
Anwendung quantenchemischer Methoden in der Thermodynamik ... · Danksagung Diese Arbeit entstand...
167
Anwendung quantenchemischer Methoden in der Thermodynamik der Stofftrennung Der Technischen Fakultät der Universität Erlangen-Nürnberg zur Erlangung des Grades DOKTOR - INGENIEUR vorgelegt von Oliver Spuhl Erlangen 2006
Transcript of Anwendung quantenchemischer Methoden in der Thermodynamik ... · Danksagung Diese Arbeit entstand...

Anwendung quantenchemischer Methoden in der
Thermodynamik der Stofftrennung
Der Technischen Fakultät der
Universität Erlangen-Nürnberg
zur Erlangung des Grades
DOKTOR - INGENIEUR
vorgelegt von
Oliver Spuhl
Erlangen 2006

Als Dissertation genehmigt von
der Technischen Fakultät der
Universität Erlangen-Nürnberg
Tag der Einreichung: 18.09.2006
Tag der Promotion: 13.10.2006
Dekan: Prof. Dr.-Ing. Alfred Leipertz
1. Berichterstatter: Prof. Dr.-Ing. Wolfgang Arlt
2. Berichterstatter Prof. Dr.-Ing. Stephan Kabelac

Danksagung Diese Arbeit entstand während meiner Tätigkeit als wissenschaftlicher Mitarbeiter am Fachgebiet
Thermodynamik und Thermische Verfahrenstechnik der TU Berlin sowie am Lehrstuhl für
Thermische Verfahrenstechnik der Friedrich-Aleander-Universität Erlangen-Nürnberg.
Mein besonderer Dank gilt Herrn Prof. Wolfgang Arlt. Er hat mich während allen Phasen meiner
Arbeit jederzeit großzügig und zuverlässig unterstützt und gefördert. Nicht zuletzt die von seiner Seite
übertragene Verantwortung und das schier endlose Vertrauen in meine Fähigkeiten war für mich ein
großer Antrieb und hat zum erfolgreichen Gelingen dieser Arbeit beigetragen.
Herrn Prof. Kableac der Universität der Bundeswehr Hamburg danke ich herzlich für die Übernahme
der Berichterstattung, Herrn Prof. König für die Übernahme des Vorsitzes im Promotionsausschuss
und Herrn Prof. Hartmaier für die Teilnahme als fachfremder Prüfer an der Promotionsprüfung.
Der Mannschaft der Firma COSMOlogic und allen voran natürlich Herrn Dr. Andreas Klamt bin ich
für die stete Bereitschaft zu Diskussionen und der schnellen Hilfe bei allen Fragen rund um COSMO-
RS sehr verbunden.
Meine Diplomarbeiter Stefanie Herzog und Matthias Buggert sowie meine studentischen Mitarbeiter
Jörg Warneke, Florian Enzenberger und Alexander Buchele haben einen großen Anteil am Entstehen
dieser Arbeit. Ihrem großen Engagement und ihren herausfordernden Diskussionen verdanke ich
wichtige Impulse.
Ein ganz besonderer Dank gebührt meinen lieben Kolleginnen und Kollegen. Das offene,
freundschaftliche und hilfsbereite Klima sowohl in Berlin als auch in Erlangen ist unübertroffen.
Feelly, Andreas, Marko, Carsten, Tobias, Matthias, Irina, Steffi, Alexander, Katharina, Mitja – der
Spaß and die angenehme Zusammenarbeit wird mir immer in Erinnerung bleiben.
Durch Dr. Joachim Groß habe ich die Freude an den theoretischen Problemen der Thermodynamik
entdeckt. Er hat mich begleitet von den Anfängen als studentische Hilfskraft über die Diplomarbeit bis
hin zur Doktorarbeit. Joachim Du bist ein wertvoller Freund geworden.
Das größte Dankeschön gebührt aber meinen Eltern, die mich auf jedem Schritt meines Lebensweges
begleitet, unterstützt und gefördert haben. Euer unermüdlicher Rückhalt hat mich hierher gebracht –
Danke.

Inhaltsverzeichnis
Inhaltsverzeichnis..................................................................................................................... IV
Symbolverzeichnis ..................................................................................................................VII
Kurzfassung..............................................................................................................................XI
Abstract ...................................................................................................................................XII
1 Einleitung und Zielsetzung dieser Arbeit........................................................................... 1
1.1 Einleitung ................................................................................................................... 1
1.2 Zielsetzung ................................................................................................................. 2
2 Grundlagen der Thermodynamik und der Quantenchemie ................................................ 4
2.1 Vorbemerkung zu den betrachteten Modellen und Methoden ................................... 4
2.2 Grundlagen der Quantenchemie................................................................................. 6
2.2.1 Ab-initio und semi-empirische Methoden.......................................................... 7
2.2.2 Dichtefunktionaltheorie...................................................................................... 8
2.2.3 Kontinuumsmodelle – Moleküle in Lösung..................................................... 11
2.3 Das COSMO-RS Modell.......................................................................................... 14
2.4 Phasengleichgewichtsbeziehungen .......................................................................... 20
2.4.1 Aktivität und Aktivitätskoeffizient................................................................... 21
2.4.2 Phasengleichgewichtsbeziehung ...................................................................... 21
2.5 Eigenschaften und Phasenverhalten von hyperverzweigten Polymeren .................. 23
2.5.1 Struktureller Aufbau und Eigenschaften hyperverzweigter Polymere............. 23
2.5.2 Phasenverhalten von hyperverzweigten Polymerlösungen .............................. 25
2.5.3 Flüssig – Flüssig – Gleichgewicht ................................................................... 27
2.5.4 Erweiterung des COSMO-RS Modells um einen Term für die Erfassung des
Free-Volume-Effektes ...................................................................................................... 31
2.6 Zustandsgleichungen und Mischungsregeln ............................................................ 33
3 Anwendbarkeit des COSMO-RS Modells zur Vorhersage von thermodynamischen
Stoffdaten in Mischungen ........................................................................................................ 40
3.1 Vorhersage von Dampf-Flüssig und Flüssig-Flüssig Phasengleichgewichten ........ 40

V
3.1.1 Vorgehen und betrachtete Systeme .................................................................. 40
3.1.2 Erstellung der Molekülgeometrien, Geometrieoptimierung und Berechnung der
Abschirmungsladungen .................................................................................................... 42
3.1.3 Dampf-Flüssig Phasengleichgewichte ............................................................. 43
3.1.4 Flüssig-Flüssig Phasengleichgewichte ............................................................. 53
4 Erweiterung des COSMO-RS Modells zur Vorhersage von Reinstoffeigenschaften und
der Identifizierung von relevanten Strukturkonformationen.................................................... 56
4.1 Die Scaled-Particle-Theorie (SPT)........................................................................... 56
4.1.1 Grundlagen zur Scaled-Particle-Theorie .......................................................... 56
4.1.2 Kritikpunkte an der Scaled-Particle-Theorie und Aspekte ihrer Anwendung . 58
4.1.3 Dampfdruck aus der Freien Enthalpie des Lösungsvorganges ........................ 59
4.1.4 Effektive Harte-Kugel-Durchmesser aus COSMO-RS.................................... 61
5 Darstellung der Ergebnisse............................................................................................... 63
5.1 Einfluss der Konformation von Molekülen auf die Berechnung der
thermodynamischen Eigenschaften...................................................................................... 63
5.1.1 Konformeranalyse ............................................................................................ 63
5.1.2 Konformeranalyse und Phasengleichgewichtsberechnungen am Beispiel des 2-
Butanols 65
5.2 Vorhersage des Phasenverhaltens von hyperverzweigten Polymeren ..................... 72
5.2.1 Einfluss der Verzweigung bzw. der Kettenlänge auf das Phasenverhalten ..... 73
5.2.2 Einfluss der Anzahl der funktionellen Gruppen auf das Phasenverhalten ....... 78
5.2.3 Abbildung der molekularen Struktur der hyperverzweigten Polymere mit
quantenchemischen Methoden ......................................................................................... 82
5.2.4 Binäre Gemische mit hyperverzweigtem Polyglycerin.................................... 88
5.2.5 Das ternäre System Ethanol/Wasser/hyperverzweigtes Polyglycerin.............. 90
5.2.6 Zusammenfassende Bemerkungen ................................................................... 91
5.3 Verknüpfung des COSMO-RS-Modells mit Zustandsgleichungen ......................... 95
5.4 Vorhersage von Reinstoffeigenschaften mit dem erweiterten COSMO-RS-Modell
102
5.4.1 Harte-Kugel-Durchmesser ............................................................................. 102
5.4.2 Dampfdrücke am Normalsiedepunkt ............................................................. 104
5.4.3 Temperaturabhängigkeit................................................................................. 108
5.4.4 Einfluss der Konformationen der Moleküle auf das Vorhersageergebnis ..... 109
5.4.5 Auswahl von Konformationen auf Basis von Dampfdrücken am

VI
Normalsiedepunkt .......................................................................................................... 111
6 Zusammenfassung.......................................................................................................... 118
7 Anhang ........................................................................................................................... 122
7.1 Übersicht der untersuchten Systeme zur Anwendbarkeit des COSMO-RS-Modells
zur Vorhersage von Dampf-Flüssig-Phasengleichgewichten ............................................ 122
7.2 Harte-Kugel-Durchmesser aus COSMO-Volumina, Dampfdrücke am
Normalsiedepunkt .............................................................................................................. 128
7.3 Ergebnisse der Konformationsanalyse, Dampfdruck am Normalsiedepunkt, Harte-
Kugel-Durchmesser von 2-Pentanol, 1-Hexanol, 1-Octanol.............................................. 141
Literaturverzeichnis................................................................................................................ 144

Symbolverzeichnis
Zeichen Bedeutung Einheit
A Oberfläche m2
a Kontaktfläche m2
a Parameter in Zustandsgleichungen -
b Parameter in Zustandsgleichungen -
b Parameter (Free Volume Modell) -
B 2. Virialkoeffizient cm3/mol
c Parameter (Free Volume Modell) -
C 3. Virialkoeffizient (cm3)2/mol2
E elektrische Feldstärke V/m
E Energiefunktional J/mol
f Fugazität -
F freie Energie J
f freie molare Energie J/mol
g partielle freie molare Enthalpie J/mol
g freie molare Enthalpie J/mol
G freie Enthalpie J
G radiale Verteilungsfunktion -
H Enthalpie J
H Hamilton Operator -
h molare Enthalpie J/mol
L Anteil linearer Einheiten -
l binärer Mischungsparameter -
k binärer Mischungsparameter -
M Molmasse g/mol
m Masse kg
n Normalenvektor -
n Stoffmenge mol
P Druck Pa
p Häufigkeitsverteilung
q molekulare Oberfläche m2
q molekulare Oberfläche der Mischung m2

VIII
R relatives van-der-Waals Gruppenvolumen -
r Molekülradius m
r molekulares Volumen m3
r molekulares Volumen der Mischung m3
r Ortsvektor -
T Energiefunktional J/mol
T Temperatur K
U Uneinheitlichkeit -
V externes Potenzial V
V Volumen m3
v molares Volumen cm3/g
v% reduziertes Volumen -
w Massenbruch -
W Wahrscheinlichkeitsfunktion -
X binärer Parameter -
x Molenbruch -
Z Zustandssumme
z Realgasfaktor
Griechische Symbole
α Parameter im NRTL Modell -
∆ Abschirmungsenergie J/mol
∆ Differenz -
γ Aktivitätskoeffizient -
δ dispersiver Anteil der Energie J/mol
ε Dielektrizitätskonstante C2 /(J m)
ε Energie J
η reduzierte Segmentzahldichte -
Θ Oberflächenanteil -
λ kombinatorischer Parameter des COSMO-RS-Modells -
Λ de-Broglie Wellenlänge m
κ Zustandsgleichungsparameter -
µ chemisches Potenzial J/mol
Π Pointing - Faktor -

IX
ρ Dichte kg/m3
ρ Elektronendichte e/Å3
σ Ladungsdichte e /Å2
σ Durchmesser m
ϕ Free-Volume Anteil -
ϕ Fugazitätskoeffizient -
ψ Wellenfunktion -
τ Parameter der lokalen zusammensetzung -
ω azentrischer Faktor
Indizes hochgestellt
COSMO-RS COSMO-RS
COSMO COSMO
cav Kavität
disp dispersiv
E Exzess
el eletrisch
fv Free-Volume
hs Harte Kugel
komb kombinatorisch
L flüssige Phase
LV Phasenübergang dampfförmig - flüssig
res residuell
rep repulsiv
sol solvation
ueg homogenes Elektronengas (uniform electron gas)
V Dampfphase
I,II,III Phasenbezeichnungen
∞ unendliche Verdünnung
* ideal
, auf die Masse bezogen
___ partiell molare Größe
0 Standardzustand
0i Reinsstoff
Indizes tiefgestellt
C kritisch

X
R reduziert
Konstanten
e Elementarladung e = 1,602177*10-19 C
R allgemeine Gaskonstante R = 8,3144 J/(mol1 K1)
k Boltzmann-Kkonstante 1,3806505*10-23 J/K
NA Avogadro-Zahl 6,0221415*1023 1/mol
h Plancksches Wirkungsquantum 6,6260693*10-34 Js
π Kreiszahl 3,14159265
Abkürzungen
COSMO Conductor-like Screening Model
COSMO-RS Conductor-like Screening Model for Real Solvents
CSM Kontinuumsolvensmodell
DFT Dichtefunktionaltheorie
LLE Flüssig – Flüssig – Gleichgewicht
MO Molekülorbital
SCRF Self Consistent Reaction Field
TZVP Triple Zeta Valence Polarization
UNIFAC UNIQUAC Functional-group Activity Coefficients
UNIFAC-FV UNIFAC – Free Volume
VLE Dampf – Flüssig – Gleichgewicht
ZGL Zustandsgleichung

Kurzfassung
In dieser Arbeit wurden die vorhandenen Methoden der Verknüpfung von Quantenchemie und statistischer Thermodynamik als Vorhersagemethode für die Phasengleichgewichts-thermodynamik betrachtet. Der Schwerpunkt der Arbeit lag auf der Untersuchung und Weiterentwicklung des COSMO-RS-Modells. Es wurden fünf Themenkomplexe behandelt und neue Lösungsmethoden vorgestellt: 1. Validierung des COSMO-RS-Modells hinsichtlich in der chemischen Verfahrenstechnik
relevanter Komponenten und ihrer binären Mischungen. Die Validierung von 136 Systemen ergab, dass COSMO-RS experimentelle Daten von Dampf-Flüssig-Phasengleichgewichten unterschiedlicher Stoffklassen in guter Übereinstimmung vorhersagen konnte.
2. Die Untersuchung des Einflusses der verschiedenen Konformationen einer Molekülstruktur auf die Vorhersagen des Phasengleichgewichtes mit dem COSMO-RS-Modell wurde am Beispiel des 2-Butanol Moleküls exemplarisch dargestellt. Für das 2-Butanol/Wasser-System wurde gezeigt, dass die Konformation mit der niedrigsten Energie nicht die besten Ergebnisse bei der Vorhersage lieferte. Vielmehr war es notwendig alle Konformationen in die Berechnung einzubeziehen.
3. Stark verzweigte Moleküle mit großer Molmasse zeigen ein spezifisches Phasenverhalten. Ein Algorithmus wurde entwickelt, der die Berechnung hyperverzweigter Makromoleküle durch Zerschneiden in Subeinheiten mit quantenchemischen Methoden zulässt. Weiterhin wurde das COSMO-RS-Modell um einen Free-Volume Term in Anlehnung an das UNIFAC-Free-Volume-Modell erweitert. Am Beispiel eines hyperverzweigten Polyglyzerinmoleküls mit 20 Hydroxylgruppen und etwa 1400g/mol wurde das Phasenverhalten im ternären System Polymer/Ethanol/Wasser vorhergesagt.
4. Für die Berechnung von Hochdruckphasengleichgewichten wurde das COSMO-RS-Modell mit der Peng-Robinson-Zustandsgleichung unter Anwendung der Wong-Sandler-Mischungsregeln kombiniert. Die Ergebnisse belegten, dass Vorhersagen auf Basis der Atomstruktur der Moleküle und der kritischen Daten bis weit in den überkritischen Bereich möglich waren.
5. Das Energiekonzept des COSMO-RS-Modells wurde um einen Term erweitert, der die Änderung der freien Enthalpie durch die Bildung einer Kavität im Lösungsmittel enthält. Es wurde ein Konzept entwickelt, dass es ermöglicht, die quantenchemischen Rechnungen zu verwenden, um einen Harte-Kugel-Durchmesser beliebiger Moleküle zu berechnen. Mit dem erweiterten Energiekonzept wurden Dampfdrücke von Komponenten der Alkane, der Alkene, der Alkohole, der Aromaten, der Ether, der Ketone, der Aldehyde und der Polyole berechnet. Die Abweichungen, verglichen mit experimentellen Daten am Normalsiedepunkt, zeigten für Alkohole eine Abweichung von durchschnittlich ca. 20 Prozent, für die anderen Stoffklassen deutlich höhere Abweichungen. Der vorhergesagte Wert des Dampfdruckes reagierte sehr sensibel auf die unterschiedlichen Konformationen einer Komponente. Er wäre somit ein Sensor, der es ermöglicht, relevante Konformationen eines Moleküls für eine Phasengleichgewichtsberechnung zu identifizieren. Berechnungen für die Stoffe 2-Pentanol, 1-Hexanol und 1-Octanol ergaben, dass Konformationen die eine gute Beschreibung des Dampfdruckes lieferten, nicht in jedem Fall auch eine gute Beschreibung in Mehrkomponentengemischen ergaben. Insgesamt kann festgehalten werden, dass alle in der Konformationsanalyse ermittelten Konformationen verwendet werden sollten. Zusätzlich ist es empfehlenswert, Konformationen mittels molekulardynamischer Rechnungen in Anwesenheit des eine Komponente umgebenden Mediums zu ermitteln.

Abstract
Existent methods which connect quantum chemistry and statistical thermodynamics to predict phase equilibria were investigated in this work. The main focus of this work laid particularly on the application and further development of the COSMO-RS model. Five groups of topics were studied and new solutions were presented: 1. Validation of the COSMO-RS model regarding the relevant components in chemical
engineering and their binary mixtures. The validation showed that predictions of the binary vapour-liquid-equilibria of 136 systems were in good agreement with the experimental data.
2. The influence of the different conformers of a molecule on the prediction of the phase behaviour was investigated. By the example of the 2-butanol/water mixture it was shown that the conformer with the lowest energy not necessarily gives the best results. In fact it was essential to include all structures in the calculation that were found in a conformer search.
3. Highly branched molecules with a large molecular weight show a specific phase behaviour. An algorithm was developed to treat hyperbranched macromolecules quantum chemically by cutting the molecules into subunits. Furthermore the COSMO-RS model was extended by a Free-Volume term following the UNIFAC-Free-Volume model to account for volume effects. By the example of a polyglycerin macromolecule having 20 hydroxyl-groups and a molecular weight of 1400 g/mol the phase behaviour of the ternary mixture polyglycerin/ethanol/water could be modelled in good agreement with the experimental data.
4. The COSMO-RS model was combined with the Peng-Robinson equation of state by applying the Wong-Sandler mixing rules. The results demonstrated that predictions on the basis of the atomic structure of the molecules and the critical data can be carried out up to the supercritical region.
5. The energy concept of the COSMO-RS model was extended by a term including the change in Gibbs free energy explicitly, when a cavity in a solvent is created. A concept was developed which makes it possible to use the results of quantum chemical calculations to derive a hard-sphere-diameter of any molecule. Vapour pressures of different components of alkanes, alkenes, alcohols, aromatic compounds, ethers, ketones, and aldehydes were calculated with the extended COSMO-RS model and compared to experimental data at the normal boiling point. The deviations for alcohols were in the range of 20%. For the other classes of components those deviations were notably larger. The calculated vapour pressure sensitively responded to the different conformers of a component. Thus the vapour pressure would be a sensor which enables the identification of the relevant conformer of a molecule for calculations of the phase behaviour. Results for the 2-pentanol, 1-hexanol, and 1-octanol, showed that conformers which deliver a good description of the vapour pressure not in any case result in a good description of the phase behaviour. It can be pointed out that all conformers found in a conformer search should be used. Additionally it is recommended that conformers should be retrieved from a molecular dynamics calculation which includes the presence of the surrounding media.

1 Einleitung und Zielsetzung dieser Arbeit
1.1 Einleitung
Die Vorhersage thermodynamischer Eigenschaften ist noch immer eine große
Herausforderung für den Verfahrensingenieur. In der chemischen und pharmazeutischen
Industrie nimmt die Vorhersage der physikalischen Eigenschaften eine stetig wachsende
Bedeutung ein, da Produktzyklen immer kürzer werden und die systematische Suche nach
innovativen Stoffen und Substanzen allein mit experimentellen Methoden nicht wirtschaftlich
ist.
Gegenwärtig haben sich zwei verschiedene Methoden zur Vorausberechnung etabliert, die es
nach der Anpassung von Reinstoff- und binären Wechselwirkungsparametern anhand
experimenteller Daten gestatten, thermodynamische Eigenschaften im allgemeinen und
Phasengleichgewichte im besonderen zu berechnen bzw. vorherzusagen. Zustandsgleichungen
haben in den letzten Jahren mit der Entwicklung leistungsfähiger Fluidmodelle eine große
Weiterentwicklung erfahren [CHAPMAN, 1989; GROSS, 2001a] und ermöglichen die
zuverlässige Beschreibung dichter Fluide. Zustandsgleichungen versagen allerdings, wenn
keine Reinstoffparameter angepasst werden können. Auf der anderen Seite steht die Gruppe
der gE-Modelle, die seit der Entwicklung der Gruppenbeitragsmethode [FREDENSLUND,
1975b] die Extrapolation auf Stoffe und Stoffgruppen ermöglicht, für die es keine oder nur
begrenzte experimentelle Daten gibt. Alle Methoden setzen eine umfangreiche Datenbasis
voraus, um die Parameter auf ein extrapolationsfähiges Level zu heben, so dass komplizierte
und zugleich wenig untersuchte Stoffe und Stoffgruppen nicht oder nur mit begrenzter
Zuverlässigkeit berechnet werden können. Gruppenbeitragsmethoden versagen, wenn keine
entsprechenden Gruppen existieren. Sie erfassen weiterhin nicht die physikalischen
Eigenschaften von Strukturisomeren nicht.
Durch die fortschreitende Entwicklung der Rechentechnik ist es in den letzten Jahren möglich
geworden, physikalische Eigenschaften von komplexen Molekülen mit quantenchemischen
Methoden immer genauer zu beschreiben. Für die thermodynamischen Eigenschaften sind
insbesondere Modelle interessant, die die Wechselwirkungseigenschaften erfassen und
berechnen. Hier gab und gibt es große Fortschritte auf dem Gebiet der Kontinuumsmodelle
für Lösungen. Bei diesen Modellen liegt der Fokus auf dem gelösten Molekül. Das
umgebende Lösungsmittel betrachtet man als Kontinuum. Kontinuumsmodelle liefern vor
allem eine genaue Beschreibung der elektrostatischen Wechselwirkungen, indem die

2
elektrostatische Abschirmung ermittelt wird. Dispersive Wechselwirkungen werden über
einen oberflächenproportionalen Ansatz erfasst. Da die Molekülgeometrie bei diesen
quantenchemischen Methoden zwangsläufig ermittelt wird, fällt die Moleküloberfläche quasi
automatisch an und ermöglicht die Berechnung der dispersiven Wechselwirkungen.
Kontinuumsmodelle haben gegenüber den herkömmlichen Methoden zur Ermittlung von
Molekülwechselwirkungen den enormen Vorteil, dass die in ihnen enthaltenen Parameter
atombasiert sind und nur einmalig an thermophysikalische Messdaten angepasst werden
müssen. Sie werden in den Modellen zu Konstanten.
1.2 Zielsetzung
Das Ziel dieser Arbeit ist es, die vorhandenen Methoden der Verknüpfung von
Quantenchemie und statistischer Thermodynamik als Vorhersagemethode für die benötigten
Größen der Phasengleichgewichtsthermodynamik zu untersuchen und zu erweitern. Hierbei
liegt das Hauptaugenmerk auf dem COSMO-RS-Modell [KLAMT, 1995b]. Dieses Modell
verbindet die Ergebnisse quantenchemischer Rechnungen von molekularen Strukturen mit der
statistischen Thermodynamik und ermöglicht aus der atomaren Struktur von Molekülen auf
ihr Wechselwirkungsverhalten in Lösung zu schließen. In verschiedenen Publikationen ist die
Anwendung auf verschiedene Fragestellungen der chemischen Industrie untersucht worden
[DIEDENHOFEN, 2003; ECKERT, 2002; ECKERT, 2003; FRANKE, 2002; JORK, 2005].
In der vorliegenden Arbeit wird das COSMO-RS-Modell auf die Anwendbarkeit hinsichtlich
der Vorausberechnung von Dampf-Flüssig- und Flüssig-Flüssig-Phasengleichgewichten
untersucht. Hierbei wird eine große Anzahl an Komponenten und ihrer binären Mischungen
über einen weiten Temperatur- und Druckbereich betrachtet.
Aufbauend auf diesen Ergebnissen soll überprüft werden, inwieweit das Modell auch für die
Vorhersage von Wechselwirkungen stark verzweigter und mit einer großen Anzahl an
funktionellen Gruppen behafteter molekularer Strukturen, den hyperverzweigten Polymeren,
geeignet ist. Dafür ist zunächst eine Methode zu entwickeln, die es erlaubt, die
quantenchemischen Rechnungen von Polymeren durchzuführen. Eine komplette
quantenchemische Geometrieoptimierung der betrachteten Strukturen ist aufgrund der Größe
der Moleküle gegenwärtig nicht möglich. Weiterhin soll das COSMO-RS-Modell für die
Betrachtung der Polymer/Lösungsmittelsysteme um einen Einflussterm erweitert werden, der
es ermöglicht, die unterschiedliche Dichteänderung von Polymer und Lösungsmittel zu

3
erfassen. Hier wird in Anlehnung an das UNIFAC-Free-Volume-Modell [OISHI, 1978] der
Free-Volume-Anteil zusätzlich zu den enthalpischen und entropischen Anteilen des
Aktivitätskoeffizienten im COSMO-RS-Modell implementiert.
Eine wichtige Einflussgröße bei der Berechnung von physikalischen Stoffeigenschaften aus
der molekularen Struktur stellt die Konformation eines Moleküls dar. In der vorliegenden
Arbeit soll untersucht werden, wie sich die verschiedenen Konformationen einer
Molekülstruktur auf den Aktivitätskoeffizienten in der Mischung auswirken und mit welchen
Methoden dem Anwender diese Konformationen zugänglich sind.
Das COSMO-RS-Modell liefert den Aktivitätskoeffizienten einer Komponente in der
Mischung. Damit sind der Anwendung bei der Berechnung von Phasengleichgewichten
Grenzen gesetzt, die sich aus den Phasengleichgewichtsbeziehungen ergeben. Die
Modellierung von Hochdruckphasengleichgewichten, wenn eine oder mehrere Komponenten
der Mischung überkritisch sind, ist mit dieser Methode nicht möglich, da es schwierig ist,
einen Standardzustand für überkritische Komponenten zu definieren. In dieser Arbeit soll auf
der Grundlage von Mischungsregeln für Parameter von Zustandsgleichungen, die auf der
Gleichheit von freien Exzessenthalpien der Zustandsgleichung und einem GE-Modell
aufbauen, das COSMO-RS-Modell verwendet werden, mit dem Ziel Mischungsparameter für
eine kubische Zustandsgleichung vorherzusagen.
Ein weiterer Schwerpunkt dieser Arbeit liegt auf der Erweiterung des Energiekonzeptes des
COSMO-RS-Modells um einen Term, der die Änderung der freien Enthalpie der Lösung
durch die Bildung einer Kavität im Lösungsmittel explizit enthält. Hierbei wird auf die
Arbeiten von Reiss [REISS, 1965] zurückgegriffen. Mit der von ihm entwickelten Scaled-
Particle-Theorie (SPT) lässt sich die Arbeit ermitteln, die notwendig ist, eine harte Kugel in
ein Fluid harter Kugeln einzufügen. Es wird dargelegt, wie sich aus dieser Arbeit die
Änderung der freien Enthalpie der Lösung darstellen lässt. Mit dem erweiterten
Energiekonzept sollen dann Reinstoffeigenschaften wie der Dampfdruck einer Komponente
abgeleitet werden. Weiterhin soll untersucht werden, ob sich aus den Ergebnissen für die
Reinstoffeigenschaften Regeln ableiten lassen, die zur Auswahl einer geeigneten
Molekülkonformation für die Mehrkomponentenphasengleichgewichte führen.

2 Grundlagen der Thermodynamik und der Quantenchemie
Thermodynamische Modelle für die zuverlässige Berechnung von Stoffeigenschaften stehen
dem Verfahrensingenieur heute zur Verfügung, wenn für die betrachteten Komponenten eine
breite Datenbasis vorhanden ist. Für die Vorhersage, insbesondere des
Wechselwirkungsverhaltens von Komponenten in Mischungen, bei geringer oder nicht
vorhandener Datenbasis bedarf es Methoden, die von der Struktur der Moleküle auf ihr
Wechselwirkungspotenzial schließen lassen. In diesem Kapitel wird das COSMO-RS Modell
[KLAMT, 1995a; KLAMT, 1998; KLAMT, 2000] vorgestellt, welches quantenchemische
Methoden in Kombination mit Ansätzen der statistischen Thermodynamik verwendet, um das
chemische Potenzial einer Komponente aus ihrer atomaren Struktur vorherzusagen. Im ersten
Unterkapitel 2.1 werden die betrachteten Methoden eingeordnet und von anderen
Entwicklungssträngen zur Vorausberechnung von Stoffeigenschaften abgegrenzt.
Unterkapitel 2.2 führt in die später benötigten Ansätze aus der Quantenchemie ein. Die
Verknüpfung von statistischen Methoden der Thermodynamik und der Quantenchemie wird
im Unterkapitel 2.3 am Beispiel des COSMO-RS Modells erläutert. Unterkapitel 2.4
diskutiert, wie Anzahl und Zustände koexistierender Phasen erfasst werden. Da sich ein Teil
der vorliegenden Arbeit mit dem Phasenverhalten am Beispiel von hyperverzweigten
Polymeren beschäftigt, werden deren Besonderheiten im Phasenverhalten in Unterkapitel 2.5
dargestellt. In Unterkapitel 2.6 werden die Grundlagen zur Verknüpfung von
Zustandsgleichungen mit Modellen für die Gibbssche Exzessenthalpie (GE-Modelle)
dargelegt.
2.1 Vorbemerkung zu den betrachteten Modellen und Methoden
Unter der Vorausberechnung von Stoffeigenschaften hat man in der phänomenologischen
Thermodynamik bis vor wenigen Jahren vor allem deren Abschätzung durch Korrelationen
erweiterter Theorien verstanden. Beispiele dafür sind Mischungsregeln für
Zustandsgleichungen oder die Anwendung von Gruppenbeitragsmethoden.
Gruppenbeitragsmethoden bilden eine große Anzahl von Molekülen auf eine kleine Anzahl
von Strukturgruppen ab. Vor allem das UNIFAC Modell [FREDENSLUND, 1975a;
FREDENSLUND, 1989] für die Berechnung der Aktivitätskoeffizienten von Komponenten in
Mischungen liefert zur Abschätzung und Vorhersage des realen Verhaltens der flüssigen
Phase für unterschiedlichste Stoffe gute Ergebnisse. Oft wird der Begriff Abschätzung und

5
Vorhersage synonym verwendet, wobei ersterer impliziert, dass er sich auf beobachtete bzw.
gemessene Daten stützt und das Ergebnis möglicherweise nur annähernd richtig sein kann.
Eine Vorhersage impliziert in jedem Fall, dass es sich um eine Berechnung bzw. Ableitung
handelt, die sich nicht auf bekannte Daten stützt.
Mit der fortschreitenden Entwicklung der Rechenkapazität ist es in den letzten Jahren
gelungen, das reale Verhalten von Reinstoffdaten und Mischungseigenschaften durch
Molekularsimulationen abzubilden und komplexe Wechselwirkungseffekte zu
berücksichtigen. Eine wichtige Stellung nimmt hierbei die Quantenmechanik ein, die
mögliche Energiezustände von mikroskopischen Systemen liefert. Sie wird in Zukunft zum
Bindeglied zwischen den molekularen Modellen und den beobachtbaren Größen werden und
sowohl in der statistischen wie auch in der phänomenologischen Thermodynamik
Modellparameter liefern. Mit ihr können für Molekularsimulationen notwendige Parameter
für Dipsersion, Kugeldurchmesser, Dipol- oder Quadrupolwechselwirkung sowie die
Positionen der Atomkerne berechnet werden [VRABEC, 2002]. In der phänomenologischen
Thermodynamik bietet die Quantenmechanik die Möglichkeit, Parameter für Modelle wie
UNIQUAC vorherzusagen [SANDLER, 2002; SUM, 1999].
Die Vorhersage von Phasengleichgewichten in Mischungen kann auf mehreren Wegen
erfolgen: Für Systeme bei moderaten Drücken und unterhalb der kritischen Punkte der
beteiligten Reinstoffe haben sich zur Berechnung Aktivitätskoeffizientenmodelle etabliert.
Mit der Entwicklung des UNIFAC Modells [FREDENSLUND, 1975a; FREDENSLUND,
1989] zur Vorhersage des Aktivitätskoeffizienten einer Komponente in einer Mischung ist es
möglich geworden, das reale Verhalten unbekannter Substanzen in der flüssigen Phase
vorherzusagen. Dies gelingt allerdings nur, wenn die Substanz aus den Strukturgruppen der
UNIFAC Matrix [WITTIG, 2003] zusammengesetzt werden kann. Bei hohen Drücken und
bei Überschreiten des kritischen Punktes der Komponenten finden Zustandsgleichungen
Anwendung. Die als thermische Zustandsgleichungen bezeichneten, liefern einen
mathematischen Zusammenhang zwischen dem Druck P, der Temperatur T und dem
bezogenen Volumen v bei vorgegebener Zusammensetzung. Ihre Zahl liegt seit der ersten
Veröffentlichung von van der Waals im Jahre 1873 heute bei mehr als Tausend. Bis es Mitte
der fünfziger Jahre des 20. Jahrhunderts zur Anwendung von leistungsfähigen Fluidtheorien
kam, waren es insbesondere semiempirisch entwickelte, kubische Zustandsgleichungen, die
bei der Modellierung des Phasenverhaltens von Mischungen angewendet wurden [PENG,
1976; SOAVE, 1972]. Insbesondere zur Beschreibung des Verhaltens von assoziierenden
Komponenten und Makromolekülen waren diese Gleichungen nicht ausreichend

6
leistungsfähig [DOHRN, 1994]. Der Ansatz, Mischungsregeln für Zustandsgleichungen auf
der Basis von Aktivitätskoeffizientenmodellen vorherzusagen und die Zustandsgleichung
damit zu einem Vorhersagemodelle für Mischungen weiterzuentwickeln [[HURON, 1979;
MICHELSEN, 1990b; VIDAL, 1978; WONG, 1992b], konnte diesen Nachteil größtenteils
aufheben.
2.2 Grundlagen der Quantenchemie
Unter dem Begriff Quantenchemie fasst man die die Anwendung der Quantenmechanik auf
chemische Probleme zusammen. Im Gegensatz zur klassischen Mechanik, in der Funktionen
beobachtbare Größen abbilden, werden in der Quantenmechanik die beobachtbaren Größen
durch Operatoren repräsentiert. Operatoren sind Anweisungen, eine bestimmte Aktion an
einer Funktion durchzuführen, beispielsweise eine Multiplikation oder eine Differentiation.
Die Quantenmechanik beruht auf einer Reihe von Postulaten, von denen das wichtigste
besagt, dass die vollständige Zustandsinformation in einer Funktion ),r,...,r,(r 21 tnΨ , der
Wellenfunktion, enthalten ist. Hierbei sind ri die Ortsvektoren der N Teilchen eines Systems
und t die Zeit. Ist die Wellenfunktion Eigenfunktion des Hamiltonoperators, dem Operator für
die Gesamtenergie des Systems, ist der dazugehörige Eigenwert die Energie des Zustandes, in
dem sich das System befindet. Das Quadrat der Wellenfunktion kann als Wahrscheinlichkeit
aufgefasst werden, dass sich Teilchen in einem differentiellen Volumenelement aufhalten.
Das Hauptaugenmerk der Quantenchemie richtet sich auf die Lösung der zeitunabhängigen
Schrödingergleichung und dabei insbesondere auf die Berechnung der Struktur von Atomen
und Molekülen. Die zeitunabhängige Lösung ist insofern ausreichend, da es sich in aller
Regel um stationäre Zustände der beschriebenen Moleküle handelt (z.B. Kernkonfiguration,
Bildungsenergie, Dipolmoment). Bei Vernachlässigung relativistischer Effekte ist die
zeitunabhängige Form der Schrödingergleichung [SCHRÖDINGER, 1926] durch folgenden
Zusammenhang gegeben:
Ψ=Ψ EH 2.1
Neben der Vernachlässigung relativistischer Effekte macht die Vernachlässigung von
Wechselwirkungen, welche mit dem Spin der Teilchen zusammenhängen, die
Schrödingergleichung zu einer Näherung. Vergleicht man Berechnungen, die auf dieser
Näherung beruhen, mit experimentellen Daten, kann sie im Rahmen der Untersuchung von
Molekülstrukturen als genau angenommen werden. Gleichung 2.1 enthält auf der linken Seite
den Hamilton Operator mit zwei Anweisungen:

7
a) die Wellenfunktion bezüglich der Raumkoordinaten zweimal in jede der
Raumrichtungen zu differenzieren und somit den Impuls der Teilchen – ihre kinetische
Energie – zu berücksichtigen
b) die Wellenfunktion mit der potenziellen Energie zu multiplizieren.
),,(2
ˆ2
2
2
2
2
2
22
zyxuzyxm
H +
∂
∂+
∂
∂+
∂
∂−=h
2.2
Auf der rechten Seite der Gleichung 2.1 steht der zugehörige Eigenwert E der Funktion Ψ . Er
kennzeichnet die Energie des Systems beschrieben durch Ψ . Löst man Gleichung 2.1 unter
Berücksichtigung von 2.2, erhält man als Lösung die Eigenfunktionen iΨ (Quantenzustand
des Systems) und die Eigenwerte Ei (Energieniveaus dieses Zustandes i) des Operators H .
Es ist nahezu unmöglich eine exakte Formulierung für die Wellenfunktionen zu finden. Kein
Teilchen führt Bewegungen aus, die unabhängig von anderen Teilchen sind. Eine
geringfügige, aber wesentliche Vereinfachung der Schrödingergleichung konnte mit
Anwendung der Born-Oppenheimer Approximation eingeführt werden [BORN, 1927]. Dabei
werden die Bewegungen von Kernen und Elektronen in Molekülen entkoppelt. Dies kann
geschehen, da die Masse eines Atomkerns erheblich größer ist als die der Elektronen. Beim
Wasserstoffproton, dem leichtesten aller Kerne, liegt ein Faktor von 1800 vor; bei einem
Kohlenstoffkern liegt das Verhältnis bei etwa 20000.
Mit guter Näherung können die Bewegungen der Kerne vernachlässigt werden, ihre
kinetische Energie ist Null – sie befinden sich an fixierten Positionen, und nur ihre repulsive
Wechselwirkung trägt zur Energie des Systems als additive Konstante bei. Die Anwendung
der Born-Oppenheimer Approximation führt zur elektronischen Schrödingergleichung. Diese
kann für Systeme mit 2 Teilchen exakt berechnet werden. Für Mehrteilchen- bzw. für
Mehrelektronensysteme müssen Näherungsverfahren angewendet werden, die die
Bestimmung der Wellenfunktion und der Lösung der elektronischen Schrödingergleichung
ermöglichen.
2.2.1 Ab-initio und semi-empirische Methoden
Je nach Tiefe der Näherung unterscheidet man zwischen ab-initio und semiempirischen
Verfahren. Ab-initio Verfahren verwenden zur Lösung der Schrödingergleichung keine
andere Information als die Werte der Fundamentalkonstanten und die Kernladungen der
beteiligten Atome. Semiempirische Verfahren beziehen experimentelle Informationen in die
Lösungsverfahren ein. Ein Beispiel für ein ab-initio Verfahren ist die so genannte Hartree-

8
Fock-Näherung (HF) [SZABO, 1989]. Hier wird die Schrödingergleichung mit Hilfe von
genäherten Wellenfunktionen (Slaterdeterminanten) iterativ gelöst, um die Energie des
Systems zu bestimmen. Näherungsweise lässt sich ein herausgegriffenes Elektron so
behandeln, als würde es sich in einem Feld der positiven Kerne und der negativen
Ladungswolke der übrigen Elektronen bewegen, wobei die Bewegungen der übrigen
Elektronen ohne Einfluss auf die Bewegung des betreffenden Elektrons sind. Die
Mehrelektronenwellenfunktion wird in Einelektronenwellenfunktionen separiert. Die
entstehenden Einelektronenwellenfunktionen werden (Spin-)Orbitale genannt. Die
Molekülorbitale werden als Linearkombinationen der Atomorbitale gebildet. Die einzelnen
linear unabhängigen Funktionen werden als Basissatz bezeichnet. Für eine korrekte
Repräsentation der Molekülorbitale ist eine unendliche Anzahl an Funktionen erforderlich.
Mit einer endlichen Anzahl erreicht man eine näherungsweise Beschreibung der
Wellenfunktion.
In der HF-Näherung wird die Korrelationsbewegung der Elektronen vernachlässigt. Die so
genannten post-HF Modelle besitzen nachgeschaltete Korrekturalgorithmen, die z.B. eine
Linearkombination aus Slaterdeterminaten verwenden [ROOS, 1992] oder die Wellenfunktion
als exponentiell entwickelte Reihe darstellen (Coupled Cluster) [BARTLETT, 1989].
Die Anwendung der HF-Näherung bedeutet noch immer einen großen Rechenaufwand. Mit
den so genannten semiempirischen Methoden, die auf demselben Grundgerüst wie die ab-
initio Methoden basieren, werden Näherungen eingeführt, die den Rechenaufwand erheblich
reduzieren. Die wird zum einen erreicht, indem ausschließlich Valenzelektronen in die
Berechnungen einbezogen werden und für diese dann ein minimaler Basissatz verwendet
wird. Zum anderen besteht ein zentraler Ansatz semiempirischer Verfahren in der
Vernachlässigung differenzieller Überlappungen. Hier wird das Produkt zweier Atomorbitale
zu Null gesetzt, wenn sie verschiedenen Atomen angehören. Die Verfahren unterscheiden sich
in der kompletten oder teilweisen Vernachlässigung der Überlappungen. Verbleibende
Überlappungsintegrale werden aus experimentellen Daten wie Bildungsenthalpien oder
Molekülgeometrien abgeschätzt. Die bekanntesten Vertreter sind MNDO [DEWAR, 1977],
AM1 [DEWAR, 1985] und PM3 [DYMEK, Jr., 1989].
2.2.2 Dichtefunktionaltheorie
Quantenmechanische Berechnungen basieren zu allererst auf der Wellenfunktion. Wenn
allerdings nach einer beobachtbaren Größe gefragt wird, dann erscheint die Wellenfunktion in
den Berechnungen in Form ihres Quadrats, einem Erwartungswert bzw. einer

9
Wahrscheinlichkeitsdichte. Die Elektronendichte wird direkt durch das Quadrat der
Wellenfunktion ausgedrückt. Diese Eigenschaft wird in der Dichtefunktionaltheorie (DFT)
angewendet. Die Dichtefunktionaltheorie beschreibt die Energie eines Atoms oder Moleküls
durch die Elektronendichte ρe(r), genauer durch ein Energiefunktional E(ρe(r)), das
ausschließlich von der Elektronendichte abhängig ist. Die DFT bietet den Vorteil, dass die
verwendete Elektronendichte eine Funktion im dreidimensionalen Raum ist, während die
Wellenfunktion der Schrödingergleichung 3n dimensional ist.
Die Grundlage der heute bekannten und angewendeten zeitunabhängigen
Dichtefunktionaltheorie (DFT) bilden die beiden Hohenberg-Kohn Theoreme (1964) [KOCH,
2002]. Das erste Hohenberg-Kohn Theorem rechtfertigt den Einsatz der Elektronendichte als
Basisvariable der DFT. Hohenberg und Kohn haben bewiesen, dass die Energie eines
quantenchemischen Systems exakt aus der Elektronendichte im Grundzustand des Atoms oder
Moleküls berechnet bzw. als Funktional der Elektronendichte ausgedrückt werden kann. Der
Grundzustand, gekennzeichnet durch die geringste Gesamtenergie, ist vom externen Potenzial
Vext, d.h. von den positiven Kernladungen, abhängig. Daraus folgt, dass die kinetische Energie
der Elektronen, die Elektron-Elektron Wechselwirkungen und die Elektronendichte ρe(r)
durch das externe Potenzial festgelegt werden bzw. dass sie sich in einem externen Potenzial
so „einstellen“, dass das Gesamtsystem im Zustand der geringsten Energie ist. Koch et al.
[KOCH, 2002] und Parr et al. [PARR, 1989] bewiesen dieses Theorem und zeigten, dass es
keine verschiedenen externen Potenziale gibt, die zur gleichen Elektronendichte des
Grundzustandes führen. Die Gesamtenergie des Systems kann in einen systemabhängigen und
einen universal gültigen Anteil, das Hohenberg – Kohn Funktional FHK, zerlegt werden:
[ ] [ ] [ ] [ ][ ]0
0 0 0 0 0
,HK
Ne ee
systemabhängig F
systemunabhängig
E E T E
ρ
ρ ρ ρ ρ
=
= + +14243 1442443
2.3
Das zweite Hohenberg-Kohn Theorem besagt, dass die Elektronendichte des Grundzustandes
mittels Variationsrechnung exakt berechnet werden kann. Für jede Elektronendichte ρ’(r)
eines n-Elektronensystems in einem externen Potenzial Vext gilt:
( )[ ] ( )[ ] ( )[ ] ( )[ ]r'r'r'r'0 ρρρρ eeNe ETEEE ++=≤ 2.4
Hierbei sind ENe die elektrostatischen attraktiven Kern – Elektron Wechselwirkungen und Eee
die repulsiven Elektron – Elektron Wechselwirkungen.
1965 stellten Kohn und Sham [KOCH, 2002] einen Weg vor, mit dem eine Annäherung an
das unbekannte universelle Funktional möglich wird. Mit der Erkenntnis, dass die
Beschreibung der kinetischen Energie nicht durch ein explizites Dichtefunktional möglich ist,

10
schlugen Kohn und Sham die Verwendung eines nicht wechselwirkenden Referenzsystems
vor. Die kinetische Energie des realen Systems wird durch die kinetische Energie eines nicht
wechselwirkenden Ein-Teilchensystems TS(ρ), das durch Orbitalfunktionale beschrieben
werden kann, und einen Korrekturterm, der die Unterschiede zwischen der realen kinetischen
Energie T und dem Ein-Teilchensystems TS umfasst, ersetzt.
( )[ ] ( )[ ] ( )[ ]rrr ρρρ CS TTT += 2.5
Der unbekannte Term der Elektron-Elektron Wechselwirkungen des Hohenberg-Kohn
Funktionals kann ebenfalls in bekannte und unbekannte Anteile zerlegt werden. Er setzt sich
zusammen aus dem Term der klassischen Wechselwirkungen zwischen zwei Elektronen EJ
und einem Korrekturterm EX, der alle nichtklassischen Anteile beinhaltet.
( )[ ] ( )[ ] ( )[ ]rrr ρρρ XJee EEE += 2.6
Durch Zusammenfassen der beiden Korrekturterme TC und EX zu
( )[ ] ( )[ ] ( )[ ]rrr ρρρ XCXC ETE += 2.7
und Einsetzen von Gl. 2.5 und Gl. 2.6 in das erste Hohenberg-Kohn Theorem (Gl. 2.3) ergibt
sich
S V J XCE T E E E= + + + 2.8
Der erste Term TS repräsentiert die kinetische Energie der Elektronen, der Zweite EV die
Wechselwirkungsenergie zwischen Elektronen und dem Atomkern. Der dritte Term EJ
berücksichtigt die Wechselwirkungen zwischen den Elektronen, unter der Annahme, dass sie
sich im elektrischen Feld des Atomkerns unabhängig voneinander bewegen. Der vierte Term
EXC stellt einen Korrekturterm dar, weil sich Elektronen im Ladungsfeld des Atomkerns
abhängig voneinander bewegen und abstoßende Effekte der Elektronen untereinander zur
Änderung der kinetischen Energie führen. Der bisher unbekannte Anteil der kinetischen
Energie TS wird mit Hilfe der HF-Theorie berechnet. Für den hier unbekannten Korrekturterm
EXC sind in der Literatur verschiedene Näherungen beschrieben [KOCH, 2002].
Die DFT hat sie sich als Standardmethode zur Berechnung von Molekülgeometrien und
Elektronendichten von Molekülen durchgesetzt. Für weiterführende Informationen zur DFT
und der enthaltenen Näherungen sei auf die Veröffentlichungen von Koch und Holthausen
[KOCH, 2002], Parr und Yang [PARR, 1989], Dreizler und Gross [DREIZLER, 1990]
hingewiesen.

11
2.2.3 Kontinuumsmodelle – Moleküle in Lösung
Mit den in den Kapiteln 2.3.1 und 2.2.2 vorgestellten Methoden betrachtet man in aller Regel
ein einzelnes Molekül in der Gasphase. Dies ist zwar ausreichend für die Berechnung der
Molekülstruktur, Wechselwirkungen in kondensierten Medien können so aber nicht
beschrieben werden. Hierfür bietet sich das Konzept der impliziten Berücksichtigung von
Molekülen an, die ein gelöstes Molekül umgeben. Diese umgebenden Molekülen werden als
Kontinuum aufgefasst und die Reaktion des gelösten Moleküls auf das umgebende
Kontinuum modelliert. Die fundamentale Größe zur Beschreibung des Lösungsvorganges ist
die Freie Enthalpie des Lösungsvorganges ∆Gsolv. Sie drückt den Unterschied der Freien
Enthalpie eines Moleküls aus, das aus der Gasphase in eine kondensierte Phase übergeht.
Ben-Naim [BEN NAIM, 1987] definiert den Lösungsvorgang wie folgt: Geht ein Molekül bei
konstanter Temperatur und konstantem Druck von einer festen Position aus der Gasphase an
eine feste Position in der fluiden oder flüssigen Phase, so nennt man diesen Vorgang
Solvation. Für theoretische Betrachtungen kann es sinnvoll sein, den Vorgang bei konstantem
Volumen anstatt bei konstantem Druck zu betrachten. Nach Ben-Naim gibt es bezüglich der
Konzentration der gelösten Substanz keine Beschränkungen. Diese kann von unendlicher
Verdünnung bis zur stark konzentrierten Lösung reichen, selbst das Lösen einer Komponente
i in sich selbst wird abgedeckt. Darüber hinaus werden keine Annahmen bezüglich der
Struktur der flüssigen Phase gemacht. Damit unterscheidet sich die Definition des
Lösungsvorganges von der in der Chemie üblichen, die nur den Bereich der stark verdünnten
Lösung abdeckt, da dort von der vollständigen Abschirmung des gelösten Moleküls vom
Lösungsmittel ausgegangen wird.
Die Änderung der freien Enthalpie G setzt sich aus zwei Anteilen zusammen:
1. Einfügen von Teilchen in das System an eine feste Position (Lösungsvorgang nach Ben-
Naim)
2. Aufheben der Beschränkung der festen Position
Das chemische Potential der Komponente i für den Lösungsvorgang kann danach formuliert
werden als:
3* ln iiii kT Λ+= ρµµ (2.9)
wobei *iµ das pseudochemische Potential der Komponente i ist, welches dadurch
gekennzeichnet ist, dass die Teilchen in ihrer Position fixiert sind. Der zweite Term auf der
rechten Seite der Gleichung stammt aus der klassischen statistischen Mechanik und beschreibt
die Differenz des chemischen Potentials von fixierten und „freien“ Teilchen. Er enthält die

12
Teilchenzahldichte ρi der Komponente i, die Boltzmann-Konstante k, die absolute Temperatur
T und 3iΛ ist die Zustandssumme des Impulses. 3
iΛ ist definiert als:
3/2
33
)2( mkT
hi
π=Λ (2.10)
Hier ist h das Plancksche Wirkungsquantum. Λ hat die Dimension einer Länge und wird auch
als de-Broglie Wellenlänge oder thermische Wellenlänge bezeichnet.
Diese Definition des Lösungsvorganges und der damit verbundenen Energieänderung des
Systems hat sich in der Thermodynamik von Lösungsvorgängen durchgesetzt [TOMASI,
1994].
Für den oben definierten Prozess kann die Änderung der freien Enthalpie des
Lösungsprozesses ∆G*sol wie folgt definiert werden:
eldisprepcavsol
GGGGG***** ∆+∆+∆+∆=∆ (2.11)
Dabei haben die einzelnen Terme folgende Bedeutung:
1. ∆G*cav ist die Änderung der freien Enthalpie, die durch das Schaffen eines Hohlraumes
(Kavität) im Lösungsmittel für die gelöste Substanz entsteht.
2. ∆G*rep
ist die Änderung der freien Enthalpie aufgrund repulsiver Wechselwirkungen.
3. ∆G*disp
ist die Änderung der freien Enthalpie aufgrund von ungerichteten, dispersiven
Wechselwirkungen.
4. ∆G*el ist die Änderung der freien Enthalpie aufgrund von elektrostatischen
Wechselwirkungen.
Kontinuumsolvensmodelle beschreiben den letzten Term ∆G*el explizit. Das Kontinuum ist
ein homogenes, isotropes elektrisches Feld, charakterisiert durch eine einzige skalare Größe,
der Dielektrizitätskonstanteε . Die Grundlagen für diese Klasse von Modellen wurden von
Born [BORN, 1920], Onsager [ONSAGER, 1936] und Kirkwoord [KIRKWOOD, 1939]
gelegt. Die Wechselwirkung eines gelösten Teilchens mit dem Lösungsmittel hängt
signifikant von seiner Ladungsverteilung und Polarisierbarkeit ab. Die Ladungsdichte des
gelösten Teilchens wird in aller Regel mit quantenmechanischen Methoden ermittelt. Das
Kontinuum wird oft auch „Reaktionsfeld“ genannt, da es eine Reaktion auf das gelöste
Teilchen hervorruft. Dieses Reaktionsfeld wird im Hamilton-Operator des gelösten Teilchens
eingebunden und ändert damit die elektronische Struktur. Dies wiederum ändert die
Polarisierung des Lösungsmittels. Die elektronische und geometrische Struktur des gelösten
Teilchens wird durch eine Iteration bis zur Selbstkonsistenz ermittelt (self consistent reaction
field, SCRF). Als Starwerte für die Iteration werden Punktladungen auf der Oberfläche des

13
Moleküls definiert. Das daraus resultierende Potenzial des Moleküls und des Reaktionsfeldes
werden in den Hamiltonoperator eingebunden. Aus der Wellenfunktion werden damit neue
Werte für die Oberflächenladungen bis zur Selbstkonsistenz berechnet.
Alle Kontinuumsmodelle definieren eine Kavität, in der sich das Teilchen befindet. Das
Volumen und Form dieser Kavität ist eine entscheidende Größe. Ist sie zu groß, wird der
Effekt des Kontinuums gedämpft, ist sie zu klein, treten Fehler in der Bestimmung der
Elektronendichte für Atome nahe der Kavitätsgrenze auf. Ist Größe und Form der Kavität
bestimmt, kann das elektrostatische Problem gelöst werden. Relativ einfach ist dies für
sphärische Kavitäten, bei denen man mit der Poisson-Gleichung der klassischen Elektrostatik
das elektrostatische Potenzial mit einer geschlossenen Funktion angeben kann. Die
Polarisationsenergie, die proportional zur Freien Enthalpie ist, erhält man aus dem
Volumenintegral über der Ladungsdichte und dem elektrostatischen Potenzial. Bei den
bekanntesten Kontinuumsmodellen, dem Polarized Continuum Model (PCM) [MIERTUS,
1981] und dem Conductor-like Screening Model (COSMO) [KLAMT, 1993] hat sich ein
Weg etabliert, der eine frei geformte Kavität konstruiert, indem überlappende Sphären um die
Atome des Moleküls gelegt werden, die einen etwa 20% größeren Durchmesser haben als die
van-der-Waals-Radien der entsprechenden Atome. Bei dieser Art von Kavitäten hat sich ein
anderes Vorgehen zur Lösung des elektrostatischen Problems etabliert. Die Poisson-
Gleichung, bzw. die Poisson-Boltzmann-Gleichung im Falle von Elektrolyten, wird nicht im
dreidimensionalen Gitter der Kavität gelöst, sondern das Problem auf die Grenzfläche
Kavität-Kontinuum abgebildet. Aus der Volumenladungsdichte wird eine
Oberflächenladungsdichte. Sowohl das oben angesprochene PCM-Modell, als auch das
COSMO-Modell basieren auf diesem Ansatz. Da das umgebende dielektrische Kontinuum als
unendlich ausgedehnt betrachtet wird, ist für die Wechselwirkung zwischen gelöstem
Teilchen und Kontinuum nur die Ladungsdichte an der Grenzfläche relevant, wiedergegeben
durch die Oberflächenladungsdichte σ(r). Die Genauigkeit der Berechnungen hängt von der
Feinheit der Einteilung der Oberfläche in Segmente ab. Die Ladungsdichte σ(r), am Punkt r
auf der Kontaktoberfläche ist eine Funktion des Normalenvektors n(r) auf der Oberfläche und
der dort herrschenden Feldstärke E(r) sowie der Dielektrizitätskonstante ε des Kontinuums
(dielektrische Randbedingung).
(r)n(r))1((r)4 E−= επεσ (2.12)
Im COSMO-Modell haben Klamt und Schüürmann das Kontinuum als perfekten Leiter mit
der Dielektrizitätskonstante ε=∞ betrachtet. Jedes Segment der Kontaktoberfläche wird einem

14
Atom explizit zugewiesen. Dem realen Lösungsmittel, bzw. seinem dielektrischen Ersatz,
wird durch Skalierung der Abschirmungsladungen des perfekten elektrischen Leiters
Rechnung getragen. Diese Skalierung beruht auf der von Kirkwood [KIRKWOOD, 1939] für
Dipole abgeleiteten Skalierungsfunktion:
211
)(+
−=
ε
εεlf
(2.13)
Der Vorteil des COSMO Ansatzes liegt in der mathematischen Vereinfachung, die eine
direkte Integration des abschirmenden Kontinuums in den Hamilton Operator, bzw. bei DFT
Rechnungen in das Energiefunktional, erlaubt. Damit ist es möglich, die Geometrie und die
Abschirmungsladungen eines Moleküls auf quantenchemisch hohem Niveau zu ermitteln.
Eine COSMO-Rechnung liefert in Anwesenheit des abschirmenden Lösungsmittels die
selbstkonsistente Geometrie einer Molekülstruktur, die dielektrische Abschirmungsenergie ∆ ,
die Ladungsdichten der in Segmente eingeteilten Teilchenoberfläche und die molekulare
Oberfläche A. Die dielektrische Abschirmungsenergie wird berechnet als Differenz des
Energiegehalts des Solvatmoleküs im perfekten elektrischen Leiter und im Vakuum und ist
ein Maß für die elektrostatischen Wechselwirkungen während des Solvatationsvorganges.
Über die Oberfläche lassen sich dispersive Wechselwirkungen beschreiben.
2.3 Das COSMO-RS Modell
Eine Erweiterung des COSMO-Modells durch Klamt [KLAMT, 1995a] ermöglicht die
Berechnung des chemischen Potenzials einer Komponente zu beliebig vielen anderen
Komponenten, d.h. in Mischungen. In dieser Erweiterung – COSMO-RS (Conductor-like
Screening Model for Real Solvents) werden Moleküle als Ansammlung von paarweise
wechselwirkenden Oberflächensegmenten behandelt. Die Wechselwirkungen der
Oberflächensegmente, die aus den COSMO-Rechnungen stammen, gehen in einem
selbstkonsistenten Ausdruck für das chemische Potenzial ein, der aus der statistischen
Thermodynamik abgeleitet ist, und ermöglichen so z.B. die Berechnung des
Aktivitätskoeffizienten einer Komponente. Zunächst werden alle beteiligten Komponenten
einer Mischung durch die auf ihrer Oberfläche ausgebildeten Abschirmungsladungsdichten,
die sich bei Einbettung in einen idealen Leiter einstellen würden, charakterisiert. Die
Häufigkeitsverteilung p(σ) der auf der Moleküloberfläche vorhandenen Ladungsdichten σ
charakterisiert jede Komponente eindeutig.
Mischungen können dargestellt werden, wenn die Verteilungsfunktion der Ladungsdichten

15
entsprechend der Zusammensetzung der Mischung gewichtet wird.
∑=i
ii pxp )()( σσ (2.14)
Das Integral von )(σip über den gesamten σ-Bereich ist die Gesamtoberfläche Ai der
Komponente i. Damit lässt sich ein normalisiertes σ –Profil angeben:
∑
==
i
ii Ax
p
A
pp
)()()('
σσσ (2.15)
Ausgehend vom Zustand des im idealen Leiter gelösten Moleküls, das als Ensemble von
Oberflächensegmenten vorliegt, kann das chemische Potenzial einer Komponente i abgeleitet
werden. In diesem Zustand hat die Komponente i die Energie des Systems S schon um die
Beiträge des Transportes aus der idealen Gasphase und – teilweisen – Etablierung in der
flüssigen Phase geändert. Diese Beiträge sind ∆G*cav, ∆G*
rep und ∆G*disp.
Das chemische Potenzial der Oberflächensegmente einer Komponente wird von Klamt aus
der Zustandssumme des Ensembles von N elektrisch geladenen Oberflächensegmenten
hergeleitet [KLAMT, 1995a].
−−= ∫ RT
eapRT
eff )',()'(exp)'('dln)(
σσσµσσσµ (2.16)
Der Ausdruck )(σµ erscheint auf beiden Seiten der Gleichung, weshalb diese iterativ gelöst
werden muss. Mit dem Startwert 0)( ≡σµ konvergiert sie schnell.
Zusammenfassend kann für das chemische Potenzial einer Komponente i folgende Gleichung
angegeben werden:
comb
iii
iS
i A µσµδµ ++−∆= )( (2.17)
Ein Vergleich mit Gleichung 2.11 zeigt, dass mit dem COSMO-RS Modell zwei Terme der
freien Enthalpie des Lösungsvorganges explizit erfasst sind, zum einen der
oberflächenproportionale Dispersionsanteil und zum anderen der elektrostatische Anteil. Der
in Gleichung 2.17 als µcomb bezeichnete kombinatorische Anteil nach Staverman-Guggenheim
berücksichtigt die verschiedene Größe und Gestalt der Moleküle. Klamt et al. haben für die
Methode Parameter mit konkreter physikalischer Bedeutung durch Anpassung an
experimentell bestimmte Gleichgewichtsdaten bestimmt. Die ausgewählten Stoffe decken
einen großen Bereich der häufigsten funktionellen Gruppen bestehend aus den Elementen H,

16
C, N, O und Cl ab. Es sind acht universelle sowie zwei elementspezifische Parameter
angepasst worden, die, einmal festgelegt, für alle beliebigen aus den genannten Elementen
bestehenden Substanzen gültig sind. Nachfolgend werden die Parameter kurz erläutert:
Die in der COSMO-Rechnung bestimmten idealen Abschirmladungsdichten werden über
Teiloberflächen gemittelt, die durch einen Radius rav bestimmt sind. Diese Mittelung ist
notwendig, da im Modell von konstanter Ladungsdichte auf den Flächensegmenten
ausgegangen wird. Die gesamte Oberfläche Ai eines Moleküls wird in n = Ai /Aeff
Kontaktoberflächen unterteilt. Der Segmentradius reff, der die effektive Kontaktfläche Aeff der
unabhängigen Oberflächensegmente bestimmt, ist ebenfalls ein anzupassender Parameter. Mit
der Anpassung eines Misfit-Energiefaktors α' wird die Näherung, ein Lösungsmittel über die
Dielektrizitätskonstante zu skalieren, korrigiert.
Obwohl der elektrostatische Anteil der Wasserstoffbrücken durch COSMO-RS gut
beschrieben wird, ist der Energiegewinn, der mit der gegenseitigen Durchdringung der
Elektronenhüllen von Donator und Akzeptor einhergeht, nicht ausreichend berücksichtigt
worden. Für Substanzen, die Wasserstoffbrücken ausbilden, wurden zwei Parameter, chb und
σhb, eingeführt. Damit wird die Wechselwirkungsenergie E(σ,σ'), die bisher nur die Misfit-
Energie charakterisierte, um den folgenden Term erweitert:
],0min[],0max[)',( hbdonhbacchbhb cE σσσσσσ +−= (2.18)
σacc und σdon geben dabei den größeren und kleineren Wert von σ bzw. σ' an. Dieser
Wasserstoffbrückenterm wird nur aktiv, wenn Oberflächenladungen von Segmenten die
Werte σacc und σdon überschreiten. Er stellt somit einen Schwellwertterm dar.
Für die Berechnung von chemischen Potenzialen in der Gasphase trägt ein Parameter ω den
an einem Ring im Molekül beteiligten Atomen Rechnung und ein Parameter η beschreibt die
Entropie in der Gasphase bezüglich eines Standardzustandes der flüssigen Phase. Die für die
Atome anzusetzenden Radien Rk bei der Beschreibung der Moleküloberfläche wurden
elementspezifisch angepasst. Ebenso stellte es sich als sinnvoll heraus, die
Dispersionskonstante δ aus Gl. 2.17 für die Elemente separat zu betrachten, so dass sich die
Dispersionskonstante eines aus k Elementen bestehenden Moleküls entsprechend ∑ kk Aδ
zusammensetzt, wobei δk die elementspezifischen Konstanten und Ak die korrespondierenden
Oberflächenanteile sind.
Abb. 2.1 gibt einen Überblick über die Schritte zur Ermittlung des Sigma-Profils einer
Komponente. Zunächst wird eine Startmolekülstruktur generiert (a), die in der
quantenmechanischen Rechnung mit der COSMO-Randbedingung optimiert wird (b). Durch

17
die Einteilung der Oberfläche der Kavität in Segmente kann jedem Segment eine
Ladungsdichte zugeordnet werden (c,d), die in einer Häufigkeitsverteilung, dem Sigma-Profil,
resultiert.
Abbildung xx. Prinzip COSMO-RS
Abb. 2.1 Herleitung der selbstkonsistenten Berechnungsgleichung für das chemische Potenzial
Ausgangspunkt ist ein Ensemble mit N Segmenten, die eine Abschirmungsladungsdichte σ
besitzen.
Ein Segment wird mit dem Index 0 bezeichnet, die restlichen mit 1 bis N-1 durchnummeriert.
Die Zustandssumme des Ausgangssystems ist Z. Es wird angenommen, dass das chemische
Potenzial µ(σ) für jeden Wert von σ bekannt ist. Es lautet:
( ))(σ
σµdN
dZkT−= (2.19)
Für ein System mit großer Anzahl N gilt:
Die Wegnahme von zwei Segmenten i und j ist gegeben durch die alte Zustandssumme,
gewichtet durch den Boltzmann Faktor:
( ) ( )
+
kTZ ii σµσµ
exp (2.20)
Die Degeneration eines Ensembles von Segmenten ist 2N/2(N/2)!, d.h. die Degeneration des
Originalensembles ist um den Faktor N größer als die des Ensembles, aus dem zwei Segmente
entfernt wurden.
Die Wahrscheinlichkeit p(0,i) eine Paarung 0,i im Originalensemble zu finden, ist gegeben

18
durch die Boltzmann gewichtete „Misfit-Energie“ von Paar 0,i, der Zustandssumme von
Ensemble ohne 0 und i und der Zunahme der Degeneration vom Startensemble verglichen mit
dem verkleinerten Ensemble:
+⋅⋅
+−⋅=
kTZ
kT
aNip iieff )()(
exp)('2/1
exp),0( 02
0 σµσµσσα (2.21)
Degeneration Misfit Wegnahme von 0 und i
Segment 0 wird nun mit allen N-1 Segmenten gepaart. Die Zustandssumme vom
Ausgangssystem ist die Summe aller N-1 Wahrscheinlichkeiten, Segment 0 in einem Paar 0,i
zu finden:
∑∑−
=
−
=
+⋅⋅
+−⋅==
1
1
02
01
1
)()(exp
)('2/1exp),0(
N
i
iieffN
i kTZ
kT
aNipZ
σµσµσσα (2.22)
Das Auflösen dieser Gleichung nach µ(σ) und gleichzeitiges Dividieren durch Z*exp(-
µ(σ0)/kT) sowie Logarithmieren führt zu:
−+−⋅−= ∑
−
=
1
1
20
0
))()('2/1(expln)(
N
i
iieff
kT
aNkT
σµσσασµ (2.23)
Für große N kann die Summe statt bis N-1 bis N ersetzt werden. Statt der Summe wird das
Integral N ∫ ')'( σσ dp geschrieben und weiterhin µ(σ0) durch µ(σ) ersetzt
−+−⋅−= ∫ '
))'()'('2/1(exp)'(ln)(
22 σ
σµσσασσµ d
kT
apNkT
eff (2.24)
Nach der Normalisierung des Ensembles auf 1mol Segmente wird mit
)(')ln()( σµσµ +−= nkT und molNNn /= (2.25)
−+−−= ∫ '
))'(')'('2/1(exp)'('ln)('
2
σσµσσα
σσµ dkT
apkT
eff (2.26)
Durch die Normalisierung hebt sich die Anzahl der Segmente N und damit auch das Quadrat
von N aus Gleichung 2.24 auf.

19
p’(σ’) bezeichnet hier die Wahrscheinlichkeit, den Wert der Ladungsdichte σ’ („’“ ist eine
allgemeine Bezeichnung für das andere ursprünglich als „i“ bezeichnete Segment.) im
normalisierten Ensemble zu finden, ausgedrückt durch „’“ in der Bezeichnung des
normalisierten Sigmaprofils:
∑==
i
iiS Ax
p
A
pp
)()()('
σσσ (2.27)
Wahrscheinlichkeit und Sigmaprofil sind Synonyme. Die Einführung des normalisierten
Ensembles erfolgt allein aus Gründen der Übersichtlichkeit.
Als Ergebnis einer COSMO-RS-Rechnung liegt das chemische Potenzial einer Komponente i
in einer Mischung vor. Der Aktivitätskoeffizient kann standardmäßig wie folgt geschrieben
werden:
−=
RT
iii
0
expµµ
γ (2.28)
Hierbei gilt zu beachten, dass der Standardzustand des COSMO-RS Modells das reine
Segmentensemble ist.

20
2.4 Phasengleichgewichtsbeziehungen
Ein Reinstoff oder eine Mischung mehrerer Komponenten in einem geschlossen System
befindet sich im Zustand des Gleichgewichts, wenn die innere Energie U des Systems als
Funktion seiner Fundamentalvariablen das Minimum erreicht hat, d.h. wenn es keine
Zustandsänderung gibt, die zu einer weiteren Abnahme der inneren Energie führt. Das System
kann dabei eine homogene Phase bilden oder in mehreren koexistierenden Phasen vorliegen.
Die Gleichgewichtsthermodynamik gibt Auskunft über die Zusammensetzung der
auftretenden Phasen und deren Anzahl bei vorgegebener Temperatur und vorgegeben Druck.
Aus dem Minimum der inneren Energie, bei konstantem Volumen V, konstanter Entropie S
und konstanter Gesamtstoffmenge können Gleichgewichtsbedingungen abgeleitet werden
(z.B. [PRAUSNITZ, 1999]). Danach definiert sich der Zustand des Gleichgewichts über die
Gleichheit der Temperaturen ϕT , der Drücke ϕP und der chemischen Potenziale ϕµ aller
Komponenten i in allen Phasenϕ .
In einem ausπ Phasen und k Komponenten bestehenden System lauten die die Beziehungen
wie folgt:
πϕTTTT
III ==== ... (2.29a)
πϕPPPP
III ==== ... (2.29b)
πϕµµµµ ii
II
i
I
i ==== ... ki ...1= (2.29c)
Man spricht auch von thermischem, mechanischem und stofflichem Gleichgewicht.
Das chemische Potenzial einer Komponente, das gleich der partiellen molaren Gibbsschen
Enthalpie ist, über die Fugazität ausgedrückt werden.
i
iiii
f
fRTPTg
00 ln),( +== µµ (2.30)
Daraus ergibt sich das Isofugazitätskriterium (Gl. 2.31), welches den vereinfachten
Ausgangspunkt für die Berechnung von Phasengleichgewichten bildet.
n
ii
II
i
I
i ffff ==== ϕ... (2.31)
Bei Mischungen, die in π Phasen und k Komponenten vorliegen, legt die Gibbssche
Phasenregel die Zahl der vorzugebenden Größen fest:
kF +−= π2 (2.32)
F Zahl der Freiheitsgrade

21
2.4.1 Aktivität und Aktivitätskoeffizient Zur Beschreibung von realen flüssigen Mischungen wird die Aktivität ai eingeführt. Sie ist
definiert als Quotient aus der Fugazität fi einer Gemischkomponente und der
Standardfugazität f0i der gleichen Komponente.
i
ii
f
fa
0
= (2.33)
Der dimensionslose Aktivitätskoeffizient γi wird durch das Verhältnis aus Aktivität ai und
einem beliebigen Konzentrationsmaß, hier dem Molenbruch xi, beschrieben.
i
ii
x
a=γ (2.34)
Als Bezugszustand für die Mischung realer Flüssigkeiten wird der Zustand des reinen Stoffes
bei Systemtemperatur und Systemdruck verwendet. Unter diesen Bedingungen kann das
chemische Potenzial als Funktion des chemischen Potenzials des reinen realen Stoffes, der
Konzentration und des Aktivitätskoeffizienten dargestellt werden.
iiii RTxRTPT γµµ lnln),(0 ++= (2.35)
2.4.2 Phasengleichgewichtsbeziehung Ausgangspunkt für die Phasengleichgewichtsberechnung ist das Isofugazitätskriterium (Gl.
2.31). Unter Verwendung des Fugazitätskoeffizienten ϕi, der als Quotient aus der Fugazität fi
und dem Partialdruck Pi der Komponente i definiert ist, wird die Fugazität V
if einer
Komponente i in der Dampfphase wie folgt beschrieben:
PxfV
i
V
i
V
i ϕ= (2.36)
Für die Beschreibung einer Komponente in der flüssigen Phase wird der Aktivitätskoeffizient
γi sowie die Standardfugazität f0i verwendet, die mit Hilfe des Dampfdruckes LV
iP0 , dem
Fugazitätskoeffizienten LV
i0ϕ des reinen Stoffes und dem Pointing-Faktor Π0i ausgedrückt
wird.
i
LV
i
LV
iiiii
L
i
L
i Pxfxf 0000 Π== ϕγγ (2.37)
Wenn zwei Phasen im Gleichgewicht stehen, von denen die eine dampfförmig (V) und die
andere flüssig (F) ist, lautet die Phasengleichgewichtsbeziehung:
i
LV
i
LV
ii
L
i
V
i
V
i PxPx 000 Π= ϕγϕ (2.38)

22
Unter der Annahme gleicher Fugazitätskoeffizienten in der Mischung und im reinen Stoff und
einer Entfernung des Systemdrucks vom Dampfdruck von kleiner als 10bar vereinfacht sich
Gl. 2.38 zu
LV
ii
L
i
V
i PxPx 0γ= (2.39)
Das Phasengleichgewicht eines Systems kann berechnet werden, wenn die
Aktivitätskoeffizienten und die Reinstoffdampfdrücke der beteiligten Komponenten bekannt
sind. Für Gemische, die über ausgeprägte Wechselwirkungen in der Dampfphase verfügen
wie zum Beispiel Systeme mit Säuren, muss der Fugazitätskoeffizient mit einer
Zustandsgleichung berechnet werden.
Stehen zwei flüssige Phasen I und II im Gleichgewicht, folgt aus Gl. 2.31 und Gl. 2.37 für die
Phasengleichgewichtsbeziehung:
II
i
II
i
I
i
I
i xx γγ = (2.40)

23
2.5 Eigenschaften und Phasenverhalten von hyperverzweigten Polymeren
Dendritische Polymere sind im Gegensatz zu linearen Polymeren stark verzweigte, mehr oder
weniger kugelförmige Makromoleküle, die sich entsprechend ihrer molekularen Struktur in
Dendrimere und hyperverzweigte Polymere unterscheiden lassen. Als Dendrimere werden
monodisperse Polymere bezeichnet, die eine perfekt verzweigte, kugelförmige Struktur mit
einer großen Anzahl funktioneller Gruppen an der Moleküloberfläche aufweisen. Im
Gegensatz dazu weisen hyperverzweigte Polymere in der dreidimensionale Molekülstruktur
Unregelmäßigkeiten auf, die durch den Syntheseprozess bedingt sind (siehe folgendes
Kapitel). Einen umfangreichen Überblick über stark verzweigte Polymere, deren molekularen
Aufbau und ihre Synthese geben Tomalia et al. [TOMALIA, 1990], Hult et al. [HULT, 1999]
und Seiler [SEILER, 2002b].
Für die Modellierung des Phasenverhaltens solcher hyperverzweigten Polymere existieren
gegenwärtig nur wenige Ansätze. Diese beruhen auf Molekularsimulationen [MANSFIELD,
1993] [LESCANEC, 1990; NAYLOR, 1989], wobei hier vor allem die regelmäßigen
Dendrimere untersucht werden, auf Gittermodellen [BAWENDI, 1987; DUDOWICZ, 1990;
FREED, 1989; FREED, 1992; FREED, 1996; JANG, 1999; JANG, 2002b; JANG, 2002a]
und auf Free-Volume Aktivitätskoeffizientenmodellen [KOUSKOUMVEKAKI, 2002;
SEILER, 2004c]. Aufgrund der komplexen dreidimensionalen Molekularstruktur der
hyperverzweigten Moleküle haben die aktuellen Ansätze zur Beschreibung
thermodynamischer Eigenschaften Struktureigenschaften noch nicht die gewünschte Güte und
Vorhersagekraft, insbesondere fehlt bei Molekularsimulationen von komplexen
dreidimensionalen Molekülen die Vorhersage des Phasengleichgewichts, bei Gittermodellen
sind viele experimentelle Daten zur Phasengleichgewichtsberechnung notwendig. In dieser
Arbeit wird der Ansatz verfolgt das COSMO-RS-Modell um einen Free-Volume-Term zu
erweitern und durch diese Kombination den Einfluss der verzweigten Struktur und der
funktionellen Gruppen auf das Phasengleichgewicht zu beschreiben. Damit ist es möglich
direkt aus der molekularen Struktur das Phasenverhalten vorherzusagen.
Zunächst werden in diesem Kapitel die Eigenschaften hyperverzweigter Polymere erläutert
und anschließend der Ansatz zur Modellierung des Phasenverhaltens vorgestellt.
2.5.1 Struktureller Aufbau und Eigenschaften hyperverzweigter Polymere
Dendrimere und hyperverzweigte Polymere werden durch Polymerisation aus Monomeren der
Struktur AnB (n ≥ 2) hergestellt. Dabei reagieren ausschließlich die Funktionalitäten A und B
miteinander. Die entstehende, hochverzweigte Polymerstruktur besitzt eine funktionelle

24
Gruppe B und viele endständige Funktionalitäten A (Abb. 2.1). Innerhalb des
Polymermoleküls treten drei sich wiederholende Einheiten auf, die ebenfalls in Abb. 2.1
dargestellt sind. Dendritische Einheiten (D) sind vollständig reagiert und ideal verzweigt, d.h.
alle Funktionalitäten des Monomers sind eine Bindung eingegangen. Lineare Einheiten (L)
besitzen eine nicht reagierte funktionelle Gruppe A, während in den endständigen terminalen
Einheiten (T) keine der Funktionalitäten A eine Bindung eingegangen ist [BURGATH, 1998].
B
A
A
B
A
A
A
A
A
A
A
A
A
A
Polymerisation
dendritische Einheit (D)
lineare Einheit (L)
terminale Einheit (T)
Abb. 2.2 Schematische Darstellung der Herstellung von hyperverzweigten Polymeren aus A2B - Monomeren und der im Polymer vorliegenden dendritischen, linearen und terminalen Einheiten [HULT, 1999].
Der Verzweigungsgrad (engl.: Degree of Branching DB) sagt aus, wie stark verzweigt das
hyperverzweigte Polymer im Vergleich zu einem ideal verzweigten Dendrimer ist.
LD
DDB
+=
22
(2.41)
D stellt den Anteil der dendritischen und L den Anteil der linearen Einheiten am
Polymermolekül dar. Beide Anteile können mittels NMR – Spektroskopie bestimmt werden
[BURGATH, 1998; HOELTER, 1997]. Der Verzweigungsgrad von Dendrimeren beträgt 1,
während lineare Moleküle einen Verzweigungsgrad von 0 besitzen. Hyperverzweigte
Polymere nehmen eine Stellung zwischen diesen beiden Grenzfällen ein.
Das Lösungsverhalten hyperverzweigter Polymere hängt sehr stark von der Art, der Anzahl
und der Orientierung der funktionellen Gruppen ab. Die Löslichkeit kann gezielt durch die
Auswahl der funktionellen Gruppen (z.B. hydrophob oder hydrophil) eingestellt werden.
Dabei ergibt sich die Orientierung der Endgruppen aus den Wechselwirkungen zwischen den
Endgruppen der Polymerketten und den Lösungsmittelmolekülen [SEILER, 2002b].
Insbesondere diese Eigenschaft macht hyperverzweigte Polymere zu interessanten
Zusatzstoffen in der thermischen Trenntechnik. Sie können einem engsiedenden oder
azeotropen Gemisch hinzu gegeben werden, um spezifische Wechselwirkungen mit einer der
Komponenten einzugehen [SEILER, 2002a].

25
2.5.2 Phasenverhalten von hyperverzweigten Polymerlösungen
Bei der Herstellung von Polymeren laufen zahlreiche Reaktionsschritte (Initialisierungs-,
Kettenwachstums- und Abbruchreaktionen) ab, die zu unterschiedlich großen Molekülen
führen. Polymere sind deshalb immer Mischungen zahlreicher Moleküle, die in Kettenlänge
und Kettenverzweigung statistisch verteilt vorliegen. Man bezeichnet diese Eigenschaft als
Polydispersität. Diese Eigenschaft lässt sich insbesondere an der Molmassenverteilung
demonstrieren.
Die Häufigkeit einer bestimmten Molmasse wird durch die Molmassenverteilung erfasst.
Lagemaßzahlen der Statistik kennzeichnen eine solche Verteilung der Molmasse. Allgemein
bezeichnet man sie auch als Momente einer Verteilung. Zur Kennzeichnung von Polymeren
haben sich das Zahlenmittel Mn, das Massenmittel Mw, das Zentrifugenmittel Mz und das
Viskositätsmittel Mη etabliert. Sie sind definiert als:
∑∑∑===
==k
i
ii
k
i
i
k
i
iin MxnMnM111
Zahlenmittel
(2.42)
Mn ist das 1. Moment oder auch der Mittelwert einer häufigkeitsgewichteten
Molmassenverteilung.
∑∑∑===
==k
i
ii
k
i
i
k
i
iiw MwmMmM111
Massenmittel (2.43)
Mw ist das 1. Moment oder auch der Mittelwert einer massengewichteten
Molmassenverteilung.
Abb. 2.3 zeigt beispielhaft eine unsymmetrische Verteilungsfunktion der Molmasse und die
dazugehörenden Lagemaßzahlen. Die Häufigkeitswerte sind mit einer Hosemann-Schramek-
Verteilungsfunktion generiert (Parameter a=1,5; b=0,0003; c=1,5).
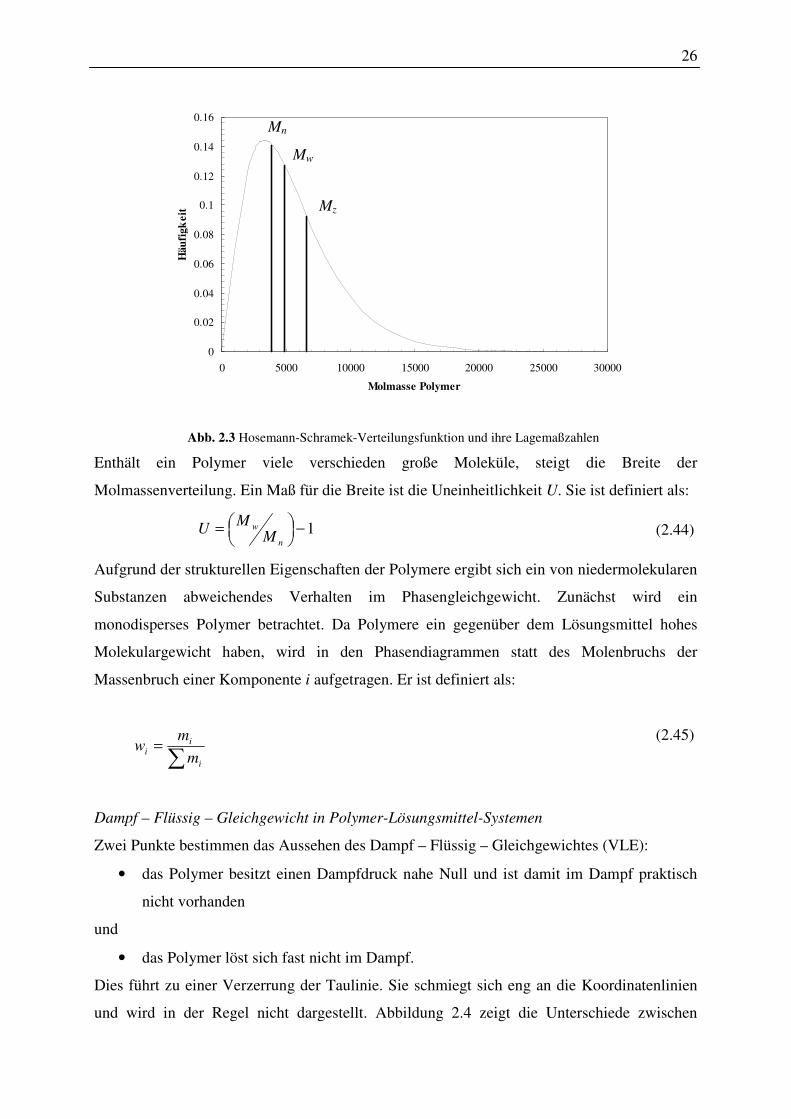
26
0
0.02
0.04
0.06
0.08
0.1
0.12
0.14
0.16
0 5000 10000 15000 20000 25000 30000
Molmasse Polymer
Häu
figk
eit
Abb. 2.3 Hosemann-Schramek-Verteilungsfunktion und ihre Lagemaßzahlen
Enthält ein Polymer viele verschieden große Moleküle, steigt die Breite der
Molmassenverteilung. Ein Maß für die Breite ist die Uneinheitlichkeit U. Sie ist definiert als:
1−
=
n
w
MM
U (2.44)
Aufgrund der strukturellen Eigenschaften der Polymere ergibt sich ein von niedermolekularen
Substanzen abweichendes Verhalten im Phasengleichgewicht. Zunächst wird ein
monodisperses Polymer betrachtet. Da Polymere ein gegenüber dem Lösungsmittel hohes
Molekulargewicht haben, wird in den Phasendiagrammen statt des Molenbruchs der
Massenbruch einer Komponente i aufgetragen. Er ist definiert als:
∑=
i
ii
m
mw
(2.45)
Dampf – Flüssig – Gleichgewicht in Polymer-Lösungsmittel-Systemen
Zwei Punkte bestimmen das Aussehen des Dampf – Flüssig – Gleichgewichtes (VLE):
• das Polymer besitzt einen Dampfdruck nahe Null und ist damit im Dampf praktisch
nicht vorhanden
und
• das Polymer löst sich fast nicht im Dampf.
Dies führt zu einer Verzerrung der Taulinie. Sie schmiegt sich eng an die Koordinatenlinien
und wird in der Regel nicht dargestellt. Abbildung 2.4 zeigt die Unterschiede zwischen
Mz
Mw
Mn
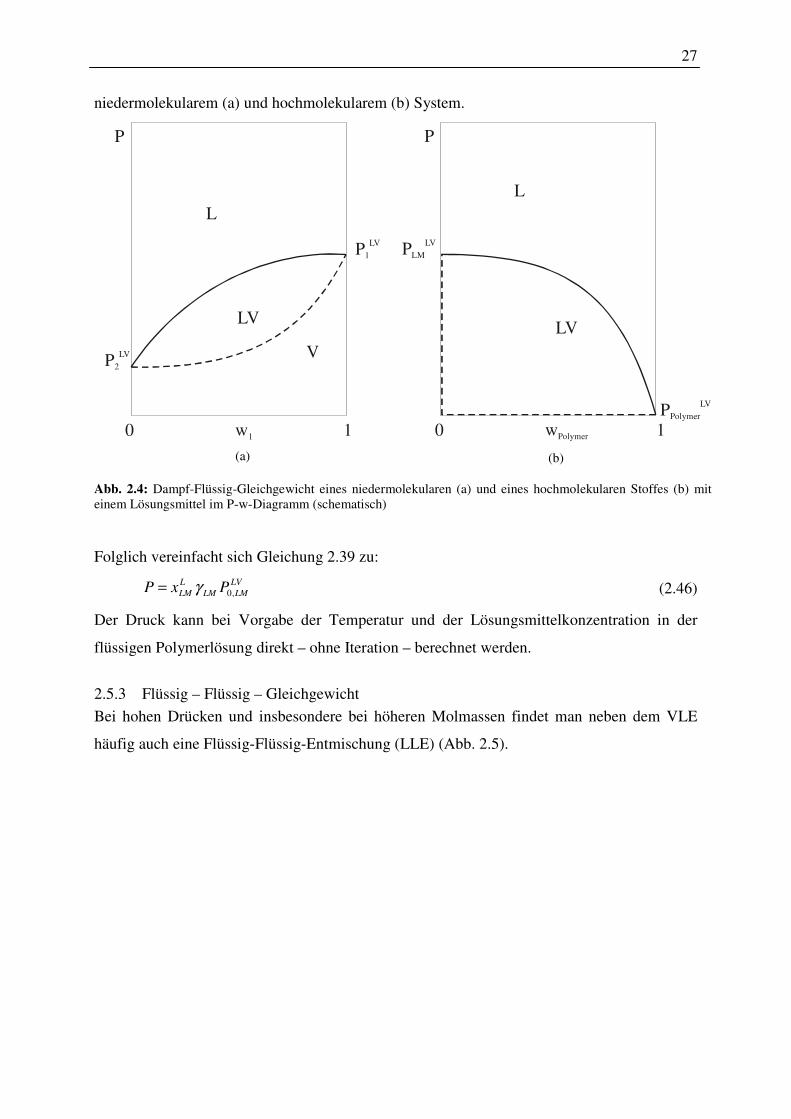
27
niedermolekularem (a) und hochmolekularem (b) System.
0 1w1
P
LV
V
L
P LV2
P LV1
(a)
0 1wPolymer
P
LV
L
P LVPolymer
P LVLM
(b)
Abb. 2.4: Dampf-Flüssig-Gleichgewicht eines niedermolekularen (a) und eines hochmolekularen Stoffes (b) mit einem Lösungsmittel im P-w-Diagramm (schematisch)
Folglich vereinfacht sich Gleichung 2.39 zu:
LV
LMLM
L
LM PxP ,0γ= (2.46)
Der Druck kann bei Vorgabe der Temperatur und der Lösungsmittelkonzentration in der
flüssigen Polymerlösung direkt – ohne Iteration – berechnet werden.
2.5.3 Flüssig – Flüssig – Gleichgewicht Bei hohen Drücken und insbesondere bei höheren Molmassen findet man neben dem VLE
häufig auch eine Flüssig-Flüssig-Entmischung (LLE) (Abb. 2.5).

28
Abb. 2.5: Flüssig-Flüssig-Phasengleichgewicht eines Polymer-Lösungsmittel-Systems im P-w-Diagramm (schematisch)
Die Löslichkeit von Polymeren in Lösungsmitteln wird sowohl durch die chemische Struktur
des Polymers, als auch durch die chemische Struktur des Lösungsmittels bestimmt. Für die
Charakterisierung des Phasenverhaltens von Polymerlösungen müssen folgende drei
Einflussfaktoren betrachtet werden [RASMUSSEN, 1989]:
1. Wechselwirkungen zwischen den Gemischkomponenten:
Hier wird zwischen gerichteten und ungerichteten Wechselwirkungen
unterschieden. Dipolkräfte und Wasserstoffbrückenbindungen sind gerichtete
Wechselwirkungen, die über große Molekülabstände hinweg wirken. Ungerichtete
Wechselwirkungen beruhen auf Dispersionskräften. Diese Kräfte werden auch als
van-der-Waals Kräfte bezeichnet.
2. Molmasse der Polymere:
Kleine Moleküle lösen sich besser als große Moleküle, so dass eine Erhöhung der
Molmasse bei konstantem Polymergehalt und konstanter Temperatur zunächst zur
Entmischung der Lösung in zwei flüssige Phasen führt.
3. Unterschiede in den freien Volumina
Durch eine unterschiedliche, temperaturabhängige Dichteänderung von Polymer
und Lösungsmittel tritt bei hohen Temperaturen (und Drücken) eine Flüssig-
Flüssig Entmischung auf [PRAUSNITZ, 1999]. Allerdings konnte gezeigt werden,

29
dass die Berücksichtigung des Effekts, der auch als Free-Volume Effekt bezeichnet
bezeichnet wird. Bei Dampf-Flüssig-Phasengleichgewichten führt ein
entsprechender Term in der Modellierung zu einer spürbaren Verbesserung in der
Modellierung [IWAI, 1981; KOUSKOUMVEKAKI, 2002; SEILER, 2004c].
Free-Volume Term für die Erfassung der Unterschiede in den freie Volumina
Die bekanntesten Aktivitätskoeffizientenmodelle zur Berücksichtigung des Free-Volume
Effektes sind das von Oishi und Prausnitz [OISHI, 1978] vorgestellte UNIFAC-FV und das
durch Elbro [ELBRO, 1990] vorgeschlagene Entropic-FV Modell. Die Aktivitätskoeffizienten
für die Lösungsmittel in einem Polymer-Lösungsmittel System werden in diesen beiden
Modellen durch Terme beschrieben, die die enthalpischen und entropischen Effekte bei der
Mischung von unterschiedlichen Komponenten berücksichtigen.
Die Beschreibung der enthalpischen Effekte erfolgt durch den residuellen Term des
UNIQUAC Modells [ABRAMS, 1975; KIKIC, 1980]. Die entropischen Effekte werden durch
zwei Terme beschrieben, wobei der kombinatorische Term aus dem ursprünglichen UNIFAC
Modell [FREDENSLUND, 1975a; FREDENSLUND, 1989] übernommen wird. Der zweite
Term zur Beschreibung des „Free-Volume“- Effektes wurde von Oishi und Prausnitz aus der
Flory-Zustandsgleichung abgeleitet.
FV
i
komb
i
res
ii γγγγ lnlnlnln ++= (2.47)
In der Flory-Zustandsgleichung beschreibt der binäre Parameter χ12 die Unterschiede in den
Wechselwirkungen, die im UNIFAC Modell durch den residuellen Term berücksichtigt
werden. Mit χ12 = 0 ergibt sich der gesuchte Beitrag zum Lösungsmittelaktivitäts-
koeffizienten [OISHI, 1978].
−
−−
−
−=
−1
3/13/1
3/1
~1
11~
~
1~1~
ln3lnim
ii
m
ii
FV
ivv
vc
v
vcγ (2.48)
Das reduzierte Volumen eines Lösungsmittels i
v% wird aus dem spezifischen Volumen vi des
Lösungsmittels und dem van-der-Waals-Volumen vi,vdW des Lösungsmittels bestimmt.
vdWi
im
bv
vv
,
~ = (2.49)
Das reduzierte Volumen der Mischung ergibt sich unter Verwendung der Massenbrüche zu:

30
∑
∑=
i
vdWii
i
i
i
mvwb
vw
v,
~ (2.50)
Das van-der-Waals Volumen einer Komponente i berechnet sich aus einem von Abrams et al.
[ABRAMS, 1975] hergeleiteten Faktor und dem Molekülvolumen ri’.
', 17,15 ivdWi rv = (2.51)
Das Molekülvolumen ergibt sich im UNIFAC Modell aus der Addition der relativen van-der-
Waals Gruppenvolumina Rk’ multipliziert mit der Häufigkeit der funktionellen Gruppe νk.
Hierbei steht der Index i für die betrachtete Komponente i und der Index k für funktionelle
Gruppen aus denen die Moleküle der Komponente i zusammengesetzt werden.
∑=k
kki Rvr '' (2.52)
Die physikalischen Bedeutungen der Parameter b und c des UNIFAC-FV Modells werden in
der Literatur diskutiert. Der geometrische Faktor b ist von Oishi und Prausnitz [OISHI, 1978]
an experimentelle Daten angepasst worden und zu b =1,28 gesetzt. Tande et al. [TANDE,
2002] verwenden für den b Parameter Werte zwischen 1,28 und 1,66 für die Modellierung
von Polymer - Lösungsmittel Systemen. Sie schlagen vor, den b-Parameter als die Fähigkeit
der Lösungsmittelmoleküle anzusehen, in das Volumen des Polymermoleküls eindringen zu
können.
Der Parameter ci wird von Beret und Prausnitz [BERET, 1975] als die Anzahl der
Freiheitsgrade eines Moleküls beschrieben, die sich durch Rotation, Translation und Vibration
ergeben. Im UNIFAC-FV Modell wird er von Oishi et al. [OISHI, 1978] für verschiedene
Lösungsmittel zu ci =1,1 gesetzt. Zur Abschätzung von physikalisch sinnvollen ci-Parametern
wird von Kontogeorgis et al. [KONTOGEORGIS, 1993] der Quotient aus UNIFAC Volumen
ri und UNIFAC Oberfläche qi des Lösungsmittels vorgeschlagen:
i
ii
q
rc = (2.53)
Eine Ausnahme stellt das technisch wichtige Lösungsmittel Wasser dar. Lindvig [LINDVIG,
2002] beschreibt, dass der „Free-Volume“-Effekt in Polymer-Wasser-Systemen klein ist, da
sich Wasser und Polymere bezüglich ihres freien Volumens ähnlich verhalten. Um den
zusätzlichen entropischen Anteil, der sich aus dem „Free-Volume“-Term ergibt, zu
vernachlässigen, wird der ci Parameter für Wasser auch in dieser Arbeit zu cWasser = 0 gesetzt.

31
2.5.4 Erweiterung des COSMO-RS Modells um einen Term für die Erfassung des Free-
Volume-Effektes
Wie im COSMO-RS Modell werden im ursprünglichen UNIFAC-Modell die
Aktivitätskoeffizienten durch einen residuellen und einen kombinatorischen Term
beschrieben. Beiden Modellen liegt ein Ensemble von paarweise wechselwirkenden
Oberflächeneinheiten zugrunde, so dass der Oberflächenanteil Θm im UNIFAC-Modell dem
σ-Profil im COSMO-RS-Modell entspricht1 [KLAMT, 2000]. Daraus ergeben sich Parallelen
zwischen dem UNIFAC-FV und dem COSMO-RS-Modell, wenn berücksichtigt wird, dass im
COSMO-RS-Modell kein Term zur Beschreibung des „Free-Volume“-Effekts vorgesehen ist.
Um aus dem „Free-Volume“-Effekt resultierende entropische Anteile an den
Aktivitätskoeffizienten beschreiben zu können, wird das COSMO-RS Modell um den im
UNIFAC-FV verwendeten FV-Term erweitert. Dazu werden die van-der-Waals-Volumina
durch die aus COSMO- Rechnungen ermittelten Molekülvolumina der Gemischkomponenten
ersetzt. Die reduzierten Volumina der Komponenten und der Mischung ergeben sich wie
folgt:
COSMOi
i
ibv
vv
,
~ = (2.54)
∑
∑=
i
COSMOii
i
i
i
mvwb
vw
v,
~ (2.55)
Dieses Vorgehen ist möglich, da das COSMO-Volumen der Moleküle dem mit dem
geometrischen Faktor b multiplizierten van-der-Waals-Volumen aus dem UNIFAC-Modell
entspricht.
vdWCOSMO bvv ≈ (2.56)
Die Ergebnisse in Tab. 2.1 zeigen, dass dieser Zusammenhang für die betrachteten Polymere
gültig ist. Für die kleineren Moleküle Ethanol und Wasser stimmt dieser Zusammenhang
näherungsweise. Aufgrund der von Tande et al. [TANDE, 2002] vorgeschlagenen Werte des
b-Parameters, für die dieser Zusammenhang nicht gilt, wird nur das van-der-Waals-Volumen
durch das COSMO-Volumen ersetzt.
1 Das Integral des Sigmaprofils ergibt die Oberfläche des Moleküls.
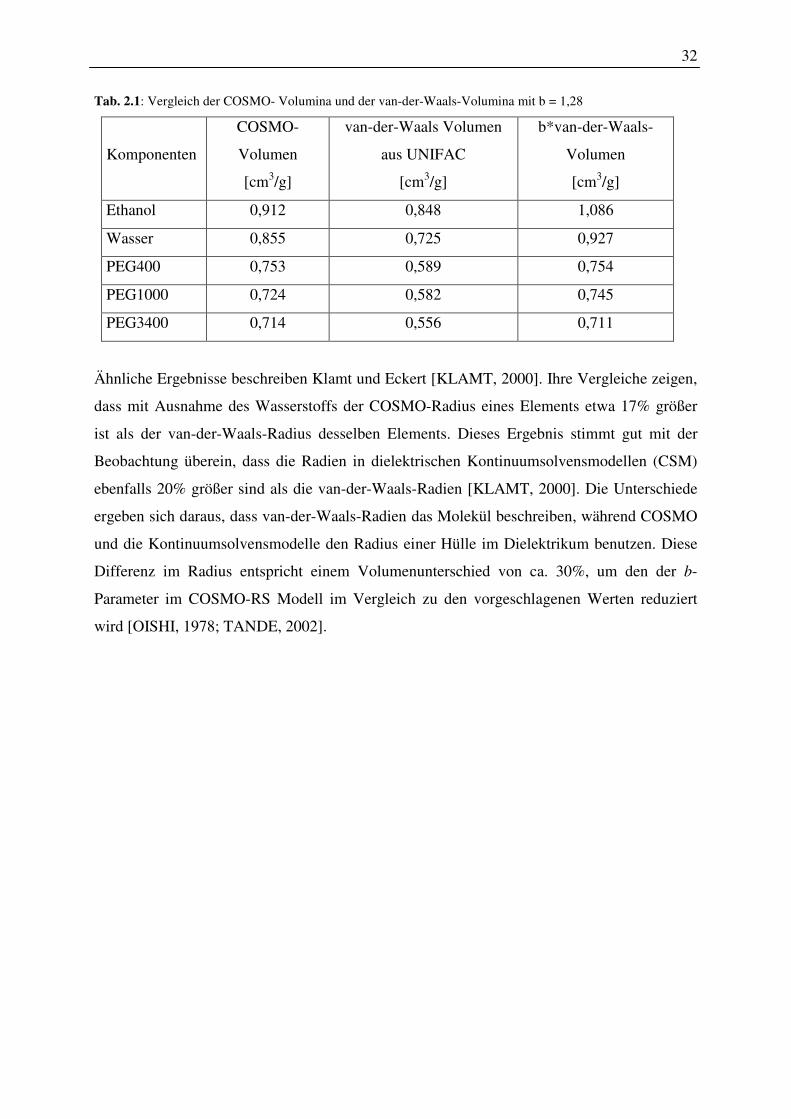
32
Tab. 2.1: Vergleich der COSMO- Volumina und der van-der-Waals-Volumina mit b = 1,28
Komponenten
COSMO-
Volumen
[cm3/g]
van-der-Waals Volumen
aus UNIFAC
[cm3/g]
b*van-der-Waals-
Volumen
[cm3/g]
Ethanol 0,912 0,848 1,086
Wasser 0,855 0,725 0,927
PEG400 0,753 0,589 0,754
PEG1000 0,724 0,582 0,745
PEG3400 0,714 0,556 0,711
Ähnliche Ergebnisse beschreiben Klamt und Eckert [KLAMT, 2000]. Ihre Vergleiche zeigen,
dass mit Ausnahme des Wasserstoffs der COSMO-Radius eines Elements etwa 17% größer
ist als der van-der-Waals-Radius desselben Elements. Dieses Ergebnis stimmt gut mit der
Beobachtung überein, dass die Radien in dielektrischen Kontinuumsolvensmodellen (CSM)
ebenfalls 20% größer sind als die van-der-Waals-Radien [KLAMT, 2000]. Die Unterschiede
ergeben sich daraus, dass van-der-Waals-Radien das Molekül beschreiben, während COSMO
und die Kontinuumsolvensmodelle den Radius einer Hülle im Dielektrikum benutzen. Diese
Differenz im Radius entspricht einem Volumenunterschied von ca. 30%, um den der b-
Parameter im COSMO-RS Modell im Vergleich zu den vorgeschlagenen Werten reduziert
wird [OISHI, 1978; TANDE, 2002].

33
2.6 Zustandsgleichungen und Mischungsregeln
Das COSMO-RS-Modell liefert zur Modellierung von Phasengleichgewichten in allererster
Linie den Aktivitätskoeffizienten einer Komponente in einer mehrkomponentigen flüssigen
Mischung. Diese Information ist ausreichend, um Flüssig-Flüssig oder Dampf-Flüssig
Phasengleichgewichte in unterkritischen Systemen zu berechnen (vgl. Kapitel 2.4). Für die
Berechnung von Hochdruckphasengleichgewichten und in Systemen mit überkritischen
Komponenten werden thermische Zustandsgleichungen zur Modellierung des
Phasenverhaltens angewendet. Für Aktivitätskoeffizientenmodelle ist es schwierig, einen
Standardzustand für überkritische Komponenten zu definieren.
Für die große Gruppe der kubischen Zustandsgleichungen soll im folgenden Abschnitt ein
Verfahren eingeführt werden, das es ermöglicht, die a-priori Fähigkeiten des COSMO-RS
Modells im Rahmen einer kubischen Zustandsgleichung zu nutzen.
Eine thermische Zustandsgleichung stellt den Zusammenhang zwischen den intensiven
Variablen Temperatur (T), Druck (P) und der bezogenen Dichte (v) in einem mathematischen
Zusammenhang für einen Reinstoff her. Unter der Vorraussetzung, dass dieselbe
Zustandsgleichung auch für die Mischung anwendbar ist, wurden Regeln eingeführt, die
Mischungsparameter liefern. Dieses Vorgehen wird als van-der-Waals Ein-Fluid Modell
bezeichnet. Eine Grenzbedingung für die Konzentrationsabhängigkeit der Parameter der
Zustandsgleichung lässt sich mit der Virialgleichung angeben. Die Virialgleichung kann mit
Hilfe der statistischen Mechanik abgeleitet werden und ist die einzige volltheoretische
Zustandsgleichung. Der Realgasfaktor Z wird als Reihenentwicklung in der Moldichte bzw.
dem spezifischem Volumen v angegeben:
...1 2 +++=v
C
v
B
RT
Pv (2.57)
Die Virialkoeffizienten B, C,… sind nur von der Temperatur abhängig. Der Zusammenhang
gilt für reale Fluide allerdings nur für ein Polynom mit unendlich vielen Gliedern. Für einen
Reinstoff ist gemäß der Phasenregel der Druck von zwei Variablen abhängig, der Temperatur
und der Dichte. Im Fall der Mischung aus j Komponenten muss der Druck eine Funktion von
j+1 Variablen sein, der Temperatur T und den volumenbezogenen Stoffmengen der
jeweiligen Komponenten i ni/V. Bei konstanter Temperatur existiert eine Polynomdarstellung
des Druckes, die nur von den ni/V Variablen abhängig ist. Es ergibt sich folgende
Konzentrationsabhängigkeit der Virialkoeffizienten:

34
∑∑∑
∑∑
=
=
i j k
ijkkji
i j
ijji
CxxxC
BxxB
(2.58)
Wird die van der Waals Zustandsgleichung
2
v
a
bv
RTP −
−= (2.59)
als Polynom in (b/v) dargestellt
RTv
a
v
b
RT
Pv
n
−
=∑
∞
=0
(2.60)
erkennt man, der Virialgleichung folgend, dass die Grenzbedingung im Limit für 0→P dem
quadratischen Zusammenhang in der Konzentration
iji j
ji
i j
ijjiRT
abxx
RT
abBxxB ∑∑∑∑
−=−== (2.61)
folgen muss. Dies gilt für alle zweiparametrigen Zustandsgleichungen der van-der-Waals-
Familie.
Im Allgemeinen haben sich die so genannten van-der-Waals Ein-Fluid-Mischungsregeln
Regeln durchgesetzt:
∑∑
∑∑
=
=
i j
ijji
i j
ijji
bxxb
axxa
(2.62)
Um die Kreuzkoeffizienten aij und bij zu erhalten, werden die Reinstoffparameter wie folgt
kombiniert:
( )
)1(2
1
ij
jjii
ij
ijjjiiij
lbb
b
kaaa
−+
=
−=
(2.63a)
(2.63b)
Die Parameter kij und lij sind binäre Wechselwirkungsparameter und werden allgemein an
experimentelle Daten binärer VLE angepasst. Der Parameter lij wird üblicherweise zu Null
gesetzt, womit sich Gleichung 2.63b zu
∑=i
iibxb (2.64)
vereinfacht. Der kij-Parameter ist für nahezu ideale Mischungen, wie z.B. Alkanmischungen
nahe Null, wohingegen er bei nichtidealen Mischungen größere Abweichungen von Null zeigt
und sich ebenfalls in Abhängigkeit von der Temperatur ändert. Zeigt eine Mischung ein
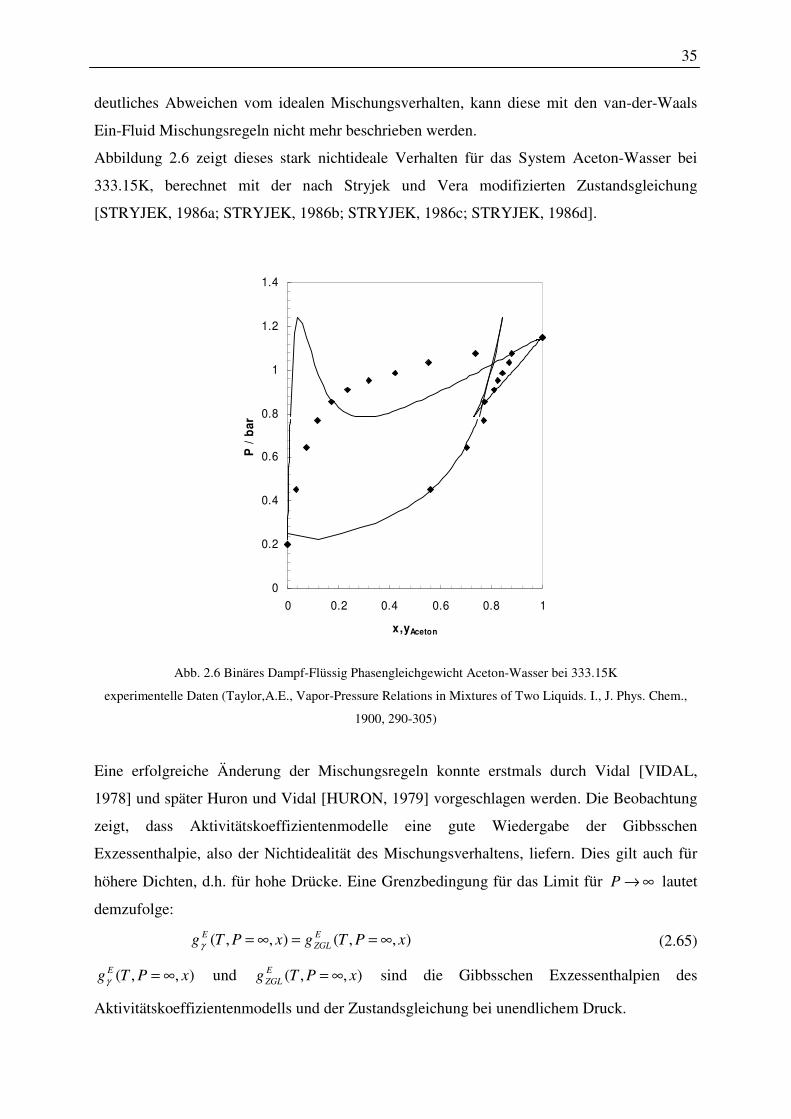
35
deutliches Abweichen vom idealen Mischungsverhalten, kann diese mit den van-der-Waals
Ein-Fluid Mischungsregeln nicht mehr beschrieben werden.
Abbildung 2.6 zeigt dieses stark nichtideale Verhalten für das System Aceton-Wasser bei
333.15K, berechnet mit der nach Stryjek und Vera modifizierten Zustandsgleichung
[STRYJEK, 1986a; STRYJEK, 1986b; STRYJEK, 1986c; STRYJEK, 1986d].
0
0.2
0.4
0.6
0.8
1
1.2
1.4
0 0.2 0.4 0.6 0.8 1
x,yAceton
P /
ba
r
Abb. 2.6 Binäres Dampf-Flüssig Phasengleichgewicht Aceton-Wasser bei 333.15K
experimentelle Daten (Taylor,A.E., Vapor-Pressure Relations in Mixtures of Two Liquids. I., J. Phys. Chem.,
1900, 290-305)
Eine erfolgreiche Änderung der Mischungsregeln konnte erstmals durch Vidal [VIDAL,
1978] und später Huron und Vidal [HURON, 1979] vorgeschlagen werden. Die Beobachtung
zeigt, dass Aktivitätskoeffizientenmodelle eine gute Wiedergabe der Gibbsschen
Exzessenthalpie, also der Nichtidealität des Mischungsverhaltens, liefern. Dies gilt auch für
höhere Dichten, d.h. für hohe Drücke. Eine Grenzbedingung für das Limit für ∞→P lautet
demzufolge:
),,(),,( xPTgxPTgE
ZGL
E ∞==∞=γ (2.65)
),,( xPTgE ∞=γ und ),,( xPTg
E
ZGL ∞= sind die Gibbsschen Exzessenthalpien des
Aktivitätskoeffizientenmodells und der Zustandsgleichung bei unendlichem Druck.

36
Mit der zusätzlichen Annahme, dass das Exzessvolumen vE bei unendlichem Druck gleich
Null ist, d.h. ∑−== ii
Ebxbv 0 , konnte folgende Mischungsregel abgeleitet werden:
−−= ∑∞
i ii
iii
E
b
ax
b
ag (2.66)
∑=i
iibxb (2.67)
Damit gelang die Berechnung von nichtidealen Systemen wie Aceton-Wasser mit der Peng-
Robinson Zustandsgleichung [PENG, 1976]. Nachteilig wirkt sich die Bedingung des
unendlichen Druckes aus. Die Gibbssche Exzessenthalpie ist eine Funktion des Druckes und
nimmt bei unendlichem Druck einen anderen Wert an als bei niedrigen Drücken. Das ist der
Grund, weshalb es unmöglich ist, Parametertabellen von GE-Modellen, die bei niedrigen
Drücken angepasst worden sind zu verwenden. Da durch die Nebenbedingung des
verschwindenden Exzessvolumens, Gleichung 2.67 angewendet werden muss, kann kein lij-
Parameter herangezogen werden, um die Mischungsgrößen zu korrigieren. Darüber hinaus
wird die quadratische Mischungsgleichung für den zweiten Virialkoeffizienten als
Grenzbedingung für niedrige Dichten nicht erreicht. Diese Grenzbedingung ist zwar zunächst
eher von theoretischem Interesse, weil sie thermodynamische Inkonsistenzen widerspiegelt.
Betrachtet man die Berechnungsgleichung für den Fugazitätskoeffizienten an,
zdVn
P
V
RT
RT
V
nVTi
i
j
ln1
ln,,
−
∂
∂−= ∫
∞
ϕ (2.68)
so fällt auf, dass die Konzentrationsabhängigkeit des Druckes bei allen Dichten – auch im
Grenzfall – den Fugazitätskoeffizienten beeinflusst. Diese Einschränkungen haben zur
Weiterentwicklung dieser so genannten GE-Mischungsregeln geführt, sowohl am unteren
Limit (P->0) als auch am oberen (P->∞) [HOLDERBAUM, 1991; MICHELSEN, 1990a;
MICHELSEN, 1990b; ORBEY, 1995a; PENELOUX, 1989; WONG, 1992b].
Wong und Sandler [WONG, 1992b; WONG, 1992a] haben eine Mischungsregel entwickelt,
die sowohl bei hohen als auch niedrigen Dichten das gewünschte Verhalten der
Zustandsgleichung ermöglicht und bestehende Parametertabellen von GE-Modellen nutzbar
macht. Sie gehen wie Huron und Vidal ebenfalls von der Gleichheit der Exzessenthalpien bei
unendlichem Druck aus, vermeiden aber deren Inkonsistenzen, indem sie ihre
Mischungsregeln zunächst in der freien Exzessenergie Ef formulieren und mit folgenden
Annahmen die Verknüpfung zur freien Exzessenthalpie gelangen:
Mit

37
EEE Pvfg += (2.69)
kann man für kleine Drücke erkennen, dass:
),,(),,( 11 xPTfxPTg bar
E
bar
E ≈ (2.70)
da der PvE-Term sehr klein ist. Die freie Exzessenergie Ef ist im Bereich von flüssigen
Dichten relativ insensitiv gegenüber dem Druck
),,(),,( 1 xPTfxPTfE
bar
E ∞→≈ (2.71)
und folglich
),,(),,( 1 xPTfxPTgE
bar
E ∞→≈ . (2.72)
Für die Gruppe der zweiparametrigen kubischen Zustandsgleichungen ergibt sich am Limit
∞→P für die freie Exzessenergie:
−=
∞→∑
i ii
ii
i
E
b
ax
b
a
RTRT
xPTf 1),,(. (2.73)
Gegenüber der gleichen Formulierung in der Gibbsschen Exzessenthalpie
∑∑ −+
−=
∞→
i
iii
i ii
iii
E
bbxRT
P
b
ax
b
a
RTRT
xPTg)(
1),,(. (2.74)
sieht man, dass die Annahme des verschwindenden Exzessvolumens im Falle der freien
Exzessenergie nicht getroffen werden muss und demzufolge auch der Freiheitsgrad in
Gleichung .6a erhalten bleibt. Für eine zweiparametrige kubische Zustandsgleichung ergibt
sich aus der Grenzbedingung für den zweiten Virialkoeffizienten (siehe Gleichung 2.61):
∑∑
−=−
i j ijji
RT
abxx
RT
ab
(2.75)
Der Ausdruck (b−a/(RT)) stellt den konzentrationsunabhängigen, zweiten
Kreuzvirialkoeffizienten dar, wobei der Kreuzterm durch
( )ijjj
jjii
iiij
kRT
ab
RT
ab
RT
ab −
−+
−=
− 1
21
(2.76)
gegeben ist und aus den Reinstoffparametern berechnet wird.
Es sei darauf hingewiesen, dass Gleichung 2.75 keine Vorschrift für die Mischungsparameter
a und b einzeln liefert, sondern diese in der Summe im Ausdruck auf der linken Seite der
Gleichung 2.75 zusammengefasst sind. Die notwendige zweite Gleichung ergibt sich aus der
Verknüpfung von Gl. 2.72 und Gl. 2.75 und gilt für ein beliebiges GE-Modell:
∑−=i i
ii
E
RTb
ax
bRT
a
CRT
Gγ
(2.77)

38
Die Konstante C ist abhängig von der gewählten Zustandsgleichung.
In verschiedenen Untersuchungen [ORBEY, 1993; WONG, 1992a] ist der Einfluss des
binären Wechselwirkungsparameters untersucht worden und es zeigte sich, dass er durch
Umformulierung des Kreuztermes der Mischungsregel eliminiert werden kann. Eine andere
Forderung ist der stetige Übergang der Mischungsregel in die van-der-Waals Ein-Fluid-
Mischungsregel für Systeme bzw. binäre Paarungen innerhalb eines Systems, die sich ideal
mischen. Orbey und Sandler [ORBEY, 1995b] haben dafür den Kreuzterm Gl. 2.75 in der
folgenden Weise umformuliert:
( ) ( )ij
ji
ii
ij
kRT
aabb
RT
ab −−+=
− 1
2
1 (2.78)
Zur Berechnung der Gibbsschen Exzessenthalpie verwenden sie das von Huron und Vidal
[HURON, 1979] modifizierte NRTL-Model:
)exp(
und
jijji
k
kik
j
jijij
i
i
E
bG
Gx
Gx
xRT
G
ατ
τγ
−=
=∑
∑∑
(2.79)
Der Unterschied zum ursprünglichen NRTL-Modell liegt in der Definition der lokalen
Zusammensetzung, welche zu der Einführung des Volumenparameters bj in der Berechnung
von Gij führt. bj ist der Volumenparameter der reinen Komponente j aus der verwendeten
kubischen Zustandsgleichung. Orbey und Sandler fanden, dass das Phasengleichgewicht mit
folgenden Annahmen und der Kenntnis des Aktivitätskoeffizienten bei unendlicher
Verdünnung vorhergesagt werden kann:
− der Parameter α wird mit einem konstanten Wert vorgegeben
− der binäre Wechselwirkungsparameter kij sei Null.
Damit ergibt sich folgende Bestimmungsgleichung für den Parameter der lokalen
Zusammensetzung τij:
)exp(ln 122
1121221 αττγτ −−= ∞
b
b (2.80)
Für die in der chemischen Industrie weit verbreitete Peng-Robinson-Zustandsgleichung
[PENG, 1976]
)()(
)(bvbbvv
Ta
bv
RTP
++−−
−= (2.81)

39
gilt für C (siehe Gl. 2.77)
)12ln(
2
1−=C
(2.82)
Genau wie die van-der-Waals-Gleichung enthält die Peng-Robinson-Zustandsgleichung neben
den Variablen der Temperatur, des Drucks und des bezogenen Volumens den Parameter a als
Maß für die anziehenden Kräfte und den Parameter b als Maß für die abstoßenden Kräfte
zwischen den Molekülen. Diese Parameter können aus den kritischen Daten der
Komponenten gewonnen werden. Für die in dieser Arbeit betrachteten nichtidealen System
wird auf die Modifikation nach Stryjek und Vera zurückgegriffen [STRYJEK, 1986a;
STRYJEK, 1986c]:
)(457235.0)(22
TP
TRTa
C
C α= (2.83a)
( )[ ]25.011)( rTT −+= κα (2.83b)
( )( )RR TT −++= 7.01 5.010 κκκ (2.83c)
320 0196554.017131848.04897153.1378893.0 ωωωκ +−+= (2.83d)
C
C
P
RTb 077796.0= (2.83e)
TC ist die kritische Temperatur, PC der kritische Druck, ω der azentrische Faktor, κ1 ein
stoffspezifische Konstante und TR die reduzierte Temperatur.
Die Einbindung des COSMO-RS-Modells erfolgt auf zwei Arten:
1. a-priori Vorhersage der Gibbsschen Exzessenthalpie für die Ermittlung der
Mischungsparameter a und b nach Gleichungen Gl. 2.75 - 2.77.
Bei diesem Vorgehen wird der binäre Wechselwirkungsparameter als anpassbarer
Parameter behandelt.
2. a-priori Vorhersage der Aktivitätskoeffizienten ∞ijγln bei unendlicher Verdünnung für
die Ermittlung der Mischungsparameter a und b nach Gleichungen Gl. 2.78 - 2.80
Hier wird der Wert des α-Parameters des modifizierten NRTL-Modells vorgegeben –
für die Breite der hier untersuchten Werte hat sich heraus gestellt, dass α =0.45 die
experimentellen Daten am besten wiedergibt. Der binäre Wechselwirkungsparameter
kij wird bei diesem Vorgehen zu Null gesetzt (kij =0)

3 Anwendbarkeit des COSMO-RS Modells zur Vorhersage von
thermodynamischen Stoffdaten in Mischungen
3.1 Vorhersage von Dampf-Flüssig und Flüssig-Flüssig Phasengleichgewichten
Wie in Kapitel 2.1 erläutert, existiert gegenwärtig eine große Anzahl an thermodynamischen
Modellen, die Interpolation und Extrapolation von experimentellen
Phasengleichgewichtsdaten erlauben und dem Prozessingenieur eine sichere Auslegung von
Verfahren ermöglichen und es gestatten, Abschätzungen im Vorfeld vorzunehmen. Sind keine
experimentellen Daten bekannt oder handelt es sich um Screening-Versuche im Vorfeld einer
Auslegung, bietet das COSMO-RS Modell eine geeignete Methode, um beispielsweise den
Einfluss von Zusatzstoffen auf das Phasengleichgewicht und die Trennoperation
vorherzusagen.
Das folgende Kapitel gibt einen Überblick über die Möglichkeiten der Vorhersage von
Dampf-Flüssig-Phasengleichgewichten und deren Genauigkeit.
3.1.1 Vorgehen und betrachtete Systeme
Mit Gleichung 2.28 liefert COSMO-RS den Aktivitätskoeffizienten einer Komponente in
einer Mischung. Die Phasengleichgewichtsbeziehung für Dampf-Flüssig
Phasengleichgewichte wird in ihrer vereinfachten Form angewendet. Assoziationen in der
Dampfphase werden vernachlässigt, der Pointing Faktor und das Verhältnis der
Fugazitätskoeffizienten werden zu Eins gesetzt.
LV
ii
L
i
V
i PxPx 0γ= (2.39)
Für Flüssig-Flüssig Phasengleichgewichte wird folgende Besziehung eingesetzt, wobei zu
beachten gilt, dass die komplette Gemischinformation vom Aktivitätskoeffizienten getragen
wird.
II
i
II
i
I
i
I
i xx γγ = (2.40)

41
In die Untersuchung der Einsetzbarkeit wurde ein breites Spektrum an Gemischen von
Komponenten betrachtet, die vor allem in der chemischen Verfahrenstechnik von Interesse
sind und einen Vergleich mit herkömmlichen Methoden zulassen. Das Hauptaugenmerk liegt
auf Mischungen der folgenden Stoffe:
• Alkane
• Alkene
• cyklische Alkane
• Alkohole
• Aromaten
• Ketone
• Ether
Thermodynamische Konsistenztests wurden für alle experimentellen Daten durchgeführt. Die
Methoden von Redlich und Kister [REDLICH, 1948] und Herrington [HERRINGTON, 1947]
sind dafür eingesetzt worden. Systeme, die diesen Konsistenztest nicht bestehen, sind nicht in
die Untersuchungen einbezogen. Die Reinstoffe sind nach Vorgabe einer Startstruktur mit der
Software HyperChem unter Anwendung der COSMO-Randbedingung mit der DFT-Methode
in der Software TURBOMOLE geometrieoptimiert (siehe auch Kapitel 3.1.2) worden.
Obwohl es möglich ist, mit dem COSMO-RS-Modell Dampfdrücke vorherzusagen, ist dieses
Vorgehen momentan noch zu ungenau und Gegenstandstand der Forschung (siehe Kapitel 1).
Da bei der Bewertung der Genauigkeit des COSMO-RS-Modells vor allem untersucht werden
soll, wie die Realität der flüssigen Phase beschrieben wird, werden für die Dampfdrücke der
reinen Komponenten sowohl experimentelle Daten als auch Korrelationen verwendet.
Experimentelle Daten werden immer dann verwendet, wenn sie durch die experimentellen
Daten vorgegeben sind und Korrelationen, falls keine experimentellen Daten vorliegen.
Tabelle 7.1 im Anhang 7.1 gibt einen Überblick über die verwendeten Systeme; die letzte
Spalte gibt Auskunft über das Vorhandensein von experimentellen Dampfdrücken.
Die Abweichung zwischen experimentellen und berechneten Drücken in Abhängigkeit von
Temperatur und Zusammensetzung, sowie die Abweichung zwischen experimentellem und
berechnetem K-Faktor wurde eingesetzt, um die Genauigkeit von COSMO-RS zu evaluieren.
Die Abweichungen sind dabei wie folgt gegeben:
[ ] 100% exp
exp
⋅−
=P
PPABW
berP (3.1)

42
[ ] 100% exp1
1exp1 ⋅
−=
K
KKABW
berK (3.2)
Der K-Faktor wird ausgedrückt als:
1
11
x
yK = (3.3)
wobei „1“ die leichter siedende Komponente im System darstellt.
3.1.2 Erstellung der Molekülgeometrien, Geometrieoptimierung und Berechnung der
Abschirmungsladungen
Alle in dieser Arbeit betrachteten Molekülstrukturen sind ausgehend von Startgeometrie unter
Nutzung der DFT Methode vollständig optimiert. Für das Erstellen der Startgeometrie wurde
die Molekülmodellierungssoftware HyperChem® Version 7 der Firma Hypercube Inc.
verwendet. Diese Software ermöglicht anhand einer zweidimensionalen Skizzenvorlage die
dreidimensionale Anordnung der Atome eines Moleküls mit Standardvorgaben für die
entsprechenden Atome von Bindungslänge, Bindungswinkel und Drehwinkel. So entsteht
zunächst eine angenäherte Geometrie, die mittels Geometrieoptimierung verfeinert wird. Für
die Geometrieoptimierung der angenäherten Struktur wurde in dieser Arbeit die
semiempirische Methode PM3 angewendet [STEWART, 1989a; STEWART, 1989b;
STEWART, 1991], die innerhalb von HyperChem arbeitet. Abb. 3.1 verdeutlicht dieses
Vorgehen.
C
C
C
C
C
C
Abb. 3.1 Erstellung von Molekülgeometrien mit HyperChem – links: zweidimensionale Skizzenvorlage, rechts:
PM3 optimierte dreidimensionale Struktur

43
Das Ergebnis der Molekülmodellierung mit HyperChem ist eine Koordinatentabelle, die die
Raumkoordinaten des betrachteten Moleküls enthält. Sie bilden die Grundlage für die
anschließende Geometrieoptimierung mit DFT. Die DFT Rechnungen wurden unter Nutzung
des Programmpakets TURBOMOLE [AHLRICHS, 1989] durchgeführt. Hierbei wurde die
die Coulomb-Wechselwirkungen berücksichtigende RI-DFT Variante [EICHKORN, 1995a;
EICHKORN, 1995b] verwendet. Eine Kombination des Austauschfunktionals von Becke
[BECKE, 1988] und dem Korrelationsfunktional nach Perdew [PERDEW, 1986],
üblicherweise als B-P Funktional bezeichnet, sind in allen Rechnungen zugrunde gelegt. Um
die Geometrien auf einem quantenmechanisch hohen Niveau zu optimieren, kam das Triple-
Zeta-Valence Potenzial (TZVP) [EICHKORN, 1997; SCHAFER, 1994] zum Einsatz. Die
Randbedingung des idealen Leiters wurde in der in TURBOMOLE implementierten Variante
von COSMO berücksichtigt [SCHAFER, 2000]. Damit werden simultan die
Abschirmungsladungen auf der Oberfläche des Moleküls zum Kontinuum, dem idealen
Leiter, berechnet.
3.1.3 Dampf-Flüssig Phasengleichgewichte
Die Ergebnisse der Vorhersagen der Dampf-Flüssig Phasengleichgewichte sind eingeteilt
nach der Art der Wechselwirkung der Moleküle bzw. Stoffe in der binären Mischung. Hier
treten nicht-assoziierende Systeme wie Alkan-Alkan Mischungen, selbst-assoziierende
Systeme wie Alkohol-Alkan Mischungen und kreuz-assoziierende Systeme wie Alkohol-
Alkohol Mischungen auf. Im Folgenden werden die Ergebnisse für jede dieser
Wechselwirkungsklasse dargestellt. Tabelle 3.1 gibt einen allgemeinen Überblick über die
berechneten Systeme, die Abweichung zu den experimentellen Daten in Prozent und die
Anzahl der zum Vergleich herangezogenen experimentellen Daten. Die mittleren
Abweichungen für alle Systeme liegen sowohl bzgl. des K-Faktors als auch bzgl. des Druckes
bei unter 4%. Abb. 3.2 zeigt die Abweichungen für Systeme mit mehr als drei Datensätzen
pro Gruppe, um einen allgemeinen Schluss ziehen zu dürfen. Die Abweichungen sind für
nicht-assoziierende kleiner als für assoziierende Systeme. Unpolare Moleküle und ihre
Mischungen zeigen eine Abweichung kleiner 4% (Alkane, Cykloalkane, Alkene, Ether). Das
Phasenverhalten wird dominiert von dispersiven Wechselwirkungen und die
Aktivitätskoeffizienten liegen im Bereich von Eins. Dieses Ergebnis könnte als trivial
bezeichnet werden, aber die Tatsache, dass in die Vorhersage des Aktivitätskoeffizienten nur
die atomare Struktur der Moleküle eingeht, hebt dieses Ergebnis als beachtlich hervor. Nicht-
assoziierende Systeme mit einer funktionellen Gruppe im Molekül zeigen eine höhere

44
Abweichung in Mischung mit unpolaren Molekülen. Die spezifischeren Wechselwirkungen
werden von COSMO-RS mit einer Abweichung von innerhalb 7% erfasst. Mischungen, die
selbst-assoziierende Moleküle enthalten, zeigen die höchsten Abweichungen in dieser
Untersuchung. Insbesondere das Phasenverhalten von Alkoholen in Mischung mit Ethern
kann nicht mit der gleichen Genauigkeit wie das der anderen Systeme vorhergesagt werden.
Als Grund kann die einfache, lineare Mischung der Sigmaprofile der Reinstoffe angesehen
werden, mit der die Mischung repräsentiert wird (vgl. Kapitel 2.3). In einem Ensemble, das
ehemals nur selbst-assoziierende und nur nicht-assoziierende Moleküle enthalten hat, führt die
lineare Mischung ohne Information von mehr oder weniger wahrscheinlichen
Wechselwirkungszentren der Oberflächenstücke offensichtlich zu Fehlinterpretationen der
Wechselwirkungsenergie.
Mischungen selbst-assoziierender Komponenten zeigen ein diversifiziertes Verhalten. Die
Aktivitätskoeffizienten in Alkohol-Alkohol Systemen sind nahe Eins. Dieses Verhalten wird
von COSMO-RS gut wiedergegeben und die Abweichung ist kleiner als 2%. Alkohole in
Mischung mit Ketonen weisen eine kreuz-assoziierende Wechselwirkung der Hydroxyl-
Gruppe mit der Keto-Gruppe auf. Das reale Verhalten zeigt höhere Aktivitätskoeffizienten als
COSMO-RS dies vorhersagt. Bei unendlicher Verdünnung führt dies zu Abweichungen von
größer als 10%. Klamt und Eckert berichten von einer Abweichung des vorhergesagten
chemischen Potenzials und des experimentellen im Bereich von etwa 1,7kJ/mol [KLAMT,
2000]. Im Bereich der unendlichen Verdünnung ist eine Evaluation möglich, da hier gilt:
∆=
∞
∞−
RT
l
i
i
RSCOSMOi µ
γ
γexp
exp,
, (3.4)
Die oben angegebene Abweichung von 10% führt damit zu Fehlern des
Aktivitätskoeffizienten bei unendlicher Verdünnung vom Faktor 2.
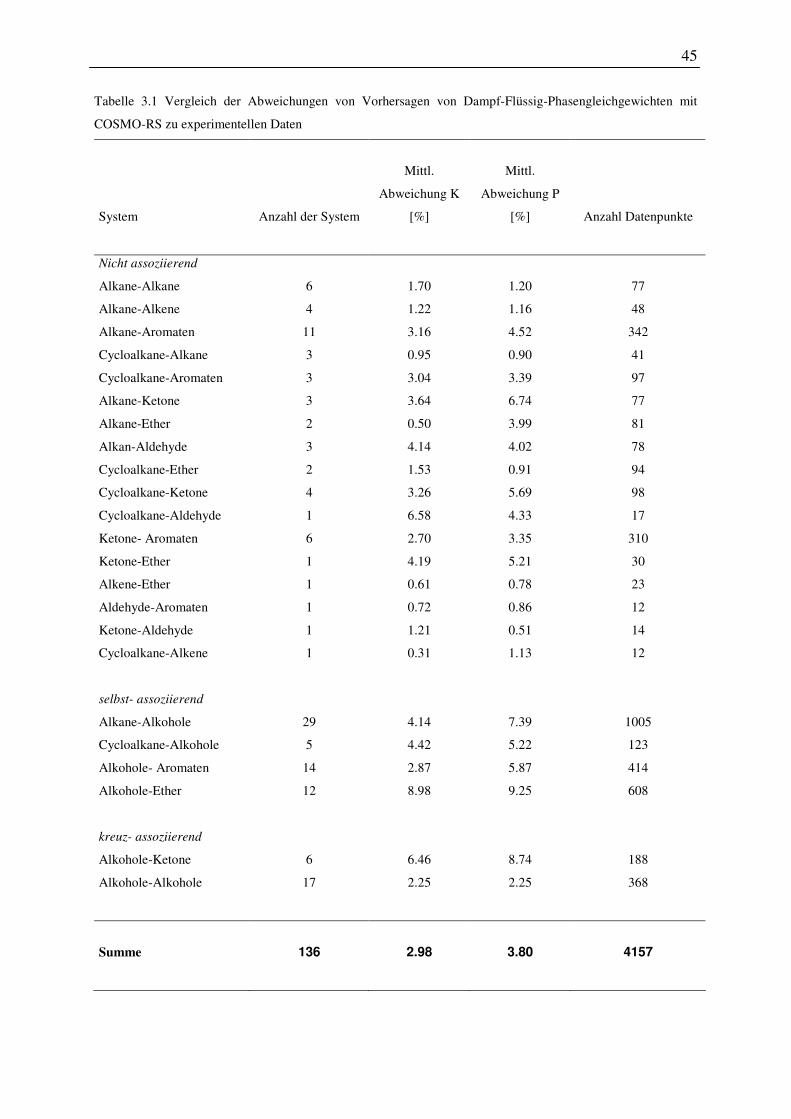
45
Tabelle 3.1 Vergleich der Abweichungen von Vorhersagen von Dampf-Flüssig-Phasengleichgewichten mit
COSMO-RS zu experimentellen Daten
System Anzahl der System
Mittl.
Abweichung K
[%]
Mittl.
Abweichung P
[%] Anzahl Datenpunkte
Nicht assoziierend
Alkane-Alkane 6 1.70 1.20 77
Alkane-Alkene 4 1.22 1.16 48
Alkane-Aromaten 11 3.16 4.52 342
Cycloalkane-Alkane 3 0.95 0.90 41
Cycloalkane-Aromaten 3 3.04 3.39 97
Alkane-Ketone 3 3.64 6.74 77
Alkane-Ether 2 0.50 3.99 81
Alkan-Aldehyde 3 4.14 4.02 78
Cycloalkane-Ether 2 1.53 0.91 94
Cycloalkane-Ketone 4 3.26 5.69 98
Cycloalkane-Aldehyde 1 6.58 4.33 17
Ketone- Aromaten 6 2.70 3.35 310
Ketone-Ether 1 4.19 5.21 30
Alkene-Ether 1 0.61 0.78 23
Aldehyde-Aromaten 1 0.72 0.86 12
Ketone-Aldehyde 1 1.21 0.51 14
Cycloalkane-Alkene 1 0.31 1.13 12
selbst- assoziierend
Alkane-Alkohole 29 4.14 7.39 1005
Cycloalkane-Alkohole 5 4.42 5.22 123
Alkohole- Aromaten 14 2.87 5.87 414
Alkohole-Ether 12 8.98 9.25 608
kreuz- assoziierend
Alkohole-Ketone 6 6.46 8.74 188
Alkohole-Alkohole 17 2.25 2.25 368
Summe 136 2.98 3.80 4157

46
0
1
2
3
4
5
6
7
8
9
10
Alkan-A
lkan
Alkan-A
lken
Cycloalka
n-Alka
n
Alkan-A
rom
at
Cycloalka
n-Aro
mat
Alkan-K
eton
Cycloalka
n-Keto
n
Alkan-A
ldehyd
Keton-
Arom
at
Alkan-A
lkohol
Cycloalka
n-Alko
hol
Alkohol-A
rom
at
Alkohol-E
ther
Alkohol-K
eton
Alkohol-A
lkohol
Ab
we
ich
un
g [
%]
K-Faktor
Druck
Abb. 3.2 Übersicht der Abweichungen für Mischungen
Im Folgenden wird näher auf die Spezifika der Wechselwirkungen und der Güte der
Vorhersagen für die einzelnen Systemklassen eingegangen.
Nicht assoziierende Mischungen
Mischungen von Alkanen zeigen ein nahezu ideales Phasenverhalten. Dies wird in allen
Fällen von COSMO-RS vorhergesagt. Mischungen von Alkanen mit Aromaten weisen eine
leichte Abweichung vom Roultschen Gesetz auf. Untersucht worden sind Mischungen mit
Benzol, Toluol, p-Xylol und Ethylbenzol, wobei die Kettenlänge der Alkane hierbei von C5
bis C10 variiert. Abb. 3.3 zeigt die Abweichungen in den untersuchten Systemen. Allgemein
ist zu erkennen, dass die Abweichung im Druck mit steigender Kettenlänge des Alkans
ebenfalls steigt. Eine Ausnahme bildet das Ethylbenzol. Bei konstanter Kettenlänge des
Alkans und unter gleichzeitigem Hinzufügen von Alkylresten zur aromatischen Struktur sinkt
die Abweichung zwischen Vorhersage und Experiment (vgl. Abb. 3.3 n-Heptan-Aromat). Der
Grund der Abweichung kann an den zu hoch vorhergesagten Aktivitätskoeffizienten beider
Komponenten in Mischung abgelesen werden. Abb. 3.4 zeigt beispielhaft die Verläufe der
Aktivitätskoeffizienten im System n-Heptan-Benzol bei 333,15K. Ähnliche Ergebnisse sind
für alle Mischungen dieser Art gefunden worden.
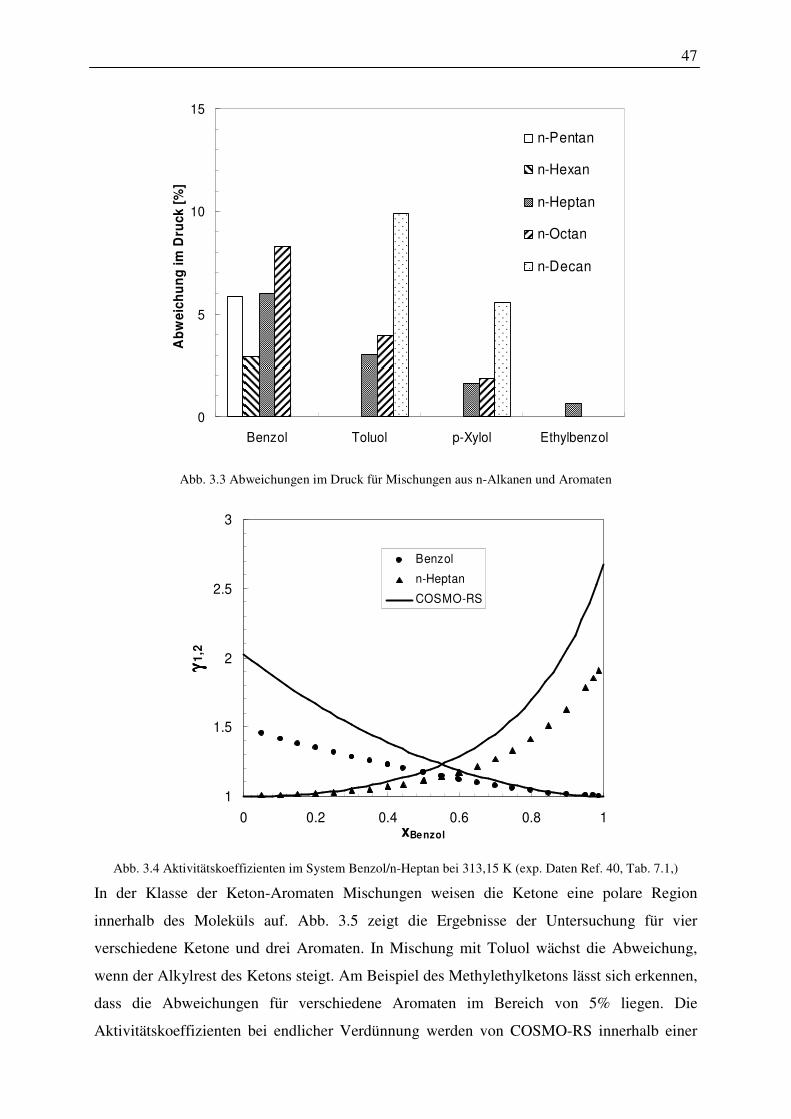
47
0
5
10
15
Benzol Toluol p-Xylol Ethylbenzol
Ab
we
ich
un
g i
m D
ruc
k [
%]
n-Pentan
n-Hexan
n-Heptan
n-Octan
n-Decan
Abb. 3.3 Abweichungen im Druck für Mischungen aus n-Alkanen und Aromaten
1
1.5
2
2.5
3
0 0.2 0.4 0.6 0.8 1xBenzol
γγ γγ1
,2
Benzol
n-Heptan
COSMO-RS
Abb. 3.4 Aktivitätskoeffizienten im System Benzol/n-Heptan bei 313,15 K (exp. Daten Ref. 40, Tab. 7.1,)
In der Klasse der Keton-Aromaten Mischungen weisen die Ketone eine polare Region
innerhalb des Moleküls auf. Abb. 3.5 zeigt die Ergebnisse der Untersuchung für vier
verschiedene Ketone und drei Aromaten. In Mischung mit Toluol wächst die Abweichung,
wenn der Alkylrest des Ketons steigt. Am Beispiel des Methylethylketons lässt sich erkennen,
dass die Abweichungen für verschiedene Aromaten im Bereich von 5% liegen. Die
Aktivitätskoeffizienten bei endlicher Verdünnung werden von COSMO-RS innerhalb einer
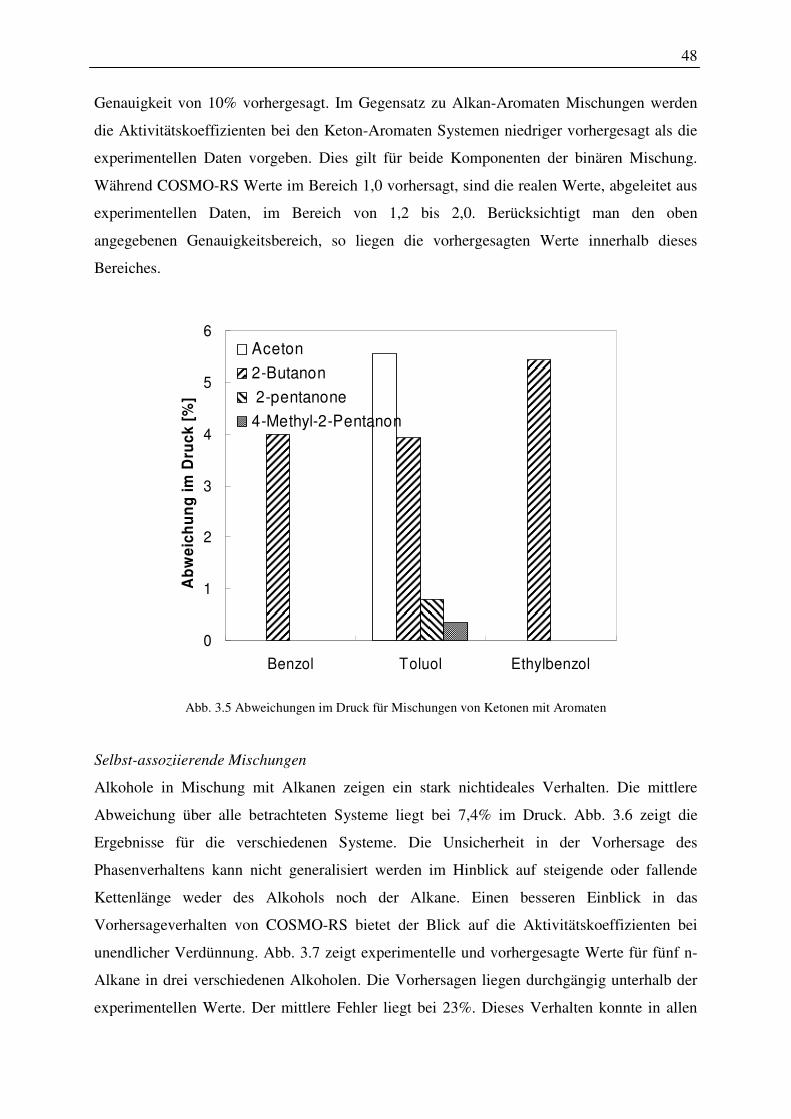
48
Genauigkeit von 10% vorhergesagt. Im Gegensatz zu Alkan-Aromaten Mischungen werden
die Aktivitätskoeffizienten bei den Keton-Aromaten Systemen niedriger vorhergesagt als die
experimentellen Daten vorgeben. Dies gilt für beide Komponenten der binären Mischung.
Während COSMO-RS Werte im Bereich 1,0 vorhersagt, sind die realen Werte, abgeleitet aus
experimentellen Daten, im Bereich von 1,2 bis 2,0. Berücksichtigt man den oben
angegebenen Genauigkeitsbereich, so liegen die vorhergesagten Werte innerhalb dieses
Bereiches.
0
1
2
3
4
5
6
Benzol Toluol Ethylbenzol
Ab
we
ich
un
g i
m D
ruc
k [
%]
Aceton
2-Butanon
2-pentanone
4-Methyl-2-Pentanon
Abb. 3.5 Abweichungen im Druck für Mischungen von Ketonen mit Aromaten
Selbst-assoziierende Mischungen
Alkohole in Mischung mit Alkanen zeigen ein stark nichtideales Verhalten. Die mittlere
Abweichung über alle betrachteten Systeme liegt bei 7,4% im Druck. Abb. 3.6 zeigt die
Ergebnisse für die verschiedenen Systeme. Die Unsicherheit in der Vorhersage des
Phasenverhaltens kann nicht generalisiert werden im Hinblick auf steigende oder fallende
Kettenlänge weder des Alkohols noch der Alkane. Einen besseren Einblick in das
Vorhersageverhalten von COSMO-RS bietet der Blick auf die Aktivitätskoeffizienten bei
unendlicher Verdünnung. Abb. 3.7 zeigt experimentelle und vorhergesagte Werte für fünf n-
Alkane in drei verschiedenen Alkoholen. Die Vorhersagen liegen durchgängig unterhalb der
experimentellen Werte. Der mittlere Fehler liegt bei 23%. Dieses Verhalten konnte in allen

49
untersuchten Alkohol-Alkan System gezeigt werden. Demgegenüber weisen die Vorhersagen
bei unendlicher Verdünnung für Alkohole in Alkanen immer größere Werte auf als die
experimentellen Daten indizieren. Abb. 3.8 zeigt dieses beispielhaft für Ethanol in vier
verschiedenen Alkanen. Die Abweichung gegenüber den experimentellen Werten liegt bei
durchschnittlich 44%. Diese Überschätzung führt zu den Abweichungen in den berechneten
Drücken der Phasengleichgewichte. Trotzdem ist die Vorhersage des engsiedenden,
azeotropen oder heteroazeotropen Verhaltens aller untersuchten Systeme gegeben.
0
2
4
6
8
10
12
14
16
18
20
Meth
anol
Ethan
ol
n-Pro
panol
Isopr
opanol
n-Butan
ol
Isobu
tanol
2-Butan
ol
tert-
Butano
l
n-Penta
nol
2-Penta
nol
3-Penta
nol
2-M
ethyl-
1-but
anol
3-M
ethyl-
1-but
anol
n-Hex
anol
∆∆ ∆∆P
/ %
Isobutan
n-Pentan
n-Hexan
n-Heptan
n-Oktan
2,2,4-Trimethylpentan
Abb. 3.6 Abweichungen im Druck für Mischungen von Alkoholen mit Alkanen
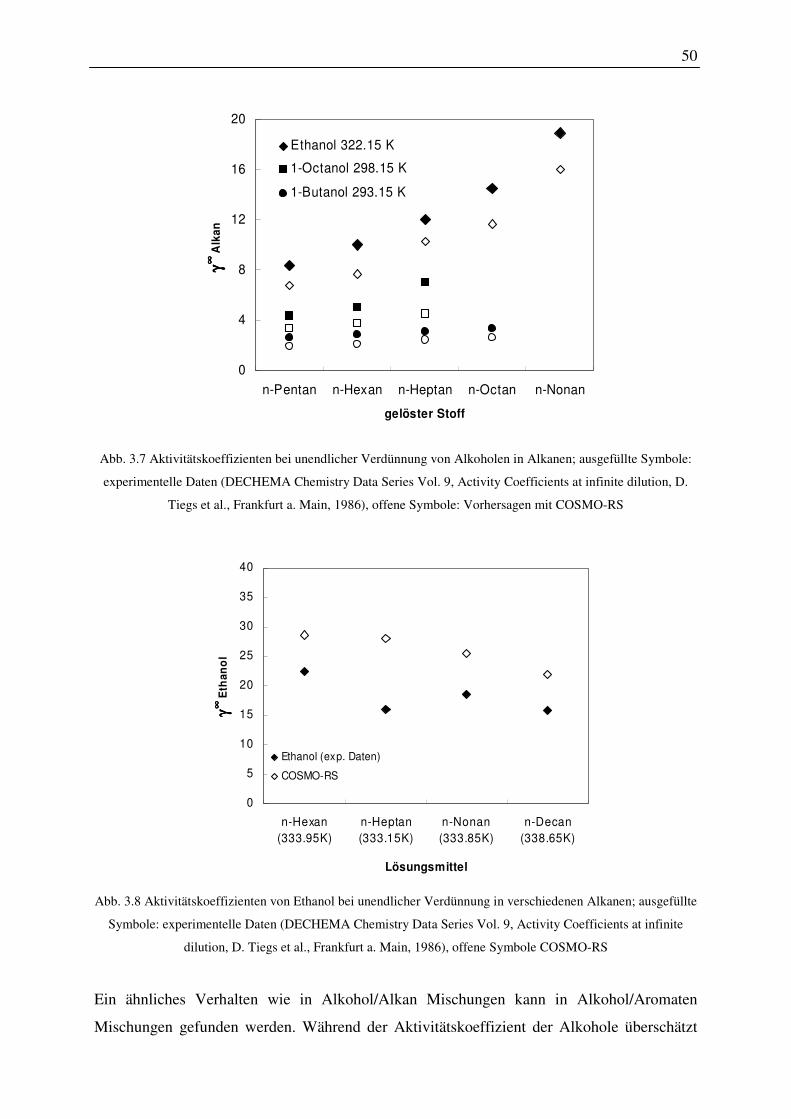
50
0
4
8
12
16
20
n-Pentan n-Hexan n-Heptan n-Octan n-Nonan
gelöster Stoff
γγ γγ∞∞ ∞∞
Alk
an
Ethanol 322.15 K
1-Octanol 298.15 K
1-Butanol 293.15 K
Abb. 3.7 Aktivitätskoeffizienten bei unendlicher Verdünnung von Alkoholen in Alkanen; ausgefüllte Symbole:
experimentelle Daten (DECHEMA Chemistry Data Series Vol. 9, Activity Coefficients at infinite dilution, D.
Tiegs et al., Frankfurt a. Main, 1986), offene Symbole: Vorhersagen mit COSMO-RS
0
5
10
15
20
25
30
35
40
n-Hexan(333.95K)
n-Heptan(333.15K)
n-Nonan(333.85K)
n-Decan(338.65K)
Lösungsmittel
γγ γγ∞∞ ∞∞
Eth
an
ol
Ethanol (exp. Daten)
COSMO-RS
Abb. 3.8 Aktivitätskoeffizienten von Ethanol bei unendlicher Verdünnung in verschiedenen Alkanen; ausgefüllte
Symbole: experimentelle Daten (DECHEMA Chemistry Data Series Vol. 9, Activity Coefficients at infinite
dilution, D. Tiegs et al., Frankfurt a. Main, 1986), offene Symbole COSMO-RS
Ein ähnliches Verhalten wie in Alkohol/Alkan Mischungen kann in Alkohol/Aromaten
Mischungen gefunden werden. Während der Aktivitätskoeffizient der Alkohole überschätzt
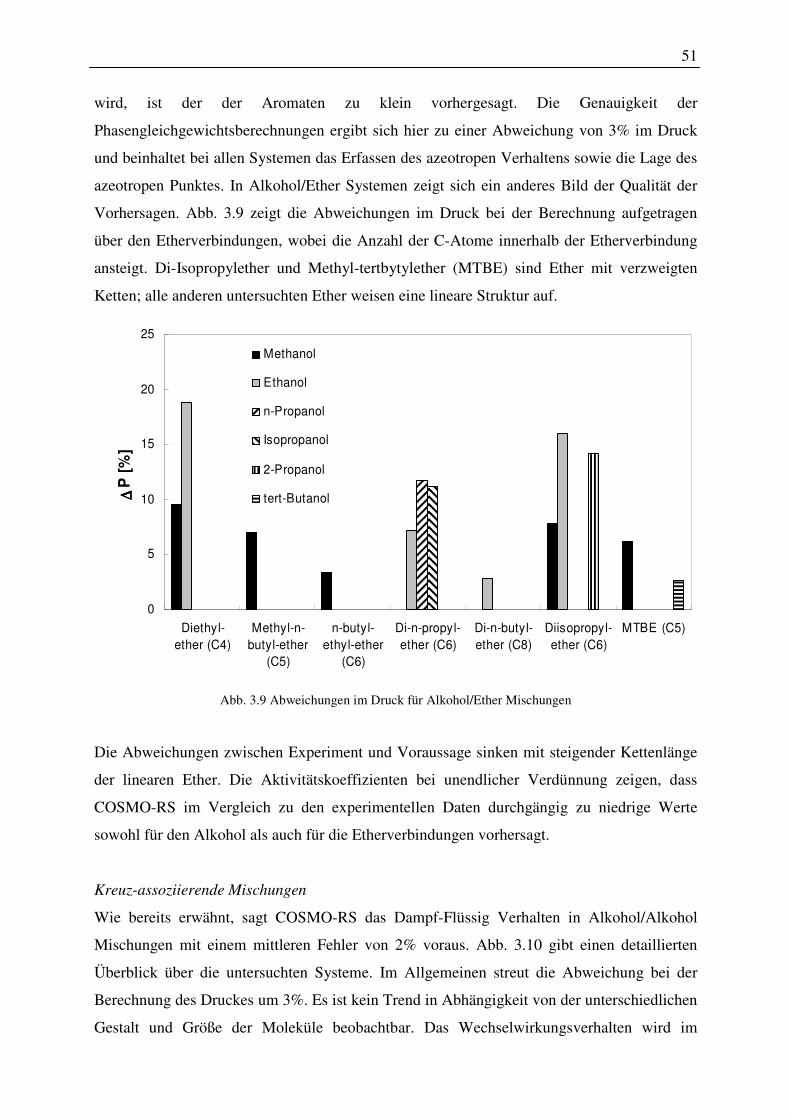
51
wird, ist der der Aromaten zu klein vorhergesagt. Die Genauigkeit der
Phasengleichgewichtsberechnungen ergibt sich hier zu einer Abweichung von 3% im Druck
und beinhaltet bei allen Systemen das Erfassen des azeotropen Verhaltens sowie die Lage des
azeotropen Punktes. In Alkohol/Ether Systemen zeigt sich ein anderes Bild der Qualität der
Vorhersagen. Abb. 3.9 zeigt die Abweichungen im Druck bei der Berechnung aufgetragen
über den Etherverbindungen, wobei die Anzahl der C-Atome innerhalb der Etherverbindung
ansteigt. Di-Isopropylether und Methyl-tertbytylether (MTBE) sind Ether mit verzweigten
Ketten; alle anderen untersuchten Ether weisen eine lineare Struktur auf.
0
5
10
15
20
25
Diethyl-ether (C4)
Methyl-n-butyl-ether
(C5)
n-butyl-ethyl-ether
(C6)
Di-n-propyl-ether (C6)
Di-n-butyl-ether (C8)
Diisopropyl-ether (C6)
MTBE (C5)
∆∆ ∆∆P
[%
]
Methanol
Ethanol
n-Propanol
Isopropanol
2-Propanol
tert-Butanol
Abb. 3.9 Abweichungen im Druck für Alkohol/Ether Mischungen
Die Abweichungen zwischen Experiment und Voraussage sinken mit steigender Kettenlänge
der linearen Ether. Die Aktivitätskoeffizienten bei unendlicher Verdünnung zeigen, dass
COSMO-RS im Vergleich zu den experimentellen Daten durchgängig zu niedrige Werte
sowohl für den Alkohol als auch für die Etherverbindungen vorhersagt.
Kreuz-assoziierende Mischungen
Wie bereits erwähnt, sagt COSMO-RS das Dampf-Flüssig Verhalten in Alkohol/Alkohol
Mischungen mit einem mittleren Fehler von 2% voraus. Abb. 3.10 gibt einen detaillierten
Überblick über die untersuchten Systeme. Im Allgemeinen streut die Abweichung bei der
Berechnung des Druckes um 3%. Es ist kein Trend in Abhängigkeit von der unterschiedlichen
Gestalt und Größe der Moleküle beobachtbar. Das Wechselwirkungsverhalten wird im
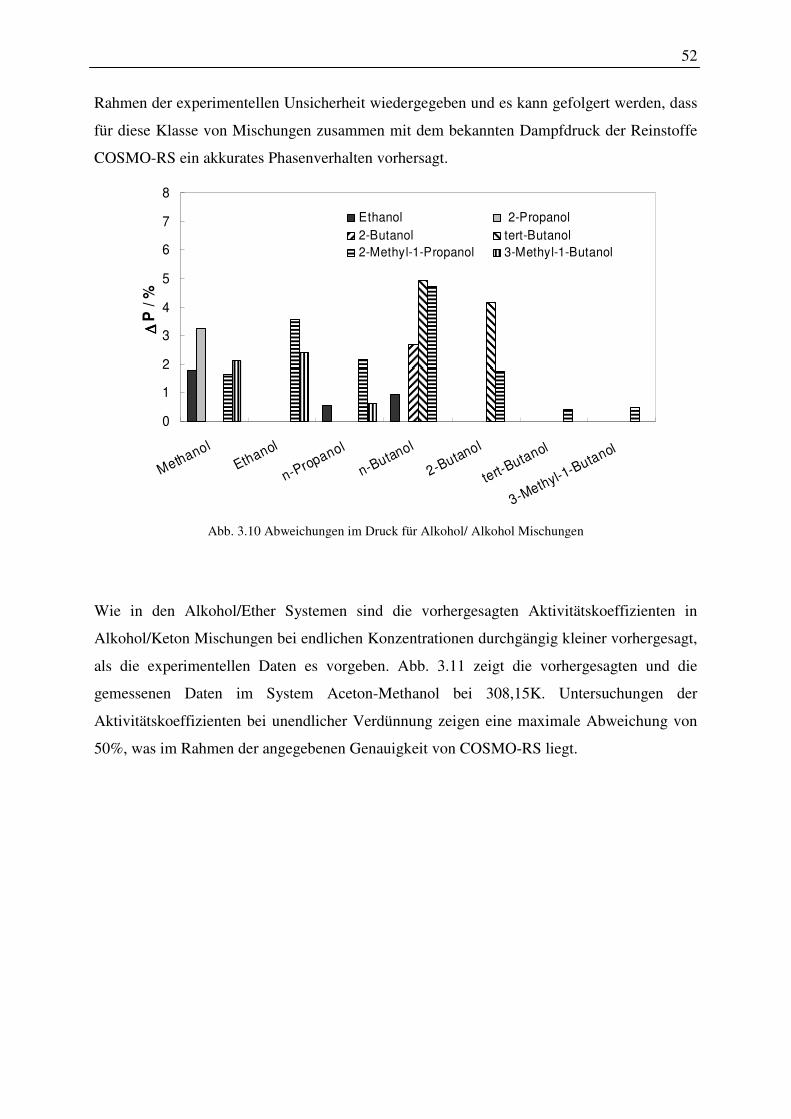
52
Rahmen der experimentellen Unsicherheit wiedergegeben und es kann gefolgert werden, dass
für diese Klasse von Mischungen zusammen mit dem bekannten Dampfdruck der Reinstoffe
COSMO-RS ein akkurates Phasenverhalten vorhersagt.
0
1
2
3
4
5
6
7
8
MethanolEthanol
n-Propanol
n-Butanol
2-Butanol
tert-Butanol
3-Methyl-1-Butanol
∆∆ ∆∆P
/ %
Ethanol 2-Propanol2-Butanol tert-Butanol2-Methyl-1-Propanol 3-Methyl-1-Butanol
Abb. 3.10 Abweichungen im Druck für Alkohol/ Alkohol Mischungen
Wie in den Alkohol/Ether Systemen sind die vorhergesagten Aktivitätskoeffizienten in
Alkohol/Keton Mischungen bei endlichen Konzentrationen durchgängig kleiner vorhergesagt,
als die experimentellen Daten es vorgeben. Abb. 3.11 zeigt die vorhergesagten und die
gemessenen Daten im System Aceton-Methanol bei 308,15K. Untersuchungen der
Aktivitätskoeffizienten bei unendlicher Verdünnung zeigen eine maximale Abweichung von
50%, was im Rahmen der angegebenen Genauigkeit von COSMO-RS liegt.
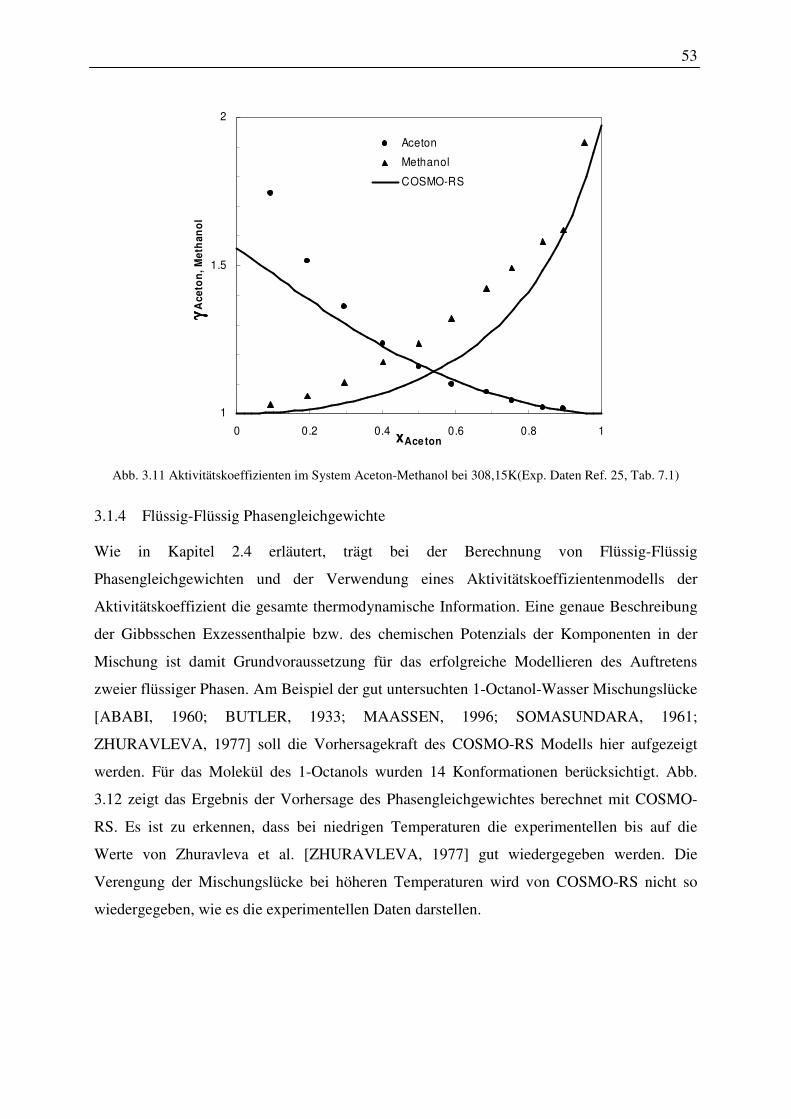
53
1
1.5
2
0 0.2 0.4 0.6 0.8 1xAceton
γγ γγA
ce
ton
, M
eth
an
ol
Aceton
Methanol
COSMO-RS
Abb. 3.11 Aktivitätskoeffizienten im System Aceton-Methanol bei 308,15K(Exp. Daten Ref. 25, Tab. 7.1)
3.1.4 Flüssig-Flüssig Phasengleichgewichte
Wie in Kapitel 2.4 erläutert, trägt bei der Berechnung von Flüssig-Flüssig
Phasengleichgewichten und der Verwendung eines Aktivitätskoeffizientenmodells der
Aktivitätskoeffizient die gesamte thermodynamische Information. Eine genaue Beschreibung
der Gibbsschen Exzessenthalpie bzw. des chemischen Potenzials der Komponenten in der
Mischung ist damit Grundvoraussetzung für das erfolgreiche Modellieren des Auftretens
zweier flüssiger Phasen. Am Beispiel der gut untersuchten 1-Octanol-Wasser Mischungslücke
[ABABI, 1960; BUTLER, 1933; MAASSEN, 1996; SOMASUNDARA, 1961;
ZHURAVLEVA, 1977] soll die Vorhersagekraft des COSMO-RS Modells hier aufgezeigt
werden. Für das Molekül des 1-Octanols wurden 14 Konformationen berücksichtigt. Abb.
3.12 zeigt das Ergebnis der Vorhersage des Phasengleichgewichtes berechnet mit COSMO-
RS. Es ist zu erkennen, dass bei niedrigen Temperaturen die experimentellen bis auf die
Werte von Zhuravleva et al. [ZHURAVLEVA, 1977] gut wiedergegeben werden. Die
Verengung der Mischungslücke bei höheren Temperaturen wird von COSMO-RS nicht so
wiedergegeben, wie es die experimentellen Daten darstellen.
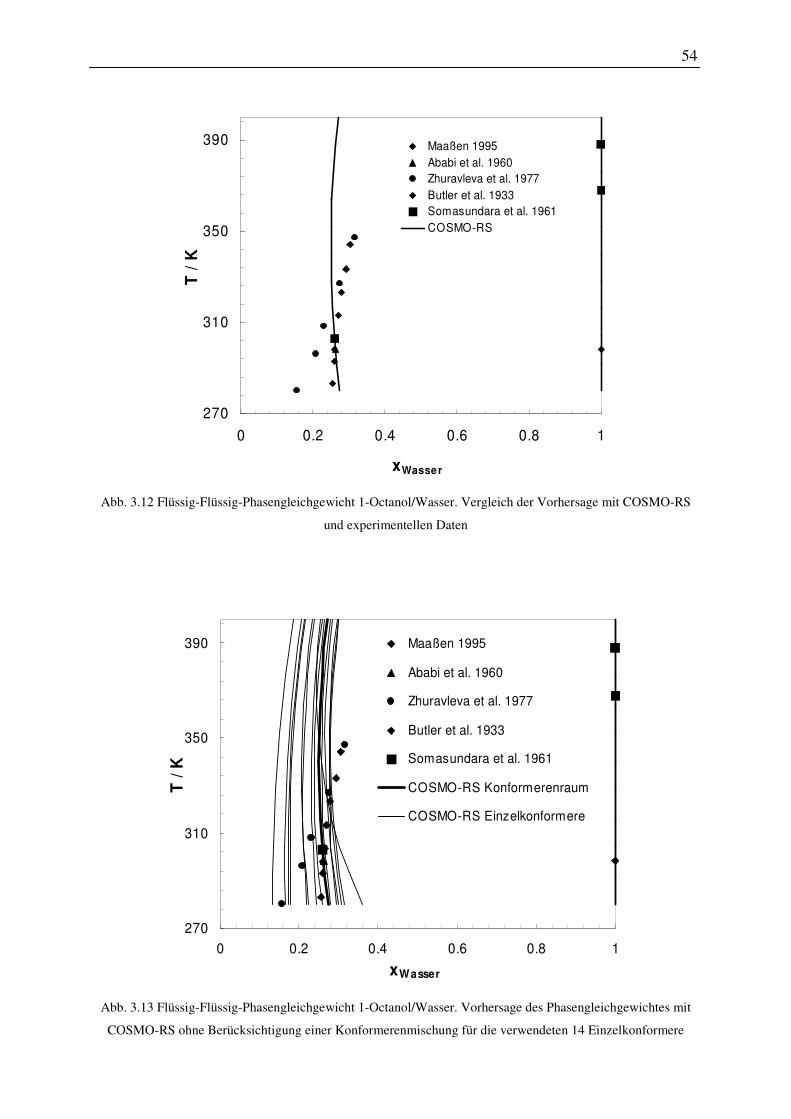
54
270
310
350
390
0 0.2 0.4 0.6 0.8 1
xWasser
T /
K
Maaßen 1995Ababi et al. 1960Zhuravleva et al. 1977Butler et al. 1933Somasundara et al. 1961COSMO-RS
Abb. 3.12 Flüssig-Flüssig-Phasengleichgewicht 1-Octanol/Wasser. Vergleich der Vorhersage mit COSMO-RS
und experimentellen Daten
270
310
350
390
0 0.2 0.4 0.6 0.8 1
xWasser
T / K
Maaßen 1995
Ababi et al. 1960
Zhuravleva et al. 1977
Butler et al. 1933
Somasundara et al. 1961
COSMO-RS Konformerenraum
COSMO-RS Einzelkonformere
Abb. 3.13 Flüssig-Flüssig-Phasengleichgewicht 1-Octanol/Wasser. Vorhersage des Phasengleichgewichtes mit
COSMO-RS ohne Berücksichtigung einer Konformerenmischung für die verwendeten 14 Einzelkonformere

55
Abb. 3.13 zeigt die Vorhersage des Flüssig-Flüssig-Phasengleichgewichtes unter Verwendung
von jeweils nur einer Konformation. Es ist zu erkennen, dass die Ergebnisse innerhalb des
Konformerenraumes stark streuen. In dem hier gewählten Beispiel wird der Einfluss
besonders in der octanolreichen Phase ersichtlich. Er ist allerdings auch in der in der Abb.
3.12 nicht aufgelösten wasserreichen Phase vorhanden. Insbesondere bei Flüssig-Flüssig
Phasengleichgewichtsberechnungen ist die Berücksichtigung aller in der
Konformationsanalyse ermittelten Konformationen von großer Bedeutung.
Zusammenfassende Betrachtungen
Die Validierung von 136 Systemen hat gezeigt, dass COSMO-RS experimentelle Daten von
Dampf-Flüssig-Phasengleichgewichtsdaten unterschiedlicher Stoffklassen in guter
Übereinstimmung vorhersagen kann, wenn der Dampfdruck der Reinstoffe bekannt ist und
das Modell als Aktivitätskoeffizientenmodell verwendet wird. Die Abweichungen zwischen
den Vorhersagen und den gemessenen Daten liegen im Vergleich zu den experimentellen
Daten für viele Systeme bei 5%. Mischungen nicht-assoziierender Komponenten werden
nahezu exakt beschrieben. Mischungen, die ein spezifischeres Wechselwirkungsverhalten
aufweisen, können nicht mit der gleichen Genauigkeit vorhergesagt werden, jedoch wird das
nichtideale Verhalten gut reproduziert; auftretende azeotrope Punkte oder heteroazeotropes
Verhalten werden erfasst.
Die Vorhersage von Flüssig-Flüssig-Phasengleichgewichten zeigt, dass es möglich ist, auf
Basis der Molekülstruktur mit COSMO-RS eine Entmischung zu modellieren. Die Güte der
Vorhersage hängt stark von der verwendeten Konformation des Moleküls ab. Aufgrund der
modellbedingten Unsicherheiten bei der Vorhersage der Aktivitätskoeffizienten der
Komponenten in der Mischung, ist eine exakte Wiedergabe der experimentellen Daten nicht
möglich.

4 Erweiterung des COSMO-RS Modells zur Vorhersage von
Reinstoffeigenschaften und der Identifizierung von relevanten
Strukturkonformationen
In Kapitel 2.3 wird gezeigt, dass im gegenwärtigen Energiekonzept von COSMO-RS die
Änderung der freien Enthalpie der Lösung durch die Bildung einer Kavität im Lösungsmittel
nicht explizit enthalten ist. Größeneffekte, bzw. entropische Effekte werden durch einen
kombinatorischen Anteil im chemischen Potential modelliert, bei dem der Staverman-
Guggenheim-Term Verwendung findet [KLAMT, 2000]. Im Folgenden wird vorgestellt, wie
dieser kombinatorische Anteil durch ein Fluidmodell für harte Kugeln ersetzt wird. Dieser
Anteil wird in der vorliegenden Arbeit mit der Scaled-Particle-Theorie (SPT) [REISS, 1965]
berechnet. Die dieser Theorie zugrunde liegenden Annahmen, beruhen auf der Ermittlung der
notwendigen Arbeit des Einfügens einer harten Kugel in ein Fluid harter Kugeln. Es wird
zunächst kurz erläutert, wie die SPT genutzt werden kann, um die Terme ∆G*cav und ∆G*
rep
zu beschreiben.
Die Scaled-Particle-Theorie für Harte Kugeln [PIEROTTI, 1976; REISS, 1959; REISS, 1971;
REISS, 1965; TULLY-SMITH, 1970] wird angewendet, um einen Ausdruck für die Terme
∆G*cav und ∆G*
rep in das COSMO-RS-Modell zu implementieren. Mit diesen Änderungen
werden Dampfdrücke bei der Normalsiedetemperatur verschiedener Substanzen vorhergesagt.
Zusätzlich werden die Vorhersagen über den Reinstoff genutzt, die relevanten
Konformationen für Phasengleichgewichtsberechnungen in Mehrstoffsystemen zu
identifizieren.
4.1 Die Scaled-Particle-Theorie (SPT)
4.1.1 Grundlagen zur Scaled-Particle-Theorie
In einer Reihe von Artikeln haben Reiss, Frisch, Lebowitz und Tully-Smith eine Fluid-
Theorie [HARRIS, 1971; LEBOWITZ, 1965; REISS, 1959; REISS, 1961; REISS, 1971;
REISS, 1960; REISS, 1965] entwickelt, die auf den Eigenschaften einer exakten radialen
Verteilungsfunktion beruht. Diese Theorie führt zu einem Näherungsausdruck für die
reversible Arbeit, die notwendig ist, um ein sphärisches Teilchen in ein Fluid aus sphärischen
Teilchen einzubringen. Es wird dort ein System aus N Teilchen mit paarweise
wechselwirkendem Potential betrachtet, in das ein Teilchen mit demselben Potential eingefügt
wird.

57
Der Grundgedanke der SPT liegt darin, dass Arbeit erforderlich ist, um Mittelpunkte von
Molekülen von einem spezifizierten Bereich im Fluid auszuschließen. Es sei angenommen,
das betrachtete Fluid bestehe aus N sphärischen, symmetrischen Molekülen mit einem
Durchmesser σ1 und einem Harte-Kugel-Potential. Weiterhin wird angenommen, dass die
Mittelpunkte aller N Moleküle von einem sphärischen Bereich des Durchmessers r im
Volumen V ausgeschlossen seien. Dieser Bereich stellt die zu betrachtende Kavität dar. Die
Wahrscheinlichkeit, dass die Kavität existiert, wird mit p0(r,ρ) bezeichnet, wobei ρ die
Anzahldichte des Fluids (N/V) ist. Die Kavität wird durch statistische Schwankungen gebildet
und kann durch die Wahrscheinlichkeit des Auftretens solcher Schwankungen durch einen
Boltzmann-Term ausgedrückt werden:
)/),(exp(),(0 kTrWrp ρρ −= (4.1)
In diesem Ausdruck (Gl. 4.1) ist W(r,ρ) die reversible Arbeit, die notwendig ist, eine Kavität
vom Radius r im Fluid zu etablieren. Das chemische Potential der in das Fluid eingebrachten
sphärischen Teilchen ist gegeben durch:
)),(ln( 0 ρµ
rpkT
−= (4.2)
Mit der SPT wird p0(r,ρ) auf der Grundlage der statistischen Mechanik und geometrischen
Überlegungen bestimmt. Der Grundgedanke zielt darauf ab, mit einer Kavität vom
Durchmesser r=0 zu starten und auf den Radius r=rKavität zu skalieren.
Zur Berechnung von W(r,ρ) ist es instruktiv, die Wahrscheinlichkeit zu betrachten, mit der
man den Mittelpunkt eines Moleküls innerhalb einer „Fluid-Schale“ zwischen r und r+dr
findet. Diese Wahrscheinlichkeit ist durch 4πr²ρG(r,ρ)dr gegeben. Die Wahrscheinlichkeit,
keinen Molekülmittelpunkt in dieser Schale anzutreffen, ist durch 1-4πr²ρG(r,ρ)dr gegeben.
Die Größe G(r,ρ) kennzeichnet den Wert der radialen Verteilungsfunktion des Fluids am
Kontaktpunkt von Kavität und Molekül. Vergleicht man die Wahrscheinlichkeit, keinen
Molekülmittelpunkt im Bereich von r=0 bis r und von r bis r+dr anzutreffen, mit dem
Ausdruck für die Wahrscheinlichkeit der Bildung der Kavität durch statistische
Schwankungen, erhält man folgenden Beziehung zwischen der reversiblen Arbeit zur
Schaffung der Kavität und der Struktur des Fluids. Sie ist gegeben durch die radiale
Verteilungsfunktion:
∫=r
drrGrkTrW0
2 ),(4/),( ρπρρ (4.3)
Reiss, Frisch, Lebowitz und Tully-Smith haben gezeigt [REISS, 1959; TULLY-SMITH,
1970], dass der Ausdruck G(r,ρ) durch eine Summe in 1/r repräsentiert werden kann:

58
∑=i
i
i rGrG )/1)((),( ρρ (4.4)
Die Faktoren Gi(ρ) müssen an exakten Ausdrücken für G(r,ρ) bestimmt werden. Aus einer
Vielzahl solcher exakten Ausdrücke für verschiedene r haben Tully-Smith und Reiss
[TULLY-SMITH, 1970] die Faktoren bis einschließlich G5 bestimmt. Es hat sich gezeigt,
dass eine Entwicklung der Art
33
2210),( rKrKrKKrW +++=ρ (4.5)
eine gute Näherung für W(r,ρ) ist. Genau wie die Faktoren Gi können mit exakten
Ausdrücken die Faktoren Ki in Gleichung 4.15 ermittelt werden, was zu folgender Form der
reversiblen Arbeit führt, die für die Schaffung der Kavität erforderlich ist [BEN AMOTZ,
1993a; BEN AMOTZ, 1993b; BEN AMOTZ, 1993c; DE SOUZA, 1994]:
)1ln()1(
)23
3(
)1(
)29
2(
)1(
2) 32
2
3
32
ηηη
η
η
ηη
η
η−−+
−
−+
−
++
−= R
RRRRR
kt
W(R,ρ (4.6)
wobei η=πρσ13/6 und R=σ2/σ1 ausdrückt. σ2 ist der Harte-Kugel-Durchmesser des gelösten
Moleküls und (σ1+σ2)/2 der Radius der Kavität. Die Größe W(r,ρ) ist gleich der partiellen
molaren freien Enthalpie cavg bzw. dem chemischen Potential der Komponente i im
Lösungsmittel S X
combSµ (siehe Gleichung 2.17). Damit kann nach Entfernen des
kombinatorischen Anteils, die Gleichung um diesen Term erweitert werden und wird zu
SPTcav
iii
iS
i gA,)( ++−∆= σµδµ (4.7)
4.1.2 Kritikpunkte an der Scaled-Particle-Theorie und Aspekte ihrer Anwendung
Die SPT ist in der Vergangenheit oft zur Berechnung von freien Lösungsenthalpien ∆G*sol
angewendet worden [BEN NAIM, 1989a; BEN NAIM, 1989b; CROVETTO, 1982; LUCAS,
1976; MOREL-DESROSIERS, 1981; PIEROTTI, 1976; POSTMA, 1982; WILHELM, 1972].
Dies liegt nicht zuletzt an ihrem vergleichsweise einfachen Aufbau, der sich dadurch
auszeichnet, dass einzig die Radien der als harte Kugeln betrachteten Spezies und deren
Konzentration bekannt sein müssen. Obwohl die Ergebnisse in der Literatur für Vorhersagen
auf Basis der Molekülgröße sehr gute Ergebnisse liefern, gibt es Einschränkungen derer man
sich bewusst sein muss.
Die Güte der Beschreibung hängt von zwei Einflussgrößen ab: Zum einen vom Hart-Kugel-
Durchmesser der betrachteten Spezies und zum anderen von der Dichte des Lösungsmittels.
Dabei ist insbesondere zu beachten, dass die aus der SPT resultierende freie Lösungsenthalpie

59
∆G*sol sehr sensitiv auf den Radius reagiert [TANG, 2000] und gleichzeitig die Bestimmung
eines Harte-Kugel-Durchmessers für reale Moleküle nicht trivial ist [BEN AMOTZ, 1990;
BEN AMOTZ, 1993c; GOGONEA, 1998; PIEROTTI, 1965]. Harte-Kugel-Durchmesser für
reale Moleküle, die aus experimentellen Daten wie Gaslöslichkeiten [PIEROTTI, 1965],
Oberflächenspannungen [MAYER, 1963], Verdampfungsenthalpien [PIEROTTI, 1976] oder
Virialkoeffizienten und Viskositäten [HIRSCHFELDER, 1964] bestimmt werden, stellen
effektive Radien dar. Sie enthalten Wechselwirkungen zwischen Lösungsmittel und gelöstem
Stoff. Andere Methoden, die van der Waals-Radien berechnen, betrachten ein Molekül ohne
die Wechselwirkung mit einem umgebenden Lösungsmittel [BONDI, 1964; EDWARD, 1970;
GOGONEA, 1998]. Tabelle 4.1 listet für das Molekül Benzol die Werte des Harte-Kugel-
Durchmessers für die unterschiedlichen Bestimmungsverfahren auf.
Tabelle 4.1 Harte-Kugeldurchmesser Benzol aus verschiedenen Bestimmungsverfahren
Durchmesser (Ǻ) Methode
5,00 Kompressibilitätsdaten [MAYER, 1963]
5,02 Oberflächenspannungen [MAYER, 1963]
5,24 Gaslöslichkeiten [PIEROTTI, 1965]
5,30 PVT-Daten [BEN AMOTZ, 1990]
5,36 van der Waals Radien [EDWARD, 1970]
Die zuverlässige Bestimmung des Hart-Kugeldurchmessers der betrachteten Stoffe ist
Voraussetzung für eine korrekte Beschreibung der freien Lösungsenthalpie ∆G*sol.
4.1.3 Dampfdruck aus der Freien Enthalpie des Lösungsvorganges
In dieser Arbeit wird die Erweiterung des Energiekonzeptes von COSMO-RS um einen
expliziten Kavitätsterm dazu verwendet, Dampfdrücke reiner Stoffe vorherzusagen. Es gibt
viele Ansätze die den Dampfdruck einer Komponente auf Basis von experimentellen Daten
interpolieren oder vorhersagen. Methoden wie die Clausius-Clapeyron-Gleichung oder
Gruppenbeitragsmethoden [POLING, 2001] benötigen in aller Regel zusätzliche Daten wie
den kritischen Druck, die kritische Temperatur oder den azentrischen Faktor und haben daher
einen limitierten Anwendungsbereich. Daneben existieren empirische Ansätze, die auf Basis
der Struktur eines Moleküls eine Beziehung zu den physikalischen Eigenschaften herstellen
(Quantitative Structure Property Relationship – QSPR). Dabei werden Dampfdrücke mittels

60
Deskriptoren von Struktureinheiten der Moleküle korreliert [BASAK, 1997; KATRITZKY,
1998; LIANG, 1998]. Um den Dampfdruck einer Komponente losgelöst von experimentellen
Daten wie den oben erwähnten vorherzusagen, soll in dieser Arbeit ein Ansatz verfolgt
werden, der den Unterschied des chemischen Potenzials eines Stoffes in der Gasphase und in
Flüssigphase auf Basis des um den Kavitätsterm der SPT erweiterten COSMO-RS-Modells
verwendet. Dabei wird auf die von Ben-Naim eingeführte Definition des Lösungsvorganges
eines Teilchens aus der Gasphase in eine kondensierte Phase zurückgegriffen (vgl. Kapitel
2.2.3).
Für reine Stoffe ist das chemische Potenzial definiert als:
PTN
G
,
∂
∂=µ (4.8)
Der Dampfdruck des Stoffes kann aus der Gleichheit der chemischen Potenziale in der
Gasphase und in der Flüssigphase bei konstanter Temperatur dargestellt werden:
),(),( 00LV
i
L
i
LV
i
V
i PTPT µµ = (4.9)
Entsprechend der Lösungstheorie nach Ben-Naim [BEN NAIM, 1987] konnte das chemische
Potenzial eines Stoffes i formuliert werden als:
3* ln iiii kt Λ+= ρµµ (2.9)
Ben-Naim definiert die freie Enthalpie des Lösungsvorganges als Differenz der
pseudochemischen Potenziale in Gas- und Flüssigphase:
V
i
L
i
solv
iG*,*, µµ −=∆ (4.10)
Zieht man auf beiden Seiten von Gleichung 4.9 den idealen Gasanteil des chemischen
Potenzials ab und setzt die Definition des chemischen Potenzials ein (Gleichung 2.9) erhält
man:
3*,3*,, ln(ln i
iG
i
IG
ii
L
i
L
i
iGV
i
V
i kTkT Λ+−Λ+=− ρµρµµµ (4.11)
+−=−
iG
i
L
iIG
i
L
i
iGV
i
V
i kTρ
ρµµµµ ln*,*,, (4.12)
Einsetzen von Gleichung 4.10 ergibt:

61
+∆=−
iG
i
L
isolv
i
iGV
i
V
i kTGρ
ρµµ ln, (4.13)
Die Differenz des chemischen Potenzials von Gasphase und idealer Gasphase wird als
Fugazitätskoeffizient ausgedrückt:
=−
LV
i
LV
iiGV
i
V
iP
PTfkT
0
0, ),(lnµµ (4.14)
damit folgt aus Gl. 4.13 und Gl. 4.14
−+∆=
kT
PkTkTG
P
PTfkT
LV
iL
i
solv
iLV
i
LV
i 0
0
0 lnln),(
ln ρ (4.15)
und somit der Dampfdruck des Stoffes i:
−+
∆=
LV
i
LV
iL
i
solv
iLV
iP
PTfkTkT
kT
GP
0
00
),(lnlnln ρ (4.16)
Der letzte Term auf der rechten Seite von Gleichung 4.16 beschreibt die Nichtidealitäten der
Gasphase. In dieser Arbeit sollen die Dampfdrücke verschiedener Komponenten am
Normalsiedepunkt betrachtet werden. Daher kann dieser Term im Folgenden vernachlässigt
werden. Für die Berechnung des Dampfdruckes sind somit die aus COSMO-RS Rechnungen
unter expliziter Berücksichtung des Kavitätstermes der SPT und Dichten der flüssigen Phase
am Normalsiedepunkt nötig. Die Dichten der betrachteten Stoffe werden aus experimentellen
Daten bezogen [DAUBERT, 1989].
4.1.4 Effektive Harte-Kugel-Durchmesser aus COSMO-RS
In Kapitel 4.1.2 wurde erläutert, dass der Radius bzw. der Durchmesser eine sensitive Größe
in der Bestimmung der freien Lösungsenthalpie darstellt. In der Literatur finden sich
verschiedene Vorgehensweisen, die Harte-Kugel-Durchmesser aus experimentellen Daten
oder den van-der-Waals-Radien (vdW Radien) der Atome der Moleküle bestimmen. In dieser
Arbeit werden die Harte-Kugel-Durchmesser aus den quantenchemischen Rechnungen
abgeleitet. Bei Anwendung der COSMO-Randbedingung wird um die Atome der Moleküle
eine Kavität konstruiert, die 17% größer ist, als der vdW Radius. Das Volumen der aus den
quantenchemischen Rechnungen wird auf das vdW-Volumen umgerechnet und als
Kugeläquivalent betrachtet. Der Vorteil dieser Methode liegt darin begründet, dass so auch

62
Unterschiede im Volumen der einzelnen Konformationen berücksichtig werden kann, für die
keine der oben erwähnten Methoden geeignet ist.
Abb. 4.1 verdeutlicht das Vorgehen.
σσ
σhs=f(σcosmo)
Abb. 4.1 Schema der Umrechnung von COSMO-Volumina in Hart-Kugel-Durchmesser
Experimentelle Untersuchungen und theoretische Abschätzungen haben gezeigt, dass der
effektive Harte-Kugel-Durchmesser eine Funktion der Temperatur ist [BEN AMOTZ, 1990;
BEN AMOTZ, 1993c]. Die Änderung des Durchmesser mit der Temperatur liegt im Bereich
4101/ −⋅−≈∂∂ Tσ bis 4105 −⋅− , was einer Verringerung des Durchmessers um einige wenige
Prozent bei einem Temperaturanstieg von 100K entspricht [BEN AMOTZ, 1990]. In dieser
Arbeit wird der Temperaturabhängigkeit durch einen Ausdruck der Störungstheorie von
Barker und Henderson [BARKER, 1967] folgend wiedergegeben. Dabei wird das Ergebnis
der Auswertung dieser Störungstheorie an Square-Well-Potenzialen durch Chen und
Kreglewski [CHEN, 1977] verwendet, welches die Temperaturabhängigkeit wie folgt
beschreibt:
−−=
kT
uT
0
0
3exp12,01()( σσ (4.17)
In dieser Gleichung ist u0 ein Energieparameter, welcher die Tiefe des Square-Well-
Potenzials beschreibt. Diese Temperaturabhängigkeit wurden in vielen Zustandsgleichungen,
die als Referenzfluid Harte Kugel oder Harte Ketten verwenden erfolgreich angewendet
[GROSS, 2000; GROSS, 2001b; HUANG, 1990; WOLBACH, 1997; WOLBACH, 1998]
In dieser Arbeit ist der Energieparameter auf einen konstanten Wert von 300J festgelegt.

5 Darstellung der Ergebnisse
5.1 Einfluss der Konformation von Molekülen auf die Berechnung der
thermodynamischen Eigenschaften
Die Konformation gibt die exakte räumliche Ausdehnung der Atome eines Moleküls an bei
vorgegebener Konstitution und Konfiguration. Hierbei bedeutet die Konstitution die Vorgabe
der Art und der Anzahl von Atomen eines Moleküls sowie deren Reihenfolge und
Verknüpfung. Auf diese Weise werden das Atomgerüst und alle Valenzbindungen
einschließlich der Bindungsanordnung beschrieben. Die Konfiguration eines Moleküls
definiert bei gegebener Konstitution die räumliche Anordnung von Atomen oder
Atomgruppen innerhalb einer Verbindung. Der Einfluss von Rotationen um
Einfachbindungen wird dabei nicht berücksichtigt. Dieser Einfluss führt zu den
unterschiedlichen Konformationen. In ihrer Gesamtheit bilden sie die Stereoisomere.
Konformationen eines Moleküls können durch Drehung um formale Einfachbindungen
theoretisch in unendlicher Anzahl existieren. Konformationen, die unterschiedlichen
Energieminima zugeordnet werden können, bezeichnet man als Konformere. Für eine
konstante Temperatur stellt sich zwischen den verschiedenen Konformeren ein Gleichgewicht
ein. Für eine gegenseitige Umwandlung ist ein Energieunterschied von etwa 16-20kJ
erforderlich [HOWLETT, 1955; WESTHEIMER, 1946].
5.1.1 Konformeranalyse
Ziel der Konformeranalyse ist die Identifizierung der Konformere die die Eigenschaften eines
Moleküls bestimmen. Die Analyse kann auf zwei grundsätzlich verschiedene Arten erfolgen:
1. Experimentelle Untersuchungen
2. Theoretische Betrachtungen unter Verwendung von quantenchemischen oder
molekular mechanischen Ansätzen
Aufgrund des hohen Aufwandes der experimentellen Techniken verwendet man diese heute
nur noch zur Validierung von theoretischen Ergebnissen. Sie sollen hier nur aufgezählt
werden. In [LAU, 1961] werden die Vor- und Nachteile, das jeweilige Anwendungsgebiet
und die erzielbaren Genauigkeiten erläutert. Hauptsächlich werden spektroskopische
Methoden angewendet wie Mikrowellen-, Infrarot-, Raman-, Ultraviolett- oder
Kernmagnetische Resonanz-Spektroskopie. Daneben werden Röntgenstrukturanalysen,
Elektronenbeugungsuntersuchen, die Bestimmung elektrischer Momente, z.B. das
Dipolmoment, Untersuchungen des optischen Drehvermögens, kalorische oder kinetische

64
Messungen angewendet.
Die deutliche Verbesserung der mathematischen Modelle (vgl. Kapitel 2.2) und die zur
Verfügung stehende Leistungsfähigkeit heutiger Computeranlagen ermöglicht eine Ermittlung
von Konformeren mit theoretischen Methoden. Dabei steht das Absuchen von Minima in der
Energiefläche der Konformationen im Mittelpunkt. Die folgenden Methoden haben sich
etabliert:
1. Systematische Suche innerhalb des Konformerenraumes: Durch Festlegen aller
möglichen Drehwinkel und Drehachsen einer Startstruktur resultieren mögliche
Stereoisomere und Konformere. Alle entstehenden Geometrien müssen
energieoptimiert werden und anschließend bzgl. ihrer Energien bewertet werden. Ein
Nachteil der Methode ist die enorme Anzahl an entstehenden Geometrien. Bei fünf
Bindungen und einem Drehwinkel von 30° liegt die Zahl der Strukturen bei etwa
300000; bei gleichem Drehwinkel und sieben Bindungen bei annähernd 36 Millionen.
Durch Verwendung von Molekülfragmenten kann die Zahl eingeschränkt werden.
2. Zufällige Suchmethoden: Hier werden zufällig neue Strukturen durch Verändern des
Drehwinkels rotierbarer Bindungen oder auch zufällige kartesische Koordinaten
generiert. Dabei springt man von einem Punkt auf der Energiefläche einer
Konstitution zum anderen. Jede neue Struktur wird mittels Energieminimierung
optimiert. Wurde sie in einem vorherigen Schritt noch nicht gefunden, wird sie
gespeichert.
3. Monte-Carlo- und Molekulardynamik-Methoden: Der Vorteil dieser Methoden liegt in
der Möglichkeit, Energiebarrieren zu überspringen, was mit den vorhergehenden
Methoden nicht möglich ist.
Einen ausführlichen Überblick über die unterschiedlichen Methoden und ihre bevorzugten
Anwendungsgebiete gibt Leach [LEACH, 2006].
In dieser Arbeit ist die Konformeranalyse der verwendeten Komponenten bzw.
Molekülstrukturen mit der kommerziellen Software HyperChem durchgeführt worden. Dabei
werden programmintern die folgenden Schritte durchgeführt:
1. Auswählen einer Startgeometrie,
2. Modifikation der Startgeometrie durch Variation von geometrischen Parametern,
3. Geometrieoptimierung der modifizierten Startgeometrie,
4. Vergleichen der aktuellen Geometrie mit denen der vorher gefundenen und Akzeptanz
falls die gefundene Geometrie einzigartig ist und ein bestimmtes Energiekriterium
erfüllt.

65
In Schritt 2., der Variation der Startgeometrie, wird die Geometrie systematisch geändert.
Dabei wird ein weiterentwickelter Monte Carlo Multiple Minimum [CHANG, 1989] Ansatz
angewendet [GOODMAN, 1991], der vor allem die Dimensionalität der systematischen
Suche reduziert, indem anfangs mit kleiner Auflösung die Energiefläche grob abgesucht wird.
Die gefundenen Variationen werden dann durch weitere systematische Geometrieänderungen
mit hoher Auflösung verfeinert. Ausgehend von einer Anfangsstruktur werden vom Benutzer
vorgegebene Drehwinkel geändert. Auf diese Weise neu gewonnene Strukturen werden
geometrieoptimiert und anschließend Auswahlkriterien unterworfen, die entweder zur
Akzeptanz oder zum Verwerfen einer Struktur führen. Dabei werden die zu vergleichenden
Strukturen überlagert und Drehwinkel sowie atomare Abstände verglichen. Ligen die Größen
innerhalb einer vorzugebenden Toleranz, wird die neue Struktur abgewiesen. Als Methode für
die Geometrieoptimierung wurde in dieser Arbeit die semiempirische Methode PM3 gewählt.
Im Folgenden soll am Beispiel des 2-Butanol Moleküls der Einfluss der Konformere auf das
Ergebnis der Modellierung mit COSMO-RS gezeigt werden.
5.1.2 Konformeranalyse und Phasengleichgewichtsberechnungen am Beispiel des 2-
Butanols
Die Startstruktur des 2-Butanolmoleküls wird mit der HyperChem-Software erstellt und
anschließend geometrieoptimiert. Wie auch bei der Geometrieoptimierung innerhalb der
Konformeranalyse wird hier die semiempirische Methode PM3 gewählt. Für diese optimierte
Struktur müssen alle zu verändernden Diederwinkel explizit definiert werden. Diese Winkel
werden dann der Konformeranalyse übergeben. Innerhalb der Konformeranalyse sind
Vorgabewerte für die Akzeptanzkriterien einer neuen Konformation zu definieren. Die
wichtigsten werden hier kurz erläutert:
• Globale Energiedifferenz: maximaler Wert in kcal/mol, der bestimmt, ob neue
Konformere relativ zum energieniedrigsten Konformer akzeptiert werden,
• Duplizitätstests:
o Minimalwert für Abstand in Angström zwischen ungebundenen Atomen,
o Winkelangabe in Grad zwischen den definierten Drehwinkeln,
o Energiedifferenz zwischen zwei Geometrien in kcal/mol,
o RMS-Fehler übereinandergelegter Atome in den zu vergleichenden
Konformationen in Angström
• Optionen für die Geometrieoptimierung:
o RMS-Energiegradient, Schwelle für Erreichen eines Energieminimums
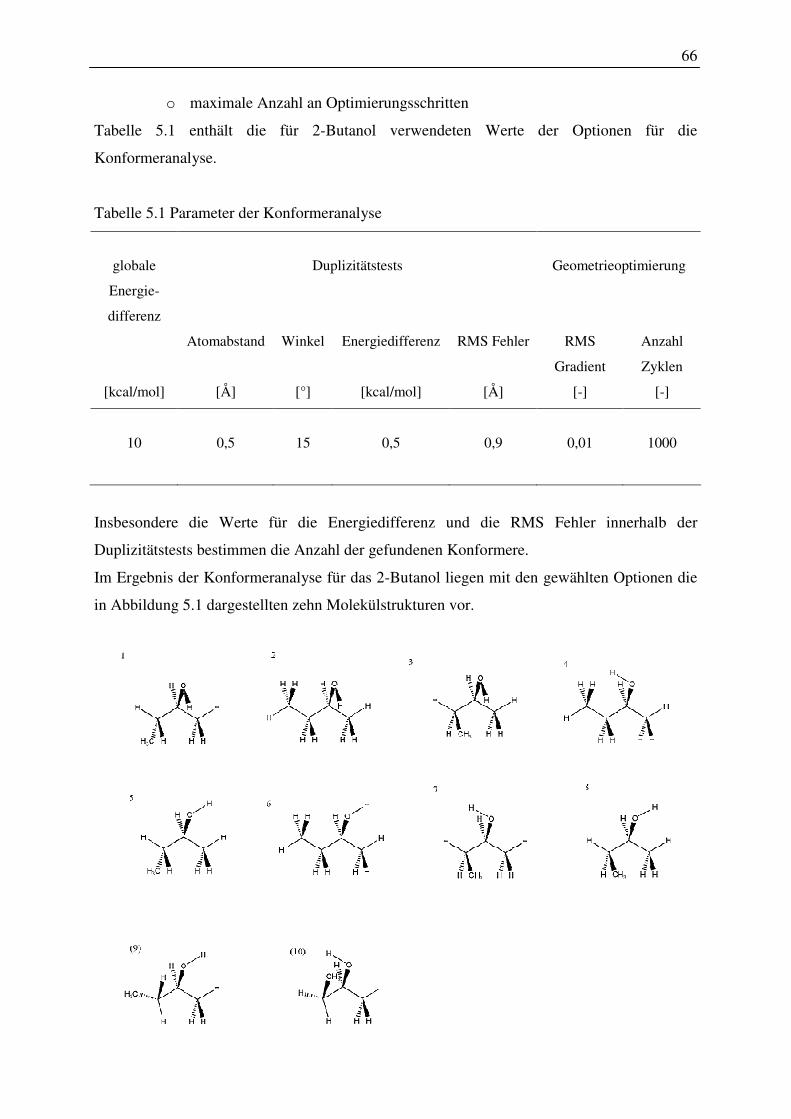
66
o maximale Anzahl an Optimierungsschritten
Tabelle 5.1 enthält die für 2-Butanol verwendeten Werte der Optionen für die
Konformeranalyse.
Tabelle 5.1 Parameter der Konformeranalyse
globale
Energie-
differenz
Duplizitätstests
Geometrieoptimierung
[kcal/mol]
Atomabstand
[Å]
Winkel
[°]
Energiedifferenz
[kcal/mol]
RMS Fehler
[Å]
RMS
Gradient
[-]
Anzahl
Zyklen
[-]
10 0,5 15 0,5 0,9 0,01 1000
Insbesondere die Werte für die Energiedifferenz und die RMS Fehler innerhalb der
Duplizitätstests bestimmen die Anzahl der gefundenen Konformere.
Im Ergebnis der Konformeranalyse für das 2-Butanol liegen mit den gewählten Optionen die
in Abbildung 5.1 dargestellten zehn Molekülstrukturen vor.

67
Abb. 5.1 Konformere des 2-Butanols aus der Konformeranalyse mit HyperChem in der Fischer-Projektion
Diese gefundenen Konformationen werden als Startgeometrien für die Geometrieoptimierung
mittels DFT und der gleichzeitigen Berechnung der Abschirmungsladungen mit dem
COSMO-Modell verwendet (vgl. Kapitel 2.3). Anhand der COSMO-Energien, der Energie
des Moleküls unter der Randbedingung des idealen Leiters, lassen sich die Konformationen
vergleichen. Tabelle 5.2 und Abb. 5.2 verdeutlichen die Ergebnisse. Die lineare Struktur
Nummer 2 ist die energieärmste und Konformation 8 besitzt die höchste COSMO-Energie.
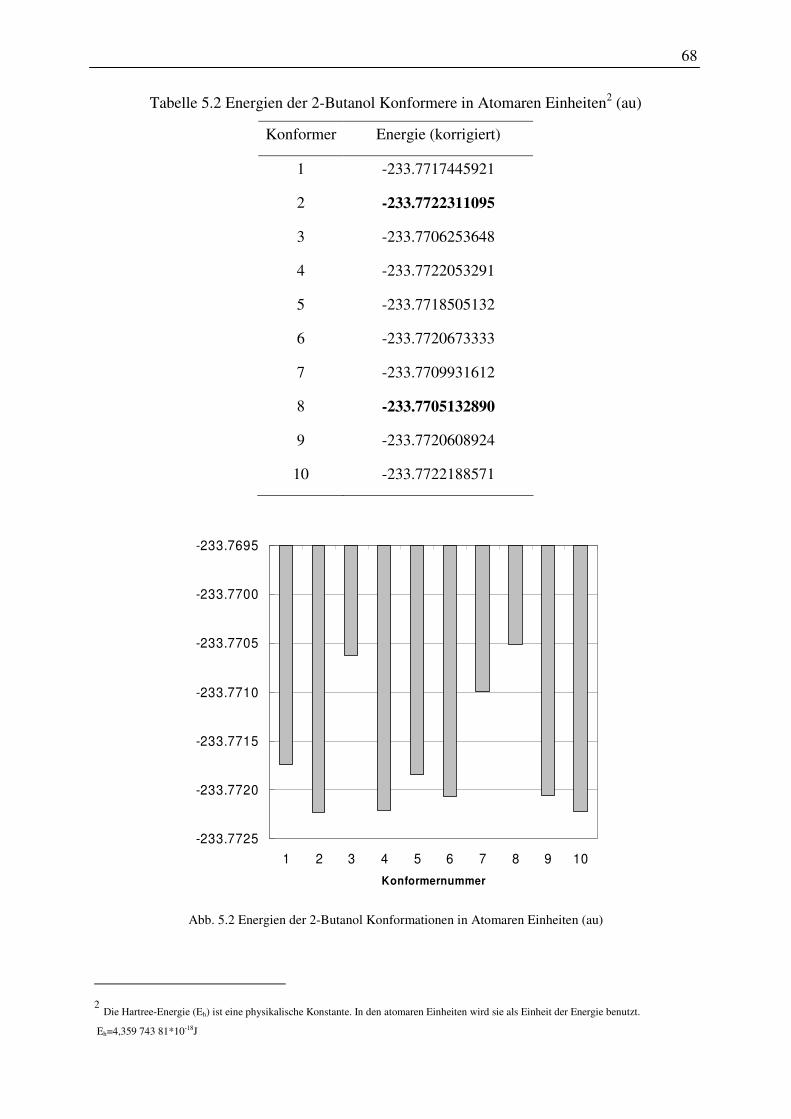
68
Tabelle 5.2 Energien der 2-Butanol Konformere in Atomaren Einheiten2 (au)
Konformer Energie (korrigiert)
1 -233.7717445921
2 -233.7722311095
3 -233.7706253648
4 -233.7722053291
5 -233.7718505132
6 -233.7720673333
7 -233.7709931612
8 -233.7705132890
9 -233.7720608924
10 -233.7722188571
-233.7725
-233.7720
-233.7715
-233.7710
-233.7705
-233.7700
-233.7695
1 2 3 4 5 6 7 8 9 10
Konformernummer
Abb. 5.2 Energien der 2-Butanol Konformationen in Atomaren Einheiten (au)
2 Die Hartree-Energie (Eh) ist eine physikalische Konstante. In den atomaren Einheiten wird sie als Einheit der Energie benutzt.
Eh=4,359 743 81*10-18J

69
Für die Untersuchung des Einflusses der verschiedenen Konformationen auf die Modellierung
des Phasengleichgewichtes werden zwei verschiedene Systemklassen untersucht. Einerseits
das binäre System 2-Butanol/Wasser bei dem experimentelle Daten bei 298,15K und 323,15K
vorliegen und andererseits die binären Systeme 2-Butanol/n-Pentan und 2-Butanol/n-Hexan.
Das 2-Butanol/Wasser-System bildet bei den gewählten Temperaturen ein Hetereoazeotrop.
Die Güte der Vorhersage wurde durch die Betrachtung der Abweichung des berechneten
Systemdrucks von experimentell ermittelten Daten bewertet. Tabelle 5.3 zeigt für die zehn
Konformationen die Abweichungen im Druck des 2-Butanol/Wasser-Systems. Die
Komponente Wasser besitzt eine Konformation. Man erkennt, dass die Abweichung in diesem
Fall für die Energiemaximum-Konformation und die Energieminimum-Konformation gleich
sind. Die geringsten Abweichungen ergeben sich für die Konformation drei.
Tabelle 5.3 Vergleich der Abweichungen im Druck für VLLE
Phasengleichgewichtsrechnungen des Systems 2-Butanol/Wasser bei 298,15 und 323,15K
Konformernummer absolute Abweichung
im Druck [%]
2 11.0
8 11.1
1 6.3
3 5.9
4 6.5
5 6.2
6 13.2
7 8.6
9 13.3
10 6.6
Weiterhin erkennt man, dass die Berechnung unter Verwendung der Konformation mit der
niedrigsten Energie und damit mit der höchsten Wahrscheinlichkeit des Auftretens dieser
Konformation nicht die geringsten Abweichungen bei der Phasengleichgewichtsberechnung
liefert. Eine Berechnung des Phasengleichgewichtes unter der Annahme, dass die reine
Komponente 2-Butanol aus einer Mischung der Konformationen dargestellt wird, bietet die
Möglichkeit den gesamten betrachteten Konformerenraum zu berücksichtigen. Die
Konformationen werden dabei hinsichtlich ihrer Energie gewichtet, wobei Konformationen

70
mit niedriger Energie wahrscheinlicher auftreten als Konformationen mit höherer Energie. Bei
Verwendung des COSMO-RS-Modells besteht die Möglichkeit, das Auftreten der
Konformere in folgender Weise zu berücksichtigen:
Die Wahrscheinlichkeit des Auftretens einer Konformation wi ist über eine Boltzman-
Verteilung derart gegeben, dass die so genannte Total Free Energy (TFE) der Konformation
in der Mischung berechnet wird und in Relation zur TFE aller, durch die Konformeranalyse
erhaltenen, Konformationen gesetzt wird:
∑ −−
−−=
onenKonformati
j
jj
iii
RTTFETFEwc
RTTFETFEwcw
]]/)(exp[[
]/)(exp[
min
min
(5.1)
Der Faktor wc in Gleichung 5.1 kennzeichnet die Symmetrie, die ausdrückt, wie oft dieselbe
molekulare Struktur durch verschiedene Drehungen innerhalb der Bindung erhalten werden
kann. Die TFE wird auf folgende Weise berechnet:
iCOSMO EETFE µ+∆+= (5.2)
Wobei ECOSMO die quantenchemische Energie des Moleküls mit der Randbedingung des
idealen Leiters darstellt, ∆E die Korrektur der Abschirmungsladungen und µi das chemische
Potenzial der betrachteten Komponente (Konformation) in der Mischung sind. Die
Wahrscheinlichkeit wi muss für jede Konzentration von i in der Mischung neu berechnet
werden, da sich das chemische Potenzial konzentrationsabhängig ändert. In Abb. 5.3 ist das
Ergebnis der Phasengleichgewichtsberechnung für das System 2-Butanol/Wasser bei einer
Temperatur von 298,15K dargestellt. Neben dem Ergebnis für die Berechnung unter
Berücksichtigung aller Konformationen sind die Ergebnisse für die Konformation mit der
niedrigsten und der höchsten COSMO-Energie aufgezeigt sowie der Konformation, die die
geringste Abweichung von den experimentellen Daten zeigt.

71
Abb. 5.3 Binäres Dampf-Flüssig-Phasengleichgewicht 2-Butanol/Wasser bei 298,15 K, Vergleich der
Vorhersage des Phasengleichgewichts mit COSMO-RS bei Verwendung verschiedener 2-Butanol-
Konformationen mit experimentellen Daten (experimentelle Daten: Gaube,J., L.Krenzer, G.Olf and R.Wendel,
Fluid Phase Equilib., 35, 1987, 279)
Für die Systeme 2-Butanol/n-Pentan und 2-Butanol/n-Hexan ergibt sich hinsichtlich der
einzelnen Konformationen im Vergleich zum 2-Butanol/Wasser-System eine andere
Reihenfolge der Abweichungen im Druck. Abb. 5.4 zeigt die gemittelten Abweichungen aller
Messpunkte. Zur besseren Übersicht sind die Daten für 2-Butanol/n-Pentan auf der
Sekundärachse aufgetragen. Man erkennt, dass innerhalb der beiden 2-Butanol/n-Alkan-
Systeme die relativen Abweichungen einer jeden Konformation gleich sind. Konformation 5
zeigt die niedrigste Abweichung und Konformation 4 die höchste. Die
Energieminimumkonformation 2 zeigt eine höhere Abweichung als die
Energiemaximumkonformation. Zusätzlich zur binären Mischung mit Alkanen sind die
Ergebnisse für das 2-Butanol/Wasser-System in das Diagramm eingetragen. Aus diesen
Ergebnissen lässt sich schließen, dass im voraus keine Aussage darüber getroffen werden
kann, welche Konformation sich am Besten eignet, experimentelle Daten wiederzugeben. Es
muss vielmehr der Konformerenraum in der oben beschriebenen Weise analysiert und
berücksichtigt werden.
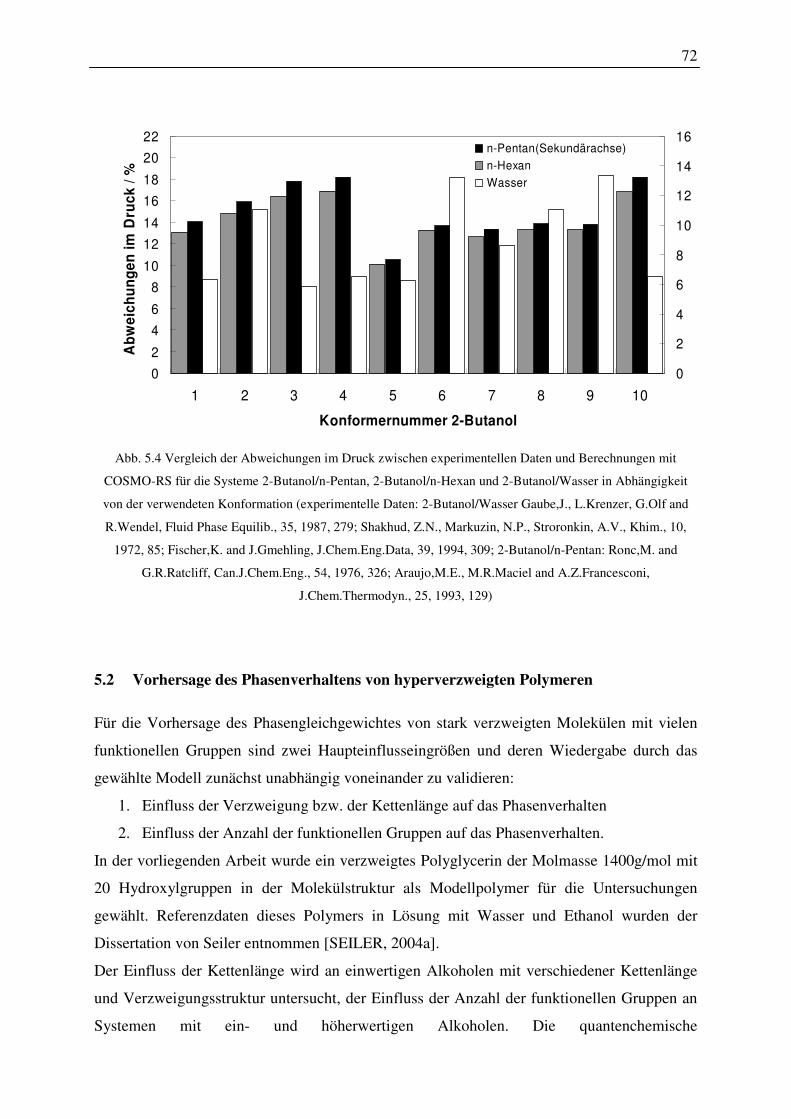
72
0
2
4
6
8
10
12
14
16
18
20
22
1 2 3 4 5 6 7 8 9 10
Konformernummer 2-Butanol
Ab
we
ich
un
ge
n i
m D
ruc
k /
%
0
2
4
6
8
10
12
14
16n-Pentan(Sekundärachse)n-HexanWasser
Abb. 5.4 Vergleich der Abweichungen im Druck zwischen experimentellen Daten und Berechnungen mit
COSMO-RS für die Systeme 2-Butanol/n-Pentan, 2-Butanol/n-Hexan und 2-Butanol/Wasser in Abhängigkeit
von der verwendeten Konformation (experimentelle Daten: 2-Butanol/Wasser Gaube,J., L.Krenzer, G.Olf and
R.Wendel, Fluid Phase Equilib., 35, 1987, 279; Shakhud, Z.N., Markuzin, N.P., Stroronkin, A.V., Khim., 10,
1972, 85; Fischer,K. and J.Gmehling, J.Chem.Eng.Data, 39, 1994, 309; 2-Butanol/n-Pentan: Ronc,M. and
G.R.Ratcliff, Can.J.Chem.Eng., 54, 1976, 326; Araujo,M.E., M.R.Maciel and A.Z.Francesconi,
J.Chem.Thermodyn., 25, 1993, 129)
5.2 Vorhersage des Phasenverhaltens von hyperverzweigten Polymeren
Für die Vorhersage des Phasengleichgewichtes von stark verzweigten Molekülen mit vielen
funktionellen Gruppen sind zwei Haupteinflusseingrößen und deren Wiedergabe durch das
gewählte Modell zunächst unabhängig voneinander zu validieren:
1. Einfluss der Verzweigung bzw. der Kettenlänge auf das Phasenverhalten
2. Einfluss der Anzahl der funktionellen Gruppen auf das Phasenverhalten.
In der vorliegenden Arbeit wurde ein verzweigtes Polyglycerin der Molmasse 1400g/mol mit
20 Hydroxylgruppen in der Molekülstruktur als Modellpolymer für die Untersuchungen
gewählt. Referenzdaten dieses Polymers in Lösung mit Wasser und Ethanol wurden der
Dissertation von Seiler entnommen [SEILER, 2004a].
Der Einfluss der Kettenlänge wird an einwertigen Alkoholen mit verschiedener Kettenlänge
und Verzweigungsstruktur untersucht, der Einfluss der Anzahl der funktionellen Gruppen an
Systemen mit ein- und höherwertigen Alkoholen. Die quantenchemische

73
Geometrieoptimierung des hyperverzweigten Polymers ist bei der verwendeten
Rechnerkonfiguration auf etwa 60 Atome pro Molekül limitiert. Das Modellpolymer besteht
aus 214 Atomen. Es wird ein Ansatz vorgestellt, der es ermöglicht, durch Zerlegung des
Moleküls in Fragmente das Sigmaprofil für das Modellpolymer zu bestimmen.
5.2.1 Einfluss der Verzweigung bzw. der Kettenlänge auf das Phasenverhalten
Im untersuchten Modellsystem Polyglycerin/Wasser/Ethanol ist die Hydroxylgruppe und ihre
Anordnung im Molekül wesentlich für das Wechselwirkungsverhalten verantwortlich.
Abschnitte im Polymer, die über wenige OH-Gruppen verfügen, werden eher unpolaren
Charakter aufweisen als solche mit vielen. Die Wiedergabe dieser prinzipiellen Eigenschaften
muss durch das verwendete Modell COMOS-RS sichergestellt werden. Zur Validierung
wurden dazu die Grenzaktivitätskoeffizienten von einwertigen Alkoholen der homologen
Reihe von Methanol bis 1-Decanol in Wasser herangezogen. Da es sich hier und auch in
Kapitel 5.2.2 um niedermolekulare Substanzen handelt, wurde der Free-Volume-Term an
dieser Stelle nicht verwendet. Abbildung 5.5 zeigt die mit COSMO-RS berechneten Verläufe
der Aktivitätskoeffizienten für verschiedene Temperaturen. Die Übereinstimmung mit den
experimentellen Daten ist gut, insbesondere wenn man die Abweichung bei den Messwerten
berücksichtigt und die Unsicherheit, mit der solche Daten gewonnen werden können. Neben
der Darstellung des Wechselwirkungsverhaltens bei unendlicher Verdünnung ist die korrekte
Beschreibung der Wechselwirkungen bei endlichen Konzentrationen wichtig. Abbildungen
5.6, 5.7 und 5.8 zeigen für die Systeme Ethanol/1-Propanol, Ethanol/Wasser und Wasser/1-
Butanol die Aktivitätskoeffizienten über den gesamten Konzentrationsbereich. Die
Vorhersage des Aktivitätskoeffizienten durch COSMO-RS gelingt für alle gezeigten Systeme
mit guter Genauigkeit. Im Alkohol/Alkohol-System ist die Abweichung der Vorhersage
gegenüber den experimentellen Daten sehr klein. Aufgrund der Ähnlichkeit der Moleküle
Ethanol und 1-Propanol in ihrer Größe und dem Wechselwirkungsverhalten ergeben sich
Aktivitätskoeffizienten nahe Eins. In den Alkohol/Wasser-System zeigt sich eine
systematische Abweichung des Aktivitätskoeffizienten des Alkohols bei hohen
Wasserkonzentrationen hin zu kleineren Werten. Dies bedeutet, dass COSMO-RS eine
weniger starke „Abneigung“ der Moleküle in der Mischung beschreibt.
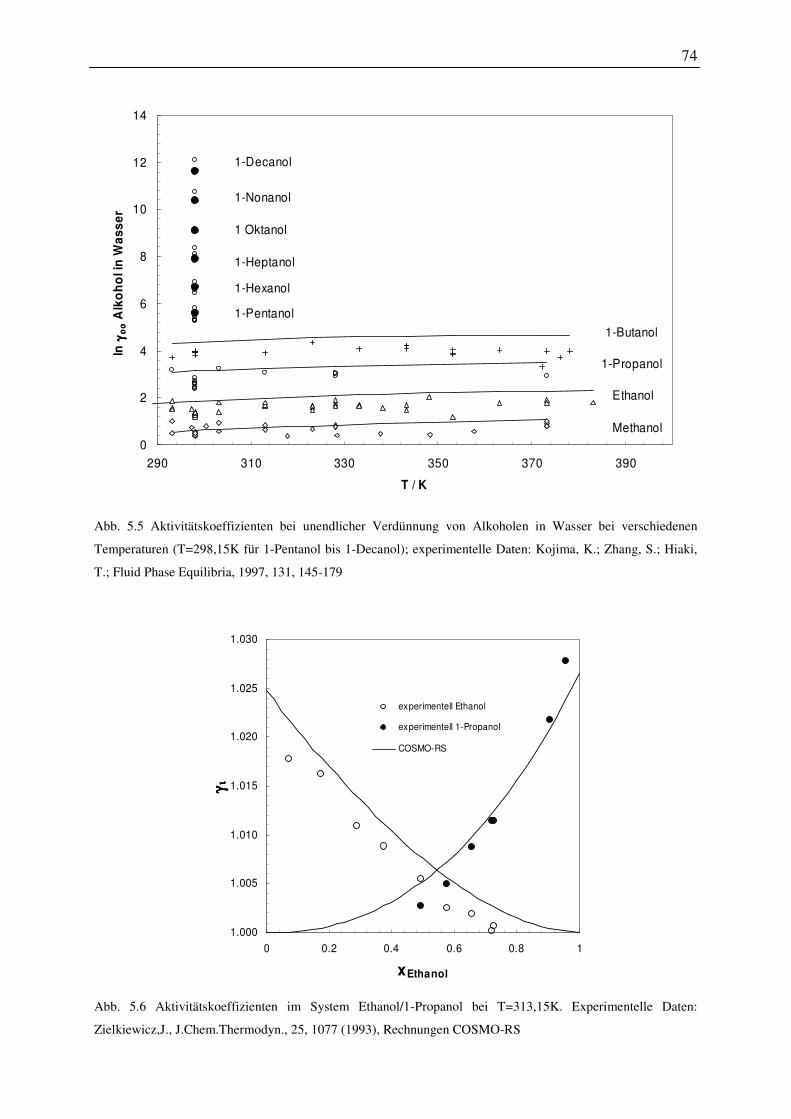
74
0
2
4
6
8
10
12
14
290 310 330 350 370 390
T / K
ln γγ γγ
οο
οο
οο
οο A
lko
ho
l in
Wa
ss
er
1-Decanol
1-Nonanol
1-Heptanol
1 Oktanol
1-Hexanol
1-Butanol
1-Propanol
Ethanol
Methanol
1-Pentanol
Abb. 5.5 Aktivitätskoeffizienten bei unendlicher Verdünnung von Alkoholen in Wasser bei verschiedenen
Temperaturen (T=298,15K für 1-Pentanol bis 1-Decanol); experimentelle Daten: Kojima, K.; Zhang, S.; Hiaki,
T.; Fluid Phase Equilibria, 1997, 131, 145-179
1.000
1.005
1.010
1.015
1.020
1.025
1.030
0 0.2 0.4 0.6 0.8 1
xEthanol
γγ γγιι ιι
experimentell Ethanol
experimentell 1-Propanol
COSMO-RS
Abb. 5.6 Aktivitätskoeffizienten im System Ethanol/1-Propanol bei T=313,15K. Experimentelle Daten:
Zielkiewicz,J., J.Chem.Thermodyn., 25, 1077 (1993), Rechnungen COSMO-RS
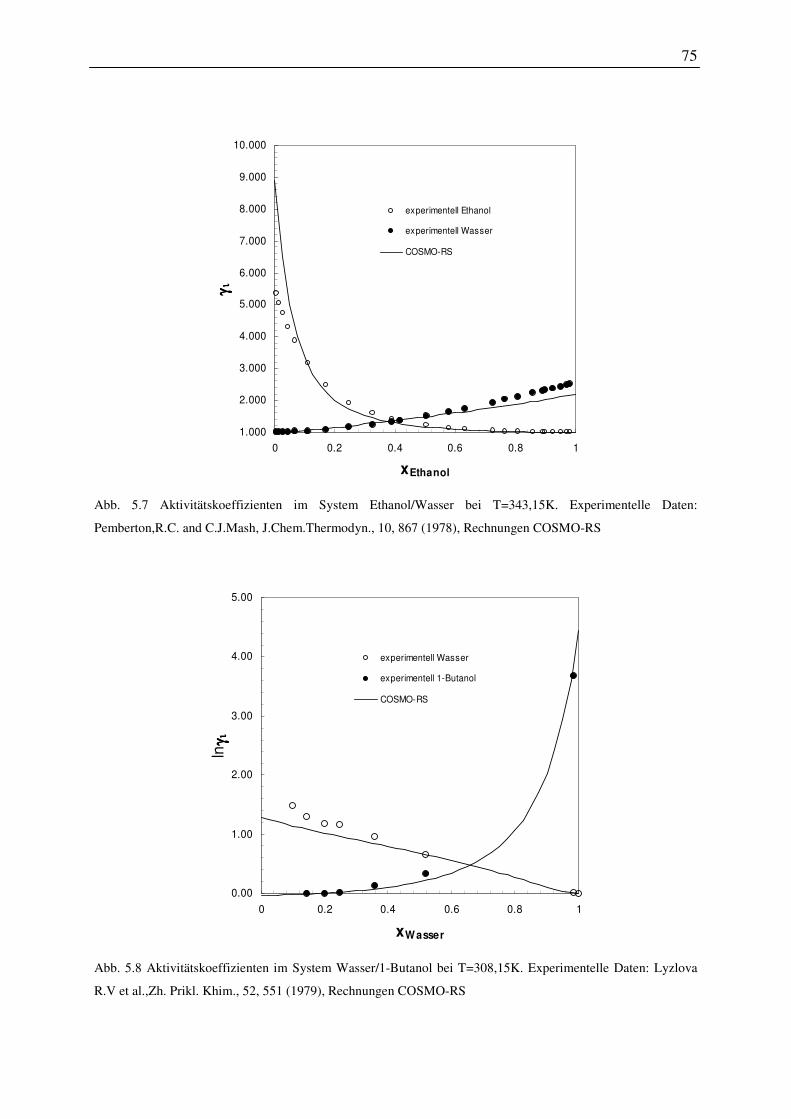
75
1.000
2.000
3.000
4.000
5.000
6.000
7.000
8.000
9.000
10.000
0 0.2 0.4 0.6 0.8 1
xEthanol
γγ γγιι ιι
experimentell Ethanol
experimentell Wasser
COSMO-RS
Abb. 5.7 Aktivitätskoeffizienten im System Ethanol/Wasser bei T=343,15K. Experimentelle Daten:
Pemberton,R.C. and C.J.Mash, J.Chem.Thermodyn., 10, 867 (1978), Rechnungen COSMO-RS
0.00
1.00
2.00
3.00
4.00
5.00
0 0.2 0.4 0.6 0.8 1
xWasser
lnγγ γγ
ιι ιι
experimentell Wasser
experimentell 1-Butanol
COSMO-RS
Abb. 5.8 Aktivitätskoeffizienten im System Wasser/1-Butanol bei T=308,15K. Experimentelle Daten: Lyzlova
R.V et al.,Zh. Prikl. Khim., 52, 551 (1979), Rechnungen COSMO-RS

76
Der Einfluss der Verzweigung auf das Phasenverhalten und seine Beschreibung durch
COSMO-RS eines Moleküls mit konstanter Anzahl an Hydroxylgruppen wurde an
Aktivitätskoeffizienten des Modellmoleküls in einer Mischung Modellmolekül/Wasser
untersucht. Ein hypothetisches Polyol mit vier Hydroxylgruppen innerhalb der Struktur ist in
drei Varianten generiert worden – eine lineare, eine vollständig verzweigte, sternförmige mit
den OH-Gruppen innen angeordnet sowie eine vollständig verzweigte, sternförmige mit den
OH-Gruppen am Rand angeordnet. Die Summenformel des Modellmoleküls lautet
C21H40OH4, sein Molgewicht beträgt 360,58 g/mol. Abb. 5.9 a-c zeigt die chemische Struktur
der drei Varianten. Enstsprechend den Standardbindungslängen und –bindungswinkeln
zwischen den Atomen wurden Startstrukturen generiert und diese unter COSMO-
Randbedingungen mittels TURBOMOLE mit DFT-Rechnungen geometrieoptimiert. Das
Ergebnis der Sigmaprofile zeigt Abb. 5.10.
Abb. 5.9 Modellmolekül mit konstanter Anzahl an Hydroxylgruppen

77
0
10
20
30
40
50
60
70
80
-4 -2 0 2 4
σσσσ / e/nm2
Hä
ufi
gk
eit
P(
σσ σσ)
OH-Gruppen außen
OH-Gruppen innen
linear
Abb. 5.10 Sigmaprofile des Modellmoleküls zur Untersuchung des Verzweigungseinflusses auf das
Phasenverhalten
Man erkennt einen deutlichen Einfluss der unterschiedlichen Molekülgeometrien bei gleicher
Summenformel. Das Sigmaprofil des linearen Modellmoleküls zeigt einen deutlichen Peak im
Bereich neutraler Ladungen und zwei definierte Spitzen im Bereich positiver und negativer
Abschirmungsladungen (vgl. Abb. 5.10). Diese Peaks der OH-Gruppen sind, da sie nicht von
anderen Atomen abgeschirmt werden, wesentlich ausgeprägter als bei den sternförmigen
Varianten. Dort ist der Bereich der neutralen Segmente auf der Moleküloberfläche nicht so
stark definiert, sondern weist eine Schulter im Bereich positiver Abschirmungsladungen auf,
was einer negativen Ladung auf der Moleküloberfläche entspricht. Im Unterschied zur
Variante mit den außenliegenden OH-Gruppen, zeigt das Molekül mit den innenliegenden
OH-Gruppen kleinere Abschirmungsladungsdichten im Bereich der Abschirmungsladungen
der OH-Gruppen, da die funktionellen Gruppen hier von anderen Atomen verdeckt werden.
Dies wirkt sich direkt auf die Aktivitätskoeffizienten der Moleküle in Lösung aus. Abb. 5.11
zeigt die theoretischen Verläufe der Aktivitätskoeffizienten von Wasser in einer Mischung
Modellmolekül/Wasser.

78
1
2
3
4
5
6
7
8
9
10
0 0.2 0.4 0.6 0.8 1xPolyol
γγ γγW
as
se
r
linear
OH-Gruppen außen
OH-Gruppen innen
Abb. 5.11 Verläufe der Aktivitätskoeffizienten des Modellmoleküls im System Modellmolekül/Wasser
Es ist zu erkennen, dass sich die Aktivitätskoeffizienten von Wasser in Mischung mit dem
verzweigten Molekül, bei dem die OH-Gruppen im Innern angeordnet und abgeschirmt sind,
deutlich von denen der Polyolstruktur unterscheiden, bei denen die OH-Gruppen nach außen
zeigen. Erwartungsgemäß läuft der Aktivitätskoeffizient hier bei unendlicher Verdünnung von
Wasser in Modellmolekül in einen Wert deutlich größer als Eins ein, da dieses Molekül einen
viel stärker unpolaren Charakter aufweist.
5.2.2 Einfluss der Anzahl der funktionellen Gruppen auf das Phasenverhalten
Im betrachteten System Wasser/Ethanol/hyperverzweigtes Polymer beeinflusst die Anzahl der
Hydroxylgruppen in der aliphatischen Struktur stark das Wechselwirkungsverhalten. Eine
hohe Anzahl Hydroxylgruppen führt zu einer Verminderung des unpolaren Charakters der C-
Kette. Die Aktivitätskoeffizienten werden mit steigender Anzahl der OH-Gruppen kleiner,
was einer verringerten Abweichung vom Roultschen Gesetz in der Mischung entspricht.
Abb. 5.12 zeigt Aktivitätskoeffizienten von 1-Propanol und 1,2-Propandiol in Wasser bei
unendlicher Verdünnung. Das Hinzufügen von einer zweiten Hydroxylgruppe bewirkt eine
starke Verringerung der Werte der Aktivitätskoeffizienten. Dieses Verhalten kann von
COSMO-RS gut wiedergegeben werden.
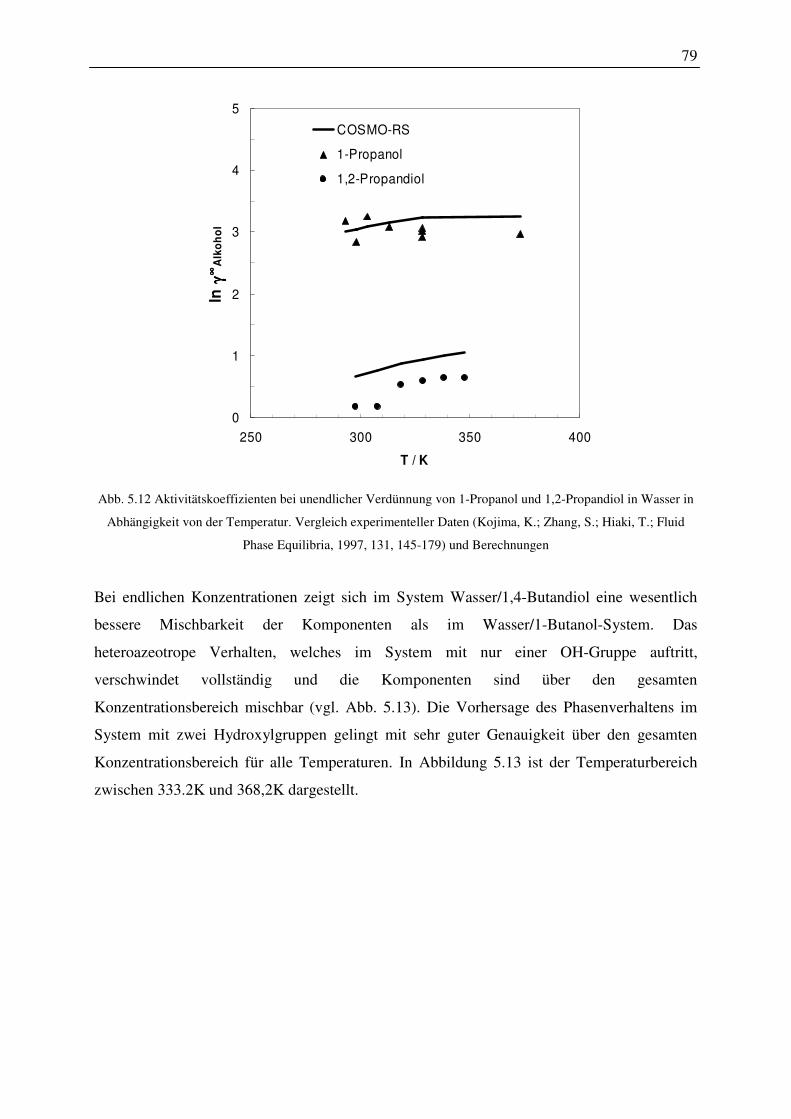
79
0
1
2
3
4
5
250 300 350 400
T / K
ln γγ γγ
∞∞ ∞∞A
lko
ho
l
COSMO-RS
1-Propanol
1,2-Propandiol
Abb. 5.12 Aktivitätskoeffizienten bei unendlicher Verdünnung von 1-Propanol und 1,2-Propandiol in Wasser in
Abhängigkeit von der Temperatur. Vergleich experimenteller Daten (Kojima, K.; Zhang, S.; Hiaki, T.; Fluid
Phase Equilibria, 1997, 131, 145-179) und Berechnungen
Bei endlichen Konzentrationen zeigt sich im System Wasser/1,4-Butandiol eine wesentlich
bessere Mischbarkeit der Komponenten als im Wasser/1-Butanol-System. Das
heteroazeotrope Verhalten, welches im System mit nur einer OH-Gruppe auftritt,
verschwindet vollständig und die Komponenten sind über den gesamten
Konzentrationsbereich mischbar (vgl. Abb. 5.13). Die Vorhersage des Phasenverhaltens im
System mit zwei Hydroxylgruppen gelingt mit sehr guter Genauigkeit über den gesamten
Konzentrationsbereich für alle Temperaturen. In Abbildung 5.13 ist der Temperaturbereich
zwischen 333.2K und 368,2K dargestellt.
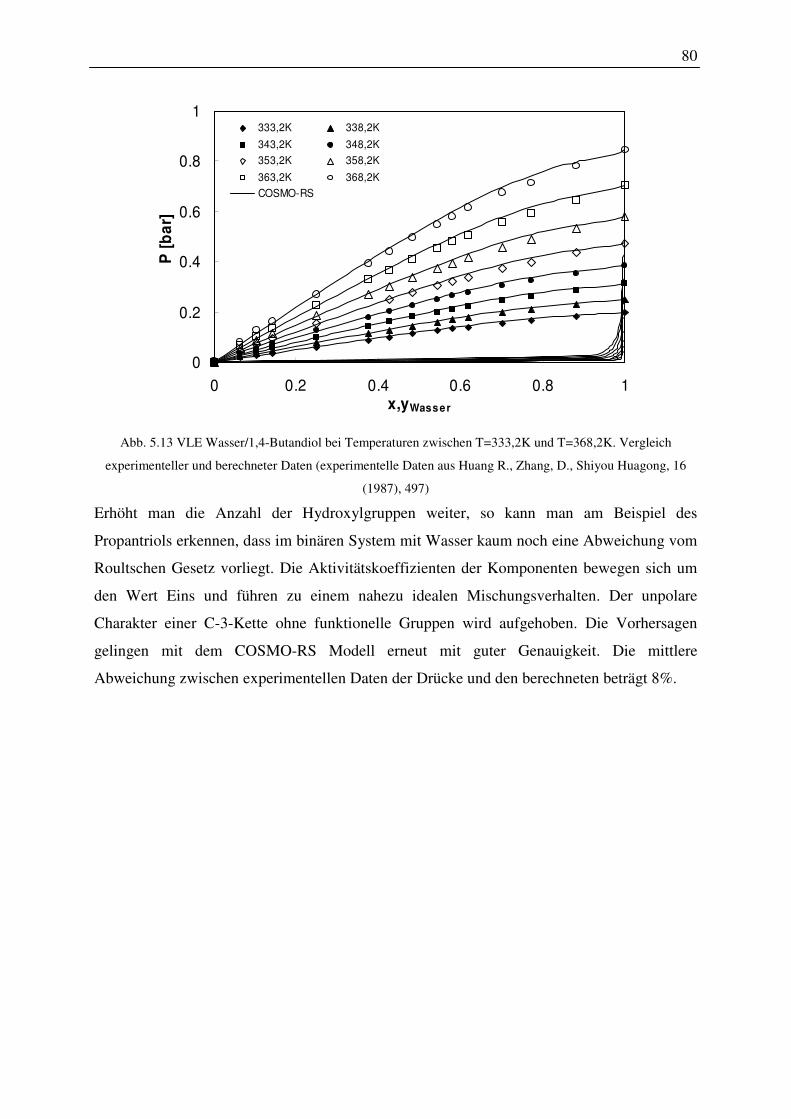
80
0
0.2
0.4
0.6
0.8
1
0 0.2 0.4 0.6 0.8 1x,yWasser
P [
ba
r]
333,2K 338,2K
343,2K 348,2K353,2K 358,2K
363,2K 368,2KCOSMO-RS
Abb. 5.13 VLE Wasser/1,4-Butandiol bei Temperaturen zwischen T=333,2K und T=368,2K. Vergleich
experimenteller und berechneter Daten (experimentelle Daten aus Huang R., Zhang, D., Shiyou Huagong, 16
(1987), 497)
Erhöht man die Anzahl der Hydroxylgruppen weiter, so kann man am Beispiel des
Propantriols erkennen, dass im binären System mit Wasser kaum noch eine Abweichung vom
Roultschen Gesetz vorliegt. Die Aktivitätskoeffizienten der Komponenten bewegen sich um
den Wert Eins und führen zu einem nahezu idealen Mischungsverhalten. Der unpolare
Charakter einer C-3-Kette ohne funktionelle Gruppen wird aufgehoben. Die Vorhersagen
gelingen mit dem COSMO-RS Modell erneut mit guter Genauigkeit. Die mittlere
Abweichung zwischen experimentellen Daten der Drücke und den berechneten beträgt 8%.

81
0
0.1
0.2
0.3
0.4
0.5
0 0.2 0.4 0.6 0.8 1x,yWasser
P [
ba
r]
323,2K
348,2K
COSMO-RS
Abb. 5.14 VLE Wasser/Propantriol bei Temperaturen von T=323,2K und T=348,2K. Vergleich experimenteller
und berechneter Daten (experimentelle Daten aus Dulitskaya, K.A.; Zh. Obshch. Khim.; 15, 9-21.1945)
In der thermischen Verfahrenstechnik kann zur Erfüllung einer Trennaufgabe das engsiedende
oder azeotrope Phasenverhalten durch Zugabe eines weiteren Stoffes selektiv geändert
werden. Ziel ist beispielsweise bei der Rektifikation, einen Zusatzstoff derart auszuwählen,
dass ein schwersiedendes Gemisch im Sumpf und eine leichtsiedende Komponente am Kopf
der Kolonne auftreten. Im System Ethanol/Wasser hat sich in der chemischen Industrie
Ethandiol als Zusatzstoff etabliert. Diese Komponente bildet mit Wasser ein schwersiedendes
binäres Gemisch. Hinsichtlich der jeweiligen Trennaufgabe sollte es möglich sein, durch
gezielte Anordnung funktioneller Gruppen innerhalb einer molekularen Struktur das
Phasenverhalten so einzustellen, dass die Trennaufgabe innerhalb der Spezifikationen erfüllt
werden kann. Die hyperverzweigten Polymere bieten hier eine hervorragende Basis, da
sowohl die Art und Anzahl der funktionellen Gruppen, als auch ihre Anordnung innerhalb der
molekularen Struktur nahezu beliebig eingestellt werden kann (vgl. [SEILER, 2004a]). Im
Hinblick auf das gewählte Testsystem und die spätere Untersuchung von verschiedenen
Polymerstrukturen gilt die Beschreibung des Systems Ethanol/Wasser/Ethandiol als
grundlegend. In Abb. 5.15 sind experimentelle Daten dieses ternären Systems dargestellt.
Aufgetragen ist die Ethanolkonzentration im Dampf und in der Flüssigkeit bei jeweils
konstanten Zugabekonzentrationen des Ethandiol. Man erkennt, dass eine Erhöhung der
Ethandiolkonzentration eine deutliche Vereinfachung der Trennaufgabe bewirkt, da das
engsiedende Verhalten aufgeweitet wird. Vorhersagen der Aktivitätskoeffizienten in diesem
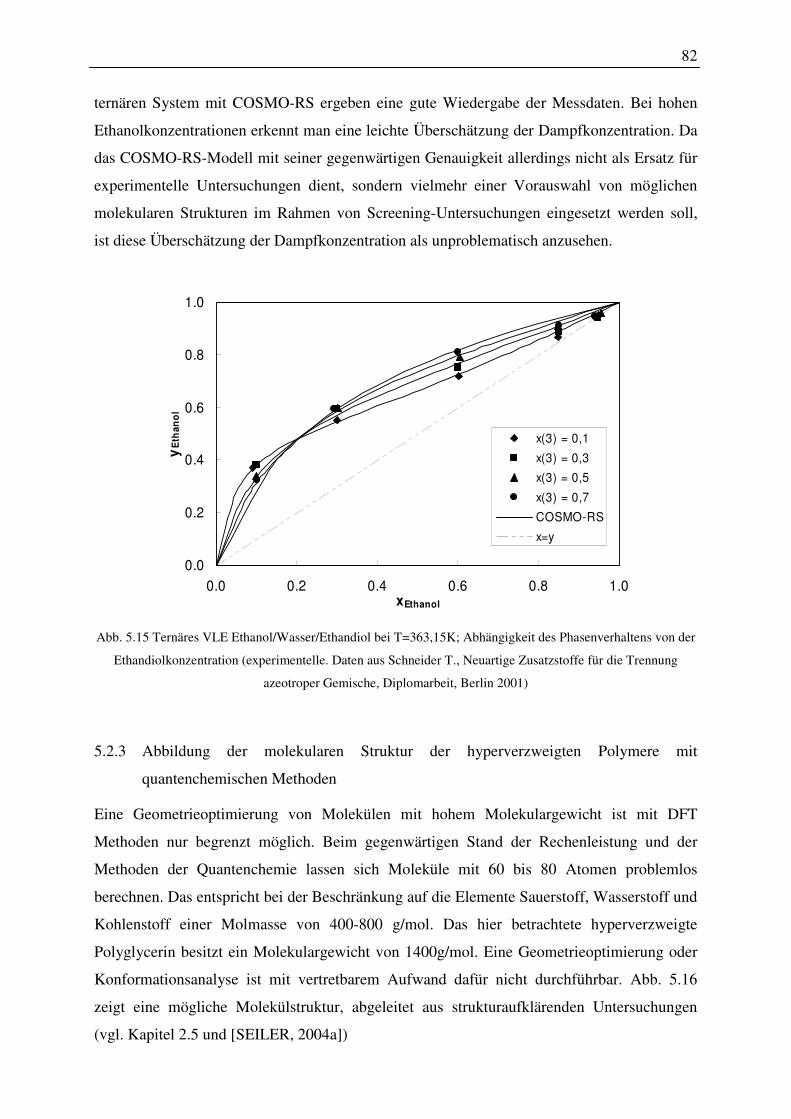
82
ternären System mit COSMO-RS ergeben eine gute Wiedergabe der Messdaten. Bei hohen
Ethanolkonzentrationen erkennt man eine leichte Überschätzung der Dampfkonzentration. Da
das COSMO-RS-Modell mit seiner gegenwärtigen Genauigkeit allerdings nicht als Ersatz für
experimentelle Untersuchungen dient, sondern vielmehr einer Vorauswahl von möglichen
molekularen Strukturen im Rahmen von Screening-Untersuchungen eingesetzt werden soll,
ist diese Überschätzung der Dampfkonzentration als unproblematisch anzusehen.
0.0
0.2
0.4
0.6
0.8
1.0
0.0 0.2 0.4 0.6 0.8 1.0xEthanol
yE
tha
no
l
x(3) = 0,1
x(3) = 0,3
x(3) = 0,5
x(3) = 0,7
COSMO-RS
x=y
Abb. 5.15 Ternäres VLE Ethanol/Wasser/Ethandiol bei T=363,15K; Abhängigkeit des Phasenverhaltens von der
Ethandiolkonzentration (experimentelle. Daten aus Schneider T., Neuartige Zusatzstoffe für die Trennung
azeotroper Gemische, Diplomarbeit, Berlin 2001)
5.2.3 Abbildung der molekularen Struktur der hyperverzweigten Polymere mit
quantenchemischen Methoden
Eine Geometrieoptimierung von Molekülen mit hohem Molekulargewicht ist mit DFT
Methoden nur begrenzt möglich. Beim gegenwärtigen Stand der Rechenleistung und der
Methoden der Quantenchemie lassen sich Moleküle mit 60 bis 80 Atomen problemlos
berechnen. Das entspricht bei der Beschränkung auf die Elemente Sauerstoff, Wasserstoff und
Kohlenstoff einer Molmasse von 400-800 g/mol. Das hier betrachtete hyperverzweigte
Polyglycerin besitzt ein Molekulargewicht von 1400g/mol. Eine Geometrieoptimierung oder
Konformationsanalyse ist mit vertretbarem Aufwand dafür nicht durchführbar. Abb. 5.16
zeigt eine mögliche Molekülstruktur, abgeleitet aus strukturaufklärenden Untersuchungen
(vgl. Kapitel 2.5 und [SEILER, 2004a])

83
H3C O O
O
OO
O O
OH
O
OHHO
HO O
OH
OH
O
OO
O
O
OH
OH
OH
O
HO
OH
OH
OOH
OHO
OHHO
OH
OH
Abb. 5.16 Molekülstruktur des hyperverzweigten Polyglycerins PG1400, abgeleitet aus experimentellen Daten
Mit NMR Untersuchungen ermittelt man den Anteil dendritischer, linearer und terminaler
Struktureinheiten, aus denen unter anderem der Verzweigungsgrad abgeleitet wird. Weiterhin
ist die Art, die Anzahl und die Lage der funktionellen Gruppen bekannt [BURGATH, 1998;
GELADE, 2001; HOELTER, 1997; VAN BENTHEM, 2001]. Die Lage der funktionellen
Gruppen und die Größe der Molekülstruktur wurden in experimentellen Untersuchungen
mittels Small-Angle-Neutron- und X-Ray-Scattering (SAXS, SANS) bestimmt [HEDDEN,
2003; SCHERRENBERG, 1998; TOPP, 1999c; TOPP, 1999a; TOPP, 1999b].
Zum gegenwärtigen Zeitpunkt gibt es prinzipiell drei Möglichkeiten, die molekularen
Strukturen für die Berechnungen mit COSMO-RS bereitzustellen:
1. Zusammensetzen der Gesamtstruktur aus kleinen Einzelfragmenten. Entsprechend den
Ergebnissen der NMR-Untersuchungen werden Basisstruktureinheiten definiert. Diese
Basiseinheiten werden an den Rändern abgesättigt und einer Geometrieoptimierung
zugeführt. Durch Mehrfachgewichtung dieser optimierten Strukturen kann das
Sigmaprofil eines großen Moleküls erzeugt werden.
2. Zusammensetzen aus möglichst großen Substrukturen. Nach Ermittlung der möglichen
Topologie der Gesamtmolekülstruktur wird diese so in Unterstrukturen aufgeteilt, dass
Abschirmungseffekte innerhalb der Molekülstruktur erfasst werden und gleichzeitig

84
eine Geometrieoptimierung mit DFT-Methoden möglich ist. Mit diesen wenigen
Unterstrukturen wird ein Sigmaprofil des Gesamtmoleküls erzeugt.
3. Berechnung der Gesamtmolekülstruktur. Die nach den vorliegenden experimentellen
Daten erzeugte Topologie wird als gesamtes Molekül einmalig der Berechnung der
Abschirmungsladungsdichte zugeführt und somit das Sigmaprofil ermittelt.
Der Vorteil der ersten Variante ist die schnelle Generierung der einzelnen, kleinen und
gleichzeitig geometrieoptimierten Fragmente. Als großer Nachteil erweist sich aber, dass
keine Informationen über Abschirmungseffekte innerhalb einer großen Struktur in das
Sigmaprofil übertragen werden können. Je nach Art der Zerlegung kann dies z.T. durch
Einsatz der Variante zwei überwunden werden. In der dritten Variante können
Abschirmungseffekte explizit erfasst werden. Befindet sich beispielsweise eine
Hydroxylgruppe im Innern des Moleküls, wird sie keinen oder nur einen geringen Beitrag zu
Abschirmungsladung auf der Oberfläche des Moleküls liefern und nur diese
Abschirmungsladungen gehen in das Wechselwirkungsverhalten ein, welches mit COSMO-
RS modelliert wird.
Zusammensetzen der Gesamtstruktur aus kleinen Einzelfragmenten.
Die Molekülstruktur des hier betrachteten Polyglycerins wurde von Hoelter et al. untersucht
[HOELTER, 1997] und es treten vier sich wiederholende Einheiten auf, eine dendritische (D),
eine terminale (T), zwei lineare (L13, L14) Einheiten und eine Starteinheit (Abb. 5.17). Ihre
Eigenschaften sind in Tabelle 5.4 zusammengestellt. Die Bindung gibt hierbei an, ob durch
eine Einheit eine Verzweigungsmöglichkeit in Form einer Bindung hinzukommt (+1), wie es
bei der Starteinheit oder bei den dendritischen Einheiten der Fall ist, bzw. ob eine
Verzweigung durch eine terminale Einheit beendet wird (-1).
Tabelle 5.4: Eigenschaften der Struktureinheiten des hyperverzweigten Polyglycerins
Einheit Molmasse
[g/mol]
OH- Gruppen
pro Einheit
Bindungen
Starteinheit 31,03 0 +1
Dendritisch D 73,07 0 +1
Linear L13 74,08 1 0
Linear L14 74,08 1 0
Terminal T 75,09 2 -1

85
O CH2
CH
O
H2C
O
O CH2
CH
O
H2C
OH
O CH2
CH
OH
H2C
OH
O CH2
CH
CH2
OH
O
H3C
O
dendritisch D linear L13
linear L14terminal T
Starteinheit
O CH2
CH
O
H2C
O
O CH2
CH
O
H2C
OH
O CH2
CH
OH
H2C
OH
O CH2
CH
CH2
OH
O
H3C
O
dendritisch D linear L13
linear L14terminal T
Starteinheit
Abb. 5.17 chemische Struktur der vier sich wiederholenden Struktureinheiten des untersuchten Polyglyzerins
Anhand der Anteile der verschiedenen Struktureinheiten und dem Molekulargewicht des
hyperverzweigten Polyglycerins kann die Anzahl der Einheiten im Molekül berechnet werden
(Tabelle 5.5). Das Ergebnis wird durch das berechnete Molekulargewicht, die Anzahl der OH-
Gruppen des Moleküls und durch die Anzahl der Bindungen überprüft. Die Anzahl der
Bindungen muss 0 ergeben, damit keine ungesättigten Bindungen im Molekül vorliegen.
Tabelle 5.5 Eigenschaften der vier sich wiederholenden Struktureinheiten des untersuchten
Polyglyzerins
Einheit Anteil
[%]
Anzahl Molmasse
[g/mol]
Anzahl
OH- Gruppen
Bindungen
Starteinheit 5 1 31,034 0 +1
Dendritisch D 25 5 365,35 0 +5
Linear L13 10 2 148,16 2 0
Linear L14 30 6 444,47 6 0
Terminal T 30 6 450,52 12 -6
gesamt 100 20 1439,53 20 0
Da mit dem COSMO-RS-Modell, im Gegensatz zu dem in Seiler et al. [SEILER, 2004b]
vorgeschlagenem Verfahren Wechselwirkungsanteile einzelner Struktureinheiten nicht

86
ausgeblendet werden können, muss an den vorliegenden Phasengleichgewichtsdaten getestet
werden, ob eine Einteilung in größere Struktureinheiten sinnvoll ist.
Zusammensetzen der Gesamtstruktur aus möglichst großen Substrukturen
Die Molekülfragmente sind so gewählt, dass sie einerseits vollständig mit DFT-Methoden
geometrieoptimiert werden können und andererseits sterische Effekte, hervorgerufen durch
Verzweigungen, berücksichtigt werden. Für dieses Vorgehen wurde das Gesamtmolekül in
vier Subeinheiten zerlegt (vgl. Abb. 5.18). Die Zusammensetzung und die Molmassen dieser
vier Subeinheiten können Tabelle 5.6 entnommen werden.
A
Starter D
D
D
D
L14
L13L14
TT
T
D
L14
L14
T
TL14
L13
L14
T
Sub4Sub3
Sub2
Sub1
B
H3C O O
O
OO
O O
OH
O
OHHO
HO O
OH
OH
O
OO
O
O
OH
OH
OH
O
HO
OH
OH
OOH
OHO
OHHO
OH
OH
A
Starter D
D
D
D
L14
L13L14
TT
T
D
L14
L14
T
TL14
L13
L14
T
Sub4Sub3
Sub2
Sub1
B
Starter D
D
D
D
L14
L13L14
TT
T
D
L14
L14
T
TL14
L13
L14
T
Sub4Sub3
Sub2
Sub1
B
H3C O O
O
OO
O O
OH
O
OHHO
HO O
OH
OH
O
OO
O
O
OH
OH
OH
O
HO
OH
OH
OOH
OHO
OHHO
OH
OH
Abb. 5.18 Molekülstruktur des hyperverzweigten Polyglycerins; A: Strukturformel des hyperverzweigten
Polyglycerins; B: Zusammensetzung des hyperverzweigten Polyglycerins aus dendritischen, linearen und
terminalen Einheiten und Zusammenfassung in große Subeinheiten zur Geometrieoptimierung
Tabelle 5.6 Zusammensetzung und Molmasse der Subeinheiten des hyperverzweigten
Polymers
Einheit S D L13 L14 T Molmasse Sub 1 0 1 1 1 2 371,40 Sub 2 1 2 0 1 1 326,34 Sub 3 0 1 1 2 1 370,39 Sub 4 0 1 0 2 2 371,40
1 5 2 6 6 gesamt
20 1439,53
Die Geometrien der vier Subeinheiten werden als vollständige Moleküle optimiert. Bei der
Optimierung muss darauf geachtet werden, dass jede Schnittstelle ausreichend abgesättigt ist.

87
Im Fall des verzweigten Polyglycerins wird diese Forderung erfüllt, indem die Schnittstellen
der Subeinheiten durch OCH2CH2OH-Gruppen oder durch CH2CH2OH-Gruppen abgesättigt
werden. Durch die Verwendung dieser zwei Endgruppen ist sichergestellt, dass jede
Schnittstelle der Subeinheiten durch eine Estergruppe (C-O-C), wie sie im hyperverzweigten
Polyglycerin vorliegt, abgesättigt wird und der Schnitt zur Erstellung der Fragmente trotz der
hohen Polarität an dieser Stelle möglich ist. Nach der Molekülgeometrieoptimierung bilden
die Subeinheiten die Grundlage für die Beschreibung des hyperverzweigten Polyglycerins.
Hierzu werden die für die Geometrieoptimierung zugefügten Gruppen ausgeblendet und
gehen nicht in die Erstellung des Sigmaprofils des hyperverzweigten Polyglycerins ein.
Berechnung der Gesamtmolekülstruktur
Eine Geometrieoptimierung ist wie oben ausgeführt für diese Molekülgröße mit der DFT
nicht möglich. Ausgehend von der Molekülstruktur nach Abb. 5.16 wurde eine Geometrie
erzeugt, die mit der semiempirischen Methode AM1 (vgl. Kap 2.2.1) optimiert worden ist.
Für diese optimierte Struktur ist in einer Single-Point-Rechnung das Sigmaprofil bestimmt
worden.
Abb. 5.19 zeigt die drei verschiedenen Sigmaprofile für die unterschiedlichen
Lösungsansätze. Die Sigmaprofile des hyperverzweigten Polyglycerins, erzeugt mit der
Zusammensetzung aus kleinen Einzelfragmenten und aus Subeinheiten, weisen einen hohen
Anteil an schwach positiv geladenen Oberflächensegmenten auf. Der Unterschied liegt in der
Anzahl der stark geladenen Segmente. Der Grund dafür ist, dass sich im Falle der
Einzelfragmente alle funktionellen Gruppen an der Moleküloberfläche befinden. Bei der
Verwendung von Subeinheiten werden einige Hydroxylgruppen abgeschirmt, was zu einer
Verringerung der Oberflächensegmente mit entsprechender Ladung führt. Aus diesem Grund
werden bei der Erstellung des Sigmaprofils des hyperverzweigten Polyglycerins mehr
Ladungen berücksichtigt als im realen Molekül einen Einfluss auf die
Ladungsdichteverteilung auf der Oberfläche haben. Das spiegelt das Sigmaprofil des
Gesamtmoleküls wider. Die Zahl der Oberflächensegmente mit positiver oder negativer
Abschirmungsladung ist deutlich reduziert. Trotz dieses Unterschiedes ist im Rahmen dieser
Arbeit die Entscheidung zugunsten einer Verwendung des Sigmaprofils aus Subeinheiten
gefallen. Die Tastsache, dass es sich um ein nicht vollständig DFT-optimiertes Molekül
handelt und dass die reale, in Lösung auftretende Struktur sich von der im Vakuum mit AM1-
optimierten unterscheiden wird, haben diese Entscheidung herbeigeführt.
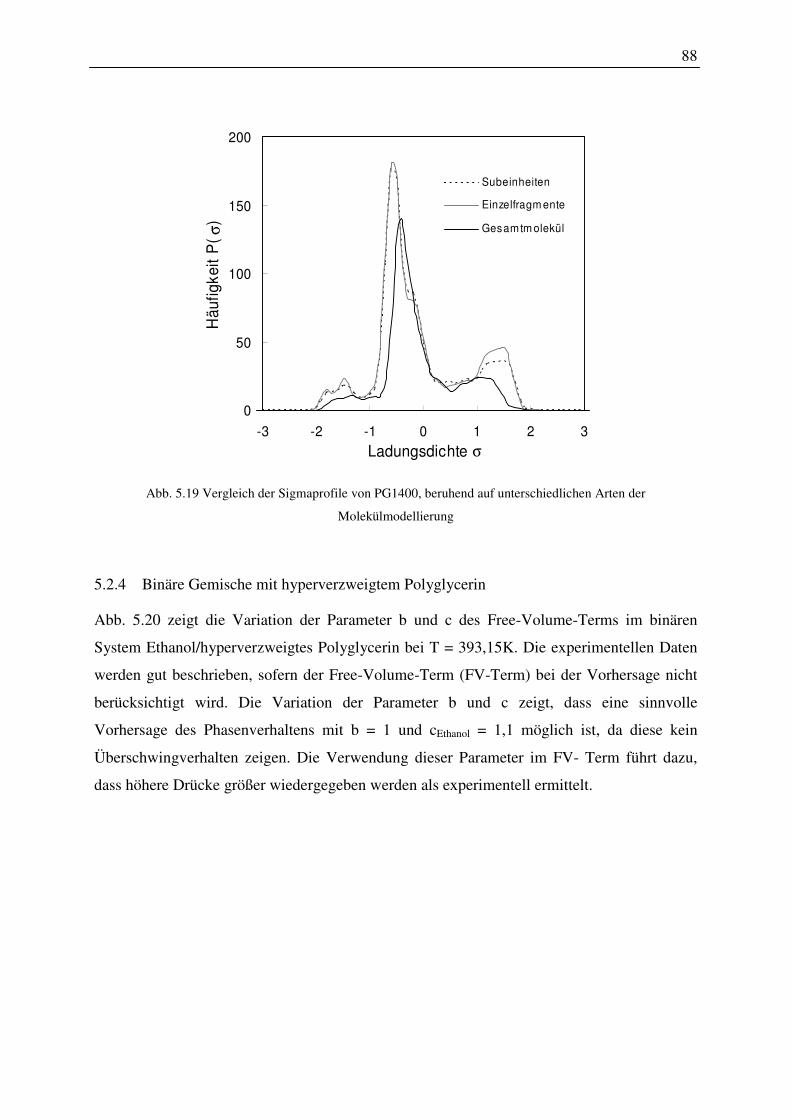
88
0
50
100
150
200
-3 -2 -1 0 1 2 3Ladungsdichte σ
Häu
figke
it P
( σ)
Subeinheiten
Einzelfragm ente
Gesamtm olekül
Abb. 5.19 Vergleich der Sigmaprofile von PG1400, beruhend auf unterschiedlichen Arten der
Molekülmodellierung
5.2.4 Binäre Gemische mit hyperverzweigtem Polyglycerin
Abb. 5.20 zeigt die Variation der Parameter b und c des Free-Volume-Terms im binären
System Ethanol/hyperverzweigtes Polyglycerin bei T = 393,15K. Die experimentellen Daten
werden gut beschrieben, sofern der Free-Volume-Term (FV-Term) bei der Vorhersage nicht
berücksichtigt wird. Die Variation der Parameter b und c zeigt, dass eine sinnvolle
Vorhersage des Phasenverhaltens mit b = 1 und cEthanol = 1,1 möglich ist, da diese kein
Überschwingverhalten zeigen. Die Verwendung dieser Parameter im FV- Term führt dazu,
dass höhere Drücke größer wiedergegeben werden als experimentell ermittelt.
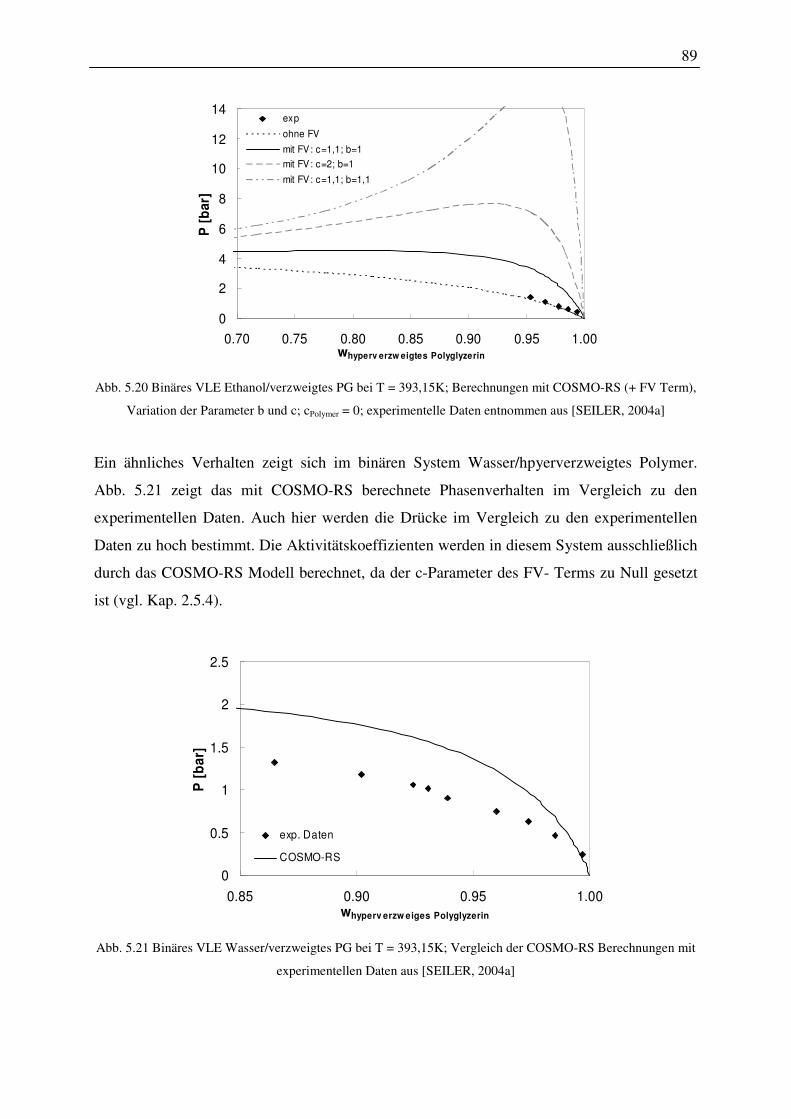
89
0
2
4
6
8
10
12
14
0.70 0.75 0.80 0.85 0.90 0.95 1.00whyperv erzw eigtes Polyglyzerin
P [
ba
r]
exp
ohne FV
mit FV: c=1,1; b=1mit FV: c=2; b=1
mit FV: c=1,1; b=1,1
Abb. 5.20 Binäres VLE Ethanol/verzweigtes PG bei T = 393,15K; Berechnungen mit COSMO-RS (+ FV Term),
Variation der Parameter b und c; cPolymer = 0; experimentelle Daten entnommen aus [SEILER, 2004a]
Ein ähnliches Verhalten zeigt sich im binären System Wasser/hpyerverzweigtes Polymer.
Abb. 5.21 zeigt das mit COSMO-RS berechnete Phasenverhalten im Vergleich zu den
experimentellen Daten. Auch hier werden die Drücke im Vergleich zu den experimentellen
Daten zu hoch bestimmt. Die Aktivitätskoeffizienten werden in diesem System ausschließlich
durch das COSMO-RS Modell berechnet, da der c-Parameter des FV- Terms zu Null gesetzt
ist (vgl. Kap. 2.5.4).
0
0.5
1
1.5
2
2.5
0.85 0.90 0.95 1.00whyperv erzw eiges Polyglyzerin
P [
ba
r]
exp. Daten
COSMO-RS
Abb. 5.21 Binäres VLE Wasser/verzweigtes PG bei T = 393,15K; Vergleich der COSMO-RS Berechnungen mit
experimentellen Daten aus [SEILER, 2004a]
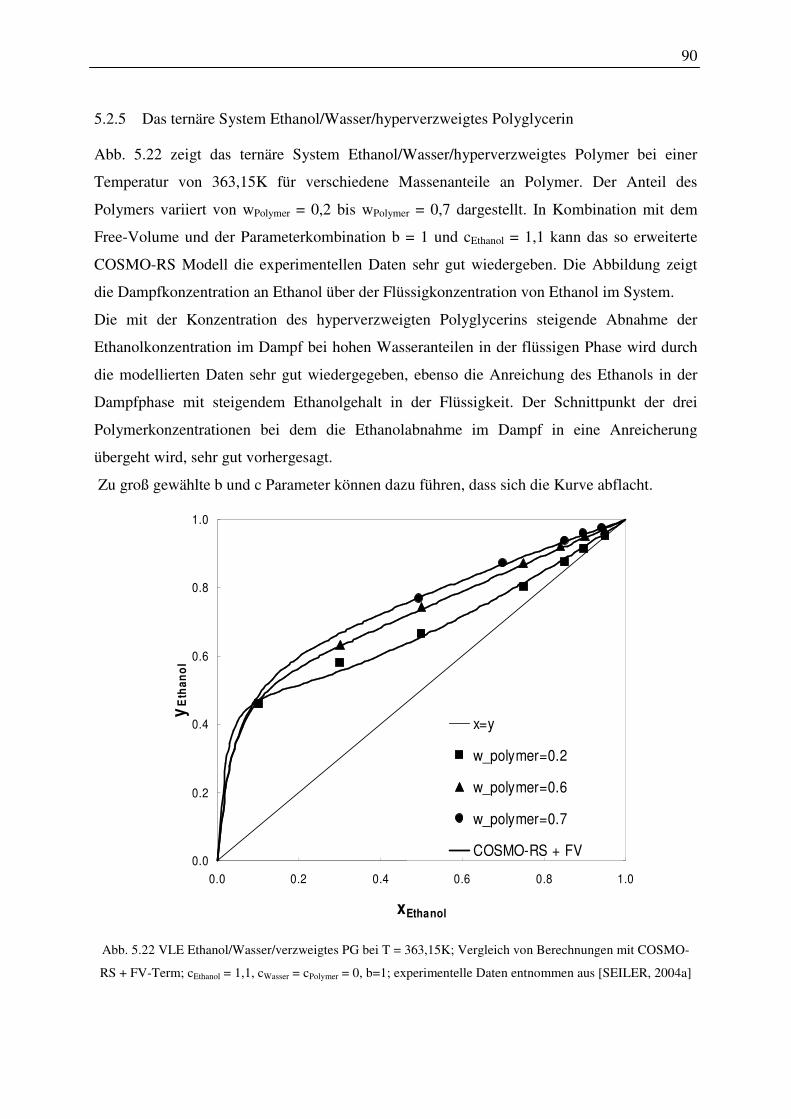
90
5.2.5 Das ternäre System Ethanol/Wasser/hyperverzweigtes Polyglycerin
Abb. 5.22 zeigt das ternäre System Ethanol/Wasser/hyperverzweigtes Polymer bei einer
Temperatur von 363,15K für verschiedene Massenanteile an Polymer. Der Anteil des
Polymers variiert von wPolymer = 0,2 bis wPolymer = 0,7 dargestellt. In Kombination mit dem
Free-Volume und der Parameterkombination b = 1 und cEthanol = 1,1 kann das so erweiterte
COSMO-RS Modell die experimentellen Daten sehr gut wiedergeben. Die Abbildung zeigt
die Dampfkonzentration an Ethanol über der Flüssigkonzentration von Ethanol im System.
Die mit der Konzentration des hyperverzweigten Polyglycerins steigende Abnahme der
Ethanolkonzentration im Dampf bei hohen Wasseranteilen in der flüssigen Phase wird durch
die modellierten Daten sehr gut wiedergegeben, ebenso die Anreichung des Ethanols in der
Dampfphase mit steigendem Ethanolgehalt in der Flüssigkeit. Der Schnittpunkt der drei
Polymerkonzentrationen bei dem die Ethanolabnahme im Dampf in eine Anreicherung
übergeht wird, sehr gut vorhergesagt.
Zu groß gewählte b und c Parameter können dazu führen, dass sich die Kurve abflacht.
0.0
0.2
0.4
0.6
0.8
1.0
0.0 0.2 0.4 0.6 0.8 1.0
xEthanol
yE
tha
no
l
x=y
w_polymer=0.2
w_polymer=0.6
w_polymer=0.7
COSMO-RS + FV
Abb. 5.22 VLE Ethanol/Wasser/verzweigtes PG bei T = 363,15K; Vergleich von Berechnungen mit COSMO-
RS + FV-Term; cEthanol = 1,1, cWasser = cPolymer = 0, b=1; experimentelle Daten entnommen aus [SEILER, 2004a]

91
5.2.6 Zusammenfassende Bemerkungen
Die Untersuchungen zum Phasenverhalten hyperverzweigter Polymere haben gezeigt, dass es
möglich ist, das Wechselwirkungsverhalten von stark verzweigten Molekülen großer
Molmasse in Lösung mit dem COSMO-RS-Modell vorherzusagen. Bei der Erstellung der
Molekülgeometrie der verzweigten Moleküle müssen mehrere Gesichtspunkte beachtet
werden. Insbesondere zu berücksichtigen ist, dass es bei starker Verzweigung zu
intramolekularen Abschirmungseffekten kommen kann.
Der in dieser Arbeit verfolgte Weg beinhaltet keine Analyse stabiler konformerer Strukturen
der Moleküle. Weiterhin wird der intermolekulare Einfluss des Lösungsmittels vernachlässigt,
da die Molekülstrukturen im Vakuum erstellt werden und durch die COSMO-Randbedingung
im idealen Leiter geometrieoptimiert werden. Ein Ansatz den Lösungsmitteleffekt zu
berücksichtigen, ist, die Molekülstruktur durch Molekularsimulationen direkt in Anwesenheit
des oder der Lösungsmittel zu generieren. Dabei wird genau wie oben beschrieben zuerst auf
experimentelle Daten über die Struktur zurückgegriffen (Verzweigungsgrad, Anzahl
funktioneller Gruppen, etc.) und eine allgemeine zweidimensionale Topologie erstellt. Hierzu
werden alle Anordnungsmöglichkeiten der dendritischen, linearen und terminalen
Strukturgruppen, die die Strukturvorgaben wiedergeben, ermittelt. Eine jeder dieser
Topologien wird mit einem Sketchingprogramm (HyperChem, Cerius2) [ACCELRYS INC.,
2005; HYPERCUBE INC., 2005] in eine dreidimensionale Molekülstruktur übertragen. Diese
Startgeometrien werden anschließend mit der Molekularmechanik-Methode
geometrieoptimiert. Hierzu werden die MM3- und MM4-Kraftfelder verwendet [ALLINGER,
1989; ALLINGER, 1996; ALLINGER, 2003; LII, 1989a; LII, 1989b]. In der Literatur wird
auch die Anwendung anderer Kraftfelder auf stark verzweigte Polymere diskutiert (PCCF
[ORTIZ, 2004], DREIDING [CAGIN, 1999; CAGIN, 2000; ORTIZ, 2004], CHARMM
[NAIDOO, 1999]). Um die elektrostatischen Energien angemessen zu berücksichtigen,
müssten jedem Atom Punktladungen zugewiesen werden, deren Stärke mit ab initio oder DFT
Rechnungen bestimmt wird. Die elektrostatische Energie ergibt sich dann als Summe der
Dipol-Dipol Wechselwirkungen. Dieser Ansatz ist in den MM3- und MM4- Kraftfeldern
integriert und parametrisiert. Als Ergebnis dieser Geometrieoptimierung liegt ein „educated
guess“ für jede der Startstrukturen vor. Um aus diesem „educated guess“ eine Geometrie zu
bestimmen, in der die elektrostatischen Wechselwirkungen der funktionellen Gruppen
angemessener berücksichtigt sind, schließt sich eine quantenmechanische
Geometrieoptimierung auf ab initio Niveau an (MP2/6-31G*). Für die Molekülstrukturen,

92
deren Größe eine ab initio Rechnung nicht zulässt, wird eine Geometrieoptimierung auf DFT
Niveau durchgeführt. Ein Vergleich der möglichen Genauigkeiten findet sich in [MONEV,
2005]. Aus quantenmechanischen Rechnung werden mittels „restrained electrostatic potential
(RESP)“- Ansatz [BAYLY, 1993; WANG, 2000] die Parameter für die atomzentrierten
Punktladungen zur Beschreibung der Coulombkräfte bestimmt. Die Molekülgeometrien
werden nun mit bekannten Parametern für die Coulomb Atom-Atom-Wechselwirkungen
mittels MM3/MM4 nochmals geometrieoptimiert, um Molekülstrukturen für die
anschließende Konformerensuche bereitzustellen.
Der Konformerenraum wird für die optimierten Startgeometrien mittels statistischer
Methoden („Conformational Space Annealing“ CSA und „Monte Carlo with minimization“
MCM) abgesucht [KIM, 2003; LEE, 1999; LEE, 2004; NAYEEM, 1991; PILLARDY, 2000;
TROSSET, 1998]. Die Suche wird unter Verwendung der zuvor benutzten MM3/MM4
Kraftfelder unter Berücksichtigung der Coulombkräfte mit dem Programmpaket TINKER
[PONDER, 2005] durchgeführt. Die gefundenen Konformationen werden im Anschluss
einem „Energieclustering“ unterzogen, um die Anzahl der Konformationen zu reduzieren und
die Rechnung mit wenigen relevanten Struktur- und Energievertretern fortzuführen. Die
Geometrie dieser Vertreter wird erneut optimiert (MM3/MM4).
Bis zu diesem Punkt sind alle Rechnungen in der Gasphase durchgeführt worden. Um den
Einfluss eines Lösemittels auf die Molekülgeometrien zu untersuchen, werden mit den
gefundenen Konformationen Molekulardynamikrechnungen durchgeführt, die das
Lösungsmittel explizit berücksichtigen. Diese Optimierungen werden ebenfalls mit
Kraftfeldern MM3/MM4 und den entsprechenden Parametersätzen für die Lösungsmittel
ausgeführt. Für die optimierten Strukturen werden die Gyrationsradien und die
hydrodynamischen Radien berechnet und mit den experimentellen Daten verglichen. Abb.
5.23 verdeutlicht das Vorgehen schematisch.
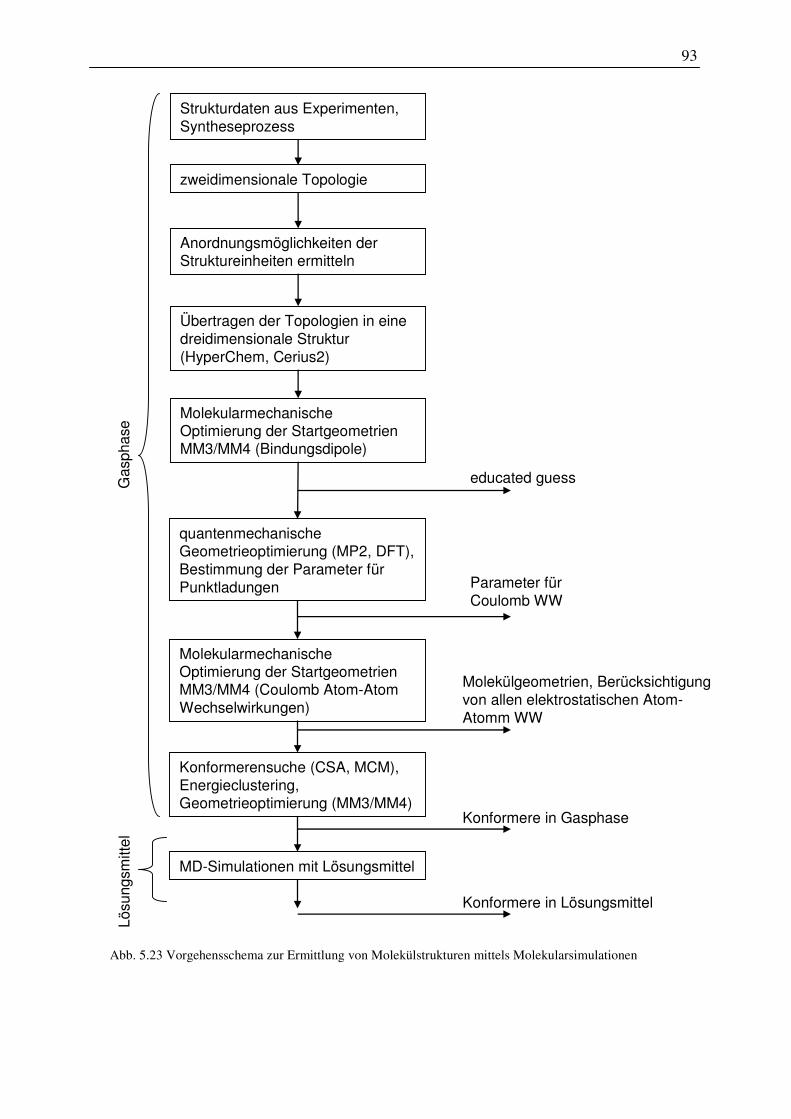
93
Strukturdaten aus Experimenten, Syntheseprozess
zweidimensionale Topologie
Anordnungsmöglichkeiten der Struktureinheiten ermitteln
Übertragen der Topologien in eine dreidimensionale Struktur (HyperChem, Cerius2)
Molekularmechanische Optimierung der Startgeometrien MM3/MM4 (Bindungsdipole)
quantenmechanische Geometrieoptimierung (MP2, DFT), Bestimmung der Parameter für Punktladungen
Molekularmechanische Optimierung der Startgeometrien MM3/MM4 (Coulomb Atom-Atom Wechselwirkungen)
Konformerensuche (CSA, MCM), Energieclustering, Geometrieoptimierung (MM3/MM4)
MD-Simulationen mit Lösungsmittel
educated guess
Parameter für Coulomb WW
Molekülgeometrien, Berücksichtigung von allen elektrostatischen Atom-Atomm WW
Konformere in Gasphase
Konformere in Lösungsmittel
Gas
phas
eLö
sung
smitt
el
Abb. 5.23 Vorgehensschema zur Ermittlung von Molekülstrukturen mittels Molekularsimulationen
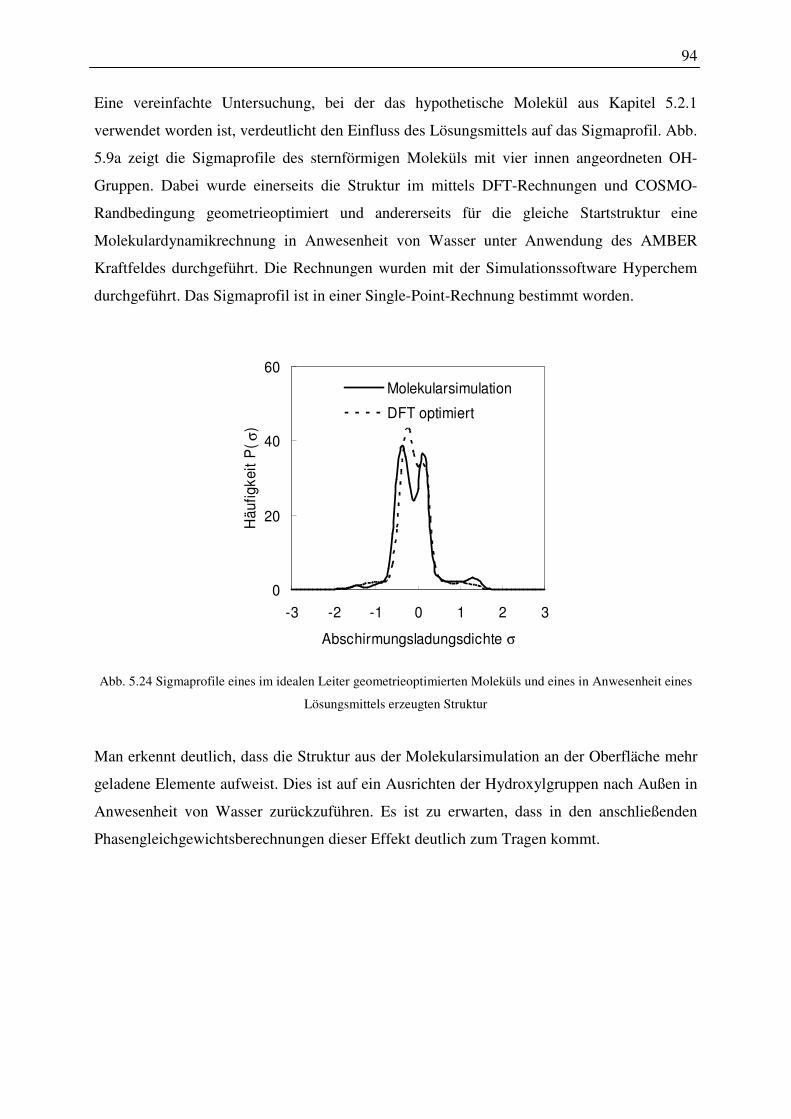
94
Eine vereinfachte Untersuchung, bei der das hypothetische Molekül aus Kapitel 5.2.1
verwendet worden ist, verdeutlicht den Einfluss des Lösungsmittels auf das Sigmaprofil. Abb.
5.9a zeigt die Sigmaprofile des sternförmigen Moleküls mit vier innen angeordneten OH-
Gruppen. Dabei wurde einerseits die Struktur im mittels DFT-Rechnungen und COSMO-
Randbedingung geometrieoptimiert und andererseits für die gleiche Startstruktur eine
Molekulardynamikrechnung in Anwesenheit von Wasser unter Anwendung des AMBER
Kraftfeldes durchgeführt. Die Rechnungen wurden mit der Simulationssoftware Hyperchem
durchgeführt. Das Sigmaprofil ist in einer Single-Point-Rechnung bestimmt worden.
0
20
40
60
-3 -2 -1 0 1 2 3
Abschirmungsladungsdichte σ
Häu
figke
it P
( σ)
Molekularsimulation
DFT optimiert
Abb. 5.24 Sigmaprofile eines im idealen Leiter geometrieoptimierten Moleküls und eines in Anwesenheit eines
Lösungsmittels erzeugten Struktur
Man erkennt deutlich, dass die Struktur aus der Molekularsimulation an der Oberfläche mehr
geladene Elemente aufweist. Dies ist auf ein Ausrichten der Hydroxylgruppen nach Außen in
Anwesenheit von Wasser zurückzuführen. Es ist zu erwarten, dass in den anschließenden
Phasengleichgewichtsberechnungen dieser Effekt deutlich zum Tragen kommt.
95
5.3 Verknüpfung des COSMO-RS-Modells mit Zustandsgleichungen
Die Kombination von COSMO-RS mit kubischen Zustandsgleichungen erfolgt auf zwei
Arten. Dabei werden die Mischungsparameter für das Ein-Fluid-Modell durch die
Anwendung der Wong-Sandler Mischungsregeln (siehe Kapitel 2.6) berechnet. Das COSMO-
RS-Modell wird genutzt, um die Gibbssche Exzessenthalpie zu berechnen.
Bei der Methode der Korrelation eines binären Wechselwirkungsparameters wird dieser an
experimentelle Phasengleichgewichtsdaten angepasst. Dabei wird das in Kapitel 2.6
beschriebene, von Huron und Vidal vorgestellte modifizierte NRTL-Modell als GE-Modell
genutzt und der Parameter τij über Gleichung 2.80 durch die Vorhersage des
Aktivitätskoeffizienten bei unendlicher Verdünnung mittels COSMO-RS berechnet.
Für die Untersuchungen zur Güte der Vorhersagbarkeit und der Korrelation bei Verwendung
des COSMO-RS-Modells wurden die in Tabelle 5.7 angegebenen Systeme betrachtet. In der
Tabelle sind weiterhin die angepassten binären Wechselwirkungsparameter für den jeweiligen
Temperaturbereich und der verwendete Alpha-Wert des NRTL-Modells vermerkt.
Tabelle 5.7 Betrachtete Systeme bei der Kombination von Peng-Robinson Zustandsgleichung und COSMO-RS;
Systemübersicht, betrachtete Temperaturbereiche, angepasste binäre Wechselwirkungsparameter und α-Wert
System Temperaturbereich
[K]
kij α
Methanol-Benzol
308,15 – 363,15
0,20
-
Ethanol-Wasser 323,15 – 363,15 0,35 0,4
523,15 0,25 0,4
598,15 0 0,4
Aceton-Wasser 373,15 – 423,15 0,23 0,4
523,15 0,18 0,4
Methanol-Wasser 298,15 – 333,15 0,2 0,4
573,15 0 0,4
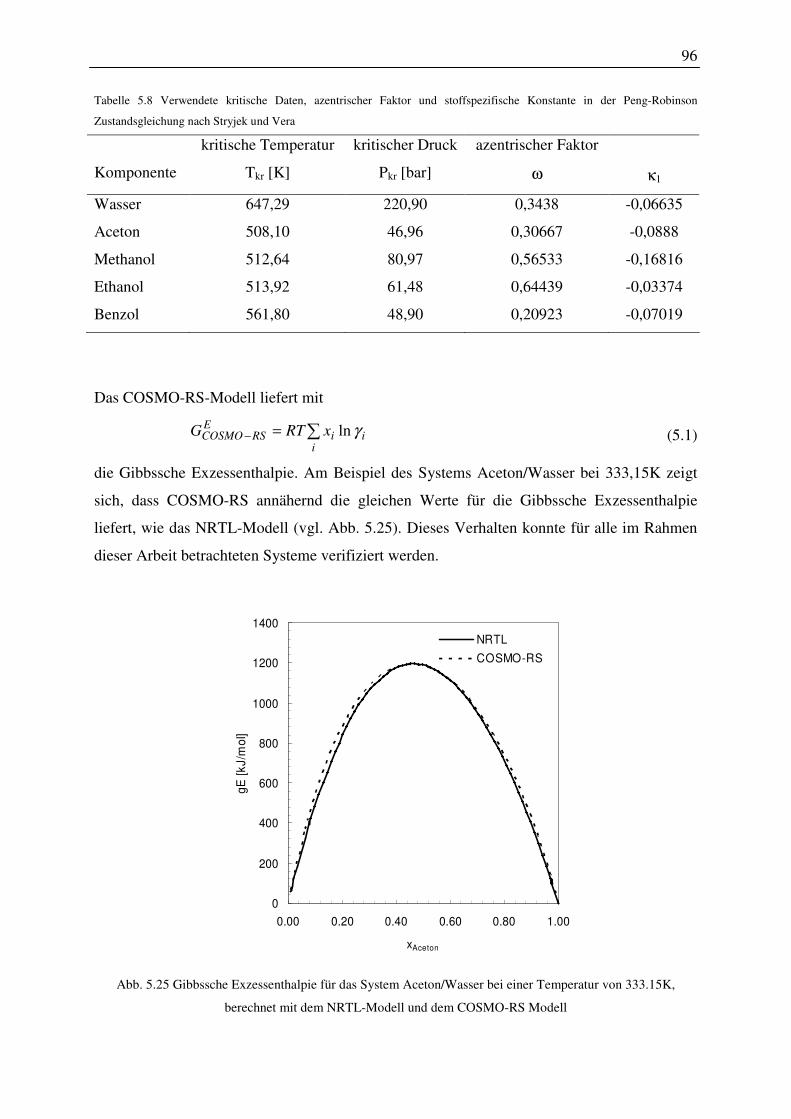
96
Tabelle 5.8 Verwendete kritische Daten, azentrischer Faktor und stoffspezifische Konstante in der Peng-Robinson
Zustandsgleichung nach Stryjek und Vera
Komponente
kritische Temperatur
Tkr [K]
kritischer Druck
Pkr [bar]
azentrischer Faktor
ω
κ1
Wasser 647,29 220,90 0,3438 -0,06635
Aceton 508,10 46,96 0,30667 -0,0888
Methanol 512,64 80,97 0,56533 -0,16816
Ethanol 513,92 61,48 0,64439 -0,03374
Benzol 561,80 48,90 0,20923 -0,07019
Das COSMO-RS-Modell liefert mit
∑=−i
iiE
RSCOSMO xRTG γln
(5.1)
die Gibbssche Exzessenthalpie. Am Beispiel des Systems Aceton/Wasser bei 333,15K zeigt
sich, dass COSMO-RS annähernd die gleichen Werte für die Gibbssche Exzessenthalpie
liefert, wie das NRTL-Modell (vgl. Abb. 5.25). Dieses Verhalten konnte für alle im Rahmen
dieser Arbeit betrachteten Systeme verifiziert werden.
0
200
400
600
800
1000
1200
1400
0.00 0.20 0.40 0.60 0.80 1.00
xAceton
gE [k
J/m
ol]
NRTL
COSMO-RS
Abb. 5.25 Gibbssche Exzessenthalpie für das System Aceton/Wasser bei einer Temperatur von 333.15K,
berechnet mit dem NRTL-Modell und dem COSMO-RS Modell
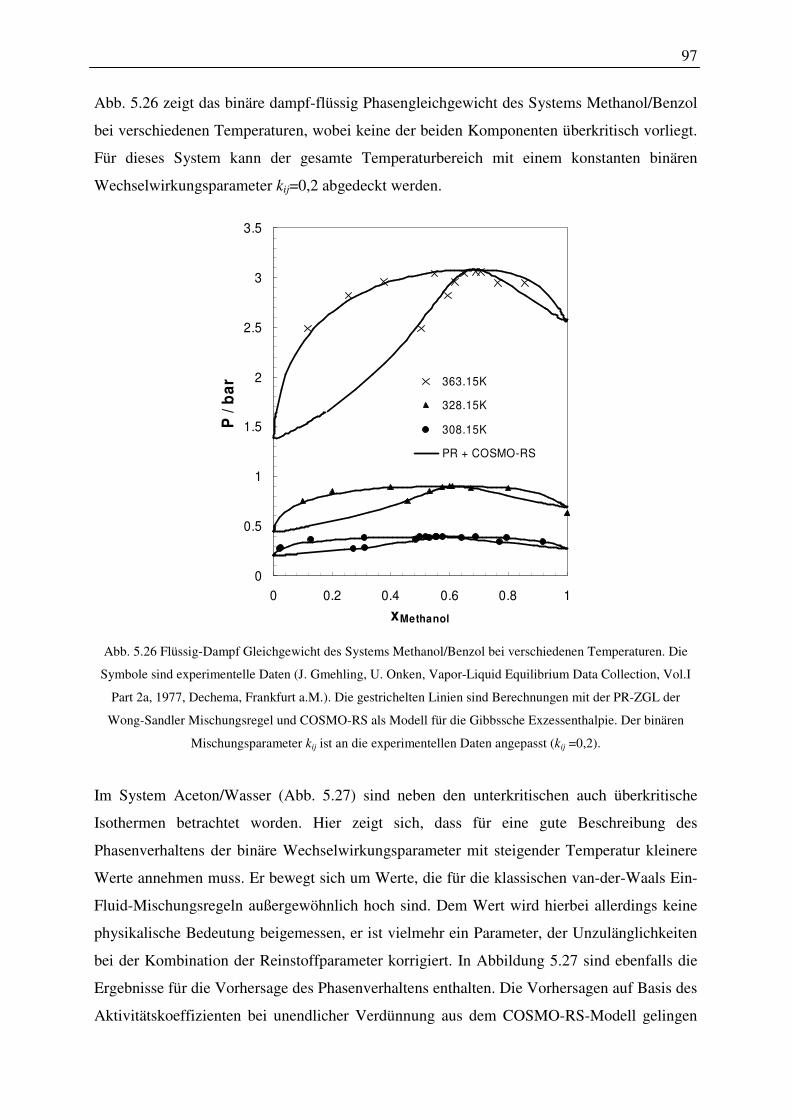
97
Abb. 5.26 zeigt das binäre dampf-flüssig Phasengleichgewicht des Systems Methanol/Benzol
bei verschiedenen Temperaturen, wobei keine der beiden Komponenten überkritisch vorliegt.
Für dieses System kann der gesamte Temperaturbereich mit einem konstanten binären
Wechselwirkungsparameter kij=0,2 abgedeckt werden.
0
0.5
1
1.5
2
2.5
3
3.5
0 0.2 0.4 0.6 0.8 1
xMethanol
P /
ba
r 363.15K
328.15K
308.15K
PR + COSMO-RS
Abb. 5.26 Flüssig-Dampf Gleichgewicht des Systems Methanol/Benzol bei verschiedenen Temperaturen. Die
Symbole sind experimentelle Daten (J. Gmehling, U. Onken, Vapor-Liquid Equilibrium Data Collection, Vol.I
Part 2a, 1977, Dechema, Frankfurt a.M.). Die gestrichelten Linien sind Berechnungen mit der PR-ZGL der
Wong-Sandler Mischungsregel und COSMO-RS als Modell für die Gibbssche Exzessenthalpie. Der binären
Mischungsparameter kij ist an die experimentellen Daten angepasst (kij =0,2).
Im System Aceton/Wasser (Abb. 5.27) sind neben den unterkritischen auch überkritische
Isothermen betrachtet worden. Hier zeigt sich, dass für eine gute Beschreibung des
Phasenverhaltens der binäre Wechselwirkungsparameter mit steigender Temperatur kleinere
Werte annehmen muss. Er bewegt sich um Werte, die für die klassischen van-der-Waals Ein-
Fluid-Mischungsregeln außergewöhnlich hoch sind. Dem Wert wird hierbei allerdings keine
physikalische Bedeutung beigemessen, er ist vielmehr ein Parameter, der Unzulänglichkeiten
bei der Kombination der Reinstoffparameter korrigiert. In Abbildung 5.27 sind ebenfalls die
Ergebnisse für die Vorhersage des Phasenverhaltens enthalten. Die Vorhersagen auf Basis des
Aktivitätskoeffizienten bei unendlicher Verdünnung aus dem COSMO-RS-Modell gelingen

98
für alle Temperaturen mit einem konstanten Alpha-Wert von α=0,4.
0
10
20
30
40
50
60
70
80
0.00 0.20 0.40 0.60 0.80 1.00
x,yAzeton
P /
bar
473.15K
373.15K
423.15K
523.15K
PR-EOS, WS+COSMO-RS mit gamma_inf
PR-EOS, WS+COSMO-RS korrel. kij
Abb. 5.27 Flüssig-Dampf Gleichgewicht des Systems Aceton/Wasser bei verschiedenen Temperaturen. Die
Symbole sind experimentelle Daten (Griswold,J.;Wong,S.Y.; Chem. Eng. Progr. Symp. Ser., 40 (1952), 18-34).
Die gestrichelten Linien sind Berechnungen mit der PR-ZGL der Wong-Sandler Mischungsregel und COSMO-
RS als Modell für die Gibbssche Exzessenthalpie. Der binären Mischungsparameter kij ist an die experimentellen
Daten angepasst (kij =0,23 für T=373,15K-473,15K und kij =0,18 für T=523,15K). Die durchgezogenen Linien
sind Vorhersagen auf Grundlage der vorhergesagten Aktivitätskoeffizienten bei unendlicher Verdünnung mit
COSMO-RS.
Für das binäre System Ethanol/Wasser sowie für das System Methanol/Wasser können im
Wesentlichen die gleichen Aussagen getroffen werden, wie schon beim Aceton/Wasser-
System. Die Ergebnisse der Berechnungen finden sich in den Abb 5.28, 5.29 und 5.30. Bei
den berechneten überkritischen Isothermen in den dargestellten Ergebnissen ist die Rechnung
jeweils bei einem maximalen Druck, der den experimentellen Daten entspricht, abgebrochen
worden. Es ist im Falle der Korrelationen ein temperaturabhängiger binärer
Wechselwirkungsparameter kij notwendig, der bei niedrigen Temperaturen nahezu konstant ist
und für höhere Temperaturen kleinere Werte annimmt. Eine Besonderheit gibt es in diesen
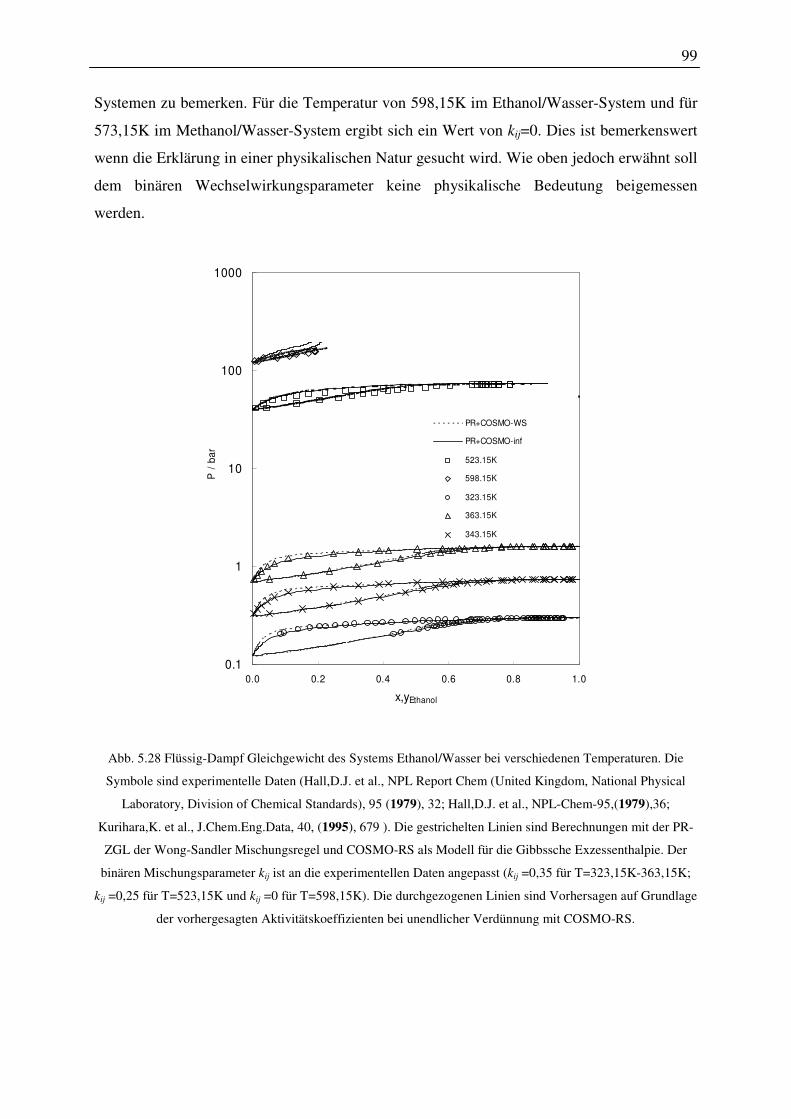
99
Systemen zu bemerken. Für die Temperatur von 598,15K im Ethanol/Wasser-System und für
573,15K im Methanol/Wasser-System ergibt sich ein Wert von kij=0. Dies ist bemerkenswert
wenn die Erklärung in einer physikalischen Natur gesucht wird. Wie oben jedoch erwähnt soll
dem binären Wechselwirkungsparameter keine physikalische Bedeutung beigemessen
werden.
0.1
1
10
100
1000
0.0 0.2 0.4 0.6 0.8 1.0
x,yEthanol
P /
bar
PR+COSMO-WS
PR+COSMO-inf
523.15K
598.15K
323.15K
363.15K
343.15K
Abb. 5.28 Flüssig-Dampf Gleichgewicht des Systems Ethanol/Wasser bei verschiedenen Temperaturen. Die
Symbole sind experimentelle Daten (Hall,D.J. et al., NPL Report Chem (United Kingdom, National Physical
Laboratory, Division of Chemical Standards), 95 (1979), 32; Hall,D.J. et al., NPL-Chem-95,(1979),36;
Kurihara,K. et al., J.Chem.Eng.Data, 40, (1995), 679 ). Die gestrichelten Linien sind Berechnungen mit der PR-
ZGL der Wong-Sandler Mischungsregel und COSMO-RS als Modell für die Gibbssche Exzessenthalpie. Der
binären Mischungsparameter kij ist an die experimentellen Daten angepasst (kij =0,35 für T=323,15K-363,15K;
kij =0,25 für T=523,15K und kij =0 für T=598,15K). Die durchgezogenen Linien sind Vorhersagen auf Grundlage
der vorhergesagten Aktivitätskoeffizienten bei unendlicher Verdünnung mit COSMO-RS.
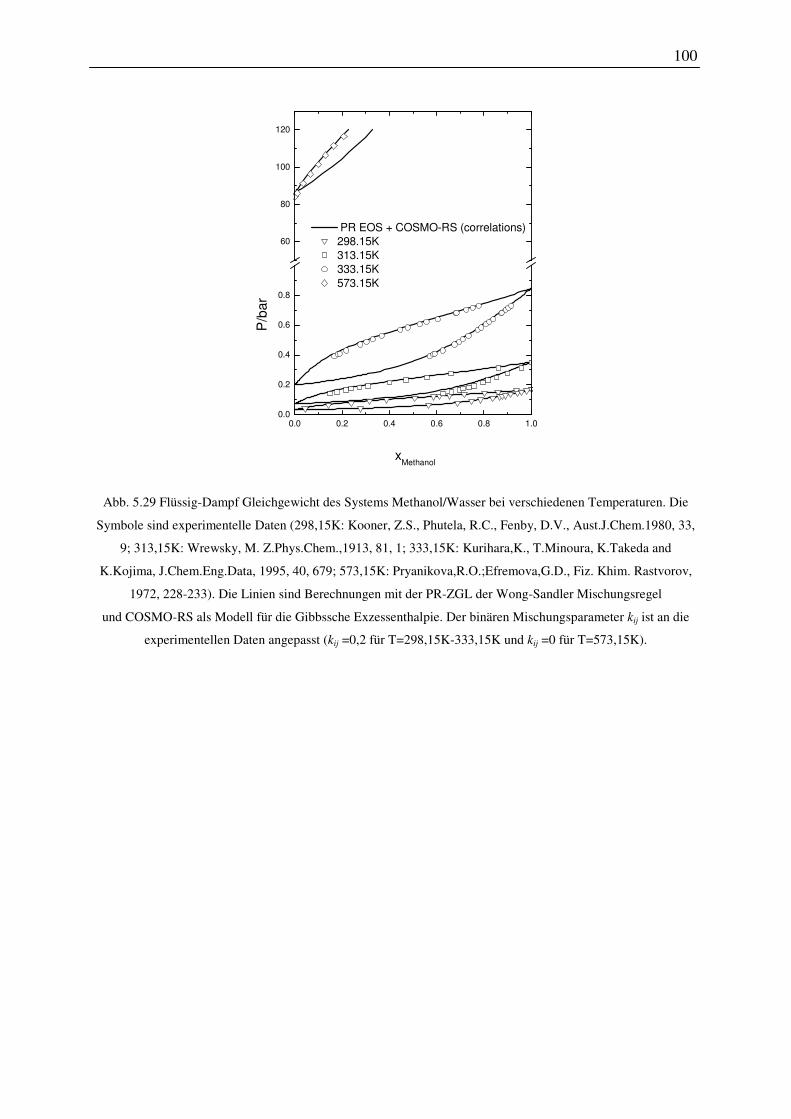
100
0.0 0.2 0.4 0.6 0.8 1.00.0
0.2
0.4
0.6
0.8
60
80
100
120
P/b
ar
xMethanol
PR EOS + COSMO-RS (correlations)298.15K313.15K333.15K573.15K
Abb. 5.29 Flüssig-Dampf Gleichgewicht des Systems Methanol/Wasser bei verschiedenen Temperaturen. Die
Symbole sind experimentelle Daten (298,15K: Kooner, Z.S., Phutela, R.C., Fenby, D.V., Aust.J.Chem.1980, 33,
9; 313,15K: Wrewsky, M. Z.Phys.Chem.,1913, 81, 1; 333,15K: Kurihara,K., T.Minoura, K.Takeda and
K.Kojima, J.Chem.Eng.Data, 1995, 40, 679; 573,15K: Pryanikova,R.O.;Efremova,G.D., Fiz. Khim. Rastvorov,
1972, 228-233). Die Linien sind Berechnungen mit der PR-ZGL der Wong-Sandler Mischungsregel
und COSMO-RS als Modell für die Gibbssche Exzessenthalpie. Der binären Mischungsparameter kij ist an die
experimentellen Daten angepasst (kij =0,2 für T=298,15K-333,15K und kij =0 für T=573,15K).
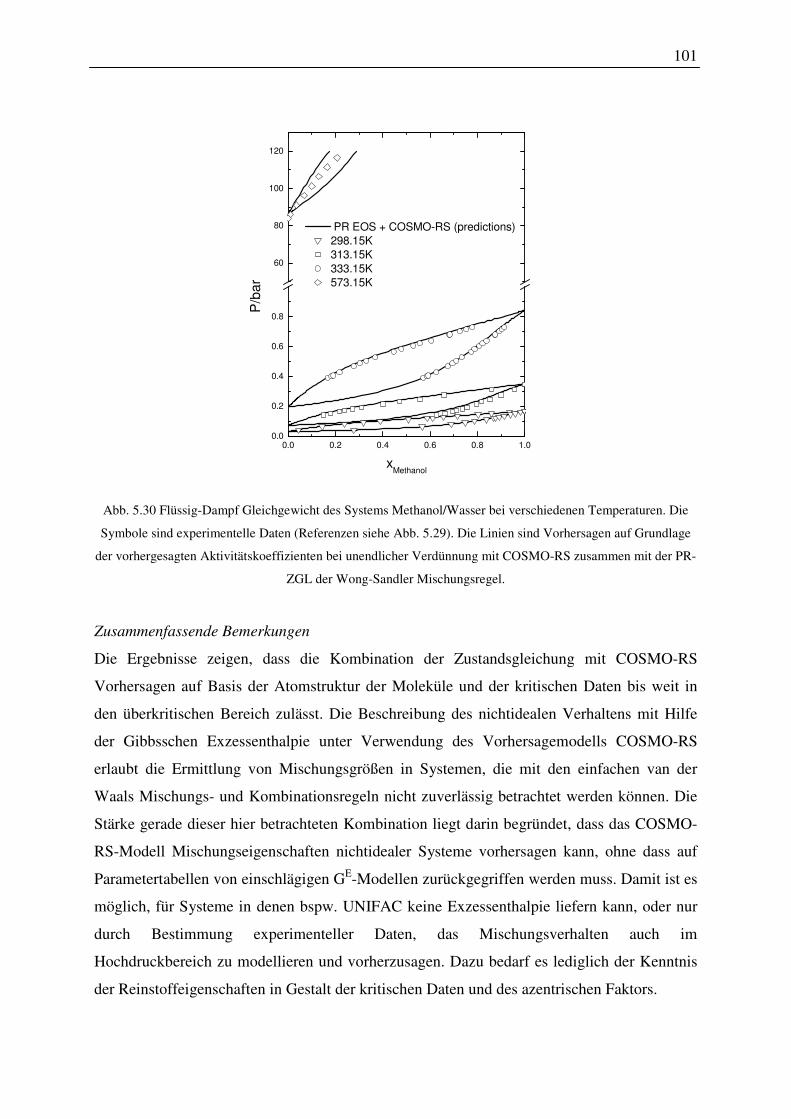
101
0.0 0.2 0.4 0.6 0.8 1.00.0
0.2
0.4
0.6
0.8
60
80
100
120
P/b
ar
xMethanol
PR EOS + COSMO-RS (predictions)298.15K313.15K333.15K573.15K
Abb. 5.30 Flüssig-Dampf Gleichgewicht des Systems Methanol/Wasser bei verschiedenen Temperaturen. Die
Symbole sind experimentelle Daten (Referenzen siehe Abb. 5.29). Die Linien sind Vorhersagen auf Grundlage
der vorhergesagten Aktivitätskoeffizienten bei unendlicher Verdünnung mit COSMO-RS zusammen mit der PR-
ZGL der Wong-Sandler Mischungsregel.
Zusammenfassende Bemerkungen
Die Ergebnisse zeigen, dass die Kombination der Zustandsgleichung mit COSMO-RS
Vorhersagen auf Basis der Atomstruktur der Moleküle und der kritischen Daten bis weit in
den überkritischen Bereich zulässt. Die Beschreibung des nichtidealen Verhaltens mit Hilfe
der Gibbsschen Exzessenthalpie unter Verwendung des Vorhersagemodells COSMO-RS
erlaubt die Ermittlung von Mischungsgrößen in Systemen, die mit den einfachen van der
Waals Mischungs- und Kombinationsregeln nicht zuverlässig betrachtet werden können. Die
Stärke gerade dieser hier betrachteten Kombination liegt darin begründet, dass das COSMO-
RS-Modell Mischungseigenschaften nichtidealer Systeme vorhersagen kann, ohne dass auf
Parametertabellen von einschlägigen GE-Modellen zurückgegriffen werden muss. Damit ist es
möglich, für Systeme in denen bspw. UNIFAC keine Exzessenthalpie liefern kann, oder nur
durch Bestimmung experimenteller Daten, das Mischungsverhalten auch im
Hochdruckbereich zu modellieren und vorherzusagen. Dazu bedarf es lediglich der Kenntnis
der Reinstoffeigenschaften in Gestalt der kritischen Daten und des azentrischen Faktors.

102
5.4 Vorhersage von Reinstoffeigenschaften mit dem erweiterten COSMO-RS-Modell
5.4.1 Harte-Kugel-Durchmesser
Wie in Kapitel 4.1.4 beschrieben, werden die Harte-Kugel-Durchmesser aus den Volumina,
die im Ergebnis der quantenchemischen Rechnungen unter der COSMO-Randbedingung
vorliegen, abgeleitet. Die Ergebnisse stimmen sehr gut mit den aus experimentellen Daten
und theoretischen Überlegungen abgeleiteten Methoden überein [BEN AMOTZ, 1990; BEN
AMOTZ, 1993c; EDWARD, 1970; GOGONEA, 1998]. In Tabelle 5.9 und Abb. 5.31 sind die
Ergebnisse für diejenigen Stoffe aufgeführt, für die Daten aller Methoden vorliegen. Eine
Übersicht der Durchmesser aller verwendeten Stoffe und ihrer Konformationen findet sich im
Anhang. Die Korrekturfunktion
COSMOharteKugel σσ 9.0= (5.3)
wurde an den Werten der Durchmesser, die Gogonea et al. [GOGONEA, 1998] aus den van-
der-Waals-Volumina der Moleküle unter Verwendung der Pauling-van-der-Waals-Radien der
Atome [PAULING, 1993] abgeleitet haben, angepasst, da deren Methode die beste
Übereinstimmung mit experimentellen Daten liefert [GOGONEA, 1998]. Man kann
erkennen, dass die Vergleichswerte von Ben-Amotz und Herschbach [BEN AMOTZ, 1990]
beispielsweise bei den Alkoholen mit kleinem Volumen (Methanol bis Propanol) eine
negative Abweichung von den aus COSMO-Rechnungen bestimmten Durchmessern zeigen
und für langkettige Alkohole (ab Hexanol) größere Werte liefern. Dahingegen zeigt der
Vergleich mit den Daten von Gogonea et al. durchgehend eine gute Übereinstimmung. Die
Abweichungen liegen im Bereich 1%.
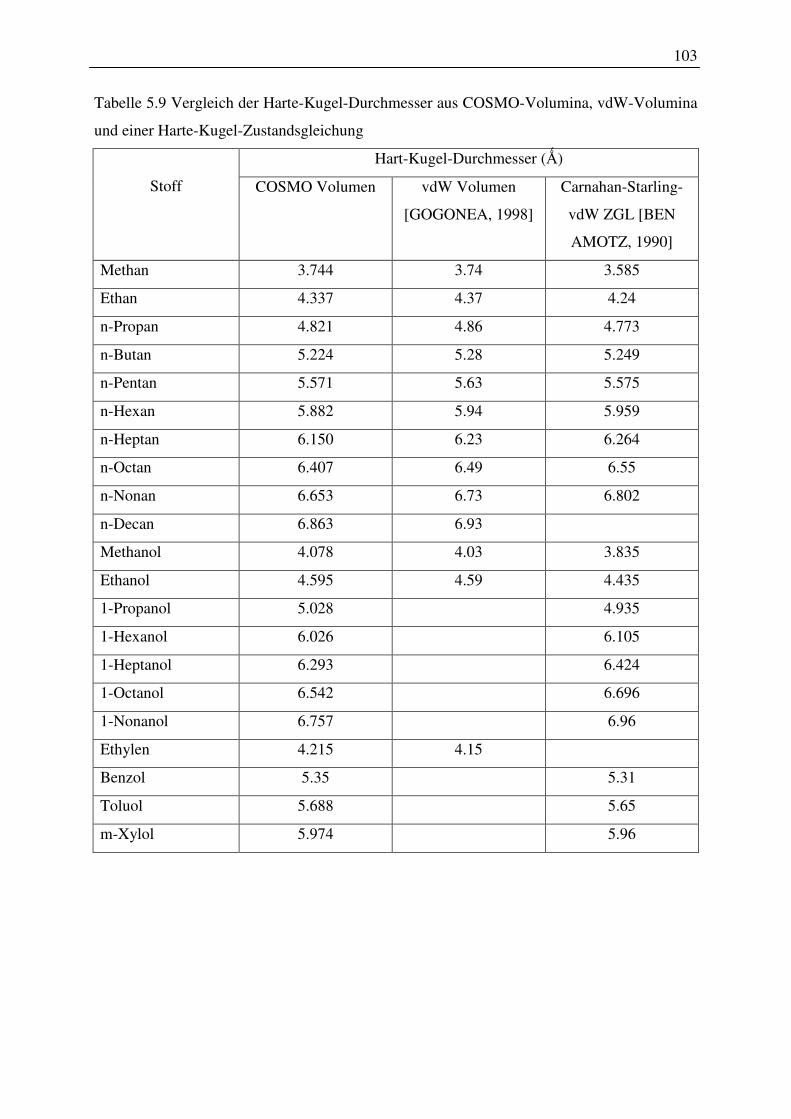
103
Tabelle 5.9 Vergleich der Harte-Kugel-Durchmesser aus COSMO-Volumina, vdW-Volumina
und einer Harte-Kugel-Zustandsgleichung
Hart-Kugel-Durchmesser (Ǻ)
Stoff COSMO Volumen vdW Volumen
[GOGONEA, 1998]
Carnahan-Starling-
vdW ZGL [BEN
AMOTZ, 1990]
Methan 3.744 3.74 3.585
Ethan 4.337 4.37 4.24
n-Propan 4.821 4.86 4.773
n-Butan 5.224 5.28 5.249
n-Pentan 5.571 5.63 5.575
n-Hexan 5.882 5.94 5.959
n-Heptan 6.150 6.23 6.264
n-Octan 6.407 6.49 6.55
n-Nonan 6.653 6.73 6.802
n-Decan 6.863 6.93
Methanol 4.078 4.03 3.835
Ethanol 4.595 4.59 4.435
1-Propanol 5.028 4.935
1-Hexanol 6.026 6.105
1-Heptanol 6.293 6.424
1-Octanol 6.542 6.696
1-Nonanol 6.757 6.96
Ethylen 4.215 4.15
Benzol 5.35 5.31
Toluol 5.688 5.65
m-Xylol 5.974 5.96
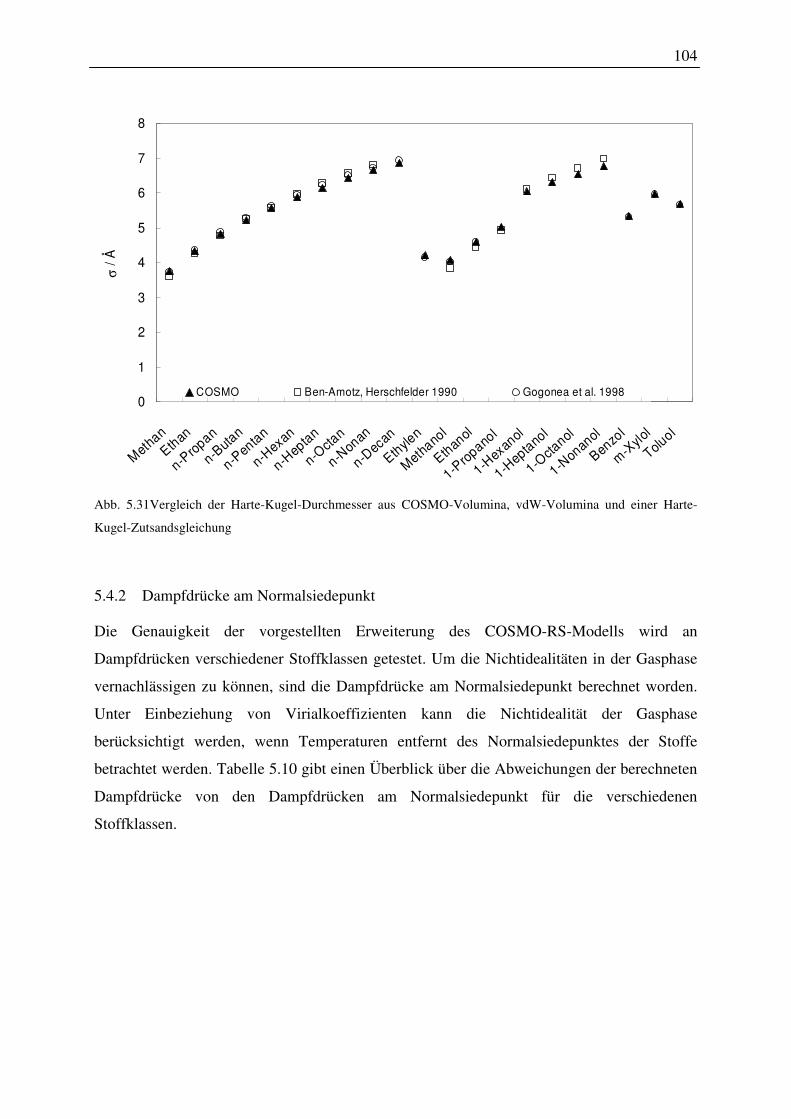
104
0
1
2
3
4
5
6
7
8
Methan
Ethan
n-Prop
an
n-Buta
n
n-Pen
tan
n-Hexa
n
n-Hep
tan
n-Octa
n
n-Non
an
n-Deca
n
Ethylen
Methanol
Ethanol
1-Prop
anol
1-Hex
anol
1-Heptanol
1-Octa
nol
1-Non
anol
Benzo
l
m-Xylo
l
Toluol
σ /
Å
COSMO Ben-Amotz, Herschfelder 1990 Gogonea et al. 1998
Abb. 5.31Vergleich der Harte-Kugel-Durchmesser aus COSMO-Volumina, vdW-Volumina und einer Harte-
Kugel-Zutsandsgleichung
5.4.2 Dampfdrücke am Normalsiedepunkt
Die Genauigkeit der vorgestellten Erweiterung des COSMO-RS-Modells wird an
Dampfdrücken verschiedener Stoffklassen getestet. Um die Nichtidealitäten in der Gasphase
vernachlässigen zu können, sind die Dampfdrücke am Normalsiedepunkt berechnet worden.
Unter Einbeziehung von Virialkoeffizienten kann die Nichtidealität der Gasphase
berücksichtigt werden, wenn Temperaturen entfernt des Normalsiedepunktes der Stoffe
betrachtet werden. Tabelle 5.10 gibt einen Überblick über die Abweichungen der berechneten
Dampfdrücke von den Dampfdrücken am Normalsiedepunkt für die verschiedenen
Stoffklassen.

105
Tabelle 5.10 Genauigkeit der Vorhersage von Dampfdrücken am Normalsiedepunkt
Stoffklasse Anzahl der
Komponenten
absolute
Abweichung (%)
Alkane 10 144
Alkene 9 75
Alkohole 16 18
Aromaten 5 21
Aldehyde 3 16
Ketone 3 25
Diole 7 38
Ether 7 38
Eine Übersicht über die einzelnen Werte der Stoffe findet sich Anhang 7.2. Abb. 5.32 zeigt
schematisch eine Übersicht über die Abweichungen der einzelnen Komponenten. Die Stoffe
sind so in das Diagramm eingetragen, dass die Anzahl der Kohlenstoffatome innerhalb einer
Stoffklasse ansteigen. Zusätzlich zu den berechneten Werten sind die Linien für den Druck
am Normalsiedepunkt der Komponenten (PLV=1,013bar) sowie eine 50prozentige
Abweichung nach oben und nach unten eingetragen. Es ist ersichtlich, dass die Vorhersage für
die Dampfdrücke der Alkane und Alkene nicht zufriedenstellend gelingt. Die Werte des
Dampfdrucks nehmen mit steigender Kettenlänge deutlich zu. Für die Stoffklasse der
Alkohole kann die Vorhersage als zufriedenstellend für diese Methode angesehen werden. Bis
auf die Vorhersagen für Methanol und Ethanol liegen alle Rechnungen innerhalb eines
Bandes von ±50% des Zielwertes von PLV=1,013bar. Ähnliches gilt für die anderen
betrachteten Stoffklassen, wobei hier die Werte deutlicher innerhalb der 50%-Banden streuen.
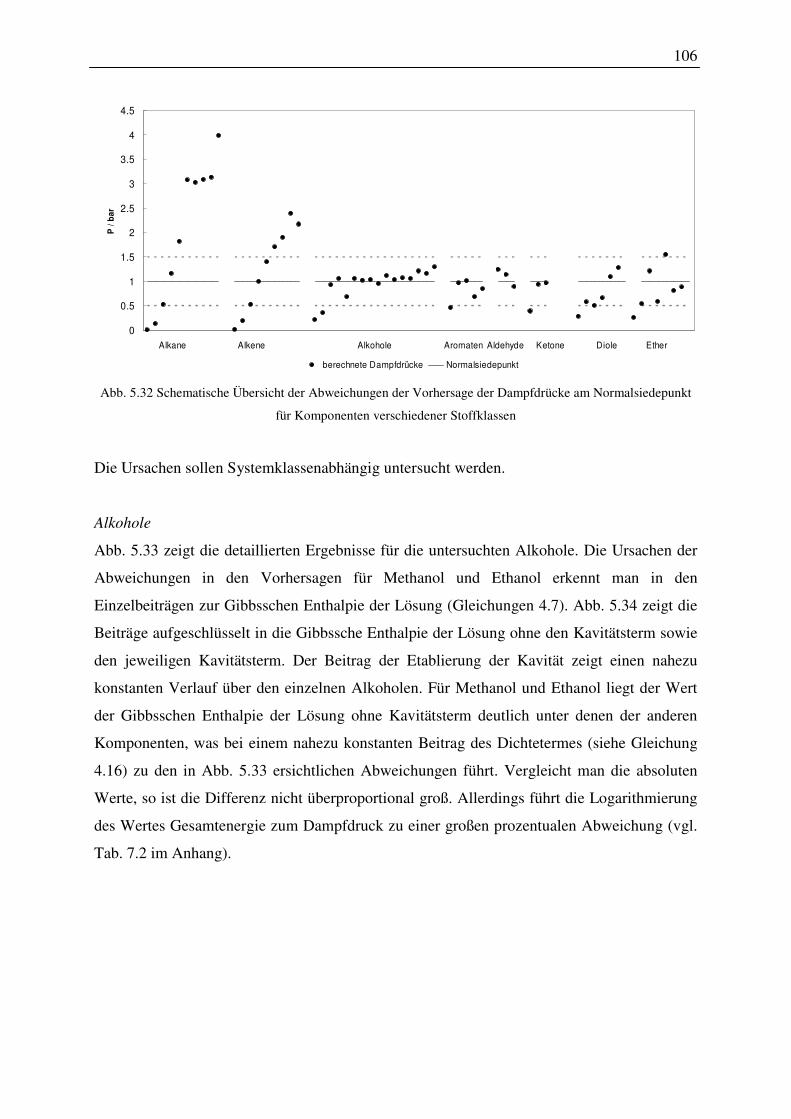
106
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5P
/ b
ar
berechnete Dampfdrücke Normalsiedepunkt
Alkane Alkene Alkohole Aromaten Aldehyde Ketone Diole Ether
Abb. 5.32 Schematische Übersicht der Abweichungen der Vorhersage der Dampfdrücke am Normalsiedepunkt
für Komponenten verschiedener Stoffklassen
Die Ursachen sollen Systemklassenabhängig untersucht werden.
Alkohole
Abb. 5.33 zeigt die detaillierten Ergebnisse für die untersuchten Alkohole. Die Ursachen der
Abweichungen in den Vorhersagen für Methanol und Ethanol erkennt man in den
Einzelbeiträgen zur Gibbsschen Enthalpie der Lösung (Gleichungen 4.7). Abb. 5.34 zeigt die
Beiträge aufgeschlüsselt in die Gibbssche Enthalpie der Lösung ohne den Kavitätsterm sowie
den jeweiligen Kavitätsterm. Der Beitrag der Etablierung der Kavität zeigt einen nahezu
konstanten Verlauf über den einzelnen Alkoholen. Für Methanol und Ethanol liegt der Wert
der Gibbsschen Enthalpie der Lösung ohne Kavitätsterm deutlich unter denen der anderen
Komponenten, was bei einem nahezu konstanten Beitrag des Dichtetermes (siehe Gleichung
4.16) zu den in Abb. 5.33 ersichtlichen Abweichungen führt. Vergleicht man die absoluten
Werte, so ist die Differenz nicht überproportional groß. Allerdings führt die Logarithmierung
des Wertes Gesamtenergie zum Dampfdruck zu einer großen prozentualen Abweichung (vgl.
Tab. 7.2 im Anhang).
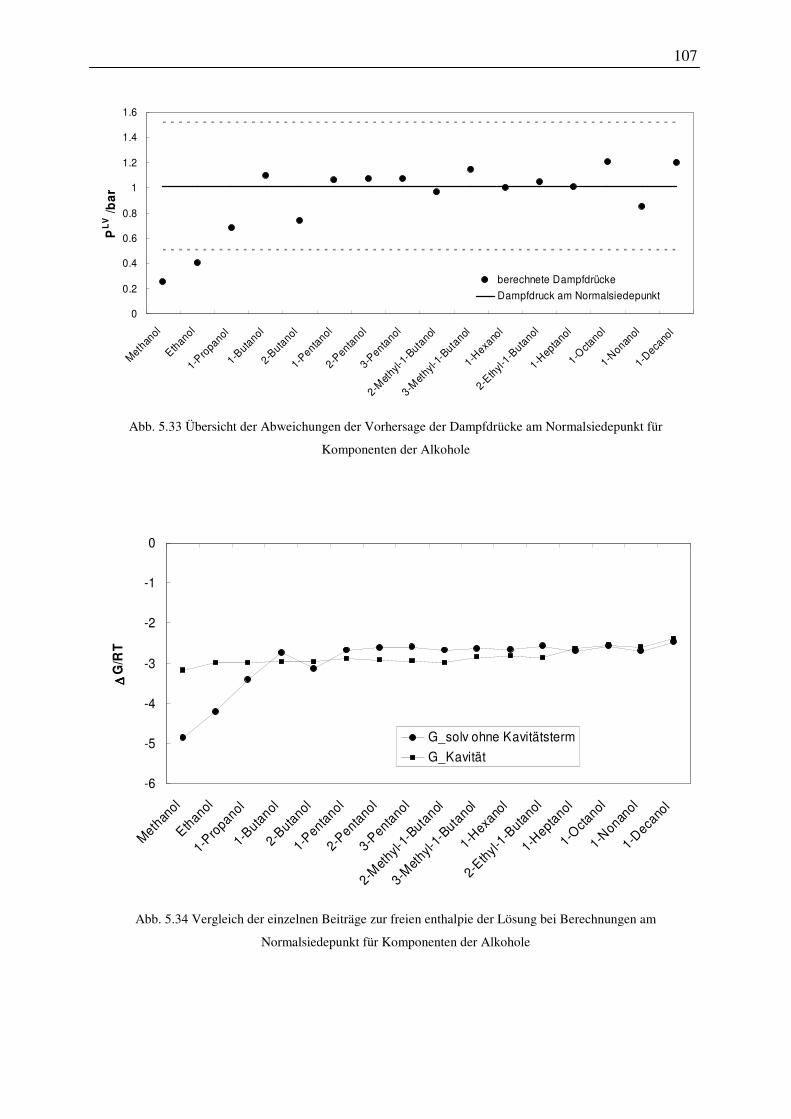
107
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
Methan
ol
Ethanol
1-Pro
panol
1-Butanol
2-Butanol
1-Pentanol
2-Pentan
ol
3-Pentanol
2-Meth
yl-1-
Butano
l
3-Meth
yl-1-
Butanol
1-Hexa
nol
2-Ethyl-
1-Butanol
1-Hep
tanol
1-Octa
nol
1-Nonan
ol
1-Deca
nol
PL
V /
ba
r
berechnete Dampfdrücke
Dampfdruck am Normalsiedepunkt
Abb. 5.33 Übersicht der Abweichungen der Vorhersage der Dampfdrücke am Normalsiedepunkt für
Komponenten der Alkohole
-6
-5
-4
-3
-2
-1
0
Methan
ol
Ethanol
1-Pro
panol
1-Butanol
2-Butanol
1-Pentanol
2-Pentanol
3-Pentanol
2-Meth
yl-1-
Butanol
3-Meth
yl-1-
Butanol
1-Hexa
nol
2-Ethyl-
1-Butanol
1-Heptanol
1-Octa
nol
1-Nonan
ol
1-Deca
nol
∆∆ ∆∆G
/RT
G_solv ohne Kavitätsterm
G_Kavität
Abb. 5.34 Vergleich der einzelnen Beiträge zur freien enthalpie der Lösung bei Berechnungen am
Normalsiedepunkt für Komponenten der Alkohole
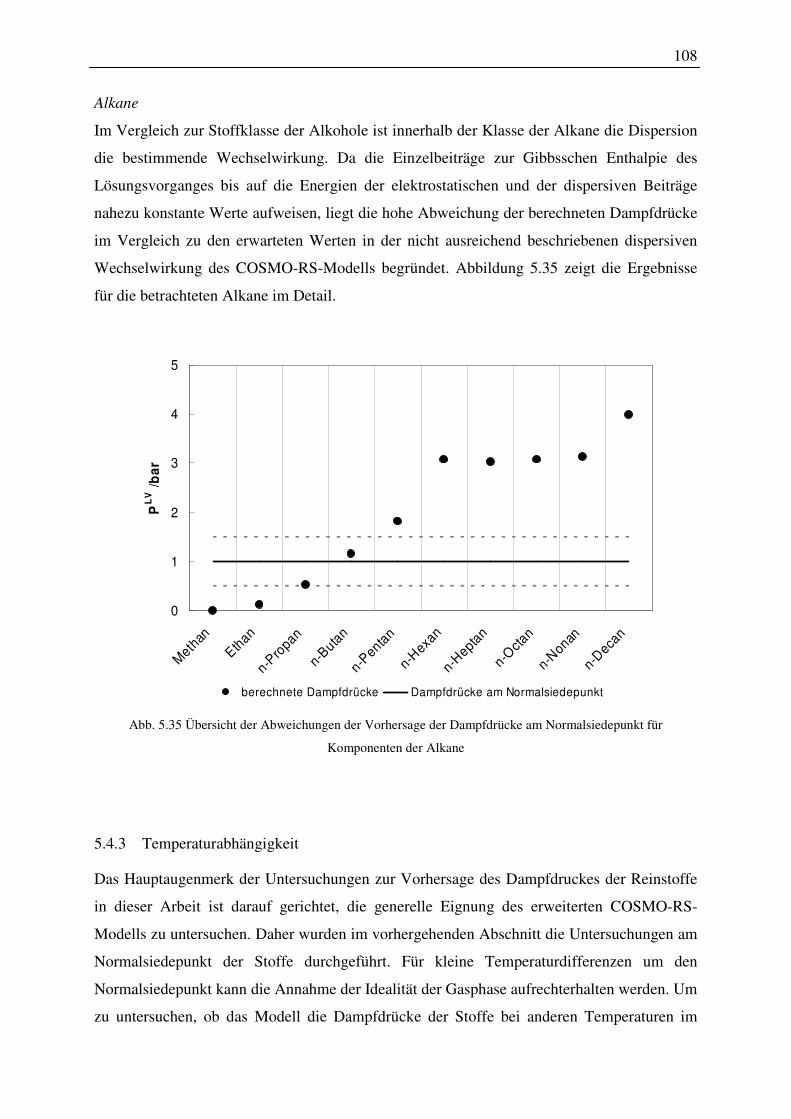
108
Alkane
Im Vergleich zur Stoffklasse der Alkohole ist innerhalb der Klasse der Alkane die Dispersion
die bestimmende Wechselwirkung. Da die Einzelbeiträge zur Gibbsschen Enthalpie des
Lösungsvorganges bis auf die Energien der elektrostatischen und der dispersiven Beiträge
nahezu konstante Werte aufweisen, liegt die hohe Abweichung der berechneten Dampfdrücke
im Vergleich zu den erwarteten Werten in der nicht ausreichend beschriebenen dispersiven
Wechselwirkung des COSMO-RS-Modells begründet. Abbildung 5.35 zeigt die Ergebnisse
für die betrachteten Alkane im Detail.
0
1
2
3
4
5
Methan
Ethan
n-Pro
pan
n-Butan
n-Pentan
n-Hexa
n
n-Heptan
n-Octa
n
n-Nonan
n-Deca
n
PL
V /b
ar
berechnete Dampfdrücke Dampfdrücke am Normalsiedepunkt
Abb. 5.35 Übersicht der Abweichungen der Vorhersage der Dampfdrücke am Normalsiedepunkt für
Komponenten der Alkane
5.4.3 Temperaturabhängigkeit
Das Hauptaugenmerk der Untersuchungen zur Vorhersage des Dampfdruckes der Reinstoffe
in dieser Arbeit ist darauf gerichtet, die generelle Eignung des erweiterten COSMO-RS-
Modells zu untersuchen. Daher wurden im vorhergehenden Abschnitt die Untersuchungen am
Normalsiedepunkt der Stoffe durchgeführt. Für kleine Temperaturdifferenzen um den
Normalsiedepunkt kann die Annahme der Idealität der Gasphase aufrechterhalten werden. Um
zu untersuchen, ob das Modell die Dampfdrücke der Stoffe bei anderen Temperaturen im

109
Rahmen der oben festgestellten Abweichung beschreiben kann, wurden Temperaturen von
jeweils 30K und 10K oberhalb und unterhalb des Normalsiedepunktes betrachtet, wobei auf
eine Einbeziehung der Alkane und Alkene wegen der großen Abweichungen verzichtet
worden ist.
Abb. 5.36 zeigt die Ergebnisse des Temperatureinflusses. Am Normalsiedepunkt liegt die
Abweichung, gemittelt über alle betrachteten Komponenten, bei 33%. Eine Änderung der
Temperatur führt relativ gesehen, zu keiner veränderten Abweichung im Vergleich zu den
experimentellen Werten. Die Vergleichsdaten der Dampfdrücke wurden über Korrelationen
bestimmt [DAUBERT, 1989].
33%
36%
33%
29%
33%
0%
5%
10%
15%
20%
25%
30%
35%
40%
TS-30K TS-10K TS TS+10K TS+30K
rel.
Fe
hle
r /
%
Abb. 5.36 Temperaturabhängigkeit der berechneten Dampfdrücke. Vergleich der Rechnungen mit den Werten
am Normalsiedepunkt (TS) sowie einer Temperaturänderung von +10K, +30K und -10K, -30K
5.4.4 Einfluss der Konformationen der Moleküle auf das Vorhersageergebnis
Wie bei den Betrachtungen zur Vorhersage von Phasengleichgewichten in
Mehrkomponentensystemen (vgl. Kapitel 3) hat die Konformation des betrachteten Stoffes
einen erheblichen Einfluss auf das Ergebnis der Vorhersage der Dampfdrücke eines
Reinstoffes.
Für die Stoffklasse der Alkohole wird dieser Einfluss hier am Beispiel von 1-Heptanol näher
erläutert. Die Konformationsanalyse ist entsprechend dem in Kapitel 5.1 beschriebenen
Vorgehen durchgeführt worden und führt zu den in Abb. 5.37 und Abb. 5.38 dargestellten
sechs Konformationen.
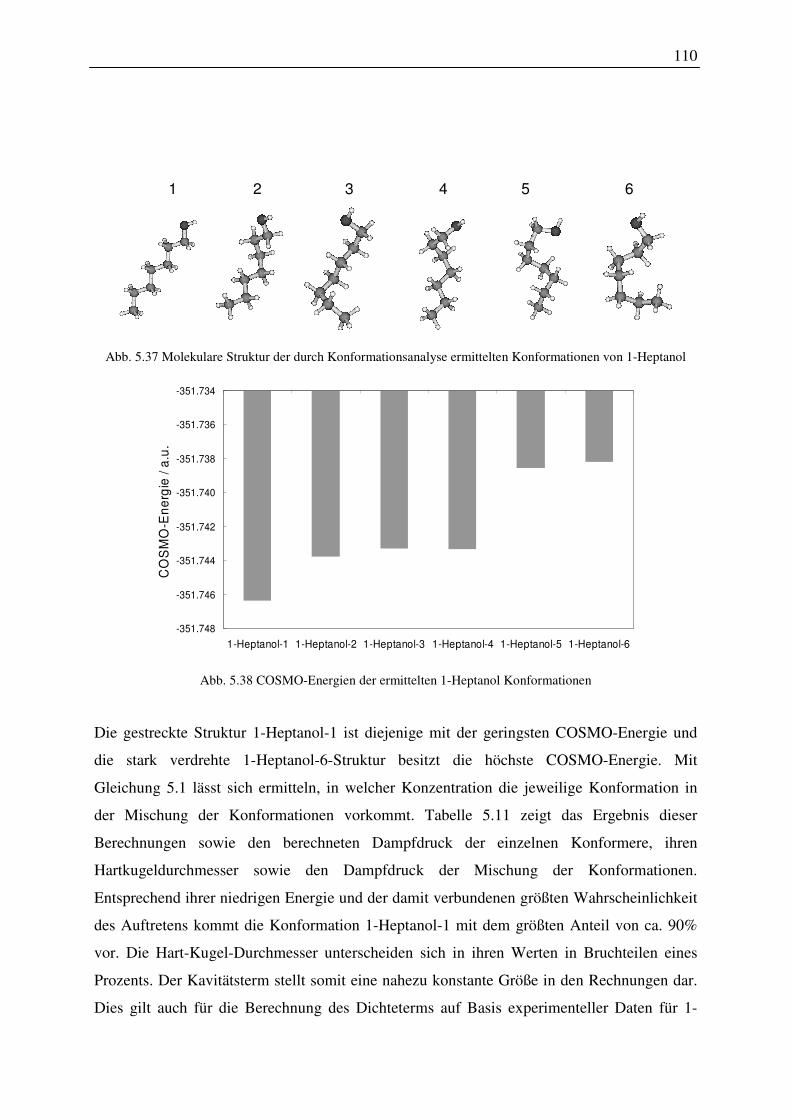
110
654321
Abb. 5.37 Molekulare Struktur der durch Konformationsanalyse ermittelten Konformationen von 1-Heptanol
-351.748
-351.746
-351.744
-351.742
-351.740
-351.738
-351.736
-351.734
1-Heptanol-1 1-Heptanol-2 1-Heptanol-3 1-Heptanol-4 1-Heptanol-5 1-Heptanol-6
CO
SM
O-E
nerg
ie /
a.u.
Abb. 5.38 COSMO-Energien der ermittelten 1-Heptanol Konformationen
Die gestreckte Struktur 1-Heptanol-1 ist diejenige mit der geringsten COSMO-Energie und
die stark verdrehte 1-Heptanol-6-Struktur besitzt die höchste COSMO-Energie. Mit
Gleichung 5.1 lässt sich ermitteln, in welcher Konzentration die jeweilige Konformation in
der Mischung der Konformationen vorkommt. Tabelle 5.11 zeigt das Ergebnis dieser
Berechnungen sowie den berechneten Dampfdruck der einzelnen Konformere, ihren
Hartkugeldurchmesser sowie den Dampfdruck der Mischung der Konformationen.
Entsprechend ihrer niedrigen Energie und der damit verbundenen größten Wahrscheinlichkeit
des Auftretens kommt die Konformation 1-Heptanol-1 mit dem größten Anteil von ca. 90%
vor. Die Hart-Kugel-Durchmesser unterscheiden sich in ihren Werten in Bruchteilen eines
Prozents. Der Kavitätsterm stellt somit eine nahezu konstante Größe in den Rechnungen dar.
Dies gilt auch für die Berechnung des Dichteterms auf Basis experimenteller Daten für 1-

111
Heptanol, der aus experimentellen Daten stammt [DAUBERT, 1989]. Die Dichteberechnung
unterscheidet nicht nach Konformationen und liefert damit einen exakt konstanten Beitrag.
Tabelle 5.11 Anteil der 1-Heptanol-Konformationen in 1-Heptanol
Konformer Anteil in % PLV(449,45K) σHS
1-Heptanol-1 89,30 0,8801 6,3001
1-Heptanol-2 4,32 1,004 6,2895
1-Heptanol-3 3,10 1,2387 6,2907
1-Heptanol-4 3,28 1,2882 6,2907
1-Heptanol-5 0,02 1,9443 6,2942
1-Heptanol-6 0,01 1,4296 6,2954
Mischung 0,91
Der berechnete Dampfdruck des mit dem um den Kavitätsterm erweiterten COSMO-RS-
Modells reagiert sehr sensitiv auf die unterschiedlichen Konformationen. Im folgenden
Kapitel wird vorgeschlagen, wie ausgehend von den Berechnungen am Normalsiedepunkt
eine Auswahl von Konformationen erfolgen kann, die für die
Phasengleichgewichtsberechnung in Mehrkomponentensystemen relevant sind.
5.4.5 Auswahl von Konformationen auf Basis von Dampfdrücken am Normalsiedepunkt
Die Untersuchungen im vorhergehenden Abschnitt haben gezeigt, dass die Güte der
Vorhersagen der Reinstoffeigenschaft Dampfdruck einer Komponente nicht die erforderliche
Genauigkeit liefert, um eine der herkömmlich verwendeten Methoden zu ersetzen. Es hat sich
allerdings auch gezeigt, dass der vorhergesagte Wert des Dampfdruckes sehr sensibel auf die
unterschiedlichen Konformationen einer Komponente reagiert. Der Dampfdruck könnte somit
ein Sensor sein, der es ermöglicht, relevante Konformationen eines Moleküls für eine
Phasengleichgewichtsberechnung zu identifizieren. Eine Konformation, die in der Vorhersage
eine geringe Abweichung vom erwarteten Dampfdruck erzielt, sollte in der
Phasengleichgewichtsberechnung ebenfalls geeignet sein, gute Ergebnisse zu erzielen.
Voraussetzung ist, dass das Wechselwirkungsverhalten der einzelnen Konformationen im
Reinstoff dem in der Mischung mit anderen Komponenten entspricht. Diese Aspekte sollen in
diesem Abschnitt diskutiert werden.

112
Als Sensormoleküle werden die Stoffe 2-Pentanol, 1-Hexanol und 1-Octanol ausgewählt. Für
diese Stoffe ist eine Konformationsanalyse entsprechend dem in Kapitel 5.1.1 beschriebenen
Vorgehen durchgeführt worden. Eine Auflistung der Konformationen mit ihren COSMO-
Energien findet sich in Anhang 7.3. Tabelle 5.12 stellt die Anzahl der gefundenen
Konformationen dar. Die hohe Zahl, verglichen mit den sechs aufgelisteten Konformationen
des 1-Heptanols ergibt sich aus einer weniger restriktiven Wahl der Randbedingungen der
Konformationsanalyse im Analysewerkzeug HyperChem. Dieses wurde mit dem Ziel, eine
feinere Unterteilung des Konformerenraumes zu erreichen, durchgeführt.
Tabelle 5.12 Anzahl der ermittelten Konformere 2-Pentanol, 1-Hexanol, 1-Octanol
2-Pentanol 1-Hexanol 1-Octanol
Anzahl der Konformationen 27 27 31
Normalsiedepunkt [K] 392,15 430,55 468,35
Für alle Konformationen ist der Dampfdruck am Normalsiedepunkt (siehe Tabelle 7.3 im
Anhang) mit der in dieser Arbeit vorgestellten Methode berechnet und für jede Konformation
der Harte-Kugel-Durchmesser aus den COSMO-Volumina berechnet worden (Gleichung 5.3).
Für die Dichten der flüssigen Phase wurden experimentelle Daten verwendet [DAUBERT,
1989]. Hier konnte nicht nach der Konformation unterschieden werden. Die Werte für den
Harte Kugel-Durchmesser und die Ergebnisse der Berechnungen des Dampfdruckes einer
jeden Konformation finden sich im Anhang 7.3.
Die Werte streuen für alle betrachteten Alkohole mit einer Genauigkeit von etwa 30% um den
erwarteten Wert von 1,013bar.
Abb. 5.39 zeigt die vorhergesagten Dampfdrücke am Normalsiedepunkt von 1-Hexanol. Dem
sind auf der Sekundärachse die Abweichungen im Druck, verglichen mit experimentellen
Daten, für das binäre System Wasser/1-Hexanol gegenübergestellt. Die Linien, die die Punkte
verbinden, sind zur Verdeutlichung des Trends eingezeichnet und stellen keine
interpolierenden Berechnungen dar. Bei der Berechnung der Phasengleichgewichte wird für
die zweite Komponente eine einzige Konformation angenommen. Bei den Betrachtungen zu
den Wasser/Alkoholsystemen ist diese Annahme trivial, da Wasser nur eine Konformation
besitzt. In den Alkan/Alkohol-Systemen ist für das Alkan die lineare, gestreckte energetisch
niedrigste Konformation gewählt worden, da an dieser Stelle insbesondere der Einfluss der
Konformationen des Alkohols untersucht worden ist.
Zunächst lässt sich erkennen, dass in der Konformationsanalyse von 1-Hexanol elf nahezu

113
identische Strukturen als stabil existierende Konformationen identifiziert werden. Sie sind
zudem diejenigen mit der geringsten COSMO-Energie. Diese Konformationen weisen mit
geringen Abweichungen denselben Wert für den vorhergesagten Dampfdruck auf. Sieben
Konformationen zeigen eine Abweichung im Dampfdruck von 3 bis 6%. Es sind dies die
Konformationen 12, 13, 14, 15, 21, 22 und 23. Für das binäre System liegen die
Abweichungen im Druck bei der Berechnung der Phasengleichgewichtsdaten mit diesen
Konformationen entsprechend der obigen Erwartung bei minimalen Werten. Allerdings macht
die Abbildung deutlich, dass nicht die Konformationen mit dem am besten vorhergesagten
Dampfdruck eine minimale Abweichung bei den Phasengleichgewichtsdaten liefert. Damit ist
der Dampfdruck kein geeigneter Sensor für die Auswahl von Konformationen. Neben diesen
Beobachtungen ist weiterhin zu erkennen, dass es einen qualitativen Trend gibt, dem folgend
stärkere negative Abweichungen im Dampfdruck zu höheren Abweichungen bei der
Phasengleichgewichtsberechnung führen und umgekehrt.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1-H
exan
ol-0
1
1-H
exan
ol-0
2
1-H
exan
ol-0
3
1-H
exan
ol-0
4
1-H
exan
ol-0
5
1-H
exan
ol-0
6
1-H
exan
ol-0
7
1-H
exan
ol-0
8
1-H
exan
ol-0
9
1-H
exan
ol-1
0
1-H
exan
ol-1
1
1-H
exan
ol-1
2
1-H
exan
ol-1
3
1-H
exan
ol-1
4
1-H
exan
ol-1
5
1-H
exan
ol-1
6
1-H
exan
ol-1
7
1-H
exan
ol-1
8
1-H
exan
ol-1
9
1-H
exan
ol-2
0
1-H
exan
ol-2
1
1-H
exan
ol-2
2
1-H
exan
ol-2
3
1-H
exan
ol-2
4
1-H
exan
ol-2
5
1-H
exan
ol-2
6
1-H
exan
ol-2
7
PL
V / b
ar
0
2
4
6
8
10
12
14
16
18
Ab
we
ich
un
g (
(Pe
xp
-Pb
er)/
Pe
xp
) / %
Dampfdruck 1-Hexanol vorhergesagt Dampfdruck Normalsiedepunkt (430.55K) Wasser-1-Hexanol
Abb. 5.39 Dampfdrücke von 1-Hexanol-Konformationen am Normalsiedepunkt, mittlere Abweichungen des
berechneten Drucks im Vergleich zu experimentellen Daten (J. Gmehling, U. Onken, J. R. Rarey-Nies. Vapor-
Liquid Equilibrium Data Collection, Aqueous systems, Supplement 2, 1988) für das System Wasser/1-Hexanol
bei 273,15K
Dieses die ursprünglich getroffenen Vermutungen bestätigende Ergebnis kann bei Vergleich
mit den Vorhersagen des binären Phasengleichgewichtes des Systems 2,3,4-
Trimethylpentan/1-Hexanol allerdings nicht verifiziert werden (Abb. 5.40). Genau jene

114
Konformationen, die im Wasser/1-Hexanol-System die geringsten Abweichungen erzielen,
zeigen im Alkan/1-Hexanol-System die größten Abweichungen. Man erkennt einen
qualitativen Trend, der entgegen dem Wasser-System exakt eine Spiegelung der
Abweichungen ist. Konformationen, die im Wasser/1-Hexanol-System geringe
Abweichungen hervorrufen, zeigen im 2,3,4-Trimethylpentan/1-Hexanol-System die höchsten
Abweichungen. Das heißt, eine Auswahl derjenigen Konformationen mit der kleinsten
Abweichung in der Vorhersage des Dampfdruckes mit dem hier vorgestellten Modell führt im
Fall der unpolaren Umgebung von 1-Hexanol zu großen Differenzen bei
Phasengleichgewichtsberechnungen.
0
2
4
6
8
10
12
14
16
18
1-Hex
anol-0
1
1-Hex
anol-0
2
1-Hexa
nol-0
3
1-Hexa
nol-0
4
1-Hexa
nol-0
5
1-Hex
anol-0
6
1-Hex
anol-0
7
1-Hexa
nol-0
8
1-Hexa
nol-0
9
1-Hexa
nol-1
0
1-Hex
anol-1
1
1-Hex
anol-1
2
1-Hexa
nol-1
3
1-Hexa
nol-1
4
1-Hexa
nol-1
5
1-Hex
anol-1
6
1-Hex
anol-1
7
1-Hexa
nol-1
8
1-Hexa
nol-1
9
1-Hexa
nol-2
0
1-Hex
anol-2
1
1-Hex
anol-2
2
1-Hexa
nol-2
3
1-Hexa
nol-2
4
1-Hexa
nol-2
5
1-Hexa
nol-2
6
1-Hex
anol-2
7
Ab
we
ich
un
g (
(Pe
xp
-Pb
er)/
Pe
xp
) / %
Wasser/1-Hexanol 2,3,4-Trimethylpentan/1-Hexanol
Abb. 5.40 Mittlere Abweichungen des berechneten Drucks im Vergleich zu experimentellen Daten (J. Gmehling,
U. Onken, J. R. Rarey-Nies. Vapor-Liquid Equilibrium Data Collection, Aqueous systems, Supplement 2, 1988)
für das System Wasser/1-Hexanol bei 273,15K und das System 2,3,4-Trimethylpentan/1-Hexanol bei 313,15K
(Heintz, A., E. Dolch und R. Lichtenthaler, Fluid Phase Equilib., 1986, 27, 61)
Für den Stoff 2-Pentanol als Sensormolekül sind dieselben Untersuchungen durchgeführt
worden. Experimentelle Daten für die binären Systeme Wasser/2-Pentanol und n-Heptan/2-
Pentanol führen, wie die Abbildungen 5.41 und 5.42 darstellen, zu ähnlichen Ergebnissen.
Geringe Abweichungen vom vorhergesagten Dampfdruck am Normalsiedepunkt einer
Konformation entsprechen nicht den niedrigsten Abweichungen der
Phasengleichgewichtsberechnungen im Vergleich zu experimentellen Daten. Der
gegenläufige Trend zwischen polarer (Wasser) und unpolarer (n-Heptan) zweiter Komponente
ist hier nicht so deutlich ausgeprägt wie im 1-Hexanol System, aber immer noch erkennbar,

115
wie Abb. 5.42 zeigt.
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.62-
Pen
tano
l-01
2-P
enta
nol-0
2
2-P
enta
nol-0
3
2-P
enta
nol-0
4
2-P
enta
nol-0
5
2-P
enta
nol-0
6
2-P
enta
nol-0
7
2-P
enta
nol-0
8
2-P
enta
nol-0
9
2-P
enta
nol-1
0
2-P
enta
nol-1
1
2-P
enta
nol-1
2
2-P
enta
nol-1
3
2-P
enta
nol-1
4
2-P
enta
nol-1
5
2-P
enta
nol-1
6
2-P
enta
nol-1
7
2-P
enta
nol-1
8
2-P
enta
nol-1
9
2-P
enta
nol-2
0
2-P
enta
nol-2
1
2-P
enta
nol-2
2
2-P
enta
nol-2
3
2-P
enta
nol-2
4
2-P
enta
nol-2
5
2-P
enta
nol-2
6
2-P
enta
nol-2
7
PL
V / b
ar
0
5
10
15
20
25
Ab
we
ich
un
g (
(Pe
xp
-Pb
er)/
Pe
xp
) / %
Dampfdruck Normalsiedepunkt (392.15K) Dampfdruck 2-Pentanol vorhergesagt Wasser/2-Pentanol
Abb. 5.41 Dampfdrücke von 2-Pentanol-Konformationen am Normalsiedepunkt, mittlere Abweichungen des
berechneten Drucks im Vergleich zu experimentellen Daten (Wolfova,J., J.Linek and I.Wichterle, Fluid Phase
Equilib., 1991, 64, 281) für das System n-Heptan/2-Pentanol bei 348,15K, 358,15K und 368,15K

116
0
5
10
15
20
25
2-P
enta
nol-0
1
2-P
enta
nol-0
2
2-P
enta
nol-0
3
2-P
enta
nol-0
4
2-P
enta
nol-0
5
2-P
enta
nol-0
6
2-P
enta
nol-0
7
2-P
enta
nol-0
8
2-P
enta
nol-0
9
2-P
enta
nol-1
0
2-P
enta
nol-1
1
2-P
enta
nol-1
2
2-P
enta
nol-1
3
2-P
enta
nol-1
4
2-P
enta
nol-1
5
2-P
enta
nol-1
6
2-P
enta
nol-1
7
2-P
enta
nol-1
8
2-P
enta
nol-1
9
2-P
enta
nol-2
0
2-P
enta
nol-2
1
2-P
enta
nol-2
2
2-P
enta
nol-2
3
2-P
enta
nol-2
4
2-P
enta
nol-2
5
2-P
enta
nol-2
6
2-P
enta
nol-2
7
Ab
we
ich
un
g (
(Pe
xp
-Pb
er)/
Pe
xp
) / %
Wasser/2-Pentanol n-Heptan/2-Pentanol
Abb. 5.42 Mittlere Abweichungen des berechneten Drucks im Vergleich zu experimentellen Daten n-Heptan/2-
Pentanol (Daten siehe Abb. 5.41) und das System Wasser/2-Pentanol bei 343,15K, 353,15K und 363,15K (J.
Gmehling, U. Onken, J. R. Rarey-Nies. Vapor-Liquid Equilibrium Data Collection, Aqueous systems,
Supplement 2, 1988)
Eine Bestätigung dieser Aussagen liefert auch das n-Hexan/1-Octanol-System. Abb. 5.43
zeigt die Ergebnisse der Betrachtungen.
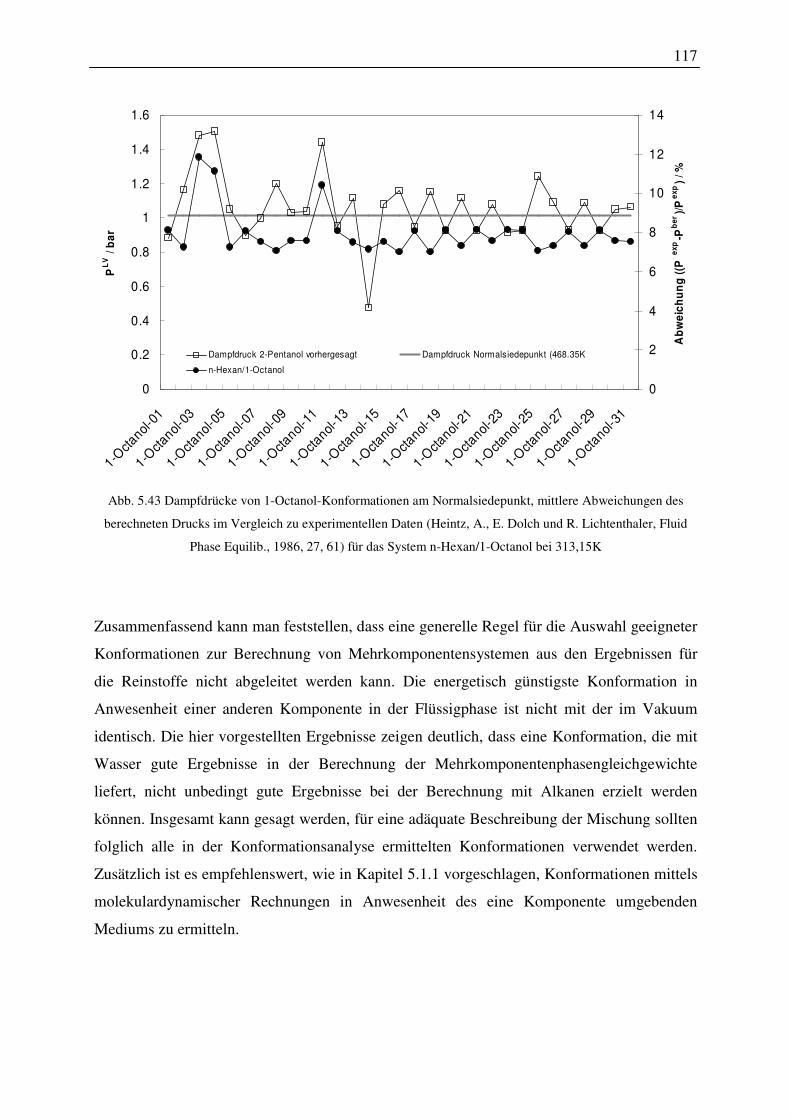
117
0
0.2
0.4
0.6
0.8
1
1.2
1.4
1.6
1-Octa
nol-01
1-Octa
nol-03
1-Octa
nol-05
1-Octa
nol-07
1-Octa
nol-09
1-Octa
nol-11
1-Octa
nol-13
1-Octa
nol-15
1-Octa
nol-17
1-Octa
nol-19
1-Octa
nol-21
1-Octa
nol-23
1-Octa
nol-25
1-Octa
nol-27
1-Octa
nol-29
1-Octa
nol-31
PL
V / b
ar
0
2
4
6
8
10
12
14
Ab
we
ich
un
g (
(Pe
xp
-Pb
er)/
Pe
xp
) / %
Dampfdruck 2-Pentanol vorhergesagt Dampfdruck Normalsiedepunkt (468.35K
n-Hexan/1-Octanol
Abb. 5.43 Dampfdrücke von 1-Octanol-Konformationen am Normalsiedepunkt, mittlere Abweichungen des
berechneten Drucks im Vergleich zu experimentellen Daten (Heintz, A., E. Dolch und R. Lichtenthaler, Fluid
Phase Equilib., 1986, 27, 61) für das System n-Hexan/1-Octanol bei 313,15K
Zusammenfassend kann man feststellen, dass eine generelle Regel für die Auswahl geeigneter
Konformationen zur Berechnung von Mehrkomponentensystemen aus den Ergebnissen für
die Reinstoffe nicht abgeleitet werden kann. Die energetisch günstigste Konformation in
Anwesenheit einer anderen Komponente in der Flüssigphase ist nicht mit der im Vakuum
identisch. Die hier vorgestellten Ergebnisse zeigen deutlich, dass eine Konformation, die mit
Wasser gute Ergebnisse in der Berechnung der Mehrkomponentenphasengleichgewichte
liefert, nicht unbedingt gute Ergebnisse bei der Berechnung mit Alkanen erzielt werden
können. Insgesamt kann gesagt werden, für eine adäquate Beschreibung der Mischung sollten
folglich alle in der Konformationsanalyse ermittelten Konformationen verwendet werden.
Zusätzlich ist es empfehlenswert, wie in Kapitel 5.1.1 vorgeschlagen, Konformationen mittels
molekulardynamischer Rechnungen in Anwesenheit des eine Komponente umgebenden
Mediums zu ermitteln.

6 Zusammenfassung
In dieser Arbeit wurden die vorhandenen Methoden der Verknüpfung von Quantenchemie
und statistischer Thermodynamik als Vorhersagemethode für die benötigten Größen der
Phasengleichgewichtsthermodynamik untersucht und erweitert. Der Schwerpunkt der Arbeit
lag auf der Untersuchung und Weiterentwicklung des COSMO-RS-Modells.
Das CSOMO-RS-Modell setzt sich aus dem COSMO-Ansatz und der Erweiterung auf Real
Solvents (RS) zusammen, die die in der COSMO-Rechnung erzeugten Oberflächensegmente
benutzt, um das chemische Potential einer Komponente zu berechnen. Der COSMO-Ansatz
selbst gehört in die Klasse der dielektrischen Kontinuumsmodelle. Im Unterschied zu den
anderen Kontinuumsmodellen geht er nicht von einem umgebenden Dielektrikum aus,
sondern betrachtet das Teilchen als in einen idealen Leiter eingebettet. In der Erweiterung
RS wird die Flüssigkeit als Ensemble aus unabhängigen, paarweise wechselwirkenden
Oberflächensegmenten aufgefasst. Mit der Kenngröße Abschirmungsladungsdichte σ eines
Segmentes lässt sich ein Molekül in seinen (elektrostatischen)
Wechselwirkungseigenschaften mit COSMO-RS beschreiben, wenn man eine
Verteilungsdichtefunktion der Abschirmungsladungen aller Segmente definiert. Ausgehend
vom Zustand des im idealen Leiters gelösten Moleküls, das als Ensemble von
Oberflächensegmenten vorliegt, wird das chemische Potential einer Komponente i aus der
Zustandssumme des Ensembles von N elektrisch geladenen Oberflächensegmenten
abgeleitet.
In der vorliegenden Arbeit wurden fünf Themenkomplexe behandelt und neue
Lösungsmethoden vorgestellt:
1. Validierung des COSMO-RS-Modells hinsichtlich in der chemischen Verfahrenstechnik
relevanten Komponenten und ihrer binären Mischungen.
Die Validierung von 136 Systemen zeigte, dass COSMO-RS experimentelle Daten von
Dampf-Flüssig-Phasengleichgewichtsdaten unterschiedlicher Stoffklassen in guter
Übereinstimmung vorhersagen konnte. Die Abweichungen zwischen den Vorhersagen
und den gemessenen Daten lagen im Vergleich zu den experimentellen Daten für viele
Systeme bei fünf Prozent. Mischungen nicht-assoziierender Komponenten wurden mit
den geringsten Abweichungen beschrieben. Mischungen, die ein spezifischeres

119
Wechselwirkungsverhalten aufweisen, konnten nicht mit der gleichen Genauigkeit
vorhergesagt werden, jedoch wurde das nichtideale Verhalten gut reproduziert.
Die Vorhersage von Flüssig-Flüssig-Phasengleichgewichten zeigt, dass auf Basis der
molekularen Struktur der Komponenten mit dem COSMO-RS-Modell eine Entmischung
zu modellieren war. Dabei hing die Genauigkeit der Vorhersage von der verwendeten
Konformation des Moleküls ab.
2. Die Untersuchung des Einflusses der verschiedenen Konformationen einer
Molekülstruktur auf die Vorhersagen des Phasengleichgewichtes mit dem COSMO-RS-
Modell wurde am Beispiel des 2-Butanol Moleküls exemplarisch dargestellt. Es wurden
zunächst die grundlegenden Methoden der Absuche des Konformerenraumes dargelegt
und anschließend mittels der kommerziellen Software HyperChem zehn repräsentative
Konformationen des 2-Butanols identifiziert. Für diese Konformationen wurde das
Phasengleichgewicht am Beispiel des 2-Butanol/Wasser-Systems vorhergesagt. Hierbei
wurde gezeigt, dass die Konformation mit der niedrigsten Energie nicht die besten
Ergebnisse bei der Vorhersage lieferte. Vielmehr war es notwendig alle Konformationen
in die Phasengleichgewichtsberechnung einzubeziehen. Dies erfolgte, indem der Anteil
einer jeden Konformationen energiegewichtet in die Berechnung des chemischen
Potenzials einer Komponente einging. Mit diesem Vorgehen lieferte die Vorhersage mit
COSMO-RS gute Ergebnisse.
3. Die Untersuchungen zum Phasenverhalten hyperverzweigter Polymere zeigten, dass das
Wechselwirkungsverhalten von stark verzweigten Molekülen großer Molmasse in
Lösung mit dem COSMO-RS-Modell vorhersagbar ist. Am Beispiel eines
hyperverzweigten Polyglyzerinmoleküls mit 20 Hydroxylgruppen und etwa 1400g/mol
wurde gezeigt, dass aus den experimentell gewonnenen Strukturdaten eine
zweidimensionale Molekülstruktur abgeleitet werden konnte. Für die
Geometrieoptimierung mit quantenchemischen Methoden musste das Gesamtmolekül in
Teile zerlegt werden, da beim gegenwärtigen Stand der Rechenleistung die
Molekülgröße auf ca. 600-800g/mol beschränkt ist. Es wurden zwei Verfahren
entwickelt, die ein großes Molekül in kleine Einheiten zerlegen. Zum einen wurde das
Polymermolekül in kleine, wiederkehrende Fragmente zerlegt. Dabei geht ein Teil des
Einflusses der intramolekularen Wechselwirkungen verloren. Das Phasenverhalten
konnte dennoch mit guter Näherung beschrieben werden. Eine geeignetere Variante war

120
das Zerlegen des Gesamtmoleküls in größere Untereinheiten, die die intramolekularen
Wechselwirkungen berücksichtigten. Mit dieser Methode konnte das Phasenverhalten
des ternären Systems Polyglyzerin/Ethanol/Wasser für verschiedene
Polymerkonzentrationen vorhergesagt werden. Für diese Berechnungen wurde das
COSMO-RS-Modell um einen Free-Volume Term in Anlehnung an das UNIFAC-Free-
Volume-Modell erweitert.
4. Für die Berechnung von Hochdruckphasengleichgewichten wurde das COSMO-RS-
Modell mit der Peng-Robinson-Zustandsgleichung unter Anwendung der Wong-Sandler-
Mischungsregeln kombiniert. Die Ergebnisse zeigten, dass Vorhersagen auf Basis der
Atomstruktur der Moleküle und der kritischen Daten bis weit in den überkritischen
Bereich möglich waren. Die Beschreibung nichtidealer binärer Systeme mit Hilfe der
Gibbsschen Exzessenthalpie unter Verwendung des Vorhersagemodells COSMO-RS
erlaubte die Ermittlung von Mischungsgrößen in Systemen, die mit den einfachen van-
der-Waals-Mischungs- und Kombinationsregeln nicht zuverlässig betrachtet werden
können. Die Stärke gerade dieser hier betrachteten Kombination liegt darin begründet,
dass das COSMO-RS-Modell Mischungseigenschaften nichtidealer Systeme vorhersagen
kann, ohne dass auf Parametertabellen von einschlägigen GE-Modellen zurückgegriffen
werden muss.
5. Das Energiekonzept des COSMO-RS-Modells wurde um einen Term erweitert, der die
Änderung der freien Enthalpie der Lösung durch die Bildung einer Kavität im
Lösungsmittel explizit enthält. Eine wesentliche Größe dieses Terms ist der Durchmesser
der harten Kugel. In Vergleichen mit anderen Methoden, die einen Harte-Kugel-
Durchmesser aus experimentellen Daten ableiten, wurde ein Konzept entwickelt, dass es
ermöglicht, die quantenchemischen Rechnungen unter der COSMO-Randbedingung zu
verwenden um einen Harte-Kugel-Durchmesser beliebiger Moleküle zu berechnen.
Mit dem erweiterten Energiekonzept wurden nach dem von Ben-Naim vorgeschlagenen
Lösungsprozess eines Moleküls, Dampfdrücke von Komponenten aus den Stoffklassen
der Alkane, der Alkene, der Alkohole, der Aromaten, der Ether, der Ketone, der
Aldehyde und der Polyole berechnet. Die Abweichungen, verglichen mit
experimentellen Daten am Normalsiedepunkt, zeigten, dass die Methode für Alkohole
innerhalb einer Fehlertoleranz von durchschnittlich ca. 20 Prozent gelingt, für die
anderen Stoffklassen aber deutlich höhere Abweichungen auftraten. Insbesondere bei den
Stoffklassen der Alkane und Alkene waren große Abweichungen zu verzeichnen. Da die

121
Einzelbeiträge zur Gibbsschen Enthalpie des Lösungsvorganges bis auf die Energien der
elektrostatischen und der dispersiven Beiträge nahezu konstante Werte aufweisen, liegt
die hohe Abweichung der berechneten Dampfdrücke im Vergleich zu den erwarteten
Werten in der nicht ausreichend beschriebenen dispersiven Wechselwirkung des
COSMO-RS-Modells begründet. Es zeigte sich allerdings, dass der vorhergesagte Wert
des Dampfdruckes sehr sensibel auf die unterschiedlichen Konformationen einer
Komponente reagiert. Der Dampfdruck wäre somit ein Sensor, der es ermöglicht,
relevante Konformationen eines Moleküls für eine Phasengleichgewichtsberechnung zu
identifizieren. Eine Konformation, die in der Vorhersage eine geringe Abweichung vom
erwarteten Dampfdruck erzielt, sollte in der Phasengleichgewichtsberechnung ebenfalls
geeignet sein, gute Ergebnisse zu erzielen. Die Berechnungen für die Stoffe 2-Pentanol,
1-Hexanol und 1-Octanol zeigten, dass Konformationen die eine gute Beschreibung des
Dampfdruckes liefern, nicht in jedem Fall auch eine gute Beschreibung in
Mehrkomponentengemischen ergaben. Eine generelle Regel für die Auswahl geeigneter
Konformationen zur Berechnung von Mehrkomponentensystemen aus den Ergebnissen
für die Reinstoffe konnte nicht abgeleitet werden. Insgesamt kann festgehalten werden,
dass für eine adäquate Beschreibung der Mischung alle in der Konformationsanalyse
ermittelten Konformationen verwendet werden sollten. Zusätzlich ist es empfehlenswert,
Konformationen mittels molekulardynamischer Rechnungen in Anwesenheit des eine
Komponente umgebenden Mediums zu ermitteln.
7 Anhang
7.1 Übersicht der untersuchten Systeme zur Anwendbarkeit des COSMO-RS-Modells
zur Vorhersage von Dampf-Flüssig-Phasengleichgewichten
Tabelle 7.1 Untersuchte Systeme zur Anwendbarkeit des COSMO-RS-Modells
Klasse Systeme Temperatur
Bereich
Datenpunkte Ref. Daten bei
xi=0 o. xi=1
Alkan-Alkan n-Pentan + n-Hexan 298.15 10 12 ja/ja
n-Hexan + 2,2-
Dimethylbutan
298.15 11 12 ja/ja
n-Hexan + n-Heptan 323.15 9 69 ja/ja
n-Heptan + n-Octan 328.15 14 21 ja/ja
n-Octan + 2-Methylpentan 293.15-293.15 12 38 nein/nein
n-Hexan + 3-Methylpentan 303.15-313.15 21 10 nein/nein
Alkan-Alken n-Butan + 1-Buten 310.93 10 20 ja/ja
n-Hexan + 1-Hexen 333.15 11 20 nein/nein
n-Heptan + 1-Hepten 328.15 15 21 ja/ja
n-Octan + 1-Hepten 328.15 12 21 ja/ja
Alkan-Alkohol Isobutan + Methanol 373.15 13 36 ja/ja
Isobutan + 1-Propanol 340.8 8 77 ja/nein
n-Pentan + Methanol 372.7-422.6 30 72 ja/ja
n-Pentan + Ethanol 372.7-422.6 31 8 ja/ja
n-Pentan + 1-Butanol 303.2 14 55 ja/ja
n-Pentan + 2-Butanol 303.15 14 55 ja/ja
n-Pentan + 1-Pentanol 303.15 14 55 ja/ja
n-Hexan + Methanol 298.15-333.15 55 30,58 ja/ja
n-Hexan + Ethanol 333.15 8 37 ja/ja
n-Hexan + 1-Butanol 323.15-332.53 39 9,27 ja/ja
n-Hexan + 2-Butanol 348.15 14 2 ja/ja
n-Hexan + Isobutanol 332.53 22 9 ja/ja
n-Hexan + 1-Pentanol 323.15 14 55 ja/ja
n-Heptan + Methanol 298.15 30 30 nein/nein
n-Heptan + Ethanol 298.15-363.15 83 30,50,55,63,79 ja/ja
n-Heptan + 1-Propanol 303.15-333.15 84 66,78 ja/ja
n-Heptan + Isopropanol 303.15-333.15 54 66 ja/ja
n-Heptan + 1-Butanol 313.15 19 80 nein/nein
n-Heptan + Isobutanol 298.15-313.15 36 27,80 nein/nein
n-Heptan + tert-Butanol 313.15 20 80 nein/nein
n-Heptan + 1-Pentanol 348.15-368.15 58 41 ja/ja
123
n-Heptan + 2-Pentanol 348.15-368.15 73 75 ja/ja
n-Heptan + 3-Pentanol 348.15-368.15 68 74 ja/ja
n-Heptan + 2-Methyl-1-
butanol
348.15-368.15 60 75 ja/ja
n-Heptan + 3-Methyl-1-
butanol
348.15-368.15 74 41 ja/ja
n-Octan + 1-Butanol 373.15 19 29 ja/ja
224-Trimethylpentan +
Ethanol
333.15 17 28 nein/nein
224-Trimethylpentan + 1-
Propanol
345.15 20 28 nein/nein
224-Trimethylpentan + 1-
Hexanol
313.15 12 27 nein/nein
Alkan-Aromat n-Pentan + Benzol 318.15 13 68 nein/nein
n-Hexan + Benzol 298.15-333.15 114 7,26,43,57 ja/ja
n-Heptan + Benzol 298.15-313.15 60 26,40 ja/ja
n-Heptan + Toluol 298.15-313.15 46 26,62 ja/ja
n-Heptan + p-Xylol 313.15 13 26 ja/ja
n-Heptan + Ethylbenzol 313.15 14 26 ja/ja
n-Octan + Benzol 313.15 14 26 ja/ja
n-Octan + Toluol 313.15-333.15 28 5,26 ja/ja
n-Octan + p-Xylol 313.15 14 26 ja/ja
n-Decan + Toluol 313.15 12 26 ja/ja
n-Decan + p-Xylol 313.15 14 26 ja/ja
Alkohol-
Alkohol
Methanol + Ethanol 298.15 15 23 ja/ja
Methanol + 2-propanol 328.15 20 24 nein/nein
Methanol + 2-Methyl-1-
Propanol
323.15-343.15 40 24 ja/ja
Methanol + 3-Methyl-1-
butanol
323.15-343.15 30 24 ja/ja
Ethanol-1-Propanol 313.15 11 79 nein/nein
Ethanol + 1-Butanol 323.15-363.15 24 23 nein/nein
Ethanol + 2-Methyl-1-
propanol
323.15-353.15 40 24 ja/ja
Ethanol + 3-Methyl-1-
butanol
232.15-353.15 40 24 ja/ja
1-Propanol + 2-Methyl-1-
propanol
343.15-353.15 20 24 ja/ja
1-Propanol + 3-Methyl-1-
butanol
353.15 10 24 ja/ja
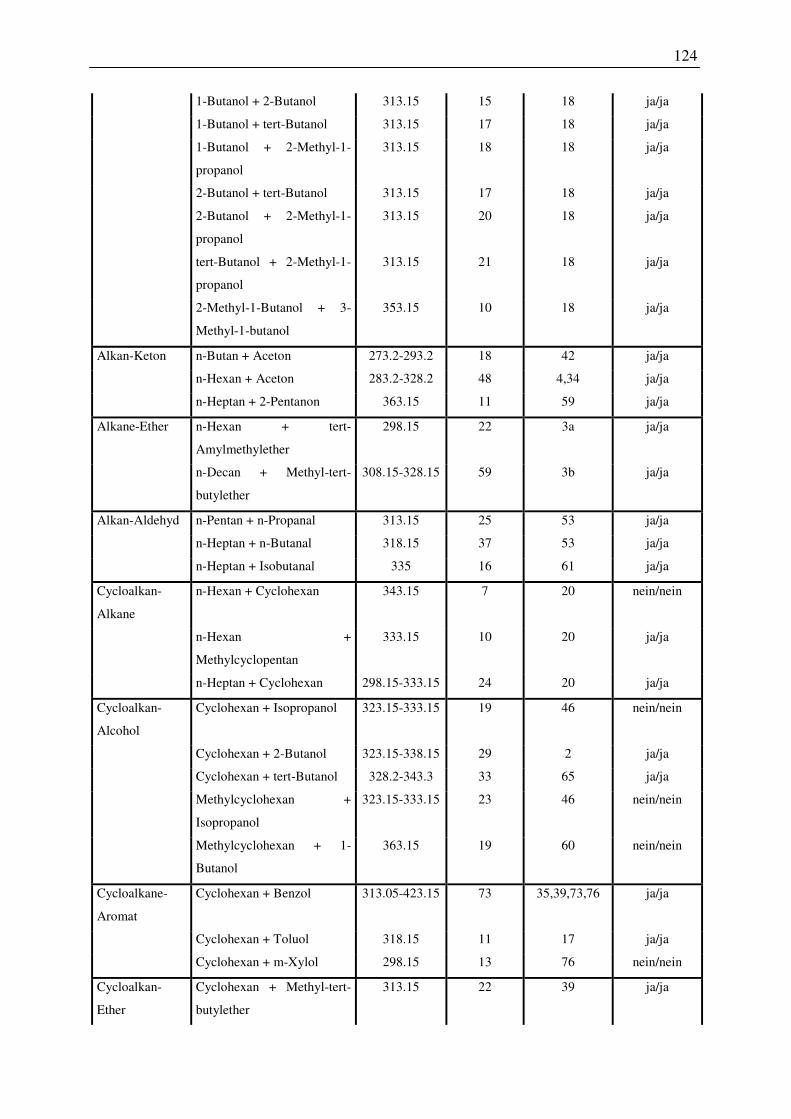
124
1-Butanol + 2-Butanol 313.15 15 18 ja/ja
1-Butanol + tert-Butanol 313.15 17 18 ja/ja
1-Butanol + 2-Methyl-1-
propanol
313.15 18 18 ja/ja
2-Butanol + tert-Butanol 313.15 17 18 ja/ja
2-Butanol + 2-Methyl-1-
propanol
313.15 20 18 ja/ja
tert-Butanol + 2-Methyl-1-
propanol
313.15 21 18 ja/ja
2-Methyl-1-Butanol + 3-
Methyl-1-butanol
353.15 10 18 ja/ja
Alkan-Keton n-Butan + Aceton 273.2-293.2 18 42 ja/ja
n-Hexan + Aceton 283.2-328.2 48 4,34 ja/ja
n-Heptan + 2-Pentanon 363.15 11 59 ja/ja
Alkane-Ether n-Hexan + tert-
Amylmethylether
298.15 22 3a ja/ja
n-Decan + Methyl-tert-
butylether
308.15-328.15 59 3b ja/ja
Alkan-Aldehyd n-Pentan + n-Propanal 313.15 25 53 ja/ja
n-Heptan + n-Butanal 318.15 37 53 ja/ja
n-Heptan + Isobutanal 335 16 61 ja/ja
Cycloalkan-
Alkane
n-Hexan + Cyclohexan 343.15 7 20 nein/nein
n-Hexan +
Methylcyclopentan
333.15 10 20 ja/ja
n-Heptan + Cyclohexan 298.15-333.15 24 20 ja/ja
Cycloalkan-
Alcohol
Cyclohexan + Isopropanol 323.15-333.15 19 46 nein/nein
Cyclohexan + 2-Butanol 323.15-338.15 29 2 ja/ja
Cyclohexan + tert-Butanol 328.2-343.3 33 65 ja/ja
Methylcyclohexan +
Isopropanol
323.15-333.15 23 46 nein/nein
Methylcyclohexan + 1-
Butanol
363.15 19 60 nein/nein
Cycloalkane-
Aromat
Cyclohexan + Benzol 313.05-423.15 73 35,39,73,76 ja/ja
Cyclohexan + Toluol 318.15 11 17 ja/ja
Cyclohexan + m-Xylol 298.15 13 76 nein/nein
Cycloalkan-
Ether
Cyclohexan + Methyl-tert-
butylether
313.15 22 39 ja/ja
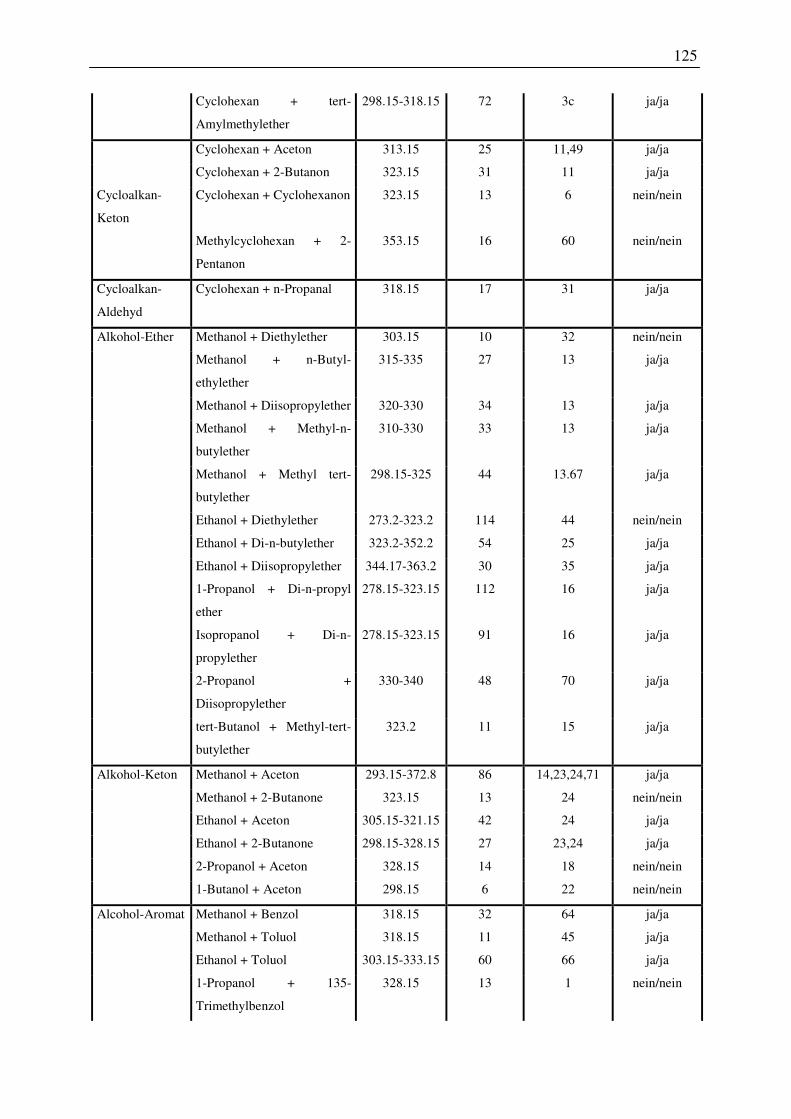
125
Cyclohexan + tert-
Amylmethylether
298.15-318.15 72 3c ja/ja
Cyclohexan + Aceton 313.15 25 11,49 ja/ja
Cyclohexan + 2-Butanon 323.15 31 11 ja/ja
Cycloalkan-
Keton
Cyclohexan + Cyclohexanon 323.15 13 6 nein/nein
Methylcyclohexan + 2-
Pentanon
353.15 16 60 nein/nein
Cycloalkan-
Aldehyd
Cyclohexan + n-Propanal 318.15 17 31 ja/ja
Alkohol-Ether Methanol + Diethylether 303.15 10 32 nein/nein
Methanol + n-Butyl-
ethylether
315-335 27 13 ja/ja
Methanol + Diisopropylether 320-330 34 13 ja/ja
Methanol + Methyl-n-
butylether
310-330 33 13 ja/ja
Methanol + Methyl tert-
butylether
298.15-325 44 13.67 ja/ja
Ethanol + Diethylether 273.2-323.2 114 44 nein/nein
Ethanol + Di-n-butylether 323.2-352.2 54 25 ja/ja
Ethanol + Diisopropylether 344.17-363.2 30 35 ja/ja
1-Propanol + Di-n-propyl
ether
278.15-323.15 112 16 ja/ja
Isopropanol + Di-n-
propylether
278.15-323.15 91 16 ja/ja
2-Propanol +
Diisopropylether
330-340 48 70 ja/ja
tert-Butanol + Methyl-tert-
butylether
323.2 11 15 ja/ja
Alkohol-Keton Methanol + Aceton 293.15-372.8 86 14,23,24,71 ja/ja
Methanol + 2-Butanone 323.15 13 24 nein/nein
Ethanol + Aceton 305.15-321.15 42 24 ja/ja
Ethanol + 2-Butanone 298.15-328.15 27 23,24 ja/ja
2-Propanol + Aceton 328.15 14 18 nein/nein
1-Butanol + Aceton 298.15 6 22 nein/nein
Alcohol-Aromat Methanol + Benzol 318.15 32 64 ja/ja
Methanol + Toluol 318.15 11 45 ja/ja
Ethanol + Toluol 303.15-333.15 60 66 ja/ja
1-Propanol + 135-
Trimethylbenzol
328.15 13 1 nein/nein
126
Isopropanol + Benzol 323.15-343.15 38 46 nein/nein
Isopropanol + Toluol 298.15-313.15 42 62 ja/ja
Isopropanol + 135-
Trimethylbenzol
353.15 16 1 nein/nein
1-Butanol + Benzol 298.15 9 54 ja/ja
1-Butanol + Toluol 363.15 17 60 nein/nein
2-Butanol + Benzol 298.15 9 54 ja/ja
tert-Butanol + Benzol 298.15 11 54 ja/ja
Isobutanol + Benzol 298.15 12 54 ja/ja
1-Pentanol + Toluol 303.15-383.15 101 56 nein/nein
3-Methyl-1-butanol + Toluol 353.15-380.15 43 51 ja/ja
Keton-Ether Aceton + Diisopropylether 343.26-363.29 33 35 ja/ja
Keton-Aromat Aceton + Toluol 308.15-328.15 44 19 nein/nein
2-Butanon + Benzol 313.15-333.15 51 19,48 ja/ja
2-Butanon + Toluol 323.15-348.15 86 19,47 ja/ja
2-Butanon + Ethylbenzol 328.15-348.15 45 19 nein/nein
2-Pentanon + Toluol 323.15 27 19 ja/ja
4-Methyl-2-pentanon +
Toluol
323.15 28 19 ja/ja
Alken-Ether Methyl-tert-butylether + 1-
Hexen
313.15 24 40 ja/ja
Aldehyd-
Aromat
Pentanal + Toluol 313.15 13 33 ja/ja
Keton-Aldehyd 2-Butanon + n-Propanal 318.15 15 19 ja/ja
Alkohol-Alken 2-Butanol + 1-Hexen 333.15 12 18 nein/nein
Alkohol-
Aldehyd
Propanal + Methanol 318.15 17 24 ja/ja
Cycloalkan-
Alken
Cyclohexan + 1-Octen 313.15 12 20 nein/nein
(1) Aguirre-Ode,F. and J.Alodney, J.Chem.Eng.Data, 34, 409 (1989). (2) Araujo,M.E., M.R.Maciel and A.Z.Francesconi,
J.Chem.Thermodyn., 25, 1295 (1993). (3a) Armando del Río, Baudilio Coto, Concepción Pando and J.A.R.Renuncio; Ind. Eng. Chem. Res.
41(2002), 1364-1369. (3b) Armando del Río, Baudilio Coto, Concepción Pando and J.A.R.Renuncio Fluid Phase Equilibria 187-188 (2001),
299-310. (3c) Armando del Río, Baudilio Coto, Concepción Pando and J.A.R.Renuncio, Ind. Eng. Chem. Res. 40 (2001), 689-695. (4)
Barraza,R.;Diaz,S.;Edwards,J.;Tapia,P.,Z. Phys. Chem. NF,117,43-54 (1979). (5) Berro,C., F.Laichoubi and E.Rauzy, J.Chem.Thermodyn.,
26, 863 (1994). (6) Boublik,T. and B.C.Lu, J.Chem.Eng.Data, 22, 331 (1977). (7) C. LI,I.P., Y.W.WONG, S.D.CHANG and B.C.-Y. LU,
J.Chem.Eng.Data, 17, 492 (1972). (8) Campbell,S.W., R.A.Wilsak and G..Thodos, J.Chem.Thermodyn., 19, 449 (1987). (9) Charles Berro,
Marek Rogalski and Andre Peneloux, J.Chem.Eng.Data, 27, 352 (1982). (10) Chun L Ho and Richard R Davison, J.Chem.Eng.Data, 24, 293
(1979). (11) Colin,A.C., A.Compostizo and M.Pena, J.Chem.Thermodyn., 16, 497 (1984). (12) S.S. Chen, Zwolinski B.J., J. Chem. Soc.
Faraday Trans., 1974 (70), 1133. (13) Farkova,J., J.Linek and I.Wichterle, Fluid Phase Equilib., 109, 53 (65). (14) Freshwater,D.C. and
K.A.Pike, J.Chem.Eng.Data, 12, 179 (1967). (15) Fuangfoo,S.;Kersting,M.;Viswanath,D.S.,J. Chem. Eng. Data,44,405-410 (1999). (16)
Garriga,R., F.Sanchez, P.Perez and M.Gracia, Fluid Phase Equilib., 138, 131 (1997). (17) Gaube,J., S.Hammer and A.Pfennig, Fluid Phase
Equilib., 123, 245 (1996). (18) Gmehling,J., Onken,U., Arlt, W., Vapor-Liquid Equilibrium Data Collection,Dechema Chemistry Data
Series, Vol.I,Part 2b (1978) Frankfurt/Main. (19) Gmehling,J., Onken,U., Arlt,W., Vapor-Liquid Equilibrium Data Collection,Dechema

127
Chemistry Data Series, Vol.I,Part 3+4 (1979) Frankfurt/Main. (20) Gmehling,J., Onken,U., Arlt,W., Vapor-Liquid Equilibrium Data
Collection,Dechema Chemistry Data Series, Vol.I,Part 6a (1980) Frankfurt/Main. (21) Gmehling,J., Onken,U., Arlt,W., Vapor-Liquid
Equilibrium Data Collection,Dechema Chemistry Data Series, Vol.I,Part 6b (1980) Frankfurt/Main. (22) Gmehling,J., Onken,U., Rarey, J.R.,
Vapor-Liquid Equilibrium Data Collection,Dechema Chemistry Data Series, Vol.I,Part 2f (1990) Frankfurt/Main. (23) Gmehling,J.,
Onken,U., Rarey-Nies, J.R., Vapor-Liquid Equilibrium Data Collection,Dechema Chemistry Data Series, Vol.I,Part 2e (1988)
Frankfurt/Main. (24) Gmehling,J., Onken,U., Vapor-Liquid Equilibrium Data Collection,Dechema Chemistry Data Series, Vol.I,Part 2a
(1977) Frankfurt/Main. (25) Gmehling,J.,AICHE Symp. Ser.,244,121-129 (1985). (26) Goral,M., Fluid Phase Equilib., 102, 275 (1994). (27)
HEINTZ,A., E.DOLCH and R.LICHTENTHALER, Fluid Phase Equilib., 27, 61 (1986). (28) Hiaki,T., K.Takahashi, T.Tsuji, M.Hongo and
K.Kojima, J.Chem.Eng.Data, 39, 605 (1994). (29) Hiaki,T.;Taniguchi,A.;Tsuji,T.;Hongo,M.,Fluid Phase Equilib.,Volume 144,145-155
(1998). (30) Hongo,M., T.Tsuji, K.Fukuchi and Y.Arai, J.Chem.Eng.Data, 39, 688 (1994). (31) Ichiro Matsunaga and Takashi Katayama, J.
Chem. Eng. Japan, 5, 397 (1973. (32) Jurgen Gmehling, Ulfert Onken and Hans-Werner Schulte, J.Chem.Eng.Data, 25, 29 (1980). (33)
KASSMANN,K.D. and H.KNAPP, Fluid Phase Equilib., 29, 241 (1986). (34) Kudryavtseva,L.S.;Susarev,M.P.,Zh. Prikl. Khim.,36,1471-
1477 (1963). (35) Lee,M.J. and C.H.Hu, Fluid Phase Equilib., 109, 83 (1995). (36) Leu,A.D. and D.B.Robinson, J.Chem.Eng.Data, 37, 10
(1992). (37) LINDBERG,G.W. and D.TASSIOS, J.Chem.Eng.Data, 16, 52 (1971). (38) Liu,Edwar.K.. and R..R..Davison, J.Chem.Eng.Data,
26, 85 (1981). (39) Lozano,L.M., E.A.Montero, M.C.Martin and M.A.Villamanan, Fluid Phase Equilib., 110, 219 (1995). (40) Lozano,L.M.,
e.A.Montero, M.C.Martin and M.A.Villamanan, Fluid Phase Equilib., 133, 155 (1997). (41) MACHOVA,I., J.LINEK and I.WICHTERLE,
Fluid Phase Equilib., 41, 257 (1988). (42) Matvienko,V.G.;Yarym-Agaev,N.L.,Zh. Prikl. Khim.,67,158-160 (1994). (43) Murray,R.S. and
M.L.Martin, J.Chem.Thermodyn., 7, 839 (1975). (44) Nagai,J.;Ishii,N.,J. Soc. Chem. Ind. Jpn.,38,86-95(1935). (45) Nagata,I.,
J.Chem.Thermodyn., 20, 467 (1988). (46) NAGATA,I., T.OHTA and Y.UCHIYAMA, J.Chem.Eng.Data, 18, 54 (1973). (47) Naumann,D.
and H.G.Wagner, J.Chem.Thermodyn., 18, 81 (1986). (48) Pena,M.D., A.C.Colin and A..Compostizo, J.Chem.Thermodyn., 10, 1101 (1978).
(49) PURI,P.S., J.POLAK and J.RUETHER, J.Chem.Eng.Data, 19, 87 (1974). (50) Ramalho,R.S. and J.Delmas, J.Chem.Eng.Data, 13, 161
(1968) . (51) Reddy,M.S. and C.V.Rao, J.Chem.Eng.Data, 10, 309 (1965). (52) Rice,Reter. and Ali.El-Nikheli, Fluid Phase Equilib., 107,
257 (1995). (53) Robert Eng and Stanley I Sandler, J.Chem.Eng.Data, 29, 156 (1984). (54) Rodriguez,V., C.Lafuente, M.Lopez, F.M.Royo
and J.S.Urieta, J.Chem.Thermodyn., 25, 679 (1993). (55) Ronc,M.;Ratcliff,G.R.,Can. J. Chem. Eng.,54,326-332 (1976). (56) SADLER
lll,L.Y., D.W.LUFF and M.D.McKINLEY, J.Chem.Eng.Data, 16, 446 (1971). (57) SAEZ,C., A.COMPOSTIZO, R.RUBIO, A.CRESPO
COLIN and M.DIAZ PENA, Fluid Phase Equilib., 24, 241 (1985). (58) Scheller,M.;Schuberth,H.;Koennecke,H.G.,J. Prakt.
Chem.,1969,974-982. (59) SCHELLER,W.A. and S.V.NARASIMHA RAO, J.Chem.Eng.Data, 18, 223 (1973). (60) Seetharamaswamy,V.,
V.Subrahmanyam, C.Chiranjivi and P.Dakshinamurty, J. Applied Chemistry, 19, 258 (1969). (61) Shealy,G.S. and S.I.Sandler,
J.Chem.Thermodyn., 17, 143 (1985). (62) Stanley J Ashcroft, Andrew D Clayton and Ronald B Shearn, J.Chem.Eng.Data, 24, 195 (1979).
(63) Tai,T.B., R.S.Ramalho and S..Kaliaguine, Can.J.Chem.Eng., 50, 771 (1972) . (64) Toghiani,H., R.K.toghiani and D.S.Viswanath,
J.Chem.Eng.Data, 39, 63 (1994). (65) Triday,J.O. and C.Veas, J.Chem.Eng.Data, 30, 171 (1985). (66) Van Ness,H.C., C.A.Soczek,
G.L.Peloquin and R.L.Machado, J.Chem.Eng.Data, 12, 217 (1967). (67) Velasco,E., M.J.Cocero and F..Mato, J.Chem.Eng.Data, 35, 21
(1990). (68) Wang,J.L. and B.C.Lu, J. Applied Chemistry, 21, 297 (1971) . (69) Smith, C.P., E.W. Engel, J. Amer. Chem. Soc., 1929 (51),
2646. (70) Wichterle,I.,ELDATA Int. Electron. J. Phys. Chem. Data,5,179-189 (1999). (71) WILSAK,R.A., S.W.CAMPBELL and
G..THODOS, Fluid Phase Equilib., 28, 13 (1986). (72) WILSAK,R.A., S.W.CAMPBELL and G..THODOS, Fluid Phase Equilib., 33, 157
(1987). (73) Wisniewska,B., J.Gregorowicz and S.Malanowski, Fluid Phase Equilib., 86, 173 (1993). (74) Wolfova,J., J.Linek and
I.Wichterle, Fluid Phase Equilib., 54, 69 (1990). (75) Wolfova,J., J.Linek and I.Wichterle, Fluid Phase Equilib., 64, 281 (1991). (76)
Young,K.L., R.A.Mentzer, R.A.Greenkorn and K.C.Chao, J.Chem.Thermodyn., 9, 979 (1977). (77) Zabaloy,M.S., H.P.gros, S.B.Bottini and
E.A.Brignole, J.Chem.Eng.Data, 39, 214 (1994). (78) Zielkiewicz,J., J.Chem.Thermodyn., 24, 455 (1992). (79) Zielkiewicz,J.,
J.Chem.Thermodyn., 25, 1077 (1993). (80) Zielkiewicz,J., J.Chem.Thermodyn., 26, 919 (1994).

128
7.2 Harte-Kugel-Durchmesser aus COSMO-Volumina, Dampfdrücke am
Normalsiedepunkt
Tabelle 7.2
Komponente Dampfdruck vorhergesagt [bar]
Abweichung Harte Kugeldurchmesser [Ǻ]
Normasiedepunkt [K]
1-butanol 1,0949 8,08489635 5,4112 390,81 1-butene 0,9357 -7,63079961 5,1323 266,9 1-decene-1 2,3385 130,848963 6,8346 443,75 1-decene-2 3,5522 250,661402 6,8366 443,75 1-decene-3 2,9875 194,916091 6,8166 443,75 1-decene-4 4,2012 314,728529 6,8286 443,75 1-decene-5 4,1158 306,298124 6,8066 443,75 1-decene-6 4,4049 334,837117 6,8236 443,75 1-heptanal-1 1,436 41,757157 6,2339 425,95 1-heptanal-11 1,5604 54,0375123 6,2219 425,95 1-heptanal-12 1,7971 77,4037512 6,2207 425,95 1-heptanal-13 1,523 50,3455084 6,2171 425,95 1-heptanal-14 1,7986 77,5518263 6,2387 425,95 1-heptanal-15 1,8923 86,8015795 6,2339 425,95 1-heptanol-01 0,8618 -14,9259625 6,3012 449,45 1-heptanol-02 0,8657 -14,5409674 6,3001 449,45 1-heptanol-03 0,8615 -14,9555775 6,3012 449,45 1-heptanol-04 0,8704 -14,076999 6,2989 449,45 1-heptanol-05 0,8613 -14,9753208 6,3012 449,45 1-heptanol-06 0,863 -14,8075025 6,3001 449,45 1-heptanol-07 0,8581 -15,2912142 6,3024 449,45 1-heptanol-08 1,0633 4,96544916 6,2989 449,45 1-heptanol-09 1,1645 14,9555775 6,3012 449,45 1-heptanol-10 0,9119 -9,98025666 6,2966 449,45 1-heptanol-11 0,8932 -11,8262586 6,2989 449,45 1-heptanol-12 0,8906 -12,082922 6,2989 449,45 1-heptanol-13 0,8858 -12,5567621 6,3001 449,45 1-heptanol-14 0,8915 -11,994077 6,3001 449,45 1-heptanol-15 1,1461 13,1391905 6,2942 449,45 1-heptanol-16 1,0885 7,45310958 6,2977 449,45 1-heptanol-17 1,2018 18,6377098 6,3012 449,45 1-heptanol-18 1,0337 2,04343534 6,286 449,45 1-heptanol-19 1,0609 4,72852912 6,2907 449,45 1-heptanol-20 1,0597 4,6100691 6,2919 449,45 1-heptanol-21 1,0441 3,07008885 6,2919 449,45 1-heptanol-22 1,4229 40,4639684 6,286 449,45 1-heptanol-23 1,2935 27,6900296 6,3036 449,45 1-heptanol-24 1,333 31,5893386 6,3106 449,45 1-heptanol-25 1,0365 2,31984205 6,2977 449,45 1-heptanol-26 1,3093 29,2497532 6,293 449,45 1-heptanol-27 1,4511 43,2477789 6,3001 449,45 1-heptanol 0,3457 -65,8736426 6,293 449,45 1-heptene 2,1675 113,968411 6,0972 366,79 1-hexanal-1 1,153 13,8203356 5,9579 401,45 1-hexanal-11 1,2369 22,1026654 5,9488 401,45 1-hexanal-12 1,7548 73,2280355 5,9435 401,45 1-hexanal-13 1,907 88,2527147 5,9448 401,45
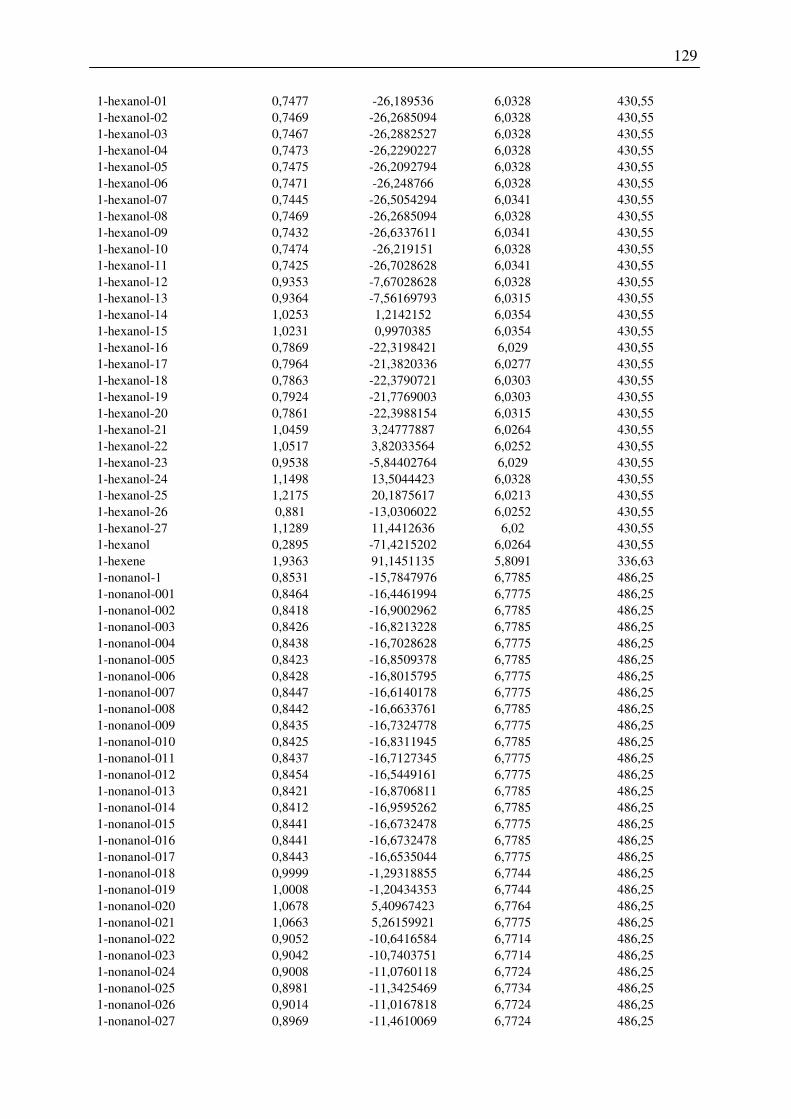
129
1-hexanol-01 0,7477 -26,189536 6,0328 430,55 1-hexanol-02 0,7469 -26,2685094 6,0328 430,55 1-hexanol-03 0,7467 -26,2882527 6,0328 430,55 1-hexanol-04 0,7473 -26,2290227 6,0328 430,55 1-hexanol-05 0,7475 -26,2092794 6,0328 430,55 1-hexanol-06 0,7471 -26,248766 6,0328 430,55 1-hexanol-07 0,7445 -26,5054294 6,0341 430,55 1-hexanol-08 0,7469 -26,2685094 6,0328 430,55 1-hexanol-09 0,7432 -26,6337611 6,0341 430,55 1-hexanol-10 0,7474 -26,219151 6,0328 430,55 1-hexanol-11 0,7425 -26,7028628 6,0341 430,55 1-hexanol-12 0,9353 -7,67028628 6,0328 430,55 1-hexanol-13 0,9364 -7,56169793 6,0315 430,55 1-hexanol-14 1,0253 1,2142152 6,0354 430,55 1-hexanol-15 1,0231 0,9970385 6,0354 430,55 1-hexanol-16 0,7869 -22,3198421 6,029 430,55 1-hexanol-17 0,7964 -21,3820336 6,0277 430,55 1-hexanol-18 0,7863 -22,3790721 6,0303 430,55 1-hexanol-19 0,7924 -21,7769003 6,0303 430,55 1-hexanol-20 0,7861 -22,3988154 6,0315 430,55 1-hexanol-21 1,0459 3,24777887 6,0264 430,55 1-hexanol-22 1,0517 3,82033564 6,0252 430,55 1-hexanol-23 0,9538 -5,84402764 6,029 430,55 1-hexanol-24 1,1498 13,5044423 6,0328 430,55 1-hexanol-25 1,2175 20,1875617 6,0213 430,55 1-hexanol-26 0,881 -13,0306022 6,0252 430,55 1-hexanol-27 1,1289 11,4412636 6,02 430,55 1-hexanol 0,2895 -71,4215202 6,0264 430,55 1-hexene 1,9363 91,1451135 5,8091 336,63 1-nonanol-1 0,8531 -15,7847976 6,7785 486,25 1-nonanol-001 0,8464 -16,4461994 6,7775 486,25 1-nonanol-002 0,8418 -16,9002962 6,7785 486,25 1-nonanol-003 0,8426 -16,8213228 6,7785 486,25 1-nonanol-004 0,8438 -16,7028628 6,7775 486,25 1-nonanol-005 0,8423 -16,8509378 6,7785 486,25 1-nonanol-006 0,8428 -16,8015795 6,7775 486,25 1-nonanol-007 0,8447 -16,6140178 6,7775 486,25 1-nonanol-008 0,8442 -16,6633761 6,7785 486,25 1-nonanol-009 0,8435 -16,7324778 6,7775 486,25 1-nonanol-010 0,8425 -16,8311945 6,7785 486,25 1-nonanol-011 0,8437 -16,7127345 6,7775 486,25 1-nonanol-012 0,8454 -16,5449161 6,7775 486,25 1-nonanol-013 0,8421 -16,8706811 6,7785 486,25 1-nonanol-014 0,8412 -16,9595262 6,7785 486,25 1-nonanol-015 0,8441 -16,6732478 6,7775 486,25 1-nonanol-016 0,8441 -16,6732478 6,7785 486,25 1-nonanol-017 0,8443 -16,6535044 6,7775 486,25 1-nonanol-018 0,9999 -1,29318855 6,7744 486,25 1-nonanol-019 1,0008 -1,20434353 6,7744 486,25 1-nonanol-020 1,0678 5,40967423 6,7764 486,25 1-nonanol-021 1,0663 5,26159921 6,7775 486,25 1-nonanol-022 0,9052 -10,6416584 6,7714 486,25 1-nonanol-023 0,9042 -10,7403751 6,7714 486,25 1-nonanol-024 0,9008 -11,0760118 6,7724 486,25 1-nonanol-025 0,8981 -11,3425469 6,7734 486,25 1-nonanol-026 0,9014 -11,0167818 6,7724 486,25 1-nonanol-027 0,8969 -11,4610069 6,7724 486,25
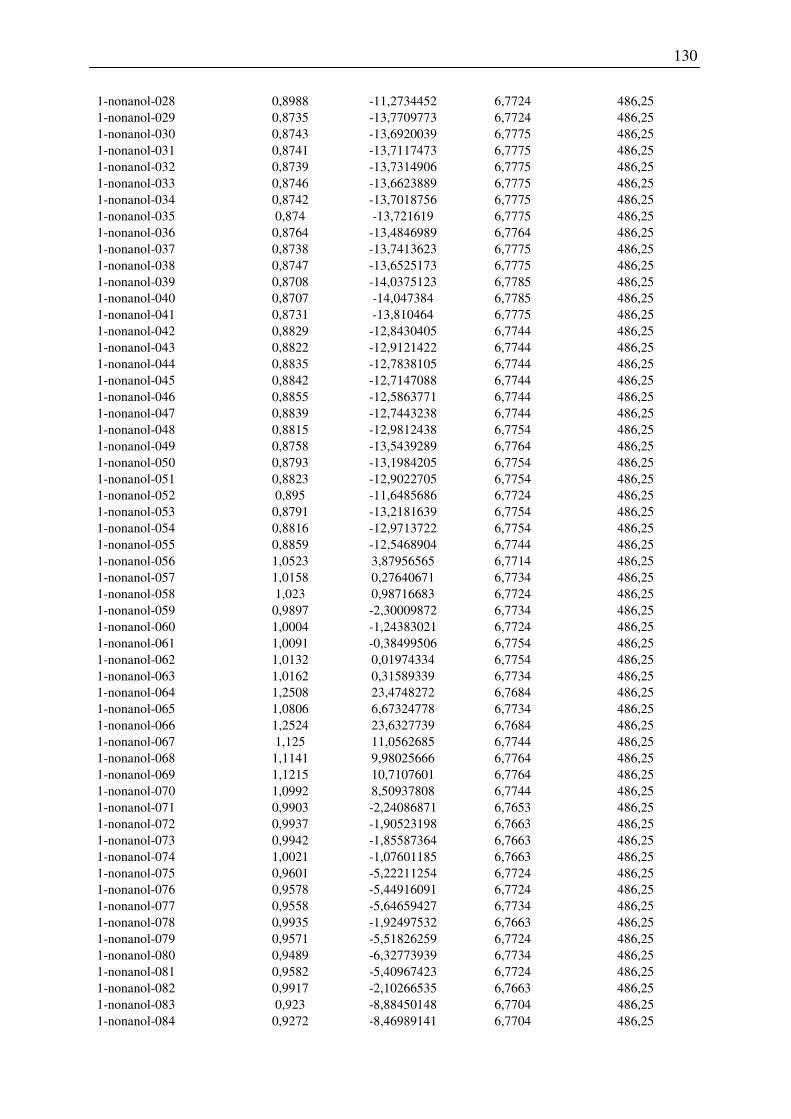
130
1-nonanol-028 0,8988 -11,2734452 6,7724 486,25 1-nonanol-029 0,8735 -13,7709773 6,7724 486,25 1-nonanol-030 0,8743 -13,6920039 6,7775 486,25 1-nonanol-031 0,8741 -13,7117473 6,7775 486,25 1-nonanol-032 0,8739 -13,7314906 6,7775 486,25 1-nonanol-033 0,8746 -13,6623889 6,7775 486,25 1-nonanol-034 0,8742 -13,7018756 6,7775 486,25 1-nonanol-035 0,874 -13,721619 6,7775 486,25 1-nonanol-036 0,8764 -13,4846989 6,7764 486,25 1-nonanol-037 0,8738 -13,7413623 6,7775 486,25 1-nonanol-038 0,8747 -13,6525173 6,7775 486,25 1-nonanol-039 0,8708 -14,0375123 6,7785 486,25 1-nonanol-040 0,8707 -14,047384 6,7785 486,25 1-nonanol-041 0,8731 -13,810464 6,7775 486,25 1-nonanol-042 0,8829 -12,8430405 6,7744 486,25 1-nonanol-043 0,8822 -12,9121422 6,7744 486,25 1-nonanol-044 0,8835 -12,7838105 6,7744 486,25 1-nonanol-045 0,8842 -12,7147088 6,7744 486,25 1-nonanol-046 0,8855 -12,5863771 6,7744 486,25 1-nonanol-047 0,8839 -12,7443238 6,7744 486,25 1-nonanol-048 0,8815 -12,9812438 6,7754 486,25 1-nonanol-049 0,8758 -13,5439289 6,7764 486,25 1-nonanol-050 0,8793 -13,1984205 6,7754 486,25 1-nonanol-051 0,8823 -12,9022705 6,7754 486,25 1-nonanol-052 0,895 -11,6485686 6,7724 486,25 1-nonanol-053 0,8791 -13,2181639 6,7754 486,25 1-nonanol-054 0,8816 -12,9713722 6,7754 486,25 1-nonanol-055 0,8859 -12,5468904 6,7744 486,25 1-nonanol-056 1,0523 3,87956565 6,7714 486,25 1-nonanol-057 1,0158 0,27640671 6,7734 486,25 1-nonanol-058 1,023 0,98716683 6,7724 486,25 1-nonanol-059 0,9897 -2,30009872 6,7734 486,25 1-nonanol-060 1,0004 -1,24383021 6,7724 486,25 1-nonanol-061 1,0091 -0,38499506 6,7754 486,25 1-nonanol-062 1,0132 0,01974334 6,7754 486,25 1-nonanol-063 1,0162 0,31589339 6,7734 486,25 1-nonanol-064 1,2508 23,4748272 6,7684 486,25 1-nonanol-065 1,0806 6,67324778 6,7734 486,25 1-nonanol-066 1,2524 23,6327739 6,7684 486,25 1-nonanol-067 1,125 11,0562685 6,7744 486,25 1-nonanol-068 1,1141 9,98025666 6,7764 486,25 1-nonanol-069 1,1215 10,7107601 6,7764 486,25 1-nonanol-070 1,0992 8,50937808 6,7744 486,25 1-nonanol-071 0,9903 -2,24086871 6,7653 486,25 1-nonanol-072 0,9937 -1,90523198 6,7663 486,25 1-nonanol-073 0,9942 -1,85587364 6,7663 486,25 1-nonanol-074 1,0021 -1,07601185 6,7663 486,25 1-nonanol-075 0,9601 -5,22211254 6,7724 486,25 1-nonanol-076 0,9578 -5,44916091 6,7724 486,25 1-nonanol-077 0,9558 -5,64659427 6,7734 486,25 1-nonanol-078 0,9935 -1,92497532 6,7663 486,25 1-nonanol-079 0,9571 -5,51826259 6,7724 486,25 1-nonanol-080 0,9489 -6,32773939 6,7734 486,25 1-nonanol-081 0,9582 -5,40967423 6,7724 486,25 1-nonanol-082 0,9917 -2,10266535 6,7663 486,25 1-nonanol-083 0,923 -8,88450148 6,7704 486,25 1-nonanol-084 0,9272 -8,46989141 6,7704 486,25
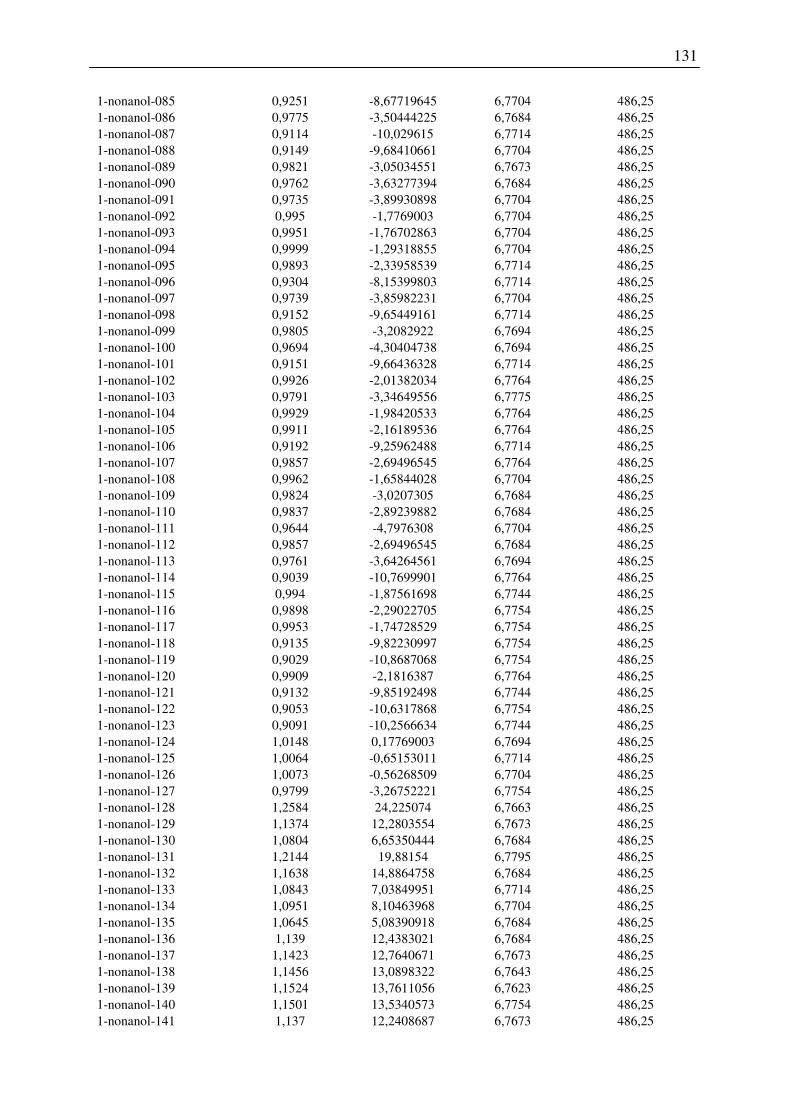
131
1-nonanol-085 0,9251 -8,67719645 6,7704 486,25 1-nonanol-086 0,9775 -3,50444225 6,7684 486,25 1-nonanol-087 0,9114 -10,029615 6,7714 486,25 1-nonanol-088 0,9149 -9,68410661 6,7704 486,25 1-nonanol-089 0,9821 -3,05034551 6,7673 486,25 1-nonanol-090 0,9762 -3,63277394 6,7684 486,25 1-nonanol-091 0,9735 -3,89930898 6,7704 486,25 1-nonanol-092 0,995 -1,7769003 6,7704 486,25 1-nonanol-093 0,9951 -1,76702863 6,7704 486,25 1-nonanol-094 0,9999 -1,29318855 6,7704 486,25 1-nonanol-095 0,9893 -2,33958539 6,7714 486,25 1-nonanol-096 0,9304 -8,15399803 6,7714 486,25 1-nonanol-097 0,9739 -3,85982231 6,7704 486,25 1-nonanol-098 0,9152 -9,65449161 6,7714 486,25 1-nonanol-099 0,9805 -3,2082922 6,7694 486,25 1-nonanol-100 0,9694 -4,30404738 6,7694 486,25 1-nonanol-101 0,9151 -9,66436328 6,7714 486,25 1-nonanol-102 0,9926 -2,01382034 6,7764 486,25 1-nonanol-103 0,9791 -3,34649556 6,7775 486,25 1-nonanol-104 0,9929 -1,98420533 6,7764 486,25 1-nonanol-105 0,9911 -2,16189536 6,7764 486,25 1-nonanol-106 0,9192 -9,25962488 6,7714 486,25 1-nonanol-107 0,9857 -2,69496545 6,7764 486,25 1-nonanol-108 0,9962 -1,65844028 6,7704 486,25 1-nonanol-109 0,9824 -3,0207305 6,7684 486,25 1-nonanol-110 0,9837 -2,89239882 6,7684 486,25 1-nonanol-111 0,9644 -4,7976308 6,7704 486,25 1-nonanol-112 0,9857 -2,69496545 6,7684 486,25 1-nonanol-113 0,9761 -3,64264561 6,7694 486,25 1-nonanol-114 0,9039 -10,7699901 6,7764 486,25 1-nonanol-115 0,994 -1,87561698 6,7744 486,25 1-nonanol-116 0,9898 -2,29022705 6,7754 486,25 1-nonanol-117 0,9953 -1,74728529 6,7754 486,25 1-nonanol-118 0,9135 -9,82230997 6,7754 486,25 1-nonanol-119 0,9029 -10,8687068 6,7754 486,25 1-nonanol-120 0,9909 -2,1816387 6,7764 486,25 1-nonanol-121 0,9132 -9,85192498 6,7744 486,25 1-nonanol-122 0,9053 -10,6317868 6,7754 486,25 1-nonanol-123 0,9091 -10,2566634 6,7744 486,25 1-nonanol-124 1,0148 0,17769003 6,7694 486,25 1-nonanol-125 1,0064 -0,65153011 6,7714 486,25 1-nonanol-126 1,0073 -0,56268509 6,7704 486,25 1-nonanol-127 0,9799 -3,26752221 6,7754 486,25 1-nonanol-128 1,2584 24,225074 6,7663 486,25 1-nonanol-129 1,1374 12,2803554 6,7673 486,25 1-nonanol-130 1,0804 6,65350444 6,7684 486,25 1-nonanol-131 1,2144 19,88154 6,7795 486,25 1-nonanol-132 1,1638 14,8864758 6,7684 486,25 1-nonanol-133 1,0843 7,03849951 6,7714 486,25 1-nonanol-134 1,0951 8,10463968 6,7704 486,25 1-nonanol-135 1,0645 5,08390918 6,7684 486,25 1-nonanol-136 1,139 12,4383021 6,7684 486,25 1-nonanol-137 1,1423 12,7640671 6,7673 486,25 1-nonanol-138 1,1456 13,0898322 6,7643 486,25 1-nonanol-139 1,1524 13,7611056 6,7623 486,25 1-nonanol-140 1,1501 13,5340573 6,7754 486,25 1-nonanol-141 1,137 12,2408687 6,7673 486,25
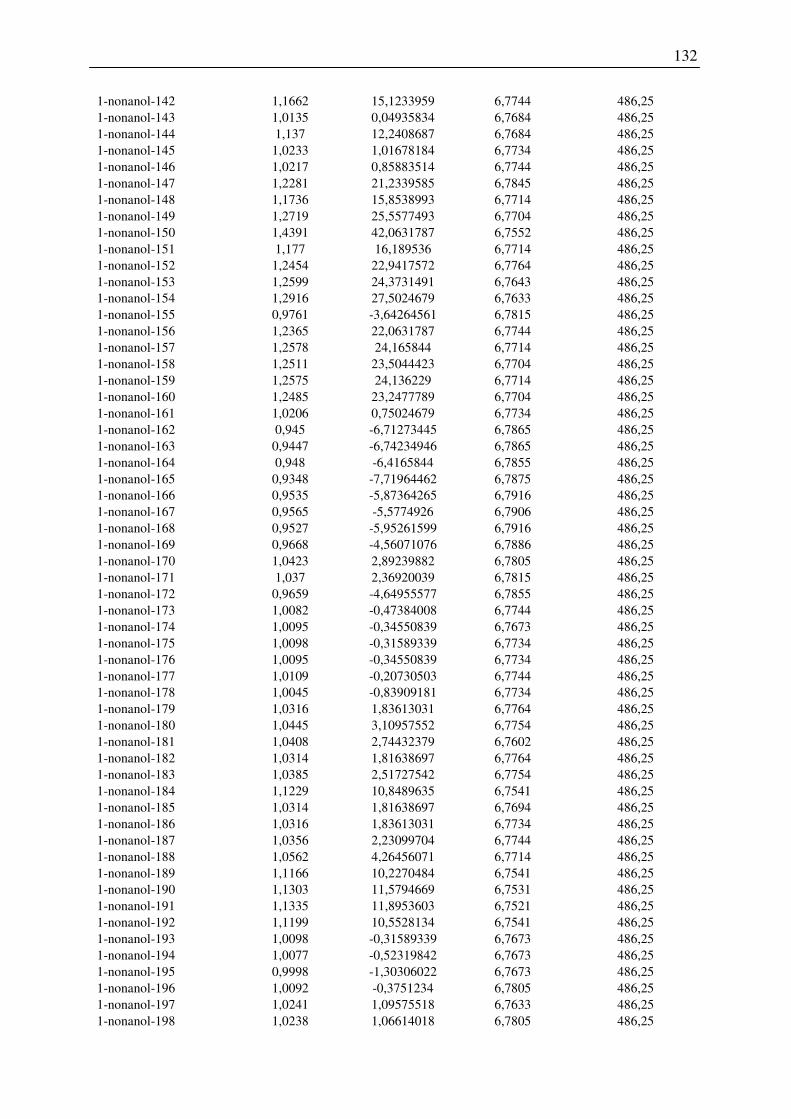
132
1-nonanol-142 1,1662 15,1233959 6,7744 486,25 1-nonanol-143 1,0135 0,04935834 6,7684 486,25 1-nonanol-144 1,137 12,2408687 6,7684 486,25 1-nonanol-145 1,0233 1,01678184 6,7734 486,25 1-nonanol-146 1,0217 0,85883514 6,7744 486,25 1-nonanol-147 1,2281 21,2339585 6,7845 486,25 1-nonanol-148 1,1736 15,8538993 6,7714 486,25 1-nonanol-149 1,2719 25,5577493 6,7704 486,25 1-nonanol-150 1,4391 42,0631787 6,7552 486,25 1-nonanol-151 1,177 16,189536 6,7714 486,25 1-nonanol-152 1,2454 22,9417572 6,7764 486,25 1-nonanol-153 1,2599 24,3731491 6,7643 486,25 1-nonanol-154 1,2916 27,5024679 6,7633 486,25 1-nonanol-155 0,9761 -3,64264561 6,7815 486,25 1-nonanol-156 1,2365 22,0631787 6,7744 486,25 1-nonanol-157 1,2578 24,165844 6,7714 486,25 1-nonanol-158 1,2511 23,5044423 6,7704 486,25 1-nonanol-159 1,2575 24,136229 6,7714 486,25 1-nonanol-160 1,2485 23,2477789 6,7704 486,25 1-nonanol-161 1,0206 0,75024679 6,7734 486,25 1-nonanol-162 0,945 -6,71273445 6,7865 486,25 1-nonanol-163 0,9447 -6,74234946 6,7865 486,25 1-nonanol-164 0,948 -6,4165844 6,7855 486,25 1-nonanol-165 0,9348 -7,71964462 6,7875 486,25 1-nonanol-166 0,9535 -5,87364265 6,7916 486,25 1-nonanol-167 0,9565 -5,5774926 6,7906 486,25 1-nonanol-168 0,9527 -5,95261599 6,7916 486,25 1-nonanol-169 0,9668 -4,56071076 6,7886 486,25 1-nonanol-170 1,0423 2,89239882 6,7805 486,25 1-nonanol-171 1,037 2,36920039 6,7815 486,25 1-nonanol-172 0,9659 -4,64955577 6,7855 486,25 1-nonanol-173 1,0082 -0,47384008 6,7744 486,25 1-nonanol-174 1,0095 -0,34550839 6,7673 486,25 1-nonanol-175 1,0098 -0,31589339 6,7734 486,25 1-nonanol-176 1,0095 -0,34550839 6,7734 486,25 1-nonanol-177 1,0109 -0,20730503 6,7744 486,25 1-nonanol-178 1,0045 -0,83909181 6,7734 486,25 1-nonanol-179 1,0316 1,83613031 6,7764 486,25 1-nonanol-180 1,0445 3,10957552 6,7754 486,25 1-nonanol-181 1,0408 2,74432379 6,7602 486,25 1-nonanol-182 1,0314 1,81638697 6,7764 486,25 1-nonanol-183 1,0385 2,51727542 6,7754 486,25 1-nonanol-184 1,1229 10,8489635 6,7541 486,25 1-nonanol-185 1,0314 1,81638697 6,7694 486,25 1-nonanol-186 1,0316 1,83613031 6,7734 486,25 1-nonanol-187 1,0356 2,23099704 6,7744 486,25 1-nonanol-188 1,0562 4,26456071 6,7714 486,25 1-nonanol-189 1,1166 10,2270484 6,7541 486,25 1-nonanol-190 1,1303 11,5794669 6,7531 486,25 1-nonanol-191 1,1335 11,8953603 6,7521 486,25 1-nonanol-192 1,1199 10,5528134 6,7541 486,25 1-nonanol-193 1,0098 -0,31589339 6,7673 486,25 1-nonanol-194 1,0077 -0,52319842 6,7673 486,25 1-nonanol-195 0,9998 -1,30306022 6,7673 486,25 1-nonanol-196 1,0092 -0,3751234 6,7805 486,25 1-nonanol-197 1,0241 1,09575518 6,7633 486,25 1-nonanol-198 1,0238 1,06614018 6,7805 486,25

133
1-nonanol-199 1,0468 3,33662389 6,7775 486,25 1-nonanol-200 1,0078 -0,51332675 6,7815 486,25 1-nonanol-201 1,0194 0,63178677 6,7684 486,25 1-nonanol-202 1,0124 -0,05923001 6,7684 486,25 1-nonanol-203 1,0193 0,6219151 6,7663 486,25 1-nonanol-204 1,0098 -0,31589339 6,7694 486,25 1-nonanol-205 1,0599 4,62981244 6,7643 486,25 1-nonanol-206 1,0695 5,5774926 6,7643 486,25 1-nonanol-207 1,0637 5,00493583 6,7643 486,25 1-nonanol-208 0,9706 -4,18558736 6,7875 486,25 1-nonanol-209 1,0275 1,43139191 6,7754 486,25 1-nonanol-210 1,0288 1,55972359 6,7744 486,25 1-nonanol-211 1,0475 3,40572557 6,7734 486,25 1-nonanol-212 1,0721 5,83415597 6,7795 486,25 1-nonanol-213 0,9701 -4,23494571 6,7865 486,25 1-nonanol-214 0,9731 -3,93879566 6,7865 486,25 1-nonanol-215 1,06 4,63968411 6,7734 486,25 1-nonanol-216 0,9765 -3,60315893 6,7875 486,25 1-nonanol-217 0,9706 -4,18558736 6,7875 486,25 1-nonanol-218 1,555 53,5044423 6,7764 486,25 1-nonanol-219 1,2725 25,6169793 6,7825 486,25 1-nonanol-220 1,1927 17,739388 6,7724 486,25 1-nonanol-221 1,3419 32,4679171 6,7602 486,25 1-nonanol-222 1,0959 8,18361303 6,7845 486,25 1-nonanol-223 1,1968 18,1441264 6,7673 486,25 1-nonanol-224 1,2001 18,4698914 6,7815 486,25 1-nonanol-225 1,1948 17,946693 6,7764 486,25 1-nonanol-226 1,4428 42,4284304 6,7541 486,25 1-nonanol-227 1,1942 17,887463 6,7825 486,25 1-nonanol-228 1,4383 41,9842053 6,7531 486,25 1-nonanol-229 1,2011 18,5686081 6,7684 486,25 1-nonanol-230 1,1529 13,810464 6,7754 486,25 1-nonanol-231 1,2422 22,6258638 6,7602 486,25 1-nonanol-232 1,2698 25,3504442 6,7744 486,25 1-nonanol-233 1,2208 20,5133268 6,7734 486,25 1-nonanol-234 1,4917 47,2556762 6,7865 486,25 1-nonanol-235 1,2335 21,7670286 6,7704 486,25 1-nonanol-236 1,158 14,3139191 6,7653 486,25 1-nonanol-237 1,3012 28,4501481 6,7491 486,25 1-nonanol-238 1,1643 14,9358342 6,7653 486,25 1-nonanol-239 1,2068 19,1312932 6,7663 486,25 1-nonanol-240 1,4741 45,5182626 6,7825 486,25 1-nonanol-241 1,2161 20,0493583 6,7643 486,25 1-nonanol-242 1,1808 16,5646594 6,7663 486,25 1-nonanol-243 1,2253 20,9575518 6,7704 486,25 1-nonanol-244 1,3309 31,3820336 6,7865 486,25 1-nonanol-245 1,2268 21,1056269 6,7795 486,25 1-nonanol-246 1,2186 20,29615 6,7785 486,25 1-nonanol-247 1,1999 18,4501481 6,7785 486,25 1-nonanol-248 1,2204 20,4738401 6,7775 486,25 1-nonanol-249 1,2063 19,0819348 6,7865 486,25 1-nonanol-250 1,402 38,4007897 6,7643 486,25 1-nonanol-251 1,3231 30,6120434 6,7724 486,25 1-nonanol-252 1,2849 26,8410661 6,7775 486,25 1-nonanol-253 1,4325 41,4116486 6,7956 486,25 1-nonanol-254 1,2494 23,3366239 6,7764 486,25 1-nonanol-255 1,4535 43,4846989 6,7936 486,25

134
1-nonanol-256 1,6052 58,4600197 6,7399 486,25 1-nonanol-257 1,2685 25,2221125 6,7795 486,25 1-nonanol-258 1,0047 -0,81934847 6,7875 486,25 1-nonanol-259 1,1157 10,1382034 6,7855 486,25 1-nonanol-260 0,9719 -4,05725568 6,7916 486,25 1-nonanol-261 0,9678 -4,46199408 6,7926 486,25 1-nonanol-262 1,0547 4,11648569 6,7734 486,25 1-nonanol-263 1,0093 -0,36525173 6,7875 486,25 1-nonanol-264 1,0041 -0,87857848 6,7875 486,25 1-nonanol-265 1,0872 7,32477789 6,7694 486,25 1-nonanol-266 1,1795 16,4363277 6,7835 486,25 1-nonanol-267 1,1605 14,5607108 6,7835 486,25 1-nonanol-268 1,1718 15,6762093 6,7835 486,25 1-nonanol-269 0,9706 -4,18558736 6,7926 486,25 1-nonanol-270 1,0969 8,28232971 6,7684 486,25 1-nonanol-271 1,0929 7,88746298 6,7734 486,25 1-nonanol-272 1,0636 4,99506417 6,7825 486,25 1-nonanol-273 1,0342 2,09279368 6,7835 486,25 1-nonanol-274 1,0315 1,82625864 6,7845 486,25 1-nonanol-275 1,1041 8,99308983 6,7724 486,25 1-nonanol-276 1,1038 8,96347483 6,7734 486,25 1-nonanol-277 1,0987 8,46001974 6,7724 486,25 1-nonanol-278 1,0655 5,18262586 6,7724 486,25 1-nonanol-279 1,0625 4,88647581 6,7744 486,25 1-nonanol-280 1,0539 4,03751234 6,7744 486,25 1-nonanol-281 1,0652 5,15301086 6,7734 486,25 1-nonanol-282 1,1147 10,0394867 6,7602 486,25 1-nonanol-283 1,1039 8,9733465 6,7714 486,25 1-nonanol-284 1,1101 9,58538993 6,7694 486,25 1-nonanol-285 1,1489 13,4155972 6,7694 486,25 1-nonanol-286 1,0975 8,34155972 6,7795 486,25 1-nonanol-287 2,0045 97,8775913 6,7875 486,25 1-nonanol-288 1,446 42,7443238 6,7855 486,25 1-nonanol-289 1,4548 43,6130306 6,7835 486,25 1-nonanol-290 1,3535 33,6130306 6,7805 486,25 1-nonanol-291 1,3624 34,4916091 6,7795 486,25 1-nonanol-292 1,3556 33,8203356 6,8086 486,25 1-nonanol-293 1,4253 40,7008885 6,7684 486,25 1-nonanol-294 1,2193 20,3652517 6,7845 486,25 1-nonanol-295 1,5227 50,3158934 6,7835 486,25 1-nonanol-296 1,1875 17,2260612 6,7886 486,25 1-nonanol-297 1,3056 28,8845015 6,7825 486,25 1-nonanol-298 1,2688 25,2517275 6,7673 486,25 1-nonanol-299 1,3139 29,70385 6,7714 486,25 1-nonanol-300 1,3524 33,5044423 6,7613 486,25 1-nonanol-301 1,2366 22,0730503 6,7734 486,25 1-nonanol-302 1,3254 30,8390918 6,7815 486,25 1-nonanol-303 1,3001 28,3415597 6,7602 486,25 1-nonanol-304 1,2883 27,1767029 6,7785 486,25 1-nonanol-305 1,4262 40,7897335 6,8106 486,25 1-nonanol-306 1,3865 36,8706811 6,7815 486,25 1-nonanol-307 1,1064 9,2201382 6,7865 486,25 1-nonanol-308 1,6188 59,8025666 6,7886 486,25 1-nonanol-309 1,3777 36,0019743 6,7855 486,25 1-nonanol-310 1,4101 39,2003949 6,7835 486,25 1-nonanol-311 1,3428 32,5567621 6,7744 486,25 1-nonanol-312 1,0434 3,00098717 6,7865 486,25
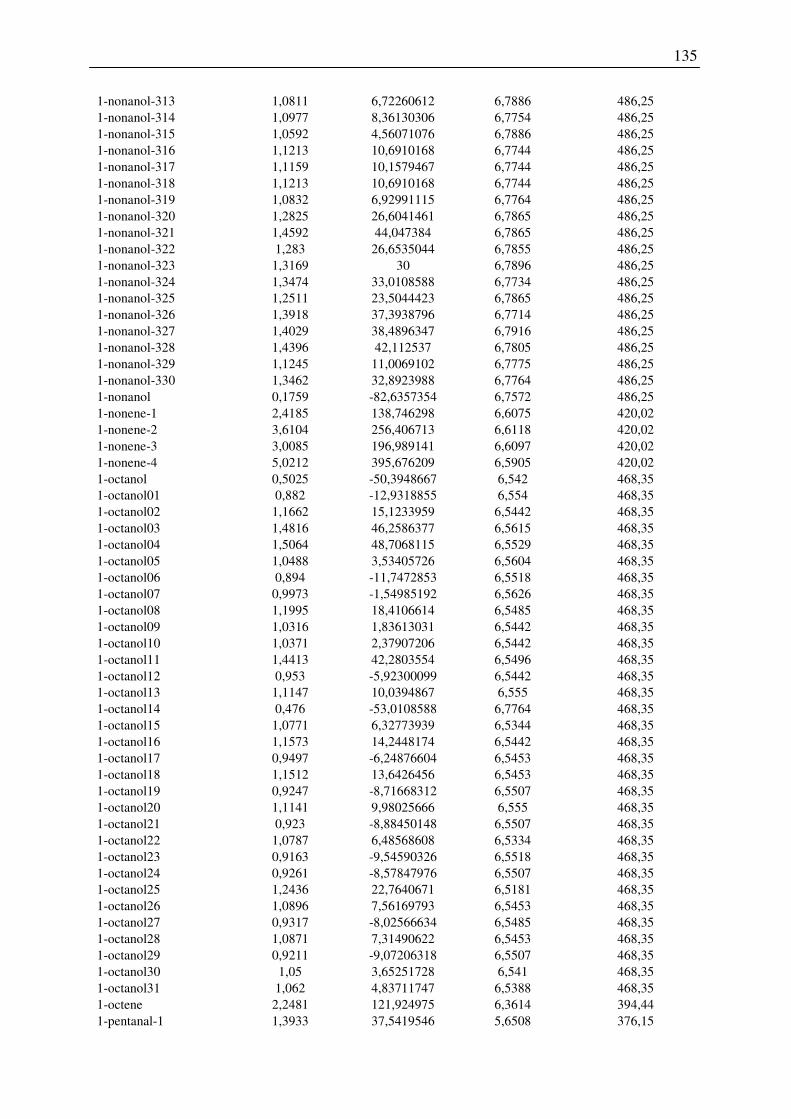
135
1-nonanol-313 1,0811 6,72260612 6,7886 486,25 1-nonanol-314 1,0977 8,36130306 6,7754 486,25 1-nonanol-315 1,0592 4,56071076 6,7886 486,25 1-nonanol-316 1,1213 10,6910168 6,7744 486,25 1-nonanol-317 1,1159 10,1579467 6,7744 486,25 1-nonanol-318 1,1213 10,6910168 6,7744 486,25 1-nonanol-319 1,0832 6,92991115 6,7764 486,25 1-nonanol-320 1,2825 26,6041461 6,7865 486,25 1-nonanol-321 1,4592 44,047384 6,7865 486,25 1-nonanol-322 1,283 26,6535044 6,7855 486,25 1-nonanol-323 1,3169 30 6,7896 486,25 1-nonanol-324 1,3474 33,0108588 6,7734 486,25 1-nonanol-325 1,2511 23,5044423 6,7865 486,25 1-nonanol-326 1,3918 37,3938796 6,7714 486,25 1-nonanol-327 1,4029 38,4896347 6,7916 486,25 1-nonanol-328 1,4396 42,112537 6,7805 486,25 1-nonanol-329 1,1245 11,0069102 6,7775 486,25 1-nonanol-330 1,3462 32,8923988 6,7764 486,25 1-nonanol 0,1759 -82,6357354 6,7572 486,25 1-nonene-1 2,4185 138,746298 6,6075 420,02 1-nonene-2 3,6104 256,406713 6,6118 420,02 1-nonene-3 3,0085 196,989141 6,6097 420,02 1-nonene-4 5,0212 395,676209 6,5905 420,02 1-octanol 0,5025 -50,3948667 6,542 468,35 1-octanol01 0,882 -12,9318855 6,554 468,35 1-octanol02 1,1662 15,1233959 6,5442 468,35 1-octanol03 1,4816 46,2586377 6,5615 468,35 1-octanol04 1,5064 48,7068115 6,5529 468,35 1-octanol05 1,0488 3,53405726 6,5604 468,35 1-octanol06 0,894 -11,7472853 6,5518 468,35 1-octanol07 0,9973 -1,54985192 6,5626 468,35 1-octanol08 1,1995 18,4106614 6,5485 468,35 1-octanol09 1,0316 1,83613031 6,5442 468,35 1-octanol10 1,0371 2,37907206 6,5442 468,35 1-octanol11 1,4413 42,2803554 6,5496 468,35 1-octanol12 0,953 -5,92300099 6,5442 468,35 1-octanol13 1,1147 10,0394867 6,555 468,35 1-octanol14 0,476 -53,0108588 6,7764 468,35 1-octanol15 1,0771 6,32773939 6,5344 468,35 1-octanol16 1,1573 14,2448174 6,5442 468,35 1-octanol17 0,9497 -6,24876604 6,5453 468,35 1-octanol18 1,1512 13,6426456 6,5453 468,35 1-octanol19 0,9247 -8,71668312 6,5507 468,35 1-octanol20 1,1141 9,98025666 6,555 468,35 1-octanol21 0,923 -8,88450148 6,5507 468,35 1-octanol22 1,0787 6,48568608 6,5334 468,35 1-octanol23 0,9163 -9,54590326 6,5518 468,35 1-octanol24 0,9261 -8,57847976 6,5507 468,35 1-octanol25 1,2436 22,7640671 6,5181 468,35 1-octanol26 1,0896 7,56169793 6,5453 468,35 1-octanol27 0,9317 -8,02566634 6,5485 468,35 1-octanol28 1,0871 7,31490622 6,5453 468,35 1-octanol29 0,9211 -9,07206318 6,5507 468,35 1-octanol30 1,05 3,65251728 6,541 468,35 1-octanol31 1,062 4,83711747 6,5388 468,35 1-octene 2,2481 121,924975 6,3614 394,44 1-pentanal-1 1,3933 37,5419546 5,6508 376,15
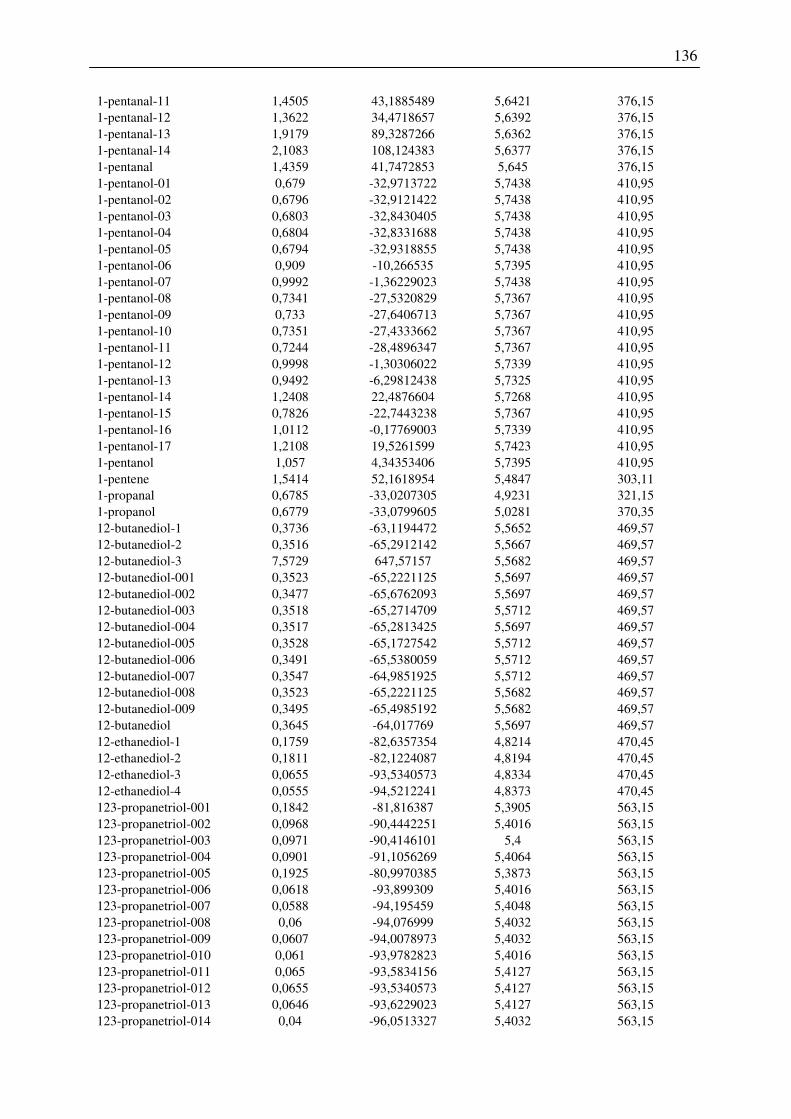
136
1-pentanal-11 1,4505 43,1885489 5,6421 376,15 1-pentanal-12 1,3622 34,4718657 5,6392 376,15 1-pentanal-13 1,9179 89,3287266 5,6362 376,15 1-pentanal-14 2,1083 108,124383 5,6377 376,15 1-pentanal 1,4359 41,7472853 5,645 376,15 1-pentanol-01 0,679 -32,9713722 5,7438 410,95 1-pentanol-02 0,6796 -32,9121422 5,7438 410,95 1-pentanol-03 0,6803 -32,8430405 5,7438 410,95 1-pentanol-04 0,6804 -32,8331688 5,7438 410,95 1-pentanol-05 0,6794 -32,9318855 5,7438 410,95 1-pentanol-06 0,909 -10,266535 5,7395 410,95 1-pentanol-07 0,9992 -1,36229023 5,7438 410,95 1-pentanol-08 0,7341 -27,5320829 5,7367 410,95 1-pentanol-09 0,733 -27,6406713 5,7367 410,95 1-pentanol-10 0,7351 -27,4333662 5,7367 410,95 1-pentanol-11 0,7244 -28,4896347 5,7367 410,95 1-pentanol-12 0,9998 -1,30306022 5,7339 410,95 1-pentanol-13 0,9492 -6,29812438 5,7325 410,95 1-pentanol-14 1,2408 22,4876604 5,7268 410,95 1-pentanol-15 0,7826 -22,7443238 5,7367 410,95 1-pentanol-16 1,0112 -0,17769003 5,7339 410,95 1-pentanol-17 1,2108 19,5261599 5,7423 410,95 1-pentanol 1,057 4,34353406 5,7395 410,95 1-pentene 1,5414 52,1618954 5,4847 303,11 1-propanal 0,6785 -33,0207305 4,9231 321,15 1-propanol 0,6779 -33,0799605 5,0281 370,35 12-butanediol-1 0,3736 -63,1194472 5,5652 469,57 12-butanediol-2 0,3516 -65,2912142 5,5667 469,57 12-butanediol-3 7,5729 647,57157 5,5682 469,57 12-butanediol-001 0,3523 -65,2221125 5,5697 469,57 12-butanediol-002 0,3477 -65,6762093 5,5697 469,57 12-butanediol-003 0,3518 -65,2714709 5,5712 469,57 12-butanediol-004 0,3517 -65,2813425 5,5697 469,57 12-butanediol-005 0,3528 -65,1727542 5,5712 469,57 12-butanediol-006 0,3491 -65,5380059 5,5712 469,57 12-butanediol-007 0,3547 -64,9851925 5,5712 469,57 12-butanediol-008 0,3523 -65,2221125 5,5682 469,57 12-butanediol-009 0,3495 -65,4985192 5,5682 469,57 12-butanediol 0,3645 -64,017769 5,5697 469,57 12-ethanediol-1 0,1759 -82,6357354 4,8214 470,45 12-ethanediol-2 0,1811 -82,1224087 4,8194 470,45 12-ethanediol-3 0,0655 -93,5340573 4,8334 470,45 12-ethanediol-4 0,0555 -94,5212241 4,8373 470,45 123-propanetriol-001 0,1842 -81,816387 5,3905 563,15 123-propanetriol-002 0,0968 -90,4442251 5,4016 563,15 123-propanetriol-003 0,0971 -90,4146101 5,4 563,15 123-propanetriol-004 0,0901 -91,1056269 5,4064 563,15 123-propanetriol-005 0,1925 -80,9970385 5,3873 563,15 123-propanetriol-006 0,0618 -93,899309 5,4016 563,15 123-propanetriol-007 0,0588 -94,195459 5,4048 563,15 123-propanetriol-008 0,06 -94,076999 5,4032 563,15 123-propanetriol-009 0,0607 -94,0078973 5,4032 563,15 123-propanetriol-010 0,061 -93,9782823 5,4016 563,15 123-propanetriol-011 0,065 -93,5834156 5,4127 563,15 123-propanetriol-012 0,0655 -93,5340573 5,4127 563,15 123-propanetriol-013 0,0646 -93,6229023 5,4127 563,15 123-propanetriol-014 0,04 -96,0513327 5,4032 563,15

137
123-propanetriol-015 0,0903 -91,0858835 5,4064 563,15 123-propanetriol-016 0,0659 -93,4945706 5,4096 563,15 123-propanetriol-017 0,036 -96,4461994 5,4143 563,15 123-propanetriol-018 0,0667 -93,4155972 5,4127 563,15 123-propanetriol-019 0,0647 -93,6130306 5,4112 563,15 123-propanetriol-020 0,0179 -98,2329714 5,4048 563,15 123-propanetriol-021 0,0904 -91,0760118 5,3969 563,15 123-propanetriol-022 0,0348 -96,5646594 5,4143 563,15 123-propanetriol-023 0,0693 -93,1589339 5,4096 563,15 123-propanetriol-024 0,0563 -94,4422507 5,4096 563,15 123-propanetriol-025 0,0667 -93,4155972 5,4064 563,15 123-propanetriol-026 0,018 -98,2230997 5,4112 563,15 123-propanetriol-027 0,0841 -91,6979269 5,4175 563,15 123-propanetriol-028 0,0525 -94,8173741 5,4175 563,15 123-propanetriol-029 0,0533 -94,7384008 5,4175 563,15 13-butanediol-1 0,1636 -83,8499506 5,5861 481,38 13-butanediol-2 0,1667 -83,5439289 5,5846 481,38 13-butanediol-3 0,4741 -53,1984205 5,5472 481,38 13-butanediol-001 0,1641 -83,8005923 5,5861 481,38 13-butanediol-002 0,1642 -83,7907206 5,5861 481,38 13-butanediol-003 0,162 -84,0078973 5,5876 481,38 13-butanediol-004 0,1654 -83,6722606 5,5846 481,38 13-butanediol-005 0,1638 -83,8302073 5,5861 481,38 13-butanediol-006 0,1644 -83,7709773 5,5861 481,38 13-butanediol-007 0,1663 -83,5834156 5,5846 481,38 13-butanediol-008 0,1654 -83,6722606 5,5846 481,38 13-butanediol-009 0,1631 -83,899309 5,5861 481,38 13-butanediol 0,0343 -96,6140178 5,5802 481,38 13-propanediol-1 0,0424 -95,8144126 5,236 460,75 14-butanediol-1 0,3196 -68,4501481 5,5817 501,15 14-butanediol-2 1,0652 5,15301086 5,5351 501,15 14-butanediol-3 0,0563 -94,4422507 5,5772 501,15 14-butanediol-001 0,3249 -67,9269497 5,5787 501,15 14-butanediol-002 0,326 -67,8183613 5,5787 501,15 14-butanediol-003 0,3234 -68,0750247 5,5802 501,15 14-butanediol-004 0,3229 -68,124383 5,5802 501,15 14-butanediol-005 0,3228 -68,1342547 5,5802 501,15 14-butanediol-006 0,3309 -67,3346496 5,5832 501,15 14-butanediol-007 0,3301 -67,4136229 5,5832 501,15 14-butanediol-008 0,3327 -67,1569595 5,5832 501,15 14-butanediol-009 0,3276 -67,6604146 5,5846 501,15 14-butanediol-010 0,3097 -69,4274432 5,5802 501,15 14-butanediol-011 0,3135 -69,0523198 5,5787 501,15 14-butanediol-012 0,3132 -69,0819348 5,5787 501,15 14-butanediol-013 0,3131 -69,0918065 5,5787 501,15 14-butanediol-014 0,6294 -37,8677196 5,5757 501,15 14-butanediol-015 0,3048 -69,911155 5,5846 501,15 14-butanediol-016 0,3055 -69,8420533 5,5846 501,15 14-butanediol-017 0,3055 -69,8420533 5,5846 501,15 14-butanediol-018 0,3056 -69,8321816 5,5757 501,15 14-butanediol-019 0,3066 -69,733465 5,5757 501,15 14-butanediol-020 0,5756 -43,1786772 5,5742 501,15 14-butanediol-021 0,7521 -25,7551826 5,5742 501,15 14-butanediol-022 0,5641 -44,3139191 5,5757 501,15 14-butanediol-023 0,2919 -71,1846002 5,5802 501,15 14-butanediol-024 0,6203 -38,7660415 5,5846 501,15 14-butanediol-025 0,6176 -39,0325765 5,5846 501,15
138
2-butanol-01 0,5365 -47,0384995 5,4159 372,7 2-butanol-02 0,5397 -46,7226061 5,4143 372,7 2-butanol-03 0,5383 -46,8608095 5,4175 372,7 2-butanol-04 0,5343 -47,2556762 5,4175 372,7 2-butanol-05 0,5387 -46,8213228 5,4175 372,7 2-butanol-06 0,5366 -47,0286278 5,4175 372,7 2-butanol-07 0,5436 -46,3376111 5,4159 372,7 2-butanol-08 0,5338 -47,3050346 5,4175 372,7 2-butanol-09 0,5345 -47,2359329 5,4175 372,7 2-butanol-10 0,5338 -47,3050346 5,4191 372,7 2-ethyl-1-butanol-1 0,7944 -21,5794669 6,02 419,65 2-ethyl-1-butanol-2 1,4105 39,2398815 6,0162 419,65 2-ethyl-1-butanol-3 1,044 3,06021718 6,0162 419,65 2-ethyl-1-butanol-4 2,4074 137,650543 6,0149 419,65 2-heptanone-1 0,9623 -5,00493583 6,2279 424,05 2-hexanone-1 0,8809 -13,0404738 5,9527 400,85 2-methyl-1-butanol-1 0,7497 -25,9921027 5,7353 401,85 2-methyl-1-butanol-2 0,9624 -4,99506417 5,7353 401,85 2-methyl-1-butanol-3 1,3595 34,2053307 5,7268 401,85 2-methyl-1-butanol 0,7719 -23,8005923 5,7325 401,85 2-methyl-2-butanol-1 0,4109 -59,4373149 5,7367 375,15 2-methyl-2-butanol-2 0,4803 -52,5863771 5,7339 375,15 2-pentanol-1 0,7785 -23,1490622 5,7353 392,15 2-pentanol-2 0,5841 -42,3395854 5,7367 392,15 2-pentanol-3 1,0725 5,87364265 5,7423 392,15 2-pentanol-4 0,6795 -32,9220138 5,7423 392,15 2-pentanol-01 0,6862 -32,260612 5,7381 392,15 2-pentanol-02 0,8122 -19,82231 5,7452 392,15 2-pentanol-03 0,5818 -42,5666338 5,7367 392,15 2-pentanol-04 0,8915 -11,994077 5,7325 392,15 2-pentanol-05 0,556 -45,1135242 5,7367 392,15 2-pentanol-06 0,653 -35,5380059 5,7409 392,15 2-pentanol-07 0,8186 -19,1905232 5,7325 392,15 2-pentanol-08 0,8186 -19,1905232 5,7325 392,15 2-pentanol-09 1,0974 8,33168806 5,7395 392,15 2-pentanol-10 1,1213 10,6910168 5,7353 392,15 2-pentanol-11 1,0182 0,51332675 5,7325 392,15 2-pentanol-12 0,7243 -28,4995064 5,748 392,15 2-pentanol-13 0,9999 -1,29318855 5,7311 392,15 2-pentanol-14 0,7004 -30,8588351 5,7423 392,15 2-pentanol-15 1,3606 34,3139191 5,724 392,15 2-pentanol-16 0,8133 -19,7137216 5,7296 392,15 2-pentanol-17 0,7998 -21,0463968 5,7282 392,15 2-pentanol-18 0,6605 -34,7976308 5,7282 392,15 2-pentanol-19 0,5523 -45,4787759 5,7325 392,15 2-pentanol-20 0,9161 -9,56564659 5,7268 392,15 2-pentanol-21 0,8027 -20,7601185 5,7311 392,15 2-pentanol-22 0,563 -44,4225074 5,7339 392,15 2-pentanol-23 0,5424 -46,4560711 5,7353 392,15 2-pentanol-24 0,6313 -37,6801579 5,7395 392,15 2-pentanol-25 0,7051 -30,3948667 5,7339 392,15 2-pentanol-26 0,7725 -23,7413623 5,7339 392,15 2-pentanol-27 0,725 -28,4304047 5,7282 392,15 2-pentanol 1,0986 8,45014808 5,7367 392,15 2-pentanone-1 0,8259 -18,4698914 5,6479 375,46 2-pentanone-2 0,9665 -4,59032577 5,6479 375,46 2-pentanone-3 1,0246 1,14511352 5,6465 375,46

139
2-pentanone-4 0,7299 -27,946693 5,661 375,46 2-pentanone-5 0,6762 -33,2477789 5,6595 375,46 2-pentanone 0,7041 -30,4935834 5,6508 375,46 23-butanediol-1 0,4596 -54,6298124 5,5712 453,85 23-butanediol-2 0,4042 -60,0987167 5,5727 453,85 23-butanediol-3 0,1921 -81,0365252 5,5802 453,85 3-methyl-1-butanol-1 0,6559 -35,2517275 5,7438 404,35 3-methyl-1-butanol-2 1,1978 18,242843 5,7438 404,35 3-methyl-1-butanol-3 1,1444 12,9713722 5,7452 404,35 3-methyl-1-butanol-4 1,2589 24,2744324 5,7367 404,35 3-pentanol-1 0,7679 -24,195459 5,7296 388,45 3-pentanol-2 0,5845 -42,3000987 5,7339 388,45 3-pentanol-3 0,524 -48,272458 5,7296 388,45 3-pentanol-4 1,0713 5,75518263 5,7183 388,45 3-pentanol-01 0,7646 -24,5212241 5,7282 388,45 3-pentanol-02 0,7535 -25,6169793 5,7296 388,45 3-pentanol-03 0,7351 -27,4333662 5,7452 388,45 3-pentanol-04 0,7151 -29,4076999 5,748 388,45 3-pentanol-05 0,7294 -27,9960513 5,7466 388,45 3-pentanol-06 0,7333 -27,6110563 5,7466 388,45 3-pentanol-07 0,7319 -27,7492596 5,7466 388,45 3-pentanol-08 0,7201 -28,9141165 5,748 388,45 3-pentanol-09 0,7309 -27,8479763 5,7466 388,45 3-pentanol-10 0,7254 -28,3909181 5,7466 388,45 3-pentanol-11 0,7288 -28,0552813 5,7466 388,45 3-pentanol-12 0,5562 -45,0937808 5,7339 388,45 3-pentanol-13 0,5473 -45,9723593 5,7353 388,45 3-pentanol-14 0,5512 -45,5873643 5,7339 388,45 3-pentanol-15 0,5518 -45,5281343 5,7339 388,45 3-pentanol-16 0,631 -37,709773 5,7423 388,45 3-pentanol 0,7813 -22,8726555 5,7296 388,45 acetone 0,4671 -53,8894373 4,9326 329,44 benzene 0,4707 -53,5340573 5,3502 353,24 di-n-butylether 1,6764 65,4886476 6,5507 413,44 di-n-propylether 1,4454 42,6850938 6,0544 362,79 diethylether 0,7419 -26,7620928 5,4048 307,58 diisopropylether-1 0,4836 -52,260612 6,0569 341,45 diisopropylether-2 0,5879 -41,964462 6,0481 341,45 diisopropylether-3 0,7532 -25,6465943 6,0162 341,45 dimethylether-1 0,4173 -58,8055281 4,6368 248,31 dimethylether-2 0,3893 -61,5695953 4,6259 248,31 ethane 0,3639 -64,076999 4,3372 184,55 ethanol 0,399 -60,6120434 4,5954 351,44 ethyl-propylether 1,0353 2,20138203 5,724 337,01 ethylbenzene 0,8012 -20,9081935 5,9879 409,35 ethylene 0,0351 -96,5350444 4,2154 169,47 m-xylene 0,906 -10,5626851 5,9736 412,27 methane 0,0017 -99,8321816 3,7442 111,66 methanol 0,249 -75,4195459 4,0777 337,85 n-butane 2,0808 105,409674 5,2241 272,65 n-butyl-ethylether 1,0506 3,71174729 6,0277 365,35 n-decane 4,3328 327,719645 6,8633 447,3 n-heptane 3,8967 284,669299 6,1504 371,58 n-hexane 4,2988 324,363277 5,8825 341,88 n-nonane 3,575 252,912142 6,6529 423,97 n-octane-1 3,7557 270,750247 6,4228 398,83 n-octane-2 4,9631 389,94077 6,4081 398,83

140
n-octane-3 4,8001 373,849951 6,4081 398,83 n-octane-4 5,9395 486,327739 6,425 398,83 n-octane-5 5,8469 477,186575 6,4171 398,83 n-octane-6 7,0337 594,343534 6,4036 398,83 n-octane 5,1251 405,932873 6,407 398,83 n-pentane 2,8258 178,953603 5,5712 309,22 n-propane 1,1504 13,5636723 4,8214 231,11 p-xylene 0,9529 -5,93287266 5,9723 411,51 propylene 0,3955 -60,9575518 4,7132 225,43
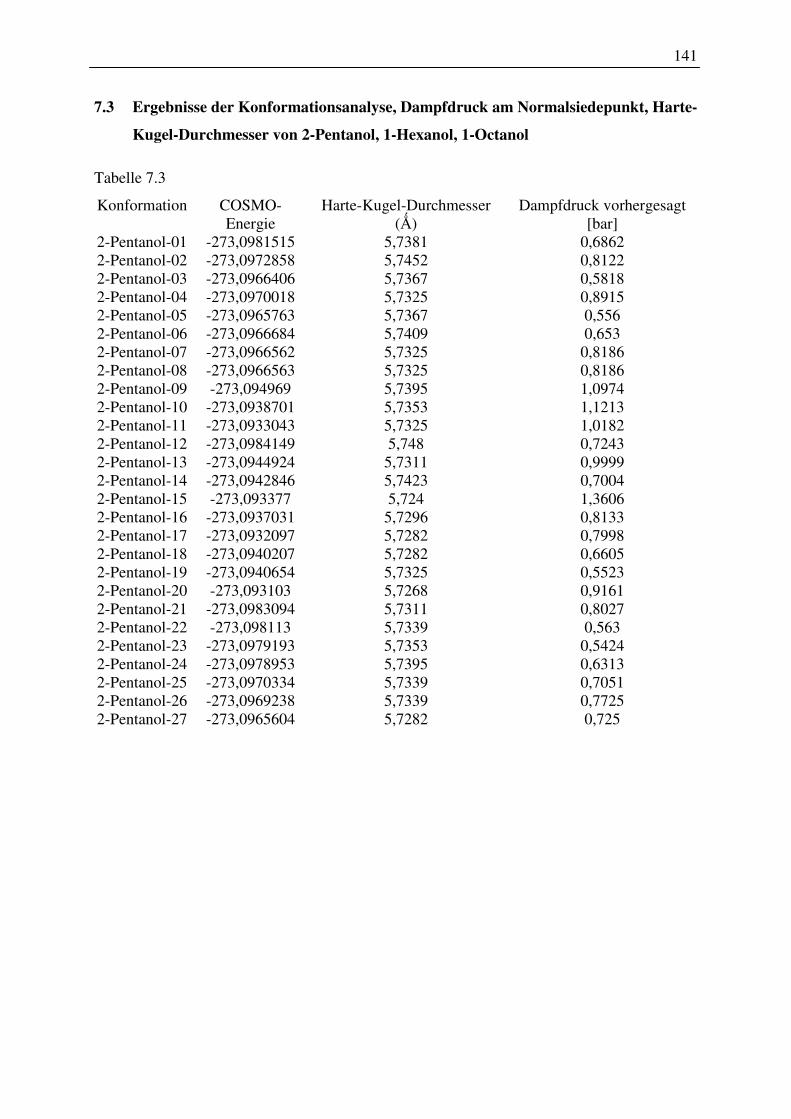
141
7.3 Ergebnisse der Konformationsanalyse, Dampfdruck am Normalsiedepunkt, Harte-
Kugel-Durchmesser von 2-Pentanol, 1-Hexanol, 1-Octanol
Tabelle 7.3
Konformation COSMO-Energie
Harte-Kugel-Durchmesser (Ǻ)
Dampfdruck vorhergesagt [bar]
2-Pentanol-01 -273,0981515 5,7381 0,6862 2-Pentanol-02 -273,0972858 5,7452 0,8122 2-Pentanol-03 -273,0966406 5,7367 0,5818 2-Pentanol-04 -273,0970018 5,7325 0,8915 2-Pentanol-05 -273,0965763 5,7367 0,556 2-Pentanol-06 -273,0966684 5,7409 0,653 2-Pentanol-07 -273,0966562 5,7325 0,8186 2-Pentanol-08 -273,0966563 5,7325 0,8186 2-Pentanol-09 -273,094969 5,7395 1,0974 2-Pentanol-10 -273,0938701 5,7353 1,1213 2-Pentanol-11 -273,0933043 5,7325 1,0182 2-Pentanol-12 -273,0984149 5,748 0,7243 2-Pentanol-13 -273,0944924 5,7311 0,9999 2-Pentanol-14 -273,0942846 5,7423 0,7004 2-Pentanol-15 -273,093377 5,724 1,3606 2-Pentanol-16 -273,0937031 5,7296 0,8133 2-Pentanol-17 -273,0932097 5,7282 0,7998 2-Pentanol-18 -273,0940207 5,7282 0,6605 2-Pentanol-19 -273,0940654 5,7325 0,5523 2-Pentanol-20 -273,093103 5,7268 0,9161 2-Pentanol-21 -273,0983094 5,7311 0,8027 2-Pentanol-22 -273,098113 5,7339 0,563 2-Pentanol-23 -273,0979193 5,7353 0,5424 2-Pentanol-24 -273,0978953 5,7395 0,6313 2-Pentanol-25 -273,0970334 5,7339 0,7051 2-Pentanol-26 -273,0969238 5,7339 0,7725 2-Pentanol-27 -273,0965604 5,7282 0,725
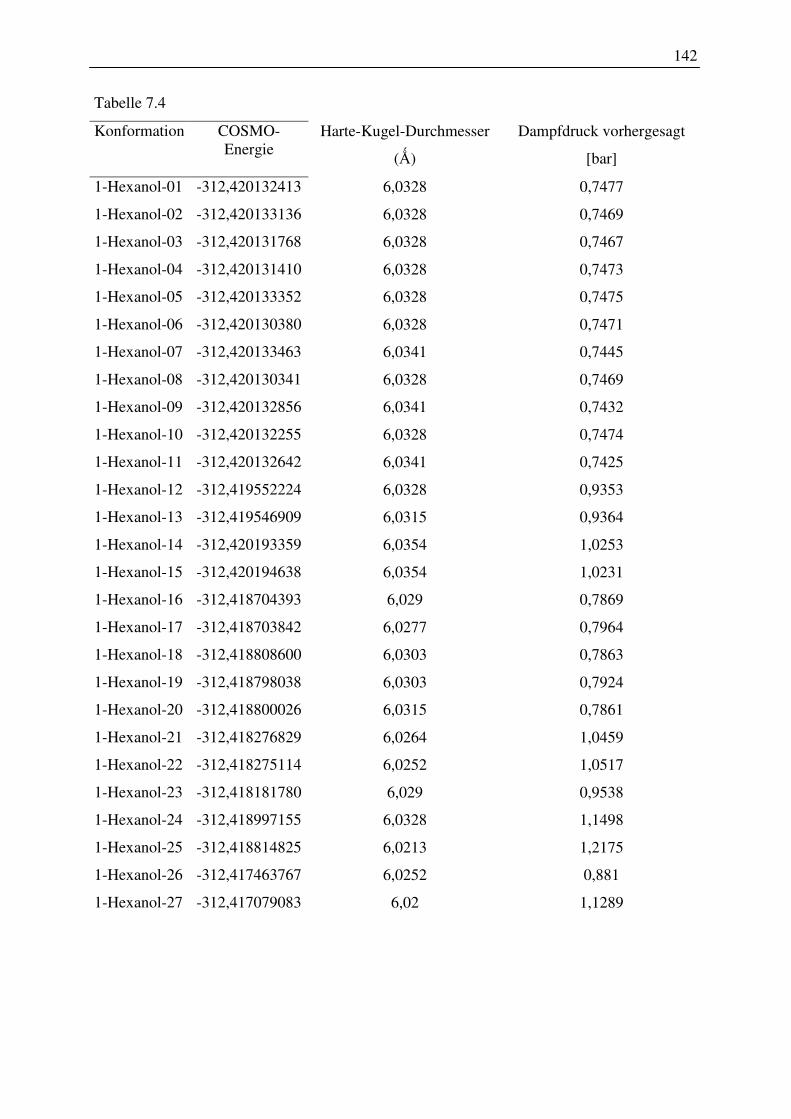
142
Tabelle 7.4
Konformation COSMO-Energie
Harte-Kugel-Durchmesser
(Ǻ)
Dampfdruck vorhergesagt
[bar]
1-Hexanol-01 -312,420132413 6,0328 0,7477
1-Hexanol-02 -312,420133136 6,0328 0,7469
1-Hexanol-03 -312,420131768 6,0328 0,7467
1-Hexanol-04 -312,420131410 6,0328 0,7473
1-Hexanol-05 -312,420133352 6,0328 0,7475
1-Hexanol-06 -312,420130380 6,0328 0,7471
1-Hexanol-07 -312,420133463 6,0341 0,7445
1-Hexanol-08 -312,420130341 6,0328 0,7469
1-Hexanol-09 -312,420132856 6,0341 0,7432
1-Hexanol-10 -312,420132255 6,0328 0,7474
1-Hexanol-11 -312,420132642 6,0341 0,7425
1-Hexanol-12 -312,419552224 6,0328 0,9353
1-Hexanol-13 -312,419546909 6,0315 0,9364
1-Hexanol-14 -312,420193359 6,0354 1,0253
1-Hexanol-15 -312,420194638 6,0354 1,0231
1-Hexanol-16 -312,418704393 6,029 0,7869
1-Hexanol-17 -312,418703842 6,0277 0,7964
1-Hexanol-18 -312,418808600 6,0303 0,7863
1-Hexanol-19 -312,418798038 6,0303 0,7924
1-Hexanol-20 -312,418800026 6,0315 0,7861
1-Hexanol-21 -312,418276829 6,0264 1,0459
1-Hexanol-22 -312,418275114 6,0252 1,0517
1-Hexanol-23 -312,418181780 6,029 0,9538
1-Hexanol-24 -312,418997155 6,0328 1,1498
1-Hexanol-25 -312,418814825 6,0213 1,2175
1-Hexanol-26 -312,417463767 6,0252 0,881
1-Hexanol-27 -312,417079083 6,02 1,1289
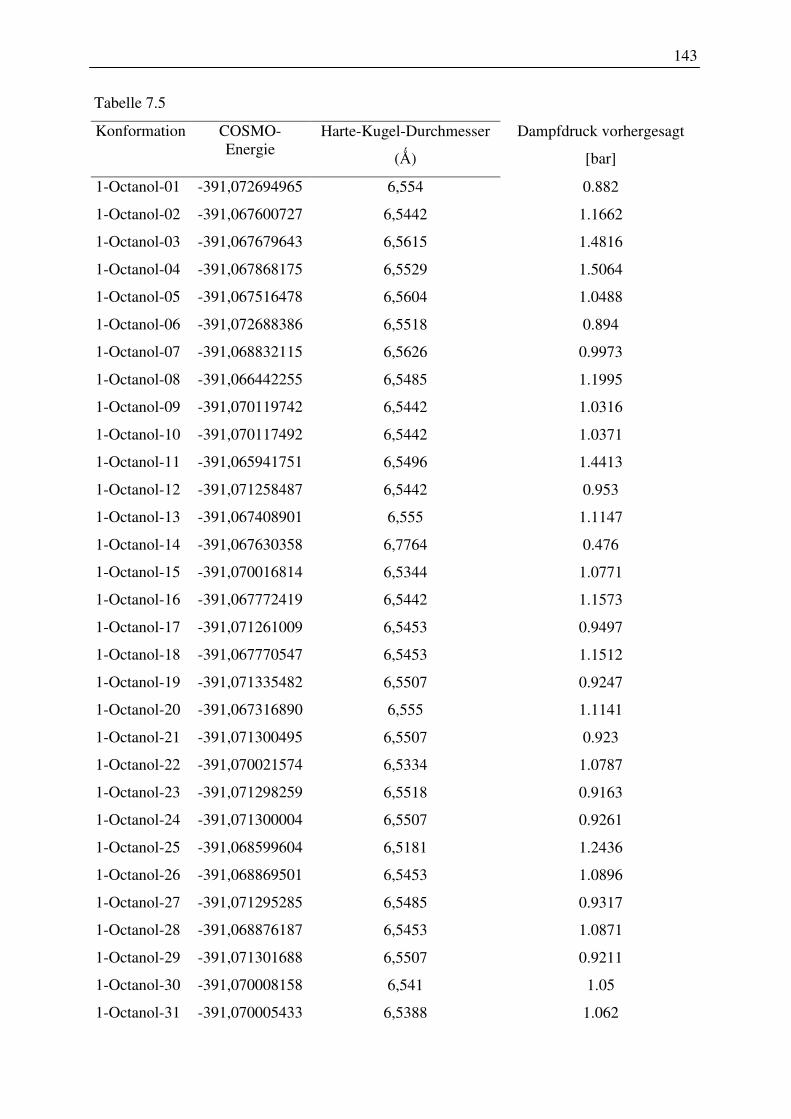
143
Tabelle 7.5
Konformation COSMO-Energie
Harte-Kugel-Durchmesser
(Ǻ)
Dampfdruck vorhergesagt
[bar]
1-Octanol-01 -391,072694965 6,554 0.882
1-Octanol-02 -391,067600727 6,5442 1.1662
1-Octanol-03 -391,067679643 6,5615 1.4816
1-Octanol-04 -391,067868175 6,5529 1.5064
1-Octanol-05 -391,067516478 6,5604 1.0488
1-Octanol-06 -391,072688386 6,5518 0.894
1-Octanol-07 -391,068832115 6,5626 0.9973
1-Octanol-08 -391,066442255 6,5485 1.1995
1-Octanol-09 -391,070119742 6,5442 1.0316
1-Octanol-10 -391,070117492 6,5442 1.0371
1-Octanol-11 -391,065941751 6,5496 1.4413
1-Octanol-12 -391,071258487 6,5442 0.953
1-Octanol-13 -391,067408901 6,555 1.1147
1-Octanol-14 -391,067630358 6,7764 0.476
1-Octanol-15 -391,070016814 6,5344 1.0771
1-Octanol-16 -391,067772419 6,5442 1.1573
1-Octanol-17 -391,071261009 6,5453 0.9497
1-Octanol-18 -391,067770547 6,5453 1.1512
1-Octanol-19 -391,071335482 6,5507 0.9247
1-Octanol-20 -391,067316890 6,555 1.1141
1-Octanol-21 -391,071300495 6,5507 0.923
1-Octanol-22 -391,070021574 6,5334 1.0787
1-Octanol-23 -391,071298259 6,5518 0.9163
1-Octanol-24 -391,071300004 6,5507 0.9261
1-Octanol-25 -391,068599604 6,5181 1.2436
1-Octanol-26 -391,068869501 6,5453 1.0896
1-Octanol-27 -391,071295285 6,5485 0.9317
1-Octanol-28 -391,068876187 6,5453 1.0871
1-Octanol-29 -391,071301688 6,5507 0.9211
1-Octanol-30 -391,070008158 6,541 1.05
1-Octanol-31 -391,070005433 6,5388 1.062

Literaturverzeichnis
Ababi, V.; Popa, A.: Equilibrium of the ternary system: lactic acid-water-organic solvent. Analele Stiint.Univ.\"A.I.Cuza,\" Iasi 6 (1960), S. 929-942
Abrams, Denis S.; Prausnitz, John M.: Statistical thermodynamics of liquid mixtures. New expression for the excess Gibbs energy of partly or completely miscible systems. AIChE
Journal 21 (1975), S. 116-128
Accelrys Inc.: Cerius2 Discover Software Programm, Accelrys Inc., San Diego CA, USA. (2005),
Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C.: Electronic-Structure Calculations on Workstation Computers - the Program System Turbomole. Chemical Physics Letters 162 (1989), S. 165-169
Allinger, Norman L.; Chen, Kuo Hsiang; Lii, Jenn Huei; Durkin, Kathleen A.: Alcohols, ethers, carbohydrates, and related compounds. I. The MM4 force field for simple compounds. Journal of Computational Chemistry 24 (2003), S. 1447-1472
Allinger, Norman L.; hen, Kuohsiang; ii, J.-H.: An improved force field (MM4) for saturated hydrocarbons. Journal of Computational Chemistry 17 (1996), S. 642-668
Allinger, Norman L.; Yuh, Young H.; Lii, Jenn Huei: Molecular mechanics. The MM3 force field for hydrocarbons. 1. Journal of the American Chemical Society 111 (1989), S. 8551-8566
Barker, John Adair; Henderson, Douglas: Perturbation theory and equation of state for fluids. Square-well potential. Journal of Chemical Physics 47 (1967), S. 2856-2861
Bartlett, R. J.: Coupled-Cluster Approach to Molecular Structure and Spectra: A Step toward Predictive Quantum Chemistry. Journal of Physical Chemistry 89 (1989), S. 1697-1708
Basak, Subhash C.;Gute, Brian D.; Grunwald, Gregory D.: Use of Topostructural, Topochemical, and Geometric Parameters in the Prediction of Vapor Pressure: A Hierarchical QSAR Approach. Journal of Chemical Information and Computer Sciences 37 (1997), S. 651-655
Bawendi, M. G.;Freed, Karl F.; Mohanty, Udayan: A lattice field theory for polymer systems with nearest-neighbor interaction energies. Journal of Chemical Physics 87 (1987), S. 5534-5540
Bayly, Christopher I.; Cieplak, Piotr; Cornell, Wendy; Kollman, Peter A.: A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. Journal of Physical Chemistry 97 (1993), S. 10269-10280
Becke, A. D.: Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Physical Review A 38 (1988), S. 3098-3100
Ben Amotz, Dor: Chemical reaction volumes in model fluid systems. 1. Hard-sphere solvation

145
and diatomic dissociation processes. Journal of Physical Chemistry 97 (1993a), S. 2314-2319
Ben Amotz, Dor; Herschbach, Dudley R.: Estimation of effective diameters for molecular fluids. Journal of Physical Chemistry 94 (1990), S. 1038-1047
Ben Amotz, Dor; Herschbach, Dudley R.: Hard fluid model for solvent-induced shifts in molecular vibrational frequencies. Journal of Physical Chemistry 97 (1993b), S. 2295-2306
Ben Amotz, Dor; Willis, Kelly G.: Molecular hard-sphere volume increments. Journal of
Physical Chemistry 97 (1993c), S. 7736-7742
Ben Naim, A.;Ting, K. L.; Jernigan, R. L.: Solvation thermodynamics of biopolymers. I. Separation of the volume and surface interactions with estimates for proteins. Biopolymers 28 (1989a), S. 1309-1325
Ben Naim, A.;Ting, K. L.; Jernigan, R. L.: Solvation thermodynamics of biopolymers. II. Correlations between functional groups. Biopolymers 28 (1989b), S. 1327-1337
Ben Naim, Arieh:Solvation thermodynamics: New York u.a.: Plenum Press, 1987
Beret, S.; Prausnitz, J. M.: Perturbed Hard-Chain Theory - Equation of State for Fluids Containing Small Or Large Molecules. AIChE Journal 21 (1975), S. 1123-1132
Bondi, A.: van der Waals volumes and radii. Journal of Physical Chemistry 68 (1964), S. 441-451
Born, M.: Volumes and hydration warmth of ions. Zeitschrift für Physik 1 (1920), S. 45-48
Born, M.; Oppenheimer, J. R.: Zur Quantentheorie der. Molekeln. Ann.Phys. 84 (1927), S. 457-
Burgath, Armin; Frey, Holger: Hyperbranched polymers. Macromolecular trees become popular. GIT Labor-Fachzeitschrift 42 (1998), S. 516-520
Butler, J. A. V.;Thomson, D. W.; Maclennan, W. H.: The free energy of the normal aliphatic alcohols in aqueous solution. I. The partial vapor pressures of aqueous solutions of methanol and propyl and butyl alcohols. II. The solubilities of some normal aliphatic alcohols in water. III. The theory of binary solutions, and its application to aqueous alcoholic solutions. Journal
of the Chemical Society (1933), S. 674-686
Cagin, Tahir; Miklis, Paul J.; Wang, Guofeng; Zamanakos, Georgios; Martin, Ryan; Li, Hao; Mainz, Daniel T.; Nagarajan, Vaidehi; Goddard, William A., III: Recent advances in simulation of dendritic polymers. Materials Research Society Symposium Proceedings 543 (1999), S. 299-310
Cagin, Tahir; Wang, Guofeng; Martin, Ryan; Breen, Nicholas; Goddard, William A., III: Molecular modelling of dendrimers for nanoscale applications. Nanotechnology 11 (2000), S. 77-84
Chang, George;Guida, Wayne C.; Still, W. Clark: An internal-coordinate Monte Carlo method for searching conformational space. Journal of the American Chemical Society 111 (1989), S. 4379-4386

146
Chapman, W. G.;Gubbins, K. E.;Jackson, G.; Radosz, M.: SAFT: equation-of-state solution model for associating fluids. Fluid Phase Equilibria 52 (1989), S. 31-38
Chen, Stephen S.; Kreglewski, Aleksander: Applications of the augmented van der Waals theory of fluids. I. Pure fluids. Berichte der Bunsen-Gesellschaft 81 (1977), S. 1048-1052
Crovetto, Rosa;Fernandez-Prini, Roberto; Japas, Maria Laura: Contribution of the cavity-formation or hard-sphere term to the solubility of simple gases in water. Journal of Physical
Chemistry 86 (1982), S. 4094-4095
Daubert, Thomas E.; Danner, Ronald P.; Design Institute for Physical Property Data; American Institute of Chemical Engineers: Physical and thermodynamic properties of pure chemicals data compilation. (1989),
de Souza, Luis E. S.;Stamatopoulou, Argyroula; Ben Amotz, Dor: Chemical potentials of hard sphere solutes in hard sphere solvents. Monte Carlo simulations and analytical approximations. Journal of Chemical Physics 100 (1994), S. 1456-1459
Dewar, Michael J. S.; Thiel, Walter: Ground states of molecules. 38. The MNDO method. Approximations and parameters. Journal of the American Chemical Society 99 (1977), S. 4899-4907
Dewar, Michael J. S.;Zoebisch, Eve G.;Healy, Eamonn F.; Stewart, James J. P.: Development and use of quantum mechanical molecular models. 76. AM1: a new general purpose quantum mechanical molecular model. Journal of the American Chemical Society 107 (1985), S. 3902-3909
Diedenhofen, Michael;Eckert, Frank; Klamt, Andreas: Prediction of Infinite Dilution Activity Coefficients of Organic Compounds in Ionic Liquids Using COSMO-RS. Journal of
Chemical and Engineering Data 48 (2003), S. 475-479
Dohrn, Ralf:Berechnung von Phasengleichgewichten: Braunschweig u.a.: Vieweg, 1994
Dreizler, Reiner M.; Gross, Eberhard K. U.:Density functional theory
an approach to the quantum many-body problem: Berlin u.a.: Springer, 1990
Dudowicz, Jacek;Freed, Karl F.; Madden, William G.: Role of molecular structure on the thermodynamic properties of melts, blends, and concentrated polymer solutions: comparison of Monte Carlo simulations with the cluster theory for the lattice model. Macromolecules 23 (1990), S. 4803-4819
Dymek, Chester J., Jr.; Stewart, James J. P.: Calculation of hydrogen-bonding interactions between ions in room-temperature molten salts. Inorganic Chemistry 28 (1989), S. 1472-1476
Eckert, F.; Klamt, A.: Fast solvent screening via quantum chemistry: COSMO-RS approach. AIChE Journal 48 (2002), S. 369-385
Eckert, Frank; Klamt, Andreas: Prediction of halocarbon thermodynamics with COSMO-RS. Fluid Phase Equilibria 210 (2003), S. 117-141
Edward, John T.: Molecular volumes and the Stokes-Einstein equation. Journal of Chemical
Education 47 (1970), S. 261-270

147
Eichkorn, Karin;Treutler, Oliver;Oehm, Holger;Haeser, Marco; Ahlrichs, Reinhart: Auxiliary basis sets to approximate Coulomb potentials. Chemical Physics Letters 240 (1995a), S. 283-290
Eichkorn, Karin;Treutler, Oliver;Oehm, Holger;Haeser, Marco; Ahlrichs, Reinhart: Auxiliary basis sets to approximate Coulomb potentials. [Erratum to document cited in CA123:93649]. Chemical Physics Letters 242 (1995b), S. 652-660
Eichkorn, Karin;Weigend, Florian;Treutler, Oliver; Ahlrichs, Reinhart: Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theoretical Chemistry Accounts 97 (1997), S. 119-124
Elbro, H. S.; Fredenslund, A.; Rasmussen, P.: A New Simple Equation for the Prediction of Solvent Activities in Polymer-Solutions. Macromolecules 23 (1990), S. 4707-4714
Franke, Robert;Krissmann, Jorg; Janowsky, Ralf: What should process engineers expect from COSMO-RS? Chemie Ingenieur Technik 74 (2002), S. 85-89
Fredenslund, A.: Unifac and Related Group-Contribution Models for Phase-Equilibria. Fluid
Phase Equilibria 52 (1989), S. 135-150
Fredenslund, A.; Jones, R. L.; Prausnitz, J. M.: Group-Contribution Estimation of Activity-Coefficients in Nonideal Liquid-Mixtures. AIChE Journal 21 (1975a), S. 1086-1099
Fredenslund, Aage;Jones, Russell L.; Prausnitz, John M.: Group-contribution estimation of activity coefficients in nonideal liquid mixtures. AIChE Journal 21 (1975b), S. 1086-1099
Freed, Karl F.; Bawendi, M. G.: Lattice theories of polymeric fluids. Journal of Physical
Chemistry 93 (1989), S. 2194-2203
Freed, Karl F.; Dudowicz, Jacek: Role of monomer structure and compressibility on the properties of multicomponent polymer blends and solutions. IV. High molecular weights, temperature dependences, and phase diagrams of binary polymer blends. Theoretica Chimica
Acta 82 (1992), S. 357-382
Freed, Karl F.; Dudowicz, Jacek: Influence of Short Chain Branching on the Miscibility of Binary Polymer Blends: Application to Polyolefin Mixtures. Macromolecules 29 (1996), S. 625-636
Gelade, Erik T. F.; Goderis, Bart; de Koster, Chris G.; Meijerink, Nico; van Benthem, Rolf A. T. M.; Fokkens, Roel; Nibbering, Nico M. M.; Mortensen, Kell: Molecular Structure Characterization of Hyperbranched Polyesteramides. Macromolecules 34 (2001), S. 3552-3558
Gogonea, Valentin;Baleanu-Gogonea, Camelia; Osawa, Eiji: Solvent hard sphere diameter from van der Waals volume. A statistical analysis of computed and solubility determined solvent diameters. THEOCHEM 432 (1998), S. 177-189
Goodman, Jonathan M.; Still, W. Clark: An unbounded systematic search of conformational space. Journal of Computational Chemistry 12 (1991), S. 1110-1117
Gross, J.; Sadowski, G.: Application of perturbation theory to a hard-chain reference fluid: an equation of state for square-well chains. Fluid Phase Equilibria 168 (2000), S. 183-199

148
Gross, Joachim:Entwicklung einer Zustandsgleichung für einfache, assoziierende und
makromolekulare Stoffe: Düsseldorf: VDI Verlag, 2001a
Gross, Joachim; Sadowski, Gabriele: Perturbed-Chain SAFT: An Equation of State Based on a Perturbation Theory for Chain Molecules. Industrial & Engineering Chemistry Research 40 (2001b), S. 1244-1260
Harris, S. J.; Tully-Smith, D. M.: Radial distribution functions and superficial quantities for rigid sphere fluids. Method of \"point\" chemical potential using scaled particle theory. Journal of Chemical Physics 55 (1971), S. 1104-1112
Hedden, Ronald C.; Bauer, Barry J.: Structure and Dimensions of PAMAM/PEG Dendrimer-Star Polymers. Macromolecules 36 (2003), S. 1829-1835
Herrington, E. F. G.: Nature (1947), S. 610-
Hirschfelder, Joseph O.;Curtis, Charles F.; Bird, R. Byron: Molecular Theory of Gases and Liquids. (1964), S. 1249-
Hoelter, D.;Burgath, A.; Frey, H.: Degree of branching in hyperbranched polymers. Acta
Polymerica 48 (1997), S. 30-35
Holderbaum, T.; Gmehling, J.: PSRK: a group contribution equation of state based on UNIFAC. Fluid Phase Equilibria 70 (1991), S. 251-265
Howlett, K. E.: The use of equilibrium constants to calculate thermodynamic quantities. II. Journal of the Chemical Society (1955), S. 1784-1789
Huang, Stanley H.; Radosz, Maciej: Equation of state for small, large, polydisperse, and associating molecules. Industrial & Engineering Chemistry Research 29 (1990), S. 2284-2294
Hult, Anders;Johansson, Mats; Malmstrom, Eva: Hyperbranched polymers. Advances in
Polymer Science 143 (1999), S. 1-34
Huron, Marie Jose; Vidal, Jean: New mixing rules in simple equations of state for representing vapor-liquid equilibriums of strongly non-ideal mixtures. Fluid Phase Equilibria
3 (1979), S. 255-271
Hypercube Inc.: HyperChem, Hypercube Inc., Florida, USA. (2005),
Iwai, Y.; Anai, Y.; Arai, Y.: Prediction of Solubilities for Volatile Hydrocarbons in Low-Density Polyethylene Using Unifac-Fv Model. Polymer Engineering and Science 21 (1981), S. 1015-1018
Jang, Jeong Gyu; Bae, Young Chan: Phase behaviors of hyperbranched polymer solutions. Polymer 40 (1999), S. 6761-6768
Jang, Jeong Gyu; Bae, Young Chan: Phase behaviors of dendrimer/solvent systems: Molecular thermodynamics approach. Journal of Chemical Physics 116 (2002a), S. 3484-3492
Jang, Jeong Gyu;Huh, Ji Yeon; Bae, Young Chan: Phase behavior of dendritic polymer/polar

149
solvent: the effects of compressibility, architecture and hydrogen bondings. Fluid Phase
Equilibria 194-197 (2002b), S. 675-688
Jork, C.;Kristen, C.;Pieraccini, D.;Stark, A.;Chiappe, C.;Beste, Y. A.; Arlt, W.: Tailor-made ionic liquids. Journal of Chemical Thermodynamics 37 (2005), S. 537-558
Katritzky, Alan R.;Wang, Yilin;Sild, Sulev;Tamm, Tarmo; Karelson, Mati: QSPR Studies on Vapor Pressure, Aqueous Solubility, and the Prediction of Water-Air Partition Coefficients. Journal of Chemical Information and Computer Sciences 38 (1998), S. 720-725
Kikic, I.;Alessi, P.;Rasmussen, P.; Fredenslund, A.: On the combinatorial part of the UNIFAC and UNIQUAC models. Canadian Journal of Chemical Engineering 58 (1980), S. 253-258
Kim, Seung Yeon; Lee, Sung Jong; Lee, Jooyoung: Conformational space annealing and an off-lattice frustrated model protein. Journal of Chemical Physics 119 (2003), S. 10274-10279
Kirkwood, John G.: The dielectric polarization of polar liquids. Journal of Chemical Physics
7 (1939), S. 911-919
Klamt, A.: Conductor-Like Screening Model for Real Solvents - A New Approach to the Quantitative Calculation of Solvation Phenomena. Journal of Physical Chemistry 99 (1995a), S. 2224-2235
Klamt, A.; Eckert, F.: COSMO-RS: a novel and efficient method for the a priori prediction of thermophysical data of liquids. Fluid Phase Equilibria 172 (2000), S. 43-72
Klamt, A.; Jonas, V.; Burger, T.; Lohrenz, J. C. W.: Refinement and parametrization of COSMO-RS. Journal of Physical Chemistry A 102 (1998), S. 5074-5085
Klamt, A.; Schuurmann, G.: Cosmo - A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. Journal of the Chemical
Society-Perkin Transactions 2 (1993), S. 799-805
Klamt, Andreas: Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. Journal of Physical Chemistry 99 (1995b), S. 2224-2235
Koch, Wolfram; Holthausen, Max C.:A chemist's guide to density functional theory: Weinheim u.a.: Wiley-VCH, 2002
Kontogeorgis, G. M.; Fredenslund, A.; Tassios, D. P.: Simple Activity-Coefficient Model for the Prediction of Solvent Activities in Polymer-Solutions. Industrial & Engineering
Chemistry Research 32 (1993), S. 362-372
Kouskoumvekaki, Irene A.;Giesen, Ralf;Michelsen, Michael L.; Kontogeorgis, Georgios M.: Free-Volume Activity Coefficient Models for Dendrimer Solutions. Industrial & Engineering
Chemistry Research 41 (2002), S. 4848-4853
Lau, Hans H.: Principles of configuration analysis. Angewandte Chemie 73 (1961), S. 423-432
Leach, Andrew R.:Molecular modelling
principles and applications: Harlow u.a.: Prentice Hall, 2006

150
Lebowitz, Joel L.;Helfand, Eugene; Praestgaard, Eigil: Scaled particle theory of fluid mixtures. Journal of Chemical Physics 43 (1965), S. 774-779
Lee, Jooyoung; Scheraga, Harold A.: Conformational space annealing by parallel computations: Extensive conformational search of met-enkephalin and of the 20-residue membrane-bound portion of melittin. International Journal of Quantum Chemistry 75 (1999), S. 255-265
Lee, Kyoungrim; Czaplewski, Cezary; Kim, Seung Yeon; Lee, Jooyoung: An efficient molecular docking using conformational space annealing. Journal of Computational
Chemistry 26 (2004), S. 78-87
Lescanec, Robert L.; Muthukumar, M.: Configurational characteristics and scaling behavior of starburst molecules: a computational study. Macromolecules 23 (1990), S. 2280-2288
Liang, Cikui; Gallagher, David A.: QSPR prediction of vapor pressure from solely theoretically-derived descriptors. Journal of Chemical Information and Computer Sciences 38 (1998), S. 321-324
Lii, Jenn Huei; Allinger, Norman L.: Molecular mechanics. The MM3 force field for hydrocarbons. 2. Vibrational frequencies and thermodynamics. Journal of the American
Chemical Society 111 (1989a), S. 8566-8575
Lii, Jenn Huei; Allinger, Norman L.: Molecular mechanics. The MM3 force field for hydrocarbons. 3. The van der Waals' potentials and crystal data for aliphatic and aromatic hydrocarbons. Journal of the American Chemical Society 111 (1989b), S. 8576-8582
Lindvig, T.; Hestkjaer, L. L.; Hansen, A. F.; Michelsen, M. L.; Kontogeorgis, G. M.: Phase equilibria for complex polymer solutions. Fluid Phase Equilibria 194 (2002), S. 663-673
Lucas, Michel: Size effect in transfer of nonpolar solutes from gas or solvent to another solvent with a view on hydrophobic behavior. Journal of Physical Chemistry 80 (1976), S. 359-362
Maassen, Siegmar:Experimentelle Bestimmung und Korrelierung von Verteilungskoeffizienten
in verdünnten Lösungen: Aachen: 1996
Mansfield, Marc L.; Klushin, Leonid I.: Monte Carlo studies of dendrimer macromolecules. Macromolecules 26 (1993), S. 4262-4268
Mayer, S. W.: A molecular parameter relation between surface tension and liquid compressibility. Journal of Physical Chemistry 67 (1963), S. 2160-2164
Michelsen, Michael L.: A method for incorporating excess Gibbs energy models in equations of state. Fluid Phase Equilibria 60 (1990a), S. 47-58
Michelsen, Michael L.: A modified Huron-Vidal mixing rule for cubic equations of state. Fluid Phase Equilibria 60 (1990b), S. 213-219
Miertus, S.;Scrocco, E.; Tomasi, J.: Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chemical Physics 55 (1981), S. 117-129

151
Monev, Valentin; Spassova, Milena; Champagne, Benoit: Charge distributions in polyacetylene chains containing a positively charged defect. International Journal of
Quantum Chemistry 104 (2005), S. 354-366
Morel-Desrosiers, Nicole; Morel, Jean Pierre: Evaluation of thermodynamic functions relative to cavity formation in liquids: uses and misuses of Scaled Particle Theory. Canadian Journal
of Chemistry 59 (1981), S. 1-7
Naidoo, Kevin J.; Hughes, Samantha J.; Moss, John R.: Computational Investigations into the Potential Use of Poly(benzyl phenyl ether) Dendrimers as Supports for Organometallic Catalysts. Macromolecules 32 (1999), S. 331-341
Nayeem, Akbar; Vila, Jorge; Scheraga, Harold A.: A comparative study of the simulated-annealing and Monte Carlo-with-minimization approaches to the minimum-energy structures of polypeptides: [Met]-enkephalin. Journal of Computational Chemistry 12 (1991), S. 594-605
Naylor, Adel M.;Goddard, William A., III;Kiefer, Garry E.; Tomalia, Donald A.: Starburst dendrimers. 5. Molecular shape control. Journal of the American Chemical Society 111 (1989), S. 2339-2341
Oishi, Takeru; Prausnitz, John M.: Estimation of solvent activities in polymer solutions using a group-contribution method. Industrial & Engineering Chemistry Process Design and
Development 17 (1978), S. 333-339
Onsager, Lars: Electric moments of molecules in liquids. Journal of the American Chemical
Society 58 (1936), S. 1486-1493
Orbey, Hasan; Sandler, Stanley I.: On the combination of equation of state and excess free energy models. Fluid Phase Equilibria 111 (1995a), S. 53-70
Orbey, Hasan; Sandler, Stanley I.: Reformulation of Wong-Sandler mixing rule for cubic equations of state. AIChE Journal 41 (1995b), S. 683-690
Orbey, Hasan;Sandler, Stanley I.; Wong, David Shan Hill: Accurate equation of state predictions at high temperatures and pressures using the existing UNIFAC model. Fluid
Phase Equilibria 85 (1993), S. 41-54
Ortiz, Wilfredo; Roitberg, Adrian E.; Krause, Jeffrey L.: Molecular Dynamics of Poly(benzylphenyl ether) Dendrimers: Effects of Backfolding on Foerster Energy-Transfer Rates. Journal of Physical Chemistry B 108 (2004), S. 8218-8225
Parr, Robert G.; Yang, Weitao:Density-functional theory of atoms and molecules: Oxford: Oxford Univ. Press u.a., 1989
Pauling, Linus:The nature of the chemical bond and the structure of molecules and crystals
an introduction to modern structural chemistry: Ithaca, New York: Cornell Univ. Press, 1993
Peneloux, Andre;Abdoul, Wahabou; Rauzy, Evelyne: Excess functions and equations of state. Fluid Phase Equilibria 47 (1989), S. 115-132
Peng, Ding Yu; Robinson, Donald B.: A new two-constant equation of state. Industrial &

152
Engineering Chemistry Fundamentals 15 (1976), S. 59-64
Perdew, J. P.: Density-Functional Approximation for the Correlation-Energy of the Inhomogeneous Electron-Gas. Physical Review B 33 (1986), S. 8822-8824
Pierotti, Robert A.: Aqueous solutions of nonpolar gases. Journal of Physical Chemistry 69 (1965), S. 281-288
Pierotti, Robert A.: A scaled particle theory of aqueous and nonaqueous solutions. Chemical
Reviews (Washington, DC, United States) 76 (1976), S. 717-726
Pillardy, Jaroslaw; Czaplewski, Cezary; Wedemeyer, William J.; Scheraga, Harold A.: Conformation-family Monte Carlo (CFMC): an efficient computational method for identifying the low-energy states of a macromolecule. Helvetica Chimica Acta 83 (2000), S. 2214-2230
Poling, Bruce E.; Prausnitz, John M.; O'Connell, John Paul:The properties of gases and
liquids: New York, NY u.a.: McGraw-Hill, 2001
Ponder, Jay: TINKER - Software Tools for Molecular Design. (2005),
Postma, Johan P. M.;Berendsen, Herman J. C.; Haak, Jan R.: Thermodynamics of cavity formation in water: a molecular dynamics study. Faraday Symposia of the Chemical Society
(1982), S. 55-67
Prausnitz, John Michael; Lichtenthaler, Ruediger N.; Azevedo, Edmundo Gomes de:Molecular thermodynamics of fluid-phase equilibria: Upper Saddle River, NJ u.a.: Prentice-Hall PTR, 1999
Rasmussen, D.; Rasmussen, P.: Phase-Equilibria in Aqueous Polymer-Solutions. Chemical
Engineering Progress 85 (1989), S. 50-56
Redlich, O.; Kister, A. T.: Ind.Eng.Chem. 40 (1948), S. 345-
Reiss, H.;Frisch, H. L.; Lebowitz, J. L.: Statistical mechanics of rigid spheres. Journal of
Chemical Physics 31 (1959), S. 369-380
Reiss, H.; Mayer, S. W.: Theory of surface tension of molten salts. Journal of Chemical
Physics 34 (1961), S. 2001-2003
Reiss, H.; Tully-Smith, D. M.: Further development of scaled particle theory for rigid spheres. Application of the statistical thermodynamics of curved surfaces. Journal of Chemical
Physics 55 (1971), S. 1674-1689
Reiss, Howard: Scaled particle methods in the statistical thermodynamics of fluids. Advances
in Chemical Physics 9 (1965), S. 1-82
Reiss, Howard;Frisch, H. L.;Helfand, E.; Lebowitz, J. L.: Aspects of the statistical thermodynamics of real fluids. Journal of Chemical Physics 32 (1960), S. 119-124
Roos, Björn O.; European Summer School:Lecture notes in quantum chemistry: Berlin u.a.: Springer, 1992

153
Sandler, Stanley I.;Lin, Shiang Tai; Sum, Amadeu K.: The use of quantum chemistry to predict phase behavior for environmental and process engineering. Fluid Phase Equilibria
194-197 (2002), S. 61-75
Schafer, A.; Huber, C.; Ahlrichs, R.: Fully Optimized Contracted Gaussian-Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. Journal of Chemical Physics 100 (1994), S. 5829-5835
Schafer, Ansgar;Klamt, Andreas;Sattel, Diana;Lohrenz, John C. W.; Eckert, Frank: COSMO Implementation in TURBOMOLE: Extension of an efficient quantum chemical code towards liquid systems. Physical Chemistry Chemical Physics 2 (2000), S. 2187-2193
Scherrenberg, Rolf;Coussens, Betty;van Vliet, Paul;Edouard, Guillaume;Brackman, Josephine;de Brabander, Ellen; Mortensen, Kell: The Molecular Characteristics of Poly(propyleneimine) Dendrimers As Studied with Small-Angle Neutron Scattering, Viscosimetry, and Molecular Dynamics. Macromolecules 31 (1998), S. 456-461
Schrödinger, E.: Quantisierung als Eigenwertproblem. Ann.Phys. 79 (1926), S. 361-
Seiler, M.;Kohler, D.; Arlt, W.: Hyperbranched polymers: new selective solvents for extractive distillation and solvent extraction. Separation and Purification Technology 29 (2002a), S. 245-263
Seiler, Matthias: Dendritic polymers - interdisciplinary research and emerging applications from unique structural properties. Chemical Engineering & Technology 25 (2002b), S. 237-253
Seiler, Matthias:Phase behavior and new applications of hyperbranched polymers in the field
of chemical engineering: 2004a
Seiler, Matthias;Rolker, Jorn;Mokrushina, Lyudmila V.;Kautz, Holger;Frey, Holger; Arlt, Wolfgang: Vapor-liquid equilibria in dendrimer and hyperbranched polymer solutions: experimental data and modeling using UNIFAC-FV. Fluid Phase Equilib. 221 (2004b), S. 83-96
Seiler, Matthias;Rolker, Jorn;Mokrushina, Lyudmila V.;Kautz, Holger;Frey, Holger; Arlt, Wolfgang: Vapor-liquid equilibria in dendrimer and hyperbranched polymer solutions: experimental data and modeling using UNIFAC-FV. Fluid Phase Equilibria 221 (2004c), S. 83-96
Soave, Giorgio: Equilibrium constants from a modified Redlich-Kwong equation of state. Chemical Engineering Science 27 (1972), S. 1197-1203
Somasundara, Rao K.; Ramana, Rao M. V.; Venkata, Rao C.: J.Sci.Ind.Res. 20b (1961), S. 283-
Stewart, James J. P.: Optimization of parameters for semiempirical methods. I. Method. Journal of Computational Chemistry 10 (1989a), S. 209-220
Stewart, James J. P.: Optimization of parameters for semiempirical methods. II. Applications. Journal of Computational Chemistry 10 (1989b), S. 221-264
Stewart, James J. P.: Optimization of parameters for semiempirical methods. III. Extension of

154
PM3 to beryllium, magnesium, zinc, gallium, germanium, arsenic, selenium, cadmium, indium, tin, antimony, tellurium, mercury, thallium, lead, and bismuth. Journal of
Computational Chemistry 12 (1991), S. 320-341
Stryjek, R.; Vera, J. H.: PRSV - an improved Peng-Robinson equation of state with new mixing rules for strongly nonideal mixtures. Canadian Journal of Chemical Engineering 64 (1986a), S. 334-340
Stryjek, R.; Vera, J. H.: PRSV2: a cubic equation of state for accurate vapor-liquid equilibria calculations. Canadian Journal of Chemical Engineering 64 (1986b), S. 820-826
Stryjek, R.; Vera, J. H.: PRSV: an improved Peng-Robinson equation of state for pure compounds and mixtures. Canadian Journal of Chemical Engineering 64 (1986c), S. 323-333
Stryjek, R.; Vera, J. H.: Vapor-liquid equilibrium of hydrochloric acid solutions with the PRSV equation of state. Fluid Phase Equilibria 25 (1986d), S. 279-290
Sum, Amadeu K.; Sandler, Stanley I.: A Novel Approach to Phase Equilibria Predictions Using Ab Initio Methods. Industrial & Engineering Chemistry Research 38 (1999), S. 2849-2855
Szabo, Attila; Ostlund, Neil S.:Modern quantum chemistry. Introduction to advanced
electronic structure theory: New York ;Hamburg: McGraw-Hill, 1989
Tande, Brian M.;Deitcher, Robert W., Jr.;Sandler, Stanley I.; Wagner, Norman J.: UNIFAC-FV Applied to Dendritic Macromolecules in Solution: Comment on \"Vapor-Liquid Equilibria for Dendritic-Polymer Solutions\" (Lieu, J. G.; Liu, M.; Frecuechet, J. M. J.; Prausnitz, J. M. J. Chem. Eng. Data 1999, 44, 613-620). Journal of Chemical and Engineering Data 47 (2002), S. 376-377
Tang, Karen E. S.; Bloomfield, Victor A.: Excluded volume in solvation: sensitivity of scaled-particle theory to solvent size and density. Biophysical Journal 79 (2000), S. 2222-2234
Tomalia, Donald A.;Naylor, Adel M.; Goddard, William A., III: Starburst dendrimers: control of size, shape, surface chemistry, topology and flexibility in the conversion of atoms to macroscopic materials. Angewandte Chemie 102 (1990), S. 119-157
Tomasi, Jacopo; Persico, Maurizio: Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chemical Reviews (Washington,
DC, United States) 94 (1994), S. 2027-2094
Topp, Andreas; Bauer, Barry J.; Klimash, June W.; Spindler, Ralph; Tomalia, Donald A.; Amis, Eric J.: Probing the Location of the Terminal Groups of Dendrimers in Dilute Solution. Macromolecules 32 (1999a), S. 7226-7231
Topp, Andreas;Bauer, Barry J.;Prosa, Ty J.;Scherrenberg, Rolf; Amis, Eric J.: Size Change of Dendrimers in Concentrated Solution. Macromolecules 32 (1999b), S. 8923-8931
Topp, Andreas; Bauer, Barry J.; Tomalia, Donald A.; Amis, Eric J.: Effect of Solvent Quality on the Molecular Dimensions of PAMAM Dendrimers. Macromolecules 32 (1999c), S. 7232-7237

155
Trosset, Jean Yves; Scheraga, Harold A.: Reaching the global minimum in docking simulations: a Monte Carlo energy minimization approach using Bezier spline. Proceedings
of the National Academy of Sciences of the United States of America 95 (1998), S. 8011-8015
Tully-Smith, D. M.; Reiss, H.: Further development of scaled particle theory of rigid sphere fluids. Journal of Chemical Physics 53 (1970), S. 4015-4025
van Benthem, Rolf A. T. M.; Meijerink, Nico; Gelade, Erik; de Koster, Chris G.; Muscat, Dirk; Froehling, Peter E.; Hendriks, Patrick H. M.; Vermeulen, Carlo J. A. A.; Zwartkruis, Theo J. G.: Synthesis and Characterization of Bis(2-hydroxypropyl)amide-Based Hyperbranched Polyesteramides. Macromolecules 34 (2001), S. 3559-3566
Vidal, Jean: Mixing rules and excess properties in cubic equations of state. Chemical
Engineering Science 33 (1978), S. 787-791
Vrabec, Jadran; Hasse, Hans: Grand equilibrium: Vapour-liquid equilibria by a new molecular simulation method. Molecular Physics 100 (2002), S. 3375-3383
Wang, Junmei; Cieplak, Piotr; Kollman, Peter A.: How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? Journal of Computational Chemistry 21 (2000), S. 1049-1074
Westheimer, F. H.; Mayer, Joseph E.: The theory of the racemization of optically active derivatives of biphenyl. Journal of Chemical Physics 14 (1946), S. 733-738
Wilhelm, Emmerich; Battino, Rubin: Solvophobic interaction. Journal of Chemical Physics
56 (1972), S. 563-566
Wittig, Roland;Lohmann, Juergen; Gmehling, Juergen: Vapor-Liquid Equilibria by UNIFAC Group Contribution. 6. Revision and Extension. Industrial & Engineering Chemistry
Research 42 (2003), S. 183-188
Wolbach, Jeffrey P.; Sandler, Stanley I.: Using Molecular Orbital Calculations To Describe the Phase Behavior of Hydrogen-Bonding Fluids. Industrial & Engineering Chemistry
Research 36 (1997), S. 4041-4051
Wolbach, Jeffrey P.; Sandler, Stanley I.: Using Molecular Orbital Calculations To Describe the Phase Behavior of Cross-associating Mixtures. Industrial & Engineering Chemistry
Research 37 (1998), S. 2917-2928
Wong, David Shan Hill;Orbey, Hasan; Sandler, Stanley I.: Equation of state mixing rule for nonideal mixtures using available activity coefficient model parameters and that allows extrapolation over large ranges of temperature and pressure. Industrial & Engineering
Chemistry Research 31 (1992a), S. 2033-2039
Wong, David Shan Hill; Sandler, Stanley I.: A theoretically correct mixing rule for cubic equations of state. AIChE Journal 38 (1992b), S. 671-680
Zhuravleva, I. K.;Zhuravlev, E. F.; Lomakina, N. G.: Phase diagrams of ternary liquid systems containing three binary phase separations with upper critical dissolution points. Zhurnal Fizicheskoi Khimii 51 (1977), S. 1700-1707


















