
Pharmakokinetisch/pharmakodynamische Untersuchung zur in ... · Referat: Bei intraabdominellen...
92
Pharmakokinetisch/pharmakodynamische Untersuchung zur in vitro Aktivtität von Levofloxacin und Moxifloxacin gegenüber B. fragilis und E. coli in Mono- und Mischkultur Dissertation zur Erlangung des akademischen Grades Dr. med. an der Medizinischen Fakultät der Universität Leipzig eingereicht von: Stefanie Rossi, geb. Hoffmann geboren am 18.03.1980 in Bad Langensalza angefertigt am: Institut für Medizinische Mikrobiologie und Infektionsepidemiologie der Universität Leipzig Betreuer: PD Dr. med. Reiner Schaumann Beschluss über die Verleihung des Doktorgrades vom: 24.09.2013
Transcript of Pharmakokinetisch/pharmakodynamische Untersuchung zur in ... · Referat: Bei intraabdominellen...

Pharmakokinetisch/pharmakodynamische Untersuchung zur in vitro
Aktivtität von Levofloxacin und Moxifloxacin gegenüber
B. fragilis und E. coli in Mono- und Mischkultur
Dissertation
zur Erlangung des akademischen Grades
Dr. med.
an der Medizinischen Fakultät
der Universität Leipzig
eingereicht von:
Stefanie Rossi, geb. Hoffmann
geboren am 18.03.1980 in Bad Langensalza
angefertigt am:
Institut für Medizinische Mikrobiologie und Infektionsepidemiologie der Universität Leipzig
Betreuer:
PD Dr. med. Reiner Schaumann
Beschluss über die Verleihung des Doktorgrades vom:
24.09.2013

"Wenn man denkt, es geht nicht mehr, hat man immer noch zwei Drittel seiner Kräfte."
Dr. Horst Köhler
Für David, Emma und Luis - Ich liebe Euch.

Inhaltsverzeichnis
I
I Bibliographische Beschreibung IV
II Abkürzungsverzeichnis V
1 Einleitung 1
1.1 Definition von Pharmakokinetik (PK) und Pharmakodynamik (PD) der
antimikrobiellen Therapie 1
1.2 Definition von Wirkmechanismus, Wirktyp, Wirkspektrum und Wirkintensität von
Antibiotika 1
1.3 Minimale Hemmkonzentration (MHK) 3
1.3.1 Methoden zur Bestimmung der MHK 3
1.3.2 Einteilung der Grenzwerte 4
1.4 Pharmakokinetisch/pharmakodynamisches Modell (PK/PD-Modell) 5
1.5 Postantibiotischer Effekt 7
1.6 Fluorchinolone 8
1.6.1 Levofloxacin 11
1.6.2 Moxifloxacin 13
1.7 Resistenzsituation, Formen und Mechanismen der Resistenz 15
1.8 Physiologische Bakterienflora des gastrointestinalen Trakts 17
1.9 Epidemiologie, Klinik und Therapie von intraabdominellen Infektionen 18
1.10 E. coli - Eigenschaften und Rolle als Krankheitserreger 21
1.11 B. fragilis - Eigenschaften und Rolle als Krankheitserreger 22
2 Zielstellung 24
3 Material 25
3.1 Geräte und Arbeitsplatz 25
3.2 Laborbedarf 25
3.3 Nährmedien zur Anzucht der verwendeten Bakterienkulturen und
Bakteriensuspensionen sowie Materialien zur Bestimmung der MHK 26

Inhaltsverzeichnis
II
4 Methoden 28
4.1 Untersuchte Bakterienstämme 28
4.2 Anzucht der Bakterienkulturen 28
4.3 Untersuchung zur Wachstumshemmung von vier E. coli Stämmen auf
supplementiertem Columbia-Gentamicin-Agar 29
4.4 Bestimmung der MHK von Levofloxacin und Moxifloxacin 29
4.5 PK/PD-Untersuchung von Levofloxacin und Moxifloxacin 30
4.6 Herstellung der Antibiotikasuspensionen 32
4.6.1 Herstellung der Inokula für die Versuche 33
4.6.2 Versuchsdurchführung 34
4.6.3 Ablesung der Versuchsergebnisse und Berechnung der Abtötungsraten 36
4.6.4 Berechnung der pharmakologischen Indizes 37
4.6.5 Statistische Datenanalyse 38
5 Ergebnisse 39
5.1 Wachstumshemmung von vier E. coli Stämmen auf supplementiertem Columbia-
Gentamicin-Agar und Beschreibung der Bakterienstämme 39
5.2 Ergebnisse der MHK-Bestimmung 40
5.3 Aktivität von Levofloxacin und Moxifloxacin im in vitro PK/PD-Modell 41
5.3.1 Aktivität von Levofloxacin gegenüber vier E. coli Stämmen in Monokultur unter
aeroben und anaeroben Bedingungen 41
5.3.2 Aktivität von Moxifloxacin gegenüber vier E. coli Stämmen in Monokultur unter
aeroben und anaeroben Bedingungen 42
5.3.3 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen in Monokultur 44
5.3.4 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm ATCC 25922 in Mischkultur 44
5.3.5 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm VA 4050 in Mischkultur 46
5.3.6 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm BK 12944 in Mischkultur 48
5.3.7 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm VA 6886 in Mischkultur 49

Inhaltsverzeichnis
III
5.3.8 Aktivität von Moxifloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm VA 4050 in Mischkultur 51
5.3.9 Aktivität von Moxifloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm BK 12944 in Mischkultur 52
5.3.10 Zusammenfassung der PK/PD-Untersuchung 54
5.3.11 Pharmakologische Indizes 54
6 Diskussion und Ausblick 57
7 Zusammenfassung der Arbeit 63
8 Literaturverzeichnis 65
9 Erklärung über die eigenständige Abfassung der Arbeit 76
10 Danksagung 77
11 Lebenslauf 78
12 Veröffentlichungen der Arbeit 80
Anhang 1-3 81

Bibliographische Beschreibung
IV
I Bibliographische Beschreibung
Stefanie Rossi, Assistenzärztin der Inneren Medizin
Pharmakokinetisch/pharmakodynamische Untersuchung zur in vitro Aktivtität von
Levofloxacin und Moxifloxacin gegenüber B. fragilis und E. coli in Mono- und Mischkultur
Universität Leipzig, Dissertation
92 S., 190 Lit., 25 Abb., 20 Tab., 3 Anlagen
Referat:
Bei intraabdominellen Infektionen findet sich häufig ein polymikrobielles Erregerspektrum
aus fakultativ und obligat anaeroben Bakterien. Natürliches Habitat der dabei am häufigsten
isolierten Erreger E. coli und B. fragilis ist der gastrointestinale Trakt. Durch Störungen der
Darmwandintegrität und nachfolgendem Eindringen der Bakterien in eine sterile Umgebung,
wie beispielsweise der Peritonealhöhle, kann es zur Infektion kommen. Die häufigste Form
der intraabdominellen Infektionen ist die sekundäre Peritonitis. Die Therapie erfolgt
chirurgisch, begleitet von einer antibiotischen Therapie. Dabei sind Kenntnisse über
Wirksamkeit und Empfindlichkeit der Erreger gegenüber den verwendeten Antibiotika von
entscheidender Bedeutung.
In dieser Arbeit wurden die MHK-Werte von vier B. fragilis und vier E. coli Stämmen für
Levofloxacin und Moxifloxacin bestimmt, wobei ein B. fragilis und drei E. coli Stämme
resistent gegenüber Levofloxacin und Moxifloxacin waren. Anschließend wurden mit einem
in vitro PK/PD-Modell die pharmakodynamischen Effekte von Levofloxacin und
Moxifloxacin in Mono- und Mischkulturen von B. fragilis und E. coli untersucht. Die
Monokulturversuche mit E. coli Stämmen wurden sowohl unter aeroben als auch anaeroben
Bedingungen durchgeführt. Durch die Untersuchung von Aktivität und Wirksamkeit von
Levofloxacin und Moxifloxacin in vitro können Aussagen über eine mögliche Verwendung
bei intraabdominellen Infektionen getroffen werden. Levofloxacin wirkte auf den sensiblen E.
coli Stamm nur unter aeroben Bedingungen bakterizid, nicht jedoch unter anaeroben
Bedingungen. Hingegen wirkte Moxifloxacin in aerober und anaerober Umgebung bakterizid
auf diesen E. coli Stamm. In den Mischkulturen zeigte Moxifloxacin eine bessere in vitro
Aktivität als Levofloxacin, wobei Moxifloxacin allerdings auch nur eine moderate Aktivität
hatte. Aufgrund der Ergebnisse könnte Moxifloxacin dennoch eine theraupeutische Option
bei der Behandlung intraabdomineller Infektionen bieten. Der Einsatz dieser Substanzen
sollte jedoch nicht unkritisch erfolgen.

Abkürzungsverzeichnis
V
II Abkürzungsverzeichnis
Abb. Abbildung
ATCC American Type Culture Collection
ATP Adenosintriphosphat
AUC Fläche unter der Kurve (Area under the curve)
CAP Amulant erworbene Pneumonie (Community acquired
pneumonia)
CLSI Clinical Laboratory Standards Institute
Cmax Maximal erreichbare Plasmakonzentration
CYP450 Cytochrom P450
DIN Deutsches Institut für Normung
DNA Desoxyribonukleinsäure (Deoxyribonucleic acid)
EARSS European Antimicrobial Resistance Surveillance System
EUCAST European Committee for Antimicrobial Susceptibility Testing
FDA US Food and Drug Administration
GABA Gamma-Aminobuttersäure
h Stunde(n)
i intermediär
i.v. intravenös
IDSA Infectious Diseases Society of America
ISO International Standard Organisation
KBE Koloniebildende Einheit
l Liter
LEV Levofloxacin
LPS Lipopolysaccharid
mg Milligramm
MHK Minimale Hemmkonzentration
MHK50 Niedrigste Konzentration, welche die Vermehrung von
mindestens 50% der untersuchten Stämme hemmt
MHK90 Niedrigste Konzentration, welche die Vermehrung von
mindestens 90% der untersuchten Stämme hemmt
ml Milliliter
MOX Moxifloxacin
n Anzahl untersuchter Stämme

Abkürzungsverzeichnis
VI
NSAR Nichtsteroidale Antirheumatika
p.o. per os
PAE Postantibiotischer Effekt
PEG Paul-Ehrlich-Gesellschaft
PK/PD Pharmakokinetisch/pharmakodynamisch
r resistent
RMA Stammbezeichnung von B. fragilis Stämmen des R. M. Alden
Research Laboratory, Santa Monica, CA, USA
RNA Ribonukleinsäure (Ribonucleic acid)
s sensibel
SIS Surgical Infection Society
spp. Species
T>MHK Zeit oberhalb der MHK
vs. versus
WAL R Stammbezeichnung von B. fragilis Stämmen aus einer
internationalen Anaerobierstudie
ZNS Zentralnervensystem

Einleitung
1
1 Einleitung
Als Grundgedanke der antiinfektiven Therapie kann das Ehrlichsche Prinzip der selektiven
Toxizität Anwendung finden. Es beschreibt den selektiven Angriff der Antiinfektiva an
Strukturen der Mikroorganismen, welche beim Wirtsorganismus (Mensch) nicht oder in
wesentlich anderer Form vorkommen [119].
1.1 Definition von Pharmakokinetik (PK) und Pharmakodynamik (PD) der
antimikrobiellen Therapie
Die Pharmakologie der antimikrobiellen Therapie wird durch die Begriffe der
Pharmakokinetik und Pharmakodynamik beschrieben.
Der 1953 erstmalig von F. H. Dost verwendete Begriff der Pharmakokinetik beschreibt die
Konzentrationsveränderungen des Antibiotikums im Organismus in Abhängigkeit von der
Zeit. Endogene Prozesse wie Resorption, Verteilung, Biotransformation und Elimination
bedingen diese Veränderungen. Die Zusammensetzung des Antibiotikums, sowie
Applikationsart und -ort fallen ebenso unter diesen Begriff [45, 185].
Die Pharmakodynamik beschreibt die Beziehungen zwischen der Konzentration des
Antibiotikums und dem hervorgerufenen antimikrobiellen Effekt. Dadurch lassen sich
Parameter wie Wirkprofil, Wirkqualität, Effektivität der Wirkung und Wirkintensität
ermitteln.
Die pharmakodynamischen Effekte der Antibiotika lassen sich in vitro anhand folgender
Parameter analysieren:
� den morphologischen Veränderungen der Bakterien
� der intitialen Abtötungsrate
� der hemmenden Wirkung auf die Vermehrung
� dem postantibiotischen Effekt
� und der effektiven Zeit des erneuten Wachstums nach Antibiotikumexposition [68,
100].
1.2 Definition von Wirkmechanismus, Wirktyp, Wirkspektrum und Wirkinten sität
von Antibiotika
In der großen Gruppe der Antibiotika können Einteilungen nach Wirkmechanismus, Wirktyp,
Wirkspektrum und Wirkintensität vorgenommen werden [67, 119].

Einleitung
2
Der Wirkmechanismus liefert Aussagen über die Angriffspunkte der Antibiotika bei
Bakterienzellen und wird unterschieden nach [74, 165]:
� Hemmung der Zellwandsynthese (z. B. Betalaktam-Antibiotika oder
Glykopeptide)
� Störung der Zellwandpermeabilität (z. B. Polypeptide)
� Hemmung des bakteriellen Stoffwechsels (z. B. Folsäureantagonisten)
� Hemmung der Proteinbiosynthese (z. B. Aminoglykoside, Makrolide)
� Eingriff in die Synthese von Nukleinsäuren oder deren Schädigung (z. B.
Fluorchinolone oder Nitroimidazole).
Beim Wirktyp wird zwischen bakteriostatischem und bakterizidem Wirktyp unterschieden.
Als Bakteriostase wird die Hemmung des Bakterienwachstums bezeichnet. Zu einer Abtötung
der Mikroorganismen kommt es hierbei nicht. Ruhende Bakterien werden nicht beeinflusst.
Bakteriostatisch wirkende Antibiotika, wie Sulfonamide, Tetrazykline oder Chloramphenicol
wirken meist über die Beeinflussung der bakteriellen Proteinbiosynthese.
Kommt es zu einer Abtötung der Bakterienzelle, wird von Bakterizidie gesprochen. Die
Zerstörung der Mikroorganismen kann sowohl in der Vermehrungsphase als auch in der
Ruhephase erfolgen. Beispiele hierfür sind Penicilline, Aminoglykoside und Fluorchinolone.
Weiterhin unterscheidet man eine konzentrationsabhängige und eine weitgehend
konzentrationsunabhängige, zeitabhängige Bakterizidie [38, 89, 119]. Neben der direkten
antibakteriellen Wirkung können bestimmte Antibiotika indirekt antibakteriell durch
Modulation des Immunsystems wirken. Beispielsweise können Makrolide und
Fluorchinolone die Zytokinbildung beeinflussen und Rifampicin kann die T-Zell-Aktivierung
supprimieren [173].
Das Wirkspektrum beschreibt die Aktivität von Antibiotika gegenüber verschiedenen
Bakterienspezies. Hier wird zwischen sogenannten Schmalspektrum- und Breitspektrum-
Antibiotika unterschieden. Schmalspektrum-Antibiotika sind gegenüber wenigen
Bakterienspezies wirksam. Breitspektrum-Antibiotika wirken gegen unterschiedliche
Bakterienspezies [166].
Die Wirkintensität eines Antibiotikums wird durch die Minimale Hemmkonzentration (MHK)
beschrieben und durch die definierten Empfindlichkeitsstufen "sensibel, intermediär und
resistent" kategorisiert.

Einleitung
3
1.3 Minimale Hemmkonzentration (MHK)
Nach DIN 58940-1 ist die MHK definiert als geringste Konzentration eines Antibiotikums
(mg/l), die unter definierten in vitro Bedingungen in einem vorgegebenen Zeitintervall (18 ±
2 h) ein visuell erkennbares mikrobielles Wachstum verhindert [6]. Die minimale bakterizide
Hemmkonzentration (MBK) ist die geringste in vitro gemessene Konzentration, welche nach
6 h zu einem Absterben der Mikroorganismen führt. Eine bakterizide Wirkung liegt bei
Abtötungsraten von mindestens 99,9% vor [6, 166]. Die MHK liefert Aussagen zur aktuellen
Empfindlichkeitssituation in vivo. Es handelt sich um einen prädiktiven in vitro bestimmten
Wert zur wahrscheinlichen Wirkung des Antibiotikums. Es erfolgt häufig eine klinische
Einstufung in die Kategorien "sensibel", "intermediär" und "resistent". Die MHK90 ist die
Antibiotikumkonzentration, welche zur Hemmung von 90% der untersuchten Stämme führt.
Die MHK50 gibt die Konzentration an, welche das Wachstum von 50% der untersuchten
Erreger hemmt [14, 133, 185].
1.3.1 Methoden zur Bestimmung der MHK
Testverfahren müssen standardisiert sein, um reproduzierbare Testergebnisse zu erhalten. Auf
Initiative des Deutschen Instituts für Normung (DIN) gibt es inzwischen eine
Standardisierungsnorm für die Bestimmung der MHK (DIN EN ISO 20776-1). Die
Mikrodilutionsmethode gilt dabei als Referenzmethode der MHK-Bestimmung [5]. Bei
Verwendung unterschiedlicher Standards können sich Diskrepanzen in den Ergebnissen
ergeben. Daher ist die Angabe des verwendeten Standards erforderlich [167]. Die
gebräuchlichsten Methoden werden im Folgenden ausführlicher erläutert.
Dilutionstest Bei diesen geometrischen Verdünnungsreihen können quantitative Aussagen bezüglich der
Empfindlichkeit von Mikroorganismen gemacht werden. Man unterscheidet den
Agardilutionstest von dem Mikro- und Makrobouillondilutionstest. Beim Agardilutionstest
werden standardisierte Bakterienmengen auf entsprechend vorbereitetes Agarmedium geimpft
und nachfolgend inkubiert. Das Agarmedium besteht aus einem geeigentem Nährmedium,
welches mit verschiedenen Konzentrationen des zu testenden Antibiotikums versetzt ist. Zur
Kontrolle wird ein Agarmedium ohne Antibiotikum mit dem standardisiertem Inokulum
beimpft [9]. Vorteil des Agardilutionstestes ist die Möglichkeit der Verwendung mehrerer
verschiedener Bakterienstämme auf einem Agarmedium.

Einleitung
4
Bei der Bouillondilutionsmethode werden mehrere Behältnisse mit gleichen Volumina einer
geeigneten Bouillon sowie gleichen Volumina einer Wirkstofflösung mit allerdings
geometrisch abnehmender Antibiotikumkonzentration mit einem definierten Inokulum
beschickt. Die MHK ist festgelegt als niedrigste Konzentration, welche noch zu einer
sichtbaren Hemmung des Erregerwachstums führt [6, 112, 153].
Gradientendiffusionstest Der Gradientendiffusionstest wurde 1991 weltweit als Epsilometer-Test (Etest®) durch AB
Biodisk eingeführt. In verschiedenen Studien konnte gezeigt werden, dass sich vergleichbare
Ergebnisse zu den Verdünnungsreihen sowie den Diffusionstesten ergeben [19, 105]. Es wird
die einfache Handhabung des Agardiffusionstestes mit der Genauigkeit der Bouillondilution
verbunden, wodurch eine direkte Bestimmung der MHK erfolgen kann. Der Etest® ist ein
vorgefertigter und stabiler Antibiotikumgradient über 15 Verdünnungsstufen auf einem
Kunststoffträgermaterial [3]. Das Antibiotikum diffundiert in das Testmedium und kann das
Bakterienwachstum hemmen. Dabei bildet sich ein ellipsenförmiger Hemmhof um den
Teststreifen, wie in Abbildung 1 dargestellt. Die Endpunkte der Hemmhofellipse ergeben den
jeweiligen MHK-Wert.
Abbildung 1: Agarmedium mit 5 Etest®-Streifen und ellipsenförmigen Hemmzonen (aus Baker et al., 1991 [19])
1.3.2 Einteilung der Grenzwerte Nach DIN 58940-1 beschreibt der Grenzwert die MHK bzw. den Hemmhofdurchmesser
(HHD), anhand derer die Erreger nach den Empfindlichkeitsprädikaten oder
Bewertungsstufen "sensibel", "intermediär" und "resistent" voneinander getrennt werden [6].

Einleitung
5
Die Einteilung basiert primär auf pharmakokinetisch/ pharmakodynamischen Beziehungen,
welche durch Studien erhoben werden. Aber auch Faktoren wie Dosierung, unerwünschte
Wirkungen, Resistenzmechanismen sowie klinisches Outcome werden berücksichtigt [84].
Um einen Abgleich der Grenzwerte innerhalb von Europa zu erzielen, wurde die
Arbeitsgruppe des European Committee for Antimicrobial Susceptibility Testing (EUCAST)
eingerichtet [85, 86]. Die EUCAST ist ein ständiges Kommittee der European Society for
Clinical Mirobiology and Infectious Diseases (ESCMID). Die Grenzwerte der EUCAST sind
im Internet unter http://www.eucast.org/ verfügbar [8]. Es gibt aber auch andere
Organisationen, die sich mit der Grenzwertfindung beschäftigen [41]. In Amerika werden die
Breakpoints durch das Clinical Laboratory Standards Institute (CLSI, vormals NCCLS =
National Committee of Clinical Laboratory Standards) herausgegeben [35]. Die EUCAST-
Grenzwerte sind häufig niedriger als die der CLSI. Teilweise sind die ausgegebenen
Grenzwerte bei beiden Komitees Bakteriengruppen-spezifisch. In Deutschland werden beide
Bewertungssysteme verwendet. Dabei ist dem klinisch tätigem Arzt meist nicht bekannt, nach
welchem System bewertet wurde [136]. Im Rahmen einer ISO-Arbeitsgruppe wurde als
weltweiter Standard zur Ermittlung der MHK die Mikrobouillondilution festgelegt [136].
"Als sensibel (s) gegen ein bestimmtes Antibiotikum wird ein Bakterienstamm dann
bezeichnet, wenn er in vitro von einer Konzentration dieses Wirkstoffs inhibiert wird, die mit
einer hohen therapeutischen Erfolgswahrscheinlichkeit assoziiert ist"[5] .
"Als intermediär (i) gegen ein bestimmtes Antibiotikum wird ein Bakterienstamm dann
bezeichnet, wenn er in vitro von einer Konzentration dieses Wirkstoffs inhibiert wird, die mit
einem unsicheren therapeutischen Ergebnis assoziiert ist. Ohne zusätzliche Berücksichtigung
weiterer Kriterien ist eine Beurteilung hinsichtlich eines Therapieerfolges durch das
entsprechende Antibiotikum nicht möglich" [5].
"Als resistent (r) gegen ein Antibiotikum wird ein Bakterienstamm dann bezeichnet, wenn er
in vitro von einer Konzentration dieses Wirkstoffs inhibiert wird, die mit einer hohen
Wahrscheinlichkeit des Therapieversagens assoziiert ist. Sind keine ausreichend hohen
Erfolgsraten durch das Antiinfektivum bei klinischen Studien zu erreichen, wird dieses
ebenfalls als "resistent" bezeichnet" [5].
1.4 Pharmakokinetisch/pharmakodynamisches Modell (PK/PD-Modell)
Die MHK ist ein wichtiger Parameter zur Quantifizierung der Aktivität eines Antibiotikums.
Da es sich um einen statischen Wert handelt, können keine Angaben über die Absterbekinetik
oder das Wachstumsverhalten der Erreger gemacht werden. Pharmakokinetische Faktoren des

Einleitung
6
Antibiotikums finden zudem keine Berücksichtigung [38, 59]. Die Interaktion zwischen
Antibiotikum und Bakterium in vitro kann zunächst durch eine Absterbekinetik simuliert
werden. Vorteil gegenüber der MHK ist die Bestimmung von Ausmaß und Geschwindigkeit
der Abtötung oder des Wachstums von Bakterien. Nachteilig ist die konstante
Antbiotikumkonzentration. Eine Möglichkeit zur vergleichenden Betrachtung von
Pharmakokinetik und Pharmakodynamik lässt sich durch PK/PD-Modelle erreichen [36].
PK/PD-Modelle bieten eine gute Möglichkeit zur Abschätzung der antimikrobiellen
Wirkintensität und Effekte durch Darstellung dynamischer Abtötungskurven. Somit lassen
sich Fragen der Dosierung und der Dosierungsintervalle (zeitabhängige vs.
konzentrationsabhängige Bakterizidie) gezielter beantworten [14, 118, 127]. Vorteile
gegenüber der alleinigen MHK-Bestimmung ergeben sich auch in Aufdeckung von
möglichen postantibiotischen Effekten. Vergleiche unterschiedlicher Antibiotika/-klassen und
die Verwendung von Mischkulturen können durch in vitro PK/PD-Modelle erfolgen [66,
106]. Zudem können pharmakodynamische Unterschiede bei Veränderungen der
Pharmakokinetik, beispielsweise Verlängerung der Halbwertszeit bei Niereninsuffizienz,
simuliert und dargestellt werden [183]. Diese Untersuchungen können in vitro oder in vivo
durch Tierversuche durchgeführt werden. Ein Vorteil der in vitro Modelle gegenüber den
Tierversuchen stellt neben ethischen Gesichtspunkten die Verwendung von Dosierungen dar,
die beim Menschen erreicht werden. Zudem bestehen Unterschiede in der Pharmakokinetik
zwischen Mensch und Tier. Weitere Vorteile der in vitro Modelle sind die meist einfacheren
und kostengünstigeren Durchführungen gegenüber in vivo Untersuchungen [36].
Trotz der vielen Vorteile können in vitro PK/PD-Modelle die Komplexität von in vivo
Infektionen nicht nachahmen: "in vitro models are neither mice or men" [183]. In vitro
PK/PD-Modelle simulieren häufig Vorgänge unter neutropenischen Bedingungen. Deshalb
können auch keine Aussagen zur Abwehrfunktion oder Beeinflussung des Immunsystems
durch Bakterien oder Antibiotika getroffen werden [32, 173].
Im Verlauf der Entwicklung von antimikrobiellen Substanzen wurden PK/PD-Indizes
eingeführt, die zum Vergleich der unterschiedlichen Wirkungen herangezogen werden
können. Dadurch können Aussagen über eine klinische Wirksamkeit getroffen werden. Diese
Indizes sind aus einem pharmakokinetischen Parameter und einem mikrobiologischen
Parameter zusammengesetzt. Die International Society of Anti-Infective Pharmacology
(ISAP), hat eine Arbeit zur einheitlichen Definition der Parameter und Indizes
herausgebracht. Nachfolgend sind die relevanten Parameter und Indizes ausgelistet [21, 116].

Einleitung
7
MHK Minimale Hemmkonzentration [mg/l oder µg/ml]
AUC Fläche unter der Kurve [mg×h/l oder µg×h/ml]
Cmax Maximal erreichbare Plasmakonzentration [mg/l oder µg/ml]
AUC/MHK Verhältnis von AUC zu MHK [dimensionslos]
Cmax/MHK Verhältnis von Cmax zu MHK [dimensionlos]
T>MHK Kumulativer Prozentsatz der Zeit , in welcher die Konzentration des
Antibiotikums oberhalb der MHK des Erregers liegt [%]
Bei der konzentrationsabhängigen Bakterizidie sind hohe Konzentrationen des Antibiotikums
für kurze Zeit nötig, um eine bestmögliche Wirkung zu erzielen. Bei den Aminoglykosiden
korreliert die Spitzenkonzentration oberhalb der MHK mit dem therapeutischen Erfolg
(Cmax/MHK). Für die Fluorchinolone ist neben dem Parameter Cmax/MHK der Parameter
AUC/MHK von besonderer Bedeutung [16]. Die zeitabhängige Bakterizidie fordert konstant
mäßig hohe Wirkstoffkonzentrationen über einen bestimmten Zeitraum zur Erzielung eines
antimikrobiellen Effektes. Der Parameter T>MHK korreliert beispielsweise bei den Betalaktam-
Antibiotika oder Makroliden am besten mit der Effektivität [38, 119]. Nach Drusano et al. hat
der Parameter T>MHK bei den Fluorchinolonen keine Bedeutung für den Erfolg der Therapie
[47].
1.5 Postantibiotischer Effekt
Der Postantibiotische Effekt (PAE) ist nach DIN 58940-1 die Wachstumshemmung für
mehrere Stunden, beziehungsweise die Verzögerung des Wachstums von Mikroorganismen
nach kurzzeitiger Einwirkung einer antimikrobiellen Substanz und deren Entfernung aus dem
Testansatz [6]. Letztlich wird eine Erholungsphase beschrieben, welche die beschädigten,
aber nicht abgetöteten Bakterien zur Regeneration benötigten. Während dieser Zeit kommt es
zu keiner erneuten Vermehrung der Mikroorganismen. Die Mikroorganismen können in
dieser Phase besser durch Leukozyten phagozytiert werden. Diese Phase kann, abhängig von
der antimikrobiellen Substanz, mehrere Stunden betragen. Bei den meisten Antibiotika lässt
sich gegenüber grampositiven Kokken ein PAE nachweisen. Gegen gramnegative Bakterien
findet sich ein PAE bei antimikrobiellen Substanzen, welche die RNA inhibieren oder die
Proteinbiosynthese der Bakterien beeinflussen. Dazu gehören beispielsweise die
Aminoglykoside und Fluorchinolone. Der durch Fluorchinolone hervorgerufene
postantibiotische Effekt gegen gramnegative Bakterien liegt zwischen einer und vier Stunden.

Einleitung
8
Klinische Relevanz ergibt sich bei der Bewertung der Dosierungsintervalle, da bei Vorliegen
eines PAE das Dosisintervall verlängert werden kann [68, 101, 102, 119, 159, 166, 190].
1.6 Fluorchinolone
Im klinischen Alltag werden die Begriffe Gyrasehemmer und Chinolone meist synonym
verwendet. Neben der DNA-Gyrase wird auch die Topoisomerase IV gehemmt, daher ist der
Begriff Gyrasehemmer nicht hinreichend. Auch haben nicht alle Substanzen die klassische
Chinolongrundstruktur. Chemisch gesehen handelt es sich um Chinoloncarbonsäuren und
Aza-Analoge, die ab der zweiten Generation fast ausnahmslos fluoriert sind und in dieser
Arbeit als Fluorchinolone bezeichnet werden [14]. Das erste Analogon dieser synthetischen
Antibiotika wurde 1962 als Nalidixinsäure für die Therapie von Harnwegsinfektionen
zugelassen. Nalidixinsäure wurde eher zufällig bei der Synthese von Chloroquin zur
Malariatherapie als Nebenprodukt entdeckt. Aufgrund sehr ungünstiger pharmakokinetischer
Eigenschaften und einer raschen Resistenzentwicklung findet Nalidixinsäure heutzutage
keine Anwendung mehr. Der Durchbruch der Therapie von Harnwegsinfekten gelang 1978
mit Norfloxacin, dem ersten Fluorchinolon. Von den inzwischen über 10000 synthetisierten
Präparaten wurden bisher nur 2% weiterentwickelt und klinisch getestet [169]. 20
Fluorchinolone sind erfolgreich auf dem Handel zugelassen. Teilweise finden die Präparate
auch Anwendung in der Veterinärmedizin [17, 147]. Die neuen Fluorchinolone unterscheiden
sich in 2 wichtigen Punkten von der Muttersubstanz Nalidixinsäure (Abb. 2): die Einführung
eines Fluoratoms an Position 6 sowie die Substitution einer Piperazinyl- oder Pyrrolidinyl-
Gruppe an Position 7 (Abb. 3). Durch diese Veränderungen besitzen die Substanzen eine
deutlich höhere antibakterielle Aktivität und ein breiteres Wirkspektrum [186] .
NN
O
COOH
C2H5
CH3
Abbildung 2: Nalidixinsäure
6
7X8
5
N12
34F
R7
R5
COOH
R2
R1
O
Abbildung 3: Fluorchinolongrundgerüst

Einleitung
9
� Einteilung
Die Paul-Ehrlich-Gesellschaft (PEG) übernahm 1998 von Naber und Adam eine Einteilung
anhand der in vitro Aktivität der Substanzen, wie in Tabelle 1 dargestellt.
Tabelle 1: Einteilung der Chinolone (nach Naber und Adam, 1998 ([120])
Gruppe Eigenschaft Beispiel
I Indikation: Harnwegsinfektion Norfloxacin
II Systemisch anwendbar mit ausgedehntem
Indikationsspektrum
Enoxacin, Ofloxacin,
Ciprofloxacin
III verbesserte Aktivität gegen grampositive und atypische
Erreger
Levofloxacin
IV verbesserte Aktivität gegen grampositive, atypische
Erreger sowie Anaerobier
Moxifloxacin
In Deutschland werden hauptsächlich 3 Substanzen aus diesen Gruppen zur Therapie
eingesetzt: Ciprofloxacin, Levofloxacin und Moxifloxacin [166].
� Wirkmechanismus
Die Fluorchinolone zeigen eine konzentrationsabhängige Pharmakodynamik und wirken
durch Hemmung der Typ II Topoisomerasen bakterizid. Zwei bakterielle Zielstrukturen
werden unterschieden: die DNA-Gyrase und die Topoisomerase IV. Topoisomerasen sind
neben anderen Enzymen notwendig für den reibungslosen Ablauf der DNA-Replikation,
indem sie superhelicale Strukturen entwinden. DNA weist in ausgestreckter Form eine 1000-
fach größere Länge als eine Bakterienzelle auf. Daher muss die DNA mittels Ausbildung von
Superhelices in eine kompakte Struktur überführt werden [165]. Die bakterielle DNA-Gyrase
ist im Gegensatz zu den Topoisomerasen der Säugetiere imstande, unter Verbrauch von ATP
negative Superhelices in ringförmige DNA-Moleküle einzufügen. Sie bestehen aus 2 A- und
2 B-Untereinheiten, gyrA und gyrB, welche durch die Gene gyrA und gyrB kodiert werden.
Die Topoisomerase IV trennt die Bakterienchromosomen nach der Replikation, welches zur
Zellteilung führt [46, 78, 186]. Die Topoisomerase IV besteht aus 2 parC- und 2 parE-
Untereinheiten, welche durch die Gene parC und parE kodiert werden. Bei gramnegativen
Bakterien wie E. coli ist die primäre Zielstruktur die DNA-Gyrase. Bei grampositiven
Bakterien einschließlich S. aureus ist die Zielstruktur die Topoisomerase IV [23]. Bei den
neueren Fluorchinolonen scheint es eine ausgeglichene Balance bezüglich der Affinität zur

Einleitung
10
Zielstruktur zu geben [170]. Eine toxische Wirkung der Fluorchinolone auf eukaryote DNA-
Gyrasen ist sehr unwahrscheinlich, da mindestens 100-fach erhöhte Konzentrationen nötig
sind, um die Aktivität der menschlichen Topoisomerasen zu inhibieren [39, 77, 186].
� Pharmakokinetik
Die Fluorchinolone haben eine lineare Pharmakokinetik [168]. Günstige pharmakokinetische
Eigenschaften führen zu einer hohen Wirksamkeit und guten Sicherheitsprofilen. Die
Plasmaproteinbindung beträgt in Abhängigkeit von der Substanz 10-40%. Bei vielen
Substanzen der Fluorchinolone ist die orale Bioverfügbarkeit sehr hoch und die Absorption
nach oraler Aufnahme erfolgt sehr schnell. Nach 1-2 h erreichen die Serumkonzentrationen
Maximalwerte. Aufgrund langer Halbwertszeiten müssen die jüngeren Fluorchinolone wie
Moxifloxacin nur einmal täglich eingenommen bzw. appliziert werden. Die Fluorchinolone
weisen ein hohes Verteilungsvolumen auf [40, 119]. Durch Veränderungen der chemischen
Strukturen werden Wirkintensität, Wirkspektrum und unerwünschte Nebenwirkungen
beeinflusst. Die DNA-Bindung wird durch die Substituenten C2, C3, C4 und C6 vermittelt.
Durch das Fluoratom an C6 kommt es zu einer verbesserten Zellwandpenetration. Der C7-
Substituent beeinflusst die Wirkintensität und das Wirkspektrum. Optimale Substituenten
scheinen dabei Aminopyrrolidine mit höchster Wirkintensität gegen grampositive Bakterien
oder Piperazine mit verbesserter Wirkung gegen gramnegative Erreger und überlegener in
vivo Aktivität zu sein. Eine Methoxy-Gruppe an C8 erweitert das Wirkspektrum auf
grampositive, atypische und obligat anaerobe Erreger und weist eine geringere Phototoxizität
auf. Durch Alkylierung an Position 7 wird eine höhere sterische Masse erreicht, dadurch
lassen sich auch unerwünschte Wirkungen auf das zentrale Nervensystem, Interaktionen mit
anderen Medikamenten und gentoxische Wirkungen deutlich reduzieren [44, 147, 161].
� Unerwünschte Wirkungen
Die Fluorchinolone haben prinzipiell ähnliche unerwünschte Wirkungen, jedoch kann sich die
Ausprägung bei den einzelnen Substanzen deutlich unterscheiden. Am häufigsten treten
gastrointestinale Beschwerden wie Übelkeit, Magenschmerzen oder Diarrhoe auf [119].
Hepatotoxische und phototoxische Reaktionen sind ebenfalls beschrieben. Sparfloxacin und
Grepafloxacin können schwere kardiale Arrhythmien (Torsade-de-Pointes) aufgrund einer
QT-Verlängerung auslösen [166]. Die zentralnervösen Nebenwirkungen wie Kopfschmerz,
Schwindel, Agitation, Schlafstörungen und in seltenen Fällen cerebrale Krämpfe sind durch
Bindung des C7-Substituenten an GABAA-Rezeptoren vermittelt. Alkylierte Seitenketten wie

Einleitung
11
beispielsweise bei Temafloxacin oder Sparfloxacin binden nur schwach an den Rezeptor.
Ciprofloxacin bindet durch eine Piperazingruppe stark an den Rezeptor [13, 44]. Frühere
Versuche an juvenilen Beagle-Hunden führten zu Knorpelschäden, daher wird empfohlen,
Fluorchinolone bei Kindern vor Abschluss des Wachstums nicht zu verwenden [160]. Jedoch
konnten Untersuchungen in den 90er Jahren keine irreversiblen Erkrankungen der kindlichen
Gelenke aufzeigen [128].
� Indikationen
Mögliche Indikationen der Fluorchinolone sind Harnwegsinfektionen (Ciprofloxacin),
Prostatitis und sexuell übertragbare Krankheiten (Ciprofloxacin, Levofloxacin),
Atemwegsinfektionen (Ciprofloxacin, Levofloxacin, Moxifloxacin), Knocheninfektionen
sowie Haut- und Weichgewebsinfektionen (Levofloxacin), intraabdominelle Infektionen und
Sepsis (Off-Label-Einsatz von Moxifloxacin) [179]. Eine Sonderindikation besteht für
Ciprofloxacin bei nachgewiesenen Infektionen mit P. aeruginosa bei Patienten mit Cystischer
Fibrose. Hier ist der Einsatz bei Kindern und Jugendlichen im Alter von 5 bis 17 Jahren
möglich [4]. In der Literatur finden sich zudem Angaben zur Prävention von Infektionen bei
neutropenischen Patienten mit akuter Leukämie[171].
� Interaktionen
Arzneimittelinteraktionen können bei gleichzeitiger Applikation der Fluorchinolone mit
NSAR oder Theophyllin auftreten. Für Levofloxacin und Moxifloxacin konnte keine
Interaktion mit Theophyllin nachgewiesen werden [114, 161]. Auf eine gleichzeitige
Einnahme von Antazida oder Eisensulfaten sollte verzichtet werden [103, 186]. H2-
Rezeptorantagonisten und Anticholinergika führen zu keiner negativen Beeinflussung der
oralen Bioverfügbarkeit [43, 161]. Vermehrte Hypoglykämieneigungen zeigten sich bei
gleichzeitiger Applikation mit Insulin oder oralen Antidiabetika wie beispielsweise
Glibenclamid [87]. Der Digoxin-Spiegel kann sich bei gleichzeitiger Gabe von Moxifloxacin
erhöhen [119].
1.6.1 Levofloxacin
Levofloxacin (LEV; Abb. 4) leitet sich von Ofloxacin ab. Ofloxacin liegt als Razemat vor.
Die linksdrehende Form hat die antibakteriellen Eigenschaften, die rechtsdrehende Form
(Dextrofloxacin) ist antibakteriell inaktiv, verursacht aber unerwünschte Wirkungen.
Levofloxacin ist nur die linksdrehende Form und kann daher höher dosiert werden [166].

Einleitung
12
N
O
O
F
N
COOH
CH3
N
H3C
Abbildung 4: Levofloxacin
� Pharmakokinetik
Levofloxacin hat eine Halbwertszeit von 7-8 h. Nach oraler Gabe von 0,25 g zeigen sich nach
1-2 h Serumspiegel von 2,5 bis 3 mg/l bzw. nach Gabe von 0,5 g Spiegel von 5 bis 6 mg/l.
Levofloxacin wird fast vollständig resorbiert. Die Plasmaeiweißbindung liegt bei 25%. Die
Ausscheidung erfolgt vorwiegend renal in aktiver Form, weshalb bei eingeschränkter
Nierenfunktion eine Dosisanpassung erfolgen sollte. Levofloxacin zeigt eine gute
Gewebepenetration, die Passage der Blut-Hirnschranke ist mäßig. Indikation und Schwere der
Erkrankung bestimmen Dosierung, Art und Dauer der Anwendung. In Deutschland ist
Levofloxacin als Tavanic® in 250 oder 500 mg Dosen zur oralen und intravenösen
Applikation erhältlich. Die maximale Tagesdosis beträgt 1000 mg (2 x 500 mg) zur Therapie
der ambulant erworbenen Pneumonie [12, 40, 166].
� Wirkspektrum
Das Wirkspektrum von Levofloxacin umfasst die meisten aeroben grampositiven und
gramnegativen Bakterien, wobei die Wirkung auf gramnegative Erreger insgesamt besser ist.
Levofloxacin erfasst auch P. aeruginosa. Zudem wirkt Levofloxacin gegen atypische Erreger
wie Legionellen oder Mycoplasmen. Verschiedene Clostridien- und Bacteroides-Arten sind
resistent. Insgesamt ist die Wirkung auf Anaerobier nur mäßig [79, 166].
� Unerwünschte Wirkungen
Als unerwünschte Wirkungen finden sich gastrointestinale Symptome wie Übelkeit,
Erbrechen, Diarrhoe oder abdominelle Schmerzen. Weiterhin können sich leichte
zentralnervöse Störungen wie Kopfschmerzen, Schwindel oder Schlafstörungen zeigen.
Selten treten schwere allergische Reaktionen oder arthropathische Beschwerden auf.
Vereinzelt wurden zudem Veränderungen des Blutbildes und Transaminansenanstiege

Einleitung
13
beschrieben. Nur selten wurden kardiotoxische Reaktionen (QT-Verlängerung) oder
phototoxische Reaktionen beschrieben [11, 79, 166].
� Indikationen und Kontraindiaktionen
Klassische Indikationen sind die bakterielle Sinusitis, exacerbierte chronische Bronchitis,
amulant erworbene Pneumonien (CAP), komplizierte Harnwegsinfektionen einschließlich
Pyelonephritis sowie Haut- und Weichgewebsinfektionen und Infektionen der Prostata.
Kontraindikationen bestehen bei Kindern, Schwangeren und Patienten mit Erkrankungen des
ZNS [12].
1.6.2 Moxifloxacin
Moxifloxacin (MOX; Abb. 5) ist in die Gruppe IV der Fluorchinolone eingeordnet. Durch
Einführung einer Methoxygruppe an Position 8, besitzt Moxifloxacin eine besonders hohe
Aktivität und geringe Phototoxizität [93].
N
O
COOHF
N
O
H3C
HN
H
H
Abbildung 5: Moxifloxacin
� Wirkspektrum
Moxifloxacin hat eine gute Wirksamkeit gegenüber Pneumokokken und fast allen anderen
Erregern von Atemwegsinfektionen einschließlich atypischer Erreger (Chlamydia spp., M.
pneumoniae und Legionella spp.). Zudem hat Moxifloxacin eine gute Aktivität gegen obligate
Anaerobier (B. fragilis, Bacteroides spp. und C. perfringens) [52, 63]. Gegenüber P.
aeruginosa ist Moxifloxacin nicht wirksam [25]. Häufig sind MRSA- und VRE-Stämme auch
gegenüber Moxifloxacin resistent [166].

Einleitung
14
� Pharmakokinetik
Nach Gabe von 0,4 g p.o. bzw. i.v. ergeben sich mittlere Serumspitzenspiegel von 3,2 mg/l
bzw. 4,4 mg/l. Die orale Bioverfügbarkeit beträgt 90%, die Einnahme kann unabhängig von
der Nahrungsaufnahme erfolgen. Die Halbwertszeit beträgt 12 h, die Plasmaeiweißbindung
wird mit durchschnittlich 40% angegeben. Aufgrund der langen Halbwertszeit wird
Moxifloxacin in Dosen von 400 mg nur einmal täglich oral oder intravenös appliziert [95,
166]. Mittels Mikrodialyse wiesen Müller et al. die gute Gewebegängigkeit von Moxifloxacin
und bevorzugte Verteilung im entzündeten Gewebe nach [117]. Experimentell passierte
Moxifloxacin bei eitriger Meningitis die Blut-Hirn-Schranke ausgezeichnet [139].
Moxifloxacin wird nach Biotransformation in Form einer Sulfo-Verbindung unverändert
renal und biliär ausgeschieden. Es kommt zu keiner Beeinflussung des CYP450-Systems. Bei
eingeschränkter Nierenfunktion muss keine Dosisanpassung erfolgen [40]. Moxifloxacin ist
in Deutschland als Avalox® 400 mg im Handel erhältlich [1].
� Unerwünschte Wirkungen
Als häufige unerwünschte Wirkungen treten gastrointestinale Symptome wie Übelkeit und
Erbrechen auf. Schwindel und Tremor wurden ebenfalls beschrieben. Im EKG können QT-
Verlängerungen auftreten. Es empfehlen sich daher regelmäßige EKG-Kontrollen unter der
Therapie mit Moxifloxacin [22, 166].
� Indikationen und Kontraindikationen
Zugelassene Einsatzgebiete in Deutschland sind die CAP sowie schwere Haut- oder
Weichgewebsinfektionen [1, 176]. In den USA besteht zusätzlich die Indikation zur Therapie
schwerer intraabdomineller Infektionen mit polymikrobiellem Keimspektrum und
Abzsessbildung [55]. In Deutschland ist die Therapie von Moxifloxacin bei
intraabdominellen Infektionen als Off-Label-Verordnung möglich [179]. Kontraindikationen
bestehen, wie auch bei Levofloxacin, bei Kindern, Schwangeren und Patienten mit
Erkrankungen des ZNS und zusätzlich bei bekanntem Long-QT-Syndrom [166].

Einleitung
15
1.7 Resistenzsituation, Formen und Mechanismen der Resistenz
Ein Bakterium ist resistent gegen ein Antibiotikum, wenn die MHK so hoch ist, dass auch bei
Verwendung der zugelassenen Höchstdosierung kein therapeutischer Erfolg zu erwarten ist
[6]. Es wird zwischen natürlicher (primärer) und erworbener (sekundärer) Resistenz
unterschieden. Die natürliche Resistenz beruht auf einer genetisch bedingten
Unempfindlichkeit der Bakterienart gegen das Antibiotikum. E. coli und andere gramnegative
Stäbchenbakterien haben eine natürliche Resistenz gegen Penicillin G, da das Antibiotikum
die Zellmembran nicht penetrieren kann. Die sekundäre Resistenz entsteht bei einer
empfindlichen Erregerart durch Selektion resistenter Stämme nach antibiotischer Einwirkung.
Die empfindlichen Bakterien der Population werden weiterhin abgetötet, während sich die
resistenten Erreger vermehren. Andererseits können durch Mutationen oder durch
Übertragung von genetischem Material resistente Bakterien entstehen, die unter Einwirkung
des Antibiotikums selektioniert werden. Spontane Chromosomenmutationen sind innerhalb
einer Bakterienpopulation selten. Da die Mutationen neben dem Resistenzerwerb auch häufig
zu Stoffwechselstörungen des Bakteriums führen, können sich die Bakterien häufig weniger
gut vermehren und die Resistenz kann nach Wegfall des Selektionsdrucks wieder
verschwinden. Der Mechanismus der übertragbaren Resistenz durch Plasmide besteht in der
Aufnahme resistenzkodierter extrachromosomaler DNA. Die plasmidvermittelte Resistenz
führt nicht zu Stoffwechselstörungen und hat damit eine größere praktische Bedeutung.
Folgende Resistenzmechanismen werden unterschieden [67, 74]:
� Bildung von inaktivierenden Enzymen: z. B. verursachen Betalaktamasen, welche den
Betalaktamring spalten, die Penicillinresistenz von Staphylococcus spp.
� Veränderung der Zielstrukturen: z. B. kann durch Alteration der A-Untereinheit der DNA-
Gyrase von E. coli die Fluorchinolon-Resistenz bedingt sein.
� Permeabilitätsbarriere: durch Änderung des D2-Porinkanals kann beispielsweise Imipenem
nicht durch die äußere Membran von P. aeruginosa eindringen.
� Aktiver Efflux: durch membranständige Effluxpumpen werden z. B. Tetrazykline schnell
aus Enterobakterien herausgepumpt und können somit keine Wirkung entfalten.
� Überproduktion des Zielmoleküls/Umgehungswege: z. B. kann durch Überexprimierung
des Zielmoleküls die erreichbare Konzentration der Folsäureantagonisten nicht ausreichen,
um eine vollständige Inhibition zu erreichen. Auch durch Aktivierung alternativer
Stoffwechselwege kann es zur Unwirksamkeit des Antibiotikums kommen.
Die Resistenz von Bakterien gegenüber den Fluorchinolonen wird durch Veränderungen der
Zielenzyme, aktiven Efflux oder reduzierte Permeabilität verursacht. Veränderungen der

Einleitung
16
Zielenzyme entstehen im Speziellen durch chromosomale Veränderungen der QRD-Regionen
(Quinolone resistance-determining regions, QRDR) der Gene, welche für die DNA-Gyrase
und Topoisomerase IV kodieren [20, 188]. Dabei führen Mutationen im gyrA- und gyrB-Gen
zur Fluorchinolon-Resistenz von gramnegativen Bakterien [164]. Insbesondere scheinen
Veränderungen im gyrA-Gen die Resistenzentstehung von obligaten Anaerobiern gegenüber
den Fluorchinolonen zu bedingen [147]. Mutationen im parC-Gen lassen sich in resistenten
grampositiven Bakterien inklusive S. aureus nachweisen [184]. Die Plasmid-vermittelte
Resistenz durch PMQR-Gene (Plasmid-mediated quinolone resistance, PMQR) bei
gramnegativen Bakterien einschließlich E. coli ist eher von untergeordneter Bedeutung, da
niedrige Resistenzlevel resultieren [143]. Innerhalb des letzten Jahrzehnts ist es in
Deutschland zu einer deutlichen Zunahme von resistenten E. coli Stämmen gegenüber den
Fluorchinolonen gekommen. 1990 lag die Anzahl von Ciprofloxacin-resistenten E. coli
Stämmen unter 1%. 2007 stieg die Resistenzrate nach Angaben der Paul-Ehrlich-Gesellschaft
auf 26,4% [129]. Insgesamt ist in Deutschland der Verbrauch an Fluorchinolonen im
ambulanten und stationären Bereich innerhalb der letzten Jahre gestiegen. Im stationären
Sektor werden die Fluorchinolone nach den Betalaktamantibiotika am zweithäufigsten bei der
antimikrobiellen Therapie verwendet [10]. Nach Angaben der PEG ist ein hoher
Selektionsdruck aufgrund des steigenden Gebrauchs der Fluorchinolone für die steigenden
Resistenzraten verantwortlich.
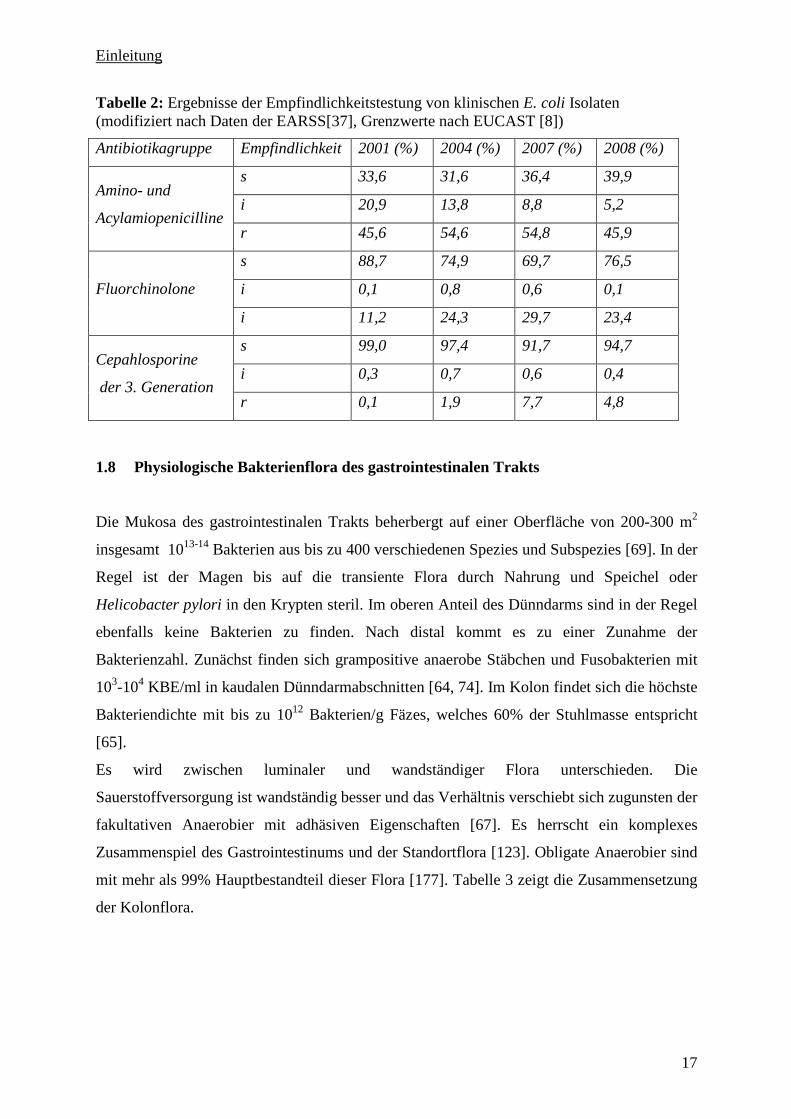
In Tabelle 2 sind die Veränderungen in der Empfindlichkeit von E. coli in Deutschland in den
Jahren 2001 bis 2008 gegenüber ausgewählten Antibiotikagruppen nach Daten des European
Antimicrobial Resistance Surveillance Network (EARS-Net) aufgelistet [37]. Steigende
Resistenzraten gegenüber den Fluorchinolonen können auch bei Bacteriodes spp. beobachtet
werden [61]. In Europa waren 9% der untersuchten Stämme (n = 1300) der B. fragilis Gruppe
resistent gegen Moxifloxacin. Außerdem waren 99% dieser Isolate resistent gegenüber
Ampicillin, wobei 96% dieser Stämme Betalaktamasen bildeten. Für Clindamycin wurden
ebenfalls steigende Resistenzraten bis 15% angegeben [72]. Gegenüber Metronidazol sind die
meisten obligaten Anaerobier weiterhin sensibel [134].
Einleitung
17
Tabelle 2: Ergebnisse der Empfindlichkeitstestung von klinischen E. coli Isolaten (modifiziert nach Daten der EARSS[37], Grenzwerte nach EUCAST [8])
Antibiotikagruppe Empfindlichkeit 2001 (%) 2004 (%) 2007 (%) 2008 (%)
s 33,6 31,6 36,4 39,9
i 20,9 13,8 8,8 5,2 Amino- und
Acylamiopenicilline r 45,6 54,6 54,8 45,9
s 88,7 74,9 69,7 76,5
i 0,1 0,8 0,6 0,1 Fluorchinolone
i 11,2 24,3 29,7 23,4
s 99,0 97,4 91,7 94,7
i 0,3 0,7 0,6 0,4 Cepahlosporine
der 3. Generation r 0,1 1,9 7,7 4,8
1.8 Physiologische Bakterienflora des gastrointestinalen Trakts
Die Mukosa des gastrointestinalen Trakts beherbergt auf einer Oberfläche von 200-300 m2
insgesamt 1013-14 Bakterien aus bis zu 400 verschiedenen Spezies und Subspezies [69]. In der
Regel ist der Magen bis auf die transiente Flora durch Nahrung und Speichel oder
Helicobacter pylori in den Krypten steril. Im oberen Anteil des Dünndarms sind in der Regel
ebenfalls keine Bakterien zu finden. Nach distal kommt es zu einer Zunahme der
Bakterienzahl. Zunächst finden sich grampositive anaerobe Stäbchen und Fusobakterien mit
103-104 KBE/ml in kaudalen Dünndarmabschnitten [64, 74]. Im Kolon findet sich die höchste
Bakteriendichte mit bis zu 1012 Bakterien/g Fäzes, welches 60% der Stuhlmasse entspricht
[65].
Es wird zwischen luminaler und wandständiger Flora unterschieden. Die
Sauerstoffversorgung ist wandständig besser und das Verhältnis verschiebt sich zugunsten der
fakultativen Anaerobier mit adhäsiven Eigenschaften [67]. Es herrscht ein komplexes
Zusammenspiel des Gastrointestinums und der Standortflora [123]. Obligate Anaerobier sind
mit mehr als 99% Hauptbestandteil dieser Flora [177]. Tabelle 3 zeigt die Zusammensetzung
der Kolonflora.

Einleitung
18
Tabelle 3: Zusammensetzung der menschlichen Kolonflora [67]
Anaerobier (99%) Fakultative Anaerobier Transiente Flora
Bacteroides spp.
Bifidobakterien
Eubakterien
(insgesamt 75%)
Clostridien
anaerobe Kokken
Fusobakterien
Laktobazillen
(insgesamt 25%)
Enterobakterien
Enterokokken
Pseudomonas spp.
Hefen
Bacillus spp.
Protozoen
1.9 Epidemiologie, Klinik und Therapie von intraabdominellen Infektionen
Unter intraabdominellen Infektionen fasst man Infektionen der Bauchorgane, wie zum
Beispiel der Gallenwege und -gänge, der Appendix vermiformis, des Peritoneums und
intraabdominelle Abszesse zusammen. Diese Infektionen können traumatisch, infektiös,
postoperativ oder im Rahmen von Tumorerkrankungen entstehen. Bei der Peritonitis lassen
sich 4 Formen unterscheiden: spontan bakterielle Peritonitis (als primäre Form) bei Ascites,
sekundäre Formen, die tertiären Formen als Rezidivinfektionen und Abszesse und
nosokomniale Peritonitis als quartäre Form. Zudem gibt es die Pilzperitonitis und die
Peritonitis bei Peritonealdialyse als Sonderformen [96]. Die sekundäre Peritonitis stellt die
häufigste Form der intraabdominellen Infektion dar und entsteht durch die Ausbreitung von
Mikroorganismen aus dem Lumen der Hohlorgane [97]. Als Ursachen der sekundären
Peritonitis finden sich Appendizitis, Hohlorganperforation, ischämische Kolitis und
postoperative Infektionen. Die Kolondivertikulitis ist die häufigste Ursache einer Perforation
[107].
Die Peritonitis stellt sich klinisch als "akutes Abdomen" dar. Kujath und Rodloff [97]
definieren den Begriff präziser als "akuten, interventionsbedürftigen Abdominalbefund", der
einen akuten abdominellen Schmerz, einen Peritonismus und eine Kreislaufreaktion
insbesondere eine Schocksymptomatik verursacht. Zur Diagnostik gehören die klinische
Untersuchung, laborchemische Untersuchungen inklusive der Abnahme von Blutkulturen und
bildgebende Verfahren wie Abdomenübersichtsaufnahmen, computertomographische oder
sonographische Untersuchungen [97]. Charakteristische Symptome einer sekundären
Peritonitis sind Fieber, diffus abdomineller Schmerz, Übelkeit und Erbrechen. Bei der
klinischen Untersuchung sind eine lokale oder diffuse Abwehrspannung und verminderte bis
aufgehobene Darmgeräusche auffällig. Laborchemisch finden sich zumeist eine Leukozytose
und ein Anstieg des C-reaktiven Proteins (CRP) [71]. In einer klinischen Studie wurde die
durchschnittliche Letalität der intraabdominellen Infektionen mit 38% angegeben. Dabei war

Einleitung
19
die Sterblichkeitsrate bei der Appendizitis mit 10% am niedrigsten und mit 60% bei der
postoperativen Peritonitis am höchsten [76].
Die Keimbesiedlung bei intraabdominellen Infektionen ist meist polymikrobiell, bestehend
aus aeroben und anaeroben Bakterien [156, 181]. Die aeroben und anaeroben Bakterien
können in ihrer Pathogenität synergistisch wirken. Dabei beschreibt der bakterielle
Synergismus die Interaktion zwischen mindestens 2 verschiedenen Bakterienstämmen, die zu
einer Erhöhung des pathogenen Effekts führen. Ein einzelner Bakterienstamm würde diese
Wirkung nicht hervorrufen [110]. Durch tierexperimentelle Versuche konnte der
synergistische Effekt von E. coli und B. fragilis bezüglich der Abzessbildung und Letalität
gezeigt werden [125, 138]. Die Art des Synergismus scheint abhängig von der Größe des E.
coli Inokulum zu sein. Hohe E. coli Inokula in Mischkultur mit B. fragilis verursachen
synergistische Letalität und niedrige Inokula von E. coli Bakterien führen zur Abzessbildung
[140]. Frühere Untersuchungen unterstützen die Hypothese, dass ein Erreger direkt das
Wachstum eines anderen anwesenden Erregers durch beispielsweise Produktion von
Wachstumsfaktoren bedingt [49]. Der Verlauf dieser Mischinfektionen ist biphasisch mit
initial diffuser Infektion und anschließender Abzessbildung [62, 174]. Es scheint zunächst zur
Vermehrung der falkultativ anaeroben Keime zu kommen. Durch den Verbrauch von
Sauerstoff wird das Redoxpotential reduziert. Dadurch entsteht ein Mileu, welches für die
Vermehrung der obligaten Anaerobier optimal ist. Nach ungefähr 20 Stunden sind diese
Anaerobier die prädominanten Keime [148]. Einige Bakterienarten wirken zudem
immunmodulierend [29, 110, 141]. In verschiedenen in vitro und in vivo Studien konnte eine
Beeinträchtigung der Phagozytosefähigkeit durch Leukozyten oder Makrophagen nach
Kontakt mit Bacteroides spp. nachgewiesen werden [82, 90, 137].
1926 stellte Martin Kirschner die noch heute gültigen Richtlinien zur chirurgischen Therapie
der sekundären Peritonitis auf. Die Behandlung erfordert nach heutigem Maßstab neben dem
chirurgischen Eingriff eine antimikrobielle Therapie sowie die intensivmedizinische
Betreuung. 2003 wurden von der IDSA, der SIS, der amerikanischen Society of Microbiology
und der Society of Infectious Disease of Pharmacists evidenzbasierte Richtlinien zur
antibiotischen Therapie von intraabdominellen Infektionen herausgegeben. Auf Basis dieser
Richtlinien wurde 2004 durch Vogel, Bodmann und eine Expertengruppe der PEG eine
entsprechende Empfehlung der Peritonitis-Therapie herausgegeben. Tabelle 4 zeigt eine
Auflistung der möglichen antibiotischen Initialtherapie der sekundären Peritonitis nach der
durch Bodmann et al. im Jahr 2010 aktualisierten Empfehlung [27]. Die verschiedenen
antibiotischen Regimes sind in ihrer Wirkintensität ähnlich und keines der genannten Regime

Einleitung
20
konnte in Untersuchungen als über- oder unterlegen herausgestellt werden [107, 135, 156,
178]. Der behandelnde Arzt sollte sich ungeachtet der Empfehlungen zur antibiotischen
Therapie durch Fachgesellschaften immer auch nach dem aktuellen Wissensstand und neuen
Erkenntnissen richten. Beispielsweise wurden 2011 die Anwendungsgebiete für Tigecyclin
aufgrund erhöhter Letalität in klinischen Studien stark eingeschränkt [132].
Tabelle 4: Empfehlungen zur Initialtherapie der verschiedenen Formen der sekundären Peritonitis (modifiziert nach Bodmann et al., 2010 [27])
Infektion Initialtherapie
lokale sekundäre Peritonitis
(z. B. frisch perforierte
Appendizitis)
Cephalosporin Gruppe 2/3a + Metronidazol
Acylaminopenicillin/Betalaktamaseinhibitor
Aminopenicillin/Betalaktamaseinhibitor
Fluorchinolon Gruppe II + Metronidazol
Carbapenem Gruppe 2
diffuse sekundäre Peritonitis
(z. B. perforierte
Sigmadivertikulitis)
Acylaminopenicillin/Betalaktamaseinhibitor
Cephalosporin Guppe 3a/4 + Metronidazol oder
Fluorchinolon Gruppe II/III + Metronidazol
Carbapenem Gruppe 1
Carbapenem Gruppe 2
Fluorchinolon Gruppe IV
Tigecyclin
postoperative Peritonitis
(z. B.
Anastomoseninsuffizienz
nach Rektumresektion)
Carbapenem Gruppe 1
Carbapenem Gruppe 2
Acylaminopenicillin/Betalaktamaseinhibitor
Fluorchinolon Gruppe IV
Tigecyclin + ggf. Pseudomonas-wirksamer Substanz
Die Dauer der Therapie richtet sich nach der zu Grunde liegenden Genese der
intraabdominellen Infektion, insbesondere der sekundären Peritonitis. Allgemein wird
empfohlen, die Therapie bis zur klinischen und paraklinischen Normalisierung der Infektion
und Infektionsparameter fortzusetzen, wobei die Dauer der Therapie bis zu 14 Tagen betragen
sollte [96, 166]. Bei den intraabdominellen Infektionen werden E. coli und B. fragilis als
häufigste Erreger isoliert [30, 111, 180].

Einleitung
21
1.10 E. coli - Eigenschaften und Rolle als Krankheitserreger
E. coli aus der Familie der Enterobacteriaceae ist ein fakultativ anaerobes, gramnegatives,
peritrich begeißeltes, nicht sporenbildendes Stäbchenbakterium. 1885 wurde es erstmals von
Theodor Escherich beschrieben. Als natürliches Reservoir dienen der menschliche und
tierische Gastrointestinaltrakt. E. coli ist überwiegend laktose-positiv, katalase-positiv und
oxidase-negativ. Laktose-negative E. coli Bakterien lassen sich auch nachweisen. Es werden
mehr als 700 verschiedenen Serotypen nach Oberflächen (O)-, Kapsel (K)- und Geißel (H)-
Antigenen unterschieden. Das O-Antigen (O-spezifische Polysaccharidkette) gehört zum
Lipopolysaccharid (LPS, Endotoxin), welches Bestandteil der Zellwand dieser gramnegativen
Bakterien ist. Lipoid A, ebenfalls Bestandteil des LPS, ist nach dem Zerfall der
Bakterienzelle für die toxische Wirkung der E. coli Bakterien verantwortlich. Durch
Bekapselung des Bakteriums besteht Schutz vor Phagozytose. Diese Antigene sind neben
Adhäsinen, Invasinen und genetischen Eigenschaften wichtige Virulenzfaktoren von E. coli.
Besondere Bedeutung kommt dabei dem K1-Antigen von E. coli zu. Mittels kultureller
Anzucht und biochemischer Identifikation ("Bunte Reihe") erfolgt die Labordiagnostik [7, 67,
74]. E. coli dient zudem als Indikator für fäkale Verunreinigung und wird häufig in der
Molekularbiologie sowie als Wirt für Klonierungsvektoren in der Bio- und Gentechnologie
verwendet [7]. Die Gattung E. coli kann in obligat und fakultativ pathogene E. coli eingeteilt
werden. Die obligat pathogenen E. coli Stämme verursachen exogen intestinale Infektionen
und lassen sich in 5 Gruppen nach verursachender Erkrankung einteilen, wie in Tabelle 5
aufgeführt [174].
Tabelle 5: Obligat pathogene E. coli Stämme und ausgelöste Erkrankung (modifiziert nach Theuretzbacher, 1999 [174])
Darmpathogene E. coli Stämme Erkrankung
EPEC (enteropathogener E. coli ) Säuglingsenteritis
ETEC (enterotoxischer E. coli ) Reisediarrhoe
EIEC (enteroinvasiver E. coli ) ruhrähnliche Erkrankung
EHEC (enterohämorrhagischer E. coli ) Diarrhoe, HUS
EAEC (enteroaggreggativer E. coli ) peristierende Enteritis
Fakultativ pathogene E. coli sind Teil der physiologischen Standortflora, wo sie
üblicherweise keine Infektionen verursachen. Erst durch Verschleppung oder Absiedlung in
andere Körperregionen kommt es zu extraintestinalen Infektionen wie urogenitalen

Einleitung
22
Infektionen, nosokomialen Infektionen, intraabdominellen Infektionen und Sepsis. Zudem
kann E. coli Pneumonien verursachen, welche meist nach Aspiration oder
beatmungsassoziiert auftreten [18, 142, 172]. E. coli ist ein häufiger Erreger der
Neugeborenen-Meningitis, welche weltweit jährlich ungefähr 50000 Todesfälle fordert. In
80% der isolierten Erreger lässt sich dabei das K1-Antigen nachweisen, welches für die
Passage der Blut-Hirn-Schranke wichtig ist [91, 92]. Einige E. coli Stämme verfügen über
Fimbrien. Dadurch können sie am Urothel haften und so aufgrund von Schmierinfektionen
aus der Analregion bei Frauen häufig zu Harnwegsinfekten führen [74]. Die Differenzierung
der Fimbrien erfolgt über eine Hämagglutinationsreaktion mit Mannose. Typ 1 Fimbrien sind
Mannose-sensibel (Hemmung der Hämagglutination durch Mannose) und verursachen
Infektionen der unteren Harnwege und Typ 2 Fimbrien sind Mannose-resistent und bedingen
Pyelonephritiden [7]. E. coli ist häufigste Erreger von Harnwegsinfektionen und zudem für
15% der nosokomialen Bakteriämien verantwortlich [10].
1.11 B. fragilis - Eigenschaften und Rolle als Krankheitserreger
B. fragilis ist ein obligat anaerobes, gramnegatives, nicht sporenbildendes, teilweise
bekapseltes und begeißeltes Stäbchen. B. fragilis gehört zur Familie der Bacteroidaceae.
Diese Familien ist sehr heterogen und in Gattungen und Arten unterteilt [2]. Die wichtigsten
Vertreter sind Bacteroides spp., Prevotella spp., Porphyrommonas spp. und Fusobacterium
spp. Tabelle 6 enthält eine Auflistung der Bacteroides Gattung und Arten [182].
Tabelle 6: Bacteroides Gattung und Arten (modifiziert nach Wexler, 2007 [182])
Gattung Art
Bacteroides acidifaciens, caccae, coprocola, coprosuis, distasonis, dorei, eggerthii,
finegoldii, fragilis , goldsteinii, helcogenes, johnsonii, intestinalis,
massiliensis, merdae, nordii, ovatus, plebeius, pyogenes, salyersai,
stercoris, thetaiotaomicron, uniformis, vulgatus
Die Bacteroides Gattung stellt mengenmäßig den größten Teil der physiologischen
Bakterienflora von Mensch und Tier dar. Allerdings stellt B. fragilis nur 0,5% der Flora im
Kolon dar [62]. B. caccae, B. distasonis, B. eggerthii, B. fragilis , B. merdae, B. ovatus, B.
stercoris, B. thetaiotaomicron, B. uniformis und B. vulgatus werden im klassischen Sinn zu
der sogenannten B. fragilis Gruppe gezählt. B. capillosus, B. coagulans, B. putredinis, B.

Einleitung
23
pyogenesa, B. tectus, B. splanchnicus, B. tectusa, B. tectum und B. ureolyticus gehören zur
non-B. fragilis Gruppe [83].
Die Lipopolysaccharide der Zellwand von Bacteroides spp. wirken weniger toxisch, da sie
sich in ihrem Aufbau erheblich von denen der Enterobakterien unterscheiden [67]. Als
Virulenzfaktoren lassen sich bei Bacteroides spp. Kapselpolysaccharide nachweisen, welche
die Abszessbildung begünstigen. Unbekapselte Bacteroides Stämme benötigen zur
Abszessbildung die Anwesenheit eines aeroben Erregers [125, 126]. Bacteroides spp. haben
für den Menschen wichtige Eigenschaften. Sie produzieren beispielsweise Glucuronidasen,
welche für den enterohepatischen Kreislauf unerlässlich sind und Butyrat zur
Enterozytenernährung. Durch die Kolonisation von Haut und Schleimhäuten scheinen die
Bakterien der Ansiedlung von pathogenen Mikroorganismen vorzubeugen, was als
"Kolonisationsresistenz" bezeichnet wird [67]. Der Nachweis der Erreger erfolgt durch
Anzucht und die Identifizierung mittels biochemischer Untersuchungen. Bei Verdacht auf
eine Anaerobierinfektion sollte die Abnahme der Probe möglichst mittels Punktion oder
intraoperativ erfolgen. Der Transport in einem geeigneten Medium sollte zügig erfolgen
[148]. B. fragilis ist ein fakultativ pathogener Erreger. Der klinische Verlauf dieser
Infektionen ist selten akut. Es überwiegen subakute und chronische Verlaufformen. Als
Ursprungsorte für eine Anaerobierinfektion finden sich gastrointestinaler und urogenitaler
Trakt sowie die Mundhöhle [74]. Es kommen Infektionen des ZNS, des Urogenital- und
Abdominalsystems sowie den Respirationstraktes vor. Dabei kann die oropharyngeale Flora
Ursache für pulmonale Infektionen mit obligaten Anaerobiern sein. Meist handelt es sich
auch hier um Mischinfektionen, welche nach Aspirationen auftreten und zu Pneumonien,
Abszessen und Empyemen führen können [28, 109]. B. fragilis ist unter den obligaten
Anaerobiern hauptverantwortlich für die Abszessbildung und Bakteriämie [88]. Bei einer B.
fragilis Bakteriämie liegt die Letalität zwischen 20 und 31% [34]. In der Geburtsmedizin ist
die Puerpuralsepsis durch B. fragilis bei einem vorzeitigen Blasensprung gefürchtet [166].

Zielstellung
24
2 Zielstellung Intraabdominelle Infektionen sind häufig bakterielle Mischinfektionen. Dabei ist die
sekundäre Peritonitis die häufigste Form. E. coli und B. fragilis werden bei diesen
polymikrobiellen Infektionen am häufigsten isoliert. Die Therapie erfordert neben der primär
chirurgischen Versorgung eine antibiotische Therapie. Dabei basiert die möglichst
zielgerechte antibiotische Therapie auf Daten von Empfindlichkeitstestungen. Die
Bestimmung der MHK liefert Hinweise zur Empfindlichkeit der Erreger, wodurch Aussagen
über eine wahrscheinliche Wirkung in vivo gemacht werden können. Die klinische Einstufung
in die Grenzwerte "sensibel", "intermediär" und "resistent" schließt aber noch weitere
Faktoren wie Dosierung, Pharmakokinetik, Pharmakodynamik sowie Ergebnisse aus in vitro
und in vivo Studien mit ein. Die Ergebnisse von Untersuchungen mittels PK/PD-Modellen
gehen unter anderem in die Bewertung von Grenzwerten ein. Sie erlauben zudem einen
Vergleich der antimikrobiellen Effekte von verschiedenen Antibiotika einer als auch
unterschiedlicher Substanzklassen.
In der vorliegenden Arbeit wird mittels eines in vitro PK/PD-Modells die Aktivität von
Levofloxacin und Moxifloxacin in Mono- und Mischkulturen gegenüber B. fragilis und E.
coli untersucht. Hierdurch sollen sich Hinweise über die Aktivität von Levofloxacin und
Moxifloxacin in Mischinfektionen wie beispielsweise intraabdominellen Infektionen ergeben
und es soll abgeleitet werden, ob sich Unterschiede der antibakteriellen Aktivität gegenüber
E. coli Stämmen unter aeroben im Vergleich mit anaeroben Bedingungen ergeben. Des
Weiteren werden PK/PD-Indizes für Levofloxacin und Moxifloxacin berechnet. Diese Werte
werden dann in Abhängigkeit mit den Ergebnissen der Untersuchung für Mono- und
Mischkulturen bewertet.
Durch die vorliegende Arbeit lassen sich Aussagen treffen, ob die MHK-Bestimmung mit
Bewertung als Therapieempfehlung bei Mischinfektionen ausreichend ist oder ob bei
polymikrobiellen Infektionen weitergehende Untersuchungen für die Therapieempfehlungen
notwendig sind.

Materialien
25
3 Material
3.1 Geräte und Arbeitsplatz
Anaerobier-Werkbank mit Brutschrank HEAREUS, Hanau, Deutschland
Beleuchtung HEAREUS, Hanau, Deutschland
Brutschrank WTB BINDER LABORTECHNIK GmbH,
Tuttlingen, Deutschland
Gefrierschrank (-80 °C) HEAREUS, Hanau, Deutschland
Kühlschrank (-20 °C) LIEBHERR, Bulle, Schweiz
Membran-Vakuumpumpe VACUUBRAND GmbH & CO.,
Wertheim, Deutschland
Werkbank HEAREUS, Hanau, Deutschland
Resazurin-Anaerobier-Indikator (The
Anaerobic Indicator)
OXOID Ltd., Basingstoke, Hampshire, GB
Ultrospec 2000 (Photometer) PHARMACIA BIOTECH, Cambridge, GB
Die anaerobe Atmosphäre in der Anaerobierwerkbank bestand aus einem Gemisch von 80%
N2, 15% CO2 und 5% H2. Mittels eines Palladiumkatalysators wurde eventuell vorhandener
Restsauerstoff zu Wasser reduziert. Durch einen Resazurin-Anaerobier-Indikator wurde die
anaerobe Atmosphäre während jeder Versuchsdurchführung überprüft.
3.2 Laborbedarf
Drygalski-Spatel, steril MERCK, Darmstadt, Deutschland
Eppendorfgefäß (1,5 ml), steril SARSTEDT, Nürnbrecht, Deutschland
Eppendorfpipette EPPENDORF, Hamburg, Deutschland
Erlenmeyerkolben (300 ml), steril SCHOTT - Duran, Mainz, Deutschland
Handschuhe Peha - soft HARTMANN, Heidenheim, Deutschland
Impfschlingen, blau, steril SARSTEDT, Nürnbrecht, Deutschland
Küvetten ( 10 × 10 × 45 mm) GREINER Bio – One GmbH,
Kremsmünster, Österreich
Levofloxacin (LEV) SANOFI-AVENTIS, Frankfurt, Deutschland
Moxifloxacin (MOX) BAYER-SCHERING PHARMA, Leverkusen,
Deutschland

Materialien
26
Pipettenspitzen (10 µl, 100 µl, 1000 µl), steril SARSTEDT, Nürnbrecht, Deutschland
Pipetus-akku ( Pipettierhilfe) HIRSCHMANN LABORGERÄTE
Gmbh & CO. KG, Heilbronn, Deutschland
Serologische Pipette (10 ml, 25 ml, 50 ml),
steril
SARSTEDT, Nürnbrecht, Deutschland
Röhrchen (13 ml, 50 ml), steril SARSTEDT, Nürnbrecht, Deutschland
Vortexer JANKE & KUNKEL GmbH & CO. KG,
Staufen, Deutschland
Waage SARTORIUS, Göttingen, Deutschland
Wattestab, steril Dr. ILONA SCHUBERT,
Laborfachhandel, Pötzschau, Deutschland
3.3 Nährmedien zur Anzucht der verwendeten Bakterienkulturen und
Bakteriensuspensionen sowie Materialien zur Bestimmung der MHK
Brucella-Bouillon
BECTON Dickinson, Heidelberg,
Deutschland
Columbia-Agar OXOID Ltd., Basingstoke, Hamsphire, GB
Endo-Agar BIOMERIEUX, Marcy l´Étoile, Frankreich
Levofloxacin- und Moxifloxacin-
Etest-Streifen
BIOMERIEUX, Marcy l´Étoile, Frankreich
Gentamicin
MERCKLE GmbH, Blaubeuren,
Deutschland
Hämin
SERVA Feinbiochemica, Heidelberg,
Deutschland
Magermilchpulver OXOID Ltd., Basingstoke, Hamsphire, GB
Müller-Hinton-Agar BECTON, Dickinson and Company, Sparks,
USA, Le Pont de Claix, Frankreich
NaHCO3
MERCK EUROLAB GmbH, Darmstadt,
Deutschland
Schafblut, defibriniert OXOID GmbH, Wesel, Deutschland
Vitamin K1
SIGMA – Aldrich, Steinheim, Deutschland

Materialien
27
Um ein optimales Nährmedium für die Anzucht der Anaerobier zu haben, wurden 2 l
Columbia-Agar mit 100 ml Schafblut, 1 mg/l Vitamin K1, 5 mg/l Hämin und 1 ml/l 0,1%
NaHCO3 supplementiert.
Für die Herstellung des Columbia-Gentamicin-Agar wurden zu 2 l supplementierten,
autoklavierten und auf 50 °C abgekühlten Columbia-Agar 80 mg Gentamicin zugegeben. Das
Agarpulver wurde nach Angaben des Herstellers in der jeweils vorgeschriebenen Menge
destilliertem Wasser gelöst und für 15 min bei einer Temperatur von 121 °C autoklaviert.
Anschließend wurde der flüssige Agar auf etwa 50 °C abgekühlt und das Flüssigmedium in
Portionen zu je 20 ml unter sterilen Bedingungen in Kunststoffpetrischalen gegossen.
2 l Brucella-Bouillon wurden mit 1 mg/l Vitamin K1, 5 mg/l Hämin und 1 ml/l 0,1% NaHCO3
supplementiert.
Im Weiteren werden die genannten supplementierten Nährmedien nur noch als
supplementierter Columbia-Agar, supplementierter Columbia-Gentamicin-Agar und
supplementierte Brucella-Bouillon bezeichnet.

Methoden
28
4 Methoden
4.1 Untersuchte Bakterienstämme
Für die Versuche wurden B. fragilis und E. coli Stämme aus der Stammsammlung des
Instituts für Medizinische Mikrobiologie der Universität Leipzig verwandt. Bei den in Tabelle
7 aufgeführten Bakterienstämmen handelt es sich um Isolate von Patientenproben und einen
Referenzstamm. Die Isolate werden in Magermilch bei -80 °C gelagert.
Tabelle 7: Verwendete Bakterienstämme und deren Herkunft
Bakterienstamm Herkunft
E. coli ATCC 25922 Referenzstamm
E. coli VA 4050 Klinisches Isolat aus Rachenabstrich, Zustand nach
Herztransplantation
E. coli BK 12944 Klinisches Isolat aus Blutkultur,
hämatologisch/onkologische Erkrankung
E. coli VA 6886 Klinisches Isolat aus Gallenflüssigkeit, Klatskin-
Carcinom
B. fragilis RMA 6791 Klinisches Isolat aus Blutkultur
B. fragilis WAL R 13267 Klinisches Isolat, Entnahmeort unbekannt
B. fragilis RMA 5120 Klinisches Isolat aus Appendix vermiformis,
Appendizitis
B. fragilis RMA 0309 Klinisches Isolat aus Abdomen
Die Stämme mit der Bezeichnung RMA wurden freundlicherweise von E. J. C. Goldstein, R.
M. Alden Research Laboratory, Santa Monica, Kalifornien, USA, zur Verfügung gestellt.
Der WAL R 13267 Stamm stammt aus einer internationalen klinischen Anaerobierstudie. Bei
den E. coli Stämmen handelt es sich um einen Referenzstamm (E. coli ATCC 25922) sowie
klinische Isolate aus Patientenproben des Institutes für Medizinische Mikrobiologie der
Universität Leipzig.
4.2 Anzucht der Bakterienkulturen
Zur Anzucht der Bakterienstämme wurden für die E. coli Stämme Endo-Agar und für die B.
fragilis Stämme supplementierter Columbia-Agar verwendet. Die Bakterien wurden aus der

Methoden
29
gefrorenen Magermilchlösung mit einer sterilen Impfschlinge auf das entsprechende
Agarmedium übertragen. Die E. coli Stämme wurden für 24 h aerob auf Endo-Agar bebrütet.
Die B. fragilis Stämme wurden für 48 h unter anaeroben Bedingungen auf supplementiertem
Columbia-Agar bebrütet. Zur Kontrolle wurde beimpfter Agar aerob bebrütet.
4.3 Untersuchung zur Wachstumshemmung von vier E. coli Stämmen auf
supplementiertem Columbia-Gentamicin-Agar
In dieser Arbeit wurden Untersuchungen mit B. fragilis und E. coli Stämmen sowohl in
Mono- als auch in Mischkulturen durchgeführt.
Bei den Mischkulturen wuchsen sowohl B. fragilis und E. coli Kolonien gemeinsam auf dem
supplementierten Columbia-Agarmedium. Um die Ergebnisse besser auswerten zu können,
wurde eine mögliche Wachtumsunterdrückung der E. coli Kolonien auf supplementiertem
Columbia-Gentamicin-Agar untersucht. Es wurden Inokula aus E. coli und B. fragilis
Stämmen hergestellt und dann zu den verschiedenen Zeitpunkten 0, 0,5, 1, 2, 4, 6 und 8 h auf
den Columbia-Gentamicin-Agarmedium und den Columbia-Agarmedium ausgestrichen und
für 48 h anaerob inkubiert.
4.4 Bestimmung der MHK von Levofloxacin und Moxifloxacin
Die MHK von Levofloxacin und Moxifloxacin der untersuchten Bakterienstämme wurde mit
dem Etest nach Angaben des Herstellers bestimmt.
Es wurden 2-3 Bakterienkolonien vom jeweiligen Agarmedium in 10 ml präreduzierte,
supplementierte Brucella-Bouillon abgeimpft. Die Suspension mit den E. coli Stämmen
wurde für 24 h aerob bei 37 °C inkubiert. Bei den B. fragilis Stämmen erfolgte die
Durchführung unter anaeroben Bedingungen mit einer Inkubationszeit von 48 h bei einer
Temperatur von 37 °C.
Nach der Bebrütung wurde für das B. fragilis Inokulum ein McFarland Standardwert 1
eingestellt, welcher einer Bakteriendichte von 3 × 108 KBE/ml entspricht. Für das E. coli
Inokulum erfolgte nach Einstellung von McFarland 1 eine 20-fache Verdünnung. Die
Beimpfung erfolgte mittels eines sterilen Wattetupfers. Dabei wurde die gesamte Oberfläche
des Agarmediums in drei Richtungen gleichmäßig ausgestrichen. Für die Etest®-Versuche der
E. coli Stämme wurde Müller-Hinton-Agar und für die B. fragilis Stämme supplementierter
Columbia-Agar verwendet, wobei eine Präreduktion des Agarmediums von ungefähr drei
Stunden erfolgte. Die Etest-Streifen von Levofloxacin und Moxifloxacin wurden danach
Methoden
30
blasenfrei und zentral mittels einer Pinzette auf das Agarmedium aufgebracht. Die Ablesung
erfolgte bei den E. coli Stämmen nach 24 h aerober Inkubation und bei den B. fragilis
Stämmen nach 48 h anaerober Inkubation bei 37 °C. Am Schnittpunkt des Hemmhofes mit
dem Teststreifen konnte die minimale Hemmkonzentration abgelesen werden.
4.5 PK/PD-Untersuchung von Levofloxacin und Moxifloxacin
Ein Dosierungsintervall bei der Gabe einer bestimmten antibiotischen Dosis pro Tag ist
Grundlage der zeitlichen Durchführung des Versuches, da dieses Intervall abhängig von der
Halbwertszeit der jeweiligen Substanz ist. Die durchschnittlichen Dosiswerte sind nach Stille
et al. [166] in Tabelle 8 aufgelistet und beziehen sich auf eine normalgewichtige,
nierengesunde Person (ca. 75 kg Körpergewicht).
Tabelle 8: Tagesdosis und Dosierungshäufigkeit der Antibiotika (nach Stille et al. [166])
Antibiotika Tagesdosis und Dosierungshäufigkeit/Tag
Levofloxacin 1 bis 2 × täglich 0,25-0,5 g p.o. oder i.v. als Kurzinfusion
Moxifloxacin 1 × täglich 0,4 g p.o. oder i.v. als Kurzinfusion
Tabelle 9 zeigt eine Auflistung der pharmakokinetischen Eigenschaften der Antibiotika nach
Werten von Stille et al. sowie der Versuchsdauer im in vitro PK/PD-Modell [166].
Tabelle 9: Pharmakokinetische Eigenschaften der Antibiotika nach Stille et al. [166] und Versuchsdauer im in vitro PK/PD-Modell
Antibiotikum i.v. Gabe von Cmax (mg/l) t ½ e (h) Versuchsdauer (h)
Levofloxacin 0,5 g in 60 min 6,2 7-8 13
Moxifloxacin 0,4 g in 30 min 4,4 12 12
Die Halbwertszeit von Moxifloxacin entspricht der Versuchsdauer. Trotz der kürzeren
Halbwertszeit von Levofloxacin wurde zur besseren Vergleichbarkeit eine ähnlich lange
Versuchsdauer wie bei Moxifloxacin gewählt. Die Plasmakonzentration des entsprechenden
Antibiotikums über die Zeit gesehen, ist abhängig von der Eliminationshalbwertszeit und
ändert sich nach folgender Formel [119]:
Ct = C0 × e – k × t
C0 = Ausgangskonzentration zum Zeitpunkt t = 0 (mg/l)
Ct = Plasmakonzentration zum Zeitpunkt t (mg/l)
Methoden
31
k = Eliminationskonstante
k = ln2 / t ½ e
t ½ e = Eliminationshalbwertszeit
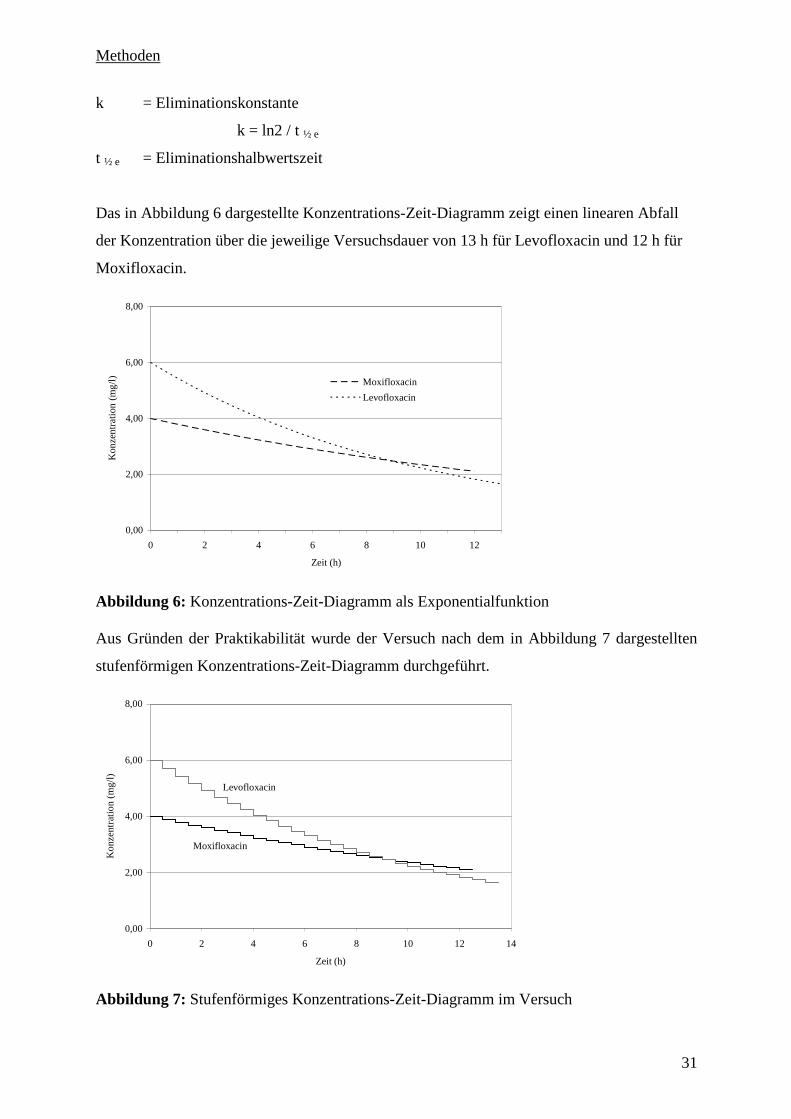
Das in Abbildung 6 dargestellte Konzentrations-Zeit-Diagramm zeigt einen linearen Abfall
der Konzentration über die jeweilige Versuchsdauer von 13 h für Levofloxacin und 12 h für
Moxifloxacin.
0,00
2,00
4,00
6,00
8,00
0 2 4 6 8 10 12
Zeit (h)
Kon
zen
tra
tion
(m
g/l)
Moxifloxacin
Levofloxacin
Abbildung 6: Konzentrations-Zeit-Diagramm als Exponentialfunktion Aus Gründen der Praktikabilität wurde der Versuch nach dem in Abbildung 7 dargestellten
stufenförmigen Konzentrations-Zeit-Diagramm durchgeführt.
0,00
2,00
4,00
6,00
8,00
0 2 4 6 8 10 12 14
Zeit (h)
Ko
nze
ntra
tion
(mg/
l)
Levofloxacin
Moxifloxacin
Abbildung 7: Stufenförmiges Konzentrations-Zeit-Diagramm im Versuch

Methoden
32
Durch entsprechende Verdünnung des Ansatzes über die Zeit wurde der Konzentrationsabfall
im in vitro PK/PD-Modell erreicht. Um die nächste Konzentrationsstufe entsprechend der
Eliminationshalbwertszeit der Antibiotika zu erreichen, wurde das Inokulum verdünnt
(Tabellen 18 und 19 im Anhang 1 und 2). Aus Gründen der Praktikabilität wurde
halbstündlich verdünnt. Für die Berechnung des jeweiligen Zugabevolumens wurde folgende
Formel verwendet:
VZU = [(VSL × CSL) / C] - VSL
VZU = zuzugebendes Volumen (ml)
VSL = eingesetztes Volumen der Stammlösung (ml)
C SL = Konzentration der Stammlösung (mg/l)
C = benötigte Konzentration (mg/l)
4.6 Herstellung der Antibiotikasuspensionen
Unter Berücksichtigung der in Tabelle 9 aufgeführten maximal erreichbaren
Plasmakonzentrationen von Levofloxacin und Moxifloxacin erfolgte die Berechnung der
Volumina für die Versuche mit den entsprechend verifizierten Antibiotikumkonzentrationen,
welche in Tabelle 10 aufgeführt sind. Die maximalen Plasmakonzentrationen wurden den
totalen Konzentrationen gleichgesetzt, da die Proteinbindung keine wesentliche
Berücksichtigung findet. Wolfson et al. konnten bei den Fluorchinolonen nur minimale
Unterschiede bei der MHK-Bestimmung in den Testmedien mit Serum im Vergleich zu
serumfreien Medien feststellen [186]. Fuhst et al. kamen zum Schluss, dass eine
Plasmaproteinbindung unter 80% die Wirkung eines Antibiotikums nicht beeinflusst. Unter
anderem konnte für Moxifloxacin gezeigt werden, dass die Wirkung der Gesamtdosis mit
Albumin identisch mit der Wirkung der berechneten freien Fraktion ist [60].
Tabelle 10: Konzentrationen der Inokulavolumen zur Herstellung der Antibiotikumsuspensionen
Antibiotikum
Konzentration im
Inokulumvolumen
(mg/l)
Lösungsmittel
Levofloxacin 60 Aqua dest.
Moxifloxacin 40 Aqua dest.

Methoden
33
Nach der Herstellung wurden die Antibiotikasuspensionen in Fraktionen zu je 1 ml aufgeteilt
und bei -80 °C tiefgefroren, um einen Aktivitätsverlust zu vermeiden.
4.6.1 Herstellung der Inokula für die Versuche Die bei der Herstellung der Inokula und im Versuch benötigte supplementierte Brucella-
Bouillon wurde immer aerob und anaerob bei 37 °C für 24 h vor Versuchsbeginn inkubiert,
um Verunreinigungen während der einzelnen Versuchsphasen auszuschließen.
Die im Versuch verwendeten supplementierten Columbia- und Columbia-Gentamicin-
Agarmedien wurden vor Versuchsbeginn für 2-3 h, die supplementierte Brucella-Bouillon für
24 h bei 37 °C in anaerober Atmosphäre präreduziert. Die Endo-Agarmedien der
Monokulturversuche wurden für 2-3 h vor Versuchsbeginn in einer aeroben Atmosphäre im
Brutschrank bei einer Temperatur von 37 °C belassen.
Für die Herstellung der Inokula wurde Endo-Agar mit den jeweiligen E. coli Stämmen unter
aeroben Bedingungen beimpft, ausgestrichen und für 24 h aerob bei 37 °C inkubiert. Für die
B. fragilis Stämme wurde supplementierter Columbia-Agar verwendet und im anaeroben
Milieu 48 h bei 37 °C inkubiert. Nach der entsprechenden Inkubationszeit wurde von den
bebrüteten Medien eine Kolonie des jeweiligen E. coli Stammes unter aeroben Bedingungen
und je eine Kolonie der B. fragilis Stämme vom entsprechenden Agarmedium unter
anaeroben Bedingungen mit einer sterilen Öse abgenommen. Diese gewonnenen Kolonien
wurden in 10 ml supplementierte Brucella-Bouillon abgeimpft. Die Bouillon mit den E. coli
Stämmen wurde 24 h aerob und die Bouillon mit den B. fragilis Stämmen 48 h anaerob bei 37
°C inkubiert. Mittels Spektrophotometer wurden die in Tabelle 11 aufgeführten
Bakteriendichten bestimmt und verdünnt, um die entsprechenden McFarland-Standards zu
erhalten.
Tabelle 11: McFarland Standard und entsprechende Bakteriendichte der verwendeten Bakterien
Bakterien Bakteriendichte (KBE/ml) McFarland Standard
B. fragilis 1,2 × 109 (entsprechend 2,4 × 108 im Inokulum) 4
E. coli 1,5 × 108 (entsprechend 3 × 107 im Inokulum) 0,5
Zu jedem Inokulum mit Antibiotikum wurde eine Kontrollkultur ohne Antibioitkum
mitgeführt. Das fehlende Antibiotikumvolumen wurde durch äquivalente Mengen
Methoden
34
supplementierter Brucella-Bouillon ergänzt. Die Ansätze wurden in sterile Röhrchen
pipettiert.
Die Vorbereitung und Versuchsdurchführung mit B. fragilis Monokulturen und
Mischkulturen erfolgten in anaerober Atmosphäre. Die Versuchsvorbereitung und
Versuchsdurchführung mit E. coli Monokulturen wurden sowohl aerob, als auch anaerob
durchgeführt. Tabellen 12 und 13 zeigen die Zusammensetzung der Versuchsinokula für
Mono- und Mischkulturen.
Tabelle 12: Pipettierschema für die Versuchsreihe mit Levofloxacin
Monokulturen
E. coli B. fragilis Mischkulturen
LEV+ LEV- LEV+ LEV- LEV+ LEV-
Brucella-Bouillon (ml) 7 8 7 8 5 6
E. coli-Suspension (ml) 2 2 0 0 2 2
B. fragilis-Suspension (ml) 0 0 2 2 2 2
Antibiotikumsuspension (ml) 1 0 1 0 1 0
Ansatzvolumen gesamt (ml) 10 10 10 10 10 10
LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin
Tabelle 13: Pipettierschema für die Versuchsreihe mit Moxifloxacin
Monokulturen
E. coli Mischkulturen
MOX+ MOX- MOX+ MOX-
Brucella-Bouillon (ml) 14 16 10 12
E. coli-Suspension (ml) 4 4 4 4
B. fragilis-Suspension (ml) 0 0 4 4
Antibiotikumsuspension (ml) 2 0 2 0
Ansatzvolumen gesamt (ml) 20 20 20 20
MOX+, mit Moxifloxacin; MOX-, Kontrolle ohne Moxifloxacin
4.6.2 Versuchsdurchführung
In Abbildung 8 ist das Pipettierschema für den Versuchsablauf bei Mischkulturen mit
entsprechendem Verdünnungsfaktor vereinfacht dargestellt. Zunächst wurden jeweils 6,6 µl
Bakteriensuspension aus den Inokula der Mischkulturen auf ein supplementiertes Columiba-

Methoden
35
Gentamicin-Agarmedium und ein Endo-Agarmedium pipettiert. Weitere 6,6 µl wurden in ein
Eppendorfgefäß mit 660 µl 0,9%iger NaCl-Lösung pipettiert und vermischt, um eine
Verdünnung der Bakterienkonzentration von 1:100 erhalten.
Im zweiten Schritt wurden dreimal jeweils 6,6 µl aus der zuvor verdünnten
Bakteriensuspension des Eppendorfgefäßes entnommen und auf ein supplementiertes
Columbia-Gentamicin-Agarmedium, ein Endo-Agarmedium und in 660 µl 0,9%ige NaCl-
Lösung gebracht. Der Inhalt des Eppendorfgefäßes wurde wieder gleichmäßig vermischt und
man erhält eine weitere Verdünnungsstufe von 1:100. Im dritten Schritt wurden erneut jeweils
6,6 µl aus dem im zweiten Schritt bearbeiteten Eppendorfgefäß entnommen und wiederum
auf ein supplementiertes Columbia-Gentamicin-Agarmedium, ein Endo-Agarmedium und in
66 µl 0,9%ige NaCl-Lösung eines Eppendorfgefäßes pipettiert. Von der zuvor hergestellten,
1:10 verdünnten Bakteriensuspension wurden 6,6 µl entnommen und auf ein supplementiertes
Columbia-Gentamicin-Agarmedium und ein Endo-Agarmedium gebracht.
Abbildung 8: Pipettierschema und Verdünnungsfaktor pro 1 ml bei Mischkulturen A, Inokulumansatz; C, Columbia-Gentamicin-Agarmedium; E, Endo-Agarmedium
Durch die einzelnen Verdünnungsstufen sollte die Vermeidung des "Drug-carry-over"-
Phänomens und eine Optimierung bei der Ablesung der Versuchsergebnisse erzielt werden.
Das "Drug-carry-over" bezeichnet die Hemmung des bakteriellen Wachstums in vitro,
bedingt durch zu hohe Antibiotikumkonzentrationen im Inokulum. Bei der Entnahme der
Verdünnungsfaktor pro 1 ml
1:151
1:151 × 102
1:151 × 104
1:151 × 105

Methoden
36
Proben kann neben Bakterien und Bouillon auch Antibiotikum mit entnommen werden. Es
kann daher zu einer weiteren Hemmung des bakteriellen Wachstums kommen. Durch die
Verdünnungen geht man von einer vernachlässigbaren Wirkung des Antibiotikums aus [104,
158].
Die zuvor auf die Agarmedien aufgebrachten Bakteriensuspensionen wurden mittels
Drygalskispatel in drei Richtungen gleichmäßig ausgestrichen. Die Endo-Agarmedien wurden
für 24 h unter aeroben Bedingungen bei 37 °C bebrütet. Die supplementierten Columbia-
Gentamicin-Agarmedien wurden für 48 h bei 37 °C in anaerober Atmosphäre inkubiert.
Um die Elimination der Antibiotika zu simulieren, wurden die Volumina der
supplementierten Brucella-Bouillon nach Vorgaben der Tabellen 18 und 19 (Anhang 1 und 2)
in die Ansätze pipettiert. Danach wurden die Inokula sofort für weitere 30 Minuten bei 37 °C
anaerob inkubiert. Insgesamt wurden alle 30 Minuten in jedem einzelnen Pipettierschritt
19,98 µl Bakteriensupsension aus den Ansatzbehältnissen entnommen, dreimal verdünnt und
jeweils gleichmäßig ausgestrichen. Die zum Auszählen der Bakterienzahlen ausgestrichenen
Anteile der Inokula von insgesamt 19,98 µl wurden bei der Berechnung der zuzugebenen
Volumina vom Gesamtvolumen abgezogen.
In der Versuchsdurchführung für die Monokulturen der E. coli und B. fragilis Stämme
wurden im ersten Verdünnungsschritt jeweils 6,6 µl des Inokulums verworfen, um gleiche
Volumenabnahmen in Mono- und Mischkulturen zu erhalten.
4.6.3 Ablesung der Versuchsergebnisse und Berechnung der Abtötungsraten Nach Inkubation der Agarmedien erfolgte die Ablesung, wobei unter günstigen
Wachstumsbedingungen jedes Bakterium eine Kolonie bildete. Ein Problem stellte die
Bildung von Bakterienverbänden dar, da jeder aus mehreren Bakterien bestehende Verband
eventuell nur eine Kolonie bilden konnte. Dieser Bakterienverband wurde deshalb als eine
Kolonie gewertet. Das Ergebnis wird als Koloniebildende Einheiten pro 1 ml angegeben und
nachfolgend als KBE/ml bezeichnet. Die Nachweisgrenze lag bei 101 KBE/ml.
Die Kolonien wurden gezählt und mit dem entsprechenden Verdünnungsfaktor multipliziert.
Um die tatsächliche Zahl der Koloniebildenden Einheiten zu erhalten, wurde folgende Formel
angewandt:
KBEt [KBE/ml] = (C0 / Ct) × KBEt(gezählt)
KBEt Anzahl der KBE zum Zeitpunkt t (KBE/ml)
C0 Bakteriendichte im Inokulum bei Versuchsbeginn (mg/l)

Methoden
37
Ct Bakteriendichte im Inokulum zum Zeitpunkt t (mg/l)
KBEt(gezählt) Anzahl der KBE im Inokulum zum Zeitpunkt t (KBE(gezählt)/ml)
Die Reduktion der KBE, welche in Log-Stufen angegeben wird, berechnet sich nach DIN
58940-7: 1994-09 nach folgender Gleichung [6]:
RKBE = Log10 (KBET0) – Log10 (KBETx)
Die prozentuale Reduktion der KBE lässt sich durch folgende Gleichung berechnen:
RKBE[%] = 100 – (KBETx/KBET0 × 100)
KBET0 KBE im Inokulum bei Versuchsbeginn in ml
KBETx KBE der Testsuspension nach Entnahme zum Zeitpunkt Tx in ml
(Tx beschreibt in diesem in vitro Modell den Endzeitpunkt des
Versuches (tLEV=13h, tMOX =12h)
Eine Reduktion der KBE um 3 Log-Stufen entspricht dabei einer Reduktion um 99,9%.
4.6.4 Berechnung der pharmakologischen Indizes
Als AUC wird die Fläche unterhalb der Konzentrations-Zeit-Kurve bezeichnet. Wenn nicht
explizit ausgeschrieben, bezieht sich die AUC auf einen Zeitraum von 24 h. Die AUC wird
mit folgender Formel als Integral über der Zeit errechnet:
∫∞
∞→ =0
0 )( dttCAUC [mg×h/l]
Bei einer linearen Kinetik lässt sich die AUC vereinfacht mit der Trapezregel berechnen,
welche nachfolgend aufgeführt ist [31]:
))(( 11
121
0 −=
−→ −+= ∑ xxx
n
ixt ttCCAUC
n
AUC Area under the curve [mg×h/l]

Methoden
38
Cx Konzentration zum Zeitpunkt tx [mg/l]
Cx-1 Konzentration zum Zeitpunkt tx-1 [mg/l]
tn letzter zu berechnender Zeitpunkt [h]
Dividiert man die AUC durch die MHK, erhält man den Index:
� MHKAUC [dimensionslos]
Das Verhältnis aus maximaler Plasmakonzentration und MHK wird durch den folgenden
Index beschrieben:
� MHKCmax [dimensionslos]
4.6.5 Statistische Datenanalyse Die errechneten KBE wurden in einer Tabelle erfasst und mit Hilfe von Graph Pad Prism5
Software (Graph Pad Software Inc., La Jolla, CA, USA) graphisch dargestellt.
Zur statistischen Analyse der Trends der Abtötungsraten über die Zeit wurde der Mann-
Trend-Test angewandt [70, 75]. Dabei wurde ein p-Wert < 0,05 als signifikant und ein p-Wert
< 0,01 als hochsignifikant gewertet. P-Werte über 0,05 wurden als nicht signifikant gewertet.
Die Versuche zur Aktivität von Moxifloxacin in Monokultur mit den B. fragilis Stämmen
wurden von Schaumann et al. durchgeführt und ausgewertet [146]. Die Werte der KBE der B.
fragilis Stämme dieser Versuche wurden für die statische Analyse der vorliegenden Arbeit
(Vergleich der Trends der Abtötungsraten in Mono- und Mischkulturen) freundlicherweise
durch Schaumann et al. zur Verfügung gestellt.

Ergebnisse
39
5 Ergebnisse
5.1 Wachstumshemmung von vier E. coli Stämmen auf supplementiertem Columbia-
Gentamicin-Agar und Beschreibung der Bakterienstämme
Auf den supplementierten Columbia-Agarmedien wachsen sowohl B. fragilis und E. coli
Stämme. Um die Auszählung der Mischkulturen zu erleichtern, wurde supplementierter
Columbia-Gentamicin-Agar mit dem Inokulum beimpft, um somit eine eventuelle
Wachstumshemmung der E. coli Stämme zu überprüfen. Die Ergebnisse sind in Tabelle 14
aufgeführt. Da sich nach der Inkubation von 48 h bei den zu verschiedenen Zeitpunkten (0,
0,5, 1, 2, 4, 6, 8 h) ausgestrichen Inokula vergleichbare Ergebnisse ergaben, sind die Zeiten
nicht extra aufgeführt.
Tabelle 14: Ergebnisse des Versuches der Wachstumshemmung auf supplementiertem Columbia-Gentamicin-Agarmedium
Stamm supplementierter
Columbia-Agar
supplementierter Columbia-
Gentamicin-Agar
E. coli ATCC 25922 Wachstum kein Wachstum
E. coli VA 4050 Wachstum Wachstum
E. coli BK 12944 Wachstum kein Wachstum
E. coli VA 6886 Wachstum kein Wachstum
Der E. coli Stamm VA 4050 ist resistent gegenüber Gentamicin. Auf den supplementierten
Columbia-Gentamicin-Agarmedien war ein uneingeschränktes Wachstum des
Bakterienstamms zu sehen. Daher wurden sowohl der E. coli Stamm VA 4050 als auch die B.
fragilis Stämme auf einem supplementierten Columbia-Gentamicin-Agarmedium ausgezählt.
Die E. coli Kolonien wurden mit denen auf Endo-Agar wachsenden E. coli Kolonien
verglichen und stimmten dabei in ihrer Zahl weitgehend überein. Alle B. fragilis Stämme
wuchsen auf supplementiertem Columbia-Gentamicin-Agarmedium.
Die E. coli Stämme bilden nach 24 h unter aeroben Bedingungen deutlich sichtbare Kolonien
auf Endo-Agar, sind lactosepositiv und haben eine glatte Oberfläche. Die E. coli Kolonien der
Stämme VA 6886 und ATCC 25922 erscheinen hierbei rund mit einem Durchmesser von
ungefähr 5 mm. Die Kolonien der E. coli Stämme VA 4050 und BK 12944 sind ebenfalls
rund mit einem Durchmesser von ungefähr 1-3 mm. Die B. fragilis Kolonien haben auf den
supplementierten Columbia-Agarmedien und Columbia-Gentamicin-Agarmedien einen

Ergebnisse
40
Durchmesser von 1-3 mm und sind glattwandig mit leicht glänzender Oberfläche und einem
grauen Kolorit. Die Kolonien des E. coli Stammes VA 4050 haben auf supplementiertem
Columbia-Gentamicin-Agarmedium einen Durchmesser von 1-3 mm mit einem
hellschimmernden beigefarbenen Hof und einem gelbfarbenen zentralen Punkt. Dadurch ist
dieser Stamm deutlich von den B. fragilis Stämmen zu unterscheiden.
5.2 Ergebnisse der MHK-Bestimmung
Bei den untersuchten Stämmen wurden die in Tabelle 15 aufgeführten MHK-Werte ermittelt.
Da diese Untersuchungen für jeden Stamm mindestens dreimal durchgeführt wurden, ergeben
sich zum Teil voneinander abweichende Werte. Die Bewertung des Grenzwertes bezüglich
der Empfindlichkeit erfolgt nach DIN 58940-4 Beiblatt 1 (Mai 2010) [5] in sensibel (s),
intermediär (i) und resistent (r) und ist angepasst an definierte Grenzwerte der EUCAST [8].
Grenzwerte Levofloxacin: ≤ 1 (s), 2 (i), > 2 (r) [5]
Grenzwerte Moxifloxacin: ≤ 0,5 (s), 1 (i), > 1 (r) [5]
Tabelle 15: MHK-Werte (mg/l) und Grenzwerte nach DIN 58940-4 [5]
Stamm Levofloxacin Moxifloxacin
E. coli ATCC 25922 0,008-0,03 (s) 0,015-0,03 (s)
E. coli VA 4050 > 32 (r) > 32 (r)
E. coli BK 12944 > 32 (r) > 32 (r)
E. coli VA 6886 > 32 (r) > 32 (r)
B. fragilis RMA 6791 1-2 (s)-(i) 0,25-0,5 (s)
B. fragilis WAL R 13267 > 32 (r) > 32 (r)
B. fragilis RMA 5120 1 (s) 0,12-0,5 (s)
B. fragilis RMA 0309 0,25-1 (s) 0,12-0,25 (s)

Ergebnisse
41
5.3 Aktivität von Levofloxacin und Moxifloxacin im in vitro PK/PD-Modell
5.3.1 Aktivität von Levofloxacin gegenüber vier E. coli Stämmen in Monokultur unter
aeroben und anaeroben Bedingungen
Im Ansatz mit Levofloxacin werden die KBE des E. coli Stammes ATCC 25922 unter
aeroben Versuchsbedingungen (Abb. 9) signifikant reduziert (p < 0,01). Levofloxacin wirkt
unter aeroben Bedingungen bakterizid (Abtötungsrate >99,9%). Bei den E. coli Stämmen VA
4050, BK 12944 und VA 6886 lassen sich unter aeroben Versuchsbedingungen keine
signifikanten Unterschiede in den Wachstumskurven mit und ohne Antibiotikum feststellen
(p > 0,05).
0 1 2 3 4 5 6 7 8 9 10 11 12 130
2
4
6
8
10
EC VA 4050 LEV+ EC VA 4050 LEV-EC BK 12944 LEV+ EC BK 12944 LEV-EC ATCC 25922 LEV+ EC ATCC 25922 LEV-EC VA 6886 LEV+ EC VA 6886 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 9: Aktivität von Levofloxacin gegenüber vier verschiedenen Stämmen von E. coli (EC) im in vitro PK/PD-Modell in Monokulturen mit und ohne Levofloxacin unter aeroben Bedingungen. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml. Auch unter anaeroben Bedingungen (Abb. 10) werden die KBE des E. coli Stammes ATCC
25922 durch Levofloxacin signifikant reduziert (p < 0,01). Dabei wirkt Levofloxacin jedoch
nicht bakterizid (Abtötungsrate 99,1%). Bei den E. coli Stämmen VA 4050, BK 12944 und
VA 6886 lassen sich auch unter anaeroben Versuchsbedingungen keine signifikanten
Unterschiede in den Wachstumskurven mit und ohne Levofloxacin feststellen (p > 0,05).
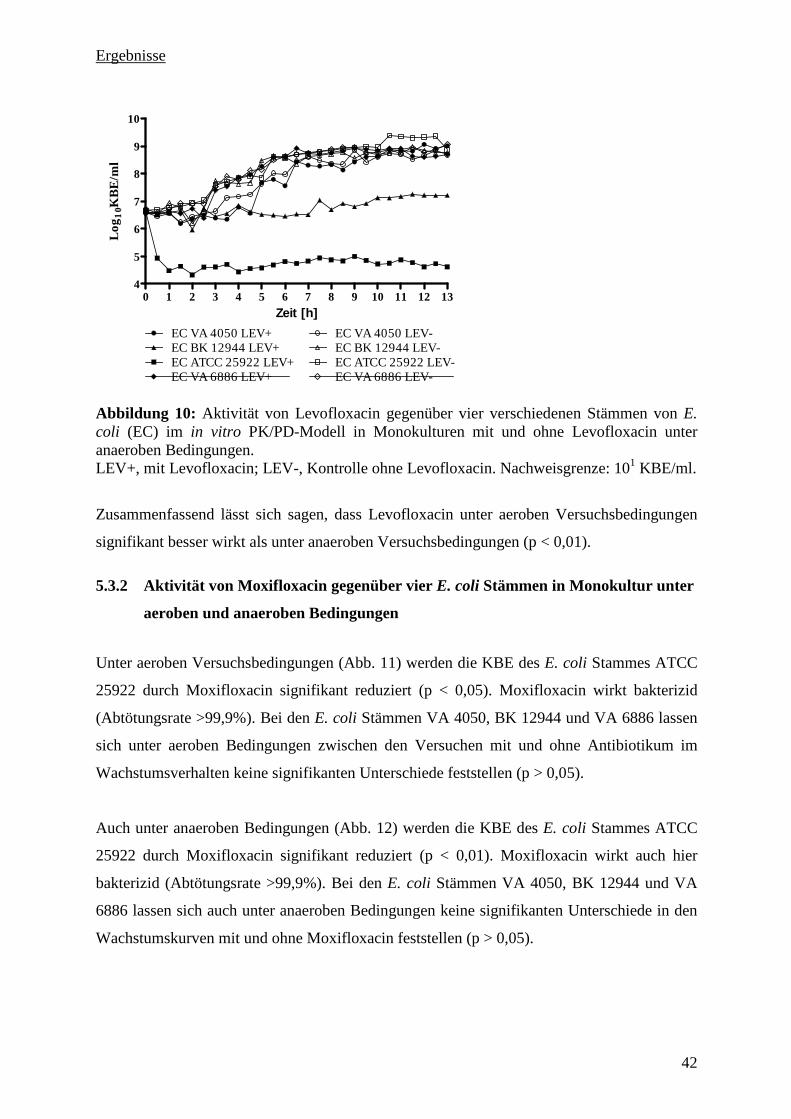
Ergebnisse
42
0 1 2 3 4 5 6 7 8 9 10 11 12 134
5
6
7
8
9
10
EC VA 4050 LEV+ EC VA 4050 LEV-EC BK 12944 LEV+ EC BK 12944 LEV-EC ATCC 25922 LEV+ EC ATCC 25922 LEV-EC VA 6886 LEV+ EC VA 6886 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 10: Aktivität von Levofloxacin gegenüber vier verschiedenen Stämmen von E. coli (EC) im in vitro PK/PD-Modell in Monokulturen mit und ohne Levofloxacin unter anaeroben Bedingungen. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.
Zusammenfassend lässt sich sagen, dass Levofloxacin unter aeroben Versuchsbedingungen
signifikant besser wirkt als unter anaeroben Versuchsbedingungen (p < 0,01).
5.3.2 Aktivität von Moxifloxacin gegenüber vier E. coli Stämmen in Monokultur unter
aeroben und anaeroben Bedingungen
Unter aeroben Versuchsbedingungen (Abb. 11) werden die KBE des E. coli Stammes ATCC
25922 durch Moxifloxacin signifikant reduziert (p < 0,05). Moxifloxacin wirkt bakterizid
(Abtötungsrate >99,9%). Bei den E. coli Stämmen VA 4050, BK 12944 und VA 6886 lassen
sich unter aeroben Bedingungen zwischen den Versuchen mit und ohne Antibiotikum im
Wachstumsverhalten keine signifikanten Unterschiede feststellen (p > 0,05).
Auch unter anaeroben Bedingungen (Abb. 12) werden die KBE des E. coli Stammes ATCC
25922 durch Moxifloxacin signifikant reduziert (p < 0,01). Moxifloxacin wirkt auch hier
bakterizid (Abtötungsrate >99,9%). Bei den E. coli Stämmen VA 4050, BK 12944 und VA
6886 lassen sich auch unter anaeroben Bedingungen keine signifikanten Unterschiede in den
Wachstumskurven mit und ohne Moxifloxacin feststellen (p > 0,05).
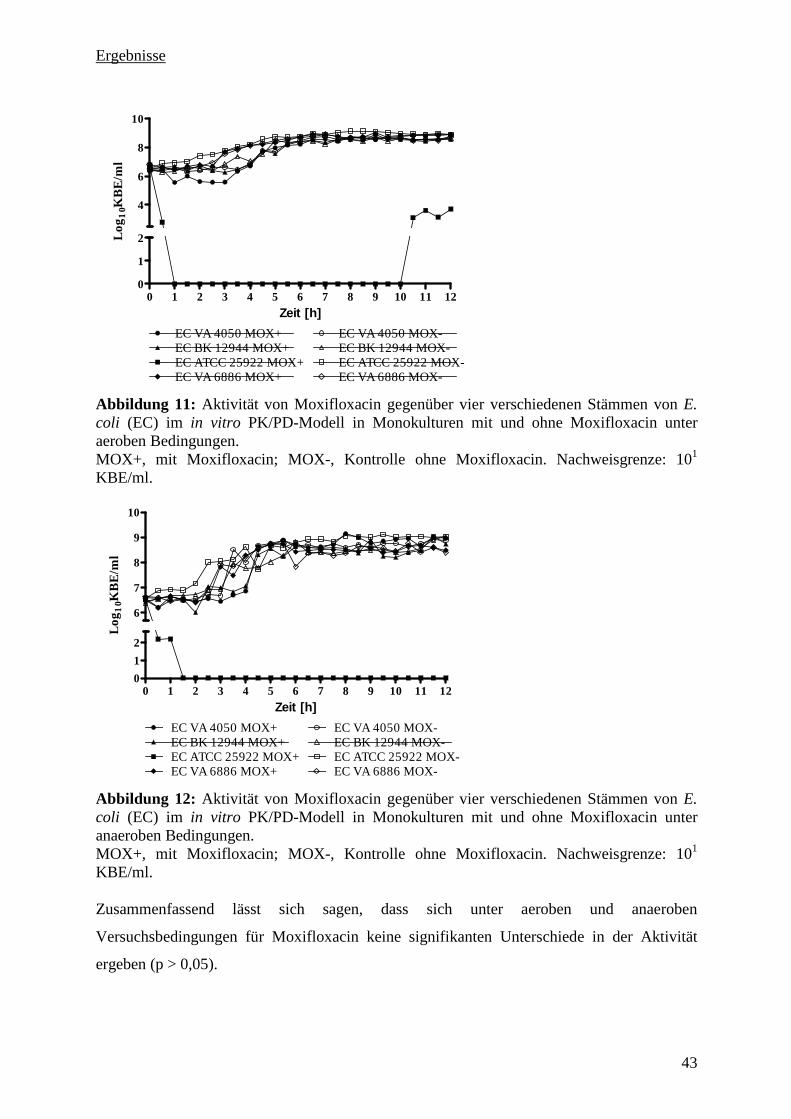
Ergebnisse
43
0 1 2 3 4 5 6 7 8 9 10 11 120
1
2
4
6
8
10
EC VA 4050 MOX+ EC VA 4050 MOX-EC BK 12944 MOX+ EC BK 12944 MOX-EC ATCC 25922 MOX+ EC ATCC 25922 MOX-EC VA 6886 MOX+ EC VA 6886 MOX-
Zeit [h]
Log
10K
BE
/ml
Abbildung 11: Aktivität von Moxifloxacin gegenüber vier verschiedenen Stämmen von E. coli (EC) im in vitro PK/PD-Modell in Monokulturen mit und ohne Moxifloxacin unter aeroben Bedingungen. MOX+, mit Moxifloxacin; MOX-, Kontrolle ohne Moxifloxacin. Nachweisgrenze: 101 KBE/ml.
0 1 2 3 4 5 6 7 8 9 10 11 120
1
2
6
7
8
9
10
EC VA 4050 MOX+ EC VA 4050 MOX-EC BK 12944 MOX+ EC BK 12944 MOX-EC ATCC 25922 MOX+ EC ATCC 25922 MOX-EC VA 6886 MOX+ EC VA 6886 MOX-
Zeit [h]
Log
10K
BE
/ml
Abbildung 12: Aktivität von Moxifloxacin gegenüber vier verschiedenen Stämmen von E. coli (EC) im in vitro PK/PD-Modell in Monokulturen mit und ohne Moxifloxacin unter anaeroben Bedingungen. MOX+, mit Moxifloxacin; MOX-, Kontrolle ohne Moxifloxacin. Nachweisgrenze: 101 KBE/ml. Zusammenfassend lässt sich sagen, dass sich unter aeroben und anaeroben
Versuchsbedingungen für Moxifloxacin keine signifikanten Unterschiede in der Aktivität
ergeben (p > 0,05).
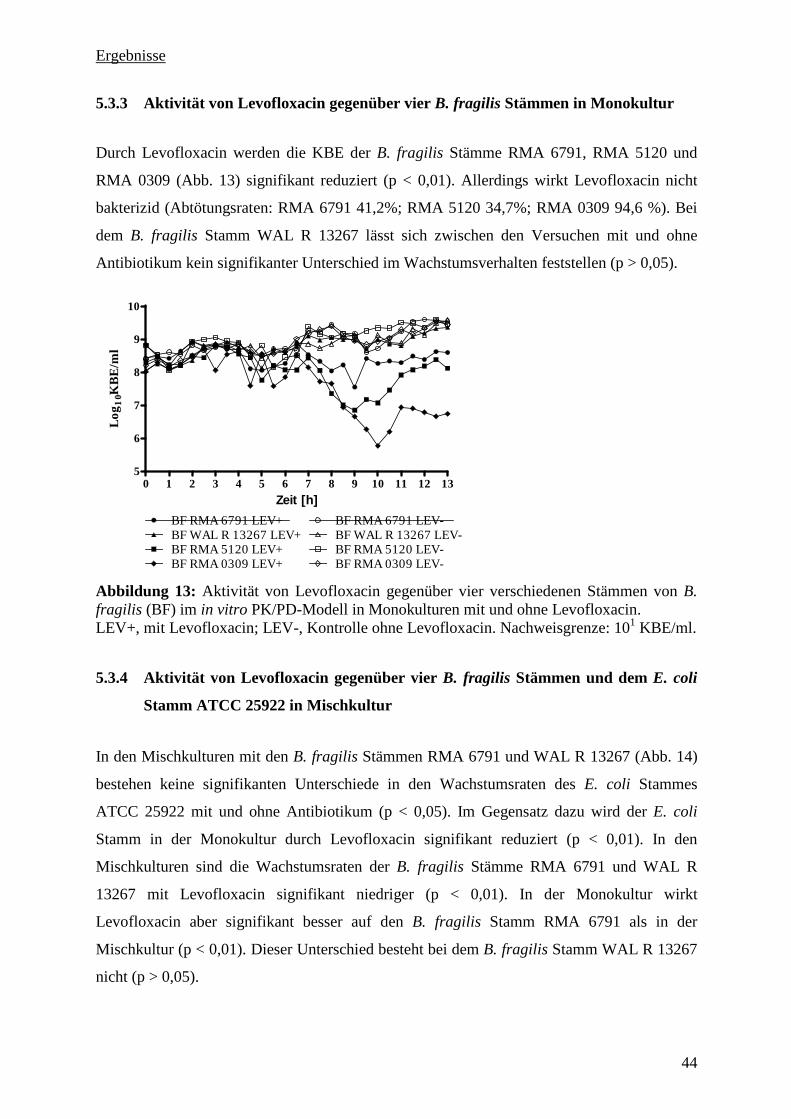
Ergebnisse
44
5.3.3 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen in Monokultur
Durch Levofloxacin werden die KBE der B. fragilis Stämme RMA 6791, RMA 5120 und
RMA 0309 (Abb. 13) signifikant reduziert (p < 0,01). Allerdings wirkt Levofloxacin nicht
bakterizid (Abtötungsraten: RMA 6791 41,2%; RMA 5120 34,7%; RMA 0309 94,6 %). Bei
dem B. fragilis Stamm WAL R 13267 lässt sich zwischen den Versuchen mit und ohne
Antibiotikum kein signifikanter Unterschied im Wachstumsverhalten feststellen (p > 0,05).
0 1 2 3 4 5 6 7 8 9 10 11 12 135
6
7
8
9
10
BF RMA 6791 LEV+ BF RMA 6791 LEV-BF WAL R 13267 LEV+ BF WAL R 13267 LEV-BF RMA 5120 LEV+ BF RMA 5120 LEV-BF RMA 0309 LEV+ BF RMA 0309 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 13: Aktivität von Levofloxacin gegenüber vier verschiedenen Stämmen von B. fragilis (BF) im in vitro PK/PD-Modell in Monokulturen mit und ohne Levofloxacin. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.
5.3.4 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm ATCC 25922 in Mischkultur
In den Mischkulturen mit den B. fragilis Stämmen RMA 6791 und WAL R 13267 (Abb. 14)
bestehen keine signifikanten Unterschiede in den Wachstumsraten des E. coli Stammes
ATCC 25922 mit und ohne Antibiotikum (p < 0,05). Im Gegensatz dazu wird der E. coli
Stamm in der Monokultur durch Levofloxacin signifikant reduziert (p < 0,01). In den
Mischkulturen sind die Wachstumsraten der B. fragilis Stämme RMA 6791 und WAL R
13267 mit Levofloxacin signifikant niedriger (p < 0,01). In der Monokultur wirkt
Levofloxacin aber signifikant besser auf den B. fragilis Stamm RMA 6791 als in der
Mischkultur (p < 0,01). Dieser Unterschied besteht bei dem B. fragilis Stamm WAL R 13267
nicht (p > 0,05).
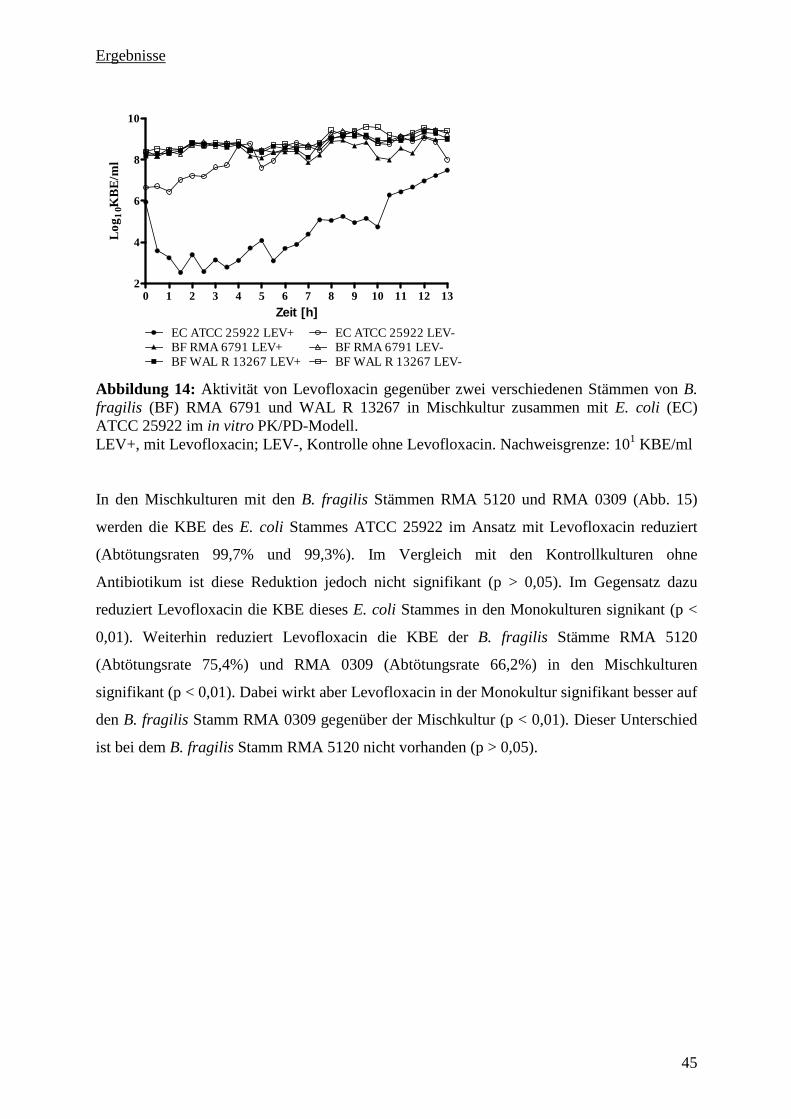
Ergebnisse
45
0 1 2 3 4 5 6 7 8 9 10 11 12 132
4
6
8
10
EC ATCC 25922 LEV+ EC ATCC 25922 LEV-BF RMA 6791 LEV+ BF RMA 6791 LEV-BF WAL R 13267 LEV+ BF WAL R 13267 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 14: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 6791 und WAL R 13267 in Mischkultur zusammen mit E. coli (EC) ATCC 25922 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml
In den Mischkulturen mit den B. fragilis Stämmen RMA 5120 und RMA 0309 (Abb. 15)
werden die KBE des E. coli Stammes ATCC 25922 im Ansatz mit Levofloxacin reduziert
(Abtötungsraten 99,7% und 99,3%). Im Vergleich mit den Kontrollkulturen ohne
Antibiotikum ist diese Reduktion jedoch nicht signifikant (p > 0,05). Im Gegensatz dazu
reduziert Levofloxacin die KBE dieses E. coli Stammes in den Monokulturen signikant (p <
0,01). Weiterhin reduziert Levofloxacin die KBE der B. fragilis Stämme RMA 5120
(Abtötungsrate 75,4%) und RMA 0309 (Abtötungsrate 66,2%) in den Mischkulturen
signifikant (p < 0,01). Dabei wirkt aber Levofloxacin in der Monokultur signifikant besser auf
den B. fragilis Stamm RMA 0309 gegenüber der Mischkultur (p < 0,01). Dieser Unterschied
ist bei dem B. fragilis Stamm RMA 5120 nicht vorhanden (p > 0,05).

Ergebnisse
46
0 1 2 3 4 5 6 7 8 9 10 11 12 13
2
4
6
8
10
EC ATCC 25922 LEV+ EC ATCC 25922 LEV-BF RMA 5120 LEV+ BF RMA 5120 LEV-BF RMA 0309 LEV+ BF RMA 0309 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 15: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 5120 und RMA 0309 in Mischkultur zusammen mit E. coli (EC) ATCC 25922 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.
5.3.5 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm VA 4050 in Mischkultur
Für den E. coli Stamm VA 4050 ergibt sich in allen Ansätzen der Mischkulturen mit den B.
fragilis Stämmen (RMA 6791, WAL R 13267, RMA 5120, RMA 0309) kein signifikanter
Unterschied der Wachstumsraten mit und ohne Antibiotikum (p > 0,05). Allerdings ist die
Wachstumsrate dieses E. coli Stammes in Monokultur gegenüber den Mischkulturen (Abb. 16
und 17) mit Levofloxacin signifikant niedriger (p < 0,01).
Die Wachstumsrate des B. fragilis Stammes RMA 6791 in der Mischkultur (Abb. 16) ist mit
Levofloxacin signifikant niedriger (p < 0,01). Allerdings wirkt Levofloxacin in der
Monokultur signifikant besser auf den B. fragilis Stammes RMA 6791 als in der Mischkultur
(p > 0,01). Im Gegensatz dazu bestehen bei dem B. fragilis Stamm WAL R 13267 mit und
ohne Levofloxacin zwischen den Misch- und Monokulturen keine signifikanten Unterschiede
im Wachstumsverhalten (p > 0,05).
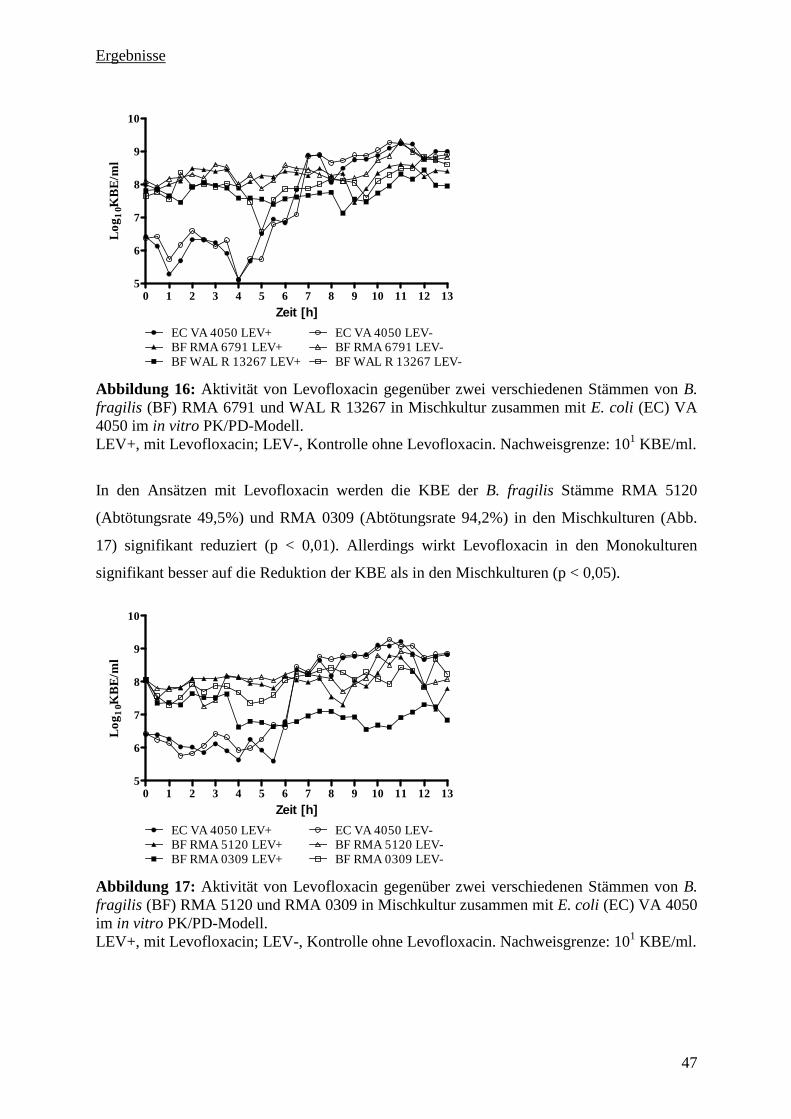
Ergebnisse
47
0 1 2 3 4 5 6 7 8 9 10 11 12 135
6
7
8
9
10
EC VA 4050 LEV+ EC VA 4050 LEV-BF RMA 6791 LEV+ BF RMA 6791 LEV-BF WAL R 13267 LEV+ BF WAL R 13267 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 16: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 6791 und WAL R 13267 in Mischkultur zusammen mit E. coli (EC) VA 4050 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.
In den Ansätzen mit Levofloxacin werden die KBE der B. fragilis Stämme RMA 5120
(Abtötungsrate 49,5%) und RMA 0309 (Abtötungsrate 94,2%) in den Mischkulturen (Abb.
17) signifikant reduziert (p < 0,01). Allerdings wirkt Levofloxacin in den Monokulturen
signifikant besser auf die Reduktion der KBE als in den Mischkulturen (p < 0,05).
0 1 2 3 4 5 6 7 8 9 10 11 12 135
6
7
8
9
10
EC VA 4050 LEV+ EC VA 4050 LEV-BF RMA 5120 LEV+ BF RMA 5120 LEV-BF RMA 0309 LEV+ BF RMA 0309 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 17: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 5120 und RMA 0309 in Mischkultur zusammen mit E. coli (EC) VA 4050 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.

Ergebnisse
48
5.3.6 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm BK 12944 in Mischkultur
In allen Ansätzen der Mischkulturen mit den B. fragilis Stämmen RMA 6791, WAL R 13267,
RMA 5120 und RMA 0309 ergeben sich für den E. coli Stamm BK 12944 keine signifikanten
Unterschiede in den Wachstumsraten mit und ohne Levofloxacin (p > 0,05). Die
Wachstumsrate dieses E. coli Stammes ist aber in den Monokulturen gegenüber den
Mischkulturen (Abb. 18 und 19) mit Levofloxacin signifikant niedriger (p < 0,01).
In der Mischkultur (Abb. 18) ist die Wachstumsrate des B. fragilis Stammes RMA 6791 mit
Levofloxacin signifikant niedriger (p < 0,01). Aber Levofloxacin wirkt in der Monokultur des
B. fragilis Stammes RMA 6791 signifikant besser im Vergleich mit der Mischkultur (p <
0,01). Im Gegensatz dazu bestehen bei dem B. fragilis Stamm WAL R 13267 mit und ohne
Levofloxacin zwischen den Misch- und Monokulturen keine signifikanten Unterschiede in
den Wachstumsraten (p > 0,05).
0 1 2 3 4 5 6 7 8 9 10 11 12 135
6
7
8
9
10
EC BK 12944 LEV+ EC BK 12944 LEV-BF RMA 6791 LEV+ BF RMA 6791 LEV-BF WAL R 13267 LEV+ BF WAL R 13267 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 18: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 6791 und WAL R 13267 in Mischkultur zusammen mit E. coli (EC) BK 12944 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml. In der Mischkultur (Abb. 19) werden die KBE des B. fragilis Stammes RMA 5120 mit
Levofloxacin moderat (Abtötungsrate 26,3%), aber im Vergleich mit der Kontrollkultur ohne
Antibiotikum nicht signifikant reduziert (p > 0,05). Im Gegensatz dazu reduziert
Levofloxacin die KBE des B. fragilis Stammes RMA 0309 in der Mischkultur signifikant
(Abtötungsrate 75,1%; p < 0,01). Levofloxacin wirkt allerdings in den Monokulturen beider
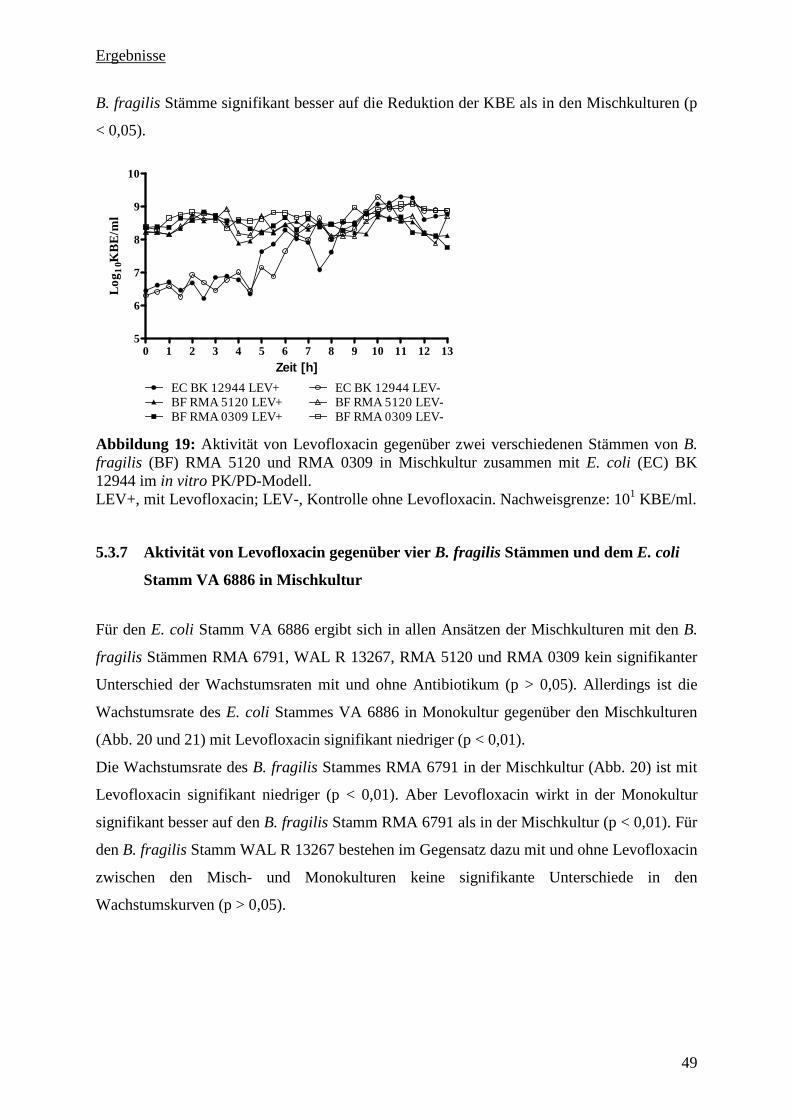
Ergebnisse
49
B. fragilis Stämme signifikant besser auf die Reduktion der KBE als in den Mischkulturen (p
< 0,05).
0 1 2 3 4 5 6 7 8 9 10 11 12 135
6
7
8
9
10
EC BK 12944 LEV+ EC BK 12944 LEV-BF RMA 5120 LEV+ BF RMA 5120 LEV-BF RMA 0309 LEV+ BF RMA 0309 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 19: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 5120 und RMA 0309 in Mischkultur zusammen mit E. coli (EC) BK 12944 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.
5.3.7 Aktivität von Levofloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm VA 6886 in Mischkultur
Für den E. coli Stamm VA 6886 ergibt sich in allen Ansätzen der Mischkulturen mit den B.
fragilis Stämmen RMA 6791, WAL R 13267, RMA 5120 und RMA 0309 kein signifikanter
Unterschied der Wachstumsraten mit und ohne Antibiotikum (p > 0,05). Allerdings ist die
Wachstumsrate des E. coli Stammes VA 6886 in Monokultur gegenüber den Mischkulturen
(Abb. 20 und 21) mit Levofloxacin signifikant niedriger (p < 0,01).
Die Wachstumsrate des B. fragilis Stammes RMA 6791 in der Mischkultur (Abb. 20) ist mit
Levofloxacin signifikant niedriger (p < 0,01). Aber Levofloxacin wirkt in der Monokultur
signifikant besser auf den B. fragilis Stamm RMA 6791 als in der Mischkultur (p < 0,01). Für
den B. fragilis Stamm WAL R 13267 bestehen im Gegensatz dazu mit und ohne Levofloxacin
zwischen den Misch- und Monokulturen keine signifikante Unterschiede in den
Wachstumskurven (p > 0,05).
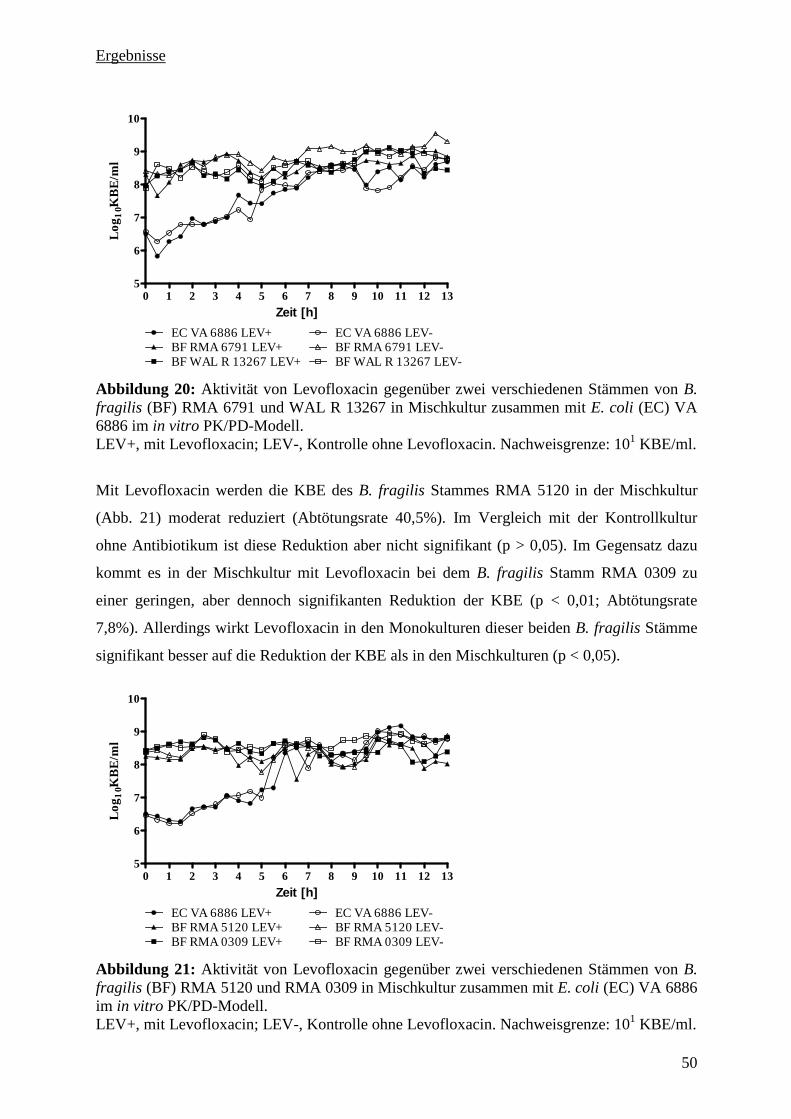
Ergebnisse
50
0 1 2 3 4 5 6 7 8 9 10 11 12 135
6
7
8
9
10
EC VA 6886 LEV+ EC VA 6886 LEV-BF RMA 6791 LEV+ BF RMA 6791 LEV-BF WAL R 13267 LEV+ BF WAL R 13267 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 20: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 6791 und WAL R 13267 in Mischkultur zusammen mit E. coli (EC) VA 6886 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.
Mit Levofloxacin werden die KBE des B. fragilis Stammes RMA 5120 in der Mischkultur
(Abb. 21) moderat reduziert (Abtötungsrate 40,5%). Im Vergleich mit der Kontrollkultur
ohne Antibiotikum ist diese Reduktion aber nicht signifikant (p > 0,05). Im Gegensatz dazu
kommt es in der Mischkultur mit Levofloxacin bei dem B. fragilis Stamm RMA 0309 zu
einer geringen, aber dennoch signifikanten Reduktion der KBE (p < 0,01; Abtötungsrate
7,8%). Allerdings wirkt Levofloxacin in den Monokulturen dieser beiden B. fragilis Stämme
signifikant besser auf die Reduktion der KBE als in den Mischkulturen (p < 0,05).
0 1 2 3 4 5 6 7 8 9 10 11 12 135
6
7
8
9
10
EC VA 6886 LEV+ EC VA 6886 LEV-BF RMA 5120 LEV+ BF RMA 5120 LEV-BF RMA 0309 LEV+ BF RMA 0309 LEV-
Zeit [h]
Log
10K
BE
/ml
Abbildung 21: Aktivität von Levofloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 5120 und RMA 0309 in Mischkultur zusammen mit E. coli (EC) VA 6886 im in vitro PK/PD-Modell. LEV+, mit Levofloxacin; LEV-, Kontrolle ohne Levofloxacin. Nachweisgrenze: 101 KBE/ml.
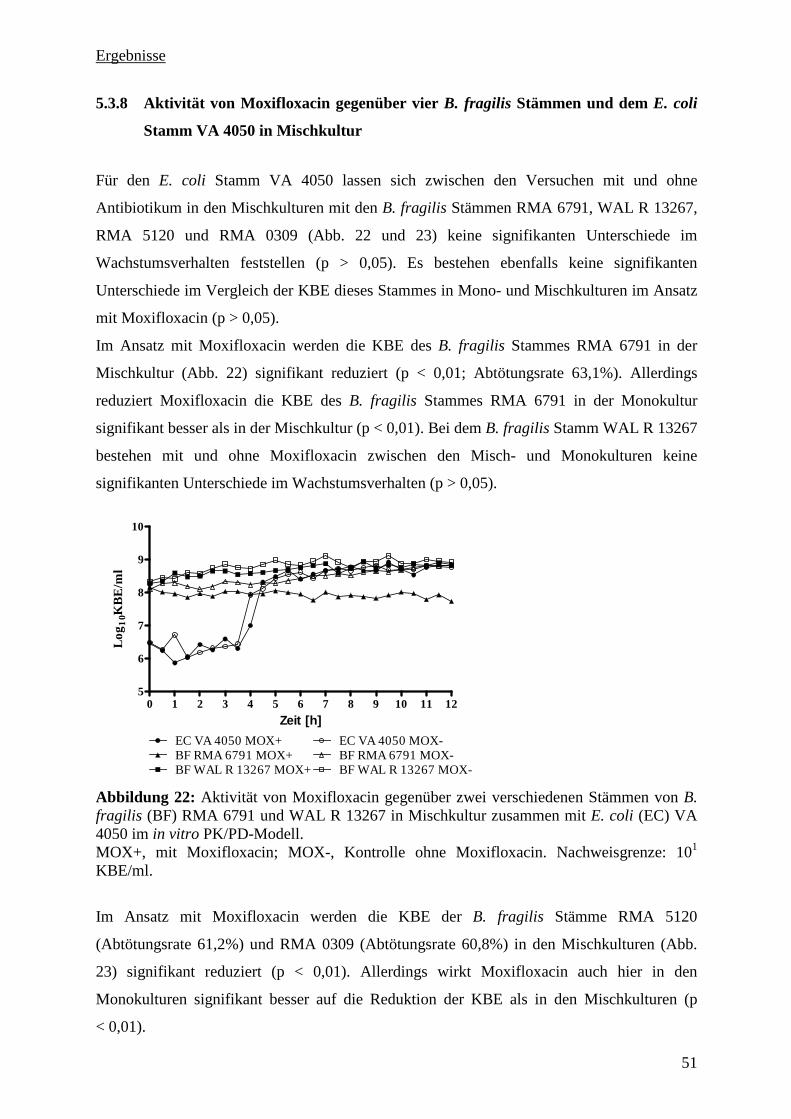
Ergebnisse
51
5.3.8 Aktivität von Moxifloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm VA 4050 in Mischkultur
Für den E. coli Stamm VA 4050 lassen sich zwischen den Versuchen mit und ohne
Antibiotikum in den Mischkulturen mit den B. fragilis Stämmen RMA 6791, WAL R 13267,
RMA 5120 und RMA 0309 (Abb. 22 und 23) keine signifikanten Unterschiede im
Wachstumsverhalten feststellen (p > 0,05). Es bestehen ebenfalls keine signifikanten
Unterschiede im Vergleich der KBE dieses Stammes in Mono- und Mischkulturen im Ansatz
mit Moxifloxacin (p > 0,05).
Im Ansatz mit Moxifloxacin werden die KBE des B. fragilis Stammes RMA 6791 in der
Mischkultur (Abb. 22) signifikant reduziert (p < 0,01; Abtötungsrate 63,1%). Allerdings
reduziert Moxifloxacin die KBE des B. fragilis Stammes RMA 6791 in der Monokultur
signifikant besser als in der Mischkultur (p < 0,01). Bei dem B. fragilis Stamm WAL R 13267
bestehen mit und ohne Moxifloxacin zwischen den Misch- und Monokulturen keine
signifikanten Unterschiede im Wachstumsverhalten (p > 0,05).
0 1 2 3 4 5 6 7 8 9 10 11 125
6
7
8
9
10
EC VA 4050 MOX+ EC VA 4050 MOX-BF RMA 6791 MOX+ BF RMA 6791 MOX-BF WAL R 13267 MOX+ BF WAL R 13267 MOX-
Zeit [h]
Log
10K
BE
/ml
Abbildung 22: Aktivität von Moxifloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 6791 und WAL R 13267 in Mischkultur zusammen mit E. coli (EC) VA 4050 im in vitro PK/PD-Modell. MOX+, mit Moxifloxacin; MOX-, Kontrolle ohne Moxifloxacin. Nachweisgrenze: 101 KBE/ml.
Im Ansatz mit Moxifloxacin werden die KBE der B. fragilis Stämme RMA 5120
(Abtötungsrate 61,2%) und RMA 0309 (Abtötungsrate 60,8%) in den Mischkulturen (Abb.
23) signifikant reduziert (p < 0,01). Allerdings wirkt Moxifloxacin auch hier in den
Monokulturen signifikant besser auf die Reduktion der KBE als in den Mischkulturen (p
< 0,01).

Ergebnisse
52
0 1 2 3 4 5 6 7 8 9 10 11 125
6
7
8
9
10
EC VA 4050 MOX+ EC VA 4050 MOX-BF RMA 5120 MOX+ BF RMA 5120 MOX-BF RMA 0309 MOX+ BF RMA 0309 MOX-
Zeit [h]
Log
10K
BE
/ml
Abbildung 23: Aktivität von Moxifloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 5120 und RMA 0309 in Mischkultur zusammen mit E. coli (EC) VA 4050 im in vitro PK/PD-Modell. MOX+, mit Moxifloxacin; MOX-, Kontrolle ohne Moxifloxacin. Nachweisgrenze: 101 KBE/ml.
5.3.9 Aktivität von Moxifloxacin gegenüber vier B. fragilis Stämmen und dem E. coli
Stamm BK 12944 in Mischkultur
0 1 2 3 4 5 6 7 8 9 10 11 125
6
7
8
9
10
EC BK 12944 MOX+ EC BK 12944 MOX-BF RMA 6791 MOX+ BF RMA 6791 MOX-BF WAL R 13267 MOX+ BF WAL R 13267 MOX-
Zeit [h]
Log
10K
BE
/ml
Abbildung 24: Aktivität von Moxifloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 6791 und WAL R 13267 in Mischkultur zusammen mit E. coli (EC) BK 12944 im in vitro PK/PD-Modell. MOX+, mit Moxifloxacin; MOX-, Kontrolle ohne Moxifloxacin. Nachweisgrenze: 101 KBE/ml.
Für den E. coli Stamm BK 12944 ergeben sich zwischen den Versuchen mit und ohne
Antibiotikum in den Mischkulturen mit den B. fragilis Stämmen RMA 6791, WAL R 13267,
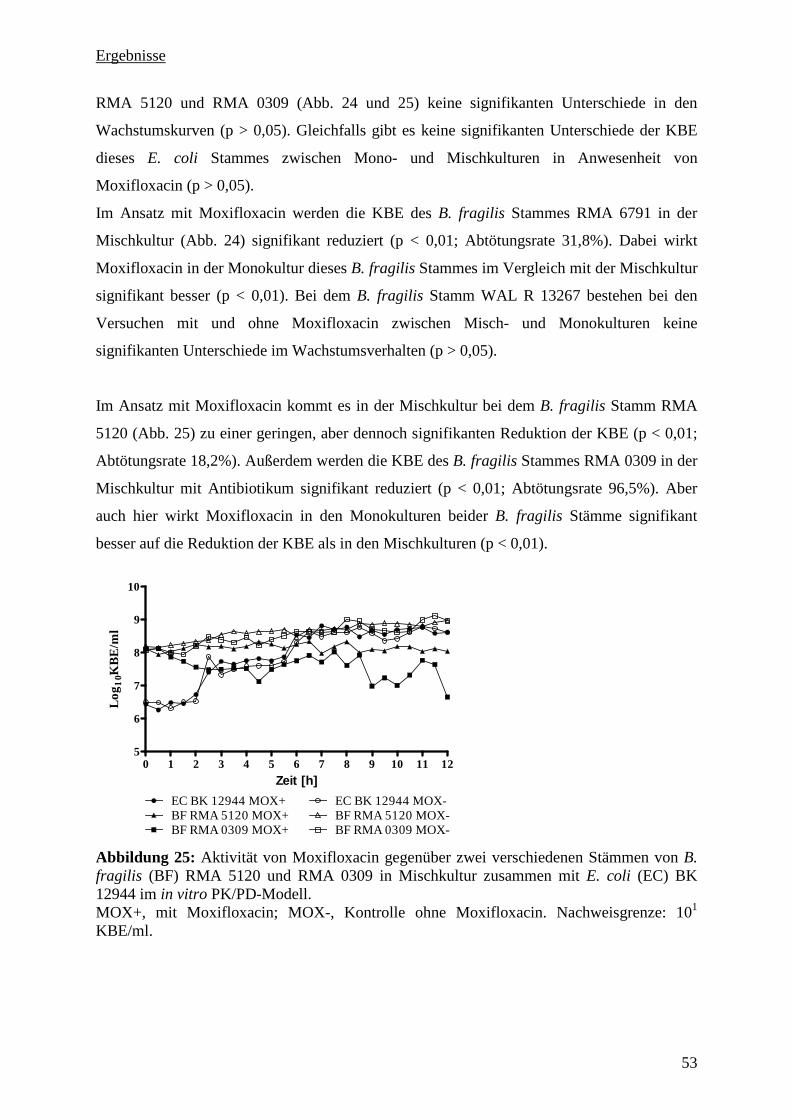
Ergebnisse
53
RMA 5120 und RMA 0309 (Abb. 24 und 25) keine signifikanten Unterschiede in den
Wachstumskurven (p > 0,05). Gleichfalls gibt es keine signifikanten Unterschiede der KBE
dieses E. coli Stammes zwischen Mono- und Mischkulturen in Anwesenheit von
Moxifloxacin (p > 0,05).
Im Ansatz mit Moxifloxacin werden die KBE des B. fragilis Stammes RMA 6791 in der
Mischkultur (Abb. 24) signifikant reduziert (p < 0,01; Abtötungsrate 31,8%). Dabei wirkt
Moxifloxacin in der Monokultur dieses B. fragilis Stammes im Vergleich mit der Mischkultur
signifikant besser (p < 0,01). Bei dem B. fragilis Stamm WAL R 13267 bestehen bei den
Versuchen mit und ohne Moxifloxacin zwischen Misch- und Monokulturen keine
signifikanten Unterschiede im Wachstumsverhalten (p > 0,05).
Im Ansatz mit Moxifloxacin kommt es in der Mischkultur bei dem B. fragilis Stamm RMA
5120 (Abb. 25) zu einer geringen, aber dennoch signifikanten Reduktion der KBE (p < 0,01;
Abtötungsrate 18,2%). Außerdem werden die KBE des B. fragilis Stammes RMA 0309 in der
Mischkultur mit Antibiotikum signifikant reduziert (p < 0,01; Abtötungsrate 96,5%). Aber
auch hier wirkt Moxifloxacin in den Monokulturen beider B. fragilis Stämme signifikant
besser auf die Reduktion der KBE als in den Mischkulturen (p < 0,01).
0 1 2 3 4 5 6 7 8 9 10 11 125
6
7
8
9
10
EC BK 12944 MOX+ EC BK 12944 MOX-BF RMA 5120 MOX+ BF RMA 5120 MOX-BF RMA 0309 MOX+ BF RMA 0309 MOX-
Zeit [h]
Log
10K
BE
/ml
Abbildung 25: Aktivität von Moxifloxacin gegenüber zwei verschiedenen Stämmen von B. fragilis (BF) RMA 5120 und RMA 0309 in Mischkultur zusammen mit E. coli (EC) BK 12944 im in vitro PK/PD-Modell. MOX+, mit Moxifloxacin; MOX-, Kontrolle ohne Moxifloxacin. Nachweisgrenze: 101 KBE/ml.

Ergebnisse
54
5.3.10 Zusammenfassung der PK/PD-Untersuchung
Mit Levofloxacin kommt es unter aeroben und anaeroben Bedingungen in den Monokulturen
zu einer signifikanten Reduktion der KBE des E. coli Stammes ATCC 25922 (p < 0,01).
Allerdings wirkt Levofloxacin nur unter aeroben Versuchsbedingungen bakterizid auf diesen
Stamm. In den Monokulturen der E. coli Stämme VA 4050, BK 12944 und VA 6886 lassen
sich unter aeroben und anaeroben Versuchsbedingungen in den Ansätzen mit Levofloxacin
keine signifikanten Unterschiede in den Wachstumsraten feststellen (p > 0,05). Des Weiteren
kommt es in den Monokulturen mit Levofloxacin zu signifikanten Reduktionen der KBE der
B. fragilis Stämme RMA 6791, RMA 5120 und RMA 0309 (p < 0,01). In den Mischkulturen
bestehen bei den Versuchen mit und ohne Levofloxacin in Bezug auf die KBE des E. coli
Stammes ATCC 25922 keine signifikanten Unterschiede (p > 0,05). Hingegen wirkt
Levofloxacin in den Monokulturen der E. coli Stämme ATCC 25922, VA 4050, BK 12944
und VA 6886 signifikant besser als in den Mischkulturen (p < 0,01). In den Mischkulturen
werden die KBE der B. fragilis Stämme RMA 5120 und RMA 0309 reduziert. Levofloxacin
wirkt aber in den Monokulturen der B. fragilis Stämme RMA 6791, RMA 5120 und RMA
0309 signifikant besser als in den Mischkulturen (p < 0,05). Bei dem B. fragilis Stamm WAL
R 13267 bestehen bei den Versuchen mit und ohne Levofloxacin zwischen Misch- und
Monokulturen keine signifikanten Unterschiede im Wachstumsverhalten (p > 0,05).
Im Gegensatz zu Levofloxacin wirkt Moxifloxacin in den Monokulturen sowohl in aerober
als auch anaerober Umgebung bakterizid auf den E. coli Stamm ATCC 25922. Bei den E. coli
Stämmen VA 4050, BK 12944 und VA 6886 bestehen zwischen den Versuchen mit und ohne
Moxifloxacin sowohl in Mono- und Mischkulturen keine signifikanten Unterschiede in den
Wachstumskurven (p > 0,05). Es besteht ebenfalls kein signifikanter Unterschied (p > 0,05)
in den Wachstumsraten der Monokulturen der E. coli Stämme VA 4050, BK 12944 und VA
6886 unter aeroben und anaeroben Bedingungen (mit und ohne Antibiotikum). Mit
Moxifloxacin werden die KBE der B. fragilis Stämme RMA 6791, RMA 5120 und RMA
0309 in den Mischkulturen signifikant reduziert (p < 0,01). Moxifloxacin wirkt auf die
Reduktion der KBE in den Monokulturen der B. fragilis Stämme RMA 6791, RMA 5120 und
RMA 0309 im Vergleich mit den Mischkulturen signifikant besser (p < 0,01).
5.3.11 Pharmakologische Indizes
Mittels Trapezregel ergibt sich bei der vorliegenden Arbeit für Levofloxacin ein Wert der
AUC von 99,8 mg/l unter Zugrundelegung einer maximalen Plasmakonzentration von 6 mg/l

Ergebnisse
55
bei zweimal täglicher Applikation. Für Moxifloxacin ergibt sich ein AUC-Wert von 55,7 mg/l
unter Annahme einer maximalen Plasmakonzentration von 4 mg/l bei einmal täglicher
Verabreichung. In den Tabellen 16 und 17 sind die berechneten Indizes von Moxifloxacin
und Levofloxacin angegeben.
Bei den gegenüber Levofloxacin resistenten E. coli Stämmen VA 4050, BK 12944, VA 6886
und dem B. fragilis Stamm WAL R 13267 ergeben sich niedrige Quotienten der AUC/MHK
von 3 und der Cmax/MHK von 0,19 für Levofloxacin, wie in Tabelle 16 aufgelistet. Im
Gegensatz dazu ergeben sich für den E. coli Stamm ATCC 25922 hohe Werte der AUC/MHK
zwischen 3326 und 12473. Für die B. fragilis Stämme RMA 5120 und 0309 liegen die
berechneten AUC/MHK-Werte zwischen 100 und 399. Bei dem B. fragilis Stamm RMA 6791
liegen die Werte der AUC/MHK zwischen 50 und 100.
Tabelle 16: Berechnete pharmakologische Indizes von Levofloxacin
Bakterienstamm MHKLEV (mg/l) AUC/MHK Cmax/MHK
E. coli ATCC 25922 0,008-0,03 3326-12473 200-750
E. coli VA 4050 > 32 3 0,19
E. coli BK 12944 > 32 3 0,19
E. coli VA 6886 > 32 3 0,19
B. fragilis RMA 6791 1-2 50-100 3-6
B. fragilis WAL R 13267 > 32 3 0,19
B. fragilis RMA 5120 1 100 6
B. fragilis RMA 0309 0,25-1 100-399 6-24
Bei den gegenüber Moxifloxacin resistenten E. coli Stämmen VA 4050, BK 12044, VA 6886
und dem B. fragilis Stamm WAL R 13267 ergeben sich ebenfalls niedrige Quotienten der
AUC/MHK von 2 und der Cmax/MHK von 0,13 (Tabelle 17). Für den E. coli Stamm ATCC
25922 lassen sich im Gegensatz dazu sehr hohe Quotienten der AUC/MHK zwischen 1857
und 3715 für Moxifloxacin berechnen. Bei den ebenfalls gegenüber Moxifloxacin sensiblen
B. fragilis Stämmen RMA 6791, RMA 5120 und RMA 0309 liegen die berechneten
AUC/MHK-Werte zwischen 111 und 464.
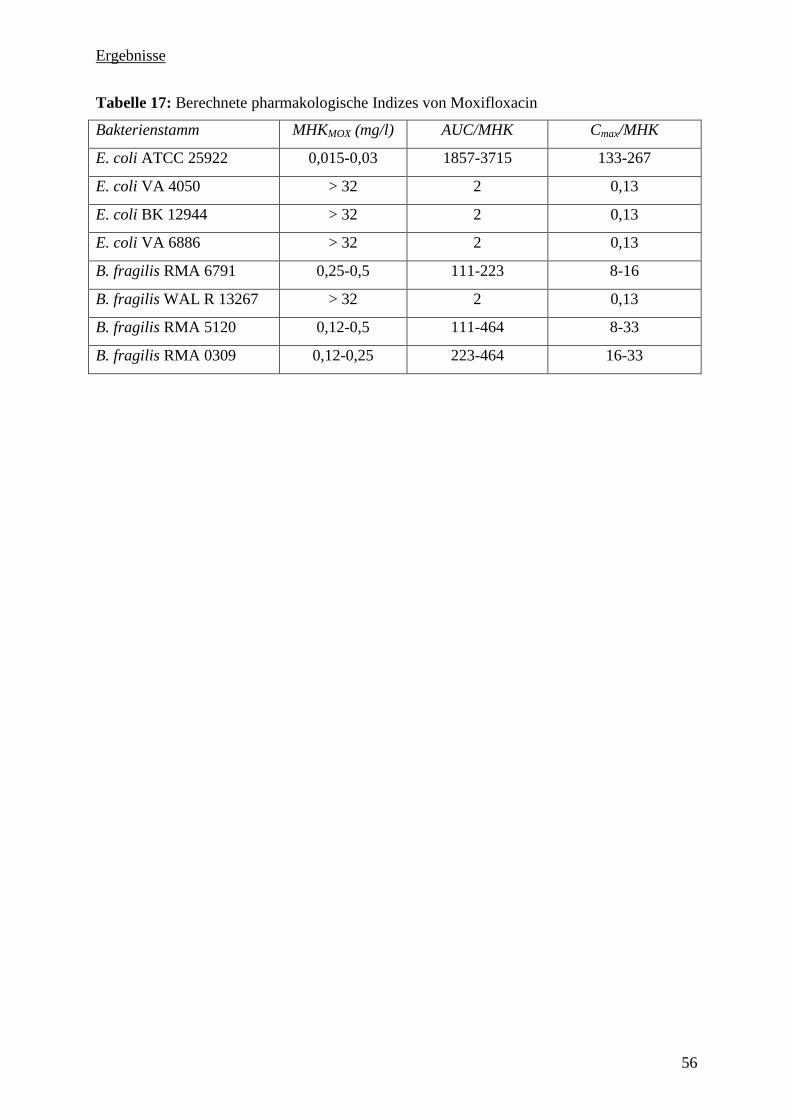
Ergebnisse
56
Tabelle 17: Berechnete pharmakologische Indizes von Moxifloxacin
Bakterienstamm MHKMOX (mg/l) AUC/MHK Cmax/MHK
E. coli ATCC 25922 0,015-0,03 1857-3715 133-267
E. coli VA 4050 > 32 2 0,13
E. coli BK 12944 > 32 2 0,13
E. coli VA 6886 > 32 2 0,13
B. fragilis RMA 6791 0,25-0,5 111-223 8-16
B. fragilis WAL R 13267 > 32 2 0,13
B. fragilis RMA 5120 0,12-0,5 111-464 8-33
B. fragilis RMA 0309 0,12-0,25 223-464 16-33

Diskussion und Ausblick
57
6 Diskussion und Ausblick
Intraabdominelle Infektionen stellen ein bedeutendes klinisches Problem dar und zählen
neben Pneumonie und Sepsis zu den wichtigsten Ursachen der infektionsbedingten
Morbidität und Letalität [26]. Da die Letalität der intraabdominellen Infektionen,
insbesondere der postoperativen Peritonitis bis zu 60% betragen kann [26], ist eine adäquate
Behandlung, bestehend aus chirurgischer Intervention und begleitender antibiotischer
Therapie, notwendig [30, 33]. Dabei ist für den Therapieerfolg von Infektionen die Kenntnis
über die Wirksamkeit und Empfindlichkeit der Erreger gegenüber dem Antibiotikum
bedeutend [50]. Im klinischen Alltag sind die Indikations- und Sicherstellung von optimalen
Therapiedosen basierend auf pharmakodynamischen Prinzipien entscheidend, um optimale
Therapieerfolge zu erzielen und das Auftreten von Resistenzen und Therapieversagen zu
mindern [133].
Durch die Bestimmung der MHK lässt sich abschätzen, ob ein Antibiotikum eine
therapeutische Option bietet [136]. Nguyen et al. konnten beispielsweise in einer
prospektiven Studie zeigen, dass zwischen in vitro Empfindlichkeit (MHK-Bestimmung) und
klinischem Outcome bei Patienten mit Bacteroides-verursachter Bakteriämie eine enge
Korrelation bestand [121]. Daraus lässt sich zunächst für die in der vorliegenden Arbeit
ermittelten MHK-Werte ableiten, dass Levofloxacin und Moxifloxacin eine Therapieoption
bei Infektionen mit B. fragilis und E. coli Stämmen darstellen können.
Mit dem vorliegenden PK/PD-Modell konnte zunächst gezeigt werden, dass es durch
Levofloxacin unter anaeroben im Gegensatz zu aeroben Bedingungen zu keiner vollständigen
Abtötung des sensiblen E. coli Stammes in der Monokultur kommt. Moxifloxacin wirkte in
beiden Versuchsdurchführungen bakterizid auf diesen Stamm. In anderen Untersuchungen
konnten im Vergleich von aerober und anaerober Atmosphäre zwar keine Unterschiede in der
maximal antibakteriellen Wirkung von verschiedenen Fluorchinolonen gegenüber fakultativ
anaeroben Bakterien nachgewiesen werden, aber die bakterizide Aktivität trat unter
anaeroben Bedingungen deutlich später auf [187, 189].
Des Weiteren ließ sich in der vorliegenden Untersuchung in den Monokulturen feststellen,
dass Levofloxacin die Erregerzahl der sensiblen B. fragilis Stämme signifikant reduzierte,
aber nicht bakterizid wirkte. Schaumann et al. konnten ebenfalls keine bakterizide Wirkung
für Moxifloxacin gegenüber diesen B. fragilis Stämmen im in vitro Modell nachweisen [146].
In anderen in vitro Untersuchungen wurden hingegen für Levofloxacin und Moxifloxacin
bakterizide Wirkungen gegenüber obligat anaeroben Bakterien nachgewiesen [122, 130, 157].
Es handelte sich aber um andere als in dieser Arbeit verwendete Stämme. In

Diskussion und Ausblick
58
Übereinstimmung mit der vorliegenden Arbeit konnten Noel et al. in einem in vitro Modell
nachweisen, dass Moxifloxacin auf obligate Anaerobier einen niedrigeren antibakteriellen
Effekt (angegeben als Log-Reduktion) hatte als auf einen E. coli Stamm [122]. In der
vorliegenden Arbeit sind die Abtötungsraten der sensiblen B. fragilis Stämme mit
Levofloxacin geringer im Vergleich mit dem sensiblen E. coli Stamm. Auch die
Abtötungsraten der B. fragilis Stämme in Monokultur mit Moxifloxacin [146] sind im
Vergleich mit dem sensiblen E. coli Stamm geringer.
Weiterhin ließ sich nachweisen, dass die in vitro Aktivitäten von Levofloxacin und
Moxifloxacin in den Mischkulturen im Vergleich mit den Monokulturen deutlich reduziert
waren. Insbesondere die Abtötungsraten der sensiblen B. fragilis Stämme waren im Vergleich
mit den Monokulturen für beide Substanzen deutlich gemindert. Levofloxacin wirkte in den
Mischkulturen auf die Reduktion der KBE des sensiblen E. coli Stammes nicht signifikant.
Durch die vorliegende Arbeit lässt sich zunächst festhalten, dass sich durch die alleinige
MHK-Bestimmung keine Rückschlüsse über die antibakteriellen Aktivitäten von
Levofloxacin und Moxifloxacin gegenüber den untersuchten Bakterienstämmen in
Mischkulturen und unter geänderten atmosphärischen Bedingungen ziehen lassen. In vitro
ließen sich reduzierte Aktivitäten der beiden Substanzen nachweisen, wobei Moxifloxacin
trotz moderater Wirkung [146] antibakteriell aktiver als Levofloxacin war. Im Gegensatz zu
den niedrigen MHK-Werten ließen sich für die B. fragilis Stämme RMA 6791, RMA 5120
und RMA 0309 im vorliegenden in vitro PK/PD-Modell weder für Levofloxacin noch für
Moxifloxacin Abtötungsraten von mehr als 99,9% feststellen. Als Konsequenz könnte dieses
bedeuten, dass es trotz niedriger MHK-Werte der einzelnen Erreger zu einem
Therapieversagen kommt, da das Erregerspektrum polymikrobiell ist [94].
Die MHK-Bestimmung erfolgt nach einer standardisierten Referenzmethode [5]. Es wird die
Empfindlichkeit eines Bakterienstamms mit standardisiertem Inokulum gegenüber einer
antibakteriellen Substanz in vitro getestet und es ergibt sich ein numerischer Wert. Dieser
wird anhand von Grenzwerten klinisch in "sensibel", "intermediär" und "resistent" eingeteilt.
Anhand dieser Bewertungen wählt der klinisch tätige Arzt ein zur Therapie geeignetes
Antibiotikum aus.
Die Grenzwerteinteilung berücksichtigt nicht nur den MHK-Wert, sondern viele weitere
Faktoren wie Dosierung, Indikation, Pharmakokinetik, Pharmakodynamik und Ergebnisse aus
Studien müssen mit einbezogen werden [27, 136].
Da bei den meisten Veröffentlichungen zu Empfindlichkeitsprüfungen MHK-Verteilungen
(MHK50 oder MHK90) angegeben werden, ist ein direkter Vergleich mit denen in der

Diskussion und Ausblick
59
vorliegenden Arbeit ermittelten MHK-Werten kritisch zu betrachten [184]. Werte der
MHK90 könnten durch resistenztragende Stämme stark ansteigen und dadurch die Verteilung
der MHK-Werte einer natürlichen Population falsch hoch darstellen [144]. MHK-
Verteilungen schließen hingegen Wildtypen ohne Resistenzmechanismen und solche Stämme
mit Resistenzmechanismen ein und sollten daher eher mit in die Bewertung der MHK
eingehen [136].
Die MHK-Bestimmung erfolgt unter Verwendung von Monokulturen. Im Gegensatz dazu
findet sich bei intraabdominellen Infektionen häufig ein polymikrobielles Erregerspektrum
und die Umgebungsbedingungen sind durch den Verbrauch von Sauerstoff zugunsten der
obligat anaeroben Bakterien verändert [148]. Zudem kommt es bei Infektionen zu
Veränderungen von pH-Wert, Sauerstoffpartialdruck (pO2) und Kohlendioxidpartialdruck
(pCO2) [94, 151]. Diese Faktoren können die antibakterielle Aktivität beeinflussen. Eine
Senkung des pH-Wertes kann eine verminderte Zellwandpermeabilität für das Antibiotikum
hervorrufen und dadurch zu einer verminderten Aktivität führen [54, 99]. Dalhoff et al.
konnten nachweisen, dass die in vitro Aktivität von Levofloxacin und Moxifloxacin bei einer
Senkung des pH-Wertes abnahm [42]. Falagas et al. wiesen nach, dass die in vitro
Empfindlichkeit von Stämmen der B. fragilis Gruppe bei Ansäuerung des Kulturmediums
auch gegenüber den Fluorchinolonen abnahm [54]. Emrich et al. konnten in einer aktuellen
Untersuchung nachweisen, dass die antibakterielle Wirkung von Levofloxacin und
Moxifloxacin bei einem pH-Wert von 5,8 gegenüber E. coli Stämmen im Vergleich zum pH-
Wert von 7,2 deutlich niedriger war [53]. Ein erniedrigter pH-Wert könnte daher ein
möglicher Einflussfaktor für die reduzierten Aktivitäten von Levofloxacin und Moxifloxacin
in der vorliegenden Arbeit sein. Daher müssen auch Infektionsart und -ort bei Bewertung der
MHK Berücksichtigung finden [115]. Aber auch die Inokulumgröße kann die antibakterielle
Aktivität insbesondere in den Mischkulturen beeinflussen [113, 152]. In den Mischkulturen
des vorliegenden PK/PD-Modells ist die Anzahl der KBE gegenüber den Monokulturen
deutlich erhöht. Stearne et al. kamen nach einer in vitro Untersuchung zu dem Schluss, dass
ein erhöhtes Inokulum ein signifikanter Faktor für die geminderte Aktivität von
Trovafloxacin gegenüber B. fragilis in Mischkulturen mit E. coli Stämmen ist [163]. Die
genannten Faktoren können in in vitro PK/PD-Modellen berücksichtigt und untersucht
werden und könnten dann mit in die Bewertung der MHK als Therapieempfehlung einfließen.
Bisher wurden aber alle Erreger nach einheitlichen Grenzwerten beurteilt. Dabei kam es
häufig zur Trennung von zusammenhängenden Erregerhäufungen, obwohl es sich um
Wildtyp-Verteilungen ohne Resistenzmechanismen handelt [136]. Durch die EUCAST

Diskussion und Ausblick
60
wurden für E. coli die Grenzwerte für Levofloxacin und Moxifloxacin Spezies-spezifisch
festgelegt. Für B. fragilis gibt es für Levofloxacin und Moxifloxacin bisher noch keine
Spezies-spezifischen Grenzwertfestlegungen durch die EUCAST [8]. Nicht Spezies-
spezifische Grenzwerte basieren auf festgelegten pharmakokinetisch/pharmakodynamischen
Daten und sind unabhängig von der MHK-Verteilung [8]. Wie in der vorliegenden Arbeit
gezeigt werden konnte, sind bei fehlenden Bakteriengruppen-spezifischen Grenzwerten
therapeutische Vorhersagen von Erfolg oder Misserfolg in Bezug auf die Interpretation der
Empfindlichkeitstestung nur eingeschränkt möglich [52]. Trotz niedriger MHK-Werte kam es
weder bei Levofloxacin noch bei Moxifloxacin [146] zu einer vollständigen Eradikation der
B. fragilis Stämme im in vitro PK/PD-Modell. Wenn die MHK-Bewertungen Spezies-
spezifisch und infektions- bzw. indikationsspezifisch sind, ergeben sich möglicherweise
höhere Grenzwerte. Um empirische Daten für die Therapieempfehlung zu erstellen, sollten
die Grenzwerteinteilungen daher Spezies-spezifisch und infektionsbezogen erfolgen [86, 115,
136, 155]. Dieses ist neben der Harmonisierung der MHK-Grenzwerte und der Bestimmung
einer gemeinsamen Referenzmethode zur MHK-Bestimmung Ziel der EUCAST [8, 27].
In der vorliegenden Arbeit konnte gezeigt werden, dass die alleinige MHK-Bestimmung bei
Monokulturen mit Bewertung zur Therapieempfehlung bei Mischinfektionen nicht
ausreichend ist. Insbesondere bei polymikrobiellen Infektionen müssen weitergehende
Untersuchungen erfolgen, um Aussagen zur antibakteriellen Aktivität bei Mischinfektionen
zu treffen, die dann mit in die Bewertung der MHK einfließen, um letztlich erfolgreich zu
therapieren.
Im Weiteren wurden in der vorliegenden Arbeit die PK/PD-Indizes AUC/MHK und
Cmax/MHK von Levofloxacin und Moxifloxacin für die untersuchten Stämme berechnet. Es
gibt zahlreiche Arbeiten und Untersuchungen, die sich mit der Entwicklung von PK/PD-
Indizes und deren Vorhersagemöglichkeiten beschäftigen [15, 16, 38, 48, 57, 58, 80]. Tabelle
20 im Anhang 3 enthält eine Auswahl an PK/PD-Indizes, vorzugsweise für Fluorchinolone,
mit den jeweils bestimmten Grenzwerten für Bakterizidie, Verbesserung des klinischen
Outcomes, Verhinderung des Auftretens von Wiederwachstum oder dem Auftreten von
Resistenzmechanismen. Schentag et al. geben für die Fluorchinolone folgende Werte an:
AUC/MHK-Quotienten > 125 führen zur Bakterizidie; AUC/MHK-Raten > 250 führen zu
einer schnellen bakteriziden Wirkung und Quotienten zwischen 30 und 60 führen zu
bakteriostatischen Effekten [150]. Wispelwey et al. fordern bei der Therapie von
gramnegativen Bakterien unabhängig von der Substanzklasse AUC/MHK-Raten > 100 für

Diskussion und Ausblick
61
einen antimikrobiellen Erfolg [185]. Thomas et al. konnten für AUC/MHK-Werte < 100 die
Förderung der Resistenzentwicklung und des Wiederwachstum nachweisen [175].
PK/PD-Indizes erlauben quantitative Vorhersagen bezüglich des Erfolgs oder des Versagens
einer antibiotischen Therapie [66]. Je kleiner der MHK-Wert ist, desto größer ist der
AUC/MHK-Quotient. Aber auch Änderungen der Pharmakokinetik führen zu Änderungen der
Quotienten. Dieses ist beispielsweise bei Vorliegen einer Niereninsuffizienz mit
eingeschränkter Clearance für ein Antibiotikum zu beachten.
In den Monokulturen des sensiblen E. coli Stammes kommt es unter aeroben Bedingungen zu
vollständigen Eradikationen, welche anhand der berechneten hohen Indizes von Levofloxacin
und Moxifloxacin auch so erwartet werden. Schon unter geänderten Umgebungsbedingungen
kommt es trotz des hohen AUC/MHK-Wertes von Levofloxacin gegenüber dem sensiblen E.
coli Stamm nicht zur Bakterizidie. Gegenüber den sensiblen B. fragilis Stämmen ist trotz
hoher Indizes keine vollständige Eradikation der Erreger festzustellen. Auch bei den
Mischkulturen geht man anhand der hohen AUC/MHK-Quotienten für die sensiblen
Bakterienstämme von guter antibakterieller Aktivität der beiden Substanzen aus, welche
anhand der ermittelten Daten so nicht vorliegen. Der von Schentag et al. angegebene
Grenzwert für eine schnelle bakterizide Wirkung (AUC/MHK > 250) ist daher in der
vorliegenden Arbeit nur für den sensiblen E. coli Stamm und unter Beachtung der
Umgebungsbedingungen zutreffend. Ein Vergleich mit der Übersichtsarbeit von Schentag et
al. ist aber auch wieder nur eingeschränkt möglich, da Erregerart und Versuchsdurchführung
unberücksichtigt bleiben [149, 150].
Welcher AUC/MHK-Wert letztlich entscheidend für den Erfolg oder das Versagen einer
Therapie ist, bedarf insbesondere für Mischkulturen und geänderten Bedingungen weiterer
Untersuchungen. Hierbei müssen unter anderem Faktoren, welche zu einer Reduktion der
antibakteriellen Aktivität führen, mit einbezogen werden. Auch durch Bestimmung von
Spezies-spezifischen Grenzwerten werden sich Änderungen der PK/PD-Indizes ergeben.
Es zeigt sich durch die vorliegende Arbeit, dass die Bestimmung der PK/PD-Indizes nicht nur
für Monokulturen, sondern auch für Mischkulturen sinnvoll ist. Möglicherweise kommt es zu
einem Therapieversagen eines Antibiotikums bei Mischinfektionen, obwohl hohe PK/PD-
Indizes und eine gute Wirksamkeit gegenüber den einzelnen Erregern vorliegen.

Diskussion und Ausblick
62
Ausblick
Die Ergebnisse der vorliegenden Arbeit zeigen, dass Moxifloxacin im Vergleich zu
Levofloxacin eine bessere in vitro Wirkung im PK/PD-Modell gegenüber Mono- und
Mischkulturen der ausgewählten B. fragilis und E. coli Stämme hat. Allerdings ist die
Aktivität von Moxifloxacin in den Mischkulturen auch nur moderat [146]. Durch
verschiedene Untersuchungen im Tiermodell und in vivo Studien konnten für Moxifloxacin
gute antibakterielle Aktivitäten bei Mischinfektionen durch fakultativ und obligate
Anaerobier gezeigt werden [51, 81, 108, 145, 162]. Die in der vorliegenden Arbeit
verwendeten Erreger stammen vornehmlich von schwerkranken Patienten und weisen deshalb
möglicherweise höhere MHK-Werte im Vergleich mit Erregern anderer Untersuchungen auf.
Auch der fehlende Einfluss des Immunsystems kann eine Erklärung für die reduzierten
antibakteriellen Aktivitäten von Levofloxacin und Moxifloxacin im in vitro PK/PD-Modell
sein [144].
Moxifloxacin ist nach aktuellen Empfehlungen zur Therapie intraabdomineller Infektionen
geeignet [27]. Für Levofloxacin wird eine Kombinationstherapie mit Metronidazol
empfohlen. In einer in vitro Studie zu Mischinfektionen mit B. fragilis und E. coli Stämmen
wirkte die Kombinationstherapie aus Levofloxacin und Metronidazol ebenso gut, wie eine
Monotherapie mit Moxifloxacin [73]. Dennoch sollte nach den Ergebnissen der vorliegenden
Arbeit eine antibiotische Therapie mit Levofloxacin oder Moxifloxacin kritisch hinterfragt
werden. Die zunehmenden Resistenzraten von E. coli oder Bacteroides spp. gegenüber den
Fluorchinolonen stellen zusätzlich ein großes Problem bei der Therapie intraabomineller
Infektionen dar [37, 61, 72, 154, 164].
Aufgrund der vorliegenden Ergebnisse ergeben sich weiterführende Untersuchungen, ob und
welche Änderungen sich für PK/PD-Indizes für ein polymikrobielles Erregerspektrum und
Veränderungen der Umgebungsbedingungen ergeben.

Zusammenfassung
63
7 Zusammenfassung der Arbeit
Dissertation zur Erlangung des akademischen Grades Dr. med.
Titel:
Pharmakokinetisch/pharmakodynamische Untersuchung zur in
vitro Aktivität von Levofloxacin und Moxifloxacin gegenüber B.
fragilis und E. coli in Mono- und Mischkultur
eingereicht von:
Stefanie Rossi, geb. Hoffmann
angefertigt am:
Institut für Medizinische Mikrobiologie und Infektionsepidemiologie
der Universität Leipzig
betreut von:
PD Dr. med. Reiner Schaumann
Monat und Jahr
der Einreichung:
08/2012
Intraabdominelle Infektionen weisen meist ein polymikrobielles Erregerspektrum aus
fakultativ und obligat anaeroben Bakterien auf. E. coli und B. fragilis werden als häufigste
Erreger bei Mischinfektionen nachgewiesen. Häufigste Form der intraabdominellen
Infektionen stellt die sekundäre Peritonitis dar, wobei die postoperative Peritonitis die höchste
Letalitätsrate aufweist. Die Therapie von intraabdominellen Infektionen erfolgt primär
chirurgisch mit begleitender antibiotischer Therapie.
Durch in vitro PK/PD-Modelle können Aussagen über das Abtötungs- oder
Wachstumsverhalten von Bakterien in Abhängigkeit von Veränderungen der
Antibiotikumkonzentration getroffen werden und dadurch Vorgänge in vivo simuliert werden.
Ziel dieser Arbeit war die Untersuchung der pharmakodynamischen Aktivitäten von
Levofloxacin und Moxifloxacin gegenüber ausgewählten B. fragilis und E. coli Stämmen in
Mono- und Mischkulturen unter Berücksichtigung pharmakokinetischer Eigenschaften. Für
die E. coli Stämme erfolgten die Monokulturenversuche unter aeroben und anaeroben
Bedingungen.

Zusammenfassung
64
Vor Versuchsbeginn erfolgte die Empfindlichkeitstestung mittels Etest. Drei der getesteten
vier B. fragilis Stämme waren sensibel gegenüber Levofloxacin und Moxifloxacin. Ein E. coli
Stamm war sensibel gegenüber Levofloxacin und Moxifloxacin, die drei anderen Stämme
waren in der Empfindlichkeitstestung resistent gegenüber beiden Substanzen.
In der vorliegenden Arbeit wirkte Levofloxacin unter aeroben Bedingungen signifikant besser
auf einen sensiblen E. coli Stamm als unter anaeroben. Für Moxifloxacin ließen sich unter
aeroben und anaeroben Bedingungen gegenüber den E. coli Stämmen keine Unterschiede in
der antibakteriellen Wirkung nachweisen. Moxifloxacin wirkte auf den sensiblen E. coli
Stamm unter aeroben und anaeroben Bedingungen bakterizid. Außerdem reduzierte
Levofloxacin signifikant die KBE der sensiblen B. fragilis Stämme in den Monokulturen,
eine Bakterizidie ließ sich nicht feststellen. In den Mischkulturen zeigten sich für
Levofloxacin und Moxifloxacin reduzierte Aktivitäten gegenüber den Monokulturen.
Moxifloxacin zeigte in dieser Untersuchung eine bessere in vitro Aktivität in Mono- und
Mischkulturen von B. fragilis und E. coli Stämmen im Vergleich mit Levofloxacin.
Es konnte in vitro gezeigt werden, dass die antibakteriellen Aktivitäten von Levofloxacin und
Moxifloxacin in den Mischkulturen und für Levofloxacin unter geänderten
Umgebungsbedingungen gemindert sind. Diese Aussage ist durch alleinige MHK-
Bestimmung mit den Monokulturen nicht möglich. Es gelten andere Bedingungen bei
Mischkulturen. Weiterführende in vitro Untersuchungen zur antibiotischen Aktivität bei
Mischinfektionen sollten daher erfolgen. Infektionsart und -ort, sowie erregerspezifische
MHK-Werte müssen ebenso wie die Daten aus in vitro PK/PD-Modellen und Ergebnissen aus
klinischen Untersuchungen in die Bewertung der MHK als Therapieempfehlungen einfließen.
Die vorliegenden Daten unterstützen die Verwendung von PK/PD-Indizes für Versuche mit
Monokulturen, da sie bei Monoinfektionen eine gute Möglichkeit zur Vorhersage des
therapeutischen Effekts bieten. Im Unterschied dazu zeigen die Versuche mit den
Mischkulturen, dass PK/PD-Indizes nicht so ohne weiteres Aussagen über einen möglichen
Therapieerfolg bei einem polymikrobiellen Erregerspektrum zulassen.

Literaturverzeichnis
65
8 Literaturverzeichnis 1. Avalox® 400 mg Filmtabletten, Fachinformation, Stand Oktober 2010. 2. Bacteroides spp. http://www.infektionsnetz.at/view.php?name=BakterienBacteroides. 3. bioMérieux. http://www.biomerieux.com. 4. Ciprobay® 250 mg/-500 mg/-750 mg Filmtabletten, Fachinformation, Stand April 2010. 5. Deutsches Institut für Normung e.V.: Beuth Verlag, Auslagestelle Budapester Straße 31,
10787 Berlin. 6. Deutsches Institut für Normung e.V.:Medizinische Mikrobiologie und Immunologie:
Diagnostische Verfahren. 4. Aufl. ed. 2004, Berlin: Beuth Verlag. 7. Escherichia coli. http://www.zct-berlin.de/klinik.praxis/Escherichia_coli.html. 8. EUCAST. http://www.eucast.org/. 9. EUCAST Definitive Document E.DEF 3.1, June 2000: Determination of minimum
inhibitory concentrations (MICs) of antibacterial agents by agar dilution. Clin Microbiol Infect 2000, 6(9):509-15.
10. GERMAP 2008 - Antibiotika-Resistenz und -Verbrauch. Bericht über den Antibiotikaverbrauch und die Verbreitung von Antibiotikaresistenzen in der Human- und Veterinärmedizin in Deutschland, ed. B.f.V.u. Lebensmittelsicherheit. 2008, Berlin, Rheinach, Freiburg: Antiinfectives Intelligence Gesellschaft für klinisch-mikrobiologische Forschung und Kommunikation mbH Rheinach.
11. Levofloxacin. http://www.infektionsnetz.at/view.php?datID=1144. 12. Tavanic® 250 mg/500 mg Filmtabletten, Fachinformation, Stand Oktober 2007. 13. Akahane, K., Sekiguchi, M., Une, T., und Osada, Y., Structure-epileptogenicity
relationship of quinolones with special reference to their interaction with gamma-aminobutyric acid receptor sites. Antimicrob Agents Chemother 1989, 33(10):1704-8.
14. Aktories, K., und Forth, W., Allgemeine und spezielle Pharmakologie und Toxikologie für Studenten der Medizin, Veterinärmedizin, Pharmazie, Chemie und Biologie sowie für Ärzte, Tierärzte und Apotheker. 9., völlig überarb. Aufl. ed. 2005, München: Urban & Fischer.
15. Ambrose, P.G., Bhavnani, S.M., und Owens, R.C., Jr., Clinical pharmacodynamics of quinolones. Infect Dis Clin North Am 2003, 17(3):529-43.
16. Ambrose, P.G., Jr., Owens, R.C., und Grasela, D., Antimicrobial pharmacodynamics. Med Clin North Am 2000, 84(6):1431-46.
17. Appelbaum, P.C., und Hunter, P.A., The fluoroquinolone antibacterials: past, present and future perspectives. Int J Antimicrob Agents 2000, 16(1):5-15.
18. Babcock, H.M., Zack, J.E., Garrison, T., Trovillion, E., Kollef, M.H., und Fraser, V.J., Ventilator-associated pneumonia in a multi-hospital system. Infection control and hospital epidemiology : the official 2003, 24(11):853-8.
19. Baker, C.N., Stocker, S.A., Culver, D.H., und Thornsberry, C., Comparison of the E Test to agar dilution, broth microdilution, and agar diffusion susceptibility testing techniques by using a special challenge set of bacteria. J Clin Microbiol 1991, 29(3):533-8.
20. Bansal, S., und Tandon, V., Contribution of mutations in DNA gyrase and topoisomerase IV genes to ciprofloxacin resistance in Escherichia coli clinical isolates. Int J Antimicrob Agents 2011, 37(3):253-5.
21. Barger, A., Fuhst, C., und Wiedemann, B., Pharmacological indices in antibiotic therapy. J Antimicrob Chemother 2003, 52(6):893-8.
22. Bichsel, A., James, C.W., und Gurk-Turner, C., Fluoroquinolone drug class update. Proc (Bayl Univ Med Cent) 2000, 13(3):289-92.

Literaturverzeichnis
66
23. Blanche, F., Cameron, B., Bernard, F.X., Maton, L., Manse, B., Ferrero, L., Ratet, N., Lecoq, C., Goniot, A., Bisch, D., und Crouzet, J., Differential behaviors of Staphylococcus aureus and Escherichia coli type II DNA topoisomerases. Antimicrob Agents Chemother 1996, 40(12):2714-20.
24. Blaser, J., Stone, B.B., Groner, M.C., und Zinner, S.H., Comparative study with enoxacin and netilmicin in a pharmacodynamic model to determine importance of ratio of antibiotic peak concentration to MIC for bactericidal activity and emergence of resistance. Antimicrob Agents Chemother 1987, 31(7):1054-60.
25. Blondeau, J.M., und Hansen, G.T., Moxifloxacin: a review of the microbiological, pharmacological, clinical and safety features. Expert Opin Pharmacother 2001, 2(2):317-35.
26. Bodmann, K.F., Complicated intra-abdominal infections: pathogens, resistance. Recommendations of the Infectliga on antbiotic therapy. Chirurg 2010, 81(1):38-49.
27. Bodmann, K.F., Grabein, B., und PEG-Expertengruppe, Empfehlungen zur kalkulierten parenteralen Initialtherapie bakterieller Erkrankungen bei Erwachsenen; Update 2010. Chemotherapie Journal 2010, 19:179-255.
28. Brook, I., Anaerobic pulmonary infections in children. Pediatr Emerg Care 2004, 20(9):636-40.
29. Brook, I., Enhancement of growth of aerobic and facultative bacteria in mixed infections with Bacteroides species. Infect Immun 1985, 50(3):929-31.
30. Brook, I., Microbiology and management of abdominal infections. Dig Dis Sci 2008, 53(10):2585-91.
31. Bruchhausen, F.v., und Hager, H., Hagers Handbuch der pharmazeutischen Praxis. 5., vollständig neubearbeitete Aufl. ed. 1990, Berlin ; New York: Springer-Verlag.
32. Budha, N.R., Lee, R.B., Hurdle, J.G., Lee, R.E., und Meibohm, B., A simple in vitro PK/PD model system to determine time-kill curves of drugs against Mycobacteria. Tuberculosis (Edinb) 2009, 89(5):378-85.
33. Caspary, W.F., Kist, M., und Stein, J., Infektiologie des Gastrointestinaltraktes. 2006, Berlin, Heidelberg: Springer Medizin Verlag Heidelberg.
34. Cheng, C.W., Lin, H.S., Ye, J.J., Yang, C.C., Chiang, P.C., Wu, T.S., und Lee, M.H., Clinical significance of and outcomes for Bacteroides fragilis bacteremia. J Microbiol Immunol Infect 2009, 42(3):243-50.
35. CLSI. www.clsi.org. 36. Points to consider on pharmacokinetics and pharmacodynamics in the development of
antibacterial medicinal products. http://www.emea.europa.eu/. 37. European Center for Disease Prevention and Control. www.ecdc.europa.eu/. 38. Craig, W.A., Pharmacokinetic/pharmacodynamic parameters: rationale for antibacterial
dosing of mice and men. Clin Infect Dis 1998, 26(1):1-10; quiz 1-2. 39. Dalhoff, A., Immunomodulatory activities of fluoroquinolones. Infection 2005, 33 Suppl
2:55-70. 40. Dalhoff, A., Pharmacokinetics of selected antibacterial agents. Antibiot Chemother 1998,
49:1-148. 41. Dalhoff, A., Ambrose, P.G., und Mouton, J.W., A long journey from minimum inhibitory
concentration testing to clinically predictive breakpoints: deterministic and probabilistic approaches in deriving breakpoints. Infection 2009, 37(4):296-305.
42. Dalhoff, A., Schubert, S., und Ullmann, U., Effect of pH on the in vitro activity of and propensity for emergence of resistance to fluoroquinolones, macrolides, and a ketolide. Infection 2005, 33 Suppl 2:36-43.
43. Deppermann, K.M., und Lode, H., Fluoroquinolones: interaction profile during enteral absorption. Drugs 1993, 45 Suppl 3:65-72.

Literaturverzeichnis
67
44. Domagala, J.M., Review: Structure-activity and structure-side-effect relationship for the quinolone antibacterials. Journal of Antimicrobial Chemotherapy 1994, 33:685-706.
45. Dost, F.H., Der Blutspiegel Kinetik der Konzentrationsabläufe in der Kreislaufflüssigkeit. 1953, Leipzig: Thieme.
46. Drlica, K., und Zhao, X., DNA gyrase, topoisomerase IV, and the 4-quinolones. Microbiol Mol Biol Rev 1997, 61(3):377-92.
47. Drusano, G.L., Johnson, D.E., Rosen, M., und Standiford, H.C., Pharmacodynamics of a fluoroquinolone antimicrobial agent in a neutropenic rat model of Pseudomonas sepsis. Antimicrob Agents Chemother 1993, 37(3):483-90.
48. Drusano, G.L., Preston, S.L., Fowler, C., Corrado, M., Weisinger, B., und Kahn, J., Relationship between fluoroquinolone area under the curve: minimum inhibitory concentration ratio and the probability of eradication of the infecting pathogen, in patients with nosocomial pneumonia. J Infect Dis 2004, 189(9):1590-7.
49. Dunn, D.L., Barke, R.A., Ewald, D.C., und Simmons, R.L., Effects of Escherichia coli and Bacteroides fragilis on peritoneal host defenses. Infect Immun 1985, 48(2):287-91.
50. Eagye, K.J., Kuti, J.L., Dowzicky, M., und Nicolau, D.P., Empiric therapy for secondary peritonitis: a pharmacodynamic analysis of cefepime, ceftazidime, ceftriaxone, imipenem, levofloxacin, piperacillin/tazobactam, and tigecycline using Monte Carlo simulation. Clin Ther 2007, 29(5):889-99.
51. Eckhardt, C., Fickweiler, K., Schaumann, R., Ackermann, G., und A, C.R., Therapeutic efficacy of moxifloxacin in a murine model of severe systemic mixed infection with Escherichia coli and Bacteroides fragilis. Anaerobe 2003, 9(4):157-60.
52. Edmiston, C.E., Krepel, C.J., Seabrook, G.R., Somberg, L.R., Nakeeb, A., Cambria, R.A., und Towne, J.B., In vitro activities of moxifloxacin against 900 aerobic and anaerobic surgical isolates from patients with intra-abdominal and diabetic foot infections. Antimicrob Agents Chemother 2004, 48(3):1012-6.
53. Emrich, N.C., Heisig, A., Stubbings, W., Labischinski, H., und Heisig, P., Antibacterial activity of finafloxacin under different pH conditions against isogenic strains of Escherichia coli expressing combinations of defined mechanisms of fluoroquinolone resistance. J Antimicrob Chemother 2010, 65(12):2530-3.
54. Falagas, M.E., McDermott, L., und Snydman, D.R., Effect of pH on in vitro antimicrobial susceptibility of the Bacteroides fragilis group. Antimicrob Agents Chemother 1997, 41(9):2047-9.
55. FDA: Avelox® www.fda.gov/. 56. Firsov, A.A., Lubenko, I.Y., Vostrov, S.N., Kononenko, O.V., Zinner, S.H., und Portnoy,
Y.A., Comparative pharmacodynamics of moxifloxacin and levofloxacin in an in vitro dynamic model: prediction of the equivalent AUC/MIC breakpoints and equiefficient doses. J Antimicrob Chemother 2000, 46(5):725-32.
57. Firsov, A.A., Zinner, S.H., Lubenko, I., und Vostrov, S.N., Gemifloxacin and ciprofloxacin pharmacodynamics in an in-vitro dynamic model: prediction of the equivalent AUC/MIC breakpoints and doses. Int J Antimicrob Agents 2000, 16(4):407-14.
58. Forrest, A., Nix, D.E., Ballow, C.H., Goss, T.F., Birmingham, M.C., und Schentag, J.J., Pharmacodynamics of intravenous ciprofloxacin in seriously ill patients. Antimicrob Agents Chemother 1993, 37(5):1073-81.
59. Frimodt-Moller, N., How predictive is PK/PD for antibacterial agents? Int J Antimicrob Agents 2002, 19(4):333-9.
60. Fuhst, C., Die Bedeutung der Plasmaproteinbindung für die Pharmakodynamik von Antibiotika. Mathematisch-Naturwissenschaftlichen Fakultät, 2004.

Literaturverzeichnis
68
61. Golan, Y., McDermott, L.A., Jacobus, N.V., Goldstein, E.J., Finegold, S., Harrell, L.J., Hecht, D.W., Jenkins, S.G., Pierson, C., Venezia, R., Rihs, J., Iannini, P., Gorbach, S.L., und Snydman, D.R., Emergence of fluoroquinolone resistance among Bacteroides species. J Antimicrob Chemother 2003, 52(2):208-13.
62. Goldstein, E.J., Intra-abdominal anaerobic infections: bacteriology and therapeutic potential of newer antimicrobial carbapenem, fluoroquinolone, and desfluoroquinolone therapeutic agents. Clin Infect Dis 2002, 35(Suppl 1):S106-11.
63. Goldstein, E.J., Citron, D.M., Warren, Y.A., Tyrrell, K.L., Merriam, C.V., und Fernandez, H., In vitro activity of moxifloxacin against 923 anaerobes isolated from human intra-abdominal infections. Antimicrob Agents Chemother 2006, 50(1):148-55.
64. Gregg, C.R., Enteric bacterial flora and bacterial overgrowth syndrome. Semin Gastrointest Dis 2002, 13(4):200-9.
65. Guarner, F., und Malagelada, J.R., Gut flora in health and disease. Lancet 2003, 361(9356):512-9.
66. Gunderson, B.W., Ross, G.H., Ibrahim, K.H., und Rotschafer, J.C., What do we really know about antibiotic pharmacodynamics? Pharmacotherapy 2001, 21(11 Pt 2):302S-18S.
67. Hahn, H., Kaufmann, S.H.E., und Suerbaum, S., Medizinische Mikrobiologie und Infektiologie. 6., komplett überarbeitete Auflage ed. 2009, Berlin, Heidelberg: Springer Berlin Heidelberg.
68. Hanberger, H., Pharmacodynamic effects of antibiotics. Studies on bacterial morphology, initial killing, postantibiotic effect and effective regrowth time. Scand J Infect Dis Suppl 1992, 81:1-52.
69. Hao, W.L., und Lee, Y.K., Microflora of the gastrointestinal tract: a review. Methods Mol Biol 2004, 268:491-502.
70. Hartung, J., und Elpelt, B., Multivariate Statistik Lehr- und Handbuch der angewandten Statistik. 6., unwesentl. veränd. Aufl. ed. 1999, München: Oldenbourg.
71. Haverkamp, W., Herth, F., und Messmann, H., Internistische Intensivmedizin, Methoden-Diagnostik-Therapie. Vol. 1. 2009, Stuttgart, New York: Georg Thieme Verlag.
72. Hedberg, M., und Nord, C.E., Antimicrobial susceptibility of Bacteroides fragilis group isolates in Europe. Clin Microbiol Infect 2003, 9(6):475-88.
73. Hermsen, E.D., Hovde, L.B., Sprandel, K.A., Rodvold, K.A., und Rotschafer, J.C., Levofloxacin plus metronidazole administered once daily versus moxifloxacin monotherapy against a mixed infection of Escherichia coli and Bacteroides fragilis in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 2005, 49(2):685-9.
74. Hof, H., Dörries, R., und Geginat, G., Medizinische Mikrobiologie [Immunologie, Virologie, Bakteriologie, Mykologie, Parasitologie, klinische Infektiologie, Hygiene]. 4., vollst. überarb. und erw. Aufl ed. 2009, Stuttgart: Thieme.
75. Hollander, M., und Wolfe, D.A., Nonparametric statistical methods. 2nd ed. 1999, New York: J. Wiley.
76. Holzheimer, R.G., und Dralle, H., Antibiotic therapy in intra-abdominal infections--a review on randomised clinical trials. Eur J Med Res 2001, 6(7):277-91.
77. Hooper, D.C., Mechanisms of action and resistance of older and newer fluoroquinolones. Clin Infect Dis 2000, 31 Suppl 2:S24-8.
78. Hoshino, K., Kitamura, A., Morrissey, I., Sato, K., Kato, J., und Ikeda, H., Comparison of inhibition of Escherichia coli topoisomerase IV by quinolones with DNA gyrase inhibition. Antimicrob Agents Chemother 1994, 38(11):2623-7.
79. Hurst, M., Lamb, H.M., Scott, L.J., und Figgitt, D.P., Levofloxacin: an updated review of its use in the treatment of bacterial infections. Drugs 2002, 62(14):2127-67.

Literaturverzeichnis
69
80. Hyatt, J.M., McKinnon, P.S., Zimmer, G.S., und Schentag, J.J., The importance of pharmacokinetic/pharmacodynamic surrogate markers to outcome. Focus on antibacterial agents. Clin Pharmacokinet 1995, 28(2):143-60.
81. Iakovlev, S.V., Ramishvili, V., Nazarov, V.V., und Eremina, L.V., Efficacy of moxifloxacin in the treatment of secondary peritonitis. Antibiot Khimioter 2006, 51(5):3-10.
82. Ingham, H.R., Sisson, P.R., Tharagonnet, D., Selkon, J.B., und Codd, A.A., Inhibition of phagocytosis in vitro by obligate anaerobes. Lancet 1977, 2(8051):1252-4.
83. Jousimies-Somer, H., und Summanen, P., Recent taxonomic changes and terminology update of clinically significant anaerobic gram-negative bacteria (excluding spirochetes). Clin Infect Dis 2002, 35(Suppl 1):S17-21.
84. Kahlmeter, G., und Brown, D.E., Harmonization of Antimicrobial Breakpoints in Europe- Can It Be Achieved? Clinical Microbiology Newsletter 2004, 26(24).
85. Kahlmeter, G., Brown, D.F., Goldstein, F.W., MacGowan, A.P., Mouton, J.W., Odenholt, I., Rodloff, A., Soussy, C.J., Steinbakk, M., Soriano, F., und Stetsiouk, O., European Committee on Antimicrobial Susceptibility Testing (EUCAST) Technical Notes on antimicrobial susceptibility testing. Clin Microbiol Infect 2006, 12(6):501-3.
86. Kahlmeter, G., Brown, D.F., Goldstein, F.W., MacGowan, A.P., Mouton, J.W., Osterlund, A., Rodloff, A., Steinbakk, M., Urbaskova, P., und Vatopoulos, A., European harmonization of MIC breakpoints for antimicrobial susceptibility testing of bacteria. J Antimicrob Chemother 2003, 52(2):145-8.
87. Karow, T., Allgemeine und spezielle Pharmakologie und Toxikologie : vorlesungsorientierte Darstellung. 2003, Puhlheim Thomas Karow.
88. Kasper, D.L., und Onderdonk, A.B., Infection with Bacteroides fragilis: pathogenesis and immunoprophylaxis in an animal model. Scand J Infect Dis Suppl 1982, 31:28-33.
89. Kayser, F.H., Medizinische Mikrobiologie verstehen, lernen, nachschlagen. 10., komplett überarb. Aufl. ed. 2001, Stuttgart: G. Thieme.
90. Kelly, M.J., The quantitative and histological demonstration of pathogenic synergy between Escherichia coli and Bacteroides fragilis in guinea-pig wounds. J Med Microbiol 1978, 11(4):513-23.
91. Khan, N.A., und Goldsworthy, G.J., Novel model to study virulence determinants of Escherichia coli K1. Infect Immun 2007, 75(12):5735-9.
92. Kim, K.S., Itabashi, H., Gemski, P., Sadoff, J., Warren, R.L., und Cross, A.S., The K1 capsule is the critical determinant in the development of Escherichia coli meningitis in the rat. J Clin Invest 1992, 90(3):897-905.
93. Moxifloxacin, ein Gyrasehemmer der vierten Generation. http://www.pharmazeutische-zeitung.de/index.php?id=22222.
94. König, C., Simmen, H.P., und Blaser, J., Effect of pathological changes of pH, pO2 and pCO2 on the activity of antimicrobial agents in vitro. Eur J Clin Microbiol Infect Dis 1993, 12(7):519-26.
95. Krasemann, C., Meyer, J., und Tillotson, G., Evaluation of the clinical microbiology profile of moxifloxacin. Clin Infect Dis 2001, 32 Suppl 1:S51-63.
96. Kujath, P., Hoffmann, M., und Rodloff, A., Antimicrobial and antimycotic therapy of intra-abdominal infections. Chirurg 2008, 79(4):295-305.
97. Kujath, P., und Rodloff, A.C., Peritonitis. Vol. 2. 2005, Bremen: UNI-Med. 98. Lee, S.Y., Fan, H.W., Sutherland, C., DeRyke, A.C., und Nicolau, D.P., Antibacterial
effects of moxifloxacin and levofloxacin simulating epithelial lining fluid concentrations against community-acquired methicillin-resistant Staphylococcus aureus. Drugs R D 2007, 8(2):69-77.

Literaturverzeichnis
70
99. Legat, F.J., Krause, R., Zenahlik, P., Hoffmann, C., Scholz, S., Salmhofer, W., Tscherpel, J., Tscherpel, T., Kerl, H., und Dittrich, P., Penetration of piperacillin and tazobactam into inflamed soft tissue of patients with diabetic foot infection. Antimicrob Agents Chemother 2005, 49(10):4368-71.
100. Levison, M.E., Pharmacodynamics of antimicrobial drugs. Infect Dis Clin North Am 2004, 18(3):451-65, vii.
101. Lode, H., Borner, K., und Koeppe, P., Pharmacodynamics of fluoroquinolones. Clin Infect Dis 1998, 27(1):33-9.
102. Löffler, G., und Petrides, P.E., Biochemie und Pathobiochemie. 8., völlig neu bearb. Aufl. ed. 2007, Berlin: Springer.
103. Lomaestro, B.M., und Bailie, G.R., Absorption interactions with fluoroquinolones. 1995 update. Drug Saf 1995, 12(5):314-33.
104. Lounis, N., Gevers, T., Van Den Berg, J., Verhaeghe, T., van Heeswijk, R., und Andries, K., Prevention of drug carryover effects in studies assessing antimycobacterial efficacy of TMC207. J Clin Microbiol 2008, 46(7):2212-5.
105. Luber, P., Bartelt, E., Genschow, E., Wagner, J., und Hahn, H., Comparison of broth microdilution, E Test, and agar dilution methods for antibiotic susceptibility testing of Campylobacter jejuni and Campylobacter coli. J Clin Microbiol 2003, 41(3):1062-8.
106. MacGowan, A., Rogers, C., und Bowker, K., In vitro models, in vivo models, and pharmacokinetics: what can we learn from in vitro models? Clin Infect Dis 2001, 33 Suppl 3:S214-20.
107. Malangoni, M.A., und Inui, T., Peritonitis - the Western experience. World J Emerg Surg 2006, 1:25.
108. Malangoni, M.A., Song, J., Herrington, J., Choudhri, S., und Pertel, P., Randomized controlled trial of moxifloxacin compared with piperacillin-tazobactam and amoxicillin-clavulanate for the treatment of complicated intra-abdominal infections. Ann Surg 2006, 244(2):204-11.
109. Marina, M., Strong, C.A., Civen, R., Molitoris, E., und Finegold, S.M., Bacteriology of anaerobic pleuropulmonary infections: preliminary report. Clin Infect Dis 1993, 16 Suppl 4:S256-62.
110. Mastropaolo, M.D., Evans, N.P., Byrnes, M.K., Stevens, A.M., Robertson, J.L., und Melville, S.B., Synergy in polymicrobial infections in a mouse model of type 2 diabetes. Infect Immun 2005, 73(9):6055-63.
111. Mazuski, J.E., Antimicrobial treatment for intra-abdominal infections. Expert Opin Pharmacother 2007, 8(17):2933-45.
112. Mendoza, M.T., What’s New in Antimicrobial Susceptibility Testing? The Philippine Journal of Microbiology and Infectious Diseases 1998, Vol. 27(3):113-5.
113. Miyazaki, H., Horii, T., Nagura, O., Suda, T., Chida, K., und Nakamura, H., Effect of the inoculum size on carbapenem susceptibilities of beta-lactamase-negative, ampicillin-resistant Haemophilus influenzae. Curr Microbiol 2009, 58(1):18-24.
114. Moise, P.A., Birmingham, M.C., und Schentag, J.J., Pharmacokinetics and metabolism of moxifloxacin. Drugs Today (Barc) 2000, 36(4):229-44.
115. Mouton, J.W., Brown, D.F., Apfalter, P., Canton, R., Giske, C.G., Ivanova, M., MacGowan, A.P., Rodloff, A., Soussy, C.J., Steinbakk, M., und Kahlmeter, G., The role of pharmacokinetics/pharmacodynamics in setting clinical MIC breakpoints: the EUCAST approach. Clin Microbiol Infect 2012, 18(3):E37-45.
116. Mouton, J.W., Dudley, M.N., Cars, O., Derendorf, H., und Drusano, G.L., Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: an update. J Antimicrob Chemother 2005, 55(5):601-7.

Literaturverzeichnis
71
117. Muller, M., Stass, H., Brunner, M., Moller, J.G., Lackner, E., und Eichler, H.G., Penetration of moxifloxacin into peripheral compartments in humans. Antimicrob Agents Chemother 1999, 43(10):2345-9.
118. Mutschler, E., Arzneimittelwirkungen kompakt Basiswissen Pharmakologie/Toxikologie. 2005, Stuttgart: WVG.
119. Mutschler, E., Mutschler Arzneimittelwirkungen Lehrbuch der Pharmakologie und Toxikologie mit einführenden Kapiteln in die Anatomie, Physiologie und Pathophysiologie. 9., vollständig neu bearb. und erw. Aufl. ed. 2008, Stuttgart: Wiss. Verl.-Ges.
120. Naber, K.G., und Adam, D., Einteilung der Fluorchinolone. Chemotherapie Journal 1998(7):66-8.
121. Nguyen, M.H., Yu, V.L., Morris, A.J., McDermott, L., Wagener, M.W., Harrell, L., und Snydman, D.R., Antimicrobial resistance and clinical outcome of Bacteroides bacteremia: findings of a multicenter prospective observational trial. Clin Infect Dis 2000, 30(6):870-6.
122. Noel, A.R., Bowker, K.E., und Macgowan, A.P., Pharmacodynamics of moxifloxacin against anaerobes studied in an in vitro pharmacokinetic model. Antimicrob Agents Chemother 2005, 49(10):4234-9.
123. O'Hara, A.M., und Shanahan, F., The gut flora as a forgotten organ. EMBO Rep 2006, 7(7):688-93.
124. Odenholt, I., und Cars, O., Pharmacodynamics of moxifloxacin and levofloxacin against Streptococcus pneumoniae, Staphylococcus aureus, Klebsiella pneumoniae and Escherichia coli: simulation of human plasma concentrations after intravenous dosage in an in vitro kinetic model. J Antimicrob Chemother 2006, 58(5):960-5.
125. Onderdonk, A.B., Bartlett, J.G., Louie, T., Sullivan-Seigler, N., und Gorbach, S.L., Microbial synergy in experimental intra-abdominal abscess. Infect Immun 1976, 13(1):22-6.
126. Onderdonk, A.B., Kasper, D.L., Cisneros, R.L., und Bartlett, J.G., The capsular polysaccharide of Bacteroides fragilis as a virulence factor: comparison of the pathogenic potential of encapsulated and unencapsulated strains. J Infect Dis 1977, 136(1):82-9.
127. Owens, R.C., Jr., und Shorr, A.F., Rational dosing of antimicrobial agents: pharmacokinetic and pharmacodynamic strategies. Am J Health Syst Pharm 2009, 66(12 Suppl 4):S23-30.
128. Patterson, D.R., Quinolone toxicity: methods of assessment. Am J Med 1991, 91(6A):35S-7S.
129. Paul-Ehrlich-Gesellschaft für Chemotherapie e.V. Arbeitsgemeinschaft "Empfindlichkeitsprüfung und Resistenz". http://www.p-e-g.org/ag_resistenz/main.htm.
130. Pendland, S.L., Diaz-Linares, M., Garey, K.W., Woodward, J.G., Ryu, S., und Danziger, L.H., Bactericidal activity and postantibiotic effect of levofloxacin against anaerobes. Antimicrob Agents Chemother 1999, 43(10):2547-9.
131. Peterson, M.L., Hovde, L.B., Wright, D.H., Brown, G.H., Hoang, A.D., und Rotschafer, J.C., Pharmacodynamics of trovafloxacin and levofloxacin against Bacteroides fragilis in an in vitro pharmacodynamic model. Antimicrob Agents Chemother 2002, 46(1):203-10.
132. Pharmakovigilanz: Tygacil. http://www.bfarm.de/SharedDocs/1_Downloads/DE/Pharmakovigilanz/roteHandBriefe/2011/rhb_tygacil.pdf;jsessionid=088285D87668549706F2269201628408.1_cid094?__blob=publicationFile.

Literaturverzeichnis
72
133. Pickerill, K.E., Paladino, J.A., und Schentag, J.J., Comparison of the fluoroquinolones based on pharmacokinetic and pharmacodynamic parameters. Pharmacotherapy 2000, 20(4):417-28.
134. Rasmussen, B.A., Bush, K., und Tally, F.P., Antimicrobial resistance in anaerobes. Clin Infect Dis. 1994, 24(Suppl 1).
135. Reinhart, K., Brunkhorst, F.M., Bone, H.G., Bardutzky, J., Dempfle, C.E., Forst, H., Gastmeier, P., Gerlach, H., Grundling, M., John, S., Kern, W., Kreymann, G., Kruger, W., Kujath, P., Marggraf, G., Martin, J., Mayer, K., Meier-Hellmann, A., Oppert, M., Putensen, C., Quintel, M., Ragaller, M., Rossaint, R., Seifert, H., Spies, C., Stuber, F., Weiler, N., Weimann, A., Werdan, K., und Welte, T., Prevention, diagnosis, therapy and follow-up care of sepsis: 1st revision of S-2k guidelines of the German Sepsis Society (Deutsche Sepsis-Gesellschaft e.V. (DSG)) and the German Interdisciplinary Association of Intensive Care and Emergency Medicine (Deutsche Interdisziplinare Vereinigung fur Intensiv- und Notfallmedizin (DIVI)). Ger Med Sci 2010, 8:Doc14.
136. Rodloff, A., Bauer, T., Ewig, S., Kujath, P., und Muller, E., Susceptible, intermediate, and resistant - the intensity of antibiotic action. Dtsch Arztebl Int 2008, 105(39):657-62.
137. Rodloff, A.C., Becker, J., Blanchard, D.K., Klein, T.W., Hahn, H., und Friedman, H., Inhibition of macrophage phagocytosis by Bacteroides fragilis in vivo and in vitro. Infect Immun 1986, 52(2):488-92.
138. Rodloff, A.C., und Hahn, H., Synergistic lethality in experimental infections with Escherichia coli and Bacteroides fragilis. Zentralbl Bakteriol Mikrobiol Hyg A 1984, 258(1):112-9.
139. Rodriguez-Cerrato, V., McCoig, C.C., Michelow, I.C., Ghaffar, F., Jafri, H.S., Hardy, R.D., Patel, C., Olsen, K., und McCracken, G.H., Jr., Pharmacodynamics and bactericidal activity of moxifloxacin in experimental Escherichia coli meningitis. Antimicrob Agents Chemother 2001, 45(11):3092-7.
140. Rotstein, O.D., und Kao, J., The spectrum of Escherichia coli-Bacteroides fragilis pathogenic synergy in an intraabdominal infection model. Can J Microbiol 1988, 34(3):352-7.
141. Rotstein, O.D., Pruett, T.L., und Simmons, R.L., Mechanisms of microbial synergy in polymicrobial surgical infections. Rev Infect Dis 1985, 7(2):151-70.
142. Ruiz, L.A., Zalacain, R., Gómez, A., Camino, J., Jaca, C., und Núñez, J.M., Escherichia coli: an unknown and infrequent cause of community acquired pneumonia. Scand J Infect Dis Suppl 2008, 40(5):424-7.
143. Sato, T., Yokota, S., Uchida, I., Okubo, T., Ishihara, K., Fujii, N., und Tamura, Y., A fluoroquinolone-resistant Escherichia coli clinical isolate without quinolone resistance-determining region mutations found in Japan. Antimicrob Agents Chemother 2011, 55(8):3964-5.
144. Schaumann, R., In vitro und in vivo Untersuchungen zur Wirksamkeit von Antibiotika gegenüber obligaten Anaerobiern in Mono- und aerob/anaeroben Mischkulturen. Habilitation. 2009.
145. Schaumann, R., Blatz, R., Beer, J., Ackermann, G., und Rodloff, A.C., Effect of moxifloxacin versus imipenem/cilastatin treatment on the mortality of mice infected intravenously with different strains of Bacteroides fragilis and Escherichia coli. J Antimicrob Chemother 2004, 53(2):318-24.
146. Schaumann, R., Goldstein, E.J., Forberg, J., und Rodloff, A.C., Activity of moxifloxacin against Bacteroides fragilis and Escherichia coli in an in vitro pharmacokinetic/pharmacodynamic model employing pure and mixed cultures. J Med Microbiol 2005, 54(Pt 8):749-53.

Literaturverzeichnis
73
147. Schaumann, R., und Rodloff, A., Activities of quinolones against obligately anaerobic bacteria. Anti-Infective Agents in Medicinal Chemistry 2007, 6(1):49-56.
148. Schaumann, R., und Rodloff, A.C., Diagnostik, Identifizierung und Empfindlichkeitsprüfung von obligaten Anaerobiern. Chemotherapie Journal 2007, 16(3):75-87.
149. Schentag, J.J., Meagher, A.K., und Forrest, A., Fluoroquinolone AUIC break points and the link to bacterial killing rates. Part 1: In vitro and animal models. Ann Pharmacother 2003, 37(9):1287-98.
150. Schentag, J.J., Meagher, A.K., und Forrest, A., Fluoroquinolone AUIC break points and the link to bacterial killing rates. Part 2: human trials. Ann Pharmacother 2003, 37(10):1478-88.
151. Simmen, H.P., Battaglia, H., Giovanoli, P., und Blaser, J., Analysis of pH, pO2 and pCO2 in drainage fluid allows for rapid detection of infectious complications during the follow-up period after abdominal surgery. Infection 1994, 22(6):386-9.
152. Singh, R., Ledesma, K.R., Chang, K.T., Hou, J.G., Prince, R.A., und Tam, V.H., Pharmacodynamics of moxifloxacin against a high inoculum of Escherichia coli in an in vitro infection model. J Antimicrob Chemother 2009, 64(3):556-62.
153. Smaill, F., Antibiotic susceptibility and resistance testing: an overview. Can J Gastroenterol 2000, 14(10):871-5.
154. Snydman, D.R., Jacobus, N.V., McDermott, L.A., Ruthazer, R., Golan, Y., Goldstein, E.J., Finegold, S.M., Harrell, L.J., Hecht, D.W., Jenkins, S.G., Pierson, C., Venezia, R., Yu, V., Rihs, J., und Gorbach, S.L., National survey on the susceptibility of Bacteroides fragilis group: report and analysis of trends in the United States from 1997 to 2004. Antimicrob Agents Chemother 2007, 51(5):1649-55.
155. Snydman, D.R., Jacobus, N.V., McDermott, L.A., Ruthazer, R., Goldstein, E., Finegold, S., Harrell, L., Hecht, D.W., Jenkins, S., Pierson, C., Venezia, R., Rihs, J., und Gorbach, S.L., In vitro activities of newer quinolones against Bacteroides group organisms. Antimicrob Agents Chemother 2002, 46(10):3276-9.
156. Solomkin, J.S., Mazuski, J.E., Bradley, J.S., Rodvold, K.A., Goldstein, E.J., Baron, E.J., O'Neill, P.J., Chow, A.W., Dellinger, E.P., Eachempati, S.R., Gorbach, S., Hilfiker, M., May, A.K., Nathens, A.B., Sawyer, R.G., und Bartlett, J.G., Diagnosis and management of complicated intra-abdominal infection in adults and children: guidelines by the Surgical Infection Society and the Infectious Diseases Society of America. Surg Infect (Larchmt) 2010, 11(1):79-109.
157. Spangler, S., Jacobs, M., und Appelbaum, P., Bactericidal activity of DU-6859a compared to activities of three quinolones, three beta-lactams, clindamycin, and metronidazole against anaerobes as determined by time-kill methodology. Antimicrobial Agents Chemotherapy. 1997, 41(4).
158. Spangler, S.K., Jacobs, M.R., und Appelbaum, P.C., Time-kill study of the activity of trovafloxacin compared with ciprofloxacin, sparfloxacin, metronidazole, cefoxitin, piperacillin and piperacillin/tazobactam against six anaerobes. J Antimicrob Chemother 1997, 39 Suppl B:23-7.
159. Spivey, J.M., The postantibiotic effect. Clin Pharm 1992, 11(10):865-75. 160. Stahlmann, R., Knorpelschädigende Wirkung der Chinolone.; Cartilage-damaging effect.
Infection 1991, 19 Suppl 1:S38-46. 161. Stass, H., und Kubitza, D., Profile of moxifloxacin drug interactions. Clin Infect Dis
2001, 32 Suppl 1:S47-50. 162. Stass, H., Rink, A.D., Delesen, H., Kubitza, D., und Vestweber, K.H., Pharmacokinetics
and peritoneal penetration of moxifloxacin in peritonitis. J Antimicrob Chemother 2006, 58(3):693-6.

Literaturverzeichnis
74
163. Stearne, L.E., Kooi, C., Goessens, W.H., Bakker-Woudenberg, I.A., und Gyssens, I.C., In vitro activity of trovafloxacin against Bacteroides fragilis in mixed culture with either Escherichia coli or a vancomycin- resistant strain of Enterococcus faecium determined by an anaerobic time-kill technique. Antimicrob Agents Chemother. 2001, 45(1):9.
164. Stein, G.E., und Goldstein, E.J., Fluoroquinolones and anaerobes. Clin Infect Dis 2006, 42(11):1598-607.
165. Steinhilber, D., Schubert-Zsilavecz, M., und Roth, H.J., Medizinische Chemie Targets und Arzneistoffe. 2005, Stuttgart: Deutscher Apotheker Verlag.
166. Stille, W., Antibiotika-Therapie Klinik und Praxis der antiinfektiösen Behandlung. 11., komplett aktualisierte und erw. Aufl. ed. 2005, Stuttgart: F. Schattauer.
167. Stock, I., und Wiedemann, B., Die Bestimmung der natürlichen Antibiotika-Empfindlichkeit. Chemotherapie Journal 1998, 4:127-35.
168. Sullivan, J.T., Woodruff, M., Lettieri, J., Agarwal, V., Krol, G.J., Leese, P.T., Watson, S., und Heller, A.H., Pharmacokinetics of a once-daily oral dose of moxifloxacin (Bay 12-8039), a new enantiomerically pure 8-methoxy quinolone. Antimicrob Agents Chemother 1999, 43(11):2793-7.
169. Takahashi, H., Hayakawa, I., und Akimoto, T., The history of the development and changes of quinolone antibacterial agents. Yakushigaku Zasshi 2003, 38(2):161-79.
170. Talan, D.A., Clinical perspectives on new antimicrobials: focus on fluoroquinolones. Clin Infect Dis 2001, 32 Suppl 1:S64-71.
171. Talbot, G.H., Cassileth, P.A., Paradiso, L., Correa-Coronas, R., und Bond, L., Oral enoxacin for infection prevention in adults with acute nonlymphocytic leukemia. The Enoxacin Prophylaxis Study Group. Antimicrob Agents Chemother 1993, 37(3):474-82.
172. Talha, K.A., Hasan, Z., Selina, F., und Palash, M.I., Organisms associated with ventilator associated pneumonia in. Mymensingh medical journal : MMJ 2009, 18(1 Suppl):S93-7.
173. Tauber, S.C., und Nau, R., Immunomodulatory properties of antibiotics. Curr Mol Pharmacol 2008, 1(1):68-79.
174. Theuretzbacher, U., Seewald, M., und Doerr, H.W., Mikrobiologie im klinischen Alltag Erreger, Diagnostik, Therapie. 1999, Stuttgart: W. Kohlhammer.
175. Thomas, J.K., Forrest, A., Bhavnani, S.M., Hyatt, J.M., Cheng, A., Ballow, C.H., und Schentag, J.J., Pharmacodynamic evaluation of factors associated with the development of bacterial resistance in acutely ill patients during therapy. Antimicrob Agents Chemother 1998, 42(3):521-7.
176. Torres, A., Garau, J., Arvis, P., Carlet, J., Choudhri, S., Kureishi, A., Le Berre, M.A., Lode, H., Winter, J., und Read, R.C., Moxifloxacin monotherapy is effective in hospitalized patients with community-acquired pneumonia: the MOTIV study--a randomized clinical trial. Clin Infect Dis 2008, 46(10):1499-509.
177. Vedantam, G., und Hecht, D.W., Antibiotics and anaerobes of gut origin. Curr Opin Microbiol 2003, 6(5):457-61.
178. Vogel, F., Bodmann, K., und PEG-Expertengruppe, Empfehlungen zur kalkulierten parenteralen Initialtherapie bakterieller Erkrankungen bei Erwachsenen. Chemotherapie Journal 2004, 13(2):46-105
179. Vogel, F., Naber, K.G., Adam, D., Bodmann, K.-F., Lebert, C., Rodloff, A.C., Sörgel, F., Graninger, W., Lehn, N., Wacha, H., und Wiedemann, B., Aktuelle Bewertung der Fluorchinolone. Arzneimitteltherapie 2005, 23(4).
180. Walker, A.P., Krepel, C.J., Gohr, C.M., und Edmiston, C.E., Microflora of abdominal sepsis by locus of infection. J Clin Microbiol 1994, 32(2):557-8.
181. Westphal, J.F., und Brogard, J.M., Biliary tract infections: a guide to drug treatment.

Literaturverzeichnis
75
Drugs 1999, 57(1):81-91. 182. Wexler, H.M., Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev
2007, 20(4):593-621. 183. White, R.L., What in vitro models of infection can and cannot do. Pharmacotherapy
2001, 21(11 Pt 2):292S-301S. 184. Wiedemann, B., und Heisig, P., Bakterielle Resistenz gegenüber Chinolonen
(Ciprofloxacin). Chemotherapie Journal 1999, 8(3):99-108. 185. Wispelwey, B., Clinical implications of pharmacokinetics and pharmacodynamics of
fluoroquinolones. Clin Infect Dis 2005, 41 Suppl 2:S127-35. 186. Wolfson, J.S., und Hooper, D.C., Fluoroquinolone antimicrobial agents. Clin Microbiol
Rev 1989, 2(4):378-424. 187. Wright, D.H., Gunderson, B.W., Hovde, L.B., Ross, G.H., Ibrahim, K.H., und
Rotschafer, J.C., Comparative pharmacodynamics of three newer fluoroquinolones versus six strains of Staphylococci in an in vitro model under aerobic and anaerobic conditions. Antimicrob Agents Chemother 2002, 46(5):1561-3.
188. Yasufuku, T., Shigemura, K., Shirakawa, T., Nakano, Y., Tanaka, K., Arakawa, S., Kinoshita, S., Nishimura, K., Kawabata, M., und Fujisawa, M., Risk factors and mechanisms of fluoroquinolone resistance in 156 Escherichia coli strains clinically isolated from urinary tract infections. Scand J Infect Dis 2011, 43(2):83-8.
189. Zabinski, R.A., Walker, K.J., Larsson, A.J., Moody, J.A., Kaatz, G.W., und Rotschafer, J.C., Effect of aerobic and anaerobic environments on antistaphylococcal activities of five fluoroquinolones. Antimicrob Agents Chemother 1995, 39(2):507-12.
190. Zhanel, G.G., Hoban, D.J., und Harding, G.K., The postantibiotic effect: a review of in vitro and in vivo data. Dicp 1991, 25(2):153-63.

Erklärung
76
9 Erklärung über die eigenständige Abfassung der Arbeit
Hiermit erkläre ich, dass ich die vorliegende Arbeit selbständig und ohne unzulässige Hilfe
oder Benutzung anderer als der angegebenen Hilfsmittel angefertigt habe. Ich versichere, dass
Dritte von mir weder unmittelbar noch mittelbar geldwerte Leistungen für Arbeiten erhalten
haben, die im Zusammenhang mit dem Inhalt der vorgelegten Dissertation stehen, und dass
die vorgelegte Arbeit weder im Inland noch im Ausland in gleicher oder ähnlicher Form einer
anderen Prüfungsbehörde zum Zweck einer Promotion oder eines anderen
Prüfungsverfahrens vorgelegt wurde. Alles aus anderen Quellen und von anderen Personen
übernommene Material, das in der Arbeit verwendet wurde oder auf das direkt Bezug
genommen wird, wurde als solches kenntlich gemacht. Insbesondere wurden alle Personen
genannt, die direkt an der Entstehung der vorliegenden Arbeit beteiligt waren.
……………………………….. ………………………………………..
Datum Unterschrift

Danksagung
77
10 Danksagung Ich möchte mich bei allen Menschen bedanken, die es mir ermöglicht haben, eine
Doktorarbeit anzufertigen und vor allem fertigzustellen.
In erster Linie möchte ich mich bei meiner großen Familie bedanken.
Lieber David, Danke, dass Du während der gesamten, wenn auch nicht immer leichten Zeit,
für mich da warst.
Liebe Mama, lieber Papa, liebe Oma, lieber Opa († 02.06.2012), Danke für Wochen, die Ihr
Emma zu Euch genommen habt, damit ich meine Arbeit schreiben konnte.
Ganz besonderer Dank gilt meiner Schwester Dana, die mich immer wieder aufgebaut hat
und mir viele hilfreiche Ratschläge gegeben hat.
Insbesondere gilt mein Dank PD Dr. med. Reiner Schaumann für die Bereitstellung des
Themas, die Unterstützung, Motivation und kritische Auseinandersetzung mit meiner Arbeit.
Ich bedanke mich bei Prof. Dr. med. Arne C. Rodloff für die Möglichkeit der Durchführung
meiner Doktorarbeit an seinem Institut.
Des Weiteren möchte ich bei den Mitarbeitern des Instituts für Medizische Mikrobiologie und
Infektionsepidemiologie der Universität Leipzig für Ihre Unterstützung und Hilfe bedanken.

Lebenslauf
78
11 Lebenslauf Persönliche Daten Stefanie Rossi geb. Hoffmann
Geboren am 18.03.1980

Lebenslauf
79

Veröffentlichung
80
12 Veröffentlichungen der Arbeit 1. Schaumann R., Hoffmann S., Hartmann J., Ackermann G., Rodloff A.C. Activity of
levofloxacin, piperacillin/tazobactam and imipenem aganist Bacteroides fragilis and
Escherichia coli in an in vitro pharmacokinetic/pharmacodynamic (PK/PD) model
employing pure and mixed cultures. ICAAC, Washington DC, 2004; Abstract A1164
2. Schaumann R., Rossi S., Rodloff A.C. Activity of levofloxacin and moxifloxacin
against B. fragilis and E. coli in an in vitro pharmacokinetic/pharmacodynamic
(PK/PD) model employing pure and mixed cultures. In Arbeit.
Anhang 1
81
Tabelle 18: Konzentrationsabfall von Levofloxacin bei einer Versuchsdauer von 13 h
Zeit (h) Konzentration
(mg/l)
Volumen
vor Abzug
(ml)
Volumen
nach
Abzug
(ml)
zugegebenes
Volumen
(ml)
neues
Volumen
(ml)
0 6 10 9,98 0 9,98
0,5 5,71 9,98 9,96 0,51 10,47
1 5,43 10,47 10,45 0,53 10,98
1,5 5,17 10,98 10,96 0,56 11,51
2 4,92 11,51 11,49 0,58 12,08
2,5 4,68 12,08 12,06 0,61 12,67
3 4,46 12,67 12,65 0,64 13,29
3,5 4,24 13,29 13,27 0,67 13,94
4 4,04 13,94 13,92 0,71 14,63
4,5 3,84 14,63 14,61 0,74 15,35
5 3,66 15,35 15,33 0,78 16,11
5,5 3,48 16,11 16,09 0,82 16,91
6 3,31 16,91 16,89 0,86 17,74
6,5 3,15 17,74 17,72 0,90 18,62
7 3,00 18,62 18,60 0,94 19,55
7,5 2,86 19,55 19,53 0,99 20,52
8 2,72 20,52 20,50 1,04 21,54
8,5 2,59 21,54 21,52 1,09 22,61
9 2,46 22,61 22,59 1,15 23,74
9,5 2,34 23,74 23,72 1,20 24,92
10 2,23 24,92 24,90 1,26 26,16
10,5 2,12 26,16 26,14 1,33 27,47
11 2,02 27,47 27,45 1,39 28,84
11,5 1,92 28,84 28,82 1,46 30,29
12 1,83 30,29 30,27 1,54 31,80
12,5 1,74 31,80 31,78 1,61 33,40
13 1,66 33,40 33,38 1,69 35,07

Anhang 2
82
Tabelle 19: Konzentrationsabfall von Moxifloxacin bei einer Versuchsdauer von 12 h
Zeit (h) Konzentration
(mg/l)
Volumen
vor Abzug
(ml)
Volumen
nach
Abzug
(ml)
zugegebenes
Volumen
(ml)
neues
Volumen
(ml)
0 4 20 19,98 0 19,98
0,5 3,89 19,98 19,96 0,54 20,50
1 3,79 20,50 20,48 0,55 21,03
1,5 3,69 21,03 21,01 0,57 21,58
2 3,60 21,58 21,56 0,58 22,14
2,5 3,50 22,14 22,12 0,60 22,72
3 3,41 22,72 22,70 0,61 23,31
3,5 3,32 23,31 23,29 0,63 23,92
4 3,23 23,92 23,90 0,65 24,55
4,5 3,15 24,55 24,53 0,66 25,19
5 3,06 25,19 25,17 0,68 25,85
5,5 2,98 25,85 25,83 0,70 26,53
6 2,90 26,53 26,51 0,72 27,23
6,5 2,83 27,23 27,21 0,74 27,94
7 2,75 27,94 27,92 0,75 28,68
7,5 2,68 28,68 28,66 0,77 29,43
8 2,61 29,43 29,41 0,79 30,20
8,5 2,54 30,20 30,18 0,82 31,00
9 2,48 31,00 30,98 0,84 31,82
9,5 2,41 31,82 31,80 0,86 32,66
10 2,35 32,66 32,64 0,88 33,52
10,5 2,29 33,52 33,50 0,91 34,40
11 2,23 34,40 34,38 0,93 35,31
11,5 2,17 35,31 35,29 0,95 36,25
12 2,11 36,25 36,23 0,98 37,20
Anhang 3
83
Tabelle 20: Auswahl an PK/PD-Indizes, Art und Ergebnisse der Untersuchungen und jeweils verwendete Antibiotika bzw. Antibiotikaklassen
PK/PD-
Index
Art der Untersuchung,
Antibiotika bzw.
Antibiotikaklassen
Ergebnisse Autor und
Jahr
Cmax/MHK in vitro Modell mit
Enoxacin und Netilmicin
bakterielles Wiederwachstum nur bei
Cmax/MHK < 8:1
Blaser,
1987 [24]
AUC/MHK verschiedene Tiermodelle
mit Fluorchinolonen
Letalität bei Pneumonie, Sepsis oder
Peritonitis >50% bei AUC/MHK
<30, sehr niedrige Letalität bei
AUC/MHK >100
Craig, 1998
[38]
Cmax/MHK Tiermodell mit
Lomefloxacin
signifikant verbessertes klinisches
Outcome bei Sepsis durch P.
aeruginosa bei Cmax/MHK 20:1
Drusano,
1993 [47]
AUC/MHK kliniche Studie mit
Levofloxacin
signifikante Eradikation von P.
aeruginosa und MRSA bei
AUC/MHK >87 bei nosokomialen
Pneumonien
Drusano,
2004 [48]
AUC/MHK in vitro Modell mit
Moxifloxacin und
Levofloxacin
AUC/MHK ~ 80 für Moxifloxacin
führt zu vergleichbaren Intensität des
antibakteriellen Effekts wie
AUC/MHK ~ 130 für Levofloxacin
Firsov,
2000 [56]
AUC/MHK in vitro Modell mit
Gemifloxacin und
Ciprofloxacin
AUC/MHK ~ 110 für Gemifloxacin
führt zu vergleichbaren Intensität des
antibakteriellen Effekts wie
AUC/MHK ~ 125 für Ciprofloxacin
Firsov,
2000 [57]
AUC/MHK klinische Studie mit
Ciprofloxacin
AUC/MHK >100 korreliert gut mit
klinischem Outcome bei pulmonalen
Infektionen
Forrest,
1993 [58]
AUC/MHK in vitro Modell mit
Levofloxacin und
Moxifloxacin
beste Eradikation von MRSA bei
AUC/MHK ~ 30
Lee, Fan,
2007 [98]

Anhang 3
84
PK/PD-
Index
Art der Untersuchung,
Antibiotika bzw.
Antibiotikaklassen
Ergebnisse Autor und
Jahr
AUC/MHK in vitro Modell mit
Moxifloxacin
99% Eradikation von E. coli bei
AUC/MHK ~ 105-148
99% Eradikation von B. fragilis bei
AUC/MHK ~ 81
Noel,
2005 [122]
AUC/MHK
Cmax/MHK
in vitro Modell mit
Levofloxacin und
Moxifloxacin
AUC/MHK ~100 und Cmax/MHK ~
10:1 beider Substanzen führten zu
maximalen Eradikationen der
untersuchten S. pneumoniae, S.
aureus, K. pneumoniae und E. coli
Stämme
Oldenholt,
Cars, 2006
[124]
AUC/MHK in vitro Modell mit
Levofloxacin und
Trovafloxacin
AUC/MHK <44 bei Levofloxacin
und Trovafloxacin fördern
Resistenzentstehung bei B. fragilis;
AUC/MHK >122 bei beiden
Substanzen führte zu vollstädiger
Eradikation und Verhinderung
Wiederwachstum von B. fragilis
Peterson,
2002 [131]
AUC/MHK in vitro Modell mit
Moxifloxacin
AUC/MHK >180 für Moxifloxacin
führte zu vollständiger Eradikation
und dauerhafter
Wachstumsunterdrückung unter
Verwendung eines hohen E. coli
Inokulums (2 x 108 KBE/ml)
Singh,
2009 [152]
AUC/MHK retrospektiv klinische
Studien mit
Cephalosporinen,
Fluorchinolonen,
Carbapenemen
AUC/MHK <100 bei den genannten
Antibiotikaklassen fördern
Resistenzentwicklung bei
nosokomialen Pneumonien mit
gramnegativen Erregern
Thomas,
1998 [175]



![Adjuvante Therapiestrategien bei der Behandlung ... · Wie von Linder et al beschrieben [28] hat sich der Mannheimer Peritonitis Index als ein zuverlässiges Messinstrument bei intraabdominellen](https://static.fdokument.com/doc/165x107/5e0cdc89d6c27a0f9030bcf8/adjuvante-therapiestrategien-bei-der-behandlung-wie-von-linder-et-al-beschrieben.jpg)














