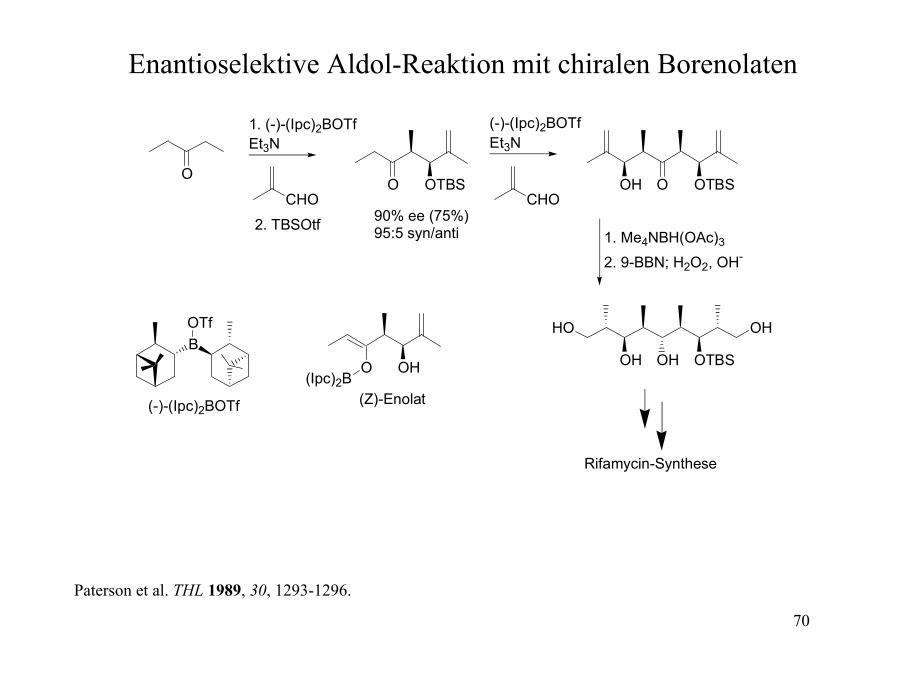
Enantioselektive Aldol-Reaktion mit chiralen Borenolaten
OO OTBS
CHO CHO
BOTf
O OH(Ipc)2B
O OTBSOH
OH OTBSOH
1. (-)-(Ipc)2BOTfEt3N
90% ee (75%)95:5 syn/anti
(-)-(Ipc)2BOTfEt3N
2. TBSOtf1. Me4NBH(OAc)3
2. 9-BBN; H2O2, OH-
(-)-(Ipc)2BOTf (Z)-Enolat
Rifamycin-Synthese
HO OH
Paterson et al. THL 1989, 30, 1293-1296.
71
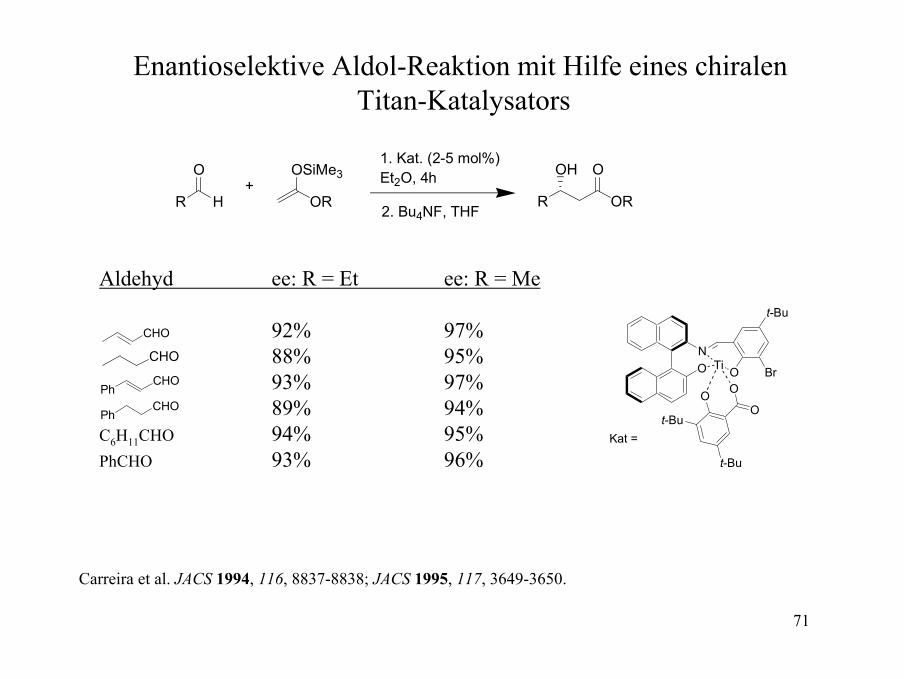
Enantioselektive Aldol-Reaktion mit Hilfe eines chiralenTitan-Katalysators
R H
O
OR
OSiMe3
R OR
OH O+
1. Kat. (2-5 mol%)Et2O, 4h
2. Bu4NF, THF
Aldehyd ee: R = Et ee: R = Me
92% 97%88% 95%93% 97%89% 94%
C6H11CHO 94% 95%PhCHO 93% 96%
CHO
CHO
PhCHO
PhCHO
NO
t-Bu
BrOTi
O
t-Bu
t-Bu
O
O
Kat =
Carreira et al. JACS 1994, 116, 8837-8838; JACS 1995, 117, 3649-3650.
72
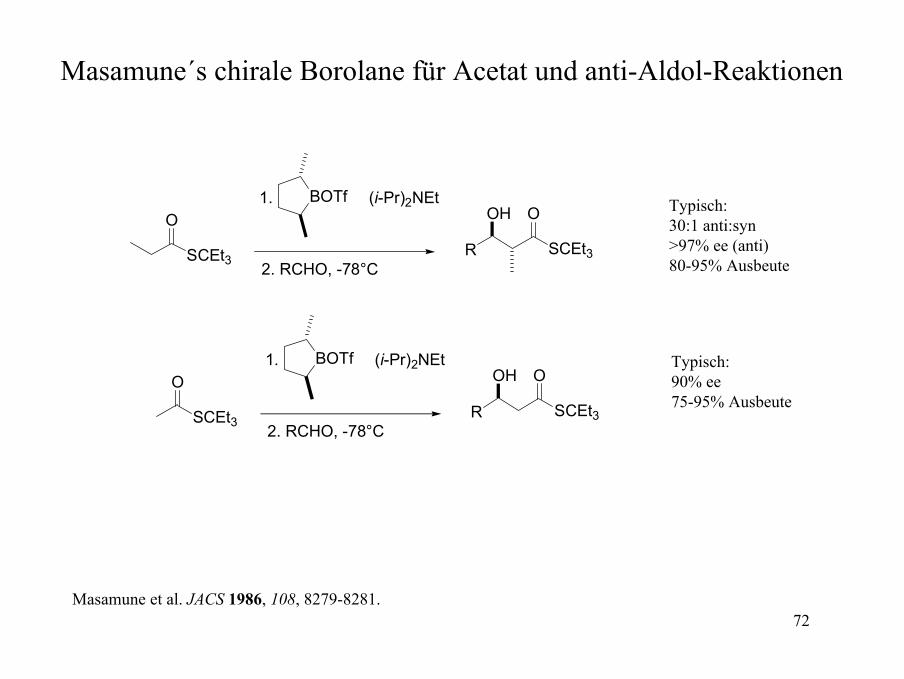
Masamune´s chirale Borolane für Acetat und anti-Aldol-Reaktionen
SCEt3
O
SCEt3
O
BOTf
BOTf
(i-Pr)2NEt
(i-Pr)2NEt
SCEt3
O
R
OH
SCEt3
O
R
OH
1.
2. RCHO, -78°C
1.
2. RCHO, -78°C
Typisch: 30:1 anti:syn>97% ee (anti)80-95% Ausbeute
Typisch: 90% ee75-95% Ausbeute
Masamune et al. JACS 1986, 108, 8279-8281.
73
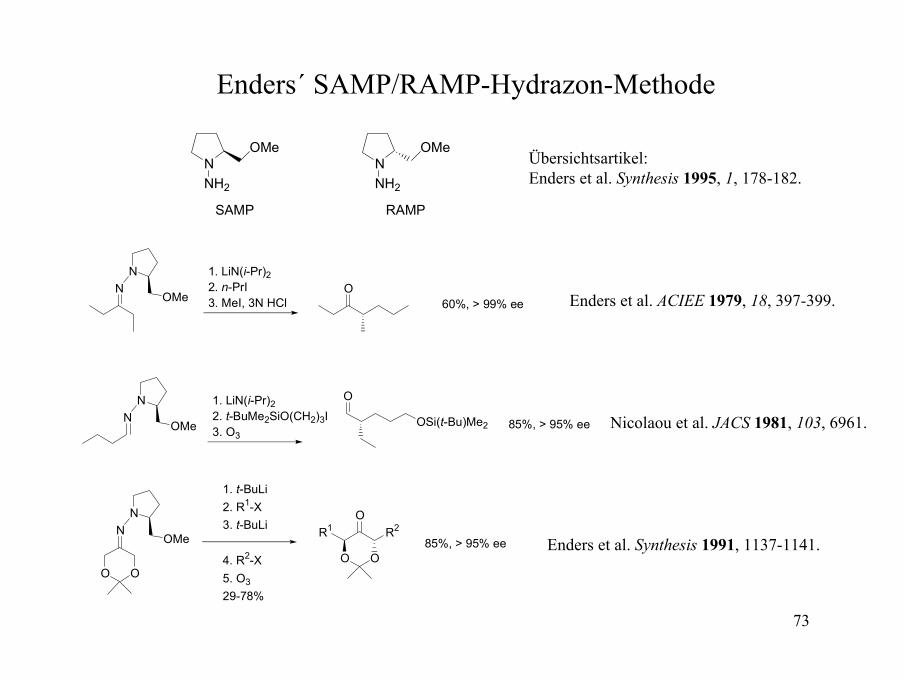
Enders´ SAMP/RAMP-Hydrazon-Methode
NNH2
OMe
SAMP
NNH2
OMe
RAMP
NN
OMeO
1. LiN(i-Pr)22. n-PrI3. MeI, 3N HCl 60%, > 99% ee
Übersichtsartikel: Enders et al. Synthesis 1995, 1, 178-182.
Enders et al. ACIEE 1979, 18, 397-399.
NN
OMe
O
OSi(t-Bu)Me2
1. LiN(i-Pr)22. t-BuMe2SiO(CH2)3I3. O3
85%, > 95% ee Nicolaou et al. JACS 1981, 103, 6961.
NN
OMe
O O
O
O O
R2R1
1. t-BuLi2. R1-X3. t-BuLi
4. R2-X5. O329-78%
85%, > 95% ee Enders et al. Synthesis 1991, 1137-1141.

74
Acyclische Stereokontrolle: Das Felkin-Anh-Modell
RH
O
R > Me
si-Angriff
re-Angriff
NuR
Nu
OHR
Nu
OH+
?
syn-Produkt anti-Produkt
S
ML
O
HNu
109°
M
SL
O
H Nu
109°Der Felkin-Anh Übergangszustand:Der Angriff des Nukleophils erfolgt aus einem 109° Winkel(Dunitz-Bürgi-Einflugschneise). Der größte Rest orientiert sich Senkrecht zur C=O Doppelbindung. Dabei ergeben sich zwei mögliche Konformationen (F und AF). Die Konformation F wird bevorzugt, da sich hierbei nur eine geringe sterische Abstoßung zwischen demNucleophil und dem Rest S ergibt. Im Fall des Übergangszustandes AF würde einegrößere Abstoßung zwischen dem Nucleophil und dem Rest M zum Tragen kommen. Es entsteht das Felkin-Produkt(syn-Produkt).
F AF

75
Acyclische Stereokontrolle: Das Cram-Chelat-Modell
R1
O
OR2
MS
Nu
M
R2O
S
O
R1
M
Nu
R1
HO NuM
SOR2
Sind am benachbarten chiralen Zentrum Substituenten mit Heteroatomen vorhanden, so besteht die Möglichkeitzur Chelatbildung mit Metallionen des Nucleophils. Werden Lithium-Organische Verbindungen oder Grignard-Verbindungen eingesetzt, so wird das Metallion zwischen dem Heteroatom und dem Carbonylsauerstoffatomchelatisiert. Dies führt zu einer Fixierung der Konformation bei der nun wiederum der Angriff des Nucleophils von der am wenigsten gehinderten Seite erfolgt.
OR1
OR1 MeMgBr
MeMgBr
+ MAT
Me
Me
R1
Me
H
O
H
R2Al
H
Me
R1
O
H
H
Me
R1
O
H
AlR2
Me
OHR1
Me
OHR1
Me
sterisch ungünstigerÜbergangszustand
Bei Verwendung von einzähnigen, sterischanspruchsvollen Lewis-Säuren kann ein völlig verändertes Bild entstehen. Zur Vermeidung von sterischen Wechsel-wirkungen befindet sich der kleinste Substituent, hier einProton, in der Nähe der Lewis-Säure. Die Lewis-Säure liegtauf der von den übrigen Substituenten abgewandten Seite. Daher erfolgt der Angriff des Nucleophils nun von der entgegengesetzten Seite.
OAl
O
Me
MAT =
76
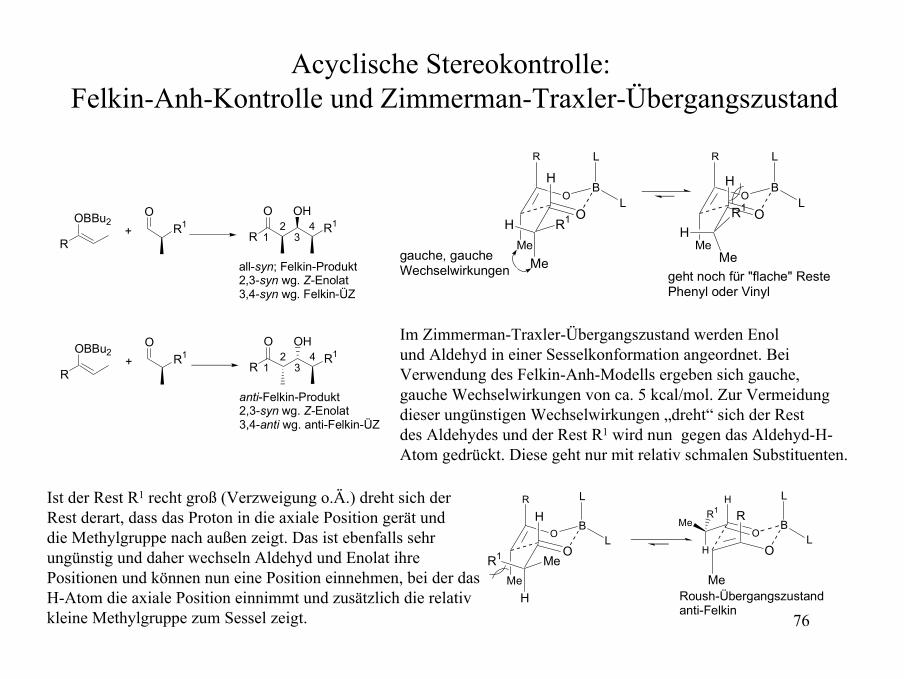
Acyclische Stereokontrolle: Felkin-Anh-Kontrolle und Zimmerman-Traxler-Übergangszustand
O
BO
Me
R L
L
Me
R1H
H
O
BO
Me
R L
L
Me
R1
H
H
gauche, gaucheWechselwirkungen geht noch für "flache" Reste
Phenyl oder Vinyl
O
BO
Me
R L
L
H
MeR1
H
O
BO
Me
H L
L
R
H
R1Me
Roush-Übergangszustandanti-Felkin
R
OBBu2
R
OBBu2
OR1
OR1
R1O OH
R1O OH
+
all-syn; Felkin-Produkt2,3-syn wg. Z-Enolat3,4-syn wg. Felkin-ÜZ
12
34
+
anti-Felkin-Produkt2,3-syn wg. Z-Enolat3,4-anti wg. anti-Felkin-ÜZ
12
34
R
R
Im Zimmerman-Traxler-Übergangszustand werden Enolund Aldehyd in einer Sesselkonformation angeordnet. BeiVerwendung des Felkin-Anh-Modells ergeben sich gauche,gauche Wechselwirkungen von ca. 5 kcal/mol. Zur Vermeidung dieser ungünstigen Wechselwirkungen „dreht“ sich der Rest des Aldehydes und der Rest R1 wird nun gegen das Aldehyd-H-Atom gedrückt. Diese geht nur mit relativ schmalen Substituenten.
Ist der Rest R1 recht groß (Verzweigung o.Ä.) dreht sich der Rest derart, dass das Proton in die axiale Position gerät und die Methylgruppe nach außen zeigt. Das ist ebenfalls sehr ungünstig und daher wechseln Aldehyd und Enolat ihrePositionen und können nun eine Position einnehmen, bei der dasH-Atom die axiale Position einnimmt und zusätzlich die relativkleine Methylgruppe zum Sessel zeigt.
77
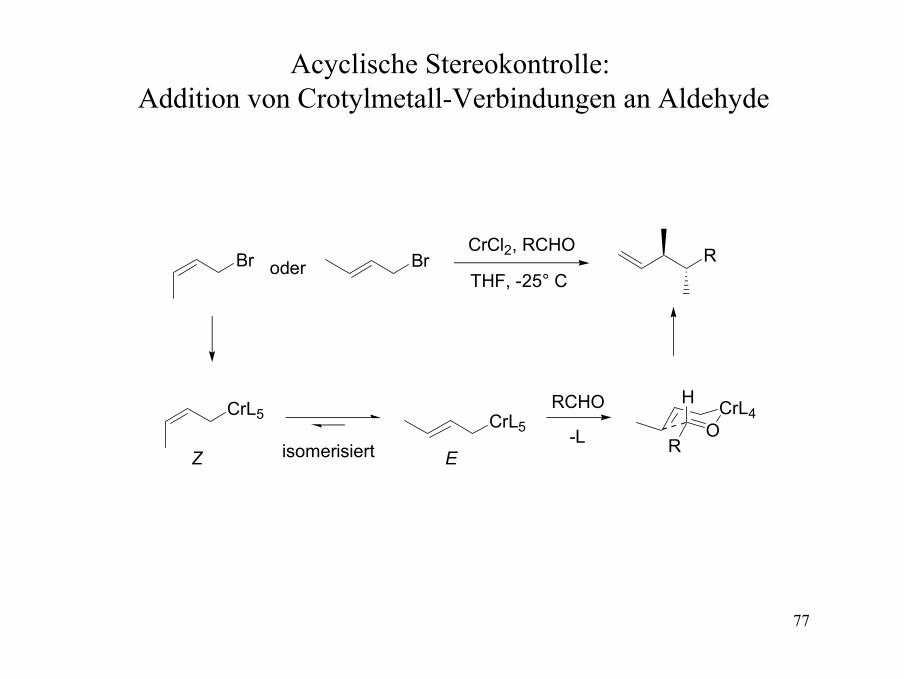
Acyclische Stereokontrolle: Addition von Crotylmetall-Verbindungen an Aldehyde
Br Br
CrL5 CrL5
RCHO
R
OCrL4
H
R
oder
-L
CrCl2, RCHO
THF, -25° C
isomerisiertZ E

78
B O
O
B O
O
RCHO
RCHO
OBH
R
O
O
OBH
R
O
O
R
OH
R
OH
antiE
Z syn
Acyclische Stereokontrolle: Crotylborierungen
H
O HB
2Ipc-Isopinocamphenyl-
+
OH
96% ee
H
O HB
2
+
OH
90% ee
H
O HB
2
+
OH
96% ee
H.C. Brown, JOC 1991, 56, 401-404
OROMe2Si B
O
OCO2iPr
CO2iPr
OH
SiMe2OR
OH
OH
+
H2O2KF, KHCO3
88% Ausbeute> 95:5 anti:syn72% ee
Roush
THL 1990, 31, 7563-7566.
CHO
NB N
Ph
PhTolO2S
SO2Tol
OH
Toluol, -78° C
97% ee, >90% Ausbeute
CoreyJACS 1989, 111, 5495-5496
79
OOR
O
OR
LUMO Dienophil
HOMO Dien
endo-ÜZ exo-ÜZ
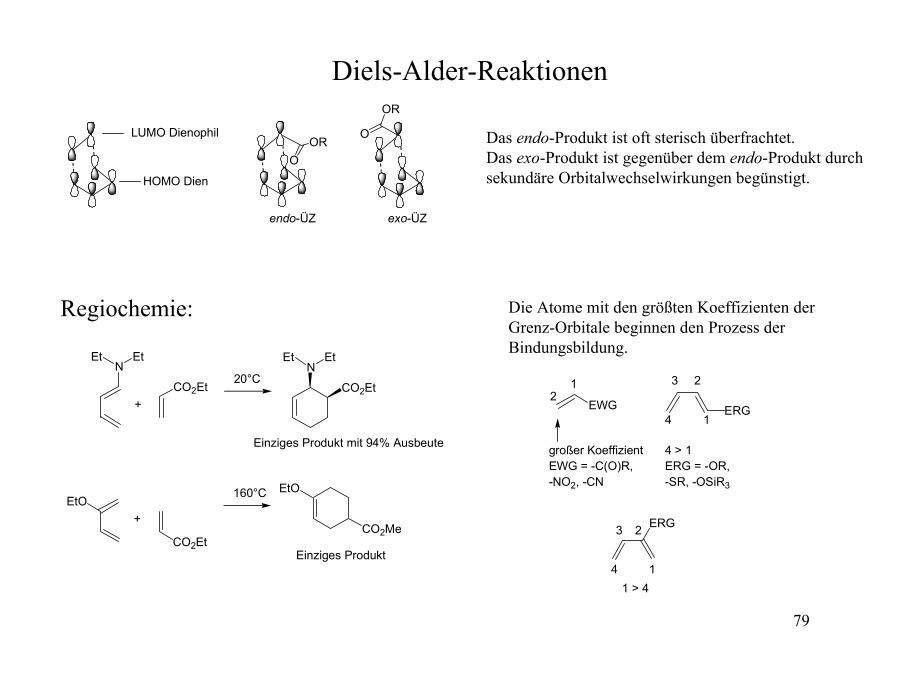
Diels-Alder-Reaktionen
Das endo-Produkt ist oft sterisch überfrachtet.Das exo-Produkt ist gegenüber dem endo-Produkt durch sekundäre Orbitalwechselwirkungen begünstigt.
Regiochemie:
EtO
NEt Et
CO2Et
CO2Et
N
CO2Et
EtEt
EtO
CO2Me
+
20°C
Einziges Produkt mit 94% Ausbeute
+
160°C
Einziges Produkt
Die Atome mit den größten Koeffizienten der Grenz-Orbitale beginnen den Prozess der Bindungsbildung.
EWG ERG
großer KoeffizientEWG = -C(O)R, -NO2, -CN
4 > 1ERG = -OR, -SR, -OSiR3
12
1
23
4
ERG
1
23
41 > 4
80
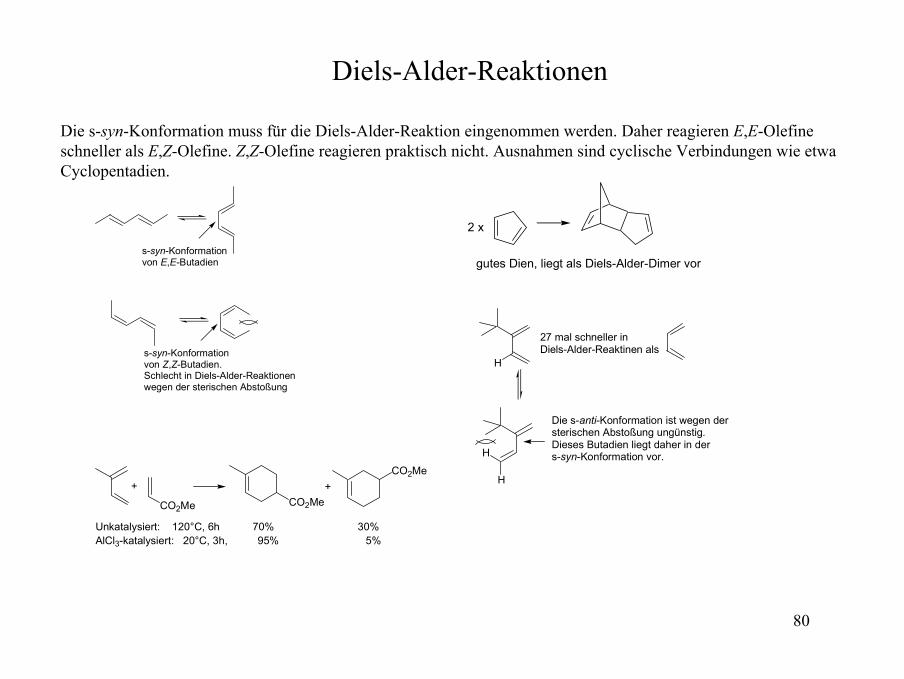
Diels-Alder-Reaktionen
Die s-syn-Konformation muss für die Diels-Alder-Reaktion eingenommen werden. Daher reagieren E,E-Olefineschneller als E,Z-Olefine. Z,Z-Olefine reagieren praktisch nicht. Ausnahmen sind cyclische Verbindungen wie etwa Cyclopentadien.
s-syn-Konformationvon E,E-Butadien
s-syn-Konformationvon Z,Z-Butadien.Schlecht in Diels-Alder-Reaktionenwegen der sterischen Abstoßung
gutes Dien, liegt als Diels-Alder-Dimer vor
2 x
27 mal schneller in Diels-Alder-Reaktinen als
Die s-anti-Konformation ist wegen dersterischen Abstoßung ungünstig.Dieses Butadien liegt daher in der s-syn-Konformation vor.
H
H
H
CO2Me CO2Me
CO2Me++
Unkatalysiert: 120°C, 6h 70% 30%AlCl3-katalysiert: 20°C, 3h, 95% 5%
81
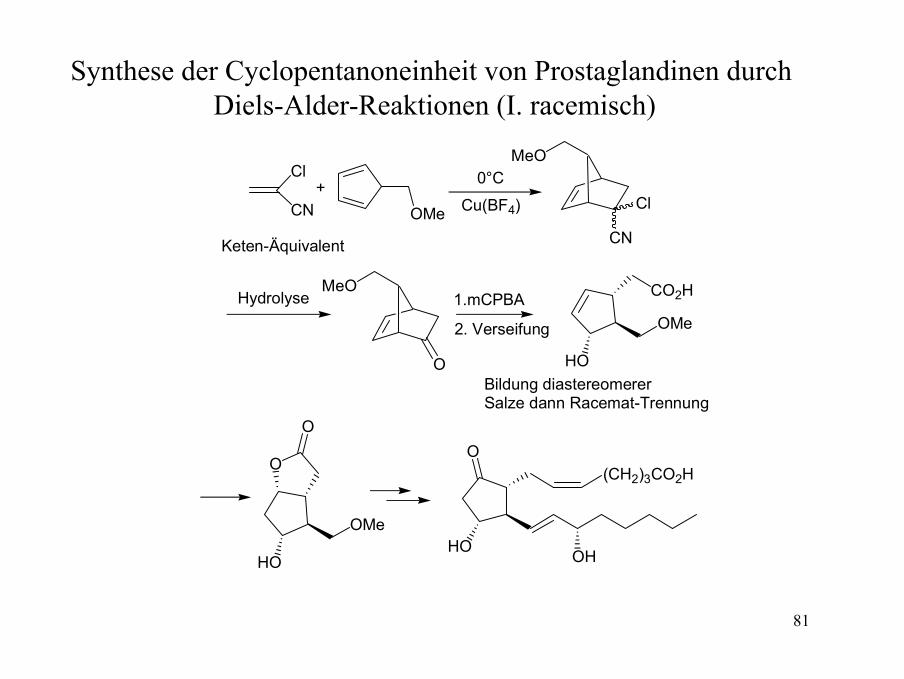
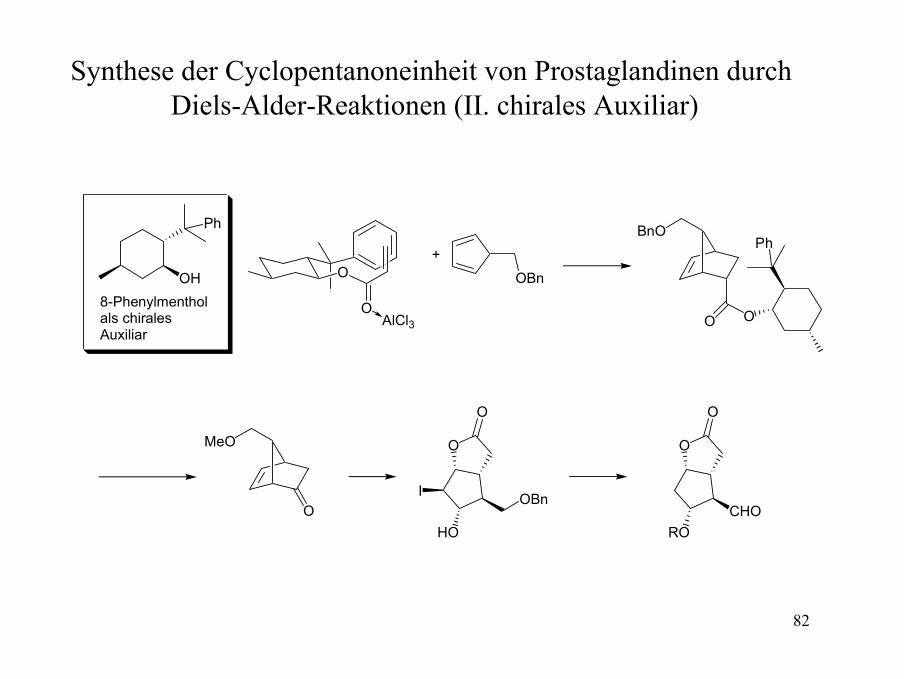
Synthese der Cyclopentanoneinheit von Prostaglandinen durch Diels-Alder-Reaktionen (I. racemisch)
Cl
CN
HO
OMe
O
O
MeO
O
OMe Cu(BF4)
O
HO
(CH2)3CO2H
OH
MeO
CN
Cl
HO
CO2H
OMe
0°C
Keten-Äquivalent
+
Hydrolyse 1.mCPBA
2. Verseifung
Bildung diastereomererSalze dann Racemat-Trennung
82
Synthese der Cyclopentanoneinheit von Prostaglandinen durch Diels-Alder-Reaktionen (II. chirales Auxiliar)
OH
Ph
MeO
O
O
OAlCl3
HO
OBn
O
O
I
OBn
ROCHO
O
O
BnO
O O
Ph+
8-Phenylmentholals chirales Auxiliar
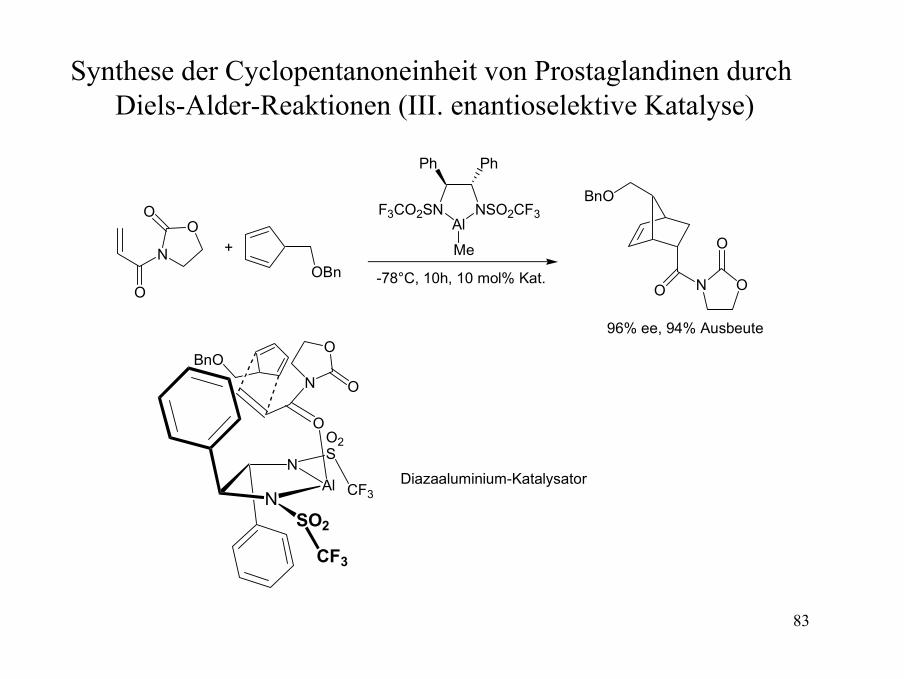
83
Synthese der Cyclopentanoneinheit von Prostaglandinen durch Diels-Alder-Reaktionen (III. enantioselektive Katalyse)
N
O
OO
OBn
F3CO2SNAl
NSO2CF3
Ph Ph
Me
N
N
O2S
Al CF3
SO2
CF3
O
N
O
O
BnO
BnO
O N O
O+
-78°C, 10h, 10 mol% Kat.
96% ee, 94% Ausbeute
Diazaaluminium-Katalysator
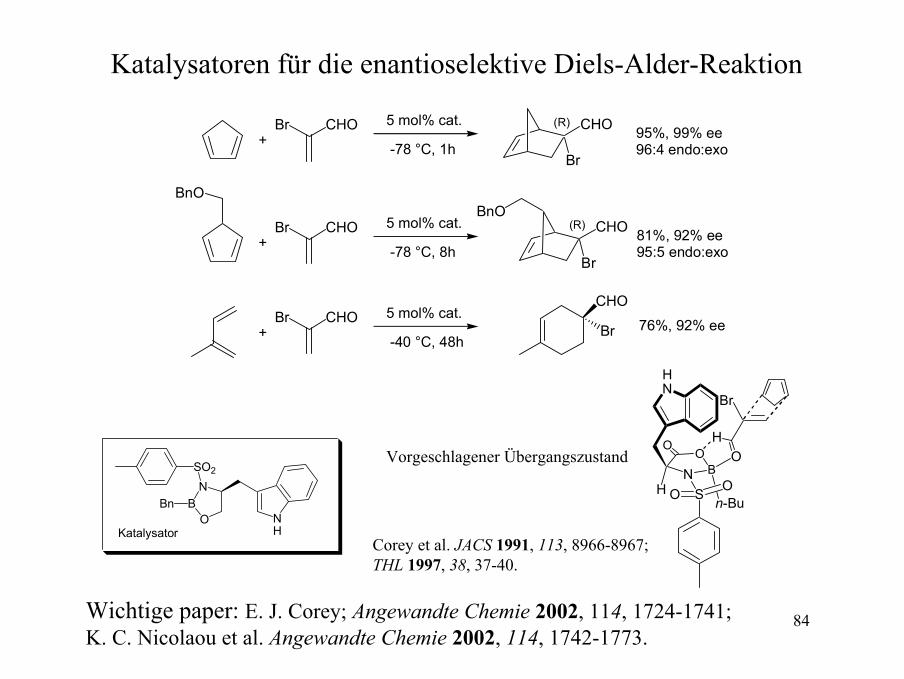
84
Katalysatoren für die enantioselektive Diels-Alder-Reaktion
Br CHO
BnO
Br CHO
Br CHO
Br
CHOBnO
Br
CHO(R)
CHO
Br
(R)
+5 mol% cat.
-78 °C, 1h95%, 99% ee96:4 endo:exo
+5 mol% cat.
-78 °C, 8h81%, 92% ee95:5 endo:exo
+5 mol% cat.
-40 °C, 48h76%, 92% ee
NH
NB
OBn
SO2
Katalysator
N BO
O
HN
H S OO n-Bu
HO
Br
Vorgeschlagener Übergangszustand
Corey et al. JACS 1991, 113, 8966-8967;THL 1997, 38, 37-40.
Wichtige paper: E. J. Corey; Angewandte Chemie 2002, 114, 1724-1741;K. C. Nicolaou et al. Angewandte Chemie 2002, 114, 1742-1773.
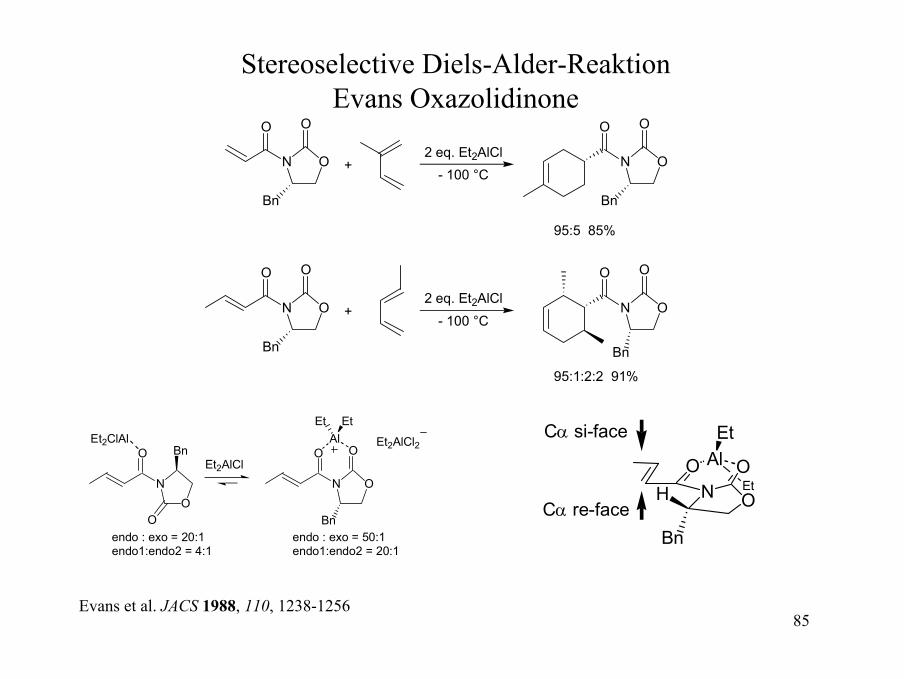
85
Stereoselective Diels-Alder-ReaktionEvans Oxazolidinone
N
O
O
O
Bn
N
O
O
O
Bn
N
O
O
O
Bn
N
O
O
O
Bn
+2 eq. Et2AlCl
- 100 °C
95:5 85%
+2 eq. Et2AlCl
- 100 °C
95:1:2:2 91%
N
O
O
Et2ClAl
O
BnEt2AlCl
N
O
O
Bn
OAl
Et Et
Et2AlCl2
endo : exo = 20:1endo1:endo2 = 4:1
endo : exo = 50:1endo1:endo2 = 20:1
ON
O
O
Al
H
Bn
Et
Et
Cα si-face
Cα re-face
Evans et al. JACS 1988, 110, 1238-1256
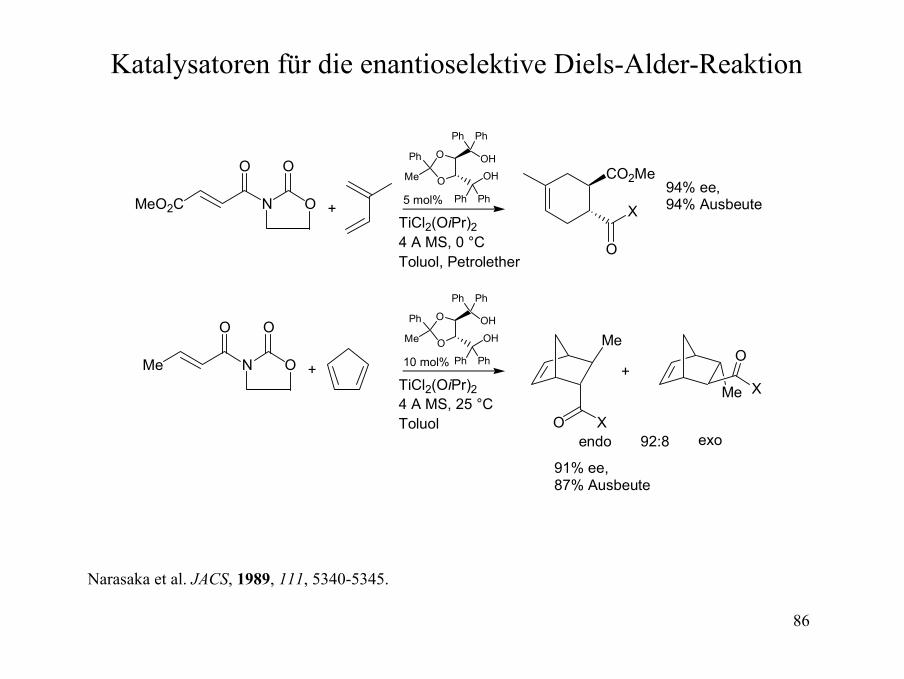
86
Katalysatoren für die enantioselektive Diels-Alder-Reaktion
MeO2C N
O O
O
O
O
Ph
Me
OHOH
Ph Ph
Ph Ph
Me N
O O
O
O
O
Ph
Me
OH
OH
Ph Ph
Ph Ph
Me
O X
CO2Me
O
X
Me
O
X
+ 5 mol%
TiCl2(OiPr)24 A MS, 0 °CToluol, Petrolether
94% ee,94% Ausbeute
+ 10 mol%
TiCl2(OiPr)24 A MS, 25 °CToluol
91% ee,87% Ausbeute
+
endo exo92:8
Narasaka et al. JACS, 1989, 111, 5340-5345.

87
Kupfer-katalysierte Hetero-Diels-Alder-Reaktionen
OEt
O
O
OEt
OEt
O
O
O
O
CO2EtMe
OEt
O
CO2EtMe
O
+10 mo% Kat.
-78 °C, THF
99,7% ee (89%)
+10 mo% Kat.
-78 °C, THF99,5% ee (89%)
N N
O O
t-But-BuCu
X X
X = OTfThorhauge, Jorgensen et al. ACIEE 1998, 37, 2404-2406.

88
Katalysatoren für die enantioselektive Diels-Alder-Reaktion
O N
O O NCu
N
OO
t-But-Bu
NCu
N
OO
t-But-Bu O O
NO
O
N O
O O
N O
O
COX
N O
O
O
H
+
2+
+ 2 SbF6-
10 mol%, CH2Cl2, -78 °C
9:1 endo:exo
99% ee
2+
+ 2 SbF6-
94% ee70% Ausbeute(endo:exo = 91:9)
93% ee90% Ausbeute(endo:exo = 95:5)
93% ee59% Ausbeute(endo:exo = 77:23)
Evans et al. ACIEE 1995, 34, 198-800; JACS 1993, 115,6460-6461.

89
8-Phenylmenthol als chirales Auxiliar
O
OPh
BnO
O
OPh
BnO
O
OPh
LiAlH4
HO
RO O
BnO
OH
BnO
+AlCl3, CH2Cl2
-55 °C, 1h, 89%
+Lewis-Säure
1,5 eq. SnCl4, 95%; 89% ee1,5 eq. TiCl4, 83%; 90% ee
+1. 1.5 eq. TiCl4
2. LiAlH4
Corey at al. JACS 1975, 97, 6908-6909.
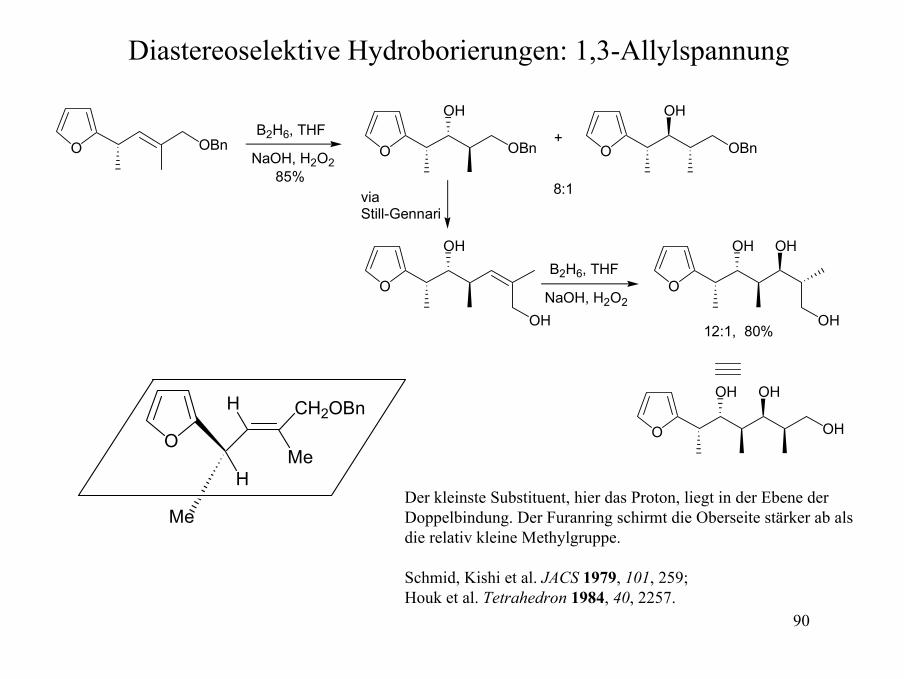
90
Diastereoselektive Hydroborierungen: 1,3-Allylspannung
OBnO OBnO
OH
O
OH
OH
OBnO
OH
O
OH OH
OH
O
OH
OH
OH
B2H6, THF
NaOH, H2O2
+
8:185%
viaStill-Gennari
B2H6, THF
NaOH, H2O2
12:1, 80%
CH2OBn
Me
H
H
Me
O
Der kleinste Substituent, hier das Proton, liegt in der Ebene der Doppelbindung. Der Furanring schirmt die Oberseite stärker ab als die relativ kleine Methylgruppe.
Schmid, Kishi et al. JACS 1979, 101, 259; Houk et al. Tetrahedron 1984, 40, 2257.
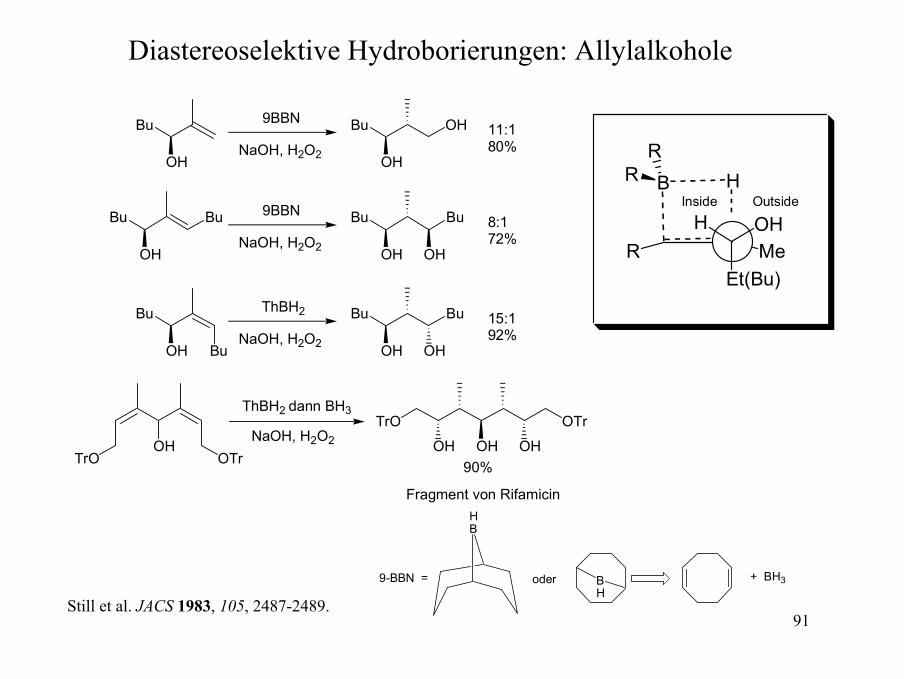
91
Diastereoselektive Hydroborierungen: Allylalkohole
Bu
OH
9BBN
NaOH, H2O2
Bu
OH
OH 11:180%
Bu
OH
9BBN Bu
OH
Bu
NaOH, H2O2
8:172%
Bu
OH
Bu
OH
ThBH2 Bu
OH
Bu
NaOH, H2O2
15:192%
Bu OH
OHOTrTrO
ThBH2 dann BH3
NaOH, H2O2 OH OH OH
OTrTrO
90%
Fragment von Rifamicin
MeOH
Et(Bu)
HR
HBR
RInside Outside
Still et al. JACS 1983, 105, 2487-2489.
9-BBN = + BH3BH
HB
oder
92
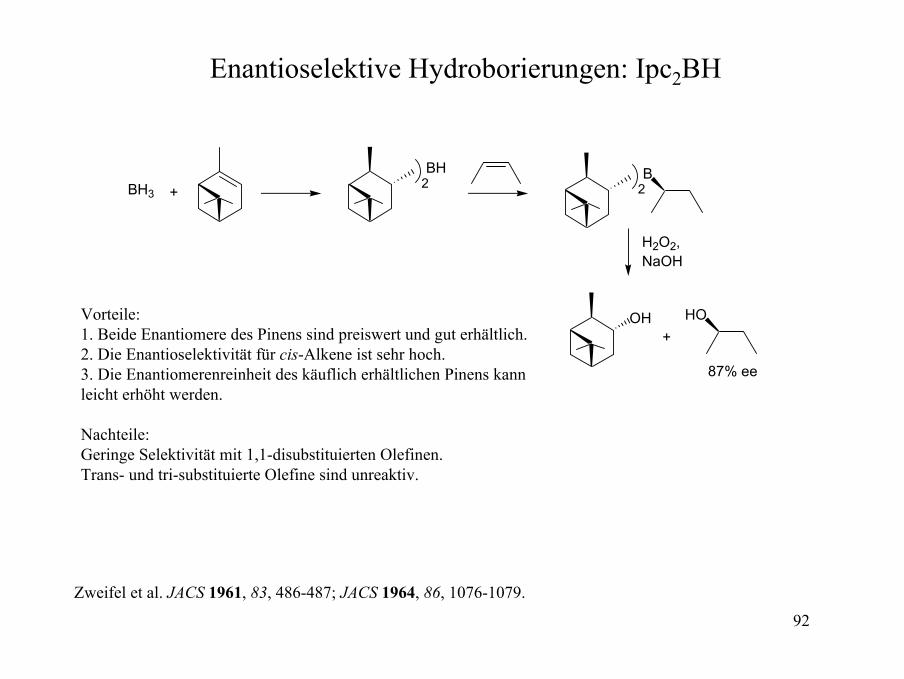
Enantioselektive Hydroborierungen: Ipc2BH
BH3 + 2BH
2B
H2O2,NaOH
OH HO
87% ee
+Vorteile:1. Beide Enantiomere des Pinens sind preiswert und gut erhältlich.2. Die Enantioselektivität für cis-Alkene ist sehr hoch.3. Die Enantiomerenreinheit des käuflich erhältlichen Pinens kannleicht erhöht werden.
Nachteile:Geringe Selektivität mit 1,1-disubstituierten Olefinen.Trans- und tri-substituierte Olefine sind unreaktiv.
Zweifel et al. JACS 1961, 83, 486-487; JACS 1964, 86, 1076-1079.
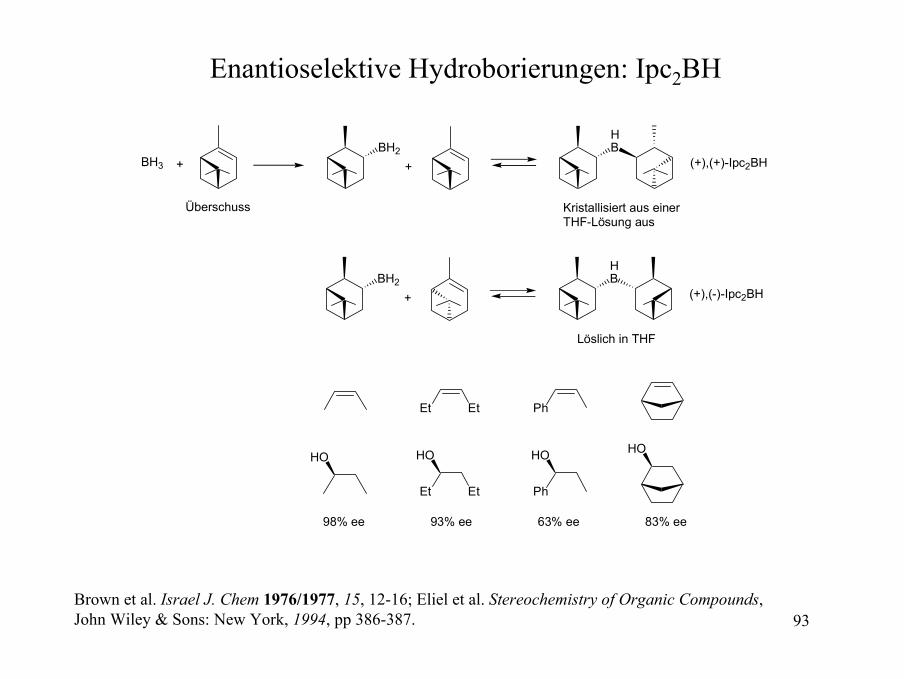
93
Enantioselektive Hydroborierungen: Ipc2BH
BH3
HO
BH2
BH2
Et Et
HO
Et Et
Ph
HO
Ph
HB
HB
HO
+
Überschuss
+ (+),(+)-Ipc2BH
Kristallisiert aus einerTHF-Lösung aus
+ (+),(-)-Ipc2BH
Löslich in THF
98% ee 93% ee 63% ee 83% ee
Brown et al. Israel J. Chem 1976/1977, 15, 12-16; Eliel et al. Stereochemistry of Organic Compounds,John Wiley & Sons: New York, 1994, pp 386-387.
94
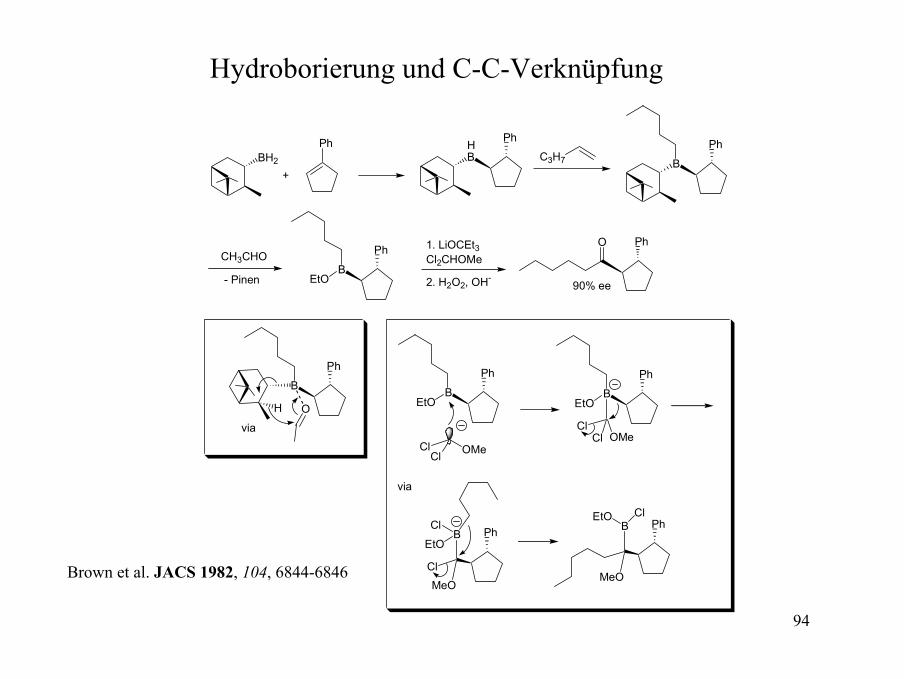
Hydroborierung und C-C-Verknüpfung
CH3CHO
BH2
Ph
B
Ph
EtO
HB
Ph
B
Ph
OHB
Ph
EtO
Cl OMeCl
B PhEtO
ClMeO
Cl
C3H7
B
Ph
EtO
ClOMeCl
B PhEtO
MeO
Cl
B
Ph
+
- Pinen
via
1. LiOCEt3Cl2CHOMe
2. H2O2, OH-
via
PhO
90% ee
Brown et al. JACS 1982, 104, 6844-6846
95
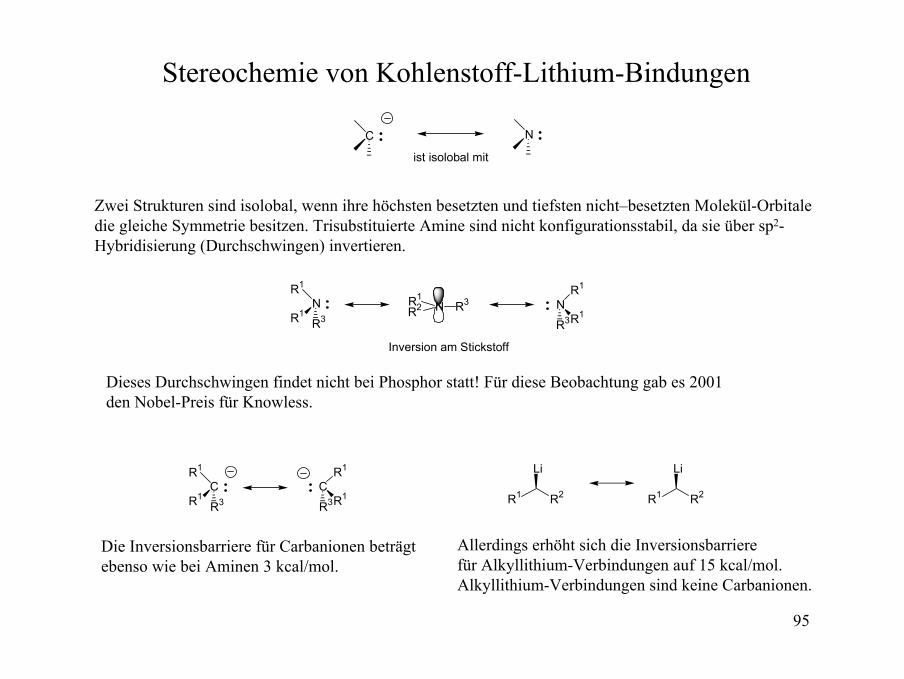
Stereochemie von Kohlenstoff-Lithium-Bindungen
C
ist isolobal mit
N
Zwei Strukturen sind isolobal, wenn ihre höchsten besetzten und tiefsten nicht–besetzten Molekül-Orbitaledie gleiche Symmetrie besitzen. Trisubstituierte Amine sind nicht konfigurationsstabil, da sie über sp2-Hybridisierung (Durchschwingen) invertieren.
R1
N
R3R1
R1
N
R3R1NR1
R2 R3
Inversion am Stickstoff
Dieses Durchschwingen findet nicht bei Phosphor statt! Für diese Beobachtung gab es 2001 den Nobel-Preis für Knowless.
R1
C
R3R1
R1
C
R3R1 R1 R2
Li
R1 R2
Li
Die Inversionsbarriere für Carbanionen beträgt ebenso wie bei Aminen 3 kcal/mol.
Allerdings erhöht sich die Inversionsbarriere für Alkyllithium-Verbindungen auf 15 kcal/mol.Alkyllithium-Verbindungen sind keine Carbanionen.

96
Enantioselektive Synthese mit chiralen Carbanionen
α-Alkoxy-Carbanionen sind stabil und können durch enantioselektive Deprotonierung hergestellt werden.
NO
OR
OH H
s-BuLi
N NN N
NO
OR
OLi HN
N
N
NH
H
NO
OR
OE H
NO
Cl
O
OHR
E H(-)-Spartein
E+ H3O+, OH-
E+ = MeI, R2CO, CO2, SiR3, D, etc.
= =
Käuflich erhältlich,allerdings nur das gezeigteEnantiomer.
Cby-Cl =
Hoppe et al. Pure Appl. Chem. 1994, 66, 1479-1486; Pure Appl. Chem. 1996, 68, 613-618.
97
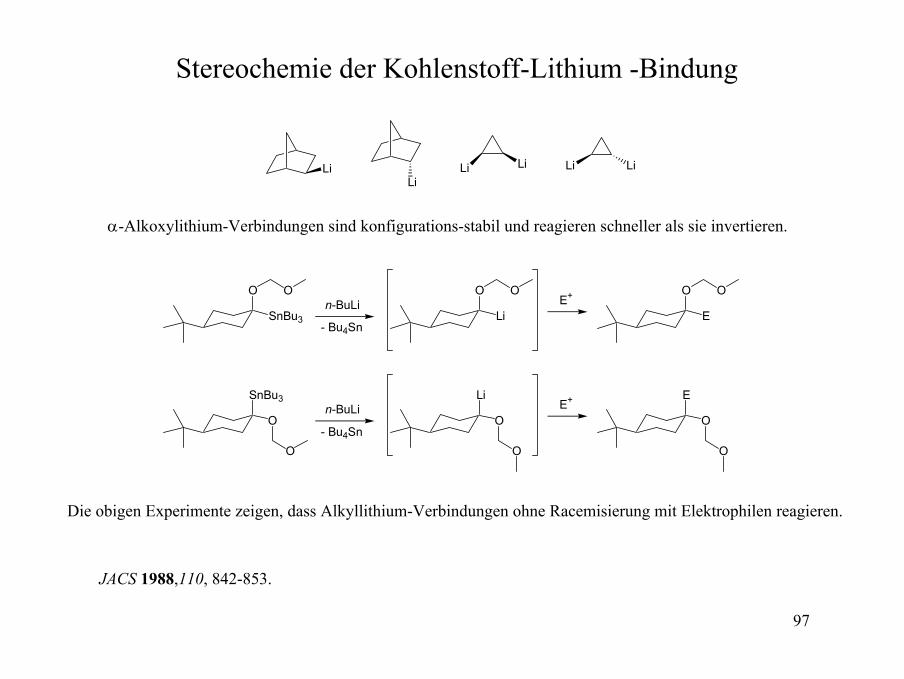
Stereochemie der Kohlenstoff-Lithium -Bindung
LiLi
LiLiLiLi
α-Alkoxylithium-Verbindungen sind konfigurations-stabil und reagieren schneller als sie invertieren.
O
SnBu3
On-BuLi
SnBu3
O
O
n-BuLiLi
O
O
O
Li
O
E
O
O
O
E
O
- Bu4Sn
E+
- Bu4Sn
E+
Die obigen Experimente zeigen, dass Alkyllithium-Verbindungen ohne Racemisierung mit Elektrophilen reagieren.
JACS 1988,110, 842-853.

98
Diastereoselektive Reduktionen: Gelenkte Hydrierungen
CH2OH
Rh(PPh3)3Cl
CH2OH
H
> 98% cis, 68% AusbeuteKOtBu
H
ORh
PPh3PPh3HH
Ph3P Rh ClPPh3
PPh3PPh2
PPh2
Rh
BF4
(C6H11)3PIr
N
PF6
Häufig verwendete Katalysatoren
Thompson et al., JACS 1974, 96, 6232-6233; Brown, ACIEE 1987, 26, 190-203; Hoveyda, Evans, Fu, Chem. Rev. 1993, 93, 1307-1370.
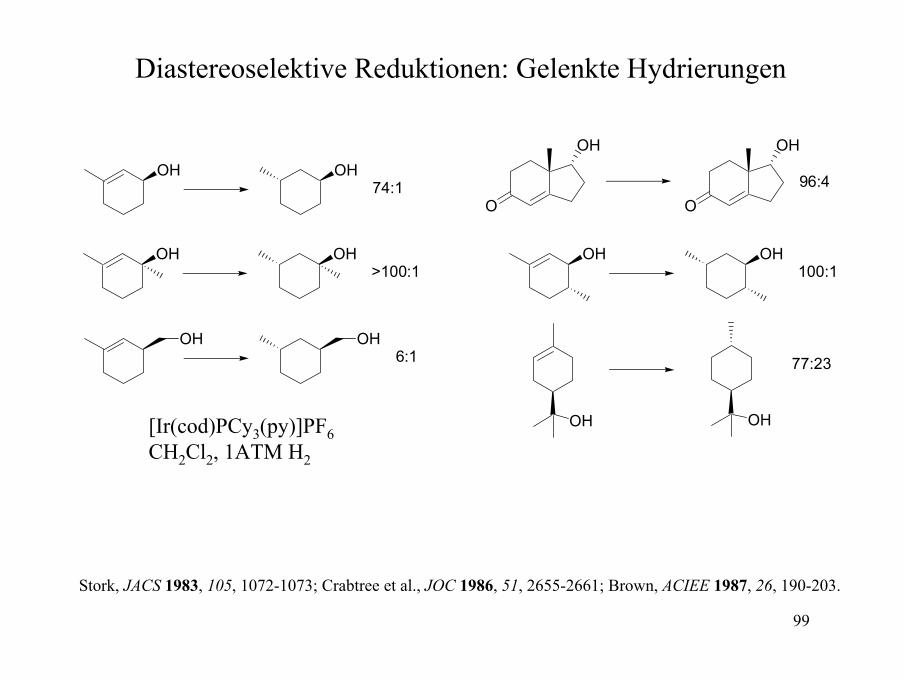
99
Diastereoselektive Reduktionen: Gelenkte Hydrierungen
OH OH74:1
OH OH>100:1
6:1OH OH
OH
O
OH
O96:4
OH OH100:1
OH OH
77:23
Stork, JACS 1983, 105, 1072-1073; Crabtree et al., JOC 1986, 51, 2655-2661; Brown, ACIEE 1987, 26, 190-203.
[Ir(cod)PCy3(py)]PF6CH2Cl2, 1ATM H2
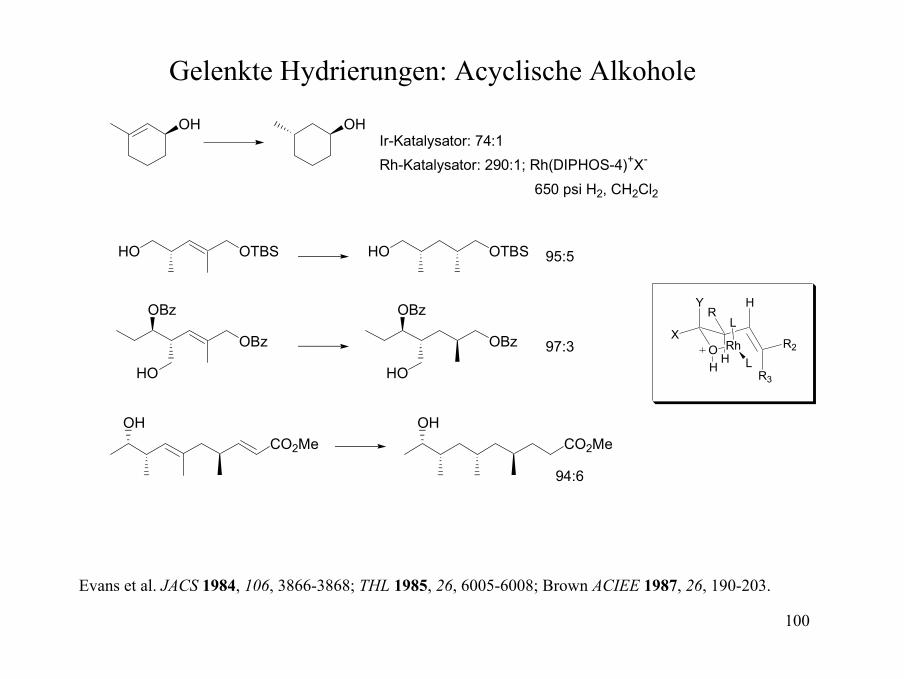
100
Gelenkte Hydrierungen: Acyclische Alkohole
OH
HO OTBS
OBz
OBz
HO
OHCO2Me
OH
OBz
HO
OBz
HO OTBS
OHCO2Me
Ir-Katalysator: 74:1Rh-Katalysator: 290:1; Rh(DIPHOS-4)+X-
650 psi H2, CH2Cl2
95:5
97:3
94:6
Evans et al. JACS 1984, 106, 3866-3868; THL 1985, 26, 6005-6008; Brown ACIEE 1987, 26, 190-203.
O Rh
RH
R2
R3
Y
X
HH
L
L
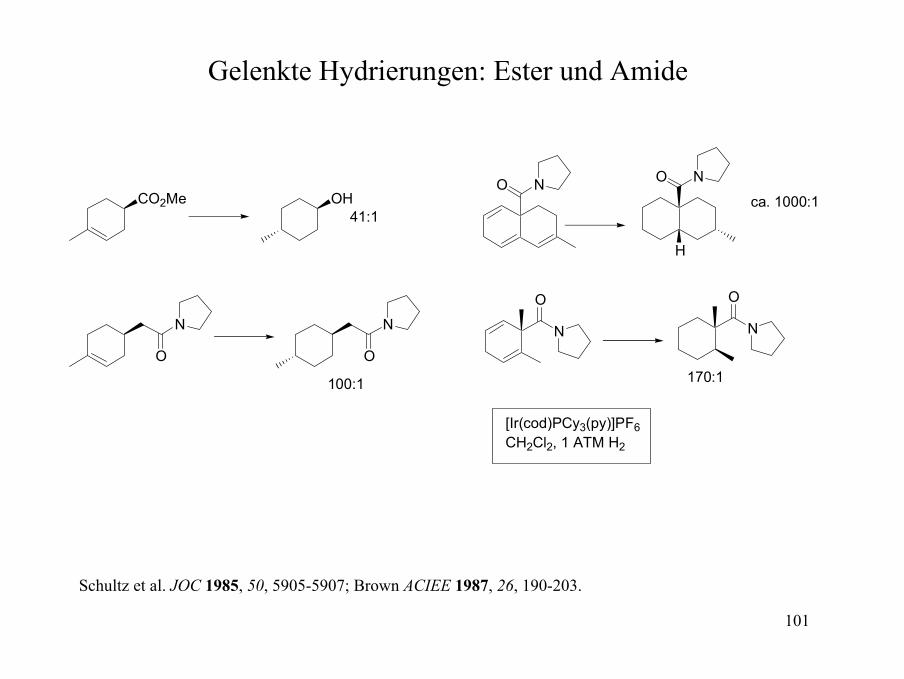
101
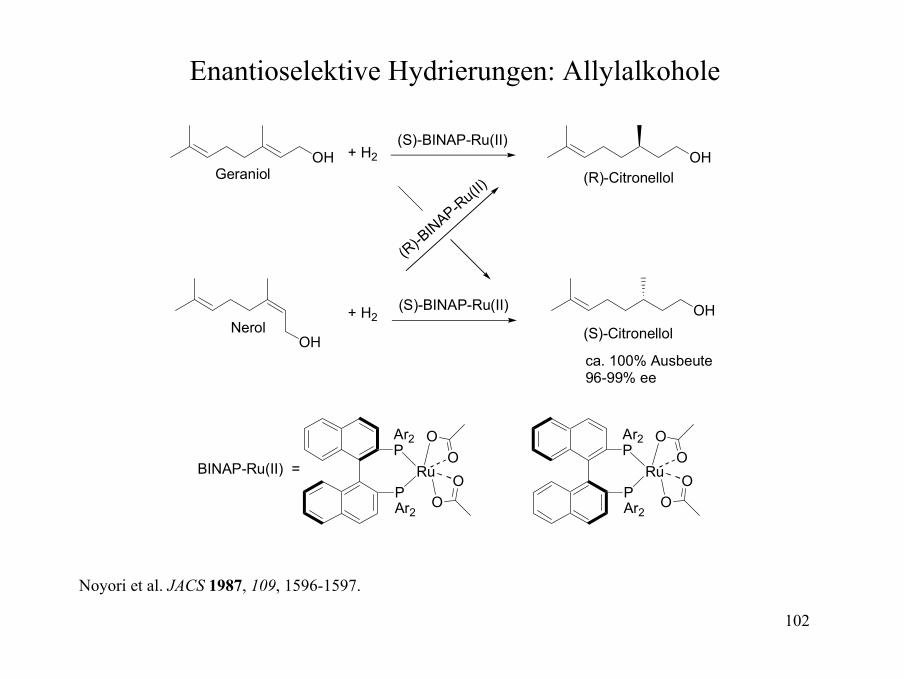
Gelenkte Hydrierungen: Ester und Amide
CO2Me OHO N
O
N
O
N N
O
O N
H
N
O
41:1
100:1
ca. 1000:1
170:1
[Ir(cod)PCy3(py)]PF6CH2Cl2, 1 ATM H2
Schultz et al. JOC 1985, 50, 5905-5907; Brown ACIEE 1987, 26, 190-203.
102
Enantioselektive Hydrierungen: Allylalkohole
OH
OH
Ar2P
PAr2
RuO
O
OO
Ar2P
PAr2
RuO
O
OO
OH
OH
+ H2
+ H2
(S)-BINAP-Ru(II)
(R)-B
INAP-Ru(II)
(S)-BINAP-Ru(II)
Geraniol (R)-Citronellol
(S)-CitronellolNerol
ca. 100% Ausbeute96-99% ee
BINAP-Ru(II) =
Noyori et al. JACS 1987, 109, 1596-1597.
103
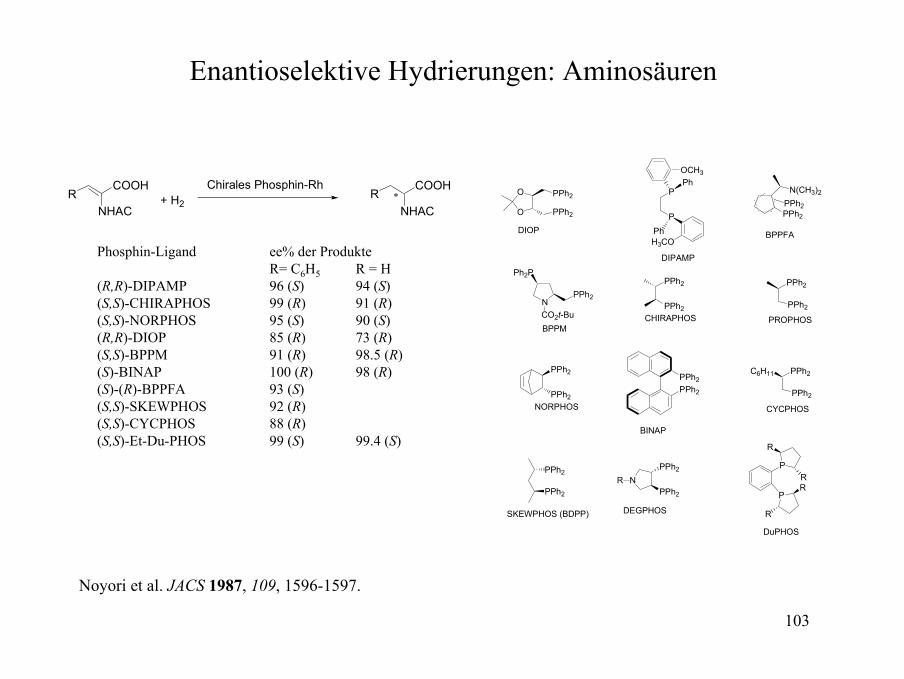
Enantioselektive Hydrierungen: Aminosäuren
O
O PPh2
PPh2
N
Ph2P
PPh2
CO2t-Bu
PPh2
PPh2
PPh2
PPh2
P
P
Ph
Ph
PPh2
PPh2
PPh2PPh2
NPPh2
PPh2
R
P
P
R
R
R
R
C6H11 PPh2
PPh2
PPh2
PPh2
OCH3
H3CO
N(CH3)2
PPh2
PPh2
DIOP
BPPM
NORPHOS
SKEWPHOS (BDPP) DEGPHOS
BINAP
CHIRAPHOS
DIPAMP
BPPFA
PROPHOS
CYCPHOS
DuPHOS
Noyori et al. JACS 1987, 109, 1596-1597.
RCOOH
NHACR ∗
COOH
NHAC+ H2
Chirales Phosphin-Rh
Phosphin-Ligand ee% der ProdukteR= C6H5 R = H
(R,R)-DIPAMP 96 (S) 94 (S)(S,S)-CHIRAPHOS 99 (R) 91 (R)(S,S)-NORPHOS 95 (S) 90 (S)(R,R)-DIOP 85 (R) 73 (R)(S,S)-BPPM 91 (R) 98.5 (R)(S)-BINAP 100 (R) 98 (R)(S)-(R)-BPPFA 93 (S)(S,S)-SKEWPHOS 92 (R)(S,S)-CYCPHOS 88 (R)(S,S)-Et-Du-PHOS 99 (S) 99.4 (S)
104
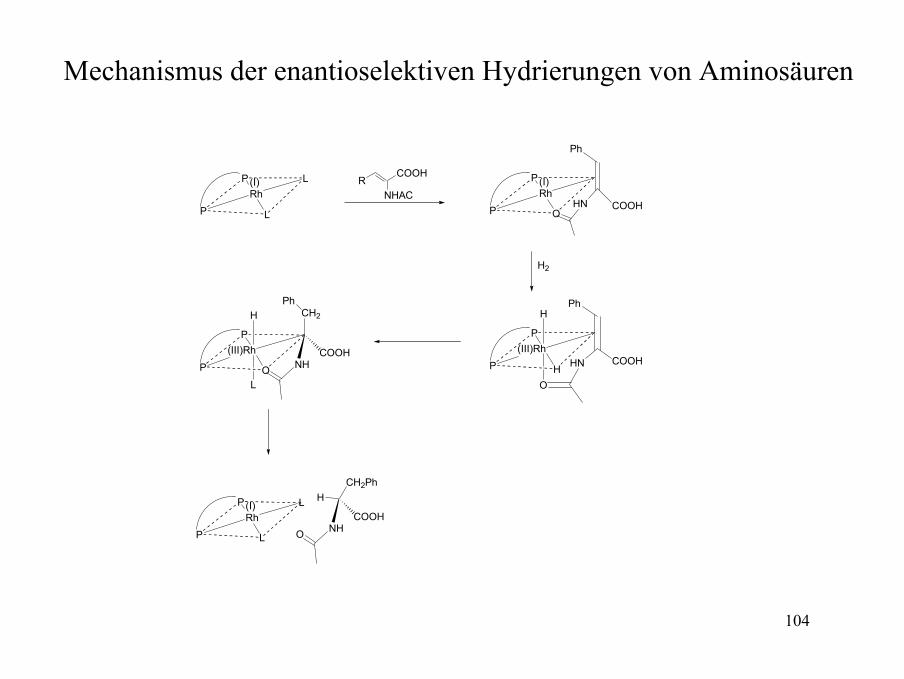
Mechanismus der enantioselektiven Hydrierungen von Aminosäuren
P
P L
L(I)Rh
RCOOH
NHAC
P
P H
(III)Rh
H
O
HN COOH
Ph
P
P
(III)Rh
H
O
CH2
NHCOOH
Ph
P
P
(I)Rh
OHN COOH
Ph
H2
L
P
P
(I)Rh
L
L
O
CH2Ph
NHCOOH
H

105
Enantioselektive Hydrierungen ungesättigter Carbonsäuren
CO2H
MeO
Me Me
COOHH
Me Me
COOHH
Ph
COOH
Ph
COOH
Me
COOHPh
COOHMe
HO
CO2H
MeO
Me
COOHPh
O O
135 atm H2; MeOH0.5% Ru(OAc)2[(S)-BINAP]
97 % ee, 100% Ausbeute
(R)-
91% ee
(R)-
93 % ee
(R)-
93 % ee
(R)-
85 % ee
Ohta, Noyori et al., JOC 1987, 52, 3174-3176.
106
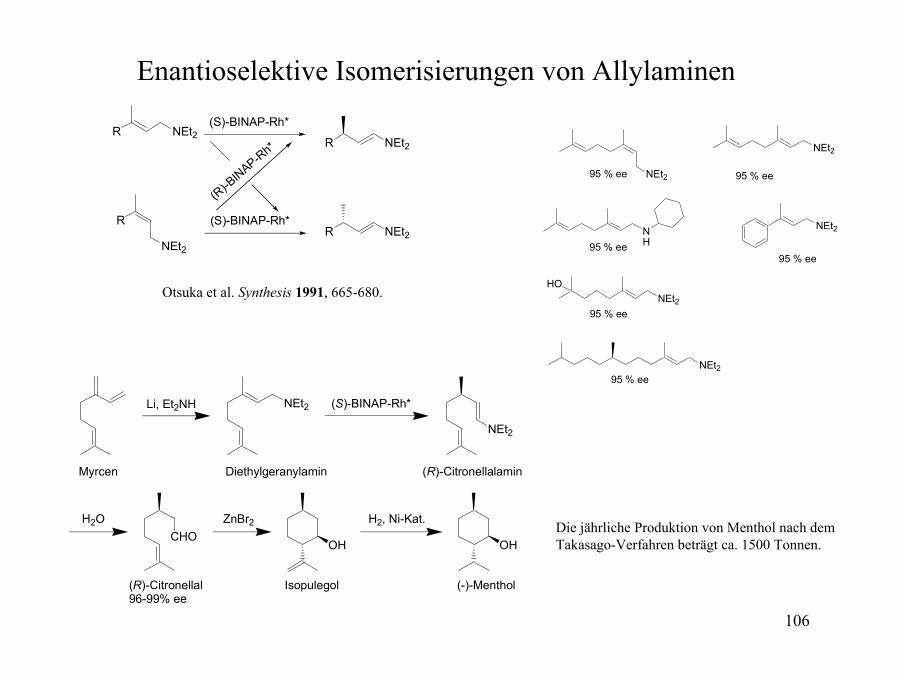
Enantioselektive Isomerisierungen von Allylaminen
R NEt2
R
NEt2R NEt2
R NEt2
(S)-BINAP-Rh*
(R)-B
INAP-Rh*
(S)-BINAP-Rh*
NEt2
NH
NEt2HO
NEt2
NEt2
NEt2
95 % ee
95 % ee
95 % ee
95 % ee
95 % ee
95 % ee
NEt2
CHOOH
ZnBr2H2O
NEt2
OH
Li, Et2NH (S)-BINAP-Rh*
H2, Ni-Kat.
Myrcen Diethylgeranylamin (R)-Citronellalamin
(R)-Citronellal96-99% ee
Isopulegol (-)-Menthol
Die jährliche Produktion von Menthol nach dem Takasago-Verfahren beträgt ca. 1500 Tonnen.
Otsuka et al. Synthesis 1991, 665-680.

107
Enantioselektive Reduktion von ungesättigten Carbonsäuren
CO2Et
CO2Et
NH
N
R R
CN
CO2Et
CO2Et
NaBH4 (2 eq.)1 mol% CoCl2EtOH/DMF, 23°C
R = CH2OSiMe2tBu
> 95 % Ausbeute
(R) 94 % ee
(S) 94 % ee
N N
H CO2Et
R1 R2
R
RN N
EtO2C H
R1 R2
R
R
Ph NMe
O
HPh N
MeO
H99 % ee
NMe
O
HN
MeO
H97 % ee
Pfaltz et al. ACIEE 1989, 28, 60-61;Acc. Chem. Res. 1993, 26, 339-345;Helv. Chim. Acta 1996, 79, 961-972.
108
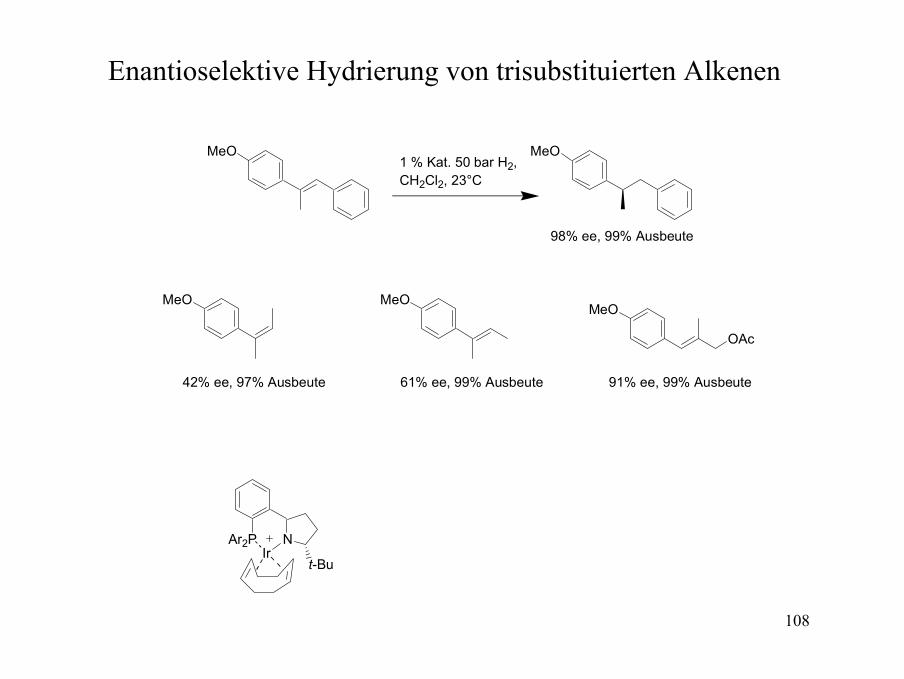
Enantioselektive Hydrierung von trisubstituierten Alkenen
MeO
MeO MeO
N
t-Bu
Ar2PIr
MeO
OAc
MeO
1 % Kat. 50 bar H2, CH2Cl2, 23°C
98% ee, 99% Ausbeute
42% ee, 97% Ausbeute 61% ee, 99% Ausbeute 91% ee, 99% Ausbeute


















