Technische Universität München - mediatum.ub.tum.de · F Fe 2O3 S SO 3 H H2O. Tabelle 1:...
165
Technische Universität München Lehrbereich Anorganische Chemie - Lehrstuhl für Bauchemie Untersuchungen zur Kinetik grundlegender Reak- tionsschritte bei der Hydratation von Portland- und Hochofenzementen Holger König Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universi- tät München zur Erlangung des akademischen Grades eines Doktors der Naturwissenschaften genehmigten Dissertation. Vorsitzender: Univ.-Prof. Dr. Klaus Köhler Prüfer der Dissertation: 1. Univ.-Prof. Dr. Johann P. Plank 2. apl. Prof. Dr. Anton Lerf Die Dissertation wurde am 31.08.2009 bei der Technischen Universität München eingereicht und durch die Fakultät für Chemie am 09.03.2010 angenommen.
Transcript of Technische Universität München - mediatum.ub.tum.de · F Fe 2O3 S SO 3 H H2O. Tabelle 1:...

Technische Universität München
Lehrbereich Anorganische Chemie - Lehrstuhl für Bau chemie
Untersuchungen zur Kinetik grundlegender Reak-
tionsschritte bei der Hydratation von Portland- und
Hochofenzementen
Holger König
Vollständiger Abdruck der von der Fakultät für Chemie der Technischen Universi-
tät München zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften
genehmigten Dissertation.
Vorsitzender: Univ.-Prof. Dr. Klaus Köhler
Prüfer der Dissertation:
1. Univ.-Prof. Dr. Johann P. Plank
2. apl. Prof. Dr. Anton Lerf
Die Dissertation wurde am 31.08.2009 bei der Technischen Universität München
eingereicht und durch die Fakultät für Chemie am 09.03.2010 angenommen.

Seite II
Die vorliegende Arbeit entstand in der Zeit von November 2003 bis August 2009
unter der Anleitung von
Prof. Dr. Johann Plank
am Lehrstuhl Bauchemie der TU München.

Seite III
Danksagung
Ich danke auf diesem Wege Prof. Dr. Wolfgang Hiller für die interessante Aufga-
benstellung und die Aufnahme an die Fakultät. Meinem ehemaligen Arbeitgeber,
Prof. Augustin Rauen danke ich für die Ermöglichung der Arbeit bei Rohrdorfer Ze-
ment. Ihm danke ich auch für die angeregten fachlichen Diskussionen und für seine
pragmatischen technischen Hinweise.
Mein besonderer Dank gilt
Prof. Dr. Johann Plank
für die fachliche Betreuung der Arbeit und die Ermöglichung der Vollendung am
Lehrstuhl für Bauchemie. Dr. Roland Sieber möchte ich für die fachlichen Diskussio-
nen sowie die intensive organisatorische Unterstützung bei der Umsetzung danken.
Den Mitarbeitern des Lehrstuhls für Bauchemie danke ich für die sehr kollegiale
Zusammenarbeit, die fachlichen Diskussionen.
Danke möchte ich auch den Mitarbeitern von Rohrdorfer Zement für die Unterstüt-
zung bei den Versuchen und die technische Betreuung der Instrumente. Hier gilt be-
sonderer Dank Herrn Peter Christlmeier für die regelmäßige kritische Hinterfragung
der gefundenen Ergebnisse sowie Dr. Franz Koubowetz für die praktischen Erklä-
rungen der Messergebnisse.
Nicht zuletzt danke ich meiner Familie und besonders meiner Frau Inna für die für
die Geduld und die moralische und organisatorische Unterstützung.

Seite IV
Inhaltsverzeichnis
Danksagung .......................................................................................................... III
Inhaltsverzeichnis.................................................................................................. IV
Abkürzungen und Symbole ................................................................................. VIII
Spezielle Nomenklatur in der Zementchemie........................................................ IX
1 Einleitung und Schrifttum............................................................................... 1
1.1 Historischer Abriss.................................................................................... 1
1.2 Modelle zur chemischen Reaktionsgeschwindigkeit ................................. 2
1.2.1 Innere Energie, Enthalpie und Reaktionswärme................................. 2
1.2.2 Treibende Kraft chemischer Reaktionen............................................. 4
1.2.3 Grundlagen der chemischen Kinetik ................................................... 5
1.2.3.1 Kinetische Kontrolle ........................................................................ 6
1.2.3.2 Diffusionskontrolle........................................................................... 9
1.3 Hydratation von Zement ......................................................................... 11
1.3.1 Hydratation der Einzelphasen........................................................... 13
1.3.1.1 Hydratation der Calciumsilikate..................................................... 13
1.3.1.2 Hydratation von Calciumaluminaten ............................................. 16
1.3.1.3 Hydratation von Calciumferrit und –alumoferrit ............................. 18
1.3.1.4 Hydratation anderer Zementbestandteile...................................... 18
1.3.2 Hydratation der komplexen Mischung Zement.................................. 19
1.3.3 Modellierung der Hydratation von Zement ........................................ 23
2 Methoden zur Erfassung der Reaktionsgeschwindigkeit .............................. 25
2.1 Stoffmengenbestimmung........................................................................ 25
2.1.1 Chemische Methoden....................................................................... 25
2.1.2 Physikalische Methoden ................................................................... 26
2.2 Indirekte Methoden ................................................................................. 26

Seite V
2.2.1 Kalorimetrie....................................................................................... 26
2.2.2 Festigkeitsprüfung ............................................................................ 27
3 Problemstellung............................................................................................ 28
4 Analytische Methoden .................................................................................. 31
4.1 Methanol-Salicylsäure-Auszug ............................................................... 31
4.2 Borsäure-Auszug .................................................................................... 31
4.3 Röntgendiffraktometrie (XRD)................................................................. 32
4.3.1 Präparation ....................................................................................... 33
4.3.2 Einfacher Scan ................................................................................. 34
4.3.3 Zyklische Langzeitmessungen.......................................................... 34
4.3.4 Quantitative Rietveldanalyse ............................................................ 37
4.3.5 Bestimmung der Reaktionsordnung.................................................. 40
4.4 Röntgenfluoreszensanalyse (XRF) ......................................................... 40
4.5 Festigkeitsprüfungen .............................................................................. 41
4.6 Kalorimetrie ............................................................................................ 41
4.7 Erstarren................................................................................................. 45
4.8 Rasterelektronenmikroskopie ................................................................. 46
4.9 Korngrößenanalyse (Lasergranulometrie) .............................................. 46
5 Charakterisierung der Ausgangsstoffe.......................................................... 48
5.1 Oxidische Zusammensetzung der Zemente ........................................... 48
5.2 Zementzusammensetzung mittels Rietveld-Analytik............................... 50
5.3 Festigkeitsentwicklung nach DIN EN 196 ............................................... 54
5.4 Erstarren................................................................................................. 54
5.5 Kornverteilung und Feinheit .................................................................... 54
6 Ergebnisse.................................................................................................... 57
6.1 Festigkeitsentwicklung der Zemente....................................................... 57

Seite VI
6.1.1 Portlandzement CEM I 42,5 R .......................................................... 59
6.1.2 Hochsulfatbeständiger Zement CEM I 42,5 R-HS ............................ 62
6.1.3 Hochofenzement............................................................................... 63
6.2 Kalorimetrie ............................................................................................ 64
6.2.1 Normaler Portlandzement (CEM I 42,5 R) ........................................ 65
6.2.2 Hochsulfatbeständiger Zement (CEM I 42,5 R-HS) .......................... 66
6.2.3 Hochofenzement CEM III/A 32,5 N-LH ............................................. 68
6.3 Mineralogische Veränderungen.............................................................. 69
6.3.1 Portlandzement CEM I 42,5 R .......................................................... 70
6.3.1.1 Portlandit....................................................................................... 72
6.3.1.2 Alit................................................................................................. 74
6.3.1.3 Ettringit.......................................................................................... 78
6.3.1.4 Anhydrit......................................................................................... 83
6.3.1.5 Zusammenfassung CEM I 42,5 R ................................................. 83
6.3.2 Hochsulfatbeständiger Zement CEM I 42,5 R-HS ............................ 85
6.3.2.1 Portlandit....................................................................................... 85
6.3.2.2 Alit................................................................................................. 86
6.3.2.3 AFt (Eisen-Ettringit)....................................................................... 89
6.3.2.4 Anhydrit......................................................................................... 92
6.3.2.5 Zusammenfassung CEM I 42,5 R-HS........................................... 92
6.3.3 Hochofenzement............................................................................... 93
6.3.3.1 Portlandit....................................................................................... 95
6.3.3.2 Alit................................................................................................. 96
6.3.3.3 Ettringit.......................................................................................... 98
6.3.3.4 Anhydrit....................................................................................... 101
6.3.3.5 Zusammenfassung CEM III/A 32,5 N-LH.................................... 102

Seite VII
6.3.4 Qualitative Rasterelektronenmikroskopie........................................ 103
7 Diskussion der Ergebnisse ......................................................................... 107
7.1 Tricalciumsilikat (Alit) ............................................................................ 109
7.2 Portlandit .............................................................................................. 111
7.3 Ettringit (AFt)......................................................................................... 114
7.4 Anhydrit ................................................................................................ 117
7.5 Praktische Aspekte ............................................................................... 119
7.5.1 Beschleunigung der Hydratation..................................................... 119
7.5.2 Verzögerung der Hydratation.......................................................... 120
7.6 Zusammenfassung und Ausblick .......................................................... 121
7.7 Summary and outlook........................................................................... 121
8 Anhang ....................................................................................................... 123
8.1 Hydratation von Portland-Zementleim .................................................. 123
8.2 Hydratation von Leim des hochsulfatbeständigen Zementes ............... 126
8.3 Hydratation von Hochofen-Zementleim ................................................ 132
8.4 Kontrolle der XRD Resultate mit REM .................................................. 136
Abbildungsverzeichnis........................................................................................ 139

Seite VIII
Abkürzungen und Symbole
AFm Calciumaluminatferritmonosulfathydrat AFt Calciumaluminatferrittrisulfathydrat bzw. beziehungsweise °C Grad Celsius ca. circa CEM I Portlandzement CEM I-HS Hochsulfatbeständiger Zement CEM III Hochofenzement DF Druckfestigkeit d.h. das heißt f() Funktion von g Gramm % Masse-Prozent h Stunden K Kelvin kJ/kg Kilojoule je Kilogramm ln() Logarithmus naturalis von µm Mikrometer m²/g Quadratmeter je Gramm mL Milliliter MPa Megapascal mW/g Milliwatt je Gramm Pkt. Punkt REM Rasterelektronenmikroskop ESEM Environmental scanning electron microscope sin() Sinus von u.a. ‚und andere(s)’ bzw. ‚unter anderem’ vgl. Vergleiche W/Z Wasser/Zement-Verhältnis XRD Röntgendiffraktometrie (x-ray diffraction) XRF Röntgenfluoreszenz (x-ray fluorescence) z.T. zum Teil << bzw. >> ‚viel kleiner als’ bzw. ‚viel größer als’ Ø Durchmesser x bis y x ‚bis’ y ∇ Laplace-Operator (dreidimensionaler Operator) ξ Nu Nusseltzahl

Seite IX
Spezielle Nomenklatur in der Zementchemie
Die Zementbestandteile stellen zum größten Teil Minerale aus den höchstwertigen
Oxiden von Hauptgruppenelementen dar. Diese Oxide unterscheiden sich in den ver-
schiedenen Mineralen stöchiometrisch nicht wesentlich. Nur das Verhältnis der Oxide
zueinander variiert. In der Zementchemie werden daher die chemischen Formeln z.T.
in einer verkürzten Form geschrieben. Hierbei werden die Oxide der Hauptbestand-
teile durch den jeweiligen Anfangsbuchstaben des Elements ersetzt.
Es bedeuten:
C CaO
S SiO2
A Al2O3
F Fe2O3
S SO3
H H2O.
Tabelle 1: Kurzschreibweise der Oxide in der Zementchemie
Aus diesen Symbolen ergeben sich die in der Silikatchemie üblichen Summenfor-
meln. Einige für diese Arbeit relevante Minerale sind hier erläutert:
Abkürzung Chemische Formel Bezeichnung
C3S 3CaO⋅SiO2 Tricalciumsilikat
C2S 2CaO⋅SiO2 Dicalciumsilikat
C3A 3CaO⋅Al2O3 Tricalciumaluminat
C4AF 4CaO⋅Al2O3⋅Fe2O3 Tetracalciumaluminatferrit
C3A⋅3CS⋅32H [Ca6Al2(OH)12](SO4)3⋅26H2O Ettringit, Calciumaluminattrisulfathydrat
C3(A,F)⋅3CS⋅32H Calciumaluminatferrattrisulfathydrat
C3(A,F)⋅CS⋅12H Calciumaluminatferratmonosulfathydrat
CH Ca(OH)2 Calciumhydroxid, Portlandit
CxSyHz xCaO⋅ySiO2⋅zH2O Calciumsilikathydrat
CxAyHz xCaO⋅yAl3O3⋅zH2O Calciumaluminathydrat

Seite X

Seite 1
1 Einleitung und Schrifttum
1.1 Historischer Abriss
Der Ursprung des Wortes „Zement“ ist bei den alten Römern zu finden. Ihre von
den Griechen übernommene Technik des Bauens mit Gussmauerwerk, dem Vorläu-
fer unseres heutigen Betons, nannten sie „Opus Caementitium“. In Frankreich ent-
stand nach dem Abzug der Römer im frühen Mittelalter aus „Caementum“ nach ver-
schiedenen Schreibweisen das Wort „Ciment“, bezeichnete jedoch nur das aus Zie-
gelbruch hergestellte feine Ziegelmehl. Im England des 18. Jahrhunderts bezeichne-
te man Trass und Puzzolane mit „Cement“. 1796 wurde durch James Parker der
„Romanzement“, ein hydraulischer Kalk, erfunden. Joseph Aspdin entwickelte 1824
den „Portland-Cement“.
Die ersten bedeutenden wissenschaftlichen Arbeiten zur Zementtechnologie leis-
tete Smeaton um 1760, welcher die Bedeutung des Tongehaltes der Rohstoffe für
die Hydraulizität der Kalke untersuchte. Wilhelm Michaelis veröffentlichte 1869 ein
umfassendes Werk über Portlandzement. Hier beschreibt er zum ersten Mal eine
Theorie zur Erhärtung von Zement [1, 2].
Michaelis ging davon aus, dass die bei der Hydratation von Zement entstehenden
Kristalle kaum zur Festigkeitsbildung beitragen. Vielmehr konzentrierte er sich auf
das entstehende „Hydrogel“. Nach seiner Theorie läuft die Reaktion zwischen dem
Wasser und den Zementbestandteilen ohne Auflösung an der Oberfläche der Körner
ab. Es bildet sich eine gelartige Phase, welche sich durch Wasserentzug nach und
nach verdichtet. Am Ende erfolgt eine Rekristallisation.
Eine weitere frühe Theorie über die Hydratation von Zement stellte Le Chatelier
vor. Er geht im Gegensatz zu Michaelis von einer Auflösung der Zementphasen und
anschließender Kristallisation der Hydratphasen aus. Le Chatelier begründet diese
Theorie auf der Tatsache, dass die Löslichkeitsprodukte der korrespondierenden
Hydrate geringer sind als die der primären Silikate. Nach seiner Auffassung bilden
die Ausgangsminerale gesättigte Lösungen, in denen die Minerale hydratisieren, und
auf Grund der geringen Löslichkeit als Kristallkeime aus der Lösung ausfallen. Durch
diesen Prozess wird die Lösung im Verhältnis zu den Ausgangsstoffen wieder unter-
sättigt, es werden neue Teilchen gelöst. Die neu gebildeten Phasen fallen als Kris-

Seite 2
tallhydrate aus, verkleben im Verlauf des Kristallwachstums miteinander und bilden
ein festes Gerüst. Nach der Theorie von Le Chatelier ist das Erstarren des Zement-
leimes mit dem bereits zu Beginn der Zementhydratation einsetzenden Kristallwachs-
tum zu erklären.
Smeschanaya und Bajkova stellen später einen moderneren Mechanismus vor.
Nach ihrer Ansicht gliedert sich die Reaktion von Zement mit Wasser in drei Etappen:
(1) Die Vermischung von Zement und Wasser – es wird eine an Mineralien ge-
sättigte Lösung gebildet (1 bis 3 Stunden).
(2) Das Wasser reagiert unmittelbar mit der Oberfläche der Zementteilchen.
Die Hydratation verläuft ohne Auflösung. Da diese Reaktion exotherm ist,
wird in dieser Etappe Wärme freigesetzt, die Reaktionsgeschwindigkeit
nimmt zu. Auf den Oberflächen der Zementteilchen bildet sich eine gelarti-
ge Haut. Durch den Verlust der Beweglichkeit der Teilchen erstarrt das Sys-
tem (bis zu 1 Tag).
(3) Es erfolgt eine Umkristallisation mit geringer Wärmeentwicklung (bis zur
vollständigen Hydratation).
Die heutigen Vorstellungen über das Abbinden und Erhärten von Zement lehnen
sich weitgehend an die Kolloidtheorie von Michaelis an [3].
1.2 Modelle zur chemischen Reaktionsgeschwindigkeit
1.2.1 Innere Energie, Enthalpie und Reaktionswärme
Die innere Energie eines Systems U ist seine gesamte Energie, welche sich aus
der kinetischen Energie (Translation, Schwingung und Rotation), der potentiellen E-
nergie (Anziehungs- und Abstoßungsenergie) aller Teilchen des Systems abzüglich
der potentiellen und kinetischen Energie des Gesamtsystems ergibt. Messbar ist nur
die Änderung der inneren Energie ∆U = U2 – U1, wobei U2 und U1 die innere Energie
in den Zuständen 2 und 1 ist.
Wenn das System mit seiner Umgebung wechselwirkt, dabei Wärmeenergie Q
und mechanische Energie (Arbeit) A übertragen werden, dann folgt aus dem ersten
Satz der Thermodynamik, dem Gesetz von der Erhaltung der Energie, dass die vom
System abgegebene oder aufgenommene Energie in Form von Wärme oder Arbeit

Seite 3
gleich der Änderung der inneren Energie des Systems beim Übergang von einem
Zustand in den anderen ist:
∆U = Q + A Gleichung 1
Mit anderen Worten: Die Änderung der inneren Energie eines Systems, welches
aus dem Zustand 1 in den Zustand 2 übergeht, ist gleich der algebraischen Summe
aller Energien, welche mit der Umgebung ausgetauscht werden.
Für eine chemische Reaktion der allgemeinen Form
aA + bB � cC + dD Gleichung 2
ist der thermische Effekt Q bei der Temperatur T die Wärmeenergie, die bei der
Wechselwirkung der Reaktionspartner A und B im stöchiometrischen Verhältnis unter
Bildung der Produkte C und D freigesetzt wird. Die Ausgangsstoffe und Reaktions-
produkte haben die gleiche Temperatur T.
Der thermische Effekt der Reaktion bei konstantem Volumen ist QV, bei konstan-
tem Druck Qp. Im Laufe der chemischen Reaktion wird Arbeit vorzugsweise gegen
die Kraft des äußeren Druckes verrichtet, der von der Änderung des Volumens des
Systems abhängt. Für einen isochoren Prozess, bei dem sich das Volumen nicht än-
dert (V = const.), ist A = 0. Folglich gilt:
∆U = QV Gleichung 3
Der thermische Effekt einer Reaktion bei konstantem Volumen und Temperatur
entspricht der Änderung der inneren Energie des Systems im Laufe der Reaktion.
Für isobare Prozesse gilt
A = -p∆V = -p(V2-V1) Gleichung 4
und folglich gilt
∆U = Qp - p∆V
oder

Seite 4
U2 - U1 = Qp – p(V2 - V1)
oder
Qp = (U2 + pV2) - (U1 + pV1) Gleichung 5
Der Term U + pV wird als Enthalpie bezeichnet. Sie ist eine Funktion des Zustan-
des und hat die Einheit von Energie.
Der thermische Effekt einer Reaktion bei konstantem Druck und Temperatur ist al-
so gleich der Änderung der Enthalpie des Systems im Laufe der Reaktion.
Qp und QV sind Funktionen des Zustandes des Systems. Für eine exotherme Re-
aktion gilt QV < 0; ∆U < 0; Qp < 0; ∆H < 0. Für eine endotherme Reaktion gilt QV > 0;
∆U > 0; Qp > 0; ∆H > 0.
Aus den vorangegangenen Gleichungen folgt
∆H = ∆U + p∆V, Gleichung 6
Qp = QV + p∆V. Gleichung 7
Bei Prozessen, an denen keine gasförmigen Stoffe teilnehmen, sind die Größen
von ∆V und p∆V sehr klein. Daher sind Qp und QV, ∆H und ∆U sehr nah.
Aus der Zustandsgleichung von Gasen pV = nRT folgt, dass p∆V = ∆nRT. Das
bedeutet, dass für Reaktionen zwischen idealen Gasen gilt:
Qp = QV + ∆nRT Gleichung 8
Dabei bedeutet ∆n die zahlenmäßige Differenz zwischen den Reaktionsprodukten
und den Ausgangsstoffen. [4]
1.2.2 Treibende Kraft chemischer Reaktionen.
Aus den Ausgangsstoffen entstehen unter bestimmten Bedingungen in einer che-
mischen Reaktion neue Stoffe mit veränderten spezifischen Eigenschaften [5, 6]. Die
erste Bedingung für eine chemische Reaktion zwischen zwei oder mehreren Teilchen

Seite 5
ist die Kollision der Teilchen und somit die Bewegung der Teilchen im Reaktions-
raum. Aus der irreversiblen Thermodynamik ist bekannt, dass die treibende Kraft für
die Bewegung der Teilchen
Kv = vµ∇− = vNlnRT ∇⋅− Gleichung 9
ist, solange die Teilchen nicht miteinander reagieren.
Teilchen reagieren chemisch miteinander, wenn bei der Reaktion Energie frei wird.
In diesem Fall ist die Änderung der freien Energie negativ. Sie berechnet sich wie
folgt:
STHG ∆⋅−∆=∆ Gleichung 10
Hierbei bedeuten
∆G die Änderung der Gibbs’schen freien Enthalpie
∆H die Reaktionsenthalpie
∆S die Reaktionsentropie
T die absolute Temperatur.
1.2.3 Grundlagen der chemischen Kinetik
Es gibt vier grundsätzliche kinetische Situationen:
a) Bei homogenen Reaktionen verlassen einzelne Teilchen infolge thermi-
scher Aktivierung ihre Positionen und hinterlassen Vakanzen, so genannte
Frenkel-Punkt-Defekte.
b) Die Konzentration von Defekten ist nach der Gibbs’schen Regel abhängig
vom thermodynamischen Zustand des Systems. So ist ihre Konzentration
an zwei gegenüberliegenden Oberflächen, bei sonst gleichen Bedingungen,
im Falle von thermodynamischen Unterschieden an den Oberflächen eben-
falls unterschiedlich. Mobile Defekte werden sich ihrem Konzentrationsgra-
dienten folgend in Bewegung setzen (Diffusion).
c) Eine Oberfläche teilt die Phasen α und β. Die Reaktion kann durch den
Fluss von Teilchen durch die stationäre Oberfläche stattfinden. Es ist aber
auch möglich, dass die Oberfläche sich zwischen den Phasen bewegt.

Seite 6
d) Im Falle der chemischen Verbindung der Phasen A und B zu AB (heteroge-
ne Reaktion) können bei Immobilität des Reaktionsproduktes AB neue
Phasengrenzen zwischen A und AB und zwischen AB und B entstehen. Die
Reaktion wird nur dann weiter ablaufen, wenn zumindest eine der Phasen
A und B durch die Phase AB diffundieren kann [7].
Für Reaktionen, die in homogenen Systemen stattfinden, entspricht die Ge-
schwindigkeit einer chemischen Reaktion der Anzahl der Akte der Wechselwirkungen
je Zeiteinheit und je Volumeneinheit bzw. wird die Kinetik durch die Anzahl der
Wechselwirkungen pro Flächeneinheit der Grenzfläche zwischen den Phasen be-
stimmt, bei Reaktionen in heterogenen Systemen.
Auf Grund dieser Sachverhalte ergeben sich für den Verlauf einer chemischen
Reaktion zwei Möglichkeiten der Limitierung der Reaktionsgeschwindigkeit:
1.2.3.1 Kinetische Kontrolle
In einem homogenen System liegen die Ausgangsstoffe gleichmäßig verteilt bzw.
gut durchmischt vor. Es stehen viele Kontaktpunkte zwischen den Reaktionspartnern
für die Wechselwirkung zur Verfügung. Bei der Reaktion von Flüssigkeiten oder
Feststoffen wird der Fortschritt der Reaktion durch die Konzentration der Ausgangs-
stoffe sowie die Temperatur bestimmt.
Für die allgemeine Reaktion zweier Komponenten A und B
A + B � ... Gleichung 11
ergibt sich die Reaktionsgeschwindigkeit als
v = k � [A]a � [B]b Gleichung 12
Im Falle der Beteiligung von Gasen an der Reaktion bestimmen die partialen Drü-
cke der gasförmigen Reagenten die Reaktionsgeschwindigkeit.
Die Konstante k ist dabei spezifisch für die jeweilige chemischen Reaktion selbst
und von der Temperatur T abhängig:
k = RT
E
0
A
ek∆
−⋅ Gleichung 13

Seite 7
Diese Gleichung wird als Arrhenius-Gleichung bezeichnet. In ihr bedeutet ∆EA die
Aktivierungsenergie. k0 ist die Konstante im Grundzustand.
Nach Gleichung 2 ist die Reaktionsgeschwindigkeit abhängig von der Konzentrati-
on der Ausgangsstoffe. Je nach Verfügbarkeit der Reaktionspartner (Aktivität, Über-
schuss, Medium, Katalysator,...) kann das Zeitgesetz folgendes Aussehen haben:
)A(ckv ⋅= Gleichung 14
)B(ckv ⋅= Gleichung 15
)B(c)A(ckv ⋅⋅= Gleichung 16
)A(ckv 2⋅= Gleichung 17
)B(c)A(ckv 2/1⋅⋅= Gleichung 18
)B(c)A(ckv nm ⋅⋅= Gleichung 19
Den Exponenten, mit dem die Konzentration eines Reaktionspartners im Zeitge-
setz auftritt, nennt man die Ordnung der Reaktion in Bezug auf diesen Partner. Hängt
die Geschwindigkeit einer Reaktion nicht von der Konzentration eines Stoffes ab,
spricht man von einer Reaktion 0-ter Ordnung in Bezug auf diesen Stoff [8].
1.2.3.1.1 Reaktionen erster Ordnung und deren Auswertung
Für den häufigsten Reaktionstyp, die Reaktion 1. Ordnung bezüglich A, nimmt das
Zeitgesetz folgende Form an:
)A(ckdt
)A(dcv ⋅=−= Gleichung 20
Zum Zeitpunkt t=0 liegt die Anfangskonzentration c0(A) vor; zum Zeitpunkt t liegt
die Konzentration c(A) vor. Unter diesen Randbedingungen lässt sich die vorliegende
Differentialgleichung durch Trennung der Variablen und anschließende Integration
leicht lösen:
∫ ∫=−)A(c
)A(c
t
00
dtkdt
)A(dc Gleichung 21
Integration zwischen den angegebenen Grenzen ergibt:

Seite 8
tk)A(cln)A(cln 0 ⋅=+− Gleichung 22
oder
tk)A(cln)A(cln 0 ⋅−=− Gleichung 23
Daraus ergibt sich:
tk
0
e)A(c)A(c ⋅−= bzw. tk
0 e)A(c)A(c ⋅−⋅= Gleichung 24
Die Abnahme der Konzentration des Reaktionspartners A mit der Zeit verläuft also
nach einer Exponentialfunktion.
1.2.3.1.2 Reaktionen nullter Ordnung und deren Auswertung
Bei einer Reaktion nullter Ordnung hängt die Reaktionsgeschwindigkeit nicht von
der Konzentration der Partner ab. Es gilt folgendes Zeitgesetz:
kdt
)A(dcv == Gleichung 25
Nach Trennung der Variablen wird zwischen den Grenzen t = 0 bis t = t und c0(A)
bis c(A) integriert:
∫∫ =−t
0
)A(c
)A(c
dtk)A(dc0
Gleichung 26
Man erhält:
tk)A(c)A(c 0 ⋅−=− bzw. tk)A(c)A(c 0 ⋅−= Gleichung 27
Diese Gleichung stellt eine Geraden-Gleichung mit dem Achsenabschnitt c0(A)
und der Steigung –k dar.

Seite 9
1.2.3.1.3 Reaktionen zweiter Ordnung und deren Auswertung
Für eine Reaktion 2. Ordnung bezüglich A nimmt das Zeitgesetz folgende Form
an:
)A(ckdt
)A(dcv 2⋅=−= Gleichung 28
Trennung der Variablen und nachfolgende Integration führt zu folgenden Bezie-
hungen:
∫∫ =−t
0
)A(c
)A(c
dtk)A²(c)A(dc
0
Gleichung 29
tk)A(c
1)A(c
1
0
⋅=− oder tk)A(c
1)A(c
1
0
⋅+= Gleichung 30
1.2.3.2 Diffusionskontrolle
Diffusion ist die Bezeichnung für jeden mit einem Masse- und/oder Ladungstrans-
port verbundenen physikalischen Ausgleichsprozess, in dessen Verlauf Teilchen (A-
tome, Moleküle, Aerosol oder Kolloidteilchen, Ionen und andere Ladungsträger) auch
gegen entgegenwirkende Kraftfelder (Schwerkraft, elektrische Felder) infolge ihrer
Wärmebewegung auf unregelmäßigen Zickzackwegen von Orten höherer Teilchen-
dichte oder Konzentration zu solchen niederer Teilchendichte oder Konzentration
gelangen, so dass allmählich ein Dichte- und Konzentrationsausgleich erfolgt.
Die Diffusion wird allgemein durch die beiden 1855 von A. Fick aufgestellten Ge-
setze beschrieben, die bei konstanter Temperatur und konstantem Druck gelten.
1. Ficksches Gesetz: Zwischen der räumlichen Änderung (dem Gradienten) der
Teilchendichte n und der Diffusionsdichte jedes als Diffusionsstrom bezeichneten
Teilchenflusses gilt die Beziehung
j = -D grad n Gleichung 31
Dennoch erfolgt der Diffusionsstrom in Richtung der abnehmenden Dichte oder
Konzentration. Der als Diffusionskoeffizient bezeichnete Proportionalitätsfaktor D hat

Seite 10
die Größenordnung 1 cm²/s für Gase, 10-4 cm²/s für Flüssigkeiten und 10-9 cm²/s für
Feststoffe.
Aus dem 1. Fickschen Gesetz und der Kontinuitätsgleichung (Erhaltung der Teil-
chenzahl)
div j + tn
∂∂
= 0 Gleichung 32
folgt als
2. Ficksches Gesetz die partielle Differenzialgleichung 2. Ordnung
tn
∂∂
= D ∆ n Gleichung 33
Ihre jeweilige Lösung beschreibt die Abhängigkeit vom Ortsvektor und der Zeit t (∆
= Laplace-Operator). Für den eindimensionalen Fall ergibt sich demnach
tn
∂∂
= D xn
2
2
∂∂
. Gleichung 34
Bei konstantem Diffusionskoeffizienten lässt sich für regelmäßige geometrische
Figuren die Gleichung lösen. Man erhält für die Verschiebung der Phasengrenze
zwischen den Phasen ξ ~ t . Somit ist auch die Kristallitgröße proportional zu t
[9].
In einem heterogenen System liegen die Ausgangsstoffe räumlich getrennt von-
einander vor. Die Reaktion findet nur an der Grenzfläche zwischen den Reaktions-
partnern statt. Um die chemische Reaktion fortschreiten zu lassen, müssen unauf-
hörlich frische Reaktionspartner zu den Kontaktstellen hin bzw. Reaktionsprodukte
von den Kontaktstellen weg transportiert werden. Die entsprechenden Teilchen dif-
fundieren durch eine oder mehrere stationäre Phasen. Je nach Zusammensetzung,
Struktur und Dicke dieser stationären Phasen ist die Transportgeschwindigkeit unter-
schiedlich. Sobald die chemische Reaktion an den Kontaktstellen schneller verläuft
als der Transport der beteiligten Stoffe erfolgt, spricht man von einer Limitierung der
Gesamtreaktion durch Diffusion [10].

Seite 11
Einen besonderen Platz nehmen mikroheterogene Reaktionen von Reaktionspart-
nern mit geringer Teilchengröße ein. Mikroheterogene Reaktionen zeichnen sich
deshalb durch intensive Diffusion aus. Dem Ausdruck für die Stoffübergangszahl
dD'Nu ⋅=β Gleichung 35
entnimmt man, dass in den Fällen, wo die Konvektion von untergeordneter Bedeu-
tung und daher die Nusseltzahl angenähert konstant ist, die Intensität des Stofftrans-
portes umgekehrt proportional der Teilchengröße ist.
Man kann die Reaktionen in mikroheterogenen Systemen in zwei Gruppen eintei-
len: Zur ersten Gruppe gehören solche Prozesse, bei denen die Mikrodiffusion des
reagierenden Stoffes an die Oberfläche der dispergierten Teilchen eine Rolle spielt;
die zweite Gruppe erfasst dagegen Prozesse, bei denen wenigstens eine Reaktions-
komponente aus einer anderen Phase herangeführt wird, so dass die Makrodiffusion
im Gesamtsystem für den Reaktionsablauf bestimmend ist [11], [12].
1.3 Hydratation von Zement
Zement ist eine Mischung aus verschiedenen Mineralien und glasartigen amor-
phen Bestandteilen. Die Teilchen besitzen sehr unterschiedliche Größen und For-
men. Es liegen sowohl gut ausgebildete Kristallite, auf andere Phasen aufgewachse-
ne Kristalle, kleinere Kristallitkeime wie auch schlecht oder nicht auskristallisierte
Phasen vor. Zement stellt demnach ein heterogenes System dar.
Unter der Hydratation von Zement versteht man die chemische Reaktion der in
Zement enthaltenen Stoffe (zumeist Mineralien) mit Wasser unter Bildung von Hydra-
ten. Die Reaktion des Gesamtsystems gliedert sich auf in viele Einzelreaktionen,
welche zum Teil nebeneinander, aber auch nacheinander ablaufen. Die im Zement
enthaltenen Phasen reagieren jede für sich mit Wasser und den im Wasser gelösten
Stoffen. Die Reaktionsprodukte dieser Reaktionen reagieren ihrerseits wiederum mit
Wasser, aber auch untereinander. Werden Wasser und Zement miteinander ver-
mischt, entsteht ein neues heterogenes System aus einer flüssigen (Wasser) und
vielen festen Phasen (Zementbestandteile). Durch Reaktion der einzelnen Phasen
miteinander verändert sich das System mit der Zeit. Die Konzentrationsverhältnisse
verschieben sich. Wärme wird freigesetzt und Feuchtigkeit wird entzogen. Gleichzei-

Seite 12
tig verändern sich auch die makroskopischen mechanischen Eigenschaften [13]. Bei
der Hydratation der Zementmineralien entstehen Salzhydrate, basische Salze, einfa-
che und komplexe Hydroxide und andere Verbindungen. Alle diese Stoffe enthalten
Wasser, welches in Form von Hydratwasser oder als OH--Ionen gebunden vorliegt
[14]. Der Zement reagiert sofort bei Kontakt mit dem Anmachwasser. Die Reaktion
läuft an der Oberfläche der Zementkörner ab. Folgender allgemeiner Mechanismus
der Hydratation ist bekannt und mit wissenschaftlich-technischen Methoden nachge-
wiesen:
(1) Das Wasser wird an der Oberfläche der Körner adsorbiert und in die Ionen H+
und OH- geteilt.
(2) Diese Ionen und ganze Wassermoleküle reagieren mit aktiven Zentren der
Kornoberfläche (nur Defekte in der Kristallstruktur sind reaktionsfähig) und bilden
Bindungen durch Chemosorption aus.
(3) Ionen werden ausgetauscht - Metallionen gehen in Lösung, Wasserstoffionen
werden gebunden. Dadurch gelangen Ca2+, Al3+und Mg2+ in das die Zementkörner
umgebende Wasser. Es bilden sich erste Kristallisationskeime der Hydratphasen.
(4) Weitere Ionen gehen in Lösung: Unter anderem Ca2+, H3SiO4- und H2SiO4
2-,
was zur Übersättigung der wässrigen Lösung führt.
(5) Aus der übersättigten Lösung kristallisieren die Hydratphasen. Man spricht
von topotaktischem Wachstum.
Die Reaktionen, die zur Bildung der Hydratphasen führen, lassen sich nicht durch
einfache stöchiometrische Gleichungen beschreiben, da vielfach Festkörperprodukte
unterschiedlicher Zusammensetzung entstehen bzw. die Umsetzungen über Zwi-
schenstufen verlaufen. Darüber hinaus hängen Art und Menge der Hydratationspro-
dukte von der chemisch-mineralogischen Zusammensetzung des Zements, die in
einem relativ breiten Bereich schwanken kann, sowie unter Umständen auch von
sogenannten latent hydraulischen Zumahlstoffen wie zum Beispiel Flugasche ab.
Daraus ergibt sich in Summe ein außerordentlich komplexes Reaktionsgeschehen,
das sich einer einfachen Beschreibung entzieht.
Bei der Hydratation von Zementen entstehen vorwiegend Teilchen mit kolloidalen
Eigenschaften (Teilchengröße im Bereich von 10-7 bis 10-9 m) [15]. Hierauf beruht die
Festigkeitsbildung der Zemente. Weniger Bedeutung für die Festigkeitsbildung haben

Seite 13
die makrokristallin gebildeten Phasen des Zementsteines wie Gips und Portlandit,
[16], [17].
1.3.1 Hydratation der Einzelphasen
Industriell hergestellter Portlandzementklinker besteht hauptsächlich aus Alit (mit
Fremdionen dotiertes Tricalciumsilikat, 3CaO�SiO2 = C3S), Belit (mit Fremdionen do-
tiertes Dicalciumsilikat, 2CaO�SiO2 = C2S), Aluminat (Tricalciumaluminat, 3CaO�SiO2
= C3A) und Ferrit (Tetracalciumaluminatferrit, 2CaO�(Al2O3)x�(Fe2O3)1-x = C4AF bzw.
C2(A,F)).
Im Portlandzement ist neben Klinker noch Calciumsulfat in Form von Gips, An-
hydrit und/oder Halbhydrat als Erstarrungsregler enthalten [18]. Daneben können
unterschiedliche Gehalte an Alkalisulfaten z.B. K2SO4 vorliegen.
1.3.1.1 Hydratation der Calciumsilikate
Die wesentliche Hydratphase, die bei der Hydratation von auf Portlandzementklin-
ker basierenden Zementen (Ausnahme Tonerdezement) entsteht, und auf der in ers-
ter Linie die Festigkeit des gebildeten Zementsteins beruht, ist die Calciumsilikathyd-
ratphase CSH. Calciumsilikathydrate entstehen durch Reaktion des Tricalciumsilikats
C3S bzw. des Dicalciumsilikats C2S mit dem zugesetzten Wasser.
2C3S + 6H � C3S2H3 + 3CH Gleichung 36
2C2S + 4H � C3S2H3 + CH Gleichung 37
Die in den Gleichungen angegebene chemische Zusammensetzung der Calcium-
silikathydrate ist lediglich beispielhaft und kann je nach Reaktionsbedingungen stark
schwanken. So hängt z.B. der Calciumgehalt der Hydratationsprodukte von der
Menge an zugefügtem Anmachwasser ab. Je mehr Wasser zum Anmachen benutzt
wurde, um so geringer ist der Calciumgehalt der Hydrate. Darüber hinaus hängt die
Zusammensetzung der Hydratphasen von einer Anzahl weiterer Einflussgrößen ab,
von denen die Temperatur, die Mahlfeinheit und natürlich die Zusammensetzung des
Zements die wichtigsten sind. Durch Austausch von H2O durch D2O fanden Thomas
et al. bei Portlandzementleimen mit einem Wasser/Zement-Verhältnis von 0,35 die
Summenformel für CxSyHz nach 28 Tagen Hydratation als C1,7SH2,1 [19], [20]. Ab-

Seite 14
weichend hiervon fanden Allen et al., dass die Menge gebundenes Wasser nur 1,8
Mol beträgt [21]. Andere Autoren ermittelten für die Zusammensetzung eines Calci-
umsilikathydrats, hergestellt aus reinem C3S bei einem Wasser/C3S-Verhältnis von
0,45, die Bruttoformel C3S2H3 [22]. Auf Basis verschiedenster experimenteller Me-
thoden wurden ebenso verschiedene Modelle zur Beschreibung der Reaktionsabläu-
fe und der Strukturbildung der CSH-Phasen aufgestellt (z.B. Powers - Wasserbin-
dung – Kolloid-Theorie, 1960; Taylor – Röntgendiffraktometrie, TGA – Tobermo-
rit/Jennite; Feldmann-Sereda – Stickstoff-Sorption, Hydratationsgrad – Schichten,
1970; Wittmann – Hydratationsgrad – Kolloid-Theorie, 1979) [23]. Nach dem heuti-
gen Kenntnisstand lässt sich die Hydratation von reinem C3S allgemein so formulie-
ren [24]:
3CaO�SiO2 + xH2O � yCaO�SiO2�(y-(3-x))H2O + (3-y)Ca(OH)2 Gleichung 38
Berliner et al. haben an synthetischem C3S Untersuchungen zum Bindungsgrad
des Anmachwassers bzw. dem Reaktionsgrad von C3S in Abhängigkeit von der Zeit
durchgeführt. Sie konnten nachweisen, dass bis ca. 15 Stunden nach dem Anmi-
schen die Reaktion durch das räumliche Wachstum der CSH-Phasen und somit kine-
tisch kontrolliert wird. Ca. 20 Stunden nach Reaktionsbeginn wird dann die Wasser-
bindung und damit die Umsetzung von C3S durch die Diffusion der Ausgangsstoffe
durch eine Schicht aus Reaktionsprodukten begrenzt [25], [26].
Nach neueren Erkenntnissen entsteht bei der Hydratation von Calciumsilikaten ein
äußeres und ein inneres Hydratationsprodukt. Während der frühen Hydratation bildet
sich CSH aus der Lösung. Es füllt die wassergefüllten Poren zwischen den festen
Teilchen der Zementpaste. Dieses äußere Hydratationsprodukt hat eine höhere Po-
rosität als das später entstehende innere Hydratationsprodukt. Letzteres entsteht
durch Diffusion von Wasser in das Innere der Calciumsilikatpartikel. Es entsteht an
der Stelle der Ausgangsstoffe. Es nimmt die Form der Calciumsilikatpartikel an und
bildet eine Hydratationsbarriere, durch die das Wasser hindurch diffundiert.
Bei den für die Herstellung von Mörtel und Beton üblichen Wassermengen
(W/Z = 0,3...0,6) liegt das Molverhältnis m (CaO/SiO2) bei 2,0 und darüber. Es ent-
stehen faserige Kristalle (sogenannte CSH(II)-Phasen). Bei m-Werten von 0,5 bis 1,5
bilden sich plättchenförmig kristallisierende Calciumsilikathydrate (CSH(I)-Phasen).
Die Bildung von CSH-Phasen mit geringem Calciumgehalt erfolgt durch Zerfall kalk-

Seite 15
reicher (CaO-gesättigter) Silikate in kalkärmere unter Abspaltung von Wasser und
Ca(OH)2. Die Struktur der im Zementstein entstehenden CxHySz-Phasen ist nicht ein-
heitlich. Taylor beschreibt die Hydrate der Calciumsilikate als ungeordnete Schicht-
strukturen aus Tobermorit und Jennite [27]. Die exotherme Umsetzung des CaO mit
Wasser unter Bildung von Calciumhydroxid ist maßgeblich verantwortlich für die
Hydratations- oder Abbindewärme. Sie ist bei der Umsetzung von C2S mit Wasser
wegen des geringeren Anteils an frei werdendem Ca(OH)2 deutlich geringer als bei
der Hydratation des C3S [28 ], [29], [30].
Die Reaktionswärme der Hydratation des C3S beträgt bis zu 500 kJ/kg. Bei unge-
nügender Wärmeableitung (z. B. in massiven Bauteilen) kann es dadurch zu einer
starken Erhitzung und somit zu thermischen Spannungen kommen. Bei normaler
Temperatur und einem Wasser/C3S-Verhältnis von 0,5 entsteht zum größten Teil
C2SH2. Bei erhöhten Konzentrationen von Alkaliionen (Na+ und K+) erhöht sich die
Basizität der Lösung, das Löslichkeitsprodukt für Ca(OH)2 wird verringert - es bildet
sich CSH(II). Dagegen reagiert C3S mit Wasser bei erhöhter Temperatur (80 bis 120
°C) zu α-C2SH(B). [15], [25], [31], [32], [33].
Die Reaktionswärme der Hydratation von C2S beträgt bis zu 250 kJ/kg. C2S oder
Belit reagiert mit Wasser unter Bildung von CSH(I). Da bei dieser Reaktion auch sehr
viel Calciumhydroxid gebildet wird, erhöht sich das C/S-Verhältnis in den Hydratati-
onsprodukten von 1 auf 1,7; bei erhöhter Temperatur erreicht das Verhältnis C/S in
den Hydraten Werte bis 1,7, es bildet sich C1,85 bis 2SH1.8. Im Laufe der Hydratation
von C2S bildet sich um die Belitkristalle eine dünne aber sehr dichte Haut aus Reak-
tionsprodukten. Dadurch wird die Reaktion stark gebremst und verläuft langsamer als
die Hydratation von Alit.
Ein weiterer Grund für die geringere Hydratationsneigung ist eine gegenüber Alit
geringere Dichte von Defekten in der Kristallstruktur des Belits. Durch Einbindung
weiterer Fremdionen (z. B. Ba2+, P5+, Cr3+, Fe3+ oder Alkalien) kann aber die Anzahl
von Defekten und somit die Aktivität angehoben werden.
Die unterschiedliche Zusammensetzung der Hydratationsprodukte von Alit und
Belit beeinflusst auch die erreichten Festigkeiten. So ist die Härte von Alitze-
mentstein und die von Belitzementstein, hergestellt bei gleichem Wasser/Silikat-
Verhältnissen, im Alter von 28 Tagen trotz unterschiedlicher Hydratationsgrade fast

Seite 16
gleich. Dadurch erklärt sich, warum kalkärmere, also belitreichere Zemente eine
stärkere Nacherhärtung, d. h. Festigkeitszuwächse nach der Normerhärtung von 28
Tagen, aufweisen. [34]
In kalorimetrischen Untersuchungen zeigten Ludwig und Singh [35], dass die
Hydratation in mehreren Stadien abläuft und dabei der geschwindigkeitsbestimmen-
de Schritt in der Reaktion wechselt. Nur in sehr frühen Abschnitten, speziell in der
Akzelerations-Periode (9 h nach dem Anmachen bei 5 °C bzw. 2 h bei 45 °C) konnte
der Mechanismus eindeutig als Reaktion 1. Ordnung bestimmt werden. Für diesen
Abschnitt wird eine Aktivierungsenergie von 54,4 kJ/mol angegeben. Später geht der
Mechanismus in eine durch Diffusion kontrollierte Reaktion über. Für diesen Ab-
schnitt wird eine Aktivierungsenergie von 17,2 kJ/mol angegeben.
In den ersten Phasen, speziell in der Akzelerationsperiode werden vor allem Pha-
sen aus den Calciumsilikaten in das Porenwasser gelöst. Aus der Porenlösung fallen
zwischen den Klinkerteilchen CSH-Phasen aus (äußeres Hydratationsprodukt). So-
bald die Poren zwischen den Klinkerteilchen mit Hydratphasen gefüllt sind, beginnt
Wasser durch Diffusion kontrolliert immer weiter in das Innere der verbliebenen aus-
gelaugten Struktur der Calciumsilikate einzudringen, das Mineral hydratisiert von in-
nen (inneres Hydratationsprodukt). Durch die unterschiedlichen Löslichkeiten von
Ca2+ und Kieselsäure werden diese ungleichmäßig in äußeres und inneres Hydrata-
tionsprodukt eingebaut. Das C/S-Verhältnis des inneren Hydratationsprodukts ist
deutlich geringer als beim äußeren Hydratationsprodukt.
Nach Paulini [36] wird in der Ruheperiode Volumenarbeit verrichtet, das System
quillt, wobei Wärme aufgenommen wird, also im Gegensatz zu den kalorimetrischen
Untersuchungen anderer Autoren die Wärmefreisetzung nicht gegen 0 zurückgeht,
sondern Wärme vom System aufgenommen wird. Paulini begründet diesen Effekt mit
Lösungsvorgängen, bei denen Wasser zwischen die Kristallgitter der Ausgangsstoffe
eingelagert wird.
1.3.1.2 Hydratation von Calciumaluminaten
C3A reagiert sehr aktiv mit Wasser unter Bildung von Calciumaluminathydraten
verschiedener Zusammensetzung. Zu Beginn der Reaktion läuft folgende Reaktion
ab, bei der C3A in eine kalkärmere und eine kalkreichere Hydratphase reagiert:

Seite 17
2C3A + 27 H2O � C2AH8 + C4AH19 + Q Gleichung 39
Die Reaktionswärme der Umwandlungen nach dieser Gleichung beträgt bis zu
1200 kJ/kg. Die Umsetzung erfolgt nicht immer einheitlich. So ist die Bildung von
C4AH19 in Verbindung mit Al(OH)3 (Gibbsit) beobachtet worden [37]. Die sich bilden-
den Kristalle der Aluminathydrate sind instabil und gehen in jedem Fall in die kubi-
sche (stabile) Form des C3AH6 (Katoit) über. Die Hülle aus plättchenförmigen Hydra-
taluminaten C2AH8 und C4AH19 ist sehr durchlässig und behindert das Vordringen
des Wassers kaum. Daher werden mit reinem C3A nach einem Tag Hydratationsgra-
de von 70 bis 80% erreicht. Bei erhöhter Temperatur bildet sich aus C3A und Was-
ser sofort kubisches C3AH6 ohne den Umweg über die schichtförmig aufgebauten
Zwischenprodukte. Durch die Temperaturerhöhung wird die Rekristallisation der A-
luminathydrate begünstigt, was zu Festigkeitsverlusten führt.
Bei der Hydratation eines C3A - Gips - Gemisches kann in den ersten 10 Minuten
nach der Wasserzugabe immer die Bildung von nadelförmigen Ettringitkristallen nach
folgender Gleichung beobachtet werden:
C3A + 3CSH2 + 26 H2O � C3A⋅3CS⋅32H (Ettringit) Gleichung 40
Ist die Sulfatkonzentration geringer als 0,889 g SO3/g C3A, so bildet sich Monosul-
fat:
C3A + CSH2 + 10 H2O � C3A⋅CS⋅12H Gleichung 41
Erst bei Unterschreitung des für diese Reaktion notwendigen stöchiometrischen
Verhältnisses von 0,296 g SO3/g C3A bilden sich entweder gemischte Hydroxidsul-
fathydrate z.B. der Zusammensetzung Ca4Al2O6(OH)0,5(SO4)�xH2O, oder die reinen
Aluminathydrate [37], [38].
Auf der Bildung von Ettringit beruht die Regulierung des Erstarrens von Zement
(sogenanntes Rücksteifverhalten) mittels der Sulfatzugabe. Durch die schnelle Reak-
tion des C3A mit Gips und Wasser werden die Klinkerteilchen mit einer Ettringit-
schicht bedeckt und an der Hydratation gehindert. Erst mit der Zeit wird diese Hülle
brüchig, so dass die Reaktion fortlaufen kann. Neuere Untersuchungen mit dem E-
SEM zeigen diese Erscheinung nicht. Vielmehr wachsen nur aus den C3A-Phasen

Seite 18
Ettringitkristalle. Das heißt, nur C3A wird in seiner Hydratation gehemmt. Nachdem
alles Sulfat aus der Lösung gebunden ist, beginnt der Ettringit sich in Monosulfat um-
zubilden [39], [40], [41].
In Untersuchungen zur Reaktion von reinem C3A mit Gips fand Tenoutasse [42],
dass die Hydratation des Calciumaluminats durch Diffusion limitiert wird. Begründet
wird diese Aussage durch die Temperaturabhängigkeit der thermischen Effekte und
der Erfüllung der Abhängigkeit gemäß der Arrhenius-Gleichung. Es wird eine Aktivie-
rungsenergie von 50 kJ/mol für das System C3A + CaSO4*2H2O angegeben.
1.3.1.3 Hydratation von Calciumferrit und –alumofer rit
Die Ferritphase liegt im Zement als Mischkristall mit den beiden Grenzzusammen-
setzungen C3A bzw. C2F vor. Die tatsächliche Zusammensetzung wird deshalb am
besten durch die Formel C2(A,F) wiedergegeben. Die für die Ferritphase häufig ver-
wendete Bezeichnung C4AF gibt somit nur einen Sonderfall wieder, bei dem exakt
die Hälfte aller Al3+-Gitterplätze durch Fe3+ besetzt ist.
Die Struktur der Calciumalumoferrite ähnelt derjenigen der Calciumaluminate. Ein-
zelne Gitterpositionen des Al2O3 sind jedoch durch Fe2O3 besetzt. Dadurch sind die
Abmessungen der Elementarzelle und die Gitterenergien verändert. Die Hydratation
dieser Minerale verläuft ähnlich derjenigen der Calciumaluminate. Tetracalciumalu-
minatferrit, C4AF, setzt sich allerdings deutlich langsamer mit Wasser um als C3A,
bildet aber ähnliche Hydratationsprodukte. Die Hydratation von Calciumaluminatferrit
wird sehr stark von Zusätzen bzw. der Zusammensetzung der wässrigen Lösung be-
stimmt. In ihnen ist das Al2O3 teilweise durch Fe2O3 ersetzt [39], [43], [44], [45], [46].
1.3.1.4 Hydratation anderer Zementbestandteile
Neben den Hauptphasen des Klinkers kommt noch eine Reihe weiterer Phasen im
Zement vor, die mit Wasser reagieren. Im Klinker liegen zum Teil geringe Mengen
freien Calcium- und Magnesiumoxides vor. Diese Oxide bilden bei Kontakt mit Was-
ser in sehr kurzer Zeit Hydroxide. Im Zement enthaltene Salze, sowohl aus dem Klin-
ker (Alkalisalze) als auch aus dem Sulfatträger (Calciumsulfat) werden in Wasser
gelöst und bei Übersättigung als Hydrate der Salze ausgefällt. Zusätze, wie glasige
Hochofenschlacke, puzzolanische Flugasche u.a. reagieren mit Wasser allein kaum
[37].

Seite 19
1.3.2 Hydratation der komplexen Mischung Zement
Bei der zeitgleichen und räumlich gemeinsamen Reaktion mehrerer Zementbe-
standteile können sich die Reaktionen gegenseitig beeinflussen. Nach dem Prinzip
von Le Chatelier können sich die Reaktionen durch Bildung gleicher Reaktionspro-
dukte gegenseitig verzögern. Durch den Verbrauch von Reaktionsprodukten durch
eine Reaktion oder eine Folgereaktion können andere Reaktionen beschleunigt wer-
den.
Bei der Hydratation der komplexen Mischung Zement haben beide Erscheinungen
ihren Platz. Alle Calciumsilikate bilden bei der Reaktion mit Wasser Calciumhydroxid.
Gleichzeitig spielt die Partikelform und -größe der Zementbestandteile eine wesentli-
che Rolle für die Geschwindigkeit der Hydratation [47]. Da die wässrige Phase
schnell mit Calciumhydroxid gesättigt ist, konkurrieren die Hydratations-Reaktionen
der Silikate miteinander. Bei der Hydratation der Calciumaluminate und –alumoferrite
wird dagegen Calciumhydroxid verbraucht. Dadurch können diese Reaktionen wie-
derum die Gesamtreaktion der Silikathydratation beschleunigen.
Rea
kti o
ns-
ge s
c hw
ind
i gke
it
Zeit (h)I II III IV V
Abbildung 1: Zeitlicher Verlauf der Wärmefreisetzung bei der Zementhydratation
nach Kondo und Ueda [48]
Zeitlich läuft die Reaktion von Zement wie in Abbildung 1 dargestellt ab. Die ein-
zelnen Perioden werden durch folgende Erscheinungen charakterisiert [49]:
I. Induktionsperiode (0 bis 0,5 h): Die Oberfläche der Klinkerteilchen überzieht
sich mit einer gelartigen Schicht aus Hydratationsprodukten. Dadurch wird der Zutritt
von Wasser behindert. Die weitere Reaktion der Zementbestandteile wird stark ver-
langsamt. Bei der Benetzung der Zementteilchen mit Wasser wird Energie freige-
setzt. Dadurch ist in dieser Zeit eine starke Wärmeentwicklung nachweisbar, die von

Seite 20
der chemisch-mineralogischen Zusammensetzung und der Gestalt und Größe der
reaktiven Oberfläche der Teilchen abhängig ist. Bei einer Modifikation des Anmach-
wassers mit z.B. Salzen wird die Benetzung und damit die Dynamik und der Betrag
der Energiefreisetzung beeinflusst.
II. Dormante Periode (0,5 bis 5 h): Die Geschwindigkeit der Reaktion geht von
einer kinetischen Kontrolle in eine Diffusionskontrolle über. Es bilden sich erste Kris-
tallisationskeime der Silikathydrate im Gel. Unterhalb einer kritischen Größe der
Keime ist ihr Zerfall thermodynamisch sinnvoller als ihr Wachstum, da in diesem Sta-
dium die Oberflächenenergie größer ist als der Energiegewinn beim Übergang in ei-
nen stabilen Zustand. Ab der kritischen Größe jedoch ist das Kristallwachstum ener-
getisch sinnvoller. Die gesamte Änderung der freien Energie des Mikrosystems ∆F
ist gleich der Differenz der Änderung der Oberflächenenergie und der Änderung der
Energie im Zusammenhang mit der Änderung der freien Energie des Volumens beim
Übergang vom flüssigen in den kristallinen Zustand:
v3
s2 fr
34
fr4F ∆π−∆π=∆ Gleichung 42
Dabei ist r der Radius des Keims; ∆fS die Änderung der freien Energie je Flächen-
einheit der Phasengrenzfläche und ∆fV die Änderung der freien Energie der Phasen-
umwandlung je Volumeneinheit. Bei der Bildung kleiner spherischer Kristallkeime
überwiegt der rechte Term. Mit dem Wachstum der Keime wird dieser Term immer
größer. Ab einer kritischen Größe jedoch beginnt der linke Term den rechten zu ü-
berwiegen – die Keime sind stabil und fangen an zu wachsen [14].
Die Kristalle wachsen, was zu Spannungen im Gel und zu Rissen führt. Dadurch
gelangt erneut Wasser an die Oberfläche der Klinkerteilchen.
III. Akzelerationsperiode (5 bis 10 h): Die Kristalle wachsen weiter und verhindern
die erneute Bildung einer dicht schließenden Gelschicht um die Zementteilchen.
IV. Dezelerationsperiode (10 bis 20 h): Langsam verringert sich die Reaktionsge-
schwindigkeit. Die Kristalle der Calciumsilikathydrate weisen eine Größe von nur 0,1
µm auf.
V. Finalperiode (20 h bis 1 Jahr): Alle Reaktionen klingen ab.

Seite 21
Die Kristallitgröße der Klinkerphasen, speziell von C3S, hat bis zu einer kritischen
Größe von ca. 30 µm keinen Einfluss auf den zeitlichen Verlauf der Reaktion. Die
Geschwindigkeit der Umsetzung in den aktiven Abschnitten korreliert dagegen mit
der Teilchengröße [50].
Nach ca. einem Jahr haben die Kristalle der silikatischen Hydratphasen noch im-
mer nur eine Größe von wenigen Mikrometern. Damit besitzen sie eine sehr große
spezifische Oberfläche (gesamte Oberfläche aller Teilchen in einem Gramm Materi-
al) von 350 bis 450 m²/g. Zum Vergleich - Zement hat eine spezifische Oberfläche
von nur 0,3 bis 0,4 m²/g. Dementsprechend sind auch die Hydrate sehr feinkörnig.
Darauf beruht auch ihre festigkeitsbildende Wirkung: Durch Verzahnen der nadelför-
migen Teilchen werden diese wie bei einem Klettverschluss miteinander verbunden.
Je kleiner und dünner die Kristallnadeln, desto größer die Festigkeit. Allerdings ist
für die Ausbildung eines festen Zementsteines die Überbrückung der Porenräume
zwischen den Teilchen notwendig. Daher steigt die Festigkeit des Zementsteines erst
mit dem Anwachsen der Kristallgröße über die Porengröße [37].
Die Hydratation der Zementbestandteile verlangsamt sich immer weiter, kommt
jedoch auch nach langer Zeit nicht zum Stillstand. Jung [51] führte Untersuchungen
zum Hydratationsgrad von Zement nach 40 Jahren Hydratationsdauer durch. Es
zeigte sich, dass auch nach dieser Zeit etwas über 2 % nicht hydratisierter Bestand-
teile im Zementstein zu finden sind [52], [53].
Die Anteile der einzelnen Zementphasen und –bestandteile bestimmen den Erhär-
tungsverlauf. Eine Erhöhung des Anteils an C2S führt zu einer deutlichen Verlangsa-
mung der Erhärtung. Dieser Einfluss ist im frühen Hydratationsstadium (1 bis 7 Tage)
stärker ausgeprägt. Zu späteren Zeitpunkten ist der Einfluss höherer Belitgehalte da-
gegen positiv. Die Kristallitgröße von Belit hat nur geringen Einfluss auf die Kinetik
des Erstarrens und der Festigkeitsentwicklung der Zemente.
Eine Erhöhung des Anteils an C3S hat deutlich komplexere Auswirkungen auf die
Hydratation des Zements. Zum einen wird bei der Reaktion von Alit mit Wasser sehr
viel Calciumhydroxid freigesetzt. Dadurch wird die flüssige Phase im Zementleim mit
Ca2+-Ionen gesättigt und übersättigt. Dies verlangsamt nach dem Prinzip von Le
Chatelier die Hydratation der anderen Klinkerphasen. Zum anderen werden bei der
Hydrolyse von C3S Silikathydrate mit ähnlicher stöchiometrischer Zusammensetzung

Seite 22
wie bei C2S, jedoch mit anderem Calcium/Silizium-Verhältnis gebildet. Weiterhin
hydratisiert C3S sehr schnell. Dadurch verlieren die Zementteilchen ihren Zusam-
menhalt, was die Zersetzung weiter beschleunigt. Somit ist in den frühen Erhär-
tungsstadien (1 bis 7 Tage) ein deutlicher Festigkeitszuwachs zu beobachten. Ande-
rerseits werden durch die schnellere Abreaktion der Zementteilchen auch die Trä-
gerpunkte des wachsenden Zementsteines, die nicht hydratisierten Zementbestand-
teile vernichtet, wodurch ein schnelleres Erstarren des Leimes ausbleibt. Im späteren
Verlauf der Erhärtung führt ein erhöhter Alitanteil in jedem Fall zu Festigkeitsgewinn.
Das schnell hydratisierende reine C3A verhält sich im Zement ähnlich. Die Kristalle
des Calciumaluminathydrats, welches sofort bei Kontakt mit Wasser gebildet wird,
binden große Mengen an Wasser durch chemische Bindung wie durch Adsorption.
Dadurch steift der Zementleim deutlich an, was gewöhnlich durch eine Erhöhung des
Wasser/Zement-Verhältnisses ausgeglichen wird. Dadurch wie durch die geringe
Eigenfestigkeit der Kristalle des Calciumaluminathydrates wird die Festigkeit des
Zements negativ beeinflusst.
Ähnlich wie erhöhte Anteile von C2S und C3S, führt auch die Erhöhung des Anteils
an C4AF zu keiner Änderung des Widerstandes beim Mischen des Leimes. Auch die
erzielten Festigkeiten in den frühen Erhärtungsstadien zeigen ähnliche Tendenzen
wie bei Zugabe von C2S und C3S. Im späteren Verlauf der Erhärtung wird die gerin-
gere Hydratationsneigung von C4AF jedoch deutlich [54].
Die Festigkeit des Zementsteins, der sich aus polymineralen Bestandteilen zu-
sammensetzt, folgt nicht dem additiven Gesetz. Man kann daher die Festigkeit der
Zemente nicht aus den Einzelfestigkeiten der einzelnen Mineralphasen berechnen.
Die Festigkeit des Zementsteines wird zusätzlich durch seine physikalische Struktur
bestimmt. Limitiert wird die Festigkeit durch die neu gebildeten festen Phasen und
vor allem durch seine Porosität, meist jedoch durch beide [16], [54], [55], [56], [57],
[58], [59], [60].
Nicht zuletzt hat das Wasser/Zement-Verhältnis wesentlichen Einfluss auf die Re-
aktion. Durch einen Verdünnungseffekt wird bei Erhöhung des Wasseranteils der
Anteil der Klinkerphasen verringert. Andererseits werden die Konzentrationen der in
der flüssigen Phase gelösten Stoffe bei Änderung des Wasseranteils im System we-

Seite 23
sentlich verändert. Folglich wird die Kinetik der Zementhydratation deutlich beein-
flusst [61].
1.3.3 Modellierung der Hydratation von Zement
Teilreaktionen der Hydratation von Zement wurden in der Literatur beschrieben.
Avrami beschrieb die Hydratation von Zement während der Dormanten Periode und
der Akzelerationsperiode als kinetisch bestimmt mit [62], [63]
m0 )]tt(k[)1ln( −=α−− Gleichung 43
Die Dezelerationsperiode wird mit der Jander-Gleichung als Diffusions-kontrolliert
beschrieben [64]:
D23/1 k])1(1[ =α−− Gleichung 44
Die Hydratation wird von Bentz auf Basis der zur Verfügung stehenden wasser-
gefüllten Poren (φw) [61]:
)t(kt w1φ=
∂α∂
Gleichung 45
Andere Modelle beziehen die Kinetik der Hydratation auf die Änderung des Radius
der idealisierten Zementpartikel oder der Partikelradienverteilung [65], [66], [67].
Odler et al. untersuchten die Porenstruktur im Laufe der Hydratation von Portland-
zementen und die daraus folgende Festigkeitsentwicklung [68]. Nonat et al. entwi-
ckelten ein räumliches Modell der Hydratationsreaktionen von Zement [69]. Geiker
modellierte das chemische Schwinden im Laufe der Hydratation von Zement [70].
Auf Basis dieser Arbeiten entwickelte Bentz eine Computersimulation der Hydrata-
tion von Portlandzement [71]. Er verarbeitet bei dieser Modellierung neben den prin-
zipiellen Mechanismen aus der ihm zur Verfügung stehenden Literatur ebenso die
Partikelgrößenverteilung der einzelnen Phasenbestandteile von Portlandzement aus
Bildern der Rasterelektronenmikroskopie [72]. In späteren Versionen bindet er weite-
re Hauptbestandteile von Zement, wie Hüttensand und Flugasche mit ein [73]. Je-
doch verwendet Bentz in seinen Modellen empirische Formeln für die Beschreibung

Seite 24
der kinetischen Abläufe, die er an zwei ausgewählten Portlandzementen mit isother-
mer Kalorimetrie kalibriert hat. Die Modellierung liefert somit plausible Beschreibun-
gen der Entwicklung der Struktur von Zementstein. Sie basiert jedoch auf der Frei-
setzung der Hydratationswärme, nicht auf den chemischen und mineralogischen
Prozessen. Die relativ gute Übereinstimmung der Ergebnisse der Modellierungen
von Bentz beruht auf einem sehr aufwendigen, zum Teil empirischen Modell. Ein
ähnliches Modell beschreiben Navi et al. in [74].
Einen anderen Weg der Modellierung der Hydratation von Zement beschreiten
Matschei et al. [75]. Auf Basis grundlegender thermodynamischer Daten, wie Lös-
lichkeiten und freie Gibbs’sche Energien, bestimmen sie die thermodynamisch stabi-
len Phasen im Gleichgewichtszustand. Durch schrittweise (empirische) Freigabe von
Teilsystemen wird der Prozess der Hydratation von Zement simuliert [76].

Seite 25
2 Methoden zur Erfassung der Reaktionsgeschwindigke it
Der zeitliche Ablauf chemischer Reaktionen kann mit Hilfe der Reaktionsdauer
charakterisiert werden. Darunter wird die Zeit verstanden, nach der eine Reaktion
nach außen hin beendet ist, bzw. nach der sich ein chemischer Gleichgewichtszu-
stand eingestellt hat. Die Reaktionsdauer kann, je nach Art der Reaktion, von ver-
schiedenen Faktoren abhängen:
� Art der beteiligten Stoffe
� Größe der reaktiven Oberfläche (Feinheit)
� Konzentration der beteiligten Stoffe
� Temperatur
� Katalysatoren
� Licht
� Druck
� Lösungsmittel
Die Reaktionsgeschwindigkeit wird als Verhältnis von Stoffmengenänderung je
Zeitintervall beschrieben [6]:
tc
V∆∆= Gleichung 46
2.1 Stoffmengenbestimmung
2.1.1 Chemische Methoden
Die Konzentration eines Stoffes kann u.a. direkt chemisch z.B. durch Titration,
Gravimetrie o.a. Methoden bestimmt werden. Allen chemischen Verfahren gemein-
sam ist, dass der zu analysierende Stoff ganz oder teilweise aus dem reagierenden
System entfernt bzw. das System aufgeteilt wird. Dadurch werden für längere Beo-
bachtungen einer Reaktion große Anzahlen von Proben bzw. große Probemengen
benötigt.
Der Zeitaufwand für chemische Analysen ist hoch. Aufgrund der in vielen Fällen
notwendigen Probenvorbereitung erfolgt die Bestimmung z.T. zeitlich sehr verzögert.
Reproduzierbarkeit und Genauigkeit sind beeinträchtigt.

Seite 26
2.1.2 Physikalische Methoden
Bei der Betrachtung dynamischer Systeme werden heute fast ausschließlich phy-
sikalische Methoden zur Bestimmung der Stoffmengenkonzentration verwendet. Alle
verwendbaren physikalischen Methoden beruhen auf der Messung physikalischer
Parameter, die in einem mathematischen Zusammenhang mit der Konzentration ste-
hen. Die Röntgenbeugungsanalyse (XRD) ist ein Beispiel für ein physikalisches Ver-
fahren zur direkten Stoffmengenbestimmung. Moderne Analysengeräte und Compu-
ter erlauben eine schnelle Messung und quantitative Auswertung von Röntgenbeu-
gungsanalysen. Dieses Verfahren eignet sich für die quantitative Verfolgung der
Hydratation von Zementen [77], [78].
2.2 Indirekte Methoden
Bei diesen Methoden wird nicht die Stoffmengenkonzentration, sondern deren Än-
derung bestimmt. Man bedient sich hierzu meist physikalischer Größen, die proporti-
onal zur Änderung der Stoffmenge einer oder mehrerer Komponenten des Systems
sind. Im Folgenden sind einige für die Zementtechnologie relevante Beispiele aufge-
führt.
2.2.1 Kalorimetrie
Wie in Pkt. 1.2.1 beschrieben zeigen die meisten Reaktionen einen thermischen
Effekt. Diese Wärmemenge ist spezifisch für jede stöchiometrische Umsetzung. Das
heißt, aus der freigesetzten oder aufgenommenen Wärme kann auf die umgesetzten
Stoffmengen, nicht jedoch direkt auf die Konzentrationen eines Stoffes geschlossen
werden. Der Wärmefluss aus einem System oder in ein System ist somit proportional
zur Reaktionsgeschwindigkeit [79], [80], [81], [82].
Die Bestandteile von Zement setzen bei ihrer Reaktion mit Wasser teilweise be-
trächtliche Wärmemengen frei. Somit ist die Kalorimetrie sehr gut geeignet, die Ge-
schwindigkeit der Hydratationsreaktion von Zement zu beschreiben. Jedoch ist die-
ses Verfahren nicht selektiv für einzelne Stoffe im Mehrstoffsystem Zement. Damit ist
diese Methode nicht geeignet, die Reaktion einzelner Bestandteile des Zements mit
Wasser im Beisein anderer Bestandteile sowie deren Hydratationsprodukte aufzu-
zeichnen.

Seite 27
2.2.2 Festigkeitsprüfung
Durch die chemische Reaktion des Zementes mit Wasser erstarrt und erhärtet das
System Zement-Wasser. Somit ist auch die Entwicklung der mechanischen Festig-
keit ein Ergebnis der Menge der umgesetzten Ausgangsstoffe.
Allerdings haben sehr viele Randbedingungen einen wesentlichen Einfluss auf die
Festigkeit des Zementsteines. So spielen physikalische Parameter, wie die Pa-
ckungsdichte des Gefüges aus Zementteilchen und Wasser, die Filmbildung des
Wassers auf der Oberfläche einzelner Teilchen und damit den mechanischen Ver-
bund zwischen Kristallen, Kapillarkräfte durch Austrocknungs- bzw. Durchfeuch-
tungsprozesse eine nicht zu vernachlässigende Rolle.
Die Größe des Einflusses dieser Parameter kann nur unzureichend kontrolliert
werden. Die Festigkeitsentwicklung ist ein Ausdruck des Verbundes innerhalb des
Gesamtsystems aller Zementbestandteile, deren Hydratationsprodukten und Wasser
(und gegebenenfalls inerter Zusätze). Sie ist also ebenfalls nicht selektiv. Mit ihrer
Hilfe kann nicht auf die Umsetzung einzelner Komponenten geschlossen werden.

Seite 28
3 Problemstellung
Die Hydratation von Zement ist ein sehr komplexer Prozess. Viele physikalische,
chemische und mineralogische Umwandlungen laufen zeitgleich wie auch nachein-
ander ab. Schon geringfügige Änderungen der inneren wie auch äußeren Reaktions-
bedingungen verändern den Verlauf und vor allem die Dynamik der Hydratation und
der Festigkeitsentwicklung des Zementsteines.
Um für den Anwender möglichst gut brauchbare Bindemittel zu erzeugen, ist es
der Wissenschaft seit Anbeginn ein Anliegen, die Mechanismen der Erhärtung von
Zement zu erforschen. Auf Grund der Konsistenz von Zementleim im Anwendungs-
zustand – einer steifen Paste, welche mit der sie umgebenden Atmosphäre in Wech-
selwirkung tritt – ist es jedoch schwierig, den Prozess der Hydratation von Zement zu
beobachten. Gleichzeitig geben aber Momentaufnahmen (unterbrochene Reaktio-
nen) nur einen sehr begrenzten Einblick in die zeitlichen Verläufe der Umwandlun-
gen. Zur Untersuchung der Umsetzungsgeschwindigkeit und –raten wurden daher
zumeist indirekte Methoden, wie die Messung des ungebundenen Wassers oder die
Wärmeentwicklung im reagierenden System, angewendet [83]. Diese Kenngrößen
ergeben sich jedoch oft aus den überlagerten Signalen mehrerer Reaktionen.
Mit Hilfe der Röntgendiffraktometrie ist es möglich, zerstörungsfrei und quantitativ
Phasengehalte von Materialien zu ermitteln. Moderne Geräte erlauben eine schnelle
Erfassung des gesamten für die Reaktion relevanten Bereichs. Mit Hilfe der Quantifi-
zierung nach Rietveld wird die Analyse deutlich sicherer als bei der Quantifizierung
mit Peak/Untergrund-Verhältnissen. Die quantitative Rietveld-Diffraktometrie ist heu-
te eine sehr zuverlässige Methode für die Verfolgung der Hydratation von Zement.
Copeland und Kantro untersuchten auf diese Weise die Reaktion von Portlandze-
menten in Abhängigkeit vom Wasser/Zement-Verhältnis im Zeitraum von 1 Tag bis
6,5 Jahre [84]. Hierbei ergab sich, dass die Umsetzungsraten bei höherem Wasser-
angebot höher lagen. Die kalorimetrische Untersuchung unterstützt diese Aussage.
Locher fand bei der Verwendung von reinem C3S jedoch, dass die Hydratation bei
geringeren Wasser/Zement-Verhältnissen schneller abläuft [85].
Diese Arbeit soll deshalb einen Beitrag zur Aufklärung des zeitlichen Verlaufs der
stofflichen Umwandlungen im erhärtenden Zementleim leisten. Ziel ist es, die Verläu-
fe der Gehalte einzelner der bei der Zementhydratation verbrauchten und gleichzeitig
Seite 29
neu gebildeten Mineralien zu bestimmen und durch Anwendung bekannter Ge-
schwindigkeitsgesetze die Reaktionskinetik mathematisch zu beschreiben. So soll
der Reaktionstyp (Ordnung) der entsprechenden Umwandlung aufgeklärt werden.
Hauptanliegen dieser Arbeit war die kinetische Betrachtung der Reaktion der einzel-
nen Bestandteile der Zemente mit XRD. Die Ergebnisse sollen mit den bekannten
stöchiometrischen Formeln und kinetischen Beschreibungen der Prozesse basierend
auf anderen Methoden (z.B. der Wärmeflusskalorimetrie) verglichen werden.
Es wird versucht, die einzelnen durch Röntgendiffraktometrie bestimmten Pha-
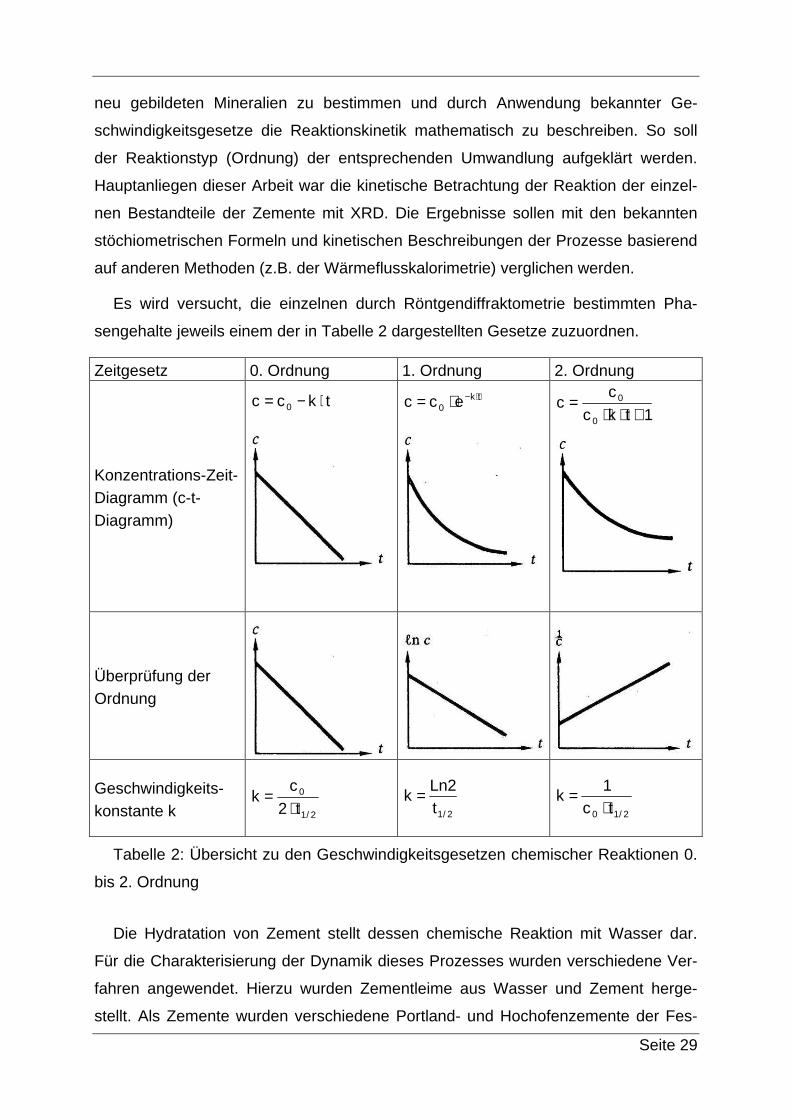
sengehalte jeweils einem der in Tabelle 2 dargestellten Gesetze zuzuordnen.
Zeitgesetz 0. Ordnung 1. Ordnung 2. Ordnung
Konzentrations-Zeit-Diagramm (c-t-Diagramm)
tkcc 0 ⋅−=
tk0 ecc ⋅−⋅=
1tkc
cc
0
0
+⋅⋅=
Überprüfung der Ordnung
Geschwindigkeits-konstante k 2/1
0
t2
ck
⋅=
2/1t2Ln
k = 2/10 tc
1k
⋅=
Tabelle 2: Übersicht zu den Geschwindigkeitsgesetzen chemischer Reaktionen 0.
bis 2. Ordnung
Die Hydratation von Zement stellt dessen chemische Reaktion mit Wasser dar.
Für die Charakterisierung der Dynamik dieses Prozesses wurden verschiedene Ver-
fahren angewendet. Hierzu wurden Zementleime aus Wasser und Zement herge-
stellt. Als Zemente wurden verschiedene Portland- und Hochofenzemente der Fes-

Seite 30
tigkeitsklassen 32,5 sowie 42,5 verwendet. Das Anmischen der Zementleime erfolgte
bei verschiedenen W/Z-Werten. Eine Aufstellung der untersuchten Systeme ist in
Tabelle 8 gegeben. Diese wurden in einen mit einer Folie verschlossenen Probenträ-
ger aus Edelstahl präpariert. Die Veränderung der Gehalte der Minerale im unter-
suchten System wurde mit Röntgendiffraktometrie durch die Folie hindurch gemes-
sen. Eine wichtige Voraussetzung für die Gültigkeit der Ergebnisse war, dass der
Zementleim einerseits kaum Wasser infolge Diffusion durch die Folie verliert und
gleichzeitig kein CO2 aus der Luft aufnehmen konnte. Demzufolge kam der Folien-
auswahl große Bedeutung zu. Als Folien wurden sowohl Mylar als auch Polycarbonat
verwendet.

Seite 31
4 Analytische Methoden
4.1 Methanol-Salicylsäure-Auszug
Bei diesem Anreicherungsverfahren für die Aluminate wird die Tatsache genutzt,
dass Calciumsilikate in Salicylsäure löslich sind, Calciumaluminate und –alumoferrite
jedoch nicht. Ebenfalls nicht in Lösung gehen Calciumsulfate. Um Verluste durch
Hydratation zu vermeiden, wird der Auszug nicht im wässrigen, sondern im alkoholi-
schen Milieu durchgeführt. Der Rückstand des Salicylsäureauszugs erlaubt meist
eine besonders genaue quantitative Bestimmung des C3A-Gehalts im Zement.
Folgende Methodik wurde angewendet:
7,500 g ± 0,001 g Klinker oder Zement werden auf der Analysenwaage eingewo-
gen und zusammen mit 270 mL ± 1 mL Methanol-Salicylsäure-Gemisch (5:1) in eine
Standard-Rührapparatur gegeben. Das Gemisch wird mittels einer Heizhaube und
Vorschaltgerät auf 60 °C ± 3 °C erhitzt und 1 Std. unter Rühren am Rückfluss ge-
kocht.
Nach Ablaufen der Zeit wird die Suspension an der Luft abgekühlt und über einen
Membranfilter aus PES (0,45 mm) in einer Saugnutsche mittels Wasserstrahl-Pumpe
abfiltriert. Hierbei wäscht man mit ca. 200 mL Methanol nach. Das Filtrat kann ver-
worfen werden.
Filterkuchen und Membranfilter werden der Nutsche entnommen und anschlie-
ßend im Exsikkator getrocknet. Am darauf folgenden Tag wird der Filterkuchen vom
Filter abgekratzt und in einem vorher auf 0,001 g genau gewogenen Gefäß ausge-
wogen [86].
4.2 Borsäure-Auszug
Calciumaluminate sind in wässriger Zuckerlösung löslich, Calciumalumoferrite je-
doch nicht. Somit können aus den nach 4.1. hergestellten Präparaten die Calciuma-
luminate abgetrennt werden.
Folgende Methodik wurde angewendet:
In einen 500 mL Stand-Rundkolben werden 100 mL ± 1 mL einer 2,5 %-igen
wässrigen Borsäurelösung vorgelegt und mit 1,000 g ± 0,001 g Material aus dem Me-
thanol-Salicylsäure-Auszug versetzt. Anschließend werden noch 250 mL ± 1 mL Bor-

Seite 32
säurelösung (2,5 %) dazu gegeben, das Gefäß mit Stopfen versehen und das Ge-
misch 1 Std. lang mit einem Schüttler durchmischt.
Danach wird das Gemisch mit einem Weißbandfilter (evtl. vorher mit Borsäurelö-
sung (2,5 %) anfeuchten) unter zu Hilfenahme einer Saugnutsche und Wasserstrahl-
pumpe abfiltriert. Mit ca. 200 mL 2,5 %-iger Borsäurelösung und ca. 200 mL 10 %-
iger Zuckerlösung wird nachgewaschen. Das Filtrat kann verworfen werden.
Filterkuchen und Filter werden der Nutsche entnommen und 1 Tag im Exsikkator
getrocknet. Nach 24 Std. wird der Filter verascht und der Rückstand bei 950 °C ge-
glüht. Das gewonnene, abgekühlte Material wird dann mit einem Achat-Mörser pul-
verisiert und in einem vorher auf 0,001 g genau gewogenen Gefäß ausgewogen und
die Ausbeute berechnet.
4.3 Röntgendiffraktometrie (XRD)
Röntgenstrahlen sind elektromagnetische Wellen, deren Wellenlänge kürzer ist als
die des sichtbaren Lichtes und in der Größenordnung atomarer Abstände in Kristal-
len liegt (ca. 100 pm). Sie bilden die Grundlage für eine Reihe verschiedener Unter-
suchungsverfahren. In dieser Arbeit wurden die Röntgenbeugungsanalyse oder
Röntgendiffraktometrie (x-ray diffraction - XRD) und die Röntgenfluoreszenzanalyse
(x-ray fluorescence – XRF, Pkt. 4.4) angewendet.
Werden Elektronen regelmäßig angeordneter Atome innerhalb eines Kristalls
durch Röntgenstrahlen zu periodischen Schwingungen angeregt, so entsteht eine
Vielzahl von Strahlungsquellen derselben Frequenz und Wellenlänge wie die einfal-
lende Strahlung. Von jedem Elektron breiten sich kugelförmig Wellenfronten aus, die
sich überlagern und Anlass zum Auftreten von Interferenz-Erscheinungen geben.
Damit bei der Beugung von Wellen an einem Gitter Verstärkung auftreten kann,
müssen ganz bestimmte geometrische Bedingungen erfüllt sein, die mit Hilfe der drei
Laue-Gleichungen oder der Bragg’schen Reflexionsgesetze beschrieben werden
können.
Bragg ging von der Voraussetzung aus, dass Kristalle aus Ebenen aufgebaut sind,
die mehr oder weniger dicht mit Atomen besetzt sind und die stets von gleichwerti-
gen parallelen Ebenen in einem konstanten Abstand d (Netzebenenabstand) beglei-
tet werden. Trifft ein Röntgenstrahl auf eine Netzebene im Kristall, so erfolgt Beu-
gung (Reflexion). Der Röntgenstrahl durchdringt im Allgemeinen einige Millionen

Seite 33
Netzebenen, bis er gänzlich absorbiert wird. An jeder einzelnen Netzebene wird da-
bei ein Teil der Strahlung reflektiert. Es tritt dadurch eine Überlagerung von Wellen
auf, wobei sich die Wellen in ganz bestimmten Richtungen, die durch geometrische
Bedingungen festgelegt sind, verstärken, während in allen anderen Richtungen Aus-
löschung erfolgt.
Nach den Gesetzen der Reflexion werden die Wellen im gleichen Winkel von der
Reflexionsebene abgestrahlt, in dem sie auf die Ebene auftreffen. Beim Auftreffen
(nahezu) paralleler gleichphasig schwingender Strahlen auf Ebenen mit konstantem
Abstand zueinander ergibt sich bei der Reflexion eine Phasenverschiebung zwischen
den Strahlen. Wenn der Phasenunterschied ein ganzzahliges Vielfaches der Wellen-
länge beträgt, schwingen die Wellen nach der Reflexion wieder gleich, die Wellen
verstärken sich. Die Bragg’schen Reflexionsbedingungen formulieren sich daher:
nλ = 2d sinΘ Gleichung 47
Der Aufbau des Kristallgitters und die Wellenlänge der Röntgenstrahlungsquelle
bestimmen somit den Beugungswinkel, unter dem es zur Interferenz kommt.
Werden feine kristalline Pulver von einem monochromatischen Röntgenstrahl ge-
troffen, so werden die Röntgenstrahlen an allen Netzebenen, für die die Bragg’sche
Beziehung erfüllt ist, gebeugt. Die gebeugten Strahlen bilden sowohl Durchstrahl- als
auch Rückstrahl-Kegel mit dem halben Öffnungswinkel 2Θ. Die Interferenzkegel
werden mit Hilfe von Filmen, Zählrohren oder Halbleiterdetektoren registriert. Bei
sehr feinen Kristallpulvern oder bei Rotation der Probe sind die Beugungskegel ho-
mogen. In diesem Fall genügt es, einen kleinen Ausschnitt (eine Linie) der Kegel zu
erfassen [87].
In der vorliegenden Arbeit wurde ein Diffraktometer vom Typ D4 Endeavor der Fa.
Bruker-AXS mit feststehender Röhre, drehbarem Probenhalter und beweglichem
LynxEye-Detektor verwendet. Das verwendete Diffraktometer ist mit einem automati-
schen Probenwechsler ausgestattet.
4.3.1 Präparation
Die Ausgangsstoffe dieser Arbeit wurden nach dem Pulver-Pressverfahren präpa-
riert. Die Materialien wurden 60 Sekunden mit 200 kPa in Stahlringe vom Typ Polysi-

Seite 34
us gepresst. Die Vorzugsorientierung einzelner Phasen wurde rechnerisch (siehe
Pkt. 4.3.4) korrigiert.
Für die Untersuchung des Reaktionsverlaufes bei der Hydratation wurde ein spe-
zielles Präparationsverfahren eingesetzt (siehe Pkt. 4.3.3).
4.3.2 Einfacher Scan
Für die quantitative Analyse der Ausgangszemente wurde jeweils ein einfacher
Scan durchgeführt. Die Peaks der für Zement und dessen Hydratphasen relevanten
Minerale liegen im Bereich zwischen 10 und 60 ° 2 Θ. Die Messungen in diesem In-
tervall erfolgten mit einer Auflösung (Schrittweite) von 0,016 ° 2 Θ (die hohe Genauig-
keit ist software-technisch bedingt) bei einer theoretischen Verweildauer von 0,5 s je
Schritt. Auf Grund der gleichzeitigen Messung eines Fensters von 6 ° 2 Θ betrug die
gesamte Messdauer jedoch nur ca. 30 Minuten je Probe.
4.3.3 Zyklische Langzeitmessungen
Zur Untersuchung des Hydratationsverlaufs der Zementpasten wurden diese zu
bestimmten Zeiten nach dem Anmischen mehrfach vermessen. Dabei wurde wie
folgt vorgegangen:
Es wurden Probenbehälter aus Edelstahl nach Abbildung 2 verwendet. Der innere
Ring wurde auf der Schmalseite mit einer Kunststofffolie überspannt und in den äu-
ßeren Ring eingesetzt. In Vorversuchen wurden verschiedene Folientypen geprüft.
Die Kunststofffolien weisen einen eigenen Diffraktionspeak auf, der das Diagramm
der darunter liegenden Probe überlagert. Die Folie Mylar zeigte einen starken Diffrak-
tionspeak im Bereich der Haupt-Klinkerphasen. Für die Versuchsserien wurde eine
5 µm dicke Polycarbonatfolie ausgewählt, deren charakteristischer Reflex nur einen
Peak von Portlandit wesentlich überlagert. Da dieser überlagerte Peak jedoch sehr
stark ausgeprägt ist, konnten die Reflexe durch rechnerische Korrektur (siehe Pkt.
4.3.4) gut voneinander getrennt werden. Die Quantifizierung wurde nicht nennens-
wert beeinträchtigt.

Seite 35
Abbildung 2: Aufbau und Beladung des Probenträgers aus Edelstahl („back loa-
ding“) für die zyklischen XRD-Langzeitmessungen während der Zementhydratation
(Großes Bild links: Probenträger komplett zerlegt; Mitte: Probenträger leer montiert;
rechts: Probenträger mit Zementpaste beladen, hintere Abdeckung offen; kleines
Bild: Probenträger gefüllt, mit Folie abgedeckt und komplett montiert in Messlage)
Für die Untersuchungen mit Röntgendiffraktometrie wurden Zementleime aus 80 g
Bindemittel und der dem vorgegebenen Wasser/Zement-Verhältnis entsprechenden
Menge entionisiertem Wasser hergestellt. Die erforderlichen Mengen der Stoffe wur-
den auf 0,01 g genau eingewogen. Die Mischung wurde 2 Minuten lang bei ca. 600
min-1 mit einem Propellerrührer in einem 120 mL Kunststoffbecher homogenisiert.
Anschließend wurde der Leim in einen Probenbehälter gemäß Abbildung 2 eingefüllt.
Bei der Befüllung zeigte die Folie nach unten. In dieser Lage wurde die Probe 2
Stunden belassen, so dass der Zementleim erstarren konnte. Ein Absenken der zu
messenden Oberfläche konnte so vermieden werden.
Das Gerät ist Bestandteil eines automatischen Labors. Über den Leitrechner des
Labors wurden die Messserien gesteuert. Nach Ablauf der vorgegebenen Erstar-
rungszeit wurden die Probenhalter mit der Folie nach oben in den Probenwechsler
des Diffraktometers eingesetzt und stündliche Messungen gestartet.

Seite 36
Bei Mylar handelt es sich um Polyethylenterephthalat-Polyester der Firma DuPont
mit einer Dicke von 6,0 µm (0,24 mil). Die Dampfdurchlässigkeit für Kohlendioxid be-
trägt 16 cc/100 in²/24 h/atm/mil entsprechend ca. 0,02 g CO2/24 h, bezogen auf die
Oberfläche des verwendeten Probenträgers, und für Wasser 1,8 g/100 in²/24 h/mil
entsprechend ca. 0,1 g/24 h, bezogen auf die Oberfläche des verwendeten Proben-
trägers. Die Versuche mit Mylar zeigten zwar gute Intensitäten (d.h. ein gutes Sig-
nal/Rauschverhältnis), jedoch überlagerten sich analytisch bedeutsame Peaks sehr
stark mit dem für das Folienmaterial typischen Diffraktionsreflex. Durch Wechsel zu
Polycarbonat-Folie konnte die Überlagerung vermieden werden. Häufig werden in
der Röntgenanalytik auch Kapton-Folien eingesetzt. Hierbei handelt es sich um ein
Polyimid der Firma DuPont. Diese Folien zeigen im üblicherweise untersuchten
Messbereich der Diffraktometrie keinen Peak. Durch die Folie wird jedoch der Un-
tergrund des Messsignals angehoben. Die Dampfdurchlässigkeit für Kohlendioxid
beträgt 45 cc/100 in²/24 h/atm/mil entsprechend ca. 0,06 g CO2/24 h, bezogen auf
die Oberfläche des verwendeten Probenträgers, und für Wasser bei einer Dicke von
2,5 µm 1,3 g/100 in²/24 h/mil entsprechend ca. 0,1 g/24 h, bezogen auf die Oberflä-
che des verwendeten Probenträgers. Kapton-Folien standen zum Zeitpunkt der Un-
tersuchungen nicht in ausreichender Menge zur Verfügung. Daher wurden die in die-
ser Arbeit beschriebenen Versuche im wesentlichen mit Polycarbonat-Folie durchge-
führt.
Der Einfluss der Folienoberfläche auf die Kristallisationsvorgänge (Keimbildung)
wurde qualitativ untersucht. Es wurden jedoch keine Anzeichen für eine bevorzugte
Kristallisation zur oder von der Folien-Oberfläche weg gefunden.
Die Messungen während der Hydratation erfolgten im Intervall von 10 bis 55 ° 2 Θ
mit einer Auflösung (Schrittweite) von 0,04 ° 2 Θ (die hohe Genauigkeit ist software-
technisch bedingt) bei einer theoretischen Verweildauer von 0,5 s je Schritt. die ge-
samte Messdauer betrug ca. 11 Minuten je Probe (gleichzeitige Messung eines
Fensters von 6 ° 2 Θ).
Die quantitative Auswertung erfolgte gemäß Pkt. 4.3.4 nach Abschluss aller Mes-
sungen durch ein Visual Basic for Applications Script. Die Aufbereitung der Daten
wurde in MS Excel durchgeführt und ist in Pkt. 4.3.5 ausführlich beschrieben.

Seite 37
4.3.4 Quantitative Rietveldanalyse
H.M. Rietveld sammelte in den 1960-er Jahren anhand von Kern- und Magnetre-
sonanzspektren die Funktionen, die die physikalischen Abhängigkeiten der Intensitä-
ten der gemessenen Strahlungen von den apparativen Gegebenheiten und der Pro-
benmatrix beschreiben. Durch Verknüpfung der Funktionen miteinander schuf er eine
mathematische Beschreibung der Intensität in Abhängigkeit von Messapparatur und
Probe. Nach diesem Modell ist es möglich, aus vorgegebenen Eigenschaften der
Messapparatur und vorgegebenen Probeneigenschaften die Intensitätsspektren zu
berechnen. Durch Variation einzelner Parameter werden nach der Methode der
kleinsten Fehlerquadrate die Ausgangsparameter so verändert, dass die gemesse-
nen Intensitätsspektren mit den berechneten übereinstimmen [88].
In der Folgezeit wurden aufbauend auf diesem Modell verschiedene Computer-
programme entwickelt, welche eine quantitative Auswertung von Diffraktogrammen
erlauben [89].
Es haben sich zwei prinzipielle Methoden etabliert:
a) die empirische Methode. Hier werden die einzelnen Peaks durch mathemati-
sche Funktionen (Gauß, PseudoVoigt u.a.) beschrieben. Detaillierte Kenntnisse über
die kristallographische Struktur der Probe wie auch den physikalischen Aufbau der
Messapparatur sind nicht erforderlich. Durch Überlagerungen und Randeinflüsse von
Peaks ergeben sich teilweise große Unsicherheiten [90].
b) die Fundamental-Parameter-Methode. Bei diesem Verfahren werden die realen
physikalisch-mathematischen Funktionen zur Beschreibung der Messapparatur wie
auch der Probe genutzt. Unter Vorgabe der genauen Parameter von Mess-Apparatur
und Probeneigenschaften lässt sich das Diffraktionsspektrum exakt berechnen. Aus
den gemessenen Spektren werden einzelne Parameter, wie Massegehalte und Git-
terparameter der Probenbestandteile als Unbekannte nach dem von Rietveld be-
schriebenen Verfahren berechnet.
In der vorliegenden Arbeit wurde das Verfahren des Fundamental-Parameter-
Ansatzes angewendet. Verwendet wurde das Programmpaket „Topas 2.1“ der Fa.
Bruker-AXS. Zur Steuerung wurden im Anhang wiedergegebene Input-Files sowie
selbst entwickelte Visual Basic for Applications Scripte verwendet. Die Steuerfiles
wurden auf Basis folgender Strukturen zusammengestellt: C3S monoklin nach Nishi;

Seite 38
C2S beta nach Mumme; C3A kubisch; C4AF nach Colville; Anhydrit und Bassanit
nach Bezou et al. [91]; Gips nach Schofield [92]; Portlandit nach Henderson und Gu-
towski [93]; Calcit nach Markgraf [94]; Ettringit nach ICSD Nummer 411451. Für die
Berechnung des Diffraktionspeaks der Polycarbonatfolie wurde eine kubische Struk-
tur der Raumgruppe p23 zu Grunde gelegt, die auf Basis einer separaten Messung
verfeinert und bei den eigentlichen Messungen fixiert wurde.
Da die Intensitäten der in-situ XRD-Messungen für die Auswertung mit Rietveld
sehr schwach waren, wurden die Parameter der Strukturen weitestgehend fixiert. Nur
Kristallitgröße und Phasenanteil wurden zur Verfeinerung freigegeben, während die
Gitterparameter (Abmessungen und Winkel der Elementarzelle) nicht freigegeben
wurden. Die rechnerische Freigabe der Gitterparameter von Ettringit ergab eine sehr
starke Korrelation mit der Kristallitgröße (R² > 98 %). Das deutet auf die unzurei-
chende Datenqualität für eine solche Verfeinerung hin. Daher wurde im weiteren auf
die Prüfung des Einbaus von Fremdatomen in Ettringit bzw. AFt im Verlauf der Reak-
tion verzichtet.
Aus den mathematischen Vorgaben des Programmpakets heraus ergibt sich eine
Normierung der kristallinen Bestandteile der Probe auf 100 %.[95] Dies entspricht
jedoch keineswegs der Realität in frischen und erhärtenden Zementleimen. Die Ge-
halte der nichtkristallinen bzw. röntgenamorphen Bestandteile lassen sich annähe-
rungsweise auf Basis stöchiometrischer Formeln (Gleichung 36 und 37) berechnen.
Auf dieser Grundlage wurde, nach einer Empfehlung von Neubauer et al. [78], der
Gehalt an CxSyHz Phase nach der Formel
2)OH(CaCSH c743,1
193c ⋅
⋅= Gleichung 48
berechnet. Im weiteren wurden die Gehalte wasserhaltiger Phasen (Calciumsulfat-
Halbhydrat, Calciumsulfatdihydrat, Calciumhydroxid, Ettringit und CxSyHz Phase) in
wasserfreie Gehalte umgerechnet:
whwh
wfwf c
MM
c ⋅= Gleichung 49
Die so erhaltenen wasserfreien Gehalte wurden auf 100 % normiert und auf den
Anteil der Feststoffeinwaage korrigiert:

Seite 39
WasserZement
Zement
j
ii mm
mc
cc
wf
wf
normiert,wf +⋅=
∑ Gleichung 50
Der Gehalt an Wasser im System ergibt sich analog aus
WasserZement
WasserWasser mm
mc
⋅= Gleichung 51
Die normierten wasserfreien Gehalte der eigentlich wasserhaltigen Phasen (Calci-
umsulfat-Halbhydrat, Calciumsulfatdihydrat, Calciumhydroxid, Ettringit und CxSyHz
Phase) wurden in die normierten wasserhaltigen Gehalte umgerechnet nach:
normiert,wfwf
whnormiert,wh c
MM
c ⋅= Gleichung 52
Der Wasseranteil der einzelnen wasserhaltigen Phasen i errechnet sich als
wfwhi iiWasser ccc −= Gleichung 53
Der Anteil freien Wassers im System errechnet sich folglich nach
∑−=jWasserWasserfreiWasser ccc Gleichung 54
Das System wird auf diese Weise zu 100 % beschrieben. Die Richtigkeit der Ana-
lyse kann nicht mit ausreichender Sicherheit überprüft werden, da sich die bekannten
analytischen Methoden (XRF, DTA, selektive Lösungsverfahren, Mikroskopie u.a.)
z.T. widersprechen. Es kann daher kein sicheres Verfahren als Referenz angegeben
werden. Für diese Arbeit ist ein systematischer Fehler in der Quantifizierung jedoch
nicht relevant. Relative Änderungen der Phasenanteile im System reichen zur Be-
schreibung der Reaktionskinetik bei der Zementhydratation aus. In der verwendeten
analytischen Strategie wird, wie im folgenden Abschnitt detailliert beschrieben, der
Korrelationsgrad der Konzentrationen und der Zeit in verschieden skalierten Syste-
men verwendet um den Reaktionsgrad zu bestimmen.

Seite 40
4.3.5 Bestimmung der Reaktionsordnung
Nach Pkt. 1.2.3.1 und Pkt. 1.2.3.2 ist für die jeweilige Reaktionsordnung ein linea-
rer Zusammenhang im System
c = k�t 0. Ordnung
ln(c) = k�t 1. Ordnung
1/c = k�t 2. Ordnung
c = k� c Diffusion
gegeben. Es wurde geprüft, ob ein funktionaler Zusammenhang nach einer dieser
Gleichungen nachgewiesen werden kann. Hierzu wurden die Datenmengen der ein-
zelnen Versuche statistisch ausgewertet.
Aus den vorhandenen Datenpunkten wurde durch lineare Regression das Be-
stimmtheitsmaß R² in den genannten Funktionen ermittelt. Die Stabilität der Mess-
werte in den einzelnen Abschnitten der Reaktionen war teilweise nicht befriedigend.
Eine rechnerische Ermittlung der Zeitpunkte der Abschnittswechsel durch Differen-
zieren war nicht möglich. Die Begrenzung der verwendeten Bereiche erfolgte daher
subjektiv visuell aus dem jeweiligen grafischen Zusammenhang der Funktion c = f(t).
In allen vier funktionalen Zusammenhängen wurde jedoch gleich derselbe Start- und
der Endpunkt für den betrachteten Abschnitt verwendet. Die ermittelten Bestimmt-
heitsmaße wurden miteinander verglichen. Der jeweils betrachteten Reaktion wurde
die Reaktionsordnung zugewiesen, deren Reaktionsgeschwindigkeitsgesetz das
höchste Bestimmtheitsmaß ergab.
4.4 Röntgenfluoreszensanalyse (XRF)
Bei der Absorption von Strahlen genügender Energie werden einzelne Elektronen
in den Atome angeregt. Die angeregten Elektronen senden beim „Zurückfallen“ in
tiefer liegende Energiezustände Röntgenstrahlen aus, die charakteristisch sind für
jedes Element. Zur Anregung verwendet man in modernen Röntgenspektrometern
Elektronenstrahlen (Elektronenstrahl-Mikrosonde, geeignet zur Untersuchung kleiner
Punkte) oder Röntgenstrahlen hoher Intensität (Kaltanregung).
Die Bestimmung der Wellenlänge der entstehenden Röntgenstrahlung ermöglicht
eine Identifizierung der vorhandenen Elemente. Die Intensität ist zwar dem Mengen-
anteil des Elementes in der Probe proportional, doch beeinflussen sich die verschie-

Seite 41
denen Atome gegenseitig. Für quantitative Analysen ist es daher notwendig, Kalib-
rierkurven aufzunehmen oder rechnerische Korrekturen durchzuführen [87].
Aus Röntgen-optischen Bedingungen müssen die zu messenden Proben eine e-
bene Oberfläche aufweisen. Es eigenen sich für die Untersuchung Presslinge aus
Pulver der zu analysierenden Substanz wie auch Gläser, die aus der zu analysieren-
den Substanz und einem geeigneten Schmelzmittel hergestellt wurden. Da die Ein-
dringtiefe der Röntgenstrahlen auf wenige µm begrenzt ist, muss die Zusammenset-
zung der zu messenden Oberfläche der der gesamten Probe entsprechen. Gläser
sind bei richtiger Herstellung sehr homogen. Glastabletten eignen sich daher sehr
gut für eine Röntgenfluoreszenzanalyse.
4.5 Festigkeitsprüfungen
Die Festigkeitsprüfungen erfolgten nach DIN EN 196. In Abweichung von dieser
Norm wurden die Proben z.T. nur über Wasser, nicht unter Wasser gelagert, um den
Einfluss der unterschiedlichen Lagerungsart zu untersuchen. Die Lagerung über
Wasser entspricht mehr den Bedingungen der Baupraxis, sie kann jedoch u.U. zu
vorzeitigem Austrocknen der Oberfläche führen.
4.6 Kalorimetrie
Beton, aber auch Mörtel stellen mikroskopisch sehr inhomogene Systeme aus
Teilchen des Zuschlags, des Bindemittels, Wasser und Luftporen dar. Auf die Wär-
mefreisetzung bei der Hydratation des Bindemittels haben alle Komponenten dieses
Systems Einfluss. Wasser und Luftporen beeinflussen die Reaktion chemisch durch
Bereitstellung oder Entzug von Ausgangsstoffen bzw. durch Abführen von Reakti-
onsprodukten. Alle beteiligten Komponenten nehmen die bei der Hydratation freige-
setzte Wärme auf. Die Temperatur der Komponenten und dadurch des gesamten
Systems erhöht sich. Um den Einfluss lokaler Inhomogenitäten auf das Messergeb-
nis gering zu halten, muss eine ausreichend große Anzahl Teilchen betrachtet wer-
den [96].
Ein weiteres Problem stellt die Wärmekapazität der Zemente und des Wassers
dar. Durch die große Wärmemenge, welche die Hydratation der Zemente freisetzt,
wird bei geringen Wasser/Zement-Verhältnissen aufgrund der adiabatischen Bedin-
gungen die Temperatur des Systems bis zum Siedepunkt der flüssigen Phase er-

Seite 42
höht. Das System wird durch Verdampfen von Wasser in seiner Zusammensetzung
verändert. Ein Rückschluss aus den Beobachtungen auf die Eigenschaften der Kom-
ponenten ist dadurch nicht möglich. Durch Zusatz inerter Komponenten, die Wärme
aufnehmen, kann die Temperaturerhöhung verringert werden.
Aus diesen Gründen wurde für die kalorimetrischen Messungen der Normmörtel
nach DIN EN 196 gewählt. Dieser Mörtel setzt sich zusammen aus 1350 g Quarz-
sand, 450 g Zement und einer für das geforderte Wasser/Zement-Verhältnis entspre-
chenden Menge entionisierten Wassers. Der nach dem in DIN EN 196 festgelegten
Mischregime hergestellte Mörtel wurde in Weißblechdosen (∅ = 100 mm; h = 100
mm) eingefüllt.
Zur Kontrolle des Wärmeabflusses wurden die gefüllten Dosen in Styropor-
Behältnissen platziert, die an allen Stellen eine Mindestdicke von 5 cm aufweisen.
Durch die Isolierung hindurch wurde ein Widerstandsthermometer PT100 mit der
Messstelle im Zentrum der Probe angeordnet. Der Temperaturverlauf im Mörtel wur-
de bis zum Ausgleich mit der Raumtemperatur aufgezeichnet.
Abbildung 3: Versuchsanordnung zur Bestimmung der Wärmefreisetzung bei der
Hydratation von Zementen
Bei der Betrachtung der Freisetzung der Hydratationswärme blieben mehrere
Punkte unberücksichtigt:
(a) Sofort nach Kontakt des Zementes mit Wasser reagieren diese beiden Stof-
fe miteinander. In dieser Anfangsreaktion wird eine sehr beträchtliche

Seite 43
Wärmemenge freigesetzt, die zur Erwärmung des Mörtels führt. Durch die
fehlende Isolierung beim Anmischen wird ein Teil dieser Wärme an die
Umgebung abgegeben und wird nicht als Temperaturdifferenz zur Raum-
temperatur erfasst.
(b) Durch die Erhöhung der Temperatur des Systems wird die Reaktion des
Zements mit dem Wasser nach dem Gesetz von Arrhenius beschleunigt.
Dadurch unterliegt die in der Auswertung ermittelte Abhängigkeit der Reak-
tionsgeschwindigkeit von der Zusammensetzung des Systems einem sys-
tematischen Fehler. Der akzeptierte Fehler stellt einen Kompromiss zu
Messungen unter konstanter Temperatur bzw. vollständiger Isolierung (adi-
abatische System) dar. Beide Systeme weisen unterschiedliche systemati-
sche Fehler auf. Ein großer Vorteil des gewählten Verfahrens besteht darin,
dass es die Reaktion in der Praxis (Massivbeton) am besten beschreibt.
(c) Die Isolierung der gewählten Versuchsanordnung weist eine geringe Wär-
medurchlässigkeit auf. Der Abfluss von Wärme durch die Isolierung ist nicht
an allen Stellen des Systems gleich groß. Dadurch entsteht eine zusätzli-
che Inhomogenität im System. Diese Ungleichmäßigkeit ist jedoch vernach-
lässigbar gering.
Die gemessenen Temperaturverläufe können unter Annahme einiger Modell-
Bedingungen als Wärmefluss angegeben werden. Hierzu wurden folgende Vereinfa-
chungen angewendet:
(a) Die Prüfanordnung besteht aus zwei Körpern: Dem äußeren Quader aus
Styropor, und dem inneren Zylinder aus dem hydratisierenden Zementleim.
Die Differenz beider Körper ergibt das Volumen des das System umhüllen-
den Styropor-Körpers.
(b) Unter Annahme einer an allen Stellen der Oberfläche des untersuchten
Systems gleichen Wanddicke ergibt sich als Gestalt für den Modell-Körper
eine Kugel, und für die Gestalt der Isolationsschicht eine Kugelhülle mit ei-
ner Wanddicke l.
(c) Die Differenz der Oberflächen des realen und des Modell-Körpers wird ver-
nachlässigt. Die Wärmeübergangswiderstände zwischen Zementleim und

Seite 44
Metalloberfläche der Dose und dieser zu Styropor sowie zwischen Styropor
und umgebender Luft werden vernachlässigt.
(d) Die spezifische Wärmekapazität von Zementleim wird in Anlehnung an [97]
mit Kkg
J920cp ⋅
= und die Wärmeleitfähigkeit des Styropor mit
KmW
02,0⋅
=λ angenommen.
Der äußere Körper hat die Gestalt eines Quaders. Sein Volumen ergibt sich aus
der Formel:
3Q m0,01010m250,0m201,0m201,0hbaV =⋅⋅=⋅⋅= . Gleichung 55
Der Radius der angenommenen äußeren Kugel errechnet sich nach der Formel:
m1341,014,34
3³m01010,04
3Vr 33 Qaußen =
⋅⋅=
π⋅⋅
= . Gleichung 56
Der innere Körper hat die Gestalt eines Zylinders. Sein Volumen errechnet sich
nach der Formel:
m³0,001298m150,04
)m105,0(14,3h
4d
hAV22
Z =⋅⋅=⋅⋅π=⋅= . Gleichung 57
Der Radius der angenommenen inneren Kugel errechnet sich nach der Formel:
m06768,014,34
3³m001298,04
3Vr 33 Zinnen =
⋅⋅=
π⋅⋅
= . Gleichung 58
Die mittlere Wanddicke der Styropor-Kugel-Hülle ergibt sich als Differenz zwi-
schen äußerem und inneren Radius. Sie beträgt 0,06641 m.
Die Richtigkeit dieses Modells wurde anhand der Abkühlung eines ausreagierten
Systems geprüft. Hierzu wurde ein System aus einem beendeten Versuch auf ca.
65 °C erhitzt. Das warme Prüfsystem wurde an die St elle eines hydratisierenden
Systems gesetzt. Der Temperaturverlauf des abkühlenden Systems wurde bis zum
Ausgleich mit der Raumtemperatur aufgezeichnet.

Seite 45
20
25
30
35
40
45
50
55
60
65
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00
Zeit [h]
Tem
pera
tur
[°C] gemessen
berechnet
Abbildung 4: Vergleich von gemessenem und berechnetem Temperaturverlauf bei
der Abkühlung eines ausreagierten Zementsystems (fast vollständige Überlagerung)
Aus der Temperatur im Inneren des Systems wurde nach der Formel für den
Wärmedurchgang:
Raum/SystemTtAl
Q ∆⋅∆⋅⋅λ= Gleichung 59
der Wärmefluss durch die Isolierung berechnet. Dieser Wärmefluss wurde als Ver-
lust im auskühlenden System verwendet, um nach der Formel:
tp TcmQ ∆∆⋅⋅= Gleichung 60
die resultierende Temperatur des Systems nach dem Zeitintervall ∆t zu berech-
nen.
Der Vergleich des gemessenen und des berechneten Temperaturverlaufes zeigen
eine gute Übereinstimmung. Das Modell stimmt mit ausreichender Genauigkeit mit
dem realen System überein.
4.7 Erstarren
Zemente ergeben mit Wasser vermischt eine sehr steife Suspension, den Zement-
leim. Durch physikalische und chemische Prozesse verändert sich die Viskosität des

Seite 46
Leims. Innerhalb weniger Stunden steift der Leim so stark an, dass er nicht mehr oh-
ne Bruch des Gefüges verformbar ist. Man spricht vom Erstarren des Leims.
Durch verschiedene Verformungsversuche kann die Viskositätsänderung (sog.
„Rücksteifen“) des Zementleims beobachtet werden. In dieser Arbeit wurde das Ver-
fahren nach Vicat gemäß DIN EN 196 angewendet. Hierzu wird ein Zementleim mit
einer in dieser Norm festgelegten Viskosität hergestellt. In Zeitabständen von 5 Minu-
ten wird eine Nadel mit einer Druckfläche von 1 mm² und einer Masse von 300 g auf
den Zementleim aufgesetzt. Es wird gemessen, wie tief die Nadel in den Leim ein-
sinkt. Als Erstarrungsbeginn wird in diesem Verfahren der Zeitpunkt angegeben, zu
dem die Nadel mindestens 1 mm weniger in den Leim eindringt, als die Höhe des
Leimkuchens beträgt. Als Erstarrungsende wird der Zeitpunkt angegeben, zu dem
die Nadel weniger als 1 mm in den Leimkuchen eindringt.
4.8 Rasterelektronenmikroskopie
Mit Hilfe eines REM vom Typ 500 der Firma Leica wurden die Hydratationsstadien
der beschriebenen Mischungen beobachtet.
Gearbeitet wurde mit Gold-bedampften Proben unter Hochvakuum. Die Proben
wurden zur Verringerung der Evakuierungszeiten im REM zuvor in der Vakuumkam-
mer eines Röntgenspektrometers schockartig im Vakuum getrocknet.
4.9 Korngrößenanalyse (Lasergranulometrie)
Die pulverförmigen Ausgangsstoffe wurden mit einem Lasergranulometer mit Tro-
ckendispergierung untersucht.
Bei diesem Analysenverfahren werden die Teilchen eines pulverförmigen Stoffes
dispergiert durch eine optische Messzelle geleitet. In der Zelle wird ein Laserstrahl an
der Oberfläche der einzelnen Körner gestreut. Auf der Rückseite der Zelle entstehen
Interferenzringe, welche durch entsprechende Fotodetektoren aufgezeichnet werden.
Unter Berücksichtigung von spezifischer Lichtabsorption des untersuchten Stoffes
wird aus der Anordnung der Interferenzringe die Korngröße des einzelnen Teilchens
bestimmt.
Von großer Bedeutung ist die Tatsache, dass es sich quasi um die eindimensiona-
le Abbildung eines 3-dimensionalen Gebildes handelt. Bei diesem Analysenverfahren
können dementsprechend z.B. längliche, plättchenförmige oder sphärische Teilchen

Seite 47
nicht voneinander unterschieden werden. Bei der Integration der Korngrößen zur
Korngrößenverteilung (nach Volumen oder unter Berücksichtigung der Reindichte
nach Masse) wird davon ausgegangen, das der Kornradius in allen Raumrichtungen
gleichgroß ist, es sich bei den Teilchen also um Kugeln handelt.
Bei sphärischen Teilchen entsprechen die gemessenen Korngrößen annähe-
rungsweise der Realität. Für die plättchenförmigen Teilchen ergibt sich eine gewisse
Unschärfe, so dass diese Ergebnisse nur einer groben Abschätzung der Korngrö-
ßenverteilung dienen können.

Seite 48
5 Charakterisierung der Ausgangsstoffe
Im Rahmen dieser Arbeit wurden 3 typische Zemente der Rohrdorfer Baustoff-
gruppe untersucht. Im einzelnen wurden ein Portlandzement CEM I 42,5 R, beste-
hend aus Portlandzement-Klinker und Sulfatträger, ein hochsulfatbeständiger Ze-
ment CEM I 42,5 R-HS aus dem Werk Eiberg, bestehend aus nahezu C3A-freiem
Klinker und Sulfatträger, sowie ein Hochofenzement CEM III/A 32,5 N-LH aus dem
Werk Rohrdorf, bestehend aus Portlandzementklinker, Sulfatträger und Hochofen-
schlacke verwendet. Alle genannten Zemente wurden der laufenden Produktion, na-
mentlich den Verkaufsilos entnommen. Es handelt sich um typische Zemente ihrer
Art.
Als Anmachwasser für die Zementleime und Mörtel wurde entionisiertes Wasser
verwendet. Für die chemischen Untersuchungen wurde destilliertes Wasser verwen-
det.
Alle übrigen verwendeten Reagenzien entsprachen in ihrer Reinheit der Qualität
„zur Analyse“.
Im folgenden wird die analytische Bestimmung der Phasenzusammensetzung die-
ser Zemente beschrieben.
5.1 Oxidische Zusammensetzung der Zemente
Die in dieser Arbeit verwendeten Zemente wurden einer Standardprüfung mittels
Röntgenfluoreszenz unterzogen. Die Bindemittel wurden zu Pulverpresslingen in
Stahlringen präpariert. In folgender Tabelle ist die oxidische Zusammensetzung der
Zemente wiedergegeben:

Seite 49
Tabelle 3: Oxidische Zusammensetzung der verwendeten Zemente, ermittelt mit-
tels Röntgenfluoreszenzanalyse
Element bzw. Oxid, (%)
CEM I 42,5 R CEM I 42,5 R-HS CEM III/A
CaO 62,85 62,87 49,15
SiO2 19,09 19,69 27,75
Al2O3 6,50 4,51 9,47
Fe2O3 2,79 6,31 1,47
SO3 3,74 3,06 3,76
MgO 3,02 2,08 6,35
K2O 0,85 0,56 0,89
Na2O wurde nasschemisch bestimmt. Der Gehalt betrug 0,29 % (Portlandzement),
0,31 % (HS-Zement) und 0,27 % (Hochofenzement). Auf eine Analyse der Spuren-
elemente wurde verzichtet, da der Einfluss derselben auf die im Rahmen dieser Ar-
beit geprüften Eigenschaften vernachlässigbar gering ist.
Die stöchiometrische Umrechnung der gemäß Tabelle 5 diffraktometrisch ermittel-
ten Sulfatträgergehalte ergibt die SO3-Gehalte, die in der folgenden Tabelle wieder-
gegeben sind:
Tabelle 4: Umrechnung der mit XRD ermittelten Sulfatträgergehalte in SO3-
Gehalte der drei Zemente
Element bzw. Oxid, (%)
CEM I 42,5 R CEM I 42,5 R-HS CEM III/A
SO3 3,60 3,46 3,67
Für den CEM I 42,5 R und Hochofenzement ergibt sich eine gute Übereinstim-
mung mit den ermittelten Calciumsulfatgehalten. Für den hochsulfatbeständigen Ze-
ment ist der Gehalt an Sulfatträger mit XRD über- oder mit XRF unterbewertet.

Seite 50
5.2 Zementzusammensetzung mittels Rietveld-Analytik
Die Ausgangszemente wie auch die erhaltenen Rückstände wurden nach Rietveld
auf ihre mineralogische Zusammensetzung untersucht. Die Ergebnisse sind in
Tabelle 5 wiedergegeben.
Tabelle 5: Mineralogische Zusammensetzung der Ausgangszemente und Auszü-
ge, ermittelt mittels Pulverdiffraktometrie und Rietveld-Verfeinerung
CEM I 42,5 R CEM I 42,5 R-HS CEM III/A Probe Mineral roh MSV1 BSV2 roh MSV BSV roh
Alit 59,73 0,27 3,45 70,51 2,54 4,17 27,77
Belit 10,07 1,08 3,18 3,39 0,37 n.n. 4,78
C3A cub. 9,75 13,39 0,05 0,42 0,52 n.n. 5,12
C3A orth. 1,95 16,23 0,48 n.n. 0,30 0,03 1,02
Brownmillerit 6,44 24,86 49,83 17,32 58,49 81,59 3,76
Periclas 3,40 9,61 14,17 1,50 4,30 4,41 1,56
Lime 0,04 0,02 0,45 0,07 0,23 0,44 n.n.
Quartz 0,13 1,36 0,06 n.n. 0,34 0,14 n.n.
Mayenite 1,22 2,82 17,31 0,33 6,42 1,32 0,71
Syngenit 0,95 11,59 4,80 n.a. 5,54 5,52 0,42
Bassanit 1,26 4,89 0,71 2,01 4,26 0,71 2,13
Anhydrit 3,53 9,72 0,44 2,75 11,56 0,79 3,22
Gips 1,79 4,13 5,06 1,58 5,14 0,87 1,30
Rest3 - - - - - - 48,21 1 Salicylsäure-Auszug; 2 Borsäure-Auszug; 3 Hüttensand, bestimmt durch Korrek-
tur mit Rutil als innerem Standard
Alle Zemente wurden zunächst unverändert („roh“) diffraktometrisch vermessen
und ihre Phasengehalte mittels Rietveld-Verfeinerung bestimmt. Die dabei erhalte-
nen Ergebnisse sind in den Spalten „roh“ von Tabelle 5 angegeben. Wie bereits vor-
her beschrieben, liefert dieses Verfahren bei Phasen mit geringem Gehalt (z.B. C3A,
C12A7 usw.) relativ ungenaue Werte.
Zur genaueren Charakterisierung der Zemente wurden einzelne Phasen aus der
Matrix selektive herausgelöst. Somit ist es möglich, im ersten Schritt die Calciumsili-
kate und im zweiten Schritt das Calciumaluminat zu lösen. Die Calciumsulfate wer-
den dabei teilweise gelöst. Auf die Quantifizierung der gelösten Calciumsulfate wurde
mit Rücksicht auf das Ziel der Analyse, der Bestimmung der Calciumaluminate und –

Seite 51
alumoferrite, an dieser Stelle verzichtet. Dadurch werden die jeweils verbleibenden
Phasen ohne störende Überlagerungen messbar. Auf diesem Wege wurden die Be-
standteile der untersuchten Zemente qualitativ und teilweise auch quantitativ unter-
sucht.
HS-Zement
BSV HS-Zement
MSV HS-Zement
Lin
(Cps
)
1500
2000
3000
4000
5000
6000
7000
� � � � � � � � � � � � �35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
Lin
(Cps
)
1500
2000
3000
4000
5000
6000
7000
�10.1 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35
Bel
ite
Alit
e
Alite + Belite
BM
BM
BM
Alit
e
Alit
e
DH
BM
AH
Alit
e
Alite + Belite
C3A
BM
Alit
eZement
Salicylsäure-Auszug
Borsäure-Auszug
Abbildung 5: Röntgenbeugungsdiffraktogramme des CEM I 42,5 R-HS: [rot] – Or-
ginalsubstanz; [grün] – Rückstand des Salicylsäure-Auszug; [blau] – Rückstand des
Borsäure-Auszug (DH – Calciumsulfatdihydrat; BM – Brownmillerit; AH – Anhydrit)
Abbildung 5 zeigt am Beispiel des CEM I 42,5 R die mit den verschiedenen Pro-
ben („roher“ Zement bzw. Rückstände des Salicylsäure- und Borsäure-Auszüge) er-
haltenen Diffraktogramme. Die für Zement typischen Mineralien sind alle gut nach-
weisbar. Vielfach überlagern sich jedoch analytisch wichtige Reflexe. Daher wurde
zur Quantifizierung das Verfahren nach Rietveld verwendet.
In Abbildung 5 ist deutlich zu erkennen, dass durch den Methanol-Salicylsäure-
Auszug die Calciumsilikate C3S und C2S nahezu vollständig gelöst werden. Dagegen
reichern sich die anderen Phasen deutlich an. Die Calciumsulfate, wie auch Mayenite
und Syngenit (K2SO4�CaSO4�H2O) werden zum Teil gelöst. Zu erkennen ist diese
Tatsache am Verlust in der Massebilanz der jeweiligen Minerale zwischen Beginn

Seite 52
und am Ende der Auszüge. Somit ist eine genauere Quantifizierung dieser Phasen
auch mit dieser Methode leider nicht möglich.
Die Auswaage der Auszüge ergab eine Zusammensetzung der Zemente, die in
Tabelle 6 dargestellt ist.
Tabelle 6: Mineralogische Zusammensetzung der Zemente nach den Lösungsver-
suchen
Probe CEM I 42,5 R CEM I 42,5 R-HS
Calciumsilikate 71,0 % 74,0 %
Calciumaluminate und z.T. Calciumsulfat
13,5 % 7,9 %
Calciumalumoferrite, Periclas, z.T. Mayenit und z.T. Calciumsulfat
15,33 % 18,1 %
CEM I 42,5 R-HS
43-0606 (I) - Calcium Sulfate - alpha-CaSO4
41-0224 (I) - Bassanite, syn - CaSO4· 0.5H2O
33-0311 (*) - Gypsum, syn - CaSO4· 2H2O
38-1429 (*) - Calcium Aluminum Oxide - Ca3Al2O6
30-0226 (*) - Brownmillerite, syn - Ca2(Al,Fe+3)2O5
06-0602 (N) - Calcium Iron Oxide - CaO· 2FeO
HS-Zement MSV - File: HS-Zement MSV.raw
HS-Zement BSV - File: HS-Zement BSV.raw
HS-Zement - File: HS-Zement.raw
� � � � � � � � � � � � �35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59
�10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34
Borsäure-Auszug
Salicylsäure-Auszug
Zement
Abbildung 6: Auszüge des CEM I 42,5 R-HS mit hinterlegten Mineralien
Der CEM III besteht aus 48 M-% Hüttensand (geprüft nach DIN EN 196 – mikro-
skopische Auszählung), Portlandzementklinker, natürlichem Anhydrit und REA-Gips.

Seite 53
Als Hochofenschlacke wurde Hüttensand der Firma Voest aus Linz zur Herstellung
des Zements verwendet. Der F-Wert1 des Hüttensandes beträgt 1,33. Es handelt
sich demnach um einen für die Anwendung in Zement wenig reaktiven Hüttensand.
Diese Ergebnisse stimmen gut mit den Ergebnissen der quantitativen Auswertung
der rohen Zemente nach Rietveld überein.
In ihrer qualitativen und quantitativen Zusammensetzung entsprechen die verwen-
deten Zemente nach dieser Untersuchung typischen Vertretern ihrer Art. Der Gehalt
an Calciumsilikaten stimmt in den zwei schlackefreien Zementen gut überein. Im
hochsulfatbeständigen Zement ist fast kein Calciumaluminat enthalten. Dagegen ist
der Gehalt an Calciumalumatferrit im CEM I 42,5 R-HS deutlich höher als im
CEM I 42,5 R. Der hochsulfatbeständige Zement enthält deutlich weniger freies Pe-
riclas als der Portlandzement. Ein direkter Einfluss auf die Reaktivität anderer Pha-
sen ist aber nicht bekannt. Daher wird dem Unterschied im MgO-Gehalt zwischen
den Zementen keine Bedeutung beigemessen. Die Gehalte der Sulfatträger (An-
hydrit, Bassanit und Gips) unterscheiden sich, da die Zemente jeweils auf ein optima-
les Erstarren eingestellt sind. Die Sulfatträger haben jedoch weniger Einfluss auf die
Reaktion der Calciumsilikate. Durch den Eintrag von Calciumionen in die Porenlö-
sung wird das Angebot von Kalk zum Einbau in die Hydratphasen beeinflusst. Im
Rahmen der vorliegenden Arbeit wird jedoch nicht weiter auf den Einfluss der ver-
schiedenen Sulfatträger auf die Silikathydratation eingegangen. Dagegen wird die
Reaktion der Calciumaluminate, - alumoferrite sowie der Schlacke von den Calcium-
sulfaten sicherlich stark qualitativ und quantitativ beeinflusst. Die Calciumsulfate tei-
len sich ungleichmäßig auf Anhydrit, Bassanit und Gips auf. Anhydrit überwiegt in
jedem Fall. Das bei der Herstellung der Zemente zugegebene Dihydrat wurde bei der
Mahlung in den Kugelmühlen teilweise zu Halbhydrat (Bassanit) entwässert. In allen
untersuchten Zementen wurden Sulfatträger derselben Quelle (natürlicher Anhydrit
und REA-Gips) verwendet. Die Zemente wurden jedoch auf verschiedenen Mühlen
gemahlen. Die Unterschiede im Sulfatangebot bei der Hydratation der Zemente be-
ruhen demnach auf der unterschiedlichen Dosierung im Zement und der unterschied-
1 F = MnOSiO
CaS5,0MgO5,0OAlCaO
2
32
+⋅+⋅++

Seite 54
lichen Entwässerung bei der Vermahlung. Es bestehen jedoch keine wesentlichen
Unterschiede in der Art und den Mengenanteilen der Sulfatträger.
5.3 Festigkeitsentwicklung nach DIN EN 196
Die verwendeten Zemente wurden nach der oben genannten Norm auf ihre Fes-
tigkeitsentwicklung untersucht. Es wurden für diese Zemente typische Werte gefun-
den. Die ermittelten Werte wurden im Rahmen der weitergehenden Untersuchungen
in dieser Arbeit verwendet und sind später als Festigkeitsentwicklung mit angegeben.
5.4 Erstarren
Die Prüfung des Erstarrens nach Vicat ergab folgende Werte:
Tabelle 7: Erstarrungszeiten in Minuten der verwendeten Zemente nach Vicat
Probe Beginn Ende
CEM I 42,5 R 155 195
CEM I 42,5 R-HS 170 230
CEM III/A 195 290
Auf Grund der gegenüber dem Normalzement CEM I 42,5 R unterschiedlichen mi-
neralogischen Zusammensetzung (C3A-Gehalt) erstarrt der CEM I 42,5 R-HS deut-
lich langsamer. Der Erstarrungsbeginn tritt nur wenig später ein als beim Portland-
zement, das Erstarrungsende verzögert sich.
Die im CEM III/A enthaltene Hochofenschlacke reagiert zu Beginn der Hydratation
nur sehr langsam. Daher ist ihr Beitrag zum Erstarren des Zementes eher gering. Es
ergibt sich ein Verdünnungseffekt im erstarrenden Zementleim. Die für die Indikation
erforderliche Viskosität des Leimes wird daher deutlich später als beim Normal- und
hochsulfatbeständigen Zement erreicht.
5.5 Kornverteilung und Feinheit
Die Ergebnisse der Analyse der Korngrößenverteilung sind in Abbildung 7 bis
Abbildung 9 wiedergegeben.
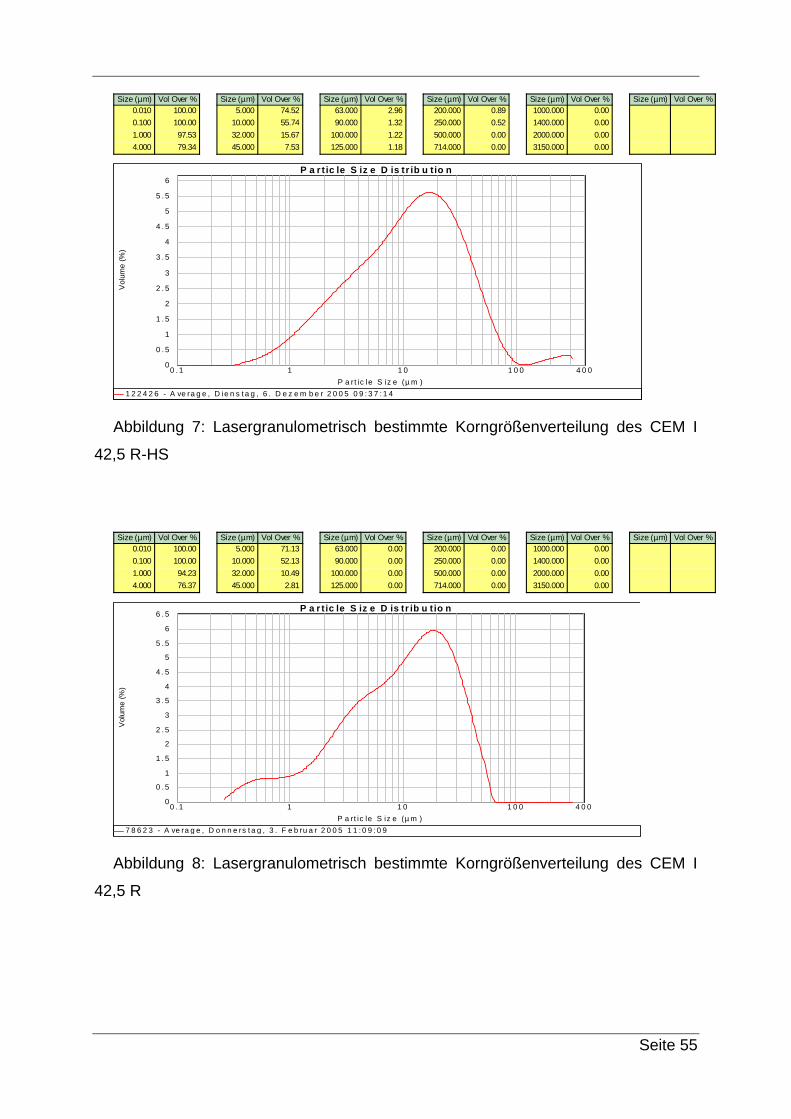
Seite 55
Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over %0.010 100.00 5.000 74.52 63.000 2.96 200.000 0.89 1000.000 0.00
0.100 100.00 10.000 55.74 90.000 1.32 250.000 0.52 1400.000 0.00
1.000 97.53 32.000 15.67 100.000 1.22 500.000 0.00 2000.000 0.00
4.000 79.34 45.000 7.53 125.000 1.18 714.000 0.00 3150.000 0.00 P a r t ic le S iz e D is t r ib u t io n
0 . 1 1 1 0 1 0 0 4 0 0
P a rt ic le S iz e (µ m )
0
0 . 5
1
1 . 5
2
2 . 5
3
3 . 5
4
4 . 5
5
5 . 5
6
Vol
ume
(%)
1 2 2 4 2 6 - A ve ra g e , D ie n s t a g , 6 . D e z e m b e r 2 0 0 5 0 9 : 3 7 : 1 4
Abbildung 7: Lasergranulometrisch bestimmte Korngrößenverteilung des CEM I
42,5 R-HS
Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over %0.010 100.00 5.000 71.13 63.000 0.00 200.000 0.00 1000.000 0.00
0.100 100.00 10.000 52.13 90.000 0.00 250.000 0.00 1400.000 0.00
1.000 94.23 32.000 10.49 100.000 0.00 500.000 0.00 2000.000 0.00
4.000 76.37 45.000 2.81 125.000 0.00 714.000 0.00 3150.000 0.00 P a r t ic le S iz e D is tr ib u t io n
0 . 1 1 1 0 1 0 0 4 0 0
P a rt ic le S iz e (µ m )
0
0 . 5
1
1 . 5
2
2 . 5
3
3 . 5
4
4 . 5
5
5 . 5
6
6 . 5
Vol
ume
(%)
7 8 6 2 3 - A ve ra g e , D o n n e rs t a g , 3 . F e b ru a r 2 0 0 5 1 1 : 0 9 : 0 9
Abbildung 8: Lasergranulometrisch bestimmte Korngrößenverteilung des CEM I
42,5 R

Seite 56
Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over % Size (µm) Vol Over %0.010 100.00 5.000 74.15 63.000 2.99 200.000 1.06 1000.000 0.00
0.100 100.00 10.000 55.66 90.000 1.28 250.000 0.67 1400.000 0.00
1.000 97.48 32.000 15.70 100.000 1.20 500.000 0.00 2000.000 0.00
4.000 79.03 45.000 7.51 125.000 1.20 714.000 0.00 3150.000 0.00 P a r t ic le S iz e D is tr ib u t io n
0 . 1 1 1 0 1 0 0 4 0 0
P a rt ic le S iz e (µ m )
0
0 . 5
1
1 . 5
2
2 . 5
3
3 . 5
4
4 . 5
5
5 . 5
6
Vol
ume
(%)
1 2 2 4 2 8 - A ve ra g e , D ie n s t a g , 6 . D e z e m b e r 2 0 0 5 0 9 : 4 1 : 4 1
Abbildung 9: Lasergranulometrisch bestimmte Korngrößenverteilung des CEM
III/A 32,5 N
Die Zemente weisen eine für ihre Art typische Korngrößenverteilung und Feinheit
auf. Der CEM I 42,5 R ist etwas feiner als die anderen zwei Zemente. Sein mittlerer
Korndurchmesser (Durchgang 50 %) beträgt 10,7 µm. Die maximale Korngröße liegt
unterhalb 63 µm. Der hochsulfatbeständige Zement ist etwas gröber. Der mittlere
Korndurchmesser beträgt 11,9 µm. Dieser Zement enthält Teilchen bis ca. 100 µm.
Der Hochofenzement entspricht in der Korngrößenverteilung dem hochsulfatbestän-
digen Zement. Der mittlere Korndurchmesser und der Durchmesser der größten ent-
haltenen Partikel entsprechen denen des CEM I 42,5 R-HS. Die Korngrößenvertei-
lungen der untersuchten Zemente stimmen relativ gut überein. Der Einfluss der
Korngrößen auf die Reaktivität der Zemente wird daher vernachlässigt.

Seite 57
6 Ergebnisse
Das Auflösen bzw. Aufbauen von Kristallen stellt einen Prozess dar, bei dem in ei-
nem binären System von einer Kristalloberfläche Teilchen in die Lösung übergehen
bzw. auf dem Kristall sich abscheiden. Mit Beginn der Reaktion gehen verschiedens-
te Ionen in Lösung. Beim Zement wird innerhalb weniger Minuten das Anmachwas-
ser mit Calciumionen übersättigt. Nach einigen Stunden geht die Konzentration an
Calciumionen in der Lösung wieder auf die Konzentration des Löslichkeitsprodukts
von Calciumhydroxid zurück. Natrium- und Kaliumionen werden in großer Menge
gelöst. Dadurch steigt der pH-Wert der Lösung auf deutlich über 12,4 (pH einer ge-
sättigten Calciumhydroxidlösung). Auch Sulfat wird in den ersten Stunden in sehr
großer Menge gelöst. Kieselsäure, Aluminium- und Eisenionen werden wenig gelöst.
Die räumliche Lage der Grenzfläche zwischen den zwei beteiligten Phasen (Fest-
stoff/Flüssigkeit) ist nicht konstant. Die zeitliche Änderung ihrer Position ist jedoch
um Größenordnungen geringer als die Transportgeschwindigkeit der Teilchen in der
flüssigen Phase. Diese wird durch Konzentrationsunterschiede (Diffusion) bestimmt.
Mit diesen Verschiebungen im System ändern sich die Gehalte der Minerale wie
auch die makroskopischen Eigenschaften des Zementleims.
6.1 Festigkeitsentwicklung der Zemente
Die Haupteigenschaft von Bindemitteln, speziell auch von Zementen, ist der Über-
gang aus einer formbaren Konsistenz in einen festen, erhärteten Zustand. Bindemit-
tel sollen andere Materialien verbinden und Kräfte übertragen. Die Fähigkeit mecha-
nische Kräfte weiterzuleiten, drückt sich in der Festigkeit eines Materials aus. Auf
Grund der relativ langsam verlaufenden chemischen Reaktion von Zement mit Was-
ser wird auch die Festigkeit über einen Zeitraum von mehreren Tagen bzw. Wochen
entwickelt. Um die großtechnische Produktion von Zement bzw. seinen Vorprodukten
zu steuern, wird seit Beginn der Zementherstellung versucht, die Festigkeitsentwick-
lung vorherzubestimmen. Hierzu werden verschiedene Ansätze verfolgt. So werden
bei relativ gleich bleibenden Zementen empirische Funktionen der Festigkeiten zu 28
Tagen und später zu den Festigkeiten nach 1 und 2 Tagen erstellt. Zum anderen
wurde von verschiedenen Autoren versucht, aus der chemischen Zusammensetzung
und/oder physikalischen Eigenschaften der untersuchten Zemente auf die Festigkei-
ten zu schließen. In diesem Zusammenhang werden je nach Verfügbarkeit z.B. die

Seite 58
Korngrößenverteilungen bzw. Feinheiten und die Zusammensetzung der Zemente
(Gehalte der Oxide der Hauptelemente) als Variablen für die empirischen Funktionen
der Festigkeitsprognosen verwendet [98], [99]. Eine mathematische Berechnung auf
Basis real im erhärtenden Zementstein bzw. Beton ablaufender Prozesse (Lösung,
chemische Umsetzung, Kristallisation, Absorptions- und Resoptionsprozesse) wurde
in der Literatur nicht gefunden [100], [101]. Es liegen jedoch allgemeine Aussagen
über die Festigkeitsentwicklung der separaten Hauptphasen des Portlandzements
vor [18], [102]. Ebenso liegen Untersuchungsergebnisse zur Korrelation zwischen
Hydratationsgrad und Festigkeit bzw. zwischen früher und später Festigkeit vor [103].
Auf Basis der Modellierung der Mikrostruktur des hydratisierenden Zementleims
(vergleiche 1.3.3) und der Gel-Poren Theorie von Powers und Brownyard entwickelte
Bentz ein Computermodell, dass die Festigkeitsentwicklung von Zement simuliert
[71]. Das Modell bezieht neben der empirischen Modellierung der einzelnen Hydrata-
tionsreaktionen auf Basis der isothermen Kalorimetrie die räumliche und mechani-
sche Struktur des Zementsteins mit ein. Dadurch gelingt es, mit diesem Modell die
Festigkeit von Zementstein und Beton in guter Übereinstimmung mit der Praxis vor-
auszusagen [104].
Die Festigkeit von zementären Systemen basiert auf dem Vorhandensein von für
die Festigkeitsbildung geeigneten festen Phasen. Hierzu zählen vor allem die CSH-
Phasen. Calciumaluminathydrat und Ettringit spielen vor allem in den früheren Sta-
dien der Hydratation eine Rolle. Portlandit und andere kristalline Phasen tragen
kaum zur Festigkeit des Zementsteins bei. Die Festigkeit des zementären Systems
hängt direkt von der Konzentration der festigkeitsbildenden Phasen ab. Die Festigkeit
steht in direktem Zusammenhang mit der Konzentration der Kraftübertragungspunk-
te:
)c(F PktDF =β Gleichung 61
Die Kraftübertragungspunkte entstehen durch die Hydratation der Zementbestand-
teile mit Wasser zu den entsprechenden Hydratphasen.
Zu Beginn der Hydratation liegt eine Paste vor, die einer Verformung kaum Wider-
stand entgegensetzt. Diese Paste erstarrt nach wenigen Stunden. Es entsteht ein

Seite 59
steifer Körper. Die Bruchfestigkeit dieses Körpers nimmt bei ungestörter Reaktion mit
der Zeit zu. Die Festigkeit von Zementstein ist abhängig von der Zeit.
)t(fDF =β Gleichung 62
Demnach gilt
)t(f)c(F Pkt = oder ))t(f('FcPkt = bzw. )t(cPkt Φ= Gleichung 63
Im folgenden Model wurde geprüft, ob ein Zusammenhang nach den Reaktions-
geschwindigkeitgesetzen vorliegt. Die Funktion nach Gleichung 58 wurde als kon-
stant vorausgesetzt. Die Druckfestigkeit wurde als Ausdruck für die Konzentration
der festigkeitsübertragenden Kontaktpunkte im Zementstein verwendet.
PktDF c→β
In den nachstehenden Bildern ist die Festigkeitsentwicklung der Zemente bei ei-
nem Wasser/Zement-Verhältnis von 0,5 wiedergegeben. Die Prüfzeitpunkte wurden
so festgelegt, dass eine möglichst gleichmäßige Verteilung der Messpunkte erhalten
wurde.
Es wurde beobachtet, dass die Erhärtung in zwei Phasen abläuft – einer schnellen
Anfangsreaktion in den ersten Stunden und, offenbar verbunden mit einem Wechsel
des Reaktionsgeschwindigkeitsgesetzes, einem langsamer verlaufenden anschlie-
ßenden Festigkeitszuwachs.
6.1.1 Portlandzement CEM I 42,5 R
Aus den stöchiometrischen Reaktionsgleichungen der Calciumsilikate (Gleichung
36 bis 38) folgt jeweils ein Reaktionsverlauf 1. Ordnung bezogen auf den Ausgangs-
stoff. Würde die Festigkeitsentwicklung des Zementleimes nur auf der Reaktion einer
einzigen Komponente beruhen, sollte die Festigkeitsentwicklung der Gleichung
tk0 e)A(c)A(c ⋅−⋅= Gleichung 64
folgen.

Seite 60
Die Erhärtung des Portlandzementes verläuft jedoch in den ersten 24 Stunden
sehr rasch. Der Anstieg der Kurve im ln(c)-ln(t)-Diagramm (Abbildung 10) ist in dieser
Zeit linear. Demnach gilt tatsächlich:
)tln(k)cln()cln( 0 ⋅+= Gleichung 65
, wobei c für die Druckfestigkeit steht.
Durch Umformen dieser Gleichung ergibt sich:
)tln(
)cln()cln(k 0−
= Gleichung 66
bzw. nach den Logarithmengesetzen [105]:
)cc
(logk0
t= bzw. 0
k
cc
t = . Gleichung 67
Daraus wiederum ergibt sich als allgemeine Formel für die Abhängigkeit der Fes-
tigkeit von der Zeit:
k0 tcc ⋅= . Gleichung 68
Der Reaktionsverlauf bezogen auf die Festigkeitsentwicklung entspricht somit ei-
ner Ordnung größer als 2 (vgl. Seite 8).
Die Geschwindigkeitskonstante k entspricht im Falle der Festigkeitsentwicklung
des Portlandzementleimes dem Anstieg der Kurve (Geraden) im ersten Abschnitt der
Hydratation (Abbildung 10). Eine Gerade wird durch mindestens zwei Punkte durch
die Formel
12
1
12
1
xx
xx
yy
yy
−−
=−
− Gleichung 69
beschrieben. Unter Einbeziehung der vorhandenen Messpunkte ergibt sich durch
Mittelung der so erhaltenen Konstanten die mittlere Geschwindigkeitskonstante der
Erhärtung von Portlandzement bei einem Wasser/Zement-Verhältnis von 0,5 zu
644,2k =

Seite 61
Gemäß diesem Ergebnis ist die Ordnung der Reaktion bei der Anfangshydratation
von Zement größer als 2. Die Festigkeitsentwicklung wird demnach durch die Reak-
tion mehrerer Minerale beeinflusst.
Die Ausgangskonzentration, also die Festigkeit zum Zeitpunkt t0, berechnet sich
zu:
0072,0c 0 = MPa
Sie ist also vernachlässigbar klein. Dieser Wert stimmt mit der Tatsache überein,
dass der Zementleim zu Beginn der Hydratation keine messbare Festigkeit aufweist,
vielmehr ist er pastös.
Nach dieser schnellen Anfangsreaktion wechselt die Kinetik der Reaktion schlag-
artig. Ab 28 Stunden verläuft der Anstieg der Festigkeit linear mit der Quadratwurzel
der Zeit (Abbildung 11). Somit liegt eine Abhängigkeit vor, die auf eine diffusionskon-
trollierte Reaktion hindeutet. Ein oder mehrere Stoffe diffundieren durch eine Schicht
eines oder mehrerer anderer Stoffe. Ob der oder die diffundierenden Stoffe den Aus-
gangsstoffen oder den Reaktionsprodukten zuzuordnen sind, kann aus diesen Er-
gebnissen nicht gefolgert werden.

Seite 62
1 2 5 10 20 36 50 100 200 500Zeit [h]
0.1
1
10
100D
ruck
fest
igke
it [M
Pa]
CEM I 42,5 RCEM I 42,5 RCEM I 42,5 R-HSCEM I 42,5 R-HSCEM III/A 32,5 N-LHCEM III/A 32,5 N-LH
Abbildung 10: Zeitlicher Verlauf der Festigkeitsentwicklung der Zemente in loga-
rithmischer Darstellung, ermittelt durch Druckfestigkeit von Normmörtel-Prismen in
Anlehnung an EN 196-1
6.1.2 Hochsulfatbeständiger Zement CEM I 42,5 R-HS
Ähnlich wie der normale Portlandzement reagiert auch der hochsulfatbeständige
Zement zu Beginn sehr schnell. Die Festigkeitsentwicklung entspricht auch bei die-
sem Zement einer Reaktion höherer Ordnung.
Unter Einbeziehung der vorhandenen Messpunkte ergibt sich durch Mittelung der
so erhaltenen Konstanten als mittlere Geschwindigkeitskonstante der Erhärtung von
hochsulfatbeständigem Zement bei einem Wasser/Zement-Verhältnis von 0,5 zu
577,2k = . Sie ist also etwas niedriger als die Konstante für normalen Portlandze-
ment.
Die Ausgangskonzentration, also die Festigkeit zum Zeitpunkt t0, berechnet sich
zu 0045,0c 0 = MPa. Sie ist ebenfalls vernachlässigbar klein.

Seite 63
Im Gegensatz zur Festigkeitsentwicklung des Normalzements verläuft der Festig-
keitszuwachs des hochsulfatbeständigen Zementes nach den ersten 24 Stunden
weder rein kinetisch kontrolliert noch rein diffusionskontrolliert. Vielmehr muss davon
ausgegangen werden, dass die Ordnung der Reaktion mehrmals wechselt. Jedoch
überwiegt im ersten Abschnitt eindeutig die kinetische Kontrolle.
12 5 10 20 36 50 100 200 500Zeit [h]
0
10
20
30
40
50
60
70
Dru
ckfe
stig
keit
[MPa
]
CEM I 42,5 RCEM I 42,5 RCEM I 42,5 R-HSCEM I 42,5 R-HSCEM III/A 32,5 N-LHCEM III/A 32,5 N-LH
Abbildung 11: Festigkeitsentwicklung der Zemente als Funktion der Quadratwurzel
der Zeit, ermittelt in Anlehnung an EN 196-1
6.1.3 Hochofenzement
Die Festigkeitsentwicklung des Hochofenzementes verläuft im logarithmischen
System nur bis 9 Stunden annähernd linear. Nach dieser Zeit ist eine lineare Abhän-
gigkeit für die Festigkeit = f(Wurzel(Zeit)) gegeben. Die Kinetik der Reaktion wechselt
also sehr früh zur Kontrolle durch Diffusion.
Die Streuung der Messwerte im Bereich der kinetischen Kontrolle ist zu groß für
verlässliche Berechnung der Geschwindigkeitskonstante.

Seite 64
Ebenso wie bei den hauptsächlich aus Klinker bestehenden Portlandzementen
kann aus den Versuchen zur Festigkeitsentwicklung des schlackehaltigen Hochofen-
zementes keine direkte Aussage über die an der Hydratation beteiligten Mineralien
(Ausgangsphasen oder bereits Reaktionsprodukte) getroffen werden.
6.2 Kalorimetrie
Zur Bestimmung der Reaktionsgeschwindigkeit wird bei der Untersuchung der
Hydratation von Zementen oft der Wärmefluss des reagierenden Systems herange-
zogen [106], [107]. So hat Kuzel ein Kalorimeter beschrieben, mit dem es möglich ist,
die Freisetzung der Hydratationswärme vom Zeitpunkt des Kontaktes des Wassers
mit dem zu untersuchenden Material zu erfassen [78], [108]. Solche Systeme sind
gut geeignet, um geringe Wärmeflüsse bei sehr geringen gerätetechnischen Mess-
unsicherheiten nachzuweisen. Als Problem erweisen sich aber bei vielen Geräten die
geringen Größen der untersuchten Systeme (Einwaagen). Die Wechselwirkung mit
der Umgebung kann ebenfalls meistens nicht ausgeschlossen werden. Aus diesen
Gründen wurde auf eine einfache Apparatur zurückgegriffen, die zwar nicht die Emp-
findlichkeit der oben beschriebenen Kalorimeter aufweist, für die kinetische Betrach-
tung des Reaktionsverlaufs aber genügen sollte.
In der Zeit des Anmischens des Mörtels finden vor allem Zerkleinerungs- und Be-
netzungsprozesse statt. Erste Lösungsvorgänge sind zu beobachten. Neubauer [78]
zeigte röntgenographisch, dass zu diesem Zeitpunkt erste Ettringitkristalle entstehen.
Diese Prozesse laufen ausnahmslos an den Oberflächen der beteiligten Komponen-
ten des Systems ab. Sie werden durch die Konzentration der beteiligten Phasen an
den Oberflächen (und der durch das Mischregime vorgegebenen Bewegung der
Phasen zueinander) bestimmt.
Die untersuchten Zemente wurden jeweils mit Wasser/Zement-Verhältnissen von
0,35; 0,40; 0,45; 0,50 und 0,55 als Mörtel nach DIN EN 196-1 (W/Z = 0,5; 450 g Ze-
ment + 1350 g Normsand + 225 g VE-Wasser) angesetzt. Die Ausgangstemperatur
aller Materialien und Geräte betrug 20 °C. Die Frei setzung der Hydratationswärme
wurde über den Verlauf der Temperatur im reagierenden System verfolgt. Über die
Charakteristik des Wärmeausgleichs mit der Umgebung wurde aus dem Tempera-
turverlauf der einzelnen Messungen der jeweilige Wärmefluss berechnet.

Seite 65
6.2.1 Normaler Portlandzement (CEM I 42,5 R)
In den ersten Minuten nach Wasserzugabe ist eine Erhöhung der Temperatur um
5 bis7 K zu beobachten. Mit fallendem Wasser/Zement-Verhältnis steigt der Betrag
des Temperaturanstiegs an (Abbildung 12).
Anschließend bleibt die Temperatur des Systems für ca. 2 Stunden konstant. Da-
nach steigt die Temperatur exponentiell mit der Zeit an. Mit fallendem Was-
ser/Zement-Verhältnis nimmt die Geschwindigkeit des Anstieges wie auch die er-
reichte Höchsttemperatur von 55 bis 60 °C zu.
20
25
30
35
40
45
50
55
60
0 6 12 18 24 30 36 42 48
Zeit [h]
Tem
pera
tur
[°C]
0,35
0,40
0,45
0,50
0,55
Abbildung 12: Zeitlicher Temperaturverlauf beim Abbinden der Mörtel aus
CEM I 42,5 R in Abhängigkeit vom Wasser/Zement-Wert
Nach 8 bis 12 Stunden endet die schnelle Wärmefreisetzung, das System kühlt
aus.
Bei bekannter Masse der Probe kann man von der Temperaturentwicklung auf
den Wärmefluss schließen. Der zeitliche Verlauf des Wärmeflusses bei der Hydrata-
tion des CEM I 42,5 R ist in Abbildung 13 dargestellt. Als Wärmeentwicklung ausge-
drückt, ergibt sich bei der Betrachtung des Temperaturverlaufes ein etwas anderes
Bild: Innerhalb der ersten Minuten ist eine sehr große Wärmefreisetzung zu beobach-
ten. Diese kann nicht genau quantifiziert werden, da ein unbestimmter Teil der Wär-
me beim Anmischen durch das Mischbehältnis, den Rührer und an die Umgebung
abgegeben wird. Nach dem Abfall der Wärmeentwicklung auf 2 bis 3 mW/g beginnt
ein langsamer Anstieg der Wärmefreisetzung. Nach 5 bis 8 Stunden (variiert in Ab-

Seite 66
hängigkeit vom W/Z) wird ein erster Wendepunkt durchlaufen. Die Wärmefreisetzung
nimmt kurz ab, anschließend nimmt sie wieder zu. Nach 7 bis 10 Stunden wird ein 2.
Maximum der Wärmefreisetzung erreicht. Mit steigendem Wasser/Zement-Verhältnis
nimmt die Zeit bis zum Erreichen dieses Maximums zu und der Wert der maximalen
Wärmefreisetzung ab.
0
5
10
15
20
25
30
35
0 6 12 18 24 30 36 42 48
Zeit [h]
Wär
mef
luß
[mW
/g]
0,35
0,40
0,45
0,50
0,55
Abbildung 13: Zeitlicher Verlauf des Wärmeflusses beim Abbinden von Mörteln
aus CEM I 42,5 R
6.2.2 Hochsulfatbeständiger Zement (CEM I 42,5 R-HS )
Wie beim CEM I 42,5 R, steigt auch bei diesem Zement die Temperatur in den
ersten Minuten um mehrere Grad K an.
Das System verharrt bei gleichen Bedingungen deutlich länger bei gleicher Tem-
peratur als das System mit CEM I 42,5 R (Abbildung 14). Die Geschwindigkeit des
Temperaturanstiegs ist ebenfalls geringer als beim normalen Portlandzement. Die
erreichten Höchsttemperaturen liegen mit ca. 50 °C deutlich niedriger. Dieses Verhal-
ten ist auf den wesentlich geringeren C3A-Gehalt des hochsulfatbeständigen Ze-
ments zurückzuführen. Die Abhängigkeit der Reaktivität vom Wasser/Zement-
Verhältnis entspricht der des Systems mit normalem Portlandzement. Jedoch folgt
die maximale Temperatur nicht dieser Abhängigkeit.

Seite 67
20
25
30
35
40
45
50
55
60
0 6 12 18 24 30 36 42 48
Zeit [h]
Tem
pera
tur
[°C]
0,35
0,40
0,45
0,50
0,55
Abbildung 14: Zeitlicher Temperaturverlauf beim Abbinden der Mörtel aus hochsul-
fatbeständigem Zement CEM I 42,5 R-HS in Abhängigkeit vom W/Z-Wert
Der Abfall der Temperatur nach Ende der schnellen Wärmefreisetzung erfolgt et-
wa gleichschnell wie im System mit CEM I 42,5 R.
0
5
10
15
20
25
30
35
0 6 12 18 24 30 36 42 48
Zeit [h]
Wär
mef
luß
[mW
/g]
0,35
0,40
0,45
0,50
0,55
Abbildung 15: Zeitlicher Verlauf des Wärmeflusses beim Abbinden von Mörteln
aus hochsulfatbeständigem Zement CEM I 42,5 R-HS
Berechnet als Wärmefluss zeigt sich ein sehr hoher Wärmefluss in den ersten Mi-
nuten der Versuche. Danach fällt die Wärmefreisetzung zurück auf ca. 2 mW/g. An-
schließend steigt der Wärmefluss aus dem untersuchten System auf bis zu 17 mW/g

Seite 68
an (dies ist nur etwa halb soviel wie beim CEM I 42,5 R) und fällt dann langsam auf
nicht mehr messbare Werte ab. Das Minimum des Wärmeflusses liegt bei ca. 2
Stunden. Dieser Punkt hängt nicht vom Wasser/Zement-Verhältnis ab. Das Maxi-
mum der Wärmeentwicklung liegt zwischen 8 Stunden und 11 Stunden. Mit zuneh-
mendem Wasser/Zement-Verhältnis nimmt auch die Zeit bis zum Erreichen des ma-
ximalen Wärmeflusses zu.
Die Messung mit W/Z=0,35 fällt aus der Reihe. Bei diesem Versuch war der frisch
hergestellte Mörtel auch bei wiederholtem Anmischen zu steif für eine ausreichende
Verdichtung, eine vollständige Entlüftung war nicht möglich. Der Wärmefluss war
durch Lufteinschlüsse gestört. Die Rohdichte dieses Mörtels war geringer als bei den
restlichen Versuchen. Dadurch ergab sich ein zu geringer Wärmefluss.
6.2.3 Hochofenzement CEM III/A 32,5 N-LH
Wie bei den zusatzstofffreien Zementen steigt auch im System mit
CEM III/A 32,5 N-LH die Temperatur in den ersten Minuten um 2 bis 5 K an
(Abbildung 16).
Die Ruheperiode ist generell nicht gut ausgeprägt. Mit steigendem Was-
ser/Zement-Verhältnis nimmt die Ausprägung der Ruheperiode noch weiter ab. Bei
W/Z = 0,30 kommt der Temperaturanstieg noch für ca. 1 Stunde zum Stillstand. Bei
W/Z = 0,35 und 0,40 ist dieser Stillstand nicht mehr zu beobachten.
20
25
30
35
40
45
50
55
60
0 6 12 18 24 30 36 42 48
Zeit [h]
Tem
pera
tur
[°C]
0,35
0,40
0,45
0,50
0,55
Abbildung 16: Zeitlicher Temperaturverlauf beim Abbinden der Mörtel aus Hoch-
ofenzement CEM III/A 32,5N-LH in Abhängigkeit vom W/Z-Wert

Seite 69
Die Geschwindigkeit des anschließenden Temperaturanstieges wie auch die ma-
ximale Temperatur (30 bis 35 °C) steigen mit fallen dem Wasser/Zement-Verhältnis.
Der in Abbildung 17 dargestellte zeitliche Verlauf des Wärmeflusses bestätigt,
dass dieser Hochofenzement im Vergleich zu den reinen Portlandzementen deutlich
langsamer hydratisiert. Dieses Ergebnis steht im völligen Einklang mit den im Pkt.
6.1.3 beschriebenen Werten für die Festigkeitsentwicklung (Abbildung 11).
0
5
10
15
20
25
30
35
0 6 12 18 24 30 36 42 48
Zeit [h]
Wär
mef
luß
[mW
/g]
0,55
0,35
0,40
0,45
0,50
Abbildung 17: Zeitlicher Verlauf des Wärmeflusses beim Abbinden von Mörteln
aus Hochofenzement
6.3 Mineralogische Veränderungen
Die Röntgendiffraktometrie (XRD) stellt ein physikalisches Verfahren dar, mit des-
sen Hilfe die Konzentration einzelner Stoffe in einem Stoffgemisch bestimmt werden
kann (vergleiche Pkt. 2.1.2).
Der Verlauf der Hydratation von Zementleimen wurde, wie in Pkt. 4.3 beschrieben,
bei Raumtemperatur mit XRD beobachtet und ausgewertet. Die Veränderung der
Masseanteile der einzelnen Minerale wurde in Abhängigkeit von der Zementart und
dem Wasser/Zement-Verhältnis betrachtet. Im Gegensatz zu den vorangegangenen
Versuchen wurden in diesen Versuchsreihen die Gehalte einzelner Komponenten
des Systems quantitativ in Abhängigkeit von der Zeit erfasst.
Ziel dieser Arbeit war u.a. die Klärung des Geschwindigkeitsgesetzes für die jewei-
lige Teilreaktion und der Reaktionsordnung bezogen auf einzelne spezifische Reakti-

Seite 70
onen. Hierzu wurde die Korrelation zwischen Gehalt einer Komponente und der Zeit
in der für die jeweilige Reaktionsordnung gültigen Funktion (vgl. Kapitel „Grundlagen
der chemischen Kinetik“) mit linearer Regression mit Microsoft Excel® untersucht. Die
Festlegung der Reaktionsordnung erfolgte über das Bestimmtheitsmaß R2 des jewei-
ligen Systems.
Folgende Kombinationen wurden betrachtet:
Nummer Zementsorte W/Z Folie
1 CEM I 42,5 R 0,45 Mylar
2 CEM I 42,5 R 0,50 Mylar
3 CEM I 42,5 R 0,60 Mylar
4 CEM III/A 32,5 N 0,30 Mylar
5 CEM III/A 32,5 N 0,40 Mylar
6 CEM III/A 32,5 N 0,35 Polycarbonat
7 CEM I 42,5 R-HS 0,30 Polycarbonat
8 CEM I 42,5 R-HS 0,35 Polycarbonat
9 CEM I 42,5 R-HS 0,40 Polycarbonat
10 CEM I 42,5 R-HS 0,50 Polycarbonat
11 CEM I 42,5 R 0,30 Polycarbonat
12 CEM I 42,5 R 0,35 Polycarbonat
13 CEM I 42,5 R 0,40 Polycarbonat
14 CEM I 42,5 R 0,50 Polycarbonat
15 CEM III/A 32,5 N 0,30 Polycarbonat
16 CEM III/A 32,5 N 0,35 Polycarbonat
17 CEM III/A 32,5 N 0,40 Polycarbonat
18 CEM III/A 32,5 N 0,45 Polycarbonat
19 CEM I 42,5 R 0,50 Polycarbonat
Tabelle 8: Auflistung der untersuchten Zemente, der jeweiligen Wasser/Zement-
Werte sowie der verwendeten Folien in Langzeit-Diffraktometrie-Versuchen
6.3.1 Portlandzement CEM I 42,5 R
Abbildung 18 zeigt die Röntgendiffraktogramme der Zementpaste des Versuches
Nr. 13, CEM I 42,5 bei W/Z=0,40, gemessen als in-situ Messung über einen Zeit-
raum von 300 h, im Winkelbereich von 27 °2 Θ bis 37 °2 Θ. In diesem Bereich sind die

Seite 71
stärksten Reflexe der Hauptklinkerminerale des Portlandzementes zu beobachten.
Es ist gut zu erkennen, dass nach einer schnellen Anfangsreaktion (hintere Diffrak-
togramme) sich die Umsetzung der Ausgangsminerale zu Hydratphasen sehr stark
verlangsamt, um anschließend mit geringer Geschwindigkeit fortzuschreiten. Weiter-
hin ist zu erkennen, dass sich die analytisch bedeutenden Peaks stark überlagern,
was eine traditionelle Quantifizierung mit Peak/Untergrund- bzw. Flächen-
Auswertung unmöglich macht.
Abbildung 18: Zeitliche Entwicklung des Röntgendiffraktogramms von Zementleim
aus CEM I 42,5 R bei W/Z = 0,40, aufgenommen über eine Hydratationszeit von 0
bis 85 h
Mit Fortschreiten der Hydratation steigt der röntgenamorphe Untergrund kontinu-
ierlich an. Hervorgerufen wird dies durch die Zunahme der Konzentration feinster,
z.T. nanoskaliger CxSyHz Phasen in der Probe. Diese Phasen weisen keine ausrei-
chend regelmäßige Struktur bzw. Kristallitgrößen auf, um mit XRD erfasst zu werden.
Abbildung 19 zeigt im Vergleich die 1. und die letzte Messung der Versuchsserie
13. Die Intensität der Reflexe der Ausgangsminerale hat im beobachteten Zeitraum
AFt CH
Alit
Alit Belit
Alit + Belit
Alit + Belit CH
C3A
Alit + Belit
AFt CH
27 29 31 33 35 37 85
0
Zeit, h
Goniometerwinkel, °2 Θ
Intensität,
counts/s

Seite 72
deutlich abgenommen. Stattdessen sind neue Reflexe durch neu gebildete Hydrat-
phasen, speziell Portlandit (33,6°; 36,1°) und Ettr ingit (28,0°; 34,5°) hinzugekommen.
Versuch 13 - Start & Ende
#117654 - Creation: 03.11.2005 23:06:39 - File: Versuch13#117654.raw
#114872 - Creation: 19.10.2005 15:06:26 - File: Versuch13#114872.raw
� � � � � � � � � � � � �27 28 29 30 31 32 33 34 35 36 37
Abbildung 19: Vergleich der Röntgendiffraktogramme von Zementleim aus
CEM I 42,5 R und W/Z = 0,40 am Beginn (t0) und am Ende (tca. 85 h) der Hydratati-
ons-Experimente
Im beobachteten Zeitraum werden nicht alle Ausgangsstoffe (-mineralien) zu
Hydratationsprodukten umgesetzt. Es verbleibt ein Anteil nicht hydratisierter Stoffe.
Die Reaktion der Klinkermineralien mit Wasser verlangsamt sich mit der Zeit.
Die Intensitäten der einzelnen Reflexe in den Diffraktogrammen verändern sich mit
unterschiedlichen Geschwindigkeiten. Daraus ist zu schließen, dass die Gehalte der
neu gebildeten Mineralien sich mit verschiedenen Geschwindigkeiten entwickeln. Im
folgenden wird deshalb der zeitliche Verlauf bei der Bildung einzelner spezifischer
Hydratphasen untersucht und dargelegt.
6.3.1.1 Portlandit
Zum Zeitpunkt t0 liegt im betrachteten System kein Portlandit vor. Sofort beim Zu-
sammentreffen des Zementes mit Wasser beginnt nach den stöchiometrischen Glei-
AFt CH
Alit
Alit
Belit
Alit + Belit
Alit + Belit CH
C3A
Alit + Belit
AFt CH
27 29 31 33 35 37 Goniometerwinkel, °2 Θ

Seite 73
chungen der Hydratation der Calciumsilikate jedoch die Bildung von Calciumhydro-
xid, welches in die wässrige Phase des Systems gelöst wird. Beim Erreichen der Sät-
tigung der wässrigen Phase mit Ca2+ und OH- beginnt die Ausfällung von Portlandit ,
welches mit XRD quantitativ erfasst werden kann. Mit fortschreitender Hydratation
wird weiter Calciumhydroxid gebildet. Parallel zur Auskristallisation der C-S-H-
Phasen wird die äquivalente Menge Portlandit nahezu gleichzeitig ausgefällt, der
Masseanteil des Portlandits im erhärtenden Zement-System wächst mit der Menge
der hydratisierten Calciumsilikate. Bei parallel dazu verlaufenden Weiterreaktionen
der Calciumsilikathydrate mit Bestandteilen der flüssigen Phase sowie bei der Hydra-
tation der Calciumaluminate und des Calciumaluminatferrits wird ungünstigerweise
ein Teil des Calciumhydroxids aus der Lösung in kalkreiches CSH(II) und im Calciu-
maluminathydrate chemisch gebunden. Daher korreliert der Gehalt des kristallinen,
röntgenographisch detektierten Calciumhydroxids nicht direkt mit dem Gehalt an C-
S-H-Phasen oder anderer Minerale im erhärteten Zement.
0
2
4
6
8
10
12
14
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
Kon
zent
ratio
n [M
-%]
0,50
0,40
0,35
0,30
W /Z-W erte
Abbildung 20: Zeitliche Entwicklung des Portlanditgehaltes im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten, ermittelt mittels Röntgendiffraktometrie

Seite 74
Der zeitliche Verlauf des Portlanditgehalts in Zementleim aus CEM I 42,5 R ist in
Abbildung 20 dargestellt. Demnach tritt bei der Bildung von Portlandit nach 10 bis 24
h ein Wechsel in der Kinetik der Portlanditbildung auf. Mit steigendem Wassergehalt
im Leim liegt der Zeitpunkt des Wechsels des Reaktionsmechanismus bei späteren
Zeitpunkten. Der maximale Gehalt an Portlandit wird im beobachteten Zeitraum
durch die Veränderung des Wasser/Zement-Verhältnisses nicht systematisch beein-
flusst.
In den ersten Stunden streuen die ermittelten Portlanditgehalte noch sehr stark.
Daher ist eine genaue Bestimmung der Reaktionsordnung für die ersten Stunden
nicht mit ausreichender Genauigkeit möglich.
6.3.1.2 Alit
Der Alit-Gehalt nimmt unabhängig vom Wasser-Gehalt im System von Beginn der
Reaktion an ab. In den ersten 10 bis 18 Stunden ist der Abfall sehr stark.
0
5
10
15
20
25
30
35
40
45
50
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
Alit
geha
lt [M
-%]
0,50
0,40
0,35
0,30
W/Z-Werte
Abbildung 21: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten, ermittelt mittels Röntgendiffraktometrie
Nach einer anfänglich schnellen Reaktion verlangsamt sich der Abfall des Gehal-
tes an Alit sehr stark. Nach ca. 18 Stunden Hydratation ist ein deutlicher Wechsel der

Seite 75
Reaktionsgeschwindigkeit erkennbar. Zu diesem Zeitpunkt wechselt der geschwin-
digkeitsbestimmende Schritt in der Reaktion.
2,5
3
3,5
4
0:00 6:00 12:00 18:00 24:00
Zeit [h]
ln(A
lit [M
-%])
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 22: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten in logarithmischer Darstellung
Die Funktion ln(cAlit)=f(t) ist im Abschnitt zwischen 2 bis 4 und 10 bis ca. 24 Stun-
den annähernd linear (Abbildung 22). Der Beginn des linearen Bereiches dieser
Funktion variiert nur sehr gering. Es ist kein systematischer Einfluss des Was-
ser/Zement-Verhältnisses erkennbar. Das Ende dieses Bereiches verlagert sich mit
steigendem W/Z-Wert zu deutlich späteren Zeiten.
Für diesen Bereich folgt der Gehalt des Alit der Funktion
tk0 e)A(c)A(c ⋅−⋅= . Gleichung 70
Es handelt sich somit um eine Reaktion erster Ordnung, bezogen auf Alit. Durch
lineare Regression im beschriebenen System wurden die Geschwindigkeitskonstan-
ten berechnet.
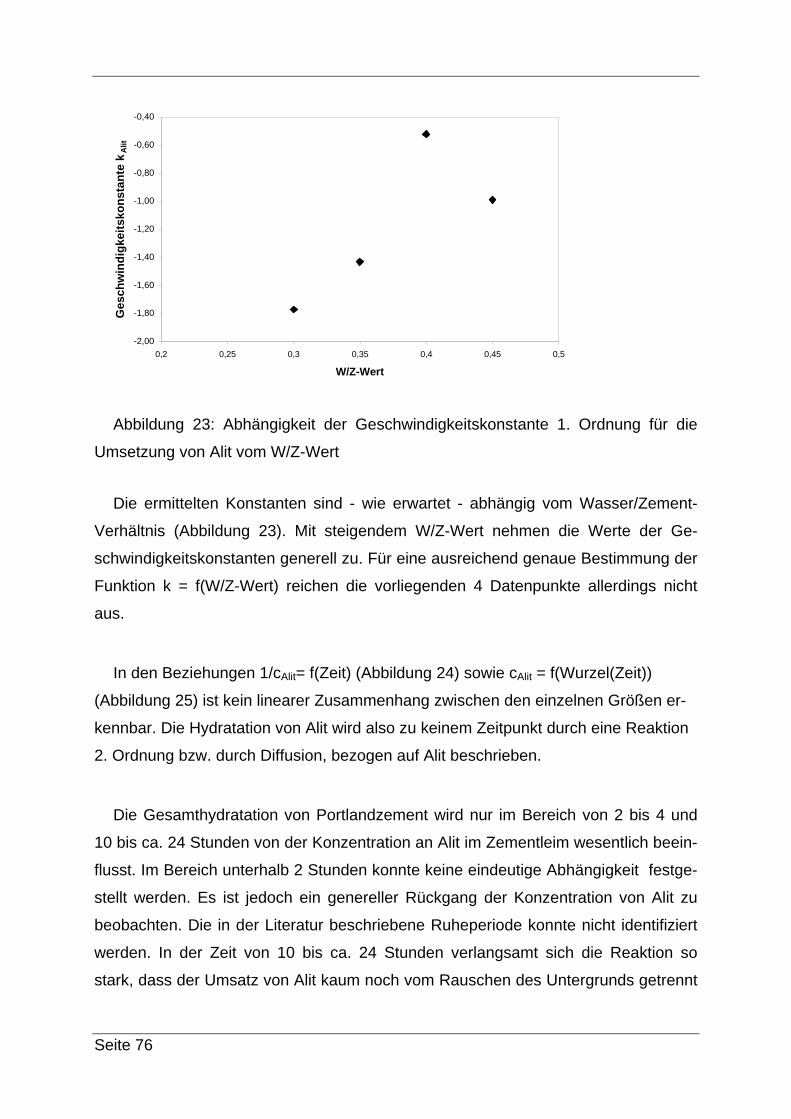
Seite 76
-2,00
-1,80
-1,60
-1,40
-1,20
-1,00
-0,80
-0,60
-0,40
0,2 0,25 0,3 0,35 0,4 0,45 0,5
W/Z-Wert
Ges
chw
indi
gkei
tsko
nsta
nte
kA
lit
Abbildung 23: Abhängigkeit der Geschwindigkeitskonstante 1. Ordnung für die
Umsetzung von Alit vom W/Z-Wert
Die ermittelten Konstanten sind - wie erwartet - abhängig vom Wasser/Zement-
Verhältnis (Abbildung 23). Mit steigendem W/Z-Wert nehmen die Werte der Ge-
schwindigkeitskonstanten generell zu. Für eine ausreichend genaue Bestimmung der
Funktion k = f(W/Z-Wert) reichen die vorliegenden 4 Datenpunkte allerdings nicht
aus.
In den Beziehungen 1/cAlit= f(Zeit) (Abbildung 24) sowie cAlit = f(Wurzel(Zeit))
(Abbildung 25) ist kein linearer Zusammenhang zwischen den einzelnen Größen er-
kennbar. Die Hydratation von Alit wird also zu keinem Zeitpunkt durch eine Reaktion
2. Ordnung bzw. durch Diffusion, bezogen auf Alit beschrieben.
Die Gesamthydratation von Portlandzement wird nur im Bereich von 2 bis 4 und
10 bis ca. 24 Stunden von der Konzentration an Alit im Zementleim wesentlich beein-
flusst. Im Bereich unterhalb 2 Stunden konnte keine eindeutige Abhängigkeit festge-
stellt werden. Es ist jedoch ein genereller Rückgang der Konzentration von Alit zu
beobachten. Die in der Literatur beschriebene Ruheperiode konnte nicht identifiziert
werden. In der Zeit von 10 bis ca. 24 Stunden verlangsamt sich die Reaktion so
stark, dass der Umsatz von Alit kaum noch vom Rauschen des Untergrunds getrennt

Seite 77
werden kann. Eine klare Abhängigkeit des Umsatzes von der Zeit konnte nicht mehr
hergestellt werden.
0,02
0,03
0,04
0,05
0,06
0:00 6:00 12:00 18:00 24:00
Zeit [h]
1/(A
lit [M
-%])
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 24: Zeitliche Entwicklung des reziproken Alit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten

Seite 78
12:00 24:00 36:00 48:00 60:00 72:00
0
5
10
15
20
25
30
35
40
45
50
√√√√t [h]
Alit
[M-%
] 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 25: Quadratwurzel der zeitlichen Entwicklung des Alit-Gehalts im Ze-
mentleim aus CEM I 42,5 R, bei verschiedenen W/Z-Werten
6.3.1.3 Ettringit
Mit Beginn der Hydratation des Portlandzementes setzt sofort die Reaktion von
C3A unter Bildung von Calciumaluminattrisulfathydrat (AFt), Ettringit, ein. Der Ettrin-
git-Gehalt steigt von ca. 5 bis 10 M-% wenige Minuten nach Reaktionsbeginn auf 15
bis 22 M-% nach 9 bis 14 Stunden. Anschließend nimmt der Gehalt langsam wieder
ab (Abbildung 26).

Seite 79
0
5
10
15
20
25
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
Ett
ringi
t [M
-%]
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 26: Zeitliche Entwicklung des Ettringit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten
Während der Anstiegsphase wechselt die Abhängigkeit der Konzentration von der
Zeit zweimal. Der Prozess der Ettringit-Bildung wird von variierenden Reaktionsbe-
dingungen stark beeinflusst. Da Ettringit aus drei Komponenten (C3A, Calciumsulfat
und Wasser) gebildet wird, kann ein Mangel oder Überschuss jeder einzelnen dieser
Phasen die Reaktionsgeschwindigkeit beeinflussen. In dieser Periode liegt Wasser
jedoch stets im Überschuss vor. Die Lösungsgeschwindigkeit von Calciumaluminat
und das Angebot an Calciumsulfat in der Lösung bestimmen somit die Bildung von
Ettringit. Die einzelnen Abschnitte der verschiedenen Reaktionsschritte können je-
doch zeitlich und mathematisch nicht genau voneinander abgegrenzt werden. Die
Übergänge sind verwischt. Das Ende der Ettringit-Bildung verlagert sich jedoch mit
zunehmenden W/Z-Werten zu späteren Zeiten.
Die in der Literatur beschriebene und in der Kalorimetrie augenscheinlich auftre-
tende Ruheperiode kann bei der Bildung von Ettringit nicht beobachtet werden. Es
wird mit Beginn der Hydratation Ettringit gebildet, wobei die Geschwindigkeit der
Ettringit-Bildung auch während der Zeit, die in der Literatur als Ruheperiode be-
schrieben wird, weiter konstant ansteigt (Abbildung 27).
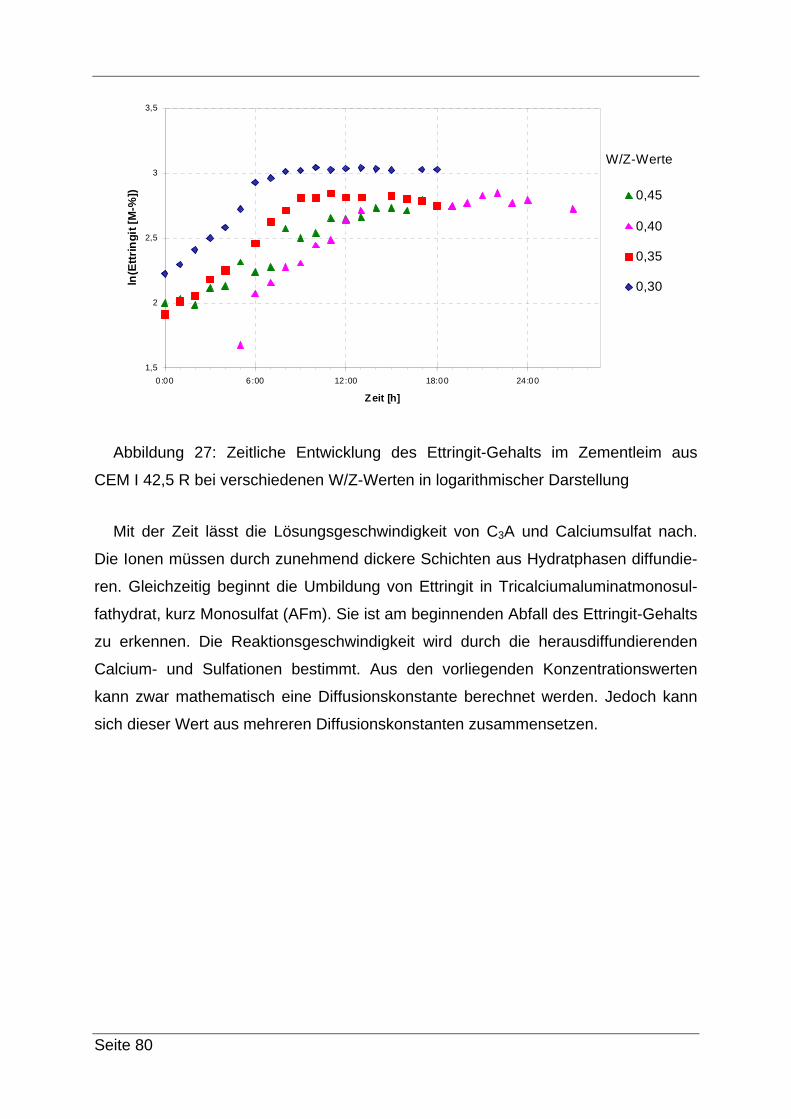
Seite 80
1,5
2
2,5
3
3,5
0:00 6:00 12:00 18:00 24:00
Zeit [h]
ln(E
ttrin
git [
M-%
]) 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 27: Zeitliche Entwicklung des Ettringit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten in logarithmischer Darstellung
Mit der Zeit lässt die Lösungsgeschwindigkeit von C3A und Calciumsulfat nach.
Die Ionen müssen durch zunehmend dickere Schichten aus Hydratphasen diffundie-
ren. Gleichzeitig beginnt die Umbildung von Ettringit in Tricalciumaluminatmonosul-
fathydrat, kurz Monosulfat (AFm). Sie ist am beginnenden Abfall des Ettringit-Gehalts
zu erkennen. Die Reaktionsgeschwindigkeit wird durch die herausdiffundierenden
Calcium- und Sulfationen bestimmt. Aus den vorliegenden Konzentrationswerten
kann zwar mathematisch eine Diffusionskonstante berechnet werden. Jedoch kann
sich dieser Wert aus mehreren Diffusionskonstanten zusammensetzen.

Seite 81
0
0,05
0,1
0,15
0,2
0:00 6:00 12:00 18:00 24:00
Zeit [h]
1/(E
ttrin
git [
M-%
]) 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 28: Zeitliche Entwicklung des reziproken Ettringit-Gehalts im Zement-
leim aus CEM I 42,5 R bei verschiedenen W/Z-Werten
Im Intervall vom Beginn der Reaktion bis 6 bzw. 14 Stunden ist je nach W/Z-Wert
die Abhängigkeit 1/cEttringit = f(Zeit) linear (Abbildung 28). Die Konzentration von
Ettringit im Zementleim folgt somit der allgemeinen Formel
tk)A(c
1)A(c
1
0
⋅+= . Gleichung 71
Die Ettringit-Bildung wird in diesem Zeitraum bestimmt durch eine Reaktion 2.
Ordnung, bezogen auf Ettringit.
Durch lineare Regression wurden die Geschwindigkeitskonstanten berechnet. Die
Geschwindigkeitskonstanten sind abhängig vom Wasser/Zement-Verhältnis
(Abbildung 29). Aus den vorliegenden 4 Datenpunkten ergibt sich eine polynomische
Funktion 2. Ordnung mit einem Minimum bei W/Z = 0,35. Dies könnte jedoch auch
auf Messungenauigkeiten beruhen (vergleiche Abbildung 23).
In den Systemen der Abhängigkeiten ln(cEttringit)=f(Zeit) (Abbildung 27) sowie
cEttringit = f(Wurzel(Zeit)) (Abbildung 30) ist kein linearer Zusammenhang zwischen
den Größen erkennbar. Die Bildung von Ettringit wird also zu keinem Zeitpunkt durch
eine Reaktion 1. Ordnung bzw. durch Diffusion, bezogen auf Ettringit , beschrieben.

Seite 82
Nur für den beschriebenen Bereich von 6 bis 14 h konnte eindeutig eine Reaktion 2.
Ordnung gefunden werden. In den anderen Abschnitten überlagern sich verschiede-
ne Reaktionen und Prozesse, woraus sich eine Reaktion höher als 2. Ordnung er-
gibt, oder die Reaktionsordnung wechselt schnell und mehrfach.
-0,25
-0,20
-0,15
-0,10
0,2 0,25 0,3 0,35 0,4 0,45 0,5
W/Z-Wert
Ges
chw
indi
gkei
tsko
nsta
nte
kE
ttrin
git
Abbildung 29: Abhängigkeit der Geschwindigkeitskonstante 2. Ordnung für die Bil-
dung von Ettringit vom W/Z-Wert
12:00 24:00 36:00 48:00 60:00 72:00
0
5
10
15
20
25
√√√√t [h]
Ettr
ingi
te [M
-%] 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 30: Quadratwurzel der zeitlichen Entwicklung des Ettringit-Gehalts im
Zementleim aus CEM I 42,5 R bei verschiedenen W/Z-Werten

Seite 83
6.3.1.4 Anhydrit
Anhydrit ist zu 3,5 % im Ausgangsstoff CEM I 42,5 R enthalten. Innerhalb der ers-
ten 6 bis 8 Stunden nach Reaktionsbeginn fällt der Anhydritanteil im Zementleim von
ca. 3,5 M-% auf Werte unter 1,5 % (Abbildung 31). Diese Werte liegen im Bereich
der analytischen Nachweisgrenze dieser Methode. Die Messwerte streuen stark. Ei-
ne Differenzierung der Abhängigkeiten der Konzentration von der Zeit ist nicht ein-
deutig möglich.
0
0,5
1
1,5
2
2,5
3
3,5
4
4,5
5
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
Anh
ydrit
[M-%
] 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 31: Zeitliche Entwicklung des Anhydritgehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten
Anhydrit, chemisch Calciumsulfat, ist wasserlöslich. Ab dem Zeitpunkt der Was-
serzugabe gehen Calcium- und Sulfationen in Lösung. Aus der Lösung werden ver-
schiedene Calcium- und sulfathaltige Phasen ausgefällt. Es stellt sich ein Gleichge-
wicht zwischen der Lösung und den festen Phasen ein. Es überlagern sich mehrere
Prozesse. Auf Grund der unsicheren Daten ist die Bestimmung der Reaktionsord-
nung weder im Anfangsstadium noch zu späteren Zeiten möglich.
6.3.1.5 Zusammenfassung CEM I 42,5 R
Bei der Hydratation von CEM I 42,5 R laufen die in der Literatur beschriebenen
mineralogischen Umwandlungen ab. Der zeitliche Verlauf der mineralogischen Ver-
änderungen stimmt jedoch nicht mit den Interpretationen der kalorimetrischen Unter-

Seite 84
suchungen in der Literatur überein. Bei der Hydratation von Zement ist eine heftige
Wärmefreisetzung zu Beginn der Reaktion zu beobachten. Es schließt sich eine Pe-
riode mit sehr geringer Wärmeentwicklung an. Nach mehreren Stunden setzt wieder
eine verstärkte Wärmeentwicklung ein. Die Wärmefreisetzung fällt je nach Zementart
nach 12 bis 30 Stunden wieder ab. Die Wärmefreisetzung wird allgemein als Reakti-
onsintensität bzw. Reaktionsgeschwindigkeit interpretiert. Daraus folgt die Annahme,
dass die Reaktionen im hydratisierenden Zement zwischen der heftigen Startreaktion
und der starken Wärmefreisetzung im mittleren Teil die Umsetzung der Ausgangs-
phasen zu Hydratphasen für mehrere Stunden fast zum Stillstand kommt (vergleiche
1.3.2). Die vorliegenden Ergebnisse zeigen ein ähnliches Bild. Die Reaktionen mit
der höchsten Wärmefreisetzung sind die Hydratation von Alit und die Hydratation von
C3A. Da eine große Menge Sulfat im Zementleim vorliegt, wird bei der Hydratation
von C3A die stöchiometrisch entsprechende Menge Ettringit gebildet. Mit Beginn der
Hydratation setzt die Reaktion von Alit mit hoher Geschwindigkeit ein. Die Reakti-
onsgeschwindigkeit nimmt in den ersten Stunden zu. 5 bis 7 Stunden nach Beginn
der Reaktion wird die maximale Reaktionsgeschwindigkeit erreicht. Anschließend
geht die Reaktionsgeschwindigkeit wieder zurück. 17 bis 20 Stunden nach Reakti-
onsbeginn kommt die Umsetzung von Alit fast zum Stillstand. Die Reaktionsge-
schwindigkeit der Bildung von Ettringit ändert sich analog. Es konnte keine ausge-
prägte Dormante Periode bei der Hydratation von Alit oder der Bildung von Ettringit
nachgewiesen werden. Die Wärmefreisetzung aus dem hydratisierenden Zementleim
korreliert demnach nicht direkt mit den Umsetzungsraten der Reaktionen mit der
höchsten spezifischen Hydratationswärme.
Aus den Messwerten wurden die Geschwindigkeitsgesetze für die Umsetzung von
Alit und für die Bildung von Ettringit aus CEM I 42,5 R bestimmt. Die Reaktionsord-
nung der Bildung von Portlandit konnte nicht bestimmt werden. Die Reaktionsglei-
chung der Hydratation von C3S bzw. Alit enthält C3S mit dem stöchiometrischen Fak-
tor 1. Portlandit wird sowohl bei der Umsetzung von Alit als auch bei der Hydratation
von Belit gebildet. Im betrachteten Zeitraum dominiert jedoch Alit stoffmengenbezo-
gen die Hydratation der Silikate. Daher ist anzunehmen, dass auch die Bildung von
Calciumhydroxid durch die Hydratation von Alit dominiert wird. Die Reaktionsordnung
entspricht somit den Angaben in der Literatur (vergleiche 1.3).

Seite 85
Die Bildung von Ettringit erfolgt nach einem Reaktionsgeschwindigkeitsgesetz
2.Ordnung. Die Reaktionsgleichung der Bildung von Ettringit nimmt daher etwa diese
Form an:
2C3A + 6CaSO4 + 64H2O � 2 C3A·3CaSO4·32H2O Gleichung 72
Die geschwindigkeitsbestimmende Teilreaktion in diesem Prozess konnte nicht
geklärt werden.
6.3.2 Hochsulfatbeständiger Zement CEM I 42,5 R-HS
6.3.2.1 Portlandit
Zu Beginn der Reaktion liegt kein Portlandit vor. Sofort nach Kontakt mit Wasser
wird die flüssige Phase mit Calciumhydroxid gesättigt, erstes Portlandit fällt aus. Die-
se Anfangsreaktion ist nach wenigen Minuten abgeschlossen (Abbildung 32). Es tritt
eine Ruhephase von mehreren Stunden ein. Nach 4 bis 6 Stunden setzt eine sehr
massive Bildung von Portlandit ein, welche allmählich abklingt. Nach ca. 48 Stunden
kommt die Reaktion scheinbar zum Stillstand, die Reaktionsgeschwindigkeit ist im
Vergleich zur beschleunigten Phase sehr stark abgefallen.
0
2
4
6
8
10
12
14
16
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
Por
tland
it [M
-%]
0,50
0,40
0,35
0,30
W/Z-Werte
Abbildung 32: Zeitliche Entwicklung des Portlanditgehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten
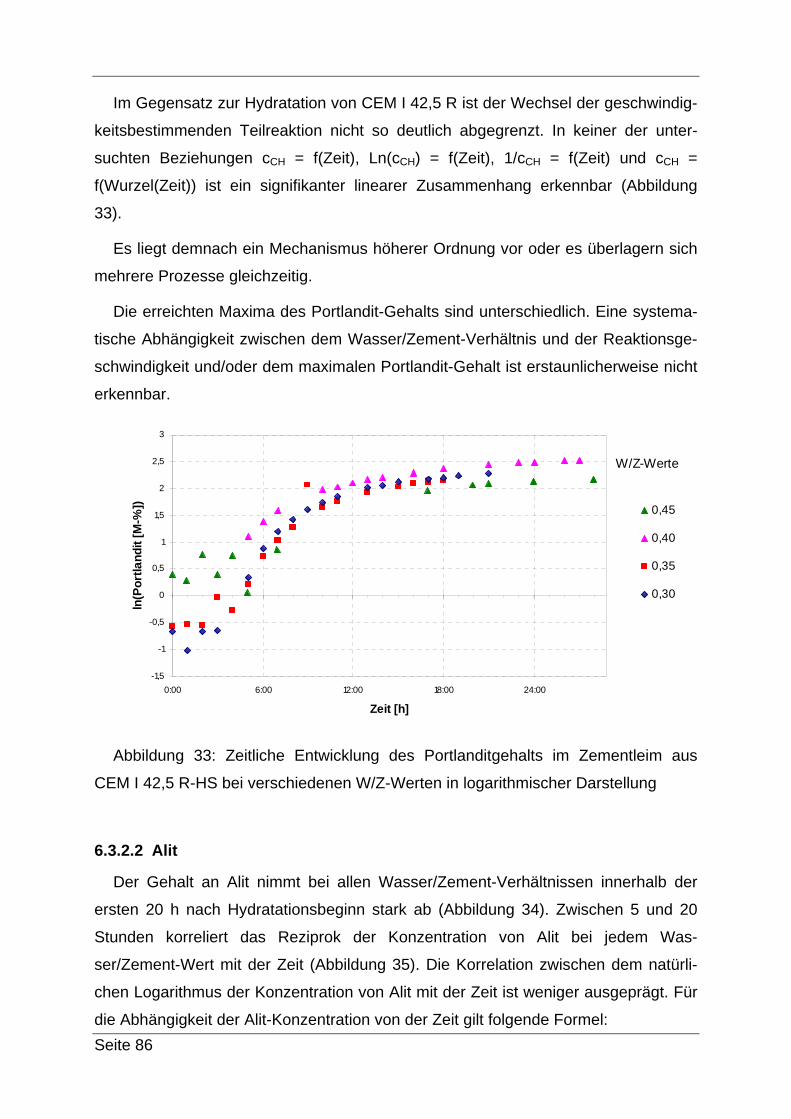
Seite 86
Im Gegensatz zur Hydratation von CEM I 42,5 R ist der Wechsel der geschwindig-
keitsbestimmenden Teilreaktion nicht so deutlich abgegrenzt. In keiner der unter-
suchten Beziehungen cCH = f(Zeit), Ln(cCH) = f(Zeit), 1/cCH = f(Zeit) und cCH =
f(Wurzel(Zeit)) ist ein signifikanter linearer Zusammenhang erkennbar (Abbildung
33).
Es liegt demnach ein Mechanismus höherer Ordnung vor oder es überlagern sich
mehrere Prozesse gleichzeitig.
Die erreichten Maxima des Portlandit-Gehalts sind unterschiedlich. Eine systema-
tische Abhängigkeit zwischen dem Wasser/Zement-Verhältnis und der Reaktionsge-
schwindigkeit und/oder dem maximalen Portlandit-Gehalt ist erstaunlicherweise nicht
erkennbar.
-1,5
-1
-0,5
0
0,5
1
1,5
2
2,5
3
0:00 6:00 12:00 18:00 24:00
Zeit [h]
ln(P
ortla
ndit
[M-%
]) 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 33: Zeitliche Entwicklung des Portlanditgehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten in logarithmischer Darstellung
6.3.2.2 Alit
Der Gehalt an Alit nimmt bei allen Wasser/Zement-Verhältnissen innerhalb der
ersten 20 h nach Hydratationsbeginn stark ab (Abbildung 34). Zwischen 5 und 20
Stunden korreliert das Reziprok der Konzentration von Alit bei jedem Was-
ser/Zement-Wert mit der Zeit (Abbildung 35). Die Korrelation zwischen dem natürli-
chen Logarithmus der Konzentration von Alit mit der Zeit ist weniger ausgeprägt. Für
die Abhängigkeit der Alit-Konzentration von der Zeit gilt folgende Formel:

Seite 87
tkc SC
⋅=3
1 Gleichung 73
Die Reaktion folgt demnach weitgehend einem Mechanismus 2. Ordnung bezogen
auf Tricalciumsilikat. Der Unterschied zum CEM I 42,5 R (Geschwindigkeitsgesetz 1.
Ordnung) ist jedoch nicht sehr deutlich. Der Korrelationskoeffizient k hängt dabei
vom Wasser/Zement-Verhältnis ab. Mit steigendem Wassergehalt im System sinkt
tendenziell die Geschwindigkeitskonstante k von 30 bei W/Z = 0,30 auf < 25 bei
W/Z = 0,50 (Abbildung 36).
0
10
20
30
40
50
60
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
Alit
[M-%
]
0,50
0,40
0,35
0,30
W/Z-Werte
Abbildung 34: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten

Seite 88
0,01
0,02
0,03
0,04
0,05
0,06
0:00 6:00 12:00 18:00 24:00
Zeit [h]
1/(A
lit [M
-%])
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 35: Zeitliche Entwicklung des reziproken Alit-Gehalts im Zementleim
aus CEM I 42,5 R-HS bei verschiedenen W/Z-Werten
0
5
10
15
20
25
30
35
0,2 0,25 0,3 0,35 0,4 0,45 0,5 0,55
W/Z-Wert
Ges
chw
indi
gkei
tsko
nsta
nte
kA
lit
Abbildung 36: Abhängigkeit der Geschwindigkeitskonstanten 2. Ordnung der
Hydratation von Alit im CEM I 42,5 R-HS vom Wasser/Zement-Verhältnis
Zu späteren Zeitpunkten (Hydratation deutlich länger als 48 h) ist der Abfall der
Konzentration von Tricalciumsilikat kaum noch messbar. In diesem Abschnitt der
Reaktion hängt demnach die Konzentration nicht mehr messbar von der Zeit ab.

Seite 89
6.3.2.3 AFt (Eisen-Ettringit)
Der vorliegende HS-Zement enthält kaum Tricalciumaluminat. Rezepturbedingt
enthält dieser Zement jedoch eine sehr hohe Menge an Brownmillerit (C4AF). Dieses
bildet nach der derzeit anerkannten Theorie bei der Hydratation ein dem Ettringit
ähnliches Mineral, die so genannte AFt-Phase. Bei diesem Mineral sind die Gitterpa-
rameter (Abmessungen und Winkel der Elementarzelle) in Abhängigkeit vom Substi-
tutionsgrad des Aluminium durch Eisen verändert. Es wurde daher geprüft, ob auf
Basis unterschiedlicher Gitterkonstanten der Einbau von Eisen in die Kristallstruktur
von AFt nachgewiesen werden kann. Hierzu wurden jeweils die Proben der Systeme
aus Zement bei einem W/Z-Wert von 0,40 nach 48 Stunden Hydratation separat
ausgewertet. In diesem Schritt wurde die Veränderung der Gitterparameter a und c
freigegeben. Die Ergebnisse dieser Untersuchungen sind in Tabelle 9 wiedergege-
ben.
Die Gitterkonstanten des Ettringits im HS-Zement unterscheiden sich nach 48
Stunden Hydratation nicht wesentlich von denen bei Portlandzement und Hochofen-
zement.
Tabelle 9: Gitterparameter von Ettringit bzw. AFt, erhalten aus verschiedenen Ze-
menten nach 48 Stunden Hydratation bei W/Z = 0,40
Gitterparameter von Ettringit Zement
a [Å] c [Å]
Portlandzement 11,15 21,53
HS-Zement 11,13 21,68
Hochofenzement 11,18 21,55
An denselben Messdaten wurde die Änderung der Gitterkonstanten mit der Zeit
untersucht. Bei allen Zementen ändert sich die Länge der Elementarzelle a mit Be-
ginn der Reaktion von ca. 11,15 Å bis zum Endwert von ca. 11,20 Å. Die Breite der
Elementarzelle c ändert sich im CEM I 42,5 R im Verlauf der Hydratation von 21,53 Å
auf ca. 21,47 Å. Im CEM I 42,5 R-HS variiert die Breite c über den gesamten Verlauf
der Hydratation unverändert zwischen 21,55 Å und 21,60 Å. Es ist keine Abhängig-
keit der Breite c von der Zeit erkennbar. Im CEM III/A 32,5 N-LH liegt der Wert der
Breite c zu Beginn der Reaktion bei ca. 21,53 Å. 72 Stunden nach Beginn der Hydra-
tation beträgt die Breite c noch ca. 21,49 Å. Die Beträge der Änderungen der Gitter-
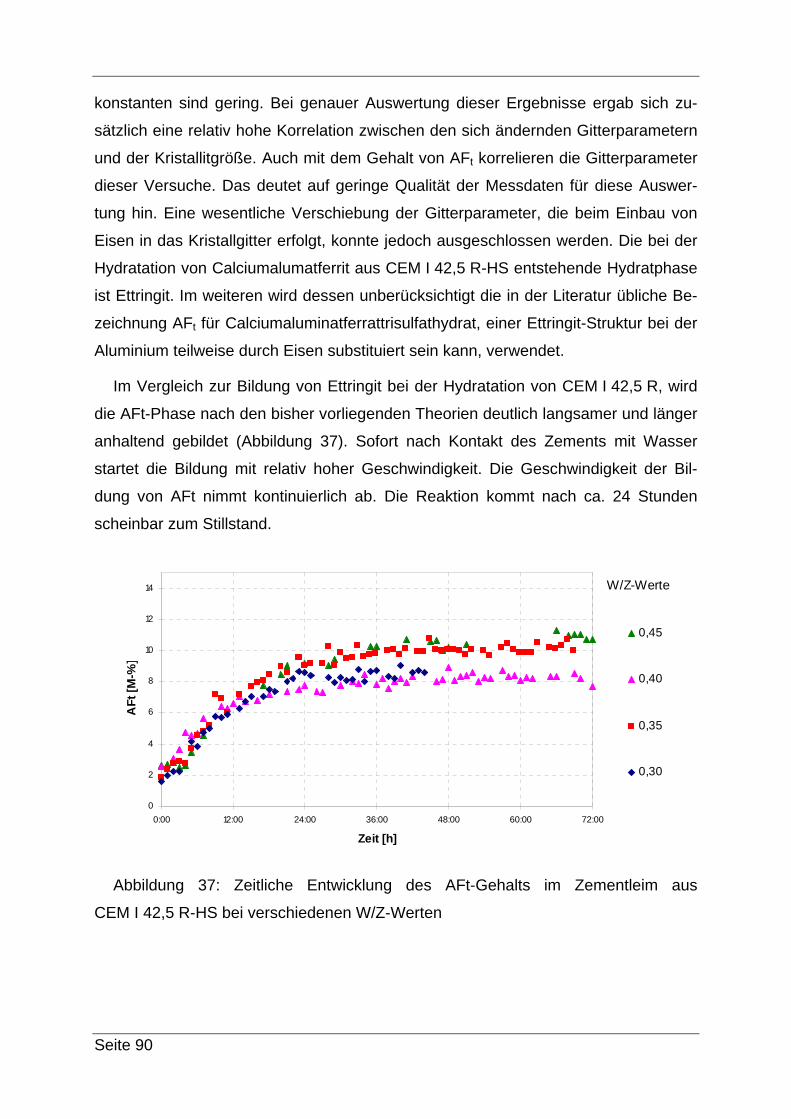
Seite 90
konstanten sind gering. Bei genauer Auswertung dieser Ergebnisse ergab sich zu-
sätzlich eine relativ hohe Korrelation zwischen den sich ändernden Gitterparametern
und der Kristallitgröße. Auch mit dem Gehalt von AFt korrelieren die Gitterparameter
dieser Versuche. Das deutet auf geringe Qualität der Messdaten für diese Auswer-
tung hin. Eine wesentliche Verschiebung der Gitterparameter, die beim Einbau von
Eisen in das Kristallgitter erfolgt, konnte jedoch ausgeschlossen werden. Die bei der
Hydratation von Calciumalumatferrit aus CEM I 42,5 R-HS entstehende Hydratphase
ist Ettringit. Im weiteren wird dessen unberücksichtigt die in der Literatur übliche Be-
zeichnung AFt für Calciumaluminatferrattrisulfathydrat, einer Ettringit-Struktur bei der
Aluminium teilweise durch Eisen substituiert sein kann, verwendet.
Im Vergleich zur Bildung von Ettringit bei der Hydratation von CEM I 42,5 R, wird
die AFt-Phase nach den bisher vorliegenden Theorien deutlich langsamer und länger
anhaltend gebildet (Abbildung 37). Sofort nach Kontakt des Zements mit Wasser
startet die Bildung mit relativ hoher Geschwindigkeit. Die Geschwindigkeit der Bil-
dung von AFt nimmt kontinuierlich ab. Die Reaktion kommt nach ca. 24 Stunden
scheinbar zum Stillstand.
0
2
4
6
8
10
12
14
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
AFt
[M-%
]
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 37: Zeitliche Entwicklung des AFt-Gehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten

Seite 91
12:00 24:00 36:00 48:00 60:00 72:00
0
2
4
6
8
10
12
14
√√√√ t [h]
AF
t [M
-%] 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 38: Entwicklung des AFt-Gehalts im Zementleim aus CEM I 42,5 R-HS
als Abhängigkeit von der Quadratwurzel der Zeit
Die Abhängigkeit cAFt = f(Wurzel(Zeit)) folgt zwischen 10 und 20 Stunden einem li-
nearen Zusammenhang (Abbildung 38). Es gilt die Formel
tkcc 0 ⋅+= . Gleichung 74
Das heißt, die Geschwindigkeit der Bildung von AFt-Phase wird bei der Hydratati-
on von HS-Zement im Gegensatz zum CEM I 42,5 R durch Diffusion kontrolliert.
Durch lineare Regression im beschriebenen System wurden die Geschwindig-
keitskonstanten berechnet. Die Geschwindigkeitskonstanten sind abhängig vom
Wasser/Zement-Verhältnis (Abbildung 39). Aus den vorliegenden 4 Datenpunkten
ergibt sich eine ungefähre lineare Abhängigkeit mit sehr starker Streuung. Für eine
ausreichend genaue Bestimmung der Funktion k=f(W/Z-Wert) reichen die vorliegen-
den 4 Datenpunkte nicht aus.

Seite 92
0
1
2
3
4
5
6
7
8
9
0,2 0,25 0,3 0,35 0,4 0,45 0,5 0,55
W/Z-Wert
Ges
chw
indi
gkei
tsko
nsta
nte
kA
Ft
Abbildung 39: Abhängigkeit der Geschwindigkeitskonstante kAFt vom W/Z-Wert bei
CEM I 42,5 R-HS
6.3.2.4 Anhydrit
Der Gehalt an Anhydrit ist im hochsulfatbeständigen Zement noch etwas geringer
als bei Portlandzement.
In den ersten 6 Stunden nimmt der Anhydritgehalt langsam ab. Danach beschleu-
nigt sich die Abnahme plötzlich. Anschließend nimmt die Umsetzungsgeschwindig-
keit nur noch allmählich ab. Nach ca. 24 Stunden kommt die Reaktion scheinbar zum
Stillstand.
Auf Grund der starken Streuung der ermittelten Werte ist eine zuverlässige Aus-
wertung der Abhängigkeit der Anhydrit-Konzentration von der Zeit nicht möglich. Es
kann daher keine Aussage über das gültigen Geschwindigkeitsgesetz getroffen wer-
den.
6.3.2.5 Zusammenfassung CEM I 42,5 R-HS
Die Reaktionen der Hydratation von CEM I 42,5 R-HS unterscheiden sich deutlich
vom Hydratationsverlauf der Reaktion von normalem CEM I 42,5 R. Die Umsetzung
von Alit folgt bei der Hydratation von CEM I 42,5 R-HS einem Geschwindigkeitsge-
setz 2. Ordnung. Die Reaktionsordnung der Bildung von Portlandit konnte nicht be-
stimmt werden, sie liegt jedoch zwischen 1 und 2. Es ergibt sich somit für die Hydra-
tation von Alit eine Reaktionsgleichung der angenäherten Form

Seite 93
2 C3S + 2 (z-1…2)H � 2 CxSyHz +(1...2) Ca(OH)2 Gleichung 75
Die stöchiometrischen Faktoren weichen von den aus der Literatur bekannten
Faktoren ab (vergleiche Gleichung 36). Der konkurrierende Prozess, der das Reakti-
onsgeschwindigkeitsgesetz im Vergleich zur Hydratation von CEM I 42,5 R verän-
dert, konnte nicht identifiziert werden.
Die Bildung von Ettringit wird bei der Hydratation von CEM I 42,5 R-HS durch Dif-
fusion bestimmt. Nach dem gefundenen Reaktionsgeschwindigkeitsgesetz diffundiert
mindestens ein Stoff bei dieser Reaktion durch eine Schicht eines anderen Stoffes.
AFt bzw. Ettringit entsteht bei der Hydratation von Calciumaluminatferrat. Da, wie in
6.3.2.3 beschrieben, kein eisenhaltiges AFt, sondern Aluminium-Ettringit entsteht,
verbleibt nach der stöchiometrischen Formel von Calciumaluminatferrat Calcium und
Eisen. Diese Elemente können in der vorliegenden Porenlösung die jeweiligen
Hydroxide Ca(OH)2 (Portlandit) und FeO(OH) (Goethit) bilden. Die Reaktionsglei-
chung der Hydratation von Brownmillerit nimmt folgende Form an:
C2(A,F) + H � AFt + CH + FH
Die stöchiometrischen Faktoren in dieser Gleichung konnten nicht bestimmt wer-
den, da die Reaktionsgeschwindigkeit durch Diffusion kontrolliert wird. Eisenhydroxid
ist in Wasser fast unlöslich. Es verbleibt daher als ungelöste Matrix bei der Hydratati-
on von Calciumaluminatferrat, durch die die Aluminium- und Calciumionen heraus
diffundieren.
6.3.3 Hochofenzement
Hochofenzement CEM III reagiert erfahrungsgemäß deutlich langsamer mit Was-
ser als CEM I 42,5 R und hochsulfatbeständiger Zement. Erklärt wird diese Tatsache
mit der geringeren Reaktivität des enthaltenen Hüttensandes.

Seite 94
Abbildung 40: Zeitliche Entwicklung des Röntgendiffraktogramms von Zementleim
aus CEM III/A 32,5 N-LH und W/Z=0,30, aufgenommen über eine Hydratationszeit
von 0 bis 66 Stunden
Die Diffraktogramme zum Reaktionsverlauf bei W/Z-Werten von 0,35; 0,40 und
0,45 sind im Anhang wiedergegeben.
Im Gegensatz zum CEM I 42,5 R und zum hochsulfatbeständigen Zement sind die
Reflexe der Klinkermineralien deutlich kleiner. Bedingt wird die geringere Intensität
der Peaks von C3S, C2S, C3A und Brownmillerit durch den mit 45 M-% deutlich ge-
ringeren Gehalt an Klinker im Zement. Die Sulfatträger sind dagegen in vergleichba-
rer Menge enthalten. Die Reflexe dieser Mineralien sind daher etwa ebenso intensiv
wie in den anderen Zementen.
AFt
CH
Folie AH
Belit Alit + Belit
Alit + Belit C3A
AFt
CH
10 20 30 40 50 55 0
66
Zeit, h
Goniometerwinkel, °2 Θ
Intensität,
counts/s
Gips
Alit
CH
CH
AFt
AFt

Seite 95
6.3.3.1 Portlandit
Bei der Hydratation der Calciumsilikate aus dem Klinker entsteht Calciumhydroxid.
Dieses wird bei Überschreiten der Sättigungskonzentration in der Porenlösung als
Portlandit ausgefällt. Bei der Hydratation von Hochofenschlacke dagegen wird Calci-
umhydroxid aus der Porenlösung verbraucht. Bei Unterschreitung der Sättigungs-
konzentration kann vorher ausgefallener Portlandit wieder gelöst werden.
0
2
4
6
8
10
12
14
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00 54:00 60:00 66:00 72:00
Zeit [h]
Por
tland
it [M
-%]
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 41: Zeitliche Entwicklung des Portlanditgehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten
In den hydratisierenden Hochofen-Zementleimen steigt die gemessene Konzentra-
tion von Portlandit in den ersten Tagen stark an. Die Geschwindigkeit der Portlandit-
bildung nimmt mit steigendem Wasser/Zement-Verhältnis ab (Abbildung 41).
Bei W/Z=0,30 steigt der Portlandit-Gehalt in den ersten 24 Stunden stark an. An-
schließend verringert sich der Anstieg. Nach 60 Stunden erreicht der Portlandit-
Gehalt einen konstanten Endwert. Bei W/Z=0,35 und W/Z= 0,40 dauert der starke
Anstieg des Portlandit-Gehalts ca. 36 Stunden. Danach ist der Gleichgewichtszu-
stand eingestellt. Bei W/Z= 0,45 dagegen wird in den ersten 12 Stunden nur wenig
Portlandit gebildet. Der nachfolgende Anstieg des Gehaltes an kristallinem Calcium-
hydroxid dauert bis 60 Stunden nach Hydratationsbeginn an.
Die Änderung der Konzentration von Portlandit verläuft in den ersten Stunden pro-
portional zur Zeit (Abbildung 41). Es gilt demnach die Funktion

Seite 96
tkcc 0 ⋅−= Gleichung 76
Die Bildungsgeschwindigkeit ist demnach unabhängig von der Konzentration des
Portlandits im untersuchten System. Es handelt sich in diesem Stadium um eine Re-
aktion nullter Ordnung bezogen auf Portlandit.
Im Laufe der Reaktion wechselt die Abhängigkeit von der Konzentration des Port-
landits. Bei Erhöhung des Wasser/Zement-Verhältnisses wird der Wechsel der Ab-
hängigkeit zu späteren Zeitpunkten verschoben (Abbildung 42).
-0,5
0
0,5
1
1,5
2
2,5
3
0:00 6:00 12:00 18:00 24:00
Zeit [h]
ln(P
ortla
ndit
[M-%
]) 0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 42: Zeitliche Entwicklung des Portlandit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten in logarithmischer Darstellung
Auf Grund des ungünstigen Verhältnisses zwischen Neigung der Kurve und
Streuung der gemessenen Intensität kann die Ordnung der Reaktion im 2. Stadium
der Reaktion nicht mehr sicher bestimmt werden.
6.3.3.2 Alit
Der Gehalt an Alit nimmt innerhalb der ersten 15 bis 20 Stunden schnell ab. In
dieser Zeit wird etwa die Hälfte des zur Verfügung stehenden C3S verbraucht. An-
schließend verlangsamt sich die Umsetzung sehr stark (Abbildung 43).
Von Beginn der Hydratation bis 6 Stunden beschleunigt die Reaktion. Von 14 bis
24 Stunden verlangsamt sich die Umsetzung wieder.

Seite 97
0
0,5
1
1,5
2
2,5
3
3,5
4
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00 54:00 60:00 66:00 72:00
Zeit [h]
ln(A
lit [M
-%])
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 43: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten in logarithmischer Darstellung
0
0,05
0,1
0,15
0,2
0,25
0,3
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00 54:00 60:00 66:00 72:00
Zeit [h]
1/(A
lit [M
-%])
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 44: Zeitliche Entwicklung des reziproken Alit-Gehalts im Zementleim
aus CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten
In keinem der untersuchten Systeme cC3S = f(Zeit) (Abbildung 73, siehe Anhang;
0-te Ordnung); ln(cC3S) = f(Zeit) (Abbildung 43; 1. Ordnung); 1/cC3S = f(Zeit)
(Abbildung 44; 2. Ordnung) und cC3S = f(Wurzel(Zeit)) (Abbildung 45; Diffusion) ist ein
eindeutig linearer Zusammenhang zu erkennen. Es ist daher kein ausschließlicher

Seite 98
Mechanismus nachweisbar. Es ist möglich, dass sich mehrere Reaktionen überla-
gern oder dass eine Reaktion höheren Typs vorliegt.
Bei Erhöhung des Wasser/Zement-Verhältnisses wird der Wechsel in der Abhän-
gigkeit des Alit-Gehalts von der Zeit zu späteren Zeitpunkten verschoben.
12:00 24:00 36:00 48:00 60:00 72:00
0
5
10
15
20
25
30
35
40
√√√√ t [h]
Alit
[M-%
]
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 45: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten als Funktion der Quadratwurzel
der Zeit
Da keine eindeutige Abhängigkeit der Alit-Konzentration von der Zeit bestimmt
werden konnte, entfällt die Bestimmung der Reaktionskonstanten und ihrer Abhän-
gigkeit vom Wasser/Zement-Verhältnis.
Es konnte keine eindeutige Abhängigkeit der Umsetzungsgeschwindigkeit vom
Wasser/Zement-Verhältnis gefunden werden.
6.3.3.3 Ettringit
Sofort nach Beginn der Hydratation von Hochofenzement beginnt auch die Bildung
von Ettringit. Die Konzentration von Ettringit nimmt im Laufe der Reaktion stetig zu.
In den ersten 9 Stunden ab Wasserzugabe beschleunigt sich die Ettringitbildung
leicht (Abbildung 46). Anschließend lässt die Bildungsgeschwindigkeit wieder leicht
nach.

Seite 99
0
5
10
15
20
25
30
35
40
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00 54:00 60:00 66:00 72:00
Zeit [h]
Ettr
ingi
t [M
-%]
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 46: Zeitliche Entwicklung des Ettringitgehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten
Die Konzentration von Ettringit im Hochofenzementleim nimmt in der Zeit zwi-
schen 14 und 50 Stunden nach Beginn der Reaktion proportional zur Wurzel der Zeit
zu (Abbildung 47). Es gilt die Formel
tkcc 0 ⋅+= . Gleichung 77
Das heißt, die Geschwindigkeit der Bildung von Ettringit wird durch Diffusion kon-
trolliert.

Seite 100
12:00 24:00 36:00 48:00 60:00 72:00
0
5
10
15
20
25
30
35
40
√√√√ t [h]
Ettr
ingi
t [M
-%]
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 47: Zeitliche Entwicklung des Ettringit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten als Funktion der Quadratwurzel
der Zeit
Durch lineare Regression im beschriebenen System wurden die Geschwindig-
keitskonstanten berechnet. Die Geschwindigkeitskonstanten sind abhängig vom
Wasser/Zement-Verhältnis (Abbildung 48). Aus den vorliegenden 4 Datenpunkten
ergibt sich eine polynomische Funktion 2. Ordnung mit einem Minimum bei W/Z =
0,40.
15
20
25
30
0,2 0,25 0,3 0,35 0,4 0,45 0,5
W/Z-Wert
Ges
chw
indi
gkei
tsko
nsta
nte
kE
ttrin
git
Abbildung 48: Abhängigkeit der Geschwindigkeitskonstanten für die Ettringit-
Bildung vom W/Z-Wert in Hochofenzementleimen

Seite 101
Die Geschwindigkeitskonstante der diffusionskontrollierten Reaktion durchläuft ein
Minimum beim W/Z-Wert von ca. 0,38. Welche Parameter im untersuchten System
(Packungsdichte, Flüssigkeitsangebot, Ionengehalte bzw. –aktivitäten, u.a.) für die
Entstehung des Minimums verantwortlich sind, konnte in den vorgestellten Versu-
chen nicht geklärt werden.
6.3.3.4 Anhydrit
Der gemessene Gehalt an Calciumsulfat (Anhydrit) liegt im Hochofenzement mit
10 bis 12 M-% (bezogen auf das Gesamtsystem inklusive Wasser) deutlich über dem
Gehalt des CEM I 42,5 R und des hochsulfatbeständigen Zements (vergleiche
Tabelle 4). Dieser hohe Wert wird jedoch durch den in der Quantifizierung mit Riet-
veld nicht erfassten amorphen Hüttensand vorgetäuscht. Der tatsächliche Gehalt
liegt, um den Hüttensandgehalt (ca. 50 M-% bezogen auf die Zementmasse) korri-
giert, wesentlich niedriger, nämlich bei nur 4 bis 5 M-%. Das Ziel der vorliegenden
Arbeit war jedoch nicht die genaue Quantifizierung der Gehalte, sondern vielmehr die
Erfassung und Beschreibung ihrer Änderungen. Daher wurde auf eine genauere Be-
stimmung der absoluten Gehalte verzichtet.
0
2
4
6
8
10
12
14
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00 54:00 60:00 66:00 72:00
Zeit [h]
Anh
ydrit
[M-%
]
0,45
0,40
0,35
0,30
W/Z-Werte
Abbildung 49: Zeitliche Entwicklung des Anhydritgehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten
Der Gehalt an Anhydrit nimmt in den ersten 36 Stunden der Hydratation stark ab
(Abbildung 49). Innerhalb der ersten ca. 8 Stunden der Hydratation des Hochofen-

Seite 102
zements wird Anhydrit nur relativ langsam abgebaut. Von 8 bis ca. 30 Stunden der
Hydratation nimmt die Konzentration an Anhydrit proportional zur Zeit ab. Es gilt an-
nähernd eine Abhängigkeit des Anhydrit-Gehalts von der Zeit der Form:
tkcc 0 ⋅−= Gleichung 78
Es handelt sich demnach tendenziell um eine Reaktion 0-ter Ordnung. Die Reakti-
onsgeschwindigkeit der Umsetzung von Anhydrit hängt somit nicht von der Konzent-
ration seines Ausgangsstoffes ab.
Bekanntermaßen wird Calciumsulfat im Anmachwasser des Zementleimes gelöst.
Je nach Ionengehalt des Anmachwassers bzw. des Porenwassers nach Beginn der
Reaktion und je nach räumlicher Verfügbarkeit und kristallographischer Modifikation
des Calciumsulfates ist die Lösungsgeschwindigkeit unterschiedlich. Löslichkeit und
Lösungsgeschwindigkeit der Calciumsulfate hängen auch von der Temperatur ab.
Durch die unterschiedlichen Angebote an Wasser in den untersuchten Systemen
ist auch die sich einstellende Ionenkonzentration in den Porenlösungen unterschied-
lich. Je mehr Wasser das System enthält, desto geringer fällt die sich einstellende
Konzentration an Calcium- und Sulfationen aus.
Bei der Bildung von Calciumaluminatsulfathydraten (Ettringit, Monosulfat) wie
auch bei der Reaktion von Hochofenschlacke mit Wasser werden Calcium- und Sul-
fationen der Lösung entzogen. Somit laufen bei der Hydratation von Zement mehrere
Reaktionen ab, an denen Calciumsulfat, speziell auch Anhydrit, beteiligt ist.
Die gemessenen Konzentrationen von Anhydrit in den hydratisierenden Hoch-
ofenzementleimen streuen, wie beim CEM I 42,5 R und hochsulfatbeständigen Ze-
ment auch, sehr stark. Aus dem Verlauf der Anhydritgehalte kann geschlossen wer-
den, dass die Ordnung der Reaktion im Verlauf der Hydratation wechselt. Der Zeit-
punkt dieses Wechsels liegt zwischen ca. 24 und 36 h. Eine systematische Abhän-
gigkeit der Reaktionsordnung vom gewählten Wasser/Zement-Verhältnis konnte
nicht festgestellt werden.
6.3.3.5 Zusammenfassung CEM III/A 32,5 N-LH
Die Reaktionsgeschwindigkeitsgesetze bei der Hydratation von CEM III/A 32,5 N-
LH unterscheiden sich wesentlich von den Reaktionen bei der Hydratation von nor-

Seite 103
malem CEM I 42,5 R. Die Reaktionsordnung der Hydratation von Alit konnte nicht
bestimmt werden. Die Bildung von Portlandit erfolgt in einer Reaktion 0. Ordnung,
d.h. die Reaktionsgeschwindigkeit der Bildung steht in keinem Zusammenhang mit
der Konzentration der Ausgangsstoffe und des gebildeten Portlandits. In CEM III/A
32,5N-LH wird demnach die Hydratation der Calciumsilikate gegenüber der Hydrata-
tion von normalem CEM I 42,5 R stark beeinflusst. Die Ettringit-Bildung verläuft deut-
lich langsamer als bei normalem CEM I 42,5 R, sie hält jedoch deutlich länger an.
Der Unterschied in den Ausgangsstoffen des CEM III/A 32,5 N-LH besteht in der zu-
sätzlich zu Klinker und Calciumsulfat vorhandenen Hochofenschlacke. Die deutliche
Änderung der Reaktionsordnungen im Hochofenzementleim zeigen, dass die Hoch-
ofenschlacke die chemischen Reaktionen stark beeinflusst. Die Annahme aus der
Literatur, dass Hochofenschlacke in den ersten Tagen nicht angegriffen wird, trifft
demnach nicht zu.
6.3.4 Qualitative Rasterelektronenmikroskopie
Die Ergebnisse der röntgendiffraktometrischen Untersuchungen wurden mittels
REM sowohl hinsichtlich der qualitativen als auch der quantitativen Phasenbildung
überprüft. Es sollte herausgefunden werden, ob zusätzliche Phasen gebildet werden.
Bei der Röntgendiffraktometrie traten innerhalb der betrachteten Phasen eine Rei-
he von Überlagerungen mehrerer Peaks auf. Teilweise sind die Hauptreflexe voll-
ständig durch die Peaks anderer Minerale überdeckt. So konnte z.B. über den ge-
samten untersuchten Zeitraum kaum Calcit detektiert werden, welches aber bei Zu-
tritt von Luft zum bei der Hydratation gebildeten Calciumhydroxid entstehen würde.
Calcit bildet jedoch im Rasterelektronenmikroskop gut erkennbare trigonale Kristalle.
Selbst bei geringer Menge an Calcit sollte dieses gut zu erkennen sein.
Daher wurden erhärtete Proben von Zementleimen aus CEM I 42,5 R mit
W/Z = 0,40 und W/Z = 0,50 nach Abschluss der jeweiligen XRD-Messreihe im REM
auf die Bildung nicht erfasster Phasen, speziell Calcit, untersucht. Die Proben wur-
den sowohl an der Oberfläche (Abbildung 50) als auch an einer Bruchfläche
(Abbildung 85, siehe Anhang) untersucht.

Seite 104
Abbildung 50: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM I 42,5 R mit W/Z = 0,40 nach 20 Tagen Hydratation - Oberfläche
Es ist gut zu erkennen, dass die neu gebildeten Phasen eine sehr feine Struktur
aufweisen. Die Matrix ist sehr dicht. Es wurden keine Separation oder größere Kris-
talle auf der Oberfläche und an den Bruchflächen gefunden, die auf Präparations-
Artefakte hindeuten würden.
Gut erkennbar sind gleichmäßig gebildete CxSyHz-Phasen mit scheinbaren Kristal-
litgrößen << 1 µm. In den Zwischenräumen haben sich nadelige Minerale, wahr-
scheinlich Ettringit, zum Teil mit Kristallitlängen bis zu 5 µm gebildet. Es konnten je-
doch bei der gegebenen Auflösung der Aufnahmen keine weiteren Phasen identifi-
ziert werden.
Zusätzlich wurde von jedem Zement eine Probe bei einem Wasser/Zement-
Verhältnis von 0,35 untersucht. Die Zementleime wurden in einen Probenträger für
die XRD präpariert. Nach 2 Tagen Lagerung bei Raumtemperatur wurden die Proben
durch Schockfrieren getrocknet. Nach einem weiteren Tag Lagerung im Exsikkator
wurden die Proben gesputtert und im Rasterelektronenmikroskop untersucht.

Seite 105
Abbildung 51: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM I 42,5 R mit W/Z = 0,35 nach 2 Tagen Hydratation – Oberfläche
Abbildung 52: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM I 42,5 R-HS mit W/Z = 0,35 nach 2 Tagen Hydratation – Oberfläche

Seite 106
Abbildung 53: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM III/A 32,5 N-LH mit W/Z = 0,35 nach 2 Tagen Hydratation – Oberfläche
Alle drei untersuchten Zementleime waren gemäß den Bildern der Rasterelektro-
nenmikroskopie gleichmäßig durchhydratisiert. Es konnten in dieser Untersuchung
keine signifikanten Segregationen festgestellt werden. Die Klinker- und Schlacketeil-
chen waren mit Hydratationsprodukten überwachsen. Deutlich zu erkennen sind na-
delige Kristalle von Ettringit, die aus den Oberflächen heraus gewachsen sind. Aus-
fällungen aus der Lösung in Form von zwischen den Partikeln liegenden Kristallen
konnten nicht beobachtet werden.
Aus den Rasterelektronenmikroskopie-Untersuchungen ergaben sich keine Hin-
weise, welche die Aussagen der Röntgendiffraktometrie gefährden. Vielmehr wurden
die Ergebnisse der Diffraktometrie, besonders die hohen Gehalte an Ettringit, qualita-
tiv bestätigt.

Seite 107
7 Diskussion der Ergebnisse
Der Verlauf der Hydratation verschiedener Zemente wurde unter dem Aspekt der
Bestimmung der Reaktionskinetik untersucht. Als Zemente wurden zwei Portland-
und ein Hochofenzement eingesetzt.
Nach der bisher anerkannten Theorie zur Hydratation von Zementen läuft die Re-
aktion in 5 Stufen ab: In einer stürmischen Anfangsreaktion (wenige Minuten, meist
während des Anmischvorgang weitgehend abgeschlossen) werden die Zementparti-
kel innerhalb kürzester Zeit von einer gelartigen Schicht aus Hydratationsprodukten
umhüllt. Da keine Barrieren zwischen den reagierenden Stoffen vorhanden sind, ver-
läuft dieser Abschnitt der Hydratation kinetisch kontrolliert. Durch den Aufbau einer
Barriere aus Hydratationsprodukten ändert sich im zweiten Abschnitt (bis ca. 0,5
Stunden) die Abhängigkeit der Umsetzungsgeschwindigkeit von der Zeit von kineti-
scher Kontrolle hin zur Diffusionskontrolle. Die Reaktion verlangsamt sich. In der an-
schließenden beschleunigten Periode (bis 8…12 Stunden) wird die Schicht aus
Hydratationsprodukten durch deren Kristallwachstum aufgebrochen. Die Umset-
zungsgeschwindigkeit steigt wieder. Diese Beobachtung wird mit einer erneut kine-
tisch bzw. Konzentrations-kontrollierten Reaktion erklärt. Im letzten Abschnitt (nach
mehreren Tagen) klingt die Reaktion wieder ab. Als Ursache wird die steigende Di-
cke der Hydratphasenschicht und die damit sinkende Diffusionsgeschwindigkeit auf-
geführt. Diese Einteilung beruht auf der Zeitabhängigkeit der Wärmefreisetzung bei
der Hydratation von C3S [48].
In vorangegangenen Arbeiten anderer Autoren wurde entweder aus der chemi-
schen Analyse der flüssigen Phase des hydratisierenden Systems oder aus physika-
lischen Erscheinungen, wie z.B. dem Wärmefluss, auf entstandene neue Phasen im
System geschlossen. Wenige Autoren befassen sich mit der in-situ Untersuchung
der Hydratation von Zement. Scrivener et al. [77] untersuchten hydratisierende Port-
landzement-Leime mit Röntgendiffraktometrie. Die vorliegende Arbeit bestätigt die
gefundenen zeitlichen Verläufe der Mineralgehalte in den Leimen. Die gefundenen
Daten wurden jedoch um den Vergleich mit hochsulfatbeständigem und Hochofen-
zement sowie um die mathematische Modellierung der Reaktionskinetik in den jewei-
ligen Hydratationsabschnitten ergänzt.

Seite 108
Es konnte gezeigt werden, dass bei der Reaktion von Zementbestandteilen mit
Wasser Perioden mit kinetischer Kontrolle durch solche mit Diffusionskontrolle abge-
löst werden. Die erzielten Ergebnisse liefern ein im Vergleich zur bisherigen Literatur
über die 5 Abschnitte unterschiedliches Bild.
Anhand der experimentell bestimmten Konzentrationsänderungen während der
Hydratation konnte die Reaktionsordnung bestimmt werden. Tabelle 10 gibt an, für
welche Mineralphasen in welchen Zementen die Konzentrationsänderungen für die
Bestimmung ausreichend waren.
Tabelle 10: Zeitabschnitte, in denen starke Konzentrationsänderungen der unter-
suchten Minerale auftreten, bei der Hydratation der drei Zemente
Zeitabschnitte (h) der Konzentrationsänderung Zement
Alit Portlandit Ettringit bzw. AFt
Anhydrit
CEM I 42,5 R 4 bis 10 bzw. 24
nicht aus-reichend
0 bis 6 bzw. 14
nicht aus-reichend
CEM I 42,5 R-HS 5 bis 20 0 bis 6 bzw.
36 10 bis 20 6 bis 24
CEM III/A 32,5 N-LH 0 bis 6
und 14 bis 24
0 bis 30 14 bis 50 8 bis 30
Mittels Röntgendiffraktometrie wurden folgende Reaktionsordnungen bezüglich
des Verbrauchs bzw. der Bildung der Phasen festgestellt.

Seite 109
Tabelle 11: Experimentell ermittelte Reaktionsordnungen für den Verbrauch wich-
tiger Zement- bzw. die Bildung wichtiger Zementhydratphasen
Reaktionsordnung beim Verbrauch bzw. der Bildung von
Zement
Alit Portlandit Ettringit bzw. AFt
Anhydrit
CEM I 42,5 R 1. ungeklärt 2. ungeklärt
CEM I 42,5 R-HS 2. nicht ein-
deutig Diffusion ungeklärt
CEM III/A 32,5 N-LH nicht ein-
deutig 0. Diffusion 0.
Zu Beginn der Reaktion finden vor allem Lösungs- und erste Kristallisationsvor-
gänge statt. Die Geschwindigkeit dieser Prozesse wird durch das Angebot an reakti-
onsfähigen Klinkerphasen begrenzt. Danach folgen Reaktionen unterschiedlicher
Natur. Es treten Reaktionen 1. und 2. Ordnung, aber auch diffusionsgesteuerte Pro-
zesse auf. Die Reaktionskinetik hängt deutlich vom Zementtyp ab. Nach 20 bis 30
Stunden haben sich ausreichend dichte Schichten aus Hydratphasen auf der Ober-
fläche der Zementbestandteile gebildet, so dass das Wasser bzw. im Wasser gelöste
Stoffe nicht mehr ungehindert zu reaktiven Zentren durchdringen können. Das Was-
ser bzw. im Wasser gelöste Stoffe müssen diese Schichten durchdringen, um mit
den reaktiven Zentren in Kontakt zu kommen. Die treibende Kraft für diese Bewe-
gung durch die Schichten aus Hydratphasen ist die Brown’sche Bewegung der Teil-
chen in der Matrix. Die Teilchen bewegen sich mit dem Gradienten der Konzentration
dieser Teilchen. Somit folgt dieser Prozess den Gesetzen der Diffusion. Die Ge-
schwindigkeit der Diffusion der mobilen Teilchen begrenzt die Geschwindigkeit des
Gesamtprozesses der Zementhydratation in dieser Phase.
Im Einzelnen reagieren die Mineralien jedoch nach unterschiedlichen Mechanis-
men und Reaktionsordnungen. Diese wird im Folgenden dargestellt.
7.1 Tricalciumsilikat (Alit)
Alit ist die für die Festigkeitsentwicklung des Zementsteins bedeutendste Phase.
Gleichzeitig hat sie im Klinker den größten Massenanteil.

Seite 110
In den verschiedenen Zementen hydratisiert das Tricalciumsilikat nach unter-
schiedlichen Mechanismen. Die höchste Reaktionsgeschwindigkeit wird im frühen
Hydratationsstadium nach einer mehrstündigen Ruhephase und bis ca. 24 bis 48
Stunden erreicht. Im CEM I 42,5 R stellt die Reaktion von Alit mit Wasser in dieser
Phase eine Reaktion 1. Ordnung dar. Die Konzentration von Alit folgt der Gleichung
tk0 e)A(c)A(c ⋅−⋅= . Gleichung 79
Je nach Wasser/Zement-Verhältnis nimmt die Reaktionsgeschwindigkeit der Um-
setzung von Alit nach ca. 12 bis 24 Stunden sehr stark ab. Die Reaktion kommt fast
zum Stillstand.
Dagegen folgt die Konzentration von Alit im hochsulfatbeständigen Zement der
Gleichung
tkcAlit
⋅=1. Gleichung 80
In diesem System reagiert Alit nach einem Mechanismus 2. Ordnung. Die Reakti-
onsgeschwindigkeit verringert sich deutlich langsamer als beim CEM I 42,5 R. Die
Umsetzung von Alit stagniert erst nach 36 bis 48 Stunden.
Im Hochofenzement kann die Reaktionsordnung nicht eindeutig bestimmt werden.
Die Reaktionsgeschwindigkeit verringert sich jedoch, wie beim CEM I 42,5 R, nach
12 bis 24 Stunden sehr stark.
Portlandzement und Hochofenzement werden aus dem gleichen Klinker herge-
stellt. Das Verhältnis der Klinkermineralien zueinander ist in beiden Zementen gleich.
Unterschiedlich ist dagegen in den untersuchten Systemen das Wasser/Klinker-
Verhältnis. Die zeitlichen Verläufe der Umsetzung von Alit unterscheiden sich in bei-
den Systemen wenig. Die Reaktionsordnung der Umsetzung von Tricalciumsilikat im
Hochofenzement konnte nicht eindeutig bestimmt werden. Es konnte somit nicht ge-
klärt werden, welche Unterschiede in den Mechanismen vorliegen.
Entgegen der in einer früheren Literatur angegebenen Reaktionsgleichung 2. Ord-
nung (Pkt. 1.3.1.1) folgt die Reaktion einem Mechanismus 1. Ordnung bezogen auf
Alit. Die Reaktionsgeschwindigkeit hängt somit von der Konzentration an Alit nach
folgender Gleichung ab:

Seite 111
Alitckv ⋅= . Gleichung 81
Dieses Ergebnis stimmt mit den Resultaten der Versuche von Ludwig und Singh
[35] überein. Diese Autoren geben für die Akzelerationsperiode ebenfalls eine Reak-
tion erster Ordnung an. Die Hydratation von Alit wird durch die Keimbildung der
CxSyHz-Phasen limitiert. Die anderen limitierenden Prozesse (Auflösungsgeschwin-
digkeit des C3S, Transport durch die Lösung, Kristallwachstum) sind sämtlich diffusi-
onskontrolliert. Sie laufen nach den vorliegenden Ergebnissen schneller ab als die
Keimbildung. Paulini legt dar, dass in der Ruheperiode das System Volumenarbeit
verrichtet (quillt) und dabei die durch chemische Reaktionen freigesetzte Wärme
durch verstärkte Lösungsvorgänge teilweise wieder aufnimmt. Dieser Effekt [36]
konnte weder bestätigt noch widerlegt werden. Die Genauigkeit der Messergebnisse
im betroffenen Bereich war nicht ausreichend, um Veränderungen der Gitterparame-
ter zuverlässig zu bestimmen.
Im Unterschied zu normalem Portland- und Hochofenzement liegt im hochsulfat-
beständigen Zement ein verändertes Verhältnis der Klinkermineralien zueinander
vor. Der hochsulfatbeständige Klinker enthält kaum C3A. Dagegen ist der Anteil an
Brownmillerit (C4AF) in diesem Zement sehr hoch.
Der Gehalt an C3S liegt bei Portland- und hochsulfatbeständigem Zement zu Be-
ginn der Reaktion und nach Abschluss des schnellen Abschnitts etwa auf gleichem
Niveau. Die Unterschiede in der Reaktionsgeschwindigkeit werden demnach nicht
durch unterschiedliche Konzentrationen von Alit in den Zementen bedingt. Da sich
Alit in beiden Zementen nicht unterscheidet, muss der Unterschied hinsichtlich der
Reaktionsordnung in einer konkurrierenden Reaktion bei der Hydratation liegen.
Welche Reaktion den Abbau von Alit verzögert oder beschleunigt, konnte nicht ge-
klärt werden.
7.2 Portlandit
Die Bildung von Portlandit verläuft weitgehend analog zur Umsetzung von Alit. Die
Reaktionsgeschwindigkeit der Bildung von Portlandit hängt direkt von der Umset-
zungsgeschwindigkeit des Alit ab. Dieser Zusammenhang wird durch die bekannten
stöchiometrischen Gleichungen zur Umsetzung von Calciumsilikaten unter Bildung

Seite 112
von Calciumhydroxid gestützt. Die Reaktionsordnungen von Alit und Portlandit stim-
men jedoch nicht überein. Das kann wie folgt begründet werden:
a) Die stöchiometrischen Koeffizienten von C3S und Calciumhydroxid
in den entsprechenden chemischen Gleichungen sind für verschie-
dene Zemente unterschiedlich.
b) Die chemischen Gleichungen beruhen auf Versuchen mit reinem
Tricalciumsilikat und nicht Alit.
c) C3S wird direkt als kristalline Phase abgebaut, Portlandit dagegen
über die Lösung ausgefällt. Beim Portlandit findet also ein zusätzli-
cher Schritt bei der Umwandlung statt.
Allerdings wird bei der Hydratation von hochsulfatbeständigem Zement tendenziell
mehr Portlandit gebildet, als bei der Reaktion von normalem Portlandzement und
Hochofenzement mit Wasser. Als mögliche Quelle für zusätzlich gebildetes Portlandit
kann die Hydratation von Calciumaluminatferrat angeführt werden, bei der neben
Aluminium-Ettringit auch Portlandit und Goethit entstehen können.
Der Unterschied zwischen den zwei verwendeten Klinkern besteht im Gehalt an
C3A und Brownmillerit. Im normalen Portlandzement und Hochofenzement wird fast
das gesamte C3A innerhalb von 48 Stunden verbraucht. Im HS-Zementleim wird da-
gegen nur ein Teil des Brownmillerits abgebaut (Abbildung 54). Dadurch verringert
sich auch die Bildung von Portlandit aus dieser Reaktion mit der Zeit.

Seite 113
Abbildung 54: Änderung der Intensität des Peaks von Brownmillerit im Laufe der
Hydratation von Zementleim aus CEM I 42,5 R-HS bei W/Z = 0,40, aufgenommen
über eine Hydratationszeit von 82 Stunden
Im Zementleim des hochsulfatbeständigen Zements läuft die Reaktion von Calci-
umsilikat unter Bildung von Calciumhydroxid ungestört ab. Im normalen Portlandze-
ment ist das Angebot an sehr reaktivem Calciumaluminat sehr hoch. Für eine voll-
ständige Bindung des Tricalciumaluminats aus dem Klinker wären im untersuchten
Portlandzement 7,3 M-% SO3 erforderlich. Das C3A aus dem Portlandzementklinker
wird sehr schnell gelöst. Neben der Bildung des gut mit XRD messbaren Ettringits
entstehen vermutlich auch schlecht kristallisiertes Monosulfat und Calciumalumi-
nathydrat. Dieser Verlauf wird von Stark et al. [109] ebenfalls angegeben. Ein direk-
ter Nachweis von Monosulfat und/oder kristallinem Calciumaluminathydrat konnte
nicht erbracht werden. Die Hydratation von Calciumaluminat kann auf diese Weise
die Umsetzung von Alit über den Verbrauch des Reaktionsproduktes Calciumhydro-
xid aus der Lösung beschleunigen.
Bei der Hydratation von Hochofenzement wird wesentlich weniger Portlandit gebil-
det als bei der Hydratation des normalen Portlandzements. In den quantitativen
Auswertungen nach Rietveld ist der amorphe Anteil, der zu Beginn der Reaktion ca.
48 M-% beträgt nicht berücksichtigt. Der Portlandit-Gehalt im Hochofenzementleim
ist deshalb um den Faktor (1 - amorpher Anteil) zu verringern.
11,6 12,5
0
82 Goniometerwinkel, ° 2 Θ
Reaktionsdauer, h

Seite 114
7.3 Ettringit (AFt)
Die Bildung von Ettringit bzw. der AFt-Phase verläuft bei den untersuchten Ze-
mentleimen sehr unterschiedlich. Die Mechanismen der Bildung und des Verbrauchs
von Ettringit im späteren Verlauf der Zementhydratation unterscheiden sich wesent-
lich voneinander.
Bei CEM I (Portlandzement) wird innerhalb der ersten 12 bis 20 Stunden sehr
schnell viel Ettringit gebildet. Dabei nimmt die gebildete Menge mit zunehmendem
Wasser/Zement-Verhältnis ab. Gleichzeitig verlängert sich die Dauer der Ettringit-
Bildung mit zunehmendem Wasser/Zement-Verhältnis. Nach Erreichen eines Maxi-
mums nimmt der Ettringit-Gehalt im Portlandzementleim langsam, aber merklich ab.
Auf Grund der Streuung der Versuchsergebnisse kann nicht genau unterschieden
werden, ob es sich um eine Reaktion 1. oder 2. Ordnung handelt. Dagegen kann Dif-
fusion ausgeschlossen werden. Die Diffusion von Ausgangsstoffen oder Hydratati-
onsprodukten durch eine Schicht spielt in der Zeit der schnellen Anfangsreaktion kei-
ne limitierende Rolle. Im nachfolgenden Abschnitt des langsamen Ettringitabbaus
kann auf Grund der geringen Änderung keine Aussage zur Reaktionskinetik getroffen
werden.
Aus der Literatur ist die These bekannt, dass in einer schnellen Anfangsreaktion
aus C3A und Calciumsulfat, dem Erstarrungsregler, sowie Wasser Ettringit gebildet
wird, der eine schützende Hülle um die Klinkerteilchen bildet und so den Wasserzu-
tritt und die weitere Hydratation behindert. Aufgrund der Versuchsergebnisse kann
jedoch Diffusion als limitierender Schritt bei der Bildung von Ettringit ausgeschlossen
werden. Somit kann diese Theorie nicht zutreffen. Dafür sprechen auch ESEM-
Untersuchungen, die am Lehrstuhl für Bauchemie der TU München unabhängig von
dieser Arbeit durchgeführt wurden. Sie zeigen, dass die Oberfläche von hydratisie-
rendem Zement keineswegs vollständig mit Ettringitkristallen bedeckt ist. Calciuma-
luminathydrat bildet Plättchen und somit räumliche Gebilde. Ettringit entwickelt nade-
lige Kristalle mit unterschiedlichem Aspektverhältnis. Dadurch ist Ettringit in der La-
ge, in Hohlräume zu wachsen und bei kurzen Nadeln oberflächliche Beläge auf den
Ausgangsphasen zu bilden. Ettringit bildet dadurch im Gegensatz zu Calciumalumi-
nathydrat weniger stabile Brücken zwischen den Zementkörnern. Somit bleiben bei
der Bildung von Ettringit statt Calciumaluminathydrat die Zementteilchen zueinander
verschiebbar.

Seite 115
Nach den vorliegenden Modellen zur Hydratation [110] wird der in den ersten 24
Stunden gebildete Ettringit anschließend in Calciumaluminatmonosulfathydrat um-
gewandelt. Der langsame Abfall des Ettringitgehalts im Portlandzementleim nach
dem Maximum bestätigt dieses Modell. Ein direkter Nachweis des Monosulfats war
mit der verwendeten Methode nicht zweifelsfrei möglich.
Bei der Hydratation von hochsulfatbeständigem Zement sollte, wie in 6.3.2.3 be-
schrieben, nicht reiner Ettringit sondern Eisenettringit (AFt-Phase) entstehen. In ihm
sollte gegenüber Ettringit ein Teil des Aluminiums durch Eisen substituiert sein. Es
entsteht jedoch, wie an der selben Stelle beschrieben, relativ reiner Aluminium-
Ettringit. Es verbleiben stöchiometrisch Calcium und Eisen, welche als Hydroxide
ausfallen [111]. Untersuchungen von Fukuhara et al. [112] und Lothenbach, welche
zu der Aussage über die Entstehung von Eisen-Ettringit bei der Hydratation von Ze-
ment führten, basieren auf isolierten C4AF-Phasen, nicht auf Zementpasten [113].
Die Bildung dieser Phase hält im Gegensatz zu Portlandzementleimen deutlich
länger an. Die Menge und die Dauer der Bildung von AFt-Phase ist dabei nicht signi-
fikant vom Wasser/Zement-Verhältnis abhängig. Die Gesamtmenge der gebildeten
AFt-Phase ist geringer als die in Portlandzementleim gebildete Ettringitmenge. Die
Bildung von AFt-Phase wird durch einen Diffusionsprozess bestimmt.
Im Gegensatz zu Portlandzementleim konnte im System aus hochsulfatbeständi-
gem Zement und Wasser nach Erreichen des maximalen AFt-Gehalts kein signifikan-
ter Abfall beobachtet werden. Die Umwandlung von Trisulfat in Monosulfat verläuft
demnach sehr langsam oder gar nicht.
Der Einbau von Eisen oder Silizium in die AFt-Phase konnte somit nicht nachge-
wiesen werden. Die gleiche Erfahrung wird in [109] geschildert. Die Autoren fanden
bei der Hydratation von Brownmillerit in Portlandzementen, dass die Körner von
C4AF als eisenangereicherte und aluminiumarme (ausgelaugte) Reste im Gerüst
verbleiben. Es bildet sich demnach trotz eines hohen Eisenangebots reiner Alumini-
um-Ettringit. Die absolute Menge an AFt-Phase ist in den HS-Zementleimen deutlich
niedriger als in Portlandzement. Die Bildung von AFt bzw. Ettringit wird jedoch nicht
vollständig vermieden.
In Hochofenzementleimen beginnt, wie in den Leimen aus Portland- und hochsul-
fatbeständigem Zement, sofort nach Kontakt mit Wasser die Bildung von Ettringit

Seite 116
(bzw. AFt-Phase). Nach 6 bis 8 Stunden beschleunigt sich die Bildung von Ettringit
und nimmt anschließend allmählich ab. Der Gehalt an Ettringit im Leim nimmt stetig
zu. Die Bildung von Ettringit wird in diesem Zeitraum durch einen Diffusionsprozess
bestimmt. Nach 48 bis 60 Stunden kommt die Reaktion zum Stillstand. Gegenüber
Portlandzement wird deutlich mehr und länger Ettringit gebildet.
Die Hydratation des Klinkers im Hochofenzement läuft prinzipiell so ab wie im
Portlandzementleim. Durch das gebildete Calciumhydroxid, die Alkalihydroxide und
das Calciumsulfat in der flüssigen Phase des Leims wird jedoch auch die glasige
Schlacke chemisch angegriffen. Aus dem Verbund des Glases werden verschiedene
Ionen herausgelöst. Diese tragen zur Bildung von Hydratphasen bei. Obwohl nach
praktischen Erfahrungen ein Beitrag der Hochofenschlacke zur Festigkeitsbildung
des Zementsteins erst zu späteren Zeitpunkten (28 Tage und später) erfolgt, nehmen
die gelösten Bestandteile des Hüttensandglases an der Bildung früher Hydratati-
onsprodukte teil. Die alkalischen und sulfatischen Bestandteile der wässrigen Lösung
im Leim lösen in frühen Hydratationsphasen bevorzugt Aluminiumionen aus dem
Glasverbund. Diese diffundieren durch das Gerüst des Glases an die Oberfläche und
bilden anschließend Ettringit. Es liegt demnach im Vergleich zu Portlandzement eine
zusätzliche Aluminiumquelle vor, die jedoch durch Diffusion limitiert Aluminium zur
Bildung von Ettringit zur Verfügung stellt.
Die Lösungsgeschwindigkeit von Aluminium aus der Hochofenschlacke ist jedoch
deutlich geringer als die Lösungsgeschwindigkeit aus Klinker. Die Bildungsge-
schwindigkeit von Ettringit ist größer als die Diffusion von Aluminium durch das
verbleibende Glasgerüst. Das Aluminium kann deshalb rasch zu Ettringit umgesetzt
werden.
Die von Tenoutasse [42] angegebene Diffusionskontrolle konnte für hochsulfatbe-
ständigen Zement und Hochofenzement bestätigt werden. Bei der Hydratation von
normalem Portlandzement wird diese Abhängigkeit nicht erfüllt. In Zementleimen aus
Portlandzement wird die Bildung von Ettringit kinetisch kontrolliert. Als Begründung
kann möglicherweise ein deutlicher Unterschied in der Zusammensetzung der flüssi-
gen Phase (Ca2+, SO42-, pH) der Systeme mit Portlandzement im Gegensatz zu Sys-
temen aus hochsulfatbeständigem Zement, Hochofenzement und reinen C3A/Gips-
Systemen angeführt werden.

Seite 117
7.4 Anhydrit
Neben Calciumsulfatdihydrat (Gips) und –Halbhydrat (Bassanit) ist Anhydrit im
Zement als Teil des Erstarrungsreglers enthalten. Nach der in der Literatur aktuell
vertretenen Theorie zum Erstarren von Zement [110] gehen das Calciumsulfat-
Halbhydrat nach der Wasserzugabe schnell, -dihydrat etwas langsamer und Anhydrit
sehr langsam in Lösung. Sie bilden mit dem Calciumaluminat aus dem Klinker Calci-
umaluminatsulfathydrat.
Für eine ausreichende Erstarrungsverzögerung von Zement sind nach praktischen
Erfahrungen 3 bis 4 M-% SO3 in Form von Calciumsulfat erforderlich. Das Verhältnis
von Anhydrit/Gips wird nach der gewünschten Verzögerungswirkung und den Früh-
festigkeiten eingestellt. Die in dieser Arbeit untersuchten Zemente enthielten An-
hydrit, Halbhydrat und Gips. Daneben liegen Alkalisulfate vor. Arcanit K2SO4 ist mit
XRD/Rietveld deutlich nachweisbar.
Tabelle 12: Sulfatphasen in den untersuchten Zementen, bestimmt mit XRD und
DTA.
Sulfatphasen [M-%] SO3-Gehalt [M-%] (berechnet) Probe
AH* HH** DH** ∑∑∑∑ AH HH DH ∑∑∑∑
CEM III/A 32,5 N-LH 2,8 0,5 2,4 8,6 1,65 0,28 1,12 3,05
CEM I 42,5 R 3,3 1,9 0,1 5,3 1,94 1,05 0,05 3,04
CEM I 42,5 R-HS 1,8 1,0 3,1 5,9 1,06 0,55 1,44 3,05
* mittels XRD ermittelt; ** - mittels DTA ermittelt
Die Probe CEM III/A 32,5 N-NA enthält noch eine geringe Menge an Syngenit (Entwässe-
rung im Intervall 260 °C -290 °C).
Der Erstarrungsbeginn der verwendeten Zemente liegt bei ca. 3 Stunden (vgl. Pkt.
5.4). Die erwünschte Wirkung der Sulfatträger in Bezug auf die Erstarrungsverzöge-
rung beschränkt sich demnach auf die ersten 3 Stunden der Reaktion von Zement
mit Wasser.

Seite 118
Die Abreaktion des Abbaus von Anhydrit aus dem Zementleim läuft dagegen über
einen deutlich längeren Zeitraum ab. Die Reaktionsordnungen konnten auf Grund
der starken Streuung der Messwerte jedoch nicht genau bestimmt werden.
Im Portlandzement wird fast unabhängig vom gewählten Wasser/Zement-
Verhältnis in den ersten 6 bis 8 Stunden fast aller Anhydrit umgesetzt. Er steht dem-
nach fast vollständig für die Erstarrungsverzögerung zur Verfügung.
Der Verbrauch von Anhydrit im hochsulfatbeständigen Zement beginnt verhalten.
Erst nach 6 bis 8 Stunden beschleunigt sich der Umsatz. Nach ca. 24 Stunden ist
Anhydrit vollständig umgewandelt. Es kommt nur ein geringer Teil des zur Verfügung
stehenden Anhydrits für die Erstarrungsverzögerung zur Wirkung. Der größte Teil
des Anhydrits reagiert erst nach dem Erstarren des Zementleims.
Im Hochofenzement hält der Abbau von Anhydrit bis ca. 30 Stunden nach Reakti-
onsbeginn an. Wie auch bei hochsulfatbeständigem Zement beginnt die Umsetzung
verhalten. Die beschleunigte Phase des Verbrauchs von Anhydrit beginnt 8 bis 12
Stunden nach dem Hydratationsbeginn. Nach 30 bis 36 Stunden ist der Anhydrit wei-
testgehend verbraucht. Eine signifikante Abhängigkeit der Reaktion vom Was-
ser/Zement-Verhältnis kann nicht festgestellt werden.
Bekanntermaßen fördert Sulfat die Festigkeitsentwicklung von Zementstein bei
Anwesenheit glasiger Hochofenschlacke. Insofern wirkt das nach dem Erstarren ver-
brauchte Sulfat lösend auf das Glas des Hüttensandes und führt zur Bildung erster
Hydratphasen aus der Hochofenschlacke.
Bei allen untersuchten Zementen verläuft der Verbrauch von Anhydrit analog zur
Bildung von Ettringit bzw. AFt-Phase im Zementleim. Es besteht demnach ein direk-
ter Zusammenhang zwischen den beiden Reaktionen.
Calciumsulfat ist in Wasser löslich. Die Menge der gelösten SO42--Ionen wird dabei
durch die Konzentration an Ca2+- und anderer Ionen aus weiteren Bestandteilen des
Zements bestimmt. Calciumaluminate und deren Hydrate sind dagegen in Wasser
weniger löslich. Bevorzugt lagern sich daher Sulfat- und Calciumionen gemeinsam
mit Wassermolekülen in das Gitter von Calciumaluminaten ein, Ettringit bzw. AFt-
Phase wachsen als Nadeln aus der Oberfläche der Klinkerteilchen heraus. In dem
Maße, in dem Sulfationen aus der Lösung zur Bildung von Calciumaluminatfer-
rathydrat verbraucht wird, kann neues Sulfat in Lösung gehen. Gleichzeitig müssen

Seite 119
die gelösten Ionen durch die an der Oberfläche des C3A bzw. des Calciumaluminat-
ferrat gebildeten Hydratphasen diffundieren.
Der Verbrauch von Calciumsulfat aus dem Zement wird nur in den ersten Minuten
durch seine Lösungsgeschwindigkeit limitiert. Nach Sättigung der flüssigen Phase
des Leims mit Calciumsulfat wird die Umsetzung des Sulfatträgers dagegen durch
die Geschwindigkeit der Neubildung von Calciumaluminat(/ferrat)-Calciumsulfat-
Hydraten limitiert. Da neben Anhydrit noch Bassanit und Gips zum Sulfatgehalt der
Lösung und der Bildung von Ettringit bzw. AFt beitragen, ist eine direkte Abhängig-
keit zwischen den Reaktionsordnungen der Bildung von Ettringit bzw. AFt und dem
Verbrauch von Anhydrit nicht zu erwarten.
Bei Hochofenzement ist eine positive Wirkung des verbleibenden Sulfatträgers zur
Aktivierung der Hochofenschlacke anzunehmen. Im hochsulfatbeständigen Zement
ist der Verbrauch des Calciumsulfates nach dem Erstarren zwar chemisch erklärbar,
ein technologischer Nutzen ist jedoch nicht erkennbar.
7.5 Praktische Aspekte
7.5.1 Beschleunigung der Hydratation
Die Verringerung der Konzentration der Hydratationsprodukte stellt eine gute Mög-
lichkeit der Beschleunigung der Hydratation dar. So werden der wässrigen Phase
des Zementleims durch Alkalien Hydroxidionen entzogen. Durch Aluminate wird Cal-
ciumhydroxid zeitweise in Calciumaluminathydraten gebunden. Nach diesem Prinzip
wirken die kommerziell erhältlichen Beschleuniger für Beton. Eine Bindung von Cal-
ciumhydroxid durch gasförmiges Kohlendioxid nach der Gleichung
Ca(OH)2 + CO2 � CaCO3 + H2O Gleichung 84
bewirkt dagegen keine Festigungssteigerung [114]. Eine beschleunigte Umset-
zung konnte auf Grund der Versuchsbedingungen (Spritzbeton) nicht nachgewiesen
werden. Trotzdem wurde dieses Verfahren von der Fa. Linde patentiert [115], [116],
[117].

Seite 120
7.5.2 Verzögerung der Hydratation
Die Umsetzung zu Hydratationsprodukten kann durch chemische oder räumliche
Blockade der Ausgangsstoffe oder durch Zugabe von Reaktionsprodukten gehemmt
werden.
Bei chemischer Blockade werden die reaktiven Zentren der Hydratation durch Re-
aktionspartner belegt, welche die reagierenden Ionen der Klinkermineralien nicht aus
ihrem Verbund, der Kristallstruktur, lösen. So wird ein Fortschreiten der Hydratati-
onsfront in das Innere der Zementpartikel unterbunden. Beispielsweise werden Alko-
hole ebenso wie Wasser von der Oberfläche der Klinkerkristalle gebunden. Eine
chemische Bindung von Alkohol durch die Klinkermineralien mit Alkohol unterbleibt
aber.
Durch Zugabe von bestimmten Reaktionspartnern können Reaktionsprodukte er-
zeugt werden, die eine schützende Schicht um die Ausgangsstoffe bilden. Nach die-
sem Prinzip wird die Verzögerung von Portlandzementklinker mit Sulfaten erklärt.
Nach neuen Erkenntnissen wird jedoch entgegen dieser Theorie nicht die komplette
Oberfläche der Klinker-Partikel bedeckt. Vielmehr wachsen AFt-Phasen gezielt auf
den reaktiven Zentren der Aluminatphasen. Dadurch werden während der Hydratati-
on nur diese Bereiche durch Sulfat-haltige Phasen in ihrer Reaktion beeinflusst.
Die sterische Abstoßung der Klinkerteilchen mit den aufwachsenden Hydratati-
onsprodukten durch Fliessmittel bewirkt dagegen keine Blockade der Hydratation.
Durch die zeitweilige Belegung von Adsorptionszentren kann aber der Wasserzutritt
zeitlich verzögert werden.

Seite 121
7.6 Zusammenfassung und Ausblick
Die prinzipielle Eignung der quantitativen Röntgenstrukturanalyse nach Rietveld
zur Beurteilung der kinetischen Gesetze bei der Hydratation von Zementen konnte
gezeigt werden. Die in der Zementanalytik vorhandenen Diffraktometer sind bei der
erforderlichen Empfindlichkeit schnell genug, um die Hydratation einzelner Zement-
phasen zeitaufgelöst beobachten zu können.
In der vorliegenden Arbeit konnten erste Vergleiche zwischen unterschiedlichen
Zementtypen hinsichtlich der Reaktionskinetik und den Geschwindigkeitsgesetzen
während der Hydratation der einzelnen Zementbestandteile getroffen werden.
Interessant erscheint die weitere Untersuchung gleicher Zementtypen aus Klinkern
mit unterschiedlichen Erhärtungs- und Erstarrungscharakteristiken. Ebenso untersu-
chenswert ist das komplette Spektrum der Zusatzmittel im Bereich der frühen Hydra-
tation. Dagegen können methodenbedingt Untersuchungen im Bereich der Startreak-
tion (Sekunden- und Minutenbereich) mit diesem Verfahren nicht durchgeführt wer-
den.
Durch die weitere Erhöhung der Empfindlichkeit der verwendeten Röntgendetekto-
ren könnten weitere Zementmineralien wie z.B. Arcanit, Syngenit u.a. in die Untersu-
chungen einbezogen werden.
7.7 Summary and outlook
The principle suitability of quantitative X-ray diffraction for discussion of kinetic re-
lations in hydration of cements was shown. The existing in cement analytics XRD
instruments are of sufficient sensitivity and allow short analyze durations to observe
the hydration of several cement phases in time.
In the current work first compares between different cement types regarding reac-
tion kinetics and reaction speed during hydration of several components of cement
are done.
It looks promising to investigate further different clinkers in comparable cement
types to find the reasons for different macroscopic cement respective clinker proper-
ties. At the same time it will be interesting to investigate the interaction of admixtures
on the clinker hydration on the early hydration of cement. Caused by methodic disad-

Seite 122
vantages the very early reaction (in the range of seconds and minutes) can’t be ana-
lyzed in quantitative way.
By further improvement of the X-ray detectors it will be possible to include addi-
tional phases as e.g. Arcanite and Syngenite into the investigations.

Seite 123
8 Anhang
8.1 Hydratation von Portland-Zementleim
0
0,5
1
1,5
2
2,5
0:00 6:00 12:00 18:00 24:00
Zeit [h]
ln(P
ortla
ndit
[%]) 0,45
0,40
0,35
0,30
Abbildung 55: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Port-
landit-Gehalts in Portlandzementleimen als Abhängigkeit ln(cCH) = f(Zeit)
0,00
0,05
0,10
0,15
0,20
0,25
0,30
0,35
0,40
0,45
0,50
0:00 6:00 12:00 18:00 24:00
Zeit [h]
1/(P
ortla
ndit
[%]) 0,45
0,40
0,35
0,30
Abbildung 56: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Port-
landit-Gehalts in Portlandzementleimen als Abhängigkeit 1/cCH = f(Zeit)
W/Z-Werte
W/Z-Werte

Seite 124
0
2
4
6
8
10
12
0,00 0,25 0,50 0,75 1,00
Wurzel(Zeit [h])
Por
tland
it [%
]
0,45
0,40
0,35
0,30
Abbildung 57: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Port-
landitgehalts in Portlandzementleimen als Abhängigkeit cCH = f(Wurzel(Zeit))
Abbildung 58: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,30 (in-situ XRD Aufnahme, durch Abrieb an den Laufrollen des Diffrakto-
meters wurde nach ca. 2/3 der Laufzeit eine Verschmutzung eingeschleppt, die je-
doch durch Rietveld herausgerechnet wurde.)
W/Z-Werte

Seite 125
Abbildung 59: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,35 (in-situ XRD Aufnahme)
Abbildung 60: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,40 (in-situ XRD Aufnahme)

Seite 126
Abbildung 61: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,50 (in-situ XRD Aufnahme)
8.2 Hydratation von Leim des hochsulfatbeständigen Zementes
0
0,5
1
1,5
2
2,5
3
0:00 6:00 12:00 18:00 24:00
Zeit [h]
1/(P
ortla
ndit
[%]) 0,45
0,40
0,35
0,30
Abbildung 62: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Port-
landit-Gehalts in HS-Zementleimen (reziprok)
W/Z-Werte

Seite 127
0
2
4
6
8
10
12
14
16
0,00 0,50 1,00 1,50 2,00 2,50 3,00
Wurzel(Zeit [h])
Por
tland
it [%
]
0,50
0,40
0,35
0,30
Abbildung 63: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Port-
landitgehalts in HS-Zementleimen als Abhängigkeit cCH = f(Wurzel(Zeit))
2,5
3,0
3,5
4,0
0:00 6:00 12:00 18:00 24:00
Zeit [h]
ln(C
3S [%
])
0,45
0,40
0,35
0,30
Abbildung 64: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Alit-
Gehalts in HS-Zementleimen als Abhängigkeit ln(cC3S) = f(Zeit)
W/Z-Werte
W/Z-Werte

Seite 128
0
10
20
30
40
50
60
0,00 0,25 0,50 0,75 1,00
Wurzel(Zeit [h])
C3S
[%]
0,45
0,40
0,35
0,30
Abbildung 65: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Alit-
Gehalts in HS-Zementleimen als Abhängigkeit cC3S = f(Wurzel(Zeit))
0
0,5
1
1,5
2
2,5
0:00 6:00 12:00 18:00 24:00
Zeit [h]
ln(A
Ft [%
])
0,45
0,40
0,35
0,30
Abbildung 66: Diffraktometrische Verfolgung der zeitlichen Entwicklung des AFt-
Gehalts in HS-Zementleimen (logarithmisch)
W/Z-Werte
W/Z-Werte

Seite 129
0,00
0,10
0,20
0,30
0,40
0,50
0,60
0,70
0:00 6:00 12:00 18:00 24:00
Zeit [h]
1/(A
Ft [%
])
0,45
0,40
0,35
0,30
Abbildung 67: Diffraktometrische Verfolgung der zeitlichen Entwicklung des AFt-
Gehalts in HS-Zementleimen (reziprok)
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
5,0
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
Anh
ydrit
[%]
0,45
0,40
0,35
0,30
Abbildung 68: Diffraktometrische Verfolgung der zeitlichen Entwicklung des An-
hydrit-Gehalts bei der Hydratation von HS-Zementleimen
W/Z-Werte
W/Z-Werte

Seite 130
Abbildung 69: Diffraktometrische Verfolgung des zeitlichen Verlaufs der Hydratati-
on des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,30 (in-situ
XRD Aufnahme)
Abbildung 70: Diffraktometrische Verfolgung des zeitlichen Verlaufs der Hydratati-
on des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,35 (in-situ
XRD Aufnahme)

Seite 131
Abbildung 71: Diffraktometrische Verfolgung des zeitlichen Verlaufs der Hydratati-
on des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,40 (in-situ
XRD Aufnahme)
Abbildung 72: Diffraktometrische Verfolgung des zeitlichen Verlaufs der Hydratati-
on des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,50 (in-situ
XRD Aufnahme)

Seite 132
8.3 Hydratation von Hochofen-Zementleim
0
5
10
15
20
25
30
35
40
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00 54:00 60:00 66:00 72:00
Zeit [h]
C3S
[%]
0,45
0,40
0,35
0,30
Abbildung 73: Diffraktometrische Verfolgung der zeitlichen Abnahme des C3S-
Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW bei ver-
schiedenen W/Z-Werten
0
0,5
1
1,5
2
2,5
3
0:00 12:00 24:00 36:00 48:00 60:00 72:00
Zeit [h]
1/(P
ortla
ndit
[%]) 0,45
0,40
0,35
0,30
Abbildung 74: Diffraktometrische Verfolgung der zeitlichen Abnahme des Portlan-
dit-Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW als Ab-
hängigkeit 1/cCH = f(Zeit)
W/Z-Werte

Seite 133
0
2
4
6
8
10
12
14
0,00 0,50 1,00 1,50 2,00 2,50 3,00
Wurzel(Zeit [h])
Por
tland
it [%
]
0,50
0,40
0,35
0,30
Abbildung 75: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Port-
landit-Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW als
Abhängigkeit cCH = f(Wurzel(Zeit))
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
0:00 6:00 12:00 18:00 24:00 30:00 36:00 42:00 48:00 54:00 60:00 66:00 72:00
Zeit [h]
ln(E
ttrin
git [
%])
0,45
0,40
0,35
0,30
Abbildung 76: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Ettrin-
git-Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW bei ver-
schiedenen W/Z-Werten (logarithmisch)
W/Z-Werte
W/Z-Werte
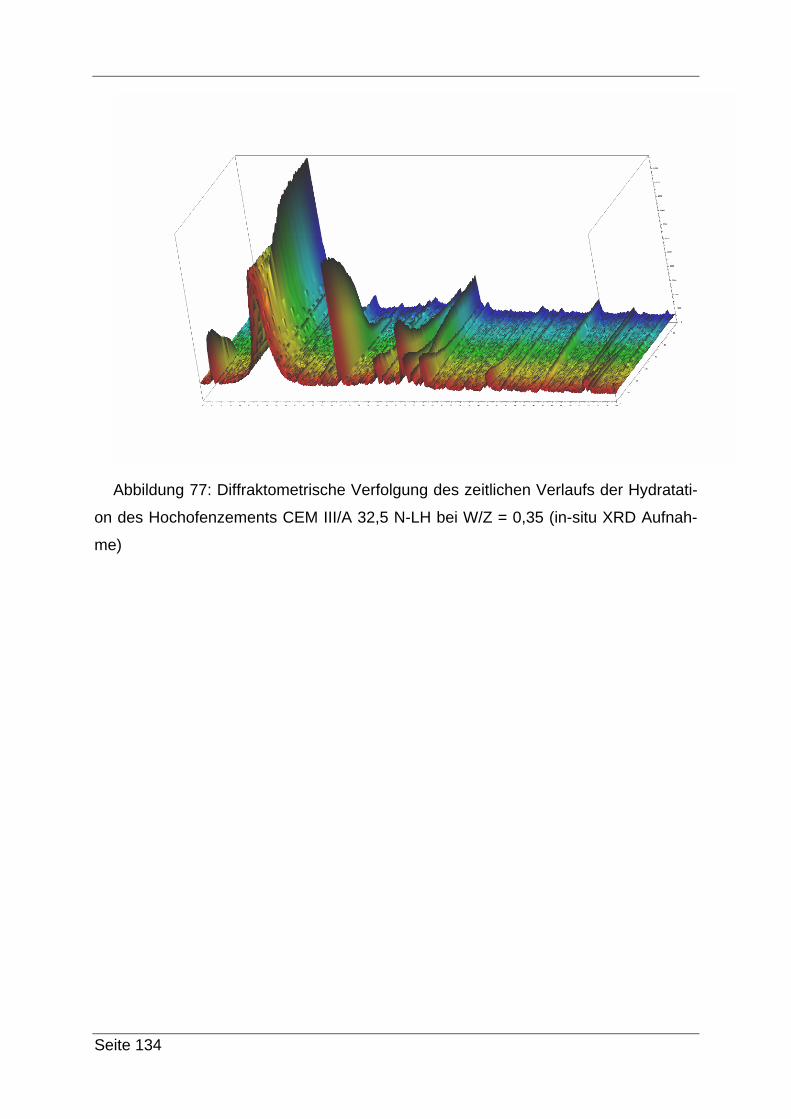
Seite 134
Abbildung 77: Diffraktometrische Verfolgung des zeitlichen Verlaufs der Hydratati-
on des Hochofenzements CEM III/A 32,5 N-LH bei W/Z = 0,35 (in-situ XRD Aufnah-
me)

Seite 135
Abbildung 78: Diffraktometrische Verfolgung des zeitlichen Verlaufs der Hydratati-
on des Hochofenzements CEM III/A 32,5 N-LH bei W/Z = 0,40 (in-situ XRD Aufnah-
me)
Abbildung 79: Diffraktometrische Verfolgung des zeitlichen Verlaufs der Hydratati-
on des Hochofenzements CEM III/A 32,5 N-LH bei W/Z = 0,45 (in-situ XRD Aufnah-
me)

Seite 136
8.4 Kontrolle der XRD Resultate mit REM
Abbildung 80: Versuch Nr. 13 (Portlandzement W/Z = 0,40) nach 20 Tagen Hydra-
tation - Oberfläche
Abbildung 81: Versuch Nr. 13 (Portlandzement W/Z = 0,40) nach 20 Tagen Hydra-
tation - Oberfläche
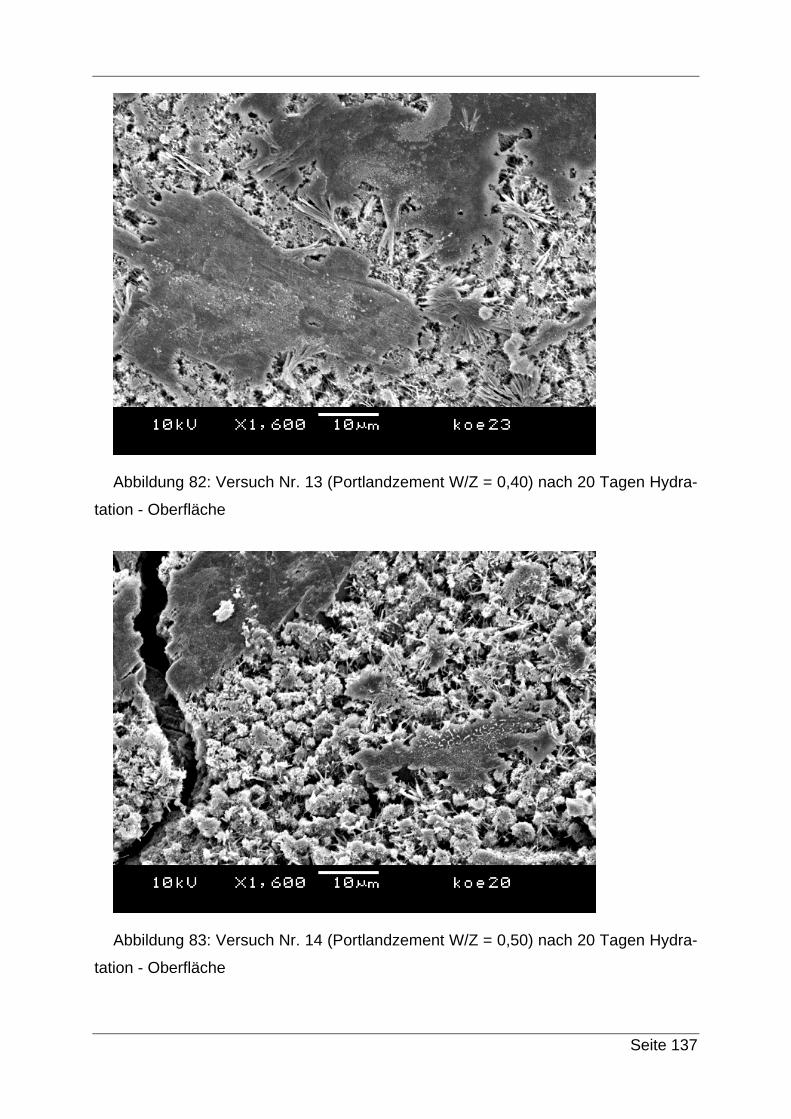
Seite 137
Abbildung 82: Versuch Nr. 13 (Portlandzement W/Z = 0,40) nach 20 Tagen Hydra-
tation - Oberfläche
Abbildung 83: Versuch Nr. 14 (Portlandzement W/Z = 0,50) nach 20 Tagen Hydra-
tation - Oberfläche

Seite 138
Abbildung 84: Versuch Nr. 14 (Portlandzement W/Z = 0,50) nach 20 Tagen Hydra-
tation - Oberfläche
Abbildung 85: Versuch Nr. 14 (Portlandzement W/Z = 0,50) nach 20 Tagen Hydra-
tation – Bruchfläche

Seite 139
Abbildungsverzeichnis
Abbildung 1: Zeitlicher Verlauf der Wärmefreisetzung bei der Zementhydratation
nach Kondo und Ueda [] ........................................................................................... 19
Abbildung 2: Aufbau und Beladung des Probenträgers aus Edelstahl („back
loading“) für die zyklischen XRD-Langzeitmessungen während der
Zementhydratation (Großes Bild links: Probenträger komplett zerlegt; Mitte:
Probenträger leer montiert; rechts: Probenträger mit Zementpaste beladen, hintere
Abdeckung offen; kleines Bild: Probenträger gefüllt, mit Folie abgedeckt und komplett
montiert in Messlage) ............................................................................................... 35
Abbildung 3: Versuchsanordnung zur Bestimmung der Wärmefreisetzung bei der
Hydratation von Zementen ....................................................................................... 42
Abbildung 4: Vergleich von gemessenem und berechnetem Temperaturverlauf bei
der Abkühlung eines ausreagierten Zementsystems (fast vollständige Überlagerung)
................................................................................................................................. 45
Abbildung 5: Röntgenbeugungsdiffraktogramme des CEM I 42,5 R-HS: [rot] –
Orginalsubstanz; [grün] – Rückstand des Salicylsäure-Auszug; [blau] – Rückstand
des Borsäure-Auszug (DH – Calciumsulfatdihydrat; BM – Brownmillerit; AH –
Anhydrit) ................................................................................................................... 51
Abbildung 6: Auszüge des CEM I 42,5 R-HS mit hinterlegten Mineralien ............ 52
Abbildung 7: Lasergranulometrisch bestimmte Korngrößenverteilung des CEM I
42,5 R-HS................................................................................................................. 55
Abbildung 8: Lasergranulometrisch bestimmte Korngrößenverteilung des CEM I
42,5 R....................................................................................................................... 55
Abbildung 9: Lasergranulometrisch bestimmte Korngrößenverteilung des CEM
III/A 32,5 N................................................................................................................ 56
Abbildung 10: Zeitlicher Verlauf der Festigkeitsentwicklung der Zemente in
logarithmischer Darstellung, ermittelt durch Druckfestigkeit von Normmörtel-Prismen
in Anlehnung an EN 196-1........................................................................................ 62
Abbildung 11: Festigkeitsentwicklung der Zemente als Funktion der Quadratwurzel
der Zeit, ermittelt in Anlehnung an EN 196-1............................................................ 63

Seite 140
Abbildung 12: Zeitlicher Temperaturverlauf beim Abbinden der Mörtel aus
CEM I 42,5 R in Abhängigkeit vom Wasser/Zement-Wert ........................................ 65
Abbildung 13: Zeitlicher Verlauf des Wärmeflusses beim Abbinden von Mörteln
aus CEM I 42,5 R ..................................................................................................... 66
Abbildung 14: Zeitlicher Temperaturverlauf beim Abbinden der Mörtel aus
hochsulfatbeständigem Zement CEM I 42,5 R-HS in Abhängigkeit vom W/Z-Wert.. 67
Abbildung 15: Zeitlicher Verlauf des Wärmeflusses beim Abbinden von Mörteln
aus hochsulfatbeständigem Zement CEM I 42,5 R-HS ............................................ 67
Abbildung 16: Zeitlicher Temperaturverlauf beim Abbinden der Mörtel aus
Hochofenzement CEM III/A 32,5N-LH in Abhängigkeit vom W/Z-Wert .................... 68
Abbildung 17: Zeitlicher Verlauf des Wärmeflusses beim Abbinden von Mörteln
aus Hochofenzement................................................................................................ 69
Abbildung 18: Zeitliche Entwicklung des Röntgendiffraktogramms von Zementleim
aus CEM I 42,5 R bei W/Z = 0,40, aufgenommen über eine Hydratationszeit von 0
bis 85 h..................................................................................................................... 71
Abbildung 19: Vergleich der Röntgendiffraktogramme von Zementleim aus
CEM I 42,5 R und W/Z = 0,40 am Beginn (t0) und am Ende (tca. 85 h) der
Hydratations-Experimente ........................................................................................ 72
Abbildung 20: Zeitliche Entwicklung des Portlanditgehaltes im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten, ermittelt mittels Röntgendiffraktometrie
................................................................................................................................. 73
Abbildung 21: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten, ermittelt mittels Röntgendiffraktometrie
................................................................................................................................. 74
Abbildung 22: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten in logarithmischer Darstellung.......... 75
Abbildung 23: Abhängigkeit der Geschwindigkeitskonstante 1. Ordnung für die
Umsetzung von Alit vom W/Z-Wert........................................................................... 76
Abbildung 24: Zeitliche Entwicklung des reziproken Alit-Gehalts im Zementleim
aus CEM I 42,5 R bei verschiedenen W/Z-Werten ................................................... 77

Seite 141
Abbildung 25: Quadratwurzel der zeitlichen Entwicklung des Alit-Gehalts im
Zementleim aus CEM I 42,5 R, bei verschiedenen W/Z-Werten .............................. 78
Abbildung 26: Zeitliche Entwicklung des Ettringit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten.......................................................... 79
Abbildung 27: Zeitliche Entwicklung des Ettringit-Gehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten in logarithmischer Darstellung.......... 80
Abbildung 28: Zeitliche Entwicklung des reziproken Ettringit-Gehalts im
Zementleim aus CEM I 42,5 R bei verschiedenen W/Z-Werten ............................... 81
Abbildung 29: Abhängigkeit der Geschwindigkeitskonstante 2. Ordnung für die
Bildung von Ettringit vom W/Z-Wert.......................................................................... 82
Abbildung 30: Quadratwurzel der zeitlichen Entwicklung des Ettringit-Gehalts im
Zementleim aus CEM I 42,5 R bei verschiedenen W/Z-Werten ............................... 82
Abbildung 31: Zeitliche Entwicklung des Anhydritgehalts im Zementleim aus
CEM I 42,5 R bei verschiedenen W/Z-Werten.......................................................... 83
Abbildung 32: Zeitliche Entwicklung des Portlanditgehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten.................................................... 85
Abbildung 33: Zeitliche Entwicklung des Portlanditgehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten in logarithmischer Darstellung.... 86
Abbildung 34: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten.................................................... 87
Abbildung 35: Zeitliche Entwicklung des reziproken Alit-Gehalts im Zementleim
aus CEM I 42,5 R-HS bei verschiedenen W/Z-Werten............................................. 88
Abbildung 36: Abhängigkeit der Geschwindigkeitskonstanten 2. Ordnung der
Hydratation von Alit im CEM I 42,5 R-HS vom Wasser/Zement-Verhältnis .............. 88
Abbildung 37: Zeitliche Entwicklung des AFt-Gehalts im Zementleim aus
CEM I 42,5 R-HS bei verschiedenen W/Z-Werten.................................................... 90
Abbildung 38: Entwicklung des AFt-Gehalts im Zementleim aus CEM I 42,5 R-HS
als Abhängigkeit von der Quadratwurzel der Zeit ..................................................... 91
Abbildung 39: Abhängigkeit der Geschwindigkeitskonstante kAFt vom W/Z-Wert bei
CEM I 42,5 R-HS...................................................................................................... 92

Seite 142
Abbildung 40: Zeitliche Entwicklung des Röntgendiffraktogramms von Zementleim
aus CEM III/A 32,5 N-LH und W/Z=0,30, aufgenommen über eine Hydratationszeit
von 0 bis 66 Stunden................................................................................................ 94
Abbildung 41: Zeitliche Entwicklung des Portlanditgehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten............................................... 95
Abbildung 42: Zeitliche Entwicklung des Portlandit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten in logarithmischer Darstellung96
Abbildung 43: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten in logarithmischer Darstellung97
Abbildung 44: Zeitliche Entwicklung des reziproken Alit-Gehalts im Zementleim
aus CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten........................................ 97
Abbildung 45: Zeitliche Entwicklung des Alit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten als Funktion der Quadratwurzel
der Zeit ..................................................................................................................... 98
Abbildung 46: Zeitliche Entwicklung des Ettringitgehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten............................................... 99
Abbildung 47: Zeitliche Entwicklung des Ettringit-Gehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten als Funktion der Quadratwurzel
der Zeit ................................................................................................................... 100
Abbildung 48: Abhängigkeit der Geschwindigkeitskonstanten für die Ettringit-
Bildung vom W/Z-Wert in Hochofenzementleimen ................................................. 100
Abbildung 49: Zeitliche Entwicklung des Anhydritgehalts im Zementleim aus
CEM III/A 32,5 N-LH bei verschiedenen W/Z-Werten............................................. 101
Abbildung 50: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM I 42,5 R mit W/Z = 0,40 nach 20 Tagen Hydratation - Oberfläche................. 104
Abbildung 51: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM I 42,5 R mit W/Z = 0,35 nach 2 Tagen Hydratation – Oberfläche .................. 105
Abbildung 52: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM I 42,5 R-HS mit W/Z = 0,35 nach 2 Tagen Hydratation – Oberfläche ............ 105

Seite 143
Abbildung 53: Rasterelektronische Aufnahme des Zementsteingefüges aus
CEM III/A 32,5 N-LH mit W/Z = 0,35 nach 2 Tagen Hydratation – Oberfläche ....... 106
Abbildung 54: Änderung der Intensität des Peaks von Brownmillerit im Laufe der
Hydratation von Zementleim aus CEM I 42,5 R-HS bei W/Z = 0,40, aufgenommen
über eine Hydratationszeit von 82 Stunden............................................................ 113
Abbildung 55: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Portlandit-Gehalts in Portlandzementleimen als Abhängigkeit ln(cCH) = f(Zeit)....... 123
Abbildung 56: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Portlandit-Gehalts in Portlandzementleimen als Abhängigkeit 1/cCH = f(Zeit)......... 123
Abbildung 57: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Portlanditgehalts in Portlandzementleimen als Abhängigkeit cCH = f(Wurzel(Zeit)) 124
Abbildung 58: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,30 (in-situ XRD Aufnahme, durch Abrieb an den Laufrollen des
Diffraktometers wurde nach ca. 2/3 der Laufzeit eine Verschmutzung eingeschleppt,
die jedoch durch Rietveld herausgerechnet wurde.)............................................... 124
Abbildung 59: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,35 (in-situ XRD Aufnahme) ................................................................... 125
Abbildung 60: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,40 (in-situ XRD Aufnahme) ................................................................... 125
Abbildung 61: Zeitlicher Ablauf der Hydratation von Portlandzement CEM I 42,5 R
bei W/Z=0,50 (in-situ XRD Aufnahme) ................................................................... 126
Abbildung 62: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Portlandit-Gehalts in HS-Zementleimen (reziprok) ................................................. 126
Abbildung 63: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Portlanditgehalts in HS-Zementleimen als Abhängigkeit cCH = f(Wurzel(Zeit)) ....... 127
Abbildung 64: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Alit-
Gehalts in HS-Zementleimen als Abhängigkeit ln(cC3S) = f(Zeit) ............................ 127
Abbildung 65: Diffraktometrische Verfolgung der zeitlichen Entwicklung des Alit-
Gehalts in HS-Zementleimen als Abhängigkeit cC3S = f(Wurzel(Zeit)) .................... 128

Seite 144
Abbildung 66: Diffraktometrische Verfolgung der zeitlichen Entwicklung des AFt-
Gehalts in HS-Zementleimen (logarithmisch) ......................................................... 128
Abbildung 67: Diffraktometrische Verfolgung der zeitlichen Entwicklung des AFt-
Gehalts in HS-Zementleimen (reziprok) ................................................................. 129
Abbildung 68: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Anhydrit-Gehalts bei der Hydratation von HS-Zementleimen ................................. 129
Abbildung 69: Diffraktometrische Verfolgung des zeitlichen Verlaufs der
Hydratation des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,30
(in-situ XRD Aufnahme).......................................................................................... 130
Abbildung 70: Diffraktometrische Verfolgung des zeitlichen Verlaufs der
Hydratation des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,35
(in-situ XRD Aufnahme).......................................................................................... 130
Abbildung 71: Diffraktometrische Verfolgung des zeitlichen Verlaufs der
Hydratation des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,40
(in-situ XRD Aufnahme).......................................................................................... 131
Abbildung 72: Diffraktometrische Verfolgung des zeitlichen Verlaufs der
Hydratation des hochsulfatbeständigen Zements CEM I 42,5 R-HS bei W/Z = 0,50
(in-situ XRD Aufnahme).......................................................................................... 131
Abbildung 73: Diffraktometrische Verfolgung der zeitlichen Abnahme des C3S-
Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW bei
verschiedenen W/Z-Werten .................................................................................... 132
Abbildung 74: Diffraktometrische Verfolgung der zeitlichen Abnahme des
Portlandit-Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW
als Abhängigkeit 1/cCH = f(Zeit)............................................................................... 132
Abbildung 75: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Portlandit-Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW
als Abhängigkeit cCH = f(Wurzel(Zeit)) .................................................................... 133
Abbildung 76: Diffraktometrische Verfolgung der zeitlichen Entwicklung des
Ettringit-Gehalts bei der Hydratation von Hochofenzement CEM III/A 32,5 N-NW bei
verschiedenen W/Z-Werten (logarithmisch)............................................................ 133

Seite 145
Abbildung 77: Diffraktometrische Verfolgung des zeitlichen Verlaufs der
Hydratation des Hochofenzements CEM III/A 32,5 N-LH bei W/Z = 0,35 (in-situ XRD
Aufnahme) .............................................................................................................. 134
Abbildung 78: Diffraktometrische Verfolgung des zeitlichen Verlaufs der
Hydratation des Hochofenzements CEM III/A 32,5 N-LH bei W/Z = 0,40 (in-situ XRD
Aufnahme) .............................................................................................................. 135
Abbildung 79: Diffraktometrische Verfolgung des zeitlichen Verlaufs der
Hydratation des Hochofenzements CEM III/A 32,5 N-LH bei W/Z = 0,45 (in-situ XRD
Aufnahme) .............................................................................................................. 135
Abbildung 80: Versuch Nr. 13 (Portlandzement W/Z = 0,40) nach 20 Tagen
Hydratation - Oberfläche ........................................................................................ 136
Abbildung 81: Versuch Nr. 13 (Portlandzement W/Z = 0,40) nach 20 Tagen
Hydratation - Oberfläche ........................................................................................ 136
Abbildung 82: Versuch Nr. 13 (Portlandzement W/Z = 0,40) nach 20 Tagen
Hydratation - Oberfläche ........................................................................................ 137
Abbildung 83: Versuch Nr. 14 (Portlandzement W/Z = 0,50) nach 20 Tagen
Hydratation - Oberfläche ........................................................................................ 137
Abbildung 84: Versuch Nr. 14 (Portlandzement W/Z = 0,50) nach 20 Tagen
Hydratation - Oberfläche ........................................................................................ 138
Abbildung 85: Versuch Nr. 14 (Portlandzement W/Z = 0,50) nach 20 Tagen
Hydratation – Bruchfläche ...................................................................................... 138

Seite 146
Literaturverzeichnis
[1] J. Stark, B. Wicht: Geschichte der Baustoffe. Wiesbaden und Berlin: Bauverlag.
(1998)
[2] R. Benedix: Bauchemie; Wiesbaden, Vieweg + Teubner. (2008)
[3] H. E. Schwiete: Zementchemische Arbeiten aus dem Institut für Gesteinshütten-
kunde. Zement-Kalk-Gips. Heft-Nr. 09/1961 S.400-409
[4] Schimanovich, Pavlovich, Tikavy, Malaschko: Obschaya Khimiya. 1.Aufl. Minsk:
Universitätskae. (1996)
[5] H. R. Christen: Grundlagen der allgemeinen und anorganischen Chemie. 2.Aufl.
Aarau: Verlag Sauerländer Aarau; Salle Verlag Frankfurt a.M.. (1969)
[6] G. Lang: Reaktionskinetik. 1. Aufl. Frankfurt am Main, Aarau: Moritz Diesterweg
GmbH, Sauerländer Verlag. (1990)
[7] H. Schmalzried: Chemical Kinetics of Solids. Weilheim. New York Basel Cam-
bridge Tokio: VCH Verlagsgesellschaft mbH. (1995)
[8] E. S. Swinbourne: Auswertung und Analyse kinetischer Messungen. Weinheim:
Verlag Chemie GmbH. (1975)
[9] W. Jost, K. Hauffe: Diffusion - Methoden der Messung und Auswertung. 2.Aufl.
Darmstadt: Dr. Dietrich Steinkopf Verlag. (1972)
[10] J. Crank: The Mathematics of Diffusion. second edition.Aufl. Oxford: Clarendon
Press. (1975)
[11] D.A. Frank-Kamenetzki: Stoff- und Wärmeübertragung in der chemischen Kine-
tik. Berlin/Göttingen/Heidelberg: Springer. (1959)
[12] B.F. Johannesson: Prestudy on diffusion and transient condensation of water
vapor in cement mortar. Cement and Concrete Research. Heft-Nr. 32/2002 pp. 955-
962

Seite 147
[13] D. P. Bentz, E. J. Garboczi, C. J. Haecker, O. M. Jensen: Effects of cement
particle size distribution on performance properties of Portland cement-based materi-
als. Cement and Concrete Research. Heft-Nr. 29/1999 pp. 1663-1671
[14] Paschenko A.A.: Physikalische Chemie der Silikate. Moskau: Vysschaya Schko-
la. (1986)
[15] V. Johansen, N.H. Christensen: Rate of formation of C3S in the system CaO-
SiO2-Al2O3-Fe2O3-MgO with addition of CaF2. Cement and Concrete Research.
Heft-Nr. 01/1979 pp. 1-5
[16] F. Häussler, M. Hempel, H. Baumbach: Long-time monitoring of the microstruc-
tural change in hardening cement paste by SANS. Advances in Cement Research.
Heft-Nr. 10/1997
[17] H. M. Jennings: A model for the microstructure of calcium silicate hydrate in
cement paste. Cement and Concrete Research. Heft-Nr. 30/2000 pp. 101-116
[18] A. Eckart; H.-M. Ludwig; J. Stark: Zur Hydratation der vier Hauptklinkerphasen
des Portlandzements. ZKG International. Heft-Nr. 8/1995 S.443-452
[19] J. J. Thomas, H. M. Jennings, A. J. Allen: Determination of the Neutron Scatter-
ing Contrast of Hydrated Portland Cement Paste using H2O/D2O Exchange. Advn.
Cem. Bas. Mat.. Heft-Nr. 07/1998 pp. 119-122
[20] S.A. FitzGerald, J.J. Thomas, D.A. Neumann, R.A. Livingston: A neutron scat-
tering study of the role of diffusion in the hydration of tricalcium silicate. Cement and
Concrete Research. Heft-Nr. 32/2002 pp. 409-413
[21] A. J. Allen, J. J. Thomas, H. M. Jennings: Composition and density of nano-
scale calcium-silicate-hydrate in cement. nature materials. Heft-Nr. vol. 6 April 2007
[22] J.Plank, C.Hirsch, D.Stephan; Construction chemicals and materials, in: Winna-
cker-Küchler: Chemische Technik (5. Auflage); Wiley-VCH; 2004
[23] K. Kurtis: Portland Cement Hydration. Atlanta: School of Civil Engineering, Lec-
tures online. (2007)
[24] F. W. Locher: Stöchiometrie der Hydratation von Tricalciumsilikat. Zement-Kalk-
Gips. Heft-Nr. 9/1967 S. 402

Seite 148
[25] R. Berliner; M. Popovici; K.W. Herwig; M. Berliner; H.M. Jennings; J.J. Thomas:
Quasielastic neutron scattering study of the effect of water-to-cement ratio on the
hydration kinetics of tricalcium silicate. Cement and concrete research. Heft-Nr.
02/1998 pp. 231-243
[26] K. Fujii; W. Kondo: Kinetics of the hydration of tricalcium silicate; J. Am. Ceram.
Soc.. Heft-Nr. 57 (1974) p. 492
[27] H.F.W. Taylor: Nanostructure of C--S--H: Current status. Adv. Cem. Based
Mat.. Heft-Nr. 1/1/1993 pp. 38-46
[28] J. S. Schweitzer, R.A. Livingston, C. Rolfs, H.-W. Becker, S. Kubsky: Ion beam
analysis of the hydration of tricalcium silicate. Nuclear Instruments and Methods in
Physics Research. Heft-Nr. B 207/2003 pp. 80-84
[29] J. Zelic, D. Rusic, D. Veza, R. Krstulovic: The role of silica fume in the kinetics
and mechanism during the early stage of cement hydration. Cement and Concrete
Research. Heft-Nr. 30/2000 pp. 1655-1662
[30] A.J. Allen, J.J. Thomas, H.M. Jennings: Composition and density of nanoscale
calcium–silicate–hydrate in cement. nature materials. Heft-Nr. VOL 6 APRIL 2007 pp.
311-316
[31] K.S. Jewstropjew, N.A. Toropow: Einführung in die Silikatchemie. Wiesbaden:
Bauverlag GmbH. (1958)
[32] K.O. Kjellsen. B. Lagerblad: Microstructure of tricalcium silicate and Portland
cement systems at middle periods of hydration-development of Hadley-grains. Ce-
ment and Concrete Research. Heft-Nr. 01/2007 pp. 13-20
[33] F. Häussler, Karen Friedmann: SANS and calorimetry - useful combinable
methods for applied basic research on hydrating C3S and cement pastes. Lacer.
Heft-Nr. 07/2002
[34] H. M. Jennings: A model for the microstructure of calcium silicate hydrate in
cement paste. Cement and Concrete Research. Heft-Nr. 30/2000 pp. 101-116
[35] U. Ludwig, N.B. Singh: Kinetics and mechanism of alite hydration. Zement-
Kalk-Gips. Heft-Nr. 12/1968 pp. 688-692

Seite 149
[36] P. Paulini: Kinetische Reaktionsmessungen der Zementhydratation mit dem
Tauchwägeverfahren. Zement-Kalk-Gips. Heft-Nr. 10/1988 pp. 525-531
[37] J. Stark, B. Wicht: Anorganische Bindemittel. Weimar: Universitätsverlag Bau-
haus-Universität Weimar. (1998)
[38] A. Eckart, J.Stark: Betrachtung der Hydratationsprodukte des Calciumaluminats
und des Calciumferrits mit ESEM-FEG; Tagungsband der 13. ibausil. Weimar: HAB
Weimar. (1997)
[39] C. Plowman, J.G. Cabrera: Mechanism and kinetics of hydration of C3A and
C4AF extracted from cement. Cement and Concrete Research. Heft-Nr. 03/1984 pp.
238-248
[40] M.R. Hartman, R. Berliner: In situ neutron powder diffraction investigation of the
hydration of tricalcium aluminate in the presence of gypsum. Journal of solid state
chemistry. Heft-Nr. 178/2005 pp. 3256-3264
[41] Katrin Bollmann: Ettringitbildung in nicht wärmebehandelten Betonen. Disserta-
tion. Aufl. Weimar: Bauhaus-Universität Weimar. (2000)
[42] N. Tenoutasse: Untersuchungen über die Kinetik der Hydratation des Tricalci-
umaluminats in Gegenwart von Calciumsulfat und Calciumchlorid. Zement-Kalk-Gips.
Heft-Nr. 10/1967 S. 435
[43] T. Liang, Y. Naru: Hydration Products of Calcium Aluminoferrite in the presence
of Gypsum. Cement and Concrete Research. Heft-Nr. 24 (1993) S. 150-158
[44] A. Emanuelson, S. Hansen, E. Henderson, A. Landa-Canovas, E. Sjöstedt:
Ferrite - Microstructure in Clinker and Hydration of Syntetic Phases and Sulfate Re-
sisting Cements. Papers of the 10. ICCC in Göteborg. Heft-Nr. Vol. 1 li060 8pp
[45] N. Meller, C. Hall, A.C. Jupe, S.L. Colston,S.D.M. Jacques, P. Barnesb, J.
Phipps: The paste hydration of brownmillerite with and without gypsum: A time re-
solved synchrotron diffraction study at 30, 70, 100 and 150 °C. Journal of Materials
Chemistry. Heft-Nr. 14/2004 pp.428-435
[46] T. Matschei, B. Lothenbach, F.P.Glasser: The AFm-phase in Portland Cement.
persönlich vom Autor - nicht veröffentlicht.

Seite 150
[47] J. W. Bullard, E. J. Garboczi: A model investigation of the influence of particle
shape on portland cement hydration. Cement and Concrete Research. Heft-Nr.
36/2006 pp. 1007-1015
[48] Kondo, R.; Ueda, S.: Kinetics and mechanism of hydration of cements. Bd. II.
Tokyo: 5. Intern. Symps. Chem. Cem. (1968)
[49] Ingo Müller: Influence of cellulose ethers on the Kinetics of early Portland ce-
ment hydration. Karlsruher Mineralogische und Geochemische Hefte, Karlsruhe: Uni-
versitätsverlag Karlsruhe. (2006)
[50] Timaschev V.V.: Einfluss der physikalischen Struktur des Zementsteines auf
seine Festigkeit. Zement. Heft-Nr. 2/1979
[51] Jung V.N.: Grundlagen der Technologie der Bindemittel. Moskau: Promstroiiz-
dat. (1951)
[52] F. Häußler, F. Eichhorn, S. Röhling, H. Baumbach: Monitoring of the hydration
process of hardening cement pastes by small-angel neutron scattering. Cement and
Concrete Research. Heft-Nr. 20/1990 pp. 644-654
[53] F. Häußler, S. Palzer, A. Eckart: Mikrostrukturuntersuchungen an hydratisie-
renden Zementklinkerphasen mittels Neutronenkleinwinkelstreuung. Thesis, Wissen-
schaftliche Zeitschrift der Bauhaus-Universität Weimar. Heft-Nr. 5/6(2001)
[54] Timaschev V.V.: Synthese und Hydratation von Bindemitteln. Moskau: Nauka.
(1986)
[55] A. Bezjak: Kinetic analysis of cement hydration including various mechanistic
concepts. Cement and Concrete Research. Heft-Nr. 05/1983 pp. 305-318
[56] S. Popovics: Model for the quantitative description of the kinetics of hardening
of portland cements. Cement and Concrete Research. Heft-Nr. 09/1987 pp. 821-838
[57] F. Häussler, M. Hempel, H. Baumbach, J. Tritthart: Nanostructural investiga-
tions of hydrating cement pastes produced from cement with different fineness levels.
Advances in Cement Research. Heft-Nr. 04/2001
[58] Qi X., J. Stark: Quantifizierung der Zementhydratation bei der Verwendung ei-
nes alkalifreien Erstarrungsbeschleunigers. ZKG International. Heft-Nr. 10/2005 pp.
68-79

Seite 151
[59] Dem Geheimnis des "schlafenden" Betons auf der Spur. VDZ-Mitteilungen.
Heft-Nr. 124 (Mai 2004)
[60] H. Hilbig, F. H. Köhler, P. Schießl: Hydratation von Hochleistungsbetonen -
NMR-spektroskopische Untersuchungen. Poster, Jahrestagung GDCh FG Bauche-
mie 2004.
[61] D. P. Bentz: Influence of the water-to-cement ratio on hydration kinetics: Simple
models based on spatial considerations. Cement and Concrete Research. Heft-Nr.
36/2006 pp. 238-244
[62] M. Avrami: J. Phys. Chem. Heft-Nr. 7, (1938) p. 1103 and 8, (1940) p. 212
[63] Thomas, Jennings: Effects of D2O and Mixing on the Early Hydration
Kinetics of Tricalcium Silicate; Chem. Mat.. Heft-Nr. 11:, 1999 pp. 1907-1914
[64] Jelenic: Kinetics of hydration of cement phases; Adv. Cem. Techn. Gosh (Ed).
Pergamon. (1987)
[65] J.M. Pommersheim, J.R. Clifton: Mathematical modelling of tricalcium silicate
hydration. Cem. Concr. Res. Heft-Nr. 9/1979 pp. 765-770
[66] T. Knudsen: The dispersion model for hydration of portland cement: 1. General
concept. Cem. Concr. Res.. Heft-Nr. 14/1984 pp. 622-630
[67] B. Osbeak, V. Johansen: Particle size distribution and rate of strength devel-
opment of portland cement. J. Am. Ceram. Soc. Heft-Nr. 72 (2/1989) pp. 197-201
[68] L. Odler, S. Abdul-Maula: Investigations on the relationship between porosity
structure and strength of hydrated Portland cement pastes III. Effect of clinker com-
position and gypsum addition. Cement and Concrete Research. Heft-Nr. 17/1987 pp.
22-30
[69] Sandrine Garrault, André Nonat: Hydrated Layer Formation on Tricalcium and
Dicalcium Silicate Surfaces: Experimental Study and Numerical Simulations. Lang-
muir. Heft-Nr. 2001, 17 pp. 8131-8138
[70] M. Geiker: Studies of Portland Cement Hydration: Measurements of Chemical
Shrinkage and a Systematic Evaluation of Hydration Curves of the Dispersion Model.
Lyngby: Ph. D Thesis, Technical University of Denmark. (1983)

Seite 152
[71] D.P. Bentz: A Three-Dimensional Cement Hydration and Microstructure Pro-
gram. I. Hydration Rate, Heat of Hydration, and Chemical Shrinkage. NISTIR 5756,
U.S. Department of Commerce. Heft-Nr. November 1995
[72] D.P. Bentz, E.J. Garboczi, H.M. Jennings, D.A. Quenard: Multi-scale digital-
image-based modelling of cement-based materials. Materials Research Society.
Heft-Nr. 370/1995 pp. 33-41
[73] D.P. Bentz: CEMHYD3D: A Three-Dimensional Cement Hydration. U.S. De-
partment of Commerce, National Institute of Standards and Technology. NISTIR.
Heft-Nr. 6485, April 2000
[74] P. Navi, C. Pignat: Simulation of cement hydration and the connectivity of the
capillary pore space. Adv. Cem. Based Mat.. Heft-Nr. 4/2/1996 pp. 58-67
[75] B. Lothenbach, F. Winnefeld: Thermodynamic modelling of the hydration of
Portland cement. Cem. Concr. Res.. Heft-Nr. 36/2006 pp. 209-226
[76] B. Lothenbach: at Nanocem workshop on "Computing Hydrate Phase Assam-
bleges of Portland Pastes". Dübendorf: November 19-20, 2007. (2007)
[77] K.L. Scrivener, T. Füllmann, E. Gallucci, G. Walenta, E. Bermejo: Quantitative
study of Portland cement hydration by X-ray diffraction/Rietveld analysis and inde-
pendent methods. Cement and Concrete Research. Heft-Nr. 34/2004 pp. 1541-1547
[78] J. Neubauer; F. Götz-Neunhoeffer: In-situ XRD-Analyse und Wärmefluss-
kalorimetrie am Beispiel der Zementhydratation. Berlin: GDCh Monographie. Heft-Nr.
Bd. 35 (2005)
[79] M. Kaszynska: Early age properties of high-strength/high-performance con-
crete. Cement & Concrete Composites. Heft-Nr. 24/2002 pp. 253-261
[80] R. Krstulovic, P. Dabic: A conceptual model of the cement hydration process.
Cement and Concrete Research. Heft-Nr. 30/2000 pp. 693-698
[81] P. Dabic, R. Krstulovic, D. Rusic: A new approach in mathematical modelling of
cement hydration development. Cement and Concrete Research. Heft-Nr. 30/2000
pp. 1017-1021
[82] W. Hemminger, Günter Höhne: Grundlagen der Kalorimetrie. Weinheim: Verlag
Chemie GmbH. (1979)

Seite 153
[83] Taylor: Cement Chemistry. 2.Aufl. London: Academic Press. (1992)
[84] L.E.Copeland, D.L. Kantro and G. Verbeck: Chemistry of Hydration of Portland-
cement, 4th International Symposium on the chemistry of cement, Vol. I, pp. 429-465,
1960
[85] Locher, F. W., Zement; Düsseldorf, 2000, Verlag Bau und Technik
[86] I. Odler, S. Abdul-Maula: Die Möglichkeiten der Trennung der einzelnen Be-
standteile des Portlandzements durch selektive Lösungsmittel. Zement-Kalk-Gips.
Heft-Nr. 10/1979
[87] Harald Krischner: Einführung in die Röntgenfeinstrukturanalyse. 2..Aufl. Braun-
schweig/Wiesbaden: Freidr. Vieweg & Sohn. (1980)
[88] H.M. Rietveld: A Profile Refinement Method for Nuclear and Magnetic Struc-
tures. J. Appl. Cryst.. Heft-Nr. 2/1969 pp. 65-71
[89] D.B. Wiles and R.A. Young: A new computer program for Rietveld analysis of
X-ray powder diffraction patterns. J. Appl. Cryst.. Heft-Nr. 14/1981 pp. 149-151
[90] G. Walenta; T. Füllmann: Advances in quantitative XRD Analysis for Clinker,
Cements and cementitious Additions. ICDD, Advences in X-ray Analysis. Heft-Nr.
47/2004 pp. 287-296
[91] C. Bezou; A. Nonat; J.C. Mutin; A.N. Norlund Christiansen; M.S. Lehmann: Of
the crystal structure of gamma-CaSO4, CaSO4*0,5(H2O), and CaSO4*0,6(H2O) by
powder diffraction methods. Journal of solid state chemistry. Heft-Nr. 117/1995 pp.
165-176
[92] Paul F. Schofield, Kevin S. Knight, Iona Stretton: Thermal expansion of gypsum
investigated by neutron powder diffraction. American Mineralogist. Heft-Nr. 81/1996
pp. 847-851
[93] Henderson D. M.: A nuclear magnetic resonance determination of the hydrogen
positions in Ca(OH)2. American Mineralogist. Heft-Nr. 47/1962 pp. 1231-1251
[94] Markgraf S.A.: High-temperature structure refinements of calcite and magnesite
American Mineralogist. Heft-Nr. 70/1985 pp. 590-600

Seite 154
[95] Rainer Schmidt: Rietveldanalyse: Stand der Technik. ZKG International. Heft-
Nr. 12/2005 pp. 62-69
[96] Wadsö L.: Application of an eight-channel isothermal conduction calorimeter for
cement hydration studies. Cement International. Heft-Nr. 5/2005 pp. 94-101
[97] Horst Kuchling: Physik. 17.Aufl. Leipzig: VEB Fachbuchverlag. (1985)
[98] MC Agrawal; Bimal K Modi: Vorhersage der Zementfestigkeit. ZKG Internatio-
nal. Heft-Nr. 6/2006 pp. 39-45
[99] Verein Deutscher Zementhersteller e.V.: Von der Korngrößenverteilung zur
Normdruckfestigkeit. VDZ-Mitteilungen - Mitteilungen des Forschungsinstituts der
Zementindustrie. Heft-Nr. Mai 2005
[100] Bunke, N.: Prüfung von Beton - Empfehlungen und Hinweise als Ergänzung
zu DIN 1048. Berlin: Beuth. (1991)
[101] Hintzen, W.: Zum Verhalten des jungen Betons unter Zwang beim Abfließen
der Hydratationswärme. Düsseldorf: Bau + Technik. (1998)
[102] Bogue, R.H.: The Chemistry of Portland Cement. 2..Aufl. New York: Reinhold
Publishing Corp.. (1955)
[103] J. Zelic, D. Rusic, R. Krstulovic: A mathematical model for prediction of com-
pressive strength in cement-silica fume blends. Cement and Concrete Research.
Heft-Nr. 34/2004 pp. 2319-2328
[104] D.P. Bentz, C.J. Haecker, X.P. Feng, P.E. Stutzman: Prediction of Cement
Physical Properties by Virtual Testing. Düsseldorf: Fifth International VDZ Congress,
Düsseldorf, September 23-27, 2002. (2003, pp. 53-63)
[105] Alfred Hilbert: Mathematik. 2.Aufl. Leipzig: VEB Fachbuchverlag. (1989)
[106] L. Buffo-Lacarriere, A. Sellier, G. Escadeillas, A. Turatsinze: Multiphasic finite
element modeling of concrete hydration. Cement and Concrete Research. Heft-Nr.
37/2007 pp. 131-138
[107] L.D. Aloia, G. Chanvillard: Determining the "apparent" activation energy of
concrete Ea-numerical simulation of the heat of hydration of cement. Cement and
Concrete Research. Heft-Nr. 32/2002 pp. 1277-1289

Seite 155
[108] H.J. Kuzel: Ein einfaches Wärmeleitungskalorimeter mit hoher Empfindlichkeit,
Fortschr. Min. 60 (1982), pp. 128-129.
[109] J. Stark, B. Möser, A. Eckart: Neue Ansätze zur Zementhydratation - Teil 1.
Zement-Kalk-Gips. Heft-Nr. 01/2001 pp. 52-60
[110] Locher, F.W.; Richardtz, W.; Sprung, S.: Erstarren von Zement - Teil I: Reak-
tion und Gefügeentwicklung. Zement-Kalk-Gips. Heft-Nr. 10/1976 pp. 435-442
[111] J. Rose, A. Bernard, A. Masion, P. Chaurand, I. Moulin, J-Y Bottero: Iron-
heavy metal interactions in cement: a positive impact for the long term release of in-
organic pollants ?. Villingen: CEMNET Workshop: Cement Research on Large-Scale
Facilities, September 10, 2007. (2007)
[112] M. Fukuhara, S. Goto, K. Asaga, M, Daimon, R. Kondo: Mechanism and kinet-
ics of C4AF hydration with gypsum. Cem. Concr. Res.. Heft-Nr. 11/1981 pp. 407-414
[113] B. Lothenbach: Diskussion zu [111]. Villingen: (2007)
[114] Schwarzenlander Johann, Linde AG: Versuche zur Beschleunigung von Beton
mit CO2. Rohrdorf: (2006)
[115] EP 1785246 ; Buinger Alexander; Kimbacher Alfred; Rebhan Dieter; Schmand
Ralf; Werthmann Eckart ; Method to dissolve carbon dioxide in water used for spay-
able concrete ; Linde AG ; 2005-12-29
[116] EP 1785245 ; Buinger Alexander, Kimbacher Alfred, Rebhan Dieter, Schmand
Ralf, Werthmann Eckart; Method to control the amount of carbon dioxide dissolved in
water for sprayable concrete ; Linde AG ; 2005-12-29
[117] DE 10310599 ; Rebhan Dieter ; Spraying concrete spray onto an object via
nozzles with addition of carbon dioxide to the concrete useful as a safety measure in
tunnel and side wall construction and for construction of mine walls ; Linde AG ;
2003-03-11