
Drucksensitive Arzneiformen für das Drug Targeting in den Darm · Drucksensitive Arzneiformen für...
149
Drucksensitive Arzneiformen für das Drug‐Targeting in den Darm Inauguraldissertation zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) der Mathematisch‐Naturwissenschaftlichen Fakultät der Ernst‐Moritz‐Arndt‐Universität Greifswald vorgelegt von Lisa Wilde geboren am 25.05.1984 in Potsdam Greifswald, 20. September 2013
Transcript of Drucksensitive Arzneiformen für das Drug Targeting in den Darm · Drucksensitive Arzneiformen für...

Drucksensitive Arzneiformen für das Drug‐Targeting in den Darm
I n a u g u r a l d i s s e r t a t i o n
zur
Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften
(Dr. rer. nat.)
der
Mathematisch‐Naturwissenschaftlichen Fakultät
der
Ernst‐Moritz‐Arndt‐Universität Greifswald
vorgelegt von Lisa Wilde geboren am 25.05.1984 in Potsdam
Greifswald, 20. September 2013

Dekan: Prof. Dr. Klaus Fesser 1. Gutachter: Prof. Dr. Werner Weitschies 2. Gutachter: Prof. Dr. Jörg Breitkreutz Tag der Promotion: 29.11.2013

Meinen Eltern


Inhaltsverzeichnis
I
Inhaltsverzeichnis
ABKÜRZUNGSVERZEICHNIS ............................................................................................................ IV
1 EINLEITUNG ............................................................................................................................... 1
1.1 PHYSIOLOGISCHE GRUNDLAGEN ..................................................................................................... 1
1.2 PRINZIPIEN DES DARM‐TARGETINGS ............................................................................................... 5
1.3 ZIELSTELLUNG ............................................................................................................................ 9
2 MATERIAL UND METHODEN .................................................................................................... 11
2.1 MATERIALIEN ZUR HERSTELLUNG DER ARZNEIFORMEN ...................................................................... 11
2.1.1 HARTFETT ..................................................................................................................................... 11
2.1.2 POLYETHYLENGLYCOL ..................................................................................................................... 11
2.1.3 TRISTEARIN ................................................................................................................................... 12
2.1.4 AGAR ........................................................................................................................................... 12
2.1.5 DICKFLÜSSIGES PARAFFIN ................................................................................................................ 13
2.1.6 NATRIUMALGINAT.......................................................................................................................... 13
2.1.7 CELLULOSEACETAT ......................................................................................................................... 13
2.1.8 ETHYLCELLULOSE ........................................................................................................................... 14
2.1.9 TRIACETIN .................................................................................................................................... 14
2.1.10 KAUGUMMI‐GRUNDMASSE ........................................................................................................... 15
2.1.11 RIBOFLAVINPHOSPHAT‐NATRIUM ................................................................................................... 16
2.1.12 PARACETAMOL ............................................................................................................................ 17
2.1.13 HYDROXYETHYLCELLULOSE ............................................................................................................ 17
2.1.14 MIKROKRISTALLINE CELLULOSE ...................................................................................................... 18
2.1.15 TALKUM ..................................................................................................................................... 18
2.1.16 HOCHDISPERSES SILICIUMDIOXID .................................................................................................... 19
2.1.17 MAGNESIUMSTEARAT ................................................................................................................... 19
2.2 HERSTELLUNGSVERFAHREN DER ARZNEIFORMEN .............................................................................. 19
2.2.1 AGAR‐KUGELN .............................................................................................................................. 19
2.2.2 HARTFETT‐W32‐ UND PEG‐1000‐KUGELN ....................................................................................... 20
2.2.3 PARAFFIN‐KAPSELN ........................................................................................................................ 24
2.2.4 TRISTEARIN‐KAPSELN...................................................................................................................... 26
2.2.5 KAUGUMMI‐MANTELTABLETTEN ...................................................................................................... 28
2.2.6 PLACEBO‐TABLETTEN...................................................................................................................... 30
2.2.7 BERECHNUNG DER OBERFLÄCHEN ..................................................................................................... 31
2.3 METHODEN ZUR UNTERSUCHUNG DER ARZNEIFORMEN ..................................................................... 32
2.3.1 GEHALTSBESTIMMUNG ................................................................................................................... 32
2.3.1.1 Paracetamol in Agar‐Kugeln ................................................................................................... 32
2.3.1.2 Riboflavinphosphat in Hartfett‐W32‐Kugeln ......................................................................... 33
2.3.1.3 Riboflavinphosphat in PEG‐1000‐Kugeln ............................................................................... 34

Inhaltsverzeichnis
II
2.3.1.4 Riboflavinphosphat in Paraffin‐Kapseln ................................................................................. 35
2.3.1.5 Paracetamol in Tristearin‐Kapseln ......................................................................................... 36
2.3.1.6 Riboflavinphosphat in Kaugummi‐Manteltabletten .............................................................. 36
2.3.2 BRUCHFESTIGKEIT MITTELS TEXTURE ANALYSER .................................................................................. 37
2.3.3 WIRKSTOFFFREISETZUNG MITTELS PADDLE‐APPARATUR ....................................................................... 38
2.3.4 STRESSTEST‐APPARATUR ................................................................................................................. 40
2.3.4.1 Prüfung der Bruchfestigkeit ................................................................................................... 41
2.3.4.2 Prüfung der Wirkstofffreisetzung ........................................................................................... 42
2.3.5 RASTERELEKTRONENMIKROSKOPIE .................................................................................................... 43
3 ERGEBNISSE ............................................................................................................................. 44
3.1 AGAR‐KUGELN ......................................................................................................................... 44
3.1.1 GEHALTSBESTIMMUNG ................................................................................................................... 44
3.1.2 BRUCHFESTIGKEIT MITTELS TEXTURE ANALYSER .................................................................................. 44
3.1.2.1 Bruchfestigkeit in Abhängigkeit von der Agar‐Konzentration ............................................... 44
3.1.2.2 Bruchfestigkeit in Abhängigkeit von der Trocknungszeit ....................................................... 46
3.1.3 BRUCHFESTIGKEIT MITTELS STRESSTEST‐APPARATUR ............................................................................ 47
3.1.3.1 Bruchfestigkeit in Abhängigkeit von der Agar‐Konzentration ............................................... 47
3.1.3.2 Bruchfestigkeit in Abhängigkeit von der Trocknungszeit ....................................................... 49
3.1.4 WIRKSTOFFFREISETZUNG MITTELS PADDLE‐APPARATUR ....................................................................... 51
3.1.5 WIRKSTOFFFREISETZUNG MITTELS STRESSTEST‐APPARATUR .................................................................. 53
3.2 HARTFETT‐W32‐KUGELN ........................................................................................................... 54
3.2.1 GEHALTSBESTIMMUNGEN ............................................................................................................... 54
3.2.2 BRUCHFESTIGKEIT MITTELS TEXTURE ANALYSER .................................................................................. 55
3.2.3 BRUCHFESTIGKEIT MITTELS STRESSTEST‐APPARATUR ............................................................................ 58
3.2.4 WIRKSTOFFFREISETZUNG MITTELS PADDLE‐APPARATUR ....................................................................... 62
3.2.5 WIRKSTOFFFREISETZUNG MITTELS STRESSTEST‐APPARATUR .................................................................. 62
3.3 PEG‐1000‐KUGELN .................................................................................................................. 64
3.3.1 GEHALTSBESTIMMUNG ................................................................................................................... 64
3.3.2 BRUCHFESTIGKEIT MITTELS TEXTURE ANALYSER .................................................................................. 64
3.3.3 BRUCHFESTIGKEIT MITTELS STRESSTEST‐APPARATUR ............................................................................ 66
3.3.4 WIRKSTOFFFREISETZUNG MITTELS PADDLE‐APPARATUR ....................................................................... 67
3.3.5 WIRKSTOFFFREISETZUNG MITTELS STRESSTEST‐APPARATUR .................................................................. 68
3.4 PARAFFIN‐KAPSELN ................................................................................................................... 69
3.4.1 GEHALTSBESTIMMUNG ................................................................................................................... 69
3.4.2 BRUCHFESTIGKEIT MITTELS TEXTURE ANALYSER .................................................................................. 69
3.4.3 BRUCHFESTIGKEIT MITTELS STRESSTEST‐APPARATUR ............................................................................ 70
3.4.4 WIRKSTOFFFREISETZUNG MITTELS PADDLE‐APPARATUR ....................................................................... 71
3.4.5 WIRKSTOFFFREISETZUNG MITTELS STRESSTEST‐APPARATUR .................................................................. 71
3.5 TRISTEARIN‐KAPSELN ................................................................................................................. 72
3.5.1 GEHALTSBESTIMMUNG ................................................................................................................... 72
3.5.2 BRUCHFESTIGKEIT MITTELS TEXTURE ANALYSER .................................................................................. 72
3.5.3 BRUCHFESTIGKEIT MITTELS STRESSTEST‐APPARATUR ............................................................................ 74
3.5.4 WIRKSTOFFFREISETZUNG MITTELS PADDLE‐APPARATUR ....................................................................... 77

Inhaltsverzeichnis
III
3.5.5 WIRKSTOFFFREISETZUNG MITTELS STRESSTEST‐APPARATUR .................................................................. 77
3.6 KAUGUMMI‐MANTELTABLETTEN .................................................................................................. 79
3.6.1 GEHALTSBESTIMMUNG ................................................................................................................... 79
3.6.2 BRUCHFESTIGKEIT MITTELS TEXTURE ANALYSER .................................................................................. 79
3.6.3 BRUCHFESTIGKEIT MITTELS STRESSTEST‐APPARATUR ............................................................................ 82
3.6.4 WIRKSTOFFFREISETZUNG MITTELS PADDLE‐APPARATUR ....................................................................... 83
3.6.5 WIRKSTOFFFREISETZUNG MITTELS STRESSTEST‐APPARATUR .................................................................. 83
4 DISKUSSION ............................................................................................................................. 87
4.1 AGAR‐KUGELN ......................................................................................................................... 88
4.2 HARTFETT‐W32‐ UND PEG‐1000‐KUGELN .................................................................................... 91
4.3 PARAFFIN‐KAPSELN ................................................................................................................. 102
4.4 TRISTEARIN‐KAPSELN ............................................................................................................... 106
4.5 KAUGUMMI‐MANTELTABLETTEN ................................................................................................ 110
5 ZUSAMMENFASSUNG ............................................................................................................ 116
LITERATURVERZEICHNIS ..................................................................................................................... 120
ANHANG ....................................................................................................................................... 128
ABBILDUNGSVERZEICHNIS .......................................................................................................................... 130
TABELLENVERZEICHNIS .............................................................................................................................. 134
GERÄTEVERZEICHNIS ................................................................................................................................. 136
CHEMIKALIENVERZEICHNIS ......................................................................................................................... 137
EIDESSTATTLICHE ERKLÄRUNG ............................................................................................................ 139
DANKSAGUNG ................................................................................................................................ 140
VERÖFFENTLICHUNGEN ..................................................................................................................... 141

Abkürzungsverzeichnis
IV
Abkürzungsverzeichnis
Abb. Abbildung
CA Celluloseacetat
CFU colony forming units (koloniebildende Einheiten)
DDRS destructive force dependent release system
EC Ethylcellulose
FAD Flavin‐adenin‐dinucleotid
FDA Food and Drug Administration (Arzneimittelzulassungsbehörde der USA)
FMN Flavin‐mononucleotid
HEC Hydroxyethylcellulose
IMMC Interdigestive Migrating Motor Complex (interdigestiver migrierender
myoelektrischer Motorkomplex)
KF Kalibrationsfaktor
KOH Kaliumhydroxid
MCC Mikrokristalline Cellulose
MW Mittelwert
PCDC pressure‐controlled colon delivery capsule
PEG Polyethylenglycol
Ph. Eur. Europäisches Arzneibuch (Pharmacopoea Europaea)
REM Rasterelektronenmikroskop
RFP Riboflavinphosphat
rpm revolutions per minute (Umdrehungen pro Minute)
SD standard deviation (Standardabweichung)
SGFsp simulated gastric fluid sine pepsin (simulierte Magenflüssigkeit ohne Pepsin)
Tab. Tabelle
UEG Untere Explosionsgrenze
USP Arzneibuch der Vereinigten Staaten von Amerika (United States Pharmacopeia)
VF Verdünnungsfaktor

1 Einleitung
1
1 Einleitung
1.1 Physiologische Grundlagen
Aufgenommene Nahrung sowie orale therapeutische Systeme durchlaufen mit ihrer Einnahme die
verschiedenen Bereiche des Gastrointestinaltraktes. Dieser besteht aus Oropharynx (Mund‐Rachen‐
Raum), Ösophagus (Speiseröhre), Magen, Dünn‐ und Dickdarm und wird gespeist durch Sekrete aus
den Ausführungsgängen verschiedener exkretorischer Drüsen. Nach der willkürlichen oralen Phase
des Schluckaktes folgt eine reflektorische Phase, in der die Nahrung oder die eingenommene feste
orale Arzneiform durch peristaltische Wellen in den Magen befördert wird. Die Geschwindigkeit der
ösophagialen Passage ist abhängig von der Konsistenz des Geschluckten und kann zwischen einer
Sekunde bei Flüssigkeiten oder zehn Sekunden bei festen Partikeln liegen [1]. Ein Verschlussdruck
von 50 – 100 mmHg am oberen Ösophagus verhindert das Eindringen von Luft und steigt nach distal
an, sodass am unteren Ösophagus Drücke zwischen 30 – 120 mmHg vorherrschen. Eine
Druckdifferenz von 15 – 25 mmHg am unteren Ösophagus‐Sphinkter zum Magenfundus verhindert
den Reflux von Mageninhalt in die Speiseröhre. Während der Mund‐Rachen‐Raum für die
Zerkleinerung der Nahrung und durch Einspeicheln für die Gleitfähigkeit durch die Speiseröhre
verantwortlich ist, dient der Magen der Speicherung, der weiteren Zerkleinerung und
Homogenisierung des Nahrungsbreis und entleert ihn nach einer Verweildauer, die abhängig von
dessen Zusammensetzung ist, über den Pylorus ins Duodenum. Der Magen, dessen Aufbau in Abb. 1
zu erkennen ist, wird untergliedert in die Abschnitte Kardia (Mageneingang), Fundus (Magenkuppel),
Korpus (Magenkörper), Antrum (Erweiterung vor dem Magenausgang) und Pylorus (Magenpförtner).
Abb. 1: Links: Anatomischer Aufbau des Magens [2]. Rechts: Querschnitt durch den Pylorus [3].
Während der Fundus und der obere Korpus eine tonische Wandspannung aufbauen, die sich dem
jeweiligen Füllungszustand des Magens anpasst, befinden sich im Hauptteil des Korpus myogene
Schrittmacherzellen, die im 20‐s‐Rhythmus Potentialwellen auslösen, die sich beim Überschreiten
einer Schwelle als peristaltische Kontraktionswellen in Richtung Pylorus ausbreiten. Die motorische

1 Einleitung
2
und sekretorische Aktivität des Gastrointestinaltraktes wird von einem eigenen enterischen
Nervensystem gesteuert. Zwischen den Mahlzeiten treten interdigestive wandernde myoelektrische
Motorkomplexe (interdigestive migrating motor complex = IMMC) auf, die teilweise ungerichtet sind,
aber durch propulsive analwärts gerichtete Kontraktionswellen für die Entleerung des Magens und
Dünndarms sorgen. Dabei werden neben Nahrungsresten auch Bakterienansammlungen und
unverdauliche Fremdkörper vor der Aktivitätsfront nach distal befördert, was zu einer Reinigung des
Magen‐Darm‐Traktes führt und unter anderem die übermäßige Keimbesiedlung des Dünndarms
verhindert [1].
Der IMMC besteht aus drei Phasen, die sich zyklisch aneinander reihen und sich in Anzahl und Stärke
der Kontraktionen unterscheiden. Phase I kennzeichnet sich durch motorische Ruhe und dauert
zwischen 10 und 15 min [4, 5]. In der sich anschließenden Phase II herrscht mittlere
Kontraktionsaktivität, während Phase III aus mehreren Minuten starker Kontraktionen besteht, die
im Antrum starten und sich progressiv bis in den Dünndarm ausbreiten. Die Aktivitätsfront eines
Zyklus des IMMC breitet sich innerhalb von 1 – 1,5 h vom Duodenum zum Ileum aus und beginnt
dann wieder von Neuem. Bei Nahrungszufuhr wird dieser Kreislauf unterbrochen und ersetzt durch
stärkere und häufigere Kontraktionen in Antrum und Dünndarm [1, 4‐6].
Während Flüssigkeiten in Abhängigkeit vom Druckgradienten zwischen proximalem Magen und
Duodenum entleert werden, erfolgt die Entleerung von festen Bestandteilen in Abhängigkeit von der
Partikelgröße. Durch vielfache Retropulsion des Nahrungsbreis, der durch peristaltische Wellen mit
einer Frequenz von 3/min Richtung Pylorus geschoben wird, werden feste Partikel zerkleinert und
durchmischt. Die sogenannte „Antrummühle“ führt zu einer Zerkleinerung der Partikel bis auf eine
Größe von < 2 mm, bei der sie dann durch synchrone Erschlaffung der Pylorusmuskulatur beim
Eintreffen antraler, peristaltischer Wellen entleert werden können. Große Partikel, die nicht
zerkleinert werden können, werden innerhalb der Phase III des IMMC aufgrund starker
Antrumkontraktionen durch den Pylorus ins Duodenum entleert [1, 6‐8]. Der Pylorus, welcher als
Schließmuskel aus einer dicken Schicht Ringmuskulatur besteht, kann sich durch Kontraktion
verschließen und somit die Durchfuhr von Nahrungsbestandteilen und unverdaulichen Festkörpern in
das Duodenum, dessen anatomischer Aufbau in Abb. 2 zu erkennen ist, steuern. Das Duodenum
(Zwölffingerdarm), welches den proximalen Teil des Dünndarms darstellt, wird nach distal gefolgt
von Jejunum (Leerdarm) und Ileum (Krummdarm), welches daran anschließend über die
Ileocaecalklappe mit dem Dickdarm verbunden ist. Der Dünndarm ist durch seine Länge von etwa
3,75 m im tonisierten und 6 ‐ 7 m im relaxierten Zustand und seine Oberflächenvergrößerung durch
Kerckringfalten, Zotten und Mikrovilli mit einer Gesamtoberfläche von 200 m2 für die Absorption von
Nahrungsbestandteilen und Arzneistoffen verantwortlich. Dagegen steht der Magen als
Resorptionsorgan nicht zur Verfügung und auch der Dickdarm ist nur für einige Wirkstoffe geeignet

1 Einleitung
3
[1, 9, 10]. Die Resorption von Monosacchariden erfolgt sehr schnell und ist im oberen Dünndarm fast
vollständig abgeschlossen. Ebenso werden die Spaltprodukte des Nahrungseiweißes zu 50 – 60 % und
die der Lipide zu 95 % im Duodenum resorbiert [1]. Einige Wirkstoffe werden bevorzugt in
bestimmten Bereichen des Gastrointestinaltraktes resorbiert und weisen wie z.B. L‐Dopa, Furosemid
und Riboflavin ein Absorptionsfenster im oberen Dünndarm auf [10, 11].
Abb. 2: Anatomischer Aufbau des Duodenums [2].
Der Dickdarm mit einer Länge von etwa 125 cm besteht aus dem Caecum (Blinddarm), dem Colon
(Grimmdarm) mit seinem aufsteigenden (Colon ascendens), quer‐verlaufenden (C. transversum),
absteigenden (C. descendens) und S‐förmigen (C. sigmoideum) Abschnitt und dem Rektum
(Enddarm). Er ist vor allem für die Rückresorption von Wasser und die damit verbundene Eindickung
des Darminhaltes verantwortlich [1, 6] und zeichnet sich durch ein besonders starkes
Bakterienwachstum aus.
Die menschliche Darmflora, deren Zusammensetzung durch Umwelteinflüsse und genotypische
Ausprägung beeinflusst wird, besteht aus etwa 500 verschiedenen bakteriellen Arten, wobei 3 – 40
Hauptspezies bis zu 99 % der Gesamtpopulation ausmachen [12, 13]. Die Bakterienbesiedlung im
Gastrointestinaltrakt unterscheidet sich hinsichtlich ihrer Ausprägung sehr zwischen den einzelnen
Abschnitten. Während im Magen nur geringe Mengen von 102 koloniebildenden Einheiten pro
Gramm (CFU/g) zu finden sind, liegt die mikrobielle Dichte im proximalen Dünndarm luminal bei
etwa 104 CFU/ml, erhöht sich im distalen Teil auf 108 Bakterien/ml und ergibt im Colon etwa 1011 –
1012 CFU/g, wobei die anaeroben Bakterien um etwa 3 Log‐Stufen häufiger vertreten sind, als die
Aerobier [12‐16]. Diese Mikroflora ist vor allem dafür verantwortlich, dass durch Fermentation
unverdaute Kohlenhydrate und Proteine abgebaut werden [15].
Mit Hilfe der sogenannten Smartpill, die 2006 von der amerikanischen
Arzneimittelzulassungsbehörde (Food and Drug Administration, FDA) für die Bewertung von
Magenentleerung und Passagezeiten durch den Gastrointestinaltrakt zugelassen wurde, wurden von
verschiedenen Arbeitskreisen Studien durchgeführt, die Erkenntnisse zu den pH‐, Temperatur‐ und
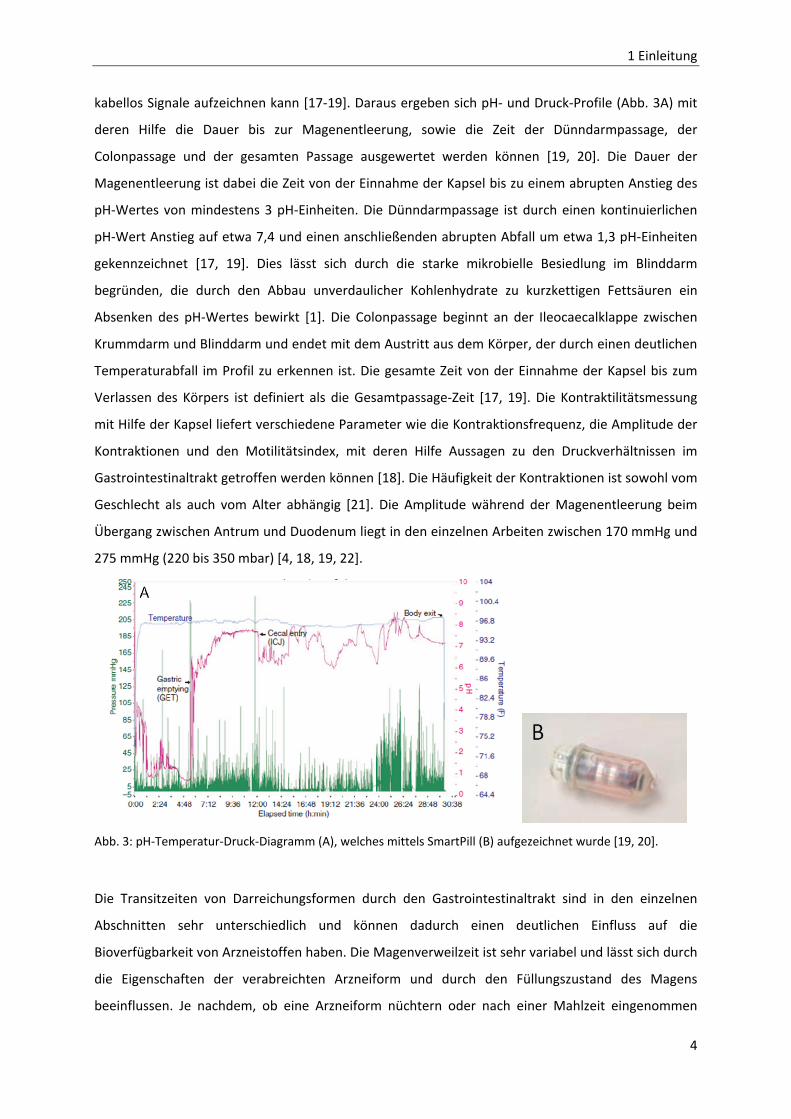
Druck‐Verhältnissen lieferten. Die Smartpill (Abb. 3B) ist eine 26 mm x 13 mm kleine unverdauliche
Kapsel, die nach Einnahme auf dem Weg durch den Gastrointestinaltrakt über bis zu fünf Tage
1 Einleitung
4
kabellos Signale aufzeichnen kann [17‐19]. Daraus ergeben sich pH‐ und Druck‐Profile (Abb. 3A) mit
deren Hilfe die Dauer bis zur Magenentleerung, sowie die Zeit der Dünndarmpassage, der
Colonpassage und der gesamten Passage ausgewertet werden können [19, 20]. Die Dauer der
Magenentleerung ist dabei die Zeit von der Einnahme der Kapsel bis zu einem abrupten Anstieg des
pH‐Wertes von mindestens 3 pH‐Einheiten. Die Dünndarmpassage ist durch einen kontinuierlichen
pH‐Wert Anstieg auf etwa 7,4 und einen anschließenden abrupten Abfall um etwa 1,3 pH‐Einheiten
gekennzeichnet [17, 19]. Dies lässt sich durch die starke mikrobielle Besiedlung im Blinddarm
begründen, die durch den Abbau unverdaulicher Kohlenhydrate zu kurzkettigen Fettsäuren ein
Absenken des pH‐Wertes bewirkt [1]. Die Colonpassage beginnt an der Ileocaecalklappe zwischen
Krummdarm und Blinddarm und endet mit dem Austritt aus dem Körper, der durch einen deutlichen
Temperaturabfall im Profil zu erkennen ist. Die gesamte Zeit von der Einnahme der Kapsel bis zum
Verlassen des Körpers ist definiert als die Gesamtpassage‐Zeit [17, 19]. Die Kontraktilitätsmessung
mit Hilfe der Kapsel liefert verschiedene Parameter wie die Kontraktionsfrequenz, die Amplitude der
Kontraktionen und den Motilitätsindex, mit deren Hilfe Aussagen zu den Druckverhältnissen im
Gastrointestinaltrakt getroffen werden können [18]. Die Häufigkeit der Kontraktionen ist sowohl vom
Geschlecht als auch vom Alter abhängig [21]. Die Amplitude während der Magenentleerung beim
Übergang zwischen Antrum und Duodenum liegt in den einzelnen Arbeiten zwischen 170 mmHg und
275 mmHg (220 bis 350 mbar) [4, 18, 19, 22].
Abb. 3: pH‐Temperatur‐Druck‐Diagramm (A), welches mittels SmartPill (B) aufgezeichnet wurde [19, 20].
Die Transitzeiten von Darreichungsformen durch den Gastrointestinaltrakt sind in den einzelnen
Abschnitten sehr unterschiedlich und können dadurch einen deutlichen Einfluss auf die
Bioverfügbarkeit von Arzneistoffen haben. Die Magenverweilzeit ist sehr variabel und lässt sich durch
die Eigenschaften der verabreichten Arzneiform und durch den Füllungszustand des Magens
beeinflussen. Je nachdem, ob eine Arzneiform nüchtern oder nach einer Mahlzeit eingenommen

1 Einleitung
5
wurde, kann sie zwischen 30 min und mehreren Stunden im Magen verbleiben [23]. Dagegen ist
unter klinischen Prüfbedingungen die Transitzeit durch den Dünndarm mit 3 h ± 1 h weitestgehend
konstant und unabhängig von den Eigenschaften einer Arzneiform oder vom Füllungszustand des
Magens [7, 9, 23, 24]. Die Dünndarmtransitzeit bei gesunden älteren Menschen unterscheidet sich
ebenfalls nicht von der gesunder junger Menschen [7] und ist auch unabhängig von der
Magenentleerung, da beide jeweils von ihren eigenen Regulationsmechanismen gesteuert werden
[9].
Während die Passage durch den Dünndarm zeitlich weitgehend vorhersehbar ist, variiert die
Passagezeit durch den Dickdarm sehr stark und hat im Vergleich zur gesamten gastrointestinalen
Transitzeit einen deutlich höheren Anteil von mehr als 20 h [23, 24]. Die für die Colonpassage
verantwortliche Schrittmacherzone befindet sich im Colon transversum und generiert
Kontraktionswellen die sowohl rückwärts Richtung Caecum als auch aboral in Richtung des Rektums
gerichtet sein können. Dadurch wird der Darminhalt für längere Zeit zwischen Caecum und Colon
ascendens zurückgehalten, sodass die Resorption von Wasser, Elektrolyten und kurzkettigen
Fettsäuren sowie der bakterielle Aufschluss nicht resorbierter Nahrungsbestandteile über einen
längeren Zeitraum ermöglicht wird [1]. Die Colonmotilität wird vorwiegend gesteuert durch myogene
Automatie. Ein Arzneistoff, der bevorzugt im proximalen Teil des Dünndarms resorbiert wird, hat auf
die gesamte Passage‐Zeit gesehen ein relativ kurzes Absorptionsfenster. Zu derartigen Arzneistoffen
gehören unter anderem Metformin, Levodopa, Gabapentin, Riboflavin oder Aciclovir [10, 23, 24].
Besonders für diese Wirkstoffe ist es von großer Bedeutung, dass sie innerhalb ihres
Absorptionsfensters möglichst schnell und komplett resorbiert werden können, um Wirkverluste
oder Nebenwirkungen aufgrund höherer Dosierungen zu vermeiden.
1.2 Prinzipien des Darm‐Targetings
Neben der lokalen Therapie von Reizdarm‐Syndrom und inflammatorischen Darmerkrankungen wie
Morbus Crohn oder Colitis ulcerosa gibt es Ansätze, das Colon auch für die Aufnahme
makromolekularer Arzneistoffe wie Proteine und Peptide zu nutzen [25]. Um einen Arzneistoff
gezielt an den gewünschten Ort der Resorption entlang des Gastrointestinaltraktes zu bringen, gibt
es verschiedene Prinzipien, derer man sich bedienen kann.
Aufgrund der physiologischen Unterschiede bezüglich Transitzeit, pH‐Wert oder bakterieller
Besiedlung zwischen den einzelnen Abschnitten ist es möglich, ausgewählte Trigger für die
Freisetzung eines Arzneistoffes einzusetzen. Niwa et al. entwickelten eine Zeit‐kontrollierte
Arzneiform für das Colon‐Targeting, die aus einer geschlossenen Ethylcellulose‐Kapsel besteht, in der
sich auf dem Boden der Kapsel eine quellbare Substanz unterhalb eines Arzneistoff‐Reservoirs

1 Einleitung
6
befindet, welche durch Mikroporen mit dem äußeren Medium in Kontakt kommen kann. Die
Quellung führt zu einer Volumenvergrößerung und dadurch zu einem Anstieg des Drucks innerhalb
der Kapsel, dem die Ethylcellulose‐Hülle ab einem bestimmten Punkt nicht mehr standhalten kann
und daraufhin aufplatzt. Durch Modifizierung der Ethylcellulose‐Schichtdicke, der Größe und Anzahl
der Mikroporen und der Menge an quellbarer Substanz kann die Zeit variiert werden, zu welcher die
Kapsel aufplatzt und den Inhalt freigibt [26].
Bott et al. kombinierten eine pH‐kontrollierte mit einer zeitabhängigen Freisetzung, um ein
Freisetzungssystem für das Colon zu entwickeln und die Variabilität der einzelnen Trigger zu
minimieren. Obwohl eine Wirkstofffreisetzung im Dünndarm für Polymere gewährleistet sein kann,
die sich bei einem pH‐Wert von 7 auflösen, kann es bei Verwendung von Polymeren, die sich bei
höheren pH‐Werten auflösen zum Versagen der Arzneiform kommen. Bei entzündlichen
Darmerkrankungen kann der pH‐Wert im Colon zu niedrig sein, um den erforderlichen Dissolutions‐
pH von > 7 zu erreichen. Zudem ist die Magenentleerung vom Füllungszustand des Magens und von
der gerade vorherrschenden Phase des IMMC im nüchternen Zustand abhängig, woraus eine
schlechte Vorhersagbarkeit des Freisetzungsortes resultiert. Zur Überwindung dieses Problems
wurde eine Arzneiform entwickelt, die aus einem Arzneistoff‐überzogenen Pellet besteht, das mit
zwei verschiedenen Überzügen versehen ist. Der innere Überzug besteht aus einer Mischung aus
Eudragit® RL und RS, die eine pH‐unabhängige verzögerte Freisetzung bewirkt, und der äußere
Überzug aus Eudragit® FS 30D, welcher sich ab einem pH‐Wert > 7 schnell auflöst. Damit wird
erreicht, dass sich der äußere, pH‐abhängige Überzug im Dünndarm auflöst und dann der Arzneistoff
durch den inneren Überzug über einen längeren Zeitraum im distalen Dünndarm und Colon
freigesetzt wird [27]. Der erforderliche pH‐Wert von > 7 kann nach Evans et al. bereits im mittleren
Dünndarm erreicht werden, sodass eine Freisetzung über 20 h, wie sie für diese Arzneiform ermittelt
wurde, gegebenenfalls zu früh und unvollständig im Colon erfolgen könnte [28].
Besonders spezifisch für das Colon‐Targeting ist auch die Verwendung von Enzymen als Trigger für
die Freisetzung von Arzneistoffen. Friend et al. untersuchten 1984 die Glykosidase‐Aktivität der
Mikroflora im Colon, und stellten fest, dass Steroidglykoside als Prodrug durch Hydrolyse spezifisch
gespalten werden und die dabei entstehenden Aglyka über die Darm‐Schleimhaut resorbiert werden
können [29]. Dies ist ein besonders interessanter Ansatz für Wirkstoffe, die bei entzündlichen
Darmerkrankungen eingesetzt werden und dabei durch Enzyme wie Azoreduktasen, Glykosidasen
oder Glucuronidasen am Wirkort in ihre aktive Form umgesetzt werden [25]. Durch Modifikation der
Steroidglykoside, der Aglyka sowie der glykosidischen Bindung können der genaue Ort und die
Freisetzungsrate beeinflusst werden [29].
Zu den im Colon vorherrschenden Bakterien gehören unter anderem Bacteroides, Bifidobacterium
und Lactobacillus, welche für die Sekretion reduktiver und hydrolytischer Enzyme wie β‐

1 Einleitung
7
Glucuronidase, β‐Galactosidase, α‐Arabinosidase und Azoreduktase verantwortlich sind, was unter
anderem zu einem Abbau von Di‐, Tri‐ und Polysacchariden führt [25]. Dies wurde in einem Drug‐
Delivery‐System aus einer Kerntablette mit mehreren Polymerüberzügen, dem sogenannten
CODES™‐System versucht zu nutzen. Die Kerntablette besteht aus dem Wirkstoff und einem
Polysaccharid, welches enzymatisch durch Enterobakterien zu organischen Säuren abgebaut wird.
Bei der Passage durch den Gastrointestinaltrakt soll die Arzneiform im Magen zunächst intakt
bleiben, bis sich im Dünndarm der äußere Überzug aus magensaftresistentem Eudragit®L löst. Da der
innere Überzug aus Eudragit® E, erst bei einem pH‐Wert von ≤ 5 löslich ist, beginnt diese innere
Schicht im Dünndarm zunächst zu quellen. Beim Übertritt in das Colon lösen sich die Polysaccharide
bzw. Zuckeralkohole im Kern, diffundieren durch den Überzug und werden dann durch die Bakterien
zu organischen Säuren abgebaut. Die daraus resultierende Erniedrigung des unmittelbar
umgebenden pH‐Wertes führt zur vollständigen Auflösung des Films und zur Freisetzung des
Wirkstoffs aus dem Tablettenkern, was durch szintigraphische Messungen im Rahmen einer In vivo‐
Studie auch gezeigt werden konnte [25, 30].
Einen Vergleich zwischen einer pH‐abhängigen und einer Enzym‐getriggerten Freisetzung stellten
McConnell et al. 2008 an, indem sie Theophyllin‐haltige Pellets einerseits mit dem
magensaftresistenten Überzug Eudragit® S und andererseits mit einer Mischung aus enzymatisch
abbaubarer Amylose und Ethylcellulose versahen und die Freisetzung des Wirkstoffs in vivo
überprüften. Während die pH‐abhängigen Pellets, die sich erst ab einem pH‐Wert von 7 auflösen
sollten, eine frühzeitige Freisetzung im Dünndarm zeigen, wird aus den Amylose‐Pellets vor Eintritt in
das Colon kein Wirkstoff freigesetzt. Obwohl beide Systeme eine ähnliche Bioverfügbarkeit zeigen,
stellte sich heraus, dass die Enzym‐getriggerte Freisetzung reproduzierbarer und verlässlicher ist [13].
Von Karrout et al. wurde das Colon‐Targeting mit Bakterien‐sensitiven Filmen untersucht, die speziell
an die pathophysiologischen Verhältnisse angepasst wurden. Die Filme bestehen aus einem
Polysaccharid, das durch die bakterielle Darmflora abgebaut werden kann und Ethylcellulose als
zusätzlichem Hilfsstoff, welcher ein vorzeitiges Auflösen und eine Quellung verhindern soll. In ihren
Untersuchungen hinsichtlich der Unterschiede zwischen der Darmflora von Gesunden und von
Patienten mit entzündlichen Darmerkrankungen zeigt sich, dass sich neben der veränderten
Gesamtmenge an Bakterienstämmen das quantitative Auftreten vereinzelter Stämme verändert.
Analog zu dieser Variabilität in der bakteriellen Besiedlung des Colons könnten sich weiterhin auch
durch mögliche Veränderungen der pH‐Werte, Passagezeiten und des Darminhaltes veränderte
Freisetzungen in vivo ergeben, die in vitro nicht abzuschätzen sind [31].
Bei den verschiedenen Ansätzen des Colon‐Targetings traten trotz Erfolgen Probleme auf, die eine
zuverlässige Freisetzung in vivo nicht garantieren könnten. Während die pH‐gesteuerte Freisetzung
durch intra‐ und interindividuelle Unterschiede und die Ähnlichkeit der pH‐Werte zwischen

1 Einleitung
8
Dünndarm und Colon problematisch ist, wird die Zeit‐gesteuerte Freisetzung trotz konstanter
Dünndarm‐Passagezeiten durch die Variabilität der Magenentleerung beeinflusst. Die Enzym‐
gesteuerte Freisetzung ist vielversprechend, da sie bei ausgewählten Polysacchariden auf das Colon
beschränkt bleibt. Allerdings ist der enzymatische Abbau ein sehr langsamer Prozess, der bis zu 12 h
andauern kann [25]. Aus diesem Grund soll im Zuge dieser Arbeit ein neuer Ansatz untersucht
werden, der sich die unterschiedlichen Druckverhältnisse im Gastrointestinaltrakt und besonders an
den Sphinkteren zu Nutzen macht. Für die Untersuchung der Kräfte im Gastrointestinaltrakt und für
die durch Druck gesteuerte Freisetzung wurden bereits verschiedene Konzepte vorgestellt.
Die „pressure‐controlled colon delivery capsule“ (PCDC) besteht aus einer mit Ethylcellulose
innenbeschichteten Gelatine‐Kapsel, die mit Polyethylenglycol 1000 befüllt wird. Bei Einnahme
dieser Kapsel löst sich die äußere Gelatine‐Hülle ab, das PEG schmilzt bei Körpertemperatur und es
wird dadurch ein Ethylcellulose‐Ballon erhalten, dessen Stabilität von der Schichtdicke abhängig ist.
Während die Kapsel im oberen Teil des Magen‐Darm‐Traktes relativ frei schwimmen kann, verdichtet
sich der Darminhalt durch verstärkte Wasserresorption im Dickdarm immer mehr und führt zum
Platzen der Kapsel. Untersuchungen mit verschiedenen Wirkstoffen, sowie mit modifizierten
Herstellungsverfahren oder Kapselfüllungen wurden durchgeführt und zeigten, dass dieses System
für das Colon‐Targeting gut geeignet ist [26, 32‐38].
Mit Hilfe eines „destructive force dependent release system“ (DDRS) untersuchten Kamba et al. die
mechanische Zerstörungskraft des Magens. Ein DDRS besteht aus einer Kerntablette mit
Modellarzneistoff, die mit einem magensaftlöslichen Überzug versehen ist. Diese überzogene
Tablette wird mit einem Teflon‐Mantel versehen, der durch Veränderung des Pressdrucks auf eine
voreingestellte Bruchfestigkeit um die Tablette gepresst werden kann. Die Hydrophobizität von
Teflon führt dazu, dass die Tablette unabhängig von der Aufenthaltsdauer in gastrointestinaler
Flüssigkeit intakt bleibt. Eine Gelatine‐Hülle um die Teflon‐Tablette schützt vor mechanischer
Belastung bei der Handhabung und beim Schlucken der Arzneiform. Nach Einnahme löst sich die
Gelatine‐Hülle auf und gibt die Tablette im Magen frei. Wenn dort die Kraft von der Magenwand
größer ist, als die voreingestellte Bruchfestigkeit des DDRS, wird die Teflon‐Schicht aufgebrochen, der
magensaftlösliche Überzug löst sich auf und gibt den Tablettenkern frei, aus dem der
Modellarzneistoff freigegeben und resorbiert werden kann. Wenn der Druck im Magen geringer als
die voreingestellte Bruchfestigkeit ist, wird die Tablette komplett ins Duodenum entleert und
Richtung Colon transportiert. Würde der Teflon‐Mantel der Tablette dann aufgrund der Verdickung
des Darminhaltes reißen, käme es aufgrund des pH‐abhängigen Überzugs, der sich nur im Sauren
auflöst nicht zur Freisetzung der Kerntablette und dadurch nicht zur Absorption. In vivo‐Studien am
Menschen ergaben, dass die Kräfte des leeren Magens bei etwa 1,5 N und beim gefüllten Magen
etwas höher zwischen 1,5 und 1,9 N liegen. Versuche mit Hunden zeigten, dass die Zerstörungskraft

1 Einleitung
9
des Magens dort unabhängig vom Füllungszustand des Magens bei etwa 3,2 N liegt. Eine
Weiterentwicklung des DDRS ist die DDRS‐SI‐Ecap, die für Untersuchungen der Zerstörungskraft des
Dünndarms entwickelt wurde. Diese besteht aus einer Kerntablette, die direkt mit Teflon ummantelt,
anschließend in einer Kapsel verpackt und dann magensaftresistent überzogen wird. Aus diesen
Untersuchungen ergab sich für den Dünndarm eine Zerstörungskraft von 1,2 N [39‐41].
Etwas niedrigere Kräfte ergaben sich bei Marciani et al. für den Magen. Sie untersuchten mit Hilfe
von Agar‐Kugeln unterschiedlicher Konzentration die Kräfte des Magens in Zusammenhang mit
unterschiedlich viskosen Flüssigmahlzeiten. Diese Kugeln haben einen Durchmesser von 1,27 cm und
können durch Veränderung der Agar‐Konzentration zwischen 0,75 % und 3 % auf eine spezielle
Bruchfestigkeit eingestellt werden. Die antralen Kräfte ergaben sich zu etwa 0,65 N, da die Kugeln
mit einer höher eingestellten Bruchfestigkeit deutlich langsamer entleert wurden, während die
weicheren Kugeln schneller zerbrachen und damit genauso schnell wie eine Flüssigmahlzeit entleert
wurden [8].
1.3 Zielstellung
Das Ziel dieser Arbeit war die Entwicklung einer drucksensitiven Darreichungsform für das Drug‐
Targeting in den Darm. Dabei sollten vor allem die hervorragenden Resorptionsbedingungen des
oberen Dünndarms Berücksichtigung finden, indem Prinzipien für Arzneiformen entwickelt werden,
die im Sinne eines „burst release“ den Arzneistoff unter Einwirkung des am Pylorus vorherrschenden
Druckes schlagartig und möglichst vollständig freisetzen. Der Wirkstoff sollte möglichst gelöst oder
suspendiert in einer flüssigen Grundlage vorliegen, um sich nach Freisetzung schnell auf der
Schleimhaut des oberen Dünndarms zu verteilen und dort durch eine verlängerte Kontaktzeit schnell
resorbiert zu werden. Die Darreichungsform sollte stabil genug sein, um den Druckverhältnissen des
oberen Gastrointestinaltraktes standzuhalten, und auch durch den sauren pH‐Wert des nüchternen
Magens unbeschädigt bleiben. Die entwickelte Arzneiform sollte dann idealerweise während der
Magenentleerung durch die im Antrum und Pylorus auftretenden hohen Drücke zerplatzen. Bei der
Entwicklung wurden zwei Prinzipien untersucht, durch die der Arzneistoff in einer flüssigen
Grundlage freigesetzt werden konnte. Zum einen wurden Arzneiformen entwickelt, die bereits einen
flüssigen bzw. halbfesten Kern in Form eines Hydrogels oder einer lipophilen flüssigen Grundlage
enthielten. Andererseits wurden Darreichungsformen entwickelt, deren Kern sich erst bei
Körpertemperatur verflüssigte und die somit bei Raumtemperatur eine feste und robuste Arzneiform
darstellten.
Zur Charakterisierung der entwickelten Darreichungsformen sollte die Bruchfestigkeit mit Hilfe des
Texture Analysers untersucht werden. Zusätzlich sollten Versuche mit einer Stresstest‐Apparatur
gemacht werden, die einerseits für Bruchfestigkeitsversuche herangezogen werden kann, aber auch

1 Einleitung
10
zur Simulation einer Freisetzung unter annähernd physiologischen Bedingungen geeignet ist [42, 43].
Eine frühzeitige Freisetzung unter Standardbedingungen und ohne Druck sollte mit Hilfe von
Freisetzungsversuchen in der Paddle‐Apparatur nach dem Amerikanischen (United States
Pharmacopeia, USP) und Europäischen Arzneibuch (Pharmacopoea Europaea, Ph. Eur.) [44, 45]
ausgeschlossen werden, die gleichzeitig die Auswirkungen einer verlängerten Verweilzeit in der
sauren Umgebung des nüchternen Magens analysieren sollten. Anhand der Ergebnisse dieser
verschiedenen Prüfungen sollte entschieden werden, ob die in dieser Arbeit entwickelten
drucksensitiven Darreichungsformen prinzipiell für das Drug‐Targeting in den oberen Teil des
Dünndarms geeignet sein könnten.

2 Material und Methoden
11
2 Material und Methoden
2.1 Materialien zur Herstellung der Arzneiformen
2.1.1 Hartfett
Hartfett ist ein Gemisch aus Mono‐, Di‐ und Triglyceriden von gesättigten Fettsäuren und wird aus
Palmkern‐ und Kokosfetten mit einem hohen Laurinsäure‐Anteil hergestellt [46, 47]. Es ist weiß,
geruch‐ und geschmacklos und besitzt aufgrund der fehlenden ungesättigten Fettsäuren nur eine
geringe Tendenz zum ranzig werden [47]. Hartfett ist wasserunlöslich, hat ein geringes Intervall
zwischen Schmelz‐ und Erstarrungspunkt und zeigt Volumenkontraktion [46, 47]. In Abhängigkeit von
der Hartfettsorte und den damit verbundenen Eigenschaften lassen sich etwa gleiche Mengen
Wasser in das geschmolzene Hartfett einarbeiten [46, 47], wobei die Emulgiereigenschaften mit
steigender Hydroxylzahl zunehmen und die Sprödigkeit abnimmt [47]. Das in dieser Arbeit
verwendete Hartfett W32 hat eine mittlere Hydroxylzahl von 40 – 50 mg KOH/g und einen
Schmelzbereich zwischen 32,0 und 33,5 °C [48]. Hartfett H15, welches für die Alginat‐Kügelchen
enthaltenden drucksensitiven Arzneiformen verwendet wurde, besitzt einen etwas höheren
Schmelzbereich zwischen 33,5 – 35,5 °C, der noch knapp unter der Körpertemperatur liegt, sodass es
nach Einnahme schmelzen würde. Beide Sorten werden in der pharmazeutischen Technologie als
Suppositorien‐Grundlage verwendet und dienten bei den entwickelten drucksensitiven Arzneiformen
als Grundlagen, die unterhalb der Körpertemperatur schmelzen und dann beim Aufplatzen der
Arzneiform flüssig zur Verfügung stehen sollten.
2.1.2 Polyethylenglycol
Polyethylenglycol (PEG, Macrogol) mit der allgemeinen Formel HO‐(CH2‐CH2‐O)n‐CH2‐CH2‐OH ist ein
Polymerisationsprodukt des Ethylenoxids [46, 47] und wie alle hochpolymeren Stoffe ein Gemisch
sehr ähnlicher Polymerhomologer, da sich beim Herstellungsprozess die Polymerisationsstufen der
einzelnen Moleküle überschneiden [49]. Mit steigender Molekülmasse nimmt die Konsistenz zu,
sodass Macrogole mit einer Molekülmasse von 200 – 600 flüssig sind, zwischen 800 und 1500
vaselinartig und zwischen 2000 und 6000 eine wachsartige Konsistenz haben [46, 47]. Die
Wasserlöslichkeit ergibt sich durch die Ausbildung von Wasserstoffbrückenbindungen zwischen dem
Ethersauerstoff und den Wassermolekülen [46] und nimmt wie die Hygroskopizität mit steigendem
Polymerisationsgrad ab [47]. Macrogol besitzt antibakterielle Eigenschaften und kann einer
autoxidativen Zersetzung unterliegen, was eine luft‐ und lichtgeschützte Aufbewahrung bedingt [46].
Höher molekulare PEG sind nicht hygroskopisch und werden als Gleit‐ oder Bindemittel
eingesetzt [49]. Des Weiteren kann PEG in der pharmazeutischen Industrie als Lösungs‐ oder

2 Material und Methoden
12
Einbettungsmittel, Weichmacher und Überzugsmaterial sowie als hydrophile Salben‐ und
Suppositoriengrundlage verwendet werden. Das in dieser Arbeit verwendete PEG 1000 besitzt einen
Erstarrungspunkt zwischen 30 und 40 °C sowie ein gutes Spreit‐ und Haftvermögen [47, 50] und
wurde deshalb als Grundlage verwendet, die unterhalb der Körpertemperatur schmelzen und ähnlich
wie das Hartfett beim Aufplatzen der Arzneiform als flüssige Grundlage zur Verfügung stehen sollte.
2.1.3 Tristearin
Tristearin (Dynasan™ 118, Sasol) ist ein Triglycerid, aus Glycerol und Stearinsäure. Es handelt sich um
ein weißes Pulver, welches unlöslich in Wasser ist und aufgrund seiner Polymorphie in verschiedenen
Modifikationen kristallisieren kann, die sich in ihren Schmelzpunkten und spektroskopischen
Eigenschaften unterscheiden [51]. Die α‐Modifikation mit dem niedrigsten Schmelzpunkt bei 55 °C
geht beim Erwärmen zunächst in die instabile β‘‐Modifikation bei 63,2 °C und anschließend in die
stabile β‐Modifikation mit dem höchsten Schmelzpunkt bei 73,5 °C über [51, 52]. Dabei ändern die
Kohlenstoffketten ihre räumliche Anordnung von einem hexagonalen über ein orthorhombisches in
ein triklines System [51]. Aufgrund seiner Wasserunlöslichkeit, seines hohen Schmelzpunktes und der
Sprödigkeit wurde es innerhalb dieser Arbeit verwendet, um in Abhängigkeit von der Schichtdicke
eine spröde und bruchsensitive Hülle für eine drucksensitive Arzneiform zu erhalten.
2.1.4 Agar
Bei Agar handelt es sich um Polysaccharide, die aus verschiedenen Spezies der Rotalgen, besonders
Gelidium, gewonnen werden. Zur Gewinnung werden die Rotalgen mit kochendem Wasser
behandelt und der dabei erhaltene Extrakt wird in heißem Zustand filtriert und anschließend
konzentriert und getrocknet [44, 53]. Agar besteht in seiner Hauptfraktion zu 70 % aus Agarose,
welche überwiegend aus 3,6‐Anhydro‐α‐L‐galactose und β‐D‐Galactose aufgebaut ist. Mit einem sehr
geringen Sulfatgehalt und dem hohen Gehalt an 3,6‐Anhydrogalactose ist es für die Gelierfähigkeit
des Agars verantwortlich. Die verbleibenden 30 % der Hauptfraktion macht Agaropektin aus, welches
durch einen Sulfatanteil von 3 – 10 % und einen geringen Anteil an 3,6‐Anhydro‐α‐L‐galactose nicht
geliert. Agar ist erhältlich als Pulver, geknäulte Streifen oder Flocken und sieht farblos oder schwach
gelblich und durchscheinend aus [44]. In kaltem Wasser führt eine Quellung zu einer 10‐fachen
Volumenvergrößerung, während in heißem Wasser ein Sol entsteht [49, 53, 54]. Schon 0,5 %ige
kolloidale Agar‐Lösungen erstarren beim Abkühlen auf 30 – 50 °C zu einem Gel und gehen erst beim
Erhitzen auf 80 °C wieder in ein Sol über [53, 54]. Dieser Sachverhalt wurde in dieser Arbeit
ausgenutzt, um aus dem heißen Agar‐Sol nach Erkalten in Abhängigkeit von der Konzentration
formstabile Arzneiformen zu erhalten.

2 Material und Methoden
13
2.1.5 Dickflüssiges Paraffin
Dickflüssiges Paraffin ist eine gereinigte Mischung aus gesättigten Kohlenwasserstoffen, die aus der
über 300 °C siedenden Destillationsfraktion des Erdöls gewonnen und durch Abkühlen von festen
Kohlenwasserstoffen befreit wird. Es ist eine transparente, ölige Flüssigkeit, die praktisch unlöslich in
Wasser, aber mischbar mit Ether und Chloroform ist und eine Viskosität von > 100 mPa*s
aufweist [44, 47]. Innerhalb dieser Arbeit wurde dickflüssiges Paraffin als lipophile, flüssige
Grundlage für eine der entwickelten Arzneiformen verwendet.
2.1.6 Natriumalginat
Bei Alginsäure handelt es sich um ein Polymer aus Mannuron‐ und Guluronsäure mit einer relativen
Molekülmasse von 120.000 – 200.000, welches aus Braunalgen der Ordnungen Laminariales und
Fucales gewonnen wird. Es wird durch Erhitzen mit Alkali und anschließendem Ausfällen mit
Salzsäure gewonnen [46, 53, 54]. Während die Natrium‐, Kalium‐, Magnesium‐ und Ammoniumsalze
der Alginsäure wasserlöslich sind, sind sie selbst und ihr Calcium‐Salz unlöslich in Wasser. Der
gelierende Effekt durch Calcium‐Ionen, der innerhalb dieser Arbeit zur Herstellung einer
drucksensitiven Arzneiform genutzt wird, ergibt sich durch die Ausbildung von Calcium‐Brücken
zwischen den Polymannuronsäure‐Ketten. Dabei entsteht aus der Knäuelformation die sogenannte
„egg box type“‐Konformation, bei der Calcium‐Ionen zwischen die stark gefalteten Guluronsäure‐
Blöcke eingelagert werden, was zur Ausbildung von Calcium‐Chelatbrücken führt [46, 53]. Für
pharmazeutische Zwecke wird überwiegend Natriumalginat verwendet, welches in Konzentrationen
zwischen 3 – 6 % salbenartige Gele bildet und im neutralen pH‐Bereich am stabilsten ist. Bei pH‐
Werten < 4,5 wird die freie Säure ausgefällt und im Basischen kommt es zur Spaltung der
glykosidischen Bindung. Seine Verwendung findet Natriumalginat überwiegend als
Viskositätserhöher, Emulsions‐ und Suspensionsstabilisator und als Binde‐ und Zerfallsmittel bei der
Tablettierung [46, 49, 54]. Innerhalb dieser Arbeit wurde Natriumalginat zur Herstellung kleiner
Alginat‐Kügelchen verwendet, die eingebettet in Hartfett und mit Celluloseacetat überzogen eine
drucksensitive Darreichungsform ergaben.
2.1.7 Celluloseacetat
Bei der teilweisen oder vollständigen O‐Acetylierung von Cellulose entsteht Celluloseacetat (CA,
Teilstruktur siehe Abb. 4) als weißliches, hygroskopisches Pulver oder Granulat. Es ist praktisch
unlöslich in Wasser, aber unter anderem löslich in Aceton [44], wodurch dieser Stoff geeignet ist für
wasserunlösliche und pH‐unabhängige Filmüberzüge und als Ausgangsprodukt für Lacke. In seltenen
Fällen wird es auch als Tablettenbindemittel verwendet [49].

2 Material und Methoden
14
Abb. 4: Teilstruktur von Celluloseacetat.
Bei der Entwicklung der drucksensitiven Arzneiformen wurde es als 5 %ige organische Lösung auf
Hartfett‐ und PEG‐Kugeln aufgesprüht und bildete dort einen spröden Überzug, der abhängig von der
Polymerbeladung bei einem bestimmten Druck brechen sollte.
2.1.8 Ethylcellulose
Ethylcellulose [EC, 48,0 – 49,5 % (m/m) Ethoxyl‐Gruppen, Teilstruktur siehe Abb. 5] ist ein weißes
oder gelblich‐weißes, geruchloses Pulver, welches unlöslich in Wasser, aber unter anderem löslich in
Ethanol ist [44, 47, 49]. In Abhängigkeit vom Substitutionsgrad nimmt die Löslichkeit zu [47]. Es
handelt sich um einen nichtionischen Celluloseether mit Molekülmassen zwischen 150.000 und
300.000 [47], bei dem die Hydroxygruppen zum Teil O‐ethyliert sind, sodass sich ein
Substitutionsgrad von 2,25 – 2,58 ergibt. Durch seine Unlöslichkeit in Wasser eignet sie sich als
Retardüberzug [49], wird aber auch als Einbettungsmaterial verwendet. Diese Eigenschaft wurde im
Zuge dieser Arbeit ausgenutzt, um einen Überzug für die Paraffin‐Kapseln zu erhalten, der im
Wässrigen unabhängig vom pH‐Wert unlöslich, spröde und drucksensitiv ist.
Abb. 5: Teilstruktur von Ethylcellulose.
2.1.9 Triacetin
Triacetin (Strukturformel siehe Abb. 6) ist ein Ester aus Glycerol und Essigsäure, der geruch‐ und
geschmacklos, nicht flüchtig und physiologisch unbedenklich ist. Als äußerer Weichmacher kann es
sich zwischen die Filmbildnermoleküle schieben und deren intermolekulare Kräfte abschwächen.

2 Material und Methoden
15
Dies führt zu einer Verbesserung der Flexibilität, der Haftfähigkeit von Filmen und einer Absenkung
der Mindestfilmbildungstemperatur [47]. Triacetin ist ein wasserlöslicher Weichmacher, der durch
Porenbildung im Film die Wirkstofffreigabe beschleunigen kann [49] und wurde innerhalb dieser
Arbeit den Überzugssuspensionen zugesetzt.
Abb. 6: Strukturformel von Triacetin.
2.1.10 Kaugummi‐Grundmasse
Die genaue Zusammensetzung von Kaugummi‐Grundmassen wird von den Herstellern als
Firmengeheimnis behandelt. Im Allgemeinen bestehen sie zu 15 – 40 % aus neutraler und
geschmackloser Kaumasse und weiteren Zutaten, die als Füllstoffe, Süßstoffe oder Weichmacher
fungieren oder geschmacks‐ bzw. konsistenzbeeinflussende Eigenschaften haben [55‐57]. Als
natürliche Kaumasse wird neben Chicle, dem eingetrockneten Latex des Breiapfelbaumes auch das
Harz Mastix verwendet, welches durch Einschnitte in den Stamm des Mastixbaumes entsteht [54].
Heutzutage werden zumeist Kaumassen synthetischen Ursprungs verwendet, die aus einer Mischung
von Elastomeren für die Elastizität, Harzen, Wachsen und Fetten als Weichmacher und Emulgatoren
bestehen. Als synthetische Elastomere werden Styrenbutadien‐Copolymere mit Polyisobuten
eingesetzt. Polyvinylacetat dient der Herabsetzung der Klebrigkeit und partiell hydrierte
Fettsäureester wie Sojabohnen‐Öl werden als Weichmacher verwendet. Aufgrund ihrer
emulgierenden Eigenschaften werden Glycerolmonostearat oder Lecithin zugesetzt, die die
Aufnahme des Speichels in den Kaugummi und damit das Herauslösen eines möglichen Wirkstoffes
während des Kauvorgangs gewährleisten sollen [55‐57]. Die innerhalb dieser Arbeit verwendete
Kaugummimasse in Form von Granulat‐Körnern (Abb. 7) der Firma Gumlink A/S (Dänemark) wurde
als wasserundurchlässiger Überzug einer Mantel‐ bzw. Mehrschichttablette verwendet. Die
Kaugummimasse konnte unter Zuhilfenahme von Magnesiumstearat als Schmiermittel direkt auf den
Tablettenkern gepresst werden und wurde außerdem ‐ in Heptan gelöst ‐ als flüssige Überzugslösung
auf die Tabletten im Tauchverfahren aufgetragen.

2 Material und Methoden
16
Abb. 7: Kaugummi‐Grundmasse als Granulat.
2.1.11 Riboflavinphosphat‐Natrium
Das in Abb. 8 dargestellte Riboflavinphosphat (RFP) ist die mono‐phosphorylierte Form des
Vitamin B2. Es ist ein gelb‐oranges, hygroskopisches und kristallines Pulver, welches löslich in Wasser
ist und in Lösung (10 g/L bei 20 °C) einen leicht sauren pH‐Wert von 5 – 6,5 aufweist [44, 58]. Es ist
lichtempfindlich [58], besitzt ein Absorptionsmaximum bei 266 nm und eine spezifische Absorption
am Absorptionsmaximum zwischen 580 und 640 [44].
N
N
NH
NH3C
H3C
O
O
O
H
OH
HHO
H
HO
P
ONaOH
O
Abb. 8: Strukturformel von Riboflavinphosphat‐Natrium.
In der Nahrung ist Vitamin B2 als Flavin‐mono‐nucleotid (FMN) und Flavin‐adenin‐dinucleotid (FAD)
enthalten und muss zunächst über Phosphatasen dephosphoryliert werden, um aus dem Dünndarm
resorbiert zu werden [59, 60]. Die Resorption geschieht abhängig von der Dosierung entweder durch
aktiven Transport bei geringen Mengen oder durch Diffusion bei höheren Mengen an Riboflavin [59,
60]. In den Mukosazellen der Dünndarmschleimhaut wird es dann an der 5‘‐Position zum
Mononucleotid phosphoryliert [1, 59, 60] und kann anschließend weiter zu FAD reagieren, sodass
70 – 90 % des Riboflavins im Organismus als FAD und der Rest als FMN vorliegen [59]. FMN und FAD
sind Coenzyme der Flavinenzyme und sind von Bedeutung für die Wasserstoffübertragung in der
Atmungskette, die Dehydrierung von Fettsäuren und die oxidative Desaminierung von Aminosäuren
[59, 60]. Im Blut werden Riboflavin und seine mono‐ und diphosphorylierten Formen an Riboflavin‐
bindende Proteine wie Albumin oder Globuline gebunden [59] und die Ausscheidung erfolgt
unverändert oder renal als Hydroxy‐ und andere Metabolite [60]. Aufgrund seiner Wasserlöslichkeit,

2 Material und Methoden
17
seiner sehr geringen Toxizität [58] und der einfachen spektroskopischen Analytik wurde es für einige
der entwickelten Arzneiformen als Modellarzneistoff verwendet.
2.1.12 Paracetamol
Bei Paracetamol, dessen Strukturformel in Abb. 9 dargestellt ist, handelt es sich um ein weißes,
kristallines Pulver mit einer geringen Wasserlöslichkeit von 14 g/L [44, 61]. Es besitzt ein
Absorptionsmaximum bei 249 nm und eine spezifische Absorption im Absorptionsmaximum
zwischen 860 und 980 [44]. Nach Aufnahme dieses nicht‐sauren Antipyretikums [60] wird es im
Organismus zu 4‐Aminophenol desacetyliert und kann im zentralen Nervensystem mit
Arachidonsäure zu einem unselektiven Cyclooxygenase‐Hemmer reagieren. Paracetamol wird schnell
und vollständig aus dem Gastrointestinaltrakt resorbiert, besitzt eine Halbwertszeit von 2 – 3 h und
wird renal in Form seiner Glucuronide und Sulfate eliminiert [59, 60]. Im Zuge dieser Arbeit wurde
Paracetamol wegen seiner guten spektroskopischen Analytik als Modellarzneistoff für zwei
entwickelte Arzneiformen ‐ Tristearin‐Kapseln und Agar‐Kugeln ‐ verwendet.
Abb. 9: Strukturformel von Paracetamol.
2.1.13 Hydroxyethylcellulose
Der nichtionogene Celluloseether Hydroxyethylcellulose [HEC, 2,0 % (m/m) TS] in Abb. 10 stellt eine
teilweise 0‐(2‐hydroxylierte) Cellulose mit einem durchschnittlichen Substitutionsgrad von 0,9 – 1 dar
und ist ein weißliches Pulver oder Granulat. Es ist sowohl in kaltem als auch in heißem Wasser löslich
und ergibt dabei kolloidale Lösungen [44], ist aber unlöslich in organischen Lösemitteln [47].
Hydroxyethylcellulose wurde innerhalb dieser Arbeit als Gelbildner für das wirkstoffhaltige Gel der
Tristearin‐Kapseln verwendet und wurde in der Pulvermischung für die Kaugummi‐Tabletten als
Quellstoff untergemischt.
Abb. 10: Teilstruktur von Hydroxyethylcellulose.

2 Material und Methoden
18
2.1.14 Mikrokristalline Cellulose
Mikrokristalline Cellulose (MCC) (Teilstruktur siehe Abb. 11) ist eine gereinigte und teil‐
polymerisierte Cellulose, die durch die Behandlung von α‐Cellulose entsteht [44]. Dabei wird
Cellulose mit Mineralsäure erwärmt und anschließend in Fragmente zerkleinert, deren Partikelgröße
zwischen wenigen bis mehreren hundert Mikrometern liegt. Durch die Behandlung wird der
Polymerisationsgrad auf Werte zwischen 200 und 300 gesenkt, was zu einer Zunahme der
Kristallinität führt [47, 49, 54]. Aufgrund des niedrigen Polymerisationsgrades weist MCC im Vergleich
zu Pulvercellulose eine gute plastische Verformbarkeit auf, die zu extrem harten Tabletten mit
geringer Friabilität führen kann [47, 49]. Das weiße, feine oder leicht körnige Pulver ist praktisch
unlöslich in Wasser [44]. Es ist ein sehr gutes Trockenbindemittel, kann für die Direktverpressung von
Tabletten verwendet werden und zeigt eine hohe Aufnahmekapazität für Wirkstoffe [49]. Im Rahmen
dieser Arbeit wurde MCC (Vivapur Typ 200) verwendet, um Placebo‐Tabletten herzustellen, die
zusammen mit den zu überziehenden Arzneiformen im Dragierkessel bzw. Mini‐Coater mit einem
Überzug versehen wurden. Außerdem war es im Pulverkern der Kaugummi‐Tabletten der Charge 2
enthalten.
Abb. 11: Teilstruktur von MCC.
2.1.15 Talkum
Talkum ist ein Magnesiumhydrosilicat der allgemeinen Formel Mg3Si4O10(OH)2 [44, 47], welches
unterschiedliche Mengen an zusätzlichen Mineralien wie Aluminium‐ und Magnesiumsilicaten
(Chlorit), Magnesiumcarbonat (Magnesit), Calciumcarbonat (Calcit) oder Calcium‐
Magnesiumcarbonat (Dolomit) enthalten kann [44]. Als Schichtsilikat besteht es aus zwei äußeren
Einzelschichten (Si2O5/OH), die von Magnesiumionen zusammengehalten werden, welche innerhalb
des Kristallaufbaus jeweils von vier Sauerstoff‐Atomen und zwei Hydroxygruppen umgeben sind. Die
Blattstrukturschichten dieses Dreischichtenminerals können leicht parallel verschoben werden,
wodurch sich die Verwendbarkeit als Gleitmittel ergibt [47]. Aufgrund seiner geringen Härte und der
Verschiebbarkeit der Schichten, fühlt sich das weißliche und in Wasser praktisch unlösliche Pulver

2 Material und Methoden
19
fettig an [44, 47, 49]. Talkum wird in der pharmazeutischen Industrie als Pudergrundlage und
Gleitmittel [47] verwendet. In dieser Arbeit wurde es als Gleitmittel bei der Herstellung der Placebo‐
Tabletten eingesetzt. Weiterhin wurde es 1 %ig den Überzugssuspensionen als Deckmittel für die
Hartfett‐ und PEG‐Kugeln sowie für die Paraffin‐Kapseln zugesetzt.
2.1.16 Hochdisperses Siliciumdioxid
Hochdisperses Siliciumdioxid, welches unter dem Handelsnamen Aerosil® erhältlich ist, wird aus
Siliciumtetrachlorid und Wasserstoff durch Flammenhydrolyse gewonnen, wobei es zunächst als
SiO2‐Aerosol vorliegt und anschließend koaguliert. Durch diese Herstellung erhält es eine sehr große
Oberfläche von 200 m2/g und einen mittleren Durchmesser von 15 nm. Aerosil® ist in der Lage bis zu
40 % Wasser aufzunehmen, ohne seine Fließfähigkeit zu verlieren und wird deshalb als Trocknungs‐
und Trockenhaltemittel eingesetzt. Durch die geringe Größe der Partikel kann es größere
Pulverpartikel umhüllen und somit die interpartikuläre Reibung herabsetzen, weshalb es als
Fließregulierungsmittel verwendet wird. Die optimale Aerosil®‐Konzentration in einer
Pulvermischung ist abhängig von der Dichte und der durchschnittlichen Teilchengröße; sie liegt meist
zwischen 0,5 und 1 % [47, 49]. Wird hochdisperses Siliciumdioxid zusammen mit Stärke in Granulaten
verarbeitet, bewirkt es einen schnellen Zerfall der Tabletten, da aufgrund der „Dochtwirkung“
leichter Wasser ins Tabletteninnere transportiert wird. Neben den bereits genannten Einsatzgebieten
wird Aerosil® weiterhin als Adsorptionsmittel, Suspensionsstabilisator, Gel‐ und Gerüstbildner
eingesetzt [47] und diente in dieser Arbeit als Fließregulierungsmittel bei der Herstellung der
Placebo‐Tabletten.
2.1.17 Magnesiumstearat
Magnesiumstearat ist ein feines, leichtes, fast geruchloses Pulver, das aus einer Mischung aus
Magnesiumstearat‐ und –palmitat besteht und in Wasser, kaltem Alkohol und Ether unlöslich ist [47,
49]. Aufgrund unterschiedlicher Herstellungsmethoden kann im Fettsäuregemisch der Stearinsäure‐
Anteil 40 – 80 % und der Palmitinsäure‐Anteil 16 – 50 % betragen und das Verhältnis von Stearin‐ zu
Palmitinsäure zwischen 8:1 und 1:1 schwanken [49]. Magnesiumstearat kann als Zusatz zu Pudern
verwendet werden oder in Konzentrationen von 0,5 bis 2 % als Schmier‐ und Formentrennmittel
eingesetzt werden [47, 49]. Im Rahmen dieser Arbeit wurde es 0,5 %ig der Pulver‐Mischung für die
Placebo‐Tabletten hinzugefügt.
2.2 Herstellungsverfahren der Arzneiformen
2.2.1 Agar‐Kugeln
In Anlehnung an die Arbeit von Marciani et al. wurden Agar‐Kugeln entwickelt, deren Bruchverhalten

2 Material und Methoden
20
durch kleine Veränderungen in der Herstellung beeinflusst werden kann. Dabei wurden die
Eigenschaften der Arzneiformen auf zwei Weisen modifiziert:
1. Verwendung unterschiedlicher Agar‐Konzentrationen
2. Verwendung einheitlicher Konzentration, aber unterschiedlicher Trocknungszeiten.
Für die Herstellung der unterschiedlich konzentrierten Agar‐Kugeln wurden je 10 Kugeln mit 1, 1,5, 2,
3, 4, 5, 6, 6,5, 7, 7,5, 8, 9 und 10 % Agar hergestellt. Zur Formgebung diente eine 2 g‐Gießform für
Vaginalglobuli, die einen Durchmesser von je 1,5 cm pro Arzneiform hatte. Zur Herstellung wurden
ca. 90 % des benötigten Wasser in einer Schottflasche mit Rührschwein auf einem Magnetrührer mit
Heizfunktion solange erwärmt, bis das Wasser eine Temperatur von 80 °C hatte. Während das
Rührschwein für eine gleichmäßige Durchmischung des Wassers sorgte, wurde die entsprechende
Menge an Agar langsam hinzugegeben, sodass eine gleichmäßige Verteilung erfolgen konnte. Danach
wurde die Heizplatte ausgeschaltet und alles solange rühren gelassen bis sich der Agar vollständig
aufgelöst hatte. Für die Herstellung wirkstoffhaltiger Darreichungsformen wurde Paracetamol im
Wasser vollständig gelöst (0,06 g/36 g Wasser), bevor Agar auf die warme Lösung aufgestreut wurde,
sodass ein 7,5 %iges Sol entstand. Anschließend wurde das verdunstete Wasser hinzugefügt, und
nach erneutem kurzem Durchmischen wurde die noch warme Agar‐Lösung mit Hilfe eines Glasstabes
in die zuvor mit Paraffin ausgepinselten Vaginalglobuli‐Gießformen gegossen. Die Gießformen
wurden im Kühlschrank vollständig erkalten gelassen und die Gießschwarte wurde vor der Entnahme
der Kugeln mit Hilfe eines Messers entfernt. Zur kurzzeitigen Aufbewahrung der Kugeln wurden sie
einzeln in Alufolie verpackt und in einer Kruke im Kühlschrank aufbewahrt.
Die Herstellung der Agar‐Kugeln, deren Eigenschaften durch definiertes Trocknen verändert werden
sollten, erfolgte analog der bereits beschriebenen Methode mit einer Agar‐Konzentration von 5 %
und Trocknungszeiten von 30 min, 1, 2 und 5 h bei 40 ± 0,5 °C im Trockenschank. Nach der Trocknung
wurden die Kugeln einzeln in Alufolie verpackt und im Kühlschrank für maximal drei Tage
aufbewahrt. Die Prüfung der Bruchfestigkeit am Texture Analyser und der Stresstest‐Apparatur fand
dementsprechend zeitnah und ohne weitere Vorbehandlung statt.
2.2.2 Hartfett‐W32‐ und PEG‐1000‐Kugeln
Als formgebende Struktur für die Herstellung der Hartfett‐W32‐ und PEG‐1000‐Kugeln wurden 2 g‐
Vaginalglobuli‐Gießformen mit einem Durchmesser von 1,5 cm verwendet, die für die Hartfett‐W32‐
Kugeln eine Masse von ca. 2 g und für die PEG‐1000‐Kugeln von ca. 2,5 g ergaben. Das verwendete
Hartfett W32 beziehungsweise das PEG 1000 wurde über dem Wasserbad bei 40 °C bis zur
Klarschmelze aufgeschmolzen, anschließend unter stetigem Rühren bis zu einem cremigen Zustand
erkalten gelassen und danach mit der entsprechenden Menge an Modellwirkstoff vermischt. Als
Modellwirkstoff für diese Arzneiformen wurde Riboflavinphosphat verwendet, welches in der

2 Material und Methoden
21
cremigen Grundlage in einer Konzentration von 5 mg/g Grundmasse suspendiert wurde um dann
unter ständigem Rühren in die Globuliformen gegossen zu werden. Nach dem vollständigen Erkalten
der somit entstandenen Kugeln wurde die Gießschwarte mit Hilfe eines Messers entfernt, und die
Kugeln konnten vorsichtig entnommen werden. In Vorbereitung auf den anschließenden Coating‐
Prozess wurden die verbleibenden Gießränder der Kugeln mit Hilfe einer warmen Platte bei etwa
40 °C entfernt und die Kugeln abgerundet, damit der Überzug gut und gleichmäßig an ihnen haften
konnte. Die verwendete Überzugslösung war eine 5 %ige Celluloseacetat‐Lösung deren
Zusammensetzung Tab. 1 zu entnehmen ist. Dabei dient Celluloseacetat als wasserunlösliches,
sprödes Polymer, das Gemisch aus Aceton und Isopropanol als dessen organisches Lösungsmittel,
Triacetin als Weichmacher zum erleichterten Auftragen der Überzugssuspension und Talkum als
Deckmittel.
Tab. 1: Zusammensetzung des 5 %igen Celluloseacetat‐Überzugs für die Hartfett‐W32‐ und PEG‐1000‐Kugeln.
Rezepturbestandteil Anteil (g)
Celluloseacetat 5,0
Talkum 1,0
Triacetin 0,5
Aceton/Isopropanol (4:1) zu 100,0
Zur Herstellung des Überzugs wurde die entsprechende Menge an Polymer, deren Berechnung in
Kapitel 2.2.7 nachzuvollziehen ist, in Aceton vollständig in einer geschlossenen Schottflasche auf
einem Magnetrührer gelöst. Anschließend wurde die entsprechende Menge Triacetin (10 % bezogen
auf die Polymermenge) in die Lösung gegeben und diese langsam mit der Hälfte des Isopropanols
versetzt bis sich die dabei gebildeten Schlieren wieder vollständig auflösten. 20 % Talkum (bezogen
auf den Polymeranteil) wurden in der restlichen Menge Isopropanol mit Hilfe des UltraTurrax fein
dispergiert. Diese Dispersion wurde anschließend zur Polymerlösung gegeben und so lange auf dem
Magnetrührer rühren gelassen bis der Feststoffanteil fein dispergiert war. Die fertige
Überzugssuspension schimmerte opaque aufgrund des fein dispergierten Talkums. Für das
Überziehen im Mini‐Coater wurden die Arzneiformen zusammen mit Placebo‐Tabletten in die
Trommel gegeben, um ein ausreichendes Füllvolumen für einen gleichmäßigen Überzug zu erreichen.
Die Trommel drehte sich mit einer konstanten Geschwindigkeit von 20 Umdrehungen pro Minute
(rpm). Bei der Arbeit mit organischen Lösemitten muss die Entwicklung explosiver Gasgemische
vermieden werden, was durch Beachtung der oberen und unteren Explosionsgrenze (UEG), die den
Zündbereich begrenzen, geschieht. Für die Sprühversuche darf die Lösemittelkonzentration den Wert
2 Material und Methoden
22
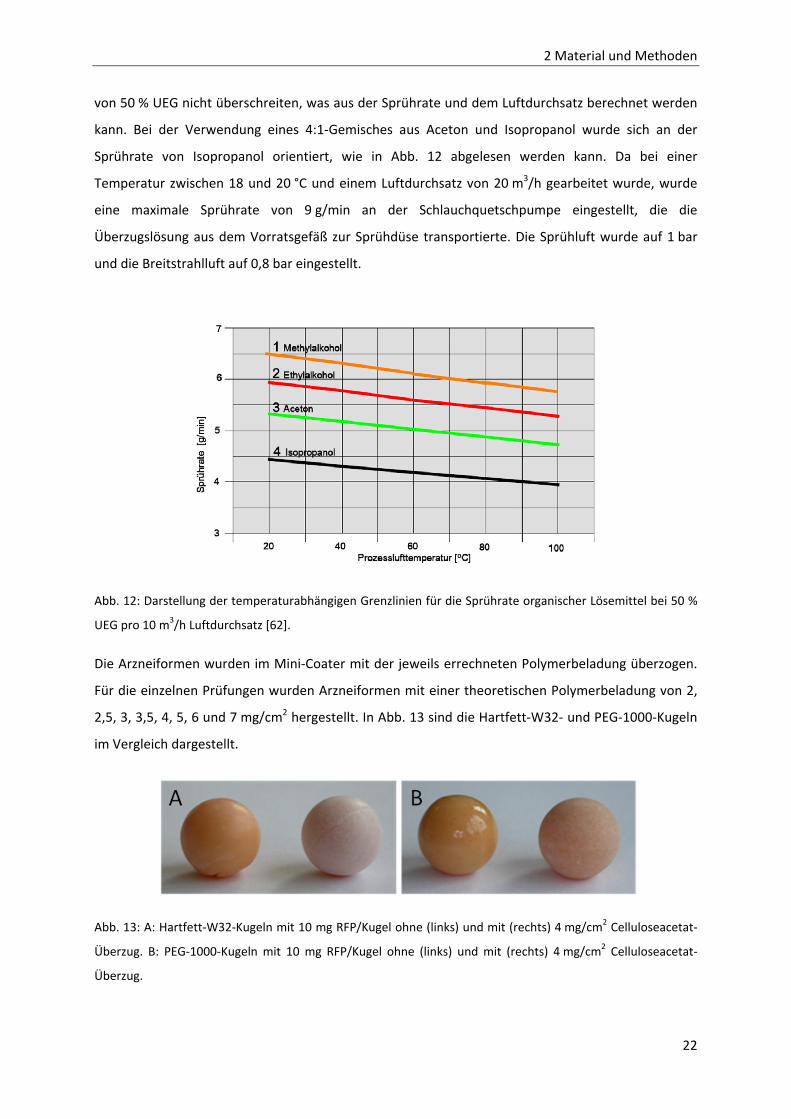
von 50 % UEG nicht überschreiten, was aus der Sprührate und dem Luftdurchsatz berechnet werden
kann. Bei der Verwendung eines 4:1‐Gemisches aus Aceton und Isopropanol wurde sich an der
Sprührate von Isopropanol orientiert, wie in Abb. 12 abgelesen werden kann. Da bei einer
Temperatur zwischen 18 und 20 °C und einem Luftdurchsatz von 20 m3/h gearbeitet wurde, wurde
eine maximale Sprührate von 9 g/min an der Schlauchquetschpumpe eingestellt, die die
Überzugslösung aus dem Vorratsgefäß zur Sprühdüse transportierte. Die Sprühluft wurde auf 1 bar
und die Breitstrahlluft auf 0,8 bar eingestellt.
Abb. 12: Darstellung der temperaturabhängigen Grenzlinien für die Sprührate organischer Lösemittel bei 50 %
UEG pro 10 m3/h Luftdurchsatz [62].
Die Arzneiformen wurden im Mini‐Coater mit der jeweils errechneten Polymerbeladung überzogen.
Für die einzelnen Prüfungen wurden Arzneiformen mit einer theoretischen Polymerbeladung von 2,
2,5, 3, 3,5, 4, 5, 6 und 7 mg/cm2 hergestellt. In Abb. 13 sind die Hartfett‐W32‐ und PEG‐1000‐Kugeln
im Vergleich dargestellt.
Abb. 13: A: Hartfett‐W32‐Kugeln mit 10 mg RFP/Kugel ohne (links) und mit (rechts) 4 mg/cm2 Celluloseacetat‐
Überzug. B: PEG‐1000‐Kugeln mit 10 mg RFP/Kugel ohne (links) und mit (rechts) 4 mg/cm2 Celluloseacetat‐
Überzug.

2 Material und Methoden
23
Gerätebedingt wurde eine vorangegangene Charge Hartfett‐W32‐Kugeln im Dragierkessel mit
Sprühpistole überzogen und enthielt in der bereits erwähnten Überzugssuspension anstelle von
Triacetin als Weichmacher 10 % Polyethylenglycol 6000 bezogen auf die Polymermenge. Die
verwendete Aufschlagsmenge auf die berechnete Polymerbeladung betrug 17 %. Dem Überzugsgut
wurden ebenfalls Placebo‐Tabletten hinzugefügt, um ein ausreichendes Füllvolumen in der Trommel
zu erreichen. Der Dragierkessel mit einem Durchmesser von 35 cm wurde mit einer
Drehgeschwindigkeit von 22 rpm betrieben. Mit diesen Kugeln, die mit theoretischen
Polymerbeladungen von 2, 2,5, 3 und 3,5 mg/cm2 hergestellt wurden, erfolgten die Prüfungen auf
Bruchfestigkeit mittels Texture Analyser und Stresstest‐Apparatur und die Freisetzung in der
Stresstest‐Apparatur. Diese Charge wird im Verlauf dieser Arbeit als Charge 1 gekennzeichnet,
während die im Mini‐Coater überzogenen Hartfett‐W32‐Kugeln als Charge 2 bezeichnet werden. Der
Austausch des zuvor verwendeten Polyethylenglycol 6000 durch Triacetin ergab sich aus
Bruchfestigkeitsversuchen mittels Texture Analyser, bei denen verschiedenen Weichmacherarten
(wasserlöslich und wasserunlöslich) und Weichmacheranteile im Überzug mit einer Polymerbeladung
von 3,5 mg/cm2 getestet wurden. Die Zusammensetzung der Überzüge sowie die dazugehörigen
Kraft‐Weg‐Diagramme finden sich im Anhang in Tab. 21 und Abb. 67.
Das Prinzip dieser Arzneiform entspricht nach Schmelzen der Grundlage bei Körpertemperatur einem
Celluloseacetat‐Ballon mit einem wirkstoffhaltigen flüssigen Kern, der in Abhängigkeit von der
Polymerbeladung bei definierten Drücken zerbrechen und den Inhalt im Sinne eines „burst release“
freigeben soll.
Als Modifizierung der Hartfett‐Kugeln wurden bei einer weiteren Charge (im Folgenden als Charge 3
bezeichnet) Alginat‐Kügelchen in das Hartfett eingebettet. Zur Herstellung der Alginat‐Kügelchen
wurde gereinigtes Wasser, welches 1,4 % Paracetamol entsprechend seiner Löslichkeit enthielt, auf
70 °C auf einem beheizbaren Magnetrührer erhitzt. Beim Erreichen der Temperatur wurde 1,5 %
Alginat eingestreut und bei ausgeschalteter Hitze 24 h rühren gelassen bis ein vollständig gelöstes Sol
entstand. Nach Auffüllen des verdunsteten Wassers, wurde das erhaltene Alginat‐Sol mit Hilfe einer
automatischen Pipette in eine 5 %ige CaCl2‐Lösung getropft. Dabei bildeten sich sofort kleine
Kügelchen, die aus der Lösung mit Hilfe eines Löffels entnommen werden konnten. Die Kügelchen
wurden sofort nach der Herstellung weiterverarbeitet und zusammen mit geschmolzenem Hartfett
H15 in 2 g‐Vaginalglobuli‐Formen ausgegossen. Dabei war zu beachten, dass sich die Kügelchen nicht
am Rand der Gießform befanden, da sie sonst nach Entnahme der Hartfett‐Kugeln mit der Luft in
Kontakt gekommen und schnell ausgetrocknet wären. Neben der Verwendung von 15 kleinen
Kügelchen (je 3 mm im Durchmesser), die in Abb. 14A zu erkennen sind, wurde auch die Möglichkeit
einer einzelnen größeren Alginat‐Kugel mit 6 mm Durchmesser in Hartfett H15 (aufgeschnitten in

2 Material und Methoden
24
Abb. 14B) untersucht. Das Tropfen des Alginat‐Sols in die CaCl2‐Lösung erfolgte dabei mit Hilfe einer
abgeschnittenen 1000 µL‐Pipettenspitze. Während die 3 mm‐Kügelchen (als fertige Arzneiform
entsprechend Charge 3a) eine durchgehende Gel‐Matrix besaßen, konnte bei den 6 mm‐Kügelchen
(als fertige Arzneiform entsprechend Charge 3b) durch Variation der Verweilzeit in der CaCl2‐Lösung
die Wandstärke der Gelmatrix beeinflusst werden. Der innere Kern blieb somit flüssig und könnte
den gelösten Wirkstoff schnell zur Verfügung stellen.
Abb. 14: A: Alginat‐Kügelchen (3 mm); B: aufgeschnittene Hartfett‐H15‐Kugel (Charge 3b) mit eingebettetem
Alginat‐Kügelchen (6 mm); Einfärbung mit blauer Lebensmittelfarbe.
Die Hartfett‐Kugeln, die die Alginat‐Kügelchen enthalten, wurden analog zur Charge 1 der Hartfett‐
W32‐Kugeln zusammen mit Placebo‐Tabletten im Dragierkessel mit Sprühpistole überzogen.
Verwendet wurde dabei der gleiche Celluloseacetat‐Überzug wie bei Charge 1 mit 20 % Talkum und
10 % PEG 6000 (beides bezogen auf den Polymer‐Anteil) in Aceton/Isopropanol als Lösungsmittel.
Die Alginat‐Kügelchen besaßen bei Verwendung von 15 Kügelchen einen theoretischen
Paracetamolgehalt von 5,9 mg und einen Durchmesser von 3 mm und bei Verwendung von einem
Kügelchen mit 6 mm Durchmesser einen theoretischen Gehalt von 2,1 mg. Dies wurde berechnet,
indem die eingewogene Menge Paracetamol bezogen auf 100 g Lösung ins Verhältnis gesetzt wurde
zur Masse von 15 bzw. einem hergestellten Alginat‐Kügelchen.
Das Freisetzungsprinzip dieser Arzneiformen entspricht ebenfalls einem bei Körpertemperatur mit
flüssigem Hartfett gefüllten Celluloseacetat‐Ballon. Die Alginat‐Kügelchen sollten eine Möglichkeit
darstellen, einen gelösten hydrophilen Wirkstoff zur Resorption im oberen Dünndarm zu bringen. Für
diese modifizierten Darreichungsformen wurde zum einen eine Gehaltsbestimmung der Alginat‐
Kügelchen und zum anderen die Bruchfestigkeitsprüfung mit dem Texture Analyser durchgeführt.
2.2.3 Paraffin‐Kapseln
Bei der Entwicklung der Paraffin‐Kapseln wurden zunächst in einem Vorversuch verschiedene Öle
und Paraffine auf ihre Verwendbarkeit als Grundlage untersucht. Dazu wurden 40 g einer 0,01 %igen

2 Material und Methoden
25
Lösung von Riboflavinphosphat in simulierter Magenflüssigkeit ohne Pepsin (SGFsp) pH 1,8 mit 40 g
des entsprechenden Öls beziehungsweise Paraffins in eine geschlossene Schottflasche gegeben und
über drei Tage unter Lichtschutz bei 37 °C und 300 Bewegungen pro Minute gerüttelt. Am Ende des
Versuchs wurde eine mögliche Umverteilung des Wirkstoffs in die lipophile Phase photographisch
festgehalten. Der Versuch wurde durchgeführt mit Rizinusöl, Erdnussöl, dickflüssigem und
dünnflüssigem Paraffin, und es zeigte sich, dass nur die Paraffine eine klare und farblose lipophile
Phase aufwiesen, während diese bei den Ölen deutlich getrübt war (Abb. 15).
Abb. 15: Ergebnis des Umverteilungsversuchs nach 72 h; von links nach rechts: dünnflüssiges Paraffin,
dickflüssiges Paraffin, Rizinusöl, Erdnussöl.
Die höhere Viskosität des dickflüssigen im Vergleich zum dünnflüssigen Paraffin führte zur
Verwendung des dickflüssigen Paraffins bei der Entwicklung dieser Arzneiform.
Als formgebende Struktur für die Paraffin‐Kapseln wurden Hartgelatine‐Steckkapseln der Größe 0
verwendet. In dem als Suspensionsgrundlage verwendeten dickflüssigen Paraffin wurde in einem
Becherglas der Modellwirkstoff Riboflavinphosphat in einer Konzentration von 10 mg/0,6 mL
suspendiert. Je 0,6 mL dieser Suspension wurden unter ständigem Rühren in die Kapselunterteile
pipettiert und anschließend mit den Kapseloberteilen verschlossen. Um ein ungewolltes Öffnen der
Arzneiformen beim anschließenden Coating‐Prozess zu verhindern, wurden die Kapseln versiegelt.
Dafür wurden die Kapseloberteile vor dem Aufsetzen mit Wasser angefeuchtet, sodass es zu einem
Anlösen der Gelatinehälften und einer Verbindung beim Trocknen kam. Im Anschluss daran konnten
die so entstandenen Arzneiformen im Mini‐Coater mit einer 5 %igen ethanolischen Ethylcellulose‐
Suspension überzogen werden, deren Zusammensetzung in Tab. 2 aufgelistet ist. Dabei dient
Ethylcellulose als wasserunlösliches, sprödes Polymer, Ethanol 99 % als dessen Lösungsmittel,
Triacetin als Weichmacher zum erleichterten Auftragen der Überzugssuspension und Talkum als
Deckmittel. Für die einzelnen Prüfungen wurden Kapseln mit einer theoretischen Polymerbeladung
von 4 mg/cm2 hergestellt.

2 Material und Methoden
26
Tab. 2: Zusammensetzung des 5 %igen Ethylcellulose‐Überzugs für die Paraffin‐Kapseln.
Rezepturbestandteil Anteil (g)
Ethylcellulose 5,0
Talkum 1,0
Triacetin 0,5
Ethanol 99 % zu 100,0
Für die Herstellung der Überzugslösung wurde die entsprechende Menge an Polymer, deren
Berechnung dem Kapitel 2.2.7 zu entnehmen ist, in ca. 60 % der Ethanol‐Menge auf einem
Magnetrührer gelöst und anschließend mit Triacetin ergänzt. Das Talkum wurde in der restlichen
Ethanol‐Menge mit Hilfe des UltraTurrax fein dispergiert und die entstandene Suspension danach
unter Rühren in die Ethylcellulose‐Lösung gegeben. Das Überziehen im Mini‐Coater erfolgte analog
zu den Hartfett‐ und PEG‐Kugeln mit Placebo‐Tabletten im Überzugsgut, um ein Mindestvolumen in
der Trommel zu erreichen. Die Einstellungen bezüglich Trommelumdrehung, Prozessluft, Sprühluft,
Breitstrahlluft, Sprührate und Temperatur wurden analog zum Auftragen des Celluloseacetat‐
Überzugs auf die beiden Kugelsorten gewählt. Abb. 16 zeigt die überzogenen Paraffin‐Kapseln mit
suspendiertem Riboflavinphosphat.
Abb. 16: Paraffin‐Kapseln mit 10 mg RFP/Kapsel und 4 mg/cm2 Ethylcellulose‐Überzug.
Direkt vor den einzelnen Prüfungen wurden radial acht Löcher unterhalb der Verschlussnaht mit Hilfe
einer Insulinspritze (Kanülendurchmesser: 0,33 mm) in die Kapsel gestochen. Diese Löcher sollten das
Eindringen von Wasser in die Kapsel ermöglichen und somit zu einem Aufweichen der Gelatine‐Hülle
zwischen den Löchern und einer Sollbruchstelle führen, sodass die Kapsel radial komplett aufreißen
und den Inhalt vollständig freigeben konnte.
2.2.4 Tristearin‐Kapseln
Für die Herstellung der Tristearin‐Kapseln wurden Hartgelatine‐Steckkapseln der Größe 0 (Abb. 17A,
Kapsel 1) als Gießform verwendet. Tristearin wurde auf einem siedenden Wasserbad aufgeschmolzen
bis es eine Temperatur von 85 ‐ 90 °C erreicht hatte. Mit Hilfe einer 1000 µL Eppendorf‐Pipette

2 Material und Methoden
27
wurde die entsprechende Menge an flüssigem Tristearin zügig in das Kapselunterteil pipettiert und
dieses dann sofort horizontal gedreht, sodass sich das flüssige Hartfett gleichmäßig auf der
Innenseite der Kapseln verteilen und dort erstarren konnte, wie in Abb. 17A (Kapsel 2) und Abb. 17B
zu erkennen ist. Sobald die Innenbeschichtung der Kapselunterteile komplett erstarrt war, wurden
sie mit einem wirkstoffhaltigen Hydrogel befüllt (Zusammensetzung siehe Tab. 3). Entsprechend
seiner Wasserlöslichkeit wurden 14 g/L Paracetamol mit Hilfe eines Magnetrührers in destilliertem
Wasser gelöst. Unter Rühren wurde dann die entsprechende Menge des Gelbildner
Hydroxyethylcellulose aufgestreut, sodass nach einstündiger Quellung und Ergänzung des
verdunsteten Wassers ein 5 %iges Gel erhalten wurde.
Tab. 3: Zusammensetzung des 5 %igen Paracetamol enthaltenden Hydrogels.
Rezepturbestandteil Anteil (g)
Paracetamol 0,399
Hydroxyethylcellulose 1,5
Gereinigtes Wasser zu 30,0
Mit Hilfe einer 1000 µL‐Pipette und einer abgeschnittenen Spitze wurde das Gel massedosiert
(0,427 g ± 0,002 g für n = 10) in das beschichtete Kapselunterteil gefüllt, sodass sich ein theoretischer
Paracetamol‐Gehalt von 5,68 mg pro Kapsel ergab. Anschließend wurden 85 µL geschmolzenes
Tristearin mit einer Eppendorf‐Pipette zügig auf das Gel pipettiert, wobei auf einen gleichmäßigen
Kontakt mit dem bestehenden Tristearin‐Rand geachtet wurde, um eine dichte Kapsel zu erhalten.
Durch das Aufbringen des unbeschichteten Kapseloberteils wurde die Kapsel verschlossen und die
fertige Arzneiform erhalten. Für die einzelnen Versuche wurden Kapseln mit einem Tristearin‐
Füllvolumen von 220, 250, 300, 330, 360, 390, 400 und 500 µL hergestellt. Zur Vorbereitung der
Kapseln auf die Testung der Bruchkraft am Texture Analyser wurde die Gelatine‐Hülle in 40 °C
warmem Wasser entfernt, damit sie keinen Einfluss auf das Testergebnis ausüben konnte, wie in
Abb. 17A (Kapsel 3) zu erkennen ist. Die Versuche im warmem Medium, also die Freisetzung in der
Paddle‐ und der Stresstest‐Apparatur sowie die Gehaltsbestimmung erforderten keine Entfernung
der äußeren Gelatine‐Hülle, was eine bessere Stabilität beim Handling gewährleisten konnte. In Abb.
17A (Kapsel 4) ist eine Kapsel mit Methylenblau‐Füllung und abgelöster Gelatine‐Hülle zu erkennen,
die die Gleichmäßigkeit der Innenbeschichtung deutlich machen soll. Die schematische Darstellung
dieser entwickelten Arzneiform mit den unterschiedlichen Schichten ist weiterhin in Abb. 17C zu
erkennen.

2 Material und Methoden
28
Abb. 17: A: Herstellungsschritte der Tristearin‐Kapsel. B: Draufsicht auf eine leere Tristearin‐Kapsel.
C: Schematischer Aufbau einer Tristearin‐Kapsel.
2.2.5 Kaugummi‐Manteltabletten
Zur Herstellung der Kaugummi‐Manteltabletten wurden zunächst Hartfettkerne mit Hilfe einer
Gießform ausgegossen. Zur Herstellung der Gießform wurden Placebo‐Tabletten entsprechender
Form und Größe hergestellt, deren Zusammensetzung in Kapitel 2.2.6 beschrieben wird und die eine
Bruchfestigkeit von mindestens 80 N aufwiesen. Diese wurden unter Verwendung von Wachs und in
einem Abstand von ca. 1 cm auf den Boden einer Petrischale mit einem Durchmesser von 11,5 cm
geklebt, sodass der Rand an der Klebefläche der Placebo‐Tabletten weitestgehend abgedichtet war.
Nach Hersteller‐Anleitung wurden die beiden flüssigen Komponenten von HS‐Dubliermasse (Z‐Dupe,
20 shore) im Masse‐Verhältnis 1:1 zügig und gleichmäßig gemischt und über die aufgeklebten
Tablettenkerne gegossen, sodass diese vollständig bedeckt waren. Nach einer Trocknungszeit von
mindestens 30 min konnte die Silikon‐Masse in einem Stück von den Tabletten und der Petrischale
gelöst werden, sodass eine Gießform mit den Negativen der Placebo‐Tabletten entstand. Es wurden
Silikon‐Gießformen unterschiedlicher Form und Größe der Vertiefungen entsprechend der
verwendeten Hartfettkerne hergestellt. Zur Herstellung der Hartfettkerne für die einzelnen
Kaugummi‐Tabletten wurden die Vertiefungen dieser Gießform mit einer Suspension aus
Hartfett W32 und Riboflavinphosphat ausgegossen, sodass sich ein Sollgehalt von 10 mg pro Kern
ergab. Dies geschah, wie schon in Kapitel 2.2.2 erwähnt, unter Verwendung des
Cremeschmelzverfahrens. Die erhärteten Kerne konnten nach Erkalten aus der Silikon‐Form
entnommen und für die Herstellung der Manteltabletten weiterverwendet werden.
Das Prinzip dieser Arzneiformen beinhaltet einen Hartfettkern, der sich bei Körpertemperatur
verflüssigen soll. Dadurch soll die Stabilität des äußeren Pulverkerns, der mit einer geringen
Presskraft um den Hartfettkern verpresst wurde, herabgesetzt werden. Ein Mantel aus hydrophober
Kaugummimasse, der als äußerste Schicht um den Tablettenkern verpresst wurde, sollte das
Eindringen von Wasser in die Arzneiform und dadurch ein frühzeitiges Aufplatzen verhindern. Dies
sollte mit Hilfe der eingefügten Sollbruchstellen erst bei Einwirken eines definierten Drucks
stattfinden.

2 Material und Methoden
29
Durch Variation von Größe, Form, Zusammensetzung und Stabilität der Kerne und des Mantels,
sowie durch das Einfügen unterschiedlicher Sollbruchstellen entstanden 50 verschiedene Chargen,
die zunächst auf ihre Beständigkeit in 37 °C warmem Wasser geprüft wurden. Auf die Angabe der
detaillierten Parameter für diese Chargen wird aus Gründen der Übersichtlichkeit verzichtet.
Während bei den Ergebnissen der Prüfung auf Bruchfestigkeit mittels Texture Analyser in Kapitel
3.6.2 interne Chargenbezeichnungen verwendet werden, wird für die drei Chargen, die weiteren
Prüfungen unterzogen wurden, eine neue, vereinfachte Bezeichnung (Charge 1 ‐ 3) verwendet. Die
Zuordnung zwischen interner und neuer Chargenbezeichnung ist in Kapitel 3.6.2 dargestellt.
Auswahlkriterien für eine weitere Prüfung waren eine Beständigkeit in 37 °C warmem Wasser von
mehr als 120 min und ein vielversprechendes Verhalten im Texture Analyser. Im Folgenden wird die
Herstellung der Kaugummi‐Tabletten der Chargen 1 – 3 erläutert, die genaue Zusammensetzung
findet sich in Tab. 4.
Tab. 4: Übersicht der geprüften Chargen der Kaugummi‐Manteltabletten.
Charge 1 Charge 2 Charge 3
Hartfettkern Ø = 9 mm; h = 3 mm bikonvex
Ø = 9 mm; h = 3 mm bikonvex
Ø = 10 mm; h = 6 mm biplan
Pulverkern Lactose 80 % HEC 20 % Ø = 10 mm; h = 6 mm bikonvex
MCC 80 % HEC 20 % Ø = 9 mm; h = 6 mm bikonvex
‐
Trennschicht ‐ NaCl ‐ Kaugummimantel
‐ gepresst
‐ Sollbruchstellen
‐ Anzahl Dippen
Ø = 13 mm bikonvex horizontal 3; 9
Ø = 12 mm bikonvex vertikal 13 Löcher 3; 9
Ø = 12 mm bikonvex vertikal gekreuzt, 4 Löcher 9
Um die Hartfettkerne der für die Chargen 1 und 2 verwendeten Tabletten mit einer Steghöhe von
3 mm wurde ein Pulvermantel gepresst, der aus einem Füllmaterial (Lactose oder MCC) und 20 %
Hydroxyethylcellulose bestand. Bei der Herstellung der Charge 2 wurde zwischen dem Hartfettkern
und dem Pulveranteil zusätzlich eine jeweils 1 mm hohe Trennschicht aus Natriumchlorid eingefügt,
welche verhindern sollte, dass das sich verflüssigende Hartfett in die Pulverschicht hineinfließt und
somit das Brechverhalten negativ beeinflussen könnte. Die Tabletten der Charge 3 bestanden aus
einem reinen Hartfettkern, auf den der Kaugummimantel direkt gepresst wurde. Das Kaugummi‐
Granulat wurde bei allen Chargen als Einzelschicht der Granulatkörner aufgepresst. Die Dicke der
Steg‐Seiten wurde durch die Verwendung unterschiedlicher Stempel für Kern und Mantel festgelegt.
Um ein einfaches Ablösen der Kaugummimasse zu gewährleisten und ein Verkleben zu verhindern,

2 Material und Methoden
30
mussten der Ober‐ und Unterstempel der Exzenterpresse beim Verpressen des Kaugummi‐Granulats
mit Hilfe von Magnesiumstearat geschmiert werden. Die aufgepressten Kaugummi‐Mäntel wurden
entsprechend ihrer Chargen unter Verwendung einer Rasierklinge und einer 0,33 mm Kanüle mit
Sollbruchstellen versehen, anschließend auf eine 0,33 mm Kanüle aufgesteckt und insgesamt drei
bzw. neun Mal in eine 20 %ige Lösung von Kaugummi‐Grundmasse in Heptan gedippt. Nach
dreimaligem Dippen erfolgte jeweils eine Trocknungszeit von 15 min. Nach vollständiger Trocknung
wurden die Tabletten von den Kanülen entfernt und die dabei entstandenen Löcher mit einem
Tropfen der Kaugummilösung verschlossen. Um einen festen und trockenen Überzug zu
gewährleisten, wurden die Tabletten mindestens 12 h bei Raumtemperatur getrocknet. Die genauen
Parameter der drei Chargen sind in Tab. 4 aufgeführt und durch die schematische Darstellung in Abb.
18 verdeutlicht.
Abb. 18: Schematische Darstellung der Kaugummi‐Manteltabletten der Chargen 1 – 3; grau: Hartfettkern,
hellblau: Trennschicht NaCl, orange: Pulvermantel, dunkelblau: Kaugummimantel mit Sollbruchstellen.
Im Folgenden werden die Kaugummi‐Tabletten mit dreimaligem Dippen als Charge 1, 2 und 3 und die
nach neumaligem Dippen als Charge 1a, 2a und 3a bezeichnet. Bis auf die unterschiedliche Anzahl
der Dippvorgänge nach Einbringen der Sollbruchstellen in den Kaugummimantel der fertigen
Tabletten gibt es zwischen den Darreichungsformen der Charge 1, 2, 3 und 1a, 2a und 3a keine
weiteren Unterschiede.
Für die Prüfungen der Gehaltsbestimmung, der Freisetzung in der Paddle‐Apparatur und den
Bruchfestigkeitsversuchen am Texture Analyser wurden Tabletten der Chargen 1, 2 und 3 verwendet.
Für die Bruchfestigkeitstests und die Freisetzung an der Stresstest‐Apparatur wurden sowohl die
Chargen 1 und 2 als auch 1a, 2a und 3a verwendet.
2.2.6 Placebo‐Tabletten
Für das Auffüllen der Trommel im Coating‐Prozess der Hartfett‐ und PEG‐Kugeln, sowie der Paraffin‐
Kapseln wurden Placebo‐Tabletten hergestellt, deren Zusammensetzung in Tab. 5 zu erkennen ist.
MCC wurde zusammen mit Aerosil® für etwa 5 min im Kubusmischer gemischt und nach Zugabe der
entsprechenden Menge an Magnesiumstearat erneut für maximal 5 min vermengt. Die so
entstandene Tabletten‐Mischung wurde dann an einer Exzenterpresse zu bikonvexen Tabletten mit

2 Material und Methoden
31
einem Durchmesser von 13 mm, einer Steghöhe von 3 mm und einer Bruchkraft von mindestens
90 N bei einer Presskraft von 7,5 Skalenteilen verpresst. Dadurch waren sie stabil genug, um den
Coating‐Prozess unbeschadet zu überstehen.
Tab. 5: Zusammensetzung der Placebo‐Tabletten.
Rezepturbestandteil Anteil (g)
Magnesiumstearat 2,5
Aerosil® 5,0
Mikrokristalline Cellulose zu 500,0
2.2.7 Berechnung der Oberflächen
Die verwendeten Kugeln hatten einen Durchmesser von 15 mm und damit eine Oberfläche von
7,07 cm2. Die Oberfläche einer einzelnen Kugel wurde mit der Anzahl der zu überziehenden Kugeln
multipliziert, um die Gesamtoberfläche zu erhalten.
Um die Oberfläche der Placebo‐Tabletten zu ermitteln, wurde zunächst anhand des Durchmessers
von 13 mm die Kreisfläche und dann anhand der Steghöhe die Mantelfläche ausgerechnet. Trotz der
Verwendung von bikonvexen Tabletten wurde bei der Berechnung zur Vereinfachung von biplanen
Tabletten ausgegangen. Demensprechend berechnete sich die Oberfläche einer Tablette zu 3,88 cm2.
Für die Berechnung der Oberfläche der Paraffin‐Kapseln, wurde vereinfacht von einem Zylinder mit
einem Kreisdurchmesser von 7,5 mm und einer Steghöhe von 23,56 mm ausgegangen, was den
Maßen einer Kapsel der Größe 0 entspricht. Als Oberfläche einer Kapsel wurden somit 5,97 cm2
angenommen.
Die somit errechneten Oberflächen des Überzugsguts wurden mit der gewünschten
Polymerbeladung (beispielhaft für 4 mg/cm2) multipliziert (Formel 1) und bei Verwendung des Mini‐
Coaters zusätzlich mit einem Aufschlag von 50 % bedacht, um das Absetzen der Polymerlösung in
Schläuchen und Kessel zu berücksichtigen. Der 50 %ige Aufschlag an Überzugspolymer ergab sich aus
vorangegangenen Untersuchungen bezüglich theoretischer und tatsächlicher Polymerbeladung.
m=4mg
cm2 ·(OTable en+OArzneiform) + 50 % Aufschlag (1)
Es wurde in beiden Fällen (Kugeln und Kapseln) mit einer 5 %igen Polymerlösung zum Überziehen
gearbeitet. Unterschiedliche Auftragsmengen ergaben sich innerhalb eines Sprühprozesses, indem
zuvor die jeweils versprühte Masse an Überzugssuspension berechnet wurde, bei der die

2 Material und Methoden
32
entsprechende Anzahl an Arzneiformen aus der Trommel entnommen wurden, während die
verbliebenen Arzneiformen weiter überzogen wurden.
2.3 Methoden zur Untersuchung der Arzneiformen
2.3.1 Gehaltsbestimmung
2.3.1.1 Paracetamol in Agar‐Kugeln
Für die Gehaltsbestimmung von Paracetamol in Agar‐Kugeln, deren Ablauf schematisch in Abb. 19
dargestellt ist, wurde von insgesamt sechs Stichproben jeweils eine in einen 200 mL‐Enghals‐
Erlenmeyerkolben mit 100,0 g SGFsp pH 1,8 gegeben. Die genaue Zusammensetzung dieses
Freisetzungsmediums ist in Kapitel 2.3.3 nachzulesen. Mit Hilfe eines am unteren Ende verdickten
Glasstabes wurde die Arzneiform eine Minute lang gründlich zerstoßen, und anschließend wurde die
Agar‐Suspension mit einem Rührfisch auf einem Magnetrührer für 30 min bei 300 rpm gerührt.
Abb. 19: Schema der Gehaltsbestimmung von Paracetamol aus Agar‐Kugeln.
Nach Filtration der Agar‐Bruchstücke durch einen Papierfilter und einer 1:10‐Verdünnung der
erhaltenen Wirkstofflösung mit SGFsp pH 1,8 wurde die Absorption photospektrometrisch bei einer
Messwellenlänge von 242 nm und einer Korrekturwellenlänge von 450 nm gegen SGFsp pH 1,8 als
Blindwert vermessen. Die Berechnung des Paracetamol‐Gehalts errechnete sich aus der Differenz der
Absorptionen (A242nm – A450nm), eingesetzt in die Geradengleichung des Mittelwertes der
Kalibrationsgeraden (Abb. 65) multipliziert mit dem Verdünnungsfaktor (VF = 10). Die Kalibration
erfolgte anhand einer Dreifachbestimmung an drei verschiedenen Tagen. Dazu wurde eine
Paracetamol‐Stammlösung (0,1 g/L SGFsp pH 1,8) hergestellt, die auf eine Verdünnungsreihe mit den
Konzentrationen 2, 4, 6, 8, 10, 12 und 14 mg/L verdünnt und bei 242 nm vermessen wurde.

2 Material und Methoden
33
2.3.1.2 Riboflavinphosphat in Hartfett‐W32‐Kugeln
Für die Gehaltsbestimmung der Hartfett‐W32‐Kugeln (Schema in Abb. 20) wurde je eine nicht‐
überzogene Kugel in einem Erlenmeyerkolben mit Rührfisch in 5 mL Diethylether innerhalb von 5 min
bei 400 rpm auf dem Magnetrührer aufgelöst. Anschließend wurden 50 mL 50 °C warmer SGFsp
pH 1,8 hinzugefügt und die Mischung erneut für 5 min bei 400 rpm gerührt. Dabei ging das zuvor in
der organischen Hartfettlösung suspendierte Riboflavinphosphat in die wässrige Phase über und
löste sich dort sofort. Mit Hilfe von Alufolie wurde unter Lichtschutz der enthaltene Ether in einem
50 °C warmen Wasserbad über 20 min abgedampft, bis eine etherfreie, wässrige Wirkstofflösung mit
dem geschmolzenen Hartfett zurück blieb.
Abb. 20: Schema der Gehaltsbestimmung von Riboflavinphosphat aus Hartfett‐W32‐Kugeln.
Die anschließende Abkühlphase auf Raumtemperatur führte dazu, dass sich das Hartfett wieder
verfestigte und die Riboflavinphosphat‐haltige Lösung mit Hilfe eines Papierfilters in einen 100,0 mL
Maßkolben abgetrennt werden konnte. Mehrmaliges Spülen des Filters in denselben Kolben sowie
anschließendes Auffüllen mit SGFsp pH 1,8 bis zur Eichmarke ergab die fertige Wirkstofflösung, die
nach zehnfacher Verdünnung photometrisch bei einer Messwellenlänge von 267 nm und einer
Korrekturwellenlänge von 600 nm gegen SGFsp pH 1,8 als Blindwert vermessen werden konnte. Der
Gehalt an Riboflavinphosphat in einer Arzneiform ergab sich dann aus der Differenz der beiden
Absorptionen (A267nm – A600nm) multipliziert mit dem Verdünnungsfaktor (VF = 10) und dem
Kalibrationsfaktor (KF = 16,81 mg/L). Der Kalibrationsfaktor ergab sich aus einer dreifachen
Kalibration an drei verschiedenen Tagen mit Hilfe einer Riboflavinphosphat‐Stammlösung (0,1 g/L),
die auf eine Verdünnungsreihe mit den Konzentrationen 2, 4, 6, 8, 10 und 12 mg/L verdünnt und bei
267 und 600 nm vermessen wurde. Das arithmetische Mittel aus den Kehrwerten der drei Anstiege
der Kalibrationsgeraden (Anhang Abb. 66) ergab den Kalibrationsfaktor von 16,81 mg/L.

2 Material und Methoden
34
Die Gehaltsbestimmung der Hartfett‐H15‐Kugeln aus Charge 3a, die als Modellarzneistoff
Paracetamol in 15 eingebetteten Alginat‐Kügelchen (Sollgehalt Paracetamol für 15 Kügelchen:
5,9 mg) enthielten, wurde zu einem früheren Zeitpunkt und dadurch vereinfacht durchgeführt. Eine
überzogene, mit einer Rasierklinge halbierte Kugel wurde mit 100,0 g 37 °C warmen Wassers bei
300 rpm für 30 min rühren gelassen. Nach der Hälfte der Zeit wurden die enthaltenen Alginat‐
Kügelchen mit Hilfe eines am unteren Ende verdickten Glasstabes zerdrückt, sodass durch eine
größere Oberfläche, das Paracetamol aus der Alginat‐Matrix in das Wasser übergehen konnte. Vor
der Absorptionsmessung bei 242 nm gegen Wasser als Blindwert wurde die erhaltene
Wirkstofflösung 1:10 verdünnt. Für die Berechnung des Paracetamol‐Gehalts wurde ein zuvor
ermittelter Kalibrationsfaktor von 15,42 mg/L verwendet, der sich aus einer Verdünnungsreihe von
Paracetamol in Wasser mit Konzentrationen zwischen 2 ‐ 14 mg/L ergab.
Die Gehaltsbestimmung der Hartfett‐H15‐Kugeln aus Charge 3b, die nur ein 6 mm großes Alginat‐
Kügelchen enthielten, erfolgte analog zu der soeben erläuterten Methode. Unterschiedlich war die
Verwendung von SGFsp pH 1,8 anstelle von Wasser und die anschließende Verdünnung der
erhaltenen Paracetamol‐Lösung von 1:2 statt 1:10. Als Kalibrationsfaktor aus einer Verdünnungsreihe
von Paracetamol in SGFsp pH 1,8 mit Konzentrationen zwischen 2 – 14 mg/L ergab sich ein Wert von
17,34 mg/L, der für die Berechnung verwendet wurde. Beide Gehaltsbestimmungs‐Methoden für die
drucksensitiven Arzneiformen mit Alginat‐Kügelchen wurden mit einer Stichprobenzahl von n = 10
durchgeführt.
2.3.1.3 Riboflavinphosphat in PEG‐1000‐Kugeln
Um den Wirkstoffgehalt der PEG‐1000‐Kugeln anhand des Schemas in Abb. 21 zu bestimmen, wurde
eine nicht‐überzogene Kugel in 75 mL SGFsp pH 1,8 bei 37 °C gegeben und mit Hilfe eines Rührfisches
für 5 min bei 600 rpm aufgelöst. Die dabei entstandene Wirkstofflösung wurde vollständig und mit
anschließendem Nachspülen des Glasgefäßes in einen 100,0 mL Maßkolben überführt, bis zur
Eichmarke aufgefüllt und nach einer zehnfachen Verdünnung bei 267 und 600 nm gegen SGFsp
pH 1,8 als Blindwert vermessen. Der Gehalt an Riboflavinphosphat in einer Arzneiform ergab sich
dann aus der Differenz der beiden Absorptionen (A267nm – A600nm) multipliziert mit dem
Verdünnungsfaktor (VF = 10) und dem Kalibrationsfaktor (KF = 16,81 mg/L), der wie zuvor bei der
Gehaltsbestimmung der Hartfettkugeln in Kapitel 2.3.1.2 beschrieben, ermittelt wurde.

2 Material und Methoden
35
Abb. 21: Schema der Gehaltsbestimmung von Riboflavinphosphat aus PEG‐1000‐Kugeln.
2.3.1.4 Riboflavinphosphat in Paraffin‐Kapseln
Analog zur Gehaltsbestimmung von Riboflavinphosphat aus Hartfett‐W32‐Kugeln, wurde auch bei
den Paraffin‐Kapseln Diethylether als apolares Lösungsmittel verwendet, um das Paraffin zu lösen
und das zuvor darin suspendierte Riboflavinphosphat in Kontakt mit dem wässrigen Medium zu
bringen. Wie in Abb. 22 zu erkennen ist, wurde je eine nicht‐überzogene Paraffin‐Kapsel mit 50 mL
50 °C warmem SGFsp pH 1,8 und einem Rührfisch in einem Erlenmeyerkolben für 1 min bei 400 rpm
gerührt, um die Auflösung der Gelatinehülle zu bewirken. Im Anschluss wurden 5 mL Diethylether
hinzugefügt und die Lösung für 10 min bei 600 rpm stark gemischt, was erst durch das kurzfristige
Lösen der Paraffin‐Tröpfchen zur vollständigen Auflösung des RFP im SGFsp pH 1,8 führte. Unter
Lichtschutz mit Hilfe von Alufolie wurde der Ether in einem 50 °C warmen Wasserbad über 20 min
abgedampft.
Abb. 22: Schema der Gehaltsbestimmung von Riboflavinphosphat aus Paraffin‐Kapseln.

2 Material und Methoden
36
Eine anschließende Abkühlung der Lösung auf Raumtemperatur ermöglichte die Abtrennung des
Paraffins mit Hilfe eines Papierfilters in einen 100,0 mL Maßkolben. Mehrfaches Spülen des
Glasgefäßes und des Filters in denselben Maßkolben und anschließendes Auffüllen bis zur Eichmarke
ergab eine Wirkstofflösung, die nach einer 1:10‐Verdünnung bei 267 und 600 nm gegen SGFsp pH 1,8
als Blindwert vermessen werden konnte. Der Gehalt an Riboflavinphosphat in einer Kapsel ergab sich
aus der Differenz der Absorptionen (A267nm – A600nm) multipliziert mit dem Verdünnungsfaktor
(VF = 10) und dem Kalibrationsfaktor (KF = 16,81 mg/L, Bestimmung siehe Kapitel 2.3.1.2).
2.3.1.5 Paracetamol in Tristearin‐Kapseln
Nach Lösen der Gelatinehülle in 37 °C warmem destilliertem Wasser erfolgte die Gehaltsbestimmung
der Tristearin‐Kapseln (Schema in Abb. 23) in einem 200 mL‐Enghals‐Erlenmeyerkolben mit 100,0 g
SGFsp pH 1,8. Die Kapsel wurde in den Kolben gegeben und mit Hilfe eines am unteren Ende
verdickten Glasstabes 1 min lang kräftig zerstoßen. Anschließend wurde die entstehende
Arzneistofflösung mittels Rührfisch auf einem Magnetrührer für 30 min bei 300 rpm gemischt, wobei
sich sowohl das Gel als auch der Wirkstoff komplett in SGFsp pH 1,8 lösen konnte. Nach einer 1:10‐
Verdünnung mit SGFsp pH 1,8 wurde die Absorption der Paracetamol‐Lösung bei einer
Messwellenlänge von 242 nm und einer Korrekturwellenlänge von 450 nm gegen SGFsp pH 1,8 als
Blindwert vermessen. Die Berechnung des Gehalts erfolgte durch Einsetzen der Differenz der
Absorptionen (A242nm – A450nm) in die Geradengleichung des Mittelwertes der Kalibrationsgeraden für
Paracetamol (siehe Kapitel 2.3.1.1) und unter Zuhilfenahme des Verdünnungsfaktors (VF = 10).
Abb. 23: Schema der Gehaltsbestimmung von Paracetamol aus Tristearin‐Kapseln.
2.3.1.6 Riboflavinphosphat in Kaugummi‐Manteltabletten
Analog zur Gehaltsbestimmung von Riboflavinphosphat aus Hartfett‐W32‐Kugeln wurde der
Wirkstoffgehalt in den Kaugummi‐Tabletten bestimmt. Dazu wurden die Tabletten waagerecht

2 Material und Methoden
37
halbiert, in einen Erlenmeyerkolben gegeben und zusammen mit 4 mL Diethylether und einem
Rührfisch für 5 min bei 400 rpm auf einem Magnetrührer gemischt. Dies führte zur Auflösung des
enthaltenen Hartfettkerns und ergab eine Suspension von Riboflavinphosphat in Ether. Nach Zugabe
von 50 mL 50 °C warmem SGFsp pH 1,8 wurde die Mischung erneut für 5 min bei 400 rpm
durchmischt, wodurch sich der suspendierte Modellarzneistoff bei Kontakt mit dem wässrigen
Medium sofort löste. Zum Abdampfen der nun wirkstofffreien Etherphase wurde der mit Alufolie
umwickelte Kolben bei 50 °C für 20 min im Wasserbad belassen. In der sich anschließenden
Abkühlungsphase erhärtete das Hartfett wieder und konnte zusammen mit den restlichen
Tablettenbestandteilen mittels Filtration abgetrennt werden. Die erhaltene Wirkstofflösung wurde
quantitativ in einen 100,0 mL Maßkolben überführt, nach mehrmaligem Spülen des Filters bis zur
Eichmarke aufgefüllt und nach einer zehnfachen Verdünnung bei einer Wellenlänge von 267 und
600 nm vermessen. Als Blindwert diente eine wirkstofffreie Kaugummi‐Tablette, die entsprechend
der verwendeten Charge in gleicher Weise hergestellt und für die Absorptionsmessung aufbereitet
wurde. Der Gehalt ergab sich aus der Differenz der beiden Absorptionen (A267nm – A600nm) multipliziert
mit dem Verdünnungfaktor (VF = 10) und dem zuvor aus drei Kalibrationen ermittelten
Kalibrationsfaktor (KF = 15,56 mg/L). Die Ermittlung des Kalibrationsfaktors erfolgte analog zu der
bereits bei den Hartfett‐Kugeln beschriebenen Methode aus drei Kalibrationen (Anhang Abb. 66) mit
einer Riboflavinphosphat‐Verdünnungsreihe von 2, 4, 6, 8, 10 und 12 mg/L. Das arithmetische Mittel
aus den Kehrwerten der Anstiege ergab einen Kalibrationsfaktor von 15,56 mg/L.
2.3.2 Bruchfestigkeit mittels Texture Analyser
Die Versuche am Texture Analyser wurden aufgrund eines Gerätewechsels an zwei verschiedenen
Modellen, dem TA.XT plus (Stable Micro Systems) und dem TAplus (Lloyd Instruments) durchgeführt,
deren gerätetechnische Unterschiede in Tab. 6 zu erkennen sind.
Tab. 6: Unterschiede der verwendeten Texture Analyser.
Parameter TA.XT plus TAplus
Kraftmesszelle 100 N 50 N
Geschwindigkeit 0,50 mm/s 1,67 mm/s
Beide Modelle sind ähnlich aufgebaut und bestehen aus einer Bodenplatte, auf die die Arzneiform
platziert wird und einem beweglichen Arm mit einer Kraftmesszelle, der auf die Bodenplatte mit
einer voreingestellten Geschwindigkeit herabfahren kann. An dem beweglichen Arm können Stempel
unterschiedlicher Größe und Form angebracht werden, um verschiedenste Proben auf ihre
physikalischen Eigenschaften wie Elastizität, Konsistenz oder Klebrigkeit zu untersuchen. Für die

2 Material und Methoden
38
Untersuchungen in dieser Arbeit wurde ein planer Stempel mit einem Durchmesser von 12 mm
verwendet, der die Kraft registrierte, mit der er auf die Arzneiform wirkte. Die verschiedenen
Arzneiformen mussten unterschiedlich auf die Prüfungen am Texture Analyser vorbereitet werden:
während die Agar‐Kugeln ohne weitere Vorbehandlung getestet werden konnten, wurde bei den
Tristearin‐Kapseln die äußere Gelatinehülle für ca. 5 min in 37 °C warmem Wasser abgelöst, da diese
sonst die Brucheigenschaften beeinflusst hätte. Sowohl die Paraffin‐Kapseln als auch die Hartfett‐
W32‐ und PEG‐1000‐Kugeln wurden vor ihrer Testung für 20 – 30 min in 37 °C warmem Wasser
belassen. Dies hatte für die Kugeln zur Folge, dass der Kern vollständig schmelzen konnte und für die
Kapseln, dass durch die radial angeordneten Löcher Wasser eindringen und die innere Gelatinehülle
auflösen konnte. Nach dieser Vorbereitung wurde die Arzneiform zügig in eine große Petrischale auf
der Bodenplatte gelegt und sofort mit der Testung begonnen. Die erhaltenen Daten wurden mit Hilfe
der zu dem jeweiligen Gerät gehörigen Software aufgezeichnet und als Kraft‐Weg‐Diagramm
ausgewertet. In diesem Diagramm ergab der erste steile Peak mit einem anschließenden steilen
Abfallen der Kurve den Punkt, an dem die Arzneiform brach und wurde zur Ermittlung der
Mittelwerte (MW) herangezogen. Tab. 7 zeigt die Arzneiformen mit ihren veränderlichen
Parametern, die für die Bruchkraftversuche am Texture Analyser verwendet wurden.
Tab. 7: Mittels Texture Analyser auf Bruchfestigkeit getestete Arzneiformen.
Arzneiform Parameter
Agar‐Kugeln Agar‐Konzentration (%) 1,5, 2, 3, 4, 5, 6, 7
Agar‐Kugeln (5 %) Trocknungszeit (h) bei 40 °C 0,5, 1, 3, 5
Hartfett‐W32‐Kugeln Polymerbeladung (mg/cm2) 3, 3,5, 4, 5, 6, 7
PEG‐1000‐Kugeln Polymerbeladung (mg/cm2) 3, 3,5, 4, 5, 6, 7
Paraffin‐Kapseln Polymerbeladung (mg/cm2) 4
Tristearin‐Kapseln Beschichtungsvolumen (µL) 250, 300, 400, 450, 500
Kaugummi‐Tabletten Charge 1, 2, 3
2.3.3 Wirkstofffreisetzung mittels Paddle‐Apparatur
Die Freisetzung in der Paddle‐Apparatur (DT 80, Erweka) wurde im Freisetzungsmedium SGFsp pH 1,8
durchgeführt, um die Verweilzeit der Arzneiformen im nüchternen Magen zu simulieren. Dazu wurde
das Medium, dessen Zusammensetzung Tab. 8 zu entnehmen ist, auf 37 °C vorgeheizt und bei einer
Rührgeschwindigkeit von 75 rpm über den gesamten Versuchszeitraum gerührt.

2 Material und Methoden
39
Tab. 8: Zusammensetzung des Mediums SGFsp pH 1,8.
Rezepturbestandteil Anteil
Konzentrierte Salzsäure 3,0 g
Natriumchlorid 2,0 g
Gereinigtes Wasser zu 1000,0 mL
pH‐Einstellung mit 1 N Natriumhydroxid‐Lösung
Die Arzneiform wurde mit Hilfe eines Metall‐Sinkers im Vessel platziert, um ein Aufschwimmen an
der Oberfläche zu verhindern. Der Freisetzungstest erfolgte in je 1000 mL SGFsp pH 1,8, und wurde
über einen Zeitraum von 3 h durchgeführt. Zu den Probenahmezeiten nach 10, 20, 30, 45, 60, 90, 120
und 180 min wurden jeweils 10 mL aus jedem Vessel mit Hilfe einer Messpipette entnommen und in
durchnummerierte Reagenzgläser überführt. Anschließend wurden die Vessel sofort mit 10 mL des
gleichen vortemperierten Mediums wieder aufgefüllt. Die gesammelten Proben wurden am Ende des
Versuchs photospektrometrisch bei der entsprechenden Messwellenlänge von 267 nm für
Riboflavinphosphat und 242 nm für Paracetamol vermessen. Mit Hilfe der zuvor ermittelten
Kalibrationsfaktoren, wie in Kapitel 2.3.1 beschrieben, wurden die freigesetzten Wirkstoffmengen
berechnet und graphisch als prozentual freigesetzte Wirkstoffmenge ausgewertet. Dabei wurden die
freigesetzten Mengen der Modellarzneistoffe jeweils auf die bei den Gehaltsbestimmungen
ermittelten Werte bezogen.
Zur optischen Verdeutlichung der Diffusionsvorgänge wurden in einem früheren Versuch Agar‐
Kugeln mit Methylorange als Farbindikator hergestellt. Dieser Azofarbstoff besitzt einen
Farbumschlag von gelborange nach rot zwischen pH‐Werten von 4,4 bis 3,0 durch Protonierung der
enthaltenen Azogruppe [63]. Durch Inkorporierung des Indikators in die Kugeln kann bei einer
Inkubation in saurem Medium das Eindringen in die Kugeln anhand eines Farbumschlages erkannt
werden. Dafür wurden 7 %ige Agar‐Kugeln hergestellt, die einerseits zusätzlich Methylorange
(Charge 1a) und andererseits neben Methylorange auch Kaliumhydroxid (KOH) enthielten (Charge
1b). Die genaue Zusammensetzung ist in Tab. 9 dargestellt. Diese Kugeln wurden über einen
Zeitraum von insgesamt 25 min in 150 mL des im Vergleich zu pH 1,8 etwas saureren SGFsp pH 1,2
belassen. Zu bestimmten Zeiten wurden die Kugeln dem Medium entnommen, mittels Rasierklinge
halbiert und photographisch festgehalten. Die Herstellung erfolgte analog zu den anderen Agar‐
Kugeln durch Suspendieren des Agars in Wasser, Erwärmen auf 90 °C bis zur vollständigen Sol‐
Bildung und anschließendem Ausgießen in 2 g‐Vaginalglobuli‐Gießformen. Das zusätzlich enthaltene
Methylorange bzw. Methylorange und KOH wurde vor Zugabe des Agars in Wasser gelöst. Die

2 Material und Methoden
40
Proben der Charge 1a färbten sich bei der Herstellung erwartungsgemäß gelborange an, bei denen
der Charge 1b ergab sich eine schwarze Färbung der gesamten Hydrogelmatrix.
Tab. 9: Zusammensetzung der Agar‐Kugeln der Chargen 1a und 1b für den Diffusionsversuch.
Rezepturbestandteil Charge 1a
Anteil (g)
Charge 1b
Anteil (g)
Agar 2,1 2,1
KOH ‐ 0,3
Methylorange 0,01 0,01
Gereinigtes Wasser zu 30,0 30,0
2.3.4 Stresstest‐Apparatur
Die Stresstest‐Apparatur, deren schematischer Aufbau in Abb. 24 dargestellt ist, ermöglicht es als
biorelevantes Freisetzungsgerät zusätzliche Faktoren zum verwendeten Medium bei der Freisetzung
zu berücksichtigen. Dazu gehören neben hydrodynamischem Stress auch der diskontinuierliche
Kontakt der Arzneiform mit dem Freisetzungsmedium und das Einwirken von Druckwellen.
Zur Realisierung dieser zusätzlichen Parameter besteht die Apparatur aus einer zentralen Achse mit
einem offenen Luftkanal, an die sieben netzartige Probekörbchen montiert sind, die an einem
Scharnier geöffnet werden können. Jedes Körbchen kann in ein separates Vessel hinabgelassen
werden, welches mit 1160 mL Freisetzungsmedium befüllt ist, sodass es eintauchen und die
Arzneiform vollständig benetzen kann. In jedem Probekörbchen ist eine Düse mit der zentralen Achse
verbunden, sodass über den Luftkanal ein Gasdurchfluss ermöglicht werden kann und ein sich an der
Düse befindlicher Ballon mit Luft befüllt und wieder entleert werden kann. Dadurch werden
peristaltische Druckwellen simuliert, deren Stärke durch ein Regulationsmodul variiert werden kann.
Des Weiteren kann die Achse über einen Schrittmotor in Rotationsbewegung versetzt werden,
sodass die Körbchen während einer vollen Rotation die Mediumsoberfläche zweimal passieren. Dies
führt zu hydrodynamischem Stress und diskontinuierlichem Mediumkontakt für die Arzneiform und
soll die physiologischen Transportvorgänge simulieren. Für eine gute Durchmischung der
Freisetzungsmedien unabhängig von der Bewegung der Probekörbchen ragt je ein Blattrührer in die
Vessel. Die Drehung der zentralen Achse, die Druckamplituden sowie die Steuerung der Druckwellen
sind Software‐kontrolliert. Durch automatische Probennahme mit Hilfe von Durchflussküvetten
erfolgt ebenfalls computergesteuert eine kontinuierliche Absorptionsmessung, die mittels Software
sofort in Form von Freisetzungsprofilen ausgewertet werden kann [43, 64, 65].

2 Material und Methoden
41
Abb. 24: Schematischer Aufbau der Stresstest‐Apparatur.
2.3.4.1 Prüfung der Bruchfestigkeit
Die Prüfung der Bruchfestigkeit mit Hilfe der Stresstest‐Apparatur erfolgte für die in Tab. 10
dargestellten Proben. Bei dem Versuch wurden die Arzneiformen für eine definierte Zeit in SGFsp
pH 1,8 bei 37 ± 0,5 °C inkubiert und im Anschluss daran mit steigendem Druck belastet. Die Agar‐
Kugeln und Tristearin‐Kapseln wurden für 5 min inkubiert, um bei den Kugeln eine
Temperaturangleichung und bei den Kapseln zusätzlich das Auflösen der äußeren Gelatinehülle zu
gewährleisten. Die Hartfett‐W32‐ und PEG‐1000‐Kugeln wurden für 25 min im Medium belassen, um
die Verweilzeit im Magen zu simulieren und die Erweichung des bei Körpertemperatur schmelzenden
Kerns zu gewährleisten. Die Paraffin‐Kapseln verblieben ebenfalls für 25 min in SGFsp pH 1,8, sodass
das Medium in die radial angeordneten Löcher der überzogenen Kapseln eindringen und die
Gelatinehülle von innen anlösen konnte, um eine Sollbruchstelle zu erzeugen. Für die Kaugummi‐
Tabletten wurde eine Inkubationszeit von 20 min gewählt, um zu gewährleisten, dass sich der
Hartfettkern vollständig verflüssigen konnte. Ohne die Proben zwischendurch auszutauschen, wirkte
je eine Druckwelle von 100, 200, 300, 350, 400, 450 und 500 mbar für 10 Sekunden auf die
Arzneiformen ein. Zwischen jeder Drucksteigerung wurde begutachtet und dokumentiert wie die
Arzneiformen auf den angelegten Druck reagierten. Sobald Arzneiformen unter der Druckeinwirkung
zerbrachen wurden sie aus der Apparatur entfernt, während die verbleibenden intakten Proben nach
erneuter fünfminütiger Inkubation weiter mit den aufsteigenden Drücken belastet wurden. Um bei
den Agar‐Kugeln mit ihrem hydrophilen Gelgerüst den Einfluss auf die Verweilzeit im
Inkubationsmedium zu untersuchen, wurden Agar‐Kugeln mit einer Konzentration von 6,5 % Agar
und 5 %ige Kugeln mit einstündiger Trocknungszeit zusätzlich für 30 min in SGFsp pH 1,8 inkubiert.
Weiterhin wurde für einige Darreichungsformen untersucht, ob die ansteigende Druckbelastung
einen Einfluss auf das Brechverhalten haben könnte, indem jeweils ausgesuchte Arzneiformen mit
einem zuvor festgelegten Druck direkt belastet wurden.

2 Material und Methoden
42
Tab. 10: Mittels Stresstest‐Apparatur getestete Arzneiformen (*30 min Inkubation).
Arzneiform Parameter
Agar‐Kugeln Agar‐Konzentration (%) 3, 5, 6, 6,5*, 7, 8, 9, 10
Agar‐Kugeln (5 %) Trocknungszeit (h) bei 40 °C 0,5, 1, 1*, 3, 5
Hartfett‐W32‐Kugeln Polymerbeladung (mg/cm2) 2, 2,5, 3, 3,5, 4
PEG‐1000‐Kugeln Polymerbeladung (mg/cm2) 2, 2,5, 3, 3,5, 4
Paraffin‐Kapseln Polymerbeladung (mg/cm2) 4
Tristearin‐Kapseln Beschichtungsvolumen (µL) 220, 250, 300, 330, 360, 390, 400
Kaugummi‐Tabletten Charge 1, 1a, 2, 2a, 3a
2.3.4.2 Prüfung der Wirkstofffreisetzung
Mit Hilfe der Stresstest‐Apparatur wurde das biorelevante Freisetzungsverhalten der in Tab. 11
aufgeführten Arzneiformen untersucht. Dazu wurden Bewegungen der Arzneiform,
diskontinuierlicher Mediumkontakt und Druckwellen additiv zur Standard‐Freisetzung in der Paddle‐
Apparatur in den Versuch integriert. Die theoretische Anwendung der entwickelten Arzneiformen
beinhaltet die Einnahme im nüchternen Zustand und die anschließende Zerstörung der Arzneiform
durch 300 mbar starke Druckwellen des Pylorus bei der Magenentleerung. Vereinfachend wurde der
Freisetzungsversuch in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C unter Verwendung des in Abb. 25
dargestellten Programmablaufs durchgeführt.
Abb. 25: Programm der Freisetzung in der Stresstest‐Apparatur.
Nach Einstellung der für die photometrische Messung benötigten Parameter, einem Funktionstest
der Ballons und der Positionierung der Arzneiformen in den einzelnen Probenkörbchen erfolgte der
Start des Freisetzungsprogramms bei kontinuierlicher Durchmischung des Freisetzungsmediums
mittels Blattrührer bei 200 rpm [66]. Anschließend an eine 30‐minütige Ruhephase, in der die
Arzneiformen in ihren Körbchen in SGFsp pH 1,8 inkubiert wurden, wirkten je drei Druckwellen mit
einer Stärke von 300 mbar für je drei Sekunden auf die Proben ein. Darauf folgte eine einminütige
Rotation der Probenkörbchen bei 100 rpm um ihre eigene Achse, wobei nach 30 Sekunden ein

2 Material und Methoden
43
Richtungswechsel erfolgte. In der darauffolgenden 30‐minütigen Ruhephase erfolgte weiterhin die
Aufzeichnung der freigesetzten Wirkstoffmenge. Nach Entnahme der Proben, wurde das Aussehen
der Arzneiformen photographisch dokumentiert, um für die Auswertung einen Zusammenhang
zwischen Aussehen und Freisetzungsprofil zu erkennen. Die Probennahme und Vermessung bei der
für den verwendeten Wirkstoff entsprechenden Wellenlänge erfolgte alle 2 min und die Kalibration
erfolgte anhand einer Ein‐Punkt‐Kalibration vor dem Start des Freisetzungsprogramms.
Tab. 11: Verwendete Arzneiformen für die Freisetzung in der Stresstest‐Apparatur.
Arzneiform Parameter
Agar‐Kugeln Agar‐Konzentration
Modellwirkstoff
7,5 %
Paracetamol
Hartfett‐W32‐Kugeln Polymerbeladung
Modellwirkstoff
3,5 mg/cm2 CA (Charge 1)
4 mg/cm2 CA (Charge 2)
Riboflavinphosphat
PEG‐1000‐Kugeln Polymerbeladung
Modellwirkstoff
4 mg/cm2 CA
Riboflavinphosphat
Paraffin‐Kapseln Polymerbeladung
Modellwirkstoff
4 mg/cm2 EC
Riboflavinphosphat
Tristearin‐Kapseln Beschichtungsvolumen
Modellwirkstoff
250 µL
Paracetamol
Kaugummi‐Tabletten
Charge
Modellwirkstoff
1, 1a, 2, 2a, 3a
Riboflavinphosphat
2.3.5 Rasterelektronenmikroskopie
Mittels Rasterelektronenmikroskopie wurden Überzüge der Hartfett‐Kugeln (2 und 3,5 mg/cm2) und
die Tristearin‐Kapseln (Innenbeschichtungsvolumen: 250 µL) untersucht. Diese wurden zunächst von
anderen Hilfsstoffen wie Hartfett und Gelresten befreit und unter Vakuum für zwei Wochen im
Exsikkator gelagert. Die Proben wurden vorbereitend für die Aufnahmen mit einer dünnen, elektrisch
leitenden Schicht im Mini Sputter Coater SC7620 überzogen. Dazu wird Argongas in einer
Vakuumkammer in einem starken elektrischen Feld ionisiert. Die entstandenen Argonionen schlagen
dabei aus einer Gold‐Palladium‐Kathode kleine Bruchstücke heraus, die sich gleichmäßig in einer
dünnen Schicht über die Probe legen [66‐68]. Anschließend konnten mit Hilfe des
Rasterelektronenmikroskops PHENOM Aufnahmen der einzelnen Proben angefertigt werden und die
Schichtdicke der untersuchten Objekte anhand von zehn Messpunkten ermittelt werden.

3 Ergebnisse
44
3 Ergebnisse
3.1 Agar‐Kugeln
3.1.1 Gehaltsbestimmung
Die Gehaltsbestimmung der Paracetamol‐haltigen 7,5 %igen Agar‐Kugeln wurde anhand von sechs
Stichproben in SGFsp pH 1,8 entsprechend der unter 2.3.1.1 beschriebenen Methode durchgeführt.
Nach Subtraktion der Absorptionen der Korrektur‐ von denen der Messwellenlänge und nach
Einsetzen in die Geradengleichung der Kalibrationen ergab sich für die Agar‐Kugeln ein mittlerer
Gehalt von 5,31 mg ± 0,11 mg Paracetamol pro Kugel, entsprechend einer prozentualen
Standardabweichung von 2 %. Für die Berechnung der prozentual freigesetzten Wirkstoffmenge in
den Freisetzungsversuchen mit der Paddle‐Apparatur und der Stresstest‐Apparatur wurde im
Folgenden ein Wirkstoffgehalt von 5,31 mg pro Arzneiform angenommen.
3.1.2 Bruchfestigkeit mittels Texture Analyser
3.1.2.1 Bruchfestigkeit in Abhängigkeit von der Agar‐Konzentration
Für die Untersuchung der Bruchfestigkeit mit Hilfe des Texture Analysers (TA.XT plus) wurden die
Arzneiformen mit einem Agar‐Anteil von 1,5, 2, 3, 4, 5, 6 und 7 % ohne weitere Vorbehandlung
geprüft. Arzneiformen mit einem Gelbildner‐Anteil von < 1,5 % konnten nicht vermessen werden, da
sie bei Entnahme aus der Gießform bereits zerbrachen. Für die Prüfung mittels Texture Analyser
ergab sich ein Kraft‐Weg‐Diagramm, wie es exemplarisch für die 7 %igen Agar‐Kugeln in Abb. 26
dargestellt ist. Dabei ist zu erkennen, dass die Graphen der einzelnen Proben einen sehr ähnlichen
Verlauf zeigen, der durch einen steilen Anstieg bis zu einem Maximum und einen anschließend
abrupten Abfall der Kurve gekennzeichnet ist. Dieses Kraftmaximum stellt das erste Brechen der
Arzneiform dar und wurde für die Mittelwertberechnung der Bruchfestigkeit herangezogen. Im
Anschluss an den Hauptpeak traten kleinere unregelmäßige Peaks auf, die ein weiteres Brechen der
Kugeln beschreiben. Nach einem zwischenzeitlichen Abfall auf Kraftwerte von annähernd 0 N, bei
denen dem Druckaufnehmer des Texture Analysers aufgrund des Brechens der Arzneiform keine
Kraft entgegen gebracht wurde, kam es zu individuellen kleineren Peaks, die durch weiteres Brechen
der entstandenen Agar‐Bruchstücke entstanden. Daran anschließend stieg die Kraft bei ca. 14 mm
Wegstrecke auf ein voreingestelltes Maximum von 5 N an. Dieses Kraftmaximum wurde erreicht,
wenn die Arzneiform vollständig zerdrückt wurde und der Stempel auf die Bodenplatte drückte.
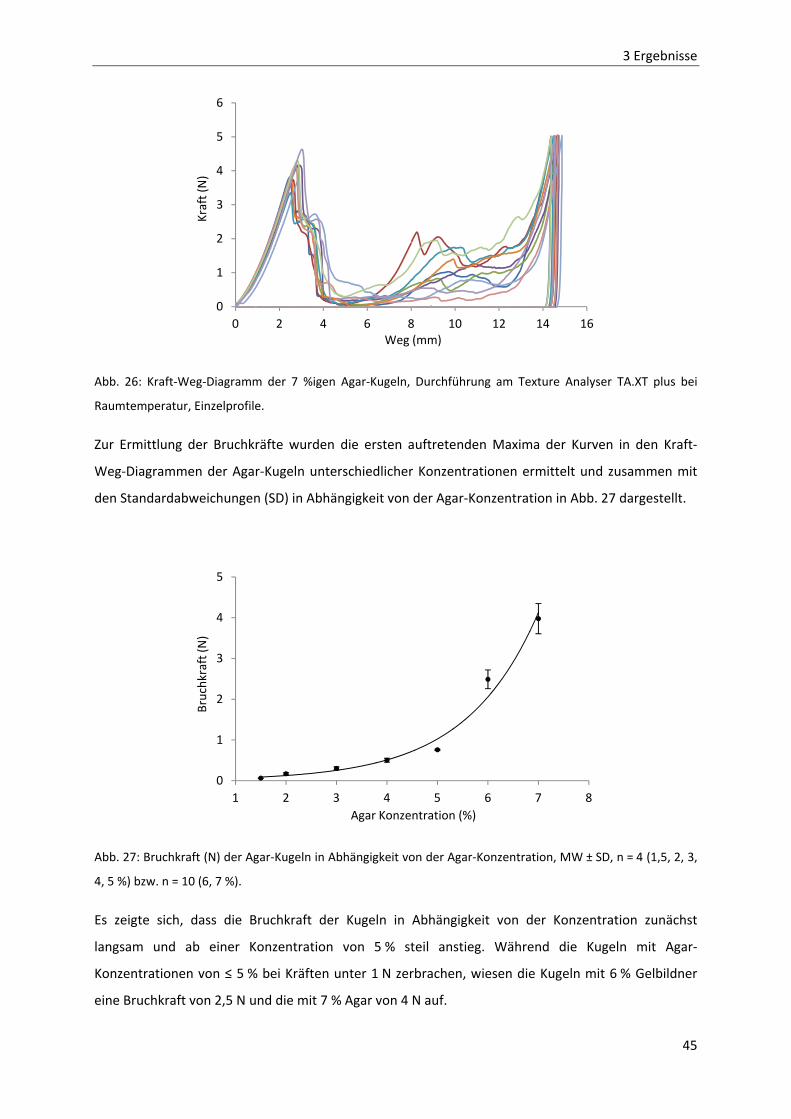
3 Ergebnisse
45
Abb. 26: Kraft‐Weg‐Diagramm der 7 %igen Agar‐Kugeln, Durchführung am Texture Analyser TA.XT plus bei
Raumtemperatur, Einzelprofile.
Zur Ermittlung der Bruchkräfte wurden die ersten auftretenden Maxima der Kurven in den Kraft‐
Weg‐Diagrammen der Agar‐Kugeln unterschiedlicher Konzentrationen ermittelt und zusammen mit
den Standardabweichungen (SD) in Abhängigkeit von der Agar‐Konzentration in Abb. 27 dargestellt.
Abb. 27: Bruchkraft (N) der Agar‐Kugeln in Abhängigkeit von der Agar‐Konzentration, MW ± SD, n = 4 (1,5, 2, 3,
4, 5 %) bzw. n = 10 (6, 7 %).
Es zeigte sich, dass die Bruchkraft der Kugeln in Abhängigkeit von der Konzentration zunächst
langsam und ab einer Konzentration von 5 % steil anstieg. Während die Kugeln mit Agar‐
Konzentrationen von ≤ 5 % bei Kräften unter 1 N zerbrachen, wiesen die Kugeln mit 6 % Gelbildner
eine Bruchkraft von 2,5 N und die mit 7 % Agar von 4 N auf.
0
1
2
3
4
5
6
0 2 4 6 8 10 12 14 16
Kraft (N)
Weg (mm)
0
1
2
3
4
5
1 2 3 4 5 6 7 8
Bruchkraft (N)
Agar Konzentration (%)

3 Ergebnisse
46
3.1.2.2 Bruchfestigkeit in Abhängigkeit von der Trocknungszeit
Für die Untersuchungen der Bruchfestigkeit in Abhängigkeit von der Trocknungszeit wurden Agar‐
Kugeln mit 5 % Gelbildner‐Anteil hergestellt und im Trockenschrank bei 40 ± 0,5 °C für 0,5, 1, 3 bzw.
5 h getrocknet. Direkt im Anschluss an die Herstellung wurde die Prüfung am Texture Analyser
durchgeführt, um eine mögliche Veränderung der Bedingungen, die durch den Trocknungsvorgang
geschaffen wurden zu vermeiden. Ohne weitere Vorbereitung konnten die Arzneiformen getestet
werden, und es ergaben sich analog zu den Diagrammen mit den unterschiedlichen Agar‐
Konzentrationen auch für die unterschiedlichen Trocknungszeiten Kraft‐Weg‐Diagramme, wie sie
beispielhaft für die einstündige Trocknung in Abb. 28 zu erkennen sind.
Abb. 28: Kraft‐Weg‐Diagramm der 5 %igen Agar‐Kugeln nach einstündiger Trocknung bei 40 ± 0,5 °C,
Durchführung am Texture Analyser TA.XT plus bei Raumtemperatur, Einzelprofile.
Die einzelnen Graphen zeigen zu Beginn auch hier wieder einen Anstieg bis zu einem Kraftmaximum,
an dem die Kurve steil abfällt und ein erster großer Peak entsteht. Nach Abfallen der Werte auf unter
1 N nimmt die Kraft erst langsam und dann sehr stark zu, bis sie bei Erreichen der zuvor eingestellten
Maximalkraft von 5 N wieder auf 0 N zurückgeht, während der Stempel von der Bodenplatte wieder
in seine Ausgangsposition zurückfährt. Die Graphen zeigen untereinander einen sehr ähnlichen
Verlauf, aber weisen im Vergleich zu den Graphen der unterschiedlichen Agar‐Konzentrationen aus
Abb. 27 ein paar Unregelmäßigkeiten besonders im Hauptpeak auf. Dies verdeutlicht sich bei der
Ermittlung der Mittelwerte der ersten Hauptpeaks, die zusammen mit den Standardabweichungen in
Abhängigkeit von der Trocknungszeit in Abb. 29 dargestellt sind. Zur Veranschaulichung des
Trocknungseinflusses wurden die Werte für die 5 %igen Agar‐Kugeln ohne Trocknungszeit im
Diagramm mit abgetragen. Es ist zu erkennen, dass die 5 %igen Agar‐Kugeln ohne Trocknung eine
Bruchkraft von unter 1 N aufweisen. Schon nach einer halbstündigen Trocknung nimmt dieser Wert
um 200 % auf 2,4 N zu. Bei Verlängerung der Trocknungsphase kommt es nach einer weiteren halben
0
1
2
3
4
5
6
0 2 4 6 8 10 12 14 16
Kraft (N)
Weg (mm)

3 Ergebnisse
47
Stunde zu keiner und nach insgesamt 3 h nur zu einer sehr geringen Erhöhung der Bruchkraft. Auch
nach fünfstündiger Trocknungszeit bei 40 ± 0,5 °C erhöht sich die aufzuwendende Bruchkraft nicht
über 3 N.
Abb. 29: Bruchkraft (N) der 5 %igen Agar‐Kugeln in Abhängigkeit von der Trocknungszeit bei 40 ± 0,5 °C,
MW ± SD, n = 4 (0 h) bzw. n = 10 (0,5, 1, 3, 5 h).
Im Vergleich zu den Arzneiformen mit unterschiedlicher Agar‐Konzentration sind die
Standardabweichungen bei den unterschiedlichen Trocknungszeiten etwas erhöht, was sich mit
zunehmender Trocknungszeit verstärkt. Neben den Veränderungen für die Bruchfestigkeit führte das
Trocknen der Arzneiformen, bedingt durch das Verdunsten des Wassers, auch zu einem Masse‐ und
Größenverlust im Durchmesser der Kugeln, der prozentual bezogen auf die 5 % Agar‐Kugeln ohne
Trocknen in Tab. 12 zu erkennen ist.
Tab. 12: Masse‐ und Größenverlust der 5 %igen Agar‐Kugeln nach 0,5, 1, 3 und 5 h Trocknung bei 40 ± 0,5 °C,
prozentuale Werte beziehen sich auf die 5 %igen Kugeln ohne Trocknung, Berechnung aus n = 6.
Zeit (h) 0,5 1 3 5
Masseverlust (%) 4,2 7,9 22,3 37,7
Größenverlust (%) 0,3 1,9 6,3 14,3
3.1.3 Bruchfestigkeit mittels Stresstest‐Apparatur
3.1.3.1 Bruchfestigkeit in Abhängigkeit von der Agar‐Konzentration
Mit Hilfe der Stresstest‐Apparatur ist es möglich, die erforderliche Bruchkraft einer Arzneiform unter
physiologischeren Bedingungen zu bestimmen. Durch Festlegung des von den Ballons ausgeübten
Druckes lässt sich somit die potentielle Eignung als drucksensitiver Arzneiform überprüfen. Anhand
0
1
2
3
4
0 1 2 3 4 5 6
Bruchkraft (N)
Trocknungszeit (h)

3 Ergebnisse
48
des zuvor erfolgten Versuches mit dem Texture Analyser wurden die Kugeln mit 1,5 und 2 % Agar für
diesen Versuch ausgeschlossen, da sie dort bereits eine zu geringe Stabilität aufwiesen. Dafür
wurden Kugeln mit höherer Konzentration bis zu 10 % Agar in den Versuch integriert. Die Ergebnisse
dieser Untersuchung sind in Tab. 13 dargestellt, und Abb. 30 zeigt den Zustand der Arzneiformen mit
unterschiedlicher Agar‐Konzentration nach dem ersten Brechen.
Tab. 13: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Agar‐Kugeln unterschiedlicher Agar‐
Konzentration: Anzahl der defekten Arzneiformen in Abhängigkeit vom angelegten Druck (mbar), n = 3 für jede
Agar‐Konzentration, 5 min Inkubation in 37 °C warmem SGFsp pH 1,8 (*30 min Inkubation).
Druck (mbar) Agar‐Konzentration (%)
3 5 6 6,5* 7 8 9 10
100 3 3 ‐ ‐ ‐ ‐ ‐ ‐
200 1 3 ‐ ‐ ‐ ‐
300 1 1 ‐ ‐ 1
350 ‐ 1 1 ‐ ‐
400 1 1 ‐ ‐ ‐
450 2 2 ‐
500 1 ‐
> 500 mbar 2
Die tabellarischen Werte (Tab. 13) lassen erkennen, dass Konzentrationen von < 6 % Agar zum
Zerbrechen der Kugeln bei bereits 100 mbar führten (siehe auch Abb. 30). Die entstandenen
Bruchstücke sind aufgrund der weichen Struktur eher klein und die Oberflächen bedingt durch das
Metallgitter der Probekörbchen porös und rissig. Die drei Arzneiformen mit 6 % des Gelbildners, die
bei Werten zwischen 200 und 400 mbar brachen, waren etwas fester und zeigten auch nach dem
Brechen in zwei fast gleich große Teile eine glatte Oberflächenstruktur. Aufgrund der 30‐minütigen
Inkubation der 6,5 %igen Agar‐Kugeln in SGFsp pH 1,8 kam es zu einer Erweichung der hydrophilen
Matrix. Daraufhin ergaben sich bei allen drei Kugeln bei einer Druckwelle von 200 mbar viele kleine,
poröse Bruchstücke. Eine Druckwelle zwischen 300 und 400 mbar hielten die 7 %igen Arzneiformen
aus bevor sich eher kleine Teile der glatten Agar‐Kugeln ablösten. Die 8 %igen Kugeln, die bei
350 mbar zum ersten Mal brachen und die 9 %igen, bei denen 450 mbar zum Brechen notwendig
waren, zeigten wieder kleinere Bruchstücke, während bei den Arzneiformen mit 10 % Agar eher ein
glatter Bruch entstand. Bei diesen Proben zerbrach nur eine Kugel bei 300 mbar, die restlichen
beiden hielten auch Drücken von 500 mbar stand.

3 Ergebnisse
49
Abb. 30: Zustand der Agar‐Kugeln nach dem ersten Zerbrechen durch Druckeinwirkung in der Stresstest‐
Apparatur in Abhängigkeit von der Konzentration: A: 3 % Agar bei 100 mbar; B: 5 % Agar bei 100 mbar; C: 6 %
Agar bei 200 mbar; D: 6,5 % Agar nach 30‐minütiger Inkubation in SGFsp pH 1,8 und bei 200 mbar; E: 7 % Agar
bei 300 mbar; F: 8 % Agar bei 350 mbar; G: 9 % Agar bei 450 mbar; H: 10 % Agar bei 300 mbar.
Für die Versuche der direkten Belastung, deren Ergebnisse in Tab. 14 dargestellt sind, wurden jeweils
individuell für die einzelnen Agar‐Konzentration die Drücke ausgewählt bei denen im
vorangegangenen Versuch die meisten Kugeln brachen oder bei breiterer Streuung der Ergebnisse
der mittlere Druck. Während bei den 3 und 5 %igen Kugeln bei 100 mbar direkt alle drei
Arzneiformen zerbrachen, blieben zwei von drei der 6 %igen Kugeln bei 300 mbar intakt. Die 7 %igen
Kugeln platzten trotz Streuung bei aufsteigender Druckbelastung bei diesem Test einheitlich bei
350 mbar direkt, während die höher konzentrierten Proben bei ihren entsprechenden Drücken nur
zu 2/3 beziehungsweise 1/3 zerbrachen. In Anbetracht dieser Ergebnisse und unter Berücksichtigung
des Verhaltens bei unterschiedlichen Inkubationszeiten in SGFsp pH 1,8 wurden für die Freisetzung in
der Stresstest‐Apparatur Arzneiformen mit 7,5 % Agar verwendet.
Tab. 14: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Agar‐Kugeln unterschiedlicher Agar‐
Konzentration bei direkter Druckeinwirkung: Anzahl der defekten Arzneiformen in Abhängigkeit vom
angelegten Druck (mbar), n = 3 für jede Agar‐Konzentration.
Agar‐Konzentration (%) 3 5 6 7 8 9 10
Druck (mbar) 100 100 300 350 400 500 500
Defekte Arzneiformen 3 3 1 3 2 2 1
3.1.3.2 Bruchfestigkeit in Abhängigkeit von der Trocknungszeit
Da in den Versuchen mit unterschiedlicher Gelbildner‐Konzentration gezeigt wurde, dass eine
Konzentration von ≤ 5 % zum Brechen der Arzneiform bei bereits 100 mbar führte, sollte in diesem

3 Ergebnisse
50
Versuch ermittelt werden, ob sich die Bruchfestigkeit durch definierte Trocknung erhöhen und auf
den gewünschten Wert von 300 mbar einstellen lassen würde. Anschließend an den Versuch mit
steigenden Drücken, wurden jeweils drei neue Kugeln der entsprechenden Trocknungszeiten
verwendet, um die direkte Druckeinwirkung zu untersuchen. Die Ergebnisse der beiden Prüfungen
sind in Tab. 15 und Tab. 16 aufgelistet und die Bilder der Zubereitungen nach dem ersten Brechen
sind in Abb. 31 dargestellt.
Tab. 15: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der 5 %igen Agar‐Kugeln bei
unterschiedlicher Trocknungszeit bei 40 ± 0,5 °C: Anzahl der defekten Arzneiformen in Abhängigkeit vom
angelegten Druck (mbar), n = 3 für jede Trocknungszeit, 5 min Inkubation in 37 °C warmem SGFsp pH 1,8
(*30 min Inkubation).
Druck (mbar) Trocknungszeit (h)
0 0,5 1 1* 3 5
100 3 ‐ ‐ ‐ ‐ ‐
200 2 ‐ 3 1 ‐
300 ‐ 3 1 ‐
350 1 ‐ 1
400 1 2
450
500
> 500
Es ist zu erkennen, dass vergleichbar mit den Ergebnissen der Bruchfestigkeitsprüfung mit dem
Texture Analyser bereits eine halbstündige Trocknung zu einer Erhöhung des erforderlichen Drucks
führt. Während die nicht getrockneten 5 %igen Agar‐Kugeln bereits bei 100 mbar zerbrachen,
platzten zwei von drei der eine halbe Stunde getrockneten Kugeln erst bei 200 mbar. Eine der Kugeln
brach in mehrere kleine Agar‐Bruchstücke, während die Zweite relativ mittig in zwei Stücke geteilt
wurde. Nach einer Stunde Trocknung brachen alle drei Kugeln einheitlich bei 300 mbar in kleine
Stücke, während eine 30‐minütige Inkubation in SGFsp pH 1,8 nach einstündiger Trocknung diesen
Wert auf 200 mbar für alle drei Proben herabsetzte. Dabei entstanden keine großen, klar
umrandeten Bruchstücke, sondern die Hydrogelmatrix wurde durch das Erweichen im Medium
großflächig zerdrückt. 3 h bei 40 ± 0,5 °C im Trockenschrank führten dazu, dass die Proben sehr
uneinheitlich zwischen 200 und 400 mbar zerbrachen, während eine fünfstündige Trocknung Kugeln
lieferte, die bei 350 und 400 mbar zerdrückt wurden.

3 Ergebnisse
51
Abb. 31: Zustand der 5 %igen Agar‐Kugeln nach dem ersten Zerbrechen durch Druckeinwirkung in der
Stresstest‐Apparatur in Abhängigkeit von der Trocknungszeit: A: 0 h bei 100 mbar; B: 0,5 h bei 200 mbar; C: 1 h
bei 300 mbar; D: 1 h bei 30‐minütiger Inkubation in 37 °C warmem SGFsp pH 1,8 und 200 mbar; E: 3 h bei
200 mbar; F: 5 h bei 350 mbar.
Der Druck für die direkte Belastung der Kugeln ergab sich aus den Ergebnissen mit den ansteigenden
Drücken. So wurde jeweils der Wert ausgewählt, bei dem die meisten Kugeln zerbrachen oder im Fall
einer breiteren Streuung, der mittlere Druck, bei dem die Kugeln nicht mehr standhalten konnten.
Bei allen Proben der unterschiedlichen Trocknungszeiten wurden alle Kugeln durch den jeweils
ausgewählten Druck zerstört, wie in Tab. 16 zu erkennen ist. Durch definiertes Trocknen wurde ein
Erhärten der Zubereitungen erreicht, welches eine Erhöhung der Bruchfestigkeit von 100 mbar ohne
Trocknung auf 300 mbar bei einstündiger und sogar 400 mbar bei fünfstündiger Trocknung zur Folge
hatte.
Tab. 16: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der 5 %igen Agar‐Kugeln bei
unterschiedlicher Trocknungszeit bei 40 ± 0,5 °C und direkter Druckeinwirkung: Anzahl der defekten
Arzneiformen in Abhängigkeit vom angelegten Druck (mbar), n = 3 für jede Trocknungszeit, 5 min Inkubation in
37 °C warmen SGFsp pH 1,8 (* 30 min Inkubation).
Trocknungszeit (h) 0 0,5 1 1* 3 5
Druck (mbar) 100 200 300 200 350 400
defekte Arzneiformen 3 3 3 3 3 3
3.1.4 Wirkstofffreisetzung mittels Paddle‐Apparatur
Mit Hilfe der Paddle‐Apparatur wurde die Freisetzung von Paracetamol aus sechs Agar‐Kugeln über
einen Zeitraum von 3 h untersucht. Um die Verweilzeit im nüchternen Magen zu simulieren, erfolgte
der Versuch in SGFsp pH 1,8 und die Messung der Absorption des Freisetzungsmediums nach

3 Ergebnisse
52
festgelegten Probenahmezeiten. Unter Berücksichtigung der ermittelten Kalibrationsgeraden
(Kapitel 2.3.1.1) und unter Einbeziehung des bei der Gehaltsbestimmung ermittelten
Wirkstoffgehalts (Kapitel 3.1.1), ergaben sich Mittelwerte der prozentualen Freisetzung mit
Standardabweichung, die graphisch in Abb. 32 zu erkennen sind.
Abb. 32: Freigesetzte prozentuale Wirkstoffmenge aus Agar‐Kugeln (7,5 %) mit 5,31 mg Paracetamol/Kugel in
1000 mL SGFsp pH 1,8 über 3 h bei 37 °C und 75 rpm; Paddle‐Apparatur, photometrische Messung bei 242 nm,
MW ± SD, n = 6.
Innerhalb der ersten 10 min kommt es zu einem kurzen steilen Anstieg in der Freisetzungskurve, der
danach etwas flacher verläuft. Für die ersten 30 min, die für die Freisetzung in der Stresstest‐
Apparatur relevant sind, ergab sich eine Freisetzung von 25 %. Im Verlauf des dreistündigen
Versuches wurden knapp 55 % des enthaltenen Paracetamols freisetzt.
Die Bestimmung der Freisetzungskinetik ergab eine Freisetzung entsprechend des Quadratwurzel‐
Zeit‐Gesetzes (Higuchi), wie Abb. 33 wiedergibt.
Abb. 33: Higuchi‐Plot für die Freisetzung von Paracetamol aus Agar‐Kugeln (7,5 %).
0
20
40
60
80
100
0 30 60 90 120 150 180
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
y = -0,4909x + 100R² = 0,953
0
20
40
60
80
100
120
0 5 10 15
100-
M/M
0
Zeit 1/2 (min1/2)
Higuchi-Plot

3 Ergebnisse
53
Die Ergebnisse der Diffusionsuntersuchungen von Agar‐Kugeln unter Verwendung eines Indikators
werden im Folgenden erläutert. Im linken Teil der Abb. 34 ist zu erkennen, dass sich bei den
Indikator‐gefärbten Kugeln (Charge 1a) bereits nach 2 min (B) ein deutlicher roter Rand im Vergleich
zu den nicht inkubierten Proben (A) gebildet hatte, was Rückschlüsse auf die Eindringtiefe des
Mediums zulässt.
Abb. 34: Links: 7 %ige Agar‐Kugeln mit 0,03 % Methylorange vor (A) und nach 2 min (B) in 150 mL SGFsp pH 1,2;
halbiert. Rechts: 7 %ige Agar‐Kugeln mit 0,03 % Methylorange und 1 % KOH vor (C) und nach 25 min (D) in
150 mL SGFsp pH 1,2; Kugeln wurden halbiert.
Die im rechten Teil der Abb. 34 gezeigten Kugeln der Charge 1b zeigen den Unterschied vor und nach
25 min Inkubation (C und D). Es ist zu erkennen, dass sich nach 25‐minütiger Inkubation in SGFsp
pH 1,2 ebenfalls ein deutlicher Rand bildet, der verglichen mit den Kugeln, die einer 2‐minütigen
Inkubationszeit unterlagen etwa die gleiche Stärke besitzt.
3.1.5 Wirkstofffreisetzung mittels Stresstest‐Apparatur
Für die Untersuchung des Freisetzungsverhaltens drucksensitiver Arzneiformen wurde neben der
Paddle‐Apparatur auch die Stresstest‐Apparatur verwendet, welche unter Einbeziehung von
Druckwellen und diskontinuierlichem Medium‐Kontakt die Simulation annähernd physiologischer
Verhältnisse ermöglicht. Anhand der Voruntersuchungen zur Bruchfestigkeit mit dem Texture
Analyser und der Stresstest‐Apparatur wurden sechs Proben 7,5 %iger Agar‐Kugeln mit einem
Wirkstoffgehalt von 5,31 ± 0,11 mg Paracetamol unter Verwendung des in Kapitel 2.3.4.2
beschriebenen Magenentleerungsprogramms auf ihre Wirkstofffreisetzung untersucht. Aus den
Freisetzungsprofilen der einzelnen Arzneiformen, die in Abb. 35 dargestellt sind, geht hervor, dass
innerhalb der ersten 30 min vor Einsetzen der drei Druckwellen gleichmäßig bis zu etwa 26 % des
Wirkstoffs freigesetzt wurden, was den Werten der Freisetzungsuntersuchungen mit der Paddle‐
Apparatur entspricht.
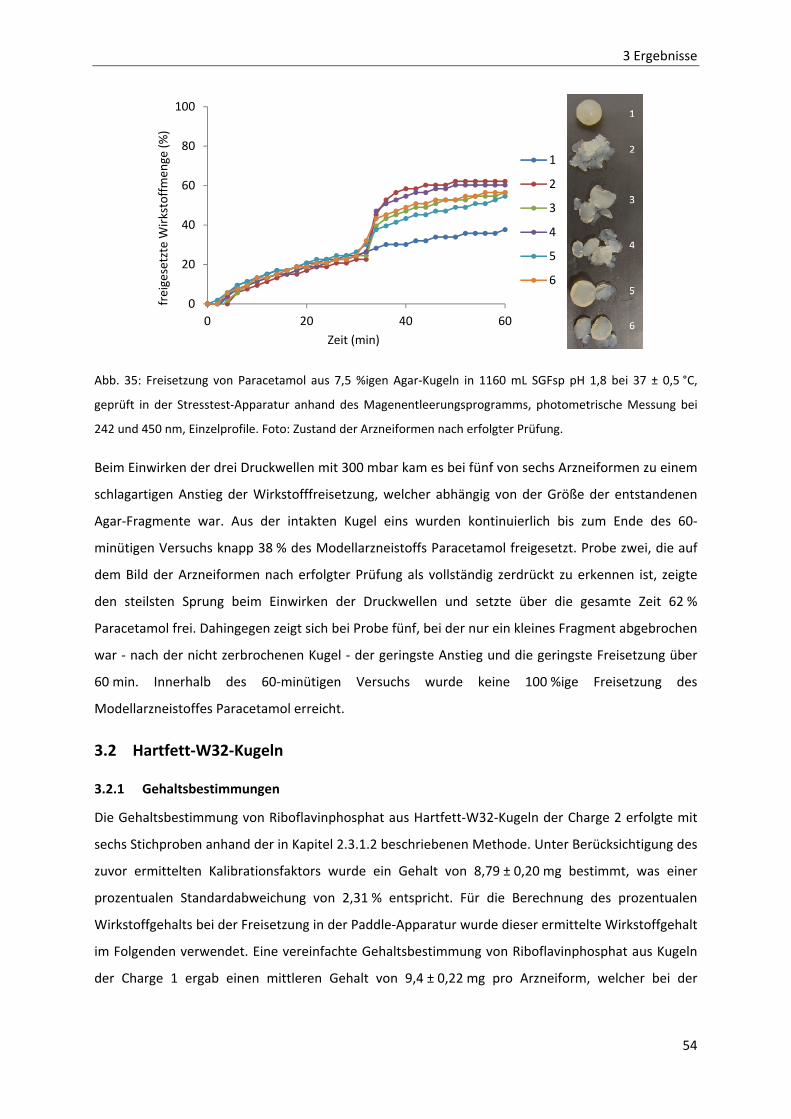
3 Ergebnisse
54
Abb. 35: Freisetzung von Paracetamol aus 7,5 %igen Agar‐Kugeln in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C,
geprüft in der Stresstest‐Apparatur anhand des Magenentleerungsprogramms, photometrische Messung bei
242 und 450 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter Prüfung.
Beim Einwirken der drei Druckwellen mit 300 mbar kam es bei fünf von sechs Arzneiformen zu einem
schlagartigen Anstieg der Wirkstofffreisetzung, welcher abhängig von der Größe der entstandenen
Agar‐Fragmente war. Aus der intakten Kugel eins wurden kontinuierlich bis zum Ende des 60‐
minütigen Versuchs knapp 38 % des Modellarzneistoffs Paracetamol freigesetzt. Probe zwei, die auf
dem Bild der Arzneiformen nach erfolgter Prüfung als vollständig zerdrückt zu erkennen ist, zeigte
den steilsten Sprung beim Einwirken der Druckwellen und setzte über die gesamte Zeit 62 %
Paracetamol frei. Dahingegen zeigt sich bei Probe fünf, bei der nur ein kleines Fragment abgebrochen
war ‐ nach der nicht zerbrochenen Kugel ‐ der geringste Anstieg und die geringste Freisetzung über
60 min. Innerhalb des 60‐minütigen Versuchs wurde keine 100 %ige Freisetzung des
Modellarzneistoffes Paracetamol erreicht.
3.2 Hartfett‐W32‐Kugeln
3.2.1 Gehaltsbestimmungen
Die Gehaltsbestimmung von Riboflavinphosphat aus Hartfett‐W32‐Kugeln der Charge 2 erfolgte mit
sechs Stichproben anhand der in Kapitel 2.3.1.2 beschriebenen Methode. Unter Berücksichtigung des
zuvor ermittelten Kalibrationsfaktors wurde ein Gehalt von 8,79 ± 0,20 mg bestimmt, was einer
prozentualen Standardabweichung von 2,31 % entspricht. Für die Berechnung des prozentualen
Wirkstoffgehalts bei der Freisetzung in der Paddle‐Apparatur wurde dieser ermittelte Wirkstoffgehalt
im Folgenden verwendet. Eine vereinfachte Gehaltsbestimmung von Riboflavinphosphat aus Kugeln
der Charge 1 ergab einen mittleren Gehalt von 9,4 ± 0,22 mg pro Arzneiform, welcher bei der
0
20
40
60
80
100
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
55
Berechnung des prozentualen Wirkstoffgehalts für die Freisetzung der Charge 1 in der Stresstest‐
Apparatur verwendet wurde.
Die vereinfachte Gehaltsbestimmung für die Hartfett‐H15‐Kugeln der Charge 3a (siehe Kapitel
2.3.1.2) ergab einen Paracetamol‐Gehalt von 3,21 ± 0,46 mg pro Arzneiform. Dies entspricht einem
prozentualen Gehalt von 54 % bei einem errechneten Sollgehalt von 5,9 mg mit einer prozentualen
Standardabweichung von 14,2 %. Bei den Darreichungsformen der Charge 3b ergab sich ein Gehalt
von 1,17 ± 0,09 mg Paracetamol, was bei einem errechneten Sollgehalt von 2,1 mg einem
prozentualen Gehalt von 56 % entspricht. In diesem Fall ist die prozentuale Standardabweichung mit
7,6 % etwa halb so groß, wie bei den Arzneiformen der Charge 3a mit 15 kleinen Alginat‐Kügelchen.
3.2.2 Bruchfestigkeit mittels Texture Analyser
Für die Prüfung der Bruchfestigkeit mittels Texture Analyser wurden Arzneiformen der Charge 1 mit
den Polymerbeladungen 2, 2,5, 3 und 3,5 mg/cm2 sowie Proben der Charge 2 mit 3, 3,5, 4, 5, 6 und
7 mg/cm2 verwendet. Alle Hartfett‐W32‐Kugeln wurden für die Prüfung vorbereitet, indem sie für
20 – 25 min in 37 ± 2 °C warmem Wasser aufbewahrt wurden, was zum vollständigen Schmelzen des
Riboflavinphosphat‐Natrium enthaltenen Hartfettkerns führte. Nach Entnahme der Proben aus dem
Wasser wurden diese sofort der Prüfung auf Bruchfestigkeit mit dem Texture Analyser (Prüfung der
Charge 1 am TA.XT plus und der Charge 2 am TAplus) zugeführt. Dabei ergaben sich Kraft‐Weg‐
Diagramme, die exemplarisch für Proben der Charge 1 mit einer Polymerbeladung von 3,5 mg/cm2 in
Abb. 36 dargestellt sind.
Abb. 36: Kraft‐Weg‐Diagramm der Hartfett‐W32‐Kugeln (Charge 1) mit 3,5 mg/cm2 Celluloseacetat‐Überzug,
Durchführung am Texture Analyser TA.XT plus nach 20‐minütiger Inkubation in 37 °C warmem Wasser,
Einzelprofile.
0
1
2
3
4
5
6
0 2 4 6 8 10 12 14 16
Kraft (N)
Weg (mm)
1
2
3
4
5
6
7
8
9
10

3 Ergebnisse
56
Die Kraft‐Weg‐Profile lassen sich nach ihrem Verlauf in drei Typen unterteilen, die sich in ihrem
Bruchverhalten unterscheiden. Bei Probe eins, zwei, vier, sechs und zehn zeigt sich ein optimaler
Verlauf, der einen konstanten Anstieg auf ein Kraftmaximum und einen steilen Abfall aufweist. Bei
den Proben drei, fünf und acht findet sich ebenfalls ein erster Peak, der aber im weiteren
Kurvenverlauf von Bereichen höherer Kräfte gefolgt wird. Bis zum Erreichen des Kraftmaximums
können mehrere kleine Kraftanstiege und –abfälle auftreten. Das Fehlen eines klaren Peaks ist bei
den Proben sieben und neun zu verzeichnen. Für die Ermittlung der Mittelwerte, die vergleichend für
Charge 1 und 2 in Abb. 37 und Abb. 38 zu erkennen sind, werden die Graphen ausgeschlossen, die
einen konstant flachen Kurvenverlauf und somit kein Aufplatzen des Überzugs aufweisen. Dies führt
innerhalb der beiden Diagramme zu einer unterschiedlichen Stichprobenzahl.
Abb. 37: Bruchkraft (N) der Hartfett‐W32‐Kugeln (Charge 1) in Abhängigkeit von der Polymerbeladung,
MW ± SD, n = 10 (für 2, 2,5 und 3 mg/cm2) bzw. n = 9 (für 3,5 mg/cm2).
Die Messwerte für Charge 1 in Abb. 37 zeigen, dass sich mit zunehmender Polymerbeladung die
erforderliche Kraft für ein Zerbrechen der Arzneiform erhöht. Bei der dünnsten Überzugsschicht liegt
die Bruchkraft bei < 1 N und erhöht sich bei 3,0 mg/cm2 auf 2,67 N wobei der Wert für 3,5 mg/cm2
mit 2,75 N nur geringfügig größer ist. Die Standardabweichung nimmt ab 3 mg/cm2 deutlich zu,
obwohl die Stichprobenzahl für die drei dünnsten Überzüge gleich ist. Auch bei Charge 2, bei der
zusätzlich Kugeln mit einer Polymerbeladung von 4, 5, 6 und 7 mg/cm2 getestet wurden, dafür aber
auf die beiden dünnsten Überzugsschichten aus Charge 1 verzichtet wurde, nimmt mit zunehmender
Polymerbeladung die Bruchkraft der Arzneiformen zu. Im direkten Vergleich der Kugeln mit
übereinstimmender Polymerbeladung (3,0 und 3,5 mg/cm2) lässt sich feststellen, dass die Werte für
Charge 2 bei 3,0 mg/cm2 um 34 % und bei 3,5 mg/cm2 sogar um 62 % bezogen auf die Werte aus
Charge 1 erhöht sind. Die Kraftänderung in Abhängigkeit von der Polymerbeladung bei Charge 2
y = 1,4749x ‐ 2,1916R² = 0,9055
0
1
2
3
4
1 2 3 4
Bruchkraft (N)
Polymerbeladung (mg/cm2)

3 Ergebnisse
57
weist einen stärker linearen Verlauf auf als bei Charge 1, was sich in einem höheren Wert für das
Bestimmtheitsmaß äußert.
Abb. 38: Bruchkraft (N) der Hartfett‐W32‐Kugeln (Charge 2) in Abhängigkeit von der Polymerbeladung,
MW ± SD, n = 5 (für 3, 3,5 und 5 mg/cm2) bzw. n = 6 (für 4, 6 und 7 mg/cm2).
Für die Untersuchungen der Hartfett‐H15‐Kugeln aus Charge 3a und 3b wurden für jede Charge
Arzneiformen mit einer Polymerbeladung von 3, 3,5 und 4 mg/cm2 hergestellt. Vorbereitend für die
Bruchfestigkeitsprüfung mittels Texture Analyser wurden sie für 25 min in 37 °C warmem,
destilliertem Wasser aufbewahrt, um ein vollständiges Schmelzen des Hartfettkerns zu ermöglichen.
Anschließend wurden sie zügig unter den Stempel des Texture Analyser (TAplus) gelegt und analog zu
den Kugeln der Charge 2 getestet. Der dünne Celluloseacetat‐Überzug der Arzneiformen mit dem
flüssigen Hartfettkern riss durch die Kraft‐Einwirkung sehr stark auf und gab dabei sowohl das
flüssige Hartfett als auch die Alginat‐Kügelchen frei, wie in Abb. 39 zu erkennen ist.
Abb. 39: Durch Krafteinwirkung aufgerissener Überzug einer Arzneiform der Charge 3a.
Dabei ergaben sich Kraft‐Weg‐Diagramme, die ähnlich zu denen der reinen Hartfett‐W32‐Kugeln im
vorderen Bereich einen steil ansteigenden und abfallenden Peak aufwiesen. Daran anschließend
traten Bereiche kleinerer Kräfte auf, bevor der Stempel bei Erreichen der zuvor eingestellten
Maximalkraft von 10 N wieder in seine Ausgangsposition zurück fuhr. Auf die graphische Darstellung
y = 1,3944x ‐ 0,5017R² = 0,9895
0
2
4
6
8
10
12
2 3 4 5 6 7 8
Bruchkraft (N)
Polymerbeladung (mg/cm2)

3 Ergebnisse
58
dieser Diagramme wird an dieser Stelle verzichtet. Für eine Stichprobenzahl von n = 6 für jede
Polymerbeladung der beiden Chargen wurden die Werte der Kraftmaxima gemittelt und in
Abhängigkeit von der Polymerbeladung in Abb. 40 abgebildet. Bei den Arzneiformen der Charge 3b
mit einer Polymerbeladung von 3 mg/cm2 konnte für eine Probe kein Kraftmaximum ermittelt
werden, weshalb sich in diesem Fall die Stichprobenzahl auf n = 5 beschränkt.
Abb. 40: Bruchkraft (N) der Hartfett‐H15‐Kugeln (Charge 3a und 3b) in Abhängigkeit von der Polymerbeladung,
MW ± SD, n = 6 bzw. n = 5 (für 3 mg/cm2 bei Charge 3b).
Bei den Proben der Charge 3a mit 15 Alginat‐Kügelchen war der Unterschied der aufzuwendenden
Kraft für das Zerbrechen der Arzneiformen zwischen den Polymerbeladungen von 3 und 3,5 mg/cm2
mit 5,2 N und 6,1 N nur gering. Bei gleichzeitig relativ großen prozentualen Standardabweichungen
von 10 und 26 % war ein signifikanter Unterschied nicht zu erkennen. Bei einer Polymerbeladung von
4 mg/cm2 Celluloseacetat stieg die Bruchkraft auf 7,4 ± 1,4 N an. Bei den Kugeln der Charge 3b waren
die Unterschiede zwischen den einzelnen Polymerbeladungen deutlicher, obwohl auch hier höhere
prozentuale Standardabweichungen von 20 % für 4 mg/cm2 und 37 % für 3,5 mg/cm2 zu verzeichnen
waren. Die Arzneiformen mit dem dünnsten Celluloseacetat‐Film von 3 mg/cm2 brachen bereits bei
2 N, während dieser Wert für die nächsthöheren Polymerbeladungen auf 4,2 N und 8,5 N anstieg.
3.2.3 Bruchfestigkeit mittels Stresstest‐Apparatur
Die Prüfung der Bruchfestigkeit mit Hilfe der Stresstest‐Apparatur wurde für die Chargen 1 und 2 der
Hartfett‐W32‐Kugeln durchgeführt. Durch das Einwirken definierter Druckwellen wurde das
Zerreißen des Celluloseacetat‐Überzugs in Abhängigkeit der Polymerbeladung untersucht. Für beide
Chargen wurden Kugeln mit einer Polymerbeladung von 2, 2,5, 3, 3,5, und 4 mg/cm2 für die Prüfung
ausgewählt, wobei für Charge 2 die Prüfung der dicksten Polymer‐Schicht wegfiel, da schon bei
3,5 mg/cm2 keine der Kugeln zerplatzte. Die für die Freisetzung der Charge 2 in der Stresstest‐
y = 2,264x ‐ 1,7018R² = 0,9903
y = 6,5541x ‐ 18,041R² = 0,9628
0
2
4
6
8
10
12
2,5 3 3,5 4 4,5
Bruchkraft (N)
Polymerbeladung (mg/cm2)
Charge 3a
Charge 3b

3 Ergebnisse
59
Apparatur verwendete Polymerbeladung von 4 mg/cm2 ergab sich aus der Tatsache, dass für diese
Kugeln Bruchfestigkeitsversuche mit einer Polymerbeladung von 4 mg/cm2 erfolgten, bei denen vier
von sechs Proben nach aufsteigenden Drücken und fünf von sechs Proben bei direkter Belastung mit
300 mbar zerbrachen. Die in Tab. 17 dargestellten Ergebnisse stammen von Arzneiformen, die mit
einer neuen Charge Hartfett W32 hergestellt werden mussten, wodurch sich Veränderungen im
Bruchverhalten ergaben. Da für die höheren Polymerbeladungen von > 5 mg/cm2 schon Kräfte von
> 6 N im Versuch mit dem Texture Analyser erreicht wurden und nicht erwartet wurde, dass diese in
der Stresstest‐Apparatur Werte unterhalb der Maximalgrenze von 500 mbar aufweisen würden,
wurden sie nicht weiter untersucht. Die Ergebnisse der Bruchkraft‐Untersuchung führten zur
Entscheidung über die verwendete Polymerbeladung für den anschließenden Freisetzungsversuch in
der Stresstest‐Apparatur. Nach 25‐minütiger Inkubation der Proben in 37 °C warmem SGFsp pH 1,8
wurden die Proben in ihren einzelnen Netzkörbchen mit aufsteigendem Druck belastet. Ein Aufreißen
der Arzneiform ließ sich neben dem akustischen Platzen vor allem an der schnellen Gelbfärbung des
Mediums durch den Modellarzneistoff Riboflavinphosphat‐Natrium erkennen. Daher mussten die
Proben anfangs nicht einzeln aus ihren Metallkörbchen entnommen werden, um einen Bruch zu
detektieren. Nach dem Notieren der defekten Kugeln konnte gleich die nächste Druckwelle angelegt
werden. Nach mehreren Prüfdurchgängen und der vermehrten Gelbfärbung des Mediums, welches
zwischendurch nicht ausgetauscht wurde, konnte ein Zerplatzen nur durch ein kurzzeitiges
Entnehmen der Proben erkannt werden. Tab. 17 zeigt für jede Charge die Anzahl der zerplatzten
Arzneiformen in Abhängigkeit von der Polymerbeladung und dem angelegten Druck. Des Weiteren ist
in Abb. 41 das Aussehen der beschädigten Arzneiformen aus Charge 1 nach Einwirken des zum
Zerplatzen geringsten notwendigen Druckes zu erkennen.
Für Charge 1 zeigte sich, dass die Hartfett‐W32‐Kugeln mit ≤ 2,5 mg/cm2 Celluloseacetat‐Überzug
bereits bei 100 mbar zerplatzen. In Abb. 41 A1 und A2 sowie B1 und B2 sind die Arzneiformen nach
der Belastung zu erkennen. Alle Proben beider Polymerbeladungen zeigen einen deutlichen und
langen Riss im Überzug, durch den der verflüssigte Inhalt heraustreten konnte. Obwohl beim
dünnsten Überzug mit einer Polymerbeladung von 2 mg/cm2 in einigen Fällen der Hartfettkern nicht
vollständig geschmolzen war, kam es trotzdem zum Aufreißen des Überzugs. Allerdings erfolgte das
Auslaufen des Inhaltes nur unvollständig und teilweise wurde der verbleibende feste Hartfett‐
Rückstand durch den zusammengedrückten Überzug darin festgehalten. Für die nächsthöhere
Polymerbeladung ergab sich ein Druck von 200 mbar, bei dem fünf von sechs Proben zerstört
wurden, während eine bereits bei 100 mbar zerbrach. Das Aussehen der Kugeln nach der 100 mbar
Druckwelle zeigt Abb. 41 C1 und C2. Während die übrigen fünf Kugeln vollständig intakt blieben, riss
eine Arzneiform weit auf und gab das flüssige Hartfett mit dem suspendierten Riboflavinphosphat
frei. Bei einer Polymerbeladung von 3,5 mg/cm2 zerplatzten alle sechs Proben gleichzeitig bei einem

3 Ergebnisse
60
Druck von 300 mbar. Bei diesem Versuch kam es bei der 100 mbar und 200 mbar Druckwelle jeweils
zum Platzen eines Ballons. Obwohl der entsprechende Druck für kurze Zeit auf die Arzneiformen
wirkte, veränderte er sich bei den anderen Ballons durch den Druckabfall in dem kaputten Ballon.
Tab. 17: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Hartfett‐W32‐Kugeln (Charge 1 und 2) in
Abhängigkeit von der Polymerbeladung: Anzahl der defekten Arzneiformen in Abhängigkeit vom angelegten
Druck (mbar), n = 6 für jede Polymerbeladung (außer n = 2 für 4 mg/cm2), 25 min Inkubation in 37 °C warmen
SGFsp pH 1,8 (*geplatzter Ballon).
Druck (mbar)
Charge 1
Polymerbeladung (mg/cm2)
Charge 2
Polymerbeladung (mg/cm2)
2 2,5 3 3,5 4 2 2,5 3 3,5
100 6 6 1 ‐* ‐ ‐ ‐ ‐ ‐
200 5 ‐* ‐ 6 2 ‐ ‐
300 6 ‐ 4 4 ‐
350 ‐ ‐ ‐
400 ‐ ‐ ‐
450 ‐ ‐ ‐
500 ‐ ‐ ‐
> 500 mbar 2 2 6
Abb. 41: Zustand der Hartfett‐W32‐Kugeln (Charge 1) nach dem ersten Zerbrechen durch Druckeinwirkung in
der Stresstest‐Apparatur in Abhängigkeit von der Polymerbeladung: A 1+2: 2 mg/cm2 CA‐Überzug bei
100 mbar; B 1+2: 2,5 mg/cm2 CA‐Überzug bei 100 mbar; C 1+2: 3 mg/cm2 CA‐Überzug bei 100 mbar; D 1+2:
3,5 mg/cm2 CA‐Überzug bei 300 mbar.
Um zu überprüfen wie das Bruchverhalten bei den Kugeln der nächsthöheren Polymerbeladung
aussah, wurden Proben aus einer anderen Charge mit 4 mg/cm2 Celluloseacetat getestet, die auch
bei 500 mbar noch vollständig intakt blieben. Dementsprechend wurden für den Freisetzungsversuch

3 Ergebnisse
61
mittels Stresstest‐Apparatur Arzneiformen mit einer Polymerbeladung von 3,5 mg/cm2 verwendet.
Die erforderlichen Drücke zum Zerplatzen der Darreichungsformen waren bei Charge 2 der Hartfett‐
Kugeln durchgehend erhöht. Während alle sechs Proben mit der kleinsten Polymerbeladung von
2 mg/cm2 erst bei 200 mbar zerbrachen, wurden bei den 2,5 mg/cm2 Kugeln vier von sechs Proben
erst bei 300 mbar zerstört. Ebenfalls vier Kugeln der Proben mit 3,0 mg/cm2 platzten bei
Druckeinwirkung von 300 mbar, während die verbleibenden zwei auch bis 500 mbar keine
Beschädigung zeigten. Die Arzneiformen mit einer Polymerbeladung von 3,5 mg/cm2 hielten den
Drücken bis 500 mbar stand und wurden am Ende des Versuches unversehrt aus dem Testgerät
entnommen.
Beispielhaft für Proben der Charge 1 mit einer Polymerbeladung von 2 und 3,5 mg/cm2 wurden REM‐
Aufnahmen (Abb. 42) erstellt, mit deren Hilfe die Schichtdicke des Überzugs aus zehn Messpunkten
ermittelt werden konnte. Dabei ergab sich für den Celluloseacetat‐Überzug mit 2 mg/cm2 eine
Schichtdicke von 19 ± 4 µm, während eine Polymerbeladung von 3,5 mg/cm2 einen Überzug mit
durchschnittlich 43 ± 7 µm lieferte.
Abb. 42: REM‐Aufnahmen des CA‐Überzugs im Querschnitt (A und B) und in der Draufsicht (C und D) mit einer
Polymerbeladung von 2 mg/cm2 (A und C) und 3,5 mg/cm2 (B und D) nach Auflösung des Hartfettkerns,
Querschnitt: 250‐fache Vergrößerung, Draufsicht: 500‐fache Vergrößerung.

3 Ergebnisse
62
Die Querschnitte der beiden Überzüge in Abb. 42 (Bild A und B) weisen eine schichtenweise
Übereinanderlagerung des Überzugsmaterial mit entstandenen Hohlräumen und eine poröse
Struktur auf, aber lassen bei gleicher Vergrößerung eine deutliche Zunahme der Schichtdicke
erkennen. Die Draufsicht auf beide Überzüge (Abb. 42 Bild C und D) lässt eine unebene
Beschaffenheit und das Vorhandensein von unterschiedlich großen Poren erkennen.
3.2.4 Wirkstofffreisetzung mittels Paddle‐Apparatur
Für die Freisetzung in der Paddle‐Apparatur wurden sechs Stichproben aus Charge 2 mit einer
Polymerbeladung von 4 mg/cm2 verwendet. Die Hartfett‐W32‐Kugeln mit Riboflavinphosphat‐
Natrium als Modellarzneistoff wurden über 3 h mit Hilfe eines Metall‐Sinkers, der ein Auftreiben der
Darreichungsform auf der Oberfläche des Freisetzungsmediums verhindern sollte, freigesetzt. Die
Parameter zu diesem Versuch sowie die Probenahmezeiten sind dem Kapitel der Freisetzung in der
Paddle‐Apparatur (2.3.3) zu entnehmen. Obwohl sich am Anfang des Versuches vereinzelt kleine
Hartfett‐Tröpfchen an der Oberfläche der Kugeln sammelten, blieb das Freisetzungsmedium bis zum
Ende des Versuches farblos und es wurde kein messbarer Wirkstoff über die 3 h freigesetzt. Aus
diesem Grund wird auf die graphische Darstellung der Ergebnisse an dieser Stelle verzichtet.
3.2.5 Wirkstofffreisetzung mittels Stresstest‐Apparatur
In Anlehnung an die Ergebnisse der Bruchfestigkeitsprüfungen wurden für die Wirkstofffreisetzung
mit Hilfe der Stresstest‐Apparatur Arzneiformen der Charge 1 mit einer Polymerbeladung von
3,5 mg/cm2 verwendet. Für diese konnte durch das verwendete Programm, welches unter 2.3.4.2
erläutert wurde, die Magenentleerung simuliert und deren Verhalten unter annähernd
physiologischen Bedingungen getestet werden. Die sechs verwendeten Proben enthielten
durchschnittlich 9,4 mg Riboflavinphosphat pro Kugel und wurden über 60 min in 37 ± 0,5 °C
warmem SGFsp pH 1,8 entsprechend des Magenentleerungsprogramms freigesetzt. Die dabei
zweiminütlich gemessenen Absorptionen wurden automatisch in Konzentrationen konvertiert und
als Freisetzungsprofile der einzelnen Proben in Abb. 43 dargestellt.
Innerhalb der ersten 30 min, die die Magenverweilzeit simulieren sollten, kam es zu keiner
Freisetzung des Modellwirkstoffes Riboflavinphosphat. In dieser Zeit waren die in den
Metallkörbchen befindlichen Proben in Ruhe und der Hartfettkern begann zu schmelzen. Mit
Einsetzen der 300 mbar Druckwelle und nach einminütiger Rotation der Körbchen um ihre Achse
stieg die prozentual freigesetzte Wirkstoffmenge durch das Einreißen des Celluloseacetat‐Überzugs
sehr steil an. Das vollständig geschmolzene Hartfett wurde durch den großen Riss im Überzug sofort
heraus gespült und das gut wasserlösliche Riboflavinphosphat ging beim Kontakt mit dem
Freisetzungsmedium sofort in Lösung und ermöglichte eine schnelle Detektion der Freisetzung.

3 Ergebnisse
63
Bereits 4 min nach der Druckwelle wurden bis auf eine Ausnahme bei Probe zwei über 79 % des
Modellwirkstoffs freigesetzt, und am Ende der 60‐minütigen Freisetzung lag die prozentuale
Freisetzungsrate bei 85 – 99 %. Das Foto in Abb. 43 zeigt die Reste des nicht detektierten
Riboflavinphosphats in den verbliebenen Überzügen nach der Freisetzung.
Abb. 43: Freisetzung von Riboflavinphosphat‐Natrium aus mit 3,5 mg/cm2 CA überzogenen Hartfett‐W32‐
Kugeln (Charge 1) in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der
Arzneiformen nach erfolgter Prüfung.
Die Durchführung dieses Versuchs für Charge 2 erfolgt anhand von sechs Proben mit einer
Polymerbeladung von 4 mg/cm2. Die Einzelprofile der Freisetzung ähneln im Verlauf denen der
Charge 1 und sind in Abb. 44 zu erkennen. Nach 30‐minütiger Ruhephase ohne Wirkstofffreisetzung
wurden mit Einsetzen der Druckwellen 60 – 100 % Riboflavinphosphat freigesetzt. Während bei
Probe eins mit der geringsten Freisetzung der verbleibende Celluloseacetat‐Überzug noch am
stärksten gelb gefärbt war, erschienen die Überzüge der Proben zwei, vier und fünf weitestgehend
weiß, was auf ein annähernd vollständiges Herauslösen des Modellarzneistoffs schließen lässt.
Obwohl der Überzug von Probe zwei sehr weiß war, wurden bis zum Ende des Versuchs nur 71 %
herausgelöst. Bei Probe drei und vier lag die prozentuale Freisetzung bei 85 – 87 %, während allein
bei Probe fünf mehr als 100 % Freisetzung erzielt wurden.
0
20
40
60
80
100
120
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
64
Abb. 44: Freisetzung von Riboflavinphosphat‐Natrium aus mit 4 mg/cm2 CA überzogenen Hartfett‐W32‐Kugeln
(Charge 2) in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der
Arzneiformen nach erfolgter Prüfung.
3.3 PEG‐1000‐Kugeln
3.3.1 Gehaltsbestimmung
Die Ermittlung des Wirkstoffgehaltes der PEG‐1000‐Kugeln wurde anhand von sechs Stichproben
entsprechend der in Kapitel 2.3.1.3 erläuterten Methode durchgeführt. Unter Berücksichtigung des
Kalibrationsfaktors wurde für diese Arzneiform ein Wirkstoffgehalt von 11,40 ± 0,18 mg
Riboflavinphosphat ermittelt, was einer prozentualen Standardabweichung von 1,59 % entspricht.
Dieser ermittelte Wirkstoffgehalt wurde in den Freisetzungsversuchen mit der Paddle‐Apparatur und
der Stresstest‐Apparatur verwendet, um die prozentuale Wirkstofffreisetzung zu bestimmen.
3.3.2 Bruchfestigkeit mittels Texture Analyser
Für die Überprüfung der Bruchfestigkeit der PEG‐1000‐Kugeln wurden Proben mit einer
Polymerbeladung von 3, 3,5, 4, 5, 6 und 7 mg/cm2 untersucht. Um ein optimales Bruchverhalten zu
erzielen, das nur von der unterschiedlichen Schichtdicke des Überzugs abhängt, wurden die
Arzneiformen vor ihrer Prüfung für 25 min in 37 °C warmem Wasser aufbewahrt. Das vollständige
Schmelzen des PEG‐Kerns wurde nach Ablauf der Inkubationszeit visuell überprüft, indem die Kugeln
aus dem Wasser herausgenommen und gegen das Licht gehalten wurden. Die Ergebnisse des
Versuchs sind beispielhaft für eine Polymerbeladung von 3,5 mg/cm2 in Abb. 45 gezeigt.
0
20
40
60
80
100
120
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
65
Abb. 45: Kraft‐Weg‐Diagramm der PEG‐1000‐Kugeln mit 3,5 mg/cm2 Celluloseacetat‐Überzug, Durchführung
am Texture Analyser TAplus nach 25‐minütiger Inkubation in 37 °C warmem Wasser, Einzelprofile.
Ähnlich den Ergebnissen der Hartfett‐W32‐Kugeln weisen die Profile im Idealfall einen konstanten
Anstieg und einen steilen Abfall der Kraft‐Werte auf, was das Brechen der Arzneiform kennzeichnet.
Durch den Gegendruck, den die Kugel im intakten Zustand dem Stempel des Texture Analyser
entgegenbringt, baut sich eine Kraft auf, die durch das Einreißen des Überzugs und dem damit
verbundenen Austreten des flüssigen Inhalts steil wieder abfällt. Mit Ausnahme einer Probe aus Abb.
45 zeigten alle Kugeln diesen Verlauf. Bei dieser Ausnahme zeigte sich ein konstanter Kraft‐Anstieg,
bei dem die Werte nicht abfielen, sondern sich in Form eines Plateaus fortpflanzten, welches
mehrere kleine Kraftabfälle und –anstiege aufwies. In diesem Fall konnte kein Kraftmaximum
ermittelt werden, sodass diese Probe der Berechnung der mittleren Kraftmaxima für die einzelnen
Polymerbeladungen entzogen wurde. Aus diesem Grund kam es zwischen den einzelnen
Polymerbeladungen trotz gleicher Anzahl geprüfter Arzneiformen zu unterschiedlichen
Stichprobenzahlen in den Ergebnissen. Die Gegenüberstellung der Bruchkräfte für die
unterschiedlichen Polymerbeladungen der PEG‐1000‐Kugeln in Abb. 46 zeigt, dass mit wachsender
Polymerbeladung auch die notwenige Kraft zum Zerdrücken der Arzneiformen zunimmt. Dies
geschieht trotz schwankender und teilweise recht hoher Standardabweichungen sehr konstant in
einem linearen Zusammenhang. Proben mit 3,0 mg/cm2 Celluloseacetat‐Überzug brechen bereits bei
knapp 3 N, während Darreichungsformen mit 7 mg/cm2 im Mittel Kräften von 11,15 N und in
Ausnahmefällen sogar 16 N standhielten.
0
1
2
3
4
5
6
7
0 5 10 15
Kraft (N)
Weg (mm)

3 Ergebnisse
66
Abb. 46: Bruchkraft (N) der PEG‐1000‐Kugeln in Abhängigkeit von der Polymerbeladung, MW ± SD, n = 5 (für 3,
3,5 und 4 mg/cm2) bzw. n = 6 (für 5, 6 und 7 mg/cm2).
3.3.3 Bruchfestigkeit mittels Stresstest‐Apparatur
Die Verwendung der Stresstest‐Apparatur für die Ermittlung der Bruchfestigkeit einer
drucksensitiven Darreichungsform ist durch das definierte Füllen der Ballons mit Druckluft sehr gut
geeignet. Somit kann die optimale Polymerbeladung der Arzneiform ermittelt werden, die beim
physiologischen Pylorus‐Druck von etwa 300 mbar zerbricht. Da in den vorangegangenen
Untersuchungen mit dem Texture Analyser die Kraftwerte ab einer Polymerbeladung von 5 mg/cm2
mit 7,53 ± 1,1 N für die Verhältnisse an der Stresstest‐Apparatur zu hoch waren, wurden für diesen
Versuch nur die geringeren Polymerbeladungen von 2, 2,5, 3, 3,5 und 4 mg/cm2 untersucht. Das
Zerbrechen einer Arzneiform konnte zum einen akustisch wahrgenommen werden und zum anderen
zu Beginn des Versuchs durch die Gelbfärbung des Prüfmediums nach einer Druckwelle. Da das
Medium zwischen den Versuchen nicht ausgetauscht wurde, erfolgte die Kontrolle später visuell
durch kurzzeitiges Öffnen der Metallkörbchen. Proben, die bei der angelegten Druckwelle zerbrachen
wurden entnommen und solche, die intakt blieben wurden sofort wieder ins Medium herabgelassen
und mit der nächsthöheren Druckwelle belastet. Die Ergebnisse in Tab. 18 zeigen, dass der
aufgewendete Druck zum Zerbrechen der Kugeln bei einer Polymerbeladung von ≤ 2,5 mg/cm2 bis
auf eine Ausnahme bei 200 mbar lag. Bei 3 mg/cm2 platzte die Hälfte der getesteten Kugeln ebenfalls
bei 200 mbar, die andere Hälfte bei 300 und 350 mbar. Ab einer Polymerbeladung von 3,5 mg/cm2
wurden die Kugeln größtenteils bei Drücken von 300 bzw. 350 mbar zum Platzen gebracht. Insgesamt
waren die Drücke, die ein Versagen der Arzneiform auslösten innerhalb einer Polymerbeladung
relativ weit gestreut und unterschieden sich um 100 bis 150 mbar.
y = 2,0013x ‐ 2,933R² = 0,989
0
5
10
15
2 3 4 5 6 7 8
Bruchkraft (N)
Polymerbeladung (mg/cm2)

3 Ergebnisse
67
Tab. 18: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der PEG‐1000‐Kugeln in Abhängigkeit von
der Polymerbeladung: Anzahl der defekten Arzneiformen in Abhängigkeit vom angelegten Druck (mbar), n = 6
für jede Polymerbeladung, 25 min Inkubation in 37 °C warmem SGFsp pH 1,8.
Druck (mbar) Polymerbeladung (mg/cm2)
2 2,5 3 3,5 4
100 ‐ ‐ ‐ ‐ ‐
200 6 5 3 ‐ ‐
300 1 1 4 3
350 2 1 2
400 1 ‐
450 1
500
> 500
Für eine vorangegangene Charge mit einer Polymerbeladung von 4 mg/cm2 ergab sich ein
Testergebnis, bei dem von sechs geprüften Darreichungsformen je eine bei 100 und 200 mbar und
drei bei 300 mbar zerbrachen. Bei einer Direktbelastung von 300 mbar zerbrachen von erneuten
sechs Proben alle Kugeln bei 300 mbar. Dies war für die Entscheidung der verwendeten
Polymerbeladung von 4 mg/cm2 für die Freisetzung mit der Stresstest‐Apparatur ausschlaggebend,
da diese noch mit Darreichungsformen der älteren PEG‐1000‐Charge durchgeführt wurde.
3.3.4 Wirkstofffreisetzung mittels Paddle‐Apparatur
Die PEG‐1000‐Kugeln wurden zu Versuchsbeginn einzeln in einem Metall‐Sinker platziert und in die
mit 1000 mL SGFsp pH 1,8 gefüllten Vessel gegeben. Entsprechend des in 2.3.3 beschriebenen
Versuchsaufbaus wurden die Proben nach Versuchsende bei 267 nm vermessen und es ergab sich
aus den Mittelwerten der sechs Proben ein Freisetzungsprofil, welches zusammen mit den
Standardabweichungen der Abb. 47 zu entnehmen ist. Innerhalb der ersten 30 min, die für den
Freisetzungsversuch in der Stresstest‐Apparatur relevant sind, wurden 7,8 % des Modellwirkstoffs
Riboflavinphosphat freigesetzt. Nach einem stetigen Anstieg der prozentual freigesetzten
Wirkstoffmenge über die dreistündige Versuchszeit ergab sich eine maximale Freisetzung von 70,3 %
bezogen auf den ermittelten Wirkstoffgehalt der Arzneiformen.

3 Ergebnisse
68
Abb. 47: Freigesetzte prozentuale Wirkstoffmenge aus PEG‐1000‐Kugeln mit 11,40 ± 0,18 mg
Riboflavinphosphat/Kugel und einer Polymerbeladung von 4 mg/cm2 Celluloseacetat in 1000 mL SGFsp pH 1,8
über 3 h bei 37 °C und 75 rpm; Paddle‐Apparatur, photometrische Messung bei 267 nm, MW ± SD, n = 6.
3.3.5 Wirkstofffreisetzung mittels Stresstest‐Apparatur
Zur Simulation der Pylorus‐Passage der PEG‐1000‐Kugeln wurde deren Wirkstofffreisetzung unter
Verwendung des Magenentleerungsprogramms in der Stresstest‐Apparatur untersucht. Die dabei
erhaltenen Freisetzungsprofile der einzelnen Darreichungsformen mit einer Polymerbeladung von
4 mg/cm2 sind zusammen mit einem Foto der Proben nach erfolgter Prüfung in Abb. 48 dargestellt.
Innerhalb der ersten 30 min, in denen das PEG schmolz, kam es bis auf eine Ausnahme bei Probe
sechs zu keiner Freisetzung des Modellarzneistoffs Riboflavinphosphat. Bei den Proben eins, drei,
vier und fünf wurden innerhalb von 6 min nach der 300 mbar Druckwelle 85 bis 92 % des Wirkstoffs
freigesetzt, was sich bis zum Ende des Versuchs nur noch geringfügig auf 87 bis 96 % erhöhte. Probe
sechs, die schon 2 min vor der eigentlichen Druckwelle eine geringe Freisetzung des Wirkstoffs
zeigte, hatte 6 min nach der Druckwelle 47 % Riboflavinphosphat freigesetzt und zeigt am Ende des
60‐minütigen Versuchs eine prozentuale Freisetzung von 56 %. Bei Probe zwei kam es während der
Druckphase zu keinem Aufreißen des Celluloseacetat‐Überzugs. Sie setzte über 60 min knapp 9 %
Riboflavinphosphat frei, was im Vergleich zur Freisetzung in der Paddle‐Apparatur nur etwa ein
Drittel ist.
0
20
40
60
80
100
0 30 60 90 120 150 180
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)

3 Ergebnisse
69
Abb. 48: Freisetzung von Riboflavinphosphat‐Natrium aus mit 4 mg/cm2 CA überzogenen PEG‐1000‐Kugeln in
1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der
Arzneiformen nach erfolgter Prüfung.
3.4 Paraffin‐Kapseln
3.4.1 Gehaltsbestimmung
Die Gehaltsbestimmung der Paraffin‐Kapseln wurde entsprechend der in Kapitel 2.3.1.4 erläuterten
Methode durchgeführt. Anhand von sechs Stichproben wurde ein mittlerer Gehalt an
Riboflavinphosphat von 9,45 ± 0,32 mg ermittelt, was einer prozentualen Standardabweichung von
3,4 % entspricht. Dieser wurde bei den Freisetzungsversuchen in der Paddle‐Apparatur und der
Stresstest‐Apparatur herangezogen, um die prozentual freigesetzte Wirkstoffmenge zu bestimmen.
3.4.2 Bruchfestigkeit mittels Texture Analyser
Die Untersuchung der Paraffin‐Kapseln auf ihre Bruchfestigkeit mittels Texture Analyser erfolgte
anhand von sechs Proben mit einer Polymerbeladung von 4 mg/cm2 Ethylcellulose, die kurz vor
Versuchsbeginn mit acht radial angeordneten Löchern der Größe 0,33 mm versehen wurden. Da das
Prinzip dieser Arzneiform von dem der anderen differierte, wurden nur Proben mit einer
Polymerbeladung, aber dafür mit unterschiedlichen Inkubationszeiten untersucht. Durch die
Verweilzeit der Kapsel in 37 °C warmem Wasser sollte dieses durch die Löcher in die Kapseln
eindringen, die innere Gelatineschicht anweichen und somit zu einer Sollbruchstelle führen, die bei
einer definierten Kraft aufplatzen sollte. Mit Hilfe dieses Versuches wurde überprüft, ob sich die
Kapseln bei einer Inkubationszeit von 5 bzw. 30 min hinsichtlich ihrer Bruchkraft unterscheiden. Die
Gegenüberstellung der beiden Diagramme bei 5 und 30 min Erweichungszeit in Abb. 49 zeigt in
keinem Diagramm das Auftreten von Peaks, die den Bruch der Darreichungsform implizieren würden.
0
20
40
60
80
100
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
70
Erst bei Erreichen der voreingestellten Maximalkraft von 20 N zeigt sich ein Kraftabfall, der durch das
Zurückfahren des Stempels in seine Ausgangsposition zu begründen ist. Das Drücken des Stempels
auf die Probe bewirkte ein Zusammendrücken dieser und das Austreten der enthaltenen
Riboflavinphosphat‐Suspension durch die Löcher der Kapsel. Die verlängerte Erweichungszeit führte
zu einem langsameren Kraftanstieg, da die Kapsel durch die verlängerte Erweichung elastischer
wurde. Es kam durch den fehlenden Innenwiderstand aufgrund der enthaltenen Löcher nicht zum
Platzen der Kapseln, sondern zu einem Zusammendrücken und damit Ausdrücken des Paraffins durch
die Löcher. Die Entstehung einer Sollbruchstelle war bei keiner der Proben zu beobachten.
Abb. 49: Kraft‐Weg‐Diagramm der Paraffin‐Kapseln mit 4 mg/cm2 Ethylcellulose‐Überzug, Durchführung am
Texture Analyser TAplus nach 5‐minütiger (links) und 25‐minütiger (rechts) Inkubation in 37 °C warmem
Wasser, Einzelprofile.
3.4.3 Bruchfestigkeit mittels Stresstest‐Apparatur
Trotz der Ergebnisse der Bruchfestigkeitsprüfung mit Hilfe des Texture Analyser wurden überzogene
Paraffin‐Kapseln mit einer Polymerbeladung von 4 mg/cm2 und jeweils acht radial angeordneten
0,33 mm großen Löchern für die Untersuchungen an der Stresstest‐Apparatur verwendet. Vor der
ersten Druckwelle von 100 mbar wurden die Kapseln in ihren Metallkörbchen für 30 min in 37 °C
warmem SGFsp pH 1,8 inkubiert. Im Anschluss an die Belastung wurde eine Beschädigung der
Kapseln visuell unter kurzzeitiger Entnahme aus den Probekörbchen beurteilt. Zeigten die Proben
keine Anzeichen eines Bruchs wurden sie wieder ins Medium hinabgelassen und mit der
nächsthöheren Druckwelle belastet. Im ersten Durchgang wurden sechs Proben aufsteigend mit 100,
200 und 300 mbar belastet und im zweiten Durchgang wurde sechs weitere Proben direkt mit einem
Druck von 300 mbar getestet. Als Ergebnis des Versuchs zeigte sich, dass die Proben bei 100 mbar
keinerlei optische Veränderungen zeigten. Nach dem zehnsekündigen Einwirken von 200 mbar
wurden zwei Kapseln leicht eingedellt und auch nach einem Druck von 300 mbar zeigten sich nur drei
Kapseln leicht eingedellt. In keinem Fall kam es zum Aufbrechen des Ethylcellulose‐Überzugs an der
radial angeordneten Sollbruchstelle. Im Durchgang mit der direkten Druckbelastung von 300 mbar
zeigte keine der sechs Proben eine optische Veränderung. Alle Kapseln wurden durch die 30‐
0
5
10
15
20
25
0 2 4 6
Kraft (N)
Weg (mm)
0
5
10
15
20
25
0 2 4 6
Kraft (N)
Weg (mm)

3 Ergebnisse
71
minütige Inkubationszeit weicher und zwei der sechs Proben zeigten speziell an der Sollbruchstelle
eine deutliche Erweichung, aber kein Aufplatzen.
3.4.4 Wirkstofffreisetzung mittels Paddle‐Apparatur
Die Freisetzung der überzogenen Paraffin‐Kapseln erfolgte anhand von sechs Stichproben mit einer
Polymerbeladung von 4 mg/cm2, die jeweils acht radial angeordnete Löcher der Größe 0,33 mm
enthielten. Die Kapseln wurden vor Versuchsbeginn einzeln in Metall‐Sinkern platziert, um ein
Aufschwimmen auf dem Freisetzungsmedium zu verhindern. Der Versuch in der Paddle‐Apparatur
fand unter den in Kapitel 2.3.3 aufgeführten Bedingungen statt. Über den Freisetzungszeitraum von
180 min wurden trotz der enthaltenen Löcher im Ethylcellulose‐Überzug weniger als 1,5 % des
enthaltenen Modellarzneistoffs freigesetzt. Auf die Darstellung der Ergebnisse wird deshalb an dieser
Stelle verzichtet.
3.4.5 Wirkstofffreisetzung mittels Stresstest‐Apparatur
Zur Simulation der Pylorus‐Passage fand die Prüfung der Wirkstofffreisetzung in der Stresstest‐
Apparatur entsprechend des in Kapitel 2.3.4.2 erläuterten Magenentleerungsprogramms statt. Die
Freisetzungsprofile der sechs Proben in Abb. 50 zeigen innerhalb der ersten 30 min keine
Wirkstoffliberation aus den Arzneiformen.
Abb. 50: Freisetzung von Riboflavinphosphat‐Natrium aus mit 4 mg/cm2 EC überzogenen Paraffin‐Kapseln in
1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der
Arzneiformen nach erfolgter Prüfung.
Die 300 mbar starken Druckwellen bewirkten bei fünf von sechs Proben eine minimale Freisetzung
von 2 – 5 % innerhalb der ersten 6 min nach Einwirken des Drucks. Bis zum Ende des 60‐minütigen
Versuchs wurden insgesamt aber nur 2 – 6 % aus den Kapseln herausgelöst. Das Foto der Proben
0
20
40
60
80
100
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
72
nach erfolgter Prüfung in Abb. 50 zeigt, dass keine der Kapseln an der Sollbruchstelle aufplatzte und
dass sich das im Paraffin sedimentierte Riboflavinphosphat an den Rändern der Kapseln absetzte. Es
kam also weder durch die Sollbruchstelle noch durch die einzelnen Löcher zu einer nennenswerten
Freisetzung des Modellarzneistoffs in das Medium.
3.5 Tristearin‐Kapseln
3.5.1 Gehaltsbestimmung
Für eine Stichprobenzahl von n = 6 wurde ein Gehalt von 5,51 ± 0,07 mg pro Kapsel bestimmt, was
einer prozentualen Standardabweichung von 1,22 % entspricht. Der ermittelte Gehalt wurde in den
Freisetzungsversuchen für die Bestimmung der prozentualen Wirkstofffreisetzung verwendet.
3.5.2 Bruchfestigkeit mittels Texture Analyser
Die Prüfung der Bruchfestigkeit mittels Texture Analyser erfolgte für Tristearin‐Kapseln mit einem
Innenbeschichtungsvolumen von 250, 300, 400 und 500 µL und 5 %igem HEC‐Gel als Füllung. Die zu
prüfende Kapsel wurde nach Ablösen der Gelatinehülle in 37 °C warmem Wasser quer zum Stempel
auf der Bodenplatte platziert, sodass der Stempel auf die Längsseite der Arzneiform drückte. Die bei
diesem Versuch erhaltenen Kraft‐Weg‐Diagramme, von denen beispielhaft das der 250 µL‐Kapsel in
Abb. 51 gezeigt ist, unterscheiden sich optisch von den Profilen der anderen Darreichungsformen.
Das Zerbrechen der Kapseln zeigte sich entlang des Profils durch mehrere kleine Peaks, da die
Tristearin‐Hülle der Kapsel sehr spröde ist und durch Einwirken des Stempels an vielen verschiedenen
Stellen brach. Im Anschluss daran traten durch das Zerdrücken der Bruchstücke erneute Kraftpeaks
auf, die aber die eigentliche Bruchkraft der Kapsel nicht überschritten. Der verlängerte Teil des
Diagramms, der zugunsten der Übersichtlichkeit nicht abgebildet ist, zeigt im Anschluss einen langen
Teil sehr geringer konstanter Kraft bevor durch weiteres Brechen der erhaltenen Tristearin‐
Bruchstücke erneut kleine Peaks auftraten. Diese wurden von einem letzten großen Peak gefolgt, der
das voreingestellte Kraftmaximum bei Annäherung an die Bodenplatte widerspiegelt, nach dessen
Erreichen der Stempel wieder in seine Ausgangsposition zurück fuhr.

3 Ergebnisse
73
Abb. 51: Kraft‐Weg‐Diagramm der Tristearin‐Kapseln mit einem Füllvolumen von 250 µL Tristearin,
Durchführung am Texture Analyser TA.XT plus nach Ablösen der Gelatinehülle in 37 °C warmem Wasser,
Einzelprofile.
Die Kraft‐Weg‐Diagramme der Kapseln mit größeren Beschichtungsvolumina zeigten einen ähnlichen
Kurvenverlauf, der sich lediglich im vorderen Teil durch eine Erhöhung der Kraftmaxima unterschied.
Aus den Profilen der unterschiedlichen Beschichtungsvolumina konnten anhand der Maximal‐Peaks
Mittelwerte berechnet werden, die die jeweilige Bruchkraft der Kapseln darstellten und zusammen
mit den jeweiligen Standardabweichungen in Abb. 52 zu erkennen sind. Zur Abschätzung der
Bruchfestigkeit in Abhängigkeit vom Beschichtungsvolumen wurden zunächst nur drei Stichproben
für die unterschiedlichen Beschichtungen untersucht. Da ein Beschichtungsvolumen von 250 µL für
das Zerbrechen bei 300 mbar am vielversprechendsten war, wurde die Stichprobenzahl für diese
Proben auf zehn erhöht.
Abb. 52: Bruchkraft (N) der Tristearin‐Kapseln in Abhängigkeit vom Tristearin‐Volumen, MW ± SD, n = 10 (für
250 µL) bzw. n = 3 (für 300, 400 und 500 µL).
0,0
0,5
1,0
1,5
2,0
2,5
0,0 0,5 1,0 1,5 2,0
Kraft (N)
Weg (mm)
y = 0,0623x ‐ 14,37R² = 0,9916
0
5
10
15
20
25
200 300 400 500
Bruchkraft (N)
Tristearin‐Volumen (µL)

3 Ergebnisse
74
Die graphische Gegenüberstellung der Bruchkräfte in Abhängigkeit von den Beschichtungsvolumina
zeigt, dass mit zunehmender Schichtdicke die aufzubringende Kraft zum Zerbrechen der Kapseln
linear steigt. Bei einem Beschichtungsvolumen von 300 µL wird eine Kraft von 3,6 ± 0,1 N zum
Zerbrechen der Kapsel benötigt, während ein Tristearin‐Füllvolumen von 500 µL zum Brechen der
Proben erst bei 17,1 ± 2,5 N führt. Die Standardabweichungen bei den größeren
Beschichtungsvolumina sind erhöht, während die Kapseln mit 250 und 300 µL ein sehr gleichmäßiges
Bruchverhalten zeigen. Da das Volumen einer Kapsel der Größe null 600 µL beträgt, füllt eine
Innenbeschichtung mit 500 µL die Kapsel schon fast vollständig aus und führt zu ungleichmäßig
beschichteten, sehr stabilen Darreichungsformen. Für die anschließenden Bruchfestigkeitsversuche
mit der Stresstest‐Apparatur wurden demnach nur Kapseln mit einem Füllvolumen von ≤ 400 µL
verwendet.
3.5.3 Bruchfestigkeit mittels Stresstest‐Apparatur
Zur erweiterten Einschätzung ‐ neben den Ergebnissen der Bruchfestigkeitsuntersuchungen am
Texture Analyser ‐ der Verwendbarkeit als drucksensitive Arzneiform wurden die Tristearin‐Kapseln
mit Hilfe der Stresstest‐Apparatur auf ihr Bruchverhalten untersucht. Durch ein unterschiedliches
Füllvolumen des Innenbeschichtungsmaterials Tristearin konnte die Bruchfestigkeit der Kapseln
verändert werden. Entsprechend den Ergebnissen vom Texture Analyser wurden für diesen Versuch
Kapseln verwendet, die eine Bruchkraft von ≤ 10 N und damit ein Füllvolumen ≤ 400 µL besaßen. Da
die Innenbeschichtung von Kapseln mit weniger als 220 µL Tristearin handwerklich nicht möglich war,
wurden für die Bruchfestigkeitsversuche mittels Stresstest‐Apparatur Arzneiformen mit einem
Füllvolumen von 220, 250, 300, 330, 360, 390 und 400 µL verwendet. Zur primären Abschätzung des
Bruchverhaltens wurden zunächst von jedem Füllvolumen je drei Kapseln getestet. Anhand dieser
Ergebnisse wurde für die mit 250 µL Tristearin innenbeschichteten Kapseln die Stichprobenzahl auf
neun erhöht, um deren Bruchverhalten genauer zu untersuchen. Im Anschluss an eine fünfminütige
Inkubation der Proben in 37 °C warmem SGFsp pH 1,8, die zum Ablösen der Gelatine und zur
Temperaturangleichung der Proben führen sollte, wurden die Kapseln mit aufsteigenden Drücken
belastet. Da es in diesem Fall aufgrund des verwendeten farblosen Modellarzneistoffs Paracetamol
zu keiner Färbung des Mediums als Sichtkontrolle kommen konnte, wurden die Metallkörbchen nach
jeder Druckwelle kurzzeitig aus dem Medium genommen und die Proben auf eine Beschädigung
untersucht. Die unbeschädigten Kapseln wurden wieder in die Probekörbchen gelegt, ins Medium
hinabgelassen und mit der nächsthöheren Druckwelle belastet. Für die direkte
Bruchfestigkeitsprüfung ohne Vorbelastung wurden nur Kapseln mit 250 µL und 300 µL verwendet,
da sie entsprechend der erhaltenen Ergebnisse für eine druckinduzierte Freisetzung am Pylorus am
ehesten in Betracht kamen. Der dabei ausgewählte Druck ergab sich aus den Ergebnissen der

3 Ergebnisse
75
ansteigenden Druckbelastung. Bei weiter Streuung der bruchinduzierenden Drücke wurde ein
mittlerer Druck ausgewählt und bei geringer bzw. keiner Streuung der Druck, bei dem die meisten
Darreichungsformen zerplatzten. Unter einer defekten Arzneiform wurde jede Beschädigung
verstanden, die das Austreten des Kapselinhalts ermöglichte, also sowohl das vollständige
Zersplittern des Mantels als auch das Abplatzen des Tristearin‐Deckels. Die Ergebnisse der
Bruchfestigkeitsprüfung für die Tristearin‐Kapseln sind in Tab. 19 dargestellt und das Aussehen
ausgewählter Proben nach dem ersten Zerplatzen zeigt Abb. 53.
Tab. 19: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Tristearin‐Kapseln in Abhängigkeit vom
Tristearin‐Volumen (µL): Anzahl der defekten Arzneiformen in Abhängigkeit vom angelegten Druck (mbar),
n = 3 für jedes Beschichtungsvolumen (außer n = 9 für 250 µL), 5 min Inkubation in 37 °C warmem SGFsp
pH 1,8.
Druck (mbar) Tristearin‐Volumen (µL)
220 250 300 330 360 390 400
100 2 ‐ ‐ ‐ ‐ ‐ ‐
200 1 ‐ ‐ ‐ ‐ ‐ ‐
300 4 ‐ ‐ ‐ ‐ ‐
350 3 ‐ ‐ ‐ ‐ ‐
400 ‐ 2 ‐ ‐ ‐ ‐
450 1 ‐ ‐ ‐ ‐ ‐
500 1 ‐ ‐ ‐ ‐ ‐
> 500 1 3 3 3 3
Anhand der Daten ist zu erkennen, dass eine Innenbeschichtung von > 300 µL zu sehr stabilen
Arzneiformen führte, die auch bei der Maximalbelastung der Stresstest‐Apparatur von 500 mbar
nicht zerbrachen. Abb. 53 D und E zeigt beispielhaft für die mit 360 und 400 µL beschichteten
Kapseln, dass diese auch nach der 500 mbar Druckwelle keine äußere Beschädigung erkennen ließen.
In Abb. 53 C sind die Proben der mit 300 µL beschichteten Tristearin‐Kapseln nach einer Druckwelle
von 400 mbar dargestellt, bei der eine Kapsel vollständig in kleine Tristearin‐Bruchstücke zerbrach,
während sich bei der zweiten der Deckel löste und nur am oberen Rand kleinere Stücke abbrachen.
Bei der direkten Belastung weiterer 300µL‐Kapseln mit 400 mbar zerbrach nur eine von drei Kapseln.
Die vier bei 300 mbar zerbrochenen Kapseln mit einer Innenbeschichtung von 250 µL sind in den
Bildern B1 und B2 zu erkennen. Bis auf den Deckel, der sich im Ganzen von den Arzneiformen löste,
zersplitterte der Rest der Kapseln in mittlere bis kleine Bruchstücke. Ohne Vorbelastung zerbrachen
von acht weiteren Proben sechs Stück bei einer Druckwelle von 350 mbar. Zwei von drei Proben mit

3 Ergebnisse
76
dem geringsten Tristearin‐Volumen von 220 µL zerbrachen bereits bei 100 mbar (Abb. 53 A),
während die dritte Kapsel erst dem Druck von 200 mbar nicht standhalten konnte.
Abb. 53: Zustand der Tristearin‐Kapseln nach dem ersten Zerbrechen durch Druckeinwirkung: A: 220 µL
Innenbeschichtung bei 100 mbar; B 1+2: 250 µL Innenbeschichtung bei 300 mbar; C: 300 µL Innenbeschichtung
bei 400 mbar; D: 360 µL Innenbeschichtung bei 500 mbar; E: 400 µL Innenbeschichtung bei 500 mbar.
Bei den meisten Proben dieser drucksensitiven Kapseln äußerten sich Beschädigungen durch die
Druckwellen in einem vollständigen Zersplittern der Tristearin‐Hülle, was das vollständige
Entweichen des Inhalts ermöglichte. Der Deckel löste sich unbeschädigt vom Körper der Kapsel ab.
Für Proben mit einem Tristearin‐Füllvolumen von 250 µL wurden REM‐Aufnahmen erstellt (Abb. 54),
mit denen anhand von zehn Messpunkten die Schichtdicke des Tristearin‐Mantels zu 461 µm ± 11µm
ermittelt werden konnte.
Abb. 54: REM‐Aufnahmen einer Tristearin‐Kapsel im Querschnitt mit einem Beschichtungsvolumen von 250 µL
nach Entfernung der Gelreste, 250‐fache Vergrößerung.
Da die Proben mit einem Tristearin‐Volumen von < 250 µL bereits bei 100 und 200 mbar brachen und
ein Innenbeschichtungsvolumen von ≥ 300 µL zu sehr stabilen Darreichungsformen führten, die bis

3 Ergebnisse
77
auf zwei Ausnahmen auch bei 500 mbar nicht zersplitterten, wurden für den Freisetzungsversuch in
der Stresstest‐Apparatur Kapseln mit einem Tristearin‐Volumen von 250 µL verwendet.
3.5.4 Wirkstofffreisetzung mittels Paddle‐Apparatur
Zur Simulation der Magenverweilzeit wurden sechs Tristearin‐Kapseln einzeln in einem Metall‐Sinker
platziert und entsprechend der in Kapitel 2.3.3 erläuterten Methode einem Freisetzungstest über
180 min unterzogen. Unter Berücksichtigung des bei der Gehaltsbestimmung ermittelten
Wirkstoffgehalts (Kapitel 3.5.1) ergab sich über die Zeit eine prozentual freigesetzte Wirkstoffmenge,
die als Mittelwert der sechs Proben mit den Standardabweichungen in Abb. 55 angegeben ist. Im
Freisetzungsprofil ist zu erkennen, dass ab der ersten Probenahme eine konstante leicht erhöhte
Absorption zu verzeichnen ist, die im Verlauf der 3 h nur um 2,4 % zunimmt. Nach Abschluss des
Versuches wurden die Tristearin‐Kapseln aus dem Freisetzungsmedium entnommen und auf optische
Beschädigungen hin untersucht. Da alle sechs Kapseln intakt waren und die gemessenen
Absorptionen über die Zeit sehr konstant blieben, wurde eine Freisetzung des Modellarzneistoffs
durch die geschlossene Kapsel ausgeschlossen.
Abb. 55: Freigesetzte prozentuale Wirkstoffmenge aus Tristearin‐Kapseln (250 µL Tristearin‐Volumen) mit
5,51 mg Paracetamol/Kapsel in 1000 mL SGFsp pH 1,8 über 3 h bei 37 °C und 75 rpm; Paddle‐Apparatur,
photometrische Messung bei 242 nm, MW ± SD, n = 6.
3.5.5 Wirkstofffreisetzung mittels Stresstest‐Apparatur
Die Prüfung der Wirkstofffreisetzung in der Stresstest‐Apparatur wurde entsprechend des in Kapitel
2.3.4.2 beschriebenen Versuchsaufbaus in SGFsp pH 1,8 bei 37 °C über einen Testzeitraum von
60 min durchgeführt. Anhand der Ergebnisse der Bruchfestigkeitsprüfungen aus Kapitel 3.5.2 und
3.5.3 wurden für diesen Versuch Kapseln mit einer Tristearin‐Innenbeschichtung von 250 µL und
einer Füllung aus Paracetamol‐haltigem HEC‐Gel verwendet. In Abb. 56 sind die Einzelprofile der
0
20
40
60
80
100
0 30 60 90 120 150 180
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)

3 Ergebnisse
78
freigesetzten Proben zu erkennen, die der Beschaffenheit der Kapseln im Anschluss an den Versuch
auf dem Foto gegenübergestellt sind.
Abb. 56: Freisetzung von Paracetamol aus Tristearin‐Kapseln mit einem Innenbeschichtungsvolumen von
250 µL in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 242 und 450 nm, Einzelprofile. Foto: Zustand der
Arzneiformen nach erfolgter Prüfung.
Anhand des Diagramms ist zu erkennen, dass innerhalb der ersten 30 min keine Freisetzung aus den
sechs Proben erfolgte. Mit Einsetzen der 300 mbar Druckwelle zeigte sich bei den Proben zwei, drei
und sechs ein Anstieg der prozentualen Wirkstofffreisetzung auf über 80 %. Durch das Lösen des im
Gel befindlichen Paracetamols im Freisetzungsmedium kam es bei diesen Proben bis zum Ende des
60‐minütigen Versuchs zu einer prozentualen Freisetzung von 96 – 105 %. Diese Proben zerbrachen
durch die Druckwelle in viele kleine Bruchstücke, sodass sich das enthaltene Gel schnell im
Freisetzungsmedium verteilen konnte. Bei Kapsel vier bewirkte der angelegte Druck neben dem
Ablösen des Tristearin‐Deckels das einseitige Abbrechen des Kapselrandes. Dies führte dazu, dass
unmittelbar nach der Druckwelle nur 38 % Paracetamol freigesetzt wurden, während sich der Rest
von insgesamt 96 % innerhalb der anschließenden halbstündigen Ruhephase ins Medium verteilte.
Die Proben eins und fünf wurden durch die Druckwelle nicht beeinflusst und konnten nach dem
Freisetzungsversuch unbeschädigt entnommen werden. Unter Berücksichtigung der
Freisetzungsprofile und der optischen Begutachtung lässt sich erkennen, dass die Wirkstoffabgabe
aus den Tristearin‐Kapseln von der Art und Weise des Zerbrechens abhängt. Zersplitterte eine Kapsel
vollständig, wurde der Wirkstoff innerhalb von wenigen Minuten freigesetzt, brach jedoch nur ein
Teil oder nur der Deckel ab, verzögerte sich die Liberation aufgrund des Diffusionsprozesses aus dem
konischen Teil der Kapsel.
0
20
40
60
80
100
120
0 20 40 60
frei
gese
tzte
Wirk
stof
fmen
ge (
%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
79
3.6 Kaugummi‐Manteltabletten
3.6.1 Gehaltsbestimmung
Die Gehaltsbestimmungen der Kaugummi‐Tabletten der Chargen 1 – 3 erfolgten anhand von jeweils
sechs Stichproben, wie in Kapitel 2.3.1.6 beschrieben. Es ergab sich für Charge 1 ein mittlerer Gehalt
an Riboflavinphosphat von 8,79 ± 0,21 mg. In Charge 2 konnte ein Gehalt von 9,87 ± 0,60 mg
bestimmt werden und Charge 3 enthielt 9,08 ± 0,45 mg des Modellarzneistoffs Riboflavinphosphat.
Die Berechnung der prozentualen Wirkstofffreisetzung bei dem Versuch in der Paddle‐Apparatur und
der Stresstest‐Apparatur erfolgte mit Hilfe dieser für die einzelnen Chargen ermittelten Werte.
3.6.2 Bruchfestigkeit mittels Texture Analyser
Die Prüfungen der Bruchfestigkeit mit Hilfe des Texture Analysers fanden für alle Chargen mit einer
Stichprobenzahl von n = 6 statt, wenn sie unbeschadet eine Verweilzeit von mindestens 120 min in
37 °C warmem Wasser überstanden. Vorbereitend für den Bruchfestigkeitsversuch am Texture
Analyser wurden alle Tabletten für 15 min in 37 °C warmem Wasser inkubiert, um ein vollständiges
Schmelzen des enthaltenen Hartfettkerns zu gewährleisten. Sofort nach Entnahme der Probe aus
dem Wasser wurden sie so unter den Stempel des Texture Analysers platziert, dass die Tablette flach
auflag und vom herabfahrenden Stempel auf der kreisrunden Seite belastet wurde. Dabei ergaben
sich Kraft‐Weg‐Diagramme, die exemplarisch in den Abb. 58 bis Abb. 60 dargestellt sind. Das
Auftreten von Kraftmaxima im Kurvenverlauf kennzeichnete das Brechen der Arzneiform. Wenn eine
Probe im Kraft‐Weg‐Diagramm keinen eindeutigen Peak aufwies und somit das Brechen dieser Probe
nicht festgestellt werden konnte, wurde diese bei der Mittelwertberechnung in Abb. 57
ausgeschlossen, weshalb sich bei einigen Chargen eine Stichprobenzahl von n = 5 ergab.
Abb. 57: Bruchkraft (N) der unterschiedlichen Chargen der Kaugummi‐Tabletten, MW ± SD, n = 6 bzw. n = 5 (für
gepr. MT 5.1, ged. MT 1.4, ged. MT 4.6, ged. MT 4.9) [* kennzeichnet die Chargen 1 (4.9), 2 (4.7) und 3 (4.10)].
0246810121416
gepr. M
T 5.1
gepr. M
T 5.2
ged. M
T 1.4
ged. M
T 1.5
ged. M
T 2.1
ged. M
T 2.2
ged. M
T 1.3
ged. M
T 3.1
ged. M
T 3.2
ged. M
T 3.3
ged. M
T 3.4
ged. M
T 4.1
ged. M
T 4.2
ged. M
T 4.3
ged. M
T 4.4
ged. M
T 4.5
ged. M
T 4.6
* ged. M
T 4.7
ged. M
T 4.8
* ged. M
T 4.9
* ged. M
T 4.10
Bruchkraft (N)

3 Ergebnisse
80
Anhand dieses Diagramms und des Verhaltens der Arzneiformen während der Testung wurden drei
Chargen ausgewählt, für die weitere Untersuchungen wie Gehaltsbestimmung,
Bruchfestigkeitsprüfung mittels Stresstest‐Apparatur sowie die Freisetzungen in der Paddle‐
Apparatur und Stresstest‐Apparatur durchgeführt wurden. Dies war namentlich „ged. MT 4.9“, die im
Folgenden als Charge 1 bezeichnet wird, „ged. MT 4.7“ entsprechend Charge 2 und „ged. MT 4.10“
die der Charge 3 entspricht [69].
Die Kurvenverläufe der Kraft‐Weg‐Diagramme aus den Untersuchungen mittels Texture Analyser für
die ausgewählten Chargen 1 – 3 sind in den Abb. 58 bis Abb. 60 zu erkennen. Allen drei Chargen ist
gemein, dass sie im Vergleich zu den anderen entwickelten Darreichungsformen eher runde
Kurvenverläufe aufwiesen und keine spitzen Peaks auftraten. Dadurch wiesen die Kraftabfälle
geringere Amplituden auf, was deren Identifizierung erschwerte.
Abb. 58: Kraft‐Weg‐Diagramm der Kaugummi‐Manteltabletten Charge 1, Durchführung am Texture Analyser
TAplus nach 15‐minütiger Inkubation in 37 °C warmem Wasser, Einzelprofile.
Im Ausschnitt des Kraft‐Weg‐Diagramms der Charge 1 in Abb. 58 sind Graphen zu erkennen, die durch
einen konstanten Kraftanstieg gekennzeichnet sind. Die Kurven zeigen in fünf von sechs Fällen
kurzzeitige Kraftabfälle, die einen Bruch kennzeichnen. Dies war während der Versuche zusätzlich
daran zu erkennen, dass Hartfett durch den aufgebrochenen Kaugummimantel herausfloss. Nach den
Kraftabfällen stieg die Kraft weiter an, während das geschmolzene Hartfett aus der Tablette
herausfloss und der Kaugummimantel konstant zusammen gedrückt wurde. Bei Erreichen des
voreingestellten Kraftmaximums von 20 N fuhr der Stempel in seine Ausgangsposition zurück, was
dazu führte, dass die aufgezeichnete Kraft steil abfiel. Als Mittelwert ergab sich eine Bruchkraft der
Charge 1 von 1,58 ± 0,32 N (n = 5).
0
1
2
3
4
5
0 1 2 3
Kraft (N)
Weg (mm)

3 Ergebnisse
81
Die Kurvenverläufe im Kraft‐Weg‐Diagramm der Charge 2 in Abb. 59 zeigen im Vergleich zur Charge 1
stärker ausgeprägte aber abgerundete Peaks.
Abb. 59: Kraft‐Weg‐Diagramm der Kaugummi‐Manteltabletten Charge 2, Durchführung am Texture Analyser
TAplus nach 15‐minütiger Inkubation in 37 °C warmem Wasser, Einzelprofile.
Nach einem kurzzeitig konstanten Kraftanstieg kam es zum Abfallen der Kraft, wobei das flüssige
Hartfett durch den an der Sollbruchstelle aufgeplatzten Mantel herausfloss. Vereinzelt wiesen die
Graphen im Anschluss daran einen zweiten kleineren Peak auf bevor der Kraftverlauf konstant bis
zum voreingestellten Kraftmaximum von 20 N anstieg. Es konnte für alle sechs Proben der Charge 2
ein Maximalpeak ermittelt werden, sodass sich für die Bruchkraft der Charge 2 ein Mittelwert von
1,95 ± 0,60 N (n = 6) ergab.
Das Kraft‐Weg‐Diagramm der Charge 3 in Abb. 60 ähnelt dem der Charge 1 und weist einen
konstanten Kraftanstieg mit kleineren und unregelmäßigeren Peaks auf. Die ersten Kraftabfälle
traten bereits bei Kräften unter 1 N auf und sind über einen konstanten Anstieg mit wellenartig
verlaufenden Peaks zwischen 2 N und 3 N verbunden bevor auch in diesem Fall das voreingestellte
Maximum von 20 N erreicht wurde und der Stempel wieder in seine Ausgangsposition zurückfuhr
(außerhalb des gezeigten Bereichs). Zur Berechnung des Mittelwertes wurden die ersten deutlichen
Peaks verwendet und es ergab sich eine mittlere Bruchkraft der Charge 3 von 1,77 ± 0,86 N (n = 6).
0
1
2
3
4
5
0 1 2 3
Kraft (N)
Weg (mm)

3 Ergebnisse
82
Abb. 60: Kraft‐Weg‐Diagramm der Kaugummi‐Manteltabletten Charge 3, Durchführung am Texture Analyser
TAplus nach 15‐minütiger Inkubation in 37 °C warmem Wasser, Einzelprofile.
Die mittleren Bruchkräfte der Kaugummi‐Tabletten aller drei Chargen lagen somit bei Werten < 2 N
und wiesen unterschiedlich große Standardabweichungen auf. Die deutlichsten Peaks waren bei
Charge 2 zu erkennen, während für Charge 1 die geringste Standardabweichung zu verzeichnen war.
3.6.3 Bruchfestigkeit mittels Stresstest‐Apparatur
Bei Verwendung der Stresstest‐Apparatur zur Beurteilung des Bruchverhaltens der Kaugummi‐
Tabletten wurden diese nach 20‐minütiger Inkubation in 37 °C warmem SGFsp pH 1,8 mit
ansteigenden Drücken von 100, 200 und 300 mbar belastet. Um ein verändertes Bruchverhalten
durch die ansteigende Belastung zu beurteilen, wurden weitere Proben direkt mit 300 mbar belastet.
Getestet wurden von den Chargen 1a, 2a und 3a nach neunmaligem Dippen in Kaugummilösung
jeweils sechs Stichproben mit ansteigender Belastung und sechs weitere mit einer direkten Belastung
von 300 mbar. Für die Chargen 1 und 2 wurden zusätzlich zuvor jeweils sechs Proben nach
dreimaligem Dippen mit ansteigendem Druck getestet. Die Ergebnisse der Versuche sind in Tab. 20
dargestellt und zeigen für die dreimal gedippten Proben der Chargen 1 und 2 für alle sechs
Stichproben bereits einen Bruch bei 100 mbar. Die Darreichungsformen der Chargen 1a, 2a und 3a,
die zur Erhöhung ihrer Bruchfestigkeit nach Einschneiden des gepressten Kaugummimantels neun
Mal in eine 20 %ige Kaugummilösung gedippt wurden, zeigen ähnliche Ergebnisse. Bei Charge 3a
zerbrachen bei 100 mbar bereits alle sechs Proben und auch für Charge 2a ergab sich keine Erhöhung
der Bruchfestigkeit durch vermehrtes Dippen in die Überzugslösung.
0
1
2
3
4
5
0 1 2 3 4
Kraft (N)
Weg (mm)

3 Ergebnisse
83
Tab. 20: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeit der Kaugummi‐Manteltabletten: Anzahl der
defekten Arzneiformen in Abhängigkeit vom angelegten Druck (mbar), n = 6 für jede Charge, 20 min Inkubation
in 37 °C warmem SGFsp pH 1,8.
Druck (mbar) Charge 1 Charge 1a Charge 2 Charge 2a Charge 3a
100 6 2 6 6 6
200 ‐ 1 ‐ ‐ ‐
300 ‐ 3 ‐ ‐ ‐
Ein verändertes Bruchverhalten zeigten allerdings die sechs Darreichungsformen der Charge 1a, bei
der ein Druck von 100 mbar nur zwei Proben zum Platzen brachte. Eine weitere Tablette brach bei
200 mbar, während die anderen drei Arzneiformen bei dem anvisierten Druck von 300 mbar zerstört
wurden und den verflüssigten Hartfettkern mit dem Modellarzneistoff freigaben. Eine direkte
Druckbelastung von 300 mbar führte bei den Chargen 1a, 2a und 3a zum Platzen aller sechs
geprüften Darreichungsformen.
3.6.4 Wirkstofffreisetzung mittels Paddle‐Apparatur
Die Versuche zur Wirkstofffreisetzung mittels Paddle‐Apparatur wurden für die Chargen 1 ‐ 3 anhand
von jeweils sechs Stichproben durchgeführt. Die Kaugummi‐Tabletten wurden jeweils in einen
Metall‐Sinker platziert und entsprechend der in Kapitel 2.3.3 erläuterten Versuchsanordnung einem
Freisetzungstest unterzogen. Bei allen drei Chargen konnte über den Zeitraum von 3 h keine
Wirkstofffreisetzung ermittelt werden. Die Darreichungsformen konnten nach dem Versuch
unversehrt aus dem noch farblosen Freisetzungsmedium entnommen werden und zeigten keine
sichtbare Veränderung, die auf eine Beschädigung zurückzuführen wäre.
3.6.5 Wirkstofffreisetzung mittels Stresstest‐Apparatur
Für die neun Mal gedippten Tabletten der drei Chargen 1a, 2a und 3a wurde die Wirkstofffreisetzung
mittels Stresstest‐Apparatur anhand des in Kapitel 2.3.4.2 erläuterten Programms zur Simulation der
Magenentleerung durchgeführt. Der Versuch wurde für jede Charge mit einer Stichprobenzahl von
n = 6 über 60 min in SGFsp pH 1,8 bei 37 °C durchgeführt. Die Tabletten der Charge 1 enthielten im
Durchschnitt 8,79 ± 0,21 mg Riboflavinphosphat, was zur Berechnung der prozentualen
Wirkstofffreisetzung herangezogen wurde. Innerhalb der ersten 30 min, in denen die Arzneiformen
in ihren Körbchen bei 37 °C inkubiert wurden, kam es zu keiner Freisetzung des Modellarzneistoffs.
Das Einwirken der 300 mbar starken Druckwellen nach 30 min führte bei allen Proben zu einem
steilen Anstieg der Kurve. Während bei Probe 1 nur 18 % Riboflavinphosphat am Ende des Versuchs
freigesetzt wurden, kam es bei Probe 4 zu einem Anstieg der Freisetzung auf 113 % bezogen auf den

3 Ergebnisse
84
durchschnittlichen Wirkstoffgehalt pro Tablette. Zwischen dieser minimalen und maximalen
Freisetzung liegen die Werte der anderen Tabletten dieser Charge bei 48 %, 61 %, 80 % und 105 %
(Abb. 61). Auf dem Foto von den Proben nach erfolgter Testung in Abb. 61 ist zu erkennen, dass die
Tabletten bei Einwirken der Druckwelle auseinander gerissen wurden. Bei Entnahme befand sich die
eine Hälfte der Arzneiform am Ballon, während die andere Hälfte in das Netz des Probenkörbchens
hinein gedrückt wurde. Das im Inneren befindliche Hartfett konnte beim Zusammenziehen der
Ballons herausgelöst werden, aber dennoch waren deutliche Reste des gelben Modellarzneistoffs in
den Resten des Kaugummimantels zu erkennen.
Abb. 61: Freisetzung von Riboflavinphosphat aus Kaugummi‐Manteltabletten der Charge 1a in 1160 mL SGFsp
pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des Magenentleerungsprogramms,
photometrische Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter
Prüfung.
Für die neun Mal gedippten Tabletten der Charge 2 mit einem durchschnittlichen Wirkstoffgehalt von
9,87 ± 0,60 mg ergaben sich die in Abb. 62 gezeigten Freisetzungsprofile. Innerhalb der ersten halben
Stunde, in der sich die Proben in Ruhe in den Körbchen befanden und in 37 °C warmem SGFsp pH 1,8
inkubierten, kam es zu keiner Freisetzung des Modellarzneistoffs. Die nach der Hälfte des
Freisetzungsversuchs ausgeübte 300 mbar Druckwelle führte dazu, dass alle Tabletten aufbrachen
und innerhalb der ersten Minuten 86 – 110 % an Riboflavinphosphat freisetzten. Bis zum Ende des
Versuches stieg dieser Wert nur noch leicht auf 91 – 115 % an. Im Mittel wurden nach dem 60‐
minütigen Versuch 101,5 ± 8,5 % aus den sechs Proben freigesetzt. Das Foto von den Arzneiformen
nach erfolgter Testung zeigt, dass auch in diesem Fall der Kaugummimantel auseinander gerissen
wurde, sodass Hartfett mit dem enthaltenen Modelarzneistoff herausfloss. Die im Kaugummimantel
verbliebenen Riboflavinphosphat‐Reste sind bei Charge 2a deutlich weniger vorhanden, was sich
auch mit den gleichmäßigeren Freisetzungsprofilen deckt.
0
20
40
60
80
100
120
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
85
Abb. 62: Freisetzung von Riboflavinphosphat aus Kaugummi‐Manteltabletten der Charge 2a in 1160 mL SGFsp
pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des Magenentleerungsprogramms,
photometrische Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter
Prüfung.
Die neunfach gedippten Kaugummi‐Tabletten der Charge 3 mit reinem Hartfettkern und einem
durchschnittlichen Wirkstoffgehalt von 9,08 ± 0,45 mg zeigten ebenfalls keine Wirkstofffreisetzung
im ersten stressfreien Teil des Versuchs, wie in Abb. 63 zu erkennen ist.
Abb. 63: Freisetzung von Riboflavinphosphat aus Kaugummi‐Manteltabletten der Charge 3a in 1160 mL SGFsp
pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des Magenentleerungsprogramms,
photometrische Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter
Prüfung.
Der Einfluss der 300 mbar starken Druckwelle im Anschluss führte zu einem steilen Anstieg der
Kurven und einer prozentualen Freisetzung an Riboflavinphosphat von 108 – 118 % bezogen auf den
ermittelten Wirkstoffgehalt. Der berechnete Mittelwert der prozentualen Freisetzung nach 60 min
0
20
40
60
80
100
120
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6
0
20
40
60
80
100
120
0 20 40 60
freigesetzte W
irkstoffmen
ge (%)
Zeit (min)
1
2
3
4
5
6

3 Ergebnisse
86
beträgt 113,4 ± 4,2 %. Auf dem Foto der Arzneiformen nach erfolgter Prüfung sind die Reste des
Kaugummimantels zu erkennen, die nach Versuchsende von den Ballons und den Probenkörbchen
entfernt wurden. Sie enthielten bei allen Proben wieder ungelöste Reste von Riboflavinphosphat, die
somit nicht der Absorptionsmessung zugeführt werden konnten.

4 Diskussion
87
4 Diskussion Mit dem Ziel der Entwicklung einer drucksensitiven Arzneiform für das Drug‐Targeting in den Darm
wurden verschiedene Ansätze untersucht, eine reproduzierbar herstellbare Darreichungsform zu
entwickeln, die ausschließlich durch einen definierten Druck zerbrechen und den Inhalt im Sinne des
„burst release“ freigeben sollte. Diese sollte nüchtern eingenommen werden und nach einer
Magenverweilzeit von ca. 30 min in der Phase III des IMMC in das Duodenum entleert werden. Die
dabei stattfindende Passage des Pylorus sollte durch die dort vorherrschenden Druckverhältnisse den
Trigger für die Freisetzung darstellen. Der in der Arzneiform enthaltene Modellarzneistoff sollte
suspendiert oder gelöst in einer flüssigen bis halbfesten Grundlage enthalten sein, sodass diese sich
sofort über die Schleimhaut verteilen könnte und der Wirkstoff durch eine verlängerte Kontaktzeit
verstärkt resorbiert werden würde. Gerade für Arzneistoffe, wie Metformin, Levodopa, Gabapentin,
Riboflavin oder Aciclovir, die ein Absorptionsfenster im oberen Dünndarm besitzen [23, 24], könnte
dies unter Umständen zu einer verbesserten und gleichmäßigeren Absorption führen.
Zur Umsetzung einer halbfesten Grundlage wurden einerseits Arzneiformen entwickelt, die wie die
Tristearin‐Kapseln mit einem hydrophilen Gel befüllt wurden oder wie die Agar‐Kugeln aus einer
hydrophilen Gelmatrix bestanden und den Modellarzneistoff in gelöster Form enthielten. Diese Gele
könnten sich auf der Schleimhaut verteilen und durch ihre halbfeste Konsistenz länger darauf haften.
Alternativ zum Hydrogel wurde bei den Paraffin‐Kapseln eine lipophile flüssige Grundlage verwendet,
die einen suspendierten Wirkstoff enthielt und unter Verwendung von Sollbruchstellen beim
Aufplatzen der Arzneiform herausgedrückt werden sollte. Als zweites Freisetzungsprinzip wurden
Hartfett und Polyethylenglycol als Grundlage verwendet, die sich bei Körpertemperatur verflüssigen
und somit erst im Körper die Eigenschaften einer drucksensitiven Darreichungsform entwickeln
sollen. Erst nach Einnahme würden diese Stoffe schmelzen, könnten nach Zerplatzen des
umgebenden Polymer‐Überzugs auf der Schleimhaut spreiten und den suspendierten Wirkstoff zur
Resorption zur Verfügung stellen. Vorteilhaft an diesem Prinzip ist die Robustheit der
Darreichungsformen beim Handling.
Voraussetzungen für eine geeignete drucksensitive Arzneiform ist das zuverlässige Aufbrechen, das
sich auch durch eine verlängerte Verweilzeit im Magen nicht verändert. Dementsprechend wurden
die entwickelten Darreichungsformen einer Freisetzung in der Paddle‐Apparatur mit SGFsp pH 1,8
unterzogen, die über den verlängerten Zeitraum von 3 h zeigen sollte, welche Menge an Arzneistoff
sich in dieser Zeit herauslösen konnte. Um dies überprüfen zu können, wurden für die einzelnen
Darreichungsformen Methoden zur Gehaltsbestimmung entwickelt, die eine gleichmäßige
Herstellung und Inkorporierung des Modellarzneistoffes nachweisen sollten. Für die Untersuchungen

4 Diskussion
88
der Bruchkraft, die für eine zuverlässige Vorhersage des Aufplatzens essentiell waren, wurden zwei
verschiedene Geräte verwendet, die auf unterschiedliche Art Aussagen über das Bruchverhalten
zuließen. Der Texture Analyser, bei dem ein Stempel senkrecht auf die Arzneiform heruntergedrückt
wird, lieferte Kraft‐Weg‐Diagramme mit charakteristischen Profilen für die einzelnen
Darreichungsformen. Diesen war trotz ihrer Unterschiede im Kraftverlauf gemein, dass sie ein
Brechen der Arzneiformen in Form eines mehr oder weniger steilen Peaks anzeigten. Mit Hilfe der
Untersuchungen des Bruchverhaltens in der Stresstest‐Apparatur konnte ermittelt werden, inwieweit
eine Arzneiform bei einem zuvor eingestellten Druck zerbricht. Die in einzelnen Probekörbchen
befindlichen Darreichungsformen wurden durch das Aufblasen von Ballons mit unterschiedlich
hohen Drücken belastet. Zuvor wurden die Arzneiformen in SGFsp pH 1,8 bei 37 °C inkubiert. Die
Feststellung eines Bruchs wurde in diesem Fall optisch und nicht anhand von Messdaten erfasst. Eine
kurzzeitige Entnahme der Arzneiform aus den Probekörbchen und bei Verwendung des
Modellarzneistoffs Riboflavinphosphat eine Färbung des Mediums ließen Beschädigungen aufgrund
der Druckeinwirkung und damit ein Aufplatzen erkennen. Bei der Freisetzung aus den
drucksensitiven Darreichungsformen mit Hilfe der Stresstest‐Apparatur wurden bei Verwendung des
Magenentleerungsprogramms die Verweilzeit im sauren Milieu des Magens, die Druckverhältnisse
des Pylorus und der dabei auftretende hydrodynamische Stress einbezogen. Die Ergebnisse dieser
Prüfung erlaubten schlussendlich eine Aussage über die Verwendbarkeit als drucksensitive
Arzneiform.
4.1 Agar‐Kugeln
Die Arbeit von Marciani et al. zur Untersuchung der vorherrschenden antralen Kräfte im Magen bei
Einnahme mit unterschiedlich viskosen Flüssigmahlzeiten [8] war die Grundlage, Agar‐Kugeln auf ihre
Eignung als drucksensitive Darreichungsform zu untersuchen. Dafür wurden Arzneiformen
hergestellt, die sich in ihrer Bruchfestigkeit unterscheiden sollten. Dies wurde einerseits realisiert,
indem unterschiedliche Konzentrationen des Gelbildners eingesetzt wurden und andererseits durch
definiertes Trocknen von 5 %igen Agar‐Kugeln bei 40 ± 0,5 °C über unterschiedlich lange Zeiträume.
Da der Modellarzneistoff in der wässrigen Grundlage entsprechend seiner Löslichkeit vollständig
gelöst wurde, bestand eine gute Dosiergenauigkeit beim Ausgießen der Arzneiformen.
Sedimentationsprozesse konnten nicht stattfinden und somit wurde der Wirkstoff bei Verwendung
gleicher Gießformen gleichmäßig unter den einzelnen Kugeln verteilt. Aufgrund des hohen
Wasseranteils ist diese Darreichungsform für eine langfristige Lagerung zumindest ohne
Konservierung problematisch.
Die Gehaltsbestimmung der 7,5 %igen Agar‐Kugeln beruhte auf dem Prinzip der Diffusion, da der
enthaltene Wirkstoff durch die hydrophile Matrix in das Medium diffundieren musste, um analytisch

4 Diskussion
89
erfasst werden zu können. Um den Weg so kurz wie möglich und die Grenzfläche zwischen Agar‐
Matrix und Medium so groß wie möglich zu gestalten, wurden die einzelnen Kugeln mit Hilfe eines
am unteren Ende verdickten Glasstabes eine Minute lang zerstoßen. Über einen Zeitraum von 30 min
konnte der Wirkstoff in das umgebende SGFsp pH 1,8 diffundieren, bevor die erhaltene wässrige
Lösung von den Agar‐Bruchstücken durch Filtration getrennt wurde. Dies geschah sehr gleichmäßig,
wie es sich in der geringen Standardabweichung von 2,1 % zeigt. Für eine noch genauere
Gehaltsbestimmung hätte die Gelmatrix komplett aufgelöst werden und die erhaltene
Wirkstofflösung in einen Maßkolben überführt werden können. Da die erhaltenen Werte aus der
gewählten Methode aber sehr gleichförmig und reproduzierbar waren, wurde eine weitere
Modifikation der Gehaltsbestimmung nicht vorgenommen.
Beim Diffusionsversuch mit dem enthaltenen Indikator war optisch zu erkennen, dass die Dicke des
gebildeten Randes bei der 2‐minütigen und 25‐minütigen Inkubation weitestgehend unabhängig von
der Inkubationszeit war. Dabei ist nicht nur Medium in die Kugeln hinein diffundiert, wie bei den
reinen Methylorange‐Kugeln am Farbumschlag zu erkennen war, sondern es wurden auch
Bestandteile heraus gelöst, wie bei den KOH enthaltenden Kugeln durch den helleren Rand deutlich
wurde. Die schwarze Farbe der KOH enthaltenden Kugeln, dürfte aus einer Reaktion zwischen KOH
und Methylorange stammen.
Die Ergebnisse der Wirkstofffreisetzung in der Paddle‐Apparatur stimmen gut mit den Ergebnissen
von El‐Helw et al. überein. In dieser Arbeit wurden Phenylbarbiturat enthaltende Agar‐Kugeln
unterschiedlicher Konzentration mittels Spritze hergestellt, indem das warme Sol in eine kalte
organische Lösung getropft wurde, wodurch sich sofort kleine Agar‐Kugeln ausbildeten. Die
Freisetzungskurven zeigen einen ähnlichen Verlauf, der sich zunächst durch eine initiale Freisetzung
des Wirkstoffs kennzeichnet, der sich an der Oberfläche und den äußeren Schichten der Kugeln
befindet und anschließend eine etwas verlangsamte Freisetzung in Abhängigkeit von der
Konzentration des Gelbildners. Auch in dieser Arbeit konnte die Freisetzungskinetik gut an das
Modell von Higuchi angepasst werden [70, 71].
In Anlehnung an die Arbeit von Marciani et. al, die Agar‐Kugeln mit einer Konzentration zwischen
0,75 und 3 % untersuchten, wurden für die Bruchfestigkeitsuntersuchungen am Texture Analyser
ähnliche Konzentrationen verwendet. Da die Kugeln mit weniger als 1,5 % Agar schon bei der
Entnahme aus der Gießform zerbrachen, wurde auf die Untersuchung geringerer Konzentrationen
verzichtet. In dieser Arbeit wurden dafür zusätzlich höhere Gelbildner‐Konzentrationen bis 10 %
untersucht, da die Drücke am Pylorus, die zum Zerbrechen der Arzneiformen führen sollten, höher
eingeschätzt wurden als die von Marciani et al. untersuchten Kräfte im Magen. Die Agar‐Kugeln
wurden vor der Testung mit dem Texture Analyser nicht in 37 °C warmem Wasser inkubiert, wie es

4 Diskussion
90
bei den anderen Arzneiformen der Fall war. Dass sich das Bruchverhalten in Abhängigkeit von der
Inkubationszeit ändern würde, war allerdings sehr wahrscheinlich und zeigte sich in den Versuchen
mit der Stresstest‐Apparatur, bei denen die Unterschiede zwischen 5‐minütiger und 30‐minütiger
Inkubation in SGFsp pH 1,8 untersucht wurden.
Durch die Krafteinwirkung entstanden kleine Bruchstücke, die in Abhängigkeit von der Agar‐
Konzentration mehr oder weniger fest waren. Die Bruchkraft in Abhängigkeit von der Agar‐
Konzentration zeigte in Abb. 27 einen exponentiellen Zusammenhang, wobei die Kraft bis zu einer
Agar‐Konzentration von 5 % fast linear verlief und es erst ab 6 % Gelbildner zu einer deutlichen
Erhöhung der erforderlichen Kraft kam. Im Gegensatz dazu ergab eine Variation der Trocknungszeit
von 5 %igen Kugeln nur geringfügige Änderungen der erforderlichen Bruchkraft. Obwohl die
aufzuwendende Kraft für eine nicht getrocknete Kugel im Vergleich zu einer Trocknungszeit von
30 min um das Dreifache geringer war, beschränkte sich der Kraft‐Unterschied nach einer
fünfstündigen Trocknung auf etwa 0,6 N. Die etwas erhöhten Standardabweichungen im Vergleich zu
den unterschiedlich konzentrierten Darreichungsformen deuten auf eine schlechtere
Reproduzierbarkeit hin. Dies könnte auf ein ungleichmäßiges Trocknen hindeuten, welches zum
Beispiel durch unterschiedliche Auflageflächen der Kugeln während des Trocknungsvorgangs und
dadurch ungleichmäßigen Feuchtigkeitsverlust zustande gekommen sein könnte. Somit wäre eine
Variation der Agar‐Konzentration in Hinblick auf Steuerbarkeit und Reproduzierbarkeit besser
geeignet, als die Variation der Trocknungszeit. Demzufolge wurden für die Freisetzungsversuche in
der Paddle‐Apparatur und der Stresstest‐Apparatur nur die Kugeln mit unterschiedlicher Agar‐
Konzentration eingesetzt.
Die Freisetzungsversuche in der Stresstest‐Apparatur zeigten das spröde Verhalten von Agar an der
Stelle der Druckeinwirkung. Trotz der vorangegangenen 30‐minütigen Inkubation und dem damit
verbundenen Erweichen der Agar‐Matrix zerbrachen die Kugeln in kleinere Bruchstücke und gaben
dann in Abhängigkeit ihrer Größe einen höheren Anteil an Paracetamol frei. Im direkten Vergleich
des Diagramms mit den Fotos der Arzneiformen nach erfolgter Prüfung lässt sich erkennen, dass mit
zunehmender Anzahl der Fragmente und mit abnehmender Größe eine erhöhte Freisetzung
aufgrund einer Oberflächenvergrößerung resultierte. Da allerdings bereits vor der ersten Druckwelle
knapp 30 % des Wirkstoffs freigegeben wurden und die Höhe dieser Freigabe stark von der
Inkubationszeit abhängig ist, sind die wirkstoffhaltigen Agar‐Kugeln als drucksensitive Arzneiformen
eher ungeeignet.
Vorteilhaft bei Verwendung von Agar ist, dass es durch Verdauungsenzyme nicht angegriffen werden
kann und somit die Arzneiform nicht verfrüht abgebaut werden würde. Es unterliegt im Darm einer
Nachquellung, was es für die Verwendung als Volumenabführmittel in Mengen von 5 – 15 g je

4 Diskussion
91
Einzeldosis geeignet macht [54]. Da pro Arzneiform allerdings nur etwa 0,16 g des Gelbildners
enthalten sind und dieser bereits gequollen vorliegt, sollte es selbst nach wiederholter Einnahme
dieser Darreichungsform zu keinem abführenden Effekt kommen.
Die Struktur einer 1 %igen Agar‐Matrix wurde von Rahbani et al. unter Verwendung von Cryo‐REM
untersucht, um Informationen über Größe und Anzahl der Poren zu erhalten. Dabei ergab sich eine
mittlerer Porengröße von 545 ± 3,7 nm, wobei die Poren mit wachsender Konzentration kleiner
wurden und das Spektrum der Porengrößen sich durch steigende Ionenstärke verbreiterte [72]. Man
könnte bei einer Agar‐Konzentration von 7,5 %, wie sie in dieser Arbeit verwendet wurde also von
deutlich kleineren Poren ausgehen, die den in Wasser gelösten Wirkstoff enthalten. Durch ein
verzweigtes und feineres Netz aus Poren könnte eine reduzierte Freisetzungsgeschwindigkeit
resultieren.
Die Hydrogel‐Matrix war sehr spröde. Dies änderte sich mit zunehmender Inkubationszeit in SGFsp
pH 1,8. Auch wenn die Kugeln bei definierter Inkubationszeit ein reproduzierbares Bruchverhalten
aufwiesen, sind sie als drucksensitive Darreichungsform weniger geeignet. Zum einen ist die
Verweilzeit im nüchternen Magen schlecht vorhersehbar, da sie von der zum Zeitpunkt der Einnahme
vorherrschenden Phase des IMMC abhängig ist. Dadurch verändert sich die Menge an Wasser die in
Abhängigkeit von der Magenverweilzeit aufgenommen werden kann, was wiederum die Konsistenz
der Darreichungsformen verändert [70]. Zum anderen variiert die Menge an Wirkstoff, die nach dem
Zerplatzen freigesetzt wird, weil es sich nicht um eine flüssige, sondern um eine gelartige Grundlage
handelt, durch die der Wirkstoff erst hindurch diffundieren muss. Für die wirkstofflose Untersuchung
von Kräften des Gastrointestinaltraktes ist diese Arzneiform aufgrund der schlechten
Vorhersagbarkeit der Verweilzeit im wässrigen Milieu des Magens nur mäßig geeignet. Sobald ein
Wirkstoff gezielt an einen bestimmten Teil des Magen‐Darm‐Traktes gelangen soll, sind die Agar‐
Kugeln aufgrund ihrer hydrophilen Matrix sogar als komplett ungeeignet einzuschätzen.
4.2 Hartfett‐W32‐ und PEG‐1000‐Kugeln
Um das Problem der Haltbarkeit und Stabilität von wässrigen Grundlagen zu umgehen, wurden in
einem weiteren Ansatz Grundlagen untersucht, die bei Raumtemperatur fest sind, sich aber bei
Körpertemperatur verflüssigen. Hierzu wurden einerseits Hartfett W32 und andererseits
Polyethylenglycol 1000 ausgewählt, da sie einen Schmelzbereich besitzen, der knapp unterhalb der
Körpertemperatur liegt [48, 50]. Die Herstellung der nicht überzogenen Kugeln unter Verwendung
von Gießformen für Vaginalglobuli mit einer Masse von 2 g war mit nur wenigen Hilfsmittel möglich
und gut reproduzierbar. Der Modellarzneistoff Riboflavinphosphat wurde in der jeweiligen Grundlage
suspendiert und unter Verwendung des Cremeschmelzverfahrens unter stetigem Rühren in die

4 Diskussion
92
Gießformen ausgegossen. Schon kleine Temperaturänderungen der geschmolzenen Grundlage oder
ungleichmäßiges Dispergieren des Modellarzneistoffs führten allerdings zu Dosierungenauigkeiten,
die sich auf die Auswertung der nachfolgenden Gehaltsbestimmung und Freisetzungsprüfungen
auswirkten. Absorptionsschwankungen können somit einerseits durch eine verstärkte Freisetzung
aus der Arzneiform aber anderseits auch durch eine ungleichmäßige Verteilung zwischen den
einzelnen Proben verursacht werden. Aus diesem Grund musste die Herstellung sehr sorgfältig
erfolgen und die gleichmäßige Verteilung des Wirkstoffs in der Grundlage mit Hilfe einer geeigneten
Gehaltsbestimmung überprüft werden.
Um aus den bei Körpertemperatur schmelzenden Grundlagen einen „Wirkstoff‐Ballon“ zu erhalten,
wurden beide Darreichungsformen mit einem spröden, wasserunlöslichen Polymer‐Überzug
versehen, der durch eine mögliche Variation der Aufenthaltszeit im nüchternen Magen nicht
beeinflusst werden sollte. Eine weitere Voraussetzung für die Verwendbarkeit als Überzug dieser
drucksensitiven Arzneiformen war ein Versprühen der Polymer‐Lösung bei Raumtemperatur, da die
Kugeln ansonsten schon beim Überziehen geschmolzen wären. Aufgrund dieser Voraussetzungen
wurde für diese Arzneiformen unter den wenigen überhaupt geeigneten Überzugsmaterialien
Celluloseacetat als Polymer ausgewählt. In Vorversuchen wurde auch mit Ethylcellulose gearbeitet,
welches als Polymer ebenfalls die genannten Voraussetzungen erfüllt, allerdings zeigte sich eine
verminderte Haftung des Überzugs auf den Kugeln. Durch die Verwendung des Mini‐Coaters konnten
unter definierten Parametern für das automatische Sprühen reproduzierbare Arzneiformen
hergestellt werden. Dagegen resultierten beim manuellen Auftragen des Überzugs mittels
Sprühpistole im Dragierkessel unterschiedlich hohe Polymerbeladungen, was sich auch in einem
geringeren Bestimmtheitsmaß der linearisierten Trendlinie für Charge 1 in Abb. 37 zeigte. Dazu
gehörte sowohl ein unterschiedliches Führen der Sprühpistole als auch der Wechsel zwischen
verschiedenen Personen, die das Überziehen durchführten. Weiterhin konnten feste Parameter
bezüglich Sprühabstand, Sprührate, Sprühdruck und Trocknungsluft nicht automatisch eingestellt
werden, was eine gute Reproduzierbarkeit erschwerte. Bereits kleine Änderungen beim Auftragen
des Überzugs dieser Darreichungsformen können ein verändertes Bruchverhalten zur Folge haben,
weshalb die Verwendung des Mini‐Coaters in diesem Fall klar zu bevorzugen ist. Weiterhin schwierig
ist auch die Berechnung der Aufschlagsmenge, um die gewünschte Polymerbeladung zu erhalten, da
leichte Veränderungen der Sprühparameter zu unterschiedlich starkem Absetzen der
Überzugssuspension führen können. Dies könnte zu einer schlechteren Reproduzierbarkeit beim
Herstellen mehrerer Chargen führen. Unter Verwendung eines automatisierten Gerätes aber konnte
die Steuerbarkeit der Bruchfestigkeit durch unterschiedliche Polymerbeladungen gut und
reproduzierbarer umgesetzt werden.

4 Diskussion
93
Die überzogenen Hartfett‐ und PEG‐Kugeln sind in ihrem festen Zustand bei Raumtemperatur sehr
robust und können über einen längeren Zeitraum in geschlossenen Gefäßen gelagert werden. Dies
wurde nicht explizit geprüft, aber Proben, die über mehrere Wochen im Labor gelagert wurden,
wiesen zumindest äußerlich keine Veränderungen auf. Trotzdem wäre es denkbar, dass die
Grundmassen während der Lagerung aufgrund der Weichmacherwirkung in den Überzug
diffundieren und die freisetzungssteuernden Eigenschaften beeinflussen könnten. Da Hartfett
aufgrund seiner Lipophilie bereits eine lange Haltbarkeit aufweist und Macrogol wenig anfällig für
mikrobiellen Befall ist, ist eine möglicherweise verringerte Haltbarkeit hauptsächlich von der
Stabilität des verwendeten Wirkstoffs abhängig. Macrogol kann eine Hydroperoxid‐induzierte
Zersetzung von Arzneistoffen zeigen und neigt zu vielen Unverträglichkeiten, weshalb die
Kompatibilität mit dem verwendeten Wirkstoff immer individuell geprüft werden sollte [73].
Die Schwierigkeit für eine Gehaltsbestimmung der Hartfett‐Kugeln ergab sich aus der Eigenschaft,
dass kleine Reste des Wirkstoffs Riboflavinphosphat im Hartfett eingeschlossen werden konnten.
Dies zeigte sich bei vorangegangenen Methoden durch eine leichte Gelbfärbung des im Anschluss an
die Gehaltsbestimmung erkalteten Hartfetts. Demzufolge wurde für die beschriebene Methode
Diethylether verwendet, welcher das Hartfett kurzfristig auflösen konnte, wodurch das gut
wasserlösliche Riboflavinphosphat die Möglichkeit hatte, bei Kontakt mit dem wässrigen Medium
sofort in Lösung zu gehen. Beim Verdampfen des Diethylethers und Erkalten des zuvor warmen
Mediums setzte sich das Hartfett an der Oberfläche ab und konnte mittels Filtration abgetrennt
werden. Das abfiltrierte und erneut mit SGFsp pH 1,8 gewaschene Hartfett war annähernd weiß, was
darauf schließen ließ, dass das gelbe Riboflavinphosphat weitestgehend vollständig in SGFsp pH 1,8
gelöst vorlag und für die Vermessung zur Verfügung stand. Die geringe prozentuale
Standardabweichung von 2,3 % für die Stichprobe von sechs Hartfett‐W32‐Kugeln lässt darauf
schließen, dass sich der suspendierte Wirkstoff bei der Herstellung gleichmäßig im geschmolzenen
Hartfett und somit in den einzelnen Arzneiformen verteilte. Die deutlich vereinfachte
Gehaltsbestimmung für Charge 3a ergab eine prozentuale Standardabweichung des Mittelwertes von
14,2 % und wich vom Sollgehalt über 50 % ab. Somit bedarf diese Methode einer Verbesserung, da
die Wirkstoffverteilung des gelösten Paracetamols in den Alginat‐Kügelchen eigentlich gleichmäßiger
sein müsste. Da Charge 3 aber nur der Versuch einer Weiterentwicklung war und sich die
anschließenden Prüfungen auf die Bruchfestigkeit am Texture Analyser beschränkten, war dieses
Ergebnis in diesem Zusammenhang unerheblich.
Der Versuch der Wirkstofffreisetzung in der Paddle‐Apparatur ergab für die sechs Proben der
Hartfett‐Kugeln (Charge 2) keine messbare Freisetzung von Riboflavinphosphat über den gesamten
Zeitraum von 3 h. Das Absetzen kleiner Hartfett‐Tröpfchen an der Kugel‐Oberfläche wies darauf hin,

4 Diskussion
94
dass der Überzug an einigen Stellen durchlässig für geschmolzenes Hartfett war. Während des
Versuchs wurde beobachtet, dass vereinzelt Tröpfchen aus geschmolzenem Hartfett austraten,
allerdings am Überzug haften blieben und die entsprechende Pore dadurch wieder verschlossen. Die
ausgetretene Menge war insgesamt sehr gering und verteilte sich nicht weiter im
Freisetzungsmedium, sodass über die gesamte Versuchszeit keine messbare Wirkstofffreisetzung
stattfand. Für die Hartfett‐Kugeln wäre somit die Voraussetzung erfüllt, während einer stressfreien
Verweilzeit im sauren Magenmilieu intakt zu bleiben und den Wirkstoff eingeschlossen zu halten. Bei
einer sehr kurzen Verweilzeit im nüchternen Magen bestünde die Gefahr, dass die Arzneiform bereits
nach wenigen Minuten entleert werden könnte, bevor der Kern komplett geschmolzen wäre. Um
dies zu umgehen wäre für die Hartfett‐Kugeln auch eine Einnahme zum Essen möglich, sodass sie auf
dem Nahrungsbrei aufliegen und dann bei der nächsten nüchternen Phase entleert werden könnten.
Da über die Zeit von 3 h keine Wirkstofffreisetzung erfolgte, bestünde dabei keine Gefahr einer
frühzeitigen ungewollten Freisetzung. Die Kontraktionsaktivität im postprandialen Magen ist
vergleichbar mit der Phase II des IMMC, bei der die Kontraktionsamplitude bei etwa 50 % des
maximalen Drucks von 300 mbar liegt [5, 74]. Die Kräfte im postprandialen Magen sind also mit etwa
150 mbar deutlich geringer als im nüchternen Magen, sodass die Arzneiform die mechanischen
Kräfte bei einer längeren Magenverweilzeit nach dem Essen überstehen sollte.
Bei den unterschiedlichen Chargen der Hartfett‐Kugeln wurden unterschiedliche Aufschlagsmengen
auf die berechnete Polymerbeladung verwendet, da das Sprühverhalten in beiden Geräten sehr
verschieden war. Die Ergebnisse der Bruchfestigkeitsuntersuchungen am Texture Analyser lieferten
unterschiedliche Kurvenverläufe für die einzelnen Proben. Ein optimaler Verlauf zeigte sich bei den
Proben eins, zwei, vier, sechs und zehn der Charge 1 (Abb. 36) mit konstantem Anstieg auf ein
Kraftmaximum und steilem Abfall, welcher sich aus dem fehlenden Gegendruck der aufgeplatzten
Arzneiform ergab. Das Durchlaufen des Maximums kennzeichnete den Bruch der Arzneiform und
wurde gefolgt von einem relativ konstanten Kurvenverlauf, der erst zum Erreichen der festgelegten
Maximalkraft wieder steil anstieg und abfiel, wenn der Stempel in seine Ausgangsposition
zurückfuhr. Bei den Proben drei, fünf und acht wurde ein erster Peak im weiteren Kurvenverlauf von
Bereichen höherer Kräfte gefolgt, die durch weitere Risse des Überzugs zustande kamen. Das Fehlen
eines klaren Peaks bei den Proben sieben und neun deutete darauf hin, dass die Arzneiform nicht
richtig aufplatzte, sondern zusammengedrückt wurde. Da der flüssige Inhalt schon vorher an einer
Stelle austreten konnte, wurde ein Druckaufbau verhindert, was bei Probe neun durch den konstant
flachen Kurvenverlauf zu erkennen war. Andererseits konnte es, wie bei Probe sieben, zu einem
langsamen Aufreißen des Überzugs kommen, sodass der Kurvenverlauf zwar keinen spitzen Peak,
aber trotzdem ein Abfallen auf niedrigere Kraftwerte zeigte.

4 Diskussion
95
Die Ergebnisse für die Chargen 1 und 2 der Hartfett‐Kugeln zeigten weiterhin, dass bei
unterschiedlichen Polymerbeladungen unterschiedlich hohe Standardabweichungen auftraten. Die
prozentualen Standardabweichungen der Charge 1 liegen dabei einigermaßen einheitlich zwischen
23 und 31 %, während bei Charge 2 die Unterschiede zwischen 11 und 45 % liegen, obwohl diese mit
Hilfe des Mini‐Coaters hergestellt wurden. Ob dies an den unterschiedlichen untersuchten
Polymerbeladungen liegt oder an guten Bedingungen beim Auftragen des Überzugs mit Hilfe der
Sprühpistole bei Charge 1 kann in diesem Zusammenhang nicht geklärt werden. Der Wechsel von
PEG 6000 (bei Charge 1) zu Triacetin (bei Charge 2) als Weichmacher erfolgte aufgrund von
Bruchfestigkeitsuntersuchungen mit verschiedenen Weichmacher‐Arten und unterschiedlich hohen
Zusätzen, deren Ergebnisse im Anhang in Tab. 21 und Abb. 67 dargestellt sind. Dazu wurden
wasserlösliche (Triacetin, Triethylcitrat) und –unlösliche (Dibutylsebacat) Weichmacher getestet, und
es ergab sich nur bei der Verwendung von 10 % Triacetin ein Überzug, der beim Aufplatzen deutliche
Peaks im Kraft‐Weg‐Diagramm erkennen ließ.
Bei der Untersuchung der Chargen 3a und 3b fiel auf, dass der Anstieg der Kraft in Abhängigkeit von
der Polymerbeladung bei Charge 3b viel ausgeprägter bei Werten zwischen 2,0 ± 0,4 N und
8,6 ± 1,7 N lag, während bei Charge 3a die Werte nur von 5,2 ± 1,3 N auf 7,4 ± 1,4 N stiegen. Das
bedeutet, dass das Vorhandensein von vielen kleinen Alginat‐Kügelchen und somit weniger Hartfett
in der gesamten Arzneiform die Bruchfestigkeit in Abhängigkeit von der Polymerbeladung weniger
beeinflusst als bei nur einer Alginat‐Kugel. Trotz guter Ergebnisse der Charge 3 bei den
Bruchfestigkeitsuntersuchungen am Texture Analyser wurden weitere Untersuchungen hinsichtlich
Freisetzung und Bruchfestigkeit an der Stresstest‐Apparatur nicht weitergeführt.
Die Ergebnisse der Bruchfestigkeitsuntersuchungen in der Stresstest‐Apparatur für Proben der
Charge 2 in Tab. 17 ergaben, dass eine Polymerbeladung von 3 mg/cm2 ideal für ein Platzen der
Arzneiform bei 300 mbar wäre. Allerdings wurde dieser Versuch mit einer neuen Hartfett‐W32‐
Charge durchgeführt, welche leicht veränderte Schmelzeigenschaften aufwies. Da mit der älteren
Hartfett‐Sorte ebenfalls Bruchfestigkeitstests mit der Stresstest‐Apparatur durchgeführt wurden, bei
denen vier von sechs Proben mit einer Polymerbeladung von 4 mg/cm2 bei ansteigender
Druckbelastung ≤ 300 mbar und fünf von sechs Proben bei direkter Druckbelastung von 300 mbar
zerbrachen, wurden für den folgenden Freisetzungsversuch der Charge 2 an der Stresstest‐Apparatur
sechs Proben mit einer Polymerbeladung von 4 mg/cm2 ausgewählt. Obwohl alle Proben der
Charge 2 bei Einwirken der Druckwelle zerbrachen, zeigte sich die prozentuale Freisetzung sehr
variabel. Dies könnte zum einen am unvollständigen Herauslösen der flüssigen Hartfett‐Suspension,
zum anderen aber auch an unterschiedlicher Wirkstoffbeladung der einzelnen Proben gelegen
haben. Bei der Herstellung einer größeren Menge an Hartfett‐Kugeln im Cremeschmelzverfahren,

4 Diskussion
96
musste die verwendete Hartfett‐Suspension trotz zügigen Ausgießens gelegentlich zwischendurch
erwärmt werden. Dadurch könnte es trotz regelmäßigen Umrührens möglich sein, dass Kugeln im
ersten Gießzyklus andere Wirkstoffbeladungen besaßen als Kugeln in einem späteren Gießzyklus.
Dies könnte erklären, warum selbst die Proben, bei denen ein annähernd weißer Polymer‐Überzug
nach dem Freisetzungsversuch verblieb nur etwa 70 % des Wirkstoffs freisetzten, obwohl bei der
Gehaltsbestimmung sehr gleichmäßige Werte erhalten wurden. Das Dispergieren des Wirkstoffs in
der lipophilen Grundlage kann ungeübt zu ungleichmäßigen Wirkstoffgehalten führen. Dahingegen
zeigte der Freisetzungsversuch für Charge 1 ein sofortiges und vollständiges Aufreißen des
Celluloseacetat‐Überzugs und eine Wirkstofffreisetzung von 85 – 99 % für alle sechs Proben. Die
beobachteten Schwankungen in der Absorption zu Beginn des Versuchs dürften auf Luftbläschen in
der Durchflussküvette zurückzuführen sein, die sich dann allerdings schnell auflösten. Durch
Sedimentation des Wirkstoffs in der geschmolzenen Hartfett‐Kugel und das Zusammendrücken der
leeren Überzugsfilme durch die Druckwelle wurde ein geringer Anteil des Modellarzneistoffs der
Detektion entzogen. Entsprechend sind auch in diesem Fall bei allen sechs Proben gelbe Rückstände
in den Resten der Überzüge zu erkennen, obwohl eine hohe prozentuale Wirkstofffreisetzung
erreicht wurde. Die ungleichmäßige Freisetzung bei Charge 2 dürfte deshalb vor allem auf ein
ungleichmäßiges Einarbeiten des Wirkstoffs bei der Herstellung der Arzneiformen zurückzuführen
sein. Trotzdem ist auch im Fall der Charge 2 gut zu erkennen, dass die 300 mbar starken Druckwellen
bei allen sechs Proben zu einem vollständigen und großflächigen Aufreißen führten und den
kompletten Inhalt sofort freigaben.
Die Hartfett‐Kugeln der Charge 3 wurden entwickelt, um durch das Lösen des Wirkstoffs im Wasser
der Alginat‐Kügelchen eine bessere Dosiergenauigkeit zu erreichen. Bei Einnahme sollte das
umgebende Hartfett schmelzen, einen Celluloseacetat‐Ballon mit dem Überzug bilden und bei
entsprechendem Druck aufplatzen. Das geschmolzene Hartfett und die Alginat‐Kügelchen könnten
sich dann über die Schleimhaut verteilen und durch verlängerte Kontaktzeit des wirkstoffhaltigen
Alginats eine verbesserte Absorption bedingen. Fraglich ist allerdings die Dauer der Kontaktzeit der
Alginat‐Kügelchen mit der Schleimhaut. Da diese sehr klein und relativ fest sind, könnten sie sich aus
der geschmolzenen Hartfett‐Grundlage heraus lösen und schnell weiter transportiert werden, was
für die gewünschte lange Verweilzeit im Bereich des Absorptionsfensters ungünstig wäre. Obwohl
Natriumalginat als anionisches Polymer eventuell mukoadhäsive Eigenschaften besitzt, könnte die
Verwendung anderer bekannt mukoadhäsiver Substanzen wie Polyacrylsäure, Carmellose oder
synthetischer Polymethacrylate gegebenenfalls zu besseren Hafteigenschaften führen [75]. Durch
eine geringere Konzentration des verwendeten Alginats könnte außerdem die Konsistenz verringert
werden, sodass weichere Kügelchen entstünden, die an die Schleimhaut gedrückt werden und dort
haften könnten.

4 Diskussion
97
Bei der versuchsweisen Herstellung größerer Alginat‐Kugeln konnte durch eine veränderte
Verweilzeit des Alginats in der CaCl2‐Lösung die Wandstärke beeinflusst werden, sodass innerhalb
einer festen Wand, ein flüssiger wirkstoffhaltiger Kern vorhanden war (Abb. 64), der sich beim
Zerplatzen auf der Schleimhaut verteilen könnte. Schwierig an dieser Arzneiform wäre allerdings
wieder ihr hydrophiler Charakter, der mit Problemen bezüglich Haltbarkeit und Verdunstung
einhergehen würden.
Abb. 64: 3 %ige Alginat‐Kugel mit flüssigem Kern, deren äußere Wand mit Hilfe einer 10 %igen CaCl2‐Lösung
erhärtet wurde.
Die Verwendung von Hartfett beschränkt sich im NRF zumeist auf Zäpfchen und Vaginalzäpfchen, die
rektal oder vaginal angewendet werden. Eine Ausnahme bilden allerdings Dronabinol‐Kapseln, die als
Antiemetikum oder Appetitstimulans oral eingenommen werden. Der lipophile und wasserunlösliche
Wirkstoff wird dabei in geschmolzenem Hartfett vollständig gelöst und in Hartgelatine‐Steckkapseln
abgefüllt. Bei dieser Arzneiform schmilzt nach Einnahme der Kapsel und deren Auflösen im Magen
der Hartfettkern und gibt den gelösten Wirkstoff frei [73]. Weitere Beispiele aus der Roten Liste für
orale Arzneiformen, die Hartfett enthalten sind Prostagutt® forte 160/120 mg Kapseln bei benigner
Prostatahyperplasie mit Extrakten aus Sägepalmenfrüchten und Brennesselwurzel und Alpan®
300 mg Weichkapseln mit Thioctsäure für die Anwendung bei Missempfindungen bei diabetischer
Polyneuropathie [76‐78].
Der Emulgatoranteil im Hartfett verbessert die Suspendierung der Wirkstoffpartikel und die
Spreitung auf der Schleimhaut [46]. Beim Aufplatzen der Arzneiform am Pylorus könnte sich die
Kontaktzeit des Hartfetts und des Arzneistoffs auf der Schleimhaut des Duodenums verlängern und
eine verbesserte Resorption wäre unter Umständen die Folge. Da die Resorption von Lipiden im
Duodenum und anfänglichen Teil des Jejunums mit über 95 % sehr effektiv ist [1], könnte das in der
Arzneiform enthaltene Hartfett gleich am Anfang des Gastrointestinaltraktes vom Körper
aufgenommen werden. Bei chronischer und häufiger täglicher Gabe der Hartfett‐W32‐Kugeln als
Arzneiform sowie verminderter Fettresorption könnte es im schlimmsten Fall zu Steatorrhö kommen

4 Diskussion
98
[60]. Dementsprechend müsste für diese Arzneiform ein Wirkstoff ausgesucht werden, bei dem eine
seltenere tägliche Gabe ausreichend wäre, um die vollständige Resorption gewährleisten zu können.
Die Durchführung einer Gehaltsbestimmung für die PEG‐1000‐Kugeln war aufgrund der
Wasserlöslichkeit der Grundlage deutlich vereinfacht, da die Arzneiformen lediglich aufgelöst und
verdünnt werden mussten, um die Absorptionen der erhaltenen Wirkstofflösungen vermessen zu
können. Es ergab sich für diese Darreichungsformen, die mit einem höheren Wirkstoffgehalt
hergestellt wurden, eine prozentuale Standardabweichung bei sechs getesteten Stichproben von nur
1,6 %, was auf eine gut reproduzierbare Herstellung hindeutet.
Die Prüfung der Wirkstofffreisetzung in der Paddle‐Apparatur zeigte nach einer Verzögerungszeit von
20 bis 30 min einen Anstieg der Freisetzung auf bis zu 72 % bis zum Ende des dreistündigen Versuchs.
Da bei den Versuchen am Texture Analyser eine Vorinkubation von 25 min nötig war, um ein
vollständiges Schmelzen des PEG‐Kerns zu garantieren, ist davon auszugehen, dass aus dem festen
oder halb geschmolzenen Kern kein Wirkstoff austreten kann. Sobald sich die Grundlage aber
verflüssigt und der wirkstoffhaltige Ballon vorliegt, diffundiert begünstigt durch den osmotischen
Effekt des Macrogols Wasser ein, welches dann wiederum Wirkstoff und Grundlage durch den
Überzug herauslöst. Dies war an dünnen Schlieren zu erkennen, die von den in Sinkern befindlichen
PEG‐Kugeln aufstiegen und sich im Medium verteilten. In Abhängigkeit von der Magenverweilzeit
könnten also unterschiedliche Mengen an Wirkstoff bereits vor dem eigentlichen Freisetzungstrigger
des Drucks abgegeben werden. Im Bereich von 25 bis 30 min wäre das unproblematisch, aber da
auch deutlich kürzere und längere Magenverweilzeiten möglich sind, sind die möglichen Folgen
schwer abzuschätzen. Eine Einnahme zum Essen wäre in Anbetracht der stark verlängerten
Magenverweilzeit aufgrund der hohen Freisetzungsrate im Gegensatz zu den Hartfett‐Kugeln nicht
sinnvoll.
Die Bruchfestigkeitsuntersuchungen am Texture Analyser ergaben wie auch bei den Hartfett‐Kugeln
unterschiedlich hohe Standardabweichungen bei den verschiedenen Polymerbeladungen. Dabei
stand die Höhe der Standardabweichung in keinem Zusammenhang mit der Höhe der
Polymerbeladung. Verglichen mit den Werten der Hartfett‐Kugeln war die aufzuwendende Kraft der
PEG‐Kugeln bei jeweils gleicher Polymerbeladung etwa gleich hoch. Die Standardabweichungen bei
den PEG‐Kugeln waren allerdings meist etwas höher, was auf ein ungleichmäßigeres Bruchverhalten
schließen lässt. Dies dürfte auf die relativ dünnen Schichtdicken der Arzneiformen und die im
Überzug vorhandenen kleinen Poren zurückzuführen sein. Die Verwendung eines wasserlöslichen
Weichmachers wie PEG 6000 oder Triacetin führte dazu, dass dieser bei Inkubation in einem
wässrigen Medium herausgelöst wurde und kleine Poren gebildet wurden. Das durch diese Poren
hinein diffundierende Wasser löste dann einen Teil des Macrogols heraus. Das dadurch veränderte

4 Diskussion
99
Volumen innerhalb des Überzugs führte dann durch unterschiedlich starke Drücke, die im Inneren
aufgebaut wurden, möglicherweise auch zu veränderten Brucheigenschaften. Zudem kann ein
vorzeitiges Austreten von geschmolzenem PEG durch die Poren dazu führen, dass sich innerhalb der
Kugel kein ausreichend hoher Druck aufbaute, der die Darreichungsformen schließlich zum Platzen
brachte. Die Kraft stieg dann bis zu einem Punkt an, an dem kontinuierlich geschmolzener Inhalt bei
einem weitestgehend konstanten Druck durch die Poren des Überzugs gedrückt wurde. Dies könnte
eine Erklärung für das bei Probe 6 im Freisetzungsversuch an der Stresstest‐Apparatur beobachtete
Freisetzungsverhalten sein. Hier wurde bereits vor der ersten Druckwelle Arzneistoff herausgelöst
und es wurde nach Anlegen von 300 mbar nochmals ein Anstieg der Wirkstofffreisetzung
beobachtet. Insgesamt kam es allerdings nur zu einer 56 %igen Wirkstofffreisetzung. Der restliche
Wirkstoff wurde beim Zusammendrücken der Kugel im Überzug festgehalten. Die Linearität des
Zusammenhangs zwischen Bruchkraft und Polymerbeladung zeigte sich bei den untersuchten
Polymerbeladungen durch ein Bestimmtheitsmaß von R2 = 0,989.
Die Bruchfestigkeitsversuche mittels Stresstest‐Apparatur für die PEG‐Kugeln zeigten eine deutliche
Streuung zwischen den angelegten Drücken innerhalb von Proben der gleichen Polymerbeladung.
Dies dürfte durch die vorstehend diskutierten Ursachen begründet sein. Bei den
Freisetzungsversuchen an der Stresstest‐Apparatur riss der Celluloseacetat‐Überzug bei drei von
sechs Proben weit auf und gab den Inhalt vollständig frei, was sich zum Teil an weißen
Überzugsresten ohne gelbe Rückstände zeigte. Durch die gute Wasserlöslichkeit der Grundlage
konnte bei diesen Proben der gesamte Inhalt herausgelöst werden. Eine Kugel riss an der Seite auf,
wurde dann aber zusammengedrückt, sodass Reste der wirkstoffhaltigen Grundlage zurückblieben.
Trotzdem wurden 87 % Riboflavinphosphat aus dieser Kugel freigesetzt. Bei zwei Proben fand kein
Einreißen des Überzugs statt, sondern lediglich ein Zusammendrücken der Arzneiform. Dies könnte
auch in diesem Fall wieder damit zusammenhängen, dass ein Teil der flüssigen Grundlage durch die
winzigen Poren entweichen konnte und sich somit der Innendruck verringerte. Besonders bei Probe
sechs ist zu erkennen, dass vor Einwirken der Druckwelle bereits knapp 18 % des enthaltenen
Riboflavinphosphats aus der Darreichungsform freigesetzt wurden, sodass bis zum Ende des
Versuchs nur gut die Hälfte des gesamten Wirkstoffs in das Freisetzungsmedium abgegeben werden
konnte. Bis auf diese zwei Proben funktioniert das Prinzip des wirkstoffhaltigen Ballons aber auch bei
Verwendung von PEG 1000 als Grundlage.
Die Verwendung von PEG als Grundlage wird als Mischung von PEG 400 und PEG 6000 im NRF in
Form von Progesteron‐Vaginalzäpfchen 25 mg eingesetzt [73]. Dabei wird das schwer wasserlösliche
Progesteron in der Schmelze der beiden PEG‐Sorten bereits unterhalb seiner eigenen
Schmelztemperatur von 130 °C gelöst. Dies wäre bei geeigneten Wirkstoffen auch für die Anwendung

4 Diskussion
100
als drucksensitive Arzneiform möglich und würde die Problematik einer ungleichmäßigen
Wirkstoffverteilung bei Suspensionszubereitungen umgehen. Außerdem könnte die Absorption nach
Zerplatzen der Arzneiform am Pylorus noch schneller erfolgen, da notwendige Lösungsschritte
wegfallen würden. Ein Wechsel von PEG 1000 zu einer 4:6‐Mischung von Macrogol 6000 und
Macrogol 400 wäre diesbezüglich denkbar, da der Schmelzpunkt dieser Mischung bei Anwendung als
Zäpfchen ebenfalls unterhalb der Körpertemperatur liegen müsste, aber die Zäpfchen bei
Raumtemperatur eine formstabile Arzneiform darstellen. Inwieweit diese Mischung dann aber auch
zum Überziehen im Coater geeignet wäre, ist unklar. PEG 400 und PEG 4000 bilden die Grundlage für
Codicaps® mono 30 mg Weichkapseln, die mit dem Wirkstoff Codein zur symptomatischen Therapie
von Reizhusten eingesetzt werden können [78]. PEG 1000 wird weiterhin als Grundlage in Setofilm
4 mg Schmelzfilm eingesetzt, der Ondansetron zur Anwendung bei Chemotherapie induzierter
Übelkeit enthält. Dieser Film wird auf die Zunge gelegt und löst sich innerhalb weniger Sekunden auf,
um eine schnelle Wirkung über die Mundschleimhaut zu erzielen [78, 79].
Erlich et al. untersuchten PEG hinsichtlich seiner Verwendbarkeit als Lösungsmittel, um schwer
wasserlösliche Wirkstoffe für den Körper schneller verfügbar zu machen. Dafür wurde Danazol, ein
Derivat des Testosterons, unter leichter Wärmezufuhr in PEG 400 gelöst und in Hartgelatinekapseln
gefüllt. Versuche an Hunden zeigten eine 3,7‐fach erhöhte Bioverfügbarkeit im Vergleich zu einer
kommerziell erhältlichen wirkstoffgleichen Danocrine®‐Kapsel [80]. Bei Verwendung eines Wirkstoffs,
der sich vollständig im PEG löst, könnte entsprechend diesem Befund zusätzlich zu einer
drucksensitiven Wirkstofffreisetzung gleichzeitig eine verbesserte Bioverfügbarkeit für schlecht
resorbierbare Wirkstoffe möglich sein.
Ein ähnliches Freisetzungsprinzip wie die entwickelten PEG‐Kugeln besitzen die PCDC’s von Matsuda
et al., die eine mit PEG 1000 gefüllte und mit Ethylcellulose innenbeschichtete Kapsel darstellen. Bei
Einnahme und Erreichen der Körpertemperatur schmilzt das PEG und es bildet sich ein Ethylcellulose‐
Ballon, der auf definierten Druck platzen soll [33, 34]. Im Vergleich zu den in dieser Arbeit
vorgestellten PEG‐Kugeln ist die Herstellung deutlich aufwändiger, weil jede Kapsel einzeln
innenbeschichtet werden muss. In weiteren Arbeiten wurde diese Herstellungsweise der PCDC’s
allerdings vereinfacht, indem die Überzugssuspension im Sprühverfahren aufgetragen wurde [36].
Die Isotropie der innerhalb dieser Arbeit entwickelten PEG‐Kugeln könnte bei der Applikation von
Vorteil sein, da die Krafteinwirkung unabhängig von der Position immer gleich sein sollte.
Polyethylenglycol wird in Form verschiedener Handelsprodukte zum Abführen bei Konstipation
eingesetzt. Dabei liegen handelsübliche Dosen für das Präparat Movicol® zwischen 6,9 g und 13,7 g
für Kinder und Erwachsene. Bei dem verwendeten PEG handelt es sich meist um Macrogol 3350
(oder Macrogol 4000 bei Laxofalk®), welches als osmotisches Laxans Wasser bindet und in den

4 Diskussion
101
Dickdarm transportiert. Die daraufhin folgende Erweichung des Stuhls und die Zunahme des
Darminhaltvolumens führen als Konsequenz zu einer erhöhten Peristaltik [60, 81‐83]. Eine Studie von
Wirz et al., die sich mit dem Einsatz von Laxantien in der palliativen Therapie tumorkranker Patienten
unter Morphintherapie befasst, zeigt, dass Macrogol 3350 sowohl prophylaktisch als auch
therapeutisch besonders erfolgreich eingesetzt werden kann [84]. Allerdings zeigte sich in einem
Bericht der Cochrane Collaboration über die Anwendbarkeit von Laxantien bei kindlicher
Konstipation, dass vereinzelt Nebenwirkungen wie Übelkeit und Erbrechen auftreten können [85].
Macrogol wird im Körper weder resorbiert noch metabolisiert, sondern verlässt den Körper
unverändert. Da die Hygroskopizität mit steigender Molekülmasse der PEG’s abnimmt, könnte für
das verwendete PEG 1000 ebenfalls ein abführender Effekt erwartet werden. Da die Menge in den
entwickelten Arzneiformen allerdings deutlich unter den verwendeten Dosierungen liegt und die
Arzneiformen für die Anwendung am Menschen ohnehin einer Größenanpassung für ein
angenehmeres Schlucken unterzogen werden müssten, sollte eine laxierende Nebenwirkung weniger
problematisch sein. Mit einer veränderten Größe müssten dann erneute Test bezüglich des
Bruchverhaltens durchgeführt werden, da sich der Druck, bei dem eine Arzneiform zerdrückt wird in
Abhängigkeit von der Fläche verändert. Bei den festen oralen Darreichungsformen findet
niedermolekulares PEG 400 vor allem in Weichkapseln als Grundlage Anwendung, während höher
molekulares wie PEG 8000 oft in Filmüberzügen, Granulaten oder Brausetabletten eingesetzt wird.
Im Vergleich zu dem von Kamba et al. vorgestellten DDRS zur Untersuchung der Druckverhältnisse im
Magen, bei dem die Arzneiform zwar durch Druck zerbricht, aber der wirkstoffhaltige Kern erst nach
Auflösen des magensaftresistenten Überzugs freigesetzt wird, stellen die Hartfett‐ und PEG‐Kugeln
den Wirkstoff direkt nach Aufplatzen des Überzugs zur Verfügung [39‐41]. Dadurch sollte für
Arzneistoffe mit einem Absorptionsfenster im oberen Dünndarm die Möglichkeit zur schnellen und
vollständigen Absorption bestehen. Des Weiteren ist die Stabilität im Vergleich zum
druckempfindlichen Teflon‐Mantel des DDRS deutlich höher, da sich die Eigenschaft als
drucksensitive Arzneiform erst im Körper bei 37 °C entwickelt. Um der Gefahr eines unvollständigen
Schmelzens der Grundlage im Falle einer verkürzten Magenverweilzeit und damit veränderten
Brucheigenschaften zu entgehen, könnten die Arzneiformen auch vor der Einnahme in 37 °C
warmem Wasser inkubiert und anschließend geschluckt werden, da sie den vorherrschenden
Drücken von 120 mbar im Ösophagus mit geschmolzenem Kern stand halten würden [1].
Verglichen mit den von Marciani et al. vorgestellten Agar‐Kugeln für die Untersuchung der antralen
Kräfte, die innerhalb dieser Arbeit für die Wirkstofffreisetzung weiter entwickelt wurden, weisen die
Hartfett‐ und PEG‐Kugeln einige Vorteile auf [8]. Obwohl durch Veränderung der Agar‐Konzentration
Arzneiformen unterschiedlicher Festigkeit hergestellt werden konnten, die für die Untersuchung der

4 Diskussion
102
Druckverhältnisse im Gastrointestinaltrakt zum Teil geeignet sind, und das Vorliegen des Wirkstoffs
in gelöster Form für die Dosiergenauigkeit von Vorteil ist, ist eine Anwendung als drucksensitive
Darreichungsform ungeeignet. Während die Bruchfestigkeit der Agar‐Kugeln sehr von der
Magenverweilzeit und der daraus resultierenden Erweichung abhängt, wird die Bruchkraft bei den
Hartfett‐ und PEG‐Kugeln hingegen durch die Schichtdicke des Polymer‐Überzugs festgelegt, der
gleichzeitig vor einer verfrühten Arzneistofffreisetzung schützen kann. Da Celluloseacetat vom pH‐
Wert des Magens nicht angegriffen wird, ist eine ungewollte frühzeitige Freisetzung zumindest für
die Hartfett‐Kugeln auszuschließen, wie sich auch in den Ergebnissen der Freisetzung in der Paddle‐
Apparatur zeigte. Die verfrühte Freisetzung des Wirkstoffs aus den PEG‐Kugeln könnte durch eine
veränderte Zusammensetzung der Überzugssuspension und damit eine höhere Dichtigkeit erreicht
werden. Allerdings könnte sich dies wieder auf das Bruchverhalten dieser Arzneiform auswirken.
4.3 Paraffin‐Kapseln
Bei der Entwicklung der Paraffin‐Kapseln wurde als Freisetzungsprinzip eine Sollbruchstelle
eingefügt. An dieser Sollbruchstelle sollte die Darreichungsform unter einem definierten Druck
aufplatzen und den Wirkstoff schlagartig freisetzen. Um eine Verteilung der wirkstoffhaltigen
Grundlage über die Schleimhaut des Duodenums gewährleisten zu können, sollte diese bereits flüssig
vorliegen. Als äußere Hülle dieser Arzneiform dienten Gelatine‐Kapseln, wodurch die Verwendung
wässriger Grundlagen ausgeschlossen war. Dementsprechend wurden verschiedene Öle und
Paraffine auf ihre Eignung hin getestet. Die Ergebnisse des Versuchs sind in dieser Arbeit nur verkürzt
in Kapitel 2.2.3 dargestellt. Dabei war es wichtig, dass der Wirkstoff unlöslich in der Grundlage war,
um einen Verlust bei der Analytik ausschließen zu können. Da bei den beiden Paraffin‐Sorten auch
nach 72 h keine Gelbfärbung oder Trübung der lipophilen Phase zu erkennen war, und sich das
dickflüssige Paraffin zusätzlich durch eine höhere Viskosität auszeichnete, wurde dieses für die
Entwicklung dieser Darreichungsform verwendet. Eine etwas höhere Konsistenz des dickflüssigen
verglichen mit dem dünnflüssigen Paraffin war wichtig, um ein vorzeitiges Ausfließen durch die
Löcher an der Sollbruchstelle auszuschließen. Unter Zuhilfenahme von Gelatine‐Steckkapseln der
Größe 0 war die Herstellung der Paraffin‐Kapseln sehr einfach und gut reproduzierbar. Nach dem
Befüllen der Kapselunterteile mit der wirkstoffhaltigen Suspension und Überziehen im Mini‐Coater
waren die Kapseln robust und konnten unter Lichtschutz gelagert werden. Allerdings zeigten sich
Sedimentationserscheinungen, die nur zum Teil wieder leicht aufschüttelbar waren. Dies war ein
deutlicher Nachteil, da die Suspension bei den Bruchfestigkeitsversuchen größtenteils nur durch die
Löcher gedrückt wurde und nicht, wie erhofft durch das Öffnen der Sollbruchstelle heraustreten
konnte. Somit war es möglich, dass zum Teil nur die Grundlage herausgedrückt wurde und der
sedimentierte Wirkstoff in der Kapsel verblieb. Dementsprechend wäre die Verwendung von

4 Diskussion
103
lipophilen Wirkstoffen vielleicht besser geeignet, um ein vollständiges Lösen in der Grundlage zu
gewährleisten und beim Herausdrücken eine gleichmäßige Wirkstoffdosierung zu erreichen.
Allerdings kann die Gleichmäßigkeit der herausgedrückten Menge nicht garantiert werden, wodurch
sich auch so Ungenauigkeiten in der Dosierung ergeben würden.
Die für die Paraffin‐Kapseln entwickelte Gehaltsbestimmung erwies sich als geeignet, da sich für die
Stichprobenzahl von n = 6 eine relativ geringe prozentuale Standardabweichung von 3,4 % ergab und
der Mittelwert mit 9,45 ± 0,32 mg recht nah am angestrebten Gehalt von 10 mg lag. Es konnte durch
Verwendung des gelben Riboflavinphosphats als Modellarzneistoff zusätzlich visuell gezeigt werden,
dass sich bei Durchführung der Gehaltsbestimmung keine Wirkstoffpartikel mehr im Paraffin
befanden, sondern diese sich komplett in der wässrigen Phase lösten.
Bei der Prüfung auf Wirkstofffreisetzung mittels Paddle‐Apparatur wurde trotz der acht radial
angeordneten Löcher über den Zeitraum von 3 h kein Wirkstoff freigesetzt. Da das dickflüssige
Paraffin vermutlich eine zu hohe Grenzflächenspannung besitzt, um aus den 0,33 mm kleinen
Löchern auszutreten und aufgrund seiner Lipophilie kein Bestreben hat, sich mit dem wässrigen
Freisetzungsmedium zu vermischen, verblieb die wirkstoffhaltige Suspension komplett in der Kapsel.
Mit diesem Verhalten wäre die Anforderung eines Rückhalts des Wirkstoffs in der Arzneiform unter
stressfreien Standardbedingungen gegeben.
Das Prinzip im Fall der entwickelten Arzneiform stellt eine überzogene Kapsel dar, die mit Löchern
versehen wurde, durch die das Freisetzungsmedium eindringen sollte, um die Gelatine‐Hülle von
innen aufzuweichen. Dabei sollte der dünne Ethylcellulose‐Überzug als spröde Barriere zwischen den
acht radial angeordneten Löchern zurück bleiben und in Abhängigkeit vom Druck aufreißen, um den
gesamten Inhalt frei zu geben. Da in den Versuchen der Bruchfestigkeit am Texture Analyser und der
Stresstest‐Apparatur trotz einer Erweichungszeit von 30 min keine einzige Kapsel aufplatzte, lässt
sich vermuten, dass das Wasser nicht gut genug durch die Löcher eindringen konnte um die Gelatine
zu erweichen. Auch der Freisetzungsversuch unter Druckeinwirkung mittels Stresstest‐Apparatur
ergab bei den 300 mbar‐Druckwellen nur eine maximale Wirkstofffreisetzung von 6 %.
Gegebenenfalls hätte eine höhere Anzahl an Löchern die Wahrscheinlichkeit des Aufbrechens erhöht,
da am Ende dieser Versuche ein leichtes Aufweichen der inneren Gelatinehülle zu erkennen war.
Ebenfalls denkbar wäre eine longitudinale Anordnung der Löcher entlang der Kapsel gewesen, um
ein längeres Aufreißen und somit eine schnellere Entleerung zu gewährleisten. Da nur die
Kapselunterteile mit der wirkstoffhaltigen Suspension befüllt werden konnten, befand sich in der
Kapsel Luft, die zusätzlich zu den Löchern einen Druckaufbau innerhalb der Kapsel verhinderte. Somit
konnte ohne ein Aufplatzen an der Sollbruchstelle nur sehr wenig Paraffin herausgedrückt werden.
Eine luftfreie Befüllung hätte unter Umständen dazu führen können, dass der Inhalt unter

4 Diskussion
104
Druckeinwirkung durch die Löcher heraus gedrückt worden wäre, aber selbst dann wäre es fraglich,
inwieweit der sedimentierte Wirkstoff vollständig durch die 0,33 mm kleinen Löcher gepresst werden
könnte.
Für das Abfüllen von Flüssigkeiten werden meist Weichgelatinekapseln verwendet, die sich von
Hartgelatinekapseln deutlich unterscheiden und als eigenständige Darreichungsform betrachtet
werden sollten. Diese Kapseln entstehen in einem Fertigungsschritt zusammen mit der Befüllung
beispielsweise unter Verwendung des Rotary‐Die‐Verfahrens nach Scherer [46, 86]. Diese
Darreichungsform wird unter anderem verwendet für wirkstoffhaltige Flüssigkeiten, wie bei Voltaren
Dolo Liquid, bei dem der Wirkstoff Diclofenac in niedermolekularem Macrogol gelöst vorliegt [87].
Auch Vitamine wie Vitamin E oder das lipophile Doxycyclin werden in pflanzlichen Ölen in
Weichgelatinekapseln abgefüllt [88, 89].
Das Abfüllen von Flüssigkeiten in Hartgelatinekapseln ist unter bestimmten Voraussetzungen auch
möglich. Dabei sollte auf den sogenannten ausbalancierten Wassergehalt (BAW = balanced amount
of water) Rücksicht genommen werden. Dieser bezeichnet den Wasseranteil in einer flüssigen
Kapselfüllung, der die mechanischen Eigenschaften des Kapselmaterials unter längerer Lagerungszeit
nicht negativ beeinflussen darf. Die Kompatibilität zwischen Kapselfüllung und Kapselmaterial ist
besonders bei amphiphilen und hydrophilen Hilfsstoffen von Bedeutung, da diese einerseits bei
einem Wassergehalt unter 10 % die Gelatinehülle austrocknen und somit brüchig machen können,
aber andererseits ein steigender Wassergehalt auf etwa 18 % zur Erweichung der Gelatinehülle
führen kann [90]. Durch das Einfüllen eines lipophilen Paraffins, ist die Wahrscheinlichkeit des
Wasserentzugs aus der Kapsel eher gering. Desweiteren bedarf es einer geeigneten Methode zur
Versiegelung der Kapselunter‐ und Kapseloberteile. Das Versiegeln der Kapselteile in dieser Arbeit
stellt eine deutlich vereinfachte Version der von Cadé et al. vorgestellten und patentierten Methode
für LICAPS™ dar [91]. Zum Schutz oxidationsempfindlicher Flüssigkeiten wie ungesättigter Fettsäuren
wurden die LICAPS™ mit einer Wasser‐Ethanol‐Mischung eingesprüht und anschließend leicht
erwärmt. Aufgrund der geringen Oberflächenspannung und der hohen Kapillarkräfte des Ethanols
dringt dieses zwischen Kapselunter‐ und Kapseloberteil ein und führt nach Erwärmen zu einer
versiegelten Arzneiform. Im Fall unserer Arzneiform wurde nur Wasser verwendet, welches die
Gelatine des Unter‐ und Oberteils anlöste und diese beim Trocknen miteinander verklebte. Da in
unserem Fall keine Stabilitätsuntersuchungen erfolgten, es sich bei flüssigem Paraffin nicht um einen
oxidationsempfindlichen Stoff handelt und aus apparativen Gründen die LICAPS™‐Technik nicht
möglich war, wurde die vereinfachte Versiegelungs‐Methode angewendet. Die Prüfung auf
Dichtigkeit, die ausschließlich visuell vorgenommen wurde, zeigte, dass sowohl vor als auch nach

4 Diskussion
105
dem Überziehen mit Ethylcellulose kein Paraffin austrat, was die vereinfachte Versiegelung innerhalb
dieser Arbeit rechtfertigte.
Bei kommerzieller GMP‐gerechter Herstellung und Versieglung von flüssigkeitsgefüllten Hartgelatine‐
Kapseln und unter Berücksichtigung geeigneter Füllungen zeigt diese Arzneiform eine
bemerkenswerte Haltbarkeit, wie Bowtle et al. 2011 zeigen konnten. Sie untersuchten dabei unter
anderem Darreichungsformen mit Lebertran, Weizenkeim‐ oder Lachsöl sowie Vitamin E‐
Zubereitungen über einen Zeitraum von 20 Jahren. Bei den Untersuchungen ergab sich eine
bemerkenswerte physikalische Stabilität hinsichtlich der Verpackung und des Kapselmaterials sowie
der Dichtigkeit der Arzneiformen. Bei entsprechender Herstellung und Versiegelung handelt es sich
bei flüssigkeitsgefüllten Hartgelatinekapseln also um äußerst robuste und langlebige
Darreichungsformen [92].
Barnwell et al. stellten 1992 eine flüssig gefüllte Hartgelatine‐Kapsel vor, die den lipophilen Wirkstoff
Propranolol mit einem hohen first‐pass‐Effekt in Ölsäure gelöst und zusammen mit Gallensäuren
enthielt. Die Gallensäuren sollten den Metabolismus absättigen und somit als „Leber‐Bypass“ eine
höhere Bioverfügbarkeit des Wirkstoffs ermöglichen. Die Kapseln wurden entsprechend der
LICAPS™‐Methode versiegelt und im Anschluss magensaftresistent überzogen [93]. In weiteren
Arbeiten wurde diese Arzneiform zu einer verzögert freisetzenden Darreichungsform entwickelt, bei
der auf den Zusatz von Gallensäuren verzichtet wurde. Dabei bestand die verzögert freisetzende
Komponente aus einer festen erodierbaren Matrix aus Gelucire®, welche nach Aktivierung des
lymphatischen Systems das enthaltene Propranolol in Ölsäure freigeben sollte. Durch dieses
biphasische System mit seiner deutlich verstärkten Bioverfügbarkeit war eine einmal tägliche Gabe
möglich [94‐96]. Der Vorteil einer Verwendung von flüssigkeitsgefüllten Arzneiformen liegt also vor
allem in einer erhöhten Bioverfügbarkeit, da Lösungsprozesse eine untergeordnete Rolle spielen und
somit die notwendige Zeit bis zur Absorption verkürzt werden kann. Wenn die Grundlage dann noch
gute Eigenschaften hinsichtlich Spreitung oder Mukoadhäsivität aufweist und somit die Kontaktzeit
zwischen Arzneistoff und Schleimhaut verlängert wäre, könnte der mitunter kurze Abschnitt mit
guten Resorptionseigenschaften unter Umständen optimal genutzt werden.
Flüssiges Paraffin wird pharmazeutisch als Laxans verwendet und ordnet sich neben den anderen
Gruppen der Quellmittel, osmotisch wirksamen und stimulierenden Laxantien als Weichmachen bei
den konventionellen Abführmitteln ein. Diese können durch Auslösung des rektokolischen Reflexes
den Darm stimulieren und somit auch bei neurologischen Erkrankungen angewendet werden [97].
Flüssiges Paraffin ist so unter anderem als Emulsion unter dem Namen Paragar® oder Paragol®N und
als Gel unter dem Namen Lansoyl® in der Schweiz erhältlich [98‐100]. Die Cochrane Collaboration hat
eine Reihe von Studien untersucht, die sich mit der Verwendung von flüssigem Paraffin als Laxans

4 Diskussion
106
und dessen Verträglichkeit befassten [85, 101]. In diesen Arbeiten wurde sowohl die Anwendbarkeit
bei kindlicher Konstipation als auch in der Palliativmedizin näher betrachtet. Die dabei verwendeten
Dosierungen lagen zumeist allerdings bei 1 mL/kg und somit weit über der Menge, die sich in einer
einzelnen der neu entwickelten Arzneiformen befindet. Nach Sharif et al. sind vereinzelt
beschriebene Nebenwirkungen wie eine verringerte Absorption von fettlöslichen Vitaminen oder gar
die Entstehung von Krebs als Folge von längerer Paraffin‐Einnahme unbegründet. Wenn es zum
Einatmen des Paraffins kommt, ist die Gefahr einer Lipid‐Pneumonie besonders bei Kindern belegt,
da die Ablagerung von Paraffin im unteren Respirationstrakt zu schweren Gewebeschädigungen
führen kann [102]. Gesundheitlich bestünde bei dieser Arzneiform also kein Problem auch wenn eine
mehrfach tägliche Einnahme über einen längeren Zeitraum erfolgen würde.
Unter den bisherigen Entwicklungen und Erfindungen findet sich keine vergleichbare Arzneiform, die
ihren Inhalt unter Druck mit Hilfe einer Sollbruchstelle freisetzen soll. Auch wenn es Patente gibt, die
sich auf Arzneiformen mit Sollbruchstellen beziehen, handelt es sich zumeist um
Wirkstoffverpackungen für Pflaster oder Ampullen [103]. Demnach wäre eine Sollbruchstelle in einer
drucksensitiven Arzneiform eine interessante Art, Wirkstoffe kontrolliert freizusetzen. Da der
bisherige Versuch des Einfügens einer Sollbruchstelle allerdings beim Freisetzungsversuch unter
physiologischen Bedingungen keine drucksensitive Wirkstofffreisetzung erkennen ließ, ist die
Paraffin‐Kapsel bislang als druckempfindliche Arzneiform ungeeignet und bedarf weiterer
Modifizierungen, die ein vollständiges Aufplatzen garantieren können. Die enthaltene Gelatinehülle
dient bei dieser Arzneiform nur der Formgebung und muss aufgrund ungünstiger physikalischer
Eigenschaften vor dem Platzen aufgelöst werden. Dementsprechend wäre es denkbar, Kapseln direkt
aus Ethylcellulose herzustellen, um ein zuverlässigeres Brechen zu gewährleisten. Da die
Ethylcellulose‐Schicht allerdings sehr dünn und fragil ist, könnten Probleme mit dem Handling
auftreten, die gegebenenfalls aber erneut durch eine äußere Gelatinehülle behoben werden könnten
4.4 Tristearin‐Kapseln
Die Herstellung der Tristearin‐Kapseln bedarf einiger Übung um reproduzierbar Arzneiformen mit
gleichem Bruchverhalten zu erzielen. Da das Beschichtungsvolumen besonders bei den 250 µL
Kapseln sehr gering ist und besonders schnell erstarrt, ist die Schmelztemperatur vor dem
Pipettieren von besonderer Bedeutung und sollte zwischen 85 und 90 °C liegen. Außerdem sollte der
Vorgang des Pipettierens sehr zügig und möglichst gleich verlaufen, da das flüssige
Beschichtungsmaterial ansonsten bereits in der Pipettenspitze erstarrt oder nur in unterschiedlich
starkem Maße wieder aus der Spitze in die Hartgelatinekapsel entleert werden kann. Schon bei
geringen Abweichungen eines dieser Parameter kann es zu ungleichmäßigen oder undichten
Tristearin‐Schichten kommen. Zur Umgehung dieses Problems und um die Herstellung auch ungeübt

4 Diskussion
107
reproduzierbarer zu machen, wäre eine automatische Innenbeschichtung denkbar. Dazu wurden
bereits Ansätze erarbeitet, die allerdings im Rahmen dieser Arbeit nicht weiter geführt werden
konnten. Vorstellbar wäre eine Apparatur, bei der sich die leere Hartgelatine‐Kapsel zwischen zwei
motorbetriebenen Rädern auf deren Lauffläche gleichmäßig dreht, während automatisch eine
Injektion des flüssigen Tristearins erfolgt. Bei konstanter Drehung kommt es somit zum
gleichmäßigen Erstarren der Innenbeschichtung. Bei Entwicklung eines solchen Modells wäre trotz
der automatischen Innenbeschichtung immer noch die Versieglung der Kapseln mit dem Tristearin‐
Deckel eine manuelle Aufgabe, da dies besondere Genauigkeit erfordert, um eine dicht
verschlossene Kapsel zu erhalten.
Nach Innenbeschichtung, Befüllung und Versiegelung dieser Arzneiform ist diese in Abhängigkeit vom
Füllmaterial unterschiedlich lange haltbar. Während die äußere Tristearin‐Schicht auch durch die
umgebende Hartgelatine‐Kapsel weitestgehend unempfindlich gegenüber mikrobiellem Befall ist und
zum Schutz der Gelatine, lediglich trocken und lichtgeschützt gelagert werden müsste, wären bei
einer wässrigen Füllung zusätzliche Maßnahmen zur Verlängerung der Haltbarkeit erforderlich. Die
innerhalb dieser Arbeit verwendete Hydrogel‐Füllung sollte nur konserviert verwendet werden, aber
ist dann trotzdem nur für sehr kurze Zeit haltbar. Besser wäre eine lipophile Füllung, in der der
Wirkstoff ebenfalls gelöst vorliegen würde. Auch eine reine Pulverfüllung wäre möglich, entspräche
aber nicht dem in dieser Arbeit zugrunde gelegten Prinzip, dass der Wirkstoff in gelöster Form
vorliegen sollte. Die Verwendung eines Lyophilisats, welches sich sehr schnell beim ersten
Flüssigkeitskontakt auflöst, wäre allerdings eine Kompromissmöglichkeit. Ebenso wäre das Abfüllen
von Grundlagen, die bei Körpertemperatur schmelzen denkbar oder die Verwendung einer 40 %igen
Hypromellose‐Haftpaste (NRF 7.8), die aufgrund des hohen Gehalts an stark quellendem Gelbildner
bei Kontakt mit der Schleimhaut an dieser haften würde [73].
Die verwendete Methode zur Gehaltsbestimmung der Tristearin‐Kapseln zeigte eine sehr geringe
prozentuale Standardabweichung von 1,2 %. Aus diesem Grund wurde auf eine mögliche
Weiterentwicklung der Gehaltsbestimmung verzichtet.
Die Prüfung auf Wirkstofffreisetzung in der Paddle‐Apparatur ergab einen Anstieg der Absorption auf
4 % nach 3 h Versuchszeit. Da alle sechs untersuchten Kapseln nach Ende des Versuchs visuell auf
Beschädigungen untersucht wurden und keine Auffälligkeiten zeigten, ist davon auszugehen, dass
aus den verschlossenen Arzneiformen in Anwesenheit des Freisetzungsmediums SGFsp pH 1,8 kein
Wirkstoff ausgetreten sein kann. Außerdem zeigte eine Untersuchung, deren Ergebnisse innerhalb
der Arbeit nicht näher dargestellt wurden, dass eine Eigenabsorption der Gelatine‐Hülle oder des
Tristearins bei der Messwellenlänge von 242 nm ebenfalls nicht vorhanden war. Da die Erhöhung der
Absorption bei allen Proben einheitlich zu erkennen war und die Befüllung in den Tristearin‐

4 Diskussion
108
Hohlkörper mit Hilfe einer automatischen Pipette erfolgte, ist das Vorhandensein von Wirkstoff an
der Außenseite einzelner Proben aus dem Herstellungsprozess auszuschließen. Somit bliebe als
Erklärung für den leichten aber kontinuierlichen Anstieg der Absorption wahrscheinlich eine
messtechnische Ungenauigkeit.
Aufgrund der sehr spröden Tristearin‐Hülle zeigten sich in den Kraft‐Weg‐Diagrammen des Texture
Analysers im Gegensatz du denen der Agar‐, Hartfett‐ oder PEG‐Kugeln sehr viele spitze Peaks, deren
Auftreten auf das mehrfache Zersplittern der Hülle hindeuten. Aus großen Bruchstücken ergaben
sich beim weiteren Herabfahren des Stempels immer mehr kleine Teile, während bei den Hartfett‐
und PEG‐Kugeln meist nur ein Hauptriss in der Hülle entstand, der sich bei weiterer Krafteinwirkung
gegebenenfalls verstärkte. Die Berechnung der Kraft‐Mittelwerte erfolgte dementsprechend nicht
anhand des ersten auftretenden Peaks, sondern anhand des Maximal‐Peaks während der Testung.
Dabei ergab sich beim Auftragen der Mittelwerte und Standardabweichungen der Kraft in
Abhängigkeit vom Füllvolumen und bei anschließender Linearisierung der Trendlinie ein
Bestimmtheitsmaß von R2 = 0,9916, welches auf einen linearen Zusammenhang zwischen
Füllvolumen und Bruchkraft der Tristearin‐Kapseln schließen lässt.
Für die Untersuchungen des Bruchverhaltens in der Stresstest‐Apparatur waren nur die Tristearin‐
Kapseln mit einem geringen Tristearin‐Füllvolumen für die Testung bis 500 mbar geeignet. Während
bei den 300 µL‐Kapseln nur zwei von drei Arzneiformen bei 400 mbar zerbrachen und die
verbleibende auch bei 500 mbar intakt blieb, wurden die Arzneiformen mit noch höheren Tristearin‐
Volumina durch keinen der angelegten Drücke beeinflusst. Auf der anderen Seite wurde die
Herstellung der Darreichungsformen durch die manuelle Umsetzung der Innenbeschichtung
begrenzt. Die Kapseln mit dem geringsten Füllungsvolumen von 220 µL zerbrachen bereits bei
Drücken unter 200 mbar. Somit begrenzte sich die Möglichkeit der hergestellten und getesteten
Kapseln auf diejenigen mit einem Tristearin‐Füllvolumen zwischen 220 µL und 300 µL. Dabei ist auch
zu beachten, dass geringfügige Volumen‐Unterschiede von 10 µL bei der Innenbeschichtung
aufgrund der schnellen Erstarrung in der Pipettenspitze mitunter schwer reproduzierbar umgesetzt
werden können. Bei der Entwicklung einer automatischen Methode zur Innenbeschichtung könnte
dieses Problem unter Umständen verringert werden. Eine Beeinflussung der Bruchkraft in
Abhängigkeit von der Inkubationszeit wurde bei den Tristearin‐Kapseln nicht untersucht, da sich
bereits bei den Ergebnissen der Freisetzung in der Paddle‐Apparatur zeigte, dass auch eine
dreistündige Verweilzeit im sauren Freisetzungsmedium äußerlich keinen Einfluss auf die
Darreichungsformen hatte. Es zeigten sich an der Tristearin‐Hülle keine Anzeichen einer Erweichung,
Quellung oder sonstiger Veränderungen, die das Bruchverhalten möglicherweise beeinflussen
könnten.

4 Diskussion
109
Trotz der leicht erhöhten Absorptionswerte bei den Freisetzungsversuchen in der Paddle‐Apparatur,
zeigte sich bei den Freisetzungsversuchen in der Stresstest‐Apparatur innerhalb der ersten dreißig‐
minütigen stressfreien Phase keine Wirkstofffreisetzung. Dies bestätigt die Annahme, dass
messtechnische Ungenauigkeiten zu diesen leicht erhöhten Werten führten, da ein solcher Effekt
sonst nie auftrat. Der Kurvenverlauf der drei Proben, die in viele kleine Bruchstücke zersplitterten
entspricht dem angestrebten Kurvenverlauf einer drucksensitiven Darreichungsform. Der Druck von
300 mbar führte neben der Ablösung des intakten Tristearin‐Deckels zum vollständigen Zerbrechen
der Kapsel‐Hülle und erlaubte somit eine annähernd 100 %ige Wirkstofffreisetzung innerhalb
weniger Minuten. Die verzögerte Freisetzung der halb zerbrochenen Kapsel könnte an der Position
innerhalb des Probenkörbchens der Stresstest‐Apparatur liegen. Da die Kapseln aufgrund der
geringeren Dichte des Hartfetts an der Oberfläche des Freisetzungsmediums schwammen, bestand
die Möglichkeit, dass der sich aufblasende Ballon seinen Druck nicht vollständig auf die Arzneiform
ausüben konnte. Dies könnte unter Umständen auch der Grund für das Versagen der verbleibenden
beiden Kapseln sein. Weiterhin könnte in diesem Fall aber auch ein erhöhtes Tristearin‐Volumen
aufgrund besonders schnellen Pipettierens die Ursache sein oder eine veränderte Position des
Tristearin‐Deckels. Dieser ist bis zu einem gewissen Maße in der Lage, die Kapsel zu stabilisieren.
Wenn zum Beispiel das Füllvolumen des inkorporierten Gels niedriger wäre und somit der Deckel
tiefer an der empfindlichen Hülle angebracht wäre, würde dies die Tristearin‐Hülle stabilisieren und
zu einem veränderten Bruchverhalten führen. Da in unserem Fall aber das enthaltene Gel
massedosiert eingefüllt wurde, ist diese Möglichkeit eher unwahrscheinlich. Trotz der zwei Versager
unter den getesteten Proben erscheint das Prinzip der Tristearin‐Kapsel in Anbetracht der Ergebnisse
aller durchgeführten Prüfungen als drucksensitive Arzneiform am besten geeignet.
Beim Arbeiten mit festen Triglyceriden wie Tristearin muss das Phänomen des Polymorphismus
berücksichtigt werden. Bei der Kristallisation von geschmolzenem Tristearin ordnen sich die Moleküle
in organisierten Kristallgittern an, deren Organisation von verschiedenen Faktoren abhängig ist. Die
Art der Kristallisation, die Zusammensetzung und Anwesenheit anderer Lipide oder Zusätze sowie die
Temperatur beim Abkühlen entscheiden darüber, ob die hexagonale α‐Form, die orthorhombische
β‘‐Form oder die triklinische β‐Form entsteht. Diese Nomenklatur wurde nach einer veränderten
Vorstellung durch Malkin et al. von Lutton et al. modifiziert und seitdem in dieser Form verwendet
[104]. Dabei ist die β‐Form aufgrund ihrer rauen und sandigen Struktur meist unerwünscht, während
sich die α‐ und β‘‐Form durch ihr besseres Erscheinungsbild und eine verbesserte Textur
auszeichnen. Diese sind allerdings thermodynamisch instabil und neigen dazu sich über eine längere
Lagerungszeit in die stabilere β‐Form umzuwandeln. Um die verschiedenen polymorphen Formen zu
erhalten, können gezielte Temperaturverläufe beim Abkühlen von geschmolzenem Tristearin
durchlaufen werden [105‐109]. Anhand dieser Angaben kann für die Herstellung der Tristearin‐

4 Diskussion
110
Kapseln zunächst die Entstehung der α‐Form vermutet werden, da von Da Silva et al. ein Abkühlen
von 90 °C auf 25 °C und bei Oh et al. ein normales Abkühlen auf Raumtemperatur zum Erhalten der
α‐Form vorgeschlagen wurde. Eine Umwandlung in die unerwünschte β‐Form ist auszuschließen, da
dies von den beiden Arbeitsgruppen durch ein 60‐minütiges Abkühlen auf 61 °C und durch eine
längere Lagerung bei 55 °C erreicht wurde [108, 109]. Außerdem konnte auch nach wochenlanger
Lagerung einzelner Proben kein sandiges Äußeres bei den fertigen Kapseln beobachtet werden. Dies
zeigte sich bei der Arbeit mit Tristearin nach der Herstellung der Kapseln nur, wenn das übrige
geschmolzene Tristearin zum Abkühlen auf dem langsam abkühlenden Wasserbad belassen wurde.
Oh et al. untersuchten außerdem die optischen Unterschiede der einzelnen Formen sowohl im
Lichtmikroskop als auch im Rasterelektronenmikroskop. Dabei zeigten besonders die REM‐
Aufnahmen deutliche Unterschiede in den Oberflächenstrukturen der einzelnen polymorphen
Formen. Während die α‐Form eine feinere Oberfläche mit kleiner Kristallgröße aufweist, kristallisiert
die β‘‐Form in spitzen Nadeln aus und die β‐Form bildet eine raue Oberfläche mit großen
sphärolitischen Kristallen [109]. Obwohl vereinzelte Proben der Tristearin‐Kapseln auch nach
mehreren Wochen keine optischen Veränderungen zeigten, müsste die Langzeitstabilität getestet
werden.
Bei Einnahme der Tristearin‐Kapseln wird der wässrige Inhalt, wie in dieser Arbeit verwendet,
komplett absorbiert. Die Bruchstücke der Hülle würden sich dagegen weiter durch den
Gastrointestinaltrakt bewegen und am Ende weitestgehend unverändert ausgeschieden werden. Zu
diesem Thema untersuchten Hamilton et al. die Absorption von Tristearin und Stearinsäure sowie
Tripalmitin und Palmitinsäure in Ratten. Dabei zeigte sich, dass die generell schlechte Absorption von
Tristearin in Anwesenheit von Triolein verbessert wurde. Sie schlossen anhand ihrer Ergebnisse
darauf, dass der limitierende Schritt für die Aufnahme des Triglycerids die mizellare Solubilisierung in
Gallensalz‐Lösungen im Darmlumen ist [110]. Für die Bruchstücke des Tristearins wird eine äußerst
geringe Absorption angenommen, die sich auch auf unveröffentlichte Versuche gründet, bei denen
eingenommene Tristearin‐Kapseln in Bruchstücken wieder entleert wurden. Eine Ansammlung des
Tristearins aufgrund von mehrfacher Einnahme sollte für den Körper nicht problematisch sein, da die
Arzneiform bei steigendem Druck zerplatzt und sich die zerbrochenen Teile unter den Chymus
mischen und normal entleert werden könnten.
4.5 Kaugummi‐Manteltabletten
Bei der Entwicklung der Kaugummi‐Tabletten wurden die lipophilen Eigenschaften der
granulatartigen Kaugummi‐Grundmasse genutzt, mit deren Hilfe die Arzneiform gegen den Einfluss
wässriger Medien geschützt werden sollte. In der Entwicklungsphase wurden fünfzig verschiedene
Chargen hergestellt, die auf ihre Eignung als drucksensitive Darreichungsform hin untersucht

4 Diskussion
111
wurden. Dabei war zunächst die Beständigkeit gegenüber wässriger Medien die wichtigste
Voraussetzung für weitere Untersuchungen. Erst wenn sie eine Zeit von 120 min in 37 °C warmem
Wasser unbeschadet überstanden, wurden sie am Texture Analyser geprüft. Die Ergebnisse dieser
Untersuchungen und der Verlauf der erhaltenen Kraft‐Weg‐Diagramme führten dann zur Auswahl
von drei verschiedenen Chargen an Kaugummi‐Tabletten, die für die weiteren Prüfungen der
Gehaltsbestimmung, der Freisetzung in der Paddle‐Apparatur und der Stresstest‐Apparatur
verwendet wurden. Die Herstellung der einzelnen Tabletten erfolgte immer durch dieselbe Person in
Handarbeit an einer Tablettenpresse. Dabei wurde versucht, mit den gegebenen Mitteln eine
gleichmäßige und reproduzierbare Herstellung zu gewährleisten, um vergleichbare Proben zu
erhalten. Die Produktion gliederte sich dabei in mehrere Schritte, die für alle drei Chargen das
Ausgießen der Hartfettkerne, das Verpressen des Kaugummimantels, das Einfügen von
Sollbruchstellen und das Dippen in die Kaugummilösung umfasste. Bei Charge 1 und 2 wurde
zusätzlich ein Zwischenschritt beim Verpressen des Pulverkerns eingefügt. Aufgrund des lipophilen
Kaugummimantels und da es sich um eine feste, wasserfreie Arzneiform handelt, sind die fertigen
Tabletten nicht besonders anfällig für mikrobiellen Befall. Der relativ dicke Mantel bietet darüber
hinaus Schutz für lichtempfindliche Wirkstoffe. Da die Langzeitstabilität der fertigen Tabletten
allerdings nicht untersucht wurde, wäre eine trockene Lagerung in geschlossenen Behältern zu
empfehlen.
Da der Wirkstoff bei allen drei Chargen der Kaugummi‐Tabletten in Hartfett suspendiert vorlag,
wurde die Gehaltsbestimmung analog zu der Methode für die Hartfett‐Kugeln durchgeführt. Dabei
lagen die Proben der Charge 2 mit 9,87 ± 0,60 mg am dichtesten am Sollgehalt von 10 mg pro
Tablette, wiesen aber die größte prozentuale Standardabweichung von 6,1 % auf. Für die Chargen 1
und 3 ergaben sich mit 8,79 ± 0,21 mg und 9,08 ± 0,45 mg zwar geringere Ist‐Gehalte, aber auch
geringere prozentuale Standardabweichungen von 2,4 % und 5,0 %.
Die Ergebnisse der Wirkstofffreisetzung in der Paddle‐Apparatur entsprechen den Ergebnissen der
Voruntersuchungen hinsichtlich der Stabilität in 37 °C warmem Wasser. Bei keiner der drei Chargen
wurde über einen Zeitraum von 3 h eine Abweichung vom gemessenen Blindwert festgestellt, und
die Beurteilung der Proben nach dem Versuch ergab keine optischen Veränderungen, die auf
mögliche Beschädigungen hindeuten könnten.
Die Kraft‐Weg‐Diagramme aus den Bruchfestigkeitsuntersuchungen am Texture Analyser
unterscheiden sich sehr deutlich von denen der anderen entwickelten Darreichungsformen. Die
Verläufe sind eher geschwungen, zum Teil flach und weisen keine deutlichen Peaks auf. So konnte
der Bruch der Proben oft nur in Zusammenhang mit den Beobachtungen bei der Durchführung der
Prüfung klar identifiziert werden. Das Austreten von geschmolzenem Hartfett in Kombination mit

4 Diskussion
112
den kleinen Kraftabfällen im Diagramm zeigte den Bruch der Arzneiformen und erlaubte die
Berechnung von Mittelwert und Standardabweichung. Anhand der Ergebnisse dieser Prüfung, die für
alle Chargen durchgeführt wurde, die eine Verweilzeit von > 120 min in 37 °C warmem Wasser
aufwiesen, erfolgte die Auswahl der weiter geprüften Chargen 1, 2 und 3. Die Mittelwerte der
Bruchkräfte entsprachen mit 1,58 ± 0,32 N für Charge 1, mit 1,95 ± 0,60 N für Charge 2 und mit
1,77 ± 0,86 N für Charge 3 in etwa den Werten der 250 µL Tristearin‐Kapseln mit 1,92 ± 0,22 N und
waren niedriger als die Werte der Hartfett‐Kugeln der Charge 1 mit 2,75 ± 0,64 N, die bei der
anschließenden Testung in der Stresstest‐Apparatur jeweils gute Ergebnisse zeigten und
weitestgehend bei 300 mbar zerbrachen. Die Ergebnisse der Bruchfestigkeitsuntersuchungen an der
Stresstest‐Apparatur für die Kaugummi‐Tabletten sind aber alle deutlich zu niedrig, da sie
größtenteils bereits bei 100 mbar zerbrachen. Aufgrund der unterschiedlichen geometrischen
Formen der verschiedenen entwickelten Arzneiformen ist ein Vergleich der Ergebnisse untereinander
sehr schwierig. Während eine Kugel aufgrund der Oberfläche die stabilste Form darstellt und
dementsprechend mehr Druck aushalten kann, ist die Stabilität bei einer Kapsel durch den
zylindrischen Körper schon etwas geringer. Bei einer Tablette mit einem geschmolzenen Kern
hingegen kann die Kraft trotz der bikonvexen Form sehr gut angreifen und führt somit zu einem
leichten Brechen der Arzneiform. Bis zu einem gewissen Punkt war dies auch das Ziel dieser
Entwicklung, allerdings wäre eine leichte Stabilisierung in diesem Fall von Vorteil, um ein vorzeitiges
Versagen zu verhindern. Da bei der Herstellung allerdings mit den gegebenen Bedingungen keine
Variation bestimmter Parameter wie Schicht‐ oder Überzugsdicke möglich war, konnte eine
ausreichende Stabilisierung nicht erreicht werden. Ansatzweise wurde dies durch eine erhöhte
Anzahl an Dippvorgängen bei der Herstellung versucht, allerdings zeigte sich nur bei Charge 1, dass
sich der erforderliche Druck von 100 mbar bei dreimaligem Dippen auf bis zu 300 mbar bei
neunmaligem Dippen erhöhte. Aufgrund der leichten Erhöhung des erforderlichen Drucks erfolgte
die Wirkstofffreisetzung in der Stresstest‐Apparatur unter Verwendung der neunmal gedippten
Chargen 1a, 2a und 3a. Weitere Modifizierungen oder noch häufigere Dippvorgänge wären eine
Möglichkeit Tabletten zu erhalten, die ausreichend stabil sind und gezielt bei 300 mbar zerbrechen.
Die Resultate der Wirkstofffreisetzung in der Stresstest‐Apparatur entsprechen den Ergebnissen der
Bruchfestigkeitsuntersuchungen. Bei allen drei Chargen zeigten die sechs Proben eine drucksensitive
Freisetzung des enthaltenen Riboflavinphosphats bei 300 mbar. Die Profile der Charge 1a zeigten
dabei allerdings eine unterschiedlich hohe freigesetzte Wirkstoffmenge, die bei den einzelnen
Proben zwischen 18 und 113 % lag. In den nach Versuchsende begutachteten Resten der
Arzneiformen befand sich noch sehr viel Riboflavinphosphat, welches trotz des Auseinanderreißens
der Tabletten aufgrund des schrumpfenden Ballons nach Druckeinwirkung in den Kaugummi‐ und
Pulverresten zurückgehalten wurde. Für diese Charge wird ein Eindringen des geschmolzenen

4 Diskussion
113
Hartfetts in den Pulverkern vermutet, bei dem auch der enthaltene Wirkstoff zwischen die
Pulverpartikel verläuft und mit der 37 °C warmen Kaugummimasse verklebt. Dieses Problem konnte
bei Charge 2a durch das Einarbeiten einer Trennschicht aus Natriumchlorid umgangen werden.
Dadurch blieb das flüssige Hartfett als innerer Kern in der Mitte der Tablette enthalten und konnte
beim Zerplatzen herausfließen und den Wirkstoff sofort an das Medium abgeben. Die
Freisetzungsprofile der Charge 2a wiesen einen viel gleichmäßigeren Verlauf auf und die
verbliebenen Reste des Kaugummimantels nach Versuchsende zeigten eine deutlich geringere
Gelbfärbung. Ein noch engerer Kurvenverlauf konnte bei Charge 3a beobachtet werden, welche
innerhalb des Kaugummimantels einen reinen Hartfettkern enthielt, der im Vergleich zu den anderen
beiden Chargen die doppelte Höhe aufwies. Durch das Schmelzen und die damit verbundene leichte
Volumenzunahme während der Inkubationszeit konnte sich ein hoher Druck aufbauen, der die
Proben zum Platzen brachte. Die verbliebenen Reste nach Versuchsende zeigten immer noch gelbe
Spuren im Kaugummimantel und waren zudem deutlich kleiner als bei Charge 2a, was auf einen
höheren Innendruck und daraus resultierend auf ein stärkeres Zerplatzen hindeuten könnte. Anhand
der Freisetzungsprofile konnte für alle drei Chargen prinzipiell eine drucksensitive
Wirkstofffreisetzung festgestellt werden, da sie unter stressfreien Bedingungen keinen Wirkstoff
freigaben, sondern erst bei Druck aufplatzten und ihren Inhalt schlagartig freisetzten. Charge 1a ist
dabei aus den genannten Gründen etwas weniger geeignet, da die genaue Menge des zur Absorption
zur Verfügung stehenden Arzneistoffs schlecht vorhergesehen werden kann.
Medizinische Kaugummis laut Europäischem Arzneibuch sind für gewöhnlich feste, einzeldosierte
Zubereitungen, die dazu bestimmt sind den enthaltenen Wirkstoff durch Kauen und nicht nach
Schlucken freizusetzen [44]. Dabei werden in Fertigarzneimitteln zum Beispiel Wirkstoffe wie Nicotin
zur Raucherentwöhnung oder Dimenhydrinat bei Reiseübelkeit verwendet, die über eine definierte
Zeit des Kauens freigesetzt und über die Mundschleimhaut resorbiert werden sollen [111, 112]. Die
Stabilität von medizinischen Kaugummis ist im Allgemeinen gut, da die Bedingungen für ein
mikrobielles Wachstum oder den chemischen Abbau der enthaltenen Stoffe schlecht sind. Der Schutz
der inkorporierten Wirkstoffe vor Licht und Sauerstoff und der sehr geringe Wassergehalt bieten
kaum Angriffsfläche für einen schnellen Verderb. Allerdings können überlagerte Kaugummis mitunter
hart werden und somit gegebenenfalls die Freigabecharakteristik verändern. Kaugummis sind
weitestgehend sehr gut verträglich und auch versehentliches Verschlucken führt kaum zu Problemen,
da der enthaltene Wirkstoff nur zu einem geringen Teil freigesetzt wird [55]. Allerdings kann sich das
Gefahrenpotential erhöhen mit einer veränderten Anwendung oder einer Anwendung bei Kindern,
die im Gebrauch von Kaugummi ungeübt sind. Bei den vorgestellten Kaugummi‐Tabletten verändert
sich die Anwendung, da sie nicht gekaut, sondern im Ganzen herunter geschluckt werden sollen, um
als drucksensitive Arzneiform zu fungieren. Da nur der äußere Mantel aus Kaugummi besteht wäre

4 Diskussion
114
die gesamte geschluckte Menge pro Einzeldosis eher gering. Diese könnte sich aber durch
Mehrfachgabe bei niedrigpotenten Wirkstoffen schnell erhöhen und gegebenenfalls zu Problemen
führen. Bei sehr häufigem Verschlucken von Kaugummis kann sich die Masse zusammen mit
Nahrungsbestandteilen zu einem Bezoar zusammenballen, der von alleine nicht entleert werden
kann, sondern manuell entfernt werden muss. Rimar et al. stellten den Fall eines 18‐jährigen
Mädchens vor, welches über Jahre mindestens fünf Kaugummis pro Tag gekaut und anschließend
geschluckt hatte. Über einen langen Zeitraum und erst in höherer Dosierung führte dies also zur
Bezoar‐Bildung, die eine gastroskopische Entfernung erforderlich machte [113]. Weitere Fälle
wurden von Milov et al. vorgestellt, bei denen Kleinkinder mit Problemen wie Konstipation und
Enkopresis (unfreiwilliges Einkoten) ins Krankenhaus kamen und zunächst mit verschiedenen
Laxantien behandelt wurden. Erst durch manuelles Ausräumen wurde erkannt, dass das
Vorhandensein vieler Kaugummis im Rektum die Ursache für die Probleme war. Es zeigte sich, dass
diese Kinder mehrere Kaugummis hintereinander und an mehreren Tagen geschluckt hatten [114].
Diese Fallbeispiele machen deutlich, dass Kaugummis zu Problemen wie Bezoar‐Bildung oder
Konstipation führen können, dies aber erst nach mehrfacher Einnahme über einen längeren Zeitraum
der Fall ist.
Bei weiteren kleinen Veränderungen dieser Arzneiformen, die ein frühzeitiges Platzen aufgrund
geringeren Drucks verhindern würden, wären die Chargen 2a und 3a aufgrund ihres
Freisetzungsverhaltens in der Stresstest‐Apparatur als drucksensitive Arzneiform geeignet.
Die Entwicklung von drucksensitiven Arzneiformen ist eine vielversprechende Methode, um
Wirkstoffe gezielt an einen Ort im Gastrointestinaltrakt zu bringen, der sich durch charakteristische
Druckverhältnisse auszeichnet. Dabei ist aber zu beachten, dass sich Anwendungsbeschränkungen
bei bestimmten gastrointestinalen Krankheiten ergeben. Für Patienten mit diffuser gastrointestinaler
Dysmotilität könnte die Anwendung drucksensitiver Arzneiformen schwierig sein, da die Krankheit
durch verzögerte Magenentleerung und abnormale Funktion des Ösophagus gekennzeichnet ist.
Dabei können Druckwellen über 230 mbar bereits im Magen auftreten und sowohl Zeit als auch Ort
des Zerplatzens beeinflussen [18]. Im Gegensatz dazu ist eine Anwendung in Gastroparese‐Patienten
mit einer verzögerten Magenentleerung durchaus möglich, da erst bei der Pylorus‐Passage erste
charakteristische Druckwellen mit 220 mbar oder mehr auftreten [19]. Dementsprechend müsste die
Anwendung drucksensitiver Darreichungsformen an den jeweiligen Krankheitszustand angepasst
werden, um ein ortsspezifisches Drug‐Targeting zu ermöglichen.
Neben dem Pylorus ist auch die Ileocaecalklappe am Übergang zwischen Dünn‐ und Dickdarm ein
interessantes Target. Während die vorgestellten entwickelten Darreichungsformen vorrangig für eine
Freisetzung am Pylorus konzipiert waren, müssten für die Anwendung im unteren Teil des

4 Diskussion
115
Gastrointestinaltraktes Modifikationen vorgenommen werden, die ein frühzeitiges Aufplatzen an den
oberen Sphinkteren verhindern könnten. Neben den Drücken entlang des Ösophagus müsste die
Arzneiform dann auch den noch höheren Drücken am Pylorus standhalten bevor sie erst an der
Ileocaecalklappe zerbricht. Dazu könnten die Darreichungsformen zum Beispiel zusätzlich mit
magensaftresistenten Überzügen versehen werden, die die Arzneiform zunächst zusätzlich
stabilisieren und sich dann erst mit Eintritt in den Dünndarm auflösen. Dies wäre eine Möglichkeit,
die für die Hartfett‐Kugeln denkbar wäre. Für die Tristearin‐Kapseln, die allgemein sehr fragil sind
und neben dem Pylorus auch durch den verdickten Darmeinhalt frühzeitig brechen könnten, wäre
eine Stabilisierung zur Anwendung an der Ileocaecalklappe eher schwierig. Diese beiden
Darreichungsformen sind unter den entwickelten aufgrund ihrer Ergebnisse bei den einzelnen
Prüfungen die vielversprechendsten und wären für eine erfolgreiche Anwendung am Menschen
durchaus denkbar.

5 Zusammenfassung
116
5 Zusammenfassung
Die zielgerichtete Steuerung einer Arzneiform zu einem bestimmten Teil des Gastrointestinaltraktes
ist ein wesentlicher Punkt, wenn es um die Erhöhung der Wirksamkeit und Bioverfügbarkeit und um
die Verringerung von möglichen Nebenwirkungen geht. Im Zuge dessen macht man sich die
physiologischen Unterschiede entlang des Verdauungstraktes zu Nutze, die unter anderem den pH‐
Wert, die Transitzeiten oder die Enzymausstattung betreffen. Verschiedene Faktoren wie
Nahrungsaufnahme oder pathophysiologische Veränderungen können allerdings dazu führen, dass
pH‐, Zeit‐ oder Enzym‐getriggerte Darreichungsformen versagen und ihren Wirkstoff entweder zu
früh, zu spät oder gar nicht freisetzen. Durch die Entwicklung neuer Arzneiformen, die ihren
Wirkstoff aufgrund eines definierten Drucks freigeben, könnten diese Probleme unter Umständen
vermieden werden. Ein weiterer Punkt, der die Bioverfügbarkeit im menschlichen Körper besonders
von schwer löslichen Wirkstoffen negativ beeinflussen kann, ist der Zeitpunkt und die Dauer der
Desintegration einer Arzneiform und das Fehlen der dafür erforderlichen Wassermengen im
Gastrointestinaltrakt. Besonders bei Wirkstoffen mit einem engen Absorptionsfenster im oberen
Dünndarm ist ein schneller Lösungsprozess des Wirkstoffs einer Arzneiform wichtig, um eine
optimale Absorption zu gewährleisten.
Aus diesem Grund bestand das Ziel der vorliegenden Arbeit in der Entwicklung neuer drucksensitiver
Darreichungsformen, die ihren Inhalt bei einem definierten Druck von 300 mbar, wie er am Pylorus
vorherrscht, schlagartig und komplett freisetzen. Dabei sollten die Arzneiformen vor ihrer Entleerung
weder durch eine verlängerte Magenverweilzeit noch durch den sauren pH‐Wert des Magens
angegriffen werden, um eine verfrühte Wirkstofffreisetzung zu vermeiden. Die entwickelten
Darreichungsformen sollten den Wirkstoff entweder in bereits gelöster Form oder dispergiert in
einer flüssigen Grundlage enthalten, die sich nach Zerplatzen über die Dünndarm‐Schleimhaut
verteilen und durch eine verlängerte Kontaktzeit die Absorptionsrate erhöhen kann. Für die
entwickelten Arzneiformen wurden Methoden zur Gehaltsbestimmung entwickelt, um die
Gleichförmigkeit und Reproduzierbarkeit einschätzen zu können. Weiterhin wurde ein dreistündiger
Freisetzungsversuch in der Paddle‐Apparatur zur Simulation einer verlängerten Magenverweilzeit
durchgeführt. Bruchfestigkeitsuntersuchungen am Texture Analyser sollten die Beeinflussung der
aufzuwendenden Kraft in Bezug zu individuellen Parametern, wie Polymerbeladung oder
Konzentration beurteilen. Mit Hilfe der Stresstest‐Apparatur wurde die direkte Einwirkung von Druck
im Bereich zwischen 100 und 500 mbar untersucht, und unter Verwendung eines Magenentleerungs‐
programms das Verhalten der Darreichungsformen im nüchternen Magen und bei der Pylorus‐
passage simuliert.

5 Zusammenfassung
117
Es wurden sechs verschiedene Prinzipien für Darreichungsformen entwickelt, deren Eignung als
drucksensitive Arzneiform anhand der genannten Prüfungen untersucht wurde.
Die Entwicklung der Agar‐Kugeln beruhte auf der Eigenschaft des Gelbildners spröde Matrizes zu
bilden, deren Bruchverhalten durch Veränderung der Konzentration oder durch eine definierte
Trocknungszeit beeinflusst werden konnte. Der Wirkstoff wurde bei der Herstellung im Wasser
gelöst, sodass sich eine gute Dosiergenauigkeit ergab. Die Versuche im Texture Analyser zeigten, dass
mit Veränderung der Konzentration des Gelbildners die gewünschten Brucheigenschaften eingestellt
werden konnten. Diese veränderten sich allerdings bei Proben der gleichen Konzentration mit
verlängerter Inkubationszeit in 37 °C warmem SGFsp pH 1,8, da das Eindringen von Wasser die
Festigkeit der Matrix verringerte, was sich in der Stresstest‐Apparatur durch einen verringerten
erforderlichen Druck zeigte. Bei den Freisetzungsversuchen war sowohl in der Paddle‐Apparatur als
auch in der Stresstest‐Apparatur eine diffusionskontrollierte Freisetzung zu erkennen, die sich bei
Druckeinwirkung in der Stresstest‐Apparatur in Abhängigkeit der entstandenen Bruchstücke erhöhte.
Als drucksensitive Arzneiform sind die Agar‐Kugeln aufgrund ihrer frühzeitigen und konstanten
Wirkstofffreisetzung und ihrer Abhängigkeit von der Inkubationszeit in 37 °C warmem SGFsp pH 1,8
eher ungeeignet.
Das Prinzip der Hartfett‐ und PEG‐Kugeln beruhte darauf, dass Hilfsstoffe, die bei Raumtemperatur
fest sind und bei Körpertemperatur schmelzen, mit einem wasserunlöslichen Polymer überzogen
wurden, der aus der Arzneiform bei Eintritt in den Körper einen flüssigkeitsgefüllten Ballon bildete.
Für beide Arzneiformen konnte eine Abhängigkeit zwischen Bruchkraft und Polymerbeladung gezeigt
werden, die sich auch mit Verlängerung der Inkubationszeit im sauren Milieu nicht veränderte.
Während die PEG‐Kugeln nach einer Verzögerungszeit von 20 – 30 min kontinuierlich Wirkstoff im
Freisetzungsversuch in der Paddle‐Apparatur freisetzten, war bei den Hartfett‐Kugeln trotz des
gleichen Überzugs keine erhöhte Absorption über 3 h zu erkennen. Bei der Simulation der
Magenentleerung konnte für beide Arzneiformen ein geeignetes Freisetzungsprofil festgestellt
werden, wobei die PEG‐Kugeln zwei Versager unter den sechs Proben enthielten. Obwohl die PEG‐
Kugeln bei einer verlängerten Magenverweilzeit vermehrt Wirkstoff freisetzen, sind im Rahmen der
gesteckten Bedingungen beide Arzneiformen sehr gut für eine drucksensitive Wirkstofffreisetzung
geeignet.
Die mit Ethylcellulose überzogenen Paraffin‐Kapseln sollten durch das Einfügen einer Sollbruchstelle
in Form von acht radial angeordneten Löchern zerplatzen und ihren Inhalt freigeben. Das wässrige
Medium sollte durch die Löcher eindringen, die innere Gelatinekapsel anlösen und somit zu einer
dünnen Polymer‐Schicht führen, die auf Druck nachgeben und den Inhalt komplett freisetzen sollte.
Die Gehaltsbestimmung ergab für die enthaltene Wirkstoffsuspension eine gute Dosiergenauigkeit.

5 Zusammenfassung
118
Auch der Freisetzungsversuch in der Paddle‐Apparatur entsprach den Anforderungen, da trotz der
eingefügten Löcher keine Wirkstofffreisetzung zu verzeichnen war. Allerdings zeigten die Kapseln
auch bei verlängerter Inkubationszeit in 37 °C warmem Wasser kein geeignetes Profil bei den
Untersuchungen am Texture Analyser und auch die simulierte Pyloruspassage an der Stresstest‐
Apparatur zeigte keine geeignete Wirkstofffreisetzung für eine druckgesteuerte Darreichungsform.
Durch Veränderung der Befüllung oder der Anordnung der Sollbruchstelle wäre es unter Umständen
möglich, die Eigenschaften der Paraffin‐Kapseln zu verbessern, aber zum jetzigen Zeitpunkt der
Entwicklung sind sie als drucksensitive Arzneiform ungeeignet.
Die entwickelten Tristearin‐Kapseln bestanden aus einer festen, spröden, wasserabweisenden Hülle
aus einem hochschmelzenden Hartfett, mit welchem in flüssiger Form handelsübliche Gelatine‐
Kapseln innenbeschichtet werden konnten und wurden mit einem wirkstoffhaltigen Hydrogel gefüllt.
Der bei der Herstellung im Wasser gelöste Wirkstoff ergab eine gute Dosiergenauigkeit. Die Festigkeit
der Tristearinhülle konnte durch eine Veränderung des Tristearinvolumens bei der
Innenbeschichtung bestimmt werden. Unter stressfreien Bedingungen erfolgte keine
Wirkstofffreisetzung aus den Kapseln über einen Versuchszeitraum von 3 h. Erst die 300 mbar
starken Druckwellen bei der Simulation der Magenentleerung führten zu einer schlagartigen und
vollständigen Wirkstofffreisetzung. Auch wenn während des Versuchs in der Stresstest‐Apparatur
zwei Versager auftraten, die trotz des angelegten Drucks intakt blieben, ist das Prinzip der Tristearin‐
Kapseln für eine drucksensitive Wirkstofffreisetzung sehr gut geeignet.
Bei den Kaugummi‐Tabletten sollte ein dünner Mantel aus lipophiler Kaugummi‐Grundmasse die
Arzneiform vor dem Einfluss wässriger Medien schützen. Ein enthaltener Kern aus Hartfett, welcher
bei Körpertemperatur schmilzt, sollte zusätzlich dafür sorgen, dass das Gerüst der Tablette instabil
wird und sie durch definierte Druckeinwirkung zum Platzen gebracht wird. Eingefügte
Sollbruchstellen sollten das Platzen steuern und ein anschließendes Dippen in eine heptanhaltige
Kaugummilösung sollte die Arzneiformen soweit stabilisieren, dass sie nicht bereits bei zu niedrigen
Drücken zerbrachen. Die Tabletten zeigten bei den Versuchen am Texture Analyser nach einer
Inkubation in 37 °C warmem Wasser ein druckgesteuertes Platzen und gaben dabei ihren flüssigen
Inhalt frei. Durch eine Erhöhung der Dippvorgänge konnte die Bruchfestigkeit in der Stresstest‐
Apparatur erhöht werden, aber trotzdem zerbrachen die meisten Proben schon bei 100 mbar. Beim
stressfreien Freisetzungsversuch in der Paddle‐Apparatur wurde für keine der drei Chargen ein
Absorptionsanstieg gemessen. Erst das Einwirken eines Drucks bei der simulierten Pyloruspassage
führte zum Aufbrechen der Tabletten und zur sofortigen Wirkstofffreisetzung. Dies funktionierte für
zwei von drei Chargen unter Abgabe der gesamten Wirkstoffmenge. Trotz des Aufbrechens bei zu
niedrigen Drücken sind die Kaugummi‐Tabletten für eine drucksensitive Freisetzung prinzipiell gut

5 Zusammenfassung
119
geeignet. Leichte Modifikationen hinsichtlich der Dippvorgänge oder der Sollbruchstellen, die zur
Stabilisierung der Tabletten führen würden, könnten eine bislang zu niedrige Bruchkraft erhöhen.

Literaturverzeichnis
120
Literaturverzeichnis
[1] Thews G, Mutschler E, Vaupel P. Anatomie, Physiologie, Pathophysiologie des
Menschen. 5. Auflage. Wissenschaftliche Verlagsgesellschaft mbH, Stuttgart, 1999.
[2] Paulsen F, Waschke J. Sobotta ‐ Atlas der Anatomie des Menschen, Innere Organe. 5.
Auflage. Urban & Fischer, München, 2010.
[3] Netter FH. Atlas der Anatomie. 5. Auflage. Urban & Fischer, München, 2011.
[4] Cassilly D, Kantor S, Knight LC, Maurer AH, Fisher RS, Semler J, Parkman HP. Gastric
emptying of a non‐digestible solid: assessment with simultaneous SmartPill pH and
pressure capsule, antroduodenal manometry, gastric emptying scintigraphy.
Neurogastroenterol Motil. 2008 Apr; 20 (4): 311‐9.
[5] Ouyang A, Sunshine AG, Reynolds JC. Caloric content of a meal affects duration but
not contractile pattern of duodenal motility in man. Dig Dis Sci. 1989 Apr; 34 (4): 528‐
36.
[6] Weitschies W. Eine Reise durch den Verdauungstrakt. Pharmazeutische Zeitung,
GOVI‐Verlag, 2001; 14/2001: 10‐6.
[7] Davis SS. The design and evaluation of controlled release systems for the
gastrointestinal tract. J Control Release. 1985; 2: 27‐38.
[8] Marciani L, Gowland PA, Fillery‐Travis A, Manoj P, Wright J, Smith A, Young P, Moore
R, Spiller RC. Assessment of antral grinding of a model solid meal with echo‐planar
imaging. Am J Physiol Gastrointest Liver Physiol. 2001 May; 280 (5): G844‐9.
[9] Davis SS, Hardy JG, Fara JW. Transit of pharmaceutical dosage forms through the
small intestine. Gut. 1986 Aug; 27 (8): 886‐92.
[10] Rouge N, Buri P, Doelker E. Drug absorption sites in the gastrointestinal tract and
dosage forms for site‐specific delivery. International Journal of Pharmaceutics. 1996
Jun 14; 136 (1‐2): 117‐39.
[11] Streubel A, Siepmann J, Bodmeier R. Drug delivery to the upper small intestine
window using gastroretentive technologies. Curr Opin Pharmacol. 2006 Oct; 6 (5):
501‐8.
[12] Diakidou A, Vertzoni M, Goumas K, Soderlind E, Abrahamsson B, Dressman J, Reppas
C. Characterization of the contents of ascending colon to which drugs are exposed
after oral administration to healthy adults. Pharm Res. 2009 Sep; 26 (9): 2141‐51.
[13] McConnell EL, Short MD, Basit AW. An in vivo comparison of intestinal pH and
bacteria as physiological trigger mechanisms for colonic targeting in man. J Control
Release. 2008 Sep 10; 130 (2): 154‐60.
[14] Poxton IR, Brown R, Sawyerr A, Ferguson A. Mucosa‐associated bacterial flora of the
human colon. J Med Microbiol. 1997 Jan; 46 (1): 85‐91.

Literaturverzeichnis
121
[15] Wilson PJ, Basit AW. Exploiting gastrointestinal bacteria to target drugs to the colon:
an in vitro study using amylose coated tablets. Int J Pharm. 2005 Aug 26; 300 (1‐2):
89‐94.
[16] Bleday R, Braidt J, Ruoff K, Shellito PC, Ackroyd FW. Quantitative cultures of the
mucosal‐associated bacteria in the mechanically prepared colon and rectum. Dis
Colon Rectum. 1993 Sep; 36 (9): 844‐9.
[17] Kuo B, McCallum RW, Koch KL, Sitrin MD, Wo JM, Chey WD, Hasler WL, Lackner JM,
Katz LA, Semler JR, Wilding GE, Parkman HP. Comparison of gastric emptying of a
nondigestible capsule to a radio‐labelled meal in healthy and gastroparetic subjects.
Aliment Pharmacol Ther. 2008 Jan 15; 27 (2): 186‐96.
[18] Tran K, Brun R, Kuo B. Evaluation of regional and whole gut motility using the
wireless motility capsule: relevance in clinical practice. Therap Adv Gastroenterol.
2012 Jul; 5 (4): 249‐60.
[19] Sarosiek I, Selover KH, Katz LA, Semler JR, Wilding GE, Lackner JM, Sitrin MD, Kuo B,
Chey WD, Hasler WL, Koch KL, Parkman HP, Sarosiek J, McCallum RW. The
assessment of regional gut transit times in healthy controls and patients with
gastroparesis using wireless motility technology. Aliment Pharmacol Ther. 2010 Jan
15; 31 (2): 313‐22.
[20] The SmartPill GI Monitoring System Brochure. SmartPill Corporation, 2009.
[21] Kloetzer L, Chey WD, McCallum RW, Koch KL, Wo JM, Sitrin M, Katz LA, Lackner JM,
Parkman HP, Wilding GE, Semler JR, Hasler WL, Kuo B. Motility of the
antroduodenum in healthy and gastroparetics characterized by wireless motility
capsule. Neurogastroenterol Motil. 2010 May; 22 (5): 527‐33, e117.
[22] Indireshkumar K, Brasseur JG, Faas H, Hebbard GS, Kunz P, Dent J, Feinle C, Li M,
Boesiger P, Fried M, Schwizer W. Relative contributions of "pressure pump" and
"peristaltic pump" to gastric emptying. Am J Physiol Gastrointest Liver Physiol. 2000
Apr; 278 (4): G604‐16.
[23] Murphy C, Pillay V, Choonara YE, du Toit LC, Ndesendo VM, Chirwa N, Kumar P.
Optimization of a dual mechanism gastrofloatable and gastroadhesive delivery
system for narrow absorption window drugs. AAPS PharmSciTech. 2012 Mar; 13 (1):
1‐15.
[24] Davis SS. Formulation strategies for absorption windows. Drug Discov Today. 2005
Feb 15; 10 (4): 249‐57.
[25] Yang L, Chu JS, Fix JA. Colon‐specific drug delivery: new approaches and in vitro/in
vivo evaluation. Int J Pharm. 2002; 235: 1‐15.
[26] Niwa K, Takaya T, Morimoto T, Takada K. Preparation and evaluation of a time‐
controlled release capsule made of ethylcellulose for colon delivery of drugs. J Drug
Target. 1995; 3 (2): 83‐9.
[27] Bott C, Rudolph MW, Schneider AR, Schirrmacher S, Skalsky B, Petereit HU, Langguth
P, Dressman JB, Stein J. In vivo evaluation of a novel pH‐ and time‐based multiunit
colonic drug delivery system. Aliment Pharmacol Ther. 2004 Aug 1; 20 (3): 347‐53.

Literaturverzeichnis
122
[28] Evans DF, Pye G, Bramley R, Clark AG, Dyson TJ, Hardcastle JD. Measurement of
gastrointestinal pH profiles in normal ambulant human subjects. Gut. 1988 Aug; 29
(8): 1035‐41.
[29] Friend DR, Chang GW. A colon‐specific drug‐delivery system based on drug glycosides
and the glycosidases of colonic bacteria. J Med Chem. 1984 Mar; 27 (3): 261‐6.
[30] Katsuma M, Watanabe S, Takemura S, Sako K, Sawada T, Masuda Y, Nakamura K,
Fukui M, Connor AL, Wilding IR. Scintigraphic evaluation of a novel colon‐targeted
delivery system (CODES (TM)) in healthy volunteers. J Pharm Sci. 2004 May; 93 (5):
1287‐99.
[31] Karrout Y, Neut C, Wils D, Siepmann F, Deremaux L, Dubreuil L, Desreumaux P,
Siepmann J. Colon targeting with bacteria‐sensitive films adapted to the disease
state. Eur J Pharm Biopharm. 2009 Sep; 73 (1): 74‐81.
[32] Takaya T, Ikeda C, Imagawa N, Niwa K, Takada K. Development of a colon delivery
capsule and the pharmacological activity of recombinant human granulocyte colony‐
stimulating factor (rhG‐CSF) in beagle dogs. J Pharm Pharmacol. 1995 Jun; 47 (6):
474‐8.
[33] Matsuda K, Takaya T, Shimoji F, Muraoka M, Yoshikawa Y, Takada K. Effect of food
intake on the delivery of fluorescein as a model drug in colon delivery capsule after
oral administration to beagle dogs. J Drug Target. 1996; 4 (2): 59‐67.
[34] Takaya T, Sawada K, Suzuki H, Funaoka A, Matsuda K, Takada K. Application of a
colon delivery capsule to 5‐aminosalicylic acid and evaluation of the pharmacokinetic
profile after oral administration to beagle dogs. J Drug Target. 1997; 4 (5): 271‐6.
[35] Hu Z, Kimura G, Ito Y, Mawatari S, Shimokawa T, Yoshikawa H, Yoshikawa Y, Takada
K. Technology to obtain sustained release characteristics of drugs after delivered to
the colon. J Drug Target. 1999; 6 (6): 439‐48.
[36] Hu Z, Kimura G, Mawatari S, Shimokawa T, Yoshikawa Y, Takada K. New preparation
method of intestinal pressure‐controlled colon delivery capsules by coating machine
and evaluation in beagle dogs. J Control Release. 1998 Dec 4; 56 (1‐3): 293‐302.
[37] Hu Z, Mawatari S, Shibata N, Takada K, Yoshikawa H, Arakawa A, Yosida Y. Application
of a biomagnetic measurement system (BMS) to the evaluation of gastrointestinal
transit of intestinal pressure‐controlled colon delivery capsules (PCDCs) in human
subjects. Pharm Res. 2000 Feb; 17 (2): 160‐7.
[38] Hu Z, Mawatari S, Shimokawa T, Kimura G, Yoshikawa Y, Shibata N, Takada K. Colon
delivery efficiencies of intestinal pressure‐controlled colon delivery capsules
prepared by a coating machine in human subjects. J Pharm Pharmacol. 2000 Oct; 52
(10): 1187‐93.
[39] Kamba M, Seta Y, Kusai A, Ikeda M, Nishimura K. A unique dosage form to evaluate
the mechanical destructive force in the gastrointestinal tract. Int J Pharm. 2000 Nov
4; 208 (1‐2): 61‐70.
[40] Kamba M, Seta Y, Kusai A, Nishimura K. Evaluation of the mechanical destructive
force in the stomach of dog. Int J Pharm. 2001 Oct 9; 228 (1‐2): 209‐17.

Literaturverzeichnis
123
[41] Kamba M, Seta Y, Kusai A, Nishimura K. Comparison of the mechanical destructive
force in the small intestine of dog and human. Int J Pharm. 2002 Apr 26; 237 (1‐2):
139‐49.
[42] Garbacz G, Klein S. Dissolution testing of oral modified‐release dosage forms. J Pharm
Pharmacol. 2012 Jul; 64 (7): 944‐68.
[43] Garbacz G, Klein S, Weitschies W. A biorelevant dissolution stress test device ‐
background and experiences. Expert Opin Drug Deliv. 2010 Nov; 7 (11): 1251‐61.
[44] Council of Europe: European Pharmacopeia Edition 7.5. Strasbourg, 2012.
[45] United States Pharmacopeial Convention: United States Pharmacopeia and National
Formulary Edition 36. Rockville, 2013.
[46] Voigt R. Pharmazeutische Technologie. 8. Auflage. Ullstein Mosby GmbH & Co. KG,
Berlin, 1995.
[47] Bauer KH, Frömming K‐H, Führer C. Lehrbuch der Pharmazeutischen Technologie. 6.
Auflage. Wissenschaftliche Verlagsgesellschaft mbH Stuttgart, Stuttgart, 1999.
[48] Product Information: Witepsol and Massa Estarinum. Sasol Germany GmbH, 2007.
[49] Bauer‐Brandl A, Ritschel WA. Die Tablette. 3. Auflage. Editio Cantor Verlag für
Medizin und Naturwissenschaften GmbH, Aulendorf, 2012.
[50] Sicherheitsdatenblatt Polyethylenglycol 1000 Ph Eur. Merck Schuchardt OHG, 2004.
[51] Belitz H‐D, Grosch W, Schieberle P. Food Chemistry. 5. Auflage. Springer Verlag,
Berlin, 2004.
[52] Product Information: Dynasan 114, 116, 118 Microcrystalline Triglycerides. Sasol
Germny GmbH, 2007.
[53] Hänsel R, Sticher O, Alban S, Franz G, Spieß E. Pharmakognosie Phytopharmazie. 9.
Auflage. Springer Medizin Verlag, Heidelberg, 2009.
[54] Teuscher E, Melzig M, Lindequist U. Biogene Arzneimittel: Ein Lehrbuch der
Pharmazeutischen Biologie. 6. Auflage. Wissenschaftliche Verlagsgesellschaft mbH,
Stuttgart, 2004.
[55] Rassing MR. Chewing gum as a drug delivery system. Adv Drug Deliv Rev. 1994; 13:
89‐121.
[56] Weinstein RD, Cushnie E, Kopec TC. Liquid and Supercritical Carbon Dioxide Loading
into Chewing Gum Base. Ind Eng Chem Res. 2003; 42: 5554‐8.
[57] Gum Base Selection and Use. J Mestres Cafosa S/A, 2000.
[58] Sicherheitsdatenblatt Riboflavinphosphat‐Natrium. Fagron GmbH & Co KG, 2010.
[59] Steinhilber D, Schubert‐Zsilavecz M, Roth HJ. Medizinische Chemie ‐ Targets,
Arzneistoffe, Chemische Biologie. 2. Auflage. Deutscher Apotheker Verlag, Stuttgart,
2010.

Literaturverzeichnis
124
[60] Mutschler E, Geisslinger G, Kroemer HK, Ruth P, Schäfer‐Korting M.
Arzneimittelwirkungen. 9. Auflage. Wissenschaftliche Verlagsgesellschaft mbH,
Stuttgart, 2008.
[61] Sicherheitsdatenblatt Paracetamol. Caesar & Loretz GmbH, 2012.
[62] Anlagendokumentation GMPC I Mini‐Coater. Glatt, 2011.
[63] Ehlers E. Analytik II. 9. Auflage. Deutscher Apotheker Verlag, Stuttgart, 1999.
[64] Dissertation Grzegorz Garbacz: Entwicklung eines Biorelevanten
Freisetzungstestgerätes zur Simulation mechanischer Stressfaktoren bei der Magen‐
Darm‐Passage von Arzneiformen. Mathematisch‐Naturwissenschaftliche Fakultät der
Ernst‐Moritz‐Universität Greifswald, 2010.
[65] Garbacz G, Wedemeyer RS, Nagel S, Giessmann T, Monnikes H, Wilson CG, Siegmund
W, Weitschies W. Irregular absorption profiles observed from diclofenac extended
release tablets can be predicted using a dissolution test apparatus that mimics in vivo
physical stresses. Eur J Pharm Biopharm. 2008 Oct; 70 (2): 421‐8.
[66] Diplom Bock, M: Neuartige Arzneiformen für das Dünndarm‐Targeting.
Mathematisch‐Naturwissenschaftliche Fakultät der Ernst‐Moritz‐Arndt‐Universität
Greifswald, 2012.
[67] Produktinformation PHENOM. LOT‐Oriel Group Europe, Darmstadt, 2010.
[68] Rein H. Rasterelektronenmikroskopie. DAZ. 2010: 50‐6.
[69] Diplom Hosang, K: Entwicklung einer Manteltablette für das Dünndarm‐Targeting.
Mathematisch‐Naturwissenschaftliche Fakultät der Ernst‐Moritz‐Arndt‐Universität
Greifswald, 2013.
[70] el‐Helw Ael R, el‐Said Y. Preparation and characterization of agar beads containing
phenobarbitone sodium. J Microencapsul. 1988 Apr‐Jun; 5 (2): 159‐63.
[71] Higuchi T. Mechanism of Sustained‐Action Medication. Theoretical Analysis of Rate of
Release of Solid Drugs Dispersed in Solid Matrices. J Pharm Sci. 1963 Dec; 52: 1145‐9.
[72] Rahbani J, Behzad AR, Khashab NM, Al‐Ghoul M. Characterization of internal
structure of hydrated agar and gelatin matrices by cryo‐SEM. Electrophoresis. 2013
Feb; 34 (3): 405‐8.
[73] Deutscher Arzneimittel‐Codex ‐ Neues Rezeptur‐Formularium (DAC NRF 1.0). Govi‐
Verlag Pharmazeutischer Verlag GmbH, Eschborn, 2011.
[74] Koziolek M, Garbacz G, Neumann M, Weitschies W. Simulating the Postprandial
Stomach: Physiological Considerations for Dissolution and Release Testing. Mol
Pharmaceut. 2013 May; 10 (5): 1610‐22.
[75] Khutoryanskiy VV. Advances in mucoadhesion and mucoadhesive polymers.
Macromol Biosci. 2011 Jun 14; 11 (6): 748‐64.
[76] Gebrauchsinformation Prostagutt forte 160/120 mg. Dr Willmar Schwabe GmbH & Co
KG, 2008.
[77] Gebrauchsinformation Alpan 300 mg. Wörwag Pharma GmbH & Co KG, 2012.

Literaturverzeichnis
125
[78] Rote Liste Online. Rote Liste Service GmbH, Frankfurt/Main, 2013.
[79] Setofilm 4 mg Schmelzfilme. Norgine GmbH, Marburg, 2010.
[80] Erlich L, Yu D, Pallister DA, Levinson RS, Gole DG, Wilkinson PA, Erlich RE, Reeve LE,
Viegas TX. Relative bioavailability of danazol in dogs from liquid‐filled hard gelatin
capsules. Int J Pharm. 1999 Mar 1; 179 (1): 49‐53.
[81] Gebrauchsinformation Movicol aromafrei. Norgine GmbH, Marburg, 2011.
[82] Gebrauchsinformation Movicol Junior Schoko. Norgine GmbH, Marburg, 2011.
[83] Gebrauchsinformation Laxofalk. Dr Falk Pharma GmbH, Freiburg, 2012.
[84] Wirz S, Klaschik E. Laxative use and efficacy in palliative care of patients with cancer
pain and morphine therapy. A retrospective study with special regard to
polyethylene glycol. Schmerz. 2003 Aug; 17 (4): 233‐9.
[85] Gordon M, Naidoo K, Akobeng AK, Thomas AG. Osmotic and stimulant laxatives for
the management of childhood constipation. Cochrane Database Syst Rev. 2012; 7:
CD009118.
[86] Keck CM, Müller RH. Moderne Pharmazeutische Technologie. 2009.
[87] Gebrauchsinformation Voltaren Dolo Liquid 12,5 mg. Novartis Consumer Health
GmbH, München, 2011.
[88] Gebrauchsinformation Doxycyclin‐ratiopharm 100 mg Weichkapseln. ratiopharm
GmbH, Ulm, 2008.
[89] Gebrauchsinformation Taxofit Vitamin E Kapseln. Divapharma GmbH, Berlin, 2009.
[90] Kuentz M, Rothlisberger D. Determination of the optimal amount of water in liquid‐
fill masses for hard gelatin capsules by means of texture analysis and experimental
design. Int J Pharm. 2002 Apr 2; 236 (1‐2): 145‐52.
[91] Cade D, Cole ET, Mayer JP, Wittwer F. Liquid filled and sealed hard gelatin capsules.
Drug Development and Industrial Pharmacy, Capsugel AG, Switzerland, 1986; 12(11‐
13): 2289‐300.
[92] Bowtle W, Kanyowa L, Mackenzie M, Higgins P. Physical stability and resistance to
peroxidation of a range of liquid‐fill hard gelatin capsule products on extreme long‐
term storage. Drug Dev Ind Pharm. 2011 Jun; 37 (6): 685‐93.
[93] Barnwell SG, Laudanski T, Story MJ, Mallinson CB, Harris RJ, Cole SK, Keating M,
Attwood D. Improved Oral Bioavailability of Propranolol in Healthy‐Human
Volunteers Using a Liver Bypass Drug Delivery System Containing Oleic‐Acid. Int J
Pharm. 1992 Dec 8; 88 (1‐3): 423‐32.
[94] Barnwell SG, Gauci L, Harris RJ, Attwood D, Littlewood G, Guard P, Pickup ME,
Barrington P. Greatly Enhanced Oral Bioavailability of Propranolol Using the Halo(Tm)
Liver‐Bypass Drug‐Delivery System. J Control Release. 1994 Jan; 28 (1‐3): 306‐9.
[95] Burns SJ, Higginbottom S, Corness D, Hay G, Whelan I, Attwood D, Barnwell SG. A
Study of Enteric‐Coated Liquid‐Filled Hard Gelatin Capsules with Biphasic Release
Characteristics. Int J Pharm. 1994 Sep 26; 110 (3): 291‐6.

Literaturverzeichnis
126
[96] Burns SJ, Higginbottom S, Whelan I, Bowtle WJ, Rosenberg E, Corness D, Hay G,
Attwood D, Barnwell SG. Formulation strategies designed to maintain the biphasic
release characteristics of liquid‐filled capsules. Int J Pharm. 1996 Sep 6; 141 (1‐2): 9‐
16.
[97] Bader S, Weber M, Becker G. Is the pharmacological treatment of constipation in
palliative care evidence based? A systematic literature review. Schmerz. 2012 Sep; 26
(5): 568‐86.
[98] Gebrauchsinformation Paragar. Spirig HealthCare AG, Egerkingen, 2005.
[99] Gebrauchsinformation Paragol. Streuli Pharma AG, Uznach, 2006.
[100] Gebrauchsinformation Lansoyl. Johnson & Johnson Consumer NV/SA, Beerse, 2011.
[101] Miles CL, Fellowes D, Goodman ML, Wilkinson S. Laxatives for the management of
constipation in palliative care patients. Cochrane Db Syst Rev. 2006; (4).
[102] Sharif F, Crushell E, O'Driscoll K, Bourke B. Liquid paraffin: a reappraisal of its role in
the treatment of constipation. Arch Dis Child. 2001 Aug; 85 (2): 121‐4.
[103] Horstmann M, Dr., Am Auge applizierbare Vorrichtung zur sicheren Dosierung
systemischer und topischer Medikation. Germany patent DE 000010246900 A1,
2002.
[104] Lutton ES. The Polymorphism of Tristearin and Some of Its Homologs. J Am Chem
Soc. 1945; 67 (4): 524‐7.
[105] Walker WW. Aging of Tristearin ‐ Comparison of Dsc and Positron Lifetime Results. J
Am Oil Chem Soc. 1987 May; 64 (5): 754‐6.
[106] Takechi C, Kawaguchi T, Kaneko F, Yamamuro O, Akita H, Ono M, Suzuki M.
Incoherent quasielastic neutron scattering study on the polymorphism of tristearin:
Dynamical properties of hydrocarbon chains. J Phys Chem B. 2007 Aug 23; 111 (33):
9706‐10.
[107] Elisabettini P, Desmedt A, Durant F. Polymorphism of stabilized and nonstabilized
tristearin, pure and in the presence of food emulsifiers. J Am Oil Chem Soc. 1996 Feb;
73 (2): 187‐92.
[108] Da Silva E, Bresson S, Rousseau D. Characterization of the three major polymorphic
forms and liquid state of tristearin by Raman spectroscopy. Chem Phys Lipids. 2009
Feb; 157 (2): 113‐9.
[109] Oh JH, McCurdy AR, Clark S, Swanson BG. Characterization and thermal stability of
polymorphic forms of synthesized tristearin. J Food Sci. 2002 Oct; 67 (8): 2911‐7.
[110] Hamilton JD, Webb JPW, Dawson AM. Absorption of Tristearin and Stearic Acid and
Tripalmitin and Palmitic Acid ‐ Studies on Rate‐Limiting Steps in Rats. Biochim
Biophys Acta. 1969; 176 (1): 27‐36.
[111] Gebrauchsinformation Superpep Reise Kaugummi‐Dragees 20 mg. Hermes
Arzneimittel GmbH, Großhesselohe, 2008.

Literaturverzeichnis
127
[112] Gebrauchsinformation Nicorette Kaugummi 4 mg whitemint. McNeil Consumer
Healthcare GmbH, Neuss, 2010.
[113] Rimar Y, Babich JP, Shaoul R. Chewing gum bezoar. Gastrointestinal Endoscopy. 2004
Jun; 59 (7): 872.
[114] Milov DE, Andres JM, Erhart NA, Bailey DJ. Chewing gum bezoars of the
gastrointestinal tract. Pediatrics. 1998 Aug; 102 (2): art. no.‐e22.

Anhang
128
Anhang
Abb. 65: Kalibrationsgerade für Riboflavinphosphat in den Hartfett‐Kugeln Charge 1 (links) und für Paracetamol
in den Agar‐Kugeln und Tristearin‐Kapseln (rechts); MW ± SD, je n = 3.
Abb. 66: Kalibrationsgerade für Riboflavinphosphat in den Hartfett‐Kugeln Charge 2, den PEG‐Kugeln und
Paraffin‐Kapseln (links) und für Riboflavinphosphat in den Kaugummi‐Tabletten (rechts); MW ± SD; je n = 3.
Tab. 21: Zusammensetzung der getesteten Überzüge Ü 1 – Ü 7 für die Entscheidung eines Weichmachers.
Rezepturbestandteil Ü 1 Ü 2 Ü 3 Ü 4 Ü 5 Ü 6 Ü 7
Celluloseacetat 10 g 10 g 10 g 10 g 10 g 10 g 10 g
Talkum 2 g 2 g 2 g 2 g 2 g 2 g 2 g
Triacetin ‐ 1 g 2 g ‐ ‐ ‐ ‐
Dibutylsebacat ‐ ‐ ‐ 1 g 0,5 g ‐ ‐
Triethylcitrat ‐ ‐ ‐ ‐ ‐ 1 g 2 g
Aceton/Isopropanol
(4:1)
ad 200 g ad 200 g ad 200 g ad 200 g ad 200 g ad 200 g ad 200 g
y = 0,0598xR² = 0,9995
0,0
0,2
0,4
0,6
0,8
1,0
0 2 4 6 8 10 12 14
Absorption
Konzentration (mg/L)
y = 0,0629x + 0,0215R² = 0,9998
0,0
0,2
0,4
0,6
0,8
1,0
0 2 4 6 8 10 12 14 16
Absorption
Konzentration (mg/L)
y = 0,0595xR² = 0,9999
0,0
0,2
0,4
0,6
0,8
1,0
0 2 4 6 8 10 12 14
Absorption
Konzentration (mg/L)
y = 0,0643xR² = 0,9992
0,0
0,2
0,4
0,6
0,8
1,0
0 2 4 6 8 10 12 14
Absorption
Konzentration (mg/L)

Anhang
129
Abb. 67: Kraft‐Weg‐Diagramme von Hartfett‐Kugeln mit einer Polymerbeladung von 3,5 mg/cm2
Celluloseacetat für die Überzüge 1 ‐ 7, Durchführung am Texture Analyser TAplus, Einzelprofile.
0
5
10
15
20
0 5 10 15 20
Kraft (N)
Weg (mm)
Überzug 1
0
5
10
15
20
0 5 10 15 20
Kraft (N)
Weg (mm)
Überzug 2
0
5
10
15
20
0 5 10 15 20
Kraft (N)
Weg (mm)
Überzug 3
0
5
10
15
20
0 5 10 15 20
Kraft (N)
Weg (mm)
Überzug 4
0
5
10
15
20
0 5 10 15 20
Kraft (N)
Weg (mm)
Überzug 5
0
5
10
15
20
0 5 10 15 20
Kraft (N)
Weg (mm)
Überzug 6
0
5
10
15
20
0 5 10 15 20
Kraft (N)
Weg (mm)
Überzug 7

Anhang
130
Abbildungsverzeichnis
Abb. 1: Links: Anatomischer Aufbau des Magens [2]. Rechts: Querschnitt durch den Pylorus
[3]. ............................................................................................................................................ 1
Abb. 2: Anatomischer Aufbau des Duodenums [2]. ............................................................................. 3
Abb. 3: pH‐Temperatur‐Druck‐Diagramm (A), welches mittels SmartPill (B) aufgezeichnet
wurde [19, 20]. ......................................................................................................................... 4
Abb. 4: Teilstruktur von Celluloseacetat. ........................................................................................... 14
Abb. 5: Teilstruktur von Ethylcellulose. .............................................................................................. 14
Abb. 6: Strukturformel von Triacetin. ................................................................................................ 15
Abb. 7: Kaugummi‐Grundmasse als Granulat. ................................................................................... 16
Abb. 8: Strukturformel von Riboflavinphosphat‐Natrium.................................................................. 16
Abb. 9: Strukturformel von Paracetamol. .......................................................................................... 17
Abb. 10: Teilstruktur von Hydroxyethylcellulose. ................................................................................ 17
Abb. 11: Teilstruktur von MCC. ............................................................................................................ 18
Abb. 12: Darstellung der temperaturabhängigen Grenzlinien für die Sprührate organischer
Lösemittel bei 50 % UEG pro 10 m3/h Luftdurchsatz [62]. .................................................... 22
Abb. 13: A: Hartfett‐W32‐Kugeln mit 10 mg RFP/Kugel ohne (links) und mit (rechts) 4 mg/cm2
Celluloseacetat‐Überzug. B: PEG‐1000‐Kugeln mit 10 mg RFP/Kugel ohne (links) und
mit (rechts) 4 mg/cm2 Celluloseacetat‐Überzug. ................................................................... 22
Abb. 14: A: Alginat‐Kügelchen (3 mm); B: aufgeschnittene Hartfett‐H15‐Kugel (Charge 3b) mit
eingebettetem Alginat‐Kügelchen (6 mm); Einfärbung mit blauer Lebensmittelfarbe. ........ 24
Abb. 15: Ergebnis des Umverteilungsversuchs nach 72 h; von links nach rechts: dünnflüssiges
Paraffin, dickflüssiges Paraffin, Rizinusöl, Erdnussöl. ............................................................ 25
Abb. 16: Paraffin‐Kapseln mit 10 mg RFP/Kapsel und 4 mg/cm2 Ethylcellulose‐Überzug. .................. 26
Abb. 17: A: Herstellungsschritte der Tristearin‐Kapsel. B: Draufsicht auf eine leere Tristearin‐
Kapsel. .................................................................................................................................... 28
Abb. 18: Schematische Darstellung der Kaugummi‐Manteltabletten der Chargen 1 – 3; grau:
Hartfettkern, hellblau: Trennschicht NaCl, orange: Pulvermantel, dunkelblau:
Kaugummimantel mit Sollbruchstellen.................................................................................. 30
Abb. 19: Schema der Gehaltsbestimmung von Paracetamol aus Agar‐Kugeln. ................................... 32
Abb. 20: Schema der Gehaltsbestimmung von Riboflavinphosphat aus Hartfett‐W32‐Kugeln. ......... 33
Abb. 21: Schema der Gehaltsbestimmung von Riboflavinphosphat aus PEG‐1000‐Kugeln. ............... 35
Abb. 22: Schema der Gehaltsbestimmung von Riboflavinphosphat aus Paraffin‐Kapseln. ................. 35
Abb. 23: Schema der Gehaltsbestimmung von Paracetamol aus Tristearin‐Kapseln. ......................... 36
Abb. 24: Schematischer Aufbau der Stresstest‐Apparatur. ................................................................. 41
Abb. 25: Programm der Freisetzung in der Stresstest‐Apparatur. ....................................................... 42

Anhang
131
Abb. 26: Kraft‐Weg‐Diagramm der 7 %igen Agar‐Kugeln, Durchführung am Texture Analyser
TA.XT plus bei Raumtemperatur, Einzelprofile. ..................................................................... 45
Abb. 27: Bruchkraft (N) der Agar‐Kugeln in Abhängigkeit von der Agar‐Konzentration,
MW ± SD, n = 4 (1,5, 2, 3, 4, 5 %) bzw. n = 10 (6, 7 %). .......................................................... 45
Abb. 28: Kraft‐Weg‐Diagramm der 5 %igen Agar‐Kugeln nach einstündiger Trocknung bei
40 ± 0,5 °C, Durchführung am Texture Analyser TA.XT plus bei Raumtemperatur,
Einzelprofile. .......................................................................................................................... 46
Abb. 29: Bruchkraft (N) der 5 %igen Agar‐Kugeln in Abhängigkeit von der Trocknungszeit bei
40 ± 0,5 °C, MW ± SD, n = 4 (0 h) bzw. n = 10 (0,5, 1, 3, 5 h). ................................................ 47
Abb. 30: Zustand der Agar‐Kugeln nach dem ersten Zerbrechen durch Druckeinwirkung in der
Stresstest‐Apparatur in Abhängigkeit von der Konzentration: A: 3 % Agar bei
100 mbar; B: 5 % Agar bei 100 mbar; C: 6 % Agar bei 200 mbar; D: 6,5 % Agar nach
30‐minütiger Inkubation in SGFsp pH 1,8 und bei 200 mbar; E: 7 % Agar bei 300 mbar;
F: 8 % Agar bei 350 mbar; G: 9 % Agar bei 450 mbar; H: 10 % Agar bei 300 mbar. .............. 49
Abb. 31: Zustand der 5 %igen Agar‐Kugeln nach dem ersten Zerbrechen durch
Druckeinwirkung in der Stresstest‐Apparatur in Abhängigkeit von der Trocknungszeit:
A: 0 h bei 100 mbar; B: 0,5 h bei 200 mbar; C: 1 h bei 300 mbar; D: 1 h bei 30‐
minütiger Inkubation in 37 °C warmem SGFsp pH 1,8 und 200 mbar; E: 3 h bei
200 mbar; F: 5 h bei 350 mbar. .............................................................................................. 51
Abb. 32: Freigesetzte prozentuale Wirkstoffmenge aus Agar‐Kugeln (7,5 %) mit 5,31 mg
Paracetamol/Kugel in 1000 mL SGFsp pH 1,8 über 3 h bei 37 °C und 75 rpm; Paddle‐
Apparatur, photometrische Messung bei 242 nm, MW ± SD, n = 6. ..................................... 52
Abb. 33: Higuchi‐Plot für die Freisetzung von Paracetamol aus Agar‐Kugeln (7,5 %). ........................ 52
Abb. 34: Links: 7 %ige Agar‐Kugeln mit 0,03 % Methylorange vor (A) und nach 2 min (B) in
150 mL SGFsp pH 1,2; halbiert. Rechts: 7 %ige Agar‐Kugeln mit 0,03 % Methylorange
und 1 % KOH vor (C) und nach 25 min (D) in 150 mL SGFsp pH 1,2; Kugeln wurden
halbiert. .................................................................................................................................. 53
Abb. 35: Freisetzung von Paracetamol aus 7,5 %igen Agar‐Kugeln in 1160 mL SGFsp pH 1,8 bei
37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 242 und 450 nm,
Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter Prüfung. ............................... 54
Abb. 36: Kraft‐Weg‐Diagramm der Hartfett‐W32‐Kugeln (Charge 1) mit 3,5 mg/cm2
Celluloseacetat‐Überzug, Durchführung am Texture Analyser TA.XT plus nach 20‐
minütiger Inkubation in 37 °C warmem Wasser, Einzelprofile. ............................................. 55
Abb. 37: Bruchkraft (N) der Hartfett‐W32‐Kugeln (Charge 1) in Abhängigkeit von der
Polymerbeladung, MW ± SD, n = 10 (für 2, 2,5 und 3 mg/cm2) bzw. n = 9 (für
3,5 mg/cm2). .......................................................................................................................... 56
Abb. 38: Bruchkraft (N) der Hartfett‐W32‐Kugeln (Charge 2) in Abhängigkeit von der
Polymerbeladung, MW ± SD, n = 5 (für 3, 3,5 und 5 mg/cm2) bzw. n = 6 (für 4, 6 und
7 mg/cm2)............................................................................................................................... 57

Anhang
132
Abb. 39: Durch Krafteinwirkung aufgerissener Überzug einer Arzneiform der Charge 3a. ................. 57
Abb. 40: Bruchkraft (N) der Hartfett‐H15‐Kugeln (Charge 3a und 3b) in Abhängigkeit von der
Polymerbeladung, MW ± SD, n = 6 bzw. n = 5 (für 3 mg/cm2 bei Charge 3b). ...................... 58
Abb. 41: Zustand der Hartfett‐W32‐Kugeln (Charge 1) nach dem ersten Zerbrechen durch
Druckeinwirkung in der Stresstest‐Apparatur in Abhängigkeit von der
Polymerbeladung: A 1+2: 2 mg/cm2 CA‐Überzug bei 100 mbar; B 1+2: 2,5 mg/cm2 CA‐
Überzug bei 100 mbar; C 1+2: 3 mg/cm2 CA‐Überzug bei 100 mbar; D 1+2:
3,5 mg/cm2 CA‐Überzug bei 300 mbar. ................................................................................. 60
Abb. 42: REM‐Aufnahmen des CA‐Überzugs im Querschnitt (A und B) und in der Draufsicht (C
und D) mit einer Polymerbeladung von 2 mg/cm2 (A und C) und 3,5 mg/cm2 (B und D)
nach Auflösung des Hartfettkerns, Querschnitt: 250‐fache Vergrößerung, Draufsicht:
500‐fache Vergrößerung. ....................................................................................................... 61
Abb. 43: Freisetzung von Riboflavinphosphat‐Natrium aus mit 3,5 mg/cm2 CA überzogenen
Hartfett‐W32‐Kugeln (Charge 1) in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in
der Stresstest‐Apparatur anhand des Magenentleerungsprogramms, photometrische
Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach
erfolgter Prüfung. .................................................................................................................. 63
Abb. 44: Freisetzung von Riboflavinphosphat‐Natrium aus mit 4 mg/cm2 CA überzogenen
Hartfett‐W32‐Kugeln (Charge 2) in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in
der Stresstest‐Apparatur anhand des Magenentleerungsprogramms, photometrische
Messung bei 267 und 600 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach
erfolgter Prüfung. .................................................................................................................. 64
Abb. 45: Kraft‐Weg‐Diagramm der PEG‐1000‐Kugeln mit 3,5 mg/cm2 Celluloseacetat‐Überzug,
Durchführung am Texture Analyser TAplus nach 25‐minütiger Inkubation in 37 °C
warmem Wasser, Einzelprofile. ............................................................................................. 65
Abb. 46: Bruchkraft (N) der PEG‐1000‐Kugeln in Abhängigkeit von der Polymerbeladung,
MW ± SD, n = 5 (für 3, 3,5 und 4 mg/cm2) bzw. n = 6 (für 5, 6 und 7 mg/cm2). .................... 66
Abb. 47: Freigesetzte prozentuale Wirkstoffmenge aus PEG‐1000‐Kugeln mit 11,40 ± 0,18 mg
Riboflavinphosphat/Kugel und einer Polymerbeladung von 4 mg/cm2 Celluloseacetat
in 1000 mL SGFsp pH 1,8 über 3 h bei 37 °C und 75 rpm; Paddle‐Apparatur,
photometrische Messung bei 267 nm, MW ± SD, n = 6. ....................................................... 68
Abb. 48: Freisetzung von Riboflavinphosphat‐Natrium aus mit 4 mg/cm2 CA überzogenen PEG‐
1000‐Kugeln in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐
Apparatur anhand des Magenentleerungsprogramms, photometrische Messung bei
267 und 600 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter
Prüfung. .................................................................................................................................. 69
Abb. 49: Kraft‐Weg‐Diagramm der Paraffin‐Kapseln mit 4 mg/cm2 Ethylcellulose‐Überzug,
Durchführung am Texture Analyser TAplus nach 5‐minütiger (links) und 25‐minütiger
(rechts) Inkubation in 37 °C warmem Wasser, Einzelprofile. ................................................ 70
Abb. 50: Freisetzung von Riboflavinphosphat‐Natrium aus mit 4 mg/cm2 EC überzogenen
Paraffin‐Kapseln in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐

Anhang
133
Apparatur anhand des Magenentleerungsprogramms, photometrische Messung bei
267 und 600 nm, Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter
Prüfung. .................................................................................................................................. 71
Abb. 51: Kraft‐Weg‐Diagramm der Tristearin‐Kapseln mit einem Füllvolumen von 250 µL
Tristearin, Durchführung am Texture Analyser TA.XT plus nach Ablösen der
Gelatinehülle in 37 °C warmem Wasser, Einzelprofile. ......................................................... 73
Abb. 52: Bruchkraft (N) der Tristearin‐Kapseln in Abhängigkeit vom Tristearin‐Volumen,
MW ± SD, n = 10 (für 250 µL) bzw. n = 3 (für 300, 400 und 500 µL). ..................................... 73
Abb. 53: Zustand der Tristearin‐Kapseln nach dem ersten Zerbrechen durch Druckeinwirkung:
A: 220 µL Innenbeschichtung bei 100 mbar; B 1+2: 250 µL Innenbeschichtung bei
300 mbar; C: 300 µL Innenbeschichtung bei 400 mbar; D: 360 µL Innenbeschichtung
bei 500 mbar; E: 400 µL Innenbeschichtung bei 500 mbar. .................................................. 76
Abb. 54: REM‐Aufnahmen einer Tristearin‐Kapsel im Querschnitt mit einem
Beschichtungsvolumen von 250 µL nach Entfernung der Gelreste, 250‐fache
Vergrößerung. ........................................................................................................................ 76
Abb. 55: Freigesetzte prozentuale Wirkstoffmenge aus Tristearin‐Kapseln (250 µL Tristearin‐
Volumen) mit 5,51 mg Paracetamol/Kapsel in 1000 mL SGFsp pH 1,8 über 3 h bei 37
°C und 75 rpm; Paddle‐Apparatur, photometrische Messung bei 242 nm, MW ± SD,
n = 6. ...................................................................................................................................... 77
Abb. 56: Freisetzung von Paracetamol aus Tristearin‐Kapseln mit einem
Innenbeschichtungsvolumen von 250 µL in 1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C,
geprüft in der Stresstest‐Apparatur anhand des Magenentleerungsprogramms,
photometrische Messung bei 242 und 450 nm, Einzelprofile. Foto: Zustand der
Arzneiformen nach erfolgter Prüfung. 78
Abb. 57: Bruchkraft (N) der unterschiedlichen Chargen der Kaugummi‐Tabletten, MW ± SD, n
= 6 bzw. n = 5 (für gepr. MT 5.1, ged. MT 1.4, ged. MT 4.6, ged. MT 4.9)
[*kennzeichnet die Chargen 1 (4.9), 2 (4.7) und 3 (4.10)]. .................................................... 79
Abb. 58: Kraft‐Weg‐Diagramm der Kaugummi‐Manteltabletten Charge 1, Durchführung am
Texture Analyser TAplus nach 15‐minütiger Inkubation in 37 °C warmem Wasser,
Einzelprofile. .......................................................................................................................... 80
Abb. 59: Kraft‐Weg‐Diagramm der Kaugummi‐Manteltabletten Charge 2, Durchführung am
Texture Analyser TAplus nach 15‐minütiger Inkubation in 37 °C warmem Wasser,
Einzelprofile. .......................................................................................................................... 81
Abb. 60: Kraft‐Weg‐Diagramm der Kaugummi‐Manteltabletten Charge 3, Durchführung am
Texture Analyser TAplus nach 15‐minütiger Inkubation in 37 °C warmem Wasser,
Einzelprofile. .......................................................................................................................... 82
Abb. 61: Freisetzung von Riboflavinphosphat aus Kaugummi‐Manteltabletten der Charge 1a in
1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 267 und 600 nm,
Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter Prüfung. ............................... 84

Anhang
134
Abb. 62: Freisetzung von Riboflavinphosphat aus Kaugummi‐Manteltabletten der Charge 2a in
1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 267 und 600 nm,
Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter Prüfung. ............................... 85
Abb. 63: Freisetzung von Riboflavinphosphat aus Kaugummi‐Manteltabletten der Charge 3a in
1160 mL SGFsp pH 1,8 bei 37 ± 0,5 °C, geprüft in der Stresstest‐Apparatur anhand des
Magenentleerungsprogramms, photometrische Messung bei 267 und 600 nm,
Einzelprofile. Foto: Zustand der Arzneiformen nach erfolgter Prüfung. ............................... 85
Abb. 64: 3 %ige Alginat‐Kugel mit flüssigem Kern, deren äußere Wand mit Hilfe einer 10 %igen
CaCl2‐Lösung erhärtet wurde. ................................................................................................ 97
Abb. 65: Kalibrationsgerade für Riboflavinphosphat in den Hartfett‐Kugeln Charge 1 (links) und
für Paracetamol in den Agar‐Kugeln und Tristearin‐Kapseln (rechts); MW ± SD, je
n = 3. .................................................................................................................................... 128
Abb. 66: Kalibrationsgerade für Riboflavinphosphat in den Hartfett‐Kugeln Charge 2, den PEG‐
Kugeln und Paraffin‐Kapseln (links) und für Riboflavinphosphat in den Kaugummi‐
Tabletten (rechts); MW ± SD; je n = 3. ................................................................................. 128
Abb. 67: Kraft‐Weg‐Diagramme von Hartfett‐Kugeln mit einer Polymerbeladung von
3,5 mg/cm2 Celluloseacetat für die Überzüge 1 ‐ 7, Durchführung am Texture
Analyser TAplus, Einzelprofile. ............................................................................................. 129
Tabellenverzeichnis
Tab. 1: Zusammensetzung des 5 %igen Celluloseacetat‐Überzugs für die Hartfett‐W32‐ und
PEG‐1000‐Kugeln. .................................................................................................................. 21
Tab. 2: Zusammensetzung des 5 %igen Ethylcellulose‐Überzugs für die Paraffin‐Kapseln. ............. 26
Tab. 3: Zusammensetzung des 5 %igen Paracetamol enthaltenden Hydrogels. ............................... 27
Tab. 4: Übersicht der geprüften Chargen der Kaugummi‐Manteltabletten. ..................................... 29
Tab. 5: Zusammensetzung der Placebo‐Tabletten. ........................................................................... 31
Tab. 6: Unterschiede der verwendeten Texture Analyser. ................................................................ 37
Tab. 7: Mittels Texture Analyser auf Bruchfestigkeit getestete Arzneiformen. ................................ 38
Tab. 8: Zusammensetzung des Mediums SGFsp pH 1,8. ................................................................... 39
Tab. 9: Zusammensetzung der Agar‐Kugeln der Chargen 1a und 1b für den Diffusionsversuch. ..... 40
Tab. 10: Mittels Stresstest‐Apparatur getestete Arzneiformen (*30 min Inkubation). ...................... 42
Tab. 11: Verwendete Arzneiformen für die Freisetzung in der Stresstest‐Apparatur......................... 43
Tab. 12: Masse‐ und Größenverlust der 5 %igen Agar‐Kugeln nach 0,5, 1, 3 und 5 h Trocknung
bei 40 ± 0,5 °C, prozentuale Werte beziehen sich auf die 5 %igen Kugeln ohne
Trocknung, Berechnung aus n = 6. ......................................................................................... 47

Anhang
135
Tab. 13: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Agar‐Kugeln
unterschiedlicher Agar‐Konzentration: Anzahl der defekten Arzneiformen in
Abhängigkeit vom angelegten Druck (mbar), n = 3 für jede Agar‐Konzentration, 5 min
Inkubation in 37 °C warmem SGFsp pH 1,8 (*30 min Inkubation). ....................................... 48
Tab. 14: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Agar‐Kugeln
unterschiedlicher Agar‐Konzentration bei direkter Druckeinwirkung: Anzahl der
defekten Arzneiformen in Abhängigkeit vom angelegten Druck (mbar), n = 3 für jede
Agar‐Konzentration. ............................................................................................................... 49
Tab. 15: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der 5 %igen Agar‐Kugeln
bei unterschiedlicher Trocknungszeit bei 40 ± 0,5 °C: Anzahl der defekten
Arzneiformen in Abhängigkeit vom angelegten Druck (mbar), n = 3 für jede
Trocknungszeit, 5 min Inkubation in 37 °C warmem SGFsp pH 1,8 (*30 min
Inkubation). ............................................................................................................................ 50
Tab. 16: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der 5 %igen Agar‐Kugeln
bei unterschiedlicher Trocknungszeit bei 40 ± 0,5 °C und direkter Druckeinwirkung:
Anzahl der defekten Arzneiformen in Abhängigkeit vom angelegten Druck (mbar),
n = 3 für jede Trocknungszeit, 5 min Inkubation in 37 °C warmen SGFsp pH 1,8
(* 30 min Inkubation). ............................................................................................................ 51
Tab. 17: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Hartfett‐W32‐Kugeln
(Charge 1 und 2) in Abhängigkeit von der Polymerbeladung: Anzahl der defekten
Arzneiformen in Abhängigkeit vom angelegten Druck (mbar), n = 6 für jede
Polymerbeladung (außer n = 2 für 4 mg/cm2), 25 min Inkubation in 37 °C warmen
SGFsp pH 1,8 (*geplatzter Ballon). ........................................................................................ 60
Tab. 18: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der PEG‐1000‐Kugeln in
Abhängigkeit von der Polymerbeladung: Anzahl der defekten Arzneiformen in
Abhängigkeit vom angelegten Druck (mbar), n = 6 für jede Polymerbeladung, 25 min
Inkubation in 37 °C warmem SGFsp pH 1,8. .......................................................................... 67
Tab. 19: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeiten der Tristearin‐Kapseln in
Abhängigkeit vom Tristearin‐Volumen (µL): Anzahl der defekten Arzneiformen in
Abhängigkeit vom angelegten Druck (mbar), n = 3 für jedes Beschichtungsvolumen
(außer n = 9 für 250 µL), 5 min Inkubation in 37 °C warmem SGFsp pH 1,8. ........................ 75
Tab. 20: Mittels Stresstest‐Apparatur ermittelte Bruchfestigkeit der Kaugummi‐
Manteltabletten: Anzahl der defekten Arzneiformen in Abhängigkeit vom angelegten
Druck (mbar), n = 6 für jede Charge, 20 min Inkubation in 37 °C warmem SGFsp
pH 1,8. .................................................................................................................................... 83
Tab. 21: Zusammensetzung der getesteten Überzüge Ü 1 – Ü 7 für die Entscheidung eines
Weichmachers. .................................................................................................................... 128

Anhang
136
Geräteverzeichnis
Geräte Hersteller / Vertreiber
Analysenwaage CPA 1003S Sartorius AG, Göttingen
Bruchfestigkeitstester TBH 225D Erweka, Heusenstamm
Dissolution‐Tester DT 80 Erweka, Heusenstamm
Dragierkessel mit Antriebsgerät AR 400 Erweka, Heusenstamm
Exzenterpresse mit Matrize und Stempel Typ KP 2 VEB Maschinen‐ und Mühlenbau, Wittenberg
Filterpapier Filtrak 390 VEB Freiberger Zellstoff‐ und Papierfabrik,
Niederschlag
Vaginalglobuliform 2 g aus Edelstahl ‐
GMPC I Mini‐Coater Glatt GmbH, Binzen
Kapseln #0 und #1 ‐
Kompressor Medic Air 300 D Silent Schneider Druckluft GmbH, Reutlingen
Kubusmischer mit Antriebsgerät AR 400 Erweka, Heusenstamm
Magnetrührer IKA® RCT basic safety control MR
3001
IKA®‐Werke GmbH & Co.KG, Staufen
Mini Sputter Coater SC 7620 Quorum Technologies Ltd, Laughton (UK)
pH Messgerät 211 Microprocessor Hanna Instruments Deutschland GmbH, Kehl
am Rhein
µL‐Pipetten Eppendorf Research mit
Kunststoffspitzen 10 ‐ 100 µL, 100 ‐ 1000 µL
Eppendorf AG, Hamburg
Präzisionswaage Acculab ATILON Sartorius AG, Göttingen
Rasterelektronenmikroskop PHENOM FEI Company, Hillboro (USA)
Sprühpistole LM 2000 HVLP SATA GmbH & Co. KG, Kornwestheim
Stresstest‐Apparatur V.2.0 Erweka, Heusenstamm
Texture Analyser TA.XT.plus Stable Micro Systems, Surrey (UK)
Texture Analyser TAplus AMETEK Lloyd Instruments, Meerbusch
UltraTurrax T25 IKA®‐Werke GmbH & Co.KG, Staufen
UV Mini 1240
UV‐Gerät Cary 50 Scan
Schimadzu Deutschland GmbH, Duisburg
Varian, Inc., Palo Alto (USA)
Wasserbad apotec® Rezeptur‐Wasserbad, Wepa Apothekenbedarf
GmbH & Co. KG, Hillscheid
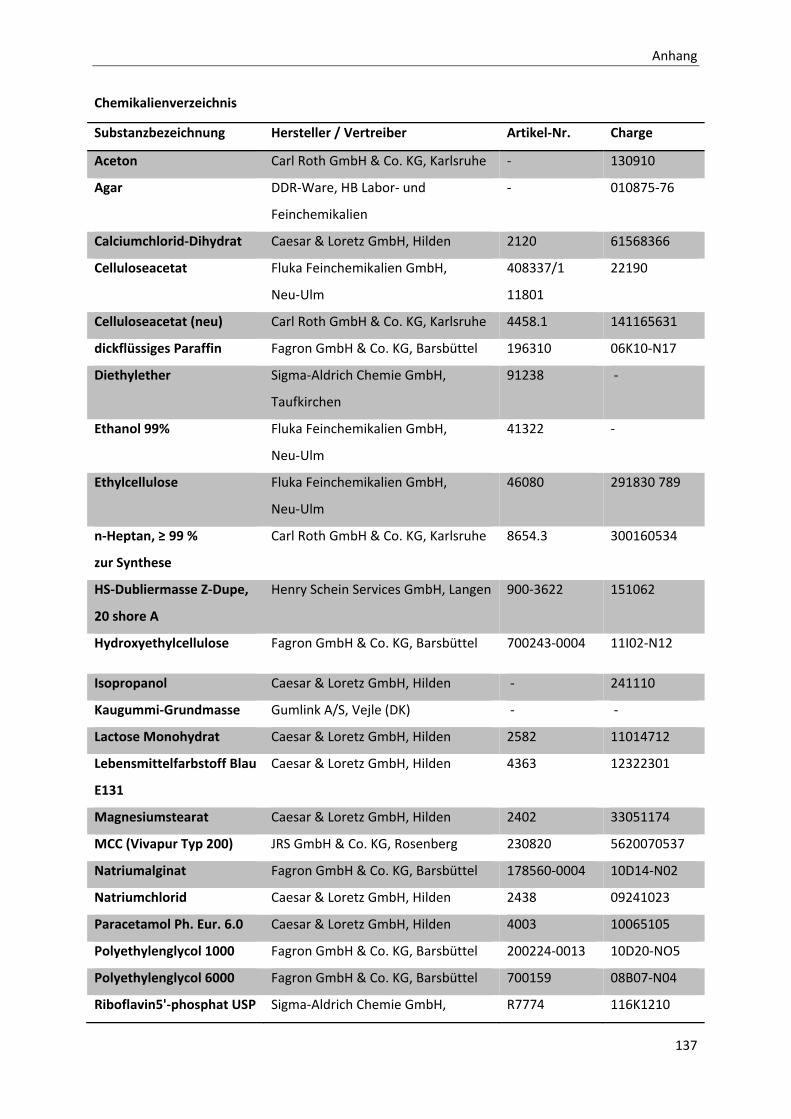
Anhang
137
Chemikalienverzeichnis
Substanzbezeichnung Hersteller / Vertreiber Artikel‐Nr. Charge
Aceton Carl Roth GmbH & Co. KG, Karlsruhe ‐ 130910
Agar DDR‐Ware, HB Labor‐ und
Feinchemikalien
‐ 010875‐76
Calciumchlorid‐Dihydrat Caesar & Loretz GmbH, Hilden 2120 61568366
Celluloseacetat Fluka Feinchemikalien GmbH,
Neu‐Ulm
408337/1
11801
22190
Celluloseacetat (neu) Carl Roth GmbH & Co. KG, Karlsruhe 4458.1 141165631
dickflüssiges Paraffin Fagron GmbH & Co. KG, Barsbüttel 196310 06K10‐N17
Diethylether Sigma‐Aldrich Chemie GmbH,
Taufkirchen
91238 ‐
Ethanol 99% Fluka Feinchemikalien GmbH,
Neu‐Ulm
41322 ‐
Ethylcellulose
Fluka Feinchemikalien GmbH,
Neu‐Ulm
46080 291830 789
n‐Heptan, ≥ 99 %
zur Synthese
Carl Roth GmbH & Co. KG, Karlsruhe 8654.3 300160534
HS‐Dubliermasse Z‐Dupe,
20 shore A
Henry Schein Services GmbH, Langen 900‐3622 151062
Hydroxyethylcellulose Fagron GmbH & Co. KG, Barsbüttel 700243‐0004 11I02‐N12
Isopropanol Caesar & Loretz GmbH, Hilden ‐ 241110
Kaugummi‐Grundmasse Gumlink A/S, Vejle (DK) ‐ ‐
Lactose Monohydrat Caesar & Loretz GmbH, Hilden 2582 11014712
Lebensmittelfarbstoff Blau
E131
Caesar & Loretz GmbH, Hilden 4363 12322301
Magnesiumstearat Caesar & Loretz GmbH, Hilden 2402 33051174
MCC (Vivapur Typ 200) JRS GmbH & Co. KG, Rosenberg 230820 5620070537
Natriumalginat Fagron GmbH & Co. KG, Barsbüttel 178560‐0004 10D14‐N02
Natriumchlorid Caesar & Loretz GmbH, Hilden 2438 09241023
Paracetamol Ph. Eur. 6.0 Caesar & Loretz GmbH, Hilden 4003 10065105
Polyethylenglycol 1000 Fagron GmbH & Co. KG, Barsbüttel 200224‐0013 10D20‐NO5
Polyethylenglycol 6000 Fagron GmbH & Co. KG, Barsbüttel 700159 08B07‐N04
Riboflavin5'‐phosphat USP Sigma‐Aldrich Chemie GmbH, R7774 116K1210
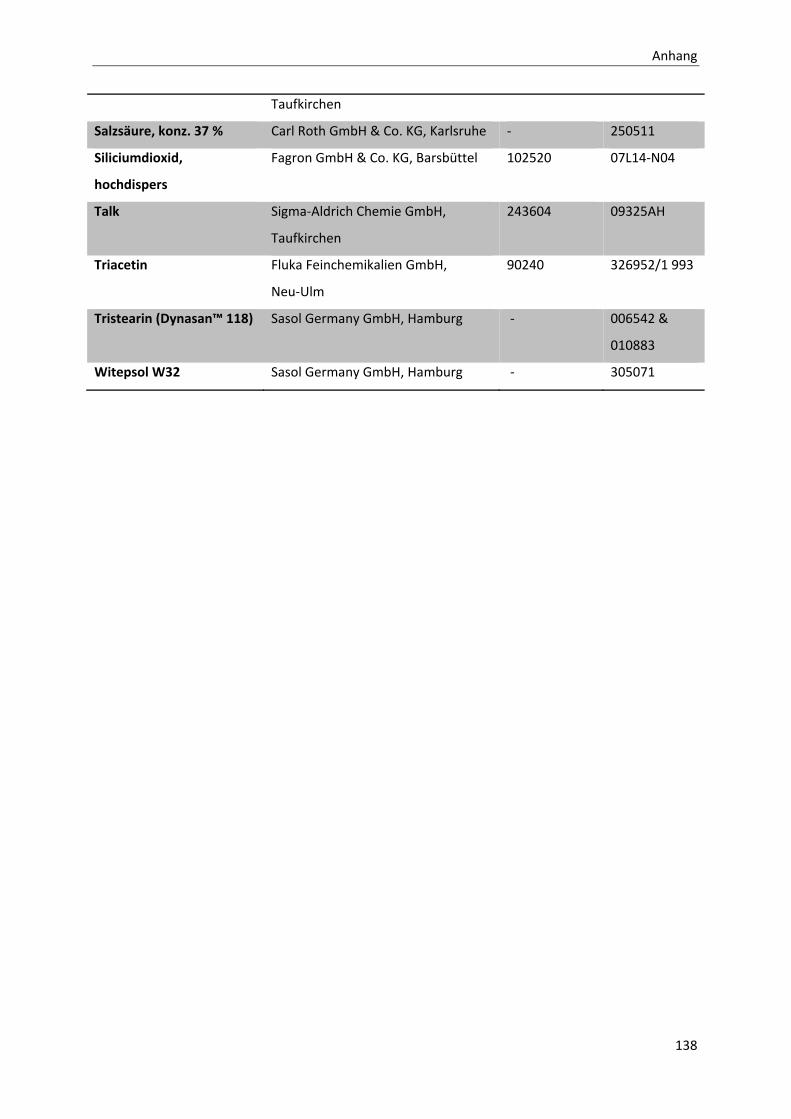
Anhang
138
Taufkirchen
Salzsäure, konz. 37 % Carl Roth GmbH & Co. KG, Karlsruhe ‐ 250511
Siliciumdioxid,
hochdispers
Fagron GmbH & Co. KG, Barsbüttel 102520 07L14‐N04
Talk Sigma‐Aldrich Chemie GmbH,
Taufkirchen
243604 09325AH
Triacetin Fluka Feinchemikalien GmbH,
Neu‐Ulm
90240 326952/1 993
Tristearin (Dynasan™ 118) Sasol Germany GmbH, Hamburg ‐ 006542 &
010883
Witepsol W32 Sasol Germany GmbH, Hamburg ‐ 305071

139
Eidesstattliche Erklärung
Hiermit erkläre ich, dass diese Arbeit bisher von mir weder an der Mathematisch‐
Naturwissenschaftlichen Fakultät der Ernst‐Moritz‐Arndt‐Universität Greifswald noch einer anderen
wissenschaftlichen Einrichtung zum Zwecke der Promotion eingereicht wurde.
Ferner erkläre ich, dass ich diese Arbeit selbständig verfasst und keine anderen als die darin
angegebenen Hilfsmittel und Hilfen benutzt und keine Textabschnitte eines Dritten ohne
Kennzeichnung übernommen habe.
Lisa Wilde

140
Danksagung
Mein besonderer Dank gilt Prof. Dr. Werner Weitschies für die Aufnahme in seinen Arbeitskreis und
die Bereitstellung des äußerst interessanten und vielversprechenden Themas. Weiterhin danke ich
ihm für sein stetes Interesse am Gelingen dieser Arbeit, für seine hilfreichen Anregungen und für
seine ständige Unterstützung.
Ich bedanke mich besonders bei Dr. Gunnar Glöckl für seine Unterstützung und vielen hilfreichen
Hinweise bei der Erarbeitung des Themas und den dabei regelmäßig aufgekommenen
Problemstellungen. Weiterhin bin ich ihm sehr dankbar für die Durchsicht von Abstracts, Postern,
Manuskripten, Diplomarbeiten und dieser Arbeit und seinen dabei entstandenen nützlichen
Kommentaren.
Ich danke Dr. Grzegorz Garbacz und Katrin Hannemann von Physiolution GmbH für die Unterstützung
bei der Testung mit der Stresstest‐Apparatur. Desweiteren gilt mein Dank Knut Seidlitz, Dr. Christiane
Schiller und Dr. Ulrike Hanke von LTS Lohmann Therapie‐Systeme AG für die technische
Unterstützung bei der Verwendung des Texture Analysers.
Für finanzielle Unterstützung möchte ich mich beim Bundesministerium für Bildung und Forschung
bedanken, welches die Arbeit im Rahmen des InnoProfile‐Projektes „Wirkstofftransport‐basierte
Konzepte und Drug‐Delivery‐Technologien zur Optimierung der klinischen Anwendung von
Arzneimitteln“ gefördert hat (FKZ: 03IP612).
Weiterhin bedanke ich mich bei Mirko Leonhardt für die Ermutigung zur Anfertigung meiner
Dissertation während meines Diploms unter seiner Betreuung. Desweiteren danke ich meinen
Diplomandinnen Mona Bock, Marieke Wolf und Katharina Hosang sowie meinen
Wahlpflichtstudenten, die durch ihren Einsatz sehr zum Vorankommen meiner Arbeit beigetragen
haben.
Für das unvergleichliche Arbeitsklima im Arbeitskreis Biopharmazie und Pharmazeutische
Technologie möchte ich mich bei allen Kollegen sehr herzlich bedanken. Sowohl die fachliche
Unterstützung und das freundschaftliche Verhältnis als auch die Auszeiten in Form von Kaffepausen,
Sommerfesten, Weihnachtsfeiern und Grillabenden haben aus der Promotion eine sehr schöne und
angenehme Zeit gemacht.
Ein weiterer Dank geht an Richard Pochepan, der mir mit den Kontrollen englischsprachlicher Texte
sehr weiter geholfen hat, sowie an Edda Schulz für die Lesekontrolle dieser Arbeit.
Ein besonderer Dank gilt meiner Familie und meinen Freunden ‐ sowohl im Arbeitskreis als auch
außerhalb ‐ für die stetige Unterstützung und den Zuspruch während meiner Studien‐ und
Promotionszeit, ohne deren Hilfe alles deutlich mühsamer gewesen wäre.
Meinem Mann Felix danke ich von Herzen für alles, was er für mich getan hat und weiterhin tut.
Ohne ihn, seine Geduld und seine grenzenlose Unterstützung wäre vermutlich schon der Beginn des
Studiums mein Ende in der Pharmazie gewesen.

141
Veröffentlichungen
Wissenschaftliche Artikel (in submission):
L. Wilde, M. Bock, M. Wolf, G. Glöckl, G. Garbacz, W. Weitschies: Development of pressure‐sensitive
dosage forms with a core liquefying at body temperature.
L. Wilde, M. Bock, G. Glöckl, G. Garbacz, W. Weitschies: Development of a pressure‐sensitive glyceryl
tristearate capsule filled with a drug‐containing hydrogel.
Posterpräsentationen:
Lisa Wilde, Ulrike Wollatz, Gunnar Glöckl, Bernd Kordaß, Werner Weitschies: Development of a
medicated chewing gum containing riboflavin phosphate. 8th World Meeting on Pharmaceutics,
Biopharmaceutics and Pharmaceutical Technology, Istanbul, 19.3.‐22.3.2012
Lisa Wilde, Ulrike Wollatz, Gunnar Glöckl, Bernd Kordaß, Werner Weitschies: Development of a
chewing gum to characterize chewing motions of patients. Symposium and workshops on behalf of
the 40. Anniversary, Greifswald, 12.9.‐13.9.2013

















