Immunoassay Puffer - applichem.com · Die Crux mit den Kreuzreaktivitäten 2 Ohne Blockierung kein...
40
Take the Pink Link! www. .com Immunoassay Puffer
Transcript of Immunoassay Puffer - applichem.com · Die Crux mit den Kreuzreaktivitäten 2 Ohne Blockierung kein...

Darmstadt hat eine weitere Topadresse:AppliChem GmbH Ottoweg 4 D - 64291 Darmstadt Fon +49 6151 9357-0 Fax +49 6151 9357-11
eMail [email protected] internet www.applichem.com
Take the Pink Link!
www. .com
Immunoassay Puffer

Take the Pink Link!
www..com
Gel Electrophoresis Size Marker
Gel Electrophoresis Size Marker
Gel Electrophoresis
Take the Pink Link!
www..com
Detergents
Take the Pink Link!
www..com
Immunoassay Buffer
Take the Pink Link!
www..com
Kontaminationen durch Nukleinsäurendurch Nukleinsäurendurch
Probleme & praktische Lösungen NukleinsäurenProbleme & praktische Lösungen Nukleinsäuren
Take the Pink Link!
www..com
WichtigesWissenswertesWunderbaresWunderbares
aus Chemie & Biologie& Biologie&
AppliCationsDer Anteil von molekularbiologischen Nachweismethoden ist in den letzten Jahren erheblich gestiegen, besonders in den Bereichen Qualitätskontrolle, Forensik, klinischer Forschung und Diagnostik – insbesondere der Infektionsdiagnostik. Gerade für diese Applikationen werden hochsensitive und gleichzeitig zuverlässige PCR-Tests benötigt. Dafür bietet AppliChem nun optimierte PCR-Kits an und widmet sich explizit der Hintergrundproblematik, die durch DNA belastete Reagenzien und Arbeitsplätze entstehen kann.
Nr.3AppliCations
Nr.3AppliCationsDNA-freie Reagenzien und Mastermixe für die PCR
AppliCationsAppliCations
Freie Nukleinsäuren verursachen als Kontaminationen große Probleme im Forschungs- und molekularbiologisch-analytischen oder klinisch-diagnostischen Labor. Durch die extrem hohe Sensitivität von DNA-Nachweistests, können kleinste Verunreinigungen in PCR-Ansätzen zusätzliche Arbeit bedeuten und im schlimm-sten Fall Ergebnisse verfälschen. Mit Derma-ExitusPlus™ (HHDK) aus der Serie von ExitusPlus™-Produkten wird erstmals ein völlig neuer Anwendungsbereich erschlossen bzw. zusätzliche Kontaminationsquellen ausgeschlossen.
Eine der Hauptquellen für Kontaminationen mit Nukleinsäuren ist der Experimentator selbst. Die Nukleinsäuren stammen z.B. aus Hautschuppen, Haaren und Speichel oder von Mikroorganismen, die seine Haut besiedeln oder z.B. beim Niesen freigesetzt werden. Gelangen diese in die PCR-Ansätze oder PCR-Reagenzien, können sie entsprechend der eingesetzten Primer (besonders 16S rDNA für Bakterien) leicht nachgewiesen werden. Ausserdem besteht die Gefahr, dass beim Öffnen und
Nr.5AppliCations
Nr.5AppliCationsDekontamination der Haut und Hände von Nukleinsäuren
AppliCations AppliCationsSize-exclusion chromatography (SEC) is a popular method to separate biomolecules based on their size. Primarily, it is applied to the separation of biopolymers such as proteins and nucleic acids, i.e. water-soluble polymers. This system is also called gel filtration, typically with beads of dextran or agarose serving as gel matrix. Smaller molecules pass significantly slower through the column than larger molecules. Not to be mixed up with gel electrophoresis, there are big differences in terms of theseparation principle. SEC does not require electric current and the sieving effect will not separate small molecules first.
It is indeed correct that smaller molecules pass more slowly through the matrix than larger molecules. This is due to the longer path the smaller molecules must travel. The longer path arises from the pores of the beads. The smaller molecules can
Keywords
No.6AppliCations
No.6AppliCationsSize-Exclusion Chromatography for purification of biomolecules
AppliCations
Take the Pink Link!
.com
Agarose-Gel-Elektrophorese AppliCations
Using ready-to-use ELISA kits from manufacturers is easy and convenient. Sometimes however, home-made ELISA is required because there is no kit available with the right antibodies or the characteristics of the available kits such as their limits of detection are not appropriate. Ready-to-use ELISA kits from good suppliers mastored for two years at 4°C without any problem. With home-made ELISA it is a completely different story. For any new measurement one has to coat a new plate, because after storage of some days the plates don’t perform as well as before.
Why is there such a great difference in storage between home-made ELISA and ELISA kits?The reason is that in professional ELISA kit production the plates are not only blocked after coating, but also stabilised. This easy to perform process has been an industry standard for thirty years. For stabilisation of a plate one has to coating stabiliser solution. It is just as simple as a “second blocking step”. But there were no such high lutions freely available in low volumes for use in research lab until now. AppliChem now offers a product for use in every researchin volumes starting as small as 50 ml, which is called the AppliCoat Plate Stabiliser (Cat. No. A7708). This stabiliser solution is easy-to-use and has a great advantage compared to almost any stabiliser used in industry. It gives better storage stability for coated antibodies and antigens than most other products do. And there is a second
Two benefits with one solutionWhen antibodies are coated onto ELISA plates, most of the antibodies are not active. When the antibodies (or any proteins) come into close contact to the plastics surface of the ELISA plate, conformational changes can occur due to surface-protein interactions. The result is that most antibodies coated on a plate are unfolded or inactive. Only around 2–8 % of all coated antibodies remain active and can bind to analytes and this is greatly variable depending on the surface characteristics of the ELISA plate, which can really differ from batch to batch or even from well to well.These differences from well to well can affect the variability of an assay, because the antibodies can be affected. If there waway of refolding antibodies and of preserving antibodies from conformational changes during storage, this could help to decrease such variabilities in assay performance. This is a key benefit of AppliCoat Plate Stabiliser. It assists antibodies and coated proteins to refold and then to preserve active conformation over a long time. Thus it has two benefits: 1. Refolding of antibody conformation of some of the coated antibodies and 2. Preserving correct conformation during storage. These benefits are used for production of high-quality ELISA kits as well as in research applications now. Even with AppliCoat Plate Stabiliser the percentage of active antibodies will still be in the range of 2–8 %. But the great difference is that the variability from well to well and from plate to plate can be minimised in most assays by using AppliCoat Plate Stabiliser. Such effects depend on the used antibodies, but when ELISA are validated (e.g. according to “Guidance for Industry: Bioanalytical Method Validation”, FDA, 2001) or according to other validation strategies, the difference can be measured in many assays.The positive effects of AppliCoat Plate Stabiliser are shown in Fig. 1. A sandwich ELISA with a monoclonal antibody has been
Keywords
Immunoassays
Antibody Stabilisation
ELISA Plates
Cross-reactivity
Interfering effects
Improving quality of ELISA
AppliCationsDie moderne Gentechnik zeigt, dass in vielen Fällen schon freie DNA-Moleküle für Infektionen, Rekombinationen oder biologische Transformationen ausreichen [1,2]. Zusätzlich werden die Nachweisverfahren für DNA-Moleküle immersensitiver. Daher wird die Detektion von Kontaminationen oder die Verhinderung von Amplifikations-Artefakten in der PCR für die Gentechnik, die Kriminalistik, die Biomedizin und die Hygiene immer wichtiger. Die vollständige Dekontamination von Geräten und Materialien von DNA-Molekülen wird so zu einem entscheidendenFaktor für die allgemeine biologische Sicherheit.
Alles oder Nichts: Erstaunliche ErkenntnisDas Mittel der Wahl zur Beseitigung von Kontaminationen durch NukleinDas Mittel der Wahl zur Beseitigung von Kontaminationen durch NukleinDas Mittel der Wahl zur Beseitigung von Kontamina säuren ist immer noch Chlorbleichlauge („bleach“) – ein Mittel das alles zerstört, nicht nur die Nukleinsäure. Dies hat uns veranlasst in Kooperation mit multiBIND Biotech, Köln, nach einer unschädlichen Alternative zu suchen und die molekulare Wirkungsweise der auf dem Markt befindlichen sonstigen DNA-Dekontaminationsmittel zu untersuchen. Hierfür wurde unter sehr hoher Belastung (großer DNA-Über-
suchen. Hierfür wurde unter sehr hoher Belastung (großer DNA-Über-suchen. Hierfür wurde unter sehr hoher Belastung (großer DNA-Überschuss) mit definierten DNA-Kontaminationen die Eigenschaften der konventionellen Mittel verglichen. Zwei Probleme werden offensichtlich: Erstens werden durch die konventionellen Mittel in keinem Fall die DNA-Moleküle effizient zerstört und zweitens enthalten diese Mittel Komponenten mit stark korrosiven oder giftigen Eigenschaften. Als Fazit daraus hat sich für uns die Notwendigkeit der Neuentwicklung einer effektiven Lösung zur DNA-Dekontamination ergeben, die wir hier als DNA-ExitusPlus™ und Autoclave-ExitusPlus™ vorstellen. Im Vergleich zu den herkömmlichen Produkten wird DNA und RNA schnell und effizient zerstört, ohne dass das Reagenz korrosive oder giftige Eigenschaften aufweist.Bei der DNA-Dekontamination unterscheidet man nach der molekularen Wirkungsweise der eingesetzten Mittel drei Grund-prinzipien zur Zerstörung oder Inaktivierung der genetischen Information: Modifikation, Denaturierung und Degradation.
prinzipien zur Zerstörung oder Inaktivierung der genetischen Information: Modifikation, Denaturierung und Degradation.
prinzipien zur Zerstörung oder Inaktivierung der genetischen Information: ModifikaJe nach Zusammensetzung der Mittel können diese drei Prinzipien einzeln oder in Kombination angewandt werden.Da nach den aktuellen Erkenntnissen zum biologischen Risikopotenzial von freien DNA-Molekülen für eine wirklich sichere DNA-Dekontamination die Zerlegung dieser DNA-Moleküle in möglichst kleine Fragmente die wirkungsvollste Methode ist, wurden die gängigen konventionellen Mittel mit unserer Neuentwicklung DNA-ExitusPlus™ im DNA-Degradationstest ver-tionellen Mittel mit unserer Neuentwicklung DNA-ExitusPlus™ im DNA-Degradationstest ver-tionellen Mittel mit unserer Neuentwicklung DNA-ExitusPlus™ im DNA-Degradationstest verglichen. Der DNA-Degradationstest erlaubt einen sensitiven, quantitativen Vergleich der Geschwindigkeit des DNA-Abbaus (Abb. 1 und 2).
Unerwarteter Weise haben wir festgestellt, dass einige der bekannten kommerziellen Mittel nur mit dem Prinzip der Modifi-kation oder Denaturierung der DNA-Moleküle arbeiten. Eine Zerlegung der DNA-Stränge erfolgt dabei nicht, sondern die genetische Information, für die diese DNA-Stränge kodieren, wird eigentlich nur maskiert. Eine genetische Information, für die diese DNA-Stränge kodieren, wird eigentlich nur maskiert. Eine genetische Informa
chemische Demaskierung der DNA-Moleküle durch Entfernung der blockierenden Gruppen würde die genetische Information wieder lesbar und am-plifizierbar machen. Nach dem heutigen Wissensstand zur Gentechnik und der Problematik der Neukombination von Erb-trägern sind solche Mittel eigentlich nicht mehr zeitgemäß. Aber auch die Mittel, die zu einer nachweisbaren Degradation
Keywords
Nukleinsäure-Dekontamination
DNA-DegradationstestPCR-Test
Autoklavieren von DNA
Nr.1AppliCations
Nr.1AppliCationsNukleinsäure-Dekontamination mit der ExitusPlus™-Technologie
AppliCations
Take the Pink Link!
www..com
TransferMembranes
Take the pink Link!
www..com
Take the pink Link!
www..com
AppliChem investiert seit den ersten Jahren der
Firmengründung intensiv in Kommunikation und Marketing.
Der frische und ungewöhnliche Auftritt hat von Anfang
an für große Aufmerksamkeit im Markt gesorgt. Unser
Wachstum bestätigt die Relevanz dieser Maßnahmen.
Wir bieten unseren Kunden eine umfassende Bibliothek
mit zahlreichen Broschüren und Appli kationen, die den
Anwendern den Alltag im Labor erleichtern.
Ein umfassender Produktkatalog mit Detail informationen,
die in keinem anderen Katalog der Branche aufgeführt
sind, erscheint in deutsch und englisch.
AppliChem bringt Vorteile durch Wissen.
„Denken ohne Wissen macht den Zufall zum Herrscher.“
Werner Kollath, deutscher Bakteriologe

Die Crux mit den Kreuzreaktivitäten 2
Ohne Blockierung kein Ergebnis 10
Tipps zur Lagerung von Antikörpern 12
Blockierer im Vergleich 14
Qualitätsverbesserung & höhere Stabilität von ELISAs 18
Produkte mit Anwendungshinweisen
Antibody Stabilizer-PBS 21
Antibody Stabilizer-Tris 21
Applicoat Plate Stabilizer 22
Blocking Buffer I 23
Blocking Buffer II EGrade 24
Blocking Buffer III BSA 25
Blocking Reagenz CA 26
Coating Buffer pH 9,6 27
Coating Buffer pH 7,4 27
CrossDown Buffer 28
CrossDown Peroxidase-Stabilizer 31
Peroxidase-Stabilizer 32
Sample Buffer T- 33
Sample Buffer T+ 33
Stripping Buffer I 34
Washing Buffer TrisT- (10X) 34
Washing Buffer TrisT+ (10X) 35
Verwandte Produkte 36
Weiterführende Literatur 36
© 2011 AppliChem • Immunoassay Puffer 1
inhalt

2 Immunoassay Puffer • AppliChem © 2011
immunoassayDie Crux mit den Kreuzreaktivitäten!Doch dieses Kreuz kann man loswerden…
Mit Antikörpern kann man viele verschiedene Substanzen einfach und spezifisch nachweisen. Dazu existieren verschiedene Methoden und Formate wie „Enzyme-linked immunosorbent assays“ (ELISA) bzw. „Enzyme immunoassays“ (EIA), „Western Blots“, „Radio immunoassays“ (RIA), „Protein-Biochips“, „Immunhistochemie“ oder auch die „Immuno-Polymerase chain reaction“ (Immuno-PCR). All diese werden als Immunoassays bezeichnet und haben leider noch eine Gemeinsamkeit – das Problem der Kreuzreaktivitäten.
Immunoassays sind heute kaum noch aus bioanaly tischen und biochemischen Laboratorien der Life Science- Branche wegzu denken. In der Forschung, der Lebens-mittelüberwachung, dem Umweltmonitoring und vor allem bei medizinischen Fragestellungen wie in der Dia-gnostik werden die verschiedenen Assays regelmäßig eingesetzt. Immunoassays sind relativ einfach durchführ-bar, in der quantitativen und qualitativen Aussage effek-tiv und nach anerkannter Lehrmeinung sehr spezifisch. Das liegt in der Gemeinsamkeit dieser Assays: Es werd
en Antikörper zum Nachweis eingesetzt und die bin-den bekanntermaßen hochspezifisch – jeder Antikör-per erkennt und bindet genau ein Antigen. Das erklärt, warum man so einfach zwischen verschiedenen Sub-stanzen unterscheiden kann.
Soviel zur Lehrbuchmeinung, doch die Praxis treibt so manchem Anwender den Schweiß auf die Stirn. Es gibt unerwartete falsche Banden im Western Blot, die Leerwert-Kontrolle im ELISA zeigt ein deutliches Signal oder der Proteinchip leuchtet flächendeckend – und
Abb. 2A unspezifischeBindungeinesmarkierten
Detektorantikörpersaneinenicht(ausreichend)blockierteOberfläche.Folge:Falsch-positiveMesssignale.
B unspezifischeBindungeinesDetektorsaneineblockierteOberfläche.TrotzBlockierungbindetderAntikörperandieProteinederBlockierung(oderanLückenindieser).Folge:Falsch-positiveMess-Signale.
C EinStörerbindetandenFc-TeildesDetektor-antikörpersundbehindertdadurchsterischdieBindungandenAnalyten.Folge:Falsch-negativeSignale.
D DerFängerantikörperbindetandenFc-TeildesgelabeltenDetektionsantikörpers.Folge:Falsch-positiveSignale.
Abb. 1DerIdealzustand.EinstörungsfreierSandwich-Assay.
Dr.WolframH.Marx,AppliChem;PDDr.Wiesmann,UniversitätMünster;Dipl.Biol.SusanneSiewert,UniversitätUlm;Dipl.Chem.NicoDankbar,UniversitätMünster;Dr.PeterRauch,CANDORBiosciennceGmbH;Dr.ChristophSpechtPARA,BioscienceGmbH
Detektorantikörper, gelabelt
Analyt
Fängerantikörper
Störende bzw. kreuzreagierende Substanz
Heterophiler Antikörper bzw. HAMA
Blockierungsprotein der Oberfläche

© 2011 AppliChem • Immunoassay Puffer 3
dies sind nur einige der unerhofften Ergebnisse, die man von Zeit zu Zeit im Laboralltag erhält. Und jedes falsche Ergebnis bedeutet Mehrarbeit, Folgekosten oder gar Fehldiagnosen mit entsprechenden Folgen für Patienten [1]. Zurück zu den Antikörpern und den Methoden: Antikörper sollen jeweils ein Antigen hoch-spe-zifisch binden. In der Praxis binden Antikörper aber häufig – wenn auch meist mit niedrigerer Affinität – auch andere Antigene, die gar nicht durch einen Assay erkannt werden sollen. Dies trifft auch auf scheinbar gut charakterisierte Antikörper mit hoher Affinität zu dem Zielanalyten zu. Die Folge sind Störun-gen oder auch Interferenzen in Assays. Ganz typisch sind hierbei unspezifische Bindungen, die zu einem hohen Hintergrund mit schlechtem Signal-Rausch-Ver-hältnis führen, Kreuzreaktivitäten und Matrixeffekte.
Störungen bei ImmunoassaysDie Störeffekte sind vielfältiger Natur. Sehr häufig tre-ten Kreuzreaktivitäten, unspezifische Bindungen und Matri xeffekte auf. Störer können in mehr oder weniger großen Konzentrationen in Realproben vorkommen und direkt mit den Analyten oder mit den Fänger- bzw. Detektorantikörpern interagieren. Vereinfacht ausge-drückt, basieren die meisten dieser Effekte auf direkter Interaktion des Analyten, des Fängerantikörpers oder des Detektorantikörpers mit fremden Substanzen oder Oberflächen. Mit der Anwendung von neuartigen Puf-fern (z.B. CrossDown Buffer) können die meisten Störeffekte ver hindert werden, indem die üblicherweise im Assay verwendeten Probenpuffer bzw. Antikörper-
verdünnungspuffer gegen CrossDown Buffer ausge-tauscht werden. Hierdurch wird die Qualität der Assays und die Effizienz der Assayentwicklung verbessert.
Um die Störungen zu ver stehen, schauen wir uns zunächst einen störungsfreien Sandwich-ELISA als Beispiel an. Danach folgen Assays mit Stör effekten, so dass die Art der jeweiligen Störung jeweils leicht erkennbar ist. Beim ungestörten Sandwich-ELISA ist der Fänger antikörper am Boden eines Wells immo-bilisiert und die restlichen Oberflächen sind aus- reichend blockiert. Ein Analyt wird vom Fänger gebun-den. Ein Detektorantikörper bindet nun wiederum den Analyten von der anderen Seite und über eine Markierung am Detektorantikörper wird die Bindung sichtbar gemacht.
Unspezifische BindungenBei unspezifischer Bindung erfolgt die Bindung an Substanzen, die entweder in weitaus höheren Konzen-trationen als der Zielanalyt vorkommen (z.B. unspezifi-sche Bindung an Albumine oder Immunglobuline), an Oberflächen (z.B. bei Western-Blot-Membranen oder ELISA-Wells) oder an Spots aus immobili sierten Anti-körpern bei Proteinchips [2]. Besonders stark davon betroffen sind Assays mit schlechter Blockierung oder in problematischen Matrices, die z.B. starke Albumin-anteile oder hohe Konzentrationen endo gener Stör-faktoren enthalten. Doch es gibt auch eine ganz andere Ursache von unspezifischen Bindungen. Antikörper werden für die Detektion häufig mit Labeln versehen. Dies können Enzyme (sehr oft alkalische Phosphatase
Abb. 3I KreuzreaktivitäteinesStörersmitFänger.
Folge:Falsch-negativeSignale.J KreuzreaktivitäteinesStörersmitDetektor.
Folge:Falsch-negativeSignale.K KreuzreaktivitäteinesStörersmitdem
DetektorunddemFänger.Folge:Falsch-positiveSignale.DiesesStörungsbildistinderPraxiszumGlückrechtseltenundeherbeiAntikörpernmitschlechterSpezifitätzufinden.EsistjedochbeiAntikör-pernanzutreffen,diegegeneinTargetgerichtetsind,daseinekonservierteAminosäuresequenzeinesProteinsdarstellt.DieseSequenzmotivekommendannggf.auchinanderenProteinen(diedadurchzumStörerwerden)vor.
Abb. 4F EineBrückenbindungdurchHAMAsbzw.
heterophileAntikörperunddadurchVerknüpfungvonFängerundDetektor.Folge:Falsch-positiveSignale.
G Anti-idiotypischerHAMAzumFänger.BindungderFab-RegiondesFängersanAnalytistnichtmehrmöglich.Folge:Falsch-negativeSignale.
H Anti-idiotypischerHAMAzumDetektor.BindungderFab-RegiondesDetektorsandenAnalytenistnichtmehrmöglich.Folge:Falsch-negativeSignale.
Abb. 5L MaskierungdesAnalytendurcheinProtein
derProbe.DasEpitopwirdsofürdenFängerblockiert,sodassdieBindungdesAnalytenentwedergarnichtoderimFalleeinersterischenBehinderungnursehrschlechterfolgenkann.Folge:Falsch-negativeSignale.

4 Immunoassay Puffer • AppliChem © 2011
immunoassayoder Peroxidase), Fluoreszenzfarbstoffe, radioaktive Isotope oder auch DNA (bei Immuno-PCR) sein. Auch hierbei können unerwünschte Effekte auftreten. Bei Fluoreszenzfarbstoffen besteht die Gefahr, dass die oft-mals hydrophoben Farbstoffe die Bindungseigenschaf-ten des Detektorantikörpers verändern, weil die Farb-stoffe selbst unerwünschte Bindungen auslösen und die Löslichkeit des gelabelten Proteins verringern. Es kann aber auch die Antigen-Anti körperbindung ver-schlechtert werden [3]. Diese Effekte können beispiels-weise dazu führen, dass der gelabelte Antikörper ver-stärkt unspezifisch an die Oberflächen (Abb. 2 A und B), an Fremdproteine der Realprobe (Abb. 2 C) oder an den Fängerantikörper (Abb. 2 D) bindet. In diesen Fällen erhält man auch in Abwesenheit des Analyten einen falsch-positiven Messwert bzw. der gesamte Assay leidet unter einem hohen Hintergrund. Bei Protein-chips werden in diesem Fall erhöhte Hintergrund-fluoreszenzen bei einzelnen Spots beobachtet oder es verschlechtert sich insgesamt das Signal-Rausch- Verhältnis. Fluoreszenzfarbstoffe können auch Proteine oder Anti körper aus Serum proben binden. So ver-ringert sich die Fluoreszenz des Farbstoffs im Extrem-fall bis hin zur vollständigen Signallöschung. Deshalb wird auch der vollständige Verzicht auf Fluoreszenzfarb-stoffe bei Proteinchips und die Benutzung anderer Label diskutiert [4]. Die Komplexität von Proteinchips ist hier-bei besonders hoch, da gleichzeitig eine Vielzahl ver-schiedener Fänger- und gelabelte Detektoranti körper in einem gemeinsamen Reaktionsvolumen verwendet wird. Dadurch erhöht sich die Gefahr von unspezi-fischen Bindungen von Proteinen der Probe oder ge-labelten Antikörpern an einzelne Spots, sowie die Gefahr von Störeffekten von Bestandteilen der Probe mit den Anti körpern [2].
KreuzreaktivitätenIm Ergebnis sehen Kreuzreaktivitäten teilweise ähnlich aus wie unspezifische Bindungen. Auch bei Kreuz-reaktivitäten erhält man Bindungen an unerwünschten Stellen bzw. an „falsche“ Substanzen. Im Unterschied zur unspezifischen Bindung spricht man von Kreuzre-aktivitäten, wenn der Kreuzreaktand bekannt ist und dessen kreuzreagierende Eigenschaft, z.B. über die Bestimmung der kompetierenden Konzentration des Kreuzreaktanden [5], nachgewiesen werden kann. Mit Kreuzreaktivität ist also die Fähigkeit des Antikörpers gemeint, auch an andere Strukturen als die des eigent-lichen Zielanalyten zu binden (Abb. 1 I, J, K). Oftmals handelt es sich um Strukturen, die eine hohe Ähnlich-keit zum Analyten haben. Beispiele hierfür sind Meta-bolite oder chemische Substanzen mit einer ähnlichen molekularen Struktur. Auch Proteine mit einer zufälli-gen Ähnlichkeit oder mit evolutionären Homologien der Aminosäuresequenz können kreuzreagieren.
Kreuzreaktivitäten spielen besonders in kompetitiven Assayformaten, wo nur ein Antikörper eingesetzt wird, eine größere Rolle [1,4]. Bei diesen Assays ist es im Regelfall Bestandteil der Validierung, mögliche kreuz-reagierende Substanzen zu identifizieren und deren Kreuzreaktivität im Experiment zu quantifizieren [5].
Doch auch bei der Detektion von Proteinen auf einem Western Blot oder bei immunhistochemischen Anwendungen können Kreuzreaktivitäten eine große Rolle spielen. Das drückt sich in einer Anfärbung weiterer Banden bzw. Zellstrukturen aus, ohne dass man immer die genauen molekularen Ursachen für das Auftreten dieser unerwünschten Bindungen kennt. Bei Western Blots handelt es sich in manchen Fällen schlicht um Proteinfragmente, die durch natürlichen Abbau oder im Rahmen der methodischen Durchfüh-rung entstanden sind. In manchen Fällen muss man jedoch das Auftreten von Kreuzreaktivitäten des Primär-antikörpers oder des Sekundärantikörpers in Betracht ziehen. Für eine ordentliche wissenschaftliche Veröffent-lichung ist es ohnehin erforderlich die Ursache zusätz-licher Banden im Western Blot zu klären.
MatrixeffekteAm wenigsten definiert ist der Begriff der Matrixeffekte. Matrixeffekte sind die Summe der Störeffekte aller Komponenten, die in einer Probe vorkommen und die Messung des Zielanalyten beeinflussen [6]. Wenn die genaue molekulare Ursache einer Störung nicht bekannt ist, aber mit der Zusammensetzung der zu vermessen-den Probe in Verbindung gebracht werden kann, spricht man im Allgemeinen von einem Matrixeffekt. Hier sind die Übergänge zu den einzelnen anderen genannten Störeffekten fließend. Für Matrixeffekte können „Anti-Animal-Antibodies“, heterophile Antikörper, endogene

© 2011 AppliChem • Immunoassay Puffer 5
Störer oder Einf lüsse der Viskosität, des pH-Werts oder der Salzkonzentration verantwortlich sein.
Es gibt auch Störeffekte, die weitgehend auf medi-zinische und diagnostische Assays beschränkt sind. Diese Störungen beruhen auf Störfaktoren, die in humanen Proben, wie Blutplasma, Serum oder Gewe-beproben zu finden sind. Da die Ergebnisse von Assays in der medizinischen Forschung oder auch der Diagno-stik häufig die Basis für spätere Therapien an Patienten bilden, sind Störungen und damit falsche Ergebnisse in derartigen Assays häufig besonders schwerwiegend. Die Erkennung einer einmal aufgetretenen Störung ist jedoch keineswegs einfacher als in anderen Assays.
„Anti-Animal-Antibodies“Humane „Anti-Animal-Antibodies“ (HAAA) können vom IgG-, IgA-, IgM- oder IgE-Typ sein und werden als Immunantwort auf den Kontakt mit Immun globulinen tierischen Ursprungs gebildet. HAAAs sind vor allem aus diagnostischen Immuno assays bekannt und einige Studien berichten von Quoten von bis zu 80 % aller Patientenproben, die HAAAs enthalten. Die Konzen-
trationen können hierbei beträchtlich sein und bis in den Bereich von einigen Milligramm pro Milliliter reichen [7].
Human-Anti-Mouse Antibodies (HAMA) sind die bekanntesten störenden Antikörper eines Immuno-assays. HAMAs sind humane Antikörper, die relativ spezifisch und mit merklicher Affinität Maus-Antikörper binden. Ursache für deren Ent stehung ist meist die Gabe von therapeutischen Antikörpern, die als Medika-mente (z.B. für Krebstherapien) eingesetzt werden. Nach therapeutischer Gabe reagiert das menschliche Immunsystem auf diese fremden Antikörper und bildet allmählich Antikörper gegen Maus-Antikörper. HAMAs stören deshalb immunologische Methoden, die mit Maus-Antikörpern arbeiten. Bei Sandwichformaten aus monoklonalen Maus-Antikörpern werden durch Brücken bildung Fänger- mit Detektorantikörper ver-knüpft (Abb. 4 F), wodurch ein falsch-positives Mess-Signal entsteht. Aufgrund von Sequenzähnlichkeit zwischen Antikörpern verschiedener Spezies sind HAMA-haltige Seren eventuell auch in der Lage, Assays zu stören, in denen Antikörper aus anderen Spezies verwendet werden.
Doch nicht nur Antikörper-Medikamente sind für die Entstehung von HAMAs verantwortlich. Der lang-jährige Umgang mit Haustieren fördert ebenfalls die Entstehung von Antikörpern, die entweder nur an Antikörper bestimmter Spezies (z.B. Kaninchen, Maus, Hund, Hamster) binden oder artenübergreifend ver-schiedene Antikörper mit unterschiedlichen Affinitäten erkennen und so Assays stören können. Manche Stör-antikörper binden nicht nur an den Fc-Abschnitt, son-dern auch direkt an die Fab-Bereiche der verwendeten Assayantikörper. Hierdurch kann die Bindung des Ana-lyten reduziert oder sogar gänzlich verhindert werden. Die Folge ist eine falsch-negative Bestimmung (Abb. 4 G und H). Wenn HAAAs an den Fc-Abschnitt eines Antikörpers binden, spricht man von Anti-isotypischen Störern. Anti-idiotypische Störer hingegen binden direkt an die hochvariable Region des Fab-Abschnitts [7].
Heterophile AntikörperHeterophile Antikörper sind laut Taber‘s Medical Dictionary „Antikörper, die andere Antigene als das spezifische Antigen binden“. Auch heterophile Antikörper können vom IgG-, IgA-, IgM- oder IgE-Typ sein. Besonders der IgM-Typ spielt eine besondere Rolle bei Seren von rheumatischen Patienten. Diese Seren enthalten sogenannte Rheumafaktoren in hoher Konzen tration. Rheumafaktoren sind IgM-Antikörper, die an Fc-Abschnitte humaner Antikörper binden und somit auch artenübergreifend an Fc-Abschnitte der im Assay verwendeten Antikörper binden können. Daher verknüpfen rheumatische Seren Fänger- mit Detektor-antikörpern mit der Folge von falsch-positiven Signa-

6 Immunoassay Puffer • AppliChem © 2011
immunoassayEin wichtiger Aspekt, der in diesem Zusammen-
hang noch erwähnt werden sollte, sind Störungen durch stark lipidhaltige Proben, da manche Analyte gut fettlöslich sind bzw. die Antikörper-Analyt-Bindung durch Lipide gestört werden kann.
Vermeidung der Stör effekte durch moderne Puffer für Immuno assays – Beispiele aus der PraxisMeist sind es also niedrig bis mittelaffine Bindungen, die dem Anwender Probleme bei Immunoassays berei-ten. Die bekannteste Strategie, die aber aus den oben genannten verschiedenen Ursachen für die Störungen heraus nicht in allen Fällen zum Ziel führen kann, ist die angepasste Blockierung. Hierfür gibt es hunderte – mitunter eher als kreativ denn als zielführend zu bezeichnende Blockierungslösungen, die in der Litera-tur beschrieben sind. Je größer der Analyt ist, desto einfacher kann mitunter auch die Blockierung sein. Kleinere Analyte erfordern meist jedoch eine sehr gute Blockierung. Ideal ist eine Blockierung, die generell bei den meisten Assays einsetzbar ist und somit die Arbeit bei der Optimierung und die Kosten vergeblicher Experimente einspart. Dies gilt insbesondere, wenn sehr teure Antikörper oder aufwändig zu gewinnende Proben benutzt werden. Die ideale Blockierung ist also universell einsetzbar und liefert bei verschiedensten Assays immer gleich bleibend gute Resultate. Casein-basierende Lösungen haben sich hierbei als besonders effizient erwiesen. Die Herstellung im Labor erfordert jedoch leider viel Zeit und auch Er fahrung, um tatsäch-lich Casein-Lösungen mit gleich bleibender Blockie-rungseffizienz zu er halten. Viele Labore haben schon negative Erfahrungen mit schwankenden Resultaten bei selbst hergestellten Casein- Blockern machen müs-sen. Die nervenaufreibende Suche nach den Ursachen ist meist das Ärgerlichste daran, zumal die Blockie-rungslösungen auf den ersten Blick immer gleich aus-sehen. Heute ermöglicht eine chemische Modifikation im industriellen Produktionsprozess die Herstellung Casein-basierter Lösungen, die sehr gut reproduzierbare Ergebnisse liefern.
Der Austausch einer nicht adäquaten Blockierung in der Immunhistochemie machte im hier gezeigten Fall die Auswertung von Versuchsreihen bei Osteo-blasten-Kulturen möglich. Eine frische Osteoblasten-Kultur zeigt am ersten Tag keine oder nur eine sehr geringe Expression des extrazellulären Matrixproteins Osteocalcin. Unter Verwendung einer modernen Blockierungs lösung (Blocking Buffer I erhältlich bei AppliChem) in Verbindung mit Anti-Osteocalcin (monoklonal, TaKaRa) wird diese Situation richtig dar-gestellt. (Abb. 6, oben links). Ein falsch positives Ergeb-nis liefert hingegen die Standard-Blockierung mit BSA (Abb. 6, oben rechts). Mit zunehmender Kulturdauer
len. Dieses ist auch der generelle Störmechanismus der heterophilen Antikörper. Die Wirkung der rheumati-schen Seren ähnelt der Wirkung der HAAAs. Der Un-terschied zu den HAAAs liegt in der Entstehung der heterophilen Antikörper: Diese werden nicht durch den Kontakt mit tierischen Immunglobulinen gebildet, sondern sind multispezifische Antikörper der frühen Immunantwort oder störende Antikörper unbekannter immunologischer Entstehungsgeschichte [7].
Störungen durch HAAAs oder durch heterophile Antikörper sind schon seit mehr als 30 Jahren bekannt. Die störenden Antikörper sind im Allgemeinen schwach bindende Antikörper [7] und stören vorwiegend Assays, die aufgrund der niedrigen Konzentration des Analyten mit geringen Verdünnungen der humanen Serum- bzw. Plasmaprobe auskommen müssen [8]. Zusätze von blockierenden Substanzen zu dem Probenpuffer – im Allgemeinen unspezifische Seren, Antikörper-Fragmente oder hohe Konzentrationen von tierischen Immunglobulinen – können durch Kompetition die Stör effekte der HAAAs bzw. heterophilen Antikörper reduzieren, aber nicht immer verhindern [7].
Endogene Störer als Bestandteile der ProbeAuch natürlich vorkommende Proteine der Realprobe können Immunoassays stören. Mitunter beruhen die Störungen auf Bindungseigenschaften von Proteinen, deren tatsächliche biologische Funktion in diesen meist niedrig bis mittelaffinen Bindungen liegt. Bekannte Störer in humanen Seren sind beispielsweise Albumine, Komplement, Lysozyme und Fibrinogen [4]. Nieder-molekulare Analyte können an Albumin binden. Doch die Zugänglichkeit des Antikörpers zu dem Analyten wird mitunter dadurch erschwert. Zahlreiche Hormone liegen an Transportproteine gebunden vor, was je nach verwendeten Antikörpern ebenfalls zu Schwierigkeiten führen kann. Die Bindefähigkeit ist z.B. wesentlicher Teil der Funktion von Proteinen wie Albumin, Kom-plement und c-reaktives Protein (CRP). Diese Proteine sind also für viele Substanzen natürliche Rezeptoren. Daher sind hier – ähnlich wie bei den Antikörpern – unspezifische Bindungen (z.B. Albumine liegen in extrem hohen Konzentrationen vor) oder sogar Kreuz-reaktionen denkbar, die die Erkennung bestimmter Analyte in einem Assay erschweren. Endogene Prote-ine können als Störer an die Assay antikörper binden (Abb. 2 C, Abb. 3 I, J, K) oder den Zielanalyten für die Assay antikörper maskieren (Abb. 5 L). Lysozyme beispiels weise binden unspezifisch an Proteine mit einem niedrigen isoelektrischen Punkt. Daher können auch Antikörper, die einen isoelektrischen Punkt von ca. 5 haben, gebunden werden und beispielsweise Brückenbildung zwischen Fänger- und Detektoranti-körper auslösen [4].
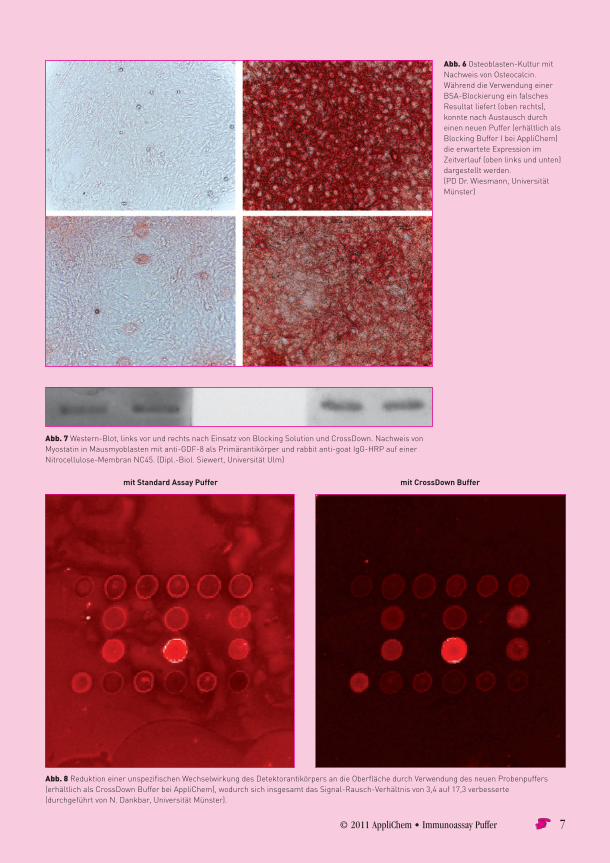
© 2011 AppliChem • Immunoassay Puffer 7
Abb. 6 Osteoblasten-KulturmitNachweisvonOsteocalcin.WährenddieVerwendungeinerBSA-BlockierungeinfalschesResultatliefert(obenrechts),konntenachAustauschdurcheinenneuenPuffer(erhältlichalsBlockingBufferIbeiAppliChem)dieerwarteteExpressionimZeitverlauf(obenlinksundunten)dargestelltwerden.(PDDr.Wiesmann,UniversitätMünster)
Abb. 7Western-Blot,linksvorundrechtsnachEinsatzvonBlockingSolutionundCrossDown.NachweisvonMyostatininMausmyoblastenmitanti-GDF-8alsPrimärantikörperundrabbitanti-goatIgG-HRPaufeinerNitrocellulose-MembranNC45.(Dipl.-Biol.Siewert,UniversitätUlm)
mit Standard Assay Puffer mit CrossDown Buffer
Abb. 8ReduktioneinerunspezifischenWechselwirkungdesDetektorantikörpersandieOberflächedurchVerwendungdesneuenProbenpuffers(erhältlichalsCrossDownBufferbeiAppliChem),wodurchsichinsgesamtdasSignal-Rausch-Verhältnisvon3,4auf17,3verbesserte(durchgeführtvonN.Dankbar,UniversitätMünster).
© 2011 AppliChem • Immunoassay Puffer 7
8 Immunoassay Puffer • AppliChem © 2011
immunoassaybauen die Osteoblasten nach und nach die extrazellu-läre Matrix auf und Osteocalcin wird synthetisiert. Die zu erwartende Zunahme der Osteocalcinexpression kann bei Verwendung der modernen Blockierung gut dargestellt werden, die Kulturen sind entsprechend mit zunehmender Kulturdauer intensiver gefärbt (Abb. 6, unten).
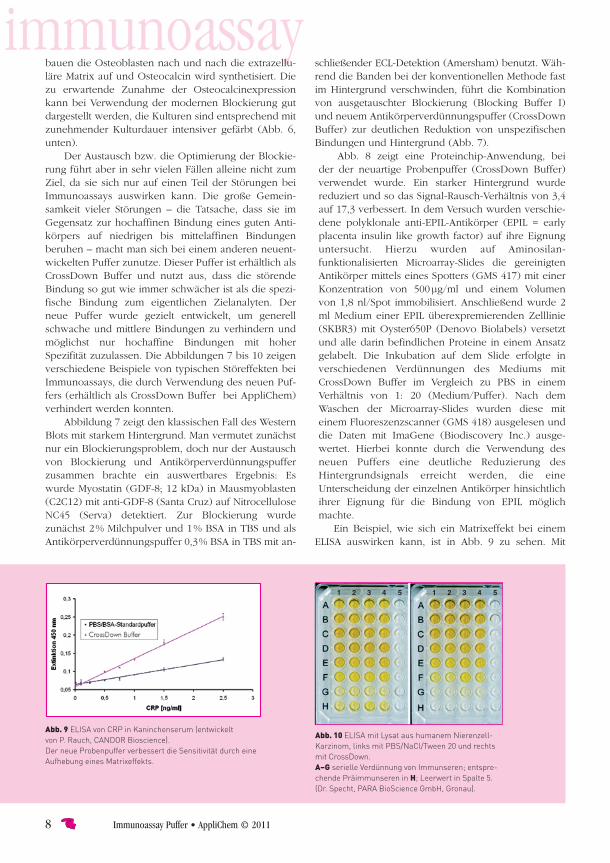
Der Austausch bzw. die Optimierung der Blockie-rung führt aber in sehr vielen Fällen alleine nicht zum Ziel, da sie sich nur auf einen Teil der Störungen bei Immunoassays auswirken kann. Die große Gemein-samkeit vieler Störungen – die Tatsache, dass sie im Gegensatz zur hochaffinen Bindung eines guten Anti-körpers auf niedrigen bis mittel affinen Bindungen beruhen – macht man sich bei einem anderen neuent-wickelten Puffer zunutze. Dieser Puffer ist erhältlich als CrossDown Buffer und nutzt aus, dass die störende Bindung so gut wie immer schwächer ist als die spezi-fische Bindung zum eigentlichen Zielanalyten. Der neue Puffer wurde gezielt entwickelt, um generell schwache und mittlere Bindungen zu verhindern und möglichst nur hochaffine Bindungen mit hoher Spezifität zuzulassen. Die Abbildungen 7 bis 10 zeigen verschiedene Beispiele von typischen Störeffekten bei Immuno assays, die durch Verwendung des neuen Puf-fers (erhältlich als CrossDown Buffer bei AppliChem) verhindert werden konnten.
Abbildung 7 zeigt den klassischen Fall des Western Blots mit starkem Hintergrund. Man vermutet zunächst nur ein Blockierungsproblem, doch nur der Austausch von Blockierung und Antikörperverdünnungspuffer zusammen brachte ein auswertbares Ergebnis: Es wurde Myostatin (GDF-8; 12 kDa) in Mausmyoblasten (C2C12) mit anti-GDF-8 (Santa Cruz) auf Nitrocellulose NC45 (Serva) detektiert. Zur Blockierung wurde zunächst 2 % Milchpulver und 1 % BSA in TBS und als Antikörperverdünnungspuffer 0,3 % BSA in TBS mit an-
schließender ECL-Detektion (Amersham) benutzt. Wäh-rend die Banden bei der konventionellen Methode fast im Hintergrund verschwinden, führt die Kombination von ausgetauschter Blockierung (Blocking Buffer I) und neuem Antikörperverdünnungspuffer (CrossDown Buffer) zur deutlichen Reduktion von unspezifischen Bindungen und Hintergrund (Abb. 7).
Abb. 8 zeigt eine Proteinchip-Anwendung, bei der der neuartige Probenpuffer (CrossDown Buffer) verwendet wurde. Ein starker Hintergrund wurde reduziert und so das Signal-Rausch-Verhältnis von 3,4 auf 17,3 verbessert. In dem Versuch wurden verschie-dene polyklonale anti-EPIL-Antikörper (EPIL = early placen ta insulin like growth factor) auf ihre Eignung untersucht. Hierzu wurden auf Aminosilan-funktionalisierten Microarray-Slides die ge reinigten Antikörper mittels eines Spotters (GMS 417) mit einer Konzentration von 500 µg/ml und einem Volumen von 1,8 nl/Spot immobilisiert. Anschließend wurde 2 ml Medium einer EPIL überexpremierenden Zelllinie (SKBR3) mit Oyster650P (Denovo Biolabels) versetzt und alle darin befindlichen Proteine in einem Ansatz gelabelt. Die Inkubation auf dem Slide erfolgte in verschiedenen Verdünnungen des Mediums mit CrossDown Buffer im Vergleich zu PBS in einem Verhältnis von 1: 20 (Medium/Puffer). Nach dem Waschen der Microarray-Slides wurden diese mit einem Fluoreszenzscanner (GMS 418) ausgelesen und die Daten mit ImaGene (Biodiscovery Inc.) ausge-wertet. Hierbei konnte durch die Verwendung des neuen Puffers eine deutliche Reduzierung des Hintergrundsignals erreicht werden, die eine Unterscheidung der einzelnen Antikörper hinsichtlich ihrer Eignung für die Bindung von EPIL möglich machte.
Ein Beispiel, wie sich ein Matrixeffekt bei einem ELISA auswirken kann, ist in Abb. 9 zu sehen. Mit
Abb. 9ELISAvonCRPinKaninchenserum(entwickeltvonP.Rauch,CANDORBioscience).DerneueProbenpufferverbessertdieSensitivitätdurcheineAufhebungeinesMatrixeffekts.
1 2 3 4 5 1 2 3 4 5
Abb. 10 ELISAmitLysataushumanemNierenzell-Karzinom,linksmitPBS/NaCl/Tween20undrechtsmitCrossDown. A–GserielleVerdünnungvonImmunseren;entspre-chendePräimmunsereninH;LeerwertinSpalte5.(Dr.Specht,PARABioScienceGmbH,Gronau).

© 2011 AppliChem • Immunoassay Puffer 9
diesem Modellassay wurde gezielt ein Matrixeffekt in-duziert. Er wurde bei einem Dienstleister für ELISA-Entwicklung und Validierung entwickelt (CANDOR Bioscience GmbH). Hierzu wurde Kaninchenserum als Matrix verwendet, das mit humanem c-reaktiven Prote-in (CRP, Biotrend) in definierten Konzentrationen ver-setzt wurde. Als Fängerantikörper wurde Clone C2 (Biotrend, 1 µg/ml Coating Konzentration in PBS), als Detektor biotinylierter Clone C6 (Biotrend, Arbeits-konzentration 2 µg/ml) verwendet. Die gespiketen Serum-Proben wurden entweder mit einem PBS-BSA-Puffer oder mit CrossDown Buffer 1:2 verdünnt und für den ELISA eingesetzt. Die Detektion erfolgte über NeutrAvidinTM-Horseradish peroxidase conjugated (Pierce, Arbeitskonzentration 0,05 µg/ml in PBS-BSA-Puffer) mit Immuno Pure®TMB Substrat (Pierce).
Ein Matrixeffekt, dessen genaue molekulare Ursache nicht bekannt ist, führt zu einer Kalibrationsgeraden mit schlechter Sensitivität. CRP ist aufgrund seiner physiologi-schen Funktion in der Lage, an sehr viele Proteine und Substanzen (Scavenger-Funktion des CRP) zu binden, wodurch wahrscheinlich in diesem Falle die Erreichbar-keit der Epitope für die Antikörper erschwert wird. Ver-mutlich liegt ein Störeffekt vor, wie er in Abb. 5 (L) sche-matisch dargestellt ist, obwohl Störeffekte, wie in Abb. 3 (I, J, K) dargestellt sind, nicht ausgeschlossen werden können. Der neue Puffer wiederum verhindert die Bindung des CRP’s an endogene Substanzen des Kanin-chenserums und verbesserte so die Sensitivität der Kalibrationsgeraden um den Faktor 3.
Der ELISA in Abb. 10 zeigt einen Fall, in dem lediglich der Proben- und Antikörperver dünnungspuffer ausge-tauscht werden musste, um ein gutes Ergebnis zu er-zielen. Im Experiment wurde als Antigen ein Lysat aus humanem Nierenzell-Karzinom immobilisiert und eine serielle Verdünnung von zwei Immunseren in Doppel-bestimmung (1:50 bis 1:36450) A-G in den Spalten 1 bis 4 aufgetragen (Abb. 10). Die entsprechenden Präimmun-seren (1:50) wurden in Reihe H pipettiert. Die Spalte 5 stellt den Leerwert dar. Das Ergebnis bei Benutzung des Standardpuffers PBS/NaCl/Tween 20 im Vergleich zu CrossDown Buffer ist eindeutig. Es ergibt sich eine bessere Sensitivität und der „Level of Detection“ (LOD) wurde von 0,051 auf 0,022 und der „Level of Quantifica-tion“ von 0,152 auf 0,065 gesenkt, bei gleichzeitig nach oben vergrößertem Messbereich. Die rein optische Begut-achtung der Konzentrationsreihe mit bloßem Auge zeigt schon, dass der neue Probenpuffer die Qualität des As-says deutlich verbessert hat. Die beschriebene Verbesse-rung ist durch Eliminierung der falsch positiven Signale bei den Präimmunseren und die Reduktion des Back-grounds zu erklären.
FazitSeit man Antikörper methodisch für bioanalytische und diagnostische Zwecke benutzt, kennt man das Phäno-men der Störeffekte und muss nach Lösungen suchen. Im Laufe der letzten 30 Jahre wurden zahlreiche mole-kulare Ursachen gefunden und deren Störungsmecha-nismen untersucht, was wiederum die Entwicklung von Vermeidungsstrategien förderte. Mit dem heutigen Stand der Technik lassen sich viele Störeffekte minimie-ren und durch die Entwicklung moderner Puffer für Immunoassays sind im wahrsten Sinne des Wortes gute Lösungen gefunden worden. Neuartig an dem hier vor-gestellten Proben- und Antikörperverdünnungspuffer ist, dass nun verschiedene Stör effekte mit unterschied-lichen molekularen Ursachen gleichzeitig minimiert werden können und ein einziger Puffer bei unter-schiedlichen Immunoassays anwendbar ist. Die hier gezeigten Ergebnisse sind nur ein Ausschnitt aus unter-schiedlichen Störeffekten bei unterschied lichen Metho-den, die mit diesem Puffer minimiert bzw. vermieden werden konnten. Ebenfalls konnten Störeffekte durch unspezifische Bindungen bei immunhistochemischen Anwendungen sowie falsch-positive Bindungen bei Immuno-PCR verhindert werden. Auch modern pro-duzierte Blockierungslösungen in reproduzierbarer Qualität mit einem sehr fein verteilten Spektrum unterschied licher Molekulargewichte für die Blockie-rung leisten einen wesentlichen Beitrag zur Sicherheit und Effizienz bei bioanalytischen Methoden. Insgesamt werden zeit- und kostenaufwändige Optimierungs-strategien verkürzt und vereinfacht, wobei gleichzeitig eine Verbesserung der Ergebniszuverlässigkeit erreicht wird.
Literatur [1] Miller, J.J. (2004) Clinical Laboratory International 28, (2), 14-17
[2] Kusnezow, W., Hoheisel, J.D. (2003) J. Mol. Recognit. 16, 165-176
[3] Patton, W.F. (2000) Electrophoresis 21, 1123-1144
[4] MacBeath, G. (2002) Nat. Genet. 32, 526-532
[5] Miller, J.J., Valdes, R.Jr. (1992) J Clin Immunoassays 15, 97-107
[6] Wood, W.G. (1991) Scand. J. Clin. Lab. Invest. Suppl. 205, 105-112
[7] Kricka, L.J. (1999) Clinical Chemistry 45,(7), 942-956
[8] Span, P.N., Grebenchtchikov N., Geurts-Moespot, J., Sweep, C.G.J. (2003)
Clinical Chemistry 49,(10), 1708-1709

10 Immunoassay Puffer • AppliChem © 2011
blockierungOhne Blockierung kein Ergebnis So kurz lässt sich zusammenfassen, wie essentiell die Blockierung bei Immunoassays ist.
Es gibt viele verschiedene Blockierungsprotokolle und sie funktionieren mal besser und mal schlech-ter in ver schiedenen Assays. Doch wirklich uni-versell einsetz bare Blockierungslösungen waren bislang schwer zu finden.
Bei allen Arten von Immunoassays muss man blockieren. Dabei versucht man, die unspezifische Bindungen der Antikörper oder von Bestandteilen der Probe an Oberflächen von ELISA-Platten oder Western-Blot-Mem branen zu verhindern. Andernfalls würden diese Bindungen zu starkem Hintergrund führen. Hierdurch wird das Ergebnis verfälscht oder sogar zunichte gemacht. Um effizient zu blockieren, darf die Blockierung also keine Lücken enthalten. So schützt sie bestmöglich vor unspezifischer Bindung. So einfach die Theorie. Doch die Praxis ist nicht ganz so trivial und der Teufel steckt häufig im Detail. Dabei wird schon bei einem Blick in die Literatur schnell klar, dass das Thema bislang nicht so einfach zu sein scheint. Immer-hin finden sich für jede Nachweismethode mit Anti-körpern hunderte verschiedener Blockierungsproto-kolle, egal ob es sich um ELISA, um Western-Blot oder die Immunhistochemie handelt. In Publikationen findet man sehr viele verschiedene Abwandlungen von Blockierungslösungen, die auf unterschiedlichen Be-standteilen basieren. Allen gemeinsam ist, dass man bestimmte Moleküle in sehr großem Überschuss zugibt, um alle Oberflächen damit zu „verkleben“. So findet man Blockierungen auf Basis von BSA, von Fisch-gelatine, von Milchpulver und Casein oder einfach auf Basis von synthetischen Substanzen. Genauso lang wie die Liste potenzieller Blockierer ist aber auch die Leidensgeschichte derer, die sie verwenden. Für jeden Assay und jeden Antikörper wird neu ausgetestet, welcher Blockierer effizient genug ist, um in diesem speziellen Fall den Hintergrund wirksam und dauer- haft zu verhindern. Gerade wenn nach der Entwick-
lung des Assays mit Realproben, also z.B. Blut- und Serum- Proben, Zell-Lysaten oder wie in der Immunhisto chemie mit ganzen Schnitten, gearbeitet wird, stoßen viele Blockierungen schnell an ihre natür-lichen Grenzen.
Die ideale BlockierungWie muss eine ideale Blockierung beschaffen sein? Sie sollte lückenlos abdecken. Dies lässt sich am effektivsten erreichen, wenn man Moleküle unter-schiedlicher Größen gleichzeitig verwendet. Große Lücken und kleine Lücken wären gleichmäßig abdeck-bar. Zusätzlich darf die Blockierung keinerlei Reaktion oder Bindung mit Bestandteilen der Probe oder mit den Antikörpern eingehen. Hier erlebt man z.B. bei BSA-Blockierungen mitunter Überraschungen. So gibt es Antikörper, bei denen zur besseren Immunisierung der Analyt vorher BSA-gekoppelt war. Kein Wunder also, dass manche Antikörper direkt und mit sehens-werter Affinität auch BSA binden. So bewirkt eine Blockierung auch manches Mal genau das Gegenteil dessen, was man bezwecken wollte. Mitunter entsteht ein Hintergrund auch dadurch, dass es zu Wechsel-wirkungen zwischen Verunreinigungen des Blockierers und der Probe kommt. Eine Kombination von hoher Blockierungseffizienz durch unterschiedlich große Mo-leküle bei gleichzeitiger Vermeidung von Verunreini-gung ließ sich im Laboralltag bislang meist nur durch Ansetzen von Casein-basierenden Blockierern erzielen, und das auch nur, wenn hochreines Casein eingesetzt wurde. Es gab jedoch immer schon einen wesentlichen Nachteil: Nur durch kontrollierte Hydrolyse über viele Stunden lässt sich mit hochreinem Casein eine sehr gute Blockierungslösung herstellen. Doch wehe dem, der nicht genügend Geduld und Fingerspitzengefühl walten lässt. Fehler führen direkt zum starken Aus-fällen des Caseins.
Dr.WolframH.Marx,AppliChem,Dr.AstridVoigt,UniversityHospitalJena,Dr.TronhungQuang,Dr.RainerKlocke,Prof.Dr.SigridNikol,UniversityHospitalMünster,Dr.ChristophSpecht,PARABioscienceGmbH
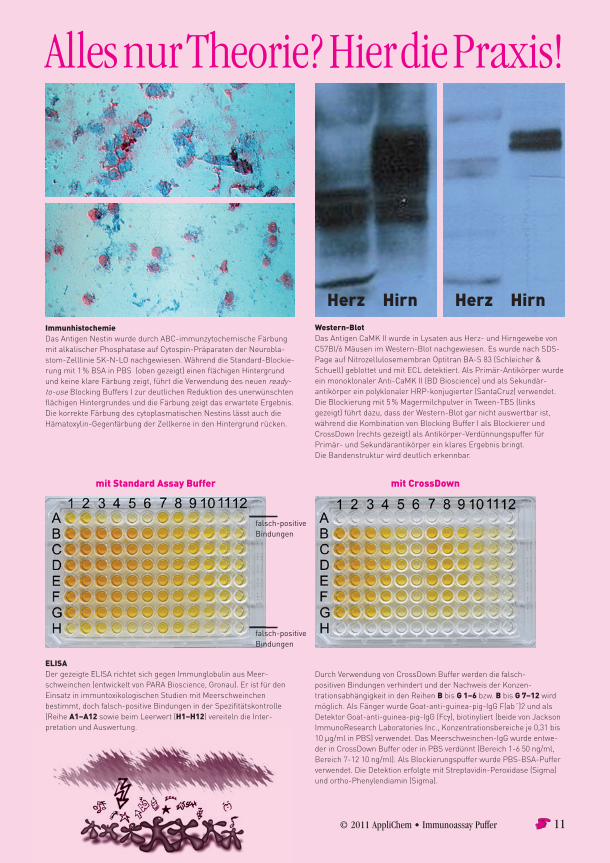
© 2011 AppliChem • Immunoassay Puffer 11
ImmunhistochemieDasAntigenNestinwurdedurchABC-immunzytochemischeFärbungmitalkalischerPhosphataseaufCytospin-PräparatenderNeurobla-stom-ZelllinieSK-N-LOnachgewiesen.WährenddieStandard-Blockie-rungmit1%BSAinPBS(obengezeigt)einenflächigenHintergrundundkeineklareFärbungzeigt,führtdieVerwendungdesneuenready-to-useBlockingBuffersIzurdeutlichenReduktiondesunerwünschtenflächigenHintergrundesunddieFärbungzeigtdaserwarteteErgebnis.DiekorrekteFärbungdescytoplasmatischenNestinslässtauchdieHämatoxylin-GegenfärbungderZellkerneindenHintergrundrücken.
Western-BlotDasAntigenCaMKIIwurdeinLysatenausHerz-undHirngewebevonC57Bl/6MäusenimWestern-Blotnachgewiesen.EswurdenachSDS-PageaufNitrozellulosemembranOptitranBA-S83(Schleicher&Schuell)geblottetundmitECLdetektiert.AlsPrimär-AntikörperwurdeeinmonoklonalerAnti-CaMKII(BDBioscience)undalsSekundär-antikörpereinpolyklonalerHRP-konjugierter(SantaCruz)verwendet.DieBlockierungmit5%MagermilchpulverinTween-TBS(linksgezeigt)führtdazu,dassderWestern-Blotgarnichtauswertbarist,währenddieKombinationvonBlockingBufferIalsBlockiererundCrossDown(rechtsgezeigt)alsAntikörper-VerdünnungspufferfürPrimär-undSekundärantikörpereinklaresErgebnisbringt.DieBandenstrukturwirddeutlicherkennbar.
Alles nur Theorie? Hier die Praxis!
ELISADergezeigteELISArichtetsichgegenImmunglobulinausMeer-schweinchen(entwickeltvonPARABioscience,Gronau).EristfürdenEinsatzinimmuntoxikologischenStudienmitMeerschweinchenbestimmt,dochfalsch-positiveBindungeninderSpezifitätskontrolle(ReiheA1–A12sowiebeimLeerwert(H1–H12)vereitelndieInter-pretationundAuswertung.
DurchVerwendungvonCrossDownBufferwerdendiefalsch-positivenBindungenverhindert undderNachweisderKonzen-trationsabhängigkeitindenReihenBbisG 1–6bzw.BbisG 7–12wirdmöglich.AlsFängerwurdeGoat-anti-guinea-pig-IgGF(ab´)2undalsDetektorGoat-anti-guinea-pig-IgG(Fcγ),biotinyliert(beidevonJacksonImmunoResearchLaboratoriesInc.,Konzentrationsbereicheje0,31bis10µg/mlinPBS)verwendet.DasMeerschweinchen-IgGwurdeentwe-derinCrossDownBufferoderinPBSverdünnt(Bereich1-650ng/ml,Bereich7-1210ng/ml).AlsBlockierungspufferwurdePBS-BSA-Pufferverwendet.DieDetektionerfolgtemitStreptavidin-Peroxidase(Sigma)undortho-Phenylendiamin(Sigma).
mit CrossDownmit Standard Assay Buffer
falsch-positive Bindungen
falsch-positive Bindungen
Herz Hirn Herz Hirn

Welche Substanzen setzt man Antikörper-Lösungen als antibakterielle Substanzen zu?Üblicherweise wird im biomedizinischen Labor Natriumazid (Endkonzentration 0,02-0,2 %) oder Thimerosal ( End konzentration 0,005 %) den Lösungen zugesetzt. Beide Substanzen sind giftig und weniger schädliche Alternativen sind gewünscht. Hier bietet sich ProClin® 300 als Alternative an, wenn es darum geht, Bakterien- und Pilzwachstum zu verhindern.
Ist es sinnvoll Albumin zuzusetzen?Ein Zusatz von Albumin ist nicht schlecht, weil Albumine Antikörper stabilisieren können. Um Antikörper für eine lange Zeit in Lösung zu stabilisieren, sind auch unsere Antibody-Stabilizer (Artikel-Nr. A7148 und A7135) gut geeignet, weil hier besondere strukturerhaltende Additive enthalten sind.
Kann man den Antikörperin 50 % Glycerin lagern?
Wenn man Antikörper wirklich einfrieren will, empfehlen wir zunächst einen Aktivitätstest nach dem Auftauen, da prak-tisch kein Antikörper ohne Aktivitätsverlust eingefroren werden kann. Für den Fall, dass die Tests positiv ausfallen, sollte der Antiköper in kleinen Portionen eingefroren werden. Nicht mehrfach einfrieren und auftauen, da mit jedem Zyklus die Aktivität sinkt! Beim Auftauen vorsichtig sein, denn zu schnelles Auftauen schädigt den gelösten Antikörper.
Die moderne Form Antikörper für lange Zeit zu lagern, ist die Lagerung in unserem Reagenz Antibody Stabilizer-PBS oder -Tris. Man kann den Antikörper direkt im Antibody Stabilizer verdünnen und direkt bei 2-8°C lagern. Die meisten Antikörper besitzen nur sehr kurze Haltbarkeiten, in Antibody
Stabilizer jedoch bis zu 5 Jahre und länger. Einer der Hauptvorteile ist, dass man vor Einlagerung nicht aliquotieren muss. Für jeden neuen Assay wird einfach nur die Antikörpermenge entnommen,
die man tatsächlich für den Versuch benötigt. Der Antikörper ist damit immer für das nächste
Experiment gebrauchsfertig und muss nicht erst langsam aufgetaut werden.
tippsblockierungBlocking Buffer I – ready-to-useAppliChem hat nun mit Blocking Buffer I eine Blockierungs lösung auf den Markt gebracht, die alle gewünschten Eigenschaften in sich vereint. Der Blocking Buffer I basiert auf hochreinem Casein, das im Rahmen der Produktion chemisch so modifi-ziert wird, dass ein Optimum in der Verteilung unter-schiedlich großer Casein-Moleküle bzw. -Fragmente entsteht. Diese neue Blockierung ist ready-to-use, daher einfach und sicher in der Anwendung. Gleichzeitig erzielt sie herausragende Blockierungs effizienz. Der Anwender braucht also nicht mehr lange nach geeig-neten Lösungen suchen. Er setzt die Blockierung von AppliChem ein und sie funktioniert direkt – ohne langes Testen und Optimieren.
Doch auch bei effektiver Blockierung kann es mitunter zu Hintergrund kommen. Dies kann z.B. auf Matrixeffekten oder Kreuzreaktivitäten beruhen (siehe Seite 2). Auch hier kann man Abhilfe schaffen. Durch den zusätzlichen Einsatz von CrossDown Buffer von AppliChem reduziert man wirksam diese Störeffekte bei verschiedenen Immunoassays – gleich ob man immunchemische Färbungen, Western-Blots oder auch ELISAs nutzt.
FazitEffiziente Blockierung und moderne Probenpuffer können Hintergrund und unerwünschte falsch-positive Signale in Immunoassays einfach und effektiv verhin-dern. Die ready-to-use-Lösungen Blocking Buffer I und CrossDown Buffer von AppliChem verbinden ohne langwieriges Optimieren eine einfache und sichere Anwendung mit optimaler Ergebniszuverlässigkeit. Bei manchen Assays sind aber auch durchaus preiswertere BSA-Blockierer einsetzbar, ohne dass Hintergrund zum Problem wird. Für diese einfachen Assays bietet AppliChem auch einen ready-to-use BSA-Blockierer an, damit die Arbeitszeit für die Wissenschaft bleibt.
12 Immunoassay Puffer • AppliChem © 2011

AppliCationsCell proliferation assays are widely used in cell biology for the study of growth factors, cytokines or media components. They are also applied in the screening of cytotoxic agents and lymphocyte activation. In order to determine the number of viable cells Cell Proliferation Kit XTT employs 2,3-Bis-(2-methoxy-4-nitro- 5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt (XTT). Only in living cells mitochondria are capable to reduce XTT to form an orange colored water soluble dye. Therefore, the concentration of the dye is proportional to the number of metabolically active cells.
The need for a reliable, sensitive and quantitative assay that would enable analysis of a large number of samples led to the development of methods, such as incorporation of radioactively labeled 3H-thymidine into DNA or the use of 5-Bromo-2‘-deoxyuridine (BrdU) as a substitute for radioactive thymidine to label DNA in living cells.The above methods have a number of disadvantages, including: use of radioactive materials and relatively complex techniques. The use of tetrazolium salts, such as MTT, commenced in the 1950s, is based on the fact that living cells reduce tetrazolium salts into colored formazan compounds.
Keywords
XTT assay
cytotoxicity testing
non radioactive assay
quantitating and viability testing of cells
No.12Cell Proliferation Assay XTT
XTT formazan
The tetrazolium salt of XTT (2,3-Bis-(2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyanilide salt) is an inner salt, that is cleaved to formazan by the succinate dehydrogenase system of the mitochondrial respiratory chain. Only living cells, possessing an intact mitochondrial membrane and also an intact cell membrane, do have active dehydrogenase. Agents that disrupt the membranes and destroy the respira tory chain will inactivate the enzyme and therefore the formation of the soluble orange formazan by reduction of the yellow tetrazolium salt. The reaction requires the presence of an electron coupling reagent, which is phenazine methosulfate, serving as an intermediate electron acceptor.
XTT tetrazolium
PMS
mitochondrial dehydrogenase
AppliCationsIn analytical laboratories, it is common practice to „seal“ bottles of HPLC solvents simply with a perforated aluminium foil or plastic cap. Connecting tubes are inserted into the bottles through the holes in the foil/cap without any additional sealing. However such basic covers do not seal the solvent containers efficiently. On entering an HPLC laboratory, one can often already smell the solvent vapors. This uncontrolled evaporation of solvents is not only a potential health hazard but also the loss of solvent by evaporation interferes with the quality and reproducibility of HPLC runs. We present here quantitative data comparing the efficiency of various sealing methods, including Applichem’s SafetyFirstCaps for HPLC.
To test for the reproducibility of the retention time and the loss of solvent by evaporation, tests were performed over a period of 31 days. The separation of three PAHs (Polycyclic Aromatic Hydrocarbons) with solvents, stored in either tightly closed bottles (SafetyFirst Caps) or bottles closed with caps with holes of different diameters, was compared. The results of the tests were dramatic.
Test Conditions
Bottle A This Bottle was closed with a SafetyFirst Cap, fitting precisely the standard GL45 glass bottle thread used.Bottle B This bottle was tightly closed with the GL45-cap provided including a Teflon-foil seal.Bottle C This bottle was closed with a cap having a 10-mm hole leaving an opening of an area of approximately 0.785 cm2.Bottle D This bottle was closed with a cap having 3 holes, 3 mm each, leaving an opening of an area of approximately 0.212 cm2.
Procedure
• At the start of the test, all 4 bottles were filled with the same mixture of Water + Methanol = 20 + 80 (w/w).• Using Bottle B as a reference, chromatograms of a mixture of the three PAHs (Naphthalene, Pyrene, Chrysene), were compared with the reference.• After the first measurement, all bottles were kept at room temperature under a fume hood with a gentle air flow for 31 days.HPLC-System HITACHI LaChrom® Elite system with Diode Array Detector under control of the EZChrom® Elite Software. Isocratic pump conditions with premixed mobile phase.HPLC-Column Purospher® RP-18e (5µ), 125 x 4 mm
Keywords
health protection
reduction of contaminant concentrations
constant HPLC-retention time
save solvents
No.10SafetyFirst Caps: Safety-Systems for HPLC
AppliCationsSeparateIT gels represent a novel gel matrix for DNA electrophoresis. Gel polymers are arranged in a conceptually different way, in accordance with a new theoretical model of gel electrophoresis. SeparateIT gels selectively retard the migration of large molecules, so that DNA bands remain sharp but are more spread out relative to each other. Thanks to this increased spacing, resolving power of SeparateIT gels is at least twice higher compared to resolving power of any other gels, including polyacrylamide gels.
The extraordinary resolving power of SeparateIT gels results from addition of a special polymer to a polymerizing solution containing a monomer and a cross-linker. While the gels with SeparateIT-like properties could be prepared with several different polymers, chemical composition and molecular weight of the polymer required a careful optimization for each particular monomer/cross-linker combination.In order to satisfy numerous requests from researchers who wish to improve resolving power of their acrylamide based gels, AppliChem now provides SeparateIT Polymer Solution. This polymer has been optimized for the gels with a ratio of acrylamide to N,N-methylene-bisacrylamide of 29 : 1. The polymer is not optimized for denaturing, urea-containing polyacrylamide gels.
SeparateIT Polymer Solution comes as a 10X solution. It should be mixed with a buffered solution of acrylamide and Bis prior to addition of TEMED and ammonium persulfate. Gels with SeparateIT Polymer Solution are prepared in the same way as any regular polyacrylamide gels. Likewise, the electrophoresis, gel staining and recording are carried out as usual. The increased resolving power of SeparateIT gels enables full separation of closely spaced bands on short gels. For example, a pair of fragments differing by 4 bp is usually resolved on less than 4 cm of gel length. Two DNA fragments in the 70 –150 bp range that differ by 1 bp can be separated on SeparateIT gels that are 8 cm long.We recommend that Mini gel cassettes, which are 8 or 10 cm long, are used for casting acrylamide-Bis gels with SeparateIT Polymer Solution. Such relatively short gels will be appropriate for the majority of demanding separations which have previously required 20 –30 cm long polyacrylamide gels. The use of shorter gels is beneficial for several reasons, including easier gel preparation and handling, lower cost of the gel materials, faster electrophoresis runs, and lower consumption of gel staining reagents. In addition, a lower amount of sample DNA needs to be loaded on a gel with a high resolving power compared to a gel with a low resolving power. This is the case because DNA bands that migrate a short distance remain sharper than the bands that migrate a long distance. It is always advantageous when closely spaced bands separate after migrating just a few centimetres.
Keywords
improved resolution
Polyacrylamide Gel Electrophoresis
SeparateIT Polymer Solution
No.7Improving Separation During Electrophoresis
AppliCationsSeparateIT gels represent a novel gel matrix for DNA electrophoresis. Gel are arranged in a conceptually different way, in accordance with a new theoretical model of gel electrophoresis. SeparateIT gels selectively retard the migration of large molecules, so that DNA bands remain sharp but are more spread out relative to each other. Thanks to this increased spacing, resolving power of SeparateIT gels is at least twice higher compared to resolving power of any other gels, including polyacrylamide gels.
The extraordinary resolving power of SeparateIT gels results from addition of a special polymer to a polymerizing solution containing a monomer and a cross-linker. While the gels with SeparateIT-like properties could be prepared with several different polymers, chemical composition and molecular weight of the polymer required a careful optimization for each particular monomer/cross-linker combination.In order to satisfy numerous requests from researchers who wish to improve resolving power of their acrylamide based gels, AppliChem now provides SeparateIT Polymer Solution. This polymer has been optimized for the gels with a ratio of acrylamide to N,N-methylene-bisacrylamide of 29 : 1. The polymer is not optimized for denaturing, urea-containing polyacrylamide gels.
SeparateIT Polymer Solution comes as a 10X solution. It should be mixed with a buffered solution of acrylamide and Bis prior to addition of TEMED and ammonium persulfate. Gels with SeparateIT Polymer Solution are prepared in the same way as any regular polyacrylamide gels. Likewise, the electrophoresis, gel staining and recording are carried out as usual. The increased resolving power of SeparateIT gels enables full separation of closely spaced bands on short gels. For example, a pair of fragments differing by 4 bp is usually resolved on less than 4 cm of gel length. Two DNA fragments in the 70range that differ by 1 bp can be separated on SeparateIT gels that are 8 cm long.We recommend that Mini gel cassettes, which are 8 or 10 cm long, are used for casting acrylamide-Bis gels with SeparateIT Polymer Solution. Such relatively short gels will be appropriate for the majority of demanding separations which have previously required 20 –30 cm long polyacrylamide gels. The use of shorter gels is beneficial for several reasons, including easier gel preparation and handling, lower cost of the gel materials, faster electrophoresis runs, and lower consumption of gel staining reagents. In addition, a lower amount of sample DNA needs to be loaded on a gel with a high resolving power compared to a gel with a low resolving power. This is the case because DNA bands that migrate a short distance remain
Improving Separation During Electrophoresis
AppliCationsSize-exclusion chromatography (SEC) is a popular method to separate biomolecules based on their size. Primarily, it is applied to the separation of biopolymers such as proteins and nucleic acids, i.e. water-soluble polymers. This system is also called gel filtration, typically with beads of dextran or agarose serving as gel matrix. Smaller molecules pass significantly slower through the column than larger molecules. Not to be mixed up with gel electrophoresis, there are big differences in terms of the separation principle. SEC does not require electric current and the sieving effect will not separate small molecules first.
It is indeed correct that smaller molecules pass more slowly through the matrix than larger molecules. This is due to the longer path the smaller molecules must travel. The longer path arises from the pores of the beads. The smaller molecules can enter the pores and go inside the beads. Entering the pores creates a longer path for the smaller molecules. There is a relationship between the delay this causes and the molecular size. The larger molecules can not enter the pores and therefore have a shorter path. The larger molecules therefore travel faster through the column than smaller molecules.Advantages of SEC/Gel FiltrationSome important features of the technique have to be mentioned. First, that the separation of the large molecules from the small ones is very effective and is performed under mild conditions does mean that biomolecules retain their biological activity. The volume of the eluate may be kept small and therefore the technique is suited for concentrated samples. The MatrixAppliChem‘s size-exclusion chromatography columns are packed with AppliXchange-G25 M, a beaded composite material composed partially of polymerized dextran. It exhibits high selectivity, high resolution and chemical stability. Molecules purified with AppliXchange-G25 M are separated according to size. The chemical interaction of gel matrix and molecules to be separated is negligible. Little or no absorption or binding takes place. Buffer and pH effects on resolution are kept minimal. Therefore, buffer conditions, pH value and temperature during the filtration process may be adapted according to the needs of the molecules but not the column material. Essential co-factors or ions necessary for the function or stability of proteins may be included in the eluent as well, since they largely don‘t interfere with the separation. The size exclusion cut-off for AppliXchange-G25 M is set at 10 kD for proteins and 10 bp for nucleic acids. Purified biomolecules are not significantly diluted when processed using AppliXchange-G25 M.Experience shows that columns may be used repeatedly, up to four times. It is essential to include an additional washing step of the columns with elution buffer after separation to remove residual molecules.The number “25” correlates to the “water regain” of the matrix. A grade “25” means that the dry matrix will take up 2.5 times its weight of water (1 g AppliXchange-G25 M absorbs 2.5 g water). This is a water regain of 2.5. Likewise, a grade of “50” gives a water regain of 5 grams (1 g AppliXchange-50 plus 5.0 g water.) The ability to absorb water is dependent on the
Keywords
Gel filtration matrix
cross-linked dextran beads
Desalting
Buffer exchange
Nucleic acid/ protein purification
Dye terminator removal
No.6Size-Exclusion Chromatography for purification of biomolecules
AppliCationsSize-exclusion chromatography (SEC) is a popular method to separate biomolecules based on their size. Primarily, it is applied to the separation of biopolymers such as proteins and nucleic acids, i.e. water-soluble polymers. This system is also called gel filtration, typically with beads of dextran or agarose serving as gel matrix. Smaller molecules pass significantly slower through the column than larger molecules. Not to be mixed up with gel electrophoresis, there are big differences in terms of the separation principle. SEC does not require electric current and the sieving effect will not separate small molecules first.
It is indeed correct that smaller molecules pass more slowly through the matrix than larger molecules. This is due to the longer path the smaller molecules must travel. The longer path arises from the pores of the beads. The smaller molecules can enter the pores and go inside the beads. Entering the pores creates a longer path for the smaller molecules. There is a relationship between the delay this causes and the molecular size. The larger molecules can not enter the pores and therefore have a shorter path. The larger molecules therefore travel faster through the column than smaller molecules.Advantages of SEC/Gel FiltrationSome important features of the technique have to be mentioned. First, that the separation of the large molecules from the small ones is very effective and is performed under mild conditions does mean that biomolecules retain their biological activity. The volume of the eluate may be kept small and therefore the technique is suited for concentrated samples. The MatrixAppliChem‘s size-exclusion chromatography columns are packed with AppliXchange-G25 M, a beaded composite material composed partially of polymerized dextran. It exhibits high selectivity, high resolution and chemical stability. Molecules purified with AppliXchange-G25 M are separated according to size. The chemical interaction of gel matrix and molecules to be separated is negligible. Little or no absorption or binding takes place. Buffer and pH effects on resolution are kept minimal. Therefore, buffer conditions, pH value and temperature during the filtration process may be adapted according to the needs of the molecules but not the column material. Essential co-factors or ions necessary for the function or stability of proteins may be included in the eluent as well, since they largely don‘t interfere with the separation. The size exclusion cut-off for AppliXchange-G25 M is set at 10 kD for proteins and 10 bp for nucleic acids. Purified biomolecules are not significantly diluted when processed using AppliXchange-G25 M.Experience shows that columns may be used repeatedly, up to four times. It is essential to include an additional washing step of the columns with elution buffer after separation to remove residual molecules.The number “25” correlates to the “water regain” of the matrix. A grade “25” means that the dry matrix will take up 2.5 times its weight of water (1 g AppliXchange-G25 M absorbs 2.5 g water). This is a water regain of 2.5. Likewise, a grade of “50” gives a water regain of 5 grams (1 g AppliXchange-50 plus 5.0 g water.) The ability to absorb water is dependent on the
Gel filtration matrix
Dye terminator removal
Size-Exclusion Chromatography for purification of biomolecules
Gel filtration matrix
Dye terminator removal
AppliCationsFreie Nukleinsäuren verursachen als Kontaminationen große Probleme im Forschungs- und molekularbiologisch-analytischen oder klinisch-diagnostischen Labor. Durch die extrem hohe Sensitivität von DNA-Nachweistests, können kleinste Verunreinigungen in PCR-Ansätzen zusätzliche Arbeit bedeuten und im schlimm-sten Fall Ergebnisse verfälschen. Mit Derma-ExitusPlus™ (HHDK) aus der Serie von ExitusPlus™-Produkten wird erstmals ein völlig neuer Anwendungsbereich erschlossen bzw. zusätzliche Kontaminationsquellen ausgeschlossen.
Eine der Hauptquellen für Kontaminationen mit Nukleinsäuren ist der Experimentator selbst. Die Nukleinsäuren stammen z.B. aus Hautschuppen, Haaren und Speichel oder von Mikroorganismen, die seine Haut besiedeln oder z.B. beim Niesen freigesetzt werden. Gelangen diese in die PCR-Ansätze oder PCR-Reagenzien, können sie entsprechend der eingesetzten Primer (besonders 16S rDNA für Bakterien) leicht nachgewiesen werden. Ausserdem besteht die Gefahr, dass beim Öffnen und Schliessen von Reaktionsgefäßen mit Nukleinsäure-Proben, wie Primern, Plasmid-DNA und genomischer DNA, die Haut und / oder der Handschuh kontaminiert werden und diese Kontamination, zusätzlich zu den endogenen Verunreinigungen (DNA, Enzyme, wie RNasen, Amylasen usw.), durch Anfassen anderer Gegenstände weiterverbreitet werden. Hier helfen – aber auch nur sehr eingeschränkt – aufwendiges Handschuhwechseln, Tragen mehrerer Handschuhe übereinander und häufiges Händewaschen. Die bisher auf dem Markt befindlichen Nukleinsäure-Dekontaminationsmittel sind aufgrund ihrer reizenden, ätzenden, giftigen oder anderweitig gesundheitsschädlichen Eigenschaften für den direkten Einsatz auf der Haut völlig ungeeignet.Mit Derma-ExitusPlus™ steht jetzt erstmalig eine dermatologisch getestete Dekontaminationslösung zur Verfügung, die in wenigen Minuten freie Nukleinsäurekontaminationen auf Haut und Händen abbaut. Basierend auf den Inhaltsstoffen der Derma-ExitusPlus™-Produkte, erfolgt der Nukleinsäure-Abbau chemisch-katalytisch. Derma-ExitusPlus™ ergänzt die Oberflächen-Dekontamination (DNA-ExitusPlus™) und den Autoklavenzusatz Autoclave-ExitusPlus™, der verhindert, dass Nukleinsäuren mit Aerosolen austreten.
Der Experimentator kommt üblicherweise mit verschiedenen Formen freier Nukleinsäuren in Berührung: Primern, Oligonukleotiden mit einer Größe von wenigen Basenpaaren, mittelgroße PCR-Fragmente oder Fragmente, die nach Restrikti-onsenzym-Verdau aus Plasmiden entstehen, bis hin zu linearisierten Plasmiden, zirkulären Plasmiden und genomischer DNA. Freie Nukleinsäuren bergen entsprechend ihrer Herkunft (z.B. virale Nukleinsäuren) bzw. ihrer Sequenz (transformierendes, krebserzeugendes Potential) nach Einschätzung der Zentralen Kommission für die Biologische Sicherheit (ZKBS) ein erhebliches Gefährdungspotential. Die Empfehlungen beim Umgang mit diesen Nukleinsäuren reichen daher von Tragen von Einmal-handschuhen bis zur Empfehlung „Personen mit erheblichen Hautverletzungen (offene Ekzeme, Wunden und Infektionen) oder mit einer ausgeprägten Verrucosis (Warzenausbildung) sollten keine Arbeiten mit diesen Nukleinsäuren durchführen“.Deshalb wurde bei der Qualitätstestung auch Wert darauf gelegt, dass lineare DNA (PCR-Bande) und ein intaktes Plasmid der Abbaureaktion unterzogen wurde.
Keywords
Nukleinsäure- Dekontamination
hautverträglich
DNA-/RNA-Abbau
Nr.5Dekontamination der Haut und Hände von Nukleinsäuren
© 2011 AppliChem • Immunoassay Puffer 13
Her damit!Her damit!Her damit!AppliCations
Der Anteil von molekularbiologischen Nachweismethoden ist in den letzten Jahren erheblich gestiegen, besonders in den Bereichen Qualitätskontrolle, Forensik, klinischer Forschung und Diagnostik – insbesondere der Infektionsdiagnostik. Gerade für diese Applikationen werden hochsensitive und gleichzeitig zuverlässige PCR-Tests benötigt. Dafür bietet AppliChem nun optimierte PCR-Kits an und widmet sich explizit der Hintergrundproblematik, die durch DNA belastete Reagenzien und Arbeitsplätze entstehen kann.
Problemstellung DNA KontaminationNukleinsäuren sind überall in der Umwelt zu finden. Daher können Sie in jedem Produktions- und Arbeitsschritt in Produkte für die PCR und in PCR-Reaktionen eingetragen werden. Es ist vielfach beschrieben worden, wie massiv PCR-Assays durch DNA-Kontaminationen gestört werden, was gerade in der klinischen Diagnostik ein schwerwiegendes Problem darstellt. Kontaminationen können also verschiedene Ursachen haben: Der Arbeitsplatz und die Gerätschaften sind mit Nukleinsäuren oder Bakterien/Viren verunreinigt (deren Nukleinsäuren in Lyseschritten freigesetzt werden können), die durch unzurei-chende oder trotz Reinigung der Oberflächen präsent sind. Nukleinsäurehaltige Aerosole, die beim Pipettieren, Zentrifugieren und vor allem beim Autoklavieren entstehen, schlagen sich nieder. Auch verkeimte Lüftungsanlagen leisten ihren unrühm-lichen Beitrag. Zuguterletzt müssen die Assay-Reagenzien genannt werden, um die es hier hauptsächlich gehen soll. Es wird geschätzt, dass sich 2 % der Publikationen mit PCR-Anwendungen mit Kontaminationen oder falsch-positiven Ergebnissen befassen. Ein Prozentsatz, der sich über die Jahre nicht verändert haben soll. Das mag daran liegen, dass sich die Zahl der Anwender erhöht hat und die Technik immer empfindlicher wird (qPCR, RT-PCR!). Wenn man „nur“ 30 PCR-Zyklen fährt, ist die Wahrscheinlichkeit eine Kontamination nachzuweisen, natürlich deutlich geringer als bei 40 oder sogar 50 Zyklen. Alle Versuche, Oberflächen und Reagenzien zu 100 % DNA-frei zu machen, sind bisher gescheitert. Es gelingt nur die Kontaminationsrate zu reduzieren (Abreicherung)!In der Literatur findet man je nach Autor die unterschiedlichsten Kontaminationen in PCR-Reagenzien: Phagen-ähnliche DNA (Newsome et al. 2004), das β-Lactamase Gen (Chiang et al. 2005), Legionellen-DNA (Evans et al. 2003), nifH und nifH-ähnliche DNA (Goto et al. 2005) und generell bakterielle DNA, wenn universelle, bakterielle 16S rDNA-Primer einge-setzt werden (Mühl et al. 2008) usw. Zum Teil wurde akribisch versucht die Kontaminationsquelle zu identifizieren, mit dem Ergebnis, dass im Prinzip alle eingesetzten Reagenzien betroffen sein können: Nukleinsäure-Isolierungsmatrizes, Taq Polymerase, Primer, Puffer, dNTPs, Wasser. Bei der Taq Polymerase erscheint das gar nicht mal so verwunderlich, denn rekombinantes und natives Enzym wird ja in Bakterien exprimiert.Eine Abreicherung der DNA-Kontamination in diesen Reagenzien erweist sich besonders bei der Taq Polymerase als problematisch. Viele Behandlungsmethoden führen zu einem Verlust der Aktivität des Enzyms (Mühl et al., 2008).
Keywords
Kontamination
DNA-freie Reagenzien
16S rDNA Nachweis
SYBR® Green/qPCR
Nr.3DNA-freie Reagenzien und Mastermixe für die PCR
Primer (besonders 16S rDNA für Bakterien) leicht nachgewiesen werden. Ausserdem besteht die Gefahr, dass beim Öffnen und Schliessen von Reaktionsgefäßen mit Nukleinsäure-Proben, wie Primern, Plasmid-DNA und genomischer DNA, die Haut und / oder der Handschuh kontaminiert werden und diese Kontamination, zusätzlich zu den endogenen Verunreinigungen (DNA, Enzyme, wie RNasen, Amylasen usw.), durch Anfassen anderer Gegenstände weiterverbreitet werden. Hier helfen – aber auch nur sehr eingeschränkt – aufwendiges Handschuhwechseln, Tragen mehrerer Handschuhe übereinander und häufiges Händewaschen. Die bisher auf dem Markt befindlichen Nukleinsäure-Dekontaminationsmittel sind aufgrund ihrer reizenden, ätzenden, giftigen oder anderweitig gesundheitsschädlichen Eigenschaften für den direkten Einsatz auf der Haut völlig Mit Derma-ExitusPlus™ steht jetzt erstmalig eine dermatologisch getestete Dekontaminationslösung zur Verfügung, die in wenigen Minuten freie Nukleinsäurekontaminationen auf Haut und Händen abbaut. Basierend auf den Inhaltsstoffen der Derma-ExitusPlus™-Produkte, erfolgt der Nukleinsäure-Abbau chemisch-katalytisch. Derma-ExitusPlus™ ergänzt die Oberflächen-Dekontamination (DNA-ExitusPlus™) und den Autoklavenzusatz Autoclave-ExitusPlus™, der verhindert, dass Nukleinsäuren mit Aerosolen austreten.
Der Experimentator kommt üblicherweise mit verschiedenen Formen freier Nukleinsäuren in Berührung: Primern, Oligonukleotiden mit einer Größe von wenigen Basenpaaren, mittelgroße PCR-Fragmente oder Fragmente, die nach Restriktionsenzym-Verdau aus Plasmiden entstehen, bis hin zu linearisierten Plasmiden, zirkulären Plasmiden und genomischer DNA. Freie Nukleinsäuren bergen entsprechend ihrer Herkunft (z.B. virale Nukleinsäuren) bzw. ihrer Sequenz (transformierendes, krebserzeugendes Potential) nach Einschätzung der Zentralen Kommission für die Biologische Sicherheit (ZKBS) ein erhebliches Gefährdungspotential. Die Empfehlungen beim Umgang mit diesen Nukleinsäuren reichen daher von Tragen von Einmalhandschuhen bis zur Empfehlung „Personen mit erheblichen Hautverletzungen (offene Ekzeme, Wunden und Infektionen) oder mit einer ausgeprägten Verrucosis (Warzenausbildung) sollten keine Arbeiten mit diesen Nukleinsäuren durchführen“.Deshalb wurde bei der Qualitätstestung auch Wert darauf gelegt, dass lineare DNA (PCR-Bande) und ein intaktes Plasmid der Abbaureaktion unterzogen wurde.
Keywords
Nukleinsäure-Dekontamination
hautverträglich
DNA-/RNA-Abbau
Der Anteil von molekularbiologischen Nachweismethoden ist in den letzten Jahren erheblich gestiegen, besonders in den Bereichen Qualitätskontrolle, Forensik, klinischer Forschung und Diagnostik – insbesondere der Infektionsdiagnostik. Gerade für diese Applikationen werden hochsensitive und gleichzeitig zuverlässige PCR-Tests benötigt. Dafür bietet AppliChem nun optimierte PCR-Kits an und widmet sich explizit der Hintergrundproblematik, die durch DNA belastete Reagenzien und Arbeitsplätze entstehen kann.
Problemstellung DNA KontaminationNukleinsäuren sind überall in der Umwelt zu finden. Daher können Sie in jedem Produktions- und Arbeitsschritt in Produkte für die PCR und in PCR-Reaktionen eingetragen werden. Es ist vielfach beschrieben worden, wie massiv PCR-Assays durch DNA-Kontaminationen gestört werden, was gerade in der klinischen Diagnostik ein schwerwiegendes Problem darstellt. Kontaminationen können also verschiedene Ursachen haben: Der Arbeitsplatz und die Gerätschaften sind mit Nukleinsäuren oder Bakterien/Viren verunreinigt (deren Nukleinsäuren in Lyseschritten freigesetzt werden können), die durch unzurei-chende oder trotz Reinigung der Oberflächen präsent sind. Nukleinsäurehaltige Aerosole, die beim Pipettieren, Zentrifugieren und vor allem beim Autoklavieren entstehen, schlagen sich nieder. Auch verkeimte Lüftungsanlagen leisten ihren unrühm-lichen Beitrag. Zuguterletzt müssen die Assay-Reagenzien genannt werden, um die es hier hauptsächlich gehen soll. Es wird geschätzt, dass sich 2 % der Publikationen mit PCR-Anwendungen mit Kontaminationen oder falsch-positiven Ergebnissen befassen. Ein Prozentsatz, der sich über die Jahre nicht verändert haben soll. Das mag daran liegen, dass sich die Zahl der Anwender erhöht hat und die Technik immer empfindlicher wird (qPCR, RT-PCR!). Wenn man „nur“ 30 PCR-Zyklen fährt, ist die Wahrscheinlichkeit eine Kontamination nachzuweisen, natürlich deutlich geringer als bei 40 oder sogar 50 Zyklen. Alle Versuche, Oberflächen und Reagenzien zu 100 % DNA-frei zu machen, sind bisher gescheitert. Es gelingt nur die Kontaminationsrate zu reduzieren (Abreicherung)!In der Literatur findet man je nach Autor die unterschiedlichsten Kontaminationen in PCR-Reagenzien: Phagen-ähnliche 2004), das β-Lactamase Gen (Chiang et al. 2005), Legionellen-DNA (Evans et al. 2003), nifH und -ähnliche DNA (Goto et al. 2005) und generell bakterielle DNA, wenn universelle, bakterielle 16S rDNA-Primer einge-et al. 2008) usw. Zum Teil wurde akribisch versucht die Kontaminationsquelle zu identifizieren, mit dem Ergebnis, dass im Prinzip alle eingesetzten Reagenzien betroffen sein können: Nukleinsäure-Isolierungsmatrizes, TaqPolymerase, Primer, Puffer, dNTPs, Wasser. Bei der Taq Polymerase erscheint das gar nicht mal so verwunderlich, denn rekombinantes und natives Enzym wird ja in Bakterien exprimiert.Eine Abreicherung der DNA-Kontamination in diesen Reagenzien erweist sich besonders bei der Taq Polymerase als problematisch. Viele Behandlungsmethoden führen zu einem Verlust der Aktivität des Enzyms (Mühl et al., 2008).
AppliCationsUsing ready-to-use ELISA kits from manufacturers is easy and convenient. Some-times however, home-made ELISA is required because there is no kit available with the right antibodies or the characteristics of the available kits such as their limits of detection are not appropriate. Ready-to-use ELISA kits from good suppliers may be stored for two years at 4°C without any problem. With home-made ELISA it is a completely different story. For any new measurement one has to coat a new plate, because after storage of some days the plates don’t perform as well as before.
Why is there such a great difference in storage between home-made ELISA and ELISA kits?The reason is that in professional ELISA kit production the plates are not only blocked after coating, but also stabilised. This easy to perform process has been an industry standard for thirty years. For stabilisation of a plate one has to incubate with a coating stabiliser solution. It is just as simple as a “second blocking step”. But there were no such high quality stabiliser so-lutions freely available in low volumes for use in research lab until now. AppliChem now offers a product for use in every research lab in volumes starting as small as 50 ml, which is called the AppliCoat Plate Stabiliser (Cat. No. A7708). This stabiliser solution is easy-to-use and has a great advantage compared to almost any stabiliser used in industry. It gives better storage stability for coated anti bodies and antigens than most other products do. And there is a second benefit with this product:Two benefits with one solutionWhen antibodies are coated onto ELISA plates, most of the antibodies are not active. When the antibodies (or any proteins) come into close contact to the plastics surface of the ELISA plate, conformational changes can occur due to surface-protein interactions. The result is that most antibodies coated on a plate are unfolded or in active. Only around 2–8 % of all coated antibodies remain active and can bind to analytes and this is greatly variable depending on the surface characteristics of the ELISA plate, which can really differ from batch to batch or even from well to well.These differences from well to well can affect the variability of an assay, because the antibodies can be affected. If there was a way of refolding antibodies and of preserving antibodies from conformational changes during storage, this could help to decrease such variabilities in assay performance. This is a key benefit of AppliCoat Plate Stabiliser. It assists antibodies and coated proteins to refold and then to preserve active conformation over a long time. Thus it has two benefits: 1. Refolding of antibody conformation of some of the coated antibodies and 2. Preserving correct conformation during storage. These bene-fits are used for production of high-quality ELISA kits as well as in research applications now. Even with AppliCoat Plate Stabiliser the percentage of active antibodies will still be in the range of 2–8 %. But the great difference is that the variability from well to well and from plate to plate can be minimised in most assays by using AppliCoat Plate Stabiliser. Such effects depend on the used antibodies, but when ELISA are validated (e.g. according to “Guidance for Industry: Bioanalytical Method Validation”, FDA, 2001) or according to other validation strategies, the difference can be measured in many assays.The positive effects of AppliCoat Plate Stabiliser are shown in Fig. 1. A sandwich ELISA with a monoclonal antibody has been
Keywords
Immunoassays
Antibody Stabilisation
ELISA Plates
Cross-reactivity
Interfering effects
No.4Improving quality of ELISA
AppliCationsThe Colorada product line is a selection of the newest fluorescent labels available today. Dye structures were carefully chosen to provide top-notch brightness perfor-mances, improving bioanalytical assays to the highest level. Eight different basic structures with precise absorption and emission maxima were selected to cover the full working range of wavelengths, starting from 400 nm up to 850 nm. The Colorada structures are based on 4 different chemical classes: coumarin, dipyrromethene borondifluoride, acenaphthopyrrole and cyanine
Coumarin dyes are chemically and photochemically robust, relatively small dyes with good absorption coefficients in the near UV / blue spectral region. Dipyrrometheneboron difluorides (BDPs) are chemically and photochemically robust, and pH insensitive. They have high absorption coefficients and excellent fluorescence quantum yields. Trimethine cyanine dyes have very high absorption coefficients, and are available in anionic (A), cationic (c) and zwitterionic (Z) form. 8-Oxo-8H-cyclopenta[a]acenaphthylene-7-carbonitriles (Acenaphthopyrrole dyes) are chemically and photochemically robust, pH insensitive dyes with high absorption coefficients and fluorescence quantum yields. Pentamethine cyanine dyes have extremely high absorption coefficients, good quantum yields of fluorescence and are available in anionic (A), cationic (C) and zwitterionic (Z) form. Heptamethine indocyanine dyes have extremely high absorption coefficients and good quan-tum yields of fluorescence in the near infrared (NIR) spectral region. They are available in anionic (A), and cationic (C).The dyes with a Large Stokes Shift (Colorada 670 LSS and Colorada 690 LSS) belong to the chemical class of heptamethine indocyanines as well. These dyes have high absorption coefficients in the far red and good quantum yields of fluorescence in the near infrared (NIR) spectral region, with a large stoke shift (> 100 nm).To meet the need of labeling the broadest range of target molecules each fluorescent compound is available with many dif-ferent reactive groups (e.g. the commonly used amine reactive and thiol reactive groups) while retaining the photophysical features of the parent dye. The reactive groups include carboxylic acid (- COOH), active ester (- COOX, NHS ester), dichloro-triazine, aliphatic amine (- NH2), azide (- N3), iodoacetamide and 2-pyridyldisulfideamine (PDA), the latter being very useful for labeling -SH in peptides or proteins.Furthermore, a new level of chemistry has been developed to provide the same fluorescent label with different charge values (anionic, cationic, zwitterionic and neutral). It is thus easy to find the ideal product among the Colorada series starting from the desired absorption/emission peak maximum and subsequently decide both the reactivity and charge properties.
Keywords
high absorption coefficients
high fluorescence quantum yields
wavelengths from 400–850 nm
coumarin
BDP
acenaphthopyrrole
cyanine
No.8Colorada: A New Generation Of Fluorescent Labels AppliCations
In analytical laboratories, it is common practice to „seal“ bottles of HPLC solvents simply with a perforated aluminium foil or plastic cap. Connecting tubes are inserted into the bottles through the holes in the foil/cap without any additional sealing. However such basic covers do not seal the solvent containers efficiently. On entering an HPLC laboratory, one can often already smell the solvent vapors. This uncontrolled evaporation of solvents is not only a potential health hazard but also the loss of solvent by evaporation interferes with the quality and reproducibility of HPLC runs. We present here quantitative data comparing the efficiency of various sealing methods, including Applichem’s SafetyFirstCaps for HPLC.
To test for the reproducibility of the retention time and the loss of solvent by evaporation, tests were performed over a period of 31 days. The separation of three PAHs (Polycyclic Aromatic Hydrocarbons) with solvents, stored in either tightly closed bottles (SafetyFirst Caps) or bottles closed with caps with holes of different diameters, was compared. The results of the tests were dramatic.
Test Conditions
Bottle A This Bottle was closed with a SafetyFirst Cap, fitting precisely the standard GL45 glass bottle thread used.
A This Bottle was closed with a SafetyFirst Cap, fitting precisely the standard GL45 glass bottle thread used.
ABottle B This bottle was tightly closed with the GL45-cap provided including a Teflon-foil seal.Bottle C This bottle was closed with a cap having a 10-mm hole leaving an opening of an area of approximately 0.785 cm2.Bottle D This bottle was closed with a cap having 3 holes, 3 mm each, leaving an opening of an area of approximately 0.212 cm2.
Procedure
• At the start of the• Using Bottle B
with the reference.• After the first measurement,
HPLC-System HITACHI LaChromIsocratic pump conditions with premixed mobile phase.HPLC-Column Purospher
Keywords
health protection
reduction of contaminant concentrations
constant HPLC-retention time
save solvents
SafetyFirst Caps: Safety-Systems for HPLC
The Colorada product line is a selection of the newest fluorescent labels available today. Dye structures were carefully chosen to provide top-notch brightness perfor-mances, improving bioanalytical assays to the highest level. Eight different basic structures with precise absorption and emission maxima were selected to cover the full working range of wavelengths, starting from 400 nm up to 850 nm. The Colorada structures are based on 4 different chemical classes: coumarin, dipyrromethene borondifluoride, acenaphthopyrrole and cyanine
Coumarin dyes are chemically and photochemically robust, relatively small dyes with good absorption coefficients in the near UV / blue spectral region. Dipyrrometheneboron difluorides (BDPs) are chemically and photochemically robust, and pH insensitive. They have high absorption coefficients and excellent fluorescence quantum yields. Trimethine cyanine dyes have very high absorption coefficients, and are available in anionic (A), cationic (c) and zwitterionic (Z) form. -cyclopenta[a]acenaphthylene-7-carbonitriles (Acenaphthopyrrole dyes) are chemically and photochemically robust, pH insensitive dyes with high absorption coefficients and fluorescence quantum yields. Pentamethine cyanine dyes have extremely high absorption coefficients, good quantum yields of fluorescence and are available in anionic (A), cationic (C) and zwitterionic (Z) form. Heptamethine indocyanine dyes have extremely high absorption coefficients and good quan-tum yields of fluorescence in the near infrared (NIR) spectral region. They are available in anionic (A), and cationic (C). (Colorada 670 LSS and Colorada 690 LSS) belong to the chemical class of heptamethine indocyanines as well. These dyes have high absorption coefficients in the far red and good quantum yields of fluorescence in the near infrared (NIR) spectral region, with a large stoke shift (> 100 nm).To meet the need of labeling the broadest range of target molecules each fluorescent compound is available with many dif-
To meet the need of labeling the broadest range of target molecules each fluorescent compound is available with many dif-
To meet the need of labeling the broadest range of target molecules each fluorescent compound is available with many different reactive groups (e.g. the commonly used amine reactive and thiol reactive groups) while retaining the photophysical features of the parent dye. The reactive groups include carboxylic acid (- COOH), active ester (- COOX, NHS ester), dichloro-), iodoacetamide and 2-pyridyldisulfideamine (PDA), the latter being very useful Furthermore, a new level of chemistry has been developed to provide the same fluorescent label with different charge values (anionic, cationic, zwitterionic and neutral). It is thus easy to find the ideal product among the Colorada series starting from the desired absorption/emission peak maximum and subsequently decide both the reactivity and charge properties.
AppliCationsEnhanced ChemiLuminescence Detection Kits for Horseradish Peroxidase in Western / Southern / Northern Blotting
The peroxidase-catalyzed oxidation of luminol produces a weak flash of light at 425 nm. The incorporation of a so-called enhancer into the buffer forces the flash signal into a glow and greatly improves the analytical characteristics of the reaction in terms of increased signal intensity and duration [1,2]. Typical enhancer compounds are substituted phenols, the most popular being p-iodophenol and p-hydroxycou-maric acid.
The enhanced, HRP catalyzed oxidation of luminol is a complex, multi-step reaction. While enhancers are useful in impro-ving enzyme turnover and increasing the equilibrium concentration of a key intermediate, the luminol radical anion, recent work [3] has shown that, by addition of a suitable catalyst, a further, large increase in light output is obtained:
As a result of this breakthrough, the family of AppliChem‘s CheLuminate-HRP chemiluminescent substrates has been developed, with specific formulations to meet the various requirements for immunoblotting (see table below for selecting the optimal substrate for your application).
Outstanding Sensitivity – The intense light output generated by the CheLuminate-HRP substrates translates into a corresponding improvement in sensitivity. Even the economical CheLuminate-HRP PicoDetect Extended formulation pro-vides a sensitivity level at least eight times higher than with phenolic enhancers, down to the low picogram (10–12) range. Detection limits are lowered further with CheLuminate-HRP FemtoDetect, to mid-femtogram (10–14), while the ultimate level in sensitivity, is achieved with CheLuminate-HRP FemtoDetect Plus, which is designed to provide femtogram (10–15)- level detection.Long Signal Duration – All substrates exhibit long light emission. However, this feature has been specifically maximized in the formulation developed for the CheLuminate-HRP FemtoDetect substrate, which offers a 24-hour light emission – at least ten times longer than with standard phenolic enhancer substrates.
Keywords
Luminol
Redox Mediator
Enhancer
Long light emission
No.9CheLuminate: Improved Chemiluminescence Keywords
XTT assay
cytotoxicity testing
non radioactive assay
quantitating and viability testing of cells
In analytical laboratories, it is common practice to „seal“ bottles of HPLC solvents
No.10
AppliCationsThe BARN PCR Blunt Cloning Systems -1 and -2 are based on the disruption of toxic barnase gene expression by cloning of PCR fragments into the multiple cloning site (MCS) of pBARN vectors. E.coli cells that contain non-recombinant vector are killed upon plating. Therefore, the blue/white screening is not required for this system. In contrast to other positive cloning systems BARN-1/-2 are not restricted to the use of special E.coli strains nor to a few restriction sites for the ligation of inserts only. In summary, pBARN cloning vectors combine advantages of positive cloning systems without the known disadvantages of other cloning systems available.
IntroductionMany cloning vectors in use today employ a blue/white screening approach to identify successful fragment insertions. DNA fragments (such as PCR products or pieces of a genome) are inserted into a multiple cloning site (MCS) within a plasmid DNA vector. Then, transformed competent E.coli cells are grown in the presence of X-gal. If the insertion was successful, the bacterial colony will be white; if not, the colony will appear in blue color. This traditional method, developed in the late seventies, has since been honed and simplified. However, it still has two major drawbacks: low efficiency of blue/white selection and high costs per cloning reaction. In addition, false-positive clones are frequently detected, making necessary sub-cloning and further time consuming investigation of candidate colonies, i.e. performing analytical restriction enzyme digest or sequencing.
To facilitate more simple and efficient high-throughput cloning several positive selection cloning systems have been developed (Table 1.). The principle of positive selection cloning is rather straightforward. Ligation of a PCR fragment into the MCS of a positive selection vector disrupts the expression/activity of the toxic gene, permitting growth of positive recom-binants (up to 100 %) upon transformation only. E.coli cells that contain non-recombinant vectors are killed upon plating. Disadvantages of known cloning systems that also employ positive selection of recombinants include: (i) The position of the poly-linker (MCS) in the middle of the toxic gene. This results in limitations regarding the choice of restriction enzymes as well as limi tations of using ‘universal sequencing primers’. (ii) Limitations for the bacterial strains to be used. (iii) Limited size of the DNA inserts. (iv) In some cases handling is cumbersome.
The pBARN PCR Blunt Cloning System is an easy-to-use, fast and highly efficient positive cloning system of blunt or blunted PCR fragments. Fragments generated by either Taq DNA polymerases or other DNA polymerases without proof-reading activity require a filling-in reaction for optimal cloning efficiency. The pBARN PCR Blunt Cloning System is based on the toxic barnase gene product, a small, highly active ribonuclease from Bacillus amyloliquefaciens (Yazynin et al. 1996; Yazynin et al., 1999; Fig. 1). pBARN-1 and -2 PCR Blunt Cloning Vectors (3.4 kb and 3.2 kb, respectively), the main component of these systems, allow direct selection of recombinants via disruption of the lethal barnase gene, once expressed. The vector contains the barnase gene under the control of the LacZ promoter, N-terminally fused to an especially designed sequence, including the multiple
Keywords
Molecular Cloning VectorPCR Blunt Cloning
Positive Selection
Barnase
No.11pBARN Cloning Vectors – Positive Selection of Recombinants
Cell proliferation assays are widely used in cell biology for the study of growth factors, cytokines or media components. They are also applied in the screening of cytotoxic agents and lymphocyte activation. In order to determine the number of employs 2,3-Bis-(2-methoxy-4-nitro-
5-sulfophenyl)-2H-tetrazolium-5-carboxanilide salt (XTT). Only in living cells mitochondria are capable to reduce XTT to form an orange colored water soluble dye. Therefore, the concentration of the dye is proportional to the number of
The need for a reliable, sensitive and quantitative assay that would enable analysis of a large number of samples led to the H-thymidine into DNA or the use of 5-Bromo-2‘-The above methods have a number of disadvantages, including: use of radioactive materials and relatively complex techniques. The use of tetrazolium salts, such as MTT, commenced in the 1950s, is based on the fact that living cells reduce
Cell Proliferation Assay XTT
The tetrazolium salt of XTT (2,3-Bis-(2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyanilide salt) is an inner salt, that is cleaved to formazan by the succinate dehydrogenase system of the mitochondrial respiratory chain. Only living cells, possessing an intact mitochondrial membrane and also an intact cell membrane, do have active dehydrogenase. Agents that disrupt the membranes and destroy the respiratory chain will inactivate the enzyme and therefore the formation of the soluble orange formazan by reduction of the yellow tetrazolium salt. The reaction requires the presence of an electron coupling reagent, which is phenazine methosulfate, serving as an intermediate electron
bioconfident grade – verlässlich biologisch sicher: Ein neu entwickeltes Expressionssystem für rekombinante Proteine aus tierischen Organismen bedient sich des pflanzlichen Gerstenkorns. Dabei werden als Expressionsplattform die Speicherzellen des Endosperms genutzt, dem Nährgewebe des pflanzlichen Samens, welche die rekombinanten Proteine in einer bio-chemisch inerten Umgebung ohne Endotoxine produzieren. Gerste ist von der FDA als sicher eingestuft worden (G.R.A.S. Generally Recognised As Safe).
AppliCationsWas bedeutet rekombinante Proteine im bioconfident grade?Üblicherweise werden Wachstumsfaktoren und Zytokine in Bakterien, Hefen oder anderen eukaryontischen Expressionssystemen (z.B. Insektenzellen, CHO-Zellen) produziert. Sowohl die Produktionsorganismen als auch die Kultivierungsreagen-zien (Medienbestandteile, Medienzusätze) bergen die Gefahr von Kontaminationen durch Bakterien, Viren und BSE. Aber auch Bestandteile davon, wie endogen pro duzierte Enzyme und andere Proteine, können als Verunreinigung auftreten. Besonders der Endotoxingehalt von bakteriell produzierten Wachstumsfaktoren ist relativ schwer zu minimieren.
Keywords
Bio-sichere Zytokine
pflanzliches Expressionssystem
Endotoxin-frei
frei von tierischen Produkten
Nr.13bioconfident grade: Zytokine & Wachstumsfaktoren
Abb. 1 Endotoxinbestimmung in Zytokinen bioconfident grade (bioconf.) im Vergleich zu einer anderen kommerziellen Quelle, gemessen durch Associates of Cape Cod Industries, Deacon Park, Knowsley, Liverpool, UK; Der Wettbewerber des IFN-a gibt als Endotoxingehalt „Weniger als 1 EU/µg rekombinanten IFN- a“ an.
1.0
0.9
0.8
0.7
0.6
0.5
0.4
0.3
0.2
0.1
0.0
[EU/µg]
0.00135
VEGF121bioconf. IFN-a
bioconf. IFN-aCommercial
0.002
1
Endotoxin levels(turbidimetric kinetic assay)
Keywords
Kontamination
DNA-freie Reagenzien
16S rDNA Nachweis
SYBR® Green/qPCR
AppliCations
Keywords
Kontamination
DNA-freie Reagenzien
16S rDNA Nachweis
SYBR
AppliCationsMany biochemical processes are markedly impaired by even small changes in the con centrations of free H+ ions. It is therefore usually necessary to stabilise the H+ concentration in vitro by adding a suitable buffer to the medium, without, however, affecting the functioning of the system under investigation. A buffer keeps the pH of a solution constant by taking up protons that are released during reactions, or by r eleasing protons when they are consumed by reactions.
This handout summarizes the most commonly used buffer substances and respec-tive physical and chemical properties.
Practical Tips – Preparing Buffer SolutionsRecommendations for the setting of the pH value of a buffer and storage conditions1. TemperatureDepending on the buffer substance, its pH may vary with temperature. It is therefore advisable, as far as possible, to set the pH at the working temperature to be used for the investigation. For instance the physiological pH value for most mammalian cells at 37°C is between 7.0 and 7.5. The temperature dependence of a buffer system is expressed as d(pKa)/dT, which describes the change of the pKa at an increase of temperature by 1°C.
2. Titration(i) Generally, the pH value is set using NaOH/KOH or HCl. Slow addition of a strong acid or base whilst stirring vigorously avoids local high concentrations of H+ or OH– ions. If this is not done, the buffer substances may undergo chemical changes that inactivate them or modify them so that they have an inhibitory action (Ellis & Morrison 1982). (ii) Under stirring CO2 dissolves in the solution. Stir solutions gently for precise measurements of the pH value. (iii) If a buffer is available in the protonised form (acid) and the non-protonised form (base), the pH value can also be set by mixing the two substances. (iv) Setting of the ionic strength of a buffer solution (if necessary) should be done in the same way as the setting of the pH value when selecting the electrolyte, since this increases depending on the electrolyte used. (v) If other components are added to the buffer (e.g. EDTA, DTT, Mg2+, b-Mercapto ethanol) changes in the pH should also be considered and pH should be retested. (vi) In the presence of divalent metal ions carbonate or phosphate buffers may form precipitates .
3. How can microbial contamination of buffer solutions be prevented?(i) Sterilization by filtration through a 0.22 µm filter unit or by autoclaving. (ii) Addition of 0.02 % (3 mM) sodium azide. (iii) Storage at +4°C. (iv) High-concen tration stock solutions.
Keywords
chemical properties
usefull pH range
buffer preparation
No.2Biological Buffers
Her damit!Keywords
chemical properties
usefull pH range
buffer preparation
AppliCationsAdvanced experiments in gene technology demonstrate that even small amounts of free DNA molecules are sufficient to cause infections, recombination or biological transformation [1,2]. The complete decontamination of equipment and surfaces from any DNA molecules is important for biological containment and safety, as well as preventing artifacts in PCR amplification experiments. Using new methods that detect extremely low levels of DNA molecules, we investigated the molecular mechanism of action of various commercially available DNA decontamination reagents. We found that when using high concentrations of DNA and short incubation times, none of the conventional reagents destroyed DNA molecules efficiently, despite their corrosive or even toxic properties.
All or Nothing at AllDestruction and elimination of nucleic acids depends to this day, on bleach, a corrosive and toxic substance. To address this issue, AppliChem partnered up with multiBIND Cologne, to develop a new and unique nucleic acid decontamination technology: the ExitusPlus™ family of products, comprising DNA-ExitusPlus™, and Autoclave-ExitusPlus™. Comparing DNA-ExitusPlus™ to conventional products, we can demonstrate that it is fast and efficient in destroying nucleic acids without harmful or toxic effects on lab workers, equipment and the environment. Most decontamination reagents are based on several molecular principles for the destruction or inactivation of genetic material: Modification and denaturation can mask, but do not destroy the genetic information encoded in DNA strands and there is the risk that they may be chemically re-activated. Thus, safe and complete DNA decontamination depends on the degradation of DNA into very small fragments. Figures 1 and 2 show the results of comparing the fragmentation process produced by the novel DNA-ExitusPlus™ with conventional reagents. Using highly sensitive detection assays, it is seen that an efficient and total fragmentation was only obtained with DNA-ExitusPlus™, whereas only partially degraded DNA pieces, some of which contained complete genetic information, were found in the decontamination products that relied on modi-fication or denaturation methodology.
Sequence-Independent Degradation of DNAOnly Applichem’s DNA-ExitusPlus™ is capable of achieving rapid and efficient degradation of nucleic acids, because its unique method of action is based on chemical and not enzymatic activity. Therefore, its effects on fragmentation are totally independent of the size and sequence of the DNA fragments. Larger plasmids require a longer incubation time than smaller ones (e.g. primers). Assuming a theoretical nicking activity of 100,000 nicks per minute, all DNA fragments will be destroyed, after several minutes, regardless of their size. Smaller fragments will disappear before the larger ones. Applying this theory to a test molecule (ccc form, 6 kb plasmid) only a small fraction of fragments with 200 to 500 bp in size will remain after 5 minutes. The nicks will be introduced statistically at any site, leaving not a single class of fragments. Remaining fragments are fully destroyed after 10 minutes.
Keywords
Nucleic acid decontamination
DNA degradation test
Autoclaving DNA
PCR test
No.1Nucleic Acid Decontamination with The ExitusPlus™ Technology
AppliChem stellt Hintergrundinformationen und Anwendungshinweise für viele Produkte zur Verfügung.
Kunden-Nummer (falls bekannt)
Titel | Vorname | Name
Institut/Abteilung
Universität/Firma
Straße
PLZ Ort
Telefon Fax
Das dürfen Sie sich nicht entgehen lassen.
Fax-Bestell-Nummer 06151/93 57 11 oder per eMail [email protected]
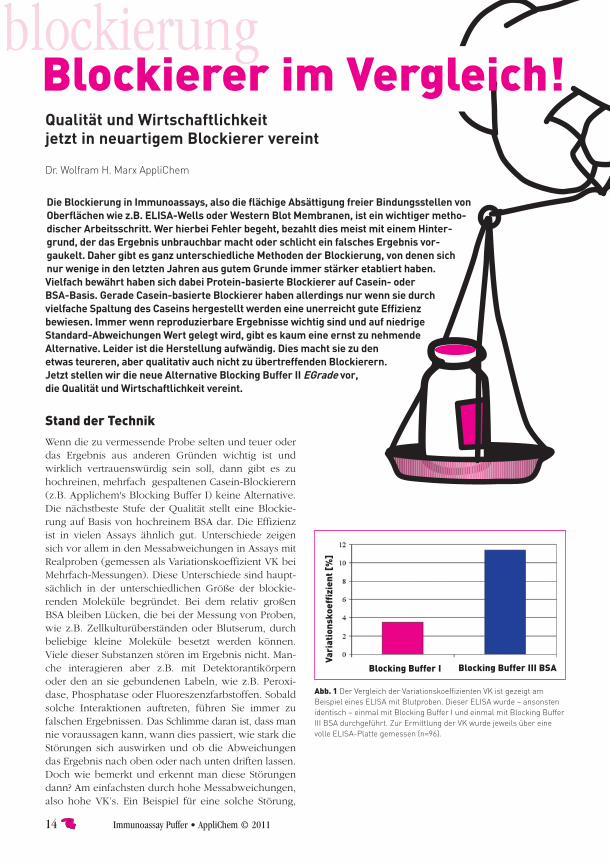
Abb. 1DerVergleichderVariationskoeffizientenVKistgezeigtamBeispieleinesELISAmitBlutproben.DieserELISAwurde–ansonstenidentisch–einmalmitBlockingBufferIundeinmalmitBlockingBufferIIIBSAdurchgeführt.ZurErmittlungderVKwurdejeweilsübereinevolleELISA-Plattegemessen(n=96).
Vari
atio
nsko
effi
zien
t [%
]
Blocking Buffer I Blocking Buffer III BSA
Stand der TechnikWenn die zu vermessende Probe selten und teuer oder das Ergebnis aus anderen Gründen wichtig ist und wirklich vertrauenswürdig sein soll, dann gibt es zu hochreinen, mehrfach gespaltenen Casein-Blockierern (z.B. Applichem's Blocking Buffer I) keine Alternative. Die nächstbeste Stufe der Qualität stellt eine Block ie-rung auf Basis von hochreinem BSA dar. Die Effizienz ist in vielen Assays ähnlich gut. Unterschiede zeigen sich vor allem in den Messabweichungen in Assays mit Realproben (gemessen als Variationskoeffizient VK bei Mehrfach-Messungen). Diese Unterschiede sind haupt-sächlich in der unterschiedlichen Größe der blockie-renden Moleküle begründet. Bei dem relativ großen BSA bleiben Lücken, die bei der Messung von Proben, wie z.B. Zellkulturüberständen oder Blutserum, durch beliebige kleine Moleküle besetzt werden können. Viele dieser Substanzen stören im Ergebnis nicht. Man-che interagieren aber z.B. mit Detektorantikörpern oder den an sie gebundenen Labeln, wie z.B. Peroxi-dase, Phosphatase oder Fluoreszenzfarbstoffen. Sobald solche Interaktionen auftreten, führen Sie immer zu falschen Ergebnissen. Das Schlimme daran ist, dass man nie voraussagen kann, wann dies passiert, wie stark die Störungen sich auswirken und ob die Abweichungen das Ergebnis nach oben oder nach unten driften lassen. Doch wie bemerkt und erkennt man diese Störungen dann? Am einfachsten durch hohe Messabweichungen, also hohe VK’s. Ein Beispiel für eine solche Störung,
14 Immunoassay Puffer • AppliChem © 2011
blockierung
Die Blockierung in Immunoassays, also die flächige Absättigung freier Bindungsstellen von Oberflächen wie z.B. ELISA-Wells oder Western Blot Membranen, ist ein wichtiger metho-discher Arbeitsschritt. Wer hierbei Fehler begeht, bezahlt dies meist mit einem Hinter-grund, der das Ergebnis unbrauchbar macht oder schlicht ein falsches Ergebnis vor-gaukelt. Daher gibt es ganz unterschiedliche Methoden der Blockierung, von denen sich nur wenige in den letzten Jahren aus gutem Grunde immer stärker etabliert haben. Vielfach bewährt haben sich dabei Protein-basierte Blockierer auf Casein- oder BSA- Basis. Gerade Casein-basierte Blockierer haben allerdings nur wenn sie durch vielfache Spaltung des Caseins hergestellt werden eine unerreicht gute Effizienz bewiesen. Immer wenn reproduzierbare Ergebnisse wichtig sind und auf niedrige Standard-Abweichungen Wert gelegt wird, gibt es kaum eine ernst zu nehmende Alternative. Leider ist die Herstellung aufwändig. Dies macht sie zu den etwas teureren, aber qualitativ auch nicht zu übertreffenden Blockierern. Jetzt stellen wir die neue Alternative Blocking Buffer II EGrade vor, die Qualität und Wirtschaftlichkeit vereint.
Dr.WolframH.MarxAppliChem
Blockierer im Vergleich!Qualität und Wirtschaftlichkeit jetzt in neuartigem Blockierer vereint

die bei Betrachtung des VK’s erst deutlich wird, ist in Abb.1 gezeigt. Es handelt sich um einen ELISA im Sandwich-Format, der zunächst auch bei Anwendung eines BSA-basierten Blockierungsreagenzes (Blocking-Buffer III BSA) gut zu funktionieren schien. Doch die VK’s sprechen ei-ne andere Sprache. Was wie ein guter Assay im Ergebnis aussieht, entpuppt sich bei genauerem Hinsehen als unzuverlässig. Erst der Vergleich des gleichen As-says mit einer Blo ckierung mit AppliChem’s Blocking Buffer I zeigt ein zuverläs-siges Ergebnis. Ein zu-nächst unscheinbarer Ef-fekt, der im Endeffekt aber über die Aussage-kraft aller dahinter lie-genden Untersuchun-gen entscheidet. Denn meistens die-nen die ELISA,
Western Blots oder andere Immunoas-
says einfach nur als Werkzeug, um tiefer liegende Hypothesen in der For-schung zu bestätigen oder zu widerlegen. Wer hierzu nun ein unzuverlässiges Werkzeug, wie im gezeigten Beispiel diesen BSA-blockierten ELISA, nutzt, muss sich durchaus gefallen lassen, wenn die erhaltenen Ergebnisse in einem wissenschaftlichen Kontext hinter-fragt oder bezweifelt werden – in diesem hier gezeig-ten Fall sind bei BSA-Blockierung Zweifel also durch-aus angebracht.Ein anderes Problem tritt seltener auf, ist aber ebenfalls nicht zu vernachlässigen. Es war über Jahre hinweg üblich, kleine Antigene – sog. Haptene – vor einer Immunisierung zur Antikörpergewinnung mit BSA als Trägermolekül zu koppeln. Dies funktioniert gut, zu-verlässig und einfach. Es hat uns nur eine Unmenge von kommerziell gehandelten sowie unter Wissen-schaftlern über Austausch verbreitete Forschungsanti-körper in die Labore gespült, die neben den gewünsch-ten Haptenen auch mit merklicher Affinität BSA binden. Denn es wurde ja auch mit bzw. gegen BSA immuni-siert. Dies wäre solange kaum ein Problem, wenn alle Produzenten von Antikörpern, sowohl die Firmen als auch die in den Instituten, die selbst Antikörper gene-rieren, diesen methodischen Schritt immer angeben. Dann wäre man gewarnt und wüsste vorab, wann man BSA-Blockierer verwenden darf und wann nicht. Doch das Gegenteil ist leider der Fall. Es hat sich seit Jahren bzw. Jahrzehnten einge bürgert bei Forschungsantikör-
pern kaum derartige wesentliche Angaben zu machen. Jeder neue Antikörper birgt daher diese experimentel-len Risiken, die dazu führen, dass man ihn nur dann wirklich mit BSA im Assay einsetzen sollte, wenn der Produzent dies auch so angibt. Dennoch bleibt es Fakt, dass BSA in vielen Fällen ein sehr guter Blockierer ist. Er ist daneben, trotz der aufwändigen BSA-Gewinnung und Aufreinigung, etwas preisgünstiger als optimale Casein-Blockierer.
Den großen Vorteilen der schon genannten guten Protein-basierten Blockierer stehen also leider auch ge-wisse Kosten gegenüber. Daher sucht man seit Jahren nach Alternativen – und es gibt viele davon. Fischgela-tine oder Molkepulver, um nur zwei Beispiele zu nen-nen. Diese führen in manchen Laboren und Assays ein Nischendasein, in vielen anderen Assays versagen sie jedoch regelmäßig. In der medizinischen Forschung, z.B. bei Pharma-Assays, bei präklinischen oder klini-schen Studien oder auch in der Diagnostik sind diese Blockierer kaum zu finden, denn die Reproduzierbar-keit ist schon aufgrund der unkontrollierbaren immer wieder schwankenden Rohstoffqualität überhaupt nicht gegeben. Wenn es auf einen Assay wirklich an-kommt z.B. weil wichtige Rückschlüsse für Zwecke der medizinischen Forschung im Rahmen von Studien ge-troffen werden sollen, kann man sie also nicht in Be-tracht ziehen. Ein Assay kann nur reproduzierbar sein, wenn die Rohstoffe reproduzierbare Qualität haben. Aus Kostengründen kommen sie also nur dort zum Ein-satz, wo man sich geringe Zuverlässigkeit leisten kann, weil es z.B. eher um die Ausbildung von Studenten geht, als um verlässliche Resultate. Hier haben diese Blockierer ihr sinnvolles Anwendungsfeld.
Alternativen sind wünschenswertDie hochwertigen Blockierer sind also mit etwas
höh eren Kosten verbunden, die anderen sind günstiger und von stark schwankender Qualität. Es liegt auf der Hand, dass man versucht hat die Vorteile beider Seiten zu verbinden. Das Ergebnis sollen die synthetischen Blockierer sein, von denen Tween® sicher der erste, bekannteste und wohl einfachste Vertreter ist. Aber auch eine Reihe anderer synthetischer Blockierer wird zu mitunter günstigen Preisen vertrieben. Die Repro-duzierbarkeit der Rohstoffe dürfte hier immer optimal sein. Doch ein Problem wurde nicht gelöst. Die Repro-duzierbarkeit der Ergebnisse. Nicht ohne Grund haben die synthetischen Blockierer die Verwendung von Pro-tein-basierten Blockierern überhaupt nicht zurückdrän-gen können, obwohl ihre Kosten nur einen Bruchteil betragen. Wenn man bedenkt, dass die Gesamtkosten der Blockierung nur im niedrigen Prozentbereich der Kosten eines gesamten Assays liegen (bei korrekter Kalkulation durch Einbeziehung der Personalkosten
© 2011 AppliChem • Immunoassay Puffer 15
Blockierer im Vergleich!Qualität und Wirtschaftlichkeit jetzt in neuartigem Blockierer vereint
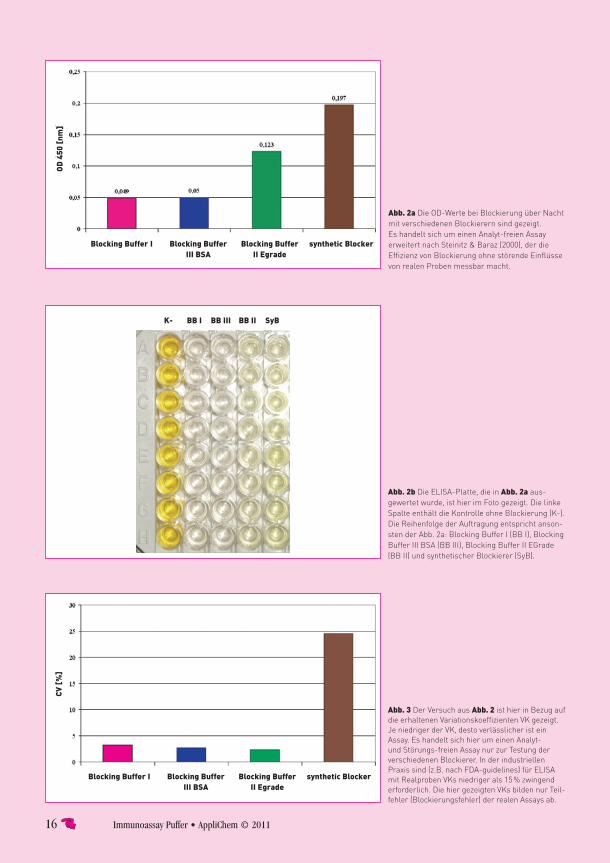
Abb. 2aDieOD-WertebeiBlockierungüberNachtmitverschiedenenBlockierernsindgezeigt.EshandeltsichumeinenAnalyt-freienAssayerweitertnachSteinitz&Baraz(2000),derdieEffizienzvonBlockierungohnestörendeEinflüssevonrealenProbenmessbarmacht.
Abb. 3DerVersuchausAbb. 2isthierinBezugaufdieerhaltenenVariationskoeffizientenVKgezeigt.JeniedrigerderVK,destoverlässlicheristeinAssay.EshandeltsichhierumeinenAnalyt-undStörungs-freienAssaynurzurTestungderverschiedenenBlockierer.InderindustriellenPraxissind(z.B.nachFDA-guidelines)fürELISAmitRealprobenVKsniedrigerals15%zwingenderforderlich.DiehiergezeigtenVKsbildennurTeil-fehler(Blockierungsfehler)derrealenAssaysab.
Abb. 2bDieELISA-Platte,diein Abb. 2aaus-gewertetwurde,isthierimFotogezeigt.DielinkeSpalteenthältdieKontrolleohneBlockierung(K-).DieReihenfolgederAuftragungentsprichtanson-stenderAbb.2a:BlockingBufferI(BBI),BlockingBufferIIIBSA(BBIII),BlockingBufferIIEGrade(BBII)undsynthetischerBlockierer(SyB).
K- BB I BB III BB II SyB
16 Immunoassay Puffer • AppliChem © 2011
CV [
%]
OD
450
[nm
]
Blocking Buffer I Blocking Buffer III BSA
Blocking Buffer II Egrade
synthetic Blocker
Blocking Buffer I Blocking Buffer III BSA
Blocking Buffer II Egrade
synthetic Blocker

© 2011 AppliChem • Immunoassay Puffer 17
Abb. 3 zeigt die Ergebnisse des gleichen Versuches, jedoch sind hier die VKs gezeigt. Hier trennt sich die Spreu vom Weizen offensichtlicher. Die in der ersten Betrachtung brauchbare synthetische Lösung zeigt, ob-wohl es sich um ein einfaches artifizielles System ohne Störkomponenten aus einer zu vermessenden Probe handelt, einen extrem hohen VK von fast 25 %. Ein solcher VK in der Blockierung überträgt sich auf den Gesamt-VK eines Assays gemäß der Fehlerfortpflan-zung der jeweiligen Einzelfehler des Assays. Es erscheint damit fraglich, ob man einen reproduzierba-ren Assay auf Basis dieser synthetischen Blockierung aufbauen kann. Sehr deutlich zeigt sich der Vorteil des neuartigen Blocking Buffer II EGrade. Die Blockierung ist sehr gut reproduzierbar. Der VK ist absolut identisch zu denen der hochwertigen Lösungen.
Durch die Kombination der Resultate von Abb. 2 und 3. lässt sich hieraus schließen, dass in jenen Assays, in denen Blocking Buffer II EGrade niedrigen Hinter-grund ermöglicht, dies auch reproduzierbar gewährleis-tet ist. Dies ist ein wesentlicher Vorteil gegenüber den sonst genannten Alternativen wie Fischgelatine, Molkepulver oder wie gezeigt auch den synthetischen Blockierern. Diese sind für die geringe Ergebnisrepro-duzierbarkeit aus den genannten Gründen bekannt.
Blocking Buffer II EGrade ermöglicht also die Kom-bination von Wirtschaftlichkeit und Reproduzierbarkeit für viele Assays. Aufgrund der gezeigten Kombination von guter Blockierung und hoher Reproduzierbarkeit gibt es damit erstmals eine echte Alternative zu den qualitativ hochwertigen Protein-Blockierern. Der er weiterte Vergleich nach Steinitz zeigt aber auch, dass selbst moderne Lösungen die Effizienz von hochwer-tigen Protein-Blockierern nicht erreichen und es daher in kritischen Anwendungen wie z.B. bei Pharma- Assays, Assays für Studien oder bei der Vermessung wertvoller Proben keine Alternativen gibt.
FazitFür viele Assays in den LifeSciences bietet AppliChem eine Alternative zu hochwertigen Protein-Blockieren. Blocking Buffer II EGrade hat das Potential, in vielen Forschungsanwendungen Wirtschaftlichkeit und gute Qualität zu verbinden.
Literatur [1] Steinitz, M. & Baraz, L. (2000) J. Immunol. Methods 238, 143-150. A
rapid method for estimating the binding of ligands to ELISA microwells.
[2] Steinitz, M. (2000) Anal. Biochem. 282, 232-238.
Quantitation of the Blocking Effect of Tween® 20 and
Bovine Serum Albumin in ELISA Mircrowells.
durch Arbeitszeiten eher im Promillebereich), dann wird klar, warum kaum einer gewillt ist, für diese Kosteneinsparungen auf Qualität zu verzichten.
Daher möchten wir im Bereich der Blockierung eine neue Möglichkeit vorstellen, auf vernünftige Weise Kosten zu sparen. Der vollkommen neuartige Blockie-rer Blocking Buffer II EGrade bietet diese Möglichkeit. Für diese Lösung aus industrieller Herstellung, von einem nach DIN ISO 9001:2000 zertifizierten Produ-zenten, die frei von jeglichen Serumproteinen und Peptid-basiert ist, ist die Herkunft der Rohstoffe kon-trolliert und die Blockierungseigenschaften von Charge zu Charge völlig einheitlich und reproduzierbar. Die Kosten sind geringer als bei Protein-basierten Blockie-rern. Die Qualität ist aber – anders als bei den synthe-tischen Blockierern – durchaus sehenswert, wenn-gleich nicht ganz so optimal wie bei Protein-basierten Blo ckierern wie Blocking Buffer I.
Analyt-unabhängiger VergleichDoch wie lässt sich nun tatsächlich die Blockierungs-effizienz und Qualität eines neuen Blockierers verläss-lich messen, so dass es nicht bei bloßen Vermutungen bleibt? Wir haben uns an den Arbeiten von Steinitz & Baraz (2000) und Steinitz (2000) methodisch orientiert, jedoch nicht die Phosphatase als Nachweisenzym, sondern die mittlerweile häufiger verwendete Peroxi-dase eingesetzt und die reine Betrachtung in Doppel-bestimmung, durch eine Achtfach-Bestimmung jedes einzelnen Messpunktes mit statistischer Betrachtung der Variationskoeffizienten (VKs) ergänzt.
Im Rahmen dieser Untersuchung wurden Blocking Buffer I als Blockierer basierend auf hochreinem und mehrfach gespaltenem Casein, Blocking Buffer III BSA als Blockierer auf BSA-Basis, der neuartige Blocking Buffer II EGrade, sowie ein kommerzieller, synthe-tischer Blockierer verglichen.
Auf jeweils blockierten Oberflächen in ELISA-Wells wurde (analog zu Steinitz (2000), jedoch mit Peroxida-se) ohne weitere Zusätze ein Detektorantikörper inku-biert. Nach Waschen wurde die Farbreaktion durchge-führt. Je geringer hiernach die optische Dichte (OD) ist, desto besser die Blockierung. Die Abb. 2. zeigt die Ergebnisse bei Blockierung über Nacht. Alle Varianten zeigen Blockierung. Die Qualität ist bei Protein-Blockierern klar überlegen, gut bei Blocking Buffer II EGrade und immer noch gut beim synthetischen Produkt. In Abb. 2a sind die ODs aufge-tragen, Abb. 2b zeigt ein Foto der blockierten ELISA-Platte. Hierbei ist in der linken Spalte die Kontrolle ohne jegliche Blockierung zu sehen. Die daneben liegenden Spalten sind analog zum Diagramm aufgetra-gen.

18 Immunoassay Puffer • AppliChem © 2011
Der Einsatz von gebrauchsfertigen, kommer-ziellen ELISA-Kits ist einfach und praktisch. Manchmal müssen aber doch selbst hergestellte ELISAs zum Einsatz kommen, weil käufliche Produkte mit den richtigen Antikörpern oder Charakte ristika, wie z.B. unpassende Detekti-onsgrenzen, nicht verfügbar sind. Die gebrauchs-fertigen ELISAs guter Lieferanten können über Jahre ohne Probleme bei 4°C gelagert werden. Mit selbst hergestellten ELISA-Kits sieht das ganz anders aus. Für jede neue Messung muss eine neue Platte beschichtet werden, da nach einigen Tagen die Qualität messbar nachlässt.
Woher kommen diese großen Unterschiede bezüglich der Lagerfähigkeit von ELISA Kits?Der Grund dafür ist in der Tatsache begründet, dass in der kommerziellen Kit-Produktion nach der Beschichtung nicht nur ein Blockierungsschritt durchgeführt wird, sondern auch eine Stabilisierung. Dieser einfach durch-zuführende Prozess wurde in der Industrie bereits vor 30 Jahren eingeführt. Zur Stabilisierung der Platten muss mit einer beschichtenden Stabilisie-rungslösung inkubiet werden. Dies entspricht quasi einem zweiten „Blockie-rungsschritt“. Bisher waren diese Stabilisierungslösungen in kleinen Mengen für den Labormaßstab nur nicht verfügbar. AppliChem bietet jetzt für alle Forschungslaboratorien diese Lösung in kleinen Abpackungen ab 50 ml an – AppliCoat Plate Stabilizer (Art.-Nr. A7708). Diese Stabilisierungslösung ist einfach zu handhaben und bietet sogar größere Vorteile im Vergleich zu irgendeinem Stabilisator aus der Industrie. Man erzielt bessere Stabilitäten während der Lagerung von Platten, beschichtet mit Antikörpern und Anti-genen, als die meisten anderen kommerziellen Produkte leisten können. Und es gibt noch einen weiteren Nutzen.
Zwei Nutzen mit einer LösungWenn Antikörper auf eine ELISA-Platte als Beschichtung aufgebracht werden, sind viele inaktiv. Wenn die Antikörper (oder irgendwelche anderen Proteine) in näheren Kontakt mit der Plastikoberfläche der ELISA-Platte geraten, kann es aufgrund der Oberflächen-Protein-Interaktion zu konformationellen Ver-änderungen kommen. Im Ergebnis werden die meisten auf der Oberfläche gebundenen Proteine entfaltet oder inaktiv. Nur 2-8 % aller Proteine erhalten nach der Beschichtung ihre Aktivität und können den Analyten binden. Dies ist in starkem Maße von den Oberflächeneigenschaften der EILSA-Platte ab-hängig und kann nicht nur von Charge zu Charge variieren, sondern sogar von Vertiefung zu Vertiefung.Gerade die Unterschiede zwischen den einzelnen Vertiefungen können durch ihre Einflüsse auf die Antikörper, den Assay beeinflussen. Wenn es die Mög-lichkeit gäbe die Antikörper zurückzufalten und den Antikörper vor kon-formationellen Änderungen während der Lagerung zu schützen, würde dies helfen die Variabilität in den Assayergebnissen zu reduzieren. Das ist der Hauptnutzen von AppliCoat Plate Stabilizer! Es unterstützt Antiköper und
Qualitätsverbesserung & höhere Stabilität von ELISAs
andere zur Beschichtung eingesetzte Proteine sich zurückzufalten und konserviert dann die aktive Konformation über einen langen Zeitraum. So gesehen hat das Reagenz also zwei Nutzen: 1. Rückfaltung der Antikörper und 2. Konservierung der richtigen Konformation während der Lagerung. Von diesen Nutzen wird in der Produktion hochwertiger ELISA-Kits Gebrauch gemacht, und das ab sofort auch in Forschungsanwendungen. Selbst mit AppliCoat Plate Stabilizer wird der Anteil aktiver Antikörper im Bereich von 2-8 % liegen, aber die großen Unterschiede von Vertiefung (well) zu Ver-tiefung werden in den meisten Assays minimiert. Die Effekte hängen von den verwendeten Antikörpern ab, aber wenn ELISAs validiert werden (z.B. nach der „Guidance for Industry: Bioanalytical Method Validation“, FDA, 2001 oder nach anderen Validierungsstrategien), können die Unterschiede in vielen Assays gemessen werden.Die positiven Effekte von AppliCoat Plate Stabilizer sind in Abb. 1 anhand eines klassischen Sandwich-ELISA-Assays mit einem monoklonalen Anti-körper dargestellt. Dieser monoklonale Antikörper hat seine Bindungsaktivi-tät nach der Beschichtung auf die Platte und Stabilisierung nur mit BSA verloren. In dieser Abbildung ist ein so genannter Hochtemperatur-„Stresstest“ bei 37°C über 84 Tage nach Zugabe der Stabilisierungslösung gezeigt. Das reale OD-Signal wurde ohne Normalisierung dargestellt, da andernfalls eine falsche Interpretation der Ergebnisse auftreten könnte. Es ist offensichtlich, dass bei Verwendung von AppliCoat Plate Stabilizer die Bindungsaktivität deutlich verbessert ist. Wenn diese Testplatte trocken bei +4°C gelagert worden wäre, würde dies einer Lagerzeit von 2 Jahren entsprechen.
Verbesserte Arbeitsabläufe für die meisten LaboratorienDurch diesen Vorteil längerer Lagerfähigkeit können neue Arbeitsabläufe in den Laboratorien eingeführt werden. Bisher mussten selbstgemachte ELISA-Platten jeweils bei Bedarf neu hergestellt werden. Das kann sich jetzt ändern. Je nachdem wieviele Platten benötigt werden, sie können alle gecoatet, geblockt und stabilisiert werden und dies für mindestens 3 bis 6 Monate. Die Platten werden bei 20°C oder 30°C für 60-120 Minuten getrocknet und kön-nen dann im Kühlschrank gelagert werden. Je nach Bedarf können sie direkt für Messungen eingesetzt werden. Dadurch, dass die Platten als ein Batch hergestellt werden, minimieren sich die Variationen von Platte zu Platte.Daher hat der neue Arbeitsablauf drei große Vorteile:1.) Weniger Arbeit, weil Beschichtung und Blockieren vieler Platten parallel
durchgeführt werden kann, statt jedesmal wieder neu anzufangen.2.) Niedrigere Messabweichungen von Platte zu Platte3.) Zeitersparnis und gegebenenfalls Verbesserung der Ergebnisqualität eines
Labors.
Industrielle Technologie jetzt auch für kleinere Forschungslabors verfügbarEntsprechend der Bedürfnisse kleinerer Forschungslaboratorien bietet AppliChem den AppliCoat Plate Stabilizer in den Packungsgrößen 50 ml, 125 ml und 500 ml an. Das Produkt kann in Kombination mit Standard-Blockierungsreagenzien wie Casein (z.B. Blocking Buffer I, A7099) oder BSA-basierten Lösungen (z.B. Blocking Buffer III BSA, A7252) und anderen Blockierern eingesetzt werden. Testen Sie es selbst!
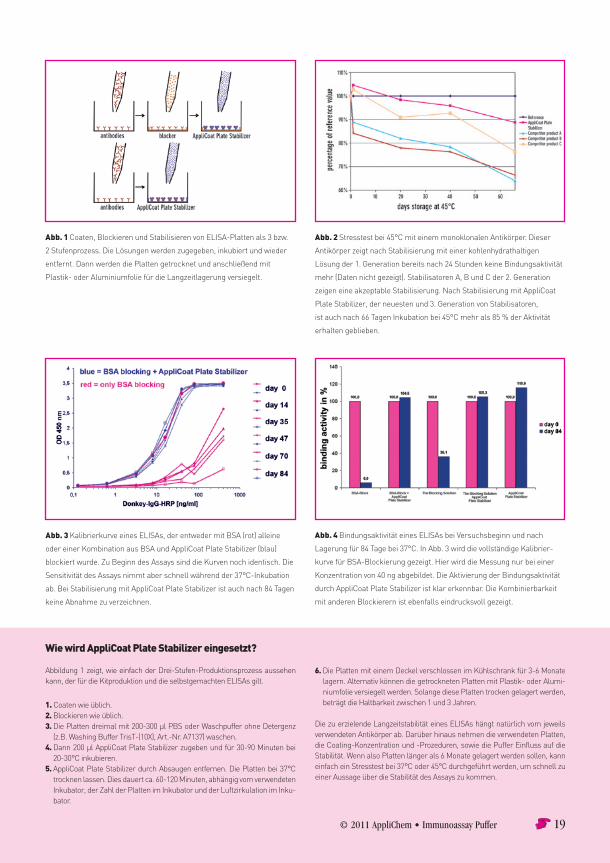
© 2011 AppliChem • Immunoassay Puffer 19
Qualitätsverbesserung & höhere Stabilität von ELISAs
Wie wird AppliCoat Plate Stabilizer eingesetzt?
Abb. 3 KalibrierkurveeinesELISAs,derentwedermitBSA(rot)alleine
odereinerKombinationausBSAundAppliCoatPlateStabilizer(blau)
blockiertwurde.ZuBeginndesAssayssinddieKurvennochidentisch.Die
SensitivitätdesAssaysnimmtaberschnellwährendder37°C-Inkubation
ab.BeiStabilisierungmitAppliCoatPlateStabilizeristauchnach84Tagen
keineAbnahmezuverzeichnen.
Abb. 1 Coaten,BlockierenundStabilisierenvonELISA-Plattenals3bzw.
2Stufenprozess.DieLösungenwerdenzugegeben,inkubiertundwieder
entfernt.DannwerdendiePlattengetrocknetundanschließendmit
Plastik-oderAluminiumfoliefürdieLangzeitlagerungversiegelt.
Abb. 4 BindungsaktivitäteinesELISAsbeiVersuchsbeginnundnach
Lagerungfür84Tagebei37°C.InAbb.3wirddievollständigeKalibrier-
kurvefürBSA-Blockierunggezeigt.HierwirddieMessungnurbeieiner
Konzentrationvon40ngabgebildet.DieAktivierungderBindungsaktivität
durchAppliCoatPlateStabilizeristklarerkennbar.DieKombinierbarkeit
mitanderenBlockierernistebenfallseindrucksvollgezeigt.
Abbildung1zeigt,wieeinfachderDrei-Stufen-Produktionsprozessaussehenkann,derfürdieKitproduktionunddieselbstgemachtenELISAsgilt.
1. Coatenwieüblich.2.Blockierenwieüblich.3.DiePlattendreimalmit200-300µlPBSoderWaschpufferohneDetergenz
(z.B.WashingBufferTrisT-(10X),Art.-Nr.A7137)waschen.4.Dann200µlAppliCoatPlateStabilizerzugebenundfür30-90Minutenbei
20-30°Cinkubieren.5.AppliCoatPlateStabilizerdurchAbsaugenentfernen.DiePlattenbei37°C
trocknenlassen.Diesdauertca.60-120Minuten,abhängigvomverwendetenInkubator,derZahlderPlattenimInkubatorundderLuftzirkulationimInku-bator.
6.DiePlattenmiteinemDeckelverschlossenimKühlschrankfür3-6Monatelagern.AlternativkönnendiegetrocknetenPlattenmitPlastik-oderAlumi-niumfolieversiegeltwerden.SolangediesePlattentrockengelagertwerden,beträgtdieHaltbarkeitzwischen1und3Jahren.
DiezuerzielendeLangzeitstabilitäteinesELISAshängtnatürlichvomjeweilsverwendetenAntikörperab.DarüberhinausnehmendieverwendetenPlatten,dieCoating-Konzentrationund-Prozeduren,sowiediePufferEinflussaufdieStabilität.WennalsoPlattenlängerals6Monategelagertwerdensollen,kanneinfacheinStresstestbei37°Coder45°Cdurchgeführtwerden,umschnellzueinerAussageüberdieStabilitätdesAssayszukommen.
Abb. 2 Stresstestbei45°CmiteinemmonoklonalenAntikörper.Dieser
AntikörperzeigtnachStabilisierungmiteinerkohlenhydrathaltigen
Lösungder1.Generationbereitsnach24StundenkeineBindungsaktivität
mehr(Datennichtgezeigt).StabilisatorenA,BundCder2.Generation
zeigeneineakzeptableStabilisierung.NachStabilisierungmitAppliCoat
PlateStabilizer,derneuestenund3.GenerationvonStabilisatoren,
istauchnach66TagenInkubationbei45°Cmehrals85%derAktivität
erhaltengeblieben.

20 Immunoassay Puffer • AppliChem © 2011
Bei uns ist weniger mehr
Der Umwelt zuliebe
Viele in der biomedizinischen Forschung einge-setzten Reagenzien sind ideale Nährlösungen für unerwünschte Keime (Bakterien, Pilze). Um deren Wachstum zu verhindern, wird autoklaviert, steril-filtriert, Antibiotika/Antimykotika oder so viel Gift zugesetzt, dass nichts mehr wachsen kann.
Einer dieser Zusätze ist Thimerosal, eine Quecksilber-haltige, umweltgefährliche Verbindung. In den Immuno-Puffern von AppliChem ist diese noch in einer Konzentration von 0,005 % enthalten.
Wir stellen auf einen ungiftigen Ersatzstoff um: ProClin® 300. Der Umwelt zuliebe.
Zu Ihrer Sicherheit
Damit Sie uns länger als Kunde erhalten bleiben, zur Information: Thimerosal ist als sehr giftig eingestuft. Die lethale Dosis (Ratte, s.c.) beträgt 98 mg/kg. Zum Vergleich, Ethidiumbromid liegt bei 110 mg/kg (Maus, s.c.).
In anderen Ländern ist Thimerosal schon als Zusatzstoff verboten, sofern es Ersatzstoffe gibt.
Für bessere Ergebnisse
Die Umstellung hat keinerlei negative Auswirkungen auf die Produktqualität.
CrossDown und die anderen Puffer aus der Immuno-Pufferlinie sorgen daher jetzt für noch mehr gute Laune. Ihre Immunoassays haben einfach eine bessere Qualität.

Bei uns ist weniger mehrprodukte
© 2011 AppliChem • Immunoassay Puffer 21
Antibody Stabilizer-PBS A7148
Stabilisierung von Antikörpern und Proteinen zur Langzeitlagerung A7148,0050 50 ml A7148,0125 125 ml
pH-Wert pH 7,4 ± 0,2Stabilisator enthält 0,1 % ProClin® 300Lagerung bei 2-8°C. Einfrieren vermeiden!*Haltbarkeit bei 2-8°C: 2 Jahre* Beim Einfrieren kann sich ein unlösliches Präzipitat bilden. Die Aktivität bleibt dadurch praktisch unbeeinflusst.
GebrauchsanweisungDer Antibody Stabilizer-PBS wurde zur Lagerung von Proteinen/Antikörpern bei 2-8°C in Lösung entwickelt. Der Puffer ist gebrauchsfertig und sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden.Der Antikörper/das Protein wird zur Lagerung mindestens 1:20 in Antibody Stabilizer-PBS verdünnt, kann aber auch deutlich stärker ver-dünnt werden. Typische Konzentrationen für gelagerte Antikörper liegen bei 400 bis 1000 ng/ml. Viele Antikörper lassen sich aber auch in sehr niedrigen Konzentrationen wie z.B. 80 ng/ml dauerhaft in Antibody Stabilizer-PBS lagern, ohne dass es zu wesentlichen Verlusten der Bindungsaktivität in den ersten Jahren kommt. Niedrige Konzentration während der Lagerung erspart wiederholte Vorverdünnung vor jeder Verwendung des Antikörpers.Bestandteile des Antibody Stabilizers-PBS haben bei Proteinen/Antikörpern strukturerhaltende Eigenschaften, die die Funktion der Proteine/Antikörper möglichst lange erhalten sollen. Die Lagerdauer der Proteine/Antikörper in dem Antibody Stabilizer-PBS hängt stark von den Eigenschaften und Konzentrationen der Proteine und Antikörper ab und kann daher nicht allgemeingültig voraus gesagt werden. Aus diesem Grund ist Antibody Stabilizer-PBS zunächst vom Anwender auf Eignung für seine speziellen Proteine/Antikörper zu testen. Konkrete Haltbarkeiten von Antikörpern lassen sich immer nur für einen speziellen Antikörper in der gewählten Konzentration angeben.Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. ProClin® ist ein eingetragenes Warenzeichen der Firma Rohm und Haas.
Antibody Stabilizer-Tris A7135
Stabilisierung von Antikörpern und Proteinen zur Langzeitlagerung A7135,0050 50 ml A7135,0125 125 ml
pH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1 % ProClin®300Lagerung bei 2-8°C. Einfrieren vermeiden!*Haltbarkeit at 2-8°C: 2 years* Beim Einfrieren kann sich ein unlösliches Präzipitat bilden. Die Aktivität bleibt dadurch praktisch unbeeinflusst.
GebrauchsanweisungDer Antibody Stabilizer-Tris wurde zur Lagerung von Proteinen/Antikörpern bei 2-8°C in Lösung entwickelt. Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Der Antikörper/das Protein wird zur Lagerung mindestens 1:20 in Antibody Stabilizer verdünnt, kann aber auch deutlich stärker verdünnt werden. Typische Konzentrationen für gelagerte Antikörper liegen bei 400 bis 1000 ng/ml.Viele Antikörper lassen sich aber auch in sehr niedrigen Konzentrationen wie z.B. 80 ng/ml dauerhaft in Antibody Stabilizer-Tris lagern, ohne dass es zu wesentlichen Verlusten der Bindungsaktivität in den ersten Jahren kommt.Niedrige Konzentration während der Lagerung erspart wiederholte Vorverdünnung vor jeder Verwendung des Antikörpers.Bestandteile des Antibody Stabilizer-Tris haben bei Proteinen/Antikörpern strukturerhaltende Eigenschaften, die die Funktion der Proteine/Antikörper möglichst lange erhalten sollen.Die Lagerdauer der Proteine/Antikörper in dem Antibody Stabilizer-Tris hängt stark von den Eigenschaften und Konzentrationen der Proteine und Antikörper ab und kann daher nicht allgemeingültig voraus gesagt werden. Aus diesem Grund ist Antibody Stabilizer zunächst vom Anwender auf Eignung für seine speziellen Proteine/Antikörper zu testen. Konkrete Haltbarkeiten von Antikörpern lassen sich immer nur für einen speziellen Antikörper in der gewählten Konzentration angeben.
ProClin® ist ein eingetragenes Warenzeichen der Firma Rohm und Haas.
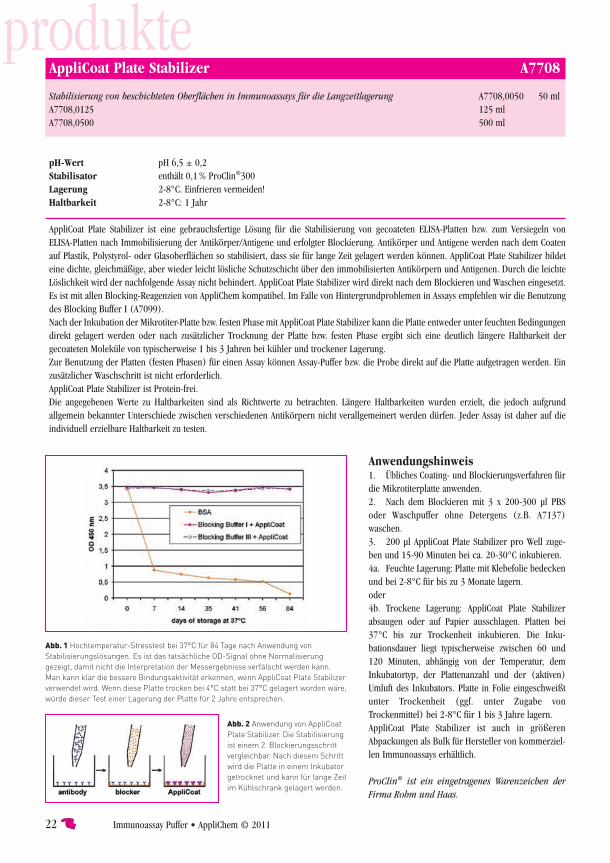
22 Immunoassay Puffer • AppliChem © 2011
AppliCoat Plate Stabilizer A7708
Stabilisierung von beschichteten Oberflächen in Immunoassays für die Langzeitlagerung A7708,0050 50 ml A7708,0125 125 ml A7708,0500 500 ml
pH-Wert pH 6,5 ± 0,2Stabilisator enthält 0,1 % ProClin®300Lagerung 2-8°C. Einfrieren vermeiden!Haltbarkeit 2-8°C: 1 Jahr
AppliCoat Plate Stabilizer ist eine gebrauchsfertige Lösung für die Stabilisierung von gecoateten ELISA-Platten bzw. zum Versiegeln von ELISA-Platten nach Immobilisierung der Antikörper/Antigene und erfolgter Blockierung. Antikörper und Antigene werden nach dem Coaten auf Plastik, Polystyrol- oder Glasoberflächen so stabilisiert, dass sie für lange Zeit gelagert werden können. AppliCoat Plate Stabilizer bildet eine dichte, gleichmäßige, aber wieder leicht lösliche Schutzschicht über den immobilisierten Antikörpern und Antigenen. Durch die leichte Löslichkeit wird der nachfolgende Assay nicht behindert. AppliCoat Plate Stabilizer wird direkt nach dem Blockieren und Waschen eingesetzt. Es ist mit allen Blocking-Reagenzien von AppliChem kompatibel. Im Falle von Hintergrundproblemen in Assays empfehlen wir die Benutzung des Blocking Buffer I (A7099).Nach der Inkubation der Mikrotiter-Platte bzw. festen Phase mit AppliCoat Plate Stabilizer kann die Platte entweder unter feuchten Bedingungen direkt gelagert werden oder nach zusätzlicher Trocknung der Platte bzw. festen Phase ergibt sich eine deutlich längere Haltbarkeit der gecoateten Moleküle von typischerweise 1 bis 3 Jahren bei kühler und trockener Lagerung. Zur Benutzung der Platten (festen Phasen) für einen Assay können Assay-Puffer bzw. die Probe direkt auf die Platte aufgetragen werden. Ein zusätzlicher Waschschritt ist nicht erforderlich.AppliCoat Plate Stabilizer ist Protein-frei. Die angegebenen Werte zu Haltbarkeiten sind als Richtwerte zu betrachten. Längere Haltbarkeiten wurden erzielt, die jedoch aufgrund allgemein bekannter Unterschiede zwischen verschiedenen Antikörpern nicht verallgemeinert werden dürfen. Jeder Assay ist daher auf die individuell erzielbare Haltbarkeit zu testen.
Abb. 1Hochtemperatur-Stresstestbei37°Cfür84TagenachAnwendungvonStabilisierungslösungen.EsistdastatsächlicheOD-SignalohneNormalisierunggezeigt,damitnichtdieInterpretationderMessergebnisseverfälschtwerdenkann.MankannklardiebessereBindungsaktivitäterkennen,wennAppliCoatPlateStabilizerverwendetwird.WenndiesePlattetrockenbei4°Cstattbei37°Cgelagertwordenwäre,würdedieserTesteinerLagerungderPlattefür2Jahreentsprechen.
Abb. 2AnwendungvonAppliCoatPlateStabilizer.DieStabilisierungisteinem2.Blockierungsschrittvergleichbar.NachdiesemSchrittwirddiePlatteineinemInkubatorgetrocknetundkannfürlangeZeitimKühlschrankgelagertwerden.
Anwendungshinweis1. Übliches Coating- und Blockierungsverfahren für die Mikrotiterplatte anwenden.2. Nach dem Blockieren mit 3 x 200-300 µl PBS oder Waschpuffer ohne Detergens (z.B. A7137) waschen.3. 200 µl AppliCoat Plate Stabilizer pro Well zuge-ben und 15-90 Minuten bei ca. 20-30°C inkubieren.4a. Feuchte Lagerung: Platte mit Klebefolie bedecken und bei 2-8°C für bis zu 3 Monate lagern.oder 4b. Trockene Lagerung: AppliCoat Plate Stabilizer absaugen oder auf Papier ausschlagen. Platten bei 37°C bis zur Trockenheit inkubieren. Die Inku-bationsdauer liegt typischerweise zwischen 60 und 120 Minuten, abhängig von der Temperatur, dem Inkubatortyp, der Plattenanzahl und der (aktiven) Umluft des Inkubators. Platte in Folie eingeschweißt unter Trockenheit (ggf. unter Zugabe von Trockenmittel) bei 2-8°C für 1 bis 3 Jahre lagern.AppliCoat Plate Stabilizer ist auch in größeren Ab packungen als Bulk für Hersteller von kommerziel-len Immunoassays erhältlich.
ProClin® ist ein eingetragenes Warenzeichen der Firma Rohm und Haas.
produkte

© 2011 AppliChem • Immunoassay Puffer 23
Blocking Buffer I A7099
Lösung zum Blockieren unspezifischer Bindungsstellen für ELISA, EIA & Western Blots A7099,0050 50 ml A7099,0125 125 ml A7099,0500 500 ml
Zusammensetzung niedermolekulares Casein mit NaCl und Tween®
pH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1% ProClin® 300Lagerung bei –20°C oder bei 2-8°C Haltbarkeit bei –20°C: 1 Jahr, wiederholtes Einfrieren ist möglich bei 2-8°C: 6 Monate
Gebrauchsanweisung
Der Blocking Buffer I dient zur Absättigung von freien Bindungsstellen von Kunststoffoberflächen oder anderen proteinbindenden Oberflächen zur Reduzierung von unspezifischen Bindungen an die Oberfläche. Durch ein besonderes Verfahren wurde das Casein des Blocking Buffers I in verschiedenen Molekulargewichten hergestellt und somit wird die Blockierungseffizienz verbessert. Die Einsatzgebiete sind neben dem ELISA, EIA oder Western Blot auch Immuno-PCR und Protein Arrays, bei denen Protein-adsorbierende Oberflächen geblockt werden müssen.
Der Puffer sollte nach dem Auftauen und unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Der Puffer ist gebrauchsfertig. Mehrfaches Einfrieren und Auftauen ist möglich. Nach der Immobilisierung des Fängerantikörpers u.ä. wird der Puffer unver-dünnt in die Wells der Mikrotiterplatte gegeben. Je nach verwendeter Oberfläche sind die nötigen Inkubationszeiten des Blockierungsschrittes unterschiedlich und sollten durch den Anwender optimiert werden. Wir empfehlen eine Blockierung über Nacht bei +4°C, doch auch kürzere Inkubationszeiten sind möglich.
Nach der Blockierung wird die Oberfläche gewaschen, um den Blocking Buffer I zu entfernen und die Oberfläche (z.B. Mikrotiterplatte oder Blotting Membran) kann für die nächsten Arbeitsschritte des Anwenders verwendet werden.
Blocking Buffer I Andere Blockierer
Reduktion des Hintergrundes Extrem effiziente Allgemein bekannte Hintergrund-Reduktion – Hintergrund-Reduktion selbst in kritischen Assays
Verwendbarkeit Für alle Immunoassays Einige Produkte nur für ELISA oder Western blotting, viele verschiedene Spezialprodukte
Einfache Anwendung Ready-to-use Für einige Produkte Vorverdünnung in anderen Puffern empfohlen
Anwendung mit Anwendbar mit allen bekannten Anwendbarkeit hängt vom Produkt ab; verschiedenen Nachweismethoden Nachweis-Methoden; negatives „Quenching“ mit Fluoreszenz-
sehr gute Ergebnisse mit Peroxidasen, Farbstoffen muss bei einigen Produkten Phosphatasen und Fluoreszenzmarkierungen vorher geprüft werden
Effekte auf Validierung Positive Effekte, reduzierte Abweichungen, Normale Effekte auf Validierung; (z.B. für neue Hintergrundeffekte werden vermieden, keine reduzierten Variationen gezeigt FDA-Richtlinien in der Industrie) Validierungskriterien werden einfach erfüllt
Lagerung und Transport Kühlung und Einfrieren für Langzeitlagerung, Kühlen und Einfrieren für wiederholtes Einfrieren und Auftauen möglich, Langzeitlagerung, aber kein Kühltransport erforderlich viele Produkte benötigen Kühltransport

Blocking Buffer II EGrade A7516
Lösung zum Blockieren unspezifischer Bindungsstellen für ELISA, EIA, RIA, A7516,0125 125 mlWestern Blots, Protein Arrays und Immuno-PCR A7516,0500 500 ml
Zusammensetzung Peptid-basierte Blockierungslösung frei von Serumproteinen, frei von BSA, Phosphat-freipH-Wert pH 7,0 ± 0,2Stabilisator enthält 0,1% ProClin® 300Lagerung bei –20°C oder bei 2-8°C Haltbarkeit bei –20°C: 1 Jahr, wiederholtes Einfrieren ist möglich bei 2-8°C: 6 Monate
Blocking Buffer II EGrade ist die ökonomischste Lösung (Economical Grade) zur Blockierung unspezifischer Bindungsstellen. Es ist DIE kostengünstige Alternative zur Casein-basierten Blocking Solution I. • effektives Blockieren • einfach und ökonomisch • BSA-frei
Gebrauchsanweisung
Blocking Buffer II EGrade verhindert in vielen Assays unspezifische und unerwünschte Bindungen an die Oberfläche. Durch Inkubation der Oberfläche (z.B. Mikrotiterplatte oder Western Blot Membran) werden freie Bindungsstellen der Oberfläche durch die Bestandteile von Blocking Buffer II EGrade abgesättigt, so dass bei den nachfolgenden Arbeitsschritten die unerwünschte Anbindung des Zielanalyten bzw. des Detektorantikörpers an die Oberfläche minimiert wird. Hierdurch wird der Hintergrund reduziert. Blocking Buffer II EGrade ist für viele Assays eine ökonomische und einfache Alternative zu BSA-haltigen Lösungen. Durch die BSA-Freiheit sind übliche Probleme wie BSA-Interaktionen und Kreuzreaktivitäten des Blockierers bei Blocking Buffer II EGrade von vorne herein ausgeschlossen.Wenn mit Blocking Buffer II EGrade keine zufriedenstellenden Ergebnisse in einem Assay erzielt werden oder häufig Realproben wie Blut, Serum und Gewebeproben vermessen werden, empfehlen wir die Verwendung unserer hocheffektiven Casein-basierten Blocking Solution I (A7099).Hintergrund und unspezifische Bindung kann nicht nur an Oberflächen auftreten, sondern auch zwischen Antikörpern und Bestandteilen der zu vermessenden Proben. In diesen Fällen kann nur durch einen Assay-Puffer, der während der immunologischen Nachweisreaktion der Antikörper eingesetzt wird, ein zufriedenstellendes Ergebnis erzielt werden. Hierfür empfehlen wir CrossDown Buffer (Art.-Nr. A6485), um optimale Ergebnisse zu erzielen. CrossDown Buffer wirkt vergleichbar einem Bindungsfilter, der hochaffine Bindungen der Antikörper unbeeinflusst zulässt und unspezifische Bindungen vermindert.Blocking Buffer II EGrade ist ein ready-to-use Reagenz und wird wie die üblichen Blockierungslösungen eingesetzt. Blocking Buffer II EGrade wird unverdünnt auf die zu blockierende Oberfläche gegeben und inkubiert.Der Puffer sollte nach dem Auftauen und unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Der Puffer ist gebrauchsfertig. Mehrfaches Einfrieren und Auftauen ist möglich.
ProClin® ist ein eingetragenes Warenzeichen der Firma Rohm und Haas.
1. Falls die Mikrotiterplatte zuvor mit detergenshaltigen Reagenzien benetzt wurde, muss die Platte 3 x mit einem detergensfreien Waschpuffer (z.B. Washing Buffer TrisT-, Art.-Nr. A7137) gewa-schen werden. Falls nur Coating Buffer (Art.-Nr. A7136 oder A7150) verwendet wurde, muss die Coating-Lösung lediglich abgesaugt oder ausgeklopft werden.
2. Zugabe von 200-300 µl Blocking Buffer II EGrade pro Well. Inkubation bei Raumtemperatur für 1-4 Stunden oder über Nacht (1 Stunde genügt häufig). Anmerkung: Durch Schütteln der Platte mit 600-900 rpm kann die Blockierungszeit je nach Anwendung weiter verringert werden. Die Blockierungszeit ist abhängig von den Eigenschaften der zu blockierenden Oberfläche und sollte daher ausgetestet werden.
3. Absaugen oder Ausklopfen des Blocking Buffer II EGrade; 3 x Waschen der Mikrotiterplatten mit einem Waschpuffer, der ein nicht ionisches Detergens enthält, z.B. Washing Buffer TrisT+ (Art.-Nr. 7158).
Absättigen / Blockieren
Mik
rotit
erpl
atte
n
Blot
ting-
Mem
bran
en 1. Falls die Blotting Membran zuvor mit detergenshaltigen Reagenzien benetzt wurde, muss die Membran 3 x mit einem detergensfreien Waschpuffer (z.B. Washing Buffer TrisT-, Art.-Nr. A7137) gewaschen werden.
2. Eintauchen der Blotting Membran in Blocking Buffer II EGrade. Inkubation bei Raumtemperatur und unter leichtem Schütteln für 1-4 Stunden oder über Nacht (1 Stunde genügt häufig). Die Blockierungszeit ist abhängig von den Eigenschaften der zu blockierenden Membran und sollte daher ausgetestet werden.
3. a) Dreimaliges Waschen der Blotting Membran mit einem Waschpuffer, der ein nicht ionisches Detergens enthält, z.B. Washing Buffer TrisT+ (Art.-Nr. 7158).
oder b) Direkte Zugabe des Detektions-Antikörpers zu Blocking Buffer
II EGrade und Fortsetzung der Inkubation zur Detektion der geblotteten Substanzen.
24 Immunoassay Puffer • AppliChem © 2011
produkte

Two – Dream Teams
© 2011 AppliChem • Immunoassay Puffer 25
Blocking Buffer III BSA A7252
Lösung zum Blockieren unspezifischer Bindungsstellen für ELISA, EIA, Immuno-PCR, A7252,0125 125 mlImmunhistochemie, Protein-Arrays & Western Blots A7252,0500 500 ml
Zusammensetzung Standard Blocking Reagenz mit BSA und Tween®
pH-Wert pH 7,4 ± 0,2Stabilisator enthält 0,1% ProClin® 300Lagerung bei –20°C oder bei 2-8°C Haltbarkeit bei –20°C: 1 Jahr, wiederholtes Einfrieren ist möglich bei 2-8°C: 6 Monate
Gebrauchsanweisung
Blocking Buffer III BSA dient zur Absättigung von freien Bindungsstellen von Kunststoffoberflächen oder anderen proteinbindenden Oberflächen zur Reduzierung von unspezifischen Bindungen an die Oberfläche. Blocking Buffer III BSA ist der Standard-Blockierer für viele Anwendungsfälle. Sofern eine Blockierung auf BSA-Basis für einen Assay die notwendige Blockierungseffizienz erreicht, ist Blocking Buffer III BSA die günstige Alternative zu universell einsetzbaren und aufwändigen Blockierern. Die Einsatzgebiete sind neben dem ELISA, EIA oder Western Blot auch Immuno-PCR, Protein Arrays und die Immunhistochemie, bei denen Protein-adsorbierende Oberflächen geblockt werden müssen.
Der Puffer sollte nach dem Auftauen und unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Der Puffer ist gebrauchsfertig. Mehrfaches Einfrieren und Auftauen ist möglich. Nach der Immobilisierung des Fängerantikörpers u.ä. wird der Puffer unverdünnt in die Wells der Mikrotiterplatte bzw. auf die jeweilige zu blockierende Oberfläche gegeben. Je nach verwendeter Oberfläche sind die nötigen Inkubationszeiten des Blockierungsschrittes unterschiedlich und sollten durch den Anwender optimiert werden. Wir empfehlen eine Blockierung über Nacht bei 4°C, doch auch kürzere Inkubationszeiten sind möglich. Nach der Blockierung wird die Oberfläche gewa-schen, um Blocking Buffer III BSA zu entfernen und die Oberfläche (z.B. Mikrotiterplatte oder Blotting Membran) kann für die nächsten Arbeitsschritte des Anwenders verwendet werden.
Sofern es trotz Verwendung von Blocking Buffer III BSA zu Hintergrund durch unspezifische Bindungen kommt, empfehlen wir den Einsatz von Blocking Buffer I (Artikel-Nr. A7099). Blocking Buffer I wird durch chemische Modifikation von hochreinem Casein gewonnen. Sie hat durch die Modifikation eine vielfach höhere Blockierungseffizienz als Standard-Blockierer und bietet damit maximale Sicherheit in der Anwendung. Blocking Buffer I ist universell für alle Assays einsetzbar, während Blocking Buffer III BSA vorwiegend bei unproblematischen Assays gute Ergebnisse erzielt.
Hintergrund und unspezifische Bindung kann nicht nur an Oberflächen auftreten, sondern auch zwischen Antikörpern und Bestandteilen der zu vermessenden Proben. In diesen Fällen kann nur durch einen Assay-Puffer, der während der immunologischen Nachweisreaktion der Antikörper eingesetzt wird, ein zufriedenstellendes Ergebnis erzielt werden. Hierfür empfehlen wir CrossDown Buffer (Artikel-Nr. A6485), um optimale Ergebnisse zu erzielen. CrossDown Buffer wirkt wie ein Bindungsfilter, der hochaffine Bindungen der Antikörper unbeeinflusst zulässt und unspezifische Bindungen vermindert.

26 Immunoassay Puffer • AppliChem © 2011
produkteBlocking Reagenz CA A3409,0010
Blocking Reagenz für Hybridisierungen und Western Blots
Lagerung: Raumtemperatur
Das Blocking Reagenz CA wird in Hybridisierungs- und Nachweisreaktionen für nicht-radioaktive Nukleinsäureproben und in Western blots eingesetzt. Wenn Immunoassays und Hybridisierungsassays, wie dot blots, Western blots, Southern blots oder Northern blots durchgeführt werden, gibt es unspezifische Bindungen, die einen unerwünschten „Hintergrund“ verursachen. Um dies unspezifische Bindung zu reduzie-ren wird das Blocking Reagenz CA eingesetzt, um freigebliebene Oberflächen nach Immobilisierung des spezifischen Proteins oder nach der Hybridisierung mit nicht-radioaktiven Proben zu blockieren. Das Blocking Reagenz CA verbessert die Sensitivität und reduziert den Hintergrund.
Achtung Magermilch hemmt die Streptavidin-Biotin Interaktion aufgrund seines Biotin-Gehalts!
Protokoll Proteine Für Blotting-Anwendungen, wie Western blots und dot blots, gebe 0,2 % (w/v) Blocking Reagenz CA in TBST oder PBST; erhitze auf 75-80°C im Wasserbad oder der Mikrowelle und rühre gut, bis das Reagenz gelöst ist. Das Blocking Reagenz CA löst sich unter Bildung einer milchig-trüben Lösung. Diese Lösung wird für die Blockierung und die Verdünnung von Antikörpern eingesetzt.
Achtung Verwende Blocking Reagenz CA in PBST nicht für alkalische Phosphatase Konjugat-Verdünnungen
Nukleinsäuren Für Hybridisierungen stelle eine 0,2 %ige Lösung (w/v) Blocking Reagenz CA in Tris-gepufferter Salzlösung (100 mM Tris · HCl pH 7,5; 600 mM NaCl) her. Erhitze auf 60-65°C im Wasserbad oder der Mikrowelle und rühre gut bis sich das Reagenz klar gelöst hat. Setze das Reagenz für die Blockierung nach den Waschschritten ein und vor der Inkubation in irgendeiner Enzyme-Konjugat-Lösung (z.B. Streptavidin-HRP, Streptavidin-AP).
Optimierung der Inkubationszeit,benötigt für die Blockierung mit Blocking Reagenz CA
1. Schneide 7 kleine Vierecke aus Nitrozellulose oder einer anderen geeigneten Membran.2. Markiere jedes Viereck mit einem Kugelschreiber in 10 Minuten Schritten (60, 50, 40, 30, 20, 10) und eines ohne
Blockierung.3. Lege das erste Viereck (60) in einige Milliliter der Blocking Reagenz CA-Lösung und gebe dann sukzessive
Vierecke in 10 Minuten Intervallen zu.4. Wasche alle Vierecke in TBST, PBST oder Tris-gepufferter Salzlösung (100 mM Tris · Cl pH 7,5; 600 mM NaCl).5. Verdünne den sekundären Antikörper oder Streptavidin (HRP-konjugiert) in Blocking Reagenz CA-Lösung.6. Inkubiere auf dem Schüttler für 1 Stunde.7. Spüle mit TBST, PBST oder Tris-gepufferter Salzlösung dreimal, jeweils 10 Minuten.8. Weise jetzt mit dem Chemilumineszenz Nachweiskit für Meerrettichperoxidase (Art.-Nr. A3417) nach.9. Evaluiere die Hintergrund-Intensität für jedes Viereck. Wähle die Inkubationszeit, die den niedrigsten Hintergrund ergibt.

© 2011 AppliChem • Immunoassay Puffer 27
Coating Buffer C pH 9.6 A7150
Puffer zur Immobilisierung von Fänger-Antikörpern und Proteinen A7150,0125 125 ml
Zusammensetzung Puffer auf Carbonat-Basis, 10-fach konzentriertpH-Wert pH 9,6 ± 0,2Stabilisator Der Puffer wird ohne bakterizide/fungizide Zusätze geliefert, damit er für eine Reihe unter- schiedlicher Systeme eingesetzt werden kann, in denen bestimmte Stabilisatoren – besonders bei der Immobilisierung – störend wirken können.Lagerung bei 2-8°C. Die Arbeitsverdünnung (s.u.) sollte sofort verwendet werden. Haltbarkeit 10-fach Konzentrat: bei 2-8° C: 1 Monat, 10-fach Konzentrat: bei –20° C: mindestens 3 Monate
GebrauchsanweisungDer Coating Buffer C pH 9,6 dient zur adsorptiven Immobilisierung von Proteinen und Antikörpern auf Kunststoffoberflächen (z.B. Mikrotiterplatten) oder anderen proteinbindenden Oberflächen. Die Einsatzgebiete sind beispielsweise ELISA, EIA und Protein Arrays.Durch die Lagerung bei 2-8°C können Salzkristalle ausfallen. Zur Herstellung der Arbeitslösung ist der Coating Buffer C pH 9,6 aus diesem Grund zuvor auf Raumtemperatur zu erwärmen, wodurch sich die ausgefallenen Salze wieder lösen. Der Puffer sollte unmittel-bar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Die Arbeitslösung wird aus der Stammlösung durch eine 1:10-Verdünnung mit destilliertem oder vollentsalztem Wasser hergestellt und sollte am selben Tag verwendet werden. Die zu immobilisie-renden Proteine/Antikörper werden nun in gewünschter Weise mit der Arbeitslösung verdünnt und nach gleichmäßiger Durchmischung für die Immobilisierung eingesetzt. Übliche Immobilisierungskonzentrationen von Fängerantikörpern bei ELISA-Anwendungen liegen zwischen 0,5 µg/ml und 2 µg/ml.Je nach verwendeter Oberfläche und je nach Art der zu immobilisierenden Proteine/Antikörper sind die nötigen Inkubationszeiten der Immobilisierung unterschiedlich und sollten durch den Anwender optimiert werden. Je nach Protein/Antikörper könnte der Coating Buffer PBS pH 7,4 (Artikel-Nr. A7136) für die Immobilisierung geeigneter sein, da der pH-Wert die räumliche Struktur und damit die Immobilisierungseigenschaften der Proteine/Antikörper beeinflusst. Für eine Optimierungsstrategie eines neu zu entwickelnden Immunoassays empfehlen wir das Testen beider Coating Buffer.
Coating Buffer PBS pH 7.4 A7136
Puffer zur Immobilisierung von Fänger-Antikörpern und Proteinen A7136,0125 125 ml
Zusammensetzung Puffer auf PBS-Basis, 10-fach konzentrierttpH-Wert pH 7,4 ± 0,2Stabilisator Der Puffer wird ohne bakterizide/fungizide Zusätze geliefert, damit er für eine Reihe unter- schiedlicher Systeme eingesetzt werden kann, in denen bestimmte Stabilisatoren – besonders bei der Immobilisierung – störend wirken können.Lagerung bei 2-8°C. Die Arbeitsverdünnung (s.u.) sollte sofort verwendet werden.Haltbarkeit 10-fach Konzentrat: bei 2-8°C: mindestens 3 Monate, 10-fach Konzentrat: bei –20°C: 12 Monate
GebrauchsanweisungDer Coating Buffer PBS pH 7,4 dient zur adsorptiven Immobilisierung von Proteinen und Antikörpern auf Kunststoffoberflächen (z.B. Mikrotiterplatten) oder anderen proteinbindenden Oberflächen. Die Einsatzgebiete sind beispielsweise ELISA, EIA und Protein Arrays.Durch die Lagerung bei 2-8°C können Salzkristalle ausfallen. Zur Herstellung der Arbeitslösung ist der Coating Buffer pH PBS 7,4 aus diesem Grund zuvor auf Raumtemperatur zu erwärmen, wodurch sich die ausgefallenen Salze wieder lösen. Der Puffer sollte unmittel-bar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Die Arbeitslösung wird aus der Stammlösung durch eine 1:10-Verdünnung mit destilliertem oder vollentsalztem Wasser hergestellt und sollte am selben Tag verwendet werden. Die zu immobilisie-renden Proteine/Antikörper werden nun in gewünschter Weise mit der Arbeitslösung verdünnt und nach gleichmäßiger Durchmischung für die Immobilisierung eingesetzt. Übliche Immobilisierungskonzentrationen von Fängerantikörpern bei ELISA-Anwendungen liegen zwischen 0,5 µg/ml und 2 µg/ml. Je nach verwendeter Oberfläche und je nach Art der zu immobilisierenden Proteine/Antikörper sind die nötigen Inkubationszeiten der Immobilisierung unterschiedlich und sollten durch den Anwender optimiert werden. Je nach Protein/Antikörper könnte der Coating Buffer C pH 9,6 (Artikel-Nr. A7150) für die Immobilisierung geeigneter sein, da der pH-Wert die räumliche Struktur und damit die Immobilisierungseigenschaften der Proteine/Antikörper beeinflusst. Für eine Optimierungsstrategie eines neu zu entwickelnden Immunoassays empfehlen wir das Testen beider Coating Buffer.

28 Immunoassay Puffer • AppliChem © 2011
produkteCrossDown Buffer A6485
Immunoassay-Puffer zur Minimierung unspezifischer Bindungen, Kreuzreaktivitäten und Matrixeffekten A6485,0050 50 ml A6485,0125 125 ml A6485,0500 500 ml
pH-Wert pH 7,2 ± 0,2 Phosphat-frei, ready-to-useStabilisator enthält 0,1 % ProClin® 300Lagerung bei –20°C oder bei 2-8°C Haltbarkeit bei –20°C: 1 Jahr, wiederholtes Einfrieren ist möglich bei 2-8°C: 6 Monate
Gebrauchsanweisung
Der CrossDown Buffer wurde zur Minimierung von unspezifischen Bindungen, Kreuzreaktivitäten und Matrixeffekten bei Immunoassays ent-wickelt. Seine Einsatzgebiete sind neben dem ELISA, EIA oder Western Blot auch Immuno-PCR, Protein Arrays und Multianalyt-Immunoassays. Bei Fluoreszenz-Immunoassays kommt es mitunter zu Quench-Effekten von Fluorescein-Farbstoffen durch Bestandteile der Immunoassays – oder möglicherweise auch durch Puffer-Bestandteile. Daher empfehlen wir die Verwendung von Oyster* – (Denovo Biolabels), CyDye** – (Amersham) oder Alexa*** – Fluoreszenzfarbstoffen (Molecular Probes).Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Der CrossDown Buffer wird an Stelle des üblichen Probenpuffers eingesetzt. Standards und Proben können beliebig, jedoch mindestens 1 : 2 in dem CrossDown Buffer verdünnt werden. Es ist jedoch darauf zu achten, dass die Standards und Proben gleich behandelt werden. Bei einigen Immunoassays könnte auch eine Vorverdünnung des CrossDown Buffer mit voll entsalztem Wasser vor Verwendung sinnvoll sein, falls bei unverdünnter Verwendung auch die spezifische Bindung reduziert wird. Dieses ist vereinzelt bei niederaffinen Antikörpern zu beobachten.Die Inkubation des Detektorantikörpers kann ebenfalls in gewünschter Verdünnung in dem Puffer durchgeführt werden. Dies ist dann empfehlenswert, wenn der Detektorantikörper im Verdacht steht, für unspezifische Bindungen bzw. Kreuzreaktivitäten verantwortlich zu sein. Bei Verwendung eines polyklonalen Detektorantikörpers, der zur Vermeidung von unspezifischen Bindungen bzw. Kreuzreaktivitäten mit CrossDown Buffer verdünnt wird, könnte es zur Verbesserung des Assays erforderlich sein, die Konzentration des Detektorantikörpers moderat zu erhöhen.Bei besonders störanfälligen und empfindlichen Assays – beispielsweise Immuno-PCR – kann der CrossDown Buffer zusätzlich auch als Waschpuffer eingesetzt werden.
CrossDown in der FACS-Analyse: CrossDown ersetzt bei der FACS-Analyse den normalerweise verwendeten Assaypuffer und wird genauso verwendet, wie der ursprüngliche Assaypuffer. Falls CrossDown „zu stark“ wirken sollte (d.h. eventuell Verringerung auch des spezifischen Signals auftritt) kann CrossDown mit dem ursprünglichen Assaypuffer verdünnt werden (1 : 2 bis 1 : 10 Verdünnung). Alternativ können ande-re physiologische Puffer wie z.B. PBS oder Hepes für die Verdünnung eingesetzt werden. Am einfachsten ist aber meistens die Verdünnung mit dem ursprünglich verwendeten Assaypuffer.
* Oyster Dye ist eingetragenes Warenzeichen der Firma Denovo Biolabels **CyDye ist eingetragenes Warenzeichen der Firma Amersham Bioscience ***Alexa Fluor Dye ist eingetragenes Warenzeichen der Firma Molecular Probes
Literatur [1] Miller, J.J. (2004) Clinical Laboratory International 28, (2), 14-17
[2] Kusnezow, W., Hoheisel, J.D. (2003) J. Mol. Recognit. 16, 165-176
[3] Patton, W.F. (2000) Electrophoresis 21, 1123-1144
[4] MacBeath, G. (2002) Nat. Genet. 32, 526-532
[5] Miller, J.J., Valdes, R.Jr. (1992) J Clin Immunoassays 15, 97-107
[6] Wood, W.G. (1991) Scand. J. Clin. Lab. Invest. Suppl. 205, 105-112
[7] Kricka, L.J. (1999) Clinical Chemistry 45,(7), 942-956
[8] Span, P.N., Grebenchtchikov N., Geurts-Moespot, J., Sweep, C.G.J. (2003)
Clinical Chemistry 49,(10), 1708-1709
FAQ

© 2011 AppliChem • Immunoassay Puffer 29
FAQWas ist das besondere an CrossDown?
CrossDown ist ein neuentwickelter Probenpuffer für Immunoassays (insbesondere ELISA, Western Blots, Proteinchips und Immuno-PCR), der unerwünschte Störeffekte vermindert bzw. verhindert. Unerwünschte Störeffekte sind beispielsweise unspezi-fische Bindungen, Kreuzreaktivitäten und Matrixeffekte. Die hoch-affine, spezifische Bindung des Antikörpers zum Analyten bleibt erhalten, während unerwünschte Bindungen des Antikörpers ver-hindert werden.
Wie muß ich den CrossDown-Puffer einsetzen? CrossDown wird als Verdünnungspuffer der Realprobe (z.B. humanes Serum oder Plasma) und/oder als Verdünnungspuffer des Detektorantikörpers verwendet. Das hängt davon ab, wo der Störeffekt vermutet wird. Falls eine unspezifische Bindung oder Kreuzreaktivität beim Detektorantikörper vermutet wird, sollte z.B. der Detektorantikörper bevorzugt in CrossDown verdünnt und eingesetzt werden.
Ersetzt CrossDown einen Blockierungspuffer für Oberflächen (z.B. Western Blot-Membranen oder ELISA-Platten)?Nein! CrossDown ist kein Blockierungspuffer von Oberflächen,
sondern sollte zur Verdünnung der Realprobe und der
Assayantikörper verwendet werden. Für die Blockierung
der Oberflächen empfehlen wir unseren Blocking Buffer I (Artikel-Nr. A7099).
Ist es egal, ob polyklonale oder monoklonale Antikörper mit
CrossDown verwendet wer-den?Ja. CrossDown kann mit poly-klonalen oder monoklona-len Antikörpern verwendet werden. Bei polyklonalen Antikörpern ist jedoch folgen-
des zu beachten: Polyklonale Antikörper sind im Prinzip eine
Mischung aus einer Vielzahl von Antikörpern mit unterschiedlichen
Affinitäten. CrossDown läßt nur hoch affine Bindungen zu, die niederaffinen Bindungen
werden weitgehend unterdrückt. Daher kann es erforderlich sein, die Konzentration des polyklonalen
Detektorantikörpers bei Verwendung von CrossDown mode-rat zu erhöhen. Durch Erhöhung der Antikörperkonzentration
erhöht sich die Konzentration der hochaffinen Antikörper, während die niederaffinen Antikörper weiterhin durch CrossDown abgefangen werden. Ob eine Erhöhung der Konzentration im Assay nötig ist, hängt demnach sehr stark von der Qualität der Antikörper ab.
Warum hilft CrossDown-Puffer bei Matrixeffekten?Ein großer Teil der sogenannten Matrixeffekte basiert auf uner-wünschten niederaffinen Bindungen von Matrixbestandteilen (z.B. Serumproteinen bei Proben aus humanem Serum) an den Analyten oder an den Antikörper. Der Analyt kann durch Proteine oder andere Bestandteile der Probenmatrix maskiert sein, so dass der Antikörper nicht an dem Zielepitop binden kann. CrossDown verhindert diese Maskierung, so dass der Antikörper wie gewünscht den Analyten bindet. Auch die Antikörper können durch Matrixbestandteile maskiert werden. Hier verhindert CrossDown die Maskierung und hebt vor-handene Maskierungen auf.
Kann ich den Puffer mehrfach einfrieren und auftauen?Es ist problemlos möglich, den Puffer mehrfach einzufrieren und wieder aufzutauen. Es ist aber wichtig, dass der Puffer vor Benutzung nochmals aufgeschüttelt wird, um eine vollständige Durchmischung des Puffers sicherzustellen.
Muß der Puffer verdünnt oder unverdünnt eingesetzt werden?CrossDown ist eine „ready-to-use“-Lösung. Die Probe bzw. der Detektorantikörper wird also direkt in den Puffer verdünnt. Lediglich in kompetitiven Assays und einigen Anwendungen der Immunhistochemie kann es sinnvoll sein, CrossDown vor der Anwendung mit Wasser oder einem physiologischen Puffer zu ver-dünnen. In diesem Fall muß der Anwender über eine entsprechende Optimierung ein ideales Mischungsverhältnis ermitteln.
Beeinflussen Pufferbestandteile die Farbreaktion?Die enzymatische Aktivität der alkalischen Phosphatase oder der Peroxidase werden durch CrossDown nicht negativ beeinflußt. Ganz im Gegenteil – Anwender berichteten von einer Erhöhung der Enzymativität, wenn der gelabelte Detektorantikörper mit CrossDown inkubiert wurde.
Kann man CrossDown mit Fluoreszenz- Farbstoffen zusammen– beispielsweise bei Proteinchip -Anwendungen – einsetzen?Ja. Für Proteinchip-Anwendungen liegen positive Ergebnisse vor. Bei Cyan-Farbstoffen (z.B. Cy5, Cy3, Oyster-Farbstoffe) wurde sogar bei einigen Anwendungen eine Erhöhung der Fluoreszenz beobachtet.
Häufig gestellte Fragen zu CrossDown Buffer
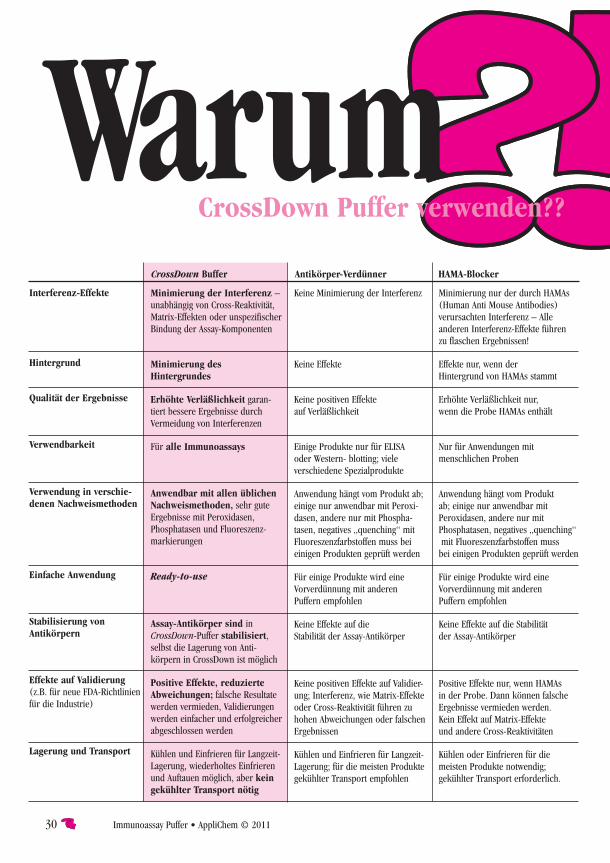
30 Immunoassay Puffer • AppliChem © 2011
Warum CrossDown Puffer verwenden??
Interferenz-Effekte
Hintergrund
Qualität der Ergebnisse
Verwendbarkeit
Verwendung in verschie-denen Nachweismethoden
Einfache Anwendung
Stabilisierung von Antikörpern
Effekte auf Validierung (z.B. für neue FDA-Richtlinien für die Industrie)
Lagerung und Transport
Minimierung der Interferenz – unabhängig von Cross-Reaktivität, Matrix-Effekten oder unspezifischer Bindung der Assay-Komponenten
Minimierung des Hintergrundes
Erhöhte Verläßlichkeit garan-tiert bessere Ergebnisse durch Vermeidung von Interferenzen
Für alle Immunoassays
Anwendbar mit allen üblichen Nachweismethoden, sehr gute Ergebnisse mit Peroxidasen, Phosphatasen und Fluoreszenz- markierungen
Ready-to-use
Assay-Antikörper sind in CrossDown-Puffer stabilisiert, selbst die Lagerung von Anti- körpern in CrossDown ist möglich
Positive Effekte, reduzierte Abweichungen; falsche Resultate werden vermieden, Validierungen werden einfacher und erfolgreicher abgeschlossen werden
Kühlen und Einfrieren für Langzeit-Lagerung, wiederholtes Einfrieren und Auftauen möglich, aber kein gekühlter Transport nötig
Keine Minimierung der Interferenz
Keine Effekte
Keine positiven Effekte auf Verläßlichkeit
Einige Produkte nur für ELISA oder Western- blotting; viele verschiedene Spezialprodukte
Anwendung hängt vom Produkt ab; einige nur anwendbar mit Peroxi- dasen, andere nur mit Phospha-tasen, negatives „quenching“ mit Fluoreszenzfarbstoffen muss bei einigen Produkten geprüft werden
Für einige Produkte wird eine Vorverdünnung mit anderen Puffern empfohlen
Keine Effekte auf die Stabilität der Assay-Antikörper
Keine positiven Effekte auf Validier- ung; Interferenz, wie Matrix-Effekte oder Cross-Reaktivität führen zu hohen Abweichungen oder falschen Ergebnissen
Kühlen und Einfrieren für Langzeit-Lagerung; für die meisten Produkte gekühlter Transport empfohlen
Minimierung nur der durch HAMAs (Human Anti Mouse Antibodies) verursachten Interferenz – Alle anderen Interferenz-Effekte führen zu flaschen Ergebnissen!
Effekte nur, wenn der Hintergrund von HAMAs stammt
Erhöhte Verläßlichkeit nur, wenn die Probe HAMAs enthält
Nur für Anwendungen mit menschlichen Proben
Anwendung hängt vom Produkt ab; einige nur anwendbar mit Peroxidasen, andere nur mit Phosphatasen, negatives „quenching“ mit Fluoreszenzfarbstoffen muss bei einigen Produkten geprüft werden
Für einige Produkte wird eine Vorverdünnung mit anderen Puffern empfohlen
Keine Effekte auf die Stabilität der Assay-Antikörper
Positive Effekte nur, wenn HAMAs in der Probe. Dann können falsche Ergebnisse vermieden werden. Kein Effekt auf Matrix-Effekte und andere Cross-Reaktivitäten
Kühlen oder Einfrieren für die meisten Produkte notwendig; gekühlter Transport erforderlich.
CrossDown Buffer Antikörper-Verdünner HAMA-Blocker

© 2011 AppliChem • Immunoassay Puffer 31
CrossDown Peroxidase-Stabilisator A8625
l Verdünnungsreagenz für Ein-Schritt-Inkubationsassays A8625,0050 50 mll Peroxidase-Konjugat-Stabilisator A8625,0125 125 mll Ersatz für HAMA-Blockierer A8625,0500 500 ml
pH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1 % ProClin®300 (ProClin® ist ein eingetragenes Warenzeichen der Firma Rohm und Haas)Lagerung 2-8°C. Beim Einfrieren kann sich ein Präzipitat bilden, das die Aktivität aber nicht beeinträchtigt.
Trotzdem: NICHT einfrieren.Haltbarkeit 24 Monate
BeschreibungEs gibt das Bestreben ELISA-Kits mit möglichst wenig Arbeitsschritten und kurzer Inkubation zu ermöglichen, besonders in der industriellen ELISA-Kit-Produktion. Ein Ansatz dafür sind Einschritt-Assays. Das bedeutet, dass die Probe direkt in der ELISA-Platte mit dem Detektor-Konjugat in einem Schritt inkubiert wird, anstelle der sequentiellen Durchführung, bei der erst die Probe in der Platte inkubiert wird. Danach wird gewaschen und erst in einem weiteren Schritt das Detektor-Konjugat zugegeben und inkubiert. Wenn man nun auf Einschritt-Assays geht, erhöht sich durch die fehlenden Waschschritte massiv das Risiko fast aller Arten von Interferenzen. Damit leidet die Robustheit und Präzision von Assays. Der CrossDown-Effekt wirkt dem nun entgegen und es lassen sich robuste und zuverlässige ELISAs im Einschritt-Verfahren durchführen. Das Detektor-Konjugat wird also in CrossDown Peroxidase-Stabilisator gelagert und dann diese Lösung direkt auf die Probe in der ELISA-Platte gegeben.
GebrauchsanweisungCrossDown Peroxidase-Stabilisator ist ein Verdünnungspuffer für die Lagerung von Peroxidase-Konjugaten. Er wird anstelle eines Stabilisators bzw. Verdünnungspuffers für Peroxidase-Konjugate eingesetzt. Zusätzlich lassen sich HAMA-Blocker ersetzen. Konjugate, die direkt in ihrer nutzbaren Endkonzentration in CrossDown Peroxidase-Stabilisator gelagert werden, können ohne weitere Verdünnungsschritte direkt im Assay eingesetzt werden. Dies können sowohl Antikörper-Peroxidase-Konjugate als auch Neutravidin- oder Strepavidin-Peroxidase-Konjugate sein. Typische Konzentrationen der Konjugate liegen zwischen 40 und 500 ng/ml.In der ELISA-Kit-Produktion dient der Stabilisator dazu, dass man das Detektor-Konjugat in geringer Endkonzentration bei 2-8°C mit dem Kit über Jahre (in der Regel werden 2 bis 2,5 Jahre angestrebt) lagern kann. Durch die geringe Konzentration des Konjugates entfällt für den Anwender des ELISA-Kits ein zusätzlicher Verdünnungsschritt vor der Verwendung des Konjugates (früher wurde das Konjugat häufig in einer Stammlösung mitgeliefert, die dann vor Durchführung des Assays mit einem Assay-Puffer verdünnt werden musste). Durch die damit automatisch bedingte höhere Konzentration des Konjugates war dieses länger haltbar. Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden.Zuverlässigkeit und Stabilität in einem: CrossDown Peroxidase-Stabilisator ist eine Kombination des CrossDown Puffers sowie eines neuarti-gen Stabilisierers für Peroxidase-Konjugate. Dadurch erhält man extrem lange Lagerstabilität zusammen mit dem CrossDown Effekt in einer Lösung.Der CrossDown Peroxidase-Stabilisator-Effekt:CrossDown Peroxidase-Stabilisator wurde zur Minimierung von unspezifischen Bindungen, Kreuzreaktivitäten und Matrixeffekten bei Immunoassays entwickelt. Störungen durch HAMAs (humane Anti-Maus Antikörper), Rheumafaktoren sowie andere endogene Störer können wirksam reduziert werden.
Auftreten von Signalreduktion:In einigen Fällen kann es zur leichten Reduktion des gewünschten Signales kommen, weil CrossDown Peroxidase-Stabilisator nieder- und mittelaffine Bindungen reduziert. Sofern nieder- bis mittelaffine Antikörper oder polyklonale Antikörper (die in der Regel auch nieder- und mittelaffin bindende Anteile enthalten) verwendet werden, kann daher eine leichte Reduktion des Signals auftreten. Im Falle von polyklonalen Antikörpern lässt sich durch moderate Erhöhung der Antikörperkonzentration auch die Anzahl hochaffiner Antikörper erhöhen, um die gewünschte Signalstärke wieder zu erreichen. Dabei bleiben die nieder- und mittelaffinen Bindungen durch CrossDown Peroxidase-Stabilisator unterdrückt.
Abb. 1: DieHaltbarkeiteinesHRP-Antikörper-KonjugateswurdeineinemELISAalsSignaldergebundenenPeroxidasegemessenundals%-WerteinesfrischverdünntenHRP-Antikörper-Konjugatesangege-ben.DasDetektor-KonjugatwurdeineinerKonzentrationvon40ng/mleingesetzt.
produkte
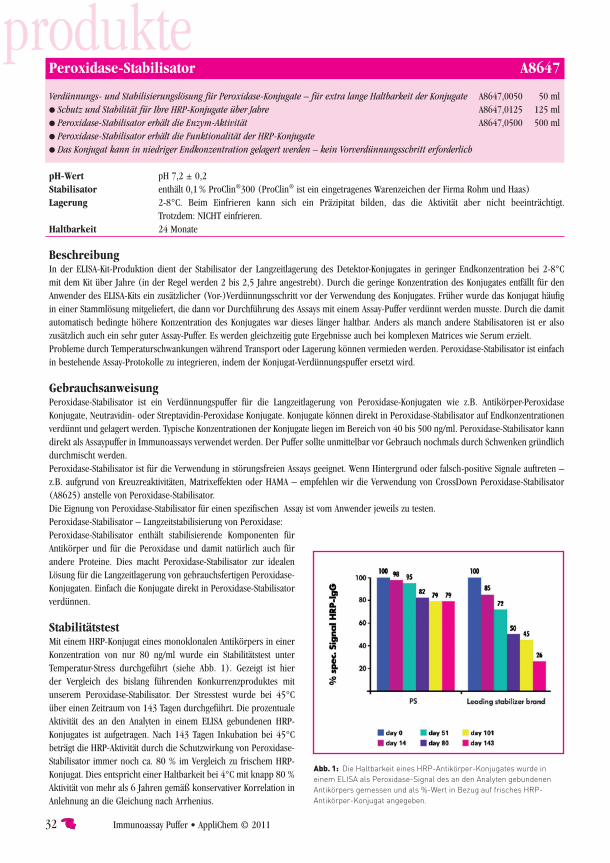
32 Immunoassay Puffer • AppliChem © 2011
Peroxidase-Stabilisator A8647
Verdünnungs- und Stabilisierungslösung für Peroxidase-Konjugate – für extra lange Haltbarkeit der Konjugate A8647,0050 50 mll Schutz und Stabilität für Ihre HRP-Konjugate über Jahre A8647,0125 125 mll Peroxidase-Stabilisator erhält die Enzym-Aktivität A8647,0500 500 mll Peroxidase-Stabilisator erhält die Funktionalität der HRP-Konjugatel Das Konjugat kann in niedriger Endkonzentration gelagert werden – kein Vorverdünnungsschritt erforderlich
pH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1 % ProClin®300 (ProClin® ist ein eingetragenes Warenzeichen der Firma Rohm und Haas)Lagerung 2-8°C. Beim Einfrieren kann sich ein Präzipitat bilden, das die Aktivität aber nicht beeinträchtigt.
Trotzdem: NICHT einfrieren.Haltbarkeit 24 Monate
BeschreibungIn der ELISA-Kit-Produktion dient der Stabilisator der Langzeitlagerung des Detektor-Konjugates in geringer Endkonzentration bei 2-8°C mit dem Kit über Jahre (in der Regel werden 2 bis 2,5 Jahre angestrebt). Durch die geringe Konzentration des Konjugates entfällt für den Anwender des ELISA-Kits ein zusätzlicher (Vor-)Verdünnungsschritt vor der Verwendung des Konjugates. Früher wurde das Konjugat häufig in einer Stammlösung mitgeliefert, die dann vor Durchführung des Assays mit einem Assay-Puffer verdünnt werden musste. Durch die damit automatisch bedingte höhere Konzentration des Konjugates war dieses länger haltbar. Anders als manch andere Stabilisatoren ist er also zusätzlich auch ein sehr guter Assay-Puffer. Es werden gleichzeitig gute Ergebnisse auch bei komplexen Matrices wie Serum erzielt. Probleme durch Temperaturschwankungen während Transport oder Lagerung können vermieden werden. Peroxidase-Stabilisator ist einfach in bestehende Assay-Protokolle zu integrieren, indem der Konjugat-Verdünnungspuffer ersetzt wird.
GebrauchsanweisungPeroxidase-Stabilisator ist ein Verdünnungspuffer für die Langzeitlagerung von Peroxidase-Konjugaten wie z.B. Antikörper-Peroxidase Konjugate, Neutravidin- oder Streptavidin-Peroxidase Konjugate. Konjugate können direkt in Peroxidase-Stabilisator auf Endkonzentrationen verdünnt und gelagert werden. Typische Konzentrationen der Konjugate liegen im Bereich von 40 bis 500 ng/ml. Peroxidase-Stabilisator kann direkt als Assaypuffer in Immunoassays verwendet werden. Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden.Peroxidase-Stabilisator ist für die Verwendung in störungsfreien Assays geeignet. Wenn Hintergrund oder falsch-positive Signale auftreten – z.B. aufgrund von Kreuzreaktivitäten, Matrixeffekten oder HAMA – empfehlen wir die Verwendung von CrossDown Peroxidase-Stabilisator (A8625) anstelle von Peroxidase-Stabilisator.Die Eignung von Peroxidase-Stabilisator für einen spezifischen Assay ist vom Anwender jeweils zu testen.Peroxidase-Stabilisator – Langzeitstabilisierung von Peroxidase:Peroxidase-Stabilisator enthält stabilisierende Komponenten für Antikörper und für die Peroxidase und damit natürlich auch für andere Proteine. Dies macht Peroxidase-Stabilisator zur idealen Lösung für die Langzeitlagerung von gebrauchsfertigen Peroxidase-Konjugaten. Einfach die Konjugate direkt in Peroxidase-Stabilisator verdünnen.
StabilitätstestMit einem HRP-Konjugat eines monoklonalen Antikörpers in einer Konzentration von nur 80 ng/ml wurde ein Stabilitätstest unter Temperatur-Stress durchgeführt (siehe Abb. 1). Gezeigt ist hier der Vergleich des bislang führenden Konkurrenzproduktes mit unserem Peroxidase-Stabilisator. Der Stresstest wurde bei 45°C über einen Zeitraum von 143 Tagen durchgeführt. Die prozentuale Aktivität des an den Analyten in einem ELISA gebundenen HRP-Konjugates ist aufgetragen. Nach 143 Tagen Inkubation bei 45°C beträgt die HRP-Aktivität durch die Schutzwirkung von Peroxidase-Stabilisator immer noch ca. 80 % im Vergleich zu frischem HRP-Konjugat. Dies entspricht einer Haltbarkeit bei 4°C mit knapp 80 % Aktivität von mehr als 6 Jahren gemäß konservativer Korrelation in Anlehnung an die Gleichung nach Arrhenius.
Abb. 1: DieHaltbarkeiteinesHRP-Antikörper-KonjugateswurdeineinemELISAalsPeroxidase-SignaldesandenAnalytengebundenenAntikörpersgemessenundals%-WertinBezugauffrischesHRP-Antikörper-Konjugatangegeben.
produkte

© 2011 AppliChem • Immunoassay Puffer 33
Sample Buffer T- A7101
Proben- und Antikörper-Verdünnungspuffer für Immunoassays speziell Immunohistochemie A7101,0050 50 ml A7101,0125 125 ml A7101,0500 500 ml
Zusammensetzung enthält keine DetergenzienpH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1 % ProClin® 300Lagerung bei –20°C oder bei 2-8°C Haltbarkeit bei –20°C: 1 Jahr, wiederholtes Einfrieren ist möglich bei 2-8°C: 6 Monate
Gebrauchsanweisung
Der Sample Buffer T- wird zur Verdünnung der Probe und/oder zur Verdünnung des Antikörpers (z.B. des Detektorantikörpers) bei Immunoassays verwendet. Er enthält keine Detergenzien und wird daher vorwiegend für die immunhistochemischen Anwendungen eingesetzt, bei denen Detergenzien stören könnten.
Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Die Primär- und Sekundärantikörper können direkt in gewünschter Weise in Sample Buffer T- verdünnt werden.
Falls es trotz Verwendung des Sample Buffers zu erhöhtem Background oder unerwünschten Kreuzreaktivitäten kommt, empfehlen wir die Verwendung des CrossDown Buffer (Artikel-Nr. A6485), der als besonderer Probenpuffer und Antikörper-Verdünnungspuffer Matrixeffekte, unspezifische Bindungen und Kreuzreaktivitäten minimiert bzw. verhindert.
Sample Buffer T+ A7134
Proben- und Antikörper-Verdünnungspuffer für Immunoassays A7134,0050 50 ml A7134,0125 125 ml A7134,0500 500 ml
Zusammensetzung enthält Tween® pH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1 % ProClin® 300Lagerung bei –20°C oder bei 2-8°C Haltbarkeit bei –20°C: 1 Jahr, wiederholtes Einfrieren ist möglich bei 2-8°C: 6 Monate
Gebrauchsanweisung
Der Sample Buffer T+ wird zur Verdünnung der Probe und/oder zur Verdünnung des Antikörpers (z.B. des Detektorantikörpers) bei Immunoassays verwendet. Seine Einsatzgebiete sind neben dem ELISA, EIA oder Western Blot auch Immuno-PCR, Protein Arrays und Multianalyt-Immunoassays.
Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Standards und Proben können direkt in gewünschter Weise in Sample Buffer T+ verdünnt werden. Es ist jedoch darauf zu achten, dass die Standards und Proben gleich behandelt werden.
Falls es trotz Verwendung des Sample Buffers T+ zu erhöhtem Background oder unerwünschten Matrixeffekten kommt, empfehlen wir die Verwendung des CrossDown Buffer (Artikel-Nr. A6485), der als besonderer Proben-und Antikörper-Verdünnungspuffer Matrixeffekte, unspezifische Bindungen und Kreuzreaktivitäten minimiert bzw. verhindert.

34 Immunoassay Puffer • AppliChem © 2011
produkteStripping Buffer I A7140
Stripping-Puffer für Western-Blots für mehrfaches Reprobing A7140,0050 50 ml A7140,0125 125 ml Zusammensetzung enthält kein β-Mercaptoethanol oder DTTpH-Wert pH 2,8 ± 0,2Stabilisator enthält 0,1% ProClin® 300Lagerung bei 2-8°C oder–20°C. Haltbarkeit bei 2-8°C: 6 Monate bei –20°C: 12 Monate
Gebrauchsanweisung
Der Stripping Buffer I ist gebrauchsfertig.
Der Stripping Buffer I dient zur Entfernung von Reaktionslösung sowie der Primär- und Sekundärantikörper bei Western Blots. Nach dem Stripping kann die Western Blot-Membran einer weiteren Detektion mit Antikörpern unterzogen werden.
Die Blot-Membran wird zur Entfernung der Antikörper in eine Schale oder ein Gefäß mit Stripping Buffer I gelegt. Die Membran wird unter leichtem Schütteln ca. 30 bis 60 Minuten bei Raumtemperatur inkubiert.
Nach dem Stripping wird die Membran mit Washing Buffer gewaschen. Bei Detektion mit alkalischer Phosphatase darf der Waschpuffer keine Phosphate enthalten. Die Zielproteine auf der Western Blot-Membran können nun erneut mit Antikörpern detektiert werden.
Wichtig: die genannten Inkubationsbedingungen sind Richtwerte, die vom Anwender angepasst werden müssen. Inkubationszeiten sind abhängig von den Eigenschaften der verwendeten Antikörper.
Washing Buffer TrisT- (10X) A7137
Waschpuffer zur Anwendung in ELISA, EIA, Western Blots und speziell Immunhistochemie A7137,0500 500 ml Zusammensetzung Puffer auf Tris-Basis ohne Detergenzien, mit 350 mM NaClpH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1% ProClin® 300Lagerung bei –20°C oder bei 2-8°C Die Arbeitsverdünnung (s.u.) sollte sofort verwendet werden.Haltbarkeit bei –20°C: 1 Jahr bei 2-8°C: 6 Monate
Gebrauchsanweisung
Der Washing Buffer TrisT- wird als Waschpuffer bei immunhistochemischen Anwendungen eingesetzt, wenn in speziellen Fällen mit einem Waschpuffer gearbeitet werden muß, der frei von Detergenzien ist.
Durch die Lagerung bei 2-8°C oder bei –20°C können Salzkristalle ausfallen. Zur Herstellung der Arbeitslösung ist der Washing Buffer TrisT- aus diesem Grund zuvor auf Raumtemperatur zu erwärmen, wodurch sich die ausgefallenen Salze wieder lösen. Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Die Arbeitslösung wird aus der Stammlösung durch eine 1:10-Verdünnung mit destilliertem oder salzarmen Wasser hergestellt und sollte am selben Tag verwendet werden.

35
Washing Buffer TrisT+ (10X) A7158
Waschpuffer zur Anwendung in ELISA, EIA und Western Blots A7158,0500 500 ml Zusammensetzung Puffer auf Tris-Basis mit Tween® und 350 mM NaClpH-Wert pH 7,2 ± 0,2Stabilisator enthält 0,1% ProClin® 300Lagerung bei –20°C oder bei 2-8°C Die Arbeitsverdünnung (s.u.) sollte sofort verwendet werden.Haltbarkeit bei –20°C: 1 Jahr bei 2-8°C: 6 Monate
Gebrauchsanweisung
Der Washing Buffer TrisT+ wird als Waschpuffer bei Immunoassays eingesetzt. Durch die Waschschritte werden bei Immunoassays unge-bundene und überschüssige Komponenten entfernt, die sonst den Assay stören würden. Die Einsatzgebiete sind neben dem ELISA, EIA oder Western Blot auch Immuno-PCR, Protein Arrays und Multianalyt-Immunoassays. Er kann in automatisierten Washern in Abhängigkeit der Salztoleranz des Washers verwendet werden.
Achtung: Unbedingt Spezifikation des jeweiligen Herstellers beachten!
Durch die Lagerung bei 2-8°C oder bei –20°C können Salzkristalle ausfallen. Zur Herstellung der Arbeitslösung ist der Washing Buffer TrisT+ aus diesem Grund zuvor auf Raumtemperatur zu erwärmen, wodurch sich die ausgefallenen Salze wieder lösen. Der Puffer sollte unmittelbar vor Gebrauch nochmals durch Schwenken gründlich durchmischt werden. Die Arbeitslösung wird aus der Stammlösung durch eine 1:10-Verdünnung mit destilliertem oder salzarmen Wasser hergestellt und sollte am selben Tag verwendet werden.
Speziell für die Immunhistochemie bieten wir den Washing Buffer TrisT- ohne Tween sowie ohne jegliche andere Detergenzien an (Artikel-Nr. A7137).

36 Immunoassay Puffer • AppliChem © 2011
Blocking-Reagenzien Artikel-Nr
Albumin acetyliert A0845Albumin Fraktion V (pH 7,0) Blotting grade A6588Denhardt's - Lösung (50X) BioChemica A2248Denhardt's-Pulvermischung (für 50X Stammlösung) BioChemica A3792Dextransulfat 500 - Natriumsalz BioChemica A2250Dextransulfat 500 - Natriumsalz für die Molekularbiologie A4970Gelatine gepulvert reinst Ph. Eur., NF A1693Heparin - Natriumsalz A3004Magermilchpulver A0830Polyvinylpyrrolidon (K90) für die Molekularbiologie A2260Salmon sperm DNA - Natriumsalz A2160Salmon sperm DNA - Natriumsalz (sonifiziert) A2159
Transfermembranen Artikel-Nr
Reprobe Nitrocellulose supported 0,22 µm Transfermembran A5237Reprobe Nitrocellulose supported 0,45 µm Transfermembran A5242Pure Nitrocellulose unsupported 0,22 µm Transfermembran A5250Pure Nitrocellulose unsupported 0,45 µm Transfermembran A5239Pure Nylon Neutral Transfermembran 0,22 µm (30 cm x 3 m) A4399Pure Nylon Neutral Transfermembran 0,45 µm A5248Reprobe Nylon Positiv-geladene Transfermembran 0,45 µm (30 cm x 3 m) A5255PVDF-Star Transfermembran 0,45 µm A5243
Broschüre Transfer Membranes (englisch)
sonstige Produkte Artikel-Nr
Chemilumineszenz-Nachweiskit für Meerrettich-Peroxidase A3417Natriumazid reinst A1430Peroxidase aus Meerrettich EIA und Immunology Grade I A3615Peroxidase aus Meerrettich EIA und Immunology Grade II A3771Streptavidin ultrapure A1495Thimerosal BioChemica A1278
Weiterführende LiteraturArnold M. Raem & Peter Rauch (Hrsg.) Immunoassays 2007 Elsevier Spektrum Akademischer Verlag (ISBN 3-8274-1636-1)
David Wild (Ed.): The Immunoassay Handbook. 3. Auflage. Elsevier Science Publishing Company, Amsterdam, Boston, Oxford 2005, ISBN 0-08-044526-8
Werner Luttmann, Kai Bratke, Michael Küpper, Daniel Myrtek: Der Experimentator: Immunologie. 2. Auflage. Spektrum Akademischer Verlag, Heidelberg 2006, ISBN 3-8274-1730-9
Verwandte Produkte

infotainingGet your free copy – www.applichem.com
TransfermembranenWir liefern eine Auswahl an Transfer-membranen, die für die Analyse von RNA, DNA und Proteinen entwickelt und getestet wurden. Alle Produkt informationen und die Protokolle finden Sie in der Broschüre „Transfer Membranes“.
AgarosenFast alle gelelektrophoretischen Anwendungen können mit Agarose-Gelen statt Acrylamid-Gelen durchgeführt werden. Egal welche DNA-Fragmentgrößen, Proteine oder RNA, ob analytische oder präparative Gele und anschließendes Blotten, mit Agarosen können alle Assays gemacht werden.
Gelelektrophorese-GrößenstandardsBei uns im Programm: gebrauchsfertige DNA- und Proteinmarker und zusätzlich lyophilisierte DNA- Marker. Alles darüber in unsrer Broschüre „Gel Electrophoresis Size Marker“.
Kostenfrei anfordern – www.applichem.com
Biologische Puffer Es gibt eigentlich kein Experiment, in dem nicht irgendeine Puffersubstanz eingesetzt wird. Die Broschüre „Biological Buffers“ gibt einen Überblick über die Eigenschaften, die Auswahlkriterien und nützliche Tipps zur Anwendung biolo gischer Puffer.
DetergenzienDetergenzien sind mehr als nur Luftblasen. Worauf es bei der Auswahl ankommt lesen Sie in unserer Broschüre „Detergents“.
Safety First: Mykoplasmen in der Zellkultur?Viele Zellkulturen sind mit Mykoplasmenkontaminiert. Wir bieten neben einemNachweis-Kit der Mykoplasmen -kontamina tion auch die Anti biotika zur Behandlung der Zellkulturen und Reagen-zien zur vorbeugenden Reinigung der CO2-Inkubatoren und Wasserbäder.

Darmstadt hat eine weitere Topadresse:AppliChem GmbH Ottoweg 4 D - 64291 Darmstadt Fon +49 6151 9357-0 Fax +49 6151 9357-11
eMail [email protected] internet www.applichem.com
4t M
atth
es +
Tra
ut ·
Darm
stadt Immunoassay
Puffer
A71,E
01/2011
A70,D